Attached files
| file | filename |
|---|---|
| EX-14.1 - EX-14.1 - BIOCEPT INC | d697720dex141.htm |
| EX-31.1 - EX-31.1 - BIOCEPT INC | d697720dex311.htm |
| EX-31.2 - EX-31.2 - BIOCEPT INC | d697720dex312.htm |
| EX-32.1 - EX-32.1 - BIOCEPT INC | d697720dex321.htm |
| EX-32.2 - EX-32.2 - BIOCEPT INC | d697720dex322.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2013
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-36284
Biocept, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 80-0943522 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 5810 Nancy Ridge Drive San Diego, California |
92121 | |
| (Address of principal executive offices) | (Zip Code) | |
(858) 320-8200
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Exchange on Which Registered | |
| Common Stock, par value $0.0001 per share | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No x
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | x | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
As of June 28, 2013, the last business day of the registrant’s most recently completed second fiscal quarter, the registrant’s common stock was not listed for trading on any exchange or over-the-counter market and there was no established public market for the common stock. The common stock began trading on The NASDAQ Capital Market on February 5, 2014. As of February 5, 2014, the aggregate market value of the registrant’s common stock held by non-affiliates was approximately $22.1 million based on the closing price for the common stock of $9.40 on that date. Shares of common stock held beneficially by Claire K.T. Reiss and by each executive officer, director, and their affiliated stockholders have been excluded from this calculation as such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of March 21, 2014, there were 4,449,594 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for the 2014 Annual Meeting of Stockholders, to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Form 10-K, are incorporated by reference in Part III, Items 10-14 of this Form 10-K. Except for the portions of the Proxy Statement specifically incorporated by reference in this Form 10-K, the Proxy Statement shall not be deemed to be filed as part hereof.
Table of Contents
1
Table of Contents
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Annual Report”) contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements included or incorporated by reference in this Annual Report other than statements of historical fact, are forward-looking statements. You can identify these and other forward-looking statements by the use of words such as “may,” “will,” “could,” “anticipate,” “expect,” “intend,” “believe,” “continue” or the negative of such terms, or other comparable terminology. Forward-looking statements also include the assumptions underlying or relating to such statements.
Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth below under the caption “Risk Factors” in Part I, Item 1A and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II, Item 7 of this Annual Report and elsewhere in this Annual Report. Moreover, we operate in an evolving environment. New risk factors and uncertainties emerge from time to time and it is not possible for us to predict all risk factors and uncertainties, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. Readers are cautioned not to place undue reliance on forward-looking statements. The forward-looking statements speak only as of the date on which they are made and we undertake no obligation to update such statements to reflect events that occur or circumstances that exist after the date on which they are made except as required by law. Readers should, however, review the factors and risks we describe in the reports and registration statements we file from time to time with the Securities and Exchange Commission, or the SEC.
2
Table of Contents
PART I
| Item 1. | Business |
Overview
We are a cancer diagnostics company that develops and commercializes proprietary circulating tumor cell, or CTC, and circulating tumor DNA, or ctDNA, tests utilizing a standard blood sample. These tests provide information to oncologists that enable them to select the most appropriate treatment for their patients based on better, timelier and more-detailed data on the characteristics of tumors. Our current OncoCEE-BR breast cancer test and our planned tests utilize our Cell Enrichment and Extraction (CEE) technology for the enumeration and analysis of CTCs, and our CEE-Selector technology for the detection and analysis of ctDNA, each performed on a standard blood sample. The CEE technology is an internally developed, microfluidics-based CTC capture and analysis platform, with enabling features that change how CTC testing can be used by clinicians by providing real-time biomarker monitoring with a standard blood sample. The CEE-Selector technology enables mutation detection with enhanced sensitivity and specificity and is applicable to nucleic acid from CTCs or other sample types, such as blood plasma for ctDNA. We believe CEE-Selector technology is an important part of certain of our pipeline CTC tests, and believe it could also be a stand-alone test for molecular analysis of biomarkers.
At our corporate headquarters facility located in San Diego, California, we operate a clinical laboratory that is certified under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and accredited by the College of American Pathologists, or CAP, and manufacture our CEE microfluidic channels, related equipment and certain reagents to perform our current breast cancer test and our planned future tests at this facility. CLIA certification and CAP accreditation are required before any clinical laboratory, including ours, may perform testing on human specimens for the purpose of obtaining information for the diagnosis, prevention, or treatment of disease, or the assessment of health. The OncoCEE-BR test and the tests we plan to offer are classified as laboratory developed tests, or LDTs, under CLIA regulations.
OncoCEE-BR is a breast cancer CTC test that is performed on a standard blood sample. It detects CTCs, which are typically very rare compared to normal blood cells, and determines the patient’s human epidermal growth factor receptor 2, or HER2, status by fluorescence in situ hybridization, or FISH. Pursuant to an agreement that we entered into with Clarient Diagnostic Services, Inc., a GE Healthcare Company, as revised in May 2013, Clarient is making OncoCEE-BR available to physicians through its sales force. (Clarient does not have the exclusive marketing rights for the test.)
We believe that the OncoCEE-BR test offers advantages over other available CTC tests, with improved sensitivity and enumeration results as well as diagnostic biomarker analyses. Competitive CTC tests rely on the expression of the epithelial cell adhesion molecule, or EpCAM, and cytokeratins for CTC capture, detection and enumeration. This approach may exclude CTCs that have undergone intrinsic modifications of their phenotype, such as the epithelial-to-mesenchymal transition, or EMT, thought to be critical for metastasis. EMT may represent a possible explanation for many patients who, despite an aggressive disease, are found to be negative
3
Table of Contents
for the presence of CTCs by current technologies. In addition to cytokeratin positive epithelial CTCs, OncoCEE™ captures and detects EpCAM and cytokeratin negative CTCs, which are more mesenchymal-like. Additionally, the OncoCEE platform enables evaluation of treatment-associated biomarkers, like HER2 status, which qualifies patients as candidates for HER2-targeted therapeutics such as Herceptin®, Perjeta®, Kadcyla® (all Genentech/Roche) and Tykerb® (GlaxoSmithKline). We plan to include immunocytochemical analysis of estrogen receptor and progesterone receptor proteins, as well as mutation analysis as appropriate, into the OncoCEE-BR test within the next year.
We anticipate launching OncoCEE-LU, a test performed on a standard blood sample for non-small cell lung cancer, or NSCLC, in the third quarter of 2014. The biomarkers to be analyzed in the OncoCEE-LU test would include EML4/ALK and ROS1 gene fusions by FISH, and the epidermal growth factor receptor, or EGFR, gene, the K-ras gene and the B-raf gene by mutation analysis, in addition to CTC enumeration. Our OncoCEE-LU test would be run on a standard blood sample. We have entered into an agreement with Life Technologies Corporation, or Life Technologies, under which we are cooperating with Life Technologies to develop, promote and commercialize our OncoCEE-LU test. Under this agreement, we would perform OncoCEE-LU tests in our laboratory and transmit the results to Life Technologies for their interpretation and reporting to healthcare professionals.
We plan to add other biomarker analyses to our OncoCEE tests as their relevance is demonstrated in clinical trials, for example, RET proto-oncogene gene fusions in NSCLC, which may indicate a particular course of therapy. In addition, we are developing a series of other CTC and ctDNA tests for different solid tumor types, including colorectal cancer, prostate cancer, gastric cancer and melanoma, each incorporating treatment-associated biomarker analyses specific to that cancer, planned to be launched over the next two to three years.

Biomarkers are molecular or cellular features of a cancer cell that indicate an abnormality. This abnormality, typically a genetic mutation or aberration, detected at either the gene, protein or metabolite level, may in fact be responsible for the transformation of the cell from a normal cell to a cancer cell. We have focused our efforts on biomarkers associated with specific targeted cancer therapeutics, or resistance to those therapeutics. Examples include an amplified HER2 gene, which is associated with HER2-targeted therapeutics like Herceptin®, Perjeta®, Kadcyla® and Tykerb® for the treatment of breast cancer, or a mutated B-raf gene, which is associated with the drugs Zelboraf® (Daiichi-Sankyo/Genentech/Roche) and Tafinlar® (GlaxoSmithKline) for the treatment of melanoma. This is important because the presence or level of these biomarkers indicates to a physician that the associated therapy is appropriate for the patient, or instead that the patient has, or has developed, resistance to that therapy.
4
Table of Contents
Biomarkers have traditionally been detected in tumor tissue after biopsy or re-section, with the analysis performed by a pathologist. We are able to perform these same analyses on CTCs or ctDNA on a standard blood sample using our CEE and CEE-Selector technology in our CLIA laboratory, meaning that the biomarkers detected in a patient’s tumor can now be monitored on a real-time basis without the need for a tissue biopsy. Because of the difficulty or inability to obtain periodic tissue biopsies, especially at the time of recurrence, this offers the physician a new source and level of information than was previously available.
We also have a research and development program focused on technology enhancements and novel platform development and a translational research group evaluating clinical applications for cancer diagnostic tests in different cancer types and clinical settings. We have the capability to offer our current and planned unique cancer diagnostic tests through our CLIA laboratory to physicians for patient care applications as well as to pharmaceutical and biopharmaceutical companies and academic centers using CTC or ctDNA testing, with biomarker analysis including genetic analysis, in their clinical trials and research efforts. CTC tests, particularly those that offer analysis of CTCs for treatment-associated biomarkers, are becoming powerful tools in the practice of personalized medicine. They enable physicians to utilize a standard blood sample as a “liquid biopsy” to assess the status of their patient’s cancer at a cellular and molecular level on an ongoing basis, and to select therapies that have the highest likelihood of benefiting their patients.
Historically, our average price received per OncoCEE-BR test performed for commercial customers has been approximately $694 and $635 for the years ended December 31, 2012 and 2013, respectively. This was heavily influenced by the fact that historically a high percentage of our sales were through our marketing partner, Clarient. We amended our arrangement with Clarient as of May 2013, and we do not expect a significant percentage of our future sales to come through Clarient. Our future average price for commercial customers could increase from our historical figure, based on recognition of the medical value of our products, publication of clinical utility study results, possible improvement of the product, introduction of additional tests, increased demand generated by our future sales and marketing efforts, and similar commercial factors. Factors that could cause pricing for commercial customers to decrease include any perceived lack of clinical utility for CTC or ctDNA testing, or increased competition from other reference labs or IVD manufacturers. Third-party governmental and private payors have reimbursement policies and fee schedules which determine the amounts, if any, we would receive for performing tests for their covered patients. Such governmental and private third-party payors frequently make determinations about how much (if anything) they are willing to pay for tests such as ours, or for components of such tests; these determinations are important to our business and can have adverse or positive effects on the price we receive for our testing. For example, private payors often look to Medicare policies and rates when setting their reimbursement rates.
In addition, our reimbursement rates can vary based on whether we are considered by private third-party payors to be an “in-network” provider, a participating provider, a covered provider or an “out-of-network” provider. These definitions can vary from insurance company to insurance company, but we are generally considered an “out-of-network” or non-participating provider by the vast majority of private third-party payors. It is not unusual for a company that offers highly specialized or unique testing to be an “out-of-network” provider. An “in-network” provider usually has a contracted arrangement with the insurance company or benefits provider. This contract governs, among other things, service-level agreements and reimbursement rates. In certain instances an insurance company may negotiate an “in-network” rate for our testing rather than pay the typical “out-of-network” rate. An “in-network” provider usually has rates that are lower per test than those that are “out-of-network”, and that rate can vary from a single digit percentage deduction discount to upwards of 25% to 30% lower than an “out-of-network” provider. The discount rate varies based on the insurance company, the testing type and often times the specifics of the patient’s insurance plan. In some plans, there is no benefit paid for out-of-network claims and our ability to collect from the patient may be hindered by the financial resources of the patient or by state laws that prohibit billing of patients for denied out-of-network claims.
We cannot predict whether, or under what circumstances, payors will reimburse for all components of our tests. Payment amounts can also vary across individual policies. Full or partial denial of coverage by payors, or
5
Table of Contents
reimbursement at inadequate levels, would have a material adverse impact on our business and on market acceptance of our tests.
To date, we have engaged in only limited sales and marketing activities. Such activities have primarily related to our OncoCEE-BR test and have been conducted pursuant to an agreement with Clarient. This agreement was revised in May 2013 and Clarient no longer has exclusive marketing rights to this test. We expect that in the future the percentage of our revenue which is generated through our arrangement with Clarient will diminish. We also have established a non-exclusive agreement with Life Technologies for the commercialization of OncoCEE-LU tests when the development and validation of the OncoCEE-LU test are completed.
Using a portion of the proceeds from our initial public offering, we plan to build an internal sales and marketing team to market and sell OncoCEE-BR and our planned future cancer diagnostic tests directly to oncologists. This team will also provide technical expertise and support for the sales representatives of our sales and marketing partners. Our plans call for starting with an initial group of 7 sales representatives, and, based on success and test volume, growing this number to 15-20 within two years.
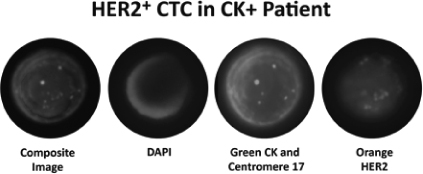
We collaborate with physicians and researchers at MD Anderson Cancer Center and plan to expand our collaborative relationships to include other key thought leaders at other institutions for the cancer types we target with OncoCEE-BR and our planned future CTC and ctDNA tests. Such relationships help us develop and validate the effectiveness and utility of OncoCEE-BR and our planned future tests in specific clinical settings and provide us access to patient samples and data. We completed a study, recently published in Cancer Medicine, utilizing our OncoCEE-BR test, and a version of this test adapted for use with bone marrow samples, with a group at MD Anderson Cancer Center comprised of breast cancer surgeons, pathologists and basic researchers. In this study, we demonstrated the ability to identify HER2 positive CTCs and disseminated tumor cells, or DTCs, seen in bone marrow in patients that had been previously classified as HER2 negative by analysis of their tumor tissue. A HER2 positive result in a patient with breast cancer provides an indication to the oncologist that there is likely to be a survival benefit from treatment with Herceptin®, which has been demonstrated in a number of large clinical studies.
We are currently involved in a new clinical study following up on this finding in CTCs, employing OncoCEE-BR tests for patient selection and monitoring. This study, led by investigators at the Dana-Farber Cancer Institute, is currently enrolling patients, and is likely to produce initial results within a year. We believe that these results will provide clinical utility data to support the wide use of OncoCEE-BR tests as a routine diagnostic test for breast cancer patients. In the screening phase of this study, we are testing in our CLIA-certified laboratory blood samples from HER2 negative patients based on standard tumor tissue analysis, to identify those patients that have HER2 positive CTCs. These patients are then being randomized to chemotherapy plus/minus Herceptin®, and followed for a period of time, with additional CTC tests, including biomarker analysis for HER2 using FISH, performed at subsequent time points.
We plan to grow our business by directly offering oncologists CTC and ctDNA tests. Based on our product development data, as well as discussions with our collaborators, we believe that our planned tests should provide important information and clinical value to oncologists. In particular, our planned CTC and ctDNA tests should deliver important, actionable information not provided by other tests. For example, the market leading clinical CTC test is the United States Food and Drug Administration, or FDA, approved CellSearch® test (Janssen Diagnostics), which provides CTC enumeration, but lacks the ability to perform biomarker analysis. We believe our ability to rapidly translate research insights about the utility of cytogenetic, immunocytochemical and molecular biomarkers to provide information to oncologists for treatment decisions in the clinical setting will improve patient treatment and management, and that these tests will become a key component in the standard of care for personalized cancer treatment.
According to the National Cancer Institute, there will be approximately 230,000 new cases of breast cancer and approximately 230,000 new cases of lung cancer diagnosed in the United States in 2013, with over 3 million patients who have had a diagnosis of these cancers and either are living with these diseases and are undergoing
6
Table of Contents
treatment or are being monitored. For example, in breast cancer, many women have been deemed cancer-free, but continue to undergo periodic monitoring to assure there has been no disease recurrence. Our OncoCEE-BR test and our planned OncoCEE-LU test only require a readily accessible standard blood sample and thus may be used to help manage these patients, including supporting the selection of appropriate treatment, at multiple time points during the course of their disease. Because our tests require only a standard blood sample, they can be particularly useful when no, old or inadequate amounts of biopsy or surgical material is available, as is often the case in lung cancer, even at the time of initial evaluation. For example, up to 25% of patients with lung cancer are not surgically treated for various reasons, including patient status (consensus statement from the American College of Chest Physicians and the Society of Thoracic Surgeons; Chest, Dec. 2012). This is also the case with breast and lung cancers once surgical resection of the tumor has taken place and treatment has been initiated. Patients with breast and lung cancer must often undergo surgical resection of their primary tumor as part of their treatment. Therefore, at the time of progression or recurrence there may be no ability to obtain a tissue biopsy. Additionally, many studies have shown that most tumors mutate during treatment and as the disease progresses, so information from the initial tumor tissue may not be relevant. Again, a significant benefit of our technology is that it allows physicians to assess the current status of the tumors on a real-time basis utilizing a standard blood sample.
We currently offer and conduct our breast cancer diagnostic tests and offer our clinical trial services at our CLIA-certified and CAP-accredited, and state-law licensed, laboratory. Our current breast cancer test and our planned near-term cancer diagnostic tests and clinical trial services include:
| • | CTC and ctDNA Testing. Our current breast cancer test and our other planned cancer diagnostic tests are based on our CEE and CEE-Selector technologies and are currently intended to be performed only in our clinical laboratory. After completing testing, we or our partner provide our customers with an easy to understand report that describes the results of the analyses performed, designed to help oncologists make better decisions about the treatment of their patients. |
| • | Clinical Trial Services. We plan to utilize our clinical laboratory and translational research capabilities to provide clinical trial and research services to pharmaceutical and biopharmaceutical companies and clinical research organizations to improve the efficiency and economic viability of their clinical trials. Our clinical trials and translational research services could leverage our knowledge of CTCs and ctDNA and our ability to develop and implement new cytogenetic, immunocytochemical and molecular diagnostic tests. Our current breast cancer test can, and our other planned cancer diagnostic tests and biomarker tests are anticipated to be able to, help optimize clinical trial patient selection, and as a result potentially improve the likelihood of success of the clinical trial. With positive results in a clinical trial, our tests would more easily then move into standard clinical practice, helping physicians select the most appropriate therapy for their patients. |
We intend to commercialize cancer diagnostic tests in the United States as LDTs performed in our CLIA-certified laboratory. We plan to evaluate potential opportunities for the commercialization of our products in other countries. We are currently exploring the possibility of introducing OncoCEE-LU technology and other offerings outside the United States as part of CE-marked IVD test kits and/or testing systems utilizing our CEE and/or CEE-Selector technologies. We also plan to evaluate this format for our other planned tests.
Our sales strategy is focused on leveraging the sales forces of partners already selling to our target markets, as well as building an internal direct sales and marketing team that can also support our partners. In both cases we plan to engage oncologists in the United States at private and group practices, hospitals and cancer centers. In addition, our internal team will market our clinical trial and research services to pharmaceutical and biopharmaceutical companies and clinical research organizations.
Our headquarters is located in San Diego, California and we had 27 full-time employees and one part-time employee as of December 31, 2013.
7
Table of Contents
Market Overview
Cancer Market Overview
Despite many advances in the treatment of cancer, it remains one of the greatest areas of unmet medical need. In 2008, the World Health Organization attributed 7.6 million deaths worldwide to cancer-related causes. The World Health Organization projects that by 2030 this number will rise to 13.1 million deaths per year. The incidence of, and deaths caused by, the major cancers are staggering. The following data published by the National Cancer Institute shows estimated new cases and deaths for 2013, and prevalence in 2010, in the United States for the major solid cancers types:
| Cancer Type |
Est. Incidence (New Cases/Year-2013) |
Est. Mortality (Deaths/ Year-2013) |
Est. Prevalence (Diagnosed and Alive as of 2010)** |
|||||||||
| Bladder |
72,570 | 15,210 | 563,640 | |||||||||
| Breast* |
232,340 | 39,620 | 2,843,629 | |||||||||
| Cervical |
12,340 | 4,030 | 249,496 | |||||||||
| Colorectal* |
142,820 | 50,830 | 1,154,481 | |||||||||
| Endometrial |
49,560 | 8,190 | 600,346 | |||||||||
| Gastric* |
21,600 | 10,990 | 72,269 | |||||||||
| Kidney |
65,150 | 13,680 | 341,505 | |||||||||
| Lung* |
228,190 | 159,480 | 399,431 | |||||||||
| Melanoma* |
76,690 | 9,480 | 921,780 | |||||||||
| Ovarian |
22,240 | 14,030 | 186,138 | |||||||||
| Pancreatic |
42,220 | 38,460 | 41,609 | |||||||||
| Prostate* |
238,590 | 29,720 | 2,617,682 | |||||||||
| Thyroid |
60,220 | 1,850 | 534,973 | |||||||||
| * | Areas where we currently have tests or active development programs. |
| ** | Includes active disease and disease-free. |
In addition to the human toll, the financial cost of cancer is overwhelming. An independent study published in 2010 and conducted jointly by the American Cancer Society and LIVESTRONG ranked cancer as the most economically devastating cause of death in the world—estimated to be as high as $895 billion globally. According to an article in the Journal of the National Cancer Institute, the direct cost of cancer deaths in the United States in 2000 was over $115 billion, and if lost wages and caregiver costs were added, the total costs increased to over $230 billion.
Cancer is a Heterogeneous Disease
Cancer constitutes a heterogeneous class of diseases, characterized by uncontrolled cell growth that results from a combination of both environmental and hereditary risk factors. Many different tissue types can become malignant, such as breast, lung, liver, and skin, and even within a particular tumor there is heterogeneity, with certain cancer cells in a patient bearing specific cellular or genetic biomarkers which others lack. It has only been in recent years that technology has progressed far enough to enable researchers to understand many cancers at a cellular and molecular level, attribute specific cancers to associated genetic changes and determine the extent to which these changes are seen in a patient’s tumor.
Cancer cells contain genetic alterations compared to normal human cells. Common genetic abnormalities correlated to cancer include gains or losses of genetic material on specific chromosomal regions, or loci, or changes in specific genes, or mutations, which ultimately result in detrimental cellular changes followed by cancerous or pre-cancerous conditions. For example, multiple gains or losses of or on various chromosomes, and the rearrangement of genetic material among chromosomes, or chromosomal translocations, have been observed in different cancer types, such as HER2 in breast cancer and EML4/ALK in NSCLC. In addition, mutations
8
Table of Contents
within gene sequences, or single nucleotide variations, can give rise to aberrant proteins that do not perform their functions correctly, leading to uncontrolled cell growth. Such genetic alterations can be a result of multiple factors, including genetic predisposition, environmental or lifestyle factors or viral infections. Importantly, these genetic changes can be used as biomarkers to help guide appropriate treatment. Detecting these biomarkers, particularly those representing drug targets, or those indicative of responsiveness or resistance of a tumor’s cells to specific therapies, helps clinicians to select drugs, design treatment regimens and optimize patient care and management. Tests that provide such predictive information have the potential to dramatically improve treatment outcomes for patients suffering from cancer.
Limitations of Traditional Cancer Diagnostic and Profiling Approaches
Cancer is difficult to diagnose and manage due to its heterogeneity at morphologic, genetic and clinical levels. Traditional methods of diagnosis for solid tumors, routinely used as the initial step in cancer detection, involve a tissue biopsy followed by a pathologist examining a thin slice of potentially cancerous tissue under a microscope. A recently obtained tissue sample is used in combination with chemical staining techniques to enable analysis of the biopsy. After staining, the pathologist determines through visual inspection whether the biopsy contains normal or cancerous cells, with those that are deemed cancerous being graded on a level of aggressiveness. Often an analysis of biomarkers relevant to that tumor type is also performed on the tissue, ranging from immunohistochemistry to FISH, to mutation analysis by various means such as microarrays and sequencing. After the diagnosis, a clinical workup is performed according to established guidelines for the specific cancer type. From there, the physician determines the stage of progression of the cancer based on a series of clinical measures, such as size, grade, metastasis rates, symptoms and patient history, and decides on a treatment plan that may include surgery, watchful waiting, radiation, chemotherapy, or stem cell transplant.
This type of analysis is dependent on the availability of a recently obtained tissue biopsy for the pathologist to analyze. Such a biopsy is often not available. A tumor may not be readily accessible for biopsy, a patient’s condition may be such that a biopsy is not advised, and for routine periodic patient monitoring to evaluate potential progression or recurrence, a biopsy is a fairly invasive procedure and not typically performed. As the length of time between when the original biopsy, diagnosis or surgery is conducted to the current evaluation of the patient increases, the likelihood that an original biopsy specimen is truly representative of the current disease condition declines, as does the usefulness of the original biopsy for making treatment decisions. This risk intensifies in situations where a drug therapy is being administered, because the drug can put selective pressure on the tumor cells to adapt and change.
Similarly, the heterogeneity referred to above means that different parts or areas of the same tumor can have different molecular features or properties. In evaluating a biopsy specimen, the pathologist will take a few thin slices of the tumor for microscopic review rather than exhaustively analyzing the whole tumor mass. The pathologist can only report on the tumor sections analyzed and if other parts of the tumor have different features, such as biomarkers corresponding to specific treatments, they can be missed. A more representative analysis of the entire tumor, as well as any metastases if they are present, is very helpful. Moreover, there are complications and morbidity associated with the invasive lung cancer biopsy procedures.
CTCs, ctDNA and Cancer
Circulating tumor cells, or CTCs, are cancer cells that have detached from the tumor matrix and invaded the patient’s blood or other bodily fluids. These cells are representative of the tumor and its metastases, and can function as their surrogates. Testing CTCs can complement pathologic information drawn from a biopsy or resected tissue sample, helping to insure that the analysis is comprehensive and not biased by tumor heterogeneity and sampling issues. They can also provide critical data when a biopsy is not possible. Clinical studies have demonstrated that the presence and number of CTCs provides information on the likely course of certain types of disease for the cancer patient, or in other words they are considered “prognostic.” Since CTCs are representative of the tumor, they can also be used for biomarker analysis, such as helping to guide therapy
9
Table of Contents
selection. Such analyses are “predictive” in that they offer insight into the likely responsiveness or resistance to particular therapies. After surgery and during any subsequent therapy or monitoring period, blood samples can periodically be drawn in a standard manner and analyzed to evaluate a therapy’s continuing effectiveness, as well as to detect other biomarkers such as new genetic mutations that may arise as a result of selection pressure by a particular therapy or by chance. Physicians can use this information to determine which therapy is most likely to benefit their patients at particular times through the course of their disease. Treatment decisions based on patient-specific information are the foundation of personalized medicine, and tests, or assays, that guide a physician in the selection of individualized therapy for a patient are termed “predictive assays.”
ctDNA is nucleic acid that is released into blood by dying tumor cells. Cell death occurs in all tissues, especially those that are rapidly dividing, and in cancer, where cell growth is not only rapid but also uncontrolled. Parts of tumors often outgrow their blood supply, resulting in cell death. Tumor cells dying as a result of therapy also release nucleic acid into blood. As a consequence, ctDNA is common in cancer patients and scientists believe that like CTCs, it may be more representative of a patient’s tumor than a few thin sections from a tissue biopsy, thus reducing the heterogeneity problem. ctDNA is found in the plasma component of blood and is readily accessible in a standard blood sample. Analyzing ctDNA for mutations that are used as biomarkers for therapy selection shows great promise. One of the strengths of this approach, in addition to not requiring a tissue biopsy, is that it is not dependent on capturing rare tumor cells from blood to provide a sample for testing. The difficulty with this approach is that the cellular context is lost since the ctDNA is mixed with a much larger amount of circulating DNA from normal cells that are continuously dying and being replaced in the body, thus making analysis challenging. This requires a mutation detection methodology with enhanced sensitivity and specificity, to distinguish mutations in particular gene regions in cancer cells from the normal gene sequence present in those same genes in normal cells which co-exist in blood as normal cells die and are replaced in the body. Our CEE-Selector technology provides this necessary sensitivity and specificity and creates an opportunity for ctDNA analysis to complement CTC analysis, or potentially to serve as the platform for stand-alone tests.
Given the incidence of cancer in the United States, with an estimated 800,000 new cases in 2013 for the major solid tumors targeted by our planned test products, the markets for our current and planned cancer diagnostic tests are very large. Furthermore, these market opportunities are even greater due to the benefits of CTC and ctDNA testing, including not only the ability to offer physicians a simple way to augment an initial tumor biopsy analysis but also to provide a means for relatively frequent monitoring of the tumor’s molecular status, utilizing a standard blood sample as a “liquid biopsy.” The latter application enables the oncologist to determine if or how a tumor is changing over time or is responding to therapy and what the next treatment should be. For example, in the United States, the incidence of new cases of breast cancer alone is estimated to be over 230,000 in 2013, and the prevalence of this disease is over 2.8 million (the number of women with a history of breast cancer in the United States, including women being treated and women who have finished treatment), with an estimated 330,000 lumpectomies performed annually in the United States. Of these lumpectomies, 20% need to be repeated because on pathological examination it is shown the procedure did not result in “clean margins,” thus suggesting not all the tumor was removed, according to a Johns Hopkins report. If a CTC test were performed at the time of initial diagnosis, at the time of surgery, or in lieu of, or as an adjunct to, a PET/CT scan (as a CTC test has the potential to identify a single tumor cell in a blood sample, while a scan requires a tumor mass of millions of cells to be detectable), to monitor disease progression or test for recurrence, thousands of tests, in breast cancer alone, could be performed per year with still relatively low market penetration.
Use of CTC- and ctDNA-Derived Biomarker Data in Cancer Treatment
CTCs and ctDNA are derived from, and are understood to be representative of, a solid tumor and its metastases and can be analyzed as adjuncts to or in place of the tumor, especially when a recent tumor biopsy is not available. In theory, almost any analysis that can be performed on tumor tissue can also be performed on CTCs, while ctDNA, because it is only nucleic acid, is more limited. We have focused our analysis of CTCs and ctDNA on known biomarkers associated with specific therapies to support treatment decisions and therapy selection made by oncologists. The biomarkers we analyze and internal to analyze consist of proteins or protein
10
Table of Contents
modifications that can be identified by immunocytochemical means, cytogenetic or chromosomal aberrations, which are detected by FISH, and gene mutations which are detected in CTCs or ctDNA by molecular diagnostic tests, including CEE-Selector techniques and gene sequencing. Specific examples include (i) for immunocytochemistry, the detection of the estrogen receptor protein in breast cancer, indicative of the likely responsiveness to hormonal therapies like tamoxifen, often sold under the trade name Nolvadex®, (ii) for FISH, the presence of an amplified HER2 gene in breast cancer, indicative of the likely responsiveness to HER2-targeted agents like trastuzumab, often sold under the trade name Herceptin®, and (iii) for mutation detection, the presence of an EGFR activating mutation in NSCLC like L858R, indicative of the likely responsiveness to EGFR-targeted agents like Tarceva®. All of these biomarkers are currently tested on tumor tissue and can be tested on CTCs, and in the latter case on ctDNA. The resulting information could then be used to guide patient care, and specifically treatment selection.
To date these types of molecular and genetic detection methods have been successfully utilized to provide predictive information for several cancers, including breast, colon, NSCLC, melanoma and others in the form of companion diagnostics, typically performed on tumor tissue. CTC and ctDNA tests, which analyze the same biomarkers but in a more convenient standard blood sample test that also permits periodic monitoring, may be used in the same way.
Our Business Strategy
We plan to provide oncologists with a straightforward means to profile and characterize their patients’ tumors on a real-time basis by analyzing CTCs and ctDNA found in standard blood draws. Biomarkers are currently detected and analyzed primarily in tissue biopsy specimens. We believe that our technology, which not only provides information on CTC enumeration but also the assessment of treatment-associated biomarkers identified within the CTCs or in ctDNA, will provide information to oncologists that improves patient treatment and management and will become a key component in the standard of care for personalized cancer treatment.
Our approach is to develop and commercialize CTC and ctDNA tests and services to enable us to offer to oncologists standard blood sample based, real-time, testing solutions for a range of solid tumor types, starting with breast cancer and progressing to future launches of tests for NSCLC, gastric cancer, colorectal cancer, prostate cancer, melanoma and others, to improve patient treatment with better prognostic and predictive tools. To achieve this, we intend to:
| • | Develop and commercialize a portfolio of proprietary CTC and ctDNA tests and services, to enable physicians to develop personalized treatment plans. We intend to continue the development of additional prognostic and predictive tests and services to provide information that is essential to personalized cancer treatment. By including predictive information on biomarkers linked to specific therapies in our analysis in addition to CTC enumeration, our tests are designed to provide a more complete profile of a patient’s disease than existing CTC tests. The biomarker information will assist physicians in selecting appropriate therapies for individual patients. Our ctDNA tests are expected to offer enhanced sensitivity and specificity based on the CEE-Selector technology, enabling earlier detection of therapy-associated mutation targets or resistance markers, again supporting treatment decisions. We have launched our first CTC test, OncoCEE-BR for breast cancer, performed in our CLIA-accredited testing facility. We are also developing a number of other CTC and ctDNA tests, including OncoCEE-LU for non-small cell lung cancer, OncoCEE-CR for colorectal cancer, OncoCEE-GATM for gastric cancer, OncoCEE-PRTM for prostate cancer and OncoCEE-METM for melanoma. We plan to perform the necessary validation studies to allow us to commercialize these tests through our clinical laboratory. |
| • | Establish our internal sales and marketing capabilities in a scalable manner. We are actively seeking additional partners to increase our market reach. We intend to build our own specialized sales force with experience in cancer diagnostic testing, focusing on key identified territories in order to provide geographic coverage throughout the United States. We plan to start with 7 sales representatives, and |
11
Table of Contents
| depending on test volume, expect to increase this group to 15-20 within two years and potentially 40-50 within five years. This team will educate physicians directly on the benefits of our tests and the clinical data supporting them, as well as provide support to and serve as technical specialists for our partners such as Life Technologies. |
| • | Develop and expand our collaborations with leading university hospitals and research centers. We collaborate with key thought leaders, physicians and clinical researchers, including those at the MD Anderson Cancer Center, Columbia University and the University of California, San Diego. Our collaborations enable us to test new technologies, validate the effectiveness and utility of our planned tests in a clinical setting and provide us access to clinically well-characterized and highly annotated patient data. These samples and data accelerate our validation process and facilitate the testing and refinement of our planned new tests. |
| • | Enhance our efforts in reaching and educating oncologists about CTC and ctDNA tests. According to the American Society for Clinical Oncology, in 2011 there were approximately 10,000 oncologists in the United States, or 12,500 if gynecologic and pediatric oncologists are included. With the support of our key thought leader collaborators, we intend to focus on oncologists by targeting our sales and marketing efforts on this important customer segment. We believe this will expand and optimize the oncology testing services and personalization of cancer treatment provided by oncologists so that they can better serve their cancer patients. |
| • | Increase our efforts to provide biopharmaceutical companies and clinical research organizations with our current and planned CTC and ctDNA tests and services. Oncology drugs have the potential to be among the most personalized of therapeutics, yet oncology drugs have one of the worst approval rates, at 11% for leading indications and 2% for secondary indications of cancer drug compounds from first administration in humans to approval (2004-2011, Biotechnology Industry Organization). In an effort to improve the outcome of clinical trials for oncology drugs, and more rapidly advance targeted therapeutics, pharmaceutical and biopharmaceutical companies are increasingly looking to companies that have cancer diagnostic tests that specifically address their needs, including the ability to characterize and monitor a patient’s tumor over time using CTC and ctDNA tests to analyze biomarkers of interest. There are over 5,000 active trials in the United States in breast, lung, colorectal, prostate and gastric cancers and melanoma according to clinicaltrials.gov. We expect to increase our sales and marketing focus in this business as well as seek additional collaborations and partnerships with pharmaceutical and biopharmaceutical companies. |
| • | Support our current and planned tests with clinical utility studies to drive adoption and facilitate reimbursement. Through our agreement with the Dana-Farber Cancer Institute, we are currently conducting testing for a study that we expect to provide clinical utility data for our OncoCEE-BR test, demonstrating that patients who are treated with targeted therapies based on biomarkers identified on their CTCs, when those biomarkers are absent on their tumor tissue, have better outcomes. In this study, we are specifically identifying patients with metastatic breast cancer that are HER2 negative, by analysis of their tumor tissue, and who have HER2 positive CTCs utilizing our OncoCEE-BR test on a standard blood sample. These patients are being randomized for treatment with chemotherapy, the current standard of care, with or without Herceptin®, and then evaluated for progression-free survival and overall survival. We intend to conduct additional studies in breast cancer, and similar studies for our NSCLC test and other CTC and ctDNA tests we plan to introduce. Clinical utility and validation studies for our planned ctDNA tests may rely on archived plasma or blood samples from clinical trials in which patient outcomes are already available, in a retrospective-prospective design that significantly shortens the length of such studies. |
| • | Continue to enhance our current and planned CTC and ctDNA tests and reduce the costs associated with providing them through internal research and development and partnering with leading technology developers and reagent suppliers. We intend to work closely with select key technology developers and suppliers to further automate the optical interpretation of our current breast cancer test and our planned additional CTC tests, including enumeration, immunocytochemical biomarker staining |
12
Table of Contents
| and FISH. We also intend to reduce the costs associated with key material components of these tests, including FISH probes. We have identified a technology group that, based on initial studies, can provide an automation system that will significantly reduce the hands-on time of our cytotechnicians for microfluidic channel analysis while increasing the uniformity, and potentially the sensitivity and quality, of the data we generate. This system is also expected to provide the ability to evaluate multiple fluorescent signals of different wavelengths simultaneously for multiplexed analysis, again enhancing efficiency. Similarly, we have identified suppliers that can provide FISH probes at reduced cost and with a broader choice of available fluors, enabling more extensive multiplexing of tests. |
Our Competitive Advantages
We believe that the competitive advantages of our tests, including our tests which are still under development, would include the following. In general, because OncoCEE-BR and our planned tests share our CEE platform, their competitive advantages would be the same.
OncoCEE-BR enables, and we anticipate our planned CTC and ctDNA tests will enable, detailed analysis of a patient’s cancer utilizing a standard blood sample, facilitating testing at any time, including when a biopsy is not available or inconclusive, offering real-time monitoring of the cancer and the response of the cancer to therapy, and allowing oncologists to select timely modifications to treatment regimens. Because CTCs and ctDNA are derived from the primary tumor or its metastases, they function as surrogates for the tumor, with the advantage of being readily accessible in a standard blood sample. This is especially important in situations where a biopsy is not available or advised. The simplicity of obtaining a standard blood sample permits repeat testing in a monitoring mode to detect recurrence or progression and to offer information on treatment modifications based on a current assessment of the cancer’s properties.
OncoCEE-BR provides, and we anticipate our planned tests will provide, more information than competitors’ existing tests, including predictive information on biomarkers linked to specific therapies. We anticipate that such additional biomarker information will enable a physician to develop a personalized treatment plan. By including biomarker information in our analysis, in addition to CTC enumeration, our current OncoCEE-BR test and our planned tests are designed to provide a more complete profile of a patient’s disease than existing CTC tests. We intend for our tests to contain actionable information to assist physicians in selecting appropriate therapies for individual patients. Our ctDNA tests are expected to offer enhanced sensitivity and specificity based on the CEE-Selector technology, enabling earlier detection of therapy-associated mutation targets or resistance markers, again supporting treatment decisions.
OncoCEE-BR and our planned CTC tests are designed to capture and detect a broader range of CTCs than existing tests and to be applicable to, or quickly modifiable for, a wide range of cancer types. Our CEE-Cap antibody capture cocktail includes antibodies targeting not only EpCAM, the traditional epithelial CTC capture antigen utilized in the CellSearch® system and in other platforms, but also other epithelial antigens as well as mesenchymal and cancer stem cell antigens, indicative of cells having undergone the epithelial-to-mesenchymal transition. These cells may be more relevant for metastasis. Our detection methods include cytokeratin staining with a broader range of cytokeratin isotypes than existing CTC tests, and we plan to introduce our CEE-Enhanced staining which would enable detection of cells specifically captured with our antibody cocktail, including EMT cells lacking cytokeratin. We believe that through our planned CEE-Enhanced staining, more CTCs and different types of CTCs will be able to be identified and potentially at earlier stages of disease, resulting in fewer non-informative cases and more information for physicians.
OncoCEE-BR is, and we anticipate our planned CTC and ctDNA tests will be, flexible and readily configurable to accommodate new biomarkers with clinical relevance as they are identified. In theory, our CEE platform permits essentially any analysis that is currently performed on tumor tissue to be performed on CTCs, including immunocytochemical staining, FISH and molecular analysis. As new therapies are approved, and to the extent that they are targeted therapies for which knowledge of a particular gene amplification event, mutation or
13
Table of Contents
presence, absence or modification, such as phosphorylation, of a protein are indicative of likely response or resistance to that therapy, we will be able to include them in our tests with minimal changes. This is attractive to pharmaceutical and biotechnology companies that are developing such therapies, or seeking ways to make their clinical trials more efficient, as this flexibility would enable them to focus on patients more likely to respond to a particular therapy and demonstrate a benefit from that therapy.
Collaborative relationships with physicians at MD Anderson Cancer Center. We have worked closely with a number of physicians at the MD Anderson Cancer Center in Houston, Texas, on various collaborative projects in different cancer types including breast, NSCLC, prostate, colorectal, ovarian, bladder, renal and endometrial. These projects provide us access to leading researchers, clinicians and key thought leaders, access to valuable patient samples and insight into clinical applications for our tests. Some of these projects have resulted in publications in leading journals, such as Cancer Discovery and Cancer Medicine, which enhances our standing in the oncology community and supports our marketing efforts.
Our planned CEE-Selector mutation tests would not be platform dependent. These tests are being designed to be able to be performed on almost any molecular instrument, which will provide flexibility in laboratory operations. To the extent we elect to develop these tests as IVDs, including pursuing CE marks for them to be marketed outside the United States, the ability to rapidly deploy them on different approved instrument platforms already in many laboratories should greatly simplify their distribution and commercialization.
Our Tests and Services
We have launched our first product, OncoCEE-BR for breast cancer, and plan to continue to launch a series of tests for CTCs in different tumor types, including NSCLC, gastric, colorectal and prostate cancers and melanoma, incorporating analyses for different biomarkers, over the next 3 years. OncoCEE-BR is and the planned tests will be based on the CEE technology platform. The CEE system isolates CTCs from blood samples of cancer patients for enumeration (or count) and genetic analysis. A sample is shipped to us in our specialized blood collection tube, called the CEE-Sure tube, for recovery and analysis of CTCs. When performing the CTC assay, the sample is processed in our laboratory. The specimen of blood is separated into its parts (red blood cells, buffy coat and plasma). The buffy coat is incubated with the antibody solution and passed through a proprietary microfluidic channel containing 9,000 microscopic posts coated with reagents to capture antibody-labeled tumor cells. The captured cells are suitable for further testing of whole cells directly in the microfluidic channel or by releasing the cells from the microfluidic channel and performing CEE-Selector or similar techniques.
Clinicians acknowledge limitations of currently available CTC test systems such as CellSearch® that rely on capture solely by anti-EpCAM antibodies and detection by anti-cytokeratin antibodies. Capture and detection based only on these two antigens is unlikely to identify all CTCs, and clinically this may result in no CTCs being detected in cases in which they are present. For example, some tumor cells that have been released into the circulatory system have undergone an EMT. These mesenchymal cells are less differentiated than epithelial cells and more similar to stem cells. OncoCEE-BR enables, and we believe our planned assays will enable, the capture of significantly more CTCs than is accomplished through the use of traditional anti-EpCAM immuno-capture alone.
In addition to enhanced capture, our technology also improves the detection of CTCs. As with EpCAM, tumor cells that have undergone EMT can down-regulate the synthesis of cytokeratin, leading to an underestimate or even an apparent absence of CTCs since their positive identification has traditionally relied on anti-cytokeratin staining. We have developed alternative methods of fluorescent cell staining that are uniquely possible within the CEE system to enhance or enable detection of CTCs with low or no cytokeratin signal. This technology is called CEE-Enhanced. We believe that the combination of specific cocktails of tumor-associated capture antibodies and more sensitive fluorescent detection of CTCs through CEE-Enhanced methodology will lead to major advances in the capture, enumeration and analysis of CTCs. CEE-Enhanced methodology is expected to be included in our commercially available tests by mid-2014.
14
Table of Contents
Analysis of CTCs performed by us incorporates both standard and proprietary methods. Immunocytochemistry which looks at proteins, analogous to the immunohistochemistry performed on tissues, can be readily applied and performed in the microfluidic channel, dependent only on suitable biomarkers. Similarly, FISH, used to evaluate cytogenetic abnormalities in cells, may be performed in our microfluidic channel using validated assays available from a number of vendors. For genetic mutation analysis, standard technologies can be applied. We have also developed proprietary CEE-Selector technology for mutation analysis in CTCs and ctDNA, with enhanced sensitivity and specificity.
CTCs are generally very rare and outnumbered many-fold by white blood cells. This complexity has been a challenge for standard technologies. We believe our CEE-Selector technology will offer enhanced specificity and sensitivity (greater than 1-in-10,000 of mutated sequence to normal sequence in a complex genetic background) compared to other approaches, and that it will potentially have broader application than just CTC analysis, including analysis of ctDNA in plasma, both in a CLIA-certified laboratory setting and as an IVD.
OncoCEE-BR is, and our planned tests would be, Laboratory Developed Tests. FDA clearance or approval is not currently required to offer these types of tests in our laboratory once they have been clinically and analytically validated. We seek licenses and approvals for our laboratory facility and for LDTs from the appropriate regulatory authorities, such as the Centers for Medicare & Medicaid Services, which oversees CLIA, and various state regulatory bodies. Certain states, such as New York, require us to obtain state licensure in order for us to perform testing on specimens taken from patients or received from ordering physicians from those states. As part of this process, the State of New York requires validation of our tests. We are currently in the process of addressing the requirements for licensure in Florida and New York, and have re-obtained all required licenses and approvals from all other states requiring licensure of out-of-state laboratories. We were required to re-license in certain states as a result of our July 2013 reincorporation to Delaware.
The following outline indicates our current (OncoCEE-BR) and planned tests and indicates the stage the product is in and the targeted date of commercialization. As discussed in “Description of the Business—Test Development Process” below, prospective assays initially begin in research (stage 1) and progress through to development (stage 2), validation (stage 3) and finally availability for commercialization (stage 4). The OncoCEE-BR test has completed all stages as to CTC and HER2 test capabilities. Our remaining identified proposed tests have completed the research stage and are at the stages shown in the table below with their respective estimated timetables for completing stage 4. As with all scientific endeavors, such timetables are only estimates; unanticipated problems might result in delays. We consider these timetables to be fairly aggressive, given the likelihood of our experiencing such unanticipated problems and associated delays.
In the development stage, there is still work to be done to finalize sensitivity and specificity of the assay. This work will vary as the assay is tested and fine-tuned in order to prepare it for validation and eventual commercial offering. In the validation stage, the assay has been fully developed and we are now able to run (or are in the process of running) a specific number of samples, both positive and negative, in order to validate that the assay results are reproducible. A validated assay is considered to have completed the availability for commercialization stage when the necessary training has been given and any necessary governmental licenses and approvals have been obtained so that we can start selling the assay through our commercial sales channel and provide patient results.
Our proposed tests have certain commonalities. For example, in each proposed test, biomarkers will be examined by one or both of FISH or CEE-Selector. Given the development, validation and commercialization of our first CTC/FISH test (OncoCEE-BR), all subsequent FISH- and Immunofluorescence-based assays have effectively been developed for the planned biomarker. Progression of these planned assays through stage 3 is largely dependent on the timing of our obtaining suitable validation specimens, although various scientific and other factors can also affect the pace of a particular proposed test’s progress through the validation stage. Thus, the OncoCEE-LU (i.e., CTC/FISH-based OncoCEE-LU), OncoCEE-GA and OncoCEE-DTC tests are targeted to be commercial in 2014. CTC-based OncoCEE-CR and OncoCEE-PR tests are targeted to be commercial in 2015 given our estimate of the timing to acquire appropriate positive and negative validation samples.
15
Table of Contents
For ctDNA based assays, CEE-Selector will be used to detect each relevant mutation, and our current estimate is that development will be completed in 2014. Biomarker mutations (such as B-raf and K-ras) are often commonly seen in different tumor types, thus, once a particular mutation assay is developed for CEE-Selector, it can be applied to any tumor type. The OncoCEE-LU ctDNA test is anticipated to be our first CEE-Selector test to undergo validation. Given the nature of a molecular based test such as CEE-Selector, specimens can be batched and tested simultaneously, thereby reducing the validation time. We are targeting the OncoCEE-LU ctDNA test to be commercial by the end of 2014. All remaining currently proposed ctDNA tests would then follow and are currently targeted to be commercial in 2015.
We currently intend to use approximately $5 million of the net proceeds of our initial public offering to fund further research and development and related activities. This includes all of the expenditures which we believe are needed to complete all four stages of development for the planned tests described below. Primarily these expenditures will be for existing and additional scientific personnel in the time periods reflected in the table below, and secondarily for obtaining a sufficient number of suitable validation specimens.
| Test Name |
Solid Tumor Type and Biomarkers |
Indication |
Status of Test or Project |
Targeted Year of Availability for | ||||
| OncoCEE-BR™ |
Breast Cancer- Enumeration; HER2 by FISH, ER, PR |
Prognosis, therapy selector, monitoring | CTC and HER2 already on the market; Validation – ER, PR | 2012 for CTC and HER2; ER Q2 2014, PR Q4 2014 | ||||
| OncoCEE-LU™ |
Lung Cancer- Enumeration; ALK and ROS1 by FISH, |
Prognosis, therapy selector, monitoring | Validation – CTC and FISH portion of assay; | 2014 /Q3 | ||||
| K-ras, B-raf and EGFR mutations by CEE- Selector™ |
Development – ctDNA portion of assay | 2014 Q4 | ||||||
| OncoCEE-GA™ |
Gastric Cancer- Enumeration; HER2 by FISH |
Prognosis, therapy selector, monitoring | Validation of CTC and FISH assay | 2014 Q2 | ||||
| OncoCEE-CR™ |
Colorectal Cancer- Enumeration; EGFR by FISH |
Prognosis, therapy selector, monitoring | Validation – CTC and FISH portion of assay | 2015 Q1 | ||||
| K-ras and B-raf by CEE-Selector™ | Development – ctDNA portion of assay | 2015 Q2 | ||||||
| OncoCEE-PR™ |
Prostate Cancer- Enumeration; PTEN deletion and AR by FISH |
Prognosis, therapy selector, monitoring | Validation – CTC and FISH assay | 2015 Q3 | ||||
| OncoCEE-ME™ |
Melanoma-Enumeration and B-raf and N-ras mutations by CEE- Selector™ |
Prognosis, therapy selector, monitoring | Development of ctDNA assays | 2015 Q2 | ||||
| OncoCEE-DTC™ |
Breast and Prostate Cancer-DTC analysis in bone marrow; HER2 and AR/PTEN by FISH, respectively |
Prognosis, therapy selector, monitoring | Validation of DTC and FISH assays | 2014 Q4 Breast 2015 Q3 Prostate | ||||
| CEE-Selector™ |
Multiple cancer types-K- ras,B-raf, EGFR and other mutations detected in plasma |
Therapy selector, monitoring | Development | 2014 Q4 | ||||
16
Table of Contents
Our Marketed OncoCEE CTC Test: OncoCEE-BR
Our OncoCEE-BR breast cancer test is the first CTC test we developed and we are currently offering it to physicians through our CLIA laboratory. It is based on a standard blood sample and can be used at the time of diagnosis and for monitoring, including at the time of progression or recurrence. This allows the physician to characterize the tumor to help define treatment options, either augmenting tissue analysis or replacing it when a tumor biopsy is not available. The test currently includes CTC enumeration and determination of HER2 status by FISH on the captured CTCs, and then more broadly to any cell captured on our CEE microfluidic channels that is not a white blood cell. HER2 status is used by oncologists to determine suitability of a patient for treatment with HER2-targeted therapeutics, which include Herceptin®, as well as Kadcyla® and Perjeta®, monoclonal antibodies directed to HER2, and Tykerb®, a kinase inhibitor with activity against HER2. We plan to add immunocytochemistry analysis of CTCs for estrogen receptor and progesterone receptor to our OncoCEE-BR test, which will provide information on suitability of breast cancer patients for endocrine or hormonal therapies such as selective estrogen receptor modulators, including tamoxifen, aromatase inhibitors that block the synthesis of estrogen, including Femara® (Novartis) and Arimidex® (AstraZeneca) or other therapeutics that block estrogen production, including Zoladex® (AstraZeneca) and Lupron® (AbbVie).
Other OncoCEE CTC Tests in Development
We are now following a similar development path for additional OncoCEE CTC tests for cancer types other than breast cancer, with a focus on large population solid tumor types, or cancers for which there are approved therapies that rely on biomarker tests we have previously developed. Examples of these tests include OncoCEE-LU for lung cancer, OncoCEE-GA for gastric cancer, OncoCEE-CRTM for colorectal cancer, OncoCEE-PR for prostate cancer, and OncoCEE-ME for melanoma, each described below.
OncoCEE-LU
Up to 25% of lung cancer patients, especially those diagnosed at Stage IIIB or Stage IV, are not treated surgically for various reasons, including tumor accessibility and status of the patient. In these cases, CTC and ctDNA tests are alternatives for obtaining more detailed information about the molecular status of the tumor that helps the physician select appropriate therapy. This is even more important as the number of targeted therapies for lung cancer with associated biomarkers increases. Our OncoCEE-LU test would include several components: CTC enumeration, FISH analysis for EML4/ALK and ROS1, and potentially for ret proto-oncogene, all linked to the drug Xalkori® (Pfizer), mutation analysis for the EGFR gene, the K-ras gene and the B-raf gene. The L858R mutation of the EGFR gene and Exon 19 deletions are activators of EGFR kinase activity and are linked to the drugs Tarceva® (Astellas/Genentech/Roche) and Iressa® (AstraZeneca). The T790M mutation of the EGFR gene is a resistance marker for EGFR tyrosine kinase inhibitors and is linked to drugs in development that address this resistance, such as Gilotrif® (Boehringer-Ingelheim) and dacomitinib (Pfizer). The codon 12 and 13 mutations of the K-ras gene are linked to non-responsiveness to the EGFR kinase inhibitors such as Tarceva® and Iressa®, and the codon 600 mutations of the B-raf gene are linked to Zelboraf® and Tafinlar®, which are both approved for melanoma and are in clinical trials for lung cancer. Our OncoCEE-LU test would be performed on a standard blood sample.
In parallel, we plan to offer ctDNA tests for mutation analysis of, for example, EGFR, K-ras and B-raf genes, to provide information in situations where CTCs are not identified. In our development of this technology platform we have generated data showing detection of the T790M mutation in ctDNA from the blood plasma of lung cancer patients progressing on tyrosine kinase inhibitors in which no CTCs were detected.
OncoCEE-GA
We are developing our OncoCEE-GA test for gastric cancer based on the identification of HER2 as a biomarker for this disease. We plan to employ our CTC HER2 FISH test, which we had previously developed for breast cancer, for the analysis of gastric cancer CTCs. The presence of HER2 positive cells is an indication for
17
Table of Contents
likely benefit from the use of Herceptin®, which has been approved for the treatment of metastatic gastric cancer. Current clinical practice relies on a biopsy for tumor tissue analysis to detect elevated HER2, in the same manner as is done for breast cancer. Our test would circumvent this need for tissue, as well as providing straightforward monitoring of HER2 status from a standard blood sample, on a real-time basis during treatment. Our OncoCEE-GA test would include CTC enumeration and HER2 analysis of CTCs by FISH.
OncoCEE-CR
Our current plan for our OncoCEE-CR test for colorectal cancer is to offer mutation testing analogous to that performed on lung cancer CTCs, namely detection of key mutations in the EGFR, K-ras and B-raf genes, along with CTC enumeration. Testing of the EGFR gene would focus on the L858R mutation and Exon 19 deletions as activators of EGFR kinase activity, and the T790M mutation as a resistance marker for certain EGFR tyrosine kinase inhibitors. Testing on the K-ras gene would focus on codons 12 and 13 mutations. Testing on the B-raf gene would focus on V600 mutations. Our OncoCEE-CR test would be run against a standard blood sample.
This testing is important because certain targeted therapies for colorectal cancer, including the monoclonal antibodies targeting EGFR, Erbitux® (Lilly/Bristol-Myers Squibb/Merck Serono) and Vectibix® (Amgen), and the kinase inhibitor Stivarga® (Onyx/Bayer) targeting vascular endothelial growth factor receptor kinases, but also ret proto-oncogene, KIT, platelet-derived growth factor receptor, or PDGF-R, and fibroblast growth factor receptor kinases, have been shown to be ineffective in patients who have a K-ras mutation, which is found in up to 40% of cases according to the National Comprehensive Cancer Network. While for each of codons 12 and 13 in K-ras, up to 15-20 mutations have been reported, there are reports in the scientific literature that patients with one particular mutation, G13D, do respond well to Erbitux®, and that there may be variability in response to different chemotherapies based on the specific K-ras mutation, suggesting that detailed information on mutation status is clinically relevant.
OncoCEE-PR
Our OncoCEE-PR test for prostate cancer would be based on the analysis of CTCs found in a standard blood sample by FISH for key biomarkers: the androgen receptor, and phosphatase and tensin homolog (PTEN). The test would also include CTC enumeration, and our CEE-Cap antibody capture cocktail would be modified from that used for breast and lung cancer to include prostate specific membrane antigen.
The androgen receptor normally binds the hormones testosterone and dihydrotestosterone, and is the target for several drug molecules, including those acting directly as antagonists for the receptor, such as Casodex® (AstraZeneca), and those acting indirectly through inhibition of androgen synthesis, such as Zytiga® (Janssen).
Phosphatase and tensin homolog, an enzyme that functions as a tumor suppressor, if mutated, deleted or otherwise functionally disrupted, removes a brake from cell replication and allows uncontrolled growth, which is seen in many cancers. If phosphatase and tensin homolog is mutated, deleted or disrupted, chemotherapy or polytherapy is usually recommended.
OncoCEE-ME
Our OncoCEE-ME melanoma test, performed on a standard blood sample, would provide information on the presence or absence and specific nature of the V600 mutation in the B-raf gene, which indicates whether the B-raf inhibitors Xelboraf® or Tafinlar® are candidate therapies for the patient. CTC enumeration would also be a component of our test.
Disseminated Tumor Cell (DTC) Assays Performed on Bone Marrow
We have shown that our CEE-Sure blood collection tubes and CEE microfluidic channels work well with bone marrow samples, and we have further demonstrated the ability to perform FISH on disseminated tumor cells, or DTCs, from bone marrow that are isolated in this way. While bone marrow biopsies are not performed
18
Table of Contents
routinely in the United States, they are utilized in Europe, especially in prostate cancer. In addition, we were involved in a study at MD Anderson Cancer Center in which bone marrow was isolated from early stage operable breast cancer patients at the time of surgery. In this later study, published in Cancer Medicine (2013, 2(2) 226-233), we found a significant percentage of patients classified as HER2 negative by their primary tumor had HER2 positive DTCs, and hence could be considered for Herceptin® therapy. DTCs provide an interesting adjunct to CTC analysis that is well suited for our technology platform, and we plan to work with collaborators and key thought leaders to determine how best to introduce a series of tests based on a bone marrow sample type.
ctDNA Tests
We plan to introduce ctDNA tests for mutation analysis performed on blood plasma isolated from a standard blood sample using the CEE-Selector technology, based on increasing interest in the research community in this type of analysis. We plan to launch the first tests, for K-ras, B-raf and EGFR mutations, in conjunction with, or as a complement to, our OncoCEE-LU test. Tests for other mutations will be added as they are developed. These tests would be similar to those performed on CTCs but would instead focus on ctDNA in plasma. These tests would lack the cellular context provided by CTCs but would not require CTC isolation and would be simpler to perform. In addition, one of the benefits of this technology is its ability to detect and identify mutations in blood plasma from cancer patients in whom we were not able to isolate CTCs. This indicates the importance of the enhanced sensitivity of the CEE-Selector technology and the ability of ctDNA tests to complement CTC tests.
Laboratory Testing
From our CLIA-certified laboratory in San Diego, California, we plan to provide test results from our current and planned CTC and ctDNA tests to oncologists in community hospitals, cancer centers, group practices and offices. At the federal level, clinical laboratories, such as ours, must be certified under CLIA in order for us to perform testing on human specimens. Our laboratory is also accredited by CAP, which is one of six accreditation organizations approved by CMS under CLIA. Our clinical laboratory is located in California and we hold the requisite license from the California Department of Public Health to operate our laboratory. In addition, Florida, Maryland, New York and Rhode Island require that we hold licenses to test specimens from patients in those states or received from ordering physicians from those states. As part of this process, the State of New York requires validation of our tests. Pennsylvania licensure or registration may be required as well, depending on the circumstances. We are currently in the process of addressing the requirements for licensure in Florida and New York, and have re-obtained all required licenses and approvals from all other states requiring licensure of out-of-state laboratories. We were required to re-license in certain states as a result of our July 2013 reincorporation to Delaware.
Clinical Trials Services
Industry research has shown many promising drugs have produced disappointing results in clinical trials. For example, a study by Princess Margaret Hospital in Toronto estimated that over a five-year study period 85% of the new therapies for solid tumors which were tested in early clinical trials in the United States, Europe and Japan failed, and that of those that survive through to Phase III trials only half will actually be approved. Given such a high failure rate of oncology drugs in clinical development, combined with constrained budgets for pharmaceutical and biopharmaceutical companies, there is a significant need for drug developers to utilize molecular diagnostics to help decrease these failure rates. For specific molecular-targeted therapeutics, the identification of appropriate biomarkers may help to optimize clinical trial patient selection and success rates by helping clinicians identify patients that are most likely to benefit from a therapy based on their individual genetic profile.
In addition to testing for oncologists and their patients, we plan to offer clinical trials testing services to help increase the efficiency and economic viability of clinical trials for pharmaceutical and biopharmaceutical companies and clinical research organizations. Our clinical trial services will be aimed at developing customizable tests and techniques utilizing CTC and ctDNA technologies to provide sensitive, real-time
19
Table of Contents
characterization of individual patient’s tumors using a standard blood sample. These tests may be useful as, and ultimately developed into, companion diagnostics associated with a specific therapeutic. Additionally, through our services we may gain further insights into biomarkers for disease progression and drug resistance, as well as those associated with current drug development efforts, which we can incorporate into tests.
Test Development Process
Our OncoCEE-BR test was, and our planned additional CTC and ctDNA tests are being, developed and validated in conjunction with leading academic and clinical research centers to ensure that the needs of the clinical community are being met with the latest research on key biomarkers that affect patient care. We utilize a research and validation process to help ensure that we are providing diagnostic, prognostic and predictive information that is clinically relevant and accurate. The time-frame for this process from design through development and market launch is dependent upon, among other things, the biomarkers in question having been discovered and validated before we incorporate them in a test, the specific clinical claims we plan to pursue, and the availability of high quality samples for validation. Our development protocol calls for us to monitor and review the process in four stages as detailed below:
| • | Stage 1, Research. We review known, validated biomarkers, preferably linked to a specific therapeutic or other high value treatment decision, and discuss with clinical collaborators and key thought leaders to characterize the opportunity, the specific clinical setting and the product profile of the candidate test. |
| • | Stage 2, Test Development. We design the test, which typically has two parts: efficient capture of CTCs and/or ctDNA from the targeted cancer type and development of the biomarker assays that will be included. For example, the first part may involve modification of the antibody capture cocktail and the second could include development of specific CEE-Selector mutation tests or testing of FISH probes. The test will be used on normal control specimens and clinical samples to assure performance and the process includes defining the performance characteristics of the test as well as developing standard protocols for our CLIA-certified laboratory, where the test will ultimately be performed. This assessment includes such features as reproducibility, accuracy, sensitivity, and specificity. |
| • | Stage 3, Clinical Validation. When the assay is performing as desired in the research laboratory, it is then transferred to the CLIA laboratory and validated on clinical samples, typically in comparison to the existing gold standard for that biomarker, which is usually tumor tissue analysis. Depending on the tumor type and specimen requirement, samples are collected from patients through collaborators, or in the case of ctDNA tests, from sample banks, where clinical information on the patients, including outcomes, is already available. |
| • | Stage 4, Availability for Commercialization. As clinical validation is completed and before launch, we take several steps to prepare a test for marketing as a LDT. We create standard operating procedures and quality assurance and quality control measures to ensure repeatability and high standards of quality. We train both our commercial and laboratory staff on the interpretation and use of the data. Licenses and approvals for our laboratory to perform or use LDTs are obtained from the appropriate regulatory authorities, such as CMS, which oversees CLIA, and different state regulatory bodies. |
Our CTC/FISH—based OncoCEE-BR test, which has already launched, is considered to have completed this test development process. All other planned tests which are mentioned in this Annual Report are all considered to currently be in Stage 2 or Stage 3 of this test development process.
As part of our long-term strategy, we may seek FDA clearance or approval to expand the commercial use of tests to other laboratories and testing sites in the United States. We will also need to complete additional activities to submit each of these tests for regulatory clearance or approval before commercialization in each of the international markets where we would plan to introduce them.
20
Table of Contents
Although the FDA maintains that it has authority to regulate the development and use of LDTs as medical devices, as a matter of enforcement discretion it has not exercised such authority with respect to most LDTs. If the FDA exercises this authority as to our current test or as to a planned test, our process would also need to allow for obtaining FDA review, clearance or approval, as applicable, which would add delay, expense and risk to our current test development process. Such an exercise of authority could arise as a result of changes in discretion on a general or particular basis, changes in applicable regulations, or changes in applicable statutes.
Research and Development
We incurred research and development expenses of $6.6 million, which represents 6010% of our net revenue, for the year ended December 31, 2012 and $3.1 million, which represents 2299% of our net revenue, for the year ended December 31, 2013. Research and development expenses represented 62% of our total operating expenses for the year ended December 31, 2012 and 38% of our total operating expenses for the year ended December 31, 2013. Major components of the research and development expenses were direct personnel costs, laboratory equipment and consumables and overhead expenses.
Technology Development
In addition to developing new CTC and ctDNA tests for different cancers to be offered through our CLIA testing laboratory, and adapting additional predictive biomarkers to these tests as their importance is demonstrated by the scientific and clinical research communities, we continue to focus on improving the base technologies underlying our tests and processes. We are exploring various ways to improve CTC capture efficiency and detection, as well as approaches to sub-categorize CTCs into different populations that may have clinical relevance. For example, by determining which antigens individual CTCs expressed that enabled their capture, we could differentiate, and enumerate, various CTC phenotypes, for example, epithelial versus mesenchymal. We are also working to simplify the test process, and in general to provide a broader range of useful data on a patient’s cancer to assist the oncologist in determining an appropriate treatment. Some of these projects and initiatives include:
| • | Improve Ability to Capture CTCs |
| • | Continued modification and optimization of our CEE microfluidic channel as a way to further enhance CTC capture efficiency. Capture efficiency directly impacts sensitivity, informative rate, and the ability to perform accurate and reliable biomarker analyses on the CTCs, all of which increase the value of our offering. We are utilizing some of our early research experience to improve CTC capture rates and reduce background contamination from normal white blood cells. |
| • | Automation of Our Test Process |
| • | Development of automation throughout the test process, but particularly at the visual evaluation steps, which include enumeration, any immunocytochemistry for biomarkers beyond those used to identify CTCs, for example protein biomarkers, and FISH analysis, is a way to drive efficiencies, reduce costs, speed up turnaround time, and generate more reliable, uniform, and in some cases more sensitive data. We have identified an automation solution for the visual analysis, which needs to be optimized and then transferred to and validated in our CLIA laboratory. We have also adapted a semi-automated system for the separation, processing and washing steps before running a sample on the microfluidic channel, which is now being used in the research laboratory and similarly needs to be transferred and validated in the CLIA laboratory. These measures will reduce costs and time as well as allow for higher-throughput as sample volumes increase. |
| • | Development of Second Generation Platform for CTC Testing |
| • | Evaluating and developing techniques for CTC capture that take advantage of our CEE-Cap antibody capture cocktail and CEE-Enhanced staining technology to modify our current CTC process to a simpler, essentially IVD, format. In addition to reducing internal costs, such an |
21
Table of Contents
| advance would offer the opportunity for us to offer a product format that enable us to access the worldwide CTC testing market. The distribution of such kits could create a new business opportunity for us. |
| • | Utilization of CEE-Selector Technology for Highly Multiplexed Mutation Testing |
| • | The CEE-Selector technology should enable us to multiplex mutation testing such that larger panels of genes can be analyzed in a single step. This should position us for the analysis at the molecular level of whole signaling pathways or enzyme cascades. We plan to take advantage of the sensitivity and specificity of the CEE-Selector technology and leverage interest in the clinical research community for detecting any actionable biomarker in a particular tumor, as opposed to only those that are known to occur at relatively higher frequencies in that type of tumor. Such multiplexed mutation tests, relying on our CEE-Selector technology, could provide a more global evaluation of a tumor through analysis of either CTCs or ctDNA. This would offer a broader range of potential treatment options as well as enable the monitoring of the effectiveness of those treatments over time. |
| • | Development of Single Cell CTC Isolation Techniques for Molecular Analysis |
| • | Tumor heterogeneity is a well-recognized problem for tissue analysis and is in part addressed by focusing on CTCs, which may provide a more universal sampling of a tumor. One result of this can be a diverse population of CTCs in a sample, with different phenotypes and genotypes represented. We are working with a collaborator on techniques for subsequent sorting of our highly enriched CTC samples released from our CEE microfluidic channels into pools of CTCs with similar phenotypes, and ultimately to single CTCs, for molecular analysis. |
Translational/Clinical Research
In the course of our research and validation studies, we have processed several hundred cancer patient samples and normal control samples for CTC enumeration and analysis. Our initial focus has been on breast cancer, where validation studies for the OncoCEE-BR test, including enumeration of CTCs compared to the CellSearch® system, and HER2 FISH performed on CTCs and compared with HER2 analysis performed on tumor tissue from the same patients, involved over 120 patient samples. The results of our validation studies, and the demonstration of a reliable and reproducible method for CTC capture and analysis using the OncoCEE platform were published in a paper entitled “Novel Platform for the Detection of Cytokeratin Positive (CK+) and Cytokeratin Negative (CK+) CTCs” appearing in the December 2011 issue of Cancer Discovery and a paper entitled “Efficient capture of circulating tumor cells with a novel immunocytochemical microfluidic device” appearing in the September 2011 issue of BioMicrofluidics.
Additional studies were conducted in breast and other tumor types, including lung, prostate and colorectal cancers, utilizing patient samples for comparison to the CellSearch® system. In head-to-head studies, the CEE system detected cytokeratin positive CTCs in comparable numbers of breast cancer patients, and in considerably more patients in the other cancer types (Cancer Discovery, December 2011). Moreover, the results clearly demonstrated that our use of the CEE-Cap capture antibody cocktail enabled recovery of more CTCs as compared to using only anti-EpCAM antibodies. This data served as a clinical validation study for CTC enumeration. When CEE-Enhanced staining is applied to detect cytokeratin-negative CTCs, we expect to see far more CTCs based on preliminary studies reported in a paper entitled “Detection of EpCAM-Negative and Cytokeratin-Negative CTCs in Peripheral Blood” appearing in the 2011 issue of the Journal of Oncology.
The CEE system has the added advantage of post-capture immunocytochemical, cytogenetic and molecular genomic analyses of the CTCs. The CEE system captured cells can be analyzed directly within the microfluidic channel, thereby removing the need to re-deposit cells on a slide, which could result in cell loss or damage. Furthermore, given the transparency of the microfluidic channel, it can be immediately analyzed on a microscope. Together these two important features allow for a very efficient process that is well suited for a LDT
22
Table of Contents
performed in a CLIA laboratory. The post-capture analyses, which focus on the evaluation of biomarkers, are particularly important and valuable to physicians and patients, as they focus on actionable information related to therapy selection. We have performed a number of clinical research studies in collaboration with MD Anderson Cancer Center investigators involving various tumor types, including breast, ovarian, endometrial, lung, colorectal, bladder and prostate cancers.
In a collaboration with physicians and researchers at MD Anderson Cancer Center, we evaluated matched samples of tumor tissue, blood for CTCs and bone marrow for DTCs in early stage breast cancer patients for evidence of HER2 amplification, which would indicate eligibility for HER2-targeted therapies like Herceptin®, a potentially life-saving treatment. These results were also presented at both the 2011 and 2012 annual meetings of the American Society of Clinical Oncology. In a study published in Cancer Medicine (2013, 2(2) 226-233) and involving 96 patients, HER2 positive CTCs and/or DTCs were identified in 18.8% of cases in which the primary tumor was HER2 negative. In the same cohort of patients, only 12.5% were HER2 positive in their primary tumor. In other words, beyond the 12 (of the 96) which traditional tumor tissue analysis had indicated could benefit from Herceptin-based therapy, the OncoCEE-BR test detected 18 (of the 96) patients who (despite the fact they were identified as being HER2 negative by primary-tumor testing) could benefit from Herceptin-based therapy. Patients classified as HER2 negative based on tumor tissue and found to have HER2 positive CTCs and/or DTCs will continue to be followed by our collaborators at MD Anderson Cancer Center to assess their overall and progression-free survival. Tumor heterogeneity is one likely cause of the discordance for HER2 status between tumor tissue and our test performed on blood and bone marrow samples. Tumor heterogeneity indicates an important clinical application for the OncoCEE-BR test, confirmation and crosschecking of the tissue analysis performed by the pathologist at the time of biopsy or surgery, especially if HER2 negative, with a CTC analysis derived from a standard blood sample.
Clinical utility studies, which demonstrate the specific clinical setting in which a particular CTC or ctDNA test is used, and how to use the information generated for medical, specifically treatment-related, decision making is a key part of our strategy and research and development plan. Data resulting from such studies is critical not only in the sales and marketing process, but also for reimbursement, as many payors now ask for peer-reviewed publications describing such studies and results before agreeing to coverage of a specific test. The study with Dana-Farber Cancer Institute is the first example of a clinical utility study for one of our tests and we plan to conduct additional studies in breast cancer and similar studies in NSCLC and other cancers for which we develop tests, including sponsoring such studies ourselves with some of the proceeds from our initial public offering.
Sales and Marketing
Our sales and marketing efforts consist of working with our partners and establishing our own direct sales force in the United States focused on selling directly to community oncologists in hospitals, cancer centers and offices as well as biopharma companies and supporting our partners as technical specialists and medical science liaisons.
To date, we have engaged in only limited sales and marketing activities, primarily through an agreement with Clarient for the OncoCEE-BR test. Under a May 2013 revision of our arrangement with Clarient, its marketing rights for OncoCEE-BR are no longer exclusive. We also have an agreement with Life Technologies Corporation for the commercialization of the OncoCEE-LU test. With the proceeds of our initial public offering we plan to build an internal sales and marketing team that will sell directly to community oncologists and serve as technical experts and clinical specialists to support the sales representatives of our partners. Under the arrangement with Clarient, as recently renegotiated, Clarient’s sales force sells the test on a nonexclusive basis, and we are responsible for performing the test, reporting the results, billing, and obtaining reimbursement for the test. Under the agreement with Life Technologies, when our OncoCEE-LU test is commercially launched, Life Technologies’ Medical Science Division sales force would sell the tests and Life Technologies’ pathologists would perform the interpretation, otherwise called the professional component of the pathology service, in Life
23
Table of Contents
Technologies’ laboratory. We would perform the technical component of the pathology service in our laboratory. Life Technologies would bill payors for the entire test, pay us for the technical component at an agreed upon rate and keep any amounts received for the professional component. Reimbursement would be based on Current Procedural Terminology, or CPT, codes. Under the Life Technologies agreement, the parties would share the payment and reimbursement risk, as we would be paid an agreed upon fee for the technical component of tests performed, and there would be a quarterly adjustment based on amounts actually received from payors. We will look to identify and engage additional groups with appropriately targeted sales efforts as partners for these and future tests and have initiated discussions with other companies.
Our plan for our sales organization calls for an initial group of 7 sales representatives placed in strategic locations around the country that have high concentrations of cancer patients, and potentially growing this number to 15-20 sales representatives within two years, and to 40-50 within five years. We have defined the initial sales territories and are targeting sales professionals with an average of 5-10 years of successful experience in clinical oncology sales or oncology diagnostic testing sales from leading biopharmaceutical, pharmaceutical or specialty reference laboratory companies. We plan on growing this specialized, oncology-focused sales force and supporting it with clinical specialists who bring significant technical knowledge in the use of CTC and ctDNA tests.
We will also be investing in sales headcount to focus on biopharma clinical trial opportunities. We plan to hire one additional representative for this initiative in the Northeast US and eventually add coverage in other areas with a large concentration of biotech and pharmaceutical companies.
Finally, we plan to invest in managed care sales and marketing in order to ensure adequate payment and coverage for our testing. The key value proposition for these customers will be focused on cost savings by offering alternatives to expensive surgeries when tumor biopsy tissue is not available.
Our sales and marketing efforts are and will be based on a five-part marketing strategy:
| • | Work with oncologists and group practices at community hospitals and cancer centers to educate them on the advantages and opportunities that CTC and ctDNA tests provide for better information, allowing them to select the most appropriate therapy for their patients, and how and when these tests are most effectively used; |
| • | Build relationships with key thought leaders in oncology, specifically in the cancers for which we are offering or plan to offer tests, to educate and support community oncologists; |
| • | Collaborate with leading research universities and institutions that enable the validation of our new tests, as well as the generation of clinical utility data; |
| • | Partner with pharmaceutical companies for clinical trial work focusing on CTC and ctDNA testing and analysis; and |
| • | Add value for the payor community by avoiding costly surgeries by providing the option of a simple blood test. |
We also take advantage of customary marketing channels commonly used by the diagnostic and pharmaceutical industries, such as medical meetings, broad-based publication of our scientific and clinical data, and the Internet. In addition, we provide easy-to-access information to our customers through our website and a data portal for physicians who wish to access test results electronically. Our customers value easily accessible information in order to quickly review their patients’ information and begin developing a treatment protocol.
Outside the United States
Outside the United States, where a central laboratory business model is less developed, we will evaluate opportunities with our existing and other partners for the conversion and/or development of our current and
24
Table of Contents
planned CTC and ctDNA tests to test systems or IVDs, and related strategies to develop and serve such regional oncology markets. We also plan to sell our clinical trial services to biopharmaceutical companies and research organizations outside the United States.
While the initial focus of our agreement with Life Technologies for OncoCEE-LU tests is on customers in the United States, the parties plan to cooperate on accessing markets internationally. We plan for this to be accomplished either through partnerships with local groups and distributors or the development of IVDs and/or test systems, including instrumentation.
Competition
As a cancer diagnostics company focused on current and planned tests for CTCs and ctDNA from standard blood samples, we rely extensively on our ability to combine novel technology and biomarker information with high-quality, state-of-the art clinical laboratory testing. We believe that we compete principally on the basis of:
| • | our ability to utilize standard blood samples, enabling testing of patients frequently through the course of their disease without a biopsy, thereby reducing cost and trauma, saving time, and providing real-time information on the current status of the tumor; |
| • | our ability to include biomarker information in our analysis, in addition to CTC enumeration, thereby providing a more complete profile of a patient’s disease than existing CTC tests can. This is actionable information that can assist physicians in selecting more personalized treatment plans for individual patients; |
| • | our current and planned CTC tests’ ability to capture and detect a broader range of CTC phenotypes than existing tests, and potentially at earlier stages of disease, resulting in fewer non-informative cases and more information for physicians. For example, our antibody capture cocktail targets not only EpCAM but also other epithelial antigens as well as mesenchymal and cancer stem cell antigens, indicative of cells having undergone the epithelial-to-mesenchymal transition. These cells may be more relevant for metastasis; |
| • | our ability to rapidly integrate new biomarkers, either validated in academic laboratories or of interest to pharmaceutical and biopharmaceutical companies in the context of their new therapies, into our current and planned tests, facilitating the expansion of actionable information for oncologists; |
| • | our research and clinical collaborations with key academic and clinical study groups, which enhance our research and development resources and, by enhancing our standing in the oncology community, support our marketing efforts; and |
| • | our planned ctDNA tests based on the CEE-Selector technology are expected to offer enhanced sensitivity and specificity in detecting mutation targets or resistance markers, again supporting treatment decisions. |
We believe that we compete favorably with respect to these factors, although we cannot assure you that we will be able to continue to do so in the future or that new products or tests that perform better than our current and planned tests and services will not be introduced. We believe that our continued success depends on our ability to:
| • | expand and enhance our current and planned OncoCEE tests to provide clinically meaningful information in additional cancers; |
| • | work with clinicians to design and implement clinical studies that demonstrate the clinical utility of our products; |
| • | continue to innovate and maintain scientifically advanced technology; |
| • | successfully market and sell tests; |
25
Table of Contents
| • | continue to comply with regulatory guidelines and obtain appropriate regulatory approvals in the United States and abroad as applicable; |
| • | continue to validate our pipeline of tests; |
| • | conduct or collaborate with clinical utility studies to demonstrate the application and medical value of our tests; |
| • | seek to obtain positive reimbursement decisions from Medicare and private third-party payors; |
| • | continue to enter into sales and marketing partnerships; |
| • | maintain existing and enter into new research and clinical collaborations with key academic and clinical study groups; |
| • | continue to attract and retain skilled scientific and clinical personnel; |
| • | continue to participate in and gain clinical trial work through biopharma partnerships; |
| • | receive payment for the testing we provide for patients; |
| • | obtain patents or other protection for our technologies, tests and services; and |
| • | obtain and maintain our clinical reference laboratory accreditations and licenses. |
Our principal competition comes from mainstream diagnostic methods, used by pathologists and oncologists for many years, which focus on tumor tissue analysis. It may be difficult to change the methods or behavior of oncologists to incorporate our CTC and ctDNA testing, including molecular diagnostic testing, into their practices in conjunction with or instead of tissue biopsies and analysis. In addition, companies offering capital equipment and kits or reagents to local pathology laboratories represent another source of potential competition. These kits are used directly by the pathologist, which can facilitate adoption. We plan to focus our marketing and sales efforts on medical oncologists rather than on pathologists.
We also face competition from companies that offer products or are conducting research to develop products for CTC or ctDNA testing in various cancers. In particular, Janssen Diagnostics, LLC markets its CellSearch® test and Atossa Genetics markets its ArgusCYTE® test, which are competitive to our OncoCEE-BR test for CTC enumeration, and HER2 analysis, respectively. However, the ArgusCYTE® test measures HER2 mRNA, which is not typically used for HER2 analysis, while we employ FISH for this analysis. FISH is generally considered to be the gold standard. CTC and ctDNA testing is a new area of science and we cannot predict what tests others will develop that may compete with or provide results similar or superior to the results we are able to achieve with the tests we develop. In addition to Janssen Diagnostics and Atossa Genetics, our competitors include public companies such as Alere (Adnagen) and Illumina as well as many private companies, including Apocell, EPIC Sciences, Clearbridge Biomedics, Cynvenio Biosystems, Fluxion Biosciences, RareCells, ScreenCell and Silicon Biosystems. Many of these groups, in addition to operating research and development laboratories, are establishing CLIA-certified testing laboratories while others are focused on selling equipment and reagents.
We expect that pharmaceutical and biopharmaceutical companies will increasingly focus attention and resources on the personalized cancer diagnostic sector as the potential and prevalence increases of molecularly targeted oncology therapies approved by the FDA along with companion diagnostics. For example, the FDA has recently approved three such agents—Xalkori® from Pfizer Inc. along with its companion anaplastic lymphoma kinase FISH test from Abbott Laboratories, Inc., Zelboraf® from Daiichi-Sankyo/Genentech/Roche along with its companion B-raf kinase V600 mutation test from Roche Molecular Systems, Inc. and Tafinlar® from GlaxoSmithKline along with its companion B-raf kinase V600 mutation test from bioMerieux. These recent FDA approvals are only the second, third and fourth instances of simultaneous approvals of a drug and companion diagnostic. The first approval was the 2010 approval of Genentech’s Herceptin® for HER2 positive breast cancer along with the HercepTest from partner Dako A/S. Our competitors may invent and commercialize technology platforms or tests that compete with ours.
26
Table of Contents
There are a number of companies which are focused on the oncology diagnostic market, such as Biodesix, Caris, Clarient, Foundation Medicine, Response Genetics, Neogenomics, Agendia, Genomic Health, and Genoptix, and which, while not currently offering CTC or ctDNA tests which are truly competitive with ours, are selling to the medical oncologists and pathologists. Large laboratory services companies, such as Sonic USA, Quest and LabCorp, provide more generalized cancer diagnostic testing.
Additionally, projects related to cancer diagnostics and genomics have received increased government funding, both in the United States and internationally. As more information regarding cancer genomics becomes available to the public, we anticipate that more products aimed at identifying targeted treatment options will be developed and that these products may compete with ours. In addition, competitors may develop their own versions of our current and planned tests in countries where we did not apply for patents or where our patents have not issued and compete with us in those countries, including encouraging the use of their test by physicians or patients in other countries.
Third-Party Suppliers and Manufacturers
Some of the components used in our current or planned products are currently sole-source, and substitutes for these components might not be able to be obtained easily or may require substantial design or manufacturing modifications. Any significant problem experienced by one of our sole source suppliers (particularly K.R. Anderson, Inc., which supplies a custom-packaged silicone compound used in our manufacturing) may result in a delay or interruption in the supply of components to us until that supplier cures the problem or an alternative source of the component is located and qualified. Any delay or interruption would likely lead to a delay or interruption in our manufacturing operations. The inclusion of substitute components must meet our product specifications and could require us to qualify the new supplier with the appropriate government regulatory authorities.
Patents and Technology
Our business is dependent upon our ability to develop and perform CTC and ctDNA tests that enable oncologists at hospitals, cancer centers and physician offices to receive information on properly characterized samples from individual cancer patients to select the most appropriate therapy for those patients. We rely on a combination of patents, patent applications, trademarks, trademark applications, trade secrets and industry know-how, in order to protect the proprietary aspects of our technology and assure that we can perform our tests.
Our patent portfolio consists of 3 issued U.S. patents, 6 pending U.S. patent applications and corresponding foreign patents and foreign patent applications. These patents and patent applications are related to various aspects of our current and planned CTC and ctDNA tests, including our CEE microfluidic channels, our CEE-Sure blood collection tubes, CEE-Cap antibody capture cocktail, CEE-Enhanced staining methodology, and CEE-Selector technology for mutation detection.
CEE Microfluidic Channels. We have three issued U.S. patents related to our current business (U.S. Patent Nos. 7,439,062, 7,695,956 and 8,158,410), and a number of additional U.S. and foreign patent applications, which cover our microfluidic channel technology. Our microfluidic channels are differentiated from other microfluidic channels used for CTC capture based on their unique geometry, particularly the arrangement of posts within the flow channel. The posts are chemically derivatized to enable capture of antibody-tagged CTCs, and are positioned to disrupt streamline or laminar flow of cells through the microfluidic channel to assure they come in contact with the posts for capture. Because the capture area of the microfluidic channel is sealed on one side with a glass cover slip, immunocytochemical and cytogenetic staining and analysis can occur within the microfluidic channel.
CEE-Sure Blood Collection Tubes. We have a U.S. patent application (13/243,432) in prosecution for our CEE-Sure blood collection tubes, which contain reagents designed to prevent clumping of blood cells and CTCs that could clog the microfluidic channels and disrupt our assays. These reagents also provide stability to the
27
Table of Contents
sample for shipping and transport, enabling blood samples to be shipped at ambient temperature from a collection site anywhere in the United States, and even outside the United States, to our laboratory in San Diego, California, and perform well in our assays for up to 96 hours after collection. DNA has been shown to be stable and accessible in cells under these conditions, and preliminary work suggests the same may be true for ctDNA, with more research required.
CEE-Cap Antibody Capture Cocktail. We have two pending U.S. patent applications (12/730,738 and 13/269,532) as well as their corresponding foreign patent applications directed to our antibody capture cocktail technology, which includes using antibodies to a number of tumor-associated antigens from cancer cells of both epithelial and mesenchymal phenotype, as well as cancer stem cells. Such technology relies on the binding of the antibodies to the target CTCs in solution, which we have shown greatly improves the capture efficiency because of superior binding kinetics and the lack of spatial constraints imposed by attachment of the antibodies to a solid surface.
CEE-Enhanced Staining. We have one U.S. pending patent application (13/241,083) as well as its corresponding foreign patent applications directed to this technology. This technology was developed to enable detection of CTCs that do not express sufficient amounts of cytokeratin, an epithelial marker that, in conjunction with DAPI and CD45 staining, is used to identify CTCs. It has made it possible to detect non-traditional CTCs, including mesenchymal types such as result from EMT, which, in conjunction with the antibody capture cocktail, has significantly increased the sensitivity of our CTC assays, and the informative rate for clinical samples.
CEE-Selector Mutation Detection Technology. This technology was developed to perform mutation analysis on CTCs, ctDNA or other sample types. It addresses the challenge of a sample in which copies of the normal gene locus vastly exceed the copies of the mutant gene locus. The technology has been demonstrated to have utility for more-sensitive mutation detection in ctDNA as well as CTC analysis. It is co-owned with Aegea Biotechnologies, Inc., with Biocept having exclusive commercial rights for clinical oncology applications, including LDTs and IVDs, where tissue, blood, bone marrow and cerebrospinal fluid are the sample types. There are two pending U.S. patent applications (13/841,842 and 61/784,101), with Aegea responsible for the prosecution of U.S. provisional application 61/784,101 and Biocept responsible for the prosecution of the other U.S. patent application. Biocept has also filed an international PCT application related to U.S. patent application 13/841,842. Lyle J. Arnold, Ph.D., our Senior Vice-President of Research & Development and Chief Scientific Officer, is the controlling person of Aegea.
In 2013, in Association for Molecular Pathology v. Myriad Genetics, the Supreme Court unanimously ruled that a “naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated,” invalidating Myriad Genetics’ patents on the BRCA1 and BRCA2 genes. This case removed some of the risk associated with testing laboratories like ours using isolated nucleic acid fragments for molecular analysis. Testing laboratories have been uncertain as to whether analysis of gene mutations covered by third party patents would violate such patents. We will continue to monitor developments in this area.
In addition to patents, we hold five U.S. registered trademarks, including a federal registration for the “CEE” mark, as well as several foreign registered trademarks and U.S. trademark applications for certain of our current and planned tests.
Through our clinical laboratory, we provide diagnostic testing and clinical services that utilize our proprietary trade secrets. In particular, we maintain trade secrets with respect to specimen accessioning, sample preparation and certain aspects of cytogenetic analysis. All of our trade secrets are kept in confidence and we take steps to ensure that our confidential information is not disseminated, including the use of non-disclosure agreements and confidentiality agreements.
Operations and Production Facilities
Our research and development laboratories, our CLIA-certified diagnostic testing laboratory and our manufacturing facility are located in our San Diego, California headquarters. The laboratories employ commercial state-of-the-art equipment as well as custom-made components specific to our CTC process that are
28
Table of Contents
generated in a small in-house engineering shop. The manufacturing facility used for the production of our CEE microfluidic channels is a Class 10,000 suite in which polydimethylsiloxane is formed into the base of our proprietary microfluidic channels in a molding process. A glass cover slip suitable for optical analysis is added to seal the channels and make them watertight by making them reactive using plasma techniques. The inside of the microfluidic channels is subsequently chemically derivatized to enable the attachment of binding elements that strongly bind to antibody-tagged or coated CTCs. Because the microfluidic channels have micrometer dimensions, and we are seeking individual cells in a blood sample to interact with the surface of the microfluidic channel, dust particles and other microscopic debris that could clog the channel needs to be avoided.
The process of performing our test is straightforward. When a health care professional takes a standard blood sample from a patient for CTC or ctDNA testing, he or she will place the blood sample in our CEE-Sure blood collection tubes, complete a requisition form, and package the specimen in our shipping kit for direct shipment to us. Once we receive the specimen at our laboratory and we enter all pertinent information about the specimen into our clinical laboratory information system, our laboratory technologists prepare the specimen for processing and analysis. Laboratory technologists, including clinical laboratory technologists and clinical laboratory scientists then conduct the analysis, including enumeration of CTCs and biomarker analysis such as FISH. The data, including images and the processed cells, are sent to our in-house or contracted pathologists or a commercialization partner’s pathologists who are experienced in the analysis and evaluation requested by the referring oncologist or pathologist.
After analysis, our in-house or contracted pathologists or a commercialization partner’s pathologists use laboratory information systems to prepare a comprehensive report, which includes selected relevant images associated with the specimen. Our Internet reporting portal allows a referring oncologist or pathologist to access his or her patient’s test results in real time in a secure manner that we believe to be compliant with HIPAA and other applicable standards. The reports are generated in industry standard .pdf formats which allows for high definition color images to be reproduced clearly.
In all cases, we provide the technical analysis, and in the case of our OncoCEE-BR test under our 2013 agreement with Clarient, we also provide the professional analysis. For our OncoCEE-LU test, while we would perform all of the technical analysis, the pathologists at our partner Life Technologies’ CLIA laboratory would provide the professional evaluation of the laboratory data. For OncoCEE-BR tests, we will send the results to the ordering oncologist and bill the payor through an arrangement we have with Xifin, Inc. For OncoCEE-LU tests, Life Technologies would send out the report and bill the appropriate parties, then pay us a predetermined fee for the technical analysis with a subsequent quarterly adjustment of that fee based on payments actually received by Life Technologies from payors.
Quality Management Program
We are committed to providing reliable and accurate diagnostic testing to our customers. Accurate specimen identification, timely communication of test results, and prompt correction of errors, is critical. We monitor and improve our performance through a variety of methods, including performance improvement indicators, internal proficiency testing and external quality audits conducted by CAP. All quality concerns and incidents are subject to review and analysis, and our procedures are designed to ensure that we are providing the best services possible to our patients and customers. Protection of patient results from misuse and improper access is imperative and electronic and paper results are guarded via password-protection and identification cards.
We have established a Quality Management Program for our laboratory designed to help ensure accurate and timely test results, a consistent high quality of our testing services. The Quality Management Program documents the quality assurance and performance improvement plans and policies, the laboratory quality assurance and quality control procedures that are necessary to ensure that we offer the highest quality of diagnostic testing services. This program is designed to satisfy all the requirements necessary for local and state licensures and accreditation for clinical diagnostic laboratories by CAP. We follow the policies and procedures
29
Table of Contents
for patient and employee safety, hazardous waste disposal and fire codes stated in the general laboratory procedure manual. We believe that all pertinent regulations of CLIA, the Occupational Safety and Health Administration, the Environmental Protection Agency and the FDA are satisfied by following the established guidelines and procedures of our Quality Management Program.
In addition to the compulsory proficiency programs and external inspections required by CMS and other regulatory agencies, we have developed a variety of internal systems and procedures to emphasize, monitor and continuously improve the quality of our operations. We maintain internal quality controls by routinely processing specimens with known diagnoses in parallel with patient specimens. We also have an internally administered proficiency program for specimen testing.
The CAP accreditation program involves unannounced on-site inspections of our laboratories. CAP is an independent, non-governmental organization of board-certified pathologists that accredits laboratories nationwide on a voluntary basis and that has been recognized by CMS as an accreditation organization to inspect laboratories to determine adherence to the CLIA standards.
Third-Party Payor Reimbursement
Revenues from our clinical laboratory testing are derived from several different sources. Depending on the billing arrangement, the instruction of the ordering physician and applicable law, parties that reimburse us for our services include:
| • | third-party payors that provide coverage to the patient, such as an insurance company, a managed care organization or a governmental payor program; |
| • | physicians or other authorized parties, such as hospitals or independent laboratories, that order the testing service or otherwise refer the services to us; |
| • | patients in cases where the patient has no insurance, has insurance that partially covers the testing, or owes a co-payment, co-insurance or deductible amount; |
| • | collaboration partners (e.g., Life Technologies for our anticipated OncoCEE-LU test); or |
| • | biopharmaceutical companies, universities or researchers for clinical trial work. |
We are reimbursed for two categories of testing, anatomic pathology, which includes cell staining and the enumeration component of CTC tests, FISH, immunocytochemistry and immunofluorescence, and molecular pathology, which includes mutation analysis. Reimbursement under the Medicare program for the diagnostic services that we offer is based on either the Medicare Physician Fee Schedule or the Medicare Clinical Laboratory Fee Schedule, each of which is subject to geographic adjustments and is updated annually. Medical services provided to Medicare beneficiaries that require a degree of physician supervision, judgment or other physician involvement, such as pathology services, are generally reimbursed under the Medicare Physician Fee Schedule, whereas clinical diagnostic laboratory tests are generally reimbursed under the Medicare Clinical Laboratory Fee Schedule. Some of the services that we provide are genetic and molecular testing, which are reimbursed as clinical diagnostic laboratory tests.
Regardless of the applicable fee schedule, Medicare payment amounts are established for each CPT code. In addition, under the Clinical Laboratory Fee Schedule, Medicare also sets a cap on the amount that it will pay for any individual test. This cap, usually referred to as the National Limitation Amount, is set at a percentage of the median of all the contractor fee schedule amounts for each billing code.
Medicare also has policies that may limit when we can bill directly for our services and when we must instead bill another provider, such as a hospital. When the testing that we perform is done on a specimen that was collected while the patient was in the hospital, as either an inpatient or outpatient, we may be required to bill the hospital for clinical laboratory services and for the technical component of pathology services. Which party is to
30
Table of Contents
be billed depends primarily on whether the service was ordered at least 14 days after the patient’s discharge from the hospital. Complying with these requirements is complex and time-consuming and may affect our ability to collect for our services. In addition, hospitals may refuse to pay our invoices or may demand pricing that negatively affects our profit margin.
Medicare requires a beneficiary to pay a 20% co-insurance amount for services billed under the Physician Fee Schedule. Medicare covers the remaining 80%. There is currently no patient co-payment or co-insurance amount applicable to testing billed under the Clinical Laboratory Fee Schedule. Patients often have supplemental insurance policies that cover the co-insurance amount for physician services.
Medicare has coverage policies that can be national or regional in scope. Coverage means that the test or assay is approved as a benefit for Medicare beneficiaries. If there is no coverage, a provider may not bill Medicare or the beneficiary for the service. There is currently no national coverage policy regarding the CTC capture/enumeration portion of our testing. The previous regional Medicare Administrative Contractor (MAC) for California, Palmetto GBA, LLC, adopted a negative coverage policy for CTC capture/enumeration (with the exception that Janssen Diagnostics, LLC’s CellSearch® test has historically been covered for CTC capture/enumeration). The current MAC for California, Noridian Healthcare Solutions, LLC, is adopting the coverage policies from Palmetto GBA. Therefore the capture/enumeration portion of our OncoCEE testing is not covered and we will receive no payment from Medicare for this service unless and until the coverage policy is changed. On November 4, 2013, we submitted a comprehensive dossier to Palmetto GBA and Noridian explaining the benefits of the capture/enumeration testing in order to seek to persuade the MACs to allow coverage for this portion of our testing. Palmetto GBA responded on November 27, 2013, denying our request for Medicare coverage for the enumeration/detection portion of our OncoCEE testing. We have not received any other indications to suggest that the negative coverage determination will be reversed. The earliest date we could submit another dossier on this matter is May 27, 2014. We intend to continue our efforts to obtain Medicare coverage for capture/enumeration. On the other hand, FISH analysis is a covered benefit for Medicare beneficiaries and accordingly we expect that the FISH portion of OncoCEE-BR and our planned tests are and will be covered and that when and as we bill Medicare we will receive payment from Medicare under the Physician Fee Schedule for FISH analysis. Molecular testing for the mutations we currently plan to test for with CEE-Selector is also a covered benefit, so we believe that CEE-Selector testing would thereby be covered and that when and as we bill Medicare we would receive payment from Medicare under the Clinical Laboratory Fee Schedule for CEE-Selector testing. As discussed above, we have not yet received from Medicare any response or adjudication regarding any of our late-2013 billings, including for the FISH portion of OncoCEE-BR testing.
Reimbursement rates paid by private third-party payors can vary based on whether we are considered to be an “in-network” provider, a participating provider, a covered provider or an “out-of-network” provider. These definitions can vary among payors, but we are generally considered an “out-of-network” or non-participating provider by the vast majority of private third-party payors. An in-network provider usually has a contract with the payor or benefits provider. This contract governs, among other things, service-level agreements and reimbursement rates. In certain instances an insurance company may negotiate an in-network rate for our testing. An in-network provider may have rates that are lower per test than those that are out-of-network, and that rate can vary widely. The rate varies based on the payor, the testing type and often the specifics of the patient’s insurance plan. If a laboratory agrees to contract as an in-network provider, it generally expects to receive quicker payment and access to additional covered patients.
Billing and Billing Codes for Third-Party Payor Reimbursement
CPT codes are the main data code set used by physicians, hospitals, laboratories and other health care professionals to report separately-payable clinical laboratory and pathology services for reimbursement purposes. The CPT coding system is maintained and updated on an annual basis by the American Medical Association. We believe there are existing codes that describe nearly all of the other steps in our testing process. We currently use a combination of different codes to bill for our testing and analysis. Many of the CPT codes used to bill for
31
Table of Contents
molecular pathology tests such as those planned in our OncoCEE-LU test were significantly revised by the CPT Code Editorial Panel effective January 1, 2013. These new codes replace the more general “stacking” codes that were previously used to bill for these services with more test-specific codes. In the Physician Fee Schedule Rule issued in November 2012, CMS stated that it had determined it would pay for the new codes as clinical laboratory tests under the Medicare Clinical Laboratory Fee Schedule. CMS has also started a process to “gapfill” the new codes. In other words, it will ask each of the MACs to determine a reasonable price for each of the new codes.
Changes in coding and reimbursement methods could have an adverse impact on our revenues going forward. However, we are currently working with our billing consultants to determine what will be required by the new coding changes. The elimination of the “stacking” codes will require us to either use the new more specific codes where applicable effective January 2013, or to use other “Not Otherwise Classified” codes when billing. The implementation of these new codes will vary from payor to payor, and it is too early to assess the impact, if any, that the migration to the new codes may have on our results of operations. The introduction of the new codes by CMS, in combination with the other actions it is considering with regard to pricing, could result in a reduction in the payments that we receive for our current breast cancer test and our planned future tests and make it more difficult to obtain coverage from Medicare or other payors. There can be no guarantees that Medicare and other payors will establish positive or adequate coverage policies or reimbursement rates.
We are moving forward with plans to obtain reimbursement coverage for the capture/enumeration components of OncoCEE-BR and our planned CTC tests. For other components and types of testing provided or anticipated to be provided by us, specific CPT codes were provided by the American Medical Association in January 2013 or we are able to utilize existing CPT codes from the Medicare Physician Fee Schedule. For these established CPT codes (for example, the codes for FISH and immunocytochemistry, or ICC), positive coverage determinations have been adopted as part of national Medicare policy or under applicable Local Coverage Determinations. Specific codes for our tests, however, do not assure an adequate coverage policy or reimbursement rate. Please see the section entitled “Legislative and Regulatory Changes Impacting Clinical Laboratory Tests” for further discussion of certain legislative and regulatory changes to these billing codes and the anticipated impact on our business.
Coverage and Reimbursement for our Current Breast Cancer Test and our Planned Future Tests
OncoCEE-BR is a new test, and because of our previous relationship with Clarient, under which Clarient had responsibility for billing and reimbursement until mid-2013, we do not have established coverage and reimbursement policies set with all third-party payors. Our Medicare Administrative Contractor has issued a negative coverage determination for the capture/enumeration component of all CTC tests (with the exception that Janssen Diagnostics, LLC’s CellSearch® test has historically been covered for CTC capture/enumeration). We have received reimbursement for the capture/enumeration component of our tests from some payors, including major private third-party payors, based on submission of standard CPT codes. FISH, ICC and Molecular Testing CPT codes are the subject of positive coverage national or local Medicare determinations. We believe these codes can be used to bill for the analysis components of our current and anticipated CTC tests.
We expect these analysis components to have a significantly greater reimbursement value than the capture/enumeration components of our current and anticipated CTC tests, based on a comparison of what we believe CellSearch® capture/enumeration reimbursement rates currently are, versus existing reimbursement rates for analysis components such as FISH and ICC analysis and molecular testing.
We believe, based on research showing that approximately 54% of new cancers occur in persons age 65 and older and that almost all Americans age 65 and older are enrolled in Medicare, that a substantial portion of the patients for whom we would expect to perform cancer diagnostic tests will have Medicare as their primary medical insurance. Only in November 2013 did we first directly bill any payor for physician-ordered testing; until May 2013, our commercialization partner Clarient was responsible for all billing associated with our tests.
32
Table of Contents
We do not have data for Clarient’s billing and collection experience with respect to our test, because Clarient paid us a contracted amount per test performed regardless of their billing and collections. From May to December 2013, we performed an average of 1-3 physician-ordered tests per month (in addition to the 20-30 tests per month which we have been performing since January 2013 for a clinical utility study with investigators at the Dana-Farber Cancer Institute). Billing for these physician-ordered tests is now handled for us by a non-Clarient billing service provider. In 2013, we invoiced, through this service provider, for 17 physician-ordered tests. Of these, 8 tests were billed to Medicare and the remainder were billed to other payors. We have been paid for one of these tests while we have not yet had any response or adjudication from any other payor as to the bills submitted in late 2013. Accordingly, we do not yet have any data regarding reimbursement history or collectability experience. In addition, we believe the sample size of 17 is too small to be the basis for any conclusion about our ongoing payor mix. We cannot assure you that, even if OncoCEE-BR and our planned tests are otherwise successful, reimbursement for the currently Medicare-covered portions of OncoCEE-BR and our planned tests would, without Medicare reimbursement for the capture/enumeration portion, produce sufficient revenues to enable us to reach profitability and achieve our other commercial objectives.
Where there is a private or governmental third-party payor coverage policy in place, we bill the payor and the patient in accordance with the established policy. Where there is no coverage policy in place, we pursue reimbursement on a case-by-case basis. Our efforts in obtaining reimbursement based on individual claims, including pursuing appeals or reconsiderations of claims denials, could take a substantial amount of time, and bills may not be paid for many months, if at all. Furthermore, if a third-party payor denies coverage after final appeal, payment may not be received at all. We are working to decrease risks of nonpayment by implementing a revenue cycle management system.
We cannot predict whether, or under what circumstances, payors will reimburse for all components of our tests. Payment amounts can also vary across individual policies. Full or partial denial of coverage by payors, or reimbursement at inadequate levels, would have a material adverse impact on our business and on market acceptance of our tests.
Legislative and Regulatory Changes Impacting Clinical Laboratory Tests
From time to time, Congress has revised the Medicare statute and the formulas it establishes for both the Medicare Clinical Laboratory Fee Schedule and the Medicare Physician Fee Schedule. The payment amounts under the Medicare fee schedules are important because they not only determine our reimbursement under Medicare, but those payment amounts are also often used as a basis for payment amounts set by other governmental and private third -party payors. For example, state Medicaid programs are prohibited from paying more than the Medicare fee schedule limit for clinical laboratory services furnished to Medicaid recipients.
Under the statutory formula for Medicare Clinical Laboratory Fee Schedule amounts, increases are made annually based on the Consumer Price Index for All Urban Consumers as of June 30 for the previous twelve-month period. From 2004-2008, Congress eliminated the Consumer Price Index for All Urban Consumers update in the Medicare Prescription Drug, Improvement and Modernization Act of 2003. In addition, for years 2009 through 2013, the Medicare Improvements for Patients and Providers Act of 2008 mandated an approximately 0.5% cut to the Consumer Price Index for All Urban Consumers update. Accordingly, the update for 2009 was reduced to 4.5% and negative 1.9% for 2010. The ACA has, among other things, imposed additional cuts to the Medicare reimbursement for clinical laboratories. The ACA replaced the 0.5% cut enacted by the Medicare Improvements for Patients and Providers Act with a “productivity adjustment” that will reduce the Consumer Price Index update in payments for clinical laboratory tests. In 2011, the productivity adjustment was -1.2%. In addition, the ACA includes a separate 1.75% reduction in the CPI update for clinical laboratories for the years 2011 through 2015. The MCTRJCA, enacted in 2012, mandated an additional change in reimbursement for clinical laboratory service programs. This legislation requires CMS to reduce the Medicare Clinical Laboratory Fee Schedule by 2% in 2013, which in turn will serve as a base for 2014 and subsequent years. CMS has projected that because of the changes required by ACA and MCTRJCA, payment for clinical laboratory services will go down by approximately 3% by 2013.
33
Table of Contents
With respect to our diagnostic services for which we expect to be reimbursed under the Medicare Physician Fee Schedule, because of the statutory formula the rates would have decreased for the past several years if Congress failed to intervene. In the past, when the application of the statutory formula results in lower payment, Congress has passed interim legislation to prevent the reductions. In November 2013, CMS issued its 2014 Physician Fee Schedule Final Rule, or the 2014 Final Rule. In the 2014 Final Rule, CMS called for a reduction of approximately 23.7% in the 2014 conversion factor that is used to calculate physician reimbursement. If in future years Congress does not adopt interim legislation to block or offset, and/or CMS does not moderate, any substantial CMS-proposed reimbursement reductions, the resulting decrease in payments from Medicare could adversely impact our revenues and results of operations. In addition, for 2012, CMS requested that the American Medical Association’s Relative Value Scale Update Committee reexamine the relative values of certain codes, including FISH codes. The Relative Value Scale Update Committee is an expert panel that provides relative value recommendations to CMS for use in annual updates to the Medicare Physician Fee Schedule. These relative values are used by CMS to determine payments, and CMS seeks to assess whether such codes are misvalued and an adjustment is necessary. In July 2013 CMS published the proposed Physician Fee Schedule for 2014. As part of that proposed rule, CMS sought to decrease payment for approximately 200 CPT codes, including those for certain anatomic and molecular pathology services, to make payments to independent laboratories and hospital outpatient departments consistent. The proposed rates were generally lower than the current rates paid to independent laboratories and physicians for the same services. For example, CMS proposed to decrease the reimbursement rate for the technical component of FISH analysis by 47%. In fact, the 2014 Final Rule as adopted left FISH reimbursement rates for independent laboratories and physicians essentially unchanged from 2013 reimbursement levels.
In addition, the 2014 Final Rule included both increases and decreases in certain relative value units and geographic adjustment factors used to determine reimbursement for a number of codes used in our current breast cancer test and our planned future tests. These codes describe services that we must perform in connection with our tests and we bill for these codes in connection with the services that we provide.
Further, with respect to the Medicare program, Congress has proposed on several occasions to impose a 20% coinsurance charge on patients for clinical laboratory tests reimbursed under the Medicare Clinical Laboratory Fee Schedule, which would require us to bill patients for these amounts. Because of the relatively low reimbursement for many clinical laboratory tests, in the event that Congress were to ever enact such legislation, the cost of billing and collecting for these services would often exceed the amount actually received from the patient and effectively increase our costs of billing and collecting.
Some of our Medicare claims may be subject to policies issued by Palmetto GBA and Noridian Healthcare Solutions, our former and current Medicare Administrative Contractor for California, respectively. Palmetto GBA, acting on behalf of many MACs, recently issued a Local Coverage Decision that affects coverage, coding and billing of many molecular diagnostic tests. Under this Local Coverage Determination, Palmetto GBA will not cover any molecular diagnostic tests, such as the capture/enumeration component of our current breast cancer test and our planned future tests, unless the test is expressly included in a National Coverage Determination issued by CMS or a Local Coverage Determination or coverage article issued by Palmetto GBA. Currently, laboratories may submit coverage determination requests to Palmetto GBA for consideration and apply for a unique billing code for each test (which is a separate process from the coverage determination). In the event that a non-coverage determination is issued, the laboratory must wait six months following the determination to submit a new request. In addition, effective January 1, 2013, Palmetto GBA implemented its new Molecular Diagnostic Services Program, under which, among other things, laboratories must use the newly-assigned billing codes specific to the test (as implemented by the American Medical Association), in order to receive the indicated reimbursement amounts. Reimbursement amounts under these new single molecular diagnostics billing codes were in some cases lower, and in some cases higher, than amounts allowed by Medicare before January 1, 2013, but most were significantly lower. Palmetto GBA currently has a negative coverage determination for the capture/enumeration component of CTC tests such as our current and anticipated CTC tests, but there is no such negative coverage determination for the analysis component of such CTC tests. Denial (or continuation of denial)
34
Table of Contents
of coverage for the capture/enumeration component of our current and anticipated CTC tests by Palmetto GBA or its successor MAC, Noridian Healthcare Solutions, or reimbursement at inadequate levels, would have a material adverse impact on our business and on market acceptance of our current breast cancer test and our planned future tests. Noridian Healthcare Solutions intends to follow, for CTC tests, the positive or negative coverage determinations which from time to time Palmetto GBA makes. Because Palmetto GBA denied on November 27, 2013 our request for coverage for the enumeration/detection portion of our OncoCEE testing, the earliest date we could submit another request on this matter is May 27, 2014. We intend to continue our efforts to obtain Medicare coverage for capture/enumeration.
Governmental Regulations
Clinical Laboratory Improvement Amendments of 1988 and State Regulation
As a provider of laboratory testing on human specimens for the purpose of diagnosis, prevention, or treatment, we are required to hold certain federal, state and local licenses, certifications and permits to conduct our business. In 1988, Congress enacted CLIA, which established quality standards for all laboratories providing testing to ensure the accuracy, reliability and timeliness of patient test results regardless of where the test was performed. Our laboratory holds a CLIA certificate of accreditation. As to state laws, we are required to meet certain laboratory licensing and other requirements. Our laboratory holds the required licenses from the applicable state agencies in which we operate. For more information on state licensing requirements, see the sections entitled see the section entitled “Description of the Business—Governmental Regulations—California State Laboratory Licensing” and “Description of the Business—Governmental Regulations—Other States’ Laboratory Licensing.”
Under CLIA, a laboratory is defined as any facility which performs laboratory testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease, or the impairment of, or assessment of health of human beings. CLIA also requires that we hold a certificate applicable to the complexity of the categories of testing we perform and that we comply with certain standards. CLIA further regulates virtually all clinical laboratories by requiring they comply with various operational, personnel, facilities administration, quality and proficiency testing requirements intended to ensure that their clinical laboratory testing services are accurate, reliable and timely. CLIA certification is also a prerequisite to be eligible to bill for services provided to state and federal health care program beneficiaries. CLIA is user-fee funded. Therefore, all costs of administering the program must be covered by the regulated facilities, including certification and survey costs.
We are subject to survey and inspection every two years to assess compliance with program standards, and may be subject to additional unannounced inspections. Laboratories performing high complexity testing are required to meet more stringent requirements than laboratories performing less complex tests. In addition, a laboratory like ours that is certified as “high complexity” under CLIA may obtain analyte specific reagents, which are used to develop LDTs.
In addition to CLIA requirements, we must comply with the standards set by CAP, which accredits our laboratory. Under CMS requirements, accreditation by CAP is sufficient to satisfy the requirements of CLIA. Therefore, because we are accredited by CAP, we are deemed to also comply with CLIA. CLIA also provides that a state may adopt laboratory regulations that are more stringent than those under federal law, and certain states have implemented their own more stringent laboratory regulatory schemes.
Federal, State and Foreign Fraud and Abuse Laws
A variety of federal and state laws prohibit fraud and abuse. These laws are interpreted broadly and enforced aggressively by various state and federal agencies, including CMS, the Department of Justice, the Office of Inspector General for HHS, and various state agencies. In addition, the Medicare and Medicaid programs increasingly use a variety of contractors to review claims data and to identify improper payments as well as fraud
35
Table of Contents
and abuse. These contractors include Recovery Audit Contractors, Medicaid Integrity Contractors and Zone Program Integrity Contractors. In addition, CMS conducts Comprehensive Error Rate Testing audits, the purpose of which is to detect improper Medicare payments. Any overpayments identified must be repaid unless a favorable decision is obtained on appeal. In some cases, these overpayments can be used as the basis for an extrapolation, by which the error rate is applied to a larger universe of claims, and which can result in even higher repayments.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, receiving, or providing remuneration, directly or indirectly, to induce or in return for either the referral of an individual, or the furnishing, recommending, or arranging for the purchase, lease or order of any health care item or service reimbursable, in whole or in part, under a federal health care program. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, credit arrangements, payments of cash, ownership interests and providing anything at less than its fair market value. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements within the health care industry, the Office of Inspector General for HHS has issued a series of regulatory “safe harbors.” These safe harbor regulations set forth certain requirements that, if met, will assure immunity from prosecution under the federal Anti-Kickback Statute. Although full compliance with these provisions ensures against prosecution under the federal Anti-Kickback Statute, the failure of a transaction or arrangement to fit within a specific safe harbor does not necessarily mean that the transaction or arrangement is illegal or that prosecution under the federal Anti-Kickback Statute will be pursued. For further discussion of the impact of federal and state health care fraud and abuse laws and regulations on our business, see the section entitled “Risk Factors—Regulatory Risks Relating to Our Business.” We are subject to federal and state health care fraud and abuse laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
In addition to the administrative simplification regulations discussed above, HIPAA also created two new federal crimes; health care fraud and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including private third-party payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from federal health care programs, such as the Medicare and Medicaid programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from federal health care programs.
Finally, another development affecting the health care industry is the increased enforcement of the federal False Claims Act and, in particular, actions brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment to the federal government. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government and permit such individuals to share in any amounts paid by the entity to the government in fines or settlement. In addition, various states have enacted false claim laws analogous to the federal False Claims Act, and some of these state laws apply where a claim is submitted to any third-party payor. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties ranging from $5,500 to $11,000 for each false claim.
Additionally, in Europe various countries have adopted anti-bribery laws providing for severe consequences, in the form of criminal penalties and/or significant fines for individuals and/or companies committing a bribery offence. Violations of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation. For instance, in the United Kingdom, under the Bribery Act 2010, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including
36
Table of Contents
any function of a public nature. Bribery of foreign public officials also falls within the scope of the Bribery Act 2010. Under the new regime, an individual found in violation of the Bribery Act 2010 faces imprisonment of up to 10 years. In addition, the individual can be subject to an unlimited fine, as can commercial organizations for failure to prevent bribery.
Physician Referral Prohibitions
Under a federal law directed at “self-referral,” commonly known as the Stark Law, there are prohibitions, with certain exceptions, on Medicare and Medicaid payments for laboratory tests referred by physicians who personally, or through a family member, have a “financial relationship”—including an investment or ownership interest or a compensation arrangement—with the clinical laboratory performing the tests. Several Stark Law exceptions are relevant to arrangements involving clinical laboratories, including: (1) fair market value compensation for the provision of items or services; (2) payments by physicians to a laboratory for clinical laboratory services; (3) certain space and equipment rental arrangements that satisfy certain requirements, and (4) personal services arrangements. The laboratory cannot submit claims to the Medicare Part B program for services furnished in violation of the Stark Law, and Medicaid reimbursements may be at risk as well. Penalties for violating the Stark Law include the return of funds received for all prohibited referrals, fines, civil monetary penalties and possible exclusion from the federal health care programs. Many states have comparable laws that are not limited to Medicare and Medicaid referrals.
Corporate Practice of Medicine
A number of states, including California, do not allow business corporations to employ physicians to provide professional services. This prohibition against the “corporate practice of medicine” is aimed at preventing corporations such as us from exercising control over the medical judgments or decisions of physicians. The state licensure statutes and regulations and agency and court decisions that enumerate the specific corporate practice rules vary considerably from state to state and are enforced by both the courts and regulatory authorities, each with broad discretion. If regulatory authorities or other parties in any jurisdiction successfully assert that we are engaged in the unauthorized corporate practice of medicine, we could be required to restructure our contractual and other arrangements. In addition, violation of these laws may result in sanctions imposed against us and/or the professional through licensure proceedings, and we could be subject to civil and criminal penalties that could result in exclusion from state and federal health care programs.
Direct Billing Laws and Other State Law Restrictions on Billing for Laboratory Services
Laws and regulations in certain states prohibit laboratories from billing physicians or other purchasers for testing that they order. Some of those laws and regulations apply only to anatomic pathology services while others extend to other types of testing. Some states may allow laboratories to bill physicians directly but may prohibit the physician (and, in some cases, other purchasers) from charging more than the purchase price for the services (or may allow only for the recovery of acquisition costs) or may require disclosure of certain information on the invoice. In some cases, and if not prohibited by law or regulation, we may bill physicians, hospitals and other laboratories directly for the services that they order. An increase in the number of states that impose similar restrictions could adversely affect us by encouraging physicians to perform laboratory services in-house or by causing physicians to refer services to other laboratories that are not subject to the same restrictions.
Physician Licensing
A number of the states where specimens originate require that the physician interpreting those specimens be licensed by that particular state. Physicians who fail to comply with these licensure requirements could face fines or other penalties for practicing medicine without a license and we could be required to pay those fines on behalf of our pathologists or subject to liability under the federal False Claims Act and similar state laws if we bill for services furnished by unlicensed pathologists. We do not believe that the services our pathologist performs constitute the practice of medicine in any state that requires out-of-state physician licensure. Our pathologist thus is not required to obtain licensure in any state where he does not reside.
37
Table of Contents
In addition, many states also prohibit the splitting or sharing of fees between physicians and non-physician entities. We do not believe that our contractual arrangements with physicians, physicians group practices or hospitals will subject us to claims under such regulations. However, changes in the laws may necessitate modifications in our relationships with our clients.
California State Laboratory Licensing
Our laboratory is licensed and in good standing under the State of California Department of Public Health standards. Our current licenses permit us to receive specimens obtained in California.
California state laws and regulations also establish standards for the day-to-day operations of clinical laboratories, including physical facility requirements and equipment, quality control and proficiency testing requirements. If we are found to be out of compliance with California statutory or regulatory standards, we may be subject to suspension, restriction or revocation of our laboratory license or assessed civil money penalties. The operator of a noncompliant laboratory may also be found guilty of a misdemeanor under California law. A finding of noncompliance, therefore, may result in harm to our business.
Other States’ Laboratory Licensing
Several states require the licensure of out-of-state laboratories that accept specimens from those states. We are currently in the process of addressing the requirements for licensure in Florida and New York, and have re-obtained all required licenses and approvals from all other states requiring licensure for out-of-state laboratories. We were required to re-license in certain states as a result of our July 2013 reincorporation to Delaware.
From time to time, other states may require out of state laboratories to obtain licensure in order to accept specimens from such states. If we identify any other state with such requirements or if we are contacted by any other state advising us of such requirements, we intend to follow instructions from the state regulators as to how we should comply with such requirements.
Other Regulatory Requirements
Our laboratory is subject to federal, state and local regulations relating to the handling and disposal of regulated medical waste, hazardous waste and biohazardous waste, including chemical, biological agents and compounds, blood and bone marrow samples and other human tissue. Typically, we use outside vendors who are contractually obligated to comply with applicable laws and regulations to dispose of such waste. These vendors are licensed or otherwise qualified to handle and dispose of such waste.
The Occupational Safety and Health Administration has established extensive requirements relating to workplace safety for health care employers, including requirements to develop and implement programs to protect workers from exposure to blood-borne pathogens by preventing or minimizing any exposure through needle stick or similar penetrating injuries.
Segment and Geographical Information
We operate in one reportable business segment and historically have derived revenues only from the United States.
Employees
As of December 31, 2013, we had a total of 27 full-time employees and one part time employee, five of whom hold doctorate degrees and seven of whom are engaged in full-time research and development activities. We plan to expand production, sales and marketing and our research and development programs, and we plan to hire additional staff as these initiatives are implemented. None of our employees is represented by a labor union.
38
Table of Contents
Available Information
Our website address is www.biocept.com. We post links to our website to the following filings as soon as reasonably practicable after they are electronically filed with or furnished to the Securities and Exchange Commission, or the SEC: annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements, and any amendments to those reports filed or furnished pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended. All such filings are available through our website free of charge. Our filings may also be read and copied at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. Information on the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330. The SEC also maintains an internet site at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
39
Table of Contents
| Item 1A. | Risk Factors |
An investment in our common stock involves a high degree of risk. You should consider carefully the risks described below, together with all of the other information included in this Annual Report, as well as in our other filings with the SEC, in evaluating our business. If any of the following risks actually occur, our business, financial condition, operating results and future prospects could be materially and adversely affected. In that case, the trading price of our common stock may decline and you might lose all or part of your investment. The risks described below are not the only ones we face. Additional risks that we currently do not know about or that we currently believe to be immaterial may also impair our business, financial condition, operating results and prospects. Certain statements below are forward-looking statements. For additional information, see the information included under the heading “Cautionary Note Regarding Forward-Looking Statements.”
Risks Relating to Our Financial Condition and Capital Requirements
We are an early stage company with a history of net losses; we expect to incur net losses in the future, and we may never achieve sustained profitability.
We have historically incurred substantial net losses, including net losses of $12.3 million in 2012 and $9.2 million in 2013, and we have never been profitable. At December 31, 2013, our accumulated deficit was approximately 122.4 million. Before 2008, we were pursuing a business plan relating to fetal genetic disorders and other fields, all of which were unrelated to cancer diagnostics. The portion of our accumulated deficit that relates to the period from inception through December 31, 2007 is approximately $66.5 million.
We expect our losses to continue as a result of costs relating to our lab operations as well as increased sales and marketing costs and ongoing research and development expenses. These losses have had, and will continue to have, an adverse effect on our working capital, total assets and stockholders’ equity. Because of the numerous risks and uncertainties associated with our commercialization efforts, we are unable to predict when we will become profitable, and we may never become profitable. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our inability to achieve and then maintain profitability would negatively affect our business, financial condition, results of operations and cash flows. Our chief executive officer Michael W. Nall, who joined us in August 2013, has not previously been the chief executive officer of a public or private company, and therefore his lack of experience may result in some of his time being spent acclimating to his new position and responsibilities. A lack of significant experience in being the chief executive officer of a public company could have an adverse effect on his ability to quickly respond to problems or effectively manage issues surrounding the operation of a public company.
We will need to raise additional capital.
We expect to need to raise additional capital to expand our business to meet our long-term business objectives. Additional financing may be from the sale of equity or convertible or other debt securities in a public or private offering, from a new credit facility or strategic partnership coupled with an investment in us or a combination of both. We may be unable to raise sufficient additional financing on terms that are acceptable to us, if at all. Failure to raise additional capital in sufficient amounts would significantly impact our ability to expand our business. For further discussion of our liquidity requirements as they relate to our long-term plans, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources.”
Risks Relating to Our Business and Strategy
If we are unable to increase sales of our OncoCEE-BR breast cancer diagnostic tests or successfully develop and commercialize other tests, our revenues will be insufficient for us to achieve profitability.
We currently derive substantially all of our revenues from sales of cancer diagnostic tests. We recently began offering our OncoCEE-BR breast cancer test through our CLIA-certified, accredited, and state-licensed
40
Table of Contents
laboratory. We are in varying stages of research and development for other cancer diagnostic tests that we may offer. If we are unable to increase sales of our OncoCEE-BR breast cancer diagnostic test or successfully develop and commercialize other cancer diagnostic tests, we will not produce sufficient revenues to become profitable.
If we are unable to execute our sales and marketing strategy for cancer diagnostic tests and are unable to gain acceptance in the market, we may be unable to generate sufficient revenue to sustain our business.
We are an early-stage company and have engaged in only limited sales and marketing activities for the OncoCEE-BR breast cancer diagnostic tests we offer through our CLIA-certified laboratory. To date, we have received very limited revenue.
Although we believe that our current test and our planned diagnostic tests represent a promising commercial opportunity, our tests may never gain significant acceptance in the marketplace and therefore may never generate substantial revenue or profits for us. We will need to establish a market for our cancer diagnostic tests and build that market through physician education, awareness programs and the publication of clinical trial results. Gaining acceptance in medical communities requires publication in leading peer-reviewed journals of results from studies using our current test and/or our planned cancer tests. The process of publication in leading medical journals is subject to a peer review process and peer reviewers may not consider the results of our studies sufficiently novel or worthy of publication. Failure to have our studies published in peer-reviewed journals would limit the adoption of our current test and our planned tests.
Our ability to successfully market the cancer diagnostic tests that we may develop will depend on numerous factors, including:
| • | conducting clinical utility studies of such tests in collaboration with key thought leaders to demonstrate their use and value in important medical decisions such as treatment selection; |
| • | whether our non-exclusive partners, Life Technologies Corporation and Clarient, vigorously support our offerings; |
| • | the success of the sales force which we intend to hire with some of the proceeds of our initial public offering; |
| • | whether healthcare providers believe such diagnostic tests provide clinical utility; |
| • | whether the medical community accepts that such diagnostic tests are sufficiently sensitive and specific to be meaningful in patient care and treatment decisions; and |
| • | whether health insurers, government health programs and other third-party payors will cover and pay for such cancer diagnostic tests and, if so, whether they will adequately reimburse us. |
Failure to achieve widespread market acceptance of our current test and our planned cancer diagnostic tests would materially harm our business, financial condition and results of operations.
If we cannot develop tests to keep pace with rapid advances in technology, medicine and science, our operating results and competitive position could be harmed.
In recent years, there have been numerous advances in technologies relating to the diagnosis and treatment of cancer. Several new cancer drugs have been approved, and a number of new drugs in clinical development may increase patient survival time. There have also been advances in methods used to identify patients likely to benefit from these drugs based on analysis of biomarkers. We must continuously develop new cancer diagnostic tests and enhance any existing tests to keep pace with evolving standards of care. Our current test and our planned tests could become obsolete unless we continually innovate and expand them to demonstrate benefit in the diagnosis, monitoring or prognosis of patients with cancer. New cancer therapies typically have only a few years of clinical data associated with them, which limits our ability to develop cancer diagnostic tests based on,
41
Table of Contents
for example, biomarker analysis related to the appearance or development of resistance to those therapies. If we cannot adequately demonstrate the applicability of our current test and our planned tests to new treatments, by incorporating important biomarker analysis, sales of our tests could decline, which would have a material adverse effect on our business, financial condition and results of operations.
If our current test and our planned tests do not continue to perform as expected, our operating results, reputation and business will suffer.
Our success depends on the market’s confidence that we can continue to provide reliable, high-quality diagnostic results. We believe that our customers are likely to be particularly sensitive to test defects and errors. As a result, the failure of our current or planned tests to perform as expected would significantly impair our reputation and the public image of our cancer tests, and we may be subject to legal claims arising from any defects or errors.
If our sole laboratory facility becomes damaged or inoperable, or we are required to vacate the facility, our ability to sell and provide cancer diagnostic tests and pursue our research and development efforts may be jeopardized.
We currently derive our revenues from our OncoCEE-BR breast cancer diagnostic tests conducted in our CLIA-certified laboratory. We do not have any clinical reference laboratory facilities outside of our facility in San Diego, California. Our facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including fire, earthquake, flooding and power outages, which may render it difficult or impossible for us to perform our diagnostic tests for some period of time. The inability to perform our current test and our planned tests or the backlog of tests that could develop if our facility is inoperable for even a short period of time may result in the loss of customers or harm to our reputation or relationships with scientific or clinical collaborators, and we may be unable to regain those customers or repair our reputation in the future. Furthermore, our facilities and the equipment we use to perform our research and development work could be costly and time-consuming to repair or replace.
The San Diego area has recently experienced serious fires and power outages, and is considered to lie in an area with earthquake risk.
Additionally, a key component of our research and development process involves using biological samples as the basis for our diagnostic test development. In some cases, these samples are difficult to obtain. If the parts of our laboratory facility where we store these biological samples were damaged or compromised, our ability to pursue our research and development projects, as well as our reputation, could be jeopardized. We carry insurance for damage to our property and the disruption of our business, but this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
Further, if our CLIA-certified laboratory became inoperable we may not be able to license or transfer our technology to another facility with the necessary state licensure and CLIA certification under the scope of which our current test and our planned cancer diagnostic tests could be performed. Even if we find a facility with such qualifications to perform our tests, it may not be available to us on commercially reasonable terms.
If we cannot compete successfully with our competitors, we may be unable to increase or sustain our revenues or achieve and sustain profitability.
Our principal competition comes from mainstream diagnostic methods, used by pathologists and oncologists for many years, which focus on tumor tissue analysis. It may be difficult to change the methods or behavior of oncologists to incorporate our CTC and ctDNA testing, including molecular diagnostic testing, in their practices in conjunction with or instead of tissue biopsies and analysis. In addition, companies offering capital equipment and kits or reagents to local pathology laboratories represent another source of potential competition. These kits are used directly by the pathologist, which can facilitate adoption. We plan to focus our marketing and sales efforts on medical oncologists rather than pathologists.
42
Table of Contents
We also face competition from companies that offer products or are conducting research to develop products for CTC or ctDNA testing in various cancers. In particular, Janssen Diagnostics, LLC markets its CellSearch® test and Atossa Genetics markets its ArgusCYTE® test, which are competitive to our OncoCEE-BR test for CTC enumeration, and HER2 analysis, respectively. CTC and ctDNA testing is a new area of science and we cannot predict what tests others will develop that may compete with or provide results similar or superior to the results we are able to achieve with the tests we develop. In addition to Janssen Diagnostics and Atossa Genetics, our competitors also include public companies such as Alere (Adnagen) and Illumina as well as many private companies, including Apocell, EPIC Sciences, Clearbridge Biomedics, Cynvenio Biosystems, Fluxion Biosciences, RareCells, ScreenCell and Silicon Biosystems. Many of these groups, in addition to operating research and development laboratories, are establishing CLIA-certified testing laboratories while others are focused on selling equipment and reagents. Our sales and distribution agreements are non-exclusive and our partners could enter into agreements with competitors.
We expect that pharmaceutical and biopharmaceutical companies will increasingly focus attention and resources on the personalized cancer diagnostic sector as the potential and prevalence of molecularly targeted oncology therapies approved by the FDA along with companion diagnostics increases. For example, the FDA has recently approved two such agents—Xalkori® from Pfizer Inc. along with its companion anaplastic lymphoma kinase FISH test from Abbott Laboratories, Inc., Zelboraf® from Daiichi-Sankyo/Genentech/Roche along with its companion B-raf kinase V600 mutation test from Roche Molecular Systems, Inc. and Tafinlar® from GlaxoSmithKline along with its companion B-raf kinase V600 mutation test from bioMerieux. These recent FDA approvals are only the second, third and fourth instances of simultaneous approvals of a drug and companion diagnostic, the first being the 2010 approval of Genentech’s Herceptin® for HER2 positive breast cancer along with the HercepTest from partner Dako A/S. Our competitors may invent and commercialize technology platforms or tests that compete with ours.
There are a number of companies which are focused on the oncology diagnostic market, such as Biodesix, Caris, Clarient, Foundation Medicine, Neogenomics, Response Genetics, Agendia, Genomic Health, and Genoptix, who while not currently offering CTC or ctDNA tests are selling to the medical oncologists and pathologists and could develop or offer CTC or ctDNA tests. Large laboratory services companies, such as Sonic USA, Quest and LabCorp, provide more generalized cancer diagnostic testing.
Additionally, projects related to cancer diagnostics and particularly genomics have received increased government funding, both in the United States and internationally. As more information regarding cancer genomics becomes available to the public, we anticipate that more products aimed at identifying targeted treatment options will be developed and that these products may compete with ours. In addition, competitors may develop their own versions of our current or planned tests in countries where we did not apply for patents or where our patents have not issued and compete with us in those countries, including encouraging the use of their test by physicians or patients in other countries.
Some of our present and potential competitors have widespread brand recognition and substantially greater financial and technical resources and development, production and marketing capabilities than we do. Others may develop lower-priced, less complex tests that payors, pathologists and oncologists could view as functionally equivalent to our current or planned tests, which could force us to lower the list price of our tests and impact our operating margins and our ability to achieve and maintain profitability. In addition, technological innovations that result in the creation of enhanced diagnostic tools that are more sensitive or specific than ours may enable other clinical laboratories, hospitals, physicians or medical providers to provide specialized diagnostic tests similar to ours in a more patient-friendly, efficient or cost-effective manner than is currently possible. If we cannot compete successfully against current or future competitors, we may be unable to increase or create market acceptance and sales of our current or planned tests, which could prevent us from increasing or sustaining our revenues or achieving or sustaining profitability.
43
Table of Contents
We expect to continue to incur significant expenses to develop and market cancer diagnostic tests, which could make it difficult for us to achieve and sustain profitability.
In recent years, we have incurred significant costs in connection with the development of cancer diagnostic tests. For the year ended December 31, 2012, our research and development expenses were $6.6 million and our sales and marketing expenses were $0.8 million. For the year ended December 31, 2013, our research and development expenses were $3.1 million and our sales and marketing expenses were $0.1 million. We expect our expenses to continue to increase for the foreseeable future as we conduct studies of our current test and our planned cancer diagnostic tests, establish a sales and marketing organization, drive adoption of and reimbursement for our diagnostic tests and develop new tests. As a result, we need to generate significant revenues in order to achieve sustained profitability.
If oncologists decide not to order OncoCEE-BR breast cancer diagnostic tests or our future cancer diagnostic tests, we may be unable to generate sufficient revenue to sustain our business.
To generate demand for our current test and our planned cancer diagnostic tests, we will need to educate oncologists, pathologists, and other health care professionals on the clinical utility, benefits and value of the tests we provide through published papers, presentations at scientific conferences, educational programs and one-on-one education sessions by members of our sales force. In addition, we need to assure oncologists of our ability to obtain and maintain adequate reimbursement coverage from third-party payors. We need to hire additional commercial, scientific, technical and other personnel to support this process. If we cannot convince medical practitioners to order our current test and our planned tests, we will likely be unable to create demand in sufficient volume for us to achieve sustained profitability.
Clinical utility studies are important in demonstrating to both customers and payors a test’s clinical relevance and value. If we are unable to identify collaborators willing to work with us to conduct clinical utility studies, or the results of those studies do not demonstrate that a test provides clinically meaningful information and value, commercial adoption of such test may be slow, which would negatively impact our business.
Clinical utility studies show when and how to use a clinical test, and describe the particular clinical situations or settings in which it can be applied and the expected results. Clinical utility studies also show the impact of the test results on patient care and management. Clinical utility studies are typically performed with collaborating oncologists at medical centers and hospitals, analogous to a clinical trial, and generally result in peer-reviewed publications. Sales and marketing representatives use these publications to demonstrate to customers how to use a clinical test, as well as why they should use it. These publications are also used with payors to obtain coverage for a test, helping to assure there is appropriate reimbursement.
We are currently conducting a clinical utility study for our OncoCEE-BR test with investigators at the Dana-Farber Cancer Institute. We will need to conduct additional studies for this test, as well as other CTC and ctDNA tests we plan to introduce, to drive test adoption in the marketplace and reimbursement. Should we not be able to perform these studies, or should their results not provide clinically meaningful data and value for oncologists, adoption of our tests could be impaired and we may not be able to obtain reimbursement for them.
We are undergoing a management transition.
Until August 26, 2013, David F. Hale, our Executive Chairman, served as our principal executive officer. On that date, Michael W. Nall began his employment with us as our Chief Executive Officer and President, with David F. Hale remaining employed as our Executive Chairman. We intend to recruit and hire other senior executives. Such a management transition subjects us to a number of risks, including risks pertaining to coordination of responsibilities and tasks, creation of new management systems and processes, differences in management style, effects on corporate culture, and the need for transfer of historical knowledge. In addition, Mr. Nall has not previously been the chief executive officer of a public or private company, and therefore his
44
Table of Contents
lack of experience may result in some of his time being spent acclimating to his new position and responsibilities. A lack of significant experience in being the chief executive officer of a public company could have an adverse effect on his ability to quickly respond to problems or effectively manage issues surrounding the operation of a public company.
The loss of key members of our executive management team could adversely affect our business.
Our success in implementing our business strategy depends largely on the skills, experience and performance of key members of our executive management team and others in key management positions, including Michael W. Nall, our Chief Executive Officer and President, William G. Kachioff, our Senior Vice-President of Finance /Chief Financial Officer, Lyle J. Arnold, Ph.D., our Senior Vice-President of Research & Development/Chief Scientific Officer, and Farideh Z. Bischoff, Ph.D., our Vice-President of Translational Research and Clinical Development. The collective efforts of each of these persons and others working with them as a team are critical to us as we continue to develop our technologies, tests and research and development and sales programs. As a result of the difficulty in locating qualified new management, the loss or incapacity of existing members of our executive management team could adversely affect our operations. If we were to lose one or more of these key employees, we could experience difficulties in finding qualified successors, competing effectively, developing our technologies and implementing our business strategy. Our Chief Executive Officer and President, Chief Financial Officer and Chief Scientific Officer have employment agreements, however, the existence of an employment agreement does not guarantee retention of members of our executive management team and we may not be able to retain those individuals for the duration of or beyond the end of their respective terms. We do not maintain “key person” life insurance on any of our employees.
In addition, we rely on collaborators, consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our collaborators, consultants and advisors are generally employed by employers other than us and may have commitments under agreements with other entities that may limit their availability to us.
The loss of a key employee, the failure of a key employee to perform in his or her current position or our inability to attract and retain skilled employees could result in our inability to continue to grow our business or to implement our business strategy.
There is a scarcity of experienced professionals in our industry. If we are not able to retain and recruit personnel with the requisite technical skills, we may be unable to successfully execute our business strategy.
The specialized nature of our industry results in an inherent scarcity of experienced personnel in the field. Our future success depends upon our ability to attract and retain highly skilled personnel, including scientific, technical, commercial, business, regulatory and administrative personnel, necessary to support our anticipated growth, develop our business and perform certain contractual obligations. Given the scarcity of professionals with the scientific knowledge that we require and the competition for qualified personnel among life science businesses, we may not succeed in attracting or retaining the personnel we require to continue and grow our operations.
Our inability to attract, hire and retain a sufficient number of qualified sales professionals would hamper our ability to increase demand for our cancer diagnostic test, to expand geographically and to successfully commercialize any other tests or products we may develop.
To succeed in selling our breast cancer diagnostic test and any other tests or products that we are able to develop, we must expand our sales force in the United States and/or internationally by recruiting additional sales representatives with extensive experience in oncology and close relationships with medical oncologists, surgeons, oncology nurses, pathologists and other hospital personnel. To achieve our marketing and sales goals, we will need to substantially build our sales and commercial infrastructure, with which to date we have had little
45
Table of Contents
experience. Sales professionals with the necessary technical and business qualifications are in high demand, and there is a risk that we may be unable to attract, hire and retain the number of sales professionals with the right qualifications, scientific backgrounds and relationships with decision-makers at potential customers needed to achieve our sales goals. We expect to face competition from other companies in our industry, some of whom are much larger than us and who can pay greater compensation and benefits than we can, in seeking to attract and retain qualified sales and marketing employees. If we are unable to hire and retain qualified sales and marketing personnel, our business will suffer.
Our dependence on commercialization partners for sales of tests could limit our success in realizing revenue growth.
We intend to grow our business through the use of commercialization partners for the sales, marketing and commercialization of our current test and our planned future tests, and to do so we must enter into agreements with these partners to sell, market or commercialize our tests. These agreements may contain exclusivity provisions and generally cannot be terminated without cause during the term of the agreement. We may need to attract additional partners to expand the markets in which we sell tests. These partners may not commit the necessary resources to market and sell our cancer diagnostics tests to the level of our expectations, and we may be unable to locate suitable alternatives should we terminate our agreement with such partners or if such partners terminate their agreement with us.
Any relationships we form with commercialization partners are subject to change over time. For example, over 75% of our revenue in 2012 was generated through our arrangement with Clarient, but Clarient is no longer marketing the OncoCEE-BR test as actively as before. In May 2013, we amended our commercialization agreement with Clarient such that Clarient is no longer the exclusive marketer of the OncoCEE-BR test. We expect that in the future the percentage of our revenue which is generated through our arrangement with Clarient will diminish. If we cannot replace any diminution in revenues we receive through Clarient, our results will be weakened.
If current or future commercialization partners do not perform adequately, or we are unable to locate commercialization partners, we may not realize revenue growth.
We depend on third parties for the supply of blood samples and other biological materials that we use in our research and development efforts. If the costs of such samples and materials increase or our third party suppliers terminate their relationship with us, our business may be materially harmed.
We have relationships with suppliers and institutions that provide us with blood samples and other biological materials that we use in developing and validating our current test and our planned future tests. If one or more suppliers terminate their relationship with us or are unable to meet our requirements for samples, we will need to identify other third parties to provide us with blood samples and biological materials, which could result in a delay in our research and development activities and negatively affect our business. In addition, as we grow, our research and academic institution collaborators may seek additional financial contributions from us, which may negatively affect our results of operations.
We currently rely on third-party suppliers for critical materials needed to perform our current test and our planned future tests and any problems experienced by them could result in a delay or interruption of their supply to us.
We currently purchase raw materials for our microfluidic channels and testing reagents under purchase orders and do not have long-term contracts with most of the suppliers of these materials. If suppliers were to delay or stop producing our materials or reagents, or if the prices they charge us were to increase significantly, or if they elected not to sell to us, we would need to identify other suppliers. We could experience delays in manufacturing the microfluidic channels or performing tests while finding another acceptable supplier, which
46
Table of Contents
could impact our results of operations. The changes could also result in increased costs associated with qualifying the new materials or reagents and in increased operating costs. Further, any prolonged disruption in a supplier’s operations could have a significant negative impact on our ability to perform cancer diagnostic tests in a timely manner.
Some of the components used in our current or planned products are currently sole-source, and substitutes for these components might not be able to be obtained easily or may require substantial design or manufacturing modifications. Any significant problem experienced by one of our sole source suppliers may result in a delay or interruption in the supply of components to us until that supplier cures the problem or an alternative source of the component is located and qualified. Any delay or interruption would likely lead to a delay or interruption in our manufacturing operations. The inclusion of substitute components must meet our product specifications and could require us to qualify the new supplier with the appropriate government regulatory authorities.
If we were sued for product liability or professional liability, we could face substantial liabilities that exceed our resources.
The marketing, sale and use of our current test and our planned future diagnostic tests could lead to the filing of product liability claims against us if someone alleges that our tests failed to perform as designed. We may also be subject to liability for errors in the test results we provide to physicians or for a misunderstanding of, or inappropriate reliance upon, the information we provide. A product liability or professional liability claim could result in substantial damages and be costly and time-consuming for us to defend.
Although we believe that our existing product and professional liability insurance is adequate, our insurance may not fully protect us from the financial impact of defending against product liability or professional liability claims. Any product liability or professional liability claim brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any product liability lawsuit could damage our reputation, result in the recall of tests, or cause current partners to terminate existing agreements and potential partners to seek other partners, any of which could impact our results of operations.
If we use biological and hazardous materials in a manner that causes injury, we could be liable for damages.
Our activities currently require the controlled use of potentially harmful biological materials and chemicals. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject to, on an ongoing basis, federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations may become significant and could have a material adverse effect on our financial condition, results of operations and cash flows. In the event of an accident or if we otherwise fail to comply with applicable regulations, we could lose our permits or approvals or be held liable for damages or penalized with fines.
We may acquire other businesses or form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, increase our debt or cause us to incur significant expense.
As part of our business strategy, we may pursue acquisitions of businesses and assets. We also may pursue strategic alliances and joint ventures that leverage our core technology and industry experience to expand our offerings or distribution. We have no experience with acquiring other companies and limited experience with forming strategic alliances and joint ventures. We may not be able to find suitable partners or acquisition candidates, and we may not be able to complete such transactions on favorable terms, if at all. If we make any
47
Table of Contents
acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. Any future acquisitions also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could have a material adverse effect on our financial condition, results of operations and cash flows. Integration of an acquired company also may disrupt ongoing operations and require management resources that would otherwise focus on developing our existing business. We may experience losses related to investments in other companies, which could have a material negative effect on our results of operations. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance or joint venture.
To finance any acquisitions or joint ventures, we may choose to issue shares of our common stock as consideration, which would dilute the ownership of our stockholders. If the price of our common stock is low or volatile, we may not be able to acquire other companies or fund a joint venture project using our stock as consideration. Alternatively, it may be necessary for us to raise additional funds for acquisitions through public or private financings. Additional funds may not be available on terms that are favorable to us, or at all.
If we cannot support demand for our current test and our planned future diagnostic tests, including successfully managing the evolution of our technology and manufacturing platforms, our business could suffer.
As our test volume grows, we will need to increase our testing capacity, implement automation, increase our scale and related processing, customer service, billing, collection and systems process improvements and expand our internal quality assurance program and technology to support testing on a larger scale. We will also need additional cytogenetic technicians, certified laboratory scientists and other scientific and technical personnel to process these additional tests. Any increases in scale, related improvements and quality assurance may not be successfully implemented and appropriate personnel may not be available. As additional tests are commercialized, we may need to bring new equipment on line, implement new systems, technology, controls and procedures and hire personnel with different qualifications. Failure to implement necessary procedures or to hire the necessary personnel could result in a higher cost of processing or an inability to meet market demand. We cannot assure you that we will be able to perform tests on a timely basis at a level consistent with demand, that our efforts to scale our commercial operations will not negatively affect the quality of our test results or that we will respond successfully to the growing complexity of our testing operations. If we encounter difficulty meeting market demand or quality standards for our current test and our planned future tests, our reputation could be harmed and our future prospects and business could suffer, which may have a material adverse effect on our financial condition, results of operations and cash flows.
We may encounter manufacturing problems or delays that could result in lost revenue.
We currently manufacture our proprietary microfluidic channels at our San Diego facility and intend to continue to do so. We believe we currently have adequate manufacturing capacity for our microfluidic channels. If demand for our current test and our planned future tests increases significantly, we will need to either expand our manufacturing capabilities or outsource to other manufacturers. If we or third party manufacturers engaged by us fail to manufacture and deliver our microfluidic channels or certain reagents in a timely manner, our relationships with our customers could be seriously harmed. We cannot assure you that manufacturing or quality control problems will not arise as we attempt to increase the production of our microfluidic channels or reagents or that we can increase our manufacturing capabilities and maintain quality control in a timely manner or at commercially reasonable costs. If we cannot manufacture our microfluidic channels consistently on a timely basis because of these or other factors, it could have a significant negative impact on our ability to perform tests and generate revenues.
48
Table of Contents
International expansion of our business would expose us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Our business strategy contemplates possible international expansion, including partnering with academic and commercial testing laboratories, and introducing OncoCEE technology outside the United States as part of CE-marked IVD test kits and/or testing systems utilizing our CEE and/or CEE-Selector technologies. Doing business internationally involves a number of risks, including:
| • | multiple, conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| • | failure by us or our distributors to obtain regulatory approvals for the sale or use of our current test and our planned future tests in various countries; |
| • | difficulties in managing foreign operations; |
| • | complexities associated with managing government payor systems, multiple payor-reimbursement regimes or self-pay systems; |
| • | logistics and regulations associated with shipping blood samples, including infrastructure conditions and transportation delays; |
| • | limits on our ability to penetrate international markets if our current test and our planned future diagnostic tests cannot be processed by an appropriately qualified local laboratory; |
| • | financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable and exposure to foreign currency exchange rate fluctuations; |
| • | reduced protection for intellectual property rights, or lack of them in certain jurisdictions, forcing more reliance on our trade secrets, if available; |
| • | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and |
| • | failure to comply with the Foreign Corrupt Practices Act, including its books and records provisions and its anti-bribery provisions, by maintaining accurate information and control over sales activities and distributors’ activities. |
Any of these risks, if encountered, could significantly harm our future international expansion and operations and, consequently, have a material adverse effect on our financial condition, results of operations and cash flows.
Declining general economic or business conditions may have a negative impact on our business.
Continuing concerns over United States health care reform legislation and energy costs, geopolitical issues, the availability and cost of credit and government stimulus programs in the United States and other countries have contributed to increased volatility and diminished expectations for the global economy. These factors, combined with low business and consumer confidence and high unemployment, precipitated an economic slowdown and recession. If the economic climate does not improve, or it deteriorates, our business, including our access to patient samples and the addressable market for diagnostic tests that we may successfully develop, as well as the financial condition of our suppliers and our third-party payors, could be adversely affected, resulting in a negative impact on our business, financial condition and results of operations.
Intrusions into our computer systems could result in compromise of confidential information.
Despite the implementation of security measures, our technology or systems that we interface with, including the Internet and related systems, may be vulnerable to physical break-ins, hackers, improper employee
49
Table of Contents
or contractor access, computer viruses, programming errors, or similar problems. Any of these might result in confidential medical, business or other information of other persons or of ourselves being revealed to unauthorized persons.
There are a number of state, federal and international laws protecting the privacy and security of health information and personal data. As part of the American Recovery and Investment Act 2009, or ARRA, Congress amended the privacy and security provisions of the Health Insurance Portability and Accountability Act, or HIPAA. HIPAA imposes limitations on the use and disclosure of an individual’s healthcare information by healthcare providers, healthcare clearinghouses, and health insurance plans, collectively referred to as covered entities. The HIPAA amendments also impose compliance obligations and corresponding penalties for non-compliance on individuals and entities that provide services to healthcare providers and other covered entities, collectively referred to as business associates. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s health information under HIPAA and extended enforcement authority to state attorneys general. The amendments also create notification requirements for individuals whose health information has been inappropriately accessed or disclosed: notification requirements to federal regulators and in some cases, notification to local and national media. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with encryption or other standards developed by the U.S. Department of Health and Human Services, or HHS. Most states have laws requiring notification of affected individuals and state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms to ensure ongoing protection of personal information. Activities outside of the United States implicate local and national data protection standards, impose additional compliance requirements and generate additional risks of enforcement for non-compliance. We may be required to expend significant capital and other resources to ensure ongoing compliance with applicable privacy and data security laws, to protect against security breaches and hackers or to alleviate problems caused by such breaches.
We depend on our information technology and telecommunications systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant aspects of our operations. In addition, our third-party billing and collections provider depends upon telecommunications and data systems provided by outside vendors and information we provide on a regular basis. These information technology and telecommunications systems support a variety of functions, including test processing, sample tracking, quality control, customer service and support, billing and reimbursement, research and development activities and our general and administrative activities. Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of our information technology or telecommunications systems or those used by our third-party service providers could prevent us from processing tests, providing test results to oncologists, pathologists, billing payors, processing reimbursement appeals, handling patient or physician inquiries, conducting research and development activities and managing the administrative aspects of our business. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business.
50
Table of Contents
Regulatory Risks Relating to Our Business
Healthcare policy changes, including recently enacted legislation reforming the U.S. health care system, may have a material adverse effect on our financial condition, results of operations and cash flows.
The 2010 Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the ACA, makes a number of substantial changes in the way health care is financed by both governmental and private insurers. Among other things, the ACA:
| • | Mandates a reduction in payments for clinical laboratory services paid under the Medicare Clinical Laboratory Fee Schedule annual Consumer Price Index update of 1.75% for the years 2011 through 2015. In addition, a permanent productivity adjustment is made to the fee schedule payment amount, which could range from 1.1% to 1.4% each year over the next 10 years. These changes in payments may apply to some or all of the tests we furnish to Medicare beneficiaries. |
| • | Establishes an Independent Payment Advisory Board to reduce the per capita rate of growth in Medicare spending if spending exceeds a target growth rate. The Independent Payment Advisory Board has broad discretion to propose policies, which may have a negative impact on payment rates for services, including clinical laboratory services, beginning in 2016, and for hospital services beginning in 2020. |
| • | Requires each medical device manufacturer to pay an excise tax equal to 2.3% of the price for which such manufacturer sells its medical devices, beginning in 2013. We believe that at this time this tax does not apply to our current cancer diagnostic test or to our products that are in development; nevertheless, this could change in the future if either the FDA or the Internal Revenue Service, which regulates the payment of this excise tax, changes its position. |
Although some of these provisions may negatively impact payment rates for clinical laboratory tests, the ACA also extends coverage to over 30 million previously uninsured people, which may result in an increase in the demand for our current test and our planned future cancer diagnostic tests. The mandatory purchase of insurance has been strenuously opposed by a number of state governors, resulting in lawsuits challenging the constitutionality of certain provisions of the ACA. In 2012, the Supreme Court upheld the constitutionality of the ACA, with the exception of certain provisions dealing with the expansion of Medicaid coverage under the law. Therefore, most of the law’s provisions will go into effect in 2013 and 2014. Congress has also proposed a number of legislative initiatives, including possible repeal of the ACA. At this time, it remains unclear whether there will be any changes made to the ACA, whether in part or in its entirety.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. The Budget Control Act of 2011, among other things, created the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers and suppliers of up to 2% per fiscal year, starting in 2013. The full impact on our business of the ACA and the sequester law is uncertain. In addition, the Middle Class Tax Relief and Job Creation Act of 2012, or MCTRJCA, mandated an additional change in Medicare reimbursement for clinical laboratory tests. This legislation requires a rebasing of the Medicare Clinical Laboratory Fee Schedule to effect a 2% reduction in payment rates otherwise determined for 2013. This will serve as a base for 2014 and subsequent years. In January 2013, as a result of the changes mandated by the ACA and MCTRJCA, the Centers for Medicare & Medicaid Services, or CMS, reduced its reimbursement for laboratory tests for 2013 by approximately 3%.
Some of our laboratory test business is subject to the Medicare Physician Fee Schedule and, under the current statutory formula, the rates for these services are updated annually. For the past several years, the application of the statutory formula would have resulted in substantial payment reductions if Congress failed to intervene. In the past, Congress passed interim legislation to prevent the decreases. On November 1, 2013, CMS issued its 2014 Physician Fee Schedule Final Rule, or the 2014 Final Rule. In the 2014 Final Rule, CMS called
51
Table of Contents
for a reduction of approximately 23.7% in the 2014 conversion factor that is used to calculate physician reimbursement. If in future years Congress does not adopt interim legislation to block or offset, and/or CMS does not moderate, any substantial CMS-proposed reimbursement reductions, the resulting decrease in payments from Medicare could adversely impact our revenues and results of operations.
In addition, many of the Current Procedure Terminology, or CPT, codes that we use to bill for cancer diagnostic tests were revised by the American Medical Association, effective January 1, 2013. In the 2013 Final Rule, CMS announced that it has decided to keep the new molecular codes on the Clinical Laboratory Fee Schedule rather than move them to the Physician Fee Schedule as some stakeholders had urged. Our reimbursement could be adversely affected by CMS’ actions. If it reduces reimbursement for the new test codes or does not pay for our codes, then our revenues would be adversely affected. There can be no guarantees that Medicare and other payors will establish positive or adequate coverage policies or reimbursement rates.
On July 9, 2013, CMS issued a proposed Physician Fee Schedule revision that would, in the aggregate, impose a 25% reduction for payments for pathology codes when services are provided by independent laboratories, to take effect beginning with calendar year 2014. The proposed cuts for certain services were drastic. For example, reimbursement for the technical component of FISH analysis would have been cut by 68%. We cannot predict the outcome of this initiative. However, the 2014 Physician Fee Schedule Final Rule issued by CMS in November 2013 left FISH reimbursement rates for independent laboratories and physicians essentially unchanged from 2013 reimbursement levels.
We cannot predict whether future health care initiatives will be implemented at the federal or state level, or how any future legislation or regulation may affect us. The expansion of government’s role in the U.S. health care industry as a result of the ACA’s implementation, and changes to the reimbursement amounts paid by Medicare and other payors for our current test and our planned future cancer diagnostic tests, may reduce our profits, if any, and have a materially adverse effect on our business, financial condition, results of operations and cash flows. Moreover, Congress has proposed on several occasions to impose a 20% coinsurance payment requirement on patients for clinical laboratory tests reimbursed under the Medicare Clinical Laboratory Fee Schedule, which would require us to bill patients for these amounts. In the event that Congress were to ever enact such legislation, the cost of billing and collecting for our tests could often exceed the amount actually received from the patient.
Our commercial success could be compromised if hospitals or other clients do not pay our invoices or if third-party payors, including managed care organizations and Medicare, do not provide coverage and reimbursement, breach, rescind or modify their contracts or reimbursement policies or delay payments for our current test and our planned future tests.
Oncologists may not order our current breast cancer test and our planned future cancer diagnostic tests unless third-party payors, such as managed care organizations and government payors (e.g., Medicare and Medicaid), pay a substantial portion of the test price. Coverage and reimbursement by a third-party payor may depend on a number of factors, including a payor’s determination that tests using our technologies are:
| • | not experimental or investigational; |
| • | medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; |
| • | supported by peer-reviewed publications; and |
| • | included in clinical practice guidelines. |
Uncertainty surrounds third-party payor reimbursement of any test incorporating new technology, including tests developed using our technologies. Technology assessments of new medical tests conducted by research
52
Table of Contents
centers and other entities may be disseminated to interested parties for informational purposes. Third-party payors and health care providers may use such technology assessments as grounds to deny coverage for a test or procedure. Technology assessments can include evaluation of clinical utility studies, which define how a test is used in a particular clinical setting or situation.
Because each payor generally determines for its own enrollees or insured patients whether to cover or otherwise establish a policy to reimburse our cancer diagnostic tests, seeking payor approvals is a time-consuming and costly process. We cannot be certain that coverage for our current test and our planned future tests will be provided in the future by additional third-party payors or that existing agreements, policy decisions or reimbursement levels will remain in place or be fulfilled under existing terms and provisions. If we cannot obtain coverage and reimbursement from private and governmental payors such as Medicare and Medicaid for our current test, or new tests or test enhancements that we may develop in the future, our ability to generate revenues could be limited, which may have a material adverse effect on our financial condition, results of operations and cash flow. Further, we have experienced in the past, and will likely experience in the future, delays and interruptions in the receipt of payments from third-party payors due to missing documentation and/or other issues, which could cause delay in collecting our revenue.
In addition, to the extent that our testing is ordered for Medicare inpatients and outpatients, only the hospital may receive payment from the Medicare program for the technical component of pathology services and any clinical laboratory services that we perform, unless the testing is ordered at least 14 days after discharge and certain other requirements are met. We therefore must look to the hospital for payment for these services under these circumstances. If hospitals refuse to pay for the services or fail to pay in a timely manner, our ability to generate revenues could be limited, which may have a material adverse effect on our financial condition, results of operations and cash flow.
We expect to depend on Medicare and a limited number of private payors for a significant portion of our revenues and if these or other payors stop providing reimbursement or decrease the amount of reimbursement for our current test and our planned future tests, our revenues could decline.
We believe, based on research showing that approximately 54% of new cancers occur in persons age 65 and older and that almost all Americans age 65 and older are enrolled in Medicare, that a substantial portion of the patients for whom we would expect to perform cancer diagnostic tests will have Medicare as their primary medical insurance. Only in November 2013 did we first directly bill any payor for physician-ordered testing; until May 2013, our commercialization partner Clarient was responsible for all billing associated with our tests. We do not have data for Clarient’s billing and collection experience with respect to our test, because Clarient paid us a contracted amount per test performed regardless of their billing and collections. From May to December 2013, we performed an average of 1-3 physician-ordered tests per month (in addition to the 20-30 tests per month which we have been performing since January 2013 for a clinical utility study with investigators at the Dana-Farber Cancer Institute). Billing for physician-ordered tests is now handled for us by a non-Clarient billing service provider. In 2013 we invoiced, through this service provider, for 17 physician-ordered tests. Of these, 8 tests were billed to Medicare and the remainder were billed to other payors. We have been paid for one of these tests while we have not yet had any response or adjudication from any other payor as to the bills submitted in late 2013. Accordingly, we do not yet have any data regarding reimbursement history or collectability experience. In addition, we believe the sample size of 17 is too small to be the basis for any conclusion about our ongoing payor mix.
Medicare and other third-party payors may change their coverage policies or cancel future contracts with us at any time, review and adjust the rate of reimbursement or stop paying for our tests altogether, which would reduce our total revenues. Payors have increased their efforts to control the cost, utilization and delivery of health care services. In the past, measures have been undertaken to reduce payment rates for and decrease utilization of the clinical laboratory testing generally. Because of the cost-trimming trends, third-party payors that currently cover and provide reimbursement for our current breast cancer test and our planned future cancer diagnostic tests may suspend, revoke or discontinue coverage at any time, or may reduce the reimbursement rates payable to us.
53
Table of Contents
Any such action could have a negative impact on our revenues, which may have a material adverse effect on our financial condition, results of operations and cash flows.
In addition, we are currently considered a “non-contracted provider” by private third-party payors because we have not entered into a specific contract to provide cancer diagnostic tests to their insured patients at specified rates of reimbursement. If we were to become a contracted provider with one more payors in the future, the amount of overall reimbursement we receive would likely decrease because we could be reimbursed less money per test performed at a contracted rate than at a non-contracted rate, which could have a negative impact on our revenues. Further, we typically are unable to collect payments from patients beyond that which is paid by their insurance and will continue to experience lost revenue as a result.
Because of certain Medicare billing policies, we may not receive complete reimbursement for tests provided to Medicare patients. Medicare reimbursement revenues are an important component of our business model, and private payors sometimes look to Medicare determinations when making their own payment determinations; therefore, incomplete or inadequate reimbursement from Medicare would negatively affect our business.
Medicare has coverage policies that can be national or regional in scope. Coverage means that the test or assay is approved as a benefit for Medicare beneficiaries. If there is no coverage, neither the supplier nor any other party, such as a reference laboratory, may bill Medicare or the beneficiary for the service. There is currently no national coverage policy regarding the CTC capture/enumeration portion of our testing. Because our laboratory is in California, the regional Medicare Administrative Contractor , or MAC, for California is the relevant MAC for all our testing. The previous MAC for California, Palmetto GBA, LLC, adopted a negative coverage policy for CTC capture/enumeration (with the exception that Janssen Diagnostics, LLC’s CellSearch® test has historically been covered for CTC capture/enumeration). The current MAC for California, Noridian Healthcare Solutions, LLC, is adopting the coverage policies from Palmetto GBA. Therefore the capture/enumeration portion of our OncoCEE testing is not currently covered and we will receive no payment from Medicare for this service unless and until the coverage policy is changed. On November 4, 2013, we submitted a comprehensive dossier explaining to Palmetto GBA and Noridian the benefits of the capture/enumeration testing in order to seek to persuade the MACs to allow coverage for this portion of our testing. Palmetto GBA responded on November 27, 2013, denying our request for Medicare coverage for the CTC capture/enumeration portion of our OncoCEE testing. We have not received any other indications to suggest that the negative coverage determination will be reversed. The earliest date we could submit another dossier on this matter is May 27, 2014. We intend to continue our efforts to obtain Medicare coverage for capture/enumeration.
We cannot assure you that, even if OncoCEE-BR and our planned tests are otherwise successful, reimbursement for the currently Medicare-covered portions of OncoCEE-BR and our planned tests would, without Medicare reimbursement for the capture/enumeration portion, produce sufficient revenues to enable us to reach profitability and achieve our other commercial objectives.
The processing of Medicare claims is subject to change at CMS’ discretion at any time. Cost containment initiatives may be a threat to Medicare reimbursement levels (including for the covered components of OncoCEE-BR and our planned tests, including FISH analysis and molecular testing) for the foreseeable future.
Long payment cycles of Medicare, Medicaid and/or other third-party payors, or other payment delays, could hurt our cash flows and increase our need for working capital.
Medicare and Medicaid have complex billing and documentation requirements that we must satisfy in order to receive payment, and the programs can be expected to carefully audit and monitor our compliance with these requirements. We must also comply with numerous other laws applicable to billing and payment for healthcare services, including privacy laws. Failure to comply with these requirements may result in non-payment, refunds, exclusion from government healthcare programs, and civil or criminal liabilities, any of which may have a material adverse effect on our revenues and earnings. In addition, failure by third-party payors to properly process our payment claims in a timely manner could delay our receipt of payment for our products and services, which may have a material adverse effect on our cash flows.
54
Table of Contents
Complying with numerous regulations pertaining to our business is an expensive and time-consuming process, and any failure to comply could result in substantial penalties.
We are subject to CLIA, a federal law regulating clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. Our clinical laboratory must be certified under CLIA in order for us to perform testing on human specimens. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. We have a current certificate of accreditation under CLIA to perform high complexity testing, and our laboratory is accredited by the College of American Pathologists, or CAP, one of six CLIA-approved accreditation organizations. To renew this certificate, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make periodic inspections of our clinical laboratory outside of the renewal process.
In addition, our laboratory is located in California and is required by state law to have a California state license; as we expand our geographic focus, we may need to obtain laboratory licenses from additional states. California laws establish standards for operation of our clinical laboratory, including the training and skills required of personnel and quality control. In addition, Florida, Maryland, New York and Rhode Island require that we hold licenses to test specimens from patients in those states or received from ordering physicians in those states. As part of this process, the State of New York requires validation of our tests. Pennsylvania licensure or registration may be required as well, depending on the circumstances. We currently do not have the necessary Florida and New York licenses, but we are in the process of addressing the requirements for licensure in those states. Other states may have similar requirements or may adopt similar requirements in the future. Finally, we may be subject to regulation in foreign jurisdictions if we seek to expand international distribution of our tests outside the United States.
If we were to lose our CLIA certification or California laboratory license, whether as a result of a revocation, suspension or limitation, we would no longer be able to offer our tests, which would limit our revenues and harm our business. If we were to lose our license in any other state where we are required to hold a license, we would not be able to test specimens from those states.
If the FDA were to begin requiring approval or clearance of our current test and our planned future tests, we could incur substantial costs and time delays associated with meeting requirements for pre-market clearance or approval or we could experience decreased demand for, or reimbursement of, our tests.
Although the FDA maintains that it has authority to regulate the development and use of laboratory developed tests, or LDTs, such as ours, as medical devices, it has not exercised its authority with respect to most LDTs as a matter of enforcement discretion. The FDA could, at any time, change its policy with regard to this matter.
We believe that our cancer diagnostic tests, as utilized in our clinical laboratory, are and would be LDTs. As a result, we believe that pursuant to the FDA’s current policies and guidance, the FDA does not require that we obtain regulatory clearances or approvals for our LDTs. The container we provide for collection and transport of blood samples from a health care provider to our clinical laboratory may be a medical device subject to the FDA regulation but is currently exempt from pre-market review by the FDA. While we believe that we are currently in material compliance with applicable laws and regulations, we cannot assure you that the FDA or other regulatory agencies would agree with our determination, and a determination that we have violated these laws, or a public announcement that we are being investigated for possible violations of these laws, could adversely affect our business, prospects, results of operations or financial condition.
Moreover, FDA policy pertaining to diagnostic testing is continuing to evolve and is subject to ongoing review and revision. A significant change in any of the laws, regulations or policies may require us to achieve regulatory compliance. At various times since 2006, the FDA has issued guidance documents or announced draft
55
Table of Contents
guidance regarding initiatives that may require varying levels of FDA oversight of our current test and our planned future tests. For example, in June 2010, the FDA announced a public meeting to discuss the agency’s oversight of LDTs prompted by the increased complexity of LDTs and their increasingly important role in clinical decision-making and disease management, particularly in the context of personalized medicine. The FDA indicated that it was considering a risk-based application of oversight to LDTs and that, following public input and discussion, it might issue separate draft guidance on the regulation of LDTs, which ultimately could require that we seek and obtain either pre-market clearance or approval of LDTs, depending upon the risk-based approach the FDA adopts. The public meeting was held in July 2010 and further public comments were submitted to the FDA through September 2010. The FDA has stated it is continuing to develop draft guidance in this area. Section 1143 of the Food and Drug Administration Safety and Innovation Act of 2012 requires the FDA to notify U.S. Congress at least 60 days before issuing a draft or final guidance regulating LDTs and to provide details of the anticipated action.
We cannot provide any assurance that FDA regulation, including pre-market review, will not be required in the future for our current test and our planned future tests, whether through additional guidance issued by the FDA, new enforcement policies adopted by the FDA or new legislation enacted by Congress. We believe it is possible that legislation will be enacted into law or guidance could be issued by the FDA which may result in increased regulatory burdens for us to continue to offer our breast cancer test or to develop and introduce new tests. Given the attention Congress continues to give to these issues, legislation affecting this area may be enacted into law and may result in increased regulatory burdens on us as we continue to offer our test and to develop and introduce new tests.
In addition, HHS requested that its Advisory Committee on Genetics, Health and Society make recommendations about the oversight of genetic testing. A final report was published in April 2008. If the report’s recommendations for increased oversight of genetic testing were to result in further regulatory burdens, they could negatively affect our business and delay the commercialization of tests in development.
The requirement of pre-market review could negatively affect our business until such review is completed and clearance to market or approval is obtained. The FDA could require that we stop selling our cancer diagnostic tests pending pre-market clearance or approval. If the FDA allows our tests to remain on the market but there is uncertainty about our tests, if they are labeled investigational by the FDA or if labeling claims the FDA allows us to make are very limited, orders from oncologists or reimbursement may decline. The regulatory approval process may involve, among other things, successfully completing additional clinical trials and making a 510(k) submission, or filing a pre-market approval application with the FDA. If the FDA requires pre-market review, our tests may not be cleared or approved on a timely basis, if at all. We may also decide voluntarily to pursue FDA pre-market review of our tests if we determine that doing so would be appropriate.
Additionally, should future regulatory actions affect any of the reagents we obtain from suppliers and use in conducting our tests, our business could be adversely affected in the form of increased costs of testing or delays, limits or prohibitions on the purchase of reagents necessary to perform our testing.
If we were required to conduct additional clinical studies or trials before continuing to offer tests that we have developed or may develop as LDTs, those studies or trials could lead to delays or failure to obtain necessary regulatory approval, which could cause significant delays in commercializing any future products and harm our ability to achieve sustained profitability.
If the FDA decides to require that we obtain clearance or approvals to commercialize our current breast cancer test or our planned future cancer diagnostic tests, we may be required to conduct additional pre-market clinical testing before submitting a regulatory notification or application for commercial sales. In addition, as part of our long-term strategy we may plan to seek FDA clearance or approval so we can sell our tests outside our CLIA laboratory; however, we would need to conduct additional clinical validation activities on our tests before we can submit an application for FDA approval or clearance. Clinical trials must be conducted in compliance with FDA regulations or the FDA may take enforcement action or reject the data. The data collected from these clinical trials may ultimately be used to support market clearance or approval for our tests. We believe it would
56
Table of Contents
likely take two years or more to conduct the clinical studies and trials necessary to obtain approval from the FDA to commercially launch our current test and our planned future tests outside of our clinical laboratory. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our test claims or that the FDA or foreign authorities will agree with our conclusions regarding our test results. Success in early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior clinical trials and studies. If we are required to conduct pre-market clinical trials, whether using prospectively acquired samples or archival samples, delays in the commencement or completion of clinical testing could significantly increase our test development costs and delay commercialization. Many of the factors that may cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to delay or denial of regulatory clearance or approval. The commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial. Moreover, the clinical trial process may fail to demonstrate that our current test and our planned future tests are effective for the proposed indicated uses, which could cause us to abandon a test candidate and may delay development of other tests.
We may find it necessary to engage contract research organizations to perform data collection and analysis and other aspects of our clinical trials, which might increase the cost and complexity of our trials. We may also depend on clinical investigators, medical institutions and contract research organizations to perform the trials properly. If these parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality, completeness or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our clinical trials may have to be extended, delayed or terminated. Many of these factors would be beyond our control. We may not be able to enter into replacement arrangements without undue delays or considerable expenditures. If there are delays in testing or approvals as a result of the failure to perform by third parties, our research and development costs would increase, and we may not be able to obtain regulatory clearance or approval for our current test and our planned future tests. In addition, we may not be able to establish or maintain relationships with these parties on favorable terms, if at all. Each of these outcomes would harm our ability to market our tests or to achieve sustained profitability.
We are subject to federal and state healthcare fraud and abuse laws and regulations and could face substantial penalties if we are unable to fully comply with such laws.
We are subject to health care fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. These health care laws and regulations include, for example:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from soliciting, receiving, offering or providing remuneration, directly or indirectly, in return for or to induce either the referral of an individual for, or the purchase order or recommendation of, any item or services for which payment may be made under a federal health care program such as the Medicare and Medicaid programs; |
| • | the federal physician self-referral prohibition, commonly known as the Stark Law, which prohibits physicians from referring Medicare or Medicaid patients to providers of “designated health services” with whom the physician or a member of the physician’s immediate family has an ownership interest or compensation arrangement, unless a statutory or regulatory exception applies; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which established federal crimes for knowingly and willfully executing a scheme to defraud any health care benefit program or making false statements in connection with the delivery of or payment for health care benefits, items or services; |
| • | federal false claims laws, which, prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, false or fraudulent claims for payment to the federal government; and |
57
Table of Contents
| • | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers. |
The risk of our being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Further, the ACA, among other things, amends the intent requirement of the federal Anti-Kickback Statute and certain criminal health care fraud statutes. Where the intent requirement has been lowered, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, because of amendments enacted in 2009 as part of the Fraud Enforcement and Recovery Act, the government may now assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Any action brought against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of these laws and regulations, we may be subject to any applicable penalty associated with the violation, including civil and criminal penalties, damages and fines, and/or exclusion from participation in Medicare, the California Medical Assistance Program (Medi-Cal—the California Medicaid program) or other state or federal health care programs. Additionally, we could be required to refund payments received by us, and we could be required to curtail or cease our operations. Any of the foregoing consequences could seriously harm our business and our financial results.
We may be required to comply with laws governing the transmission, security and privacy of health information that require significant compliance costs, and any failure to comply with these laws could result in material criminal and civil penalties.
Under the administrative simplification provisions of HIPAA, HHS has issued regulations which establish uniform standards governing the conduct of certain electronic health care transactions and protecting the privacy and security of Protected Health Information used or disclosed by health care providers and other covered entities.
The privacy regulations regulate the use and disclosure of Protected Health Information by health care providers engaging in certain electronic transactions or “standard transactions.” They also set forth certain rights that an individual has with respect to his or her Protected Health Information maintained by a covered health care provider, including the right to access or amend certain records containing Protected Health Information or to request restrictions on the use or disclosure of Protected Health Information. The HIPAA security regulations establish administrative, physical and technical standards for maintaining the integrity and availability of Protected Health Information in electronic form. These standards apply to covered health care providers and also to “business associates” or third parties providing services involving the use or disclosure of Protected Health Information. The HIPAA privacy and security regulations establish a uniform federal “floor” and do not supersede state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing Protected Health Information. As a result, we may be required to comply with both HIPAA privacy regulations and varying state privacy and security laws.
Moreover, the Health Information Technology for Economic and Clinical Health Act, or HITECH, among other things, established certain health information security breach notification requirements. In the event of a breach of unsecured Protected Health Information, a covered entity must notify each individual whose Protected Health Information is breached, federal regulators and in some cases, must publicize the breach in local or national media. Breaches affecting 500 individuals or more are publicized by federal regulators who publicly identify the breaching entity, the circumstances of the breach and the number of individuals affected.
These laws contain significant fines and other penalties for wrongful use or disclosure of Protected Health Information. Given the complexity of HIPAA and HITECH and their overlap with state privacy and security laws, and the fact that these laws are rapidly evolving and are subject to changing and potentially conflicting
58
Table of Contents
interpretation, our ability to comply with the HIPAA, HITECH and state privacy requirements is uncertain and the costs of compliance are significant. Adding to the complexity is that our operations are evolving and the requirements of these laws will apply differently depending on such things as whether or not we bill electronically for our services, or provide services involving the use or disclosure of Protected Health Information and incur compliance obligations as a business associate. The costs of complying with any changes to the HIPAA, HITECH and state privacy restrictions may have a negative impact on our operations. Noncompliance could subject us to criminal penalties, civil sanctions and significant monetary penalties as well as reputational damage.
Clinical research is heavily regulated and failure to comply with human subject protection regulations may disrupt our research program leading to significant expense, regulatory enforcement, private lawsuits and reputational damage.
Clinical research is subject to federal, state and, for studies conducted outside of the United States, international regulation. At the federal level, the FDA imposes regulations for the protection of human subjects and requirements such as initial and ongoing institutional review board review; informed consent requirements, adverse event reporting and other protections to minimize the risk and maximize the benefit to research participants. Many states impose human subject protection laws that mirror or in some cases exceed federal requirements. HIPAA also regulates the use and disclosure of Protected Health Information in connection with research activities. Research conducted overseas is subject to a variety of national protections such as mandatory ethics committee review, as well as laws regulating the use, disclosure and cross-border transfer of personal data. The costs of compliance with these laws may be significant and compliance with regulatory requirements may result in delay. Noncompliance may disrupt our research and result in data that is unacceptable to regulatory authorities, data lock or other sanctions that may significantly disrupt our operations.
Violation of a state’s prohibition on the corporate practice of medicine could result in a material adverse effect on our business.
A number of states, including California, do not allow business corporations to employ physicians to provide professional services. This prohibition against the “corporate practice of medicine” is aimed at preventing corporations such as us from exercising control over the medical judgments or decisions of physicians. The state licensure statutes and regulations and agency and court decisions that enumerate the specific corporate practice rules vary considerably from state to state and are enforced by both the courts and regulatory authorities, each with broad discretion. If regulatory authorities or other parties in any jurisdiction successfully assert that we are engaged in the unauthorized corporate practice of medicine, we could be required to restructure our contractual and other arrangements. In addition, violation of these laws may result in sanctions imposed against us and/or the professional through licensure proceedings, and we could be subject to civil and criminal penalties that could result in exclusion from state and federal health care programs.
Intellectual Property Risks Related to Our Business
Our collaborators may assert ownership or commercial rights to inventions we develop from our use of the biological materials which they provide to us, or otherwise arising from the collaboration.
We collaborate with several institutions, physicians and researchers in scientific matters. We do not have written agreements with certain of such collaborators (including the MD Anderson Cancer Center, Columbia University and the University of California, San Diego), or the written agreements we have do not cover intellectual property rights. Also, we rely on numerous third parties to provide us with blood samples and biological materials that we use to develop tests. If we cannot successfully negotiate sufficient ownership and commercial rights to any inventions that result from our use of a third party collaborator’s materials, or if disputes arise with respect to the intellectual property developed with the use of a collaborator’s samples, or data developed in a collaborator’s study, we may be limited in our ability to capitalize on the market potential of these inventions or developments.
59
Table of Contents
If we are unable to maintain intellectual property protection, our competitive position could be harmed.
Our ability to protect our discoveries and technologies affects our ability to compete and to achieve sustained profitability. Currently, we rely on a combination of U.S. and foreign patents and patent applications, copyrights, trademarks and trademark applications, confidentiality or non-disclosure agreements, material transfer agreements, licenses, consulting agreements, work-for-hire agreements and invention assignment agreements to protect our intellectual property rights. We also maintain certain company know-how, trade secrets and technological innovations designed to provide us with a competitive advantage in the market place as trade secrets. Currently, we own 3 issued U.S. patents, 6 pending U.S. patent applications and their corresponding foreign patents and patent applications, relevant to our cancer diagnostics business, as well as 2 pending U.S. patent applications and their corresponding foreign patent applications we jointly own with Aegea Biotechnologies, Inc. (for which we have the exclusive rights for specified fields of use). While we intend to pursue additional patent applications, it is possible that our pending patent applications and any future applications may not result in issued patents. Even if patents are issued, third parties may independently develop similar or competing technology that avoids our patents. Further, we cannot be certain that the steps we have taken will prevent the misappropriation of our trade secrets and other confidential information as well as the misuse of our patents and other intellectual property, particularly in foreign countries where we have not filed for patent protection. In addition, if Aegea Biotechnologies, Inc. were to challenge the scope of our rights under or attempt to terminate its Assignment and Exclusive Cross-License Agreement with us, our ability to use the technologies we in-license from Aegea, or to prevent others from using them in the fields of use for which we have an exclusive license, could be compromised.
From time to time the U.S. Supreme Court, other federal courts, the U.S. Congress or the U.S. Patent and Trademark Office, or USPTO, may change the standards of patentability and any such changes could have a negative impact on our business. For instance, in 2008, the Court of Appeals for the Federal Circuit issued a decision that methods or processes cannot be patented unless they are tied to a machine or involve a physical transformation. The U.S. Supreme Court later reversed that decision in Bilski v. Kappos, finding that the “machine-or-transformation” test is not the only test for determining patent eligibility. The Court, however, declined to specify how and when processes are patentable. In 2012, in the case Mayo Collaborative Services v. Prometheus Laboratories, Inc., the U.S. Supreme Court reversed the Federal Circuit’s application of Bilski and invalidated a patent focused on a diagnostic process because the patent claim embodied a law of nature. It is unclear at this time whether the USPTO will amend its patent prosecution guidelines for determining patentability of diagnostic or other processes, and how lower courts will implement the decision. Some aspects of our technology involve processes that may be subject to this evolving standard and we cannot guarantee that any of our pending process claims will be patentable as a result of such evolving standards.
In 2013, in Association for Molecular Pathology v. Myriad Genetics, the Supreme Court unanimously ruled that, “A naturally occurring DNA segment is a product of nature and not patent eligible merely because it has been isolated,” invalidating Myriad Genetics’ patents on the BRCA1 and BRCA2 genes. However, the Supreme Court also held that manipulation of a gene to create something not found in nature, such as a strand of synthetically-produced complementary DNA, could still be eligible for patent protection. The Supreme Court noted that method patents, which concern technical procedures for carrying out a certain process, are not affected by the ruling.
It should also be noted that in 2010, the Secretary’s Advisory Committee on Genetics, Health and Society voted to approve a report entitled “Gene Patents and Licensing Practices and Their Impact on Patient Access to Genetic Tests.” That report defines “patent claims on genes” broadly to include claims to isolated nucleic acid molecules as well as methods of detecting particular sequences or mutations. The report also contains six recommendations, including the creation of an exemption from liability for infringement of patent claims on genes for anyone making, using, ordering, offering for sale or selling a test developed under the patent for patient care purposes, or for anyone using the patent-protected genes in the pursuit of research. The report also recommended that HHS should explore, identify and implement mechanisms that will encourage more voluntary adherence to current guidelines that promote nonexclusive in-licensing of diagnostic genetic and genomic technologies. It is unclear whether HHS will act upon these recommendations, or if the recommendations would result in a change in law or process that could negatively impact our patent portfolio or future research and development efforts.
60
Table of Contents
We may face intellectual property infringement claims that could be time-consuming and costly to defend, and could result in our loss of significant rights and the assessment of treble damages.
From time to time we may face intellectual property infringement or misappropriation claims from third parties. Some of these claims may lead to litigation. The outcome of any such litigation can never be guaranteed, and an adverse outcome could affect us negatively. For example, were a third-party to succeed on an infringement claim against us, we may be required to pay substantial damages, including treble damages if such infringement were found to be willful. In addition, we could face an injunction, barring us from conducting the allegedly infringing activity. The outcome of the litigation could require us to enter into a license agreement which may not be pursuant to acceptable or commercially reasonable or practical terms or which may not be available at all.
It is also possible that an adverse finding of infringement against us may require us to dedicate substantial resources and time in developing non-infringing alternatives, which may or may not be possible. In the case of diagnostic tests, we would also need to include non-infringing technologies which would require us to re-validate the test. Any such re-validation, in addition to being costly and time consuming, may be unsuccessful.
Finally, we may initiate claims to assert or defend our own intellectual property against third parties. Any intellectual property litigation, irrespective of whether we are the plaintiff or the defendant, and regardless of the outcome, is expensive and time-consuming, and could divert our management’s attention from our business and negatively affect our operating results or financial condition.
Risks Relating to Our Common Stock
The price of our common stock may be volatile.
Before our recently completed initial public offering, there was no public market for our common stock. Market prices for securities of early-stage life sciences companies have historically been particularly volatile. The factors that may cause the market price of our common stock to fluctuate include, but are not limited to:
| • | progress, or lack of progress, in developing and commercializing our current breast cancer test and our planned future cancer diagnostic tests; |
| • | favorable or unfavorable decisions about our tests from government regulators, insurance companies or other third-party payors; |
| • | our ability to recruit and retain qualified research and development personnel; |
| • | changes in investors’ and securities analysts’ perception of the business risks and conditions of our business; |
| • | changes in our relationship with key collaborators; |
| • | changes in the market valuation or earnings of our competitors or companies viewed as similar to us; |
| • | changes in key personnel; |
| • | depth of the trading market in our common stock; |
| • | termination of the lock-up agreements or other restrictions on the ability of our existing stockholders to sell shares after our initial public offering; |
| • | changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| • | the granting or exercise of employee stock options or other equity awards; |
| • | realization of any of the risks described under this section entitled “Risk Factors”; and |
| • | general market and economic conditions. |
61
Table of Contents
In addition, the equity markets have experienced significant price and volume fluctuations that have affected the market prices for the securities of newly public companies for a number of reasons, including reasons that may be unrelated to our business or operating performance. These broad market fluctuations may result in a material decline in the market price of our common stock and you may not be able to sell your shares at prices you deem acceptable. In the past, following periods of volatility in the equity markets, securities class action lawsuits have been instituted against public companies. Such litigation, if instituted against us, could result in substantial cost and the diversion of management attention.
Our failure to meet the continued listing requirements of The NASDAQ Capital Market could result in a de-listing of our common stock.
If we fail to satisfy the continued listing requirements of The NASDAQ Capital Market, such as the corporate governance requirements or the minimum closing bid price requirement, NASDAQ may take steps to de-list our common stock. Such a de-listing would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a de-listing, we would take actions to restore our compliance with NASDAQ’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below the NASDAQ minimum bid price requirement or prevent future non-compliance with NASDAQ’s listing requirements.
If our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not obtain or retain a listing on The NASDAQ Capital Market and if the price of our common stock is less than $5.00, our common stock will be deemed a penny stock. The penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser’s written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks; and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
Our quarterly operating results may fluctuate significantly.
We expect our operating results to be subject to quarterly fluctuations. Our net loss and other operating results will be affected by numerous factors, including:
| • | the rate of adoption and/or continued use of our current breast cancer test and our planned future tests by healthcare practitioners; |
| • | variations in the level of expenses related to our development programs; |
| • | addition or reduction of resources for sales and marketing; |
| • | addition or termination of clinical utility studies; |
| • | any intellectual property infringement lawsuit in which we may become involved; |
| • | third party payor determinations affecting our tests; and |
| • | regulatory developments affecting our tests. |
62
Table of Contents
If our quarterly operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially.
Future sales of our common stock, or the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is doing well.
Sales of substantial amounts of our common stock, or the perception that these sales may occur, could materially and adversely affect the price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. We have outstanding 185,550 shares of common stock as of December 31, 2013, all of which are restricted securities that may be sold only in accordance with the resale restrictions under Rule 144 of the Securities Act of 1933, as amended. In addition, as of December 31, 2013, we had outstanding options to purchase 333,106 shares of our common stock and outstanding warrants to purchase shares of our common and Series A preferred stock overlying an estimated aggregate of 1,029,152 common stock equivalents. We plan to register for offer and sale the shares of common stock that are reserved for issuance pursuant to outstanding options. Shares covered by such registration statements upon the exercise of stock options generally will be eligible for sale in the public market, except that affiliates will continue to be subject to volume limitations and other requirements of Rule 144 under the Securities Act. The issuance or sale of such shares could depress the market price of our common stock.
In the future, we also may issue our securities if we need to raise additional capital. The number of new shares of our common stock issued in connection with raising additional capital could constitute a material portion of the then-outstanding shares of our common stock.
If securities or industry analysts do not publish research or reports or publish unfavorable research or reports about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us, our business, our market or our competitors. We do not currently have and may never obtain research coverage by securities and industry analysts. If no securities or industry analysts commence coverage of our company, the trading price for our stock would be negatively impacted. In the event we obtain securities or industry analyst coverage, if one or more of the analysts who covers us downgrades our stock, our stock price would likely decline. If one or more of these analysts ceases to cover us or fails to regularly publish reports on us, interest in our stock could decrease, which could cause our stock price or trading volume to decline.
Our largest stockholder continues to have substantial influence over us and could delay or prevent a change in corporate control.
Claire K. T. Reiss beneficially owns approximately 69% and 42% of our common stock at December 31, 2013 and immediately after our initial public offering, respectively, assuming the conversion of underlying notes and preferred stock into shares of common stock, excluding warrants, restricted stock units and stock options. Mrs. Reiss has significant influence over the outcome of matters submitted to our stockholders for approval, including the election of directors and any merger, consolidation or sale of all or substantially all of our assets. Accordingly, this concentration of ownership might harm the market price of our common stock by:
| • | delaying, deferring or preventing a change in control; |
| • | impeding a merger, consolidation, takeover or other business combination involving us; or |
| • | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
63
Table of Contents
If we are unable to favorably assess the effectiveness of our internal control over financial reporting, investors may lose confidence in our financial reporting and our stock price could be materially adversely affected.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404(a) of the Sarbanes-Oxley Act, or the subsequent testing by our independent registered public accounting firm conducted in connection with Section 404(b) of the Sarbanes-Oxley Act after we no longer qualify as an “emerging growth company,” may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses or that may require prospective or retroactive changes to our consolidated financial statements or identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
We are required to disclose changes made in our internal control procedures on a quarterly basis and our management is required to assess the effectiveness of these controls annually. However, for as long as we are an “emerging growth company” under the recently enacted JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404. We could be an “emerging growth company” for up to five years. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
We are an “emerging growth company,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an emerging growth company, as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements and exemptions from the requirements of holding nonbinding advisory votes on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our common stock held by non-affiliates exceeds $700 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three year period before that time, we would cease to be an emerging growth company immediately. Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company” which would allow us to take advantage of many of the same exemptions from disclosure requirements including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies. As a
64
Table of Contents
result, changes in rules of U.S. generally accepted accounting principles or their interpretation, the adoption of new guidance or the application of existing guidance to changes in our business could significantly affect our financial position and results of operations.
We will incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, the listing requirements of The NASDAQ Stock Market and other applicable securities rules and regulations. Compliance with these rules and regulations has increased and will continue to increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly, and increase demand on our systems and resources. The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. In order to maintain and, if required, improve our disclosure controls and procedures and internal control over financial reporting to meet this standard, significant resources and management oversight may be required. As a result, management’s attention may be diverted from other business concerns, which could harm our business and operating results. Further, there are significant corporate governance and executive compensation related provisions in the Dodd-Frank Wall Street Reform and Consumer Protection Act, enacted in 2010, that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access. Recent legislation permits smaller “emerging growth companies” to implement many of these requirements over a longer period and up to five years from the pricing of our initial public offering. We intend to take advantage of this new legislation but cannot guarantee that we will not be required to implement these requirements sooner than budgeted or planned and thereby incur unexpected expenses. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
As a public company that is subject to these rules and regulations, we may find it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to incur substantial costs to maintain our current levels of such coverage. We cannot predict or estimate the amount or timing of additional costs we may incur to respond to these requirements. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
Existing stockholders may view our initial public offering unfavorably.
The process of effecting an initial public offering has taken considerable time and is associated with several personnel and other changes, including a reverse common stock split. Some of our current stockholders have
65
Table of Contents
invested in our securities at prices which are at or above the initial public offering price per share. No assurances can be given as to whether any stockholders will seek to take actions against our company or the board with respect to our initial public offering process.
Anti-takeover provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board and management.
Certain provisions of our amended certificate of incorporation and amended and restated bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our board of directors. (For example, Delaware law provides that if a corporation has a classified board of directors, stockholders cannot remove any director during his or her term without cause.) These provisions also could limit the price that investors might be willing to pay in the future for our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. These provisions, among other things:
| • | classify our board of directors into three classes of equal (or roughly equal) size, with all directors serving for a three-year term and the directors of only one class being elected at each annual meeting of stockholders, so that the terms of the classes of directors are “staggered”; |
| • | allow the authorized number of directors to be changed only by resolution of our board of directors; |
| • | authorize our board of directors to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the board of directors and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that our board of directors does not approve; |
| • | establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be acted on at stockholder meetings; and |
| • | limit who may call a stockholders meeting. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, or DGCL, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
Because we do not expect to pay cash dividends for the foreseeable future, you must rely on appreciation of our common stock price for any return on your investment. Even if we change that policy, we may be restricted from paying dividends on our common stock.
We do not intend to pay cash dividends on shares of our common stock for the foreseeable future. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend upon results of operations, financial performance, contractual restrictions, restrictions imposed by applicable law and other factors our board of directors deems relevant. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our common stock. Investors seeking cash dividends in the foreseeable future should not purchase our common stock.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Our ability to utilize our federal net operating loss, carryforwards and federal tax credit may be limited under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended. The limitations apply if an “ownership change,” as defined by Section 382, occurs. Generally, an ownership change occurs if the percentage
66
Table of Contents
of the value of the stock that is owned by one or more direct or indirect “five percent shareholders” increases by more than 50 percentage points over their lowest ownership percentage at any time during the applicable testing period, typically three years. If we have experienced an “ownership change” at any time since our formation, we may already be subject to limitations on our ability to utilize our existing net operating losses and other tax attributes to offset taxable income. In addition, future changes in our stock ownership, which may be outside of our control, may trigger an “ownership change” and, consequently, Section 382 and 383 limitations. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards and other tax attributes to offset United States federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. As of December 31, 2013, we had federal and state net operating loss carryforwards of approximately $111.7 million and $97.7 million, respectively, and federal and California research and development credits of $3.1 million and $3.0 million, respectively, which could be limited if we experience an “ownership change.”
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because early-stage life sciences companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
| Item 1B. | Unresolved Staff Comments. |
Not applicable.
| Item 2. | Properties. |
We have a lease for approximately 48,000 square feet of space in San Diego, California for use as a clinical reference laboratory and corporate headquarters, including manufacturing and research laboratories. The average rent for the remaining lease period is approximately $106,500 per month. This lease expires in 2020.
In September 2013, we entered into an amendment of the lease, extending the term for 21 months so that it now ends on July 31, 2020 and providing for five months of free base rent (August 2013—December 2013). In return, we agreed, among other things, to forfeit our security deposit and to issue common stock warrants to the landlord. We issued warrants for an aggregate of 50,260 shares of our common stock at an exercise price of $10.00, determined by dividing the warrant coverage amount of $502,605, which is 100% of the five months of base rent forgone, by the exercise price, which was set at the price per share of our common stock sold in our initial public offering.
Immediately following the execution of such amendment, we paid all amounts due under our lease. As of December 31, 2013, we did not have any rent past due.
In September 2012, in connection with an amendment of the lease, which included a rent deferral through November 30, 2012, we issued to our landlord warrants which now are exercisable through September 2019 for 1,587 shares of our common stock at an exercise price of $25.20 per share.
| Item 3. | Legal Proceedings. |
In the normal course of business, we may be involved in legal proceedings or threatened legal proceedings. We are not party to any legal proceedings or aware of any threatened legal proceedings which are expected to have a material adverse effect on our financial condition, results of operations or liquidity.
| Item 4. | Mine Safety Disclosures. |
Not applicable.
67
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock began trading on The NASDAQ Capital Market on February 5, 2014 under the symbol “BIOC.” Before such time, there was no public market for our common stock.
The last sale price for our common stock as reported by The NASDAQ Capital Market on March 21, 2014 was $7.70 per share.
Holders of Record
As of March 21, 2014, there were approximately 217 holders of record of our common stock. The actual number of common stockholders is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Dividend Policy
We have never declared dividends on our equity securities, and currently do not plan to declare dividends on shares of our common stock in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our board of directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our board of directors.
Securities Authorized for Issuance under Equity Compensation Plans
Information about our equity compensation plans is incorporated herein by reference to Part III, Item 12 of this Annual Report.
Repurchases of Equity Securities
In July, 2013, we repurchased 711 shares of common stock in connection with the settlement of a shareholder lawsuit.
Use of Proceeds
Our initial public offering of common stock was effected through a Registration Statement on Form S-1 (File No. 333-191323), which was declared effective by the Securities and Exchange Commission on February 4, 2013. On February 4, 2014, additional shares of our common stock were registered through a Registration Statement on Form S-1 (File No. 333-193760) filed pursuant to Rule 462(b) under the Securities Act. On February 10, 2014, a total of 1,900,000 shares of common stock were sold on our behalf at an initial public offering price of $10.00 per share, for aggregate gross offering proceeds of $19 million, managed by Aegis Capital Corp.
We paid to the underwriters underwriting discounts totaling approximately $1.3 million in connection with the offering. In addition, we incurred additional costs of approximately $1.0 million in connection with the offering, which when added to the underwriting discounts paid by us, amounts to total costs of approximately $2.3 million. Thus, the net offering proceeds to us, after deducting underwriting discounts and offering expenses,
68
Table of Contents
were approximately $16.7 million. No offering expenses were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning ten percent or more of any class of our equity securities or to any other affiliates.
There has been no material change in the expected use of the net proceeds from our initial public offering as described in our registration statement on Form S-1.
| Item 6. | Selected Financial Data |
Not applicable.
69
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
The following discussion and analysis of our financial condition and results of operations should be read together with “Selected Financial Data” in Part II, Item 6 of this Report and our financial statements and related notes in Part II, Item 8 of this Report. This discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. You should review the sections entitled “Risk Factors” and “Cautionary Note Regarding Forward Looking Statements” for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements.
Overview
We are an early-stage cancer diagnostics company that develops and commercializes proprietary circulating tumor cell, or CTC, and circulating tumor DNA, or ctDNA, tests utilizing a standard blood sample. Our current CTC breast cancer test provides, and our planned future tests would provide, information to oncologists that enable them to select appropriate personalized treatment for their patients based on better, timelier and more-detailed data on the characteristics of tumors.
Our current breast cancer test and our planned future tests utilize our Cell Enrichment and Extraction (CEE) technology for the enumeration and analysis of CTCs, and our CEE-Selector technology for the detection and analysis of ctDNA, each performed on a standard blood sample. The CEE technology is an internally developed, microfluidics-based CTC capture and analysis platform, with enabling features that change how CTC testing can be used by clinicians by providing real-time biomarker monitoring with only a standard blood sample. The CEE-Selector technology enables mutation detection with enhanced sensitivity and specificity and is applicable to nucleic acid from CTCs or other samples types, such as blood plasma for ctDNA. We believe the CEE-Selector technology is an important part of certain of our pipeline CTC tests and will be a stand-alone test for molecular analysis of biomarkers.
At our corporate headquarters facility located in San Diego, California, we operate a clinical laboratory that is certified under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and accredited by the College of American Pathologists, or CAP. We manufacture our CEE microfluidic channels, related equipment and certain reagents to perform our current breast cancer test and our planned future tests at this facility. CLIA certification and CAP accreditation are required before any clinical laboratory, including ours, may perform testing on human specimens for the purpose of obtaining information for the diagnosis, prevention, or treatment of disease or the assessment of health. The tests we offer and intend to offer are classified as laboratory developed tests, or LDTs, under CLIA regulations.
We are in the process of commercializing our first test, OncoCEE-BR, for breast cancer, and anticipate launching an OncoCEE-LU test for non-small cell lung cancer, or NSCLC, in the third quarter of 2014. These tests utilize our CEE technology platform and provide CTC enumeration as well as biomarker analysis from a standard blood sample. In the case of the OncoCEE-BR test, biomarker analysis involves fluorescence in situ hybridization, or FISH, for the detection and quantitation of the human epidermal growth factor receptor 2, or HER2, gene copy number. We plan to include immunocytochemical analysis of estrogen receptor and progesterone receptor proteins in the OncoCEE-BR test within the next year. The OncoCEE-LU test’s biomarker analysis would include FISH for EML4/ALK and ROS1 gene fusions, as well as mutation analysis for the epidermal growth factor receptor, or EGFR, gene, the K-ras gene and the B-raf gene.
The L858R mutation of the EGFR gene and Exon 19 deletions as activators of EGFR kinase activity are linked to the drugs Tarceva® and Iressa® (AstraZeneca). The T790M mutation of the EGFR gene as a resistance marker for EGFR tyrosine kinase inhibitors is linked to drugs in clinical development that address this resistance such as Gilotrif® (Boehringer-Ingelheim) and dacomitinib (Pfizer). The codon 12 and 13 mutations of the K-ras gene are linked to non-responsiveness to the EGFR kinase inhibitors, and the codon 600 mutations of the B-raf gene are linked to Zelboraf® and Tafinlar®, which are both approved for melanoma and are in clinical trials for lung cancer. Our OncoCEE-LU test would be performed on a standard blood sample.
70
Table of Contents
We plan to add other biomarker analyses to our current breast cancer test and our planned future OncoCEE tests as their relevance is demonstrated in clinical trials, for example, ret proto-oncogene gene fusions in NSCLC, which may indicate a particular course of therapy, and NRAS for melanoma, which may predict therapy resistance. In addition, we are developing a series of other CTC and ctDNA tests for different solid tumor types, including colorectal cancer, prostate cancer, gastric cancer and melanoma, each incorporating treatment-associated biomarker analyses specific to that cancer, planned to be launched over the next two to three years.
Key Factors Affecting our Results of Operations and Financial Condition
Our overall long-term growth plan depends on our ability to develop and commercialize tests through our CLIA laboratory. We have the OncoCEE-BR test available as a commercial product and we plan to enhance revenue for this product through the efforts of a sales and marketing organization we plan to hire. We are developing additional OncoCEE tests for non-small cell lung, colorectal, gastric and prostate cancers and melanoma that we expect to make available to physicians over the next three years. To facilitate market adoption of our tests, we anticipate having to successfully complete additional clinical utility studies with clinical samples to generate clinical utility data and then publish our results in peer-reviewed scientific journals. Our ability to complete such clinical studies is dependent upon our ability to leverage our collaborative relationships with leading institutions to facilitate our research, to conduct the appropriate clinical studies and to obtain favorable clinical data.
We believe that the factors discussed in the following paragraphs have had and are expected to continue to have a material impact on our results of operations and financial condition.
Revenues
Almost all of our revenues in 2012 and 2013 were generated by our OncoCEE-BR test. Over 75% of annual revenue was generated through our arrangement with Clarient in 2012, and over 75% of annual revenue was generated through our relationship with Dana Farber in 2013. The clinical laboratory industry is highly competitive, and our relationships and our partners’ relationships with decision-makers at hospitals, cancer centers or physician offices is a critical component of securing their business. Consequently, our ability to establish and manage partnerships with groups that have sales and marketing capabilities in our target markets and attract and maintain productive sales personnel that have and can grow these relationships will largely determine our ability to grow our clinical services revenue.
In 2012, $67,000, which represented the majority of our revenue for that year, was billed to our commercial partner, Clarient, which until May 2013 had responsibility for billing the third-party payors. Because Clarient paid us a contracted amount per test performed regardless of their billing and collections, we do not have data about the payor mix, reimbursement history and collectability experience for the tests performed under such arrangement. Clarient has paid us for all tests that we conducted under our arrangement in 2012. In the May 2013 revision of our arrangements with Clarient, we undertook responsibility for billing the payors and for reporting the results of the tests to the ordering physicians, and the exclusivity of Clarient’s marketing partner rights for OncoCEE-BR ended. The May 2013 revision of our arrangements with Clarient will, in general, have the effect of delaying the timing of revenue recognition (see the “Revenue Recognition” paragraph of Note 3 of the notes to our audited financial statements) and adding uncertainty to the collectability of our accounts receivable.
We expect that in the future the percentage of our revenue which is generated through our arrangement with Clarient will diminish. Since May 2013, the number of tests performed under our agreement with Clarient has decreased significantly.
In 2013, approximately $104,000 was billed to our clinical partner, Dana Farber, which represented the majority of our revenue for that year.
71
Table of Contents
In November 2013 we first directly billed payors for physician-ordered testing; until May 2013, our commercialization partner Clarient was responsible for all billing associated with our tests. We do not have data for Clarient’s billing and collection experience with respect to our test, because Clarient paid us a contracted amount per test performed regardless of their billing and collections. From May to December 2013, we performed an average of 1-3 physician-ordered tests per month (in addition to 20-30 tests per month performed since January 2013 under our development collaboration program with the Dana-Farber Cancer Institute). Billing for physician-ordered tests is now handled for us by a non-Clarient billing service provider. In November and December 2013 we invoiced, through this service provider, for 13 physician-ordered tests. Of these tests, 8 were billed to private third-party payors and 5 were billed to Medicare. We have not yet had any response or adjudication from the payors as to these bills, and accordingly we do not yet have any data as to reimbursement history or collectability experience. In addition, we believe the sample size of 13 is too small to be the basis for any conclusion about our ongoing payor mix.
The transition period to the new billing service provider was lengthened due to our focus on other priorities, as we knew the amounts for the small number of unbilled physician-ordered tests were immaterial. The transition of the billing function to our billing service provider was completed in December 2013. Our small backlog of unbilled tests has now been billed, and all future tests will be billed in a timely manner.
Cost of Revenues
Our cost of revenues consists principally of personnel costs, laboratory and manufacturing supplies and overhead. We are pursuing various strategies to reduce and control our cost of revenues, including automating aspects of our processes, developing more efficient technology and methods, attempting to negotiate improved terms with our suppliers and exploring relocating our operations to a lower-cost facility.
Operating Expenses
We classify our operating expenses into three categories: research and development, sales and marketing, and general and administrative. Our operating expenses principally consist of personnel costs, outside services, laboratory consumables and overhead, development costs, and legal and accounting fees.
Research and Development Expenses. We incur research and development expenses principally in connection with our efforts to develop and improve our tests. Our primary research and development expenses consist of direct personnel costs, laboratory equipment and consumables and overhead expenses. We anticipate that research and development expenses will increase in the near-term, principally as a result of hiring additional personnel to develop and validate tests in our pipeline and to perform work associated with clinical utility studies and development collaborations. In addition, we expect that our costs related to collaborations with research and academic institutions will increase. All research and development expenses are charged to operations in the periods in which they are incurred.
Sales and Marketing Expenses. Our sales and marketing expenses consist principally of personnel and related overhead costs for our sales team and their support personnel, travel and entertainment expenses, and other selling costs including sales collaterals and trade shows. We expect our sales and marketing expenses to increase significantly now that we have completed our initial public offering as we hire additional sales and marketing personnel and launch new tests.
General and Administrative Expenses. General and administrative expenses consist principally of personnel-related expenses, professional fees, such as legal, accounting and business consultants, occupancy costs, and other general expenses. We expect that our general and administrative expenses will increase as we expand our business operations. When we begin billing a significant number of tests, bad debt is expected to become a greater expense. We further expect that general and administrative expenses will increase significantly due to increased information technology, legal, insurance, accounting and financial reporting expenses associated with being a public company.
72
Table of Contents
Seasonality
We expect our test volume to decrease during vacation and holiday seasons, when patients are less likely to visit their health care providers. We expect this trend in seasonality to continue for the foreseeable future.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of our financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates based on historical experience and make various assumptions, which management believes to be reasonable under the circumstances, which form the basis for judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
The notes to our audited and unaudited financial statements, which are included elsewhere in this prospectus, contain a summary of our significant accounting policies. We consider the following accounting policies critical to the understanding of the results of our operations:
| • | Revenue recognition; |
| • | Accounts receivable and bad debts; |
| • | Stock-based compensation; |
| • | Common stock valuation; and |
| • | Warrant liability. |
Revenue Recognition
We recognize revenue in accordance with ASC 605, Revenue Recognition, and ASC 954-605, Health Care Entities, Revenue Recognition which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred and title and the risks and rewards of ownership have been transferred to the client or services have been rendered; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. For contract partners, revenue is recorded based upon the contractually agreed upon fee schedule. When assessing collectability, we consider whether we have sufficient payment history to reliably estimate a payor’s individual payment patterns. For new tests where there is limited evidence of payment history at the time the tests are completed, we recognize revenue equal to the amount of cash received until such time as reimbursement experience can be established.
Our primary source of revenue for the year ended December 31, 2013 was Dana Farber Cancer Institute, a collaboration partner. This revenue was derived from clinical laboratory testing performed in our laboratories under our collaboration agreement. As there was a contractually agreed upon price under our collaboration agreement as in effect until May 2013, and collectability from our collaboration partner is reasonably assured, revenues for these tests under our collaboration agreement as in effect until May 2013 is earned at the time the test is completed and the results are delivered to the third party.
Accounts Receivable and Bad Debts
We carry accounts receivable at original invoice amounts, less an estimate for doubtful receivables, based on a review of all outstanding amounts on a periodic basis. The estimate for doubtful receivables is determined from an analysis of the accounts receivable on a quarterly basis, and is recorded as bad debt expense. Since we only recognize revenue to the extent we expect to collect such amounts, bad debt expense related to receivables
73
Table of Contents
from patient service revenue is recorded in general and administrative expense in the statements of operations. Accounts receivable are written off when deemed uncollectible. Recoveries of accounts receivable previously written off are recorded when received.
Stock-Based Compensation Expense
We account for stock-based compensation under the provisions of ASC Topic 718, Compensation—Stock Compensation, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees and directors based on estimated fair values on the grant date. We estimate the fair value of stock option awards on the date of grant using the Black-Scholes option pricing model, or Black-Scholes valuation model. The fair value of restricted stock unit awards is determined by the price of the Company’s common stock on the date of grant. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. We estimate forfeitures at the time of grant and revise our estimates in subsequent periods if actual forfeitures differ from those estimates. At December 31, 2013, we had unrecognized compensation cost related to nonvested stock options and restricted stock units of approximately $861,000 and $135,000, respectively, which amounts are expected to be recognized over the next 2.81 years and 1.56 years, respectively.
We account for stock-based compensation awards to non-employees in accordance with ASC Topic 505-50, Equity-Based Payments to Non-Employees. Under ASC 505-50, we determine the fair value of the warrants or stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. All issuances of equity instruments issued to non-employees as consideration for goods or services received by us are accounted for based on the fair value of the equity instruments issued. These awards are recorded in expense and additional paid-in capital in stockholders’ equity over the applicable service periods based on the fair value of the options at the end of each period.
Calculating the fair value of stock-based awards requires the input of highly subjective assumptions into the Black-Scholes valuation model. Stock-based compensation expense is calculated using our best estimate, which involves inherent uncertainties, and the application of our management’s judgment. Significant estimates include the fair value of our common stock at the date of grant, the expected life of the stock option, stock price volatility, risk-free interest rate and forfeiture rates.
Common Stock Valuation
In the absence of a public trading market, our board of directors determined a reasonable estimate of the then-current fair value of our common stock for purposes of granting stock-based compensation based on input from management and valuation reports prepared by an independent third-party valuation specialist. We determined the fair value of our common stock utilizing methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants Practice Aid, “Valuation of Privately-Held-Company Equity Securities Issued as Compensation,” which we refer to as the AICPA Practice Aid. In addition, we exercised judgment in evaluating and assessing the foregoing based on several factors including:
| • | the nature and history of our business; |
| • | our historical operating and financial results; |
| • | the market value of companies that are engaged in a similar business to ours; |
| • | the lack of marketability of our common stock; |
| • | the price at which shares of our equity instruments have been sold; |
74
Table of Contents
| • | our progress in developing our technology; |
| • | the overall inherent risks associated with our business at the time stock option grants or warrants were approved; and |
| • | the overall equity market conditions and general economic trends. |
Warrant Liability
Warrants for shares that are contingently redeemable and for which the exercise price is not fixed are classified as liabilities on the accompanying balance sheets and carried at their estimated fair value, determined through use of a probability-weighted Black-Scholes valuation model. At the end of each reporting period, any changes in fair value are recorded as a component of total other income/(expense). We continued to adjust the carrying value of the warrants until the completion of our initial public offering on February 10, 2014, at which time the exercise price was fixed and the fair value of those warrants was reclassified to shareholders’ deficit.
Results of Operations
Years Ended December 31, 2013 and 2012
The following table sets forth certain information concerning our results of operations for the periods shown:
| Year Ended December 31, | Change | |||||||||||||||
| 2012 | 2013 | $ | % | |||||||||||||
| (dollars in thousands) | ||||||||||||||||
| Revenue |
$ | 109 | $ | 134 | $ | 25 | 23 | % | ||||||||
| Cost of revenues |
1,202 | 2,330 | 1,128 | 94 | % | |||||||||||
| Research and development expenses |
6,562 | 3,086 | (3,476 | ) | (53 | %) | ||||||||||
| General and administrative expenses |
2,063 | 2,513 | 450 | 22 | % | |||||||||||
| Sales and marketing expenses |
785 | 149 | (636 | ) | (81 | %) | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total Operating Loss |
(10,503 | ) | (7,944 | ) | 2,559 | (24 | %) | |||||||||
| Interest income/(expense), net |
(2,187 | ) | (2,070 | ) | 117 | (5 | %) | |||||||||
| Change in fair value of warrant liability |
454 | 782 | 328 | 72 | % | |||||||||||
| Other income/(expense) |
(23 | ) | — | 23 | (100 | %) | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Income/(loss) before income taxes |
(12,259 | ) | (9,232 | ) | 3,027 | (25 | %) | |||||||||
| Income tax expense |
1 | 1 | — | — | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (12,260 | ) | $ | (9,233 | ) | $ | 3,027 | (25 | %) | ||||||
|
|
|
|
|
|
|
|||||||||||
Revenue
Revenues were approximately $134,000 for the year ended December 31, 2013, compared with approximately $109,000 for the year ended December 31, 2012, an increase of approximately $25,000, or 23%. The increase was primarily related to clinical trial testing services for our development collaboration program with the Dana-Farber Cancer Institute, partially offset by a decrease in revenues from Clarient. The average price per commercial test decreased from $694 for the year ended December 31, 2012 to an average of $635 for the year ended December 31, 2013. The average price per clinical test was $400 for the year ended December 31, 2013.
Cost of Revenues
Cost of revenues was $2.3 million for the year ended December 31, 2013, compared with $1.2 million for the year ended December 31, 2012, an increase of $1.1 million, or 94%. The increase was primarily related to the volume of clinical tests performed, which increased from zero for the year ended December 31, 2012 to 258 for
75
Table of Contents
the year ended December 31, 2013. The volume of commercial tests performed decreased from 130 for the year ended December 31, 2012 to 23 for the year ended December 31, 2013. The net volume increase was due to clinical tests performed under our 2013 development collaboration program with the Dana-Farber Cancer Institute, partially offset by fewer tests performed under our arrangement with Clarient for the year ended December 31, 2013 as compared to 2012.
Operating Expenses
Research and Development Expenses. Research and development expenses were $3.1 million for the year ended December 31, 2013, compared with $6.6 million for the year ended December 31, 2012, a decrease of $3.5 million, or 53%.The decrease was primarily due to a $1.8 million decrease in personnel expenses relating to a reduction in research and development headcount from an average of 16 for the year ended December 31, 2012 to an average of 9 for the same period in 2013, and a $1.1 million decrease in research and development expenses due to the allocation of lab expenses to cost of revenues based on the number of samples processed.
General and Administrative Expenses. General and administrative expenses were $2.5 million for the year ended December 31, 2013, compared with $2.1 million for the year ended December 31, 2012, an increase of $0.4 million, or 22%. The increase was primarily due to an increase of $266,000 in legal fees, particularly fees pertaining to our patent portfolio, as well as an increase in stock-based compensation expense of $358,000, partially offset by a $310,000 decrease in personnel-related expenses resulting from a reduction in general and administrative headcount from an average of 8 for the year ended December 31, 2012 to an average of 6 for the same period in 2013.
Sales and Marketing Expenses. Sales and marketing expenses were $0.1 million for the year ended December 31, 2013, compared with $0.8 million for the year ended December 31, 2012, a decrease of $0.7 million, or 81%.The decrease was primarily due to a decrease in personnel-related expenses resulting from a reduction in sales and marketing headcount from an average of 2 for the year ended December 31, 2012 to an average of 1 for the same period in 2013.
Interest Income and Expense
Interest expense was $2.1 million for the year ended December 31, 2013, compared with $2.2 million for the year ended December 31, 2012, with the $0.1 million decrease primarily related to lower average debt balances, partially offset by increased debt discount amortization as a result of entering into new financing arrangements with associated discounts during 2013.
Change in Fair Value of Warrant Liability
The change in the fair value of warrant liability was $0.8 million for the year ended December 31, 2013 compared with $0.5 million for the year ended December 31, 2012, an increase of $0.3 million, or 72%. The increase is due to a larger decline in the price of the shares underlying warrants during the year ended December 31, 2013 as compared to the year ended December 31, 2012.
Income Taxes
Over the past several years we have generated operating losses in all jurisdictions in which we may be subject to income taxes. As a result, we have accumulated significant net operating losses and other deferred tax assets. Because of our history of losses and the uncertainty as to the realization of those deferred tax assets, a full valuation allowance has been recognized. We do not expect to report a provision for income taxes until we have a history of earnings, if ever, that would support the realization of our deferred tax assets.
76
Table of Contents
We have not completed a study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since our formation, due to the complexity and cost associated with such a study, and the fact that there may be additional ownership changes in the future. We estimate that if such a change did occur, the federal and state net operating loss carryforwards and research and development credits that can be utilized in the future will be significantly limited.
Liquidity and Capital Resources
We are actively working to improve our financial position and enable the growth of our business, by raising new capital and resolving our outstanding debt.
Pursuant to a note and warrant purchase agreement executed as of June 28, 2013 to reflect certain prior and possible future borrowings under a series of notes, we borrowed an aggregate of $5.0 million through December 31, 2013 (including $0.7 million borrowed under this arrangement during fiscal year 2012) and in January 2014 borrowed an additional $0.2 million under this arrangement. The principal amount of and accrued interest on the notes automatically converted into an aggregate of 547,794 shares of our common stock upon the closing of our initial public offering, at a conversion price equal to the price per share of our common stock sold in our initial public offering. The number of shares underlying the associated common stock warrants were fixed so that such warrants became exercisable, at $10.00 per share for an aggregate of 258,249 shares of common stock.
In June 2013, we arranged the conversion of all outstanding indebtedness under our May 2010 amended and restated loan agreement, our February 2011 note and warrant purchase agreement and our January 2012 note and warrant purchase agreement. In this series of transactions, promissory notes with outstanding principal totaling $20,231,000 and accrued interest of approximately $2,581,000 were converted into 42,245,834 shares of Series A preferred stock. The conversion included the issuance of 41,694,760 shares of Series A preferred stock to directors and their affiliates and other related parties. All of the converted notes and interest were in default and classified as current as of December 31, 2012.
In connection with the conversion of the debt outstanding under the May 2010 amended and restated loan agreement, we issued 23,809 common stock warrants to Goodman Co. Ltd., a 5% shareholder.
In July 2013, we amended a secured promissory note with a principal balance of $1.4 million, held by a trust affiliated with Claire K. T. Reiss, a 5% shareholder and at the time a director, to provide that all principal of and accrued interest on the note would automatically convert into common stock upon the closing of an initial public offering, at the price per share at which common stock is sold in such initial public offering. This amendment was not related to Mrs. Reiss’ later decision to resign from the board of directors.
In July 2013, we entered into a revolving line of credit with UBS Bank USA in the initial amount of $1.5 million. The maximum amount of this line of credit has subsequently been increased to approximately $2.6 million. Interest accrues daily on the outstanding balance and is paid monthly at a variable rate which is currently 2.75% over the 30 day LIBOR rate or a current effective annual interest rate of 2.92%. UBS Bank USA has the right to terminate the revolving line of credit at any time, and if it does, all amounts drawn under the revolving line of credit would be immediately payable. An affiliate of our director David F. Hale, and an affiliate of Claire K. T. Reiss, a 5% shareholder and at the time a director, an affiliate of our director Edward Neff, an affiliate of our director Bruce E. Gerhardt, and an affiliate of our director Ivor Royston guaranteed the loan and pledged financial assets to UBS Bank USA to secure their guaranties. In return, we issued common stock warrants to the guarantors. The number of shares subject to the common stock warrants will be determined by dividing the warrant coverage amount, which is 50% of the fair market value of the collateral provided by the respective guarantors to secure their respective guaranty obligations to UBS Bank USA, by the exercise price, which was set at the price per share of our common stock sold in our initial public offering. The number of shares underlying the associated common stock warrants were fixed so that such warrants became exercisable at $10.00 per share for an aggregate of 128,903 shares of common stock. We have entered into an agreement with the guarantors that provides for us to reimburse them for any amounts paid by them on such guaranties. This reimbursement obligation is secured by a security interest in our assets.
77
Table of Contents
In September 2013, we entered into an amendment of the lease for our headquarters/laboratory building in San Diego, California, extending the term through July 31, 2020 and providing for five months of free base rent (August 2013—December 2013). In return, we agreed, among other things, to forfeit our security deposit and to issue common stock warrants to the landlord. We issued warrants for an aggregate of 50,260 shares of our common stock at an exercise price of $10.00, determined by dividing the warrant coverage amount of $502,605, which is 100% of the five months of base rent forgone, by the exercise price, which was set at the price per share of our common stock sold in our initial public offering.
Cash Flows
Our net cash flow from operating, investing and financing activities for the periods below were as follows:
| Year Ended | ||||||||
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| (dollars in thousands) | ||||||||
| Cash provided by (used in): |
||||||||
| Operating activities |
$ | (8,607 | ) | $ | (6,202 | ) | ||
| Investing activities |
(8 | ) | (1 | ) | ||||
| Financing activities |
8,365 | 6,087 | ||||||
|
|
|
|
|
|||||
| Net increase (decrease) in cash and cash equivalents |
$ | (250 | ) | $ | (116 | ) | ||
|
|
|
|
|
|||||
Cash Used in Operating Activities. Net cash used in operating activities was $6.2 million for the year ended December 31, 2013, compared to net cash used in operating activities of $8.6 million for the year ended December 31, 2012. In all periods the primary use of cash was to fund our net loss.
Cash Used in Investing Activities. Cash used in investing activities was $1,000 for the year ended December 31, 2013, compared to $8,000 for the year ended December 31, 2012. The cash used in investing activities in 2012 was primarily used to acquire laboratory equipment and software.
Cash Provided by Financing Activities. Net cash provided by financing activities was $6.1 million for the year ended December 31, 2013, compared to net cash provided by financing activities of $8.4 million for the year ended December 31, 2012. Our primary source of financing in all periods consisted of loans received from our major shareholder and members of our board of directors and their affiliates, in exchange for convertible promissory notes and warrants.
Capital Resources and Expenditure Requirements
We expect to continue to incur substantial operating losses in the future. It may take several years to achieve positive operational cash flow or we may not ever achieve positive operational cash flow. We expect that we will use a portion of the net proceeds from our initial public offering and our revenues from operations to hire sales and marketing personnel, support increased sales and marketing activities, fund further research and development, clinical utility studies and future enhancements of our tests, acquire equipment, implement automation and scale our capabilities to prepare for significant test volume, for general corporate purposes and to fund ongoing operations and the expansion of our business, including the increased costs associated with being a public company. We may also use a portion of the net proceeds of our initial public offering to acquire or invest in businesses, technologies, services or products, although we do not have any current plans to do so.
As of December 31, 2013, our cash and cash equivalents totaled approximately $69,000. While we currently are in the commercialization stage of operations, we have not yet achieved profitability and anticipate that we will continue to incur net losses for the foreseeable future. On February 10, 2014, we received proceeds of approximately $16,673,000 as a result of the closing of our initial public offering, net of underwriting discounts and additional costs incurred. We believe that our cash resources should be sufficient to support currently
78
Table of Contents
forecasted operations through at least the next twelve months. However, we operate in a market that makes its prospects difficult to evaluate, and the likelihood that we will need additional debt or equity financing in the future to execute on our current or future business strategies beyond the next twelve months is probable. We can provide no assurances that any sources of a sufficient amount of financing will be available to us on favorable terms, if at all. If we are unable to raise a sufficient amount of financing in a timely manner, we would likely need to scale back our general and administrative activities and certain of our research and development activities. Our forecast pertaining to our current financial resources and the costs to support our general and administrative and research and development activities are forward-looking statements and involve risks and uncertainties. Actual results could vary materially and negatively as a result of a number of factors, including:
| • | our ability to secure financing and the amount thereof; |
| • | the costs of operating and enhancing our laboratory facilities; |
| • | the costs of developing our anticipated internal sales and marketing capabilities; |
| • | the scope, progress and results of our research and development programs, including clinical utility studies; |
| • | the scope, progress, results, costs, timing and outcomes of the clinical utility studies for our cancer diagnostic tests; |
| • | our ability to manage the costs for manufacturing our microfluidic channels; |
| • | the costs of maintaining, expanding and protecting our intellectual property portfolio, including potential litigation costs and liabilities; |
| • | our ability to obtain adequate reimbursement from governmental and other third-party payors for our tests and services; |
| • | the costs of additional general and administrative personnel, including accounting and finance, legal and human resources, as a result of becoming a public company; |
| • | our ability to collect revenues; and |
| • | other risks discussed in the section entitled “Risk Factors”. |
As of December 31, 2013, we had approximately $9.1 million of outstanding indebtedness (including $0.5 million of interest accrued thereon and excluding $1.2 million of associated debt discounts), $6.9 million of which converted to equity as a result of our initial public offering on February 10, 2014. A portion of the proceeds from our initial public offering were subsequently used to repay substantially all of our outstanding indebtedness.
We believe that our cash resources should be sufficient to support currently forecasted operations through at least the next twelve months. However, we operate in a market that makes its prospects difficult to evaluate, and the likelihood that we will need additional debt or equity financing in the future to execute on its current or future business strategies beyond the next twelve months is probable. Furthermore, we may need to raise additional capital to expand our business to meet our long-term business objectives. We expect that our operating expenses and capital expenditures will increase in the future as we expand our business. We plan to increase our sales and marketing headcount to promote our current breast cancer test and our planned future cancer diagnostic tests and our research and development headcount to validate the tests currently in our pipeline. These headcount increases are aimed to expand our pipeline and to perform work associated with our research collaborations. Until we can generate a sufficient amount of revenues to finance our cash requirements, which we may never do, we may need to continue to raise additional capital to fund our operations.
We may raise additional capital to fund our current operations and to fund expansion of our business to meet our long-term business objectives through public or private equity offerings, debt financings, borrowings or strategic partnerships coupled with an investment in our company or a combination thereof. If we raise additional funds through the issuance of convertible debt securities, or other debt securities, these securities could be
79
Table of Contents
secured and could have rights senior to those of our common stock. In addition, any new debt incurred by us could impose covenants that restrict our operations. The issuance of any new equity securities will also dilute the interest of our current stockholders. Given the risks associated with our business, including our unprofitable operating history and our ability or inability to develop additional tests, additional capital may not be available when needed on acceptable terms, or at all. If adequate funds are not available, we will need to curb our expansion plans or limit our research and development activities, which would have a material adverse impact on our business prospects and results of operations.
Off-Balance Sheet Arrangements
We have not engaged in any off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
Not applicable.
80
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
Biocept, Inc.
Index to Financial Statements
81
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Biocept, Inc.
We have audited the accompanying balance sheets of Biocept, Inc. as of December 31, 2013 and 2012, and the related statements of operations and comprehensive loss, shareholders’ deficit and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Biocept, Inc. as of December 31, 2013 and 2012, and the results of its operations and its cash flows for the years then ended in conformity with accounting principles generally accepted in the United States of America.
/s/ Mayer Hoffman McCann P.C.
San Diego, California
March 28, 2014
82
Table of Contents
Biocept, Inc.
| December 31, | December 31, | Pro Forma December 31, |
||||||||||
| 2012 | 2013 | 2013 | ||||||||||
| (unaudited) | ||||||||||||
| Current assets: |
||||||||||||
| Cash & cash equivalents |
$ | 185,256 | $ | 69,178 | $ | 69,178 | ||||||
| Accounts receivable |
18,885 | 9,200 | 9,200 | |||||||||
| Inventories, net |
61,283 | 92,823 | 92,823 | |||||||||
| Prepaid expenses and other current assets |
310,442 | 799,131 | 260,813 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current assets |
575,866 | 970,332 | 432,014 | |||||||||
| Fixed assets, net |
624,730 | 358,887 | 358,887 | |||||||||
| Other non-current assets |
269,083 | 500 | 500 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total assets |
$ | 1,469,679 | $ | 1,329,719 | $ | 791,401 | ||||||
|
|
|
|
|
|
|
|||||||
| Current liabilities: |
||||||||||||
| Accounts payable |
$ | 1,387,677 | $ | 1,540,618 | $ | 1,540,618 | ||||||
| Accrued liabilities |
3,346,806 | 2,242,058 | 1,745,899 | |||||||||
| Line of credit |
— | 1,981,000 | 1,665,757 | |||||||||
| Notes payable |
21,631,427 | 5,200,599 | — | |||||||||
| Warrant liability |
981,747 | 2,140,532 | — | |||||||||
| Supplier financings |
251,146 | 218,925 | 218,925 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total current liabilities |
27,598,803 | 13,323,732 | 5,171,199 | |||||||||
| Notes payable, net of current portion |
745,000 | — | — | |||||||||
| Deferred rent |
510,771 | 462,001 | 462,001 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total liabilities |
28,854,574 | 13,785,733 | 5,633,200 | |||||||||
| Commitments and contingencies (see Note 16) |
||||||||||||
| Shareholders’ deficit: |
||||||||||||
| Series A convertible preferred stock, $0.0001 par value, 36,460,000 authorized; 27,175,213 issued and outstanding at December 31, 2012; 100,000,000 authorized; 69,421,047 issued and outstanding at December 31, 2013; liquidation preference of $16,305,127 at December 31, 2012 and $41,652,628 at December 31, 2013 (see Note 9); 5,000,000 shares authorized, no shares issued and outstanding on a pro forma basis at December 31, 2013 (see Unaudited Pro Forma Information paragraphs in Note 3). |
2,718 | 6,942 | — | |||||||||
| Common stock, $0.0001 par value, 14,600,000 authorized; 160,393 issued and outstanding at December 31, 2012; 53,000,000 authorized; 185,550 issued and outstanding at December 31, 2013 (see Note 9); 40,000,000 authorized; 2,600,162 issued and outstanding on a pro forma basis at December 31, 2013 (see Unaudited Pro Forma Information paragraphs in Note 3). |
16 | 19 | 260 | |||||||||
| Additional paid-in capital |
85,800,164 | 109,958,001 | 118,453,075 | |||||||||
| Accumulated deficit |
(113,187,793 | ) | (122,420,976 | ) | (123,295,134 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Total shareholders’ deficit |
(27,384,895 | ) | (12,456,014 | ) | (4,841,799 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Total liabilities and shareholders’ deficit |
$ | 1,469,679 | $ | 1,329,719 | $ | 791,401 | ||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements
83
Table of Contents
Biocept, Inc.
Statements of Operations and Comprehensive Loss
| For the year ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Revenues |
$ | 109,289 | $ | 134,245 | ||||
| Cost of revenues |
1,201,694 | 2,329,900 | ||||||
|
|
|
|
|
|||||
| Gross profit/(loss) |
(1,092,405 | ) | (2,195,655 | ) | ||||
| Operating expenses |
||||||||
| Research and development expenses |
6,562,152 | 3,086,737 | ||||||
| General and administrative expenses |
2,063,199 | 2,513,136 | ||||||
| Sales and marketing expenses |
785,319 | 148,903 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(10,503,075 | ) | (7,944,431 | ) | ||||
| Other income/(expense) |
||||||||
| Interest expense, net |
(2,187,499 | ) | (2,070,064 | ) | ||||
| Change in fair value of warrant liability |
454,389 | 782,112 | ||||||
| Other income/(expense) |
(22,541 | ) | — | |||||
|
|
|
|
|
|||||
| Total other income/(expense) |
(1,755,651 | ) | (1,287,952 | ) | ||||
|
|
|
|
|
|||||
| Loss before income taxes |
(12,258,726 | ) | (9,232,383 | ) | ||||
| Income tax expense |
800 | 800 | ||||||
|
|
|
|
|
|||||
| Net loss & comprehensive loss |
$ | (12,259,526 | ) | $ | (9,233,183 | ) | ||
|
|
|
|
|
|||||
| Weighted-average shares outstanding used in computing net loss per share attributable to common shareholders: |
||||||||
| Basic |
160,393 | 181,762 | ||||||
|
|
|
|
|
|||||
| Diluted |
160,393 | 181,762 | ||||||
|
|
|
|
|
|||||
| Net loss per common share: |
||||||||
| Basic |
$ | (76.43 | ) | $ | (50.80 | ) | ||
|
|
|
|
|
|||||
| Diluted |
$ | (76.43 | ) | $ | (50.80 | ) | ||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements
84
Table of Contents
Biocept, Inc.
Statements of Shareholders’ Deficit
| Series A Preferred Stock | Common Stock | Additional Paid-in Capital |
Accumulated Deficit |
Total | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
| Balance at December 31, 2011 |
27,175,213 | $ | 2,718 | 160,393 | $ | 16 | $ | 85,548,030 | $ | (100,928,267 | ) | $ | (15,377,503 | ) | ||||||||||||||
| Stock-based compensation expense |
— | — | — | — | 252,134 | — | 252,134 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (12,259,526 | ) | (12,259,526 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2012 |
27,175,213 | 2,718 | 160,393 | 16 | 85,800,164 | (113,187,793 | ) | (27,384,895 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Stock-based compensation expense |
— | — | — | — | 952,521 | — | 952,521 | |||||||||||||||||||||
| Stock issuance for RSU |
— | — | 21,846 | 2 | (2 | ) | — | — | ||||||||||||||||||||
| Exercise of stock options |
— | — | 4,021 | 1 | 20,104 | — | 20,105 | |||||||||||||||||||||
| Repurchase of common shares |
— | — | (710 | ) | — | (4,111 | ) | — | (4,111 | ) | ||||||||||||||||||
| Shares issued for conversion of notes payable and accrued interest of $20.2 million and $2.6 million, respectively |
42,245,834 | 4,224 | — | — | 22,808,180 | — | 22,812,404 | |||||||||||||||||||||
| Reclassification of warrant liability derivative due to triggering event |
— | — | — | — | 381,145 | — | 381,145 | |||||||||||||||||||||
| Net loss |
— | — | — | — | — | (9,233,183 | ) | (9,233,183 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at December 31, 2013 |
69,421,047 | $ | 6,942 | 185,550 | $ | 19 | $ | 109,958,001 | $ | (122,420,976 | ) | $ | (12,456,014 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
The accompanying notes are an integral part of these financial statements
85
Table of Contents
Biocept, Inc.
| For the year ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Cash Flows From Operating Activities |
||||||||
| Net loss |
$ | (12,259,526 | ) | $ | (9,233,183 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization |
365,568 | 266,554 | ||||||
| Inventory reserve |
56,004 | (70,004 | ) | |||||
| Stock-based compensation |
252,134 | 952,521 | ||||||
| Non-cash interest expense related to convertible debt and other financing activities |
2,159,234 | 2,066,287 | ||||||
| Change in fair value of warrant liabilities |
(454,389 | ) | (782,112 | ) | ||||
| Increase/(decrease) in cash resulting from changes in: |
||||||||
| Accounts receivable |
(13,634 | ) | 9,685 | |||||
| Inventory |
(117,287 | ) | 38,464 | |||||
| Prepaid expenses and other current assets |
77,654 | (37,691 | ) | |||||
| Other non-current assets |
— | 268,583 | ||||||
| Accounts payable |
354,553 | (175,280 | ) | |||||
| Accrued liabilities |
730,836 | 233,852 | ||||||
| Deferred rent |
241,837 | 259,961 | ||||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(8,607,016 | ) | (6,202,363 | ) | ||||
| Cash Flows From Investing Activities |
||||||||
| Purchases of fixed assets |
(8,046 | ) | (711 | ) | ||||
|
|
|
|
|
|||||
| Net cash used in investing activities |
(8,046 | ) | (711 | ) | ||||
| Cash Flows From Financing Activities |
||||||||
| Proceeds from exercise of stock options |
— | 20,105 | ||||||
| Payments for repurchase of shares |
— | (4,111 | ) | |||||
| Payments on supplier and other third party financings |
(164,974 | ) | (154,998 | ) | ||||
| Proceeds from borrowings on line of credit |
— | 1,981,000 | ||||||
| Proceeds from issuance of notes payable |
5,960,000 | — | ||||||
| Proceeds from issuance of convertible notes and warrants |
2,570,000 | 4,245,000 | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
8,365,026 | 6,086,996 | ||||||
|
|
|
|
|
|||||
| Net decrease in Cash and Cash Equivalents |
(250,036 | ) | (116,078 | ) | ||||
| Cash and Cash Equivalents at Beginning of Period |
435,292 | 185,256 | ||||||
|
|
|
|
|
|||||
| Cash and Cash Equivalents at End of Period |
$ | 185,256 | $ | 69,178 | ||||
|
|
|
|
|
|||||
| Supplemental Disclosures of Cash Flow Information: |
||||||||
| Cash paid during the period for: |
||||||||
| Interest |
$ | 28,276 | $ | 3,777 | ||||
|
|
|
|
|
|||||
| Taxes |
$ | 800 | $ | 800 | ||||
|
|
|
|
|
|||||
Non-cash Investing and Financing Activities:
For the years ended December 31, 2012 and 2013, the Company financed insurance premiums of $128,929 and $122,777, respectively, through third party financings. Such financings occur on an annual basis during the three months ended December 31 of each year.
During the year ended December 31, 2013, 21,846 shares of common stock, with a par value of $0.0001, were issued for restricted stock units.
86
Table of Contents
During the year ended December 31, 2013, convertible notes with a principal balance of $20,231,000 and accrued interest of approximately $2,581,000 were converted into 42,245,834 shares of preferred stock with a par value of $0.0001. In conjunction with this conversion, $236,799 of derivative warrant liabilities were reclassified to additional paid-in capital, as the underlying exercise prices on the warrants were determined by the debt conversion. In addition, during the year ended December 31, 2013, an additional $144,346 of derivative warrant liabilities were reclassified to additional paid-in capital when their underlying exercise price was fixed.
During the year ended December 31, 2013, the Company issued to its landlord a warrant to purchase common shares with a warrant coverage amount of $502,605 and an exercise price equal to the price per share of the Company’s common stock sold in an initial public offering under the Securities Act (“IPO”). The fair value of the warrant as calculated under the Company’s probability weighted Black-Scholes valuation model was approximately $309,000 at issuance in September 2013 (see Note 7), and is recorded on the balance sheet as a component of deferred rent and warrant liability.
During the year ended December 31, 2013, the Company incurred $538,318 in costs directly associated with its anticipated IPO, which are reflected on the balance sheet as a component of prepaid expenses and other current assets at December 31, 2013. A liability of $328,221 for associated unpaid invoices is recorded as a component of accounts payable at December 31, 2013.
The accompanying notes are an integral part of these financial statements
87
Table of Contents
BIOCEPT, INC.
1. The Company and Business Activities
Biocept, Inc. (“the Company”) was founded in California in May 1997 and is a commercial-stage cancer diagnostics company developing and commercializing proprietary circulating tumor cell (CTC) and circulating tumor DNA (ctDNA) tests utilizing a standard blood sample to improve the treatment that oncologists provide to their patients by providing better, more detailed information on the characteristics of their tumor.
The Company operates a clinical laboratory that is CLIA-certified (under the Clinical Laboratory Improvement Amendment of 1988) and CAP-accredited (by the College of American Pathologists), and manufactures CEE microfluidic channels, related equipment and certain reagents to perform the Company’s diagnostic tests in a facility located in San Diego, California. CLIA certification and accreditation are required before any clinical laboratory may perform testing on human specimens for the purpose of obtaining information for the diagnosis, prevention, treatment of disease, or assessment of health. The tests the Company offers are classified as laboratory developed tests (LDTs), under the CLIA regulations.
In July 2013, the Company effected a reincorporation to Delaware by merging itself with and into Biocept, Inc., a Delaware corporation, which had been formed to be and was a wholly-owned subsidiary of the Company since July 23, 2013.
2. Liquidity
At December 31, 2012 and 2013, the Company had accumulated deficits of approximately $113,188,000 and $122,421,000, respectively. For the years ended December 31, 2012 and 2013, the Company incurred net losses of approximately $12,260,000 and $9,233,000, respectively. In addition, as of December 31, 2013, the Company had notes payable and amounts outstanding under a line of credit due within one year, gross of applicable debt discounts, totaling approximately $8,371,000. The Company borrowed a total of $8,530,000, and $6,226,000 during the years ended December 31, 2012 and 2013, respectively, under note agreements with certain shareholders and a line of credit. While the Company is currently in the commercialization stage of operations, the Company has not yet achieved profitability and anticipates that it will continue to incur net losses in the foreseeable future.
Historically, the Company’s principal sources of cash have included revenues from clinical laboratory testing through contracted partners, proceeds from the issuance of common and preferred stock and proceeds from the issuance of debt. The Company’s principal uses of cash have included cash used in operations, payments relating to purchases of property and equipment and repayments of borrowings. The Company expects that the principal uses of cash in the future will be for hiring of sales and marketing personnel and increased sales and marketing activities, funding of research and development, capital expenditures, and general working capital requirements. The Company expects that, as revenues grow, sales and marketing and research and development expenses will continue to grow, albeit at a slower rate and, as a result, the Company will need to generate significant net revenues to achieve and sustain income from operations.
On February 10, 2014, the Company received net proceeds of approximately $16,673,000 as a result of the closing of its IPO, net of underwriting discounts and additional costs incurred (see Note 18). Management believes that its cash resources should be sufficient to support currently forecasted operations through at least the next twelve months. However, the Company operates in a market that makes its prospects difficult to evaluate, and the likelihood that the Company will need additional debt or equity financing in the future to execute on its current or future business strategies beyond the next twelve months is probable. Management also believes that, if necessary, it can implement plans in the short term to conserve existing cash should additional financing activities be delayed. Capital outlays and operating expenditures may occur over the next twelve months as the Company expands its infrastructure, commercialization, and research and development activities.
88
Table of Contents
3. Summary of Significant Accounting Policies
Basis of Presentation
The financial statements and accompanying notes are prepared in accordance with accounting principles generally accepted in the United States of America.
Certain prior period amounts have been reclassified to conform to the current period presentation. Such reclassifications did not affect the Company’s Balance Sheets, Results of Operations or Cash Flows for the years ended December 31, 2012 and 2013.
Use of Estimates
The preparation of financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. On an ongoing basis, management evaluates these estimates and judgments, including those related to inventories, long-lived assets, convertible debt, derivative liabilities, income taxes, and stock-based compensation. The Company bases its estimates on various assumptions that it believes are reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
Unaudited Pro Forma Information
The unaudited pro forma balance sheet information as of December 31, 2013 gives effect to (i) the automatic conversion of all outstanding shares of the Company’s Series A preferred stock into 1,652,851 shares of common stock, (ii) the conversion of convertible promissory notes and accrued interest of approximately $6,886,000 (as of December 31, 2013) into an aggregate of 688,610 shares of the Company’s common stock in connection with the closing of the Company’s IPO, (iii) the write-off of $874,158 to interest expense for the unamortized debt discount on notes payable, (iv) the reclassification to line of credit of $315,243 for the unamortized debt discount previously classified against notes payable, (v) the issuance of an estimated 73,151 shares of common stock upon such IPO pursuant to the settlement of certain restricted stock units (which are currently expressed in shares of preferred stock) in accordance with their terms, (vi) the termination of certain warrants upon the closing of the Company’s IPO in accordance with their terms and (vii) the reclassification to shareholders’ deficit of the fair value of certain warrants the exercise price and/or exercisability period length of which will be fixed upon the closing of the Company’s IPO in accordance with their terms, assuming for all such items an IPO price of $10.00 per share.
The unaudited pro forma balance sheet information as of December 31, 2013 assumes that the completion of the Company’s IPO had occurred as of December 31, 2013, and excludes shares of common stock issued in the IPO and any related net proceeds. In October 2013 the Board of Directors approved an amendment of the Company’s certificate of incorporation, to be filed in connection with the Company’s IPO, which would decrease the number of common shares authorized to 40,000,000 and decrease the number of preferred shares authorized to 5,000,000.
Reverse Stock Split and Change in Par Value of Common Stock and Preferred Stock
In November 2011, the Company effected a 1:3 reverse stock split of the Company’s common shares. In addition, in July 2013, in conjunction with its reincorporation in the state of Delaware, the Company initiated par values for preferred and common shares equal to $0.0001. On November 1, 2013, the Company effected a 1:14 reverse stock split for all common shares. All references to share and per share amounts in the financial statements and accompanying notes to the financial statements have been retroactively restated to reflect the 1:14 reverse stock split and the change in par value.
89
Table of Contents
Revenue Recognition
Revenue is recognized in accordance with the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 605, Revenue Recognition, and ASC 954-605 Health Care Entities, Revenue Recognition which requires that four basic criteria must be met before revenue can be recognized: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred and title and the risks and rewards of ownership have been transferred to the client or services have been rendered; (3) the price is fixed or determinable; and (4) collectability is reasonably assured. For contract partners, revenue is recorded based upon the contractually agreed upon fee schedule. When assessing collectability, the Company considers whether there is sufficient payment history to reliably estimate a payor’s individual payment patterns. For new tests where there is limited evidence of payment history at the time the tests are completed, the Company recognizes revenue equal to the amount of cash received until such time as reimbursement experience can be established.
The Company’s main source of revenue for the years ended December 31, 2012 and 2013 is through contracted partners. This revenue is derived from clinical laboratory testing performed in the Company’s laboratories under agreements with such partners. As there is a contractually agreed upon price, and collectability from the partners is reasonably assured, revenues for these tests are earned at the time the test is completed and the results are delivered to the partners or a third party.
Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities of three months or less to be cash equivalents. The Company places its cash and cash equivalents with reputable financial institutions that are insured by the Federal Deposit Insurance Corporation (FDIC). At times, deposits held may exceed the amount of insurance provided by the FDIC. The Company has not experienced any losses in its cash and cash equivalents and believes they are not exposed to any significant credit risk.
Fair Value Measurement
The Company uses a three-tier fair value hierarchy to prioritize the inputs used in the Company’s fair value measurements. These tiers include: Level 1, defined as observable inputs such as quoted prices in active markets for identical assets; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions. The Company believes the carrying amount of cash and cash equivalents, accounts receivable, accounts payable and accrued expenses approximate their estimated fair values due to the short-term maturities of these financial instruments.
As of December 31, 2012 and 2013, the Company classified the fair value measurements of the Company’s warrant liability derivative as Level 3. See Note 7 for further details about the inputs and assumptions used to determine the fair value of the warrant liability at each balance sheet date.
The values attributed to such warrants as of December 31, 2012 and 2013 were as follows:
| Fair Value Measurements Using | ||||||||||||
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||
| Liabilities |
||||||||||||
| Warrant Liability at December 31, 2012 |
— | — | $ | 981,747 | ||||||||
| Warrant Liability at December 31, 2013 |
— | — | $ | 2,140,532 | ||||||||
90
Table of Contents
The following table includes a summary of changes in the fair value of the warrants for the years ended December 31, 2012 and 2013:
| Fair Value Measurements at Reporting Date Using Significant Unobservable Inputs (Level 3) |
||||
| Balance at December 31, 2011 |
923,325 | |||
| Warrant liability incurred in 2012 |
512,811 | |||
| Change in fair value in 2012 |
(454,389 | ) | ||
|
|
|
|||
| Balance at December 31, 2012 |
981,747 | |||
| Warrant liability incurred in 2013 |
2,322,042 | |||
| Warrant liability reclassified to additional paid-in capital in 2013 |
(381,145 | ) | ||
| Change in fair value in 2013 |
(782,112 | ) | ||
|
|
|
|||
| Balance at December 31, 2013 |
$ | 2,140,532 | ||
|
|
|
|||
Concentration of Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of temporary cash investments. The Company has not experienced losses in such accounts. Management believes that the Company is not exposed to any significant credit risk with respect to its cash and cash equivalents.
In 2012, the Company launched commercial operations in partnership with a commercial partner, Clarient Diagnostic Services, Inc. (“Clarient”), a GE Healthcare Company. For the years ended December 31, 2012 and 2013, 79% and 10%, respectively, of the revenue earned was billed through this relationship. In addition, at December 31, 2012, 100% of the receivables were due from Clarient. In 2013, the Company entered into a research support agreement with a not-for-profit tax-exempt organization, Dana Farber Partners Cancer Care, Inc. (“Dana Farber”). For the year ended December 31, 2013, 77% of the revenue earned was billed through this relationship. In addition, 100% of the receivables were due from Dana Farber at December 31, 2013. For the year ended December 31, 2013, three customers made up 78%, 11% and 10% of total revenues.
All of the Company’s sales for all periods presented were generated in the United States of America.
Certain components used in the Company’s current or planned products are available from only one supplier, and substitutes for these components cannot be obtained easily or would require substantial design or manufacturing modifications or identification and qualification of alternative sources.
Accounts Receivable
Accounts receivable are carried at original invoice amounts, less an estimate for doubtful receivables, based on a review of all outstanding amounts on a periodic basis. The estimate for doubtful receivables is determined from an analysis of the accounts receivable on a quarterly basis, and is recorded as bad debt expense. As the Company only recognizes revenue to the extent collection is expected and reasonably assured, bad debt expense related to receivables from patient service revenue is recorded in general and administrative expense in the statement of operations and comprehensive loss. Accounts receivable are written off when deemed uncollectible. Recoveries of accounts receivable previously written off are recorded when received. As of December 31, 2012 and 2013, management determined that all of the amounts recorded as accounts receivable were collectible, and no allowance for doubtful accounts was needed.
Inventories
Inventories are valued at the lower of cost or market value. Cost is determined by the average cost method. The Company records adjustments to its inventory for estimated obsolescence or diminution in market value equal to
91
Table of Contents
the difference between the cost of the inventory and the estimated market value. At the point of loss recognition, a new cost basis for that inventory is established, and subsequent changes in facts and circumstances do not result in the restoration or increase in that newly established cost basis.
Fixed Assets
Fixed assets consist of machinery and equipment, furniture and fixtures, computer equipment and software, leasehold improvements, capital leased equipment and construction in process. Fixed assets are stated at cost less accumulated depreciation and amortization. Additions, improvements, and major renewals are capitalized. Maintenance, repairs, and minor renewals are expensed as incurred. Depreciation is determined using the straight-line method over the estimated useful lives of the assets, which range from three to five years. Leasehold improvements are amortized over the life of the lease or the asset, whichever is shorter. Depreciation expense for the years ended December 31, 2012 and 2013 was approximately $366,000 and $267,000, respectively.
Upon sale, retirement or disposal of fixed assets, the accounts are relieved of the cost and the related accumulated depreciation or amortization with any gain or loss recorded to the statement of operations.
Fixed assets are reviewed for impairment whenever changes in circumstances indicate that the carrying amount of an asset may not be recoverable. These computations utilize judgments and assumptions inherent in the estimates of future cash flows to determine recoverability of these assets. If the assumptions about these assets were to change as a result of events or circumstances, the Company may be required to record an impairment loss.
Warrant Liability
Warrants for shares that are contingently redeemable and for which the exercise price is not fixed are classified as liabilities on the accompanying balance sheets and carried at their estimated fair value, determined through use of a Black-Scholes valuation model. As of and for the years ended December 31, 2012 and 2013, the Company evaluated and concluded that the fair value obtained from the Black-Scholes method of valuing the warrant liability does not materially differ from the valuation of such warrants using the Monte Carlo or binomial lattice simulation models, and therefore the use of the Black-Scholes valuation model was considered a reasonable method to value the warrants. At the end of each reporting period, any changes in fair value are recorded as a component of other income (expense). The Company will continue to adjust the carrying value of the warrants until the completion of its IPO on February 10, 2014, at which time the exercise price was fixed and the fair value of those warrants was reclassified to shareholders’ deficit.
Stock-based Compensation
The Company accounts for stock-based compensation under the provisions of FASB ASC Topic 718, Compensation—Stock Compensation, which requires the measurement and recognition of compensation expense for all stock-based awards made to employees and directors based on estimated fair values on the grant date. The Company estimates the fair value of stock-based awards on the date of grant using the Black-Scholes option pricing model (“Black-Scholes valuation model”). The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods using the straight-line method. The Company estimates forfeitures at the time of grant and revises these estimates in subsequent periods if actual forfeitures differ from those estimates. See additional information in Note 10.
The Company accounts for stock-based compensation awards to non-employees in accordance with FASB ASC Topic 505-50, Equity-Based Payments to Non-Employees (“ASC 505-50”). Under ASC 505-50, the Company determines the fair value of the warrants or stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. All issuances of equity instruments issued to non-employees as consideration for goods or services
92
Table of Contents
received by the Company are accounted for based on the fair value of the equity instruments issued. These awards are recorded in expense and additional paid-in capital in shareholders’ equity over the applicable service periods based on the fair value of the options at the end of each period.
Calculating the fair value of stock-based awards requires the input of highly subjective assumptions into the Black-Scholes valuation model. Stock-based compensation expense is calculated using the Company’s best estimates, which involves inherent uncertainties, and the application of management’s judgment. Significant estimates include the fair value of the Company’s common stock at the date of grant, the expected life of the stock option, stock price volatility, risk-free interest rate and forfeiture rates.
Research and Development
Research and development costs are expensed as incurred. The amounts expensed in the years ended December 31, 2012 and 2013 were approximately $6,562,000 and $3,087,000, respectively, which includes salaries of research and development personnel.
Income Taxes
The Company provides for income taxes utilizing the liability method. Under the liability method, current income tax expense or benefit is the amount of income taxes expected to be payable or refundable for the current year. A deferred income tax asset or liability is computed for the expected future impact of differences between the financial reporting and tax bases of assets and liabilities and for the expected future tax benefit to be derived from tax credits. Tax rate changes are reflected in the computation of the income tax provision during the period such changes are enacted.
Deferred tax assets are reduced by a valuation allowance when, in management’s opinion, it is more likely than not that some portion or all of the deferred tax assets will not be realized. The Company considers the scheduled reversal of deferred tax liabilities, projected future taxable income, and tax planning strategies in making this assessment. The Company’s valuation allowance is based on available evidence, including its current year operating loss, evaluation of positive and negative evidence with respect to certain specific deferred tax assets including evaluation sources of future taxable income to support the realization of the deferred tax assets. The Company has established a full valuation allowance on the deferred tax assets as of December 31, 2012 and 2013, and therefore has not recognized any income tax benefit or expense in the periods presented.
ASC 740, Income Taxes (“ASC 740”), clarifies the accounting for uncertainty in income taxes recognized in the financial statements. ASC 740 provides that a tax benefit from uncertain tax positions may be recognized when it is more-likely-than- not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, based on the technical merits of the position. Income tax positions must meet a more-likely-than-not recognition threshold to be recognized. ASC 740 also provides guidance on measurement, derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
The Company recognizes interest and/or penalties related to income tax matters in income tax expense. There is no accrual for interest or penalties for income taxes on the balance sheets at December 31, 2012 and 2013, and the Company has not recognized interest and/or penalties in the statements of operations for the years ended December 31, 2012 and 2013.
Recent Accounting Pronouncements
In July 2013, the FASB issued authoritative guidance which requires netting unrecognized tax benefits against deferred tax assets for a loss or other carryforward that would apply in settlement of uncertain tax positions. This guidance will be effective for annual reporting periods beginning after December 15, 2013. The Company does not believe that adoption of this guidance will have a material impact on the Company’s financial statements or disclosures.
93
Table of Contents
4. Balance Sheet Details
The following provides certain balance sheet details:
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Fixed Assets |
||||||||
| Machinery and equipment |
$ | 2,761,560 | $ | 2,761,560 | ||||
| Furniture and office equipment |
209,844 | 209,844 | ||||||
| Computer equipment and software |
681,508 | 681,508 | ||||||
| Leasehold improvements |
373,653 | 373,653 | ||||||
| Capital lease equipment |
677,000 | 677,000 | ||||||
| Construction in process |
11,588 | 12,299 | ||||||
|
|
|
|
|
|||||
| 4,715,153 | 4,715,864 | |||||||
| Accumulated depreciation and amortization |
4,090,423 | 4,356,977 | ||||||
|
|
|
|
|
|||||
| Total fixed assets, net |
$ | 624,730 | $ | 358,887 | ||||
|
|
|
|
|
|||||
| Accrued Liabilities |
||||||||
| Accrued interest |
$ | 1,963,007 | $ | 524,885 | ||||
| Accrued payroll |
185,150 | 125,299 | ||||||
| Deferred wages |
972,405 | 1,377,987 | ||||||
| Accrued vacation |
224,187 | 213,601 | ||||||
| Other |
2,057 | 286 | ||||||
|
|
|
|
|
|||||
| Total accrued liabilities |
$ | 3,346,806 | $ | 2,242,058 | ||||
|
|
|
|
|
|||||
During the year ended December 31, 2013, the Company incurred $538,318 in costs directly associated with its anticipated IPO, which are reflected on the balance sheet as a component of prepaid expenses and other current assets at December 31, 2013. A liability of $328,221 for associated unpaid invoices is recorded as a component of accounts payable at December 31, 2013.
As of December 31, 2012 other non-current assets of $269,000 consisted solely of deposits for the San Diego building, which is leased under a non-cancelable operating lease. During the year ended December 31, 2013, the Company amended its lease agreement and forfeited the balance.
5. Line of Credit
In July 2013, the Company entered into a revolving line of credit with UBS Bank USA in the initial amount of $1.5 million. Interest accrues daily on the outstanding balance and is paid monthly at a variable rate which, as of December 31, 2013, was 2.75% over the 30 day LIBOR rate or a nominal annual interest rate of 2.92%. As of December 31, 2013, the amount outstanding under this revolving line of credit is approximately $2.0 million. Three of the Company’s related parties guaranteed the loan and pledged financial assets to the bank to secure their guaranties, as approved by the Company’s board of directors. In return, the Company issued common stock warrants to the guarantors. The number of shares subject to the common stock warrants will be determined by dividing the warrant coverage amount, which is 50% of the fair market value of the collateral provided by the respective guarantors to secure their respective guaranty obligations to the bank, by the exercise price, which will be set at the price per share of the Company’s common stock sold in its IPO. See Note 7 for further discussion of the warrant liabilities. The Company has entered into an agreement with the guarantors that provides for reimbursement of any amounts paid by them on their guaranties. This reimbursement obligation is secured by a security interest in the Company’s assets.
94
Table of Contents
6. Notes Payable
The following is a summary of the Company’s short-term and long-term debt obligations:
| December 31, | ||||||||
| 2012 | 2013 | |||||||
| Note payable to shareholder; principal and interest payable in quarterly installments until maturity on April 2015, bearing interest at a per annum fixed rate of 3.25%. As of June 28, 2013, the note payable was converted into preferred shares. (“Goodman Note”) (See Note 7) |
$ | 1,935,000 | $ | — | ||||
| Secured convertible note to a major shareholder. (“2008 Convertible Note”) (See Note 7) |
1,400,000 | 1,400,000 | ||||||
| Secured convertible notes. Total includes convertible notes due to a major shareholder of $11,250,000 at December 31, 2012. As of June 28, 2013, the notes payable were converted into preferred shares. (“2011 Convertible Bridge Notes”) (See Note 7) |
12,336,427 | — | ||||||
| Notes payable to shareholders issued in 2012. Includes notes of $5,810,000 to a major shareholder at December 31, 2012. As of June 28, 2013, the notes payable were converted into preferred shares. (“2012 Revolver Notes”) (See Note 7) |
5,960,000 | — | ||||||
| Unsecured convertible notes, issued under a note and warrant purchase agreement dated as of June 28, 2013, net of discounts related to warrants aggregating $0 and $874,158 at December 31, 2012 and 2013, respectively. Includes notes of $720,000 and $2,505,000 to a major shareholder at December 31, 2012 and 2013, respectively. (“2013 Convertible Bridge Notes”) (See Note 7) |
745,000 | 4,115,842 | ||||||
| Other debt discount (See Notes 5 and 7) |
— | (315,243 | ) | |||||
|
|
|
|
|
|||||
| Total notes payable |
22,376,427 | 5,200,599 | ||||||
| Less current portion |
21,631,427 | 5,200,599 | ||||||
|
|
|
|
|
|||||
| Long-term portion |
$ | 745,000 | $ | — | ||||
|
|
|
|
|
|||||
Except for the non-current balance of the 2013 Convertible Bridge Notes, all outstanding notes payable and convertible notes payable were classified as current as of December 31, 2012, as the Company was unable to make principal and interest payments on these notes during the year ended December 31, 2012, or prior to the conversion of certain of the notes as of June 28, 2013. None of the lenders had sought any remedy for this default as of December 31, 2012 or prior to the conversion of the notes as of June 28, 2013.
On June 28, 2013, approximately $20,231,000 of outstanding notes payable and $2,581,000 of accrued interest were converted into 42,245,834 preferred shares, in accordance with the provisions of the debt conversion agreements of that date. As of December 31, 2013, all remaining principal payments for outstanding notes payable and convertible notes are due within one year.
Total interest expense incurred for all notes, convertible notes, and the line of credit, including amortization of debt discounts, for the years ended December 31, 2012 and 2013 was approximately $2,125,000 and $1,964,000, respectively, of which approximately $1,957,000 and $516,000 was recorded as accrued interest as of December 31, 2012 and 2013, respectively.
7. Convertible Notes and Warrants
Outstanding Warrants—Preferred Shares
Goodman Note
During April 2005, the Company entered into an unsecured loan agreement for $15,000,000. The note required interest payments and principal settlement upon maturity at the earliest of (a) April 20, 2010, (b) the Company being acquired, or (c) the Company having a change in control, other than through the sale of preferred shares.
95
Table of Contents
During January 2009, the Company entered into an amendment and restatement of the unsecured amended loan, whereby the parties agreed that the principal amount would be reduced to $3,000,000. The amended and restated unsecured note bears interest at a variable rate per annum based on prime plus 25 basis points. 25% of the accrued interest was due and payable quarterly in arrears on the last business day of each three-month quarter beginning February 1, 2009. The remaining 75% of the accrued interest was not to be compounded by becoming part of the principal, and was due and payable in a lump-sum payment on the maturity date. The principal and any interest amounts that remain outstanding was set to mature at the earlier of (a) April 20, 2010, or (b) the date immediately prior to the Company’s closing of an acquisition or asset transfer as defined by the Company’s amended and restated articles of incorporation.
In conjunction with the 2009 amendment, the Company issued a warrant to purchase preferred shares issued in the first equity financing to occur subsequent to the execution of the note, and in which the Company receives at least $2,000,000 in gross aggregate proceeds. The exercise price of the warrant is equal to the per share price of preferred shares sold in that equity financing, and the number of shares that may be exercised is equal to 10% of the principal amount of the convertible loan divided by the exercise price. Early termination of the warrant can occur upon an IPO, or if the Company is acquired. The holder of the warrant is to be given 20 days advance notice of such an event, and the warrant will terminate if not exercised before the date of the event.
A qualifying equity financing occurred during February 2009, which set the warrant exercise price at $0.60 per share.
During May 2010, the Company entered into a second amendment and restatement of the Goodman Note in order to extend the maturity date and amend the timing of payments to be made to the lender and to secure the Company’s obligations under the note. The secured amended and restated note bears interest at a per annum fixed rate of 3.25% and is due and payable quarterly in arrears on the last business day of each three-month quarter beginning May 1, 2010. On the effective date of the second amendment, the Company paid the lender $750,000 which was applied to the principal balance of $3,000,000. Beginning May 1, 2010, principal payments are due and payable quarterly in advance. For principal payments due and payable during the period of May 1, 2010 through January 31, 2011, the quarterly principal payment was equal to $45,000; for principal payments due and payable during the period of February 1, 2012 through January 31, 2014, the quarterly principal payment is equal to $90,000; and for principal payments due and payable during the period of February 1, 2014 through the maturity date, the quarterly principal payment is equal to $150,000. In addition to the $750,000 principal paid on the effective date of the amendment, the Company paid principal payments of $135,000 and $180,000 during the years ended December 31, 2010 and 2011, respectively. No principal payments were made during the years ended December 31, 2012 or 2013.
As of June 28, 2013 the holder of the Goodman Note agreed to convert the total principal balance owed under the Goodman Note of $1,935,000 and accrued interest of approximately $105,000 into 3,777,324 preferred shares at a conversion price of $0.54 per share. Although the conversion price of the debt was greater than the value of the preferred shares at the time of conversion, the Company did not record a gain on the conversion under the troubled debt restructuring accounting guidance since the transaction occurred between related parties, and thus, was treated as a capital transaction.
In July 2013, in connection with this conversion, the Company issued to such beneficial owner a warrant to purchase 23,809 shares of common stock at an exercise price which will be set at the price per share of the Company’s common stock sold in the Company’s IPO. The warrants will be exercisable for a two-year period beginning with the closing of the Company’s IPO. In accordance with guidance applicable to accounting for derivative financial instruments that are accounted for as liabilities, the warrants are initially recorded at their fair value and are then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For the warrants for common shares issued under the Goodman Note agreement, the Company used a probability weighted Black-Scholes valuation model. The fair value of the Goodman Note warrants was approximately $62,000 and is included in warrant liabilities at December 31, 2013.
96
Table of Contents
2008 Convertible Note
In December 2008, the Company issued a convertible note in the principal amount of $1,400,000 which is secured by all assets of the Company to an affiliate of a major shareholder. The 2008 Convertible Note bears interest at a variable rate based on prime per annum payable at maturity, and matures at the earliest occurrence of, (a) the passing of 48 months from inception of the note, (b) the closing date of an acquisition or asset transfer as defined by the note, or (c) the closing date of the issuance and sale of shares of common stock of the Company in the Company’s IPO.
Upon the closing of a sale by the Company of its preferred shares in which the Company receives an aggregate of at least $20,000,000 in cumulative gross proceeds, including conversion of the convertible loan amount before the maturity date, the unpaid principal and accrued interest shall automatically be converted into the number of preferred shares, of the series sold by the Company in such sale, equal to the unpaid principal and accrued interest divided by the per share purchase price of the preferred shares in such sale. The 2008 Convertible Note may also be converted before the maturity date at the option of the holder at the closing of an equity financing involving the sale of the Company’s preferred shares in which the Company receives an aggregate of at least $2,000,000 in cumulative gross proceeds, with a conversion price equal to the per share price included in that equity financing. In July 2013, the Company amended the 2008 Convertible Note to provide that all principal and accrued interest on the note would automatically convert into common stock upon the closing of an IPO at the price per share at which common stock is sold in such IPO.
Issued with the 2008 Convertible Note was a warrant to purchase preferred shares issued in the first equity financing to occur subsequent to the execution of the 2008 Convertible Note, and in which the Company receives at least $2,000,000 in gross aggregate proceeds. The exercise price of the warrant is equal to the per share price of preferred shares sold in that equity financing, and the number of shares that may be exercised is equal to 10% of the principal amount of the convertible loan divided by the exercise price. Early termination of the warrant can occur upon an IPO or if the Company is acquired. The holder of the warrant is to be given 20 days advance notice of such an event, and the warrant will terminate if not exercised before the date of the event.
A qualifying equity financing occurred during February 2009, which set the 2008 Convertible Note conversion price and the warrant exercise price at $0.60 per share. The 2008 Convertible Note remains outstanding at December 31, 2012 and 2013.
2011 Convertible Bridge Notes
In February 2011, the Company executed a note and warrant purchase agreement with a major shareholder’s affiliates. In exchange for a series of loans in an aggregate amount equal to $5,000,000 over a period through September 1, 2011, the Company issued secured convertible promissory notes and warrants to purchase preferred shares. The aggregate amount was subsequently raised to $6,000,000 and then $15,000,000 during the year and the funding period was first extended to February 2012 and then to December 2012. Other investors, including related parties, also became party to this arrangement and purchased 2011 Convertible Bridge Notes and warrants.
All unpaid principal and interest outstanding was initially payable on December 31, 2011. During 2012, the maturity date was extended to December 31, 2012. The 2011 Convertible Bridge Notes are secured by virtually all of the assets of the Company. The 2011 Convertible Bridge Notes bear interest at 8%, payable at maturity. The number of preferred shares for which the warrants are exercisable is determined by dividing the warrant coverage amount, which is 20% of the principal amount of the notes issued under the agreement, by the exercise price.
Upon the closing of the sale by the Company of its preferred stock in which the Company receives an aggregate of at least $20,000,000 in cumulative gross proceeds, including conversion of the 2011 Convertible Bridge Notes,
97
Table of Contents
before the maturity date, the unpaid principal and accrued interest shall automatically be converted into the number of preferred shares, of the series sold by the Company in such sale, equal to the unpaid principal and accrued interest divided by the per share purchase price of the preferred shares in such sale. At any time before the maturity date the investor may elect to convert all or any amount of the unpaid principal and accrued interest into the Company’s Series A preferred shares at $0.54 per share. Early termination of the warrants can occur upon an IPO or if the Company is acquired. The holders of the warrants are to be given 20 days advance notice of such an event, and the warrants will terminate if not exercised before the date of the event.
In accordance with guidance applicable to accounting for derivative financial instruments that are accounted for as liabilities, the warrants are initially recorded at their fair value and are then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For stock-based derivative financial instruments issued under the note and warrant purchase agreement dated February 2011, the Company used the Black-Scholes valuation model. The Company recorded approximately $1,400,000 related to the fair value of the warrants at the date of issuance, as a discount to the carrying value of the 2011 Convertible Bridge Notes, accreted as interest expense over the life of the debt. The Company valued the warrants at the date of each issuance using the Black-Scholes valuation model with the following underlying assumptions: contractual term of 5 years, an underlying preferred share price between $0.25 and $0.54, an exercise price of $0.54, an average risk-free interest rate between 0.70% and 2.26%, a dividend yield of 0%, and volatilities between 100.0% and 105.0%. Approximately $302,000 related to accretion of the discount was recognized as interest expense during the year ended December 31, 2012. The discount was fully accreted as of December 31, 2012.
As of December 31, 2012, the Company had issued the 2011 Convertible Bridge Notes with an aggregate principal amount of approximately $12,336,000. No further note or warrant issuances were made under this agreement during the year ended December 31, 2013. As of December 31, 2012, the Company was in default for payment on the 2011 Convertible Bridge Notes, and no principal payments were made in 2013 prior to their conversion. As of June 28, 2013 the investors under these notes elected to convert the total principal balance owed under the 2011 Convertible Bridge Notes of approximately $12,336,000 and accrued interest of approximately $1,832,000 into 26,237,611 preferred shares at a conversion price of $0.54 per share. Upon the conversion, the exercise price of the related warrants was set at $0.54 per share, and the $236,799 fair value of the warrants was reclassified into additional paid-in capital as of June 28, 2013. Although the conversion price of the debt was greater than the value of the preferred shares at the time of conversion, the Company did not record a gain on the conversion under the troubled debt restructuring accounting guidance since the transaction occurred between related parties, and thus, was treated as a capital transaction.
2012 Revolver Notes
On January 13, 2012, the Company executed a note and warrant purchase agreement with several shareholders, including a major shareholder, calling for (in addition to the issuance of certain related warrants) the issuance of a series of notes to be issued between January 13, 2012 and April 5, 2012 totaling up to $1,750,000, with an original maturity date in April 2012. The 2012 Revolver Notes were amended on April 5, 2012 to extend the maturity date to May 31, 2012 or July 31, 2012, depending on certain milestones, and to allow the Company to issue up to $5,000,000 in notes payable under this agreement, as needed. The 2012 Revolver Notes were amended again on November 8, 2012 to increase the amount of notes payable the Company can issue to $8,000,000, and to provide that all notes issued under this agreement shall have the same maturity date of either November 30, 2012 or December 31, 2012, depending on certain milestones. The 2012 Revolver Notes bear interest at 10%, payable at maturity.
Beginning on the closing of the sale by the Company of its preferred shares in which the Company receives an aggregate of at least $20,000,000 in cumulative gross proceeds, the warrants are exercisable for preferred shares of the series sold by the Company in such sale, at an exercise price equal to the purchase price per share of the preferred shares sold by the Company in such sale. The number of preferred shares for which the warrants are exercisable is determined by dividing the warrant coverage amount, which is 20% of the principal amount of the
98
Table of Contents
notes issued under the agreement on the issuance date of such 2012 Revolver Notes, by the exercise price. At any time prior to the maturity date, the investor may elect to convert all or any amount of the unpaid principal and accrued interest into the Company’s Series A preferred stock at $0.54 per share, or if a qualified financing has occurred, at the purchase price per share of the preferred shares sold by the Company in such qualified financing. Early termination of the warrant can occur upon an IPO, or if the Company is acquired. The holders of the warrants are to be given 20 days advance notice of such an event, and the warrants will terminate if not exercised before the date of the event.
In accordance with guidance applicable to accounting for derivative financial instruments that are accounted for as liabilities, the warrants are initially recorded at their fair value and are then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For the 2012 Revolver Notes and warrants issued under the note and warrant purchase agreement dated January 13, 2012, the Company used the Black-Scholes valuation model. The Company recorded approximately $396,000 related to the fair value of the warrants issued, as a discount to the carrying value of the debt, accreted as interest expense over the life of the debt. The Company valued the warrants at the date of each issuance using the Black-Scholes valuation model with the following underlying assumptions: contractual term of 5 years, an underlying preferred share price between $0.24 and $0.30, an exercise price of $0.54, an average risk-free interest rate between 0.62% and 1.02%, a dividend yield of 0%, and volatility of 105.0%. Approximately $396,000 related to accretion of the discount was recognized as interest expense during the year ended December 31, 2012. The discount was fully accreted as of December 31, 2012.
As of December 31, 2012, the Company had issued $5,960,000 in notes payable under the 2012 Revolver Notes agreement. The Company was in default for payment of these notes as of December 31, 2012, and no principal payments were made in 2013 prior to conversion. As of June 28, 2013 the investors under the 2012 Revolver Notes elected to convert the total principal balance of approximately $5,960,000 owed under the 2012 Revolver Notes and accrued interest of approximately $645,000 into 12,230,899 preferred shares at a conversion price of $0.54 per share, pursuant to note conversion agreements of that date. Although the conversion price of the debt was greater than the value of the preferred shares at the time of conversion, the Company did not record a gain on the conversion under the troubled debt restructuring accounting guidance since the transaction occurred between related parties, and thus, was treated as a capital transaction. On September 13, 2013, the exercise price of the warrants was fixed at $0.54 per share, and the fair value of the warrant liability of approximately $144,000 on that date was reclassified to additional paid-in capital.
Other
On September 10, 2012, the Company issued a warrant to its landlord in exchange for a rent deferral through November 30, 2012. The number of Series A preferred shares exercisable under the warrant agreement is determined by dividing the warrant coverage amount of $40,000 by the exercise price. The exercise price of the warrants is $0.60, or, upon the closing of the sale by the Company of its preferred stock in which the Company receives an aggregate of at least $15,000,000 in cumulative gross proceeds, the warrant’s exercise price will be the price per share for which the Company sells its preferred shares in such sale. The term of the warrant is seven years. Early termination of the warrant can occur if the Company is acquired. The holder of the warrant is to be given 20 days advance notice of such an event, and the warrant will terminate if not exercised before the date of the event. The fair value of the preferred warrant due to the landlord at December 31, 2012 and 2013 is not material to the financial statements.
As of December 31, 2012, warrants to purchase preferred stock are reflected as a liability on the balance sheet, which is adjusted to estimated fair value at the end of each reporting period over the term of the warrants. These warrants were reclassified to additional paid-in capital during the year ended December 31, 2013. The fair value of the warrant liability for warrants to purchase preferred stock as of December 31, 2012 of approximately $982,000 was estimated using the Black-Scholes valuation model with the following assumptions: contractual term between 3.08 and 4.92 years, an underlying preferred share price of $0.25, an exercise price of $0.54, an average risk-free interest rate between 0.35% and 0.70%, a dividend yield of 0%, and volatility of 105.0%.
99
Table of Contents
Outstanding Warrants—Common Shares
2013 Convertible Bridge Notes
The Company executed a convertible note and warrant purchase agreement as of June 28, 2013 with several shareholders, including a major shareholder, relating to the Company’s borrowing as needed of, and issuance of a series of unsecured convertible notes for, up to $7,000,000. The Company had borrowed $745,000 and $4,990,000 as of December 31, 2012 and 2013, respectively, against the 2013 Convertible Bridge Notes, including $720,000 and $2,505,000, respectively, from a major shareholder. The maturity date of the 2013 Convertible Bridge Notes is May 31, 2014 and may be extended at the option of the respective note holders for two successive six month periods. The 2013 Convertible Bridge Notes bear interest at 8.0% per annum, payable at maturity.
The 2013 Convertible Bridge Notes automatically convert into the Company’s common stock upon the closing of an IPO of at least $8,000,000 in cumulative gross proceeds, at a price equal to the price per share of the Company’s common stock sold in the IPO. The number of common shares for which the warrants are exercisable is determined by dividing the warrant coverage amount, which is 50% of the principal amount of the notes issued under the agreement, by the exercise price, which is the price per share of the Company’s common stock sold in the IPO. The warrants will be exercisable for a five-year period beginning with the closing of the Company’s IPO. Early termination of the warrants can occur upon any capital reorganization, any reclassification of the capital stock, or an asset transfer or acquisition of the Company. The holders of the warrants are to be given 20 days advance notice of such an event, and the warrants will terminate if not exercised prior to the date of the event.
In accordance with guidance applicable to accounting for derivative financial instruments that are accounted for as liabilities, the warrants are initially recorded at their fair value and are then re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For the warrants for common shares issued under the 2013 Convertible Bridge Notes agreement, the Company used a probability weighted Black-Scholes valuation model. The Company recorded approximately $1,559,000 related to the fair value of the warrants issued, as a discount to the carrying value of the debt, accreted to interest expense using the effective interest method from the date of issuance over the life of the debt. These warrants to purchase common stock were valued as of their date of issuance, using the following assumptions: exercise price of between $1.48 and $14.28 per share, contractual term of 5 years, a risk-free interest rate between 1.38% and 1.73%, a dividend yield of 0%, and volatility between 100.0%—105.0%. The value of the warrants using the probability weighted Black-Scholes valuation model accounted for a probability between 75% and 80%, while a fair value of $0 was weighted between 20% and 25%. The fair value of the warrants is recorded as a liability of approximately $1,399,000 at December 31, 2013. Approximately $685,000 related to accretion of the discount was recognized as interest expense during the year ended December 31, 2013, with approximately $874,000 remaining unamortized and reflected as a discount to the debt.
Line of Credit
Three of the Company’s related parties guaranteed the Company’s Line of Credit (see Note 5) and pledged financial assets to the bank to secure their guaranties, as approved by the Company’s board of directors. In return, the Company issued common stock warrants to the guarantors. The fair market value of the collateral provided by the respective guarantors at December 31, 2013 was $2,178,000. The number of shares subject to the common stock warrants will be determined by dividing the warrant coverage amount, which is 50% of the fair market value of the collateral provided by the respective guarantors to secure their respective guaranty obligations to the bank, by the exercise price, which will be set at the price per share of the Company’s common stock sold in its IPO. The warrants will be exercisable for a two-year period beginning with the closing of the Company’s IPO.
In accordance with guidance applicable to accounting for derivative financial instruments that are accounted for as liabilities, the warrants are initially recorded at their fair value and are then re-valued at each reporting date,
100
Table of Contents
with changes in the fair value reported in the statements of operations. For the warrants for common shares issued in connection with the Company’s Line of Credit, the Company used a probability weighted Black-Scholes valuation model. The Company recorded approximately $454,000 related to the fair value of the warrants issued, as a discount to the carrying value of the debt, accreted to interest expense on a straight line basis from the date of issuance over the life of the debt. These warrants to purchase common stock were valued as of their date of issuance, using the following assumptions: exercise price between $1.48 and $14.28 per share, contractual term of 2 years, a risk-free interest rate between 0.38% and 1.38%, a dividend yield of 0%, and volatility between 90.0% and 105.0%. The value of the warrants using the probability weighted Black-Scholes valuation model accounted for a probability of 75%, while a fair value of $0 was weighted 25%. The fair value of the warrants is recorded as a liability of approximately $390,000 at December 31, 2013. Approximately $139,000 related to accretion of the discount was recognized as interest expense during the year ended December 31, 2013, with approximately $315,000 remaining unamortized and reflected as a discount to outstanding debt.
Other
On September 10, 2013, the Company, as part of a lease amendment for its non-cancellable operating lease for its office, laboratory, and warehouse space at its San Diego, California facility, issued a warrant to its landlord. The warrant coverage amount was $502,605. The number of shares subject to the common stock warrants will be determined by dividing the warrant coverage amount by the exercise price, which will be set at the price per share of the Company’s common stock sold in its IPO. The warrants will be exercisable for a five-year period beginning with the closing of the Company’s IPO.
In accordance with guidance applicable to accounting for derivative financial instruments that are accounted for as liabilities, the warrant is initially recorded at fair value and then is re-valued at each reporting date, with changes in the fair value reported in the statements of operations. For the warrant for common shares issued to the landlord, the Company used a probability weighted Black-Scholes valuation model. The Company recorded approximately $309,000 related to the fair value of the warrant issued at issuance in September 2013, as a reduction in deferred rent liability, accreted to rent expense on a straight line basis from the date of issuance over the term of the amended lease. The warrant was valued as of the date of issuance, using the following assumptions: exercise price of between $3.08 and $14.28 per share, contractual term of 5 years, a risk-free interest rate of 1.38%, a dividend yield of 0%, and volatility of 105.0%. The value of the warrant using the probability weighted Black-Scholes valuation model accounted for a probability of 75%, while a fair value of $0 was weighted 25%. The fair value of the warrant is recorded as a liability of approximately $282,000 at December 31, 2013.
As of December 31, 2013, warrants to purchase common stock are reflected as a liability on the balance sheet, which is adjusted to estimated fair value at the end of each reporting period over the term of the warrants. The aggregate fair value of the warrant liability for warrants to purchase common stock as of December 31, 2013 of approximately $2,132,000 was estimated using a probability weighted Black-Scholes valuation model with the following assumptions for both the five-year and two-year common stock warrant terms separately:
| Five-year term | Two-year term | |||||||
| Stock price |
$ | 1.48 – 7.69 | $ | 1.48 – 7.69 | ||||
| Exercise price |
$ | 1.48 – 7.69 | $ | 1.48 – 7.69 | ||||
| Expected dividend yield |
0 | % | 0 | % | ||||
| Discount rate-bond equivalent yield |
1.73 | % | 0.38 | % | ||||
| Expected life (in years) |
5.0 | 2.0 | ||||||
| Expected volatility |
100.0 | % | 90.0 | % | ||||
At December 31, 2013 the values of both the five-year and two-year common stock warrants using the probability weighted Black-Scholes valuation models accounted for a probability of 75%, while a fair value of $0 was weighted 25%.
101
Table of Contents
Change in estimated fair value of warrant liability
The change in the estimated fair value of the total liability outstanding for all outstanding warrants of approximately $454,000 and $782,000 was recognized as a non-cash gain and included in total other income/(expense) in the Company’s statements of operations and comprehensive loss for the years ended December 31, 2012 and 2013, respectively.
8. Supplier Financing
In 2011, the Company purchased certain laboratory equipment under financing agreements with a supplier, a business owned by a member of the Company’s board of directors, totaling approximately $256,000. Financing was granted for the purchase of the equipment at a stated interest rate of 0.0%. The Company has utilized its average interest rate for 2012 and 2013 of 8.0% to amortize the payments and record interest expense of approximately $17,000 and $5,000 for the years ended December 31, 2012 and 2013, respectively, utilizing the effective interest expense method. The remaining balance owed under these financing agreements was approximately $60,000 and $66,000 as of December 31, 2012 and 2013, respectively. The remaining balance owed under these financing agreements was due in 2013 (see Note 18).
In 2011, the Company purchased laboratory software under a financing agreement with a supplier for approximately $177,000. This software financing agreement bears an interest rate of 7.4% per annum. The balance owed under these financing agreements was approximately $62,000 at both December 31, 2012 and 2013.
In 2012 and 2013, the Company obtained third-party financing for certain business insurance premiums. The financing bears an interest rate of 5.95% per annum, and all financing is due within one year. The balances due under these annual financing arrangements were approximately $129,000 and $91,000 as of December 31, 2012 and 2013, respectively.
9. Shareholders’ Deficit
(a) Common Stock
In November of 2011, the Company amended and restated its articles of incorporation to decrease the number of authorized shares of common stock from 44,260,000 to 14,600,000. The authorized number shares of common stock at December 31, 2011 and 2012 was 14,600,000. In conjunction with the November 2011 amendment, the Company declared a 1:3 reverse stock split for all common shares. On November 1, 2013, the Company effected a 1:14 reverse stock split for all common shares. All references to share and per share amounts in the financial statements and accompanying notes to the financial statements have been retroactively restated to reflect the 1:14 reverse stock split.
On July 22, 2013, the Company amended its articles of incorporation to increase the number of authorized shares of common stock from 14,600,000 to 53,000,000. The authorized number of shares of common stock at December 31, 2013 was 53,000,000. In addition, on July 30, 2013, the Company assigned a par value to its common shares of $0.0001 in conjunction with its reincorporation in Delaware. The new par value per common share has been retroactively reflected in the financial statements for all periods presented.
(b) Preferred Stock
In November of 2011, the Company amended and restated its articles of incorporation so that each share of the issued and outstanding Series AA preferred stock and each share of the issued and outstanding Series BB preferred stock of the Company was converted into one share of Series A preferred stock. As of December 31, 2011 and 2012, all 36,460,000 authorized shares of preferred stock are designated as Series A preferred stock. On July 22, 2013, the Company amended its articles of incorporation to increase the number of authorized preferred shares from 14,600,000 to 100,000,000. In addition, on July 30, 2013, the Company assigned a par value to its preferred shares of $0.0001 in conjunction with its reincorporation in Delaware. The new par value per preferred share has been retroactively reflected in the financial statements for all periods presented.
102
Table of Contents
Holders of the Company’s preferred shares are entitled to receive, when and as declared by the board of directors and in preference to common shareholders, non-cumulative cash dividends at the rate of 8% per annum of the applicable original issue price on each outstanding preferred share. The original issue price of each share of Series A preferred stock was $0.60. No dividends were declared during 2011 or 2012 or in the first nine months of 2013. Dividends cannot be granted for common shareholders while shares of preferred stock remain outstanding.
The holders of preferred shares have the right to one vote for each common share into which the preferred shares are convertible. Upon the liquidation, dissolution, or winding up of the Company, either voluntary or involuntary, the preferred shareholders will be paid out an amount equal to the original issue price plus all declared and unpaid dividends. If, upon any liquidation, distribution, or winding up of the Company, and the assets of the Company are insufficient to make payment in full to all holders of preferred shares of the liquidation preference, then such assets shall be distributed among the holders of preferred shares ratably in proportion to the full amounts to which they would be entitled.
The convertible preferred shares may be converted into common shares at any time at the option of the holder utilizing the then effective Series A preferred conversion price. All preferred shares shall be automatically converted into common shares utilizing the then effective Series A preferred conversion price upon a) the election of the holders of a majority of the outstanding shares of Series A preferred stock, or b) the closing of a firmly underwritten public offering pursuant to an effective registration statement under the Securities Act of 1933 covering the sale of the Company’s common stock if gross proceeds are at least $20,000,000 and the per share price is at least $25.20.
The effective conversion price is equal to the original issue price divided by $25.20 as may be adjusted for dilutive issuances of common shares, common share rights or options, common share splits and combinations, dividends, and distributions. The effective conversion rate is not adjusted for issuances of common share options, warrants or rights to employees, directors, or non-employee service providers.
During the year ended December 31, 2013, 42,245,834 shares of Series A preferred stock were issued for the conversion of approximately $20,231,000 of debt and $2,581,000 of accrued interest, primarily to related parties (see Notes 6 and 7).
10. Accounting for Stock-Based Compensation Expense
2007 Equity Incentive Plan
The 2007 Equity Incentive Plan (“2007 Plan”) authorizes the grant of the following types of awards: (i) nonstatutory stock options, or NSOs, (ii) incentive stock options, or ISOs, (iii) restricted stock awards, (iv) restricted stock unit awards, or RSUs, (v) stock appreciation rights, or SARs, (vi) performance awards, and (vii) other stock awards. Awards may be granted to employees, officers, non-employee board members, consultants, and other service providers of the Company. However, ISOs may not be granted to non-employees. Prior to November 2011, the Company was authorized to issue 7,500,000 options under the 2007 Plan. In conjunction with the 1:3 reverse common stock split in November 2011, the number of shares authorized under the 2007 Plan decreased to 2,500,000 shares and further reduced to 178,571 shares as a result of the 1:14 reverse split in November 2013. As of December 31, 2012 and 2013, shares available for grant under the 2007 Plan were 50,127 and 77,061, respectively.
2013 Equity Incentive Plan
In July 2013, the Company adopted a new stock-based compensation plan entitled the 2013 Equity Incentive Plan (“2013 Plan”). The 2013 Plan authorizes the grant of the following types of awards: (i) nonstatutory stock options, (ii) ISOs, (iii) restricted stock awards, (iv) restricted stock unit awards, (v) stock appreciation rights, and (vi) performance compensation awards. Awards may be granted to employees, officers, non-employee board members, consultants, and other service providers of the Company. However, ISOs may not be granted to non-employees. The Company has authorized a total of 403,571 shares of common stock for issuance pursuant to all
103
Table of Contents
awards granted under the 2013 Equity Incentive Plan, subject to an increase of 800,000 shares upon the completion of an IPO, and subject to additional increases every January 1 equal to the lesser of (i) 5% of the Company’s outstanding common stock on such January 1, or (ii) a number of shares determined by the Company’s board of directors in its discretion for use on such particular January 1. As of December 31, 2013, 401,640 stock options and RSUs have been granted under the 2013 Plan, and 1,931 shares are available for grant under the 2013 Plan. On February 10, 2014, in connection with the closing of the Company’s IPO, the number of shares of common stock covered by the 2013 Plan increased by 800,000. Additionally, 335,798 options were granted under the 2013 Plan in February and March 2014 (see Note 18).
Options granted under either plan vest over a maximum period of four years and expire ten years from the date of grant. Options generally vest either (i) over four years, 25% on the one year anniversary of the date of grant and monthly thereafter for the remaining three years; or (ii) over four years, monthly vesting beginning month-one after the grant and monthly thereafter. Certain options have been granted which vest 50% on the grant date and monthly thereafter for the remaining two years.
The fair value of stock options is determined on the date of grant using the Black-Scholes valuation model. Such value is recognized as expense over the requisite service period, net of estimated forfeitures, using the straight-line method. The determination of the fair value of stock options is affected by the Company’s stock price, as well as assumptions regarding a number of complex and subjective variables. The volatility assumption is based on a combination of the historical volatility of the Company’s common stock and the volatilities of similar companies over a period of time equal to the expected term of the stock options. The volatilities of similar companies are used in conjunction with the Company’s historical volatility because of the lack of sufficient relevant history for the Company’s common stock equal to the expected term. The expected term of employee stock options represents the weighted-average period the stock options are expected to remain outstanding. The expected term assumption is estimated based primarily on the options’ vesting terms and remaining contractual life and employees’ expected exercise and post-vesting employment termination behavior. The risk-free interest rate assumption is based upon observed interest rates on the grant date appropriate for the term of the employee stock options. The dividend yield assumption is based on the expectation of no future dividend payouts by the Company.
The assumptions used in the Black-Scholes pricing model for options granted during the years ended December 31, 2012 and 2013 are as follows:
| For the years ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Volatility |
96.8 | % | 105.0 | % | ||||
| Risk-free interest rate |
0.79% – 1.15 | % | 1.38 – 1.69 | % | ||||
| Dividend yield |
0.00 | % | 0.00 | % | ||||
| Expected term (years) |
6.08 | 5.26 – 6.02 | ||||||
| Expected forfeiture rate |
0.00 | % | 0.00 – 5.00 | % | ||||
Using the assumptions described above, the weighted-average estimated fair value of options granted in 2012 and 2013 were approximately $1.82 and $4.43, respectively.
104
Table of Contents
A summary of stock option activity for 2012 and 2013 is as follows:
| Number of Shares |
Weighted Average Exercise Price Per Share |
Average Remaining Contractual Term in Years |
||||||||||
| Outstanding at December 31, 2011 |
78,987 | $ | 4.90 | 8.3 | ||||||||
| Granted |
330 | 4.62 | ||||||||||
| Exercised |
— | — | ||||||||||
| Cancelled/forfeited/expired |
(15,799 | ) | 4.92 | |||||||||
|
|
|
|||||||||||
| Outstanding at December 31, 2012 |
63,518 | $ | 4.97 | 6.2 | ||||||||
|
|
|
|||||||||||
| Granted |
300,438 | 5.18 | ||||||||||
| Exercised |
(4,021 | ) | 5.00 | |||||||||
| Cancelled/forfeited/expired |
(26,829 | ) | 5.20 | |||||||||
|
|
|
|||||||||||
| Outstanding at December 31, 2013 |
333,106 | $ | 5.14 | 9.3 | ||||||||
|
|
|
|||||||||||
| Vested and unvested expected to vest, December 31, 2013 |
331,540 | $ | 5.14 | 9.3 | ||||||||
|
|
|
|||||||||||
The intrinsic value of options exercised during the year ended December 31, 2013 was $3,450. The intrinsic value of options outstanding and vested and unvested expected to vest at December 31, 2013 was $8,204 and $8,192, respectively.
The Company received $20,105 in proceeds from stock options exercised during the year ended December 31, 2013. The tax benefit related to stock options exercised during the year ended December 31, 2013 was not significant.
Further information about the options outstanding and exercisable at December 31, 2012 and 2013 is as follows:
| Options Outstanding and Exercisable at December 31, 2012 |
||||||||||||
| Weighted Average Exercise Price |
Total Shares Outstanding |
Weighted Average Contractual Life (in years) |
Total Shares Exercisable |
|||||||||
| $4.62 |
33,031 | 7.2 | 16,173 | |||||||||
| $5.04 |
30,408 | 5.1 | 28,244 | |||||||||
| $125.58 |
79 | 1.1 | 79 | |||||||||
|
|
|
|
|
|||||||||
| 63,518 | 44,496 | |||||||||||
|
|
|
|
|
|||||||||
| Options Outstanding and Exercisable at December 31, 2013 |
||||||||||||
| Weighted |
Total Shares Outstanding |
Weighted Average Contractual Life (in years) |
Total Shares Exercisable |
|||||||||
| $4.62 |
20,208 | 7.3 | 13,731 | |||||||||
| $5.04 |
12,460 | 5.5 | 12,455 | |||||||||
| $5.18 |
300,438 | 9.6 | 110,825 | |||||||||
|
|
|
|
|
|||||||||
| 333,106 | 137,011 | |||||||||||
|
|
|
|
|
|||||||||
The intrinsic value of options exercisable at December 31, 2013 was $5,575.
Restricted Stock Units (“RSUs”)
In November 2010, the Company issued to a member of the board of directors a restricted stock unit award for 390,000 shares of Series BB preferred stock. In November 2011, these RSUs were modified to be redeemable for Series A preferred stock under the same terms and conditions of the original grant. The shares will not vest
105
Table of Contents
unless a change in control, as defined, or IPO occurs within 10 years of the vesting commencement date of October 2010. There will be no expense to record for these awards unless and until it becomes probable that the award will vest. As of December 31, 2012 and 2013, it was not probable that these awards will vest and therefore, no expense was recorded during the years ended December 31, 2012 or 2013.
In March 2011, the Company awarded a restricted stock unit award to a member of the board of directors for 428,597 shares of Series BB preferred stock. Also in March 2011, the Company awarded an additional performance-based restricted stock unit award for an estimated 574,108 shares of Series BB preferred stock to the same member. In November 2011, these RSUs were modified to be redeemable for Series A preferred stock under the same terms and conditions of the original grant. The number of shares in the restricted stock units is based on certain milestones to be achieved. None of the shares under either award vest unless a change in control or IPO occurs within 10 years after January 1, 2011. There will be no expense to record for these awards until it becomes probable that an award will vest. As of December 31, 2012 and 2013, it was not probable that these awards will vest and therefore, no expense was recorded during the years ended December 31, 2012 or 2013.
The board of directors approved a resolution in December 2010, that each January 1 each person (other than two identified individuals) who is serving as a non-employee director on such January 1 shall be automatically granted an annual restricted stock unit award covering a number of common shares equal to 0.25% of the fully diluted outstanding common stock of the Company as of the December 31 immediately preceding such January 1. These restricted stock unit awards will be granted automatically on each January 1 and will vest in equal monthly installments over 12 months from the date of the grant. Additionally, in January 2012, each person (other than two identified individuals) who is serving as a non-employee director is to be granted a “true up grant” in addition to the annual grant covering a number of common shares equal to 0.25% of the fully diluted outstanding common shares of the Company as of the immediately preceding December 31. These grants will vest 100% on the date of the grant. In January 2012, five restricted stock unit awards for a total of 20,930 common shares were granted in accordance with this resolution. In addition, on January 1, 2012, an additional five restricted stock unit awards were granted to non-employee directors for a total of 20,930 common shares, vesting immediately upon grant. Although vested, shares are only delivered on the earlier of (i) the date that is 10 years from the grant date, (ii) the date of a change in control, (iii) the date of termination of the holder from the Company, (iv) the date of death or disability, or (v) the date of an unforeseeable emergency as described in Internal Revenue Code section 409A.
The RSU awards due to be granted on January 1, 2013 were not granted during the year ended December 31, 2013. In lieu of this issuance, RSU awards for 8,735 shares of common stock each were granted to three directors and an RSU award for 14,285 shares of common stock was granted to another director, on July 31, 2013. All RSUs issued in July 2013 vest in equal monthly installments over five months beginning August 1, 2013. The shares underlying the 2013 awards will be distributed no later than August 20, 2014.
In August 2013, 60,712 RSU awards were granted to certain executive employees. These awards vest 50% on the date of grant, with the remaining 50% vesting in equal monthly installments over twenty-four months beginning August 31, 2013.
106
Table of Contents
Stock-based Compensation Expense
The following table presents the effects of stock-based compensation related to equity awards to employees and nonemployees on the statement of operations during the periods presented:
| For the years ended December 31, |
||||||||
| 2012 | 2013 | |||||||
| Stock Options |
||||||||
| Research and development expenses |
$ | 32,210 | $ | 298,618 | ||||
| General and administrative expenses |
22,530 | 221,726 | ||||||
| Sales and marketing expenses |
3,994 | — | ||||||
|
|
|
|
|
|||||
| Total expenses related to stock options |
58,734 | 520,344 | ||||||
| RSUs |
||||||||
| Research and development expenses |
— | 72,500 | ||||||
| General and administrative expenses |
193,400 | 359,677 | ||||||
|
|
|
|
|
|||||
| Total stock-based compensation |
$ | 252,134 | $ | 952,521 | ||||
|
|
|
|
|
|||||
As of December 31, 2013, total unrecognized share-based compensation expense related to nonvested stock option and RSU awards, adjusted for estimated forfeitures, was approximately $861,000 and $135,000, respectively, and is expected to be recognized over a weighted-average period of approximately 2.8 years and 1.6 years, respectively.
11. Net Loss per Common Share
Basic and diluted net loss per common share is determined by dividing net loss applicable to common shareholders by the weighted-average common shares outstanding during the period. Because there is a net loss attributable to common shareholders for the years ended December 31, 2012 and 2013, the outstanding shares of Series A preferred stock, RSUs, convertible debt, warrants, and common stock options have been excluded from the calculation of diluted loss per common share because their effect would be anti-dilutive. Therefore, the weighted-average shares used to calculate both basic and diluted loss per share are the same.
In November 2013, the Company effected a 1:14 reverse stock split of all common shares outstanding. The calculation of weighted-average shares outstanding has been adjusted for this reverse split as if it had occurred on January 1, 2012.
The following potentially dilutive securities have been excluded from the computations of diluted weighted-average shares outstanding for the periods presented, as they would be anti-dilutive:
| For the years ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Series A preferred (number of common stock equivalents) |
647,007 | 1,652,851 | ||||||
| Preferred warrants outstanding (number of common stock equivalents) |
192,262 | 192,262 | ||||||
| Notes payable convertible into preferred shares (number of common stock equivalents) |
599,466 | — | ||||||
| Preferred share RSUs (number of common stock equivalents) |
33,158 | 89,647 | ||||||
| Common warrants outstanding |
— | 836,890 | ||||||
| Notes payable convertible into common shares |
665,178 | 1,110,649 | ||||||
| Common share RSUs |
54,615 | 133,971 | ||||||
| Common options outstanding |
63,518 | 333,106 | ||||||
|
|
|
|
|
|||||
| Total anti-dilutive common share equivalents |
2,255,204 | 4,349,376 | ||||||
|
|
|
|
|
|||||
107
Table of Contents
12. 401(k) Plan
The Company sponsors a 401(k) savings plan for all eligible employees. The Company may make discretionary matching contributions to the plan to be allocated to employee accounts based upon employee deferrals and compensation. To date, the Company has not made any matching contributions into the savings plan.
13. Income Taxes
For the year ended December 31, 2012 and 2013, the provision for income taxes was calculated as follows:
| For the years ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Current: |
||||||||
| Federal |
$ | — | $ | — | ||||
| State |
800 | 800 | ||||||
|
|
|
|
|
|||||
| Total |
800 | 800 | ||||||
|
|
|
|
|
|||||
| Deferred |
||||||||
| Federal |
— | — | ||||||
| State |
— | — | ||||||
|
|
|
|
|
|||||
| Total |
— | — | ||||||
|
|
|
|
|
|||||
| Provision for income tax |
$ | 800 | $ | 800 | ||||
|
|
|
|
|
|||||
The following table provides a reconciliation between income taxes computed at the federal statutory rate and the Company’s provision for income taxes:
| For the years ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Income tax at statutory rate |
$ | (4,167,967 | ) | $ | (3,139,368 | ) | ||
| State liability |
(602,296 | ) | (321,058 | ) | ||||
| Permanent items |
3,164 | 6,932 | ||||||
| Stock Compensation |
19,298 | 171,003 | ||||||
| Nondeductible Interest |
521,531 | 395,089 | ||||||
| Expiration of net operating losses |
146,175 | 188,316 | ||||||
| Other |
80 | (6,723 | ) | |||||
| Research and development credit |
(215,502 | ) | (103,500 | ) | ||||
| Valuation allowance |
4,296,317 | 2,810,109 | ||||||
|
|
|
|
|
|||||
| Provision for income tax |
$ | 800 | $ | 800 | ||||
|
|
|
|
|
|||||
Deferred income taxes are provided for temporary differences in recognizing certain income and expense items for financial and tax reporting purposes. The deferred tax assets consisted primarily of the income tax benefits from net operating loss carryforwards, deferred rent, and research and development credits. Valuation allowances have been recorded to fully offset deferred tax assets at December 31, 2012 and 2013, as it is more likely than not that the assets will not be utilized.
At December 31, 2013, the Company has federal net operating loss carryforwards of approximately $111,673,000 expiring beginning in 2020 and California net operating loss carryforwards of approximately $97,656,000 expiring beginning in 2014. Additionally, at December 31, 2013, the Company has research and development credits of approximately $3,132,000 and $3,004,000 for federal and California purposes, respectively. The federal research and development tax credits will begin to expire in 2018. The California research and development tax credits do not expire.
108
Table of Contents
For the years ended December 31, 2012 and 2013, the Company has evaluated the various tax positions reflected in their income tax returns for both federal and state jurisdictions, and all open tax years in these jurisdictions, to determine if the Company has any uncertain tax positions on the historical tax returns. The Company recognizes the impact of an uncertain tax position on an income tax return at the largest amount that the relevant taxing authority is more-likely-than not to sustain upon audit. The Company does not recognize uncertain income tax positions if they have less than 50 percent likelihood of being sustained. Based on this assessment, the Company believes there are no tax positions for which a liability for unrecognized tax benefits should be recorded as of December 31, 2012 or 2013. The Company is subject to taxation in the United States and California. The Company’s federal filings prior to 2010 and the Company’s California filings prior to 2009 are no longer subject to examination. The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. Due to the existence of the valuation allowance, future changes in unrecognized tax benefits will not impact the Company’s effective tax rate. The Company is currently not under examination by any taxing authorities and does not believe its unrecognized tax benefits will significantly change in the next twelve months.
The tax effects of carryforwards that give rise to deferred tax assets consist of the following:
| For the years ended December 31, | ||||||||
| 2012 | 2013 | |||||||
| Net operating loss carryforward |
$ | 41,100,511 | $ | 43,666,636 | ||||
| Research and development credits |
4,898,055 | 5,114,652 | ||||||
| Accruals and other |
688,089 | 742,045 | ||||||
| Deferred rent |
203,463 | 176,893 | ||||||
|
|
|
|
|
|||||
| 46,890,118 | 49,700,226 | |||||||
| Less valuation allowance |
(46,890,118 | ) | (49,700,226 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The Company has not completed a study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since its formation, due to the complexity and cost associated with such a study, and the fact that there may be additional ownership changes in the future. Based on preliminary assessments, the Company believes that ownership changes occurred in 2010 and 2013. The Company estimates that if such a change did occur, the federal and state net operating loss carryforwards and research and development credits that can be utilized in the future will be significantly limited.
On September 13, 2013, the U.S. Treasury Department released final income tax regulations on the deduction and capitalization of expenditures related to tangible property. These final regulations apply to tax years beginning on or after January 1, 2014, and may be adopted in earlier years. The Company does not intend to early adopt the tax treatment of expenditures to improve tangible property and the capitalization of inherently facilitative costs to acquire tangible property as of January 1, 2013. The tangible property regulations will require the Company to make additional tax accounting method changes as of January 1, 2014; however, management does not anticipate the impact of these changes to be material to the Company’s consolidated financial position, its results of operations and its footnote disclosures.
14. Collaborative Agreements
On August 17, 2011, the Company entered into a three year exclusive collaboration agreement with Clarient Diagnostic Services, Inc. to collaborate to promote and maximize the commercialization of the Company’s or jointly developed diagnostic tests (together, the “Diagnostic Tests”) in the United States. Clarient is responsible for marketing, providing customer service, and for third party billing on all Diagnostic Tests performed under the agreement, and for performing the professional component of the Diagnostic Tests. The Company is responsible for promoting sales of the Diagnostic Tests in the United States, as well as performing all technical components of all Diagnostic Tests sold by either party.
109
Table of Contents
Under this agreement, the Company invoices Clarient for the performance of each of the Diagnostic Tests at a contractually agreed-upon rate. Clarient is responsible for billing the patient, provider and/or payer for each completed test, and bears all collection risk related to such billings. Sales of Diagnostic Tests under this agreement did not commence until 2012. The total amount of revenue the Company earned under this agreement was approximately $86,000 and $14,000 for the years ended December 31, 2012 and 2013, respectively.
The agreement was replaced as of May 2013 to remove exclusivity provisions and to modify the performance obligations of the parties. As a result of the replacement agreement, the Company will be responsible for billing third party payors for tests performed under the Clarient agreement. Revenue derived from the Clarient arrangement after the replacement date is recognized as collected, provided all other revenue recognition criteria are met.
In January 2013, the Company entered into a research support agreement with Dana Farber, a not-for-profit tax-exempt organization. The Company is responsible for performing all technical components of the diagnostic tests as ordered by Dana Farber and recognizes revenue as collected, provided all other revenue recognition criteria are met. The total amount of revenue the Company earned under this agreement was approximately $104,000 for the year ended December 31, 2013.
15. Related Party Transactions
During 2005, the Company executed the Goodman Note in favor of an investor which became a beneficial owner of more than 5% of the Company’s common stock. As of December 31, 2012, the Company had $1,935,000 outstanding on this note. In June 2013, the investor converted the entire principal amount of $1,935,000 and accrued interest of approximately $105,000 due on the Goodman Note into 3,777,324 shares of Series A preferred stock.
During 2008, the Company executed the 2008 Convertible Note with an affiliate of a major shareholder who is a member of the board of directors in the amount of $1,400,000. A warrant to purchase preferred shares was issued along with the convertible promissory note (see Notes 6 and 7). In July 2013, the Company amended the 2008 Convertible Note with a principal balance of $1,400,000, held by a related party, to provide that all principal of and accrued interest on the note would automatically convert into common stock upon the closing of an IPO at the price per share at which common stock is sold in such IPO.
As of December 31, 2012 and 2013, the Company had $17,780,000 and $3,905,000, respectively, of notes payable outstanding to affiliates of a major shareholder who is a member of the board of directors under several note and warrant purchase agreements (see Notes 6 and 7). As of June 28, 2013, $17,060,000 of principal and $2,339,000 of interest due on a portion of these notes was converted into shares of 35,923,845 Series A preferred stock.
As of December 31, 2012 and 2013, the Company had approximately $1,000,000 and $1,479,000, respectively, of notes payable outstanding with other board members under several different note and warrant purchase agreements (see Notes 6 and 7). As of June 28, 2013, approximately $975,000 of principal and $101,000 of interest due on a portion of these notes were converted into 1,993,591 preferred shares.
In September and December 2013, the Company issued common stock warrants to three shareholders in conjunction with their guarantees on the Company’s borrowings under the Company’s line of credit (see Notes 5 and 7).
During 2011, the Company entered into two supplier financing arrangements with a business owned by a member of the board of directors totaling $256,000, of which $60,000 and $66,000 is outstanding as of December 31, 2012 and 2013, respectively (see Note 18).
110
Table of Contents
A member of the Company’s management is the controlling person of Aegea Biotechnologies, Inc. (“Aegea”). On September 2, 2012, the Company entered into an Assignment and Exclusive Cross-License Agreement with Aegea Biotechnologies, Inc. The total amount of invoices received by the Company from Aegea during the year ended December 31, 2013 was approximately $2,000, which are unpaid and recorded in accounts payable at December 31, 2013.
The Company believes that these transactions were on terms at least as favorable to the Company as could have been obtained from unrelated third parties.
16. Commitments and Contingencies
Operating Leases
The Company leases office, laboratory, and warehouse space at its San Diego, California facility under a non-cancelable operating lease. The initial lease was for an eight-year term expiring in 2012. In November 2011, the Company extended the lease term through October 31, 2018 and expanded the original premises by 9,849 square feet. Under the amended lease, the landlord delivered the expanded premises in May 2013. The Company records rent expense on a straight-line basis over the life of the lease and records the excess of expense over the amounts paid as deferred rent.
For the years ended December 31, 2012 and 2013, rent expense was approximately $937,000 and $1,143,000, respectively. As of December 31, 2012 the Company owed rent in arrears of approximately $185,000. As of December 31, 2013, the Company owed no rent in arrears. This amount is included in accounts payable on the balance sheet.
In September 2013, the Company amended its non-cancellable operating lease for its office, laboratory, and warehouse space at its San Diego, California facility. The amendment extends the maturity date of the lease through July 31, 2020. As part of this amendment, the landlord waived the lease payments due from August 1, 2013 through December 31, 2013 of approximately $503,000, and the Company forfeited its long-term deposit of approximately $269,000. In conjunction with this amendment, the Company granted to the landlord a warrant to purchase common shares with a warrant coverage amount of $502,605 and an exercise price equal to the price per share of the Company’s common stock sold in the Company’s IPO (see Note 7).
The future minimum lease payments under the amended lease agreement as December 31, 2013 are as follows:
| 2014 |
$ | 1,233,846 | ||
| 2015 |
1,270,861 | |||
| 2016 |
1,308,987 | |||
| 2017 |
1,348,257 | |||
| 2018 |
1,388,705 | |||
| Thereafter |
2,285,501 | |||
|
|
|
|||
| Total |
$ | 8,836,157 | ||
|
|
|
Employment Agreements
Under the terms of certain employment agreements with executive officers, the Company would incur additional cash compensation expense of $150,000 immediately, and $225,000 annually, upon the closing of an IPO or the Company’s receipt of aggregate proceeds of $15,000,000 or more from the sales of equity securities. All payments required under these agreements as a result of the closing of the Company’s IPO on February 10, 2014 have been subsequently made in February and March 2014.
111
Table of Contents
Legal Proceedings
In the normal course of business, the Company may be involved in legal proceedings or threatened legal proceedings. The Company is not party to any legal proceedings or aware of any threatened legal proceedings which are expected to have a material adverse effect on its financial condition, results of operations or liquidity.
The Company’s former Vice President of Operations filed an administrative proceeding against the Company with the California Labor Commissioner in April 2013, seeking damages for alleged unpaid wages and penalties. A hearing was held on August 19, 2013 which resulted in a finding against the Company for approximately $65,000, of which $40,000 was paid during the year ended December 31, 2013 and $25,000 was accrued as of December 31, 2013 (see Note 18).
17. Selected Quarterly Financial Data (Unaudited)
The following is selected quarterly financial data as of and for the periods ending:
| First Quarter | Second Quarter | Third Quarter | Fourth Quarter | |||||||||||||
| December 31, 2012 |
||||||||||||||||
| Balance sheet data: |
||||||||||||||||
| Cash & cash equivalents |
$ | 82,486 | $ | 361,062 | $ | 72,483 | $ | 185,256 | ||||||||
| Total assets |
1,544,311 | 1,870,220 | 1,265,874 | 1,469,679 | ||||||||||||
| Total non-current liabilities |
2,034,960 | 1,918,018 | 1,816,319 | 1,255,771 | ||||||||||||
| Total shareholders’ deficit |
(18,973,870 | ) | (21,646,012 | ) | (24,477,238 | ) | (27,384,895 | ) | ||||||||
| Statement of operations and comprehensive loss data: |
||||||||||||||||
| Revenues |
$ | 10,373 | $ | 54,020 | $ | 23,949 | $ | 20,947 | ||||||||
| Gross profit/(loss) |
(145,056 | ) | (255,677 | ) | (267,449 | ) | (424,223 | ) | ||||||||
| Research and development expenses |
2,253,303 | 1,544,206 | 1,506,935 | 1,257,708 | ||||||||||||
| General and administrative expenses |
623,018 | 542,116 | 447,586 | 450,479 | ||||||||||||
| Sales and marketing expenses |
212,447 | 189,360 | 201,739 | 181,773 | ||||||||||||
| Loss from operations |
(3,233,824 | ) | (2,531,359 | ) | (2,423,709 | ) | (2,314,183 | ) | ||||||||
| Net loss |
$ | (3,717,239 | ) | $ | (2,725,279 | ) | $ | (2,869,883 | ) | $ | (2,947,125 | ) | ||||
| Net loss per common share:1 |
||||||||||||||||
| Basic |
$ | (23.18 | ) | $ | (16.99 | ) | $ | (17.89 | ) | $ | (18.37 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted |
$ | (23.18 | ) | $ | (16.99 | ) | $ | (17.89 | ) | $ | (18.37 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted-average shares outstanding used in computing net loss per share attributable to common shareholders: |
||||||||||||||||
| Basic |
160,393 | 160,393 | 160,393 | 160,393 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted |
160,393 | 160,393 | 160,393 | 160,393 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| 1 | Basic and diluted net loss per common share are computed independently for each of the components and quarters presented. Therefore, the sum of quarterly basic and diluted per share information may not equal annual basic and diluted net loss per common share. |
112
Table of Contents
| First Quarter | Second Quarter | Third Quarter | Fourth Quarter | |||||||||||||
| December 31, 2013 |
||||||||||||||||
| Balance sheet data: |
||||||||||||||||
| Cash & cash equivalents |
$ | 17,964 | $ | 4,483 | $ | 302,908 | $ | 69,178 | ||||||||
| Total assets |
1,095,023 | 991,576 | 1,083,089 | 1,329,719 | ||||||||||||
| Total non-current liabilities |
1,252,921 | 508,527 | 167,291 | 462,001 | ||||||||||||
| Total shareholders’ deficit |
(29,300,361 | ) | (8,215,261 | ) | (10,272,840 | ) | (12,456,014 | ) | ||||||||
| Statement of operations and comprehensive loss data: |
||||||||||||||||
| Revenues |
$ | 35,154 | $ | 48,369 | $ | 31,922 | $ | 18,800 | ||||||||
| Gross profit/(loss) |
(512,097 | ) | (544,868 | ) | (587,158 | ) | (551,532 | ) | ||||||||
| Research and development expenses |
710,206 | 690,582 | 975,104 | 710,845 | ||||||||||||
| General and administrative expenses |
451,157 | 478,163 | 806,872 | 776,944 | ||||||||||||
| Sales and marketing expenses |
96,404 | 27,932 | 5,342 | 19,225 | ||||||||||||
| Loss from operations |
(1,769,864 | ) | (1,741,545 | ) | (2,374,476 | ) | (2,058,546 | ) | ||||||||
| Net loss |
$ | (1,925,974 | ) | $ | (1,975,009 | ) | $ | (2,860,191 | ) | $ | (2,472,009 | ) | ||||
| Net loss per common share:1 |
||||||||||||||||
| Basic |
$ | (10.67 | ) | $ | (10.83 | ) | $ | (15.72 | ) | $ | (13.57 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted |
$ | (10.67 | ) | $ | (10.83 | ) | $ | (15.72 | ) | $ | (13.57 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted-average shares outstanding used in computing net loss per share attributable to common shareholders: |
||||||||||||||||
| Basic |
180,540 | 182,304 | 181,954 | 182,203 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Diluted |
180,540 | 182,304 | 181,954 | 182,203 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| 1 | Basic and diluted net loss per common share are computed independently for each of the components and quarters presented. Therefore, the sum of quarterly basic and diluted per share information may not equal annual basic and diluted net loss per common share. |
18. Subsequent Events
The Company has evaluated all events or transactions that occurred after the balance sheet dates of December 31, 2013 and 2012, through March 28, 2014.
Subsequent to December 31, 2013, the Company has borrowed approximately $175,000 under the 2013 Convertible Bridge Notes.
Subsequent to December 31, 2013, the Company repaid in full the remaining amounts outstanding of approximately $70,000 due for laboratory equipment under financing agreements with a supplier, which is a business owned by a member of the Company’s board of directors.
Subsequent to December 31, 2013, the maximum amount of the Company’s line of credit discussed in Note 5 above was increased to approximately $2.6 million and common stock warrants were issued to four shareholders in conjunction with their guarantees on the Company’s additional borrowings under the line of credit. On February 10, 2014, the current outstanding balance under the line of credit of $2,346,000 plus accrued interest of $27,043 was paid in full.
Pursuant to an underwriting agreement dated February 4, 2014 between the Company and Aegis Capital Corp. (“Aegis”), as representative of the several underwriters named therein, an IPO of 1,900,000 shares of common stock at $10.00 per share was effected on February 5, 2014. The closing of the sale of these shares to the
113
Table of Contents
underwriters occurred on February 10, 2014. The Company received, after deducting underwriting discounts and additional costs incurred, approximately $16.7 million from the sale of these 1,900,000 shares. The underwriters had the option, through March 21, 2014, to purchase up to 285,000 shares of common stock at $9.30 per share to cover overallotments, which was not exercised. In addition, designees of Aegis were issued warrants to buy (in the aggregate) up to 95,000 shares of common stock at $12.50 per share.
On February 4, 2014, as contemplated by the registration statement covering the IPO, 69,421,047 shares of outstanding Series A Preferred Stock were converted into 1,652,851 shares of common stock and the Company’s certificate of incorporation was amended to provide for an authorized capitalization of 40,000,000 shares of common stock and 5,000,000 shares of preferred stock.
In connection with the closing of the Company’s IPO on February 10, 2014, (i) the $1,400,000 principal amount and $233,982 of accrued interest related to the 2008 Convertible Note were converted at $10.00 per share into a total of 163,399 shares of common stock, (ii) the $5,165,000 principal amount and $313,017 of accrued interest related to the 2013 Convertible Bridge Notes were converted at $10.00 per share into a total of 547,794 shares of common stock, (iii) the exercise price of the warrants associated with the 2013 Bridge Notes was fixed at $10.00 per share for an aggregate of 258,249 shares of common stock, (iv) the exercise price of the warrants associated with the $2,578,104 of collateral provided to secure the Company’s Line of Credit was fixed at $10.00 per share for an aggregate of 128,903 shares of common stock, (v) 73,151 shares of common stock vested as settlement of certain restricted stock units (which were previously expressed in shares of preferred stock) and became issuable subsequent to the expiration of the 180 day lock-up period, (vi) the Company’s Executive Chairman ceased to be an employee and continues to serve as (non-executive) Chairman, (vii) the number of shares of common stock covered by the 2013 Equity Incentive Plan increased by 800,000, (viii) the preferred warrants previously outstanding were canceled due to early termination clauses associated with the IPO, and (ix) the exercise prices of common warrants previously outstanding were fixed, whereby the carrying amount of the associated liability was adjusted to fair value and then reclassified to shareholders’ deficit.
On February 13, 2014, the Compensation Committee of the Company’s Board of Directors approved the payment of $1,009,552 in deferred salary obligations, including contractual interest, to current and former named executive officers pursuant to previously existing agreements. An additional $172,089 in deferred salary obligations and interest thereon was paid to former employees other than named executive officers. Also on February 13, 2014, the Company’s Board of Directors approved annual cash retainers to non-employee directors, and granted 238,500 stock options under the 2013 Equity Incentive Plan to non-employee directors. Subsequently in February and March 2014, the Company’s Board of Directors approved grants of 97,298 stock options to employees and a non-employee director of the company.
On February 25, 2014, the aforementioned administrative proceeding filed with the California Labor Commissioner by the Company’s former Vice President of Operations was settled in full following payment of the remaining $25,000 due (see Note 16).
114
Table of Contents
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. |
Not applicable.
| Item 9A. | Controls and Procedures. |
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our periodic and current reports that we file with the SEC is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable and not absolute assurance of achieving the desired control objectives. In reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. In addition, the design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, control may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
As of December 31, 2013, we carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Based on this evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of December 31, 2013.
Management’s Report on Internal Control over Financial Reporting
This Annual Report does not include a report of management’s assessment regarding internal control over financial reporting due to a transition period established by the rules of the SEC for newly public companies.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting identified in management’s evaluation pursuant to Rules 13a-15(d) of the Exchange Act during the quarter ended December 31, 2013 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
Not applicable.
115
Table of Contents
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information required by this item and not set forth below will be set forth in the sections entitled “Election of Directors” and “Executive Officers” in our Proxy Statement for our 2014 Annual Meeting of Stockholders, or Proxy Statement, to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2013, and is incorporated herein by reference.
We have adopted a code of ethics that applies to our Chief Executive Officer and other senior financial officers (our Chief Financial Officer, Controller and other senior financial officers performing similar functions), which we refer to as the Code of Business Conduct and Ethics. The Code of Business Conduct and Ethics is available on our website at www.biocept.com under the Corporate Governance section of the Investor Relations portion of the website. Our Code of Business Conduct and Ethics is designed to meet the requirements of Section 406 of Regulation S-K and the rules promulgated thereunder. We will promptly disclose on our website (i) the nature of any amendment to the Code of Business Conduct and Ethics that applies to any covered person, and (ii) the nature of any waiver, including an implicit waiver, from a provision of the Code of Business Conduct and Ethics that is granted to one of the covered persons.
| Item 11. | Executive Compensation |
The information required by this item will be set forth in the section entitled “Executed Compensation” in our Proxy Statement and is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item will be set forth in the sections entitled “Security Ownership of Certain Beneficial Owners and Management” and “Executive Compensation” in our Proxy Statement and is incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this item will be set forth in the section entitled “Transactions with Related Persons” in our Proxy Statement and is incorporated herein by reference.
| Item 14. | Principal Accounting Fees and Services |
The information required by this item will be set forth in the section entitled “Ratification of Selection of Independent Registered Public Accounting Firm” in our Proxy Statement and is incorporated herein by reference.
116
Table of Contents
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
(a) The following documents are filed as part of this Report:
1. Financial Statements. The following documents are included in Part II, Item 8 of this Report and are incorporated by reference herein:
2. Financial Statement Schedules.
3. Exhibits.
EXHIBITS
| Exhibit No. |
Description of Exhibit | |
| 3.1###### | Certificate of Amendment of Certificate of Incorporation. | |
| 3.2# | Amended and Restated Bylaws. | |
| 4.1## | Specimen Common Stock certificate of Biocept, Inc. | |
| 4.2## | Form of Representative’s Warrant (dated February 10, 2014). | |
| 10.1+# | 2007 Equity Incentive Plan. | |
| 10.1.1+# | Form of Stock Option Grant Notice and Option Agreement under 2007 Equity Incentive Plan. | |
| 10.1.2+# | Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Agreement under 2007 Equity Incentive Plan. | |
| 10.2+### | 2013 Equity Incentive Plan. | |
| 10.2.1+# | Form of Notice of Stock Option Grant under 2013 Equity Incentive Plan. | |
| 10.2.2+# | Form of Stock Option Agreement under 2013 Equity Incentive Plan. | |
| 10.2.3+# | Form of Restricted Stock Unit Agreement under 2013 Equity Incentive Plan. | |
| 10.2.4+# | Form of Restricted Stock Unit Agreement under 2013 Equity Incentive Plan (for senior officers: as used August 8, 2013). | |
| 10.2.5+## | Form of Restricted Stock Unit Agreement under 2013 Equity Incentive Plan (for non-employee directors: as used August 8, 2013). | |
| 10.3+# | Form of Indemnification Agreement between us and our officers and directors. | |
| 10.4+# | Form of Indemnity Agreement between Biocept, Inc., a California corporation, and its officers and directors. | |
117
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.5+# | Employment Agreement, between us and David F. Hale, dated March 10, 2011. | |
| 10.6+# | Employment Agreement, between us and Michael W. Nall, effective as of August 26, 2013. | |
| 10.7+# | Employment Agreement, between us and Lyle J. Arnold, dated April 30, 2011. | |
| 10.8+# | Employment Agreement, between us and William G. Kachioff, dated August 1, 2011. | |
| 10.10+# | Form of Amended and Restated Salary Reduction and Contingent Payment Agreement. | |
| 10.11## | Lease, between us and Nexus Equity VIII LLC, dated March 31, 2004. | |
| 10.11.1# | First Amendment to Lease, between us and ARE-SD Region No. 18, LLC, dated November 1, 2011. | |
| 10.11.2# | Second Amendment to Lease, between us and ARE-SD Region No. 18, LLC, dated September 10, 2012. | |
| 10.11.3# | Warrant to Purchase Preferred Stock, dated September 10, 2012, issued by us in favor of ARE-SD Region No. 18, LLC. | |
| 10.11.4# | Third Amendment to Lease, between us and ARE-SD Region No. 18, LLC, dated as of January 31, 2013, and effective as of January 1, 2013. | |
| 10.11.5# | Fourth Amendment to Lease, between us and ARE-SD Region No. 18, LLC, dated as of September 10, 2013, and effective as of August 1, 2013. | |
| 10.11.6# | Warrant to Purchase Common Stock, dated September 10, 2013, issued by us in favor of ARE-SD Region No. 18, LLC. | |
| 10.12# | Amended and Restated Investor Rights Agreement, dated as of October 31, 2011, among us and certain investors named therein. | |
| 10.13†##### | Collaboration Agreement dated as of November 2, 2012 between us and Life Technologies Corporation. | |
| 10.14#### | Collaboration Agreement dated as of August 17, 2011 between us and Clarient Diagnostic Services, Inc. | |
| 10.14.1# | Laboratory Services Agreement dated July 29, 2013, effective as of May 1, 2013, between us and Clarient Diagnostic Services, Inc. | |
| 10.15†## | Master Laboratory Research Support and Services Agreement dated as of July 9, 2012 between us and Dana Farber Partners Cancer Care, Inc. | |
| 10.16# | Note and Warrant Purchase Agreement dated as of December 22, 2008 between us and The Reiss Family GST Exempt Marital Deduction Trust. | |
| 10.16.1# | Secured Convertible Promissory Note (original principal amount of $1,400,000), dated December 22, 2008, issued by us in favor of The Reiss Family GST Exempt Marital Deduction Trust. | |
| 10.16.1.1# | Amendment of Secured Convertible Promissory Note, dated July 15, 2013. | |
| 10.17# | Amended and Restated Loan Agreement dated as of May 18, 2010 between us and Goodman Co. Ltd. | |
| 10.17.1# | Warrant to Purchase Preferred Stock dated as of January 21, 2009, issued by us in favor of Goodman Co. Ltd. | |
| 10.17.2# | Loan Conversion Agreement dated as of June 28, 2013 between us and Goodman Co. Ltd. [Pursuant to this Agreement, indebtedness was converted into 3,777,324 shares of our Series A Preferred Stock.] | |
118
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.17.3# | Warrant to Purchase Common Stock dated as of July 31, 2013, issued by us in favor of Goodman Co. Ltd. | |
| 10.18# | Note and Warrant Purchase Agreement dated as of February 1, 2011 between us and various investors. | |
| 10.18.1## | First Amendment to Note and Warrant Purchase Agreement, dated as of July 1, 2011. | |
| 10.18.2# | Second Amendment to Note and Warrant Purchase Agreement, dated as of August 1, 2011. | |
| 10.18.3## | Omnibus Amendment Agreement, dated as of September 30, 2011. | |
| 10.18.4# | Amendment to Note and Warrant Purchase Agreement, dated as of June 23, 2012. | |
| 10.18.5# | Amendment to Note and Warrant Purchase Agreement, dated as of November 8, 2012. | |
| 10.18.6# | Form of Secured Convertible Promissory Note, issued by us in favor of various investors under the Note and Warrant Purchase Agreement dated as of February 1, 2011. | |
| 10.18.6.1## | The several Note Conversion Agreements, each dated as of June 28, 2013. [Pursuant to such Agreements, indebtedness was converted into 2,234,922 shares of our Series A Preferred Stock.] | |
| 10.18.6.2# | Note Conversion Agreement dated as of June 28, 2013, among us, The Reiss Family Survivor’s Trust UDT dated December 19, 1988 and The Reiss Family GST Exempt Marital Deduction Trust. [Pursuant to this Agreement, indebtedness was converted into 35,923,845 shares of our Series A Preferred Stock.] | |
| 10.19# | Note and Warrant Purchase Agreement dated as of January 13, 2012 between us and various investors. | |
| 10.19.1# | Omnibus Amendment Agreement dated as of November 8, 2012 between us and various investors. | |
| 10.19.2# | Form of Promissory Note, issued by us in favor of various investors under the Note and Warrant Purchase Agreement dated as of January 13, 2012. | |
| 10.19.2.1## | The several Note Conversion Agreements, each dated as of June 28, 2013. [Pursuant to such Agreements, indebtedness was converted into 309,743 shares of our Series A Preferred Stock.] | |
| 10.19.2.2# | Note Conversion Agreement dated as of June 28, 2013, among us, The Reiss Family Survivor’s Trust UDT dated December 19, 1988 and The Reiss Family GST Exempt Marital Deduction Trust. (Included as Exhibit 10.18.6.2.) | |
| 10.19.3# | Form of Warrant to Purchase Preferred Stock, issued by us in favor of various investors under the Note and Warrant Purchase Agreement dated as of January 13, 2012. | |
| 10.19.4# | Form of Amendment of Warrant to Purchase Preferred Stock, dated as of September 13, 2013. | |
| 10.20# | Form of Note and Warrant Purchase Agreement dated as of June 28, 2013 between us and various investors. | |
| 10.20.1# | Form of Convertible Promissory Note, issued by us in favor of various investors under the Note and Warrant Purchase Agreement dated as of June 28, 2013. | |
| 10.20.2# | Form of Warrant to Purchase Common Stock, issued by us in favor of various investors under the Note and Warrant Purchase Agreement dated as of June 28, 2013. | |
119
Table of Contents
| Exhibit No. |
Description of Exhibit | |
| 10.21# | Reimbursement Agreement dated as of July 11, 2013 among us, The Reiss Family Survivor’s Trust UDT dated December 19, 1988, Edward Neff and Hale Biopharma Ventures, LLC. | |
| 10.21.1# | Form of Warrant to Purchase Common Stock, issued by us in favor of various guarantors under the Reimbursement Agreement dated as of July 11, 2013. | |
| 10.21.2# | Subordination Agreement dated as of July 11, 2013 between us and The Reiss Family GST Exempt Marital Deduction Trust UDT dated December 19, 1988. | |
| 10.22##### | Assignment and Exclusive Cross-License Agreement between us and Aegea Biotechnologies, Inc. dated June 2, 2012. | |
| 10.23+# | Restricted Stock Unit Grant Notice / Agreement between us and Ivor Royston, dated as of November 8, 2010, as amended on February 15, 2012. | |
| 10.24###### | Underwriting Agreement between us and Aegis Capital Corp., dated February 4, 2014. | |
| 14.1 | Code of Ethics for Chief Executive Officer and Other Senior Financial Officers | |
| 31.1 | Certification of Michael Nall, Chief Executive Officer, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of William Kachioff, Chief Financial Officer, pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1* | Certification of Michael Nall, Chief Executive Officer, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2* | Certification of William Kachioff, Chief Financial Officer, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| # | Filed as an exhibit to the registrant’s Registration Statement on Form S-1 ( File No. 333-191323) on September 23, 2013 and incorporated herein by reference. |
| ## | Filed with the registrant’s November 5, 2013 Amendment No. 2 to Registration Statement on Form S-1, and incorporated herein by reference. |
| ### | Filed with the registrant’s November 20, 2013 Amendment No. 3 to Registration Statement on Form S-1, and incorporated herein by reference. |
| #### | Filed with the registrant’s January 8, 2014 Amendment No. 6 to Registration Statement on Form S-1, and incorporated herein by reference. |
| ##### | Filed with the registrant’s January 30, 2014 Amendment No. 8 to Registration Statement on Form S-1, and incorporated herein by reference. |
| ###### | Filed with the registrant’s Current Report on Form 8-K filed on February 14, 2014 and incorporated herein by reference. |
| + | Indicates management contract or compensatory plan. |
| † | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment pursuant to Rule 406 under the Securities Act of 1933. |
| * | This certification is not deemed “filed” for purposes of Section 18 of the Securities Exchange Act, or otherwise subject to the liability of that section. Such certification will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except to the extent that the registrant specifically incorporates it by reference. |
120
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Biocept, Inc. | ||||||
| Dated: March 28, 2014 | By: | /s/ Michael W. Nall | ||||
| Michael W. Nall | ||||||
| President, Chief Executive Officer and Director | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Michael W. Nall Michael W. Nall |
President, Chief Executive Officer and Director (Principal Executive Officer) | March 28, 2014 | ||
| /s/ William G. Kachioff William G. Kachioff |
Chief Financial Officer and Senior Vice President of Finance (Principal Financial and Accounting Officer) | March 28, 2014 | ||
| /s/ David F. Hale David F. Hale |
Chairman and Director | March 28, 2014 | ||
| /s/ M. Faye Wilson M. Faye Wilson |
Director | March 28, 2014 | ||
| /s/ Marsha A. Chandler Marsha A. Chandler |
Director | March 28, 2014 | ||
| /s/ Bruce E. Gerhardt Bruce E. Gerhardt |
Director | March 28, 2014 | ||
| /s/ Bruce A. Huebner Bruce A. Huebner |
Director | March 28, 2014 | ||
| /s/ Edward Neff Edward Neff |
Director | March 28, 2014 | ||
| /s/ Ivor Royston Ivor Royston |
Director | March 28, 2014 | ||
121