Attached files
| file | filename |
|---|---|
| EX-10.8 - EX-10.8 - IMMUNIC, INC. | d543159dex108.htm |
| EX-23.1 - EX-23.1 - IMMUNIC, INC. | d543159dex231.htm |
| EX-10.6 - EX-10.6 - IMMUNIC, INC. | d543159dex106.htm |
| EX-23.2 - EX-23.2 - IMMUNIC, INC. | d543159dex232.htm |
Table of Contents
As filed with the Securities and Exchange Commission on April 3, 2014
Registration No. 333-191711
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 5
to
FORM S-1
REGISTRATION STATEMENT
Under
The Securities Act of 1933
Vital Therapies, Inc.
(Exact name of Registrant as specified in its charter)
| Delaware | 2834 | 56-2358443 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
15010 Avenue of Science, Suite 200
San Diego, CA 92128
(858) 673-6840
(Address, including zip code, and telephone number, including area code, of Registrant’s principal executive offices)
Terence E. Winters, Ph.D.
Co-Chairman of the Board &
Chief Executive Officer
Vital Therapies, Inc.
15010 Avenue of Science, Suite 200
San Diego, CA 92128
(858) 673-6840
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Martin J. Waters Robert F. Kornegay Anthony G. Mauriello Wilson Sonsini Goodrich & Rosati, Professional Corporation 12235 El Camino Real, Suite 200 San Diego, CA 92130-3002 (858) 350-2300 |
Michael V. Swanson Chief Financial Officer Vital Therapies, Inc. 15010 Avenue of Science, Suite 200 San Diego, CA 92128 (858) 673-6840 |
Thomas A. Coll (858) 550-6000 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after this registration statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | x (Do not check if a smaller reporting company) | Smaller reporting company | ¨ |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment that specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any state or other jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION DATED APRIL 3, 2014
Preliminary Prospectus
Shares

Common Stock
This is the initial public offering of shares of our common stock. Prior to this offering, there has been no public market for our common stock. The initial public offering price of our common stock is expected to be between $ and $ per share.
We have been approved to list our common stock on the NASDAQ Global Market under the symbol “VTL.”
The underwriters have an option to purchase a maximum of additional shares of common stock from us.
We are an “emerging growth company” under applicable Securities and Exchange Commission rules and we have elected to comply with reduced public company reporting requirements.
Our business and an investment in our common stock involve significant risks. These risks are described under the caption “Risk Factors” beginning on page 10 of this prospectus.
| Price to Public | Underwriting Discounts and Commissions(1) |
Proceeds
to Vital Therapies, Inc. |
||||||||||
| Per Share |
$ | $ | $ | |||||||||
| Total |
$ | $ | $ | |||||||||
| (1) | We refer you to “Underwriting” beginning on page 135 of this prospectus for additional information regarding total underwriting compensation. |
Certain of our directors, officers, existing investors and their affiliated entities have indicated an interest in purchasing an aggregate of approximately $20.0 million in shares of our common stock in this offering at the initial public offering price. However, because indications of interest are not binding agreements or commitments to purchase, these persons and entities may determine to purchase fewer shares than they indicate an interest in purchasing or not to purchase any shares in this offering. It is also possible that these persons and entities could purchase more shares of our common stock. The underwriters will receive the same underwriting discount on any shares purchased by these persons and entities as they will on any other shares sold to the public in this offering.
Delivery of the shares of common stock will be made on our about , 2014.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
| BofA Merrill Lynch | Credit Suisse |
| William Blair |
Canaccord Genuity |
The date of this prospectus is , 2014
Table of Contents
For investors outside the United States: neither we nor the underwriters have done anything that would permit this offering or possession or distribution of this prospectus or any free writing prospectus we may provide to you in connection with this offering in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus and any such free writing prospectus outside of the United States.
| Page | ||||
| 1 | ||||
| 10 | ||||
| 39 | ||||
| 41 | ||||
| 42 | ||||
| 43 | ||||
| 45 | ||||
| 48 | ||||
| MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
50 | |||
| 69 | ||||
| 97 | ||||
| 107 | ||||
| 117 | ||||
| 121 | ||||
| 123 | ||||
| 129 | ||||
| MATERIAL U.S. FEDERAL INCOME AND ESTATE TAX CONSEQUENCES TO NON-U.S. HOLDERS |
132 | |||
| 135 | ||||
| 142 | ||||
| 142 | ||||
| 142 | ||||
| F-1 | ||||
We have not, and the underwriters have not, authorized any other person to provide you with different information than that contained in this prospectus and any related free writing prospectus that we may provide to you in connection with this offering. If anyone provides you with different or inconsistent information, you should not rely on it. We are not, and the underwriters are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
Until , 2014, all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This requirement is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
Table of Contents
This summary provides an overview of selected information contained elsewhere in this prospectus and does not contain all of the information you should consider before investing in our common stock. You should carefully read this prospectus and the registration statement of which this prospectus is a part in their entirety before investing in our common stock, including the information discussed under “Risk Factors” beginning on page 10 and our financial statements and notes thereto that appear elsewhere in this prospectus. As used in this prospectus, the terms “we,” “our,” “us,” “Vital Therapies,” or “the Company” refer to Vital Therapies, Inc. and its subsidiaries, taken as a whole, unless the context otherwise requires it.
Overview
We are a biotherapeutic company focused on developing a cell-based therapy targeting the treatment of all forms of acute liver failure. Our product candidate, the ELAD System, is a human cell-based bio-artificial liver support system that operates outside the body, or extracorporeal, and is designed to allow the patient’s own liver to regenerate to a healthy state, or to stabilize the patient until transplant. We believe the ELAD System has the potential to be a life-saving therapy in patients suffering from acute liver failure. The ELAD System has received orphan designation in the United States and Europe for the treatment of patients with acute liver failure. This designation provides tax credits for qualified clinical testing, seven years of market exclusivity in the United States, and ten years of market exclusivity in Europe for the first approved orphan drug for a given indication. However, orphan designation does not alter the standard regulatory requirements or the process for obtaining marketing approval.
Acute liver failure, including acute-on-chronic, surgically-induced and fulminant liver failures, represents a serious unmet medical need affecting at least 30,000 patients annually in the United States with similar incidence rates in Europe. Except for liver transplant, which is limited by a shortage of donor organs, standard-of-care treatment focuses on the management of disease complications, does not restore lost liver function, and is associated with high mortality. We believe the ELAD System holds considerable therapeutic promise because it has shown trends indicating the potential to increase survival rates in patients with acute liver failure. Prior to the initiation of our ongoing Phase 3 clinical trial program discussed below, more than 145 subjects have received the ELAD System therapy in seven clinical trials and through a compassionate use program, which we believe collectively show a promising therapeutic profile.
The ELAD System is an allogeneic cellular therapy system incorporating our human liver-derived cells, or VTL C3A cells, contained in four hollow fiber cartridges that are combined with single use customized disposable sets and a reuseable bedside unit to provide extracorporeal circulation of blood plasma to the VTL C3A cells and the return of treated plasma back to the patient. The VTL C3A cells remain within these four ELAD cartridges during the treatment session and only the treated plasma, which is later reconstituted with the patient’s blood cells, is returned to the patient. We have customized our human liver-derived C3A cell line to create an optimized bank of cells for use in the ELAD System that we culture and expand using proprietary techniques. These cells have been shown to retain many key synthetic and metabolic processes of normal human hepatocytes, the primary functional cell of the liver. The four ELAD cartridges collectively contain 440 grams, or approximately one pound, of VTL C3A cells. The patient’s blood plasma is treated by our VTL C3A cells in a single session of continuous therapy lasting between three and ten days. We believe that the ELAD System therapy facilitates the recovery of liver function and has the potential to improve clinical outcomes and increase the likelihood of survival in patients with acute liver failure.
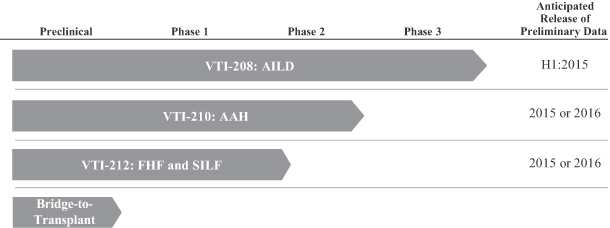
We are currently enrolling patients in one Phase 3 clinical trial and have regulatory allowance to begin enrolling patients in a second Phase 3 trial, each in various forms of acute liver failure. In March 2013, we initiated VTI-208, a Phase 3 randomized, controlled clinical trial in 200 subjects with alcohol-induced liver decompensation, or AILD. As of April 2, 2014, 90 subjects had been enrolled in this trial and 47 clinical sites were open for subject enrollment. In addition, we have obtained regulatory allowance in the United States, United Kingdom, Spain and Australia to begin enrolling patients and have initiated clinical sites in a second Phase 3 randomized, controlled clinical trial, VTI-210, in 120 subjects with severe acute alcoholic hepatitis, or
1
Table of Contents
AAH, which is a subset of AILD. We expect the enrollment of subjects in VTI-210 to begin in the first half of 2014. These studies are designed to complement each other and confirm study outcomes, and may be combined to support a biologics license application, or BLA, in the United States and a marketing authorization application in the European Union, or E.U. In addition, based upon discussions with United States and European regulatory authorities, we believe our VTI-208 and VTI-210 clinical trials, if successful from both a statistical and clinical standpoint, may support product registration on a stand-alone basis. According to the Food and Drug Administration, or the FDA, a second confirmatory clinical trial that substantiates positive results in VTI-208 may be necessary to support a BLA. The European Medicines Agency, or EMA, has informed us that, if deemed successful, VTI-210 will be sufficient to support product registration in the E.U. Based on assumptions derived from our earlier clinical trials and the medical literature, we have designed each of our VTI-208 and VTI-210 clinical trials with 95% power to achieve statistical significance for their primary endpoint of overall survival.
In the second half of 2014, we also expect to initiate enrollment of VTI-212, a Phase 2/3 clinical program in subjects with either fulminant hepatic failure, or FHF, or surgery-induced acute liver failure, or SILF. We plan to begin this program with a Phase 2 single-arm component enrolling 40 subjects, which may later be followed by a randomized, controlled Phase 3 component the size of which has not yet been determined. Results from the single-arm component will be compared with historical or case-matched controls, and we currently anticipate Phase 2 data in 2015 or 2016. Since FHF and SILF have high mortality rates and affect a very small number of patients for which there is currently no satisfactory therapeutic intervention available, the results from the Phase 2 single arm component may provide support for an expedited regulatory approval pathway. However, regulatory agreement on an expedited approval pathway has not yet been sought and may never be granted. In the event that randomized data are necessary for approval in FHF and SILF, we expect to perform the randomized Phase 3 portion of the program, the design of which would be finalized upon analysis of the Phase 2 component. Data from VTI-212 may also be used to support our planned marketing applications for AILD and AAH.
The following table lists our pivotal clinical trials for the ELAD System in AILD and AAH, our Phase 2 clinical trial in FHF and SILF, and a future indication expansion opportunity in bridge-to-transplant (anticipated release dates assuming funding from this offering):
At our facility we manufacture the ELAD cartridges containing the VTL C3A cells, repackage the disposable sets, and assemble the ELAD System bedside units. We believe that our internal manufacturing infrastructure will allow us to complete clinical development of the ELAD System and support worldwide commercialization efforts. We intend to build a targeted sales force to commercialize the ELAD System in the United States and Europe. In the United States, we expect to focus our efforts on approximately 100 liver transplant centers and an equal number of other specialist intensive care centers. We expect to target a similar number of institutions in Europe. We also intend to opportunistically pursue markets outside the United States and Europe either through direct sales or collaborations.
2
Table of Contents
We own exclusive worldwide commercial rights to the ELAD System free of royalties and our key U.S. patent will remain in effect until 2027 or later if extended. In addition, we have developed proprietary methods and know-how for growing, storing and optimizing the function of our VTL C3A cells. This know-how includes proprietary techniques for culturing and expanding our VTL C3A cells. We have assembled this know-how through our ELAD System product development and believe that our proprietary banks of VTL C3A cells and manufacturing expertise create significant barriers to entry.
If approved for marketing, the ELAD System will be eligible for 12 years of data exclusivity in the United States under the Biologics Price Competition and Innovation Act of 2012. Upon approval, if any, our orphan designation for the ELAD System for the treatment of patients with acute liver failure will provide market exclusivity for seven years in the United States and ten years in Europe.
The ELAD System
The key to the performance of the ELAD System is our VTL C3A cell bank. Our VTL C3A cells are distinct from publicly available C3A cells and have been optimized for the ELAD System therapy. These cells are immortal, which means that they can be made to propagate for prolonged periods, and we believe this will permit cell production at commercial scale. In addition, they have been shown to retain many of the specific metabolic processes and pathways of human liver cells. These functions include an active cytochrome P450 enzyme system, involved in the metabolism of many compounds such as drugs, toxins and hormones, as well as the production of liver-specific proteins such as albumin, anti-thrombin III, alpha-fetoprotein, C3 complement, Factor V, transferrin, alpha-1-antichymotrypsin and alpha-1-antitrypsin.
The ELAD System therapy uses VTL C3A cells from the same source to treat all patients. This process is known as allogeneic cellular therapy. In contrast, autologous cellular therapy uses a patient’s own cells, which are manipulated in individual production batches, a costly and complex process. As a result, the production and logistics of the ELAD System therapy do not face the challenges commonly associated with autologous cellular therapies.
Differentiating Factors of the ELAD System
Unlike other potential therapies developed for acute liver failure in the past, we believe the ELAD System holds a unique combination of attributes:
| • | Biologically active. The ELAD System is designed to mimic liver function and improve survival in patients with acute liver failure by replicating key biologic processes performed by human hepatocytes, which are believed to be responsible for 500 or more biologic processes. |
| • | Human cellular therapy. The ELAD System therapy uses allogeneic human cells to avoid the safety, technical and immunologic risks associated with animal cell therapies. We are not aware of any FDA-approved animal-based cellular therapy for use in patients. |
| • | Immortal human liver-derived C3A cells. Our VTL C3A cells are immortal, retain many key functions of hepatocytes and can be expanded to adequate treatment amounts. |
| • | Commercially scalable. We have developed scalable production processes and manufacturing capabilities sufficient to support the development and commercialization of the ELAD System. |
| • | Ease of use. The ELAD System is an allogeneic cellular therapy designed for convenient production and administration. Our VTL C3A cells usually remain alive for the duration of a normal patient therapy without the need for replacement, enhancing ease-of-use and reducing cost-of-goods. |
3
Table of Contents
The ELAD System Clinical Development
A full description of the ELAD System’s clinical development can be found in the section entitled “Business.” Prior to the initiation of our ongoing Phase 3 clinical trial program, over 145 subjects have received therapy with the ELAD System at multiple clinical sites in the United States, Europe, Asia and the Middle East in six randomized, controlled, clinical trials, one single-arm trial and a compassionate-use program. Certain results from selected trials are described below:
| • | Phase 2b AILD Trial. Between 2009 and 2011, 37 subjects with AILD were randomized into a pre-defined cohort of a controlled Phase 2b clinical trial in the United States and Europe comparing the ELAD System plus standard-of-care to standard-of-care alone. After the elimination of eight subjects under pre-defined criteria, the per protocol analysis revealed that the ELAD System-treated subjects experienced a 58% increase in 90-day overall survival relative to control subjects. In addition, the ELAD System-treated subjects experienced improvements in serum levels of bilirubin, sodium and creatinine, which are pertinent to our understanding of the mechanism of action of the ELAD System in these subjects, and which we believe are consistent with improvement in liver function. |
| • | China Pivotal Trial in Acute Flare of Viral Hepatitis. Between 2006 and 2007, 69 subjects with acute liver failure were randomized in a controlled clinical trial at two hospitals in Beijing, China comparing the ELAD System plus standard-of-care to standard-of-care alone. The trial was stopped by the lead hospital’s ethics committee after an interim intent-to-treat analysis of the first 49 subjects revealed a 72% increase in 28-day transplant-free survival relative to control subjects. Improvements were also seen in serum bilirubin and serum sodium for subjects treated with the ELAD System. Subsequent follow-up at three years and five years showed durable improvements in transplant-free survival. |
| • | Pilot Trials in FHF. Between 1999 and August 2003, our predecessor company enrolled 44 subjects with FHF in two separate randomized clinical trials comparing the ELAD System plus standard-of-care to standard-of-care alone. A post-hoc meta-analysis of the two trials limited to the 26 subjects who were listed for liver transplant at the time of enrollment revealed a 50% increase in 30-day overall survival relative to control subjects. |
Our Strategy
Our goal is to become the leading biotherapeutic company developing and marketing cell-based therapies for acute liver failure. Key elements of our strategy to achieve this goal are:
| • | Successfully complete the ELAD System’s clinical development in acute liver failure; |
| • | Obtain regulatory approval for the ELAD System in the United States and Europe; |
| • | Maximize the commercial potential of the ELAD System in the United States and Europe by establishing a targeted sales force focused on liver transplant and specialist intensive care centers; |
| • | Opportunistically explore commercial opportunities for the ELAD System in other international markets; |
| • | Continue to technically develop and improve our ELAD System and explore other uses for human liver-derived C3A cells, including the potential commercialization of proteins and other compounds produced by human liver-derived C3A cells; and |
| • | Pursue development of the ELAD System in additional indications. |
4
Table of Contents
Our Risks
Our business is subject to numerous risks, which are highlighted in the section entitled “Risk Factors” immediately following this prospectus summary. These risks represent challenges to the successful implementation of our strategy and to the growth and future profitability of our business. Some of these risks are:
| • | We are a clinical-stage company with no approved products, which makes assessment of our future viability difficult; |
| • | We are totally dependent upon the success of the ELAD System, our sole product candidate, which is a combination product that may increase the complexity of both biologic and medical device regulatory issues; |
| • | We cannot give any assurance that we will successfully complete the ELAD System’s clinical development, or that it will receive regulatory approval in a timely manner or at all; |
| • | If we fail to obtain regulatory approval in the United States and Europe, our business would be materially and adversely harmed; |
| • | If we are able to secure marketing approval, our commercial success will be determined by our ability to obtain acceptable pricing and reimbursement for the ELAD System therapy; |
| • | We have limited experience in conducting pivotal clinical trials used to support regulatory approval and prior clinical trials of the ELAD System did not demonstrate a statistically significant improvement in survival, the primary endpoint that is needed to support regulatory approval; |
| • | If we fail to select appropriate subjects for our clinical programs or if these subjects do not progress as expected, it will be difficult for us to demonstrate the statistically significant efficacy of the ELAD System therapy necessary to gain approval; |
| • | Random variations in the standard-of-care could cause our clinical trials to fail; |
| • | Ethical considerations require us to conduct open-label clinical trials of the ELAD System where control subjects do not receive a sham treatment and this may introduce unacceptable bias into our trial results; |
| • | Our patent rights may prove to be an inadequate barrier to competition and we do not hold any patents covering our VTL C3A cells or the production processes we use to grow the VTL C3A cells in the ELAD cartridges; |
| • | We rely on third party suppliers, and in some instances, a single third party supplier, for critical components of the ELAD System and these suppliers could cease to manufacture the components, go out of business or otherwise not perform as anticipated; |
| • | Cellular therapy is complex and we do not have a complete understanding of the mechanism of action of the ELAD System; |
| • | If we are unable to implement our sales, marketing, distribution, training and support strategies, we will not be able to effectively commercialize the ELAD System and may not reach profitability; and |
| • | We will need to raise additional capital. |
Implications of Being an Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and, for as long as we continue to be an “emerging growth company,” we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies but not to “emerging growth companies,” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, reduced disclosure obligations
5
Table of Contents
regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an “emerging growth company” for up to five fiscal years following the effective date of this prospectus, or until the earliest of (i) the last day of the first fiscal year in which our annual gross revenues exceed $1 billion, (ii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, which would occur on the last day of the fiscal year that the market value of our common stock held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter, or (iii) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three-year period. We are choosing to “opt out” of the extended transition periods available under the JOBS Act for complying with new or revised accounting standards, and intend to take advantage of the other exemptions.
Corporate Information
We were incorporated in California in May 2003 as Vitagen Acquisition Corp., changed our name to Vital Therapies, Inc. in June 2003, and reincorporated in Delaware in January 2004. Our principal executive offices are located at 15010 Avenue of Science, Suite 200, San Diego, CA 92128. Our telephone number is (858) 673-6840. Our website address is http://www.vitaltherapies.com. Information contained on our website is not incorporated by reference into this prospectus, and should not be considered to be part of this prospectus. You should not rely on our website or any such information in making your decision whether or not to purchase our common stock.
“Vital Therapies” and “ELAD” are registered trademarks of Vital Therapies and the Vital Therapies logo is a trademark of Vital Therapies. Other service marks, trademarks, and tradenames referred to in this prospectus are the property of their respective owners. Except as set forth above and solely for convenience, the trademarks and tradenames in this prospectus are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto.
6
Table of Contents
The Offering
| Common stock offered by us |
shares |
| Common stock to be outstanding after this offering |
shares |
| Underwriters’ option to purchase additional shares |
shares |
| Use of proceeds |
We intend to use the net proceeds from this offering to fund the clinical development of the ELAD System, and for working capital and other general corporate purposes. See “Use of Proceeds.” |
| Risk factors |
See the section entitled “Risk Factors” and other information included in this prospectus for a discussion of factors you should consider carefully before deciding to invest in shares of our common stock. |
| NASDAQ Global Market symbol |
VTL |
The number of shares of common stock to be outstanding following this offering is based on 16,615,661 shares of our common stock outstanding as of February 28, 2014 (including preferred stock on an as-converted basis), and excludes:
| • | 3,136,269 shares of our common stock issuable upon the exercise of options outstanding as of February 28, 2014, with a weighted-average exercise price of $6.71 per share; |
| • | 250,646 shares of our common stock issuable upon the exercise of warrants outstanding as of February 28, 2014, with a weighted-average exercise price of $95.21 per share; and |
| • | 91,796 shares of our common stock reserved for future issuance as of February 28, 2014, under our stock-based compensation plans, including our 2012 Stock Plan and our 2014 Equity Incentive Plan, which will become effective in connection with this offering and contains provisions that will automatically increase its share reserve on the date of this prospectus and on each annual anniversary of the date of this prospectus, as more fully described in “Executive and Director Compensation—Equity Incentive Plans”. |
Unless otherwise noted, the information in this prospectus reflects and assumes the following:
| • | the conversion of all outstanding shares of our preferred stock into an aggregate of 16,009,423 shares of common stock upon the closing of this offering; |
| • | the filing of our amended and restated certificate of incorporation immediately prior to the closing of this offering; and |
| • | no exercise by the underwriters of their option to purchase additional shares. |
7
Table of Contents
Summary Consolidated Financial Data
The consolidated statements of operations data for the years ended December 31, 2012 and 2013 are derived from our audited consolidated financial statements included elsewhere in this prospectus. Our historical results are not necessarily indicative of the results that may be expected in the future. You should read this information together with our audited consolidated financial statements and related notes appearing elsewhere in this prospectus and the information under the captions “Selected Consolidated Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
| Years Ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| (In thousands, except share |
||||||||
| Consolidated Statements of Operations Data: |
||||||||
| Operating expenses: |
||||||||
| Research and development |
$ | 5,097 | $ | 21,787 | ||||
| General and administrative |
4,483 | 9,615 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
9,580 | 31,402 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(9,580 | ) | (31,402 | ) | ||||
| Total other income (expense) |
2,879 | (1,316 | ) | |||||
|
|
|
|
|
|||||
| Net loss |
(6,701 | ) | (32,718 | ) | ||||
| Amortization of deemed dividend |
— | (64 | ) | |||||
| Accretion to redemption value of senior redeemable convertible preferred stock |
(942 | ) | (6,303 | ) | ||||
|
|
|
|
|
|||||
| Net loss attributable to common stockholders |
$ | (7,643 | ) | $ | (39,085 | ) | ||
|
|
|
|
|
|||||
| Net loss per share attributable to common stockholders, basic and diluted(1) |
$ | (17.89 | ) | $ | (74.86 | ) | ||
|
|
|
|
|
|||||
| Weighted-average common shares outstanding, basic and diluted(1) |
427,117 | 522,102 | ||||||
|
|
|
|
|
|||||
| Pro forma net loss per share attributable to common stockholders, basic and diluted (unaudited)(1) |
$ | (3.14 | ) | |||||
|
|
|
|||||||
| Pro forma weighted-average common shares outstanding, basic and diluted (unaudited)(1) |
10,008,616 | |||||||
|
|
|
|||||||
| (1) | Please refer to Note 2 of our consolidated financial statements for an explanation of the method used to calculate the historical and pro forma net loss per share attributable to common stockholders and the number of shares used in the computation of the per share amounts. |
8
Table of Contents
| Actual As of December 31, 2013 |
Pro Forma- Private Offerings(1) |
Pro Forma- Converted(2) |
Pro Forma As Adjusted-IPO(3)(4) | |||||||||||
| (In thousands) | ||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||
| Cash and cash equivalents |
$ | 38,186 | $ | 56,554 | $ | 56,554 | ||||||||
| Working capital |
36,409 | 54,777 | 57,377 | |||||||||||
| Total assets |
46,585 | 64,953 | 64,953 | |||||||||||
| Future purchase rights |
2,600 | 2,600 | — | |||||||||||
| Preferred stock |
83,475 | 101,843 | — | |||||||||||
| Total stockholders’ (deficit) equity |
(44,657 | ) | (44,657 | ) | 59,786 | |||||||||
| (1) | The Pro Forma-Private Offerings balance sheet data in the table above reflects the sale and issuance of 2,296,016 shares of our senior redeemable convertible preferred stock at $8.00 per share in January 2014 and, pursuant to a preemptive rights offering in February 2014, and the receipt of proceeds of $18.4 million. |
| (2) | The Pro Forma-Converted balance sheet data in the table above reflects the 2014 private offerings referred to in (1) above plus the conversion of all outstanding shares of preferred stock as of December 31, 2013 and the senior preferred stock issued in the 2014 private offerings into an aggregate of 16,009,423 shares of our common stock and the reclassification of the future purchase rights liabilities to additional paid-in capital, a component of stockholders’ (deficit) equity. |
| (3) | The Pro Forma As Adjusted-IPO balance sheet data in the table above reflects the 2014 private offerings and pro forma conversion and reclassification described in (1) and (2) above plus the sale of shares of our common stock in this offering and the application of the net proceeds at an assumed initial public offering price of $ per share, the midpoint of the price range set forth on the cover of this prospectus, after deducting the estimated underwriting discount and estimated offering expenses payable by us. |
| (4) | A $1.00 increase (decrease) in the assumed initial public offering price of $ per share, the midpoint of the range set forth on the cover of this prospectus, would increase (decrease) cash and cash equivalents and each of working capital, total assets and total stockholders’ equity by $ , assuming that the number of shares offered by us, as set forth on the cover of this prospectus, remains the same, and after deducting the estimated underwriting discount. Each increase (decrease) of 1.0 million shares in the number of shares offered by us would increase (decrease) cash and cash equivalents and each of working capital, total assets and total stockholders’ equity by approximately $ , assuming an initial public offering price of $ , the midpoint of the price range set forth on the cover of this prospectus, and after deducting the estimated underwriting discount. The Pro Forma As Adjusted-IPO information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
Cash and Cash Equivalents at February 28, 2014
At February 28, 2014, we held cash and cash equivalents of $51.3 million, of which $1.2 million was restricted.
9
Table of Contents
Investing in our common stock involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information in this prospectus, including our consolidated financial statements and related notes, before deciding whether to purchase shares of our common stock. If any one or more of the following risks are realized, our business, financial condition, results of operations and prospects could be materially and adversely affected. In that event, the price of our common stock could decline and you could lose part or all of your investment.
Risks Related to Our Business
We are a clinical-stage company with no approved products, which makes assessment of our future viability difficult.
We are a clinical-stage company and we have no approved products or revenues from the sale of products. Our operations to date have been limited to organizing, staffing and financing our company, applying for patent rights, manufacturing on a clinical scale, undertaking clinical trials of our product candidate, and engaging in research and development. We have not yet demonstrated an ability to obtain regulatory approval, manufacture commercial-scale products, or conduct the sales and marketing activities necessary for successful product commercialization. As a result, there is limited information about us for investors to use when assessing our future viability and our potential to successfully develop product candidates, conduct clinical trials, manufacture our products on a commercial scale, obtain regulatory approval and profitably commercialize any approved products.
We are totally dependent upon the success of the ELAD System, our sole product candidate.
The ELAD System is designed to improve survival rates of patients with acute liver failure. The ELAD System is a novel product candidate whose safety, efficacy and other attributes have not been demonstrated in well-designed, large scale, clinical trials and are not fully understood. As a cell-based therapy, the ELAD System’s mechanism-of-action is complex and we cannot be certain that our currently-targeted indications of AILD, AAH, FHF and SILF in the United States and Europe, and viral hepatitis (predominantly hepatitis B) in China represent suitable applications for the ELAD System, or even ones where the ELAD System therapy can or will ultimately be shown to be safe and effective in well-designed clinical trials necessary to support regulatory approval in any jurisdiction. For example, the FDA has expressed concern about the open-label design of study VTI-208, our pivotal study in AILD, and the need to apply a consistent standard-of-care and to standardize post-discharge care, both being issues that could significantly confound the study results, impact morbidity and mortality and cause the FDA or other regulatory authorities to require that we repeat clinical trials with different trial designs. Finally, even if the ELAD System is proven to be safe and effective and ultimately receives regulatory approval, there is no guarantee that its commercialization will be successful. If the ELAD System should fail at any stage in our clinical trials or at the marketing stage, our business and operating results and financial condition will be materially and adversely affected.
We cannot give any assurance that we will successfully complete the ELAD System’s clinical development, or that the ELAD System will receive regulatory approval in a timely fashion or at all.
We must be evaluated in light of the uncertainties and complexities affecting a clinical-stage combination product biologic and medical device company. We have not completed clinical development for any of the ELAD System’s potential indications in the United States or Europe where the ELAD System is regulated as a combination biologic and medical device, and a combined somatic cell Advanced Therapy Medicinal Product, respectively. We expect to conduct two Phase 3 clinical trials and a Phase 2/3 clinical program designed to establish the safety and efficacy of the ELAD System and to support approval in the United States and Europe. These clinical trials are expected to be performed in subjects with AILD, AAH, FHF and SILF. Any additional indications we elect to pursue will require the initiation and completion of additional Phase 3 clinical trials demonstrating safety and efficacy for each such indication. For example, the FDA has noted its view that
10
Table of Contents
preliminary clinical evidence, at this time, does not indicate that the ELAD System may demonstrate a substantial improvement over standard-of-care. There is no guarantee that our clinical trials will be completed in a timely fashion or succeed. Our ability ultimately to reach profitability is critically dependent on our future success in obtaining regulatory approval for the ELAD System. However, there is no guarantee that our clinical trials will be successful, or that regulators will approve the ELAD System in a timely manner, or at all.
If we fail to obtain regulatory approval as anticipated in the United States and Europe, our business would be harmed.
We require regulatory approval for each indication we are seeking before we can market and sell the ELAD System in a particular jurisdiction, for such indication. Our ability to obtain regulatory approval of the ELAD System depends on, among other things, successful completion of clinical trials, and demonstrating efficacy with statistical significance and safety in humans. The results of our current and future clinical trials may not meet the FDA, the European Medicines Agency, or EMA, or other regulatory agencies’ requirements to approve the ELAD System for marketing under any specific indication, and these regulatory agencies may otherwise determine that our manufacturing processes or facilities are insufficient to support approval. For example, the FDA has noted its view that preliminary clinical evidence, at this time, does not indicate that the ELAD System may demonstrate a substantial improvement over standard-of-care. As such, we may need to conduct more clinical trials than we currently anticipate and upgrade our manufacturing processes and facilities, which may require significant additional time and expense, which could delay or prevent approval. If we fail to obtain regulatory approval in a timely manner, our commercialization of the ELAD System would be delayed and our business would be harmed.
If we are able to secure marketing approval, our commercial success will be determined by our ability to obtain acceptable pricing and reimbursement for the ELAD System therapy.
Therapies such as the ELAD System are paid for primarily by private and government insurance, although in some markets payment may be made by private individuals and their families. The payment for medical products is a highly bureaucratic, politicized and regulated process and considers factors such as cost effectiveness and patient benefit. Furthermore, there are no therapies approved to restore liver function and the lack of an established reimbursement structure introduces additional uncertainty with regard to reimbursement for the ELAD System. There is great pressure to reduce costs. We will have no control over the pricing that is set by the government and private insurers if we are able to secure marketing approval for the ELAD System. In markets where payment will be made by private individuals and their families, we cannot predict if such private payors will be prepared to pay an acceptable price.
If we are unable to implement our sales, marketing, distribution, training and support strategies or enter into agreements with third parties to perform these functions in markets outside of the United States and Europe, we will not be able to effectively commercialize the ELAD System and may not reach profitability.
Our technology is new and complex, and potential customers will have limited knowledge of, or experience with, the ELAD System. In addition, we have no ELAD System-related sales and marketing experience either domestically or abroad. We have not commercialized the ELAD System anywhere and do not plan to introduce the ELAD System, if approved, into the United States or other foreign jurisdictions until late 2016 at the earliest. Our commercial success will depend on our ability to market and receive adequate reimbursement of the ELAD System. This success will also depend on our ability to obtain and maintain adequate pricing for the ELAD System.
We do not have a sales or marketing infrastructure and have no experience in the sale, marketing or distribution of biologic products and medical devices. To achieve commercial success for the ELAD System, if and when we obtain marketing approval, we will need to establish a sales and marketing organization.
In the future, we expect to build a targeted sales, marketing, training and support infrastructure to market the ELAD System in the United States and Europe and to establish collaborations opportunistically to market,
11
Table of Contents
distribute and support the ELAD System outside of the United States and Europe. There are risks involved with establishing our own sales, marketing, distribution, training and support capabilities. For example, recruiting and training sales and marketing personnel and personnel necessary to initially provide on-site device support and later device training to end-users is expensive and time consuming and could delay any product launch. If the commercial launch of the ELAD System is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales, marketing, training and support personnel.
Factors that may inhibit our efforts to commercialize the ELAD System on our own include:
| • | our inability to recruit, train and retain adequate numbers of effective sales, marketing, training and support personnel; |
| • | the inability of sales personnel to obtain access to physicians, including key opinion leaders, or to persuade adequate numbers of physicians to use the ELAD System; |
| • | our inability to properly support the ELAD System therapy with our own qualified personnel at each customer site or our inability to properly train and support our customers to use the ELAD System effectively on their own; |
| • | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive or integrated product offerings; and |
| • | unforeseen costs and expenses associated with creating an independent sales, marketing, training and support organization. |
If we are unable to establish our own sales, marketing, distribution, training and support capabilities and instead enter into arrangements with third parties to perform these services, our product revenues and our profitability, if any, are likely to be lower than if we were to market, sell and distribute the ELAD System ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell, market and distribute the ELAD System or may be unable to do so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to commercialize the ELAD System effectively. If we do not establish sales, marketing, distribution, training and support capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing the ELAD System and achieving profitability, and our business would be harmed.
We have incurred losses since our inception and expect to incur significant losses in the foreseeable future and may never become profitable. Even if we ultimately achieve profitability, it may not be sustained and we may require additional capital.
We are a clinical-stage company and clinical development of a novel therapy is a highly speculative undertaking. We have incurred significant losses in each fiscal year since our inception, including net losses of $6.7 million, and $32.7 million during 2012 and 2013, respectively. As of December 31, 2013, we had a deficit accumulated during the development stage of $103.2 million. We expect to spend a considerable amount of our resources on the completion of our clinical programs and the work necessary to submit and gain approval, on the production of the ELAD cartridges and bedside units, on investment in production facilities, and on the commercial launch and sales and marketing of the ELAD System. We also expect to expend considerable resources on research and development to develop new and improved products and to understand the mechanism of action of the ELAD System. We do not expect to earn revenues until late 2016 at the earliest, and anticipate incurring additional losses and negative cash flow from operations for at least the next several years. Even if we do achieve profitability in the future, there is no guarantee that we will be able to sustain this profitability in subsequent periods and we may need to raise additional capital.
12
Table of Contents
Our ability to use our net operating losses to offset future taxable income may be subject to certain limitations.
As of December 31, 2013, we had net operating loss, or NOL, carryforwards of approximately $37.3 million and $35.8 million, net of estimated limitations caused by certain ownership changes under Section 382 of the Internal Revenue Code for federal and state income tax purposes, respectively. In general, under Section 382, a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its pre-change NOLs to offset future taxable income. We believe our existing NOLs are subject to limitations arising from previous ownership changes, and if we undergo an ownership change in connection with or after this offering, our ability to utilize NOLs could be further limited. Future changes in our stock ownership, some of which are outside of our control, could also result in additional ownership changes under Section 382. Furthermore, our ability to utilize NOLs of companies that we may acquire in the future may be subject to limitations. For these reasons, we may not be able to utilize a material portion of the NOLs, even if we attain profitability.
Our internal computer systems, or those used by our clinical investigators, contract research organizations or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of development programs for the ELAD System.
We rely on information technology systems to keep financial records, maintain laboratory and corporate records, communicate with staff and external parties and operate other critical functions. Despite the implementation of security measures, our internal computer systems and those used by our clinical investigators, contract research organizations, or CROs, and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. The techniques that could be used by criminal elements or foreign governments to attack these computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. Activities in China may be particularly at risk. As a result, we may not be able to address these techniques proactively or implement adequate preventative measures. While, to our knowledge, we have not experienced any such system failure, theft of information, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our clinical development activities. For example, the loss of clinical trial data from ongoing or future clinical trials could result in delays in regulatory approval efforts and significantly increase costs to recover or reproduce the data. To the extent that any disruption, theft of information, or security breach were to result in a loss of or damage to data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the clinical development and the future development of the ELAD System could be delayed.
Risks Related to the ELAD System’s Clinical Development
We have limited experience in conducting pivotal clinical trials used to support regulatory approval and our prior clinical trials of the ELAD System did not demonstrate a statistically significant improvement in survival, the primary endpoint that is needed to support regulatory approval.
We are currently undertaking our first pivotal clinical trials for the ELAD System. While the endpoints and populations for the pivotal trials are derived from results of initial studies and medical literature, in none of those prior studies have we demonstrated an effect in the population and on the endpoints prospectively described in the study plan. The pivotal trials are primarily based on trends derived from post-hoc, retrospective analyses of data subsets. Our prior clinical trials of the ELAD System in AILD were not powered to, and did not demonstrate, statistically significant improvement over the standard-of-care in the primary endpoint of 90-day survival. Similarly, our prior clinical trials of the ELAD System in FHF did not demonstrate statistically significant improvement in the primary endpoint of 28-day survival. The lack of statistical significance could be attributed to various factors including the lack of power to demonstrate significance, the design of the studies or the lack of an ELAD System treatment benefit. Although we did complete a pivotal clinical program in acute liver failure in China in 2007, the underlying clinical trial was terminated early by the lead hospital due to achievement of safety and efficacy goals and a determination that it was unethical to continue. We are now in the
13
Table of Contents
process of conducting a pivotal program for the United States and Europe. We have not yet completed a pivotal clinical trial program of the size and complexity of our currently planned pivotal program for the United States, Europe, and Australia and we cannot provide any guarantee that we will successfully complete such a program. If this pivotal program is completed, there can be no assurance that the data generated can be used to support marketing approval for any indication in the United States or Europe. If our Phase 3 clinical trials do not achieve statistical significance for the primary endpoint, we will not receive marketing approval and we will not be able to commercialize the ELAD System.
The results of previous clinical trials may not be predictive of future results.
Positive results from our prior clinical trials, including either statistical significance in some endpoints or trends towards statistical significance in other endpoints, should not be relied upon as evidence that our current or future clinical trials will necessarily succeed. While we believe that we have learned valuable lessons from the results of prior trials and have attempted to use these lessons to guide our design of current and future clinical trials, there can be no guarantee that these lessons are correct or that we will effectively incorporate them into the design of current and future clinical trials. For example, our primary endpoint in VTI-208, 90-day survival, is based on the results of a subset of subjects in VTI-206. Though that subset showed a trend toward increased survival at 90-days, it consisted of only 29 subjects. The FDA has noted its belief that this preliminary clinical evidence does not indicate that our product may demonstrate a substantial improvement over the current standard-of-care. We cannot provide any guarantee that our current and future clinical trials will provide statistically significant data sufficient to support regulatory approval.
If we fail to select appropriate subjects for our Phase 3 clinical trials or if these subjects do not progress as expected, it will be difficult for us to demonstrate the statistically significant efficacy of the ELAD System therapy necessary to gain approval.
We have designed VTI-208 and VTI-210 in accordance with input provided by regulatory authorities that we must demonstrate a statistically significant improvement in a survival endpoint. VTI-208 and VTI-210 will include concurrent control subjects in a 1:1 ratio with treated subjects and all subjects will be included in the statistical analysis. Also, each study is designed to enroll subjects with an expected death rate between 50 and 75% in 30 to 90 days without the ELAD System therapy. It is necessary to select subjects with these death rates in order to be able to determine whether the ELAD System has an effect on treated subjects with a manageable number of subjects in the clinical trial. If we do not succeed in selecting appropriate subjects or if the subjects we select do not progress as expected, we may not be able to be demonstrate statistically significant efficacy of the ELAD System therapy to gain approval.
Random variation or changes in standard-of-care could cause our clinical trials to fail.
Regulatory authorities worldwide have adopted the standard that, to gain marketing approval, clinical trials should produce a result that has less than a 5% probability of being due to random variation. There is no assurance that any of our clinical trials will meet that standard. In addition, we have designed all of our clinical trials to be judged by a survival primary endpoint, which may be difficult to achieve for many reasons, including unanticipated survival rates of control subjects due to random variations, deficiencies in our exclusion and inclusion criteria, and the standard-of-care of the subjects, which may vary from site to site and country to country and is continuously evolving. For example, FDA has expressed concern that the VTI-208 study may not be adequately designed to provide convincing evidence of efficacy if there are significant differences in how the ELAD System subjects and controls are treated during the treatment period and after hospital discharge. Variations in length of hospital stay, rates of hospital re-admission, alcohol recidivism rates, nutritional support, and concomitant medications, which are not within our control, could significantly confound the study results and call into question whether any difference in survival is due to the ELAD System or to these factors. Any of these factors, which are beyond our control, could materially and adversely affect the results of our Phase 3 clinical trials and prevent us from gaining regulatory approval of our ELAD System therapy. In addition, even if the results of our clinical programs are positive, our inability to control or adequately account for these factors
14
Table of Contents
between treatment arms could cause the FDA or other regulatory authorities to determine that the results are not adequate to support marketing approval.
The ELAD System treatment could result in significant clinical risks to the patient, including death.
The ELAD System therapy is targeted towards very sick patients who are likely to die if left untreated. Patients in acute liver failure quickly develop failure of other organs including lungs, kidney, brain, and blood coagulation systems. Patients who receive the ELAD System therapy may die due to other serious health problems even if the ELAD System is effective.
All extracorporeal therapy systems cause a decline in blood platelets, which can lead to coagulation problems and uncontrolled bleeding because platelets are critical to the formation of blood clots. Patients with acute liver failure generally have serious blood clotting problems since the liver produces most of the body’s blood clotting proteins. These patients therefore have wide variations in their ability to coagulate their blood. To minimize blood clotting issues, some patients require an infusion of small amounts of anti-coagulant therapy, which can aggravate bleeding. Because every patient is different, the need for anti-coagulant therapy is not predictable and must be established during therapy, a process that can affect the course of the therapy. The risk of uncontrolled bleeding may be addressed during the ELAD System therapy by administering platelet transfusions to patients whose platelets drop below a safe level. However, there have been cases of uncontrolled bleeding during and after the ELAD System therapy. Additionally, some patients have abnormal red blood cells, which have weakened cell walls subject to rupture by physical force, a process known as hemolysis. The physical force exerted on the red blood cells by the ultrafiltrate generator in the ELAD System line can, in some cases, be enough to cause hemolysis which, if not arrested, can be fatal. The incidence of hemolysis is about 1-2% in the acute liver failure patients enrolled in our trials to date.
Human liver-derived C3A cells have been shown in animal studies to have the capacity to grow into a tumor mass under certain conditions. Although this has not been seen in the subjects treated with the ELAD System to date, it is possible that some VTL C3A cells could escape from the ELAD cartridges and cause tumors in patients or produce substances that could lead to the development of malignant tumors. These or other adverse events, even those that are currently unforeseen, could significantly affect our development and commercialization efforts, cause the regulatory authorities to place our clinical trials on hold or to refuse to grant or maintain the marketing approval or result in withdrawal of the ELAD System from the market.
Ethical considerations require us to conduct open-label clinical trials of the ELAD System where control subjects do not receive a sham treatment and this could introduce unacceptable bias into our trial results.
We are not conducting any of our clinical trials with a sham control extracorporeal circuit that includes empty cartridges. This is due to the potential harm that the extracorporeal circuit can cause to control subjects without the potential for any benefit, which makes it unethical to subject the controls to a sham. Although regulatory agencies agree that, due to the nature of the ELAD System therapy, it is not possible to conduct a blinded study, they have expressed concern that the open-label nature of the study may introduce significant bias in the treatment of the ELAD System or control subjects, since the study subject, physicians and caregivers know who has, and has not received the ELAD System therapy. We have developed a protocol that attempts to minimize this bias to the extent possible, including defining a protocol-specific standard of care, specifying steroid treatment, standardizing the discharge criteria for both the ELAD System and control subjects, requiring that follow-up visits are conducted by a blinded reviewer, ensuring home healthcare nurses and other clinical personnel are unaware of treatment assignment, educating subjects not to reveal treatment assignment to their caregivers and monitoring concomitant medications, alcohol recidivism and interaction with the healthcare system to provide evidence that there is no meaningful difference between the groups that could significantly confound the trial data. However, there is no guarantee that bias will not enter into the trial, affect the results or cause regulatory agencies to refuse marketing approval of the ELAD System.
15
Table of Contents
If we encounter difficulties enrolling subjects in our clinical trials, our clinical trials could be delayed or otherwise adversely affected.
Clinical trials for the ELAD System require us to identify and enroll a large number of subjects that meet all of the entry criteria set forth in our protocols, including having the disease under investigation. We may not be able to enroll a sufficient number of subjects who meet our protocol requirements in a timely manner. Subject enrollment is affected by numerous factors, many of which fall outside our control, including:
| • | timeliness of contracting with clinical trial sites, and obtaining approval of the trial by the institutional review boards, or IRBs, at each site; |
| • | lack of a sufficient number of subjects who meet the enrollment criteria for our clinical trials; |
| • | perceived risks and benefits of the product candidate under study; |
| • | availability of competing therapies and clinical trials; |
| • | efforts to facilitate timely enrollment in clinical trials; |
| • | scheduling conflicts with participating clinicians; and |
| • | proximity and availability of clinical trial sites for prospective subjects. |
Additionally, even if we are able to identify an appropriate subject population for a clinical trial, there can be no assurance that the subjects will complete the study.
If we have difficulty enrolling a sufficient number of subjects to conduct our clinical trials as planned or if enrolled subjects fail to complete the study or comply with our protocols, particularly with regard to follow-up appointments, the completion of our clinical trials will be delayed and our business would be harmed.
We may face delays in completing our clinical trials, and we may be required to suspend, repeat or terminate our clinical trials if they are not conducted in accordance with applicable regulatory requirements, the results are negative or inconclusive, or the clinical trials are not well-designed or executed as expected.
Our current and future clinical trials must be conducted in accordance with regulations governing clinical studies, and are subject to oversight by the FDA, foreign governmental agencies, ethics committees and IRBs at the medical institutions where the clinical trials are conducted. In addition, clinical trials may require large numbers of test subjects. Changes in regulatory requirements may occur at any time and we may need to amend clinical trial protocols to reflect such changes. Amendments may require us to resubmit our clinical trial protocols to ethics committees for reexamination, which may impact the costs, timing or successful completion of the underlying trial.
Our current and future clinical trials may require amendment or be delayed, unsuccessful or terminated as a result of many factors, including:
| • | delays or failures in designing an appropriate clinical trial protocol with sufficient statistical power and in reaching agreement on trial design with investigators and regulatory authorities; |
| • | delays or failure in reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| • | delays or failure by CROs, investigators and clinical trial sites in ensuring the proper and timely conduct of our clinical trials; |
| • | delays or failure by us in manufacturing sufficient quantities of the ELAD System pursuant to required quality standards for use in our clinical trials and by third-party manufacturers in supplying necessary and suitable components for the system; |
16
Table of Contents
| • | delays or failure in transporting the ELAD System to clinical trial sites with sufficient rapidity to enable treatment to begin early enough to have an opportunity for clinical benefit; |
| • | delays or failure in completing data analysis and achieving primary and secondary endpoints; |
| • | regulators or clinical site Ethics Committees or IRBs may suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or concerns about patient safety; |
| • | we may suspend or terminate our clinical trials if we believe the ELAD System is exposing the participating subjects to unacceptable health risks or for other reasons; |
| • | subjects may not complete our clinical trials due to safety issues, adverse events, inconvenience or other reasons; |
| • | subjects in our clinical trials may die or suffer other adverse events for reasons that may be either related or unrelated to the ELAD System, particularly given the critically ill nature of these subjects; |
| • | we may have difficulty in maintaining contact with subjects after treatment, preventing us from collecting the data required by our study protocol; and |
| • | final analysis of the data of our clinical trials may conclude that the ELAD System lacks sufficient clinical efficacy or presents unacceptable safety risks. |
Should any of our clinical trials fail to provide evidence of safety and efficacy sufficient to satisfy the requirements of the regulatory authorities, the ELAD System will not be approved. If we experience delays in the completion of, or termination of, any clinical trial of the ELAD System, the commercial prospects of the ELAD System will be harmed, and our ability to generate revenues will be delayed or eliminated. In addition, any delays in completing our clinical trials will increase our costs, slow down our development and approval process and delay or jeopardize our ability to commercialize the ELAD System. Any of these occurrences may harm our business, financial condition and prospects significantly.
Risks Related to Regulatory Matters
The FDA regulatory approval process is complex, time-consuming and unpredictable.
In the United States, the ELAD System is regulated as a combination biologic and medical device. Before the ELAD System can be marketed in the United States, we must submit and the FDA must approve a BLA. In addition, the device components of the ELAD System must be found acceptable as part of the BLA. Because the ELAD System is a novel therapy involving a combination biologic and medical device, the regulatory review process is complex, time-consuming and unpredictable. As a result, our development costs, timelines and approvals are not readily predictable.
The time required to obtain approval by the FDA to market a new therapy is unpredictable but typically takes many years and depends upon many factors, including the substantial discretion of the regulatory authorities.
The ELAD System could fail to receive regulatory approval for many reasons, including the following:
| • | the FDA may disagree with the design or implementation of our clinical trials or study endpoints. For example, it has expressed concern about the open-label design and multiplicity of confounding variables, including the need for delineating the standard-of-care that both treatment and controls will receive during our studies; |
| • | we may be unable to demonstrate to the satisfaction of the FDA that the ELAD System is safe and effective for its proposed indications or that the ELAD System provides significant clinical benefits; |
| • | the results of our clinical trials may not meet the level of statistical significance required by the FDA for approval or may not support approval of a label that could command a price sufficient for us to be profitable; |
17
Table of Contents
| • | the FDA may disagree with our interpretation of data from preclinical studies or clinical trials. For example, the FDA has stated there are insufficient preclinical and clinical data to determine whether the ELAD System has the potential to provide a clinically meaningful improvement in liver function; |
| • | the opportunity for bias in the clinical trials as a result of the open-label design may not be adequately handled and may cause our trial to fail; |
| • | the ELAD System may be subject to an FDA advisory committee review, which is triggered by an FDA request, which is solely within the FDA’s discretion, which may result in unexpected delays or hurdles to approval; |
| • | the FDA may determine that the manufacturing processes at our facilities or facilities of third party manufacturers with which we contract for clinical and commercial supplies are inadequate; |
| • | even if VTI-208 is successful in demonstrating a statistically significant improvement over standard-of-care, in light of the fact that certain confounding factors may be viewed by the FDA as limiting the persuasiveness of the study results, a single Phase 3 clinical trial may not be sufficient to provide the substantial evidence of effectiveness necessary to support regulatory approval, and therefore we may need more than one Phase 3 clinical trial to secure regulatory approval; |
| • | the FDA has commented that even if one of our Phase 3 clinical trials, including VTI-208, is a statistical and clinical success, a second confirmatory trial that substantiates positive results may be necessary to support a BLA; and |
| • | the approval policies or regulations of the FDA may significantly change in a manner rendering our clinical data insufficient for approval. |
The FDA has expressed concern that the VTI-208 study may not be adequately designed to provide convincing evidence of efficacy if there are significant differences in how the ELAD System subjects and control subjects are treated during the study and after discharge from the hospital. Differences in length of hospital stay, rates of hospital re-admission, alcohol recidivism rates, nutritional support, and concomitant medications could significantly confound the study results.
In addition, even if we were to obtain approval, the FDA may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve the ELAD System with a label that does not include the labeling claims necessary or desirable for successful commercialization of the ELAD System. Any of the above could materially harm the ELAD System’s commercial prospects.
The regulatory approval processes of foreign regulatory authorities are lengthy, time-consuming and inherently unpredictable.
Outside the United States, our ability to market the ELAD System is contingent upon receiving marketing authorizations from appropriate regulatory authorities. If our clinical programs are successful, we currently anticipate submitting applications for marketing authorization to the European Medicines Agency in the European Union. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country and we may be unable to meet such requirements. If the regulatory authority is satisfied that adequate evidence of safety, efficacy, and quality has been presented, a marketing authorization will be granted. The foreign regulatory approval process involves all of the risks associated with FDA approval.
Even if the ELAD System receives regulatory approval, we will be subject to ongoing regulatory requirements and may face regulatory or enforcement action.
If any ELAD System product receives regulatory approval, we will be subject to significant ongoing regulation by the FDA and other regulatory authorities, including regulation of our manufacturing operations, and any third-party manufacturing operations for compliance with applicable current Good Manufacturing Practices, or cGMP, and/or Quality System Regulation, or QSR, post-approval clinical data, adverse event reporting and complaint
18
Table of Contents
handling, and advertising and promotional activities. Failure to comply with regulatory requirements may subject us to sanctions. These may include warning letters, adverse publicity, civil and criminal penalties, injunctions, product seizures or detention, and refusal to approve pending product marketing applications.
Risks Related to the Medical Device Components of the ELAD System
If we or our third-party manufacturers fail to comply with the Quality System Regulation in the United States or Medical Device Directives and Standards in Europe, our business would suffer.
We are required to demonstrate and maintain compliance with applicable regulations for the manufacturing of combination biologic products, including specified parts of the QSR and European Medical Device Directives, or MDD. Our third-party medical device manufacturers, are required to demonstrate and maintain compliance with the QSR and MDD. The QSR and MDD are complex regulatory schemes that cover the methods and documentation of the design, testing, control, manufacturing, labeling, quality assurance, packaging, storage and shipping of the ELAD System. Regulatory agencies enforce the QSR and MDD through periodic inspections. Prior to approval of the ELAD System, our manufacturing facility will be subject to a preapproval inspection to determine compliance with the applicable regulations, including cGMPs, parts of the QSR, the European drug cGMP regulations, and the MDD. In addition, our third-party medical device component manufacturers will be subject to a preapproval inspection to determine compliance with QSR and MDD requirements. Our failure, or the failure of our third-party manufacturers, to pass a preapproval inspection, or take satisfactory and prompt corrective action in response to an adverse inspection, could prevent or significantly delay approval of the ELAD System.
The ELAD System bedside unit is based on a cardiopulmonary bypass system that is no longer commercially available.
Until recently the ELAD System bedside unit was based exclusively on the Sorin Stöckert Perfusion System S3 Double Head Pump Module, a medical device indicated for use during cardiopulmonary bypass surgery. Our recent clinical trials have been carried out using an ELAD System bedside unit based on Sorin’s S3 system. However, Sorin recently withdrew the S3 system from the market and replaced it with an updated S5 system. We have carried out testing of an ELAD System bedside unit based on the S5 and we believe that the S3 and S5 systems are equivalent and interchangeable from a clinical and regulatory perspective. We have submitted information to both the U.S. and the European regulatory authorities to support equivalence. There can be no assurance that regulatory authorities will view the S3 and S5 systems interchangeably, or that Sorin will cooperate with us or provide us with the documentation necessary to obtain regulatory approval or commercialize the device.
One of the ELAD System component suppliers is subject to an FDA consent decree which, if not lifted, would force us to find another supplier for these components.
One of the components of the ELAD System bedside unit is manufactured by Terumo Cardiovascular Systems, or Terumo. In March 2011, Terumo entered into a consent decree with the FDA which limits its ability to ship products from certain of its manufacturing facilities including the one that manufactures the component we use. We have signed a Certificate of Medical Necessity that allows us to continue to use those components we already own while Terumo works to resolve the issues associated with the consent decree. Once the consent decree is lifted Terumo has indicated that it will resume shipping of this component. Should Terumo not be able to fulfill the requirements of the consent decree, we will have to source these components from an alternative supplier. There is no guarantee that Terumo will be able to fulfill the requirements of the consent decree, or that an alternative supplier can be found or will agree to acceptable terms.
Changes in any of the device components could affect our ability to obtain and maintain approval and commercialization efforts.
The device components of the ELAD System will be reviewed as part of the BLA for the ELAD System. If the manufacturers of those components make modifications, or if we elect to change a component, we will need to perform validation testing and obtain FDA and other regulatory approval prior to using the modified
19
Table of Contents
component. If FDA or any other regulatory body fails to approve use of those modified devices or take significant enforcement action against the manufacturer, we would not be able to market or may have to suspend marketing the ELAD System in certain jurisdictions.
We may be unable to demonstrate that devices cleared for different uses may be safe and effectively used in the ELAD System.
Most device components of the ELAD System have been previously cleared for use by the FDA or other regulatory authorities. However, we will be using the components for purposes outside the scope of the cleared indications. Other device components have no regulatory approvals. We will need to conduct additional bench testing to bridge the differences between the cleared indications for use and the proposed use in the ELAD System or to obtain approval. The failure to provide adequate bridging information or to obtain approval for these device components could delay or prevent approval of the ELAD System.
Risks Related to the Cellular Component of ELAD System and Related Components
If we fail to comply with cGMPs our business will suffer.
We are required to demonstrate and maintain compliance with cGMPs. The cGMPs describe the methods to be used in, and the facilities or controls to be used for, the manufacture, processing, packing, or holding of a biologic to assure the biologic meets the requirements for safety, and has the quality, purity, and potency characteristics that it purports or is represented to possess. Regulatory agencies enforce these requirements through periodic inspections. Prior to approval of the ELAD System, our manufacturing facilities will be subject to a preapproval inspection to determine compliance with U.S. and European cGMPs and applicable QSR and MDD requirements. Our failure to pass such an inspection, or take satisfactory and prompt corrective action in response to an adverse inspection, could prevent or significantly delay approval of the ELAD System.
We rely on third party suppliers, and in some instances, a single third party supplier, for critical components of the ELAD System and these suppliers could cease to manufacture the components, go out of business or otherwise not perform as anticipated.
While the growing of our VTL C3A cells is under our control, the manufacture of all of the other parts and pieces of the ELAD System are undertaken by third party suppliers. We currently rely on a single source of supply for many critical components, including components of the ELAD System bedside unit, the ultrafiltrate generator cartridges, the media we use to grow and ship our VTL C3A cells, the cartridges in which our VTL C3A cells are grown and the bioreactors that have been developed to grow and store the ELAD cartridges. We are currently developing additional sources of supply for these components sufficient to support future clinical development and, ultimately, commercialization of the ELAD System. If we were to fail to do so, and a single source of supply of a critical component of the ELAD System were to become unavailable, our ability to continue clinical development or to initiate commercialization of the ELAD System would be severely compromised. In addition, we rely on third party suppliers for the safety of products of human and animal origin that are incorporated in the ELAD System production process and these suppliers could cease to manufacture the components, inadequately test these components, go out of business or otherwise not perform as anticipated. We do not have long-term agreements with our suppliers, and we purchase components on a purchase order basis. For components that are not readily available from other sources, we are subject to the risks that our suppliers will raise their prices or impose other terms or conditions that are less favorable or unacceptable to us.
For instance, newborn calf serum, which is a component of the cell growth media, is used in the manufacture of the ELAD System. It is obtained from an outside supplier. We are wholly reliant on the guarantee of our supplier that the calf serum used in our manufacturing procedures is free of transmitted animal viruses and other pathogens. Should the source of supply become infected, or the supplier become unable to continue to supply calf serum of the quality necessary to support human use, or the regulations change such that the calf serum cannot be used for human use, we would have to find alternative sources of supply and manufacturing methods, for which there is no guarantee of success.
20
Table of Contents
Human albumin and Trypsin-EDTA are also used in the ELAD System manufacture and are each provided by a single supplier. In addition, while these products are tested to be free of contamination by the supplier, we cannot guarantee that will continue to be the case.
If our facility becomes inoperable, we will be unable to continue manufacturing our product candidate and as a result, our business will be harmed until we are able to secure a new facility.
We manufacture and assemble the ELAD System at our facility in San Diego, California. No other manufacturing or assembly facilities are currently available to us, and any additional manufacturing or assembly facilities that we use will need to be approved by regulatory authorities prior to our use. Our facility and the equipment we use to manufacture the ELAD System would be costly to replace and could require substantial lead-time to repair or replace. The facility may be harmed or rendered inoperable by natural or man-made disasters, including fire, earthquakes, flooding and power outages, which may render it difficult or impossible for us to perform our research, development and manufacturing for some period of time. The inability to perform our research, development and manufacturing activities, combined with our limited inventory of reserve raw materials and manufactured supplies, may result in the delay of clinical trials or loss of customers or harm our reputation, and we may be unable to reestablish relationships with those customers in the future. Although we possess insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
We may be unable to manage our anticipated manufacturing growth to support our clinical development activities and long-term commercial demand for the ELAD System.
In order to support our ongoing clinical programs, we will need to increase production of our ELAD System. Similarly, if and when the ELAD System is approved for sale, we will need to expand our manufacturing space in San Diego and build new manufacturing facilities to meet anticipated demand for the ELAD System in the United States and abroad. These activities involve significant expense, including the construction of new clean rooms and bioreactors, the movement and installation of key manufacturing equipment and the modification of manufacturing processes. In addition, we must also notify, and in some cases obtain approval from, the FDA and other regulatory authorities of any changes or modifications to our manufacturing facilities and processes, and there can be no assurance that they will authorize us to proceed. If we are not able to expand our manufacturing capacity to meet future demand, our business would be harmed.
Further, our anticipated growth will place additional strain on our organization, employees and third-party suppliers, resulting in an increased need for us to carefully monitor quality assurance. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals.
We forecast the requirements for components and materials used in the ELAD System, and if our forecasts are incorrect, we may experience delays in shipments or increased inventory costs.
We keep limited materials, components and finished product on hand. To manage our manufacturing operations with our suppliers, we forecast anticipated product orders and material requirements to predict our future inventory needs and to enter into purchase orders on the basis of these requirements. Our limited historical experience may not provide us with enough data to accurately predict future demand. If our business expands, our demand for components and materials would increase and our suppliers may be unable to meet our demand. Many of our components are medical devices, which have fixed future expiration dates. If we overestimate our component and material requirements, we will have excess inventory, which may have to be disposed of if it exceeds approved expiration dates, which would increase our expenses. If we underestimate our component and material requirements, we may have inadequate inventory, which could interrupt, delay or prevent delivery of the ELAD System to our customers. Any of these occurrences would negatively affect our financial performance and the level of satisfaction our customers have with our business.
21
Table of Contents
We may not be able to grow our VTL C3A cells reliably and cost-effectively.
Operations with human cells, even a stable, immortal cell line such as the VTL C3A cells used in the ELAD System, can be subject to conditions and influences that we may not be able to control. Although our VTL C3A cells are stored at three separate locations in the United States and the United Kingdom, it is possible that all three locations could be destroyed and we will lose all or a portion of our cell banks. It is also possible that the cells will simply cease to function. While we take precautions to prevent this from happening, the ELAD System employs new technologies and we could encounter unforeseen complications. To date, we have only produced the small number of the ELAD cartridges required to support our clinical trials. As we increase production to support Phase 3 clinical trials and long-term commercial demand, we may experience significant scale-up issues, which may cause quality and cost problems. If we cannot produce the required number of the ELAD cartridges in a cost-effective manner, our business could be materially harmed.
Cellular therapy is complex and we do not have a complete understanding of the mechanism of action of the ELAD System.
Cellular therapy is a complex treatment with multiple variables that are not fully understood. Our VTL C3A cells used in the ELAD cartridges produce hundreds of metabolites. Likewise, the plasma ultrafiltrate formed from blood, which has been treated by our VTL C3A cells in our ELAD cartridges, is a similarly complex material. The composition and stability of the treated blood can be affected by the conditions of its generation in the ELAD System bedside unit and could affect treatment outcomes. For instance, while patients treated with the ELAD System typically only require a single set of cartridges, some patients require more than one set during their three to ten day treatment period, which may have implications for not only efficacy, but also cost-of-goods. While we believe that we have identified the key parameters of the ELAD System VTL C3A cartridges and set them in an appropriate range, it is possible that there are other variables that are important to safety and efficacy that have not been anticipated. We believe that we have set these parameters at realistic levels that can be controlled by the specification set for a supplier and confirmed by us in our quality control procedures, but it is possible that unanticipated complications will emerge.
Risks Related to the ELAD System’s Future Commercialization
It is difficult to forecast future performance; our financial results may fluctuate unpredictably.
Our limited operating history makes it difficult for us to predict our future commercialization efforts. A number of factors, over which we have limited or no control, may contribute to fluctuations in our financial results, such as:
| • | delays in receipt of anticipated purchase orders; |
| • | our ability to recruit, train and retain sales, marketing, training and support personnel; |
| • | our inability to educate physicians about the ELAD System and drive the adoption of the ELAD System therapy for any approved indications; |
| • | performance of our targeted sales force in the United States and Europe and future partners in other markets; |
| • | results of clinical trials evaluating the ELAD System therapy; |
| • | positive or negative media coverage of the ELAD System or products of our competitors or our industry; |
| • | our ability to obtain further regulatory clearances or approvals, including for other indications; |
| • | delays in, or failure of, product and component deliveries by our subcontractors and suppliers; |
| • | changes in the length of the sales process; |
| • | changes in healthcare coverage and reimbursement policies; |
22
Table of Contents
| • | customer response to the introduction of new product offerings; and |
| • | fluctuations in foreign currencies. |
The human clinical trial results may not be representative of the results that are obtained after the ELAD System product launch.
Human clinical trials are very complicated undertakings and working with subjects in acute liver failure is particularly difficult because of the serious nature of the disease and the co-morbidities experienced by the subjects. Not enough is known about the function of the liver to understand the progression of liver disease and any single patient can react differently to the ELAD System therapy. This means that clinical trials done at different times in different groups of subjects may obtain different results. Safety risks not identified in our clinical trials may first appear after we obtain approval and commercialize the ELAD System. Any new post-marketing adverse events may significantly impact our ability to market the ELAD System and may require that we recall and discontinue commercialization of the product. Any of these events will harm our business.
The ELAD System is a very complicated therapy and will need to be delivered by well-trained staff. There is no guarantee that we will be able to implement such training and find sufficient numbers of people to enable us to grow at an acceptable rate.
In the initial commercialization period, it will be essential for us to have our own trained staff present during the delivery of the ELAD System therapy. This may entail the construction and operation of training centers and will require the hiring of personnel of appropriate ability to be adequately trained. The differences in language and culture may make this a difficult undertaking. If we cannot recruit, train and retain significant numbers of physicians and nurses, our ability to grow will be restrained and we may find that the ELAD System therapy is being delivered by people with a substandard level of training, and with potentially material adverse results. If the ELAD System therapy is delivered improperly or the bedside device or the ELAD cartridges are not properly maintained by our customers, the ELAD System may not provide the intended benefit or could harm patients. This may in turn result in perceptions, even if unfounded, that the ELAD System is ineffective or that our bedside device or the ELAD cartridges are defective, which could materially harm our reputation and ability to market the ELAD System effectively.
We could lose our valuable employees and thereby lose our advantage in the marketplace.
We are highly dependent on the efforts of our key employees, including senior management and senior scientific, clinical, regulatory, operational and other personnel. The development of new therapeutic products requires expertise from a number of different disciplines, some of which are not widely available.
Our key employees have a significant amount of know-how and experience in our company and the loss of one or more of them could have a material and adverse effect on our operations. While we have taken steps to incentivize and to retain our employees, including the granting of stock options, paying competitive salaries and implementing appropriate bonus programs, these factors may not be enough to retain the key employees that we need.
The loss of the services of existing personnel, the failure to recruit additional key scientific, managerial, clinical, regulatory, operational and other personnel in a timely manner, and the loss of our employees to our competitors would harm our research and development programs and our business. We may experience difficulty in hiring and retaining highly skilled employees with appropriate qualifications. If we fail to attract new personnel or fail to retain and motivate our current personnel, our business and future growth prospects would be harmed.
Competitive products could be developed which make the ELAD System obsolete.
The biotherapeutic and medical device industries are highly competitive and we face potential competition from pharmaceutical companies, specialty pharmaceutical, medical device and biotechnology companies worldwide. Given the significant unmet medical need for novel therapies to treat acute liver failure, many companies, universities and research organizations are actively engaged in the discovery, research and
23
Table of Contents
development of potential therapies in this field. Several of these entities are engaged in research on cell-based approaches to acute liver failure. Although we are not aware of any ongoing human clinical trials involving potentially competitive product candidates, such trials could be taking place or could begin in the near future. We are not aware of any company that is in human clinical trials with a human cell-based product for the treatment of acute liver failure. At least four companies have prior research work on various human hepatocyte cell lines including Exten Industries, Hepalife Technologies, Fresenius, and Hybrid Organ GmbH. In addition, the University College London, and the University of Amsterdam and its spinout Hep-Art Medical Devices are actively pursuing animal research in this area. Several companies have also attempted to develop extracorporeal therapy based upon primary porcine hepatocytes, although ongoing research in this area is difficult to ascertain. Two commercially available liver dialysis systems, from Gambro and Fresenius, have undergone extensive clinical development, although both have failed to show an improvement in long-term survival among patients with acute liver failure. Both rely on not only traditional dialysis circuits to remove water-soluble toxins, but also albumin dialysis circuits to remove albumin-bound molecules. In addition, there are several drugs available to treat symptoms associated with acute liver failure, including steroids, pentoxifylline and N-acetylcysteine. These three drugs, alone or in combination, are used frequently in patients with acute liver failure. While we are not aware of any of these other entities being close to undergoing human clinical trials with a human cell-based product for the treatment of acute liver failure, it is possible that these trials are going on without our knowledge and that such a product may get to market much faster than we expect, which could harm our business.
The coverage and reimbursement status of new therapies is uncertain, and failure to obtain adequate coverage and reimbursement for the ELAD System therapy could limit our ability to generate revenue and become profitable.
There is significant uncertainty surrounding the third-party coverage and reimbursement of novel and newly approved therapies, particularly for indications for which there is no current effective treatment or standard- of- care is relatively inexpensive. Due to the novel nature of the ELAD System and the potential for it to offer therapeutic benefit after a single administration of continuous therapy lasting three to ten days, we face additional uncertainty related to coverage and reimbursement. We will depend in large part on the availability of coverage and the establishment of adequate reimbursement levels for the ELAD System from third-party payors, including government payors, such as the Medicare and Medicaid programs, and managed care organizations.
Third-party payors are increasingly focused on containing healthcare costs by limiting both coverage and the level of reimbursement for new therapies and, as a result, they may not cover or provide adequate payment for the ELAD System. Obtaining adequate coverage and reimbursement approval for a product from a third-party payor is a time-consuming, costly and sometimes unpredictable process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of the ELAD System. However, we cannot guarantee that we will be able to provide data sufficient to gain acceptance with respect to adequate coverage and reimbursement. Payors may conclude that the ELAD System is less safe, less effective or less cost-effective than existing or later introduced therapies, and third-party payors may not approve the ELAD System for coverage and reimbursement or may cease providing or provide inadequate coverage and reimbursement. Coverage and reimbursement determinations are made on a payor-by-payor basis. Obtaining acceptable coverage and reimbursement from one payor does not guarantee the Company will obtain similar acceptable coverage or reimbursement from another payor. As there is a large number of third-party payors, obtaining coverage and reimbursement in the United States and internationally will consume significant time and resources. A third-party payor’s decision to provide coverage does not imply that an adequate reimbursement rate will be approved. There can be no assurance that our clinical data will allow for satisfactory pricing of the ELAD System and the failure to obtain coverage and adequate reimbursement for the ELAD System would materially and adversely affect our business. Moreover, healthcare cost containment initiatives that limit or deny reimbursement for the ELAD System would also materially and adversely affect our business.
24
Table of Contents
Our relationships with investigators, healthcare professionals, institutional providers, consultants, third-party payors, and customers are subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to penalties, including without limitation, civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations.
Healthcare providers, physicians and others play a primary role in the recommendation and prescribing of any product candidates for which we may obtain marketing approval. In the United States, our current business operations and future arrangements with investigators, healthcare professionals, institutional providers, consultants, third-party payors and customers, may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we research, market, sell and distribute our products that obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations, include, but are not limited to, the following:
| • | the federal healthcare program anti-kickback statute prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or paying any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return, for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, service or item for which payment is made, in whole or in part, under a federal healthcare programs; |
| • | the federal civil and criminal false claims laws and civil monetary penalties laws, including civil whistleblower or qui tam actions, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, to the federal government, claims for payment or approval that are false or fraudulent or from knowingly making a false statement to improperly avoid, decrease or conceal an obligation to pay money to the federal government; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly or willfully falsifying, concealing, or covering up by any trick, scheme or device a material fact or making any materially false statement in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, and as amended again by the final HIPAA omnibus rule, Modifications to the HIPAA Privacy, Security, Enforcement, and Breach Notification Rules Under HITECH and the Genetic Information Nondiscrimination Act; Other Modifications to HIPAA, published in January 2013, imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization by entities subject to the rule, such as health plans, clearinghouses and healthcare providers; |
| • | the federal transparency law, enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the ACA), and its implementing regulations, require manufacturers of drugs, devices, biologicals and medical supplies to report to the U.S. Department of Health and Human Services information related to payments and other transfers of value made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; and |
| • | analogous state laws and regulations, including but not limited to: state anti-kickback and false claims laws, which may apply to our business practices, including but not limited to, research, distribution, |
25
Table of Contents
| sales and marketing arrangements and claims involving healthcare items or services reimbursed by state governmental and non-governmental third-party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; and state laws and regulations that require manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these or any other health regulatory laws or any other governmental regulations that may apply to us, we may be subject to penalties, including without limitation, civil, criminal and administrative penalties, damages, monetary fines, disgorgement, enhanced government reporting and oversight under a corporate integrity agreement or other similar arrangement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare program, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses or divert our management’s attention from the operation of our business. If any of the physicians or other providers or entities with whom we expect to do business are found to be not in compliance with applicable healthcare laws, they also may be subject to similar penalties.
Healthcare policy changes, including recent laws to reform the U.S. healthcare system, may have a material adverse effect on us.
In the United States and in other countries, there have been and we expect there will continue to be a number of legislative and regulatory proposals to change the healthcare system in ways that could significantly and adversely affect the business of developing and marketing new therapies by reducing the costs paid for medical products and services. For instance, the U.S. government and other governments have shown significant interest in pursuing healthcare reform, as evidenced by the passing of the ACA. Such government-adopted reform measures may adversely impact the pricing of healthcare products and services in the United States or internationally and the amount of reimbursement available from third-party payors. For instance, under the ACA, there is a new 2.3% U.S. federal excise tax on the sale of certain medical devices. While we do not believe the tax will be applicable to us, the U.S. may seek to enforce the tax on us. In addition, in some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell the ELAD System profitably, if it is ultimately approved. The continuing efforts of U.S. and other governments, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce healthcare costs may adversely affect the prices we are able to charge for the ELAD System, if approved, and our ability to generate revenues and achieve and maintain profitability.
Risks Related to Doing Business Internationally
We plan to do business internationally, which may prove to be difficult and fraught with economic, regulatory and political issues.
We may commercialize the ELAD System in countries where the business, economic and political climates are very different from those of the United States. We may not be aware of some of these issues and it may be difficult for a U.S. company to overcome these issues and ultimately become profitable. For instance, we completed our Chinese pivotal clinical trial in 2007 and submitted our data to the China FDA, or CFDA, showing a statistically significant improvement in transplant-free survival among the ELAD System-treated subjects compared with control subjects. However, in the past six years this application has been neither approved nor rejected and the timing and nature of any potential decision is highly uncertain. Moreover, currency controls are
26
Table of Contents
in effect in many foreign countries and could become much tighter in the future, which will hinder our ability to repatriate any profits or capital. These foreign countries may also favor businesses that are owned by nationals of those countries as opposed to foreign-owned businesses operating locally. As a small company, we may not have the resources to engage in the negotiation and time-consuming work needed to overcome some of these potential issues.
In the event that we receive marketing approval in foreign countries outside of the United States and Europe, we currently anticipate creating wholly-owned subsidiaries in those countries. These subsidiaries will need to build an effective sales, marketing, distribution, training and support staff and system, find an effective marketing partner or both. Any internal sales, marketing, training and support capabilities of the subsidiaries will need to be developed by these subsidiaries and will need to be built from scratch. The culture and accepted practices related to selling medical products in many foreign countries are unique and it is possible that we will not be able to successfully penetrate these markets. A similar consideration applies to selling in the United States since each medical system is very different and requires a different strategic approach. We cannot guarantee that our approach to the U.S., European, Chinese or any other international market will be effective.
The medical systems in many foreign countries are very different from that of the United States and could cause significant problems for the ELAD System.
The medical systems in many countries around the world pose challenges to the commercialization of the ELAD System. For instance, most medical care in China is delivered on a private pay basis and it may be difficult to receive payment for the ELAD System therapy delivered or the price of our product, which we expect to be relatively high, may prove to be beyond the capability of the targeted Chinese patient to pay. Further, as we have encountered in our clinical trials, the standard and the operation of the delivery of care in China are different, causing problems with the operation of the ELAD System therapy. These issues include the withholding of necessary medicines, the inadequate staffing of Chinese hospitals, the shortage of blood products, the differing practice of delivery of extracorporeal therapies, and the attitude of physicians and nurses. These issues and others are likely to occur in other countries around the world and there is no assurance that we will overcome these challenges or succeed in commercializing the ELAD System in foreign countries.
We face increased risks of doing business due to the extent of our operations internationally.
We currently anticipate our foreign commercialization efforts will be through wholly-owned, foreign domiciled subsidiaries. Our efforts to expand internationally pose risks that could adversely affect our business. These risks include, among others, the effects of:
| • | fluctuations in foreign currency exchange rates and controls; |
| • | competitive disadvantages to established foreign businesses with significant current market share and business and customer relationships; |
| • | nationalization; |
| • | tax and regulatory policies of local governments and the possibility of trade embargoes; |
| • | political instability, war or other hostilities; and |
| • | laws and policies of the United States and foreign governments affecting foreign trade and investment. |
Any of these risks could cause significant interruptions in our operations, which would adversely affect our ability to commercialize the ELAD System internationally and our financial condition, results of operations and business.
Revenues, profits and cash flows derived in foreign countries by foreign subsidiaries may be denominated in foreign currency. The value of this currency may be controlled or adjusted periodically by foreign governments, and may be subject to changes in the political and economic conditions.
27
Table of Contents
Foreign economic, political and social conditions and government policies could materially and adversely affect our business.
A significant portion of our operations may be conducted in foreign countries and it is anticipated that a significant percentage of our revenues may be derived from these countries. Accordingly, our results of operations, financial condition and prospects are subject, to a significant degree, to economic, political, legal and social developments around the world. The economies of many of these countries differ from the economy of the United States in many respects, including:
| • | level of government involvement; |
| • | economic structure; |
| • | allocation of resources; |
| • | level of development; |
| • | inflation rates; |
| • | growth rate; and |
| • | control of foreign exchange. |
The legal systems in many foreign countries have inherent uncertainties that could limit the legal protections available to us.
We are subject to the laws and regulations of foreign governments, including those applicable to foreign investment and, in particular, laws applicable to wholly foreign-owned enterprises. Any litigation in these countries may be protracted and may result in substantial costs and diversion of resources and management attention. For example, in 2007, one of our clinical sites in China was sued in connection with the death of a subject of our clinical trial. An expert panel concluded that neither the ELAD System nor the clinical site was at fault and dismissed the lawsuit. Nevertheless, we were later informed that the subject’s family had been awarded approximately $100,000 in a subsequent civil proceeding brought against the clinical site. We ultimately decided to reimburse the clinical site for $100,000, which was partially insured. In addition, these countries may enact new laws or amend current laws that may be detrimental to us, which may have a material adverse effect on our business operations.
We have limited business insurance coverage internationally.
The insurance industry in many parts of the world is still in an early stage of development. Insurance companies in many countries offer only limited business insurance options. As a result, we have not maintained, and currently do not maintain, any liability, hazard or other insurance covering our services, business, operations, errors, acts or omissions, personnel or properties in China, or in any other countries where we may ultimately commercialize the ELAD System. To the extent that we are unable to recover from others for any uninsured losses, such losses could result in a loss of capital and significant harm to our business. If any action, suit, or proceeding is brought against us and we are unable to pay a judgment rendered against us or defend ourselves against such action, suit, or proceeding, our business, financial condition and operations could be negatively affected.
We must comply with the U.S. Foreign Corrupt Practices Act.
The U.S. Foreign Corrupt Practices Act, to which we are subject, prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. Other countries, such as the United Kingdom and China, have similar laws with which we must comply. Although we attempt to rigidly adhere to the requirements of the U.S. Foreign Corrupt Practices Act and all similar laws to which we are subject, there remains the risk that an
28
Table of Contents
employee or agent of ours could be accused of violating one or more of these laws, particularly in geographies where significant overlap exists between local government and healthcare industries. Such an accusation, even if unwarranted, could prove disruptive to our developmental and commercialization efforts.
We could be subject to additional income and other tax liabilities.
We are subject to income and other taxes in the United States and may be subject to income and other taxes in various other foreign jurisdictions. Significant planning is required in evaluating a worldwide provision for income and other taxes. During the ordinary course of business, there may be transactions for which the ultimate tax determination is uncertain. We may be subject to audit in various jurisdictions and such jurisdictions may assess additional income or other tax against us. Although we believe our tax positions are reasonable, the final determination of tax audits and any related litigation could be materially different from our historical income tax provisions and accruals. The results of an audit or litigation could have a material and adverse effect on our operating results or cash flows in the period or periods for which that determination is made.
Risks Related to Intellectual Property
Our patent rights may prove to be an inadequate barrier to competition.
We hold a patent in the United States which claims a method of using C3A cells to treat a patient’s blood, which we believe covers the ELAD System therapy. In addition, we have been granted a patent with claims covering an extracorporeal device configuration, which we believe includes our ELAD System, independent of cell-type used. Foreign counterparts of these patents have been issued in Australia, Canada, Indonesia, Israel, Japan, Mexico, New Zealand, Singapore, South Africa, South Korea and Taiwan and remain under review in certain other jurisdictions, including Europe, Brazil, China, India and the Philippines. In addition to these two U.S. patents, as of December 31, 2013, we hold two additional patents in the U.S., and additional patent applications related to developments in the ELAD System are pending. However, the lifespan of any one patent is limited, and each of these patents will ultimately expire and we cannot be sure that pending applications will be granted, or that we will discover new inventions which we can successfully patent. Moreover, any of our granted patents may be held invalid by a court of competent jurisdiction, and any of these patents may also be construed narrowly by a court of competent jurisdiction in such a way that it is held to not directly cover the ELAD System. Furthermore, even if our patents are held to be valid and broadly interpreted, third parties may find legitimate ways to compete with the ELAD System by inventing around our patent. Finally, the process of obtaining new patents is lengthy and expensive, as is the process for enforcing patent rights against an alleged infringer. Any such litigation could take years, cost large sums of money and pose a significant distraction to management. Indeed, certain jurisdictions outside of the United States and Europe where we hope to commercialize the ELAD System have a history of inconsistent, relatively lax or ineffective enforcement of patent rights. In such jurisdictions, even a valid patent may have limited value. Our failure to effectively prosecute our patents would have a harmful impact on our ability to commercialize the ELAD System in these jurisdictions.
We do not hold any patents covering our VTL C3A cells or the production processes we use to grow the VTL C3A cells in the ELAD cartridges.
C3A cells are publicly available and the proprietary methods and production process that we use to grow our VTL C3A cells in the ELAD cartridges are our trade secrets, but they are not currently covered by a patent and no patents are pending. Although we have sought patent protection for certain aspects of our technology, such as our method of using human liver-derived C3A cells to treat a patient’s blood, and we have obtained orphan designation in the United States and Europe for the use of C3A cells to treat acute liver failure, we have not sought patent protection for the proprietary methods we use to grow VTL C3A cells in our facility. Although we believe that some of these methods may be patentable, we prefer to avoid the disclosure requirements inherent in the patenting process, as such disclosure could provide competitors with insights that allow them to invent around any granted patents. We believe that this concern is particularly appropriate since C3A cells are now publicly available, and have been available for research purposes for more than twenty years. Despite this availability, we are not aware of any third parties who have either demonstrated an ability to grow C3A cells in
29
Table of Contents
the quantities we do, or succeeded in treating a human subject with such cells. In addition, patent protection expires 20 years after the application’s priority date which does not apply to trade secret protection. In light of the foregoing, we do not currently contemplate seeking patent protection for our production methods and instead intend to keep our production methods protected as trade secrets, which does not require us to publicly disclose these methods and which is not subject to a formal expiration date. However, trade secrets are vulnerable to inadvertent disclosure and misappropriation. In addition, independent discovery and publication of these methods by third parties, which is now more feasible given the public availability of C3A cells, would also destroy their trade secret protection. If any of these were to occur, our business may be harmed.
We protect much of our intellectual property as trade secrets. Confidentiality agreements with employees and third parties may not prevent unauthorized disclosure of trade secrets and other proprietary information.
Trade secrets offer a relatively limited form of protection as they do not create any barrier for third-parties who independently develop this information and who may even patent the information. In the course of our research and development activities and our business activities, we often rely on confidentiality agreements to protect our proprietary information. Such confidentiality agreements may be used, for example, when we talk to vendors of laboratory or clinical development services or potential strategic partners. In addition, each of our employees is required to sign a confidentiality agreement upon joining us. We take steps to protect our proprietary information, and our confidentiality agreements are carefully drafted to protect our proprietary interests. Nevertheless, there can be no assurance that an employee or an outside party will not make an unauthorized disclosure of our proprietary confidential information. This might happen intentionally or inadvertently. It is possible that a competitor will make use of such information, and that our competitive position will be compromised, in spite of any legal action we might take against persons making such unauthorized disclosures. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States sometimes are less willing than U.S. courts to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how, which would harm our business.
If our ELAD cartridges or our VTL C3A cells are stolen, misappropriated or reverse engineered, others could produce competing products.
Third parties, including those involved in shipping our ELAD System cartridges or in any manufacturing abroad that we may undertake, often have custody or control of our ELAD cartridges. If our ELAD cartridges, or VTL C3A cells from our proprietary VTL C3A cell bank that are stored to grow in these cartridges, were stolen, misappropriated or reverse engineered, they could be used by other parties who may be able to reproduce these cartridges for their own commercial gain. If this were to occur, it would be difficult for us to challenge this type of use, especially in countries with limited intellectual property protection or in countries in which we do not have patents covering the misappropriated the ELAD cartridges. In such instance, our business would be harmed.
Ownership of our intellectual property may be claimed by others.
The ELAD System has been under development for over 20 years and certain of our predecessor companies have filed for reorganization and bankruptcy. We were founded in 2003 by acquisition of the assets of a prior company after a bankruptcy. While we believe we have performed extensive diligence on the ownership of the intellectual property rights and have developed our own innovative technology which is independent of prior intellectual property rights, there could be claims by parties associated with the prior entities that could lead to costly and time consuming legal actions. In addition, we have engaged in collaborations with third parties where intellectual property has been developed. In one instance, we were engaged in a dispute over the ownership of intellectual property when a collaborator of ours pursued patent rights over technology which we believe we may have held rights to under the collaboration agreement. Although a patent which claims a different configuration than our ELAD System was ultimately issued in the United States to our former collaborator, we do not hold any rights to this patent. We are unaware of any active development with respect to the claimed system. Other such disputes could arise in the future or emerge from past activities which could lead others to claim our intellectual property.
30
Table of Contents
We may be involved in future costly intellectual property litigation, which could impact our future business and financial performance.
Our industry has been characterized by frequent intellectual property litigation. Our competitors or other patent holders may assert that our ELAD System and the methods we employ are covered by their patents. For instance, we are aware of other patents issued in the liver support field which we believe do not cover our ELAD System or its use. If our ELAD System or methods are found to infringe any valid patents, we could be prevented from marketing our ELAD System. In addition, we do not know whether our competitors or potential competitors have applied for, or will apply for or obtain, patents that will prevent, limit or interfere with our ability to make, use, sell, import or export our ELAD System.
Litigation related to infringement and other intellectual property claims, with or without merit, is unpredictable, can be expensive and time-consuming and could divert management’s attention from our core business. If we lose this kind of litigation, a court could require us to pay substantial damages, and prohibit us from using technologies essential to our ELAD System, any of which would have a material adverse effect on our business, results of operations and financial condition. We do not know whether necessary licenses would be available to us on satisfactory terms, or whether we could redesign our ELAD System or processes to avoid infringement.
Competing products may also appear in other countries in which our patent coverage might not exist or be as strong. If we lose a foreign patent lawsuit, we could be prevented from marketing our ELAD System in one or more countries.
In addition, we may hereafter become involved in litigation to protect our trademark rights associated with our company name or the names used with our ELAD System. Names used with our ELAD System and procedures may be claimed to infringe names held by others or to be ineligible for proprietary protection. If we have to change the name of our company or our ELAD System, we may experience a loss in goodwill associated with our brand name, customer confusion and a loss of sales.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets owned by third parties.
Many of our employees were previously employed at universities or other life science companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other confidential or proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key personnel could hamper our ability to develop and commercialize the ELAD System, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Our Capital Requirements and Finances
Our future capital needs are uncertain and we will need to raise additional funds in the future.
We will need to raise substantial additional capital to:
| • | complete our ongoing and future clinical trials and related regulatory applications; |
| • | fund our operations; |
| • | commence and expand the commercialization of our products; and |
| • | further our research and development. |
Our future funding requirements will depend on many factors, including:
| • | market acceptance of our products; |
31
Table of Contents
|
|
| • | the cost of our research and development activities; |
| • | the cost and timing of our clinical development activities, in particular the rate of approval of our clinical trial applications, the rate of initiation of our clinical sites and the rate of enrollment of our clinical trials; |
| • | the cost of filing and prosecuting patent applications; |
| • | the cost of defending, in litigation or otherwise, any claims that we infringe third-party patents or violate other intellectual property rights; |
| • | the cost and timing of regulatory clearances or approvals, if any; |
| • | the cost and timing of establishing additional sales, marketing and distribution capabilities; |
| • | the cost and timing of establishing additional technical support capabilities; |
| • | the effect of competing technological and market developments; and |
| • | the extent to which we acquire or invest in businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions. |
We cannot assure you that we will be able to obtain additional funds on acceptable terms, or at all. If we raise additional funds by issuing equity securities, our stockholders will experience dilution. Debt financing, if available, may involve covenants restricting our operations or our ability to incur additional debt. Any debt or additional equity financing that we raise may contain terms that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, which we have no prior experience in, it may be necessary to relinquish some rights to our technologies or our products, or grant licenses on terms that are not favorable to us. If we are unable to raise adequate funds, we may have to liquidate some or all of our assets, or delay, reduce the scope of or eliminate some or all of our development programs.
If we do not have, or are not able to obtain, sufficient funds, we may have to delay development or commercialization of our products or license to third parties the rights to commercialize products or technologies that we would otherwise seek to commercialize. We also may have to reduce marketing, customer support or other resources devoted to our products or cease operations. Any of these factors could harm our operating results.
Any acquisitions that we make could disrupt our business and harm our financial condition.
We expect to evaluate potential strategic acquisitions of complementary businesses, products or technologies. We may also consider joint ventures, licensing and other collaborative projects. We may not be able to identify appropriate acquisition candidates or strategic partners, or successfully negotiate, finance or integrate acquisitions of any businesses, products or technologies. Furthermore, the integration of any acquisition and management of any collaborative project may divert our management’s time and resources from our core business and disrupt our operations. We do not have any experience with acquiring companies or products. Any cash acquisition we pursue would diminish the proceeds from this offering available to us for other uses, and any stock acquisition would dilute our stockholders’ ownership. While we from time to time evaluate potential collaborative projects and acquisitions of businesses, products and technologies, and anticipate continuing to make these evaluations, we have no present understandings, commitments or agreements with respect to any acquisitions or collaborative projects.
Risks Related to Being a Public Company
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain executive management and qualified board members.
As a public company, we will be subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, the Sarbanes-Oxley Act, the Dodd-Frank Act, the listing requirements of the Nasdaq Global Market and other applicable securities rules and regulations. Compliance with these rules and
32
Table of Contents
regulations will increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources, particularly after we are no longer an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, or the JOBS Act. The Exchange Act requires, among other things, that we file annual, quarterly and current reports with respect to our business and operating results. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight may be required. As a result, management’s attention may be diverted from other business concerns, which could adversely affect our business and operating results. Although we have already hired additional employees to assist us in complying with these requirements, we may need to hire more employees in the future or engage outside consultants, which will increase our costs and expenses.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us and our business may be adversely affected.
However, for as long as we remain an “emerging growth company,” we may take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation and financial statements in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.”
We would cease to be an “emerging growth company” upon the earliest of: (1) the first fiscal year following the fifth anniversary of this offering, (2) the first fiscal year after our annual gross revenue is $1.0 billion or more, (3) the date on which we have, during the previous three-year period, issued more than $1.0 billion in non-convertible debt securities and (4) as of the end of any fiscal year in which the market value of our common stock held by non-affiliates exceeded $700 million as of the end of the second quarter of that fiscal year.
We also expect that being a public company will make it more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of our board of directors, particularly to serve on our audit committee and compensation committee, and qualified executive officers.
We cannot predict if investors will find our common stock less attractive if we choose to rely on these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our common stock and our stock price may decline or be more volatile.
Under Section 107(b) of the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected
33
Table of Contents
not to avail our company of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
As a result of becoming a public company, we will be obligated to develop and maintain proper and effective internal control over financial reporting. We may not complete our analysis of our system of internal control over financial reporting in a timely manner, or these internal controls may not be determined to be designed or operating effectively, which may adversely affect investor confidence in our company and, as a result, the value of our common stock.
We will be required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting for the first fiscal year beginning after the closing of this offering. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting.
We are in the very early stages of the costly and challenging process of compiling the system and processing documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing or any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal controls are effective, which could result in a loss of investor confidence in the accuracy and completeness of our financial reports. This could cause the price of our common stock to decline, and we may be subject to investigation or sanctions by the Securities and Exchange Commission, or SEC.
We will be required to disclose changes made in our internal control and procedures on a quarterly basis. However, our independent registered public accounting firm will not be required to report on the effectiveness of our internal control over financial reporting pursuant to Section 404 until the later of the year following our first annual report required to be filed with the SEC, and the date we are no longer an “emerging growth company” if we take advantage of the exemptions contained in the JOBS Act. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied that our internal controls over financial reporting are designed and operating effectively to prevent or detect a material misstatement to the financial statements. To comply with the requirements of being a public company, we may need to undertake various actions, such as implementing new internal controls and procedures or hiring accounting or internal audit staff.
If we do not remediate material weaknesses in our internal control over financial reporting, the accuracy and timeliness of our financial reporting may be adversely affected.
We have not maintained an effective control environment to ensure that the design and execution of our controls has consistently resulted in effective review of our financial statements and supervision by appropriate individuals. As a result of these factors, certain misstatements in our annual financial statements were identified and brought to the attention of management by our independent registered public accounting firm for correction. We and our independent registered public accounting firm concluded that these control deficiencies constituted a material weakness in our internal control over financial reporting. A material weakness is a control deficiency, or a combination of control deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
We are in the process of implementing measures designed to improve our internal control over financial reporting to remediate the control deficiencies that led to our material weakness. We cannot assure you that the measures we have taken to date, or any measures we may take in the future, will be sufficient to remediate the control deficiencies that led to our material weakness in our internal control over financial reporting or to avoid potential future material weaknesses. In addition, neither our management nor independent registered public accounting firm has ever performed an evaluation of our internal control over financial reporting in accordance
34
Table of Contents
with the provisions of the Sarbanes-Oxley Act because no such evaluation has been required. Had we or our independent registered public accounting firm performed an evaluation of our internal control over financial reporting in accordance with the provisions of the Sarbanes-Oxley Act, additional significant deficiencies or material weaknesses may have been identified. If we are unable to successfully remediate any significant deficiency or material weakness in our internal control over financial reporting, or identify any additional significant deficiencies or material weaknesses that may exist, the accuracy and timing of our financial reporting may be adversely affected, we may be unable to maintain compliance with securities law requirements regarding timely filing of periodic reports in addition to applicable stock exchange listing requirements, investors may lose confidence in our financial reporting, and our stock price may decline as a result.
Risks Related to This Offering and Ownership of our Common Stock
There has been no prior public market for our common stock and an active trading market may not develop.
Prior to this offering, there has been no public market for our common stock. An active trading market may not develop following closing of this offering or, if developed, may not be sustained. The lack of an active market may impair the value of your shares and your ability to sell your shares at the time you wish to sell them. An inactive market may also impair our ability to raise capital by selling shares and may impair our ability to acquire other companies, products or technologies by using our shares as consideration.
If securities or industry analysts do not publish research or publish unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will rely in part on the research and reports that equity research analysts publish about us and our business. We do not currently have and may never obtain research coverage by equity research analysts. Equity research analysts may elect not to provide research coverage of our common stock after the closing of this offering, and such lack of research coverage may adversely affect the market price of our common stock. In the event we obtain equity research analyst coverage, we will not have any control of the analysts or the content and opinions included in their reports. The price of our stock could decline if one or more equity research analysts downgrade our stock or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of our company or fails to publish reports on us regularly, demand for our stock could decrease, which in turn could cause our stock price or trading volume to decline.
We expect that the price of our common stock will fluctuate substantially and you may not be able to sell the shares you purchase in this offering at or above the offering price.
The initial public offering price for the shares of our common stock sold in this offering will be determined by negotiation between representatives of the underwriters and us. This price may not reflect the market price of our common stock following this offering. In addition, the market price of our common stock is likely to be highly volatile and may fluctuate substantially due to many factors, including:
| • | clinical data and government approvals relating to the ELAD System; |
| • | volume and timing of sales of the ELAD System; |
| • | the introduction of new products or product enhancements by us or our competitors; |
| • | disputes or other developments with respect to our intellectual property rights or the intellectual property rights of others; |
| • | our ability to develop, obtain regulatory clearance or approval for and market new and enhanced products on a timely basis; |
| • | product liability claims or other litigation; |
| • | quarterly variations in our or our competitors’ results of operations; |
35
Table of Contents
| • | sales of large blocks of our common stock, including sales by our executive officers and directors; |
| • | developments in our industry; |
| • | media exposure of the ELAD System or products of our competitors; |
| • | changes in governmental regulations or in the status of our regulatory approvals or applications; |
| • | changes in earnings estimates or recommendations by securities analysts; |
| • | our ability to meet investors expectations regarding our future operating performance; and |
| • | general market conditions and other factors, including factors unrelated to our operating performance or the operating performance of our competitors. |
These and other factors may make the price of our stock volatile and subject to unexpected fluctuations.
New investors in our common stock will experience immediate and substantial dilution after this offering.
If you purchase shares of our common stock in this offering, you will incur immediate and substantial dilution in pro forma net tangible book value. If the holders of outstanding options exercise those options, you will incur further dilution. See “Dilution.”
A sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
We expect that significant additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities. If we sell common stock, convertible securities or other equity securities, your investment in our common stock will be diluted. These sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders. In addition, if our stockholders sell substantial amounts of our common stock in the public market after this offering, including shares issued upon the exercise of outstanding options or warrants, the market price of our common stock could decline. These sales also might make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate. See “Shares Eligible for Future Sale.”
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
After this offering, our officers, directors and principal stockholders and their affiliates collectively will control approximately % of our outstanding common stock, and in particular, one stockholder and his affiliates will control approximately % of our outstanding common stock. This ownership does not reflect potential purchases of shares by certain of our officers, directors, existing investors and their affiliates in this offering and any such purchases would result in our officers, directors and principal stockholders and their affiliated entities owning additional common stock after this offering. See “Underwriting.” As a result, these stockholders, if they act together, will be able to exert substantial influence over the management and affairs of our company and most matters requiring stockholder approval, including the election of directors. This concentration of ownership may have the effect of delaying or preventing a change in control and might adversely affect the market price of our common stock. This concentration of ownership may not be in the best interests of our other stockholders.
We have broad discretion in the use of proceeds of this offering for working capital and general corporate purposes.
The net proceeds of this offering will be allocated to research and development activities, preparation for commercialization and general corporate purposes. Our management will have broad discretion over the use and investment of the net proceeds of this offering within those categories, and accordingly investors in this offering
36
Table of Contents
will need to rely upon the judgment of our management with respect to the use of proceeds, with only limited information concerning management’s specific intentions. See “Use of Proceeds.”
Anti-takeover provisions in our amended and restated certificate of incorporation, amended and restated bylaws, and Fourth Amended and Restated Investors’ Rights Agreement, as well as Delaware law, could discourage a takeover.
Our amended and restated certificate of incorporation, bylaws, Fourth Amended and Restated Investors’ Rights Agreement, and Delaware law, contain provisions that might enable our management to resist a takeover, and might make it more difficult for an investor to acquire a substantial block of our common stock. These provisions:
| • | authorize our board of directors to issue, without further action by our stockholders, up to 20,000,000 shares of undesignated preferred stock; |
| • | require that any action to be taken by our stockholders be effected at a duly called annual or special meeting and not by written consent; |
| • | specify that special meetings of our stockholders can be called only by a supermajority (75%) vote of our directors then in office; |
| • | specify that our board of directors may amend or repeal our bylaws only pursuant to a supermajority (75%) vote of our directors then in office; |
| • | specify that our stockholders may amend or repeal our bylaws only pursuant to a supermajority (75% and majority of the minority, if applicable) vote of the outstanding shares of our capital stock; |
| • | require in general the approval of a supermajority (75% and majority of the minority, if applicable) vote of our outstanding shares of capital stock to amend or repeal certain provisions of our certificate of incorporation; |
| • | require the approval of a supermajority (75% and majority of the minority, if applicable) vote of our outstanding shares of capital stock to approve the sale or liquidation of the company; |
| • | establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors; |
| • | provide that directors may be removed only for cause by a supermajority (75%) vote of our outstanding shares of capital stock; |
| • | provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though less than a quorum; |
| • | provide that in general the number of directors on our board may only be fixed from time to time by a supermajority (75%) vote of our directors then in office; |
| • | establish that our board of directors is divided into three classes, Class I, Class II and Class III, with each class serving staggered terms; and |
| • | provide that certain stockholders affiliated with Muneer A. Satter, referred to as the Satter Investors, have rights to nominate up to 40% of our directors. |
These provisions might discourage, delay or prevent a change in control of our company or a change in our management. The existence of these provisions could adversely affect the voting power of holders of common stock and limit the price that investors might be willing to pay in the future for shares of our common stock.
Our certificate of incorporation also contains a provision that provides us with protections similar to Section 203 of the Delaware General Corporation Law and will prevent us from engaging in a business combination with a person who acquires at least 15% of our common stock for a period of three years from the
37
Table of Contents
date such person acquired such common stock, except for certain of our current stockholders, including Mr. Satter and entities affiliated with him, and, in certain instances, persons who purchase common stock from certain of our current stockholders, and unless board or stockholder approval is obtained prior to the acquisitions. These anti-takeover provisions and other provisions under Delaware law could discourage, delay or prevent a transaction involving a change in control of our company, even if doing so would benefit our stockholders. These provisions could also discourage proxy contests and make it more difficult for you and other stockholders to elect or remove directors of your choosing and to cause us to take other corporate actions you desire. For additional information, including with respect to the specific voting requirements in our amended and restated certificate of incorporation, see “Description of Capital Stock.”
We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of our stock.
We have never paid cash dividends on our common stock and do not anticipate paying cash dividends on our common stock in the foreseeable future. The payment of dividends on our common stock will depend on our earnings, financial condition and other business and economic factors affecting us at such time as our board of directors may consider relevant. If we do not pay dividends, our stock may be less valuable because a positive return on your investment will only occur if our stock price appreciates.
38
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that are based on our management’s beliefs and assumptions and on information currently available to our management. The forward-looking statements are contained principally in “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business.” Forward-looking statements include information concerning our possible or assumed future results of operations, business strategies, financing plans, competitive position, industry environment, potential growth opportunities and the effects of competition. Forward-looking statements include all statements that are not historical facts and can be identified by terms such as “anticipates,” “believes,” “could,” “seeks,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “projects,” “should,” “will,” “would” or similar expressions and the negatives of those terms.
These forward-looking statements include, among other things, statements about:
| • | the initiation, cost and timing of our clinical programs for the ELAD System; |
| • | the timing of, and our ability to obtain and maintain regulatory approvals for the ELAD System; |
| • | regulatory developments in the U.S. and foreign countries; |
| • | the potential market for the ELAD System; |
| • | the rate and degree of market acceptance and clinical utility of the ELAD System; |
| • | our commercialization, marketing and manufacturing capabilities and strategy; |
| • | our plans to improve the ELAD System; |
| • | our plans to explore other uses for our VTL C3A cells; |
| • | our plans to obtain funding for our operations; |
| • | the performance of third parties in connection with the development of the ELAD System, including third parties involved in our clinical trials and third-party suppliers; |
| • | the development, regulatory approval, efficacy and commercialization of competing products; |
| • | our ability to retain key scientific or management personnel; |
| • | our intellectual property position; |
| • | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
| • | our anticipated use of the net proceeds in the offering; and |
| • | our ability to achieve and maintain effective internal control over financial reporting. |
Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements including those described in “Risk Factors” and elsewhere in this prospectus. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our management’s beliefs and assumptions only as of the date of this prospectus. You should read this prospectus and the documents that we have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
39
Table of Contents
This prospectus also contains estimates and other information concerning our industry, which is based on government and industry publications. This information involves a number of assumptions and limitations, and you are cautioned not to give undue weight to these estimates. These government and industry publications generally indicate that their information has been obtained from sources believed to be reliable.
40
Table of Contents
We estimate that the net proceeds from our sale of shares of common stock in this offering at an assumed initial public offering price of $ per share, the midpoint of the price range set forth on the cover of this prospectus, after deducting the estimated underwriting discount and estimated offering expenses payable by us, will be approximately $ , or $ if the underwriters’ option to purchase additional shares is exercised in full. A $1.00 increase (decrease) in the assumed initial public offering price would increase (decrease) the net proceeds to us from this offering by $ , assuming the number of shares offered by us, as set forth on the cover of this prospectus, remains the same and after deducting the estimated underwriting discount. Similarly, each increase or decrease of 1.0 million in the number of shares of common stock offered by us would increase or decrease the net proceeds that we receive from this offering by approximately $ , assuming the assumed initial public offering price remains the same and after deducting the estimated underwriting discount.
The principal purposes of this offering are to obtain additional capital for the development and possible approval of the ELAD System, create a public market for our common stock, and facilitate our future access to the public equity markets. We intend to use approximately $ of the net proceeds to fund the clinical development of the ELAD System and the remainder for working capital and other general corporate purposes. However, as of the date of this prospectus, we have not specifically allocated amounts of the net proceeds among such purposes or for specific development programs.
We believe that the net proceeds from this offering and our existing cash and cash equivalents will be sufficient to fund our operations through at least the next months. In particular, we believe that the net proceeds from this offering and our existing cash and cash equivalents will be sufficient to fund development through receipt of initial data from our leading Phase 3 clinical trials.
The expected use of the net proceeds from this offering represents our intentions based upon our current plans and business conditions. As of the date of this prospectus, we cannot predict with certainty all of the particular uses for the net proceeds to be received upon the completion of this offering or the amounts that we will actually spend on the uses set forth above. The amounts and timing of our actual expenditures depend on numerous factors, including the rate of subject enrollment in our clinical trials, filing requirements with various regulatory agencies, clinical trial results, and any unforeseen cash needs. Our management will retain broad discretion over the allocation of the net proceeds from this offering and could spend the proceeds in ways that do not improve our results of operations or enhance the value of our stock. Pending use of the proceeds from this offering, we intend to invest the proceeds in a variety of short-term investment-grade and interest-bearing instruments, direct or guaranteed obligations of the U.S. government, or certificates of deposits.
41
Table of Contents
We have never declared or paid cash dividends on our common stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any dividends on our common stock in the foreseeable future. Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant.
42
Table of Contents
The following table sets forth our consolidated cash and cash equivalents and capitalization:
| • | on an actual basis as of December 31, 2013; |
| • | on a pro forma-private offerings basis to reflect the sale and issuance of 2,296,016 shares of our senior redeemable convertible preferred stock at $8.00 per share in January 2014 and, pursuant to a preemptive rights offering, in February 2014, and the receipt of proceeds of $18.4 million, or the 2014 private offerings; |
| • | on a pro forma-converted basis to reflect the 2014 private offerings, the conversion of all outstanding shares of our preferred stock as of December 31, 2013 and the senior preferred stock issued in the 2014 private offerings into an aggregate of 16,009,423 shares of our common stock, the reclassification of the future purchase rights liabilities to additional paid-in capital, a component of stockholders’ (deficit) equity, and the filing of our amended and restated certificate of incorporation upon the closing of this offering; and |
| • | on a pro forma as adjusted-IPO basis to further reflect the 2014 private offerings and pro forma conversion and reclassification described above plus our receipt of the net proceeds from our sale of shares of our common stock in this offering at an assumed initial public offering price of $ per share, the midpoint of the price range set forth on the cover of this prospectus, after deducting the estimated underwriting discount and estimated offering expenses payable by us. |
The above pro forma items are presented in the table below to give effect to such events as if they occurred on December 31, 2013:
| Actual As of December 31, 2013 |
Pro Forma- Private Offerings |
Pro Forma- Converted |
Pro Forma As Adjusted-IPO | |||||||||||
| (In thousands, except share and per share amounts) | ||||||||||||||
| Cash and cash equivalents |
$ | 38,186 | $ | 56,554 | $ | 56,554 | ||||||||
|
|
|
|
|
|
|
| ||||||||
| Future purchase rights liabilities |
$ | 2,600 | $ | 2,600 | $ | — | ||||||||
| Junior convertible preferred stock, 3,501,401 shares designated, 3,501,400 shares issued and outstanding, liquidation preference of $1,500,000, actual; no shares authorized, issued and outstanding, pro forma as adjusted-IPO |
1,359 | 1,359 | — | |||||||||||
| Senior redeemable convertible preferred stock, 17,000,000 shares designated, 10,212,007 shares issued and outstanding, liquidation preference of $126,886,137 actual; no shares authorized, issued and outstanding, pro forma as adjusted-IPO |
82,116 | 100,484 | — | |||||||||||
| Stockholders’ (deficit) equity: |
||||||||||||||
| Preferred stock, $0.0001 par value: 25,000,000 shares authorized (4,498,599 undesignated), and no shares issued or outstanding, actual and pro forma as adjusted-IPO |
— | — | — | |||||||||||
| Common stock, $0.0001 par value: 29,250,000 shares authorized, actual; 130,000,000 shares authorized pro forma as adjusted-IPO; 606,238 shares issued and outstanding, actual; and shares issued and outstanding, pro forma as adjusted-IPO |
— | — | 2 | |||||||||||
| Additional paid-in capital |
58,413 | |
58,413 |
|
162,854 | |||||||||
| Accumulated other comprehensive income |
96 | 96 | 96 | |||||||||||
| Deficit accumulated during development stage |
(103,166 | ) | (103,166 | ) | (103,166 | ) | ||||||||
|
|
|
|
|
|
|
| ||||||||
| Total stockholders’ (deficit) equity |
(44,657 | ) | (44,657 | ) | 59,786 | |||||||||
|
|
|
|
|
|
|
| ||||||||
| Total capitalization |
$ | 41,418 | $ | 59,786 | $ | 59,786 | ||||||||
|
|
|
|
|
|
|
| ||||||||
43
Table of Contents
The number of shares of common stock to be outstanding following this offering is based on 14,319,645 shares of our common stock outstanding as of December 31, 2013 (including preferred stock on an as-converted basis) and 2,296,016 shares of senior preferred stock sold in January 2014 and, pursuant to a preemptive rights offering, in February 2014, on an as-converted basis, but excludes:
| • | 3,098,573 shares of our common stock issuable upon the exercise of options outstanding as of December 31, 2013, with a weighted-average exercise price of $6.71 per share; |
| • | 250,646 shares of our common stock issuable upon the exercise of warrants outstanding as of December 31, 2013, with a weighted-average exercise price of $95.21 per share; and |
| • | 29,492 shares of our common stock reserved for future issuance as of December 31, 2013 under our stock-based compensation plans, including our 2012 Stock Plan and our 2014 Equity Incentive Plan, which will become effective in connection with this offering and contains provisions that will automatically increase its share reserve on the date of this prospectus and on each annual anniversary of the date of this prospectus, as more fully described in “Executive and Director Compensation—Equity Incentive Plans”. |
44
Table of Contents
If you invest in our common stock, your ownership interest will be diluted to the extent of the difference between the amount per share paid by purchasers of shares of our common stock in this initial public offering and the pro forma as adjusted net tangible book value per share of our common stock immediately after closing of this offering.
Our historical net tangible book deficit is the amount of our total tangible assets less our total liabilities. Historical net tangible book deficit per share is our historical net tangible book deficit divided by the number of shares of common stock outstanding as of December 31, 2013. Our net tangible book deficit as of December 31, 2013 was $44.7 million, or $73.66 per share.
Pro forma net tangible book value gives effect to (i) the sale of 2,296,016 shares of our senior redeemable convertible preferred stock at $8.00 per share and the receipt of proceeds of $18.4 million in January and February 2014; and (ii) the conversion of all outstanding shares of our preferred stock (including the senior preferred stock sold in January 2014 and, pursuant to a preemptive rights offering, in February 2014) into an aggregate of 16,009,423 shares of our common stock, which will occur immediately prior to the closing of this offering, and the reclassification of our future purchase rights liabilities to additional paid-in capital, a component of stockholders’ (deficit) equity. Our pro forma net tangible book value as of December 31, 2013 would have been approximately $59.8 million, or $3.60 per share.
Pro forma as adjusted net tangible book value is our pro forma net tangible book value, after giving effect to the sale of shares of our common stock in this offering at an assumed initial public offering price of $ per share, which is the midpoint of the price range set forth on the cover of this prospectus, after deducting the estimated underwriting discount and estimated offering expenses payable by us. Our pro forma as adjusted net tangible book value as of December 31, 2013 would have been approximately $ , or $ per share. This amount represents an immediate increase to pro forma net tangible book value of $ per share to our existing stockholders, and an immediate dilution of $ per share to new investors participating in this offering. Dilution per share to new investors is determined by subtracting pro forma as adjusted net tangible book value per share after this offering from the initial public offering price per share paid by new investors.
The following table illustrates this dilution on a per share basis:
| Assumed initial public offering price per share |
||||||
| Historical net tangible book deficit per share as of December 31, 2013 |
$(73.66) | |||||
| Pro forma increase in net tangible book value per share attributable to the senior redeemable convertible preferred stock issued in January and February 2014 |
— | |||||
| Pro forma increase in net tangible book value per share attributable to the conversion of convertible preferred stock and the reclassification of the future purchase rights to equity |
77.26 | |||||
|
|
|
|||||
| Pro forma net tangible book value per share as of December 31, 2013 |
3.60 | |||||
| Increase to pro forma net tangible book value per share attributable to new investors |
||||||
|
|
|
|||||
| Pro forma as adjusted net tangible book value per share, after giving effect to this offering |
||||||
|
| ||||||
| Dilution of pro forma as adjusted net tangible book value per share to new investors |
||||||
|
|
A $1.00 increase in the assumed public offering price to $ per share would increase our pro forma as adjusted net tangible book value per share by $ , and would increase dilution per share to new investors in this offering by $ , assuming that the number of shares offered by us, as set forth on the cover of this prospectus, remains the same and after deducting the estimated underwriting discount and estimated offering
45
Table of Contents
expenses payable by us. A $1.00 decrease in the assumed public offering price to $ per share would decrease our pro forma as adjusted net tangible book value per share by $ , and decrease dilution per share to new investors in this offering by $ . We may also increase or decrease the number of shares we are offering. An increase of 1.0 million shares in the number of shares offered by us at $ per share would increase pro forma as adjusted net tangible book value by approximately $ and the dilution per share to new investors in this offering would be $ . Similarly, a decrease of 1.0 million shares in the number of shares offered by us at $ per share would decrease pro forma ad adjusted net tangible book value by $ and the dilution per share to new investors in this offering would be $ . The pro forma as adjusted information discussed above is illustrative only and will be adjusted based on the actual initial public offering price, the number of shares we sell in this offering and other terms of this offering.
In addition, to the extent any outstanding options or warrants to purchase common stock are exercised, new investors would experience further dilution. If the underwriters exercise their option to purchase additional shares in full, the pro forma as adjusted net tangible book value per share after giving effect to this offering at an assumed public offering price of $ per share would be approximately $ per share, and the dilution in pro forma net tangible book value per share to new investors in this offering would be approximately $ per share.
The following table summarizes, on a pro forma as adjusted basis as of December 31, 2013, the total number of shares of common stock purchased from us, the total consideration paid to us and the average price per share paid to us by existing stockholders (including senior preferred stock purchased in January 2014 and, pursuant to a preemptive rights offering, in February 2014) and by new investors purchasing shares of common stock in this offering at an assumed initial public offering price of $ , the midpoint of the price range set forth on the cover of this prospectus, before deducting the estimated underwriting discount and estimated offering expenses payable by us:
| Shares Purchased | Total Consideration | Average Price Per Share |
||||||||||||||||||
| Number | Percent | Amount | Percent | |||||||||||||||||
| Existing stockholders |
16,615,661 | % | $169,618,569 | % | $10.21 | |||||||||||||||
| New investors |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Totals |
100 | % | 100 | % | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
Each $1.00 increase or decrease in the assumed initial public offering price of per share would increase or decrease, as applicable, the total consideration paid by new investors and total consideration paid by all stockholders by approximately $ assuming that the number of shares offered by us, as set forth on the cover of this prospectus, remains the same.
In addition, to the extent any outstanding options or warrants to purchase common stock are exercised or any outstanding shares of restricted stock or restricted stock units that we may grant in the future vest, new investors will experience further dilution.
If the underwriters exercise their option to purchase additional shares in full, our existing stockholders would own % and our new investors would own % of the total number of shares of our common stock outstanding after this offering.
The number of shares of common stock to be outstanding following this offering is based on 14,319,645 shares of our common stock outstanding as of December 31, 2013 (including preferred stock on an as-converted basis) plus, on a pro forma basis, 2,296,016 shares of senior preferred stock sold in January 2014 and, pursuant to a preemptive rights offering, in February 2014 on an as-converted basis, but excludes:
| • | 3,098,573 shares of our common stock issuable upon the exercise of options outstanding as of December 31, 2013, with a weighted-average exercise price of $6.71 per share; |
46
Table of Contents
| • | 250,646 shares of our common stock issuable upon the exercise of warrants outstanding as of December 31, 2013, with a weighted-average exercise price of $95.21 per share; and |
| • | 29,492 shares of our common stock reserved for future issuance as of December 31, 2013, under our stock-based compensation plans, including our 2012 Stock Plan and our 2014 Equity Incentive Plan, which will become effective in connection with this offering and contains provisions that will automatically increase its share reserve on the date of this prospectus and on each anniversary of the date of this prospectus, as more fully described in “Executive and Director Compensation—Equity Incentive Plans”. |
The foregoing discussion and tables do not reflect potential purchases by certain of our officers, directors, existing investors and their affiliated entities who have indicated an interest in purchasing shares in this offering as described in “Underwriting”.
47
Table of Contents
SELECTED CONSOLIDATED FINANCIAL DATA
The following table summarizes our selected consolidated financial data for the periods and as of the dates indicated. Our selected consolidated statements of operations data for each of the years ended December 31, 2012 and 2013, and our selected consolidated balance sheet data as of December 31, 2012 and 2013, have been derived from our audited consolidated financial statements and related notes included elsewhere in this prospectus. Our selected consolidated financial data should be read together with the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and with our audited consolidated financial statements and their related notes, which are included elsewhere in this prospectus.
| Years Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| (In thousands, except share and per share amounts) |
||||||||
| Consolidated Statement of Operations Data: |
||||||||
| Operating expenses: |
||||||||
| Research and development |
$ | 5,097 | $ | 21,787 | ||||
| General and administrative |
4,483 | 9,615 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
9,580 | 31,402 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(9,580 | ) | (31,402 | ) | ||||
| Other income (expense): |
||||||||
| Interest income |
4 | 5 | ||||||
| Interest expense |
(413 | ) | — | |||||
| Other (expense) income, net |
7 | (15 | ) | |||||
| Revaluation of preferred stock warrant liabilities |
180 | — | ||||||
| Revaluation of future purchase rights liabilities |
3,101 | (1,306 | ) | |||||
|
|
|
|
|
|||||
| Total other income (expense) |
2,879 | (1,316 | ) | |||||
|
|
|
|
|
|||||
| Net loss |
(6,701 | ) | (32,718 | ) | ||||
| Amortization of deemed dividend |
— | (64 | ) | |||||
| Accretion to redemption value of senior redeemable convertible preferred stock |
(942 | ) | (6,303 | ) | ||||
|
|
|
|
|
|||||
| Net loss attributable to common stockholders |
$ | (7,643 | ) | $ | (39,085 | ) | ||
|
|
|
|
|
|||||
| Net loss per share attributable to common stockholders, basic and diluted(1) |
$ | (17.89 | ) | $ | (74.86 | ) | ||
|
|
|
|
|
|||||
| Weighted-average common shares outstanding, basic and diluted(1) |
427,117 | 522,102 | ||||||
|
|
|
|
|
|||||
| Pro forma net loss per share attributable to common stockholders, basic and diluted (unaudited)(1) |
$ | (3.14 | ) | |||||
|
|
|
|||||||
| Pro forma weighted-average common shares outstanding, basic and diluted (unaudited)(1) |
10,008,616 | |||||||
|
|
|
|||||||
| (1) | Please refer to Note 2 of our consolidated financial statements for an explanation of the method used to calculate the historical and pro forma net loss per share attributable to common stockholders and the number of shares used in the computation of the per share amounts. |
48
Table of Contents
| As of December 31, | ||||||||
| 2012 | 2013 | |||||||
| (In thousands) | ||||||||
| Consolidated Balance Sheet Data: |
||||||||
| Cash, cash equivalents and short-term investments |
$ | 18,473 | $ | 38,186 | ||||
| Working capital |
17,403 | 36,409 | ||||||
| Total assets |
20,332 | 46,585 | ||||||
| Future purchase rights |
— | 2,600 | ||||||
| Preferred stock |
26,176 | 83,475 | ||||||
| Deficit accumulated during development stage |
(70,448 | ) | (103,166 | ) | ||||
| Total stockholders’ deficit |
(7,632 | ) | (44,657 | ) | ||||
As of February 28, 2014 we had cash and cash equivalents of $51.3 million, of which $1.2 million was restricted.
49
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with the section entitled “Selected Consolidated Financial Data” and our consolidated financial statements and related notes included elsewhere in this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. See “Special Note Regarding Forward-Looking Statements.” Our actual results may differ materially from those described below. You should read the “Risk Factors” section of this prospectus for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a biotherapeutic company focused on developing a cell-based therapy targeting the treatment of all forms of acute liver failure. Our product candidate, the ELAD System, is an extracorporeal bio-artificial liver therapy designed to allow the patient’s own liver to regenerate to a healthy state, or to stabilize the patient until transplant. The ELAD System is the only bio-artificial liver support system containing immortal human liver-derived cells, or C3A cells, to enter Phase 3 clinical trials. We designed the ELAD System to supplement key aspects of normal liver function to improve patient survival. We estimate that at least 30,000 patients annually in the United States experience forms of acute liver failure, such as acute-on-chronic, surgically-induced and fulminant liver failures, for which the ELAD System may be a life-saving therapy. Outside of liver transplant, which is severely limited by availability of organs and not available to many patients, the current standard-of-care for acute liver failure is primarily focused on the management of complications, which does not restore lost liver function and is associated with a high rate of mortality. The ELAD System has received orphan designation in the United States and Europe for the treatment of patients with acute liver failure. This designation provides tax credits for qualified clinical testing, seven years of market exclusivity in the United States, and ten years of market exclusivity in Europe for the first approved orphan drug for a given indication. However, orphan designation does not alter the standard regulatory requirements or the process for obtaining marketing approval.
We are currently enrolling patients in one Phase 3 clinical trial, have regulatory allowance to begin enrolling patients in a second Phase 3 trial, and also expect to initiate later this year a Phase 2/3 program, each in forms of acute liver failure. In March 2013, we initiated VTI- 208, a Phase 3 randomized, controlled clinical trial in 200 subjects with AILD. In addition, we have obtained regulatory allowance in the United States, United Kingdom, Spain and Australia to begin enrolling patients and have initiated clinical sites in a second Phase 3 randomized, controlled clinical trial, VTI-210, in 120 subjects with severe AAH. We expect the enrollment of subjects in VTI-210 to begin in the first half of 2014. Finally, we also expect to initiate enrollment in the Phase 2 component of VTI-212, a single-arm clinical trial in 40 subjects with FHF or SILF, in the second half of 2014.
Vital Therapies, Inc. was formed in May 2003 to acquire the assets of VitaGen (formerly Hepatix) in a bankruptcy proceeding. Our predecessor companies developed the ELAD System, completing two pilot trials in acute liver failure and two randomized, controlled Phase 1 and Phase 2 trials in FHF, but failed to attract funds sufficient to continue development of the system. Beginning in June 2003, we refocused the company to pursue regulatory approval and commercialize the ELAD System in China. In 2007, we completed a pivotal trial in acute liver failure in subjects with viral hepatitis in China, and we submitted an application for marketing in China. Our application is still under review in China and we do not expect approval in China until we have approval in the United States.
We restarted our clinical program in the United States and Europe in 2008. Since then, we have run two Phase 2 trials and selected AILD and AAH as indications for our Phase 3 pivotal trial program in the United States and Europe. We have also made significant improvements in the ELAD System bedside unit and our
50
Table of Contents
proprietary cartridge cell growth production process, including (i) the incorporation of an updated version of the cardiovascular base unit that has improved features and functionalities and reliability; (ii) new and improved cartridges for ultrafiltrate, cell filters and the ELAD cartridges; (iii) tubing sets that have been optimized to recirculate smaller volumes of ultrafiltrate and blood through the system to reduce the risk of clotting and other
potential adverse side effects; and (iv) improvements to our cell culture and growth processes to reduce cost and increase manufacturing efficiency and yield.
We are a development stage company and have incurred net losses since inception of $103.2 million through December 31, 2013, including $6.7 million and $32.7 million for the years ended December 31, 2012 and 2013, respectively. We anticipate that we will continue to incur increasing losses for at least the next several years. Due to the uncertainties involved with biological product development and the clinical trial process, we cannot predict the timing or accuracy of future expenses, when product approval for the ELAD System might occur, or when profitability may be achieved or sustained.
Financial Operations Overview
Operating Expenses
The following table summarizes our operating expenses for the years ended December 31, 2012 and 2013.
| Years Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| (In thousands) | ||||||||
| Operating expenses: |
||||||||
| Research and development |
$ | 5,097 | $ | 21,787 | ||||
| General and administrative |
4,483 | 9,615 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
$ | 9,580 | $ | 31,402 | ||||
|
|
|
|
|
|||||
Research and Development Expenses
Research and development expenses relate to the development of the ELAD System and are expensed as incurred. Our research and development expenses consist primarily of:
| • | expenses incurred under agreements with clinical sites, clinical research organizations, or CROs, and statistical and regulatory consultants that assist us with our clinical trials; |
| • | employee-related expenses, which include salaries, benefits, travel and stock-based compensation; |
| • | the cost of acquiring and manufacturing clinical trial materials; |
| • | facilities, depreciation, and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities and equipment, and depreciation of fixed assets; and |
| • | costs associated with other research and regulatory activities. |
We anticipate that our research and development expenses will increase substantially as we conduct our Phase 3 clinical trials of the ELAD System and begin the regulatory approval process for commercialization over the next several years.
We do not track our employee and facility related research and development costs by clinical trial, as we typically use our employee and infrastructure resources across multiple clinical trials and the allocation of such costs would be arbitrary and would not provide a meaningful assessment. During the second half of 2012, we began tracking direct costs related to VTI-208 to prepare for the commencement of that Phase 3 clinical trial in
51
Table of Contents
2013. Prior to that time, clinical trial costs for VTI-208 were not significant. Since 2012, clinical trial costs associated with VTI-208 have been our largest research related expenditure.
The costs of clinical trials may vary significantly over the life of a project owing to, but not limited to, the following:
| • | per subject trial costs; |
| • | the number of sites included in the trials; |
| • | the countries in which the trial is conducted; |
| • | the number of subjects that participate in the trials; |
| • | continuing quality assurance activities and standards consistent with FDA and other regulatory requirements; |
| • | potential additional safety monitoring or other studies requested by regulatory agencies; and |
| • | the frequency and duration of subject follow-up visits. |
A change in the outcome of any of these variables could result in a significant change in the costs and timing associated with the development of the ELAD System. For example, if the FDA or other regulatory authority were to require us to conduct clinical trials beyond those which we currently anticipate will be required for the completion of clinical development of the ELAD System or if we experience significant delays in enrollment in any clinical trial, we could be required to expend significant additional financial resources and time on the completion of the clinical development of the ELAD System.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive, finance, information technology, marketing, and legal functions. Other general and administrative expenses include facility costs, stock-based compensation, and professional fees for legal, consulting, accounting and tax services.
We anticipate that our general and administrative expenses will increase for primarily the following reasons:
| • | increased payroll, expanded infrastructure and higher consulting, legal, accounting and investor relations costs, and director and officer insurance premiums associated with being a public company; |
| • | expansion of our facilities to support our growth; |
| • | to support our business activities, which we expect to expand as we continue the development of the ELAD System; and |
| • | to build a marketing, reimbursement, training and support team before we receive regulatory approval of the ELAD System in anticipation of commercial launch. |
Other Income (Expense)
Interest Income
Our cash, cash equivalents, and short-term investments are invested primarily in money market funds and U.S. treasury bills, which generate a small amount of interest income, but in our opinion provide liquidity and protection from loss of principal. We expect to continue to make similar investments as we obtain additional financing proceeds.
52
Table of Contents
Interest Expense
Interest expense has consisted primarily of interest paid on debt and an equipment lease line, and non-cash interest associated with amortization of debt discount and financing costs. As of December 31, 2012, we no longer have any debt for which we record interest expense.
Revaluation of Preferred Stock Warrant Liabilities
Warrants for our Series A, Series B, Series C, and Series D redeemable convertible preferred stock were accounted for as liabilities and their value was remeasured at the end of each financial reporting period. The value of these preferred stock warrant liabilities is primarily related to the fair value of our common stock. We are required to recognize a gain or loss on these preferred stock warrant liabilities depending in part upon fluctuations in the fair value of our common stock. As a result of the junior preferred stock offering in February 2012, we recognized a final valuation adjustment for these preferred stock warrant liabilities prior to their conversion into common stock warrants.
Revaluation of Future Purchase Rights Liabilities
Future rights to purchase shares of our senior redeemable convertible preferred stock that were granted in connection with our senior preferred stock financing are accounted for as liabilities and their value is remeasured at the end of each financial reporting period. The value of these future purchase rights liabilities will fluctuate in conjunction with increases or decreases in the fair value of our common stock and the number of preferred and common shares and future purchase rights outstanding relative to our enterprise value at each reporting date. We recognize a gain or loss based on fluctuations in the fair value of these future purchase rights liabilities.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. On an on-going basis, management makes its best estimate of the ultimate outcome for these items based on historical trends and other information available when the financial statements are prepared. Changes in estimates are typically recognized in the period when new information regarding estimates becomes available to management. Actual results could differ from those estimates.
Our significant accounting policies are described in more detail in the notes to our financial statements appearing elsewhere in this prospectus. However, we believe that the following accounting policies are the most critical for fully understanding and evaluating our financial condition and results of operations.
Clinical Trial Accruals
As part of the process of preparing our financial statements, we are required to estimate our accrued expenses. Our clinical trial accrual process seeks to account for expenses resulting from our obligations under agreements with clinical sites, CROs, vendors, and consultants in connection with conducting our clinical trials. We account for these expenses according to the progress of each trial as measured by subject enrollment, the timing of various aspects of the trial and if available, information from our service providers. During the course of a clinical trial, we adjust our rate of clinical expense recognition if actual results differ from our estimates. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and could result in us reporting amounts that are over or understated for a particular period and adjustments to our research and development expenses may be necessary in future periods. Through
53
Table of Contents
December 31, 2013, there have been no material adjustments to our prior period estimates of accrued expenses for clinical trials.
Future Purchase Rights Liabilities
In September 2012, we entered into a senior preferred stock purchase agreement (as amended in December 2013, the Senior Preferred Purchase Agreement) that authorizes the multi-stage issuance of 14,765,632 shares of our senior redeemable convertible preferred stock for $8.00 per share. As of December 31, 2013, 10,212,007 shares have been issued under the Senior Preferred Purchase Agreement and 4,553,625 shares are subject to future purchase rights, as described below. Pursuant to the Senior Preferred Purchase Agreement, we granted to the investors in the senior preferred stock financing the right, subject to the satisfaction of certain conditions, to purchase additional shares of senior preferred stock for a purchase price of $8.00 per share at multiple subsequent closings in accordance with a schedule provided in the Senior Preferred Purchase Agreement. These future purchase rights terminate upon certain events, including the closing of this offering. Upon termination, the future purchase rights liabilities, as discussed below, would be reclassified to additional paid-in capital, a component of stockholders’ (deficit) equity.
These future purchase rights for additional shares of our senior preferred stock are legally detachable from the shares of the underlying senior preferred stock and, as a result, are considered freestanding instruments that are accounted for separately from such shares of senior preferred stock. As the future purchase rights are exercisable for shares of our redeemable convertible preferred stock, the future purchase rights are instruments that embody obligations to transfer assets and are classified as liabilities under the accounting guidance that distinguishes liabilities from equity.
Our future purchase rights liabilities are recorded at their estimated fair value on the date of issuance as a discount on the underlying preferred stock and are remeasured to reflect changes in the estimated fair value at each reporting date, with any decrease or increase in the estimated fair value being recorded as other income or expense, respectively. The fair value of these liabilities is estimated using a binomial lattice model that is based on the characteristics of the common and preferred stock on the valuation date, probabilities related to our operations and clinical development, as well as assumptions for volatility, remaining expected life, risk-free interest rate and, in some cases, credit spread. Changes in the fair value of the future purchase rights will fluctuate in conjunction with increases or decreases in the fair value of our common stock, and the number of preferred and common shares and future purchase rights outstanding relative to our enterprise value at each reporting date.
Stock-based Compensation
We measure and recognize compensation expense for all stock-based payments made to employees, directors, and consultants based on estimated fair value, net of an estimated forfeiture rate. Currently, our stock-based awards consist only of stock options; however, future grants under our equity compensation plans may consist of shares of restricted stock and restricted stock units. We estimate the fair value of stock options granted and stock purchase rights using the Black-Scholes-Merton, or BSM, option pricing model, which requires the use of estimates to value employee stock-based compensation at the date of grant.
We recognize stock-based compensation cost for employees and directors on a straight-line basis over the requisite service period of the award. Stock-based compensation expense is recognized only for those awards that are ultimately expected to vest. We estimate forfeitures based on an analysis of our historical employee turnover and will continue to evaluate the appropriateness of the forfeiture rate based on actual forfeiture experience, analysis of employee turnover and other factors. We will revise the forfeiture estimate, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Changes in forfeiture estimates, which have not been material to date, impact compensation cost in the period in which the change in estimate occurs.
The fair value of options granted to nonemployees is estimated using the BSM option pricing model and is remeasured at each reporting date until vested with changes in fair value recognized as expense in the consolidated statements of operations.
54
Table of Contents
The BSM option pricing model requires the input of highly subjective assumptions, including the estimated fair value of our common stock, the risk-free interest rate, the expected dividend yield of our common stock, the expected volatility of the price of our common stock, and the expected term of the option. These estimates involve inherent uncertainties, the application of management’s judgment and are highly complex and subjective. If factors change and different assumptions are used, our stock-based compensation expense could be materially different in the future. These assumptions are estimated as follows:
Risk-free Interest Rate
We base the risk-free interest rate assumption on zero-coupon U.S. treasury instruments appropriate for the expected term of the stock option grants.
Expected Dividend Yield
We base the expected dividend yield assumption on the fact that we have never paid cash dividends and have no present intention to pay cash dividends. Consequently, we used an expected dividend yield of zero.
Expected Volatility
As we do not have a trading history for our common stock, the expected stock price volatility for our common stock is estimated based on volatilities of a peer group of similar companies by taking the average historic price volatility for these peers for a period equivalent to the expected term of the stock option grants. The peer group was developed based on companies in the biotechnology industry whose shares are publicly traded.
Expected Term
The expected term represents the period of time that options are expected to be outstanding. As we do not have sufficient historical experience for determining the expected term of the stock option awards granted we determined the expected life assumption using either the simplified method, which is an average of the contractual term of the option and its ordinary vesting period, or the comparable average expected term utilizing those companies in the peer group noted above, as applicable.
The range of assumptions we used to determine the fair value of employee and nonemployee stock options granted in each period were as follows:
| Years Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| Risk-free interest rate |
0.2%—1.0% | 0.1%—1.1% | ||||||
| Expected dividend yield |
0% | 0% | ||||||
| Expected volatility |
100.0% | 90%—100% | ||||||
| Expected term of options (years) |
1.0—6.0 | 0.5—6.0 | ||||||
We estimate the level of forfeitures expected to occur and record stock-based compensation expense only for those awards that we ultimately expect will vest. Through December 31, 2013, actual forfeitures have not been material.
55
Table of Contents
The following table summarizes the allocation of stock-based compensation expense to employees and nonemployees (in thousands):
| Years Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| Research and development |
$ | 50 | $ | 411 | ||||
| General and administrative |
94 | 537 | ||||||
|
|
|
|
|
|||||
| Totals |
$ | 144 | $ | 948 | ||||
|
|
|
|
|
|||||
Common Stock Valuation
Due to the absence of a public trading market for our common stock, it is necessary to estimate the fair value of the common stock underlying our stock-based awards when performing fair value calculations using the BSM option pricing model. The fair value of the common stock underlying our stock-based awards was assessed on each grant date by our board of directors. All options to purchase shares of our common stock have been granted with an exercise price per share no less than the fair value per share of our common stock underlying those options on the date of grant.
For stock option grants our board of directors considered various objective and subjective factors to determine the fair value of our common stock, including: external market conditions affecting the biopharmaceutical industry, trends within the biopharmaceutical industry, the superior rights and preferences of the junior and senior preferred stock relative to our common stock at the time of each grant, our results of operations and financial position, status of our research and development efforts, our stage of development and business strategy, the lack of an active public market for our common stock and our junior and senior preferred stock, and the likelihood of achieving a liquidity event such as an initial public offering (IPO) or sale of our company in light of prevailing market conditions. Upon completion of our initial public offering, our common stock will be valued by reference to the publicly traded price of our common stock and not on highly complex estimates based on a range of objectives and subjective factors, as described above.
April 2012 Grants
Our board of directors determined that no internal or external value generating factors had occurred since the February 3, 2012 closing of the junior preferred stock financing; as such, our board of directors utilized the junior preferred stock issuance price of $0.43 per share as the basis for establishing the exercise price of our April 2012 grants.
September 2012 Grant
On September 25, 2012, we completed the first closing of our senior preferred stock financing. At this time our board of directors determined that other than the senior preferred stock financing no additional internal or external value generating factors had occurred and, therefore, established the exercise price of the September 26, 2012 grants at $8.00 per share, the issuance price of the senior preferred stock and the related rights.
December 2012 Grant
Our board of directors determined that no internal or external value generating factors had occurred between the September 25, 2012 financing and the December 7, 2012 grants; as such, our board of directors determined the exercise price of the December 2012 grants to be $8.00.
March, May, June, and July 2013 Grants
Through June 2013, we completed three additional closings of our senior preferred stock at $8.00 per share and our investors increased their total purchase commitments by $22.7 million to approximately $108.8 million.
56
Table of Contents
At the time of each of the 2013 grants, our board of directors determined that these events did not increase the fair value of our common stock beyond that of the issuance price of our senior preferred stock and the related rights and, therefore, determined the exercise price of the March, May, June, and July 2013 grants to be $8.00 per share.
September 2013 Grants
On September 9, 2013 and September 18, 2013, our board of directors granted common stock options with an exercise price of $10.50 per share. In setting the exercise price, our board of directors considered a valuation analysis prepared as of September 3, 2013, reflecting a fair value of our common stock of $9.94 per share; however, the board of directors elected to set the exercise price of the stock options granted above the fair value of the common stock. In addition, the board of directors determined no internal or external value generating events had taken place from September 4, 2013 through September 18, 2013 that would have impacted the assumptions utilized in the September 3, 2013 valuation analysis.
December 2013 Grants
On December 9, 2013, our board of directors granted common stock options with an exercise price of $8.00 per share. In setting the exercise price, our board of directors considered a valuation analysis prepared as of December 1, 2013, reflecting a fair value of our common stock of $6.47 per share; however, our board of directors elected to set the exercise price of the stock options granted above the fair value of the common stock. In addition, our board of directors determined no internal or external value generating events had taken place from December 1, 2013 through December 9, 2013 that would have impacted the assumptions utilized in the December 1, 2013 valuation analysis.
February 2014 Grants
On February 3, 2014, our board of directors granted common stock options with an exercise price of $8.00 per share. In setting the exercise price, our board of directors considered the January 24, 2014 financing in which our senior redeemable convertible preferred stock was sold for $8.00 per share to new investors in an arms-length transaction and determined the fair value of our common stock was no greater than $8.00 per share.
The following table summarizes, by grant date, the number of stock options granted since January 1, 2012 and the associated per share exercise price for each grant:
| Option Grant Dates |
Number of Shares Underlying Options |
Exercise Price Per Share |
||||||
| April 1, 2012 |
655,043 | $ | 0.43 | |||||
| April 25, 2012 |
60,695 | 0.43 | ||||||
| September 26, 2012 |
1,535,463 | 8.00 | ||||||
| December 7, 2012 |
87,448 | 8.00 | ||||||
| March 4, 2013 |
121,854 | 8.00 | ||||||
| May 13, 2013 |
262,343 | 8.00 | ||||||
| June 30, 2013 |
133,222 | 8.00 | ||||||
| July 15, 2013 |
50,175 | 8.00 | ||||||
| September 9, 2013 |
187,190 | 10.50 | ||||||
| September 18, 2013 |
75,000 | 10.50 | ||||||
| December 9, 2013 |
141,608 | 8.00 | ||||||
| February 12, 2014 |
61,653 | 8.00 | ||||||
57
Table of Contents
On February 3, 2014, all options granted by us in September 2013 and still outstanding at that time were re-priced from $10.50 to $8.00. In setting the exercise price, our board of directors considered the January 24, 2014 financing in which our senior redeemable convertible preferred stock was sold for $8.00 per share to new investors in an arms-length transaction and determined that the fair value of our common stock was no greater than $8.00 per share.
As discussed below, the common stock fair value per share for the April 1 and April 25, 2012 grants was $0.43, for the September 26 and December 7, 2012 grants was $1.17, for the March 4, 2013 grant was $6.85, for the May 13, June 30, and July 15, 2013 grants was $6.82, for the September 9 and September 18, 2013 grants was $9.94, and for the December 9, 2013 grants was $6.77. The intrinsic value of all outstanding options as of December 31, 2013, would have been $ , based on the IPO price per share of $ , the midpoint of the price range set forth on the cover of this prospectus, of which approximately $ and $ relates to vested and unvested options, respectively.
In connection with the preparation of the consolidated financial statements, we determined the estimated fair value of our common stock using methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants, or AICPA, Audit and Accounting Practice Aid Series: Valuation of Privately Held Company Equity Securities Issued as Compensation, or the AICPA Practice Guide. Many objective and subjective factors, along with significant judgment by our board of directors, were utilized to determine the fair value of our common stock. The key factors primarily consisted of:
| • | the prices at which we sold shares of preferred stock; |
| • | the superior rights, privileges and preferences of the preferred stock relative to our common stock at the time of each grant; |
| • | the lack of an active public market for our common and our preferred stock; |
| • | the stage of clinical development of the ELAD System; |
| • | the financial forecasts utilized to determine future cash balances and expected near-term and long-term capital requirements; |
| • | the selection of appropriate market comparable transactions; |
| • | an assessment of current financing and biopharmaceutical industry environments, |
| • | the probability and timing of various possible liquidity events; |
| • | the estimated weighted-average cost of capital; and |
| • | the discount for lack of marketability of our common stock. |
There is inherent uncertainty in these estimates and if we had made different assumptions that those described above, the fair value of the underlying common stock and amount of our stock-based compensation expense, net loss and net loss per share amounts would have differed.
December 31, 2012 Valuation Analysis
During the first quarter of 2013, we commenced an analysis to determine the enterprise value of our company and the fair value of our common stock as of December 31, 2012, which utilized an Option Pricing Model, or OPM. OPM is appropriate to use when the range of possible future outcomes is so difficult to predict that any such forecast would be highly speculative, or there is a substantially contemporaneous sale of stock to a third party. OPM treats common stock and convertible preferred stock as call options on the enterprise value, with exercise prices based on the liquidation preference of the convertible preferred stock. Therefore, the common stock has value only if the funds available for distribution to the stockholders exceed the value of the
58
Table of Contents
liquidation preference to be received by holders of our redeemable convertible preferred stock at the time of a liquidity event, such as a merger, sale or IPO, assuming the enterprise has funds available to make a liquidation preference meaningful and collectible by the stockholders. The common stock is modeled to be a call option with a claim on the enterprise at an exercise price equal to the remaining value immediately after the convertible preferred stock is liquidated. OPM uses the BSM option pricing model to price the call option.
The OPM considered the relative rights and preferences of the various securities, including the seniority of the liquidation preferences for preferred equity, and the potential for dilution caused by the conversion of the preferred equity. Additionally, our preferred stockholders have various rights that give them greater control and influence over future liquidity, financing and other decisions relating to our company than the holders of our common stock. The OPM applied a percentage of participation for each class of shares for each valuation interval based on BSM option pricing models, and a 25.4% discount for lack of marketability was applied for the common stockholders. The resulting implied per share value of our common stock was $1.17 per share as of December 31, 2012, which was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in September and December 2012 at an exercise price of $8.00 per share.
2013 Valuation Analyses
Due to our management’s and board of directors’ decision to pursue an IPO, coupled with our belief that we could reasonably estimate the form and timing of potential liquidity events, we utilized a Probability Weighted Expected Return Method, or PWERM, for our 2013 valuations. Under this method, the implied fair value of our common stock is estimated based upon an analysis of future values assuming various outcomes. The value is based on the probability-weighted present value of expected future investment returns considering each of the possible outcomes available to us as well as the rights of each share class. The possible outcomes considered are based upon an analysis of future scenarios as described below:
| • | closing of an initial public offering; |
| • | sale to a strategic acquirer; |
| • | continuation as a private company with subsequent liquidation event; and |
| • | dissolution. |
Critical assumptions required to perform the PWERM include the following:
| • | Scenarios: Expected future events were identified. |
| • | Scenario probabilities: The probabilities of occurrence of each event were estimated. |
| • | Valuation: Expected future values under each scenario were estimated. |
| • | Timing: Expected timing to the event under each scenario were estimated. |
| • | Risk adjusted discount rates: Risk-adjusted discount rates were selected for each equity class based on the rights and preferences of each equity class and market data. |
| • | Discounts: Appropriate minority or marketability discounts, if any, required to estimate the per share value of the various equity classes were determined. |
In determining the implied fair value of our common stock in the IPO scenario at each valuation date, we assumed that the redeemable convertible preferred stock then outstanding would be converted into common stock. In allocating value to our common stock in the merger or sale scenario, we first allocated to our outstanding shares of redeemable convertible preferred stock the greater of the liquidation preference of the redeemable convertible preferred stock and the amount that would have been payable had all such shares of redeemable convertible preferred stock been converted to common stock.
59
Table of Contents
March 31, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by December 31, 2013 |
25 | % | ||
| Sale by December 31, 2013 |
20 | % | ||
| Private company |
30 | % | ||
| Dissolution |
25 | % | ||
A 13% discount for lack of marketability was applied for common stockholders. The resulting implied fair value of $6.85 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in March 2013 at an exercise price of $8.00 per share.
June 30, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by November 15, 2013 |
35 | % | ||
| Sale by November 15, 2013 |
10 | % | ||
| Private company |
30 | % | ||
| Dissolution |
25 | % | ||
A 9% discount for lack of marketability was applied for common stockholders. The resulting implied fair value of $6.82 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in May, June, and July 2013 at an exercise price of $8.00 per share. The decrease in implied fair value of our common stock from March 31, 2013, was primarily due to dilution from the issuance of substantially more shares of our senior redeemable convertible preferred stock during the second quarter of 2013 that have superior rights to our common stock.
September 3, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by November 15, 2013 |
50 | % | ||
| Sale by November 15, 2013 |
10 | % | ||
| Private company |
25 | % | ||
| Dissolution |
15 | % | ||
A 7% discount for lack of marketability was applied for common stockholders. The resulting implied fair value of $9.94 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in September 2013 at an exercise price of $10.50 per share. The implied fair value of our common stock increased from June 30, 2013, primarily due to the increased likelihood of an IPO scenario as a result of progress made toward a public offering. In addition, we decreased our discount for lack of marketability reflecting a decrease in the expected time to liquidity.
60
Table of Contents
September 30, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by November 15, 2013. |
50 | % | ||
| Sale by November 15, 2013 |
10 | % | ||
| Private company |
25 | % | ||
| Dissolution |
15 | % | ||
A 6% discount for lack of marketability was applied for common stockholders, which resulted in an implied fair value of $10.02 per share. This increase in implied fair value of our common stock from September 3, 2013 was associated with a slight decrease in our discount for lack of marketability due to a decrease in the expected time to liquidity. There were no changes to our probability-weighted scenarios as no significant changes occurred from our September 3, 2013 valuation analysis.
December 1, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by May 15, 2014 |
20 | % | ||
| Sale by September 30, 2015 |
10 | % | ||
| Private company |
50 | % | ||
| Dissolution |
20 | % | ||
A discount for lack of marketability was applied for common stockholders of 10%, 21% and 28% for the IPO, sale and private company scenarios, respectively. The resulting implied fair value of $6.77 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in December 2013 at an exercise price of $8.00 per share. The decrease in fair value of our common stock from September 30, 2013 was primarily related to the increases in discount for lack of marketability due to an increase in the expected time to a liquidity event associated with the projected timing of an initial public offering or a sale of our company.
December 31, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by May 15, 2014 |
20 | % | ||
| Sale by September 30, 2015 |
10 | % | ||
| Private company |
50 | % | ||
| Dissolution |
20 | % | ||
A discount for lack of marketability was applied for common stockholders of 9%, 20% and 28% for the IPO, sale and private company scenarios, respectively, which resulted in an implied fair value of $5.93 per share. The decrease in fair value of our common stock from December 1, 2013 was primarily related to dilution from the issuance of additional shares of our senior redeemable convertible preferred stock in December 2013, partially offset by a slight decrease in discount for lack of marketability for the IPO and sale scenarios reflecting a decrease in the expected time to liquidity.
Income Taxes
Income taxes are provided for tax effects of transactions reported in the financial statements and consist of income taxes currently due plus deferred income taxes related to temporary differences between the basis of certain
61
Table of Contents
assets and liabilities for financial statement purposes and for income tax reporting purposes. Deferred taxes are determined based on the difference between the financial statement value and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse. A valuation allowance is provided if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. Based on our analysis of both positive and negative factors, we have determined that it is more likely than not that we will not be able to realize our deferred tax assets, and therefore we have recorded a full valuation allowance against our deferred tax assets. Our analysis included an assessment of our lack of profitability and our future projections of forecasted revenue and expense levels.
As of December 31, 2013, we had federal and state net operating losses, or NOLs, of approximately $37.3 million and $35.8 million, respectively, which begin to expire in 2023 and 2014, respectively, for federal and state tax purposes.
Under Section 382 of the Internal Revenue Code, or the Code, substantial changes in our ownership may limit the amount of NOLs that could be utilized annually in the future to offset taxable income, if any. Specifically, this limitation may arise in the event of a cumulative change in ownership of our company of more than 50% within a three-year period as determined under the Code, which we refer to as an ownership change. Any such annual limitation may significantly reduce the utilization of these NOLs before they expire. We believe that a change in ownership, as defined in Section 382, occurred in February 2012. As a result, the deferred tax asset associated with our federal and state net operating loss carryovers and federal research credits has been reduced based on the Section 382 limitations. The amount of the reduction in our deferred tax assets is based on the estimated amount of the net operating loss carryover and federal research credits we believe cannot be used as a result of estimated Section 382 limitations. As a result, we have reduced our deferred tax assets by $27.7 million and have estimated that approximately $58.7 million and $59.6 million of our federal and state NOLs, respectively, cannot be used in future years as a result of this change in ownership. Additionally, we have estimated that approximately $2.2 million and $1.6 million of our federal and state research credits carryover, respectively, cannot be used in future years. Exact computations of limitations under Section 382 have not been completed, but the amounts reflect our best estimate of limitations and net operating losses and credits available in future years. We have not determined if other changes under Section 382 subsequent to the February 2012 have occurred. We expect the issuance of common stock in this offering, together with the issuance of common stock in any other transactions involving our common stock, may result in an additional ownership change, which could further limit the amount of the NOLs we may use to offset future taxable income, if any.
Results of Operations
Comparison of Years Ended December 31, 2012 and 2013
The $16.7 million increase in research and development expense during the year ended December 31, 2013, as compared to the year ended December 31, 2012, was primarily attributable to increases of:
| • | $6.7 million in fees paid to CROs, clinical sites, and other costs related to our VTI-208 trial activities, principally due to patient enrollment that commenced in March 2013; |
| • | $4.5 million in salaries and wages, stock-based compensation, and other compensation related costs due to an increase in headcount from 26 employees in 2012 to 57 employees in 2013; |
| • | $2.5 million in manufacturing supplies primarily related to lab supplies and consumables associated with the VTI-208 trial; |
| • | $1.7 million in consulting fees related to professional services associated with the VTI-208 trial; |
| • | $0.7 million in travel costs to support our clinical trial activities; and |
| • | $0.4 million of allocated facilities costs attributable to additional clinical office space in 2013. |
62
Table of Contents
The $5.1 million increase in general and administrative expense during the year ended December 31, 2013, as compared to the year ended December 31, 2012, was primarily attributable to increases of:
| • | $2.1 million in salaries and wages, stock-based compensation, and other compensation related expenses due to increased headcount to support our operations; |
| • | $1.0 million in consulting fees primarily related to professional services for accounting, marketing and information technology consultants; |
| • | $0.7 million in expenses related to executive and board member recruitment; and |
| • | $0.5 million in facilities related to additional office space in 2013. |
The $413,000 decrease in interest expense for 2013, as compared to 2012, was attributable to no outstanding convertible notes or loans during 2013. Our convertible notes were converted into shares of our senior preferred stock in September 2012 and our term loan was repaid in October 2012.
The $180,000 decrease in revaluation of preferred stock warrant liabilities was attributable to the final valuation adjustment of our preferred stock warrant liabilities in 2012, as these warrants were converted to common stock warrants in connection with the junior preferred stock offering in February 2012.
The $1.3 million recognized for the revaluation of future purchase rights liabilities for the year ended December 31, 2013 was the result of an increase in the estimated fair value of future purchase rights liabilities associated with our senior preferred stock financings and an increase during 2013 in the number of shares available under such rights from December 31, 2012 to December 31, 2013. The increase in the estimated fair value of the future purchase rights was primarily the result of an increase to our enterprise value offset in part by an increase in the number of shares of senior preferred stock outstanding as of December 31, 2013 as compared to the prior year.
Liquidity and Capital Resources
To date, we have not generated significant revenues attributable to the ELAD System. We have a history of incurring losses and negative cash flows from operations and, as of December 31, 2013, we had a deficit accumulated during the development stage of $103.2 million. We expect that our research and development and general and administrative expenses will continue to increase and, as a result, we will need additional capital to fund our operations, which we may seek to obtain through a combination of equity offerings, debt financings, government or other third-party financing, marketing and distribution arrangements and other collaborations, strategic alliances and licensing arrangements.
Since our inception through December 31, 2013, we have funded our operations principally with net proceeds of $140.3 million from the issuance of convertible notes, preferred stock, common stock, and warrants. As of December 31, 2013, we had cash and cash equivalents of approximately $38.2 million. Cash in excess of immediate requirements is invested in accordance with our investment policy, primarily with an intent to maximize liquidity and preserve capital. As of December 31, 2013, such funds were held in cash and money market funds.
The following table shows a summary of our cash flows for each of the two years ended December 31, 2012 and 2013.
| Year Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| (In thousands) | ||||||||
| Cash (used in) provided by: |
||||||||
| Operating activities |
$ | (9,244 | ) | $ | (28,648 | ) | ||
| Investing activities |
(14,588 | ) | 11,909 | |||||
| Financing activities |
27,501 | 50,445 | ||||||
63
Table of Contents
Net cash used in operating activities
During the year ended December 31, 2013, operating activities used $28.6 million of cash. The use of cash primarily related to our net loss of $32.7 million, partially offset by $3.0 million of non-cash charges related to the revaluation of future purchase rights, depreciation, deferred rent, and stock-based compensation and $1.0 million of net changes in our operating assets and liabilities. Net cash provided by changes in our operating assets and liabilities during the year ended December 31, 2013 consisted primarily of an increase of $2.2 million in accounts payable and accrued liabilities, reflecting an increase in clinical activities and the timing of payments made by us to vendors, partially offset by an increase in prepaid clinical costs of $841,000, an increase in lease deposits of $198,000, and an increase of $102,000 related to other assets.
Net operating cash used in the year ended December 31, 2012 was $9.2 million. The use of cash during 2012 primarily related to our net loss of $6.7 million, $473,000 of net changes in our operating assets and liabilities, and adjustments to our net loss of $180,000 due to the noncash revaluation of the preferred stock warrant liabilities, partially offset by $1.2 million of noncash charges related to depreciation, interest, and stock-based compensation. Changes in our operating assets and liabilities consisted primarily of a decrease of $231,000 in accounts payable and accrued liabilities, due primarily to the timing of payments made by us to vendors, and an increase in prepaid expenses of $242,000 due primarily to increases in prepaid clinical costs of $54,000.
Net cash (used in) provided by investing activities
During the year ended December 31, 2013, investing activities provided $11.9 million of cash, primarily related to sales of short-term investments of $17.0 million, partially offset by $3.0 million of purchases of short-term investments, as well as $1.5 million in purchases of capital equipment and a $608,000 increase in restricted cash requirements primarily related to our clinical trial obligations and lease arrangements.
During the year ended December 31, 2012, investing activities used $14.6 million of cash due to the purchase of $14.0 million in short-term investments, capital equipment purchases of $261,000, and an increase in restricted cash requirements of $355,000 associated with clinical trial obligations and potential corporate wind-down and employee related expenses.
Net cash provided by financing activities
During the year ended December 31, 2013, financing activities provided $50.4 million of cash, compared to $27.5 million during the year ended December 31, 2012. The increase of cash during 2013 primarily related to the sale of additional senior preferred stock for $53.2 million, net of offering costs. These increases were partially offset by deferred financing costs of $3.1 million. Our 2012 financing activities included the sale of senior preferred stock for $21.1 million, net of offering costs, and proceeds of $6.9 million from a loan that converted to senior preferred stock during 2012. These financings were partially offset by principal payments of $533,000 on our term loan.
Funding Requirements
We have not completed development of the ELAD System. We expect to continue to incur significant expenses and increasing operating losses for at least the next few years.
As of December 31, 2013, we had $38.2 million in cash and cash equivalents. In January 2014, we issued 2.1 million shares of our senior redeemable convertible preferred stock for aggregate proceeds of $16.4 million and, in February 2014, we issued 241,016 shares of our senior redeemable convertible preferred stock pursuant to a preemptive rights offering for aggregate proceeds of $1.9 million. As of February 28, 2014, we had $51.3 million in cash and cash equivalents, of which $1.2 million was restricted. We believe that our existing cash and cash equivalents as of February 28, 2014 will be sufficient to meet our anticipated cash requirements for at least the next 13 months. However, our forecast of the period of time through which our financial resources
64
Table of Contents
will be adequate to support our operations is a forward-looking statement that involves risks and uncertainties, and actual results could vary materially.
Our future capital requirements are difficult to forecast and will depend on many factors, including:
| • | the scope, progress, results and costs of research and development and clinical trials related to the ELAD System or any future product candidates; |
| • | the cost of scaling up and validating the manufacturing process for the ELAD System or any other product candidates for commercialization; |
| • | the cost of commercialization activities, including reimbursement, marketing, sales and distribution costs, both before and after product approval (if any); |
| • | our ability to maintain existing collaborations and to establish new collaborations, licensing or other arrangements and the financial terms of such agreements; |
| • | the number and characteristics of any future product candidates we pursue; |
| • | the costs involved with being a public company; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patents, including litigation costs and the outcome of such litigation; and |
| • | the timing, receipt and amount of sales of, or royalties on the ELAD System and any future product candidates. |
Contractual Obligations and Commitments
Our most significant clinical trial expenditures are to investigative sites and to CROs. The agreements are cancellable by either party at any time upon written notice, but obligate us to reimburse the providers for any time or costs incurred through the date of termination and do not have any cancellation penalties. These items are not included in the table below. We lease office and manufacturing space in San Diego, California. The following table summarizes our contractual obligations at December 31, 2013 and the effect such obligations are expected to have on our cash flow in future periods:
| Payments Due by Period | ||||||||||||||||||||
| Total | Less Than 1 Year |
2-3 Years |
3-5 Years |
More Than 5 Years |
||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Operating lease obligations |
$ | 3,023 | $ | 840 | $ | 1,692 | $ | 491 | $ | — | ||||||||||
| Purchase obligations |
2,410 | 2,410 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total contractual obligations |
$ | 5,433 | $ | 3,250 | $ | 1,692 | $ | 491 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
As of December 31, 2013, our purchase obligations included existing purchase commitments for future minimum payments of $192,000 with a vendor for raw materials that will be manufactured and utilized on an as needed basis. During the years ended December 31, 2012 and 2013, we purchased $462,000 and $724,000, respectively, of materials from this vendor.
As of December 31, 2013, our purchase obligations included a purchase order with a vendor for cartridges that will be manufactured and delivered on an agreed upon schedule during 2014 for future payments of $1.5 million, exclusive of prepayments totaling $95,000. If we cancel any future shipment, we would be required to pay 50% of the scheduled invoice amount for that shipment.
As of December 31, 2013, we had a purchase order with a vendor for $693,000 in equipment to be received and paid for in 2014. Orders in process may be canceled only with vendor’s consent and upon payment of the vendor’s cancellation charges.
65
Table of Contents
Quantitative and Qualitative Disclosures About Market Risk
Interest Rate Sensitivity
We had cash and cash equivalents of $38.2 million at December 31, 2013, which was held for working capital purposes. We do not enter into investments for trading or speculative purposes. We do not believe that we have any material exposure to changes in the fair value of these investments as a result of changes in interest rates due to their short-term nature. Declines in interest rates, however, will reduce future investment income.
Foreign Currency Risk
To date, our international agreements have been denominated almost exclusively in U.S. dollars. Accordingly, we have a limited, but increasing, exposure to foreign currency exchange rates. To date, we have not entered into, and do not have any current plans to enter into, any foreign currency hedging transactions or derivative financial transactions. We expect our transactions outside of the United States in the near-term will primarily entail payments for clinical trials, and for vendors and consultants supporting those trials within Europe and Australia, which may increase our exposure to foreign currency risk in future periods. We do not expect to maintain any significant amount of assets outside of the United States.
The functional currency of our foreign subsidiaries are the local currencies. Accordingly, the effects of exchange rate fluctuations on the net assets of these operations are accounted for as translation gains or losses in accumulated other comprehensive income within stockholders’ equity. We do not believe that a change of 10% in such foreign currency exchange rates would have a material impact on our financial position or results of operations.
Our balance sheet as of December 31, 2013 includes cash and cash equivalent balances of $127,000 denominated in renminbi at our Chinese subsidiary, VTI China. The majority of VTI China’s operational activities are denominated and transacted in renminbi with the exception of intercompany investments and loans that are transacted in U.S. dollars. Exchange rate losses recognized for all periods presented were insignificant. We plan to contract directly or through wholly-owned foreign subsidiaries with clinical investor sites, CROs and consultants outside the United States based primarily on tax and liability considerations. Accordingly, we may be subject to fluctuations in foreign currency rates in connection with certain of these agreements. Transactions denominated in currencies other than the functional currency are recorded based on exchange rates at the time such transactions arise. As of December 31, 2012 and 2013, substantially all of our total liabilities were denominated in the functional currency.
Effects of Inflation
We do not believe that inflation and changing prices had a significant impact on our results of operations for any periods presented herein.
Recently Issued and Adopted Accounting Pronouncements
In February 2013, the FASB issued ASU No. 2013-02, Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income. ASU 2013-02 requires an entity to provide information about the amounts reclassified out of accumulated other comprehensive income by component. In addition, an entity is required to present, either on the face of the statement where net income is presented or in the notes, significant amounts reclassified out of accumulated other comprehensive income by the respective line items of net income but only if the amount reclassified is required under GAAP to be reclassified to net income in its entirety in the same reporting period. ASU 2013-02 is effective prospectively for reporting periods beginning after December 15, 2012. We adopted ASU 2013-02 as of January 1, 2013 and the adoption did not have a material impact on our financial condition or results of operations.
66
Table of Contents
In July 2013, the FASB issued ASU No. 2013-11, Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists, which requires an unrecognized tax benefit, or a portion of an unrecognized tax benefit, to be presented in the financial statements as a reduction to a deferred tax asset for a net operating loss (NOL) carryforward, a similar tax loss, or a tax credit carryforward. If an NOL carryforward, a similar tax loss, or a tax credit carryforward is not available at the reporting date under the tax law of the applicable jurisdiction to settle any additional income taxes that would result from the disallowance of a tax position or the tax law of the applicable jurisdiction does not require the entity to use, and the entity does not intend to use, the deferred tax asset for such purpose, the unrecognized tax benefit should be presented in the financial statements as a liability and should not be combined with deferred tax assets. The new guidance is effective for fiscal years beginning after December 15, 2013 and earlier adoption is permitted. We are currently evaluating the impact of the new guidance; however, management does not believe it will have a material effect on our financial position or results of operations.
Internal Controls and Procedures
In connection with past audits of our financial statements, our independent registered public accounting firm identified and reported adjustments to management. Certain of these adjustments were deemed to be the result of internal control deficiencies that constitute material weaknesses in our internal control over financial reporting. If one or more material weaknesses persist or if we fail to establish and maintain effective internal control over financial reporting, our ability to accurately report our financial results could be adversely affected.
We have not maintained an effective control environment in that the design and execution of our controls was limited due to the lack of proper segregation of duties resulting from inadequate staffing levels, the ineffective review over financial transactions and the maintenance of our books and records. The lack of adequate staffing levels resulted in insufficient time spent on review and approval of certain information used to prepare our financial statements and the maintenance of effective controls to adequately monitor and review significant transactions for financial statement completeness and accuracy. Such examples include correct financial statement classification, the valuation of financing transactions entered into during the period, and maintenance of documentation in support of such transactions. These control deficiencies, although varying in severity, contributed to the material weaknesses in the control environment noted by our independent registered public accounting firm.
Although remediation efforts are still in progress, management is taking steps to address the causes of our audit adjustments and to improve our internal control over financial reporting, including the implementation of new accounting processes and control procedures and the identification of gaps in our skills base and expertise of the staff required to meet the financial reporting requirements of a public company. We have hired additional accounting personnel who are degreed accountants, which has enabled us to expedite our month-end close process, thereby facilitating the timely preparation of financial reports and strengthen our segregation of duties. We intend to hire incremental qualified staff as part of a comprehensive review of our internal controls and formalization of our review and approval processes.
We may identify additional control deficiencies, which could give rise to significant deficiencies and other material weaknesses in addition to the material weaknesses previously identified.
We will be required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting for the first fiscal year beginning after the closing of this offering. This assessment will need to include disclosure of any material weaknesses identified by management over our internal control over financial reporting.
We are in the very early stages of the costly and challenging process of compiling the system and processing documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing or any required remediation in a timely fashion. During the evaluation and
67
Table of Contents
testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal controls are designed and operating effectively, which could result in a loss of investor confidence in the accuracy and completeness of our financial reports. This could cause the price of our common stock to decline and we may be subject to investigation or sanctions by the SEC.
We will be required to disclose changes made in our internal controls and procedures on a quarterly basis. However, our independent registered public accounting firm will not be required to report on the effectiveness of our internal control over financial reporting pursuant to Section 404 until the later of the year following our first annual report required to be filed with the SEC or the date we are no longer an “emerging growth company” if we take advantage of the exemptions contained in the JOBS Act. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied that our internal controls over financial reporting are designed and operating effectively to prevent or detect a material misstatement to the financial statements. To comply with the requirements of being a public company, we may need to undertake various actions, such as implementing new internal controls and procedures or hiring accounting or internal audit staff.
Off Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under SEC rules.
68
Table of Contents
Overview
We are a biotherapeutic company focused on developing a cell-based therapy targeting treatment of all forms of acute liver failure. Our product candidate, the ELAD System, is an extracorporeal bio-artificial liver therapy designed to allow the patient’s own liver to regenerate to a healthy state or to stabilize the patient until transplant. The ELAD System is the only bio-artificial liver support system containing immortal human liver-derived cells, or C3A cells, to enter Phase 3 clinical trials. We designed the ELAD System to supplement key aspects of normal liver function to improve patient survival. We estimate that at least 30,000 patients annually in the United States experience forms of acute liver failure, such as acute-on-chronic, surgery-induced and fulminant liver failures, for which the ELAD System may be a life-saving therapy. Outside of liver transplant, which is severely limited by availability of organs and not available to many patients, the current standard-of-care for acute liver failure is primarily focused on the management of complications, which does not restore lost liver function and is associated with a high rate of mortality. The ELAD System has received orphan designation in the United States and Europe for the treatment of patients with acute liver failure. This designation provides tax credits for qualified clinical testing, seven years of market exclusivity in the United States, and ten years of market exclusivity in Europe for the first approved orphan drug for a given indication. However, orphan designation does not alter the standard regulatory requirements or the process for obtaining marketing approval.
We are currently enrolling patients in one Phase 3 clinical trial, and have regulatory allowance to begin enrolling patients in a second Phase 3 trial, each in forms of acute liver failure. In March 2013, we initiated VTI-208, a Phase 3 randomized, controlled clinical trial in 200 subjects with AILD. In addition, we have obtained regulatory allowance in the United States, United Kingdom, Spain and Australia to begin enrolling patients and have initiated clinical sites in a second Phase 3 randomized, controlled clinical trial, VTI-210, in 120 subjects with severe AAH, which is a subset of AILD. We expect the enrollment of subjects in VTI-210 to begin in the first half of 2014.
These studies are designed to complement each other and confirm study outcomes, and may be combined to support product registration in the United States and the European Union, or E.U. In addition, based upon discussions with United States and European regulatory authorities, we believe the VTI-208 and VTI-210 clinical trials, if successful from both a statistical and clinical standpoint, may support product registration on a stand-alone basis. According to the FDA, a second confirmatory clinical trial that substantiates positive results may be necessary to support a Biologics License Application, or BLA. EMA has informed us that the VTI-210 trial, if deemed successful, will be sufficient to support product registration in the E.U. We designed VTI-208 with input from the FDA to support product registration in the United States. Similarly, we designed VTI-210 with input from the EMA to support product registration in Europe. We currently anticipate having Phase 3 clinical trial data from VTI-208 in the first half of 2015.
In the second half of 2014, we also expect to initiate enrollment of VTI-212, a Phase 2/3 clinical program in subjects with either FHF or SILF. We plan to begin this program with a Phase 2 single-arm component enrolling 40 subjects, which may later be followed by a randomized, controlled Phase 3 component the size of which has not yet been determined. Results from the single-arm component will be compared with historical or case-matched controls, and we currently anticipate Phase 2 data in 2015 or 2016. Since FHF and SILF have high mortality rates and affect a very small number of patients for which there is currently no satisfactory therapeutic intervention available, the results from the Phase 2 single arm component may provide support for an expedited regulatory approval pathway. However, regulatory agreement on an expedited regulatory pathway has not yet been sought and may never be granted. In the event that randomized data are necessary for approval in FHF and SILF, we expect to perform the randomized Phase 3 portion of the program, the design of which would be finalized upon analysis of the Phase 2 component. Data from VTI-212 may also be used to support our planned marketing applications for AILD and AAH.
The ELAD System consists of customized disposable sets and a reusable bedside unit attached to four disposable cartridges containing our human liver-derived C3A cells, or VTL C3A cells. The bedside unit is based
69
Table of Contents
on a cardio-pulmonary bypass machine, which we have configured for the ELAD System therapy. This bedside unit and customized disposable sets are attached to the cartridges where the patient’s blood plasma is treated by our VTL C3A cells before being returned to the patient. The four ELAD cartridges collectively contain 440 grams, or approximately one pound, of VTL C3A cells from our proprietary cell bank. We employ proprietary methods designed to manufacture large quantities of these cells consistently and cost-effectively. These cells have been shown to retain many key synthetic and metabolic processes of normal human hepatocytes, the primary functional cell of the liver.
During the ELAD System therapy, blood plasma is drawn from the patient through a central venous line and then passes into the cartridges via our bedside unit. Therapy is expected to consist of a single session of continuous allogeneic cellular therapy lasting between three and ten days. As the treatment with our VTL C3A cells is not patient-specific, we avoid the costly logistical and production challenges typically associated with autologous cellular therapies.
The ELAD System has shown trends indicating the potential to increase survival rates in acute liver failure. Prior to the initiation of our ongoing Phase 3 clinical trial programs, over 145 subjects have received the ELAD System therapy, including earlier iterations of the ELAD System, in seven clinical trials and through a compassionate use program. Data from a 69 subject randomized, controlled clinical trial performed at two hospital centers in Beijing, China showed a statistically significant difference in 28-day and 56-day transplant-free survival rates in the ELAD System-treated group as compared with the control group using the pre-defined logrank test, a statistical test that compares the survival distributions of the two study groups. This technique is widely used in clinical trials to document the efficacy of a new treatment compared to a control treatment when the measurement is the time to event (such as the time from initial treatment to death, or to a liver transplant). Data from a subset comprising the first 49 subjects in this clinical trial revealed a statistically significant difference in 84-day survival using a more sensitive analytical technique, the Wilcoxon rank-sum test. This test is another statistical method used in the analysis of clinical trial data that compares two populations, for example, in respect to their survival times. Additionally, subsets of data from other Phase 1 and Phase 2 clinical trials showed a trend indicating that the ELAD System may increase survival rates.
Early clinical trials of the ELAD System carried out prior to the current pivotal trial program were pilot in nature and designed to identify patient populations and clinical trial designs that were appropriate and necessary to pursue in pivotal clinical studies to support regulatory approval in the United States and Europe. Although they demonstrated trends towards increased survival, to date none of our prior trials were designed to adequately power or demonstrate statistically significant increased survival and the FDA has noted its view that our preliminary clinical evidence, at this time, does not indicate that the ELAD System may demonstrate a substantial improvement over the current standard-of-care. For each of the Phase 3 clinical trials to demonstrate efficacy, a statistical plan has been generated, and the studies have been powered with a sufficient number of subjects, such that, should the survival benefits be consistent with those from prior smaller studies, the sample size will be adequate to prove efficacy within statistical criteria traditionally acceptable to the FDA. We have also designed these studies with guidance from the FDA and with the intent of minimizing bias, to the extent possible, in an open label study design. These plans have been shared with the FDA.
Other artificial liver devices to reach late-stage clinical trials have been either acellular or contained pig cells. We believe that human liver cells are necessary to allow an artificial liver to replicate key aspects of the intricate biology of normal human liver function.
We own exclusive worldwide commercial rights to the ELAD System free of royalties. If our marketing applications are approved, we intend to commercialize the ELAD System in the United States and Europe with a targeted sales force. We intend to opportunistically pursue markets outside the United States and Europe either through direct sales or collaborations. We also believe that the ELAD System may have potential use for viral hepatitis, liver resection support and liver transplant support, although we have generated limited clinical data to support these indications.
70
Table of Contents
We are currently the owner of record of four U.S. patents and over a dozen issued or allowed foreign patents, including in Europe and Australia. One granted U.S. patent claims a method of using C3A cells to treat a patient’s blood and is set to expire in April 2027 unless extended further. In addition, a second U.S. patent claims an extracorporeal device configuration, which we believe to include our ELAD System, independent of cell-type used and is set to expire in May 2025, unless extended further. Moreover, if approved, the ELAD System will be eligible for 12 years of data exclusivity in the United States under the Biologics Price Competition and Innovation Act of 2012. Finally, orphan designation provides market exclusivity for seven years in the United States and ten years in Europe upon regulatory approval.
Our Strategy
Our goal is to become the leading biotherapeutic company developing and marketing cell-based therapies targeting the treatment of acute liver failure. Key elements of our strategy to achieve this goal are:
| • | Successfully complete the ELAD System’s clinical development in acute liver failure. In March 2013, we began enrollment in VTI-208 for AILD and we expect to begin enrollment in VTI-210 for AAH in the first half of 2014 and VTI-212 for FHF and SILF in the second half of 2014. We anticipate having data from the VTI-208 Phase 3 trial for AILD in the first half of 2015. |
| • | Obtain regulatory approval for the ELAD System in the United States and Europe. If our VTI-208 or VTI-210 Phase 3 clinical trials are statistically and clinically successful, we plan to submit a BLA to the FDA and a Marketing Authorization Application, or an MAA, to the EMA, in 2016 for the indications of AILD and AAH, respectively. If the Phase 2 and or Phase 3 component of VTI-212 provides compelling evidence of the safety and efficacy of the ELAD System in FHF and SILF we plan to seek agreement from the regulatory authorities regarding the next steps to be taken in order to enable marketing authorization, which may or may not involve an additional randomized controlled Phase 3 clinical trial either prior to or after contingent marketing approval. |
| • | Maximize the commercial potential of the ELAD System in the United States and Europe. If approved, we intend to directly commercialize the ELAD System in the United States and Europe with a targeted sales force focusing on liver transplant and other specialist intensive care centers. We believe that we can price the ELAD System in a range consistent with other currently marketed life saving therapies, such as left ventricular assist devices, orphan biologic medications for hereditary metabolic diseases, and monoclonal antibody medications for cancer. |
| • | Opportunistically explore commercial opportunities for the ELAD System in other international markets. We have completed a trial for the ELAD System that may be considered pivotal in China. The study predominantly enrolled subjects with viral hepatitis, and our application for marketing approval, filed in 2007, is still under review. However, we do not currently anticipate receiving approval in China prior to our receipt of FDA approval, if any, in the United States. We plan to address the commercial opportunity in China following approval in the United States and Europe. We also plan to evaluate commercial opportunities in Australia, Japan, India, other Asian markets, the Middle East, Brazil, and Africa. |
| • | Pursue development of the ELAD System in additional indications. If the ELAD System is approved for use in treating AILD, AAH, FHF or SILF, we anticipate pursuing the ELAD System’s clinical development in viral hepatitis and bridge-to-transplant. |
| • | Technical Improvements and New Applications. We plan to continue our development of the ELAD System with the incorporation of technical improvements to our bedside unit, customized disposable sets, and cells. In addition, we plan to explore the development of next generation systems and other uses for VTL C3A cells, including the potential commercialization of proteins and other C3A-produced compounds. |
71
Table of Contents
Our ELAD System Product Candidate
The ELAD System is a Phase 3 investigational, extracorporeal bio-artificial liver therapy designed to supplement hepatic function in order to improve survival rates among patients with acute liver failure. The ELAD System consists of a reusable bedside unit attached to four disposable cartridges, which collectively contain thousands of hollow fibers and approximately one pound of VTL C3A cells from our proprietary cell bank.
The below figure is a schematic of the ELAD System:
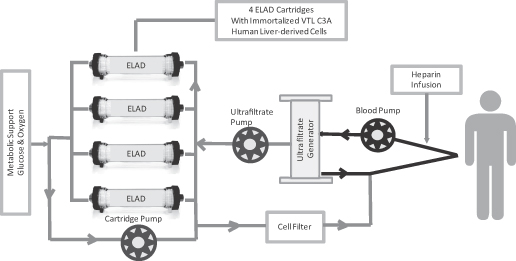
During ELAD System therapy, blood is drawn from the patient via a central venous line and then passes into the bedside unit where plasma ultrafiltrate is isolated by an ultrafiltrate generator. The patient’s plasma ultrafiltrate then passes into the four cartridges where it contacts our VTL C3A cells after passing through fibers which allow appropriate two-way transfer of toxins, metabolites and nutrients, mimicking liver function. The fibers, made of a semi-permeable membrane, permit passage of macromolecules and other substances from our VTL C3A cells to the patient’s plasma ultrafiltrate. At the same time, these fibers permit the passage of toxins such as ammonia and bilirubin and nutrients such as glucose and oxygen from the plasma ultrafiltrate to our VTL C3A cells. Heparin, a widely-available anti-coagulant drug, is administered during the ELAD System therapy in some patients to prevent blood-clotting in the ultrafiltrate cartridge.
Treated plasma ultrafiltrate is then filtered, reconstituted with blood cells and returned to the patient via the central venous line. Meanwhile, the bedside unit monitors and adjusts glucose and oxygen concentrations in the plasma ultrafiltrate as well as temperature in order to maintain the cells’ viability. Therapy is expected to consist of a single session of continuous treatment lasting between three and ten days, as determined by the treating physician.
Our Proprietary VTL C3A Cell Bank
The key to the performance of the ELAD System is our VTL C3A cell bank. The bank contains human liver-derived cells that can be grown in almost unlimited quantities, stored and shipped worldwide. These properties enable continuous patient treatment for ten days usually without changing the cartridges. Moreover, the immortal VTL C3A cells mimic certain functions of human liver cells and have been shown to retain many of the specific metabolic processes and pathways of hepatocytes. These functions include an active P450 enzyme system as well as the production of liver-specific proteins such as albumin, anti-thrombin III, alpha-fetoprotein, C3 complement, Factor V, transferrin, alpha-1-antichymotrypsin and alpha-1-antitrypsin.
Past attempts to develop a liver assist product based on human hepatocytes faced two problems: human hepatocytes could not be expanded in the large quantities needed for a commercial product and the cells lost their
72
Table of Contents
hepatocyte functions in a short time. This meant that products based on human hepatocytes were not scalable and could not provide the prolonged treatment necessary for liver support. In the late 1980’s, scientists at Baylor College of Medicine in Houston set out to develop a human hepatocyte cell line that could be grown in commercial quantities, ceased cell division on filling space available, retained key hepatocyte functions and survived therapy in human blood. Baylor entered into a cooperative program with The Wistar Institute to develop a new cell line. The parties recognized that the cell properties they sought would likely be found in a tumor cell and worked with the HepG2 cell line derived from a human hepatoblastoma, a mildly metastatic human liver tumor taken from a young person. This cell line was not homogeneous and they searched for a clone of HepG2, which displayed the properties they sought. The C3A cell satisfied most of the criteria that they had specified and they began work with C3A cells in a specially-designed bedside delivery system.
The C3A cell line has been studied by independent investigators who have confirmed its hepatocyte properties, such as the presence of a functional cytochrome P450 toxin-processing enzyme system and the production of many liver-specific proteins. However, the C3A cell line does not entirely mimic the behavior of primary hepatocytes. For instance, it has not been shown to process ammonia as effectively as primary hepatocytes do and C3A produces large amounts of alpha-fetoprotein, or AFP. We believe these issues are manageable since excess ammonia can be removed by dialysis if needed, and AFP is a non-toxic fetal analogue of albumin.
We have customized the now-publicly available C3A cell line we originally acquired from Baylor University to create a proprietary VTL C3A cell bank for use in the ELAD System, which we culture and expand through proprietary techniques. This cell bank represents a well-characterized stock of original cells that have been subjected to rigorous testing for viruses, pathogens and other contaminants in order to allow them to be used in humans. This bank contains enough cells to enable our clinical development and commercialization. We own this VTL C3A cell bank exclusively and on a royalty-free basis. In addition, we have developed proprietary methods for growing, storing and optimizing the function of these cells.
The ELAD System therapy is not patient specific and our VTL C3A cells, which are derived from a single source, are used to treat all patients. This process is known as allogeneic cellular therapy. In contrast, autologous cellular therapy uses a patient’s own cells, which are manipulated in individual production batches, and is a costly and complex process. As a result, the production and logistics of treatment with our VTL C3A cells does not face the challenges commonly associated with autologous cellular therapies.
Our proprietary VTL C3A cell bank is stored in three separate locations around the world for security purposes and is used as the basis for growing approximately one pound of cells needed for each patient at our production facility in San Diego, California.
Differentiating Factors of the ELAD System
Unlike other potential therapies developed for acute liver failure in the past, we believe the ELAD System has a unique combination of attributes:
| • | Biologically active. The ELAD System is a biologically active system that is designed to replicate many liver functions. The functional unit of the liver, the hepatocyte, is thought to be responsible for 500 or more biologic processes necessary for human life. Moreover, many processes of the liver occur at ever-fluctuating rates in response to the liver’s constantly changing biologic environment. As a result, we believe that an acellular solution to liver failure is unlikely to effectively replace lost liver function. We believe that a cellular approach, capable of replicating key biologic processes performed by a human hepatocyte, is best able to provide the requisite flexibility and breadth of function to sufficiently supplement liver function and improve survival in patients with acute liver failure. |
| • | Human cellular therapy. The ELAD System is based on human cells which confer a considerable advantage over non-human, animal-based cell therapies. Given the widespread availability of animal tissues, much work has been done on the use of animal liver cells, often derived from pigs, to treat |
73
Table of Contents
| humans with liver failure. While immunological risk is always present in cellular therapy, the use of non-human animal tissues presents greater immunological risk compared to human cellular therapy. Humans possess naturally occurring antibodies that react with antigens on porcine cell surfaces. These antibodies can mount an immediate attack in the presence of porcine cells, causing these cells to rapidly lose function and die. Moreover, repeated treatments with a porcine cell may cause subsequent immune responses to become increasingly severe. The infusion of porcine enzymes into a patient’s blood stream also poses immunologic risk. We are not aware of any FDA-approved non-human animal-based cellular therapy for use in patients. |
| • | Immortal human liver-derived C3A cells. Our VTL C3A cells used in the ELAD System are derived from a human hepatoblastoma and are immortal. They can be expanded and can survive a session of up to ten days of the ELAD System therapy, usually without needing to be replaced during treatment. Hepatocytes are the main cell in the liver and deteriorate rapidly when removed from the body. Hepatocytes cannot be grown outside the body as they de-differentiate and die in a short period of time. The inability to expand makes hepatocytes unsuitable for a liver assist device which requires large amounts of hepatocytes. |
| • | Commercially scalable. We have developed a process to grow our VTL C3A cell line in our facility in San Diego, California that is designed for scalable production. Each patient set of four cartridges contains a total of about one pound of cells and is grown in a production process that takes about six weeks. The process is carefully controlled and is performed under ultra-clean conditions to avoid contamination in our cGMP compliant production plant. The process is scalable by modular units. |
| • | Minimal manipulation needed by site. Prior to shipment, the ELAD cartridges are put into a dormant state and can survive up to 60 hours before being used for treatment. When the hospital receives the cartridges, they are unpacked by our ELAD System specialists on site, placed on the bedside unit, flushed with saline and are ready to be used for patient therapy. Since our VTL C3A cells are immortal, they can remain alive for the duration of anticipated patient therapy and the ELAD cartridges do not usually need to be replaced, thus enhancing ease-of-use and reducing cost-of-goods. |
Liver Failure
The liver performs a wide variety of vital life functions including metabolic, regulatory, detoxification and synthetic activities. The primary liver cell, the hepatocyte, is thought to be responsible for approximately 500 or more specific biologic processes. In addition, the liver also serves as a reservoir for immune cells which clear the blood of pathogens. As a result, the liver’s failure to perform its normal role can have devastating or fatal consequences. Causes of liver failure are numerous, and the condition is typically described in terms of rapidity of onset. The two main categories are acute liver failure and chronic liver failure. Liver disease represents the twelfth leading cause of death in the United States. In China, where viral hepatitis is endemic, liver disease represents the fifth leading cause of death and over 1 million cases of liver failure are estimated to occur each year in urban areas alone.
Acute Liver Failure
Acute liver failure refers to the rapid onset of liver dysfunction in a patient without chronic liver disease. The most common early features include yellowing of the skin and eyes, mental status changes and bleeding. Acute liver failure may occur from a variety of causes such as acute flares of viral hepatitis, paracetamol or acetaminophen overdose, surgical interventions, idiosyncratic drug reactions and excessive alcohol intake. Despite improvements in intensive care management, mortality continues to be unacceptably high. Most acute liver failure patients, except active drinkers of alcohol, are considered suitable for liver transplant. When acute liver failure occurs in the presence of underlying liver disease, such as with AILD, the condition is sometimes referred to as acute-on-chronic liver failure. Other forms of acute liver failure include FHF and SILF.
74
Table of Contents
Alcohol-Induced Liver Decompensation
AILD is a life-threatening form of acute liver failure precipitated by the recent ingestion of alcohol. AILD can occur with or without chronic underlying liver disease and can manifest itself in several clinical patterns depending on the acute response of the liver to alcohol’s toxic effect. One common and well-recognized form of AILD is AAH, which is characterized by inflammation and enlargement of the liver. Most subjects with AAH are thought to have some underlying chronic changes to the liver as a result of longer-term alcohol use, although these changes may be reversible on abstention. Because of the frequency with which the AILD population has been found to have some degree of pre-existing liver disease, this condition is also often referred to as acute-on-chronic liver failure, or acute-on-chronic hepatitis.
A second form of AILD, which we refer to as non-AAH AILD, occurs in subjects with underlying stable chronic liver disease who suffer from an episode of acute liver failure due to excessive consumption of alcohol. Current standard-of-care for AILD is defined by the treating facility and typically includes the use of anti-inflammatory drugs, such as corticosteroids, and the treatment of secondary complications such as bleeding, kidney failure and hepatic coma and may include admission to an intensive care unit. Since abstinence from alcohol for six months is generally considered a pre-requisite for inclusion on a liver transplant list, organ transplantation is usually not available for these patients.
The Department of Health and Human Services in the United States estimates that for 2011 the number of hospital admissions related to AAH in the United States was approximately 99,000, with approximately 14,000 of these admissions identifying AAH as the primary diagnosis. In addition, approximately 300,000 hospital admissions occurred in 2011 related to alcoholic cirrhosis, alcohol liver damage not-otherwise-specified or alcoholic fatty liver, with approximately 50,000 hospital admissions identifying these conditions as the primary diagnosis. We believe that a subset of these patients have a form of non-AAH AILD that may be treatable with the ELAD System. Incidence rates for both AAH and non-AAH AILD appear to be similar in Europe.
Fulminant Hepatic Failure
A second form of acute liver failure is FHF, a relatively rare condition characterized by a rapid deterioration of liver function with altered mental state and coagulopathy in individuals without known pre-existing liver disease. The most frequent causes include drug or toxin-induced liver injury, viral hepatitis, autoimmune disease and hypoperfusion. Two thousand cases of FHF are estimated to occur in the United States each year. The standard-of-care includes liver transplantation, although these patients tend to progress very rapidly and may succumb to their disease before a suitable organ is made available. We believe the ELAD System may provide patients with a bridge-to-transplant, or potentially, recovery without transplantation.
Surgery-Induced Liver Failure
Another form of acute liver failure is SILF, which is comprised of three varieties, as follows:
| 1. | Primary Graft Non-Function, which occurs when a newly transplanted liver fails to function. This is a life threatening medical emergency, and can lead to death if a new organ does not become available quickly. We believe the ELAD System may provide patients with a bridge-to-transplant until a second liver becomes available. |
| 2. | Small-For-Size or Split Liver Transplant occurs when the transplanted liver is functioning, but may be too small to sustain the patient, either because only a small donor liver was available, or because a live person donated a portion of their liver for transplantation. We believe the ELAD System may be able to support the patient’s liver function until the donated organ regenerates to a size large enough to become independent of external support. Moreover the ELAD System may also enable transplantation of smaller liver fragments than typically used, potentially expanding the available pool of donor organs. |
| 3. | Other forms of surgery-induced liver failure. Although liver resection can provide a cure for primary liver cancer, resection of too much tissue can lead to rapidly progressing liver failure. Currently, surgeons will typically only resect up to 50% of the liver in order to avoid death from liver failure. |
75
Table of Contents
| However, more extensive resections occasionally occur, and resection of smaller portions can also lead to liver failure. We believe the ELAD System may be able to support these patients and also enable surgeons to perform larger tumor resections. |
We estimate that the first two categories of SILF account for several hundred patients a year in the United States, while the third category could represent an annual population of 10,000 or more cases a year in the United States.
Chronic Liver Failure
Chronic liver failure refers to a gradual loss of liver function and is usually characterized by the presence of widespread cirrhosis, which refers to the replacement of normal liver tissue by fibrosis, scar tissue and regenerative nodules. As normal liver is destroyed, the organ gradually fails to perform its normal metabolic, regulatory and synthetic functions. Unfortunately, damage from cirrhosis cannot be reversed and lost liver function can only be regained through transplantation. For this reason, we do not anticipate that the ELAD System will be effective for cirrhotic patients outside of the potential to bridge these patients to transplant.
Limitations of Currently Available Treatment Options for Acute Liver Failure
Given the liver’s complexity, there are no simple or widely effective medical solutions to acute liver failure. The only long-term cure for acute liver failure is surgical transplantation. In 2012, according to the United Network for Organ Sharing, fewer than 6,500 liver transplants were performed in the United States due to an insufficient number of available donor organs. According to the United Network for Organ Sharing, the average billable charge for a liver transplant in 2011, including the one month before surgery and six months after surgery, was $577,100. There are approximately 16,000 patients currently on the transplant waiting list and approximately 1,500 patients die while waiting each year. Similarly, there are approximately 6,000 liver transplants performed per year in Europe. Outside of transplant, current therapy is defined by the treating facility and is mostly supportive and designed to manage the symptoms and complications associated with acute liver failure.
Pharmaceuticals
N-acetylcysteine is approved by the FDA for the prevention of acute liver injury following the ingestion of toxic amounts of acetaminophen. Other treatments, including steroids and pentoxifylline, are often used off-label to manage symptoms associated with acute liver failure, although steroids in particular have been shown to increase risk of potentially fatal infections. Despite the availability of these treatments, the mortality rate for acute liver failure remains above approximately 40%. There are no known mechanisms for pharmacologically addressing liver failure specifically or restoring lost liver function.
Liver Support Devices
There are no medical devices approved for liver support that have been proven to improve survival in patients with acute liver failure. Clinical trials have been conducted in recent years involving two acellular liver support devices that filter toxic metabolites from the blood. In 2004, Gambro acquired the Molecular Adsorbents Recirculation System, or MARS, a liver dialysis system. However, in 2010, a randomized, controlled clinical trial comparing MARS to standard-of-care among 189 subjects with AILD and other forms of liver failure failed to show any improvement in 28-day survival for subjects treated with MARS. Likewise, in 2010 a similar liver support device, Prometheus, developed by Fresenius SE & Co. KGaA, also failed to show an improvement in either 28-day or 90-day survival in a randomized, controlled trial among 145 subjects with AILD and other forms of liver failure. MARS is currently cleared as a device in the United States for use in hepatic encephalopathy, drug overdoses and poisoning cases. MARS and Prometheus are both commercialized in Europe under CE marks.
In 1999-2000, Circe Biomedical, Inc. ran a 171-subject randomized, controlled clinical trial in acute liver failure using a liver support device that incorporated live pig liver cells. However, the trial did not meet its 30-day survival endpoint.
76
Table of Contents
Clinical Experience with the ELAD System in Acute Liver Failure
Prior to the initiation of our ongoing Phase 3 clinical trial program, over 145 subjects have received the ELAD System therapy, including earlier iterations of the ELAD System, at multiple clinical sites in the United States, Europe, Asia and the Middle East in six randomized, controlled, clinical trials, one single-arm trial and a compassionate-use program. Currently, we have a Phase 3 randomized, controlled clinical trial underway and a second Phase 3 clinical trial is anticipated to begin enrolling subjects in the first half of 2014. We also expect to begin a Phase 2 clinical trial of the ELAD System in patients with FHF or SILF in the second half of 2014. Two early studies in acute liver failure were conducted using a different configuration of the ELAD System which involved circulation of whole blood to the VTL C3A cartridges rather than blood plasma. In these studies, 36 subjects were enrolled, of which 23 received the ELAD System treatment and 13 were controls. The data from these studies led to the development of the next configuration of the ELAD System utilizing blood plasma and informed the design of the next clinical trials in acute liver failure.
The following table summarizes subsequent clinical trials as well as currently planned clinical trials using the later configuration of the ELAD System involving the treatment of blood plasma:
| Trial |
Date | Study Design | Indication(s) | Sites* | Location(s) | Total Subjects Enrolled | ||||||
| VTI-208 (Phase 3) |
Commenced March 2013 |
Randomized, controlled |
AILD | 40+ planned | U.S., Europe, Australia |
200 planned | ||||||
| VTI-210 (Phase 3) |
Expected to begin enrollment in 2014 |
Randomized, controlled |
AAH | 25+ planned | U.S., Europe | 120 planned | ||||||
| VTI-212 (Phase 2/3) |
Expected to begin enrollment in 2014 |
Single-arm in Phase 2 component |
FHF and SILF |
15+ planned | U.S. | 40 planned | ||||||
| VTI-206 (Phase 2b) |
2009 - 2011 | Randomized, controlled |
AILD and other |
26 | U.S., Europe | 62 | ||||||
| Compassionate- use program |
2008 - 2010 | Single-arm | Various | 6 | U.S., U.K., Singapore, Saudi Arabia |
16 | ||||||
| VTI-201 (Phase 2a) |
2008 - 2009 | Randomized, controlled |
AILD and other |
6 | U.S. | 18 | ||||||
| VTIC-301 (Pivotal) |
2006 - 2007 | Randomized, controlled |
Various, primarily viral hepatitis |
2 | China | 69 | ||||||
| CR-202 (Phase 2) |
2002 - 2003 | Randomized, controlled |
FHF | 8 | U.S., U.K. | 19 | ||||||
| PS-0698 (Phase 1) |
1999 - 2000 | Randomized, controlled |
FHF | 6 | U.S., U.K. | 25 | ||||||
| * | Represents peak numbers of clinical sites which will fluctuate throughout the trials in part based on resources and competition between trials for patients. |
The ELAD System’s Clinical Development in Alcohol-Induced Liver Decompensation
VTI-206: Phase 2b AILD Trial. Between 2009 and 2011, we enrolled 62 subjects in a randomized, controlled Phase 2b clinical trial of the ELAD System therapy in AILD and non-AILD subjects, which were two
77
Table of Contents
pre-defined and separately randomized cohorts. The clinical trial was conducted at 26 clinical sites in the United States and Europe. After discussions with the FDA, and to better inform our Phase 3 clinical trial, we adopted a 90-day overall survival endpoint. In each of the pre-defined cohorts, subjects were randomized in a 1:1 ratio to either the ELAD System therapy plus standard therapy, or medical treatment in accordance with the standard therapy of the institution.
In January 2011, the trial’s Data Safety and Monitoring Board, or DSMB, reported that only the AILD cohort had the possibility of showing any treatment effect and therefore recommended that we discontinue the non-AILD cohort. This second cohort contained subjects with severe forms of chronic liver disease who would not be eligible for the ELAD System therapy under our Phase 3 clinical trial criteria. Discussion with our FDA reviewer indicated the statistical analysis would be affected by closing the non-AILD arm, and therefore the study would not be able to serve as a pivotal trial. It was decided to terminate VTI-206 and design a new trial in AILD subjects. There were no unexpected ELAD System-related safety issues, as the DSMB did not detect any differences in the rates of serious events between the treated and control groups. Generally, the serious adverse events reported in this study were reflective of the severity of disease and co-morbidities present in the patient population. There were 67 serious adverse events reported by 35 subjects in this study, of which 6 events were reported as possibly or probably treatment-related, and consisted of gastrointestinal bleeding (1 subject), vomiting blood (1 subject), kidney failure (1 subject), reaction to blood infection (1 subject), vaginal bleeding (1 subject) and breakdown of red blood cells (1 subject). No serious adverse events were reported as definitely treatment-related.
The trial was terminated in April 2011 when 37 subjects had been enrolled in the predefined AILD cohort, which resulted in 29 subjects in the per protocol analysis due to elimination of eight subjects under predefined criteria. This subset showed a non-significant trend (p=0.27) toward improved 90-day survival of 69.2% in the ELAD System-treated group versus 43.8% in the control group. This trend is depicted in the figure below:
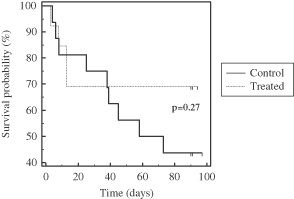
Several clinical laboratory values, including bilirubin, creatinine, and sodium, were monitored for safety in this study. We believe that these parameters are also biomarkers pertinent to our understanding of the mechanism of action of the ELAD System in these subjects. As such, we conducted a post-hoc analysis evaluating the effect of the ELAD System and standard-of-care versus standard-of-care alone on these biomarkers. These data showed subjects treated with the ELAD System demonstrated positive trends which we believe are consistent with improvements in liver function. A fourth biomarker also included in liver failure algorithms, INR, is not presented because it is transiently and artificially impacted by the administration of anti-coagulant drugs used during the ELAD System therapy. The other biomarkers are:
| • | Bilirubin. Bilirubin is a degradation product of hemoglobin that is normally processed and excreted by the liver. In liver failure, bilirubin accumulates in the body and is responsible for the yellow color of jaundice. In VTI-206, the ELAD System-treated subjects experienced a statistically significant |
78
Table of Contents
| reduction in serum bilirubin levels over the five days of therapy when compared with baseline, while control subjects did not. |
| • | Creatinine. Serum creatinine is a biomarker of kidney function. Kidney failure frequently is induced by liver failure. In VTI-206, the ELAD System-treated subjects demonstrated a trend towards reduction in creatinine in the first six days of the trial, while control subjects did not. |
| • | Sodium. Sodium is an electrolyte normally found in blood in a relatively narrow concentration band. Reductions in serum sodium frequently occur in subjects with acute liver failure. In VTI-206, the ELAD System-treated subjects experienced an increase in serum sodium over the first six days, while control subjects experienced a decrease. |
The below charts depict trends of several biomarkers in VTI-206:
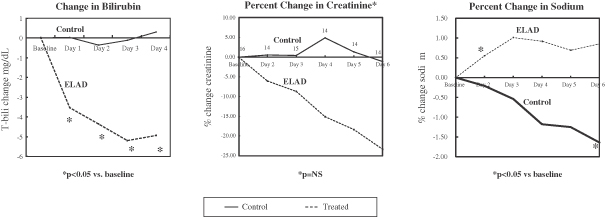
VTI-201: Phase 2a AILD and Non-AILD Trial. In preparation for VTI-206, we completed a small, randomized, controlled clinical study, enrolling 18 subjects with AILD and non-AILD, of which 16 participated in the trial. The study was intended to establish the safety of moving forward with our modified ELAD System, to define enrollment criteria for future studies in the United States and Europe and to assess the logistics of carrying out a large study in the United States and Europe. This trial was performed in 2008 and 2009, demonstrated no unexpected safety issues, and provided what we believe is preliminary evidence of the ELAD System’s potential efficacy in this population. Generally, the serious adverse events reported in this study were reflective of the severity of disease and co-morbidities present in the patient population. There were 39 serious adverse events reported by 11 subjects in this study, of which only 2 events, reduction in the number of red blood cells (1 subject) and reduction in the number of blood platelets (1 subject), were reported as possibly treatment-related. No serious adverse events were reported as definitely treatment-related.
Phase 3 Plan in AILD and AAH. Since January 2011, we have engaged in dialogue with both the FDA and EMA on the design of our Phase 3 program. We used the input we received from both authorities to design VTI-208, which is primarily intended to support potential FDA approval in AILD, and VTI-210, which is primarily intended to support potential EMA approval in AAH.
In March 2013, we enrolled the first subject in VTI-208, a 1:1 randomized, controlled pivotal trial designed to enroll 200 subjects with AILD. This trial is being conducted at over 40 sites in the United States, Europe and Australia. The study’s primary endpoint is overall survival through up to at least study day 91 using a Kaplan-Meier survival analysis. VTI-208’s trial design is similar to that of VTI-206, although VTI-208 contains several elements designed to better focus the study on what we believe to be our addressable patient population. These changes include the addition of criteria intended to exclude subjects who are either too unstable to survive the first five days post-randomization, who have non-regenerable livers, or who have criteria suggesting that they are already
79
Table of Contents
recovering in the immediate pre-randomization period. Assuming a response pattern similar to the per-protocol subset analysis in VTI-206, the trial is 95% powered to reach a p-value of 0.05. A p-value of less than or equal to 0.05 means that there is at least a 95% probability that increased survival in the ELAD System treatment group is due to the ELAD System treatment and not chance. As of April 2, 2014, 90 subjects had been enrolled in this trial and 47 clinical sites were actively enrolling subjects. The FDA has expressed concern that the VTI-208 study may not be adequately designed to provide convincing evidence of efficacy if there are significant differences in how the ELAD System subjects and controls are treated during the treatment period and after hospital discharge. Variations in length of hospital stay, rates of hospital re-admission, alcohol recidivism rates, nutritional support, and concomitant medications, which are not within our control, could significantly confound the study results and call into question whether any demonstrated difference in survival is due to the ELAD System or to these factors. We have developed a protocol that attempts to minimize this bias to the extent possible, including defining a protocol-specific standard of care, specifying steroid treatment, standardizing the discharge criteria for both the ELAD System and control subjects, requiring that follow-up visits are conducted by a blinded reviewer, ensuring home health care nurses and other clinical personnel are unaware of treatment assignment, educating subjects not to reveal treatment assignment to their caregivers and monitoring concomitant medications, alcohol recidivism and interaction with the health care system to provide evidence that there is no meaningful difference between the groups that could significantly confound the trial data.
We have obtained regulatory allowance in the United States, United Kingdom, Spain and Australia to begin enrolling patients and have initiated clinical sites in VTI-210, a 1:1 randomized, controlled pivotal trial designed to enroll 120 subjects with AAH who have failed seven days of steroid therapy according to predefined criteria. The study’s primary endpoint is overall survival through study day 91 using a Kaplan-Meier survival analysis. Assuming a median survival of 45 days for the control group and 90 days for the ELAD System-treated group, the trial is 95% powered to reach a p-value of 0.05. We expect to enroll our first patient in this trial in the first half of 2014.
We expect to report preliminary data from VTI-208 in the first half of 2015 and VTI-210 in 2015 or 2016.
The ELAD System’s Clinical Development in Fulminant Hepatic Failure
Phase 1 and 2 Trials of Fulminant Hepatic Failure. Our predecessor companies, Hepatix and VitaGen, tested earlier versions of the ELAD System in four clinical trials among subjects with FHF.
PS-0698 and CR-202. Between May 1999 and August 2000, VitaGen enrolled 25 human subjects with FHF in an open-label, randomized Phase 1 study, titled PS-0698, at six clinical sites in the United States and the United Kingdom comparing the ELAD System therapy to medical treatment in accordance with the standard therapy of the institution. The ELAD System-treated subjects received either two or four cartridges, depending on body weight, with each cartridge containing 80 to 100 grams of VTL C3A cells. Of the 25 subjects enrolled, the first six subjects received the ELAD System therapy, with the following 19 subjects randomized to 10 ELAD System treatment (of which nine were treated) and nine controls. Between March 2002 and January 2003, VitaGen enrolled 19 human subjects with FHF in a Phase 2 open-label clinical study, titled CR-202, at eight clinical sites in the United States, with 13 subjects randomized to the ELAD System treatment (of which 11 were treated) and six subjects randomized to standard-of-care. In CR-202, all the ELAD System-treated subjects received four ELAD cartridges containing 80 to 100 grams of VTL C3A cells.
Clinical study PS-0698 was designed to evaluate the safety of the ELAD System in FHF subjects. Clinical study CR-202 was designed to help inform future studies of the ELAD System in FHF. We believe the data from these preliminary studies are useful in evaluating the ELAD System’s potential as a treatment for FHF.
We believe the data from study PS-0698 suggest that the principal effect of the ELAD System therapy is to allow severely ill patients with FHF to survive the first few days following study entry, so that they can receive a liver transplant. All of the control subjects who died did so within the first five days following their enrollment. All transplant subjects received their donor organs during this same time period. The ELAD System did not
80
Table of Contents
affect any other study variables associated with hepatic function or dysfunction. There were no serious adverse events reported that were considered by the trial investigator to be treatment-related.
In study CR-202, no intergroup differences in mortality were observed. Only two subjects, one treated and one control, died during the study. The proportion of subjects who received transplants was higher for treated subjects (54.5%) compared with control subjects (16.7%). Of the six subjects in the ELAD System group who received a transplant, three were not on the transplant list, unlike the single transplanted control who was listed. Generally, the serious adverse events reported in this study were reflective of the severity of disease and co-morbidities present in the patient population. There were 11 serious adverse events reported by seven subjects in this study. Three serious adverse events were determined by the trial investigator to be definitely treatment-related, which consisted of reduction in the number of blood platelets (1 subject), device malfunction (1 subject), and low blood pressure (1 subject). Five serious adverse events were determined by the trial investigator to be possibly or probably treatment-related, which consisted of reduction in the number of blood platelets (3 subjects), reduced body temperature (1 subject), and an increase in lactic acid (1 subject).
Neither study PS-0698 nor study CR-202 was designed to demonstrate a statistically significant improvement in overall survival or bridge-to-transplant/recovery (BTT/R). In neither case was a statistically significant improvement demonstrated. However, because inclusion criteria for these studies were similar and the endpoints were identical, we performed a post-hoc meta-analysis to explore the impact of transplant-listing on treatment outcome. While there was no evidence of an ELAD System treatment effect in the subset of subjects not listed for transplant, in the subset of subjects prospectively listed for transplant (N=26), this meta-analysis revealed positive survival trends. Moreover, as suggested by the study PS-0698 data alone, we observed additional benefit in the group treated with the ELAD System therapy in BTT/R. While BTT/R is not an acceptable regulatory endpoint, we believe that it is clinically important, and these data have helped inform the design of our Phase 3 clinical trial in FHF.
In summary this meta-analysis revealed the following:
| • | thirty-day survival rates were 75% for the ELAD System-treated subjects and 50% for the control subjects (p=0.12) |
| • | an analysis of Kaplan-Meier curves comparing success at a BTT/R endpoint revealed additional benefits for ELAD System-treated subjects (p=0.06) |
| • | a chi-squared analysis of the BTT/R status of subjects at the end of the study showed a statistically significant advantage for the ELAD System-treated subjects versus control subjects (p=0.03) |
The overall survival trend of the meta-analysis is demonstrated in the following Kaplan-Meier chart on the left and the bridge-to-transplant or recovery of the meta analysis is demonstrated in the following Kaplan-Meier chart on the right:
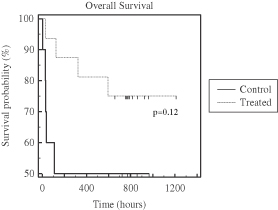
|
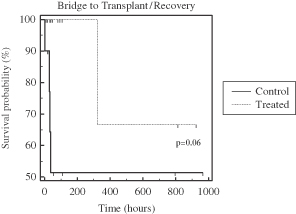
|
81
Table of Contents
Statistically significant differences were not observed for other outcome variables, including those related to hepatic function, in either the overall population or the subset.
Phase 2/3 Program in Fulminant Hepatic Failure and Surgery Induced Liver Failure. In the second half of 2014, we also expect to initiate enrollment of VTI-212, a Phase 2/3 clinical program in subjects with either FHF or SILF. We plan to begin this program with a Phase 2 single-arm component enrolling 40 subjects, which may later be followed by a randomized, controlled Phase 3 component the size of which has not yet been determined. Results from the initial Phase 2 single-arm component will be compared with historical or case-matched controls, and we currently anticipate Phase 2 data in 2015 or 2016.
Since FDA guidelines provide for flexibility in review of marketing applications for life-threatening conditions, such as FHF and SILF, and because these conditions affect a small number of patients it may be possible to seek marketing approval in FHF and SILF based on results from the Phase 2 single-arm component. However, regulatory agreement on an expedited approval pathway has not yet been sought and may never be granted. In the event that randomized data are necessary for approval in FHF and SILF, we expect to perform the randomized Phase 3 portion of the program, the design of which would be finalized upon analysis of the Phase 2 component. Data from VTI-212 may also be used to support our planned marketing applications for AILD and AAH.
This trial is expected to be conducted primarily in the United States. The symptoms of FHF and SILF are similar and we currently expect to use similar inclusion criteria for either population.
The ELAD System’s Clinical Development in Acute Flare of Viral Hepatitis
VTIC-301. Between 2006 and 2007, we enrolled 69 subjects with acute liver failure in a randomized, controlled open label trial at two hospitals in Beijing. Inclusion criteria focused the trial’s enrollment on subjects anticipated to have a 50% chance of death by 84 days, and the majority of enrolled subjects were experiencing an acute flare of viral hepatitis. The study was designed to enroll 120 subjects but was terminated early by one of the hospital’s ethics committee because, in light of the results discussed below, it would have been unethical to continue to treat control subjects with standard-of-care alone. Endpoints included survival at 14, 28, 56 and 84 days, as analyzed using a logrank method.
A significant protocol amendment was enacted after the enrollment of the first 49 subjects, in which inclusion criteria were changed, reducing the severity of disease, and a shorter ELAD System treatment time was recommended. This change in study design resulted in far fewer deaths or transplants in the second subset of 19 ELAD System treated treatment and control subjects. A revised statistical plan was prepared to accommodate these differences in subject populations. Separate analyses were performed on the 49-subject subset and the full 68-subject population, and additional statistical analysis techniques were proposed, such as the use of Wilcoxon rank-sum techniques to analyze continuous variables such as survival time.
Analysis of the first 49 subjects (32 subjects randomized to be treated with the ELAD System for 3 days along with standard-of-care for the treating institution and 17 subjects randomized to be treated with standard-of-care alone) revealed the following:
| • | significant differences in 28- and 56-day survival using the logrank test (p=0.015 and 0.026 respectively); |
| • | significant differences in 84-day survival using the Wilcoxon test (p=0.049) (logrank was not significant at 14 and 84 days, p=0.074 and 0.058, respectively); and |
| • | no unexpected safety issues. |
Generally, the serious adverse events reported in this study were reflective of the severity of disease and co-morbidities present in the patient population. There were 16 post-treatment adverse events in 8 of the 32 treated subjects that the investigators reported as possibly or probably treatment related.
82
Table of Contents
These efficacy results are depicted in the below Kaplan-Meier curve:
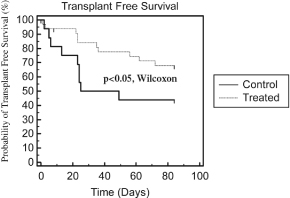
Analysis of all 68 subjects treated (44 subjects randomized to be treated with the ELAD System for 1 to 3 days along with standard-of-care for the treating institution and 24 subjects randomized to be treated with standard of care alone) revealed the following:
| • | Significant differences in 28-day survival using the logrank test (p=0.015); |
| • | No significant differences in 14, 56 and 84-day survival using the logrank test; and |
| • | No unexpected safety issues. |
Based on these results, it was concluded that the Wilcoxon test is a more sensitive technique to elucidate differences between groups in the ELAD System clinical trials, and that a more severely diseased population and more extended treatment times should be evaluated in future clinical studies. Future clinical trials may be analyzed using the more conservative logrank technique.
One further observation from the 49 subjects in the first part of this study was that the ELAD System can significantly decrease bilirubin levels, an important biomarker of liver function in patients with acute liver failure. The overall magnitude of the decrease in mean end of treatment total bilirubin in the ELAD group was 25.1% (17.5 to 13.1 mg/dL) compared to an increase in the control group of 36.8% (17.1 to 23.4 mg/dL). Serum sodium, another biomarker associated with survival in subjects with acute liver failure, also was significantly improved in the subjects treated with the ELAD System relative to control subjects.
These China pivotal trial data formed the basis of a submission for marketing approval to the China FDA, or CFDA, in September 2007. It should be noted that this study was not designed, and will not be used, as a pivotal trial to support approval of the ELAD System in the United States and Europe.
Subsequent to the completion of the VTIC-301 clinical study, an additional protocol was prepared by the treating physicians to explore the long-term survival of subjects enrolled in this study. Following the grant of informed consent, subjects enrolled in VTIC-301 were contacted and invited to return to the treating hospital for examination for recurrence of liver disease or the incidence of cancer. This study was carried out in 23 and 22 subjects, respectively, 3 and 5 years following initial randomization.
83
Table of Contents
These data from the first 49 subjects suggest that the survival benefit (statistically significant at 3 years and 5 years, p<0.05, logrank) afforded to those subjects treated with the ELAD System is maintained over a 3 and 5 year period relative to those subjects in the control group. These trends are depicted in the Kaplan-Meier curves below:
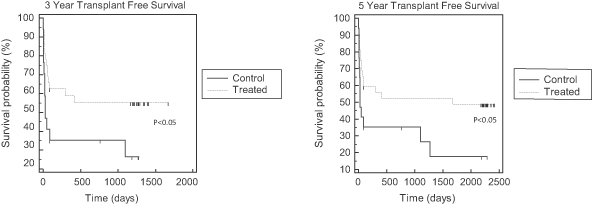
While these follow-up analyses were not prospectively defined in the VTIC-301 protocol, we believe they provide valuable information on the long-term survival of this group of patients.
The results of VTIC-301 were submitted to the CFDA, for marketing approval in September 2007. However, a regulation enacted in 2009 prevents the approval of novel foreign medical products until they are approved in their home markets first. Our application in China is pending.
Future Clinical Development Plans
In the future, we anticipate pursuing clinical development of the ELAD System in additional indications, including bridge-to-transplant.
Future Commercialization Opportunity
The VTL C3A cells which we grow in our facility produce large quantities of proteins and other cell products, such as albumin, alpha-fetoprotein, alpha-1 antichymotrypsin, alpha-1 antitrypsin, C3 complement, anti-thrombin 3, factor V, factor VII, fibrinogen, and transferrin. Many of these compounds have known or potential industrial and/or therapeutic applications. We plan to explore the commercialization potential for these compounds, although we do not expect to generate revenues in this area in the near-term.
Commercialization Strategy and Organization
Given our stage of development, we have not yet established a commercial organization or distribution capabilities. If approved, we intend to directly commercialize the ELAD System in the United States and Europe with a targeted hospital sales force and to either commercialize directly or utilize a variety of types of collaboration in other markets.
Sales, Marketing and Reimbursement
Following FDA and/or EMA approval, if any, we intend to launch the ELAD System commercially in the United States and Europe, respectively. We expect to direct our initial sales and marketing efforts at those sites which will have participated in our Phase 3 clinical trials, which we anticipate exceed 40 in number. Subsequently, we plan to gradually expand our focus in the United States to approximately 100 liver-transplant centers, as well as
84
Table of Contents
to another 100 specialist intensive care centers with a similar penetration in Europe. We expect that our commercial infrastructure would be comprised of a targeted hospital sales force led by several experienced sales management personnel, an internal marketing and medical affairs staff, and a reimbursement support team. We currently intend to focus our initial commercial efforts on the United States and European markets, which we believe represent the largest and most readily addressable market opportunities for the ELAD System. In addition, we believe that Australia, China, Japan, India, other Asian markets, the Middle East, Brazil, and Africa represent significant opportunities because of the prevalence of liver disease in these geographies, and we intend to pursue the commercialization of the ELAD System in certain of these markets through collaborations.
We expect the ELAD System to be reimbursed as an in-patient drug in the United States. For patients eligible for Medicare, who we anticipate to constitute a minority of the ELAD System-addressable patients, reimbursement is expected to occur under a diagnosis related group, which may be eligible for a New Technology Add-on Payment and outlier payments. Beginning prior to regulatory approval in the United States, we plan to request appropriate codes and groupings for the ELAD System therapy from the Centers for Medicare & Medicaid Services, which may take several years to complete, if successful. We also expect to work with private payers to develop appropriate case-rates for the ELAD System reimbursement. We believe that we will ultimately be able to price the ELAD System in a range consistent with other currently marketed life saving therapies, such as left ventricular assist devices, orphan biologic medications for hereditary metabolic diseases, and monoclonal antibody medications for cancer.
Manufacturing and Supply
Our manufacturing facility is licensed as a drug and medical device manufacturer by the California Food and Drug Bureau. This facility was recently remodeled in order to expand manufacturing capacity to support worldwide clinical trials and early marketing demands for the ELAD System. This remodel increased manufacturing clean room, quality control laboratory, and warehouse and refrigeration space. The increased clean room space will allow modular increases in the ELAD cartridge manufacture.
At our facility, we manufacture the ELAD System, which is comprised of our proprietary VTL C3A cells, cartridges and the ELAD System bedside unit. The system contains both reusable and disposable medical device components. We source certain components of the ELAD System from third-party suppliers. Most components of our ELAD System are FDA-cleared and CE marked. In a few cases we manufacture a device or a device component ourselves. Based on discussions with the pertinent regulatory authorities, we have determined that all components will have to be submitted for approval for use with the ELAD System as part of the BLA.
Training and Support
We also expect to deploy a training and support team at the liver transplant and specialist intensive care centers that our sales and marketing team are expected to target. During the initial commercialization period, it will be essential for us to have our own trained staff present during the delivery of the ELAD System therapy. This may entail the construction and operation of training centers and will require the hiring of personnel of appropriate ability to be adequately trained.
All biopharmaceutical production activities are conducted under cGMP, the standards established by the FDA for pharmaceutical and biologics production. Medical devices are manufactured in accordance with pertinent device regulations. The equipment used in the manufacturing process is based on designs typically encountered in the production of other biotechnology products, and has been customized to tailor their use to the ELAD System production. The ELAD cartridges and bedside units are tested according to the FDA’s and other applicable regulatory bodies’ standards before they can be released for use in humans. All device components shipped from us to our investigational sites are subject to quality control inspection.
85
Table of Contents
Intellectual Property
We believe that we have a patent portfolio and substantial know-how relating to the ELAD System. Our patent portfolio includes claims directed to our ELAD System, specific clonal cells and cell-lines derived from human liver-derived C3A cells, as well as methods of growing these cells. We are currently the owner of record of four issued U.S. patents and over a dozen issued or allowed foreign patents. One granted U.S. patent claims a method of using C3A cells to treat a patient’s blood and is currently set to expire in April 2027, unless extended further. In addition, a second granted U.S. patent claims covering an extracorporeal device configuration, which we believe to include our ELAD System, independent of cell-type used is set to expire in May 2025, unless extended further. Foreign counterparts of one or more of these patents have been issued in Australia, Canada, Indonesia, Israel, Japan, Mexico, New Zealand, Singapore, South Africa, South Korea and Taiwan and remain under review in certain other jurisdictions including Europe, Brazil, China, India and the Philippines.
We strive to protect the proprietary technology that underlies the ELAD System. We seek patent protection in the United States and internationally for the ELAD System, its methods of use and processes of manufacture, and any other technology to which we have rights, where available and when appropriate. We also rely on trade secrets that may be important to the development of our business.
A predecessor company initially developed the ELAD System after the technology was spun out of Baylor College of Medicine in 1990. In 2003, we acquired substantially all of the assets of the predecessor, including trade secrets, know how, clinical experience and key employees and facilities. Among those assets was a U.S. patent, which we had exclusively licensed from Baylor, which is now expired.
Our success will depend on the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to our business, the validity and enforceability of our patents, the continued confidentiality of our trade secrets as well as our ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may own or license in the future, nor can we be sure that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors—Risks Relating to Intellectual Property.”
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing the non-provisional priority application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office, or PTO, in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent.
The term of a U.S. patent that covers an FDA-approved biologic may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Amendments permit a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the biologic is under regulatory review. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved biologic may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of our BLA, we expect to apply for patent term extensions.
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. For example, significant aspects of our proprietary technology are based on unpatented trade secrets and
86
Table of Contents
know-how. This includes our methods of expanding, culturing and optimizing the performance of the human VTL C3A cell line.
Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
We seek trademark protection in the United States and outside of the United States where available and when appropriate. We have registered trademark rights in the Vital Therapies and the ELAD System marks in the United States.
Competitive Environment
The biotherapeutic and medical device industries are highly competitive and rapidly evolving, and we face potential competition from major pharmaceutical companies, specialty pharmaceutical companies, medical device developers and biotechnology companies worldwide, many of which have longer more established operating histories, and significantly greater financial, technical, marketing, sales and distribution and other resources than us. Given the significant unmet medical need for novel therapies to treat acute liver failure, many companies, public and private universities and research organizations are actively engaged in the discovery and research and development of potential products in this field. Several of these entities are engaged in research on cell-based approaches to acute liver failure. Although we are not aware of any ongoing human clinical trials involving potentially competitive product candidates, such trials could be taking place or could begin in the near future. In addition, these entities compete with us for limited resources including personnel, trial sites and potential complementary assets.
We are not aware of any company that is in human clinical trials with a human cell-based product for the treatment of patients with acute liver failure. At least four companies have conducted prior research on various human hepatocyte cell lines including Exten Industries, Hepalife Technologies, Fresenius, and Hybrid Organ GmbH. In addition, the University College London, and the University of Amsterdam and its spinout Hep-Art Medical Devices are actively pursuing animal research in this area.
Several companies have also attempted to develop extracorporeal therapy based upon primary porcine hepatocytes, although ongoing research in this area is difficult to ascertain.
Two commercially available liver dialysis systems, from Gambro and Fresenius, have undergone extensive clinical development, although both have failed to show an improvement in long-term survival among patients with acute liver failure. Both rely on not only traditional dialysis circuits to remove water-soluble toxins, but also albumin dialysis circuits to remove albumin-bound molecules.
In addition, there are several drugs used to treat symptoms associated with acute liver failure, including steroids, pentoxifylline and N-acetylcysteine. These drugs, alone or in combination, are used frequently in patients with acute liver failure.
87
Table of Contents
Government Regulation
We operate in a highly regulated industry that is subject to significant federal, state, local and foreign regulation. Our present and future business has been, and will continue to be, subject to a variety of laws including, the Federal Food, Drug, and Cosmetic Act, or FDC Act, and the Public Health Service Act, or PHS Act, among others. Biologics and medical devices are subject to regulation under the PHS Act and FDC Act.
Regulation of Combination Products
The FDA has specified a definition for the term “combination product,” which includes: (1) A product comprised of two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity; (2) Two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products; (3) A drug, device, or biological product packaged separately that according to its investigational plan or proposed labeling is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of the approved product would need to be changed, e.g., to reflect a change in intended use, dosage form, strength, route of administration, or significant change in dose; or (4) Any investigational drug, device, or biological product packaged separately that according to its proposed labeling is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
The FDA is divided into various Centers by product type. Different Centers typically review drug, biologic, or device applications. In order to review an application for a combination product, the FDA must decide which Center should be responsible for the review. FDA regulations require that the FDA determine the combination product’s primary mode of action, or PMOA, which is the single mode of a combination product that provides the most important therapeutic action of the combination product. The Center that regulates that portion of the product that generates the PMOA becomes the lead evaluator. If there are two independent modes of action, neither of which is subordinate to the other, the FDA makes a determination as to which Center to assign the product based on consistency with other combination products raising similar types of safety and effectiveness questions or to the Center with the most expertise in evaluating the most significant safety and effectiveness questions raised by the combination product. When evaluating an application, a lead Center may consult other Centers but still retain complete reviewing authority, or it may collaborate with another Center, by which the Center assigns review of a specific section of the application to another Center, delegating its review authority for that section. Typically, the FDA requires a single marketing application submitted to the Center selected to be the lead evaluator, although the agency has the discretion to require separate applications to more than one Center. One reason to submit multiple evaluations is if the applicant wishes to receive some benefit that accrues only from approval under a particular type of application, like new drug product exclusivity. If multiple applications are submitted, each may be evaluated by a different lead Center.
The ELAD System is regulated as a combination biologic/device in the United States. Based upon the proposed mechanism of action, the primary Center within the FDA responsible for its regulation is the Center for Biologics Evaluation and Research, or CBER. The CBER office responsible for review is the Office of Cellular, Tissue and Gene Therapies, and the marketing application will be a BLA. CBER will consult with the Center for Devices and Radiological Health, or CDRH, in reviewing the device components of the ELAD System.
FDA Approval Process
In the United States, pharmaceutical and biological products and medical devices are subject to extensive regulation by the FDA. The FDC Act, PHS Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export
88
Table of Contents
of these products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending license applications, warning and other letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Preclinical Studies
Biological product development in the United States typically involves preclinical laboratory and animal tests. Preclinical tests include laboratory evaluation of product chemistry, formulation, and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an investigational new drug application, or IND, along with other information, including information about product chemistry, manufacturing and controls, and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has not objected to the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical Studies
Clinical trials involve the administration of the investigational biologic to healthy volunteers or subjects under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations and good clinical practices, or GCP, an international standard meant to protect the rights and health of subjects and to define the roles of clinical trial sponsors, administrators, and monitors, as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. subjects and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial subjects. The clinical trial protocol, and protocol amendments, and informed consent information for subjects in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of investigational products are required to register on clinicaltrials.gov, a National Institute of Health website registry database, and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly-available information to gain knowledge regarding the progress of development programs.
Marketing Approval
Clinical trials to support BLAs, which are applications for marketing approval, are typically conducted in three sequential Phases, but the Phases may overlap. In Phase 1, the initial introduction of the investigational biologic candidate into healthy human subjects, the investigational biologic is tested to assess metabolism,
89
Table of Contents
pharmacokinetics, pharmacological actions, side effects associated with increasing doses and, if possible, early evidence on effectiveness. Phase 2 usually involves trials in a limited subject population, to determine the effectiveness of the investigational biologic for a particular indication or indications, dosage tolerance and optimum dosage, and identify common adverse effects and safety risks. In the case of product candidates for severe or life-threatening diseases such as cancer, the initial human testing is often conducted in patients rather than in healthy volunteers.
If an investigational biologic demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 clinical trials are undertaken to obtain additional information about clinical efficacy and safety in a larger number of subjects, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the investigational drug and to provide adequate information for its labeling. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the efficacy and safety of the biologic for use in a specific indication or population. A single Phase 3 clinical trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multi-center trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity, or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible.
After completion of the required clinical testing, a BLA is prepared and submitted to the FDA. FDA approval of the BLA is required before marketing of the product may begin in the United States. The BLA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product’s manufacture and controls. The cost of preparing and submitting a BLA is substantial. The submission of most BLAs is subject to a substantial application fee and the manufacturer or sponsor of an approved BLA is also subject to annual product and establishment user fees.
The FDA has 60 days from its receipt of a BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of BLAs. Most such applications for standard review biologics products are reviewed within twelve months of submission; most applications for priority review biologics are reviewed within eight months of submission. Priority review for biologics is limited to those products intended to treat a serious or life-threatening disease with unmet medical need relative to the currently approved products. The review process may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission.
The FDA may also refer applications for novel biologics products or biologics products that present difficult questions of safety or efficacy, to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the biologic product is manufactured. The FDA will not approve the BLA unless compliance with current good manufacturing practice, or cGMP, is satisfactory, including the ELAD System compliance with applicable parts of the device QSR regulations as defined for combination products, and the BLA contains data that provide substantial evidence that the biologic is safe, pure and potent in the indication studied. Manufacturers of biologics also must comply with the FDA’s general biological product standards.
After the FDA evaluates the BLA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter outlines the deficiencies in the submission and may require substantial additional testing, including additional large-scale clinical testing or information in order for the FDA to reconsider the application. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the BLA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included.
90
Table of Contents
An approval letter authorizes commercial marketing and distribution of the biologic with specific prescribing information for specific indications. As a condition of BLA approval, the FDA may require substantial post-approval testing and surveillance to monitor the product’s safety or efficacy and may impose other conditions, including labeling restrictions, which can materially affect the product’s potential market and profitability. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems or safety issues are identified following initial marketing.
Changes to some of the conditions established in an approved application, including changes in indications, labeling, device components or manufacturing processes or facilities, require submission and FDA approval of a new BLA or BLA supplement before the change can be implemented. A BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing BLA supplements as it does in reviewing BLAs.
Post-Approval Requirements
Once a BLA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of biologics, including standards and regulations, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet.
Biologics may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic reports is required following FDA approval of a BLA. The FDA also may require post-marketing testing, known as Phase 4 testing, Risk Evaluation and Mitigation Strategies, or REMS, and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control as well as product manufacturing, packaging and labeling procedures must continue to conform to cGMP’s after approval. Manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA during which the agency inspects manufacturing facilities to assess compliance with applicable regulations such as cGMPs and the QSR. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMP’s. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Exclusivity and Approval of Competing Products
Biosimilar Exclusivity
The Biologics Price Competition and Innovation Act of 2009, or BPCIA, creates an abbreviated approval pathway for biological products shown to be highly similar to or interchangeable with an FDA-licensed reference biological product. Biosimilarity sufficient to reference a prior FDA-approved product requires that there be no differences in conditions of use, route of administration, dosage form, and strength, and no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency. Biosimilarity must be shown through analytical studies, animal studies, and at least one clinical study, absent a waiver. A biosimilar product may be deemed interchangeable with a prior approved product if it meets the higher hurdle of demonstrating that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. No biosimilar or interchangeable products have been approved under the BPCIA to date. Complexities associated with the larger, and often more complex, structures of biological products, as well
91
Table of Contents
as the process by which such products are manufactured, pose significant hurdles to implementation which are still being evaluated by the FDA.
A reference biologic is granted twelve years of exclusivity from the time of first licensure of the reference product, and no application for a biosimilar can be submitted for four years from the date of licensure of the reference product. The first biologic product submitted under the abbreviated approval pathway that is determined to be interchangeable with the reference product has exclusivity against a finding of interchangeability for other biologics for the same condition of use for the lesser of (i) one year after first commercial marketing of the first interchangeable biosimilar, (ii) eighteen months after the first interchangeable biosimilar is approved if there is no patent challenge, (iii) eighteen months after resolution of a lawsuit over the patents of the reference biologic in favor of the first interchangeable biosimilar applicant, or (iv) 42 months after the first interchangeable biosimilar’s application has been approved if a patent lawsuit is ongoing within the 42-month period.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biologic intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug or biologic for this type of disease or condition will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. Orphan drug exclusivity does not prevent FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the BLA application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Federal and State Fraud and Abuse, Privacy and Transparency Laws
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of federal and state laws have been applied to restrict certain business operations and activities in the biopharmaceutical and medical device industries in recent years. These laws that may affect our ability to operate include, but are not limited to:
The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return, for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, service or item for which payment is made, in whole or in part, under a federal health care program. The federal healthcare program anti-kickback statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution, the exceptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing, or recommending may be subject to scrutiny if they do not qualify for a statutory exception or a
92
Table of Contents
regulatory safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal healthcare program anti-kickback statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to, or approval by, the federal government, or knowingly making, using, or causing to be made or used, a false record or statement material to a false or fraudulent claim to the federal government. Recently, the civil False Claims Act has been used to assert liability on the basis of kickbacks and improper referrals, improperly reported government pricing metrics such as Medicaid Best Price or Average Manufacturer Price, improper use of supplier or provider Medicare numbers when detailing a provider of services, improper promotion of drugs or off-label uses not expressly approved by the FDA in a drug’s label, and misrepresentations with respect to the services rendered or items provided. The federal criminal false claims law prohibits, among other things, at any time knowingly and willingly making, or causing to be made, any false statement or representation of a material fact for use in determining rights to a benefit or payment under a federal healthcare program.
Many states also have statutes or regulations similar to the federal fraud and abuse laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor (e.g. private payors). Sanctions under federal, and state healthcare fraud and abuse laws may include, without limitation, civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare program, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations.
Additionally, the civil monetary penalties statute, which, among other things, imposes fines against any person who is determined to have presented, or caused to be presented, claims to a federal healthcare program that the person knows, or should know, is for an item or service that was not provided as claimed or is false or fraudulent. The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, including private third party payors and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services relating to healthcare matters. Many states have similar fraud and abuse statutes and regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, private payors. In addition, we may be subject to, or our marketing activities may be limited by, data privacy and security regulation by both the federal government and the states in which we conduct our business.
The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Moreover, recent healthcare reform legislation has strengthened many of these laws. For example, the ACA, among other things, amends the intent requirement of the federal healthcare program anti-kickback statute to a stricter standard such that a person or entity does not need to have actual knowledge of the federal healthcare program anti-kickback statute or specific intent to violate it in order to have committed a violation. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act. Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors, it is possible that some of our business activities may not satisfy the statutory exceptions or regulatory safe harbors and we could be subject to challenge under one or more of such laws. State law equivalents to these federal laws may also apply. Such a challenge could have a material adverse effect on our business, financial condition and results of operations.
93
Table of Contents
Additionally, the federal Physician Payments Sunshine Act within the ACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biological, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program (with certain exceptions) to report to the Centers for Medicare & Medicaid Services, or CMS, information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals. The Physician Payments Sunshine Act provisions were recently implemented in final regulation that requires applicable manufacturers to report annually to CMS certain ownership and investment interests held by physicians and their immediate family members, with data collection beginning on August 1, 2013. Manufacturers are required to report such data to CMS by March 13, 2014 (and by the 90th day of each subsequent calendar year). Other state laws require pharmaceutical companies to adopt and or disclose specific compliance policies to regulate the Company’s interactions with healthcare professionals. Moreover, some states, such as Minnesota and Vermont, also impose an outright ban on certain gifts to physicians.
Violations of some of these laws may result in substantial fines. These laws affect our promotional activities by limiting the kinds of interactions we may have with hospitals, physicians or other potential purchasers or users of our products. Both the disclosure laws and gift bans impose additional administrative and compliance burdens on us. Although we seek to structure our interactions in compliance with all applicable requirements, these laws are broadly written, and it is often difficult to determine precisely how a law will be applied in specific circumstances. If an employee were to offer an inappropriate gift to a customer, we could be subject to a claim under an applicable state law. Similarly if we fail to comply with a reporting requirement, we could be subject to penalties under applicable federal or state laws including, without limitation, civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and the curtailment or restructuring of our operations. In addition to the federal and state disclosure and gift ban laws, certain countries outside of the United States have similarly enacted disclosure laws for which the company in its activities may be subjected to from time to time.
Regulation in the European Union
Biologics and medical devices are subject to extensive regulation outside of the United States. In the European Union, for instance, a centralized approval procedure may be used to authorize the marketing of a product in all countries of the European Union, which includes most major European markets. However, for certain products, if this procedure is not used, approval in one country of the European Union can be used to obtain approval in a second country of the European Union under two simplified application processes, either the mutual recognition procedure or the decentralized procedure. Both of these procedures rely on the principle of mutual recognition. In addition to regulatory approval, pricing and reimbursement approvals are also required in most countries.
In Europe, the ELAD System is regulated as a Combination Somatic Cell Advanced Therapy Medicinal Product, or ATMP. The primary regulatory license application in Europe (a Marketing Authorization Application, MAA) will be made to The Committee for Advanced Therapies, or CAT, and the Committee for Human Medicinal Products, or CHMP, which are the committees at the European Medicines Agency, or EMA, that are responsible for assessing the quality, safety and efficacy of ATMPs. Marketing Authorization Applications for ATMPs can only be filed using the Centralized Procedure. The CHMP and the CAT (Co)-Rapporteurs and CHMP Coordinators liaise closely together to prepare the ATMP Assessment Reports. The draft opinion prepared by the CAT is issued on any scientific assessment of ATMPs necessary to draw up the scientific opinions by the CHMP relating to granting, variation, suspension or revocation of an authorization to place ATMPs on the market in accordance with Regulation (EC) No 1394/2007 and pharmacovigilance. The CAT has also established collaborations with Notified Bodies, or NBs, in Europe in order to review the device components of combination device products, and we anticipate that the device components of our submission will be reviewed by one of those NBs. During the clinical trial phase in Europe, the company is granted authorization to conduct clinical studies through local regulatory agencies in each country, each of which has its own format and regulation for the issuance of Clinical Trial
94
Table of Contents
Authorizations, or CTAs. In general, it is necessary to obtain separate authorizations in each country for each clinical trial protocol from the medicines and device agencies as there is yet to be developed a procedure for dealing with combination products like the ELAD System. These authorizations have been granted by the UK’s Medicines and Healthcare Products Regulatory Agency, or MHRA, and are being sought in other European countries though no assurance can be made that we will obtain additional authorizations. The EMA has provisions for providing companies with scientific advice through the Scientific Advice Working Party, or SAWP, and we have sought and obtained advice on our clinical development program through the SAWP process. We anticipate obtaining further guidance and advice through future interactions with the SAWP.
In other jurisdictions we anticipate that there will be different requirements for authorization for clinical trials and ultimately marketing of the ELAD System due to the complex nature of the combination of biological and device components of our ELAD System.
Other Regulations
We are also subject to numerous, federal, state, local and foreign laws and regulations relating to such matters as safe working conditions, manufacturing practices, fire hazard control, environmental protection and the disposal of hazardous and potentially hazardous substances and biological materials. We may incur significant costs to comply with such laws and their related regulations now or in the future. In addition, we are also subject to laws and regulations in foreign countries outside of the United States and Europe where we may seek to commercialize the ELAD System. In certain cases, these foreign laws and regulations may change at inopportune times and prevent the ELAD System’s timely commercialization. For example, several years after we submitted our 2007 regulatory package in China, we were notified of a then-newly-enacted 2009 regulation which prohibits the ELAD System’s approval in China until it is first approved in the United States. As such, we now do not expect regulatory consideration of approval in China until after approvals are received in the United States.
Legal Proceedings
We are not currently a party to any litigation, and we are not aware of any pending or threatened litigation against us that we believe would adversely affect our business, operating results, financial condition or cash flows. Our industry is characterized by frequent claims and litigation, including claims regarding patent and other intellectual property rights as well as product liability. As a result, in the future, we may be involved in various legal proceedings from time to time.
Employees
As of December 31, 2013, we had 70 employees, 11 of whom held Ph.D. or M.D. degrees. Of our employees, 46 were engaged in research and development, 11 in manufacturing and 13 in administration. None of our employees is represented by a labor organization or under any collective bargaining arrangement, and we have never had a work stoppage. We consider our employee relations to be good. We have also engaged numerous consultants.
Facilities
We lease approximately 39,100 square feet in San Diego in two different facilities under leases that expire in June 2017. Our corporate headquarters are in a 19,100 square foot facility, and we lease a 20,000 square foot facility for our manufacturing operations, which also includes laboratories for research and development. We believe that our current facilities are adequate to meet our ongoing needs and that additional facilities are available for lease to meet our future needs.
95
Table of Contents
Corporate Information
Vital Therapies, Inc. was formed in May 2003 to acquire the assets of VitaGen (formerly Hepatix) in a bankruptcy proceeding. Our predecessor companies developed the ELAD System, completing two pilot trials in acute liver failure and two randomized, controlled Phase 1 and Phase 2 trials in FHF, but failed to attract funds sufficient to continue development of the system. Beginning in June 2003, we refocused the company to pursue regulatory approval and commercialize the ELAD System in China. In 2007, we completed a pivotal trial in acute liver failure in subjects with viral hepatitis in China, and we submitted a marketing application to the CFDA. Our application is still under review in China and we do not expect approval in China until we have approval in the United States. We restarted our clinical program in the United States and Europe in 2009. Since then, we have run two Phase 2 trials and selected AILD and FHF as indications for our Phase 3 pivotal trial programs in the United States and Europe. We have also made significant improvements in the ELAD System bedside unit and our proprietary cartridge cell growth production process.
96
Table of Contents
Executive Officers and Directors
The following table sets forth the names, ages and positions of our executive officers and directors as of March 11, 2013:
| Name |
Age | Position | ||||
| Terence E. Winters, Ph.D. |
71 | Co-Chairman and Chief Executive Officer | ||||
| Robert A. Ashley, M.A. |
56 | Executive Vice President, Chief Technical Officer | ||||
| Duane Nash, M.D., J.D. |
43 | Executive Vice President, Chief Business Officer | ||||
| Michael V. Swanson, M.B.A. |
59 | Chief Financial Officer | ||||
| Aron P. Stern |
60 | Chief Administrative Officer, Secretary | ||||
| Andrew Henry |
49 | Vice President, Clinical Operations | ||||
| Andrea Loewen |
47 | Vice President, Regulatory Affairs and Quality Assurance | ||||
| Richard Murawski |
65 | Vice President, Manufacturing | ||||
| Muneer A. Satter, J.D., M.B.A(2)(3) |
53 | Co-Chairman and Lead Director | ||||
| Jean-Jacques Bienaimé(2) |
60 | Director | ||||
| Philip M. Croxford, M.B.A.(4) |
53 | Director | ||||
| Douglas E. Godshall, M.B.A.(1) |
49 | Director | ||||
| Errol R. Halperin, J.D., L.L.M.(1)(3) |
73 | Director | ||||
| J. Michael Millis, M.D.(3)(4) |
55 | Director | ||||
| Lowell E. Sears, M.B.A.(1)(2) |
63 | Director | ||||
| Randolph C. Steer, M.D., Ph.D.(2)(3)(4) |
64 | Director | ||||
| (1) | Member of the Audit Committee. |
| (2) | Member of the Compensation Committee. |
| (3) | Member of Nominating and Governance Committee. |
| (4) | Member of Quality and Innovation Committee. |
Executive Officers
Terence E. Winters, Ph.D. has served as the Chairman of our board of directors from June 2003 to March 2013. Dr. Winters became Co-Chairman of our board of directors in March 2013 and currently serves as such. Dr. Winters has served as our Chief Executive Officer since June 2003. Dr. Winters is also a Special Limited Partner of Valley Ventures, an investor in Vital Therapies since May 2003. He was formerly a General Partner of Columbine Venture Funds and Vice President of DS Ventures, a venture capital subsidiary of Diamond Shamrock Corp., a chemical, life science and petroleum company. Dr. Winters was previously a director of three public companies: CollaGenex Pharmaceuticals, Inc., a developer and marketer of proprietary medical therapies to the dermatology market, Orthologic Corp., a biotechnology company focused on development and commercialization of novel synthetic peptides for tissue repair and healing, and Clinuvel Pharmaceuticals, a global biopharmaceutical company committed to developing drugs for the treatment of a range of severe skin disorders. Dr. Winters has also served as a director of over 20 private companies. He earned a B.Sc. as well as a Ph.D. in chemistry from the University of Wales, U.K. He also completed a post-doctoral fellowship at the University of California, Los Angeles.
We believe Dr. Winters is qualified to serve on our board of directors based on his experience with corporate governance as a director of many private and public life science companies and over 30 years of experience as a general partner of venture capital funds with investments in life sciences and medical technology companies.
Robert A. Ashley, M.A. has served as our Executive Vice President and Chief Technical Officer since September 2013. Between May 2008 and September 2013 he served as our Vice President and Chief Operating Officer. Mr. Ashley’s career in the pharmaceutical industry extends for 34 years. He was formerly Chairman, President and Chief Executive Officer of AmpliMed Corporation, a privately-held cancer drug development
97
Table of Contents
company, from January 2004 to March 2007, and Senior Vice President of Commercial Development at CollaGenex Pharmaceuticals, Inc., a publicly-held pharmaceutical company, from September 1994 to December 2003. Prior to that he held positions of increasing responsibility at Bristol-Myers Squibb from January 1989 to September 1994, and with Amersham International from 1979 to 1989. He earned a Masters Degree in Biochemistry from Oxford University. Mr. Ashley is the inventor of several issued and pending patents, as well as the author of several scientific papers. He serves on the Board of Directors of Rowpar Pharmaceuticals, a privately-held manufacturer of proprietary dental pharmaceuticals.
Duane Nash, M.D., J.D. has served as our Executive Vice President since May 2013 and as our Chief Business Officer since March 2012. Between March 2012 and May 2013, he also served as our Medical Director. Dr. Nash completed his internship in general surgery at the University of California at San Francisco during which he served as a member of the liver transplant team. Dr. Nash also practiced as an attorney from November 2002 to February 2008, most recently at the law firm of Davis Polk, where he focused on intellectual property litigation and corporate matters. Dr. Nash joined Vital Therapies from Wedbush PacGrow Life Sciences where he was employed from March 2009 to March 2012 serving most recently as Vice President in Equity Research. Before that he was a research analyst at Pacific Growth Equities from April 2008 through March 2009, which was subsequently acquired by Wedbush Securities, Inc. Dr. Nash has served on the board of directors of Akebia Therapeutics, Inc., a publicly-traded biotech company focused on the treatment of anemia and vascular disease, since May 2013, and on the board of directors of Aerpio Therapeutics Inc., a clinical-stage biopharmaceutical company focused on advancing innovative therapies for vascular diseases, since September 2012. Dr. Nash earned a B.A. in biology from Williams College, an M.D. from Dartmouth Medical School, a J.D. from the University of California, Berkeley, and an M.B.A. from the University of Oxford.
Michael V. Swanson, M.B.A. joined us in August 2013 as our Chief Financial Officer. Mr. Swanson has over 20 years of experience in senior financial positions in both public and private life sciences companies. Mr. Swanson was Chief Financial Officer of Amira Pharmaceuticals, Inc., a pharmaceutical company focused on the discovery and early development of drugs to treat inflammatory and fibrotic diseases, from May 2008 until the company was acquired in September 2011, and of Panmira Pharmaceuticals, LLC, a spin out from Amira from September to December 2011. Since January 2012, Mr. Swanson has been providing financial consulting services to development stage companies. From July 2000 to April 2008, Mr. Swanson served in senior finance positions including Senior Vice President, Finance and Chief Financial Officer at Prometheus Laboratories Inc., a specialty pharmaceutical company marketing and selling pharmaceutical products and diagnostic testing services for gastrointestinal diseases and disorders. Previously, Mr. Swanson was Senior Vice President and Chief Financial Officer of Advanced Tissue Sciences, Inc., a publicly-traded biomedical company, where he served in senior financial positions for over ten years. Mr. Swanson also served as Director of Finance of the Fisher Scientific Group, Inc., a health and scientific technology company, and its parent, The Henley Group, Inc., a widely diversified holding company. Mr. Swanson began his career working approximately nine years with the public accounting firm of Deloitte Haskins & Sells, now Deloitte & Touche LLP. Mr. Swanson earned a B.S. in business administration from the California Polytechnic State University at San Luis Obispo and an M.B.A. from the University of Southern California. He is also a Certified Public Accountant (inactive).
Aron P. Stern, M.B.A. has served as our Chief Administrative Officer since August 2013 and as our Secretary since October 2005. Between June 2003 and August 2013, Mr. Stern served as our Treasurer, Vice President and Chief Financial Officer. Mr. Stern has over 20 years of experience in capital formation, acquisitions, financial strategy and financial and operational management in growth-stage high technology and biotechnology companies. He previously was Chief Financial Officer at each of Protein Polymer Technologies, Inc., a developer of a protein-based technology, 4-D Neuroimaging, a medical equipment manufacturing company, and VitaGen, Inc., our predecessor company. Mr. Stern also held positions at Apple Computer and Isis Pharmaceuticals, a developer of antisense drugs. Mr. Stern earned a B.S. in economics and business administration and an M.B.A. in finance and marketing from the University of California, Berkeley.
Andrew Henry has served as our Vice President of Clinical Operations since April 2013. Mr. Henry is responsible for the global implementation of our clinical program. Mr. Henry has 25 years of experience
98
Table of Contents
managing clinical trials in life science companies with roles at Schering-Plough Oncology, Novartis Oncology and MedImmune. Between January 2009 and February 2013, Mr. Henry served as Senior Director of Clinical Trial Management and Senior Director of Global Clinical Operational Strategy of MedImmune, a biopharmaceutical company that is under AstraZeneca’s biologics division. At MedImmune Mr. Henry oversaw operations of all clinical studies being performed by the company across all therapeutic areas. From November 1997 to August 2008, Mr. Henry held roles as Senior Clinical Research Scientist and Head Clinical Resources and Development Director at Novartis Oncology, an ethical pharmaceutical company, where he oversaw clinical studies and the Department of Clinical Research Scientists/Clinical Trial Heads. Mr. Henry earned a B.S. in biology and biopsychology from William Paterson University.
Andrea Loewen has served as our Vice President of Regulatory Affairs and Quality since July 2013 and oversees our quality and regulatory systems and develops regulatory strategies. Ms. Loewen has 24 years of experience in regulatory and quality management roles, including positions at Baxter Healthcare, Biogen Idec, and Shire Pharmaceuticals. From June 2009 to July 2013, she served as the Head of Regulatory Affairs for Shire Pharmaceuticals Regenerative Medicine business unit, where she was responsible for global regulatory strategy and filings for development stage and commercial combination products. Between March 2008 and June 2009, Ms. Loewen served as Senior Director of Regulatory Affairs for Ceregene, Inc., a development stage biotech company, and was responsible for global regulatory strategy and filings for combination products. Ms. Loewen earned her B.A. in biology from Gustavus Adolphus College.
Richard Murawski has served as our Vice President of Manufacturing since July 2013. Mr. Murawski has more than 40 years of experience in manufacturing facility design, construction, start-up, validation, and supply chain management, both domestically and internationally, including 17 major plant start-ups. From February 2013 to July 2013, Mr. Murawski was self-employed as a consultant. From June 2010 to February 2013, Mr. Murawski served as the Vice President/General Manager for Dendreon Corporation, a biotech manufacturing company, with responsibility for, among others, manufacturing, engineering, materials management, and facilities. Between June 2008 and July 2010, Mr. Murawski served as Chief Executive Officer of Murawski and Associates, a biotech consulting company, where he consulted companies on managing operations and biopharmaceutical facilities. From June 2002 to July 2008, Mr. Murawski served as Senior Vice President of Operations and Corporate Officer of Favrille, Inc., a biotech manufacturing company, and was responsible for manufacturing, engineering, materials management, facilities, technical services, and EHS functions. Mr. Murawski earned his B.S. in chemical engineering from the Newark College of Engineering at the New Jersey Institute of Technology.
Board of Directors
Muneer A. Satter, J.D., M.B.A. has served on our board of directors since March 2013 and is our Co-Chairman and Lead Director. Mr. Satter manages Satter Investment Management, or SIM, a private investment firm and family office with significant investments in several life sciences and medical technology companies. He has also managed the Satter Foundation, a private family foundation since 1997. Mr. Satter has served as Chairman or Co-Chairman of Akebia Therapeutics, a publicly-traded biotech company focused on the treatment of anemia and vascular disease, since May 2013. Since October 2013, he has served as Co-Chairman of Aerpio Therapeutics, a biotech company focused on diabetic macular edema and vascular leaks. Mr. Satter is Co-Founder and has served as Chairman of Restorsea, a company which holds exclusive rights to a unique enzyme for an anti-aging cream since May 2013. He has served as Co-Chairman of Linq3, a unique secure payment platform for lottery transactions since May 2013. He is Vice Chairman of the Board of the Goldman Sachs Foundation and GS Gives, where he is also Chairman of the Investment Committee overseeing $1.2 billion of assets. He is on the Board of the Nature Conservancy where he is Chairman of the Finance Committee overseeing a $1.6 billion endowment. He is also on the Board of Trustees of Northwestern University. He is on the Board of Advisors of the American Enterprise Institute. He is a member of the Council on Foreign Relations and is on the board of World Business Chicago, which is chaired by Chicago’s Mayor Rahm Emanuel. Mr. Satter
is a retired partner at Goldman Sachs, where he was at the firm for twenty-four years and a partner of the firm for
99
Table of Contents
sixteen years in the Merchant Banking Division. He was the Global Head of the Mezzanine Group, where he raised and managed over $30 billion of assets. He was also a senior member of the Investment Committee and Chairman of the Risk Committee for the Merchant Banking Division, which at the time had over $80 billion of assets under management. Additionally, he was former Co-Chairman of Room to Read, which builds 2,000 libraries and schools per year in developing countries. Mr. Satter earned a B.A. from Northwestern University and a J.D. and M.B.A. from Harvard Law School and Harvard Business School.
We believe Mr. Satter is qualified to serve on our board of directors because of his extensive experience in merchant banking and venture capital investments and his experience as an investor and director of other companies in the life sciences and medical technology industries.
Jean-Jacques Bienaimé has served on our board of directors since September 2013. Since May 2005, Mr. Bienaimé has served as the chief executive officer and as a director of BioMarin Pharmaceutical Inc., a publicly-traded company that develops and commercializes innovative pharmaceuticals for serious diseases and medical conditions. From November 2002 to April 2005, Mr. Bienaimé served as chairman, chief executive officer and president of Genencor, a biotechnology company focused on industrial bioproducts and targeted cancer biotherapeutics. Prior to joining Genencor, Mr. Bienaimé was chairman, president and chief executive officer of SangStat Medical Corporation, another biotechnology company. He became president of SangStat Medical Corporation, a global pharmaceutical company, in 1998 and chief executive officer in 1999. Prior to joining SangStat Medical Corporation, Mr. Bienaimé held various management positions from 1992 to 1998 with Rhône-Poulenc Rorer Pharmaceuticals (now known as Sanofi-Aventis), including Senior Vice President of Corporate Marketing and Business Development, and Vice President and General Manager of the Advanced Therapeutic and Oncology division. Mr. Bienaimé currently serves on the boards of Intermune, Inc., a biotechnology company, Portola Pharmaceuticals and The Biotech Industry Organization. He earned an M.B.A. from the Wharton School at the University of Pennsylvania and an B.S. in economics from the Ecole Superieure de Commerce de Paris.
We believe that Mr. Bienaimé is qualified to serve on our board of directors based on his extensive experience in the management of biotechnology organizations, business development, and sales and marketing of both biotechnology and pharmaceutical products.
Philip M. Croxford, M.B.A. has served on our board of directors since 2009. Mr. Croxford has been Senior Vice President of Global Operations for LifeCell Corporation, a tissue biotechnology engineering company, since September 2013, and is a member of its Executive Leadership Team. Prior to his promotion, Mr. Croxford was Senior Vice President of Global Commercial Operations for LifeCell. From June 2008 to December 2012 he was President and Chief Executive Officer of ArjoHuntleigh, North America, a member of The Getinge AB Group, a global medical and safety technology company listed on the OMX Nordic Exchange. From February 2006 to October 2007, Mr. Croxford was President and Chief Executive Officer and member of the board of directors of Draeger Medical, Inc., a private medical device company. Mr. Croxford also served as Group Vice President and Worldwide General Manager of Arrow International, Inc. and as an Operating Company Board Member and Vice President for the Worldwide Wound Management Business of Ethicon, a Johnson & Johnson Company. Mr. Croxford also held other executive management positions with Johnson & Johnson Company. He earned a B.Sc. (Honors) degree in pharmacy from the University of Manchester, School of Pharmacy, in England. Mr. Croxford is also a licensed member of the Royal Pharmaceutical Society of Great Britain. He earned a Post Graduate Diploma in marketing from the Royal Chartered Institute of Marketing, and is a Chartered Member of the Institute. He earned his M.B.A. from Heriot Watt University, Edinburgh Business School, United Kingdom.
We believe Mr. Croxford is qualified to serve on our board of directors based on his significant operational and leadership experience and extensive experience in the life sciences and medical technology industries.
Douglas E. Godshall, M.B.A. has served on our board of directors since May 2013. Mr. Godshall has been the Chief Executive Officer of HeartWare International, Inc., a developer of a range of implantable mechanical
100
Table of Contents
circulatory assist devices, since September 2006 and a director since October 2006. Prior to joining HeartWare, Mr. Godshall served in various executive and managerial positions at Boston Scientific Corporation, a manufacturer and a developer of medical supplies and medical devices in a variety of fields, including as a member of Boston Scientific’s Operating Committee and President, Vascular Surgery. Previously, Mr. Godshall spent five years as Vice President, Business Development, at Boston Scientific, where he was focused on acquisition strategies for the cardiology, electrophysiology, neuroradiology and vascular surgery divisions. In March 2012, Mr. Godshall was appointed a director of pSivida Corp., a developer of drug delivery products for treatment of back-of-the-eye diseases. Mr. Godshall earned a B.A. in business from Lafayette College and an M.B.A from Northeastern University.
We believe that Mr. Godshall is qualified to serve on our board of directors based on his experience as an executive officer and director of several life sciences and medical technology companies.
Errol R. Halperin, J.D., L.L.M. has served on our board of directors since December 2012. Mr. Halperin has been a senior partner of DLA Piper since January 1979 until December 2013. At the present time, Mr. Halperin is a senior strategic advisor to DLA Piper (U.S.). In his practice, Mr. Halperin has provided general business advice to clients in a broad range of sectors, including the manufacturing, real estate industry and real estate investment trust industry. Mr. Halperin’s practice is concentrated in the areas of mergers and acquisitions, corporate law, international transactions, real estate law and federal income tax law. Mr. Halperin has also served on the board of Equity Residential, a public REIT, and private company boards. Currently, he is a director of Elkay Manufacturing Company, a privately held corporation, and Pangea Properties, a real estate investment and management company. He also currently serves as a director of other late stage venture companies. Before joining DLA Piper, from June 1972 to January 1979 he was legislation counsel of the Joint Committee on Taxation of the United States Congress; and was an assistant branch chief of the Legislation and Regulations Division of the Chief Counsel for the Internal Revenue Service from June 1968 to June 1972. Mr. Halperin earned a B.S. and a J.D. from DePaul University and an L.L.M. in taxation from New York University.
We believe that Mr. Halperin is qualified to serve on our board of directors based on his experience as an attorney practicing for a nationally known firm in the areas of mergers and acquisitions and corporate transactions and his involvement in advising venture capital backed companies.
J. Michael Millis, M.D. has served on our board of directors since 2006. Dr. Millis is a Professor of Surgery and Chief of the Section of Transplantation at the University of Chicago. Dr. Millis currently serves on the medical staff at the University of Chicago Hospital. His current research explores the application of cellular technology on patient care, including how hepatocyte transplantation, extracorporeal assist technology and stem cells can assist in the care of patients with liver disease or liver tumors. Dr. Millis has been associated with the ELAD System since 1994, and was an investigator on the Phase 1 clinical trial. He has been the chairman of the Vital Therapies Clinical Advisory Board since 2003 and chairman of the China Clinical Advisory Board since 2004. Dr. Millis currently serves or has served in the past on a variety of committees, such as the American Association for the Study of Liver Diseases’ Ethics Committee, the American Society of Transplant Surgeons, Studies of Pediatric Liver Transplantation Scientific Advisory Board and Publication and Research Committee, and the ROBI Liver Intestinal Subcommittee, for which he is the former chairman. Dr. Millis is also a member of the Medical Advisory Committee of the American Liver Foundation Illinois Chapter. He also serves on the board of directors of Gift of Hope (the organ procurement organization for Illinois) and the Editorial Board of the American Journal of Transplantation, Transplantation, and Liver Transplantation. He earned a B.A. in chemistry and political science from Emory University and an M.D. with high honors from the University of Tennessee. He completed his surgical residency, clinical and research fellowship in liver transplantation at the University of California, Los Angeles.
We believe that Dr. Millis is qualified to serve on our board of directors based on his participation on multiple advisory committees and boards and his research in the treatment of liver disease.
101
Table of Contents
Lowell E. Sears, M.B.A. has served on our board of directors since May 2013. Mr. Sears is the Chairman and CEO of Sears Capital Management, a venture investment and portfolio management firm specializing in life sciences. He has served on the board of directors of Cellerant Therapeutics, Inc., a clinical stage biotechnology company focused on the regulation of the hematopoietic, or blood-forming, system, since February 2012 and has been Chairman of the Board since June 2012. Mr. Sears has also served on the board of directors of SymBio Pharmaceuticals, KK, Ltd., a biotechnology company that is engaged in identifying and development of therapeutics for the treatment of leukemia, multiple myeloma and lymphoma, since September 2005. From 1986 until 1994, Mr. Sears was a part of the senior management team of Amgen, Inc., a developer and manufacturer of therapeutics targeting cancers, kidney ailments, inflammatory disorders, and metabolic diseases, where he was Chief Financial Officer as well as the Senior Vice President responsible for the Asia Pacific Region. Prior to joining Amgen, Mr. Sears held senior planning and financial positions with Atlantic Richfield Company, an oil company from 1976 until 1986, including Chief Financial Officer for its Ventures Division. He earned a B.A. in economics from Claremont McKenna College and an M.B.A. from the Stanford University Graduate School of Business.
We believe that Mr. Sears is qualified to serve on our board of directors based on his experience as a senior manager and director of private and publicly traded companies, including several in the life sciences industry.
Randolph C. Steer, M.D., Ph.D. has served on our board of directors since May 2005. Dr. Steer has been an independent pharmaceutical, biotechnology and medical devices consultant since 1989. Dr. Steer has served as Associate Director of Medical Affairs at Marion Laboratories, a then-public pharmaceutical company; Medical Director at Ciba Consumer Pharmaceuticals (a division of Ciba-Geigy Corporation, a then-public pharmaceutical company); Vice President, Senior Vice President and Member of the Executive Committee at Physicians World Communications Group; Chairman, President and Chief Executive Officer of Advanced Therapeutics Communications International, a drug regulatory group serving the United States, Mexico, Latin America, the Pacific Rim, Europe and Japan; Chairman and Chief Executive Officer of Vicus.com, Inc.; and President and Chief Operating Officer of Capstone Therapeutics Corp., a public biotechnology company. Dr. Steer is a member of the board of directors of Techne Corporation, a public biotechnology company, and the Board of Trustees of the Mayo Clinic. He earned his B.A. in Physiology from University of Minnesota. He earned an M.D. from the Mayo Medical School and a Ph.D. from the University of Minnesota where he also completed a residency and subspecialty training in clinical and chemical pathology. He is a Fellow of the American College of Clinical Pharmacology.
We believe that Dr. Steer is qualified to serve on our board of directors based on his experience as a senior manager in multiple biotechnology and medical device companies.
Family Relationships
There are no family relationships among any of our directors and executive officers.
Board Composition
Our board of directors is currently composed of nine members. At each annual meeting of stockholders following the offering, a class of directors will be elected for a three-year term to succeed the class whose terms are then expiring. The terms of the directors will expire upon the election and qualification of successor directors at the annual meeting of stockholders to be held during the years 2015 for the Class I directors, 2016 for the Class II directors and 2017 for the Class III directors.
The Class I directors will be Mr. Croxford and Drs. Steer and Winters.
The Class II directors will be Messrs. Bienaimé, Godshall and Satter.
The Class III directors will be Messrs. Halperin and Sears and Dr. Millis.
102
Table of Contents
The division of our board of directors into three classes with staggered three-year terms may delay or prevent a change of our board of directors or a change in control. See the sections of this prospectus captioned “Description of Capital Stock—Anti-Takeover Effects of Delaware Law and Our Certificate of Incorporation, Bylaws and Fourth Amended and Restated Investors’ Rights Agreement” for a discussion of other anti-takeover provisions found in the certificate of incorporation.
Following this offering, certain of our stockholders affiliated with Mr. Satter, referred to as the Satter Investors, will retain contractual rights to nominate up to 40% of our directors, as provided in our Fourth Amended and Restated Investors’ Rights Agreement, dated as of August 28, 2013, as amended. To date, the Satter Investors have not exercised their rights to designate any candidates for nomination but have reserved the right to do so in the future. The rights of the Satter Investors to nominate directors are described under the caption “Description of Capital Stock—Anti-Takeover Effects of Delaware Law and Our Certificate of Incorporation, Bylaws and Fourth Amended and Restated Investors’ Rights Agreement.”
Director Independence
Upon the closing of this offering, we anticipate that our common stock will be listed on The NASDAQ Global Market, or NASDAQ. Under the rules of NASDAQ, independent directors must comprise a majority of a listed company’s board of directors within a specified period of the closing of this offering. In addition, the rules of NASDAQ require that, subject to specified exceptions, each member of a listed company’s audit, compensation and nominating and governance committees be independent. Audit committee members and compensation members must also satisfy separate independence criteria set forth in Rule 10A-3 and Rule 10C-1, respectively, under the Securities Exchange Act of 1934, as amended. Under the rules of NASDAQ, a director will only qualify as an “independent director” if, among other things, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
To be considered independent for purposes of Rule 10A-3 and under the rules of NASDAQ, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, our board of directors, or any other board committee: (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries; or (2) be an affiliated person of the listed company or any of its subsidiaries.
To be considered independent under the rules of NASDAQ, a member of a compensation committee of a listed company may not, other than in his or her capacity as a member of the compensation committee, our board of directors, or any other board committee accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries. Additionally, in determining whether a director is eligible to serve on the compensation committee, the listed company’s board must also consider the affiliate status of the director and whether such affiliation would impair the director’s judgment as a member of the compensation committee.
Our board of directors undertook a review of its composition, the composition of its committees and the independence of our directors and considered whether any director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our board of directors has determined that none of Drs. Millis and Steer and Messrs. Croxford, Godshall, Halperin, Satter, Bienaimé and Sears, representing eight of our nine directors, has a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the rules of NASDAQ.
103
Table of Contents
In making this determination, our board of directors considered the relationships that each nonemployee director has with us and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director.
Lead Director
Our board of directors has established certain corporate governance principles in connection with this offering. Our board of directors determined as part of our bylaws that one of our directors should serve as a lead director. Mr. Satter has been appointed to serve as our Lead Director and Co-Chairman. As Lead Director, Mr. Satter will, among other responsibilities, coordinate the scheduling of, and preparing the agenda for, meetings of our board of directors, stockholders and independent directors, preside (or designate another director to preside) over such meetings, and define the scope, quality, quantity and timeliness of the flow of information between management and our board of directors that is necessary for our board of directors to effectively and responsibly perform its duties. While we have offered Mr. Satter the same compensation for his service as a member of our board of directors and as Lead Director as we pay to each other nonemployee member of our board of directors, Mr. Satter has declined to receive (and has not received) any compensation for his service on our board of directors or as Lead Director.
Our Fourth Amended and Restated Investors’ Rights Agreement, dated August 28, 2013, as amended, or the Senior Preferred IRA, provides that for so long as Mr. Satter and Dr. Winters both serve as members of our board of directors, each shall serve as Co-Chairman of our board of directors and Mr. Satter shall serve as our Lead Director. In the event that Mr. Satter serves as a member of our board of directors at a time when Dr. Winters does not, Mr. Satter will serve as our Chairman of the board and Lead Director. This provision survives the closing of this offering. Dr. Winters will serve as Co-Chairman only so long as he is both a director and Chief Executive Officer. As the Satter Investors’ ownership percentage decreases, their rights to nominate directors similarly decreases. The rights of the Satter Investors to nominate directors are described in more detail under the caption “Description of Capital Stock—Anti-Takeover Effects of Delaware Law and Our Certificate of Incorporation, Bylaws and Fourth Amended and Restated Investors’ Rights Agreement.”
Board Committees
Our board of directors has an audit committee, a compensation committee, a nominating and governance committee and a quality and innovation committee, each of which has the composition and the responsibilities described below.
Audit Committee
Our audit committee oversees our corporate accounting and financial reporting process and assists our board of directors in monitoring our financial systems and our legal and regulatory compliance. Our audit committee will also:
| • | oversee the work of our independent auditors; |
| • | approve the hiring, discharging and compensation of our independent auditors; |
| • | approve engagements of the independent auditors to render any audit or permissible non-audit services; |
| • | review the qualifications, independence and performance of the independent auditors; |
| • | review our financial statements and review our critical accounting policies and estimates; |
| • | review the adequacy and effectiveness of our internal controls; |
| • | review our policies with respect to risk assessment and risk management; |
| • | review and monitor our policies and procedures relating related person transactions; and |
| • | review and discuss with management and the independent auditors the results of our annual audit, our quarterly financial statements and our publicly filed reports. |
104
Table of Contents
The members of our audit committee are Messrs. Godshall, Halperin and Sears. Mr. Sears serves as the chairman. Our board of directors has determined that each of the members of our audit committee is independent under Rule 10A-3 and the rules of NASDAQ and financially literate, and that Mr. Sears qualifies as an audit committee financial expert within the meaning of the rules and regulations of the SEC.
Compensation Committee
Our compensation committee oversees our corporate compensation programs. The compensation committee will also:
| • | review and recommend for approval by the independent members of our board of directors policies, plans and arrangements relating to compensation and benefits of our officers and employees; |
| • | review and recommend for approval by the independent members of our board of directors corporate goals and objectives relevant to compensation of our Chief Executive Officer and other executive officers; |
| • | evaluate the performance of our executive officers in light of established goals and objectives; |
| • | recommend compensation of our executive officers based on its evaluations; |
| • | administer the issuance of stock options and other awards under our equity compensation plans; |
| • | review and discuss with management the compensation discussion and analysis required by SEC rules; |
| • | engage a compensation consultant, legal counsel or other advisors (other than in-house counsel) to advise on executive compensation and assess the independence of each in accordance with NASDAQ; |
| • | evaluate whether any compensation consultant, legal counsel or other advisor (other than in-house legal counsel) has a conflict of interest in accordance with the SEC rules; and |
| • | prepare the annual compensation committee report required by SEC rules. |
The members of our compensation committee are Messrs. Bienaimé, Satter and Sears and Dr. Steer. Dr. Steer is the chairman of our compensation committee. Our board of directors has determined that each member of our compensation committee is independent under the current rules of NASDAQ.
Nominating and Governance Committee
Our nominating and governance committee oversees and assists our board of directors in reviewing and recommending nominees for election as directors. The nominating and governance committee will also:
| • | evaluate and make recommendations regarding the organization and governance of our board of directors and its committees; |
| • | assess the performance of members of our board of directors and make recommendations regarding committee and chair assignments; |
| • | recommend desired qualifications for board of director membership and conduct searches for potential members of our board of directors; and |
| • | review and make recommendations with regard to our corporate governance guidelines. |
The members of our nominating and governance committee are Messrs. Halperin and Satter and Drs. Millis and Steer. Mr. Halperin serves as the chairman. Our board of directors has determined that each member of our nominating and governance committee is independent under the current rules of NASDAQ.
105
Table of Contents
Quality and Innovation Committee
Our quality and innovation committee provides assistance to our board of directors in its oversight of product quality and safety, manufacturing practices, scientific and technical direction and human and animal studies. The members of our quality and innovation committee are Drs. Millis and Steer and Mr. Croxford. Dr. Millis is the chairman of our quality and innovation committee.
Committee Charters
Our board of directors has adopted charters for each of the audit committee, the compensation committee, the nominating and governance committee and the quality and innovation committee. These charters will be available on the corporate governance section of our website, which is located at www.vitaltherapies.com.
Compensation Committee Interlocks and Insider Participation
None of our executive officers serves, or in the past has served, as a member of our board of directors or compensation committee, or other committee serving an equivalent function, of any entity that has one or more executive officers who serve as members of our board of directors or our compensation committee. None of the members of our compensation committee is, or has ever been, an officer or employee of our company.
Code of Business Conduct and Ethics
Our board of directors has adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. Following the closing of this offering, a copy of the code of business conduct will be available on the corporate governance section of our website, which is located at www.vitaltherapies.com. If we make any substantive amendments to, or grant any waivers from, the code of business conduct and ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website.
106
Table of Contents
EXECUTIVE AND DIRECTOR COMPENSATION
Summary Compensation Table
The following table summarizes the compensation paid to our Chief Executive Officer and our two other highest paid executive officers during the year ended December 31, 2012 and 2013. We refer to these executive officers as our named executive officers.
| Name and Principal Position |
Year | Salary ($) |
Bonus ($) |
Option Awards ($)(1) |
All Other Compensation ($)(5) |
Total ($) |
||||||||||||||||||
| Terence E. Winters, Ph.D.(2) |
2013 | 418,125 | 110,719 | — | 17,946 | 546,790 | ||||||||||||||||||
| Co-Chairman and Chief Executive Officer |
2012 | 325,000 | — | 266,174 | — | 591,174 | ||||||||||||||||||
| Michael V. Swanson(3) |
2013 | 94,791 | 20,854 | 657,900 | 3,260 | 776,805 | ||||||||||||||||||
| Chief Financial Officer |
||||||||||||||||||||||||
| Andrew Henry(4) |
2013 | 201,875 | 35,076 | 281,554 | 84,139 | 602,644 | ||||||||||||||||||
| Vice President, Clinical Operations |
||||||||||||||||||||||||
| (1) | The amounts in the “Option Awards” column reflect the aggregate grant date fair value of stock options granted during 2012 computed in accordance with the provisions of Accounting Standards Codification (ASC) 718, Compensation—Stock Compensation. The assumptions that we used to calculate these amounts are discussed in Note 9 to our financial statements appearing at the end of this prospectus. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the stock options, the exercise of the stock options, or the sale of the common stock underlying such stock options. |
| (2) | Dr. Winters became a full-time employee in May 2012. |
| (3) | Mr. Swanson became a full-time employee in August 2013. |
| (4) | Mr. Henry became a full-time employee in April 2013. |
| (5) | Includes $70,202 in relocation expenses paid to Mr. Henry and company paid premiums on health plans and group term life insurance for each of our executive officers. |
Outstanding Equity Awards at Fiscal Year-End for Fiscal 2013
The following table sets forth certain information concerning outstanding equity for our named executive officers awards at fiscal year-end December 31, 2013:
| Name |
Vesting Commencement Date(2) |
Number of Securities Underlying Unexercised Options Exercisable(1) |
Number of Securities Underlying Unexercised Options Unexercisable |
Option Exercise Price |
Option Expiration Date |
|||||||||||||||
| Terence E. Winters, Ph.D. |
2/8/2012 | (3) | 128,394 | — | $ | 0.43 | 3/31/2022 | |||||||||||||
| Co-Chairman and Chief Executive Officer |
9/13/2012 | (4) | 386,672 | — | 8.00 | 9/25/2022 | ||||||||||||||
| Michael V. Swanson |
9/9/2013 | (5) | 90,000 | — | 10.50 | 9/8/2023 | ||||||||||||||
| Chief Financial Officer |
||||||||||||||||||||
| Andrew Henry |
5/13/2013 | (6) | 57,343 | — | 8.00 | 5/12/2023 | ||||||||||||||
| Vice President, Clinical Operations |
||||||||||||||||||||
| (1) | The options listed above are subject to an early exercise right and may be exercised in full prior to the vesting of the shares underlying such options. Any shares purchased by early exercising unvested options are subject to repurchase by the company in event the optionee ceases providing services to the company and are released from this repurchase right in accordance with the options original vesting schedule. Vesting of all options or early exercise shares is subject to continued service on the applicable vesting date. |
107
Table of Contents
| (2) | All options listed above vest in equal monthly installments over the four year period following the vesting commencement date. |
| (3) | 85,596, or 46%, of the shares subject to these options were vested as of December 31, 2013 and 58,361 of these have been exercised as of December 31, 2013. |
| (4) | 120,834, or 31%, of the shares subject to these options were vested as of December 31, 2013. |
| (5) | 5,624, or 6%, of the shares subject to these options were vested as of December 31, 2013. Subsequent to year end, this option grant was re-priced to $8.00. |
| (6) | 8,361, or 15%, of the shares subject to these options were vested as of December 31, 2013. |
Executive Employment Arrangements
Terence E. Winters
We entered into an employment letter agreement, dated October 31, 2013, with Dr. Winters, which sets forth the terms and conditions of his employment with us. The employment letter agreement has no specific term and provides for at-will employment. This agreement supersedes all existing agreements he may have with us concerning his employment relationship. The agreement provides that Dr. Winters’ current annual base salary is $450,000 and he is eligible for an annual bonus equal to 40% of his base salary.
Michael V. Swanson
We entered into an employment letter agreement, dated August 26, 2013, with Mr. Swanson, our current Chief Financial Officer, which sets forth the terms and conditions of his employment with us. The employment letter agreement has no specific term and provides for at-will employment. Mr. Swanson’s current annual base salary is $325,000 and his annual bonus opportunity is 25% of his annual base salary. In addition, Mr. Swanson received an option grant to purchase 90,000 shares of our common stock on September 9, 2013 at an exercise price per share of $10.50 (re-priced to $8.00 per share in February 2014) that vests over a period of four years from his employment start date in equal monthly installments, subject to Mr. Swanson continuing to provide services to us.
Andrew Henry
We entered into an employment letter agreement, dated April 8, 2013, with Mr. Henry, our current Vice President of Clinical Operations, which sets forth the terms and conditions of his employment with us. The employment letter agreement has no specific term and provides for at-will employment. Mr. Henry’s current annual base salary is $285,000 and his annual bonus opportunity is 25% of his annual base salary. In addition, Mr. Henry received an option grant to purchase 57,343 shares of our common stock on May 13, 2013 at an exercise price per share of $8.00 that vests over a period of four years from his employment start date in equal monthly installments, subject to Mr. Henry continuing to provide services to us.
Executive Change of Control and Severance Agreements
We expect that each of our executive officers will enter into a change of control and severance agreement, which provides for the severance and change of control benefits described below. Each change of control and severance agreement will become effective immediately prior to the effectiveness of the registration statement to which this prospectus forms a part and will supersede any existing agreement or arrangement the executive may have with us that provides for severance and/or change of control payments or benefits.
108
Table of Contents
If prior to the two-month period before or after the twelve-month period following a change of control (such period, the “Change of Control Period”), an executive officer’s employment is terminated without “cause” or an executive officer resigns for “good reason” (as such terms are defined in the change of control and severance agreement), such officer will be eligible to receive the following benefits if such officer timely signs and does not revoke a release of claims:
| • | continued payment of base salary for a period of six months (12 months in the case of Dr. Winters); and |
| • | reimbursement by us for up to six months (12 months in the case of Dr. Winters) of COBRA premiums to continue health insurance coverage for such officer and such officer’s eligible dependents, or taxable monthly payments for the equivalent period in the event payment for COBRA premiums would violate applicable law. |
If, within the Change of Control Period, such officer’s employment is terminated without cause or such officer resigns for good reason, such officer will be entitled to the following benefits if such officer timely signs a release of claims:
| • | a lump sum payment equal to (x) 12 months (18 months in the case of Dr. Winters) his annual base salary (for the year of the change of control or such officer’s termination, whichever is greater), plus (y) 1x (1.5x in the case of Dr. Winters) the greater of: (A) such officer’s target annual bonus (for the year of the change of control or such officer’s termination, whichever is greater) or (B) such officer’s actual bonus for performance relating to the calendar year immediately prior to the calendar year of such officer’s termination; |
| • | reimbursement by us for up to 12 months (18 months in the case of Dr. Winters) of COBRA premiums to continue health insurance coverage for such officer and such officer’s eligible dependents, or taxable monthly payments for the equivalent period in the event payment for COBRA premiums would violate applicable law; and |
| • | 100% accelerated vesting of all outstanding equity awards. |
In addition, in the event any of the amounts provided for under these agreements or otherwise payable to our executive officers would constitute “parachute payments” within the meaning of Section 280G of the Internal Revenue Code and could be subject to the related excise tax, the executive officer would be entitled to receive either full payment of benefits under this agreement or such lesser amount which would result in no portion of the benefits being subject to the excise tax, whichever results in the greater amount of after-tax benefits to the executive officer. The agreements do not require us to provide any tax gross-up payments.
Director Compensation
The following table summarizes compensation paid to our nonemployee directors during or with respect to the fiscal year ended December 31, 2013. Messrs. Bienaimé, Godshall, Satter and Sears were not elected to our board of directors until 2013.
| Name |
Fees Earned or Paid in Cash ($) |
Option Awards ($)(1) |
Total ($) | |||||||||
| Jean-Jacques Bienaimé |
11,750 | 548,250 | (3) | 560,000 | ||||||||
| Philip M. Croxford |
34,750 | 18,413 | (4) | 53,163 | ||||||||
| Douglas E. Godshall |
29,875 | 399,797 | (5) | 429,672 | ||||||||
| J. Michael Millis, M.D. |
60,355 | (2) | — | (6) | 60,355 | |||||||
| Randolph C. Steer, M.D., Ph.D. |
39,750 | — | (7) | 39,750 | ||||||||
| Errol R. Halperin |
34,000 | 347,834 | (8) | 381,834 | ||||||||
| Lowell E. Sears |
38,500 | 399,797 | (9) | 438,297 | ||||||||
109
Table of Contents
| (1) | The amounts in the “Option Awards” column reflect the aggregate grant date fair value of stock options granted during the fiscal year computed in accordance with the provisions of ASC 718. The assumptions that we used to calculate these amounts are discussed in Note 9 to our financial statements appearing at the end of this prospectus. These amounts do not reflect the actual economic value that will be realized by the director upon the vesting of the stock options, the exercise of the stock options, or the sale of the common stock underlying such stock options. |
| (2) | Dr. Millis’ fees earned or paid in cash include $25,500 in fees earned from services as Chair of our Clinical Advisory Board. |
| (3) | Mr. Bienaimé has a total of 75,000 stock options outstanding as of December 31, 2013. Subsequent to December 31, 2013, this option grant was re-priced from $10.50 to $8.00. |
| (4) | Mr. Croxford had a total of 37,584 stock options outstanding as of December 31, 2013. |
| (5) | Mr. Godshall has a total of 81,425 stock options outstanding as of December 31, 2013. |
| (6) | Dr. Millis had a total of 33,834 stock options outstanding as of December 31, 2013. |
| (7) | Dr. Steer had a total of 50,175 stock options outstanding as of December 31, 2013. |
| (8) | Mr. Halperin has a total of 30,842 stock options outstanding as of December 31, 2013. |
| (9) | Mr. Sears has a total of 81,425 stock options outstanding as of December 31, 2013. |
Director Compensation Policy
Beginning April 1, 2012, we paid our nonemployee directors (other than Mr. Satter who has not received any compensation for his service on our board of directors) an annual fee of $20,000 each as compensation for their service on our board of directors. These fees are payable quarterly ($5,000 per quarter). For each board meeting exceeding four per year, an additional $2,500 are paid for each in person meeting and $500 for each telephonic meeting. In 2012, we also granted to each of Mr. Croxford and Drs. Millis and Steer 50,175 non-qualified common stock options, vesting monthly over four years, subject to the director’s continued service with us.
On May 13, 2013, our board of directors approved a new compensation policy for our nonemployee directors (other than Mr. Satter, who has elected not to receive any compensation for his service as Co-Chairman or a director) which provides for the following compensation to our non-employee directors:
| • | each nonemployee director will receive an annual base retainer of $30,000; |
| • | in addition to the $30,000 annual base retainer, the chairman of our audit committee will receive an annual fee of $15,000 and other members of our audit committee will receive an annual fee of $7,500; |
| • | in addition to the $30,000 annual base fee, the chairman of our other committees will receive an annual fee of $10,000 and other members of our other committees will receive an annual fee of $5,000; |
| • | in addition to the fees listed above, each director shall receive $2,500 for each meeting in excess of four meetings per year and $500 for each telephonic meeting. |
Pursuant to this policy, except as approved by our board of directors, each new director will receive an initial grant of 50,175 non-qualified common stock options at a strike price not less than fair market value of a share of our common stock on the date of grant with vesting monthly over forty-eight months, subject to the director’s continued service with us.
Dr. Winters is not eligible to receive any board compensation because he is an employee. Mr. Satter has declined board compensation. We reimburse our directors for their reasonable expenses incurred in connection with attending board and committee meetings (other than Mr. Satter, who is not seeking reimbursement for such expenses).
In June 2013, each of our nonemployee directors was offered the opportunity to participate in our Senior Preferred Stock financing and to receive one non-qualified option to purchase our common stock for each share of Senior Preferred Stock purchased in the financing, up to a total investment of $500,000. On September 18, 2013, in connection with his appointment to our board of directors, Mr. Bienaimé was granted an option to
110
Table of Contents
purchase 75,000 shares. As of December 31, 2013, Messrs. Croxford, Godshall, Halperin and Sears had been granted stock options exercisable at $8.00 per share for 3,750, 31,250, 20,667, and 31,250 shares of common stock, respectively, in connection with the purchase of Senior Preferred Stock. While the Satter Investors have participated in the Senior Preferred Stock financing and purchased shares of our Senior Preferred Stock, Mr. Satter has declined to receive (and has not received) any stock options.
In February 2014, Messrs. Croxford, Godshall, Halperin and Sears were granted stock options exercisable at $8.00 per share for 1,607, 13,393, 8,857 and 13,393 shares of common stock, respectively.
Equity Incentive Plans
2014 Equity Incentive Plan
Our board of directors has adopted, and we will ask our stockholders to approve, our 2014 Equity Incentive Plan, which we refer to as the 2014 Plan. The 2014 Plan will become effective as of the business day immediately prior to the effective date of this offering. Our 2014 Plan provides for the grant of incentive stock options, within the meaning of Section 422 of the Internal Revenue Code of 1986, as amended, or the Code, to our employees and any parent and subsidiary corporations’ employees, and for the grant of nonstatutory stock options, stock appreciation rights, restricted stock, restricted stock units, performance units and performance shares to our employees, directors and consultants and our parent and subsidiary corporations’ employees and consultants.
Share Reserve. The shares reserved for issuance under the 2014 Plan will initially include (a) those shares reserved but unissued under our 2012 Stock Option Plan, or the 2012 Plan, as of the effective date described above and (b) shares subject to awards granted under the 2012 Plan that expire, terminate, or are forfeited (provided that the maximum number of shares that may be added to the 2014 Plan pursuant to (a) and (b) is 3,200,000 shares). In addition, the 2014 Plan provides for annual increases in the number of shares available for issuance thereunder beginning upon the effectiveness of this offering, and on each annual anniversary of the effectiveness of this offering thereafter, equal to the least of:
| • | 1,200,000 shares of our common stock; |
| • | 3% of the outstanding shares of our common stock on the second-to-the-last day prior to each anniversary date of the effectiveness of this offering; or |
| • | an amount as our board of directors may determine. |
Administration. Our board of directors or a committee of our board of directors will administer our 2014 Plan. In the case of awards intended to qualify as “performance based compensation” within the meaning of Section 162(m) of the Code, the committee will consist of two (2) or more “outside directors” within the meaning of Section 162(m) of the Code. In addition, if we determine it is desirable to qualify transactions under the 2014 Plan as exempt under Rule 16b-3 of the Securities Exchange Act of 1934, as amended, or Rule 16b-3, such transactions will be structured to satisfy the requirements for exemption under Rule 16b-3. Subject to the provisions of the 2014 Plan, the administrator will have the power to determine the terms of the awards, including the exercise price, the number of shares subject to each such award, the exercisability of the awards and the form of consideration payable upon exercise. The administrator also will have the authority to institute, without stockholder approval, an exchange program whereby the exercise prices of outstanding awards may be increased or reduced, outstanding awards may be surrendered or cancelled in exchange for awards of the same type (which may have higher or lower exercise prices), awards of a different type or cash, or outstanding awards may be transferred to a third party.
Stock Options. Our 2014 Plan provides for the grant of incentive stock options and nonstatutory stock options. The grant of incentive stock options that qualify under Section 422 of the Code may only be made to our employees and those of any parent or subsidiary of ours. Nonstatutory stock options may be granted to our
111
Table of Contents
employees, directors, consultants, independent contractors and advisors. The exercise price of options granted under our 2014 Plan must at least be equal to the fair market value of our common stock on the date of grant. The term of an incentive stock option may not exceed ten (10) years, except that with respect to any participant who owns ten percent (10%) of the voting power of all classes of our outstanding stock as of the grant date, the term must not exceed five (5) years, and the exercise price must equal at least one hundred ten percent (110%) of the fair market value on the grant date. Subject to the provisions of our 2014 Plan, the administrator will determine the terms of all other options.
After termination of an employee, director or consultant, he or she may exercise his or her option, to the extent vested, for the period of time specified in the option agreement. In the absence of a specified time in the option agreement, the option generally will remain exercisable for twelve (12) months following a termination due to death or disability, and for three (3) months in all other cases. However, an option may not be exercised later than the expiration of its term.
Stock Appreciation Rights. Our 2014 Plan provides for the grant of stock appreciation rights. Stock appreciation rights allow the recipient to receive the appreciation in the fair value of our common stock between the exercise date and the date of grant. The administrator will determine the terms of stock appreciation rights, including when such rights become exercisable and whether to pay the increased appreciation in cash or with shares of our common stock, or a combination thereof, except that the per share exercise price for the shares to be issued pursuant to the exercise of a stock appreciation right will be no less than one hundred percent (100%) of the fair value per share on the date of grant. Stock appreciation rights expire under the same rules that apply to stock options.
Restricted Stock Awards. Our 2014 Plan provides for the grant of restricted stock awards. Restricted stock awards are shares of our common stock that vest in accordance with terms and conditions established by the administrator. The administrator will determine the number of shares of restricted stock granted to any employee. The administrator may impose whatever conditions to vesting it determines to be appropriate (for example, the administrator may set restrictions based on the achievement of specific performance goals or continued service to us); provided, however, that the administrator, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed. Recipients of restricted stock awards generally will have voting and dividend rights with respect to such shares upon grant without regard to vesting, unless the administrator provides otherwise. Shares of restricted stock that do not vest are subject to our right of repurchase or forfeiture.
Restricted Stock Units. Our 2014 Plan provides for the grant of restricted stock units. Restricted stock units are bookkeeping entries representing an amount equal to the fair value of one share of our common stock. Subject to the provisions of our 2014 Plan, the administrator determines the terms and conditions of restricted stock units, including the vesting criteria, which may include accomplishing specified performance criteria or continued service to us and the form and timing of payment. Notwithstanding the foregoing, the administrator, in its sole discretion, may accelerate the time at which any restrictions will lapse or be removed.
Performance Units and Shares. Our 2014 Plan provides for the grant of performance units and performance shares. Performance units and performance shares are awards that will result in a payment to a participant only if the vesting criteria established by the administrator are achieved or the awards otherwise vest. The administrator will establish organizational or individual performance goals in its discretion, which, depending on the extent to which they are met, will determine the number and the value of performance units and performance shares to be paid out to participants. Performance units shall have an initial dollar value established by the administrator prior to the grant date. Performance shares shall have an initial value equal to the fair value of our common stock on the grant date. Payment for performance units and performance shares may be made in cash or in shares of our common stock with equivalent value, or in some combination, as determined by the administrator.
Nonemployee Directors. Our 2014 Plan provides that all nonemployee directors will be eligible to receive all types of awards (except for incentive stock options) under the 2014 Plan. In connection with this offering, we intend
112
Table of Contents
to implement a formal policy pursuant to which our nonemployee directors will be eligible to receive equity awards under the 2014 Plan. Our 2014 Plan provides that the number of shares subject to awards granted to a nonemployee director in a given fiscal year will not be greater than 100,000, increased to 150,000 in the fiscal year of his or her initial service as a nonemployee director.
Transferability of Awards. Unless the administrator provides otherwise, our 2014 Plan generally does not allow for the transfer of awards and only the recipient of an option or stock appreciation right may exercise such an award during his or her lifetime.
Certain Adjustments. In the event of certain changes in our capitalization, to prevent diminution or enlargement of the benefits or potential benefits available under the 2014 Plan, the administrator will make adjustments to one or more of the number and class of shares that may be delivered under the 2014 Plan or the number, class and price of shares covered by each outstanding award and the numerical share limits contained in the 2014 Plan. In the event of our proposed liquidation or dissolution, the administrator will notify participants as soon as practicable and all awards, to the extent that they have not been previously exercised, will terminate immediately prior to the consummation of such proposed transaction.
Change in Control Transactions. Our 2014 Plan provides that in the event of a merger or change in control, as defined in the 2014 Plan, each outstanding award will be treated as the administrator determines, including, without limitation, that awards may be assumed or substituted for by the acquiring or succeeding corporation, awards may be terminated immediately prior to the consummation of the merger or change in control, awards may vest in whole or in part prior to or upon consummation of the merger or change in control and, to the extent the administrator determines, terminate on or immediately prior to the effectiveness of the merger or change in control, or awards may be terminated in exchange for cash or property or replaced with other rights or property. If a successor corporation does not assume or substitute an equivalent award for any outstanding award, then such award will fully vest, all restrictions on such award will lapse and all performance goals or other vesting criteria applicable to such award will be deemed achieved at one hundred percent (100%) of target levels. Additionally, if a successor corporation does not assume or substitute an option or stock appreciation right, the administrator will notify the participant in writing or electronically that such award will be exercisable for a specified period of time determined by the administrator prior to the transaction, and such award will then terminate upon the expiration of such period. The administrator will not be required to treat all awards similarly in the event of a merger or change in control. In addition, in the event of a change of control, options, stock appreciation rights, restricted stock, and restricted stock units held by our nonemployee directors, if any, will vest fully and become immediately exercisable, all restrictions on his or her restricted stock will lapse, and all performance goals or other vesting requirements for his or her performance shares and units will be deemed achieved at one hundred percent (100%) of target levels, and all other terms and conditions met.
Plan Amendments and Termination. Our 2014 Plan will automatically terminate in 2024, unless we terminate it sooner. In addition, our board of directors has the authority to amend, suspend or terminate the 2014 Plan provided such action does not impair the rights of any participant unless mutually agreed to in writing by the participant and us.
2012 Stock Option Plan
Our 2012 Plan was adopted by our board of directors in April 2012 and approved by our stockholders in September 2012. Our 2012 Plan provides for the grant of incentive stock options, within the meaning of Section 422 of the Internal Revenue Code of 1986, as amended, to our employees and any parent and subsidiary corporations’ employees, and for the grant of nonstatutory stock options and stock purchase rights to our employees, directors and consultants and any parent and subsidiary corporations’ employees and consultants. We will not grant any additional awards under our 2012 Plan following this offering; instead, we will grant awards under our 2013 Plan. However, our 2012 Plan will continue to govern the terms and conditions of outstanding awards granted thereunder.
113
Table of Contents
Share Reserve. As of December 31, 2013, under the 2012 Plan, options to purchase 139,071 shares of common stock had been exercised, options to purchase 3,098,573 shares of common stock were outstanding, and 29,492 shares were available for future grant under our 2012 Plan, for an aggregate of 3,128,065 total shares of our common stock reserved for issuance pursuant to our 2012 Plan. Subsequent to December 31, 2013, our board of directors approved an increase of 100,000 to the number of shares reserved for issuance under the 2012 Plan.
Administration. Our board of directors currently administers our 2012 Plan. Under our 2012 Plan, the administrator has the power to determine the terms of the awards, including the employees, directors and consultants who will receive awards, the exercise price, the number of shares subject to each award, the vesting schedule and exercisability of awards and the form of consideration payable upon exercise.
Stock Options. With respect to all incentive stock options granted under the 2012 Plan, the exercise price must at least be equal to the fair value of our common stock on the date of grant. With respect to all nonstatutory stock options granted under the 2012 Plan, the exercise price must at least be equal to eighty-five percent (85%) of the fair value of our common stock on the date of grant. However, with respect to any participant who owns ten percent (10%) of the voting power of all classes of our outstanding stock as of the grant date, the exercise price of the option must equal at least one hundred ten percent (110%) of the fair value on the grant date. The term of an option may not exceed ten (10) years, except that with respect to any participant who owns ten percent (10%) of the voting power of all classes of our outstanding stock as of the grant date, the term of an incentive stock option must not exceed five (5) years.
All options granted under the Plan are exercisable immediately subject to which we have a repurchase right that lapses as the option vests.
Unless otherwise provided by our board of directors in the option agreement, after termination of an employee, director or consultant (other than due to death or disability), he or she may exercise his or her option, to the extent vested, for a period of three (3) months following such termination. If termination is due to death or disability, the option will remain exercisable for a period of twelve (12) months following such termination, or such longer period of time as approved by our board of directors and specified in the stock option agreement. However, an option may not be exercised later than the expiration of its term.
Transferability. Unless the administrator provides otherwise, our 2012 Plan generally does not allow for the transfer of awards under the 2012 Plan other than by will, the laws of descent and distribution, and only the recipient of an option may exercise an award during his or her lifetime.
Certain Adjustments. In the event of certain changes in our capitalization, to prevent diminution or enlargement of the benefits or potential benefits available under the 2012 Plan, our board of directors will make adjustments to the number, class and price of shares covered by each outstanding award.
Change in Control Transactions. Our 2012 Plan provides that in the event of a change in control, as defined in the 2012 Plan, the surviving, continuing, successor, or purchasing corporation or other business entity or parent thereof may assume or substitute an equivalent award for each outstanding option under the 2012 Plan. If there is no assumption or substitution of outstanding options, such options will terminate upon the expiration of such stated notice period.
In addition, pursuant to their stock option agreements, certain optionees, including our named executive officers, are eligible for full vesting acceleration of their outstanding options in the event their service is terminated other than for cause or they resign from their service for good reason, in either case, within twelve months following a change in control.
Plan Amendments and Termination. Our board of directors has the authority to amend, alter, suspend or terminate the 2012 Plan, provided (i) such action does not impair the rights of any participant unless mutually
114
Table of Contents
agreed to in writing by the participant and us or (ii) such action is required to enable an option designated as an incentive stock option to qualify as an incentive stock option or is necessary to comply with any applicable law, regulation or rule. We intend to terminate the 2012 Plan as to new awards as of the effective date of this offering.
Executive Incentive Compensation Plan
Our Executive Incentive Compensation Plan, or Incentive Plan, was adopted by our board of directors on July 8, 2013. The Incentive Plan allows a committee appointed by the board to provide cash incentive awards to selected employees, including our named executive officers, based upon performance goals established by our board of directors or its committee.
Under the Incentive Plan, a committee appointed by the board determines the performance goals applicable to any award, which goals may include, without limitation, the attainment of research and development milestones, sales bookings, business divestitures and acquisitions, cash flow, cash position, earnings (which may include earnings before interest and taxes, earnings before taxes and net earnings), earnings per share, net income, net profit, net sales, operating cash flow, operating expenses, operating income, operating margin, overhead or other expense reduction, product defect measures, product release timelines, productivity, profit, return on assets, return on capital, return on equity, return on investment, return on sales, revenue, revenue growth, sales results, sales growth, stock price, time to market, total shareholder return, working capital, and individual objectives such as peer reviews or other subjective or objective criteria. Performance goals that include the Company’s financial results may be determined in accordance with generally accepted accounting principles, or GAAP, or such financial results may consist of non-GAAP financial measures. The performance goals may be on an individual, divisional, business unit or Company-wide basis. The performance goals may differ from participant to participant and from award to award.
The committee appointed by the board may, in its sole discretion and at any time, increase, reduce or eliminate a participant’s actual award, or increase, reduce or eliminate the amount allocated to the bonus pool for a particular performance period. The actual award may be below, at or above a participant’s target award, in the discretion of the committee appointed by the board. The committee appointed by the board may determine the amount of any reduction on the basis of such factors as it deems relevant, and it is not required to establish any allocation or weighting with respect to the factors it considers.
Actual awards are paid in cash only after they are earned, which usually requires continued employment through the date of payment of the award. Payment of bonuses occurs as soon as administratively practicable after they are earned, but no later than the dates set forth in the Incentive Plan.
Our board of directors has the authority to amend, alter, suspend or terminate the Incentive Plan provided such action does not impair the existing rights of any participant with respect to any earned bonus.
Limitation on Liability and Indemnification Matters
The amended and restated certificate of incorporation to be in effect upon the closing of this offering, will limit or eliminate the personal liability of our directors to the fullest extent permitted by Delaware law. Consequently, our directors will not be personally liable to us or our stockholders for monetary damages for any breach of their fiduciary duties as directors, except liability for:
| • | any breach of the director’s duty of loyalty to us or our stockholders; |
| • | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| • | unlawful payments related to dividends or unlawful stock repurchases, redemptions, or other distributions as provided in Section 174 of the Delaware General Corporation Law; or |
| • | any transaction from which the director derived an improper personal benefit. |
115
Table of Contents
The amended and restated bylaws to be in effect upon the closing of this offering provide that we indemnify our directors and officers to the fullest extent permitted by Delaware law. These amended and restated bylaws also provide that we shall advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity, regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law.
We have entered and expect to continue to enter into agreements to indemnify our directors, executive officers and other employees as determined by our board of directors. With certain exceptions, these agreements provide for indemnification for related expenses including, among others, attorneys’ fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers. We also maintain directors’ and officers’ liability insurance.
The limitation of liability and indemnification provisions in our amended and restated certificate of incorporation and amended and restated bylaws, which will become effective upon the closing of this offering, may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duty of care. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Further, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers. At present, there is no pending litigation or proceeding involving any of our directors, officers or employees for which indemnification is sought, and we are not aware of any threatened litigation that may result in claims for indemnification.
116
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
The following is a summary of transactions since January 1, 2010 to which we have been a party in which the amount involved exceeded $120,000 and in which any of our executive officers, directors, promoters or beneficial holders of more than 5% of our capital stock had or will have a direct or indirect material interest, other than compensation arrangements which are described under the section of this prospectus titled “Executive and Director Compensation.”
Senior Preferred Stock Financing
Effective as of June 5, 2013, we entered into the Fourth Amended and Restated Senior Preferred Stock Purchase Agreement, as amended in December 2013, or the Senior Preferred Purchase Agreement, with Muneer A. Satter Revocable Trust and certain affiliated trusts and other entities, or the Satter Investors, certain of our directors and officers and certain unaffiliated third party investors, all of which are collectively referred to herein as the Investors. Muneer A. Satter is a member of our board of directors and is the trustee of the Muneer A. Satter Revocable Trust and the trustee, investment advisor or manager of each other Satter Investor. In his capacity as trustee, investment advisor or manager as applicable, Mr. Satter has sole voting and dispositive power over all shares held by the Satter Investors. The Senior Preferred Purchase Agreement contemplates the issuance by us of an aggregate of approximately $118.1 million of our senior redeemable convertible preferred stock for the purchase price of $8.00 per share to the Investors. As of February 28, 2014, we had issued an aggregate of 12,508,023 shares of our senior redeemable convertible preferred stock for an aggregate purchase price of $100.1 million. Upon closing of this offering, all rights and obligations under the Senior Preferred Purchase Agreement with respect to the issuance and sale of additional shares of our senior redeemable convertible preferred stock will terminate automatically and be of no further force or effect.
The following table presents the number of shares issued to our directors, officers and holders of more than 5% of our capital stock or entities affiliated with them pursuant to the Senior Preferred Purchase Agreement as of February 28, 2014.
| Purchaser |
Shares of Senior Preferred Stock |
Aggregate Purchase Price |
||||||
| Trusts and Other Entities Affiliated with Muneer A. Satter(1) |
3,550,638 | $ | 28,405,101 | |||||
| BSL Associates LLC and an Affiliated Entity(2) |
625,000 | 5,000,000 | ||||||
| Douglas E. Godshall(3) |
54,018 | 357,143 | ||||||
| Lowell E. Sears |
44,643 | 357,143 | ||||||
| Errol R. Halperin(4) |
39,417 | 315,336 | ||||||
| Jean-Jacques Bienaimé |
12,500 | 100,000 | ||||||
| Philip M. Croxford |
7,250 | 58,000 | ||||||
| Terence E. Winters, Ph.D.(5) |
6,250 | 50,000 | ||||||
| J. Michael Millis, M.D. |
1,250 | 10,000 | ||||||
| Randolph C. Steer, M.D., Ph.D. |
1,250 | |
10,000 |
| ||||
| Aron P. Stern |
378 | 3,025 | ||||||
| (1) | Includes shares held by Muneer A. Satter Revocable Trust and various other trusts and other entities for which Mr. Satter serves as trustee, investment advisor or manager and, in such capacity, has sole voting and dispositive control over such shares. |
| (2) | Consists of shares held by BSL Associates LLC and NP1 LLC for which Ralph Finerman serves as sole manager, and, in such capacity, has sole voting and dispositive control over such shares. As of February 28, 2014, BSL Associates LLC and NP1 LLC together no longer beneficially own more than 5% of our capital stock. |
| (3) | Includes 9,375 shares acquired by Douglas E. Godshall in connection with his exercise of preemptive rights under the terms of the Fourth Amended and Restated Investor Rights Agreement, dated August 28, 2013. |
117
Table of Contents
| (4) | Consists of 39,417 shares held by Errol R. Halperin and Libby G. Halperin. |
| (5) | Consists of 6,250 shares held by Terence E. Winters and Eileen Y. Winters. |
The Senior Preferred Purchase Agreement provides to certain of the Investors (including the Satter Investors) certain rights relating to the delivery of certain financial statements and other information. In addition, the Senior Preferred Purchase Agreement restricts the issuance of stock options or any comparable equity or equity-based incentives to any person who was a director, officer or other member of senior management as of September 25, 2012 unless such person shall have received a material promotion and commensurate increase in duties and responsibilities. While the information rights described above will be suspended for so long as we are subject to and remain in compliance with the periodic reporting requirements of the SEC, the restrictions on future equity awards described in this paragraph, as well as certain other covenants and agreements contained in the Senior Preferred Purchase Agreement, will survive completion of the offering and remain in full force and effect in accordance with their terms.
Senior Preferred Investors’ Rights Agreement
We and certain of our directors and stockholders, including the Satter Investors, are parties to a Fourth Amended and Restated Investors’ Rights Agreement, dated August 28, 2013, or the Senior Preferred IRA. The Senior Preferred IRA contains customary preemptive rights in favor of our stockholders party thereto, as well as customary registration rights and related provisions, including customary market standoff provisions. All preemptive rights in favor of our stockholders under the Senior Preferred IRA will terminate upon the closing of this offering.
The Senior Preferred IRA also provides that, for so long as the Satter Investors hold at least 30% of our outstanding common stock, the Satter Investors will have the right to nominate 40% of our directors (rounded up to the nearest whole number). If the Satter Investors hold less than 30% (but at least 20%) of our outstanding common stock, they will have the right to nominate 30% of our directors (rounded up to the nearest whole number). If the Satter Investors hold less than 20% (but at least 10%) of our outstanding common stock, they will have the right to nominate 20% of our directors (rounded up to the nearest whole number). If the Satter Investors hold less than 10% (but at least 2%) of our outstanding common stock, they will have the right to nominate 10% of our directors (rounded up to the nearest whole number). For so long as the Satter Investors hold less than 2% of our outstanding common stock, they will not have the contractual right to nominate any representatives to our board of directors. These rights survive the closing of this offering. To date the Satter Investors have not exercised their rights to nominate any directors, but they have reserved the right to do so in the future.
The Senior Preferred IRA provides that for so long as Mr. Satter and Dr. Winters both serve as members of our board of directors, each shall serve as co-chairman of our board of directors and Mr. Satter shall serve as our Lead Director. In the event that Mr. Satter serves as a member of our board of directors at a time when Dr. Winters does not, Mr. Satter will serve as our chairman of the board and Lead Director. This provision survives the closing of this offering. Dr. Winters will serve as Co-Chairman only so long as he is both a director and Chief Executive Officer.
Convertible Debt Financing
Between May 2012 and September 2012, we sold an aggregate of $7.2 million in secured convertible promissory notes to certain of the Satter Investors. In September 2012, these secured convertible promissory notes converted into an aggregate of 911,949 shares of our senior redeemable convertible preferred stock pursuant to the terms of the notes.
Junior Preferred Securities Purchase Agreement
We entered into a securities purchase agreement in February 2012, or the Junior Preferred Purchase Agreement, in connection with the sale of Junior Preferred Stock. Pursuant to the Junior Preferred Purchase
118
Table of Contents
Agreement, we sold an aggregate of 3,501,400 shares of our Junior Preferred Stock. The purchase included a holder of more than 5% of our capital stock, a director and an officer.
| Purchaser |
Shares of Junior Preferred Stock |
Aggregate Purchase Price |
||||||
| Trusts and Other Entities Affiliated with Muneer A. Satter(1) |
2,589,764 | $ | 1,109,455 | |||||
| Terence E. Winters, Ph.D.(2) |
116,713 | 50,000 | ||||||
| Aron P. Stern |
4,669 | 2,000 | ||||||
| (1) | Includes shares held by Muneer A. Satter Revocable Trust and various other trusts and other entities for which Mr. Satter serves as trustee, investment advisor or manager and, in such capacity, has sole voting and dispositive control over such shares. |
| (2) | Consists of 116,713 shares held by Terence E. Winters and Eileen Y. Winters. |
The Junior Preferred Purchase Agreement contains various negative and affirmative covenants, including those requiring us to deliver specified financial statements and annual budgets to certain investors, and other covenants rights with respect to the management of our company. This agreement will be terminated in full at or prior to closing of this offering.
Junior Preferred Investors’ Rights Agreement
Our company and certain of the holders of our junior preferred stock are parties to an investors’ rights agreement, dated February 23, 2012, or the Junior Preferred IRA. The Junior Preferred IRA contains customary preemptive rights in favor of our stockholders party to the agreement, as well as certain restrictions on transfer, registration rights, market standoff provisions and various affirmative and negative covenants with respect to management of our company. Upon the closing of this offering, all covenants in this agreement, except for the rights relating to the registration of shares under the Securities Act, will terminate. For a description of these registration rights, see the section of this prospectus entitled “Description of Capital Stock—Registration Rights.”
Series D Investors’ Rights Agreement
We and certain of our directors and stockholders, including the Satter Investors, are parties to an investors’ rights agreement, dated June 7, 2011, or the Series D IRA. The Series D IRA contains restrictions on transfer (for compliance with applicable securities laws) and customary registration rights. Upon the closing of this offering, all covenants in this agreement, except for the rights relating to the registration of shares under the Securities Act, will terminate. For a description of these registration rights, see the section of this prospectus entitled “Description of Capital Stock—Registration Rights.”
Indemnification of Officers and Directors
We have also entered into indemnification agreements with each of our directors, executive officers and certain controlling persons. The indemnification agreements and our amended restated certificate of incorporation and amended and restated bylaws require us to indemnify our directors, executive officers and certain controlling persons to the fullest extent permitted by Delaware law. See “Executive and Director Compensation—Limitations on Liability and Indemnification Matters” above.
Related-Person Transactions Policy
We have adopted a written Related-Person Transactions Policy that sets forth our policies and procedures regarding the identification, review, consideration, approval and oversight of “related-person transactions.” For purposes of our policy only, a “related-person transaction” is a past, present or future transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we and any “related person” are participants, the amount involved exceeds $120,000 and a related person has a direct or indirect material
119
Table of Contents
interest. Various transactions are not covered by this policy, including transactions involving compensation for services provided to us as an employee, director, consultant or similar capacity by a related person, equity and debt financing transactions with a related person that are approved by the Board, and other transactions not otherwise discloseable under Item 404 of Regulation S-K. A “related person,” as determined since the beginning of our last fiscal year, is any executive officer, director or nominee to become director, a holder of more than 5% of our common stock, including any immediate family members of such persons. Any related-person transaction may only be consummated if approved or ratified by the affirmative vote of seventy-five percent (75%) of our dis-interested directors then in office in accordance with the policy guidelines set forth below.
Under the policy, where a transaction has been identified as a related-person transaction, management must present information regarding the proposed related-person transaction to our audit committee for review and recommendation for approval to our board of directors. In considering related-person transactions, our audit committee and board of directors take into account the relevant available facts and circumstances including, but not limited to whether the terms of such transaction are no less favorable than terms generally available to an unaffiliated third-party under the same or similar circumstances and the extent of the related person’s interest in the transaction. In the event a director has an interest in the proposed transaction, the director must recuse himself or herself from the deliberations and approval process. We did not previously have a formal policy concerning transactions with related persons.
120
Table of Contents
The following table sets forth the beneficial ownership of our common stock as of February 28, 2014 by:
| • | each person, or group of affiliated persons, who we know to beneficially own more than 5% of our common stock; |
| • | each of our named executive officers; |
| • | each of our directors; and |
| • | all of our executive officers and directors as a group. |
The percentage ownership information prior to the offering shown in the table is based on an aggregate of 16,615,661 shares of our common stock and is comprised of 606,238 shares of our common stock outstanding as of February 28, 2014 and 16,009,423 shares of our common stock issuable upon conversion of our outstanding shares of preferred stock. The percentage ownership information after the offering assumes the issuance of shares of common stock in this offering. The table does not reflect any potential purchases of shares by certain of our officers, directors, existing investors and their affiliates, including entities affiliated with one of our directors, who have indicated an interest in purchasing shares in this offering as described in “Underwriting.”
We have determined beneficial ownership in accordance with the rules of the Securities and Exchange Commission. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options and warrants that are either immediately exercisable or exercisable on or before April 29, 2014, which is 60 days after February 28, 2014. These shares are deemed to be outstanding and beneficially owned by the person holding those options and warrants for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
Unless otherwise noted below, the address of each of the individuals and entities named in the table below is c/o Vital Therapies, Inc., 15010 Avenue of Science, Suite 200, San Diego, California 92128. Beneficial ownership representing less than 1% is denoted with an asterisk (*).
| Number of Shares of Common Stock Beneficially Owned |
Percentage of Common Stock Beneficially Owned | |||||||||
| Before Offering |
After Offering | |||||||||
| 5% Stockholders: |
||||||||||
| Trusts and Other Entities Affiliated with Muneer A. Satter(1) |
6,409,167 | 38.28 | % | |||||||
| Named Executive Officers and Directors: |
||||||||||
| Terence E. Winters, Ph.D.(2) |
698,572 | 4.08 | % | |||||||
| Michael V. Swanson(3) |
90,000 | * | ||||||||
| Andrew Henry(3) |
57,343 | * | ||||||||
| Muneer A. Satter(1) |
6,409,167 | 38.28 | % | |||||||
| Jean-Jacques Bienaimé(4) |
87,500 | * | ||||||||
| Douglas E. Godshall(5) |
148,836 | * | ||||||||
| Philip M. Croxford(6) |
62,782 | * | ||||||||
| J. Michael Millis, M.D.(7) |
51,513 | * | ||||||||
| Randolph C. Steer, M.D., Ph.D.(8) |
51,861 | * | ||||||||
| Errol R. Halperin(9) |
119,116 | * | ||||||||
| Lowell E. Sears(10) |
139,461 | * | ||||||||
| All directors and executive officers as a group (16 people)(11) |
8,891,215 | 47.31 | % | |||||||
121
Table of Contents
| (1) | Consists of 6,282,519 shares and warrants to acquire 126,648 shares that are held by Muneer A. Satter Revocable Trust and various other trusts and other entities for which Mr. Satter serves as trustee, investment advisor or manager and, in such capacity, has sole voting and dispositive control over all such shares. |
| (2) | Consists of 59,877 shares held by Terence E. Winters, 123,202 shares held by Terence E. Winters and Eileen Y. Winters, 427 shares that may be acquired pursuant to the exercise of warrants held of record by Terence E. Winters, and options to purchase 515,066 shares of common stock. |
| (3) | Consists of options to purchase shares of common stock. |
| (4) | Consists of 12,500 shares held and options to purchase 75,000 shares of common stock. |
| (5) | Consists of 54,018 shares held and options to purchase 94,818 shares of common stock. |
| (6) | Consists of 23,591 shares held and options to purchase 39,191 shares of common stock. |
| (7) | Consists of 17,679 shares held and options to purchase 33,834 shares of common stock. |
| (8) | Consists of 1,549 shares held, 137 shares that may be acquired pursuant to the exercise of warrants, and options to purchase 50,175 shares of common stock. |
| (9) | Consists of 40,000 shares held by Errol R. Halperin, 39,417 shares held by Errol R. Halperin and Libby G. Halperin, and options to purchase 39,699 shares of common stock. |
| (10) | Consists of 44,643 shares held and options to purchase 94,818 shares of common stock. |
| (11) | Consists of 6,714,514 shares held or beneficially owned, 127,258 shares that may be acquired pursuant to the exercise of warrants, and options to purchase 2,049,433 shares of common stock. |
122
Table of Contents
The following is a summary of all material characteristics of our capital stock as set forth in our amended and restated certificate of incorporation and amended and restated bylaws, which will become effective upon closing of this offering. The summary does not purport to be complete and is qualified in its entirety by reference to our amended and restated certificate of incorporation and amended and restated bylaws, all of which are incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and the applicable provisions of Delaware law.
General
Upon the closing of this offering and the filing of the amended and restated certificate of incorporation to be effective upon closing of this offering, our authorized capital stock will consist of 130,000,000 shares of common stock, par value $0.0001 per share, and 20,000,000 shares of preferred stock, par value $0.0001 per share.
Upon the closing of this offering, all the outstanding shares of our existing preferred stock will automatically convert into an aggregate of 16,009,423 shares of our common stock.
Common Stock
Outstanding Shares
Based on 606,238 shares of common stock outstanding as of February 28, 2014, the conversion of preferred stock outstanding as of February 28, 2014 into 16,009,423 shares of common stock upon the closing of this offering, the issuance of shares of common stock in this offering, and no exercise of options or warrants, there will be shares of common stock outstanding upon the closing of this offering. As of February 28, 2014, assuming the conversion of all outstanding convertible preferred stock into common stock upon the closing of this offering, we had approximately 185 record holders of our common stock.
As of February 28, 2014, there were 250,646 shares of common stock subject to outstanding warrants, and 3,136,269 shares of common stock subject to outstanding options.
Voting
Each holder of common stock is entitled to one vote for each share on all matters submitted to a vote of the stockholders, including the election of directors. Our amended and restated certificate of incorporation and amended and restated bylaws do not provide for cumulative voting rights. Because of this absence of cumulative voting, the holders of a majority of the shares of common stock entitled to vote in any election of directors can elect all of the directors standing for election, if they should so choose. In addition, our amended and restated certificate of incorporation also provides that our directors may be removed only for cause by the affirmative vote of the holders of at least 75% of the combined voting power of all our stockholders entitled to vote on the election of directors, voting together as a single class.
Subject to supermajority votes for some matters, matters shall be decided by the affirmative vote of our stockholders having a majority in voting power of the votes cast by the stockholders present or represented and voting on such matter, provided that the holders of our common stock are not allowed to vote on any amendment to our certificate of incorporation that relates solely to the terms of one or more series of preferred stock if the holders of such affected series are entitled, either separately or together with the holders or one or more such series, to approve such amendment. The affirmative vote of the holders of at least 75% of the votes that all of our stockholders would be entitled to cast in any annual election of directors and, in some cases, the affirmative vote of a majority of minority stockholders entitled to vote in any annual election of directors are required to amend or repeal our bylaws, amend or repeal certain provisions of our certificate of incorporation, approve certain
123
Table of Contents
transactions with certain affiliates, or approve the sale or liquidation of the company. The vote of a majority of minority of stockholders applies when an individual or entity and its affiliates or associates together own more than 50% of the voting power of our then outstanding capital stock, excluding any such persons that own 15% or more of our outstanding voting stock immediately prior to the closing of this offering, and such a vote would require the approval of the majority of our voting stock, excluding the voting stock held by such a majority holder.
Dividends
Subject to preferences that may be applicable to any then outstanding preferred stock, holders of common stock are entitled to receive ratably those dividends, if any, as may be declared from time to time by our board of directors out of legally available funds. For more information, see “Dividend Policy.”
Liquidation
In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preferences that may be granted to the holders of any then outstanding shares of preferred stock.
Rights and Preferences
Holders of common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences and privileges of the holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock, which we may designate and issue in the future.
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued pursuant to this offering, when paid for, will be fully paid and nonassessable.
Preferred Stock
As of February 28, 2014, there were (i) 12,508,023 shares of our senior preferred stock outstanding and (ii) 3,501,400 shares of our junior preferred stock outstanding. All outstanding shares of our preferred stock will convert into shares of our common stock upon closing of this offering.
Upon the closing of this offering, our board of directors will have the authority, without further action by the stockholders, to issue up to 20,000,000 shares of preferred stock in one or more series and to fix the rights, preferences, privileges and restrictions thereof. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, redemption rights, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of such series, any or all of which may be greater than the rights of common stock.
Following closing of this offering, our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control of our company and may adversely affect the market price of the common stock and the voting and other rights of the holders of common stock. Upon the closing of this offering, no shares of preferred stock will be outstanding, and we have no present plan to issue any shares of preferred stock.
124
Table of Contents
Stock Options
As of February 28, 2014, we had outstanding options to purchase an aggregate of 3,136,269 shares of our common pursuant to our 2012 Stock Plan, at a weighted-average exercise price of $6.71. As of February 28, 2014, 91,796 shares of our common stock remain available for future grant or issuance under our 2012 Stock Plan.
Warrants
As of February 28, 2014, we had outstanding warrants to purchase an aggregate of 250,646 shares of our common stock at a weighted-average exercise price of $95.21.
Registration Rights
Under our investors’ rights agreements, following the closing of this offering, the holders of approximately 17,886,697 shares of common stock (including the shares underlying the warrants described in “Shares Eligible for Future Sale—Warrants” and shares of common stock issuable upon exercise of certain outstanding options) or their transferees, have the right to require us to register the offer and sale of their shares, or to include their shares in any registration statement we file, in each case as described below.
Senior Preferred Investors’ Rights Agreement
Pursuant to our Fourth Amended and Restated Investors’ Rights Agreement, dated August 28, 2013, as amended, or the Senior Preferred IRA, the holders of 16,288,941 shares of common stock, including shares of common stock issuable upon exercise of certain outstanding warrants and options, or their transferees, will be entitled to certain rights with respect to the registration of such shares under the Securities Act. Beginning on the date that is 180 days following the effective date of this prospectus, and subject to limitations in the Senior Preferred IRA agreement, including our ability to delay registration in certain circumstances, the holders of at least 25% of these securities then outstanding may demand on three occasions, that we use our reasonable best efforts to register these securities using a long form registration statement for public resale if the anticipated aggregate offering price, net of underwriting discounts and commissions, would exceed $15 million. If we register any of our common stock either for our own account or for the account of other security holders, the holders of these securities are entitled to include their shares of common stock in that registration, subject to the ability of the underwriters to limit the number of shares included in the offering. After closing of this offering, we will be obligated to use our reasonable best efforts to make short form registration statements available, and after such time the holders of at least 25% of these securities then outstanding may also demand, but not more than two times in any 12-month period, that we register all or a portion of these securities using a short form registration statement, provided, among other limitations, that the proposed aggregate selling price is at least $15 million. We will be responsible for paying all registration expenses, including the reasonable fees of legal counsel for the selling holders, and the holders selling their shares will be responsible for paying all selling expenses.
Registration rights under the Senior Preferred IRA terminate, as to a given holder of registration rights, when such holder and such holder’s affiliates can sell all of their registrable securities in a three-month period pursuant to Rule 144.
Junior Preferred Investors’ Rights Agreement
Pursuant to our investors’ rights agreement, dated February 23, 2012, or the Junior Preferred IRA, the holders of 705,048 shares of common stock, including shares of common stock issuable upon exercise of certain outstanding warrants or options, or their transferees will be entitled to certain rights with respect to the registration of such shares under the Securities Act. After the earlier of the third anniversary of the Junior Preferred IRA or six months after the effective date of this prospectus, and subject to limitations in the agreement, including our ability to delay registration in certain circumstances, the holders of at least 25% of
125
Table of Contents
these securities then outstanding may require, on two occasions, that we use our best efforts to register these securities using a long form registration statement for public resale if the anticipated aggregate offering price, net of underwriting discounts and commissions, would exceed $7 million. Following our initial public offering, if we register any of our common stock either for our own account or for the account of other security holders, the holders of these securities are entitled to include their shares of common stock in that registration, subject to the ability of the underwriters to limit the number of shares included in the offering to as few as 45% of the offering. After closing of this offering, the holders of these securities then outstanding may also require us, but not more than one time in any 12-month period, to register all or a portion of these securities using a short form registration statement when the use of that form becomes available to us, provided, among other limitations, that the proposed aggregate selling price is at least $1 million. We will be responsible for paying all registration expenses, including the reasonable fees of legal counsel for the selling holders, and the holders selling their shares will be responsible for paying all selling expenses.
Registration rights under Junior Preferred IRA terminate upon the earliest of (i) the date that is five years after the effective date of this prospectus, or (ii) as to a given holder of registration rights, when such holder and such holder’s affiliates can sell all of their registrable securities in a three-month period pursuant to Rule 144.
Series D Investors’ Rights Agreement
Pursuant to our investors’ rights agreement, dated June 7, 2011, or the Series D IRA, the holders of 892,708 shares of common stock, including shares of common stock issuable upon exercise of certain outstanding warrants or options, or their transferees will be entitled to certain rights with respect to the registration of such shares under the Securities Act. After the earlier of the third anniversary of the Series D IRA or six months after the effective date of this prospectus, and subject to limitations in the agreement, including our ability to delay registration in certain circumstances, the holders of at least 25% of these securities then outstanding may require, on two occasions, that we use our best efforts to register these securities using a long-form registration statement for public resale if the anticipated aggregate offering price, net of underwriting discounts and commissions, would exceed $7 million. Following our initial public offering, if we register any of our common stock either for our own account or for the account of other security holders, the holders of these securities are entitled to include their shares of common stock in that registration, subject to the ability of the underwriters to limit the number of shares included in the offering to as few as 45% of the offering. After closing of this offering, the holders of these securities then outstanding may also require us, but not more than one time in any 12-month period, to register all or a portion of these securities using a short form registration statement when the use of that form becomes available to us, provided, among other limitations, that the proposed aggregate selling price is at least $1 million. We will be responsible for paying all registration expenses, including the reasonable fees of legal counsel for the selling holders, and the holders selling their shares will be responsible for paying all selling expenses.
Registration rights under the Series D IRA terminate upon the earliest of (i) the date that is five years after the effective date of this prospectus, or (ii) as to a given holder of registration rights, when such holder and such holder’s affiliates can sell all of such holder’s registrable securities in a three month-period pursuant to Rule 144.
Anti-Takeover Effects of Delaware Law and Our Certificate of Incorporation, Bylaws and Fourth Amended and Restated Investors’ Rights Agreement
Certain provisions of Delaware law, our amended and restated certificate of incorporation and amended and restated bylaws that will become effective upon closing of this offering, and the Senior Preferred IRA contain provisions that could have the effect of delaying, deferring or discouraging another party from acquiring control of us. These provisions, which are summarized below, are expected to discourage certain types of coercive takeover practices and inadequate takeover bids. These provisions are also designed in part to encourage anyone seeking to acquire control of us to first negotiate with our board of directors. We believe that the advantages gained by protecting our ability to negotiate with any unsolicited and potentially unfriendly acquirer outweigh the disadvantages of discouraging such proposals, including those priced above the then-current market value of our common stock, because, among other reasons, the negotiation of such proposals could improve their terms.
126
Table of Contents
Certificate of Incorporation and Bylaws
Our amended and restated certificate of incorporation and amended and restated bylaws that will become effective upon closing of this offering include provisions that:
| • | authorize our board of directors to issue, without further action by the stockholders, up to 20,000,000 shares of undesignated preferred stock; |
| • | require that any action to be taken by our stockholders be effected at a duly called annual or special meeting and not by written consent; |
| • | specify that special meetings of our stockholders can be called only by a supermajority (75%) vote of our directors then in office; |
| • | our board of directors may amend or repeal our bylaws only pursuant to a supermajority (75%) vote of our directors then in office; |
| • | our stockholders may amend or repeal our bylaws only pursuant to a supermajority (75% and majority of the minority, if applicable) vote of the outstanding shares of our capital stock; |
| • | require in general the approval of a supermajority (75% and majority of the minority, if applicable) vote of our outstanding shares of capital stock to amend or repeal certain provisions of our certificate of incorporation; |
| • | require the approval of a supermajority (75% and majority of the minority, if applicable) vote of our outstanding shares of capital stock to approve the sale or liquidation of the company; |
| • | establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors; |
| • | provide that directors may be removed only for cause by a supermajority (75%) vote of our outstanding shares of capital stock; |
| • | provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though less than a quorum; |
| • | provide that in general the number of directors on our board may only be fixed from time to time by a supermajority (75%) vote of our directors then in office; and |
| • | establish that our board of directors is divided into three classes, Class I, Class II and Class III, with each class serving staggered terms. |
Senior Preferred Investors’ Rights Agreement
The Senior Preferred IRA also provides that, for so long as the Satter Investors hold at least 30% of our outstanding common stock, the Satter Investors will have the right to nominate 40% of our directors (rounded up to the nearest whole number). If the Satter Investors hold less than 30% (but at least 20%) of our outstanding common stock, they will have the right to nominate 30% of our directors (rounded up to the nearest whole number). If the Satter Investors hold less than 20% (but at least 10%) of our outstanding common stock, they will have the right to nominate 20% of our directors (rounded up to the nearest whole number). If the Satter Investors hold less than 10% (but at least 2%) of our outstanding common stock, they will have the right to nominate 10% of our directors (rounded up to the nearest whole number). For so long as the Satter Investors hold less than 2% of our outstanding common stock, they will not have the contractual right to nominate any representatives to our board of directors. These rights survive the closing of this offering. To date the Satter Investors have not exercised their rights to nominate any directors, but they have reserved the right to do so in the future.
The Senior Preferred IRA provides that for so long as Mr. Satter and Dr. Winters both serve as members of our board of directors, each shall serve as Co-Chairman of the board of directors and Mr. Satter shall serve as our Lead Director. In the event that Mr. Satter serves as a member of our board of directors at a time when
127
Table of Contents
Dr. Winters does not, Mr. Satter will serve as our Chairman of the board and Lead Director. This provision survives the closing of this offering. Dr. Winters will serve as Co-Chairman only so long as he is both a director and Chief Executive Officer.
Delaware Anti-Takeover Statute
We will elect in our amended and restated certificate of incorporation not to be subject to Section 203 of the Delaware General Corporation Law, an anti-takeover law. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a business combination, such as a merger, with a person or group owning 15% or more of the corporation’s voting stock for a period of three years following the date the person became an interested stockholder, unless (with certain exceptions) the business combination or the transaction in which the person became an interested stockholder is approved in a prescribed manner. Accordingly, we are not subject to any anti-takeover effects of Section 203. However, our amended and restated certificate of incorporation contains provisions that have the same effect as Section 203, except that they provide that certain of our current stockholders, including Mr. Satter and entities affiliated with him, and any persons to whom certain of our current stockholders sell their common stock will be deemed to have been approved by our board of directors, and thereby not subject to the restrictions set forth in Section 203.
Listing
Our common stock has been approved for listing on The NASDAQ Global Market under the symbol “VTL.”
Transfer Agent and Registrar
Upon the closing of this offering, the transfer agent and registrar for common stock will be American Stock Transfer & Trust Company, LLC, or AST. The transfer agent and registrar’s address is 6201 15th Avenue Brooklyn, New York 11219.
128
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no public market for our common stock, and although we expect that our common stock will be approved for listing on the NASDAQ Global Market we cannot assure you that there will be an active public market for our common stock following this offering. We cannot predict what effect, if any, sales of our shares in the public market or the availability of shares for sale will have on the market price of our common stock. Future sales of substantial amounts of common stock in the public market, including shares issued upon exercise of outstanding options, or the perception that such sales may occur, however, could adversely affect the market price of our common stock and also could adversely affect our future ability to raise capital through the sale of our common stock or other equity-related securities of ours at times and prices we believe appropriate.
Upon closing of this offering, based on our shares outstanding as of February 28, 2014 and after giving effect to the conversion of all outstanding shares of our convertible preferred stock, shares of our common stock will be outstanding, or shares of common stock if the underwriters exercise their option to purchase additional shares in full. All of the shares of common stock expected to be sold in this offering will be freely tradable without restriction or further registration under the Securities Act unless held by our “affiliates,” as that term is defined in Rule 144 under the Securities Act. The remaining outstanding shares of our common stock will be deemed “restricted securities” as that term is defined under Rule 144. Restricted securities may be sold in the public market only if their offer and sale is registered under the Securities Act or if the offer and sale of those securities qualify for an exemption from registration, including exemptions provided by Rules 144 and 701 under the Securities Act, which are summarized below.
As a result of the lock-up agreements described below and the provisions of Rules 144 or 701 and assuming no exercise of the underwriters’ option to purchase additional shares in full and no purchases of shares by existing investors or their affiliates as described in “Underwriting”, the shares of our common stock that will be deemed “restricted securities” will be available for sale in the public market following the closing of this offering as follows:
| • | The shares expected to be sold in this offering will be eligible for immediate sale in the public market; and |
| • | approximately 16,700,000 shares will be eligible for sale upon expiration of the lock-up agreements and market stand-off provisions described below and a similar lock-up period existing investors have agreed to with us, beginning more than 180 days after the date of this prospectus subject in some cases to applicable volume limitations under Rule 144. |
We may issue shares of our common stock from time to time for a variety of corporate purposes, including in capital raising activities through future public offerings or private placements, in connection with the exercise of stock options, vesting of restricted stock units and other issuances relating to our employee benefit plans, and as consideration for future acquisitions, investments or other purposes. The number of shares of our common stock that we may issue may be significant, depending on the events surrounding such issuances. In some cases, the shares we issue may be freely tradable without restriction or further registration under the Securities Act. In other cases, we may grant registration rights covering the shares issued in connection with these issuances, in which case the holders of the common stock will have the right, under certain circumstances, to cause us to register any resale of such shares to the public.
Lock-Up Agreements
Our officers and directors and the holders of substantially all of our outstanding shares of capital stock have agreed with the underwriters, subject to certain exceptions, not to dispose of or hedge any of their common stock or securities convertible into or exchangeable or exercisable for shares of common stock for a period through the date that is 180 days after the date of this prospectus, except with the prior written consent of the representatives of the underwriters on behalf of the underwriters, as described below.
129
Table of Contents
The representatives may, at any time or from time to time before the termination of the lock-up period release all or any portion of the securities subject to lock-up agreements; provided, however, that, subject to limited exceptions, if such release is for the securities of any of our officers or directors, at least three business days before the release, the representatives must notify us of the impending release and we will announce the impending release through a major news service at least two business days before the effective date of the release.
Rule 144
Subject to the lock-up agreements described above, in general, under Rule 144, beginning 90 days after the date of this prospectus, a person who is not our affiliate and has not been our affiliate for purposes of the Securities Act at any time during the preceding three months will be entitled to sell any shares of our common stock that such person has beneficially owned for at least six months, including the holding period of any prior owner, other than one of our affiliates, without being required to comply with the notice or manner of sale or volume limitation provisions of Rule 144. Sales of our common stock by any such person would be subject to the availability of current public information about us if the shares to be sold were beneficially owned by such person for less than one year.
In addition, under Rule 144, a person may sell shares of our common stock acquired from us immediately upon the closing of this offering, without regard to the registration requirements of the Securities Act or the availability of public information about us, if:
| • | the person is not our affiliate and has not been our affiliate at any time during the preceding three months; and |
| • | the person has beneficially owned the shares to be sold for at least one year, including the holding period of any prior owner other than one of our affiliates. |
Beginning 90 days after the date of this prospectus, our affiliates who have beneficially owned shares of our common stock for at least six months, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell within any three-month period a number of shares that does not exceed the greater of:
| • | 1% of the number of shares of our common stock then outstanding, which will equal approximately shares immediately after this offering; and |
| • | the average weekly trading volume in our common stock during the four calendar weeks preceding the date of filing of a notice on Form 144 with respect to the sale. |
Sales under Rule 144 by our affiliates are also subject to manner of sale provisions and notice requirements and to the availability of current public information about us. To the extent that shares were acquired from one of our affiliates, a person’s holding period for the purpose of effecting a sale under Rule 144 would commence on the date of transfer from the affiliate.
Rule 701
In general, under Rule 701 any of our employees, consultants or advisors who purchased shares of our common stock pursuant to a written compensatory plan or contract before the effective date of this offering may sell these shares beginning 90 days after the date of this prospectus in reliance upon Rule 144, but without compliance with certain restrictions, including the holding period, contained in Rule 144.
As of February 28, 2014, 139,071 shares of our outstanding common stock had been issued in reliance on Rule 701 as a result of exercises of stock options. All of these shares, however, are subject to lock-up agreements or market stand-off provisions as discussed above, and, as a result, these shares will only become eligible for sale at the earlier of the expiration of the lock-up period or upon obtaining the consent of the representatives on behalf of the underwriters to release all or any portion of these shares from the lock-up agreements.
130
Table of Contents
Stock Options
As of February 28, 2014, options to purchase an aggregate 3,136,269 shares of our common stock were outstanding. We intend to file one or more registration statements on Form S-8 under the Securities Act to register the offer and sale of all shares of our common stock subject to outstanding stock options and all shares reserved for issuance under our equity compensation plans. We expect to file the registration statement covering these shares after the date of this prospectus, which will permit the resale of such shares in the public market, subject, with respect to certain of the shares, to the provisions of the lock-up agreements. We expect to file this registration statement as soon as permitted under the Securities Act.
Warrants
Upon closing of this offering, warrants entitling holders to purchase an aggregate of 250,646 shares of our common stock at a weighted-average exercise price of $95.21 per share will be outstanding. See “Description of Capital Stock” for additional information. Such shares issued upon exercise of the warrants may be able to be sold after the expiration of the lock-up period described above subject the requirements of Rule 144 described above.
Registration Rights
Upon closing of this offering, the holders of approximately 17,886,697 shares of our common stock (including the certain shares of common stock underlying the warrants described in “Warrants” above and certain shares of common stock issuable upon exercise of outstanding options), will be eligible to exercise certain rights to cause us to register their shares for resale under the Securities Act, subject to various conditions and limitations. These registration rights are described under the caption “Description of Capital Stock.” Upon the effectiveness of a registration statement covering these shares, the shares would become freely tradable, and a large number of shares may be sold into the public market. If that occurs, the market price of our common stock could be adversely affected.
131
Table of Contents
MATERIAL U.S. FEDERAL INCOME AND ESTATE TAX CONSEQUENCES TO NON-U.S. HOLDERS
The following is a summary of the material U.S. federal income tax and estate tax consequences of the ownership and disposition of our common stock to non-U.S. holders, but does not purport to be a complete analysis of all the potential tax considerations relating thereto. This summary is based upon the provisions of the Internal Revenue Code of 1986, as amended, or the Code, Treasury regulations promulgated thereunder, administrative rulings and judicial decisions, all as of the date hereof. These authorities may be changed, possibly retroactively, so as to result in U.S. federal income tax or estate tax consequences different from those set forth below.
This summary does not address the tax considerations arising under the laws of any U.S. state or local or any non-U.S. jurisdiction, the potential application of the Medicare contribution tax or under U.S. federal gift and estate tax laws, except to the limited extent indicated below. In addition, this discussion does not address tax considerations applicable to an investor’s particular circumstances or to investors that may be subject to special tax rules, including, without limitation:
| • | banks, insurance companies or other financial institutions; |
| • | persons subject to the alternative minimum tax; |
| • | tax-exempt organizations; |
| • | dealers in securities or currencies; |
| • | traders in securities that elect to use a mark-to-market method of accounting for their securities holdings; |
| • | persons that own, or are deemed to own, more than five percent of our common stock (except to the extent specifically set forth below); |
| • | certain former citizens or long-term residents of the United States; |
| • | persons who hold our common stock as a position in a hedging transaction, “straddle,” “conversion transaction” or other risk reduction transaction; |
| • | persons who do not hold our common stock as a capital asset within the meaning of Section 1221 of the Code (generally, for investment purposes); or |
| • | persons deemed to sell our common stock under the constructive sale provisions of the Code. |
In addition, if a partnership or entity classified as a partnership for U.S. federal income tax purposes holds our common stock, the tax treatment of a partner generally will depend on the status of the partner and upon the activities of the partnership. Accordingly, partnerships that hold our common stock, and partners in such partnerships, should consult their tax advisors.
You are urged to consult your tax advisor with respect to the application of the U.S. federal income tax laws to your particular situation, as well as any tax consequences of the purchase, ownership and disposition of our common stock arising under the U.S. federal estate or gift tax rules or under the laws of any U.S. state or local or any non-U.S. or other taxing jurisdiction or under any applicable tax treaty.
Non-U.S. Holder Defined
For purposes of this discussion, you are a non-U.S. holder if you are any holder (other than a partnership or entity classified as a partnership for U.S. federal income tax purposes) that is not:
| • | an individual citizen or resident of the U.S.; |
| • | a corporation or other entity taxable as a corporation created or organized in the U.S. or under the laws of the United States or any political subdivision thereof; |
132
Table of Contents
| • | an estate whose income is subject to U.S. federal income tax regardless of its source; or |
| • | a trust (x) whose administration is subject to the primary supervision of a U.S. court and which has one or more U.S. persons who have the authority to control all substantial decisions of the trust or (y) which has made an election to be treated as a U.S. person. |
Distributions
If we make distributions on our common stock, those payments will constitute dividends for U.S. tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent those distributions exceed both our current and our accumulated earnings and profits, they will constitute a return of capital and will first reduce your basis in our common stock, but not below zero, and then will be treated as gain from the sale of common stock.
Any dividend paid to you generally will be subject to U.S. withholding tax either at a rate of 30% of the gross amount of the dividend or such lower rate as may be specified by an applicable income tax treaty. In order to receive a reduced treaty rate, you must provide us with an Internal Revenue Service, or IRS, Form W-8BEN or other appropriate version of IRS Form W-8 certifying qualification for the reduced rate.
Dividends received by you that are effectively connected with your conduct of a U.S. trade or business are taxed at the same graduated rates applicable to U.S. persons, net of certain deductions and credits, subject to an applicable income tax treaty providing otherwise. In addition, if you are a corporate non-U.S. holder, dividends you receive that are effectively connected with your conduct of a U.S. trade or business may also be subject to a branch profits tax at a rate of 30% or such lower rate as may be specified by an applicable income tax treaty. Payments of effectively connected dividends that are included in the gross income of a non-U.S. holder generally are exempt from withholding tax. In order to obtain this exemption, you must provide us with an IRS Form W-8 ECI or other applicable IRS Form W-8 properly certifying such exemption.
If you are eligible for a reduced rate of withholding tax pursuant to a tax treaty, you may be able to obtain a refund of any excess amounts currently withheld if you timely file an appropriate claim for refund with the IRS.
Gain on Disposition of Common Stock
You generally will not be required to pay U.S. federal income tax on any gain realized upon the sale or other disposition of our common stock unless:
| • | the gain is effectively connected with your conduct of a U.S. trade or business (and, if an income tax treaty applies, the gain is attributable to a permanent establishment maintained by you in the U.S.), in which case you will be required to pay tax on the net gain derived from the sale (net of certain deductions or credits) under regular graduated U.S. federal income tax rates, and for a non-U.S. holder that is a corporation, such non-U.S. holder may also be subject to a branch profits tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty; |
| • | you are an individual who is present in the U.S. for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met, in which case you will be required to pay a flat 30% tax on the gain derived from the sale, which tax may be offset by U.S. source capital losses (even though you are not considered a resident of the U.S.) subject to applicable income tax or other treaties providing otherwise; or |
| • | our common stock constitutes a U.S. real property interest by reason of our status as a “U.S. real property holding corporation” for U.S. federal income tax purposes (a USRPHC) at any time within the shorter of the five-year period preceding the disposition or your holding period for our common stock. We believe that we are not currently and will not become a USRPHC. However, because the |
133
Table of Contents
| determination of whether we are a USRPHC depends on the fair market value of our U.S. real property relative to the fair market value of our other business assets, there can be no assurance that we will not become a USRPHC in the future. |
Federal Estate Tax
Our common stock held (or treated as held) by an individual non-U.S. holder at the time of death will be included in such holder’s gross estate for U.S. federal estate tax purposes, unless an applicable estate tax treaty provides otherwise, and therefore may be subject to U.S. federal estate tax.
Backup Withholding and Information Reporting
Generally, we must report annually to the IRS the amount of dividends paid to you, your name and address, and the amount of tax withheld, if any. A similar report will be sent to you. Pursuant to applicable income tax treaties or other agreements, the IRS may make these reports available to tax authorities in your country of residence.
Payments of dividends or of proceeds on the disposition of common stock made to you may be subject to additional information reporting and backup withholding at a current rate of 28% unless you establish an exemption, for example by properly certifying your non-U.S. status on a Form W-8BEN or another appropriate version of IRS Form W-8. Notwithstanding the foregoing, backup withholding and information reporting may apply if either we or our paying agent has actual knowledge, or reason to know, that you are a U.S. person.
Backup withholding is not an additional tax; rather, the U.S. income tax liability of persons subject to backup withholding will be reduced by the amount of tax withheld. If withholding results in an overpayment of taxes, a refund or credit may generally be obtained from the IRS, provided that the required information is furnished to the IRS in a timely manner.
Foreign Account Tax Compliance Act (FATCA)
FATCA imposes a U.S. federal withholding tax of 30% on dividends and the gross proceeds of a disposition of our common stock to a “foreign financial institution” (as specifically defined for this purpose) unless such institution enters into an agreement with the U.S. government to withhold on certain payments and to collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which includes certain equity and debt holders of such institution, as well as certain account holders that are foreign entities with U.S. owners). A U.S. federal withholding tax of 30% generally applies to dividends and the gross proceeds of a disposition of our common stock to a non-financial foreign entity unless such entity provides the withholding agent with either a certification that it does not have any substantial direct or indirect U.S. owners or provides information regarding direct and indirect U.S. owners of the entity. Under the final Treasury Regulations, the withholding provisions described above will generally apply to payments of dividends on our common stock made on or after July 1, 2014 and to payments of gross proceeds from a sale or other disposition of such common stock on or after January 1, 2017. You should consult your tax advisors regarding these withholding provisions.
The preceding discussion of U.S. federal tax considerations is for general information only. It is not tax advice. Each prospective investor should consult its own tax advisor regarding the particular U.S. federal, state and local and non-U.S. tax consequences of purchasing, holding and disposing of our common stock, including the consequences of any proposed change in applicable laws.
134
Table of Contents
Under the terms and subject to the conditions contained in an underwriting agreement dated 2014, we have agreed to sell to the underwriters named below, for whom Merrill Lynch, Pierce, Fenner & Smith Incorporated and Credit Suisse Securities (USA) LLC are acting as representatives, the following respective numbers of shares of common stock:
| Name |
Number of Shares | |
| Merrill Lynch, Pierce, Fenner & Smith Incorporated |
||
| Credit Suisse Securities (USA) LLC |
||
| William Blair & Company, L.L.C. |
||
| Canaccord Genuity Inc. |
||
|
| ||
|
|
The underwriting agreement provides that the underwriters are obligated to purchase all the shares of common stock in the offering if any are purchased, other than those shares covered by the option to purchase additional shares described below. The underwriting agreement also provides that if an underwriter defaults, the purchase commitments of the non-defaulting underwriters may be increased or the offering may be terminated.
We have granted to the underwriters a 30-day option to purchase on a pro rata basis up to an aggregate of additional shares at the initial public offering price less the underwriting discounts and commissions.
The underwriters are offering the shares, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel including the validity of the shares, and subject to other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officer’s certificates and legal opinions. The offering of the shares by the underwriters is also subject to the underwriters’ right to reject any order in whole or in part.
The underwriters propose to offer the shares of common stock initially at the public offering price on the cover page of this prospectus and to selling group members, if any, at that price less a selling concession of up to $ per share. After the initial public offering the representatives may change the public offering price and selling concession.
The following table shows the public offering price, underwriting discount and proceeds before expenses to us. The information assumes either no exercise or full exercise by the underwriters of their option to purchase additional shares.
| Per Share | Without Option |
With Option |
||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discount |
$ | $ | $ | |||||||||
| Proceeds, before expenses, to Vital Therapies, Inc. |
$ | $ | $ | |||||||||
We estimate that our out of pocket expenses for this offering (not including any underwriting discounts and commissions) will be approximately $ million.
The representatives have informed us that they do not expect sales to accounts over which the underwriters have discretionary authority to exceed 5% of the shares of common stock being offered.
We have agreed that we will not (i) offer, sell, issue, contract to sell, pledge or otherwise dispose of, (ii) offer, sell, issue, contract to sell, contract to purchase or grant any option, right or warrant to purchase, (iii) enter into any
135
Table of Contents
swap, hedge or any other agreement that transfers, in whole or in part, the economic consequences of ownership, (iv) establish or increase a put equivalent position or liquidate or decrease a call equivalent position or (v) file with the Securities and Exchange Commission a registration statement under the Securities Act relating to, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, or publicly disclose the intention to take any such action, without the prior written consent of the representatives, for 180 days after the date of this prospectus, subject to certain exceptions.
The restrictions in the foregoing paragraph do not apply to (i) the sale of shares of common stock to the underwriters pursuant to the underwriting agreement, (ii) the issuance by us of shares of common stock pursuant to the conversion or exchange of convertible or exchangeable securities or the exercise of warrants, in each case outstanding on the date of this prospectus, (iii) the grant of stock options, restricted stock or other equity-based compensation awards pursuant to the terms of a plan disclosed in this prospectus, or (iv) the issuance of common stock upon exercise of such options or equity-based compensation awards pursuant to the terms of such plan disclosed in this prospectus.
Our executive officers, our directors and substantially all of our existing security holders have agreed that they will not offer, sell, assign, transfer, pledge, contract to sell, or otherwise dispose of or announce the intention to otherwise dispose of, or enter into any swap, hedge or similar agreement or arrangement that transfers, in whole or in part, the economic consequence of ownership of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock without, in each case, the prior written consent of the representatives, for a period of 180 days after the date of this prospectus, subject to certain exceptions.
Certain of our directors, officers, existing investors and their affiliated entities have indicated an interest in purchasing an aggregate of approximately $ of our common stock in this offering at the initial public offering price. However, because indications of interest are not binding agreements or commitments to purchase, these persons and entities may determine to purchase fewer shares than they indicate an interest in purchasing or not to purchase any shares in this offering. It is also possible that these persons and entities could purchase more shares of our common stock. The underwriters will receive the same underwriting discount on any shares purchased by these persons and entities as they will on any other shares sold to the public in this offering. Shares purchased will be subject to the lock-up agreements described above.
We have agreed to indemnify the several underwriters against liabilities under the Securities Act, or contribute to payments that the underwriters may be required to make in that respect.
We have been approved to list our common stock on the NASDAQ Global Market under the symbol ‘‘VTL.’’
Prior to the offering, there has been no public market for our common stock. The initial public offering price will be determined by negotiation between us and the representatives. In addition to prevailing market conditions, the principal factors that will be considered in determining the initial public offering price will include:
| • | the history of, and prospects for, our company and the industry in which we compete; |
| • | our past and present financial information; |
| • | an assessment of our management; |
| • | its past and present operations, and the prospects for, and timing of, our future revenues; |
| • | the present state of our development; |
| • | the above factors in relation to market values and various valuation measures of other companies engaged in activities similar to ours; and |
136
Table of Contents
| • | other information included in this prospectus and otherwise available to the underwriters. |
We cannot assure you that the initial public offering price will correspond to the price at which our common stock will trade in the public market subsequent to this offering or that an active trading market for our common stock will develop and continue after this offering.
The underwriters and their respective affiliates are full-service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The underwriters and their respective affiliates may in the future provide financial advisory or investment banking services to us from time to time for which they expect to receive customary compensation and expense reimbursement.
In addition, in the ordinary course of their business activities, the underwriters and their affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers. Such investments and securities activities may involve securities and/or instruments of ours or our affiliates. The underwriters and their affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or financial instruments and may hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
In connection with the offering the underwriters may engage in stabilizing transactions, short sales and syndicate covering transactions, penalty bids and passive market making in accordance with Regulation M under the Exchange Act.
| • | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. |
| • | Syndicate covering transactions involve purchases of our common stock in the open market to cover syndicate short positions. Sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the short position is not greater than the amount of additional shares for which the underwriters’ option described above may be exercised. The underwriters may close out any covered short position by either exercising their option to purchase additional shares and/or purchasing shares in the open market. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through their option to purchase additional shares. In a naked short position, the short position is greater than the amount of additional shares for which their option to purchase additional shares may be exercised. A naked short position can only be closed out by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. |
| • | Penalty bids permit the representatives to reclaim a selling concession from a syndicate member when the common stock originally sold by the syndicate member is purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
| • | In passive market making, market makers in the common stock who are underwriters or prospective underwriters may, subject to limitations, make bids for or purchases of our common stock until the time, if any, at which a stabilizing bid is made. |
These stabilizing transactions, short sales and syndicate covering transactions, penalty bids and passive market making may have the effect of raising or maintaining the market price of our common stock or preventing
137
Table of Contents
or retarding a decline in the market price of our common stock. Neither we nor any of the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. These transactions may be effected on the NASDAQ Global Market or otherwise and, if commenced, may be discontinued at any time.
A prospectus in electronic format may be made available on the web sites maintained by one or more of the underwriters, or selling group members, if any, participating in this offering and one or more of the underwriters participating in this offering may distribute prospectuses electronically. In connection with the offering, certain of the underwriters or securities dealers may distribute prospectuses by electronic means, such as e-mail. The representatives may agree to allocate a number of shares to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make internet distributions on the same basis as other allocations.
Notice to Prospective Investors in the European Economic Area
In relation to each Member State of the European Economic Area (each, a “Relevant Member State”), no offer of shares may be made to the public in that Relevant Member State other than:
| A. | to any legal entity which is a qualified investor as defined in the Prospectus Directive; |
| B. | to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives; or |
| C. | in any other circumstances falling within Article 3(2) of the Prospectus Directive, |
provided that no such offer of shares shall require us or the representatives to publish a prospectus pursuant to Article 3 of the Prospectus Directive or supplement a prospectus pursuant to Article 16 of the Prospectus Directive.
Each person in a Relevant Member State who initially acquires any shares or to whom any offer is made will be deemed to have represented, acknowledged and agreed that it is a “qualified investor” within the meaning of the law in that Relevant Member State implementing Article 2(1)(e) of the Prospectus Directive. In the case of any shares being offered to a financial intermediary as that term is used in Article 3(2) of the Prospectus Directive, each such financial intermediary will be deemed to have represented, acknowledged and agreed that the shares acquired by it in the offer have not been acquired on a non-discretionary basis on behalf of, nor have they been acquired with a view to their offer or resale to, persons in circumstances which may give rise to an offer of any shares to the public other than their offer or resale in a Relevant Member State to qualified investors as so defined or in circumstances in which the prior consent of the representatives has been obtained to each such proposed offer or resale.
We, the representatives and their affiliates will rely upon the truth and accuracy of the foregoing representations, acknowledgements and agreements.
This prospectus has been prepared on the basis that any offer of shares in any Relevant Member State will be made pursuant to an exemption under the Prospectus Directive from the requirement to publish a prospectus for offers of shares. Accordingly any person making or intending to make an offer in that Relevant Member State of shares which are the subject of the offering contemplated in this prospectus may only do so in circumstances in which no obligation arises for us or any of the underwriters to publish a prospectus pursuant to Article 3 of the Prospectus Directive in relation to such offer. Neither we nor the underwriters have authorized, nor do we authorize, the making of any offer of shares in circumstances in which an obligation arises for us or the underwriters to publish a prospectus for such offer.
138
Table of Contents
For the purpose of the above provisions, the expression “an offer to the public” in relation to any shares in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and the shares to be offered so as to enable an investor to decide to purchase or subscribe the shares, as the same may be varied in the Relevant Member State by any measure implementing the Prospectus Directive in the Relevant Member State and the expression “Prospectus Directive” means Directive 2003/71/EC (including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member States) and includes any relevant implementing measure in the Relevant Member State and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
Notice to Prospective Investors in the United Kingdom
In addition, in the United Kingdom, this document is being distributed only to, and is directed only at, and any offer subsequently made may only be directed at persons who are “qualified investors” (as defined in the Prospectus Directive) (i) who have professional experience in matters relating to investments falling within Article 19 (5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005, as amended (the “Order”) and/or (ii) who are high net worth companies (or persons to whom it may otherwise be lawfully communicated) falling within Article 49(2)(a) to (d) of the Order (all such persons together being referred to as “relevant persons”). This document must not be acted on or relied on in the United Kingdom by persons who are not relevant persons. In the United Kingdom, any investment or investment activity to which this document relates is only available to, and will be engaged in with, relevant persons.
Notice to Prospective Investors in Switzerland
The shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, us, or the shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of shares will not be supervised by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes (“CISA”). The investor protection afforded to acquirers of interests in collective investment schemes under the CISA does not extend to acquirers of shares.
Notice to Prospective Investors in the Dubai International Financial Centre
This prospectus relates to an Exempt Offer in accordance with the Offered Securities Rules of the Dubai Financial Services Authority (“DFSA”). This prospectus is intended for distribution only to persons of a type specified in the Offered Securities Rules of the DFSA. It must not be delivered to, or relied on by, any other person. The DFSA has no responsibility for reviewing or verifying any documents in connection with Exempt Offers. The DFSA has not approved this prospectus nor taken steps to verify the information set forth herein and has no responsibility for the prospectus. The shares to which this prospectus relates may be illiquid and/or subject to restrictions on their resale. Prospective purchasers of the shares offered should conduct their own due diligence on the shares. If you do not understand the contents of this prospectus you should consult an authorized financial advisor.
Notice to Prospective Investors in Australia
No placement document, prospectus, product disclosure statement or other disclosure document has been lodged with the Australian Securities and Investments Commission (“ASIC”), in relation to the offering. This
139
Table of Contents
prospectus does not constitute a prospectus, product disclosure statement or other disclosure document under the Corporations Act 2001 (the “Corporations Act”), and does not purport to include the information required for a prospectus, product disclosure statement or other disclosure document under the Corporations Act.
Any offer in Australia of the shares may only be made to persons (the “Exempt Investors”) who are “sophisticated investors” (within the meaning of section 708(8) of the Corporations Act), “professional investors” (within the meaning of section 708(11) of the Corporations Act) or otherwise pursuant to one or more exemptions contained in section 708 of the Corporations Act so that it is lawful to offer the shares without disclosure to investors under Chapter 6D of the Corporations Act.
The shares applied for by Exempt Investors in Australia must not be offered for sale in Australia in the period of 12 months after the date of allotment under the offering, except in circumstances where disclosure to investors under Chapter 6D of the Corporations Act would not be required pursuant to an exemption under section 708 of the Corporations Act or otherwise or where the offer is pursuant to a disclosure document which complies with Chapter 6D of the Corporations Act. Any person acquiring shares must observe such Australian on-sale restrictions.
This prospectus contains general information only and does not take account of the investment objectives, financial situation or particular needs of any particular person. It does not contain any securities recommendations or financial product advice. Before making an investment decision, investors need to consider whether the information in this prospectus is appropriate to their needs, objectives and circumstances, and, if necessary, seek expert advice on those matters.
Notice to Prospective Investors in Hong Kong
The shares have not been offered or sold and will not be offered or sold in Hong Kong, by means of any document, other than (a) to “professional investors” as defined in the Securities and Futures Ordinance (Cap. 571) of Hong Kong and any rules made under that Ordinance; or (b) in other circumstances which do not result in the document being a “prospectus” as defined in the Companies Ordinance (Cap. 32) of Hong Kong or which do not constitute an offer to the public within the meaning of that Ordinance. No advertisement, invitation or document relating to the shares has been or may be issued or has been or may be in the possession of any person for the purposes of issue, whether in Hong Kong or elsewhere, which is directed at, or the contents of which are likely to be accessed or read by, the public of Hong Kong (except if permitted to do so under the securities laws of Hong Kong) other than with respect to the shares which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” as defined in the Securities and Futures Ordinance and any rules made under that Ordinance.
Notice to Prospective Investors in Japan
The shares have not been and will not be registered under the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948, as amended) and, accordingly, will not be offered or sold, directly or indirectly, in Japan, or for the benefit of any Japanese Person or to others for re-offering or resale, directly or indirectly, in Japan or to any Japanese Person, except in compliance with all applicable laws, regulations and ministerial guidelines promulgated by relevant Japanese governmental or regulatory authorities in effect at the relevant time. For the purposes of this paragraph, “Japanese Person” shall mean any person resident in Japan, including any corporation or other entity organized under the laws of Japan.
Notice to Prospective Investors in Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the shares may not be circulated or distributed, nor may the shares be
140
Table of Contents
offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the “SFA”), (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275, of the SFA, or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA.
Where the shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
| (a) | a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or |
| (b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor, |
securities (as defined in Section 239(1) of the SFA) of that corporation or the beneficiaries’ rights and interest (howsoever described) in that trust shall not be transferred within six months after that corporation or that trust has acquired the shares pursuant to an offer made under Section 275 of the SFA except:
| (a) | to an institutional investor or to a relevant person defined in Section 275(2) of the SFA, or to any person arising from an offer referred to in Section 275(1A) or Section 276(4)(i)(B) of the SFA; |
| (b) | where no consideration is or will be given for the transfer; |
| (c) | where the transfer is by operation of law; |
| (d) | as specified in Section 276(7) of the SFA; or |
| (e) | as specified in Regulation 32 of the Securities and Futures (Offers of Investments) (Shares and Debentures) Regulations 2005 of Singapore. |
141
Table of Contents
The validity of the shares of common stock offered hereby will be passed upon for us by Wilson Sonsini Goodrich & Rosati, Professional Corporation, San Diego, California. Cooley LLP, San Diego, California is representing the underwriters in the offering.
The consolidated financial statements as of December 31, 2013 and 2012 and for each of the two years in the period ended December 31, 2013, and cumulatively, for the period January 1, 2009 to December 31, 2013 included in this prospectus have been so included in reliance on the report of PricewaterhouseCoopers LLP, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
The consolidated statements of operations and comprehensive loss (not presented separately herein), redeemable convertible preferred stock and stockholders’ deficit and cash flows (not presented separately herein) of Vital Therapies, Inc. (a development stage enterprise) for the period from May 23, 2003 (Inception) to December 31, 2008, have been audited by Hein & Associates LLP, independent registered public accounting firm, as set forth in their report thereon appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act with respect to the shares of common stock that we are offering. The registration statement, including the attached exhibits and schedules, contains additional relevant information about us and our common stock. This prospectus does not contain all of the information set forth in the registration statement and the exhibits and schedules thereto. The rules and regulations of the SEC allow us to omit from this prospectus certain information included in the registration statement.
For further information about us and our common stock, you may inspect a copy of the registration statement and the exhibits and schedules to the registration statement without charge at the offices of the SEC at 100 F Street, N.E., Washington, D.C. 20549. You may obtain copies of all or any part of the registration statement from the Public Reference Section of the SEC, 100 F Street, N.E., Washington, D.C. 20549 upon the payment of the prescribed fees.
You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements and other information regarding registrants like us that file electronically with the SEC. You can also inspect our registration statement on this website.
Upon closing of this offering, we will become subject to the reporting and information requirements of the Exchange Act and we will file reports, proxy statements and other information with the SEC.
142
Table of Contents
VITAL THERAPIES, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Vital Therapies, Inc.
In our opinion, based on our audits and the report of other auditors, the accompanying consolidated balance sheets as of December 31, 2013 and 2012 and the related consolidated statements of operations, of comprehensive loss, of redeemable convertible preferred stock and stockholders’ deficit and of cash flows for the years then ended present fairly, in all material respects, the financial position of Vital Therapies, Inc. and its subsidiaries (a development stage company) at December 31, 2013 and 2012, and the results of their operations and their cash flows for the years then ended and cumulatively for the period from May 23, 2003 (date of inception) to December 31, 2013 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We did not audit the financial statements of the Company for the period from May 23, 2003 (date of inception) to December 31, 2008. Those financial statements, which reflect a deficit of $34 million accumulated during the development stage, were audited by other auditors whose report, dated July 16, 2009, except with respect to the effect of the May 2011 and March 2012 stock splits described in Note 1, as to which the date is July 22, 2013, has been furnished to us, and our opinion expressed herein, insofar as it relates to the cumulative amounts for the period from May 23, 2003 (date of inception) to December 31, 2008, is based solely on the report of other auditors. We conducted our audits of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits and the report of other auditors provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
San Diego, California
March 11, 2014
F-2
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Vital Therapies, Inc.
We have audited the consolidated statements of operations, comprehensive loss, redeemable convertible preferred stock and stockholders’ deficit and cash flows of Vital Therapies, Inc. and Subsidiaries (a development stage enterprise) for the period from May 23, 2003 (inception) to December 31, 2008. The consolidated statements of operations, comprehensive loss and cash flows are not presented separately herein. The consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated results of operations and cash flows of Vital Therapies, Inc. (a development stage enterprise) for the period from May 23, 2003 (inception) to December 31, 2008 (not presented separately herein) in conformity with accounting principles generally accepted in the United States of America.
/s/ HEIN & ASSOCIATES LLP
Irvine, California
July 16, 2009 (July 22, 2013 as to the effect of the May 2011 and March 2012 stock splits described in Note 1)
F-3
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
(In thousands, except share and per share amounts)
| December 31, | Pro Forma December 31, 2013 |
|||||||||||
| 2012 | 2013 | |||||||||||
| (unaudited) | ||||||||||||
| Assets |
||||||||||||
| Current assets: |
||||||||||||
| Cash and cash equivalents |
$ | 4,477 | $ | 38,186 | $ | 38,186 | ||||||
| Restricted cash |
355 | 963 | 963 | |||||||||
| Short-term investments |
13,996 | — | — | |||||||||
| Deferred financing costs |
— | 3,506 | 3,506 | |||||||||
| Other current assets and prepaid expenses |
320 | 1,200 | 1,200 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current assets |
19,148 | 43,855 | 43,855 | |||||||||
| Property and equipment, net |
1,184 | 2,467 | 2,467 | |||||||||
| Other assets |
— | 263 | 263 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total assets |
$ | 20,332 | $ | 46,585 | $ | 46,585 | ||||||
|
|
|
|
|
|
|
|||||||
| Liabilities, Redeemable Convertible Preferred Stock and Stockholders’ (Deficit) Equity |
||||||||||||
| Current liabilities: |
||||||||||||
| Accounts payable |
$ | 1,144 | $ | 1,224 | $ | 1,224 | ||||||
| Accrued expenses |
601 | 3,395 | 3,395 | |||||||||
| Stock option repurchase liability |
— | 227 | 227 | |||||||||
| Future purchase rights liabilities |
— | 2,600 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current liabilities |
1,745 | 7,446 | 4,846 | |||||||||
|
|
|
|
|
|
|
|||||||
| Other long-term liabilities |
43 | 321 | 321 | |||||||||
|
|
|
|
|
|
|
|||||||
| Commitments and contingencies |
||||||||||||
| Convertible preferred stock (Junior); $0.0001 par value, 3,501,401 shares designated at December 31, 2012 and 2013; 3,501,400 shares issued and outstanding at December 31, 2012 and 2013; $1,500 liquidation preference at December 31, 2013; no shares issued or outstanding pro forma at December 31, 2013 |
1,342 | 1,359 | — | |||||||||
| Redeemable convertible preferred stock (Senior); $0.0001 par value, 10,768,199 and 17,000,000 shares designated at December 31, 2012 and 2013, respectively; 3,518,199 and 10,212,007 shares issued and outstanding at December 31, 2012 and 2013, respectively; $126,886 liquidation preference at December 31, 2013; no shares issued or outstanding pro forma at December 31, 2013 |
24,834 | 82,116 | — | |||||||||
| Stockholders’ (deficit) equity: |
||||||||||||
| Preferred stock, $0.0001 par value; 19,000,000 and 25,000,000 authorized (4,730,400 and 4,498,599 undesignated) at December 31, 2012 and 2013, respectively, and no shares issued or outstanding at December 31, 2012 and 2013 |
|
— |
|
— | — | |||||||
| Common stock, $0.0001 par value; 24,000,000 and 29,250,000 shares authorized at December 31, 2012 and 2013, respectively; 467,167 and 606,238 shares issued and outstanding at December 31, 2012 and 2013, respectively; 14,319,645 shares issued and outstanding pro forma at December 31, 2013 |
— | — | 1 | |||||||||
| Additional paid-in capital |
62,728 | 58,413 | 144,487 | |||||||||
| Accumulated other comprehensive income |
88 | 96 | 96 | |||||||||
| Deficit accumulated during development stage |
(70,448 | ) | (103,166 | ) | (103,166 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Total stockholders’ (deficit) equity |
(7,632 | ) | (44,657 | ) | 41,418 | |||||||
|
|
|
|
|
|
|
|||||||
| Total liabilities, redeemable convertible preferred stock, and stockholders’ (deficit) equity |
$ | 20,332 | $ | 46,585 | $ | 46,585 | ||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
F-4
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Consolidated Statements of Operations
(In thousands, except share and per share amounts)
| Years Ended December 31, | Period From May 23, 2003 (inception) to December 31, 2013 |
|||||||||||
| 2012 | 2013 | |||||||||||
| Revenues |
$ | — | $ | — | $ | 22 | ||||||
| Cost of revenues |
— | — | 13 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
— | — | 9 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating expenses: |
||||||||||||
| Research and development |
5,097 | 21,787 | 62,979 | |||||||||
| General and administrative |
4,483 | 9,615 | 41,315 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
9,580 | 31,402 | 104,294 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(9,580 | ) | (31,402 | ) | (104,285 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Other income (expense): |
||||||||||||
| Interest income |
4 | 5 | 767 | |||||||||
| Interest expense |
(413 | ) | — | (14,298 | ) | |||||||
| Other income (expense), net |
7 | (15 | ) | 271 | ||||||||
| Revaluation of preferred stock warrant liabilities |
180 | — | 12,579 | |||||||||
| Revaluation of future purchase rights liabilities |
3,101 | (1,306 | ) | 1,795 | ||||||||
|
|
|
|
|
|
|
|||||||
| Total other income (expense) |
2,879 | (1,316 | ) | 1,114 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss before cumulative effect of change in accounting principle |
(6,701 | ) | (32,718 | ) | (103,171 | ) | ||||||
| Cumulative effect of change in accounting principle |
— | — | 5 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
(6,701 | ) | (32,718 | ) | (103,166 | ) | ||||||
| Amortization of deemed dividend |
— | (64 | ) | (64 | ) | |||||||
| Accretion to redemption value of redeemable convertible preferred stock |
(942 | ) | (6,303 | ) | (8,690 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss attributable to common stockholders |
$ | (7,643 | ) | $ | (39,085 | ) | $ | (111,920 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per share attributable to common stockholders, basic and diluted |
$ | (17.89 | ) | $ | (74.86 | ) | ||||||
|
|
|
|
|
|||||||||
| Weighted-average common shares outstanding, basic and diluted |
427,117 | 522,102 | ||||||||||
|
|
|
|
|
|||||||||
| Pro forma net loss per share attributable to common stockholders, basic and diluted (unaudited) |
$ | (3.14 | ) | |||||||||
|
|
|
|||||||||||
| Pro forma weighted-average common shares outstanding, basic and diluted (unaudited) |
10,008,616 | |||||||||||
|
|
|
|||||||||||
The accompanying notes are an integral part of these financial statements.
F-5
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Consolidated Statements of Comprehensive Loss
(In thousands)
|
Years Ended |
Period From May 23, 2003 (inception) to December 31, 2013 |
|||||||||||
| 2012 | 2013 | |||||||||||
| Net loss |
$ | (6,701 | ) | $ | (32,718 | ) | $ | (103,166 | ) | |||
| Other comprehensive income: |
||||||||||||
| Foreign currency translation |
5 | 8 | 96 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total comprehensive loss |
$ | (6,696 | ) | $ | (32,710 | ) | $ | (103,070 | ) | |||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
F-6
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Consolidated Statements of Redeemable Convertible Preferred Stock and Stockholders’ Deficit
(In thousands, except shares)
| Series A-D, Junior Convertible, and Senior Redeemable Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Note Receivable from Officer |
Accumulated Other Comprehensive Income |
Deficit Accumulated During Development Stage |
Total Stockholders’ Deficit |
||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| Balance at May 23, 2003 (inception) |
— | $ | — | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (967 | ) | (967 | ) | |||||||||||||||||||||||||
| Issuance of restricted common stock |
— | — | 125 | — | 4 | (2 | ) | — | — | 2 | ||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2003 |
— | — | 125 | — | 4 | (2 | ) | — | (967 | ) | (965 | ) | ||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (2,482 | ) | (2,482 | ) | |||||||||||||||||||||||||
| Private placement—from February 2 to May 24, 2004 and conversion of loan |
986 | 2,899 | 1,472 | — | 442 | — | — | — | 442 | |||||||||||||||||||||||||||
| Proceeds from note receivable from founders |
— | — | — | — | — | 1 | — | — | 1 | |||||||||||||||||||||||||||
| Exercise of stock options |
— | — | 18 | — | 1 | — | — | — | 1 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 2 | — | — | — | 2 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 83 | — | — | — | — | — | (83 | ) | (83 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2004 |
986 | 2,982 | 1,615 | — | 449 | (1 | ) | — | (3,532 | ) | (3,084 | ) | ||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (5,278 | ) | (5,278 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 1 | — | 1 | |||||||||||||||||||||||||||
| Private placement—from October 24 to December 23, 2005 and conversion of loan |
5,994 | 8,568 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Warrant issued in connection with the Series B preferred stock financing |
— | — | — | — | 82 | — | — | — | 82 | |||||||||||||||||||||||||||
| Conversion of loan |
877 | 2,981 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Proceeds from note receivable from founders |
— | — | — | — | — | 1 | — | — | 1 | |||||||||||||||||||||||||||
| Exercise of stock options |
— | — | 246 | — | 8 | — | — | — | 8 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 9 | — | — | — | 9 | |||||||||||||||||||||||||||
| Repurchase unvested restricted stock from founder |
— | — | (5 | ) | — | — | — | — | — | — | ||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 98 | — | — | — | — | — | (98 | ) | (98 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2005 |
7,857 | 14,629 | 1,856 | — | 548 | — | 1 | (8,908 | ) | (8,359 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (6,106 | ) | (6,106 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 4 | — | 4 | |||||||||||||||||||||||||||
| Reclassification of warrant to liabilities |
— | — | — | — | (82 | ) | — | — | — | (82 | ) | |||||||||||||||||||||||||
| Exercise of stock options |
— | — | 274 | — | 39 | — | — | — | 39 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 12 | — | — | — | 12 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 139 | — | — | — | — | — | (139 | ) | (139 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2006 |
7,857 | 14,768 | 2,130 | — | 517 | — | 5 | (15,153 | ) | (14,631 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (8,165 | ) | (8,165 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 22 | — | 22 | |||||||||||||||||||||||||||
| Private placement—September 2007 and conversion of loan |
18,800 | 27,915 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Exercise of stock options |
— | — | 902 | — | 128 | — | — | — | 128 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 29 | — | — | — | 29 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 136 | — | — | — | — | — | (136 | ) | (136 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2007 |
26,657 | 42,819 | 3,032 | — | 674 | — | 27 | (23,454 | ) | (22,753 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (10,294 | ) | (10,294 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 42 | — | 42 | |||||||||||||||||||||||||||
| Exercise of stock options |
— | — | 150 | — | 21 | — | — | — | 21 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 54 | — | — | — | 54 | |||||||||||||||||||||||||||
F-7
Table of Contents
| Series A-D, Junior Convertible, and Senior Redeemable Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Note Receivable from Officer |
Accumulated Other Comprehensive Income |
Deficit Accumulated During Development Stage |
Total Stockholders’ Deficit |
||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| Sale of common stock for cash in connection with consulting services |
— | — | 59 | — | 9 | — | — | — | 9 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 127 | — | — | — | — | — | (127 | ) | (127 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2008 |
26,657 | 42,946 | 3,241 | — | 758 | — | 69 | (33,875 | ) | (33,048 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (12,198 | ) | (12,198 | ) | |||||||||||||||||||||||||
| Other comprehensive loss |
— | — | — | — | — | — | (2 | ) | — | (2 | ) | |||||||||||||||||||||||||
| Exercise of stock options |
— | — | 4 | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 51 | — | — | — | 51 | |||||||||||||||||||||||||||
| Conversion of preferred stock to common stock |
(5,852 | ) | (9,368 | ) | 5,852 | — | 9,368 | — | — | — | 9,368 | |||||||||||||||||||||||||
| Adjust previously recorded accretion |
— | — | — | — | (583 | ) | — | — | 583 | — | ||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 182 | — | — | (182 | ) | — | — | — | (182 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2009 |
20,805 | 33,760 | 9,097 | — | 9,412 | — | 67 | (45,490 | ) | (36,011 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (12,378 | ) | (12,378 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 3 | — | 3 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 43 | — | — | — | 43 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 142 | — | — | (142 | ) | — | — | — | (142 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2010 |
20,805 | 33,902 | 9,097 | — | 9,313 | — | 70 | (57,868 | ) | (48,485 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (5,879 | ) | (5,879 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 13 | — | 13 | |||||||||||||||||||||||||||
| Private placement—from February 23 to June 7, 2011 and conversion of loans |
69,462 | 20,218 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 36 | — | — | — | 36 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 538 | — | — | (538 | ) | — | — | — | (538 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2011 |
90,267 | 54,658 | 9,097 | — | 8,811 | — | 83 | (63,747 | ) | (54,853 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (6,701 | ) | (6,701 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 5 | — | 5 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 144 | — | — | — | 144 | |||||||||||||||||||||||||||
| Conversion of Series A-D preferred stock to common stock |
(90,267 | ) | (54,715 | ) | 458,070 | — | 54,715 | — | — | — | 54,715 | |||||||||||||||||||||||||
| Private placement of junior convertible preferred stock—from February 3 to March 5, 2012 |
3,501,400 | 1,326 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Private placement senior redeemable convertible preferred stock—from September 25 to October 29, 2012 and conversion of loans |
3,518,199 | 23,965 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 942 | — | — | (942 | ) | — | — | — | (942 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2012 |
7,019,599 | 26,176 | 467,167 | — | 62,728 | — | 88 | (70,448 | ) | (7,632 | ) | |||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | (32,718 | ) | (32,718 | ) | |||||||||||||||||||||||||
| Other comprehensive income |
— | — | — | — | — | — | 8 | — | 8 | |||||||||||||||||||||||||||
| Exercise of stock options, net of repurchase liability |
— | — | 139,071 | — | 135 | — | — | — | 135 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 948 | — | — | — | 948 | |||||||||||||||||||||||||||
| Private placement senior redeemable convertible preferred stock—from February 28 to December 18, 2013 |
6,693,808 | 50,996 | — | — | 905 | — | — | — | 905 | |||||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 6,303 | — | — | (6,303 | ) | — | — | — | (6,303 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2013 |
13,713,407 | $ | 83,475 | 606,238 | $ | — | $ | 58,413 | $ | — | $ | 96 | $ | (103,166 | ) | $ | (44,657 | ) | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-8
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Consolidated Statement of Cash Flows
(In thousands)
|
Years Ended |
Period From May 23, 2003 (inception) to December 31, 2013 |
|||||||||||
| 2012 | 2013 | |||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (6,701 | ) | $ | (32,718 | ) | $ | (103,166 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
651 | 799 | 5,986 | |||||||||
| Stock-based compensation |
144 | 948 | 1,327 | |||||||||
| Noncash interest expense |
382 | — | 13,517 | |||||||||
| Revaluation of preferred stock warrant liabilities |
(180 | ) | — | (12,584 | ) | |||||||
| Revaluation of future purchase rights liabilities |
(3,101 | ) | 1,306 | (1,795 | ) | |||||||
| Deferred rent |
43 | (16 | ) | 27 | ||||||||
| Gain on sale of equipment |
(7 | ) | — | (25 | ) | |||||||
| Other |
(2 | ) | (1 | ) | 2 | |||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Other assets and prepaid expenses |
(242 | ) | (1,141 | ) | (1,214 | ) | ||||||
| Accounts payable |
(219 | ) | (91 | ) | 560 | |||||||
| Accrued expenses |
(12 | ) | 2,266 | 2,849 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(9,244 | ) | (28,648 | ) | (94,516 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Purchases of short-term investments |
(13,992 | ) | (2,999 | ) | (16,991 | ) | ||||||
| Sales of short-term investments |
— | 17,000 | 17,000 | |||||||||
| Proceeds from sale of equipment |
20 | — | 140 | |||||||||
| Increase in restricted cash |
(355 | ) | (608 | ) | (963 | ) | ||||||
| Purchases of property and equipment |
(261 | ) | (1,484 | ) | (4,669 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) provided by investing activities |
(14,588 | ) | 11,909 | (5,483 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from debt, net of issuance costs |
6,934 | — | 30,277 | |||||||||
| Deferred financing costs |
— | (3,112 | ) | (3,367 | ) | |||||||
| Principal payments on term loan |
(533 | ) | — | (2,154 | ) | |||||||
| Proceeds from issuance of common stock |
— | — | 62 | |||||||||
| Proceeds from issuance of preferred stock, net of issuance costs |
21,100 | 53,195 | 112,717 | |||||||||
| Proceeds from exercise of stock options |
— | 135 | 334 | |||||||||
| Proceeds from early exercise of stock options |
— | 227 | 227 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
27,501 | 50,445 | 138,096 | |||||||||
|
|
|
|
|
|
|
|||||||
| Effect of exchange rate changes on cash and cash equivalents |
5 | 3 | 89 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net change in cash and cash equivalents |
3,674 | 33,709 | 38,186 | |||||||||
| Cash and cash equivalents, beginning of period |
803 | 4,477 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents, end of period |
$ | 4,477 | $ | 38,186 | $ | 38,186 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental cash flow information: |
||||||||||||
| Cash paid for interest |
$ | 32 | $ | — | $ | 518 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental disclosure of noncash investing and financing activities: |
||||||||||||
| Deferred offering costs included in liabilities |
$ | — | $ | 394 | $ | 394 | ||||||
|
|
|
|
|
|
|
|||||||
| Purchase of property and equipment through liabilities |
$ | 502 | $ | 170 | $ | 672 | ||||||
|
|
|
|
|
|
|
|||||||
| Leasehold improvements paid for by landlord |
$ | — | $ | 478 | $ | 478 | ||||||
|
|
|
|
|
|
|
|||||||
| Purchase of assets through debt |
$ | — | $ | — | $ | 2,807 | ||||||
|
|
|
|
|
|
|
|||||||
| Issuance of Series D warrant in connection with Series D redeemable convertible preferred stock |
$ | — | $ | — | $ | 3,265 | ||||||
|
|
|
|
|
|
|
|||||||
| Conversion of debt for redeemable convertible preferred stock |
$ | 7,296 | $ | — | $ | 33,996 | ||||||
|
|
|
|
|
|
|
|||||||
| Conversion of redeemable convertible preferred stock to common stock |
$ | 54,715 | $ | — | $ | 64,083 | ||||||
|
|
|
|
|
|
|
|||||||
| Valuation of future purchase rights upon issuance |
$ | 3,101 | $ | 1,294 | $ | 4,395 | ||||||
|
|
|
|
|
|
|
|||||||
| Beneficial conversion underlying the senior preferred stock |
$ | — | $ | 969 | $ | 969 | ||||||
|
|
|
|
|
|
|
|||||||
| Accretion to redemption value of redeemable convertible preferred stock |
$ | 942 | $ | 6,303 | $ | 8,690 | ||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
F-9
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements
1. Description of Business and Basis of Financial Statements
Description of Business
We began operations as a California corporation on May 23, 2003 through the acquisition of the assets and business of VitaGen, Inc. and in June 2003 changed our name to Vital Therapies, Inc. In January 2004, we were re-incorporated in Delaware. We are a biotherapeutic company focused on developing a cell-based therapy targeting the treatment of all forms of acute liver failure. Our product candidate, currently in Phase 3 clinical trials, the ELAD® System, or ELAD, is an extracorporeal human cell-based bio-artificial liver therapy designed to allow the patient’s own liver to regenerate to a healthy state, or to stabilize the patient until transplant. Since inception, we have devoted essentially all of our efforts to product development, clinical testing and pilot manufacturing and have not realized revenues from our planned principal operations. Accordingly, we are considered to be a development stage enterprise. Our headquarters are located in San Diego, California.
Basis of Presentation and Consolidation
The accompanying consolidated financial statements have been prepared in conformity with U.S. generally accepted accounting principles, or GAAP, and include the accounts of Vital Therapies, Inc. and its wholly-owned subsidiaries located in the United Kingdom (currently inactive) and China. All intercompany accounts and transactions have been eliminated in consolidation. We manage our operations as a single segment for the purposes of assessing performance and making operating decisions.
In May 2011, we executed a 1 for 10 reverse stock split on all outstanding shares of common and preferred stock. In March 2012, we executed a 1 for 340 reverse stock split on all outstanding shares of common and junior preferred stock. All share and per share amounts have been retroactively restated to reflect these reverse stock splits.
Liquidity
We have incurred losses since inception and negative cash flows from operating activities for the year ended December 31, 2013. As of December 31, 2013, we had working capital of $36.4 million and a deficit accumulated during the development stage of $103.2 million. We anticipate that we will continue to incur net losses into the foreseeable future as we work toward completing the ELAD System’s clinical development through the clinical trial process and expand our corporate and manufacturing infrastructure.
We plan to continue to fund our losses from operations and capital funding needs through future debt and equity financings. If we are not able to secure adequate additional capital funding, we may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, or suspend or curtail planned programs. Any of these actions could materially harm our business, results of operations, and future prospects.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with GAAP requires us to make certain estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates and assumptions.
F-10
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Unaudited Pro Forma Balance Sheet Information
The accompanying unaudited pro forma balance sheet as of December 31, 2013, has been prepared to give effect to the automatic conversion upon the closing of a qualified initial public offering of all outstanding shares of senior redeemable convertible preferred stock and convertible junior preferred stock as of December 31, 2013 into 13,713,407 shares of common stock and the reclassification of our future purchase rights liabilities to additional paid-in capital, a component of stockholders’ (deficit) equity, as though the proposed initial public offering had occurred on December 31, 2013.
Unaudited Pro Forma Net Loss Per Share
The following table presents the computation of our unaudited pro forma net loss per share (in thousands, except share and per share amounts):
| Year Ended December 31, 2013 |
||||
| (unaudited) |
||||
| Numerator: |
||||
| Net loss attributable to common stockholders. |
$ | (39,085 | ) | |
| Less (Add): |
||||
| Change in fair value of future purchase rights liabilities |
1,306 | |||
| Accretion of redeemable convertible preferred stock |
6,303 | |||
| Amortization of deemed dividend |
64 | |||
|
|
|
|||
| $ | (31,412 | ) | ||
|
|
|
|||
| Denominator: |
||||
| Weighted-average common shares outstanding, basic and diluted |
522,102 | |||
| Pro forma adjustments to reflect assumed weighted-average effect of conversion of convertible preferred stock |
9,486,514 | |||
|
|
|
|||
| Pro forma weighted-average common shares outstanding, basic and |
10,008,616 | |||
|
|
|
|||
| Pro forma net loss per share attributable to common stockholders, basic and diluted |
$ | (3.14 | ) | |
|
|
|
|||
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and highly-liquid investments with original maturities of three months or less when acquired and are stated at cost, which approximates market value.
Restricted Cash
Restricted cash relates to amounts reserved for various clinical trial obligations, lease arrangements and certain provisions of the junior preferred stock agreement.
F-11
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Short-term Investments
Short-term investments consist of highly liquid investments with an original maturity at the time of purchase of greater than three months and consist of obligations of the U.S. government.
Investments are classified as available-for-sale and stated at fair value. The net unrealized gains or losses on available-for-sale securities are reported as a component of other comprehensive income (loss). The specific identification method is used to compute the realized gains and losses on investments. Investments are periodically reviewed for impairment. If the carrying value of an investment exceeds its fair value and the decline in value is determined to be other-than-temporary, an impairment loss is recognized for the difference.
Fair Value of Financial Instruments
Fair value is defined as the price that would be received to sell an asset or be paid to transfer a liability in an orderly transaction between market participants on the measurement date. Accounting guidance establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. The standard describes three levels of inputs that may be used to measure fair value:
Level 1—Quoted prices in active markets for identical assets or liabilities. Our Level 1 assets consisted of U.S. treasuries and money market funds for the periods presented. We had no Level 1 liabilities for any period presented.
Level 2—Observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities, quoted prices in markets with insufficient volume or infrequent transactions (less active markets), or model-derived valuations in which all significant inputs are observable or can be derived principally from or corroborated with observable market data for substantially the full term of the assets or liabilities. We had no Level 2 assets or liabilities for any period presented.
Level 3—Unobservable inputs to the valuation methodology that are significant to the measurement of the fair value of assets or liabilities. Our Level 3 liabilities included preferred stock warrants and future purchase rights liabilities during the periods presented. We had no Level 3 assets in any period presented. We estimated the fair value of the preferred stock warrants and the future purchase rights using a binomial lattice model depending on the underlying attributes of the preferred stock warrants or future purchase rights, as applicable.
The carrying value of cash and cash equivalents, restricted cash, other current assets and prepaid expenses, accounts payable, and accrued expenses approximate fair value due to the short period of time to maturity.
The future purchase rights were recorded at their estimated fair value at the date of issuance. The binomial lattice model used in the determination of the estimated fair value of the future purchase rights is based on various assumptions, including the estimated fair value of our senior preferred and common stock. We record subsequent adjustments to reflect the increase or decrease in estimated fair value at each reporting date as a gain or loss.
Deferred Financing Costs
Deferred financing costs represent direct costs associated with the future issuance of our corporate securities. Direct costs include, but are not limited to the legal, accounting and printing costs. Indirect costs,
F-12
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
associated with the future issuance of corporate securities are expensed as incurred. Upon the completion of the proposed issuance, the deferred financing costs will be offset against the proceeds from the security issuance. If the proposed issuance is not completed, the deferred financing costs will be charged to expense.
Property and Equipment, Depreciation and Amortization
Property and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets (generally three to five years). Leasehold improvements are stated at cost and amortized on a straight-line basis over the lesser of the remaining term of the related lease or the estimated useful lives of the assets. Construction in progress is not depreciated until the underlying asset is placed in service. Repairs and maintenance costs are charged to expense as incurred.
Impairment of Long-lived Assets
Long-lived assets consist primarily of property and equipment. An impairment loss is recorded if and when events and circumstances indicate that assets might be impaired and the undiscounted cash flows estimated to be generated by those assets are less than the carrying amount of those assets. While our current and historical operating losses and negative cash flows are indicators of impairment, we believe that future cash flows to be received support the carrying value of our long-lived assets and, accordingly, have not recognized any impairment losses through December 31, 2013.
Redeemable Convertible Preferred Stock
Our Series A, Series B, Series C, and Series D redeemable convertible preferred stock and junior convertible and senior redeemable convertible preferred stock are classified as mezzanine equity instead of a component of stockholders’ deficit in accordance with authoritative guidance for the classification and measurement of potentially redeemable securities, as the preferred stock is conditionally redeemable at the holder’s option or upon certain change in control events that are outside our control, including liquidation, sale, or transfer of control of the company.
Warrants to Purchase Redeemable Convertible Preferred Stock
We classify our preferred stock warrants that are either puttable or that are redeemable as liabilities on the consolidated balance sheets at fair value. At the end of each reporting period, changes in fair value during the period are recorded as a component of other income (expense), net. In February 2012, a final valuation adjustment was recognized for these preferred stock warrants prior to their conversion to common stock warrants in conjunction with the junior preferred stock offering.
Future Purchase Rights Liabilities
Our future purchase rights liabilities are recorded at their estimated fair value on the date of issuance as a discount on the underlying preferred stock and are remeasured to reflect changes in the estimated fair value at each reporting date, with any decrease or increase in the estimated fair value being recorded as other income or expense, respectively. The fair value of these liabilities is estimated using a binomial lattice model that is based on the characteristics of the common and preferred stock on the valuation date, probabilities related to our
F-13
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
operations and clinical development, as well as assumptions for volatility, remaining expected life, risk-free interest rate and, in some cases, credit spread. Changes in the fair value of the future purchase rights fluctuate in conjunction with increases or decreases in the fair value of our common stock, and the number of preferred and common shares and future purchase rights outstanding relative to our enterprise value at each reporting date.
Revenues and Cost of Revenues
Revenues to date relate to compassionate use treatment for patients and are recognized when the product is shipped and the risks and rewards of ownership have transferred to the hospital administering the treatments. Shipping charges billed to hospitals are included in revenues and the related shipping costs are included in cost of revenues. Cost of revenues consists of direct materials costs incurred during the manufacturing of the ELAD cartridges. Revenues and cost of revenues have not been significant to date.
Research and Development
Research and development costs consist primarily of employee-related expenses, contractors, clinical trial sites and contract research organizations engaged in the development of the ELAD System, expenses associated with obtaining regulatory approvals, and the cost of acquiring and manufacturing clinical trial materials. All research and development costs are expensed as incurred.
Stock-based Compensation
We measure and recognize compensation expense for all stock-based payments made to employees and directors based on estimated fair value, net of an estimated forfeiture rate, and to consultants based on estimated fair value. Currently, our stock-based awards consist only of stock options; however, future grants under our equity compensation plans may consist of shares of restricted stock and restricted stock units. We estimate the fair value of stock options granted using the Black-Scholes-Merton, or BSM, option pricing model, which requires the use of estimates to value employee stock-based compensation at the date of grant.
We recognize stock-based compensation cost for employees and directors on a straight-line basis over the requisite service period of the award. Stock-based compensation expense is recognized only for those awards that are ultimately expected to vest. We estimate forfeitures based on an analysis of our historical employee turnover and will continue to evaluate the appropriateness of the forfeiture rate based on actual forfeiture experience, analysis of employee turnover and other factors. We will revise the forfeiture estimate, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Changes in forfeiture estimates, which have not been material to date, impact compensation cost in the period in which the change in estimate occurs.
The fair value of options granted to consultants is estimated using the BSM option pricing model and is remeasured at each reporting date with changes in fair value recognized as expense in the consolidated statements of operations.
F-14
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
The BSM option pricing model requires the input of highly subjective assumptions, including the risk-free interest rate, the expected dividend yield of our common stock, the expected volatility of the price of our common stock, and the expected term of the option. These estimates involve inherent uncertainties and the application of management’s judgment. If factors change and different assumptions are used, our stock-based compensation expense could be materially different in the future. These assumptions are estimated as follows:
Risk-free Interest Rate
We base the risk-free interest rate assumption on zero-coupon U.S. treasury instruments appropriate for the expected term of the stock option grants.
Expected Dividend Yield
We base the expected dividend yield assumption on the fact that we have never paid cash dividends and have no present intention to pay cash dividends. Consequently, we used an expected dividend yield of zero.
Expected Volatility
As we do not have a trading history for our common stock, the expected stock price volatility for our common stock is estimated based on volatilities of a peer group of similar companies by taking the average historic price volatility for these peers for a period equivalent to the expected term of the stock option grants. The peer group was developed based on companies in the biotechnology industry whose shares are publicly-traded.
Expected Term
The expected term represents the period of time that options are expected to be outstanding. As we do not have sufficient historical experience for determining the expected term of the stock option awards granted we determined the expected life assumption using either the simplified method, which is an average of the contractual term of the option and its ordinary vesting period, or the comparable average expected term utilizing those companies in the peer group noted above, as applicable.
Common Stock Valuation
Due to the absence of a public market trading our common stock, it is necessary to estimate the fair value of the common stock underlying our stock-based awards when performing fair value calculations using the BSM option pricing model. The fair value of the common stock underlying our stock-based awards was assessed on each grant date by our board of directors. All options to purchase shares of our common stock have been granted with an exercise price per share no less than the fair value per share of our common stock underlying those options on the date of grant.
In the absence of a public trading market for our common stock, we determined the estimated fair value of our common stock using methodologies, approaches, and assumptions consistent with the American Institute of Certified Public Accountants, or AICPA, Audit and Accounting Practice Aid Series: Valuation of Privately Held Company Equity Securities Issued as Compensation, or the AICPA Practice Aid.
F-15
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Leases
We lease all of our office space and enter into various other operating lease agreements in conducting our business. At the inception of each lease, we evaluate the lease agreement to determine whether the lease is an operating or capital lease. Some of our lease agreements may contain renewal options, tenant improvement allowances, rent holidays or rent escalation clauses. When such items are included in a lease agreement, we record a deferred rent asset or liability on the consolidated balance sheets equal to the difference between the rent expense and future minimum lease payments due. The rent expense related to operating leases is recognized on a straight-line basis in the statements of operations over the terms of the leases.
Comprehensive Loss
Comprehensive loss is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. Comprehensive loss has been reflected in the consolidated statements of comprehensive loss and as a separate component of the consolidated statements of redeemable convertible preferred stock and stockholders’ deficit for all periods presented.
Foreign Currency Translation and Transactions
The functional currencies of each our subsidiaries in the United Kingdom (currently inactive) and China is the local currency. Assets and liabilities of the subsidiaries are translated at the rate of exchange at the balance sheet date. Expenses are translated at the average rate of exchange rates in effect during the reporting period. Gains and losses resulting from foreign currency translation are included in accumulated other comprehensive income in the accompanying consolidated balance sheets. Gains and losses resulting from foreign currency transactions are included in the results of operations, which to date, have not been significant.
Income Taxes
We account for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the consolidated financial statements. Under this method, deferred tax assets and liabilities are determined on the basis of the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.
We recognize net deferred tax assets to the extent we believe these assets are more likely than not to be realized. In making such a determination, management considers all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax-planning strategies, and results of recent operations. If management determines that we would be able to realize our deferred tax assets in the future in excess of their net recorded amount, management would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes.
We record uncertain tax positions in accordance with ASC 740 on the basis of a two-step process whereby (1) management determines whether it is more likely than not that the tax positions will be sustained on the basis of the technical merits of the position and (2) for those tax positions that meet the more-likely-than-not recognition threshold, management recognizes the largest amount of tax benefit that is more than 50 percent likely to be realized upon ultimate settlement with the related tax authority. We recognize interest and penalties related to unrecognized tax benefits, if any, within income tax expense and any accrued interest and penalties are included within the related tax liability line.
F-16
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
2. Summary of Significant Accounting Policies (Continued)
Net Loss Per Share
Basic net loss per share attributable to common stockholders is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding for the period, without consideration for common stock equivalents. Excluded from the weighted-average number of shares outstanding are shares that have been issued upon the early exercise of stock options and are subject to future vesting, which was a total of 45,159 shares as of December 31, 2013. Diluted net loss per share attributable to common stockholders is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method. Common stock equivalents are comprised of redeemable convertible preferred stock, warrants for the purchase of common stock, and options outstanding under our stock option plan. For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding due to our net loss position.
Potentially dilutive securities not included in the calculation of diluted net loss per share attributable to common stockholders because to do so would be anti-dilutive are as follows (in common stock equivalent shares):
| As of December 31, | ||||||||
| 2012 | 2013 | |||||||
| Redeemable convertible preferred stock |
7,019,599 | 13,713,407 | ||||||
| Options to purchase common stock |
2,336,314 | 3,098,573 | ||||||
| Warrants to purchase common stock |
250,646 | 250,646 | ||||||
Recently Issued and Adopted Accounting Pronouncements
In February 2013, the FASB issued ASU No. 2013-02, Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income. ASU 2013-02 requires an entity to provide information about the amounts reclassified out of accumulated other comprehensive income by component. In addition, an entity is required to present, either on the face of the statement where net income is presented or in the notes, significant amounts reclassified out of accumulated other comprehensive income by the respective line items of net income but only if the amount reclassified is required under GAAP to be reclassified to net income in its entirety in the same reporting period. ASU 2013-02 is effective prospectively for reporting periods beginning after December 15, 2012. We adopted ASU 2013-02 as of January 1, 2013 and the adoption did not have a material impact on our financial condition or results of operations.
In July 2013, the FASB issued ASU No. 2013-11, Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists, which requires an unrecognized tax benefit, or a portion of an unrecognized tax benefit, to be presented in the financial statements as a reduction to a deferred tax asset for a net operating loss, or NOL, carryforward, a similar tax loss, or a tax credit carryforward. If an NOL carryforward, a similar tax loss, or a tax credit carryforward is not available at the reporting date under the tax law of the applicable jurisdiction to settle any additional income taxes that would result from the disallowance of a tax position or the tax law of the applicable jurisdiction does not require the entity to use, and the entity does not intend to use, the deferred tax asset for such purpose, the unrecognized tax benefit should be presented in the financial statements as a liability and should not be combined with deferred tax assets. The new guidance is effective for fiscal years beginning after December 15, 2013 and earlier adoption is permitted. We are currently evaluating the impact of the new guidance; however, management does not believe it will have a material effect on our financial position or results of operations.
F-17
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
3. Other Financial Information
Short-term Investments
The estimated fair value of our available-for-sale securities was as follows at December 31, 2012 (in thousands):
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value |
|||||||||||||
| U.S. treasury securities |
$ | 13,996 | $ | — | $ | — | $ | 13,996 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
There were no impairments considered other-than-temporary for any period presented. Gross realized gains and losses on sales of marketable securities were not significant for any period presented.
Property and Equipment
Property and equipment, leasehold improvements, and related accumulated depreciation and amortization were as follows (in thousands):
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Manufacturing and laboratory equipment |
$ | 3,191 | $ | 3,229 | ||||
| Office furniture and equipment |
47 | 112 | ||||||
| Clinical equipment |
726 | 1,606 | ||||||
| Computer equipment and software |
95 | 122 | ||||||
| Leasehold improvements |
2,029 | 2,830 | ||||||
| Construction in progress |
— | 323 | ||||||
|
|
|
|
|
|||||
| 6,088 | 8,222 | |||||||
| Less: accumulated depreciation and amortization |
(4,904 | ) | (5,755 | ) | ||||
|
|
|
|
|
|||||
| Total |
$ | 1,184 | $ | 2,467 | ||||
|
|
|
|
|
|||||
Depreciation and amortization expense for the years ended December 31, 2012 and 2013 was $651,000 and $851,000, respectively.
In cases where our lessor grants to us leasehold improvement allowances that reduce our rent expense, we capitalize the improvements as incurred and recognize deferred rent, which is amortized over the shorter of the lease term or the expected useful life of the improvements. During the year ended December 31, 2013, we capitalized $478,000 in such leasehold improvements.
F-18
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
3. Other Financial Information (Continued)
Accrued Expenses
Accrued expenses consist of (in thousands):
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Accrued clinical costs |
$ | 312 | $ | 2,067 | ||||
| Accrued offering costs |
28 | 496 | ||||||
| Accrued compensation and related taxes |
137 | 512 | ||||||
| Deferred Rent, current |
8 | 142 | ||||||
| Accrued other |
116 | 178 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 601 | $ | 3,395 | ||||
|
|
|
|
|
|||||
4. Debt
Convertible Loans
Series A
In May 2003, we received a total of $645,000 in the form of convertible loans, with an 8.0% annual interest rate. In October 2005, the total principal of $645,000 and accrued interest of $124,000 converted into Series A preferred stock (Note 7).
From June 2003 to January 2004, we received a total of $1.0 million in the form of convertible loans, with an 8.0% annual interest rate. In May 2004, the total principal of $1.0 million and accrued interest of $14,000 converted into Series A preferred stock (see Note 7).
Series B
From January to October 2005, we received a total of $1.9 million in the form of convertible loans, with an 8.0% annual interest rate. From October through December 2005, the total principal of $1.9 million and accrued interest of $71,000 converted into Series B preferred stock (see Note 7).
Series C
In November 2006, we received a total of $3.5 million in the form of convertible loans, with an 8.0% annual interest rate. In June 2007, we received a total of $1.7 million in the form of convertible loans, with an 8.0% annual interest rate. During September 2007, the total principal of $5.2 million and accrued interest of $262,000 converted into Series C preferred stock (see Note 7).
Series D
From September 2009 through May 2011, we received a total of $16.2 million in the form of convertible loans, with an 8.0% annual stated interest rate, which could increase to 12.0% upon certain events of default. In May 2011, the total principal of $16.2 million and accrued interest of $1.2 million converted into Series D preferred stock (see Note 7). The holders of the Series D loans received warrants to purchase Series D preferred stock. The number of securities was variable depending on the valuation of the Series D preferred stock. The warrants were not initially exercisable, but became exercisable upon conversion of the related loan into our
F-19
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
4. Debt (Continued)
Series D preferred stock. In connection with the Series D loans we paid legal fees of $122,000, which were recorded as deferred financing costs and amortized to interest expense over the life of the Series D loans.
From May to September 2012, we received a total of $7.2 million in the form of convertible loans. Interest on the loans accrued at 10% per year compounded and was added to the principal amount of the convertible loans at the end of each calendar quarter. Accrued interest was payable on the maturity date in September 2012. These convertible loans automatically converted in conjunction with a qualified equity financing at a conversion price equal to the price per share paid in the financing. We issued 911,949 shares of senior preferred stock upon the conversion of a these loans for $7.2 million of principal and $146,000 of accrued interest (see Note 7).
Term Loan and Troubled Debt Restructuring
In September 2008, we entered into a term loan with CIT Healthcare LLC (the Term Loan) at a fixed interest rate of approximately 10.6% for a period of up to four years that provided a maximum of $4.0 million to be used for capital expenditures. In connection with the Term Loan, we paid a loan origination fee and legal fees totaling $80,000 and issued a warrant to purchase 94 shares of our Series C preferred stock at $1,496 per share. The loan origination and legal fees were recorded as deferred financing costs and were amortized over the life of the loan to interest expense. Additionally, we recorded $42,000 for the estimated fair value (as determined utilizing a BSM valuation model) of the warrants as debt discount, which was amortized over the life of the loan to interest expense.
In May 2012, CIT Healthcare LLC agreed to modify the scheduled payment terms of the Term Loan and deferred 50% of the remaining monthly payments to a final balloon payment due at the end of the original loan term. These modifications were determined to meet the definition of a troubled debt restructuring. The outstanding debt and accrued interest were paid in full in accordance with the amended provisions in October 2012.
5. Commitments and Contingencies
Operating Leases
We lease office, manufacturing and research and development facilities, and equipment under various non-cancellable operating lease agreements through 2017. Facility leases generally provide for periodic rent increases and many contain escalation clauses and renewal options. Certain leases require us to pay property taxes and routine maintenance.
Future minimum annual obligations under all non-cancellable operating lease commitments, including the facility leases described above, at December 31, 2013 are as follows (in thousands):
| Year Ending December 31, 2013 | ||||||||||||||||||||||||
| Total | 2014 | 2015 | 2016 | 2017 | 2018 | |||||||||||||||||||
| Operating lease obligations |
$ | 3,023 | $ | 840 | $ | 839 | $ | 853 | $ | 491 | $ | — | ||||||||||||
Total rent expense under our operating leases was $459,000 and $659,000 during the years ended December 31, 2012 and 2013, respectively.
We recognize rent expense for our facility operating leases on a straight-line basis. We account for the difference between the minimum lease payments and the straight-line amount as deferred rent. Current and
F-20
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
5. Commitments and Contingencies (Continued)
long-term deferred rent totaled $8,000 and $43,000 at December 31, 2012, respectively, and $142,000 and $321,000 at December 31, 2013, respectively.
Purchase Commitments
The following table summarizes our purchase obligations at December 31, 2013 (in thousands):
| Payments Due by Period | ||||||||||||||||||||
| Total | Less Than 1 Year |
2-3 Years |
3-5 Years |
More Than 5 Years |
||||||||||||||||
| Purchase obligations |
$ | 2,410 | $ | 2,410 | $ | — | $ | — | $ | — | ||||||||||
As of December 31, 2013, our purchase obligations include existing purchase commitments for future minimum payments of $192,000 with a vendor for raw materials that will be manufactured and utilized on an as needed basis. During the years ended December 31, 2012 and 2013, we purchased $462,000 and $724,000, respectively, of materials from this vendor.
As of December 31, 2013, our purchase obligations include a purchase order with a vendor for cartridges that will be manufactured and delivered on an agreed upon schedule during 2014 for future payments of $1.5 million, exclusive of prepayments totaling $95,000. If we cancel any future shipment, we would be required to pay 50% of the scheduled invoice amount for that shipment.
As of December 31, 2013, we had a purchase order with a vendor for $693,000 in equipment to be received and paid for in 2014. Orders in process may be canceled only with vendor’s consent and upon payment of the vendor’s cancellation charges.
Our most significant clinical trial expenditures are to investigative sites and to contract research organizations. The agreements are cancellable by either party at any time upon written notice, but obligate us to reimburse the providers for any time or costs incurred through date or termination and do not have any cancellation penalties.
Legal Proceedings
We are not currently a party to any litigation, nor are we aware of any pending or threatened litigation against us that we believe would materially affect our business, operating results, financial condition or cash flows. Our industry is characterized by frequent claims and litigation, including claims regarding patent and other intellectual property rights, as well as for product liability. As a result, in the future, we may be involved in various legal proceedings from time to time.
F-21
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
6. Fair Value
The following fair value hierarchy table presents information about each major category of our financial assets and liabilities measured at fair value on a recurring basis (in thousands):
| Fair Value Measurement at December 31, 2012 | ||||||||||||||||
| Fair Value | Level 1 | Level 2 | Level 3 | |||||||||||||
| Assets: |
||||||||||||||||
| Cash equivalents: |
||||||||||||||||
| U.S. treasuries |
$ | 2,000 | $ | 2,000 | $ | — | $ | — | ||||||||
| Money market funds |
1,580 | 1,580 | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total cash equivalents |
$ | 3,580 | $ | 3,580 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Short-term investments: |
||||||||||||||||
| U.S. treasuries |
$ | 13,996 | $ | 13,996 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Fair Value Measurement at December 31, 2013 | ||||||||||||||||
| Fair Value | Level 1 | Level 2 | Level 3 | |||||||||||||
| Assets |
||||||||||||||||
| Cash equivalents: |
||||||||||||||||
| Money market funds |
$ | 37,158 | $ | 37,158 | $ | — | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Liabilities |
||||||||||||||||
| Future purchase rights |
$ | 2,600 | $ | — | $ | — | $ | 2,600 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
We report the change in fair value during each period as a nonoperating gain or loss. The following table summarizes the changes in Level 3 preferred stock warrant liabilities measured at fair value on a recurring basis for the year ended December 31, 2012 (in thousands):
| Fair Value of Preferred Stock Warrant Liabilities |
||||
| Balance at January 1, 2012 |
$ | 180 | ||
| Revaluation of warrants |
(180 | ) | ||
|
|
|
|||
| Balance at December 31, 2012 |
$ | — | ||
|
|
|
|||
The preferred stock warrants were converted to common stock warrants in February 2012 (see Note 2).
F-22
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
6. Fair Value (Continued)
The following table summarizes the changes in Level 3 future purchase rights liabilities measured at fair value on a recurring basis for the years ended December 31, 2012 and 2013 (in thousands):
| Fair Value of Future Purchase Rights Liabilities |
||||
| Balance at January 1, 2012 |
$ | — | ||
| Initial valuation of future purchase rights |
3,101 | |||
| Revaluation of future purchase rights |
(3,101 | ) | ||
|
|
|
|||
| Balance at December 31, 2012 |
— | |||
| Initial valuation of additional future purchase rights |
1,294 | |||
| Revaluation of future purchase rights |
1,306 | |||
|
|
|
|||
| Balance at December 31, 2013 |
$ | 2,600 | ||
|
|
|
|||
We valued the future purchase rights liabilities in the periods ended December 31, 2012 and 2013 using a binomial lattice option pricing model with the following assumptions:
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Common stock fair value |
$ | 1.17 | $ | 5.93 | ||||
| Preferred stock price |
$ | 8.00 | $ | 8.00 | ||||
| Volatility |
80.0 | % | 85.0 | % | ||||
| Risk-free interest rate |
0.36 | % | 0.38 | % | ||||
| Contractual life (years) |
3.08 | 2.08 | ||||||
| Number of nodes |
37 | 25 | ||||||
| Dividend yield |
0.0 | % | 0.0 | % | ||||
7. Redeemable Convertible Preferred Stock
Series A Redeemable Convertible Preferred Stock
In May 2004, we issued 688 shares of Series A preferred stock, or Series A, for proceeds of $2.3 million, net of issuance costs of $60,000. We also issued 298 shares of Series A in conjunction with the conversion of a $1.0 million loan and $14,000 of accrued interest.
In October 2005, we issued 877 shares of Series A in conjunction with the conversion of a $645,000 loan and $124,000 of accrued interest.
In February 2012, all of the Series A converted into 22,686 shares of common stock in conjunction with our junior preferred stock purchase agreement.
Series B Redeemable Convertible Preferred Stock
In December 2005, we completed the issuance of 5,994 shares of Series B preferred stock, or Series B, for proceeds of $8.6 million, net of issuance costs of $317,000, and including the conversion of a $1.9 million loan
F-23
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
and $71,000 of accrued interest. Each Series B investor received a warrant to purchase 1.152 additional shares of Series B preferred stock. In conjunction with the junior preferred stock financing in February 2012, all of the Series B and warrants for the purchase of Series B converted into 45,350 shares of common stock and warrants for the purchase of 1,771 shares of common stock, respectively.
Series C Redeemable Convertible Preferred Stock
In September 2007, we issued 18,800 shares of Series C preferred stock, or Series C, for proceeds of $27.9 million, net of issuance costs of $210,000 and including the conversion of a $5.2 million loan and $262,000 of accrued interest. Each Series C investor received a warrant to purchase one additional share of Series C preferred stock. In conjunction with the junior preferred stock financing in February 2012, all of the Series C and warrants for the purchase of Series C converted into 136,055 shares of common stock and warrants for the purchase of 9,922 shares of common stock, respectively.
Series D Redeemable Convertible Preferred Stock
During May and June 2011, we issued 69,462 shares of Series D preferred stock, or Series D, for proceeds of $6.0 million, net of issuance costs of $134,000, and for the conversion of a $16.2 million loan and $1.2 million of accrued interest. We issued a total of 65,807 warrants to Series D investors of which 47,773 were previously committed in connection with the issuance of a loan. We recognized $8.7 million as the fair value of the warrants to purchase 47,773 shares of Series D, which was recorded as interest expense upon the conversion of the loan to Series D. The remaining 18,034 warrants, for which the initial fair value was determined utilizing a Binomial Monte-Carlo Cliquet option pricing model, totaled $3.2 million and was recorded as a discount to Series D. The aggregate value of the warrants issued in the amount of $11.9 million was recorded as the fair value of the preferred stock warrant liabilities at the time of issuance. In conjunction with the junior preferred stock financing in February 2012, all of the Series D and warrants for the purchase of Series D converted into 253,979 shares of common stock and warrants for the purchase of 240,615 shares of common stock, respectively.
F-24
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
The following is a rollforward of our Series A, Series B, Series C, and Series D preferred stock activity (in thousands, except share amounts):
| Series A Redeemable and Convertible Preferred Stock |
Series B Redeemable and Convertible Preferred Stock |
Series C Redeemable and Convertible Preferred Stock |
Series D Redeemable and Convertible Preferred Stock |
|||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
| Balance at May 23, 2003 (inception) |
— | $ | — | — | $ | — | — | $ | — | — | $ | — | ||||||||||||||||||||
| Issuance of preferred stock |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2003 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Private placement—from February 2 to May 24, 2004 and conversion of loan |
986 | 2,899 | — | — | — | — | — | — | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 83 | — | — | — | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2004 |
986 | 2,982 | — | — | — | — | — | — | ||||||||||||||||||||||||
| Private placement—from October 24 to December 23, 2005 and conversion of loan |
— | — | 5,994 | 8,568 | — | — | — | — | ||||||||||||||||||||||||
| Conversion of loan |
877 | 2,981 | — | — | — | — | — | — | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 85 | — | 13 | — | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2005 |
1,863 | 6,048 | 5,994 | 8,581 | — | — | — | — | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 59 | — | 80 | — | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2006 |
1,863 | 6,107 | 5,994 | 8,661 | — | — | — | — | ||||||||||||||||||||||||
| Private placement—September 2007 and conversion of loan |
— | — | — | — | 18,800 | 27,915 | — | — | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 53 | — | 72 | — | 11 | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2007 |
1,863 | 6,160 | 5,994 | 8,733 | 18,800 | 27,926 | — | — | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 36 | — | 49 | — | 42 | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2008 |
1,863 | 6,196 | 5,994 | 8,782 | 18,800 | 27,968 | — | — | ||||||||||||||||||||||||
| Conversion of preferred stock to common stock |
(322 | ) | (1,095 | ) | (183 | ) | (273 | ) | (5,347 | ) | (8,000 | ) | — | — | ||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 54 | — | 53 | — | 75 | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2009 |
1,541 | 5,155 | 5,811 | 8,562 | 13,453 | 20,043 | — | — | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 39 | — | 63 | — | 40 | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2010 |
1,541 | 5,194 | 5,811 | 8,625 | 13,453 | 20,083 | — | — | ||||||||||||||||||||||||
| Series D Financing for cash and debt conversion |
— | — | — | — | — | — | 69,462 | 20,218 | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | 39 | — | 63 | — | 40 | — | 396 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2011 |
1,541 | 5,233 | 5,811 | 8,688 | 13,453 | 20,123 | 69,462 | 20,614 | ||||||||||||||||||||||||
| Accretion to redemption value of redeemable convertible preferred stock |
— | — | — | — | — | — | — | 57 | ||||||||||||||||||||||||
| Conversion of Series A-D preferred stock to common stock |
(1,541 | ) | (5,233 | ) | (5,811 | ) | (8,688 | ) | (13,453 | ) | (20,123 | ) | (69,462 | ) | (20,671 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2012 |
— | $ | — | — | $ | — | — | $ | — | — | $ | — | ||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
F-25
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
Issuance of Junior Convertible Preferred Stock
In February 2012, we entered into a securities purchase agreement for the sale of junior convertible preferred stock. In conjunction with the junior preferred stock financing, the previously issued Series A, Series B, Series C, and Series D preferred stock converted into shares of common stock at the following conversion ratios: Series A: 14.72155 for 1; Series B: 7.8041 for 1; Series C: 10.1141 for 1; and Series D: 3.65637 for 1. As a result, we reclassified the aggregate value of the Series A, Series B, Series C, and Series D preferred stock of $54.7 million to stockholders’ (deficit) equity.
In connection with the junior preferred stock financing, we entered into an Investors’ Rights Agreement, dated February 23, 2012 (the Junior Preferred IRA) with certain of the purchasers of junior preferred stock. The Junior Preferred IRA contains customary preemptive rights in favor of the stockholders party to such agreement, as well as certain restrictions on transfer, registration rights, market standoff provisions and various affirmative and negative covenants with respect to management of our company.
The junior preferred stock financing totaled $1.5 million of convertible, but not redeemable, preferred stock at approximately $0.43 a share, for an aggregate of 3,501,400 shares. During February and March 2012, we received proceeds of $1.3 million, net of issuance costs of $40,000.
Issuance of Senior Redeemable Convertible Preferred Stock
In September 2012, we entered into a senior preferred stock purchase agreement (as amended in December 2013, the Senior Preferred Purchase Agreement) that authorizes the multi-stage issuance of 14,765,632 shares of our senior redeemable convertible preferred stock for $8.00 per share. As of December 31, 2013, 10,212,007 shares have been issued under the Senior Preferred Purchase Agreement and 4,553,625 shares are subject to future stock purchase rights, as described below. Pursuant to the Senior Preferred Purchase Agreement, we granted the investors in the senior preferred stock financing the right, subject to the satisfaction of certain conditions, to purchase additional shares of senior preferred stock for a purchase price of $8.00 per share at multiple subsequent closings in accordance with a schedule provided in the Senior Preferred Purchase Agreement. If an investor does not participate in subsequent closings, such investor does not have the right to participate in future closings. The purchase price has been allocated to the future purchase rights and any related stock options at their estimated fair value, as described under “Future Purchase Rights” below.
In connection with the senior preferred stock financing, we have entered into a Fourth Amended and Restated Investors’ Rights Agreement, dated August 28, 2013 (the Senior Preferred IRA). The Senior Preferred IRA contains customary preemptive rights in favor of the stockholders party to such agreement, as well as customary registration rights and related provisions, including customary market standoff provisions.
From September to October 2012, we issued 2,606,250 shares of senior preferred stock for proceeds of $19.8 million, net of issuance costs of $240,000. We also issued 911,949 shares of senior preferred stock upon the conversion of a loan entered into during 2012 that included $7.2 million of principal and $146,000 of accrued interest.
In February 2013, we closed an additional senior preferred stock financing and issued 305,571 shares of senior preferred stock for proceeds of $2.3 million, net of issuance costs of $134,000 incurred by us for services rendered by a third-party professional services firm that is also utilized by certain of our investors.
F-26
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
In May 2013, we closed an additional senior preferred stock financing and issued 158,150 shares of senior preferred stock for proceeds of $1.2 million, net of issuance costs of $28,000.
In June 2013, we closed an additional senior preferred stock financing and issued 3,830,050 shares of senior preferred stock for proceeds of $30.6 million, net of issuance costs of $90,000.
In December 2013, we amended the terms of the Senior Preferred Stock Purchase Agreement to provide for a partial acceleration of existing future preferred stock financing closings under the Senior Preferred Stock Purchase Agreement and the sale of additional shares of senior preferred stock to certain of our existing investors and to certain of our directors and officers (the December 2013 Amendment). No rights to purchase additional shares of senior preferred stock were received by any existing or new investor, and no changes were made to any future preferred stock financing closing under the Senior Preferred Stock Purchase Agreement that was not accelerated. Pursuant to the Senior Preferred Stock Purchase Agreement, as amended by the December 2013 Amendment, we issued 2.4 million and 555,000 shares of senior preferred stock for proceeds of $19.1 million, net of issuance costs of $102,000, and $4.4 million in December 2013 and January 2014, respectively.
Future Purchase Rights
Pursuant to the Senior Preferred Purchase Agreement, we granted to the investors in the senior preferred stock financing the right, subject to the satisfaction of certain conditions, to purchase additional shares of senior preferred stock for a purchase price of $8.00 per share at multiple subsequent closings in accordance with a schedule provided in the Senior Preferred Purchase Agreement. These future purchase rights terminate automatically upon certain events, including the closing of this offering. Upon termination, the future purchase rights liabilities, as discussed below, would be reclassified to additional paid-in capital, a component of stockholders’ (deficit) equity.
These future purchase rights for additional shares of our senior preferred stock are legally detachable from the shares of the underlying senior preferred stock and, as a result, are considered freestanding instruments that are accounted for separately from such shares of senior preferred stock. As the future purchase rights are exercisable for shares of our redeemable convertible preferred stock, the future purchase rights are instruments that embody obligations to transfer assets and are classified as liabilities under the accounting guidance that distinguishes liabilities from equity.
The following table summarizes the number of shares subject to purchase under future purchase rights initially granted, the fair value per share of the senior preferred stock subject to the rights as initially granted, the initial grant date fair value recorded, the fair value of our common stock used in determining such value, and beneficial conversion amounts underlying the preferred stock that were recorded in connection with certain purchases of such preferred stock under the Senior Preferred Purchase Agreement and the related stock options granted to certain members of our board of directors (in thousands except share, rights, and per share and rights amounts):
| Issuance Date |
Number of Shares Subject to Future Purchase Rights Granted |
Fair Value per Share of Rights |
Grant Date Fair Value Recorded |
Common Stock Value Used in Determining Fair Value |
Beneficial Conversion on Underlying Preferred Stock/ Options |
|||||||||||||||
| September 25, 2012 |
3,750,000 | $ | 2.80 | $ | 3,101 | $ | 2.30 | — | ||||||||||||
| February 28, 2013 |
891,250 | $ | 2.83 | $ | 864 | $ | 6.85 | $ | 513 | |||||||||||
| April 30, 2013 |
461,271 | $ | 2.40 | $ | 379 | $ | 6.85 | $ | 192 | |||||||||||
| June 26, 2013 |
66,250 | $ | 0.76 | $ | 50 | $ | 6.82 | $ | 264 | |||||||||||
F-27
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
In conjunction with the issuance of senior preferred stock during the year ended December 31, 2013, we estimated the fair value of the future purchase rights for the shares issued to be $1.3 million. The future purchase rights were recorded at their estimated fair value, with the remaining amount of the proceeds allocated to the senior preferred stock, resulting in the senior preferred stock being recorded at an amount per share less than the fair value of our common stock at that time. Accordingly, we recorded an aggregate beneficial conversion amount underlying the senior preferred stock of $705,000, an amount equal to the number of shares of senior preferred stock sold on that date multiplied by the difference between the estimated fair value of the underlying common stock and the value allocated to the senior preferred stock on that date. The beneficial conversion amount was recorded as an offset to additional paid-in capital and is being amortized as a deemed dividend over the redemption period using an effective interest rate method. For the year ended December 31, 2013, we recognized a deemed dividend of $50,000.
In connection with the issuance of senior preferred stock in June 2013 discussed above, we also granted to certain members of our board of directors who participated in the senior preferred stock financings one common stock option for each share of preferred stock purchased through June 2013. An aggregate of 86,917 common stock options were granted to these board members, which were valued at $4.91 per share utilizing the BSM option pricing model. After allocation of the proceeds to the underlying future purchase rights as noted above, the remaining amount of the proceeds were allocated between the common stock options and the senior preferred stock acquired using the relative fair value method. As the allocated value per share of the senior preferred stock acquired was less than the fair value of our common stock on such date, we recorded a beneficial conversion associated with the options granted, on the underlying senior preferred stock of $264,000. This beneficial conversion was also recorded as an offset to additional paid-in capital and is being amortized as a deemed dividend over the redemption period using an effective interest rate method. For the year ended December 31, 2013, we recognized a deemed dividend of $14,000.
Certificate of Incorporation
The material terms of our restated certificate of incorporation are as follows:
Authorized Shares
The certificate of incorporation was amended authorizing the company to issue 54,250,000 shares of stock consisting of 29,250,000 shares of common stock with a par value of $0.0001 per share and 25,000,000 shares of preferred stock, of which 17,000,000 shares are designated as senior preferred stock and 3,501,401 shares are designated as junior preferred stock, both with a par value at $0.0001 per share. Together, the senior preferred stock and the junior preferred stock are collectively referred to as the Preferred Stock.
Dividends
Dividends on each share of senior preferred stock shall accrue on a daily basis, compounding annually, at the rate of 8.0% per annum of the sum of (i) original purchase price and (ii) cumulative unpaid dividends, whether or not declared. Junior preferred stock only receives dividends if and when declared by our board of directors. The dividends on the preferred stock do not convert to common stock upon the completion of a Qualified Public Offering or a sale of our company.
F-28
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
Liquidation Preference
In the event of any liquidation, dissolution or winding up, whether voluntary or involuntary, the senior preferred stock has a first liquidation preference equivalent to 150% of the original purchase price per share plus all cumulative unpaid dividends, whether or not declared. Once the senior preferred stock has been paid, junior preferred stock has a liquidation preference equal to the original purchase price plus all declared and unpaid dividends. If, upon any such liquidation event, our assets available to be distributed to the holders of the senior preferred stock are insufficient, payment shall be distributed pro rata.
Redemption
Holders may require us to redeem all, but not less than all, the Preferred Stock any time after September 2022. The redemption price is equal to the greater of (i) the liquidation value plus all cumulative unpaid dividends, whether or not declared thereon, and (ii) the fair value of the common stock issuable upon conversion of such share of the Preferred Stock and will occur in three equal installments over three years. Redemption requires a majority vote of the senior preferred stock investors. We must notify the junior preferred stock investors of the redemption within five days of receiving the redemption request. Junior preferred stock investors then have 20 days to redeem all (but not less than all) of the outstanding shares of junior preferred stock.
We are accreting the carrying amounts of the redeemable convertible preferred stock, redeemable anytime after September 2022, to the redemption amount using an effective interest rate method.
Conversion Rights
Each share of the Preferred Stock shall be convertible, at the option of the holder thereof, at any time and into common stock at a conversion price of $8.00 for each share of senior preferred stock and $0.4284 for each share of junior preferred stock. The conversion prices of the Preferred Stock are subject to anti-dilution protection. Each share of the senior preferred stock will automatically convert into shares of common stock at the then applicable conversion price immediately upon the earliest of (i) the closing of a qualified public offering (ii) a sale of our company, or (iii) the date specified by the holders of not less than two-thirds of the outstanding shares of senior preferred stock.
Voting Rights
The holder of each share of the Preferred Stock shall have the right to one vote for each share of common stock into which the Preferred Stock could then be converted (with any fractional share determined on an aggregate conversion basis being rounded to the nearest whole share, with one-half being rounded up to one). With respect to such vote, such holder shall have full voting rights and powers equal to the voting rights and powers of the holders of common stock. The holder of each share of the Preferred Stock is entitled to notice of any stockholders’ meeting in accordance with our bylaws, and is entitled to vote, together as a single class with holders of common stock with respect to any question upon which holders of common stock have the right to vote.
Subject to supermajority votes for some matters, matters shall be decided by the affirmative vote of our stockholders having a majority in voting power of the votes cast by the stockholders present or represented and voting on such matter, provided that the holders of our common stock are not allowed to vote on any amendment
F-29
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
7. Redeemable Convertible Preferred Stock (Continued)
to our certificate of incorporation that relates solely to the terms of one or more series of preferred stock if the holders of such affected series are entitled, either separately or together with the holders or one or more such series, to approve such amendment. The affirmative vote of the holders of at least 75% of the votes that our stockholders would be entitled to cast in any annual election of directors and, in some cases, the affirmative vote of a majority of minority shareholders entitled to vote in any annual election of directors, is required to amend or repeal our bylaws, amend or repeal certain provisions of our certificate of incorporation, approve certain transactions with certain affiliates, or approve of the sale or liquidation of the company. In addition, our amended and restated certificate of incorporation also provides that our directors may be removed only for cause by the affirmative vote of the holders of at least 75% of the combined voting power of all our stockholders entitled to vote on the election of directors, voting together as single class.
Warrants
We issued warrants to purchase redeemable convertible preferred stock in connection with financing activities or for consulting services. In connection with the junior preferred stock financing in February 2012, all warrants to purchase Series B, Series C, and Series D preferred stock converted to common stock warrants. The common stock warrants outstanding as of December 31, 2013, have a weighted-average exercise price of $95.21 and expire between February 2016 and September 2019. Our warrant activity is as follows:
| Number Outstanding |
Weighted- average Exercise Price |
|||||||
| Balance outstanding—January 1, 2012 |
252,308 | $ | 95.84 | |||||
| Expiration of warrants |
(1,662 | ) | $ | 191.69 | ||||
|
|
|
|||||||
| Balance outstanding—December 31, 2012 and 2013 |
250,646 | $ | 95.21 | |||||
|
|
|
|||||||
8. Common Stock
Stock Reserved for Future Issuance
Shares reserved for future issuance at December 31, 2013 are as follows:
| Number of Shares |
||||
| Conversion of the Preferred Stock |
13,713,407 | |||
| Exercise of common stock warrants |
250,646 | |||
| Common stock options outstanding |
3,098,573 | |||
| Common stock options available for future grant |
29,492 | |||
|
|
|
|||
| Total common shares reserved for future issuance |
17,092,118 | |||
|
|
|
|||
9. Stock Compensation Plans
Equity Incentive Plans
Our 2012 Stock Option Plan, or the 2012 Plan, as approved by our board of directors and stockholders, provides for the grant of stock options, restricted stock, restricted stock units, stock purchase rights, and
F-30
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
9. Stock Compensation Plans (Continued)
performance awards to employees, directors, and consultants. Option grants under the 2012 Plan generally have a ten-year term and vest over four years. The 2012 Plan also provides that options granted under the 2012 Plan are exercisable immediately, subject to a repurchase right that lapses as the option vests. As of December 31, 2013, under the 2012 Plan, the aggregate number of shares of common stock that may be issued under the 2012 Plan is 3,128,065, which is comprised of 3,098,573 options outstanding and 29,492 available for future grant. Subsequent to December 31, 2013, our board of directors approved an increase of 100,000 shares in the number of shares reserved for issuance under the 2012 Plan.
As of December 31, 2013, options for the purchase of 139,071 shares of our common stock have been exercised, of which 45,159 are unvested and subject to repurchase. Under the authoritative guidance, early exercise is not considered an exercise for accounting purposes and, therefore, any potential payment for unvested shares is recognized as a liability at the exercise price. As of December 31, 2013, we have not repurchased any shares related to these early exercises and our repurchase liability is $227,000.
Prior to the adoption of the 2012 Plan, we granted incentive awards under the 2004 Stock Option Plan. In February 2012 and in connection with the junior preferred stock financing, 27,230 outstanding options from the 2004 Stock Option Plan were canceled, which represented all options outstanding at the time of cancellation. In conjunction with the cancellation of options, we expensed previously unrecognized compensation cost of $27,000.
The following table summarizes stock option activity:
| Options | Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding as of January 1, 2012 |
27,230 | $ | 44.20 | |||||||||||||
| Granted |
2,338,649 | $ | 5.68 | |||||||||||||
| Forfeited or cancelled |
(29,565 | ) | $ | 40.74 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding as of December 31, 2012 |
2,336,314 | $ | 5.69 | 9.6 | $ | 529,060 | ||||||||||
| Granted |
971,392 | $ | 8.67 | |||||||||||||
| Exercised |
(139,071 | ) | $ | 2.61 | ||||||||||||
| Forfeited |
(70,062 | ) | $ | 8.14 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding as of December 31, 2013 |
3,098,573 | $ | 6.71 | 8.8 | $ | 3,379,947 | ||||||||||
|
|
|
|||||||||||||||
| Options vested as of December 31, 2013 |
920,684 | $ | 5.38 | 8.7 | $ | 1,784,873 | ||||||||||
|
|
|
|||||||||||||||
| Options vested and expected to vest as of December 31, 2013 |
3,087,209 | $ | 6.70 | 8.8 | $ | 3,373,152 | ||||||||||
|
|
|
|||||||||||||||
| Options exercisable as of December 31, 2013 |
3,098,573 | $ | 6.71 | 8.8 | $ | 3,379,947 | ||||||||||
|
|
|
|||||||||||||||
The aggregate intrinsic value of options is calculated as the difference between the exercise price of the options and the deemed fair value of our common stock for those shares that had exercise prices lower than the deemed fair value of our common stock. The number of options vested and expected to vest is calculated as the total number of options vested plus the number of unvested options remaining after applying our estimated forfeiture rate. The total fair value of options that vested during the years ended December 31, 2012 and 2013
F-31
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
9. Stock Compensation Plans (Continued)
was $102,000 and $835,000, respectively. The total fair value of options exercised during the year ended December 31, 2013 was $230,000. We have not capitalized or recognized an income tax benefit from the exercise of any stock options as we continue to record a full valuation allowance on our deferred tax assets.
Stock-based Compensation Expense
The weighted-average grant date fair value of stock options granted during the years ended December 31, 2012 and 2013 were $0.47, and $5.53, respectively. The following are the ranges of underlying assumptions used to determine the fair value of stock options granted to employees and nonemployees:
| Years Ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Risk-free interest rate |
0.2% - 1.0 | % | 0.1% - 1.1 | % | ||||
| Expected dividend yield |
0 | % | 0 | % | ||||
| Expected volatility |
100.0 | % | 90% -100.0 | % | ||||
| Expected term of options (years) |
1.0 - 6.0 | 0.5 - 6.0 | ||||||
December 31, 2012 Valuation Analysis
During the first quarter of 2013, we commenced an analysis of the enterprise value of our company and the fair value of our common stock as of December 31, 2012, which utilized an Option Pricing Model, or OPM. OPM is appropriate to use when the range of possible future outcomes is so difficult to predict that any such forecast would be highly speculative, or there is a substantially contemporaneous sale of stock to a third party. OPM treats common stock and convertible preferred stock as call options on the enterprise value, with exercise prices based on the liquidation preference of the convertible preferred stock. Therefore, the common stock has value only if the funds available for distribution to the stockholders exceed the value of the liquidation preference to be received by holders of our redeemable convertible preferred stock at the time of a liquidity event, such as a merger, sale or an initial public offering, or IPO, assuming the enterprise has funds available to make a liquidation preference meaningful and collectible by the stockholders. The common stock is modeled to be a call option with a claim on the enterprise at an exercise price equal to the remaining value immediately after the convertible preferred stock is liquidated. OPM uses the BSM option pricing model to price the call option.
The OPM considered the relative rights and preferences of the various securities, including the seniority of the liquidation preferences for preferred equity, and the potential for dilution caused by the conversion of the preferred equity. Additionally, our preferred stockholders have various rights that give them greater control and influence over future liquidity, financing and other decisions relating to our company than the holders of our common stock. The OPM applied a percentage of participation for each class of shares for each valuation interval based on BSM option pricing models and a 25.4% discount for lack of marketability was applied for the common stockholders. The resulting implied per share value of our common stock was $1.17 per share as of December 31, 2012, which was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in September and December 2012 at an exercise price of $8.00 per share.
F-32
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
9. Stock Compensation Plans (Continued)
2013 Valuation Analyses
Due to our management’s and board of directors’ decision to pursue an IPO, coupled with our belief that we could reasonably estimate the form and timing of potential liquidity events, we utilized a Probability Weighted Expected Return Method, or PWERM, for our 2013 valuations. Under this method, the implied fair value of our common stock is estimated based upon an analysis of future values assuming various outcomes. The value is based on the probability-weighted present value of expected future investment returns considering each of the possible outcomes available to us as well as the rights of each share class. The possible outcomes considered are based upon an analysis of future scenarios as described below:
| • | closing of an initial public offering; |
| • | sale to a strategic acquirer; |
| • | continuation as a private company with subsequent liquidation event; and |
| • | dissolution. |
Critical assumptions required to perform the PWERM include the following:
| • | Scenarios: Expected future events were identified. |
| • | Scenario probabilities: Estimates of the probability of occurrence of each event were identified. |
| • | Valuation: Expected future values under each scenario were estimated. |
| • | Timing: Expected timing to the event under each scenario were estimated. |
| • | Risk adjusted discount rates: Risk-adjusted discount rates were selected for each equity class based on the rights and preferences of each equity class and market data. |
| • | Discounts: Appropriate minority or marketability discounts, if any, required to estimate the per share value of the various equity classes were determined. |
In determining the implied fair value of our common stock in the IPO scenario, we assumed that the redeemable convertible preferred stock then outstanding would be converted into common stock. In allocating value to our common stock in the merger or sale scenario, we first allocated to our outstanding shares of redeemable convertible preferred stock the greater of the liquidation preference of the redeemable convertible preferred stock and the amount that would have been payable had all such shares of redeemable convertible preferred stock been converted to common stock.
There is inherent uncertainty in these estimates and, if we had made different assumptions, the fair value of the underlying common stock and amount of our stock-based compensation expense, net loss and net loss per share amounts would have differed.
March 31, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by December 31, 2013 |
25 | % | ||
| Sale by December 31, 2013 |
20 | % | ||
| Private company |
30 | % | ||
| Dissolution |
25 | % | ||
F-33
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
9. Stock Compensation Plans (Continued)
A 13% discount for lack of marketability was applied for common stockholders. The resulting implied fair value of $6.85 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in March 2013 at an exercise price of $8.00 per share.
June 30, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by November 15, 2013 |
35 | % | ||
| Sale by November 15, 2013 |
10 | % | ||
| Private company |
30 | % | ||
| Dissolution |
25 | % | ||
A 9% discount for lack of marketability was applied for common stockholders. The resulting implied fair value of $6.82 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in May, June, and July 2013 at an exercise price of $8.00 per share. The decrease in implied fair value of our common stock from March 31, 2013 was primarily due to dilution from the issuance of substantially more shares of our redeemable convertible senior preferred stock during the second quarter of 2013 that have superior rights to our common stock.
September 3, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by November 15, 2013 |
50 | % | ||
| Sale by November 15, 2013 |
10 | % | ||
| Private company |
25 | % | ||
| Dissolution |
15 | % | ||
A 7% discount for lack of marketability was applied for common stockholders. The resulting implied fair value of $9.94 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in September 2013 at an exercise price of $10.50 per share. The implied fair value of our common stock increased from June 30, 2013 primarily due to the increased likelihood of an IPO scenario as a result of progress made toward a public offering. In addition, we decreased our discount for lack of marketability reflecting a decrease in the expected time to liquidity.
September 30, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by November 15, 2013. |
50 | % | ||
| Sale by November 15, 2013 |
10 | % | ||
| Private company |
25 | % | ||
| Dissolution |
15 | % | ||
F-34
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
9. Stock Compensation Plans (Continued)
A 6% discount for lack of marketability was applied for common stockholders, which resulted in an implied fair value of $10.02 per share. This increase in implied fair value of our common stock from September 3, 2013 was associated with a slight decrease in our discount for lack of marketability due to a decrease in the expected time to liquidity. There were no changes to our probability-weighted scenarios as no significant changes occurred from our September 3, 2013 valuation analysis.
December 1, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by May 15, 2014 |
20 | % | ||
| Sale by September 30, 2015 |
10 | % | ||
| Private company |
50 | % | ||
| Dissolution |
20 | % | ||
A discount for lack of marketability was applied for common stockholders of 10%, 21% and 28% for the IPO, sale and private company scenarios, respectively. The resulting implied fair value of $6.77 per share was utilized to determine the BSM fair value for the purpose of calculating stock-based compensation expense related to the options granted in December 2013 at an exercise price of $8.00 per share. The decrease in fair value of our common stock from September 30, 2013 was primarily related to the increases in discount for lack of marketability due to an increase in the expected time to a liquidity event associated with the projected timing of an IPO or a sale of our company.
December 31, 2013 Valuation Analysis
Our analysis considered the following probability-weighted scenarios:
| Scenario |
Weight | |||
| IPO by May 15, 2014 |
20 | % | ||
| Sale by September 30, 2015 |
10 | % | ||
| Private company |
50 | % | ||
| Dissolution |
20 | % | ||
A discount for lack of marketability was applied for common stockholders of 9%, 20% and 28% for the IPO, sale and private company scenarios, respectively, which resulted in an implied fair value of $5.93 per share. The decrease in fair value of our common stock from December 1, 2013 was primarily related to dilution from the issuance of additional shares of our redeemable convertible senior preferred stock in December 2013, partially offset by a slight decrease in discount for lack of marketability for the IPO and sale scenarios reflecting a decrease in the expected time to liquidity.
F-35
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
9. Stock Compensation Plans (Continued)
Total noncash stock-based compensation expense for all stock awards recognized in our consolidated statements of operations is as follows (in thousands):
| Years Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| Research and development |
$ | 50 | $ | 411 | ||||
| General and administrative |
94 | 537 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 144 | $ | 948 | ||||
|
|
|
|
|
|||||
As of December 31, 2013, there was $4.5 million of total compensation cost related to unvested stock option awards not yet recognized, which is expected to be recognized over a remaining weighted-average vesting period of 2.9 years.
10. Income Taxes
Our net loss before income tax was subject to tax in the following jurisdictions for the following periods (in thousands):
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| United States |
$ | (6,321 | ) | $ | (32,524 | ) | ||
| Foreign |
(380 | ) | (194 | ) | ||||
|
|
|
|
|
|||||
| $ | (6,701 | ) | $ | (32,718 | ) | |||
|
|
|
|
|
|||||
Our rate reconciliation consists of the following:
| Year Ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| Federal statutory rate |
35.0% | 35.0% | ||||||
| State tax (net of federal benefit) |
5.8% | 5.3% | ||||||
| Warrants/purchase rights liabilities |
19.9% | (1.6% | ) | |||||
| Non-deductible interest expense and fees |
(1.4% | ) | — | |||||
| Research and development credit |
1.8% | 3.9% | ||||||
| Other permanent differences |
(0.8% | ) | (0.8% | ) | ||||
| Change in valuation allowance |
355.5% | (40.0% | ) | |||||
| Foreign net operating losses |
(0.6% | ) | (1.2% | ) | ||||
| 382 limited net operating losses and credits |
(415.2% | ) | (0.6% | ) | ||||
|
|
|
|
|
|||||
| Effective tax rate |
0.0% | 0.0% | ||||||
|
|
|
|
|
|||||
F-36
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
10. Income Taxes (Continued)
Significant components of our deferred tax assets are shown below. A valuation allowance has been established as realization of such deferred tax assets has not met the more likely-than-not threshold requirement. If our judgment changes and it is determined that we will be able to realize these deferred tax assets, the tax benefits relating to any reversal of the valuation allowance on deferred tax assets will be accounted for as a reduction to income tax expense.
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| (in thousands) | ||||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 3,521 | $ | 15,073 | ||||
| Research and development tax credit |
123 | 1,426 | ||||||
| Foreign net operating loss carryforwards |
813 | 504 | ||||||
| Other, net |
706 | 1,247 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
5,163 | 18,250 | ||||||
| Less valuation allowance |
(5,163 | ) | (18,250 | ) | ||||
|
|
|
|
|
|||||
| $ | — | $ | — | |||||
|
|
|
|
|
|||||
We have a history of incurring net operating losses each year since inception that is due to our history as a development stage company with no realized revenues from our planned principal operations. These cumulative operating losses provide significant negative evidence in the determination of whether or not we will be able to realize our deferred tax assets such as our net operating losses and other favorable temporary differences. As a result, we have maintained a full valuation allowance against the entire balance of deferred tax assets since the date of inception. The valuation allowance decreased by $23.8 million and increased by $13.1 million for the years ended December 31, 2012 and 2013, respectively.
As of December 31, 2013, we had available net operating loss, or NOL, carryforwards of approximately $37.3 million and $35.8 million for federal and state income tax purposes, respectively. The federal and state NOLs begin to expire in 2023 and 2014, respectively. As of December 31, 2013, we have federal and state research and development tax credits available for income tax purposes of approximately $1.0 million and $608,000, respectively. The federal research and development credits begin to expire in 2023 and the state research and development tax credits do not expire. These carryforwards are net of the Section 382 and 383 limitations discussed below.
As of December 31, 2013, we also have available NOLs from our Chinese subsidiary of approximately $2.0 million. The Chinese NOLs begin to expire in 2014.
Sections 382 and 383 of the Internal Revenue Code (the IRC) limit a company’s ability to utilize certain net operating losses and tax credit carryforwards in the event of a cumulative change in ownership in excess of 50%, as defined. We believe that a change in ownership, as defined in Section 382, occurred in February 2012. As a result, the deferred tax asset associated with our federal and state net operating loss carryforwards and federal and state research credits have been reduced based on the Section 382 limitations. The amount of the reduction in our deferred tax assets is based on the estimated amount of the NOL carryforwards and federal and state research credits we believe cannot be used based on the estimated amount of our Section 382 annual limitation. We have reduced our deferred tax assets by $27.7 million and have estimated that approximately $58.7 million and $59.6 million of our federal and state NOLs, respectively, cannot be used in future years as a result of this change in
F-37
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
10. Income Taxes (Continued)
ownership. Additionally, we have estimated that approximately $2.2 million and $1.6 million of our federal and state research credits, respectively, cannot be used in future years. Exact computations of limitations under Section 382 have not been completed, but the amounts reflect our best estimate of limitations and net operating losses and credits available in future years. We have not determined if other changes under Section 382 subsequent to the February 2012 estimated change date have occurred. If additional limitations against the utilization of net operating losses and credits exist, it could impact our future cash flows, but will not impact our 2013 consolidated financial statements, due to the existence of a full valuation allowance against our deferred tax assets.
The following table summarizes the activity related to our unrecognized tax benefits (in thousands):
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Balance at beginning of year |
$ | — | $ | — | ||||
| Additions based on tax positions related to the current year |
— | 337 | ||||||
| Additions for tax positions of prior years |
— | 70 | ||||||
|
|
|
|
|
|||||
| Balance at end of year |
$ | — | $ | 407 | ||||
|
|
|
|
|
|||||
We do not anticipate any significant changes in the amount of unrecognized tax benefits over the next twelve months. Due to the full valuation allowance we have on our net deferred tax asset balance, there are no unrecognized tax benefits that would impact the effective tax rate if recognized.
We are subject to U.S. federal, California and various other states and Chinese income taxes. We are no longer subject to U.S. federal or state income tax examination by tax authorities for tax returns filed for the years ended on or before December 31, 2008. However, to the extent allowed by law, the taxing authorities may have the right to examine the period from 2003 through 2013 where NOLs were generated and carried forward, and make adjustments to the amount of the NOL carryforward amount. We are not currently under examination by any federal or state jurisdictions.
11. Related Party Transactions
Investors
During the year ended December 31, 2012, we incurred an aggregate of $1.2 million of costs on behalf of certain of our stockholders, directors and officers and their respective affiliates for third-party professional services incurred in connection with the closing of our junior and senior preferred stock financings and convertible loans.
Directors
During the years ended December 31, 2012 and 2013, we paid an aggregate of $7,000 and $39,000, respectively, to a member of our board of directors for services rendered as chair of our Clinical Advisory Board.
One member of our board of directors was a partner and is no longer practicing law as of the end of 2013 with a firm that provides certain legal services to us. For the years ended December 31, 2012 and 2013, we incurred an aggregate of $73,000 and $100,000, respectively, in fees for these services. As of December 31, 2012, accounts payable included $6,000 related to these services.
F-38
Table of Contents
VITAL THERAPIES, INC.
(A Development Stage Company)
Notes to Consolidated Financial Statements (Continued)
11. Related Party Transactions (Continued)
Clinical Advisory Board
Certain members of our Clinical Advisory Board hold positions at medical institutions where we conduct business. During the year ended December 31, 2013, we incurred an aggregate of $216,000 in fees to these institutions, primarily for clinical trial costs. As of December 31, 2013, accounts payable included $5,000 due to these institutions.
12. Subsequent Events
For our financial statements as of December 31, 2013 and for the year then ended, we evaluated subsequent events through March 11, 2014, the date on which those financial statements were available to be issued.
In January 2014, we issued 555,000 shares of senior redeemable convertible preferred stock at $8.00 per share for proceeds of $4.4 million in connection with the December 2013 Amendment (see Note 7).
In January 2014, we completed a private placement to new investors for the sale of 1.5 million shares of our senior redeemable convertible preferred stock at a price of $8.00 per share for proceeds of $12.0 million.
In February 2014, we completed a pre-emptive rights offering, triggered by the private placement to new investors in January 2014, for 241,016 shares of our senior redeemable convertible preferred stock at a price of $8.00 per share for proceeds of $1.9 million.
In February 2014, options to purchase 257,987 shares of common stock originally granted in September 2013 were re-priced from an exercise price of $10.50 per share to an exercise price of $8.00 per share.
F-39
Table of Contents

Table of Contents
PART II
INFORMATION NOT REQUIRED IN THE PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution. |
Estimated expenses, other than underwriting discounts and commissions, payable by the Registrant in connection with the sale of the common stock being registered under this registration statement are as follows:
| SEC registration fee |
$ | 11,700 | ||
| FINRA filing fee |
14,200 | |||
| Listing fee |
25,000 | |||
| Printing and engraving expenses |
500,000 | |||
| Legal fees and expenses |
3,085,000 | |||
| Accounting fees and expenses |
960,000 | |||
| Blue Sky fees and expenses (including legal fees) |
15,000 | |||
| Transfer agent and registrar fees and expenses |
20,000 | |||
| Miscellaneous |
24,100 | |||
|
|
|
|||
| Total |
$ | 4,655,000 | ||
|
|
|
| Item 14. | Indemnification of Directors and Officers. |
On closing of this offering, the Registrant’s amended and restated certificate of incorporation will contain provisions that eliminate, to the maximum extent permitted by the General Corporation Law of the State of Delaware, the personal liability of the Registrant’s directors and executive officers for monetary damages for breach of their fiduciary duties as directors or officers. The Registrant’s amended and restated certificate of incorporation and amended and restated bylaws will provide that the Registrant must indemnify its directors and executive officers and may indemnify its employees and other agents to the fullest extent permitted by the General Corporation Law of the State of Delaware.
Sections 145 and 102(b)(7) of the General Corporation Law of the State of Delaware provide that a corporation may indemnify any person made a party to an action by reason of the fact that he or she was a director, executive officer, employee or agent of the corporation or is or was serving at the request of a corporation against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by him or her in connection with such action if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful, except that, in the case of an action by or in right of the corporation, no indemnification may generally be made in respect of any claim as to which such person is adjudged to be liable to the corporation.
The Registrant has entered into indemnification agreements with its directors and executive officers, in addition to the indemnification provided for in its amended and restated certificate of incorporation and amended and restated bylaws, and intends to enter into indemnification agreements with any new directors and executive officers in the future.
The Registrant has purchased and intends to maintain insurance on behalf of any person who is or was a director or officer of the Registrant against any loss arising from any claim asserted against him or her and incurred by him or her in any such capacity, subject to certain exclusions.
The Underwriting Agreement (Exhibit 1.1 hereto) provides for indemnification by the underwriters of the Registrant and its executive officers and directors, and by the Registrant of the underwriters, for certain liabilities, including liabilities arising under the Securities Act.
See also the undertakings set out in response to Item 17 herein.
II-1
Table of Contents
| Item 15. | Recent Sales of Unregistered Securities. |
Since January 1, 2010, the Registrant has issued and sold the following securities:
On May 19, 2011 the Registrant raised $17.5 million through conversion of outstanding Convertible Notes and accrued interest in a private placement of 51,453 shares of Series D Preferred Stock and Warrants to purchase Series D Preferred Stock at a purchase price of $340.00 per share for each share of Series D Preferred Stock purchased (with no additional consideration for any warrants issued with the Series D Preferred Stock purchased). Warrants to purchase 47,773 shares of Series D Preferred Stock at $340.00 per share were issued to the Convertible Notes holders. The securities issued in the private placement were sold exclusively to accredited investors. Such transactions were exempt from registration under Sections 4(2) and 4(6) of the Securities Act and under Rule 506 of Regulation D.
Between May 11 and June 7, 2011, the Registrant raised an additional $6.1 million in cash in a private placement of 18,008 shares of Series D Preferred Stock and warrants to purchase Series D Preferred Stock at a purchase price of $340.00 per share of Series D Preferred Stock purchased (with no additional consideration for any warrants issued with the Series D purchased). The securities issued in the private placement were sold exclusively to accredited investors. Such transactions were exempt from registration under Sections 4(2) and 4(6) of the Securities Act and under Rule 506 of Regulation D.
On or about February 2, 2012, after the Registrant’s Certificate of Incorporation was amended to set forth new conversion rates for each then existing series of preferred stock, the holders of a majority of the then outstanding shares of preferred stock consented in writing to the conversion of all then outstanding shares of preferred stock causing their automatic conversion to common stock. The shares of common stock issuable upon conversion were issued with for no additional consideration with the number of shares of common stock receivable for each share of preferred stock being 14.72155, 7.80410, 10.11410 and 3.65637, for each share of Series A Preferred Stock, Series B Preferred Stock, Series C Preferred Stock and Series D Preferred Stock, respectively. Subsequently, the Company effected a 1 for 340 reverse stock split. All holders of preferred stock who received the common stock upon conversion were accredited investors and any offering was done on a private placement basis. Such transactions were exempt under Sections 4(2) and 4(6) of the Securities Act and under Rule 506 of Regulation D.
During February and March 2012, the Registrant raised $1.5 million in a private placement of 3,501,400 shares of Junior Preferred Stock at a price $0.4284 per share. The securities issued in the private placement were sold exclusively to accredited investors. Such transactions were exempt from registration under Sections 4(2), 4(6) and/or 3(a)(9) of the Securities Act and under Rule 506 of Regulation D.
Between May 14, 2012 and October 29, 2012, the Registrant raised $28.1 million in private placements, consisting of the issuance and sale of 2,606,250 shares of Senior Preferred Stock at a price of $8.00 per share for aggregate proceeds of $20.9 million and the issuance and sale of $7.2 million in secured convertible loans, the outstanding principal amount of which (together with all accrued and unpaid interest) was subsequently converted into 911,949 shares of Senior Preferred Stock at $8.00 per share. The securities issued in the private placement were sold exclusively to accredited investors. Such transactions were exempt under Sections 4(2) and 4(6) of the Securities Act and under Rule 506 of Regulation D.
Between February and June 26, 2013, the Registrant raised an aggregate of $34.4 million in private placements in the issuance and sale of 4,293,771 shares of Senior Convertible Preferred Stock at a price of $8.00 per share. The securities issued in the private placements were sold exclusively to accredited investors. Such transactions were exempt under Sections 4(2) and 4(6) of the Securities Act and under Rule 506 of Regulation D.
Between December 2013 and February 2014, the Registrant raised an aggregate of $37.6 million in private
placements through the issuance and sale of 4,696,053 shares of Senior Convertible Preferred Stock at a price of $8.00 per share. The securities issued in the private placements were sold exclusively to accredited investors. Such transactions were exempt under Sections 4(2) and 4(6) of the Securities Act and under Rule 506 of Regulation D.
II-2
Table of Contents
Since January 1, 2010 to February 28, 2014, the Registrant granted stock options under the 2012 Plan to purchase an aggregate of 3,275,340 shares of common stock, net of cancellations, at a weighted-average exercise price of $6.56 per share, to certain employees, consultants, and directors. Such transactions were exempt from registration under Sections 4(2) of the Securities Act and/or Rule 701 of the Securities Act.
Since January 1, 2010 to February 28, 2014, the Registrant issued and sold to certain employees an aggregate of 139,071 shares of common stock upon the exercise of options under the 2012 Plan at a weighted-average exercise price of $2.61 per share. Such transactions were exempt from registration under Sections 4(2) of the Securities Act and/or Rule 701 of Securities Act.
There were no underwriters employed in connection with any of the transactions set forth above.
| Item 16. | Exhibits and Financial Statement Schedules. |
| (a) | Exhibits: |
| Exhibit Number |
Exhibit Title | |
| 1.1* | Form of Underwriting Agreement. | |
| 3.1^ | Amended and Restated Certificate of Incorporation of the Registrant, filed October 12, 2013. | |
| 3.1.1^ | Certificate of Amendment of Amended and Restated Certificate of Incorporation, filed December 26, 2013. | |
| 3.2^ | Form of Amended and Restated Certificate of Incorporation of the Registrant, to be effective upon closing of the offering. | |
| 3.3^ | Amended and Restated Bylaws of the Registrant, adopted September 25, 2012. | |
| 3.4^ | Form of Second Amended and Restated Bylaws of the Registrant, to be effective upon closing of the offering. | |
| 4.1^ | Specimen Common Stock Certificate of the Registrant. | |
| 4.2^ | Fourth Amended and Restated Investors’ Rights Agreement, dated August 28, 2013. | |
| 4.3^ | Investors’ Rights Agreement, dated February 23, 2012. | |
| 4.4^ | Amended and Restated Investors’ Rights Agreement, dated June 7, 2011. | |
| 5.1^ | Opinion of Wilson Sonsini Goodrich & Rosati, Professional Corporation. | |
| 10.1+^ | Form of Indemnification Agreement between the Registrant and its directors and officers. | |
| 10.2+^ | Employment Letter Agreement between the Registrant and Duane Nash, dated October 30, 2013. | |
| 10.3+^ | Employment Letter Agreement between the Registrant and Robert A. Ashley, dated October 30, 2013. | |
| 10.4+^ | Employment Letter Agreement between the Registrant and Terence E. Winters, dated October 31, 2013. | |
| 10.5+^ | Employment Letter Agreement between the Registrant and Michael V. Swanson, dated August 30, 2013. | |
| 10.6+ | Employment Letter Agreement between the Registrant and Andrew Henry, dated October 30, 2013. | |
| 10.7+^ | Employment Letter Agreement between the Registrant and Aron P. Stern, dated October 30, 2013. | |
| 10.8+ | Employment Letter Agreement between the Registrant and Andrea Loewen, dated October 30, 2013. | |
| 10.9+^ | Employment Letter Agreement between the Registrant and Richard Murawski, dated October 30, 2013. | |
| 10.10+^ | 2012 Stock Option Plan and form of agreements. | |
| 10.11+^ | 2014 Equity Incentive Plan and form of agreements. | |
| 10.12+^ | Executive Incentive Compensation Plan. | |
II-3
Table of Contents
| Exhibit Number |
Exhibit Title | |
| 10.13+^ | Form Change of Control and Severance Agreement. | |
| 10.14+^ | Non-Employee Director Compensation Policy. | |
| 10.15^ | Standard Industrial/Commercial Multi-Tenant Lease and Addendum between DermTech International and R.E. Hazard Contracting Company, dated April 5, 2001, as amended. | |
| 10.16^ | Standard Office Lease between Arden Realty Limited Partnership and the Registrant, dated May 7, 2013. | |
| 21.1^ | List of subsidiaries of the Registrant. | |
| 23.1 | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm. | |
| 23.2 | Consent of Hein & Associates LLP, Independent Registered Public Accounting Firm. | |
| 23.3^ | Consent of Wilson Sonsini Goodrich & Rosati, Professional Corporation (included in Exhibit 5.1). | |
| 24.1^ | Power of Attorney (included on the signature page of the registration statement on Form S-1 filed with the SEC on October 11, 2013). | |
| ^ | Previously filed. |
| + | Indicates a management contract or compensatory plan. |
| * | To be filed by amendment. |
(b) Financial Statement Schedules. All financial statement schedules are omitted because they are not applicable or the information is included in the Registrant’s consolidated financial statements or related notes.
| Item 17. | Undertakings. |
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreements, certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
The undersigned hereby undertakes that:
| (1) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (2) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-4
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of San Diego, State of California, on April 3, 2014.
| VITAL THERAPIES, INC. | ||
| By: |
/s/ Terence E. Winters, Ph.D. | |
| Terence E. Winters, Ph.D. | ||
| Co-Chairman and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities indicated below:
| Signature |
Title |
Date | ||
| /s/ Terence E. Winters, Ph.D. Terence E. Winters, Ph.D. |
Co-Chairman and Chief Executive Officer (Principal Executive Officer) | April 3, 2014 | ||
| /s/ Michael V. Swanson Michael V. Swanson |
Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | April 3, 2014 | ||
| * Muneer A. Satter |
Co-Chairman and Lead Director | April 3, 2014 | ||
| * Jean-Jacques Bienaimé |
Director | April 3, 2014 | ||
| * Philip M. Croxford |
Director | April 3, 2014 | ||
| * Douglas E. Godshall |
Director | April 3, 2014 | ||
| * Errol R. Halperin |
Director | April 3, 2014 | ||
| * J. Michael Millis, M.D. |
Director | April 3, 2014 | ||
| * Lowell E. Sears |
Director | April 3, 2014 | ||
| * Randolph C. Steer, M.D., Ph.D.
* Pursuant to power of attorney |
Director | April 3, 2014 | ||
| /s/ Terence E. Winters, Ph.D. Name: Terence E. Winters, Ph.D. Title: Attorney-in-Fact |
||||
II-5
Table of Contents
| Exhibit |
Exhibit Title | |
| 1.1* | Form of Underwriting Agreement. | |
| 3.1^ | Amended and Restated Certificate of Incorporation of the Registrant, filed October 12, 2013. | |
| 3.1.1^ | Certificate of Amendment of Amended and Restated Certificate of Incorporation, filed December 26, 2013. | |
| 3.2^ | Form of Amended and Restated Certificate of Incorporation of the Registrant, to be effective upon closing of the offering. | |
| 3.3^ | Amended and Restated Bylaws of the Registrant, adopted September 25, 2012. | |
| 3.4^ | Form of Second Amended and Restated Bylaws of the Registrant, to be effective upon closing of the offering. | |
| 4.1^ | Specimen Common Stock Certificate of the Registrant. | |
| 4.2^ | Fourth Amended and Restated Investors’ Rights Agreement, dated August 28, 2013. | |
| 4.3^ | Investors’ Rights Agreement, dated February 23, 2012. | |
| 4.4^ | Amended and Restated Investors’ Rights Agreement, dated June 7, 2011. | |
| 5.1^ | Opinion of Wilson Sonsini Goodrich & Rosati, Professional Corporation. | |
| 10.1+^ | Form of Indemnification Agreement between the Registrant and its directors and officers. | |
| 10.2+^ | Employment Letter Agreement between the Registrant and Duane Nash, dated October 30, 2013. | |
| 10.3+^ | Employment Letter Agreement between the Registrant and Robert A. Ashley, dated October 30, 2013. | |
| 10.4+^ | Employment Letter Agreement between the Registrant and Terence E. Winters, dated October 31, 2013. | |
| 10.5+^ | Employment Letter Agreement between the Registrant and Michael V. Swanson, dated August 30, 2013. | |
| 10.6+ | Employment Letter Agreement between the Registrant and Andrew Henry, dated October 30, 2013. | |
| 10.7+^ | Employment Letter Agreement between the Registrant and Aron P. Stern, dated October 30, 2013. | |
| 10.8+ | Employment Letter Agreement between the Registrant and Andrea Loewen, dated October 30, 2013. | |
| 10.9+^ | Employment Letter Agreement between the Registrant and Richard Murawski, dated October 30, 2013. | |
| 10.10+^ | 2012 Stock Option Plan and form of agreements. | |
| 10.11+^ | 2014 Equity Incentive Plan and form of agreements. | |
| 10.12+^ | Executive Incentive Compensation Plan. | |
| 10.13+^ | Form Change of Control and Severance Agreement. | |
| 10.14+^ | Non-Employee Director Compensation Policy. | |
| 10.15^ | Standard Industrial/Commercial Multi-Tenant Lease and Addendum between DermTech International and R.E. Hazard Contracting Company, dated April 5, 2001, as amended. | |
| 10.16^ | Standard Office Lease between Arden Realty Limited Partnership and the Registrant, dated May 7, 2013. | |
| 21.1^ | List of subsidiaries of the Registrant. | |
| 23.1 | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm. | |
| 23.2 | Consent of Hein & Associates LLP, Independent Registered Public Accounting Firm. | |
| 23.3^ | Consent of Wilson Sonsini Goodrich & Rosati, Professional Corporation (included in Exhibit 5.1). | |
| 24.1^ | Power of Attorney (included on the signature page of the registration statement on Form S-1 filed with the SEC on October 11, 2013). | |
| ^ | Previously filed. |
| + | Indicates a management contract or compensatory plan. |
| * | To be filed by amendment. |