Attached files
| file | filename |
|---|---|
| EX-23.1 - EX-23.1 - LDR HOLDING CORP | d698395dex231.htm |
Table of Contents
As filed with the Securities and Exchange Commission on April 2, 2014
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
LDR Holding Corporation
(Exact name of registrant as specified in its charter)
| Delaware |
3841 |
20-3933262 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
LDR Holding Corporation
13785 Research Boulevard, Suite 200, Austin, Texas 78750
(512) 344-3333
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Scott Way
General Counsel
LDR Holding Corporation
13785 Research Boulevard, Suite 200, Austin, Texas 78750
(512) 344-3333
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Carmelo M. Gordian Ted A. Gilman Andrews Kurth LLP 111 Congress, Suite 1700 Austin, TX 78701 (512) 320-9200 |
B. Shayne Kennedy Chase C. Leavitt Latham & Watkins LLP 650 Town Center Drive, 20th Floor Costa Mesa, CA 92626 (714) 540-1235 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (check one) ¨
| Large Accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | x (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
CALCULATION OF REGISTRATION FEE
|
| ||||||||
| Title of securities to be registered | Amount to be registered |
Proposed maximum offering price per share |
Proposed maximum aggregate offering price |
Amount of registration fee | ||||
| Primary Offering |
||||||||
| Common Stock, par value $0.001 per share |
(1) | (1) | $ 25,000,000(2) | $ 3,220(3) | ||||
| Secondary Offering |
||||||||
| Common Stock, par value $0.001 per share |
3,800,000(4) | $31.61(5) | $120,118,000(5) | $15,172(5) | ||||
| Total |
$18,692 | |||||||
|
| ||||||||
|
| ||||||||
| (1) | Omitted pursuant to Rule 457(o) under the Securities Act of 1933, as amended. |
| (2) | Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) under the Securities Act. |
| (3) | Calculated pursuant to Rule 457(o) based on an estimate of the proposed maximum aggregate offering price. |
| (4) | Includes shares of common stock that the underwriters have the option to purchase pursuant to their overallotment option. |
| (5) | Estimated solely for the purpose of calculating the registration fee in accordance with Rule 457(c) under the Securities Act of 1933, as amended, based on the average of the high and low prices of the registrant’s common stock as reported on the NASDAQ Global Select Market on March 27, 2014, which is within five business days of the filing of this registration statement. |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant files a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this prospectus is not complete and may be changed. Neither we nor the selling stockholders may sell these securities until the Securities and Exchange Commission declares our registration statement effective. This prospectus is not an offer to sell these securities and neither we nor the selling stockholders are soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to completion, dated April 2, 2014
Shares
LDR HOLDING CORPORATION
Common Stock

$ per share
| • LDR Holding Corporation is offering shares and selling stockholders are offering shares. We will not receive any proceeds from the sale of shares by selling stockholders |
• The last reported sale price our common stock on , 2014 was $ per share.
• Trading symbol: NASDAQ Global Select Market LDRH |
This investment involves risk. See ‘‘Risk Factors’’ beginning on page 12.
We are an “emerging growth company” as defined by the Jumpstart Our Business Startups Act of 2012 and, as such, we have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
| Per Share | Total | |||||||
| Public offering price |
$ | $ | ||||||
| Underwriting discount |
$ | $ | ||||||
| Proceeds, before expenses, to LDR Holding Corporation |
$ | $ | ||||||
| Proceeds to Selling Stockholders |
$ | $ | ||||||
The underwriters have a 30-day option to purchase up to additional shares of common stock from the selling stockholders to cover over-allotments, if any.
Neither the Securities and Exchange Commission nor any state securities commission has approved of anyone’s investment in these securities, or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Piper Jaffray | William Blair |
| Cowen and Company | RBC Capital Markets | |||
| JMP Securities | Stephens Inc. | |||
| Bryan, Garnier & Co. | ||||
The date of this prospectus is , 2014.
Table of Contents
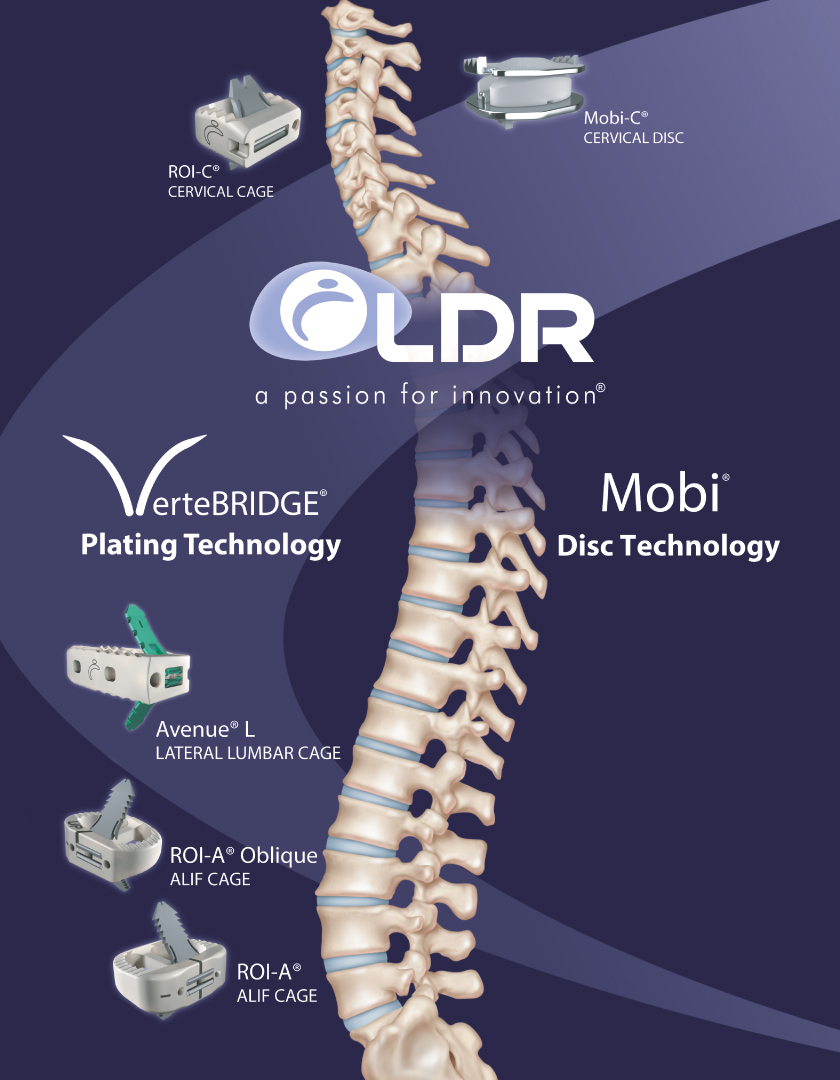
Table of Contents
| Page | ||||
| 1 | ||||
| 8 | ||||
| 10 | ||||
| 12 | ||||
| 55 | ||||
| 56 | ||||
| 57 | ||||
| 57 | ||||
| 58 | ||||
| 59 | ||||
| 60 | ||||
| 62 | ||||
| Management’s Discussion and Analysis of Financial Condition and Results of Operations |
64 | |||
| 79 | ||||
| 110 | ||||
| 120 | ||||
| 139 | ||||
| 145 | ||||
| 148 | ||||
| 152 | ||||
| Material United States Federal Tax Considerations for Non-United States Holders |
154 | |||
| 159 | ||||
| 167 | ||||
| 167 | ||||
| 167 | ||||
| F-1 | ||||
You should rely only on the information contained in this prospectus or any related free writing prospectus we may authorize to be delivered to you. Neither we, the selling stockholders nor the underwriters have authorized anyone to provide you with information different from, or in addition to, that contained in this prospectus and any related free writing prospectus. We, the selling stockholders and the underwriters take no responsibility for, and can provide no assurances as to the reliability of, any information that others may give you. This prospectus is not an offer to sell, nor is it seeking an offer to buy, these securities in any jurisdiction where the offer or sale is not permitted. The information contained in this prospectus is only accurate as of the date of this prospectus, regardless of the time or delivery of this prospectus and any sale of our common stock.
i
Table of Contents
The items in the following summary are described in more detail later in this prospectus. This summary provides an overview of selected information and does not contain all of the information you should consider. Before investing in our common stock, you should read the entire prospectus carefully, including the information set forth under the headings “Risk Factors” and the consolidated financial statements and related notes included in this prospectus.
Unless the context otherwise requires, references in this prospectus to “LDR Holding,” “LDR,” “we,” “our,” “us,” and the “company” refer to LDR Holding Corporation together with its subsidiaries.
Overview
We are a global medical device company focused on designing and commercializing novel and proprietary surgical technologies for the treatment of patients suffering from spine disorders. Our primary products are based on our VerteBRIDGE fusion and Mobi non-fusion platforms, both of which are designed for applications in the cervical and lumbar spine. We believe our VerteBRIDGE and Mobi platforms enable products that are less invasive, provide greater intra-operative flexibility, offer simplified surgical techniques and promote improved clinical outcomes for patients as compared to existing alternatives. In August 2013, we received approval from the U.S. Food and Drug Administration, or FDA, for the Mobi-C cervical disc replacement device, the first and only cervical disc replacement device to receive FDA approval to treat both one-level and two-level cervical disc disease. Industry sources expect that the cervical disc replacement market, which is currently relatively small, will be one of the fastest growing segments of the U.S. spine implant market.
For the years ended December 31, 2013, 2012 and 2011, our total revenues were $111.6 million, $90.9 million and $78.0 million, respectively, and we had net losses of $27.9 million, $9.7 million and $1.8 million in 2013, 2012 and 2011, respectively. We expect to continue to incur losses in the near term. For the past three calendar years, revenue from our VerteBRIDGE and Mobi platform products has grown at a compounded annual growth rate, or CAGR, of 30% and represented approximately 83% of our revenue in 2013. Through December 31, 2013, we had an accumulated deficit of $83.8 million.
Our highly differentiated VerteBRIDGE fusion platform targets the cervical and lumbar spine fusion markets. Our VerteBRIDGE products are designed around our proprietary plating technology that enables surgeons to implant VerteBRIDGE devices with direct visualization of the disc and to affix the devices to the vertebrae from inside the spinal disc space. We believe that VerteBRIDGE is the first platform of interbody devices with integrated fixation to market that may be implanted without screws, resulting in a zero-profile construct without hardware protruding outside of the vertebral body. We believe these differentiated features offer distinct advantages over other interbody devices, including simplified and less invasive surgical techniques for both cervical and lumbar fusion procedures. While these products only address a specific segment of the overall spine implant market, our VerteBRIDGE products have been used in more than 50,000 device implantations since the introduction of the VerteBRIDGE platform in 2007.
Our Mobi non-fusion platform is highlighted by Mobi-C, a cervical disc replacement device with a patented mobile bearing core that is designed to replicate the natural anatomical movement of the spine by facilitating independent bending and twisting similar to a healthy disc. Mobi-C is the first and only cervical disc replacement device to have received FDA approval for both one-level and two-level cervical disc replacement procedures. In addition, Mobi-C is the only cervical disc replacement device that has demonstrated overall clinical superiority in an FDA pivotal clinical trial as compared to two-level traditional fusion. Mobi-C is implanted without the use of keels or screws, allowing for what we believe
1
Table of Contents
is a simplified, less invasive and bone sparing surgical approach as compared to other existing cervical disc replacement devices. To our knowledge, only one potentially competitive two-level cervical disc replacement device, the Medtronic Prestige LP, is currently in development. We have no knowledge of, nor can we predict, when this device or any other potentially competitive device might be available for sale in the United States. We estimate that 30% of U.S. patients indicated for surgery with symptomatic cervical disc disease may be candidates for two-level cervical disc replacement procedures. As a result, we believe the FDA approval of Mobi-C, its overall clinical superiority as compared to two-level traditional fusion procedures and its innovative design features will allow us to penetrate, and drive growth in, the U.S. cervical disc replacement market, which we believe has the potential to grow relative to the total U.S. spine market. Currently, the market for FDA approved two-level devices is new with Mobi-C being the first-to-market cervical disc device approved for one-level and two-level cervical disc procedures. Accordingly, we need to further develop the market for such procedures, which will require investment in time and capital to advocate and support the launch of Mobi-C in the United States. To support this launch, we intend to leverage our extensive international experience with Mobi-C, including clinical and commercial knowledge accumulated from more than 17,000 Mobi-C implantations outside the United States. While the cervical disc market in the United States is currently relatively small, we believe that clinical data from second generation products such as Mobi-C will help drive the growth of the market.
We market and sell our products globally through a highly adaptable worldwide sales organization that allows us to continually expand our sales channels and rapidly penetrate new geographic markets. Our U.S. sales organization consists of sales managers that recruit, develop and lead direct sales representatives and a broad network of independent sales agencies. We estimate that non-captive independent sales agencies drive approximately 35% of total U.S. spine implant market revenue, and Mobi-C is the only cervical disc replacement device currently available to this large independent sales agency network. We believe this market position will facilitate the recruitment of high-quality direct sales representatives and independent sales agencies. We currently generate revenue in more than 25 countries globally, and revenue from outside of the United States represented approximately 26% of our total revenue in 2013. Our international sales organization includes sales managers and direct sales representatives located in various countries, including France, Germany, Spain, South Korea, China and Brazil. We also sell to distributors in certain international markets. A majority of our U.S. and international independent sales agencies and international distributors have agreed to offer one or more of our VerteBRIDGE and/or Mobi-C products on an exclusive basis. We expect to continue to make investments in our sales organization by broadening our relationships with independent sales agencies, expanding exclusivity commitments among these agencies and international distributors and increasing our number of direct sales representatives.
We were founded in 2000 in France, one of the world’s leading regions for spine surgery advancements. We believe our product development capabilities are differentiated by our extensive spine industry experience and our ability to leverage close collaboration with thought leaders in spine surgery. Our capabilities allow us to gain significant design feedback and clinical experience, which facilitates our ability to affix a CE mark to our products or gain regulatory clearances and market registrations globally. Our product development experience, incorporating input from both European and U.S. thought leaders in spine surgery, is a key component of our ability to design highly differentiated and clinically beneficial technologies. In addition, we have a robust product pipeline that we expect will enhance our product platforms and enable us to continue to penetrate the fastest growing segments of the global spine implant market.
Market Opportunity
Our VerteBRIDGE and Mobi platform products incorporate design advancements that provide us with competitive advantages in the estimated $9.6 billion global spine implant market. Industry sources
2
Table of Contents
estimate that the U.S. spine implant market represented approximately 73% of the global spine market in 2012, or $7.0 billion, and that it is expected to grow to approximately $9.0 billion by 2018. These sources estimate that the U.S. cervical disc replacement market, which is currently relatively small, will grow from $147 million in 2012 to $601 million by 2018, representing a CAGR of 26%, making it one of the fastest growing segments of the U.S. spine implant market. We believe several factors will influence growth in the spine implant market segments in which we compete, including patients’ and surgeons’ demand for technologies that allow for less invasive treatment options, the availability of clinical safety and efficacy data demonstrating improved patient outcomes provided by new technologies and an aging global population leading to continued growth in the number of spinal procedures performed.
Treatment Alternatives for Degenerative Conditions of the Spine
Treatment for degenerative conditions of the spine usually begins with medical management in the form of lifestyle changes, such as exercise, weight loss and anti-inflammatory and pain medication. While these treatments can be successful, clinical management of the degenerative process is challenging, and many patients with chronic symptoms that have failed these conservative treatments ultimately require spine surgery.
Fusions are the most common instrumented spine surgical procedure. Fusions are performed in the lumbar spine as well as the cervical spine. Fusion in the cervical spine is referred to as anterior cervical discectomy and fusion, or ACDF. Fusion procedures attempt to alleviate back and neck pain by removing diseased disc material and inserting bone grafts, interbody devices or fixation systems, including rods, screws and plates to restore disc height and permanently join, or fuse, two or more contiguous vertebrae. Traditional fusion procedures are considered highly invasive and are associated with several limitations, including high rates of subsequent surgery, loss of motion, disruption of bone and soft tissues, lengthy surgical procedure times and prolonged patient recovery times.
Recently, there has been a progression toward less invasive fusion procedures, which include stand-alone fusion devices. Stand-alone fusion devices are interbody devices that are placed into the disc space and allow surgeons to restore disc height and eliminate motion at diseased discs that do not require external fixation implants affixed to the vertebrae. Rather, the fixation is integrated into the implant device itself. More recently, non-fusion alternatives, such as cervical disc replacements, have been the subject of extensive clinical research. The goals of cervical disc replacement are to restore natural disc height, thereby decompressing the nerves causing pain, while also maintaining normal spinal motion at the affected vertebral segments.
Our Solutions
We believe that our VerteBRIDGE fusion platform addresses many of the limitations of traditional fusion and existing stand-alone implants by offering the following benefits:
| • | less invasive access; |
| • | simplified surgical procedure; |
| • | zero-profile design; and |
| • | integrated, screwless, self-locking plating system with extensive implant selection. |
We believe that our Mobi-C cervical disc replacement device addresses many of the limitations of traditional ACDF procedures and offers the following benefits:
| • | overall clinical superiority as compared to two-level ACDF; |
| • | preservation of motion; |
3
Table of Contents
| • | reduced rate of adjacent level disc degeneration; |
| • | lower rates of subsequent surgery; and |
| • | simplified, less invasive surgical procedure. |
In addition to addressing many of the limitations associated with ACDF, we believe that our Mobi-C cervical disc replacement device also addresses many of the limitations associated with existing cervical disc replacement devices and offers the following benefits:
| • | first-to-market in the United States with FDA approval for both a one- and two-level cervical disc replacement; |
| • | simplified, less invasive and bone sparing surgical approach; |
| • | advanced mobile bearing technology; and |
| • | clinical data demonstrating overall clinical superiority as compared to two-level ACDF. |
Although we believe our VerteBRIDGE fusion platform and Mobi-C cervical disc device provide numerous clinical benefits, not all cervical spine patients may be suitable candidates.
Our Competitive Strengths
We focus on providing novel and proprietary technologies for both fusion and non-fusion applications in the cervical and lumbar spine. Our founders and executive management team collectively have over 100 years of experience in developing and commercializing innovative spine surgery products. We believe that this focus and experience, combined with the following competitive strengths, will allow us to continue to grow our revenue and rapidly increase our presence in the fastest growing segments of the spine implant market:
| • | first-to-market with an FDA approved one-level and two-level cervical disc replacement device; |
| • | PMA approval process creates a barrier to entry to the cervical disc replacement market; and |
| • | highly differentiated technology platforms. |
While we believe that we have designed our VerteBRIDGE and Mobi platform products with characteristics and attributes that provide them with competitive advantages over currently existing alternatives, spine surgeons may not adopt our devices for a variety of reasons, including lack of experience with us or our products, existing relationships with competitors and distributors that sell our competitors’ products and the time commitment required for training to be able to use our products.
Our Strategy
Our goal is to strengthen our position as a global innovator of spine technologies and become a leader in the spine implant market. To achieve our goal, we are pursuing the following strategies:
| • | invest in our U.S. infrastructure to capitalize on the FDA approval of Mobi-C; |
| • | promote the differentiated features of our VerteBRIDGE fusion platform to further penetrate the large global spine fusion market; |
| • | leverage our culture of innovation to expand our product portfolio; and |
| • | invest in our international infrastructure to broaden our global market presence. |
4
Table of Contents
Our Products
Our VerteBRIDGE fusion and Mobi non-fusion platform products for both cervical and lumbar applications are characterized by patented technologies and innovative design features that we believe offer distinct advantages compared to other available products. We refer to these products in our financial reporting as our exclusive technology products. We also offer fusion technologies that are not significantly differentiated from competitive devices from a feature or clinical benefit perspective but that augment our VerteBRIDGE and Mobi technologies and allow us to offer our customers comprehensive surgical solutions. We refer to these products in our financial reporting as our traditional fusion products. In 2013, revenue from our exclusive technology products represented approximately 83% of our total revenue, and revenue from our traditional fusion products represented approximately 17% of our total revenue.
Our VerteBRIDGE platform is designed to provide fusion without screws or external plates, thereby providing a zero-profile construct. We believe that the integrated titanium plates that comprise the integrated fixation solution in our VerteBRIDGE fusion products are distinct in their ability to be implanted directly in the plane of the surgical exposure, with a minimally invasive surgical technique and provide screwless fixation intra-discally through the interbody device. VerteBRIDGE plating increases segmental stability, and its multiple sizing options are designed to provide significant intra-operative flexibility. The products in our VerteBRIDGE platform are composed of poly-ether-ether-ketone, or PEEK, a high performance polymer. Some surgeons may choose to use implants composed of alternative implant materials, such as allograft bone or titanium. VerteBRIDGE is applicable for one- or two-level cervical and lumbar fusion with or without supplemental fixation. Our VerteBRIDGE platform products consist of ROI-C, ROI-A, ROI-A Oblique and Avenue L.
Our Mobi non-fusion platform utilizes an ultra-high molecular weight polyethylene core and cobalt chromium alloy superior and inferior endplates. The Mobi platform can be used in cervical and lumbar procedures, although the highlight of our Mobi platform is our Mobi-C cervical disc replacement device. Mobi-C endplates incorporate short, lateral, inclined teeth that provide initial stability and fixation. The endplates are titanium plasma sprayed and coated with hydroxyapatite, an essential ingredient of normal bone, that helps promote bone ongrowth and long-term fixation. The mobile bearing core is designed to move within the prosthesis as the neck bends and twists, thereby simulating the movement of a healthy disc. The mobile core feature is also designed to minimize stresses in the implant-to-bone interface. The surgical technique to implant Mobi-C avoids the need to cut into the vertebral bone above and below the implant or to perform significant endplate preparation. We believe that Mobi-C is further differentiated by its ability to be easily adjusted, removed or repositioned intra-operatively. Mobi-C is indicated in the United States for the treatment of symptomatic cervical disc disease with radiculopathy or myelopathy at one or two contiguous levels.
We also have a broad portfolio of traditional fusion products. Our traditional fusion products include Easyspine, MC+, ROI, ROI-T, SpineTune and C-Plate. Unlike the products in our VerteBRIDGE platform or Mobi-C non-fusion platform, which offer highly differentiated features and benefits not found in competitive devices, our traditional fusion products are those devices that we offer for sale, but which cannot be significantly differentiated from competitive devices from a feature or clinical benefit perspective. Some industry experts may describe such traditional fusion products as spine commodity products. Examples include pedicle screw systems, anterior cervical plates and interbody cages absent integrated fixation.
Risks Affecting Our Business
Our business is subject to a number of risks, including risks that may prevent us from achieving our business objectives or may adversely affect our business, financial condition, results of operations, cash
5
Table of Contents
flows and prospects, that you should consider before making a decision to invest in our common stock. These risks are discussed more fully in the “Risk Factors” section of this prospectus, beginning on page 12.
These risks include, but are not limited to, the following:
| • | we have incurred losses in the past and may not be able to achieve or sustain profitability in the future; |
| • | our VerteBRIDGE and Mobi platform products represent a highly concentrated product offering that may limit our ability to grow our revenue and compete effectively against other companies, and our business would be adversely affected in the event we are unable to sell one or more of our products; |
| • | to be commercially successful, we must convince spine surgeons that our spine technologies are attractive alternatives to our competitors’ products; |
| • | we only recently received approval from the FDA for our Mobi-C cervical disc replacement device and have limited experience selling it in the United States, and even if we are successful launching Mobi-C in the U.S. market, its commercial success will depend on surgeon acceptance and third-party payor coverage and reimbursement; |
| • | if reimbursement from third-party payors for our products significantly declines, surgeons, hospitals and other healthcare providers may be reluctant to use our products and our sales may decline; |
| • | if we are unable to maintain and expand our network of direct sales representatives, independent sales agencies and distributors, we may not be able to generate anticipated sales; |
| • | we operate in a very competitive business environment and if we are unable to compete successfully against our existing or potential competitors, our sales and operating results may be negatively affected and we may not grow; |
| • | the safety and efficacy of our products is not yet supported by long-term clinical data, which could limit sales, and our products might therefore prove to be less safe or effective than initially thought; |
| • | if we or our sales representatives fail to comply with U.S. federal and state fraud and abuse laws, we could be subject to civil and criminal penalties, which could adversely impact our reputation and business operations; and |
| • | if we are unable to protect our intellectual property rights or if we are accused of infringing on the intellectual property rights of others, our competitive position could be harmed or we could be required to incur significant expenses to enforce or defend our rights. |
Corporate Information
LDR Holding Corporation was incorporated in 2006 as LDR Holding Corporation in the State of Delaware. Our principal executive office is located at 13785 Research Boulevard, Suite 200, Austin, Texas 78750 and our telephone number is (512) 344-3333. Our website address is http://www.ldrholding.com. We completed our initial public offering in October 2013, and our common stock is listed on The NASDAQ Global Select Market under the symbol “LDRH.”
We currently have a subsidiary, LDR Médical S.A.S., a French société par actions simplifiée, which we refer to as Médical, whose capital stock is currently held by LDR Holding. Certain of our employees
6
Table of Contents
hold warrants to purchase Class A Stock of Médical, which, unless noted otherwise, are included in the disclosures regarding outstanding options under our equity plans throughout this prospectus. Pursuant to the terms of the second amended and restated put-call agreement, as amended, between LDR Holding, Médical and Médical’s warrant holders, upon the exercise of such warrants, the shares of Class A Stock of Médical issued upon exercise of such warrants shall automatically be exchanged for 5.80087 shares of our common stock.
The “LDR Holding” name, the “LDR” name, the “Mobi,” “Mobi-C,” “Mobidisc,” “Avenue,” “C-Plate,” “VerteBRIDGE,” “ROI,” “Spinetune,” “Easyspine,” “MC+,” “ROI-A,” “ROI-C” and “ROI-T” names, and the phrase “a passion for innovation” and related images, logos and symbols appearing in this prospectus are our properties, trademarks and service marks. Other marks appearing in this prospectus are the property of their respective owners.
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart our Business Startups Act of 2012, or the JOBS Act. An emerging growth company may take advantage of specified reduced reporting requirements and is relieved of certain other significant requirements that are otherwise generally applicable to public companies. As an emerging growth company,
| • | we may present only two years of audited financial statements and only two years of related Management’s Discussion & Analysis of Financial Condition and Results of Operations; |
| • | we are exempt from the requirement to obtain an attestation and report from our auditors on the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002; |
| • | we are permitted to provide less extensive disclosure about our executive compensation arrangements; |
| • | we are not required to give our stockholders non-binding advisory votes on executive compensation or golden parachute arrangements; and |
| • | we have elected to use an extended transition period for complying with new or revised accounting standards. |
We may take advantage of these provisions until the last day of our fiscal year following the fifth anniversary of the date of the first sale of our common equity securities pursuant to an effective registration statement under the Securities Act of 1933, as amended, or the Securities Act, which such fifth anniversary will occur in 2018. However, if certain events occur prior to the end of such five-year period, including if we become a “large accelerated filer,” our annual gross revenues exceed $1.0 billion or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to the end of such five-year period.
7
Table of Contents
| Shares of common stock offered by us |
shares. |
| Shares of common stock offered by the Selling Stockholders |
shares. |
| Shares of our common stock outstanding after this offering |
shares. |
| Option to purchase additional shares |
The Selling Stockholders have granted the underwriters a 30-day option to purchase up to additional shares of our common stock to cover over-allotments, if any. |
| Use of proceeds |
We intend to use the net proceeds of this offering to (i) continue expanding our sales and marketing efforts in the U.S. and internationally, including our continued launch of Mobi-C in the U.S. market, and (ii) make continued investments in research and development. In addition, we may also use a portion of our net proceeds to acquire and invest in complementary products, technologies or businesses; however, we currently have no agreements or commitments to complete any such transaction and are not involved in negotiations to do so. We intend to use any remaining net proceeds from this offering for working capital general corporate purposes. We will not receive any of the proceeds from the sale of shares of common stock by the selling stockholders. See “Use of Proceeds.” |
| NASDAQ Global Select Market symbol |
LDRH |
| Risk Factors |
Investment in our common stock involves substantial risks. You should read this prospectus carefully, including the section entitled “Risk Factors” and the financial statements and the related notes to those statements included in this prospectus, before investing in our common stock. |
The number of shares of common stock outstanding after this offering is based on 24,076,667 shares of our common stock outstanding as of December 31, 2013, and excludes:
| • | 1,572,582 shares of common stock issuable upon exercise of options outstanding at December 31, 2013, at a weighted-average exercise price of $6.17 per share; |
| • | 1,816,623 shares of common stock reserved for future issuance under our 2013 Equity Incentive Plan, including 963,066 shares of common stock that were automatically added to our 2013 Equity Incentive Plan as of January 1, 2014; and |
| • | 351,877 shares of common stock reserved for future issuance under our Amended and Restated 2013 Employee Stock Purchase Plan, including 240,766 shares of common stock that were automatically added to our Amended and Restated 2013 Employee Stock Purchase Plan as of January 1, 2014. |
8
Table of Contents
Except as otherwise noted, all information in this prospectus:
| • | assumes no exercise of the underwriters’ over-allotment option to purchase additional shares; and |
| • | assumes no exercise of the outstanding options described above. |
9
Table of Contents
SUMMARY CONSOLIDATED FINANCIAL INFORMATION
The following tables summarize our consolidated financial and operating data for the periods indicated. The summary consolidated statement of comprehensive loss data for the years ended December 31, 2013, 2012 and 2011 have been derived from our audited consolidated financial statements included elsewhere in this prospectus. Our historical results are not necessarily indicative of the results that may be expected in the future.
| Year Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands, except per share data) | ||||||||||||
| Consolidated Statement of Comprehensive Loss Data: |
||||||||||||
| Revenue |
$ | 111,594 | $ | 90,918 | $ | 77,975 | ||||||
| Cost of goods sold |
17,947 | 14,770 | 12,564 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
93,647 | 76,148 | 65,411 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
9,380 | 10,751 | 9,040 | |||||||||
| Sales and marketing |
67,676 | 51,846 | 42,270 | |||||||||
| General and administrative |
18,915 | 14,825 | 11,121 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
95,971 | 77,422 | 62,431 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating income (loss) |
(2,324 | ) | (1,274 | ) | 2,980 | |||||||
| Other income (expenses), net |
(23,940 | ) | (7,248 | ) | (3,480 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes |
(26,264 | ) | (8,522 | ) | (500 | ) | ||||||
| Income tax expense |
(1,671 | ) | (1,179 | ) | (1,328 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (27,935 | ) | $ | (9,701 | ) | $ | (1,828 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per common share: |
||||||||||||
| Basic and diluted |
$ | (3.09 | ) | $ | (2.10 | ) | $ | (0.41 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average number of shares outstanding: |
||||||||||||
| Basic and diluted |
9,040,567 | 4,615,352 | 4,490,481 | |||||||||
|
|
|
|
|
|
|
|||||||
| Year Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
| Other Financial Data: |
||||||||||||
| Depreciation and amortization |
$ | 4,024 | $ | 3,133 | $ | 2,387 | ||||||
| Adjusted EBITDA(1) |
2,969 | 2,154 | 5,551 | |||||||||
10
Table of Contents
| As of December 31, 2013 |
||||
| (in thousands) | ||||
| Consolidated Balance Sheet Data: |
||||
| Cash and cash equivalents |
$ | 56,678 | ||
| Working capital |
55,376 | |||
| Total assets |
127,993 | |||
| Line of credit, net of discounts |
18,162 | |||
| Long-term debt, net of discounts and current portion |
2,758 | |||
| Total liabilities |
50,316 | |||
| Total stockholders’ equity |
77,677 | |||
| (1) | We define Adjusted EBITDA as operating income (loss) plus depreciation and amortization and stock-based compensation expense. We present Adjusted EBITDA because we believe it is a useful indicator of our operating performance. Our management uses Adjusted EBITDA principally as a measure of our operating performance and believes that Adjusted EBITDA is useful to our investors because it is frequently used by securities analysts, investors and other interested parties in their evaluation of the operating performance of companies in industries similar to ours. Our management also uses Adjusted EBITDA for planning purposes, including the preparation of our annual operating budget and financial projections. |
| Adjusted EBITDA should not be considered in isolation or as a substitute for a measure of our liquidity or operating performance prepared in accordance with U.S. generally accepted accounting principles, or GAAP, and is not indicative of net income (loss) from operations as determined under GAAP. Adjusted EBITDA and other non-GAAP financial measures have limitations that should be considered before using these measures to evaluate our liquidity or financial performance. Adjusted EBITDA does not include certain expenses that may be necessary to review our operating results and liquidity requirements. Our definition and calculation of Adjusted EBITDA may differ from that or other companies. |
| The following table presents a reconciliation of operating income (loss) to Adjusted EBITDA for the periods presented: |
| Year Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
| Operating income (loss), as reported |
$ | (2,324 | ) | $ | (1,274 | ) | $ | 2,980 | ||||
| Add back: |
||||||||||||
| Depreciation and amortization |
4,024 | 3,133 | 2,387 | |||||||||
| Stock-based compensation |
1,269 | 295 | 184 | |||||||||
|
|
|
|
|
|
|
|||||||
| Adjusted EBITDA |
$ | 2,969 | $ | 2,154 | $ | 5,551 | ||||||
|
|
|
|
|
|
|
|||||||
11
Table of Contents
Investing in our common stock involves a high degree of risk. You should carefully consider the following risks and other information contained in this prospectus before you decide whether to buy our common stock. If any of the events contemplated by the following discussion of risks should occur, our business, results of operations and financial condition could suffer significantly. As a result, the market price of our common stock could decline, and you may lose all or part of your investment in our common stock.
Risks Related to Our Business
We have incurred losses in the past and may not be able to achieve or sustain profitability in the future.
We have incurred significant losses in most fiscal years since inception. We incurred net losses of $1.8 million, $9.7 million and $27.9 million in 2011, 2012 and 2013, respectively. As a result of ongoing losses, we had an accumulated deficit of $83.8 million at December 31, 2013. We expect to continue to incur significant product development, clinical and regulatory, sales and marketing and other expenses. Additionally, we expect that our general and administrative expense will increase due to the additional operational and reporting costs associated with being a public company. We will need to generate significant additional revenue to achieve and maintain profitability, and even if we achieve profitability, we cannot be sure that we will remain profitable for any substantial period of time. Our failure to achieve or maintain profitability could negatively impact the value of our common stock.
Our VerteBRIDGE and Mobi platform products represent highly concentrated product offerings that may limit our ability to grow our revenue and compete effectively against other companies, and our business would be adversely affected in the event we are unable to sell one or more of our products.
We offer novel and proprietary surgical technologies for use in both the fusion and non-fusion spine implant markets. Our VerteBRIDGE fusion platform and Mobi non-fusion platform products are designed for both cervical and lumbar applications. Our VerteBRIDGE and Mobi platform products address only a portion of the types of products used in spine implant surgeries. We do not offer surgeons a full range of products to meet all of their needs in spine implant surgeries and product clearances vary by country. For example, our product offering does not include a portfolio of osteobiologic solutions, minimally invasive posterior instrumentation or posterior cervical instrumentation. Our competitors’ products are often used in the same surgery as our products, and this may limit our ability to compete effectively against our competitors, limiting our revenue growth.
In addition, a substantial amount of our revenue is generated by just a few products. For example, in 2013, 83% of our revenue came from our VerteBRIDGE and Mobi platform products, most of which came from products incorporating our VerteBRIDGE plating technology. These categories of products are dominated by ROI-A and Avenue L, in the lumbar product portfolio, and ROI-C, in the cervical product portfolio. In the event we are unable to market and sell these devices or any future device that represents a substantial amount of our revenue, our results of operations and financial condition will be materially adversely affected.
To be commercially successful, we must convince spine surgeons that our VerteBRIDGE and Mobi platform products are attractive alternatives to our competitors’ products.
Spine surgeons play a significant role in determining the course of treatment and, ultimately, the type of product that will be used to treat a patient, so we rely on effectively marketing to them. Acceptance of our products depends on educating spine surgeons as to the distinctive characteristics, perceived clinical benefits, safety and cost-effectiveness of our products as compared to our competitors’ products and on training spine surgeons in the proper application of our products. If we are not successful in convincing spine surgeons of the merits of our products or educating them on the use of our products, they may not
12
Table of Contents
use our products and we will be unable to increase our sales and sustain growth or reach profitability. Our exclusive technology products represented approximately 74%, 80% and 83% of our sales for the years ended December 31, 2011, 2012 and 2013, respectively. As a result, continued market acceptance of those products is critical to our continued success. If the volume of sales of these products declines, our business, financial position and results of operations could be materially and adversely affected.
Furthermore, we believe spine surgeons will not widely adopt our products unless they determine, based on experience, clinical data and published peer-reviewed journal articles, that our products and the techniques to implant them provide benefits or are attractive alternatives to our competitors’ products. Surgeons may be hesitant to change their medical treatment practices for the following reasons, among others:
| • | lack of experience with our products; |
| • | existing relationships with competitors and distributors that sell their products; |
| • | lack or perceived lack of evidence supporting additional patient benefits; |
| • | perceived liability risks generally associated with the use of new products and procedures; |
| • | less favorable coverage and reimbursement amount compared to other products and techniques; and |
| • | the time commitment that may be required for training. |
In addition, we believe recommendations and support of our products by influential spine surgeons are essential for market acceptance and adoption. If we do not receive support from such surgeons or long-term data does not show the benefits of using our products, surgeons may not use our products. In such circumstances, we may not achieve expected sales and may be unable to achieve profitability.
We only recently received approval from the U.S. Food and Drug Administration for our Mobi-C device and have limited experience selling it in the United States.
Our future growth is significantly dependent on the success of Mobi-C, our cervical disc replacement device, in the U.S. market. While we have been selling Mobi-C internationally since 2004, we did not obtain approval for Mobi-C from the U.S. Food and Drug Administration, or FDA, until August 2013 and have only recently begun the commercial launch of the product in the United States. As a result, we have limited history selling Mobi-C in the United States upon which you can evaluate its prospects. We cannot assure you that our launch of Mobi-C in the U.S. market will be successful, and our future growth may be limited.
Even if we are successful with Mobi-C’s launch in the U.S. market, its commercial success depends on surgeon acceptance and third-party payor coverage and reimbursement.
The current standard of care for the treatment of cervical disc disease is anterior cervical discectomy and fusion, or ACDF. The market for cervical disc replacements in the United States is still developing. While we believe that this market will grow in the near future as a result of the introduction of a new generation of advanced cervical disc replacements and broader coverage by commercial payors, there can be no assurance that surgeons will widely adopt cervical disc replacement as an alternative to ACDF, and even if they do, that they will elect to use Mobi-C and not a product, or combination of products, offered by one of our competitors.
The success of Mobi-C depends significantly on the availability and level of government and third-party payor coverage and reimbursement amount for procedures using our Mobi-C device. Many payors, including Medicare contractors, state Medicaid programs and private insurers may not reimburse for use of cervical disc replacement devices, including our Mobi-C device. While we believe there is a trend for
13
Table of Contents
broader coverage of cervical disc replacement devices generally in the United States, we cannot assure you that this coverage will continue to improve or that payors will not cease reimbursing for procedures using cervical disc replacement devices altogether. Additionally, even where coverage for procedures using cervical disc replacement devices exists, it is typically limited to treatment of cervical disc disease in one vertebral level, or one-level disease. Furthermore, surgeons are reimbursed at a significantly lower amount for performing a procedure using a cervical disc replacement device than traditional ACDF. In addition to its approval for treatment of one-level disease, Mobi-C is the first cervical disc replacement device approved to treat cervical disc disease in two adjacent vertebral levels, or two-level disease. As a result, we will need to work with payors to obtain coverage for the use of Mobi-C to treat two-level cervical disc disease. In February 2014, the American Medical Association Current Procedural Terminology (CPT) Editorial Panel approved a new CPT code as an add-on code to report the performance of total disc arthroplasty at an additional level, effective January 1, 2015. Even with the new code, however, payors may nonetheless conclude that Mobi-C for the treatment of two-level disease is experimental or investigational, may defer coverage pending additional long-term data or may deny coverage altogether based on a combination of these or other factors. Furthermore, even if government and third-party payors cover procedures using Mobi-C for the treatment of two-level cervical disc disease, it is possible that these payors may also cover procedures using our competitors’ cervical disc replacement devices when used to treat two-level cervical disc disease, even without our competitors’ products having FDA approval for such treatment. If the trend towards coverage and reimbursement amount of procedures involving cervical disc replacement devices does not continue, slows or stops altogether, our ability to market and sell Mobi-C would be significantly limited and our business and operations would suffer. Additionally, if we establish coverage and reimbursement amount for procedures using Mobi-C for treatment of two-level cervical disc disease, and our competitors’ products receive the same treatment without FDA approval for such treatment, our competitive advantage in the United States would be limited, which would significantly limit our ability to grow our business.
If coverage and reimbursement amount from third-party payors for our products significantly decline, surgeons, hospitals and other healthcare providers may be reluctant to use our products and our sales may decline.
In the United States, surgeons, hospitals and other healthcare providers who purchase medical devices like our products generally rely on third-party payors, principally Medicare, Medicaid and private health insurance plans, to pay for all or a portion of the costs and fees associated with the spinal surgery and the products utilized in the procedure, including the cost of our products. Our customers are generally reimbursed by governmental payors, such as Medicare, in a single bundled payment for all items and services provided to the patient, regardless of whether our products are used in the procedure. Our customers’ access to adequate coverage and reimbursement for the procedures performed using our products by government and third-party payors is central to the acceptance of our current and future products. We may be unable to sell our products on a profitable basis if government and third-party payors deny coverage or reduce their current levels of reimbursement. To contain costs of new technologies, governmental healthcare programs and third-party payors are increasingly scrutinizing new and even existing treatments by requiring extensive evidence of favorable clinical outcomes and cost effectiveness. Surgeons, hospitals and other healthcare providers may not purchase our products if they do not receive satisfactory reimbursement from these third-party payors for the cost of the procedures using our products. Payors continue to review their coverage policies carefully for existing and new therapies and can, without notice, deny coverage for treatments that include the use of our products. If we are not successful in reversing existing non-coverage policies, if other third-party payors issue similar policies or if our customers are not able to be reimbursed at cost-effective levels, this could have a material adverse effect on our business and operations.
For example, in August 2013, Aetna Inc., a large private insurance provider, issued a policy bulletin that classified PEEK interbody cervical cages such as ROI-C as experimental and investigational. Such a
14
Table of Contents
change may impact whether Aetna will continue to cover procedures utilizing such cages, and it may encourage other private insurance providers and government payors to evaluate their own coverage of PEEK interbody cervical cages.
In addition, some healthcare providers in the United States have adopted or are considering a managed care system in which providers contract to provide comprehensive healthcare for a fixed cost per person. Healthcare providers may attempt to control costs by authorizing fewer surgical procedures or by requiring the use of the least expensive implant available. Additionally, as a result of reform of the U.S. healthcare system, changes in reimbursement policies or healthcare cost containment initiatives may limit or restrict coverage and reimbursement for our products and cause our revenue to decline.
Outside of the United States, reimbursement systems vary significantly by country. Many foreign markets have government-managed healthcare systems that govern reimbursement for spine implants and procedures. Additionally, some foreign reimbursement systems provide for limited payments in a given period and therefore result in extended payment periods. If adequate levels of reimbursement from third-party payors outside of the United States are not obtained, international sales of our products may decline.
New products may impact the growth in sales of our existing products.
As an innovative company, we plan to continue to introduce new products that may be indicated for uses similar to our existing products. Growth in sales of our existing products may be negatively impacted due to the introduction of such new products. For example, while we have limited experience selling Mobi-C and Avenue-L in the United States, growth in sales of ROI-C and ROI-A may be impacted by Mobi-C and Avenue-L, respectively.
If we are unable to maintain and expand our network of direct sales representatives, independent sales agencies and international distributors, we may not be able to generate anticipated sales.
We first began operations in 2000 and have operated in the United States since 2005. We have limited experience marketing and selling our Mobi-C cervical disc replacement device, for which we received FDA approval in August 2013, and we have limited experience marketing that device in the United States. As of December 31, 2013, our sales organization consists of regional sales managers that lead direct sales representatives and over 190 independent sales agencies that employ over 600 sales representatives. As of December 31, 2013, our U.S. sales organization includes 40 sales managers. For the year ending December 31, 2013, the vast majority of our U.S. sales were from independent sales agencies. Outside of the United States, we sell through direct sales representatives and distributors that are often managed by our direct sales managers located in our foreign offices. Our international operations consists of over 160 employees and 55 distributors, which together had sales in over 27 countries in 2013. Our operating results are directly dependent upon the sales and marketing efforts of not only our employees, but also our independent sales agencies and distributors. We expect our direct sales representatives, independent sales agencies and distributors to develop long-lasting relationships with the surgeons they serve. If our direct sales representatives, independent sales agencies or distributors fail to adequately promote, market and sell our products, our sales could significantly decrease.
We face significant challenges and risks in managing our geographically dispersed distribution network and retaining the individuals who make up that network. If any of our direct sales representatives were to leave us, or if any of our independent sales agencies or distributors were to cease to do business with us, our sales could be adversely affected. Some of our independent sales agencies and distributors account for a significant portion of our sales volume, and if any such independent sales agencies or distributors were to cease to distribute our products, our sales could be adversely affected. In such a situation, we may need to seek alternative independent sales agencies or distributors or increase our reliance on our direct sales representatives, which we may be unable to do in a timely and efficient manner, if at all.
15
Table of Contents
While a majority of our independent sales agencies and distributors have contractual exclusivity, that contractual exclusivity is often limited to one or more of our products. Where some level of exclusivity exists, these independent sales agencies and distributors are permitted to offer a competitor’s products that do not compete directly with our products. Our competitors may require that these sales agents and distributors cease doing business with us. In addition, we may not be able to rely on our independent sales agencies or distributors to distribute new products that we introduce that compete with products of our competitors that they also represent. If a direct sales representative, independent sales agency or distributor were to depart and be retained exclusively by one of our competitors, we may be unable to prevent them from helping competitors solicit business from our existing customers, which could further adversely affect our sales. Because of the intense competition for their services, we may be unable to recruit or retain additional qualified independent sales agencies or distributors or to hire additional direct sales representatives to work with us. We may not be able to enter into agreements with them on favorable or commercially reasonable terms, if at all. Failure to hire or retain qualified direct sales representatives or independent sales agencies or distributors would prevent us from expanding our business and generating sales.
As we launch new products and increase our marketing efforts with respect to existing products, we will need to expand the reach of our marketing and sales networks. Our future success will depend largely on our ability to continue to hire, train, retain and motivate skilled sales managers, direct sales representatives and independent sales agencies and distributors with significant technical knowledge in various areas, such as spinal care practices, spine injuries and disease and spinal health. New hires require training and take time to achieve full productivity. If we fail to train new hires adequately, or if we experience high turnover in our sales force in the future, we cannot be certain that new hires will become as productive as may be necessary to maintain or increase our sales.
If we are unable to expand our sales and marketing capabilities domestically and internationally, we may not be able to effectively commercialize our products, which would adversely affect our business, results of operations and financial condition.
We operate in a very competitive business environment and if we are unable to compete successfully against our existing or potential competitors, our sales and operating results may be negatively affected and we may not grow.
The spine industry is intensely competitive, subject to rapid change and highly sensitive to the introduction of new products or other market activities of industry participants. Our ability to compete successfully will depend on our ability to develop future products that reach the market in a timely manner, are well adopted by surgeon customers and receive adequate coverage and reimbursement from third-party payors. Because of the size of the potential market, we anticipate that companies will dedicate significant resources to developing products competitive to ours.
We have numerous competitors in the spine market, which include Medtronic Spine and Biologics, DePuy Synthes Spine (a division of Johnson & Johnson), Globus Medical, NuVasive and Stryker, which together represent a significant portion of the spine market. We also compete with many smaller spine market participants such as Alphatec Spine, Biomet Spine, Integra, Orthofix International and Zimmer. At any time, these or other industry participants may develop alternative treatments, products or procedures for the treatment of spine disorders that compete directly or indirectly with our products. They may also develop and patent processes or products earlier than we can or obtain regulatory clearance, approvals or CE Certificates of Conformity for competing products more rapidly than we can, which could impair our ability to develop and commercialize similar processes or products. If alternative treatments are, or are perceived to be, superior to our spine surgery products, sales of our products could be negatively affected and our results of operations could suffer.
16
Table of Contents
Many of our larger competitors are either publicly traded or divisions or subsidiaries of publicly traded companies. These competitors enjoy several competitive advantages over us, including:
| • | greater financial, human and other resources for product research and development, sales and marketing and litigation; |
| • | significantly greater name recognition; |
| • | established relationships with spine surgeons, hospitals and other healthcare providers; |
| • | large and established sales and marketing and distribution networks; |
| • | products supported by long-term clinical data; |
| • | greater experience in obtaining and maintaining regulatory clearances, approvals or CE Certificates of Conformity for products and product enhancements; |
| • | more expansive portfolios of intellectual property rights; and |
| • | broader product portfolios affording them greater ability to cross-sell their products or to incentivize hospitals or surgeons to use their products. |
The spine industry is becoming increasingly crowded with new participants. Many of these new competitors specialize in a specific product or focus on a particular market segment, making it more difficult for us to increase our overall market position. The frequent introduction by competitors of products that are, or claim to be, superior to our products, or that are alternatives to our existing or planned products may also create market confusion that may make it difficult to differentiate the benefits of our products over competing products. In addition, the entry of multiple new products and competitors may lead some of our competitors to employ pricing strategies that could adversely affect the pricing of our products and pricing in the spine market generally.
As a result, without the timely introduction of new products and enhancements, our products may become obsolete over time. If we are unable to develop innovative new products, maintain competitive pricing and offer products that spine surgeons perceive to be as reliable as those of our competitors, our sales or margins could decrease, thereby harming our business.
Our reliance on third-party suppliers, including limited source and single source suppliers, for our implants and instruments could harm our ability to meet demand for our products in a timely and cost effective manner.
We rely on third-party suppliers, almost all of which are located in Europe, to manufacture and supply our implants and instruments. We currently rely on a number of limited or single source suppliers, such as Greatbatch, which supplies Mobi-C cervical disc replacement devices for the U.S. market, In’Tech Medical, which supplies a majority of our surgical instrumentation, and CF Plastiques, which is one of our interbody device suppliers. We generally do not have long-term contracts with our suppliers, and, as a result, our suppliers generally are not required to provide us with any guaranteed minimum production levels. As a result, there can be no assurances that we will be able to obtain sufficient quantities of key components or products in the future, which could have a material adverse effect on our business.
For us to be successful, our suppliers must be able to provide us with products and components in substantial quantities, in compliance with regulatory requirements, in accordance with agreed upon specifications, at acceptable costs and on a timely basis. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured products ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility that products will not be delivered on a timely basis, the possibility of increases in pricing for our products, the possibility of breach of the manufacturing agreement by the third party and the possibility of termination or non-renewal of the agreement by the third party.
17
Table of Contents
In addition, our reliance on third-party suppliers involves a number of risks, including, among other things:
| • | suppliers may fail to comply with regulatory requirements or make errors in manufacturing components that could negatively affect the efficacy or safety of our products or cause delays in shipments of our products; |
| • | we or our suppliers may not be able to respond to unanticipated changes in customer orders, and if orders do not match forecasts, we or our suppliers may have excess or inadequate inventory of materials and components; |
| • | we may be subject to price fluctuations due to a lack of long-term supply arrangements for key components; |
| • | we may experience delays in delivery by our suppliers due to changes in demand from us or their other customers; |
| • | we or our suppliers may lose access to critical services and components, resulting in an interruption in the manufacture, assembly and shipment of our systems; |
| • | fluctuations in demand for products that our suppliers manufacture for others may affect their ability or willingness to deliver components to us in a timely manner; |
| • | we may be required to obtain regulatory approvals related to any change in our supply chain; |
| • | our suppliers may wish to discontinue supplying components or services to us for risk management reasons; |
| • | we may not be able to find new or alternative components or reconfigure our system and manufacturing processes in a timely manner if the necessary components become unavailable; and |
| • | our suppliers may encounter financial or other hardships unrelated to our demand for components, which could inhibit their ability to fulfill our orders and meet our requirements. |
If any of these risks materialize, it could significantly increase our costs and impact our ability to meet demand for our products and could have a material adverse effect on our business. If we are unable to satisfy commercial demand for our products in a timely manner, our ability to generate revenue would be impaired, market acceptance of our products could be adversely affected and customers may instead purchase or use our competitors’ products. In addition, we could be forced to secure new or alternative components through a replacement supplier. Securing a replacement supplier could be difficult, especially for complex components that are manufactured in accordance with our custom specifications.
There are a limited number of suppliers and third-party manufacturers that operate under the FDA’s current Good Manufacturing Practices, maintain certifications from the International Organization for Standardization that are recognized as harmonized standards in the EEA, and that have the necessary expertise and capacity to manufacture our products. As a result, it may be difficult for us to locate manufacturers for our anticipated future needs, and our anticipated growth could strain the ability of our current suppliers to deliver products, materials and components to us. If we are unable to arrange for third-party manufacturing of our products, or to do so on commercially reasonable terms, we may not be able to complete development of, market and sell our new products.
The introduction of new or alternative components may require design changes to our system that are subject to FDA and other regulatory clearances, approvals or new CE Certificates of Conformity. We may also be required to assess the new manufacturer’s compliance with all applicable regulations and
18
Table of Contents
guidelines, which could further impede our ability to manufacture our products in a timely manner. As a result, we could incur increased production costs, experience delays in deliveries of our products, suffer damage to our reputation and experience an adverse effect on our business and financial results.
We may in the future elect to manufacture certain new products developed or certain existing products without the assistance of third parties. However, in order to make that election, we will need to invest substantial additional funds and recruit qualified personnel in order to operate our development manufacturing facility on a commercial basis. There can be no assurance that we will successfully manufacture our own products and if we are not able to make or obtain adequate supplies of our products, it will be more difficult for us to launch new products and compete effectively.
Pricing pressure from hospital customers and our competitors may impact our ability to sell our products at prices necessary to support our current business strategies.
The spine market has attracted numerous new companies and technologies, and encouraged more established companies to intensify competitive pricing pressure. As a result of this increased competition, we believe there will be increased pricing pressure in the future. Because the hospital and other healthcare provider customers that purchase our products typically bill various third-party payors to cover all or a portion of the costs and fees associated with the procedures in which our products are used, including the cost of the purchase of our products, changes in the amount such payors are willing to reimburse our customers for procedures using our products could create pricing pressure for us. If competitive forces drive down the prices we are able to charge for our products, our profit margins will shrink, which will adversely affect our ability to invest in and grow our business.
Much of the pricing competition relates to traditional fusion products, such as pedicle screws, cervical plates and traditional cages. We may not be successful in convincing third-party payors of the superiority of our innovative products as compared to traditional products, and reimbursement for these products may be significantly impacted. Additionally, as more competitors introduce innovative fusion products, we may face additional pricing pressure for our products that will impact our future results.
Moreover, many hospital customers, through the contracting process, limit the number of spine manufacturers that may sell to their institution. As we have a limited number of products to sell, hospitals may choose to contract with our competitors who have a broader range of products that may be used in a wider variety of spine procedures, regardless of the differentiated attributes of our products or local surgeon preference for our products. In addition, our competitors may actively position their broader product portfolios against us during the hospital contracting process. Limitations on the number of hospitals to which we can sell may significantly restrict our ability to grow.
We obtain some of our products through private-label distribution agreements that subject us to minimum performance requirements and other criteria. Our failure to satisfy those criteria could cause us to lose those rights of distribution.
We have entered into private-label distribution agreements with manufacturers of Spinetune and C-Plate. These manufacturers brand their products according to our specifications, and we may have exclusive rights in certain fields of use and territories to sell these products subject to minimum purchase or other performance criteria. Though these agreements do not individually or in the aggregate represent a material portion of our business, if we do not meet these performance criteria, or if we fail to renew these agreements, we may lose exclusivity in a field of use or territory or cease to have any rights to these products, which could have an adverse effect on our sales and our business. Furthermore, some of these manufacturers are smaller, undercapitalized companies that may not have sufficient resources to continue operations or to continue to supply us sufficient product without additional access to capital.
19
Table of Contents
If our private label manufacturers fail to provide us with sufficient supply of their products, or if their supply fails to meet appropriate quality requirements, our business could suffer.
Our private-label manufacturers are sole source suppliers of the products we purchase from them. We may not be able to locate or establish additional or replacement manufacturers of these products in a timely manner. Moreover, these private-label manufacturers typically own the intellectual property associated with their products, and even if we could find a replacement manufacturer for the product, we may not have sufficient rights to enable the replacement party to manufacture the product. While we have entered into agreements with our private-label manufacturers to provide us sufficient quantities of products, we cannot assure you that they will do so, or that any products they do provide us will not contain defects in quality. Our private-label manufacturing agreements have expiration terms and the manufacturers may not choose to renew them. The agreements also include provisions allowing for termination under certain circumstances such as either party’s uncured material breach, either party’s insolvency or failure to make minimum purchases.
We also rely on these private-label manufacturers to comply with the regulations and requirements of the FDA, the competent authorities or notified bodies of the EEA countries, or foreign regulatory authorities and their failure to comply with strictly enforced regulatory requirements could expose us to regulatory action including warning letters, product recalls, termination of distribution, product seizures or civil and criminal penalties. Any quality control problems that we experience with respect to products manufactured by our private-label manufacturers, any inability by us to provide our customers with sufficient supply of products or any investigations or enforcement actions by the FDA, the competent authorities or notified bodies of the EEA countries or other foreign regulatory authorities could adversely affect our reputation or commercialization of our products and adversely and materially affect our business and operating results.
If we do not successfully implement our business strategy, our business and results of operations will be adversely affected.
We formed our business strategy based on assumptions about the spine market that might prove wrong. We believe that various demographics and industry-specific trends, including the aging of the general population, increasingly active lifestyles, our products, and increasing acceptance of cervical disc replacements, will help drive growth in the spine market and our business, but these demographics and trends are uncertain. Actual demand for our products could differ materially from projected demand if our assumptions regarding these factors prove to be incorrect or do not materialize, or if alternative treatments to those offered by our products gain widespread acceptance.
We may not be able to successfully implement our business strategy. To implement our business strategy, we need to, among other things, establish our Mobi-C cervical disc replacement device as a standard of care for cervical disc replacement, leverage our VerteBRIDGE fusion platform to further penetrate the global spine fusion market, expand the size and productivity of our U.S. and international sales organization and expand our technology portfolio. Our strategy of focusing exclusively on the spine market may limit our ability to grow. In addition, we are seeking to increase our sales and, in order to do so, will need to commercialize additional products and expand the number of our direct sales representatives, independent sales agencies and international distributors in new and existing territories, all of which could result in our becoming subject to additional or different foreign and domestic regulatory requirements with which we may not be able to comply. Moreover, even if we successfully implement our business strategy, our operating results may not improve or may decline. We may decide to alter or discontinue aspects of our business strategy and may adopt different strategies due to business or competitive factors not currently foreseen, such as new medical technologies that would make our products obsolete. Any failure to implement our business strategy may adversely affect our business, results of operations and financial condition.
20
Table of Contents
The proliferation of physician-owned distributorships could result in increased pricing pressure on our products or harm our ability to sell our products to physicians who own or are affiliated with those distributorships.
Physician-owned distributorships, or PODs, are medical device distributors that are owned, directly or indirectly, by physicians. These physicians derive revenue from selling or arranging for the sale of medical devices for use in procedures they perform on their own patients at hospitals or ambulatory surgical centers, or ASCs, that agree to purchase from or through the POD, or that otherwise furnish ordering physicians with income that is based directly or indirectly on those orders of medical devices.
These companies and the physicians who own, or partially own, them have significant market knowledge and access to the surgeons who use our products and the hospitals that purchase our products, and growth in this area may reduce our ability to compete effectively for business from surgeons who own such distributorships. On March 26, 2013, the U.S. Department of Health and Human Services Office of Inspector General issued a special fraud alert on PODs and stated that it views PODs as inherently suspect under the Federal Anti-Kickback Statute and is concerned about the proliferation of PODs. Nonetheless, the number of PODs in the spine industry may continue to grow as economic pressures increase throughout the industry, hospitals, insurers and physicians search for ways to reduce costs, and, in the case of the physicians, search for ways to increase their incomes.
If we are unable to train surgeons on the safe and appropriate use of our products, we may be unable to achieve our expected growth.
An important part of our sales process includes the ability to train surgeons on the safe and appropriate use of our products. If we become unable to attract potential new surgeon customers to our training programs, or if we are unable to attract existing customers to training programs for future products, we may be unable to achieve our expected growth.
There is a learning process involved for spine surgeons to become proficient in the use of our products. It is critical to the success of our commercialization efforts to train a sufficient number of spine surgeons and to provide them with adequate instruction in the use of our products via trade shows and leads generated by our sales force. This training process may take longer than expected and may therefore affect our ability to increase sales. Following completion of training, we rely on the trained surgeons to advocate the benefits of our products in the broader marketplace. Convincing surgeons to dedicate the time and energy necessary for adequate training is challenging, and we cannot assure you we will be successful in these efforts. If surgeons are not properly trained, they may misuse or ineffectively use our products. This may also result in unsatisfactory patient outcomes, patient injury, negative publicity or lawsuits against us, any of which could have an adverse effect on our business.
Although we believe our training methods for surgeons are conducted in compliance with FDA and other applicable regulations developed both nationally and in third countries, if the FDA or other competent authority determines that our training constitutes promotion of an unapproved use or promotion of an intended purpose not covered by the current CE mark affixed to our product, they could request that we modify our training or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalty.
We have a limited operating history and may face difficulties encountered by early stage companies in new and rapidly evolving markets.
We began operations in 2000. Accordingly, we have a limited operating history upon which to base an evaluation of our business and prospects. In assessing our prospects, you must consider the risks and difficulties frequently encountered by early stage companies in new and rapidly evolving markets,
21
Table of Contents
particularly companies engaged in the development and sales of medical devices. These risks include our ability to:
| • | manage rapidly changing and expanding operations; |
| • | establish and increase awareness of our brand and strengthen customer loyalty; |
| • | grow our direct sales force and increase the number of our independent sales agents and distributors to expand sales of our products in the United States and in targeted international markets; |
| • | implement and successfully execute our business and marketing strategy; |
| • | respond effectively to competitive pressures and developments; |
| • | continue to develop and enhance our products and products in development; |
| • | obtain regulatory clearance, approval or CE Certificate of Conformity to commercialize new products and enhance our existing products; |
| • | expand our presence and commence operations in international markets; |
| • | perform clinical research and trials on our existing products and current and future product candidates; and |
| • | attract, retain and motivate qualified personnel. |
We can also be negatively affected by general economic conditions. Because of our limited operating history, we may not have insight into trends that could emerge and negatively affect our business.
As a result of these or other risks, our business strategy might not be successful.
Our business could suffer if we lose the services of key members of our senior management, key advisors or personnel.
We are dependent upon the continued services of key members of our executive management and a limited number of key advisors and personnel. In particular, we are highly dependent on the skills and leadership of our founders and executive management team. The loss of any one of these individuals could disrupt our operations or our strategic plans. Additionally, our future success will depend on, among other things, our ability to continue to hire and retain the necessary qualified scientific, technical, sales, marketing and managerial personnel, for whom we compete with numerous other companies, academic institutions and organizations. The loss of members of our management team, key advisors or personnel, or our inability to attract or retain other qualified personnel or advisors, could have a material adverse effect on our business, results of operations and financial condition.
The safety and efficacy of some of our products is not yet supported by long-term clinical data, which could limit sales, and our products might therefore prove to be less safe or effective than initially thought.
Our VerteBRIDGE platform products that we market in the United States either have received premarket clearance under Section 510(k) of the FDCA, or are exempt from premarket review. The FDA’s 510(k) clearance process requires us to show that our proposed product is “substantially equivalent” to another 510(k)-cleared product. This process is shorter and typically requires the submission of less supporting documentation than other FDA approval processes and does not always require long-term clinical studies. In the EEA, manufacturers of medical devices are required by the Council Directive 93/42/EEC concerning medical devices, known as the Medical Devices Directive, to collect post-marketing clinical data in relation to their CE marked medical devices. Post-market surveillance includes the conduct of post-market clinical follow-up studies permitting manufacturers to gather information concerning
22
Table of Contents
quality, safety or performance of medical devices after they have been placed on the market in the EEA. All information collected as part of the post-market surveillance process must be reviewed, investigated and analyzed on a regular basis in order to determine whether trending conclusions can be made concerning the safety or performance of the medical device and decisions must be taken in relation to the continued marketing of medical devices currently on the market. We expect to incur ongoing costs to comply with these post-market clinical obligations in France and other EEA markets for so long as we continue to market and sell products in those markets. We anticipate that these costs will be immaterial going forward. Except for Mobi-C, we lack the breadth of published long-term clinical data supporting the safety and efficacy of our cervical technology products or our lumbar technology products and the benefits they offer that might have been generated in connection with other approval processes. For these reasons, spine surgeons may be slow to adopt our products, we may not have comparative data that our competitors have or are generating, and we may be subject to greater regulatory and product liability risks. Further, future patient studies or clinical experience, including those that we are starting in France, may indicate that treatment with our products does not improve patient outcomes. Such results would slow the adoption of our products by spine surgeons, would significantly reduce our ability to achieve expected sales and could prevent us from achieving and maintaining profitability. Moreover, if future results and experience indicate that our products cause unexpected or serious complications or other unforeseen negative effects, we could be subject to mandatory product recalls, suspension or withdrawal of FDA or other government’s clearance or approval, CE Certificates of Conformity, significant legal liability or harm to our business reputation.
If we do not enhance our product offerings through our research and development efforts, we may be unable to compete effectively.
In order to increase our market share in the spine market, we must enhance and broaden our product offerings in response to changing customer demands and competitive pressures and technologies. We might not be able to successfully develop, obtain regulatory approval, clearance or CE Certificates of Conformity to market new products, and our future products might not be accepted by the surgeons or the third-party payors who reimburse for many of the procedures performed with our products. The success of any new product offering or enhancement to an existing product will depend on numerous factors, including our ability to:
| • | properly identify and anticipate surgeon and patient needs; |
| • | develop and introduce new products or product enhancements in a timely manner; |
| • | adequately protect our intellectual property and avoid infringing upon the intellectual property rights of third parties; |
| • | demonstrate the safety and efficacy of new products through the conduct of clinical investigations or the collection of existing relevant clinical data; and |
| • | obtain the necessary regulatory clearances, approvals or CE Certificates of Conformity for new products or product enhancements. |
If we do not develop and obtain regulatory clearance, approval or CE Certificates of Conformity for new products or product enhancements in time to meet market demand, or if there is insufficient demand for these products or enhancements, our results of operations will suffer. Our research and development efforts may require a substantial investment of time and resources before we are adequately able to determine the commercial viability of a new product, technology, material or other innovation. In addition, even if we are able to develop enhancements or new generations of our products successfully, these enhancements or new generations of products may not produce sales in excess of the costs of development and they may be quickly rendered obsolete by changing customer preferences or the introduction by our competitors of products embodying new technologies or features.
23
Table of Contents
If we fail to manage our anticipated growth properly, our business could suffer.
Our growth has placed, and, if it continues, will continue to place, a significant strain on our management and our operational and financial resources and systems. Failure to manage our growth effectively could cause us to over-invest or under-invest in infrastructure, and result in losses or weaknesses in our infrastructure, which could materially adversely affect us. Additionally, our anticipated growth will increase the demands placed on our suppliers, resulting in an increased need for us to monitor our suppliers carefully for quality assurance. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals.
We may seek to grow our business through acquisitions of or investments in new or complementary businesses, products or technologies, and the failure to manage any acquisitions or investments, or the failure to integrate them with our existing business, could have a material adverse effect on us.
From time to time we expect to consider opportunities to acquire or make investments in other technologies, products and businesses that may enhance our capabilities, complement our current products or expand the breadth of our markets or customer base. Potential and completed acquisitions and strategic investments involve numerous risks, including:
| • | problems assimilating the purchased technologies, products or business operations; |
| • | issues maintaining uniform standards, procedures, controls and policies; |
| • | unanticipated costs associated with acquisitions; |
| • | diversion of management’s attention from our core business; |
| • | adverse effects on existing business relationships with suppliers and customers; |
| • | risks associated with entering new markets in which we have limited or no experience; |
| • | potential loss of key employees of acquired businesses; and |
| • | increased legal and accounting compliance costs. |
We have no current commitments with respect to any acquisition or investment, and we have limited experience acquiring technology. We do not know if we will be able to identify suitable acquisitions, complete any such acquisitions on favorable terms or at all, successfully integrate any acquired business, product or technology into our business or retain any key personnel, suppliers or international distributors. Our ability to grow through acquisitions successfully depends upon our ability to identify, negotiate, complete and integrate suitable target businesses and to obtain any necessary financing. These efforts could be expensive and time-consuming, and may disrupt our ongoing business and prevent management from focusing on our operations. If we are unable to integrate any acquired businesses, products or technologies effectively, our business, results of operations and financial condition will be materially adversely affected.
We are required to maintain high levels of inventory, which could consume a significant amount of our resources and reduce our cash flows.
As a result of the need to maintain substantial levels of inventory, we are subject to the risk of inventory obsolescence. Many of our products come in sets that feature components in a variety of sizes allowing the implant or device to be customized to the patient’s needs. In order to market our products effectively, we often must maintain and provide surgeons and hospitals with implant sets, back-up products and products of different sizes. For each surgery, fewer than all of the components of the set are used, and therefore certain portions of the set may become obsolete before they can be used. In the event that a substantial portion of our inventory becomes obsolete, it could have a material adverse effect on our
24
Table of Contents
earnings and cash flows due to the resulting costs associated with the inventory impairment charges and costs required to replace such inventory.
We provide our customers with the use of instrument sets for each surgery performed using our products, which could consume a significant amount of our resources and reduce our cash flows.
We provide our customers with the use of instrument sets for each surgery performed using our products. As a result, we are required to maintain significant levels of instrument sets. The amount of this investment is driven by the number of surgeons or hospitals using our products, and as the number of different surgeons and hospitals that use our products increases, the number of instrument sets required to meet this demand will increase. Because we do not have the sales volume of some larger companies, we may not be able to utilize our instrument sets as often and our return on assets may be lower when compared to such companies. In addition, as we introduce new products, new instrument sets may be required, with a significant initial investment required to accommodate the launch of the product.
Consolidation in the healthcare industry could lead to demands for price concessions or to the exclusion of some suppliers from certain of our markets, which could have an adverse effect on our business, results of operations or financial condition.
Because healthcare costs have risen significantly over the past decade, numerous initiatives and reforms initiated by legislators, regulators and third-party payors to curb these costs have resulted in a consolidation trend in the healthcare industry to aggregate purchasing power. As the healthcare industry consolidates, competition to provide products and services to industry participants has become and will continue to become more intense. This in turn has resulted and will likely continue to result in greater pricing pressures and the exclusion of certain suppliers from important market segments as group purchasing organizations, independent delivery networks and large single accounts continue to use their market power to consolidate purchasing decisions for hospitals. We expect that market demand, government regulation, third-party coverage and reimbursement policies and societal pressures will continue to change the worldwide healthcare industry, resulting in further business consolidations and alliances among our customers, which may reduce competition, exert further downward pressure on the prices of our products and may adversely impact our business, results of operations or financial condition.
Our sales volumes and our operating results may fluctuate over the course of the year.
Our business is generally not seasonal in nature. However, our sales may be influenced by summer vacation and winter holiday periods, during which we have experienced fewer spine surgeries taking place. We have experienced and continue to experience meaningful variability in our sales and gross profit among quarters, as well as within each quarter, as a result of a number of factors, including, among other things:
| • | the number of products sold in the quarter; |
| • | the number of surgical procedures performed during the quarter; |
| • | the demand for, and pricing of our products and the products of our competitors; |
| • | the timing of or failure to obtain regulatory clearances, approvals or CE Certificates of Conformity for products; |
| • | costs, benefits, and timing of new product introductions; |
| • | increased competition; |
| • | the availability and cost of components and materials; |
| • | the number of selling days in the quarter; |
25
Table of Contents
| • | fluctuation and foreign currency exchange rates; and |
| • | impairment and other special charges. |
If we experience material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately report our financial condition or results of operations which may adversely affect investor confidence in us and, as a result, the value of our common stock.
As a result of becoming a public company, we will be required, under Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting beginning with our Annual Report on Form 10-K for the year ended December 31, 2014. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency or combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of a company’s annual and interim financial statements will not be detected or prevented on a timely basis.
We are further enhancing the computer systems processes and related documentation necessary to perform the evaluation needed to comply with Section 404. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal controls are effective. The effectiveness of our controls and procedures may be limited by a variety of factors, including:
| • | faulty human judgment and simple errors, omissions or mistakes; |
| • | fraudulent action of an individual or collusion of two or more people; |
| • | inappropriate management override of procedures; and |
| • | the possibility that any enhancements to controls and procedures may still not be adequate to assure timely and accurate financial control. |
If we are unable to conclude that our internal control over financial reporting is effective, we could lose investor confidence in the accuracy and completeness of our financial reports, which would likely cause the price of our common stock to decline.
When we cease to be an “emerging growth company” under the federal securities laws, our auditors will be required to express an opinion on the effectiveness of our internal controls. If we are unable to confirm that our internal control over financial reporting is effective, or if our auditors are unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, which would cause the price of our common stock to decline.
We expect to incur significant additional costs as a result of being a public company, which may adversely affect our operating results and financial condition.
We expect to incur costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as rules implemented by the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, or the Dodd-Frank Act, the Securities and Exchange Commission, or the SEC, and NASDAQ. These rules and regulations are expected to increase our accounting, legal and financial compliance costs and make some activities more time-consuming and costly. In addition, we will incur additional costs associated with our public company reporting requirements and we expect those costs to increase in the future. For example, we will be required to devote significant resources to
26
Table of Contents
complete the assessment and documentation of our internal control system and financial process under Section 404 of the Sarbanes-Oxley Act, including an assessment of the design of our information systems associated with our internal controls. We will incur significant costs to remediate any material weaknesses we identify through these efforts. We also expect these rules and regulations to make it more expensive for us to maintain directors’ and officers’ liability insurance and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees, or as executive officers. We cannot predict or estimate the amount of additional costs we may incur or the timing of such costs.
New laws and regulations, as well as changes to existing laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act, the Dodd-Frank Act and rules adopted by the SEC and NASDAQ, would likely result in increased costs to us as we respond to their requirements, which may adversely affect our operating results and financial condition.
If we experience significant disruptions in our information technology systems, our business may be adversely affected.
We depend on our information technology systems for the efficient functioning of our business, including accounting, data storage, compliance, purchasing and inventory management. Our current systems are not fully redundant; however, we are in the process of upgrading them. We expect these upgrades to take two to three years to complete. While we will attempt to mitigate interruptions, we may experience difficulties in implementing some upgrades which would impact our business operations, or experience difficulties in operating our business during the upgrade, either of which could disrupt our operations, including our ability to timely ship and track product orders, project inventory requirements, manage our supply chain and otherwise adequately service our customers. In the event we experience significant disruptions as a result of the current implementation of our information technology systems, we may not be able to repair our systems in an efficient and timely manner. Accordingly, such events may disrupt or reduce the efficiency of our entire operation and have a material adverse effect on our results of operations and cash flows.
We are increasingly dependent on sophisticated information technology for our infrastructure. Our information systems require an ongoing commitment of significant resources to maintain, protect and enhance existing systems. Failure to maintain or protect our information systems and data integrity effectively could have a materially adverse effect on our business. For example, third parties may attempt to hack into systems and may obtain our proprietary information.
Fluctuations in insurance cost and availability could adversely affect our profitability or our risk management profile.
We hold a number of insurance policies, including product liability insurance, directors’ and officers’ liability insurance, general liability insurance, property insurance and workers’ compensation insurance. If the costs of maintaining adequate insurance coverage increase significantly in the future, our operating results could be materially adversely affected. Likewise, if any of our current insurance coverage should become unavailable to us or become economically impractical, we would be required to operate our business without indemnity from commercial insurance providers. If we operate our business without insurance, we could be responsible for paying claims or judgments against us that would have otherwise been covered by insurance, which could adversely affect our results of operations or financial condition.
27
Table of Contents
Risks Related to our Legal and Regulatory Environment
Our medical device products and operations are subject to extensive governmental regulation both in the United States and abroad, and our failure to comply with applicable requirements could cause our business to suffer.
Our medical device products and operations are subject to extensive regulation by the FDA and various other federal, state and foreign governmental authorities, such as the competent authorities of EEA countries. Government regulation of medical devices is meant to assure their safety and effectiveness, and includes regulation of, among other things:
| • | design, development and manufacturing; |
| • | testing, labeling, content and language of instructions for use and storage; |
| • | clinical trials; |
| • | product safety; |
| • | marketing, sales and distribution; |
| • | regulatory clearances and approvals including premarket clearance and approval; |
| • | conformity assessment procedures; |
| • | product traceability and record keeping procedures; |
| • | advertising and promotion; |
| • | product complaints, complaint reporting, recalls and field safety corrective actions; |
| • | post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; |
| • | post-market studies; and |
| • | product import and export. |
The regulations to which we are subject are complex and have tended to become more stringent over time. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated sales.
Before we can market or sell a new regulated product or a significant modification to an existing product in the United States, we must obtain either clearance under Section 510(k) of the FDCA or an approval of a premarket approval, or PMA, application unless the device is specifically exempt from premarket review. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence. In the PMA approval process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices for which the 510(k) process cannot be used and that are deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. Modifications to products that are approved through a PMA application generally need FDA approval. Similarly, some modifications made to products cleared through a 510(k) may require a new 510(k). The FDA’s 510(k) clearance process usually takes from three to 12 months, but may last longer. The process of obtaining a PMA is much more costly and uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is submitted to the FDA until an approval is obtained.
28
Table of Contents
In the United States, other than Mobi-C, our currently commercialized products either have received premarket clearance under Section 510(k) of the FDCA or we have determined that they are exempt from premarket review. The FDA could disagree with our determination that a product is exempt and require the product to go through premarket review. In addition, the FDA could determine that a product requires a Class III designation rather than Class II. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to existing products than we had expected, our product introductions or modifications could be delayed or canceled, which could cause our sales to decline. The FDA may demand that we obtain a PMA prior to marketing certain of our future products. In addition, if the FDA disagrees with our determination that a product we currently market is subject to an exemption from premarket review, the FDA may require us to submit a 510(k) or PMA in order to continue marketing the product. Further, even with respect to those future products where a PMA is not required, we cannot assure you that we will be able to obtain the 510(k) clearances with respect to those products.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
| • | we may not be able to demonstrate to the FDA’s satisfaction that our products are safe and effective for their intended uses; |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and |
| • | the manufacturing process or facilities we use may not meet applicable requirements. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions that may prevent or delay approval or clearance of our products under development or impact our ability to modify our currently approved or cleared products on a timely basis. For example, in 2011, the FDA announced a Plan of Action to modernize and improve the FDA’s premarket review of medical devices, and has implemented, and continues to implement, reforms intended to streamline the premarket review process. In addition, as part of the Food and Drug Administration Safety and Innovation Act of 2012, or FDASIA, Congress enacted several reforms entitled the Medical Device Regulatory Improvements and additional miscellaneous provisions which will further affect medical device regulation both pre- and post-approval. Any change in the laws or regulations that govern the clearance and approval processes relating to our current and future products could make it more difficult and costly to obtain clearance or approval for new products, or to produce, market and distribute existing products.
The FDA could also reclassify some or all of our products that are currently classified as Class II to Class III requiring additional controls, clinical studies and submission of a PMA for us to continue marketing and selling those products. Under new changes instituted by FDASIA, the FDA may now change the classification of a medical device by administrative order instead of by regulation. Although the revised process is simpler, the FDA must still publish a proposed order in the Federal Register, hold a device classification panel meeting and consider comments from affected stakeholders before issuing the reclassification order. We cannot guarantee that the FDA will not reclassify any of our Class II devices into Class III and require us to submit a PMA for FDA review and approval of the safety and effectiveness of our products.
Any delay in, or failure to receive or maintain, clearance or approval for our products under development could prevent us from generating revenue from these products or achieving profitability. Additionally, the FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny on us, could dissuade some surgeons from using our products and adversely affect our reputation and the perceived safety and efficacy of our products.
29
Table of Contents
In addition, even after we have obtained the proper regulatory clearance or approval to market a product, the FDA has the power to require us to conduct postmarketing studies. For example, as a condition of approval, we are required to conduct a post-approval study of Mobi-C, as well as an enhanced surveillance study. The post-approval study will take the form of continued follow-up for the existing pivotal clinical trial subjects to evaluate overall success rate over a total of seven years. This study is expected to cost approximately $1.35 million annually and should be completed in the first half of 2015. The enhanced surveillance study will be a post-market surveillance system designed primarily to identify any adverse events, device malfunctions or complaints for patients implanted with the device during the first ten years of commercial use in the United States, with an expected cost of approximately $150,000 per year. Failure to conduct required studies in a timely manner could result in the revocation of the 510(k) clearance or PMA approval for the product that is subject to such a requirement and could also result in the recall or withdrawal of the product, which would prevent us from generating sales from that product in the United States.
In the EEA, our medical devices must comply with the Essential Requirements laid down in Annex I to the EEA Medical Devices Directive. Compliance with these requirements is a prerequisite to be able to affix the CE mark of conformity to our medical devices, without which they cannot be marketed or sold in the EEA. To demonstrate compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive we must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements laid down in Annex I to the Medical Devices Directive, a conformity assessment procedure requires the intervention of a third party organization designated by competent authorities of a EEA country to conduct conformity assessments, known as a Notified Body. The Notified Body would typically audit and examine products’ Technical File and the quality system for the manufacture, design and final inspection of our devices before issuing a CE Certificate of Conformity demonstrating compliance with the relevant Essential Requirements laid down in Annex I to the Medical Devices Directive.
Medical device manufacturers must carry out a clinical evaluation of their medical devices to demonstrate conformity with the relevant Essential Requirements laid down in Annex I to the Medical Device Directive covering safety and performance. This clinical evaluation is part of the product’s Technical File. A clinical evaluation includes an assessment of whether a medical device’s performance is in accordance with its intended use, and that the known and foreseeable risks linked to the use of the device under normal conditions are minimized and acceptable when weighed against the benefits of its intended purpose. The clinical evaluation conducted by the manufacturer must also address any clinical claims, the adequacy of the device labeling and information (particularly claims, contraindications, precautions/warnings) and the suitability of related Instructions for Use. This assessment must be based on clinical data, which can be obtained from (i) clinical studies conducted on the devices being assessed; (ii) scientific literature from similar devices whose equivalence with the assessed device can be demonstrated; or (iii) both clinical studies and scientific literature.
With respect to implantable devices or devices classified as Class III in the EEA, the manufacturer must conduct clinical studies to obtain the required clinical data, unless relying on existing clinical data from similar devices can be justified. As part of the conformity assessment process, depending on the type of devices, the Notified Body will review the manufacturer’s clinical evaluation process, assess the clinical evaluation data of a representative sample of the devices’ subcategory or generic group (for Class IIa and IIb devices), or assess all the clinical evaluation data, verify the manufacturer’s assessment of that data and assess the validity of the clinical evaluation report and the conclusions drawn by the manufacturer (for implantable and Class III devices). The conduct of clinical studies to obtain clinical data that might be required as part of the described clinical evaluation process can be expensive and time-consuming.
30
Table of Contents
Even after we receive a CE Certificate of Conformity enabling us to affix the CE mark on our products and to sell these products in the EEA countries, a Notified Body or a competent authority may require post-marketing studies of our products. In fact, in March of 2010, the French government requested us to start such studies on our products sold in France. Failure to comply with such requests in a timely manner could result in the withdrawal of our CE Certificate of Conformity and the recall or withdrawal of the subject product from the EEA market, which would prevent us from generating sales for that product in the EEA. Moreover, each CE Certificate of Conformity is valid for a maximum of five years, more commonly three years. At the end of each period of validity we are required to apply to the Notified Body for a renewal of the CE Certificates of Conformity. There may be delays in the renewal of the CE Certificates of Conformity or the Notified Body may require modifications to our products or to the related Technical Files before it agrees to issue the new CE Certificates of Conformity.
On September 26, 2012, the European Commission adopted a package of legislative proposals designed to replace the existing regulatory framework for medical devices in the EEA. These proposals are intended to strengthen the medical devices rules in the EEA countries. On October 22, 2013, the European Parliament voted in favor of an amended draft of the Regulation. The proposed text is currently being discussed by the Council of the European Union. If and when adopted the proposed new legislation may prevent or delay the CE marking of our products under development or impact our ability to modify our currently CE marked products on a timely basis.
To market and sell our products in other countries, we must seek and obtain regulatory approvals, certifications or registrations and comply with the laws and regulations of those countries. These laws and regulations, including the requirements for approvals, certifications or registrations and the time required for regulatory review, vary from country to country. Obtaining and maintaining foreign regulatory approvals, certifications or registrations are expensive, and we cannot be certain that we will receive regulatory approvals, certifications or registrations in any foreign country in which we plan to market our products. If we fail to obtain or maintain regulatory approvals, certifications or registrations in any foreign country in which we plan to market our products, our ability to generate revenue will be harmed.
Failure to comply with applicable laws and regulations could jeopardize our ability to sell our products and result in enforcement actions such as:
| • | warning letters; |
| • | fines; |
| • | injunctions; |
| • | civil penalties; |
| • | termination of distribution; |
| • | recalls or seizures of products; |
| • | delays in the introduction of products into the market; |
| • | total or partial suspension of production; |
| • | refusal of the FDA or other regulator to grant future clearances or approvals; |
| • | withdrawals or suspensions of current clearances or approvals, resulting in prohibitions on sales of our products; |
| • | withdrawal or suspension of the CE Certificates of Conformity granted by the Notified Body or delay in obtaining these certificates; and/or |
| • | in the most serious cases, criminal penalties. |
31
Table of Contents
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and have a material adverse effect on our reputation, business, results of operations and financial condition.
There is no guarantee that the FDA will grant 510(k) clearance or PMA approval of our future products, and failure to obtain necessary clearances or approvals for our future products would adversely affect our ability to grow our business.
Some of our new products will require FDA clearance of a 510(k) or FDA approval of a PMA. The FDA may not approve or clear these products for the indications that are necessary or desirable for successful commercialization. Indeed, the FDA may refuse our requests for 510(k) clearance or premarket approval of new products.
The FDA recently issued guidance documents intended to explain the procedures and criteria the FDA will use in assessing whether a 510(k) submission meets a minimum threshold of acceptability and should be accepted for substantive review. Under the “Refuse to Accept” guidance, the FDA conducts an early review against specific acceptance criteria to inform 510(k) and PMA submitters if the submission is administratively complete, or if not, to identify the missing element(s). Submitters are given the opportunity to provide the FDA with the identified information. If the information is not provided within a defined time, the submission will not be accepted for FDA review. Significant delays in receiving clearance or approval, or the failure to receive clearance or approval for our new products would have an adverse effect on our ability to expand our business.
Modifications to our products may require new 510(k) clearances, premarket approvals, or revisions to our existing CE Certificates of Conformity, or may require us to cease marketing or recall the modified products until clearances or approvals are obtained.
Any modification to a 510(k)-cleared device that could significantly affect its safety or effectiveness, including significant design and manufacturing changes, or that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, approval of a PMA. The FDA requires every manufacturer to make this determination in the first instance, but the FDA may review any manufacturer’s decision. The FDA may not agree with our decisions regarding whether new clearances or approvals are necessary. We have modified some of our 510(k) cleared products, and have determined based on our review of the applicable FDA guidance that in certain instances the changes did not require new 510(k) clearances or PMA approval. If the FDA disagrees with our determination and requires us to seek new 510(k) clearances or PMA approval for modifications to our previously cleared products for which we have concluded that new clearances or approvals are unnecessary, we may be required to cease marketing or distribution of our products or to recall the modified product until we obtain clearance or approval, and we may be subject to significant regulatory fines or penalties.
Furthermore, potential changes to the 510(k) program may make it more difficult for us to make modifications to our previously cleared products, by either imposing more strict requirements on when a new 510(k) clearance for a modification to a previously cleared product must be submitted, or applying more onerous review criteria to such submissions. In July and December 2011, respectively, the FDA issued draft guidance documents addressing when to submit a new 510(k) clearance due to modifications to 510(k)-cleared products and the criteria for evaluating substantial equivalence. The July 2011 draft guidance document was ultimately withdrawn as the result of the FDASIA. As a result, the FDA’s original guidance document regarding 510(k) modifications, which dates back to 1997, remains in place. It is uncertain when the FDA will seek to issue new guidance on product modifications. Any efforts to do so could result in a more rigorous review process and make it more difficult to obtain clearance for device modifications.
In the EEA, we must inform the Notified Body that carried out the conformity assessment of the medical devices we market or sell in the EEA of any planned substantial changes to our quality system or changes
32
Table of Contents
to our devices which could affect compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive or the devices’ intended purpose. The Notified Body will then assess the changes and verify whether they affect the products’ conformity with the Essential Requirements laid down in Annex I to the Medical Devices Directive or the conditions for the use of the device. If the assessment is favorable, the Notified Body will issue a new CE Certificate of Conformity or an addendum to the existing CE Certificate of Conformity attesting compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive. If it is not, we may not be able to continue to market and sell the product in the EEA.
Modifications to our class III products, such as the Mobi-C, may require new PMAs or PMA supplements, or even require us to cease promoting or to recall the modified products until such approvals are obtained, and the FDA may not agree with our conclusions regarding whether new approvals are required.
Any modification to a device approved through the PMA pathway that impacts the safety or effectiveness of the device requires submission to the FDA and FDA approval of a PMA supplement or, in some cases, a new PMA application. In the future, we may submit PMA supplements for changes to the Mobi-C. Certain changes to a device approved through the PMA pathway do not require submission and approval of a new PMA or PMA supplement and may only require notice to FDA in a PMA Annual Report. In the future, we may make changes to the Mobi-C that we may determine do not require approval of a new PMA or PMA supplement. The FDA may not agree with our decisions not to seek approval of a new PMA or PMA supplement.
If the FDA disagrees with us and requires us to submit a new PMA or PMA supplement for modifications we have made to the Mobi-C for which we did not seek approval of a PMA or PMA supplement, we may be required to cease promoting or to recall the modified product until we obtain approval. In addition, we could be subject to significant regulatory fines or other penalties. Furthermore, our products could be subject to recall if the FDA determines, for any reason, that our products are not safe or effective or that appropriate regulatory submissions were not made. Delays in receipt or failure to receive approvals, the loss of previously received approvals, or the failure to comply with any other existing or future regulatory requirements, could reduce our sales, profitability and future growth prospects.
We may fail to obtain or maintain foreign regulatory approvals to market our products in other countries.
We currently market our products internationally and intend to expand our international marketing. International jurisdictions require separate regulatory approvals and compliance with numerous and varying regulatory requirements. For example, outside of the United States and the EEA, we intend to continue to seek regulatory clearance to market our primary products in China, Brazil, Japan, South Korea and other key markets. The approval procedures vary among countries and may involve requirements for additional testing, and the time required to obtain approval may differ from country to country and from that required to obtain clearance or approval in the United States and the necessary CE Certificates of Conformity in the EEA countries.
Clearance or approval in the United States and/or a CE Certificate of Conformity in the EEA countries does not ensure approval or certification by regulatory authorities in other countries or jurisdictions, and approval or certification by one foreign regulatory authority does not ensure approval or certification by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval or certification process may include all of the risks associated with obtaining FDA clearance or approval. In addition, some countries only approve or certify a product for a certain period of time, and we are required to re-approve or re-certify our products in a timely manner prior to the expiration of our prior approval or certification. We may not obtain foreign regulatory approvals on a timely basis, if at all. We
33
Table of Contents
may not be able to file for regulatory approvals or certifications and may not receive necessary approvals to commercialize our products in any market. If we fail to receive necessary approvals or certifications to commercialize our products in foreign jurisdictions on a timely basis, or at all, or if we fail to have our products re-approved or re-certified, our business, results of operations and financial condition could be adversely affected.
These and other factors may have a material adverse effect on our international operations or on our business, results of operations and financial condition generally.
If we or our suppliers fail to comply with ongoing EEA and FDA or other foreign regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain clearance or approval, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA and other domestic and foreign regulatory bodies. In particular, we and our third-party suppliers are required to comply with the FDA’s Quality System Regulation, or QSR. In the EEA countries, compliance with harmonized standards is also recommended as this is interpreted as a presumption of conformity with the relevant Essential Requirements laid down in Annex I to the Medical Devices Directive or the quality system requirements laid down in the other Annexes to the Directive. These FDA regulations and EU standards cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. Compliance with harmonized standards in the EEA is also subject to regular review through the conduct of inspection by Notified Bodies or other certification bodies. If we, or our manufacturers, fail to adhere to QSR requirements in the United States or other harmonized standards in the EEA, this could delay production of our products and lead to fines, difficulties in obtaining regulatory clearances, CE Certificate of Conformity, recalls, enforcement actions, including injunctive relief or consent decrees, or other consequences, which could, in turn, have a material adverse effect on our financial condition or results of operations.
In addition, the FDA audits compliance with the QSR through periodic announced and unannounced inspections of manufacturing and other facilities. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in any of the following enforcement actions:
| • | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | unanticipated expenditures to address or defend such actions; |
| • | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| • | operating restrictions or partial suspension or total shutdown of production; |
| • | refusing or delaying our requests for 510(k) clearance or premarket approval of new products or modified products; |
| • | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| • | refusal to grant export approval for our products; or |
| • | criminal prosecution. |
34
Table of Contents
Any of these sanctions could have a material adverse effect on our reputation, business, results of operations and financial condition. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements, which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
Outside the EEA and the United States our products and operations are also often required to comply with standards set by foreign regulatory bodies, and those standards, types of evaluation and scope of review differ among foreign regulatory bodies. We intend to comply with the standards enforced by such foreign regulatory bodies as needed to commercialize our products. If we fail to comply with any of these standards adequately, a foreign regulatory body may take adverse actions similar to those within the power of a Notified Body or competent authority or the FDA. Any such action may harm our reputation and business, and could have an adverse effect on our business, results of operations and financial condition.
If our products, or malfunction of our products, cause or contribute to a death or a serious injury, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. All manufacturers placing medical devices in the market in the EEA are legally bound to report any serious or potentially serious incidents involving devices they produce or sell to the competent authority in whose jurisdiction the incident occurred. Were this to happen to us, the relevant competent authority would file an initial report, and there would then be a further inspection or assessment if there were particular issues. Either this would be carried out either by the competent authority or it could require that the Notified Body carry out the inspection or assessment.
Any such adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Adverse events involving our products have been reported to us in the past, and we cannot guarantee that they will not occur in the future. Any corrective action, whether voluntary or involuntary, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
In the EEA, we must comply with the EU Medical Device Vigilance System. Under this system, incidents must be reported to the relevant authorities of the EEA countries, and manufacturers are required to take Field Safety Corrective Actions, or FSCAs, to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market. An incident is defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient or user or of other persons or to a serious deterioration in their state of health. An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device. FSCAs must be communicated by the manufacturer or its European Authorized Representative to its customers and/or to the end users of the device through Field Safety Notices.
Our products may in the future be subject to product recalls. A recall of our products, either voluntarily or at the direction of the FDA or another governmental authority, including a third country authority, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In this
35
Table of Contents
case, the FDA, the authority to require a recall must be based on an FDA finding that there is reasonable probability that the device would cause serious injury or death. In addition, foreign governmental bodies have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacture. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. The FDA requires that certain classifications of recalls be reported to the FDA within 10 working days after the recall is initiated. A government-mandated or voluntary recall by us or one of our international distributors could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be subject to liability claims, be required to bear other costs, or take other actions that may have a negative impact on our future sales and our ability to generate profits. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA or another third country competent authority. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA or another third country competent authority. If the FDA, another third country competent authority, or our Notified Body disagrees with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA or another third country competent authority could take enforcement action for failing to report the recalls when they were conducted and our Notified Body could suspend or withdraw our CE Certificate of Conformity as a result.
Further, under the FDA’s medical device reporting, or MDR, regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Repeated product malfunctions may result in a voluntary or involuntary product recall, which could divert managerial and financial resources, impair our ability to manufacture our products in a cost-effective and timely manner, and have an adverse effect on our reputation, results of operations and financial condition. We are also required to follow detailed recordkeeping requirements for all firm-initiated medical device corrections and removals, and to report such corrective and removal actions to FDA if they are carried out in response to a risk to health and have not otherwise been reported under the MDR regulations. In addition, in December of 2012, the FDA issued a draft guidance intended to assist the FDA and industry in distinguishing medical device recalls from product enhancements. Per the guidance, if any change or group of changes to a device addresses a violation of the FDCA, that change would generally constitute a medical device recall and require submission of a recall report to the FDA.
If the third parties on which we rely to conduct our clinical trials and to assist us with pre-clinical development do not perform as contractually required or expected, we may not be able to obtain regulatory clearance, approval or a CE Certificate of Conformity for or commercialize our products.
We often must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct our clinical trials and prepare our PMA submissions. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval or a CE Certificate of Conformity for, or successfully commercialize, our products on a timely basis, if at all, and our business, operating results and prospects may be adversely affected. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control.
36
Table of Contents
The results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
Our ongoing research and development, pre-clinical testing and clinical trial activities are subject to extensive regulation and review by numerous governmental authorities both in the United States and abroad. We are currently conducting pre-and post-market clinical studies of some of our products to gather information about these products’ safety, efficacy or optimal use. In the future we may conduct clinical trials to support approval of new products. Clinical studies must be conducted in compliance with FDA regulations or the FDA may take enforcement action. The data collected from these clinical studies may ultimately be used to support market clearance for these products. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product candidate claims or that the FDA or foreign authorities and Notified Bodies will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile.
We may be subject to enforcement action if we engage in improper marketing or promotion of our products.
Our promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition of the promotion of unapproved, or off-label, use. Surgeons may use our products off-label, as the FDA and equivalent third country agencies do not restrict or regulate a surgeon’s choice of treatment within the practice of medicine. However, if the FDA or an equivalent third country agency determines that our promotional materials or training constitutes promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an off-label use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of the products could be impaired. Although our policy is to refrain from statements that could be considered off-label promotion of our products, the FDA or another regulatory agency could disagree and conclude that we have engaged in off-label promotion. In addition, the off-label use of our products may increase the risk of product liability claims. Product liability claims are expensive to defend and could divert our management’s attention, result in substantial damage awards against us, and harm our reputation.
Further, the advertising and promotion of our products is subject to EEA Member States laws implementing the Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other EEA Member State legislation governing the advertising and promotion of medical devices. In addition, we are subject to EU and national Codes of Conduct. These laws and Codes of Conduct may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
37
Table of Contents
If we or our sales representatives fail to comply with U.S. federal and state fraud and abuse laws, we could be subject to civil and criminal penalties, which could adversely impact our reputation and business operations.
There are numerous U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback, false claims, physician self-referral and physician transparency laws. Our relationships with surgeons, hospitals, group purchasing organizations and our independent sales agencies are subject to scrutiny under these laws. Violations of these laws are punishable by criminal and civil sanctions, including significant monetary penalties and, in some instances, imprisonment and exclusion from participation in federal and state healthcare programs, including the Medicare, Medicaid and Veterans Administration health programs.
Healthcare fraud and abuse regulations are complex, and even minor irregularities can potentially give rise to claims that a statute or prohibition has been violated. The laws that may affect our ability to operate include:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as Medicare and Medicaid; |
| • | the federal False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid or other third-party payors that are false or fraudulent; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, as amended, which created federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| • | the Federal Trade Commission Act and similar laws regulating advertisement and consumer protections; |
| • | the federal Foreign Corrupt Practices Act of 1997, which prohibits corrupt payments, gifts or transfers of value to foreign officials; and |
| • | foreign and/or U.S. state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
Further, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or, collectively, the PPACA, among other things, amends the intent requirements of the federal Anti-Kickback Statute and the criminal statute governing healthcare fraud. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. In addition, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act or federal civil money penalties statute. Possible sanctions for violation of laws include monetary fines, civil and criminal penalties, exclusion from government healthcare programs such as Medicare and Medicaid and forfeiture of amounts collected in violation of such prohibitions. Moreover, while we do not submit claims and our customers make the ultimate decision on how to submit claims, from time-to-time, we may provide reimbursement guidance to our customers. If a government authority were to conclude that we provided improper advice to our customers and/or encouraged the submission of false claims for reimbursement, we could face action against us by government authorities. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our reputation, business, results of operations and financial condition.
38
Table of Contents
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under such laws, it is possible that some of our business activities, including our relationship with surgeons, hospitals, group purchasing organizations and our independent sales agencies, could be subject to challenge under one or more of such laws. We have entered into consulting agreements and royalty agreements with surgeons, including some who order and use our products in procedures they perform. While these transactions were structured to comply with all applicable laws, including state and federal anti-kickback laws, to the extent applicable, regulatory agencies may view these transactions as prohibited arrangements that must be restructured, or discontinued, or for which we could be subject to significant penalties, including debarment. We could be adversely affected if regulatory agencies interpret our financial relationships with spine surgeons who order our products to be in violation of applicable laws. This could subject us to civil and criminal penalties for non-compliance, the cost of which could be substantial.
To enforce compliance with the federal laws, the U.S. Department of Justice, or DOJ, has recently increased its scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Dealing with investigations can be time- and resource-consuming and can divert management’s attention from the business. Additionally, settlements with the DOJ or other law enforcement agencies have forced healthcare providers to agree to additional onerous compliance and reporting requirements as part of a consent decree or corporate integrity agreement. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
In certain cases, federal and state authorities pursue actions for false claims on the basis that manufacturers and distributors are promoting off-label uses of their products. Pursuant to FDA regulations, we can only market our products for cleared or approved uses. Although surgeons are permitted to use medical devices for indications other than those cleared or approved by the FDA, we are prohibited from promoting products for off-label uses. We market our products and provide promotional materials and training programs to surgeons regarding the use of our products. If it is determined that our marketing, promotional materials or training programs constitute promotion of unapproved uses, we could be subject to significant fines in addition to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure and criminal penalty.
Most of these laws apply to not only the actions taken by us, but also actions taken by our independent sales agencies. We have limited knowledge and control over the business practices of our independent sales agencies, and we may face regulatory action against us as a result of their actions which could have a material adverse effect on our reputation, business, results of operations and financial condition.
The PPACA, among other things, also imposed new reporting requirements on device manufacturers for payments by them and in some cases their distributors to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members (commonly known as the Physician Payments Sunshine Act or Open Payments). Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission.
The period between August 1, 2013 and December 31, 2013 was the first reporting period under the Open Payments system. Starting February 18, 2014, Open Payments registration and data submission for the Federal Physician Payments Sunshine Act opened with a two-phased approach for reporting the applicable 2013 payments. Phase 1 (February 18, 2014—March 31, 2014) included user registration in CMS’ Enterprise Portal and submission of corporate profile information and aggregate payment data from the first reporting period. Phase 2 (begins in May and extends for at least 30 days) includes industry registration in the Open Payments system, submission of detailed payment data from the first reporting
39
Table of Contents
period, and legal attestation to the accuracy of the data. Thereafter, device manufacturers must submit reports by the 90th day of each subsequent calendar year. Due to the difficulty in complying with Physician Payments Sunshine Act and the nature of our U.S. sales force concentrated with independent sales agencies, we cannot assure you that we will successfully report all transfers of value by us and our independent sales agencies, and any failure to comply could result in significant fines and penalties.
In addition, there has been a recent trend of increased state regulation of payments made to physicians. Certain states mandate implementation of commercial compliance programs, impose restrictions on device manufacturer marketing practices and require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare providers. A similar trend is observed in foreign jurisdictions such as France. In France, a recently adopted law and a decree require companies working in the health sector to publicly disclose direct or indirect benefits granted to, and agreements entered into with, physicians and other healthcare professionals. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may violate one or more of the requirements.
The scope and enforcement of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Federal or state regulatory authorities might challenge our current or future activities under these laws. Any such challenge, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business, either of which could have a material adverse effect on our reputation, business, results of operations and financial condition. Any state or federal regulatory review of us, regardless of the outcome, would be costly and time-consuming. Additionally, we cannot predict the impact of any changes in these laws, whether or not retroactive.
Similar laws are increasingly being introduced in the individual EEA countries. The provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medical devices is prohibited. The provision of benefits or advantages to physicians is also governed by the national antibribery laws of the EEA countries. One such example is the UK Bribery Act. Payments made to physicians in certain EEA countries must also be publically disclosed. Moreover, agreements with physicians must often be the subject of prior notification and approval by the physician’s employer, his/her competent professional organization, and/or the competent authorities of the EEA countries. These requirements are provided in the national laws, industry codes, or professional codes of conduct applicable in the EEA countries. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
Legislative or regulatory healthcare reforms may make it more difficult and costly for us to obtain regulatory clearance or approval of our products and to produce, market and distribute our products after clearance or approval is obtained.
Recent political, economic and regulatory influences are subjecting the healthcare industry to fundamental changes. The sales of our products depend in part on the availability of coverage and reimbursement from third-party payors such as government health programs, private health insurers, health maintenance organizations and other healthcare-related organizations. Both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation and regulations designed to contain or reduce the cost of healthcare. Such legislation and regulations may result in decreased reimbursement for medical devices and/or the procedures in which they are used, which may further exacerbate industry-wide pressure to reduce the prices charged for medical devices. This could harm our ability to market our products and generate sales.
40
Table of Contents
In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our products. Delays in receipt of, or failure to receive, regulatory clearances or approvals for our new products would have a material adverse effect on our business, results of operations and financial condition.
Federal and state governments in the United States have recently enacted legislation to overhaul the nation’s healthcare system. While the goal of healthcare reform is to expand coverage to more individuals, it also involves increased government price controls, additional regulatory mandates and other measures designed to constrain medical costs. The PPACA significantly impacts the medical device industry. Among other things, the PPACA:
| • | imposes an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States (described in more detail below); |
| • | establishes a new Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; |
| • | implements payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models; and |
| • | creates an independent payment advisory board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate. |
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. On August 2, 2011, the President signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes reductions to Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
Our financial performance may be adversely affected by medical device tax provisions in the healthcare reform laws.
The PPACA imposes, among other things, an excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States, which began in 2013. Under these provisions, the Congressional Research Service predicts that the total cost to the medical device industry may be up to $20 billion over the next decade. The Internal Revenue Service issued final regulations implementing the tax in December 2012 which requires, among other things, bi-monthly payments and quarterly reporting. We are subject to this excise tax on our sales of certain medical devices we import into the United States. We anticipate that primarily all of our sales of medical devices in the United States will be subject to this 2.3% excise tax. During the year ended December 31, 2013, we recognized $0.7 million in tax expense associated with the medical device tax in the United States, which is included in general and administrative expenses.
41
Table of Contents
Risks Related to our Financial Results and Need for Financing
Our quarterly revenue and operating results are unpredictable and may fluctuate significantly from quarter to quarter due to factors outside our control, which could adversely affect our business, consolidated financial statements and the trading price of our common stock.
Our revenue and operating results may vary significantly from quarter to quarter and year to year due to a number of factors, many of which are outside of our control and any of which may cause our stock price to fluctuate. Our sales and results of operations will be affected by numerous factors, including:
| • | our ability to drive increased sales of our cervical technology products and our lumbar technology products; |
| • | our ability to establish and maintain an effective and dedicated sales organization, including independent sales agencies and distributors; |
| • | pricing pressure applicable to our products, including adverse third-party coverage and reimbursement outcomes; |
| • | results of clinical research and trials on our existing products and products in development; |
| • | the mix of our products sold because profit margins differ amongst our products; |
| • | timing of new product offerings, acquisitions, licenses or other significant events by us or our competitors; |
| • | the ability of our suppliers to timely provide us with an adequate supply of products; |
| • | the evolving product offerings of our competitors; |
| • | regulatory approvals and legislative changes affecting the products we may offer or those of our competitors; |
| • | interruption in the manufacturing or distribution of our products; |
| • | the effect of competing technological, industry and market developments; |
| • | changes in our ability to obtain regulatory clearance or approval for our products or to obtain or maintain our CE Certificates of Conformity for our products; and |
| • | our ability to expand the geographic reach of our sales and marketing efforts. |
Many of the products we may seek to develop and introduce in the future will require FDA approval or clearance before commercialization in the United States, and commercialization of such products outside of the United States would likely require additional regulatory approvals, CE Certificates of Conformity and import licenses. As a result, it will be difficult for us to forecast demand for these products with any degree of certainty. In addition, we will be increasing our operating expenses as we expand our commercial capabilities. Accordingly, we may experience significant, unanticipated quarterly losses. If our quarterly or annual operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly or annual fluctuations in our operating results may, in turn, cause the price of our common stock to fluctuate substantially. We believe that quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
We might require additional capital to support business growth, and this capital might not be available on terms favorable to us or at all.
We intend to continue to make investments to support our business growth and may require additional funds to respond to business challenges, including the need to develop new technologies or enhance our existing technologies and platform, upgrade our operating infrastructure and acquire complementary
42
Table of Contents
businesses and technologies. Accordingly, we may need to engage in equity or debt financings to secure additional funds. If we raise additional funds through further issuances of equity or convertible debt securities, our existing stockholders could suffer significant dilution, and any new equity securities we issue could have rights, preferences and privileges superior to those of holders of our common stock, including shares of common stock sold in this offering. Any debt financing obtained by us in the future would likely be senior to our common stock, would likely cause us to incur interest expenses, and could involve restrictive covenants relating to our capital raising activities and other financial and operational matters, which may increase our expenses and make it more difficult for us to obtain additional capital and to pursue business opportunities, including potential acquisitions. We may also be required to secure any such debt obligations with some or all of our assets. In addition, we may not be able to obtain additional financing on terms favorable to us, if at all. It is also possible that we may allocate significant amounts of capital toward solutions or technologies for which market demand is lower than anticipated and, as a result, abandon such efforts. If we are unable to obtain adequate financing or financing on terms satisfactory to us when we require it, or if we expend capital on projects that are not successful, our ability to continue to support our business growth and to respond to business challenges could be significantly limited, or we may even have to scale back our operations. Any of these negative developments could have a material adverse effect on our business, operating results, financial condition and common stock price.
Continuing worldwide economic instability, including challenges faced by the Eurozone countries, could adversely affect our revenues, financial condition or results of operations.
Since fiscal year 2008, the global economy has been impacted by the sequential effects of an ongoing global financial crisis. This global financial crisis, including the European sovereign debt crisis, has caused extreme disruption in the financial markets, including severely diminished liquidity and credit availability. Substantially all of our suppliers are located in Europe, and many of our distribution agreements outside of the United States are denominated in Euros. There can be no assurance that there will not be further deterioration in the global economy and in Europe. Our customers and suppliers may experience financial difficulties or be unable to borrow money to fund their operations, which may adversely impact their ability to purchase our products or to pay for our products on a timely basis, if at all. As with our customers and suppliers, these economic conditions make it more difficult for us to accurately forecast and plan our future business activities. In light of the current economic state of many countries outside the United States, we continue to monitor their creditworthiness. Failure to receive payment of all or a significant portion of these receivables could adversely affect our results of operations. Further, there are concerns for the overall stability and suitability of the Euro as a single currency, given the economic and political challenges facing individual Eurozone countries. Continuing deterioration in the creditworthiness of the Eurozone countries, the withdrawal of one or more member countries from the EU, or the failure of the Euro as a common European currency could adversely affect our revenues, financial condition or results of operations.
Our existing revolving credit facility contains restrictive covenants that may limit our operating flexibility.
Our existing revolving credit facility contains certain restrictive covenants that limit our ability to transfer or dispose of assets, merge with other companies or consummate certain changes of control, acquire other companies, pay dividends, incur additional indebtedness and liens, experience changes in management and enter into new businesses. We therefore may not be able to engage in any of the foregoing transactions unless we obtain the consent of the lender or terminate the revolving credit facility. There is no guarantee that we will be able to generate sufficient cash flow or sales to meet the financial covenants or pay the principal and interest on any such debt. Furthermore, there is no guarantee that future working capital, borrowings or equity financing will be available to repay or refinance any such debt.
43
Table of Contents
Risks Related to our Intellectual Property and Potential Litigation
If we are unable to protect our intellectual property rights, our competitive position could be harmed or we could be required to incur significant expenses to enforce our rights.
We rely primarily on patent, copyright, trademark and trade secret laws, know-how and continuing technological innovation, as well as confidentiality and non-disclosure agreements and other methods, to protect our proprietary technologies and know-how. However, these legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. As of December 31, 2013, we owned over 330 issued patents globally, of which 27 were issued U.S. patents. As of December 31, 2013, we owned over 80 patent applications pending globally, of which 28 were patent applications pending in the United States. As of December 31, 2013, 20 of our U.S. issued patents have pending continuations or divisionals which may provide additional intellectual property protection if those continuations and divisionals issue as U.S. patents. Our issued patents expire between 2020 and 2031, subject to payment of required maintenance fees, annuities and other charges. As of December 31, 2013, we also owned 15 U.S. trademark registrations and 59 foreign trademark registrations, as well as five pending U.S. trademark registrations and 14 pending foreign trademark registrations.
We have applied for patent protection relating to certain existing and proposed products and processes. While we generally apply for patents in those countries where we intend to make, have made, use or sell patented products, we may not accurately predict all of the countries where patent protection will ultimately be desirable. If we fail to timely file a patent application in any such country, we may be precluded from doing so at a later date. Furthermore, we cannot assure you that any of our patent applications will issue as patents. The rights granted to us under our patents, including prospective rights sought in our pending patent applications, may not be meaningful or provide us with any commercial advantage and they could be opposed, contested or circumvented by our competitors or be declared invalid or unenforceable in judicial or administrative proceedings. In addition, our pending patent applications include claims to material aspects of our products and procedures that are not currently protected by issued patents. The process of applying for patent protection itself is time consuming and expensive. The failure of our patents to protect our technology adequately might make it easier for our competitors to offer the same or similar products or technologies. Competitors may be able to design around our patents or develop products that provide outcomes that are comparable to ours without infringing on our intellectual property rights.
We have entered into confidentiality agreements and intellectual property assignment agreements with many of our officers, employees, consultants and advisors regarding our intellectual property and proprietary technology. In the event of unauthorized use or disclosure or other breaches of such agreements, we may not be provided with meaningful protection for our intellectual property and proprietary technology.
Due to differences between foreign and U.S. patent laws, our patented intellectual property rights may not receive the same degree of protection in foreign countries as they would in the United States. Even if patents are granted outside the United States, effective enforcement in those countries may not be available, and the scope of protection may vary significantly from country to country. We have filed patent applications only in the United States, some countries in Europe and less than a dozen other countries, and we therefore lack any patent protection in all other countries. In countries where we do not have significant patent protection, we may not be able to stop a competitor from marketing products in such countries that are the same as or similar to our products.
We rely on our trademarks, trade names and brand names to distinguish our products from the products of our competitors, and have registered or applied to register many of these trademarks. We cannot assure you that our trademark applications will be approved. Third parties may also oppose our
44
Table of Contents
trademark applications, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. Further, we cannot assure you that competitors will not infringe upon our trademarks, or that we will have adequate resources to enforce our trademarks.
If a competitor infringes one of our patents, trademarks or other intellectual property rights, enforcing those patents, trademarks and other rights may be difficult, time consuming or unsuccessful. Even if successful, litigation to defend our patents and trademarks against challenges or enforce our intellectual property rights could be expensive and time consuming and could divert management’s attention from managing our business. Litigation to defend our patents and trademarks against challenges or enforce our intellectual property rights could provoke significant retaliatory litigation, which could be costly, result in the diversion of management’s time and efforts, require us to pay damages and other amounts or prevent us from marketing our existing or future products. Moreover, we may not have sufficient resources or desire to defend our patents or trademarks against challenges or to enforce our intellectual property rights.
The medical device industry is characterized by patent litigation and we could become subject to litigation that could be costly, result in the diversion of management’s time and efforts, require us to pay damages or prevent us from marketing our existing or future products.
Our commercial success will depend in part on not infringing the patents or violating the other proprietary rights of others. Significant litigation regarding patent rights occurs in our industry. Our competitors in both the United States and abroad, many of which have substantially greater resources and have made substantial investments in patent portfolios and competing technologies, may have applied for or obtained or may in the future apply for and obtain, patents that will prevent, limit or otherwise interfere with our ability to make, use and sell our products. Generally, we do not conduct independent reviews of patents issued to third parties. In addition, patent applications in the United States and elsewhere can be pending for many years before issuance, so there may be applications of others now pending of which we are unaware that may later result in issued patents that will prevent, limit or otherwise interfere with our ability to make, use or sell our products. The large number of patents, the rapid rate of new patent applications and issuances, the complexities of the technology involved and the uncertainty of litigation increase the risk of business assets and management’s attention being diverted to patent litigation. We have received in the past, and expect to receive in the future, particularly as a public company, communications from various industry participants alleging our infringement of their patents, trade secrets or other intellectual property rights and/or offering licenses to such intellectual property. Any lawsuits resulting from such allegations could subject us to significant liability for damages and invalidate our proprietary rights. Any potential intellectual property litigation also could force us to do one or more of the following:
| • | stop making, selling or using products or technologies that allegedly infringe the asserted intellectual property; |
| • | lose the opportunity to license our technology to others or to collect royalty payments based upon successful protection and assertion of our intellectual property rights against others; |
| • | incur significant legal expenses; |
| • | pay substantial damages or royalties to the party whose intellectual property rights we may be found to be infringing; |
| • | pay the attorney fees and costs of litigation to the party whose intellectual property rights we may be found to be infringing; |
45
Table of Contents
| • | redesign those products that contain the allegedly infringing intellectual property, which could be costly, disruptive and/or infeasible; or |
| • | attempt to obtain a license to the relevant intellectual property from third parties, which may not be available on reasonable terms or at all. |
Any litigation or claim against us, even those without merit, may cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our core business and harm our reputation. Further, as the number of participants in the spine industry grows, the possibility of intellectual property infringement claims against us increases. If we are found to infringe the intellectual property rights of third parties, we could be required to pay substantial damages (which may be increased up to three times of awarded damages) and/or substantial royalties and could be prevented from selling our products unless we obtain a license or are able to redesign our products to avoid infringement. Any such license may not be available on reasonable terms, if at all, and there can be no assurance that we would be able to redesign our products in a way that would not infringe the intellectual property rights of others. If we fail to obtain any required licenses or make any necessary changes to our products or technologies, we may have to withdraw existing products from the market or may be unable to commercialize one or more of our products, all of which could have a material adverse effect on our business, results of operations and financial condition.
In addition, we generally indemnify our customers and international distributors with respect to infringement by our products of the proprietary rights of third parties. Third parties may assert infringement claims against our customers or distributors. These claims may require us to initiate or defend protracted and costly litigation on behalf of our customers or distributors, regardless of the merits of these claims. If any of these claims succeed, we may be forced to pay damages on behalf of our customers or distributors or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, our customers may be forced to stop using our products.
We may be subject to damages resulting from claims that we, our employees, our independent sales agencies or our international distributors have wrongfully used or disclosed alleged trade secrets of our competitors or are in breach of non-competition or non-solicitation agreements with our competitors.
Many of our employees were previously employed at other medical device companies, including our competitors or potential competitors, in some cases until recently. Many of our independent sales agencies and distributors sell, or in the past have sold, products of our competitors. We may be subject to claims that we, our employees or our independent sales agencies and distributors have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of these former employers or competitors. In addition, we have been and may in the future be subject to claims that we caused an employee to breach the terms of his or her non-competition or non-solicitation agreement. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. If our defense to those claims fails, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Any litigation or the threat thereof may adversely affect our ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize product candidates, which could have an adverse effect on our business, results of operations and financial condition.
If product liability lawsuits are brought against us, our business may be harmed, and we may be required to pay damages that exceed our insurance coverage.
Our business exposes us to potential product liability claims that are inherent in the testing, manufacture and sale of medical devices for spine surgery procedures. Spine surgery involves significant risk of serious complications, including bleeding, nerve injury, paralysis and even death. Furthermore, if spine surgeons
46
Table of Contents
are not sufficiently trained in the use of our products, they may misuse or ineffectively use our products, which may result in unsatisfactory patient outcomes or patient injury. We could become the subject of product liability lawsuits alleging that component failures, malfunctions, manufacturing flaws, design defects or inadequate disclosure of product-related risks or product-related information resulted in an unsafe condition or injury to patients.
We have had, and continue to have, a small number of product liability claims relating to our products, none of which either individually, or in the aggregate, have resulted, or do we believe will result, in a material negative impact on our business. In the future, we may be subject to additional product liability claims, some of which may have a negative impact on our business.
Regardless of the merit or eventual outcome, product liability claims may result in:
| • | decreased demand for our products; |
| • | injury to our reputation; |
| • | significant litigation costs; |
| • | substantial monetary awards to or costly settlements with patients; |
| • | product recalls; |
| • | loss of revenue; |
| • | the inability to commercialize new products or product candidates; and |
| • | diversion of management attention from pursuing our business strategy and may be costly to defend. |
Our existing product liability insurance coverage may be inadequate to protect us from any liabilities we might incur. If a product liability claim or series of claims is brought against us for uninsured liabilities or in excess of our insurance coverage, our business could suffer. Any product liability claim brought against us, with or without merit, could result in the increase of our product liability insurance rates or the inability to secure coverage in the future. In addition, a recall of some of our products, whether or not the result of a product liability claim, could result in significant costs and loss of customers.
In addition, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts or scope to protect us against losses. Any claims against us, regardless of their merit, could severely harm our financial condition, strain our management and other resources and adversely affect or eliminate the prospects for commercialization or sales of a product or product candidate that is the subject of any such claim.
We may be subject to various litigation claims and legal proceedings.
We, as well as certain of our officers and distributors, may be subject to other claims or lawsuits. These lawsuits may result in significant legal fees and expenses and could divert management’s time and other resources. If the claims contained in these lawsuits are successfully asserted against us, we could be liable for damages and be required to alter or cease certain of our business practices or product lines. Any of these outcomes could cause our business, financial performance and cash position to be negatively impacted.
Risks Related to Our International Operations
We are exposed to risks related to our international operations and failure to manage these risks may adversely affect our operating results and financial condition.
We sell our products globally and have a number of offices around the world. During the years ended December 31, 2011, 2012 and 2013, approximately 33%, 29% and 26% of our revenue, respectively,
47
Table of Contents
was attributable to our international customers. As of December 31, 2013, approximately 52% of our employees were located abroad. In addition, substantially all of our manufacturing is conducted in France and a substantial portion of our suppliers are located in Europe. The sale and shipment of our products across international borders, as well as the purchase of components and products from international sources, subjects us to extensive U.S., French and EEA and foreign governmental trade, import and export and customs regulations and laws. Compliance with these regulations and laws is costly and exposes us to penalties for non-compliance. We expect that our international activities will be dynamic over the foreseeable future as we continue to pursue opportunities in international markets. Therefore, we are subject to risks associated with having worldwide operations. These international operations will require significant management attention and financial resources.
International operations are subject to inherent risks and our future results could be adversely affected by a number of factors, including:
| • | requirements or preferences for domestic products or solutions, which could reduce demand for our products; |
| • | differing existing or future regulatory and certification requirements; |
| • | management communication and integration problems related to entering new markets with different languages, cultures and political systems; |
| • | greater difficulty in collecting accounts receivable and longer collection periods; |
| • | difficulties in enforcing contracts; |
| • | difficulties and costs of staffing and managing foreign operations; |
| • | the uncertainty of protection for intellectual property rights in some countries; |
| • | potentially adverse tax consequences, including regulatory requirements regarding our ability to repatriate profits to the United States; |
| • | tariffs and trade barriers, export regulations and other regulatory and contractual limitations on our ability to sell our solutions in certain foreign markets; and |
| • | political and economic instability and terrorism. |
Additionally, our international operations expose us to risks of fluctuations in foreign currency exchange rates. To date, a significant portion of our international sales have been denominated in Euros. In addition, a substantial portion of our manufacturing costs are also denominated in Euros. As a result, a decline in the value of the Euro against the U.S. dollar could have a material adverse effect on the gross margins and profitability of our international operations if it is not offset by a decline in manufacturing costs as a result of the decline in the Euro. In addition, sales to countries that do not utilize the Euro could decline as the cost of our products to our customers in those countries increases. In addition, because our financial statements are denominated in U.S. dollars, a decline in the Euro would negatively impact our overall revenue as reflected in our financial statements. To date, we have not used risk management techniques to hedge the risks associated with these fluctuations. Even if we were to implement hedging strategies, not every exposure can be hedged and, where hedges are put in place based on expected foreign currency exchange exposure, they are based on forecasts that may vary or that may later prove to have been inaccurate. As a result, fluctuations in foreign currency exchange rates or our failure to successfully hedge against these fluctuations could have a material adverse effect on our operating results and financial condition.
48
Table of Contents
Failure to comply with the United States Foreign Corrupt Practices Act, or the FCPA, and similar laws associated with our activities outside the United States could subject us to penalties and other adverse consequences.
We are subject to the FCPA and other anti-bribery legislation around the world. The FCPA generally prohibits covered entities and their intermediaries from engaging in bribery or making other prohibited payments, offers or promises to foreign officials for the purpose of obtaining or retaining business or other advantages. In addition, the FCPA imposes recordkeeping and internal controls requirements on publicly traded corporations and their foreign affiliates, which are intended to, among other things, prevent the diversion of corporate funds to the payment of bribes and other improper payments, and to prevent the establishment of “off books” slush funds from which such improper payments can be made. As a substantial portion of our revenue is, and we expect will continue to be, from jurisdictions outside of the United States, we face significant risks if we fail to comply with the FCPA and other laws that prohibit improper payments, offers or promises of payment to foreign governments and their officials and political parties by us and other business entities for the purpose of obtaining or retaining business or other advantages. In many foreign countries, particularly in countries with developing economies, some of which represent significant markets for us, it may be a local custom that businesses operating in such countries engage in business practices that are prohibited by the FCPA or other laws and regulations. Although we have implemented a company policy requiring our employees and consultants to comply with the FCPA and similar laws, such policy may not be effective at preventing all potential FCPA or other violations. Although our agreements with our international distributors clearly state our expectations for our distributors’ compliance with U.S. laws, including the FCPA, and provide us with various remedies upon any non-compliance, including the ability to terminate the agreement, we also cannot guarantee our distributors’ compliance with U.S. laws, including the FCPA. Therefore there can be no assurance that none of our employees and agents, or those companies to which we outsource certain of our business operations, have not and will not take actions that violate our policies or applicable laws, for which we may be ultimately held responsible. As a result of our focus on managing our growth, our development of infrastructure designed to identify FCPA matters and monitor compliance is at an early stage. Any violation of the FCPA and related policies could result in severe criminal or civil sanctions, which could have a material and adverse effect on our reputation, business, operating results and financial condition.
New regulations related to “conflict minerals” may force us to incur additional expenses, may make our supply chain more complex and may result in damage to our reputation with customers.
The Dodd-Frank Act requires the SEC to establish new disclosure and reporting requirements for those companies who use certain minerals and metals mined in the Democratic Republic of Congo and adjoining countries in their products, known as conflict minerals, whether or not these products are manufactured by third parties. The new rule could affect sourcing at competitive prices and availability in sufficient quantities of certain minerals used in the manufacture of our products, including tantalum. The number of suppliers who provide conflict-free minerals may be limited. In addition, there may be material costs associated with complying with the disclosure requirements, such as costs related to determining the source of certain minerals used in our products, as well as costs of possible changes to products, processes or sources of supply as a consequence of such verification activities. Since our supply chain is complex, we may not be able to verify the origins for these minerals used in our products sufficiently through the due diligence procedures that we implement, which may harm our reputation. In addition, we may encounter challenges to satisfy those customers who require that all of the components of our products be certified as conflict-free, which could place us at a competitive disadvantage if we are unable to do so.
49
Table of Contents
Risks Related to Our Common Stock
Our stock price may be volatile, and you may not be able to resell shares of our common stock at or above the price you paid.
Our stock price may be highly volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include those discussed in this “Risk Factors” section of this prospectus and others such as:
| • | a slowdown in the medical device industry or the general economy; |
| • | actual or anticipated quarterly or annual variations in our results of operations or those of our competitors; |
| • | changes in accounting principles or changes in interpretations of existing principles, which could affect our financial results; |
| • | actual or anticipated changes in our growth rate relative to our competitors; |
| • | changes in earnings estimates or recommendations by securities analysts; |
| • | fluctuations in the values of companies perceived by investors to be comparable to us; |
| • | announcements by us or our competitors of new products or services, significant contracts, commercial relationships, capital commitments or acquisitions; |
| • | competition from existing technologies and products or new technologies and products that may emerge; |
| • | the entry into, modification or termination of agreements with our sales representatives or distributors; |
| • | developments with respect to intellectual property rights; |
| • | sales, or the anticipation of sales, of our common stock by us, our insiders or our other stockholders, including upon the expiration of contractual lock-up agreements; |
| • | sales, or the anticipation of sales, by our employees following any purchase of shares pursuant to our equity incentive plans; |
| • | our ability to develop and market new and enhanced products on a timely basis; |
| • | successful adoption of our Mobi-C cervical disc replacement device in the United States and positive and negative coverage and reimbursement policies by insurance companies and other third-party payors; |
| • | our commencement of, or involvement in, litigation; |
| • | additions or departures of key management or technical personnel; and |
| • | changes in laws or governmental regulations applicable to us. |
In recent years, the stock markets generally have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors may significantly affect the market price of our common stock, regardless of our actual operating performance.
In addition, in the past, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Securities litigation brought against us following volatility in our stock price, regardless of the merit or ultimate results of such litigation, could result in substantial costs, which would hurt our financial condition and operating results and divert management’s attention and resources from our business.
50
Table of Contents
Securities analysts may not publish favorable research or reports about our business or may publish no information at all, which could cause our stock price or trading volume to decline.
The trading market for our common stock will be influenced to some extent by the research and reports that industry or financial analysts publish about us and our business. We do not control these analysts. As a newly public company, we may be slow to attract research coverage and the analysts who publish information about our common stock will have had relatively little experience with our company, which could affect their ability to accurately forecast our results and could make it more likely that we fail to meet their estimates. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us provide inaccurate or unfavorable research or issue an adverse opinion regarding our stock price, our stock price could decline. If one or more of these analysts cease coverage of our company or fail to publish reports covering us regularly, we could lose visibility in the market, which in turn could cause our stock price or trading volume to decline.
We are an “emerging growth company” and we cannot be certain if the reduced disclosure requirements applicable to “emerging growth companies” will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not “emerging growth companies.” In particular, while we are an “emerging growth company” (i) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (ii) we will be exempt from any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotations or a supplement to the auditor’s report on financial statements, (iii) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and (iv) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved. In addition, while we are an “emerging growth company” we will not be required to comply with any new financial accounting standard until such standard is generally applicable to private companies.
As a result, our financial statements may not be comparable to companies that are not “emerging growth companies” or elect not to avail themselves of this provision. We may remain an “emerging growth company” until as late as December 31, 2018 (the fiscal year-end following the fifth anniversary of the completion of our initial public offering), though we may cease to be an “emerging growth company” earlier under certain circumstances, including (i) if the market value of our common stock that is held by nonaffiliates exceeds $700 million as of any June 30, in which case we would cease to be an “emerging growth company” as of the following December 31, or (ii) if our gross revenue exceeds $1 billion in any fiscal year.
The exact implications of the JOBS Act are still subject to interpretations and guidance by the SEC and other regulatory agencies, and we cannot assure you that we will be able to take advantage of all of the benefits of the JOBS Act. In addition, investors may find our common stock less attractive if we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline and/or become more volatile.
Our management might apply the proceeds of this offering in ways that do not increase the value of your investment.
Our management will have broad discretion as to the use of the net proceeds of this offering and you will be relying on the judgment of our management regarding the application of these proceeds. We might
51
Table of Contents
apply the net proceeds of this offering in ways with which you do not agree, or in ways that do not yield a favorable return. If our management applies these proceeds in a manner that does not yield a significant return, if any, on our investment of these net proceeds, it would adversely affect the market price of our common stock. For more information on our management’s planned use of proceeds, please read “Use of Proceeds” elsewhere in this prospectus. Pending their use, we may also invest the net proceeds from this offering in a manner that does not produce income or that loses value.
A significant portion of our total outstanding shares of common stock is restricted from immediate resale but may be sold into the market in the near future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares of common stock intend to sell shares, could reduce the market price of our common stock. As of March 24, 2014 and assuming the sale of shares contemplated by this offering, entities affiliated with Austin Ventures, Keensight Capital, Telegraph Hill Partners and PTV Sciences and our founders, Christophe Lavigne, Hervé Dinville and Patrick Richard, beneficially owned, collectively, approximately % of our outstanding common stock. If one or more of them were to sell a substantial portion of the shares they hold, it could cause our stock price to decline. As of March 24, 2014 and assuming the sale of shares contemplated by this offering, we had million outstanding shares of common stock. Approximately million shares of common stock are subject to a 60-day contractual lock-up with the underwriters of this offering, which such 60-day contractual lock-up is set to expire on , 2014. The underwriters may, in their sole discretion and without notice, release all or any portion of the shares from these lock-up arrangements, and the lock-up agreements are subject to certain exceptions.
As of March 24, 2014, and assuming the sale of shares contemplated by this offering, holders of an aggregate of approximately million shares of our common stock have rights, subject to some conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders.
In addition, as of March 24, 2014, there were 2,072,331 shares subject to outstanding options granted under our equity incentive plans and 19,531 shares subject to outstanding options granted outside our existing equity incentive plans that will become eligible for sale in the public market to the extent permitted by any applicable vesting requirements, the lock-up agreements with the underwriters described above and Rules 144 and 701 under the Securities Act of 1933. We have also registered shares of common stock that may be issued pursuant to our 2013 Equity Incentive Plan and Amended and Restated 2013 Employee Stock Purchase Plan, which shares will become freely tradable in the public market upon issuance and once vested.
If our executive officers, directors and principal stockholders choose to act together, they will be able to exert significant influence over us and our significant corporate decisions and may act in a manner that advances their best interests and not necessarily those of other stockholders.
As of March 24, 2014 and assuming the sale of shares completed by this offering, our executive officers, directors, and beneficial owners of 5% or more of our outstanding common stock and their affiliates beneficially own approximately % of our outstanding common stock. As a result, these persons, acting together, will have the ability to significantly influence the outcome of all matters requiring stockholder approval, including the election and removal of directors and any merger, consolidation, or sale of all or substantially all of our assets, and they may act in a manner that advances their best interests and not necessarily those of other stockholders, by among other things:
| • | delaying, deferring or preventing a change in control of us; |
| • | entrenching our management and/or our board of directors; |
52
Table of Contents
| • | impeding a merger, consolidation, takeover or other business combination involving us; |
| • | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us; or |
| • | causing us to enter into transactions or agreements that are not in the best interests of all stockholders. |
We do not anticipate paying any cash dividends in the foreseeable future, and accordingly, stockholders must rely on stock appreciation for any return on their investment.
We do not anticipate declaring any cash dividends to holders of our common stock in the foreseeable future. In addition, our ability to pay cash dividends is currently prohibited by the terms of our existing loan agreement and may be prohibited by future loan agreements. Consequently, investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investment.
Anti-takeover provisions contained in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could impair a takeover attempt.
Our amended and restated certificate of incorporation and our amended and restated bylaws contain provisions that could delay or prevent a change in control of our company. These provisions could also make it more difficult for stockholders to elect directors and take other corporate actions. These provisions include:
| • | providing for a classified board of directors with staggered, three-year terms; |
| • | prohibiting cumulative voting in the election of directors; |
| • | providing that our directors may be removed only for cause; |
| • | authorizing the board of directors to issue, without stockholder approval, preferred stock with rights senior to those of our common stock; |
| • | authorizing the board of directors to change the authorized number of directors and to fill board vacancies; |
| • | requiring the approval of our board of directors and/or the holders of a supermajority of our outstanding shares of capital stock to amend our bylaws and certain provisions of our amended and restated certificate of incorporation; |
| • | requiring stockholders that seek to present proposals before, or to nominate candidates for election of directors at, a meeting of stockholders to provide advance written notice of such proposals or nominations; |
| • | prohibiting stockholder action by written consent; |
| • | limiting the liability of, and providing indemnification to, our directors and officers; and |
| • | prohibiting our stockholders from calling a special stockholder meeting. |
We are subject to Section 203 of the Delaware General Corporation Law, which, subject to some exceptions, prohibits “business combinations” between a Delaware corporation and an “interested stockholder,” which is generally defined as a stockholder who becomes a beneficial owner of 15% or more of a Delaware corporation’s voting stock, for a three-year period following the date that the stockholder became an interested stockholder.
These and other provisions in our amended and restated certificate of incorporation and our amended and restated bylaws discourage potential takeover attempts, reduce the price that investors might be
53
Table of Contents
willing to pay in the future for shares of our common stock and result in the market price of our common stock being lower than it would be without these provisions. Please read “Description of Capital Stock—Anti-Takeover Effects of Delaware Law and Certain Provisions of our Certificate of Incorporation and Amended and Restated Bylaws.”
Challenges to our tax structure by tax authorities may adversely affect our financial position.
We have and will continue to have subsidiaries organized under the laws of various jurisdictions. Such subsidiaries are and will be subject to the tax laws of such jurisdictions. If the tax authorities of any or all of these jurisdictions were to successfully challenge our tax position with respect to income tax treaties and transfer prices affecting transactions between our subsidiaries or between our subsidiaries and us, we could be subject to increased taxes and possibly penalties, which could have a material adverse effect on our financial position.
Each of our non-U.S. subsidiaries intends to structure its operations with the goal that each such entity would not be subject to U.S. corporate income tax on the basis that it is not engaged in a trade or business in the United States. Nevertheless, there is a risk that the U.S. Internal Revenue Service may successfully assert that one or more of such entities is engaged in a trade or business in the United States, in which case these entities would be subject to U.S. tax at regular corporate rates on income that is effectively connected with the conduct of a U.S. trade or business, plus an additional 30% (or a lower applicable treaty rate, if any) “branch profits” tax on the dividend equivalent amount, which is generally effectively connected income with certain adjustments, deemed withdrawn from the United States. Any such tax would result in an effective tax rate that is higher than anticipated.
54
Table of Contents
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements. The forward-looking statements are contained principally in the sections entitled “Prospectus Summary,” “Risk Factors,” “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and “Business.” All statements other than statements of historical facts contained in this prospectus, including statements regarding our future results of operations or financial condition, business strategy and plans and objectives of management for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “believe,” “will,” “may,” “estimate,” “continue,” “anticipate,” “intend,” “should,” “plan,” “expect,” “predict,” “could,” “potentially” or other similar expressions. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy and financial needs.
Forward-looking statements include, but are not limited to, statements about:
| • | our expectations regarding our revenue, expenses, sales, effective tax rates and operations; |
| • | our ability to manage growth; |
| • | anticipated trends, developments and challenges in our business and the markets in which we operate; |
| • | our ability to develop and market future products; |
| • | our ability to compete in our markets, expand into new markets and innovation by our competitors; |
| • | our ability to stay abreast of new or modified laws and regulations that currently apply or become applicable to our business; |
| • | our ability to establish and maintain intellectual property protection for our products or avoid claims of infringement; |
| • | our ability to hire and retain key personnel; |
| • | our expectations regarding the use of proceeds from this offering; |
| • | our anticipated cash needs and our estimates regarding our capital requirements; and |
| • | the future trading prices of our common stock and the impact of securities analysts’ reports on these prices. |
These statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results of operation, financial condition, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that the expectations reflected in the forward-looking statements contained in this prospectus are reasonable, we caution you that these statements are based on a combination of facts and factors currently known by us and our projections of the future, about which we cannot be certain. Moreover, we operate in a competitive and rapidly changing industry in which new risks may emerge from time to time, and it is not possible for management to predict all risks.
You should refer to the section of this prospectus entitled “Risk Factors” for a discussion of important risks that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors and new risks that may emerge in the future, we cannot assure you that the forward-looking statements in this prospectus will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. We do not undertake to update any of the forward-looking statements after the date of this prospectus, except to the extent required by law.
55
Table of Contents
This prospectus contains estimates, information and data concerning the spine industry, including estimates and forecasts of market sizes and growth rates, that are based on industry publications and reports, including those generated by GlobalData and iData Research, Inc., as described further below. Although we have not independently verified the data contained in these industry publications and reports, we believe based on our industry experience that these publications are reliable and that the conclusions they contain are reasonable. However, you should not give undue weight to the information contained in these reports because such information involves a number of assumptions and limitations and if any of these assumptions turn out to be incorrect, actual results may differ from the projections based on these assumptions. The markets described in these reports may not grow at the rates projected or at all, which could have a material adverse effect on our business and on the market price of our common stock.
The GlobalData report described herein, “Cervical Total Disc Replacement—Global Analysis and Market Forecasts”, January 2013, analyzes the global cervical disc replacement industry, including projected market and financial performance in major global markets, such as the United States, the European Union, Brazil, India and China, as well as a strengths, weaknesses, opportunities and threats for selected spine implant companies. The GlobalData report speaks as of its original publication date and not as of the date of this prospectus, and the opinions expressed in the GlobalData report are subject to change without notice.
The iData Research report described herein, “U.S. Market for Spinal Implants &VCF”, May 2013, analyzes specific spine industry segments in the United States and provides a trend analysis for such industry segments. The iData Research report speaks as of its original publication date and not as of the date of this prospectus, and the opinions expressed in the iData Research report are subject to change without notice.
The industry in which we operate is subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the “Risk Factors” section of this prospectus. These and other factors could cause results to differ materially from those expressed in the estimates made by the publishers of these industry publications and reports by us.
56
Table of Contents
We estimate that our net proceeds to us from the sale of shares of our common stock in this offering will be approximately $ million based on an assumed public offering price of $ per share, which was the closing price of our common stock as reported on the NASDAQ Global Select Market on , 2014, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Each $1.00 increase or decrease in the assumed public offering price would increase or decrease, as applicable, our net proceeds by approximately $ million, assuming that the number of shares offered by us, as listed on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase or decrease of 1.0 million in the number of shares we are offering would increase or decrease, as applicable, the net proceeds to us from this offering by approximately $ million, after deducting the estimated underwriting discounts and estimated offering expenses payable by us, assuming the assumed public offering price stays the same. We will not receive any of the proceeds from the sale of shares of common stock by the selling stockholders. If the underwriters exercise their option to purchase additional shares, there will be no change in total proceeds to us, as any additional shares would be sold by the selling stockholders.
We intend to use the net proceeds of this offering to (i) continue expanding our sales and marketing efforts in the U.S. and internationally, including our continued launch of Mobi-C in the U.S. market, and (ii) make continued investments in research and development. In addition, we may also use a portion of our net proceeds to acquire and invest in complementary products, technologies or businesses; however, we currently have no agreements or commitments to complete any such transaction and are not involved in negotiations to do so. We intend to use any remaining net proceeds from this offering for working capital general corporate purposes. The principal purposes of this offering are to facilitate an orderly distribution of shares by the selling stockholders and to increase the public float of our common stock.
Pending the uses mentioned above, we intend to invest the net proceeds of this offering in short-term, interest-bearing, investment-grade securities. Our management will have broad discretion in the application of the net proceeds to us from this offering, and investors will be relying on the judgment of our management regarding the application of the proceeds.
Our common stock has been listed on the NASDAQ Global Select Market under the symbol “LDRH” since October 9, 2013. Prior to that date, there was no public trading market for our common stock. Our common stock priced at $15.00 per share in our initial public offering on October 8, 2013. The following table sets forth for the periods indicated the high and low intra-day sale prices per share of our common stock as reported on the NASDAQ Global Select Market:
| High | Low | |||||||
| Fourth Quarter 2013 (from October 9, 2013) |
$ | 26.99 | $ | 17.79 | ||||
| First Quarter 2014 |
$ | 40.39 | $ | 23.34 | ||||
| Second Quarter 2014 (through , 2014) |
$ | $ | ||||||
On , 2014, the last reported sale price of our common stock on the NASDAQ Global Select Market was $ per share. As of March 24, 2014, we had 119 holders of record of our common stock. The actual number of holders of common stock is greater than these numbers of record holders and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and nominees. The number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
57
Table of Contents
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain any future earnings to fund the development and expansion of our business, and therefore we do not anticipate paying cash dividends on our common stock in the foreseeable future. Any future determination to pay dividends will be at the discretion of our board of directors and will depend on our results of operations, financial condition capital requirements and other factors deemed relevant by our board of directors. In addition, our credit agreement with Comerica Bank includes limitations on our ability to pay dividends.
58
Table of Contents
The following table sets forth our capitalization as of December 31, 2013:
| • | on an actual basis; and |
| • | on an as-adjusted basis to reflect the sale by us of shares of common stock in this offering at an assumed public offering price of $ per share, which was the closing price of our common stock as reported on the NASDAQ Global Select Market on , 2014, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
You should read the information below in conjunction with the financial statements and the related notes thereto and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus.
| As of December 31, 2013 | ||||||||
| Actual | As
Adjusted(1) |
|||||||
| (Unaudited) (in thousands) |
||||||||
| Long-term debt, net of discounts and current portion |
$ | 2,758 | $ | |||||
| Stockholders’ equity: |
||||||||
| Common stock, $0.001 par value; 107,000,000 shares authorized, 24,076,667 shares issued and outstanding, actual; 107,000,000 shares authorized, shares issued and outstanding, as adjusted |
24 | |||||||
| Preferred stock, $0.001 par value; 5,000,000 shares authorized, no shares issued and outstanding, actual and as adjusted |
— | — | ||||||
| Additional paid-in capital |
161,216 | |||||||
| Accumulated other comprehensive loss |
198 | |||||||
| Accumulated deficit |
(83,761 | ) | ||||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
77,677 | |||||||
|
|
|
|
|
|||||
| Total capitalization |
$ | 80,435 | $ | |||||
|
|
|
|
|
|||||
| (1) | Each $1.00 increase or decrease in the assumed public offering price would increase or decrease, as applicable, our cash and cash equivalents, additional paid-in capital, total stockholders’ equity and total capitalization by approximately $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase or decrease of 1.0 million in the number of shares we are offering would increase or decrease, as applicable, our cash and cash equivalents, additional paid-in capital, total stockholders’ equity and total capitalization by approximately $ million, after deducting the estimated underwriting discounts and estimated offering expenses payable by us, assuming the assumed public offering price stays the same. |
The table above excludes each of the following as of December 31, 2013:
| • | 1,572,582 shares of common stock issuable upon exercise of options outstanding at December 31, 2013, at a weighted-average exercise price of $6.17 per share; |
| • | 1,816,623 shares of common stock to be reserved for future issuance under our 2013 Equity Incentive Plan, including 963,066 shares of common stock that were automatically added to our 2013 Equity Incentive Plan as of January 1, 2014; and |
| • | 351,877 shares of common stock to be reserved for future issuance under our Amended and Restated 2013 Employee Stock Purchase Plan, including 240,766 shares of common stock that were automatically added to our Amended and Restated 2013 Employee Stock Purchase Plan as of January 1, 2014. |
59
Table of Contents
If you invest in our common stock in this offering, your interest will be diluted to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock after this offering. As of December 31, 2013, our historical net tangible book value was $68.0 million, or $2.82 per share. Our historical net tangible book value represents total tangible assets less total liabilities, all divided by the number of shares of common stock outstanding on December 31, 2013.
After giving effect to the sale of shares of common stock offered by us in this offering at an assumed public offering price of $ per share, the last reported sale price of our common stock on the NASDAQ Global Select Market on , 2014, and after deducting underwriting discounts and estimated offering expenses payable by us, our adjusted net tangible book value as of December 31, 2013 would have been $ million, or $ per share. This represents an immediate increase in net tangible book value of $ per share to existing stockholders and an immediate dilution of $ per share to new investors, or approximately % of the assumed public offering price of $ per share. The following table illustrates this dilution on a per share basis:
| Assumed public offering price per share |
$ | |||
| Historical net tangible book value per share as of December 31, 2013 |
2.82 | |||
| Increase in net tangible book value per share attributable to this offering |
||||
| As adjusted net tangible book value per share after giving effect to this offering |
||||
|
|
|
|||
| Dilution per share to new investors in this offering |
$ | |||
|
|
|
A $1.00 increase or decrease in the assumed public offering price, would increase or decrease, as applicable, our as adjusted net tangible book value per share after this offering by $ per share and the dilution in as adjusted net tangible book value to new investors by $ per share, assuming the number of shares offered by us, as set forth on the cover of this prospectus, remains the same and after deducting underwriting discounts and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase or decrease of 1.0 million in the number of shares we are offering would increase or decrease, as applicable, our as adjusted net tangible book value after this offering by approximately $ million, or $ per share, after deducting the estimated underwriting discounts and estimated offering expenses payable by us, assuming the assumed public offering price stays the same
The following table summarizes, on an as adjusted basis as of December 31, 2013 the differences between existing stockholders and new investors in this offering with respect to the number of shares of common stock purchased from us, the total consideration paid to us, and the average price per share paid. The calculation below is based on an assumed public offering price of $ per share, the last reported sale price of our common stock on the NASDAQ Global Select Market on , 2014, before deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
| Shares Purchased | Total Consideration | Average Price Per Share |
||||||||||||||||
| Number |
Percent | Amount | Percent | |||||||||||||||
| Existing stockholders |
% | $ | % | $ | ||||||||||||||
| New investors |
||||||||||||||||||
| Total |
100.0 | % | $ | 100.0 | % | $ | ||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||
60
Table of Contents
The foregoing tables and calculations exclude, as of December 31, 2013:
| • | 1,572,582 shares of common stock issuable upon exercise of options outstanding at December 31, 2013, at a weighted-average exercise price of $6.17 per share; |
| • | 1,816,623 shares of common stock reserved for future issuance under our 2013 Equity Incentive Plan, including 963,066 shares of common stock that were automatically added to our 2013 Equity Incentive Plan as of January 1, 2014; and |
| • | 351,877 shares of common stock reserved for future issuance under our Amended and Restated 2013 Employee Stock Purchase Plan, including 240,766 shares of common stock that were automatically added to our Amended and Restated 2013 Employee Stock Purchase Plan as of January 1, 2014. |
To the extent any of these options or warrants are exercised, there will be further dilution to new investors.
61
Table of Contents
SELECTED CONSOLIDATED FINANCIAL INFORMATION
The following tables present our selected consolidated financial and operating data for the periods indicated. The summary consolidated statement of comprehensive loss data for the years ended December 31, 2013, 2012 and 2011 and the summary consolidated balance sheet data as of December 31, 2013 and 2012 have been derived from our audited consolidated financial statements included elsewhere in this prospectus. The selected consolidated balance sheet data as of December 31, 2011 have been derived from our audited consolidated financial statements, which are not included in this prospectus. Our historical results are not necessarily indicative of the results that may be expected in the future. The summary financial information below should be read in conjunction with the information contained in “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” the consolidated financial statements and notes thereto, and other financial information included elsewhere in this prospectus.
| Year Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands, except per share data) | ||||||||||||
| Consolidated Statement of Comprehensive Loss Data: |
||||||||||||
| Revenue |
$ | 111,594 | $ | 90,918 | $ | 77,975 | ||||||
| Cost of goods sold |
17,947 | 14,770 | 12,564 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
93,647 | 76,148 | 65,411 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
9,380 | 10,751 | 9,040 | |||||||||
| Sales and marketing |
67,676 | 51,846 | 42,270 | |||||||||
| General and administrative |
18,915 | 14,825 | 11,121 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
95,971 | 77,422 | 62,431 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating income (loss) |
(2,324 | ) | (1,274 | ) | 2,980 | |||||||
| Other income (expenses), net |
(23,940 | ) | (7,248 | ) | (3,480 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes |
(26,264 | ) | (8,522 | ) | (500 | ) | ||||||
| Income tax expense |
(1,671 | ) | (1,179 | ) | (1,328 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (27,935 | ) | $ | (9,701 | ) | $ | (1,828 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per common share: |
||||||||||||
| Basic and diluted |
$ | (3.09 | ) | $ | (2.10 | ) | $ | (0.41 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average number of shares outstanding: |
||||||||||||
| Basic and diluted |
9,040,567 | 4,615,352 | 4,490,481 | |||||||||
|
|
|
|
|
|
|
|||||||
| Year Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
| Other Financial Data: |
||||||||||||
| Depreciation and amortization |
$ | 4,024 | $ | 3,133 | $ | 2,387 | ||||||
| Adjusted EBITDA(1) |
2,969 | 2,154 | 5,551 | |||||||||
62
Table of Contents
| As of December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||
| Cash and cash equivalents |
$ | 56,678 | $ | 19,135 | $ | 5,599 | ||||||
| Working capital |
55,376 | 33,959 | 17,120 | |||||||||
| Total assets |
127,993 | 81,591 | 57,890 | |||||||||
| Line of credit, net of discounts |
18,162 | 18,985 | 7,201 | |||||||||
| Long-term debt, net of discounts and current portion |
2,758 | 30,326 | 14,375 | |||||||||
| Total liabilities |
50,316 | 77,222 | 45,060 | |||||||||
| Total redeemable preferred stock |
— | 35,000 | 35,000 | |||||||||
| Total stockholders’ equity (deficit) |
77,677 | (30,631 | ) | (22,170 | ) | |||||||
| (1) | See footnote (1) to the table in the section of this prospectus titled “Summary Consolidated Financial Information” for a definition of Adjusted EBITDA and reconciliation to operating income (loss). |
63
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with the consolidated financial statements and the notes to those statements included in this prospectus. This discussion and analysis may contain forward-looking statements that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, such as those set forth under heading “Risk Factors,” and elsewhere in this prospectus.
Overview
We are a global medical device company focused on designing and commercializing novel and proprietary surgical technologies for the treatment of patients suffering from spine disorders. Our primary products are based on our VerteBRIDGE fusion and Mobi non-fusion platforms, both of which are designed for applications in the cervical and lumbar spine. We believe our VerteBRIDGE and Mobi platforms enable products that are less invasive, provide greater intra-operative flexibility, offer simplified surgical techniques and promote improved clinical outcomes for patients as compared to existing alternatives.
Our revenue is generated from sales to two types of customers: hospitals and stocking distributors. Revenue from sales to hospitals is recognized when we are notified the product has been used or implanted and a valid purchase order has been received. Product sales to hospitals are billed to and paid by the hospital as part of their normal payment processes with payment received by us in the form of an electronic transfer, check or credit card. Revenue from our stocking distributors is generally recognized at the time the product is shipped to the distributor. Product sales to stocking distributors are billed to and paid by the distributor as part of their normal payment processes with payment received by us in the form of an electronic transfer.
Our VerteBRIDGE and Mobi platform products are used in the fastest growing segments of the global spine implant market, including the lumbar fusion segment, the cervical fusion segment and the motion preservation segment. In August 2013, we received approval from the U.S. Food and Drug Administration, or FDA, for the Mobi-C cervical replacement device, the first and only cervical disc replacement device to receive FDA approval to treat both one-level and two-level cervical disc disease. We expect sales of Mobi-C in the United States to account for a significant portion of our expected increase in total revenue, primarily in 2014 and beyond. We expect that sales of Mobi-C in the United States will result in increases to our sales and marketing expenses as we increase our manufacturing and sales capabilities to handle the expected increase in demand for Mobi-C. In the event that demand for Mobi-C in the United States is not at the levels we anticipate, this would delay the anticipated increase in our total revenue and would cause us to defer related increases in sales and marketing expenses, although certain sales and marketing expenses related to training surgeons, seeking hospital approvals and pursuing third-party payor coverage and reimbursement would still be incurred. In addition, we anticipate that sales of our VerteBRIDGE products will continue to increase due to continued market penetration in key global markets.
Components of our Results of Operations
Revenue
We generate revenue from the sales of implants designed around our proprietary VerteBRIDGE fusion and Mobi non-fusion platform technologies. In addition, we market numerous traditional fusion implants so that combined we can offer to spine surgeons comprehensive spine surgical solutions. We sell our implants primarily to hospitals, for use by spine surgeons to treat spine disorders. We expect to
64
Table of Contents
increase revenue by establishing Mobi-C as a standard of care for cervical disc disease, leveraging our VerteBRIDGE platform and broadening our portfolio of products to further penetrate the fastest growing segments of the global spine implant market. We also expect to increase our revenue by expanding our geographic presence in the United States and other countries.
Cost of Goods Sold
We rely on third-party suppliers to manufacture our products. Our cost of goods sold primarily consist of costs of products purchased from our third-party suppliers, excess and obsolete inventory charges, royalties, shipping, inspection and related costs incurred in making our products available for sale or use. We expect our cost of goods sold to continue to increase in absolute dollars due primarily to increased sales volume.
Research and Development Expenses
Our research and development expenses primarily consist of engineering, product development, clinical and regulatory expenses, consulting services, outside prototyping services, outside research activities, materials, depreciation and other costs associated with development of our products. Research and development expenses also include related personnel and consultants’ compensation and stock-based compensation expense. We expense research and development costs as they are incurred. We expect research and development expense to continue to increase in absolute dollars as we develop new products to expand our product pipeline, add research and development personnel and undergo clinical activities, including clinical studies to gain additional regulatory clearances.
Sales and Marketing Expenses
Sales and marketing expenses primarily consist of salaries, benefits and other related costs, including stock-based compensation, for personnel employed in sales, marketing, medical education and training departments. In addition, our sales and marketing expenses include commissions and bonuses, generally based on a percentage of sales, to our sales managers, independent sales agencies and direct sales representatives. We provide our implants in kits that consist of a range of implant sizes and include a separate instrument set necessary to complete the surgical procedure. We generally consign our instrument sets to our sales organization or our hospital customers that purchase the implants used in spine surgery. Our sales and marketing expenses include depreciation of these instrument sets. We expect our sales and marketing expenses to continue to increase in absolute dollars with the commercialization of our current and pipeline products and continued investment in our global sales organization, including broadening our relationships with independent sales agencies, expanding exclusivity commitments among our independent sales agencies and international distributors and increasing the number of our direct sales representatives.
General and Administrative Expenses
General and administrative expenses primarily consist of salaries, benefits and other related costs, including stock-based compensation, for personnel employed in finance, legal, compliance, administrative, information technology and human resource departments. General and administrative expenses include facility costs and the 2.3% excise tax on the sale of medical devices in the United States that came into effect on January 1, 2013. General and administrative expenses also include legal expenses related to the development and protection of our intellectual property portfolio. We expect our general and administrative expenses to continue to increase in absolute dollars as we hire additional personnel to support the growth of our business and U.S. medical device excise taxes to increase as our sales volumes increase in the United States. Additionally, we expect to incur increased expenses as a result of being a public company.
65
Table of Contents
Income Tax Expense
We are taxed at the rates applicable within each jurisdiction in which we operate, primarily in the United States, France and Brazil. The composite income tax rate, tax provisions, deferred tax assets and deferred tax liabilities will vary according to the jurisdiction in which profits arise. Tax laws are complex and subject to different interpretations by management and the respective governmental taxing authorities, and require us to exercise judgment in determining our income tax provision, our deferred tax assets and liabilities and the valuation allowance recorded against our net deferred tax assets. Deferred tax assets and liabilities are determined using the enacted tax rates in effect for the years in which those tax assets are expected to be realized. A valuation allowance is established when it is more likely than not that the future realization of all or some of the deferred tax assets will not be achieved. We have recorded a full valuation allowance on our net deferred tax assets in the United States as of December 31, 2013 and 2012 due to uncertainties related to our ability to utilize our deferred tax assets in the foreseeable future.
Critical Accounting Policies and Estimates
The preparation of our consolidated financial statements requires us to make assumptions, estimates and judgments that affect the reported amounts of assets and liabilities, the disclosures of contingent assets and liabilities as of the date of the consolidated financial statements, and the reported amounts of sales and expenses during the reporting periods. Certain of our more critical accounting policies require the application of significant judgment by management in selecting the appropriate assumptions for calculating financial estimates. By their nature, these judgments are subject to an inherent degree of uncertainty. On an ongoing basis, we evaluate our judgments, including those related to inventories, recoverability of long-lived assets and the fair value of our common stock. We use historical experience and other assumptions as the basis for our judgments and making these estimates. Because future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Any changes in those estimates will be reflected in our consolidated financial statements as they occur. As an “emerging growth company,” we have elected to delay the adoption of new or revised accounting standards until those standards would otherwise apply to private companies. As a result, our financial statements may not be comparable to those of other public companies. While our significant accounting policies are more fully described in Note 2 to our consolidated financial statements, we believe that the following accounting policies and estimates are most critical to a full understanding and evaluation of our reported financial results. The critical accounting policies addressed below reflect our most significant judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
Our revenue is generated from sales to two types of customers: hospitals and stocking distributors. Sales to hospitals represent over 85% of our revenue. We utilize a network of direct sales representatives and independent sales agencies for sales in the United States and a combination of independent sales agencies and distributors for sales outside the United States. We recognize revenue when persuasive evidence of an arrangement exists, product delivery has occurred, pricing is fixed or determinable and collection is reasonably assured. We generate a significant portion of our revenue from inventory maintained at hospitals or with our direct sales representatives or independent sales agencies. For these products, we recognize revenue at the time we are notified the product has been used or implanted and a valid purchase order has been received. For all other transactions, we recognize revenue when title to the goods and risk of loss transfer to customers, provided there are no remaining performance obligations that will affect the customer’s final acceptance of the sale. We generally recognize revenue from sales to distributors at the time the product is shipped to the distributor. Distributors, who sell the products to their customers, take title to the products and assume all risks of ownership at time of shipment. Our distributors are obligated to pay within specified terms regardless of when, if ever, they sell the products.
66
Table of Contents
Our policy is to classify shipping and handling costs billed to customers as revenue and the related expenses as cost of goods sold. In general, our customers do not have any rights of return or exchange.
Accounts Receivable and Allowance for Doubtful Accounts
The majority of our accounts receivable is composed of amounts due from hospitals. Accounts receivable are carried at cost less an allowance for doubtful accounts. On a regular basis, we evaluate accounts receivable and estimate an allowance for doubtful accounts, as needed, based on various factors such as customers’ current credit conditions, length of time past due and the general economy as a whole. We write off receivables against the allowance when they are deemed uncollectible.
Excess and Obsolete Inventory
We state inventories at the lower of cost or market. We determine cost on a weighted average basis. The majority of our inventory is finished goods, because we primarily utilize third-party suppliers to source our products. We evaluate the carrying value of our inventories in relation to the estimated forecast of product demand, which takes into consideration the estimated life cycle of product releases. A significant decrease in demand could result in an increase in the amount of excess inventory quantities on hand. When quantities on hand exceed estimated sales forecasts, we record a reserve for excess inventories, which results in a corresponding charge to cost of goods sold. Charges incurred for excess and obsolete inventory were $0.6 million, $1.6 million and $0.6 million for the years ended December 31, 2013, 2012 and 2011, respectively.
The need to maintain substantial levels of inventory impacts the risk of inventory obsolescence. Many of our products come in kits, which feature implants in a variety of sizes so that the implant or device may be customized to the patient’s needs. In order to market our products effectively, we often must maintain and provide surgeons and hospitals with consignment implant sets, back-up products and products of different sizes. For each surgery, fewer than all of the components of the set are used, and therefore certain portions of the set may become obsolete before they can be used. Additionally, our industry is characterized by regular new product development that could result in an increase in the amount of obsolete inventory quantities on hand. For example, as we introduce new products or next-generation products, we may be required to take charges for excess and obsolete inventory that could have a significant impact on the value of our inventory or our operating results.
Goodwill and Intangible Assets
Goodwill represents the excess purchase price over the fair value of the net tangible and identifiable intangible assets acquired by us. Goodwill is tested for impairment at a minimum on an annual basis. Goodwill is tested for impairment at the reporting unit level by comparing the reporting unit’s carrying amount, to the fair value of the reporting unit. The fair values are estimated using an income and discounted cash flow approach. We completed our annual goodwill impairment test in the fourth quarter of 2013 and determined that there was no impairment as the fair value of the reporting unit to which goodwill is allocated was substantially in excess of the carrying value.
We were formed in 2006, principally as a financing vehicle associated with the reorganization of our two principal subsidiaries, LDR Spine USA, Inc. (Spine) and Médical. In connection with our formation, the stockholders of Spine and Médical agreed to contribute their ownership interests to us in exchange for our preferred stock and/or common stock. For accounting purposes, Médical was deemed to be the acquirer of Spine. The goodwill that was recorded represents the excess of the fair value of the capital that was exchanged in the transaction and the fair value of the identifiable assets and assumed liabilities of Spine that were acquired by Médical as of the date of the transaction. As of such date, we concluded that the goodwill should be assigned to the Spine reporting unit, as the U.S. operations were expected to benefit from the synergies of the business combination as contemplated in the transaction.
67
Table of Contents
The impairment evaluation related to goodwill requires the use of considerable management judgment to determine discounted future cash flows, including estimates and assumptions regarding the amount and timing of cash flows, cost of capital and growth rates. Cash flow assumptions used in the assessment are estimated using assumptions in our annual operating plan as well as our five-year strategic plan. Our annual operating plan and strategic plan contain revenue assumptions that are derived from existing technologies as well as future revenues attributed to in-process technologies and the associated launch, growth and decline assumptions normal for the life cycle of those technologies. In addition, management considers relevant market information, peer company data and historical financial information. We also considered our historical operating losses in assessing the risk related to our future cash flow estimates and attempted to reflect that risk in the development of our weighted average cost of capital.
Intangible assets with finite useful lives are amortized over the period of estimated benefit using the straight-line method and estimated useful lives ranging from three to ten years. Intangible assets are tested for impairment annually or whenever events or circumstances indicate that a carrying amount of an asset (asset group) may not be recoverable. If impairment is indicated, we measure the amount of the impairment loss as the amount by which the carrying amount exceeds the fair value of the asset. Fair value is generally determined using a discounted future cash flow analysis.
Long-Lived Assets
We evaluate the recoverability of the carrying amount of long-lived assets, which include property and equipment, whenever events or changes in circumstances indicate that the carrying amount of an asset may not be fully recoverable. We assess impairment when the undiscounted future cash flows from the use and eventual disposition of an asset are less than its carrying value. If impairment is indicated, we measure the amount of the impairment loss as the amount by which the carrying amount exceeds the fair value of the asset. We base our fair value methodology on quoted market prices, if available. If quoted market prices are not available, we estimate fair value based on prices of similar assets or other valuation techniques including present value techniques.
Our long-lived assets consist of instruments, which are handheld devices used by spine surgeons during surgical procedures to facilitate the implantation of our products. There are no contractual terms with respect to the usage of our instruments by our customers. Surgeons are under no contractual commitment to use our instruments. We maintain ownership of these instruments and, when requested, we allow the surgeons to use the instruments to facilitate implantation of our related products. We do not currently charge for the use of our instruments and there are no minimum purchase commitments of our products. As our surgical instrumentation is used numerous times over several years, often by many different customers, instruments are recognized as long-lived assets once they have been placed in service. Instruments, and instrument parts, that have not been placed in service are carried at cost, net of allowances for excess and obsolete instruments, and are included as surgical instruments within property and equipment, net on the consolidated balance sheets. Once placed in service, instruments are carried at cost, less accumulated depreciation. Depreciation is computed using the straight-line method based on average estimated useful lives. Estimated useful lives are determined principally in reference to associated product life cycles, and average five years. As instruments are used as tools to assist surgeons, depreciation of instruments is recognized as a sales and marketing expense. Instrument depreciation expense was $2.6 million, $2.1 million and $1.6 million for the years ended December 31, 2013, 2012 and 2011, respectively.
We review instruments for impairment whenever events or changes in circumstances indicate that the carrying value of the assets may not be recoverable. An impairment loss would be recognized when estimated future undiscounted cash flows relating to the assets are less than the asset’s carrying amount. An impairment loss is measured as the amount by which the carrying amount of an asset exceeds its fair value.
68
Table of Contents
Income Taxes
We recognize deferred tax assets and liabilities for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. We measure deferred tax assets and liabilities using enacted tax rates expected to apply to taxable income in the year in which such items are expected to be received or settled. We recognize the effect on deferred tax assets and liabilities of a change in tax rates in the period that includes the enactment date.
We establish a valuation allowance to offset any deferred tax assets if, based upon available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
While we believe that our tax positions are fully supportable, there is a risk that certain positions could be challenged successfully. In these instances, we look to establish reserves. If we determine that a tax position is more likely than not of being sustained upon audit, based solely on the technical merits of the position, we recognize the benefit. We measure the benefit by determining the amount that has likelihood greater than 50% of being realized upon settlement. We presume that all tax positions will be examined by a taxing authority with full knowledge of all relevant information. We regularly monitor our tax positions, tax assets and tax liabilities. We reevaluate the technical merits of our tax positions and recognize an uncertain tax benefit or reverse a previously recorded tax benefit when (i) a tax audit is completed, (ii) applicable tax law, including a tax case or legislative guidance, changes or (iii) the statute of limitations expires. Significant judgment is required in accounting for tax reserves.
Stock-Based Compensation
We apply the fair value recognition provisions of Financial Accounting Standards Board Accounting Standards Codification Topic 718, Compensation-Stock Compensation, which we refer to as ASC 718. Determining the amount of stock-based compensation to be recorded requires us to develop estimates of the fair value of stock options as of their grant date. Stock-based compensation expense is recognized ratably over the requisite service period, which in most cases is the vesting period of the award. Calculating the fair value of stock-based awards requires that we make highly subjective assumptions. We use the Black-Scholes option pricing model to value our stock option awards. Use of this valuation methodology requires that we make assumptions as to the volatility of our common stock, the expected term of our stock options, the risk free rate of return for a period that approximates the expected term of our stock options and our expected dividend yield. Because we are a newly public company with a limited operating history, we utilize the historical stock price volatility from a representative group of public companies to estimate expected stock price volatility. We selected companies from the medical device industry, specifically those who are focused on the design, development and commercialization of products for the treatment of spine disorders, and who have similar characteristics to us, such as stage of life cycle and size. We intend to continue to utilize the historical volatility of the same or similar public companies to estimate expected volatility until a sufficient amount of historical information regarding the price of our publically traded stock becomes available. We use the simplified method as prescribed by the Securities and Exchange Commission Staff Accounting Bulletin No. 107, Share-based Payment, to calculate the expected term of stock option grants to employees as we do not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term of stock options granted to employees. We utilize a dividend yield of zero because we have never paid cash dividends and have no current intention to pay cash dividends. The risk-free rate of return used for each grant is based on the U.S. Treasury yield curve in effect at the time of grant for instruments with a similar expected life.
69
Table of Contents
We estimated the fair value of options granted using a Black-Scholes option pricing model with the following assumptions:
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Expected dividend yield |
— | % | — | % | — | % | ||||||
| Volatility |
52.02 | 54.77 | 70.19 | |||||||||
| Risk-free rate of return |
0.17 | 0.44 | 0.34 | |||||||||
| Forfeiture rate |
7.90 | 4.30 | 4.31 | |||||||||
| Expected life |
6 years | 6 years | 6 years | |||||||||
The estimated fair value of stock-based awards for employee and non-employee director services are expensed over the requisite service period. Option awards issued to non-employees, excluding non-employee directors, are recorded at their fair value as determined in accordance with authoritative guidance, are periodically revalued as the options vest and are recognized as expense over the related service period. As a result, the charge to operations for non-employee awards with vesting conditions is affected each reporting period by changes in the fair value of our common stock.
Stock-based compensation expense related to employee stock options and ESPP awards totaled $1.3 million, $0.3 million and $0.2 million for the years ended December 31, 2013, 2012 and 2011, respectively. As of December 31, 2013, we had $2.8 million of total unrecognized stock-based compensation expense, which we expect to recognize over a weighted-average remaining vesting period of approximately 4 years. While our stock-based compensation for stock options granted to employees to date has not been material to our financial results, we expect the impact to grow in future periods due to the potential increases in the value of our common stock and headcount.
Under ASC 718, we are required to estimate the level of forfeitures expected to occur and record stock-based compensation expense only for those awards that we ultimately expect will vest. We estimate our forfeiture rate based on the type of award, employee class and historical experience. Through December 31, 2013, actual forfeitures have not been material.
Results of Operations
The following table sets forth, for the periods indicated, our results of operations and our results of operations as a percentage of revenue:
| Years Ended December 31, | ||||||||||||||||||||||||
| 2013 | 2012 | 2011 | ||||||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||
| Revenue |
$ | 111,594 | 100 | % | $ | 90,918 | 100 | % | $ | 77,975 | 100 | % | ||||||||||||
| Cost of goods sold |
17,947 | 16 | 14,770 | 16 | 12,564 | 16 | ||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Gross profit |
93,647 | 84 | 76,148 | 84 | 65,411 | 84 | ||||||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| Research and development |
9,380 | 8 | 10,751 | 12 | 9,040 | 12 | ||||||||||||||||||
| Sales and marketing |
67,676 | 61 | 51,846 | 57 | 42,270 | 54 | ||||||||||||||||||
| General and administrative |
18,915 | 17 | 14,825 | 16 | 11,121 | 14 | ||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Total operating expenses |
95,971 | 86 | 77,422 | 85 | 62,431 | 80 | ||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Operating income (loss) |
(2,324 | ) | (2 | ) | (1,274 | ) | (1 | ) | 2,980 | 4 | ||||||||||||||
| Total other income (expense), net |
(23,940 | ) | (21 | ) | (7,248 | ) | (8 | ) | (3,480 | ) | (4 | ) | ||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Loss before income taxes |
(26,264 | ) | (24 | ) | (8,522 | ) | (9 | ) | (500 | ) | (1 | ) | ||||||||||||
| Income tax expense |
(1,671 | ) | (1 | ) | (1,179 | ) | (1 | ) | (1,328 | ) | (2 | ) | ||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
| Net loss |
$ | (27,935 | ) | (25 | )% | $ | (9,701 | ) | (11 | )% | $ | (1,828 | ) | (2 | )% | |||||||||
|
|
|
|
|
|
|
|||||||||||||||||||
70
Table of Contents
Year Ended December 31, 2013 Compared to the Year Ended December 31, 2012
Revenue
The following table sets forth, for the periods indicated, our revenue by product category and geography expressed as dollar amounts and the changes in revenue between the specified periods expressed in dollar amounts and as percentages:
| Years Ended December 31, |
Change 2012/2013 | |||||||||||||||
| 2013 | 2012 | $ | % | |||||||||||||
| (in thousands) | ||||||||||||||||
| Exclusive technology products |
$ | 92,818 | $ | 72,805 | $ | 20,013 | 27 | % | ||||||||
| Traditional fusion products |
18,776 | 18,113 | 663 | 4 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenue |
$ | 111,594 | $ | 90,918 | $ | 20,676 | 23 | % | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Years Ended December 31, |
Change 2012/2013 | |||||||||||||||
| 2013 | 2012 | $ | % | |||||||||||||
| (in thousands) | ||||||||||||||||
| United States |
$ | 82,307 | $ | 64,801 | $ | 17,506 | 27 | % | ||||||||
| International |
29,287 | 26,117 | 3,170 | 12 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenue |
$ | 111,594 | $ | 90,918 | $ | 20,676 | 23 | % | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The increase in total revenue in the year ended December 31, 2013 compared to the year ended December 31, 2012 was primarily driven by an increase in revenue from our exclusive technology products in the United States, including sales of our Avenue L product, which was launched in September 2012, and increased penetration in existing markets through the expansion of our network of independent sales agencies and direct sales representatives. Revenue growth from our exclusive technology products was driven by a 22% increase in revenue from our exclusive cervical products and by a 39% increase in revenue from our exclusive lumbar products. Sales of our exclusive cervical and lumbar products represented 64% and 36% of total exclusive technology revenue, respectively, for the year ended December 31, 2013.
International revenue increased as a result of increased market penetration in existing territories as well as expansion into one new sales territory.
Cost of Goods Sold
Cost of goods sold was $17.9 million in the year ended December 31, 2013 compared to $14.8 million in the year ended December 31, 2012, an increase of $3.2 million or 22%. The increase was primarily due to an increase in sales volume.
Research and Development Expenses
Research and development expenses were $9.4 million in the year ended December 31, 2013 compared to $10.8 million in the year ended December 31, 2012, a decrease of $1.4 million or 13%. The decrease was primarily due to a $2.2 million reduction in costs associated with our FDA pivotal clinical trial for Mobi-C and a $0.4 million increase in research and development tax credits. The decrease was partially offset by our continued investment in research and development, including an increase of $0.9 million associated with salaries and benefits of additional research and development personnel.
71
Table of Contents
Sales and Marketing Expenses
Sales and marketing expenses were $67.7 million in the year ended December 31, 2013 compared to $51.8 million in the year ended December 31, 2012, an increase of $15.8 million or 31%. The increase was primarily due to a $6.7 million increase in commissions as a result of the increase in sales volume, a $4.8 million increase in compensation costs associated with our investments in our sales organization, including hiring of additional direct sales management and marketing personnel, a $2.4 million increase in medical training workshops and product launch initiatives, a $0.7 million increase in depreciation expense associated with the increase in the deployment of instruments sets and cases and a $0.7 million increase in other sales and marketing related expenses, including travel and entertainment and other costs.
General and Administrative Expenses
General and administrative expenses were $18.9 million in the year ended December 31, 2013 compared to $14.8 million in the year ended December 31, 2012, an increase of $4.1 million or 28%. The increase was primarily due to a $3.1 million increase in employee salaries and benefits associated with the increase in general and administrative personnel, a $0.7 million increase in tax associated with the medical device excise tax in the United States and a $1.0 million increase in rent expense and depreciation expense associated with our office expansion. The increase was partially offset by a $0.6 million decrease in product liability insurance associated with our new policy that went into effect on January 1, 2013.
Total Other Income (Expense), net
Total other income (expense), net of $(23.9) million in the year ended December 31, 2013 primarily consisted of $7.4 million in expense related to the beneficial conversion feature of our promissory notes, $6.9 million of accretion related to warrants and discounts on long-term debt, $3.3 million in interest expense associated with our long-term debt, a $5.6 million increase in the fair value of our warrant liability and a $0.8 million loss due to the effect of changes in foreign exchange rates on payables and receivables held in currencies other than their functional (local) currency. Total other income (expense), net of $(7.2) million in the year ended December 31, 2012 primarily consisted of $3.2 million in interest expense associated with our long-term debt, a $1.6 million increase in the fair value of our warrant liability, $1.4 million of accretion related to warrants and discounts on long-term debt and a $1.0 million loss due to the effect of changes in foreign exchange rates on payables and receivables held in currencies other than their functional (local) currency.
Income Tax Expense
Income tax expense was $1.7 million in the year ended December 31, 2013 compared to $1.2 million in the year ended December 31, 2012. Our effective tax rate calculated as a percentage of income before income taxes was (6.4)% for the year ended December 31, 2013 and (13.8)% for the year ended December 31, 2012. The change in the effective tax rate was a result of the increase in our overall loss position.
72
Table of Contents
Year Ended December 31, 2012 Compared to the Year Ended December 31, 2011
Revenue
The following table sets forth, for the periods indicated, our revenue by product category and geography expressed as dollar amounts and the changes in revenue between the specified periods expressed in dollar amounts and as percentages:
| Years Ended December 31, |
Change 2011/2012 | |||||||||||||||
| 2012 | 2011 | $ | % Change | |||||||||||||
| (in thousands) | ||||||||||||||||
| Exclusive technology products |
$ | 72,805 | $ | 57,450 | $ | 15,355 | 27 | % | ||||||||
| Traditional fusion products |
18,113 | 20,525 | (2,412 | ) | 12 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenue |
$ | 90,918 | $ | 77,975 | $ | 12,943 | 17 | % | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Years Ended December 31, |
Change 2011/2012 | |||||||||||||||
| 2012 | 2011 | $ | % Change | |||||||||||||
| (in thousands) | ||||||||||||||||
| United States |
$ | 64,801 | $ | 52,062 | $ | 12,739 | 24 | % | ||||||||
| International |
26,117 | 25,913 | 204 | 1 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenue |
$ | 90,918 | $ | 77,975 | $ | 12,943 | 17 | % | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The increase in total revenue from 2011 to 2012 was primarily driven by an increase in revenue from our exclusive technology products in the United States. Revenue growth in the United States was primarily due to the increase in sales from our exclusive technology products, including sales of our Avenue L product, which was launched in September 2012, and increased penetration in existing markets through the expansion of our network of direct sales representatives, independent sales agencies and international distributors. Revenue growth from our exclusive technology products was driven by a 25% increase in revenue from our exclusive cervical products and by a 32% increase in revenue from our exclusive lumbar products. Sales of our exclusive cervical and lumbar products represented 67% and 33% of our total exclusive technology revenue, respectively, in 2012.
Cost of Goods Sold
Cost of goods sold was $14.8 million in 2012 compared to $12.6 million in 2011, an increase of $2.2 million or 18%. The increase was primarily due to a $1.2 million increase attributable to increased sales volume with the remaining $1.0 million increase attributable to an increase in inventory reserves and other costs.
Research and Development Expenses
Research and development expenses were $10.8 million in 2012 compared to $9.0 million in 2011, an increase of $1.7 million or 19%. The increase was primarily due to a $1.2 million increase in employee salaries and benefits and travel expenses associated with the increase in research and development personnel, a $0.2 million reduction in research and development tax credits and a $0.2 million increase in professional fees associated with our quality system software implementation and surgeon consulting fees associated with product development.
73
Table of Contents
Sales and Marketing Expenses
Sales and marketing expenses were $51.8 million in 2012 compared to $42.3 million in 2011, an increase of $9.6 million or 23%. The increase was primarily due to a $4.6 million increase in compensation costs associated with our investment in our sales organization, including hiring of additional direct sales representatives and marketing personnel, a $3.5 million increase in commissions as a result of the increase in sales volume, a $1.2 million increase in other sales and marketing related expenses, including travel and entertainment and other costs, a $0.6 million increase in medical training workshops and product launch initiatives and a $0.4 million increase in depreciation expense associated with the increase in the deployment of instruments sets and cases. The increase was partially offset by a $0.7 million decrease in professional fees mainly associated with product registrations in new territories.
General and Administrative Expenses
General and administrative expenses were $14.8 million in 2012 compared to $11.1 million in 2011, an increase of $3.7 million or 33%. The increase was primarily due to a $1.1 million increase in employee salaries and benefits associated with the increase in general and administrative personnel, a $0.8 million increase in rent expense associated with our new corporate headquarters, a $0.8 million increase in outside accounting, consulting and other professional fees, a $0.6 million increase in bad debt expense and product liability insurance associated with the increase in sales volume and a $0.4 million increase in depreciation expense and software maintenance fees.
Total Other Income (Expense), net
Total other income (expense), net of $(7.2) million in 2012 primarily consisted of $4.6 million in interest expense associated with our long-term debt, a $1.6 million increase in the fair value of our warrant liability and a $1.0 million loss due to the effect of changes in foreign exchange rates on payables and receivables held in currencies other than their functional (local) currency. Total other income (expense), net of $(3.5) million in 2011 primarily consisted of $2.6 million in interest expense associated with our long-term debt, a $0.7 million increase in the fair value of our warrant liability and a $0.3 million loss due to the effect of changes in foreign exchange rates on payables and receivables held in currencies other than their functional (local) currency.
Income Tax Expense
Income tax expense was $1.2 million in 2012 compared to $1.3 million in 2011. Our effective tax rate calculated as a percentage of income before income taxes was (13.8)% for 2012 and (265.6)% for 2011. The change in the effective tax rate was due to relatively consistent tax expense in the foreign jurisdictions where we are profitable and our inability to record a deferred tax benefit for pre-tax losses in the United States as a result of our full valuation allowance against net operating losses.
Liquidity and Capital Resources
Since our inception in 2000, we have incurred significant losses, and as of December 31, 2013, we had an accumulated deficit of $83.8 million. We have not yet achieved profitability, and anticipate that we will continue to incur losses in the near term. We expect that research and development, sales and marketing and general and administrative expenses will continue to grow, and, as a result, we will need to generate significant revenues to achieve profitability. To date, we have funded our operations primarily with proceeds from our initial public offering of common stock, the sale of preferred stock, various credit facilities, a convertible debt issuance and cash flow from operations. As of December 31, 2013, we had outstanding indebtedness of $25.3 million, working capital of $55.4 million and cash and cash equivalents of $56.7 million, including $6.1 million of cash and cash equivalents held by our non-U.S. subsidiaries. If these funds are repatriated to the United States or used for U.S. operations, they may be subject to U.S. tax. Outstanding indebtedness as of December 31, 2013 includes an $18.2 million
74
Table of Contents
outstanding balance on our line of credit with Comerica Bank which expires in April 2014. Negotiations with Comerica Bank and other financial institutions regarding the refinancing of our credit facility are on-going.
In October 2013, we completed our initial public offering which generated net proceeds of approximately $77.5 million, after deducting underwriting discounts and expenses of approximately $8.7 million. With the proceeds of our initial public offering, we (i) paid approximately $10.0 million under our loan facility with Escalate Capital Partners, (ii) paid approximately $17.1 million to satisfy obligations to the holders of our Series C preferred stock under the Series C Voting Agreement in which such holders agreed to vote all of their shares of preferred stock in favor of the conversion of preferred stock to common stock in connection with our initial public offering, and (iii) repaid approximately $2.8 million for the portion of our convertible notes that were not converted into shares of our common stock upon the consummation of our initial public offering.
We believe that our existing cash and cash equivalents will be sufficient to fund our operations and satisfy our current cash requirements for at least the next 12 months. From time to time, we may explore additional financing sources to meet our working capital requirements, make continued investment in research and development and make capital expenditure needed for us to maintain and expand our business. We may not be able to obtain additional financing on terms favorable to us, if at all. It is also possible that we may allocate significant amounts of capital toward solutions or technologies for which market demand is lower than anticipated and, as a result, abandon such efforts. If we are unable to obtain adequate financing or financing on terms satisfactory to us when we require it, or if we expend capital on projects that are not successful, our ability to continue to support our business growth and to respond to business challenges could be significantly limited, or we may even have to scale back our operations. If we raise additional funds through further issuances of equity or convertible debt securities, our existing stockholders could suffer significant dilution, and any new equity securities we issue could have rights, preferences and privileges superior to those of holders of our common stock.
The following table summarizes, for the periods indicated, cash flows from operating, investing and financing activities:
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (in thousands) | ||||||||||||
| Net cash used in operating activities |
$ | (5,770 | ) | $ | (4,363 | ) | $ | (2,693 | ) | |||
| Net cash used in investing activities |
(4,689 | ) | (5,870 | ) | (4,663 | ) | ||||||
| Net cash provided by financing activities |
47,771 | 23,868 | 5,387 | |||||||||
| Effect of exchange rate on cash |
231 | (99 | ) | (70 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net change in cash and cash equivalents |
$ | 37,543 | $ | 13,536 | $ | (2,039 | ) | |||||
|
|
|
|
|
|
|
|||||||
Cash Used in Operating Activities
Net cash used in operating activities was $5.8 million in the year ended December 31, 2013, compared to $4.4 million in the year ended December 31, 2012 an increase of $1.4 million. The increase in net cash used in operating activities was primarily attributable to an $18.2 million increase in net loss, a $3.2 million increase in accounts receivable and a $2.8 million increase in prepaid expenses and other current assets. The increase was partially offset by a $10.4 million net increase in non-cash charges, which includes accretion related to warrants and discounts on long-term debt, depreciation of surgical instruments and cases, provision for excess and obsolete inventories, non-cash interest expense and changes in fair value of common stock warrants. The increase in net cash used in operating activities was further offset by a $4.3 million decrease in the change in inventory and an $1.0 million decrease in the change in restricted cash.
75
Table of Contents
Net cash used in operating activities was $4.4 million in 2012 compared to $2.7 million in 2011, an increase of $1.7 million. The increase in net cash used in operating activities was primarily attributable to a $7.9 million increase in net loss and a $2.5 million increase in the change in inventory. The increase was partially offset by a $5.1 million net increase in non-cash charges, which includes depreciation of surgical instruments and cases, provision for excess and obsolete inventories, non-cash interest expense and changes in fair value of common stock warrants, a $2.6 million increase in the change in accounts payable and accrued expenses and a $1.3 million decrease in the change in prepaid and other current assets.
Cash Used in Investing Activities
Net cash used in investing activities was $4.7 million in the year ended December 31, 2013 compared to $5.9 million in the year ended December 31, 2012, a decrease of $1.2 million. The decrease in net cash used in investing activities was primarily attributable to a $1.3 million decrease in purchases of instruments and cases during the year ended December 31, 2013.
Net cash used in investing activities was $5.9 million in 2012 compared to $4.7 million in 2011, an increase of $1.2 million. The increase in net cash used in investing activities was primarily attributable to a $1.1 million increase in purchases of instruments and cases to support our increased volume of sales and the launch of new products.
Cash Provided by Financing Activities
Net cash provided by financing activities was $47.8 million in the year ended December 31, 2013 compared to $23.9 million in the year ended December 31, 2012, an increase of $23.9 million. The increase was primarily attributable to the $86.2 million raised during our initial public offering in October 2013, offset by a $16.3 million decrease in proceeds from long-term debt, an $11.4 million decrease in borrowings under our line of credit, increase in payments of $9.1 million on long-term debt, payments of $17.1 million made as dividends to holders of Series C Preferred Stock and $8.7 million in stock issuance costs associated with our initial public offering.
Net cash provided by financing activities was $23.9 million in 2012 compared to $5.4 million in 2011, an increase of $18.5 million. The increase was primarily attributable to a $10.5 million increase in proceeds from long-term debt and a $9.8 million increase in borrowings under our line of credit. The increase was partially offset by an increase in payments on long-term debt of $3.0 million.
Contractual Obligations and Commitments
The following table summarizes our outstanding contractual obligations as of December 31, 2013.
| Total | Payments Due by Period | |||||||||||||||||||
| Less than 1 year |
1-3 Years | 4-5 Years | After 5 Years |
|||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Long term debt obligations(1) |
$ | 7,162 | $ | 4,404 | $ | 1,903 | $ | 758 | $ | 97 | ||||||||||
| Operating lease obligations |
14,564 | 3,066 | 5,069 | 4,399 | 2,030 | |||||||||||||||
| Purchase obligations(2) |
13,599 | 7,411 | 6,188 | — | — | |||||||||||||||
| (1) | Long-term debt obligations consisted of outstanding principal and exclude scheduled interest payments of $361 as follows: $146 in less than 1 year, $154 in 1-3 years, $56 in 4-5 years and $5 after 5 years. |
| (2) | Reflects minimum annual volume commitments to purchase inventory under certain of our supplier contracts. |
76
Table of Contents
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Seasonality
Our business is generally not seasonal in nature. However, our sales may be influenced by summer vacation and timing of holiday periods during which we have experienced a reduction in spine surgeries taking place. In addition, product registrations in certain countries and orders associated therewith may result in seasonality not typical of our ongoing operations.
Recent Issued Accounting Pronouncements
Section 107 of the JOBS Act provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We are electing to delay such adoption of new or revised accounting standards, and as a result, we may not comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. As a result of this election, our financial statements may not be comparable to the financial statements of other public companies. We may take advantage of these reporting exemptions until we are no longer an “emerging growth company.”
Quantitative and Qualitative Disclosure About Market Risk
We are exposed to various market risks, which may result in potential losses arising from adverse changes in market rates, such as interest rates and foreign exchange rates. We do not enter into derivatives or other financial instruments for trading or speculative purposes and do not believe we are exposed to material market risk with respect to our cash and cash equivalents.
Interest Rate Risk
We are exposed to interest rate risk in connection with any future borrowings under our Comerica loan agreements, which bears interest at a floating rate based on Comerica’s prime rate plus an applicable borrowing margin. For variable rate debt, interest rate changes generally do not affect the fair value of the debt instrument, but do impact future earnings and cash flows, assuming other factors are held constant. In the ordinary course of business, we may enter into contractual arrangements to reduce our exposure to interest rate risks.
Foreign Exchange Risk
We operate in countries other than the United States, and, therefore, we are exposed to foreign currency risks. Revenue from sales outside of the United States represented approximately 26% of our total revenue in 2013. We bill most direct sales outside of the United States in local currencies, which are comprised of the Euro and the Brazilian Real. Operating expenses related to these sales are largely denominated in the same respective currency, thereby limiting our transaction risk exposure. We therefore believe that the risk of a significant impact on our operating income from foreign currency fluctuations is not significant. Additionally, we have intercompany foreign transactions between our subsidiaries, which are denominated in currencies other than their functional currency. Fluctuations from the beginning to the end of any given reporting period result in the remeasurement of our intercompany foreign transactions generating transaction gains or losses in the respective period and are reported in total other income (expense), net in our consolidated financial statements. The monetary assets and liabilities of our foreign subsidiaries denominated in other currencies are translated into U.S. dollars at each balance sheet date resulting in a foreign currency translation adjustment reflected in accumulated other comprehensive loss. We recorded foreign currency translation losses of $655,000 and $742,000 in
77
Table of Contents
2013 and 2012, respectively. We do not currently hedge our exposure to foreign currency exchange rate fluctuations; however, we may choose to hedge our exposure in the future.
Controls and Procedures
Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with GAAP. We are currently in the process of reviewing, documenting and testing our internal control over financial reporting.
We have not performed an evaluation of our internal control over financial reporting, such as required by Section 404 of the Sarbanes-Oxley Act, nor have we engaged an independent registered public accounting firm to perform an audit of our internal control over financial reporting as of any balance sheet date or for any period reported in our financial statements. Presently, we are not an accelerated filer, as such term is defined by Rule 12b-2 of the Securities Exchange Act of 1934, as amended, and therefore, our management is not presently required to perform an annual assessment of the effectiveness of our internal control over financial reporting. This requirement will first apply to our Annual Report on Form 10-K for the year ending December 31, 2014. Our independent registered public accounting firm will first be required to attest to the effectiveness of our internal control over financial reporting for our Annual Report on Form 10-K for the first year we are no longer an “emerging growth company.”
78
Table of Contents
Overview
We are a global medical device company focused on designing and commercializing novel and proprietary surgical technologies for the treatment of patients suffering from spine disorders. Our primary products are based on our VerteBRIDGE fusion and Mobi non-fusion platforms, both of which are designed for applications in the cervical and lumbar spine. We believe our VerteBRIDGE and Mobi platforms enable products that are less invasive, provide greater intra-operative flexibility, offer simplified surgical techniques and promote improved clinical outcomes for patients as compared to existing alternatives. As one example, in August 2013 we received approval from the U.S. Food and Drug Administration, or FDA, for the Mobi-C cervical disc replacement device, the first and only cervical disc replacement device to receive FDA approval to treat both one-level and two-level cervical disc disease and the only device technology to demonstrate overall clinical superiority (as compared to traditional fusion for two cervical vertebral levels) in an FDA pivotal clinical trial. Industry sources expect that the cervical disc replacement market, which is currently relatively small, will be one of the fastest growing segments of the U.S. spine implant market.
For the years ended December 31, 2013, 2012 and 2011, our total revenues were $111.6 million, $90.9 million and $78.0 million, respectively, and we had net losses of $27.9 million, $9.7 million and $1.8 million in 2013, 2012 and 2011, respectively. We expect to continue to incur losses in the near term. For the past three calendar years, revenue from our VerteBRIDGE and Mobi platform products has grown at a compounded annual growth rate, or CAGR, of 30% and represented approximately 83% of our revenue in 2013. Through December 31, 2013, we had an accumulated deficit of $83.8 million.
Our VerteBRIDGE and Mobi platform products incorporate design advancements that provide us with competitive advantages, discussed in detail below, in the estimated $9.6 billion global spine implant market. Industry sources estimate that the U.S. spine implant market represented approximately 73% of the global spine market in 2012, or $7.0 billion, and that it is expected to grow to approximately $9.0 billion by 2018. These sources estimate that the U.S. cervical disc replacement market will grow from $147 million in 2012 to $601 million by 2018, representing a CAGR of 26%. The U.S. cervical disc replacement market is one of the fastest growing segments of the U.S. spine implant market. We believe several factors will influence growth in the spine implant market segments in which we compete, including patients’ and surgeons’ demand for technologies that allow for less invasive treatment options, the availability of clinical safety and efficacy data demonstrating improved patient outcomes provided by new technologies and an aging global population leading to continued growth in the number of spinal procedures performed.
Our highly differentiated VerteBRIDGE fusion platform targets the cervical and lumbar spine fusion markets. Our VerteBRIDGE products are designed around our proprietary, less invasive plating technology that enables surgeons to implant VerteBRIDGE devices with direct visualization of the disc and to affix the devices to the vertebrae from inside the spinal disc space. VerteBRIDGE is the first platform of interbody devices with integrated fixation to market that may be implanted without screws, resulting in a zero-profile construct without hardware protruding outside of the vertebral body. We believe these differentiated features offer distinct advantages over other interbody devices, including simplified and less invasive surgical techniques for both cervical and lumbar fusion procedures. While these products only address a specific segment of the overall spine implant market, our VerteBRIDGE products have been used in more than 50,000 device implantations since the introduction of the VerteBRIDGE platform in 2007.
Our Mobi non-fusion platform is highlighted by Mobi-C, a cervical disc replacement device with a patented mobile bearing core that is designed to replicate the natural anatomical movement of the spine
79
Table of Contents
by facilitating independent bending and twisting. Mobi-C is the first and only cervical disc replacement device to have received FDA approval for both one-level and two-level cervical disc replacement procedures. In addition, Mobi-C is the only cervical disc replacement device that has demonstrated overall clinical superiority in an FDA pivotal clinical trial as compared to two-level traditional fusion. Mobi-C is implanted without the use of keels or screws, allowing for what we believe is a simplified, less invasive and bone sparing surgical approach as compared to other existing cervical disc replacement devices. We estimate that 30% of U.S. patients indicated for anterior surgery with symptomatic cervical disc disease may be candidates for two-level cervical disc replacement procedures. As a result, we believe the FDA approval of Mobi-C, its overall clinical superiority as compared to two-level traditional fusion procedures and its innovative design features will allow us to penetrate, and drive our growth in, the U.S. cervical disc replacement market, which we believe has the potential to grow relative to the total U.S. spine market. Currently, the market for FDA approved two-level devices is new with Mobi-C being the first-to-market cervical disc device approved for one-level and two-level cervical disc procedures. Accordingly, we need to grow the market for such procedures, which will require investment in time and capital to advocate and support the launch of Mobi-C in the United States. To support this launch of Mobi-C, we intend to leverage our extensive international experience with Mobi-C, including clinical and commercial knowledge accumulated from more than 17,000 Mobi-C implantations outside the United States. While the cervical disc market in the United States is currently relatively small, we believe that clinical data from second generation products such as Mobi-C will help drive the growth of the market.
We market and sell our products globally through a highly adaptable worldwide sales organization that allows us to continually expand our sales channels and rapidly penetrate new geographic markets. Our U.S. sales organization consists of sales managers that recruit, develop and lead direct sales representatives and a broad network of independent sales agencies. We estimate that non-captive independent sales agencies drive approximately 35% of total U.S. spine implant market revenue, and Mobi-C is the only cervical disc replacement device currently available to this large independent sales agency network. We believe this market position will facilitate the recruitment of high-quality direct sales representatives and independent sales agencies. We currently generate revenue in more than 25countries globally, and revenue from outside of the United States represented approximately 26% of our total revenue in 2013. Our international sales organization includes sales managers and direct sales representatives located in various countries, including France, Germany, Spain, South Korea, China and Brazil. We also sell to independent stocking distributors in certain international markets. A majority of our U.S. and international independent sales agencies and distributors have agreed to offer one or more of our VerteBRIDGE and/or Mobi-C products on an exclusive basis. We expect to continue to make investments in our sales organization by broadening our relationships with independent sales agencies, expanding exclusivity commitments among these agencies and international distributors and increasing our number of direct sales representatives.
We were founded in 2000 in France, one of the world’s leading regions for spine surgery advancements. We believe our product development capabilities are differentiated by our extensive spine industry experience and our ability to leverage close collaboration with thought leaders in spine surgery. Our capabilities allow us to gain significant design feedback and clinical experience, which facilitates our ability to affix a CE mark to our products or gain regulatory clearances and market registrations globally. Our product development experience, incorporating input from both European and U.S. thought leaders in spine surgery, is a key component of our ability to design highly differentiated and clinically beneficial technologies. In addition, we have a robust product pipeline that we expect will enhance our product platforms and enable us to continue to penetrate the fastest growing segments of the global spine implant market.
80
Table of Contents
Industry Background
Spine Anatomy
The spine is a column of bone and cartilage that extends from the base of the skull to the pelvis and acts as the core of the human skeleton, supporting the body and the head. The spine consists of approximately 24 interlocking bones called vertebrae that are stacked on top of one another. There are seven cervical vertebrae in the neck, 12 thoracic vertebrae in the central portion of the spine and five lumbar vertebrae in the lower back. The bottom of the spine consists of the sacrum and finally the coccyx. Vertebrae are paired into motion segments, or levels, that move by means of two facet joints that provide stability and enable the spine to bend and twist, with the neck being the most mobile region of the spine. The back, or posterior, part of each vertebra is comprised of a bony arch called the lamina that protects the spinal cord. Soft tissues, including ligaments, tendons and muscles, are attached to the laminae, which provide stability to the spinal column and facilitate movement of the spine. The front, or anterior, part of each vertebra is referred to as the vertebral body. The majority of the load in the spine is supported by this anterior part of the spine. Between each pair of vertebrae is an intervertebral disc, a disc-shaped pad of fibrous cartilage with a jelly-like core that facilitates motion and also absorbs stresses during movement. The spine encloses and protects the spinal cord, a column of nerve tracts that run from every area of the body to the brain, through openings between the vertebrae called foramen, and which deliver sensation and control to the entire body.
81
Table of Contents
The figures below illustrate the spinal anatomy as well as various disorders of the spine.
Disorders of the Spine
Given the complexity of its structure, the spine is susceptible to various ailments that can lead to instability and significant pain for a patient. Back pain affects an estimated 31 million people in the United States and is a leading cause of healthcare expenditures globally. Spine disorders are primarily caused by degenerative disease, deformity, tumors and trauma.
Degenerative disc disease accounts for the large majority of operative conditions affecting the spine and typically results from repetitive stresses experienced during the normal aging process. The disease progression involves gradual weakening and thinning of the shock absorbing intervertebral discs. This condition can occur at any level of the spine, though it most commonly occurs in the cervical and lumbar regions. The progressive changes in the discs can lead to a host of conditions, including:
| • | osteoarthritis, which is the breakdown of the tissue or cartilage that protects and cushions joints; |
| • | herniated disc, which is an abnormal bulge or breaking open of a spinal disc; and |
| • | spinal stenosis, which is the narrowing of the space around the nerves in the spine. |
82
Table of Contents
Typical symptoms, depending on the location of the condition, include neck or back pain, pain radiating in the arm or leg, numbness, loss of motor and reflex functions and lack of flexibility. Over time, the progression of these conditions can be compounded because damage to one vertebral segment will place additional stress on the vertebral levels adjacent to the affected vertebral segment, including the level above, or superior level, or level below, or inferior level, leading to multilevel disc disease. The vast majority of patients that require surgical spine interventions suffer from disease at either one or two contiguous vertebral levels.
Treatment Alternatives for Degenerative Conditions of the Spine
Treatment for degenerative conditions of the spine usually begins with medical management in the form of lifestyle changes such as exercise and weight loss, and anti-inflammatory and pain medication. While these treatments can be successful, clinical management of the degenerative process is challenging, and many patients with chronic symptoms that have failed these conservative treatments ultimately require spine surgery.
Spine surgery is typically performed utilizing either non-instrumented procedures that do not use implants or instrumented procedures that use implants. Access to the spine to perform such procedures can be accomplished through several anatomical areas: posterior, or directly through the patient’s back; anterior, or through the patient’s front or stomach; and lateral, or through the patient’s side. Non-instrumented procedures include discectomies and laminectomies, which involve the removal of part of a damaged disc or lamina in order to relieve pressure on the nerve roots or spinal cord. Non-instrumented procedures are generally performed when conservative therapies are no longer effective at treating the pain associated with disc disease. When non-instrumented procedures fail or in cases where the disease is more advanced prior to the first surgery, patients must turn to instrumented surgical procedures that involve the implantation of one or more devices to restore disc height and to either eliminate mobility of the affected vertebral segments or stabilize normal motion of the spine.
Lumbar Spine Treatment
Fusion is the most common instrumented lumbar spine procedure. These procedures attempt to alleviate back pain by removing damaged disc material and permanently joining, or fusing, two or more contiguous vertebrae. The goal of fusion is to decompress spinal nerves that are causing pain, restore the appropriate space between the vertebrae surrounding the diseased disc and eliminate mobility of the affected vertebral segment. By restoring disc height and eliminating motion, fusion attempts to prevent the pinching of the nerves exiting the spine, thereby reducing pain. Traditional fusion procedures typically involve an incision in the skin and cutting of muscle or movement of organs to gain access to the spine. The surgeon then removes bone or diseased disc material that is believed to be the source of pain, and inserts implants to the spine in order to stabilize the diseased vertebrae. Examples of implants include interbody devices that replace the diseased disc material, as well as metal rods, screws and plates that provide stability while the vertebrae fuse together during the six to 18 months following surgery. Traditional fusion procedures are considered highly invasive and are associated with several limitations, including high rates of subsequent surgery, loss of motion, disruption to soft tissues, lengthy surgical procedure times and prolonged patient recovery times. Recently, there has been significant surgeon and patient interest in less invasive fusion procedures, which include stand-alone fusion devices. Stand-alone fusion devices are interbody devices that are placed into the disc space and allow surgeons to restore disc height and eliminate motion at diseased discs, but do not require additional fixation, such as plates, rods and screws affixed to the vertebrae. Rather, the fixation is integrated into the implant device itself.
83
Table of Contents
While stand-alone fusion procedures have been developed to address many of the limitations of traditional fusion procedures, existing stand-alone fusion devices continue to be associated with the following limitations:
| • | Extended Retraction and Soft Tissue Disruption. Current stand-alone fusion devices require the creation of a pathway from the skin to the diseased disc that is large enough to allow for the implantation of the integrated fixation at significant angles. Extended retraction is often necessary, thereby resulting in additional soft tissue disruption. |
| • | Stand-Alone Implants may Incorporate Integrated Fixation Resulting in an External Profile on the Vertebrae. Some current stand-alone fusion devices are designed with integrated fixation, including plates and screws, that are affixed to the exterior of the vertebral body. These fixation methods can result in additional surgical morbidity, including irritation of the major blood vessels of the body or other soft tissues, dysphagia, or difficulty swallowing, or significant bruising. |
Non-fusion procedures, including lumbar disc replacement, are available but have not achieved significant market adoption in lumbar spine surgeries. We believe that the lack of adoption of non-fusion procedures for lumbar spine applications is due to limited clinical evidence demonstrating its superiority over spinal fusion, as well as the difficulty in identifying patients with lumbar disc disease that are appropriately indicated for non-fusion procedures.
Cervical Spine Treatment
Anterior cervical discectomy and fusion, or ACDF, is a current standard of care for the treatment of cervical disc disease and remains the most common instrumented cervical spine procedure. ACDF surgery involves decompressing the spinal nerves by removing the diseased disc material, and then stabilizing the vertebral segment by inserting bone grafts, interbody devices, or fixation systems including screws and plates. In cervical spine fusion, we believe there has been a progression toward less invasive and zero-profile stand-alone interbody devices that allow surgeons to restore disc height and eliminate the motion of diseased cervical discs with fixation integrated into the implant.
More recently, non-fusion procedures, including cervical disc replacements, have been the subject of extensive clinical research. The goals of cervical disc replacement are to restore natural disc height, thereby decompressing the nerves causing pain, while also maintaining normal spinal motion at the affected vertebral segments.
While cervical disc replacement devices have been developed to overcome limitations of traditional ACDF procedures, existing cervical disc replacement devices developed prior to Mobi-C have been associated with the following limitations:
| • | Approved Devices have Limited Indications. Current cervical disc replacement devices, other than Mobi-C, have indications limited in the United States to treat only one level of the cervical spine. |
| • | Other than Mobi -C, Existing Devices Require More Invasive Procedures. Current cervical disc replacement devices have cumbersome designs that require invasive implant procedures, including more extensive retraction resulting in soft tissue disruption and significant cuts into the vertebral bone above and below the implant. |
| • | Complex Surgical Technique. Surgical techniques to implant current cervical disc replacement devices are associated with challenging surgical steps in order to facilitate implantation or to provide additional fixation. |
84
Table of Contents
| • | Lack of Clinical Data Demonstrating Superiority as Compared to ACDF. We believe that the lack of peer-reviewed clinical data demonstrating superiority as compared to ACDF has limited the adoption of current disc replacement devices and thereby impeded market growth of cervical disc replacement devices. |
Market Opportunity
Industry sources estimate that the global spine implant market was valued at approximately $9.6 billion in 2012. The United States represented approximately 73% of the global spine implant market in 2012, or $7.0 billion, and is expected to grow to $9.0 billion by 2018. The spine implant market is a subset of the total spine market and consists of seven segments: traditional thoracolumbar fixation, traditional interbody fusion, traditional cervical fixation, minimally invasive surgical devices, or MIS, motion preservation, vertebral compression fracture, or VCF, and spinal electrical stimulation.
The chart below illustrates key components of the 2012 global spine implant market:
2012 Spine Implant Market
($ in millions)
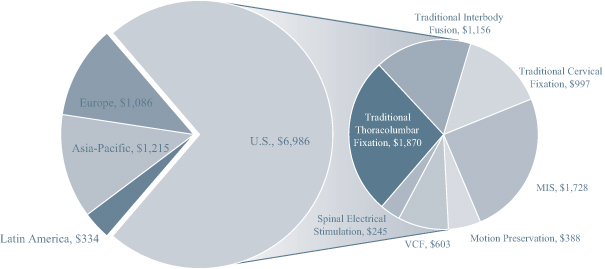
We compete in both the largest and fastest growing segments of the global spine market, including traditional thoracolumbar fixation, traditional interbody fusion, traditional cervical fixation, MIS and motion preservation.
U.S. Lumbar Fusion Market
The U.S. lumbar spinal fusion market is comprised of the traditional thoracolumbar fixation, traditional interbody fusion and MIS markets. The U.S. lumbar spinal fusion market was valued at $4.8 billion in 2012. The traditional lumbar interbody device market includes the market for anterior, posterior, transforaminal and lateral interbody devices, or ALIF, PLIF, TLIF and LLIF, respectively. These markets were valued at $233 million, $176 million, $272 million and $186 million, respectively, in 2012. Moreover, LLIF procedures are expected to be the fastest growing interbody device segment and the market for LLIF procedures is estimated to grow to $309 million by 2018. The market for stand-alone lumbar interbody fusion was valued at $78 million in 2012, representing approximately 34% of the 2012 ALIF market. We believe that stand-alone lumbar interbody devices will continue to rapidly penetrate into the U.S. ALIF market.
85
Table of Contents
U.S. Cervical Fusion Market
The U.S. cervical fusion market is comprised of anterior and posterior fixation devices. Approximately 316,000 cervical fusion procedures were performed in the United States in 2012, of which approximately 88% were anterior fusion procedures. The market for cervical fusion procedures was valued at $1.0 billion in 2012. The cervical fusion market is expected to remain relatively constant due to the increased adoption of cervical disc replacement devices. The traditional interbody fusion device market also includes cervical interbody fusion devices that are typically implanted in ACDF procedures. The cervical interbody fusion device market was valued at $209 million in 2012. The market for stand-alone cervical interbody fusion devices was valued at $78 million in 2012, representing approximately 37% of the cervical interbody fusion device market. We believe that stand-alone cervical devices will continue to rapidly penetrate into the U.S. cervical interbody fusion market.
U.S. Motion Preservation Market
The U.S. motion preservation market consists of cervical and lumbar disc replacement, dynamic stabilization, annular closure, nucleus replacement and facet arthroplasty devices. The U.S. motion preservation market, while currently representing only a small portion of the U.S. spine market, represents the fastest growing segment of the U.S. spine implant market, and is projected to grow from $388 million in 2012 to $783 million by 2018, representing a CAGR of 12%. The market for cervical disc replacement is estimated to be the primary growth driver for the U.S. motion preservation market. The U.S. cervical disc replacement market was valued at $147 million in 2012 and is projected to grow to $601 million by 2018, representing a CAGR of 26%. Within the population of patients indicated for surgery with symptomatic cervical disc disease, approximately 42% of patients are indicated for a one-level cervical disc replacement procedure, and approximately 30% are indicated for a two-level procedure. We estimate that the on-label market revenue potential for two-level cervical disc replacement is approximately 55% of the total cervical disc replacement market, due to the use of multiple devices in the two-level procedure.
Growth Drivers
We expect several factors will continue to influence growth in the spine implant market segments in which we compete. First, we believe that patients and surgeons will continue to demand new surgical technologies that allow for less invasive access, provide greater intra-operative flexibility and promote improved clinical outcomes. Second, we believe that as clinical evidence is published demonstrating the superiority of new technologies, such as cervical disc replacement devices, over traditional treatments, patients and surgeons will become increasingly aware of the clinical benefits of these technologies and more rapidly adopt them as an alternative to traditional surgical techniques. Finally, we believe that overall improvement to the standard of care resulting from the introduction of new products to the United States will increase global demand for these new technologies outside of the United States.
Our Solutions
Our VerteBRIDGE fusion platform and Mobi non-fusion platform products are designed for applications in the cervical and lumbar spine. We believe these platforms enable products that are less invasive, provide greater intra-operative flexibility, offer simplified surgical techniques and promote improved clinical outcomes for patients as compared to existing alternatives.
VerteBRIDGE Fusion Platform
Our highly differentiated VerteBRIDGE fusion platform is designed around our proprietary, less invasive plating technology. Our proprietary plating technology consists of curved titanium plates that allow VerteBRIDGE devices to be implanted with direct visualization of the disc and affixed to the vertebrae from inside the spinal disc space without screws. These differentiated features allow surgeons to perform a wide range of cervical and lumbar fusion procedures using our VerteBRIDGE devices.
86
Table of Contents
We believe that our VerteBRIDGE fusion platform addresses many of the limitations of traditional fusion and existing stand-alone implants by offering the following benefits:
| • | Less Invasive Access. The surgical incision and retraction required to insert a VerteBRIDGE device and secure it to the vertebral body is no larger than that required to complete the initial discectomy. As a result, the procedure requires less retraction than traditional fusion procedures and many existing stand-alone implants. This can lead to decreased disruption of soft tissue and support structures. |
| • | Simplified Surgical Procedure. Once the VerteBRIDGE device is in place, the surgeon can quickly affix it to vertebral bodies by inserting the plates through the implant holder. We believe this results in a simplified surgical procedure with fewer steps as compared to existing stand-alone devices, which can result in shorter operating times. |
| • | Zero-profile Design. The design of VerteBRIDGE devices does not require external screws or plates on the anterior surfaces of the spine, which enables self-stabilizing implant products with zero profile on the exterior of the vertebral body. We believe that this distinct feature can help avoid the risks associated with hardware protruding beyond the vertebral body into the major blood vessels and soft tissue, thereby reducing surgical morbidity. |
| • | Integrated, Screwless, Self-Locking Plating System with Extensive Implant Selection. VerteBRIDGE devices have a differentiated self-locking design and extension implant selection enabling them to be closely matched to the patient’s anatomy and securely implanted in multiple footprints and heights, thereby ensuring appropriate sizing and reducing the risk of implant migration. |
Not all spine patients may be suitable candidates for using our VerteBRIDGE products, and some may be more appropriately treated by other existing procedures or by new procedures that are developed in the future. For example, certain patients with severe instability of the spine may be better candidates for treatment with an anterior plate and/or pedicle screws than with our VerteBRIDGE fusion platform products when used on a stand-alone basis. In addition, some surgeons may not prefer our products when treating patients with poor bone quality.
Mobi Non-fusion Platform
Our Mobi non-fusion platform consists of disc replacement devices with a patented mobile bearing core that facilitates independent bending and twisting in a manner similar to a healthy disc. The highlight of our Mobi platform is the Mobi-C cervical disc replacement device which targets an emerging, but rapidly growing, segment of the U.S. spine implant market. Mobi-C has a zero-profile design and is implanted without the use of keels or screws, allowing for what we believe to be a simplified, less invasive and bone sparing surgical approach as compared to existing cervical disc replacement devices. We believe that Mobi-C is further differentiated by its ability to be easily adjusted, removed or repositioned intra-operatively. This flexible surgical implant procedure is particular to our Mobi platform. Mobi-C is the first and only cervical disc replacement device that is FDA approved for both one- and two-level implantation. In addition, Mobi-C demonstrated overall clinical superiority at all time points throughout its FDA pivotal clinical trial as compared to two-level ACDF.
We believe that our Mobi-C cervical disc replacement device addresses many of the limitations of traditional ACDF procedures and offers the following benefits:
| • | Overall Clinical Superiority as Compared to Two-level ACDF. In its FDA pivotal clinical trial at two-year follow up, Mobi-C demonstrated overall clinical superiority as compared to two-level ACDF with an overall success factor of 70% at two years as compared to an overall success factor of 37% with ACDF. In the FDA pivotal clinical trial, overall success was defined as improvement in Neck Disability Index, or NDI, no subsequent surgeries, no |
87
Table of Contents
| adverse event considered a major complication, no deterioration from pre-operative neurological states and radiographic success. |
| • | Preservation of Motion. Mobi-C incorporates an advanced mobile bearing core that is designed to provide more normal physiologic motion in one- and two-level cervical disc treatment, allowing patients, on average, to maintain or improve their range of motion at two years for the treated levels. |
| • | Reduced Rate of Adjacent Level Disc Degeneration. Mobi-C has demonstrated significantly lower rates of adjacent level disc degeneration as compared to ACDF in one- and two-level cervical disc treatment. In the FDA pivotal clinical trial, patients treated with Mobi-C at two-levels were statistically significantly less likely to demonstrate radiographic evidence of the advancement of disc disease to vertebral levels above or below the treated segments as compared to ACDF. |
| • | Lower Rates of Subsequent Surgery. Mobi-C has demonstrated statistically significantly lower rates of subsequent surgery as compared to ACDF in one- and two-level cervical disc treatment. The likelihood of subsequent surgery for the two-level Mobi-C patients was approximately one-third that of the two-level ACDF patients. Two-level rates of subsequent surgery at two years were 3.1% with Mobi-C as compared to 11.4% with ACDF. |
| • | Simplified, Less Invasive Surgical Procedure. Mobi-C simplifies surgery with innovative instrumentation that, we believe, allows the implantation to be performed in fewer steps, less retraction resulting in reduced disruption to bone and soft tissue as compared to other cervical discs and in a manner substantially consistent with proven surgical techniques. |
In addition to addressing many of the limitations associated with ACDF, we believe that our Mobi-C cervical disc replacement device also addresses many of the limitations associated with existing cervical disc replacement devices and offers the following benefits:
| • | First-to-market in the United States with FDA Approval for Both a One- and Two-Level Cervical Disc Replacement. Mobi-C is the only cervical disc replacement device to have FDA approval for both one-level and two-level cervical disc replacement procedures. In addition, Mobi-C is the only cervical disc replacement device that has demonstrated overall clinical superiority as compared to two-level ACDF. |
| • | Simplified, Less Invasive and Bone Sparing Surgical Approach. Mobi-C has a minimally invasive design, resulting in less disruption to bone and soft tissue. Mobi-C does not require invasive fixation into the vertebral bone, which results in a bone sparing implant procedure. Mobi-C is also available in a wide range of implant sizes, which is intended to enable a more accurate anatomical fit. |
| • | Advanced Mobile Bearing Technology. Mobi-C uses an advanced mobile bearing core technology, which facilitates independent bending and twisting similar to a healthy disc. The mobile bearing core technology is designed to replicate the natural anatomical movement of the spine, which can minimize the stress placed on the implant-bone interface. |
| • | Clinical Data Demonstrating Overall Clinical Superiority as Compared to Two-level ACDF. Mobi-C has extensive clinical data from two arms of its 575 patients randomized in the FDA pivotal clinical trials that demonstrated overall clinical superiority over ACDF for two-level cervical disc replacement, although the clinical trials were not powered to demonstrate one-level superiority with respect to one-level disc replacement. We believe that this superiority claim will drive adoption of the technology and overall market growth of non-fusion devices. |
88
Table of Contents
Not all cervical spine patients may be suitable candidates for using our Mobi-C products. In addition, some cervical discs on the market may have a more constrained articulation that could be more suitable for some patients, some surgeons may prefer cervical discs with greater fixation features than Mobi-C, and some cervical discs have been studied for use adjacent to a prior fusion whereas this was not part of the Mobi-C clinical study.
The following table identifies the characteristics of our Mobi-C cervical disc replacement device and, to the best of our knowledge, the characteristics of the other cervical disc replacement devices currently approved for sale in the United States:
| Company/Product |
LDR Mobi-C | DePuy Synthes Prodisc-C |
Medtronic Prestige ST |
Medtronic Bryan |
Globus Secure-C |
NuVasive PCM | ||||||
| One-Level Indication |
Yes | Yes | Yes | Yes | Yes | Yes | ||||||
| Two-Level Indication |
Yes | No | No | No | No | No | ||||||
| Primary Fixation | Inclined Lateral Teeth |
Central Keels | Anterior Screws | Press Fit | Central Keels | Serrations | ||||||
| Superiority |
Yes (Two-Level Only) |
No | No | No | Yes (One-Level Only) |
No | ||||||
| Heights |
5-7mm | 5-7mm | 6-7mm | 6mm | 7-12mm | 6.5-8mm | ||||||
Our Competitive Strengths
We focus on providing novel and proprietary technologies for both fusion and non-fusion applications in the cervical and lumbar spine. Our founders and executive management team collectively have over 100 years of experience in developing and commercializing innovative spine surgery products. We believe that this focus and experience, combined with the following competitive strengths, will allow us to continue to grow our revenue and rapidly increase our presence in the fastest growing segments of the spine implant market:
| • | First-to-market with an FDA Approved One-level and Two-level Cervical Disc Replacement Device. Mobi-C is the only cervical disc replacement device to have FDA approval for both one-level and two-level cervical disc replacement procedures. The emerging U.S. cervical disc replacement market is expected to grow 26% per year from $147 million in 2012 to $601 million by 2018. We estimate that 30% of patients undergoing anterior cervical surgery are indicated for two-level procedures; however, all cervical disc replacement devices currently available in the market are approved for only one-level procedures. We believe our approval for two-level cervical disc replacement procedures creates a significant on-label opportunity for us to penetrate into and drive growth of the cervical disc replacement market. |
| • | PMA Approval Process Creates a Barrier to Entry to the Cervical Disc Replacement Market. The risk, uncertainty, duration and expense of the premarket approval, or PMA, approval process creates a significant barrier to entry. As a Class III medical device, any entrant into the two-level cervical disc replacement market would be required to undergo a lengthy and costly FDA clinical trial and PMA approval process. As a result, any potential competitive entrants could not rely on our Mobi-C device for predicate-based 510(k) clearance and would be required to generate new clinical data comparing their device to either our Mobi-C device or ACDF. Potential competitive entrants may have difficulty enrolling patients to satisfy clinical trials given the presence of our approved and available device with clinical superiority compared to two-level ACDF. We believe this regulatory barrier to entry creates a significant competitive advantage for our Mobi-C device in the two-level cervical disc replacement market. |
89
Table of Contents
| • | Highly Differentiated Technology Platforms. Both our VerteBRIDGE fusion platform and Mobi non-fusion platform incorporate technological advancements that we believe provide us with competitive advantages in the marketplace and provide benefits to both surgeons and patients as compared to existing treatment alternatives. These benefits include less invasive access, simplified surgical procedures, bone-sparing surgical techniques and zero profile implant designs. In addition, we believe we are the only spine implant device company to provide all of our products in sterile packaging, which enhances implant traceability and potentially reduces infection rates and hospital costs. |
While we believe that we have designed our VerteBRIDGE and Mobi platform products with characteristics and attributes that provide them with competitive advantages over currently existing alternatives, spine surgeons may not adopt our devices for a variety of reasons, including lack of experience with us or our products, existing relationships with competitors and distributors that sell our competitors’ products and the time commitment required for training to be able to use our products.
Our Strategy
Our goal is to strengthen our position as a global innovator of spine technologies and become a leader in the spine implant market. To achieve our goal, we are pursuing the following strategies:
| • | Invest in our U.S. Infrastructure to Capitalize on the FDA Approval of Mobi-C. We plan to continue to invest in our U.S. infrastructure, including increasing the size and geographic breadth of our U.S. sales organization, developing comprehensive marketing support materials and surgeon training and education programs, executing upon our reimbursement strategies and building sufficient inventory of implants and instrument sets to capitalize on the FDA approval of Mobi-C. We intend to leverage these continued investments and the clinical benefits of our Mobi-C cervical disc replacement device, including the first and only FDA approval for both one- and two-level cervical disc replacement, overall clinical superiority as compared to two-level ACDF and a simplified, less invasive bone sparing surgical approach to establish Mobi-C as a standard of care for the treatment of cervical disc disease and rapidly penetrate the U.S. market for cervical disc replacements. |
| • | Promote the Differentiated Features of our VerteBRIDGE Fusion Platform to Further Penetrate the Large Global Spine Fusion Market. We plan to dedicate significant resources to promote the differentiated features and related benefits of our VerteBRIDGE technology platform to continue to penetrate the global cervical and lumbar spine fusion markets rapidly. |
| • | Leverage our Culture of Innovation to Expand our Product Portfolio. We believe our clinical experience and collaboration with surgeons enables us to innovate, design, develop and market new clinically beneficial technologies rapidly. We intend to continue to leverage our culture of innovation to expand our product portfolio by investing in internal research and development of new technologies, commercializing products currently in development and pursuing licensing activities and strategic acquisitions as the opportunities may arise. In addition, we intend to continue to expand our intellectual property portfolio to protect our innovations. As of December 31, 2013, our intellectual property portfolio consisted of over 330 issued patents and over 80 patent applications pending globally. |
| • | Invest in our International Infrastructure to Broaden our Global Market Presence. We intend to expand our presence in new and existing international markets through ongoing investment in our international infrastructure. |
90
Table of Contents
Our Products
Our VerteBRIDGE fusion and Mobi non-fusion platform products for both cervical and lumbar applications are characterized by patented technologies and innovative design features that we believe offer distinct advantages compared to other available products. We refer to these products in our financial reporting as our exclusive technology products. We also offer fusion technologies that are not significantly differentiated from competitive devices from a feature or clinical benefit perspective but that augment our VerteBRIDGE and Mobi technologies and allow us to offer our customers comprehensive surgical solutions. We refer to these products in our financial reporting as our traditional fusion products. In 2013, revenue from our exclusive technology products represented approximately 83% of our total revenue, and revenue from our traditional fusion products represented approximately 17% of our total revenue.
VerteBRIDGE Fusion Platform Products
Our VerteBRIDGE platform is designed to provide fusion without screws or external plates, thereby providing a zero-profile construct. The integrated titanium plates that comprise the integrated fixation solution in our VerteBRIDGE fusion products are distinct in their ability to be implanted directly in the plane of the surgical exposure, with a minimally invasive surgical technique, and provide screwless fixation intra-discally through the interbody device. VerteBRIDGE plating increases segmental stability, and multiple sizing options provide significant intra-operative flexibility. VerteBRIDGE is applicable for one-level cervical and lumbar fusion with or without supplemental fixation.
The figure below demonstrates the VerteBRIDGE technology:
VerteBRIDGE Products – Cervical Spine VerteBRIDGE Products – Lumbar Spine
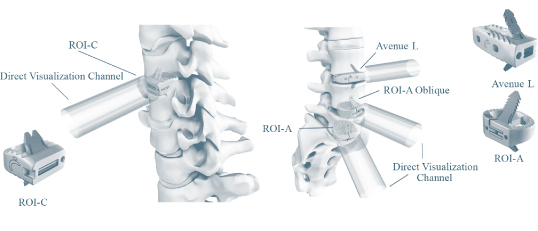
91
Table of Contents
Since introduction of the VerteBRIDGE platform in 2007, there have been over 50,000 implantations using VerteBRIDGE technology. Our VerteBRIDGE platform products and their launch dates are presented in the table below:
| Selected Product |
Description |
Region |
Launch | |||||
| ROI-C |
Anterior cervical fusion Poly-ether-ether-ketone, or PEEK, interbody device. Provided in anatomic and lordotic options. | United States International |
|
2009 2008 |
| |||
| ROI-A |
Anterior lumbar fusion PEEK interbody device. | United States International |
|
2008 2007 |
| |||
| ROI-A Oblique |
Oblique lumbar fusion PEEK interbody device. | United States International |
|
2011 2009 |
| |||
| Avenue L |
Lateral lumbar fusion PEEK interbody device. | United States International |
|
2012 2010 |
| |||
Mobi Non-fusion Platform Products
Our Mobi non-fusion platform utilizes an ultra-high molecular weight polyethylene core and cobalt chromium alloy superior and inferior endplates. The Mobi platform can be used in cervical and lumbar procedures, although the highlight of our Mobi platform is our Mobi-C cervical disc replacement device. Mobi-C endplates incorporate short, lateral, inclined teeth that provide initial stability and fixation. The endplates are titanium plasma sprayed and coated with hydroxyapatite, an essential ingredient of normal bone, that helps promote bone ongrowth and long-term fixation. The mobile bearing core is designed to move within the prosthesis as the neck bends and twists, thereby simulating the movement of a healthy disc. The mobile core feature is also designed to minimize stresses in the implant-to-bone interface. The surgical technique to implant Mobi-C avoids the need to cut into the vertebral bone above and below the implant or to perform significant endplate preparation. Mobi-C is indicated in the United States for the treatment of symptomatic cervical disc disease with radiculopathy or myelopathy at one or two contiguous levels.
The figure below demonstrates our Mobi-C cervical disc replacement device:
| The Mobi-C Cervical Disc | Anterior View of Mobi-C Implanted at Two Contiguous Levels |

92
Table of Contents
Over 17,000 Mobi-C implants have been implanted globally to date. Our Mobi platform products and their launch dates are presented in the table below:
| Selected Product |
Description |
Region |
Launch | |||||
| Mobi-C |
Cobalt Chromium alloy and ultra-high molecular weight polyethylene, mobile-bearing cervical disc replacement device. | United States International |
|
2013 2004 |
| |||
| Mobidisc |
Cobalt Chromium alloy and ultra-high molecular weight polyethylene, mobile-bearing lumbar disc replacement device. | International | 2004 | |||||
Traditional Fusion Products
We also have a broad portfolio of traditional fusion products. Our traditional fusion products include Easyspine, MC+, ROI, ROI-T, SpineTune and C-Plate. Unlike the products in our VerteBRIDGE platform or Mobi-C non-fusion platform, which offer highly differentiated features and benefits not found in competitive devices, our traditional fusion products are those devices that we offer for sale, but which cannot be significantly differentiated from competitive devices from a feature or clinical benefit perspective. Some industry experts may describe such traditional fusion products as spine commodity products. Examples include pedicle screw systems, anterior cervical plates and interbody cages absent integrated fixation. Our traditional fusion products are presented in the table below:
| Selected Product |
Description |
Region |
Launch | |||||
| Easyspine |
Titanium alloy pedicle screw system incorporating a flattened rod offering variable stiffness with a constant diameter and a pre-assembled closure mechanism enhancing simplicity. | United States International |
|
2005 2002 |
| |||
| MC+ |
PEEK cervical interbody device with a single, straight titanium plate to provide fixation to the vertebral bone inferior to the interbody device. MC+ was our first product to offer intra-discal fixation. | United States International |
|
2005 2003 |
| |||
| ROI |
PEEK interbody device designed to be implanted bilaterally from a posterior lumbar surgical approach. Open design provides stability of similar closed interbody devices but with larger graft area. | United States International |
|
2005 2002 |
| |||
| ROI-T |
Curved PEEK interbody device designed to be implanted unilaterally from a posterior lumbar approach. Tapered tip eases insertion. | United States International |
|
2007 2007 |
| |||
| SpineTune |
Pedicle screw fixation system for the thoracic and lumbar spine comprised of a titanium, in-line, open, polyaxial screws. | United States International |
|
2010 2010 |
| |||
| C-Plate |
Anterior cervical plate with a narrow, low-profile that incorporates an integrated, zero-step flexible locking ring and variable and semi-fixed screw options. | International | 2010 | |||||
Mobi-C Clinical Data Summary
We have established a significant body of clinical evidence demonstrating the safety and efficacy of Mobi-C. We conducted two concurrent FDA pivotal clinical trials studying Mobi-C for treatment of one-level and two-level cervical disc disease. Data from these trials was submitted to the FDA to support PMA approval of Mobi-C for use in the treatment of one-level and two-level cervical disc disease. Mobi-C is the only device to have FDA approval for treatment of two-level cervical disc disease. The data from these trials demonstrated that Mobi-C results in clinically superior outcomes as compared to ACDF for treatment of two-level cervical disc disease.
93
Table of Contents
Mobi-C FDA Pivotal Clinical Trials (Mobi-C vs. ACDF)
To support our Mobi-C PMA application, we conducted two concurrent, randomized, prospective, multi-center, non-inferiority studies, with preplanned tests for superiority that were designed to establish the safety and effectiveness of Mobi-C as a treatment for patients with symptomatic cervical disc disease with radiculopathy or myeloradiculopathy at one or two contiguous levels. The FDA pivotal clinical trial concluded that Mobi-C is safe and effective for treatment of patients with symptomatic cervical disc disease in one or two contiguous levels. Additionally, Mobi-C demonstrated further advantages as compared to ACDF in several areas, including:
| • | statistically significant superiority of two-level overall success at all time points of data measurement through two years; |
| • | decreased incidence of continued adjacent level degeneration for both one- and two-levels; and |
| • | lower rates of subsequent surgery. |
Patients enrolled in the trials presented without prior cervical fusion and either were unresponsive to non-operative conservative treatment for six weeks after symptom onset, or had the presence of progressive symptoms or had signs of nerve or spinal cord compression. Patients were followed post-operatively at six weeks, and three, six, 12, 18 and 24 months. The one-level trial included 245 patients and the two-level trial included 330 patients, each enrolled with 2:1 randomization against ACDF across 24 sites with 24 months of follow-up, and encompassed the ability to test for superiority as compared to ACDF if non-inferiority was achieved. Subsequent to the required 24 month follow-up, patients were followed at 36 and 48 months.
The primary objective of both trials was to evaluate the overall success rate of Mobi-C as compared to ACDF. The studies evaluated multiple criteria in order to determine overall success over a two year period. All control and Mobi-C patients had to satisfy each of the following criteria to be considered an overall success:
| • | improvement in Neck Disability Index, or NDI; |
| • | no subsequent surgery; |
| • | no adverse events that were considered major complications; |
| • | no deterioration from pre-operative neurologic status; and |
| • | radiographic success. |
The NDI is a standard metric for measuring patient disability due to neck pain. It is derived from a patient reported questionnaire and is based on a 50 point scale, which is sometimes reported as point or percentage. In our trial, patients had to have at least a 15 point improvement in the raw NDI score to be considered an NDI success.
The results from the primary endpoint are summarized in the graphs below:
Overall success is reported as the percentage of patients that met all pre-defined criteria for success. In the one-level trial, Mobi-C patients achieved a higher level of overall success at early time points, including six months, and maintained a higher level of overall success as compared to ACDF at all time points. The data was sufficient to demonstrate that treatment with Mobi-C was not inferior to ACDF. In the two-level trial, Mobi-C patients on average achieved a statistically superior level of overall success as compared to ACDF at all time points.
94
Table of Contents
Mobi-C Overall Success as compared to ACDF
| One-Level | Two-Level |
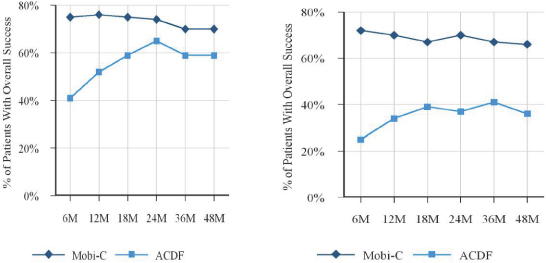
The charts below demonstrate the level of NDI success and subsequent surgery rates for patients in the two-level trial. In this trial, Mobi-C patients on average achieved a statistically significant improvement of their NDI score as compared to ACDF at all time points. In addition, patients had to be free from any trial-defined subsequent surgeries in order to be considered a clinical success. A subsequent surgery is defined as any reoperation, removal, revision or supplemental fixation at the treated spine level. In the two-level trial, patients were nearly three and half times more likely to have a subsequent surgery when treated with ACDF as compared to those treated with Mobi-C.
| Mobi-C NDI Success as Compared to ACDF
Two-Level |
Mobi-C Subsequent Surgery Rates as Compared to ACDF
Two-Level |
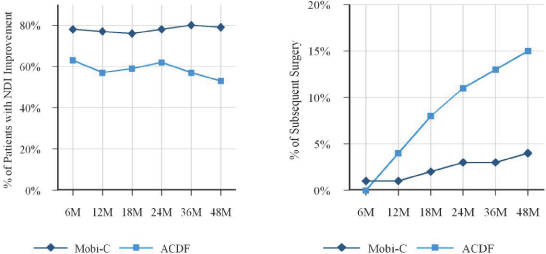
In addition to the primary endpoints that were components of the overall success, the FDA pivotal clinical trials analyzed other secondary endpoints, including adjacent segment degeneration. Adjacent segment degeneration, which is an indicator of disease progression, was measured radiographically,
95
Table of Contents
utilizing the standardized Kellgren-Lawrence scale. A patient’s pre-operative level of degeneration was compared to the level of degeneration at annual time points at both the superior and inferior vertebral segments to the treated levels. In the two-level trial, patients treated with Mobi-C demonstrated statistically significantly lower rates of adjacent segment degeneration at 12, 24, 36 and 48 months as compared to patients treated with ACDF.
Mobi-C Adjacent Segment Degeneration as compared to ACDF (Two-Level)
| Superior-Level | Inferior-Level |
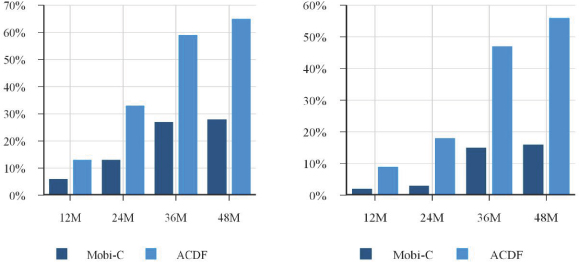
Overall Trial Conclusions
The FDA pivotal clinical trial concluded that Mobi-C is safe and effective for treatment of patients with symptomatic cervical disc disease with radiculopathy or myelopathy at one or two contiguous levels. Additionally, Mobi-C demonstrated overall clinical superiority to traditional fusion in two-level applications. Mobi-C is the first and only cervical disc replacement device to demonstrate overall clinical superiority as compared to traditional fusion for treatment at two-levels.
Other Ongoing Clinical Trials
As a condition of approval of the Mobi-C cervical disc replacement device by the FDA, we are required to follow all patients who received Mobi-C prior to FDA premarket approval for a period of seven years from treatment. Five-year results from the follow-up of FDA pivotal clinical trial patients are expected to be published in 2015. We are also conducting research regarding the cost effectiveness and cost utility of outcomes for patients treated with Mobi-C to support our reimbursement coverage efforts.
In addition, we continue to conduct clinical studies on a smaller scale to evaluate clinical efficacy of our portfolio of VerteBRIDGE and Mobi platform products and support respective global marketing efforts and product clearance, approval, CE mark and registrations.
Coverage and Reimbursement Overview
Private insurance coverage for one-level and/or two-level cervical disc replacement has been steadily improving for the last several years with positive coverage policies issued by several national and regional payors. As of December 2013, there were approximately 122 million U.S. members with coverage for one-level and/or two-level cervical disc replacement from a variety of insurers including Cigna, United Healthcare, Aetna, Blue Cross Blue Shield Louisiana and others. We intend to actively engage in
96
Table of Contents
activities to support increasingly broad, positive coverage. Peer-reviewed publications of the clinical data from the Mobi-C FDA pivotal clinical trial by the surgeon investigators will be an important, supportive component of our effort, as will support from the relevant physician specialty societies.
For payors who currently cover one-level and/or two-level cervical disc replacement, we plan to communicate the FDA approval of Mobi-C for both one- and two-level cervical disc replacement and approved indications, and provide our published peer-reviewed evidence in our request to have Mobi-C added to their existing coverage policy. For payors who do not yet cover one-level and/or two-level cervical disc replacement, we believe that the FDA approval for both one- and two-level cervical disc replacement and published evidence will add to the substantial and growing library of clinical evidence, including long term evidence from multiple FDA pivotal clinical trials detailing the safety and efficacy of cervical disc replacement. In addition, effective January 1, 2015, surgeons will be able to use a new CPT code as an add-on code to report the performance of total disc arthroplasty at an additional level. Although we anticipate that the trend toward broader coverage for one-level and two-level disc replacement will continue as payors have the opportunity to review the most recently available clinical evidence, including the data from the Mobi-C FDA pivotal clinical trial, we can provide no assurances that payors will continue to broaden coverage for such procedures.
Because Mobi-C was just recently approved and is the only device to be approved by the FDA for both one- and two-level cervical disc replacement, only a small number of commercial payors have published positive coverage policies for two-level applications and consequently, most still do not cover two-level cases as a matter of policy. We intend to utilize published, peer-reviewed evidence from the two-level Mobi-C FDA pivotal clinical trial, including the conclusion of overall clinical superiority to ACDF and further longer-term evidence, to petition in support of positive coverage for Mobi-C for two-level cervical disc replacement. We believe that the demonstration of overall clinical superiority of Mobi-C as compared to traditional fusion will be an important and compelling component of our activities supporting positive coverage. In addition, we feel that the substantial and growing library of evidence for one-level cervical disc replacement, including long term evidence, further supports cervical disc replacement as a safe and effective procedure. Finally, we are continuing to conduct clinical trials evaluating the cost effectiveness and cost utility of two-level cervical disc replacement. We believe that this research will further support increasingly broad coverage for one and two-level cervical disc replacement as safe, effective and cost-effective treatments for patients suffering from cervical disc disease.
Product Development and Research
We were founded in France, one of the world’s leading regions for spine surgery advancements. Our founders began with the goal of leveraging their significant experience in the spine market to innovate technologies that meet future spine market needs and address those needs while remaining compatible with current surgical techniques. We have developed a highly effective product development department that leverages our design and development expertise, in close collaboration with thought leaders in spine surgery, to design, develop and launch innovative technologies.
We also have dedicated clinical and regulatory personnel who work in parallel with our product development team to facilitate regulatory clearances, market registrations and the CE marking of our products. Our research and development team engages in close collaboration with physician advisory groups consisting of active spine surgeons. The historical relationship of our design team with surgeons enables our design engineers to engage in frequent dialogue with these surgeons both in and out of the operating room, and allows us to gain significant design feedback and clinical experience rapidly. Our product development team also includes a U.S. market team focused on interacting with U.S. surgeons, designing technologies specific to the U.S. market and supporting U.S. product introductions and product management. Our product development experience, incorporating both European and U.S. inputs, is a key component of our ability to design highly differentiated and clinically beneficial technologies for the
97
Table of Contents
global spine implant market. In addition, we have a robust product pipeline that will enhance our product platforms and enable us to continue to penetrate the fastest growing segments of the global spine implant market. For the years ended December 31, 2011, 2012 and 2013, we spent $9.0 million, $10.8 million and $9.4 million, respectively on research and development.
Sales and Marketing
We market and sell our products globally through a highly adaptable sales organization. Our U.S. sales organization is comprised of a team of experienced sales managers employed by us that recruit, develop and lead direct sales representatives and a broad network of over 190 independent sales agencies that employ over 600 sales representatives. Our U.S. sales organization includes 40 sales managers and our international sales organization includes management and direct sales personnel in various countries, including France, Germany, Spain, South Korea, China and Brazil. We also sell to distributors in certain international markets. We currently generate revenue in more than 25 countries globally. Our international capabilities allow us to gather meaningful marketing experience with new technologies prior to introduction in the United States and other countries with stringent regulatory requirements. For example, over 17,000 Mobi-C implantations were achieved prior to its introduction in the United States in 2013.
We believe our sales organization provides us with broad geographic coverage and allows us to maintain the flexibility to expand our sales channels and penetrate new geographic markets rapidly. Our sales organization consists of sales professionals that are experienced in the spine industry, the vast majority of whom have demonstrated previous sales success working with other spine industry manufacturers. In addition to general sales and marketing training, we provide our sales organization, including our independent sales agencies, with comprehensive, hands-on cadaveric and dry-lab training sessions on the clinical benefits of our products. We believe our robust training and professional development programs have been an important component of our success to date and will help support our anticipated future growth.
Our independent sales agencies are compensated solely on commission, with some having an opportunity to receive stock options if they meet a minimum dollar amount of sales. Our sales managers and direct sales representatives have compensation arrangements that include base salaries, bonuses, commissions and stock options. A majority of our independent sales agencies and our international distributors have agreed to offer one or more of our products on an exclusive basis. We believe that the continued growth of our portfolio of VerteBRIDGE and Mobi platform products and the launch of the Mobi-C cervical disc replacement device in the United States will support the expansion of our sales organization. For example, we estimate that non-captive independent sales agencies drive 35% of total spine market revenue. As most other currently available cervical disc replacement devices are sold by their manufacturers through a direct sales force, our Mobi-C cervical disc replacement device is the only cervical disc replacement device available to this large independent sales agency network. We believe the approval of Mobi-C represents a compelling opportunity for us to attract additional high quality agencies and expand exclusive commitments to our portfolio. We expect that Mobi-C will also provide the opportunity for us to recruit additional high quality direct sales representatives to supplement our independent sales agency network and broaden our geographic coverage and market penetration.
Spine Community Involvement, Education and Training
We market our technologies to spine surgeons globally, and our active involvement within the global spine surgeon community is a key element of our strategy to broaden the use of our products. We are committed to advancing the science of our technologies and we focus our efforts within the spine community to provide quality surgeon education and training, implement surgeon feedback into our product development process and obtain support from various spine societies. We believe that our success has been, and will continue to be driven by the quality of our products and reputation within the spine surgeon community.
98
Table of Contents
We believe the most effective way to introduce and build market demand for our products is by training active spine surgeons in the use of our products, and we devote significant resources to surgeon training and education. We conduct a significant number of national and regional cadaveric training labs in the United States and internationally where surgeons have the opportunity to learn from other surgeons who have significant clinical experience utilizing our products. This education includes approved patient indications and contra-indications for our products, overviews of the features and clinical benefits of our products and review of clinical examples of procedures. We also provide proctored, hands-on training on the safe and effective implantation of our devices. For many of our international markets, these events represent extremely valuable training opportunities for surgeons for whom training on advanced techniques is not available in their local markets. As of the date of this filing, we have trained thousands of spine surgeons in the use of our VerteBRIDGE and Mobi platform technologies worldwide. In addition, we have trained more than 650 U.S. surgeons on the use of Mobi-C subsequent to it receiving FDA approval for one and two-level indications in August 2013.
In addition to surgeon training and education, we consult with surgeon experts and actively solicit their feedback throughout the entire product development process. We also work with surgeons and other healthcare professionals in the area of clinical research in order to gain a better understanding of the safety and efficacy of our products, and support the necessary requirements for product clearances, CE marking and registrations internationally. We also conduct post-market research to gather clinical data and to ensure that the implants and instruments that we provide meet the intended use of our products.
Moreover, we are an active participant within the spine industry, and regularly support and maintain a presence at numerous national and regional professional society congresses, such as the North American Spine Society, the Cervical Spine Research Society, EuroSpine and the AANS/CNS Joint Section on Spine. At these meetings, we meet with current and potential surgeon users, demonstrate the clinical benefits of our products and generate awareness among these societies as to the clinical benefits of our products.
Suppliers
We rely on a network of over 20 third parties, primarily located in Europe, to manufacture, package and sterilize all of our products. For us to be successful, our suppliers must be able to provide us with products and components in substantial quantities, in compliance with regulatory requirements, in accordance with agreed upon specifications, at acceptable costs and on a timely basis. We generally do not have long-term contracts with our suppliers and they are not required to provide us with any guaranteed minimum production levels. However, we do have single or limited source contracts with certain suppliers, including CF Plastiques, Invibio and Greatbatch Medical, each of which have terms in excess of one year. None of these single or limited source contracts may be terminated by the supplier other than for cause. We do not have any material contracts with any suppliers other than the agreements that we have with CF Plastiques, Invibio and Greatbatch Medical.
Our third-party manufacturers meet FDA, International Organization for Standardization, or ISO, and other quality standards supported by our internal policies and procedures. We believe these manufacturing relationships allow us to work with suppliers who have well-developed specific competencies while minimizing our capital investment, controlling costs and shortening cycle times, all of which we believe allows us to compete with larger volume manufacturers of spine surgery products.
There are a limited number of suppliers and third-party manufacturers that operate under the FDA’s current Good Manufacturing Practices, or cGMP, maintain ISO certifications and have the necessary expertise and capacity to manufacture our products. We select our suppliers carefully and frequently conduct on-site audits to ensure that our suppliers meet our internal quality control standards. Our internal quality assurance group conducts comprehensive examinations of prospective supplier facilities. In addition, we and our suppliers are subject to periodic unannounced inspections by U.S. and international regulatory authorities to ensure compliance with quality system regulations.
99
Table of Contents
In most cases, we have redundant manufacturing capabilities for each of our products to ensure our inventory needs are met while maintaining high quality. However, we currently rely on a small number of limited or single source suppliers, such as Greatbatch, which supplies our Mobi-C cervical disc replacement device for the U.S. market, In’Tech Medical, which supplies a majority of our surgical instrumentation, and CF Plastiques, which is one of our interbody device suppliers. To date, we have not experienced any difficulty in locating and obtaining the materials necessary to meet demand for our products, and we believe manufacturing capacity is sufficient to meet global market demand for our products for the foreseeable future.
Competition
The medical device industry and the spine implant market in particular, are highly competitive, subject to rapid technological change and significantly affected by new product introductions and market activities of other participants. Our currently marketed products are, and any future products we commercialize will be, subject to intense competition. We believe that our significant competitors include Medtronic Spine and Biologics, DePuy Synthes Spine (a division of Johnson & Johnson), Globus Medical, NuVasive and Stryker, which together represent a significant portion of the spine market. Each of these significant competitors compete in both the traditional and non-traditional product markets. In particular, each of these significant competitors have products that directly compete with our VerteBRIDGE fusion platform. Several of these significant competitors also have products that directly compete with our Mobi non-fusion platform, including Prodisc-C from DePuy Synthes Spine, Prestige ST and Bryan from Medtronic, Secure-C from Globus Medical and PCM from NuVasive. To our knowledge, only one potentially competitive two-level cervical disc replacement device, the Medtronic Prestige LP, is currently in development. We have no knowledge of, nor can we predict, when this device or any other potentially competitive device might be available for sale in the United States.
We also compete with many other spine market participants such as Alphatec Spine, Biomet Spine, Integra, Orthofix International and Zimmer, whose products generally have a smaller market share than the significant competitors listed above but a larger market share than our products. At any time, these and other potential market entrants may develop new devices or treatment alternatives that may compete directly with our products. In addition, they may gain a market advantage by developing and patenting competitive products or processes earlier than we can or by obtaining regulatory clearances, CE Certificates of Conformity or market registrations more rapidly than we can.
Many of our current and potential competitors have significantly greater financial, technical, marketing and other resources than we do and may be able to devote greater resources to the development, promotion, sale and support of their products. Our competitors may also have more extensive customer bases and broader customer relationships than we do, including relationships with our potential customers. In addition, many of these companies have longer operating histories and greater brand recognition than we do. Because of the size of the spine market and the high growth profile of the segments in which we compete, we anticipate that companies will dedicate significant resources to developing competing products. We believe that the principal competitive factors in our markets include:
| • | improved outcomes for medical conditions affecting the spine; |
| • | acceptance by spine surgeons; |
| • | ease of use and reliability; |
| • | product price and qualification for reimbursement; |
| • | technical leadership and superiority; |
| • | effective marketing and distribution; and |
| • | speed to market. |
100
Table of Contents
We cannot assure you that we will be able to compete effectively against our competitors in regard to any one or all of these factors. Our ability to compete effectively will depend on the acceptance of our products by surgeons, hospitals and customers, and our ability to achieve better clinical outcomes than products developed by our existing or future competitors. In addition, many of our competitors could use their superior financial resources to develop products that have features or clinical outcomes similar to our products, which would harm our ability to successfully compete.
Regulatory Matters
Our business is subject to federal, state, local and foreign regulations, such as ISO 13485, FDA 21 CFR Part 820 and the Medical Devices Directive. While some laws and regulations are clear, some of the pertinent laws have not been definitively interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of subjective interpretations. In some cases there are industry guidelines or draft guidelines, however these too are subject to interpretation. In addition, these laws and their interpretations are subject to change.
Competent authorities in the European Economic Area, or EEA, which is made of the 28 EU Member States, Iceland, Liechtenstein and Norway, countries and U.S. federal as well as state governmental agencies continue to subject the healthcare industry to intense regulatory scrutiny, including heightened civil and criminal enforcement efforts. We believe that we have structured our business operations and relationships with our customers to comply with all applicable legal requirements. However, it is possible that governmental entities or other third parties could interpret these laws differently and assert otherwise. We discuss below the statutes and regulations that are most relevant to our business.
European Economic Area (EEA)
In the EEA, our devices are required to comply with the Essential Requirements laid down in Annex I to the Council Directive 93/42/EEC concerning medical devices, known as the Medical Devices Directive. Compliance with these requirements entitles us to affix the CE mark of conformity to our medical devices, without which they cannot be commercialized in the EEA. To demonstrate compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive and obtain the right to affix the CE mark of conformity to our medical devices, we must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements laid down in the Medical Devices Directive, a conformity assessment procedure requires the intervention of a Notified Body, which is an organization designated by the competent authorities of a EEA country to conduct conformity assessments. The Notified Body would typically audit and examine products’ Technical File and the quality system for the manufacture, design and final inspection of our devices before issuing a CE Certificate of Conformity demonstrating compliance with the relevant Essential Requirements laid down in Annex I to the Medical Devices Directive. Following the issuance of this CE Certificate of Conformity, we can draw up an EC Declaration of Conformity and affix the CE mark to the products covered by this CE Certificate of Conformity and the EC Declaration of Conformity. We have now successfully passed several Notified Body audits since our original certification in April 30, 2002. Following these audits, our Notified Body issued ISO Certificates and CE Certificates of Conformity allowing us to draw up an EC Declaration of Conformity and affix the CE mark of conformity to certain of our devices.
After the product has been CE marked and placed on the market in the EEA, we must comply with a number of regulatory requirements relating to:
| • | registration of medical devices in individual EEA countries; |
| • | pricing and reimbursement of medical devices; |
101
Table of Contents
| • | establishment of post-marketing surveillance and adverse event reporting procedures; |
| • | Field Safety Corrective Actions, including product recalls and withdrawals; and |
| • | interactions with physicians. |
Failure to comply with these requirements may result in enforcement measures being taken against us by the competent authorities of the EEA countries. These can include fines, administrative penalties, compulsory product withdraws, injunctions and criminal prosecution. Such enforcement measures would have an adverse effect on our capacity to market our products in the EEA and, consequently, on our business and financial position.
Further, the advertising and promotion of our products in the EEA is subject to the provisions of the Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other national legislation in the EEA countries governing the advertising and promotion of medical devices. These laws may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
Sales of medical devices are subject to foreign government regulations, which vary substantially from country to country. In order to market our products outside the United States, we must obtain regulatory approvals or CE Certificates of Conformity and comply with extensive safety and quality regulations. The time required to obtain approval by a foreign country or to obtain a CE Certificate of Conformity may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ. In the EEA, we are required to obtain Certificates of Conformity before drawing up an EC Declaration of Conformity and affixing the CE mark of conformity to our medical devices. Many other countries, such as Australia, India, New Zealand, Pakistan and Sri Lanka, accept CE Certificates of Conformity or FDA clearance or approval although others, such as Brazil, Canada and Japan require separate regulatory filings.
U.S. Food and Drug Administration Regulation
Our products are medical devices subject to extensive regulation by the FDA and other federal, state, local and foreign regulatory bodies. FDA regulations govern, among other things, the following activities that we or our partners perform and will continue to perform:
| • | product design and development; |
| • | product testing; |
| • | product manufacturing; |
| • | product safety; |
| • | post-market adverse event reporting, including reporting of deaths, serious injuries or device malfunctions; |
| • | post-market surveillance; |
| • | complaint handling; |
| • | repair or recall of products; |
| • | product storage; |
| • | record keeping; |
| • | premarket clearance or approval; |
102
Table of Contents
| • | post-market approval studies; |
| • | advertising and promotion; and |
| • | product marketing sales and distribution. |
FDA’s Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device we wish to distribute commercially in the United States will require either prior 510(k) clearance or prior approval of a PMA application from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose low to moderate risk are placed in either class I or II, which, absent an exemption, requires the manufacturer to submit to the FDA a premarket notification requesting permission for commercial distribution. This process is known as 510(k) clearance. Some low risk devices are exempt from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device are placed in class III, requiring approval of a PMA application. Our currently marketed products include Class I devices marketed pursuant to an exemption from 510(k) premarket clearance, Class II devices marketed under FDA 510(k) premarket clearance and Class III devices requiring premarket approval. Both premarket clearance and PMA applications are subject to the payment of user fees, paid at the time of submission for FDA review. The FDA can also impose restrictions on the sale, distribution or use of devices at the time of their clearance or approval, or subsequent to marketing.
510(k) Clearance Pathway
To obtain 510(k) clearance, we must submit a premarket notification demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of PMA applications. The FDA’s 510(k) clearance pathway usually takes from three to 12 months from the date the application is completed, but it can take significantly longer and clearance is never assured. Although many 510(k) premarket notifications are cleared without clinical data, in some cases, the FDA requires significant clinical data to support substantial equivalence. In reviewing a premarket notification, the FDA may request additional information, including clinical data, which may significantly prolong the review process. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will require a new 510(k) clearance or, depending on the modification, could require a PMA application. The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination regarding whether a new premarket submission is required for the modification of an existing device, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or approval of a PMA application is obtained. If the FDA requires us to seek 510(k) clearance or approval of a PMA application for any modifications to a previously cleared product, we may be required to cease marketing or recall the modified device until we obtain this clearance or approval. In addition, in these circumstances, we may be subject to significant regulatory fines or penalties for failure to submit the requisite PMA application(s). We have made and plan to continue to make minor additional product enhancements that we believe do not require new 510(k) clearances. In addition, the FDA is currently evaluating the 510(k) process and may make substantial changes to industry requirements.
Premarket Approval Pathway
A PMA application must be submitted if the device cannot be cleared through the 510(k) process. The PMA application process is generally more costly and time consuming than the 510(k) process and requires proof of the safety and effectiveness of the device to the FDA’s satisfaction. Accordingly, a PMA
103
Table of Contents
application must be supported by extensive data including, but not limited to, technical information regarding device design and development, pre-clinical and clinical trials, data and manufacturing and labeling to support the FDA’s determination that the device is safe and effective for its intended use. After a PMA application is complete, the FDA will accept the application and begins an in-depth review of the submitted information. By statute, the FDA has 180 days to review the “accepted application,” although, generally, review of the application takes between one and three years, but may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with Quality System Regulations, or QSRs, which impose elaborate design development, testing, control, documentation and other quality assurance procedures in the design and manufacturing process. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution and collection of long-term follow-up data from patients in the clinical study that supported approval. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including the loss or withdrawal of the approval. New PMA applications or PMA application supplements are required for significant modifications to the manufacturing process, labeling and design of a device that is approved through the PMA modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel, depending on the nature of the proposed change.
Clinical Trials
A clinical trial is almost always required to support a PMA application and may be required for a 510(k) premarket notification. In the United States, absent certain limited exceptions, human clinical trials intended to support product clearance or approval require an Investigational Device Exemption, or IDE, application. Some types of studies deemed to present “non-significant risk” are deemed to have an approved IDE once certain requirements are addressed and IRB approval is obtained. If the device presents a “significant risk” to human health, as defined by the FDA, the Sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to evaluate the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of subjects, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the responsible institutional review boards at the clinical trial sites. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials. Additionally, after a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to unacceptable health risks that outweigh the benefits of participation in the study. During a study, we are required to comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting, record keeping and prohibitions on the promotion of investigational devices or making safety or efficacy claims for them. We are also responsible for the appropriate labeling and distribution of investigational devices. Our clinical trials must be conducted in accordance with FDA regulations and federal and state regulations concerning human subject protection, including informed consent and healthcare privacy. The investigators must also obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices and comply with all reporting and recordkeeping requirements. The FDA’s grant
104
Table of Contents
of permission to proceed with clinical testing does not constitute a binding commitment that the FDA will consider the study design adequate to support clearance or approval. In addition, there can be no assurance that the data generated during a clinical study will meet chosen safety and effectiveness endpoints or otherwise produce results that will lead the FDA to grant marketing clearance or approval. Similarly, in the EEA, the clinical study must be approved by the local ethics committee and in some cases, including studies of high-risk devices, by the Ministry of Health in the applicable country.
Pervasive and Continuing FDA Regulation
After a device is placed on the market, regardless of its classification or premarket pathway, numerous regulatory requirements apply. These include, but are not limited to:
| • | establishing establishment registration and device listings with the FDA; |
| • | Quality System Regulation, or QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, process control, documentation and other quality assurance procedures; |
| • | labeling regulations, which prohibit the promotion of products for uncleared or unapproved, or off-label, uses and impose other restrictions on labeling; |
| • | clearance or approval of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use; |
| • | medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
| • | corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the U.S. Federal Food, Drug, and Cosmetic Act, or FDCA, that may present a risk to health. In addition, FDA may order a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; and |
| • | post-approval restrictions or conditions, including requirements to conduct post-market surveillance studies to establish continued safety data. |
The FDA has broad post-market and regulatory enforcement powers. The Agency may conduct unannounced inspections to determine compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of subcontractors. Failure by us or our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in sanctions and related consequences including, but not limited to:
| • | untitled letters or warning letters; |
| • | fines, injunctions, consent decrees and civil penalties; |
| • | recall, detention or seizure of our products; |
| • | operating restrictions, partial suspension or total shutdown of production; |
| • | refusal of or delay in granting our requests for 510(k) clearance or premarket approval of new products or modified products; |
| • | withdrawing 510(k) clearance or premarket approvals that are already granted; |
| • | refusal to grant export approval for our products; |
105
Table of Contents
| • | criminal prosecution; and |
| • | unanticipated expenditures to address or defend such actions. |
We are subject to unannounced device inspections by the FDA, the Office of Compliance, the Center for Devices and Radiological Health and the Center for Biologics Evaluation and Research, as well as other regulatory agencies overseeing the implementation and adherence of applicable state and federal tissue licensing regulations. These inspections may include our suppliers’ facilities.
Third-Party Coverage and Reimbursement
Healthcare providers in the United States generally rely on third-party payors, principally private insurers and governmental payors such as Medicare and Medicaid, to cover and reimburse all or part of the cost of a spine surgery in which our products are used. We expect that sales volumes and prices of our products will continue to depend in large part on the availability of coverage and reimbursement amounts from such third-party payors. Third party payors perform analyses on new technologies to determine if they are medically necessary, before providing coverage for them. These third-party payors may still deny reimbursement on covered technologies if they determine that a device used in a procedure was not used in accordance with the payor’s coverage policy. Particularly in the United States, third-party payors continue to carefully review, and increasingly challenge, the utilization of specific technologies in spine procedures.
Medicare coverage and reimbursement policies are developed by the Centers for Medicare & Medicaid Services, or CMS, the federal agency responsible for administering the Medicare program, and its contractors. CMS establishes Medicare coverage and reimbursement policies for medical products and procedures and such policies are periodically reviewed and updated. While private payors vary in their coverage and payment policies, most look upon the coverage and payment levels established by Medicare as a benchmark by which to make their own coverage decisions. Medicare reimbursement rates for the same or similar procedures vary due to geographic location, nature of the facility in which the procedure is performed (i.e., teaching or community hospital) and other factors. We cannot assure you that government or private third-party payors will cover and reimburse the procedures using our products in whole or in part in the future or that payment rates will be adequate.
A key component in ensuring whether the appropriate payment amount is received for physician and other services, including those procedures using our products, is the existence of a CPT code. To receive payment, healthcare practitioners must submit claims to insurers using these codes for payment for medical services. CPT codes are assigned, maintained and annually updated by the American Medical Association and its CPT Editorial Board. The lack of a CPT code that describes procedures performed using our products, or a change to an existing code that describes such procedures, may adversely affect reimbursement for these procedures. For example, two-level cervical disc replacement procedures currently require the use of Category III “T-Code” 0092T. T-codes are tracking codes for new technologies for which no specific physician payment has been assigned. However, the Current Procedural Terminology Editorial Panel recently accepted the addition of Category I code 22858X as an add-on code to support two-level cervical disc replacement procedures. The code will become effective January 1, 2015.
In the United States, a large percentage of insured individuals receive their medical care through managed care programs, which monitor and often require pre-approval of the services that a member will receive. Some managed care programs pay their providers on a per capita basis, which puts the providers at financial risk for the services provided to their patients by paying these providers a predetermined payment per member per month and, consequently, may limit the willingness of these providers to use our products.
106
Table of Contents
We believe that the overall escalating cost of medical products and services has led to, and will continue to lead to, increased pressures on the healthcare industry to reduce the costs of products and services. We cannot assure you that government or private third-party payors will cover and reimburse the procedures using our products in whole or in part in the future or that payment rates will be adequate. In addition, it is possible that future legislation, regulation or reimbursement policies of third-party payors will adversely affect the demand for our procedures and products or our ability to sell them on a profitable basis. The unavailability or inadequacy of third-party payor coverage or reimbursement could have a material adverse effect on our business, operating results and financial condition.
Internationally, reimbursement and healthcare payment systems vary substantially from country to country and include single-payor, government-managed systems as well as systems in which private payors and government managed systems exist side-by-side. Our ability to achieve market acceptance or significant sales volume in international markets we enter will be dependent in large part on the availability of reimbursement for procedures performed using our products under the healthcare payment systems in such markets. A small number of countries may require us to gather additional clinical data before recognizing coverage and reimbursement for our products. It is our intent to complete the requisite clinical studies and obtain coverage and reimbursement approval in countries where it makes economic sense to do so.
Healthcare Fraud and Abuse
Federal healthcare fraud and abuse laws most frequently apply to our business when a customer submits a claim for an item or service that is reimbursed under Medicare, Medicaid or most other federally funded healthcare programs. The federal Anti-Kickback Statute prohibits unlawful inducements for the referral of business reimbursable under federal healthcare programs, such as remuneration provided to physicians to induce them to use certain tissue products or medical devices reimbursable by Medicare or Medicaid. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham consultant arrangements with physicians. Further, the recently enacted Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively, the PPACA, among other things, amends the intent requirements of the federal Anti-Kickback Statute and the criminal statutes governing healthcare fraud, which prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. A person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them. In addition, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act or federal civil money penalties statute. The majority of states also have anti-kickback laws which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigations of healthcare providers and suppliers throughout the country for a wide variety of Medicare billing practices, and has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and suppliers’ compliance with the healthcare coverage and reimbursement rules and fraud and abuse laws.
107
Table of Contents
The PPACA, among other things, also imposed new reporting requirements on device manufacturers for payments by them and in some cases their distributors to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members (commonly known as the Physician Payments Sunshine Act or Open Payments). Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission.
The period between August 1, 2013 and December 31, 2013 was the first reporting period under the Open Payments system. Starting February 18, 2014, Open Payments registration and data submission for the Federal Physician Payments Sunshine Act opened with a two-phased approach for reporting the applicable 2013 payments. Phase 1 (February 18, 2014—March 31, 2014) includes user registration in CMS’ Enterprise Portal and submission of corporate profile information and aggregate payment data from the first reporting period. Phase 2 (begins in May and extends for at least 30 days) included industry registration in the Open Payments system, submission of detailed payment data from the first reporting period, and legal attestation to the accuracy of the data. Thereafter, device manufacturers must submit reports by the 90th day of each subsequent calendar year. Due to the difficulty in complying with Physician Payment Sunshine Act and the nature of our U.S. sales force concentrated with independent sales agencies, we cannot assure you that we will successfully report all transfers of value by us and our independent sales agencies, and any failure to comply could result in significant fines and penalties.
In addition, there has been a recent trend of increased state regulation of payments made to physicians. Certain states mandate implementation of commercial compliance programs, impose restrictions on device manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare providers. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may violate one or more of the requirements.
If a governmental authority were to conclude that we are not in compliance with applicable laws and regulations, we and our officers and employees could be subject to severe criminal and civil penalties, including exclusion from participation as a supplier of product to beneficiaries covered by Medicare or Medicaid.
Similar laws are increasingly being introduced in the individual EEA countries. The provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medical devices is prohibited. The provision of benefits or advantages to physicians is also governed by the national antibribery laws of the EEA countries. One such example is the UK Bribery Act. Payments made to physicians in certain EEA countries must also be publically disclosed. Moreover, agreements with physicians must often be the subject of prior notification and approval by the physician’s employer, his/her competent professional organization, and/or the competent authorities of the EEA countries. These requirements are provided in the national laws, industry codes, or professional codes of conduct applicable in the EEA countries. Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
Intellectual Property
Our success depends upon our ability to protect our intellectual property. For protection of our intellectual property, we rely on a combination of intellectual property rights, including patents, trade secrets, copyrights and trademarks, as well as customary confidentiality and other contractual
108
Table of Contents
protections. As of December 31, 2013, we owned over 330 issued patents globally, of which 27 were issued U.S. patents. As of December 31, 2013, we owned over 80 patent applications pending globally, of which 28 were patent applications pending in the United States. As of December 31, 2013, 20 of our U.S. issued patents have pending continuations or divisionals in process which may provide additional intellectual property protection if issued as U.S. patents. Our issued patents expire between 2020 and 2031, subject to payment of required maintenance fees, annuities and other charges. As of December 31, 2013, we also owned 15 U.S. trademark registrations and 59 foreign trademark registrations, as well as five pending U.S. trademark registrations and 14 pending foreign trademark registrations.
We also rely upon trade secrets, know-how, continuing technological innovation, and may in the future rely upon licensing opportunities, to develop and maintain our competitive position. We protect our proprietary rights through a variety of methods, including confidentiality agreements and proprietary information agreements with suppliers, employees, consultants and others who may have access to proprietary information.
Employees
As of December 31, 2013, we had 323 employees, 183 of whom were primarily engaged in sales and marketing, 64 of whom were primarily engaged in product development and research, and 76 of whom were primarily engaged in administration and finance. A majority of these employees are located outside the United States. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good
Facilities
Our principal offices are located in Austin, Texas and Troyes, France. Our offices in Austin, Texas occupy approximately 67,410 square feet of leased space in Austin, Texas. The lease term expires in 2019. We may extend our lease an additional five years or choose to let the initial lease expire and find alternative office space to purchase or lease. Our offices in Troyes, Frances occupy approximately 23,099 square feet of leased space. The initial lease expires in 2015. We may extend our lease until 2018 or choose to let the initial lease expire and find additional office space to purchase or lease.
We also maintain sales, distribution or development offices in France, Brazil, China and Korea. We believe that our current facilities are suitable and adequate to meet our current needs. We intend to add new facilities or expand existing facilities as we add employees, and we believe that suitable additional or substitute space will be available as needed to accommodate any such expansion of our operations.
Legal Proceedings
We are not aware of any pending or threatened legal proceeding against us that could have a material adverse effect on our business, operating results or financial condition. The medical device industry is characterized by frequent claims and litigation, including claims regarding patent and other intellectual property rights as well as improper hiring practices. As a result, we may be involved in various additional legal proceedings from time to time.
109
Table of Contents
Executive Officers and Directors
The following table sets forth information about our directors, executive officers and other key employees as of March 24, 2014.
| Name |
Age | Position(s) | ||||
| Executive Officers |
||||||
| Christophe Lavigne |
46 | President, Chief Executive Officer and Chairman of the Board of Directors | ||||
| Hervé Dinville |
62 | Executive Vice President of Research and Development, Médical | ||||
| Patrick Richard |
50 | Executive Vice President of European Sales, Médical | ||||
| Robert E. McNamara |
57 | Executive Vice President, Chief Financial Officer | ||||
| Scott Way |
42 | Executive Vice President, General Counsel, Compliance Officer and Secretary | ||||
| Andre Potgieter |
46 | Executive Vice President, U.S. Sales, LDR Spine | ||||
| James Burrows |
44 | Executive Vice President, Chief Operating Officer, LDR Spine | ||||
| G. Joseph Ross |
46 | Executive Vice President, Global Marketing, LDR Spine | ||||
| Non-Employee Directors |
||||||
| Joseph Aragona(1) |
57 | Director | ||||
| Kevin Lalande(1)(2)(3) |
41 | Director | ||||
| Stefan Widensohler(1)(2)(3) |
54 | Director | ||||
| William Burke(2)(3) |
54 | Director | ||||
| (1) | Member of our Nominating and Corporate Governance Committee |
| (2) | Member of our Audit Committee |
| (3) | Member of our Compensation Committee |
Executive Officers
Christophe Lavigne, 46, is one of our co-founders and has served as Chairman of the Board of Directors, President and Chief Executive Officer since 2006. Additionally, Mr. Lavigne has served as Chairman of the Board of Directors and President of Médical since 2002 and President of its predecessor entity since its founding in 2000. Mr. Lavigne has more than 20 years working for spinal device companies in roles of increasing responsibility, culminating as Sales and Marketing Director for JBS, a privately held medical device manufacturer, and as Sales and Marketing Neuro-Spine Director for Aesculap, Inc., a medical device manufacturer and a division of B. Braun Melsungen AG. Mr. Lavigne won the French prize of Ernst & Young’s “Entrepreneur of the Year” award in 2006 and the Ernst & Young “Entrepreneur of the Year” award for the central Texas region in 2011. Mr. Lavigne holds a degree in mechanical engineering from the IUT Dijon, France, a degree in business from the ESARC Paris business school and a masters in marketing (DEESMA) from the European Business School Federation—Zurich. Mr. Lavigne was selected to serve on our Board of Directors because of his substantial knowledge of the Company he has as our co-founder, Chairman and Chief Executive Officer.
Hervé Dinville, 62, is one of our co-founders, has served as Executive Vice President of Research and Development since January 2013 and as our head of Research and Development and a director of LDR Médical since our founding in 2000. For the past 25 years, he has worked extensively in clinical and developmental settings with prominent spine surgeons to design novel implantable devices and to improve surgical procedures. From 1997 to 2000, Mr. Dinville was the Research and Development
110
Table of Contents
Manager for Aesculap, Inc., a medical device manufacturer and a division of B. Braun Melsungen AG. From 1988 to 1997, Mr. Dinville was in charge of the development of the orthopedic and spine ranges for JBS, a privately held medical device manufacturer.
Patrick Richard, 50, is one of our co-founders, has served as Executive Vice President since January 2013 and as our head of European Sales and a director of LDR Médical since our founding in 2000. For the past 20 years, Mr. Richard has worked with spinal device companies in Europe to introduce numerous new procedures and technologies in the European market, including minimally invasive anterior spine surgery, sequential loading and correction for scoliosis applications and cervical and lumbar disc replacement devices. Mr. Richard served as regional sales manager for JBS, a privately held medical device manufacturer and as sales director of France for Aesculap, Inc., a medical device manufacturer and a division of B. Braun Melsungen AG.
Robert E. McNamara, 57, has served as our Executive Vice President since January 2013 and as our Chief Financial Officer since April 2012. From September 2010 to April 2012, Mr. McNamara served as a financial consultant, working primarily in the medical device and biotechnology industries. From May 2009 to September 2010, he served as Chief Financial Officer of Purfresh, Inc., a privately held clean technology company. From December 2004 to September 2008, Mr. McNamara was the Senior Vice President and Chief Financial Officer of Accuray, Inc., a publicly traded medical device manufacturer. In addition, Mr. McNamara has served as the Senior Vice President and Chief Financial Officer for publicly traded medical device companies, Somnus Medical Technologies and Target Therapeutics, was a member of the Board of Directors of Northstar Neurosciences and is the former Mayor of Menlo Park, California. Mr. McNamara holds a B.S. in Accounting from the University of San Francisco and an M.B.A. in Finance from The Wharton School of the University of Pennsylvania.
Scott Way, 42, has served as our Executive Vice President since January 2013 and as our General Counsel and Compliance Officer since January 2011, and as our Secretary since May 2012. From November 2007 to January 2011, Mr. Way served as Corporate Counsel for a division of DJO Incorporated, a medical device manufacturer, with primary responsibility for all legal matters related to its surgical implant and durable medical goods divisions. From December 2004 to November 2007, he was Corporate Counsel for Encore Medical, Inc., a public company and manufacturer of medical devices that was acquired by the Blackstone Group in a going private transaction in 2006, and later merged in November of 2007 with DJO. Mr. Way holds a B.A. in Political Science from Kenyon College and a J.D. from The University of Texas School of Law.
André Potgieter, 46, has served as our Executive Vice President, U.S. Sales since January 2013 and as our Vice President, U.S. Sales from October 2007 to January 2013. He has spent 20 years in the spinal implant industry, including eight years in Sales, Regional Sales Management and Director of Sales positions with Kyphon Global, Inc., a provider of minimally invasive spinal surgery solutions and a division of Medtronic Spine LLC, and seven years as a Spine Consultant with Synthes Spine, a provider of orthopedic and neurological solutions acquired by Johnson & Johnson. Mr. Potgieter holds a B.A. in Finance from the University of Tennessee.
James Burrows, 44, has served as our Executive Vice President since January 2013 and as our Chief Operating Officer since December 2009. Mr. Burrows joined LDR in January 2005. From 2001 to 2004, Mr. Burrows was the Director of Product Marketing for Centerpulse Orthopedics, a provider of musculoskeletal health solutions that was acquired by Zimmer Orthopedics in May 2003. Mr. Burrows has over 20 years of experience in the orthopedic and spine industry, working for Intermedics Orthopedics, Sulzer Medica and Centerpulse and has served in various leadership roles including strategic, marketing, technical and logistical duties. He holds six U.S. patents on medical device product designs. Mr. Burrows holds a B.S. in Engineering Technology and an M.B.A. from Texas State University.
111
Table of Contents
G. Joseph Ross, 46, has served as our Executive Vice President of Global Marketing since January 2013 and as our Vice President of U.S. Marketing since 2010. From 1995 to 2007, Mr. Ross served in a variety of roles of increasing responsibility for DePuy Spine, a division of Johnson & Johnson, including sales, marketing and lastly as Worldwide Vice President of New Business Development. Mr. Ross has 18 years of experience in the spine medical device industry and holds multiple patents for spine implant and instrument designs. Mr. Ross holds a BBA in Marketing from the University of Notre Dame and an MS in Management from Troy University. He also served five years as an officer in the United States Navy.
Non-Employee Directors
Joseph Aragona, 57, has served on our Board of Directors since March 2006. Mr. Aragona is one of the founders of Austin Ventures, a venture capital firm, and has served as general partner since 1982. He has invested in a broad range of industries, including medical device, software, services, industrial products and special situations. Mr. Aragona serves on the boards of directors of a number of private companies. Mr. Aragona holds an A.B. in Economics from Harvard University and an M.B.A. from the Harvard Business School. Mr. Aragona was selected to serve on our Board of Directors because of his substantial experience as a venture capitalist and as a director of a number of privately held companies.
Kevin M. Lalande, 41, has served on our board of directors since December 2005. Mr. Lalande is one of the founders and a Managing Director of Santé Ventures, a venture capital firm. Prior to founding Santé Ventures in 2006, Mr. Lalande spent seven years with Austin Ventures. Before joining Austin Ventures, Mr. Lalande was a management consultant with McKinsey & Company. Previously, Mr. Lalande co-founded and sold three companies: NetProfit, sold to a privately held advertising agency in 1996; Serus, sold to Netopia in 1998; and TimeMarker, sold to PrimeHoldings in 2001. Mr. Lalande holds a B.S. in Electrical and Computer Engineering from Brigham Young University and an M.B.A. with Highest Distinction from the Harvard Business School. Mr. Lalande was selected to serve on our Board of Directors because of his substantial experience as a venture capitalist and as a director of a number of privately held companies.
Stefan Widensohler, 53, has served on our board of directors since May 2006. Since 1992, Mr. Widensohler has been President, Chief Executive Officer, Principal and Proprietor of KRAUTH medical KG, a privately held company in Hamburg, Germany. Until 2012, KRAUTH medical KG was a medical device distribution and service company. KRAUTH medical KG sold its medical device distribution and service business and now focuses on investing in healthcare start-up companies. In 1996, Mr. Widensohler also co-founded Invatec SpA, an Italian medical device company that specializes in interventional cardiology and peripheral vascular products, which was acquired by Medtronic, Inc. in 2010. Mr. Widensohler joined Medtronic following the acquisition and served as Vice President, Global Sales for the acquired business until 2012 to assist with the integration of Invatec into Medtronic. Since July 2013, Mr. Widensohler has served as a director of St. Jude Medical, a publicly traded medical device company. Since January 2011, Mr. Widensohler has served as a director of MyoPowers Medical Technologies SA, a privately held medical device company. He serves as Member of the Advisory Board of TowerBrook Capital Partners L.P., a private equity fund. He also serves as deputy chairman of the Board of BVMed, the German Health Industry Manufacturers Association. Mr. Widensohler is an Economics graduate from the Private Academy of Bad Harzburg, Germany. Mr. Widensohler was selected to serve on our Board of Directors because of his substantial experience as an investor in the healthcare market and as a director of a number of privately and publicly held companies.
William W. Burke, 54, has served on our board of directors since October 2013. Mr. Burke served as Executive Vice President & Chief Financial Officer of IDEV Technologies, a developer of next generation medical devices for use by interventional radiologists, vascular surgeons and cardiologists, from November 2009 to December 2013. IDEV was acquired by Abbott Laboratories, a publicly traded healthcare company, in August 2013 and operates as a wholly owned subsidiary of Abbott. He also
112
Table of Contents
serves as a member of the board of directors of Medical Action Industries, a manufacturer of disposable medical products. From August 2004 through December 2007, he served as Executive Vice President & Chief Financial Officer of ReAble Therapeutics, a diversified orthopedic device company. ReAble was sold to The Blackstone Group in a going private transaction in 2006 and subsequently merged with DJO Incorporated in late 2007. Subsequent to the completion of the transaction with DJO, Mr. Burke served as a consultant to Blackstone/DJO from December 2007 until June 2008. From 2001 to 2004, he served as Chief Financial Officer of Cholestech Corporation, a medical diagnostics products company. From 1985 to 2001, he was employed as a senior investment banker by several firms, with a primary focus on medical technology companies. Mr. Burke received a B.B.A. in Finance from The University of Texas at Austin and an MBA from The Wharton School of the University of Pennsylvania. Mr. Burke was selected to serve on our Board of Directors because of his knowledge of accounting matters, his business experience with other medical technology companies, and his experience as the chief financial officer of other companies, including other public companies.
Board Composition
Our Board of Directors currently consists of five members. In accordance with our Amended and Restated Certificate of Incorporation and our Amended and Restated Bylaws, our Board of Directors is divided into three classes, Class I, Class II, and Class III, with each class containing, as nearly as possible, one-third of the total number of directors and serving staggered three-year terms. Directors in a particular class are elected for three-year terms at the annual meeting of stockholders in the year in which their terms expire. The classification of our Board of Directors may have the effect of delaying or preventing changes in our management or a change in control of our company.
The authorized number of directors may be changed only by our Board of Directors. Vacancies on the Board of Directors may be filled only by persons elected by a majority of the remaining directors. A director elected by the Board of Directors to fill a vacancy in a class, including vacancies created by an increase in the number of directors, serves for the remainder of the full term of that class or until the director’s successor is duly elected and qualified. Any increase or decrease in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors.
Our directors may be removed only for cause by the affirmative vote of the holders of two thirds of our outstanding voting stock.
Board Independence
We completed our initial public offering in October 2013, and our common stock trades on the NASDAQ Global Select Market, or NASDAQ. Under NASDAQ rules, independent directors must comprise a majority of a listed company’s board of directors within twelve months from the date of listing. In addition, NASDAQ rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation and nominating and governance committees be independent within twelve months from the date of listing. Audit committee members must also satisfy additional independence criteria, including those set forth in Rule 10A-3 promulgated under the Exchange Act. Under NASDAQ rules, a director will qualify as an “independent director” only if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. In order to be considered independent for purposes of Rule 10A-3 under the Exchange Act, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries, other than compensation for board service; or (2) be an affiliated person of the listed company or any of its subsidiaries.
113
Table of Contents
Our Board of Directors has undertaken a review of its composition and that of its committees as well as the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our Board of Directors has determined that each of Messrs. Burke, Lalande and Widensholder is “independent” in accordance with NASDAQ rules and Rule 10A-3 under the Exchange Act.
Board Committees
Our board of directors has an audit committee, a compensation committee, and a nominating and corporate governance committee.
Audit Committee
Our audit committee consists of William Burke (chair), Kevin Lalande and Stefan Widensohler. The functions of the audit committee include:
| • | appointing our independent registered public accounting firm and being directly responsible for the compensation, retention and oversight of the independent registered public accounting firm, who will report directly to the audit committee; |
| • | reviewing and pre-approving the engagement of our independent registered public accounting firm to perform audit services and any permissible non-audit services, including the fees to be paid for those services; |
| • | reviewing our annual and quarterly financial statements and reports and discussing the financial statements and reports with our independent registered public accounting firm and management; |
| • | reviewing and approving all related person transactions; |
| • | reviewing with our independent registered public accounting firm and management significant issues that may arise regarding accounting principles and financial statement presentation, as well as matters concerning the scope, adequacy and effectiveness of our internal controls over financial reporting; |
| • | establishing procedures for the receipt, retention and treatment of complaints received by us regarding internal controls over financial reporting, accounting or auditing matters; and |
| • | preparing the audit committee report for inclusion in our proxy statement for our annual meeting. |
All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and NASDAQ.
Our board of directors has determined that Mr. Burke qualifies as an audit committee financial expert within the meaning of applicable SEC regulations. In making this determination, our board of directors considered the nature and scope of experience that Mr. Burke has previously had with public reporting companies, including service as Chief Financial Officer of public companies and service on public company audit committees. Our board of directors has determined that all of the current members of our audit committee satisfy the relevant independence requirements for service on the audit committee set forth in the rules of NASDAQ and applicable SEC rules. Both our independent registered public accounting firm and management periodically meet privately with our audit committee.
Our board of directors has adopted an audit committee charter. We believe that the composition of our audit committee, and our audit committee’s charter and functioning, comply with the applicable requirements of NASDAQ and SEC rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
114
Table of Contents
The full text of our audit committee charter can be found on the investor relations portion of our website at http://ir.ldr.com under the heading “Corporate Governance.” Information contained in, or accessible through, our website is not a part of this prospectus.
Compensation Committee
Our compensation committee consists of Kevin Lalande (chair), William Burke and Stefan Widensohler. The functions of the compensation committee include:
| • | determining the compensation and other terms of employment of our Chief Executive Officer and other executive officers and reviewing and approving our performance goals and objectives relevant to that compensation; |
| • | administering and implementing our incentive compensations plans and equity-based plans, including approving option grants, restricted stock and other awards; |
| • | evaluating our incentive compensation plans, equity-based plans and similar programs advisable for us, as well as modifications or terminations of our existing plans and programs; |
| • | reviewing and approving the terms of any employment-related agreements, severance arrangements, change-in-control and similar agreements or provisions with our Chief Executive Officer and other executive officers; |
| • | approving, authorizing and providing oversight over a sub-committee to grant equity awards to non-executive officer employees, subject to grant guidelines approved by our compensation committee; |
| • | reviewing and discussing the Compensation Discussion & Analysis required in our annual report and proxy statement with management and determining whether to include the Compensation Discussion & Analysis in the annual report or proxy; and |
| • | preparing a report on executive compensation for inclusion in our proxy statement for our annual meeting. |
Each member of our compensation committee is a non-employee director, as defined in Rule 16b-3 promulgated under the Exchange Act, and an outside director, as defined pursuant to Section 162(m) of the Internal Revenue Code of 1986. Furthermore, our board of directors has determined that each of Messrs. Lalande, Burke and Widensohler satisfy the independence standards for compensation committee membership established by the SEC and the listing rules of NASDAQ, as applicable.
Our board of directors has adopted a compensation committee charter. We believe that the composition of our compensation committee, and our compensation committee’s charter and functioning, comply with the applicable requirements of NASDAQ and SEC rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
The full text of our compensation committee charter can be found on the investor relations portion of our website at http://ir.ldr.com under the heading “Corporate Governance.” Information contained in, or accessible through, our website is not a part of this prospectus.
115
Table of Contents
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Joseph Aragona (chair), Kevin Lalande and Stefan Widensohler. The functions of the nominating and corporate governance committee include:
| • | evaluating director performance on the board of directors and applicable committees of the board of directors; |
| • | identifying, recruiting, evaluating and recommending individuals for membership on our board of directors, the audit committee and the compensation committee; |
| • | considering questions of independence or possible conflicts of interest (other than related person transactions) of members of our board of directors or our executive officers; |
| • | evaluating nominations by stockholders of candidates for election to our board of directors; |
| • | reviewing and recommending to our board of directors any amendments to our corporate governance documents; and |
| • | making recommendations to our board of directors regarding management succession planning. |
Our board of directors has determined that Messrs. Lalande and Widensohler each satisfy the independence standards for nominating and corporate governance committee members established by the SEC and NASDAQ listing standards, as applicable. Mr. Aragona does not qualify as an independent director; however, pursuant to NASDAQ listing standards, he may remain on the nominating and corporate governance committee until one year from the listing of our common stock on NASDAQ.
Our board of directors has adopted a nominating and corporate governance committee charter. We believe that the composition of our nominating and corporate governance committee, and our nominating and corporate governance committee’s charter and functioning, comply with the applicable requirements of NASDAQ and SEC rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
The full text of our nominating and corporate governance committee charter can be found on the investor relations portion of our website at http://ir.ldr.com under the heading “Corporate Governance.” Information contained in, or accessible through, our website is not a part of this prospectus.
Compensation Committee Interlocks and Insider Participation
None of our executive officers serve as a member of the board of directors or compensation committee of any entity that has one or more of its executive officers serving as a member of our board of directors or our compensation committee.
Code of Conduct
Our board of directors has adopted a Code of Conduct. The Code of Conduct applies to all of our employees, officers (including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions), agents and representatives, including directors and consultants.
Our board of directors has also adopted an additional Code of Ethics for our Chief Executive Officer, Chief Financial Officer and Principal Accounting Officer. This Code of Ethics, applicable to the specified officers, contains additional requirements including a prohibition on personal loans from the company (except where permitted by, and disclosed pursuant to, applicable law), and a requirement to review reports to be filed with SEC.
116
Table of Contents
We intend to disclose future amendments to certain provisions of our Code of Conduct and our Code of Ethics for our Chief Executive Officer, Chief Financial Officer and Principal Accounting Officer, or waivers of those provisions, applicable to any principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions or our directors on our website identified below. The full text of our Code of Conduct and our Code of Ethics for our Chief Executive Officer, Chief Financial Officer and Principal Accounting Officer can be found in the Investor Relations section of our website at http://ir.ldr.com under the heading “Corporate Governance.” Information contained in, or accessible through, our website is not a part of this prospectus.
Director Compensation
Prior to our October 2013 initial public offering, we did not compensate non-employee directors for their service on our board of directors. We also have not previously, and do not now, provide separate compensation to any director who is also our employee, including Mr. Lavigne, our President and Chief Executive Officer. In addition, we do not compensate any non-employee directors that are not independent directors, such as Mr. Aragona, an affiliate of Austin Ventures.
Following our initial public offering, we implemented a compensation program for independent non-employee directors under which each independent non-employee director receives an annual fee of $30,000. Independent non-employee directors receive an additional $10,000 annually for serving on the audit committee of our board of directors, an additional $7,500 annually for serving on the compensation committee of our board of directors and an additional $5,000 for serving on the nominating and corporate governance committee of our board of directors. The chairman of our audit committee receives an additional $10,000 annually, the chairman of our compensation committee receives an additional $7,500 annually and the chairman of our nominating and corporate governance committee receives an additional $3,000 annually.
According to our Non-Employee Independent Director Compensation Policy adopted by our board of directors on September 11, 2013, each independent member of our board of directors who is not our employee or an employee of any of our subsidiaries may be eligible to receive cash compensation or equity awards. So long as an independent, non-employee director continues to serve as an independent, non-employee director following our annual stockholders’ meeting, whether or not such individual is standing for re-election at such meeting, such independent, non-employee director shall be granted an equity award in the form of an option to purchase 622 shares of our common stock (as adjusted to reflect stock, dividends, stock splits, combinations, recapitalizations and the like with respect to such shares). Such shares shall vest in 12 equal monthly installments over the 12-month period measured from the grant date for so long as such independent, non-employee director remains in our service through each vesting date, provided, however, that if our next regular annual stockholder’s meeting occurs prior to the end of the 12-month vesting period for such annual grant, any unvested option shares subject to such annual grant shall become vested immediately prior to such meeting if such non-employee director serves until such meeting.
The compensation paid to each of our independent non-employee directors in 2013 is set forth below. Christophe Lavigne, our President and Chief Executive Officer, and Joseph Aragona, who the board of directors has determined is not an independent director due to his relationship with Austin Ventures, receive no separate compensation for serving as directors.
| Name |
Fees earned or paid in cash ($)(1) |
Option Awards ($)(2)(3) |
Total ($) |
|||||||||
| William W. Burke |
$ | 14,375 | $ | 22,219 | $ | 36,594 | ||||||
| Kevin M. Lalande |
14,375 | 22,219 | 36,594 | |||||||||
| Stefan Widensohler |
13,125 | 22,219 | 35,344 | |||||||||
117
Table of Contents
| (1) | Cash fees paid for board of directors and/or committee service reflect a partial year of service beginning upon the completion of our initial public offering in October 2013. |
| (2) | Amounts represent the aggregate grant date fair market value of stock options granted during 2013, computed in accordance with FASB ASC Topic 718. Assumptions used in calculating the grant date fair value of the stock options reported in this column are set forth in Note 11 to the consolidated financial statements of LDR Holding Corporation and subsidiaries, included in our Annual Report on Form 10-K for the year ended December 31, 2013 filed with the Securities and Exchange Commission on March 4, 2014. |
| (3) | As of December 31, 2013, the aggregate number of shares subject to outstanding equity awards held by our non-employee directors was: |
| Name |
Stock Options | |||
| William W. Burke |
3,111 | |||
| Kevin M. Lalande |
3,111 | |||
| Stefan Widensohler |
3,111 | |||
We provide reimbursement to our non-employee directors (including Mr. Aragona) for their reasonable expenses incurred in attending meetings of our board of directors and committees of our board of directors. Directors are also entitled to the protection provided by their indemnification agreements and the indemnification provisions in our amended and restated certificate of incorporation and bylaws.
Limitations on Liability and Indemnification Agreements
As permitted by Delaware law, provisions in our amended and restated certificate of incorporation and amended and restated bylaws, limit or eliminate the personal liability of directors for a breach of their fiduciary duty of care as a director. The duty of care generally requires that, when acting on behalf of the corporation, a director exercise an informed business judgment based on all material information reasonably available to him or her. Consequently, a director will not be personally liable to us or our stockholders for monetary damages or breach of fiduciary duty as a director, except for liability for:
| • | any breach of the director’s duty of loyalty to us or our stockholders; |
| • | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| • | any act related to unlawful stock repurchases, redemptions or other distributions or payments of dividends; or |
| • | any transaction from which the director derived an improper personal benefit. |
These limitations of liability do not limit or eliminate our rights or any stockholder’s rights to seek non-monetary relief, such as injunctive relief or rescission. These provisions will not alter a director’s liability under other laws, such as the federal securities laws or other state or federal laws. Our amended and restated certificate of incorporation that will become effective upon the completion of this offering also authorizes us to indemnify our officers, directors and other agents to the fullest extent permitted under Delaware law.
As permitted by Delaware law, our amended and restated bylaws to be effective upon the consummation of this offering will provide that:
| • | we will indemnify our directors, officers, employees and other agents to the fullest extent permitted by law; |
| • | we must advance expenses to our directors and officers, and may advance expenses to our employees and other agents, in connection with a legal proceeding to the fullest extent permitted by law; and |
| • | the rights provided in our amended and restated bylaws are not exclusive. |
118
Table of Contents
If Delaware law is amended to authorize corporate action further eliminating or limiting the personal liability of a director or officer, then the liability of our directors or officers will be so eliminated or limited to the fullest extent permitted by Delaware law, as so amended. Our bylaws also permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in connection with their services to us, regardless of whether our bylaws permit such indemnification. We have obtained such insurance.
In addition to the indemnification that will be provided for in our amended and restated certificate of incorporation and amended and restated bylaws, we will enter into separate indemnification agreements with each of our directors and executive officers, which may be broader than the specific indemnification provisions contained in the Delaware General Corporation Law. These indemnification agreements may require us, among other things, to indemnify our directors and executive officers for some expenses, including attorneys’ fees, expenses, judgments, fines and settlement amounts incurred by a director or executive officer in any action or proceeding arising out of his service as one of our directors or executive officers or any other company or enterprise to which the person provides services at our request. We believe that these provisions and agreements are necessary to attract and retain qualified individuals to serve as directors and executive officers.
This description of the indemnification provisions of our amended and restated certificate of incorporation, our amended and restated bylaws and our indemnification agreements is qualified in its entirety by reference to these documents, each of which is attached as an exhibit to the registration statement of which this prospectus forms a part.
Insofar as indemnification for liabilities arising under the Securities Act of 1933, as amended, or the Securities Act, may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act, and is, therefore, unenforceable.
There is no pending litigation or proceeding naming any of our directors or officers as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director or officer.
119
Table of Contents
Summary Compensation Table
The following table shows the compensation awarded or paid to, or earned by, our Chief Executive Officer and our two other most highly compensated executive officers for the fiscal years ended December 31, 2013 and 2012. We refer to these three executive officers in this prospectus as our named executive officers.
| Name and Principal Position |
Year | Salary ($) |
Bonus ($) |
Option Awards ($)(1) |
Nonequity Incentive Plan Compensation ($)(2) |
All Other Compensation ($)(3) |
Total ($) |
|||||||||||||||||||||
| Christophe Lavigne, |
2013 | 450,000 | 0 | 603,134 | 172,305 | 27,522 | 1,252,961 | |||||||||||||||||||||
| President and Chief |
2012 | 440,000 | 0 | 104,274 | 137,394 | 129,774 | 811,442 | |||||||||||||||||||||
| Executive Officer |
||||||||||||||||||||||||||||
| Robert McNamara, |
2013 | 300,000 | 75,000 | 1,857,863 | 0 | 6,008 | 2,238,871 | |||||||||||||||||||||
| Executive Vice |
2012 | 206,923 | 71,250 | 119,970 | 0 | 53,458 | 451,601 | |||||||||||||||||||||
| President and Chief |
||||||||||||||||||||||||||||
| Financial Officer |
||||||||||||||||||||||||||||
| Patrick Richard, |
2013 | 243,024 | (4) | 26,560 | (4) | 452,347 | 0 | 19,771 | 741,702 | |||||||||||||||||||
| Executive Vice |
||||||||||||||||||||||||||||
| President of European |
||||||||||||||||||||||||||||
| Sales, Médical |
||||||||||||||||||||||||||||
| (1) | We calculate the values of option awards based on the aggregate grant date fair market value of stock options, computed in accordance with FASB ASC Topic 718. Assumptions used in calculating the fair market value of stock options are described in Note 11 to the consolidated financial statements of LDR Holding Corporation and subsidiaries, included in our Annual Report on Form 10-K for the year ended December 31, 2013 filed with the Securities and Exchange Commission. |
| (2) | These amounts represent payments approved by our compensation committee and our Board of Directors and paid to the named executive officers under our 2012 Executive Bonus Plan and 2013 Executive Bonus Plan, each as described below. |
| (3) | Includes (a) for Mr. Lavigne and Mr. McNamara, payments related to mobile phone usage and an executive wellness examination, (b) in the case of Mr. Lavigne, (i) the cost to us for leasing Mr. Lavigne an automobile, which was $19,855 for 2013 and $21,120 for 2012, (ii) a payment of $46,619 approved by our compensation committee, representing a reimbursement for the exercise price of 25,114 stock options in 2012, (iii) a payment of $16,375 approved by our compensation committee, representing a gross-up for the estimated alternative minimum tax incurred by Mr. Lavigne in connection with the exercise of 25,114 stock options in 2012, and (iv) a payment of $36,131 approved by our compensation committee, representing amounts associated with the withholding taxes associated with the payments made in 2012 described above, (c) in the case of Mr. McNamara, relocation expenses and temporary living and travel expenses incurred by him from May 1, 2012 until his relocation to Austin, Texas (Mr. McNamara was hired in April 2012 and his amounts reflect payments received in 2012 following his start date), and (d) in the case of Mr. Richard, a car allowance totaling $6,907, and unemployment insurance premiums totaling $12,864, paid on Mr. Richard’s behalf. |
| (4) | Mr. Richard’s base salary and bonus is calculated based on Euros. The amount reported in the table represents the U.S. dollar equivalent based on the exchange rate of 1.328, calculated by averaging the monthly average exchange rate for each month in calendar year 2013. |
Cash Awards under the 2012 Executive Compensation Plan
Mr. Lavigne participated in, and was eligible for cash awards under our 2012 Executive Compensation Plan, or our 2012 ECP. The 2012 ECP was a performance-based compensation program adopted in December 2011 by our Board of Directors upon the recommendation of our compensation committee. Payment of cash awards under the 2012 ECP was based on the achievement of certain pre-determined performance or financial objectives designated by the compensation committee of our Board of Directors.
The 2012 ECP was developed by our compensation committee, which, prior to our initial public offering, was composed of Matthew Crawford, Kevin Lalande, Christophe Lavigne, Pierre Remy and
120
Table of Contents
Robert Shepler. Each member of the compensation committee had substantial experience with healthcare companies, and some served as directors of similarly situated companies, which provided them with invaluable insight into current market trends with respect to executive compensation in our industry. The compensation committee determined the appropriate compensation for each of our executive officers based on such collective industry knowledge. The compensation committee did not utilize third-party industry compensation reports for determining compensation for our executive officers under the 2012 ECP.
Mr. Lavigne’s payments under our 2012 ECP were based on our satisfaction of three different performance criteria: (1) sales growth, (2) operating results and (3) our ratio of receivables and inventory to sales. Mr. Lavigne’s target sales growth bonus under the 2012 ECP was $85,800, based on the following formula:
| 2012 Target Sales |
$ | 95,900,000 | ||
| 2011 Base Sales |
$ | 78,000,000 | ||
|
|
|
|||
| Target Sales Growth |
$ | 17,900,000 | ||
| Bonus Multiplier |
0.4793 | % | ||
|
|
|
|||
| Target Sales Growth Bonus |
$ | 85,800 | * |
| * | Rounded to the nearest hundred. |
The bonus multiplier was determined by dividing the Target Sales Growth Bonus by the Target Sales Growth ($85,800/$17,900,000=0.004793). In 2012, our adjusted 2012 sales were $93,363,568. Accordingly, the sales growth portion of Mr. Lavigne’s 2012 ECP payment was calculated as follows:
| 2012 Adjusted Sales |
$ | 93,363,568 | ||||
| 2011 Base Sales |
- | $ | 78,000,000 | |||
|
|
|
|
||||
| Actual Sales Growth |
$ | 15,363,658 | ||||
| Bonus Multiplier |
x | 0.4793 | % | |||
|
|
|
|
||||
| Actual Sales Growth Bonus |
$ | 73,638 |
For 2012, Mr. Lavigne earned $73,638 as his sales growth bonus.
Mr. Lavigne’s target operating results bonus was $46,200, based on the following formula:
| 2012 Target Operating Results |
$ | 2,111,000 | ||||
| Bonus Multiplier |
x | 2.1885 | % | |||
|
|
|
|
||||
| Target Operating Results Bonus |
$ | 46,200 | * |
| * | Rounded to the nearest dollar. |
The bonus multiplier was determined by dividing the Target Operating Results Bonus by the 2012 Target Operating Results ($46,200/$2,111,000=0.021885). In 2012, our adjusted 2012 operating results were $2,112,976. As a result, the operating results portion of Mr. Lavigne’s 2012 ECP payment was calculated as follows:
| 2012 Adjusted Operating Results |
$ | 2,112,976 | ||||
| Bonus Multiplier |
x | 2.1885 | % | |||
|
|
|
|
||||
| Adjusted Operating Results Actual Bonus |
$ | 46,242 |
For 2012, Mr. Lavigne earned $46,242 as his operating results bonus.
121
Table of Contents
The ratio of receivables and inventory to sales portion of Mr. Lavigne’s payment under our 2012 ECP is equal to $5,000 for each percent by which the average ratio of our receivables plus inventory over the latest twelve months of sales, or the RIS ratio, calculated as of June 30, 2012 and as of December 31, 2012, decreased below Mr. Lavigne’s 2012 target RIS ration of 40.64% (rounded). In 2012, the RIS ratio decreased to 37.14% (rounded). Mr. Lavigne’s RIS ratio bonus was calculated as follows: $5,000 x (0.4064-0.3714) x 100. Accordingly, Mr. Lavigne earned $17,514 as part of his RIS ratio bonus.
Cash Award under Robert McNamara’s 2012 Offer Letter
Pursuant to the Offer Letter dated April 2, 2012 to Robert McNamara, Mr. McNamara was eligible to receive an annual bonus of up to twenty-five percent (25%) of his annual base salary in the discretion of the compensation committee based upon the chief executive officer’s evaluation of Mr. McNamara’s performance in 2012. For 2012, Mr. McNamara’s annual base salary was $300,000. In February 2013, the compensation committee of the Board of Directors approved a bonus payment of approximately $71,250 to Mr. McNamara.
The target amounts to be paid to each of Messrs. Lavigne and McNamara pursuant to the 2012 ECP and Mr. McNamara’s Offer Letter, as applicable, and the actual amounts paid to such persons in respect of 2012 performance are set forth in the table below.
| Named Executive Officer |
Target Cash Incentive ($) |
Actual Amount Paid ($) |
||||||
| Christophe Lavigne |
132,000 | 137,394 | ||||||
| Robert McNamara |
75,000 | 71,250 | ||||||
Cash Awards under the 2013 Executive Compensation Plan
Other than Patrick Richard, each of our named executive officers participated in, and was eligible for cash awards under our 2013 Executive Compensation Plan, or our 2013 ECP. The 2013 ECP was a performance-based compensation program adopted in February 2013 by our compensation committee. Payment of cash awards under the 2013 ECP was based on the achievement of certain pre-determined performance or financial objectives designated by the compensation committee of our Board of Directors, in the case of Mr. Lavigne, or Mr. Lavigne in the case of our other named executive officers.
The 2013 ECP was developed by our compensation committee, which, prior to our initial public offering, was composed of Matthew Crawford, Kevin Lalande, Christophe Lavigne, Pierre Remy and Robert Shepler. Each member of the compensation committee had substantial experience with healthcare companies, and some served as directors of similarly situated companies, which provided them with invaluable insight into current market trends with respect to executive compensation in our industry. The compensation committee determined the appropriate compensation for each of our executive officers based on such collective industry knowledge. The compensation committee did not utilize third-party industry compensation reports for determining compensation for our executive officers under the 2013 ECP.
122
Table of Contents
Mr. Lavigne’s payments under our 2013 ECP were based on our satisfaction of three different performance criteria: (1) sales growth, (2) operating results and (3) our ratio of receivables and inventory to sales. Mr. Lavigne’s target sales growth bonus under the 2013 ECP was $97,500, based on the following formula:
| 2013 Target Sales |
$ | 112,097,000 | ||
| 2012 Adjusted Base Sales |
$ | 90,958,000 | ||
|
|
|
|||
| Target Sales Growth |
$ | 21,139,000 | ||
| Bonus Multiplier |
0.004612 | |||
|
|
|
|||
| Target Sales Growth Bonus |
$ | 97,500 | * |
| * | Rounded to the nearest hundred. |
The bonus multiplier was determined by dividing the Target Sales Growth Bonus by the Target Sales Growth ($97,500/$21,139,000=0.004612). In 2013, our adjusted 2013 sales were $113,048,000. Accordingly, the sales growth portion of Mr. Lavigne’s 2013 ECP payment was calculated as follows:
| 2013 Adjusted Sales |
$ | 113,048,000 | ||||
| 2012 Base Sales |
- | $ | 90,917,000 | |||
|
|
|
|
||||
| Actual Sales Growth |
$ | 22,131,000 | ||||
| Bonus Multiplier |
x | 0.004612 | ||||
|
|
|
|
||||
| Actual Sales Growth Bonus |
$ | 102,068 |
For 2013, Mr. Lavigne earned $102,068 as his sales growth bonus.
Mr. Lavigne’s target operating results bonus was $52,500, based on the following formula:
| 2013 Target Operating Results |
$ | 4,073,000 | ||||
| Bonus Multiplier |
x | 0.012889762 | ||||
|
|
|
|
||||
| Target Operating Results Bonus |
$ | 52,500 | * |
| * | Rounded to the nearest hundred. |
The bonus multiplier was determined by dividing the Target Operating Results Bonus by the 2013 Target Operating Results ($52,500/$4,357,094=0.0120493). In 2013, our adjusted 2013 operating results were $4,393,000. As a result, the operating results portion of Mr. Lavigne’s 2013 ECP payment was calculated as follows:
| 2013 Adjusted Operating Results |
$ | 4,393,000 | ||||
| Bonus Multiplier |
x | 0.012889762 | ||||
|
|
|
|
||||
| Adjusted Operating Results Actual Bonus |
$ | 56,625 |
For 2013, Mr. Lavigne earned $56,625 as his operating results bonus.
The ratio of receivables and inventory to sales portion of Mr. Lavigne’s payment under our 2013 ECP is equal to $5,000 for each percent by which the average ratio of our receivables plus inventory over the latest twelve months of sales, or the RIS ratio, calculated as of June 30, 2013 and as of December 31, 2013, decreased below Mr. Lavigne’s 2013 target RIS ration of 38.3% (rounded). In 2013, the RIS ratio decreased to 35.58% (rounded). Mr. Lavigne’s RIS ratio bonus was calculated as follows: $5,000 x (0.383-0.3558) x 100. Accordingly, Mr. Lavigne earned $13,613 as part of his RIS ratio bonus.
123
Table of Contents
Under the 2013 ECP, Mr. McNamara’s target bonus was $75,000, payable in the discretion of the compensation committee based upon the chief executive officer’s evaluation of Mr. McNamara’s performance in 2013. Mr. McNamara received a bonus of $75,000 in 2013.
Bonus Payment to Patrick Richard
Mr. Richard’s 2013 bonus payment was paid at the discretion of the chief executive officer, based upon his evaluation of Mr. Richard’s performance during 2013. Mr. Richard received a bonus payment of €20,000, which is equal to approximately $26,560, based on an assumed exchange rate of 1.328 U.S. Dollars per Euro.
Option Grants to Our Named Executive Officers in Fiscal 2013
In September 2013, our Board of Directors approved, conditioned on and effective immediately prior to the effectiveness of our registration statement related to our initial public offering, option grants to a number of our executive officers and directors, including certain of our named executive officers. The options have an exercise price equal to $15.00 per share, equal to the per share fair market value of our common stock on the date of our initial public offering price per share of our common stock. The options granted in October 2013 vest in 48 equal monthly installments from the applicable vesting commencement date, subject to the recipient’s continued service with us on each vesting date, and subject to certain accelerated vesting provisions described below under “Potential Payments Upon Termination or Change in Control.” The options granted in October 2013 expire ten years from the date of grant in October 2023.
The applicable vesting commencement date, exercise price and the number of shares underlying the options received by each named executive officer is set forth below.
| Named Executive Officer |
Exercise Price |
Number of Options |
Vesting Commencement Date |
|||||||||
| Christophe Lavigne |
$ | 15.00 | (1) | 82,962 | 9/11/2013 | |||||||
| Robert McNamara |
$ | 15.00 | (1) | 85,185 | 9/11/2013 | |||||||
| Patrick Richard |
$ | 15.00 | (1) | 62,221 | 9/11/2013 | |||||||
| (1) | Equal to our initial public offering price per share of our common stock. |
These grants were made under our 2013 Equity Incentive Plan, which is described below under “Stock Plans—2013 Equity Incentive Plan.”
124
Table of Contents
Outstanding Equity Awards at Fiscal Year-End
The following table below sets forth information regarding the outstanding equity awards held by our named executive officers at December 31, 2013. Please refer to “Potential Payments Upon Termination or a Change in Control” for a description of certain vesting acceleration provisions applicable to certain of these option grants.
| Name |
Date of Grant |
Vesting Commencement Date |
Number of Securities Underlying Unexercised Options (#) Exercisable |
Number of Securities \Underlying Unexercised Options (#) Unexercisable |
Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Options (#) Unearned |
Option Exercise Price ($) |
Option Expiration Date |
|||||||||||||||||||||
| Christophe Lavigne(1) |
4/1/2009 | 4/1/2009 | 11,111 | (3) | — | — | $ | 1.9305 | 4/1/2014 | |||||||||||||||||||
| 11/19/2010 | 1/1/2011 | 20,056 | (4) | — | — | $ | 3.4155 | 11/19/2015 | ||||||||||||||||||||
| 7/10/2012 | 7/10/2012 | 35,053 | (4) | — | — | $ | 6.534 | 7/10/2022 | ||||||||||||||||||||
| 10/8/2013 | 9/11/2013 | 5,185 | (5) | 77,777 | (5) | $ | 15.00 | 10/08/2023 | ||||||||||||||||||||
| Robert McNamara(1) |
5/21/2012 | 4/23/2012 | 66,666 | (6) | — | — | $ | 4.19 | 5/21/2022 | |||||||||||||||||||
| 10/8/2013 | 9/11/2013 | 4,166 | (7) | 62,500 | (7) | — | $ | 15.00 | 10/08/2023 | |||||||||||||||||||
| 10/8/2013 | 9/11/2013 | 1,157 | (8) | 17,361 | (8) | — | $ | 15.00 | 10/08/2023 | |||||||||||||||||||
| Patrick Richard(2) |
12/07/2007 | 12/7/2007 | 84,374 | (9) | — | — | $ | 2.07 | 12/31/2020 | |||||||||||||||||||
| 4/1/2009 | 7/17/2009 | 11,109 | (10) | — | — | $ | 1.76 | 6/30/2022 | ||||||||||||||||||||
| 11/19/2010 | 1/1/2011 | 14,189 | (11) | — | — | $ | 3.11 | 6/30/2022 | ||||||||||||||||||||
| 7/10/2012 | 7/10/2012 | 32,862 | (12) | — | — | $ | 5.94 | 6/30/2022 | ||||||||||||||||||||
| 10/08/2013 | 10/9/2013 | 3,888 | (13) | 58,333 | (13) | — | $ | 15.00 | 6/30/2023 | |||||||||||||||||||
| (1) | All options granted to Messrs. Lavigne and McNamara were granted under the 2007 Stock Option/Stock Issuance Plan. The options described in the this table may be exercised prior to vesting with the stock acquired on exercise being subject to repurchase rights of the company at the lesser of fair market value or the exercise price, with the repurchase rights lapsing when the underlying options would have vested. All options listed in this table held by Mr. Lavigne and McNamara are entitled to acceleration of vesting if the named executive officer is involuntarily terminated upon or following a change in control of us. Certain of these options are also subject to limited acceleration upon the named executive officer’s change in control of us. Certain of these options are also subject to limited acceleration upon the named executive officer’s termination without cause or resignation for good reason. Please see “Potential Payments upon Termination or a Change in Control” for more information. |
| (2) | Mr. Richard has been granted warrants to purchase Class A Stock of Médical. Upon exercise of the such warrants, each share of Médical received by Mr. Richard will be automatically exchanged for 5.80087 shares of our common stock, pursuant to the terms of the Second Amended and Restated Put-Call Agreement, dated August 6, 2013, by and among us and certain other parties listed therein. The exercise price per share of Class A Stock of Médical issuable upon exercise of the warrants is payable in Euros, calculated by using the U.S. Dollar to Euro currency exchange rate in effect on the date of grant. |
| (3) | All of the options are vested on the vesting commencement date. |
| (4) | One fourth of the total number of shares vested on the first anniversary of the vesting commencement date, and an additional 1/48th of the total number of shares vest each month thereafter, subject to continuous service. |
| (5) | Represents options granted in connection with the consummation of our initial public offering. Subject to continuous service, the shares shall vest in equal monthly installments over the 48 months following the vesting commencement date. |
| (6) | Represents options granted in connection with the commencement of the respective named executive officer’s employment with us. One fourth of the total number of shares vested on the first anniversary of the vesting commencement date, and an additional 1/48th of the total number of shares vest each month thereafter, subject to continuous service. |
| (7) | Represents options granted pursuant to the terms of the respective named executive officer’s employment offer letter, which provided for such grant in connection with the consummation of our initial public offering. Subject to continuous service, the shares shall vest in equal monthly installments over the 48 months following the vesting commencement date upon completion of each additional month of service. |
| (8) | Represents options granted in connection with the consummation of our initial public offering. Subject to continuous service, the shares shall vest in equal monthly installments over the 48 months following the vesting commencement date. |
footnotes continued on following page
125
Table of Contents
| (9) | All of the shares of our common stock exchangeable for the Class A Stock of Médical issuable upon exercise of the warrants to purchase such Class A Stock are vested on the vesting commencement date. |
| (10) | All of the shares of our common stock exchangeable for the Class A Stock of Médical issuable upon exercise of the warrants to purchase such Class A Stock are vested on the vesting commencement date. |
| (11) | The shares of our common stock exchangeable for the Class A Stock of Médical issuable upon exercise of the warrants to purchase such Class A Stock were subject to performance-based vesting, with performance requirements determined by the Chief Executive Officer. |
| (12) | One fourth of the total number of our common stock exchangeable for the Class A Stock of Médical issuable upon exercise of the warrants to purchase such Class A Stock vested on the first anniversary of the vesting commencement date, and an additional 1/48th of the total number of shares vest each month thereafter, subject to continuous service. |
| (13) | Represents the number of shares of our common stock exchangeable for the Class A Stock of Médical issuable upon exercise of warrants to purchase such Class A Stock granted in connection with the consummation of our initial public offering. Subject to continuous service, the shares of our common stock exchangeable for the Class A Stock of Médical issuable upon exercise of warrants to purchase such Class A Stock shall vest in equal monthly installments over the 48 months following the vesting commencement date. |
Option Exercises in 2013
On February 5, 2013, Mr. Lavigne exercised options to acquire 22,222 shares of our common stock. Neither Mr. McNamara or Mr. Richard exercised any option awards or warrants to purchase Class A Stock of Médical in the 2013 fiscal year.
Pension Benefits
We do not provide any pension benefits to our named executive officers, except as described below under “Retirement Plans.”
Other Compensatory Benefits
We provide the following benefits to our named executive officers, generally on the same basis provided to all of our employees, except that Messrs. Lavigne and McNamara also are entitled to reimbursement for annual wellness examinations and have certain rights to reimbursement of health insurance costs following separation from service as described in “Potential Payments upon Termination or a Change in Control,” Mr. Lavigne receives a corporate car, and Mr. Richard receives a car allowance:
| • | medical, dental and vision insurance; |
| • | 401(k) plan (see “Retirement Plans” below for a description of our 401(k) plan); |
| • | employee assistance program; |
| • | short- and long-term disability, life insurance, accidental death and dismemberment insurance; |
| • | health and dependent care flexible spending accounts; |
| • | 2013 Equity Incentive Plan (see “Stock Plans—2013 Equity Incentive Plan below for a description of the 2013 Equity Incentive Plan); and |
| • | 2013 Employee Stock Purchase Plan (see “Stock Plans—2013 Employee Stock Purchase Plan below for a description of the 2013 Employee Stock Purchase Plan). |
Retirement Plans
We maintain a 401(k) retirement plan which is intended to be a tax qualified defined contribution plan under Section 401(k) of the Internal Revenue Code. All of our U.S. employees are eligible to participate on the first day of the month following the completion of certain eligibility requirements, which include three months of service. The 401(k) plan includes a salary deferral arrangement pursuant to which participants may elect to reduce their current compensation up to the statutorily prescribed limit, which was equal to $17,500 in 2013 and is equal to $17,500 in 2014 (catch up contributions for employees over 50 allow for an additional $5,500 each year), and have the amount of their compensation reduction contributed to the 401(k) plan.
126
Table of Contents
Employment Agreements with our Named Executive Officers
We are party to certain employment agreements or offer letters with our President and Chief Executive Officer, Christophe Lavigne, our Executive Vice President, Chief Financial Officer, Robert McNamara and our Executive Vice President—LDR Médical, Patrick Richard. Each named executive officer, like substantially all of our employees, has executed an employee proprietary information agreement as a condition to his employment, which includes non-solicit, non- compete and confidentiality provisions.
The following chart shows the annual base salaries for our named executive officers as of December 31, 2013 and option grants issued during 2013 pursuant to employment offer letters as applicable.
| Named Executive Officer |
Base Salary as of December 31, 2013 ($) |
Offer Letter Option Grant (# of shares)(1) |
Option Exercise Price per Share ($) |
Vesting Commencement Date |
||||||||||||
| Christophe Lavigne |
450,000 | (2) | — | — | — | |||||||||||
| Robert McNamara |
300,000 | 66,666 | (3) | 15.00 | 9/11/2013 | |||||||||||
| Patrick Richard |
243,024 | — | — | — | ||||||||||||
| (1) | See the “Outstanding Equity Awards at Fiscal Year-End” table and the accompanying footnotes above for more information regarding the initial option grants to each named executive officer, including information regarding the exercise price and vesting provisions thereof. |
| (2) | Mr. Lavigne’s base salary increased to $500,000 for 2014. |
| (3) | Granted in connection with our initial public offering pursuant to the terms of the Offer Letter, dated April 2, 2012 with Mr. McNamara. |
Employment Agreements with our Chief Executive Officer and Chief Financial Officer
These agreements were each entered into on June 10, 2013. Each agreement provides for “at will” employment of the respective named executive officer, which means that the employment relationship can be ended by the named executive officer or by us at any time, subject to the executive’s potential right to certain severance benefits as described below under “Potential Payments upon Termination or a Change in Control.” Each agreement provides for an annual salary, subject to increase pursuant to our policies in effect from time to time.
Mr. Lavigne’s employment agreement additionally provides for an automobile benefit, similar to what he has received in the past, pursuant to which we pay for the lease of Mr. Lavigne’s automobile, and we will pay all reasonable costs of maintaining Mr. Lavigne’s immigration status in the United States to allow him to continue working in the United States. Each of Messrs. Lavigne and McNamara is also entitled to certain benefits upon termination of employment under certain circumstances, including in connection with a change in control of us. These benefits are described below in “Potential Payments upon Termination or a Change in Control.”
The foregoing description of the employment agreements of Mr. Lavigne and Mr. McNamara is a summary only and is qualified in its entirety by reference to the employment agreements of such named executive officers, all of which have been filed with the SEC as exhibits to most recent our Annual Report on Form 10-K.
Potential Payments upon Termination or a Change in Control
General Severance Benefits
The employment agreements of Messrs. Lavigne and McNamara, which are described above under “Employment Agreements with Named Executive Officers,” include certain severance benefits payable to Messrs. Lavigne and McNamara in the event that such named executive officer is involuntarily terminated by us without cause or he resigns his position with us for good reason (“cause” and “good reason” as defined under these employment agreements are described below). In that situation, each such
127
Table of Contents
named executive officer would be entitled to the following severance benefits: (i) the payment of a lump-sum severance payment in an amount equal to the sum of (x) such named executive officer’s annual base salary as in effect through the date of his termination for the severance term (“severance term” as defined under these employment agreements as described below), as applicable, plus (y) any unpaid bonus through the date of such named executive officer’s termination (any such bonus is presumed to have been earned, and any bonus that is paid on a quarterly basis or annual basis will be paid on a pro rata basis for the number of days through the date of such named executive officer’s termination for the applicable period); and (ii) provided that the named executive officer properly elects COBRA continuation coverage, the payment of COBRA premiums for health care coverage for such named executive officer, his spouse and his covered dependents for the severance term, as applicable. In addition, each of the employment agreements provides that such named executive officer’s outstanding equity awards will become vested and exercisable immediately prior to the effective time of the termination of his employment with respect to that number of additional shares that would have become vested during the severance term, as applicable, immediately following the date of such named executive officer’s termination, as if he has remained employed with us through such period. However, in the case of Mr. McNamara, the 66,666 options to purchase shares of our common stock granted to him on May 21, 2012 as part of his initial option grant will become fully vested and exercisable as of his termination date. Furthermore, Mr. Lavigne’s employment agreement provides that he would be entitled to the reimbursement of all of his reasonable, documented costs associated with the relocation of his family to France, so long as such relocation occurs within 12 months following his termination date.
Messrs. Lavigne and McNamara are entitled to accelerated vesting of their stock options and other equity awards granted under our 2007 Stock Option/Stock Issuance Plan and 2013 Equity Incentive Plan in the event they are terminated in connection with a change in control of us, as described below under “Stock Plans—2007 Stock Option/Stock Issuance Plan” and “Stock Plans—2013 Equity Incentive Plan.”
Additional Benefits upon Termination in Connection with a Change in Control
In addition to the severance benefits described above under “General Severance Benefits,” the employment agreements with Messrs. Lavigne and McNamara, which are described above under “Employment Agreements with Named Executive Officers,” include certain benefits payable to Messrs. Lavigne and McNamara in the event of a termination in connection with a change in control (“change in control” as defined under these employment agreements as described below). In the event a change in control occurs during the term of any such named executive officer’s employment with us and (a) such named executive officer is not offered continued employment by the acquiring company and in connection therewith, such named executive officer is terminated without cause or he terminates his employment for good reason, or (b) such named executive officer is offered continuing employment, but he is terminated without cause or he terminates his employment for good reason, in either event, within 12 months of the change in control event, then, in addition to any other accrued amounts payable to such named executive officer through the date of termination of his employment, the named executive officer will be entitled to the following benefits: (i) the payment of a lump-sum payment in an amount equal to the sum of (x) such named executive officer’s annual base salary as in effect through the date of his termination for the severance term, as applicable, plus (y) any unpaid bonus through the date of such named executive officer’s termination (any such bonus is presumed to have been earned, and any bonus that is paid on a quarterly basis or annual basis will be paid on a pro rata basis for the number of days through the date of such named executive officers termination for the applicable period); plus (z) in the case of Mr. Lavigne, 200% of Mr. Lavigne’s target annual bonus for the fiscal year in which the termination occurs, or in the case of Mr. McNamara, 100% of such named executive officer’s target annual bonus for the fiscal year in which the termination occurs (provided, in the case of Mr. McNamara, that we have completed a firm commitment underwritten public offering, pursuant to an effective registration statement under the Securities Act, covering the offer and sale of our common stock in the aggregate amount of at least $20 million); and (ii) provided that such named executive officer properly elects COBRA continuation coverage, the payment of COBRA premiums for health care
128
Table of Contents
coverage for such named executive officer, his spouse and his covered dependents for the severance term, as applicable. In addition, each of the outstanding equity awards held by Mr. Lavigne will become fully vested and exercisable immediately prior to the effective time of the termination of such named executive officer in connection with a change in control, and each of the equity awards held by Mr. McNamara, whether he is offered continued employment with the acquiring company or not, will become fully vested and exercisable immediately prior to the effective time of the change in control. Except as set forth in the employment agreement, Messrs. Lavigne and McNamara are not entitled to any payments, COBRA benefits or additional equity vesting in the event that such named executive officer is offered continued employment by the acquiring company following the change in control event with the acquiring company assuming the employment agreement, or entering into an agreement substantially similar to the employment agreement, provided that Mr. McNamara will be entitled to the additional vesting upon a change in control as described in the preceding sentence.
Benefits upon Disability or Death
In addition to the severance benefits described above under “General Severance Benefits” and the additional benefits described above under “Additional Benefits upon Termination in Connection with a Change in Control,” the employment agreements with Messrs. Lavigne and McNamara, which are described above under “Employment Agreements with Named Executive Officers,” include certain benefits payable to the named executive officer in the event of death or disability (“disability” as defined under these employment agreements as described below). In the event of a termination of a named executive officer due to death or disability, each of such named executive officer’s outstanding equity awards will become fully vested and exercisable immediately prior to the date of such named executive officer’s disability or death. In addition, we will pay the COBRA premium for health coverage for each of the named executive officers (in the event of their disability), their spouse and covered dependents, to the extent eligible. With respect to Messrs. Lavigne and McNamara, the payment of such COBRA benefits in the event of death or disability will be for the applicable severance term, immediately following the date of their termination. In no event will any named executive officer or such named executive officer’s estate or beneficiaries be entitled to any severance payments described above under “General Severance Benefits.”
The benefits described above under “General Severance Benefits,” “Additional Benefits upon Termination in Connection with a Change in Control” and “Benefits upon Disability of Death” are conditioned on Mr. Lavigne and Mr. McNamara, as applicable, signing and returning to us a non-revocable general release of claims providing for a release of all claims relating to his employment or the his employment agreement against us or our successor, our subsidiaries and our parent, as applicable, and their respective directors, officers and stockholders, in a form satisfactory to us, provided that such release becomes effective and irrevocable no later than 60 days following such named executive officer’s termination of employment date or such earlier date required by the release. In no event is the named executive officer entitled to any of the benefits described above under “General Severance Benefits,” “Additional Benefits upon Termination in Connection with a Change in Control” and “Benefits upon Disability of Death” in the event of a termination of the named executive officer’s employment by us with cause or by the named executive officer without good reason.
In addition, our 2007 Stock Option/Stock Issuance Plan provides for full acceleration of the vesting of any unvested stock options or stock purchase rights, including those held by our named executive officers, in the event that they are not otherwise assumed or substituted with new equivalent options or shares in a merger or change in control of us. See “Stock Plans—2007 Stock Option/Stock Issuance Plan” for more information.
Definitions of “Cause,” “Good Reason,” “Change in Control,” “Disability,” and “Severance Term”
The employment agreements of Messrs. Lavigne and McNamara define “cause” and “good reason” in substantially the same manner.
129
Table of Contents
“Cause” is generally defined as:
| (a) | the named executive officer’s willful, knowing or grossly negligent failure or refusal to perform his duties or to follow the reasonable directions of our Chief Executive Officer (or in the case of Mr. Lavigne, our Board of Directors), which has continued for 30 days following written notice of such failure from our Chief Executive Officer (or in the case of Mr. Lavigne, our Board of Directors); |
| (b) | the named executive officer’s breach of any fiduciary duty owed to us; |
| (c) | material and willful misfeasance or malfeasance by the named executive officer in connection with the performance of his duties under his employment agreement; |
| (d) | the named executive officers’ commission of an act which is a fraud or embezzlement or a crime involving moral turpitude; |
| (e) | the conviction of the named executive officer for, or a plea of guilty or nolo contendre by the named executive officer to, a criminal act which is a felony; |
| (f) | a material breach or default by the named executive officer of any provision of his employment agreement which has continued for 30 days following notice of such breach or default from the Chief Executive Officer (or in the case of Mr. Lavigne, our Board of Directors); or |
| (g) | the named executive officer’s abuse of drugs or alcohol to our detriment. |
“Good reason” is generally defined as the occurrence of any one or more of the following events without the named executive officer’s prior written consent, unless we fully correct the circumstances constituting “good reason” within 30 days after notice from the named executive officer that “good reason” exists:
| (a) | a material reduction of the named executive officer’s duties and responsibilities under his employment agreement; |
| (b) | a relocation of the named executive officer’s workplace more than 50 miles outside the workplace the named executive officer has been assigned to work over the prior six month period; |
| (c) | our reduction of the named executive officer’s annual base salary, other than in the event of a reduction in compensation of all our executive officers, generally, so long as the named executive officer’s reduction is no more than the average reduction; |
| (d) | if in connection with a change in control the acquiring company does not expressly assume the named executive officer’s employment agreement or offer to enter into an agreement with the named executive officer on terms substantially similar to the named executive officer’s employment agreement in all material respects; or |
| (e) | our breach of the named executive officer’s employment agreement. |
In order for the named executive officer’s resignation with good reason to be effective, the named executive officer must deliver to us written notice of his resignation for good reason within 30 days after the date the named executive officer first knows or should reasonably know of the occurrence constituting good reason.
“Change in control” is generally defined as a change in ownership or control of us effected through any of the following transactions:
| (a) | a transaction or series of transactions (other than an offering of our common stock to the general public through a registration statement filed with the SEC) whereby any “person” or related “group” of “persons” (as such terms are used in Sections 13(d) and |
130
Table of Contents
| 14(d)(2) of the Exchange Act) (other than us, any of our subsidiaries, an employee benefit plan maintained by us or any of our subsidiaries or a “person” that, prior to such transaction, directly or indirectly controls, is controlled by, or is under common control with, us) directly or indirectly acquires beneficial ownership (within the meaning of Rule 13d-3 under the Exchange Act) of our securities possessing more than 50% of the total combined voting power of our securities outstanding immediately after such acquisition; |
| (b) | during any period of two consecutive years, individuals who, at the beginning of such period, constitute our Board of Directors, together with any new director(s) (other than a director designated by a person who shall have entered into an agreement with us to effect a transaction described in clause (a) above or clause (c) below) whose election by our Board of Directors or nomination for election by our stockholders was approved by a vote of at least two-thirds of the directors then still in office who either were directors at the beginning of the two-year period or whose election or nomination for election was previously so approved, cease for any reason to constitute a majority our Board of Directors; |
| (c) | the consummation by us (whether directly involving us or indirectly involving us through one or more intermediaries) of (x) a merger, consolidation, reorganization, or business combination or (y) a sale or other disposition of all or substantially all of our assets in any single transaction or series of related transactions or (z) the acquisition of assets or stock of another entity, in each case other than a transaction: |
| (i) | which results in our voting securities outstanding immediately before the transaction continuing to represent (either by remaining outstanding or by being converted into our voting securities or the person that, as a result of the transaction, controls, directly or indirectly, us or owns, directly or indirectly, all or substantially all of the our assets or otherwise succeeds to the our business, or a successor entity), directly or indirectly, at least a majority of the combined voting power of the successor entity’s outstanding voting securities immediately after the transaction, and |
| (ii) | after which no person or group beneficially owns voting securities representing 50% or more of the combined voting power of the successor entity; provided, however, that no person or group shall be treated for purposes of this clause (c)(ii) as beneficially owning 50% or more of combined voting power of the successor entity solely as a result of the voting power held in the us prior to the consummation of the transaction; or |
| (d) | our stockholders approve our liquidation or dissolution. |
“Disability” is generally defined as any illness or any physical or mental impairment which substantially limits a major life activity and that, with or without reasonable accommodation by us:
| (a) | renders it impossible or impracticable for the named executive officer to perform his duties and responsibilities hereunder for a continuous period of at least three months, or |
| (b) | prevents the named executive officer from performing his essential duties and responsibilities hereunder for more than six months during any 12-month period as determined by mutual agreement of a physician selected by us or our insurers and a physician selected by the named executive officer; provided, however, that if the opinion of the our physician and the named executive officer’s physician conflict, our physician and the named executive officer’s physician shall together agree upon a third physician, whose opinion shall be binding. |
131
Table of Contents
“Severance term” is generally defined as (i) with respect to Mr. Lavigne, 24 months; and (ii) with respect to Mr. McNamara, (a) six months, in the event we have not completed a firm commitment underwritten public offering, pursuant to an effective registration statement under the Securities Act, covering the offer and sale of our common stock in the aggregate amount of at least $20 million, or (b) 12 months, in the event we have not completed such public offering.
However, if the payments and benefits due to Mr. Lavigne and Mr. McNamara under their respective employment agreements or otherwise would be subject to Section 280G of the Internal Revenue Code of 1986, or the Code, the payments and benefits will be reduced to the extent necessary so that no portion thereof shall be subject to the excise tax imposed by Section 4999 of the Code, but only if such reduction places Mr. Lavigne or Mr. McNamara, as applicable, in a better after-tax position than if such reduction were not made.
Employment Agreement with our Executive Vice President—LDR Médical
Patrick Richard, our Executive Vice President—LDR Médical, and Médical are parties to an employment agreement (which has been suspended while Mr. Richard serves as a director of Médical), which was entered into on January 1, 2001. The employment agreement provides for an annual salary, subject to increase pursuant to our policies in effect from time to time. Mr. Richard’s compensation is subject to deductions for social security, including his social security contribution, additional retirement and social welfare contributions, medical insurance and unemployment insurance, as provided by French law. Médical provides Mr. Richard a car allowance as reimbursement for the various tangible and intangible expenses incurred by Mr. Richard in connection with the use of his private vehicle for business matters. Mr. Richard’s employment agreement provides for non-compete obligations on the part of Mr. Richard, for a maximum period of two years following the termination of Mr. Richard’s employment. In consideration for the non-compete obligation, if Mr. Richard voluntarily terminates his employment with Médical, he will receive a payment equal to 50% of his annual fixed salary, bonus and the value of his other perquisites (for example, his car allowance) then in effect, provided, however, if Mr. Richard is involuntarily terminated, he will receive a payment equal to 60% of his annual fixed salary, bonus and the value of his other perquisites then in effect. Notwithstanding the foregoing, Médical is entitled to waive the non-compete obligation upon notice to Mr. Richard, which will terminate Médical’s obligation to pay the non-compete payments described above. Furthermore, any failure by Mr. Richard to observe the non-compete obligation will also release Médical from its obligation to pay the non-compete payments. In addition the payments described above, upon the termination of his employment, Mr. Richard would be the beneficiary of certain unemployment insurance payments, which would be paid by a third party private insurance provider. Médical has historically paid the premiums associated with such unemployment insurance on behalf of Mr. Richard, who is not entitled to public unemployment insurance payments because he serves as a director of Médical. Mr. Richard participates in permanent and private medical insurance arrangements in France and is entitled to a death in-service benefit. In the event of Mr. Richard’s death or disability, Mr. Richard and/or his family would receive the same benefits as other employees of Médical.
Stock Plans
2007 Stock Option/Stock Issuance Plan
Our 2007 Stock Option/Stock Issuance Plan was initially adopted by our Board of Directors in September 2007, and subsequently approved by our stockholders, and was amended in February 2009, July 2009, December 2010 and April 2012. After the completion of our initial public offering, no further equity awards have been or will be issued under the 2007 Stock Option/Stock Issuance Plan.
Our 2007 Stock Option/Stock Issuance Plan provides for the grant of nonstatutory stock options, incentive stock options and stock purchase rights to our employees, directors and consultants. As of December 31, 2013, options to purchase 1,200,293 shares of common stock were outstanding, including (i) 19,531 shares of common stock issuable outside the plan, and (ii) 509,062 shares of common stock
132
Table of Contents
issuable upon the exchange of Class A Stock of Médical issuable upon exercise of warrants to purchase such Class A Stock, which such shares have been reserved against the available number of shares under the plan. Awards that expire, become unexercisable or are forfeited become available for future grant under the 2013 Equity Incentive Plan.
The standard form of option agreement under the 2007 Stock Option/Stock Issuance Plan provides that options will vest 25% on the first anniversary of the vesting start date with the remainder vesting ratably over the next 36 months, subject to continued service through each applicable vesting date. Under our 2007 Stock Option/Stock Issuance Plan, our Board of Directors, or a committee designated by our Board of Directors, has the authority to grant options with early exercise rights, subject to our repurchase right that lapses as the shares vest on the original vesting schedule, and to provide for accelerated vesting. The term of an option issued pursuant to the plan may not exceed ten years and the exercise price of an option may not be less than the fair market value on the grant date.
Shares of common stock issued pursuant to the early exercise of an option granted under the 2007 Stock Option/Stock Issuance Plan continue to vest in accordance with the vesting schedule that applied to such option. The standard form of stock purchase agreement for the purchase of shares of common stock pursuant to options granted under the 2007 Stock Option/Stock Issuance Plan also restricts the transfer of shares of our common stock issued pursuant to an award for the period specified by the representative of the underwriters not to exceed 180 days following the effective date of the registration statement related to this offering.
The term of an incentive stock option issued pursuant to the plan may not exceed ten years and the exercise price of an incentive stock option may not be less than the fair market value on the grant date, except that with respect to any optionee who owned 10% of the voting power of all classes of our outstanding stock or 10% of the voting power of all classes of the outstanding stock of any of our subsidiaries as of the grant date, the term may not exceed five years and the exercise price of the incentive stock option must equal at least 110% of the fair market value on the grant date.
After the termination of service of an employee, director or consultant, he or she may exercise his or her options to the extent vested for the period of time stated in his or her option agreement. Generally, if termination is due to disability or death, the option will remain exercisable for one year. In all other cases, the option will generally remain exercisable for three months following the termination of service. However, in no event may an option be exercised later than the expiration of its term.
Generally, our 2007 Stock Option/Stock Issuance Plan does not allow for the transfer of awards and only the recipient of an award may exercise an award during his or her lifetime. However, non-incentive stock options may be assigned during the recipient’s lifetime to one or more of the recipients family members (or to a trust for the benefit of the recipient or the recipient’s family members) in connection with estate planning or pursuant to a domestic relations order.
Pursuant to our 2007 Stock Option/Stock Issuance Plan, outstanding option awards (or unvested stock) at the time of a change in control automatically vest so that each option becomes, immediately prior to the change in control, exercisable by the holder (or vested in the case of unvested stock). However, our 2007 Stock Option/Stock Issuance Plan provides that, in the event of a change in control, outstanding options (or unvested stock) do not vest on an accelerated basis if and to the extent that (i) the outstanding option (or unvested stock) is assumed by the successor corporation or otherwise continued in full force and effect (and any repurchase rights are assigned to the successor corporation), (ii) the outstanding option is replaced with a cash retention program which preserves the spread existing on the unvested option shares at the time of the change in control and provides for the subsequent payout of that spread in accordance with the same vesting schedule as the underlying unvested option or (iii) the acceleration of such option is subject to other limitations imposed by the administrator of the 2007 Stock Option/Stock Issuance Plan at the time of the grant.
133
Table of Contents
Under our 2007 Stock Option/Stock Issuance Plan, “change in control” is defined as a change in ownership or control of us, effected through any of the following:
| • | a merger, consolidation or other reorganization approved by our stockholders, unless securities representing more than 50% of the total combined voting power of the voting securities of the successor corporation are immediately thereafter beneficially owned by the persons or beneficially owned our securities immediately prior to such transaction; |
| • | a stockholder-approved sale, transfer or other disposition of all or substantially all of our assets in liquidation or dissolution of us; or |
| • | the acquisition, directly or indirectly, by any person or group, of securities possessing more than 50% of the total combined voting power of our outstanding securities pursuant to a tender or exchange offer made directly to our stockholders. |
The 2007 Stock Option/Stock Issuance Plan is administered by our Board of Directors. Our Board of Directors is permitted to delegate the administration of the 2007 Stock Option/Stock Issuance Plan to a committee. The administrator of the 2007 Stock Option/Stock Issuance Plan is empowered to adjust the terms of the options, including the exercise price, in the event of a dividend, recapitalization, stock split, merger, consolidation, repurchase, share exchange or other change in our corporate structure in order to prevent diminution or enlargement of the benefits intended to be granted to the optionees. Our Board of Directors also has the authority to amend or modify the 2007 Stock Option/Stock Issuance Plan, as long as the amendment of modification does not adversely affect the rights and obligations of any participant without the consent of that participant. In addition, certain amendments may require stockholder approval pursuant to applicable laws and regulations.
2013 Equity Incentive Plan
Our 2013 Equity Incentive Plan, or our 2013 Plan, was adopted by our Board of Directors in August 2013, and became effective on the date immediately preceding the consummation of our initial public offering offering. Our 2013 Plan serves as the successor equity incentive program to our 2007 Stock Option/Stock Issuance Plan, as amended. As of December 31, 2013, 853,557 shares of common stock were available for issuance pursuant to our 2013 Equity Incentive Plan.
The shares available for issuance under our 2013 Plan also includes (i) any shares of common stock issued pursuant to the 2007 Stock Option/Stock Issuance Plan that are forfeited or repurchased by us in accordance with the terms of the 2007 Stock Option/Stock Issuance Plan, (ii) any shares of common stock that are issuable upon exercise of awards granted pursuant to the 2007 Stock Option/Stock Issuance Plan that expire or become unexercisable for any reason without having been exercised, and (iii) any shares of common stock that are otherwise returned or restored in accordance with the terms of the 2007 Stock Option/Stock Issuance Plan.
In addition, this share reserve automatically increases on January 1, 2014 and each subsequent anniversary through January 1, 2023, by an amount equal to the smaller of (i) 4% of the number of shares of our common stock issued and outstanding on the immediately preceding December 31; and (ii) an amount determined by the compensation committee of our Board of Directors. For January 1, 2014, 963,066 shares of common stock were added to our 2013 Equity Incentive Plan pursuant to this evergreen provision.
Appropriate adjustments will be made in the number of authorized shares and other numerical limits in our 2013 Plan and in outstanding awards to prevent dilution or enlargement of participants’ rights in the event of a stock split or other change in our capital structure. Shares subject to awards granted under our 2013 Plan which expire, are repurchased, or are cancelled or forfeited will again become available for issuance under our 2013 Plan. The shares available will not be reduced by awards settled in cash or by shares withheld to satisfy tax withholding obligations. Only the net number of shares issued upon the exercise of stock appreciation rights or options exercised by means of a net exercise will be deducted from the shares available under our 2013 Plan.
134
Table of Contents
Awards may be granted under our 2013 Plan to our employees, including officers, directors, or consultants, and our present or future affiliated entities. While we may grant incentive stock options only to employees, we may grant nonstatutory stock options, stock appreciation rights, restricted stock purchase rights or bonuses, restricted stock units, performance shares, performance units and cash-based awards, or other stock-based awards to any eligible participant.
Our 2013 Plan is generally administered by the compensation committee of our Board of Directors. Subject to the provisions of the 2013 Plan, the compensation committee determines in its discretion the persons to whom and the times at which awards are granted, the sizes of such awards and all of their terms and conditions. The compensation committee has the authority to construe and interpret the terms of our 2013 Plan and awards granted under it.
Notwithstanding the foregoing, and to the extent permitted by applicable law, the Board of Directors or the compensation committee may, in its discretion, delegate to a committee comprised of one or more officers and/or directors, the authority to grant one or more awards of options or stock appreciation rights, without further approval of the Board of Directors or compensation committee, to any employee, other than a person who, at the time of such grant, is subject to Section 16 of the Exchange Act or is a “covered employee” under Section 162(m) of the Internal Revenue Code, and to exercise such other powers under our 2013 Plan, provided, however, that (i) no employee may be granted pursuant to such delegation one or more such awards in any fiscal year of the Company for more than 29,630 shares of our common stock (subject to adjustment as described above), (ii) the exercise price per share of such awards shall be not less than the fair market value per share of stock on the effective date of grant, (iii) each such award shall be subject to the terms and conditions of the appropriate stand for of award agreement approved by the Board of Directors of the compensation committee and shall conform to the provisions of our 2013 Plan, and (iv) each such award shall conform to guidelines as shall be established from time to time by resolution of the Board of Directors of the compensation committee. Pursuant to this power, our Board of Directors has created a Non-Executive Stock Option Committee, consisting of Christophe Lavigne, the Chairman of our Board of Directors and our President and Chief Executive Officer. The Non-Employee Stock Option Committee also has the ability to grant options to sales agents of the Company, subject to certain parameters specified by the Board of Directors.
As described above, awards may be granted under our 2013 Plan to our employees, officers, directors or consultants or those of any present or future parent or subsidiary corporation or other affiliated entity. All awards will be evidenced by a written agreement between us and the holder of the award and may include any of the following:
| • | Stock Options. We may grant nonstatutory stock options or incentive stock options (as described in Section 422 of the Internal Revenue Code), each of which gives its holder the right, during a specified term (not exceeding 10 years) and subject to any specified vesting or other conditions, to purchase a number of shares of our common stock at an exercise price per share determined by the administrator, which may not be less than the fair market value of a share of our common stock on the date of grant. |
| • | Stock Appreciation Rights. A stock appreciation right gives its holder the right, during a specified term (not exceeding 10 years) and subject to any specified vesting or other conditions, to receive the appreciation in the fair market value of our common stock between the date of grant of the award and the date of its exercise. We may pay the appreciation in shares of our common stock or in cash, except that a stock appreciation right granted in tandem with a related option is payable only in stock. |
| • | Restricted Stock. The administrator may grant restricted stock awards either as a bonus or as a purchase right at such price as the administrator determines. Shares of restricted stock remain subject to forfeiture until vested, based on such terms and conditions as the administrator specifies. Holders of restricted stock will have the right to vote the shares and |
135
Table of Contents
| to receive any dividends paid, except that the dividends may be subject to the same vesting conditions as the related shares. |
| • | Restricted Stock Units. Restricted stock units represent rights to receive shares of our common stock (or their value in cash) at a future date without payment of a purchase price (unless required under applicable state corporate laws), subject to vesting or other conditions specified by the administrator. Holders of restricted stock units have no voting rights or rights to receive cash dividends unless and until shares of common stock are issued in settlement of such awards. However, the administrator may grant restricted stock units that entitle their holders to dividend equivalent rights. |
| • | Performance Shares and Performance Units. Performance shares and performance units are awards that will result in a payment to their holder only if specified performance goals are achieved during a specified performance period. Performance share awards are rights denominated in shares of our common stock, while performance unit awards are rights denominated in dollars. The administrator establishes the applicable performance goals based on one or more measures of business performance enumerated in the 2013 Plan, such as net revenues, gross margin, net income or total stockholder return. To the extent earned, performance share and unit awards may be settled in cash or in shares of our common stock. Holders of performance shares or performance units have no voting rights or rights to receive cash dividends unless and until shares of common stock are issued in settlement of such awards. However, the administrator may grant performance shares that entitle their holders to dividend equivalent rights. |
| • | Cash-based Awards and Other Stock-based Awards. The administrator may grant cash-based awards that specify a monetary payment or range of payments or other stock-based awards that specify a number or range of shares or units that, in either case, are subject to vesting or other conditions specified by the administrator. Settlement of these awards may be in cash or shares of our common stock, as determined by the administrator. Their holder will have no voting rights or right to receive cash dividends unless and until shares of our common stock are issued pursuant to the award. The administrator may grant dividend equivalent rights with respect to other stock-based awards. |
Our 2013 Plan authorizes the compensation committee, without further stockholder approval, to provide for the cancellation of stock options or stock appreciation rights with exercise prices in excess of the fair market value of the underlying shares of common stock in exchange for new options with exercise prices equal to the fair market value of the underlying common stock, other equity awards, or a cash payment.
In the event of a change in control as described in our 2013 Plan, the acquiring or successor entity may assume or continue all or any awards outstanding under our 2013 Plan or substitute substantially equivalent awards. Any awards which are not assumed or continued in connection with a change in control or are not exercised or settled prior to the change in control will terminate effective as of the time of the change in control. The compensation committee may provide for the acceleration of vesting of any or all outstanding awards upon such terms and to such extent as it determines, except that the vesting of all awards held by members of our Board of Directors who are not employees will automatically be accelerated in full. Our 2013 Plan also authorizes the compensation committee, in its discretion and without the consent of any participant, to cancel each or any outstanding award denominated in shares upon a change in control in exchange for a payment to the participant with respect to each share subject to the cancelled award of an amount equal to the excess of the consideration to be paid per share of common stock in the change in control transaction over the exercise price per share, if any, under the award.
Our 2013 Plan continues in effect until it is terminated by the administrator, provided, however, that all awards will be granted, if at all, within 10 years of its effective date. The administrator may amend,
136
Table of Contents
suspend or terminate our 2013 Plan at any time, provided that without stockholder approval, the plan cannot be amended to increase the number of shares authorized, change the class of persons eligible to receive incentive stock options, or effect any other change that would require stockholder approval under any applicable law or listing rule.
2013 Employee Stock Purchase Plan
Our Board of Directors approved our 2013 Employee Stock Purchase Plan, or our 2013 ESPP, in August 2013 and amended and restated the 2013 ESPP in October 2013.
A total of 111,111 shares of our common stock was initially authorized and reserved for sale under our 2013 ESPP. In addition, our 2013 ESPP provides for an automatic annual increase in the number of shares available for issuance under the plan on January 1 of each year beginning in 2014 and continuing through and including January 1, 2023 equal to the lesser of (i) 1% of our then issued and outstanding shares of common stock on the immediately preceding December 31, or (ii) a number of shares as our Board of Directors may determine. For January 1, 2014, 240,766 shares of common stock were added to our 2013 ESPP pursuant to this evergreen provision.
The compensation committee of our Board of Directors administers our 2013 ESPP. The compensation committee has the authority to construe and interpret the terms of our 2013 ESPP and awards granted under it.
Our employees and employees of any parent or subsidiary corporation designated by our compensation committee are eligible to participate in our 2013 ESPP if they are customarily employed by us for more than 20 hours per week and more than five months in any calendar year. However, an employee may not be granted a right to purchase stock under our 2013 ESPP if: (i) the employee immediately after such grant would own stock possessing 5% or more of the total combined voting power or value of all classes of our capital stock or of any parent or subsidiary corporation, or (ii) the employee’s rights to purchase stock under all of our employee stock purchase plans would accrue at a rate that exceeds $25,000 in value for each calendar year of participation in such plans.
Our 2013 ESPP is generally designed to comply with the provisions of Section 423 of the Internal Revenue Code and will typically be implemented through a series of sequential offering periods, generally six months in duration, as established by the compensation committee. In addition, our compensation committee may establish an offering period to commence on the effective date of our 2013 ESPP of such duration as the compensation committee may determine (subject to restrictions imposed by applicable law and the terms of our 2013 ESPP described in the following sentence). Our compensation committee is authorized to establish additional or alternative concurrent, sequential, or overlapping offering periods and offering periods having a different duration or different starting or ending dates, provided that no offering period may have a duration exceeding 27 months.
Amounts accumulated for each participant, generally through payroll deductions, are credited toward the purchase of shares of our common stock at the end of each offering period at a price generally equal to 85% of the lower of the fair market value of our common stock at the beginning of the offering period or on the purchase date (which will typically be at the end of an offering period).
No participant may purchase under our 2013 ESPP in any calendar year shares having a value of more than $25,000 measured by the fair market value per share of our common stock on the first day of the applicable offering period. Prior to the beginning of any offering period, our compensation committee may alter the maximum number of shares that may be purchased by any participant during the offering period or specify a maximum aggregate number of shares that may be purchased by all participants in the offering period. If insufficient shares remain available under the plan to permit all participants to purchase the number of shares to which they would otherwise be entitled, our compensation committee
137
Table of Contents
will make a pro rata allocation of the available shares. Any amounts withheld from participants’ compensation in excess of the amounts used to purchase shares will be refunded, without interest.
In the event of a change in control, an acquiring or successor corporation may assume our rights and obligations under our 2013 ESPP. If the acquiring or successor corporation does not assume such rights and obligations, then the purchase date of the offering periods then in progress will be accelerated to a date prior to the change in control as specified by our compensation committee, but the number of shares subject to outstanding purchase rights shall not be adjusted.
Our compensation committee has the authority to amend, suspend or terminate our 2013 ESPP, except that, subject to certain exceptions described in the 2013 ESPP, no such action may adversely affect any outstanding rights to purchase stock under our 2013 ESPP.
Equity Compensation Plan Information
The following table includes information as of December 31, 2013 for equity compensation plans:
| Plan Category |
Number
of securities to be issued upon exercise of outstanding options |
Weighted- average exercise price of outstanding options |
Number
of securities remaining available for future issuance under equity compensation plans |
|||||||||
| Equity compensation plans approved by security holders(1) |
1,553,051 | $ | 6.23 | 964,668 | (3) | |||||||
| Equity compensation plans not approved by security holders(2) |
19,531 | $ | 1.69 | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Total |
1,572,582 | $ | 6.17 | 964,668 | ||||||||
| (1) | Includes securities issuable under our 2007 Stock Option/Stock Issuance Plan, our 2013 Equity Incentive Plan and our 2013 Employee Stock Purchase Plan. |
| (2) | Includes securities issuable upon exercise of stock options granted to Jean Blanchet by the our board of directors on November 17, 2006. |
| (3) | Includes (i) 853,557 shares of common stock available for issuance under our 2013 Equity Incentive Plan and (ii) 111,111 shares of common stock available for issuance under our 2013 Employee Stock Purchase Plan. No shares are reserved for future issuance under our amended and restated 2007 Stock Option/Stock Issuance Plan other than shares issuable upon exercise of equity awards outstanding under such plan. |
Beginning in 2014, the number of shares of common stock reserved under the 2013 Equity Incentive Plan automatically increases on January 1st of each year by an amount described above under “—Stock Plans—2013 Equity Incentive Plan.” The annual increase for January 1, 2014 was 963,066 shares. Beginning in 2014, the number of shares of common stock reserved under our 2013 Employee Stock Purchase Plan automatically increases on January 1st of each year by an amount described above under “—Stock Plans—2013 Employee Stock Purchase Plan.” The annual increase for January 1, 2014 was 240,766 shares.
Other Compensation Policies
Recovery of Compensation
Currently, we have not implemented a policy regarding retroactive adjustments to any cash or equity-based incentive compensation paid to our named executive officers or other employees where the payments were predicated upon the achievement of financial results that were subsequently the subject of a financial restatement. We expect that the compensation committee will adopt, or recommend that our Board of Directors adopt, a compensation recovery policy consistent with the requirements of regulations promulgated under Section 954 of the Dodd–Frank Wall Street Reform and Consumer Protection Act, if and when such regulations are enacted.
138
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
The following is a description of transactions or series of transactions since January 1, 2011, to which we were or will be a party, in which:
| • | the amount involved in the transaction exceeds $120,000; and |
| • | any of our executive officers, directors and principal stockholders, including their immediate family members, had or will have a direct or indirect material interest. |
Compensation arrangements for our named executive officers and our directors are described elsewhere in this prospectus under “Management—Director Compensation” and “Executive Compensation.”
Convertible Note Financing
In April 2012 and May 2012, LDR Holding and Médical issued and sold an aggregate of approximately $15,000,000 of subordinated secured convertible promissory notes, which we refer to as the “convertible notes,” which bore an interest rate of 6%. The convertible notes issued by Médical, in an aggregate face amount of $5,227,134, were denominated in Euros. Any payments under the Médical convertible notes were made in Euros.
Certain of the convertible notes were sold to entities affiliated with members of our board of directors and other holders of more than 5% of a class of our voting securities. We believe that the terms obtained and consideration received in the convertible note financing were comparable to terms and consideration we could have received in an arms’ length transaction. Our full board of directors reviewed and approved the terms of our convertible note financing.
Upon the completion of our initial public offering, the convertible notes became due and payable. At each holder’s option, a holder was entitled to receive either (i) an amount of cash equal to one and one-half times all unpaid principal and accrued but unpaid interest on such holder’s convertible notes or (ii) a number of shares of common stock equal to (a) the unpaid principal and accrued but unpaid interest on such holder’s convertible notes, divided by (b) 50% of the price per share of common stock sold in the public offering.
The table below and the accompanying footnotes summarize this sale and subsequent conversion and repayment of the convertible notes in connection with our initial public offering:
| Purchaser |
Convertible Notes Purchased |
Number of Shares of Common Stock Issued Upon Completion of the Initial Public Offering |
Cash Paid Upon Completion of Initial Public Offering |
|||||||||
| Austin Ventures VIII, L.P.(1) |
$ | 3,558,085 | 516,950 | $ | * | |||||||
| Entities affiliated with Telegraph Hill Partners(2) |
$ | 3,356,060 | 487,595 | $ | * | |||||||
| PTV Sciences II, L.P.(3) |
$ | 2,130,496 | 309,537 | $ | * | |||||||
| Verwaltungsgesellschaft AD. KRAUTH(4) |
$ | 728,225 | 105,803 | $ | * | |||||||
| Entities affiliated with Paris Orléans SCA(5) |
$ | 948,950 | 137,450 | $ | * | |||||||
| Entities affiliated with Keensight Capital(6) |
$ | 3,420,164 | 247,695 | $ | 2,786,592 | |||||||
| Dahlia A Sicar SCA(7) |
$ | 858,020 | 124,279 | $ | * | |||||||
| Total |
$ | 15,000,000 | 1,929,309 | $ | 2,786,658 | |||||||
| (1) | Austin Ventures VIII, L.P. is a holder of greater than 5% of our common stock. Joseph Aragona, an affiliate of Austin Ventures VIII, L.P. is a member of our board of directors. |
footnotes continued on following page
139
Table of Contents
| (2) | Entities affiliated with Telegraph Hill Partners are, collectively, holders of greater than 5% of our common stock. Robert G. Shepler and Dr. Thomas A. Raffin, affiliates of Telegraph Hill Partners, were members of our board of directors prior to the completion of our initial public offering. |
| (3) | Entities affiliated with PTV Sciences II, L.P. are, collectively, holders of greater than 5% of our common stock. Matthew Crawford, an affiliate of PTV Sciences II, L.P., was a member of our board of directors prior to the completion of our initial public offering. |
| (4) | Verwaltungsgesellschaft AD. KRAUTH was, prior to the completion of our initial public offering, a holder of greater than 5% of a class of our voting stock. Stefan Widensohler, an affiliate of Verwaltungsgesellschaft AD. KRAUTH, is a member of our board of directors. |
| (5) | Entities affiliated with the Paris Orléans SCA were, collectively, prior to the completion of our initial public offering, holders of greater than 5% of a class of our voting stock. Pierre Rémy, who was an affiliate of Paris Orléans SCA at such time, was a member of our board of directors prior to the completion of our initial public offering. Until November 2013, Keensight Capital was a majority-owned subsidiary of Paris Orléans SCA, and was known as R Capital Management. In November 2013, R Capital Management ceased to be a subsidiary of Paris Orleans SCA and changed its name to Keensight Capital. |
| (6) | Entities affiliated with Keensight Capital are, collectively, holders of greater than 5% of our common stock. Pierre Rémy, an affiliate of Keensight Capital, was a member of our board of directors prior to the completion of our initial public offering. Until November 2013, Keensight Capital was a majority-owned subsidiary of Paris Orléans SCA, and was known as R Capital Management. In November 2013, R Capital Management ceased to be a subsidiary of Paris Orleans SCA and changed its name to Keensight Capital. |
| (7) | Dahlia A Sicar SCA was, prior to the completion of our initial public offering, a holder of greater than 5% of a class of our voting stock. |
| * | Indicates less than $100. |
Amended and Restated Put-Call Agreement
In connection with our convertible note financing, we entered into an amendment to our amended and restated put-call agreement, with the holders of the capital stock of Médical, other than LDR Holding (which now owns 100% of the outstanding capital stock of Médical), including Messrs. Lavigne, Dinville and Richard, Dahlia A Sicar SCA (a holder of greater than 5% of a class of our voting stock prior to the completion of our initial public offering), and Jean-Louis Médus (one of our directors prior to the completion of our initial public offering).
In August 2013, we and the holders of the capital stock of Médical entered into a second amended and restated put-call agreement that provides in the event that either (i) a public offering, including our initial public offering, is consummated or (ii) a transaction constituting a liquidation event, as defined in LDR Holding’s amended and restated certificate of incorporation (as then in effect), is approved pursuant to the amended and restated voting agreement, described below, LDR Holding could require the holders of Class A Stock of Médical (other than holders of warrants to purchase Class A Stock of Médical) to exchange their shares of Class A Stock of Médical for shares of LDR Holding common stock in the ratio of one share of Médical’s Class A Stock for 5.80087 shares of LDR Holding common stock. In addition, prior to the completion of our initial public offering, each holder of Médical’s Class A Stock, other than LDR Holding, was entitled to, at such holder’s discretion, exchange all, but not less than all, of the holder’s Class A stock of Médical for common stock of LDR Holding at the same exchange ratio.
In October 2013, in connection with the consummation of our initial public offering, all of the outstanding shares of Class A Stock of Médical (other than warrants to purchase Class A Stock of Médical) were converted into shares of LDR Holding pursuant to the second amended and restated put-call agreement. The following table sets forth the number of shares of LDR Holding common stock received by each of the related parties pursuant to such conversion.
| Médical Shareholders |
Common Stock | |||
| Jean-Louis Médus |
33,645 | |||
| Christophe Lavigne |
1,161,335 | |||
| Hervé Dinville |
1,161,335 | |||
| Patrick Richard |
580,668 | |||
140
Table of Contents
Pursuant to the second amended and restated put-call agreement, following the completion of our initial public offering, in the event that any holder of a warrant to purchase Class A Stock of Médical exercises his or her warrant, the shares of Médical received by such holder will be converted into shares of LDR Holding’s common stock at the exchange ratio listed above. As of December 31, 2013, Hervé Dinville held warrants to purchase Class A Stock of Médical that, if exercised, would convert into an aggregate of 205,376 shares of common stock (some of which are subject to vesting conditions) and Patrick Richard held warrants to purchase Class A Stock of Médical that, if exercised, would convert into an aggregate of 205,376 shares of common stock (some of which are subject to vesting conditions), pursuant to the amended and restated put-call agreement.
Amended and Restated Investors’ Rights Agreement
In connection with our convertible note financing, we entered into an amendment to our amended and restated investors’ rights agreement with certain of our stockholders, including Austin Ventures VIII, L.P., entities affiliated with Telegraph Hill Partners, entities affiliated with PTV Sciences, Verwaltungsgesellschaft AD. KRAUTH, entities affiliated with the Paris Orléans SCA (which, at such time, included entities affiliated with Keensight Capital), entities affiliated with Path4 Ventures (a holder of greater than 5% of a class of our voting stock prior to the completion of our initial public offering) and Dahlia A Sicar SCA. The amended and restated investors’ rights agreement, as amended, among other things:
| • | grants such stockholders certain registration rights with respect to shares of our common stock, including shares of common stock issued or issuable upon conversion of our convertible preferred stock and our convertible notes; |
| • | obligated us to deliver periodic financial statements to any stockholder who holds at least 2,000,000 shares of our then outstanding preferred stock or 148,148 shares of our common stock, which we refer to as a “qualified holder;” and |
| • | granted a right of first offer with respect to sales of our shares by us, subject to specified exclusions, to qualified holders. |
For more information regarding the registration rights provided in this agreement, please refer to the section of this prospectus titled “Description of Capital Stock—Registration Rights.”
Certain provisions of this agreement, other than those relating to registration rights, terminated automatically upon completion of our initial public offering.
Amended and Restated Voting Agreement
In connection with our sale of Series C preferred stock in September of 2007, we entered into our amended and restated voting agreement with certain of our stockholders, including Austin Ventures VIII, L.P., entities affiliated with Telegraph Hill Partners, entities affiliated with PTV Sciences, Verwaltungsgesellschaft AD. KRAUTH, entities affiliated with the Paris Orléans SCA (which at such time included entities affiliated with Keensight Capital), entities affiliated with Path4 Ventures and Dahlia A Sicar SCA. Our amended and restated voting agreement provided, among other things:
| • | for the voting of shares with respect to the constituency of our board of directors; |
| • | the right of Verwaltungsgesellschaft AD. KRAUTH to appoint a non-voting observer to our board of directors; and |
| • | requirements for voting of shares of capital stock in the event of an acquisition of the Company. |
141
Table of Contents
The amended and restated voting agreement terminated automatically upon completion of our initial public offering.
Amended and Restated Right of First Refusal and Co-Sale Agreement
In connection with our sale of Series C preferred stock in September of 2007, we entered into our amended and restated right of first refusal and co-sale agreement with certain of our stockholders, including Austin Ventures VIII, L.P., entities affiliated with Telegraph Hill Partners, entities affiliated with PTV Sciences, Verwaltungsgesellschaft AD. KRAUTH, entities affiliated with the Paris Orléans SCA (which at such time included entities affiliated with Keensight Capital), entities affiliated with Path4 Ventures and Dahlia A Sicar SCA. The amended and restated right of first refusal and co-sale agreement among other things:
| • | granted our investors certain rights of first refusal and co-sale with respect to proposed transfers of our securities by certain stockholders; and |
| • | granted us certain rights of first refusal with respect to proposed transfers of our securities by certain stockholders. |
The amended and restated right of first refusal and co-sale agreement terminated automatically upon completion of our initial public offering.
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers. These agreements, among other things, require us to indemnify each director and executive officer to the fullest extent permitted by Delaware law, including indemnification expenses such as attorneys’ fees, judgments, fines and settlement amounts incurred by the director or executive officer in any action or proceeding, including any action or proceeding by or in right of us, arising out of the person’s services as a director or executive officer. These indemnification agreements are described in more detail under “Management—Limitations on Liability and Indemnification Agreements.”
Loan to Christophe Lavigne
In February 2007, we lent Christophe Lavigne, our President and Chief Executive Officer, $200,000 in connection with Mr. Lavigne’s purchase of a primary residence in Austin, Texas. The loan to Mr. Lavigne bore interest at the greater of (i) 6.0% per year or (ii) the short-term applicable federal rate. Interest payments under the loan were due quarterly, however, our board of directors subsequently deferred interest payments until the maturity date, and the principal balance was due in February 2014. The principal payment was subject to acceleration, among other events, five days before the date that we filed a registration statement with the Securities and Exchange Commission. Mr. Lavigne repaid this loan in full in August 2013.
Series C Voting Agreement
Our certificate of incorporation in existence prior to the completion of our initial public offering provided that all outstanding shares of our preferred stock would convert into shares of common stock upon the closing of a “qualified public offering”, which was defined as a firm commitment underwritten public offering by a nationally recognized underwriter pursuant to an effective registration statement under the Securities Act of 1933, as amended, with aggregate gross proceeds to us and the selling stockholders therein of at least $50 million at a per share price not less than $23.625 per share. Our initial public offering was not a qualified public offering.
Our certificate of incorporation in effect prior to our initial public offering provided that all outstanding shares of our preferred stock would convert into shares of common stock on the date specified by the
142
Table of Contents
written consent or agreement of (x) the holders of at least 66 2/3% of the then outstanding shares of Series A preferred stock and Series B preferred stock, voting together as a single class on an as-converted basis, and (y) the holders of at least 66 2/3% of the then outstanding shares of Series C preferred stock.
Following our receipt of indications regarding valuation from the underwriters prior to our initial public offering, the holders of the requisite percentage of our Series C preferred stock, through their representatives on our board of directors, asked us for cash compensation in order to agree to convert their preferred stock to common stock and to relinquish the rights, preferences and privileges associated with those preferred shares, including a liquidation preference. As a result of these negotiations, the holders of Series C preferred stock agreed to accept a payment from us in an aggregate amount equal to approximately $17.5 million, or 50% of the liquidation preference of the Series C preferred stock, to vote in favor of conversion. The holders of our Series C preferred stock owned the necessary percentage of our Series A and Series B preferred stock to convert all outstanding shares of our preferred stock pursuant to our then-existing certificate of incorporation.
In August 2013, we entered into a voting agreement with the holders of our Series C preferred stock, which agreement was subsequently amended and restated in September 2013, or the Series C Voting Agreement. Pursuant to the Series C Voting Agreement, we paid the holders of our Series C preferred stock the amounts set forth in the table below in October 2013, following the completion of our initial public offering.
| Recipients |
Amounts Paid | |||
| Entities affiliated with Telegraph Hill Partners |
$ | 10,086,909 | ||
| Entities affiliated with PTV Sciences |
$ | 3,635,210 | ||
| Entities affiliated with Paris Orléans SCA |
$ | 1,000,000 | ||
| Entities affiliated with Keensight Capital |
$ | 250,000 | ||
| Dahlia A Sicar SCA |
$ | 1,027,881 | ||
| Austin Ventures VIII, L.P |
$ | 1,500,000 | ||
| Comerica Ventures Incorporated |
$ | 42,814 | ||
| Total |
$ | 17,542,814 | ||
Joseph Aragona, one of our current directors, is affiliated with Austin Ventures; Matthew Crawford, one of our directors prior to the completion of our initial public offering, is affiliated with PTV Sciences; Thomas Raffin and Robert G. Shepler, each one of our directors prior to the completion of our initial public offering, are affiliated with Telegraph Hill Partners; and Pierre Rémy, one of our directors prior to the completion of our initial public offering, was affiliated with Paris Orléans SCA and is affiliated with Keensight Capital. Until November 2013, Keensight Capital was a majority-owned subsidiary of Paris Orléans SCA, and was known as R Capital Management. In November 2013, R Capital Management ceased to be a subsidiary of Paris Orleans SCA and changed its name to Keensight Capital.
Participation in our Initial Public Offering
The table below sets forth shares of our common stock that were purchased by our principal stockholders in our initial public offering, which was completed in October 2013.
| Purchaser |
Shares Purchased in Initial Public Offering |
|||
| Entities affiliated with Telegraph Hill Partners |
50,000 | |||
| Austin Ventures VIII, L.P |
50,000 | |||
| Entities affiliated with PTV Sciences |
50,000 | |||
| Entities affiliated with Keensight Capital |
10,000 | |||
143
Table of Contents
Policies and Procedures for Related Person Transactions
As provided by our audit committee charter and corporate governance guidelines, as in effect upon the completion of our initial public offering, our audit committee must review and approve in advance any related person transaction, and all of our directors, officers and employees are required to report to our audit committee any such related person transaction prior to its completion. Prior to the completion of our initial public offering, our board of directors reviewed related person transactions. Each of the related person transactions described above was submitted to and approved by our board of directors.
144
Table of Contents
PRINCIPAL AND SELLING STOCKHOLDERS
The following table sets forth, as of March 24, 2014, information regarding the beneficial ownership of our common stock by:
| • | each person, or group of affiliated persons, who is known by us to be the beneficial owner of five percent or more of our outstanding common stock (on an as-converted to common stock basis); |
| • | each of our directors; |
| • | each of our named executive officers; |
| • | all our directors and executive officers as a group (12 persons); and |
| • | all selling stockholders. |
The information in the following table is calculated based on 24,078,191 shares of common stock outstanding before this offering and shares of common stock outstanding after this offering. The number of shares outstanding is based on the number of shares of common stock outstanding on March 24, 2014 as adjusted to give effect to the sale of shares of common stock in this offering.
Except as otherwise indicated below, the address of each officer, director and five percent stockholder listed below is c/o LDR Holding Corporation, 13785 Research Boulevard, Suite 200, Austin, Texas 78750.
We have determined beneficial ownership in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options and other equity awards that are either immediately exercisable or exercisable within 60 days of March 24, 2014. These shares are deemed to be outstanding and beneficially owned by the person holding those options for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them.
145
Table of Contents
| Shares of Common Stock Beneficially Owned Prior to this Offering |
Shares Being Offered |
Shares of Common Stock Beneficially Owned After this Offering |
||||||||||||||
| Number | Percent | Number | Percent | |||||||||||||
| 5% Stockholders |
||||||||||||||||
| Austin Ventures VIII, L.P.(1) |
3,286,800 | 13.7 | % | % | ||||||||||||
| Entities Affiliated with Telegraph Hill Partners(2) |
3,103,014 | 12.9 | % | % | ||||||||||||
| Entities Affiliated with Keensight Capital(3) |
2,872,119 | 11.9 | % | |||||||||||||
| Entities affiliated with PTV Sciences(4) |
2,039,942 | 8.5 | % | % | ||||||||||||
| Gilder, Gagnon, Howe & Co. LLC(5) |
1,211,058 | 5.0 | % | % | ||||||||||||
| Directors and Executive Officers |
||||||||||||||||
| Christophe Lavigne(6) |
1,351,832 | 5.6 | % | % | ||||||||||||
| Hervé Dinville(7) |
1,314,860 | 5.4 | % | % | ||||||||||||
| Patrick Richard(8) |
734,193 | 3.0 | % | % | ||||||||||||
| Robert McNamara(9) |
82,321 | * | * | |||||||||||||
| Joseph C. Aragona(1) |
3,286,800 | 13.7 | % | % | ||||||||||||
| William W. Burke(10) |
604 | * | * | |||||||||||||
| Kevin M. Lalande(11) |
604 | * | * | |||||||||||||
| Stefan Widensohler(12) |
663,073 | 2.8 | % | % | ||||||||||||
| All executive officers and directors as a group (12 persons)(13) |
4,481,139 | 18.0 | % | % | ||||||||||||
| * | Percentage of shares beneficially owned does not exceed 1%. |
| (1) | Number of shares prior to this offering consists of shares held of record by Austin Ventures VIII, L.P. (“AV VIII”). AV Partners VIII, L.P. (“AVP VIII”) is the general partner of AV VIII and may be deemed to have sole voting and investment power over the shares held by AV VIII. Joseph C. Aragona, Kenneth P. DeAngelis, John D. Thornton and Christopher A. Pacitti are the general partners of AVP VIII and share voting and/or dispositive power over the shares held by AVP VIII. The address for Austin Ventures VIII, L.P. is 300 West Sixth Street, Suite 2300, Austin, Texas 78701. |
| (2) | Number of shares prior to this offering consists of (a) 69,671 shares of common stock held by Telegraph Hill Partners, L.P. (“THP I”), (b) 16,149 shares of common stock held by THP Affiliates Fund, LLC (“THP I AFF”), (c) 375,320 shares of common stock held by Telegraph Hill Partners SBIC, L.P.(“THP SBIC”), (d) 2,584,683 shares of common stock held by Telegraph Hill Partners II, L.P. (“THP II”), and (e) 56,891 shares of common stock held by THP II Affiliates Fund, LLC (“THP II AFF”). Telegraph Hill Partners Investment Management, LLC (“THP IM”) is the general partner of THP I and the manager of THP I AFF. Telegraph Hill Partners II Investment Management, LLC (“THP II IM”) is the general partner of THP II and the manager of THP II AFF. Telegraph Hill Partners Management Company, LLC (“THPMC”) is the manager of both THP IM and THP II IM. Robert G. Shepler, J. Matthew Mackowski, Dr. Thomas A. Raffin and Deval Lashkari are each managers of THPMC and are deemed to have beneficial ownership of the shares held by THP I, THP II, THP Affiliates and THP II Affiliates. Telegraph Hill Partners SBIC, LLC (“THP SBIC, LLC”) is the general partner of THP SBIC. Robert Shepler, Dr. Thomas Raffin, J. Matthew Mackowski and Deval Lashkari are the managers of THP SBIC, LLC and are deemed to have beneficial ownership of the shares held by THP SBIC. The address for Telegraph Hill Partners is 360 Post Street, Suite 601, San Francisco, California 94108. |
| (3) | Number of shares prior to this offering consists of (a) 2,262,584 shares of common stock held by R Capital Technologies FCPR (“RCT”), (b) 361,840 shares of common stock held by R Capital Prive Technologies FCPR (“RCPT”), and (c) 247,695 shares of common stock held by R Capital III FCPR (“RCIII”). Keensight Capital is the managing company of each of RCT, RCPT and RCIII. Until November 2013, Keensight Capital was a majority-owned subsidiary of Paris Orleans SCA, and was known as R Capital Management. In November 2013, R Capital Management ceased to be a subsidiary of Paris Orleans SCA and changed its name to Keensight Capital. Jean-Michel Beghin, Jerome Pujol and Pierre Remy are the managing partners of Keensight and each is deemed to be the beneficial owner of the shares held by RCT, RCPT and RCIII. The address for Keensight Capital is 64 rue de Lisbonne, 75008 Paris, France. |
| (4) | Number of shares prior to this offering consists of (a) 941,171 shares of common stock held by PTV Sciences II, L.P. (“PTVS II”), and (b) 1,098,771 shares of common stock held by Pinto Technology Ventures, L.P. (“PTV”). Pinto Technology Ventures GP, L.P. (“PTV GP”) is general partner of PTV. Pinto TV GP Company LLC (“TV GP”) is the general partner of PTV GP. Pinto Technology Ventures GP II, L.P. (“PTV GP II”) is the general partner of PTVS II. TV GP is the general partner of PTV GP II. Each of Matthew Crawford and Rick Anderson is a manager of TV GP and is deemed to have beneficial ownership of the shares held by PTV and PTVS II. The address for PTV Sciences is 3600 N. Capital of Texas Highway, Building B, Suite 245, Austin, Texas 78746. |
footnotes continued on following page
146
Table of Contents
| (5) | Based on information in a Schedule 13G filed on February 12, 2014. The address for Gilder, Gagnon, Howe & Co. LLC is 3 Columbus Circle, 26th Floor, New York, New York 10019. |
| (6) | Includes 73,516 shares subject to options that are currently exercisable or will become exercisable within 60 days of March 24, 2014. |
| (7) | Includes warrants to purchase shares of Médical that are convertible into 153,525 shares of common stock pursuant to the Second Amended and Restated Put-Call Agreement and which currently exercisable or will become exercisable within 60 days of March 24, 2014. See Certain Relationships and Related Person Transactions—Second Amended and Restated Put-Call Agreement.” |
| (8) | Includes warrants to purchase shares of Médical that are convertible into 153,525 shares of common stock pursuant to the Second Amended and Restated Put-Call Agreement and which currently exercisable or will become exercisable within 60 days of March 24, 2014. See Certain Relationships and Related Person Transactions—Second Amended and Restated Put-Call Agreement.” |
| (9) | Includes 82,321 shares subject to options that are currently exercisable or will become exercisable within 60 days of March 24, 2014. |
| (10) | Includes 604 shares subject to options that are currently exercisable or will become exercisable within 60 days of March 24, 2014. |
| (11) | Includes 604 shares subject to options that are currently exercisable or will become exercisable within 60 days of March 24, 2014. |
| (12) | Includes 604 shares subject to options that are currently exercisable or will become exercisable within 60 days of March 24, 2014. |
| (13) | Includes 796,189 shares subject to options or warrants to purchase shares of Médical (that are exercisable for shares of common stock) that are currently exercisable or will become exercisable within 60 days of March 24, 2014. |
147
Table of Contents
The following is a description of our capital stock and certain provisions of our amended and restated certificate of incorporation and amended and restated bylaws. This description is intended as a summary and is qualified in its entirety by the provisions of our amended and restated certificate of incorporation and amended and restated bylaws, which have been filed with the SEC and are incorporated by reference to the registration statement of which this prospectus is a part.
Outstanding Shares
Our authorized capital stock consists of 107,000,000 shares of common stock, $0.001 par value per share, and 5,000,000 shares of preferred stock, $0.001 par value per share. As of December 31, 2013, assuming the completion of this offering, we will have shares of common stock issued and outstanding. We have approximately 119 stockholders of record. In addition, as of December 31, 2013, options to purchase 1,572,582 shares of common stock were outstanding.
Common Stock
Dividends
Subject to preferences that may be applicable to any then outstanding preferred stock, holders of common stock are entitled to receive ratably those dividends, if any, as may be declared from time to time by the board of directors out of legally available funds. After the completion of this offering, we do not anticipate declaring any cash dividends to the holders of our common stock for the foreseeable future.
Voting Rights
Each holder of our common stock is entitled to one vote for each share on all matters submitted to a vote of the stockholders, including the election of directors. Under our amended and restated certificate of incorporation and amended and restated bylaws, our stockholders do not have cumulative voting rights. Because of this, the holders of a majority of the shares of common stock entitled to vote in any election of directors can elect all of the directors standing for election, if they should so choose.
Liquidation
In the event of our liquidation, dissolution or winding up, holders of common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities and the satisfaction of any liquidation preference granted to the holders of any outstanding shares of preferred stock.
Rights and Preferences
Holders of common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to the common stock. The rights, preferences and privileges of the holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock that we may designate in the future.
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued pursuant to this offering will be, fully paid and nonassessable.
Preferred Stock
The board of directors has the authority, without further action by the stockholders, to issue up to 5,000,000 shares of preferred stock in one or more series, to establish from time to time the number of
148
Table of Contents
shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon, and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding. The board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in control of us and may adversely affect the market price of the common stock and the voting and other rights of the holders of common stock.
Options
As of December 31, 2013, options to purchase 1,572,582 shares of common stock were outstanding, at an average exercise price of $6.17 per share.
Registration Rights
Demand Registration Rights
Pursuant to our amended and restated investors’ rights agreement dated September 11, 2007, the holders of at least (1) a majority of the registrable shares of our common stock issued upon conversion of preferred stock, convertible notes and certain warrants, if the Company has not effected a demand registration, or (2) one-third of the then-outstanding registrable shares, if the Company has effected a demand registration, can request that we file up to two registration statements registering all or a portion of their registrable shares. As of March 24, 2014, and assuming the sale of shares contemplated by this offering, the holders of approximately million shares of our common stock, which were issued upon conversion of preferred stock, convertible notes and certain warrants have demand registration rights. Under specified circumstances, we also have the right to defer filing of a requested registration statement for a period of not more than 90 days, which right may not be exercised more than once during any period of 12 consecutive months. These registration rights are subject to additional conditions and limitations, including the right of the underwriters to limit the number of shares included in any such registration under certain circumstances.
Form S-3 Registration Rights
Pursuant to the amended and restated investors’ rights agreement, if we are eligible to file a registration statement on Form S-3, the holders of at least 25% of the registrable shares of common stock issued upon conversion of preferred stock, convertible notes and certain warrants (or 50% of the registrable shares of common stock issued upon the conversion of our Series C preferred stock) have the right to demand that we file additional registration statements, including a shelf registration statement, for such holders on Form S-3.
Piggyback Registration Rights
Pursuant to the amended and restated investors’ rights agreement, whenever we propose to file a registration statement under the Securities Act, other than with respect to a registration related to employee benefit or similar plans, a registration on any form which does not include substantially the same information as would be required to be included in this registration statement, or a registration in which the only common stock being registered is common stock issuable upon conversion of debt securities which are also being registered, the holders of registrable shares of common stock issued upon conversion of preferred stock, convertible notes and certain warrants are entitled to notice of the registration and have the right to include their registrable shares in such registration. As of March 24, 2014, and assuming the sale of shares contemplated by this offering, the holders of approximately million shares of our common stock will be entitled to notice of this registration and will be entitled to include their shares of common stock in the registration statement. The underwriter(s) of any
149
Table of Contents
underwritten offering will have the right to limit the number of shares having registration rights to be included in the registration statement. The Company intends to obtain waivers of these piggyback registration rights with respect to this offering from the requisite holders.
Expenses of Registration
We are required to pay all expenses relating to any demand, Form S-3 or piggyback registration, other than underwriting discounts and commissions, subject to certain limited exceptions. We will not pay for any expenses of any demand registration if the request is subsequently withdrawn by the holders of a majority of the shares requested to be included in such a registration statement, subject to limited exceptions.
Expiration of Registration Rights
The registration rights described above will expire for each holder upon the earlier of (i) October 15, 2020 or (ii) the date after this offering on which the holder may sell all of his, her or its shares under Rule 144 under the Securities Act during any 90-day period without regard to current public information, volume limitations, manner of sale restrictions or Form 144 filing requirements. For a description of Rule 144, see “Shares Eligible for Future Sale—Rule 144.”
Holders of approximately 15.2 million of our shares with these registration rights have signed or are expected to sign agreements with the underwriters prohibiting the exercise of their registration rights for 60 days following the date of this prospectus. These agreements are described below under “Underwriting.”
Anti-Takeover Effects of Delaware Law and Certain Provisions of our Certificate of Incorporation and Amended and Restated Bylaws
Delaware Law
We are governed by Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. A “business combination” includes mergers, asset sales or other transactions resulting in a financial benefit to the stockholder. An “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years, did own, 15% or more of the corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring or preventing a change in our control
Amended and Restated Certificate of Incorporation and Amended and Restated Bylaw Provisions
Our amended and restated certificate of incorporation and amended and restated bylaws:
| • | provide for a classified board of directors; |
| • | permit our board of directors to issue shares of preferred stock, with any rights, preferences and privileges as they may designate, including the right to approve an acquisition or other change in our control; |
| • | provide that the authorized number of directors may be changed only by resolution of the board of directors; |
| • | provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| • | require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent; |
150
Table of Contents
| • | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner, and also specify requirements as to the form and content of a stockholder’s notice; |
| • | prohibit cumulative voting (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); |
| • | provide that special meetings of our stockholders may be called only by the chairman of the board of directors, our Chief Executive Officer or president (in the absence of a Chief Executive Officer) or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors; and |
| • | provide that stockholders will be permitted to amend our amended and restated bylaws and certain provisions of our amended and restated certificate of incorporation only upon receiving at least sixty-six and two thirds percent of the votes entitled to be cast by holders of all outstanding shares then entitled to vote generally in the election of directors, voting together as a single class. |
These and other provisions contained in our certificate of incorporation and bylaws could delay or discourage some types of transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares over then current prices, and may limit the ability of stockholders to remove current management or approve transactions that stockholders may deem to be in their best interests and, therefore, could adversely affect the price of our common stock.
Stock Exchange Listing
Our common stock is listed on the NASDAQ Global Select Market under the trading symbol “LDRH.”
Transfer Agent and Registrar
The Transfer Agent and Registrar for our common stock is American Stock Transfer & Trust Company, LLC.
151
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Future sales of substantial amounts of our common stock in the public market could reduce prevailing market prices. Furthermore, since a substantial number of shares will be subject to contractual and legal restrictions on resale as described below, sales of substantial amounts of our common stock in the public market after these restrictions lapse could adversely affect the prevailing market price and our ability to raise equity capital in the future
Based on the number of shares outstanding as of March 24, 2014, upon completion of this offering, shares of our common stock will be outstanding, assuming no exercise of options or warrants. 5,590,000 of the shares sold in our initial public offering are freely tradable and all of the shares sold in this offering by us, plus any shares sold upon exercise of the underwriters’ option to purchase additional shares, will be freely tradable, except for any shares acquired by our “affiliates,” as that term is defined in Rule 144 under the Securities Act. Except as set forth below, the remaining shares of common stock outstanding after this offering will be restricted as a result of securities laws or lock-up agreements. Restricted shares may be sold in the public market only (i) upon expiration of any applicable lock-up agreement and (ii) if registered or if their resale qualifies for an exemption from registration under Rule 144 or Rule 701 under the Securities Act, each as described below
As a result of the contractual restrictions described below and the provisions of Rules 144 and 701, the restricted shares will be available for sale in the public market as follows:
| • | shares will be eligible for sale upon completion of this offering; and |
| • | shares will be eligible for sale upon the expiration of the lock-up agreements executed in connection with this offering, described below, beginning 60 days after the date of this prospectus. |
In addition, of the 2,091,862 shares of our common stock that were subject to stock options outstanding as of March 24, 2014, options to purchase 1,352,929 shares were vested as of March 24, 2014. Shares issued upon the exercise of such vested options will be eligible for sale upon completion of this offering.
Lock-Up Agreements
We, the selling stockholders, and all of our directors and executive officers have agreed not to sell or otherwise transfer or dispose of any of our securities for a period of 60 days from the date of this prospectus, subject to certain exceptions. Piper Jaffray & Co. may permit early release of shares subject to the lock-up agreements. Please read “Underwriting” for a description of these lock-up provisions.
Rule 144
In general, under Rule 144 as currently in effect, a person who is not deemed to have been one of our affiliates for purposes of the Securities Act at any time during 90 days preceding a sale and who has beneficially owned the shares proposed to be sold for at least six months, including the holding period of any preceding holders that are not affiliates of us, is entitled to sell such shares, subject only to the availability of current public information about us and subject to the lock-up agreements described above. If such a non-affiliated person has beneficially owned the shares proposed to be sold for at least one year, including the holding period of any preceding holders that are not affiliates of us, then such person is entitled to sell such shares without regard to the limitations of Rule 144.
152
Table of Contents
In general, under Rule 144 as currently in effect, our affiliates or persons selling shares on behalf of our affiliates are entitled to sell upon expiration of the lock-up agreements described above, within any three-month period, a number of shares that does not exceed the greater of:
| • | 1% of the number of shares of our common stock then outstanding, which will equal approximately shares immediately after this offering; or |
| • | the average weekly trading volume of our common stock during the four calendar weeks preceding the filing of notice of the sale. |
Sales under Rule 144 by our affiliates or persons selling shares on behalf of our affiliates are also subject to certain manner of sale provisions and notice requirements and to the availability of current public information about us.
Rule 701
In general, under Rule 701 of the Securities Act, any of our employees, directors, officers or natural person consultants or advisors who purchased shares from us in connection with a compensatory stock or option plan or other written compensatory agreement before the date of this prospectus is eligible to resell those shares 90 days after the date of this prospectus in reliance on Rule 144 without having to comply with the holding period requirement of Rule 144 and, in the case of non-affiliates, without having to comply with the public information, volume limitation or notice filing provisions of Rule 144.
Stock Issued Under Compensatory Plans
We filed with the SEC a registration statement on Form S-8 under the Securities Act to register common stock issuable under our 2004 Stock Option/Stock Issuance Plan, our amended and restated 2007 Stock Option/Stock Issuance Plan, our 2013 Equity Incentive Plan and our 2013 Employee Stock Purchase Plan. Accordingly, shares registered under such registration statement are available for sale in the open market, unless such shares are subject to vesting restrictions with us, Rule 144 restrictions applicable to our affiliates or the lock-up restrictions described above.
Registration Rights
As of March 24, 2014, and assuming the sale of shares contemplated by this offering, the holders of approximately million shares of our common stock, or their transferees, are entitled to rights with respect to the registration of their shares under the Securities Act. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates, immediately upon the effectiveness of this registration. For more information about these registration rights, please read “Description of Capital Stock—Registration Rights.”
153
Table of Contents
MATERIAL UNITED STATES FEDERAL TAX CONSIDERATIONS FOR NON-UNITED STATES HOLDERS
The following is a summary of the material United States federal income and estate tax consequences of the acquisition, ownership and disposition of shares of our common stock purchased pursuant to this offering by a non-United States holder, as we define that term below. This summary is based upon current provisions of the Internal Revenue Code of 1986, as amended, or the Code, Treasury regulations promulgated thereunder, judicial opinions, administrative pronouncements and published rulings of the United States Internal Revenue Service, or IRS, all as in effect as of the date hereof. These authorities may be changed, possibly retroactively, resulting in United States federal tax consequences different from those set forth below. We have not sought, and will not seek, any ruling from the IRS with respect to the statements made in the following summary, and there can be no assurance that the IRS will not take a position contrary to such statements or that any such contrary position taken by the IRS would not be sustained.
This summary is limited to non-United States holders who purchase shares of our common stock issued pursuant to this offering and who hold our common stock as a capital asset within the meaning of Section 1221 of the Code (generally property held for investment) for United States federal income tax purposes. This summary does not purport to be complete and does not address the tax considerations arising under the laws of any state, local or non-United States jurisdiction, or under United States federal estate or gift tax laws, except as specifically described below. In addition, this summary does not address tax considerations that may be applicable to an investor’s particular circumstances (including the impact of the unearned income Medicare contribution tax), nor does it address the special tax rules applicable to special classes of non-United States holders, including, without limitation:
| • | banks, insurance companies or other financial institutions; |
| • | partnerships or other entities treated as partnerships for United States federal income tax purposes; |
| • | holders subject to the alternative minimum tax; |
| • | United States expatriates; |
| • | tax-exempt organizations; |
| • | tax-qualified retirement plans; |
| • | brokers or dealers in securities or currencies; |
| • | real estate investment trusts; |
| • | regulated investment companies; |
| • | mutual funds; |
| • | “controlled foreign corporations,” passive foreign investment companies,” and corporations that accumulate earnings to avoid United States federal income tax; |
| • | traders in securities that elect to use a mark-to-market method of accounting for their securities holdings; |
| • | persons that will hold common stock as a position in a hedging transaction, “straddle,” “conversion,” or other integrated transaction for tax purposes; or |
| • | persons who acquired ordinary shares through the exercise or cancellation of employee stock options or otherwise as compensation for their services. |
154
Table of Contents
If a partnership, including any entity treated as a partnership for United States federal income tax purposes, is a holder of shares of our common stock, the tax treatment of a partner in the partnership will generally depend upon the status of the partner, the activities of the partnership and certain determinations made at the partner level. A holder of shares of our common stock that is a partnership, and partners in such partnership, are urged to consult their tax advisors regarding the tax consequences of the acquisition, ownership and disposition of shares of our common stock.
For purposes of this discussion, a “non-United States holder” is a beneficial owner of our common stock that is neither a United States person nor a partnership. A United States person means a person who is for United States federal income tax purposes:
| • | an individual who is a citizen or resident of the United States; |
| • | a corporation, including any entity treated as a corporation for United States federal income tax purposes, created or organized in or under the laws of the United States, any state within the United States, or the District of Columbia; |
| • | an estate the income of which is subject to United States federal income taxation regardless of its source; or |
| • | a trust (1) if it is subject to the primary supervision of a court within the United States and one or more United States persons have the authority to control all substantial decisions of the trust or (2) that has a valid election in effect under applicable Treasury regulations to be treated as a United States person. |
An individual is generally treated as a resident of the United States in any calendar year for United States federal income tax purposes if the individual is present in the United States for at least 31 days in that calendar year and for an aggregate of at least 183 days during the three-year period ending on the last day of the current calendar year. For purposes of the 183-day calculation, all of the days present in the current year, one-third of the days present in the immediately preceding year and one-sixth of the days present in the second preceding year are counted. Residents are generally taxed for United States federal income tax purposes as if they were United States citizens.
You are urged to consult your tax advisor with respect to the application of the United States federal income tax laws to your particular situation as well as any tax consequences arising under the United States federal estate or gift tax rules or under the laws of any state, local, non-United states or other taxing jurisdiction or under any applicable tax treaty.
Distributions on Shares of Our Common Stock
As described in the section entitled “Dividend Policy,” we do not anticipate declaring or paying dividends to holders of our common stock in the foreseeable future. However, if distributions are paid on shares of our common stock, the distributions will constitute dividends for United States federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under United States federal income tax principles. To the extent a distribution exceeds our current and accumulated earnings and profits, it will constitute a return of capital that is applied against and reduces, but not below zero, the adjusted tax basis of your shares in our common stock. Any remainder will be treated as capital gain from the sale of shares of our common stock and will be treated as described below under “—Gain on Disposition.” Dividends paid to a non-United States holder generally will be subject to withholding of United States federal income tax at the rate of 30% or such lower rate as may be specified by an applicable income tax treaty, the benefits of which a non-United States holder is eligible. However, if the dividend is effectively connected with the non-United States holder’s conduct of a trade or business in the United States and, where required by an applicable income tax treaty, is attributable to a United States permanent establishment or fixed based maintained by such
155
Table of Contents
non-United States holder, the dividend will not be subject to any withholding tax, provided certain certification and disclosure requirements are met, as described below, but will be subject to United States federal income tax imposed on net income on the same basis that applies to United States persons generally at the regular graduated rates. A corporate non-United States holder under certain circumstances also may be subject to a branch profits tax equal to 30%, or such lower rate as may be specified by an applicable income tax treaty, the benefits of which a non-United States holder is eligible, on a portion of its effectively connected earnings and profits for the taxable year, as adjusted for certain items. Non-United States holders are urged to consult their tax advisors regarding the potential applicability of any income tax treaty.
To claim the benefit of an applicable income tax treaty or to claim exemption from withholding because the income is effectively connected with the conduct of a trade or business in the United States, a non-United States holder must provide to the applicable withholding agent a properly executed IRS Form W-8BEN for treaty benefits or W-8ECI for effectively connected income, or such successor forms as the IRS designates, and certify under penalties of perjury that such holder is not a United States person prior to the payment of distributions on our common stock. These forms must be periodically updated. Special certification and other requirements apply to certain non-United States holders that are pass-through entities and non-United States holders whose stock is held through certain foreign intermediaries. Non-United States holders may obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim with the IRS.
Gain on Disposition
A non-United States holder generally will not be subject to United States federal income tax, including withholding, on gain recognized on a sale or other disposition of shares of our common stock unless any one of the following is true:
| • | the gain is effectively connected with the non-United States holder’s conduct of a trade or business in the United States and, where an applicable income tax treaty applies, is attributable to a United States permanent establishment or fixed base maintained by such non-United States holder; |
| • | the non-United States holder is a nonresident alien individual present in the United States for 183 days or more in the taxable year of the disposition and certain other requirements are met; or |
| • | our common stock constitutes a United States real property interest by reason of our status as a “United States real property holding corporation,” or USRPHC, for United States federal income tax purposes at any time during the shorter of (1) the period during which you hold our common stock and (2) the five-year period ending on the date you dispose of our common stock. |
Unless an applicable income tax treaty provides otherwise, gain described in the first bullet point above will be subject to the United States federal income tax imposed on net income on the same basis that applies to United States persons generally, at the regular graduated rates, but will generally not be subject to withholding. Corporate non-United States holders also may be subject to a branch profits tax at a rate of 30% (or such lower rate specified by an applicable income tax treaty) on such gain, as adjusted for certain items. Gain described in the second bullet point above will be subject to a flat 30% United States federal income tax (or such lower rate specified by an applicable income tax treaty), which may be offset by certain United States source capital losses (even though the individual is not considered a resident of the United States) provided the non-United States holder has timely filed United States federal income tax returns with respect to such losses.
156
Table of Contents
With respect to the third bullet point, we believe we are not currently, and will not become, a USRPHC for United States federal income tax purposes. However, because the determination of whether we are a USRPHC depends on the fair market value of our United States real property interests relative to the fair market value of our other business assets, we cannot assure you that we will not become a USRPHC in the future. If we are or were to become a USRPHC, as long as our common stock is “regularly traded,” as defined by applicable Treasury regulations, on an established securities market, it will not be treated as a United States real property interest with respect to any non-United States holder that holds, actually or constructively, no more than 5% of such regularly traded common stock throughout the shorter of the five-year period ending on the date of the sale or other disposition or the non-United States holder’s holding period for such stock, and any gain arising from any such sale or taxable disposition will not be subject to United States federal income tax. If we are determined to be a USRPHC and the foregoing exception does not apply, among other things, a purchaser may be required to withhold 10% of the proceeds payable to a non-United States holder from a disposition of shares of our common stock, and the non-United States holder generally will be taxed on its net gain derived from the disposition at the graduated United States federal income tax rates applicable to United States persons.
Non-United States holders are urged to consult any potentially applicable income tax treaties that may provide for different rules.
Legislation Involving Payments to Certain Foreign Entities
A 30% withholding tax applies to any dividends paid on shares of our common stock to a foreign financial institution or non-financial foreign entity (including, in some cases, when such foreign financial institution or entity is acting as an intermediary), and on the gross proceeds of the sale or other disposition of shares of our common stock, unless (i) in the case of a foreign financial institution, such institution enters into an agreement with the U.S. government to withhold on certain payments, and to collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which includes certain equity and debt holders of such institution, as well as certain account holders that are foreign entities with U.S. owners), (ii) in the case of a non-financial foreign entity, such entity either certifies it does not have any substantial United States owners or provides the withholding agent with a certification identifying the substantial direct and indirect U.S. owners of the entity or (iii) the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from these rules. Under certain circumstances, a non-United States holder might be eligible for refunds or credits of such taxes. Foreign financial institutions located in jurisdictions that have an intergovernmental agreement with the United States governing these withholding and reporting requirements may be subject to different rules.
Under the applicable Treasury regulations and IRS guidance, these withholding and reporting requirements will apply to payments of dividends on our common stock made on or after July 1, 2014 and to payments of gross proceeds from the sale or other disposition of such stock on or after January 1, 2017. Non-United States holders are encouraged to consult their tax advisors regarding the possible implications of this legislation on an investment in shares of our common stock.
United States Federal Estate Taxes
Shares of our common stock owned or treated as owned by an individual who at the time of death is a non-United States holder are considered United States situs assets and will be included in the individual’s estate for United States federal estate tax purposes, unless an applicable estate tax treaty provides otherwise.
157
Table of Contents
Information Reporting and Backup Withholding
Information reporting and backup withholding (currently at a 28% rate) generally will apply to dividends paid with respect to our common stock. In certain circumstances, provided the applicable withholding agent does not have knowledge or reason to know the holder is a United States person, non-United States holders may avoid information reporting and backup withholding if they provide to the applicable withholding agent a properly executed IRS Form W-8BEN for treaty benefits or W-8ECI for effectively connected income, or such successor forms as the IRS designates, and certify under penalties of perjury as to their status as non-United States holders or otherwise establish an exemption and certain other requirements are met. Copies of information returns may also be made available to the tax authorities in the country in which the non-United States holder resides or is established under the provisions of an applicable income tax treaty. Non-United States holders are urged to consult their tax advisors regarding the application of the information reporting and backup withholding rules to them.
The gross proceeds from the disposition of shares of our common stock may be subject to information reporting and backup withholding. If a non-United States holder sells shares of our common stock outside the United States through a non-United States office of a non-United States broker and the sales proceeds are paid to such holder outside the United States, then the United States backup withholding and information reporting requirements generally will not apply to that payment. However, United States information reporting will (although backup withholding will not) generally apply to a payment of sale proceeds, even if that payment is made outside the United States, if a non-United States holder sells shares of our common stock through a non-United States office of a broker that:
| • | is a United States person for United States federal tax purposes; |
| • | is a foreign person that derives 50% or more of its gross income in specific periods from the conduct of a trade or business in the United States; |
| • | is a “controlled foreign corporation” for United States tax purposes; or |
| • | is a foreign partnership, if at any time during its tax year (1) one or more of its partners are United States persons who in the aggregate hold more than 50% of the income or capital interests in the partnership; or (2) the foreign partnership is engaged in a United States trade or business, |
unless the broker has documentary evidence in its files that the non-United States holder is not a United States person and certain other conditions are met, or the non-United States holder otherwise establishes an exemption. In such circumstances, backup withholding will not apply unless the broker has actual knowledge or reason to know the non-United States holder is not a non-United States person.
If a non-United States holder receives payments of the proceeds of a sale of shares of our common stock to or through a United States office of a broker, the payment is subject to both backup withholding and information reporting unless such non-United States holder provides to the applicable withholding agent a properly executed IRS Form W-8BEN (or valid substitute or successor form) certifying under penalties of perjury that such holder is not a United States person or otherwise establishes an exemption.
Backup withholding is not an additional tax. Amounts withheld under the backup withholding rules from a payment to a non-United States holder may be refunded in instances of excess withholding or credited against the non-United States holder’s United States federal income tax liability, if any, provided an appropriate claim is timely filed with the IRS.
158
Table of Contents
Piper Jaffray & Co. is acting as representative of each of the underwriters named below. Subject to the terms and conditions set forth in an underwriting agreement among us, the selling stockholders and the underwriters, we and the selling stockholders have agreed to sell to the underwriters, and each of the underwriters has agreed, severally and not jointly, to purchase from us and the selling stockholders, the number of shares of our common stock set forth opposite its name below.
| Name |
Number of Shares | |
| Piper Jaffray & Co. |
||
| William Blair & Company, L.L.C. |
||
| Cowen and Company, LLC |
||
| RBC Capital Markets, LLC |
||
| JMP Securities LLC |
||
| Stephens Inc. |
||
| Bryan, Garnier & Co. |
||
| Total |
||
|
|
Subject to the terms and conditions set forth in the underwriting agreement, the underwriters have agreed, severally and not jointly, to purchase all of the shares sold under the underwriting agreement if any of these shares are purchased. If an underwriter defaults, the underwriting agreement provides that the purchase commitments of the nondefaulting underwriters may be increased or the underwriting agreement may be terminated.
We and the selling stockholders have agreed to indemnify the several underwriters against certain liabilities, including liabilities under the Securities Act relating to losses or claims resulting from material misstatements in or omissions from this prospectus, the registration statement of which this prospectus is a part, certain free writing prospectuses that may be used in the offering and in any marketing materials used in connection with this offering and to contribute to payments the underwriters may be required to make in respect of those liabilities.
The underwriters are offering the shares, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel, including the validity of the shares, and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officers’ certificates and legal opinions. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
Commissions and Discounts
The representative has advised us and the selling stockholders that the underwriters propose initially to offer the shares to the public at the public offering price set forth on the cover page of this prospectus and to dealers at that price less a concession not in excess of $ per share. After the initial offering, the public offering price, concession or any other term of this offering may be changed.
159
Table of Contents
The following table shows the public offering price, underwriting discount and proceeds before expenses to us and the selling stockholders. The information assumes either no exercise or full exercise by the underwriters of their overallotment option.
| Per Share | Without Option |
With Option |
||||||||||
| Public offering price |
$ | $ | $ | |||||||||
| Underwriting discounts and commissions |
$ | $ | $ | |||||||||
| Proceeds, before expenses, to us |
$ | $ | $ | |||||||||
| Proceeds, before expenses, to the selling stockholders |
$ | $ | $ | |||||||||
The underwriting agreement provides that the obligations of the several underwriters to pay for and accept delivery of the shares of common stock offered by this prospectus are subject to the approval of certain legal matters by their counsel and to certain other conditions. The underwriters are obligated to take and pay for all of the shares of common stock offered by this prospectus if any such shares are taken. However, the underwriters are not required to take or pay for the shares covered by the underwriters’ over-allotment option described below. If an underwriter defaults, the underwriting agreement provides that the purchase commitments of the non-defaulting underwriters may be increased.
The underwriters initially propose to offer part of the shares of common stock directly to the public at the offering price listed on the cover page of this prospectus and part to certain dealers. After the initial offering of the shares of common stock, the offering price and other selling terms may from time to time be varied by the representative.
The selling stockholders have granted to the underwriters an option, exercisable for 30 days from the date of this prospectus, to purchase up to additional shares of common stock at the public offering price listed on the cover page of this prospectus, less underwriting discounts and commissions. The underwriters may exercise this option solely for the purpose of covering over-allotments, if any, made in connection with the offering of the shares of common stock offered by this prospectus. To the extent the option is exercised, each underwriter will become obligated, subject to certain conditions, to purchase about the same percentage of the additional shares of common stock as the number listed next to the underwriter’s name in the preceding table bears to the total number of shares of common stock listed next to the names of all underwriters in the preceding table.
The estimated offering expenses payable by us, exclusive of the underwriting discounts and commissions, are approximately $ million. We have also agreed to reimburse the underwriters for certain of their expenses in an amount up to $ as set forth in the underwriting agreement.
The underwriters have informed us that they do not intend sales to discretionary accounts to exceed 5% of the total number of shares of common stock offered by them.
Our common stock is listed on the NASDAQ Global Select Market under the trading symbol “LDRH”.
No Sales of Similar Securities
We, the selling stockholders, our executive officers and directors and our other existing security holders have agreed not to sell or transfer any shares of our common stock or securities convertible into, exchangeable for, exercisable for, or repayable with shares of our common stock, for 60 days after the date of this prospectus without first obtaining the written consent of Piper Jaffray. Specifically, we and these other persons have agreed, with certain limited exceptions, not to directly or indirectly:
| • | offer, pledge, announce the intention to sell, sell or contract to sell any shares of our common stock; |
160
Table of Contents
| • | sell any option or contract to purchase any shares of our common stock; |
| • | purchase any option or contract to sell any shares of our common stock; |
| • | grant any option, right or warrant for the sale of any shares of our common stock; |
| • | dispose of or otherwise transfer any shares of our common stock; |
| • | demand that we file a registration statement related to our common stock; or |
| • | enter into any swap or other agreement that transfers, in whole or in part, the economic consequence of ownership of any shares of our common stock whether any such swap or transaction is to be settled by delivery of shares or other securities, in cash or otherwise. |
The restrictions in the preceding paragraph do not apply to transfers of securities:
| • | that are acquired in open market transactions after the completion of the offering; |
| • | as a bona fide gift or gifts or charitable contribution(s); |
| • | to an immediate family member or any trust for the direct or indirect benefit of stockholder or the immediate family of the stockholder; |
| • | if the stockholder is a corporation partnership, limited liability company, trust or other business entity (1) transfers to another corporation, partnership, limited liability company, trust or other business entity that is a direct or indirect affiliate of the stockholder or (2) distributions of shares of common stock or any security convertible into or exercisable for common stock to limited partners, limited liability company members or stockholders of the stockholder; |
| • | if the stockholder is a trust, to the beneficiary of such trust; |
| • | by testate succession or intestate succession;, |
| • | to us in a transaction exempt from Section 16(b) of the Exchange Act upon a vesting event of the stockholder’s securities or upon the exercise of options or warrants to purchase our securities on a “cashless” or “net exercise” basis or to cover tax withholding obligations of the stockholder in connection with such vesting or exercise (but for the avoidance of doubt, excluding all manners of exercise that would involve a sale in the open market of any securities relating to such options or warrants, whether to cover the applicable aggregate exercise price, withholding tax obligations or otherwise); |
| • | to us (1) in connection with the termination of employment or other termination of a service provider and pursuant to agreements whereby we have the option to repurchase such shares or securities, (2) pursuant to option agreements relating to the early exercise by a stockholder of unvested options issued pursuant to our equity incentive plans or (3) pursuant to agreements under which we have a right of first refusal with respect to transfers of such shares or securities; |
| • | pursuant to a bona fide third party tender offer, merger, consolidation or other similar transaction made to all holders of our capital stock involving a change of control of the Company; or |
| • | pursuant to the underwriting agreement; |
provided, in the case of a transfer described in bullets two through six above, that such transfer shall not involve a disposition for value and each transferee agrees to be subject to the restrictions described in the immediately preceding paragraph and; and provided, further, in the case of a transfer described in bullets one through eight above, that no filing by any party under Section 16(a) of the Exchange Act, shall be required or shall be made voluntarily in connection with such transfer.
161
Table of Contents
In addition, the transfer restrictions described above do not apply to:
| • | the exercise of stock options granted pursuant to our equity plans; |
| • | the conversion of exercise of warrants into common stock or any other security convertible or exchangeable for common stock; |
| • | the sale by a stockholder of common stock purchased pursuant to our 2013 Employee Stock Purchase Plan; or |
| • | the sale by a stockholder of common stock pursuant to an existing 10b5-1 plan existing on the date hereof, provided that sales under such plans shall not occur prior to second business day following our earnings announcement for the three months ended March 31, 2014. |
This lock-up provision applies to shares of our common stock and to securities convertible into or exchangeable or exercisable for or repayable with shares of our common stock. It also applies to shares of our common stock owned now or acquired later by the person executing the agreement or for which the person executing the agreement later acquires the power of disposition.
Price Stabilization, Short Positions and Penalty Bids
Until the distribution of the shares is completed, SEC rules may limit underwriters and selling group members from bidding for and purchasing shares of our common stock. However, the representatives may engage in transactions that stabilize the price of our common stock, such as bids or purchases to peg, fix or maintain that price.
In connection with this offering, the underwriters may purchase and sell shares of our common stock in the open market. These transactions may include short sales, purchases on the open market to cover positions created by short sales and stabilizing transactions. Short sales involve the sale by the underwriters of a greater number of shares than they are required to purchase in this offering. “Covered” short sales are sales made in an amount not greater than the underwriters’ overallotment option described above. The underwriters may close out any covered short position by either exercising their overallotment option or purchasing shares in the open market. In determining the source of shares to close out the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the overallotment option. “Naked” short sales are sales in excess of the overallotment option. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of our common stock in the open market after pricing that could adversely affect investors who purchase in this offering. Stabilizing transactions consist of various bids for or purchases of shares of our common stock made by the underwriters in the open market prior to the closing of this offering.
The underwriters may also impose a penalty bid. This occurs when a particular underwriter repays to the underwriters a portion of the underwriting discount received by it because the representatives have repurchased shares sold by or for the account of such underwriter in stabilizing or short covering transactions.
Similar to other purchase transactions, the underwriters’ purchases to cover the syndicate short sales may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. The underwriters may conduct these transactions on NASDAQ, in the over-the-counter market or otherwise.
162
Table of Contents
Neither we nor any of the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common stock. In addition, neither we nor any of the underwriters make any representation that the representatives will engage in these transactions or that these transactions, once commenced, will not be discontinued without notice.
Electronic Offer, Sale and Distribution of Shares
In connection with this offering, certain of the underwriters or securities dealers may distribute prospectuses by electronic means, such as e-mail. In addition, one or more of the underwriters may facilitate Internet distribution for this offering to certain of their Internet subscription customers. Any such underwriter may allocate a limited number of shares for sale to its online brokerage customers. An electronic prospectus is available on the Internet websites maintained by any such underwriter. Other than the prospectus in electronic format, the information on the websites of any such underwriter is not part of this prospectus.
Other Relationships
The underwriters and their respective affiliates are full service financial institutions engaged in various activities, which may include securities trading, commercial and investment banking, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. Certain of the underwriters and their affiliates have engaged in, and may in the future engage in, investment banking and other commercial dealings in the ordinary course of business with us or our affiliates. They have received, or may in the future receive, customary fees and commissions for these transactions.
In the ordinary course of their various business activities, the underwriters and their respective affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments of the issuer. The underwriters and their respective affiliates may also make investment recommendations and/or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
Bryan, Garnier & Co. is not a U.S. registered broker-dealer; therefore, it will effect orders in the offering outside of the United States or within the United States to the extent permitted by Rule 15a-6 under the Exchange Act.
Selling Restrictions
European Economic Area
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”) an offer to the public of any shares of our common stock may not be made in that Relevant Member State, except that an offer to the public in that Relevant Member State of any shares of our common stock may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
| (a) | to any legal entity which is a qualified investor as defined in the Prospectus Directive; |
| (b) | to fewer than 100 or, if the Relevant Member State has implemented the relevant provision of the 2010 PD Amending Directive, 150, natural or legal persons (other than qualified investors as defined in the Prospectus Directive), as permitted under the Prospectus Directive, subject to obtaining the prior consent of the representatives for any such offer; or |
163
Table of Contents
| (c) | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares of our common stock shall result in a requirement for the publication by us or any underwriter of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer to the public” in relation to any shares of our common stock in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any shares of our common stock to be offered so as to enable an investor to decide to purchase any shares of our common stock, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State, the expression “Prospectus Directive” means Directive 2003/71/EC (and amendments thereto, including the 2010 PD Amending Directive, to the extent implemented in the Relevant Member State), and includes any relevant implementing measure in the Relevant Member State, and the expression “2010 PD Amending Directive” means Directive 2010/73/EU.
United Kingdom
Each underwriter has represented and agreed that:
| (a) | it has only communicated or caused to be communicated and will only communicate or cause to be communicated an invitation or inducement to engage in investment activity (within the meaning of Section 21 of the Financial Services and Markets Act 2000 (the “FSMA”)) received by it in connection with the issue or sale of the shares of our common stock in circumstances in which Section 21(1) of the FSMA does not apply to us; and |
| (b) | it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares of our common stock in, from or otherwise involving the United Kingdom. |
Canada
The common shares may be sold only to purchasers purchasing as principal that are both “accredited investors” as defined in National Instrument 45-106 Prospectus and Registration Exemptions and “permitted clients” as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the common shares must be made in accordance with an exemption from the prospectus requirements and in compliance with the registration requirements of applicable securities laws.
Hong Kong
The common shares may not be offered or sold in Hong Kong by means of any document other than (i) in circumstances which do not constitute an offer to the public within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong), or (ii) to “professional investors” within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder, or (iii) in other circumstances which do not result in the document being a “prospectus” within the meaning of the Companies Ordinance (Cap. 32, Laws of Hong Kong) and no advertisement, invitation or document relating to the shares may be issued or may be in the possession of any person for the purpose of issue (in each case whether in Hong Kong or elsewhere), which is directed at, or the contents of which are likely to be accessed or read by, the public in Hong Kong (except if permitted to do so under the laws of Hong Kong) other than with respect to common shares which are or are intended to be disposed of only to persons outside Hong Kong or only to “professional investors” within the meaning of the Securities and Futures Ordinance (Cap. 571, Laws of Hong Kong) and any rules made thereunder.
164
Table of Contents
Singapore
This prospectus has not been registered as a prospectus with the Monetary Authority of Singapore. Accordingly, this prospectus and any other document or material in connection with the offer or sale, or invitation for subscription or purchase, of the common shares may not be circulated or distributed, nor may the common shares be offered or sold, or be made the subject of an invitation for subscription or purchase, whether directly or indirectly, to persons in Singapore other than (i) to an institutional investor under Section 274 of the Securities and Futures Act, Chapter 289 of Singapore (the “SFA”), (ii) to a relevant person pursuant to Section 275(1), or any person pursuant to Section 275(1A), and in accordance with the conditions specified in Section 275 of the SFA or (iii) otherwise pursuant to, and in accordance with the conditions of, any other applicable provision of the SFA, in each case subject to compliance with conditions set forth in the SFA.
Where the common shares are subscribed or purchased under Section 275 of the SFA by a relevant person which is:
| (a) | a corporation (which is not an accredited investor (as defined in Section 4A of the SFA)) the sole business of which is to hold investments and the entire share capital of which is owned by one or more individuals, each of whom is an accredited investor; or |
| (b) | a trust (where the trustee is not an accredited investor) whose sole purpose is to hold investments and each beneficiary of the trust is an individual who is an accredited investor, |
shares, debentures and units of shares and debentures of that corporation or the beneficiaries’ rights and interest (howsoever described) in that trust shall not be transferred within six months after that corporation or that trust has acquired the common shares pursuant to an offer made under Section 275 of the SFA except:
| (a) | to an institutional investor (for corporations, under Section 274 of the SFA) or to a relevant person defined in Section 275(2) of the SFA, or to any person pursuant to an offer that is made on terms that such shares, debentures and units of shares and debentures of that corporation or such rights and interest in that trust are acquired at a consideration of not less than S$200,000 (or its equivalent in a foreign currency) for each transaction, whether such amount is to be paid for in cash or by exchange of securities or other assets, and further for corporations, in accordance with the conditions specified in Section 275 of the SFA; |
| (b) | where no consideration is or will be given for the transfer; or |
| (c) | where the transfer is by operation of law. |
Switzerland
The common shares may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (the “SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering or marketing material relating to the common shares or the offering may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering or marketing material relating to the offering, or the common shares have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of common shares will not be supervised
165
Table of Contents
by, the Swiss Financial Market Supervisory Authority FINMA, and the offer of common shares has not been and will not be authorized under the Swiss Federal Act on Collective Investment Schemes (“CISA”). Accordingly, no public distribution, offering or advertising, as defined in CISA, its implementing ordinances and notices, and no distribution to any non-qualified investor, as defined in CISA, its implementing ordinances and notices, shall be undertaken in or from Switzerland, and the investor protection afforded to acquirers of interests in collective investment schemes under CISA does not extend to acquirers of common shares.
United Arab Emirates
This offering has not been approved or licensed by the Central Bank of the United Arab Emirates (the “UAE”), Securities and Commodities Authority of the UAE and/or any other relevant licensing authority in the UAE including any licensing authority incorporated under the laws and regulations of any of the free zones established and operating in the territory of the UAE, in particular the Dubai Financial Services Authority (“DFSA”), a regulatory authority of the Dubai International Financial Centre (“DIFC”). The offering does not constitute a public offer of securities in the UAE, DIFC and/or any other free zone in accordance with the Commercial Companies Law, Federal Law No 8 of 1984 (as amended), DFSA Offered Securities Rules and NASDAQ Dubai Listing Rules, accordingly, or otherwise. The common shares may not be offered to the public in the UAE and/or any of the free zones.
The common shares may be offered and issued only to a limited number of investors in the UAE or any of its free zones who qualify as sophisticated investors under the relevant laws and regulations of the UAE or the free zone concerned.
France
This prospectus (including any amendment, supplement or replacement thereto) is not being distributed in the context of a public offering in France within the meaning of Article L. 411-1 of the French Monetary and Financial Code (Code monétaire et financier).
This prospectus has not been and will not be submitted to the French Autorité des marchés financiers (the “AMF”) for approval in France and accordingly may not and will not be distributed to the public in France.
Pursuant to Article 211-3 of the AMF General Regulation, French residents are hereby informed that:
1. the transaction does not require a prospectus to be submitted for approval to the AMF;
2. persons or entities referred to in Point 2°, Section II of Article L.411-2 of the Monetary and Financial Code may take part in the transaction solely for their own account, as provided in Articles D. 411-1, D. 734-1, D. 744-1, D. 754-1 and D. 764-1 of the Monetary and Financial Code; and
3. the financial instruments thus acquired cannot be distributed directly or indirectly to the public otherwise than in accordance with Articles L. 411-1, L. 411-2, L. 412-1 and L. 621-8 to L. 621-8-3 of the Monetary and Financial Code.
This prospectus is not to be further distributed or reproduced (in whole or in part) in France by the recipients of this prospectus. This prospectus has been distributed on the understanding that such recipients will only participate in the issue or sale of our common stock for their own account and undertake not to transfer, directly or indirectly, our common stock to the public in France, other than in compliance with all applicable laws and regulations and in particular with Articles L. 411-1 and L. 411-2 of the French Monetary and Financial Code.
166
Table of Contents
The validity of the shares of common stock offered hereby and certain other legal matters will be passed upon for us by Andrews Kurth LLP, Austin, Texas. Certain attorneys of Andrews Kurth LLP own, in the aggregate, 10,700 shares of common stock. Certain legal matters will be passed upon for the underwriters by Latham & Watkins LLP, Costa Mesa, California.
The consolidated financial statements of LDR Holding Corporation and its subsidiaries as of December 31, 2013 and 2012 and for each of the years in the three-year period ended December 31, 2013 have been included herein and in the registration statement in reliance upon the report of KPMG LLP, independent registered public accounting firm, appearing elsewhere herein and upon the authority of said firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement, of which this prospectus is a part, on Form S-1 with the SEC relating to this offering. This prospectus does not contain all of the information in the registration statement and the exhibits and financial statements included with the registration statement. References in this prospectus to any of our contracts, agreements or other documents are not necessarily complete, and you should refer to the exhibits attached to the registration statement for copies of the actual contracts, agreements or documents.
Our filings with the SEC are available to the public on the SEC’s website at http://www.sec.gov. You may also read and copy, at SEC prescribed rates, any document we file with the SEC, including the registration statement (and its exhibits) of which this prospectus is a part, at the SEC’s Public Reference Room located at 100 F Street, N.E., Washington D.C. 20549. You can call the SEC at 1-800-SEC-0330 to obtain information on the operation of the Public Reference Room.
We are not, and the underwriters are not, making an offer to sell these securities in any jurisdiction where an offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate as of the date on the front cover of this prospectus only. Our business, financial condition, results of operations and prospects may have changed since that date.
Following the completion of this offering, we will be subject to the informational reporting requirements of the Exchange Act and, in accordance with the Exchange Act, will file periodic reports, proxy and information statements and other information with the SEC. Such annual, quarterly and current reports; proxy and information statements; and other information can be inspected and copied at the locations set forth above. We also expect to make our periodic reports and other information filed with or furnished to the SEC available, free of charge, through our website at http://www.ldrholding.com, as soon as reasonably practicable after those reports and other information are electronically filed with or furnished to the SEC. The information contained on or accessible through our corporate web site or any other web site that we may maintain is not part of this prospectus or the registration statement of which this prospectus is a part. You may also request a copy of these filings, at no cost to you, by writing or telephoning us at the following address:
LDR Holding Corporation
13785 Research Boulevard, Suite 200
Austin, Texas 78750
Attention: Chief Financial Officer
(512) 344-3333
167
Table of Contents
We will report our financial statements on a year ended December 31. We intend to furnish or make available to our stockholders annual reports containing consolidated financial statements audited by our independent registered public accounting firm. We also intend to furnish or make available to our stockholders our quarterly reports containing unaudited interim financial information for each of the first three quarters of each fiscal year.
168
Table of Contents
LDR Holding Corporation
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-7 | ||||
| F-9 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
LDR Holding Corporation and Subsidiaries:
We have audited the accompanying consolidated balance sheets of LDR Holding Corporation and subsidiaries as of December 31, 2013 and 2012, and the related consolidated statements of comprehensive loss, stockholders’ equity (deficit) and cash flows for each of the years in the three-year period ended December 31, 2013. These consolidated financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of LDR Holding Corporation and subsidiaries as of December 31, 2013 and 2012, and the results of their operations and their cash flows for each of the years in the three year period ended December 31, 2013, in conformity with U.S. generally accepted accounting principles.
/s/ KPMG LLP
Austin, Texas
March 4, 2014
F-2
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
(in thousands, except share and per share amounts)
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| ASSETS |
| |||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 56,678 | $ | 19,135 | ||||
| Accounts receivable, net |
22,193 | 16,309 | ||||||
| Inventory, net |
17,690 | 16,772 | ||||||
| Other current assets |
4,780 | 3,768 | ||||||
| Prepaid expenses |
1,593 | 806 | ||||||
|
|
|
|
|
|||||
| Total current assets |
102,934 | 56,790 | ||||||
| Property and equipment, net |
12,695 | 12,296 | ||||||
| Goodwill |
6,621 | 6,621 | ||||||
| Intangible assets, net |
3,073 | 2,619 | ||||||
| Restricted cash |
2,000 | 2,000 | ||||||
| Related party notes receivable |
— | 270 | ||||||
| Deferred tax assets |
513 | 554 | ||||||
| Other assets |
157 | 441 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 127,993 | $ | 81,591 | ||||
|
|
|
|
|
|||||
| LIABILITIES, REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 8,128 | $ | 7,855 | ||||
| Accrued expenses |
16,324 | 10,759 | ||||||
| Line of credit, net of discount |
18,162 | — | ||||||
| Short-term financing |
2,641 | 1,772 | ||||||
| Current portion of long-term debt |
1,763 | 1,903 | ||||||
| Deferred tax liabilities |
540 | 542 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
47,558 | 22,831 | ||||||
| Line of credit, net of discount |
— | 18,985 | ||||||
| Long-term debt, net of discount and current portion |
2,758 | 30,326 | ||||||
| Warrant liability |
— | 4,167 | ||||||
| Other long-term liabilities |
— | 913 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
50,316 | 77,222 | ||||||
| Commitments and contingencies |
||||||||
| Series C redeemable convertible preferred stock |
— | 35,000 | ||||||
| Stockholders’ equity (deficit): |
||||||||
| Series A-1 convertible preferred stock; $0.001 par value; zero and 11,120,119 shares authorized, issued and outstanding, liquidation preference of zero and $10,000 at December 31, 2013 and December 31, 2012, respectively |
— | 11 | ||||||
| Series A-2 convertible preferred stock; $0.001 par value; zero and 18,097,848 shares authorized, issued and outstanding, liquidation preference of zero and $3,145 at December 31, 2013 and December 31, 2012, respectively |
— | 18 | ||||||
| Series B convertible preferred stock; $0.001 par value; zero and 14,838,368 shares authorized, issued and outstanding, liquidation preference of zero and $14,000 at December 31, 2013 and December 31, 2012, respectively |
— | 15 | ||||||
| Common stock; $0.001 par value; 107,000,000 and 18,167,361 shares authorized at December 31, 2013 and December 31, 2012, respectively; 24,076,667 and 4,642,143 shares issued and outstanding at December 31, 2013 and December 31, 2012, respectively |
24 | 5 | ||||||
| Additional paid-in capital |
161,216 | 25,603 | ||||||
| Accumulated other comprehensive income (loss) |
198 | (457 | ) | |||||
| Accumulated deficit |
(83,761 | ) | (55,826 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity (deficit) |
77,677 | (30,631 | ) | |||||
|
|
|
|
|
|||||
| Total liabilities, redeemable preferred stock and stockholders’ equity (deficit) |
$ | 127,993 | $ | 81,591 | ||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
F-3
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(in thousands, except share and per share amounts)
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Revenue |
$ | 111,594 | $ | 90,918 | $ | 77,975 | ||||||
| Cost of goods sold |
17,947 | 14,770 | 12,564 | |||||||||
|
|
|
|
|
|
|
|||||||
| Gross profit |
93,647 | 76,148 | 65,411 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
9,380 | 10,751 | 9,040 | |||||||||
| Sales and marketing |
67,676 | 51,846 | 42,270 | |||||||||
| General and administrative |
18,915 | 14,825 | 11,121 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
95,971 | 77,422 | 62,431 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating income (loss) |
(2,324 | ) | (1,274 | ) | 2,980 | |||||||
| Other operating income (expense): |
||||||||||||
| Other expense |
(771 | ) | (987 | ) | (252 | ) | ||||||
| Interest income |
10 | 26 | 29 | |||||||||
| Interest expense |
(3,273 | ) | (3,240 | ) | (2,369 | ) | ||||||
| Accretion related to warrants and discounts on long-term debt |
(6,900 | ) | (1,404 | ) | (236 | ) | ||||||
| Expense related to beneficial conversion of promissory notes |
(7,413 | ) | — | — | ||||||||
| Change in fair value of common stock warrants |
(5,593 | ) | (1,643 | ) | (652 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Total other income (expense), net |
(23,940 | ) | (7,248 | ) | (3,480 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Loss before income taxes |
(26,264 | ) | (8,522 | ) | (500 | ) | ||||||
| Income tax expense |
(1,671 | ) | (1,179 | ) | (1,328 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
(27,935 | ) | (9,701 | ) | (1,828 | ) | ||||||
| Other comprehensive income (loss): |
||||||||||||
| Foreign currency translation |
655 | 742 | (585 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive loss |
$ | (27,280 | ) | $ | (8,959 | ) | $ | (2,413 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per common share: |
||||||||||||
| Basic and diluted |
$ | (3.09 | ) | $ | (2.10 | ) | $ | (0.41 | ) | |||
| Weighted average number of shares outstanding (see Note 15): |
||||||||||||
| Basic and diluted |
9,040,567 | 4,615,352 | 4,490,481 | |||||||||
See accompanying notes to consolidated financial statements.
F-4
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
(in thousands, except share data)
| Series A-1 Preferred Stock |
Series A-2 Preferred Stock |
Series
B Preferred Stock |
Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total Stockholders’ Equity (Deficit) |
|||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
|||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2010 |
11,120,119 | $ | 11 | 15,435,248 | $ | 15 | 14,838,368 | $ | 15 | 4,480,604 | $ | 5 | $ | 26,251 | $ | (614 | ) | $ | (44,297 | ) | $ | (18,614 | ) | |||||||||||||||||||||||||
| Exercise of preferred stock warrants |
— | — | 2,662,600 | 3 | — | — | — | — | 15 | — | — | 18 | ||||||||||||||||||||||||||||||||||||
| Modification of common stock warrants |
— | — | — | — | — | — | — | — | (1,522 | ) | — | — | (1,522 | ) | ||||||||||||||||||||||||||||||||||
| Exercises of common stock options |
— | — | — | — | — | — | 92,285 | — | 176 | — | — | 176 | ||||||||||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | 184 | — | — | 184 | ||||||||||||||||||||||||||||||||||||
| Issuance of common stock |
— | — | — | — | — | — | 4,351 | — | 1 | — | — | 1 | ||||||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (1,828 | ) | (1,828 | ) | ||||||||||||||||||||||||||||||||||
| Currency translation adjustment |
— | — | — | — | — | — | — | — | — | (585 | ) | — | (585 | ) | ||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
| Balance at December 31, 2011 |
11,120,119 | 11 | 18,097,848 | 18 | 14,838,368 | 15 | 4,577,240 | 5 | 25,105 | (1,199 | ) | (46,125 | ) | (22,170 | ) | |||||||||||||||||||||||||||||||||
| Exercises of common stock options |
— | — | — | — | — | — | 63,598 | — | 132 | — | — | 132 | ||||||||||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | 295 | — | — | 295 | ||||||||||||||||||||||||||||||||||||
| Issuance of common stock |
— | — | — | — | — | — | 1,305 | 71 | — | — | 71 | |||||||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (9,701 | ) | (9,701 | ) | ||||||||||||||||||||||||||||||||||
| Currency translation adjustment |
— | — | — | — | — | — | — | — | — | 742 | — | 742 | ||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
| Balance at December 31, 2012 |
11,120,119 | $ | 11 | 18,097,848 | $ | 18 | 14,838,368 | $ | 15 | 4,642,143 | $ | 5 | $ | 25,603 | $ | (457 | ) | $ | (55,826 | ) | $ | (30,631 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
F-5
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)—(Continued)
(in thousands, except share data)
| Series A-1 Preferred Stock |
Series A-2 Preferred Stock |
Series
B Preferred Stock |
Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total Stockholders’ Equity (Deficit) |
|||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
Number of Shares |
Par Value |
|||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2012 |
11,120,119 | $ | 11 | 18,097,848 | $ | 18 | 14,838,368 | $ | 15 | 4,642,143 | $ | 5 | $ | 25,603 | $ | (457 | ) | $ | (55,826 | ) | $ | (30,631 | ) | |||||||||||||||||||||||||
| Issuance of common stock in a public offering |
— | — | — | — | — | — | 5,750,000 | 6 | 86,244 | — | — | 86,250 | ||||||||||||||||||||||||||||||||||||
| Underwriting discount and issuance costs |
— | — | — | — | — | — | — | — | (8,744 | ) | — | — | (8,744 | ) | ||||||||||||||||||||||||||||||||||
| Conversion of preferred stock to common stock in connection with the initial public offering |
(11,120,119 | ) | (11 | ) | (18,097,848 | ) | (18 | ) | (14,838,368 | ) | (15 | ) | 10,977,667 | 11 | 35,033 | — | — | 35,000 | ||||||||||||||||||||||||||||||
| Dividends on Series C Preferred Stock |
— | — | — | — | — | — | — | — | (17,143 | ) | — | — | (17,143 | ) | ||||||||||||||||||||||||||||||||||
| Conversion of convertible notes |
— | — | — | — | — | — | 1,929,309 | 2 | 28,955 | — | — | 28,957 | ||||||||||||||||||||||||||||||||||||
| Reclassification of warrant liability to equity |
— | — | — | — | — | — | — | — | 9,760 | — | — | 9,760 | ||||||||||||||||||||||||||||||||||||
| Exercise of preferred stock warrant |
— | — | — | — | — | — | 10,889 | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Exercise of common stock warrant |
— | — | — | — | — | — | 650,669 | — | 4 | — | — | 4 | ||||||||||||||||||||||||||||||||||||
| Exercises of common stock options |
— | — | — | — | — | — | 115,990 | — | 235 | — | — | 235 | ||||||||||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | 1,269 | — | — | 1,269 | ||||||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (27,935 | ) | (27,935 | ) | ||||||||||||||||||||||||||||||||||
| Currency translation adjustment |
— | — | — | — | — | — | — | — | — | 655 | — | 655 | ||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
| Balance at December 31, 2013 |
— | $ | — | — | $ | — | — | $ | — | 24,076,667 | $ | 24 | $ | 161,216 | $ | 198 | $ | (83,761 | ) | $ | 77,677 | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-6
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Operating activities: |
||||||||||||
| Net loss |
(27,935 | ) | (9,701 | ) | (1,828 | ) | ||||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Bad debt expense |
517 | 587 | 187 | |||||||||
| Provision for excess and obsolete inventories |
641 | 1,565 | 586 | |||||||||
| Depreciation and amortization |
4,024 | 3,133 | 2,387 | |||||||||
| Stock-based compensation |
1,269 | 295 | 184 | |||||||||
| Accretion related to warrants and discounts on long-term debt |
6,900 | 1,404 | 236 | |||||||||
| Change in fair value of common stock warrants |
5,593 | 1,643 | 652 | |||||||||
| Beneficial conversion related to promissory notes |
7,413 | — | — | |||||||||
| Deferred income tax expense (benefit) |
37 | (250 | ) | (46 | ) | |||||||
| Loss on disposal of assets |
57 | 163 | 83 | |||||||||
| Unrealized foreign currency loss |
713 | 817 | — | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Cash restricted for line of credit agreement |
— | (1,000 | ) | — | ||||||||
| Accounts receivable |
(6,313 | ) | (3,114 | ) | (2,756 | ) | ||||||
| Prepaid expenses and other current assets |
(2,141 | ) | 660 | (672 | ) | |||||||
| Inventory |
(1,231 | ) | (5,504 | ) | (2,986 | ) | ||||||
| Other assets |
215 | 99 | (21 | ) | ||||||||
| Accounts payable |
16 | 2,027 | 1,275 | |||||||||
| Accrued expenses |
4,505 | 1,921 | 26 | |||||||||
| Other long-term liabilities |
(50 | ) | 892 | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(5,770 | ) | (4,363 | ) | (2,693 | ) | ||||||
| Investing activities: |
||||||||||||
| Proceeds from sale of property and equipment |
54 | 50 | — | |||||||||
| Purchase of intangible assets |
(780 | ) | (675 | ) | (505 | ) | ||||||
| Purchase of property and equipment |
(3,963 | ) | (5,245 | ) | (4,158 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(4,689 | ) | (5,870 | ) | (4,663 | ) | ||||||
| Financing activities: |
||||||||||||
| Proceeds from issuance of common stock |
86,250 | 71 | 1 | |||||||||
| Issuance costs from initial public offering |
(8,744 | ) | — | — | ||||||||
| Proceeds from Employee Stock Purchase Plan |
976 | — | — | |||||||||
| Exercise of stock options |
235 | 132 | 176 | |||||||||
| Dividends on Series C Preferred Stock |
(17,143 | ) | — | — | ||||||||
| Payments on capital leases |
(22 | ) | (163 | ) | (228 | ) | ||||||
| Net proceeds (payments) on short-term financings |
759 | 551 | (844 | ) | ||||||||
| Proceeds from line of credit |
376 | 11,767 | 2,000 | |||||||||
| Payments on line of credit |
(1,209 | ) | — | — | ||||||||
| Proceeds from long-term debt |
— | 16,311 | 5,823 | |||||||||
| Payments on long-term debt |
(13,711 | ) | (4,603 | ) | (1,559 | ) | ||||||
| Debt issuance costs |
— | (198 | ) | — | ||||||||
| Proceeds from exercise of common stock warrants |
4 | — | — | |||||||||
| Proceeds from exercise of preferred stock warrants |
— | — | 18 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
47,771 | 23,868 | 5,387 | |||||||||
F-7
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS—(Continued)
(in thousands)
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Effect of exchange rate on cash |
231 | (99 | ) | (70 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net change in cash and cash equivalents |
37,543 | 13,536 | (2,039 | ) | ||||||||
| Cash and cash equivalents, beginning of period |
19,135 | 5,599 | 7,638 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents, end of period |
$ | 56,678 | $ | 19,135 | $ | 5,599 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental disclosure of interest and income taxes paid |
||||||||||||
| Cash paid for interest |
$ | 2,234 | $ | 2,246 | 2,224 | |||||||
| Cash paid for taxes |
1,599 | 1,414 | 2,988 | |||||||||
| Supplemental disclosure of noncash investing and financing activities |
||||||||||||
| Conversion of convertible notes to common stock |
$ | 14,470 | $ | — | $ | — | ||||||
| Reclassification of warrant liability |
(9,760 | ) | — | — | ||||||||
| Issuance of warrants for long-term debt |
— | — | 350 | |||||||||
See accompanying notes to consolidated financial statements.
F-8
Table of Contents
LDR HOLDING CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | ORGANIZATION AND BUSINESS DESCRIPTION |
Description of Business
LDR Holding Corporation (Holding), a Delaware corporation, and its subsidiaries, LDR Spine USA, Inc. (Spine), LDR Medical, SAS (Medical) and LDR Brazil, LTDA (LDR Brazil) (collectively, the Company), operates as a medical device company that designs and commercializes novel and proprietary surgical technologies for the treatment of patients suffering from spine disorders. The Company’s primary products are based on the VerteBRIDGE fusion platform and Mobi non-fusion platform, both of which are designed for applications in the cervical and lumbar spine for both fusion and nonfusion surgical treatments. The Company has offices in Troyes, France; Santo Andre, Brazil; Beijing and Shanghai, China and in Austin, Texas, which serves the U.S. market and is the corporate headquarters. The primary markets for products are the U.S. and Western Europe.
Initial Public Offering
On October 15, 2013, the Company completed its initial public offering (IPO) of 5,750,000 shares of common stock at a price of $15.00 per share including 750,000 shares sold to underwriters for the exercise of their over-allotment option to purchase additional shares. The IPO generated net proceeds of approximately $77.5 million, after deducting underwriting discounts and expenses of approximately $8.7 million.
Reverse Stock Split Ratio
On October 3, 2013, the Company effected a reverse stock split of the Company’s common stock such that each 6.75 shares of issued common stock were reclassified into one share of common stock. All common stock share and per-share amounts for all periods presented in these financial statements have been adjusted retroactively to reflect the reverse stock split.
Reclassifications
Certain reclassifications have been made to prior periods’ consolidated financial statements to conform to the current period presentation. These reclassifications did not result in any change in previously reported net loss, total assets or stockholders’ deficit.
| 2. | SIGNIFICANT ACCOUNTING POLICIES |
(a) Principles of Consolidation
The accompanying consolidated financial statements include the results of Holding and its subsidiaries, Spine, Medical and LDR Brazil. All significant intercompany accounts and transactions have been eliminated in consolidation.
Prior to the IPO in October 2013, Holding owned 100% of the outstanding stock and voting rights of Spine and LDR Brazil and 52.95% of the outstanding stock and voting rights of Medical. The remaining 47.05% of the outstanding shares of Medical were subject to the Escrow Agreement discussed in note 10. The shares subject to the Escrow Agreement have been considered as if converted; thus, giving Holding 100% effective ownership of all subsidiaries.
After the IPO, Holding owns 100% of the outstanding stock and voting rights of Spine, Medical and LDR Brazil.
F-9
Table of Contents
(b) Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Actual results are likely to differ from those estimates, and such differences may be material to the financial statements.
(c) Foreign Currency Translation and Other Comprehensive Loss
The functional currency of the Company’s foreign subsidiaries is as follows: Medical is the euro and LDR Brazil is the real. Both companies translate monetary assets and liabilities denominated in other currencies into U.S. dollars at exchange rates in effect at each balance sheet date. Revenues and expenses are translated at the average of the exchange rates in effect during the period, and nonmonetary items are translated at historical rates. The resulting translation adjustments are included in other comprehensive loss. The accumulated foreign currency translation adjustments are reflected as accumulated other comprehensive loss, a component of stockholders’ equity (deficit). Gains and losses arising from intercompany foreign transactions are included in other income (expense) on the consolidated statements of comprehensive loss. The Company recognized foreign exchange losses in other income (expense) of approximately $764,000, $965,000 and $221,000 for the years ended December 31, 2013, 2012 and 2011, respectively.
(d) Cash and Cash Equivalents
The Company considers all highly liquid investments with a remaining maturity of three months or less at date of purchase to be cash equivalents.
(e) Restricted Cash
The Company’s restricted cash secures certain obligations under the Line of Credit (note 8).
(f) Accounts Receivable
The Company generally extends credit to certain customers without requiring collateral; others are required to provide a letter of credit. The Company provides for an allowance for doubtful accounts based on management’s evaluations of the collectability of accounts receivable. Trade receivables are written off when the Company has determined amounts are uncollectible. Management has recorded an allowance for doubtful accounts of $1,493,000 and $1,388,000 as of December 31, 2013 and 2012, respectively.
The changes in the allowance for doubtful accounts during the years ended December 31, 2013 and 2012 are as follows (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Balance, beginning of period |
$ | 1,388 | $ | 1,050 | ||||
| Additions |
517 | 587 | ||||||
| Deductions/adjustments |
(412 | ) | (249 | ) | ||||
|
|
|
|
|
|||||
| Balance, end of period |
$ | 1,493 | $ | 1,388 | ||||
|
|
|
|
|
|||||
F-10
Table of Contents
(g) Inventory
Inventory is carried at the lower of cost or market using the weighted average method, net of an allowance for excess and obsolete inventory. The components of inventory, net of allowance, as of December 31, 2013 and 2012 are as follows (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Finished goods |
$ | 14,452 | $ | 13,230 | ||||
| Work in process |
2,737 | 3,205 | ||||||
| Raw materials |
501 | 337 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 17,690 | $ | 16,772 | ||||
|
|
|
|
|
|||||
As of December 31, 2013 and 2012, inventory held by hospitals and sales agents on behalf of the Company was $5,796,000 and $4,778,000, respectively.
The Company reviews the components of its inventory on a periodic basis for excess, obsolete or impaired inventory and records a reserve for items identified. The Company recorded an allowance for excess and obsolete inventory of $3,477,000 and $2,792,000 as of December 31, 2013 and 2012, respectively. The changes in the allowance for excess and obsolete inventory during the years ended December 31, 2013 and 2012 are as follows (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Balance, beginning of period |
$ | 2,792 | $ | 1,975 | ||||
| Additions |
641 | 1,565 | ||||||
| Deductions/adjustments |
44 | (748 | ) | |||||
|
|
|
|
|
|||||
| Balance, end of period |
$ | 3,477 | $ | 2,792 | ||||
|
|
|
|
|
|||||
(h) Property and Equipment
Property and equipment are carried at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the related assets. Useful lives are as follows: two to three years for computer equipment, five to ten years for furniture and equipment and surgical instruments, and the shorter of their estimated useful life or the term of the related lease for leasehold improvements. Upon retirement or sale, the cost of assets disposed and the related accumulated depreciation are removed from the accounts, and any resulting gain or loss is credited or charged to operations. Major renewals and betterments are capitalized. Repairs and maintenance and minor replacements are charged to expense as incurred.
(i) Goodwill and Other Intangible Assets
Goodwill is not amortized, but is tested annually for impairment or more frequently if impairment indicators exist. Such indicators could include (1) a significant adverse change in legal factors or in business climate, (2) unanticipated competition, or (3) an adverse action or assessment by a regulator.
In January 2012, the Company adopted new accounting guidance related to annual and interim goodwill impairment tests. The updated accounting guidance allows entities to first assess qualitative factors before performing a quantitative assessment of the fair value of a reporting unit. If it is determined on the basis of qualitative factors that the fair value of the reporting unit is more-likely than-not less than the carrying amount, the existing quantitative impairment test is required. The Company’s evaluation of
F-11
Table of Contents
goodwill completed during the years ended December 31, 2013 and 2012 resulted in no impairment losses.
Definite lived intangible assets consists of patents and software licenses. The Company capitalizes third party legal fees and application costs related to its internally developed patents. Legal costs incurred in the defense of the Company’s patents are expensed as incurred.
Definite lived intangibles are amortized over their estimated useful lives: ten years for patents and three years for software licenses. Patents and software licenses are amortized on a straight-line basis and are stated net of accumulated amortization. The Company recorded no impairment loss during the years ended December 31, 2013 and 2012.
(j) Valuation of Long-Lived Assets
Long-lived assets are monitored and reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of any such asset may not be recoverable. The determination of recoverability is based on an estimate of undiscounted cash flows expected to result from the use of an asset and its eventual disposition. The estimate of undiscounted cash flows is based upon, among other things, certain assumptions about expected future operating performance, growth rates and other factors. The Company’s estimates of undiscounted cash flows may differ from actual cash flows due to, among other things, technological changes, economic conditions, changes to its business model or changes in its operating performance. If the sum of the undiscounted cash flows is less than the carrying value of the asset, an impairment charge is recognized, measured as the amount by which the carrying value exceeds the fair value of the asset. The Company did not record an impairment of the Company’s long-lived assets for any of the periods presented.
(k) Revenue Recognition
Revenue is recognized when evidence of an arrangement exists, fees are fixed or determinable, collection of the fees is reasonably assured, and delivery or customer acceptance of the product has occurred and no other significant obligations remain. Further, for direct markets (U.S., France and Germany), the Company recognizes revenue on its products when the spinal implant is used in surgery and a valid purchase order has been received. The Company recognizes revenue on sales to distributors worldwide when no right of return exists for the distributors, and the Company has no further obligation.
(l) Shipping and Handling
Shipping and handling costs are included in cost of goods sold, when related to revenue producing activities and were immaterial for the periods presented.
(m) Advertising Costs
The Company expenses advertising costs as incurred. The Company incurred approximately $268,000, $141,000 and $115,000 in advertising costs during the years ended December 31, 2013, 2012 and 2011, respectively.
(n) Research and Development
The Company expenses research and development costs as incurred.
(o) Fair Value of Financial Instruments
The fair value of the Company’s financial instruments reflects the amounts that the Company estimates to receive in connection with the sale of an asset or paid in connection with the transfer of a liability in
F-12
Table of Contents
an orderly transaction between market participants at the measurement date (exit price). The fair value hierarchy that prioritizes the use of inputs used in valuation techniques is as follows:
Level 1—quoted prices in active markets for identical assets and liabilities;
Level 2—observable inputs other than quoted prices in active markets, such as quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets and liabilities in markets that are not active, or other inputs that are observable or can be corroborated by observable market data;
Level 3—unobservable inputs reflecting management’s assumptions, consistent with reasonably available assumptions made by other market participants. These valuations require significant judgment.
The carrying amounts of the Company’s financial instruments, which primarily include cash and cash equivalents, accounts receivable, accounts payable and accrued expenses, approximate their fair values due to their short maturities. The carrying amount of the Company’s long-term debt approximates its fair value due to the relatively recent issuances and short maturities. The common stock warrant liability, further discussed in footnotes 9(c) and 10(h), is classified as a Level 3 liability under the fair value hierarchy. The common stock warrants were exercised in December 2013. The fair value measurement of the financial liability was as follows as of December 31, 2012 (in thousands):
| Balance at December 31, 2012 | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Balance | |||||||||||||
| Warrant liability |
$ | — | $ | — | $ | 4,167 | $ | 4,167 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | — | $ | — | $ | 4,167 | $ | 4,167 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The following table presents a reconciliation of the liability measured at fair value on a recurring basis using significant unobservable inputs (Level 3) as of December 31, 2013 and 2012 (in thousands).
| Fair Value Measurements |
||||
| Balance as of December 31, 2011 |
$ | 2,524 | ||
| Warrants issued |
— | |||
| Total change in fair value |
1,643 | |||
|
|
|
|||
| Balance as of December 31, 2012 |
4,167 | |||
| Warrants issued |
— | |||
| Total change in fair value |
5,593 | |||
| Reclassification from liability to equity |
(9,760 | ) | ||
|
|
|
|||
| Balance as of December 31, 2013 |
$ | — | ||
|
|
|
|||
The valuation of the warrant liability is discussed in note 10.
(p) Stock-Based Compensation
The Company recognizes compensation costs for all stock-based payment awards made to employees based upon each award’s estimated grant date fair value. The Company utilizes the Black-Scholes option pricing model, which requires a number of assumptions to determine the fair value of the awards. Compensation cost is recognized on a straight-line basis over the requisite service period of the award, net of an estimated forfeiture rate. Adjustments for forfeitures are made in the period in which they occur.
F-13
Table of Contents
(q) Net Loss Per Share
The Company computes basic net loss per common share by dividing net loss attributable to common stockholders by the weighted average common shares outstanding for the period, which includes shares of Medical that are subject to the Escrow Agreement discussed in note 10 prior to the IPO. During periods of income, the Company allocates participating securities a proportional share of income determined by dividing total weighted average participating securities by the sum of the total weighted average common shares and participating securities (the two-class method). The Company’s preferred stock participates in any dividends declared by the Company and are therefore considered to be participating securities. During periods of loss, the Company allocates no loss to participating securities because they have no contractual obligation to share in the losses of the Company. Warrants that are liability classified are excluded from the calculation of basic net loss per share. The Company computes diluted net loss per common share after giving consideration to the dilutive effect of the Company’s convertible preferred stock, stock options and warrants that are outstanding during the period and the conversion of the Company’s convertible debt into shares of common stock, except where such would be anti-dilutive. Because the Company reported losses for the periods presented, all potentially dilutive common shares consisting of preferred stock, stock options, warrants and convertible debt are antidilutive. Refer to note 15 for the Company’s calculation of net loss per share for the periods presented.
(r) Income Taxes
The Company accounts for income taxes using the asset and liability method whereby deferred tax asset and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws expected to be in effect when the asset or liability is expected to be realized or settled. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amount that is deemed more likely than not to be realized.
In the ordinary course of business, there are many transactions for which the ultimate tax outcome is uncertain. The Company assesses uncertain tax positions in each of the tax jurisdictions in which it has operations and accounts for the related financial statement implications. Unrecognized tax benefits are reported using the two step approach under which tax effects of a position are recognized only if it is more likely than not to be sustained and the amount of the tax benefit recognized is equal to the largest tax benefit that is greater than fifty percent likely of being realized upon ultimate settlement of the tax position. Determining the appropriate level of unrecognized tax benefits requires the Company to exercise judgment regarding the uncertain application of tax law. The amount of unrecognized tax benefits is adjusted when information becomes available or when an event occurs indicating a change is appropriate. The Company includes interest and penalties related to its uncertain tax positions as part of income tax expense, if any.
(s) Interest Expense
The Company amortizes discounts associated with long-term debt obligations using the effective interest rate method.
| 3. | RECENT ACCOUNTING PRONOUNCEMENTS |
In March 2013, the FASB issued new accounting guidance clarifying the accounting for the release of cumulative translation adjustment into net income when a parent either sells part or all of its investment in a foreign entity or no longer holds a controlling interest in a subsidiary or group of assets that is a nonprofit or a business within a foreign entity. The new standard is effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2013. The Company does not
F-14
Table of Contents
anticipate that this adoption will have a significant impact on the Company’s financial position, results of operations or cash flows.
In July 2013, the FASB issued new accounting guidance that requires that unrecognized tax benefits be classified as an offset to deferred tax assets to the extent of any net operating loss carryforwards, similar tax loss carryforwards, or tax credit carryforwards available at the reporting date in the applicable tax jurisdiction to settle any additional income taxes that would result from the disallowance of a tax position. An exception would apply if the tax law of the tax jurisdiction does not require the Company to use, and it does not intend to use, the deferred tax asset for such purpose. This guidance is effective for reporting periods beginning after December 15, 2013. The Company does not expect the adoption of these provisions to have a material effect on the consolidated financial statements.
| 4. | CONCENTRATION OF CREDIT RISK |
Financial instruments that potentially subject the Company to a concentration of credit risk principally consist of cash and cash equivalents and accounts receivable. While the Company’s cash and cash equivalents are on deposit with high quality FDIC insured financial institutions, at times, such deposits exceed insured limits. The Company has not experienced any losses in such accounts.
The Company believes that the concentration of credit risk in its accounts receivable is substantially mitigated by the Company’s evaluation process, relatively short collection terms and the high level of creditworthiness of its customers. The Company evaluates the status of each of its customers, but generally requires no collateral. The Company has not experienced any significant losses in such accounts. The Company maintains reserves for credit losses.
The Company had no customers that represented greater than 10% of the Company’s trade receivable or revenue balances as of December 31, 2013 and 2012, and for the years ended December 31, 2013, 2012 and 2011.
| 5. | PROPERTY AND EQUIPMENT |
Property and equipment consist of the following (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Furniture and equipment |
$ | 3,798 | $ | 3,033 | ||||
| Surgical instruments |
19,345 | 16,470 | ||||||
| Leasehold improvements |
608 | 497 | ||||||
|
|
|
|
|
|||||
| 23,751 | 20,000 | |||||||
| Less accumulated depreciation and amortization |
(11,056 | ) | (7,704 | ) | ||||
|
|
|
|
|
|||||
| $ | 12,695 | $ | 12,296 | |||||
|
|
|
|
|
|||||
Included in property and equipment are instrument sets used by surgeons during the implant process. Depreciation on the instrument sets commences once the instrument set has been placed into service. As of December 31, 2013 and 2012, the Company had $2.0 million and $3.3 million, respectively, in instrument sets that had not yet been placed into service.
The Company had approximately $690,000 and $661,000 in instrument sets and equipment under capital lease as of December 31, 2013 and 2012, respectively, and accumulated amortization thereon of approximately $660,000 and $500,000, respectively.
F-15
Table of Contents
For the years ended December 2013, 2012 and 2011, the Company recorded depreciation expense of $3.5 million, $2.7 million and $2.0 million, respectively, including amortization of instrument sets and equipment under capital leases of $133,000, $129,000 and $181,000, respectively.
| 6. | INTANGIBLE ASSETS |
Intangible assets consist of the following (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Patents and trademarks |
$ | 5,423 | $ | 4,513 | ||||
| Software licenses |
414 | 224 | ||||||
|
|
|
|
|
|||||
| 5,837 | 4,737 | |||||||
| Less accumulated amortization |
(2,764 | ) | (2,118 | ) | ||||
|
|
|
|
|
|||||
| $ | 3,073 | $ | 2,619 | |||||
|
|
|
|
|
|||||
The Company recorded amortization expense of $528,000, $445,000 and $395,000 during the years ended December 31, 2013, 2012 and 2011, respectively.
The expected amortization for each of the next five years as of December 31, 2013 is as follows (in thousands):
| 2014 |
$ | 606 | ||
| 2015 |
509 | |||
| 2016 |
438 | |||
| 2017 |
396 | |||
| 2018 |
327 | |||
| Thereafter |
797 | |||
|
|
|
|||
| $ | 3,073 | |||
|
|
|
| 7. | ACCRUED EXPENSES |
Accrued expenses consist of the following (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Compensation and other employee related costs |
$ | 8,715 | $ | 6,216 | ||||
| Contributions withheld for Employee Stock Purchase Plan |
976 | — | ||||||
| Royalties |
1,184 | 909 | ||||||
| Clinical and regulatory costs |
327 | 338 | ||||||
| Government grants |
974 | 933 | ||||||
| Rent |
1,893 | 1,250 | ||||||
| Other |
2,255 | 1,113 | ||||||
|
|
|
|
|
|||||
| $ | 16,324 | $ | 10,759 | |||||
|
|
|
|
|
|||||
| 8. | LINE OF CREDIT |
In February 2011, the Company entered into an amended loan agreement with a bank under which it may make periodic borrowings (the Line of Credit). Under the terms of the Line of Credit, Spine and Holding may make aggregate borrowings equal to the lesser of the amount available based on a borrowing base
F-16
Table of Contents
calculation or $14.0 million. The Line of Credit bears interest at the bank’s prime rate plus 1.5%, is secured by substantially all of Spine’s tangible assets, requires the Company to maintain a minimum cash balance of $1.0 million and was originally set to expire on November 23, 2012. In April 2012, the Company amended its existing Line of Credit. The amended agreement (1) increased the borrowing base calculation from $14.0 million to $19.0 million, (2) amended the interest rate from the bank’s prime rate plus 1.5% to the bank’s prime rate plus 2.0% (5.25% at December 31, 2013 and 2012), (3) increased the required minimum cash balance to be maintained with the bank from $1.0 million to $2.0 million and (4) extended the maturity date from November 23, 2012 to April 25, 2014.
As of December 31, 2013 and 2012, the Company had outstanding principal totaling $18.2 million and $19.0 million, respectively. The Line of Credit contains certain financial covenant requirements. The Company was in compliance with all covenants at December 31, 2013 and 2012.
In connection with the initial Line of Credit, the Company issued warrants to purchase an aggregate of 154,506 shares of Holding’s Series C preferred stock to the bank, which were immediately vested and exercisable. The warrants were valued at $155,000 based on the fair value of the Company’s Series C preferred stock on the issuance date of $1.165 per share using the Black Scholes model with the following assumptions: risk free interest rate of 3.0%; dividend yield of 0%; weighted average expected life of the warrant of seven years; and a 75% volatility factor. The warrants were recorded as a debt discount and an increase in additional paid in capital and were being amortized over the term of the Line of Credit. Upon the closing of the Company’s IPO in October 2013, these warrants were net exercised for 10,889 shares of common stock.
| 9. | LONG-TERM DEBT |
Long-term debt consists of the following (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Short-term financing |
$ | 2,641 | $ | 1,772 | ||||
| Various notes payable |
4,521 | 6,235 | ||||||
| Loan facility |
— | 9,998 | ||||||
| Convertible notes |
— | 22,500 | ||||||
|
|
|
|
|
|||||
| Total long-term debt |
7,162 | 40,505 | ||||||
| Less current portion of long-term debt and short-term financing |
(4,404 | ) | (3,675 | ) | ||||
|
|
|
|
|
|||||
| Long-term debt |
2,758 | 36,830 | ||||||
| Less unamortized discounts |
— | (6,504 | ) | |||||
|
|
|
|
|
|||||
| Long-term debt, net of discount and current portion |
$ | 2,758 | $ | 30,326 | ||||
|
|
|
|
|
|||||
(a) Short-Term Financing
Medical borrows funds from various financial institutions in France on a short-term basis. The funds are typically repaid within 90 days and are collateralized by certain assets of Medical, including accounts receivable. The weighted average interest rate for the short-term balances outstanding at December 31, 2013 and 2012 was 4.8%.
(b) Various Notes Payable
Medical has loan agreements with six and seven different entities as of December 31, 2013 and December 31, 2012, respectively. The amounts of the outstanding loans vary from approximately
F-17
Table of Contents
$109,000 to $1.1 million at December 31, 2013, and from approximately $34,000 to $1.3 million at December 31, 2012, and bear interest at rates varying between 2.53% and 4.65% at December 31, 2013 and 2012. Maturity dates for these loans vary from 2014 to 2019, and the loans are secured by certain assets of Medical.
(c) Loan Facility
In 2007, the Company entered into a loan facility with a third party under which the Company could make maximum aggregate borrowings of up to $15.0 million, with an initial maturity date of January 31, 2011. A total of $5.0 million was initially drawn on the loan facility (Loan A) in January 2007 and an additional $1.5 million was subsequently drawn in July 2007. Loan A had a fixed interest rate of 12.0%, accruing through December 31, 2009. Prior to October 13, 2008, the date of the third amendment to the financing agreement, Loan A included “payment in kind” (PIK) interest of 3.0% in addition to a 9.0% fixed rate. Subsequent to the third amendment, the PIK interest was added to the outstanding principal amount of the loan and was payable at the earlier of maturity of the loan or repayment of the loan in full. As of January 29, 2010, and for the next 24 months, the loan was payable in 24 equal payments of principal, plus any remaining PIK interest. The lender received warrants exercisable for 153,397 shares of the Company’s common stock in connection with the initial draw (First Warrant) and the second draw (Second Warrant). All warrants related to this financing were immediately exercisable at $0.00675 per share. The First and Second Warrants were originally exercisable through January 31, 2014. The estimated fair value of the First and Second Warrants was $248,000 as determined using a Black Scholes valuation model, and was deemed equity classified.
In 2008, the Company entered into a third amendment to the loan facility. This amendment reduced the maximum aggregate borrowings to $12.5 million and advanced the Company an additional $6.0 million (Loan B). Loan B had an initial maturity date of September 28, 2012, bore interest at 12.5% and required interest only payments through December 31, 2009. Principal payments under Loan B commenced on January 29, 2010 with 25% of the outstanding principal payable in 12 equal monthly payments, plus accrued and unpaid interest, 35% payable over the following 12 months and 40% payable over the final 9 months. The debt was collateralized by all assets of the Company, including intellectual property, through a subordination agreement. The debt also contained certain financial covenant requirements. The Company issued a warrant to purchase 313,823 shares of common stock to the lender in connection with the issuance of Loan B (Third Warrant). The Third Warrant was immediately exercisable at $0.00675 per share through October 13, 2015. The estimated fair value of the Third Warrant was $625,000 as determined using a Black Scholes valuation model, and was deemed equity classified.
In May 2009, the Company entered into a fourth amendment to the loan facility. This amendment altered the covenant requirements with respect to foreign exchange considerations.
In November 2009, the Company entered into a fifth amendment to the loan facility. The amendment combined Loans A and B (Modified Loans) and set a new maturity date of July 31, 2012. The amendment deferred principal repayment until January 1, 2011. A $3.0 million payment of outstanding advances on the Modified Loans was required prior to the date of amendment. The lender received an additional warrant exercisable for 24,074 shares of the Company’s common stock in connection with the November 23, 2009 amendment (Fourth Warrant). The Fourth Warrant was immediately exercisable at $0.00675 per share through November 23, 2016. The estimated fair value of the Fourth Warrant was $633,000 as determined using a Black Scholes valuation model, and was deemed equity classified.
In February 2011, the Company amended the loan facility to revise the terms of the Modified Loans and to obtain an additional loan of $3.2 million (Loan C). The Modified Loans and Loan C had interest rates of 13.5% and 12.5%, respectively. Interest payments are due monthly commencing February 28, 2011.
F-18
Table of Contents
Principal payments were due monthly commencing January 31, 2013. The amended loan facility was originally schedule to mature on December 31, 2014.
In connection with Loan C, the Company modified the First, Second, Third and Fourth Warrants (collectively, the Old Warrants) and issued new warrants to the lender exercisable for a total of 604,002 shares of the Company’s common stock (2011 Warrants). The 2011 Warrants are immediately exercisable at $0.00675 per share through February 11, 2018. The 2011 Warrants contained an anti dilution provision whereby the number of shares issuable upon exercise would be increased by a number equal to 3.5% of equity securities that may be issued in one or more transactions occurring from February 11, 2011 until the Company has raised $10.0 million in gross proceeds from such transactions, excluding any shares that may have been issued or sold in connection with an acquisition or an initial public offering of the Company’s common stock. The Company recorded a liability for the estimated fair value of the 2011 Warrants on the date of issue of $1.9 million. In connection with the warrant modification, the Company reduced additional paid in capital by $1.5 million, representing the estimated fair value of the Old Warrants at the modification date, and recorded an additional debt discount of $350,000. As of December 31, 2012, the fair value of the warrant liability was $4.2 million.
Upon the conversion of the Promissory Notes discussed below in October 2013, the anti-dilution provision of the 2011 Warrants resulted in the number of shares subject to the warrant increasing by 46,667.
In April 2012, the Company entered into an amendment agreement to its existing loan facility which required an immediate principal payment of $3.0 million and delayed the remaining monthly principal payments from commencing January 31, 2013 to January 31, 2014.
As of December 31, 2012, the Company had an outstanding principal balance on the Loan Facility of approximately $10.0 million and unamortized discounts of $252,000. The company was in compliance with all covenants as of December 31, 2012.
In October 2013, in connection with the Company’s IPO, the Loan Facility was repaid in full, resulting in zero balances for the outstanding principal balance and unamortized discount as of December 31, 2013. In December 2013, the 2011 Warrants were exercised for 650,669 shares of the Company’s common stock at a total exercise price of $4,392.
(d) Convertible Notes
In May 2012, the Company issued Subordinated Secured Convertible Promissory Notes (Promissory Notes) with an original face amount of $22.5 million in exchange for $15.0 million, resulting in a debt discount of $7.5 million. The Promissory Notes included an original interest rate of 6% per year with the face amount and accrued interest due and payable upon the earliest of April 25, 2016 or an initial public offering with aggregate gross proceeds to the Company of at least $50.0 million. The resulting $7.5 million discount on the Promissory Notes was originally scheduled to be accreted over the term of the debt until April 25, 2016, which when coupled with the stated interest rate on the notes, yielded an effective rate of approximately 18%. Upon the consummation of a change of control or initial public offering, the holders of the Promissory Notes originally had an option to elect to convert some or all of the Promissory Notes into shares of the Company’s common stock equal to the original principal amount of $15.0 million and accrued interest divided by (i) the price per share of the common stock in a public offering or received in a change of control times (ii) a 50% discount. Upon the Company’s IPO, and election by certain note-holders to convert their Promissory Notes into 1,929,309 shares of common stock, the Company recorded $7.4 million in additional interest expense related to this beneficial conversion feature. One note-holder elected to receive $2.8 million in cash instead of exercising the
F-19
Table of Contents
conversion feature embedded in the Promissory Notes. As of December 31, 2013, all Promissory Notes have been converted or repaid, resulting in a zero balance.
As of December 31, 2012, the Company had an outstanding principal balance on the Promissory Notes totaling $22.5 million, unamortized discounts of $6.3 million and accrued interest of $904,000, which is included in other long-term liabilities on the consolidated balance sheets. The Company was in compliance with all covenants as of December 31, 2012.
(e) Future Minimum Principal Payments
Future minimum principal payments of long-term debt by year as of December 31, 2013 are as follows (in thousands):
| 2014 |
$ | 4,404 | ||
| 2015 |
1,145 | |||
| 2016 |
758 | |||
| 2017 |
538 | |||
| 2018 |
220 | |||
| Thereafter |
97 | |||
|
|
|
|||
| $ | 7,162 | |||
|
|
|
| 10. | STOCKHOLDERS’ EQUITY |
Authorized Stock
The Company’s amended and restated certificate of incorporation, effective upon the completion of the IPO, authorizes the Company to issue 112,000,000 shares of common and preferred stock, consisting of 107,000,000 shares of common stock with $0.001 par value and 5,000,000 shares of preferred stock with $0.001 par value.
Prior to the IPO in October 2013, the Company was authorized to be issue was 92,723,696 shares of stock, consisting of 18,167,361 shares of common stock with $0.001 par value and 74,556,335 shares of preferred stock with $0.001 par value, of which 11,120,119 shares were designated as Series A-1 Convertible Preferred Stock (Series A-1), 18,097,848 shares were designated as Series A-2 Convertible Preferred Stock (Series A-2), 14,838,368 shares were designated Series B Convertible Preferred Stock (Series B) and 30,500,000 shares were designated as redeemable Series C Convertible Preferred Stock (Series C).
Initial Public Offering
On October 15, 2013, the Company completed its IPO of 5,750,000 shares of common stock at a price of $15.00 per share including 750,000 shares sold to underwriters for the exercise of their over-allotment option to purchase additional shares. The IPO generated net proceeds of approximately $77.5 million, after deducting underwriting discounts and expenses of approximately $8.7 million.
Upon the IPO, the previously outstanding shares of convertible preferred stock and redeemable convertible preferred stock were converted on a 6.75-to-one basis into shares of common stock, resulting in all of the outstanding shares of Series A-1, Series A-2, Series B and Series C preferred stock being converted into 10,977,667 shares of common stock. Following the closing of the IPO, there were no shares of preferred stock outstanding.
F-20
Table of Contents
With the proceeds of the IPO, the Company paid its Series C Stockholders an aggregate of $17.1 million in exchange for agreeing to vote in favor of the conversion of the Series C preferred stock to common stock.
In conjunction with the merger of Spine and Medical in 2006, Holding and the individual Medical stockholders entered into an Escrow Agreement in which the individual Medical stockholders delivered their shares to an escrow agent, and Holding delivered its stock certificates to be held in escrow. In October 2013, upon closing of the Company’s IPO, each share of Medical’s Class A stock was automatically exchanged for 5.80087 shares of common stock, or an aggregate of 3,110,024 shares.
| 11. | STOCK-BASED COMPENSATION |
(a) Stock Option Activity
A summary of the stock option activity for the Company for the years ended December 31, 2013, 2012 and 2011 is as follows:
| Shares | Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| (Years) | ($000’s) | |||||||||||||||
| Outstanding—December 31, 2012 |
1,253,909 | $ | 3.29 | 7.45 | $ | 3,835 | ||||||||||
| Granted |
461,600 | 13.36 | ||||||||||||||
| Exercised |
(81,155 | ) | 2.90 | |||||||||||||
| Forfeited |
(61,728 | ) | 5.73 | |||||||||||||
|
|
|
|
|
|||||||||||||
| Outstanding—December 31, 2013 |
1,572,626 | $ | 6.17 | 7.17 | $ | 27,413 | ||||||||||
|
|
|
|
|
|||||||||||||
| Vested and expected to vest: |
||||||||||||||||
| At December 31, 2012 |
1,233,131 | $ | 3.29 | 7.45 | $ | 3,806 | ||||||||||
| At December 31, 2013 |
1,519,889 | 6.17 | 7.17 | 26,716 | ||||||||||||
| Exercisable: |
||||||||||||||||
| At December 31, 2012 |
1,253,909 | $ | 3.29 | 7.45 | $ | 3,835 | ||||||||||
| At December 31, 2013 |
1,223,533 | 3.65 | 7.17 | 24,306 | ||||||||||||
In 2007, the Company adopted the LDR Holding Corporation 2007 Stock Option/Stock Issuance Plan (the 2007 Plan). Concurrent with the IPO in October 2013, the Company adopted the LDR Holding Corporation 2013 Equity Incentive Plan (the 2013 Plan) and terminated the 2007 Plan. No further grants will be made under the 2007 Plan. Any shares of common stock that are subject to outstanding awards under the 2007 Plan which are forfeited or lapse for any reason prior to exercise or settlement and would otherwise have returned to the share reserve under the 2007 Plan, instead will be available for issuance under the 2013 Plan.
The purpose of the 2007 Plan and the 2013 Plan (collectively, the Plans) is to provide eligible persons employed by or providing services to the Company with the opportunity to acquire or increase their equity interest in the Company.
2007 Plan
Under the 2007 Plan, incentive stock options may only be granted to Company employees and shall be issued at an exercise price not less than 100% of the fair market value of the Company’s common stock
F-21
Table of Contents
at the grant date, as determined by the Company’s Board of Directors, except for incentive stock option grants to a stockholder that owns greater than 10% of the Company’s outstanding stock or 10% of the voting power of all classes of the outstanding stock of any of our subsidiaries, in which case the exercise price per share is not less than 110% of the fair market value of the Company’s common stock at the date of grant. Nonstatutory stock options may be granted to Company employees, members of the Board of Directors and consultants at an exercise price determined by the Board of Directors. Options granted under the Plans are vested no later than ten years from the date of grant. At the time of grant, the Company’s Board of Directors determines the exercise price and vesting schedule. Generally, 25% of each option was vested one year from the vesting commencement date, as defined in the option agreement, and then the option vested ratably over each of the next 36 months. Under the Plans, stock options may be exercised before they became fully vested. In the event of termination of service, shares issued from the exercise of unvested stock options are subject to repurchase by the Company at the original purchase price until they vest. The shares subject to each option outstanding shall automatically become vested shares upon a change in ownership or control of the Company. Each such option shall, immediately prior to the consummation of a change in control, become exercisable for all of the shares of common stock at that time subject to that option.
2013 Plan
Under the 2013 Plan, incentive stock options may only be granted to Company employees and shall be issued at an exercise price not less than 100% of the fair market value of the Company’s common stock at the grant date, except for incentive stock options grants to a stockholder that owns greater than 10% of the combined voting power of all classes of stock of the Company, in which case the exercise price per share is not less than 110% of the fair value of the Company’s common stock at the date of grant. Nonstatutory stock options, stock appreciation rights, restricted stock rights or bonuses, restricted stock units, performance shares, performance units and cash-based awards, or other stock-based awards may be granted to Company employees, officers, directors or consultants or those of any present or future parent or subsidiary corporation or other affiliated entity.
As of December 31, 2013, the Company has 853,557 shares of common stock reserved for issuance under the 2013 Plan. This share reserve will automatically increase on January 1, 2014 and each subsequent anniversary through January 1, 2023, by an amount equal to the smaller of (i) 4% of the number of shares of the Company’s common stock issued and outstanding on the immediately preceding December 31; and (ii) an amount determined by the compensation committee of the Company’s board of directors. Shares subject to awards granted under the 2013 Plan which expire, are repurchased, or are canceled or forfeited will again become available for issuance under the 2013 Plan. The shares available will not be reduced by awards settled in cash or by shares withheld to satisfy tax withholding obligations. Only the net number of shares issued upon the exercise of stock appreciation rights or options exercised by means of a net exercise will be deducted from the shares available under the 2013 Plan.
Awards Granted to LDR Medical Employees
Included in the above table as of December 31, 2013 and 2012, are options to purchase 509,062 and 390,549 shares, respectively, of Holdings common stock, which relate to options held by Medical employees to purchase Medical Class A common stock. Prior to the IPO in October 2013, those options were subject to the Escrow Agreement (Note 10). As such, when exercised, the Medical common stock become subject to the Escrow Agreement, under which the Medical shares were converted to Company common stock upon certain future events, as defined in the Escrow Agreement. As the shares subject to the Escrow Agreement were accounted for as if converted, these instruments were valued using the same assumptions as were other Company stock options and have been included within the roll forward of Company stock options above. After the IPO, upon exercise for Medical shares, the Medical shares are required to be converted to the Company’s common stock.
F-22
Table of Contents
Awards Granted Outside the Plans
As of December 31, 2013 and 2012, there were 19,531 shares of Holdings common stock issuable upon exercise of options issued outside the Plans at an exercise price of $1.69 per share.
Performance Share Awards
In March 2013, the Company awarded options for the purchase of 81,841 shares of performance awards under the 2007 Plan. These awards vested or were cancelled on December 31, 2013 depending on whether certain performance goals were achieved. During the year ended December 31, 2013, the Company recognized $413,000 in compensation expense related to the performance share awards.
(b) Employee Stock Purchase Program
Concurrent with the IPO in October 2013, the Company established the LDR Holding Corporation 2013 Employee Stock Purchase Plan (ESPP), a qualified plan under Section 423 of the Code. The purpose of the ESPP is to advance the interests of the Company by providing an incentive to attract, retain and reward employees and to motivate employees to contribute to the growth and profitability of the Company. The compensation committee of the Company’s board of directors administers the ESPP.
A total of 111,111 shares of common stock are initially authorized and reserved for sale under the ESPP. The ESPP provides for an automatic increase in the number of shares available for issuance under the plan on January 1 of each year beginning in 2014 and continuing through and including January 1, 2023 equal to the lesser of (i) 1% of the Company’s issued and outstanding shares of common stock on the immediately preceding December 31, or (ii) a number of shares as determined by the Company’s board of directors.
The ESPP is generally designed to comply with the provisions of Section 423 of the Internal Revenue Code and will be typically implemented through a series of sequential offering periods, generally of a six-month duration, as established by the compensation committee.
Under the ESPP, eligible employees can purchase shares of the Company’s common stock at 85% of the lower of the fair market value on (i) the first trading day of the offering period and (ii) the purchase date, which is typically the end of an offering period. Amounts accumulated for each participant, generally through payroll deductions, are credited toward the purchase of the Company common stock on the purchase date.
No participant may purchase under the ESPP in any calendar year shares having a value of more than $25,000 measured by the fair market value per share of the Company’s common stock on the first day of the applicable offering period. Any amounts withheld from participants’ compensation in excess of the amounts used to purchase shares will be refunded, without interest.
(c) Accounting for Stock-Based Compensation
The Company uses the Black-Scholes option pricing model in valuing its stock awards. The Black-Scholes model requires estimates regarding dividend yield, volatility, risk-free rate of return, estimated forfeitures during the service period and the expected term of the award.
The expected dividend yield assumption is based on the Company’s expectation of future dividend payouts. The volatility assumption is based on the historical volatilities of comparable public companies. The risk free rate of return assumption is based upon observed interest rates appropriate for the expected term of stock awards. The expected term is derived using the simplified method and represents the weighted average period that the stock awards are expected to remain outstanding.
F-23
Table of Contents
The fair value of stock option grants has been estimated at the date of grant using the Black-Scholes option pricing model with the following weighted average assumptions:
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Expected dividend yield |
— | % | — | % | — | % | ||||||
| Volatility |
52.02 | 54.77 | 70.19 | |||||||||
| Risk-free rate of return |
0.17 | 0.44 | 0.34 | |||||||||
| Forfeiture rate |
7.90 | 4.30 | 4.31 | |||||||||
| Expected life |
6 years | 6 years | 6 years | |||||||||
The weighted average grant date fair value per share for options granted during the years ended December 31, 2013, 2012 and 2011 was $7.08, $2.80 and $1.96, respectively. The total intrinsic value of options exercised during the years ended December 31, 2013, 2012 and 2011 was approximately $1,680,000, $188,000 and $214,000, respectively. The unrecognized compensation expense related to unvested options granted under the Plans was $2,836,000 at December 31, 2013 and $1,072,000 at December 31, 2012, and is expected to be recognized over a period of approximately 4 years.
The Company’s stock-based compensation expense related to employee stock options and ESPP awards for the years ended December 31, 2013, 2012 and 2011 was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Research and development |
$ | 153 | $ | 48 | $ | 28 | ||||||
| Sales and marketing |
786 | 181 | 114 | |||||||||
| General and administrative |
330 | 66 | 42 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 1,269 | $ | 295 | $ | 184 | ||||||
|
|
|
|
|
|
|
|||||||
| 12. | RETIREMENT SAVINGS PLANS |
(a) U.S.-Based Employees
The Company maintains an employee savings and retirement plan (the 401(k) Plan) covering all of its employees. The 401(k) Plan permits but does not require matching contributions by the Company on behalf of participants. The Company has not made matching contributions in any of the periods presented in these financial statements. Starting in January 2014, the Company will match 50% of employee contributions for the first 6% contributed by each employee.
(b) France-Based Employees
The Company provides retirement indemnities for employees located in France commensurate with other similar companies in that region. Accruals for these employee retirement obligations amounted to $230,000 and $172,000 at December 31, 2013 and 2012, respectively. Expenses related to these programs were not material in the periods presented.
| 13. | RELATED PARTY TRANSACTIONS |
In 2007, the Company made a loan to an officer of $200,000, with a maturity date of February 2014. Interest accrued at a rate of 6% per annum. The original terms called for interest only payments due quarterly over the term of the note; however, the Board of Directors subsequently deferred interest payments until the maturity date. The balance of the loan, including accrued interest, was $0 and $270,000 at December 31, 2013 and 2012, respectively.
F-24
Table of Contents
In February 2013, the Company made a loan to an officer for $54,000 in connection with the exercise of 22,222 stock options. Interest accrued at a rate of 2.5% per annum with unpaid principal and interest due on August 5, 2014. The loan was secured by 22,222 shares of the Company’s common stock that are held by the officer. The balance of the loan, including accrued interest, was $0 at December 2013.
The Company recognized $8,000, $12,000 and $12,000 and in related party interest income during the years ended December 31, 2013, 2012, and 2011, respectively.
| 14. | INCOME TAXES |
The components of loss before income taxes are as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Domestic |
$ | (27,460 | ) | $ | (11,723 | ) | (3,539 | ) | ||||
| Foreign |
1,196 | 3,201 | 3,039 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | (26,264 | ) | $ | (8,522 | ) | (500 | ) | ||||
|
|
|
|
|
|
|
|||||||
The components of the provision for income taxes are as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Current taxes: |
||||||||||||
| United States |
$ | 57 | $ | 32 | 165 | |||||||
| International |
1,577 | 1,397 | 1,209 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current tax expense |
1,634 | 1,429 | 1,374 | |||||||||
| Deferred taxes: |
||||||||||||
| United States |
5 | 5 | 5 | |||||||||
| International |
32 | (255 | ) | (51 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Total deferred tax expense (benefit) |
37 | (250 | ) | (46 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Income tax expense |
$ | 1,671 | $ | 1,179 | 1,328 | |||||||
|
|
|
|
|
|
|
|||||||
F-25
Table of Contents
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred taxes at December 31, 2013 and 2012 are as follows (in thousands):
| Years Ended December 31, | ||||||||
| 2013 | 2012 | |||||||
| Deferred tax assets: |
||||||||
| Current deferred tax assets: |
||||||||
| Inventory |
$ | 1,649 | $ | 1,375 | ||||
| Reserves and allowances |
296 | 206 | ||||||
| Other |
5 | 60 | ||||||
| Valuation allowance |
(1,383 | ) | (1,250 | ) | ||||
|
|
|
|
|
|||||
| Total current deferred tax assets |
567 | 391 | ||||||
| Noncurrent deferred tax assets: |
||||||||
| Net operating loss and tax credits carryforwards |
24,311 | 19,535 | ||||||
| Amortization |
149 | 169 | ||||||
| Other |
749 | 1,230 | ||||||
|
|
|
|
|
|||||
| Gross noncurrent tax assets |
25,209 | 20,934 | ||||||
| Valuation allowance |
(23,942 | ) | (19,116 | ) | ||||
|
|
|
|
|
|||||
| Total noncurrent deferred tax assets |
$ | 1,267 | $ | 1,818 | ||||
|
|
|
|
|
|||||
| Deferred tax liabilities: |
||||||||
| Current deferred tax liabilities: |
||||||||
| Prepaid and other |
$ | 1,107 | $ | 933 | ||||
|
|
|
|
|
|||||
| Total current deferred tax liabilities |
1,107 | 933 | ||||||
| Noncurrent deferred tax liabilities: |
||||||||
| Depreciation |
754 | 1,164 | ||||||
| Interest |
— | 100 | ||||||
|
|
|
|
|
|||||
| Total noncurrent deferred tax liabilities |
$ | 754 | $ | 1,264 | ||||
|
|
|
|
|
|||||
The Company has established a full valuation allowance relating to substantially all of its U.S. net deferred tax assets due to uncertainties regarding the realization of the deferred tax assets based on the Company’s lack of earnings history in the U.S. The valuation allowance increased by $5.0 million and $2.7 million during the years ended December 31, 2013 and 2012, respectively.
As of December 31, 2013 and 2012, the Company has no accrued interest or penalties associated with uncertain tax positions. The jurisdictions in which the Company files income taxes include the U.S., France and Brazil. The Company’s returns are not currently under examination by the Internal Revenue Service or other taxing authorities. The Company is subject to income tax examinations for our U.S. federal and state and local income taxes for 2004 and subsequent years and foreign tax examinations for 2006 and subsequent years.
F-26
Table of Contents
A reconciliation of the statutory U.S. federal tax rate to the Company’s effective rate is as follows:
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Statutory U.S. federal tax rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State taxes |
(0.2 | ) | 0.5 | (33.1 | ) | |||||||
| Valuation allowance |
(15.5 | ) | (45.4 | ) | (335.5 | ) | ||||||
| Permanent items |
(24.7 | ) | (2.0 | ) | (22.3 | ) | ||||||
| Prior year true-up |
0.2 | — | 84.7 | |||||||||
| Foreign tax rate differentials |
(0.2 | ) | (0.9 | ) | 6.6 | |||||||
|
|
|
|
|
|
|
|||||||
| Total |
(6.4 | )% | (13.8 | )% | (265.6 | )% | ||||||
|
|
|
|
|
|
|
|||||||
The Company pays income taxes in France related to intercompany sales in the year the sale occurs; however the recognition of tax expense related to intercompany sales is deferred in the consolidated financial statements until the product is sold to an unrelated third party. The deferred income tax charge is included in other current assets in the consolidated balance sheets. As of December 31, 2013 and 2012, the deferred income tax charge was $1.5 million and $2.2 million, respectively.
As of December 31, 2013, the Company had federal net operating loss carryforwards of approximately $64.5 million, which will begin to expire in 2024, if not utilized prior to that time. Under the provisions of the Internal Revenue Code, certain substantial changes in the Company’s ownership may result in a limitation on the amount of net operating loss carryforwards which can be used in future years.
Earnings occurring outside the U.S. are deemed to be indefinitely reinvested outside of the U.S. to support our foreign operations. As a result, we continue to accumulate earnings overseas for investment in our business outside the U.S. and to use cash generated from U.S. operations and short- and long-term borrowings to meet our U.S. cash needs. Should we require more capital in the U.S. than is generated by our domestic operations, we could elect to repatriate earnings from our non-U.S. subsidiaries or raise additional capital in the U.S. through debt or equity issuances. These alternatives could result in higher effective tax rates, increased interest expense, or other dilution of our earnings. The Company has not estimated the deferred tax liabilities associated with our permanently reinvested earnings as it is impractical to do so.
| 15. | NET LOSS PER SHARE |
As further discussed in note 2, the Company computes net loss per share using a methodology that gives effect to the impact of outstanding participating securities (the two-class method). As there were net losses recognized during the periods presented, there was no income allocation required under the two-class method or dilution attributed to the weighted average shares outstanding in the calculation of diluted loss per share.
F-27
Table of Contents
Net loss per share for the years ended December 31, 2013, 2012 and 2011 was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Numerator |
||||||||||||
| Net loss attributable to common stockholders |
$ | (27,935 | ) | $ | (9,701 | ) | $ | (1,828 | ) | |||
|
|
|
|
|
|
|
|||||||
| Denominator |
||||||||||||
| Weighted average shares outstanding—basic |
9,041 | 4,615 | 4,490 | |||||||||
| Dilutive effect of preferred stock, warrant options and convertible debt |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Weighted average shares outstanding—diluted |
9,041 | 4,615 | 4,490 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss per common share—basic and diluted |
$ | (3.09 | ) | $ | (2.10 | ) | $ | (0.41 | ) | |||
| Potentially dilutive securities(1) |
||||||||||||
| Redeemable convertible preferred stock |
— | 4,451 | 4,451 | |||||||||
| Convertible preferred stock |
— | 6,527 | 6,527 | |||||||||
| Common stock warrants |
— | 604 | 604 | |||||||||
| Preferred stock warrants |
— | 23 | 23 | |||||||||
| Stock options |
1,573 | 1,254 | 962 | |||||||||
| (1) | The impact of potentially dilutive securities on earnings per share is anti-dilutive in a period of net loss. |
Due to the timing of the completion of the Company’s IPO on October 15, 2013, the weighted average number of shares outstanding for 2013 only reflects the additional shares of common stock issued as a result of (i) the conversion of the Company’s preferred stock, (ii) the conversion of the Company’s convertible debt, and (iii) the shares issued upon warrant exercise for approximately 2.5 months. As of December 31, 2013, the Company actually had in excess of 24 million shares of common stock outstanding, which if used in the computation of loss per share, would have significantly reduced the actual loss per share for both basic and diluted purposes.
| 16. | COMMITMENTS AND CONTINGENCIES |
(a) Lease Commitments
The Company leases office space, equipment and vehicles under noncancelable operating leases which expire by 2019. Certain of these leases contain rent holidays and scheduled rent increases which are included in the Company’s rent expense and recognized on a straight-line basis. Total rent expense under these operating leases was $2.4 million, $1.9 million and $1.6 million for the years ended December 31, 2013, 2012 and 2011, respectively.
Future minimum lease payments under noncancelable operating leases and (with initial remaining lease terms in excess of one year) as of December 31, 2013 are as follows (in thousands):
| Operating Leases |
||||
| Year: |
||||
| 2014 |
$ | 3,066 | ||
| 2015 |
2,834 | |||
| 2016 |
2,235 | |||
| 2017 |
2,219 | |||
| 2018 |
2,180 | |||
| Thereafter |
2,030 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 14,564 | ||
|
|
|
|||
F-28
Table of Contents
(b) Government Grants
The Company receives grants from the French government to pursue research and development of its French products. These grants are refundable when and if the products become technologically feasible. The Company has recorded grants received of $974,000 and $933,000 in accrued expenses in the consolidated balance sheets as of December 31, 2013 and December 31, 2012, respectively. There are potential contingent gains on grants recorded as liabilities related to products that will not be viable.
(c) Litigation
From time to time, the Company may be involved in litigation relating to claims arising out of its ordinary course of business. Management believes that there are no claims or actions pending or threatened against the Company, the ultimate disposition of which would have a material impact on the Company’s financial position, results of operations or cash flows.
| 17. | SEGMENT AND GEOGRAPHIC INFORMATION |
Operating segments are defined as components of an enterprise for which separate financial information is available and evaluated regularly by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company globally manages the business within one reportable segment. Segment information is consistent with how management reviews the business, makes investing and resource allocation decisions and assesses operating performance. The Company’s products are principally sold in the United States and France.
The following table represents total sales by geographic area, based on the location of the customer (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| United States |
$ | 82,307 | $ | 64,801 | $ | 52,062 | ||||||
| France |
11,233 | 9,388 | 9,128 | |||||||||
| Other Countries(1) |
18,054 | 16,729 | 16,785 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 111,594 | $ | 90,918 | $ | 77,975 | ||||||
|
|
|
|
|
|
|
|||||||
| (1) | No additional locations are individually significant. |
The Company classifies its products into two categories: exclusive technology and traditional fusion products. The following table represents total sales by product category (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Exclusive technology products |
$ | 92,818 | $ | 72,805 | $ | 57,450 | ||||||
| Traditional fusion products |
18,776 | 18,113 | 20,525 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | 111,594 | $ | 90,918 | $ | 77,975 | ||||||
|
|
|
|
|
|
|
|||||||
F-29
Table of Contents
The following table represents long-lived assets by geographic area (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| United States |
$ | 7,446 | $ | 6,020 | ||||
| France |
3,615 | 4,700 | ||||||
| Other Countries(1) |
1,634 | 1,576 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 12,695 | $ | 12,296 | ||||
|
|
|
|
|
|||||
| (1) | No additional locations are individually significant. |
| 18. | SUBSEQUENT EVENTS |
In January 2014, common stock reserved for issuance under the 2013 Plan increased by 963,066 to 1,816,623 shares. In January 2014, common stock reserved for sale under the ESPP increased by 240,766 to 351,877 shares. These annual automatic share increases are part of the 2013 Plan and ESPP as discussed in Notes 11 and 10, respectively.
| 19. | QUARTERLY FINANCIAL DATA (UNAUDITED) |
The following table sets forth our unaudited quarterly condensed consolidated statements of operations data for each of the eight quarters ended December 31, 2013. The data has been prepared on the same basis as the audited consolidated financial statements and related notes included herein and you should read the following tables in conjunction with such financial statements. The table includes all necessary adjustments, consisting only of normal recurring adjustments that we consider necessary for a fair presentation of this data. The results of historical periods are not necessarily indicative of future results.
| December 31, 2013 |
September 30, 2013 |
June 30, 2013 |
March 31, 2013 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenue |
$ | 31,978 | $ | 27,195 | $ | 26,579 | $ | 25,842 | ||||||||
| Gross profit |
26,592 | 23,102 | 22,410 | 21,543 | ||||||||||||
| Operating loss |
(905 | ) | (906 | ) | (235 | ) | (278 | ) | ||||||||
| Net loss |
(15,105 | ) | (7,984 | ) | (2,959 | ) | (1,887 | ) | ||||||||
| Net loss per common share |
$ | (0.69 | ) | $ | (1.68 | ) | $ | (0.62 | ) | $ | (0.41 | ) | ||||
| December 31, 2012 |
September 30, 2012 |
June 30, 2012 |
March 31, 2012 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Revenue |
$ | 24,966 | $ | 20,948 | $ | 22,379 | $ | 22,625 | ||||||||
| Gross profit |
20,905 | 17,371 | 18,937 | 18,935 | ||||||||||||
| Operating income (loss) |
180 | (764 | ) | (743 | ) | 53 | ||||||||||
| Net loss |
(2,426 | ) | (3,050 | ) | (2,099 | ) | (2,126 | ) | ||||||||
| Net loss per common share |
$ | (0.52 | ) | $ | (0.66 | ) | $ | (0.45 | ) | $ | (0.46 | ) | ||||
F-30
Table of Contents

Table of Contents
Shares
LDR HOLDING CORPORATION
Common Stock

PROSPECTUS
Until , 2014 all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
| Piper Jaffray | William Blair | |||||
| Cowen and Company | RBC Capital Markets | |||||
| JMP Securities | Stephens Inc. | |||||
| Bryan, Garnier & Co. | ||||||
, 2014
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution |
The following table sets forth the estimated costs and expenses, other than underwriting discounts and commissions, to be paid by us in connection with the sale of the shares of common stock being registered hereby.
| SEC registration fee |
$ | |||
| FINRA filing fee |
* | |||
| Listing fee |
* | |||
| Printing and engraving expenses |
* | |||
| Legal fees and expenses |
* | |||
| Accounting fees and expenses |
* | |||
| Blue Sky fees and expenses (including legal fees) |
* | |||
| Transfer agent and registrar fees and expenses |
* | |||
| Miscellaneous |
* | |||
|
|
|
|||
| Total |
* | |||
|
|
|
| * | To be provided by amendment. |
| Item 14. | Indemnification of Directors and Officers |
Section 145 of the Delaware General Corporation Law authorizes a court to award, or a corporation’s board of directors to grant, indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities, including reimbursement for expenses incurred, arising under the Securities Act of 1933, as amended, or the Securities Act. Our amended and restated certificate of incorporation provides for indemnification of our directors, officers, employees and other agents to the maximum extent permitted by the Delaware General Corporation Law, and our amended and restated bylaws provide for indemnification of our directors, officers, employees and other agents to the maximum extent permitted by the Delaware General Corporation Law. In addition, we have entered into indemnification agreements with our directors, officers and some employees containing provisions that may be in some respects broader than the specific indemnification provisions contained in the Delaware General Corporation Law. The indemnification agreements may require us, among other things, to indemnify our directors against certain liabilities that may arise by reason of their status or service as directors, officers and employees and to advance their expenses incurred as a result of any proceeding against them as to which they could be indemnified.
We maintain a directors’ and officers’ insurance policy.
The underwriting agreement to be entered into in connection with this offering will provide that the underwriters will indemnify us, our directors and certain of our officers against liabilities resulting from information furnished by or on behalf of the underwriters specifically for use in the Registration Statement.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. Please read “Item 17. Undertakings” for more information on the SEC’s position regarding such indemnification provisions.
II-1
Table of Contents
| Item 15. | Recent Sales of Unregistered Securities |
Since January 1, 2011, we have made sales of the following unregistered securities:
| • | Between January 1, 2011 and October 10, 2013, we granted stock options under our 2007 Stock Option/Stock Issuance Plan and the 2013 Equity Incentive Plan to purchase an aggregate of 866,468 shares of our common stock at exercise prices ranging between $3.105 and $15.00 to a total of 109 employees, directors and consultants. Of these, stock options to purchase an aggregate of 37,715 shares have been cancelled without being exercised, 30,941 have been exercised and 797,812 remain outstanding. |
| • | Since January 1, 2011, we issued and sold an aggregate of 237,044 shares of our common stock to employees, directors and consultants at exercise prices ranging between $0.607 and $4.185 upon the exercise of stock options granted under our 2004 Stock Option/Stock Issuance Plan and our 2007 Stock Option/Stock Issuance Plan. Of these, none have been repurchased and all shares remain outstanding. |
| • | Between April 2012 and May 2012, LDR Holding and Médical issued and sold an aggregate of approximately $15,000,000 of subordinated secured convertible promissory notes, which bore an interest rate of 6%. In October 2013, we issued an aggregate of 1,929,309 shares of common stock pursuant to the conversion of the subordinated secured convertible promissory notes into common stock in connection with our initial public offering. |
| • | In October 2013, we issued (1) an aggregate of 3,110,024 shares of common stock in connection with the exchange of shares of capital stock of Médical into shares of our common stock pursuant to the second amended and restated put-call agreement and in connection with our initial public offering, and (2) 10,889 shares of Common Stock pursuant to the exercise of a warrant, which shares were issued in accordance with the net exercise provision of the warrant. |
| • | In December 2013, we issued an aggregate of 650,669 shares of our common stock pursuant to the exercise of warrants issued February 11, 2011, for aggregate proceeds of $4,392.02 |
Unless otherwise stated, the sales of the above securities were deemed to be exempt from registration under the Securities Act in reliance upon Section 4(a)(2) of the Securities Act or Regulation D promulgated thereunder, or Rule 701 promulgated under Section 3(b) of the Securities Act as transactions by an issuer not involving any public offering or pursuant to benefit plans and contracts relating to compensation as provided under Rule 701. The recipients of the securities in each of these transactions represented their intentions to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were placed upon the stock certificates issued in these transactions.
| Item 16. | Exhibits and Financial Statement Schedules |
(a) Exhibits
| Incorporated by Reference | ||||||||||||
| Exhibit Number |
Exhibit Description |
Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||
| 1.1* | Form of Underwriting Agreement | |||||||||||
| 3.1 | Amended and Restated Certificate of Incorporation of LDR Holding Corporation, adopted on October 15, 2013. | 10-Q | 001-36095 | 11/7/2013 | 3.1 | |||||||
II-2
Table of Contents
| Incorporated by Reference | ||||||||||||
| Exhibit Number |
Exhibit Description |
Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||
| 3.2 | Amended and Restated Bylaws of LDR Holding Corporation, adopted on October 15, 2013. | 10-Q | 001-36095 | 11/7/2013 | 3.2 | |||||||
| 4.1.1 | Amended and Restated Investors’ Rights Agreement, dated September 11, 2007. | S-1 | 333-190829 | 8/26/2013 | 4.2 | |||||||
| 4.1.2 | Amendment No. 1 to Investors’ Rights Agreement, dated April 25, 2012. | S-1 | 333-190829 | 8/26/2013 | 4.3 | |||||||
| 4.1.3 | Amendment No. 2 to Investors’ Rights Agreement, dated September 12, 2013. | S-1/A | 333-190829 | 09/19/2013 | 4.3.1 | |||||||
| 4.2 | Second Amended and Restated Put-Call Agreement, dated August 6, 2013, by and among the Registrant, LDR Médical S.A.S. and the individuals listed on Schedule A thereto. | S-1/A | 333-190829 | 09/19/2013 | 4.4 | |||||||
| 4.3 | Warrant to Purchase Series C Convertible Preferred Stock issued to Comerica Bank on November 23, 2009. | S-1 | 333-190829 | 8/26/2013 | 4.7 | |||||||
| 4.4 | Amendment No. 1 to Warrant to Purchase Series C Convertible Preferred Stock, dated September 12, 2013. | S-1/A | 333-190829 | 09/19/2013 | 4.7.1 | |||||||
| 4.5 | Amended and Restated Warrant to Purchase Stock issued to Escalate Capital I, L.P. on February 11, 2011. | S-1 | 333-190829 | 8/26/2013 | 4.8 | |||||||
| 5.1* | Opinion of Andrews Kurth LLP | |||||||||||
| 10.1 | Form of Amended and Restated Indemnification Agreement for directors and officers of the Registrant, as currently in effect. | S-1/A | 333-190829 | 09/19/2013 | 10.1 | |||||||
| 10.2 | 2007 Stock Option/Stock Issuance Plan, as amended. | S-1 | 333-190829 | 8/26/2013 | 10.2 | |||||||
| 10.3 | Form of Stock Option Agreement for 2007 Stock Option/Stock Issuance Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.4 | |||||||
| 10.4 | 2013 Equity Incentive Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.5 | |||||||
| 10.5 | Form of Stock Option Agreement for 2013 Equity Incentive Plan. | 10-K | 001-36095 | 03/04/2014 | 10.5 | |||||||
II-3
Table of Contents
| Incorporated by Reference | ||||||||||||
| Exhibit Number |
Exhibit Description |
Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||
| 10.6 | Form of Notice of Grant-Standard Exercise for 2013 Equity Incentive Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.7 | |||||||
| 10.7 | Form of Restricted Stock Unit Agreement for 2013 Equity Incentive Plan. | 10-K | 001-36095 | 03/04/2014 | 10.7 | |||||||
| 10.8 | Form of Stock Option Agreement-International for 2013 Equity Incentive Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.8 | |||||||
| 10.9 | Amended and Restated 2013 Employee Stock Purchase Plan. | S-1/A | 333-190829 | 10/08/2013 | 10.9 | |||||||
| 10.10 | Employment Terms Letter Agreement, dated June 10, 2013, between the Registrant and Christophe Lavigne. | S-1 | 333-190829 | 8/26/2013 | 10.10 | |||||||
| 10.11 | Employment Terms Letter Agreement, dated June 10, 2013, between the Registrant and Robert McNamara. | S-1 | 333-190829 | 8/26/2013 | 10.11 | |||||||
| 10.12 | Employment Terms Letter Agreement, dated June 10, 2013, between the Registrant and Andre Potgieter. | S-1 | 333-190829 | 8/26/2013 | 10.12 | |||||||
| 10.13 | Form of Employment, Confidential Information, and Invention Assignment Agreement. | S-1/A | 333-190829 | 09/19/2013 | 10.13 | |||||||
| 10.14 | Lease Agreement, dated August 10, 2011, by and between LDR Spine USA, Inc. and 13875 Research Blvd, LLC. | S-1 | 333-190829 | 8/26/2013 | 10.14 | |||||||
| 10.15 | First Amendment to Lease Agreement, dated November 5, 2012, by and between LDR Spine USA, Inc. and 13785 Research Blvd, LLC. | S-1 | 333-190829 | 8/26/2013 | 10.15 | |||||||
| 10.16 | English translation of Lease Agreement, dated June 24, 2013, by and between LDR Médical S.A.S. and Departement de L’Aube. | S-1 | 333-190829 | 8/26/2013 | 10.16 | |||||||
| 10.17 | Loan and Security Agreement, dated November 23, 2009, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.17 | |||||||
II-4
Table of Contents
| Incorporated by Reference | ||||||||||||
| Exhibit Number |
Exhibit Description |
Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||
| 10.18 | First Amendment to Loan and Security Agreement, dated June 30, 2010, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.18 | |||||||
| 10.19 | Second Amendment to Loan and Security Agreement, dated September 30, 2010, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.19 | |||||||
| 10.20 | Third Amendment to Loan and Security Agreement, dated February 10, 2011, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.20 | |||||||
| 10.21 | Fourth Amendment to Loan and Security Agreement dated as of April 2011, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.21 | |||||||
| 10.22 | Fifth Amendment to Loan and Security Agreement dated as of April 25, 2012, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.22 | |||||||
| 10.23 | Amended and Restated Loan and Security Agreement, dated February 11, 2011, by and between Escalate Capital I, L.P., on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.23 | |||||||
| 10.24 | First Amendment to Loan and Security Agreement, dated April 25, 2012, by and between Escalate Capital I, L.P., on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.24 | |||||||
II-5
Table of Contents
| Incorporated by Reference | ||||||||||||
| Exhibit Number |
Exhibit Description |
Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||
| 10.25# | English translation of Supply Agreement, dated May 29, 2011, by and between CF Plastiques and LDR Médical S.A.S. | S-1 | 333-190829 | 8/26/2013 | 10.26 | |||||||
| 10.26# | English translation of Amendment to Supply Agreement between LDR Holding Corporation and CF Plastiques, entered into on December 6, 2013. | 8-K/A | 001-36095 | 12/19/2013 | 10.1 | |||||||
| 10.27# | Supply Agreement, dated January 31, 2003, by and between Invibio Inc. and LDR Médical S.A.S., as amended by those certain Letters of Amendment dated November 10, 2004, November 23, 2004, July 9, 2008, June 17, 2009 and December 7, 2011. | S-1 | 333-190829 | 8/26/2013 | 10.27 | |||||||
| 10.28# | English translation of Subcontracting Agreement, effective November 28, 2012, by and between Greatbatch Medical SAS and LDR Médical S.A.S. | S-1/A | 333-190829 | 10/08/2013 | 10.28 | |||||||
| 10.29 | Non-Employee Independent Director Compensation Policy. | S-1/A | 333-190829 | 10/08/2013 | 10.29 | |||||||
| 21.1 | Subsidiaries of the Registrant. | S-1 | 333-190829 | 8/26/2013 | 21.1 | |||||||
| 23.1 | Consent of KPMG LLP, independent registered public accounting firm. | X | ||||||||||
| * | To be filed by Amendment |
| # | Confidential treatment has been requested for portions of this exhibit. These portions have been omitted from the exhibit filed with the Securities and Exchange Commission and submitted separately to the Securities and Exchange Commission. |
| Item 17. | Undertakings |
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement, certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
II-6
Table of Contents
The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-7
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, LDR Holding Corporation has duly caused this registration statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Austin, State of Texas, on the 2nd day of April, 2014.
| LDR HOLDING CORPORATION | ||
| By: |
/s/ Christophe Lavigne | |
| Christophe Lavigne President and Chief Executive Officer | ||
SIGNATURES AND POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Christophe Lavigne, Robert McNamara and Scott Way, and each of them, either of whom may act without the joinder of the other, as his true and lawful attorneys-in-fact and agents with full power of substitution and re-substitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments (including post-effective amendments) to this registration statement, and to sign any registration statement for the same offering covered by the registration statement that is to be effective upon filing pursuant to Rule 462(b) promulgated under the Securities Act, and all post-effective amendments thereto, and to file the same, with all exhibits thereto and all documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or any of them, or his or their substitute or substitutes, may lawfully do or cause to be done or by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, as amended this registration statement has been signed by the following persons in the capacities indicated on the 2nd day of April, 2014.
| Signature |
Title | |
| /s/ Christophe Lavigne |
President, Chief Executive Officer and | |
| /s/ Robert McNamara |
Executive Vice President, Chief Financial Officer (Principal Financial Officer) | |
| /s/ Denise Cruz |
Controller (Principal Accounting Officer) | |
| /s/ Joseph Aragona |
Director | |
| /s/ Kevin Lalande |
Director | |
| /s/ William W. Burke |
Director | |
| /s/ Stefan Widensohler |
Director | |
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Exhibit Description |
Incorporated by Reference | ||||||||||
| Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||||
| 1.1* | Form of Underwriting Agreement | |||||||||||
| 3.1 | Amended and Restated Certificate of Incorporation of LDR Holding Corporation, adopted on October 15, 2013. | 10-Q | 001-36095 | 11/7/2013 | 3.1 | |||||||
| 3.2 | Amended and Restated Bylaws of LDR Holding Corporation, adopted on October 15, 2013. | 10-Q | 001-36095 | 11/7/2013 | 3.2 | |||||||
| 4.1.1 | Amended and Restated Investors’ Rights Agreement, dated September 11, 2007. | S-1 | 333-190829 | 8/26/2013 | 4.2 | |||||||
| 4.1.2 | Amendment No. 1 to Investors’ Rights Agreement, dated April 25, 2012. | S-1 | 333-190829 | 8/26/2013 | 4.3 | |||||||
| 4.1.3 | Amendment No. 2 to Investors’ Rights Agreement, dated September 12, 2013. | S-1/A | 333-190829 | 09/19/2013 | 4.3.1 | |||||||
| 4.2 | Second Amended and Restated Put-Call Agreement, dated August 6, 2013, by and among the Registrant, LDR Médical S.A.S. and the individuals listed on Schedule A thereto. | S-1/A | 333-190829 | 09/19/2013 | 4.4 | |||||||
| 4.3 | Warrant to Purchase Series C Convertible Preferred Stock issued to Comerica Bank on November 23, 2009. | S-1 | 333-190829 | 8/26/2013 | 4.7 | |||||||
| 4.4 | Amendment No. 1 to Warrant to Purchase Series C Convertible Preferred Stock, dated September 12, 2013. | S-1/A | 333-190829 | 09/19/2013 | 4.7.1 | |||||||
| 4.5 | Amended and Restated Warrant to Purchase Stock issued to Escalate Capital I, L.P. on February 11, 2011. | S-1 | 333-190829 | 8/26/2013 | 4.8 | |||||||
| 5.1* | Opinion of Andrews Kurth LLP | |||||||||||
| 10.1 | Form of Amended and Restated Indemnification Agreement for directors and officers of the Registrant, as currently in effect. | S-1/A | 333-190829 | 09/19/2013 | 10.1 | |||||||
| 10.2 | 2007 Stock Option/Stock Issuance Plan, as amended. | S-1 | 333-190829 | 8/26/2013 | 10.2 | |||||||
Table of Contents
| Exhibit Number |
Exhibit Description |
Incorporated by Reference | ||||||||||
| Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||||
| 10.3 | Form of Stock Option Agreement for 2007 Stock Option/Stock Issuance Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.4 | |||||||
| 10.4 | 2013 Equity Incentive Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.5 | |||||||
| 10.5 | Form of Stock Option Agreement for 2013 Equity Incentive Plan. | 10-K | 001-36095 | 03/04/2014 | 10.5 | |||||||
| 10.6 | Form of Notice of Grant-Standard Exercise for 2013 Equity Incentive Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.7 | |||||||
| 10.7 | Form of Restricted Stock Unit Agreement for 2013 Equity Incentive Plan. | 10-K | 001-36095 | 03/04/2014 | 10.7 | |||||||
| 10.8 | Form of Stock Option Agreement-International for 2013 Equity Incentive Plan. | S-1/A | 333-190829 | 09/19/2013 | 10.8 | |||||||
| 10.9 | Amended and Restated 2013 Employee Stock Purchase Plan. | S-1/A | 333-190829 | 10/08/2013 | 10.9 | |||||||
| 10.10 | Employment Terms Letter Agreement, dated June 10, 2013, between the Registrant and Christophe Lavigne. | S-1 | 333-190829 | 8/26/2013 | 10.10 | |||||||
| 10.11 | Employment Terms Letter Agreement, dated June 10, 2013, between the Registrant and Robert McNamara. | S-1 | 333-190829 | 8/26/2013 | 10.11 | |||||||
| 10.12 | Employment Terms Letter Agreement, dated June 10, 2013, between the Registrant and Andre Potgieter. | S-1 | 333-190829 | 8/26/2013 | 10.12 | |||||||
| 10.13 | Form of Employment, Confidential Information, and Invention Assignment Agreement. | S-1/A | 333-190829 | 09/19/2013 | 10.13 | |||||||
| 10.14 | Lease Agreement, dated August 10, 2011, by and between LDR Spine USA, Inc. and 13875 Research Blvd, LLC. | S-1 | 333-190829 | 8/26/2013 | 10.14 | |||||||
| 10.15 | First Amendment to Lease Agreement, dated November 5, 2012, by and between LDR Spine USA, Inc. and 13785 Research Blvd, LLC. | S-1 | 333-190829 | 8/26/2013 | 10.15 | |||||||
| 10.16 | English translation of Lease Agreement, dated June 24, 2013, by and between LDR Médical S.A.S. and Departement de L’Aube. | S-1 | 333-190829 | 8/26/2013 | 10.16 | |||||||
Table of Contents
| Exhibit Number |
Exhibit Description |
Incorporated by Reference | ||||||||||
| Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||||
| 10.17 | Loan and Security Agreement, dated November 23, 2009, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.17 | |||||||
| 10.18 | First Amendment to Loan and Security Agreement, dated June 30, 2010, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.18 | |||||||
| 10.19 | Second Amendment to Loan and Security Agreement, dated September 30, 2010, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.19 | |||||||
| 10.20 | Third Amendment to Loan and Security Agreement, dated February 10, 2011, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.20 | |||||||
| 10.21 | Fourth Amendment to Loan and Security Agreement dated as of April 2011, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.21 | |||||||
| 10.22 | Fifth Amendment to Loan and Security Agreement dated as of April 25, 2012, by and between Comerica Bank, on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.22 | |||||||
| 10.23 | Amended and Restated Loan and Security Agreement, dated February 11, 2011, by and between Escalate Capital I, L.P., on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.23 | |||||||
Table of Contents
| Exhibit Number |
Exhibit Description |
Incorporated by Reference | ||||||||||
| Form | File No. | Filing Date | Exhibit Number |
Provided Herewith | ||||||||
| 10.24 | First Amendment to Loan and Security Agreement, dated April 25, 2012, by and between Escalate Capital I, L.P., on the one hand, and LDR Holding Corporation and LDR Spine USA, Inc., on the other hand. | S-1 | 333-190829 | 8/26/2013 | 10.24 | |||||||
| 10.25# | English translation of Supply Agreement, dated May 29, 2011, by and between CF Plastiques and LDR Médical S.A.S. | S-1 | 333-190829 | 8/26/2013 | 10.26 | |||||||
| 10.26# | English translation of Amendment to Supply Agreement between LDR Holding Corporation and CF Plastiques, entered into on December 6, 2013. | 8-K/A | 001-36095 | 12/19/2013 | 10.1 | |||||||
| 10.27# | Supply Agreement, dated January 31, 2003, by and between Invibio Inc. and LDR Médical S.A.S., as amended by those certain Letters of Amendment dated November 10, 2004, November 23, 2004, July 9, 2008, June 17, 2009 and December 7, 2011. | S-1 | 333-190829 | 8/26/2013 | 10.27 | |||||||
| 10.28# | English translation of Subcontracting Agreement, effective November 28, 2012, by and between Greatbatch Medical SAS and LDR Médical S.A.S. | S-1/A | 333-190829 | 10/08/2013 | 10.28 | |||||||
| 10.29 | Non-Employee Independent Director Compensation Policy. | S-1/A | 333-190829 | 10/08/2013 | 10.29 | |||||||
| 21.1 | Subsidiaries of the Registrant. | S-1 | 333-190829 | 8/26/2013 | 21.1 | |||||||
| 23.1 | Consent of KPMG LLP, independent registered public accounting firm. | X | ||||||||||
| * | To be filed by Amendment |
| # | Confidential treatment has been requested for portions of this exhibit. These portions have been omitted from the exhibit filed with the Securities and Exchange Commission and submitted separately to the Securities and Exchange Commission. |