Attached files
| file | filename |
|---|---|
| EX-1.1 - EX-1.1 - GENOCEA BIOSCIENCES, INC. | a2220737zex-1_1.htm |
| EX-5.1 - EX-5.1 - GENOCEA BIOSCIENCES, INC. | a2220737zex-5_1.htm |
| EX-23.1 - EX-23.1 - GENOCEA BIOSCIENCES, INC. | a2220652zex-23_1.htm |
Use these links to rapidly review the document
TABLE OF CONTENTS
As filed with the Securities and Exchange Commission on July 14, 2014
Registration No. 333-197247
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 1
TO
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
GENOCEA BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
2836 (Primary Standard Industrial Classification Code Number) |
51-0596811 (I.R.S. Employer Identification Number) |
100 Acorn Park Drive 5th Floor
Cambridge, MA 02140
(617) 876-8191
(Address, including zip code, and telephone number, including area code, of registrant's principal executive offices)
William D. Clark
President & Chief Executive Officer
100 Acorn Park Drive 5th Floor
Cambridge, MA 02140
(617) 876-8191
(Name, address, including zip code, and telephone number, including area code, of agent for service)
| Copies to: | ||
Marc Rubenstein, Esq. Ropes & Gray LLP Prudential Tower 800 Boylston Street Boston, MA 02199 (617) 951-7000 |
Barbara Kosacz, Esq. Marc Recht, Esq. Nicole Brookshire, Esq. Cooley LLP 500 Boylston Street, 14th Floor Boston, MA 02116-3736 (617) 937-2357 |
|
Approximate date of commencement of proposed sale to public:
As soon as practicable after this Registration Statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer o | Non-accelerated filer ý (Do not check if a smaller reporting company) |
Smaller reporting company o |
CALCULATION OF REGISTRATION FEE
|
||||||||
Class of Securities to be Registered
|
Number of Shares to be Registered(1) |
Proposed Maximum Offering Price Per Share(2) |
Proposed Maximum Aggregate Offering Price |
Amount of Registration Fee(3) |
||||
|---|---|---|---|---|---|---|---|---|
Common Stock, $0.001 par value per share |
3,910,000 | $15.25 | $59,627,500 | $7,681 | ||||
|
||||||||
- (1)
- Estimated
solely for the purpose of calculating the registration fee in accordance with Rule 457(a) under the Securities Act of 1933, as amended.
Includes shares that the underwriters have the option to purchase.
- (2)
- Estimated
solely for the purpose of calculating the registration fee pursuant to Rule 457(c) under the Securities Act of 1933, as amended and based
on the average of the high and low sales price of the registrant's common stock as reported on The NASDAQ Global Market on July 11, 2014.
- (3)
- Previously paid on July 3, 2014.
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. This preliminary prospectus is not an offer to sell these securities and we are not soliciting offers to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED JULY 14, 2014
PRELIMINARY PROSPECTUS

3,400,000 Shares
Common Stock
$ per share
We are selling 3,400,000 shares of our common stock.
We have granted the underwriters an option to purchase up to 510,000 additional shares of common stock.
Our common stock is listed on the NASDAQ Global Market under the symbol "GNCA". On July 10, 2014, the last sale price on our common stock was $15.31 per share.
We are an "emerging growth company" as that term is used in the Jumpstart Our Business Startups Act of 2012 and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
Investing in our common stock involves risk. See "Risk Factors" beginning on page 11.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed on the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
| |
Per share | Total | |||||
|---|---|---|---|---|---|---|---|
Public Offering Price |
$ | $ | |||||
Underwriting Discounts and Commissions(1) |
$ | $ | |||||
Proceeds to Genocea (before expenses) |
$ | $ | |||||
- (1)
- We refer you to "Underwriting" beginning on page 127 for additional information regarding underwriting compensation.
The underwriters expect to deliver the shares of common stock to investors on or about , 2014 through the book entry facilities of The Depositary Trust Company.
| Citigroup | Cowen and Company |
Stifel
Needham & Company
, 2014
We are responsible for the information contained in this prospectus and in any free-writing prospectus we prepare or authorize. We have not authorized anyone to provide you with different information, and we take no responsibility for any other information others may give you. We are not, and the underwriters are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should not assume that the information contained in this prospectus is accurate as of any date other than the date on the cover of this prospectus.
i
This summary highlights information contained in other parts of this prospectus or incorporated by reference into this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2013 and our other filings with the Securities and Exchange Commission (the "SEC") listed in the section of this prospectus entitled "Incorporation of Documents By Reference" and does not contain all of the information that you should consider in making your investment decision. Before investing in our common stock, you should also consider, among other things, the matters described under "Risk Factors" and "Management's Discussion and Analysis of Financial Condition and Results of Operations," in each case appearing elsewhere in this prospectus, in our Annual Report on Form 10-K for the year ended December 31, 2013 or in our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, incorporated by reference herein. Unless the context requires otherwise, references in this prospectus to "Genocea", "we", "us" and "our" refer to Genocea Biosciences, Inc.
We are a clinical stage biotechnology company that discovers and develops novel vaccines and immunotherapies to address diseases with significant unmet medical needs. We use our proprietary discovery platform, ATLAS, to rapidly design products that act through T cell (or cellular) immune responses, in contrast to approved products, which are designed to act primarily through B cell (or antibody) immune responses. We believe that by harnessing T cells we can develop first-in-class products to treat or prevent diseases where T cells are central to the control of the disease.
In September 2013, we announced human proof-of-concept data for GEN-003, a candidate therapeutic vaccine, or immunotherapy, that we are developing to treat herpes simplex virus-2, or HSV-2, infections. These data from our double-blind, placebo-controlled, dose-escalating Phase 1/2a trial represented the first reported instance of a therapeutic vaccine working against an infectious disease. We have now completed the follow-up review of patients for 12 months after their last dose of vaccine. Final analysis of the data showed that for the best performing 30µg dose group, there was a sustained reduction in the viral shedding rate. After completion of dosing for this dose group, the viral shedding rate fell by 52% versus baseline and, at six months after the final dose, the shedding rate remained at 40% below baseline. At 12 months, the viral shedding rate returned to baseline for this dose group. The reduction in the genital lesion rate after completion of the third dose was greatest for the 30µg dose group, at 48%. After six months, the reduction from baseline in genital lesion rate for this dose group was 65% and, after 12 months, the genital lesion rate was 42% lower than baseline. GEN-003 was safe and well tolerated over the 12 months of this trial. We believe the six-month duration of reduced viral shedding and genital lesion rates may be clinically meaningful. If GEN-003 successfully completes clinical development and is approved, we believe it would represent an important new treatment option for patients with HSV-2.
We are also developing a second T cell-stimulating vaccine candidate, GEN-004, a potential universal vaccine against pneumococcus, a leading cause of infectious disease mortality worldwide. In June 2014, we announced top line data from a Phase 1 clinical trial for GEN-004. This trial met its safety, tolerability and immunogenicity goals. We plan to advance GEN-004 into a Phase 2a trial in the third quarter of 2014.
Vaccine and Immunotherapy Overview
Vaccines represent a major healthcare success story, having eradicated or significantly reduced the global prevalence of many infectious diseases. Today, there are vaccines approved to protect against approximately 30 infectious diseases. Total global vaccine revenues in 2012 were $27 billion.
Vaccines work by training the immune system to respond to an infectious pathogen by exposing it to that pathogen, or a component of that pathogen, in a controlled way. Such components are often
1
immunogenic molecules of a pathogen, called antigens. Vaccines rely on an ability of the human immune system, called adaptive immunity, to "remember" an invading organism and develop an immune response to it. The adaptive immune system consists of two main components: the B cell arm and the T cell arm. To date, all approved vaccines have been developed primarily to elicit B cell responses. B cells produce antibodies, which identify and initiate processes to kill foreign organisms by binding to one or more structures, such as antigens, on the pathogen surface. B cell responses are effective against organisms in the bloodstream, but are generally ineffective against those that reside primarily in host cells or mucosal surfaces such as those of the genitalia, nose and throat. For these organisms, no vaccines or vaccines with limited effectiveness exist. To address these pathogens, vaccines that engage the T cell immune system may represent the optimal solution.
Immunotherapies are designed to augment or boost the immune system to allow it to better protect the body against disease. In the case of infectious disease, currently approved immunotherapies aim to treat an infection rather than prevent it. A well-known example of an early immunotherapy is the use of existing interferon-alfa 2a to treat infections caused by hepatitis C virus, or HCV. Immunotherapy approaches against cancer have also been developed, with limited success. As with vaccines, we believe immunotherapies that engage the T cell immune system may represent an optimal solution to treat, and potentially prevent, disease.
Limitations and Challenges of Current Vaccine and Immunotherapy Discovery
We believe T cell target discovery has been particularly challenging for two reasons. The first is the diversity of human T cell responses. B cell responses to a particular antigen are generally more uniform across all humans than T cell responses. The specific antigens that elicit T cell responses vary across humans—people from different genetic groups can have different T cell responses to the same invading organism. Traditional discovery involves testing antigens in animals which are typically bred from a single genetic lineage and cannot effectively account for the diversity of human T cell responses that is necessary to optimize vaccine design.
The second challenge to T cell target discovery relates to the number of target candidates. Antibodies typically target proteins on a pathogen's surface. For B cell vaccines eliciting an antibody response, the number of potential targets has typically been small, limiting the number and combination of targets that need to be tested to find a protective vaccine. By contrast, the potential targets for T cell responses include every protein in the pathogen, including proteins that are not just on the surface of the pathogen, which can number in the thousands. The number and combination of candidate T cell targets, therefore, increases exponentially with pathogen size. For many larger organisms, the complexities associated with the pathogen size have presented a fundamental barrier to the discovery of effective T cell vaccines using traditional vaccine discovery tools, which usually rely on empirically selecting the potential targets from the proteins of a pathogen and iteratively testing them in animal models. This process is slow and labor intensive and can take many years.
The ATLAS Discovery Platform: A Novel Approach to Vaccine and Immunotherapy Discovery
We have developed a proprietary technology platform that is designed to overcome the challenges associated with developing products that stimulate targeted T cell immunity. ATLAS, our AnTigen Lead Acquisition System, allows us to mimic ex vivo, or outside the body, the T cell responses of human populations exposed to an infectious pathogen by using human blood samples from those populations. We use ATLAS as a high throughput engine to rapidly screen T cells from hundreds of human subjects against every protein in a pathogen, and use the pattern of responses for each subject to determine which pathogen proteins are associated with protective responses. By comparing antigens identified in individuals who naturally control their infection with those who do not, we can select the antigens that may have the best likelihood of inducing protective T cell immune responses.
2
We believe that, by identifying T cell antigens in this way, ATLAS will help create vaccines and immunotherapies against pathogens that are generally inaccessible to antibodies and have therefore not been addressed successfully by B cell vaccines. By identifying the targets of human T cell responses ex vivo from human samples, rather than in animal models, we account for the diversity of human T cell responses. We also can screen every protein in a pathogen to choose from all possible antigens. We believe these factors will significantly increase our likelihood of success in efficiently discovering T cell-based vaccines against diseases associated with unmet needs.
GEN-003: A Therapeutic Vaccine Candidate for HSV-2
HSV-2 is a sexually transmitted disease affecting approximately 16% of the United States population between the ages of 14 to 49, and more than 500 million people worldwide, according to the Centers for Disease Control and Prevention and the World Health Organization. HSV-2 is a chronic, lifelong infection for which there is no cure. The virus persists in two states: inactive, or latent, and active. During latent states, patients have no symptoms or manifestations of disease. Intermittently, the virus activates, spreading to the skin and mucous membranes of the genital region. In active states, the virus can be detected by laboratory tests, and at these times the person is said to be shedding virus. Shedding lasts hours to several days or longer and is believed to be controlled eventually by the immune system. In general, when shedding is of short duration symptoms may not be present (so-called asymptomatic shedding). When shedding persists, ulcers may develop (clinical or symptomatic shedding). Sexual contact during either symptomatic or asymptomatic shedding events can lead to disease transmission. There is no known cure for HSV-2. For patients experiencing outbreaks, oral antiviral drugs are the only treatment option, but they are of limited effectiveness in reducing viral shedding, the risk of transmission from viral shedding and symptomatic outbreaks. We used ATLAS to design GEN-003 as a therapeutic vaccine, or immunotherapy, and are developing GEN-003 to treat people with HSV-2 infections.
In our Phase 1/2a trial, which followed patients for 12 months after their third vaccine dose, we have generated human proof-of-concept data in patients with moderate-to-severe infections. Final analysis of the data showed that for the best performing 30µg dose group, there was a sustained reduction in the viral shedding rate. After completion of dosing for this dose group, the viral shedding rate fell by 52% versus baseline and, at six months after the final dose, the shedding rate remained 40% below baseline. At 12 months, the viral shedding rate returned to baseline for this dose group. The reduction in the genital lesion rate after completion of the third dose was greatest for the 30µg dose group at 48%. After six months, the reduction from baseline in genital lesion rate for this dose group was 65% and after 12 months, the genital lesion rate was 42% lower than baseline. GEN-003 was safe and well tolerated over the 12 months of this trial.
We believe that these initial clinical trial results demonstrate that GEN-003 has the potential to be a first-in-class immunotherapy to treat HSV-2. These data also suggest that GEN-003 may become the first immunotherapy to effectively treat an infection that is controlled significantly by the T cell immune system. We expect to initiate a Phase 2 trial in mid-2014 to optimize the vaccine dose and potentially to improve upon either the magnitude and/or durability of the viral shedding and genital lesion rate reductions we have observed to date. This trial will study six combinations of protein and adjuvant doses and is designed around the 30µg per protein/50µg Matrix-M dose of GEN-003, which was the dose that drove the largest reductions in viral shedding and lesion rates in the Phase 1/2a trial.
Based on market research surveys conducted on our behalf with more than 400 patients with HSV-2 infections and more than 300 physicians who treat patients with HSV-2 infections, we believe that, if approved, GEN-003 could be used either as monotherapy or in combination with oral antiviral medication. We anticipate that, since the mechanisms of action for GEN-003 and oral antiviral medication may complement each other, the control against symptoms and disease transmission risk
3
offered by the combination could exceed that of either therapy alone. We therefore believe GEN-003 can be an important treatment option for people infected with HSV-2.
GEN-004: A Prophylactic Vaccine Candidate for Pneumococcus
Our second program derived from ATLAS is GEN-004, a T cell vaccine that we are developing to protect against all strains of the bacteria pneumococcus. Pneumococcus is the most common cause of bacterial pneumonia in the world, and kills more children under age five globally than any other organism. Pneumococcus often resides harmlessly in the nose and throat, but can also spread to other parts of the body and cause disease. There are safe and effective vaccines that induce antibodies against pneumococcus, including Prevnar from Pfizer, which achieved global revenue of $4.0 billion in 2013. However, Prevnar and other available vaccines protect against only a small number of the more than 90 pneumococcus serotypes known to cause disease, meaning that a universal pneumococcus vaccine would address a significant unmet need.
We have designed GEN-004 as a potential universal vaccine to fight all serotypes of pneumococcus, and to do so through a T cell-based mechanism of action that complements existing vaccines. Using ATLAS, we have identified three protein antigens that associate highly with a protective T cell response against pneumococcus in humans. Moreover, as these proteins are conserved in all sequenced strains of pneumococci, we believe GEN-004 may be able to help protect against invasive disease caused by any pneumococcal serotype. We have demonstrated in mice that GEN-004 clears pneumococcus from the nose and throat through a T cell-mediated mechanism of action. We announced top line data in June 2014 from a Phase 1 clinical trial of GEN-004. This trial met its safety, tolerability and immunogenicity goals, including measurable increases in the blood of T helper 17 (TH17) cells, a rare cell type that provides immunity at epithelial and mucosal surfaces. We plan to advance GEN-004 into a Phase 2a trial in the third quarter of 2014. If successful, we believe it could be the first clinical trial in which a vaccine induces a T cell response to reduce colonization by pneumococcus in the nasopharnyx, a necessary precursor to pneumococcal disease.
Other Opportunities
We have a number of other non-clinical stage research programs intended to address other areas of high unmet clinical need, all discovered using our ATLAS platform. Our chlamydia and HSV-2 prophylaxis programs have achieved promising non-clinical study results from candidates generated using ATLAS. We are collaborating with the Bill & Melinda Gates Foundation on malaria vaccine research. We also believe ATLAS may offer utility in the discovery of new treatments for cancer.
In March 2014, we announced a joint research collaboration with the Dana-Farber Cancer Institute and Harvard Medical School to characterize anti-tumor T cell responses in melanoma patients. This collaboration extends the use of our proprietary ATLAS platform for the potential rapid discovery of T cell antigens to cancer immunotherapy approaches.
Our Product Candidate Pipeline
The following table describes our current development programs:
Vaccine Candidate
|
Program | Stage of Development |
Next Milestone | Anticipated Timeline |
||||
|---|---|---|---|---|---|---|---|---|
GEN-003 |
HSV-2 Therapeutic | Phase 2 | Complete Phase 2 dose optimization | Mid-2015 | ||||
GEN-004 |
Pneumococcus Prophylaxis | Phase 2a | Complete Phase 2a | Mid-2015 | ||||
GEN-001 |
Chlamydia Prophylaxis | Pre-clinical | File IND | 2017 | ||||
GEN-002 |
HSV-2 Prophylaxis | Pre-clinical | File IND | 2017 | ||||
GEN-005 |
Malaria Prophylaxis | Research | Initiate pre-clinical studies | Second half of 2015 |
4
Our Team
Our management and scientific teams possess considerable experience in vaccine and anti-infective research, manufacturing, clinical development and regulatory matters. We have also assembled a team of leading advisors, led by George Siber, M.D., to guide the further development of our programs. Previously, Dr. Siber was the Chief Scientific Officer of Wyeth Vaccines, where he led the development of several first-in-class vaccines including Prevnar. He is also an inventor of Respigam and Cytogam, antibodies to treat and protect against respiratory syncytial virus and cytomegalovirus, respectively. Dr. Siber is one of our directors and chairs our Scientific Advisory Board.
Our Strategy
Our objective is to be the leading T cell vaccine company. Key components of our strategy are to:
- •
- Continue to rapidly advance our lead vaccine candidate,
GEN-003. GEN-003 is a potential first-in-class therapeutic vaccine candidate we are developing to treat HSV-2 infections, for which we
have successfully completed a Phase 1/2a trial and expect to commence a Phase 2 dose optimization trial in mid-2014. We intend to commence a further Phase 2 trial in mid-2015 to
optimize the dosing regimen. We retain all rights to GEN-003 and plan to advance this program through regulatory approval and commercialize this vaccine through a focused commercial effort in the
United States. Outside the United States, we intend to evaluate partnerships for GEN-003 opportunistically.
- •
- Advance GEN-004 into human proof-of-concept clinical
trials. Our second clinical-stage product candidate is GEN-004, a vaccine candidate designed to prevent infections caused by all strains
of pneumococcus. We have demonstrated proof-of-concept of GEN-004 in mice. We announced positive top line data for our Phase 1 clinical trial in June 2014 and plan to commence our
Phase 2a human proof-of-concept trial in the third quarter of 2014 with results from this trial expected to be available in mid-2015. We believe this trial could provide the first evidence in
humans that a T cell vaccine, with the potential to become a universal vaccine, can reduce colonization by pneumococcus. We retain all rights to this program, other than certain rights we have granted
in developing countries, and intend to opportunistically partner this program.
- •
- Advance our discovery stage and non-clinical novel vaccine
programs. We expect similarly to advance our novel non-clinical prophylactic vaccine programs against chlamydia, HSV-2 and malaria
through human proof-of-concept. We will seek partnerships opportunistically for late-stage development and commercialization of such programs. We will also continue to investigate, either alone, or
through partnerships, the applicability of ATLAS to the development of cancer immunotherapies.
- •
- Utilize ATLAS, our vaccine discovery platform, to develop additional T cell vaccine candidates. We intend to continue to use ATLAS to discover and advance novel T cell vaccines. Since we begin our vaccine candidate discovery process by profiling human populations exposed to a pathogen, and use these subjects' own cells to comprehensively screen the entire proteome of the pathogen, we believe we have a better chance of identifying vaccines likely to protect against pathogens of interest. We intend to opportunistically expand our pipeline using ATLAS to discover T cell vaccines against pathogens for which B cell vaccines are ineffective or non-existent.
Risk Factors
An investment in our common stock involves a high degree of risk. These risks are discussed more fully in the "Risk Factors" section of this prospectus immediately following this prospectus summary and in our Annual Report on Form 10-K for the year ended December 31, 2013 and in our Quarterly
5
Report on Form 10-Q for the quarter ended March 31, 2014, both of which are incorporated by reference herein. These risk factors include, among others:
- •
- We have incurred significant losses since our founding in 2006, which we anticipate will continue for the foreseeable
future. We have never generated revenue from product sales and may never generate revenue from product sales.
- •
- Failure to obtain additional funding when needed would force us to delay, limit, reduce or terminate our development or
commercialization efforts of our product candidates.
- •
- Our product candidates, including GEN-003 and GEN-004, are designed to work by eliciting T cell responses, which is
a novel approach for vaccines and medical treatments and therefore could produce unexpected adverse clinical outcomes or result in delays in our obtaining regulatory approval.
- •
- If our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities we may
incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
- •
- Our product candidates, including GEN-003, may include one or more novel vaccine adjuvants, which may make it difficult
for us to predict the requirements the United States Food and Drug Administration, or FDA, or other regulatory agencies may impose to demonstrate the safety of the product candidate.
- •
- We expect to rely on third parties to conduct the majority of our product manufacturing and clinical development of our
product candidates. If they fail to meet deadlines or perform in an unsatisfactory manner, our business could be harmed.
- •
- Our future commercial success depends upon attaining significant market acceptance of our product candidates, if approved,
among patients, physicians, third-party payors and others in the medical community.
- •
- If we are unable to obtain or protect intellectual property rights related to our product candidates, we may not be able to compete effectively in our markets.
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenue during our most recently completed fiscal year, we qualify as an "emerging growth company" as defined in Section 2(a) of the Securities Act of 1933, as amended, which we refer to as the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. As an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable, in general, to public companies that are not emerging growth companies. These provisions include:
- •
- reduced disclosure about our executive compensation arrangements;
- •
- no non-binding shareholder advisory votes on executive compensation or golden parachute arrangements;
- •
- exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting; and
- •
- reduced disclosure of financial information in this prospectus, including two years of audited financial information and two years of selected financial information.
We may take advantage of these exemptions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company if we have more than $1.0 billion in annual revenues as of the end of a fiscal year, have more than
6
$700.0 million in market value of our capital stock held by non-affiliates or if we issue more than $1.0 billion of non-convertible debt over a three year period. We may choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of some reduced reporting burdens in this prospectus. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock.
The JOBS Act permits an emerging growth company to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. We have irrevocably elected not to avail ourselves of this extended transition period and, as a result, we will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth public companies.
Corporate Information
We were incorporated in the state of Delaware in August 2006 as Genocea, Inc., and we subsequently changed our name to Genocea Biosciences, Inc. Our principal executive offices are located at Cambridge Discovery Park, 100 Acorn Park Drive, 5th Floor, Cambridge, Massachusetts 02140, and our telephone number is (617) 876-8191. Our Internet website is www.genocea.com. We have included our website address in this prospectus solely as an inactive textual reference. The information on, or that can be accessed through, our website is not part of this prospectus, and you should not rely on any such information in making the decision whether to purchase our common stock.
Genocea® and the Genocea logo are our registered trademarks. The other trademarks, trade names and service marks appearing in this prospectus are the property of their respective owners.
7
Common stock offered by us |
3,400,000 shares | |
Common stock to be outstanding immediately following this offering |
20,806,977 shares |
|
Option to purchase additional shares |
The underwriters have an option for a period of 30 days to purchase up to 510,000 additional shares of our common stock. |
|
Use of proceeds |
We currently estimate that we will use the net proceeds from this offering, together with our existing cash and cash equivalents and future available borrowings under our credit facility, (1) to further fund clinical development and manufacturing of GEN-003 into a Phase 3 clinical study, (2) to further fund clinical development and manufacturing of GEN-004, (3) to fund research and development and manufacturing of our prophylactic chlamydia, HSV-2 and malaria programs through filing of an investigational new drug, or IND, application and (4) for working capital and other general corporate purposes, including funding the costs of operating as a public company. See "Use of Proceeds". |
|
Risk factors |
You should read the "Risk Factors" section of this prospectus for a discussion of factors to consider carefully before deciding to invest in shares of our common stock. |
|
NASDAQ Global Market symbol |
GNCA |
The number of shares of common stock to be outstanding after this offering is based on 17,406,977 shares of common stock outstanding at June 30, 2014 and excludes the following:
- •
- 2,233,769 shares of common stock issuable upon exercise of stock options outstanding at June 30, 2014 at a
weighted-average exercise price of $6.68 per share;
- •
- 3,878 shares of common stock issuable upon the exercise of warrants outstanding at June 30, 2014 at a
weighted-average exercise price of $7.74 per share;
- •
- 2,596,921 shares of common stock reserved for future issuance under our 2014 Equity Incentive Plan, which was adopted in
January 2014; and
- •
- 200,776 shares of common stock reserved for future issuance under our 2014 Employee Stock Purchase Plan, which was adopted in January 2014.
Unless otherwise indicated, all information in this prospectus reflects or assumes no issuance or exercise of stock options or warrants on or after June 30, 2014 and no exercise by the underwriters of their option to purchase up to an additional 510,000 shares of common stock in this offering.
8
The following summary financial data for the years ended December 31, 2012 and 2013 are derived from our audited financial statements incorporated by reference in this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2013. The summary financial data as of March 31, 2014 and for the three months ended March 31, 2013 and 2014 have been derived from our unaudited financial statements incorporated by reference in this prospectus from our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014. These unaudited financial statements have been prepared on a basis consistent with our audited financial statements and, in our opinion, contain all adjustments, consisting only of normal and recurring adjustments, necessary for a fair presentation of such financial data. You should read this data together with our financial statements and related notes and the information under the captions "Selected Financial Data" and "Management's Discussion and Analysis of Financial Condition and Results of Operations" incorporated by reference in this prospectus. For more details on how you can obtain the documents incorporated by reference in this prospectus, see "Where You Can Find More Information" and "Incorporation of Documents By Reference" appearing elsewhere in this prospectus. Our historical results are not necessarily indicative of our future results, and our operating results for the three months ended March 31, 2014 are not necessarily indicative of the results that may be expected for the year ending December 31, 2014 or any other interim periods or any future year or period.
| |
Year Ended December 31, |
Three Months Ended March 31, |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands, except per share data)
|
2012 | 2013 | 2013 | 2014 | |||||||||
Statement of Operations Data: |
|||||||||||||
Grant revenue |
$ | 1,977 | $ | 731 | $ | 259 | $ | — | |||||
Operating expenses: |
|||||||||||||
Research and development |
11,240 | 15,695 | 3,980 | 4,407 | |||||||||
General and administrative |
3,690 | 4,961 | 810 | 1,966 | |||||||||
| | | | | | | | | | | | | | |
Total operating expenses |
14,930 | 20,656 | 4,790 | 6,373 | |||||||||
| | | | | | | | | | | | | | |
Loss from operations |
(12,953 | ) | (19,925 | ) | (4,531 | ) | (6,373 | ) | |||||
Other income (expense): |
|||||||||||||
Change in fair value of warrant |
93 | (222 | ) | (6 | ) | (725 | ) | ||||||
Loss on debt extinguishment |
— | (200 | ) | — | — | ||||||||
Interest expense, net |
(507 | ) | (459 | ) | (127 | ) | (231 | ) | |||||
| | | | | | | | | | | | | | |
Other expense |
(414 | ) | (881 | ) | (133 | ) | (956 | ) | |||||
| | | | | | | | | | | | | | |
Net loss |
$ | (13,367 | ) | $ | (20,806 | ) | $ | (4,664 | ) | $ | (7,329 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Reconciliation of net loss to net loss attributable to common stockholders: |
|||||||||||||
Net loss |
$ | (13,367 | ) | $ | (20,806 | ) | $ | (4,664 | ) | $ | (7,329 | ) | |
Accretion of redeemable convertible preferred stock to redemption value |
(1,781 | ) | (1,605 | ) | (395 | ) | (180 | ) | |||||
| | | | | | | | | | | | | | |
Net loss attributable to common stockholders |
$ | (15,148 | ) | $ | (22,411 | ) | $ | (5,059 | ) | $ | (7,509 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Net loss per share attributable to common stockholders—basic and diluted(1) |
$ | (51.35 | ) | $ | (75.46 | ) | $ | (17.09 | ) | $ | (0.76 | ) | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Weighted-average number of common shares used in net loss per share attributable to common stockholders—basic and diluted(1) |
295 | 297 | 296 | 9,859 | |||||||||
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
9
| |
As of March 31, 2014 | ||||||
|---|---|---|---|---|---|---|---|
(in thousands)
|
Actual | As Adjusted(2)(3) | |||||
Balance Sheet Data: |
|||||||
Cash and cash equivalents |
$ | 65,839 | $ | 114,220 | |||
Working capital |
62,395 | 110,776 | |||||
Total assets |
68,213 | 116,594 | |||||
Common stock and additional paid-in-capital |
142,833 |
191,214 |
|||||
Total stockholders' equity |
55,373 | 103,754 | |||||
- (1)
- See
Note 2 within the notes to our financial statements incorporated by reference in this prospectus for a description of the method used to
calculate basic and diluted net loss per common share.
- (2)
- As
adjusted to reflect the the sale of shares of our common stock in this offering at an assumed public offering price of $15.31 per share (the last
reported price of our common stock on the NASDAQ Global Market on July 10, 2014) after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
- (3)
- A $1.00 increase (decrease) in the assumed public offering price of $15.31 per share (the last reported price of our common stock on the NASDAQ Global Market on July 10, 2014) would increase (decrease) the as adjusted amount of each of cash and cash equivalents and total stockholders' equity by approximately $3.2 million, assuming that the number of shares offered by us, as set forth on the cover of this prospectus, remains the same and after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
10
Investing in our common stock involves a high degree of risk. You should carefully consider the risks and uncertainties described below together with all of the other information contained in this prospectus, including our financial statements and the related notes appearing at the end of this prospectus, before deciding to invest in our common stock. If any of the following risks actually occurs, our business, prospectus, operating results and financial condition could suffer materially, the trading price of our common stock could decline and you could lose all or part of your investment. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently believe to be immaterial may also adversely affect our business.
Risks Related to Our Financial Position and Need for Additional Capital
We have incurred significant losses since our founding in 2006 and anticipate that we will continue to incur significant losses for the foreseeable future and may never achieve or maintain profitability.
We are a clinical-stage biotechnology company, and we have not yet generated significant revenues. We have incurred net losses each year since our inception, including net losses of $13.4 million and $20.8 million for the years ended December 31, 2012 and 2013, respectively, and $4.7 million and $7.3 million for the three months ended March 31, 2013 and 2014, respectively. As of March 31, 2014, we had accumulated a deficit of $87.5 million. To date, we have not commercialized any products or generated any revenues from the sale of products and have financed our operations primarily through private placements of our preferred stock and our initial public offering completed in February 2014, and we do not expect to generate any product revenues in the foreseeable future. We do not know whether or when we will generate product revenues or become profitable.
We have devoted most of our financial resources to research and development, including our clinical and non-clinical technology development and development activities. To date, we have financed our operations primarily through the sale of equity securities and debt facilities and, to a lesser extent, through grants from governmental agencies and a private not-for-profit organization. The amount of our future net losses will depend, in part, on the rate of our future expenditures and our ability to obtain funding through equity or debt financings, strategic collaborations or additional grants. We have not completed pivotal clinical studies for any product candidate and it will be several years, if ever, before we have a product candidate ready for commercialization. Even if we obtain regulatory approval to market a product candidate, our future revenues will depend upon the size of any markets in which our product candidates have received approval, our ability to achieve sufficient market acceptance, reimbursement from third-party payors and other factors.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We anticipate that our expenses will increase significantly if and as we:
- •
- commence a planned Phase 2 clinical trial in mid-2014 to optimize the vaccine dose of GEN-003, our most advanced
product candidate that we are developing for the treatment of HSV-2 infections, and commence a planned Phase 2 clinical trial in mid-2015 to optimize the dosing regimen;
- •
- commence a planned Phase 2a clinical trial of GEN-004 in the third quarter of 2014, our second most advanced
product candidate that we are developing to prevent infections caused by all strains of pneumococcus;
- •
- initiate additional non-clinical, clinical or other studies for our other product candidates;
- •
- manufacture material for clinical trials and for commercial sale;
- •
- seek regulatory approvals for our product candidates that successfully complete clinical trials;
11
- •
- establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain
marketing approval;
- •
- seek to discover and develop additional product candidates;
- •
- acquire or in-license other product candidates and technologies;
- •
- make royalty milestone or other payments under any in-license agreements;
- •
- maintain, protect and expand our intellectual property portfolio;
- •
- attract and retain skilled personnel; and
- •
- create additional infrastructure to support our operations as a public company and our product development and planned future commercialization efforts.
The net losses we incur may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline.
To become and remain profitable, we must succeed in developing and eventually commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing non-clinical studies and clinical trials of our product candidates, discovering additional product candidates, obtaining regulatory approval for these product candidates and manufacturing, marketing and selling any products for which we may obtain regulatory approval. We are only in the preliminary stages of most of these activities. We may never succeed in these activities and, even if we do, may never generate revenues that are significant enough to achieve profitability.
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. If we are required by the FDA or the European Medicines Agency to perform studies in addition to those currently expected, or if there are any delays in completing our clinical trials or the development of any of our product candidates, our expenses could increase.
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our company and could impair our ability to raise capital, expand our business, maintain our research and development efforts, diversify our product offerings or even continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We will require substantial additional financing to achieve our goals, and a failure to obtain this necessary capital when needed would force us to delay, limit, reduce or terminate our product development or commercialization efforts.
As of March 31, 2014, our cash and cash equivalents were $65.8 million and the net proceeds from our initial public offering completed in February 2014 was $61.4 million, excluding offering expenses payable by us. We believe that we will continue to expend substantial resources for the foreseeable future developing GEN-003, GEN-004 and our pre-clinical product candidates. These expenditures will include costs associated with research and development, potentially acquiring new technologies, potentially obtaining regulatory approvals and manufacturing products, as well as marketing and selling products approved for sale, if any. In addition, other unanticipated costs may arise. Because the outcome of our planned and anticipated clinical trials is highly uncertain, we cannot reasonably
12
estimate the actual amounts necessary to successfully complete the development and commercialization of our product candidates.
Our future capital requirements depend on many factors, including:
- •
- the progress, results and costs of our planned Phase 2 dose optimization clinical trial and our planned
Phase 2 dose regimen clinical trial of GEN-003;
- •
- the scope, progress, results and costs of non-clinical development, laboratory testing and clinical trials for our other
product candidates, including our planned Phase 2a clinical trial of GEN-004;
- •
- the number and development requirements of other product candidates that we pursue;
- •
- the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates if clinical trials are
successful and the outcome of regulatory review of our product candidates;
- •
- the cost and timing of future commercialization activities for our products, if any of our product candidates are approved
for marketing, including product manufacturing, marketing, sales and distribution costs;
- •
- The cost of our general and administrative functions;
- •
- the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval;
- •
- the cost of manufacturing our product candidates for clinical trials in preparation for regulatory approval and in
preparation for commercialization;
- •
- our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of
such agreements;
- •
- the costs involved in preparing, filing, prosecuting patent applications, maintaining, defending and enforcing our
intellectual property rights, including litigation costs and the outcome of such litigation;
- •
- the timing, receipt, and amount of sales of, or royalties or milestone payments on, our future products, if any; and
- •
- the extent to which we acquire or in-license other products or technologies.
Based on our current operating plan, we believe that the net proceeds we received from our initial public offering completed in February 2014 together with this offering and our existing cash and cash equivalents will be sufficient to fund our projected operating expenses and capital expenditure requirements through at least the first quarter of 2017, by which time we anticipate that we will have completed our Phase 2 program for GEN-003 and have conducted our end of Phase 2 meeting with the FDA. However, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us when needed, we would be required to delay, limit, reduce or terminate non-clinical studies, clinical trials or other development activities for one or more of our product candidates or delay, limit, reduce or terminate our establishment of sales and marketing capabilities or other activities that may be necessary to commercialize our product candidates.
13
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates on unfavorable terms to us.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings and license and development agreements with strategic partnerships with third parties. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take certain actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through strategic partnerships with third parties, we may have to relinquish valuable rights to our technologies or product candidates, future revenue streams, research programs or product candidates or grant licenses on terms that are not favorable to us. If we are unable to raise additional when needed, we would be required to delay, limit, reduce or terminate our product development or commercialization efforts for GEN-003, GEN-004 or our non-clinical product candidates, or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Risks Related to Clinical Development, Regulatory Review and Approval of Our Product Candidates
Because our product candidates are in an early stage of development, there is a high risk of failure, and we may never succeed in developing marketable products or generating product revenue.
Our early encouraging non-clinical and clinical results for GEN-003 and for GEN-004 are not necessarily predictive of the final results of our ongoing or future clinical trials. Success in non-clinical studies may not be predictive of similar results in humans during clinical trials, and successful results from early or small clinical trials of a vaccine candidate may not be replicated in later and larger clinical trials. If the results of our ongoing or future clinical trials are inconclusive with respect to the efficacy of our product candidates or if we do not meet our clinical endpoints with statistical significance or if there are safety concerns or adverse events associated with our product candidates, we may be prevented or delayed in obtaining marketing approval for our product candidates. Alternatively, even if we obtain regulatory approval, that approval may be for indications or patient populations that are not as broad as intended or desired or may require labeling that includes significant use or distribution restrictions or safety warnings. We may also be required to perform additional or unanticipated clinical trials to obtain approval or be subject to additional post-marketing testing requirements to maintain regulatory approval. In addition, regulatory authorities may withdraw their approval of the product or impose restrictions on its distribution in the form of a modified risk evaluation and mitigation strategy.
If we do not obtain regulatory approval for our current and future product candidates, our business will be adversely affected.
Our product candidates are subject to extensive governmental regulations relating to, among other things, research, clinical trials, manufacturing, import, export and commercialization. In order to obtain regulatory approval for the commercial sale of any product candidate, we must demonstrate through extensive non-clinical studies and clinical trials that the product candidate is safe and effective for use in each target indication. Clinical trials are expensive, time-consuming and uncertain as to outcome. We may gain regulatory approval for GEN-003, GEN-004 or our other non-clinical product candidates in some but not all of the territories available or some but not all of the target indications, resulting in limited commercial opportunity for the approved vaccine, or we may never obtain regulatory approval for these product candidates for any indication in any jurisdiction.
14
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on the speed at which we can recruit patients to participate in testing our product candidates. If patients are unwilling to participate in our studies because of negative publicity from adverse events in the biotechnology industries or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting studies and obtaining regulatory approval of potential products may be delayed or prevented. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology or termination of the clinical trials altogether.
Additionally, in order to identify vaccine candidates using our ATLAS platform, we need to collect and process blood samples from human cohorts exposed to a pathogen. If we are unable to collect blood from a sufficient cohort for an indication we may be unable to identify additional product candidates.
We may not be able to identify, recruit and enroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a study, to complete our clinical trials in a timely manner. Patient enrollment is affected by factors including:
- •
- severity of the disease under investigation;
- •
- design of the study protocol;
- •
- size of the patient population;
- •
- eligibility criteria for the trial in question;
- •
- perceived risks and benefits of the product candidate under study;
- •
- proximity and availability of clinical trial sites for prospective patients;
- •
- availability of competing therapies and clinical trials;
- •
- efforts to facilitate timely enrollment in clinical trials;
- •
- patient referral practices of physicians; and
- •
- ability to monitor patients adequately during and after treatment.
We may not be able to initiate or continue clinical trials if we cannot enroll a sufficient number of eligible patients to participate in the clinical trials required by regulatory agencies. If we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate on-going or planned clinical trials, any of which would have an adverse effect on our business.
We may not be able to comply with requirements of foreign jurisdictions in conducting trials outside of the United States.
To date, we have not conducted any clinical trials outside of the United States. Our ability to successfully initiate, enroll and complete a clinical trial in any foreign country, should we attempt to do so, is subject to numerous risks unique to conducting business in foreign countries, including:
- •
- difficulty in establishing or managing relationships with contract research organizations, or CROs, and physicians;
- •
- different standards for the conduct of clinical trials;
- •
- our inability to locate qualified local consultants, physicians and partners;
15
- •
- the potential burden of complying with a variety of foreign laws, medical standards and regulatory requirements, including
the regulation of pharmaceutical and biotechnology products and treatment; and
- •
- the acceptability of data obtained from studies conducted outside the United States to the FDA in support of a Biologics License Application, or BLA.
If we fail to successfully meet requirements for the conduct of clinical trials outside of the United States, we may be delayed in obtaining, or be unable to obtain, regulatory approval for our product candidates in the United States or in countries outside of the United States.
We may encounter substantial delays in our clinical trials or we may fail to demonstrate safety and efficacy to the satisfaction of applicable regulatory authorities.
Before obtaining marketing approval from regulatory authorities for the sale of our product candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates for the intended indications. Clinical testing is expensive, time-consuming and uncertain as to outcome. We cannot guarantee that clinical trials will be conducted as planned or completed on schedule, if at all. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
- •
- delays by us in reaching a consensus with regulatory agencies on trial design;
- •
- delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites;
- •
- delays in obtaining required Institutional Review Board, or IRB, approval at each clinical trial site;
- •
- imposition of a clinical hold by regulatory agencies for any reason, including safety concerns raised by other clinical
trials of similar vaccines that may reflect an unacceptable risk with GEN-003 or after an inspection of clinical operations or trial sites;
- •
- failure to perform in accordance with the FDA's good clinical practices, or GCP, or applicable regulatory guidelines in
other countries;
- •
- delays in the testing, validation, manufacturing and delivery of the product candidates to the clinical sites;
- •
- delays caused by patients not completing participation in a trial or not returning for post-treatment follow-up;
- •
- clinical trial sites or patients dropping out of a trial or failing to complete dosing;
- •
- occurrence of serious adverse events in clinical trials that are associated with the product candidates that are viewed to
outweigh its potential benefits; or
- •
- changes in regulatory requirements and guidance that require amending or submitting new clinical protocols.
Delays, including delays caused by the above factors, can be costly and could negatively affect our ability to complete a clinical trial. Our IND for GEN-003 was subject to a clinical hold from January 2012 to July 2012. In our original IND submission, we described a finding of osteonecrosis (microscopic evidence of bone and bone marrow death) in a toxicity study of GEN-003 conducted in mice. Because this finding was not present in toxicity studies conducted in other species, we reasoned that this was a mouse-specific finding and did not indicate a risk to humans in clinical trials. However, the FDA instituted a clinical hold and provided us with several options that would resolve this issue to their satisfaction. We selected the option to conduct an additional toxicity study in a highly relevant species (non-human primate) that would be more representative of a risk to humans. The study was conducted,
16
no bone or bone marrow toxicity was observed, and the FDA subsequently lifted the clinical hold, allowing us to proceed with the first study in humans of GEN-003.
We cannot give any assurance that we will be able to resolve any future clinical holds imposed by the FDA or other regulatory authorities outside of the United States, or any delay caused by other factors described above or any other factors, on a timely basis or at all. If we are not able to successfully initiate and complete subsequent clinical trials, we will not be able to obtain regulatory approval and will not be able to commercialize our product candidates.
Our product candidates, including GEN-003 and GEN-004, are based on T cell activation, which is a novel approach for vaccines, immunotherapies and medical treatments. Consequently, it may be difficult for us to predict the time and cost of product development. Unforeseen problems with the T cell approach to vaccines may prevent further development or approval of our product candidates. Because of the novelty of this approach, there may be unknown safety risks associated with the vaccines and immunotherapies that we develop. Regulatory agencies such as the FDA may require us to conduct extensive safety testing prior to approval to demonstrate a low risk of rare and severe adverse events caused by the vaccines and immunotherapies. If approved, the novel mechanism of action of the vaccines and immunotherapies may adversely affect physician and patient perception and uptake of our products.
We have concentrated our research and development efforts on T cell vaccine and immunotherapy technology, and our future success is highly dependent on the successful development of T cell vaccines and immunotherapies in general, and our product candidates in particular. There can be no assurance that any development problems we or others researching T cell vaccines and immunotherapies may experience in the future will not cause significant delays or unanticipated costs, or that such development problems can be solved.
Public perception of vaccine safety issues, including adoption of novel vaccine mechanisms of action, may adversely influence willingness of subjects to participate in clinical trials, or if approved, of physicians to prescribe and of patients to receive novel vaccines. For example, GEN-004 is being developed for prevention of Pneumococcal infections, and parental aversion to new vaccines or vaccines in general may adversely influence later stage clinical trials of this product candidate or, if approved, its commercial success.
GEN-003 includes a novel vaccine adjuvant and our other product candidates may include one or more novel adjuvants, which may make it difficult for us to predict the time and cost of product development as well as the requirements the FDA or other regulatory agencies may impose to demonstrate the safety of the product candidate.
Novel vaccine adjuvants, included in some of our product candidates, may pose an increased safety risk to patients. Adjuvants are compounds that are added to vaccine antigens to enhance the activation and improve immune response and efficacy of vaccines. Development of vaccines with novel adjuvants requires evaluation in larger numbers of patients prior to approval than would be typical for therapeutic drugs. Guidelines for evaluation of vaccines with novel adjuvants have been established by the FDA and other regulatory bodies and expert committees. Our product candidates, including GEN-003, may include one or more novel vaccine adjuvants. Any vaccine, because of the presence of an adjuvant, may have side effects considered to pose too great a risk to patients to warrant approval of the vaccine. Traditionally, regulatory authorities have required extensive study of novel adjuvants because vaccines typically get administered to healthy populations, in particular infants, children and the elderly, rather than in people with disease. Such extensive study has often included long-term monitoring of safety in large general populations that has at times exceeded 10,000 subjects. This contrasts with the few thousand subjects typically necessary for approval of novel therapeutics. Although GEN-003 is being developed as a treatment, and therefore is not expected to be administered to uninfected subjects, regulators nonetheless may require us to amass a prophylactic vaccine-like safety
17
database. To date, the FDA and other major regulatory agencies have only approved vaccines containing five adjuvants, which makes it difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates in the United States or elsewhere.
If we fail to obtain regulatory approval in jurisdictions outside the United States, we will not be able to market our products in those jurisdictions.
We intend to market our product candidates, if approved, in international markets. Such marketing will require separate regulatory approvals in each market and compliance with numerous and varying regulatory requirements. The approval procedures vary among countries and may involve requirements for additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. In addition, in many countries outside the United States, a vaccine must be approved for reimbursement before it can be approved for sale in that country. In some cases, the price that we intend to charge for our vaccine is also subject to approval. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our vaccines in any market.
Even if we receive regulatory approval for our product candidates, such vaccines and immunotherapies will be subject to ongoing regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions, and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
Any regulatory approvals that we receive for our product candidates may also be subject to limitations on the approved indications for which the product may be marketed or to conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the vaccine or immunotherapy potentially over many years. In addition, if the FDA approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing practice, or cGMP, and GCP, for any clinical trials that we conduct post-approval.
Later discovery of previously unknown problems with an approved product, including adverse events of unanticipated severity or frequency, or with manufacturing operations or processes, or failure to comply with regulatory requirements, may result in, among other things:
- •
- restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or
mandatory product recalls;
- •
- fines, warning letters, or holds on clinical trials;
- •
- refusal by the FDA to approve pending applications or supplements to approved applications filed by us, or suspension or
revocation of product license approvals;
- •
- product seizure or detention, or refusal to permit the import or export of products; and
- •
- injunctions or the imposition of civil, criminal and/or administrative penalties, damages, monetary fines, disgorgement, exclusion from participation in Medicare, Medicaid and other federal health care programs, and curtailment or restructuring of our operations.
18
The FDA's policies may change and additional government regulations may be enacted that could affect regulatory approval that we have received for our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or not able to maintain regulatory compliance, we may lose any marketing approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
The statistical methods for the analysis of the data generated from our clinical trials may become subject to re-evaluation and modifications and may cause data that was previously thought to be statistically significant to be less so or to not be determined to be statistically significant.
We rely on statistical methods for the analysis of the data generated from our clinical trials that are generally accepted for the diseases for which we are developing product candidates. Despite their acceptance as standard models, these methods may become subject to re-evaluation and modification after we have received the results of these clinical trials and may cause data that was previously thought to be statistically significant to be less so or to not be determined to be statistically significant. Recently, the third party that had performed the analysis of the data from our Phase 1/2a GEN-003 trial, using a widely accepted and applied model for HSV-2 treatment-related data over the past several years by us and most others, informed us that it was re-evaluating the suitability of the model used to perform this analysis. This accepted model was previously used in determining that certain observed results after the third dose and after six months in our Phase 1/2a GEN-003 trial are statistically significant. As a result, we may determine that a modified or different model is more appropriate and that certain of these results, when analyzed using such a modified or different model, may be shown to be less statistically significant or not statistically significant. Although we do not believe that any change in the analysis of these results will affect our anticipated development activities with respect to GEN-003, it is possible that a change in the methodology used to analyze data from future trials, and in particular pivotal trials for which we expect to seek approval to market our product candidates, could affect the development of our product candidates or our ability to obtain approval for our product candidates. In addition, such a change in methodology may cause us to need to restate or correct information that we had previously disclosed regarding the statistical significance of our clinical trial results.
Risks Related to Our Reliance on Third Parties
We rely on third parties to conduct non-clinical studies and clinical trials for our product candidates, and if they do not properly and successfully perform their obligations to us, we may not be able to obtain regulatory approvals for our product candidates or may otherwise make incorrect public disclosures.
We rely on third party CROs and other third parties to assist in managing, monitoring and otherwise carrying out our GEN-003 and GEN-004 clinical trials. We further rely on these third parties to report on and provide statistical analysis of the GEN-003 and GEN-004 clinical trial data and results. We expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to conduct our clinical trials. We compete with many other companies for the resources of these third parties. The third parties on whom we rely generally may terminate their engagements at any time, and having to enter into alternative arrangements would delay development and commercialization of our product candidates.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, the FDA and foreign regulatory authorities require compliance with regulations and standards, including GCP, for designing, conducting, monitoring, recording, analyzing, and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the rights, integrity and confidentiality of trial
19
participants are protected. Although we rely on third parties to conduct our clinical trials, we are responsible for ensuring that each of these clinical trials is conducted in accordance with its general investigational plan and protocol. In addition, our reliance on the reports and analyses provided by third parties for research and development activities may cause us to make incorrect disclosures of information to the public.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their duties under their agreements, if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to clinical trial protocols or to regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, the clinical trials of our product candidates may not meet regulatory requirements. If clinical trials do not meet regulatory requirements or if these third parties need to be replaced, non-clinical development activities or clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates on a timely basis or at all.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We intend to rely on third parties to conduct some or all aspects of our product manufacturing, and these third parties may not perform satisfactorily.
We do not have any manufacturing facilities or personnel. We do not expect to independently conduct all aspects of our product manufacturing. We currently rely, and expect to rely, on third parties with respect to manufacturing. For example, we rely on third party suppliers and manufacturers to manufacture and supply vaccines for our initial GEN-003 and GEN-004 clinical trials. This reliance on third parties increases the risk that we will not have sufficient quantities of our product candidates or products or such quantities at an acceptable cost or quality, which could delay, prevent or impair our development or commercialization efforts.
Any of these third parties may terminate their engagement with us at any time. If we need to enter into alternative arrangements, it could delay our product development activities. Our reliance on these third parties for manufacturing activities will reduce our control over these activities but will not relieve us of our responsibility to ensure compliance with all required regulations regarding manufacturing.
Reliance on third party manufacturers entails risks to which we would not be subject if we manufactured the product candidates ourselves, including:
- •
- the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms;
- •
- reduced control as a result of using third party manufacturers for all aspects of manufacturing activities, including
regulatory compliance and quality assurance;
- •
- termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or
damaging to us;
- •
- the possible misappropriation of our proprietary information, including our trade secrets and know-how or infringement of
third party intellectual property rights by our contract manufacturers; and
- •
- disruptions to the operations of our third party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier.
20
Any of these events could lead to clinical trial delays or failure to obtain regulatory approval, or affect our ability to successfully commercialize future products. Some of these events could be the basis for FDA action, including injunction, recall, seizure or total or partial suspension of production.
Third party manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States. Our failure, or the failure of our third party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our products.
Our product candidates and any products that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Any performance failure on the part of our existing or future manufacturers could delay clinical development or marketing approval. We do not currently have arrangements in place for redundant supply or a second source for bulk drug substance. If our current contract manufacturers cannot perform as agreed, we may be required to replace such manufacturers. Although we believe that there are several potential alternative manufacturers who could manufacture our product candidates, we may incur added costs and delays in identifying and qualifying any such replacement.
Our current and anticipated future dependence upon others for the manufacture of our product candidates or products may adversely affect our future profit margins and our ability to commercialize any products that receive marketing approval on a timely and competitive basis.
If we are unable to manufacture our products in sufficient quantities, or at sufficient yields, or are unable to obtain regulatory approvals for a manufacturing facility for our products, we may experience delays in product development, clinical trials, regulatory approval and commercial distribution.
Completion of our clinical trials and commercialization of our product candidates require access to, or development of, facilities to manufacture our product candidates at sufficient yields and at commercial-scale. We have no experience manufacturing, or managing third parties in manufacturing, any of our product candidates in the volumes that will be necessary to support large-scale clinical trials or commercial sales. Efforts to establish these capabilities may not meet initial expectations as to scheduling, scale-up, reproducibility, yield, purity, cost, potency or quality.
We expect to rely on third parties for the manufacture of clinical and, if necessary, commercial quantities of our product candidates. These third-party manufacturers must also receive FDA approval before they can produce clinical material or commercial products. Our products may be in competition with other products for access to these facilities and may be subject to delays in manufacture if third parties give other products greater priority. We may not be able to enter into any necessary third-party manufacturing arrangements on acceptable terms, or on a timely basis. In addition, we may have to enter into technical transfer agreements and share our know-how with the third-party manufacturers, which can be time-consuming and may result in delays.
Our reliance on contract manufacturers may adversely affect our operations or result in unforeseen delays or other problems beyond our control. Because of contractual restraints and the limited number of third-party manufacturers with the expertise, required regulatory approvals and facilities to manufacture our bulk vaccines on a commercial-scale, replacement of a manufacturer may be expensive and time-consuming and may cause interruptions in the production of our vaccine. A third-party manufacturer may also encounter difficulties in production. These problems may include:
- •
- difficulties with production costs, scale-up and yields;
21
- •
- unavailability of raw materials and supplies;
- •
- insufficient quality control and assurance;
- •
- shortages of qualified personnel;
- •
- failure to comply with strictly enforced federal, state and foreign regulations that vary in each country where product
might be sold; and
- •
- lack of capital funding.
As a result, any delay or interruption could have a material adverse effect on our business, financial condition, results of operations and cash flows.
We may not be successful in establishing and maintaining strategic partnerships, which could adversely affect our ability to develop and commercialize products.
A part of our strategy is to evaluate and, as deemed appropriate, enter into partnerships in the future when strategically attractive, including potentially with major biotechnology or pharmaceutical companies. We face significant competition in seeking appropriate partners for our product candidates, and the negotiation process is time-consuming and complex. In order for us to successfully partner our product candidates, potential partners must view these product candidates as economically valuable in markets they determine to be attractive in light of the terms that we are seeking and other available products for licensing by other companies. Even if we are successful in our efforts to establish strategic partnerships, the terms that we agree upon may not be favorable to us, and we may not be able to maintain such strategic partnerships if, for example, development or approval of a product is delayed or sales of an approved product are disappointing. Any delay in entering into strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates and reduce their competitiveness even if they reach the market.
In addition, our strategic partners may breach their agreements with us, and we may not be able to adequately protect our rights under these agreements. Furthermore, our strategic partners will likely negotiate for certain rights to control decisions regarding the development and commercialization of our product candidates, if approved, and may not conduct those activities in the same manner as we would do so.
If we fail to establish and maintain strategic partnerships related to our product candidates, we will bear all of the risk and costs related to the development of any such product candidate, and we may need to seek additional financing, hire additional employees and otherwise develop expertise which we do not have and for which we have not budgeted. This could negatively affect the development of any unpartnered product candidate.
Risks Related to Our Intellectual Property
If we are unable to obtain or protect intellectual property rights related to our product candidates, we may not be able to compete effectively in our markets.
We rely upon a combination of patents, patent applications, know how and confidentiality agreements to protect the intellectual property related to our platform technology and product candidates. The patent position of biotechnology companies is generally uncertain because it involves complex legal and factual considerations. The standards applied by the United States Patent and Trademark Office, or U.S. PTO, and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology patents. The patent applications that we own or in-license may fail to result in issued patents with claims that cover our discovery platform or product candidates in the United States or in other countries. There is no assurance that all
22
potentially relevant prior art relating to our patents and patent applications or those of our licensors has been found, and prior art that we have not disclosed could be used by a third party to invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue and even if such patents cover our discovery platform or product candidates, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications, or those of our licensors, may not adequately protect our platform technology, provide exclusivity for our product candidates, prevent others from designing around our patents with similar products, or prevent others from operating in jurisdictions in which we did not pursue patent protection. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
If patent applications we hold or have in-licensed with respect to our platform or product candidates fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our product candidates or ATLAS discovery platform, it could dissuade companies from collaborating with us. We or our licensors have filed several patent applications covering aspects of our product candidates. We cannot offer any assurances about which, if any, patents will issue, the breadth of any such patents or whether any issued patents will be found invalid and unenforceable or will be challenged by third parties. Any successful opposition to these patent applications, or patents that may issue from them, or to any other patent applications or patents owned by or licensed to us, could deprive us of rights necessary for the successful commercialization of any product candidate that we may develop. Since patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we or our licensors were the first to file a patent application relating to any particular aspect of a product candidate. Furthermore, if third parties have filed such patent applications, an interference proceeding in the United States can be initiated by such third party, or by the U.S. PTO itself, to determine who was the first to invent any of the subject matter covered by the patent claims of our applications.
In the United States, for patent applications filed prior to March 16, 2013, assuming the other requirements for patentability are met, the first to invent is entitled to the patent, while outside the United States, the first to file a patent application is entitled to the patent. On March 16, 2013, the United States transitioned to a 'first to file' system more like that in the rest of the world in that the first inventor to file a patent application is entitled to the patent. Under either the prior system or current one, third parties are allowed to submit prior art prior to the issuance of a patent by the U.S. PTO, and may become involved in opposition, derivation, reexamination, inter partes review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, which could adversely affect our competitive position with respect to third parties.
In addition, patents have a limited lifespan. In most countries, including the United States, the natural expiration of a patent is 20 years from the date it is filed. Various extensions of patent term may be available in particular countries, however in all circumstances the life of a patent, and the protection it affords, has a limited term. If we encounter delays in obtaining regulatory approvals, the period of time during which we could market a product under patent protection could be reduced. We expect to seek extensions of patent terms where these are available in any countries where we are prosecuting patents. Such possible extensions include those permitted under the Drug Price Competition and Patent Term Restoration Act of 1984 in the United States, which permits a patent term extension of up to five years to cover an FDA-approved product. The actual length of the extension will depend on the amount of patent term lost while the product was in clinical trials. However, the applicable authorities, including the FDA in the United States, and any equivalent regulatory authority in other countries, may not agree with our assessment of whether such extensions
23
are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and non-clinical data, and then may be able to launch their product earlier than might otherwise be the case.
Any loss of, or failure to obtain, patent protection could have a material adverse impact on our business. We may be unable to prevent competitors from entering the market with a product that is similar to or the same as our products.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or misappropriate or otherwise violate our intellectual property rights. To counter infringement or unauthorized use, litigation may be necessary to enforce or defend our intellectual property rights, to protect our trade secrets and/or to determine the validity and scope of our own intellectual property rights or the proprietary rights of others. Such litigation can be expensive and time consuming. Many of our current and potential competitors have the ability to dedicate substantially greater resources to litigate intellectual property rights than we can. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results. In addition, in an infringement proceeding, a court may decide that a patent owned by or licensed to us is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Third-party claims of intellectual property infringement or misappropriation may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our ability to develop, manufacture, market and sell our product candidates, and to use our or our licensors' proprietary technologies without infringing the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions, reexamination, and inter partes review proceedings before the U.S. PTO and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the fields in which we are developing and may develop our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims for example to materials, formulations, methods of manufacture, methods of analysis, and/or methods for treatment related to the use or manufacture of our products or product candidates. In some cases, we may have failed to identify relevant such third-party patents or patent application. For example, applications filed before November 29, 2000 and certain applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Except for the preceding exceptions, patent applications in the United States and elsewhere are generally published only after a waiting period of approximately 18 months after the earliest filing. Therefore, patent applications covering our platform
24
technology or our products or product candidates could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our platform technologies, our products or product candidates and/or the use, analysis, and/or manufacture of our product candidates.
If any third-party patents were held by a court of competent jurisdiction to cover aspects of our materials, formulations, methods of manufacture, methods of analysis, and/or methods for treatment, the holders of any such patents would be able to block our ability to develop and commercialize the applicable product candidate until such patent expired or unless we obtain a license. Such licenses may not be available on acceptable terms, if at all. Even if we were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates. Defending against claims of patent infringement or misappropriation of trade secrets could be costly and time consuming, regardless of the outcome. Thus, even if we were to ultimately prevail, or to settle at an early stage, such litigation could burden us with substantial unanticipated costs. In addition, litigation or threatened litigation could result in significant demands on the time and attention of our management team, distracting them from the pursuit of other company business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys' fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
We may face a claim of misappropriation if a third party believes that we inappropriately obtained and used trade secrets of such third party. If we are found to have misappropriated a third party's trade secrets, we may be prevented from further using such trade secrets, limiting our ability to develop our product candidates, and we may be required to pay damages. During the course of any patent or other intellectual property litigation, there could be public announcements of the results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these announcements as negative, the perceived value of our products, programs, or intellectual property could be diminished. Accordingly, the market price of our common stock may decline.
We have in-licensed a portion of our intellectual property, and, if we fail to comply with our obligations under these arrangements, we could lose such intellectual property rights or owe damages to the licensor of such intellectual property.
We are a party to a number of license agreements that are important to our business, and we may enter into additional license agreements in the future. Our discovery platform is built, in part, around patents exclusively in-licensed from academic or research institutions. Certain of our in-licensed intellectual property also covers, or may cover, GEN-003 and other product candidates. See "Business—In-License Agreements" and "Business—Other Collaborations" for a description of our license agreements or other collaborations with The Regents of the University of California, President and Fellows of Harvard College, University of Washington, Children's Medical Center Corporation, Isconova AB (now Novavax), and the Dana-Farber Cancer Institute and Harvard Medical School.
25
Our existing license agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty and other obligations on us. If there is any conflict, dispute, disagreement or issue of non-performance between us and our licensing partners regarding our rights or obligations under the license agreements, including any such conflict, dispute or disagreement arising from our failure to satisfy payment obligations under any such agreement, we may owe damages, our licensor may have a right to terminate the affected license, and our ability to utilize the affected intellectual property in our drug discovery and development efforts, and our ability to enter into collaboration or marketing agreements for an affected product candidate, may be adversely affected.
Confidentiality agreements with employees and third parties may not prevent unauthorized disclosure of proprietary information.
In addition to the protection afforded by patents, we rely on confidentiality agreements to protect proprietary know-how that may not be patentable or that we may elect not to patent, processes for which patents are difficult to enforce and any other elements of our platform technology and discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, and outside scientific advisors, contractors and collaborators. Although we use reasonable efforts to protect our know-how, our employees, consultants, contractors, or outside scientific advisors might intentionally or inadvertently disclose our know-how information to competitors. In addition, competitors may otherwise gain access to our know-how or independently develop substantially equivalent information and techniques.
Enforcing a claim that a third party illegally obtained and is using any of our know-how is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States sometimes are less willing than U.S. courts to protect know-how. Misappropriation or unauthorized disclosure of our know-how could impair our competitive position and may have a material adverse effect on our business.
Risks Related to Commercialization of Our Product Candidates
Our future commercial success depends upon attaining significant market acceptance of our product candidates, if approved, among physicians, patients, third-party payors and others in the medical community.
Even if we obtain marketing approval for GEN-003, GEN-004 or any other products that we may develop or acquire in the future, the product may not gain market acceptance among physicians, third-party payors, patients and others in the medical community. For example, we currently expect that GEN-003 will be required to be administered by injection initially and with boosters. Physicians or patients may not accept this product as a result of this anticipated dosing requirement. In addition, market acceptance of any approved products depends on a number of other factors, including:
- •
- the efficacy and safety of the product, as demonstrated in clinical trials;
- •
- the clinical indications for which the product is approved and the label approved by regulatory authorities for use with
the product, including any warnings that may be required on the label;
- •
- acceptance by physicians and patients of the product as a safe and effective treatment and the willingness of the target
patient population to try new therapies and of physicians to prescribe new therapies;
- •
- the cost, safety and efficacy of treatment in relation to alternative treatments;
- •
- the availability of reimbursement for an adequate course of treatment by third-party payors and government authorities;
26
- •
- relative convenience and ease of administration;
- •
- the prevalence and severity of adverse side effects;
- •
- the effectiveness of our sales and marketing efforts; and
- •
- the restrictions on the use of our products together with other medications, if any.
Market acceptance is critical to our ability to generate significant revenue. Any product candidate, if approved and commercialized, may be accepted in only limited capacities or not at all. If any approved products are not accepted by the market to the extent that we expect, we may not be able to generate significant revenue and our business would suffer.
If we are unable to establish sales, marketing and distribution capabilities, we may not be successful in commercializing our product candidates if and when they are approved.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any product for which we have obtained marketing approval, we will need to establish a sales and marketing organization.
In the future, we expect to build a focused sales and marketing infrastructure to market or co-promote some of our product candidates in the United States, if and when they are approved. There are risks involved with establishing our own sales, marketing and distribution capabilities. For example, recruiting and training a sales force is expensive and time consuming and could delay any product launch. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our products on our own include:
- •
- our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
- •
- the inability of sales personnel to obtain access to physicians;
- •
- the lack of adequate numbers of physicians choosing to prescribe any future products;
- •
- the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage
relative to companies with more extensive product lines; and
- •
- unforeseen costs and expenses associated with creating an independent sales and marketing organization.
If we are unable to establish our own sales, marketing and distribution capabilities and enter into arrangements with third parties to perform these services, our product revenues and our profitability, if any, are likely to be lower than if we were to market, sell and distribute any products that we develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute our product candidates or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales, marketing and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
Coverage and reimbursement may be limited or unavailable in certain market segments for our product candidates, which could make it difficult for us to sell our products profitably.
Market acceptance and sales of any approved products will depend significantly on the availability of adequate coverage and reimbursement from third-party payors and may be affected by existing and
27
future health care reform measures. Third-party payors, such as government health care programs, private health insurers and health maintenance organizations, decide which drugs they will provide coverage for and establish reimbursement levels. Coverage and reimbursement decisions by a third-party payor may depend upon a number of factors, including the third-party payor's determination that use of a product is:
- •
- a covered benefit under its health plan;
- •
- safe, effective and medically necessary;
- •
- appropriate for the specific patient;
- •
- cost-effective; and
- •
- neither experimental nor investigational.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling health care costs. Coverage and reimbursement can vary significantly from payor to payor. As a result, obtaining coverage and reimbursement approval for a product from each government and other third-party payor will require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payor separately, with no assurance that we will be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. We cannot be sure that coverage or adequate reimbursement will be available for any of our product candidates. Also, we cannot be sure that coverage determinations or reimbursement amounts will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only to limited levels, we may not be able to commercialize certain of our products. In addition, in the United States third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drugs. As a result, significant uncertainty exists as to whether and how much third-party payors will reimburse patients for their use of newly approved drugs, which in turn will put pressure on the pricing of drugs.
Price controls may be imposed, which may adversely affect our future profitability.
In international markets, reimbursement and health care payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. In some countries, particularly member states of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on coverage, prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after coverage and reimbursement has been obtained. Reference pricing used by various European Union member states and parallel distribution, or arbitrage between low-priced and high-priced member states, can further reduce prices. In some countries, we may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidates to other available vaccines in order to obtain or maintain coverage, reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. There can be no assurance that our vaccine candidates will be considered cost-effective by third-party payors, that an adequate level of reimbursement will be available or that the third-party payors' reimbursement policies will not adversely affect our ability to sell our products profitably. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
28
The impact of recent health care reform legislation and other changes in the health care industry and in health care spending on us is currently unknown, and may adversely affect our business model.
In the United States, and in some foreign jurisdictions, the legislative landscape continues to evolve. Our revenue prospects could be affected by changes in health care spending and policy in the United States and abroad. We operate in a highly regulated industry and new laws or judicial decisions, or new interpretations of existing laws or decisions, related to health care availability, the method of delivery or payment for health care products and services could negatively impact our business, operations and financial condition. There is significant interest in promoting health care reform, as evidenced by the enactment in the United States of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act in 2010. It is likely that federal and state legislatures within the United States and foreign governments will continue to consider changes to existing health care legislation. We cannot predict the reform initiatives that may be adopted in the future or whether initiatives that have been adopted will be repealed or modified. The continuing efforts of the government, insurance companies, managed care organizations and other payors of health care services to contain or reduce costs of health care may adversely affect:
- •
- the demand for any drug products for which we may obtain regulatory approval;
- •
- our ability to set a price that we believe is fair for our products;
- •
- our ability to obtain coverage and reimbursement approval for a product;
- •
- our ability to generate revenues and achieve or maintain profitability; and
- •
- the level of taxes that we are required to pay.
We face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully, than we do.
The development and commercialization of new drug products is highly competitive. Our future success depends on our ability to demonstrate and maintain a competitive advantage with respect to the design, development and commercialization of our product candidates. Our objective is to design, develop and commercialize new products with superior efficacy, convenience, tolerability and safety. In many cases, the products that we commercialize will compete with existing, market-leading products.
Oral antivirals, such as valacyclovir and famciclovir, are products currently approved to treat patients with HSV-2. GEN-003, our lead product candidate, will compete with these products, if approved. In addition, one or more products not currently approved for the treatment of HSV-2, including pritelivir (AiCuris) and HerpV (Agenus) and other vaccines in development by Coridon Pty, Ltd and Vical Incorporated may in the future be granted marketing approval for the treatment of HSV-2 or other conditions for which GEN-003 might be approved.
Many of our potential competitors have significantly greater financial, manufacturing, marketing, drug development, technical and human resources than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing, including recruiting patients, obtaining regulatory approvals, recruiting patients and in manufacturing pharmaceutical products. Large and established companies such as Merck & Co., Inc., GlaxoSmithKline plc, Novartis, Inc., Sanofi Pasteur, SA, Pfizer Inc. and MedImmune, LLC (a subsidiary of AstraZeneca PLC), among others, compete in the vaccine market. In particular, these companies have greater experience and expertise in securing government contracts and grants to support their research and development efforts, conducting testing and clinical trials, obtaining regulatory approvals to market products, manufacturing such products on a broad scale and marketing approved products. These companies also have significantly greater research and marketing capabilities than we do and may also have products that have been approved or are in late stages of development, and have collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies
29
may also invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product that we develop obsolete. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or FDA approval or discovering, developing and commercializing products before we do. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. If we are not able to compete effectively against potential competitors, our business will not grow and our financial condition and operations will suffer.
Our products may cause undesirable side effects or have other properties that delay or prevent their regulatory approval or limit their commercial potential.
Undesirable side effects caused by our products or even competing products in development that utilize a common mechanism of action could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities and potential product liability claims. We expect to commence a Phase 2 dose optimization clinical trial for GEN-003 in mid-2014. Serious adverse events deemed to be caused by our product candidates could have a material adverse effect on the development of our product candidates and our business as a whole. The most common adverse events to date in the clinical trial evaluating the safety and tolerability of GEN-003 have been fatigue, myalgia (muscle pain), pain tenderness and induration (inflammatory hardening of the skin). Our understanding of the relationship between GEN-003 and these events, as well as our understanding of adverse events in future clinical trials of other product candidates, may change as we gather more information, and additional unexpected adverse events may be observed.
If we or others identify undesirable side effects caused by our product candidates either before or after receipt of marketing approval, a number of potentially significant negative consequences could result, including:
- •
- our clinical trials may be put on hold;
- •
- we may be unable to obtain regulatory approval for our vaccine candidates;
- •
- regulatory authorities may withdraw approvals of our vaccines;
- •
- regulatory authorities may require additional warnings on the label;
- •
- a medication guide outlining the risks of such side effects for distribution to patients may be required;
- •
- we could be sued and held liable for harm caused to patients; and
- •
- our reputation may suffer.
Any of these events could prevent us from achieving or maintaining market acceptance of our products and could substantially increase commercialization costs.
Risks Related to Our Indebtedness
Our level of indebtedness and debt service obligations could adversely affect our financial condition, and may make it more difficult for us to fund our operations.
In September 2013 we entered into a secured credit facility pursuant to a working capital term loan facility with Ares Capital Corporation providing for term loans of up to an aggregate of $10.0 million. On September 30, 2013, we drew down an initial $3.5 million under our secured credit facility and paid off our then existing secured credit facility. We drew down the remaining $6.5 million in December 2013. All obligations under our secured credit facility are secured by substantially all of our existing property and assets, excluding our intellectual property and licensed-in technology. This
30
indebtedness may create additional financing risk for us, particularly if our business or prevailing financial market conditions are not conducive to paying off or refinancing our outstanding debt obligations at maturity. This indebtedness could also have important negative consequences, including:
- •
- we will need to repay our indebtedness by making payments of interest and principal, which will reduce the amount of money
available to finance our operations, our research and development efforts and other general corporate activities; and
- •
- our failure to comply with the restrictive covenants in our secured credit facility could result in an event of default that, if not cured or waived, would accelerate our obligation to repay this indebtedness, and the lender could seek to enforce its security interest in the assets securing such indebtedness.
To the extent additional debt is added to our current debt levels, the risks described above could increase.
We may not have cash available to us in an amount sufficient to enable us to make interest or principal payments on our indebtedness when due.
Failure to satisfy our current and future debt obligations under our secured credit facility could result in an event of default and, as a result, our lender could accelerate all of the amounts due. In the event of an acceleration of amounts due under our secured credit facility as a result of an event of default, we may not have sufficient funds or may be unable to arrange for additional financing to repay our indebtedness. In addition, our lender could seek to enforce its security interests in the assets securing such indebtedness.
We are subject to certain restrictive covenants which, if breached, could have a material adverse effect on our business and prospects.
Our secured credit facility imposes operating and other restrictions on us. Such restrictions will affect, and in many respects limit or prohibit, our ability and the ability of any future subsidiary to, among other things:
- •
- dispose of certain assets;
- •
- change our lines of business;
- •
- engage in mergers or consolidations;
- •
- incur additional indebtedness;
- •
- create liens on assets;
- •
- pay dividends and make distributions or repurchase our capital stock; and
- •
- engage in certain transactions with affiliates.
Risks Related to Our Business and Industry
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop our products, conduct our clinical trials and commercialize our product candidates.
We are highly dependent on members of our senior management, including William Clark, our President and Chief Executive Officer, Seth Hetherington, M.D., our Chief Medical Officer, Jonathan Poole, our Chief Financial Officer, Jessica Flechtner, Ph.D., our Senior Vice President of Research, and Paul Giannasca, Ph.D., our Vice President, Biopharmaceutical Development and Production. The loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives. We have employment agreements with each of these members of senior management and we maintain a keyman insurance policy on Mr. Clark for $2.0 million.
31
Recruiting and retaining qualified scientific, clinical, manufacturing, sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
Our employees, independent contractors, principal investigators, consultants, commercial partners, and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraudulent or other illegal activity by our employees, independent contractors, principal investigators, consultants, commercial partners, and vendors. Misconduct by these parties could include intentional, reckless and/or negligent conduct that fails: to comply with the laws of the FDA and similar foreign regulatory bodies; provide true, complete and accurate information to the FDA and similar foreign regulatory bodies; to comply with manufacturing standards we have established; to comply with federal, state and foreign health care fraud and abuse laws and regulations; to report financial information or data accurately; or to disclose unauthorized activities to us. In particular, the promotion, sale and marketing of health care items and services, as well as certain business arrangements in the health care industry are subject to extensive laws and regulations intended to prevent misconduct, including fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and, structuring and commission(s), certain customer incentive programs and other business arrangements. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials. It is not always possible to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
32
Our relationships with health care professionals, institutional providers, principal investigators, consultants, customers (actual and potential) and third-party payors are, and will continue to be, subject, directly and indirectly, to federal and state health care fraud and abuse, false claims, marketing expenditure tracking and disclosure, government price reporting, and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face penalties, including, without limitation, civil, criminal, and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations.
Our business operations and activities may be directly or indirectly subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and the federal False Claims Act. If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. These laws may impact, among other things, our current activities with principal investigators and research subjects, as well as proposed and future sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by the federal government and state governments in which we conduct our business. The laws that may affect our ability to operate include, but are not limited to:
- •
- the federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, receiving,
offering or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an
individual, or the purchase, lease, order or recommendation of any good, facility, item or service for which payment may be made, in whole or in part, under a federal health care program, such as the
Medicare and Medicaid programs;
- •
- federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things,
individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other third-party payors that are false or fraudulent or
knowingly making a false statement to improperly avoid, decrease or conceal an obligation to pay money to the federal government;
- •
- the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal
statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any health care benefit program or obtain, by means of false or fraudulent pretenses,
representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, regardless of the payor (e.g., public or private) and
knowingly and willfully falsifying, concealing, or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for,
health care benefits, items or services relating to health care matters;
- •
- HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and their respective implementing regulations, which impose requirements on certain covered health care providers, health plans, and health care clearinghouses as well as their respective business associates that perform services for them that involve the use, or disclosure of, individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization;
33
- •
- the federal Physician Payments Sunshine Act, created under Section 6002 of the Patient Protection and Affordable
Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, collectively, ACA, and its implementing regulations requires manufacturers of drugs, devices, biologicals and medical
supplies for which payment is available under Medicare, Medicaid or the Children's Health Insurance Program (with certain exceptions) to report annually to the United States Department of Health and
Human Services information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching
hospitals, as well as ownership and investment interests held by physicians and their immediate family members, with data collection required beginning August 1, 2013 and reporting to the
Centers for Medicare & Medicaid Services required by March 31, 2014 and by the 90th day of each subsequent calendar year;
- •
- federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that
potentially harm consumers;
- •
- federal government price reporting laws, changed by ACA to, among other things, increase the minimum Medicaid rebates owed
by most manufacturers under the Medicaid Drug Rebate Program and offer such rebates to additional populations, that require us to calculate and report complex pricing metrics to government programs,
where such reported prices may be used in the calculation of reimbursement and/or discounts on our marketed drugs (participation in these programs and compliance with the applicable requirements may
subject us to potentially significant discounts on our products, increased infrastructure costs, and potentially limit our ability to offer certain marketplace discounts);
- •
- the Foreign Corrupt Practices Act, a United States law which regulates certain financial relationships with foreign
government officials (which could include, for example, certain medical professionals); and
- •
- state law equivalents of each of the above federal laws, such as anti-kickback, false claims, consumer protection and unfair competition laws which may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements as well as submitting claims involving health care items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restricts payments that may be made to health care providers; state laws that require drug manufacturers to file reports with states regarding marketing information, such as the tracking and reporting of gifts, compensations and other remuneration and items of value provided to health care professionals and entities (compliance with such requirements may require investment in infrastructure to ensure that tracking is performed properly, and some of these laws result in the public disclosure of various types of payments and relationships, which could potentially have a negative effect on our business and/or increase enforcement scrutiny of our activities); and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, with differing effects.
In addition, the regulatory approval and commercialization of any of our product candidates outside the United States will also likely subject us to foreign equivalents of the health care laws mentioned above, among other foreign laws.
Efforts to ensure that our business arrangements will comply with applicable health care laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other health care laws and regulations. If our operations are
34
found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, civil, criminal, and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations.
We may encounter difficulties in managing our growth and expanding our operations successfully.
As we seek to advance our product candidates through clinical trials and commercialization, we will need to expand our development, regulatory, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to manage additional relationships with various strategic partners, suppliers and other third parties. Future growth will impose significant added responsibilities on members of management. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively and hire, train and integrate additional management, administrative and, if necessary, sales and marketing personnel. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing our company.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
- •
- decreased demand for any product candidates or products that we may develop;
- •
- injury to our reputation and significant negative media attention;
- •
- withdrawal of clinical trial participants;
- •
- significant costs to defend the related litigations;
- •
- a diversion of management's time and our resources;
- •
- substantial monetary awards to trial participants or patients;
- •
- product recalls, withdrawals, or labeling, marketing or promotional restrictions;
- •
- loss of revenue;
- •
- the inability to commercialize any product candidates that we may develop; and
- •
- a decline in our stock price.
Failure to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. We currently carry product liability insurance covering our clinical trials in the amount of
35
$5.0 million in the aggregate. Although we maintain product liability insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We must comply with environmental laws and regulations, and failure to comply with these laws and regulations could expose us to significant liabilities.
We use hazardous chemicals and radioactive and biological materials in certain aspects of our business and are subject to a variety of federal, state and local laws and regulations governing the use, generation, manufacture, distribution, storage, handling, treatment and disposal of these materials. We cannot eliminate the risk of accidental injury or contamination from the use, manufacture, distribution, storage, handling, treatment or disposal of hazardous materials. In the event of contamination or injury, or failure to comply with environmental, occupational health and safety and export control laws and regulations, we could be held liable for any resulting damages and any such liability could exceed our assets and resources. We are uninsured for third-party contamination injury.
We may not be able to win government, academic institution or non-profit contracts or grants.
From time to time, we may apply for contracts or grants from government agencies, non-profit entities and academic institutions. Such grants have been our only source of revenue to date. Such contracts or grants can be highly attractive because they provide capital to fund the on-going development of our technologies and product candidates without diluting our stockholders. However, there is often significant competition for these contracts or grants. Entities offering contracts or grants may have requirements to apply for or to otherwise be eligible to receive certain contracts or grants that our competitors may be able to satisfy that we cannot. In addition, such entities may make arbitrary decisions as to whether to offer contracts or make grants, to whom the contracts or grants will be awarded and the size of the contracts or grants to each awardee. Even if we are able to satisfy the award requirements, there is no guarantee that we will be a successful awardee. Therefore, we may not be able to win any contracts or grants in a timely manner, if at all.
Risks Related to Our Common Stock and This Offering
We are eligible to be treated as an "emerging growth company" as defined in the Jumpstart Our Business Startups Act of 2012, and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an "emerging growth company", as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
- •
- being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim
financial statements, with correspondingly reduced "Management's Discussion and Analysis of Financial Condition and Results of Operations" disclosure;
- •
- not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting;
36
- •
- not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board
providing for supplemental auditor's reports for additional information about the audit and the financial statements;
- •
- reduced disclosure obligations regarding executive compensation; and
- •
- exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved.
We have taken advantage of reduced reporting burdens in this prospectus. For example, we have not included all of the executive compensation related information that would be required if we were not an emerging growth company. We cannot predict whether investors will find our common stock less attractive if we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile. In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of these accounting standards until they would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our common stock held by non-affiliates exceeds $700.0 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three-year period before that time, we would cease to be an emerging growth company immediately. Even after we no longer qualify as an emerging growth company, we may still qualify as a "smaller reporting company" if the market value of our common stock held by non-affiliates is below $75.0 million as of June 30 in any given year, which would allow us to take advantage of many of the same exemptions from disclosure requirements, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements.
We cannot predict what the market price of our common stock will be and, as a result, it may be difficult for you to sell your shares of our common stock.
An inactive market may impair our ability to raise capital by selling shares of our common stock and may impair our ability to enter into strategic partnerships or acquire companies or products by using our shares of common stock as consideration. We cannot predict the prices at which our common stock will trade. It is possible that in one or more future periods our results of operations may be below the expectations of public market analysts and investors and, as a result of these and other factors, the price of our common stock may fall.
The market price of our common stock may be highly volatile, and you may not be able to resell your shares at or above the offering price in this transaction and you may incur substantial losses.
Our stock price is likely to be volatile. The stock market in general and the market for biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, our
37
stockholders could incur substantial losses. The market price for our common stock may be influenced by many factors, including:
- •
- the success of competitive products or technologies;
- •
- results of clinical trials of our product candidates;
- •
- the timing of the release of results of our clinical trials;
- •
- results of clinical trials of our competitors' products;
- •
- regulatory actions or legal developments with respect to our products or our competitors' products;
- •
- developments or disputes concerning patent applications, issued patents or other proprietary rights;
- •
- the results of our efforts to discover, develop, acquire or in-license additional product candidates or products;
- •
- actual or anticipated fluctuations in our financial condition and operating results;
- •
- publication of research reports by securities analysts about us or our competitors or our industry;
- •
- our failure or the failure of our competitors to meet analysts' projections or guidance that we or our competitors may
give to the market;
- •
- additions and departures of key personnel;
- •
- strategic decisions by us or our competitors, such as acquisitions, divestitures, spin-offs, joint ventures, strategic
investments or changes in business strategy;
- •
- the passage of legislation or other regulatory developments affecting us or our industry;
- •
- fluctuations in the valuation of companies perceived by investors to be comparable to us;
- •
- sales of our common stock by us, our insiders or our other stockholders;
- •
- speculation in the press or investment community;
- •
- announcement or expectation of additional financing efforts;
- •
- changes in accounting principles;
- •
- terrorist acts, acts of war or periods of widespread civil unrest;
- •
- natural disasters and other calamities;
- •
- changes in market conditions for biopharmaceutical stocks; and
- •
- changes in general market and economic conditions.
In addition, the stock market has recently experienced significant volatility, particularly with respect to pharmaceutical, biotechnology and other life sciences company stocks. The volatility of pharmaceutical, biotechnology and other life sciences company stocks often does not relate to the operating performance of the companies represented by the stock. As we operate in a single industry, we are especially vulnerable to these factors to the extent that they affect our industry or our products, or to a lesser extent our markets. In the past, securities class action litigation has often been initiated against companies following periods of volatility in their stock price. This type of litigation could result in substantial costs and divert our management's attention and resources, and could also require us to make substantial payments to satisfy judgments or to settle litigation.
38
Our executive officers, directors and principal stockholders own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As of June 30, 2014, our executive officers and directors, combined with our stockholders who owned more than 5% of our outstanding common stock, beneficially own shares representing approximately 70.6% of our capital stock. We expect that, after this offering, that same group will continue to beneficially hold at least 59.6% of our outstanding common stock. Accordingly, even after this offering, these stockholders will be able to exert a significant degree of influence over our management and affairs and over matters requiring stockholder approval, including the election of our board of directors and approval of significant corporate transactions. This concentration of ownership could have the effect of entrenching our management and/or the board of directors, delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could have a material and adverse effect on the fair market value of our common stock.
A significant portion of our total outstanding shares may be sold into the public market in the near future, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time after the expiration of the lock-up agreements described in the "Underwriting" section of this prospectus. These sales, or the market perception that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. After this offering, we will have 20,806,977 shares of common stock outstanding based on the number of shares outstanding as of June 30, 2014. Of these shares, 9,507,115 shares of common stock, including the 5,500,000 shares sold in our initial public offering, and all of the shares of common stock to be sold in this offering, may be resold in the public market immediately. The remaining 11,299,862 shares, or 54.3% of our outstanding shares after this offering, are currently restricted as a result of securities laws or lock-up agreements but will be able to be sold, subject to any applicable volume limitations under federal securities laws with respect to affiliate sales, in the near future as set forth below.
In addition, as of June 30, 2014, there were 2,233,769 shares subject to outstanding options that will become eligible for sale in the public market to the extent permitted by any applicable vesting requirements, the lock-up agreements and Rules 144 and 701 under the Securities Act. Moreover, after this offering, holders of an aggregate of 11,476,987 shares of our common stock and holders of warrants to purchase 3,878 shares of our common stock will have rights, subject to some conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. If such holders, by exercising their registration rights, cause a large number of securities to be registered and sold into the public market, these sales could have an adverse effect on the market price for our common stock. We also intend to register all shares of common stock that we may issue under our employee benefit plans, including our 2014 Equity Incentive Plan. Once we register these shares and they are issued in accordance with the terms of the plans, they can be freely sold in the public market upon issuance, subject to the lock-up agreements and the restrictions imposed on our affiliates under Rule 144. For more information, see "Shares Eligible for Future Sale—Rule 144".
You will incur immediate and substantial dilution as a result of this offering.
The public offering price of our common stock will be substantially higher than the net tangible book value per share of our common stock. Therefore, if you purchase common stock in this offering, you will pay a price per share that substantially exceeds our pro forma adjusted net tangible book value per share after giving effect to this offering. To the extent shares subsequently are issued under options or warrants, you will incur further dilution. Based on the public offering price of $15.31 per share (the
39
last reported price of our common stock on the NASDAQ Global Market on July 10, 2014), you will incur immediate and substantial dilution of $10.30 per share, representing the difference between our pro forma net tangible book value per share, after giving effect to this offering, based on the number of shares outstanding as of March 31, 2014.
We have had a material weakness in internal control over financial reporting in the past and cannot assure you that additional material weaknesses will not be identified in the future. Our failure to implement and maintain effective internal control over financial reporting could result in material misstatements in our financial statements which could require us to restate financial statements, cause investors to lose confidence in our reported financial information and have a negative effect on our stock price.
As reported in our Quarterly Report on Form 10-Q filed with the SEC on May 9, 2014, during the quarter ended March 31, 2014, management and our independent registered public accounting firm identified a material weakness in our internal control over financial reporting (as defined in the Public Company Accounting Oversight Board's Auditing Standard No. 5) related to the accounting for a non-cash stock compensation expense for a milestone-based stock option award. We have implemented a remediation plan to address this material weakness, as described in our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014.
We cannot assure you that additional material weaknesses or significant deficiencies in our internal control over financial reporting will not be identified in the future. Any failure to maintain or implement required new or improved controls, or any difficulties we encounter in their implementation, could result in additional significant deficiencies or material weaknesses, cause us to fail to meet our periodic reporting obligations or result in material misstatements in our financial statements. Any such failure could also adversely affect the results of periodic management evaluations regarding the effectiveness of our internal control over financial reporting. The existence of a material weakness could result in errors in our financial statements that could result in a restatement of financial statements, cause us to fail to meet our reporting obligations and cause investors to lose confidence in our reported financial information, leading to a decline in our stock price.
We have broad discretion in the use of net proceeds from this offering and may not use them effectively.
We currently intend to use the net proceeds from this offering, together with our existing cash and cash equivalents and future available borrowings under our credit facility, (1) to further fund clinical development and manufacturing of GEN-003 into a Phase 3 clinical study, (2) to further fund clinical development and manufacturing of GEN-004, (3) to fund research and development and manufacturing of our prophylactic chlamydia, HSV-2 and malaria programs through filing of an IND application and (4) for working capital and other general corporate purposes, including funding the costs of operating as a public company. See the section of this prospectus entitled "Use of Proceeds." Although we currently intend to use the net proceeds from this offering in such a manner, we will have broad discretion in the application of the net proceeds. Our failure to apply these funds effectively could affect our ability to continue to develop and commercialize our product candidates. Pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or loses value.
We incur significant costs as a result of being a public company and our management expects to devote substantial time to public company compliance programs.
As a public company, we incur significant legal, insurance, accounting and other expenses that we did not incur as a private company. In addition, our administrative staff are required to perform additional tasks. For example, in anticipation of becoming a public company, we adopted additional internal controls and disclosure controls and procedures and bear all of the internal and external costs of preparing and distributing periodic public reports in compliance with our obligations under the
40
securities laws. We invest resources to comply with evolving laws, regulations and standards, and this investment could result in increased general and administrative expenses and may divert management's time and attention from product development activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed. In connection with our initial public offering, we increased our directors' and officers' insurance coverage, which increased our insurance cost. In the future, it will be more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of our board of directors, particularly to serve on our audit committee and compensation committee, and qualified executive officers.
In addition, in order to comply with the requirements of being a public company, we have and may need to undertake various actions, including implementing new internal controls and procedures and hiring new accounting or internal audit staff. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file with the SEC is recorded, processed, summarized and reported within the time periods specified in the SEC's rules and forms, and that information required to be disclosed in reports under the Securities Exchange Act of 1934 as amended, or the Exchange Act, is accumulated and communicated to our principal executive and financial officers. Any failure to develop or maintain effective controls could adversely affect the results of periodic management evaluations. In the event that we are not able to demonstrate compliance with the Sarbanes-Oxley Act, that our internal control over financial reporting is perceived as inadequate, or that we are unable to produce timely or accurate financial statements, investors may lose confidence in our operating results and the price of our ordinary shares could decline. In addition, if we are unable to continue to meet these requirements, we may not be able to remain listed on the NASDAQ Global Market.
Since becoming a public company, we are required to comply with certain of the SEC's rules that implement Section 404 of the Sarbanes-Oxley Act, which require management to certify financial and other information in our quarterly and annual reports and provide an annual management report on the effectiveness of our internal control over financial reporting commencing with our second annual report. This assessment must include the disclosure of any material weaknesses in our internal control over financial reporting identified by our management or our independent registered public accounting firm. To achieve compliance with Section 404 within the prescribed period, we engage in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404. If we identify one or more material weaknesses, it could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statement.
Our independent registered public accounting firm will not be required to formally attest to the effectiveness of our internal control over financial reporting until the later of our second annual report or the first annual report required to be filed with the SEC following the date we are no longer an "emerging growth company" as defined in the JOBS Act. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal controls in the future.
41
Provisions in our charter documents and under Delaware law have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders, and prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and amended and restated by-laws contain provisions that may have the effect of discouraging, delaying or preventing a change in control of us or changes in our management. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions:
- •
- authorize "blank check" preferred stock, which could be issued by our board of directors without stockholder approval and
may contain voting, liquidation, dividend and other rights superior to our common stock;
- •
- create a classified board of directors whose members serve staggered three-year terms;
- •
- specify that special meetings of our stockholders can be called only by our board of directors, the chairperson of our
board of directors, our chief executive officer or our president;
- •
- prohibit stockholder action by written consent;
- •
- establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our
stockholders, including proposed nominations of persons for election to our board of directors;
- •
- provide that our directors may be removed only for cause;
- •
- provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though
less than a quorum;
- •
- specify that no stockholder is permitted to cumulate votes at any election of directors;
- •
- expressly authorize our board of directors to modify, alter or repeal our amended and restated by-laws; and
- •
- require supermajority votes of the holders of our common stock to amend specified provisions of our amended and restated by-laws.
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in our management.
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Any provision of our amended and restated certificate of incorporation, our amended and restated by-laws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
42
Our ability to use net operating losses to offset future taxable income may be subject to certain limitations.
In general, under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, a corporation that undergoes an "ownership change" is subject to limitations on its ability to utilize its pre-change net operating losses, or NOLs, to offset future taxable income. Our existing NOLs may be subject to substantial limitations arising from previous ownership changes, and if we undergo an ownership change in connection with or after this offering, our ability to utilize NOLs could be further limited by Section 382 of the Code. In addition, future changes in our stock ownership, many of which are outside of our control, could result in an ownership change under Section 382 of the Code. Our NOLs may also be impaired under state law. Accordingly, we may not be able to utilize a material portion of our NOLs. Furthermore, our ability to utilize our NOLs is conditioned upon our attaining profitability and generating U.S. federal taxable income. We have incurred net losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future; thus, we do not know whether or when we will generate the U.S. federal taxable income necessary to utilize our NOLs.
Our amended and restated certificate of incorporation designates the state or federal courts located in the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders' ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated certificate of incorporation provides that, subject to limited exceptions, the state and federal courts located in the State of Delaware will be the sole and exclusive forum for (1) any derivative action or proceeding brought on our behalf, (2) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders, (3) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law, our amended and restated certificate of incorporation or our amended and restated by-laws or (4) any other action asserting a claim against us that is governed by the internal affairs doctrine. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock shall be deemed to have notice of and to have consented to the provisions of our amended and restated certificate of incorporation described above. This choice of forum provision may limit a stockholder's ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and employees. Alternatively, if a court were to find these provisions of our amended and restated certificate of incorporation inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business and financial condition.
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be your sole source of gain.
You should not rely on an investment in our common stock to provide dividend income. We do not anticipate that we will pay any cash dividends to holders of our common stock in the foreseeable future. Instead, we plan to retain any earnings to maintain and expand our operations. In addition, our ability to pay cash dividends is currently prohibited by the terms of our debt financing arrangement, and any future debt financing arrangement may contain terms prohibiting or limiting the amount of dividends that may be declared or paid on our common stock. Accordingly, investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any return on their investment. As a result, investors seeking cash dividends should not purchase our common stock.
43
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference into this prospectus contain forward-looking statements. Forward-looking statements are neither historical facts nor assurances of future performance. Instead, they are based on our current beliefs, expectations and assumptions regarding the future of our business, future plans and strategies, our clinical results and other future conditions. The words "anticipate", "believe", "contemplate", "continue", "could", "estimate", "expect", "forecast", "goal", "intend", "may", "plan", "potential" "predict", "project", "should", "target", "will", "would", or the negative of these terms or other similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words.
The forward-looking statements in this prospectus include, among other things, statements about:
- •
- the timing of results of our ongoing and planned clinical trials;
- •
- our planned clinical trials for GEN-003 and GEN-004;
- •
- our estimates regarding the amount of funds we require to complete our planned Phase 2 clinical trials for GEN-003
and planned Phase 2a trial and potential future clinical trials for GEN-004;
- •
- our estimate for when we will require additional funding;
- •
- our plans to commercialize GEN-003 and our other vaccine candidates;
- •
- the timing of, and our ability to, obtain and maintain regulatory approvals for our product candidates;
- •
- the rate and degree of market acceptance and clinical utility of any approved product candidate;
- •
- the potential benefits of strategic partnership agreements and our ability to enter into selective strategic partnership
arrangements;
- •
- our ability to quickly and efficiently identify and develop product candidates;
- •
- our commercialization, marketing and manufacturing capabilities and strategy;
- •
- our intellectual property position; and
- •
- our estimates regarding expenses, future revenues, capital requirements, the sufficiency of our current and expected cash resources and our need for additional financing.
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements included in this prospectus, particularly in the "Risk Factors" section and other documents incorporated by reference herein, that we believe could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments we may make or collaborations or strategic partnerships we may enter into.
You should read this prospectus, other documents incorporated by reference herein and the documents that we have filed as exhibits to the registration statement of which this prospectus is a part completely and with the understanding that our actual future results may be materially different from what we expect. The forward-looking statements represent our views as of the date of this prospectus. We anticipate that subsequent events and developments will cause our views to change. However, while we may elect to update these forward-looking statements at some point in the future, we have no current intention of doing so except to the extent required by applicable law. You should, therefore, not
44
rely on these forward-looking statements as representing our views as of any date subsequent to the date of this prospectus.
This prospectus and the other documents incorporated by reference herein include statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third party research, surveys and studies are reliable, we have not independently verified such data. With respect to the information from IMS Health, this represents MIDAS Ex-Manufacturer levels sales data. This information is an estimate derived from the use of information under license from the following IMS Health information service: MIDAS Sales Data. IMS Health expressly reserves all rights.
45
The net proceeds of the sale of 3,400,000 shares of common stock in this offering will be approximately $48.4 million at the assumed public offering price of $15.31 per share (the last reported priced of our common stock on the NASDAQ Global Market on July 10, 2014) after deducting underwriting discounts and commissions and estimated offering expenses payable by us. If the underwriters exercise their option to purchase additional share of common stock in full, we estimate that the net proceeds will be approximately $7.3 million, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Each $1.00 increase or decrease in the assumed public offering price of $15.31 per share (the last reported price of our common stock on the NASDAQ Global Market on July 10, 2014) would increase or decrease our net proceeds by approximately $3.2 million, assuming the number of shares offered by us, as set forth on the cover of this prospectus, remains the same and after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
As of March 31, 2014, we had cash and cash equivalents of $65.8 million. We intend to use the net proceeds from this offering, together with our existing cash and cash equivalents as follows:
- •
- approximately $25 million to fund research, manufacturing and clinical development in connection with GEN-003. We
expect that these funds will be sufficient to enable us to complete our Phase 2 program and to have conducted an end of Phase 2 meeting with the FDA relating to this program;
- •
- approximately $10 million to fund research and development expenses in connection with GEN-004. We expect to
complete our planned Phase 2a trial and to be able to initiate a further Phase 2 clinical trial in a target patient population;
- •
- approximately $8 million to fund research and development and manufacturing of our prophylactic chlamydia, HSV-2
and malaria programs to finalize the vaccine candidates, advance the candidates through non-clinical toxicology and file an IND; and
- •
- the remainder for working capital and other general corporate purposes.
Our expected use of net proceeds from this offering represents our current intentions based upon our present plans and business condition, which could change in the future as our plans and business conditions evolve. As of the date of this prospectus, we cannot predict with certainty all of the particular uses for the net proceeds to be received upon the completion of this offering or the amounts that we will actually spend on the uses set forth above. We may also use a portion of the net proceeds to in-license, acquire or invest in complementary technologies, products or assets.
The amount and timing of our actual expenditures will depend upon numerous factors, including the results of our research and development efforts, the timing and success of non-clinical studies, our ongoing clinical trials or clinical trials we may commence in the future and the timing of regulatory submissions. As a result, our management will have broad discretion over the use of the net proceeds from this offering.
Pending the use of the proceeds from this offering, we intend to invest the net proceeds in short-term, interest-bearing, investment-grade securities, certificates of deposit or government securities.
46
MARKET PRICE OF OUR COMMON STOCK
Our common stock has been listed on the NASDAQ Global Market under the symbol "GNCA" since February 5, 2014. Prior to that, there was no public market for our common stock. The following table sets forth for the periods indicated the high and low sales prices per share of our common stock as reported on the NASDAQ Global Market:
Year ending December 31, 2014
|
High | Low | |||||
|---|---|---|---|---|---|---|---|
First Quarter(1): |
$ | 23.99 | $ | 10.90 | |||
Second Quarter: |
$ | 23.99 | $ | 16.76 | |||
Third Quarter (through July 10, 2014): |
$ | 20.00 | $ | 15.15 | |||
- (1)
- Represents the period from February 5, 2014, the date on which our common stock first began to trade on the NASDAQ Global Market after the pricing of our initial public offering, through March 31, 2014, the end of our first fiscal quarter.
On July 10, 2014, the last reported closing price for our common stock was $15.31 per share. Computershare Trust Company, N.A. is the transfer agent and registrar for our common stock. As of June 30, 2014, there were 55 holders of record of our common stock.
47
We have never declared or paid cash dividends on our common stock. We currently intend to retain all available funds and any future earnings, if any, to fund the development and expansion of our business and we do not anticipate paying any cash dividends in the foreseeable future. Any future determination to pay dividends will be made at the discretion of our board of directors. Additionally, our ability to pay dividends on our common stock is limited by restrictions under the terms of the agreements governing our secured credit facility. Payment of future cash dividends, if any, will be at the discretion of the board of directors after taking into account various factors, including our financial condition, operating results, current and anticipated cash needs, the requirements of current or then-existing debt instruments and other factors the board of directors deems relevant.
48
The following table sets forth our cash and cash equivalents and capitalization as of March 31, 2014:
- •
- on an actual basis; and
- •
- on an as adjusted basis to give further effect to the sale of shares of our common stock offered in this offering at an assumed public offering price of $15.31 per share (the last reported price of our common stock on the NASDAQ Global Market on July 10, 2014), after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
You should read this information together with our unaudited financial statements and related notes and the "Management's Discussion and Analysis of Financial Condition and Results of Operations" in our Annual Report on Form 10-K for the year ended December 31, 2013 and the Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, which are incorporated by reference in this prospectus.
| |
As of March 31, 2014 | ||||||
|---|---|---|---|---|---|---|---|
(in thousands, except per share data)
|
Actual | As Adjusted | |||||
Cash and cash equivalents |
$ | 65,839 | $ | 114,220 | |||
| | | | | | | | |
| | | | | | | | |
Stockholders' (deficit) equity: |
|||||||
Common stock, $0.001 par value; 191,690 shares authorized, 17,322 shares issued, and 17,299 shares outstanding as of March 31, 2014, and 191,690 shares authorized and 20,722 shares issued and 20,699 outstanding, as adjusted |
17 | 20 | |||||
Additional paid-in capital |
142,816 | 191,194 | |||||
Deficit accumulated during the development stage |
(87,460 | ) | (87,460 | ) | |||
| | | | | | | | |
Total stockholders' equity |
55,373 | 103,754 | |||||
| | | | | | | | |
Total capitalization |
$ | 55,373 | $ | 103,754 | |||
| | | | | | | | |
| | | | | | | | |
The table above does not include:
- •
- 2,002,806 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2014 at a
weighted average exercise price of $4.93 per share;
- •
- 61,832 shares of common stock issuable upon the exercise of outstanding warrants as of March 31, 2014 at a
weighted-average exercise price of $6.95 per share;
- •
- 2,646,788 shares of common stock reserved for future issuance as of March 31, 2014 under our 2014 Equity Incentive
Plan which was adopted in January 2014; and
- •
- 200,776 shares of common stock reserved for future issuance under our 2014 Employee Stock Purchase Plan, which was adopted in January 2014.
49
If you invest in our common stock in this offering, your ownership interest will be diluted immediately to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock after this offering.
Our historical net tangible book value as of March 31, 2014 was $55.4 million, or $3.20 per share of our common stock. Historical net tangible book value per share represents the amount of our total tangible assets less total liabilities, divided by the number of shares of our common stock outstanding as of March 31, 2014.
After giving effect to our issuance and sale of 3,400,000 shares of our common stock in this offering at an assumed public offering price of $15.31 per share, which was the last reported sale price of our common stock on the NASDAQ Global Market on July 10, 2014, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us, our as adjusted net tangible book value as of March 31, 2014 would have been $103.8 million, or $5.01 per share. This represents an immediate increase in as adjusted net tangible book value per share of $1.81 to existing stockholders and immediate dilution of $10.30 in as adjusted net tangible book value per share to new investors purchasing common stock in this offering. Dilution per share to new investors is determined by subtracting as adjusted net tangible book value per share after this offering from the public offering price per share paid by new investors. The following table illustrates this dilution on a per share basis:
Assumed public offering price per share |
$ | 15.31 | |||||
Historical net tangible book value per share as of March 31, 2014 |
$ | 3.20 | |||||
Increase in net tangible book value per share attributable to new investors |
1.81 | ||||||
| | | | | | | | |
As adjusted net tangible book value per share after this offering |
5.01 | ||||||
| | | | | | | | |
Dilution per share to new investors |
$ | 10.30 | |||||
If the underwriters exercise their option to purchase additional shares or if any additional shares are issued in connection with the exercise of options, you will experience further dilution.
50
Overview
We are a clinical stage biotechnology company that discovers and develops novel vaccines and immunotherapies to address diseases with significant unmet needs. We use our proprietary discovery platform, ATLAS, to rapidly design vaccines and immunotherapies that act through T cell (or cellular) immune responses, in contrast to approved vaccines and immunotherapies, which are designed to act primarily through B cell (or antibody) immune responses. We believe that by harnessing T cells we can develop first-in-class vaccines and immunotherapies to address infectious diseases where T cells are central to the control of the disease.
In September 2013, we announced human proof-of-concept data for GEN-003, a candidate therapeutic vaccine, or immunotherapy, that we are developing to treat herpes simplex virus-2, or HSV-2, infections. These data from our double-blind, placebo-controlled, dose-escalating Phase 1/2a trial represented the first reported instance of a therapeutic vaccine working against an infectious disease. We have now completed the follow-up review of patients for 12 months after their last dose of vaccine. Final analysis of the data showed that for the best performing 30µg dose group, there was a sustained reduction in the viral shedding rate. After completion of dosing for this dose group, the viral shedding rate fell by 52% versus baseline and, at six months after the final dose, the shedding rate remained at 40% below baseline. At 12 months, the viral shedding rate returned to baseline for this dose group. The reduction in the genital lesion rate after completion of the third dose was greatest for the 30µg dose group at 48%. After six months, the reduction from baseline in genital lesion rate for this dose group was 65% and, after 12 months, the genital lesion rate was 42% lower than baseline. GEN-003 was safe and well tolerated over the 12 months of this trial. We believe the six-month duration of reduced viral shedding and genital lesion rates may be clinically meaningful. If GEN-003 successfully completes clinical development and is approved, we believe it would represent a first-in-class vaccine for patients with HSV-2.
We are also developing a second T cell-simulating vaccine candidate, GEN-004, a potential universal Streptococcus pneumoniae, or pneumococcus, vaccine to protect against a leading cause of infectious disease mortality worldwide. In June 2014, we announced top line data from a Phase 1 clinical trial for GEN-004. This trial met its safety, tolerability and immunogenicity goals including measurable increases in the blood of T helper 17 (TH17) cells, a rare cell type that provides immunity at epithelial and mucosal surfaces. We plan to advance GEN-004 into a Phase 2a trial in the third quarter of 2014 to seek to demonstrate that GEN-004 can reduce colonization by pneumococcus in a human challenge model by mid-2015.
Vaccines represent a major healthcare success story, having eradicated or significantly reduced the global prevalence of many infectious diseases. To date, all approved vaccines have been developed primarily to elicit B cell responses. However, there remain many infections for which no effective vaccines or only partially effective vaccines exist. A major reason is that the organisms that cause these infections largely evade the antibody immune response generated by B cells, which can generally only address pathogens in the bloodstream. Such organisms may reside in host cells or mucosal surfaces of the nose and throat. To address these pathogens, vaccines targeting responses from the T cell arm of the immune system may present the solution.
We believe T cell target discovery has been particularly challenging for two reasons. First, the diversity of human T cell responses contrasts with the generally uniform B cell responses in humans. Second, the number of candidate targets for T cell responses can be exponentially greater than for B cell responses. These complexities represent fundamental barriers that traditional vaccine discovery tools, which rely largely on empirically selecting the potential targets from the proteins of a pathogen and iteratively testing them in animal models, have not been able to address.
51
We have designed the ATLAS platform to overcome these T cell target discovery challenges. We believe ATLAS represents the most comprehensive high throughput system for T cell vaccine and immunotherapy discovery in the biopharmaceutical industry. ATLAS is designed to mimic one important part of the human immune system in a laboratory setting. Using ATLAS, we are able to measure T cell responses to the entire set of protein targets for a specific pathogen in blood samples from large, genetically diverse populations, allowing us to identify vaccine and immunotherapy targets associated with protective T cell responses to disease. By comparing antigens identified in individuals who naturally control their infection with those who do not, we can select the antigens that may have the best likelihood of inducing protective T cell immune responses.
We have generated human proof-of-concept data for our lead product candidate, GEN-003, which we designed using ATLAS. GEN-003 is a therapeutic vaccine, or immunotherapy, candidate we are developing to treat people with HSV-2 infections. In our Phase 1/2a trial, which followed patients for 12 months after their third vaccine dose, we have generated human proof-of-concept data in patients with moderate-to-severe infections. Final analysis of the data showed that for the best performing 30µg dose group, there was a sustained reduction in the viral shedding rate. After completion of dosing for this dose group, the viral shedding rate fell by 52% versus baseline and, at six months after the final dose, the shedding rate remained at 40% below baseline. At 12 months, the viral shedding rate returned to baseline for this dose group. The reduction in the genital lesion rate after completion of the third dose was greatest for the 30µg dose group, at 48%. After six months, the reduction from baseline in genital lesion rate for this dose group was 65%, and after 12 months, the genital lesion rate was 42% lower than baseline. GEN-003 was safe and well tolerated over the 12 months of this trial.
We believe this represents the first time an immunotherapy has demonstrated activity against HSV-2 in humans. We also believe it represents the first time anti-viral efficacy has been observed for an immunotherapy designed primarily to elicit T cell responses to address an infectious pathogen for which T cell immunity is considered central to the control of the disease. Viral shedding is an important marker of the disease, indicating that the virus has been released to skin cells, potentially resulting in symptomatic outbreaks and/or to transmission through sexual contact. The genital lesion rate is a manifestation of the HSV-2 infection. We expect to initiate a Phase 2 trial in mid-2014 to optimize the dose of GEN-003 and potentially to improve upon either the magnitude and/or durability of the viral shedding and genital lesion rate reductions we have observed to date. This trial will study six combinations of protein and adjuvant doses and is designed around the 30µg per protein/50µg Matrix-M dose of GEN-003, which was the dose that drove the largest reductions in viral shedding and genital lesion rates in the Phase 1/2a trial.
Our second program derived from ATLAS is GEN-004, a universal pneumococcal T cell vaccine that we are developing to protect against all strains of pneumococcus, the most common cause of bacterial pneumonia in the world. We announced top line data in June 2014 from a Phase 1 clinical trial of GEN-004. This trial met its safety, tolerability and immunogenicity goals including measurable increases in the blood of T helper 17 (TH17) cells, a rare cell type that provides immunity at epithelial and mucosal surfaces. We plan to advance GEN-004 into a Phase 2a trial in the third quarter of 2014.
We believe we are a leader in the field of T cell vaccine and immunotherapy discovery and development. Our management and scientific teams possess considerable experience in vaccine, immunotherapy and anti-infective research, manufacturing, clinical development and regulatory matters. We have also assembled a team of leading advisors, led by George Siber, M.D., to guide the further development of our programs. Previously, Dr. Siber was the Chief Scientific Officer of Wyeth Vaccines, where he led the development of several first-in-class vaccines including the pneumococcal vaccine, Prevnar, the top selling vaccine in the world by value. He is also an inventor of Respigam and Cytogam, the first antibodies approved to protect against respiratory syncytial virus and cytomegalovirus, respectively. Dr. Siber is one of our directors and chairs our Scientific Advisory Board.
52
Since our inception and through June 30, 2014, we have received an aggregate of $158.0 million in gross proceeds from the issuance of equity securities and gross proceeds from debt facilities and an aggregate of $6.7 million in grant revenue. We estimate that we have spent approximately $45.0 million on research and development from 2011 through March 31, 2014.
Our Strategy
Our objective is to be the leading T cell vaccine company. Key components of our strategy are:
- •
- Continue to rapidly advance our lead vaccine candidate,
GEN-003. GEN-003 is a potential first-in-class therapeutic vaccine candidate we are developing to treat HSV-2 infections. We expect to
initiate a Phase 2 dose optimization trial in mid-2014, and plan to commence a further Phase 2 trial in mid-2015 to optimize the dosing regimen. We retain all rights to GEN-003 and plan
to advance this program through regulatory approval and commercialize this vaccine through a focused commercial effort in the United States. Outside the United States, we intend to evaluate
partnerships for GEN-003 opportunistically.
- •
- Advance GEN-004 into human proof-of-concept clinical
trials. Our second clinical-stage product candidate is GEN-004, a vaccine candidate designed to prevent infections caused by all strains
of pneumococcus. We have demonstrated proof-of-concept of GEN-004 in mice. We announced positive Phase 1 safety, tolerability and immunogenicity data for GEN-004 in June 2014. We plan to
commence a Phase 2a trial in the third quarter of 2014 to seek to demonstrate human proof-of-concept with GEN-004 by mid-2015. We believe this trial could provide the first evidence in humans
that a T cell vaccine, with potential to become a universal vaccine, can reduce colonization by pneumococcus. We retain all rights to this program, other than certain rights we have granted in
developing countries, and intend to opportunistically partner this program.
- •
- Advance our discovery stage and non-clinical novel vaccine
programs. We expect similarly to advance our novel non-clinical prophylactic vaccine programs against chlamydia, HSV-2 and malaria
through human proof of concept. We will seek partnerships opportunistically for late-stage development and commercialization of such programs. We will also continue to investigate, either alone or
through partnerships, the applicability of ATLAS to the development of cancer immunotherapies.
- •
- Utilize ATLAS, our vaccine discovery platform, to develop additional T cell vaccine candidates. We intend to continue to use ATLAS to discover and advance novel T cell vaccines. Since we begin our vaccine candidate discovery process by profiling human populations exposed to a pathogen, and use these subjects' own cells to comprehensively screen the entire proteome of the pathogens, we believe we have a better chance of identifying vaccines likely to protect against pathogens of interest. We intend to opportunistically expand our pipeline using ATLAS to discover T cell vaccines against pathogens for which B cell vaccines are ineffective or non-existent.
Vaccine Overview
Vaccines represent a major healthcare success story. They have eradicated smallpox and dramatically reduced the mortality and morbidity associated with many other infectious diseases, such as diphtheria, measles, polio and tetanus. Today, there are vaccines approved to treat and protect against approximately 30 infectious diseases. Total global vaccine revenues in 2012 were $27 billion.
Vaccines trace their roots to the smallpox vaccine, first tested in 1796 by Edward Jenner. Dr. Jenner demonstrated that he could protect subjects against smallpox by inoculating them with cow pox, a similar virus. More than 200 years later, the concept of a vaccine remains the same: training the immune system to respond to an infectious pathogen by exposing it to that pathogen, or a component
53
of that pathogen, in a controlled way. Most vaccines are prophylactic, preventing an invading organism from causing disease. A vaccine can also be therapeutic, fighting an existing infection.
How Vaccines and Immunotherapies Work
Vaccines rely on an ability of the human immune system called adaptive immunity to "remember" an invading organism and develop an immune response to it. When confronted with a new organism, the immune system first seeks to eliminate the pathogen through an initial response of the so-called "innate immune system" and then generates immunological memory, or adaptive immunity, in which the immune system recognizes and "remembers" the invasive pathogen in order to combat it in the future. A vaccine introduces a pathogen or a specific portion of a pathogen to the adaptive immune system in a controlled manner in order to invoke acquired immunity against the specific pathogen it is designed to address.
The adaptive immune system consists of two main components: the B cell arm, and the T cell arm. B cells and T cells are types of white blood cells, or lymphocytes. To date, vaccines have been thought to work primarily by harnessing the B cell arm of the adaptive immune system. The main function of B cells is to produce antibodies, a special type of protein that identifies and initiates processes to kill foreign organisms. Antibodies bind to one or more structures on the pathogen surface. These structures may be proteins or complex sugars, called polysaccharides, or other molecules, which are specific to the organism. Some B cells turn into so-called memory B cells following exposure to an organism, ensuring that the immune system will recognize the same pathogen in the future.
Immunotherapies are designed to augment or boost the immune system to allow it to better protect the body against disease. In the case of infectious disease, currently approved immunotherapies aim to treat an infection rather than prevent it. A well known example of an early immunotherapy is the use of interferon-alfa 2a to treat infections caused by hepatitis C virus, or HCV. Immunotherapy approaches against cancer have also been developed, with limited success. As with vaccines, we believe immunotherapies that engage the T cell immune system may represent an optimal solution to treat, and potentially prevent, disease.
Current Target Discovery
Vaccines available today have been developed to stimulate the production of antibodies and therefore protect against invading organisms that are primarily controlled by the B cell arm of the immune system. This type of immunity is effective against organisms that mediate disease in locations, primarily the bloodstream, that are accessible to antibodies and/or cells that kill organisms with the help of antibodies.
Scientists have employed two alternative approaches for designing vaccines to induce antibody responses. The first approach has been to present a modified version of the whole pathogen to the immune system. In this approach, the vaccine is either an inactivated, or killed, pathogen or an attenuated pathogen, where the pathogen is live but rendered far less infectious. The advantage of this approach is that it enables vaccine development without knowing the specific surface structure of the pathogen that antibodies target for response and immunological memory. There are also significant disadvantages to this approach. Inactivating or attenuating pathogens in a large-scale, reproducible way is challenging, and there is a concern that attenuated pathogens could reactivate and cause the diseases they were designed to prevent. Another limitation is the potential that side effects of the vaccine may be more severe than when only part of the organism is used as the vaccine. A recent example is the pertussis, or whooping cough, vaccine that was originally developed as a whole killed vaccine, but later changed to a subunit, or purified protein, vaccine because of the rare but severe side effects of the whole cell vaccine. Due to these challenges, and the resultant regulatory hurdles, vaccines are increasingly designed using a second, and more targeted, approach.
54
The second approach to design vaccines to induce an antibody response is to immunize with specific antigens, or immunogenic proteins, from the pathogen. Such antigens often are paired with either (1) an adjuvant that gives the immune system a "danger signal" to enhance the ability of the immune system to recognize the proteins as foreign substances or (2) a vector, such as a virus that is used to deliver the antigens to the immune system to enhance the response, or some combination of an adjuvant and vector are utilized. These so-called subunit, or purified protein, vaccines, while generally easier to produce than whole pathogen vaccines, pose a different challenge: selecting the optimal antigen or antigens from the pathogen to elicit the desired immune response.
Modern vaccine antigen discovery largely consists of the search for the optimal antigens for immunological, and primarily B cell, responses. To date, this process has largely been empirical, meaning that it has required the testing of each potential antigen in animal models of disease to determine its ability to be recognized by the immune system. There is considerable time and cost associated with testing each antigen, singly and in various combinations, to determine which antigens can elicit the desired immune response. However, these hurdles have been somewhat mitigated by the fact that, for most pathogens currently addressed by vaccines, there is a small number of candidate antigens.
Limitations and Challenges of Current Target Discovery
Despite more than 200 years of vaccine history, there remain many organisms for which effective or comprehensive vaccines do not exist. These include viruses such as HSV-2, cytomegalovirus, and Epstein-Barr virus, which causes mononucleosis, and bacteria that include pneumococcus, Chlamydia trachomatis, or chlamydia, and Staphylococcus aureus, or staphylococcus, which causes a wide range of soft tissue, organ and blood infections. Parasites such as Plasmodium falciparum, which causes much of the world's malaria, also have yet to be addressed with vaccines. Collectively, these organisms are responsible for millions of deaths and morbidity for millions more people annually.
Vaccines that elicit B cell responses generally do not work for these pathogens, in part because the organisms evade B cell-mediated immunity. Some pathogens, such as HSV-2 and chlamydia, spend most of their life cycles sequestered within host cells and are inaccessible to antibodies that primarily reside in the bloodstream. Mucosal surfaces of the nasopharynx (nose and throat), gastrointestinal tract and genitalia, are also less accessible to antibodies in the bloodstream and harbor pathogens such as pneumococcus and staphylococcus. To address these pathogens, vaccines that engage the T cell immune system may represent the optimal solution.
T cells, like B cells, are a type of white blood cell, of the immune system. They are generally classified as CD8+ cytotoxic T lymphocytes, or CTL, or killer T cells, and CD4+, or helper T cells. Killer T cells recognize and eliminate pathogen-infected host cells. On the other hand, helper T cells produce compounds called cytokines that stimulate other immune cells to help fight infection. To initiate T cell responses to an infection, another type of specialized white blood cell, called antigen-presenting cells, or APCs, engulf invading pathogens. APCs process pathogen-derived protein antigens into smaller pieces, or epitopes, and place them on their surface as epitopes for recognition by killer T cells or helper T cells. Upon recognition, T cells activate to help eliminate the infection. Activated T cells can also become long-lived memory T cells that respond to infection should the host contact the infectious agent again, thus providing long-term protective immunity.
As with B cell vaccine development, there are two potential approaches to developing vaccines that induce T cell immune responses. The first approach would be to develop an attenuated or inactivated pathogen vaccine. As discussed, such a vaccine may present significant manufacturing, safety and regulatory challenges. To date, no whole pathogen vaccine has been developed to induce T cell responses.
55
The second potential approach would be to develop a subunit vaccine. However, there have been relatively few advances toward identifying target antigens that will elicit T cell responses, and, without the right antigen or antigens, a vaccine will not elicit the optimal immune response.
Discovering T cell antigens is particularly challenging due to the human diversity of T cell response and to pathogen size. Humans can belong to one of nine different genetic supertypes that influence how epitopes are presented to T cells, and hence the set of proteins that make up a pathogen can range into the thousands. These challenges represent fundamental barriers to the development of vaccines against infectious organisms for which T cell immunity is critical for effective control.
Challenge #1: Diversity of human T cell responses. B cell responses to a particular antigen are generally more uniform across all humans than T cell responses. As a result, a vaccine designed to elicit a B cell response generally works across broad populations. However, the T cell arm of the immune system poses a complexity challenge. In contrast with a fairly uniform antibody response, each person has one of nine human leukocyte antigen, or HLA, supertypes that govern, among other things, the specific targets of T cell responses. A person belonging to one supertype may mount a T cell response to a different protein epitope—or an entirely different protein—than someone with a different supertype. Given these different HLA supertypes, modeling diseases in animals, which are typically bred from a single genetic lineage, cannot effectively account for or produce a vaccine candidate intended to address the human diversity in T cell responses.
Challenge #2: Complexity of target selection due to pathogen size. Antibodies produced by B cells typically target proteins on a pathogen's surface. For B cell vaccines targeting surface proteins, the number of potential targets has typically been limited. For example, the hepatitis B virus, addressable by two approved vaccines, consists of four proteins. Choosing the vaccine antigen from this small candidate list required testing only these four proteins, singly and in combination to find the most protective formulation. Here again, the T cell arm of the immune system works differently. It is not just surface proteins of a pathogen that can be targets for a vaccine, but rather every pathogen protein, collectively its full "proteome", can be a target of T cell responses. The number of candidate antigens, therefore, increases substantially based on the genetic complexity of the pathogen. For example, for HSV-2 the proteome comprises nearly 80 proteins, substantially increasing the complexity associated with target antigen selection, as the number of potential antigen combinations increases exponentially. The chlamydia proteome exceeds 900 proteins and the proteome for Plasmodium falciparum, a parasite that causes malaria, exceeds 5,000 proteins. In the case of such organisms, testing each protein in animals, singly and in various combinations to identify candidate antigens, could take many years. For many organisms, the complexities associated with the pathogen size have presented a fundamental barrier to discovering effective T cell vaccines.
The combination of these two challenges renders discovery of T cell antigens by traditional empirical methods exceedingly difficult. We believe these challenges explain why no approved vaccines have been developed on the basis of T cell responses.
The ATLAS Discovery Platform: A Novel Approach to Vaccine and Immunotherapy Discovery
We have developed a proprietary technology platform that is designed to overcome the challenges associated with developing vaccines that stimulate T cell immunity. We have engineered this technology into a high throughput discovery platform we call ATLAS, our AnTigen Lead Acquisition System. This system mimics part of the human T cell immune system ex vivo, or outside the body. By comparing antigens identified in individuals who naturally control their infection with those who do not, we can select the antigens that may have the best likelihood of inducing protective T cell immune responses. We believe that this enables ATLAS to rapidly identify targets of T cell responses that are applicable to broad populations, over the range of HLA supertypes and represents a comprehensive throughput system designed for T cell antigen discovery in the biopharmaceutical industry.
56
To use ATLAS, we collect T cells and APCs from hundreds of human donors who were naturally exposed to the disease-causing pathogen of interest. We segregate these donors into cohorts based on their clinical status. At one end of the spectrum are those exposed subjects who remained uninfected despite contact. At the other end of the spectrum, we include subjects who were unable to clear their infection or control their disease without significant intervention. If applicable, we also include subject cohorts between these ends of the spectrum, such as those with mild infections.
We also create a library of every protein in the proteome of the pathogen of interest. We express each individual protein in bacterial hosts, which are cultured with APCs from each human donor. As each donor's APCs ingest the complete proteomic library, they present peptide epitopes from each protein on their surface. These epitopes can be recognized by T cells derived from the same donor. If the T cell recognizes the epitope on the surface of the APC, which it will do if has seen the epitope before and is a memory T cell for that particular epitope, it will be activated. The level of activation can be quantified by the amount of interferon gamma, or IFN-g, a cytokine produced by the T cell. We use the pattern of responses for each subject to infer which pathogen proteins are associated with productive, non-productive or even deleterious immune responses. The diagram below illustrates the process by which we use ATLAS to identify pathogens to elicit a T cell response.
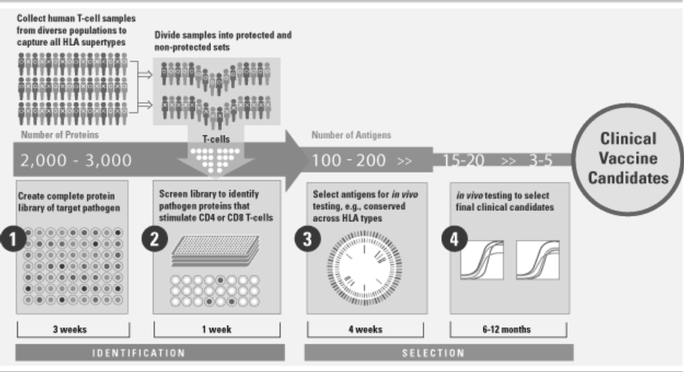
We use ATLAS as a high throughput engine to comprehensively and rapidly screen human T cells to identify potentially relevant T cell vaccine antigens. Furthermore, ATLAS allows us to screen large proteomes in an efficient manner to identify antigens likely to best stimulate the T cell immune system, a process that is otherwise slow and labor intensive. By comparing antigens identified in individuals who control their infection with those who do not, we can select the antigens that may have the best likelihood of inducing protective immune responses. Since we discover the target antigens from human responses rather than animal responses, we believe we can use the targets to produce vaccine and immunotherapy candidates that have a high probability of generating protective immunity in humans. To date, we have applied this platform to identify human T cell antigens from several viral and microbial proteomes, with sizes ranging from several dozen, as with HSV-2, to a few thousand expressed proteins, as with pneumococcus and chlamydia.
57
In summary, we believe that ATLAS offers all of the following important advantages over other approaches to vaccine design and discovery:
- •
- Enables vaccine and immunotherapy discovery for pathogens that are
generally inaccessible to antibodies. For pathogens that reside in human cells or otherwise generally evade antibody responses, and
which, as a result have not successfully been addressed by B cell vaccines, ATLAS represents a means to identify targets of effective T cell responses. This pathogen list includes dozens of bacteria,
viruses and parasites that collectively account for millions of deaths and morbidity for millions more annually.
- •
- Decreases the risk of vaccine and immunotherapy discovery failure by
identifying targets of T cell responses in humans. By comprehensively screening the T cell responses of persons who have mounted
effective immune responses to infectious disease pathogens, and comparing these responses to those who have not, ATLAS identifies antigens that associate with protection in humans. By identifying the
targets of human T cell responses ex vivo from human samples, rather than in animal models, we both account for diversity of human T cell responses and
avoid being misled by discovery in animals.
- •
- Selects targets relevant to broad
populations. We believe ATLAS is highly efficient and can analyze T cells from a large number of individuals. Traditional analog vaccine
antigen discovery necessarily focuses on the identification of epitopes that are able to be presented by APCs for only a minority of the target population. In contrast, we can process blood samples
from hundreds of ethnically diverse subjects and therefore can ensure, from analyzing across the range of HLA supertypes, that our antigens are broadly relevant. As a result, we anticipate that both
GEN-003 and GEN-004 will stimulate T cell responses across broad HLA types.
- •
- Reduces the time and cost of vaccine discovery. As we have demonstrated in both our HSV-2 and pneumococcus programs, after we collect blood samples from human cohorts exposed to a pathogen, we believe we can identify vaccine candidates in less than one year and for a few million dollars, compared to the industry norms of up to 10 years and $100 million to discover B cell vaccines, according to GlaxoSmithKline.
We believe that our discovery platform can enable vaccine and immunotherapy discovery for a wide range of infectious disease pathogens, in addition to our clinical stage vaccine and immunotherapy. We have identified antigens that appear to associate with protective human responses in our prophylactic HSV-2 and chlamydia programs and demonstrated subsequently that these antigens can protect against disease in accepted animal models. We have also embarked upon a program to discover protective T cell antigens from Plasmodium falciparum, a causative agent of malaria under a program funded in part by an investment from the Bill & Melinda Gates Foundation, or the Gates Foundation. Many other pathogens evade antibody responses and therefore may be tractable to ATLAS, including those that cause tuberculosis, gonorrhea, and dengue fever.
We also believe ATLAS may offer utility in the discovery of new treatments for cancer. In recent years, new cancer immunotherapies such as Yervoy (ipilimumab; Bristol-Myers Squibb) have successfully delivered improved outcomes against cancers such as melanoma by reversing the inhibitive effect that cancer cells can have on T cell immune responses. Recruiting T cells to drive the containment of cancerous cells holds promise as a new approach to cancer treatment.
Knowing the target or targets of the T cell responses may enable the development of next-generation immunotherapies with greater specificity that, in theory, could offer further protection against cancer.
In March 2014, we announced a joint research collaboration with the Dana-Farber Cancer Institute and Harvard Medical School to characterize anti-tumor T cell responses in melanoma patients. This
58
collaboration extends the use of our proprietary ATLAS platform for the rapid discovery of T cell antigens to cancer immunotherapy approaches.
Antigen Discovery Using ATLAS—A Vignette from the Discovery of GEN-003 to Treat HSV-2
Strong evidence for the role of T cells in controlling an HSV-2 infection emerged when a researcher at the University of Washington, Christine Posavad, Ph.D., identified a previously unknown and relevant patient population. These people were each in a sexual relationship with someone that had an HSV-2 infection, but had no evidence of infection by culture of, or measurable antibody response to, HSV-2. However, these individuals had evidence of T cell memory against HSV-2, indicating previous contact with HSV-2. In these patients, Dr. Posavad concluded that T cells are the driver of the protective response, but she could not comprehensively screen for the specificity of T cells that drove this response.
Based in part on Dr. Posavad's observations and other emerging evidence of the role of T cells in controlling HSV-2 infection, we decided to use ATLAS to identify T cell stimulating antigens for HSV-2. We started by collecting blood from 195 people exposed to, or infected with, HSV-2. For each person, we documented the infection severity based on clinical records and assigned the subjects to a cohort according to this. Crucially, we included 43 subjects of the type identified by Dr. Posavad. We chose our sample size to enable statistical comparisons within and across cohorts. We also recruited genetically and ethnically diverse individuals to ensure broad HLA supertype coverage. The table below provides further details on the patients:
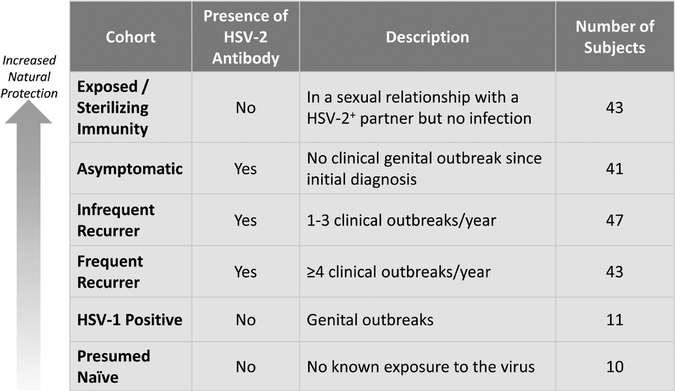
We also built two copies of a library consisting of each protein in the HSV-2 proteome. Since both killer and helper T cells are thought likely to play a role in controlling an HSV-2 infection, we believed that measuring both T cell responses would be necessary to optimize the design of a candidate vaccine. Research has shown that one cytokine T cells use to defend against HSV-2 is IFN-g. Therefore, for each subject in the study, we separately measured the IFN-g responses of helper T cell and killer T cells to each HSV-2 protein. An example of the output from our assay measuring killer T cells for one subject is below. We generate similar assays for all subjects for both killer and helper T cells.
59
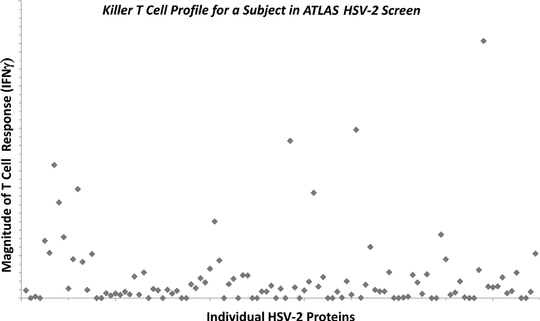
ATLAS enables the generation of the above outputs. In this particular subject, the responses to many proteins hovered at a low level, while several proteins elicited relatively strong T cell responses.
Analyzing the experimental results of the 195 ethnically diverse subjects has enabled us to associate T cell responses to individual proteins with better control or improved outcomes of HSV-2 infection. Using statistical analyses to identify commonalities and differences within the clinical cohorts and across them, we identified a small group of candidate antigens associated with protective T cell responses to HSV-2 in humans. We further produced and tested the selected antigens in animal models to arrive at the two proteins to be included in GEN-003. We believe that because we collected samples from ethnically diverse subjects, GEN-003 should work across patients regardless of HLA supertype. The entire process, including devising clinical cohorts, collecting the blood from 195 subjects, building two copies of the protein library, running proteins through ATLAS and determining priority candidate antigens took 15 months.
Our Product Candidate Pipeline
The following table describes our current development programs:
Vaccine Candidate
|
Program | Stage of Development |
Next Milestone | Anticipated Timeline |
||||
|---|---|---|---|---|---|---|---|---|
GEN-003 |
HSV-2 Therapeutic | Phase 2 | Complete Phase 2 dose optimization trial | Mid-2015 | ||||
GEN-004 |
Pneumococcus Prophylaxis | Phase 2a | Complete Phase 2a trial | Mid-2015 | ||||
GEN-001 |
Chlamydia Prophylaxis | Pre-clinical | File IND | 2017 | ||||
GEN-002 |
HSV-2 Prophylaxis | Pre-clinical | File IND | 2017 | ||||
GEN-005 |
Malaria Prophylaxis | Research | Initiate pre-clinical studies | Second half of 2015 |
GEN-003 Market Opportunity
Herpes Simplex Virus—2 (HSV-2)
We are developing our lead product candidate, GEN-003, to treat patients with HSV-2 infections. GEN-003 consists of two protein antigens. The first antigen is ICP4.2, a large fragment of the protein ICP4 that we discovered in ATLAS screens to be a T cell antigen associated with protection from infection or with less severe infection. The second antigen is glycoprotein D2, or gD2, a B cell antigen
60
that is the target of antibodies that provide anti-viral activity during the time in the life cycle of HSV-2 where the pathogen is susceptible to inactivation by antibodies. gD2 was also a target of T cells in our ATLAS screens and was selected based on such ATLAS screens as ATLAS prioritized gD2 as the B cell antigen most associated with T cell responses. We pair the antigens with Matrix-M2, a novel adjuvant that we have licensed exclusively for this indication from Novavax, Inc., or Novavax. See "—Other Collaborations—Isconova AB".
HSV-2 is a sexually transmitted disease. HSV-2 infections have become an epidemic, spreading to approximately 16% of the United States population between the ages of 14 and 49, and more than 500 million people worldwide, according to the Centers for Disease Control and Prevention, or CDC, and the World Health Organization, or WHO.
For infected individuals, the disease can manifest in a number of ways, with so-called viral shedding as the common element. For some of the virus' life cycle, it lies dormant within nerve cells near the spine. Although there may be no visible sign of infection, the virus lives within these nerve cells. Periodically, the virus reactivates and virus travels to skin cells of the genitalia where they are released. The release of the virus is called viral shedding and can be detected by swabbing the genital area and testing the swab for the presence of viral DNA. For reasons not completely understood, reactivation of the virus within the nerve cells may occur, resulting in a large amount of virus shedding from skin and mucus membranes. If the replication is maintained for a long enough period of time and at a high enough level, the virus destroys the cells it inhabits and causes ulcers to form on the skin. Patients experiencing such visible ulcers are considered symptomatic patients. It is generally believed that the immune system responds to episodes of HSV-2 outbreaks by activating T cells that reduce viral replication and destroy infected cells, allowing healing and resolution of genital ulcers, usually after a few days, although for many patients ulcers return at variable intervals. Patients may also experience periodic, low-frequency viral shedding. Because the shedding at these times does not lead to the development of ulcers, these episodes are called asymptomatic shedding. These asymptomatic patients continue to pose a disease transmission risk through sexual contact while shedding virus.
Some people, approximately 60% of those infected, are asymptomatic or fail to recognize or seek medical attention for an initial mild outbreak of ulcers. According to the New England Journal of Medicine, roughly 40% of persons infected with HSV-2 experience visible symptoms. It has been reported in the Annals of Internal Medicine that approximately 70% of the people with visible symptoms experience three or more outbreaks per year, which we consider to be moderate-to-severe disease. Patients with HSV-2 experience significant distress because of the potential negative impact on their ability to form and maintain sexual relationships. Infection with HSV-2 can involve substantial risks in addition to the infection itself. For example, persons with HSV-2 infection have a threefold increased risk for human immunodeficiency virus, or HIV, acquisition. Additionally, pregnant women can transmit HSV-2 to infants in childbirth, which can result in severe brain damage or death.
61
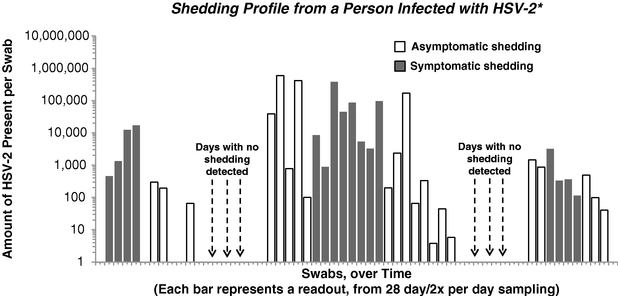
- *
- Note: Each bar represents 1 swab; 2 swabs collected per day; the absence of a bar means no shedding was detected on the swab on a particular day.
The total number of days during a month that HSV-2 virus can be detected in the genital area with or without visible ulcers is called the shedding frequency. A pattern of shedding and outbreak for one person is illustrated in the graph above. Viral shedding is measured by collecting swabs of the genital area, following a protocol that has been used in decades of studies of HSV-2 viral shedding. In the example shown above, the subject collected swabs twice daily for 28 days. HSV-2 DNA was detectable in approximately 66% of the collected swabs, meaning the patient's shedding frequency is 66% for the period measured. Some swabs had no detectable viral DNA, meaning the subject did not shed virus at the time of sample collection (exemplified by the blank areas of the above graph). The magnitude of viral shedding varied widely from day to day and only sometimes resulted in clinical symptoms such as visible genital ulcers. Ulcers generally appear after several days of asymptomatic shedding and at times when the magnitude of shedding is highest. The extent, frequency, and duration of shedding vary from person to person, but the pattern is relatively consistent for each person.
Limitations of Current HSV-2 Treatment Options
There is no known cure for HSV-2. For patients infected with HSV-2, oral antiviral drugs are the only treatment option. The most commonly prescribed treatment is valacyclovir including Valtrex, marketed by GlaxoSmithKline. Other medications available are acyclovir (Zovirax, marketed by GlaxoSmithKline) and famciclovir (Famvir, marketed by Novartis). These drugs all work by limiting the ability of the virus to replicate when it emerges from latency. Sales for these oral antivirals totaled $1.6 billion globally in 2012, including nearly $700 million in the United States, according to IMS Health.
Some patients treat their disease episodically. At the onset of outbreaks, or in the case of some patients, at the onset of prodrome, a tingling sensation that may precede an outbreak, patients take antiviral medication to reduce the duration and severity of the outbreak. According to the approved Valtrex prescribing information, episodic treatment only reduces the duration of outbreaks by up to 50% when compared to placebo. Patients treating their symptoms episodically are not protected against asymptomatic viral shedding and, therefore, have no reduced risk of transmission of infection to an uninfected sexual partner while asymptomatic.
Some patients treat their infection with daily antiviral medication. This approach is called chronic suppressive therapy, and has been shown to reduce—but not eliminate—viral shedding, the frequency
62
of symptomatic outbreaks of genital ulcers, and the risk of transmission of the infection to an uninfected sexual partner. Even on chronic suppressive therapy, based on the valacyclovir prescribing information, 35% of patients taking chronic suppressive therapy suffer outbreaks within six months after initiation of treatment and 46% of patients suffer outbreaks within 12 months. Patients taking chronic suppressive therapy reduce their disease transmission risk only by as much as 52%.
Due to the limited effectiveness of oral antiviral therapy, there remains a significant unmet medical need, against both the symptoms of HSV-2 and disease transmission risk from viral shedding.
GEN-003: An Immunotherapy Candidate for HSV- 2
We have shown that GEN-003 is the first immunotherapy known to have demonstrated a statistically significant reduction in viral shedding rate and the signs of clinical genital herpes disease as measured by genital lesion rates. See "—Clinical Development—GEN-003-001—Our Phase 1/2a Clinical Trial". Moreover, we have shown that the effect on the genital lesion rate persists for 12 months after administration. The initial, immediate-post-doing data were presented in a late-breaker presentation at the Interscience Conference on Antimicrobial Agents and Chemotherapy in September 2013. We believe that these initial clinical results demonstrate that GEN-003 has the potential to be a first-in-class immunotherapy to treat HSV-2.
We believe that, if approved for the treatment of HSV-2 infections, GEN-003 could address the unmet needs of patients in several ways. For patients taking episodic therapy, GEN-003 could offer reduced symptomatic and asymptomatic viral shedding, potentially reducing disease transmission risk. Since episodic therapy offers no protection against disease transmission during asymptomatic shedding, these patients and their sexual partners are unprotected when the infected partner is not taking anti-viral medication.
For patients on chronic suppressive therapy, we believe GEN-003 may provide both improved outcomes and increased convenience. For some patients, we anticipate that physicians will prescribe GEN-003 as baseline therapy. Such patients may still take oral antivirals in case of an outbreak to further control symptoms. Replacing daily therapy may offer convenience to these patients. For other patients, we anticipate that physicians may prescribe GEN-003 alongside chronic suppressive therapy. This combination therapy approach mirrors the treatment practice of other chronic viral infections such as HIV and hepatitis C virus. We anticipate that, since the mechanisms of action for GEN-003 and oral antiviral medication should complement each other, the control against symptoms and disease transmission risk offered by the combination would exceed that of either therapy alone. In a market research survey conducted on our behalf with more than 400 patients with HSV-2 infections in the United States, the United Kingdom, France and Germany, and more than 300 physicians who treat patients with HSV-2 infections, 56% of patients on chronic suppressive therapy indicated an intent to use GEN-003 in combination with other therapies and 37% of such patients indicated an intent to use GEN-003 on its own, if it were approved; 30% of patients on episodic therapy indicated an intent to use GEN-003 in combination with other therapies and 60% of such patients indicated an intent to use GEN-003 on its own, if it were approved; and 15% of patients not taking any HSV-2 therapy indicated an intent to use GEN-003 in combination with other therapies and 65% of such patients indicated an intent to use GEN-003 on its own, if it were approved. This was a limited survey and may or may not be representative of how patients might ultimately use GEN-003, if at all, if GEN-003 successfully completes clinical development and is approved by regulatory authorities.
Non-clinical Evaluation of Our GEN-003 Product Candidate
We tested GEN-003 in the guinea pig therapy model, the standard animal model of recurrent disease. Guinea pigs are used because the course of infection in the animal closely mirrors that of humans, with an initial outbreak that resolves, followed by frequent and periodic recurrences that last a few days. GEN-003 decreased ulcers over time by up to 55% versus placebo, measured over 63 days
63
after initial immunization. This is the standard interval across which to measure impact on ulcers in the guinea pig model. Additionally, the vaccine reduced viral shedding significantly. In the period after completing immunization, from days 37-63, GEN-003 almost completely eliminated viral shedding. We are unaware of any other vaccine demonstrating similar impact either on clinical symptoms or on viral shedding in this model.
Clinical Development
GEN-003-001—Our Phase 1/2a Clinical Trial
We have completed a Phase 1/2a trial, testing the safety, T and B cell immunogenicity, and impact on viral shedding of GEN-003 in subjects with documented recurrent HSV-2 infection. We also measured, as an exploratory endpoint, the effect of GEN-003 on the genital lesion rate. The trial was conducted at seven sites in the United States, including some of the leading institutions for scientific and clinical research of HSV-2. The trial was double-blind, placebo-controlled and dose-escalating. We enrolled subjects between 18 and 50 years of age. An independent Data Safety Monitoring Board monitored the safety of subjects enrolled in the clinical trial.
This trial enrolled 143 otherwise healthy subjects with a history of three to nine genital herpes outbreaks per year when not on suppressive therapy. Subjects were randomized into one of three dose cohorts. Within each cohort, subjects were randomized in a 3:1:1 ratio, whereby for every three subjects receiving GEN-003, one would receive placebo and one would receive the ICP4.2 and gD2 proteins without the Matrix-M2 adjuvant. We included this last cohort to demonstrate that Matrix-M2 was necessary to achieve the desired biological responses. There were three vaccine dose groups, based on the amount of protein. The lowest dose group subjects received 10µg of each protein; in the middle dose group, the protein doses increased to 30µg, and in the high dose group the protein dose was 100µg. For all subjects receiving GEN-003 (proteins plus adjuvant), the Matrix M2 dose was 50µg. Subjects received three vaccinations, on days zero, 21 and 42. The diagram below illustrates the dosing and swabbing regimen in the trial and the points in time at which data was gathered.
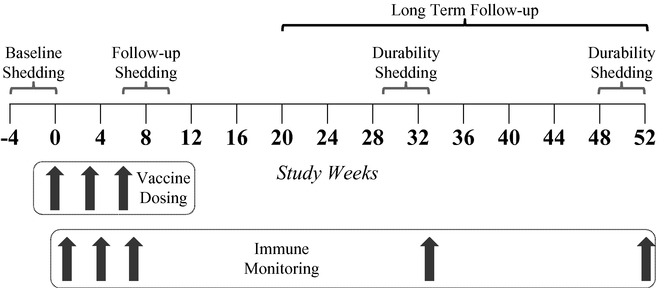
The primary objective of this trial was to monitor the safety profile of the proposed vaccine. Overall, GEN-003 was well-tolerated. During the seven days following each injection, side-effects were generally those considered typically associated with vaccines, such as fatigue, site injection pain, tenderness and swelling. Among all vaccine dose groups, the frequency of adverse events, or AEs, appeared greater among those subjects given the 10µg dose. In the 30µg and 100µg dose cohorts, the AE rate was lower than that of the 10µg cohort. In addition, the frequency of AEs appeared to
64
diminish with subsequent doses. Beyond the week following vaccination with the GEN-003 immunotherapy, the AE types and frequencies appeared similar to those following vaccination with placebo. The AEs have been transient, resolved over a few days and resulted in only two subjects discontinuing further vaccinations: one for a combination of symptoms (myalgia and fatigue; and pain and tenderness at the injection site) and one for injection site pain.
Additionally, we measured the immunotherapy-induced T cell and B cell immune responses. We structured and statistically powered the trial to measure the proposed immunotherapy's impact on the viral shedding rate, an important marker of virus activity. We selected this endpoint because of the connection between shedding, symptomatic outbreaks, and risk of transmission of virus by sexual contact. Every subject in the study swabbed their genitalia twice per day for 28 days before receiving the first assigned treatment injection, and after treatment, using the standard protocol that has been used for many clinical trials of HSV-2 shedding.
We measured immunotherapy activity in two ways: the impact on viral shedding and the impact on signs of clinical genital herpes disease as measured by genital lesion rates, defined as the total days in which a patient reported the presence of a visible genital lesion during swabbing days, divided by the total number of swabbing days. The impact on viral shedding was determined by viral DNA present in swabs from subjects over the 28-day measurement period before receiving the assigned treatment and immediately after completing the three-dose regimen and again at six months and 12 months after the final dose. The genital lesion rate was measured at the same time points.
Final analysis of the data showed that for the best performing 30µg dose group, there was a sustained reduction in the viral shedding rate. After completion of dosing for this dose group, the viral shedding rate fell by 52% versus baseline and, at six months after the final dose, the shedding rate remained at 40% below baseline. At 12 months, the viral shedding rate returned to baseline for this dose group. The reduction in the genital lesion rate after completion of the third dose was greatest for the 30µg dose group, at 48%. After six months, the reduction from baseline in genital lesion rate for this dose group was 65% and after 12 months the genital lesion rate was 42% lower than baseline.
The predefined statistical analysis methodology that was used for this trial was consistent with methodologies previously applied by others to all data sets from trials of therapeutic interventions against HSV-2 that we are aware of as published in major medical journals. This analysis showed that the reductions observed in viral shedding and genital lesion rates for the 30µg dose group after the third vaccine dose and after six months were statistically significant. Lower patient numbers at the 12 month analysis point meant that statistical analysis was not possible. The suitability of this statistical method for the analysis of HSV-2 trial data is currently under review.
The following tables summarize the data demonstrating the reduction in the frequency of viral shedding and the genital lesion rate observed following administration of GEN-003 vaccine.
Viral Shedding Frequency
| |
After Dose 3 | After 6 months | After 12 months | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Treatment Group
|
# of Subjects |
Change from Baseline |
# of Subjects |
Change from Baseline |
# of Subjects |
Change from Baseline |
|||||||||||||
Placebo |
26 | 3 | % | 23 | 34 | % | 13 | -1 | % | ||||||||||
Proteins only |
26 | 35 | % | 22 | 16 | % | 15 | 95 | % | ||||||||||
GEN-003 (10 µg) |
27 |
0 |
% |
26 |
60 |
% |
0 |
— |
|||||||||||
GEN-003 (30 µg) |
27 | -52% | * | 19 | -40% | * | 20 | -8 | % | ||||||||||
GEN-003 (100 µg) |
26 | -31% | * | 24 | -17 | % | 20 | -26 | % | ||||||||||
65
Genital Lesion Rate
| |
After Dose 3 | After 6 months | After 12 months | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Treatment Group
|
# of Subjects |
Change from Baseline |
# of Subjects |
Change from Baseline |
# of Subjects |
Change from Baseline |
|||||||||||||
Placebo |
26 | 26 | % | 23 | 28 | % | 13 | -44 | % | ||||||||||
Proteins only |
26 | -29 | % | 22 | -29 | % | 15 | -73 | % | ||||||||||
GEN-003 (10 µg) |
27 |
-39 |
% |
26 |
-24 |
% |
0 |
— |
|||||||||||
GEN-003 (30 µg) |
27 | -48% | * | 19 | -65% | * | 20 | -42 | % | ||||||||||
GEN-003 (100 µg) |
26 | -46% | * | 24 | -32 | % | 20 | -15 | % | ||||||||||
Note: *statistically significant change from baseline p<0.05
We are unaware of any other vaccine that has demonstrated the effects that we observed of GEN-003 on HSV-2 genital lesion rate or viral shedding in humans. While we have not yet tested any booster regimen, based on the durability of response to date, we anticipate booster doses, if necessary, would be administered at intervals of six months or more. Based on the market research conducted on our behalf, a product profile consistent with the data from the GEN-003 30µg dose group with a booster dose at six months was highly attractive to patient survey participants. This was a limited survey and may or may not be representative of how patients might ultimately or desire to use GEN-003, if at all, if GEN-003 successfully completes clinical development and is approved by regulatory authorities.
Our data have also demonstrated that GEN-003 induced a broad immune response in vaccinated subjects at all dose levels. T cell responses increased from baseline 21-fold to ICP4.2 and 10-fold to gD2. Subjects also experienced strong increases in antibody response to ICP4.2 and gD2, as measured by immunoglobulin G, or IgG, a standard measure of antibody response. The antibodies generated in response to the vaccine are able to prevent the virus from infecting new cells, as measured by a standard assay for evaluating the ability of the virus to infect cells in vitro. The chart below shows the T cell immune response aggregated across all dose levels.
Fold Increase in T Cell Response from Baseline by Treatment Group
66
Next Steps: Phase 2 Dose Optimization and Dose Regimen Trials for GEN-003
We expect to initiate a Phase 2 dose ranging trial of GEN-003 in mid-2014 and anticipate data by mid-2015. The primary trial objective is to optimize the vaccine dose. We are comparing two protein levels, a 30µg and a 60µg per protein dose, each combined with 25µg, 50µg or 75µg of Matrix-M2, for a total of six dose cohorts. We anticipate the trial to enroll approximately 300 patients in total with similar or identical enrollment criteria and endpoints as the Phase 1/2a trial. Following completion of this Phase 2 dose ranging trial, we intend to complete a Phase 2 dose regimen trial where we will seek to optimize our dosing regimen, or the number of doses and the interval between doses. We anticipate that clinical trial enrollment criteria and endpoints for both of these trials will be similar or identical to those of the preceding trials.
Potential for GEN-003 to Treat HSV-1 Infection
We anticipate that GEN-003 may also help a patient's immune system fight herpes simplex virus type-1, or HSV-1. HSV-1 is most commonly identified with cold sores and has infected approximately 60% of Americans, according to the CDC. Increasingly, HSV-1 has been associated with outbreaks of genital ulcers, though the frequency and severity of such outbreaks generally is less than those associated with HSV-2. HSV-1 and HSV-2 are related viruses and the proteins in GEN-003 are present in, and nearly identical to, those found in HSV-1. Consequently, we believe that GEN- 003 may be active against HSV-1 and thus intend to study the potential for GEN-003 to combat HSV-1.
The Opportunity to Prevent HSV-2 Infections
In addition to treating HSV-2 infection with GEN-003, we believe that ATLAS may help to develop a vaccine that can prevent HSV-2 from infecting healthy persons. We believe that a vaccine that has therapeutic effect may be the foundation for a preventative vaccine. Since there will not likely be pre-existing immune responses to build upon in uninfected subjects, the preventative vaccine may include additional or different antigens than those in GEN-003 to be fully protective. Using data from the same ATLAS screening effort with which we designed GEN-003, we identified eight additional candidate antigens that could be added to GEN-003 or included in another vaccine for prophylaxis of HSV-2 infections. We have already demonstrated that several of the eight candidate antigens can provide some protection against infection in initial studies in mice. A prophylactic vaccine may be an important step in halting the epidemic, and could be used to treat uninfected partners of HSV-2 infected subjects to prevent them from acquiring the disease. The vaccine could also be used more broadly as a preventative measure. We intend to pursue development of a prophylactic HSV-2 vaccine and anticipate that we would partner this program at the appropriate point of clinical development.
GEN-004 Market Opportunity
Pneumococcal Disease
We are developing GEN-004 to prevent infections caused by pneumococcus. The Gates Foundation has noted that pneumococcus kills more children under age five globally than any other organism. GEN-004 consists of three whole Pneumococcal T cell protein antigens, SP0148, SP1912 and SP2108, combined with the adjuvant Alhydrogel, a form of alum that is available in several approved vaccines.
There are more than 90 serotypes, or strains, of pneumococcus known to exist. Each strain differs slightly in the composition of the polysaccharide capsule, a sugar-based component that covers the bacterial cell. These differences have likely arisen as the organism has evolved to evade human antibody responses. Pneumococcus is a bacterium that often resides harmlessly in the nose and throat but can cause otitis media, or middle ear infection, as well as pneumonia, an infection in the lungs. Such consequences of infection are considered non-invasive Pneumococcal disease, or NIPD.
67
Invasive Pneumococcal Disease, or IPD, arises when pneumococcus enters the bloodstream and potentially spreads to other organs. The consequences of IPD can be severe and, according to the CDC, 10% of patients with IPD die. IPD is classified into three categories. Bacteremic pneumonia is an infection in one or both lungs with pneumococcus also in the bloodstream. It is generally a more severe infection than pneumonia that is not invasive. Other examples of IPD include sepsis, the presence of bacterial infection in the blood along with symptoms such as fever, elevated heart rate and respiratory rate, and high or low white blood cell count, and meningitis, an inflammation of the brain and spinal column.
Limitations of Current Pneumococcal Vaccines
Global revenue exceeded $5.0 billion in 2012, of which more than 70% came from Prevnar-13, marketed by Pfizer, which is named for the 13 capsular polysaccharides types, derived from 13 strains of pneumococcus, included in the vaccine. The Prevnar family achieved global revenue of $4.0 billion in 2013. Other Pneumococcal vaccines include Synflorix, marketed by GlaxoSmithKline, and Pneumovax-23, marketed by Merck. These vaccines have dramatically reduced IPD caused by the serotypes addressed by the vaccines.
The predecessor vaccine to Prevnar-13, Prevnar-7, led to the dramatic reduction of IPD caused by the seven vaccine serotypes of pneumococcus that are addressed by the vaccine. According to the CDC, the hospitalization rates due to IPD infection from these strains fell after the introduction of Prevnar-7, from 80 cases per 100,000 children in 2000 to less than 1 per 100,000 by 2007. In pre-approval randomized trials, Prevnar-7 was demonstrated to be safe and highly efficacious against IPD, moderately efficacious against pneumonia, and somewhat effective in reducing middle ear infection episodes and related office visits. The expectation is that Prevnar-13, introduced in 2010, will result in similar benefit against the seven serotypes covered by Prevnar-7 plus the additional six serotypes included in that vaccine.
Nevertheless, significant limitations exist with this and other pneumococcal vaccines. As noted previously, there are more than 90 known serotypes of pneumococcus. Prevnar-13 covers only 13 of these serotypes. Incidence of invasive disease caused by the 75+ serotypes not included in that vaccine are rapidly increasing. As a consequence, Pfizer is believed to be working on a third generation Prevnar vaccine. Already a complex vaccine, each of the polysaccharide shells included in Prevnar-13, representing 13 of the most common disease-causing serotypes of pneumococcus, is conjugated, or chemically linked, to a protein carrier. It is believed that there are limits to how many polysaccharides that physically can be included in the vaccine. Moreover, the protective capacity per serotype appears to diminish as new polysaccharides are added to the vaccine. Still, other large companies, including GlaxoSmithKline, Merck, and Sanofi Pasteur, are also believed to be working on new vaccines against pneumococcus. To our knowledge, all of these companies' product candidates are being developed to elicit a B cell response.
GEN-004: A Prophylactic Vaccine Candidate for Pneumococcus
We have designed GEN-004 to fight more than 90 serotypes of pneumococcus, and to do so through a T cell-based mechanism of action that complements existing vaccines. Since 2009, we have collaborated with Rick Malley, M.D., of Boston Children's Hospital, a leading researcher on host immunity to pneumococcus. He was the first person to demonstrate that Pneumococci are rapidly cleared from the nose, before they can get into the lungs and bloodstream, by a type of helper T cell called T17 cells. This is important because before pneumococci can cause IPD, they need to take up residence inside the nose, known as colonization. If the immune system could be taught to make T17 cells against pneumococci in sufficient quantities, then the bacteria will not have the ability to colonize, thus reducing or eliminating IPD occurrence. The majority of healthy adults are not colonized with pneumococcus, presumably due to T17 responses that they have generated through natural
68
exposure. We believe a vaccine that stimulates T17 cells to reduce or prevent colonization of the nasopharynx by pneumococcus could be highly effective against all forms of pneumococcal disease including IPD and NIPD infections.
Guided by this insight, we used ATLAS to design a novel pneumococcal vaccine, GEN-004. Since adults are generally "protected" against colonization by pneumococcus, we screened the blood of 50 healthy, ethnically diverse adults using ATLAS. We collected their APCs and T cells and screened the entire pneumococcus proteome, which consists of more than 2,200 proteins, to identify proteins associated with a strong T17 T cell response, as measured by their induction of the cytokine IL-17A, the predominant cytokine secreted by T17 cells. Based on these studies, we identified three protein antigens that associate highly with a protective T cell response against pneumococcus in humans. Moreover, as these proteins are conserved in all sequenced strains of Pneumococci, we believe GEN-004 may be able to help protect against invasive Pneumococcal disease caused by any Pneumococcal serotype, including those covered by the Prevnar franchise.
We have demonstrated proof-of-concept of GEN-004 in a mouse model of nasal colonization, as demonstrated below. In this model, mice are immunized with the antigens adsorbed to ahydrogel and then challenged intranasally with live pneumococci. After 10 days, the nasal cavity is washed with saline, and the numbers of pneumococcal bacteria that colonized the nose are counted. We and others have shown that the prevention of colonization in this model is due to IL-17A secretion from helper T cells.
Clinical Development of GEN-004
In June 2014, we announced positive top line data from a Phase 1 clinical trial in the United States to evaluate the safety of, and immune response to, GEN-004. The Phase 1 clinical trial met its safety, tolerability and immunogenicity goals, including measurable increases in the blood of T helper 17 (TH17) cells.
69
The Phase 1 clinical trial was a randomized, double-blind, dose-escalation, placebo-controlled clinical trial that enrolled 90 healthy adult volunteers. Serum IgG titers increased in a dose-dependent manner to each of the antigens included in GEN-004 and measurable increases in peripheral TH17 responses were seen among subjects receiving the highest dose (100µg) with adjuvant. There were no serious adverse events related to the vaccine.
Based on these data, we plan to advance GEN-004 into a Phase 2a trial in the third quarter of 2014. We anticipate data from our Phase 2a trial in mid-2015. Subjects in the clinical trial will receive either GEN-004 or placebo, and then be "challenged" intranasally with live pneumococcus, much like in the mouse colonization model. This means that pneumococcus will be introduced to the nasal cavity. We expect to enroll as many as 90 healthy adults in this trial. We will monitor AEs, antibody and T-cell immune responses as determined by IgG and IL-17A, and incidence of post-challenge colonization. We will follow these patients for a year and expect the initial results will be available in mid-2015. If successful, we believe this has the potential to be the first time a protein subunit vaccine will have directly demonstrated a reduction in nasopharyngeal colonization in humans.
Our Chlamydia Program
Chlamydia is the most commonly reported bacterial sexually transmitted disease in the United States. According to the CDC, an estimated 2.9 million infections occur annually in the United States. Despite the widespread availability of antibiotics that are effective against Chlamydia trachomatis, the pathogen that causes chlamydia infections, incidence has increased at greater than 5% per year over the past decade, according to the CDC. A key reason for this is that chlamydia is often an asymptomatic infection, so infected individuals do not seek treatment, which can result in severe consequences, particularly in women, such as pelvic inflammatory disease, infertility and serious neonatal infections.
Despite the need, vaccine development to combat chlamydia has been virtually non-existent. There has not been a chlamydia vaccine clinical trial since the 1960s, in which an attenuated pathogen vaccine demonstrated no lasting protection and showed hints of disease exacerbation. Antibodies appear to be unlikely to protect against infection as the pathogen is intracellular for much of its life cycle. Additionally, as a large genome pathogen, Chlamydia trachomatis represents a large T cell antigen discovery challenge. For these reasons, we believe that chlamydia is a particularly attractive pathogen for use of ATLAS to identify a vaccine candidate.
We have achieved promising non-clinical results from candidates generated using ATLAS. We collected blood from 144 subjects spanning multiple clinical cohorts, ranging from subjects whose infections spontaneously cleared, representing a putative natural protection cohort, to subjects with infertility caused by chlamydia infection. From the more than 900 proteins in the Chlamydia trachomatis proteome, we identified 22 novel proteins associated with a protective response. From these we have demonstrated that three proteins, when given in an animal model of infection and when paired with the Matrix-M2 adjuvant can significantly reduce infection risk.
If the program were to reach the clinic, we believe it would be the first vaccine against chlamydia to be in clinical trials in more than 50 years. If it can successfully prevent chlamydia infections, we believe it would address a major unmet clinical need. As resources permit, we intend to opportunistically pursue development of this program.
Our Malaria Program
Malaria is one of the deadliest infectious diseases in the world. Approximately 600 thousand to one million people died in 2010 due to malaria, primarily in the developing world. There is no vaccine to prevent malaria, an infection caused by the plasmodium parasites transmitted by mosquitoes. We previously collaborated with the Naval Medical Research Center, or NMRC, and recently initiated a
70
second collaboration with the Gates Foundation for which malaria is a priority infectious disease. When the parasite is injected into the blood through the bite of an infected mosquito, it rapidly travels to the liver where it replicates exponentially, is released into the bloodstream, and causes sickness. T cells in the liver could potentially be used to kill the cells in which the parasite is hiding, before the parasite is able to replicate itself, and could therefore protect against blood infection. Both the Gates Foundation and NMRC have sponsored several studies investigating killed or attenuated whole organism vaccines, which induce immunity, but are impractical to manufacture due to the fact that the vaccines are based on irradiated parasites grown within the salivary glands of mosquitoes.
We are in the process of collecting blood samples from subjects immunized with the killed organism and who were either protected or not protected after live parasite challenge to use ATLAS to identify the protein antigens that are associated with protective T cell responses. The identification of the protein targets of the T cell responses can enable the generation of a protein plus adjuvant vaccine designed to induce liver T cell responses and prevent malaria disease in a safe, scalable and affordable way.
Competition
The biotechnology and pharmaceutical industries are characterized by intense and rapidly changing competition to develop new technologies and proprietary products. While we believe that our proprietary patent portfolio and T cell vaccine expertise provide us with competitive advantages, we face potential competition from many different sources, including larger and better-funded pharmaceutical companies. Not only must we compete with other vaccine companies but any products that we may commercialize will have to compete with existing therapies and new therapies that may become available in the future.
There are other organizations working to improve existing therapies or to develop new vaccines or therapies for our initially selected indications. Depending on how successful these efforts are, it is possible they may increase the barriers to adoption and success of our GEN-003 and GEN-004 product candidates, if approved. These efforts include the following:
- •
- HSV-2: The current standard of care for the treatment of HSV-2 is valacyclovir, an oral antiviral medicine. Other currently approved oral antiviral medications include acyclovir and famciclovir. AiCuris, a private company based in Germany, is developing a new oral antiviral, pritelivir, and has advanced the compound into Phase 2 testing. We understand the company will pursue once-weekly dosing with this drug. We believe that GEN-003 may offer advantages in terms of improved symptom control, reduced disease transmission risk and improved compliance when compared to oral antivirals.
- •
- Pneumococcus: The current standard of care for the prevention of pneumococcus is Prevnar-7/Prevenar-13, marketed by Pfizer. In select countries, Synflorix, marketed by GlaxoSmithKline, is also widely accepted. Additionally, Pneumovax-23, marketed by Merck, is labeled by use for persons over 65. We believe that each of these companies is seeking to develop improvements to their product. We believe these represent incremental improvements,
There are also several companies attempting to develop new therapeutic vaccines against HSV-2, including Agenus Inc., Coridon Pty Ltd, Sanofi Pasteur and Vical Incorporated. We believe GEN-003 has advantages against each of the vaccines being developed by these companies based on the screens of human protection that we have conducted using ATLAS that include these competitors' antigens, published reports of non-clinical vaccine efficacy, announced clinical results in the case of Agenus, Inc. and our own clinical results to date. However, there can be no assurance that one or more of these companies or other companies will not achieve similar or superior clinical results in the future as compared to GEN-003 or that our future clinical trials will be successful.
71
adding a few additional strains to their coverage. In addition, we are aware of a pneumococcus vaccine that Sanofi Pasteur has taken into Phase 1 trials. This is a protein subunit vaccine designed to cover all strains of pneumococcus, but was designed to induce B cell responses. For many pneumococcal strains with dense sugars on their surface, the protein targets of the antibodies induced by the vaccine will be blocked by sugars that cover them. We believe that by covering all known pneumococcus serotypes, with a T cell-based mechanism of action that complements existing vaccines, GEN-004 may offer broader protection than existing vaccines. However, there can be no assurance that one or more of these companies or other companies will not achieve similar or superior clinical results in the future as compared to GEN-004 or that our ongoing and future clinical trials of GEN-004 will be successful.
Many of our competitors, such as Merck, GlaxoSmithKline, and Sanofi Pasteur, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of vaccines and the commercialization of those vaccines. Accordingly, our competitors may be more successful than us in obtaining approval for vaccines and achieving widespread market acceptance. Our competitors' vaccines may be more effective, or more effectively marketed and sold, than any vaccine we may commercialize and may render our vaccines obsolete or non-competitive.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. We expect any vaccines that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our products, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic products.
Intellectual Property
We strive to protect and enhance the proprietary technology, inventions and improvements that are commercially important to our business, including seeking, maintaining, and defending patent rights, whether developed internally or licensed from third parties. We also rely on trade secrets relating to our proprietary technology platform and on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain our proprietary position in the vaccine field. We additionally rely on regulatory protection afforded through data exclusivity, market exclusivity and patent term extensions where available. Still further, we utilize trademark protection for our company name, and expect to do so for products and/or services as they are marketed.
72
Our commercial success may depend in part on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business; defend and enforce our patents; preserve the confidentiality of our trade secrets; and operate without infringing the valid enforceable patents and proprietary rights of third parties. Our ability to stop third parties from making, using, selling, offering to sell or importing our products may depend on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. With respect to both licensed and company-owned intellectual property, we cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our commercial products and methods of manufacturing the same.
We have developed or in-licensed numerous patents and patent applications and possess substantial know-how and trade secrets relating to the development and commercialization of vaccine products. As of the date of this prospectus, our patent portfolio includes the following:
ATLAS
Our discovery platform patent portfolio includes three patent families, currently comprising four issued U.S. patents and two pending U.S. applications. We hold an exclusive license from The Regents of the University of California to the first patent family, including U.S. Patent 6,004,815 and the related U.S. Patents 6,287,556 and 6,599,502. This first family includes claims to fundamental aspects of the ATLAS platform, developed by our scientific founder, Darren Higgins, Ph.D. while he was employed at the University of California, Berkeley. Patents in this family have a patent term until August 2018. We hold a further exclusive license from President and Fellows of Harvard College to the second patent family, which covers methods related to the ATLAS discovery platform. This second patent family includes a pending U.S. application and corresponding applications in Europe, Canada and Australia. Patents issuing from these applications are expected to expire in 2027. We wholly own the third patent family, which is specifically directed to the ATLAS platform as utilized by us. This third patent family includes U.S. Patent 8,313,894, a pending U.S. patent application, and corresponding pending applications in Europe, Canada and Australia. Patents issuing from applications in this family are expected to have a patent term until at least July 2029; issued U.S. Patent 8,313,894 has a term that includes Patent Term Adjustment and extends until at least June 2030.
GEN-003 (HSV-2)
We wholly own a portfolio of patent applications directed to HSV-2 vaccines, including GEN-003. This portfolio includes two patent families covering HSV-2 vaccine compositions and methods for inhibiting or treating HSV-2 infections. The first patent family includes U.S. Patent 8,617,564. A U.S. application and applications in Europe, Canada, Australia, Japan, Brazil, Russia, India, China and nine additional foreign jurisdictions are pending in the first patent family. A U.S. application and applications in Europe, Canada, Australia and Japan are pending in the second family. Patents that issue from applications in these families are expected to expire in 2030 and 2031; issued U.S. Patent 8,617,564 has a term that includes Patent Term Adjustment and extends until at least January 2031. We own a further patent family covering follow-on HSV-2 vaccine compositions.
We hold a license from the University of Washington to a patent family that includes U.S. Patent 8,197,824 and European Patent No. 2263686 covering compositions of certain HSV-2 proteins and methods for treating HSV infections. This family includes pending applications in the United States, Europe and Canada. This patent family has a patent term until at least July 2023.
We hold a license from Isconova AB (now Novavax) to two patent families covering Matrix-M2, the adjuvant used in GEN-003. Both patent families include issued patents in Europe; the first patent
73
family also includes an issued patent in Japan. Applications in the United States, Canada, Australia, Japan (for the second family) and three additional foreign jurisdictions. These patent families have patent terms until at least July 2023 and July 2024.
GEN-004 (Pneumococcus)
We co-own with Children's Medical Center Corporation, or Childrens, a patent portfolio of patent applications directed to pneumococcus vaccines, including GEN-004. This patent portfolio includes two patent families covering pneumococcal vaccine compositions and methods for inhibiting or treating pneumococcal infections. A U.S. application and applications in Europe, Canada, Australia, Japan, Brazil, Russia, India, China and nine additional foreign jurisdictions are pending in the first patent family. A U.S. application and applications in Europe, Australia, Japan, Brazil, Russia, India, China and nine additional foreign jurisdictions are pending in the second patent family. Patents that issue from applications in these patent families are expected to have patent terms until at least 2030 and 2032, respectively. We hold an exclusive license to Childrens' interest in these patent rights. We co-own with Childrens two further patent families covering follow-on pneumococcal vaccine compositions, and Childrens' interest in these patents is also exclusively licensed to us.
GEN-001 (Chlamydia)
Our chlamydia patent portfolio includes four patent families (one of which overlaps with the ATLAS portfolio). We hold an exclusive license from President and Fellows of Harvard College to three of these four patent families. We wholly own the fourth patent family. The patent families cover chlamydia vaccine and immunogenic compositions and methods for inhibiting or treating chlamydia infections. A European Patent is issued in the first patent family; a U.S. application and applications in Canada and Australia are pending. A U.S. application and applications in Europe, Canada, Australia and Japan are pending in the second patent family. The third patent family includes U.S. Patent 8,637,053. A U.S. application and applications in Canada, Australia and Japan are pending in the third patent family. A U.S. application and applications in Europe, Canada, Australia and Japan are pending in the fourth patent family. Patents issuing from applications in these four patent families are expected to expire between 2027 and 2031.
In addition to the above, we have established expertise and development capabilities focused in the areas of non-clinical research and development, manufacturing and manufacturing process scale-up, quality control, quality assurance, regulatory affairs and clinical trial design and implementation. We believe that our focus and expertise will help us develop products based on our proprietary intellectual property.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing the non-provisional application. In the United States, a patent's term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the United States Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent.
The term of a patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration of a United States patent as compensation for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Moreover, a patent can only be extended once, and thus, if a single patent is applicable to multiple products, it can only be
74
extended based on one product. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of a new drug application, or NDA, we expect to apply for patent term extensions for patents covering our product candidates and their methods of use.
In-License Agreements
University of California
In August 2006, we entered into an exclusive license agreement with The Regents of the University of California, or UC, granting us an exclusive, royalty-bearing sublicensable license to a patent family that includes claims to fundamental aspects of the ATLAS platform, to make, use, offer for sale, import and sell licensed products and services, and to practice licensed methods in all fields of use in the United States. This patent family consists entirely of issued United States patents with a patent term until August 2018. UC retains the right to practice and to allow other educational and non-profit institutions to practice, the licensed intellectual property licensed under the agreement for educational and research purposes.
Until first commercial sale of a licensed product or service, we are obligated to pay UC an annual license maintenance fee in the low five figures. Upon commercialization of our products and services covered by the licensed patents, we are obligated to pay UC royalties in the low single digits, subject to a minimum annual royalty in the low five figures, on the net sales of such products and services sold by us or our affiliates for the life of any licensed patents covering the products or services. The royalties payable to UC are subject to reduction for any third party payments required to be made, with a minimum floor in the low single digits. In addition, we agreed to pay UC a flat royalty in the low single digits on net sales of products sold by us or our affiliates which include a polypeptide, nucleotide sequence, biological organism or chemical entity identified in the practice of a licensed method or service, but not otherwise covered, by the licensed patent for the life of the licensed patents. If we receive any revenue (cash or non-cash) from any sublicensees, we must pay UC a percentage of such revenue, excluding certain categories of payments but including royalties on net sales by sublicensees, varying in the low-double digits for any sublicense depending on the scope of the license. Under the terms of the agreement, we are obligated to pay UC a specified development milestone payment and a specified commercial milestone payment up to $500 thousand in the aggregate for the first licensed product covered by the licensed patents, plus up to an additional $250 thousand if specified development and commercial milestones are met for each subsequent licensed product covered by the licensed patents. As of June 30, 2014, we have not made any milestone payments.
We are required to diligently develop and market licensed products, services and methods. If we are unable to meet our diligence obligations, even after any extension thereof, UC has the right, depending on the number of years the agreement has been effective, to either terminate the agreement or convert our exclusive license to a non-exclusive license.
Unless earlier terminated, the agreement with UC will remain in effect until the expiration of the last-to-expire patent under the licensed patent rights. We may terminate the agreement at any time by giving UC advance written notice. The agreement may also be terminated by UC in the event of a material breach by us that remains uncured after a specified period of time.
Harvard University
In November 2007, we entered into an exclusive license agreement with President and Fellows of Harvard College, or Harvard, granting us an exclusive, worldwide, royalty-bearing, sublicensable license to three patent families, to develop, make, have made, use, market, offer for sale, sell, have sold and import licensed products and to perform licensed services. This agreement was amended and restated
75
in November 2012. The Harvard intellectual property covers methods related to the ATLAS discovery platform, as well as certain chlamydia immunogenic compositions and methods for inhibiting or treating chlamydia infections. Any patents within this portfolio that have issued or may be issued will expire normally in 2027 and 2028. Harvard retains the right to make and use, and to grant licenses to other not-for-profit research organizations to make and use, the licensed intellectual property for internal research, teaching and other educational purposes.
We are obligated to pay Harvard an annual license maintenance fee ranging from the low five figures to the mid five figures depending on the type of product and the number of years after the effective date of the agreement. For products covered by the licensed patent rights, we are obligated to pay Harvard milestone payments up to $1.8 million in the aggregate upon the achievement of certain development and regulatory milestones. For products discovered using the licensed methods, we are obligated to pay Harvard milestone payments up to $600 thousand in the aggregate for each of the first three products and up to $300 thousand in the aggregate for each additional product under the agreement upon the achievement of certain development and regulatory milestones. As of June 30, 2014, we have paid $66 thousand in aggregate milestone payments. Upon commercialization of our products covered by the licensed patent rights or discovered using the licensed methods, we are obligated to pay Harvard royalties on the net sales of such products and services sold by us, our affiliates and our sublicensees. This royalty varies depending on the type of product or service but is in the low single digits. The royalty based on sales by our sublicensees is the greater of the applicable royalty rate or a percentage in the high single digits or the low double digits of the royalties we receive from such sublicensee depending on the type of product. Depending on the type of commercialized product or service, royalties are payable until the expiration of the last-to-expire valid claim under the licensed patent rights or for a period of 10 years from first commercial sale of such product or service. The royalties payable to Harvard are subject to reduction, capped at a specified percentage, for any third party payments required to be made. In addition to the royalty payments, if we receive any additional revenue (cash or non-cash) under any sublicense, we must pay Harvard a percentage of such revenue, excluding certain categories of payments, varying from the low single digits to up to the low double digits depending on the scope of the license that includes the sublicense.
We are required to use commercially reasonable efforts to develop licensed products, introduce them into the commercial market and market them, in compliance with an agreed upon development plan. We are also obligated to achieve specified development milestones. If we are unable to meet our development milestones for any type of product or service, absent any reasonable proposed extension or amendment thereof, Harvard has the right, depending on the type of product or service, to terminate this agreement with respect to such products or to convert the license to a non-exclusive, non-sublicensable license with respect to such products and services.
Our agreement with Harvard will expire on a product-by-product or service-by-service and country-by-country basis until the expiration of the last-to-expire valid claim under the licensed patent rights. We may terminate the agreement at any time by giving Harvard advance written notice. Harvard may also terminate the agreement in the event of a material breach by us that remains uncured; in the event of our insolvency, bankruptcy, or similar circumstances; or if we challenge the validity of any patents licensed to us.
76
University of Washington
In January 2010, we entered into a patent license agreement with the University of Washington, or UW, which was subsequently amended and partially terminated with respect to specified patent rights in July 2012 and was further amended in September 2012 and November 2013. The agreement grants a worldwide, sublicensable, co-exclusive license to certain patent rights, and an exclusive license to certain other patent rights, to manufacture, have manufactured on our behalf, use, offer to sell or sell, offer to lease or lease, import, or otherwise offer to dispose or dispose of licensed products to prevent or treat HSV-2. Patents within the remaining licensed patent rights include claims to compositions of certain HSV-2 proteins and methods for treating HSV infections, with a patent term until at least July 2023. UW retains the right for itself and the Fred Hutchinson Cancer Research Center to make and use products and processes covered by the licensed patent rights for academic research, teaching and any other academic purpose.
Until the first commercial sale of a licensed product, we are obligated to pay UW an annual license maintenance fee in the low five figures. For each product covered by the licensed patent rights, we are obligated to pay UW milestone payments up to $750 thousand in the aggregate upon the achievement of certain development and commercial milestones. As of June 30, 2014, we have paid $25 thousand in milestone payments. Upon commercialization of our licensed products covered by the licensed patent rights, we are obligated to pay UW royalties in the low single digits on the net sales of such products sold by us, our affiliates and our sublicensees, subject to a minimum annual royalty payment in the low five figures following the first commercial sale of a licensed product. Royalties are payable on a country-by-country and licensed product-by-licensed product basis until the earlier of the termination of this agreement, or the date on which the manufacture, importation, use or sale of the licensed product is no longer covered by a valid claim of a licensed patent in such country. The royalties payable to UW are subject to reduction, capped at a specified percentage, for any third-party payments required to be made. In addition to the royalty payments, if we receive any additional revenue (cash or non-cash) under any sublicense, we must pay UW a percentage of such revenue, excluding certain categories of payments and payments made in consideration of additional intellectual property rights that are necessary or useful for commercialization of the licensed product, varying from the mid-single digits to the low double digit range depending on whether certain clinical study milestones have been achieved at the time the sublicense was granted.
We are required to use commercially reasonable efforts to commercialize the inventions covered by the licensed patents and to make and sell the licensed products within a reasonable period of time. We are also obligated to achieve specified development and regulatory performance milestones.
Our agreement with UW will expire on the date on which no valid claim in a licensed patent is pending or subsisting in any country worldwide. We may terminate the agreement on a licensed product-by-licensed product basis or in its entirety at any time by giving UW advance written notice. UW may also terminate the agreement in the event of a material breach by us that remains uncured within a specified timeframe; in the event of our insolvency, bankruptcy, or similar circumstances; or if we challenge the validity of the licensed patents.
Other Collaborations
Dana-Farber Cancer Institute and Harvard Medical School
In March 2014, we announced a joint research collaboration with Dana-Farber Cancer Institute and Harvard Medical School to characterize anti-tumor T cell responses in melanoma patients. This collaboration extends the use of our proprietary ATLAS platform for the rapid discovery of T cell antigens to cancer immunotherapy approaches.
77
Children's Medical Center Corporation
In September 2008, we entered into a collaborative research agreement with Childrens that was funded by PATH Vaccine Solutions, or PATH. The collaborative research project led to the identification of certain highly conserved pneumococcal antigens that are able to protect against colonization. The intellectual property covering these antigens is co-owned by us and Childrens and covers pneumococcal vaccine compositions and methods for inhibiting or treating pneumococcus infections. In February 2010, we entered into an exclusive license agreement with Childrens, which was amended and restated in March 2012. This agreement grants us an exclusive, worldwide, sublicensable license under Childrens' rights to the jointly-owned intellectual property to make, have made, use, sell, offer for sale, import and export licensed products and to practice licensed processes for the prevention and treatment of Streptococcus pneumoniae. Childrens retains the right to practice and use, and to allow academic non-profit research organizations to practice and use, the licensed intellectual property for research, educational, clinical and charitable purposes. Under the terms of the agreement, our license from Childrens is subject to PATH's separate non-exclusive, royalty-free license from Childrens to develop pneumococcal T cell-based protein vaccines worldwide and to market and sell such vaccines in developing countries.
For products covered by the licensed patent rights, we are obligated to pay Childrens milestone payments up to $390 thousand in the aggregate upon the achievement of certain development and commercial milestones. As of June 30, 2014 we have not made any milestone payments. Upon commercialization of our products, we are obligated to pay Childrens royalties in the low single digits on the net sales of licensed products sold by us, our affiliates and our sublicensees. The royalties payable to Childrens are subject to reduction for any third-party payments required to be made, with a minimum floor in the low single digits. Royalties are payable for the term of the license agreement, which is 15 years from the effective date of the amended and restated agreement or until expiration of the last-to-expire patent under the licensed patent rights, whichever period is longer. If we receive any additional revenue (cash or non-cash) under any sublicense, we must pay Childrens a percentage of such income varying from the mid-single digits to low double digits depending on the clinical stage of development of the product, provided that such percentage may increase to match our financial obligations to third parties.
We are required to use commercially reasonable efforts to bring at least one licensed product to market as soon as reasonably practical, consistent with sound and legal business practices and judgment and to accomplish the objectives set forth in an agreed upon development plan. If we are unable to meet our diligence obligations, even after any extensions thereof, Childrens has the right to terminate in this agreement in whole or in part.
Unless earlier terminated, the agreement with Childrens will remain in effect until the later of 15 years from the effective date of the amended and restated agreement or the expiration of the last to expire patent under the licensed patent rights. We may terminate the agreement in its entirety or on a country-by-country and licensed product-by-licensed product basis, at any time by giving Childrens advance written notice. Childrens may terminate the agreement in the event of our bankruptcy, insolvency or similar circumstances; if we use confidential information to formally challenge Childrens' joint ownership of the licensed patent rights; or if we materially breach the agreement and do not cure such breach within a specified time period.
Isconova AB
In August 2009, we entered into an exclusive license and collaboration agreement with Isconova AB, now Novavax. The agreement grants us a worldwide, sublicensable, exclusive license to two patent families, to import, make, have made, use, sell, offer for sale and otherwise exploit licensed vaccine products containing an adjuvant which incorporates or is developed from Matrix-A, Matrix-C and/or
78
Matrix-M technology, in the fields of HSV and chlamydia, and the time-limited exclusive fields of Neisseria gonorrhoeae, cytomegalovirus, or CMV, and Mycobacterium tuberculosis. After a specified period of time, the license grant to us in the time-limited exclusive fields will convert to a non-exclusive license with respect to all licensed intellectual property rights that were not jointly invented by us and Novavax under the collaboration. Under the terms of this agreement, Novavax also grants us a worldwide, sublicensable, non- exclusive license under such licensed intellectual property rights to import, make, have made, use, sell, offer for sale and otherwise exploit licensed products in the field of Streptococcus pneumoniae. Our rights in the field of Streptococcus pneumoniae are exclusive with respect to all intellectual property rights jointly invented by us and Novavax under the collaboration. The agreement further grants us certain limited rights to use Novavax trademarks.
For licensed products in each unique disease field under the agreement, we are obligated to pay Novavax milestone payments up to approximately $3 million in the aggregate upon the achievement of certain development and commercial milestones. As of June 30, 2014, we have paid $100 thousand in aggregate milestone payments. Upon commercialization of our products, we are obligated to pay Novavax royalties on the net sales of licensed products sold by us, our affiliates and our sublicensees. The royalties payable to Novavax are in the low single digits and vary on a country-by-country and licensed product-by-licensed product basis based on the amount of net sales and the nature and timing of the licensed product's development. The royalties payable to Novavax are subject to reduction if the licensed product is not covered by one or more valid claims of the licensed patent rights, or if we are required to make any third-party payments. Royalties are payable for 10 years from first commercial sale in any particular country or until the date on which offer for sale of a licensed product is no longer covered by a valid claim of the licensed patent rights in such country, whichever period is longer. In addition to the royalty payments, if we receive any additional revenue (cash or non-cash) under any sublicenses, we must pay Novavax a percentage of such revenue, up to the low double digits.
We are required to use commercially reasonable efforts to perform specified research activities in accordance with an agreed-upon research plan. We are also obligated to use commercially reasonable efforts consistent with prudent business judgment and business and market conditions to research, develop and carry out the commercialization of licensed products in HSV and chlamydia.
Our agreement with Novavax will expire on a country-by-country and licensed product-by-licensed product basis on the date of the expiration of the royalty term with respect to such licensed product in such country. We may terminate the agreement on a country-by-country and licensed product-by-licensed product basis or in its entirety at any time by giving Novavax advance written notice. Both parties may also terminate the agreement in the event of a material breach by the other party that remains uncured or for bankruptcy, insolvency or similar circumstances. Novavax may terminate this agreement if we challenge the validity of any patents licensed to us.
Trade Secrets
We may rely, in some circumstances, on trade secrets to protect our technology. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants, contractors or collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
79
United States Government Regulation
Biological products such as vaccines are subject to regulation under the Federal Food, Drug, and Cosmetic Act, or FD&C Act, and the Public Health Service Act, or PHS Act, and other federal, state, local and foreign statutes and regulations. Both the FD&C Act and the PHS Act and their corresponding regulations govern, among other things, the testing, manufacturing, safety, efficacy, labeling, packaging, storage, record keeping, distribution, reporting, advertising and other promotional practices involving biological products. FDA reviews the research plan and study protocols before clinical testing of biological products begins and can delay initiation of such testing until it is satisfied that the product is appropriate for human testing. FDA approval also must be obtained before marketing of biological products. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources and we may not be able to obtain the required regulatory approvals.
United States Biological Products Development Process
The process required by the FDA before a biological product may be marketed in the United States generally involves the following:
- •
- completion of nonclinical laboratory tests and animal studies according to good laboratory practices, or GLPs, and
applicable requirements for the humane use of laboratory animals or other applicable regulations;
- •
- submission to the FDA of an application for an IND, which must become effective before human clinical trials may begin;
- •
- performance of adequate and well-controlled clinical trials according to the FDA's regulations commonly referred to as
good clinical practices, or GCPs, and any additional requirements for the protection of human research subjects and their health information, to establish the safety and efficacy of the proposed
biological product for its intended use;
- •
- submission to the FDA of a Biologics License Application, or BLA, for marketing approval that includes substantial
evidence of safety, purity, and potency from results of nonclinical testing and clinical trials;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is
produced to assess compliance with GMP, to assure that the facilities, methods and controls are adequate to preserve the biological product's identity, strength, quality and purity and, if applicable,
the FDA's current good tissue practices, or GTPs, for the use of human cellular and tissue products;
- •
- potential FDA audit of the nonclinical and clinical trial sites that generated the data in support of the BLA; and
- •
- FDA review and approval, or licensure, of the BLA.
Before testing any biological product candidate in humans, the product candidate enters the preclinical study stage. Preclinical studies, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the preclinical studies must comply with federal regulations and requirements including GLPs.
The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical studies may continue even after the
80
IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, studies may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such studies.
Clinical trials involve the administration of the biological product candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor's control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain AEs should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA's regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical trial must be reviewed and approved by an IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical studies are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
- •
- Phase 1. The biological product is initially
introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically
administer to healthy volunteers, the initial human testing is often conducted in patients.
- •
- Phase 2. The biological product is evaluated in a
limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage
tolerance, optimal dosage and dosing schedule.
- •
- Phase 3. Clinical studies are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical studies are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling.
Post-approval clinical studies, sometimes referred to as Phase 4 clinical studies, may be conducted after initial marketing approval. These clinical studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. Annual progress reports detailing the results of the clinical studies must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA, the NIH and the investigators for serious and unexpected AEs, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information
81
qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor's initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical studies may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB's requirements or if the biological product has been associated with unexpected serious harm to patients.
Concurrent with clinical studies, companies usually complete additional animal studies and must also develop additional information about the physical characteristics of the biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with GMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHS Act emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
United States Review and Approval Processes
After the completion of clinical trials of a biological product, FDA approval of a BLA must be obtained before commercial marketing of the biological product. The BLA must include results of product development, laboratory and animal studies, human studies, information on the manufacture and composition of the product, proposed labeling and other relevant information. In addition, under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain data to assess the safety and effectiveness of the biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each BLA must be accompanied by a significant user fee. PDFUA also imposes an annual product fee for biologics and an annual establishment fee on facilities used to manufacture prescription biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business.
Within 60 days following submission of the application, the FDA reviews the BLA to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe and potent, or effective, for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with GMP to assure and preserve the product's identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel
82
that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the biological product approval process, the FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the biological product. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS; the FDA will not approve the BLA without a REMS, if required.
Before approving a BLA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND study requirements and GCP requirements. To assure GMP, GTP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA in its present form, the FDA will issue a complete response letter that usually describes all of the specific deficiencies in the BLA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific indications for use and dosages and administration regimens, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a REMS, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product's safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized.
One of the performance goals agreed to by the FDA under the PDUFA is to review 90% of standard BLAs in 10 months from filing and 90% of priority BLAs in six months from filing, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the BLA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
Federal and State Fraud and Abuse, Transparency and Privacy Laws
In the United States, our business activities are subject to numerous other laws by federal and state authorities, in addition to the FDA, including but not limited to, the United States Federal Communications Commission, the United States Department of Health and Human Services, or HHS, and its various divisions, including but not limited to, the Centers for Medicare & Medicaid Services, or CMS. These laws are enforced by various federal and state enforcement authorities, including but not
83
limited to, the United States Department of Justice, and individual United States Attorney offices within the Department of Justice, HHS' various enforcement divisions, including but not limited to, the Office of Inspector General, or OIG, the Office for Human Research Protections, or OHRP, and the Office of Research Integrity, or ORI, and other state and local government agencies.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, purchasing, leasing, ordering, or arranging for or recommending, the purchase lease, or order of any good, facility, service or item for which payment is made, in whole or in part, under a federal health care program, such as Medicare. The federal Anti-Kickback Statute has been interpreted to apply to arrangements between manufacturers on one hand and prescribers, purchasers and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to, or approval by, the federal government, or knowingly making, using, or causing to be made or used, a false record or statement material to a false or fraudulent claim to the federal government. Recently, the civil False Claims Act has been used to assert liability on the basis of kickbacks and improper referrals, improperly reported government pricing metrics such as Medicaid Best Price or Average Manufacturer Price, improper use of supplier or provider Medicare numbers when detailing a provider of services, improper promotion of drugs or off-label uses not expressly approved by the FDA in a drug's label, and misrepresentations with respect to the services rendered or items provided.
Additionally, the civil monetary penalties statute, among other things, imposes fines against any person who is determined to have presented, or caused to be presented, claims to a federal health care program that the person knows, or should know, is for an item or service that was not provided as claimed or is false or fraudulent.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of, or payment for, health care benefits, items or services relating to health care matters.
Many states have similar fraud and abuse statutes and regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, private payors.
Additionally, the federal Physician Payments Sunshine Act within the Health Care and Education Reconciliation Act, or Health Care Reform Law, and its implementing regulations, require that certain manufacturers of drugs, devices, biological and medical supplies for which payment is available under Medicare, Medicaid or the Children's Health Insurance Program (with certain exceptions) to report to CMS, information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually to CMS certain ownership and investment interests held by physicians and their immediate family members.
84
In addition, we may be subject to, or our marketing activities may be limited by, data privacy and security regulation by both the federal government and the states in which we conduct our business.
If our operations are found to be in violation of any of the health regulatory laws described above, or any other laws that apply to us, we may be subject to penalties, including, without limitation, civil, criminal, and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal health care programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations.
Reimbursement
In both domestic and foreign markets, the commercial success of any approved products will depend, in part, on the availability of coverage and adequate reimbursement of such products from third-party payors, such as government health care programs, commercial insurance and managed care organizations. Patients who are provided vaccinations, and providers providing vaccinations, generally rely on third-party payors to reimburse all or part of the associated health care costs. Sales of any approved vaccines will therefore depend substantially, both domestically and abroad, on the extent to which the costs of our approved vaccines will be paid by third-party payors. These third-party payors are increasingly challenging the prices charged for medical products and services and imposing controls to manage costs. The containment of health care costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the FDA-approved products for a particular indication. In addition, there is significant uncertainty regarding the reimbursement status of newly approved health care products. Third-party payors are increasingly examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. If third-party payors do not consider our products to be cost-effective compared to other therapies, the payors may not cover our products after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
Within the United States, if we obtain appropriate approval in the future to market any of our current product candidates, we may seek approval and coverage for those products under Medicaid, Medicare and the Public Health Service, or PHS, pharmaceutical pricing program and also seek to sell the products to federal agencies.
Medicaid is a joint federal and state program that is administered by the states for low income and disabled beneficiaries. Under the Medicaid Drug Rebate Program, manufacturers are required to pay a rebate for each unit of product reimbursed by the state Medicaid programs. The amount of the rebate for each product is set by law and may be subject to an additional discount if certain pricing increases more than inflation.
Medicare is a federal program administered by the federal government that covers individuals age 65 and over as well as those with certain disabilities. Medicare Part D provides coverage to enrolled Medicare patients for self-administered drugs (i.e., drugs that do not need to be administered by a physician). Medicare Part D is administered by private prescription drug plans approved by CMS and each drug plan establishes its own Medicare Part D formulary for prescription drug coverage and pricing, which the drug plan may modify from time-to-time.
85
Medicare Part B covers most injectable drugs given in an in-patient setting, and some drugs administered by a licensed medical provider in hospital outpatient departments and doctors' offices. Medicare Part B is administered by Medicare Administrative Contractors, which generally have the responsibility of making coverage decisions. Subject to certain payment adjustments and limits, Medicare generally pays for Part B covered drugs based on a percentage of manufacturer-reported average sales prices.
Drug products are subject to discounted pricing when purchased by federal agencies via the Federal Supply Schedule, or FSS. FFS participation is required for a drug product to be covered and paid for by certain federal agencies and for coverage under Medicaid, Medicare Part B and the PHS pharmaceutical pricing program. FSS pricing is negotiated periodically with the Department of Veterans Affairs. FSS pricing is intended to not exceed the price that a manufacturer charges its most-favored non-federal customer for its product. In addition, prices for drugs purchased by the Veterans Administration, Department of Defense (including drugs purchased by military personnel and dependents through the TRICARE retail pharmacy program), Coast Guard, and PHS are subject to a cap on pricing (known as the "federal ceiling price") and may be subject to an additional discount if pricing increases more than inflation.
To maintain coverage of drugs under the Medicaid Drug Rebate Program, manufacturers are required to extend discounts to certain purchasers under the PHS pharmaceutical pricing program. Purchasers eligible for discounts include hospitals that serve a disproportionate share of financially needy patients, community health clinics and other entities that receive health services grants from the PHS.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the HHS, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a trial. It is also possible that comparative effectiveness research demonstrating benefits in a competitor's product could adversely affect the sales of any of our approved products. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The United States and state governments continue to propose and pass legislation designed to reduce the cost of health care. In March 2010, the United States Congress enacted the Health Care Reform Law which has the potential to change health care financing by both governmental and private payors. In the future, there may continue to be additional proposals relating to the reform of the United States health care system, some of which could further limit the prices we are able to charge, or the amounts of reimbursement available for our vaccine candidates once they are approved.
Outside the United States, ensuring adequate coverage and payment for our products will face challenges. In international markets, reimbursement and health care payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. Pricing of prescription pharmaceuticals is subject to governmental control in many countries. Pricing negotiations with governmental authorities can extend well beyond the receipt of regulatory marketing approval for a product and may require us to conduct a clinical trial that compares the cost effectiveness of our product candidates or products to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in our commercialization efforts. Third-party payors are challenging the prices charged for medical products and services, and many third-party payors limit
86
reimbursement for newly-approved health care products. Recent budgetary pressures in many European Union countries are also causing governments to consider or implement various cost-containment measures, such as price freezes, increased price cuts and rebates. If budget pressures continue, governments may implement additional cost-containment measures. Cost-control initiatives could decrease the price we might establish for products that we may develop or sell, which would result in lower product revenues or royalties payable to us. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates. Whether or not we obtain FDA approval for a product candidate, we must obtain approval from the comparable regulatory authorities of foreign countries or economic areas, such as the European Union, before we may commence clinical trials or market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Certain countries outside of the United States have a process that requires the submission of a clinical trial application much like an IND prior to the commencement of human clinical trials. In Europe, for example, a clinical trial application, or CTA, must be submitted to the competent national health authority and to independent ethics committees in each country in which a company intends to conduct clinical trials. Once the CTA is approved in accordance with a country's requirements, clinical trial development may proceed in that country. In all cases, the clinical trials must be conducted in accordance with good clinical practices, or GCPs and other applicable regulatory requirements.
Under European Union regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure is compulsory for medicinal products produced by biotechnology or those medicinal products containing new active substances for specific indications such as the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, viral diseases and designated orphan medicines, and optional for other medicines which are highly innovative. Under the centralized procedure, a marketing application is submitted to the European Medicines Agency where it will be evaluated by the Committee for Medicinal Products for Human Use and a favorable opinion typically results in the grant by the European Commission of a single marketing authorization that is valid for all European Union member states within 67 days of receipt of the opinion. The initial marketing authorization is valid for five years, but once renewed is usually valid for an unlimited period. The decentralized procedure provides for approval by one or more "concerned" member states based on an assessment of an application performed by one member state, known as the "reference" member state. Under the decentralized approval procedure, an applicant submits an application, or dossier, and related materials to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state's assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
Manufacturing
We do not have any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates for non-clinical studies and clinical trials, as
87
well as for commercial manufacture if our product candidates receive marketing approval. To date, we have obtained materials for GEN-003 and GEN-004 from third-party manufacturers who are sole source suppliers to us. For both product candidates, we intend to identify and qualify contract manufacturers to provide the protein process development, protein production and adjuvant production and fill-and-finish services prior to submission of an NDA to the FDA.
Employees
As of June 30, 2014, we had 51 full time employees. Of these 51 employees, 41 employees are engaged in research, development and clinical activities and 10 employees are engaged in finance, human resources, facilities and business and general management. We have no collective bargaining agreements with our employees and we have not experienced any work stoppages. We consider our relations with our employees to be good.
Facilities
Our principal executive offices are located at 100 Acorn Park Drive, Cambridge, Massachusetts 02140, where we occupy approximately 23,666 square feet of laboratory and office space. Our lease term expires on February 28, 2017.
Legal Proceedings
From time to time, we are subject to various legal proceedings and claims that arise in the ordinary course of our business activities. Although the results of litigation and claims cannot be predicted with certainty, as of the date of this prospectus, we do not believe we are party to any claim or litigation, the outcome of which, if determined adversely to us, would individually or in the aggregate be reasonably expected to have a material adverse effect on our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
88
Executive Officers, Significant Employees and Directors
Below is a list of the names, ages as of June 30, 2014 and positions, and a brief account of the business experience of the individuals who serve as our executive officers and directors as of the date of this prospectus.
Name
|
Age | Position | |||
|---|---|---|---|---|---|
William Clark |
45 | President and Chief Executive Officer; Director (Class III) | |||
Seth Hetherington, M.D. |
61 | Chief Medical Officer | |||
Jonathan Poole |
39 | Chief Financial Officer | |||
Robert E. Farrell Jr., CPA |
48 | Vice President of Finance and Administration | |||
Jessica Baker Flechtner, Ph.D. |
42 | Senior Vice President of Research | |||
Paul Giannasca, Ph.D. |
50 | Vice President, Biopharmaceutical Development and Production | |||
George Siber, M.D. |
69 | Director (Class III) | |||
Kevin Bitterman, Ph.D. |
37 | Director (Class I) | |||
Katrine Bosley |
46 | Director (Class II) | |||
Simeon J. George, M.D. |
35 | Director (Class I) | |||
Stephen J. Hoffman, M.D., Ph.D. |
60 | Director (Class II) | |||
William Clark has served as our President and Chief Executive Officer since February 2011. Previously he served as our Chief Business Officer from August 2010 to February 2011. Mr. Clark has served on our board of directors since February 2011. Prior to joining our Company, he served as Chief Business Officer at Vanda Pharmaceuticals, Inc., or Vanda, a biopharmaceutical company he co-founded in 2004. While at Vanda, he lead the company's strategic and business development activities, and played a central role in raising more than $220 million in multiple public and private financings. Prior to Vanda, Mr. Clark was a principal at Care Capital, LLC, a venture capital firm investing in biopharmaceutical companies, after serving in a variety of commercial and strategic roles at SmithKline Beecham (now GlaxoSmithKline). Mr. Clark holds a B.A. from Harvard University and an M.B.A. from The Wharton School at the University of Pennsylvania. We believe that Mr. Clark's operation and historical experience with our Company gained from serving as our Chief Executive Officer, President and member of our board of directors, combined with his prior experience at Vanda and in the venture capital industry focusing on biopharmaceutical companies qualify him to serve as a member of our board of directors.
Seth Hetherington, M.D. has served as our Chief Medical Officer since joining our Company in January 2011. Prior to joining our Company, Dr. Hetherington served as Senior Vice President of Clinical and Regulatory Affairs at Icagen, Inc., or Icagen, from May 2006 through December 2010. Prior to Icagen, Dr. Hetherington served as Vice President, Clinical Development and Chief Medical Officer at Inhibitex Inc. from June 2002 through April 2005 and held various positions of increasing responsibility in clinical drug development at GlaxoSmithKline from 1995 through June 2002. Dr. Hetherington has also served as a faculty member at the University of North Carolina School of Medicine and held appointments at several leading academic medical centers, including the University of Tennessee, St. Jude Children's Research Hospital in Memphis and Albany Medical College. Dr. Hetherington earned his B.S. at Yale University and his M.D. at the University of North Carolina, Chapel Hill. He completed his postgraduate training in pediatrics and pediatric infectious diseases at the University of North Carolina and the University of Minnesota, respectively. Dr. Hetherington has published extensively in medical and scientific literature, and is board certified in both pediatrics and pediatric infectious diseases. He also served as the industry representative to the Vaccines and Related Blood Products Advisory Committee of the FDA. He currently serves as the industry representative on the National Vaccine Advisory Committee of the U.S. Department of Health and Human Services.
89
Jonathan Poole has served as our Chief Financial Officer since joining our Company in April 2014. Prior to joining our Company, Mr. Poole served as Senior Vice President of Finance for Pipeline and Technical Operation at Shire plc ("Shire") from June 2013 through March 2014, leading finance support for Shire's global business development, research and development and technical operations activities. Mr. Poole previously served as divisional Chief Financial Officer of Shire HGT, Shire's rare disease division, from May 2010 through June 2013 and held various positions of increasing responsibility in finance at Shire from 2006 through May 2010. He began his career in the United Kingdom in investment banking at UBS Warburg and ING Barings and also worked as an investment manager for Avanti Capital plc, a United Kingdom private equity investment firm. Mr. Poole has a MBA from London Business School and a BSc in biological sciences from Durham University in the United Kingdom.
Robert E. Farrell Jr., CPA has served as our Vice President of Finance and Administration since joining our Company in May 2009. Prior to joining our Company, he served as Senior Director of Finance at Magen Biosciences, Inc., or Magen, from September 2008 to May 2009. In that position, he was responsible for all finance and administrative functions and he played a key role in the acquisition of Magen by PPD, Inc. Prior to Magen, Mr. Farrell held senior level financial positions at Oscient Pharmaceuticals Corp. and NeoGenesis Pharmaceuticals, Inc. where he built and directed all financial reporting efforts and helped guide the company through an initial public offering. Mr. Farrell is a licensed certified public accountant and holds a B.S. degree in Accounting from Bentley University.
Jessica Baker Flechtner, Ph.D. has held multiple scientific roles since joining our Company in March 2007 and has served as our Senior Vice President of Research since February 2014, Vice President of Research from January 2010 to February 2014 and Senior Director of Research from March 2007 to January 2010. Prior to joining our Company, Dr. Flechtner was an Immunology Consultant at BioVest International, Inc. from June 2006 to March 2007, where she guided the development of assays to evaluate the success of the company's autologous Follicular (Non-Hodgkin's) Lymphoma vaccine in patients. As a researcher at Mojave Therapeutics, Inc., or Mojave, and Antigenics Inc. (now Agenus), which acquired Mojave's intellectual property, from 2001 to 2005, Dr. Flechtner developed protein and peptide-based vaccines and immunotherapies for cancer, infectious disease, autoimmunity and allergy. She is an inventor on 10 pending or issued patents and has multiple peer-reviewed scientific publications. Dr. Flechtner performed her post-doctoral work in the laboratory of Dr. Harvey Cantor at the Dana Farber Cancer Institute and Harvard Medical School and holds a Ph.D. in Cellular Immunology and B.S. in Animal Science from Cornell University. She is a member of the American Association of Immunologists and the American Society for Microbiology.
Paul Giannasca, Ph.D. has served as our Vice President, Biopharmaceutical Development & Production since joining our Company in January 2010. Prior to joining our Company, Dr. Giannasca served as Vice President, Development at Acambis (now Sanofi Pasteur) from 2004 to 2010. He also served as Project Leader of the Clostridium difficile program and R&D Franchise Head for Nosocomials at Acambis/Sanofi Pasteur. Prior to Acambis, he was a senior scientist at OraVax from 1995 to 1999, where he contributed to the company's research initiatives for several vaccines, focusing on evaluating vaccine adjuvants and elucidating mechanisms of vaccine-induced protection. Dr. Giannasca holds multiple patents covering active and passive immunization against Clostridium difficile disease and has published more than 25 papers in the areas of infectious diseases, vaccine-induced protection and vaccine development. Dr. Giannasca received his B.S. in Biology from Fairleigh Dickinson University and his Ph.D. in Molecular and Cellular Biology from the University of Massachusetts-Amherst. He completed his post-doctoral training at Harvard Medical School/Children's Hospital Boston.
George Siber, M.D. has served as a member of our board of directors since 2007. From 1996 to 2007, Dr. Siber served as Executive Vice President and Chief Scientific Officer of Wyeth Vaccines, or Wyeth. While at Wyeth, Dr. Siber oversaw the development and approval of multiple widely-used
90
childhood vaccines, including Prevnar, a pneumococcal vaccine which has achieved multibillion dollar revenues; Acel-Imune, an acellular pertussis vaccine; and Meningitec, a meningococcal meningitis vaccine. Prior to Wyeth, Dr. Siber was Director of the Massachusetts Public Health Biologic Laboratories and a Harvard Medical School Associate Professor of Medicine at Dana Farber Cancer Institute. During this time, Dr. Siber led the research and manufacturing of multiple vaccines and immune globulins including Respigam, a human immune globulin against respiratory syncytial virus. Since 2007, Dr. Siber has served on the boards of directors of several vaccine companies, including Crucell, Selecta Biosciences, Vedantra Pharmaceuticals and Affinivax Inc., and as a consultant or scientific advisory board member of ClearPath Vaccines Company, of which he is currently the Chief Scientific Officer, PaxVax, Vaxess Technologies, Inc., the Bill & Melinda Gates Foundation, PATH, the Wellcome Trust, the European Commission (on vaccinations), the National Institutes of Health, or NIH, and the Korean FDA. Dr. Siber serves as a member of the Board of Trustees of the International Vaccine Institute. Dr. Siber holds an MD degree from McGill University in Canada, received post-doctoral training in Internal Medicine at Rush-Presbyterian Hospital in Chicago and Beth Israel Hospital in Boston and Infectious Disease and vaccinology training at Children's Hospital and Beth Israel Hospital, Harvard Medical School Boston. We believe that Dr. Siber's experience in life sciences and vaccine industries and his experience overseeing the development of multiple vaccines qualifies him to serve as a member of our board of directors.
Kevin Bitterman, Ph.D. has served as a member of our board of directors since August 2006. Dr. Bitterman serves as a partner at Polaris Partners, or Polaris, where he has been employed since 2004 and where he focuses on investments in life sciences companies. Prior to joining Polaris, Dr. Bitterman completed his Ph.D. in genetics at Harvard Medical School. His doctoral research focused on the molecular regulation of caloric restriction and on modulation of a novel class of protein deacetylases. Dr. Bitterman is a cofounder of Sirtris Pharmaceuticals, Inc. acquired by GlaxoSmithKline and was the founding CEO at Visterra Inc. and Editas Medicine Inc. In additional to representing Polaris as a director of our Company, he currently represents Polaris as a director of Editas Medicine Inc., InSeal Medical, Kala Pharmaceuticals, Neuronetics, Inc., Visterra, Inc., TARIS Biomedical, and Vets First Choice. He received a Ph.D. in Genetics from Harvard Medical School and a Bachelor's in Biology from Rutgers College. We believe that Dr. Bitterman's extensive experience investing in, guiding and leading start-up and early phase companies, as well as his experience as a director of other companies, qualifies him to serve as a member of our board of directors.
Katrine Bosley has served as a member of our board of directors since March 2013 and as our chairperson since August 2013. Ms. Bosley is the Chief Executive Officer of Editas Medicine Inc., or Editas, a position to which she was appointed in June 2014. Prior to Editas, Ms. Bosley was the Entrepreneur-in-Residence at The Broad Institute from September 2013 to May 2014. She served as Chief Executive Officer of Avila Therapeutics Inc., or Avila, from May 2009 to March, 2012, when Avila was acquired by Celgene Corporation. Before Avila, she was Vice President, Strategic Operations at Adnexus, a Bristol-Myers Squibb Company and was Vice President, Business Development at Adnexus Therapeutics Inc., or Adnexus, before that. She joined Adnexus from Biogen Idec where she held roles in business development, commercial operations, and portfolio strategy in the United States and Europe and led the in-licensing of Tysabri (natalizumab) among a number of other transactions. Earlier, she was part of the healthcare team at the venture firm Highland Capital Partners from 1993 to 1995. In addition to serving as a director of our Company, Ms. Bosley currently serves as a director of Galapagos NV, Scholar Rock LLC, and Coco Therapeutics Ltd. Ms. Bosley graduated from Cornell University with a Bachelor of Arts degree in biology. We believe that Ms. Bosley's experience as a chief executive officer of a biotechnology company and her breadth of experience in creating strategic and business development value qualifies her to serve as a member of our board of directors.
Simeon J. George, M.D. has served as a member of our board of directors since February 2009. Since 2007, Dr. George has served as partner of S.R. One, Limited, or S.R. One, and leads S.R. One's
91
west coast investment activities. Prior to joining S.R. One, Dr. George was a consultant at Bain & Company from October 2006 to August 2007. In addition to serving as a director of our Company, Dr. George currently serves as a director of RuiYi, Auxogyn, Inc., eFFECTOR, Inc., HTG Molecular and Principia Biosciences. He received his BA in Neuroscience from the Johns Hopkins University, where he graduated Phi Beta Kappa, and received his MD from the University of Pennsylvania School of Medicine and his MBA (Mayer Scholar) from the Wharton School of the University of Pennsylvania. We believe that Dr. George's experience in the venture capital industry, particularly with biotechnology and pharmaceutical companies, as well as his experience as a director of other companies, qualifies him to serve as a member of our board of directors.
Stephen J. Hoffman, M.D., Ph.D. has served as a member of our board of directors since December 2010. Dr. Hoffman has been a Senior Advisor to PDL BioPharma, Inc. since February 2014. Prior to that, Dr. Hoffman served as a managing director at Skyline Ventures, a venture capital firm, since May 2007. From January 2003 to March 2007, Dr. Hoffman was a general partner at TVM Capital, a venture capital firm. Prior to that, he served as President, Chief Executive Officer and a director of Allos Therapeutics, Inc., or Allos, a biopharmaceutical company, from 1994 to 2002, and as Chairman of the Board until 2012. From 1990 to 1994, Dr. Hoffman completed a fellowship in clinical oncology and a residency/fellowship in dermatology, both at the University of Colorado. Dr. Hoffman was the scientific founder of Somatogen Inc., a biotechnology company that was acquired by Baxter International, Inc., a global medical products and services company, in 1998, where he held the position of Vice President of Science and Technology from 1987 until 1990. In addition to serving as a director of our Company, he currently serves as a director of several biopharmaceutical companies, including AcelRx, Inc., Concert Pharmaceuticals, Inc., Collegium Pharmaceuticals, Inc., Dicerna Pharmaceuticals, Inc. and Proteon Therapeutics, Inc. Previously, Dr. Hoffman served on the board of directors of Sirtris Pharmaceuticals, Inc., a pharmaceutical company that was acquired by GlaxoSmithKline, in 2008. Dr. Hoffman holds a Ph.D. in bio-organic chemistry from Northwestern University and an M.D. from the University of Colorado School of Medicine. We believe that Dr. Hoffman's scientific, financial and business expertise, including his diversified background as an executive officer and investor in public pharmaceutical companies as well as a director of a public pharmaceutical company, qualifies him to serve as a member of our board of directors.
Board Composition and Election of Directors
Board Composition
Our board of directors is currently comprised of six members. Our board of directors has determined that each of Dr. Bitterman, Ms. Bosley, Dr. George and Dr. Hoffman is independent for NASDAQ purposes. The members of our board of directors were elected in compliance with the provisions of the voting agreement among us and our major stockholders. The voting agreement terminated upon the completion of the initial public offering on February 10, 2014 and we have no further contractual obligations regarding the election of our directors. See "Certain Relationships and Related Party Transactions." Our directors hold office until their successors have been elected and qualified or until their earlier death, resignation or removal. There are no family relationships among any of our directors or executive officers.
Our amended and restated certificate of incorporation and amended and restated by-laws provide that the authorized number of directors may be changed only by resolution of our board of directors. Our amended and restated certificate of incorporation and amended and restated by-laws also provide that our directors may be removed only for cause by the affirmative vote of the holders of at least 75% of the votes that all our stockholders would be entitled to cast in an annual election of directors, and that any vacancy on our board of directors, including a vacancy resulting from an enlargement of our board of directors, may be filled only by vote of a majority of our directors then in office.
92
In accordance with the terms of our certificate of incorporation and by-laws that, our board of directors are divided into three classes, class I, class II and class III, with members of each class serving staggered three-year terms. The members of the classes are divided as follows:
- •
- the class I directors are Dr. Bitterman and Dr. George, whose terms expire in 2015;
- •
- the class II directors are Ms. Bosley and Dr. Hoffman, whose terms expire in 2016; and
- •
- the class III directors are Mr. Clark and Dr. Siber, whose terms expire in 2017.
Upon the expiration of the term of a class of directors, directors in that class will be eligible to be elected for a new three-year term at the annual meeting of stockholders in the year in which their term expires.
We have no formal policy regarding board diversity. Our priority in selection of board members is identification of members who will further the interests of our stockholders through his or her established record of professional accomplishment, the ability to contribute positively to the collaborative culture among board members, knowledge of our business and understanding of the competitive landscape.
Director Independence
Applicable NASDAQ rules require a majority of a listed company's board of directors to be comprised of independent directors within one year of listing. In addition, the NASDAQ rules require that, subject to specified exceptions, each member of a listed company's audit, compensation and nominating and corporate governance committees be independent and that audit committee members also satisfy independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Under applicable NASDAQ rules, a director will only qualify as an "independent director" if, in the opinion of the listed company's board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. In order to be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or otherwise be an affiliated person of the listed company or any of its subsidiaries.
In November 2013, our board of directors undertook a review of the composition of our board of directors and its committees and the independence of each director. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our board of directors has determined that Dr. Bitterman, Ms. Bosley, Dr. George and Dr. Hoffman are "independent directors" as defined under applicable NASDAQ rules. In making such determination, our board of directors considered the relationships that each such non-employee director has with our company and all other facts and circumstances that our board of directors deemed relevant in determining his or her independence, including the beneficial ownership of our capital stock by each non-employee director. Mr. Clark is not an independent director under these rules because he is our Chief Executive Officer and Dr. Siber is not an independent director under these rules because of his consulting relationship with us. Please see the section of this prospectus titled "Certain Relationships and Related Party Transactions".
There are no family relationships among any of our directors or executive officers.
Board Committees
Our board of directors has three standing committees: the audit committee, the compensation committee and the nominating and corporate governance committee.
93
Audit Committee
Our audit committee is composed of Ms. Bosley, Dr. George and Dr. Hoffman, with Dr. Hoffman serving as chairman of the committee. Our board of directors has determined that each member of the audit committee meets the independence requirements of Rule 10A-3 under the Exchange Act and the applicable listing standards of NASDAQ. Our board of directors has determined that Ms. Bosley and Dr. Hoffman are "audit committee financial experts" within the meaning of the SEC regulations and applicable listing standards of NASDAQ. The audit committee's responsibilities include:
- •
- appointing, approving the compensation of, and assessing the qualifications, performance and independence of our
independent registered public accounting firm;
- •
- pre-approving audit and permissible non-audit services, and the terms of such services, to be provided by our independent
registered public accounting firm;
- •
- reviewing the internal audit plan with the independent registered public accounting firm and members of management
responsible for preparing our financial statements;
- •
- reviewing and discussing with management and the independent registered public accounting firm our annual and quarterly
financial statements and related disclosures as well as critical accounting policies and practices used by us;
- •
- reviewing the adequacy of our internal control over financial reporting;
- •
- establishing policies and procedures for the receipt and retention of accounting-related complaints and concerns;
- •
- recommending, based upon the audit committee's review and discussions with management and the independent registered
public accounting firm, whether our audited financial statements shall be included in our Annual Report on Form 10-K;
- •
- monitoring our compliance with legal and regulatory requirements as they relate to our financial statements and accounting
matters;
- •
- preparing the audit committee report required by the rules of the SEC to be included in our annual proxy statement;
- •
- viewing all related party transactions for potential conflict of interest situations and approving all such transactions;
and
- •
- reviewing and discussing with management and our independent registered public accounting firm our earnings releases and scripts.
Compensation Committee
Our compensation committee is composed of Dr. Bitterman and Dr. George, with Dr. Bitterman serving as chairman of the committee. Our board of directors has determined that each member of the compensation committee is "independent" as defined under the applicable listing standards of NASDAQ. The compensation committee's responsibilities include:
- •
- annually reviewing and approving corporate goals and objectives relevant to the compensation of our chief executive
officer;
- •
- evaluating the performance of our chief executive officer in light of such corporate goals and objectives and determining
and approving the compensation of our chief executive officer;
- •
- reviewing and approving the compensation of our other executive officers;
94
- •
- appointing, compensating and overseeing the work of any compensation consultant, legal counsel or other advisor retained
by the compensation committee;
- •
- conducting the independence assessment outlined in NASDAQ rules with respect to any compensation consultant, legal counsel
or other advisor retained by the compensation committee;
- •
- annually reviewing and reassessing the adequacy of the committee charter in its compliance with the listing requirements
of NASDAQ;
- •
- reviewing and establishing our overall management compensation philosophy and policy;
- •
- overseeing and administering our equity compensation and other compensatory plans;
- •
- reviewing and approving our equity and incentive policies and procedures for the grant of equity-based awards and
approving the grant of such equity-based awards;
- •
- reviewing and making recommendations to the board of directors with respect to director compensation; and
- •
- reviewing and discussing with management the compensation discussion and analysis to be included in our annual proxy statement or Annual Report on Form 10-K.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee is composed of Dr. Bitterman, Ms. Bosley and Dr. Hoffman, with Ms. Bosley serving as chairman of the committee. Our board of directors has determined that each member of the nominating and corporate governance committee is "independent" as defined under the applicable listing standards of NASDAQ. The nominating and corporate governance committee's responsibilities include:
- •
- developing and recommending to the board of directors criteria for board and committee membership;
- •
- establishing procedures for identifying and evaluating board of director candidates, including nominees recommended by
stockholders;
- •
- identifying individuals qualified to become members of the board of directors;
- •
- recommending to the board of directors the persons to be nominated for election as directors and to each of the board's
committees;
- •
- developing and recommending to the board of directors a set of corporate governance principles;
- •
- articulating to each director what is expected, including reference to the corporate governance principles and directors'
duties and responsibilities;
- •
- reviewing and recommending to the board of directors practices and policies with respect to directors;
- •
- reviewing and recommending to the board of directors the functions, duties and compositions of the committees of the board
of directors;
- •
- reviewing and assessing the adequacy of the committee charter and submitting any changes to the board of directors for
approval;
- •
- consider and report to the board of directors any questions of possible conflicts of interest of board of directors members;
95
- •
- provide for new director orientation and continuing education for existing directors on a periodic basis;
- •
- performing an evaluation of the performance of the committee;
- •
- overseeing the evaluation of the board of directors and management; and
- •
- our board of directors may establish other committees from time to time.
Compensation Committee Interlocks and Insider Participation
None of our executive officers serves as a member of the board of directors or compensation committee, or other committee serving an equivalent function, of any other entity that has one or more of its executive officers serving as a member of our board of directors or compensation committee. None of the members of our compensation committee has ever been employed by us. For a description of transactions between us and members of our compensation committee and affiliates of such members, please see the section of this prospectus titled "Certain Relationships and Related Party Transactions".
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that applies to all of our employees, officers and directors, including those officers responsible for financial reporting. Our code of business conduct and ethics is available on our website. We intend to disclose any amendments to the code, or any waivers of its requirements, on our website.
96
EXECUTIVE AND DIRECTOR COMPENSATION
Overview
The following discussion relates to the compensation of our President and Chief Executive Officer, William Clark, and our two most highly compensated executive officers (other than our Chief Executive Officer), Seth Hetherington, M.D., our Chief Medical Officer, and Jessica Flechtner, Ph.D., our Senior Vice President of Research. These three executives are collectively referred to in this prospectus as our named executive officers. Each year, the compensation committee of our board of directors and our board of directors review and determine the compensation of our named executive officers.
Elements of Executive Compensation
The compensation of our named executive officers consists of base salary, annual cash bonuses and equity awards as well as employee benefits that are made available to substantially all salaried employees. Our named executive officers are also entitled to certain compensation and benefits upon certain terminations of employment and change of control transactions pursuant to employment letter agreements.
Base Salaries. Base salaries for our named executive officers are reviewed annually by our compensation committee and are set by our board of directors. When making its base salary recommendations to our board of directors, our compensation committee takes factors into account such as each executive's experience and individual performance, the company's performance as a whole, data from surveys of compensation paid by comparable companies, cost of living increases and general industry conditions, but does not assign any specific weighting to any factor. Our board of directors determines each named executive officer's base salary after reviewing the compensation committee's recommendation. In fiscal 2013, on the recommendation of our compensation committee, our board of directors approved a base salary of $335 thousand for Mr. Clark, $333 thousand for Dr. Hetherington and $242 thousand for Dr. Flechtner, representing an increase of 2.0%, 2.0% and 10.0%, respectively, from the base salary for each such executive in 2012.
Annual Cash Bonuses. Our annual cash bonus program promotes and rewards the achievement of key strategic business goals and individual performance goals. For fiscal 2013, the target annual bonus as a percentage of base salary for each of Mr. Clark, Dr. Hetherington and Dr. Flechtner was 40%, 30% and 25%, respectively. In the case of Mr. Clark, 100% of his annual bonus was based on the achievement of pre-established corporate performance goals and, in the case of Drs. Hetherington and Flechtner, 50% of the executive's respective annual bonus was based on the achievement of pre-established corporate performance goals and 50% was based on a quantitative and qualitative assessment of pre-established individual performance goals.
At the beginning of fiscal 2013, our compensation committee established the corporate performance goals for 2013, each having a designated weighting. These corporate performance goals included key strategic and financial goals related to business development and grant funding, maintenance of a certain level of cash reserves, the development and commencement of certain clinical and commercial programs, the completion of research reports, and other strategic objectives related to our clinical pipeline. Also at the beginning of fiscal 2013, our chief executive officer, working with each of Dr. Hetherington and Dr. Flechtner, established each executive's individual performance goals and their weightings. These goals included objectives related to the oversight of clinical activities for compliance with laws, developing and conducting clinical programs and studies, research and development, managing studies according to schedule and within budgets, business and corporate development and demonstrating leadership with respect to direct reports.
In February 2014, our compensation committee met to evaluate the extent to which the performance goals for 2013 were achieved. In determining the level of corporate performance for the
97
year, our compensation committee evaluated the company's performance against the pre-established corporate performance goals for such year and took into account our chief executive officer's assessment of his own performance and his assessment of the extent to which the corporate performance goals were achieved. Based on its evaluation, our compensation committee determined that 80% of the corporate performance goals were achieved. Our board of directors reviewed the compensation committee's recommendation and approved the achievement of the corporate performance goals at this level.
With respect to Dr. Hetherington's and Dr. Flechtner's individual performance goals, Mr. Clark evaluated each executive's performance during the year against his or her pre-established individual performance goals, taking into consideration an assessment by the executive of his or her own performance. Based on this evaluation, Mr. Clark determined that, for each executive, 98% of his or her individual performance goals were achieved. After reviewing Mr. Clark's determination of individual goal achievement and determining the level of corporate goal achievement, our compensation committee recommended, and our board of directors approved, an annual bonus for 2013 of $107,320, for Mr. Clark, $88,989, for Dr. Hetherington, and $54,087 for Dr. Flechtner.
Equity Awards. Our named executive officers participate in the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan, which we refer to as the "2007 Equity Plan". Our named executive officers are also eligible to participate in the Genocea Biosciences, Inc. 2014 Equity Incentive Plan, which we refer to as the "2014 Equity Plan". Our 2014 Equity Plan was adopted by our board of directors in connection with our initial public offering and following our initial public offering, all equity-based awards, including awards to our named executive officers, will be made under our 2014 Equity Plan.
Initial awards of stock options granted to our named executive officers generally vest as to 25% of the shares subject to the stock option 10 to 12 months from the date of grant and thereafter continue to vest in monthly installments over the following 36 months, generally subject to the executive's continued employment. Other time-vesting stock option awards generally vest in 48 equal monthly installments, generally subject to the executive's continued employment.
In July 2013, we granted time-vesting stock options to each of our named executive officers. Mr. Clark received a stock option to purchase 133,602 shares of our common stock, Dr. Hetherington received a stock option to purchase 57,017 shares of our common stock, and Dr. Flechtner received a stock option to purchase 56,378 shares of our common stock. These stock option awards vested as to 1/8th of the shares subject to the stock option on the date of grant and continue to vest in equal monthly installments over 42 months, generally subject to the executive's continued employment. Mr. Clark also received a performance-vesting stock option to purchase 81,670 shares of our common stock that vested in full upon the completion of our initial public offering on February 10, 2014. In connection with our initial public offering, on February 4, 2014, Dr. Flechtner received a stock option to purchase 11,402 shares of our common stock and, on February 20, 2014, she received an additional stock option to purchase 14,397 shares of our common stock. These stock option awards vest in equal monthly installments over 48 months, generally subject to the executive's continued employment.
Stock option awards serve to align the interests of our named executive officers with our shareholders because no value is created unless the value of our common stock appreciates after grant. Stock option awards also encourage retention through the use of time-based vesting and the achievement of key strategic goals through the use of performance-based vesting. Pursuant to agreements with our named executive officers, all of each executive's stock option awards will vest automatically upon certain terminations of employment following a change of control of our company. See "—Employment Letter Agreements" below for additional details about these agreements.
98
Benefits. We provide modest benefits to our named executive officers, which are limited to participation in our 401(k) plan and basic health and welfare benefit coverage. These benefits are available to substantially all of our salaried employees.
Employment Letter Agreements. We have entered into an amended and restated employment letter agreement with each of our named executive officers that, in each case, includes severance and change of control protections. Our named executive officers are also subject to restrictive covenants, covering noncompetition, nonsolicitation and confidentiality.
Summary Compensation Table
The following table sets forth information about certain compensation awarded or paid to our named executive officers for fiscal years 2012, in the case of Mr. Clark and Dr. Hetherington, and 2013, in the case of all of our named executive officers.
Name and principal position
|
Year | Salary ($)(2) |
Option awards ($)(3) |
Nonequity incentive plan compensation ($)(4) |
Total ($) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
William Clark, |
2013 | 334,280 | 413,842 | 107,320 | 855,442 | |||||||||||
President and Chief Executive Officer |
2012 | 327,921 | — | 105,242 | 433,163 | |||||||||||
Seth Hetherington, M.D., |
2013 | 331,459 | 176,616 | 88,989 | 597,064 | |||||||||||
Chief Medical Officer |
2012 | 324,776 | — | 84,895 | 409,671 | |||||||||||
Jessica Flechtner, Ph.D., |
2013 | 238,333 | 174,637 | 54,087 | 467,057 | |||||||||||
Senior Vice President, Research(1) |
||||||||||||||||
- (1)
- Dr. Flechtner
was not a named executive officer in fiscal year 2012 and, as a result, no amounts with respect to fiscal year 2012 have been included
for Dr. Flechtner in the table above.
- (2)
- Salaries
include amounts contributed by the named executive officer to our 401(k) plan.
- (3)
- Amounts
shown reflect the aggregate grant date fair value of time-vesting stock options awarded in fiscal 2013, computed in accordance with FASB ASC Topic
718 and exclude the value of estimated forfeitures. Assumptions used in the calculation of these amounts are included in Note 12 to our annual financial statements, which are incorporated
herein by reference. Mr. Clark was also granted a performance-vesting stock option in 2013. The grant date fair value of the performance-vesting stock option granted to Mr. Clark in
fiscal year 2013 is based on the probable outcome of the performance conditions associated with the stock option as of the date of grant. No amount was included in the table above for this stock
option since the performance conditions were not considered probable of occurring on the date of grant. The aggregate grant date fair value of the performance-vesting stock option assuming that the
highest levels of performance conditions are achieved is $252,981. No stock options were awarded to our named executive officers in fiscal 2012.
- (4)
- Amounts shown reflect the cash amount paid to the named executive officer for the relevant fiscal year that was earned based on the achievement of company performance goals, in the case of Mr. Clark, and company and individual performance goals, in the case of Drs. Hetherington and Flechtner.
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information regarding equity awards held by our named executive officers as of December 31, 2013. Our named executive officers do not hold any equity awards other than stock options.
99
Name
|
Number of securities underlying unexercised options (#) exercisable |
Number of securities underlying unexercised options (#) unexercisable |
Equity incentive plan awards: number of securities underlying unexercised unearned options (#) |
Option Exercise Price ($)(7) |
Option Expiration Date(8) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
William Clark, |
33,172 | (1) | 6,635 | (1) | — | 2.86 | 8/19/2020 | |||||||||
President and Chief Executive Officer |
18,537 | (2) | 6,180 | (2) | — | 2.86 | 12/17/2020 | |||||||||
|
— | — | 39,807 | (3) | 2.86 | 12/17/2020 | ||||||||||
|
241,232 | (2) | 99,337 | (2) | — | 2.02 | 2/17/2021 | |||||||||
|
30,616 | (4) | 102,986 | (4) | 3.45 | 7/25/2023 | ||||||||||
|
— | — | 81,670 | (3) | 3.45 | 7/25/2023 | ||||||||||
Seth Hetherington, M.D., |
55,474 | (1) | 18,493 | (1) | — | 2.02 | 2/17/2021 | |||||||||
Chief Medical Officer |
25,551 | (3) | — | — | 2.02 | 2/17/2021 | ||||||||||
|
13,066 | (4) | 43,951 | (4) | 3.45 | 7/25/2023 | ||||||||||
Jessica Flechtner, Ph.D., |
1,680 | (1) | — | — | 1.67 | 5/15/2017 | ||||||||||
Senior Vice President, Research |
420 | (1) | — | — | 1.67 | 8/20/2017 | ||||||||||
|
840 | (1) | — | — | 1.67 | 1/23/2018 | ||||||||||
|
8,403 | (5) | — | — | 2.38 | 6/30/2019 | ||||||||||
|
2,941 | (3) | — | — | 2.38 | 6/30/2019 | ||||||||||
|
4,376 | (2) | 191 | (2) | — | 2.86 | 3/28/2019 | |||||||||
|
10,333 | (2) | 4,256 | (2) | — | 2.02 | 2/17/2021 | |||||||||
|
5,462 | (6) | — | — | 2.02 | 2/17/2021 | ||||||||||
|
12,919 | (4) | 43,459 | (4) | — | 3.45 | 7/25/2023 | |||||||||
- (1)
- Reflects
time-based stock options to purchase shares of our common stock that generally vest as to 25% of the shares subject to the stock option 10 to
12 months from the date of grant and thereafter vest in equal monthly installments on the last day of each calendar month over the following 36 months, generally subject to the
executive's continued employment. In the case of the stock option granted to Dr. Flechtner on August 20, 2007, the stock option vested as to 25% of the shares subject to the stock option
on the date of grant and thereafter vested in equal monthly installments on the last day of each calendar month over the following 36 months.
- (2)
- Reflects
time-based stock options to purchase shares of our common stock that generally vest in 48 equal monthly installments, generally on each
monthly anniversary of the date of grant or the last day of each calendar month following the date of grant, generally subject to the executive's continued employment. In the case of the stock option
granted to Dr. Flechtner on March 28, 2010, the first installment vested on March 18, 2010.
- (3)
- Reflects
performance-based stock options to purchase shares of our common stock that vest as to 100% of the shares subject to the stock option, in the case
of Mr. Clark, upon the company's achievement of specified strategic financing or development milestones, in the case of Dr. Hetherington, upon the company's achievement of a milestone
related to the initiation of a clinical trial, and in the case of Dr. Flechtner, upon the company's achievement of a specified financial goal, in each case, generally subject to the executive's
continued employment. The performance-based stock option awarded to Mr. Clark on July 25, 2013 vested in full upon the completion of our initial public offering on February 10,
2014.
- (4)
- Reflects time-based stock options to purchase shares of our common stock that vested as to 1/8th of the shares subject to the stock option on the date of grant and that continue to vest in
100
equal monthly installments over 42 months following the date of grant, generally subject to the executive's continued employment.
- (5)
- Reflects
a time-based stock option to purchase shares of our common stock that vested as to 6.25% of the shares subject to the stock option on the date of
grant and that continues to vest as to 2.0833% of the shares subject to the stock option on the last day of each calendar month thereafter, generally subject to the executive's continued employment.
- (6)
- Reflects
a time-based stock option to purchase shares of our common stock that vested as to 2,162 shares subject to the stock option on the date of grant
and that continued to vest in equal monthly installments over the following 29 months, generally subject to the executive's continued employment.
- (7)
- The
exercise price of the stock options is not less than the fair market value of a share of our common stock, as determined by our board of directors
based, in part, on an independent third party valuation.
- (8)
- All stock options have a 10-year term measured from the date of grant, except in the case of the stock option awarded to Dr. Flechtner on March 28, 2010, which has a term of nine years from the date of grant.
Retirement Benefits
We do not maintain any qualified or non-qualified defined benefit plans or supplemental executive retirement plans that cover our named executive officers. We offer a tax-qualified retirement plan, which we refer to as our 401(k) plan, to eligible employees, including our named executive officers. Our 401(k) plan permits eligible employees to defer their annual eligible compensation subject to the limitations imposed by the Internal Revenue Service. We may, but are not required to, make discretionary profit-sharing contributions on behalf of eligible employees under this plan. We did not make any contributions on behalf of eligible employees in fiscal year 2013.
Employment Letter Agreements
Mr. Clark, Dr. Hetherington and Dr. Flechtner
On January 16, 2014, we entered into an amended and restated employment letter agreement with each of Mr. Clark, Dr. Hetherington and Dr. Flechtner, which became effective prior to the completion of our initial public offering. Each employment letter agreement provides for an initial base salary of $399,433, in the case of Mr. Clark, $369,458 in the case of Dr. Hetherington, and $287,012 in the case of Dr. Flechtner, as well as a discretionary performance-based bonus, with a target, as a percentage of base salary, of 50%, 35% and 30% for each of Mr. Clark, Dr. Hetherington and Dr. Flechtner, respectively. Each agreement also provides for severance payments and benefits upon certain terminations of the executive's employment as described below.
Termination of Employment without Cause or for Good Reason Following a Change of Control. If, within 12 months after a change of control (as defined in the executive's employment letter agreement), the executive's employment is terminated by us without cause or the executive terminates his or her employment for good reason (as such terms are defined in the executive's employment letter agreement), all stock options or other equity awards then held by the executive will fully vest. In addition, the executive will be entitled to receive base salary and payment of COBRA premiums for 18 months, in the case of Mr. Clark, 15 months, in the case of Dr. Hetherington, and 12 months, in the case of Dr. Flechtner, following such termination of employment.
Termination of Employment without Cause or for Good Reason. If the executive's employment is terminated by us without cause or the executive terminates his or her employment for good reason (as
101
such terms are defined in the executive's employment letter agreement) other than following a change of control as described above, the executive will be entitled to receive base salary and payment of COBRA premiums for 12 months, in the case of Mr. Clark, nine months, in the case of Dr. Hetherington, and six months, in the case of Dr. Flechtner, following such termination of employment.
Termination of Employment Due to Death or Disability. If the executive's employment is terminated by us due to the executive's disability or is terminated due to the executive's death, we will pay the executive a portion of the executive's target annual cash bonus for the year in which such termination of employment occurs, prorated based on the number of days the executive was employed during such year until the date of such termination.
Severance Subject to Release of Claims. Our obligation to provide the executive with any severance payments or other benefits under the executive's employment letter agreement is conditioned on the executive signing and not revoking an effective release of claims in our favor.
Other Termination of Employment. If the executive's employment is terminated for any reason other than by us without cause, by the executive for good reason, or due to the executive's death or disability, the executive will only be entitled to receive earned but unpaid base salary and any accrued but not used vacation as of the termination date.
280G Better-of Provision. In the event of a change in ownership or control of our company under Section 280G of the Internal Revenue Code of 1986, as amended, or the Code, and the regulations thereunder, if any portion of the payments made pursuant to the executive's employment letter agreement (or otherwise) constitutes an "excess parachute payment" within the meaning of Section 280G of the Code, the executive will be entitled to receive an amount of such payments reduced so that no portion of the payments would constitute an excess parachute payment, or the amount otherwise payable to the executive under the employment letter agreement (or otherwise) reduced by all applicable taxes, including the excise tax, whichever amount results in the greater amount payable to the executive.
Employment Conditioned on Restrictive Covenants. As a condition to the executive's employment with us, the executive was required to sign and must comply with the terms of an At-Will Employment, Confidential Information, Invention Assignment and Non-Competition Agreement, pursuant to which the executive has agreed not to compete with us for a period of 12 months following the termination of his or her employment and not to solicit our employees or independent contractors for a period of 36 months following the termination of his or her employment. Each executive has also agreed to covenants relating to the use and disclosure of confidential information and the assignment of inventions.
Mr. Poole
We entered into an employment letter agreement with Jonathan Poole, who began serving as our Chief Financial Officer on April 7, 2014. Mr. Poole's employment letter agreement provides for an initial base salary of $320,000, a discretionary, performance-based bonus, with a target, as a percentage of base salary, of 35%, and a cash signing bonus of $50,000. Pursuant to the employment letter agreement, we granted Mr. Poole a stock option to purchase 200,726 shares of our common stock under the 2014 Equity Plan. Mr. Poole's employment letter agreement also provides for severance payments and benefits upon certain terminations of employment as described below.
If, within 12 months after a change of control (as defined in Mr. Poole's employment letter agreement), Mr. Poole's employment is terminated by us without cause or by him for good reason (as such terms are defined in his employment letter agreement), all stock options or other equity awards then held by him will fully vest. In addition, Mr. Poole will be entitled to receive base salary and
102
payment of COBRA premiums for 15 months following such termination of employment. If Mr. Poole's employment is terminated by us without cause or by him for good reason other than following a change of control, he will be entitled to receive base salary and payment of COBRA premiums for nine months following such termination of employment. In the event Mr. Poole's employment is terminated by us due to his disability or upon his death, we will pay Mr. Poole a portion of his target annual cash bonus for the year in which such termination of employment occurs, prorated based on the number of days he was employed during such year until the date of such termination. Upon a termination of employment other than by us without cause, or by Mr. Poole for good reason, or due to his death or disability, Mr. Poole will only be entitled to receive earned but unpaid base salary and any accrued but not used vacation as of the termination date.
Mr. Poole's employment letter agreement also contains provisions that condition any severance payments or other benefits on his execution and non-revocation of an effective release of claims in our favor and provisions regarding Section 280G of the Code and restrictive covenants similar to those provisions that are included in the employment letter agreements with our named executive officers, as described above.
2013 Director Compensation
The following table sets forth information concerning the compensation earned by our directors during 2013. In 2013, Dr. Siber and Ms. Bosley were the only directors who were compensated for service on our board of directors. Mr. Clark receives no additional compensation for his service as a director, and, consequently, is not included in this table. The compensation received by Mr. Clark as our chief executive officer during 2013 is included in the "Summary Compensation Table" above.
103
Name
|
Fees Earned or Paid in Cash ($)(1) |
Option Awards ($)(2) |
Total ($) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
George Siber, M.D. |
124,992 | 54,263 | 179,255 | |||||||
Katrine Bosley |
32,500 | 192,355 | 224,855 | |||||||
- (1)
- Amounts
represent annual director and, in the case of Dr. Siber, consulting fees, for services rendered by Dr. Siber and Ms. Bosley.
Amounts paid to Dr. Siber were paid in equal bi-monthly installments and amounts paid to Ms. Bosley were paid quarterly in arrears.
- (2)
- Amounts represent the aggregate grant date fair value of awards of time-vesting stock options granted to Dr. Siber and Ms. Bosley in fiscal 2013. These amounts were computed in accordance with FASB ASC Topic 718 and exclude the value of estimated forfeitures. Assumptions used in the calculation of these amounts are included in Note 12 to our annual financial statements, which are incorporated herein by reference. Dr. Siber was also granted a performance-vesting stock option in fiscal 2013 that vested upon the completion of our initial public offering on February 10, 2014. The grant date fair value of the performance-vesting stock option granted to Dr. Siber in 2013 is based on the probable outcome of the performance conditions associated with this stock option as of the date of grant. No amount is included in the table above for this stock option since the performance conditions were not considered probable of occurring on the date of grant. The aggregate grant date fair value of this performance-vesting stock option assuming that the highest levels of performance conditions are achieved is $32,908.
As of December 31, 2013, our directors held the following aggregate number of options to purchase shares of our common stock: Dr. Siber held options to purchase 133,672 shares of our common stock, Ms. Bosley held options to purchase 36,966 shares of our common stock and Dr. Bitterman, Dr. George and Dr. Hoffman held no options to purchase shares of our common stock. As of December 31, 2013, Ms. Bosley held 24,615 restricted shares, which she received upon the exercise of the option granted to her on February 4, 2013.
Non-Employee Director Compensation Policy
Our board of directors has adopted a non-employee director compensation policy, which became effective prior to the completion of our initial public offering and is designed to enable us to attract and retain, on a long-term basis, highly qualified non-employee directors. Under the policy, all non-employee directors are paid cash compensation as set forth in the following table:
| |
Annual Retainer |
|||
|---|---|---|---|---|
Board of Directors: |
||||
All non-employee members |
$ | 35,000 | ||
Additional retainer for chair |
$ | 25,000 | ||
Audit Committee: |
||||
Members |
$ | 7,500 | ||
Additional retainer for chair |
$ | 7,500 | ||
Compensation Committee: |
||||
Members |
$ | 5,000 | ||
Additional retainer for chair |
$ | 5,000 | ||
Nominating and Corporate Governance Committee: |
||||
Members |
$ | 3,500 | ||
Additional retainer for chair |
$ | 3,500 | ||
104
Under our non-employee director compensation policy, each individual who is not an employee who is initially appointed or elected to our board of directors will be eligible to receive a grant of stock options to purchase 10,084 shares of our common stock under our 2014 Equity Plan at the time of his or her initial appointment or election to our board of directors, which will vest annually in equal installments over a three-year period. In addition, each continuing non-employee director will be eligible to receive, on the first business day following January 1st of each calendar year, an annual stock option grant to purchase 5,042 shares of our common stock, which will vest in full on the first anniversary of the date of grant. The stock options will be granted with an exercise price equal to the fair market value of a share of our common stock on the date of grant and have a 10-year term.
Director Agreements
Dr. Siber
We have entered into a consulting agreement with Dr. Siber dated May 16, 2007, as amended on June 30, 2009, December 16, 2010, June 15, 2011 and June 5, 2013, providing for a consulting fee of $10 thousand per month, for consulting services performed by Dr. Siber related to strategic scientific and business development as well as for his service as the chairman of our board of directors. Dr. Siber was also entitled to receive grants of restricted stock and stock options in connection with his service to us. All stock options granted to Dr. Siber pursuant to the consulting agreement will fully vest if, within 12 months following a change of control, either we (or our successor) terminate the consulting agreement without cause (as such term is defined in the consulting agreement), subject to Dr. Siber's continued service to the company, or we (or our successor) do not offer to extend the term of the agreement. As of September 19, 2013, Dr. Siber ceased being the chairman of our board of directors and assumed the role of executive director and chairman of our scientific advisory board.
Dr. Siber has agreed not to solicit our employees, contractors, and customers for a period of 12 months following the termination of the consulting agreement and is subject to covenants relating to the use and disclosure of confidential information and the assignment of inventions. Unless extended or earlier terminated, the term of the consulting agreement will expire on June 17, 2015.
In 2013, performance-vesting and time-vesting stock options were granted to Dr. Siber. The performance-vesting stock option generally vests upon the company's achievement of certain financial goals, and vested in full upon the completion of our initial public offering on February 10, 2014. The time-vesting stock option generally vests in equal monthly installments over 48 months and vests in full upon a change of control of our company, generally subject to Dr. Siber's continued service to our company.
Ms. Bosley
We entered into a letter agreement with Ms. Bosley, the chair of our board of directors, dated as of February 4, 2013, the date she was appointed to serve on our board of directors. Pursuant to the letter agreement, Ms. Bosley was entitled to an annual fee for board meeting attendance of $30,000 per year (which annual fee was subsequently increased to $50,000 per year) and, on February 4, 2013, we granted Ms. Bosley an initial stock option award that vests ratably over 48 months, subject to Ms. Bosley's continued service on our board of directors on the applicable vesting date, and vests as to all of the shares subject to the stock option immediately prior to the occurrence of a covered transaction (as defined in the 2007 Equity Plan). Ms. Bosley subsequently exercised this stock option and, with respect to the portion of the stock option that was not vested on the date it was exercised, received shares of restricted stock that vest on the same schedule as the stock option. Effective as of the completion of our initial public offering, this letter agreement terminated and Ms. Bosley is eligible to participate in our non-employee director compensation policy described above on the same terms as other directors.
105
On September 19, 2013, Ms. Bosley assumed the role of chair of our board of directors. In connection with such appointment, on October 21, 2013 she was granted a stock option award that vests as to 25% of the shares subject to the stock option on the vesting commencement date and thereafter continues to vest in monthly installments over the following 36 months, subject to Ms. Bosley's continued service on our board of directors. All of the shares subject to the stock option will vest immediately prior to the occurrence of a covered transaction (as defined in the 2007 Equity Plan).
106
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
The following is a description of transactions, since January 1, 2012, to which we have been a party, in which the amount involved exceeded or will exceed $120 thousand, and in which any related person had a direct or indirect material interest.
Series C Preferred Stock Financing
In September 2012 and June 2013, we issued and sold an aggregate of 52,586,206 shares of our Series C preferred stock at a purchase price of $0.58 per share for an aggregate purchase price of $30.5 million. The following table sets forth the number of shares of our Series C preferred stock that we issued to our 5% stockholders and their affiliates in this transaction:
Investor
|
Shares of Series C Preferred Stock |
Purchase Price ($) |
|||||
|---|---|---|---|---|---|---|---|
CVF, LLC |
12,931,034 | 7,500,000 | |||||
Bill & Melinda Gates Foundation |
8,620,690 | 5,000,000 | |||||
Polaris Venture Partners and related funds |
6,075,152 | 3,523,588 | |||||
Lux Ventures, and related funds |
5,295,318 | 3,071,284 | |||||
S.R. One, Limited |
4,851,958 | 2,814,135 | |||||
Johnson & Johnson Development Corporation |
4,187,214 | 2,428,584 | |||||
Skyline Venture Partners V, L.P. |
3,140,414 | 1,821,440 | |||||
Cycad Group, LLC |
2,514,096 | 1,458,176 | |||||
Auriga Ventures, III FCPR |
2,425,980 | 1,407,068 | |||||
Participation in our Initial Public Offering
Certain holders of more than 5% of our voting securities purchased an aggregate of 1,105,675 shares of our common stock in our initial public offering completed on February 10, 2014.
Indemnification Agreements
In connection with our initial public offering, we entered into indemnification agreements with each of our directors and executive officers. These agreements will require us to indemnify these individuals and, in certain cases, affiliates of such individuals, to the fullest extent permissible under Delaware law against liabilities that may arise by reason of their service to us or at our direction, and to advance expenses incurred as a result of any proceeding against them as to which they could be indemnified.
Registration Rights Agreement
In connection with our Series C preferred stock financing, on September 28, 2012, we entered into an amended and restated registration rights agreement with the holders of all of our then-outstanding shares of preferred stock including certain of our named executive officers and entities with which certain of our directors are affiliated. The agreement provides that these holders have the right to demand that we file a registration statement with respect to the common stock issued upon conversion of our preferred stock. These holders may also request that shares of common stock held by them be included in certain registration statements that we are otherwise filing. See "Description of Capital Stock—Registration Rights".
Right of First Refusal and Co-Sale Agreement
In connection with our Series C preferred stock financing, on September 28, 2012, we entered into an amended and restated right of first refusal and co-sale agreement with the holders of all of our
107
then-outstanding shares of preferred stock including certain of our named executive officers and entities with which certain of our directors are affiliated. Pursuant to the terms of this agreement, in the event of a proposed sale of shares of our common or preferred stock, the seller was required to first offer such shares to the company and to the other investors, subject to certain conditions and restrictions. This agreement terminated upon the completion of our initial public offering on February 10, 2014.
Voting Agreement
In connection with our Series C preferred stock financing on September 28, 2012, we entered into an amended and restated voting agreement with the holders of all of our then-outstanding shares of preferred stock including certain of our named executive officers and entities with which certain of our directors are affiliated, with respect to the election of directors and certain other matters. All of our current directors were elected pursuant to the terms of this agreement. This agreement terminated upon the completion of our initial public offering on February 10, 2014.
Investor Rights Agreement
In connection with our Series C preferred stock financing, on September 28, 2012, we entered into an amended and restated investor rights agreement with the holders of all of our then-outstanding shares of preferred stock including certain of our named executive officers and entities with which certain of our directors are affiliated. Pursuant to the terms of this agreement, we granted our investors certain information rights as well as the right to participate pro rata in any future private financing rounds. This agreement terminated upon the completion of our initial public offering on February 10, 2014.
Transactions with Our Executive Officers, Directors and 5% Stockholders
On May 16, 2007, we entered into a consulting agreement with Dr. George Siber, a member of our board of directors. The consulting agreement was amended on each of June 30, 2009, December 16, 2010, June 15, 2011 and June 5, 2013 and is in effect through June 17, 2015. Pursuant to the consulting agreement, Dr. Siber performs various consulting services for us, including determining our general scientific and business direction, recruitment of scientific advisory board members and consultants, recruitment of full-time management and scientific personnel and identifying and reviewing scientific developments and intellectual property. Since the beginning of our last fiscal year, Dr. Siber has been paid approximately $10 thousand per month under the consulting agreement. See "Executive and Director Compensation—Director Agreements—Dr. Siber" for further details on compensation paid to Dr. Siber under the consulting agreement.
Related Person Transactions Policy
We have adopted a related person transaction approval policy that governs the review of related person transactions. Pursuant to this policy, if we want to enter into a transaction with a related person or an affiliate of a related person, our Vice President of Finance and Administration will review the proposed transaction to determine, based on applicable NASDAQ and SEC rules, if such transaction requires pre-approval by the audit committee and/or board of directors. If pre-approval is required, such matters will be reviewed at the next regular or special audit committee and/or board of directors meeting. We may not enter into a related person transaction unless our Vice President of Finance and Administration has either specifically confirmed in writing that no further reviews are necessary or that all requisite corporate reviews have been obtained.
108
The following table sets forth information relating to the beneficial ownership of our common stock as of June 30, 2014, by: each person, or group of affiliated persons, known by us to beneficially own more than 5% of our outstanding shares of common stock; each of our directors; each of our named executive officers; and all directors and executive officers as a group.
The number of shares beneficially owned by each entity, person, director or executive officer is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any shares over which the individual has sole or shared voting power or investment power as well as any shares that the individual has the right to acquire within 60 days of June 30, 2014 through the exercise of any stock options, warrants or other rights. Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all shares of common stock held by that person.
The percentage of shares beneficially owned is computed on the basis of 17,406,977 shares of our common stock outstanding as of June 30, 2014. Shares of our common stock that a person has the right to acquire within 60 days of June 30, 2014 are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all directors and executive officers as a group. Unless otherwise indicated below, the address for each beneficial owner listed is c/o Genocea Biosciences, Inc., Cambridge Discovery Park, 100 Acorn Park Drive, Cambridge, MA 02140.
109
| |
|
Percentage of Shares Beneficially Owned |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Name and Address of Beneficial Owner
|
Number of Shares Beneficially Owned |
Before Offering |
After Offering |
|||||||
5% or greater stockholders: |
||||||||||
FMR LLC, and related funds(1) |
2,165,362 | 12.4 | % | 10.4 | % | |||||
245 Summer Street |
||||||||||
Boston, MA 02210 |
||||||||||
Polaris Venture Partners, and related funds(2) |
2,128,682 | 12.2 | % | 10.2 | % | |||||
650 East Kendall Street, 4th Floor |
||||||||||
Cambridge, MA 02142 |
||||||||||
S.R. One, Limited(3) |
1,671,668 | 9.6 | % | 8.0 | % | |||||
c/o Corporation Service Company |
||||||||||
2595 Interstate Drive, Suite 103 |
||||||||||
Harrisburg, PA 17110 |
||||||||||
Johnson & Johnson Development Corporation(4) |
1,556,554 | 8.9 | % | 7.5 | % | |||||
410 George Street |
||||||||||
New Brunswick, NJ 08901 |
||||||||||
Skyline Venture Partners V, L.P.(5) |
1,292,415 | 7.4 | % | 6.2 | % | |||||
525 University Avenue, Suite 610 |
||||||||||
Palo Alto, CA 94301 |
||||||||||
CVF, LLC(6) |
1,218,740 | 7.0 | % | 5.9 | % | |||||
222 N. LaSalle Street, Suite 2000 |
||||||||||
Chicago, IL 60601 |
||||||||||
Lux Ventures, and related funds(7) |
1,098,329 | 6.3 | % | 5.3 | % | |||||
295 Madison Avenue, 24th Floor |
||||||||||
New York, NY 10017 |
||||||||||
Cycad Group, LLC(8) |
876,494 | 5.0 | % | 4.2 | % | |||||
1270 Coast Village Circle |
||||||||||
Santa Barbara, CA 93108 |
||||||||||
Directors and Named Executive Officers: |
||||||||||
William Clark(9) |
494,498 | 2.8 | % | 2.3 | % | |||||
Seth Hetherington(10) |
114,381 | * | * | |||||||
Jessica Baker Flechtner(11) |
62,618 | * | * | |||||||
George Siber, M.D.(12) |
98,345 | * | * | |||||||
Kevin Bitterman, Ph.D.(13) |
2,128,682 | 12.2 | % | 10.2 | % | |||||
Katrine Bosley(14) |
31,092 | * | * | |||||||
Simeon J. George, M.D.(15) |
1,671,668 | 9.6 | % | 8.0 | % | |||||
Stephen J. Hoffman, M.D., Ph.D. |
— | — | — | |||||||
All executive officers and directors as a group (11 persons)(16) |
4,703,128 | 25.7 | % | 21.7 | % | |||||
- *
- Represents
beneficial ownership of less than one percent of our outstanding common stock.
- (1)
- Fidelity Management & Research Company ("Fidelity"), 245 Summer Street, Boston, Massachusetts 02210, a wholly-owned subsidiary of FMR LLC and an investment adviser registered under Section 203 of the Investment Advisers Act of 1940, is the beneficial owner of 1,460,819 shares of common stock as a result of acting as investment adviser to various investment companies registered under Section 8 of the Investment Company Act of 1940. Fidelity SelectCo, LLC ("SelectCo"), 1225 17th Street, Suite 1100, Denver, Colorado 80202, a wholly-owned subsidiary of FMR LLC and an investment adviser registered under Section 203 of the Investment Advisers Act of 1940, is the beneficial owner of 652,879 shares of common stock as a
110
result of acting as investment adviser to various investment companies registered under Section 8 of the Investment Company Act of 1940 (the "SelectCo Funds"). Fidelity Management Trust Company, 245 Summer Street, Boston, Massachusetts 02210, a wholly-owned subsidiary of FMR LLC and a bank as defined in Section 3(a)(6) of the Exchange Act, is the beneficial owner of 51,664 shares of common stock as a result of its serving as investment manager of the institutional accounts (the "institutional accounts"). Edward C. Johnson 3d, Chairman of FMR LLC, and FMR LLC, through its control of Fidelity, and the funds each has sole power to dispose of the 1,460,819 shares owned by the funds. Edward C. Johnson 3d and FMR LLC, through its control of SelectCo, and the SelectCo Funds each has sole power to dispose of the 652,879 owned by the SelectCo Funds. Edward C. Johnson 3d and FMR LLC, through its control of Fidelity Management Trust Company, each has sole dispositive power over 51,664 shares and sole power to vote or to direct the voting of 51,664 shares of common stock owned by the institutional accounts. Members of the family of Edward C. Johnson 3d, Chairman of FMR LLC, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. For information regarding FMR LLC and its affiliates, we have relied on the Schedule 13G filed by FMR LLC with the SEC on May 12, 2014.
- (2)
- Consists
of (i) 2,062,206 shares of common stock held by Polaris Venture Partners V, L.P., (ii) 35,653 shares of common stock held by
Polaris Venture Partners Entrepreneurs' Fund, L.P., (iii) 12,531 shares of common stock held by Polaris Venture Partners Founders' Fund V, L.P., and (iv) 18,292 shares of
common stock held by Polaris Venture Partners Special Founders' Fund V, L.P. (together with Polaris Venture Partners V, L.P., Polaris Venture Partners Entrepreneurs' Fund, L.P.
and Polaris Venture Partners Founders' Fund V, L.P., the Polaris Funds). North Star Venture Management 2000, LLC directly or indirectly provides investment advisory services to
various venture capital funds, including the Polaris Funds. Jonathan Flint and Terrance McGuire, managing members of North Star Venture Management 2000, LLC, exercise voting and investment
power with respect to North Star Venture Management, 2000. Each of the Polaris Funds has the sole voting and investment power with respect to the shares of the Company directly held by the applicable
Polaris Fund. The respective general partners of the Polaris Funds may be deemed to have sole voting and investment power with respect to the shares held by such funds. The respective general partners
disclaim beneficial ownership of all the shares held by the Polaris Funds except to the extent of their proportionate pecuniary interests therein. The members of North Star Venture Management
2000, LLC (the Polaris Management Members) are also members of Polaris Venture Management Co., V, L.L.C. (the general partner of each of the Polaris Funds). Jonathan Flint and Terrance
McGuire, managing members of Polaris Venture Management Co. V, L.L.C., exercise voting and investment power with respect to Polaris Venture Management Co. V, L.L.C. As members of the
general partner and North Star Venture Management 2000, LLC, the Polaris Management Members may be deemed to share voting and investment powers for the shares held by the Polaris Funds. The
Polaris Management Members disclaim beneficial ownership of all such shares held by the funds except to the extent of their proportionate pecuniary interests therein. Kevin Bitterman, a director of
the Company, has an assignee interest in Polaris Venture Management Co. V, L.L.C. To the extent that he is deemed to share voting and investment powers with respect to the shares held by the
Polaris Funds, Dr. Bitterman disclaims beneficial ownership of all the shares held by the funds except to the extent of his proportionate pecuniary interest therein.
- (3)
- Consists of 1,617,668 shares of common stock held by S.R. One, Limited, an indirect, wholly-owned subsidiary of GlaxoSmithKline plc. Simeon J. George is a Vice President at S.R. One, Limited and an employee of GlaxoSmithKline LLC, a wholly-owned subsidiary of GlaxoSmithKline plc. Dr. George disclaims beneficial ownership of all the shares held by S.R. One, Limited except to the extent of his proportionate pecuniary interest therein.
111
- (4)
- Consists
of 1,556,554 shares of common stock held by Johnson & Johnson Development Corporation.
- (5)
- Consists
of 1,292,415 shares of common stock held by Skyline Venture Partners V, L.P. The general partner of Skyline Venture Partners V, L.P.
is Skyline Venture Management V, LLC. John G. Freund and Yasunori Kaneko are Managers of Skyline Venture Management V, LLC and hereby disclaim beneficial ownership of all the shares held
by Skyline Venture Partners V, L.P. except to the extent of his proportionate pecuniary interest therein.
- (6)
- Consists
of 1,218,740 shares of common stock. Richard H. Robb, manager of CVF, LLC, exercises voting and investment power with respect to shares held
by CVF, LLC. Mr. Robb disclaims beneficial ownership of all shares held by CVF, LLC except to the extent of his pecuniary interest therein.
- (7)
- Consists
of (i) 1,050,432 shares of common stock held by Lux Ventures II, L.P. ("LV-II") and (ii) 47,897 shares of common stock held by
Lux Ventures II Sidecar, L.P. ("Sidecar") (together with LV-II and Sidecar, the "Lux Funds"). Lux Venture Partners II, L.P. ("LVP-II") is (i) the general partner of the Lux Funds,
Robert Paull, Joshua Wolfe and Peter Hebert are the individual managers of LCM LLC (the "Individual Managers"). LVP II and LCM LLC disclaim beneficial ownership of such shares, except to
the extent of their pecuniary interest therein. LCM LLC, as sole member, may be deemed to share voting and investment powers for the shares held by the Lux Funds. As one of three individual
managers, each of the Individual Managers disclaims beneficial ownership over the shares reported herein, and in all events disclaims beneficial ownership except to the extent of his pecuniary
interest therein.
- (8)
- Consists
of 876,494 shares of common stock. K. Leonard Judson (Managing Director and President) and Paul F. Glenn (Chairman) are the sole managers and
directors of Cycad Group, LLC (the "Cycad Directors"). The Cycad Directors have shared voting and investment power with respect to the shares held by Cycad Group, LLC, and may be deemed
beneficial owners of the shares held by Cycad Group, LLC. Mr. Judson and Mr. Glenn disclaim beneficial ownership of the shares beneficially owned by Cycad Group, LLC except
to the extent of their pecuniary interest therein.
- (9)
- Consists
of 472,567 shares of common stock that can be acquired upon the exercise of outstanding options and 21,931 shares of common stock that can be
acquired upon the exercise of options within 60 days of June 30, 2014.
- (10)
- Consists
of 108,923 shares of common stock that can be acquired upon the exercise of outstanding options and 5,458 shares of common stock that can be
acquired upon the exercise of options within 60 days of June 30, 2014.
- (11)
- Consists
of 58,586 shares of common stock that can be acquired upon the exercise of outstanding options and 4,032 shares of common stock that can be
acquired upon the exercise of options within 60 days of June 30, 2014.
- (12)
- Consists
of 2,016 shares of common stock, 95,047 shares of common stock that can be acquired upon the exercise of outstanding options and 1,282 shares of
common stock that can be acquired upon the exercise of options within 60 days of June 30, 2014.
- (13)
- Consists
of shares held by Polaris Venture Partners or related funds. By virtue of the relationships described in footnote 2 above, Dr. Bitterman
may be deemed to share beneficial ownership in the shares held by Polaris Venture Partners or related funds. Dr. Bitterman disclaims beneficial ownership of the shares referred to in footnote 2
above.
- (14)
- Consists of 31,092 shares of common stock held by Katrine Bosley.
112
- (15)
- Consists
of shares held by S.R. One, Limited. By virtue of the relationships described in footnote 3 above, Dr. George may be deemed to share
beneficial ownership in the shares held by S.R. One, Limited. Dr. George disclaims beneficial ownership of the shares referred to in footnote 3 above.
- (16)
- Consists of 3,833,458 shares of common stock, 831,785 shares of common stock that can be acquired upon the exercise of outstanding options and 37,885 shares of common stock that can be acquired upon the exercise of options within 60 days of June 30, 2014.
113
General
The following description of our capital stock is intended as a summary only and is qualified in its entirety by reference to our fifth amended and restated certificate of incorporation and amended and restated by-laws and the applicable provisions of the Delaware General Corporation Law. We refer in this section to our fifth amended and restated certificate of incorporation as our certificate of incorporation, and we refer to our amended and restated by-laws as our by-laws. Our authorized capital stock consists of 175,000,000 shares of our common stock, par value $0.001 per share.
As of June 30, 2014, we had issued and outstanding:
- •
- 17,406,977 shares of our common stock;
- •
- options to purchase a total of 2,233,769 shares of our common stock with a weighted average exercise price of $6.68 per
share.
- •
- 3,878 shares of common stock issuable upon the exercise of warrants outstanding at June 30, 2014 at a weighted-average exercise price of $7.74 per share; and
As of June 30, 2014, we had 55 stockholders of record.
Common Stock
Dividend Rights. Subject to preferences that may apply to shares of preferred stock outstanding at the time, holders of outstanding shares of common stock are entitled to receive dividends out of assets legally available at the times and in the amounts as the board of directors may from time to time determine.
Conversion or Redemption Rights. Our common stock is neither convertible nor redeemable.
Liquidation Rights. Upon our liquidation, dissolution or winding up, the holders of our common stock are entitled to receive pro rata our assets which are legally available for distribution, after payment of all debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any then-outstanding shares of preferred stock.
Rights and Preferences. Holders of common stock have no preemptive, conversion or subscription rights and there are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences and privileges of the holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate in the future.
Preferred Stock
Our board of directors have the authority, without further action by our stockholders, to issue up to 25,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon, and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of our common stock. The purpose of authorizing our board of directors to issue preferred stock and determine its rights and preferences is to eliminate delays associated with a stockholder vote on specific issuances. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other
114
corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control of us and may adversely affect the market price of our common stock and the voting and other rights of the holders of our common stock. It is not possible to state the actual effect of the issuance of any shares of preferred stock on the rights of holders of common stock until the board of directors determines the specific rights attached to that preferred stock.
We have no present plans to issue any shares of preferred stock.
Registration Rights
We are party to an amended and restated registration rights agreement with the holders of approximately 11,474,454 million shares of our common stock.
Under the amended and restated registration rights agreement, holders of registrable shares (other than warrant shares) can demand that we file a registration statement or request that their shares be included on a registration statement that we are otherwise filing, in either case, registering the resale of their shares of common stock. These registration rights are subject to conditions and limitations, including the right, in certain circumstances, of the underwriters of an offering to limit the number of shares included in such registration and our right, in certain circumstances, not to effect a requested S-1 or S-3 registration within 90 days before or 180 days following the Company's estimated date of filing of a registration statement pertaining to an underwritten public offering of securities for the account of the Company offering of our securities, including this offering.
Demand Registration Rights
Following the six-month anniversary of the completion of our initial public offering on February 10, 2014, the holders of at least a majority of the registrable shares (other than warrant shares) may require us to file a registration statement under the Securities Act at our expense with respect to the resale of their registrable shares, and we are required to use our best efforts to effect the registration.
Piggyback Registration Rights
If we propose to register any of our securities under the Securities Act for our own account or the account of any other holder, the holders of registrable shares are entitled to notice of such registration and to request that we include registrable shares for resale on such registration statement, subject to the right of any underwriter to limit the number of shares included in such registration. We will pay all registration expenses, other than underwriting discounts and commissions, related to any demand or piggyback registration. The amended and restated registration rights agreement contains customary cross-indemnification provisions, pursuant to which we are obligated to indemnify the selling stockholders, in the event of misstatements or omissions in the registration statement attributable to us except in the event of fraud and they are obligated to indemnify us for misstatements or omissions attributable to them.
Form S-3 Registration Rights
After the expiration of the 180-day period following the completion of our initial public offering on February 10, 2014, the holders of approximately 11,474,454 shares of our common stock will be entitled to certain Form S-3 registration rights if we are eligible to file a registration statement on Form S-3. As a result, holders owning a certain percentage of our capital stock and certain other identified holders will have the right to demand that we file a registration statement on Form S-3 so long as the aggregate value of the securities to be sold under the registration statement is at least $3 million, subject to certain exceptions.
115
Expenses of Registration
We will pay all expenses relating to any demand, piggyback, or Form S-3 registration, other than underwriting discounts and commissions, subject to specified conditions and limitations.
Termination of Registration Rights
The registration rights granted to any holder of registrable shares will terminate when all such holder's registrable securities could be sold or no longer qualify as registrable shares.
Anti-Takeover Effects of Our Certificate of Incorporation and Our By-Laws
Our certificate of incorporation and by-laws contain certain provisions that are intended to enhance the likelihood of continuity and stability in the composition of the board of directors and which may have the effect of delaying, deferring or preventing a future takeover or change in control of the company unless such takeover or change in control is approved by the board of directors.
These provisions include:
Classified Board. Our certificate of incorporation provides that our board of directors is divided into three classes of directors, with the classes as nearly equal in number as possible. As a result, approximately one-third of our board of directors will be elected each year. The classification of directors will have the effect of making it more difficult for stockholders to change the composition of our board. Our certificate of incorporation also provides that, subject to any rights of holders of preferred stock to elect additional directors under specified circumstances, the number of directors will be fixed exclusively pursuant to a resolution adopted by our board of directors. Following the completion of our initial public offering on February 10, 2014, our board of directors consists of six members.
Action by Written Consent; Special Meetings of Stockholders. Our certificate of incorporation provides that stockholder action can be taken only at an annual or special meeting of stockholders and cannot be taken by written consent in lieu of a meeting. Our certificate of incorporation and the by-laws also provide that, except as otherwise required by law, special meetings of the stockholders can be called only by or at the direction of the board of directors pursuant to a resolution adopted by a majority of the total number of directors. Except as described above, stockholders are not be permitted to call a special meeting or to require the board of directors to call a special meeting.
Removal of Directors. Our certificate of incorporation provides that our directors may be removed only for cause by the affirmative vote of at least 75% of the voting power of our outstanding shares of capital stock, voting together as a single class. This requirement of a supermajority vote to remove directors could enable a minority of our stockholders to prevent a change in the composition of our board.
Advance Notice Procedures. Our by-laws establish an advance notice procedure for stockholder proposals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to the board of directors. Stockholders at an annual meeting are only be able to consider proposals or nominations specified in the notice of meeting or brought before the meeting by or at the direction of the board of directors or by a stockholder who was a stockholder of record on the record date for the meeting, who is entitled to vote at the meeting and who has given our Secretary timely written notice, in proper form, of the stockholder's intention to bring that business before the meeting. Although the by-laws do not give the board of directors the power to approve or disapprove stockholder nominations of candidates or proposals regarding other business to be conducted at a special or annual meeting, the by-laws may have the effect of precluding the conduct of certain business at a meeting if the proper procedures are not followed or may discourage or deter a potential acquirer
116
from conducting a solicitation of proxies to elect its own slate of directors or otherwise attempting to obtain control of the company.
Super Majority Approval Requirements. The Delaware General Corporation Law generally provides that the affirmative vote of a majority of the shares entitled to vote on any matter is required to amend a corporation's certificate of incorporation or by-laws, unless either a corporation's certificate of incorporation or by-laws requires a greater percentage. Our certificate of incorporation and by-laws provide that the affirmative vote of holders of at least 75% of the total votes eligible to be cast in the election of directors is required to amend, alter, change or repeal the by-laws. This requirement of a supermajority vote to approve amendments to our by-laws could enable a minority of our stockholders to exercise veto power over any such amendments.
Authorized but Unissued Shares. Our authorized but unissued shares of common stock and preferred stock are available for future issuance, including this offering, without stockholder approval. These additional shares may be utilized for a variety of corporate purposes, including future public offerings to raise additional capital, corporate acquisitions and employee benefit plans. The existence of authorized but unissued shares of common stock and preferred stock could render more difficult or discourage an attempt to obtain control of a majority of our common stock by means of a proxy contest, tender offer, merger or otherwise.
Exclusive Forum. Our certificate of incorporation provides that, subject to limited exceptions, the state or federal courts located in the State of Delaware are the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees to us or our stockholders, (iii) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law, our certificate of incorporation or our by-laws, or (iv) any other action asserting a claim against us that is governed by the internal affairs doctrine. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock shall be deemed to have notice of and to have consented to the provisions of our certificate of incorporation described above. Although we believe these provisions benefit us by providing increased consistency in the application of Delaware law for the specified types of actions and proceedings, the provisions may have the effect of discouraging lawsuits against our directors and officers. The enforceability of similar choice of forum provisions in other companies' certificates of incorporation has been challenged in legal proceedings, and it is possible that, in connection with one or more actions or proceedings described above, a court could find the choice of forum provisions contained in our certificate of incorporation to be inapplicable or unenforceable.
Section 203 of the Delaware General Corporation Law
We are subject to the provisions of Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a publicly-held Delaware corporation from engaging in a "business combination" with an "interested stockholder" for a three-year period following the time that this stockholder becomes an interested stockholder, unless the business combination is approved in a prescribed manner. A "business combination" includes, among other things, a merger, asset or stock sale or other transaction resulting in a financial benefit to the interested stockholder. An "interested stockholder" is a person who, together with affiliates and associates, owns, or did own within three years prior to the determination of interested stockholder status, 15% or more of the corporation's voting stock.
Under Section 203, a business combination between a corporation and an interested stockholder is prohibited unless it satisfies one of the following conditions: before the stockholder became interested, the board of directors approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; upon consummation of the transaction which
117
resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding, shares owned by persons who are directors and also officers, and employee stock plans, in some instances; or at or after the time the stockholder became interested, the business combination was approved by the board of directors of the corporation and authorized at an annual or special meeting of the stockholders by the affirmative vote of at least two-thirds of the outstanding voting stock which is not owned by the interested stockholder.
A Delaware corporation may "opt out" of these provisions with an express provision in its original certificate of incorporation or an express provision in its certificate of incorporation or by-laws resulting from a stockholders' amendment approved by at least a majority of the outstanding voting shares. We have not opted out of these provisions. As a result, mergers or other takeover or change in control attempts of us may be discouraged or prevented.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare Trust Company, N.A. The transfer agent and registrar's address is 144 Fernwood Ave, Edison, New Jersey 08837.
Listing
Our common stock is listed on the NASDAQ Global Market under the symbol "GNCA".
118
SHARES ELIGIBLE FOR FUTURE SALE
Future sales of our common stock, including shares issued upon the exercise of outstanding options or warrants, in the public market after this offering, or the perception that those sales may occur, could cause the prevailing market price for our common stock to fall or impair our ability to raise equity capital in the future. As described below, only a limited number of shares of our common stock will be available for sale in the public market for a period of several months after completion of this offering due to contractual and legal restrictions on resale described below. Future sales of our common stock in the public market either before (to the extent permitted) or after restrictions lapse, or the perception that those sales may occur, could adversely affect the prevailing market price of our common stock at such time and our ability to raise equity capital at a time and price we deem appropriate.
Sale of Restricted Shares
As of June 30, 2014, based on the number of shares of our common stock then outstanding, assuming (1) the closing of this offering, (2) no exercise of the underwriters' option to purchase additional shares of common stock, and (3) no exercise of outstanding options or warrants, we would have had outstanding an aggregate of approximately 20,806,977 shares of common stock, including the 5,500,000 shares sold in our initial public offering, and all of the shares of common stock to be sold in this offering, and any shares sold upon exercise of the underwriters' option to purchase additional shares, will be freely tradable in the public market without restriction or further registration under the Securities Act of 1933, as amended, or the Securities Act, unless the shares are held by any of our "affiliates" as such term is defined in Rule 144 of the Securities Act. The remaining shares of common stock are "restricted securities" as such term is defined in Rule 144 or subject to lock up agreements in effect in connection with the initial public offering or entered into in connection with this offering (as described below). These restricted securities were issued and sold by us, or will be issued and sold by us, in private transactions and are eligible for public sale only if registered under the Securities Act or if they qualify for an exemption from registration under the Securities Act, including the exemptions provided by Rule 144 or Rule 701, which rules are summarized below.
As a result of the lock-up agreements referred to below and the provisions of Rule 144 and Rule 701 under the Securities Act, the shares of our common stock (excluding the shares sold in this offering) that will be available for sale in the public market are as follows:
Approximate Number of Shares
|
First Date Available for Sale into Public Market | |
|---|---|---|
| 6,801,513 shares, or 39.1% | August 3, 2014 due to lock up agreements in effect in connection with our initial public offering. However, the representatives of the underwriters can waive the provisions of these lock-up agreements and allow these stockholders to sell their shares at any time. | |
4,498,349 shares, or 25.8% |
90 days after the date of this prospectus upon expiration of the lock-up agreements referred to below, subject in some cases to applicable volume limitations under Rule 144 |
Lock-up Agreements
In connection with this offering, we, our directors, our officers and stockholders beneficially owning approximately 32.8% of our shares of common stock outstanding as of June 30, 2014, have agreed with the underwriters not to dispose of or hedge any shares of our common stock or securities convertible into or exchangeable for shares of common stock during the period from the date of the lock-up agreement continuing through the date 90 days after the date of this prospectus, except with the prior written consent of Citigroup Global Markets Inc. and Cowen and Company, LLC, the
119
representatives of the underwriters. The representatives of the underwriters have advised us that they have no current intent or arrangement to release any of the shares subject to the lock-up agreements prior to the expiration of the lock-up period.
The lock-up agreements do not contain any pre-established conditions to the waiver by Citigroup Global Markets Inc. and Cowen and Company, LLC on behalf of the underwriters of any terms of the lock-up agreements. Any determination to release shares subject to the lock-up agreements would be based on a number of factors at the time of determination, including but not necessarily limited to the market price of the common stock, the liquidity of the trading market for the common stock, general market conditions, the number of shares proposed to be sold, contractual obligations to release certain shares subject to the lock-up agreements in the event any such shares are released, subject to certain specific limitations and thresholds, and the timing, purpose and terms of the proposed sale.
Following the lock-up periods set forth in the agreements described above, and assuming that the representatives of the underwriters do not release any parties from these agreements, all of the shares of our common stock that are restricted securities or are held by our affiliates as of the date of this prospectus will be eligible for sale in the public market in compliance with Rule 144 under the Securities Act.
Rule 144
In general, persons who have beneficially owned restricted shares of our common stock for at least six months, and any affiliate of the Company who owns either restricted shares of our common stock, are entitled to sell their securities without registration with the SEC under an exemption from registration provided by Rule 144 under the Securities Act.
Non-Affiliates
Any person who is not deemed to have been one of our affiliates at the time of, or at any time during the three months preceding, a sale may sell an unlimited number of restricted securities under Rule 144 if:
- •
- the restricted securities have been held for at least six months, including the holding period of any prior owner other
than one of our affiliates;
- •
- we have been subject to the Exchange Act periodic reporting requirements for at least 90 days before the sale; and
- •
- we are current in our Exchange Act reporting at the time of sale.
Any person who is not deemed to have been an affiliate of ours at the time of, or at any time during the three months preceding, a sale and has held the restricted securities for at least one year, including the holding period of any prior owner other than one of our affiliates, will be entitled to sell an unlimited number of restricted securities without regard to the length of time we have been subject to Exchange Act periodic reporting or whether we are current in our Exchange Act reporting.
Affiliates
Persons seeking to sell restricted securities who are our affiliates at the time of, or any time during the three months preceding, a sale would be subject to the restrictions described above. They are also subject to additional restrictions, by which such person would be required to comply with the manner
120
of sale and notice provisions of Rule 144 and would be entitled to sell within any three-month period only that number of securities that does not exceed the greater of either of the following:
- •
- 1% of the number of shares of our common stock then outstanding, which will equal approximately 208,070 shares immediately
after the completion of this offering based on the number of shares outstanding as of June 30, 2014; or
- •
- the average weekly trading volume of our common stock on the NASDAQ Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale.
Rule 701
In general, under Rule 701 a person who purchased shares of our common stock pursuant to a written compensatory plan or contract and who is not deemed to have been one of our affiliates during the immediately preceding 90 days may sell these shares in reliance upon Rule 144, but without being required to comply with the notice, manner of sale or public information requirements or volume limitation provisions of Rule 144. Rule 701 also permits affiliates to sell their Rule 701 shares under Rule 144 without complying with the holding period requirements of Rule 144. All holders of Rule 701 shares, however, are required to wait until 90 days after the date of this prospectus before selling such shares pursuant to Rule 701. Substantially all Rule 701 shares are subject to lock-up agreements as described below and in the section of this prospectus titled "Underwriting" and will become eligible for sale upon the expiration of the restrictions set forth in those agreements.
Registration Rights
The holders of 11,476,987 shares of our common stock are entitled to specified rights with respect to the registration of the offer and sale of their shares under the Securities Act. Registration of the offer and sale of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act immediately upon the effectiveness of the registration. See the section of this prospectus titled "Description of Capital Stock—Registration Rights" for additional information.
Stock Options and Form S-8 Registration Statement
As of June 30, 2014, we had outstanding options to purchase an aggregate of 2,233,769 shares of our common stock, of which options to purchase 1,085,684 shares were vested. We have filed a registration statement on Form S-8 under the Securities Act to register all of the shares of our common stock subject to outstanding options and options and other awards issuable pursuant to our 2007 Equity Plan and our 2014 Equity Plan. Accordingly, shares of our common stock registered under the registration statements will be available for sale in the open market, subject to Rule 144 volume limitations applicable to affiliates, and subject to any vesting restrictions and lock-up agreements applicable to these shares.
121
MATERIAL UNITED STATES FEDERAL INCOME AND ESTATE TAX
CONSIDERATIONS FOR NON-U.S. HOLDERS
The following discussion describes the material U.S. federal income and estate tax considerations relating to the purchase, ownership and disposition of our common stock by Non-U.S. Holders (defined below). This summary does not address all aspects of U.S. federal income and estate taxes, does not discuss the potential application of the alternative minimum tax or the 3.8% Medicare tax on net investment income and does not deal with state, local or non-U.S. tax consequences that may be relevant to Non-U.S. Holders of our common stock. This discussion is based upon Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, the Treasury regulations promulgated or proposed thereunder and administrative and judicial interpretations thereof, in effect and available as of the date hereof and all of which are subject to differing interpretations and to change, revocation or repeal at any time, possibly on a retroactive basis.
This summary assumes that shares of our common stock are held as "capital assets" within the meaning of Section 1221 of the Code (generally, property held for investment). This summary does not purport to deal with all aspects of U.S. federal income and estate taxation that might be relevant to particular Non-U.S. Holders in light of their particular investment circumstances or status, nor does it address specific tax considerations that may be relevant to particular persons (including, for example, financial institutions, broker-dealers and traders in securities, insurance companies, partnerships or other pass-through entities or entities that are treated as disregarded entities for U.S. federal income tax purposes (regardless of their places of organization or formation), certain U.S. expatriates, tax-exempt organizations, pension plans, "controlled foreign corporations", "passive foreign investment companies", corporations that accumulate earnings to avoid U.S. federal income tax, persons in special situations, such as those who have elected to mark securities to market or those who hold common stock as part of a straddle, hedge, conversion transaction, synthetic security or other integrated investment or risk reduction strategy). In addition, except as explicitly addressed herein with respect to estate tax, this summary does not address estate and gift tax considerations or considerations under the tax laws of any state, local or non-U.S. jurisdiction.
For purposes of this summary, a "Non-U.S. Holder" means a beneficial owner of common stock that for U.S. federal income tax purposes is not classified as a partnership and is not:
- •
- an individual who is a citizen or resident of the United States;
- •
- a corporation or any other organization taxable as a corporation for U.S. federal income tax purposes, created or
organized in or under the laws of the United States, any state thereof or the District of Columbia;
- •
- an estate, the income of which is included in gross income for U.S. federal income tax purposes regardless of its source;
or
- •
- a trust if (1) a U.S. court is able to exercise primary supervision over the trust's administration and one or more U.S. persons have the authority to control all of the trust's substantial decisions or (2) the trust has a valid election in effect under applicable Treasury regulations to be treated as a U.S. person.
If an entity that is treated as a partnership for U.S. federal income tax purposes holds our common stock, the tax treatment of persons treated as its partners for U.S. federal income tax purposes will generally depend upon the status of the partner and the activities of the partnership. Partnerships and other entities that are classified as partnerships for U.S. federal income tax purposes and persons holding our common stock through a partnership or other entity treated as a partnership for U.S. federal income tax purposes are urged to consult their own tax advisors.
122
There can be no assurance that the Internal Revenue Service ("IRS") will not challenge one or more of the tax consequences described herein, and we have not obtained, nor do we intend to obtain a ruling from the IRS with respect to the U.S. federal income or estate tax consequences to a Non-U.S. Holder of the purchase, ownership or disposition of our common stock.
THIS SUMMARY IS FOR GENERAL INFORMATION ONLY AND IS NOT INTENDED TO BE TAX ADVICE. NON-U.S. HOLDERS ARE URGED TO CONSULT THEIR TAX ADVISORS CONCERNING THE U.S. FEDERAL INCOME AND ESTATE TAXATION, STATE, LOCAL AND NON-U.S. TAXATION AND OTHER TAX CONSEQUENCES TO THEM OF THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR COMMON STOCK.
Distributions on Our Common Stock
As discussed under "Dividend Policy" above, we do not anticipate paying any cash dividends in the foreseeable future. In the event that we do make a distribution of cash or property (other than certain stock distributions) with respect to our common stock (or in the case of certain redemptions that are treated as distributions with respect to our common stock), any such distributions generally will constitute dividends for U.S. federal income tax purposes to the extent of our current and accumulated earnings and profits, if any, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will constitute a return of capital and will first reduce the holder's adjusted tax basis in our common stock, but not below zero. Any remaining excess will be treated as capital gain, subject to the tax treatment described below in "—Gain on Sale, Exchange or Other Disposition of Our Common Stock". Any such distribution would also be subject to the discussion below under the sections titled "—Additional Withholding and Reporting Requirements" and "—Backup Withholding and Information Reporting".
Dividends paid to a Non-U.S. Holder generally will be subject to a 30% U.S. federal withholding tax unless such Non-U.S. Holder provides us or our agent, as the case may be, with the appropriate IRS Form W-8, such as:
- •
- IRS Form W-8BEN or Form W-8BEN-E (or successor forms) certifying, under penalties of perjury, a reduction in
withholding under an applicable income tax treaty, or
- •
- IRS Form W-8ECI (or successor form) certifying that a dividend paid on common stock is not subject to withholding tax because it is effectively connected with a trade or business in the United States of the Non-U.S. Holder (in which case such dividend generally will be subject to regular graduated U.S. tax rates as described below).
The certification requirement described above must be provided to us or our agent prior to the payment of dividends and must be updated periodically. The certification also may require a Non-U.S. Holder that provides an IRS form or that claims treaty benefits to provide its U.S. taxpayer identification number. Special certification and other requirements apply in the case of certain Non-U.S. Holders that hold shares of our common stock through intermediaries or are pass-through entities for U.S. federal income tax purposes.
Each Non-U.S. Holder is urged to consult its own tax advisor about the specific methods for satisfying these requirements. A claim for exemption will not be valid if the person receiving the applicable form has actual knowledge or reason to know that the statements on the form are false.
If dividends are effectively connected with a trade or business in the United States of a Non-U.S. Holder (and, if required by an applicable income tax treaty, attributable to a U.S. permanent establishment or fixed base), the Non-U.S. Holder, although exempt from the withholding tax described above (provided that the certifications described above are satisfied), generally will be subject to U.S. federal income tax on such dividends on a net income basis in the same manner as if it were a resident of the United States. In addition, if a Non-U.S. Holder is treated as a corporation for U.S. federal
123
income tax purposes, the Non-U.S. Holder may be subject to an additional "branch profits tax" equal to 30% (unless reduced by an applicable income treaty) of its earnings and profits in respect of such effectively connected dividend income.
Non-U.S. Holders that do not timely provide us or our agent with the required certification, but which are eligible for a reduced rate of U.S. federal withholding tax pursuant to an income tax treaty, may obtain a refund or credit of any excess amount withheld by timely filing an appropriate claim for refund with the IRS.
Gain on Sale, Exchange or Other Disposition of Our Common Stock
Subject to the discussion below under the sections titled "—Additional Withholding and Reporting Requirements" and "—Backup Withholding and Information Reporting", in general, a Non-U.S. Holder will not be subject to U.S. federal income tax or withholding tax on gain realized upon such holder's sale, exchange or other disposition of shares of our common stock unless (i) such Non-U.S. Holder is an individual who is present in the United States for 183 days or more in the taxable year of disposition, and certain other conditions are met; (ii) we are or have been a "United States real property holding corporation", as defined in the Code (a "USRPHC"), at any time within the shorter of the five-year period preceding the disposition and the Non-U.S. Holder's holding period in the shares of our common stock, and certain other requirements are met; or (iii) such gain is effectively connected with the conduct by such Non-U.S. Holder of a trade or business in the United States (and, if required by an applicable income tax treaty, is attributable to a permanent establishment or fixed base maintained by such Non-U.S. Holder in the United States).
If the first exception applies, the Non-U.S. Holder generally will be subject to U.S. federal income tax at a rate of 30% (or at a reduced rate under an applicable income tax treaty) on the amount by which such Non-U.S. Holder's capital gains allocable to U.S. sources exceed capital losses allocable to U.S. sources during the taxable year of the disposition. If the third exception applies, the Non-U.S. Holder generally will be subject to U.S. federal income tax with respect to such gain on a net income basis in the same manner as if it were a resident of the United States and a Non-U.S. Holder that is a corporation for U.S. federal income tax purposes may also be subject to a branch profits tax with respect to any earnings and profits attributable to such gain at a rate of 30% (or at a reduced rate under an applicable income tax treaty).
Regarding the second exception, generally, a corporation is a USRPHC only if the fair market value of its U.S. real property interests (as defined in the Code) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests plus its other assets used or held for use in a trade or business. Although there can be no assurance in this regard, we believe that we are not, and do not anticipate becoming, a USRPHC. However, because the determination of whether we are a USRPHC depends on the fair market value of our U.S. real property interests relative to the fair market value of other business assets, there can be no assurance that we have not been a USRPHC in the past and will not become a USRPHC in the future. Even if we became a USRPHC, a Non-U.S. Holder would not be subject to U.S. federal income tax on a sale, exchange or other disposition of our common stock by reason of our status as USRPHC so long as our common stock is regularly traded on an established securities market and such Non-U.S. Holder does not own and is not deemed to own (directly, indirectly or constructively) more than 5% of our common stock at any time during the shorter of the five year period ending on the date of disposition and the Non-U.S. Holder's holding period. However, no assurance can be provided that our common stock will be regularly traded on an established securities market for purposes of the rules described above. Prospective investors are encouraged to consult their own tax advisors regarding the possible consequences to them if we are, or were to become, a USRPHC.
124
Additional Withholding and Reporting Requirements
Legislation enacted in March 2010 and related guidance (commonly referred to as "FATCA") will impose, in certain circumstances, U.S. federal withholding at a rate of 30% on payments of (a) dividends on our common stock on or after July 1, 2014, and (b) gross proceeds from the sale or other disposition of our common stock on or after January 1, 2017. In the case of payments made to a "foreign financial institution" as defined under FATCA (including, among other entities, an investment fund), the tax generally will be imposed, subject to certain exceptions, unless such institution (i) enters into (or is otherwise subject to) and complies with an agreement with the U.S. government (a "FATCA Agreement") or (ii) complies with an intergovernmental agreement between the United States and a foreign jurisdiction (an "IGA"), in either case to, among other things, collect and provide to the U.S. or other relevant tax authorities certain information regarding U.S. account holders of such institution. In the case of payments made to a foreign entity that is not a foreign financial institution, the tax generally will be imposed, subject to certain exceptions, unless such foreign entity provides the withholding agent with a certification that it does not have any "substantial U.S. owner" (generally, any specified U.S. person that directly or indirectly owns more than a specified percentage of such entity) or that identifies its substantial U.S. owners. If our common stock is held through a foreign financial institution that enters into (or is otherwise subject to) a FATCA Agreement, such foreign financial institution (or, in certain cases, a person paying amounts to such foreign financial institution) generally will be required, subject to certain exceptions, to withhold such tax on payments of dividends and proceeds described above made to (x) a person (including an individual) that fails to comply with certain information requests or (y) a foreign financial institution that has not entered into (and is not otherwise subject to) a FATCA Agreement and is not required to comply with FATCA pursuant to applicable foreign law enacted in connection with an IGA.
Prospective investors should consult their own tax advisors regarding the possible impact of these rules on their investment in our common stock, and the entities through which they hold our common stock, including, without limitation, the process and deadlines for meeting the applicable requirements to prevent the imposition of this 30% withholding tax under FATCA.
Backup Withholding and Information Reporting
We must report annually to the IRS and to each Non-U.S. Holder the gross amount of the distributions on our common stock paid to the holder and the tax withheld, if any, with respect to the distributions. Non-U.S. Holders may have to comply with specific certification procedures to establish that the holder is not a United States person (as defined in the Code) in order to avoid backup withholding at the applicable rate, currently 28%, with respect to dividends on our common stock. Dividends paid to Non-U.S. Holders subject to the U.S. withholding tax, as described above under the section titled "—Distributions on Our Common Stock", generally will be exempt from U.S. backup withholding.
Information reporting and backup withholding will generally apply to the proceeds of a disposition of our common stock by a Non-U.S. Holder effected by or through the U.S. office of any broker, U.S. or foreign, unless the holder certifies its status as a Non-U.S. Holder and satisfies certain other requirements, or otherwise establishes an exemption. Generally, information reporting and backup withholding will not apply to a payment of disposition proceeds to a Non-U.S. Holder where the transaction is effected outside the United States through a non-U.S. office of a broker. However, for information reporting purposes, dispositions effected through a non-U.S. office of a broker with substantial U.S. ownership or operations generally will be treated in a manner similar to dispositions effected through a U.S. office of a broker. Prospective investors should consult their own tax advisors regarding the application of the information reporting and backup withholding rules to them.
125
Copies of information returns may be made available to the tax authorities of the country in which the Non-U.S. Holder resides or, in which the Non- U.S. Holder is incorporated, under the provisions of a specific treaty or agreement.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules from a payment to a Non-U.S. Holder can be refunded or credited against the Non-U.S. Holder's U.S. federal income tax liability, if any, provided that an appropriate claim is timely filed with the IRS.
Federal Estate Tax
Common stock owned (or treated as owned) by an individual who is not a citizen or a resident of the United States (as defined for U.S. federal estate tax purposes) at the time of death will be included in the individual's gross estate for U.S. federal estate tax purposes unless an applicable estate or other tax treaty provides otherwise, and therefore, may be subject to U.S. federal estate tax. The test for whether an individual is a resident of the United States for federal estate tax purposes differs from the test used for U.S. federal income tax purposes. Some individuals, therefore, may be "Non-U.S. Holders" for U.S. federal income tax purposes, but not for U.S. federal estate tax purposes, and vice versa.
126
Citigroup Global Markets Inc. and Cowen and Company, LLC are acting as joint book-running managers of the offering and as representatives of the underwriters named below. Subject to the terms and conditions stated in the underwriting agreement dated the date of this prospectus, each underwriter named below has severally agreed to purchase, and we have agreed to sell to that underwriter, the number of shares set forth opposite the underwriter's name.
Underwriter
|
Number of Shares |
|||
|---|---|---|---|---|
Citigroup Global Markets Inc. |
||||
Cowen and Company, LLC |
||||
Stifel, Nicolaus & Company, Incorporated |
||||
Needham & Company, LLC |
||||
| | | | | |
Total |
3,400,000 | |||
| | | | | |
| | | | | |
The underwriting agreement provides that the obligations of the underwriters to purchase the shares included in this offering are subject to approval of legal matters by counsel and to other conditions. The underwriters are obligated to purchase all the shares (other than those covered by the over-allotment option described below) if they purchase any of the shares.
Shares sold by the underwriters to the public will initially be offered at the price set forth on the cover of this prospectus. Any shares sold by the underwriters to securities dealers may be sold at a discount from the initial public offering price not to exceed $ per share. If all the shares are not sold at the initial offering price, the underwriters may change the offering price and the other selling terms. The representatives have advised us that the underwriters do not intend to make sales to discretionary accounts.
If the underwriters sell more shares than the total number set forth in the table above, we have granted to the underwriters an option, exercisable for 30 days from the date of this prospectus, to purchase up to 510,000 additional shares at the public offering price less the underwriting discount. To the extent the option is exercised, each underwriter must purchase a number of additional shares approximately proportionate to that underwriter's initial purchase commitment. Any shares issued or sold under the option will be issued and sold on the same terms and conditions as the other shares that are the subject of this offering.
We, our officers and directors, certain of our employees and our other stockholders have agreed that, for a period of 90 days from the date of this prospectus, we and they will not, without the prior written consent of Citigroup Global Markets Inc. and Cowen and Company, LLC, dispose of or hedge any shares or any securities convertible into or exchangeable for our common stock. Citigroup Global Markets Inc. and Cowen and Company, LLC in their sole discretion may release any of the securities subject to these lock-up agreements at any time, which, in the case of officers and directors, shall be with notice.
Our common stock is listed on the NASDAQ Global Market under the symbol "GNCA".
The following table shows the underwriting discounts and commissions that we are to pay to the underwriters in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the underwriters' over-allotment option.
| |
Paid by Genocea | ||||||
|---|---|---|---|---|---|---|---|
| |
No Exercise | Full Exercise | |||||
Per share |
|||||||
Total |
|||||||
127
We estimate that our portion of the total expenses of this offering will be $550,000. We have agreed to reimburse the underwriters for certain expenses in an amount up to $30,000.
In connection with the offering, the underwriters may purchase and sell shares in the open market. Purchases and sales in the open market may include short sales, purchases to cover short positions, which may include purchases pursuant to the over-allotment option, and stabilizing purchases.
- •
- Short sales involve secondary market sales by the underwriters of a greater number of shares than they are required to
purchase in the offering.
- •
- "Covered" short sales are sales of shares in an amount up to the number of shares represented by the underwriters'
over-allotment option.
- •
- "Naked" short sales are sales of shares in an amount in excess of the number of shares represented by the underwriters'
over-allotment option.
- •
- Covering transactions involve purchases of shares either pursuant to the underwriters' over-allotment option or in the
open market in order to cover short positions.
- •
- To close a naked short position, the underwriters must purchase shares in the open market. A naked short position is more
likely to be created if the underwriters are concerned that there may be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who
purchase in the offering.
- •
- To close a covered short position, the underwriters must purchase shares in the open market or must exercise the
over-allotment option. In determining the source of shares to close the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the
open market as compared to the price at which they may purchase shares through the over-allotment option.
- •
- Stabilizing transactions involve bids to purchase shares so long as the stabilizing bids do not exceed a specified maximum.
Purchases to cover short positions and stabilizing purchases, as well as other purchases by the underwriters for their own accounts, may have the effect of preventing or retarding a decline in the market price of the shares. They may also cause the price of the shares to be higher than the price that would otherwise exist in the open market in the absence of these transactions. The underwriters may conduct these transactions on the NASDAQ Global Market, in the over-the-counter market or otherwise. If the underwriters commence any of these transactions, they may discontinue them at any time.
Other Relationships
The underwriters are full-service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, principal investment, hedging, financing and brokerage activities. Certain of the underwriters and their respective affiliates may, from time to time, engage in transactions with and perform services for us in the ordinary course of their business for which they may receive customary fees and reimbursement of expenses. In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (which may include bank loans and/or credit default swaps) for their own account and for the accounts of their customers and may at any time hold long and short positions in such securities and instruments. Such investments and securities activities may involve securities and/or instruments of ours or our affiliates. The underwriters and their affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or financial instruments and may hold,
128
or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, or to contribute to payments the underwriters may be required to make because of any of those liabilities.
Notice to Prospective Investors in the European Economic Area
In relation to each member state of the European Economic Area that has implemented the Prospectus Directive (each, a relevant member state), with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state (the relevant implementation date), an offer of shares described in this prospectus may not be made to the public in that relevant member state other than:
- •
- to any legal entity which is a qualified investor as defined in the Prospectus Directive;
- •
- to fewer than 100 or, if the relevant member state has implemented the relevant provision of the 2010 PD Amending
Directive, 150 natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of
the relevant Dealer or Dealers nominated by us for any such offer; or
- •
- in any other circumstances falling within Article 3(2) of the Prospectus Directive,
provided that no such offer of shares shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive.
For purposes of this provision, the expression an "offer of securities to the public" in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the shares to be offered so as to enable an investor to decide to purchase or subscribe for the shares, as the expression may be varied in that member state by any measure implementing the Prospectus Directive in that member state, and the expression "Prospectus Directive" means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the relevant member state) and includes any relevant implementing measure in the relevant member state. The expression 2010 PD Amending Directive means Directive 2010/73/EU.
The sellers of the shares have not authorized and do not authorize the making of any offer of shares through any financial intermediary on their behalf, other than offers made by the underwriters with a view to the final placement of the shares as contemplated in this prospectus. Accordingly, no purchaser of the shares, other than the underwriters, is authorized to make any further offer of the shares on behalf of the sellers or the underwriters.
Notice to Prospective Investors in the United Kingdom
This prospectus is only being distributed to, and is only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive that are also (i) investment professionals falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (the Order) or (ii) high net worth entities, and other persons to whom it may lawfully be communicated, falling within Article 49(2)(a) to (d) of the Order (each such person being referred to as a "relevant person"). This prospectus and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other persons in the United Kingdom. Any person in the United Kingdom that is not a relevant person should not act or rely on this document or any of its contents.
129
Notice to Prospective Investors in France
Neither this prospectus nor any other offering material relating to the shares described in this prospectus has been submitted to the clearance procedures of the Autorité des Marchés Financiers or of the competent authority of another member state of the European Economic Area and notified to the Autorité des Marchés Financiers. The shares have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France. Neither this prospectus nor any other offering material relating to the shares has been or will be:
- •
- released, issued, distributed or caused to be released, issued or distributed to the public in France; or
- •
- used in connection with any offer for subscription or sale of the shares to the public in France.
Such offers, sales and distributions will be made in France only:
- •
- to qualified investors (investisseurs qualifiés) and/or to
a restricted circle of investors (cercle restreint d'investisseurs), in each case investing for their own account, all as defined in, and in accordance
with articles L.411-2, D.411-1, D.411-2, D.734-1, D.744-1, D.754-1 and D.764-1 of the French Code monétaire et financier;
- •
- to investment services providers authorized to engage in portfolio management on behalf of third parties; or
- •
- in a transaction that, in accordance with article L.411-2-II-1°-or-2°-or 3° of the French Code monétaire et financier and article 211-2 of the General Regulations (Règlement Général) of the Autorité des Marchés Financiers, does not constitute a public offer (appel public à l'épargne).
The shares may be resold directly or indirectly, only in compliance with articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the French Code monétaire et financier.
Notice to Prospective Investors in Australia
No prospectus or other disclosure document (as defined in the Corporations Act 2001 (Cth) of Australia ("Corporations Act")) in relation to the common stock has been or will be lodged with the Australian Securities & Investments Commission ("ASIC"). This document has not been lodged with ASIC and is only directed to certain categories of exempt persons. Accordingly, if you receive this document in Australia:
- (a)
- you
confirm and warrant that you are either:
- (i)
- a
"sophisticated investor" under section 708(8)(a) or (b) of the Corporations Act;
- (ii)
- a
"sophisticated investor" under section 708(8)(c) or (d) of the Corporations Act and that you have provided an accountant's certificate to
us which complies with the requirements of section 708(8)(c)(i) or (ii) of the Corporations Act and related regulations before the offer has been made;
- (iii)
- a
person associated with the company under section 708(12) of the Corporations Act; or
- (iv)
- a
"professional investor" within the meaning of section 708(11)(a) or (b) of the Corporations Act, and to the extent that you are unable to
confirm or warrant that you are an exempt sophisticated investor, associated person or professional investor under the Corporations Act any offer made to you under this document is void and incapable
of acceptance; and
- (b)
- you warrant and agree that you will not offer any of the common stock for resale in Australia within 12 months of that common stock being issued unless any such resale offer is exempt
130
from the requirement to issue a disclosure document under section 708 of the Corporations Act.
Notice to Prospective Investors in Hong Kong
The shares may not be offered or sold in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), or (ii) to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a "prospectus" within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to shares which are or are intended to be disposed of only to persons outside Hong Kong or only to "professional investors" within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
Notice to Prospective Investors in Japan
The shares offered in this prospectus have not been and will not be registered under the Financial Instruments and Exchange Law of Japan. The shares have not been offered or sold and will not be offered or sold, directly or indirectly, in Japan or to or for the account of any resident of Japan (including any corporation or other entity organized under the laws of Japan), except (i) pursuant to an exemption from the registration requirements of the Financial Instruments and Exchange Law and (ii) in compliance with any other applicable requirements of Japanese law.
Notice to Prospective Investors in Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the shares may not be circulated or distributed, nor may the shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the SFA), (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with conditions set forth in the SFA.
Where the shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
- •
- a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of
which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or
- •
- a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor,
shares, debentures and units of shares and debentures of that corporation or the beneficiaries' rights and interest (howsoever described) in that trust shall not be transferred within six months after that
131
corporation or that trust has acquired the shares pursuant to an offer made under Section 275 of the SFA except:
- •
- to an institutional investor (for corporations, under Section 274 of the SFA) or to a relevant person defined in
Section 275(2) of the SFA, or to any person pursuant to an offer that is made on terms that such shares, debentures and units of shares and debentures of that corporation or such rights and
interest in that trust are acquired at a consideration of not less than $0.2 million (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in
cash or by exchange of securities or other assets, and further for corporations, in accordance with the conditions specified in Section 275 of the SFA;
- •
- where no consideration is or will be given for the transfer; or
- •
- where the transfer is by operation of law.
132
The validity of the issuance of our common stock offered in this prospectus will be passed upon for us by Ropes & Gray LLP, Boston, Massachusetts. Certain legal matters in connection with this offering will be passed upon for the underwriters by Cooley, LLP, Boston, Massachusetts.
The financial statements of Genocea Biosciences, Inc. appearing in Genocea Biosciences, Inc. Annual Report (Form 10-K) for the year ended December 31, 2013, have been audited by Ernst & Young LLP, independent registered public accounting firm, as set forth in its report thereon, included therein, and incorporated herein by reference. Such financial statements are incorporated herein by reference in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of common stock offered hereby. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement or the exhibits and schedules filed therewith. For further information with respect to us and the common stock offered hereby, reference is made to the registration statement and the exhibits and schedules filed therewith. Statements contained in this prospectus regarding the contents of any contract or any other document that is filed as an exhibit to the registration statement are not necessarily complete, and each such statement is qualified in all respects by reference to the full text of such contract or other document filed as an exhibit to the registration statement. A copy of the registration statement and the exhibits and schedules filed therewith may be inspected without charge at the public reference room maintained by the SEC, located at 100 F Street N.E., Washington, D.C. 20549, and copies of all or any part of the registration statement may be obtained from such offices upon the payment of the fees prescribed by the SEC. Please call the SEC at 1-800-SEC-0330 for further information about the public reference room. The SEC also maintains a website that contains reports, proxy and information statements and other information regarding registrants that file electronically with the SEC. The address is www.sec.gov.
We are subject to the information and periodic reporting requirements of the Exchange Act and, in accordance therewith, will file periodic reports, proxy statements and other information with the SEC. Such periodic reports, proxy statements and other information are available for inspection and copying at the public reference room and website of the SEC referred to above.
We also maintain a website at http://www.genocea.com and make available free of charge through this website our Annual Report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act. We make these reports available through our website as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the SEC. The information contained on, or that can be accessed through, our website is not a part of this prospectus. The reference to our web address does not constitute incorporation by reference of the information contained in, or that can be accessed through, our website.
INCORPORATION OF DOCUMENTS BY REFERENCE
The SEC allows us to "incorporate by reference" information from other documents that we file with it, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus.
133
Information in this prospectus supersedes information incorporated by reference that we filed with the SEC prior to the date of this prospectus.
We incorporate by reference into this prospectus and the registration statement of which this prospectus is a part the information or documents listed below that we have filed with the SEC.
- •
- our Annual Report on Form 10-K for the fiscal year ended December 31, 2013 filed on March 21, 2014;
- •
- our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 filed on May 9, 2014;
- •
- our Current Reports on Form 8-K filed on February 12, 2014, February 25, 2014, March 3, 2014
(only the portion of the Form 8-K disclosing Item 1.01) and April 8, 2014; and
- •
- the description of our common stock contained in our Registration Statement on Form 8-A filed on January 30, 2014, including any amendment or report filed for the purpose of updating such description. Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus will be deemed modified, superseded or replaced for purposes of this prospectus to the extent that a statement contained in this prospectus modifies, supersedes or replaces such statement.
You may request, orally or in writing, a copy of any or all of the documents incorporated herein by reference. These documents will be provided to you at no cost, by contacting: Investor Relations, Genocea Biosciences, Inc. 100 Acorn Park Drive, 5th Floor, Cambridge, Massachusetts 02140, (617) 876-8191 email address: ir@genocea.com. In addition, copies of any or all of the documents incorporated herein by reference may be accessed at our website at www.genocea.com. The information contained in, or accessible through, our website does not constitute part of this prospectus.
134
3,400,000 Shares
Genocea Biosciences, Inc.
Common Stock
PRELIMINARY PROSPECTUS
, 2014
| Citigroup | Cowen and Company |
Stifel
Needham & Company
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth the costs and expenses, other than the underwriting discounts and commissions, payable by the registrant in connection with the sale of common stock being registered. All amounts are estimates except for the SEC registration fee, the Financial Industry Regulatory Authority, or FINRA, filing fee and NASDAQ listing fee.
Item
|
Amount to be paid |
|||
|---|---|---|---|---|
SEC registration fee |
$ | 7,681 | ||
FINRA filing fee |
11,018 | |||
Blue Sky fees and expenses |
10,000 | |||
Printing and engraving expenses |
75,000 | |||
Legal fees and expenses |
300,000 | |||
Accounting fees and expenses |
100,000 | |||
Transfer Agent fees and expenses |
5,000 | |||
Miscellaneous expenses |
41,301 | |||
Total |
$ | 550,000 | ||
| | | | | |
Item 14. Indemnification of Directors and Officers
Section 145 of the General Corporation Law of the State of Delaware provides as follows:
A corporation shall have the power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys' fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interest of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful. The termination of any action, suit or proceeding by judgment, order, settlement, conviction or upon a plea of nolo contendere or its equivalent shall not, of itself, create a presumption that the person did not act in good faith and in a manner which the person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had reasonable cause to believe that his conduct was unlawful.
A corporation shall have the power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses (including attorneys' fees) actually and reasonably incurred by him in connection with the defense or settlement of such action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation and except that no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or the court in which such action or suit was brought
II-1
shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
As permitted by the Delaware General Corporation Law, we have included in our certificate of incorporation a provision to eliminate the personal liability of our directors for monetary damages for breach of their fiduciary duties as directors, subject to certain exceptions. In addition, our certificate of incorporation and by-laws provide that we are required to indemnify our officers and directors under certain circumstances, including those circumstances in which indemnification would otherwise be discretionary, and we are required to advance expenses to our officers and directors as incurred in connection with proceedings against them for which they may be indemnified.
We entered into indemnification agreements with our directors and officers that provide broader indemnity rights than those provided under the Delaware General Corporation Law and our certificate of incorporation. The indemnification agreements are not intended to deny or otherwise limit third-party or derivative suits against us or our directors or officers, but to the extent a director or officer were entitled to indemnity or contribution under the indemnification agreement, the financial burden of a third-party suit would be borne by us, and we would not benefit from derivative recoveries against the director or officer. Such recoveries would accrue to our benefit but would be offset by our obligations to the director or officer under the indemnification agreement.
The underwriting agreement provides that the underwriters are obligated, under certain circumstances, to indemnify our directors, officers and controlling persons against certain liabilities, including liabilities under the Securities Act. Reference is made to the form of underwriting agreement filed as Exhibit 1.1 hereto.
We maintain directors' and officers' liability insurance for the benefit of our directors and officers.
Item 15. Recent Sales of Unregistered Securities
In the three years preceding the filing of this registration statement, we have issued the following securities that were not registered under the Securities Act.
Sales of Capital Stock
On June 24, 2013, we issued 26,293,103 shares of Series C preferred stock at a price per share of $0.58 for total consideration of $15,250,000 to 21 investors.
On September 28, 2012, we issued 26,293,103 shares of Series C preferred stock at a price per share of $0.58 for total consideration of $15,250,000 to 21 investors.
Issuances of preferred stock were exempt pursuant to Rule 506 and Section 4(a)(2) of the Securities Act.
Sales of Warrants
On September 30, 2013, in connection with the working capital term loan facility with Ares Capital Corporation, we issued warrants to purchase 689,655 shares of our Series C preferred stock at an exercise price of $0.58 per share to Ares Capital Corporation. Upon completion of the initial public offering on February 10, 2014, these Series C preferred stock warrants automatically converted into warrants exercisable for 57,954 shares of common stock at an exercise price of $6.90 per share.
On January 3, 2011, we issued warrants to purchase 517,242 shares of our Series B preferred stock at an exercise price of $0.58 per share to Lighthouse Capital Partners VI, L.P. Upon completion of the initial public offering on February 10, 2014, these Series B preferred stock warrants automatically
II-2
converted into warrants exercisable for 43,465 shares of common stock at an exercise price of $6.90 per share.
Sales of warrants were exempt pursuant to Rule 506 and Section 4(a)(2) of the Securities Act.
Grants and Exercises of Stock Options
From January 1, 2014 through June 30, 2014 we granted options to purchase a total of 762,503 shares of our common stock to employees and non-employees, at a weighted average price of $14.42 per share. During the same period, we issued 59,275 shares of common stock upon the exercise of options to purchase such shares of common stock at a weighted average price of $2.28 per share.
In 2013, we granted options to purchase a total of 559,742 shares of our common stock to employees and non-employees, at a weighted average price of $3.41 per share. During the same period, we issued 31,809 shares of common stock upon the exercise of options to purchase such shares of common stock at a weighted average price of $1.33 per share.
In 2012, we granted options to purchase a total of 54,879 shares of our common stock to employees and consultants, at a weighted average exercise price of $1.79 per share. In 2012, we issued 545 shares of common stock upon the exercise of options to purchase such shares of common stock at a weighted average price of $2.24 per share.
In 2011, we granted options to purchase a total of 617,504 shares of our common stock to employees and consultants, at a weighted average exercise price of $2.08 per share. In 2011, we issued 2,386 shares of common stock upon the exercise of options to purchase such shares of common stock at a weighted average price of $2.26 per share.
Option grants and the issuances of common stock upon exercise of such options were exempt pursuant to Rule 701 and Section 4(2) of the Securities Act.
Item 16. Exhibits and financial statement schedules
(a) Exhibits
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 1.1 | Form of Underwriting Agreement | ||
| 3.1 | Fifth Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Company's Current Report on Form 8-K, File No. 001-36289, filed on February 12, 2014) | ||
| 3.2 | Amended and Restated By-laws (incorporated by reference to Exhibit 3.2 to the Company's Current Report on Form 8-K, File No. 001-36289, filed on February 12, 2014) | ||
| 4.1 | Form of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.2 | Form of Warrant to Purchase Preferred Stock, dated January 7, 2008 (incorporated by reference to Exhibit 4.2 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.3 | Preferred Stock Purchase Warrant, dated October 25, 2011, issued to Lighthouse Capital Partners VI, L.P. (incorporated by reference to Exhibit 4.3 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
II-3
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 4.4 | Preferred Stock Purchase Warrant, dated September 30, 2013, issued to Ares Capital Corporation (incorporated by reference to Exhibit 4.3 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.5 | Fourth Amended and Restated Registration Rights Agreement (incorporated by reference to Exhibit 4.5 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 5.1 | Opinion of Ropes & Gray LLP | ||
| 10.1 | Form of Director Indemnification Agreement (incorporated by reference to Exhibit 10.1 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.2 | + | Amended and Restated Exclusive License Agreement between Children's Medical Center Corporation and Genocea Biosciences, Inc., dated March 23, 2012 (incorporated by reference to Exhibit 10.2 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.3 | + | Amended and Restated License Agreement between Genocea Biosciences, Inc. and President and Fellows of Harvard College, dated November 19, 2012 (incorporated by reference to Exhibit 10.3 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.4 | + | License and Collaboration Agreement between Genocea Biosciences, Inc. and Isconova AB, dated August 5, 2009, as amended on March 19, 2010, June 18, 2010, August 17, 2010, October 19, 2011 and February 6, 2012 (incorporated by reference to Exhibit 10.4 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.5 | + | Exclusive License Agreement for Escherichia Coli K12 to Deliver Protein to the Macrophage Cytosol between Genocea Biosciences, Inc. and The Regents of the University of California, dated August 18, 2006 (incorporated by reference to Exhibit 10.5 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.6 | + | Patent License Agreement between Genocea Biosciences, Inc. and University of Washington dated January 27, 2010, as amended on July 19, 2012 (incorporated by reference to Exhibit 10.6 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.7 | Loan and Security Agreement, dated September 30, 2013, by and between Ares Capital Corporation and Genocea Biosciences, Inc. (incorporated by reference to Exhibit 10.7 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.8 | Lease, dated as of July 3, 2012, between TBCI, LLC and Genocea Biosciences, Inc. (incorporated by reference to Exhibit 10.8 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.9 | Agreement Regarding Sublease, dated as of July 9, 2012, by TBCI, LLC, FoldRx Pharmaceuticals, Inc., Pfizer Inc. and Genocea Biosciences, Inc. (incorporated by reference to Exhibit 10.9 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
II-4
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 10.10 | Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan, as amended on June 24, 2013 (incorporated by reference to Exhibit 10.10 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.11 | † | Consulting Agreement between Genocea Biosciences, Inc. and George Siber, dated May 16, 2007, as amended on June 30, 2009, December 16, 2010, June 15, 2011 and June 5, 2013 (incorporated by reference to Exhibit 10.11 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.12 | † | Amended and Restated Employment Letter Agreement between William Clark and Genocea Biosciences, Inc., dated January 16, 2014 (incorporated by reference to Exhibit 10.12 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.13 | † | Amended and Restated Employment Letter Agreement between Seth Hetherington, M.D. and Genocea Biosciences, Inc., dated January 16, 2014 incorporated by reference to Exhibit 10.13 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.14 | † | Amended and Restated Employment Letter Agreement between Jessica Flechtner, Ph.D. and Genocea Biosciences, Inc., dated January 16, 2014 (incorporated by reference to Exhibit 10.14 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.15 | † | Letter Agreement, dated April 7, 2014, by and between Genocea Biosciences, Inc. and Jonathan Poole (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K, File No. 001-36289, filed on April 8, 2014) | |
| 10.16 | † | Genocea Biosciences, Inc. 2014 Equity Incentive Plan (incorporated by reference to Exhibit 10.15 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.17 | † | Genocea Biosciences, Inc. Cash Incentive Plan (incorporated by reference to Exhibit 10.16 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.18 | † | Genocea Biosciences, Inc. Cash Bonus Program for Fiscal Years 2012, 2013 and 2014 (incorporated by reference to Exhibit 10.17 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.19 | + | Amendment No. 2 to Patent License Agreement between Genocea Biosciences, Inc. and University of Washington dated September 12, 2012 (incorporated by reference to Exhibit 10.18 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.20 | + | Amendment No. 3 to Patent License Agreement between Genocea Biosciences, Inc. and University of Washington dated November 7, 2013 (incorporated by reference to Exhibit 10.19 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.21 | † | Form of Nonstatutory Stock Option Granted under the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.20 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
II-5
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 10.22 | † | Form of Incentive Stock Option Granted under the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.21 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.23 | † | Form of Incentive Stock Option under the Genocea Biosciences, Inc. 2014 Equity Incentive Plan (incorporated by reference to Exhibit 10.21 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.24 | † | Form of Nonstatutory Stock Option under the Genocea Biosciences, Inc. 2014 Equity Incentive Plan (incorporated by reference to Exhibit 10.23 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.25 | † | Letter Agreement, dated January 9, 2014 terminating the Letter Agreement between Genocea Biosciences, Inc. and Katrine Bosley, dated February 4, 2013 (incorporated by reference to Exhibit 10.24 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.26 | † | Restricted Stock Agreement between Genocea Biosciences, Inc. and Katrine Bosley, dated November 7, 2013 (incorporated by reference to Exhibit 10.25 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.27 | † | Genocea Biosciences, Inc. 2014 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.26 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.28 | † | Nonstatutory Stock Option granted under the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan to Katrine Bosley, dated May 13, 2013 (incorporated by reference to Exhibit 10.28 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.29 | + | Bioprocessing Services Agreement between Genocea Biosciences, Inc. and FUJIFILM Diosynth Biotechnologies U.S.A., Inc. dated February 26, 2014 (incorporated by reference to Exhibit 10.1 to the Company's Form 10-Q, File No. 001-36289, filed on May 9, 2014) | |
| 23.1 | Consent of Ernst & Young LLP | ||
| 23.2 | Consent of Ropes & Gray LLP (included in Exhibit 5.1) | ||
| 24.1 | Power of attorney (previously filed) | ||
- †
- Indicates
a management contract or compensatory plan.
- +
- Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment and this exhibit has been submitted separately to the SEC.
(b) Financial statement schedules
Schedules not listed above have been omitted because the information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
The undersigned registrant hereby undertakes to deliver or cause to be delivered with the prospectus, to each person to whom the prospectus is sent or given, the latest annual report to security holders that is incorporated by reference in the prospectus and furnished pursuant to and meeting the
II-6
requirements of Rule 14a-3 or Rule 14c-3 under the Securities Exchange Act of 1934; and, where interim financial information required to be presented by Article 3 of Regulation S-X are not set forth in the prospectus, to deliver, or cause to be delivered to each person to whom the prospectus is sent or given, the latest quarterly report that is specifically incorporated by reference in the prospectus to provide such interim financial information.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
The undersigned Registrant hereby undertakes:
(1) That for purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) That for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-7
Pursuant to the requirements of the Securities Act of 1933, as amended, the Registrant has duly caused this Amendment No. 1 to it's Registration Statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of Cambridge, Commonwealth of Massachusetts, on July 14, 2014.
| GENOCEA BIOSCIENCES, INC. | ||||
By: |
/s/ WILLIAM CLARK |
|||
| William Clark President and Chief Executive Officer |
||||
Pursuant to the requirements of the Securities Act of 1933, Amendment No. 1 to the Registration Statement on Form S-1 has been signed by the following persons in the capacities and on the dates indicated.
Signature
|
Title
|
Date
|
||||
|---|---|---|---|---|---|---|
| /s/ WILLIAM CLARK William Clark |
Chief Executive Officer, President and Director (Principal Executive Officer) | July 14, 2014 | ||||
/s/ JONATHAN POOLE Jonathan Poole |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |
July 14, 2014 |
||||
* Kevin Bitterman, Ph.D. |
Director |
July 14, 2014 |
||||
* Katrine Bosley |
Director |
July 14, 2014 |
||||
* Simeon J. George, M.D. |
Director |
July 14, 2014 |
||||
* Stephen J. Hoffman, M.D., Ph.D. |
Director |
July 14, 2014 |
||||
* George Siber, M.D. |
Director |
July 14, 2014 |
||||
*By: |
/s/ JONATHAN POOLE Jonathan Poole Attorney-in-Fact |
|||||
II-8
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 1.1 | Form of Underwriting Agreement | ||
| 3.1 | Fifth Amended and Restated Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Company's Current Report on Form 8-K, File No. 001-36289, filed on February 12, 2014) | ||
| 3.2 | Amended and Restated By-laws (incorporated by reference to Exhibit 3.2 to the Company's Current Report on Form 8-K, File No. 001-36289, filed on February 12, 2014) | ||
| 4.1 | Form of Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.2 | Form of Warrant to Purchase Preferred Stock, dated January 7, 2008 (incorporated by reference to Exhibit 4.2 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.3 | Preferred Stock Purchase Warrant, dated October 25, 2011, issued to Lighthouse Capital Partners VI, L.P. (incorporated by reference to Exhibit 4.3 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.4 | Preferred Stock Purchase Warrant, dated September 30, 2013, issued to Ares Capital Corporation (incorporated by reference to Exhibit 4.3 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 4.5 | Fourth Amended and Restated Registration Rights Agreement (incorporated by reference to Exhibit 4.5 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 5.1 | Opinion of Ropes & Gray LLP | ||
| 10.1 | Form of Director Indemnification Agreement (incorporated by reference to Exhibit 10.1 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.2 | + | Amended and Restated Exclusive License Agreement between Children's Medical Center Corporation and Genocea Biosciences, Inc., dated March 23, 2012 (incorporated by reference to Exhibit 10.2 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.3 | + | Amended and Restated License Agreement between Genocea Biosciences, Inc. and President and Fellows of Harvard College, dated November 19, 2012 (incorporated by reference to Exhibit 10.3 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.4 | + | License and Collaboration Agreement between Genocea Biosciences, Inc. and Isconova AB, dated August 5, 2009, as amended on March 19, 2010, June 18, 2010, August 17, 2010, October 19, 2011 and February 6, 2012 (incorporated by reference to Exhibit 10.4 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
II-9
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 10.5 | + | Exclusive License Agreement for Escherichia Coli K12 to Deliver Protein to the Macrophage Cytosol between Genocea Biosciences, Inc. and The Regents of the University of California, dated August 18, 2006 (incorporated by reference to Exhibit 10.5 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.6 | + | Patent License Agreement between Genocea Biosciences, Inc. and University of Washington dated January 27, 2010, as amended on July 19, 2012 (incorporated by reference to Exhibit 10.6 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.7 | Loan and Security Agreement, dated September 30, 2013, by and between Ares Capital Corporation and Genocea Biosciences, Inc. (incorporated by reference to Exhibit 10.7 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.8 | Lease, dated as of July 3, 2012, between TBCI, LLC and Genocea Biosciences, Inc. (incorporated by reference to Exhibit 10.8 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.9 | Agreement Regarding Sublease, dated as of July 9, 2012, by TBCI, LLC, FoldRx Pharmaceuticals, Inc., Pfizer Inc. and Genocea Biosciences, Inc. (incorporated by reference to Exhibit 10.9 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.10 | Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan, as amended on June 24, 2013 (incorporated by reference to Exhibit 10.10 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | ||
| 10.11 | † | Consulting Agreement between Genocea Biosciences, Inc. and George Siber, dated May 16, 2007, as amended on June 30, 2009, December 16, 2010, June 15, 2011 and June 5, 2013 (incorporated by reference to Exhibit 10.11 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.12 | † | Amended and Restated Employment Letter Agreement between William Clark and Genocea Biosciences, Inc., dated January 16, 2014 (incorporated by reference to Exhibit 10.12 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.13 | † | Amended and Restated Employment Letter Agreement between Seth Hetherington, M.D. and Genocea Biosciences, Inc., dated January 16, 2014 incorporated by reference to Exhibit 10.13 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.14 | † | Amended and Restated Employment Letter Agreement between Jessica Flechtner, Ph.D. and Genocea Biosciences, Inc., dated January 16, 2014 (incorporated by reference to Exhibit 10.14 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
| 10.15 | † | Letter Agreement, dated April 7, 2014, by and between Genocea Biosciences, Inc. and Jonathan Poole (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K, File No. 001-36289, filed on April 8, 2014) | |
II-10
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 10.16 | † | Genocea Biosciences, Inc. 2014 Equity Incentive Plan (incorporated by reference to Exhibit 10.15 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.17 | † | Genocea Biosciences, Inc. Cash Incentive Plan (incorporated by reference to Exhibit 10.16 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.18 | † | Genocea Biosciences, Inc. Cash Bonus Program for Fiscal Years 2012, 2013 and 2014 (incorporated by reference to Exhibit 10.17 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.19 | + | Amendment No. 2 to Patent License Agreement between Genocea Biosciences, Inc. and University of Washington dated September 12, 2012 (incorporated by reference to Exhibit 10.18 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.20 | + | Amendment No. 3 to Patent License Agreement between Genocea Biosciences, Inc. and University of Washington dated November 7, 2013 (incorporated by reference to Exhibit 10.19 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.21 | † | Form of Nonstatutory Stock Option Granted under the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.20 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.22 | † | Form of Incentive Stock Option Granted under the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan (incorporated by reference to Exhibit 10.21 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.23 | † | Form of Incentive Stock Option under the Genocea Biosciences, Inc. 2014 Equity Incentive Plan (incorporated by reference to Exhibit 10.21 to the Company's Registration Statement on Form S-1, File No. 333-193043, filed on December 23, 2013) | |
| 10.24 | † | Form of Nonstatutory Stock Option under the Genocea Biosciences, Inc. 2014 Equity Incentive Plan (incorporated by reference to Exhibit 10.23 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.25 | † | Letter Agreement, dated January 9, 2014 terminating the Letter Agreement between Genocea Biosciences, Inc. and Katrine Bosley, dated February 4, 2013 (incorporated by reference to Exhibit 10.24 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.26 | † | Restricted Stock Agreement between Genocea Biosciences, Inc. and Katrine Bosley, dated November 7, 2013 (incorporated by reference to Exhibit 10.25 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.27 | † | Genocea Biosciences, Inc. 2014 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.26 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 23, 2014) | |
II-11
| Exhibit Number |
Exhibit Index | ||
|---|---|---|---|
| 10.28 | † | Nonstatutory Stock Option granted under the Genocea Biosciences, Inc. Amended and Restated 2007 Equity Incentive Plan to Katrine Bosley, dated May 13, 2013 (incorporated by reference to Exhibit 10.28 to the Company's Registration Statement on Form S-1, File No. 333-193043, as amended on January 13, 2014) | |
| 10.29 | + | Bioprocessing Services Agreement between Genocea Biosciences, Inc. and FUJIFILM Diosynth Biotechnologies U.S.A., Inc. dated February 26, 2014 (incorporated by reference to Exhibit 10.1 to the Company's Form 10-Q, File No. 001-36289, filed on May 9, 2014) | |
| 23.1 | Consent of Ernst & Young LLP | ||
| 23.2 | Consent of Ropes & Gray LLP (included in Exhibit 5.1) | ||
| 24.1 | Power of attorney (previously filed) | ||
- †
- Indicates
a management contract or compensatory plan.
- +
- Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment and this exhibit has been submitted separately to the SEC.
II-12