Attached files
| file | filename |
|---|---|
| EX-23.1 - EX-23.1 - Intra-Cellular Therapies, Inc. | d592775dex231.htm |
| EX-23.2 - EX-23.2 - Intra-Cellular Therapies, Inc. | d592775dex232.htm |
Table of Contents
As filed with the Securities and Exchange Commission on December 9, 2013
Registration No. 333-191238
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 2
to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Intra-Cellular Therapies, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 36-4742850 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
3960 Broadway
New York, New York 10032
(212) 923-3344
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Sharon Mates, Ph.D.
Chairman, President and Chief Executive Officer
Intra-Cellular Therapies, Inc.
3960 Broadway
New York, New York 10032
(212) 923-3344
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
William C. Hicks, Esq.
Scott A. Samuels, Esq.
John P. Condon, Esq.
Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C.
One Financial Center
Boston, MA 02111
(617) 542-6000
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||||||
| Non-accelerated filer | ¨ | (Do not check if a smaller reporting company) | Smaller reporting company | x | ||||||
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this preliminary prospectus is not complete and may be changed. The selling stockholders named in this prospectus may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and the selling stockholders named in this prospectus are not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to Completion, Dated December 9, 2013
PROSPECTUS

21,961,496 Shares of Common Stock
This prospectus relates to the offering and resale by the selling stockholders identified herein of up to 21,961,496 shares of our common stock, par value $0.0001 per share. These shares were privately issued to the selling stockholders on August 29, 2013 in exchange for shares of Intra-Cellular Therapies, Inc., a Delaware corporation, which is now our wholly-owned subsidiary and which has assumed the name ITI, Inc. We will not receive any proceeds from the sale of these shares by the selling stockholders. The selling stockholders may sell the shares as set forth herein under “Plan of Distribution.” For a list of the selling stockholders, see the section entitled “Selling Stockholders” on page 99. We have borne and will continue to bear the costs relating to the registration of these shares.
There is not currently, and there has never been, any public market for any of our securities. Our securities are not currently eligible for trading on any national securities exchange, including the NASDAQ Stock Market, or any over-the-counter markets, including the OTC Markets—OTCQB tier, or OTCQB. We cannot assure you that our securities will become eligible for trading on any exchange or market. In connection with this offering, we have arranged for a registered broker-dealer to apply to have our common stock quoted on the OTCQB or another over-the-counter system. Until such time as our common stock is quoted on the OTCQB or another public trading market otherwise develops, the selling stockholders identified herein may only sell their shares of our common stock pursuant to this prospectus at a fixed price of $6.3528 per share. At and after such time, the selling stockholders may sell all or a portion of their shares through public or private transactions at prevailing market prices or at privately negotiated prices.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
We are an “emerging growth company” as defined under the federal securities laws, and, as such, are eligible for reduced public company reporting requirements. See “Prospectus Summary—Implications of Being an Emerging Growth Company.”
Investment in our common stock involves risks. See “Risk Factors” beginning on page 10 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2013
Table of Contents
| 1 | ||||
| 8 | ||||
| 9 | ||||
| 10 | ||||
| 30 | ||||
| 32 | ||||
| UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL INFORMATION |
34 | |||
| 36 | ||||
| MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
59 | |||
| 70 | ||||
| 76 | ||||
| 85 | ||||
| 89 | ||||
| 95 | ||||
| 95 | ||||
| 95 | ||||
| 95 | ||||
| SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT |
96 | |||
| 99 | ||||
| 108 | ||||
| 110 | ||||
| 114 | ||||
| 116 | ||||
| 116 | ||||
| 116 | ||||
ABOUT THIS PROSPECTUS
You should rely only on the information contained in this prospectus or contained in any prospectus supplement or free writing prospectus filed with the Securities and Exchange Commission. Neither we nor the selling stockholders have authorized anyone to provide you with additional information or information different from that contained in this prospectus filed with the Securities and Exchange Commission. The selling stockholders are offering to sell, and seeking offers to buy, shares of our common stock only in jurisdictions where offers and sales are permitted. The information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or of any sale of shares of our common stock. Our business, financial condition, results of operations and prospects may have changed since that date.
For investors outside the United States: Neither we nor the selling stockholders have done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of the shares of common stock and the distribution of this prospectus outside the United States.
i
Table of Contents
The following summary highlights selected information contained elsewhere in this prospectus. This summary is not complete and does not contain all the information that should be considered before investing in our common stock. Before making an investment decision, investors should carefully read the entire prospectus, paying particular attention to the risks referred to under the headings “Risk Factors” and “Cautionary Statement Regarding Forward-Looking Statements” and our financial statements and the notes to those financial statements.
As used in this prospectus, unless the context requires otherwise, the terms “Company,” “we,” “our” and “us” refer to Intra-Cellular Therapies, Inc. and our wholly-owned operating subsidiary, ITI, Inc.
Overview
We are a biopharmaceutical company focused on the discovery and clinical development of innovative, small molecule drugs that address underserved medical needs in neuropsychiatric and neurological disorders by targeting intracellular signaling mechanisms within the central nervous system, or CNS. Our lead product candidate, ITI-007, is in clinical development as a first-in-class treatment for schizophrenia. Current medications available for the treatment of schizophrenia do not adequately address the broad array of symptoms associated with this CNS disorder. Use of these current medications also is limited by their substantial side effects. ITI-007 is designed to be effective across a wider range of symptoms, treating both the acute and residual phases of schizophrenia, with improved safety and tolerability.
ITI-007 exhibited antipsychotic efficacy in a randomized, double-blind, placebo and active controlled Phase 2 clinical trial in patients with an acutely exacerbated episode of schizophrenia. In December 2013, we announced the clinical results from this Phase 2 trial. In this Phase 2 trial, 335 patients were randomized to receive one of four treatments: 60 mg of ITI-007, 120 mg of ITI-007, 4 mg of risperidone (active control) or placebo in a 1:1:1:1 ratio, orally once daily for 28 days. The primary endpoint for this clinical trial was change from baseline to Day 28 on the Positive and Negative Syndrome Scale, or PANSS, total score. In this study, ITI-007 met the trial’s pre-specified primary endpoint, improving symptoms associated with schizophrenia as measured by a statistically significant and clinically meaningful decrease in the PANSS total score. The trial also met key secondary outcome measures related to efficacy on PANSS subscales and safety. Additional data from the Phase 2 trial are set forth below in the section of this prospectus entitled “Description of Our Business—Our Clinical Programs—ITI-007 Program—ITI-007 for the treatment of exacerbated and residual schizophrenia—Phase 2 Clinical Trial (ITI-007-005). We plan to request a meeting with the U.S. Food and Drug Administration, or FDA, to discuss our clinical development plan for ITI-007, including our plan to conduct separate, but overlapping, well-controlled clinical efficacy trials in schizophrenia and bipolar disorder.
We are also pursing clinical development of ITI-007 for the treatment of additional CNS diseases and disorders. At the lowest doses, ITI-007 has been demonstrated to act primarily as a potent 5-HT2A serotonin receptor antagonist. As the dose is increased, additional benefits are derived from the engagement of additional drug targets, including modest dopamine receptor modulation and modest inhibition of serotonin transporters. We believe that combined interactions at these receptors may provide additional benefits above and beyond selective 5-HT2A antagonism for treating agitation, aggression and sleep disturbances in diseases that include dementia, Alzheimer’s disease and autism spectrum disorders, while avoiding many of the side effects associated with more robust dopamine receptor antagonism. As the dose of ITI-007 is further increased, leading to moderate dopamine receptor modulation, inhibition of serotonin transporters, and indirect glutamate modulation, these actions complement the complete blockade of 5-HT2A serotonin receptors. In this dose range, we believe that ITI-007 will be useful in treating the symptoms associated with schizophrenia, bipolar disorder, major depressive disorder and other neuropsychiatric diseases.
Given the potential utility for ITI-007 and follow-on compounds to treat these additional indications, we may investigate, either on our own or with a partner, agitation, aggression and sleep disturbances in diseases that
1
Table of Contents
include dementia, Alzheimer’s disease and autism spectrum disorders; major depressive disorder; intermittent explosive disorder; non-motor symptoms and motor complications associated with Parkinson’s disease; and post-traumatic stress disorder. We hold exclusive, worldwide commercialization rights to ITI-007 and a family of compounds from Bristol-Myers Squibb Company pursuant to an exclusive license.
We have a second major program that has yielded a portfolio of compounds that selectively inhibits the enzyme phosphodiesterase 1, or PDE1. PDE1 helps regulate brain activity related to cognition, memory processes and movement/coordination. We have licensed the lead compound in this portfolio, ITI-214, and other compounds in this portfolio, to Takeda Pharmaceutical Company Limited, or Takeda. ITI-214 is the first compound in its class to successfully advance into Phase 1 clinical trials and is being developed for the treatment of cognitive impairment associated with schizophrenia, or CIAS, and other disorders. The results of our first Phase 1 clinical trial in 70 subjects in a randomized, double-blind, placebo-controlled study indicate that ITI-214 was safe and well-tolerated across a broad range of single oral doses. Other compounds in the PDE1 portfolio outside the Takeda collaboration are being advanced for the treatment of other indications, including non-CNS therapeutic areas.
Our pipeline also includes pre-clinical programs that are focused on advancing drug candidates for the treatment of cognitive dysfunction, in both schizophrenia and Alzheimer’s disease, and for disease modification and the treatment of neurodegenerative disorders, including Alzheimer’s disease.
According to the National Institute of Mental Health, over 1% of the world’s population suffers from schizophrenia, and more than 3 million Americans suffer from the illness in any given year. Worldwide sales of antipsychotic drugs used to treat schizophrenia and other CNS related disorders exceeded $40 billion in 2012. These drugs have been increasingly used by physicians to address a range of disorders in addition to schizophrenia, including bipolar disorder and a variety of psychoses and related conditions in elderly patients. Despite their commercial success, current antipsychotic drugs have substantial limitations, including inadequate efficacy and severe side effects.
We have assembled a management team with significant industry experience to lead the discovery and development of our product candidates. We complement our management team with a group of scientific and clinical advisors that includes recognized experts in the fields of schizophrenia and other central nervous system disorders, including Nobel Laureate, Dr. Paul Greengard, one of our co-founders.
Reverse Merger and Selling Stockholders
Pursuant to an Agreement and Plan of Merger dated August 23, 2013, or the Merger Agreement, by and among Oneida Resources Corp., which we refer to as the Company, we, our and us; ITI, Inc., a Delaware corporation and wholly-owned subsidiary of the Company, or Merger Sub; and Intra-Cellular Therapies, Inc., a Delaware corporation, which we refer to as ITI; Merger Sub merged with and into ITI, with ITI remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. This transaction is referred to throughout this prospectus as the “Merger.” The Merger was effective on August 29, 2013, upon the filing of a Certificate of Merger with the Secretary of State of the State of Delaware. As part of the Merger, ITI changed its name to ITI, Inc. A copy of the Merger Agreement is filed as Exhibit 2.1 to the registration statement of which this prospectus forms a part.
Immediately following the Merger, a newly organized wholly-owned subsidiary of the Company named “Intra-Cellular Therapies, Inc.”, or Name Change Merger Sub, merged with and into the Company, leaving the Company as the surviving corporation. We refer to this transaction as the “Name Change Merger.” In connection with the Name Change Merger, we relinquished our corporate name “Oneida Resources Corp.” and assumed in its place the name “Intra-Cellular Therapies, Inc.” The Name Change Merger and name change became effective
2
Table of Contents
on August 29, 2013, upon the filing of a Certificate of Ownership and Merger with the Secretary of State of the State of Delaware. The Certificate of Ownership and Merger is filed as Exhibit 3.3 to the registration statement of which this prospectus forms a part.
At the effective time of the Merger, or the Effective Time, the legal existence of Merger Sub ceased and each share of ITI common stock and each share of ITI preferred stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock, which we refer to as the Exchange. We issued an aggregate of 22,134,647 shares of our common stock upon such exchange of the outstanding shares of ITI common stock and preferred stock. In addition, at the Effective Time, we assumed ITI’s 2003 Equity Incentive Plan, as amended, or the 2003 Equity Incentive Plan, and all options to purchase ITI common stock then outstanding under the 2003 Equity Incentive Plan, and such options became exercisable for an aggregate of 1,462,380 shares of our common stock, subject to the vesting and other terms of such options. The vesting of such options was not accelerated as a result of the Merger. At the Effective Time, we also assumed the outstanding warrant to purchase ITI common stock, and such warrant became exercisable for 1,822 shares of our common stock.
Immediately following the Effective Time, pursuant to the terms of a Redemption Agreement dated August 29, 2013, or the Redemption Agreement, by and among the Company and its then-current sole stockholder, we completed the closing of a redemption of 5,000,000 shares of our common stock, or the Redemption, from our then-current sole stockholder in consideration of $60,000, plus professional costs related to the transaction not to exceed $20,000. The 5,000,000 shares constituted all of the issued and outstanding shares of our capital stock, on a fully-diluted basis, immediately prior to the Merger. A copy of the Redemption Agreement is filed as Exhibit 10.17 to the registration statement of which this prospectus forms a part.
Upon completion of the Merger and the Redemption, the former stockholders of ITI held 100% of the outstanding shares of our capital stock.
The issuance of shares of our common stock in the Merger was exempt from registration under Section 4(a)(2) of the Securities Act of 1933, as amended, or the Securities Act, and Rule 506(b) of Regulation D promulgated thereunder. This prospectus relates to the sale or other disposition from time to time of up to 21,961,496 shares of our common stock issued in the Merger that are held by the selling stockholders named in this prospectus.
3
Table of Contents
Our Product Candidates
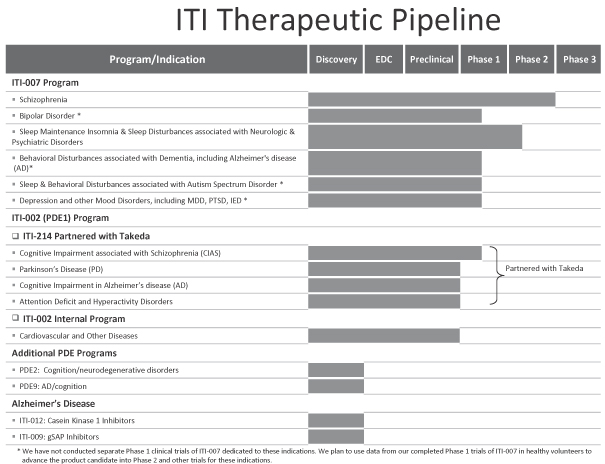
Our pipeline includes two product candidates in clinical development and two product candidates in advanced pre-clinical testing. We believe that our product candidates offer innovative therapeutic approaches and may provide significant advantages relative to current therapies. The following table summarizes our product candidates and programs:
Our Strategy
Our goal is to discover and develop novel small molecule therapeutics for the treatment of CNS diseases in order to improve the lives of people suffering from such illnesses. Using our key understanding of intracellular signaling, we seek to accomplish our goal, using our in-house expert drug discovery and clinical development teams, in two ways:
| • | we seek to have the capability to develop first-in-class medications with novel mechanisms that have the potential to treat CNS diseases for which there are no previously marketed drugs; and |
| • | we seek to develop drugs that can differentiate themselves in competitive markets by addressing aspects of CNS disease which are either not treated by currently marketed drugs or can be effective with fewer side effects. |
4
Table of Contents
The key elements of our strategy are to:
| • | complete the development of ITI-007 for its lead indication, treatment of acute symptoms in schizophrenia, and for additional neuropsychiatric indications, such as bipolar disorder and residual symptoms in schizophrenia; |
| • | expand the commercial potential of ITI-007 by investigating its usefulness in neurological areas, such as behavioral disturbances in dementia, including Alzheimer’s disease and autism spectrum disorder, and in additional neuropsychiatric indications, such as sleep disorders associated with neuropsychiatric and neurological disorders and major depressive disorder; |
| • | continue to develop with our collaboration partner, Takeda, PDE inhibitor compounds, such as ITI-214, for CNS indications such as CIAS; and |
| • | advance earlier stage product candidates in our pipeline. |
Risks Relating to Our Business
We are a biopharmaceutical company, and our business and ability to execute our business strategy are subject to a number of risks of which you should be aware before you decide to buy our common stock. In particular, you should consider the following risks, which are discussed more fully in the section of this prospectus entitled “Risk Factors”:
| • | We currently do not have, and may never have, any products that generate significant revenues. |
| • | There is no guarantee that our planned clinical trials for ITI-007 in acute schizophrenia or in other indications will be successful. |
| • | Preliminary and interim data from our clinical studies that we may announce or publish from time to time may change as more patient data become available. |
| • | If the FDA does not agree with our clinical development plans to advance ITI-007 for the treatment of schizophrenia and bipolar disorder with separate, but overlapping, well-controlled clinical trials in both indications, our development of ITI-007 may be delayed and the costs of our development of ITI-007 would increase. |
| • | Safety issues with our product candidates, or with product candidates or approved products of third parties that are similar to our product candidates, could give rise to delays in the regulatory approval process, restrictions on labeling or product withdrawal after approval. |
| • | We expect our net losses to continue for at least several years and are unable to predict the extent of future losses or when we will become profitable, if ever. |
| • | We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations. |
| • | Our lead product candidate, ITI-007, is only part way through the clinical trials we anticipate needing to complete before we may be able to submit an NDA to the FDA. Clinical trials are long, expensive and unpredictable, and there is a high risk of failure. |
| • | Delays, suspensions and terminations in our clinical trials could result in increased costs to us, delay our ability to generate product revenues and therefore may have a material adverse effect on our business, results of operations and future growth prospects. |
| • | We rely on third parties to conduct our clinical trials and perform data collection and analysis, which may result in costs and delays that prevent us from successfully commercializing our product candidates. |
| • | Even if we successfully complete the clinical trials of one or more of our product candidates, the product candidates may fail for other reasons. |
5
Table of Contents
| • | Following regulatory approval of any of our drug candidates, we will be subject to ongoing regulatory obligations and restrictions, which may result in significant expense and limit our ability to commercialize our potential products. |
| • | Relying on third-party manufacturers may result in delays in our clinical trials, regulatory approvals and product introductions. |
| • | We will need to continue to manage our organization and we may encounter difficulties with our staffing and any future transitions, which could adversely affect our results of operations. |
| • | Our ability to compete may be undermined if we do not adequately protect our proprietary rights. |
| • | Many of our competitors have greater resources and capital than us, putting us at a competitive disadvantage. If our competitors develop and market products that are more effective than our product candidates, they may reduce or eliminate our commercial opportunity. |
| • | There is currently no market for our common stock and there can be no assurance that any market will ever develop. You may therefore be unable to re-sell shares of our common stock at times and prices that you believe are appropriate. |
| • | Our common stock may not be eligible for listing or quotation on any securities exchange. |
| • | The price of our common stock could be subject to volatility related or unrelated to our operations. |
| • | Management and certain members of our board of directors beneficially own a substantial amount of our outstanding equity securities and will be able to exert substantial control over us. |
| • | Because we became a reporting company under the Exchange Act by means other than a traditional underwritten initial public offering, we may not be able to attract the attention of research analysts at major brokerage firms. |
Implications of Being an Emerging Growth Company
As a company with less than $1.0 billion in revenue during our last fiscal year, we qualify as an “emerging growth company” as defined in the Jumpstart Our Business Startups, or JOBS, Act enacted in April 2012. An emerging growth company may take advantage of reduced reporting requirements that are otherwise applicable to public companies. These provisions include, but are not limited to:
| • | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes- Oxley Act of 2002, or Sarbanes-Oxley Act; |
| • | reduced disclosure obligations regarding executive compensation in our periodic reports, proxy statements and registration statements; and |
| • | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We may take advantage of these provisions for up to five years after the first sale of our common equity securities pursuant to an effective registration statement under the Securities Act, which we expect will be pursuant to the registration statement of which this prospectus forms a part or a registration statement on Form S-8 that we may file in the future. However, if certain events occur prior to the end of such five year period, including if we become a “large accelerated filer,” our annual gross revenues exceed $1 billion or we issue more than $1 billion of non-convertible debt in any three year period, we would cease to be an emerging growth company prior to the end of such five year period.
We may choose to take advantage of some but not all of these reduced burdens. We have taken advantage of certain of the reduced disclosure obligations regarding executive compensation in this registration statement and
6
Table of Contents
may elect to take advantage of other reduced burdens in future filings. As a result, the information that we provide to our stockholders may be different than you might receive from other public reporting companies in which you hold equity interests.
Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. However, we have irrevocably elected not to avail ourselves of this extended transition period for complying with new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We are also a “smaller reporting company” as defined in Rule 12b-2 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and have elected to take advantage of certain of the scaled disclosure available to smaller reporting companies.
Our Corporate Information
We were originally incorporated in the State of Delaware in August 2012 under the name “Oneida Resources Corp.” Prior to the Merger, Oneida Resources Corp. was a “shell” company registered under the Exchange Act with no specific business plan or purpose until it began operating the business of ITI through the Merger transaction on August 29, 2013. ITI was incorporated in Delaware in May 2001 to focus primarily on the development of novel drugs for the treatment of neuropsychiatric and neurologic diseases and other disorders of the central nervous system. Effective upon the Merger, a wholly-owned subsidiary of the Company merged with and into ITI, and ITI continues as the operating subsidiary of the Company. As used herein, the words the “Company,” “we,” “us,” and “our” refer to the current Delaware corporation operating the business of ITI as a wholly-owned subsidiary, which business will continue as the business of the Company.
Our corporate headquarters and laboratory are located at 3960 Broadway, New York, New York, and our telephone number is (212) 923-3344. We also have an office in Towson, Maryland. We maintain a website at www.intracellulartherapies.com, to which we regularly post copies of our press releases as well as additional information about us. Our filings with the Securities and Exchange Commission, or SEC, will be available free of charge through the website as soon as reasonably practicable after being electronically filed with or furnished to the SEC. Information contained in our website does not constitute a part of this prospectus or our other filings with the SEC.
All brand names or trademarks appearing in this prospectus are the property of their respective holders. Use or display by us of other parties’ trademarks, trade dress, or products in this prospectus is not intended to, and does not, imply a relationship with, or endorsements or sponsorship of, us by the trademark or trade dress owners.
7
Table of Contents
| Common stock offered by selling stockholders |
21,961,496 shares |
| Common stock outstanding |
22,134,647 shares |
| Use of proceeds |
We will not receive any proceeds from the sale of the shares of common stock offered by the selling stockholders. |
| Offering price |
The selling stockholders may only sell their shares of our common stock pursuant to this prospectus at a fixed price of $6.3528 per share until such time as our common stock is quoted on the OTCQB or another public trading market for our common stock otherwise develops. At and after such time, the selling stockholders may sell all or a portion of their shares through public or private transactions at prevailing market prices or at privately negotiated prices. |
| Risk factors |
You should read the “Risk Factors” section of this prospectus for a discussion of factors to consider carefully before deciding to invest in shares of our common stock. |
| Market for our shares |
There is not now and never has been any market for our securities and an active market may never develop. In connection with this offering, we have arranged for a broker-dealer to apply to have our common stock quoted on the OTCQB or another over-the-counter system. In the future, we intend to seek to have our common stock quoted on a national securities exchange. However, we may not be successful in having our shares quoted on an over-the-counter market or listed on a national securities exchange. |
The number of shares of common stock outstanding is based on an aggregate of 22,134,647 shares outstanding as of December 1, 2013, and excludes:
| • | 1,427,025 shares of common stock issuable upon exercise of outstanding options as of December 1, 2013, at a weighted average exercise price of $1.96 per share, of which 1,085,030 shares were vested as of such date; |
| • | 1,822 shares of common stock issuable upon the exercise of a warrant outstanding as of December 1, 2013, at an exercise price of $6.0264 per share; and |
| • | 799,934 shares of common stock reserved for future issuance under our 2013 Equity Incentive Plan, or the 2013 Plan, plus up to an additional maximum of 1,462,380 shares which may be issued solely after the cancellation or expiration of any unexercised stock options that we assumed in the Merger, plus any future increases in the number of shares of common stock reserved for issuance under the 2013 Plan pursuant to evergreen provisions. |
Unless otherwise indicated in this prospectus, all share and per share figures reflect the exchange of each share of ITI common stock and each share of ITI preferred stock then outstanding for 0.5 shares of our common stock upon the Effective Time of the Merger on August 29, 2013; however, the share and per share numbers in the audited financial statements of ITI for the years ended December 31, 2012 and 2011 included in this prospectus are not adjusted to give effect to the Merger.
8
Table of Contents
The following tables summarize our financial data for the periods presented and should be read together with the sections of this prospectus entitled “Risk Factors,” “Unaudited Pro Forma Condensed Combined Financial Information,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and our financial statements and related notes appearing elsewhere in this prospectus. The statements of operations for the years ended December 31, 2011 and 2012 included in this prospectus have been derived from our audited financial statements and footnotes included elsewhere in this prospectus. The following summary statements of operations for the nine months ended September 30, 2012 and 2013 and the balance sheet data as of December 31, 2012 and September 30, 2013 have been derived from our audited and unaudited financial statements and footnotes, respectively, included elsewhere in this prospectus. We have prepared the unaudited financial statements on the same basis as the audited financial statements and have included all adjustments, consisting only of normal recurring adjustments, which in our opinion are necessary to state fairly the financial information set forth in those statements. Our historical results are not necessarily indicative of the results we expect in the future, and our interim results should not necessarily be considered indicative of results we expect for the full year.
| Years Ended December 31, | Nine-Months Ended September 30, | |||||||||||||||
| 2012 | 2011 | 2013 | 2012 | |||||||||||||
| (Audited) | (Audited) | (Unaudited) | (Unaudited) | |||||||||||||
| Statements of Operations: |
||||||||||||||||
| Revenues |
$ | 3,117,991 | $ | 23,361,959 | $ | 1,909,471 | $ | 2,449,055 | ||||||||
| Costs and expenses: |
||||||||||||||||
| Research and development |
15,486,476 | 7,654,546 | 16,897,903 | 14,969,710 | ||||||||||||
| General and administrative |
4,034,925 | 4,612,450 | 3,245,585 | 2,916,139 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total costs and expenses |
19,521,401 | 12,266,996 | 20,143,488 | 17,885,849 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(16,403,410 | ) | 11,094,963 | (18,234,017 | ) | (15,436,794 | ) | |||||||||
| Interest expense |
(193,498 | ) | (15 | ) | (604,960 | ) | — | |||||||||
| Interest income |
39,002 | 62,315 | 11,589 | 29,730 | ||||||||||||
| Income taxes |
(32,921 | ) | (64,834 | ) | — | (24,690 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net (loss) income |
(16,590,827 | ) | 11,092,429 | (18,827,388 | ) | (15,431,754 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net (loss) income per common share: |
||||||||||||||||
| Basic |
$ | (2.96 | ) | $ | 1.98 | $ | (2.43 | ) | $ | (2.75 | ) | |||||
| Diluted |
$ | (2.96 | ) | $ | 1.68 | $ | (2.43 | ) | $ | (2.75 | ) | |||||
| Weighted average number of common shares: |
||||||||||||||||
| Basic |
5,607,539 | 5,601,495 | 7,737,250 | 5,603,575 | ||||||||||||
| Diluted |
5,607,539 | 6,595,238 | 7,737,250 | 5,603,575 | ||||||||||||
| September 30, 2013 |
December 31, 2012 |
|||||||
| (Unaudited) | (Audited) | |||||||
| Balance Sheet data: |
||||||||
| Cash and cash equivalents |
$ | 44,072,012 | $ | 15,645,528 | ||||
| Total assets |
47,336,046 | 19,823,680 | ||||||
| Total liabilities |
7,774,662 | 2,839,595 | ||||||
| Accumulated deficit |
(49,523,687 | ) | (30,696,299 | ) | ||||
| Total stockholders’ equity |
39,561,384 | 16,984,085 | ||||||
9
Table of Contents
Investing in our common stock involves a high degree of risk. In addition to the information, documents or reports included or incorporated by reference in this prospectus and, if applicable, any prospectus supplement or other offering materials, you should carefully consider the risks described below in addition to the other information contained in this prospectus, before making an investment decision. Our business, financial condition or results of operations could be harmed by any of these risks. As a result, you could lose some or all of your investment in our common stock. The risks and uncertainties described below are not the only ones we face. Additional risks not currently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Risks Related to Our Business
We currently do not have, and may never have, any products that generate significant revenues.
We have a limited operating history on which to evaluate our business and prospects. To date, we have not generated any product revenues from our product candidates currently in development. We cannot guarantee that any of our product candidates currently in development will ever become marketable products.
We must demonstrate that our drug candidates satisfy rigorous standards of safety and efficacy for their intended uses before the U.S. Food and Drug Administration, or FDA, and other regulatory authorities in the European Union and elsewhere will approve them for commercialization. Significant additional research, preclinical testing and clinical testing is required before we can file applications with the FDA or other regulatory authorities for premarket approval of our drug candidates. In addition, to compete effectively, our drugs must be easy to administer, cost-effective and economical to manufacture on a commercial scale. We may not achieve any of these objectives. ITI-007, our most advanced drug candidate, has just completed a Phase 2 clinical trial and ITI-214 is currently in Phase 1 clinical trials. We cannot be certain that the clinical development of these or any other drug candidates in preclinical testing or clinical development will be successful, that we will receive the regulatory approvals required to commercialize them or that any of our other research and drug discovery programs will yield a drug candidate suitable for investigation through clinical trials. Our commercial revenues from our product candidates currently in development, if any, will be derived from sales of drugs that will not become marketable for several years, if at all.
There is no guarantee that our planned clinical trials for ITI-007 in acute schizophrenia or in other indications will be successful.
In our Phase 1 and Phase 2 clinical trials, our lead product candidate, ITI-007, has demonstrated improved sleep maintenance, antipsychotic efficacy, and clinical signals consistent with reduction in negative symptoms associated with schizophrenia, depression and anxiety, and other symptoms associated with impaired social function. ITI-007 exhibited antipsychotic efficacy in a randomized, double-blind, placebo and active controlled Phase 2 clinical trial in patients with an acutely exacerbated episode of schizophrenia. Our preclinical studies and initial clinical trials demonstrate that ITI-007 has shown evidence of addressing the symptoms of schizophrenia without causing cardiovascular and metabolic abnormalities, or motor impairments. Further, ITI-007 was shown effective at a dose that did not cause adverse effects displayed by existing antipsychotic drugs that tend to lead to high rates of noncompliance by the patients who most need these drugs. We are currently planning confirmatory later-stage clinical trials.
The historical rate of failures for product candidates in clinical development and late-stage clinical trials is high. While we plan to conduct further clinical studies in patients with acute schizophrenia and other indications, there is no guarantee that we will have the same level of success in these trials as we have had in our earlier clinical trials, or be successful at all. We may need to conduct additional clinical trials before we are able to advance ITI-007 into Phase 3 clinical trials in patients with acute schizophrenia.
10
Table of Contents
In addition, although we believe that ITI-007 and follow-on compounds may also have clinical utility in indications other than acute schizophrenia, such as behavioral disturbances in dementia, bipolar disorder, intermittent explosive disorder, non-motor disorders associated with Parkinson’s disease, obsessive compulsive disorder and anxiety disorders and post-traumatic stress disorder, we have never tested ITI-007 in Phase 2 clinical trials in the patient population for these other indications.
If we do not successfully complete clinical development of ITI-007, we will be unable to market and sell products derived from it and to generate product revenues. Even if we do successfully complete clinical trials for ITI-007 in patients with acute schizophrenia, those results are not necessarily predictive of results of future pivotal trials that may be needed before we may submit a New Drug Application, or NDA, to the FDA for the initial or other future indications. Of the vast number of drugs in development, only a small percentage result in the submission of an NDA to the FDA, and even less result in the NDA ultimately being approved by the FDA for commercialization.
Preliminary and interim data from our clinical studies that we may announce or publish from time to time may change as more patient data become available.
From time to time, we may announce or publish preliminary or interim data from our clinical studies. Preliminary and interim results of a clinical trial are not necessarily predictive of final results. Preliminary and interim data are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. As a result, preliminary and interim data should be viewed with caution until the final data are available. Material adverse changes in the final data compared to the interim data could significantly harm our business prospects.
If the FDA does not agree with our clinical development plans to advance ITI-007 for the treatment of schizophrenia and bipolar disorder with separate, but overlapping, well-controlled clinical trials in both indications, our development of ITI-007 may be delayed and the costs of our development of ITI-007 would increase.
Subject to discussions with the FDA, we intend to initiate Phase 3 trials in schizophrenia in the second half of 2014 and plan to initiate separate additional trials in bipolar disorder in 2015. We expect that the planned trials in bipolar disorder will overlap in time with the clinical conduct of the planned trials in schizophrenia. We have not yet discussed our plans to develop ITI-007 for the treatment of bipolar disorder with the FDA. With the recent completion of the ITI-007-005 Phase 2 trial in schizophrenia, we plan to request a meeting with the FDA to discuss our clinical development plan for ITI-007, including our plan to conduct separate, but overlapping, well-controlled clinical efficacy trials in schizophrenia and bipolar disorder. The FDA may not agree with our clinical development plans to advance ITI-007 for the treatment of schizophrenia and bipolar disorder with separate, but overlapping, well-controlled clinical trials in both indications. Our clinical plans may change based on any discussions with the FDA. If the FDA does not agree with our clinical development plans for ITI-007, our development of ITI-007 may be delayed and the costs of our development of ITI-007 would increase, which may have an adverse effect on our business, financial condition and results of operations.
Safety issues with our product candidates, or with product candidates or approved products of third parties that are similar to our product candidates, could give rise to delays in the regulatory approval process, restrictions on labeling or product withdrawal after approval.
Problems with product candidates or approved products marketed by third parties that utilize the same therapeutic target or that belong to the same therapeutic class as our product candidates could adversely affect the development, regulatory approval and commercialization of our product candidates. In 2012, the FDA released draft guidance recommending that prospective suicidality assessments be performed in clinical trials of any drug being developed for a psychiatric indication. Our development programs are focused on psychiatric indications. Our PDE1 program is a novel target and may have unexpected safety effects that do not appear until late in clinical development or after commercial approval. To date, we have not experienced any treatment-related
11
Table of Contents
serious adverse effects, or SAEs, in clinical trials for any of our product candidates; however, some approved products marketed by third parties for psychiatric indications that utilize different therapeutic targets or are in a different therapeutic class have experienced SAEs. As we continue the development and clinical trials of our product candidates, there can be no assurance that our product candidates will not experience any SAEs.
Discovery of previously unknown class effect problems may prevent or delay clinical development and commercial approval of product candidates or result in restrictions on permissible uses after their approval, including withdrawal of the medicine from the market. Many drugs acting on the central nervous system, or CNS, include boxed warnings and precautions related to suicidal behavior or ideation, driving impairment, somnolence/sedation and dizziness, discontinuation, weight gain, non-insulin dependent (type II) diabetes, cardiovascular side effects, sleep disturbances, and motor disturbances. If we or others later identify undesirable side effects caused by the mechanisms of action or classes of our product candidates or specific product candidates:
| • | we may be required to conduct additional clinical trials or implement a Risk Evaluation and Mitigation Strategies program prior to approval; |
| • | regulatory authorities may not approve our product candidates or, as a condition of approval, require specific warnings and contraindications; |
| • | regulatory authorities may withdraw their approval of the product and require us to take our drug off the market; |
| • | we may have limitations on how we promote our drugs; |
| • | sales of products may decrease significantly; |
| • | we may be subject to litigation or product liability claims; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase our commercialization costs and expenses, which, in turn, could delay or prevent us from generating significant revenues from its sale.
Finally, if the FDA determines that a drug may present a risk of substance abuse, it can recommend to the Drug Enforcement Administration that the drug be scheduled under the Controlled Substances Act. Any failure or delay in commencing or completing clinical trials or obtaining regulatory approvals for our product candidates would delay commercialization of our product candidates, and severely harm our business and financial condition.
If we seek to enter into strategic alliances for our drug candidates, but fail to enter into and maintain successful strategic alliances, we may have to reduce or delay our drug candidate development or increase our expenditures.
An important element of a biotechnology company’s strategy for developing, manufacturing and commercializing its drug candidates may be to enter into strategic alliances with pharmaceutical companies or other industry participants to advance its programs and enable it to maintain its financial and operational capacity. We may face significant competition in seeking appropriate alliances. If we seek such alliances, we may not be able to negotiate alliances on acceptable terms, if at all. In addition, these alliances may be unsuccessful. If we seek such alliances and then fail to create and maintain suitable alliances, we may have to limit the size or scope of, or delay, one or more of our drug development or research programs. If we elect to fund drug development or research programs on our own, we will have to increase our expenditures and will need to obtain additional funding, which may be unavailable or available only on unfavorable terms.
To the extent we are able to enter into collaborative arrangements or strategic alliances, we will be exposed to risks related to those collaborations and alliances.
We are currently party to a license and collaboration agreement with Takeda Pharmaceutical Company Limited, or Takeda. Biotechnology companies at our stage of development sometimes become dependent upon
12
Table of Contents
collaborative arrangements or strategic alliances to complete the development and commercialization of drug candidates, particularly after the Phase 2 stage of clinical testing. If we elect to enter into collaborative arrangements or strategic alliances, these arrangements may place the development of our drug candidates outside our control, may require us to relinquish important rights or may otherwise be on terms unfavorable to us.
Dependence on collaborative arrangements or strategic alliances would subject us to a number of risks, including the risk that:
| • | we may not be able to control the amount and timing of resources that our collaborators may devote to the drug candidates; |
| • | our collaborators may experience financial difficulties; |
| • | we may be required to relinquish important rights, such as marketing and distribution rights; |
| • | business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness or ability to complete its obligations under any arrangement; |
| • | a collaborator could independently move forward with a competing drug candidate developed either independently or in collaboration with others, including our competitors; and |
| • | collaborative arrangements are often terminated or allowed to expire, which would delay the development and may increase the cost of developing our drug candidates. |
We expect our net losses to continue for at least several years and are unable to predict the extent of future losses or when we will become profitable, if ever.
We have experienced significant net losses since our inception. As of September 30, 2013, we had an accumulated deficit of approximately $49.5 million. We expect to incur net losses over the next several years as we advance our programs and incur significant clinical development costs. We have not received, and do not expect to receive for at least the next several years, any revenues from the commercialization of our product candidates. Substantially all of our revenues to date were from our license and collaboration agreement with Takeda and our agreements with various U.S. governmental agencies and other parties, including our research and development grants. We anticipate that our collaborations, which provide us with research funding and potential milestone payments, will continue to be our primary sources of revenues for the next several years. We cannot be certain that the milestones required to trigger payments under our existing collaborations will be achieved or that we will enter into additional collaboration agreements. To obtain revenues from our product candidates, we must succeed, either alone or with others, in developing, obtaining regulatory approval for, and manufacturing and marketing drugs with significant market potential. We may never succeed in these activities, and may never generate revenues that are significant enough to achieve profitability.
We will require substantial additional funding, which may not be available to us on acceptable terms, or at all, and, if not so available, may require us to delay, limit, reduce or cease our operations.
We have consumed substantial amounts of capital since our inception. Our cash, cash equivalents and investment securities totaled $46.1 million at September 30, 2013. On August 29, 2013, immediately prior to the Merger, ITI received proceeds of approximately $60.0 million from the closing of its private placement of ITI common stock, which included approximately $15.3 million in principal and $0.8 million in accrued interest from the conversion of ITI’s then outstanding convertible promissory notes, and which resulted in net proceeds, after expenses, of approximately $40.0 million. While we believe that our existing cash resources and anticipated payments from our existing collaborations will be sufficient to fund our cash requirements through at least the third quarter of 2014, we will require significant additional financing in the near future to continue to fund our operations, to complete clinical development and commercialize ITI-007, and to conduct the research and development and clinical and regulatory activities necessary to bring other product candidates to market. Our future capital requirements will depend on, and could increase significantly as a result of, many factors, including:
| • | the progress in, and the costs of, our preclinical studies and clinical trials and other research and development programs; |
13
Table of Contents
| • | the scope, prioritization and number of our research and development programs; |
| • | the ability of our collaborators and us to reach the milestones, and other events or developments, triggering payments under our collaboration agreements or to otherwise make payments under these agreements; |
| • | our ability to enter into new, and to maintain existing, collaboration and license agreements; |
| • | the extent to which our collaborators are obligated to reimburse us for clinical trial costs under our collaboration agreements; |
| • | the costs involved in filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; |
| • | the costs of securing manufacturing arrangements for clinical or commercial production; |
| • | the costs of preparing applications for regulatory approvals for our product candidates; |
| • | the costs of establishing, or contracting for, sales and marketing capabilities if we obtain regulatory clearances to market our product candidates; and |
| • | the costs associated with litigation. |
Some of these factors are outside of our control. Our existing cash resources will not be sufficient to complete advanced clinical development of any of our product candidates. Accordingly, we will continue to require substantial additional capital to continue our clinical development and commercialization activities. Because successful development of our product candidates is uncertain, we are unable to estimate the actual funds we will require to complete research and development and commercialize our product candidates under development.
Until we can generate significant continuing revenues, we expect to satisfy our future cash needs through our existing cash, cash equivalents and investment securities, strategic collaborations, private or public sales of our securities, debt financings, grant funding, or by licensing all or a portion of our product candidates or technology. Turmoil and volatility in the financial markets have adversely affected the market capitalizations of many biotechnology companies, and generally made equity and debt financing more difficult to obtain. This, coupled with other factors, may limit our access to additional financing. This could have a material adverse effect on our ability to access sufficient funding. We cannot be certain that additional funding will be available to us on acceptable terms, or at all. If we do obtain additional funding through equity offerings, the ownership of our existing stockholders will be diluted, and the terms of any financing may adversely affect the rights of our stockholders. In addition, the issuance of additional shares by us, or the possibility of such issuance, may cause the market price of our shares to decline. If funds are not available, we will be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or our commercialization efforts. We also could be required to seek funds through arrangements with collaboration partners or otherwise that may require us to relinquish rights to some of our technologies or product candidates or otherwise agree to terms unfavorable to us.
Our management has broad discretion over the use of our cash and we may not use our cash effectively, which could adversely affect our results of operations.
Our management has significant flexibility in applying our cash resources and could use these resources for corporate purposes that do not increase our market value, or in ways with which our stockholders may not agree. We may use our cash resources for corporate purposes that do not yield a significant return or any return at all for our stockholders, which could adversely affect our future growth prospects.
14
Table of Contents
Our lead product candidate, ITI-007, is only part way through the clinical trials we anticipate needing to complete before we may be able to submit an NDA to the FDA. Clinical trials are long, expensive and unpredictable, and there is a high risk of failure.
Preclinical testing and clinical trials are long, expensive and unpredictable processes that can be subject to delays. It may take several years to complete the preclinical testing and clinical development necessary to commercialize a drug, and delays or failure can occur at any stage. Interim results of clinical trials do not necessarily predict final results, and success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials.
In connection with clinical trials, we face risks that a product candidate may not prove to be efficacious; patients may die or suffer other adverse effects for reasons that may or may not be related to the product candidate being tested; the results may not confirm the positive results of our earlier preclinical studies and clinical trials; and the results may not meet the level of statistical significance required by the FDA or other regulatory agencies. If we do not successfully complete preclinical and clinical development, we will be unable to market and sell products derived from our product candidates and to generate product revenues. Even if we do successfully complete clinical trials, those results are not necessarily predictive of results of additional trials that may be needed before an NDA may be submitted to the FDA or the FDA may approve the NDA.
Delays, suspensions and terminations in our clinical trials could result in increased costs to us, delay our ability to generate product revenues and therefore may have a material adverse effect on our business, results of operations and future growth prospects.
The commencement of clinical trials can be delayed for a variety of reasons, including: delays in demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial; reaching agreement on acceptable terms with prospective contract research organizations and clinical trial sites; manufacturing sufficient quantities of a product candidate; obtaining clearance from the FDA to commence clinical trials pursuant to an Investigational New Drug, or IND; obtaining institutional review board approval to conduct a clinical trial at a prospective clinical trial site; and patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical trial sites, the availability of effective treatments for the relevant disease and the eligibility criteria for the clinical trial.
Once a clinical trial has begun, it may be delayed, suspended or terminated due to a number of factors, including: ongoing discussions with regulatory authorities regarding the scope or design of our clinical trials or requests by them for supplemental information with respect to our clinical trial results; failure to conduct clinical trials in accordance with regulatory requirements; lower than anticipated screening or retention rates of patients in clinical trials; serious adverse events or side effects experienced by participants; and insufficient supply or deficient quality of product candidates or other materials necessary for the conduct of our clinical trials.
Many of these factors may also ultimately lead to denial of regulatory approval of a current or potential product candidate. If we experience delays, suspensions or terminations in a clinical trial, the commercial prospects for the related product candidate will be harmed, and our ability to generate product revenues will be delayed.
We rely on third parties to conduct our clinical trials and perform data collection and analysis, which may result in costs and delays that prevent us from successfully commercializing our product candidates.
Although we design and manage our current preclinical studies and clinical trials, we do not now have the ability to conduct clinical trials for our product candidates on our own. In addition to our collaborators, we rely on contract research organizations, medical institutions, clinical investigators, and contract laboratories to perform data collection and analysis and other aspects of our clinical trials. In addition, we also rely on third parties to assist with our preclinical studies, including studies regarding biological activity, safety, absorption, metabolism, and excretion of product candidates.
15
Table of Contents
Our preclinical activities or clinical trials may be delayed, suspended, or terminated if: the quality or accuracy of the data obtained by the third parties on whom we rely is compromised due to their failure to adhere to our clinical protocols or regulatory requirements or if for other reasons, these third parties do not successfully carry out their contractual duties or fail to meet regulatory obligations or expected deadlines, or these third parties need to be replaced.
If the third parties on whom we rely fail to perform, our development costs may increase, our ability to obtain regulatory approval, and consequently, to commercialize our product candidates may be delayed or prevented altogether. We currently use several contract research organizations to perform services for our preclinical studies and clinical trials. While we believe that there are numerous alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without delays or incurring additional expenses.
Even if we successfully complete the clinical trials of one or more of our product candidates, the product candidates may fail for other reasons.
Even if we successfully complete the clinical trials for one or more of our product candidates, the product candidates may fail for other reasons, including the possibility that the product candidates will:
| • | fail to receive the regulatory approvals required to market them as drugs; |
| • | be subject to proprietary rights held by others requiring the negotiation of a license agreement prior to marketing; |
| • | be difficult or expensive to manufacture on a commercial scale; |
| • | have adverse side effects that make their use less desirable; or |
| • | fail to compete with product candidates or other treatments commercialized by our competitors. |
If we are unable to receive the required regulatory approvals, secure our intellectual property rights, minimize the incidence of any adverse side effects or fail to compete with our competitors’ products, our business, financial condition, and results of operations could be materially and adversely affected.
Following regulatory approval of any of our drug candidates, we will be subject to ongoing regulatory obligations and restrictions, which may result in significant expense and limit our ability to commercialize our potential products.
With regard to our drug candidates, if any, approved by the FDA or by another regulatory authority, we are held to extensive regulatory requirements over product manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping. Regulatory approvals may also be subject to significant limitations on the indicated uses or marketing of the drug candidates. Potentially costly follow-up or post-marketing clinical studies may be required as a condition of approval to further substantiate safety or efficacy, or to investigate specific issues of interest to the regulatory authority. Previously unknown problems with the drug candidate, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
In addition, the law or regulatory policies governing pharmaceuticals may change. New statutory requirements may be enacted or additional regulations may be enacted that could prevent or delay regulatory approval of our drug candidates. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or elsewhere. If we are not able to maintain regulatory compliance, we might not be permitted to market our drugs and our business could suffer.
16
Table of Contents
Our product candidates may not gain acceptance among physicians, patients, or the medical community, thereby limiting our potential to generate revenues, which will undermine our future growth prospects.
Even if our product candidates are approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product candidate by physicians, health care professionals and third-party payors, and our profitability and growth will depend on a number of factors, including:
| • | the ability to provide acceptable evidence of safety and efficacy; |
| • | pricing and cost effectiveness, which may be subject to regulatory control; |
| • | our ability to obtain sufficient third-party insurance coverage or reimbursement; |
| • | effectiveness of our or our collaborators’ sales and marketing strategy; |
| • | relative convenience and ease of administration; |
| • | the prevalence and severity of any adverse side effects; and |
| • | availability of alternative treatments. |
If any product candidate that we develop does not provide a treatment regimen that is at least as beneficial as the current standard of care or otherwise does not provide some additional patient benefit over the current standard of care, that product will not achieve market acceptance and we will not generate sufficient revenues to achieve profitability.
The failure to attract and retain skilled personnel and key relationships could impair our drug development and commercialization efforts.
We are highly dependent on our senior management and key clinical development, scientific and technical personnel. Competition for these types of personnel is intense. The loss of the services of any member of our senior management, clinical development, scientific or technical staff may significantly delay or prevent the achievement of drug development and other business objectives and could have a material adverse effect on our business, operating results and financial condition. We also rely on consultants and advisors to assist us in formulating our strategy. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, that may affect their ability to contribute to us. We intend to expand and develop new drug candidates, and will need additional funding to grow our business. We will need to hire additional employees in order to continue our research and clinical trials and to market our drugs when approved. This strategy will require us to recruit additional executive management and clinical development, regulatory, scientific, technical and sales and marketing personnel. There is currently intense competition for skilled executives and employees with relevant clinical development, scientific, technical and sales and marketing expertise, and this competition is likely to continue. The inability to attract and retain sufficient clinical development, scientific, technical and managerial personnel, due to intense competition and our limited resources, would limit or delay our product development efforts, which would adversely affect the development of our drug candidates and commercialization of our potential drugs and growth of our business.
We may not be able to continue or fully exploit our partnerships with outside scientific and clinical advisors, which could impair the progress of our clinical trials and our research and development efforts.
We work with scientific and clinical advisors at academic and other institutions who are experts in the field of CNS disorders. They advise us with respect to our clinical trials. These advisors are not our employees and may have other commitments that would limit their future availability to us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services, which may impair our reputation in the industry and delay the development or commercialization of our product candidates.
17
Table of Contents
Relying on third-party manufacturers may result in delays in our clinical trials, regulatory approvals and product introductions.
We have no manufacturing facilities and do not have extensive experience in the manufacturing of drugs or in designing drug-manufacturing processes. We have contracted with third-party manufacturers to produce, in collaboration with us, our product candidates, including ITI-007, for clinical trials. If any of our product candidates are approved by the FDA or other regulatory agencies for commercial sale, we may need to amend our contract with our current manufacturer or contract with another third party to manufacture them in larger quantities. While we believe that there are alternative sources available to manufacture our product candidates, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without delays or additional expenditures. We cannot estimate these delays or costs with certainty but, if they were to occur, they could cause a delay in our development and commercialization efforts. We have not entered into long-term agreements with our current third-party manufacturers or with any alternate suppliers. Although we intend to do so prior to any commercial launch of a product that is approved by the FDA in order to ensure that we maintain adequate supplies of commercial drug product, we may be unable to enter into such agreements or do so on commercially reasonable terms, which could delay a product launch or subject our commercialization efforts to significant supply risk.
The manufacturers of our product candidates are obliged to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs. In addition, the facilities used by our contract manufacturers or other third party manufacturers to manufacture our product candidates must be approved by the FDA pursuant to inspections that will be conducted after we request regulatory approval from the FDA. A failure of any of our current or future contract manufacturers to establish and follow cGMPs and to document their adherence to such practices may lead to significant delays in clinical trials or in obtaining regulatory approval of product candidates or the ultimate launch of products based on our product candidates into the market. Failure by our current or future third-party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of the government to grant pre-market approval of drugs, delays, suspension or withdrawal of approvals, seizures or recalls of products, operating restrictions, and criminal prosecutions.
We will need to continue to manage our organization and we may encounter difficulties with our staffing and any future transitions, which could adversely affect our results of operations.
We will need to effectively manage our operations and facilities in order to advance our drug development programs (including ITI-007, ITI-214 and those compounds covered by our collaboration with Takeda), achieve milestones under our license and collaboration agreement with Takeda, facilitate additional collaborations, and pursue other development activities. It is possible that our infrastructure may be inadequate to support our future efforts and growth. In particular, we may have to develop internal sales, marketing, and distribution capabilities if we decide to market any drug that we may successfully develop. We may not successfully manage our operations and, accordingly, may not achieve our research, development, and commercialization goals.
Our ability to generate product revenues will be diminished if our products do not receive coverage from payors or sell for inadequate prices, or if patients are unable to obtain adequate levels of reimbursement.
Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Adequate coverage and reimbursement from governmental health care programs, such as Medicare and Medicaid, and commercial payors is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Even if we obtain coverage for any approved products, the resulting reimbursement payment rates might not be adequate or may require co-payments that patients find unacceptably high. Patients are unlikely to use any products we may market unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of those products.
18
Table of Contents
In addition, the market for any products for which we may receive regulatory approval will depend significantly on access to third-party payors’ drug formularies, or lists of medications for which third-party payors provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payors may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling health care costs. In addition, in the United States, no uniform policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products candidates to each payor separately, with no assurance that coverage will be obtained. If we are unable to obtain coverage of, and adequate payment levels for, our products from third-party payors, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize any approved products and thereby adversely impact our profitability, results of operations, financial condition, and future success.
In the future, if we have products that are approved, health care legislation may make it more difficult to receive revenues from those products.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals in recent years to change the health care system in ways that could impact our ability to sell our products profitably. In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, PPACA, became law in the United States. PPACA substantially changed the way health care is financed by both governmental and private insurers and significantly affects the health care industry. Among the provisions of PPACA of importance to our potential product candidates are the following:
| • | imposition of an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government health care programs; |
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, retroactive to January 1, 2010, to 23% and 13% of the average manufacturer price for most branded and generic drugs, respectively; |
| • | expansion of health care fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals beginning in 2014 and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the Federal Poverty Level, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
19
Table of Contents
| • | new requirements to report certain financial arrangements with physicians and teaching hospitals, as defined in PPACA and its implementing regulations, including reporting any “payments or transfers of value” made or distributed to prescribers, teaching hospitals and other health care providers and reporting any ownership and investment interests held by physicians and their immediate family members and applicable group purchasing organizations during the preceding calendar year, with data collection to be required beginning August 1, 2013 and reporting to the Centers for Medicare & Medicaid Services, or CMS, to be required by March 31, 2014 and by the 90th day of each subsequent calendar year; |
| • | a new requirement to annually report drug samples that manufacturers and distributors provide to physicians; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
Many of the details regarding the implementation of PPACA are yet to be determined and, at this time, it remains unclear what the full effect that PPACA would have on our business. On June 28, 2012, the U.S. Supreme Court upheld the constitutionality of PPACA, excepting certain provisions that would have required each state to expand its Medicaid programs or risk losing all of the state’s Medicaid funding. At this time, it remains unclear whether there will be any further changes made to PPACA, whether in part or in its entirety. Some states have indicated that they intend not to implement certain sections of PPACA, and some members of the U.S. Congress are still working to repeal PPACA. We cannot predict whether these challenges will continue or other proposals will be made or adopted, or what impact these efforts may have on us.
In addition, in many foreign countries, particularly the countries of the European Union, the pricing of prescription drugs is subject to government control. In some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. We may face competition from lower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, there may be importation of foreign products that compete with any products we may market, which could negatively impact our profitability.
We expect that PPACA, as well as other health care reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we may receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other health care reforms may prevent us from being able to generate revenue, attain profitability, or commercialize any products for which we receive regulatory approval.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market any products we may develop, we may not be able to generate product revenue.
We do not currently have an organization for the sales, marketing or distribution of pharmaceutical products. In order to market any products that may be approved by the FDA, we must build our sales, marketing, managerial, and related capabilities or make arrangements with third parties to perform these critical commercial services. If we are unable to establish adequate sales, marketing, and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable.
20
Table of Contents
Risks Related to Our Intellectual Property
Our ability to compete may be undermined if we do not adequately protect our proprietary rights.
Our commercial success depends on obtaining and maintaining proprietary rights to our product candidates and technologies and their uses, as well as successfully defending these rights against third-party challenges. We will only be able to protect our product candidates, proprietary technologies, and their uses from unauthorized use by third parties to the extent that valid and enforceable patents, or effectively protected trade secrets, cover them. We have patent rights under issued patents in many cases covering our ITI-007 and ITI-002 development programs. Nonetheless, the issued patents and patent applications covering our primary technology programs remain subject to uncertainty and continuous monitoring and action by us due to a number of factors, including:
| • | we may not have been the first to make the inventions covered by our pending patent applications or issued patents; |
| • | we may not have been the first to file patent applications for our product candidates or the technologies we rely upon; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | our disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability; |
| • | any or all of our pending patent applications may not result in issued patents; |
| • | we may not seek or obtain patent protection in all countries that will eventually provide a significant business opportunity; |
| • | any patents issued to us or our collaborators may not provide a basis for commercially viable products, may not provide us with any competitive advantages or may be challenged by third parties; |
| • | our proprietary technologies may not be patentable; |
| • | others may design around our patent claims to produce competitive products which fall outside of the scope of our patents; |
| • | others may identify prior art which could invalidate our patents; or |
| • | changes to patent laws that limit the exclusivity rights of patent holders. |
Even if we have or obtain patents covering our product candidates or technologies, we may still be barred from making, using and selling our product candidates or technologies because of the patent rights of others. Others have or may have filed, and in the future are likely to file, patent applications covering compounds, assays, genes, gene products or therapeutic products that are similar or identical to ours. There are many issued U.S. and foreign patents relating to genes, nucleic acids, polypeptides, chemical compounds or therapeutic products, and some of these may encompass reagents utilized in the identification of candidate drug compounds or compounds that we desire to commercialize. Numerous U.S. and foreign issued patents and pending patent applications owned by others exist in the area of central nervous system disorders and the other fields in which we are developing products. These could materially affect our ability to develop our product candidates or sell our products. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, that may later result in issued patents that our product candidates or technologies may infringe. These patent applications may have priority over patent applications filed by us.
We regularly conduct searches to identify patents or patent applications that may prevent us from obtaining patent protection for our proprietary compounds or that could limit the rights we have claimed in our patents and patent applications. Disputes may arise regarding the ownership or inventorship of our inventions. It is difficult to determine how such disputes would be resolved. Others may challenge the validity of our patents. If our patents are found to be invalid, we will lose the ability to exclude others from making, using or selling the inventions claimed therein.
21
Table of Contents
Some of our academic institutional licensors, research collaborators and scientific advisors have rights to publish data and information to which we have rights. If we cannot maintain the confidentiality of our technology and other confidential information in connection with our collaborations, then our ability to receive patent protection or protect our proprietary information will be impaired. Additionally, any employee whose employment with us terminates, whether voluntarily by the employee or by us in connection with restructurings or otherwise, may seek future employment with our competitors. Although each of our employees is required to sign a confidentiality agreement with us at the time of hire, we cannot guarantee that the confidential nature of our proprietary information will be maintained in the course of such future employment. In addition, technology that we may license-in may become important to some aspects of our business. We generally will not control the patent prosecution, maintenance or enforcement of in-licensed technology.
Confidentiality agreements with employees and others may not adequately prevent disclosure of our trade secrets and other proprietary information and may not adequately protect our intellectual property, which could limit our ability to compete.
Because we operate in the highly technical field of drug discovery and development of small molecule drugs, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We enter into confidentiality and intellectual property assignment agreements with our corporate partners, employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Enforcing a claim that a party illegally obtained and is using our trade secrets is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. The failure to obtain or maintain trade secret protection could adversely affect our competitive position.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in our industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our product candidates, technologies or activities infringe the intellectual property rights of others. If our drug development activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. We may need to resort to litigation to enforce a patent issued to us, protect our trade secrets or determine the scope and validity of third-party proprietary rights. From time to time, we may hire scientific personnel formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of their prior affiliations. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. We also may not be able to afford the costs of litigation.
The patent applications of pharmaceutical and biotechnology companies involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. The U.S. Patent and Trademark Office’s, or USPTO’s, standards are uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination
22
Table of Contents
proceedings in the USPTO (and foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent office), which proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. Similarly, opposition or invalidity proceedings could result in loss of rights or reduction in the scope of one or more claims of a patent in foreign jurisdictions. In addition, such interference, reexamination and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States and foreign countries may permit others to use our discoveries or to develop and commercialize our technology and products without providing any compensation to us or may limit the number of patents or claims we can obtain. In particular, there have been proposals to shorten the exclusivity periods available under U.S. patent law that, if adopted, could substantially harm our business. The product candidates that we are developing are protected by intellectual property rights, including patents and patent applications. If any of our product candidates becomes a marketable product, we will rely on our exclusivity under patents to sell the compound and recoup our investments in the research and development of the compound. If the exclusivity period for patents is shortened, then our ability to generate revenues without competition will be reduced and our business could be materially adversely impacted. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws, and those countries may lack adequate rules and procedures for defending our intellectual property rights. For example, some countries, including many in Europe, do not grant patent claims directed to methods of treating humans and, in these countries, patent protection may not be available at all to protect our product candidates. In addition, U.S. patent laws may change, which could prevent or limit us from filing patent applications or patent claims to protect our products and/or technologies or limit the exclusivity periods that are available to patent holders. For example, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was recently signed into law and includes a number of significant changes to U.S. patent law. These include changes to transition from a “first-to-invent” system to a “first-to-file” system and to the way issued patents are challenged. These changes may favor larger and more established companies that have more resources to devote to patent application filing and prosecution. The USPTO has been in the process of implementing regulations and procedures to administer the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act may affect our ability to obtain, enforce or defend our patents. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will ultimately have on the cost of prosecuting our patent applications, our ability to obtain patents based on our discoveries and our ability to enforce or defend our issued patents.
If we fail to obtain and maintain patent protection and trade secret protection of our product candidates, proprietary technologies and their uses, we could lose our competitive advantage and competition we face would increase, reducing our potential revenues and adversely affecting our ability to attain or maintain profitability.
Risks Related to Our Industry
We will be subject to stringent regulation in connection with the marketing of any products derived from our product candidates, which could delay the development and commercialization of our products.
The pharmaceutical industry is subject to stringent regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Neither we nor our collaborators can market a pharmaceutical product in the United States until it has completed rigorous preclinical testing and clinical trials and an extensive regulatory clearance process implemented by the FDA. Satisfaction of regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product, and requires substantial resources. Even if regulatory approval is obtained, it may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, and/or marketing of such products, and requirements for post-approval studies, including additional research and development and clinical trials. These limitations may limit the size of the market for the product or result in the incurrence of additional costs. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues and continue our business.
23
Table of Contents
Outside the United States, the ability to market a product is contingent upon receiving approval from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing, and reimbursement vary widely from country to country. Only after the appropriate regulatory authority is satisfied that adequate evidence of safety, quality, and efficacy has been presented will it grant a marketing authorization. Approval by the FDA does not automatically lead to the approval by regulatory authorities outside the United States and, similarly, approval by regulatory authorities outside the United States will not automatically lead to FDA approval.
Many of our competitors have greater resources and capital than us, putting us at a competitive disadvantage. If our competitors develop and market products that are more effective than our product candidates, they may reduce or eliminate our commercial opportunity.
Competition in the pharmaceutical and biotechnology industries is intense and increasing. We face competition from pharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies, both in the United States and abroad. Some of these competitors have products or are pursuing the development of drugs that target the same diseases and conditions that are the focus of our drug development programs.
For example, our potential products for the treatment of acute schizophrenia would compete with, among other branded products, Abilify®, marketed jointly by Bristol-Myers Squibb and Otsuka Pharmaceutical, Fanapt®, marketed by Novartis Pharmaceuticals, Seroquel XR®, marketed by AstraZeneca, Invega®, marketed by Janssen, and Latuda®, marketed by Sunovion. In addition, our products will compete with, among other generic antipsychotic drugs, haloperidol, risperidone, quetiapine, olanzapine and clozapine.
Many of our competitors and their collaborators have significantly greater experience than we do in the following:
| • | identifying and validating targets; |
| • | screening compounds against targets; |
| • | preclinical studies and clinical trials of potential pharmaceutical products; and |
| • | obtaining FDA and other regulatory approvals. |
In addition, many of our competitors and their collaborators have substantially greater capital and research and development resources, manufacturing, sales and marketing capabilities, and production facilities. Smaller companies also may prove to be significant competitors, particularly through proprietary research discoveries and collaboration arrangements with large pharmaceutical and established biotechnology companies. Many of our competitors have products that have been approved or are in advanced development and may develop superior technologies or methods to identify and validate drug targets and to discover novel small molecule drugs. Our competitors, either alone or with their collaborators, may succeed in developing drugs that are more effective, safer, more affordable, or more easily administered than ours and may achieve patent protection or commercialize drugs sooner than us. Our competitors may also develop alternative therapies that could further limit the market for any drugs that we may develop. Our failure to compete effectively could have a material adverse effect on our business.
Any claims relating to improper handling, storage, or disposal of biological, hazardous, and radioactive materials used in our business could be costly and delay our research and development efforts.
Our research and development activities involve the controlled use of potentially harmful hazardous materials, including volatile solvents, biological materials such as blood from patients that have the potential to transmit disease, chemicals that cause cancer, and various radioactive compounds. Our operations also produce hazardous waste products. We face the risk of contamination or injury from the use, storage, handling or disposal
24
Table of Contents
of these materials. We are subject to federal, state and local laws and regulations governing the use, storage, handling, and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations could be significant, and current or future environmental regulations may impair our research, development, or production efforts. If one of our employees were accidentally injured from the use, storage, handling, or disposal of these materials, the medical costs related to his or her treatment would be covered by our workers’ compensation insurance policy. However, we do not carry specific biological or hazardous waste insurance coverage and our general liability insurance policy specifically excludes coverage for damages and fines arising from biological or hazardous waste exposure or contamination. Accordingly, in the event of contamination or injury, we could be subject to criminal sanctions or fines or be held liable for damages, our operating licenses could be revoked, or we could be required to suspend or modify our operations and our research and development efforts.
Consumers may sue us for product liability, which could result in substantial liabilities that exceed our available resources and damage our reputation.
Researching, developing, and commercializing drug products entail significant product liability risks. Liability claims may arise from our and our collaborators’ use of products in clinical trials and the commercial sale of those products. Consumers may make these claims directly and our collaborators or others selling these products may seek contribution from us if they receive claims from consumers. We have obtained limited product liability insurance coverage for our clinical trials. Our product liability insurance coverage for clinical trials is currently limited to an aggregate of $10 million. As such, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Although we currently have product liability insurance that covers our clinical trials, we will need to increase and expand this coverage as we commence larger scale trials and if our product candidates are approved for commercial sale. This insurance may be prohibitively expensive or may not fully cover our potential liabilities. Inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of products that we or our collaborators develop. Product liability claims could have a material adverse effect on our business and results of operations. Our liability could exceed our total assets if we do not prevail in a lawsuit from any injury caused by our drug products.
Risks Relating to Owning Our Common Stock
There is currently no market for our common stock and there can be no assurance that any market will ever develop. You may therefore be unable to re-sell shares of our common stock at times and prices that you believe are appropriate.
Our common stock is not listed on a national securities exchange, an over-the-counter market or any other exchange. Therefore, there is no trading market, active or otherwise, for our common stock and our common stock may never be included for trading on any stock exchange, automated quotation system or any over-the-counter market. Accordingly, our common stock is highly illiquid and you will likely experience difficulty in re-selling such shares at times and prices that you may desire.
Our common stock may not be eligible for listing or quotation on any securities exchange.
We do not currently meet the initial listing standards of any national securities exchange and our common stock is not quoted for sale on any over-the-counter trading system. We cannot assure you that we will be able to meet the initial listing standards of any national securities exchange, or, if we do meet such initial listing standards, that we will be able to maintain any such listing. Further, the national securities exchanges have adopted so-called “seasoning” rules that require that we meet certain requirements, including prescribed periods of time trading over-the-counter and minimum filings of periodic reports with the Securities and Exchange Commission, or SEC, before we are eligible to apply for listing on such national securities exchanges. We intend to contact an authorized market maker for an over-the-counter quotation system for sponsorship of our common
25
Table of Contents
stock, but we cannot guarantee that such sponsorship will be approved and our common stock listed and quoted for sale. Even if our common stock is quoted for sale on an over-the-counter quotation system, buyers may be insufficient in numbers to allow for a robust market and it may prove impossible to sell your shares. In addition, an investor may find it difficult to obtain accurate quotations as to the market value of our common stock. In addition, if we fail to meet the criteria set forth in SEC regulations, various requirements would be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling our common stock, which may further affect its liquidity. This would also make it more difficult for us to raise additional capital.
The price of our common stock could be subject to volatility related or unrelated to our operations.
If a market for our common stock develops, its market price could fluctuate substantially due to a variety of factors, including market perception of our ability to meet our growth projections and expectations, quarterly operating results of other companies in the same industry, trading volume in our common stock, changes in general conditions in the economy and the financial markets or other developments affecting our business and the business of others in our industry. In addition, the stock market itself is subject to extreme price and volume fluctuations. This volatility has had a significant effect on the market price of securities issued by many companies for reasons related and unrelated to their operating performance and could have the same effect on our common stock.
The designation of our common stock as a “penny stock” would limit the liquidity of our common stock.
Our common stock may be deemed a “penny stock” (as that term is defined under Rule 3a51-1 of the Securities Exchange Act of 1934, as amended, or the Exchange Act) in any market that may develop in the future. Generally, a “penny stock” is a common stock that is not listed on a securities exchange and trades for less than $5.00 per share. Prices often are not available to buyers and sellers and the market may be very limited. Penny stocks in start-up companies are among the riskiest equity investments. Broker-dealers who sell penny stocks must provide purchasers of these stocks with a standardized risk-disclosure document prepared by the SEC. The document provides information about penny stocks and the nature and level of risks involved in investing in the penny stock market. A broker must also provide purchasers with bid and offer quotations and information regarding broker and salesperson compensation and make a written determination that the penny stock is a suitable investment for the purchaser and obtain the purchaser’s written agreement to the purchase. Many brokers choose not to participate in penny stock transactions. Because of the penny stock rules, there may be less trading activity in penny stocks in any market that develops for our common stock in the future and stockholders are likely to have difficulty selling their shares.
Management and certain members of our board of directors beneficially own a substantial amount of our outstanding equity securities and will be able to exert substantial control over us.
Our executive officers and directors beneficially own a substantial percentage of the outstanding equity securities of the Company. Accordingly, if they act as a group, the executive officers and directors of the Company will be able to make all business decisions, including with respect to such matters as amendments to the Company’s charter, other fundamental corporate transactions, such as mergers, asset sales and the sale of the Company, and otherwise will be able to direct the Company’s business and affairs.
We will incur increased costs and demands upon management as a result of complying with the laws and regulations affecting public companies, which could harm our operating results.
As a public company, we will incur significant legal, accounting and other expenses, including costs associated with public company reporting requirements. We will also incur substantial expenses in connection with the preparation and filing of the registration statement for resale of our common stock of which this
26
Table of Contents
prospectus forms a part, and responding to SEC comments in connection with its review of such registration statement. We will also incur costs associated with current corporate governance requirements, including requirements under Section 404 and other provisions of the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, as well as rules implemented by the SEC or any stock exchange or inter-dealer quotations system on which our common stock may be listed in the future. The expenses incurred by public companies for reporting and corporate governance purposes have increased dramatically in recent years. We expect these rules and regulations to substantially increase our legal and financial compliance costs and to make some activities more time-consuming and costly. We are unable currently to estimate these costs with any degree of certainty. We also expect that these new rules and regulations may make it difficult and expensive for us to retain our director and officer liability insurance, and if we are able to retain such insurance, we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage available to privately-held companies. As a result, it may be more difficult for us to attract and retain qualified individuals to serve on our board of directors or as our executive officers.
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and investors’ views of us.
We will be required to comply with Section 404 of the Sarbanes-Oxley Act. Section 404 of the Sarbanes-Oxley Act requires public companies to conduct an annual review and evaluation of their internal controls and attestations of the effectiveness of internal controls by independent auditors. Ensuring that we have adequate internal financial and accounting controls and procedures in place so that we can produce accurate financial statements on a timely basis is a costly and time-consuming effort that will need to be evaluated frequently. Our failure to maintain the effectiveness of our internal controls in accordance with the requirements of the Sarbanes-Oxley Act could have a material adverse effect on our business. We could lose investor confidence in the accuracy and completeness of our financial reports, which could have an adverse effect on the price of our common stock. In addition, if our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
We are an emerging growth company and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an emerging growth company under the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies, which may include, but are not limited to, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, exemptions from the requirements of holding a nonbinding advisory stockholder vote on executive compensation and any golden parachute payments not previously approved, and exemption from the requirement of auditor attestation in the assessment of our internal control over financial reporting. If we do, the information that we provide stockholders may be different than what is available with respect to other public companies. We cannot predict if investors will find our common stock less attractive because we will rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Under the JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. However, we have irrevocably elected not to avail ourselves of this extended transition period for complying with new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We will remain an emerging growth company until the earliest of (1) the end of the fiscal year in which the market value of our common stock that is held by non-affiliates exceeds $700 million as of the end of the second
27
Table of Contents
fiscal quarter, (2) the end of the fiscal year in which we have total annual gross revenues of $1 billion or more during such fiscal year, (3) the date on which we issue more than $1 billion in non-convertible debt in a three-year period or (4) the end of the fiscal year following the fifth anniversary of the date of the first sale of our common stock pursuant to an effective registration statement filed under the Securities Act of 1933, as amended, or the Securities Act. Decreased disclosures in our SEC filings due to our status as an “emerging growth company” may make it harder for investors to analyze our results of operations and financial prospects.
Because we became a reporting company under the Exchange Act by means other than a traditional underwritten initial public offering, we may not be able to attract the attention of research analysts at major brokerage firms.
Because we did not become a reporting company by conducting an underwritten initial public offering, or IPO, of our common stock, and because we will not be listed on a national securities exchange, security analysts of brokerage firms may not provide coverage of our company. In addition, investment banks may be less likely to agree to underwrite secondary offerings on our behalf than they might if we became a public reporting company by means of an IPO because they may be less familiar with our company as a result of more limited coverage by analysts and the media, and because we became public at an early stage in our development. The failure to receive research coverage or support in the market for our shares will have an adverse effect on our ability to develop a liquid market for our common stock.
The resale of shares covered by the registration statement of which this prospectus forms a part could adversely affect the market price of our common stock in the public market, should one develop, which result would in turn negatively affect our ability to raise additional equity capital.
The sale, or availability for sale, of our common stock in the public market may adversely affect the prevailing market price of our common stock and may impair our ability to raise additional capital by selling equity or equity-linked securities. We filed the registration statement of which this prospectus forms a part with the SEC to register the resale of substantially all of the shares of our common stock issued in connection with the Merger. Once effective, the registration statement will permit the resale of these shares at any time, subject to applicable lock-up restrictions described in the “Certain Relationships and Related Person Transactions—Agreements with Stockholders—Lock-Up Provisions in Registration Rights Agreement” section. The resale of a substantial number of shares of our common stock in the public market could adversely affect the market price for our common stock and make it more difficult for you to sell shares of our common stock at times and prices that you feel are appropriate. Furthermore, we expect that, because there will be a large number of shares registered pursuant to the registration statement, selling stockholders will continue to offer shares covered by the registration statement of which this prospectus forms a part for a significant period of time, the precise duration of which cannot be predicted. Accordingly, the adverse market and price pressures resulting from an offering pursuant to the registration statement may continue for an extended period of time and continued negative pressure on the market price of our common stock could have a material adverse effect on our ability to raise additional equity capital.
If securities or industry analysts do not publish, or cease publishing, research or reports about us, our business or our market, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
If a trading market for our common stock develops, the trading market for our common stock will be influenced by whether industry or securities analysts publish research and reports about us, our business, our market or our competitors and, if any analysts do publish such reports, what they publish in those reports. We may not obtain analyst coverage in the future. Any analysts that do cover us may make adverse recommendations regarding our stock, adversely change their recommendations from time to time, and/or provide more favorable relative recommendations about our competitors. If any analyst who may cover us in the future were to cease
28
Table of Contents
coverage of our company or fail to regularly publish reports on us, or if analysts fail to cover us or publish reports about us at all, we could lose, or never gain, visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
We do not anticipate paying cash dividends in the foreseeable future.
We currently intend to retain any future earnings for funding growth. We do not anticipate paying any dividends in the foreseeable future. As a result, you should not rely on an investment in our securities if you require dividend income. Capital appreciation, if any, of our shares may be your sole source of gain for the foreseeable future. Moreover, you may not be able to re-sell your shares at or above the price you paid for them.
29
Table of Contents
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference in this prospectus include forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Exchange Act that relate to future events or our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to differ materially from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. Words such as, but not limited to, “believe,” “expect,” “anticipate,” “estimate,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “targets,” “likely,” “will,” “would,” “could,” “should,” “continue,” and similar expressions or phrases, or the negative of those expressions or phrases, are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Although we believe that we have a reasonable basis for each forward-looking statement contained in this prospectus, we caution you that these statements are based on our projections of the future that are subject to known and unknown risks and uncertainties and other factors that may cause our actual results, level of activity, performance or achievements expressed or implied by these forward-looking statements, to differ. The sections in this prospectus entitled “Description of Our Business,” “Risk Factors,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” as well as other sections in this prospectus, discuss some of the factors that could contribute to these differences. These forward-looking statements include, among other things, statements about:
| • | the initiation, cost, timing, progress and results of our development activities, preclinical studies and clinical trials; |
| • | the timing of and our ability to obtain and maintain regulatory approval of our existing product candidates, any product candidates that we may develop, and any related restrictions, limitations, and/or warnings in the label of any approved product candidates; |
| • | our plans to research, develop and commercialize our future product candidates; |
| • | our collaborators’ election to pursue research, development and commercialization activities; |
| • | our ability to obtain future reimbursement and/or milestone payments from our collaborators; |
| • | our ability to attract collaborators with development, regulatory and commercialization expertise; |
| • | our ability to obtain and maintain intellectual property protection for our product candidates; |
| • | our ability to successfully commercialize our product candidates; |
| • | the size and growth of the markets for our product candidates and our ability to serve those markets; |
| • | the rate and degree of market acceptance of any future products; |
| • | the success of competing drugs that are or become available; |
| • | regulatory developments in the United States and other countries; |
| • | the performance of our third-party suppliers and manufacturers and our ability to obtain alternative sources of raw materials; |
| • | our ability to obtain additional financing; |
| • | our use of the proceeds from our recently completed private placement; |
| • | the accuracy of our estimates regarding expenses, future revenues, capital requirements and the need for additional financing; and |
| • | our ability to attract and retain key scientific or management personnel. |
We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements
30
Table of Contents
we make. We have included important cautionary statements in this prospectus, particularly in the “Risk Factors” section, that we believe could cause actual results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments we may make.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement of which this prospectus forms a part, completely and with the understanding that our actual future results may be materially different from what we expect. The forward-looking statements contained in this prospectus are made as of the date of this prospectus, and we do not assume, and specifically disclaim, any obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise.
31
Table of Contents
Pursuant to an Agreement and Plan of Merger dated August 23, 2013, or the Merger Agreement, by and among Oneida Resources Corp., which we refer to as the Company, we, our and us; ITI, Inc., a Delaware corporation and wholly-owned subsidiary of the Company, or Merger Sub; and Intra-Cellular Therapies, Inc., a Delaware corporation, which we refer to as ITI; Merger Sub merged with and into ITI, with ITI remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. This transaction is referred to throughout this prospectus as the “Merger.” The Merger was effective on August 29, 2013, upon the filing of a Certificate of Merger with the Secretary of State of the State of Delaware. As part of the Merger, ITI changed its name to ITI, Inc.
Immediately following the Merger, a newly organized wholly-owned subsidiary of the Company named “Intra-Cellular Therapies, Inc.”, or Name Change Merger Sub, merged with and into the Company, leaving the Company as the surviving corporation. We refer to this transaction as the “Name Change Merger.” In connection with the Name Change Merger, we relinquished our corporate name “Oneida Resources Corp.” and assumed in its place the name “Intra-Cellular Therapies, Inc.” The Name Change Merger and name change became effective on August 29, 2013, upon the filing of a Certificate of Ownership and Merger with the Secretary of State of the State of Delaware.
At the effective time of the Merger, or the Effective Time, the legal existence of Merger Sub ceased and each share of ITI common stock and each share of ITI preferred stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock, which we refer to as the Exchange. We issued an aggregate of 22,134,647 shares of our common stock upon such exchange of the outstanding shares of ITI common stock and preferred stock. In addition, at the Effective Time, we assumed ITI’s 2003 Equity Incentive Plan, as amended, or the 2003 Equity Incentive Plan, and all options to purchase ITI common stock then outstanding under the 2003 Equity Incentive Plan, and such options became exercisable for an aggregate of 1,462,380 shares of our common stock, subject to the vesting and other terms of such options. The vesting of such options was not accelerated as a result of the Merger. At the Effective Time, we also assumed the outstanding warrant to purchase ITI common stock, and such warrant became exercisable for 1,822 shares of our common stock.
Immediately following the Effective Time, pursuant to the terms of a Redemption Agreement dated August 29, 2013, or the Redemption Agreement, by and among the Company and its then-current sole stockholder, we completed the closing of a redemption of 5,000,000 shares of our common stock, or the Redemption, from our then-current sole stockholder in consideration of $60,000, plus professional costs related to the transaction not to exceed $20,000. The 5,000,000 shares constituted all of the issued and outstanding shares of our capital stock, on a fully-diluted basis, immediately prior to the Merger.
Upon completion of the Merger and the Redemption, the former stockholders of ITI held 100% of the outstanding shares of our capital stock. Unless otherwise indicated in this prospectus, all share and per share figures reflect the exchange of each share of ITI common stock and each share of ITI preferred stock then outstanding for 0.5 shares of our common stock at the Effective Time of the Merger; however, the share and per share numbers in the financial statements of ITI included in this prospectus are not adjusted to give effect to the Merger.
As a condition to the Merger, we entered into an Indemnity Agreement with our former sole officer and director, or the Indemnity Agreement, pursuant to which we agreed to indemnify such former officer and director for actions taken by him in his official capacities relating to the consideration, approval and consummation of the Merger and certain related transactions.
The Merger is being accounted for as a capital transaction. Upon the effectiveness of the Merger, the Company’s business became the operation of ITI and its business. Immediately following the Effective Time, our
32
Table of Contents
board of directors, which immediately prior to the Effective Time consisted of Samir N. Masri as our sole director, appointed Sharon Mates, Ph.D., who was Chairman, President and Chief Executive Officer of ITI, as our Chairman, President and Chief Executive Officer, to serve on our board of directors with Mr. Masri. At the Effective Time, Mr. Masri resigned from all of his positions as an officer of the Company. In addition, immediately following the Effective Time, our board of directors appointed Lawrence J. Hineline, who was the Vice President of Finance, Chief Financial Officer and Secretary of ITI, as our Vice President of Finance, Chief Financial Officer and Secretary; Allen A. Fienberg, Ph.D., who was the Vice President of Business Development of ITI, as our Vice President of Business Development; Lawrence P. Wennogle, Ph.D., who was the Vice President, Drug Discovery of ITI, as our Vice President, Drug Discovery; and Kimberly E. Vanover, Ph.D., who was the Vice President, Clinical Development of ITI, as our Vice President, Clinical Development. On September 9, 2013, which was the eleventh day following the date that we filed with the SEC and transmitted to our sole stockholder prior to the Merger, a Schedule 14f-1 reporting a change in the majority of our directors, Christopher Alafi, Ph.D., Richard Lerner, M.D., Joel S. Marcus and Sir Michael Rawlins, M.D., FRCP, FMedSci, were appointed to our board of directors to serve on our board of directors with Dr. Mates, and Mr. Masri resigned from our board of directors as of such date. Each of Dr. Mates, Dr. Alafi, Dr. Lerner, Mr. Marcus, and Sir Michael were directors of ITI immediately prior to the Merger.
Prior to the Merger, ITI sold to accredited investors approximately $60.0 million of its shares of common stock, or 18,889,307 shares at a price of $3.1764 per share, which included $15.3 million in principal and $0.8 million in accrued interest from the conversion of ITI’s then outstanding convertible promissory notes, or Notes. We refer to this transaction as the Private Placement and the number of shares stated in the preceding sentence does not reflect the Exchange in the Merger. The price per share in the Private Placement, as adjusted for the Exchange in the Merger, would be $6.3528 per share of our post-Merger common stock. Also, ITI granted the investors in the Private Placement registration rights requiring ITI or any successor to register those shares of ITI common stock (which were exchanged for shares of our common stock, along with the rest of the outstanding shares of ITI capital stock, except for dissenting shares, at the Effective Time) for public resale, as described in more detail below. The then existing stockholders of ITI who agreed to become parties to the registration rights agreement also became entitled to such registration rights, subject to specified differences in the agreement between the rights of new investors and existing stockholders. The existing Second Amended and Restated Investor Rights Agreement, by and among ITI and the investors listed therein, dated as of October 25, 2007, as amended, was terminated at the Effective Time. The Private Placement closed immediately prior to the filing of a Certificate of Merger with the Secretary of State of the State of Delaware, on August 29, 2013.
The Merger Agreement has been filed as Exhibit 2.1 to the registration statement of which this prospectus forms a part to provide investors and security holders with information regarding its terms. It is not intended to provide any other factual information about the Company or ITI. The representations, warranties and covenants contained in the Merger Agreement were made only for the purposes of such agreement and as of specified dates, were solely for the benefit of the parties to such agreement, and may be subject to limitations agreed upon by the contracting parties. The representations and warranties may have been made for the purposes of allocating contractual risk between the parties to the agreement instead of establishing these matters as facts, and may be subject to standards of materiality applicable to the contracting parties that differ from those applicable to investors. Investors are not third-party beneficiaries under the Merger Agreement and should not rely on the representations, warranties and covenants or any descriptions thereof as characterizations of the actual state of facts or condition of the Company, ITI or any of their respective subsidiaries or affiliates. In addition, the assertions embodied in the representations and warranties contained in the Merger Agreement are qualified by information in a confidential disclosure schedule provided by ITI, which is not being filed with the registration statement of which this prospectus forms a part as permitted by the SEC’s rules and regulations. Accordingly, investors should not rely on the representations and warranties as characterizations of the actual state of facts, since (i) they were made only as of the date of such agreement or a prior, specified date, (ii) in some cases they are subject to qualifications with respect to materiality, knowledge and/or other matters, and (iii) they may be modified in important part by the underlying disclosure schedule. Moreover, information concerning the subject matter of the representations and warranties may change after the date of the Merger Agreement, which subsequent information may or may not be fully reflected in the Company’s public disclosures.
33
Table of Contents
UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL INFORMATION
The following unaudited pro forma condensed combined financial statements are based on the historical financial statements of Intra-Cellular Therapies, Inc., or ITI, and the historical financial statements of Oneida Resources Corp., or the Company. The historical financial statements of ITI and the historical financial statements of the Company are included elsewhere in this prospectus. The unaudited pro forma condensed combined statements of operations for the nine months ended September 30, 2013 and the twelve months ended December 31, 2012 give effect to the Merger, which was consummated on August 29, 2013, as if it had occurred on January 1, 2012.
Unaudited Pro Forma Condensed Combined Statement of Operations
for the Nine Months Ended September 30, 2013
of
Intra-Cellular Therapies, Inc. and Oneida Resources Corp.
| Intra-Cellular Therapies, Inc. |
Oneida Resources Corp. |
Pro Forma Adjustments |
Pro Forma Combined |
|||||||||||||
| Total revenues |
$ | 1,909,471 | $ | — | $ | — | $ | 1,909,471 | ||||||||
| Total costs and expenses |
20,143,488 | 52,227 | (52,227 | )(a) | 20,143,488 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(18,234,017 | ) | (52,227 | ) | 52,227 | (18,234,017 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Other expense |
(593,371 | ) | (65 | ) | 65 | (a) | (593,371 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (18,827,388 | ) | $ | (52,292 | ) | $ | 52,292 | $ | (18,827,388 | ) | |||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per common share—basic and diluted |
$ | (2.43 | ) | $ | (0.01 | ) | $ | — | $ | (2.43 | ) | |||||
| Weighted-average common shares outstanding—basic and diluted |
7,737,250 | 5,000,000 | — | 7,737,250 | ||||||||||||
Unaudited Pro Forma Condensed Combined Statement of Operations
for the Period Ended December 31, 2012
of
Intra-Cellular Therapies, Inc. and Oneida Resources Corp.
| Intra-Cellular Therapies, Inc. |
Oneida Resources Corp. |
Pro Forma Adjustments |
Pro Forma Combined |
|||||||||||||
| Total revenues |
$ | 3,117,991 | $ | — | $ | — | $ | 3,117,991 | ||||||||
| Total costs and expenses |
19,521,401 | (337 | ) | 337 | (a) | 19,521,401 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(16,403,410 | ) | 337 | (337 | ) | (16,403,410 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Other expense |
(187,417 | ) | — | — | (247,417 | ) | ||||||||||
| (60,000 | )(b) | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (16,590,827 | ) | $ | 337 | $ | (60,337 | ) | $ | (16,650,827 | ) | |||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per common share—basic and diluted |
$ | (2.96 | ) | $ | — | $ | — | $ | (2.97 | ) | ||||||
| Weighted-average common shares outstanding—basic and diluted |
5,607,539 | 2,800,000 | — | 5,607,539 | ||||||||||||
34
Table of Contents
Notes to Unaudited Pro Forma Condensed Combined Statements of Operations
as of September 30, 2013 and December 31, 2012
of
Intra-Cellular Therapies, Inc. and Oneida Resources Corp.
(1) DESCRIPTION OF TRANSACTIONS AND BASIS OF PRESENTATION:
Pursuant to the Merger Agreement, Merger Sub merged with and into ITI, with ITI remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. The Merger was effective on August 29, 2013, upon the filing of a Certificate of Merger with the Secretary of State of the State of Delaware. As part of the Merger, ITI changed its name to ITI, Inc.
At the Effective Time of the Merger, the legal existence of Merger Sub ceased and each share of ITI common stock and each share of ITI preferred stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of the Company’s common stock. The Company issued an aggregate of 22,134,647 shares of common stock upon such exchange of the outstanding shares of ITI common stock and preferred stock.
Further, ITI’s officers and directors became the officers and directors of the Company and the Company adopted the business plan of ITI. ITI is the accounting acquirer (legal acquiree) and the Company is the accounting acquiree (legal acquirer).
Since at completion of the Merger, the Company was a shell corporation, the transaction is being accounted for as a capital transaction. In addition, on August 29, 2013, ITI completed the Private Placement, in which ITI issued 18,889,307 shares of ITI common stock resulting in net proceeds of approximately $40 million. The Statements of Operations for the year ended December 31, 2012 and the nine months ended September 30, 2013 are presented as if the Merger and Private Placement occurred at the beginning of the period.
(2) PRO FORMA ADJUSTMENTS:
(a) To record the elimination of the Company’s operating expenses.
(b) To record the payment of $60,000 to redeem the shares of common stock of the Company outstanding immediately prior to the Merger.
35
Table of Contents
Overview
We were originally incorporated in the State of Delaware in August 2012 under the name “Oneida Resources Corp.” Prior to the Merger, Oneida Resources Corp. was a “shell” company registered under the Exchange Act, with no specific business plan or purpose until it began operating the business of ITI through the Merger transaction on August 29, 2013. ITI was incorporated in Delaware in May 2001 to focus primarily on the development of novel drugs for the treatment of neuropsychiatric and neurologic diseases and other disorders of the central nervous system. Effective upon the Merger, a wholly-owned subsidiary of the Company merged with and into ITI, and ITI continues as the operating subsidiary of the Company. As used herein, the words the “Company,” “we,” “us,” and “our” refer to the current Delaware corporation operating the business of ITI as a wholly-owned subsidiary, which business will continue as the business of the Company.
We are a biopharmaceutical company focused on the discovery and clinical development of innovative, small molecule drugs that address underserved medical needs in neuropsychiatric and neurological disorders by targeting intracellular signaling mechanisms within the central nervous system, or CNS. Our lead product candidate, ITI-007, is in clinical development as a first-in-class treatment for schizophrenia. Current medications available for the treatment of schizophrenia do not adequately address the broad array of symptoms associated with this CNS disorder. Use of these current medications also is limited by their substantial side effects. ITI-007 is designed to be effective across a wider range of symptoms, treating both the acute and residual phases of schizophrenia, with improved safety and tolerability.
ITI-007 exhibited antipsychotic efficacy in a randomized, double-blind, placebo and active controlled Phase 2 clinical trial in patients with an acutely exacerbated episode of schizophrenia. In December 2013, we announced the clinical results from this Phase 2 trial. In this Phase 2 trial, 335 patients were randomized to receive one of four treatments: 60 mg of ITI-007, 120 mg of ITI-007, 4 mg of risperidone (active control) or placebo in a 1:1:1:1 ratio, orally once daily for 28 days. The primary endpoint for this clinical trial was change from baseline to Day 28 on the Positive and Negative Syndrome Scale, or PANSS, total score. In this study, ITI-007 met the trial’s pre-specified primary endpoint, improving symptoms associated with schizophrenia as measured by a statistically significant and clinically meaningful decrease in the PANSS total score. The trial also met key secondary outcome measures related to efficacy on PANSS subscales and safety. Additional data from the Phase 2 trial are set forth below in the section entitled “—Our Clinical Programs—ITI-007 Program—ITI-007 for the treatment of exacerbated and residual schizophrenia—Phase 2 Clinical Trial (ITI-007-005).” We plan to request a meeting with the FDA to discuss our clinical development plan for ITI-007, including our plan to conduct separate, but overlapping, well-controlled clinical efficacy trials in schizophrenia and bipolar disorder.
We are also pursuing clinical development of ITI-007 for the treatment of additional CNS diseases and disorders. At the lowest doses, ITI-007 has been demonstrated to act primarily as a potent 5-HT2A serotonin receptor antagonist. As the dose is increased, additional benefits are derived from the engagement of additional drug targets, including modest dopamine receptor modulation and modest inhibition of serotonin transporters. We believe that combined interactions at these receptors may provide additional benefits above and beyond selective 5-HT2A antagonism for treating agitation, aggression and sleep disturbances in diseases that include dementia, Alzheimer’s disease and autism spectrum disorders, while avoiding many of the side effects associated with more robust dopamine receptor antagonism. As the dose of ITI-007 is further increased, leading to moderate dopamine receptor modulation, inhibition of serotonin transporters, and indirect glutamate modulation, these actions complement the complete blockade of 5-HT2A serotonin receptors. In this dose range, ITI-007 has been shown effective in treating the symptoms associated with schizophrenia, and we believe will be useful for the treatment of bipolar disorder, major depressive disorder and other neuropsychiatric diseases.
Given the potential utility for ITI-007 and follow-on compounds to treat these additional indications, we may investigate, either on our own or with a partner, agitation, aggression and sleep disturbances in diseases that include dementia, Alzheimer’s disease and autism spectrum disorders; major depressive disorder; intermittent explosive disorder; non-motor symptoms and motor complications associated with Parkinson’s disease; and
36
Table of Contents
post-traumatic stress disorder. We hold exclusive, worldwide commercialization rights to ITI-007 and a family of compounds from Bristol-Myers Squibb Company pursuant to an exclusive license.
We have a second major program that has yielded a portfolio of compounds that selectively inhibits the enzyme phosphodiesterase 1, or PDE1. PDE1 helps regulate brain activity related to cognition, memory processes and movement/coordination. We have licensed the lead compound in this portfolio, ITI-214, and other compounds in this portfolio, to Takeda Pharmaceutical Company Limited, or Takeda. ITI-214 is the first compound in its class to successfully advance into Phase 1 clinical trials and is being developed for the treatment of cognitive impairment associated with schizophrenia, or CIAS, and other disorders. The results of our first Phase 1 clinical trial in 70 subjects in a randomized, double-blind, placebo-controlled study indicate that ITI-214 was safe and well-tolerated across a broad range of single oral doses. Other compounds in the PDE1 portfolio outside the Takeda collaboration are being advanced for the treatment of other indications, including non-CNS therapeutic areas.
Our pipeline also includes pre-clinical programs that are focused on advancing drug candidates for the treatment of cognitive dysfunction, in both schizophrenia and Alzheimer’s disease, and for disease modification and the treatment of neurodegenerative disorders, including Alzheimer’s disease.
We have assembled a management team with significant industry experience to lead the discovery and development of our product candidates. We complement our management team with a group of scientific and clinical advisors that includes recognized experts in the fields of schizophrenia and other central nervous system disorders, including Nobel Laureate, Dr. Paul Greengard, one of our co-founders.
Our corporate headquarters and laboratory are located at 3960 Broadway, New York, New York, and our telephone number is (212) 923-3344. We also have an office in Towson, Maryland. We maintain a website at www.intracellulartherapies.com, to which we regularly post copies of our press releases as well as additional information about us. Our filings with the SEC will be available free of charge through the website as soon as reasonably practicable after being electronically filed with or furnished to the SEC. Information contained in our website does not constitute a part of this prospectus or our other filings with the SEC.
All brand names or trademarks appearing in this prospectus are the property of their respective holders. Use or display by us of other parties’ trademarks, trade dress, or products in this prospectus is not intended to, and does not, imply a relationship with, or endorsements or sponsorship of, us by the trademark or trade dress owners.
Our Strategy
Our goal is to discover and develop novel small molecule therapeutics for the treatment of CNS diseases in order to improve the lives of people suffering from such illnesses. Using our key understanding of intracellular signaling, we seek to accomplish our goal, using our in-house expert drug discovery and clinical development teams, in two ways:
| • | we seek to have the capability to develop first-in-class medications with novel mechanisms that have the potential to treat CNS diseases for which there are no previously marketed drugs; and |
| • | we seek to develop drugs that can differentiate themselves in competitive markets by addressing aspects of CNS disease which are either not treated by currently marketed drugs or can be effective with fewer side effects. |
The key elements of our strategy are to:
| • | complete the development of ITI-007 for its lead indication, treatment of acute symptoms in schizophrenia, and for additional neuropsychiatric indications, such as bipolar disorder and residual symptoms in schizophrenia; |
37
Table of Contents
| • | expand the commercial potential of ITI-007 by investigating its usefulness in neurological areas, such as behavioral disturbances in dementia, including Alzheimer’s disease and autism spectrum disorder, and in additional neuropsychiatric indications, such as sleep disorders associated with neuropsychiatric and neurological disorders and major depressive disorder; |
| • | continue to develop with our collaboration partner, Takeda, PDE inhibitor compounds, such as ITI-214, for CNS indications such as CIAS; and |
| • | advance earlier stage product candidates in our pipeline. |
Our Drug Discovery Platform and Capabilities
Based on the pioneering efforts of ITI co-founder and Nobel laureate, Dr. Paul Greengard, we have developed a detailed understanding of intracellular signaling pathways and intracellular targets. We have used that knowledge to develop several state of the art technology platforms, including one called CNSProfileTM. This technology monitors the phosphoprotein changes elicited by major psychotropic drug classes and subclasses, and generates a unique molecular signature for drug compounds. By monitoring how the levels of these phosphoproteins change in vivo, we identify intracellular signaling pathways through which several major drug classes operate. Along with what we believe to be state of the art drug discovery efforts, we have used, and may continue to use, this information as a tool to validate our selection of preclinical candidate molecules.
During the years ended December 31, 2012 and 2011, we incurred $15.5 million and $7.7 million in research and development expenses, respectively.
Disease and Market Overview
Our programs for small molecule therapeutics are designed to address various CNS diseases that we believe are underserved or unmet by currently available therapies and that represent large potential commercial market opportunities for us. Background information on the CNS diseases and related commercial markets that may be addressed by our programs is set forth below.
Schizophrenia
Schizophrenia is a disabling and chronic mental illness that is characterized by multiple symptoms during an acute phase of the disorder that can include so-called “positive” symptoms, such as hearing voices, grandiose beliefs and suspiciousness or paranoia. These symptoms can be accompanied by additional, harder to treat symptoms, such as social withdrawal, blunted emotional response and speech deficits, collectively referred to as “negative” symptoms, difficulty concentrating and disorganized thoughts, or cognitive impairment, depression and insomnia. Such residual symptoms often persist even after the acute positive symptoms subside, and contribute substantially to the social and employment disability associated with schizophrenia. Current antipsychotic medications provide some relief for the symptoms associated with the acute phase of the disorder, but they do not effectively treat the residual phase symptoms associated with chronic schizophrenia. Currently available medications used to treat acute schizophrenia are limited in their use due to side effects that can include movement disorders, weight gain, metabolic disturbances, and cardiovascular disorders. There is an unmet medical need for new therapies that have improved side effect and efficacy profiles.
According to the National Institute of Mental Health, over 1% of the world’s population suffers from schizophrenia, and more than 3 million Americans suffer from the illness in any given year. Worldwide sales of antipsychotic drugs used to treat schizophrenia and other CNS related disorders exceeded $40 billion in 2012. These drugs have been increasingly used by physicians to address a range of disorders in addition to schizophrenia, including bipolar disorder and a variety of psychoses and related conditions in elderly patients. Despite their commercial success, current antipsychotic drugs have substantial limitations, including inadequate efficacy and severe side effects.
38
Table of Contents
The first-generation, or typical, antipsychotics that were introduced in the late-1950s block dopamine receptors. While typical antipsychotics are effective against positive symptoms of schizophrenia in many patients, these drugs often induce disabling motor disturbances, and they fail to address or worsen most of the negative symptoms and cognitive disturbances associated with schizophrenia.
Most schizophrenia patients in the United States are treated today with second-generation, or atypical, antipsychotics, which induce fewer motor disturbances than typical antipsychotics, but still fail to address most of the negative symptoms of schizophrenia and other symptoms associated with social function impairment. Many patients with schizophrenia have deficits in social function. Social function is the ability to recognize, understand, process and use external cues to solve problems, maintain work performance, and conduct interpersonal relationships. Deficits in social function often remain after positive symptoms, such as hallucinations and delusions, have resolved in these patients. In addition, currently prescribed treatments do not effectively address or may exacerbate cognitive disturbances associated with schizophrenia. It is believed that the efficacy of atypical antipsychotics is due to their interactions with dopamine and 5-HT2A receptors. The side effects induced by the atypical agents may include weight gain, non-insulin dependent (type II) diabetes, cardiovascular side effects, sleep disturbances, and motor disturbances. We believe that these side effects generally arise either from non-essential receptor interactions or from excessive dopamine blockade.
The limitations of currently available antipsychotics result in poor patient compliance. A landmark study funded by the National Institute of Mental Health, the Clinical Antipsychotic Trials of Intervention Effectiveness, also referred to as CATIE, which was published in The New England Journal of Medicine in September 2005, found that 74% of patients taking typical or atypical antipsychotics discontinued treatment within 18 months because of side effects or lack of efficacy. We believe there is a large underserved medical need for new therapies that have improved side effect and efficacy profiles.
Bipolar Disorder
Bipolar disorder, commonly referred to as manic-depressive illness, is characterized by extreme shifts in mood. Individuals with bipolar disorder may experience intense feelings of over-excitement, irritability, and impulsivity with grandiose beliefs and racing thoughts, referred to as a manic episode. Symptoms of depression may include feeling tired, hopeless and sad, with difficulty concentrating and thoughts of suicide. Some people experience both types of symptoms in the same “mixed” episode. Severe symptoms of bipolar disorder can be associated with hallucinations or delusions, otherwise referred to as psychosis.
Bipolar disorder affects 4.4% of the adult United States population, or approximately 13 million adults, with a worldwide prevalence of 2.4%. In 2012, therapeutics used to treat bipolar disorder had global sales of approximately $6 billion.
Bipolar disorder is often treated with antipsychotic medications alone or in combination with mood stabilizers. The side effects and safety risks associated with antipsychotic drugs in patients with bipolar disorder are similar to those experienced by patients with schizophrenia. Moreover, a large national research program conducted from 1998 to 2005 called the Systematic Treatment Enhancement Program for Bipolar Disorder, or STEP-BD, followed 4,360 patients with bipolar disorder long term and showed that about half of patients who were treated for bipolar disorder still experienced lingering and recurrent symptoms, indicating a clear need for improved treatments.
Alzheimer’s Disease
Alzheimer’s disease is a progressive neurodegenerative disorder that slowly destroys memory and thinking skills, and eventually even the ability to carry out simple tasks. Its symptoms include cognitive dysfunction, memory abnormalities, progressive impairment in activities of daily living, and a host of behavioral and neuropsychiatric symptoms. Alzheimer’s disease primarily affects older people and, in most cases, symptoms first appear after age 60. Alzheimer’s disease gets worse over time and is fatal.
39
Table of Contents
The market for Alzheimer’s disease therapeutics is categorized into two segments: acetylcholinesterase inhibitors and NMDA receptor antagonists, which include Aricept®, Namenda®, Exelon® and Ebixa®. Acetylcholinesterase inhibitors, which account for 40% of the total worldwide market, had total sales of $4.1 billion in 2011. In 2012, global sales of CNS therapeutics for dementia and Alzheimer’s disease reached $8 billion.
According to the Alzheimer’s Association, 5.2 million people in the United States are living with Alzheimer’s disease, and it is currently the fifth leading cause of death for people age 65 and older. It has been estimated that 35.6 million people worldwide were living with dementia in 2010. This number is expected to nearly double to 65.7 million by 2030 and to 115.4 million by 2050. While the diagnostic criteria for Alzheimer’s disease mostly focus on the related cognitive deficits, it is often the behavioral and psychiatric symptoms that are most troublesome for caregivers and lead to poor quality of life for patients. These symptoms include agitation, aggressive behaviors, and psychosis. Studies have suggested that approximately 20% to 51% of Alzheimer’s disease patients may develop psychosis, commonly consisting of hallucinations and delusions. The diagnosis of Alzheimer’s disease psychosis is associated with more rapid cognitive and functional decline and institutionalization.
The FDA has not approved any drug to treat the behavioral symptoms of Alzheimer’s disease. As symptoms progress and become more severe, physicians often resort to off-label use of antipsychotic medications in these patients. Current antipsychotic drugs are associated with a number of side effects, which can be problematic for elderly patients with Alzheimer’s disease. In addition, antipsychotic drugs may exacerbate the cognitive disturbances associated with Alzheimer’s disease. Current antipsychotic drugs also have a boxed warning for use in elderly patients with dementia-related psychosis due to increased mortality and morbidity. There is a large unmet medical need for a safe and effective therapy to treat the behavioral symptoms in patients with Alzheimer’s disease.
Parkinson’s Disease
Parkinson’s disease is a chronic and progressive neurodegenerative disorder that involves malfunction and death of neurons in a region of the brain that controls movement. This neurodegeneration creates a shortage of an important brain signaling chemical, or neurotransmitter, known as dopamine, thereby rendering patients unable to direct or control their movements in a normal manner. Parkinson’s disease is characterized by well-known motor symptoms, including tremors, limb stiffness, slowness of movements, and difficulties with posture and balance, as well as by non-motor symptoms, which include sleep disturbances, mood disorders, cognitive impairment and psychosis. Parkinson’s disease progresses slowly in most people and the severity of symptoms tends to worsen over time.
Parkinson’s disease is the second most common neurodegenerative disorder after Alzheimer’s disease. According to the National Parkinson Foundation, about 1 million people in the United States and from approximately 4 to 6 million people worldwide suffer from this disease. Parkinson’s disease is more common in people over 60 years of age, and the prevalence of this disease is expected to increase significantly as the average age of the population increases. Parkinson’s disease patients are commonly treated with dopamine replacement therapies, such as levodopa, commonly referred to as L-DOPA, which is metabolized to dopamine, and dopamine agonists, which are molecules that mimic the action of dopamine. Sales of therapeutics such as L-DOPA and dopamine agonists used to treat the motor symptoms of the disease reached $2.5 billion in 2012.
Non-motor symptoms can be particularly distressing and even more troublesome to patients with Parkinson’s disease than the primary motor disturbances. Non-motor symptoms substantially contribute to the burden of Parkinson’s disease and deeply affect the quality of life of patients and their caregivers. Non-motor symptoms of Parkinson’s disease are associated with increased caregiver stress and burden, nursing home placement, and increased morbidity and mortality.
Treatment of non-motor symptoms associated with Parkinson’s disease poses a challenge to physicians. Current dopamine replacement drugs used to treat the motor symptoms of Parkinson’s disease do not help, and sometimes worsen, the non-motor symptoms. No drugs are currently approved by the FDA for treating the broad non-motor symptoms associated with Parkinson’s disease, and this remains a large unmet medical need.
40
Table of Contents
Depression
Major depressive disorder, or MDD, is a brain disorder that can be associated with symptoms of sadness, hopelessness, helplessness, feelings of guilt, irritability, loss of interest in formerly pleasurable activities, cognitive impairment, disturbed sleep patterns, and suicide ideation or behavior. Different people may experience different symptoms, but everyone with major depression experiences symptoms that are severe enough to interfere with everyday functioning, such as the ability to concentrate at work or school, social interactions, eating and sleeping. Sometimes the depressive episode can be so severe it is accompanied by psychosis (hallucinations and delusions). According to the National Institute of Mental Health, approximately 3% of teenagers and approximately 7% of adults experience MDD each year. Worldwide sales of antidepressant drugs reached $11.9 billion in 2011. The antidepressant market is primarily composed of selective serotonin reuptake inhibitors such as Lexapro® (marketed by Forest Laboratories and Lundbeck) and selective norepinephrine reuptake inhibitors, or SNRIs, such as Cymbalta® (marketed by Eli Lilly). Antipsychotics such as Seroquel® (marketed by Astrazeneca) and Abilify® (marketed jointly by Bristol Myers Squibb and Otsuka Pharmaceutical) are also used as adjunctive treatments with antidepressant treatment. The National Institute of Mental Health-funded Sequenced Treatment Alternatives to Relieve Depression, or STAR*D, study showed that only one-third of treated patients experience complete remission of depressive symptoms. Nearly two-thirds of patients were considered treatment-resistant.
Our Clinical Programs
Our pipeline includes two product candidates in clinical development and two product candidates in advanced pre-clinical testing. We believe that our product candidates offer innovative therapeutic approaches and may provide significant advantages relative to current therapies. The following table summarizes our product candidates and programs:
41
Table of Contents
ITI-007 Program
Our lead product candidate, ITI-007, possesses mechanisms of action that we believe have the potential to yield a first-in-class antipsychotic therapy. In our pre-clinical and clinical trials to date, ITI-007 combines potent serotonin 5-HT2A receptor antagonism, dopamine receptor phosphoprotein modulation (DPPM), glutamatergic modulation and serotonin reuptake inhibition into a single drug candidate for the treatment of acute and residual schizophrenia. At dopamine D2 receptors, ITI-007 has been demonstrated to have dual properties and to act as both a post-synaptic antagonist and a pre-synaptic partial agonist. ITI-007 has also been demonstrated to stimulate phosphorylation of glutamatergic NMDA NR2B, or GluN2B, receptors in a mesolimbic specific manner. We believe that this regional selectivity in brain areas thought to mediate the efficacy of antipsychotic drugs, together with serotonergic, glutamatergic, and dopaminergic interactions, may result in antipsychotic efficacy for positive, negative, affective and cognitive symptoms associated with schizophrenia. The serotonin reuptake inhibition could allow for antidepressant activity for the treatment of schizoaffective disorder, co-morbid depression, and/or as a stand-alone treatment for major depressive disorder. We believe ITI-007 may also be useful for the treatment of bipolar disorder and other psychiatric and neurodegenerative disorders, particularly behavioral disturbances associated with dementia, autism and other CNS diseases.
We believe these features of ITI-007 may be able to improve the quality of life of patients with schizophrenia and enhance social function to allow them to more fully integrate into their families and their workplace. In addition, ITI-007 may be shown to treat disorders at either low-doses (e.g., sleep, aggression and agitation) or high-doses (e.g., acute exacerbated and residual schizophrenia, bipolar disorders, and mood disorders).
Phase 1 studies to support multiple clinical indications
We have conducted a series of Phase 1 safety studies of ITI-007 in Europe and the United States during the period from 2007 to 2011. All of the studies conducted to date in the United States have been conducted under an Investigational New Drug, or IND, filed in 2007 by ITI. Data from these studies are being used to support the clinical development of ITI-007 in multiple indications, including acute exacerbated schizophrenia, sleep disorders in neuropsychiatric and neurodegenerative disease, major depressive disorders, bipolar disorders, behavioral disturbances in dementia and Alzheimer’s disease, autism, posttraumatic stress disorder, or PTSD, and intermittent explosive disorder, or IED. We have completed the following three Phase 1 trials in healthy volunteers:
| • | A Phase 1, double-blind placebo controlled, single ascending dose study in 40 healthy volunteers in Europe in 2007. ITI-007 was generally well tolerated at all doses. Most adverse events, or AEs, were mild to moderate and all treatment related AEs resolved. The most frequent AE was headache. |
| • | A Phase 1, placebo controlled multiple ascending dose study in 25 healthy volunteers in Europe from 2007 to 2008. ITI-007 was generally well tolerated at all doses. Most AEs were mild to moderate and all treatment related AEs resolved. |
| • | A Phase 1, open-label positron emission tomography, or PET, study to demonstrate receptor occupancy, safety, tolerability and pharmacokinetics after single oral dose administration of ITI-007 in 16 healthy male volunteers. This study was conducted in the United States from 2007 to 2009. ITI-007 was well tolerated, all AEs were of mild or moderate intensity and all treatment related AEs resolved. Dose related increases in receptor occupancy at dopamine D2 receptors in the striatum were demonstrated after ITI-007 administration. Brain occupancy at 5-HT2A and serotonin reuptake transporters also was demonstrated after singles doses of ITI-007. |
We continued Phase 1 development of ITI-007 in patients with schizophrenia in order to advance ITI-007 in this target therapeutic indication. Specifically, we conducted the following additional studies:
| • | A Phase1b/2, placebo controlled multiple ascending dose study in 45 patients with stable schizophrenia in the United States during 2009 to 2010. ITI-007 was generally well tolerated at all doses. All AEs were mild to moderate and all treatment related AEs resolved. The overall percentage of patients reporting |
42
Table of Contents
| treatment related AEs was similar for those treated with ITI-007 (83.3% to 100%, across dose groups) and placebo (72.7%). The majority of the treatment related AEs that occurred at the commencement of the study decreased in terms of frequency and/or severity with repeated administration. We observed signs consistent with clinical efficacy in stable patients with schizophrenia in this study. |
| • | A Phase 1, randomized study to determine the tolerability, safety and pharmacokinetics of ITI-007 using different dosing regimens in 11 patients with schizophrenia. This study was conducted in the United States in 2011. In this study, we showed that administration of ITI-007 in a capsule dosage form taken with food reduced the incidence of treatment related AEs and all treatment related AEs resolved. The most commonly reported treatment related AE in this study was somnolence, commonly known as drowsiness. |
ITI-007 for the treatment of exacerbated and residual schizophrenia
In multiple clinical trials of ITI-007 in patients with schizophrenia, the drug candidate has demonstrated clinical signals consistent with reductions in psychosis, depression and insomnia. Reductions in psychosis are consistent with the potential to treat acute schizophrenia, whereas reductions in depression and insomnia are consistent with the potential to treat residual phase schizophrenia. ITI-007 has been shown to be safe and well-tolerated across a wide range of doses in these studies. Further, at doses that have demonstrated clinical activity, ITI-007 has caused fewer adverse effects than those typically associated with antipsychotic drug treatment, such as impaired motor function. These adverse side effects can be a major cause of patient noncompliance with current antipsychotic therapies.
Phase 2 Clinical Trial (ITI-007-005)
ITI-007 exhibited antipsychotic efficacy in ITI-007-005, a randomized, double-blind, placebo and active controlled Phase 2 clinical trial in patients with an acutely exacerbated episode of schizophrenia. In December 2013, we announced the clinical results from this Phase 2 trial. In this Phase 2 trial, 335 patients were randomized to receive one of four treatments: 60 mg of ITI-007, 120 mg of ITI-007, 4 mg of risperidone (active control) or placebo in a 1:1:1:1 ratio. Patients received study treatment orally once daily in the morning for 28 days. Of those randomized, 311 patients were included in the intent-to-treat primary analysis. Subject participation lasted approximately 7 to 8 weeks, including a one week screening period, a four week treatment period followed by stabilization on standard of care, and a safety follow up visit approximately two weeks after stabilization. The primary endpoint for this clinical trial was change from baseline to Day 28 on the Positive and Negative Syndrome Scale, or PANSS, total score. The PANSS is a well-validated 30-item rating scale that measures the ability of a drug to reduce schizophrenia symptom severity. The PANSS measures positive symptoms, such as delusions, suspiciousness, and hallucinations; negative symptoms, such as blunted affect, social and emotional withdrawal, and stereotyped thinking; and general psychopathology, such as anxiety, tension, depression, and active social avoidance.
Secondary endpoints in this trial included weekly assessments of the PANSS total score as well as its subscales (Positive Symptom Subscale, Negative Symptom Subscale, and General Psychopathology Subscale) and the Negative Symptom Factor (based on a subset of PANSS questions), individual item response on the PANSS, and the Calgary Depression Scale for Schizophrenia. Safety and tolerability were also assessed.
In December 2013, we announced that topline results from the ITI-007-005 study indicated that ITI-007 met the trial’s pre-specified primary endpoint, improving symptoms associated with schizophrenia as measured by a statistically significant and clinically meaningful decrease in the PANSS total score. The trial also met key secondary outcome measures related to efficacy on PANSS subscales and safety.
Many patients with schizophrenia have deficits in social function. Social function is the ability to recognize, understand, process and use external cues to solve problems, maintain work performance and conduct interpersonal relationships. Deficits in social function often remain after positive symptoms, such as
43
Table of Contents
hallucinations and delusions, have resolved in these patients. In the Phase 2 trial, ITI-007 exhibited a differentiating response profile across a broad range of symptoms that we believe is consistent with improvements in these social functioning deficits. The study also showed that ITI-007 was well-tolerated at the tested doses. ITI-007 demonstrated a favorable safety profile in the study without characteristic antipsychotic drug side effects or any serious adverse events.
ITI-007 at a dose of 60 mg demonstrated a statistically significant improvement in psychosis (p = 0.017) on the trial’s pre-specified primary endpoint, which was change from baseline on the PANSS total score, compared to placebo. The primary statistical analysis was pre-specified and used a Mixed-Effect Model Repeated Measure method for handling missing data in the intent-to-treat (ITT) study population and a Bonferroni procedure to correct for multiple two-sided comparisons (each dose of ITI-007 compared to placebo). The trial’s pre-specified sensitivity analysis on the primary endpoint used the analysis of covariance (ANCOVA) model and last observation carried forward (LOCF) method for handling missing data for the ITT population and confirmed the positive outcome with statistically significant improvements compared to placebo in patients receiving the 60 mg dose of ITI-007 (p = 0.011). ITI-007 at a dose of 60 mg also significantly improved the positive symptom subscale (p < 0.05) and the general psychopathology subscale (p < 0.05) on the PANSS after 28 days of treatment using the ANCOVA-LOCF on the ITT population.
The improvement in the PANSS total score in the 120 mg dose group did not reach statistical significance. We believe that it is possible that sedation, the most frequent side effect in the 120 mg dose group, interfered with the ability to detect an efficacy signal at this dose administered once daily in the morning. Approximately 32.5% of subjects randomized to 120 mg ITI-007 experienced sedation/somnolence, compared to 21% of subjects randomized to risperidone, 17% of subjects randomized to 60 mg ITI-007, and 13% randomized to placebo. We believe that nighttime administration may be more appropriate for testing the effectiveness of the 120 mg dose of ITI-007 in this patient population. In the trial, the 60 mg dose of ITI-007 was effective when administered once daily in the morning.
Consistent with preliminary indications from the interim analysis and with the drug’s pharmacological profile, ITI-007 at a dose of 60 mg significantly improved certain items on the negative symptom and general psychopathology subscales consistent with improved social function. The study was statistically powered only on the primary endpoint. ITI-007 did significantly improve many secondary endpoints, although the study was not designed for significance on secondary endpoints and was not powered to detect statistical differences in subgroup analyses. Additional secondary endpoints, including depression, continue to be analyzed, and we expect that data will be presented at upcoming scientific conferences.
A high percentage (74%) of randomized subjects completed trial participation. Only 19% of subjects discontinued from study treatment during the 28 day study treatment period, and an additional 7% of subjects completed study treatment but were lost to follow up.
In the Phase 2 trial, ITI-007 was well-tolerated. The most frequent adverse event was sedation, as described above. There were no serious adverse events related to ITI-007. There were no clinically meaningful changes in safety measures with ITI-007. Notably, ITI-007 demonstrated a favorable metabolic profile with no increase of blood levels of glucose, insulin, cholesterol or triglycerides over a four week treatment period. Moreover, in contrast to risperidone, 60 mg of ITI-007 was effective with no difference from placebo on weight change parameters, prolactin levels, extrapyramidal symptoms (EPS) or akathisia. ITI-007 was not associated with EPS as measured by the Simpson-Angus Scale, Barnes Akathisia Rating Scale, or Abnormal Involuntary Movement Scale. There was no increase in suicidal ideation or behavior with ITI-007.
Subject to discussions with the FDA, we intend to initiate Phase 3 trials in schizophrenia in the second half of 2014 and plan to initiate separate additional trials in bipolar disorder in 2015. We expect that the planned trials in bipolar disorder will overlap in time with the clinical conduct of the planned trials in schizophrenia. We have not yet discussed our plans to develop ITI-007 for the treatment of bipolar disorder with the FDA. With the
44
Table of Contents
recent completion of the ITI-007-005 Phase 2 trial in schizophrenia, we plan to request a meeting with the FDA to discuss our clinical development plan for ITI-007, including our plan to conduct separate, but overlapping, well-controlled clinical efficacy trials in schizophrenia and bipolar disorder. The FDA may not agree with our clinical development plans to advance ITI-007 for the treatment of schizophrenia and bipolar disorder with separate, but overlapping, well-controlled clinical trials in both indications. Our clinical plans may change based on any discussions with the FDA. If the FDA does not agree with our clinical development plans for ITI-007, our development of ITI-007 may be delayed and the costs of our development of ITI-007 would increase, which may have an adverse effect on our business, financial condition and results of operations.
ITI-007 for the treatment of bipolar disorder
The pharmacological profile of ITI-007 offers the potential to treat bipolar mania, depression, and mixed symptoms at doses similar to those targeted for the treatment of schizophrenia. We believe that ITI-007 may be effective alone or in combination with mood stabilizers. Given that many patients with bipolar disorder also experience disturbed sleep and cognitive impairment similar to that observed in schizophrenia, we believe that ITI-007 may treat a wide array of symptoms in patients with bipolar disorder, including improvement of cognition and sleep. We expect that data from our completed Phase 1 studies and data from our Phase 2 trial in acute exacerbated schizophrenia will be used to advance ITI-007 directly into well-controlled clinical trials for the treatment of bipolar disorder. Based on the successful completion of our Phase 2 trial in acute exacerbated schizophrenia, subject to discussions with the FDA, we intend to initiate Phase 3 trials in schizophrenia in the second half of 2014 and plan to initiate separate additional trials in bipolar disorder in 2015. We expect that the planned trials in bipolar disorder will overlap in time with the clinical conduct of the planned trials in schizophrenia. We have not yet discussed our plans to develop ITI-007 for the treatment of bipolar disorder with the FDA. With the recent completion of the ITI-007-005 Phase 2 trial in schizophrenia, we plan to request a meeting with the FDA to discuss our clinical development plan for ITI-007, including our plan to conduct separate, but overlapping, well-controlled clinical efficacy trials in schizophrenia and bipolar disorder.
The FDA may not agree with our clinical development plans to advance ITI-007 for the treatment of schizophrenia and bipolar disorder with separate, but overlapping, well-controlled clinical trials in both indications. Our clinical plans may change based on any discussions with the FDA. If the FDA does not agree with our clinical development plans for ITI-007, our development of ITI-007 may be delayed and the costs of our development of ITI-007 would increase, which may have an adverse effect on our business, financial condition and results of operations.
ITI-007 for the treatment of sleep disturbances associated with neurologic and psychiatric disorders
In a Phase 2 double-blind, placebo controlled cross-over clinical trial conducted in 19 patients at low doses completed in 2008 and conducted in Europe. The primary outcome measure was slow wave sleep as determined by polysomnography. ITI-007 demonstrated a dose-related statistically significant increase in slow wave sleep. Secondary measures were consistent with improvement of sleep maintenance in patients with primary insomnia, indicated by decreased waking after sleep onset, increased total sleep time, and no increase in latency to sleep onset. At these low doses ITI-007 did not induce sleep, but rather helped maintain sleep once sleep had been initiated. In addition, ITI-007 was not associated with next day cognitive impairment, or “hang-over” effects. We believe that ITI-007 may be particularly useful in the treatment of sleep disorders that accompany neuropsychiatric and neurologic disorders, including schizophrenia, autism spectrum disorder, or ASD, Parkinson’s disease and dementia. Previous work has suggested that selective 5-HT2A receptor antagonists increase deep, slow wave sleep in both humans and animals. We believe, however, that other neuropharmacological mechanisms, in addition to 5-HT2A receptor antagonism, such as engaging some dopamine modulation, may be beneficial for the successful treatment of sleep maintenance insomnia, or SMI, in humans. We believe that ITI-007 represents a new approach to the treatment of sleep maintenance insomnia because of its unique pharmacology and neuropharmacological interactions beyond selective 5-HT2A receptor antagonism. We believe that ITI-007 offers a potentially new approach to the treatment of sleep maintenance
45
Table of Contents
disorders, particularly in those disorders that accompany neuropsychiatric and neurologic disorders. Many of these disorders are accompanied by profound sleep deficits, which impair daytime functioning including cognition, exacerbate disease symptoms and increase the cost of care. We are presently exploring clinical designs to incorporate the examination of sleep disturbances in one or more of these indications. There is no assurance that any such design would be sufficient for an FDA approval for this indication.
ITI-007 for the treatment of behavioral disturbances associated with dementia, including Alzheimer’s disease
Behavioral disturbances are common in dementia and Alzheimer’s disease. These disturbances are a major component of the burden to caregivers, and often lead to institutionalization. Although currently available treatments for patients with dementia mainly address cognitive disturbances, behavioral disturbances are considerably more problematic and likely more amenable to drug treatment. Several behavioral symptoms are quite prevalent in patients with dementia, including patients with Alzheimer’s disease. Rates of depression in Alzheimer’s disease are estimated to be up to 87%, although most estimates are between 30% and 50%. Agitation and aggression are present in approximately 60% of patients. Sleep disturbances, particularly as an increased likelihood of day-night reversal, are present in up to approximately 60% of patients. In view of the potential multiple effects of ITI-007 on aggression, agitation, sleep disorders and depression, and its safety profile to date, we believe that ITI-007 may provide a novel therapy for treating the behavioral disturbances accompanying dementia, including Alzheimer’s disease. Clinical trials are planned to address the therapeutic utility of ITI-007 for the treatment of behavioral disturbances in dementia and Alzheimer’s disease. We believe that our completed Phase 1 studies support advancing ITI-007 into Phase 2 trials in this patient population, although we may conduct additional Phase 1 studies to better define the dose range in elderly subjects and patients with dementia.
ITI-007 for the treatment of sleep and behavioral disturbances associated with autism spectrum disorder
Sleep problems are common in patients with autism spectrum disorder, or ASD, and are not adequately treated by currently available interventions. Approximately two thirds of children and adolescents with ASD experience sleep problems, higher than the rate of sleep problems in age-matched developmentally typical children. Moreover, individuals with ASD suffer from behavioral disturbances, including aggression, irritability, anxiety and depression. With its multiple pathway mechanism of action, we believe that ITI-007 could address the multi-faceted behavioral symptoms associated with ASD. 5-HT2A receptor antagonism is predicted to increase slow wave sleep, improve sleep maintenance and reduce aggression. D2 receptor modulation is predicted to improve sleep maintenance and reduce irritability and aggression. Serotonin reuptake inhibition is predicted to reduce anxiety and depression. Accordingly, we believe that ITI-007 could improve sleep maintenance, reduce behavioral disturbances and enhance social interaction in patients with ASD. We believe that our completed Phase 1 studies support advancing ITI-007 into Phase 2 trials in this patient population, and we are presently exploring the feasibility of such trials.
ITI-007 for the treatment of depression and other mood disorders
As a potent 5-HT2A receptor antagonist and serotonin reuptake inhibitor, we believe that ITI-007 could improve symptoms of depression with fewer side effects than selective serotonin reuptake inhibitors, or SSRIs. Dopamine modulation by ITI-007 may reduce irritability and aggression that can accompany many mood disorders. As such, ITI-007 may be effective for the treatment of mood disorders including major depressive disorder, or MDD, posttraumatic stress disorder, or PTSD, and intermittent explosive disorder, or IED. We are presently exploring the feasibility of clinical studies in these indications.
ITI-002 (PDE1) Program
We have a second major program, called our ITI-002 program, that has generated a portfolio of compounds that have demonstrated the ability to modulate CNS pathways that are critical to controlling cognition and motor
46
Table of Contents
behavior through the inhibition of an important intracellular enzyme, PDE1. In March 2011, we entered into a license and collaboration agreement with Takeda to develop and commercialize selected PDE1 inhibitors in our ITI-002 program for the treatment of CIAS and other disorders, including Parkinson’s disease, cognitive impairment in Alzheimer’s disease, and Attention Deficit Hyperactivity Disorder. Cognitive deficits are believed to underlie much of the significant functional impairments observed in patients with schizophrenia. One of these portfolio compounds, ITI-214, has advanced into Phase 1 clinical studies. In the first quarter of 2013, we announced the completion by Takeda of a single ascending dose Phase 1 study in 70 healthy volunteers in the United States under an IND filed by Takeda in 2012. Takeda will be solely responsible for development, manufacturing and commercialization of PDE1 inhibitors. The results of this randomized, double-blind, placebo-controlled Phase 1 study indicated that ITI-214 was safe and well-tolerated across a broad range of single oral doses. Moreover, the study demonstrated a favorable pharmacokinetic profile of ITI-214 consistent with once-a-day dosing. We believe that this study represents a significant milestone as the first use of a potent and highly specific PDE1 inhibitor in humans. We have worked closely with Takeda since 2011 to advance ITI-214 into clinical development and to optimize select backup/follow-on compounds for treating other CNS diseases, including Parkinson’s disease, cognitive impairment in Alzheimer’s disease and attention deficit and hyperactivity disorders. We believe that inhibition of PDE1 may also be beneficial in a number of therapeutic indications outside of CNS diseases, such as pulmonary arterial hypertension, heart failure, muscular dystrophy and inflammatory disease. We are pursuing additional ITI-002 PDE1 inhibitor compounds outside the scope of the Takeda collaboration for the treatment of cardiovascular and other disorders.
Additional PDE Programs
There are multiple forms and isoforms of PDE with distinct roles in intracellular signaling. We have developed strong internal expertise in the design and synthesis of inhibitors specific for individual PDE isoforms. Based on our understanding of the expression and functions of these isoforms in the CNS, we have identified PDE2 and PDE9 as compelling targets for drug discovery. We believe that inhibitors of these PDEs may be useful in treating neurodegeneration and bioenergetic failure in a variety of CNS diseases.
Alzheimer’s disease—ITI-012 (Casein Kinase 1 Inhibitors) and ITI-009 (gSAP Inhibitors)
We are pursuing early stage drug discovery programs targeting two different pathways thought to be involved in the pathogenesis of Alzheimer’s disease. The first program targets the enzyme casein kinase 1, or CK1, the misregulation of which in Alzheimer’s disease may provoke misfolding of a neuronal protein, tau, which has been linked to cellular loss in the brains of patients with Alzheimer’s disease. We are currently optimizing our CK1 inhibitors in anticipation of advancing them into preclinical development. We have a second program targeting the protein Gamma Secretase Activating Protein, or gSAP. We have demonstrated in preclinical models that inhibiting gSAP lowers the level of a toxic protein located in the brain called Abeta. Scientists in the field of dementia and Alzheimer’s disease believe that inhibiting the accumulation of Abeta may slow the onset of Alzheimer’s disease. The discovery of gSAP was made by ITI in collaboration with Dr. Paul Greengard, Nobel Laureate and ITI co-founder. The preclinical characterization of this class of molecules is ongoing. We believe that these compounds have the potential to provide novel, disease-modifying treatments for Alzheimer’s disease and related disorders.
Intellectual Property
Our Patent Portfolio
As of December 1, 2013, we owned or controlled approximately 60 patent families filed in the United States and other major markets worldwide, including approximately 29 issued or allowed U.S. patents, 43 pending U.S. patent applications, 98 issued foreign patents, and 332 foreign patent applications, directed to novel compounds, formulations, methods of treatment, synthetic methods, and platform technologies.
47
Table of Contents
Our ITI-007 program on novel compounds for neuropsychiatric and neurodegenerative diseases includes patents exclusively in-licensed from Bristol Myers Squibb on families of compounds, including the ITI-007 lead molecule. We have extensively characterized this lead and filed additional patent applications on polymorphs, formulations, additional indications, derivatives and additional compounds. The ITI-007 lead molecule has composition of matter protection through 2025 and additional Orange Book-listable protection to 2034. Additionally, we expect to have data exclusivity in the European Union for up to 11 years from launch. We also have a follow-on program, directed to compounds structurally related to the ITI-007 lead, but having composition of matter protection beyond 2031.
Our program on PDE1 inhibitors for cognition and dopamine-mediated disorders, such as Parkinson’s disease, includes patent protection for the lead, ITI-214, as well as a wide range of filings on other proprietary compounds and indications. Certain PDE1 inhibitors are being developed under a joint development agreement with Takeda, under which we received an upfront cash payment and are eligible to receive payments for development and sales, as well as royalty payments. We also have an option to co-promote with Takeda in the U.S., and we retain certain rights to PDE1 inhibitor compounds and uses outside the scope of that collaboration. The ITI-214 lead molecule has composition of matter protection to 2029, with possible extensions and additional Orange Book-listable protection to 2034. Additionally, we expect to have data exclusivity in the European Union for up to 11 years from commercial launch. We are also evaluating potential follow-on compounds for ITI-214 which would have patent protection beyond 2030.
We have also filed patent applications on novel proprietary targets and lead compounds for Alzheimer’s disease, which would provide compound protection beyond 2028 or beyond 2034, depending on which compound is ultimately selected for development.
License Agreement
The Bristol-Myers Squibb License Agreement
On May 31, 2005 we entered into a world-wide, exclusive License Agreement with Bristol-Myers Squibb Company, or BMS, pursuant to which we hold a license to certain patents and know-how of BMS relating to ITI-007 and other specified compounds. The agreement was amended on November 3, 2010. The licensed rights are exclusive, except BMS retains rights in specified compounds in the fields of obesity, diabetes, metabolic syndrome and cardiovascular disease. However, BMS has no right to use, develop or commercialize ITI-007 and other specified compounds in any field of use. We have the right to grant sublicenses of the rights conveyed by BMS. We are obliged under the license to use commercially reasonable efforts to develop and commercialize the licensed technology. We are also prohibited from engaging in the clinical development or commercialization of specified competitive compounds.
Under the agreement, we made an upfront payment of $1.0 million to BMS. Under the agreement, we may be obliged to make milestone payments to BMS for each licensed product of up to an aggregate of approximately $14.8 million, which includes a milestone payment of $1.25 million that we intend to make in connection with our recently completed Phase 2 trial. We are also obliged to make tiered single digit percentage royalty payments on sales of licensed products. We are obliged to pay to BMS a percentage of non-royalty payments made in consideration of any sublicense.
The agreement extends, and royalties are payable, on a country-by-country and product-by-product basis, through the later of ten years after first commercial sale of a licensed product in such country, expiration of the last licensed patent covering a licensed product, its method of manufacture or use, or the expiration of other government grants providing market exclusivity, subject to certain rights of the parties to terminate the agreement on the occurrence of certain events. On termination of the agreement, we may be obliged to convey to BMS rights in developments relating to a licensed compound or licensed product, including regulatory filings, research results and other intellectual property rights.
48
Table of Contents
Collaboration Agreement
The Takeda Pharmaceutical License and Collaboration Agreement
On February 25, 2011, we entered into a license and collaboration agreement with Takeda Pharmaceutical Company Limited under which we agreed to collaborate to research, develop and commercialize our proprietary compound ITI-214 and other selected compounds that selectively inhibit PDE1 for use in the prevention and treatment of human diseases. As part of the agreement, we assigned to Takeda certain patents owned by us that claim ITI-214 and granted Takeda an exclusive license to develop and commercialize compounds identified in the conduct of the research program that satisfy specified criteria. However, we have retained rights to all compounds that do not meet the specified criteria and we continue to develop PDE1 inhibitors outside the scope of the agreement.
Under the terms of the agreement, we are conducting a research program with an initial term of three years to identify and characterize compounds that meet certain specified criteria sufficient for further development by Takeda. We were responsible for our expenses incurred in the conduct of certain research activities specified in the research plan. Takeda has agreed to reimburse us for expenses we incur in conducting additional research activities.
Takeda is obliged to use commercially reasonable efforts to develop and commercialize licensed compounds at its expense, and has agreed to reimburse us for the costs and expenses of development activities we may perform. We have formed a joint steering committee with Takeda to coordinate and oversee activities on which we collaborate under the agreement. We have the option to co-promote any licensed product in the United States by assuming responsibility for a certain percentage of the detailing activity with respect to that product.
We are responsible for supplying Takeda with ITI-214 for nonclinical activities and phase 1 clinical trials at our expense. Takeda is responsible, at its expense, for the manufacture and supply of compounds that it develops and commercializes under the agreement for all other activities.
Upon execution of the agreement, Takeda made a nonrefundable payment to us. We are eligible to receive payments of approximately $500,000,000 in the aggregate upon achievement of certain development milestones and up to an additional $250,000,000 in the aggregate upon achievement of certain sales-based milestones, along with tiered royalty payments ranging from the high single digits to the low teens in percent based on net sales by Takeda.
The agreement extends, on a country-by-country and product-by-product basis, through the later of expiration of the last licensed patent covering a licensed product, its method of manufacture or use, the expiration of other government grants providing market exclusivity or ten years after first commercial sale of a licensed product in such country, subject to rights of the parties to sooner terminate the agreement on certain events and the right of Takeda to unilaterally terminate the agreement upon a specified number of days’ prior notice. Upon the termination of the agreement, Takeda is obliged to assign to us the patents covering ITI-214 assigned to Takeda upon the execution of the agreement, to grant us a license to develop and commercialize licensed compounds developed by Takeda and to transfer to us certain materials, information and regulatory materials reasonably necessary for us to continue the development and commercialization of those compounds.
Manufacturing
We do not own or operate manufacturing facilities for the production of any of our product candidates, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently rely on one third-party contract manufacturer for all of our required raw materials, active pharmaceutical ingredient, or API, and finished product for our preclinical research and clinical trials, including the Phase 2 trial for ITI-007 for the treatment of schizophrenia. We believe that we would be able to contract with another third-party contract manufacturer to obtain API if our existing source of API was no longer available, but there is no assurance that API would be available from another third-party manufacturer on acceptable terms, on the timeframe that our business
49
Table of Contents
would require, or at all. We do not have long-term agreements with our existing third-party contract manufacturer. We also do not have any current contractual relationships for the manufacture of commercial supplies of any of our product candidates if they are approved. As ITI-007 and any of our other product candidates continue to progress towards potential regulatory approval, we intend to enter into agreements with a third-party contract manufacturer and one or more back-up manufacturers for the commercial production of those products. Development and commercial quantities of any products that we develop will need to be manufactured in facilities, and by processes, that comply with the requirements of the FDA and the regulatory agencies of other jurisdictions in which we are seeking approval. We currently employ internal resources to manage our manufacturing contractors.
Sales and Marketing
We currently have no marketing, sales or distribution capabilities. In order to commercialize any of our product candidates, we must develop these capabilities internally or through collaboration with third parties. In selected therapeutic areas where we feel that our product candidates can be commercialized by a specialty sales force that calls on a limited and focused group of physicians, we may plan to participate in the commercialization of our product candidates in the United States. In therapeutic areas that require a large sales force selling to a large and diverse prescribing population, we may elect to commercialize through, or in collaboration with, strategic partners. We may choose to commercialize our products in markets outside of the United States by establishing one or more strategic alliances in the future.
Competition
We face, and will continue to face, intense competition from pharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies, both in the United States and abroad. We compete, or will compete, with existing and new products being developed by our competitors. Some of these competitors are pursuing the development of pharmaceuticals that target the same diseases and conditions that our research and development programs target.
Even if we are successful in developing our product candidates, the resulting products would compete with a variety of established drugs in the areas of our targeted CNS therapeutic indications. Our potential products for the treatment of schizophrenia and bipolar disorder would compete with, among other branded products, Abilify®, marketed jointly by Bristol-Myers Squibb and Otsuka Pharmaceutical; Fanapt®, marketed by Novartis Pharmaceuticals; Seroquel XR®, marketed by AstraZeneca; Invega®, marketed by Janssen; and Latuda®, marketed by Sunovion. In addition, our product candidates, if approved, will compete with, among other generic antipsychotic products, haloperidol, risperidone, quetiapine, olanzapine and clozapine.
In addition, the companies described above and other competitors may have a variety of drugs in development or awaiting FDA approval that could reach the market and become established before we have a product to sell. Our competitors may also develop alternative therapies that could further limit the market for any drugs that we may develop. Many of our competitors are using technologies or methods different or similar to ours to identify and validate drug targets and to discover novel small molecule drugs. Many of our competitors and their collaborators have significantly greater experience than we do in the following:
| • | identifying and validating targets; |
| • | screening compounds against targets; |
| • | preclinical and clinical trials of potential pharmaceutical products; and |
| • | obtaining FDA and other regulatory clearances. |
In addition, many of our competitors and their collaborators have substantially greater advantages in the following areas:
| • | capital resources; |
| • | research and development resources; |
50
Table of Contents
| • | manufacturing capabilities; and |
| • | sales and marketing. |
Smaller companies also may prove to be significant competitors, particularly through proprietary research discoveries and collaborative arrangements with large pharmaceutical and established biotechnology companies. Many of our competitors have products that have been approved by the FDA or are in advanced development. We face competition from other companies, academic institutions, governmental agencies and other public and private research organizations for collaborative arrangements with pharmaceutical and biotechnology companies, in recruiting and retaining highly qualified scientific and management personnel and for licenses to additional technologies. Our competitors, either alone or with their collaborators, may succeed in developing technologies or drugs that are more effective, safer, and more affordable or more easily administered than ours and may achieve patent protection or commercialize drugs sooner than us. Developments by others may render our product candidates or our technologies obsolete. Our failure to compete effectively could have a material adverse effect on our business.
Government Regulation
United States—FDA Process
The research, development, testing, manufacture, labeling, promotion, advertising, import and export, distribution and marketing, among other things, of drug products are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, fines, civil penalties, product recalls, product seizures, total or partial suspension of production or distribution, injunctions and/or criminal prosecution.
Drug Approval Process. None of our drug product candidates may be marketed in the United States until the drug has received FDA approval. Such approval can take many years to obtain and may be rejected by the FDA at a number of steps. The steps required before a drug may be marketed in the United States generally include the following:
| • | completion of extensive pre-clinical laboratory tests, animal studies, and formulation studies in accordance with the FDA’s Good Laboratory Practice, or GLP, regulations; |
| • | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each proposed indication; |
| • | submission to the FDA of an NDA after completion of all clinical trials; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with current Good Manufacturing Practices, or cGMPs; |
| • | satisfactory completion of FDA inspections of clinical trial sites to assure that data supporting the safety and effectiveness of product candidates has been generated in compliance with Good Clinical Practices; and |
| • | FDA review and approval of the NDA prior to any commercial marketing or sale of the drug in the United States. |
The development and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
51
Table of Contents
Pre-clinical tests include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies. The conduct of the pre-clinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the pre-clinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about the conduct of the trial, such as whether human research subjects will be exposed to an unreasonable health risk. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. We cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin.
Clinical trials involve administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be provided to the FDA as part of a separate submission to the IND. Further, an Institutional Review Board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the study protocol and informed consent information for study subjects for any clinical trial before it commences at that center, and the IRB must monitor the study until it is completed. There are also requirements governing reporting of on-going clinical trials and clinical trial results to public registries. Study subjects must sign an informed consent form before participating in a clinical trial.
Clinical trials necessary for product approval typically are conducted in three sequential phases, but the phases may overlap.
| • | Phase 1 usually involves the initial introduction of the investigational drug into a limited population, typically healthy humans, to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness. |
| • | Phase 2 usually involves trials in a limited patient population to (i) evaluate dosage tolerance and appropriate dosage; (ii) identify possible adverse effects and safety risks; and (iii) evaluate preliminarily the efficacy of the drug for specific targeted indications. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| • | Phase 3 trials, commonly referred to as pivotal studies, are undertaken in an expanded patient population at multiple, geographically dispersed clinical trial centers to further evaluate clinical efficacy and test further for safety by using the drug in its final form. There can be no assurance that Phase 1, Phase 2 or Phase 3 testing will be completed successfully within any specified period of time, if at all. Furthermore, we, the FDA or an IRB may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Moreover, the FDA may approve an NDA for a product candidate, but require that the sponsor conduct additional clinical trials to further assess the drug after NDA approval under a post-approval commitment. Post-approval trials are typically referred to as Phase 4 clinical trials. |
During the development of a new drug, sponsors are given an opportunity to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach an agreement on the next phase of development. Sponsors typically use the end of Phase 2 meeting to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support approval of the new drug. A sponsor may request a Special Protocol Assessment, or SPA, to reach an agreement with the FDA that the protocol design, clinical endpoints, and statistical analyses are acceptable to support regulatory approval of the product candidate with respect to effectiveness in the indication
52
Table of Contents
studied. If such an agreement is reached, it will be documented and made part of the administrative record, and it will be binding on the FDA except in limited circumstances, such as if the FDA identifies a substantial scientific issue essential to determining the safety or effectiveness of the product after clinical studies begin, or if the sponsor fails to follow the protocol that was agreed upon with the FDA. There is no guarantee that a study will ultimately be adequate to support an approval even if the study is subject to an SPA.
Concurrent with clinical trials, companies usually complete additional animal safety studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and the manufacturer must develop methods for testing the quality, purity and potency of the final drugs. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
Assuming successful completion of the required clinical testing, the results of pre-clinical studies and of clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. An NDA must be accompanied by a significant user fee, which is waived for the first NDA submitted by a qualifying small business. In July 2012, the Food and Drug Administration Safety and Innovation Act, or FDASIA, was signed into law. Among other things, FDASIA reauthorizes the FDA’s authority to collect user fees from industry participants to fund reviews of innovator drugs.
The testing and approval process requires substantial time, effort and financial resources. The FDA will review the NDA and may deem it to be inadequate to support approval, and we cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but it typically follows such recommendations.
Before approving an NDA, the FDA inspects the facility or the facilities at which the drug and/or its active pharmaceutical ingredient is manufactured and will not approve the product unless the manufacturing is in compliance with cGMPs. If the FDA evaluates the NDA and the manufacturing facilities are deemed acceptable, the FDA may issue an approval letter, or in some cases a Complete Response Letter. The approval letter authorizes commercial marketing of the drug for specific indications. As a condition of NDA approval, the FDA may require post-marketing testing and surveillance to monitor the drug’s safety or efficacy, or impose other conditions. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or additional clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, pre-clinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials is not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Alternatively, the FDA could also approve the NDA with a Risk Evaluation and Mitigation Strategy to mitigate risks of the drug, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. Once the FDA approves a drug, the FDA may withdraw product approval if on-going regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase 4 clinical trials, and surveillance programs to monitor the safety effects of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
Post-Approval Requirements. After a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. In addition, certain changes to an approved product, such as adding new indications, making certain manufacturing
53
Table of Contents
changes, or making certain additional labeling claims, are subject to further FDA review and approval. Before a company can market products for additional indications, it must obtain additional approvals from the FDA, typically a new NDA. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. A company cannot be sure that any additional approval for new indications for any product candidate will be approved on a timely basis, or at all.
If post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA are required to (i) report certain adverse reactions to the FDA and maintain pharmacovigilance programs to proactively look for these adverse events; (ii) comply with certain requirements concerning advertising and promotional labeling for their products; and (iii) continue to have quality control and manufacturing procedures conform to cGMPs after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities, which includes assessment of on-going compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. We intend to use third-party manufacturers to produce our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA, including recall of the product from the market or withdrawal of approval of the NDA for that drug.
Patent Term Restoration and Marketing Exclusivity. Depending upon the timing, duration and specifics of FDA approval of the use of our drugs, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the extension must be requested prior to expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Data and market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent data exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct, or obtain a right of reference to all of the pre-clinical studies, adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
54
Table of Contents
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials and approval of foreign countries or economic areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
In the European Economic Area, or EEA, which is comprised of the 27 member states of the European Union, or Member States, plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of MAs:
| • | Community MAs—These are issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and are valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA; for products that constitute a significant therapeutic, scientific or technical innovation; or for products that are in the interest of public health in the European Union. |
| • | National MAs—These are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, and are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure, an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State. The competent authority of the Reference Member State prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling or packaging proposed by the Reference Member State, the product is subsequently granted a National MA in all the Member States (i.e., in the Reference Member State and the Member States Concerned). |
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA assess the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
As in the United States, it may be possible in foreign countries to obtain a period of market and/or data exclusivity that would have the effect of postponing the entry into the marketplace of a competitor’s generic product. For example, if any of our products receive marketing approval in the EEA, we expect they will benefit from eight years of data exclusivity and ten years of marketing exclusivity. An additional non-cumulative one-year period of marketing exclusivity is possible if during the data exclusivity period (the first eight years of the 10-year marketing exclusivity period), we obtain an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies. The data exclusivity period begins on the date of the product’s first marketing authorization in the European Union and prevents generics from relying on the marketing authorization holder’s pharmacological, toxicological and clinical data for a period
55
Table of Contents
of eight years. After eight years, a generic product application may be submitted and generic companies may rely on the marketing authorization holder’s data. However, a generic cannot launch until two years later (or a total of 10 years after the first marketing authorization in the European Union of the innovator product), or three years later (or a total of 11 years after the first marketing authorization in the European Union of the innovator product) if the marketing authorization holder obtains marketing authorization for a new indication with significant clinical benefit within the eight-year data exclusivity period. In Japan, our products may be eligible for eight years of data exclusivity. There can be no assurance that we will qualify for such regulatory exclusivity, or that such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies.
When conducting clinical trials in the European Union, we must adhere to the provisions of the European Union Clinical Trials Directive and the laws and regulations of the European Union Member States implementing them. These provisions require, among other things, that the prior authorization of an Ethics Committee and the competent Member State authority is obtained before commencing the clinical trial.
Pricing and Reimbursement
In the United States and internationally, sales of products that we market in the future, and our ability to generate revenues on such sales, are dependent, in significant part, on the availability of adequate coverage and reimbursement from third-party payors, such as state and federal governments, managed care providers and private insurance plans. Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and the out-of-pocket obligations of member patients for such products. We may need to conduct pharmacoeconomic studies to demonstrate the cost-effectiveness of our products for formulary coverage and reimbursement. Even with such studies, our products may be considered less safe, less effective or less cost-effective than existing products, and third-party payors may not provide coverage and reimbursement for our product candidates, in whole or in part.
In addition, particularly in the United States and increasingly in other countries, we are required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our products that are reimbursed by such entities. It is possible that future legislation in the United States and other jurisdictions could be enacted to potentially impact reimbursement rates for the products we are developing and may develop in the future and could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market.
Political, economic and regulatory influences are subjecting the health care industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the health care system in ways that could significantly affect our future business. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, the PPACA, enacted in March 2010, substantially changes the way health care is financed by both governmental and private insurers. Among other cost containment measures, PPACA establishes:
| • | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; |
| • | a new Medicare Part D coverage gap discount program, in which pharmaceutical manufacturers who wish to have their drugs covered under Part D must offer discounts to eligible beneficiaries during their coverage gap period, or the donut hole; and |
| • | a new formula that increases the rebates a manufacturer must pay under the Medicaid Drug Rebate Program. |
56
Table of Contents
In addition, other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. The ATRA, among other things, also reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Sales and Marketing
The FDA regulates all advertising and promotion activities for products under its jurisdiction prior to and after approval, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to collect additional data or conduct additional pre-clinical studies and clinical trials. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, and often reflect a physician’s belief that the off-label use is the best treatment for the patient. The FDA does not regulate the behavior of physicians in their choice of treatments, but FDA regulations do impose stringent restrictions on manufacturers’ communications regarding off-label uses. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
Outside the United States, our ability to market a product is contingent upon obtaining marketing authorization from the appropriate regulatory authorities. The requirements governing marketing authorization, pricing and reimbursement vary widely from country to country.
We may also be subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations and very few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented, for payment to third-party payors (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws.
Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal health care programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties also may be imposed upon executive
57
Table of Contents
officers and employees, including criminal sanctions against executive officers under the so-called “responsible corporate officer” doctrine, even in situations where the executive officer did not intend to violate the law and was unaware of any wrongdoing. Given the penalties that may be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the government was to allege or convict us or our executive officers of violating these laws, our business could be harmed. In addition, private individuals have the ability to bring similar actions. Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. Further, there are an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state authorities.
Employees
As of December 1, 2013, we employed 21 employees, 20 of whom were full-time. To successfully develop our drug candidates, we must be able to attract and retain highly skilled personnel. We anticipate hiring additional employees for research and development, clinical and regulatory affairs and general and administrative activities over the next few years. In addition, we intend to use clinical research organizations and third parties to perform our clinical studies and manufacturing.
Properties
Our headquarters are located at 3960 Broadway, New York, New York 10032, where we occupy approximately 13,000 square feet of office and laboratory space. The term of the lease expires September 30, 2014, and we have the option to extend the term of the lease for one additional year, until September 30, 2015. We also lease office space in Towson, Maryland on a month to month basis.
Legal Proceedings
We are not currently involved in any material legal proceedings.
58
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL
CONDITION AND RESULTS OF OPERATIONS
The following discussion of the financial condition and results of operations of Intra-Cellular Therapies, Inc. and its wholly-owned subsidiary should be read in conjunction with the financial statements and the notes to those statements appearing at the end of this prospectus. Some of the information contained in this discussion and analysis or set forth elsewhere in this prospectus, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. You should read the “Risk Factors” section of this prospectus for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
Effective as of August 29, 2013, we consummated a reverse merger with the privately held biopharmaceutical company formerly named “Intra-Cellular Therapies, Inc.,” and changed our name from “Oneida Resources Corp.” to “Intra-Cellular Therapies, Inc.” The privately held company survived the merger as a wholly owned subsidiary of ours and changed its name to ITI, Inc.
We are a biopharmaceutical company focused on the discovery and clinical development of innovative, small molecule drugs that address underserved medical needs in neuropsychiatric and neurological disorders by targeting intracellular signaling mechanisms within the central nervous system. Our lead product candidate, ITI-007, is in Phase 2 clinical trials as a first-in-class treatment for schizophrenia. Results from the Phase 2 trial are included in “Description of Our Business—Our Clinical Programs—ITI-007 Program.” Subject to discussions with the FDA, we intend to initiate Phase 3 trials in schizophrenia in the second half of 2014 and plan to initiate separate additional trials in bipolar disorder in 2015. We expect that the planned trials in bipolar disorder will overlap in time with the clinical conduct of the planned trials in schizophrenia. We believe that ITI-007 and follow-on compounds have utility to treat additional indications, which we may investigate, either on our own or with a partner. We hold exclusive, worldwide commercialization rights to ITI-007 and a family of related compounds from Bristol-Myers Squibb Company.
We have a second major program called ITI-002 that has yielded a portfolio of compounds that selectively inhibits the enzyme PDE1. We have licensed the lead compound in the ITI-002 portfolio, ITI-214, and other compounds in that portfolio, to Takeda. ITI-214 is the first compound in its class to successfully advance into Phase 1 clinical trials and is being developed for the treatment of cognitive impairment associated with schizophrenia and other disorders.
Our pipeline also includes preclinical programs that are focused on advancing drugs for the treatment of cognitive dysfunction, in both schizophrenia and Alzheimer’s disease, and for disease modification and the treatment of neurodegenerative disorders, including Alzheimer’s disease.
Since inception, we have devoted all of our efforts and resources to our research and development activities. We have incurred significant net losses since inception. As of September 30, 2013, our accumulated deficit was $49.5 million. We expect to continue incurring substantial losses for the next several years as we continue to develop our clinical and pre-clinical drug candidates and programs. Our operating expenses are comprised of research and development expenses and general and administrative expenses.
We have not generated any revenue from product sales to date and we do not expect to generate revenues from product sales for at least the next several years. Our revenues for the fiscal years ended December 31, 2012 and 2011 have been primarily from a license and collaboration agreement with Takeda, and, to a much lesser extent, from grants from U.S. government agencies and foundations in 2011 only. Prior to 2011, our revenue was entirely from grants from these agencies and foundations.
Our corporate headquarters and research facility are located in New York, New York.
59
Table of Contents
Recent Developments
Private Placement
Prior to the Merger, which we describe below, on August 29, 2013, ITI sold to accredited investors approximately $60.0 million of its shares of common stock, or 18,889,307 shares, at a price of $3.1764 per share, which included approximately $15.3 million in principal and $0.8 million in accrued interest from the conversion of ITI’s then outstanding convertible promissory notes, or Notes, and which resulted in net proceeds, after expenses, of approximately $40.0 million. We refer to this transaction as the Private Placement. Also, ITI granted the investors in the Private Placement registration rights requiring ITI or any successor to register those shares of ITI common stock (which were exchanged for shares of our common stock, along with the rest of the outstanding shares of ITI capital stock, except for dissenting shares, at the Effective Time) for public resale, as described in more detail below. The then existing stockholders of ITI who agreed to become parties to the registration rights agreement also became entitled to such registration rights, subject to specified differences in the agreement between the rights of new investors and existing stockholders. The existing Second Amended and Restated Investor Rights Agreement, by and among ITI and the investors listed therein, dated as of October 25, 2007, as amended, was terminated at the Effective Time. The Private Placement closed immediately prior to the filing of a Certificate of Merger with the Secretary of State of the State of Delaware on August 29, 2013.
Reverse Merger
On August 29, 2013, pursuant to the Merger Agreement, Merger Sub merged with and into ITI, with ITI remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. The Merger was effective on August 29, 2013, upon the filing of a Certificate of Merger with the Secretary of State of the State of Delaware. At the Effective Time of the Merger, the name of ITI was changed to ITI, Inc. Immediately following the Effective Time, the Company completed the Name Change Merger by which the Company changed its name to “Intra-Cellular Therapies, Inc.”
At the Effective Time, the legal existence of Merger Sub ceased and each share of ITI common stock and each share of ITI preferred stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock. We issued an aggregate of 22,134,647 shares of our common stock upon such exchange of the outstanding shares of ITI common stock and preferred stock. In addition, at the Effective Time, we assumed ITI’s 2003 Equity Incentive Plan, as amended, or the 2003 Equity Incentive Plan, and all options to purchase ITI common stock then outstanding under the 2003 Equity Incentive Plan, and such options became exercisable for an aggregate of 1,462,380 shares of our common stock, subject to the vesting and other terms of such options. The vesting of such options was not accelerated as a result of the Merger. At the Effective Time, we also assumed the outstanding warrant to purchase ITI common stock, and such warrant became exercisable for 1,822 shares of our common stock.
Immediately following the Effective Time, pursuant to the terms of the Redemption Agreement, we completed the closing of a redemption of 5,000,000 shares of our common stock from our then-current sole stockholder in consideration of $60,000, plus professional costs related to the transaction that were approximately $20,000. The 5,000,000 shares constituted all of the issued and outstanding shares of our capital stock, on a fully-diluted basis, immediately prior to the Merger.
In accordance with Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 805, Business Combinations, ITI is considered the acquirer for accounting purposes, and will account for the transaction as a capital transaction, because ITI’s former stockholders received 100% of the voting rights in the combined entity and ITI’s senior management represents all of the senior management of the combined entity. Consequently, the assets and liabilities and the historical operations that will be reflected in our consolidated financial statements will be those of ITI and will be recorded at the historical cost basis of the Company.
60
Table of Contents
Results of Operations
Revenues
The following discussion summarizes the key factors our management believes are necessary for an understanding of our financial statements.
We have not generated any revenue from product sales to date and we do not expect to generate revenues from product sales for at least the next several years. Our revenues for fiscal years ended December 31, 2012 and 2011 have been primarily from the license and collaboration agreement with Takeda, and, to a much lesser extent, from grants from U.S. government agencies and foundations in 2011 only. Prior to 2011, our revenue was entirely from grants from these agencies and foundations.
The revenue from Takeda has been comprised primarily of an upfront payment, a milestone payment and reimbursements for costs incurred in the development of and patent prosecutions for compounds subject to the collaboration. The upfront payment was evaluated and it was determined that there were separate units of accounting for the deliverables that are provided for in the license and collaboration agreement. A larger portion of the upfront payment was considered a license fee, and the remaining portion was deemed to be related to the performance of agreed upon activities under the collaboration component of the license and collaboration agreement. We determined this amount in accordance with ASC Topic 605-25, Revenue Recognition, using best estimate of selling price, for the work that we would be required to perform. We considered multiple factors in estimating this amount, including, but not limited to, direct external expenses and internal costs for salary and related fringes, among others. The straight line method of amortization with a three-year schedule was used and revenue was and will be recognized equally for the years 2011 through 2013. Revenue from the license payment was recognized as earned when received. Revenue from milestone payments is recognized when all of the following conditions are met: (1) the milestone payments are non-refundable, (2) the achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement, (3) substantive effort on our part is involved in achieving the milestone, (4) the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (5) a reasonable amount of time passes between the up-front license payment and the first milestone payment. Reimbursement revenue is recognized when the costs are incurred and the services have been performed.
We expect our revenues for the next several years to consist of limited reimbursable costs incurred for patent prosecutions, amortized revenue in 2013 related to the upfront payment made by Takeda, and reimbursements related to our collaboration with Takeda under the license and collaboration agreement. In addition, we expect to receive possible milestone payments under the license and collaboration agreement, but these are not assured at this time and would not be significant enough to fund operations for a meaningful period of time.
Expenses
The process of researching and developing drugs for human use is lengthy, unpredictable and subject to many risks. We are unable with any certainty to estimate either the costs or the timelines in which those costs will be incurred. We have one project, ITI-007 for the treatment of schizophrenia, which consumes a large proportion of our current, as well as projected, resources. We intend to pursue other disease indications that ITI-007 may address, but there are large costs associated with pursuing FDA approval for those indications, which would include the cost of additional clinical trials. Our other projects, exclusive of the Takeda collaboration, are still in the preclinical stages, and will require extensive funding not only to complete preclinical testing, but to enter into and complete clinical trials. Expenditures that we incur on these projects will be subject to availability of funding in addition to the funding required for the advancement of ITI-007. Any failure or delay in the advancement of ITI-007 could require us to re-allocate resources from our other projects to the advancement of ITI-007, which could have a significant material adverse impact on the advancement of these other projects and on our operations.
61
Table of Contents
Our operating expenses are comprised of (i) research and development expenses and (ii) general and administrative expenses. Our research and development costs are comprised of:
| • | internal recurring costs, such as labor and fringe benefits, materials and supplies, facilities and maintenance costs; and |
| • | fees paid to external parties who provide us with contract services, such as preclinical testing, manufacturing and related testing, and clinical trial activities. |
General and administrative expenses are incurred in three major categories:
| • | salaries and related benefit costs; |
| • | patent, legal and professional costs; and |
| • | office and facilities overhead. |
We expect that our general and administrative costs will increase substantially from prior periods due to the increased costs associated with being a public reporting entity.
The following table sets forth our revenues and operating expenses for the fiscal years ended December 31, 2012 and 2011, and for the three and nine month periods ended September 30, 2013 and 2012:
| For the Year Ended December 31 |
Three Months Ended September 30 |
Nine Months Ended September 30 |
||||||||||||||||||||||
| 2012 | 2011 | 2013 | 2012 | 2013 | 2012 | |||||||||||||||||||
| (Audited) | (Unaudited) | (Unaudited) | ||||||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||
| Revenues |
$ | 3,118 | $ | 23,362 | $ | 668 | $ | 378 | $ | 1,909 | $ | 2,449 | ||||||||||||
| Expenses |
||||||||||||||||||||||||
| Research and Development |
15,486 | 7,655 | 4,158 | 1,797 | 16,898 | 14,970 | ||||||||||||||||||
| General and Administrative |
4,035 | 4,612 | 1,295 | 802 | 3,246 | 2,916 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| 19,521 | 12,267 | 5,453 | 2,599 | 20,144 | 17,886 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net Income (Loss) |
$ | (16,591 | ) | $ | 11,092 | $ | (4,912 | ) | $ | (2,223 | ) | $ | (18,827 | ) | $ | (15,432 | ) | |||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
Comparison of Years Ended December 31, 2012 and December 31, 2011
Revenues
Revenue decreased for the year ended December 31, 2012 as compared to the year ended December 31, 2011 by approximately $20.2 million due primarily to milestone and upfront revenue earned from Takeda in 2011 and, to a much lesser extent, greater reimbursable costs from Takeda and grant revenue in 2011, and no corresponding milestone or upfront revenue or grant revenue in 2012.
Research and Development Expenses
Total research and development expenses were approximately $15.5 million for the fiscal year ended December 31, 2012, as compared to $7.7 million for the fiscal year ended December 31, 2011. This increase of $7.8 million in total research and development expenses is due primarily to an increase of $9.1 million in direct costs for clinical trials. Clinical trial costs for the fiscal year ended December 31, 2011 were $1.5 million. This increase in clinical trial costs was offset in part by lower costs during the fiscal year ended December 31, 2012 to manufacture drug product required for clinical trials and testing, and by lower costs associated with non-clinical drug testing.
The research and development expenses incurred for amounts payable to external parties started to become a larger component of our research and development costs during the fiscal year ended December 31, 2011, and
62
Table of Contents
comprise a significant portion of our research and development spending during the fiscal year ended December 31, 2012, due primarily to the preparation for and commencement of our Phase 2 clinical trial for ITI-007. We incurred expenses of approximately $12.2 million and $4.2 million during the years ended December 31, 2012 and 2011, respectively, for amounts payable to external parties who manufactured, tested and performed clinical trial activities for all of our projects. During the same periods, our internal research and development expenses were approximately $3.3 million and $3.5 million during the years ended December 31, 2012 and 2011, respectively. As of December 1, 2013, we employed 13 full time personnel in our research and development group.
The clinical development work related to ITI-007 requires the largest portion of our resources and, consequently, comprises the majority of our spending. We spent approximately $11.3 million and $2.9 million on direct external costs for the development of ITI-007, exclusive of internal labor and fringes, during the periods ended December 31, 2012 and 2011, respectively. As development of ITI-007 progresses, we anticipate costs for ITI-007 to increase considerably in the next several years as we complete the ongoing Phase 2 clinical trial for ITI-007 and begin other clinical trials. We are also required to complete non-clinical testing to obtain FDA approval and manufacture material needed for clinical trial use, which includes non-clinical testing of the drug product and the creation of an inventory of drug product in anticipation of possible FDA approval.
We currently have several projects in addition to ITI-007 that are in the research and development stages. These are in the areas of cognitive dysfunction and the treatment of neurodegenerative diseases, including Alzheimer’s disease, among others. We have used internal resources and incurred expenses not only in relation to the development of ITI-007 but on these additional projects as well. We have not, however, reported these costs on a project by project basis, as they are broadly spread among these projects. The external costs for these projects have been minimal and are reflected in the amounts discussed in this section “—Research and Development Expenses.” During the years ended December 31, 2012 and 2011, we also incurred costs that were both reimbursable and non-reimbursable under the license and collaboration agreement with Takeda. We incurred approximately $700,000 and $320,000 on direct costs that were billable to Takeda for the years ended December 31, 2012 and 2011, respectively. We anticipate that these costs will be reduced significantly as the research portion of the license and collaboration agreement concludes in February 2014.
General and Administrative Expenses
Salaries and related benefit costs for our executive, finance and administrative functions for 2012 and 2011 constituted slightly less than half of the total general and administrative costs. The next major categories of expenses are patent costs, some of which are reimbursed by Takeda, legal, accounting and other professional fees and, to a lesser extent, facilities and general office-related overhead. We expect all of these costs to increase significantly as we expand our operations and become subject to the reporting requirements of a public company. General and administrative expenses were $4.0 million for the fiscal year ended December 31, 2012 compared to $4.6 million for the fiscal year ended December 31, 2011. The decrease is the result of higher legal, patent and personnel costs in 2011 related to consummating the license and collaboration agreement, offset in part by a decrease in patent costs for other non-Takeda related products.
Comparison of Three and Nine Month Periods Ended September 30, 2013 and September 30, 2012
Revenues
Revenue decreased for the nine months ended September 30, 2013 as compared to the nine months ended September 30, 2012 by approximately $540,000 due primarily to lower reimbursable third party manufacturing costs offset partially by an increase in reimbursable patent costs both associated with the Takeda agreement. Revenue increased for the three months ended September 30, 2013 as compared to the three months ended September 30, 2012 by approximately $290,000 due to similar increases in both the reimbursable third party manufacturing costs and patent costs associated with the Takeda agreement.
63
Table of Contents
Research and Development Expenses
Research and development expenses increased for both the three and nine month periods ended September 30, 2013 as compared to the three and nine month periods ended September 30, 2012 by approximately $2.4 million and $1.9 million, respectively. The increase in the three month period ended September 30, 2013 as compared to the three month period ended September 30, 2012 is due almost exclusively to outside clinical testing. The increase in the nine month period ended September 30, 2013 as compared to the nine month period ended September 30, 2012 is due to approximately $3.2 million of increased spending for outside clinical testing, which is primarily the result of an increase in the number of clinical trial subjects for our Phase 2 trial of ITI-007 in patients with schizophrenia, and is offset partially by lower non-clinical testing of $0.9 million and $0.3 million of lower labor and related costs.
General and Administrative Expenses
General and administrative expenses increased for both the three and nine month periods ended September 30, 2013 as compared to the three and nine month periods ended September 30, 2012 by approximately $493,000 and $330,000, respectively. The increase of $493,000 for the three month period ended September 30, 2013 as compared to the three month period ended September 30, 2012 is primarily the result of an approximately $182,000 increase in patent filing costs, with the remainder comprised primarily of higher professional fees. The $330,000 increase for the nine month period ended September 30, 2013 as compared to the nine month period ended September 30, 2012 is due primarily to a $146,000 increase in professional fees and a $170,000 increase in patent filing costs. Professional fee increases in 2013 are due to the activity associated with being a public reporting company.
Liquidity and Capital Resources
Through September 30, 2013, we have funded our operations with approximately $149.2 million of cash that has been obtained from the following main sources: $40.0 million of net proceeds from the Private Placement which closed on August 29, 2013 (net of $0.5 million of unpaid costs); $25.4 million from other sales of equity; $0.4 million from the exercise of stock options; $15.3 million in sales of convertible promissory notes; $40.6 million from grants from government agencies and foundations; and $27.9 million in total payments received under the license and collaboration agreement with Takeda, including approximately $1.7 million for reimbursement of development costs incurred from 2011 through September 30, 2013, and $1.8 million for patent costs incurred during the same time period. During the nine months ended September 30, 2013, we did not receive any funding through grants. We do not believe that grant revenue will be a significant source of funding in the near future. We expect that reimbursements of our development costs by Takeda will decline going forward, and we do not expect such reimbursements to be a significant source of funding in the future. We also expect the reimbursements for patent filing costs will remain at the same level, but because reimbursements will be offset by the actual expenditures incurred, reimbursements do not represent a net source of funding for us.
In October and November 2012, we issued convertible promissory notes, or Notes, having an aggregate principal amount of approximately $15.2 million. We issued additional Notes having an aggregate principal amount of $0.1 million in March 2013. The Notes were unsecured and accrued interest at a rate of 6% per year, and were originally scheduled to mature on April 25, 2013, but maturity was extended until October 25, 2013. The principal amount of the Notes, together with the accrued interest thereon, converted into shares of ITI common stock at the closing of the private placement described above under “Recent Developments—Private Placement.”
As of December 31, 2012, we had a total of $19.1 million in cash, cash equivalents and certificates of deposit, or CDs, and approximately $2.8 million of short term liabilities from operations. As of September 30, 2013, we had a total of $46.1 million in cash, cash equivalents and CDs, and approximately $7.8 million of short term liabilities consisting of short term liabilities from operations. Excluding the increase in net cash of approximately $40.0 million from the Private Placement which closed in August 2013 (net of $0.5 million of
64
Table of Contents
unpaid costs) and the conversion of $15.2 million of convertible notes outstanding at December 31, 2012, we used $17.3 million in working capital for the nine months ended September 30, 2013. This reduction in working capital is due primarily to the funding of the Phase 2 clinical trial for ITI-007, our lead drug candidate, and to a lesser extent to fund recurring operating expenses, the preparation for additional clinical trials and non-clinical testing. We expect to consume working capital of approximately $7.0 million to $8.5 million for the fourth quarter of 2013. This is expected to be due primarily to recurring expenses and for costs incurred for the completion of the Phase 2 clinical trial and the preparations for additional trials and non-clinical testing related to the development of ITI-007.
We expect that cash to be used in the fourth quarter of 2013 will be between $10.0 million and $13.0 million as we pay down a significant portion of our accounts payable and accrued expenses recorded at September 30, 2013 and use funds for ongoing operations. In connection with the completion of our Phase 2 clinical trial of ITI-007 in patients with acutely exacerbated schizophrenia, we intend to make a milestone payment of $1.25 million to BMS.
Our cash, cash equivalents and investment securities totaled $46.1 million at September 30, 2013. On August 29, 2013, immediately prior to the Merger, ITI sold approximately $60.0 million of its common stock, which included approximately $15.3 million in principal and $0.8 million in accrued interest from the conversion of its then outstanding convertible promissory notes, and which resulted in net proceeds, after expenses, of approximately $40.0 million (net of $0.5 million of unpaid costs). While we believe that our existing cash resources and anticipated payments from our existing collaborations will be sufficient to fund our cash requirements through at least the third quarter of 2014, we will require significant additional financing in the future to continue to fund our operations.
We have incurred losses in every year since inception with the exception of the fiscal year ended December 31, 2011. These losses have resulted in significant cash used in operations. During the fiscal year ended December 31, 2012, our cash used in operations was approximately $18.9 million. During the fiscal year ended December 31, 2011, if we exclude the upfront fee and milestone payments from Takeda, our cash used in operations would have been $7.8 million. This increase of cash used during the fiscal year ended December 31, 2012 is primarily due to the clinical development and clinical trial activities for ITI-007. While we have several research and development programs underway, the ITI-007 program has advanced the furthest and will continue to consume increasing amounts of cash for conducting clinical trials and the testing and manufacturing of product material. As we continue to conduct these activities necessary to pursue FDA approval of ITI-007 and our other product candidates, we expect the cash needed to fund operations to increase significantly over the next several years.
We seek to balance the level of cash, cash equivalents and marketable securities on hand with our projected needs and to allow us to withstand periods of uncertainty relative to the availability of funding on favorable terms. Until we can generate significant revenues from operations, we will need to satisfy our future cash needs through public or private sales of our equity securities, sales of debt securities, the incurrence of debt from commercial lenders, strategic collaborations, licensing a portion or all of our product candidates and technology and, to a lesser extent, grant funding. We cannot be sure that future funding will be available to us when we need it on terms that are acceptable to us, or at all. We sell securities and incur debt when the terms of such transactions are deemed favorable to us and as necessary to fund our current and projected cash needs. The amount of funding we raise through sales of our common stock or other securities depends on many factors, including, but not limited to, the status and progress of our product development programs, projected cash needs, availability of funding from other sources, our stock price and the status of the capital markets. Due to the recent volatile nature of the financial markets and, in particular, the adverse impact on market capitalization and valuation of biotechnology companies, equity and debt financing may be difficult to obtain. In addition, any unfavorable development or delay in the progress for our ITI-007 program could have a material adverse impact on our ability to raise additional capital.
65
Table of Contents
To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our existing stockholders will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of our stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring debt, making capital expenditures or declaring dividends.
If adequate funds are not available to us on a timely basis, we may be required to: (1) delay, limit, reduce or terminate preclinical studies, clinical trials or other clinical development activities for one or more of our product candidates, including our lead candidate ITI-007 as well as our other preclinical stage product candidates; (2) delay, limit, reduce or terminate our discovery research or preclinical development activities; or (3) enter into licenses or other arrangements with third parties on terms that may be unfavorable to us or sell, license or relinquish rights to develop or commercialize our product candidates, technologies or intellectual property at an earlier stage of development and on less favorable terms than we would otherwise agree.
Our cash is maintained in money market accounts and, to a lesser extent, in CDs at major financial institutions. Due to the current low interest rates available for these instruments, we are earning limited interest income. Our investment portfolio has not been adversely impacted by the problems in the credit markets that have existed over the last several years, but there can be no assurance that our investment portfolio will not be adversely affected in the future.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires management to make estimates and assumptions that affect reported amounts of assets and liabilities as of the date of the balance sheet and reported amounts of revenues and expenses for the periods presented. Judgments must also be made about the disclosure of contingent liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Management makes estimates and exercises judgment in revenue recognition and stock-based compensation. Actual results may differ from those estimates and under different assumptions or conditions.
We believe that the following critical accounting policies affect management’s more significant judgments and estimates used in the preparation of our financial statements:
Revenue Recognition
Revenue is recognized when all terms and conditions of the agreements have been met, including persuasive evidence of an arrangement, delivery has occurred or services have been rendered, price is fixed or determinable and collectability is reasonably assured. We are reimbursed for certain costs incurred on specified research projects under the terms and conditions of grants, collaboration agreements, and awards. We record the amount of reimbursement as revenues on a gross basis in accordance with ASC Topic 605-45, Revenue Recognition/Principal Agent Considerations. We are the primary obligor with respect to purchasing goods and services from third-party suppliers, are obligated to compensate the service provider for the work performed, and have discretion in selecting the supplier. Provisions for estimated losses on research grant projects and any other contracts are made in the period such losses are determined.
Effective January 1, 2011, we adopted a new accounting standard that amends the guidance on the accounting for arrangements involving the delivery of more than one element. Pursuant to the new standard, each
66
Table of Contents
required deliverable is evaluated to determine whether it qualifies as a separate unit of accounting. For us, this determination is generally based on whether the deliverable has “stand-alone value” to the customer. We adopted this new accounting standard on a prospective basis for all Multiple-Deliverable Revenue Arrangements, or MDRAs, entered into on or after January 1, 2011, and for any MDRAs that were entered into prior to January 1, 2011, but materially modified on or after that date.
For MDRAs entered into prior to January 1, 2011 (pre-2011 arrangements) and not materially modified thereafter, we continue to apply our prior accounting policy with respect to such arrangements. Under this policy, in general, revenue from non-refundable, up-front fees related to intellectual property rights/licenses, where we have continuing involvement and where standalone value could not be determined under the previous guidance, is recognized ratably over the estimated period of ongoing involvement. In general, the consideration with respect to the other deliverables is recognized when the goods or services are delivered.
The adoption of this accounting standard did not have a material impact on our results of operations for the three and nine months ended September 30, 2013 and years ended December 31, 2012 and 2011, or on our financial positions as of those dates.
In January 2011, we adopted ASC Topic 605-28, Milestone Method. Under this guidance, we recognize revenue contingent upon the achievement of a substantive milestone in its entirety in the period the milestone is achieved. Substantive milestone payments are recognized upon achievement of the milestone only if all of the following conditions are met:
| • | the milestone payments are non-refundable; |
| • | achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; |
| • | substantive effort on our part is involved in achieving the milestone; |
| • | the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and |
| • | a reasonable amount of time passes between the up-front license payment and the first milestone payment, as well as between each subsequent milestone payment. |
Determination as to whether a payment meets the aforementioned conditions involves management’s judgment. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone, and therefore, the resulting payment would be considered part of the consideration for the single unit of accounting and be recognized as revenue as such performance obligations are performed under either the proportional performance or straight-line methods, as applicable. In addition, the determination that one such payment was not a substantive milestone could prevent us from concluding that subsequent milestone payments were substantive milestones and, as a result, any additional milestone payments could also be considered part of the consideration for the single unit of accounting and would be recognized as revenue as such performance obligations are performed under either the proportional performance or straight-line methods, as applicable.
Stock-Based Compensation
Stock-based payments are accounted for in accordance with the provisions of ASC Topic 718, Compensation—Stock Compensation. The fair value of share-based payments is estimated, on the date of grant, using the Black-Scholes-Merton option-pricing model, or the Black-Scholes model. The resulting fair value is recognized ratably over the requisite service period, which is generally the vesting period of the option.
For all time vesting awards granted, expense is amortized using the straight-line attribution method. For awards that contain a performance condition, expense is amortized using the accelerated attribution method. As
67
Table of Contents
stock-based compensation expense recognized in the statements of operations for the three and nine months ended September 30, 2013 and 2012 and the fiscal years ended December 31, 2012 and 2011 is based on share-based awards ultimately expected to vest, it has been reduced for estimated forfeitures. ASC Topic 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Pre-vesting forfeitures are based on our historical experience for the three- and nine- month periods ended September 30, 2013 and for the fiscal years ended December 31, 2012 and 2011, and have not been material.
We utilize the Black-Scholes model for estimating fair value of our stock options granted. Option valuation models, including Black-Scholes model, require the input of subjective assumptions, and changes in the assumptions used can materially affect the grant date fair value of an award. These assumptions include the risk-free rate of interest, expected dividend yield, expected volatility and the expected life of the award.
Expected volatility rates are based on historical volatility of the common stock of comparable publicly traded entities and other factors due to the lack of historic information of our common stock. The expected life of stock-based options is the period of time for which the stock-based options are expected to be outstanding. Given the lack of historic exercise data, the expected life is determined using the “simplified method” which is defined as the midpoint between the vesting date and the end of the contractual term.
The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. We have not paid dividends to our stockholders since our inception and do not plan to pay cash dividends in the foreseeable future. Therefore, we have assumed an expected dividend rate of zero.
Given the absence of an active market for our common stock, the exercise price of the stock options on the date of grant was determined and approved by the board of directors using several factors, including progress and milestones achieved in our business development and performance, the price per share of our convertible preferred stock offerings and general industry and economic trends. In establishing the estimated fair value of our common stock, we considered the guidance set forth in American Institute of Certified Public Accountants Practice Guide, “Valuation of Privately-Held-Company Equity Securities Issued as Compensation.”
Under ASC Topic 718, the cumulative amount of compensation cost recognized for instruments classified as equity that ordinarily would result in a future tax deduction under existing tax law shall be considered to be a deductible difference in applying ASC Topic 740, Income Taxes. The deductible temporary difference is based on the compensation cost recognized for financial reporting purposes; however, these provisions currently do not impact us, as all the deferred tax assets have a full valuation allowance.
Since we had net operating loss carry-forwards as of September 30, 2013 and December 31, 2012 and 2011, no excess tax benefits for the tax deductions related to share-based awards were recognized in the statements of operations.
Equity instruments issued to non-employees are accounted for under the provisions of ASC Topic 718 and ASC Topic 505-50, Equity/Equity-Based Payments to Non-Employees. Accordingly, the estimated fair value of the equity instrument is recorded on the earlier of the performance commitment date or the date the services required are completed and are marked to market during the service period.
Recently Issued Accounting Pronouncements
We review new accounting standards to determine the expected financial impact, if any, that the adoption of each such standard will have. For the recently issued accounting standards that we believe may have an impact on our financial statements, see “Notes to Condensed Consolidated Financial Statements (Unaudited)—Note 2—Summary of Significant Accounting Policies” included in this prospectus.
68
Table of Contents
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Effective at the Effective Time of the Merger, Raich Ende Malter & Co. LLP, or REM, was dismissed as the independent registered public accounting firm that audits the financial statements of the Company. Effective as of the Effective Time, our board of directors engaged Ernst & Young LLP, or E&Y, as the independent registered public accounting firm to audit the Company’s financial statements for the fiscal year ended December 31, 2013.
REM’s audit report on the Company’s financial statements for the period from August 29, 2012 (inception) through March 31, 2013, did not contain an adverse opinion or a disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles.
During the Company’s most recent fiscal year (since inception) and any subsequent interim period prior to the date of this prospectus, there were no disagreements with REM on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure, which disagreements, if not resolved to the satisfaction of REM, would have caused it to make reference to the subject matter thereof in connection with its report.
During the Company’s most recent fiscal year (since inception) and any subsequent interim period prior to the date of this prospectus, neither the Company nor anyone acting on its behalf consulted E&Y regarding the application of accounting principles to a specified transaction, either completed or proposed or the type of audit opinion that might be rendered on the Company’s financial statements.
The Company has provided REM with a copy of this prospectus prior to the filing of the registration statement of which this prospectus forms a part, and has requested that REM furnish to the Company a letter addressed to the Securities and Exchange Commission stating whether REM agrees with the statements made by the Company in this prospectus. REM has furnished such letter, which letter is filed as Exhibit 16.1 to the registration statement of which this prospectus forms a part, as required by Item 304(a)(3) of Regulation S-K.
69
Table of Contents
DIRECTORS AND EXECUTIVE OFFICERS
The following table sets forth certain information concerning our executive officers and directors as of December 1, 2013:
| Name |
Age | Position | ||
| Executive Officers |
||||
| Sharon Mates, Ph.D. |
60 | Chairman, President and Chief Executive Officer | ||
| Lawrence J. Hineline |
57 | Vice President of Finance, Chief Financial Officer and Secretary | ||
| Allen A. Fienberg, Ph.D. |
53 | Vice President of Business Development | ||
| Lawrence P. Wennogle, Ph.D. |
63 | Vice President, Drug Discovery | ||
| Kimberly E. Vanover, Ph.D. |
47 | Vice President, Clinical Development | ||
| Non-Employee Directors |
||||
| Christopher Alafi, Ph.D. |
50 | Director | ||
| Richard Lerner, M.D. |
75 | Director | ||
| Joel S. Marcus |
66 | Director | ||
| Sir Michael Rawlins, M.D., FRCP, FMedSci |
72 | Director | ||
Executive Officers
Sharon Mates, Ph.D. Dr. Mates has been the Chairman of the board of directors, President and Chief Executive Officer of ITI since June 2002. Dr. Mates co-founded ITI in May 2002. Prior to co-founding ITI, Dr. Mates was a co-founder of Functional Genetics, and served as its Chairman and Chief Executive Officer from December 2000 until August 2003. From 1989-1998 Dr. Mates was the President and a board member of North American Vaccine Inc. and its predecessor companies. She has served on several boards, and recently completed a board membership and a two-year chairmanship of the Board of the New York Biotechnology Association. Dr. Mates has also served on the Advisory Council of the Center for Society and Health at the Harvard School of Public Health, the Board of Visitors of the Biotechnology Institute of the University of Maryland and the board of directors of Gilda’s Club of New York. Earlier in her career, Dr. Mates spent several years as a research analyst and investment banker, and as an advisor to the life sciences industry. Dr. Mates received her B.S. from the Ohio State University and her Ph.D. from the University of Washington, and completed her postdoctoral fellowships at The Massachusetts General Hospital and Harvard Medical School.
We believe that Dr. Mates possesses specific attributes that qualify her to serve as chairman of our board of directors, including the perspective and experience she brings as the co-founder, President and Chief Executive of ITI, which brings historic knowledge, operational expertise and continuity to our board of directors, and her industry expertise, including over 24 years of experience leading both private and public companies.
Lawrence J. Hineline, CPA. Mr. Hineline has served as Vice President of Finance, Chief Financial Officer and Secretary of ITI since June 2002. From December 2000 to November 2003, Mr. Hineline was the Vice-President of Finance and Chief Financial Officer of Functional Genetics, Inc. Prior to that, Mr. Hineline served as the Vice President of Finance of North American Vaccine, Inc. and its predecessor companies from 1993 to 2000, and he served as Corporate Controller from 1989 to 1993. During this time, Mr. Hineline oversaw the growth of the accounting function and its systems for the company that emerged as a start-up and was later acquired by Baxter Health Care. Mr. Hineline is a licensed CPA in the State of Maryland and received his Bachelor’s Degree from the University of Maryland Baltimore County.
Allen A. Fienberg, Ph.D. Dr. Fienberg has served as Vice President of Business Development of ITI since June 2002. He co-founded ITI in May 2002. Dr. Fienberg received his A.B. degree in Genetics from the University of California, Berkeley and his Ph.D. in Human Genetics from Yale University. He completed post-doctoral studies at The Rockefeller University under the direction of Dr. Paul Greengard from 1991-1999.
70
Table of Contents
From 1999-2001, Dr. Fienberg was a staff scientist at the Genomics Institute of the Novartis Research Foundation and was appointed a Research Assistant Professor at The Rockefeller University from 2001-2002.
Lawrence P. Wennogle, Ph.D. Dr. Wennogle has served as Vice President, Drug Discovery of ITI since January 2003. For the past 33 years, Dr. Wennogle has been involved in research and development in the pharmaceutical industry aimed at the discovery of novel pharmaceutical entities for human diseases. He was a Staff Scientist and Principal Research Fellow at Ciba-Geigy and Novartis Pharmaceutical Corporation for 19 years, where he led drug discovery programs for CNS disorders, cardiovascular diseases, diabetes and inflammation. Dr. Wennogle received his B.A. from Ithaca College and his Ph.D. in Biochemistry from the University of Colorado, Boulder. He then completed two post-doctoral positions, one at the University of Colorado and the second at the Pasteur Institute in Paris, France, working under Jean-Pierre Changeux on the structure-function of the nicotinic acetylcholine receptor.
Kimberly E. Vanover, Ph.D. Dr. Vanover joined ITI in March 2007 and has been Vice President, Clinical Development of ITI since January 2011. Previously, she was Executive Director, Clinical Development of ITI from January 2008 to December 2010 and Senior Director, Clinical Development of ITI from March 2007 to December 2007. She has spent over 20 years on the discovery and development of small molecule drugs for the treatment of neuropsychiatric and neurodegenerative diseases. Dr. Vanover was Postdoctoral Research Scientist at Lederle Laboratories from 1992 to 1994, Postdoctoral Research Trainee in the Department of Psychiatry at the University of California San Diego from 1994 to 1995, Senior Scientist and Group Leader at CoCensys from 1995 to 2000 and held positions as Group Leader and Director at ACADIA Pharmaceuticals from 2000 to 2007. In these positions, Dr. Vanover participated in the discovery and development of a broad range of new CNS therapeutics, including drugs to treat psychosis, insomnia, cognitive impairment, movement disorders, acute and neuropathic pain, anxiety, epilepsy, and drug abuse. Dr. Vanover received her B.A. in Psychology from the University of Missouri and her Ph.D. in Biopsychology from the University of Chicago.
Non-Employee Directors
Christopher Alafi, Ph.D. Dr. Alafi has served on the board of directors of ITI since January 2013. Dr. Alafi has been a General Partner of Alafi Capital Company, LLC, a venture capital firm, since 1995. He was previously a Physiology and Anatomy teacher at Santa Monica College, a visiting scholar in the Department of Chemistry at Stanford University and a researcher at DNAX. Dr. Alafi currently serves as a director of ISTO Technologies, Inc. and has previously served as a director of Coley Pharmaceutical Group, Inc., CyberGold, Inc. and Stereotaxis, Inc. Dr. Alafi received a B.A. in Biology from Pomona College and a D.Phil. in Biochemistry from the University of Oxford.
We believe that Dr. Alafi possesses specific attributes that qualify him to serve as a member of our board of directors, including the perspective and experience he brings as a General Partner of Alafi Capital Company, LLC.
Richard Lerner, M.D. Dr. Lerner has served on the board of directors of ITI since 2002. Dr. Lerner served as President of the Scripps Research Institute, a private, non-profit biomedical research organization from 1986 to January 2012, and since then has served and continues to serve as Institute Professor. Dr. Lerner received the Wolf Prize in Chemistry in 1994, the California Scientist of the Year Award in 1996, the Paul Ehrlich and Ludwig Darmstaedter Prize in 2003, and the Prince of Asturias Award in 2012 for his achievements in the development of catalytic antibodies and combinatorial antibody libraries. Dr. Lerner is a member of the National Academy of Sciences and the Royal Swedish Academy of Sciences. Dr. Lerner served as a director of Kraft Foods, Inc. from 2005 to March 2012 and currently serves as a director of Opko Health, Inc., Teva Pharmaceutical Industries Ltd., and Sequenom, Inc. Dr. Lerner received his M.D. from Stanford Medical School.
We believe that Dr. Lerner possesses specific attributes that qualify him to serve as a member of our board of directors, including his service as a director of other public companies, combined with his business acumen and judgment provide our board of directors with valuable scientific and operational expertise and leadership skills.
71
Table of Contents
Joel S. Marcus J.D., CPA. Mr. Marcus has served on the board of directors of ITI since April 2006. Mr. Marcus co-founded Alexandria Real Estate Equities, Inc. in 1994, Alexandria Venture Investments in 1996, and the annual Alexandria Summit in 2011. He has served as Chairman of the Board of Directors of Alexandria Real Estate Equities, Inc. since May 2007, Chief Executive Officer since March 1997, President since February 2009, and a director since the company’s inception in 1994. From 1986 to 1994, Mr. Marcus was a partner at the law firm of Brobeck, Phleger & Harrison LLP, specializing in corporate finance and capital markets, venture capital, and mergers and acquisitions. From 1984 to 1994, he also served as General Counsel and Secretary of Kirin-Amgen, Inc., a joint venture that financed the development of, and owned patents to, two multi-billion dollar genetically engineered biopharmaceutical products. Mr. Marcus was formerly a practicing certified public accountant and tax manager with Arthur Young & Co. specializing in the financing and taxation of REITs. He received his undergraduate and Juris Doctor degrees from the University of California, Los Angeles. In addition to ITI, Mr. Marcus serves on the boards of the Accelerator Corporation, of which he was one of the original architects and co-founders, Foundation for the National Institutes of Health (FNIH), Multiple Myeloma Research Foundation (MMRF), and the Partnership for New York City. Mr. Marcus also served on the Board of Trustees of PennyMac Mortgage Investment Trust, a publicly traded mortgage REIT, from August 2009 to August 2012. Mr. Marcus received the Ernst & Young 1999 Entrepreneur of the Year Award (Los Angeles—Real Estate).
We believe that Mr. Marcus possesses specific attributes that qualify him to serve as a member of our board of directors, including his many years of experience in the life sciences industry and his extensive experience serving as a director and an executive officer of other public companies.
Sir Michael Rawlins, M.D., FRCP, FMedSci. Sir Michael has served on the board of directors of ITI since May 2013. Sir Michael is known for his long standing leadership of the United Kingdom’s National Institute for Clinical Excellence, or NICE, which he led from its inception in 1999 through March 2013. Recently in July 2012, Sir Michael was appointed as the President of the United Kingdom’s Royal Society of Medicine, a center for education and scholarship both in the UK and globally. Sir Michael was a professor of clinical pharmacology and a general physician at the University of Newcastle upon Tyne from 1973 to 2006. He received the Prince Mahidol Award for Medicine in 2012, the Galen Medal in 2010, and the Hutchinson Medal in 2003. Sir Michael was appointed Knight Bachelor in 1999.
We believe that Sir Michael possesses specific attributes that qualify him to serve as a member of our board of directors, including his extensive experience in areas of health policy and economics.
Scientific Advisory Board
We have a Scientific Advisory Board which is chaired by Paul Greengard, Ph.D., one of our founders. Dr. Greengard received his Ph.D. in biophysics from Johns Hopkins University in 1953. After postgraduate work in England, he served for nine years as director of biochemical research at the Geigy Research Laboratories. In 1968, he was appointed Professor of Pharmacology at Yale University. In 1983, he was appointed the Vincent Astor Professor at The Rockefeller University, where he founded the Laboratory of Molecular and Cellular Neuroscience.
Dr. Greengard is a pioneer in the field of neuronal signal transduction and his seminal discoveries over the years have provided a framework by which to understand the complexity of how neurotransmitters function in the brain. Dr. Greengard’s many awards and honors include the CIBA-Geigy Drew Award in Biomedical Research (1979), the New York Academy of Sciences Award in Biological and Medical Sciences (1980), the Andrew D. White Professorship-at-Large of Cornell University (1981-87), the Pfizer Biomedical Research Award (1987), the Ralph W. Gerard Prize in Neuroscience, Society for Neuroscience (1994), the Charles A. Dana Award for Pioneering Achievements in Health (1997), and the Nobel Prize in Physiology or Medicine (2000). Dr. Greengard has also been a consultant to major pharmaceutical companies and a Chairman and member of the scientific advisory boards of numerous biotechnology companies.
72
Table of Contents
We have additional members of our Scientific Advisory Board who change from time to time, with whom we consult on an as-needed basis.
Medical Advisory Board
Carol A. Tamminga, M.D. is the Chair of our Medical Advisory Board. Dr. Tamminga is the Chair of the Psychiatry Department at the University of Texas Southwestern School of Medicine. She holds the McKenzie Foundation Chair in Psychiatry, the Communities Foundation of Texas, Inc. Chair in Brain Science and is the Chief of Translational Neuroscience Research in Schizophrenia.
Jeffrey Lieberman, M.D. is the Lawrence C. Kolb Professor and Chairman of Psychiatry, at the Columbia University College of Physicians and Surgeons; and Director, of the New York State Psychiatric Institute; Psychiatrist-in-Chief, New York Presbyterian Hospital-Columbia University Medical Center.
John M. Kane, M.D. is Professor and Chairman of Psychiatry at The Hofstra North Shore-LIJ School of Medicine and Vice President for Behavioral Health Services at The North Shore-LIJ Health System.
Christoph U. Correll, M.D. is Professor of Psychiatry and Molecular Medicine, Hofstra North Shore LIJ School of Medicine; Medical Director, Recognition and Prevention (RAP) Program, The Zucker Hillside Hospital, North Shore Long Island Jewish Health System.
Donald Goff, M.D. is Professor and Vice Chair for Research in the Department of Psychiatry at New York University Langone Medical Center (NYULMC) and Director of the Nathan S. Kline Institute for Psychiatric Research.
Board Composition and Election of Directors
Terms of Office
Our restated certificate of incorporation and our restated bylaws provide that, subject to any applicable rights of holders of any preferred stock then outstanding, the authorized number of directors may be changed only by resolution of our board of directors. We currently have authorized five directors. In accordance with our restated certificate of incorporation and restated bylaws, our board of directors is divided into three classes with staggered three-year terms. At each annual meeting of stockholders commencing with the meeting in 2014, the successors to the directors whose terms then expire will be elected to serve until the third annual meeting following the election. Our directors are divided among the three classes as follows:
| • | the Class I directors are Richard Lerner, M.D. and Sir Michael Rawlins, M.D., FRCP, FMedSci, and their terms will expire at the annual meeting of stockholders to be held in 2014; |
| • | the Class II directors are Christopher Alafi, Ph.D. and Joel S. Marcus, and their terms will expire at the annual meeting of stockholders to be held in 2015; and |
| • | the Class III director is Sharon Mates, Ph.D. and her term will expire at the annual meeting of stockholders to be held in 2016. |
Any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that each class will consist of approximately one-third of the directors.
Director Independence
Our securities are not listed on a national securities exchange or on any inter-dealer quotation system which has a requirement that a majority of directors be independent. We evaluate independence, however, by the standards for director independence set forth in the NASDAQ Marketplace Rules. Under Rules 5605 and 5615 of
73
Table of Contents
the NASDAQ Marketplace Rules, a majority of a listed company’s board of directors must be comprised of independent directors, subject to certain phase-in exceptions. In addition, NASDAQ Marketplace Rules require that, subject to specified exceptions, each member of a listed company’s audit, compensation and governance and nominating committees be independent and that audit committee members also satisfy independence criteria set forth in Rule 10A-3 under the Exchange Act. Under Rule 5605(a)(2) of the NASDAQ Marketplace Rules, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Based upon information requested from and provided by each director concerning their background, employment and affiliations, including family relationships, our board of directors has determined that none of Dr. Alafi, Dr. Lerner, Mr. Marcus or Sir Michael, representing four out of our five directors, has a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under Rule 5605(a)(2) of the NASDAQ Marketplace Rules. Dr. Mates is employed by the Company and is therefore not independent under NASDAQ Marketplace Rules.
Board of Directors’ Meetings
During the fiscal year ended December 31, 2012, there were five meetings of ITI’s board of directors, and the compensation committee, which was the only standing committee of ITI’s board of directors, met one time. No director of ITI attended fewer than 75% of the total number of meetings of ITI’s board of directors and of committees of its board of directors on which he or she served during fiscal 2012, except for David Kipnis, M.D., a former director of ITI, who attended three of the six meetings of ITI’s board of directors and committees of its board of directors during fiscal 2012. In addition, Christopher Alafi, Ph.D. and Sir Michael Rawlins, M.D., FRCP, FMedSci were not elected as directors of ITI until 2013. Our board of directors intends to adopt a policy under which each member of our board of directors is strongly encouraged but not required to attend each annual meeting of our stockholders. Neither we nor ITI held an annual meeting of stockholders in 2012.
Committees of the Board of Directors
Our board of directors does not currently have an audit committee, a compensation committee or a nominating and governance committee. Our board of directors intends to establish an audit committee, a compensation committee and a nominating and corporate governance committee. Each committee will operate under a charter to be approved by our board of directors.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors and executive officers, and persons who own more than ten percent of a registered class of our equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock and other equity securities. Officers, directors and greater than ten percent stockholders are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file.
To our knowledge, based solely on a review of the copies of such reports furnished to us and written representations regarding the filing of required reports, we believe that all Section 16(a) filing requirements applicable to our directors, executive officers and greater-than-ten-percent beneficial owners with respect to fiscal 2012 were met, as our directors, executive officers and greater-than-ten percent beneficial owners were not required to file reports under Section 16(a) before the Company became an Exchange Act reporting company in fiscal 2013.
74
Table of Contents
Code of Ethics
Prior to the Merger, the Company had adopted a code of conduct and ethics. We intend to adopt an amended and restated code of conduct and ethics that will apply to all of our employees, including our principal executive officer and our principal financial and accounting officer, and plan to post a copy of such code of conduct and ethics on our website at www.intracellulartherapies.com. Once we adopt the amended and restated code of conduct and ethics, disclosure regarding any amendments to, or waivers from, provisions of the amended and restated code of conduct and ethics that we intend to adopt that apply to our directors, principal executive and financial officers will be included in a Current Report on Form 8-K within four business days following the date of the amendment or waiver, unless website posting or the issuance of a press release of such amendments or waivers is then permitted by any applicable stock exchange.
Board Leadership Structure and Role on Risk Oversight
Our board of directors does not have a policy regarding the separation of the roles of Chief Executive Officer and Chairman of the board of directors, as our board of directors believes it is in the best interest of the Company to make that determination based on the position and direction of the Company and the membership of the board of directors. Our board of directors has determined that having an employee director serve as Chairman is in the best interest of the Company’s stockholders at this time because of the efficiencies achieved in having the role of Chief Executive Officer and Chairman combined, and because the detailed knowledge of our day-to-day operations and business that the Chief Executive Officer possesses greatly enhances the decision-making processes of our board of directors as a whole. We have a strong governance structure in place, including independent directors, to ensure the powers and duties of the dual role are handled responsibly. We do not have a lead independent director.
The Chairman of the board of directors and the other members of the board of directors work in concert to provide oversight of our management and affairs. Our board of directors encourages communication among its members and between management and the board of directors to facilitate productive working relationships. Working with the other members of the board of directors, Dr. Mates also strives to ensure that there is an appropriate balance and focus among key board responsibilities such as strategic development, review of operations and risk oversight.
75
Table of Contents
Unless we specifically indicate otherwise, all share and per share numbers included in this “Executive Compensation” section have been adjusted as necessary to reflect the exchange of shares in the Merger.
Summary Compensation Table
The following table shows the total compensation paid or accrued during the last two fiscal years ended December 31, 2012 and 2011 to (1) our President and Chief Executive Officer and (2) our two next most highly compensated executive officers who earned more than $100,000 during the fiscal year ended December 31, 2012 and were serving as executive officers as of such date.
| Name and Principal Position |
Year | Salary ($) |
Bonus ($) |
Option Awards ($)(1) |
All Other Compensation ($)(2) |
Total ($) |
||||||||||||||||||
| Sharon Mates, Ph.D. |
2012 | 588,400 | 117,700 | 100,000 | 7,750 | 813,850 | ||||||||||||||||||
| Chairman, President and Chief Executive Officer |
2011 | 565,800 | 313,200 | (3) | — | 7,600 | 886,600 | |||||||||||||||||
| Lawrence J. Hineline |
2012 | 250,000 | 17,500 | 20,000 | 7,750 | 295,250 | ||||||||||||||||||
| Vice President of Finance, Chief Financial Officer and Secretary |
2011 | 237,600 | 30,900 | — | 7,378 | 275,878 | ||||||||||||||||||
| Allen A. Fienberg, Ph.D. |
2012 | 250,400 | 8,800 | 20,000 | 7,750 | 286,950 | ||||||||||||||||||
| Vice President of Business Development |
2011 | 243,100 | 17,000 | — | 7,543 | 267,643 | ||||||||||||||||||
| (1) | These amounts represent the aggregate grant date fair value for option awards granted to our named executive officers, computed in accordance with FASB ASC Topic 718. See Note 5 to our audited financial statements for the fiscal years ended December 31, 2012 and 2011 included in this prospectus for details as to the assumptions used to calculate the fair value of the option awards. See also our discussion of stock-based compensation under “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Estimates.” |
| (2) | Consists of $250 in life insurance premiums we paid for a term life insurance policy to benefit the executive officer with a face value of $150,000, and the balance in matching contributions under our 401(k) plan. |
| (3) | Dr. Mates received a bonus of $113,200 for her performance during the fiscal year ended December 31, 2011 plus an additional bonus of $200,000 for her performance in connection with our entering into the license and collaboration agreement with Takeda Pharmaceutical Company Limited. |
2012 Fiscal Year Grants of Plan-Based Awards
The following table shows information regarding grants of equity awards that we made during the fiscal year ended December 31, 2012 to each of our executive officers named in the Summary Compensation Table. We did not grant any non-equity incentive plan awards during the fiscal year ended December 31, 2012.
| Name |
Compensation Committee Approval(1) |
Grant Date(1) |
All Other Option Awards: Number of Securities Underlying Options (#)(2) |
Exercise or Base Price of Option Awards ($/Sh)(3) |
Grant Date Fair Value of Stock and Option Awards ($)(4) |
|||||||||||||||
| Sharon Mates, Ph.D. |
12/20/2011 | 5/1/2012 | 50,000 | 2.84 | 100,000 | |||||||||||||||
| Lawrence J. Hineline |
12/20/2011 | 5/1/2012 | 10,000 | 2.84 | 20,000 | |||||||||||||||
| Allen A. Fienberg, Ph.D. |
12/20/2011 | 5/1/2012 | 10,000 | 2.84 | 20,000 | |||||||||||||||
| (1) | On December 20, 2011, the compensation committee of ITI approved these option grants to be granted following the completion of a valuation of ITI’s common stock. Following the completion of the valuation of ITI’s common stock, on May 1, 2012 the board of directors of ITI approved these grants at an exercise price of $2.84 per share. In addition, on December 20, 2012, the compensation committee approved grants |
76
Table of Contents
| of 50,000 options to Dr. Mates, 10,000 options to Mr. Hineline and 7,500 options to Dr. Fienberg (the “Additional Grants”) to be granted following the completion of a valuation of ITI’s common stock and an increase in the number of shares reserved under ITI’s 2003 Equity Incentive Plan. Following the completion of the valuation of ITI’s common stock and the increase in the number of shares reserved under ITI’s 2003 Equity Incentive Plan, on May 31, 2013 the board of directors of ITI approved the Additional Grants at an exercise price of $3.26 per share. |
| (2) | These awards are subject to vesting, as described in detail under “—Outstanding Equity Awards at 2012 Fiscal Year-End” below. |
| (3) | The 2003 Equity Incentive Plan provides that the exercise price shall be determined by using the fair market value of our common stock, which is defined under the 2003 Equity Incentive Plan as the value of our common stock determined in good faith by our board of directors. |
| (4) | These amounts represent the aggregate grant date fair value for option awards granted to our named executive officers, computed in accordance with FASB ASC Topic 718. See Note 5 to our audited financial statements for the fiscal years ended December 31, 2012 and 2011 included in this prospectus for details as to the assumptions used to calculate the fair value of the option awards. See also our discussion of stock-based compensation under “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Estimates.” |
Narrative Disclosure to Summary Compensation Table and Grants of Plan-Based Awards Table
Sharon Mates, Ph.D. We entered into an employment agreement with Dr. Mates in February 2008, who has been our President and Chief Executive Officer since 2003. The agreement provides for a salary of $503,000 effective in February 2008, subject to our annual review and adjustment in the discretion of our board of directors, and that Dr. Mates is eligible for bonus payments and stock options as may be awarded by our board of directors. The most recent adjustment, effective on January 1, 2013, increased Dr. Mates’ salary to $611,900. In 2012, she was awarded a bonus of $117,700, which represented a bonus of 20% of her then current base salary of $588,400. In addition, her employment agreement provides that we will pay the premium on a life insurance policy in an amount equal to one and one half times her base salary; however, we paid a premium in the amount of $250 on a life insurance policy with a face value of $150,000, to which she assented. For 2012, we also paid $7,500 in matching contributions under our 401(k) plan. The employment agreement also provides that Dr. Mates is entitled to participate in our benefit plans on the same basis as other executive level employees as well as long-term disability insurance and reimbursement for reasonable business expenses. The initial term of the agreement was three years and will be renewed for successive one year terms, unless we or Dr. Mates provides notice that we or she, as the case may be, does not wish to renew the agreement or wishes to renew the agreement on different terms than those contained in the agreement.
If Dr. Mates’ employment is terminated for any reason, she will be entitled to compensation and benefits through the last day of her employment, including accrued but untaken vacation. If her employment is terminated due to her death or disability, we will also pay her or her estate the compensation which would otherwise have been payable to her through the end of the month in which such termination occurs as well as payment for any accrued but untaken vacation. If her employment is terminated without cause by us or she terminates her employment for good reason, she will receive the following severance benefits following her employment termination, on condition that she executes a general release in our favor: (a) payment of 12 months of her then current base salary and the pro rata portion of an amount equal to the bonus she was awarded for the previous year, if any, which severance payments will be paid in one lump sum on the date the general release she executes becomes effective; (b) payment for 12 months of the portion of the COBRA premiums that we paid prior to her termination; and (c) all of her unvested stock options will become fully vested and exercisable. Dr. Mates will also be entitled to such severance benefits if we elect not to renew her employment agreement for reasons other than death, disability or cause, but (i) such severance benefits are conditioned on Dr. Mates executing a general release in favor of us, returning all our property, and complying with her employment agreement, proprietary information, inventions, and non-competition agreement, and the general release and (ii) Dr. Mates will not be eligible for such severance benefits if she or we wish to renew the agreement on different terms than those
77
Table of Contents
contained in her employment agreement. In the event of a change of control, all of her unvested stock options and restricted stock will immediately vest. If her employment is terminated for reasons other than death or disability within three months before or 12 months following a change of control, she terminates her employment for good reason during such period, or she terminates her employment for any reason within one month following a change of control, she will be eligible for the following severance benefits following her employment termination: (a) payment of 18 months of her then current base salary and the pro rata portion of an amount equal to the bonus she was awarded for the previous year, which severance payments will be paid in one lump sum on the eighth day following the effective date of the general release, and (b) payment for 18 months of the portion of the COBRA premiums that we paid prior to her termination. Such severance benefits following a change of control are payable on condition that she executes a general release in favor of us, returns all our property and complies with her post-termination obligations under her employment agreement, her proprietary information, inventions, and non-competition agreement, and her general release.
Pursuant to her proprietary information, inventions, and non-competition agreement, Dr. Mates has agreed to not (i) solicit customers, consultants, contractors or employees of ours for a period of one year after the termination of her employment or (ii) compete with us for a period of one year after the later of the termination of her employment or the date a court of competent jurisdiction enters an order enforcing the non-competition provision.
Lawrence J. Hineline. We entered into an employment agreement with Mr. Hineline in February 2008, who has been our Vice President of Finance, Chief Financial Officer and Secretary since 2002. The agreement provides for a salary of $216,400 effective in February 2008, subject to our annual review and adjustment in the discretion of our board of directors, and that Mr. Hineline is eligible for bonus payments and stock options as may be awarded by our board of directors. The most recent adjustment, effective on January 1, 2013, increased Mr. Hineline’s salary to $257,500. In 2012, he was awarded a bonus of $17,500, which represented a bonus of 7% of his then current base salary of $250,000. In addition, his employment agreement provides that we will pay the premium on a life insurance policy in an amount equal to one and one half times his base salary; however, we paid a premium in the amount of $250 on a life insurance policy with a face value of $150,000, to which he assented. For 2012, we also paid $7,500 in matching contributions under our 401(k) plan. The employment agreement also provides that Mr. Hineline is entitled to participate in our benefit plans on the same basis as other executive level employees as well as long-term disability insurance and reimbursement for reasonable business expenses. The initial term of the agreement was three years and will be renewed for successive one year terms, unless we or Mr. Hineline provides notice that we or he, as the case may be, does not wish to renew the agreement or wishes to renew the agreement on different terms than those contained in the agreement.
If Mr. Hineline’s employment is terminated for any reason, he will be entitled to compensation and benefits through the last day of his employment, including accrued but untaken vacation. If his employment is terminated due to his death or disability, we will also pay him or his estate the compensation which would otherwise have been payable to him through the end of the month in which such termination occurs as well as payment for any accrued but untaken vacation. If his employment is terminated without cause by us or he terminates his employment for good reason, he will receive the following severance benefits following his employment termination, on condition that he executes a general release in our favor: (a) payment of 12 months of his then current base salary and the pro rata portion of an amount equal to the bonus he was awarded for the previous year, if any, which severance payments will be paid in one lump sum on the date the general release he executes becomes effective; (b) payment for 12 months of the portion of the COBRA premiums that we paid prior to his termination; and (c) all of his unvested stock options will become fully vested and exercisable. Mr. Hineline will also be entitled to such severance benefits if we elect not to renew his employment agreement for reasons other than death, disability or cause, but (i) such severance benefits are conditioned on Mr. Hineline executing a general release in our favor, returning all our property, and complying with his employment agreement, proprietary information, inventions, and non-competition agreement, and the general release and (ii) Mr. Hineline will not be eligible for such severance benefits if he or we wish to renew the agreement on different terms than those contained in his employment agreement. In the event of a change of control, all of his
78
Table of Contents
unvested stock options and restricted stock will immediately vest. If his employment is terminated for reasons other than death or disability within three months before or 12 months following a change of control, he terminates his employment for good reason during such period, or he terminates his employment for any reason within one month following a change of control, he will be eligible for the following severance benefits following his employment termination: (a) payment of 18 months of his then current base salary and the pro rata portion of an amount equal to the bonus he was awarded for the previous year, which severance payments will be paid in one lump sum on the eighth day following the effective date of the general release, and (b) payment for 18 months of the portion of the COBRA premiums that we paid prior to his termination. Such severance benefits following a change of control are payable on condition that he executes a general release in favor of us, returns all our property and complies with his post-termination obligations under his employment agreement, his proprietary information, inventions, and non-competition agreement, and his general release.
Pursuant to his proprietary information, inventions, and non-competition agreement, Mr. Hineline has agreed to not (i) solicit customers, consultants, contractors or employees of ours for a period of one year after the termination of his employment or (ii) compete with us for a period of one year after the later of the termination of his employment or the date a court of competent jurisdiction enters an order enforcing the non-competition provision.
Allen A. Fienberg, Ph.D. We entered into an employment agreement with Dr. Fienberg in February 2008, who has been our Vice President of Business Development since 2002. The agreement provides for a salary of $221,400 effective in February 2008, subject to our annual review and adjustment in the discretion of our board of directors, and that Dr. Fienberg is eligible for bonus payments and stock options as may be awarded by our board of directors. The most recent adjustment, effective on January 1, 2013, increased Dr. Fienberg’s salary to $257,900. In 2012, he was awarded a bonus of $8,800, which represented a bonus of 3.5% of his then current base salary of $250,400. In addition, his employment agreement provides that we will pay the premium on a life insurance policy in an amount equal to one and one half times his base salary; however, we paid a premium in the amount of $250 on a life insurance policy with a face value of $150,000, to which he assented. For 2012, we also paid $7,500 in matching contributions under our 401(k) plan. The employment agreement also provides that Dr. Fienberg is entitled to participate in our benefit plans on the same basis as other executive level employees as well as long-term disability insurance and reimbursement for reasonable business expenses. The initial term of the agreement was three years and will be renewed for successive one year terms, unless we or Dr. Fienberg provides notice that we or he, as the case may be, does not wish to renew the agreement or wishes to renew the agreement on different terms than those contained in the agreement.
If Dr. Fienberg’s employment is terminated for any reason, he will be entitled to compensation and benefits through the last day of his employment, including accrued but untaken vacation. If his employment is terminated due to his death or disability, we will also pay him or his estate the compensation which would otherwise have been payable to him through the end of the month in which such termination occurs as well as payment for any accrued but untaken vacation. If his employment is terminated without cause by us or he terminates his employment for good reason, he will receive the following severance benefits following his employment termination, on condition that he executes a general release in our favor: (a) payment of 12 months of his then current base salary and the pro rata portion of an amount equal to the bonus he was awarded for the previous year, if any, which severance payments will be paid in one lump sum on the date the general release he executes becomes effective; (b) payment for 12 months of the portion of the COBRA premiums that we paid prior to his termination; and (c) all of his unvested stock options will become fully vested and exercisable. Dr. Fienberg will also be entitled to such severance benefits if we elect not to renew his employment agreement for reasons other than death, disability or cause, but (i) such severance benefits are conditioned on Dr. Fienberg executing a general release in our favor, returning all our property, and complying with his employment agreement, proprietary information, inventions, and non-competition agreement, and the general release and (ii) Dr. Fienberg will not be eligible for such severance benefits if he or we wish to renew the agreement on different terms than those contained in his employment agreement. In the event of a change of control, 75% of his unvested stock options and restricted stock will immediately vest.
79
Table of Contents
Pursuant to his proprietary information, inventions, and non-competition agreement, Dr. Fienberg has agreed to not (i) solicit customers, consultants, contractors or employees of ours for a period of one year after the termination of his employment or (ii) compete with us for a period of one year after the later of the termination of his employment or the date a court of competent jurisdiction enters an order enforcing the non-competition provision.
The meanings of the terms “cause,” “good reason,” “disability” and “change of control” for purposes of these employment agreements are described below under “Potential Payments upon Termination or Change in Control.”
Outstanding Equity Awards at 2012 Fiscal Year-End
The following table shows grants of stock options and grants of unvested stock awards outstanding on the last day of the fiscal year ended December 31, 2012, including both awards subject to performance conditions and non-performance-based awards, to each of the executive officers named in the Summary Compensation Table.
| Name(1) |
Number of Securities Underlying Unexercised Options (#)(2) Exercisable |
Number of Securities Underlying Unexercised Options (#)(2) Unexercisable |
Option Exercise Price ($) |
Option Expiration Date |
||||||||||||
| Sharon Mates, Ph.D. |
50,000 | 0 | $ | 0.30 | 12/16/2013 | |||||||||||
| 50,000 | 0 | $ | 0.50 | 12/19/2014 | ||||||||||||
| 25,000 | 0 | $ | 0.60 | 12/14/2015 | ||||||||||||
| 25,000 | 0 | $ | 1.36 | 12/5/2016 | ||||||||||||
| 37,500 | 0 | $ | 1.50 | 12/12/2017 | ||||||||||||
| 50,000 | 0 | $ | 1.50 | 12/18/2018 | ||||||||||||
| 50,000 | 0 | $ | 2.74 | 6/10/2020 | ||||||||||||
| 33,333 | 16,667 | $ | 2.74 | 12/21/2020 | ||||||||||||
| 16,666 | 33,334 | $ | 2.84 | 4/30/2022 | ||||||||||||
| Lawrence J. Hineline |
50,000 | 0 | $ | 0.30 | 12/16/2013 | |||||||||||
| 37,500 | 0 | $ | 0.50 | 12/19/2014 | ||||||||||||
| 12,500 | 0 | $ | 0.60 | 12/14/2015 | ||||||||||||
| 12,500 | 0 | $ | 1.36 | 12/5/2016 | ||||||||||||
| 12,500 | 0 | $ | 1.50 | 12/12/2017 | ||||||||||||
| 10,000 | 0 | $ | 1.50 | 12/18/2018 | ||||||||||||
| 10,000 | 0 | $ | 2.74 | 6/10/2020 | ||||||||||||
| 6,666 | 3,334 | $ | 2.74 | 12/21/2020 | ||||||||||||
| 3,333 | 6,667 | $ | 2.84 | 4/30/2022 | ||||||||||||
| Allen A. Fienberg, Ph.D. |
37,500 | 0 | $ | 0.30 | 12/16/2013 | |||||||||||
| 37,500 | 0 | $ | 0.50 | 12/19/2014 | ||||||||||||
| 12,500 | 0 | $ | 0.60 | 12/14/2015 | ||||||||||||
| 12,500 | 0 | $ | 1.36 | 12/5/2016 | ||||||||||||
| 12,500 | 0 | $ | 1.50 | 12/12/2017 | ||||||||||||
| 10,000 | 0 | $ | 1.50 | 12/18/2018 | ||||||||||||
| 10,000 | 0 | $ | 2.74 | 6/10/2020 | ||||||||||||
| 6,666 | 3,334 | $ | 2.74 | 12/21/2020 | ||||||||||||
| 3,333 | 6,667 | $ | 2.84 | 4/30/2022 | ||||||||||||
| (1) | On December 20, 2012, the compensation committee of ITI approved grants of 50,000 options to Dr. Mates, 10,000 options to Mr. Hineline and 7,500 options to Dr. Fienberg (the “Additional Grants”) to be granted |
80
Table of Contents
| following the completion of a valuation of ITI’s common stock and an increase in the number of shares reserved under ITI’s 2003 Equity Incentive Plan. Following the completion of the valuation of ITI’s common stock and the increase in the number of shares reserved under ITI’s 2003 Equity Incentive Plan, on May 31, 2013 the board of directors of ITI approved the Additional Grants at an exercise price of $3.26 per share, which are not reflected in this table. |
| (2) | Unless otherwise indicated, each option to purchase our common stock vests as to 1/3 of the shares on the first anniversary of the grant date, 1/3 of the shares on the second anniversary of the grant date, and 1/3 of the shares on the third anniversary of the grant date. Each option to purchase our common stock that expires on April 30, 2022 vests as to 1/3 of the shares on December 20, 2012, 1/3 of the shares on December 20, 2013 and 1/3 of the shares on December 20, 2014. Each of these options has a ten year term from the date of grant. |
Option Exercises and Stock Vested in 2012
During the fiscal year ended December 31, 2012, none of our named executive officers exercised any options.
Pension Benefits
We do not have any qualified or non-qualified defined benefit plans.
Nonqualified Deferred Compensation
We do not have any nonqualified defined contribution plans or other deferred compensation plans.
Potential Payments upon Termination or Change in Control
Upon termination of employment without cause or a resignation for good reason, each as defined below, our named executive officers are entitled to receive severance payments. Severance for termination without cause or termination for good reason, each as defined below, for named executive officers is 12 months of base salary plus the pro rata portion of an amount equal to the bonus awarded to such named executive officer for the previous year, if any. In addition, each named executive officer is entitled to payment of 12 months of the portion of the premiums for medical insurance coverage under COBRA that we paid prior to such named executive officer’s termination. Payment of these severance benefits is conditioned on the named executive officer signing a general release in our favor.
The table below summarizes the potential payments and benefits to each of our named executive officers assuming a termination without cause or resignation for good reason had occurred as of December 31, 2012.
| Name |
Severance Payments(1) |
Bonus Payments(2) |
Post-Termination Benefits(3) |
Total Benefits |
||||||||||||
| Sharon Mates, Ph.D. |
$ | 588,400 | $ | 113,200 | $ | 14,976 | $ | 716,576 | ||||||||
| Lawrence J. Hineline |
$ | 250,000 | $ | 30,900 | $ | 14,976 | $ | 295,876 | ||||||||
| Allen A. Fienberg, Ph.D. |
$ | 250,400 | $ | 17,000 | $ | 14,976 | $ | 282,376 | ||||||||
| (1) | The severance agreements for our named executive officers are set forth in their respective employment agreements. |
| (2) | Reflects a pro rata portion of the named executive officer’s 2011 bonus based on the period from January 1, 2012 through December 31, 2012, which equals the full amount of the named executive officer’s 2011 bonus. However, the 2012 bonus had already been paid to such named executive officers prior to December 31, 2012 in the amounts set forth in the “Summary Compensation Table” above, so we would not have paid the amounts set forth in this column at December 31, 2012 in addition to the 2012 bonus payments already made. |
81
Table of Contents
| (3) | Represents premiums that would be payable by us for continuation of the executive’s medical and dental insurance coverage, assuming a termination without cause or resignation for good reason had occurred as of December 31, 2012. |
The table below summarizes the potential payments and benefits to each of our named executive officers assuming a termination following a change in control had occurred at December 31, 2012. Each of our named executive officers has agreed in writing that the Merger does not constitute a change in control under their respective employment agreements.
| Name |
Severance Payments(1) |
Bonus Payments(1)(2) |
Value of Additional Vested Option Awards(1)(3) |
Post- Termination Benefits(1)(4) |
Total Benefits |
|||||||||||||||
| Sharon Mates, Ph.D. |
$ | 882,600 | $ | 113,200 | $ | 22,668 | $ | 22,464 | $ | 1,040,932 | ||||||||||
| Lawrence J. Hineline |
$ | 375,000 | $ | 30,900 | $ | 4,533 | $ | 22,464 | $ | 432,897 | ||||||||||
| Allen A. Fienberg, Ph.D. |
N/A | N/A | $ | 3,400 | N/A | $ | 3,400 | |||||||||||||
| (1) | Each of our named executive officers, except for Dr. Fienberg, shall, if the executive’s employment is terminated for reasons other than death or disability within three months before or 12 months following a change of control, the executive terminates his or her employment for good reason during such period, or the executive terminates his or her employment for any reason within one month following a change of control, be entitled to (a) payment of 18 months of the executive’s then current base salary and the pro rata portion of an amount equal to the bonus the executive was awarded for the previous year, if any, and (b) payment by us of 18 months of the portion of the premiums for medical insurance coverage under COBRA that we paid prior to such executive’s termination. Such severance benefits following a change of control are payable on condition that the executive executes a general release in favor of us, returns all our property and complies with his or her post-termination obligations under his or her employment agreement, proprietary information, inventions, and non-competition agreement, and general release. In addition, in the event of a change of control, any unvested stock options or restricted stock awarded to Dr. Mates or Mr. Hineline will immediately vest and become exercisable and 75% of any unvested stock options or restricted stock awarded to Dr. Fienberg will immediately vest and become exercisable. |
| (2) | Reflects a pro rata portion of the named executive officer’s 2011 bonus based on the period from January 1, 2012 through December 31, 2012, which equals the full amount of the named executive officer’s 2011 bonus. However, the 2012 bonus had already been paid to such named executive officers prior to December 31, 2012 in the amounts set forth in the “Summary Compensation Table” above, so we would not have paid the amounts set forth in this column at December 31, 2012 in addition to the 2012 bonus payments already made. |
| (3) | This represents the intrinsic value of the number of option shares that would vest, assuming a change of control termination had occurred at December 31, 2012. |
| (4) | Represents premiums that would be payable by us for continuation of the executive’s medical and dental insurance coverage, assuming a change of control termination had occurred at December 31, 2012. |
The table below summarizes the potential payments and benefits to each of our named executive officers assuming a change in control without termination had occurred at December 31, 2012.
| Name |
Severance Payments |
Bonus Payments |
Value of Additional Vested Option Awards(1)(2) |
Post- Termination Benefits |
Total Benefits |
|||||||||||||||
| Sharon Mates, Ph.D. |
N/A | N/A | $ | 22,668 | N/A | $ | 22,668 | |||||||||||||
| Lawrence J. Hineline |
N/A | N/A | $ | 4,533 | N/A | $ | 4,533 | |||||||||||||
| Allen A. Fienberg, Ph.D. |
N/A | N/A | $ | 3,400 | N/A | $ | 3,400 | |||||||||||||
82
Table of Contents
| (1) | In the event of a change of control, any unvested stock options or restricted stock awarded to Dr. Mates or Mr. Hineline will immediately vest and become exercisable and 75% of any unvested stock options or restricted stock awarded to Dr. Fienberg will immediately vest and become exercisable. |
| (2) | This represents the intrinsic value of the number of option shares that would vest, assuming a change of control without termination had occurred at December 31, 2012. |
For purposes of severance payments, “good reason” is defined as an executive resigning after the occurrence of one of the following events without the executive’s written consent:
| • | The assignment to the executive of any duties or responsibilities which result in the material diminution of the executive’s position; |
| • | a reduction by the Company in the executive’s annual base salary of 5% or greater with respect to Dr. Mates and Mr. Hineline, and of greater than 5% with respect to Dr. Fienberg; |
| • | a material change in the geographic location at which the executive is required to perform services; or |
| • | material breach by the Company of any material provision of the executive’s employment agreement. |
The executive must provide us with written notice within 60 days after the occurrence of a good reason event, and we have 30 days to correct the event after receipt of the notice.
For purposes of severance payments, “cause” is defined as a termination by us after the occurrence of one of the following events:
| • | a good faith finding by the Company that the executive has engaged in gross negligence or gross misconduct that is materially injurious to the Company; |
| • | the executive’s conviction of a felony or crime involving fraud or embezzlement of Company property; |
| • | the executive’s material breach of the executive’s employment agreement which, if curable, has not been cured by the executive within 60 days after he or she receives written notice from the Company stating with reasonable specificity the nature of the breach; |
| • | material breach of fiduciary duty; or |
| • | refusal to follow or implement a clear and reasonable directive of our board of directors as a whole (or an officer of the Company, in the case of Mr. Hineline and Dr. Fienberg), provided that such directive is ethical and legal and which, if curable, has not been cured by the executive within 60 days after he or she receives written notice from the Company stating with reasonable specificity the nature of such refusal. |
For purposes of severance payments, the determination of “disability” will occur when the executive is unable due to a physical or mental condition to perform the essential functions of his or her position with or without reasonable accommodation for 90 consecutive days, or 180 days in the aggregate whether or not consecutive, during any 360-day period, or based on the written certification by a licensed physician of the likely continuation of such condition for such period.
For purposes of severance payments, a “change in control” means:
| • | a sale, lease or other disposition of all or substantially all of the assets of the Company; |
| • | a consolidation or merger of the Company with or into any other corporation or other entity or person, or any other corporate reorganization, in which the stockholders of the Company immediately prior to such consolidation, merger or reorganization, own less than 50% of the outstanding voting power of the surviving entity (and its parent) following the consolidation, merger or reorganization; or |
| • | any transaction (or series of related transactions involving a person or entity, or a group of affiliated persons or entities) in which in excess of 50% of the Company’s outstanding voting power is transferred. |
83
Table of Contents
Notwithstanding the foregoing, a “change in control” will not be deemed to occur on account of the sale or acquisition of the Company’s capital stock by institutional investors or venture capital firms for the primary purpose of obtaining financing for the Company.
Director Compensation
The following table shows the total compensation paid or accrued during the fiscal year ended December 31, 2012 to each of our directors, other than Dr. Mates who does not receive compensation for her service as a director.
| Name |
Fees Earned or Paid in Cash ($) |
Stock Awards ($) |
Option Awards(1) ($) |
Non-Equity Incentive Plan Compensation ($) |
Change in Pension Value and Nonqualified Deferred Compensation Earnings |
All Other Compensation ($) |
Total ($) |
|||||||||||||||||||||
| David Kipnis, M.D.(2)(3) |
N/A | N/A | 24,500 | N/A | N/A | N/A | 24,500 | |||||||||||||||||||||
| Richard Lerner, M.D.(4) |
N/A | N/A | 24,500 | N/A | N/A | N/A | 24,500 | |||||||||||||||||||||
| Joel S. Marcus(5) |
N/A | N/A | 24,500 | N/A | N/A | N/A | 24,500 | |||||||||||||||||||||
| (1) | These amounts represent the aggregate grant date fair value for option awards granted to our named executive officers, computed in accordance with FASB ASC Topic 718. See Note 5 to our audited financial statements for the fiscal years ended December 31, 2012 and 2011 included in this prospectus for details as to the assumptions used to calculate the fair value of the option awards. See also our discussion of stock-based compensation under “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Estimates.” |
| (2) | Dr. Kipnis resigned from the board of directors effective December 31, 2012. Effective January 1, 2013, the board of directors appointed Christopher Alafi, Ph.D. as a director, and effective May 10, 2013, the board of directors appointed Sir Michael Rawlins as a director. |
| (3) | As of December 31, 2012, Dr. Kipnis held 102,500 options to purchase shares of our common stock, of which 96,250 options were vested. |
| (4) | As of December 31, 2012, Dr. Lerner held 110,000 options to purchase shares of our common stock, of which 103,750 options were vested. |
| (5) | As of December 31, 2012, Mr. Marcus held 77,500 options to purchase shares of our common stock, of which 71,250 options were vested. |
Director Compensation Policy
As compensation to our non-employee directors for the year ending December 31, 2013 and the years ended December 31, 2012 and 2011, we granted options to purchase 20,000 shares, 12,500 shares and 12,500 shares of our common stock, respectively, to each of our non-employee directors serving during such years. We granted any non-employee director who resigned from or joined the ITI board of directors during such years the pro rata portion of the annual option grant representing the portion of such year during which such non-employee director served. We intend to adopt a non-employee director compensation policy designed to ensure that the compensation aligns the directors’ interests with the long-term interests of the stockholders, that the structure of the compensation is simple, transparent and easy for stockholders to understand and that our directors are fairly compensated.
84
Table of Contents
EQUITY COMPENSATION PLAN INFORMATION
The following table provides certain aggregate information with respect to all of our equity compensation plans in effect as of December 31, 2012.
| Plan category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights |
Weighted-average exercise price of outstanding options, warrants and rights |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||||
| Equity compensation plans approved by security holders(1)(2) |
1,707,113 | $ | 1.38 | 44,867 | (1) | |||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
| Total |
1,707,113 | (2) | $ | 1.38 | (2) | 44,867 | (2) | |||||
| (1) | This plan consists of the 2003 Equity Incentive Plan. The 2003 Equity Incentive Plan terminated by its terms in July 2013. As a result of such termination, no additional awards may be granted under the 2003 Equity Incentive Plan, but equity awards previously granted under the 2003 Equity Incentive Plan will remain outstanding and continue to be governed by the terms of the 2003 Equity Incentive Plan. |
| (2) | The table above does not include shares that are reserved for issuance under the 2013 Equity Incentive Plan, which was adopted in connection with the Merger, which consists of 799,934 shares reserved plus up to an additional 1,462,380 shares reserved solely after the cancellation or expiration of any unexercised stock options that we assumed in the Merger, subject to adjustment as provided in the plan. |
2003 Equity Incentive Plan
The ITI 2003 Equity Incentive Plan, as amended, was adopted by the board of directors of ITI in July 2003 and by the stockholders of ITI in September 2003. The 2003 Equity Incentive Plan was subsequently amended in January 2006, February 2010 and December 2012, and expired by its terms in July 2013. As a result of such expiration, no additional awards may be granted under the 2003 Equity Incentive Plan, but equity awards previously granted under the 2003 Equity Incentive Plan will remain outstanding and continue to be governed by the terms of the 2003 Equity Incentive Plan. In connection with the Merger, we assumed the options then outstanding under the 2003 Equity Incentive Plan, and immediately following the Merger on August 29, 2013, the only outstanding awards under the 2003 Equity Incentive Plan were options to purchase 1,462,380 shares of our common stock. The 2003 Equity Incentive Plan is administered by our board of directors.
If we are acquired, the surviving or acquiring company may assume or continue the outstanding options by substituting either (a) the consideration payable with respect to the outstanding shares of common stock in connection with the acquisition or (b) shares of stock of the successor or acquiring company. If the surviving or acquiring company does not assume or continue the outstanding options, the outstanding options will be accelerated in full prior to the effective time of the acquisition and will terminate if not exercised at or prior to such effective time.
2013 Equity Incentive Plan
In August 2013, our board of directors approved the 2013 Equity Incentive Plan. The 2013 Equity Incentive Plan became effective on November 7, 2013, the date that is 20 days after we mailed the definitive information statement on Schedule 14C to our sole stockholder prior to the Merger. Unless sooner terminated by our board of directors or our stockholders, the 2013 Equity Incentive Plan will expire 10 years from its date of effectiveness. Under our 2013 Equity Incentive Plan, we may grant incentive stock options, nonstatutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights and other stock awards to our employees, directors and consultants.
85
Table of Contents
The maximum number of shares of our common stock that may be delivered in satisfaction of awards under the 2013 Equity Incentive Plan is 799,934 shares, plus up to an additional maximum of 1,462,380 shares which may be issued solely after the cancellation or expiration of any unexercised stock options that we assumed in the Merger. These numbers are subject to adjustment in the event of a stock split, stock dividend or other change in our capitalization.
In addition, the 2013 Equity Incentive Plan contains an “evergreen” provision, which allows for an annual increase in the number of shares of our common stock available for issuance under the 2013 Equity Incentive Plan on January 1 of each year commencing on January 1, 2014 and ending upon expiration of the 2013 Equity Incentive Plan. The annual increase in the number of shares shall be equal to the lesser of:
| • | 800,000 shares of our common stock; |
| • | 4% of the number of shares of our common stock outstanding as of such date; and |
| • | such lesser number of shares as determined by our board of directors prior to the applicable January 1st date. |
Shares of our common stock to be issued under the 2013 Equity Incentive Plan may be authorized but unissued shares of our common stock or previously issued shares acquired by us. Any shares of our common stock underlying awards that otherwise expire, terminate, or are forfeited or reacquired by us will again be available for issuance under the 2013 Equity Incentive Plan.
The 2013 Equity Incentive Plan will be administered by our board of directors until we establish a compensation committee. Our board of directors, or compensation committee once established, will have full power and authority to determine the terms of awards granted pursuant to this plan, including:
| • | which employees, directors and consultants shall be granted awards; |
| • | the type of award to be granted; |
| • | the terms and conditions of each award, including the schedule upon which the participant may exercise or otherwise receive common stock under the award; and |
| • | all other terms and conditions upon which each award may be granted in accordance with the 2013 Equity Incentive Plan. |
However, at such time as the Company may be subject to Section 162(m) of the Internal Revenue Code of 1986, as amended, or the Code, a maximum of 200,000 shares of our common stock subject to options, stock appreciation rights and other awards whose value is determined by reference to an increase over an exercise or strike price of at least 100% of the fair market value on the date the award is granted may be granted to any one participant during any one calendar year.
Our board of directors may amend or discontinue the 2013 Equity Incentive Plan at any time and may amend any outstanding award. No such amendment may materially impair the rights under any outstanding award without the holder’s consent. Stockholder approval will be required for any amendment to the 2013 Equity Incentive Plan to the extent such approval is required by law, including the Code or applicable stock exchange requirements.
If we are acquired, our board of directors (or compensation committee, once established) will (i) arrange for the surviving corporation or acquiring corporation (or the surviving or acquiring corporation’s parent company) to assume or continue the award or to substitute a similar award for the award; (ii) cancel or arrange for the cancellation of the award, to the extent not vested or not exercised prior to the effective time of the transaction, in exchange for such cash consideration, if any, as our board of directors in its sole discretion, may consider appropriate; and (iii) make a payment, in such form as may be determined by our board of directors equal to the excess, if any, of (A) the value of the property the holder would have received upon the exercise of the award immediately prior to the effective time of the transaction, over (B) any exercise price payable by such holder in
86
Table of Contents
connection with such exercise. In addition in connection with such transaction, our board of directors may accelerate the vesting, in whole or in part, of the award (and, if applicable, the time at which the award may be exercised) to a date prior to the effective time of such transaction and may arrange for the lapse, in whole or in part, of any reacquisition or repurchase rights held by the Company with respect to an award.
Indemnification of Directors and Officers
Our restated certificate of incorporation and our restated bylaws provide that each person who was or is made a party or is threatened to be made a party to or is otherwise involved (including, without limitation, as a witness) in any action, suit or proceeding, whether civil, criminal, administrative or investigative, by reason of the fact that he or she is or was one of our directors or officers or is or was serving at our request as a director, officer, or trustee of another corporation, or of a partnership, joint venture, trust or other enterprise, including service with respect to an employee benefit plan, whether the basis of such proceeding is alleged action in an official capacity as a director, officer or trustee or in any other capacity while serving as a director, officer or trustee, shall be indemnified and held harmless by us to the fullest extent authorized by the Delaware General Corporation Law against all expense, liability and loss (including attorneys’ fees, judgments, fines, ERISA excise taxes or penalties and amounts paid in settlement) reasonably incurred or suffered by such.
Section 145 of the Delaware General Corporation Law permits a corporation to indemnify any director or officer of the corporation against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, if he or she had no reasonable cause to believe his or her conduct was unlawful. In a derivative action (i.e., one brought by or on behalf of the corporation), indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or suit if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the Delaware Chancery Court or the court in which the action or suit was brought shall determine that such person is fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
Pursuant to Section 102(b)(7) of the Delaware General Corporation Law, Article Eighth of our restated certificate of incorporation eliminates the liability of a director to us or our stockholders for monetary damages for such a breach of fiduciary duty as a director, except for liabilities arising:
| • | from any breach of the director’s duty of loyalty to us or our stockholders; |
| • | from acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; |
| • | under Section 174 of the Delaware General Corporation Law; and |
| • | from any transaction from which the director derived an improper personal benefit. |
We have entered into indemnification agreements with our directors and certain officers, in addition to the indemnification provided in our restated certificate of incorporation and our restated bylaws, and intend to enter into indemnification agreements with any new directors and executive officers in the future. We have purchased and intend to maintain insurance on behalf of any person who is or was a director or officer against any loss arising from any claim asserted against him or her and incurred by him or her in any such capacity, subject to certain exclusions.
87
Table of Contents
In addition, as a condition to the Merger, we also entered into an indemnity agreement with the former officer and director of the Public Shell pursuant to which we agreed to indemnify such former officer and director for actions taken by him in his official capacity relating to the consideration, approval and consummation of the Merger and certain related transactions.
The foregoing discussion of our restated certificate of incorporation, restated bylaws, indemnification agreements, indemnity agreement, and Delaware law is not intended to be exhaustive and is qualified in its entirety by such restated certificate of incorporation, restated bylaws, indemnification agreements, indemnity agreement, or law.
Compensation Committee Interlocks and Insider Participation
We currently do not have a compensation committee. While ITI had a compensation committee comprised of all directors serving on the ITI board of directors, no member of ITI’s compensation committee has at any time been an employee of ours or ITI other than Dr. Mates, who did not vote on her own compensation. None of our executive officers serves as a member of the board of directors or compensation committee of any other entity that has one or more executive officers serving as a member of our board of directors or compensation committee.
88
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
Since January 1, 2011, ITI has engaged in the following transactions with its directors, executive officers and holders of more than 5% of its voting securities, which we refer to as our principal stockholders, and affiliates or immediate family members of our directors, executive officers and principal stockholders. We believe that all of these transactions were on terms as favorable as could have been obtained from unrelated third parties.
As described above, the following executive officers and directors held the following positions at ITI prior to the Merger:
| • | Sharon Mates, Ph.D., our President, Chief Executive Officer and Chairman of our board of directors, was the President, Chief Executive Officer and Chairman of the board of directors of ITI prior to the Merger. |
| • | Lawrence J. Hineline, our Vice President of Finance, Chief Financial Officer and Secretary, was the Vice President of Finance, Chief Financial Officer and Secretary of ITI prior to the Merger. |
| • | Allen A. Fienberg, Ph.D., our Vice President of Business Development, was the Vice President of Business Development of ITI prior to the Merger. |
| • | Lawrence P. Wennogle, Ph.D., our Vice President, Drug Discovery, was the Vice President, Drug Discovery of ITI prior to the Merger. |
| • | Kimberly E. Vanover, Ph.D., our Vice President, Clinical Development, was the Vice President, Clinical Development of ITI prior to the Merger. |
| • | Our directors Christopher Alafi, Ph.D., Richard Lerner, M.D., Joel S. Marcus and Sir Michael Rawlins, M.D., FRCP, FMedSci were each directors of ITI prior to the Merger. |
Some of our directors are affiliated with our principal stockholders as indicated in the table below:
| Director |
Affiliation with Principal Stockholder | |
| Christopher Alafi, Ph.D. | Dr. Alafi is a General Partner of Alafi Capital Company, LLC. | |
| Joel S. Marcus | Mr. Marcus is co-founder, Chairman of the board of directors, Chief Executive Officer, President and a director of Alexandria Real Estate Equities, Inc., which is the managing member of Alexandria Equities, LLC. | |
The directors of ITI were previously selected as directors of ITI in accordance with the terms of ITI’s then existing restated certificate of incorporation, which is no longer effective following the Merger, and ITI’s Second Amended and Restated Voting Agreement effective as of October 25, 2007, as amended, which was terminated immediately prior to the Effective Time of the Merger.
89
Table of Contents
Convertible Promissory Notes Issued in 2012 and 2013
In October 2012, ITI entered into a convertible note purchase agreement with certain investors pursuant to which ITI issued convertible promissory notes having an aggregate principal amount of approximately $15.3 million, which were issued on October 25, 2012, November 14, 2012 and March 20, 2013. Certain of these convertible promissory notes were purchased by our principal stockholders in the following amounts and on the following dates:
| Name of Beneficial Owner(1) |
Original Principal Amount of Convertible Notes |
Issuance Date | ||||||
| Alafi Capital Company, LLC |
$ | 6,423,419 | October 25, 2012 | |||||
| Alexandria Equities, LLC |
$ | 1,812,307 | October 25, 2012 | |||||
| Sosland Family Trust B Partnership |
$ | 4,783,094 | October 25, 2012 | |||||
| (1) | Does not include the convertible promissory note having a principal amount of $124,975 held by J.D.F. Holdings Ltd., in which Allen A. Fienberg, Ph.D., our Vice President of Business Development, holds a 20% ownership interest. Dr. Fienberg has no voting or investment control with respect to any of the securities owned by J.D.F. Holdings Ltd. |
The convertible promissory notes were unsecured, accrued interest at the rate of 6% per year and had a maturity date of October 25, 2013. The convertible promissory notes converted into shares of ITI common stock in connection with the Private Placement discussed in “—Common Stock Issued in Private Placement in 2013” below. In addition, ITI paid $7,200 to the purchasers’ legal counsel for fees and expenses of purchasers’ legal counsel incurred in connection with the convertible promissory note financing.
Common Stock Issued in Private Placement in 2013
The following table summarizes ITI’s sales of its common stock on August 29, 2013 in the Private Placement to our officers, directors and beneficial owners of more than five percent of any class of our voting securities. The purchase price of $3.1764 per share (as adjusted to $6.3528 after giving effect to the Merger) was the fair market value as determined by arms-length negotiations between sophisticated investors and ITI’s management and board of directors. In addition, the holders of the convertible promissory notes elected to convert the aggregate principal amount plus accrued interest on all of ITI’s outstanding convertible promissory notes into shares of ITI common stock at the purchase price of $3.1764 per share (as adjusted to $6.3528 after giving effect to the Merger). ITI received no additional consideration from the conversion of the convertible promissory notes.
| Name of Beneficial Owner(1) |
Purchase Price of ITI Common Stock |
Principal Plus Accrued Interest of Convertible Notes Through Date of Conversion |
Shares of ITI Common Stock Issued(5) |
|||||||||
| Sharon Mates, Ph.D.(2) |
$ | 24,998 | — | 7,870 | ||||||||
| Joel S. Marcus and Barbara A. Marcus Family Trust(3) |
$ | 100,006 | — | 31,484 | ||||||||
| Alafi Capital Company, LLC |
$ | 4,747,498 | $ | 6,748,637 | 3,619,234 | |||||||
| Moshe Alafi(4) |
$ | 100,006 | — | 31,484 | ||||||||
| Alexandria Equities, LLC |
$ | 1,339,462 | $ | 1,904,064 | 1,021,133 | |||||||
| Entities affiliated with Fidelity Investments |
$ | 12,700,003 | — | 3,998,238 | ||||||||
| David N. Sosland Trust A |
$ | 900,004 | — | 283,341 | ||||||||
| Sosland Family Trust B Partnership |
$ | 400,006 | $ | 5,025,263 | 1,707,993 | |||||||
| The Sosland Foundation |
$ | 2,235,128 | — | 703,667 | ||||||||
| (1) | Does not include 104,301 shares of ITI common stock issued to J.D.F Holdings Ltd., in which Allen A. Fienberg, Ph.D., our Vice President of Business Development, holds a 20% ownership interest. Dr. Fienberg has no voting or investment control with respect to any of the shares owned by J.D.F. Holdings Ltd. |
90
Table of Contents
| (2) | Dr. Mates is our Chairman, President and Chief Executive Officer. |
| (3) | Mr. Marcus is one of our directors. Mr. Marcus may also be deemed to beneficially own the shares held by Alexandria Equities, LLC set forth in the table. |
| (4) | Moshe Alafi may also be deemed to beneficially own the shares held by Alafi Capital Company, LLC set forth in the table. |
| (5) | Does not reflect the adjustment in the number of shares as a result of the Merger. |
At the Effective Time of the Merger, on August 29, 2013, each share of ITI preferred stock and ITI common stock outstanding immediately prior to the Effective Time was exchanged for 0.5 shares of the Company’s common stock. The following table summarizes the exchange of the outstanding shares of ITI preferred stock and common stock at the Effective Time by our officers, directors and beneficial owners of more than five percent of any class of our voting securities.
| Name of Beneficial Owner |
Number of Shares of ITI Preferred Stock Held Immediately Prior to Exchange |
Number of Shares of ITI Common Stock Held Immediately Prior to Exchange |
Number of Shares of Company Common Stock Held Immediately Following Exchange |
|||||||||
| Sharon Mates, Ph.D.(1) |
— | 2,107,870 | 1,053,935 | |||||||||
| Lawrence J. Hineline(2) |
— | 100,000 | 50,000 | |||||||||
| Allen A. Fienberg, Ph.D.(3) |
— | 475,000 | 237,500 | |||||||||
| Lawrence P. Wennogle(4) |
— | 200,000 | 100,000 | |||||||||
| Christopher Alafi, Ph.D.(5) |
1,007,505 | 0 | 503,753 | |||||||||
| Richard Lerner, M.D.(6) |
— | 75,000 | 37,500 | |||||||||
| Joel S. Marcus and Barbara A. Marcus Family Trust(7) |
— | 31,484 | 15,742 | |||||||||
| Alafi Capital Company, LLC |
3,466,535 | 3,619,234 | 3,542,885 | |||||||||
| Moshe Alafi(8) |
— | 31,484 | 15,742 | |||||||||
| Alexandria Equities, LLC |
1,546,579 | 1,021,133 | 1,283,856 | |||||||||
| Entities affiliated with Fidelity Investments |
— | 3,998,238 | 1,999,120 | |||||||||
| Paul Greengard, Ph.D.(9) |
— | 2,262,500 | 1,131,250 | |||||||||
| David N. Sosland Trust A |
1,131,233 | 283,341 | 707,287 | |||||||||
| The Sosland Family Trust B Partnership |
2,189,115 | 1,707,993 | 1,948,554 | |||||||||
| The Sosland Foundation |
761,429 | 703,667 | 732,548 | |||||||||
| (1) | Dr. Mates is our Chairman, President and Chief Executive Officer. |
| (2) | Mr. Hineline is our Vice President of Finance, Chief Financial Officer and Secretary. |
| (3) | Dr. Fienberg is our Vice President of Business Development. Does not include: (i) 311,745 shares of ITI preferred stock and 104,301 shares of ITI common stock held by J.D.F. Holdings Ltd., in which Dr. Fienberg holds a 20% ownership interest; or (ii) 100,000 shares of ITI common stock held by two trusts for the benefit of members of Dr. Fienberg’s family, which were exchanged for an aggregate of 258,023 shares of our common stock at the Effective Time. Dr. Fienberg has no voting or investment control with respect to any of the shares owned by J.D.F. Holdings Ltd. or held in the trusts. |
| (4) | Dr. Wennogle is our Vice President, Drug Discovery. |
| (5) | Dr. Alafi is one of our directors. Consists of shares held by a trust for the benefit of members of Dr. Alafi’s family. Dr. Alafi may also be deemed to beneficially own the shares held by Alafi Capital Company, LLC set forth in the table. Does not include 1,007,550 shares of ITI preferred stock, which were exchanged for 503,776 shares of our common stock at the Effective Time, held by two other trusts for the benefit of members of Dr. Alafi’s family, as Dr. Alafi does not have voting or investment control over the shares held by those trusts. |
| (6) | Dr. Lerner is one of our directors. Consists of shares held by the Lerner Family Trust UAD 11/14/94, or the Lerner Family Trust. Dr. Lerner shares voting and investment control with respect to the shares held by the Lerner Family Trust. |
91
Table of Contents
| (7) | Mr. Marcus is one of our directors. Mr. Marcus may also be deemed to beneficially own the shares held by Alexandria Equities, LLC set forth in the table. |
| (8) | Moshe Alafi may also be deemed to beneficially own the shares held by Alafi Capital Company, LLC set forth in the table. |
| (9) | Does not include 3,000,000 shares of ITI common stock, which were exchanged for 1,500,000 shares of our common stock at the Effective Time, held by six trusts for the benefit of members of Dr. Greengard’s family, as the trustee of these trusts, Ursula von Rydingsvard, who is Dr. Greengard’s spouse, has sole voting and investment control over the shares held by the trusts. |
Assumption of Outstanding Stock Options in Merger
At the Effective Time, on August 29, 2013, the Company assumed all options to purchase ITI common stock then outstanding under the ITI 2003 Equity Incentive Plan, and such options became exercisable for an aggregate of 1,462,380 shares of Company common stock, subject to the vesting and other terms of such options. The vesting of such options was not accelerated as a result of the Merger. The following table provides the number of outstanding options and the weighted average exercise price of such options under the 2003 Equity Incentive Plan held by our officers, directors and beneficial owners of more than five percent of any class of our voting securities that the Company assumed from ITI in connection with the Merger, as adjusted for the exchange ratio in the Merger:
| Name of Beneficial Owner |
Number of Shares of the Company’s Common Stock Underlying Outstanding Options |
Weighted Average Exercise Price Per Share |
||||||
| Sharon Mates, Ph.D. |
387,500 | $ | 2.02 | |||||
| Lawrence J. Hineline |
125,000 | $ | 1.54 | |||||
| Allen A. Fienberg, Ph.D. |
122,500 | $ | 1.51 | |||||
| Lawrence P. Wennogle, Ph.D. |
130,000 | $ | 1.34 | |||||
| Kimberly E. Vanover, Ph.D. |
49,750 | $ | 2.38 | |||||
| Christopher Alafi, Ph.D. |
29,375 | $ | 3.26 | |||||
| Richard Lerner, M.D. |
105,000 | $ | 2.49 | |||||
| Joel S. Marcus |
110,000 | $ | 2.42 | |||||
| Sir Michael Rawlins, M.D., FRCP, FMedSci |
27,000 | $ | 3.26 | |||||
The Redemption
Immediately following the Effective Time, pursuant to the terms of a Redemption Agreement dated August 29, 2013 by and among the Company and its then-current sole stockholder, we completed the closing of a redemption of 5,000,000 shares of Company common stock from our then-current sole stockholder in consideration of $60,000, plus professional costs related to the transaction not to exceed $20,000. The 5,000,000 shares constituted all of the issued and outstanding shares of the Company’s capital stock, on a fully-diluted basis, immediately prior to the Merger.
Agreements with Stockholders
Termination of Existing Stockholder Agreements
In connection with ITI’s Series C preferred stock financing in 2007 and 2010, ITI entered into various stockholder agreements with the holders of its common stock and preferred stock relating to voting rights, information rights and registration rights, among other things. The stockholder agreements terminated immediately prior to the Effective Time of the Merger. Parties to the existing registration rights agreement,
92
Table of Contents
however, as well as our directors and executive officers and all of our other stockholders, were provided with an opportunity to become parties to the registration rights agreement that ITI entered into in connection with the Private Placement, as discussed below under “—Registration Rights Agreement.”
Registration Rights Agreement
At the closing of the Private Placement, ITI entered into a registration rights agreement with the investors in the Private Placement and also the existing stockholders of ITI who agreed to become parties to certain provisions of the agreement or who choose to become parties in the future, which covers substantially all of our outstanding shares of common stock as of December 1, 2013. We assumed the registration rights agreement in connection with the Merger. Pursuant to the registration rights agreement and subject to the rules and regulations of the SEC, we have agreed to file a shelf registration statement covering the resale of the shares of our common stock held by the investors in the Private Placement and the shares of our common stock held by the former stockholders of ITI who are parties to the agreement. We were required to file the shelf registration statement within 45 days of the date of the registration rights agreement (October 13, 2013), which we filed on September 18, 2013. The registration statement of which this prospectus forms a part is the shelf registration statement that we are required to file under the registration rights agreement. In the event fewer than all of our outstanding shares of common stock can be registered pursuant to the so-called Rule 415 doctrine, priority will be given to the shares issued in the Private Placement, but in such event at least 25% of the shares registered shall be those held by ITI’s existing stockholders prior to the Private Placement.
We will be liable to each investor in the Private Placement (but not to the former stockholders of ITI who are parties to the agreement) for liquidated damages, on a 30-day basis, equal to 1.0% of the aggregate purchase price paid by the investor for the registrable shares of our common stock then held by the investor, subject to an overall cap of 5%, (i) if we fail to file the registration statement on time, (ii) if the registration statement is not declared effective within 150 days from the date of the registration rights agreement (January 26, 2014), (iii) if we suspend (subject to limited blackout periods described below) or terminate the registration statement prior to the date which is the earlier of (x) the third anniversary of its effectiveness (or the third anniversary of the date on which all registrable shares are included therein, if later) and (y) the date on which all of the registrable shares cease to be registrable shares, or (iv) in the event one or more suspensions of the effectiveness of the registration statement exceeds 60 days in the aggregate during any 12-month period. We will be permitted to suspend the registration statement one or more times during any 12-month period provided such suspensions do not exceed 30 consecutive days or 60 days in the aggregate in any 12-month period. Any suspension associated with our filing of an annual, periodic or current report, as required by the Exchange Act, will be permitted and will not be counted against the 60 day limitation. Any shares not registered due to the Rule 415 doctrine will not be subject to liquidated damages. Expenses with respect to the filing and effectiveness of such registration statement (but not selling expenses, or underwriter or agent compensation) will be paid by us, including expenses of one counsel for the selling stockholders.
Lock-Up Provisions in Registration Rights Agreement
One of the provisions of the registration rights agreement that is applicable to the former stockholders of ITI who are parties to the agreement, other than the investors in the Private Placement, who hold an aggregate of approximately 12,603,527 shares of common stock, is a lock-up provision pursuant to which these stockholders agreed, subject to specified exceptions, not to sell, transfer, dispose of, contract to sell, sell any option or contract to purchase, or otherwise transfer or dispose of, directly or indirectly, without the written consent of Leerink Swann LLC or, in certain circumstances, the two lead institutional investors in the Private Placement, any shares of our common stock or any securities convertible into or exercisable or exchangeable for shares of our common stock until the earlier of (a) up to 180 days (as requested by the lead underwriter) after the date on which our shares of common stock are listed for trading on a national securities exchange in connection with a firm commitment underwritten public offering by us with gross proceeds to us of at least $40 million, or (b) the date that is 18 months following the date of the Merger (February 28, 2015). These lock-up provisions will not apply to, among other things, shares of common stock acquired in connection with any follow-on securities offerings
93
Table of Contents
by us or in open market transactions, or upon the exercise of stock options granted pursuant to our equity incentive plans, so long as the shares acquired upon exercise remain subject to the lock-up provisions in the agreement, or certain gifts and other transfers for estate-planning purposes or by stockholders who are entities to their limited partners, members or stockholders, as specified in the agreement. In the event that a former stockholder of ITI was also an investor in the Private Placement, then these lock-up provisions in the agreement will only apply with respect to the shares held by such stockholder that were not purchased in the Private Placement. Under the registration rights agreement, we did not enter into any lock-up agreement or other restriction on the transfer or registration of the shares of our common stock acquired by the investors in the Private Placement (other than those imposed by non-disclosure agreements and the requirement that each investor continue to hold 200 shares of ITI common stock (which were exchanged for 100 shares of our common stock in the Merger) until the earlier of (y) the date on which our shares are listed on a national securities exchange or (z) the date 18 months after the closing of the Private Placement (February 28, 2015)).
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and certain of our officers. The indemnification agreements, our restated certificate of incorporation and our restated bylaws require us to indemnify our directors and officers to the fullest extent permitted by Delaware law. In addition, as a condition to the Merger, we also entered into an indemnity agreement with the former sole officer and director of the Company pursuant to which we agreed to indemnify him for actions taken by him in his official capacities relating to the consideration, approval and consummation of the Merger and certain related transactions. See “Indemnification of Directors and Officers.”
Indemnity Agreement
As a condition to the Merger, we entered into an Indemnity Agreement with our former sole officer and director pursuant to which we agreed to indemnify such former officer and director for actions taken by him in his official capacities relating to the consideration, approval and consummation of the Merger and certain related transactions.
Policy for Approval of Related Person Transactions
We do not currently have a policy for the review and approval of related person transactions. We intend to adopt such a policy when we adopt an audit committee charter and establish an audit committee, which we expect will be responsible for reviewing and approving all transactions in which we are a participant and in which any parties related to us, including our executive officers, our directors, beneficial owners of more than 5% of our securities, immediate family members of the foregoing persons and any other persons whom our board of directors determines may be considered related parties under Item 404 of Regulation S-K, has or will have a direct or indirect material interest.
Legal Proceedings
We are not aware of any material proceedings in which any of our directors, executive officers or affiliates, any owner of record or beneficially of more than 5% of our common stock, or any associate of any such director, officer, affiliate or security holder is a party adverse to us or any of our subsidiaries or has a material interest adverse to us.
94
Table of Contents
We are filing the registration statement of which this prospectus forms a part to permit holders of the shares of our common stock described in the section entitled “Selling Stockholders” to resell such shares. We will not receive any proceeds from the resale of any shares offered by this prospectus by the selling stockholders.
We have never paid cash dividends on any of our capital stock and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. We do not intend to pay cash dividends to holders of our common stock in the foreseeable future.
DETERMINATION OF OFFERING PRICE
The selling stockholders may only sell their shares of our common stock pursuant to this prospectus at a fixed price of $6.3528 per share until such time as our common stock is quoted on the OTCQB or another public trading market for our common stock otherwise develops. At and after such time, the selling stockholders may sell all or a portion of their shares through public or private transactions at prevailing market prices or at privately negotiated prices. The fixed price of $6.3528 at which the selling stockholders may sell their shares pursuant to this prospectus was determined based upon the purchase price per share of ITI common stock in the Private Placement, as adjusted for the Exchange of our common stock in the Merger, which was completed on August 29, 2013. We have included a fixed price at which selling stockholders may sell their shares pursuant to this prospectus prior to the time there is a public market for our stock in order to comply with the rules of the SEC that require that, if there is no market for the shares being registered, the registration statement must include a price at which the shares may be sold. All shares being offered pursuant to this prospectus will be sold by existing stockholders without our involvement.
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
There is not currently, and there has never been, any market for any of our securities. Our securities are not eligible for trading on any national securities exchange and are not quoted for sale on any over-the-counter markets, including the OTCQB.
As of December 1, 2013, we had 22,134,647 outstanding shares of common stock and no outstanding shares of preferred stock. As of December 1, 2013, there were 258 holders of record of our common stock.
95
Table of Contents
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth the number of shares of our common stock beneficially owned as of December 1, 2013, by (i) each of our current directors and named executive officers, (ii) all executive officers and directors as a group, and (iii) each person known by us to be the beneficial owner of more than 5% of the outstanding shares of our common stock. Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them based on information provided to us by these stockholders, subject to community property laws, where applicable. Percentage of ownership is based on 22,134,647 shares of common stock outstanding on December 1, 2013, after giving effect to the Private Placement and Merger on August 29, 2013. Unless otherwise noted below, the address of each stockholder below is c/o Intra-Cellular Therapies, Inc., 3960 Broadway, New York, New York 10032.
| Beneficial Owner |
Title |
Shares of Common Stock Beneficially Owned (#)(1) |
Percentage of Common Stock Beneficially Owned (%)(1) |
|||||||
| Directors and Named Executive Officers | ||||||||||
| Sharon Mates, Ph.D.(2) |
Chairman, President and Chief Executive Officer | 1,391,430 | 6.2 | % | ||||||
| Lawrence J. Hineline(3) |
Vice President of Finance, Chief Financial Officer and Secretary | 164,999 | * | |||||||
| Allen A. Fienberg, Ph.D.(4) |
Vice President of Business Development | 351,665 | 1.6 | % | ||||||
| Christopher Alafi, Ph.D.(5) |
Director | 4,046,638 | 18.3 | % | ||||||
| Richard Lerner, M.D.(6) |
Director | 116,250 | * | |||||||
| Joel S. Marcus(7) |
Director | 1,383,348 | 6.2 | % | ||||||
| Sir Michael Rawlins, M.D., FRCP, FMedSci |
Director | — | * | |||||||
| All current executive officers and directors as a group (9 persons)(8) |
7,717,411 | 33.5 | % | |||||||
| Other 5% or More Stockholders | ||||||||||
| Alafi Capital Company, LLC and Moshe Alafi(9) |
3,558,627 | 16.1 | % | |||||||
| Alexandria Real Estate Equities, Inc.(10) |
1,283,856 | 5.8 | % | |||||||
| Entities affiliated with Fidelity Investments(11) |
1,999,120 | 9.0 | % | |||||||
| Paul Greengard, Ph.D.(12) |
1,131,250 | 5.1 | % | |||||||
| Morton I. Sosland(13) |
3,388,389 | 15.3 | % | |||||||
| * | Represents beneficial ownership of less than 1% of the shares of common stock. |
| (1) | Beneficial ownership is determined in accordance with SEC rules, and includes any shares as to which the stockholder has sole or shared voting power or investment power, and also any shares which the stockholder has the right to acquire within 60 days of December 1, 2013, whether through the exercise or conversion of any stock option, convertible security, warrant or other right. The indication herein that shares are beneficially owned is not an admission on the part of the stockholder that he, she or it is a direct or indirect beneficial owner of those shares. |
| (2) | Consists of 1,053,935 shares of common stock and options to purchase 337,495 shares of common stock which are exercisable within 60 days of December 1, 2013. |
| (3) | Consists of 50,000 shares of common stock and options to purchase 114,999 shares of common stock which are exercisable within 60 days of December 1, 2013. |
| (4) | Consists of 237,500 shares of common stock and options to purchase 114,165 shares of common stock which are exercisable within 60 days of December 1, 2013. Does not include: (i) 208,023 shares of |
96
Table of Contents
| common stock held by J.D.F. Holdings Ltd., in which Dr. Fienberg holds a 20% ownership interest; and (ii) 50,000 shares of common stock held by two trusts for the benefit of members of Dr. Fienberg’s family. Dr. Fienberg has no voting or investment control with respect to any of the shares owned by J.D.F. Holdings Ltd. or held in the trusts. |
| (5) | Consists of 3,542,885 shares of common stock held by Alafi Capital Company, LLC, or Alafi Capital, and 503,753 shares of common stock held by a trust for the benefit of members of the Alafi family. Dr. Alafi is a managing partner of Alafi Capital and has shared voting and investment power with respect to the shares owned by Alafi Capital and full voting and investment power with respect to shares owned by the trust. Does not include 503,776 shares held by two other trusts for the benefit of members of the Alafi family for which Dr. Alafi does not have voting or investment control. The address for Dr. Alafi is c/o Alafi Capital Company, LLC, 8 Admiral Drive, Suite 324, Emeryville, CA 94608. |
| (6) | Consists of options to purchase 78,750 shares of common stock held by Dr. Lerner which are exercisable within 60 days of December 1, 2013, and 37,500 shares of common stock held by the Lerner Family Trust UAD 11/14/94, or the Lerner Family Trust. Dr. Lerner shares voting and investment control with respect to the shares held by the Lerner Family Trust. |
| (7) | Consists of (i) 1,283,856 shares of common stock held by Alexandria Equities, LLC, (ii) 15,742 shares of common stock held by the Joel S. Marcus and Barbara A. Marcus Family Trust, and (iii) options to purchase 83,750 shares of common stock held by Mr. Marcus, which are exercisable within 60 days of December 1, 2013. Mr. Marcus is the Chairman, CEO and Founder of Alexandria Real Estate Equities, Inc., which is the managing member of Alexandria Equities, LLC, which has full voting and investment power with respect to the shares owned by Alexandria Equities, LLC. As an officer of Alexandria Real Estate Equities, Inc., Mr. Marcus may be deemed to have voting and investment power with respect to the shares owned by Alexandria Equities, LLC. Mr. Marcus disclaims beneficial ownership of the shares held by Alexandria Equities, LLC, except to the extent of his underlying pecuniary interest therein. The address for Mr. Marcus is c/o Alexandria Real Estate Equities, Inc., 385 East Colorado Boulevard, Suite 299, Pasadena, CA 91101. |
| (8) | See footnotes 2 through 7. Also includes 100,000 shares of common stock and options to purchase 123,332 shares of common stock held by Lawrence P. Wennogle, Ph.D., Vice President, Drug Discovery, which are exercisable within 60 days of December 1, 2013, and options to purchase 39,749 shares of common stock, which are exercisable within 60 days of December 1, 2013, held by Kimberly E. Vanover, Ph.D., Vice President, Clinical Development. |
| (9) | Consists of 3,542,885 shares of common stock held by Alafi Capital and 15,742 shares of common stock held by Moshe Alafi. Christopher Alafi, Ph.D., one of our directors, and Moshe Alafi are each managing partners of Alafi Capital and share voting and investment power with respect to the shares owned by Alafi Capital. The address for Moshe Alafi and Alafi Capital is 8 Admiral Drive, Suite 324, Emeryville, CA 94608. |
| (10) | Consists of 1,283,856 shares of common stock held by Alexandria Equities, LLC. Joel S. Marcus, one of our directors, is the Chairman, CEO and Founder of Alexandria Real Estate Equities, Inc., which is the managing member of Alexandria Equities, LLC, which has full voting and investment power with respect to the shares owned by Alexandria Equities, LLC. As an officer of Alexandria Real Estate Equities, Inc., Mr. Marcus may be deemed to have voting and investment power with respect to the shares owned by Alexandria Equities, LLC. Mr. Marcus disclaims beneficial ownership of the shares held by Alexandria Equities, LLC, except to the extent of his underlying pecuniary interest therein. The address for Alexandria Equities, LLC is c/o Alexandria Real Estate Equities, Inc., 385 East Colorado Boulevard, Suite 299, Pasadena, CA 91101. |
| (11) | Fidelity Management & Research Company, or Fidelity, 82 Devonshire Street, Boston, Massachusetts 02109, a wholly-owned subsidiary of FMR LLC and an investment adviser registered under Section 203 of the Investment Advisers Act of 1940, is the beneficial owner of 1,999,120 shares of common stock as a result of acting as investment adviser to various investment companies registered under Section 8 of the Investment Company Act of 1940. Edward C. Johnson 3d and FMR LLC, through its control of Fidelity, and the funds each has sole power to dispose of the 1,999,120 shares of common stock owned by the Funds. Members of the family of Edward C. Johnson 3d, Chairman of FMR LLC, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting |
97
Table of Contents
| power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. Neither FMR LLC nor Edward C. Johnson 3d, Chairman of FMR LLC, has the sole power to vote or direct the voting of the shares owned directly by the Fidelity Funds, which power resides with the Funds’ Boards of Trustees. Fidelity carries out the voting of the shares under written guidelines established by the Funds’ Boards of Trustees. |
| (12) | Consists of 1,131,250 shares of common stock held by Dr. Greengard. Does not include 1,500,000 shares of common stock held by six trusts for the benefit of members of Dr. Greengard’s family, as the trustee of these trusts, Ursula von Rydingsvard, Dr. Greengard’s spouse, has sole voting and investment control over the shares held by the trusts. The address for Dr. Greengard and the trusts is Dr. Paul Greengard, c/o TAG Associates, 75 Rockefeller Plaza, 9th Floor, New York, NY 10019. |
| (13) | Consists of 707,287 shares of common stock held by David N. Sosland Trust A; 1,948,554 shares of common stock held by The Sosland Family Trust B Partnership; and 732,548 shares of common stock held by The Sosland Foundation. Morton I. Sosland is Trustee of the David N. Sosland Trust A, Managing Partner of The Sosland Family Trust B Partnership and Vice Chairman of The Sosland Foundation, which we refer to collectively as the Sosland Holders. As such, Mr. Sosland has sole voting and investment power with respect to the shares held by the Sosland Holders. The address for Mr. Sosland and the Sosland Holders is 4800 Main Street, Suite 100, Kansas City, MO 64112. |
98
Table of Contents
This prospectus covers the resale by the selling stockholders identified below of 21,961,496 shares of our common stock. The selling stockholders acquired our securities pursuant to the Exchange in the Merger. None of our selling stockholders received any of our securities as compensation for underwriting services. We will not receive any proceeds from the resale of the common stock by the selling stockholders.
Except as disclosed in the footnotes below, none of the selling stockholders has been an officer or director of ours or any of our predecessors or affiliates within the past three years. Except as disclosed in the footnotes below, no selling stockholder had a material relationship with the company or any of its affiliates within the last three years. Except as disclosed in the footnotes below, none of the selling stockholders is affiliated with a broker dealer.
The following table and the accompanying footnotes are based in part on information supplied to us by the selling stockholders. The table and footnotes assume that the selling stockholders will sell all of the shares listed. However, because the selling stockholders may sell all or some of their shares under this prospectus from time to time, or in another permitted manner, we cannot assure you as to the actual number of shares that will be sold by the selling stockholders or that will be held by the selling stockholders after completion of any sales. We do not know how long the selling stockholders will hold the shares before selling them.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC and includes voting or investment power with respect to our common stock. Shares of our common stock subject to options or warrants that are currently exercisable or exercisable within 60 days of December 1, 2013 are considered outstanding and beneficially owned by the person holding the options or warrants for the purpose of calculating the percentage ownership of that person but not for the purpose of calculating the percentage ownership of any other person. Except as otherwise noted in the footnotes below, we believe the persons and entities in this table have sole voting and investing power with respect to all of the shares of our common stock beneficially owned by them, subject to community property laws, where applicable. The inclusion of any shares in this table does not constitute an admission of beneficial ownership by the persons named below. The beneficial owners listed below are sorted alphabetically by first name.
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| 3MD Partnership Ltd. |
4,619 | * | 4,619 | — | * | |||||||||||||||
| Abbot A. Thayer |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Achyuet Sahasrabudhe |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Akinori Nishi |
2,500 | * | 2,500 | — | * | |||||||||||||||
| Alafi Capital Company, LLC(4) |
3,542,885 | 16.0 | 3,542,885 | — | * | |||||||||||||||
| Alan Mindel |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Alexander Pancoe Trust DTD 9/2/86 Mariann Pancoe & Gladys Pancoe TTEES |
12,000 | * | 12,000 | — | * | |||||||||||||||
| Alexandria Equities, LLC(5) |
1,283,856 | 5.8 | 1,283,856 | — | * | |||||||||||||||
| Allen Fienberg, Ph.D.(6) |
351,665 | 1.6 | 237,500 | 114,165 | * | |||||||||||||||
| Allen M. Demby |
4,723 | * | 4,723 | — | * | |||||||||||||||
| Alzheimer Drug Discovery Foundation, Inc. |
20,047 | * | 18,225 | 1,822 | * | |||||||||||||||
| Andrew D. and Gloria Wahl |
3,750 | * | 3,750 | — | * | |||||||||||||||
| Andrew Koopman |
1,575 | * | 1,575 | — | * | |||||||||||||||
99
Table of Contents
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| Andrew Rosen |
5,000 | * | 5,000 | — | * | |||||||||||||||
| Angus Nairn |
7,500 | * | 7,500 | — | * | |||||||||||||||
| Anthony Behette |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Aron Galinovsky |
1,800 | * | 1,800 | — | * | |||||||||||||||
| Art II w/r/t Gwendoline Hoguet F/B/O Geoffrey R. Hoguet |
82,876 | * | 82,876 | — | * | |||||||||||||||
| Arthur A. Feder |
5,250 | * | 3,750 | 1,500 | * | |||||||||||||||
| Arthur Pancoe Trust DTD 9/14/90 Arthur Pancoe Trustee |
38,000 | * | 38,000 | — | * | |||||||||||||||
| Ashnik Management, LLC |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Avraham and Susan Tahari |
6,055 | * | 6,055 | — | * | |||||||||||||||
| Barry Fienberg |
3,935 | * | 3,935 | — | * | |||||||||||||||
| Barry Levine |
1,600 | * | 1,600 | — | * | |||||||||||||||
| BB Trust |
4,000 | * | 4,000 | — | * | |||||||||||||||
| BGF World HealthScience Fund(7) |
174,133 | * | 174,133 | — | * | |||||||||||||||
| BlackRock Health Sciences Opportunities Portfolio, a series of BlackRock Funds(8) |
434,754 | 2.0 | 434,754 | — | * | |||||||||||||||
| BlackRock Health Sciences Trust(9) |
38,681 | * | 38,681 | — | * | |||||||||||||||
| Brad Deering |
3,463 | * | 3,463 | — | * | |||||||||||||||
| Brian Leentjes |
3,463 | * | 3,463 | — | * | |||||||||||||||
| Brian Skillern |
2,310 | * | 2,310 | — | * | |||||||||||||||
| Britton Joint Recovable Trust |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Broadfin Healthcare Master Fund, Ltd |
425,009 | 1.9 | 425,009 | — | * | |||||||||||||||
| Bruce and Bobra Locker Jt. WROS |
2,951 | * | 2,951 | — | * | |||||||||||||||
| Carlos Bermudez |
6,296 | * | 6,296 | — | * | |||||||||||||||
| Carol M. Mates |
3,935 | * | 3,935 | — | * | |||||||||||||||
| Charles E. Schroeder |
6,055 | * | 6,055 | — | * | |||||||||||||||
| Charles H. Scholpp |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Charles Sebestyen |
2,000 | * | 2,000 | — | * | |||||||||||||||
| Christopher D. Alafi as Trustee of The Moshe H. Alafi and Margaret E. Alafi Generation-Skipping Trust(10) |
503,753 | 2.3 | 503,753 | — | * | |||||||||||||||
| Claude Greengard |
1,968 | * | 1,968 | — | * | |||||||||||||||
| Dan Coombs and Mary Ellen Coombs |
4,619 | * | 4,619 | — | * | |||||||||||||||
| Daryl R. Schaller |
5,000 | * | 5,000 | — | * | |||||||||||||||
| David A. Miratsky |
2,755 | * | 2,755 | — | * | |||||||||||||||
| David Fyfe |
3,000 | * | 3,000 | — | * | |||||||||||||||
| David Kipnis(11) |
96,250 | * | 10,000 | 86,250 | * | |||||||||||||||
| David M. Schwaber |
41,726 | * | 41,726 | — | * | |||||||||||||||
| David N. Sosland Trust A(12) |
707,287 | 3.2 | 707,287 | — | * | |||||||||||||||
| David T. Quinby |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Deerfield Special Situations Fund, L.P. |
319,749 | 1.4 | 319,749 | — | * | |||||||||||||||
| Deerfield Special Situations International Master Fund, L.P. |
262,672 | 1.2 | 262,672 | — | * | |||||||||||||||
| Dennis C. Dean |
3,150 | * | 3,150 | — | * | |||||||||||||||
| Donald Wameldorph Jr. |
2,754 | * | 2,754 | — | * | |||||||||||||||
| Douglas P. Baker |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Duncan M. Scott |
3,028 | * | 3,028 | — | * | |||||||||||||||
| Edward Cronin |
1,750 | * | 1,750 | — | * | |||||||||||||||
100
Table of Contents
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| Edward Lopez |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Ellis R. Geulbeau |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Eric J. Hinds |
3,935 | * | 3,935 | — | * | |||||||||||||||
| Eric Nestler |
7,500 | * | 7,500 | — | * | |||||||||||||||
| Erin Coffey |
2,951 | * | 2,951 | — | * | |||||||||||||||
| Fidelity Advisor Series VII: Fidelity Advisor Biotechnology Fund(13) |
99,050 | * | 99,050 | — | * | |||||||||||||||
| Fidelity Mt. Vernon Street Trust: Fidelity Growth Company Fund(13) |
999,560 | 4.5 | 999,560 | — | * | |||||||||||||||
| Fidelity Select Portfolios: Biotechnology Portfolio(13) |
900,510 | 4.1 | 900,510 | — | * | |||||||||||||||
| Florence Kaufman |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Franklin M. Berger |
317,170 | 1.4 | 317,170 | — | * | |||||||||||||||
| G. Kenneth Baum Revocable Trust, under agreement dated Feb. 28, 1989, as amended |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Gary Grenley |
4,619 | * | 4,619 | — | * | |||||||||||||||
| Gary L. Sturm |
5,509 | * | 5,509 | — | * | |||||||||||||||
| GC&H Investments |
13,933 | * | 13,933 | — | * | |||||||||||||||
| GC&H Investments LLC |
135,347 | * | 135,347 | — | * | |||||||||||||||
| George O’Conner Trust |
5,509 | * | 5,509 | — | * | |||||||||||||||
| George R. Martin |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Gerrard A. Rutter |
1,800 | * | 1,800 | — | * | |||||||||||||||
| Gilberto Fisone |
2,500 | * | 2,500 | — | * | |||||||||||||||
| Greg Ryan |
5,250 | * | 5,250 | — | * | |||||||||||||||
| Gregory Hurley |
6,297 | * | 6,297 | — | * | |||||||||||||||
| Gregory J. Coffey |
2,951 | * | 2,951 | — | * | |||||||||||||||
| Gretchen Snyder |
39,999 | * | 12,500 | 27,499 | * | |||||||||||||||
| Guy Brad Brown and Beth Sutton |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Hannah Gabrielle Pancoe Trust DTD 9/24/87 Michael S. Pancoe, Gladys Pancoe & Eleanor Pancoe TTEES |
12,000 | * | 12,000 | — | * | |||||||||||||||
| Harold Melcher Revocable Trust |
3,148 | * | 3,148 | — | * | |||||||||||||||
| Harold O. LaFlash and Greta G. LaFlash Revocable Trust |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Henry Stronski and Jami L. Stronski |
2,951 | * | 2,951 | — | * | |||||||||||||||
| Hongwen Zhu |
3,583 | * | 3,583 | — | * | |||||||||||||||
| Howard K. Fuguet |
2,755 | * | 2,755 | — | * | |||||||||||||||
| Howard Wengrow |
3,936 | * | 3,936 | — | * | |||||||||||||||
| Hudson Asset Partners, LLC |
25,000 | * | 25,000 | — | * | |||||||||||||||
| ING BlackRock Health Sciences Opportunities Portfolio(14) |
52,912 | * | 52,912 | — | * | |||||||||||||||
| J. Michael Bowman |
1,750 | * | 1,750 | — | * | |||||||||||||||
| J.D.F. Holdings Ltd.(15) |
208,023 | * | 208,023 | — | * | |||||||||||||||
| James and Jana Grinnan |
2,755 | * | 2,755 | — | * | |||||||||||||||
| James Bibb |
2,500 | * | 2,500 | — | * | |||||||||||||||
| James Colthurst |
3,028 | * | 3,028 | — | * | |||||||||||||||
| James O’Callaghan |
5,000 | * | 5,000 | — | * | |||||||||||||||
| James S. Murday |
1,750 | * | 1,750 | — | * | |||||||||||||||
| James Surmeier |
7,500 | * | 7,500 | — | * | |||||||||||||||
101
Table of Contents
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| James Vann Cunningham |
1,725 | * | 1,725 | — | * | |||||||||||||||
| Janice Blustein |
1,968 | * | 1,968 | — | * | |||||||||||||||
| Jay Miselis |
2,000 | * | 2,000 | — | * | |||||||||||||||
| Jeff Wilmes |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Jeffrey Spotz |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Jill and Chris Manning |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Joe and Loretta Pillari Jt. WROS |
2,676 | * | 2,676 | — | * | |||||||||||||||
| Joel S. Marcus and Barbara A. Marcus Family Trust(16) |
15,742 | * | 15,742 | — | * | |||||||||||||||
| John C. Tomesch and Kristine K. Tomesch |
22,770 | * | 22,770 | — | * | |||||||||||||||
| John R Bartos |
5,500 | * | 5,500 | — | * | |||||||||||||||
| John S. and Linda McPhee |
6,968 | * | 6,968 | — | * | |||||||||||||||
| Joseph C. and Jennifer J. Garone |
5,000 | * | 5,000 | — | * | |||||||||||||||
| Joseph H. McCall |
5,000 | * | 5,000 | — | * | |||||||||||||||
| Joseph Haim Heskel |
15,742 | * | 15,742 | — | * | |||||||||||||||
| Joseph P. Hendrick, Jr. |
37,499 | * | 12,500 | 24,999 | * | |||||||||||||||
| Joseph P. Slattery |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Joyce Bielski |
3,149 | * | 3,149 | — | * | |||||||||||||||
| Judy Reed Smith |
5,903 | * | 5,903 | — | * | |||||||||||||||
| Jules P. Devigne |
5,903 | * | 5,903 | — | * | |||||||||||||||
| Jules P. Devigne and Deborah G. Devigne |
5,903 | * | 5,903 | — | * | |||||||||||||||
| Julia Lee Pancoe Trust DTD 2/25/83 E. Michael Pancoe, Gladys Pancoe & Eleanor Pancoe TTEES—FBO Julia Lee Pancoe |
12,000 | * | 12,000 | — | * | |||||||||||||||
| Karl Seck |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Kenneth S. Pizzo Jr. |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Kent Sullivan |
3,463 | * | 3,463 | — | * | |||||||||||||||
| Kevin C. McDonough |
3,850 | * | 3,850 | — | * | |||||||||||||||
| Kevin Gabrik |
3,149 | * | 3,149 | — | * | |||||||||||||||
| Kevin J. Surace |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Klondike Resources, Inc. |
163,123 | * | 163,123 | — | * | |||||||||||||||
| Larry Gelbfish |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Las Lomas Trust |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Lawrence Goodman |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Lawrence Hineline(17) |
164,999 | * | 50,000 | 114,999 | * | |||||||||||||||
| Lawrence P. Wennogle(18) |
223,332 | 1.0 | 100,000 | 123,332 | * | |||||||||||||||
| Leonard DeOliverra |
3,900 | * | 3,900 | — | * | |||||||||||||||
| Lerner Family Trust UAD 11/14/94(19) |
37,500 | * | 37,500 | — | * | |||||||||||||||
| Leslie F. Greengard |
1,968 | * | 1,968 | — | * | |||||||||||||||
| Leyla Greengard |
1,968 | * | 1,968 | — | * | |||||||||||||||
| Llano Resources, Inc. |
2,823 | * | 2,823 | — | * | |||||||||||||||
| Lloyd and Deborah Schill Jt. WROS |
3,500 | * | 3,500 | — | * | |||||||||||||||
| Louis Fienberg |
3,935 | * | 3,935 | — | * | |||||||||||||||
| Luigi Maneini |
2,750 | * | 2,750 | — | * | |||||||||||||||
| Lynn Frances Melcher Trust |
3,148 | * | 3,148 | — | * | |||||||||||||||
| Madeleine Behette |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Marc Bielski and Heather Gordon-Bielski |
3,149 | * | 3,149 | — | * | |||||||||||||||
| Marc E. Jaffe |
2,500 | * | 2,500 | — | * | |||||||||||||||
| Marc Flajolet |
8,750 | * | 8,000 | 750 | * | |||||||||||||||
102
Table of Contents
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| Marguerite Behette Hart |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Mariann Pancoe Trust |
8,080 | * | 8,080 | — | * | |||||||||||||||
| Marilyn Blond Melcher Revocable Trust |
3,148 | * | 3,148 | — | * | |||||||||||||||
| Mark Cook |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Mark Flaming |
2,000 | * | 2,000 | — | * | |||||||||||||||
| Marvin Mermelstein |
1,817 | * | 1,817 | — | * | |||||||||||||||
| Mary and David Boies |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Mary Osbakken |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Matthew Ross |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Max Flaming |
2,000 | * | 2,000 | — | * | |||||||||||||||
| Max G. Johnson TTEE / Max Johnson Living Trust U/A 1/7/99 |
2,750 | * | 2,750 | — | * | |||||||||||||||
| MCK Corporation |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Michael Hafke |
5,902 | * | 5,902 | — | * | |||||||||||||||
| Michael J. and Jane M. Sullivan |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Michael Pancoe Living Trust |
8,080 | * | 8,080 | — | * | |||||||||||||||
| Michael T. Smith |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Moe Leichter |
3,936 | * | 3,936 | — | * | |||||||||||||||
| Mohammed S. Rais |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Moshe Alafi(20) |
15,742 | * | 15,742 | — | * | |||||||||||||||
| Neponsit Properties LLC |
7,870 | * | 7,870 | — | * | |||||||||||||||
| New Ventures I, LLC |
212,384 | 1.0 | 212,384 | — | * | |||||||||||||||
| New York Small Business Venture Fund II LLC |
283,725 | 1.3 | 283,725 | — | * | |||||||||||||||
| New York Small Business Venture Fund III LLC |
233,371 | 1.1 | 233,371 | — | * | |||||||||||||||
| NJTC Venture Fund SBIC, L.P. |
935,390 | 4.2 | 935,390 | — | * | |||||||||||||||
| Paul Breglio |
6,233 | * | 6,233 | — | * | |||||||||||||||
| Paul Greengard, Ph.D.(21) |
1,131,250 | 5.1 | 1,131,250 | — | * | |||||||||||||||
| Paul Greengard Trust DTD 3/22/07 FBO Emerson Greengard Greeve(22) |
250,000 | 1.1 | 250,000 | — | * | |||||||||||||||
| Paul Greengard Trust DTD 10/31/01 FBO Delfine van Haarlem Greeve(22) |
250,000 | 1.1 | 250,000 | — | * | |||||||||||||||
| Paul Greengard Trust DTD 11/11/96 FBO Anne Blustein Greengard(22) |
250,000 | 1.1 | 250,000 | — | * | |||||||||||||||
| Paul Greengard Trust DTD 11/11/96 FBO Daniel Bijan Greengard(22) |
250,000 | 1.1 | 250,000 | — | * | |||||||||||||||
| Paul Greengard Trust DTD 11/11/96 FBO Philip Ramin Greengard(22) |
250,000 | 1.1 | 250,000 | — | * | |||||||||||||||
| Paul Greengard Trust DTD 12/7/98 FBO Natasha Marieke Greeve(22) |
250,000 | 1.1 | 250,000 | — | * | |||||||||||||||
| Paul Stern |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Pedro M. Cuatrecasas |
7,084 | * | 7,084 | — | * | |||||||||||||||
| Peng Li |
26,999 | * | 4,000 | 22,999 | * | |||||||||||||||
| Per Svenningson |
5,000 | * | 5,000 | — | * | |||||||||||||||
| Peter A. Feinstein MD PC Profit Sharing PL FBO Peter A. Feinstein MD |
5,903 | * | 5,903 | — | * | |||||||||||||||
| Peter F. Stewart |
1,750 | * | 1,750 | — | * | |||||||||||||||
103
Table of Contents
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| Peter G. Schultz |
7,500 | * | 7,500 | — | * | |||||||||||||||
| Philip F. Palmedo L.P. |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Philip H. Lippel |
1,750 | * | 1,750 | — | * | |||||||||||||||
| R. Lea Bone |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Rahul Auradkar and Umarani Bangalore |
2,310 | * | 2,310 | — | * | |||||||||||||||
| Ralph Gitz |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Ralph Glasgal |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Ralph T. Wood |
6,925 | * | 6,925 | — | * | |||||||||||||||
| Randolph C. Metcalfe Revocable Living Trust |
4,000 | * | 4,000 | — | * | |||||||||||||||
| Randy Perillo |
5,902 | * | 5,902 | — | * | |||||||||||||||
| Ray Crespo |
1,500 | * | 1,500 | — | * | |||||||||||||||
| Richard A. Melcher Revocable Trust |
3,148 | * | 3,148 | — | * | |||||||||||||||
| Richard H. Scheller |
7,500 | * | 7,500 | — | * | |||||||||||||||
| Robert C. Lannert Trust |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Robert C. Brown |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Robert Davis |
45,000 | * | 25,000 | 20,000 | * | |||||||||||||||
| Robert E. Cawthorn |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Robert Garcia |
2,078 | * | 2,078 | — | * | |||||||||||||||
| Robert Kaufman |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Rong Zheng |
3,463 | * | 3,463 | — | * | |||||||||||||||
| Rory Riggs |
11,806 | * | 11,806 | — | * | |||||||||||||||
| Rossi Motors Inc. Ronald G. Rossi |
6,926 | * | 6,926 | — | * | |||||||||||||||
| Ruby Elizabeth Koch-Fienberg Trust dated December 21st 2007, made by Allen A. Fienberg as grantor with Louis Fienberg trustee(23) |
25,000 | * | 25,000 | — | * | |||||||||||||||
| Samuel Rosenberg |
2,919 | * | 2,919 | — | * | |||||||||||||||
| Scott R. Runyan |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Scott R. Swix and Kim M. Swix |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Sharad and Chandrika Patel |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Sharon Mates, Ph.D.(24) |
1,391,430 | 6.2 | 1,053,935 | 337,495 | 1.5 | |||||||||||||||
| Shireen Michele Alafi, as Trustee of the Astrid Sophie Hovmoller Alafi GST Exempt Trust under The Christopher D. Alafi Generation-Skipping Trust under agreement dated October 12, 2012(25) |
251,888 | 1.1 | 251,888 | — | * | |||||||||||||||
| Shireen Michele Alafi, as Trustee of the Caroline Charlotte Hovmoller Alafi GST Exempt Trust under The Christopher D. Alafi Generation-Skipping Trust under agreement dated October 12, 2012(25) |
251,888 | 1.1 | 251,888 | — | * | |||||||||||||||
| Simon and Enas Muzio |
2,310 | * | 2,310 | — | * | |||||||||||||||
| So Young Jang |
2,417 | * | 2,417 | — | * | |||||||||||||||
| Sooner Ranch Investments, LP |
2,500 | * | 2,500 | — | * | |||||||||||||||
| Spencer A. and Susan L. Joyner Revocable Trust |
5,509 | * | 5,509 | — | * | |||||||||||||||
| Stefan Nowina |
4,619 | * | 4,619 | — | * | |||||||||||||||
| Stephen E. Budorick |
3,935 | * | 3,935 | — | * | |||||||||||||||
| Steve Kontos |
3,250 | * | 3,250 | — | * | |||||||||||||||
| Steven B. Nakovich, Jr., Insurance Trust Dated 3-6-1981 |
108,176 | * | 108,176 | — | * | |||||||||||||||
104
Table of Contents
| Shares Beneficially Owned Before the Offering |
Shares Being Offered (#) |
Shares Beneficially Owned After the Offering |
||||||||||||||||||
| Name of Beneficial Owner |
(#) | (%)(1) | (#)(1)(2)(3) | (%)(1)(2) | ||||||||||||||||
| Steven K. and Deborah S. Nelson |
5,000 | * | 5,000 | — | * | |||||||||||||||
| Steven Moger |
6,926 | * | 6,926 | — | * | |||||||||||||||
| Sunstone Partners LLC |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Tamas Bartfai |
2,500 | * | 2,500 | — | * | |||||||||||||||
| Tanjn and Tina S. Obut |
5,500 | * | 5,500 | — | * | |||||||||||||||
| The Benjamin Trust |
3,148 | * | 3,148 | — | * | |||||||||||||||
| The George K. Baum Family Foundation |
7,870 | * | 7,870 | — | * | |||||||||||||||
| The Rockefeller University |
400,000 | 1.8 | 400,000 | — | * | |||||||||||||||
| The Sosland Family Trust B Partnership(12) |
1,948,554 | 8.8 | 1,948,554 | — | * | |||||||||||||||
| The Sosland Foundation(12) |
732,548 | 3.3 | 732,548 | — | * | |||||||||||||||
| Theo Vafakos |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Thomas and Joy Licata |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Thomas D. Paul |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Thomas J. Jones III |
6,925 | * | 6,925 | — | * | |||||||||||||||
| Thomas Jefferson Jones |
6,925 | * | 6,925 | — | * | |||||||||||||||
| Thomas Mayberry |
5,500 | * | 5,500 | — | * | |||||||||||||||
| Thomas Price and Lyn Dowdle Price |
3,150 | * | 3,150 | — | * | |||||||||||||||
| Timothy Brickle |
2,500 | * | 2,500 | — | * | |||||||||||||||
| Todd H. Tracey |
1,575 | * | 1,575 | — | * | |||||||||||||||
| Tom Giftos |
4,722 | * | 4,722 | — | * | |||||||||||||||
| Trevor P. Castor |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Ursula Von Rydingsvard |
3,935 | * | 3,935 | — | * | |||||||||||||||
| Vincent Latour |
4,523 | * | 4,523 | — | * | |||||||||||||||
| Vinit Shah |
2,333 | * | 2,333 | — | * | |||||||||||||||
| Visium Balanced Master Fund, Ltd. |
700,479 | 3.2 | 700,479 | — | * | |||||||||||||||
| Vita Pure |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Weeks & Leo Co. Inc. |
7,870 | * | 7,870 | — | * | |||||||||||||||
| Wei Tong Ma |
7,250 | * | 7,250 | — | * | |||||||||||||||
| William Huff |
2,310 | * | 2,310 | — | * | |||||||||||||||
| William Ristvedt |
2,000 | * | 2,000 | — | * | |||||||||||||||
| William W. Crossman |
1,750 | * | 1,750 | — | * | |||||||||||||||
| Wing Real Estate LLC |
7,250 | * | 7,250 | — | * | |||||||||||||||
| Xiao Tao Du and LiLi |
7,250 | * | 7,250 | — | * | |||||||||||||||
| Yona Sofia Koch-Fienberg Trust dated December 21st 2007, made by Allen A. Fienberg as grantor with Louis Fienberg trustee(23) |
25,000 | * | 25,000 | — | * | |||||||||||||||
| Yonghong Guo |
6,833 | * | 6,833 | — | * | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total |
22,837,306 | — | 21,961,496 | — | — | |||||||||||||||
| * | Less than 1% |
| (1) | Applicable percentage ownership is based on 22,134,647 shares of our common stock outstanding as of December 1, 2013. |
| (2) | Assumes the sale of all shares offered in this prospectus. |
| (3) | Consists of options to purchase shares of common stock exercisable within 60 days of December 1, 2013 held by the selling stockholder, except for Alzheimer Drug Discovery Foundation, Inc., which consists of a warrant to purchase 1,822 shares of common stock which is immediately exercisable. |
| (4) | Christopher Alafi, Ph.D., one of our directors, and Moshe Alafi are managing partners of Alafi Capital Company, LLC, or Alafi Capital, and have shared voting and investment power with respect to the shares owned by Alafi Capital. |
105
Table of Contents
| (5) | Joel S. Marcus, one of our directors, is the Chairman, CEO and Founder of Alexandria Real Estate Equities, Inc., which is the managing member of Alexandria Equities, LLC, which has full voting and investment power with respect to the shares owned by Alexandria Equities, LLC. Alexandria Real Estate Equities, Inc. is a reporting company under the Exchange Act. As an officer of Alexandria Real Estate Equities, Inc., Mr. Marcus may be deemed to have voting and investment power with respect to the shares owned by Alexandria Equities, LLC. Mr. Marcus disclaims beneficial ownership of the shares held by Alexandria Equities, LLC, except to the extent of his underlying pecuniary interest therein. |
| (6) | Allen A. Fienberg, Ph.D., is our Vice President of Business Development. Consists of 237,500 shares of common stock and options to purchase 114,165 shares of common stock which are exercisable within 60 days of December 1, 2013. Does not include: (i) 208,023 shares of common stock held by J.D.F. Holdings Ltd., in which Dr. Fienberg holds a 20% ownership interest; and (ii) 50,000 shares of common stock held by two trusts for the benefit of members of Dr. Fienberg’s family. Dr. Fienberg has no voting or investment control with respect to any of the shares owned by J.D.F. Holdings Ltd. or held in the trusts. |
| (7) | BlackRock, Inc. is the ultimate parent holding company of BlackRock Investment Management, LLC, the Investment Adviser of BGF World HealthScience Fund (the “BlackRock Fund I”). On behalf of BlackRock Investment Management, LLC, the Investment Adviser of the BlackRock Fund I, Erin Xie, as a Managing Director of BlackRock Investment Management, LLC, has voting and investment power over the referenced securities held by such fund. Erin Xie expressly disclaims beneficial ownership of all shares held by the BlackRock Fund I. |
| (8) | BlackRock, Inc. is the ultimate parent holding company of BlackRock Advisors, LLC, the Investment Manager of BlackRock Health Sciences Opportunities Portfolio, a series of BlackRock Funds (the “BlackRock Fund II”). On behalf of BlackRock Advisors, LLC, the Investment Manager of the BlackRock Fund II, Erin Xie, as a Managing Director of BlackRock Advisors, LLC, has voting and investment power over the referenced securities held by such fund. Erin Xie expressly disclaims beneficial ownership of all shares held by the BlackRock Fund II. |
| (9) | BlackRock, Inc. is the ultimate parent holding company of BlackRock Advisors, LLC, the Investment Adviser of BlackRock Health Sciences Trust (the “BlackRock Fund III”). On behalf of BlackRock Advisors, LLC, the Investment Adviser of the BlackRock Fund III, Erin Xie, as a Managing Director of BlackRock Advisors, LLC, has voting and investment power over the referenced securities held by such fund. Erin Xie expressly disclaims beneficial ownership of all shares held by the BlackRock Fund III. |
| (10) | Christopher Alafi, Ph.D. is trustee of this trust and has full voting and investment power with respect to shares owned by the trust. |
| (11) | Dr. Kipnis was a director of ITI from May 2002 until December 2012. |
| (12) | Morton I. Sosland is Trustee of the David N. Sosland Trust A, Managing Partner of The Sosland Family Trust B Partnership and Vice Chairman of The Sosland Foundation, which we refer to collectively as the Sosland Holders. As such, Mr. Sosland has sole voting and investment power with respect to the shares held by the Sosland Holders. |
| (13) | Fidelity Management & Research Company, or Fidelity, 82 Devonshire Street, Boston, Massachusetts 02109, a wholly-owned subsidiary of FMR LLC and an investment adviser registered under Section 203 of the Investment Advisers Act of 1940, is the beneficial owner of 1,999,120 shares of common stock as a result of acting as investment adviser to various investment companies registered under Section 8 of the Investment Company Act of 1940. Edward C. Johnson 3d and FMR LLC, through its control of Fidelity, and the funds each has sole power to dispose of the 1,999,120 shares of common stock owned by the Funds. Members of the family of Edward C. Johnson 3d, Chairman of FMR LLC, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. Neither FMR LLC nor Edward C. Johnson 3d, Chairman of FMR LLC, has the sole power to |
106
Table of Contents
| vote or direct the voting of the shares owned directly by the Fidelity Funds, which power resides with the Funds’ Boards of Trustees. Fidelity carries out the voting of the shares under written guidelines established by the Funds’ Boards of Trustees. |
| (14) | BlackRock, Inc. is the ultimate parent holding company of BlackRock Advisors, LLC, the Sub-Adviser of ING BlackRock Health Sciences Opportunities Portfolio (the “BlackRock Fund IV”). On behalf of BlackRock Advisors, LLC, the Sub-Adviser of the BlackRock Fund IV, Erin Xie, as a Managing Director of BlackRock Advisors, LLC, has voting and investment power over the referenced securities held by such fund. Erin Xie expressly disclaims beneficial ownership of all shares held by the BlackRock Fund IV. |
| (15) | Allen A. Fienberg, Ph.D., our Vice President of Business Development, holds a 20% ownership interest in shares held by J.D.F. Holdings Ltd. Dr. Fienberg has no voting or investment control with respect to any of the shares owned by J.D.F. Holdings Ltd. |
| (16) | Consists of 15,742 shares of common stock held by the Joel S. Marcus and Barbara A. Marcus Family Trust. Mr. Marcus, who is one of our directors, may also be deemed to beneficially own shares held by Alexandria Equities, LLC described in footnote 5 above, as well as options to purchase 83,750 shares of common stock held by Mr. Marcus, which are exercisable within 60 days of December 1, 2013. |
| (17) | Lawrence J. Hineline is our Vice President of Finance, Chief Financial Officer and Secretary. Consists of 50,000 shares of common stock and options to purchase 114,999 shares of common stock, which are exercisable within 60 days of December 1, 2013. |
| (18) | Lawrence P. Wennogle, Ph.D. is our Vice President, Drug Discovery. Consists of 100,000 shares of common stock and options to purchase 123,332 shares of common stock, which are exercisable within 60 days of December 1, 2013. |
| (19) | Richard Lerner, M.D., one of our directors, shares voting and investment control with respect to the shares held by the Lerner Family Trust UAD 11/14/94. |
| (20) | Mr. Alafi may also be deemed to beneficially own the shares held by Alafi Capital described above. Mr. Alafi is a managing partner of Alafi Capital and shares voting and investment power with respect to the shares owned by Alafi Capital. |
| (21) | Paul Greengard, Ph.D. is one of our founders and principal stockholders. Does not include 1,500,000 shares of common stock held by six trusts for the benefit of members of Dr. Greengard’s family, as the trustee of these trusts, Ursula von Rydingsvard, Dr. Greengard’s spouse, has sole voting and investment control over the shares held by the trusts. |
| (22) | See footnote 21. |
| (23) | See footnote 6. |
| (24) | Sharon Mates, Ph.D. is our Chairman, President and Chief Executive Officer. Consists of 1,053,935 shares of common stock and options to purchase 337,495 shares of common stock, which are exercisable within 60 days of December 1, 2013. |
| (25) | This trust is for the benefit of members of the Alafi family. Neither Christopher Alafi, Ph.D., one of our directors, nor Moshe Alafi has voting or investment control with respect to shares owned by this trust. |
107
Table of Contents
The selling stockholders, which as used herein includes donees, pledgees, transferees or other successors-in-interest selling shares of common stock or interests in shares of common stock received after the date of this prospectus from a selling stockholder as a gift, pledge, partnership distribution or other transfer, may, from time to time, sell, transfer or otherwise dispose of any or all of their shares of common stock or interests in shares of common stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These dispositions may be at fixed prices, at prevailing market prices at the time of sale, at prices related to the prevailing market price, at varying prices determined at the time of sale, or at negotiated prices.
The selling stockholders may use any one or more of the following methods when disposing of shares or interests therein:
| • | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| • | block trades in which the broker-dealer will attempt to sell the shares as agent, but may position and resell a portion of the block as principal to facilitate the transaction; |
| • | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| • | an exchange distribution in accordance with the rules of the applicable exchange; |
| • | privately negotiated transactions; |
| • | short sales; |
| • | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| • | broker-dealers may agree with the selling stockholders to sell a specified number of such shares at a stipulated price per share; |
| • | a combination of any such methods of sale; and |
| • | any other method permitted pursuant to applicable law. |
The selling stockholders may, from time to time, pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock, from time to time, under this prospectus, or under an amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The selling stockholders also may transfer the shares of common stock in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
In connection with the sale of our common stock or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the common stock in the course of hedging the positions they assume. The selling stockholders may also sell shares of our common stock short and deliver these securities to close out their short positions, or loan or pledge the common stock to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The aggregate proceeds to the selling stockholders from the sale of the common stock offered by them will be the purchase price of the common stock less discounts or commissions, if any. Each of the selling stockholders
108
Table of Contents
reserves the right to accept and, together with their agents from time to time, to reject, in whole or in part, any proposed purchase of common stock to be made directly or through agents. We will not receive any of the proceeds from this offering.
The selling stockholders and any underwriters, broker-dealers or agents that participate in the sale of the common stock or interests therein may be “underwriters” within the meaning of Section 2(11) of the Securities Act. Any discounts, commissions, concessions or profit they earn on any resale of the shares may be underwriting discounts and commissions under the Securities Act. Selling stockholders who are “underwriters” within the meaning of Section 2(11) of the Securities Act will be subject to the prospectus delivery requirements of the Securities Act.
To the extent required, the shares of our common stock to be sold, the names of the selling stockholders, the respective purchase prices and public offering prices, the names of any agents, dealer or underwriter, any applicable commissions or discounts with respect to a particular offer will be set forth in an accompanying prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes this prospectus.
In order to comply with the securities laws of some states, if applicable, the common stock may be sold in these jurisdictions only through registered or licensed brokers or dealers. In addition, in some states the common stock may not be sold unless it has been registered or qualified for sale or an exemption from registration or qualification requirements is available and is complied with.
We have advised the selling stockholders that the anti-manipulation rules of Regulation M under the Exchange Act may apply to sales of shares in the market and to the activities of the selling stockholders and their affiliates. In addition, we will make copies of this prospectus (as it may be supplemented or amended from time to time) available to the selling stockholders for the purpose of satisfying the prospectus delivery requirements of the Securities Act. The selling stockholders may indemnify any broker-dealer that participates in transactions involving the sale of the shares against certain liabilities, including liabilities arising under the Securities Act.
We have agreed to indemnify the selling stockholders against liabilities, including liabilities under the Securities Act and state securities laws, relating to the registration of the shares offered by this prospectus.
We have agreed with the selling stockholders to keep the registration statement of which this prospectus constitutes a part effective until the earlier of the third anniversary of the date the registration statement is declared effective by the SEC (or, if later, the third anniversary of the date that all of the shares required to be registered by us have been included in the registration statement) and such time as all of the shares covered by this prospectus have been disposed of pursuant to and in accordance with the registration statement.
109
Table of Contents
The following statements are qualified in their entirety by reference to the detailed provisions of our restated certificate of incorporation and restated bylaws.
Capital Structure
We currently have authorized capital stock of 105,000,000 shares, of which 100,000,000 are designated as common stock, par value $0.0001 per share, and 5,000,000 shares are designated as preferred stock, par value $0.0001 per share. As of December 1, 2013, 22,134,647 shares of our common stock and no shares of our preferred stock were issued and outstanding.
Common Stock
The holders of our common stock are entitled to one vote per share on matters on which our stockholders vote. There are no cumulative voting rights. Subject to any preferential dividend rights of any outstanding shares of preferred stock, holders of our common stock are entitled to receive dividends, if declared by our board of directors, out of funds that we may legally use to pay dividends. If we liquidate or dissolve, holders of our common stock are entitled to share ratably in our assets once our debts and any liquidation preference owed to any then-outstanding preferred stockholders are paid. Our restated certificate of incorporation does not provide our common stock with any redemption, conversion or preemptive rights.
Preferred Stock
If we issue preferred stock in the future, such preferred stock would have priority over common stock with respect to dividends and other distributions, including the distribution of assets upon liquidation. Our board of directors has the authority, without further stockholder authorization, to issue from time to time up to 5,000,000 shares of preferred stock in one or more series and to fix the terms, limitations, voting rights, relative rights and preferences and variations of each series. Although we have no present plans to issue any shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing a change of control of us or an unsolicited acquisition proposal.
Warrant
As of December 1, 2013, we had one warrant outstanding to purchase 1,822 shares of our common stock at an exercise price of $6.0264 per share, which expires on April 19, 2023.
Registration Rights
On August 29, 2013, ITI entered into a registration rights agreement with the investors in the Private Placement and also the existing stockholders of ITI who agreed to become parties to certain provisions of the agreement or who choose to become parties in the future, which covers substantially all of our outstanding shares of common stock as of December 1, 2013. We assumed the registration rights agreement in connection with the Merger.
Resale Registration Rights
Pursuant to the registration rights agreement and subject to the rules and regulations of the SEC, we have agreed to file a shelf registration statement covering the resale of the shares of our common stock held by the investors in the Private Placement and the shares of our common stock held by the former stockholders of ITI who are parties to the agreement. We were required to file the shelf registration statement within 45 days of the date of the registration rights agreement (October 13, 2013), which we filed on September 18, 2013. The
110
Table of Contents
registration statement of which this prospectus forms a part is the shelf registration statement that we are required to file under the registration rights agreement. In the event fewer than all of our outstanding shares of common stock can be registered pursuant to the so-called Rule 415 doctrine, priority will be given to the shares issued in the Private Placement, but in such event at least 25% of the shares registered shall be those held by ITI’s existing stockholders prior to the Private Placement.
Registration of these shares under the Securities Act would result in the shares becoming saleable under the Securities Act immediately upon the effectiveness of such registration. Any sales of securities by holders of these shares could adversely affect the trading prices, if any, of our common stock.
We will be liable to each investor in the Private Placement (but not to the former stockholders of ITI who are parties to the agreement) for liquidated damages, on a 30-day basis, equal to 1.0% of the aggregate purchase price paid by the investor for the registrable shares of our common stock then held by the investor, subject to an overall cap of 5%, (i) if we fail to file the registration statement on time, (ii) if the registration statement is not declared effective within 150 days from the date of the registration rights agreement (January 26, 2014), (iii) if we suspend (subject to limited blackout periods described below) or terminate the registration statement prior to the date which is the earlier of (x) the third anniversary of its effectiveness (or the third anniversary of the date on which all registrable shares are included therein, if later) and (y) the date on which all of the registrable shares cease to be registrable shares, or (iv) in the event one or more suspensions of the effectiveness of the registration statement exceeds 60 days in the aggregate during any 12-month period. We will be permitted to suspend the registration statement one or more times during any 12-month period provided such suspensions do not exceed 30 consecutive days or 60 days in the aggregate in any 12-month period. Any suspension associated with our filing of an annual, periodic or current report, as required by the Exchange Act, will be permitted and will not be counted against the 60 day limitation. Any shares not registered due to the Rule 415 doctrine will not be subject to liquidated damages. Expenses with respect to the filing and effectiveness of such registration statement (but not selling expenses, or underwriter or agent compensation) will be paid by us, including expenses of one counsel for the selling stockholders.
Form S-3 Demand Registration Rights
Pursuant to the registration rights agreement, at any time after we become eligible to file a registration statement on Form S-3, subject to specified limitations set forth in the registration rights agreement, the holders of at least 12% of the registrable shares of common stock then outstanding may request that we register on Form S-3 all or a portion of the registrable shares so long as the total amount of the shares being registered have an anticipated aggregate offering price, net of selling expenses, of at least $7,500,000.
“Piggyback” Registration Rights
Pursuant to the registration rights agreement, if we propose to register any of our common stock in a firm commitment underwritten offering, the holders of registrable shares of our common stock will be entitled to notice of the registration and have the right to require us to register all or a portion of the registrable shares then held by them, subject to our right and the right of our underwriters to reduce the number of shares proposed to be registered in view of market conditions.
Expenses of Registration
We have agreed to pay all fees and expenses relating to the registration statement of which this prospectus forms a part, as well as all Form S-3 demand registrations and piggyback registrations, including up to $25,000 in fees of one special counsel of the investors in connection with the filing of the registration statement of which this prospectus forms a part.
111
Table of Contents
Expiration of Registration Rights
The resale registration rights described above shall terminate upon the earlier of (1) the date on which all registrable shares have been effectively registered under the Securities Act and disposed of in accordance with such registration statement, and (2) the later of the third anniversary of the date (A) the registration statement of which this prospectus forms a part is declared effective and (B) all registrable shares have been registered in the registration statement of which this prospectus forms a part.
Lock-Up Provisions in Registration Rights Agreement
The registration rights agreement contains lock-up provisions applicable to holders of our common stock. See “Certain Relationships and Related Person Transactions—Agreements with Stockholders—Lock-Up Provisions in Registration Rights Agreement.”
Anti-Takeover Effects of Delaware Law and Our Restated Certificate of Incorporation and Restated Bylaws
The provisions of Delaware law and our restated certificate of incorporation and restated bylaws could discourage or make it more difficult to accomplish a proxy contest or other change in our management or the acquisition of control by a holder of a substantial amount of our voting stock. It is possible that these provisions could make it more difficult to accomplish, or could deter, transactions that stockholders may otherwise consider to be in their best interests or in our best interests. These provisions are intended to enhance the likelihood of continuity and stability in the composition of our board of directors and in the policies formulated by the board of directors and to discourage certain types of transactions that may involve an actual or threatened change of our control. These provisions are designed to reduce our vulnerability to an unsolicited acquisition proposal and to discourage certain tactics that may be used in proxy fights. Such provisions also may have the effect of preventing changes in our management.
Delaware Statutory Business Combinations Provision
We are subject to the anti-takeover provisions of Section 203 of the Delaware General Corporation Law. Section 203 prohibits a publicly-held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is, or the transaction in which the person became an interested stockholder was, approved in a prescribed manner or another prescribed exception applies. For purposes of Section 203, a “business combination” is defined broadly to include a merger, asset sale or other transaction resulting in a financial benefit to the interested stockholder, and, subject to certain exceptions, an “interested stockholder” is a person who, together with his or her affiliates and associates, owns, or within three years prior, did own, 15% or more of the corporation’s voting stock.
Classified Board of Directors; Removal of Directors for Cause
Pursuant to our restated certificate of incorporation and restated bylaws, our board of directors is divided into three classes, with the term of office of the first class to expire at the first annual meeting of stockholders following the initial classification of directors, the term of office of the second class to expire at the second annual meeting of stockholders following the initial classification of directors, and the term of office of the third class to expire at the third annual meeting of stockholders following the initial classification of directors. At each annual meeting of stockholders, directors elected to succeed those directors whose terms expire, other than directors elected by the holders of any series of preferred stock under specified circumstances, will be elected for a three-year term of office. All directors elected to our classified board of directors will serve until the election and qualification of their respective successors or their earlier resignation or removal. Members of the board of directors may only be removed for cause and only by the affirmative vote of 80% of our outstanding voting stock. These provisions are likely to increase the time required for stockholders to change the composition of the board of directors. For example, at least two annual meetings will be necessary for stockholders to effect a change in a majority of the members of the board of directors.
112
Table of Contents
Advance Notice Provisions for Stockholder Proposals and Stockholder Nominations of Directors
Our restated bylaws provide that, for nominations to the board of directors or for other business to be properly brought by a stockholder before a meeting of stockholders, the stockholder must first have given timely notice of the proposal in writing to our Secretary. For an annual meeting, a stockholder’s notice generally must be delivered not less than 90 days nor more than 120 days prior to the first anniversary of the previous year’s annual meeting date. For a special meeting, the notice must generally be delivered not earlier than the 90th day prior to the meeting and not later than the later of (1) the 60th day prior to the meeting or (2) the 10th day following the day on which public announcement of the meeting is first made. Detailed requirements as to the form of the notice and information required in the notice are specified in the restated bylaws. If it is determined that business was not properly brought before a meeting in accordance with our bylaw provisions, such business will not be conducted at the meeting.
Special Meetings of Stockholders
Special meetings of the stockholders may be called only by our board of directors pursuant to a resolution adopted by a majority of the total number of directors.
No Stockholder Action by Written Consent
Any action to be effected by our stockholders must be effected at a duly called annual or special meeting of the stockholders.
Super Majority Stockholder Vote Required for Certain Actions
The Delaware General Corporation Law provides generally that the affirmative vote of a majority of the shares entitled to vote on any matter is required to amend a corporation’s certificate of incorporation or bylaws, unless the corporation’s certificate of incorporation or bylaws, as the case may be, requires a greater percentage. Our restated certificate of incorporation requires the affirmative vote of the holders of at least 80% of our outstanding voting stock to amend or repeal any of the provisions discussed in this section of this prospectus entitled “Anti-Takeover Effects of Delaware Law and Our Restated Certificate of Incorporation and Restated Bylaws.” This 80% stockholder vote would be in addition to any separate class vote that might in the future be required pursuant to the terms of any preferred stock that might then be outstanding. An 80% vote is also required for any amendment to, or repeal of, our restated bylaws by the stockholders. Our restated bylaws may be amended or repealed by a simple majority vote of the board of directors.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare Trust Company, N.A., with offices at 250 Royall Street, Canton, Massachusetts 02021.
113
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Future sales of substantial amounts of shares of our common stock, including shares issued upon the exercise of outstanding options, in the public market or the possibility of these sales occurring could cause the prevailing market price for our common stock to fall or impair our ability to raise equity capital in the future. As of December 1, 2013, we had outstanding 22,134,647 shares of common stock. All of these shares are restricted securities under Rule 144, in that they were issued in a private transaction not involving a public offering.
Restrictions on the Use of Rule 144 by Shell Companies or Former Shell Companies
Rule 144 is not available for the resale of securities initially issued by companies that are, or previously were, blank check companies like us, to their promoters or affiliates despite technical compliance with the requirements of Rule 144. Rule 144 also is not available for resale of securities issued by any shell companies (other than business combination-related shell companies) or any issuer that has been at any time previously a shell company. The SEC has provided an exception to this prohibition, however, if the following conditions are met:
| • | the issuer of the securities that was formerly a shell company has ceased to be a shell company; |
| • | the issuer of the securities is subject to the reporting requirements of Section 13 or 15(d) of the Exchange Act; |
| • | the issuer of the securities has filed all Exchange Act reports and materials required to be filed, as applicable, during the preceding 12 months (or such shorter period that the issuer was required to file such reports and materials), other than Form 8-K reports; and |
| • | at least one year has elapsed from the time that the issuer filed current Form 10 type information with the SEC reflecting its status as an entity that is not a shell company. |
As a result, none of our stockholders is currently able to sell shares of our common stock in reliance on Rule 144. Assuming we continue to meet the requirements set forth above, Rule 144 will become available to our stockholders on September 5, 2014. Our stockholders may currently resell their shares of our common stock only pursuant to a registration statement that has been declared effective under the Securities Act or pursuant to another exemption from registration.
Lock-Up Provisions in Registration Rights Agreement
The registration rights agreement contains lock-up provisions applicable to holders of our common stock. See “Certain Relationships and Related Person Transactions—Agreements with Stockholders—Lock-Up Provisions in Registration Rights Agreement.”
Registration Rights
The holders of an aggregate of 21,961,496 shares of our common stock, or their permitted transferees, are entitled to rights with respect to the registration of these shares under the Securities Act. Registration of these shares under the Securities Act would result in these shares becoming fully tradable without restriction under the Securities Act immediately upon the effectiveness of the registration statement, except for shares held by affiliates. See “Description of Capital Stock—Registration Rights” for additional information.
Stock Options
As of December 1, 2013, we had outstanding options to purchase 1,427,025 shares of our common stock at a weighted-average exercise price of $1.96 per share, of which 1,085,030 shares were vested as of such date. We intend to file a registration statement on Form S-8 under the Securities Act covering all of the shares of common
114
Table of Contents
stock subject to equity grants outstanding or reserved under the ITI 2003 Equity Incentive Plan, which we assumed in the Merger, and our 2013 Equity Incentive Plan. Accordingly, shares of our common stock issued under the 2003 Equity Incentive Plan and 2013 Equity Incentive Plan will be eligible for sale in the public market, subject to vesting restrictions. However, resales of certain shares held by our affiliates registered on the Form S-8 will be subject to volume limitations, manner of sale, notice and public information requirements of Rule 144. See “Equity Compensation Plan Information—2003 Equity Incentive Plan” and “Equity Compensation Plan Information—2013 Equity Incentive Plan” for additional information regarding these plans.
Warrant
As of December 1, 2013, we had one warrant outstanding to purchase 1,822 shares of our common stock at an exercise price of $6.0264 per share, which expires on April 19, 2023. Any shares purchased pursuant to this warrant will be “restricted shares” and may be sold in the public market only if they are registered under the Securities Act or qualify for an exemption from such registration.
115
Table of Contents
The validity of the shares of common stock being offered by this prospectus will be passed upon for us by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., Boston, Massachusetts.
Ernst & Young LLP, independent registered accounting firm, has audited our financial statements at December 31, 2012 and 2011, and for the years then ended, as set forth in their report. We have included our financial statements in the prospectus and elsewhere in the registration statement in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
Raich Ende Malter & Co. LLP, independent registered accounting firm, has audited the financial statements of Oneida Resources Corp. for the period from August 29, 2012 (inception) to March 31, 2013. We have included the financial statements of Oneida Resources Corp. in the prospectus and elsewhere in the registration statement in reliance on Raich Ende Malter & Co. LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We are subject to the information requirements of the Exchange Act, and are required to file reports, proxy statements, information statements and other information with the SEC. You may read and copy this information, for a copying fee, at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549 on official business days during the hours of 10:00 a.m. to 3:00 p.m. Please call the SEC at 1-800-SEC-0330 for more information on its Public Reference Room. Our SEC filings will also be available to the public from commercial document retrieval services, and at the website maintained by the SEC at http://www.sec.gov.
Our Internet website address is http://www.intracellulartherapies.com. Information contained on the website does not constitute part of this prospectus. We have included our website address in this prospectus solely as an inactive textual reference. When the registration statement of which this prospectus forms a part is effective, we will make available, through a link to the SEC’s website, electronic copies of the materials we file with the SEC, including annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, the Section 16 reports filed by our executive officers, directors and 10% stockholders and amendments to those reports.
This prospectus is part of a registration statement that we filed with the SEC. The registration statement contains more information than this prospectus regarding us and the securities, including exhibits and schedules. You can obtain a copy of the registration statement from the SEC at any address listed above or from the SEC’s website.
116
Table of Contents
INTRA-CELLULAR THERAPIES, INC.
| Page | ||||
| Audited Financial Statements of Intra-Cellular Therapies, Inc. for the Years ended December 31, 2012 and 2011 |
||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| Statements of Redeemable Convertible Preferred Stock and Stockholders’ Deficit |
F-5 | |||
| F-6 | ||||
| F-7 | ||||
| Unaudited Condensed Consolidated Financial Statements of Intra-Cellular Therapies, Inc. as of September 30, 2013 and December 31, 2012 and for the Three- and Nine-Months Ended September 30, 2013 and 2012 |
||||
| F-22 | ||||
| F-23 | ||||
| F-24 | ||||
| F-25 | ||||
ONEIDA RESOURCES CORP.
INDEX TO FINANCIAL STATEMENTS
| Page | ||||
| Audited Financial Statements of Oneida Resources Corp. for the Period from August 29, 2012 (Inception) to March 31, 2013 |
||||
| F-35 | ||||
| F-36 | ||||
| F-37 | ||||
| F-38 | ||||
| F-39 | ||||
| F-40 | ||||
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors of
Intra-Cellular Therapies, Inc.
We have audited the accompanying balance sheets of Intra-Cellular Therapies, Inc. as of December 31, 2012 and 2011, and the related statements of operations, comprehensive income, redeemable convertible preferred stock and stockholders’ deficit, and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Intra-Cellular Therapies, Inc. at December 31, 2012 and 2011, and the results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
McLean, VA
June 19, 2013
F-2
Table of Contents
Intra-Cellular Therapies, Inc.
Balance Sheets
| December 31 | ||||||||
| 2012 | 2011 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 15,645,528 | $ | 13,693,215 | ||||
| Certificates of deposit |
3,500,000 | 9,200,123 | ||||||
| Accounts receivable |
300,429 | 349,063 | ||||||
| Prepaid expenses and other current assets |
188,702 | 114,468 | ||||||
|
|
|
|
|
|||||
| Total current assets |
19,634,659 | 23,356,869 | ||||||
| Property and equipment, net |
58,266 | 67,056 | ||||||
| Other assets |
130,755 | 170,800 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 19,823,680 | $ | 23,594,725 | ||||
|
|
|
|
|
|||||
| Liabilities, redeemable convertible preferred stock and stockholders’ deficit |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 41,608 | $ | 595,864 | ||||
| Accrued liabilities |
588,065 | 1,103,486 | ||||||
| Accrued employee benefits |
726,657 | 669,739 | ||||||
| Deferred revenue—short-term |
1,666,674 | 1,666,666 | ||||||
| Convertible promissory notes |
15,173,013 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
18,196,017 | 4,035,755 | ||||||
| Deferred revenue—long-term |
— | 1,666,667 | ||||||
| Series A Redeemable Convertible Preferred Stock, $0.001 par value: |
6,755,992 | 6,459,992 | ||||||
| Series B Redeemable Convertible Preferred Stock, $0.001 par value: |
8,936,955 | 8,475,905 | ||||||
| Series C Redeemable Convertible Preferred Stock, $0.001 par value: |
15,141,345 | 14,205,340 | ||||||
| Stockholders’ deficit: |
||||||||
| Common stock, $.001 par value: 30,000,000 shares authorized; 11,269,530 and 11,202,990 shares issued and outstanding at December 31, 2012 and 2011, respectively |
11,270 | 11,202 | ||||||
| Additional paid-in capital |
1,478,400 | 2,845,336 | ||||||
| Accumulated deficit |
(30,696,299 | ) | (14,105,472 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ deficit |
(29,206,629 | ) | (11,248,934 | ) | ||||
|
|
|
|
|
|||||
| Total liabilities, redeemable convertible preferred stock and stockholders’ deficit |
$ | 19,823,680 | $ | 23,594,725 | ||||
|
|
|
|
|
|||||
See accompanying notes.
F-3
Table of Contents
Intra-Cellular Therapies, Inc.
Statements of Operations
| Year Ended December 31 | ||||||||
| 2012 | 2011 | |||||||
| Revenues: |
||||||||
| License and collaboration revenue |
$ | 3,117,991 | $ | 22,327,464 | ||||
| Grant revenue |
— | 1,034,495 | ||||||
|
|
|
|
|
|||||
| Total revenues |
3,117,991 | 23,361,959 | ||||||
|
|
|
|
|
|||||
| Costs and expenses: |
||||||||
| Research and development |
15,486,476 | 7,654,546 | ||||||
| General and administrative |
4,034,925 | 4,612,450 | ||||||
|
|
|
|
|
|||||
| Total costs and expenses |
19,521,401 | 12,266,996 | ||||||
|
|
|
|
|
|||||
| (Loss) income from operations |
(16,403,410 | ) | 11,094,963 | |||||
| Interest expense |
(193,498 | ) | (15 | ) | ||||
| Interest income |
39,002 | 62,315 | ||||||
| Income taxes |
(32,921 | ) | (64,834 | ) | ||||
|
|
|
|
|
|||||
| Net (loss) income |
(16,590,827 | ) | 11,092,429 | |||||
| Cumulative dividends on redeemable convertible preferred stock |
(1,672,223 | ) | (1,669,786 | ) | ||||
|
|
|
|
|
|||||
| Net (loss) income attributable to common stockholders |
(18,263,050 | ) | 9,422,643 | |||||
|
|
|
|
|
|||||
| Net (loss) income per common share: |
||||||||
| Basic |
$ | (1.63 | ) | $ | 0.39 | |||
| Dilutive |
(1.63 | ) | 0.33 | |||||
| Weighted average number of common shares: |
||||||||
| Basic |
11,215,077 | 11,202,990 | ||||||
| Dilutive |
11,215,077 | 13,190,476 | ||||||
See accompanying notes.
F-4
Table of Contents
Intra-Cellular Therapies, Inc.
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Deficit
| Series A Redeemable Convertible Preferred Stock |
Series B Redeemable Convertible Preferred Stock |
Series C Redeemable Convertible Preferred Stock |
Common Stock | Additional Paid-in |
Accumulated | Total Stockholders’ |
||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||||||||||||||
| Balance at December 31, 2010 |
3,700,000 | $ | 6,163,992 | 3,631,898 | $ | 8,005,827 | 5,762,765 | $ | 13,269,334 | 11,202,990 | $ | 11,202 | $ | 4,266,968 | $ | (25,197,901 | ) | $ | (20,919,731 | ) | ||||||||||||||||||||||||
| Share-based compensation |
— | — | — | — | — | — | — | — | 280,452 | — | 280,452 | |||||||||||||||||||||||||||||||||
| Accretion of issuance costs |
— | — | — | 11,466 | — | 20,832 | — | — | (32,298 | ) | — | (32,298 | ) | |||||||||||||||||||||||||||||||
| Dividends on redeemable convertible preferred stock |
— | 296,000 | — | 458,612 | — | 915,174 | — | — | (1,669,786 | ) | — | (1,669,786 | ) | |||||||||||||||||||||||||||||||
| Net income |
— | — | — | — | — | — | — | — | — | 11,092,429 | 11,092,429 | |||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
| Balance at December 31, 2011 |
3,700,000 | 6,459,992 | 3,631,898 | 8,475,905 | 5,762,765 | 14,205,340 | 11,202,990 | 11,202 | 2,845,336 | (14,105,472 | ) | (11,248,934 | ) | |||||||||||||||||||||||||||||||
| Exercise of stock options |
— | — | — | — | — | — | 66,540 | 68 | 31,013 | — | 31,081 | |||||||||||||||||||||||||||||||||
| Share-based compensation |
— | — | — | — | — | — | — | — | 295,106 | — | 295,106 | |||||||||||||||||||||||||||||||||
| Accretion of issuance costs |
— | — | — | — | — | 20,832 | — | — | (20,832 | ) | — | (20,832 | ) | |||||||||||||||||||||||||||||||
| Dividends on redeemable convertible preferred stock |
— | 296,000 | — | 461,050 | — | 915,173 | — | — | (1,672,223 | ) | — | (1,672,223 | ) | |||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | (16,590,827 | ) | (16,590,827 | ) | |||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
| Balance at December 31, 2012 |
3,700,000 | $ | 6,755,992 | 3,631,898 | $ | 8,936,955 | 5,762,765 | $ | 15,141,345 | 11,269,530 | $ | 11,270 | $ | 1,478,400 | $ | (30,696,299 | ) | $ | (29,206,629 | ) | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
See accompanying notes.
F-5
Table of Contents
Intra-Cellular Therapies, Inc.
Statements of Cash Flows
| Year Ended December 31 | ||||||||
| 2012 | 2011 | |||||||
| Operating activities |
||||||||
| Net (loss) income |
$ | (16,590,827 | ) | $ | 11,092,429 | |||
| Adjustments to reconcile net loss (income) to net cash (used in) provided by operating activities: |
||||||||
| Depreciation |
47,747 | 189,186 | ||||||
| Share-based compensation expense |
295,106 | 280,452 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Accounts receivable |
48,634 | (349,063 | ) | |||||
| Prepaid expenses and other assets |
(34,189 | ) | 521,245 | |||||
| Accounts payable |
(554,256 | ) | 321,426 | |||||
| Accrued liabilities and employee benefits |
(448,493 | ) | 1,016,953 | |||||
| Deferred revenue |
(1,666,659 | ) | 3,132,038 | |||||
|
|
|
|
|
|||||
| Net cash (used in) provided by operating activities |
(18,902,937 | ) | 16,204,666 | |||||
| Investing activities |
||||||||
| Purchases of investments |
(12,000,000 | ) | (7,200,000 | ) | ||||
| Maturities of investments |
17,700,122 | 2,850,000 | ||||||
| Purchase of property and equipment |
(38,957 | ) | (17,825 | ) | ||||
|
|
|
|
|
|||||
| Net cash provided by (used in) investing activities |
5,661,165 | (4,367,825 | ) | |||||
| Financing activities |
||||||||
| Proceeds from issuance of Series C Redeemable Convertible Preferred Stock, net of offering costs |
— | — | ||||||
| Proceeds from issuance of convertible promissory notes, net |
15,163,004 | — | ||||||
| Proceeds from stock option exercises |
31,081 | — | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
15,194,085 | — | ||||||
|
|
|
|
|
|||||
| Net increase (decrease) in cash and cash equivalents |
1,952,313 | 11,836,841 | ||||||
| Cash and cash equivalents at beginning of year |
13,693,215 | 1,856,374 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at end of year |
$ | 15,645,528 | $ | 13,693,215 | ||||
|
|
|
|
|
|||||
| Cash paid for interest |
$ | — | $ | 15 | ||||
|
|
|
|
|
|||||
| Cash paid for taxes |
$ | 13,857 | $ | 30,589 | ||||
|
|
|
|
|
|||||
See accompanying notes.
F-6
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements
December 31, 2012
1. Organization
Intra-Cellular Therapies, Inc. (ITI or the Company) was incorporated in the state of Delaware on May 22, 2001 and commenced operations in June 2002. The Company was founded to discover and develop drugs for the treatment of neurological and psychiatric disorders. The Company’s technology is built on a unique and proprietary understanding of the intracellular effects of neurotransmitters. This know-how has allowed ITI to develop new drugs based on novel drug targets and to create unique molecular signatures for known neurotransmitters and drugs. This technology has also allowed ITI to screen potential lead compounds in more specific ways than are currently available. The Company’s technology addresses diseases of the central nervous system, including schizophrenia, cognition, Parkinson’s disease, anxiety, depression, Alzheimer’s disease, sleep, and those related to women’s health.
The Company earns its license and collaboration revenue from its significant partnership with Takeda Pharmaceutical Company Limited (Takeda). For the year ended December 31, 2011, the Company earned grant revenue under grants awarded by U.S. government agencies and foundations. In order to further its research projects and support its collaborations, the Company will require additional financing until such time that revenue streams are sufficient to generate consistent positive cash flow from operations. Possible sources of funds are strategic alliances, additional equity offerings, grants and contracts, and research and development funding from third parties.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Although actual results could differ from those estimates, management does not believe that such differences would be material.
Cash and Cash Equivalents
The Company considers all highly liquid investments with a maturity of three months or less from the date of purchase to be cash equivalents. Cash and cash equivalents consist of certificates of deposit with commercial banks and financial institutions. Certificates of deposit with a maturity date of more than three months are classified separately on the balance sheet. Their carrying values approximate the fair market value.
Fair Value Measurements
The Company applies the fair value method under ASC 820, Fair Value Measurements and Disclosures. ASC 820 defines fair value, establishes a fair value hierarchy for assets and liabilities measured at fair value and requires expanded disclosures about fair value measurements. The ASC 820 hierarchy ranks the quality and reliability of inputs, or assumptions, used in the determination of fair value and requires assets and liabilities carried at fair value to be classified and disclosed in one of the following categories based on the lowest level input used that is significant to a particular fair value measurement:
| • | Level 1—Fair value is determined by using unadjusted quoted prices that are available in active markets for identical assets and liabilities. |
| • | Level 2—Fair value is determined by using inputs other than Level 1 quoted prices that are directly or indirectly observable. Inputs can include quoted prices for similar assets and liabilities in active |
F-7
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies (continued)
| markets or quoted prices for identical assets and liabilities in inactive markets. Related inputs can also include those used in valuation or other pricing models, such as interest rates and yield curves that can be corroborated by observable market data. |
| • | Level 3 – Fair value is determined by inputs that are unobservable and not corroborated by market data. Use of these inputs involves significant and subjective judgments to be made by a reporting entity—e.g., determining an appropriate adjustment to a discount factor for illiquidity associated with a given security. |
The Company evaluates financial assets and liabilities subject to fair value measurements on a recurring basis to determine the appropriate level at which to classify them each reporting period. This determination requires the Company to make subjective judgments as to the significance of inputs used in determining fair value and where such inputs lie within the ASC 820 hierarchy.
The Company has no assets or liabilities that were measured using quoted prices for similar assets and liabilities or significant unobservable inputs (Level 2 and Level 3 assets and liabilities, respectively) as of December 31, 2012. The carrying value of cash held in money market funds of approximately $1.2 million as of December 31, 2012, is included in cash and cash equivalents and approximates market value based on quoted market price or Level 1 inputs.
Financial Instruments
The Company considers the recorded costs of its financial assets and liabilities, which consist of cash equivalents, accounts receivable, accounts payable and accrued liabilities, to approximate their fair value because of their relatively short maturities at December 31, 2012 and 2011. Management believes that the risks associated with its financial instruments are minimal as the counterparties are various corporations, financial institutions and government agencies of high credit standing.
Concentration of Credit Risk
Cash equivalents are held with major financial institutions in the United States. Certificates of deposit held with banks may exceed the amount of insurance provided on such deposits. Generally, these deposits may be redeemed upon demand and, therefore, bear minimal risk.
Accounts Receivable
Accounts receivable that management has the intent and ability to collect are reported in the balance sheets at outstanding amounts, less an allowance for doubtful accounts. The Company writes off uncollectible receivables when the likelihood of collection is remote.
The Company evaluates the collectability of accounts receivable on a regular basis. The allowance, if any, is based upon various factors including the financial condition and payment history of customers, an overall review of collections experience on other accounts and economic factors or events expected to affect future collections experience. No allowance was recorded as of December 31, 2012, as the Company has a history of collecting on all accounts including government agencies and collaborations funding its research.
F-8
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies (continued)
Property and Equipment
Property and equipment is stated at cost and depreciated on a straight-line basis over estimated useful lives ranging from three to five years. Leasehold improvements are amortized using the straight-line method over the shorter of the estimated useful life of the assets or the term of the related lease. Expenditures for maintenance and repairs are charged to operations as incurred.
When indicators of possible impairment are identified, the Company evaluates the recoverability of the carrying value of its long-lived assets based on the criteria established in ASC 360, Property, Plant and Equipment. The Company considers historical performance and anticipated future results in its evaluation of potential impairment. The Company evaluates the carrying value of those assets in relation to the operating performance of the business and undiscounted cash flows expected to result from the use of those assets. Impairment losses are recognized when carrying value exceeds the undiscounted cash flows then management must determine the fair value of the underlying asset. No such impairment losses have been recognized to date.
Revenue Recognition
Revenue is recognized when all terms and conditions of the agreements have been met, including persuasive evidence of an arrangement, delivery has occurred or services have been rendered, price is fixed or determinable and collectability is reasonably assured. The Company is reimbursed for certain costs incurred on specified research projects under the terms and conditions of grants, collaboration agreements, and awards. The Company records the amount of reimbursement as revenues on a gross basis in accordance with ASC 605-45, Revenue Recognition/Principal Agent Considerations. The Company is the primary obligor with respect to purchasing goods and services from third-party suppliers, is obligated to compensate the service provider for the work performed, and has discretion in selecting the supplier. Provisions for estimated losses on research grant projects and any other contracts are made in the period such losses are determined.
Effective January 1, 2011, the Company adopted a new accounting standard that amends the guidance on the accounting for arrangements involving the delivery of more than one element. Pursuant to the new standard, each required deliverable is evaluated to determine whether it qualifies as a separate unit of accounting. For ITI this determination is generally based on whether the deliverable has “stand-alone value” to the customer. The Company adopted this new accounting standard on a prospective basis for all Multiple-Deliverable Revenue Arrangements (MDRAs) entered into on or after January 1, 2011, and for any MDRAs that were entered into prior to January 1, 2011, but materially modified on or after that date.
For MDRAs entered into prior to January 1, 2011 (pre-2011 arrangements) and not materially modified thereafter, we continue to apply our prior accounting policy with respect to such arrangements. Under this policy, in general, revenue from non-refundable, up-front fees related to intellectual property rights/licenses, where we have continuing involvement and where standalone value could not be determined under the previous guidance, is recognized ratably over the estimated period of ongoing involvement. In general, the consideration with respect to the other deliverables is recognized when the goods or services are delivered.
The adoption of this accounting standard did not have a material impact on our results of operations for the years ended December 31, 2012 and 2011, or on our financial positions as of December 31, 2012 and 2011. Our results of operations for the year ended December 31, 2010 also would not have been materially impacted if the accounting standard had been adopted on January 1, 2010.
F-9
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies (continued)
In January 2011, the Company adopted ASC Topic 605-28, Milestone Method. Under this guidance, we recognize revenue contingent upon the achievement of a substantive milestone in its entirety in the period the milestone is achieved. Substantive milestone payments are recognized upon achievement of the milestone only if all of the following conditions are met:
| • | The milestone payments are non-refundable; |
| • | Achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; |
| • | Substantive effort on our part is involved in achieving the milestone; |
| • | The amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and |
| • | A reasonable amount of time passes between the up-front license payment and the first milestone payment, as well as between each subsequent milestone payment. |
Determination as to whether a payment meets the aforementioned conditions involves management’s judgment. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone, and therefore, the resulting payment would be considered part of the consideration for the single unit of accounting and be recognized as revenues in accordance with the revenue models described above. In addition, the determination that one such payment was not a substantive milestone could prevent us from concluding that subsequent milestone payments were substantive milestones and, as a result, any additional milestone payments could also be considered part of the consideration for the single unit of accounting and would be recognized as revenue as such performance obligations are performed under either the proportional performance or straight-line methods, as applicable.
Deferred Revenue
Cash received as prepayment on future services is deferred and recognized as revenue as the services are performed. The Company must remit interest on any deferred revenue related to a governmental agency. As of December 31, 2012 and 2011, no interest was due as the Company did not have any deferred revenue from a government agency.
Research and Development
Except for payments made in advance of services, the Company expenses its research and development costs as incurred. For payments made in advance, the Company recognizes research and development expense as the services are rendered. Research and development costs primarily consist of salaries and related expenses for personnel and resources and the costs of clinical trials. Other research and development expenses include preclinical analytical testing, outside services, providers, materials and consulting fees.
Income Taxes
Income taxes are accounted for using the liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and its respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which those temporary differences are expected to be recovered or settled.
F-10
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies (continued)
The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when necessary to reduce net deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable for the period and the change during the period in deferred tax assets and liabilities.
The Company accounts for uncertain tax positions pursuant to ASC 740 (previously included in Financial Accounting Standards Board (FASB) Interpretation No. 48, Accounting for Uncertainty in Income Taxes—an Interpretation of FASB Statement No. 109). Financial statement recognition of a tax position taken or expected to be taken in a tax return is determined based on a more-likely-than-not threshold of that position being sustained. If the tax position meets this threshold, the benefit to be recognized is measured as the tax benefit having the highest likelihood of being realized upon ultimate settlement with the taxing authority. The Company recognizes interest accrued related to unrecognized tax benefits and penalties in the provision for income taxes.
Comprehensive Income (Loss)
ASC 220-10, Reporting Comprehensive Income, requires the presentation of the comprehensive income or loss and its components as part of the financial statements. For the years ended December 31, 2012, 2011 and 2010, the Company’s net (loss) income equals comprehensive (loss) income.
Share-Based Compensation
Share-based payments are accounted for in accordance with the provisions of ASC 718, Compensation—Stock Compensation (ASC 718). The fair value of share-based payments is estimated, on the date of grant, using the Black-Scholes-Merton option-pricing model (the Black-Scholes model). The resulting fair value is recognized ratably over the requisite service period, which is generally the vesting period of the option.
For all time vesting awards granted, expense is amortized using the straight-line attribution method. For awards that contain a performance condition, expense is amortized using the accelerated attribution method. As share-based compensation expense recognized in the statements of operations for the years ended December 31, 2012 and 2011, is based on share-based awards ultimately expected to vest, it has been reduced for estimated forfeitures.
ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Pre-vesting forfeitures are based on the Company’s historical experience for the years ended December 31, 2012, 2011 and 2010, and have not been material.
The Company utilizes the Black-Scholes model for estimating fair value of its stock options granted. Option valuation models, including Black-Scholes model, require the input of subjective assumptions, and changes in the assumptions used can materially affect the grant date fair value of an award. These assumptions include the risk-free rate of interest, expected dividend yield, expected volatility and the expected life of the award.
Expected volatility rates are based on historical volatility of the common stock of comparable publicly traded entities and other factors due to the lack of historic information of the Company’s common stock. The expected life of stock-based options is the period of time for which the stock-based options are expected to be outstanding. Given the lack of historic exercise data, the expected life is determined using the “simplified method” which is defined as the midpoint between the vesting date and the end of the contractual term.
F-11
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies (continued)
The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. The Company has not paid dividends to its stockholders since its inception and does not plan to pay cash dividends in the foreseeable future. Therefore, the Company has assumed an expected dividend rate of zero.
Given the absence of an active market for the Company’s common stock, the exercise price of the stock options on the date of grant was determined and approved by the board of directors using several factors, including progress and milestones achieved in the Company’s business development and performance, the price per share of its convertible preferred stock offerings and general industry and economic trends. In establishing the estimated fair value of the common stock, the Company considered the guidance set forth in American Institute of Certified Public Accountants Practice Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation.
Under ASC 718, the cumulative amount of compensation cost recognized for instruments classified as equity that ordinarily would result in a future tax deduction under existing tax law shall be considered to be a deductible difference in applying ASC 740, Income Taxes. The deductible temporary difference is based on the compensation cost recognized for financial reporting purposes; however, these provisions currently do not impact the Company, as all the deferred tax assets have a full valuation allowance.
Since the Company had net operating loss carryforwards as of December 31, 2012 and 2011, no excess tax benefits for the tax deductions related to share-based awards were recognized in the statements of operations.
Equity instruments issued to non-employees are accounted for under the provisions of ASC 718 and ASC 505-50, Equity/Equity-Based Payments to Non-Employees. Accordingly, the estimated fair value of the equity instrument is recorded on the earlier of the performance commitment date or the date the services required are completed and are marked to market during the service period.
(Loss) Earnings Per Share
(Loss) Earnings per share is calculated under the two-class method under which all earnings (distributed and undistributed) are allocated to each class of common stock and participating securities based on their respective rights to receive dividends. In the event that the Board of Directors shall declare a dividend payable in cash or other property on the then-outstanding shares of common stock, the holders of the Redeemable Preferred Series A, B, and C convertible preferred stock shall be entitled to receive the amount of dividends per share of Preferred Stock that would be payable on the largest number of whole shares of Common Stock into which each share of Preferred Stock could then be converted. Therefore, the Redeemable Preferred Series A, B, and C Preferred Stock are participating securities.
F-12
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
2. Summary of Significant Accounting Policies (continued)
Basic net (loss) income per common share is determined by dividing the net (loss) income allocable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted net (loss) income per share is computed by dividing the net (loss) income allocable to common stockholders by the weighted-average number of common stock equivalents outstanding for the period. The treasury stock method is used to determine the dilutive effect of the Company’s stock option grants and the if-converted method is used to determine the dilutive effect of the Company’s Redeemable Preferred Series A, B, and C convertible preferred stock.
| Year Ended December 30 | ||||||||
| 2012 | 2011 | |||||||
| Basic (loss) income per common share |
||||||||
| Net (loss) income |
$ | (16,590,827 | ) | $ | 11,092,429 | |||
| Less: Undistributed (loss) earnings allocated to participating securities |
(1,672,223 | ) | (6,747,903 | ) | ||||
|
|
|
|
|
|||||
| Net (loss) earnings allocable to common shares |
$ | (18,263,050 | ) | $ | 4,344,526 | |||
|
|
|
|
|
|||||
| Basic weighted average common shares outstanding |
11,215,077 | 11,202,990 | ||||||
|
|
|
|
|
|||||
| Basic (loss) earnings per common share |
$ | (1.63 | ) | $ | 0.39 | |||
| Diluted (loss) earnings per common share |
||||||||
| Net (loss) earnings |
$ | (16,590,827 | ) | $ | 11,092,429 | |||
| Less: Undistributed (loss) earnings allocated to participating securities |
(1,672,223 | ) | (6,747,903 | ) | ||||
|
|
|
|
|
|||||
| Net (loss) earnings allocable to common shares |
$ | (18,263,050 | ) | $ | 4,344,526 | |||
|
|
|
|
|
|||||
| Basic weighted average common shares outstanding |
11,215,077 | 11,202,990 | ||||||
| Effect of dilutive options |
— | 1,987,486 | ||||||
|
|
|
|
|
|||||
| Diluted weighted average common shares outstanding |
11,215,077 | 13,190,476 | ||||||
|
|
|
|
|
|||||
| Diluted (loss) earnings per common share |
$ | (1.63 | ) | $ | 0.33 | |||
The following common stock equivalents were excluded in the calculation of diluted (loss) earnings per share because their effect would be anti-dilutive as applied to the loss from operations as of December 31, 2012:
| Year Ended December 31 | ||||||||
| 2012 | 2011 | |||||||
| Series A, B, and C Preferred Stock |
13,094,663 | 13,094,663 | ||||||
| Stock options |
2,132,194 | — | ||||||
| Convertible promissory notes |
834,106 | — | ||||||
Recently Issued Accounting Pronouncements
In June 2011, the FASB issued ASU 2011-05, Presentation of Comprehensive Income. ASU 2011-05 revises the manner in which entities present comprehensive income in their financial statements. The recent guidance removes the presentation options in ASC 220 and requires entities to report components of comprehensive income in either (1) a continuous statement of comprehensive income or (2) two separate but consecutive statements. ASU 2011-05 did not change the items that must be reported in other comprehensive income. The Company adopted the provisions of ASU 2011-05 for the year ended December 31, 2012 and elected the second option. However, for the years ended December 31, 2012 and 2011, the Company’s net (loss) income equals comprehensive (loss) income, and, therefore, a separate statement of other comprehensive income was not necessary.
F-13
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
3. Property and Equipment
Property and equipment consist of the following:
| December 31 | ||||||||
| 2012 | 2011 | |||||||
| Computer equipment |
$ | 92,318 | $ | 107,940 | ||||
| Furniture and fixtures |
42,736 | 42,736 | ||||||
| Scientific equipment |
2,824,076 | 2,786,539 | ||||||
| Leasehold improvements |
319,553 | 319,553 | ||||||
|
|
|
|
|
|||||
| 3,278,683 | 3,256,768 | |||||||
| Less accumulated depreciation |
(3,220,417 | ) | (3,189,712 | ) | ||||
|
|
|
|
|
|||||
| $ | 58,266 | $ | 67,056 | |||||
|
|
|
|
|
|||||
Depreciation expense for the years ended December 31, 2012, 2011 and 2010 was $47,747, $189,186 and $386,992, respectively.
4. Redeemable Convertible Preferred Stock
In 2002, the Company issued 2,900,000 shares of Series A redeemable convertible preferred stock (Series A Preferred Stock) for proceeds of $2,828,322. In 2003, the Company issued 1,250,000 shares of Series A Preferred Stock for proceeds of $1,250,000.
In 2006, the Company issued a total of 5,000,216 shares of Series B redeemable convertible preferred stock (Series B Preferred Stock) for proceeds of $7,863,725, net of $58,850 in offering costs.
In 2007, the Company issued a total of 4,030,024 shares of Series C redeemable convertible preferred stock (Series C Preferred Stock) for proceeds of $7,935,898, net of $64,102 in offering costs. In 2010, the Company issued a total of 2,687,915 shares of Series C Preferred Stock for proceeds of $5,274,886, net of $60,894 in offering costs.
In connection with the Series C Preferred Stock offering, approximately $450,000 of Series A Preferred Stock, $2,173,451 of Series B Preferred Stock and $1,896,116 of Series C Preferred Stock were converted to 2,773,492 shares of common stock in accordance with their respective conversion ratios.
Dividends are cumulative and accrue on each outstanding share of Series A, Series B and Series C Preferred Stock at an 8% rate per annum. These dividends would be paid when and if declared by the Board of Directors. At December 31, 2012 and 2011, accrued and unpaid dividends for each respective preferred stock issuance were as follows:
| December 31 | ||||||||
| 2012 | 2011 | |||||||
| Series A Preferred Stock |
$ | 3,325,667 | $ | 3,029,667 | ||||
| Series B Preferred Stock |
3,807,154 | 3,346,402 | ||||||
| Series C Preferred Stock |
4,099,657 | 3,184,484 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 11,232,478 | $ | 9,560,553 | ||||
|
|
|
|
|
|||||
F-14
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
4. Redeemable Convertible Preferred Stock (continued)
The Series A, Series B and Series C Preferred Stock have a liquidation preference senior to that of the common stock.
Series A Preferred Stock has a liquidation preference of $6,755,992 and $6,459,992 for December 31, 2012 and 2011, respectively. Series B Preferred Stock has a liquidation preference of $8,936,955 and $8,475,905 for December 31, 2012 and 2011, respectively. Series C Preferred Stock has a liquidation preference of $15,141,345 and $14,205,340 for December 31, 2012 and 2011, respectively.
The Company is obligated to redeem shares of Series A, Series B and Series C Preferred Stock, if requested, by the majority of the holders. The beginning redemption of Series A, B and C Preferred Stock is February 26, 2016. The redemption for the Series A, Series B and Series C Preferred Stock, if requested, would take place in three equal installments over a two-year period.
The redemption price shall be equal to $1.00 per share, $1.58 per share and $1.99 per share plus all accrued and unpaid dividends for the Series A, Series B and Series C Preferred Stock, respectively, subject to certain equity adjustments for specified anti-dilutive transactions as defined.
The holders of the Series A, Series B and Series C Preferred Stock have the right to convert such shares, at their option and at any time, into shares of common stock at the then applicable conversion rate, as defined. The initial conversion rate is one common share for each preferred share, which is adjusted for specified anti-dilutive transactions, as defined. At December 31, 2012, the Company has reserved 3,700,000 shares, 3,631,898 shares and 5,762,765 shares of common stock for conversion of Series A, Series B and Series C Preferred Stock, respectively.
The Series A, Series B and Series C Preferred Stock will automatically convert into common stock at the then applicable conversion rate upon a majority vote of the Series A, Series B and Series C Preferred Stockholders or upon a public offering of the Company’s common stock, resulting in aggregate proceeds to the Company of at least $20 million and a price per share of at least $5.00.
The holders of Series A, Series B and Series C Preferred Stock are entitled to the whole number of votes equal to the number of shares of common stock into which such shares could be converted.
5. Share-Based Compensation
The Company sponsors the Intra-Cellular Therapies, Inc. 2003 Equity Incentive Plan (the Plan) to provide for the granting of stock awards, such as stock options, restricted common stock and stock appreciation rights to employees, directors and other individuals as determined by the Board of Directors. The Company reserved 3,700,000 shares of common stock for issuance under the Plan. In December 2012, the Company increased the number of shares of common stock reserved for issuance under the plan to 5,700,000.
Stock options granted under the Plan may be either incentive stock options (ISOs) as defined by the Internal Revenue Code, or non-qualified stock options. The Board of Directors determines who will receive options, the vesting periods (which are generally two to three years) and the exercise prices. Options have a maximum term of 10 years. The exercise price of ISOs granted under the Plan must be at least equal to the fair market value of the common stock on the date of grant.
F-15
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
5. Share-Based Compensation (continued)
Total stock-based compensation expense, related to all of the Company’s share-based awards to employees, directors and non-employees recognized during the years ended 2012 and 2011, was comprised of the following:
| Year Ended December 31 | ||||||||
| 2012 | 2011 | |||||||
| Research and development |
$ | 111,206 | $ | 113,534 | ||||
| General and administrative |
183,900 | 166,918 | ||||||
|
|
|
|
|
|||||
| Total share-based compensation expense |
$ | 295,106 | $ | 280,452 | ||||
|
|
|
|
|
|||||
The following table describes the weighted-average assumptions used for calculating the value of options granted for the years ended December 31:
| 2012 | 2011 | |||||||
| Dividend yield |
0.0 | % | — | % | ||||
| Expected volatility |
79.7 | % | — | |||||
| Weighted-average risk-free interest rate |
1.2 | % | — | |||||
| Expected term |
6.3 years | — | ||||||
Information regarding the stock options activity including employees, directors and non-employees as of December 31, 2012, and changes during the year then ended, are summarized as follows:
| Number of Shares |
Weighted- Average Exercise Price |
Weighted- Average Contractual Life |
||||||||||
| Outstanding at December 31, 2011 |
3,175,567 | $ | 0.61 | 5.0 years | ||||||||
| Options granted |
322,200 | 1.42 | 6.3 years | |||||||||
| Options exercised |
(66,540 | ) | 0.47 | 3.0 years | ||||||||
| Options canceled or expired |
(17,000 | ) | 1.37 | 7.9 years | ||||||||
|
|
|
|
|
|||||||||
| Outstanding at December 31, 2012 |
3,414,227 | 0.66 | 4.4 years | |||||||||
|
|
|
|
|
|||||||||
| Vested or expected to vest at December 31, 2012 |
3,416,227 | 0.69 | ||||||||||
|
|
|
|
|
|||||||||
| Exercisable at December 31, 2012 |
3,151,353 | $ | 0.58 | 3.9 years | ||||||||
|
|
|
|
|
|||||||||
The weighted-average grant date fair value for awards granted during the year ended December 31, 2012, was $0.99. Total intrinsic value of the options exercised was approximately $63,000 and $35,000 in the year ended December 31, 2012. The total fair value of shares vested in the years ended December 31, 2012 and 2011, was approximately $332,000 and $182,000 respectively.
During 2012, the Company granted options to certain scientific advisory board members of the Company to purchase 39,000 shares of common stock at an average exercise price of $1.42. There were no options granted during 2011. The options vest ratably over a period of 12 to 24 months. Stock compensation related to these grants will fluctuate with any changes in the underlying value of the Company’s common stock, as the performance period is not fixed.
F-16
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
5. Share-Based Compensation (continued)
The unrecognized share-based compensation expense related to employee stock option awards at December 31, 2012, is $285,327 and will be recognized over a weighted-average period of 1.9 years. The unrecognized share-based compensation expense related to employee stock option awards at December 31, 2011, is $259,899 and will be recognized over a weighted-average period of 1.5 years.
6. Income Taxes
The provision (benefit) for income taxes consists of:
| December 31 | ||||||||
| 2012 | 2011 | |||||||
| Current |
$ | 32,921 | $ | 64,834 | ||||
| Deferred |
(6,378,456 | ) | 4,359,809 | |||||
| Valuation allowance |
6,378,456 | (4,359,809 | ) | |||||
|
|
|
|
|
|||||
| Provision (benefit) for income taxes |
$ | 32,921 | $ | 64,834 | ||||
|
|
|
|
|
|||||
The deferred tax provision has been entirely offset by a valuation allowance because the Company is currently utilizing the underlying tax benefits generated in previous years. The difference between the amounts of income tax benefit that would result from applying domestic federal statutory tax rates to the net loss relates to certain nondeductible expenses, state income taxes and the valuation allowance.
The Company’s deferred tax assets and liabilities were as follows:
| December 31 | ||||||||
| 2012 | 2011 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 8,418,507 | $ | 1,118,070 | ||||
| Accrued expenses |
215,865 | 416,268 | ||||||
| Accrued employee benefits |
282,268 | 259,638 | ||||||
| Capitalized research and development costs |
27,516 | 108,138 | ||||||
| Research and development credit |
1,928,714 | 1,612,459 | ||||||
| Deferred revenue |
643,669 | 1,287,333 | ||||||
| Deferred tax liabilities: |
||||||||
| Depreciation |
130,017 | 147,165 | ||||||
|
|
|
|
|
|||||
| Net deferred tax asset |
11,646,556 | 4,949,071 | ||||||
| Valuation allowance |
(11,646,556 | ) | (4,949,071 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax asset |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The net operating loss carryforwards of approximately $21.8 million will begin to expire in the year 2030 if unused. The use of the Company’s net operating loss carryforwards may be restricted due to changes in Company ownership.
F-17
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
7. Collaborations and License Agreements
Takeda Pharmaceutical Company Limited
On February 25, 2011, ITI entered into a license and collaboration agreement with Takeda Pharmaceutical Company Limited (Takeda) to develop and commercialize selective phosphodiesterase type 1 (PDE1) inhibitors, discovered by ITI, for the treatment of cognitive impairment associated with schizophrenia. This agreement is targeted worldwide, but ITI has retained the option to co-promote with Takeda in the United States.
Upon execution of the agreement, Takeda made a nonrefundable payment to the Company. ITI is eligible to receive payments of approximately $500 million in the aggregate upon achievement of certain development milestones and up to an additional $250 million in the aggregate upon achievement of certain sales-based milestones, along with tiered royalty payments based on net sales by Takeda. Takeda will be solely responsible for development, manufacturing and commercialization of PDE1 inhibitors. ITI and Takeda have formed a joint steering committee to coordinate and oversee activities on which the two companies collaborate under the agreement. ITI has the right, but not the obligation, to sit on the joint steering committee. There are no performance, cancellation, termination, or refund provisions in the arrangement that contain material financial consequences to the Company.
The Company evaluates all deliverables within an arrangement to determine whether or not they provide value on a stand-alone basis. The Company identified two deliverables in the arrangement, (1) a license to the Company’s intellectual property, and (2) research and development services (“R&D services”). Based on this evaluation, the deliverables were separated into units of accounting. The arrangement consideration that is fixed or determinable at the inception of the arrangement was allocated to the separate units of accounting based on their relative selling prices. We may exercise significant judgment in determining whether a deliverable is a separate unit of accounting, as well as in estimating the selling prices of such unit of accounting.
To determine the selling price of a separate deliverable, we use the hierarchy as prescribed in ASC Topic 605-25 based on vendor-specific objective evidence (VSOE), third-party evidence (TPE) or best estimate of selling price (BESP). VSOE is based on the price charged when the element is sold separately and is the price actually charged for that deliverable. TPE is determined based on third-party evidence for a similar deliverable when sold separately and BESP is the price at which we would transact a sale if the elements of collaboration and license arrangements were sold on a stand-alone basis. We were not able to establish VSOE or TPE for the deliverables within collaboration and license arrangements, as we do not have a history of entering into such arrangements or selling the individual deliverables within such arrangements separately. In addition, there may be significant differentiation in these arrangements, which indicates that comparable third-party pricing may not be available. We determined that the selling price for the deliverables within collaboration and license arrangements should be determined using BESP. The process for determining BESP involved significant judgment on our part and included consideration of multiple factors such as prices offered by third parties, estimated direct expenses and other costs, and available data. The Company was able to determine the BESP for the license and R&D services, and thus, allocated the consideration in this arrangement based on relative selling price of each deliverable. The revenue allocated to the license was recognized upon the execution of the agreement as Takeda obtained the right to use the license upon execution of the agreement. The revenue for R&D services is being recognized over the estimated service period of 3 years.
During the years ended December 31, 2012 and December 31, 2011, the Company recognized revenue of $3.1 million and $22.3 million under this agreement, respectively. At December 31, 2012 and 2011, $1.7 million and $3.3 million of revenue was deferred under this agreement.
F-18
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
7. Collaborations and License Agreements (continued)
Beginning in 2003, the Company entered into several cooperative agreements and grants with the U.S. Army and the National Institutes of Health. Under these research agreements, the Company uses its patented technology to examine and characterize the effects of various agents and drugs on the signaling pathways in the brain and the biochemical mechanisms associated with various diseases of the brain. These agreements were originally from one to three years in length. The Company has not received any funding from these agreements since 2010. For the years ended December 31, 2012 and 2011, the Company recognized revenue of approximately $0 million and $1.03 million, respectively.
In May 2002, the Company entered into a license agreement (the License) and research agreement with a university. Under the provisions of the License, the Company is entitled to use this organization’s patented technology and other intellectual property relating to diagnosis and treatment of central nervous system disorders.
The License expires upon expiration of the patent rights or 15 years subsequent to the first sale of products developed through this License. The Company is required to make future milestone payments for initiation of clinical trials and approval of a New Drug Application (NDA). Should the Company commercialize the technology related to this License, the Company would be required to make royalty payments, and would also be required to pay fees under any sublicense agreements with third parties.
In connection with the License, the Company issued 800,000 shares of common stock to the organization. Upon issuance of the shares, the Company recorded the estimated fair value of the shares issued, approximately $120,000, as research and development expense.
In addition, the Company is required to use at least $1 million annually of its resources for the development and commercialization of the technology until the Company submits a NDA. The Company met its spending requirements in 2012, 2011 and 2010. There were no other payments made or required for the years ended December 31, 2012 and 2011.
In May 2005, the Company entered into a license agreement (the Agreement) with a company for the use of this company’s patented compounds. ITI intends to test and use the compounds in its research and development program as candidates for potential new drugs.
The Agreement expires on the later of 10 years after the first commercial sale of a product developed using the licensed compound or upon expiration of the patent rights. The Company is required to make future milestone payments for commencement of certain clinical trials and filings with the U.S. Food and Drug Administration. Should the Company sell products covered by the Agreement, the Company would be required to make royalty payments. There were no payments under this Agreement for the years ended December 31, 2012 and 2011.
8. Convertible Promissory Notes
In October 2012, the Company entered into an agreement with existing investors to obtain $15.2 million in exchange for convertible promissory notes net of $26,888 of offering costs. The proceeds will be used to finance research studies. The debt plus 6% accrued interest will be converted to 5,051,960 shares at ($3.01/share) of Series D Preferred Stock at the maturity date of October 25, 2013 or later if amended. The Company has amortized fees associated with the debt issuance and the balance remaining as of December 31, 2012 is $16,880.
F-19
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Financial Statements (continued)
9. Commitments and Contingencies
The Company currently has operating lease agreements with commitments for $605,000 through 2013 for laboratory and office facilities. Rent expense for the years ended December 31, 2012 and 2011 was $809,332 and $691,823, respectively.
10 . Employee Benefit Plan
The Company sponsors a defined contribution 401(k) plan covering all full-time employees. Participants may elect to contribute up to 15% of their annual pre-tax earnings up to the federally allowed maximum limits. The Company makes a matching contribution of 50% on the first 6% of contributions made by participants. Participant and Company contributions vest immediately. During the years ended December 31, 2012 and 2011, the Company recorded matching contribution expense of $79,656 and $76,231, respectively.
11. Subsequent Events
The Company evaluated subsequent events through June 19, 2013, the date these financial statements were issued.
F-20
Table of Contents
| CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
Intra-Cellular Therapies, Inc. As of September 30, 2013 and December 31, 2012 and for the Three- and Nine-Months Ended September 30, 2013 and 2012 |
F-21
Table of Contents
Intra-Cellular Therapies, Inc. and Subsidiaries
Condensed Consolidated Balance Sheets
| September 30, 2013 |
December 31, 2012 |
|||||||
| (Unaudited) | (Audited) | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 44,072,012 | $ | 15,645,528 | ||||
| Certificates of deposit |
2,000,000 | 3,500,000 | ||||||
| Accounts receivable |
251,291 | 300,429 | ||||||
| Prepaid expenses and other current assets |
806,288 | 188,702 | ||||||
|
|
|
|
|
|||||
| Total current assets |
47,129,591 | 19,634,659 | ||||||
| Property and equipment, net |
75,700 | 58,266 | ||||||
| Other assets |
130,755 | 130,755 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 47,336,046 | $ | 19,823,680 | ||||
|
|
|
|
|
|||||
| Liabilities, redeemable convertible preferred stock, and stockholders’ deficit |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 4,027,097 | $ | 41,608 | ||||
| Accrued and other current liabilities |
2,571,126 | 404,656 | ||||||
| Accrued employee benefits |
759,757 | 726,657 | ||||||
| Deferred revenue |
416,682 | 1,666,674 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
7,774,662 | 2,839,595 | ||||||
| Stockholders’ equity (deficit): |
||||||||
| Common stock, $.0001 par value: 100,000,000 shares authorized; 22,134,647 and 14,599,612 shares issued and outstanding at September 30, 2013 and December 31, 2012, respectively |
2,213 | 1,460 | ||||||
| Additional paid-in capital |
89,082,858 | 47,678,924 | ||||||
| Accumulated deficit |
(49,523,687 | ) | (30,696,299 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
39,561,384 | 16,984,085 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 47,336,046 | $ | 19,823,680 | ||||
|
|
|
|
|
|||||
See accompanying notes.
F-22
Table of Contents
Intra-Cellular Therapies, Inc. and Subsidiaries
Condensed Consolidated Statements of Operations
| Three-Months Ended September 30, | Nine-Months Ended September 30, | |||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| (Unaudited) | (Unaudited) | (Unaudited) | (Unaudited) | |||||||||||||
| Revenues |
$ | 667,955 | $ | 377,911 | $ | 1,909,471 | $ | 2,449,055 | ||||||||
| Costs and expenses: |
||||||||||||||||
| Research and development |
4,157,742 | 1,797,429 | 16,897,903 | 14,969,710 | ||||||||||||
| General and administrative |
1,295,571 | 802,053 | 3,245,585 | 2,916,139 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total costs and expenses |
5,453,313 | 2,599,482 | 20,143,488 | 17,885,849 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(4,785,358 | ) | (2,221,571 | ) | (18,234,017 | ) | (15,436,794 | ) | ||||||||
| Interest expense |
(131,888 | ) | — | (604,960 | ) | — | ||||||||||
| Interest income |
5,626 | 6,656 | 11,589 | 29,730 | ||||||||||||
| Income taxes |
— | (8,230 | ) | — | (24,690 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
(4,911,620 | ) | (2,223,145 | ) | (18,827,388 | ) | (15,431,754 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per common share: |
||||||||||||||||
| Basic |
$ | (0.42 | ) | $ | (0.40 | ) | $ | (2.43 | ) | $ | (2.75 | ) | ||||
| Dilutive |
(0.42 | ) | (0.40 | ) | (2.43 | ) | (2.75 | ) | ||||||||
| Weighted average number of common shares: |
||||||||||||||||
| Basic & Dilutive |
11,779,745 | 5,607,022 | 7,737,250 | 5,603,575 | ||||||||||||
See accompanying notes.
F-23
Table of Contents
Intra-Cellular Therapies, Inc. and Subsidiaries
Condensed Consolidated Statements of Cash Flows
| Nine-Months Ended September 30 | ||||||||
| 2013 | 2012 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Cash flows provided by (used in) operating activities |
||||||||
| Net loss |
$ | (18,827,388 | ) | $ | (15,431,754 | ) | ||
| Adjustments to reconcile net loss to net cash provided by operating activities: |
||||||||
| Depreciation |
15,821 | 39,413 | ||||||
| Share-based compensation expense |
282,450 | 282,629 | ||||||
| Changes in operating assets and liabilities: |
||||||||
| Accounts receivable |
49,138 | 241,534 | ||||||
| Prepaid expenses and other assets |
(617,586 | ) | 2,099 | |||||
| Accounts payable |
3,985,489 | 2,078,182 | ||||||
| Accrued and other current liabilities and employee benefits |
2,398,611 | (738,321 | ) | |||||
| Deferred revenue |
(1,249,992 | ) | (1,249,995 | ) | ||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(13,963,457 | ) | (14,776,213 | ) | ||||
| Cash flows provided by (used in) investing activities |
||||||||
| Purchases of investments |
— | (1,000,000 | ) | |||||
| Maturities of investments |
1,500,000 | 5,950,123 | ||||||
| Purchase of property and equipment |
(33,255 | ) | (38,957 | ) | ||||
|
|
|
|
|
|||||
| Net cash provided by investing activities |
1,466,745 | 4,911,166 | ||||||
| Cash flows provided by (used in) financing activities |
||||||||
| Proceeds from stock option exercises |
316,827 | 7,022 | ||||||
| Proceeds from stock subscription |
109,834 | — | ||||||
| Gross proceeds of public offering |
43,941,850 | — | ||||||
| Payment of costs of public offering |
(3,445,315 | ) | — | |||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
40,923,196 | 7,022 | ||||||
|
|
|
|
|
|||||
| Net increase (decrease) in cash and cash equivalents |
28,426,484 | (9,858,025 | ) | |||||
| Cash and cash equivalents at beginning of period |
15,645,528 | 13,693,215 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at end of period |
$ | 44,072,012 | $ | 3,835,190 | ||||
|
|
|
|
|
|||||
| Cash paid for interest |
$ | 3,317 | $ | — | ||||
| Cash paid for taxes |
$ | 13,437 | $ | 13,857 | ||||
See accompanying notes.
F-24
Table of Contents
Intra-Cellular Therapies, Inc.
Notes to Condensed Consolidated Financial Statements (Unaudited)
September 30, 2013
1. Organization
Intra-Cellular Therapies, Inc. (the Company), through its wholly-owned operating subsidiary, ITI, Inc. (ITI), is a biopharmaceutical company focused on the discovery and clinical development of innovative, small molecule drugs that address underserved medical needs in neuropsychiatric and neurological disorders by targeting intracellular signaling mechanisms within the central nervous system (CNS). The Company’s lead product candidate, ITI-007, is in Phase 2 clinical trials as a first-in-class treatment for schizophrenia.
ITI was incorporated in the State of Delaware on May 22, 2001 under the name “Intra-Cellular Therapies, Inc.” and commenced operations in June 2002. ITI was founded to discover and develop drugs for the treatment of neurological and psychiatric disorders.
On August 29, 2013, ITI completed a reverse merger (the Merger) with a public shell company named Oneida Resources Corp. (Oneida). Oneida was formed in August 2012 as a vehicle to investigate and, if such investigation warranted, acquire a target company or business seeking the perceived advantages of being a publicly held corporation. In the Merger, each outstanding share of capital stock of ITI was exchanged for 0.5 shares of common stock of Oneida, and each outstanding option and outstanding warrant of ITI was assumed by Oneida and became exercisable for 0.5 shares of Oneida common stock. As a result of the Merger and related transactions, ITI survived as a wholly-owned subsidiary of Oneida, Oneida changed its fiscal year end from March 31 to December 31, and Oneida changed its name to Intra-Cellular Therapies, Inc. (the Company). In addition, the Company began operating ITI and its business, and therefore ceased being a shell company. Following the Merger and the redemption of all then outstanding shares of Oneida at the closing of the Merger, the former shareholders of ITI owned 100% of the shares of the Company’s outstanding capital stock.
Immediately prior to the Merger, on August 29, 2013, ITI sold to accredited investors approximately $60.0 million of its shares of common stock, or 18,889,307 shares at a price of $3.1764 per share (the Private Placement), which included $15.3 million in principal and $0.8 million in accrued interest from the conversion of ITI’s then outstanding convertible promissory notes (the Notes).
In accordance with Financial Accounting Standards Board (FASB), Accounting Standards Codification (ASC) Topic 805, Business Combinations, ITI is considered the acquirer for accounting purposes, and will account for the transaction as a capital transaction, because ITI’s former stockholders received 100% of the voting rights in the combined entity and ITI’s senior management represents all of the senior management of the combined entity. Consequently, the assets and liabilities and the historical operations that will be reflected in our consolidated financial statements will be those of ITI and will be recorded at the historical cost basis of the Company. All share and per share amounts in the consolidated financial statements and related notes have been retrospectively adjusted to reflect the one for 0.5 shares common stock exchange as well as the conversion of the Notes and Redeemable Preferred Series A, B, and C convertible preferred stock.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Although actual results could differ from those estimates, management does not believe that such differences would be material.
F-25
Table of Contents
Cash and Cash Equivalents
The Company considers all highly liquid investments with a maturity of three months or less from the date of purchase to be cash equivalents. Cash and cash equivalents consist of money market investments and certificates of deposit with commercial banks and financial institutions. Certificates of deposit with a maturity date of more than three months are classified separately on the balance sheet. Their carrying values approximate the fair market value.
Fair Value Measurements
The Company applies the fair value method under ASC Topic 820, Fair Value Measurements and Disclosures. ASC Topic 820 defines fair value, establishes a fair value hierarchy for assets and liabilities measured at fair value and requires expanded disclosures about fair value measurements. The ASC Topic 820 hierarchy ranks the quality and reliability of inputs, or assumptions, used in the determination of fair value and requires assets and liabilities carried at fair value to be classified and disclosed in one of the following categories based on the lowest level input used that is significant to a particular fair value measurement:
| • | Level 1—Fair value is determined by using unadjusted quoted prices that are available in active markets for identical assets and liabilities. |
| • | Level 2—Fair value is determined by using inputs other than Level 1 quoted prices that are directly or indirectly observable. Inputs can include quoted prices for similar assets and liabilities in active markets or quoted prices for identical assets and liabilities in inactive markets. Related inputs can also include those used in valuation or other pricing models, such as interest rates and yield curves that can be corroborated by observable market data. |
| • | Level 3—Fair value is determined by inputs that are unobservable and not corroborated by market data. Use of these inputs involves significant and subjective judgments to be made by a reporting entity – e.g., determining an appropriate adjustment to a discount factor for illiquidity associated with a given security. |
The Company evaluates financial assets and liabilities subject to fair value measurements on a recurring basis to determine the appropriate level at which to classify them each reporting period. This determination requires the Company to make subjective judgments as to the significance of inputs used in determining fair value and where such inputs lie within the ASC Topic 820 hierarchy.
The Company has no assets or liabilities that were measured using quoted prices for similar assets and liabilities or significant unobservable inputs (Level 2 and Level 3 assets and liabilities, respectively) as of September 30, 2013 and December 31, 2012. The carrying value of cash held in money market funds of approximately $21.2 million as of September 30, 2013 and approximately $1.2 million as of December 31, 2012, is included in cash and cash equivalents and approximates market value based on quoted market price or Level 1 inputs.
Financial Instruments
The Company considers the recorded costs of its financial assets and liabilities, which consist of cash equivalents, accounts receivable, accounts payable and accrued liabilities, to approximate their fair value because of their relatively short maturities at September 30, 2013 and December 31, 2012. Management believes that the risks associated with its financial instruments are minimal as the counterparties are financial institutions of high credit standing.
Concentration of Credit Risk
Cash equivalents are held with major financial institutions in the United States. Certificates of deposit held with banks may exceed the amount of insurance provided on such deposits. Generally, these deposits may be redeemed upon demand and, therefore, bear minimal risk.
F-26
Table of Contents
Accounts Receivable
Accounts receivable that management has the intent and ability to collect are reported in the balance sheets at outstanding amounts, less an allowance for doubtful accounts. The Company writes off uncollectible receivables when the likelihood of collection is remote.
The Company evaluates the collectability of accounts receivable on a regular basis. The allowance, if any, is based upon various factors including the financial condition and payment history of customers, an overall review of collections experience on other accounts and economic factors or events expected to affect future collections experience. No allowance was recorded as of September 30, 2013 and December 31, 2012, as the Company has a history of collecting on all accounts including, but not limited to, collaborations funding its research.
Property and Equipment
Property and equipment is stated at cost and depreciated on a straight-line basis over estimated useful lives ranging from three to five years. Leasehold improvements are amortized using the straight-line method over the shorter of the estimated useful life of the assets or the term of the related lease. Expenditures for maintenance and repairs are charged to operations as incurred.
When indicators of possible impairment are identified, the Company evaluates the recoverability of the carrying value of its long-lived assets based on the criteria established in ASC Topic 360, Property, Plant and Equipment. The Company considers historical performance and anticipated future results in its evaluation of potential impairment. The Company evaluates the carrying value of those assets in relation to the operating performance of the business and undiscounted cash flows expected to result from the use of those assets. Impairment losses are recognized when carrying value exceeds the undiscounted cash flows then management must determine the fair value of the underlying asset. No such impairment losses have been recognized to date.
Revenue Recognition
The Company earns its license and collaboration revenue from its significant partnership with Takeda Pharmaceutical Company Limited (Takeda). In order to further its research projects and support its collaborations, the Company will require additional financing until such time that revenue streams are sufficient to generate consistent positive cash flow from operations. Possible sources of funds include strategic alliances, additional equity offerings, grants and contracts, and research and development funding from third parties.
Revenue is recognized when all terms and conditions of the agreements have been met, including persuasive evidence of an arrangement, delivery has occurred or services have been rendered, price is fixed or determinable and collectability is reasonably assured. The Company is reimbursed for certain costs incurred on specified research projects under the terms and conditions of grants, collaboration agreements, and awards. The Company records the amount of reimbursement as revenues on a gross basis in accordance with ASC Topic 605-45, Revenue Recognition/Principal Agent Considerations. The Company is the primary obligor with respect to purchasing goods and services from third-party suppliers, is obligated to compensate the service provider for the work performed, and has discretion in selecting the supplier. Provisions for estimated losses on research grant projects and any other contracts are made in the period such losses are determined.
The Company engages in transactions with delivery of more than one element. Each required deliverable is evaluated to determine whether it qualifies as a separate unit of accounting. For the Company, this determination is generally based on whether the deliverable has “stand-alone value” to the customer. The Company adopted accounts for all Multiple-Deliverable Revenue Arrangements (MDRAs) in accordance with ASC Topic 605-25, Revenue Recognition—Multiple Element Arrangements.
F-27
Table of Contents
The Company accounts for milestone revenue in accordance with ASC Topic 605-28, Milestone Method. Under this guidance, the Company recognizes revenue contingent upon the achievement of a substantive milestone in its entirety in the period the milestone is achieved. Substantive milestone payments are recognized upon achievement of the milestone only if all of the following conditions are met:
| • | The milestone payments are non-refundable; |
| • | Achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; |
| • | Substantive effort on our part is involved in achieving the milestone; |
| • | The amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and |
| • | A reasonable amount of time passes between the up-front license payment and the first milestone payment, as well as between each subsequent milestone payment. |
Determination as to whether a payment meets the aforementioned conditions involves management’s judgment. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone, and therefore, the resulting payment would be considered part of the consideration for the single unit of accounting and be recognized as revenue in accordance with the revenue models described above. In addition, the determination that one such payment was not a substantive milestone could prevent us from concluding that subsequent milestone payments were substantive milestones and, as a result, any additional milestone payments could also be considered part of the consideration for the single unit of accounting and would be recognized as revenue as such performance obligations are performed under either the proportional performance or straight-line methods, as applicable.
Deferred Revenue
Cash received as prepayment on future services is deferred and recognized as revenue as the services are performed. The Company must remit interest on any deferred revenue related to a governmental agency. As of September 30, 2013 and December 31, 2012, no interest was due as the Company did not have any deferred revenue from a government agency.
Research and Development
Except for payments made in advance of services, the Company expenses its research and development costs as incurred. For payments made in advance, the Company recognizes research and development expense as the services are rendered. Research and development costs primarily consist of salaries and related expenses for personnel and resources and the costs of clinical trials. Other research and development expenses include preclinical analytical testing, outside services, providers, materials and consulting fees.
Income Taxes
Income taxes are accounted for using the liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and its respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which those temporary differences are expected to be recovered or settled.
The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when necessary to reduce net deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable for the period and the change during the period in deferred tax assets and liabilities.
F-28
Table of Contents
The Company accounts for uncertain tax positions pursuant to ASC Topic 740 (previously included in FASB Interpretation No. 48, Accounting for Uncertainty in Income Taxes–an Interpretation of FASB Statement No. 109). Financial statement recognition of a tax position taken or expected to be taken in a tax return is determined based on a more-likely-than-not threshold of that position being sustained. If the tax position meets this threshold, the benefit to be recognized is measured as the tax benefit having the highest likelihood of being realized upon ultimate settlement with the taxing authority. The Company recognizes interest accrued related to unrecognized tax benefits and penalties in the provision for income taxes.
Comprehensive Income (Loss)
ASC Topic 220-10, Reporting Comprehensive Income, requires the presentation of the comprehensive income or loss and its components as part of the financial statements if comprehensive income (loss) differs from net income (loss). For the three- and nine-months ended September 30, 2013 and the year ended December 31, 2012, the Company’s net loss equals comprehensive loss.
Share-Based Compensation
Share-based payments are accounted for in accordance with the provisions of ASC Topic 718, Compensation—Stock Compensation. The fair value of share-based payments is estimated, on the date of grant, using the Black-Scholes-Merton option-pricing model (the Black-Scholes model). The resulting fair value is recognized ratably over the requisite service period, which is generally the vesting period of the option.
For all time vesting awards granted, expense is amortized using the straight-line attribution method. For awards that contain a performance condition, expense is amortized using the accelerated attribution method. As share-based compensation expense recognized in the statements of operations for the three- and nine-months ended September 30, 2013 and 2012 and the year ended December 31, 2012, is based on share-based awards ultimately expected to vest, it has been reduced for estimated forfeitures.
ASC Topic 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Pre-vesting forfeitures are based on the Company’s historical experience for the three- and nine-months ended September 30, 2013 and 2012 and the year ended December 31, 2012, and have not been material.
The Company utilizes the Black-Scholes model for estimating fair value of its stock options granted. Option valuation models, including the Black-Scholes model, require the input of subjective assumptions, and changes in the assumptions used can materially affect the grant date fair value of an award. These assumptions include the risk-free rate of interest, expected dividend yield, expected volatility and the expected life of the award.
Expected volatility rates are based on historical volatility of the common stock of comparable publicly traded entities and other factors due to the lack of historic information of the Company’s common stock. The expected life of stock-based options is the period of time for which the stock-based options are expected to be outstanding. Given the lack of historic exercise data, the expected life is determined using the “simplified method” which is defined as the midpoint between the vesting date and the end of the contractual term.
The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. The Company has not paid dividends to its stockholders since its inception and does not plan to pay cash dividends in the foreseeable future. Therefore, the Company has assumed an expected dividend rate of zero.
Given the absence of an active market for the Company’s common stock, the exercise price of the stock options on the date of grant was determined and approved by the board of directors using several factors, including progress and milestones achieved in the Company’s business development and performance, the price per share
F-29
Table of Contents
of its convertible preferred stock offerings and general industry and economic trends. In establishing the estimated fair value of the common stock, the Company considered the guidance set forth in American Institute of Certified Public Accountants Practice Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation.
Under ASC Topic 718, the cumulative amount of compensation cost recognized for instruments classified as equity that ordinarily would result in a future tax deduction under existing tax law shall be considered to be a deductible difference in applying ASC Topic 740, Income Taxes. The deductible temporary difference is based on the compensation cost recognized for financial reporting purposes; however, these provisions currently do not impact the Company, as all the deferred tax assets have a full valuation allowance.
Since the Company had net operating loss carryforwards as of September 30, 2013 and December 31, 2012, no excess tax benefits for the tax deductions related to share-based awards were recognized in the statements of operations.
Equity instruments issued to non-employees are accounted for under the provisions of ASC Topic 718 and ASC Topic 505-50, Equity/Equity-Based Payments to Non-Employees. Accordingly, the estimated fair value of the equity instrument is recorded on the earlier of the performance commitment date or the date the services required are completed and are marked to market during the service period.
Loss Per Share
Loss per share is calculated under the two-class method under which all earnings (distributed and undistributed) are allocated to each class of common stock and participating securities based on their respective rights to receive dividends.
Basic net loss per common share is determined by dividing the net loss allocable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted net loss per share is computed by dividing the net loss allocable to common stockholders by the weighted-average number of common stock equivalents outstanding for the period. The treasury stock method is used to determine the dilutive effect of the Company’s stock option grants.
The following common stock equivalents were excluded in the calculation of diluted loss per share because their effect would be anti-dilutive as applied to the loss from operations as of the three- and nine- months ended September 30, 2013 and 2012:
| Three-Months Ended September 30 |
Nine-Months Ended September 30 |
|||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| Stock options |
712.525 | 901,210 | 710,819 | 901,210 | ||||||||||||
Recently Issued Accounting Pronouncements
In April 2013, FASB issued Accounting Standards Update (ASU) 2013-02, Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income, which amended interim and annual reporting requirements about accumulated other comprehensive income (AOCI). In interim periods, companies are required to report information about reclassifications out of AOCI and changes in AOCI balances. The provision of ASU 2013-02 became effective for the first quarter of 2013. The adoption of ASU 2103-02 did not have a material effect on the Company’s consolidated results of operations, financial position or liquidity.
F-30
Table of Contents
3. Property and Equipment
Property and equipment consist of the following:
| September 30, 2013 |
December 31, 2012 |
|||||||
| Computer equipment |
$ | 93,915 | $ | 92,318 | ||||
| Furniture and fixtures |
46,523 | 42,736 | ||||||
| Scientific equipment |
2,851,947 | 2,824,076 | ||||||
| Leasehold improvements |
319,553 | 319,553 | ||||||
|
|
|
|
|
|||||
| 3,311,938 | 3,278,683 | |||||||
| Less accumulated depreciation |
(3,236,238 | ) | (3,220,417 | ) | ||||
|
|
|
|
|
|||||
| $ | 75,700 | $ | 58,266 | |||||
|
|
|
|
|
|||||
Depreciation expense for the three- and nine-months ended September 30, 2013 was $4,729, and $15,821 respectively.
4. Share-Based Compensation
At the Effective Time of the Merger, the Company assumed all stock options then outstanding under ITI’s 2003 Equity Incentive Plan (the 2003 Plan). The 2003 Plan provided for the granting of stock awards, such as stock options, restricted common stock and stock appreciation rights to employees, directors and other individuals as determined by the Board of Directors. The 2003 Plan expired by its terms in July 2013 and no new awards may be granted. As of September 30, 2013, the only outstanding awards under the 2003 Plan were options to purchase 1,462,380 shares of common stock.
Stock options granted under the 2003 Plan may be either incentive stock options (ISOs) as defined by the Internal Revenue Code of 1986, as amended (the Code), or non-qualified stock options. The Board of Directors determined who received options as well as the vesting periods (which are generally two to three years) and exercise prices of options. Options have a maximum term of ten years. The exercise price of ISOs granted under the 2003 Plan must be at least equal to the fair market value of the common stock on the date of grant.
In addition, in August 2013, the Board of Directors approved the 2013 Equity Incentive Plan (the 2013 Plan). The Company expects the 2013 Plan will be effective on November 7, 2013 upon the effectiveness of stockholder approval of the plan by the Company’s sole stockholder prior to the Merger. The maximum number of shares of common stock that may be delivered in satisfaction of awards under the 2013 Plan is 799,934 shares, plus up to an additional maximum of 1,462,380 shares which may be issued solely after the cancellation or expiration of any unexercised stock options under the 2003 Plan that the Company assumed in the Merger. In addition, the 2013 Plan contains an “evergreen” provision, which allows for an annual increase in the number of shares of common stock available for issuance under the 2013 Plan on January 1 of each year commencing on January 1, 2014 and ending upon expiration of the 2013 Plan. The annual increase in the number of shares shall be equal to the lesser of: 800,000 shares of common stock; 4% of the number of shares of common stock outstanding as of such date; and such lesser number of shares as determined by the Board of Directors prior to the applicable January 1st date.
These numbers are subject to adjustment in the event of a stock split, stock dividend or other change in the Company’s capitalization. Unless sooner terminated by the Board of Directors or stockholders, the 2013 Plan will expire 10 years from its date of effectiveness. Under the 2013 Plan, the Company may grant ISOs, nonstatutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights and other stock awards to its employees, directors and consultants. As of September 30, 2013, no awards have been made under the 2013 Plan.
F-31
Table of Contents
Total stock-based compensation expense, related to all of the Company’s share-based awards to employees, directors and consultants recognized during three- and nine-months ended September 30, 2013 and 2012, was comprised of the following:
| Three-Months Ended September 30 |
Nine-Months Ended September 30 |
|||||||||||||||
| 2013 | 2012 | 2013 | 2012 | |||||||||||||
| Research and development |
$ | 38,856 | $ | 32,200 | $ | 96,943 | $ | 86,845 | ||||||||
| General and administrative |
80,361 | 65,261 | 185,507 | 195,784 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total share-based compensation expense |
$ | 119,217 | $ | 97,461 | $ | 282,450 | $ | 282,629 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The following table describes the weighted-average assumptions used for calculating the value of options granted during the nine-months ended September 30, 2013:
| 2013 | ||
| Dividend yield |
0% | |
| Expected volatility |
80% | |
| Weighted-average risk-free interest rate |
1.40% - 1.80% | |
| Expected term |
6.2 years | |
Information regarding the stock options activity including employees, directors and consultants as of September 30, 2013, and changes during the period then ended, are summarized as follows:
| Number of Shares |
Weighted- Average Exercise Price |
Weighted- Average Contractual Life |
||||||||||
| Outstanding at December 31, 2012 (audited) |
1,707,114 | $ | 1.3802 | 4.4 years | ||||||||
| Options granted (unaudited) |
247,600 | $ | 3.2600 | 6.2 years | ||||||||
| Options exercised (unaudited) |
(489,667 | ) | $ | 0.6470 | 1.3 years | |||||||
| Options canceled or expired (unaudited) |
(2,667 | ) | $ | 2.9725 | 8.7 years | |||||||
|
|
|
|
|
|||||||||
| Outstanding at September 30, 2013 (unaudited) |
1,462,380 | $ | 1.9410 | 5.5 years | ||||||||
|
|
|
|
|
|||||||||
| Vested or expected to vest at September 30, 2013 (unaudited) |
1,462,380 | $ | 1.9410 | 5.5 years | ||||||||
|
|
|
|
|
|||||||||
| Exercisable at September 30, 2013 (unaudited) |
1,118,156 | $ | 1.5801 | 6.9 years | ||||||||
|
|
|
|
|
|||||||||
5. Collaborations and License Agreements
The Bristol-Myers Squibb License Agreement
On May 31, 2005 the Company (through its wholly owned operating subsidiary, ITI) entered into a world-wide, exclusive License Agreement with Bristol-Myers Squibb Company (BMS), pursuant to which the Company holds a license to certain patents and know-how of BMS relating to ITI-007 and other specified compounds. The agreement was amended on November 3, 2010. The licensed rights are exclusive, except BMS retains rights in specified compounds in the fields of obesity, diabetes, metabolic syndrome and cardiovascular disease. However, BMS has no right to use, develop or commercialize ITI-007 and other specified compounds in any field of use. The Company has the right to grant sublicenses of the rights conveyed by BMS. The Company is obliged under the license to use commercially reasonable efforts to develop and commercialize the licensed technology. The Company is also prohibited from engaging in the clinical development or commercialization of specified competitive compounds.
F-32
Table of Contents
Under the agreement, the Company made an upfront payment of $1.0 million to BMS, and may be obliged to make milestone payments for each licensed product of up to an aggregate of approximately $14.8 million. The Company is also obliged to make tiered single digit percentage royalty payments on sales of licensed products. The Company is obliged to pay to BMS a percentage of non-royalty payments made in consideration of any sublicense.
The agreement extends, and royalties are payable, on a country-by-country and product-by-product basis, through the later of ten years after first commercial sale of a licensed product in such country, expiration of the last licensed patent covering a licensed product, its method of manufacture or use, or the expiration of other government grants providing market exclusivity, subject to certain rights of the parties to terminate the agreement on the occurrence of certain events. On termination of the agreement, the Company may be obliged to convey to BMS rights in developments relating to a licensed compound or licensed product, including regulatory filings, research results and other intellectual property rights.
The Takeda License and Collaboration Agreement
On February 25, 2011, the Company (through its wholly owned operating subsidiary, ITI) entered into a license and collaboration agreement with Takeda Pharmaceutical Company Limited (Takeda) under which the Company agreed to collaborate to research, develop and commercialize its proprietary compound ITI-214 and other selected compounds that selectively inhibit PDE1 for use in the prevention and treatment of human diseases. As part of the agreement, the Company assigned to Takeda certain patents owned by the Company that claim ITI-214 and granted Takeda an exclusive license to develop and commercialize compounds identified in the conduct of the research program that satisfy specified criteria. However, the Company has retained rights to all compounds that do not meet the specified criteria and the Company continues to develop PDE1 inhibitors outside the scope of the agreement.
Under the terms of the agreement, the Company is conducting a research program with an initial term of three years to identify and characterize compounds that meet certain specified criteria sufficient for further development by Takeda. The Company is responsible for the Company’s expenses incurred in the conduct of certain research activities specified in the research plan. Takeda has agreed to reimburse the Company for expenses the Company incurs in conducting additional research activities.
Takeda is obliged to use commercially reasonable efforts to develop and commercialize licensed compounds at its expense, and has agreed to reimburse the Company for the costs and expenses of development activities the Company may perform. The Company has formed a joint steering committee with Takeda to coordinate and oversee activities on which the Company and Takeda collaborate under the agreement. The Company has the option to co-promote any licensed product in the United States by assuming responsibility for a certain percentage of the detailing activity with respect to that product.
The Company is responsible for supplying Takeda with ITI-214 for nonclinical activities and Phase 1 clinical trials at the Company’s expense. Takeda is responsible, at its expense, for the manufacture and supply of compounds that it develops and commercializes under the agreement for all other activities.
Upon execution of the agreement, Takeda made a nonrefundable payment to the Company. The Company is eligible to receive payments of approximately $500 million in the aggregate upon achievement of certain development milestones and up to an additional $250 million in the aggregate upon achievement of certain sales-based milestones, along with tiered royalty payments ranging from the high single digits to the low teens in percent based on net sales by Takeda.
The agreement extends, on a country-by-country and product-by-product basis, through the later of expiration of the last licensed patent covering a licensed product, its method of manufacture or use, the expiration of other government grants providing market exclusivity or ten years after first commercial sale of a licensed product in such country, subject to rights of the parties to sooner terminate the agreement on certain events and the right of
F-33
Table of Contents
Takeda to unilaterally terminate the agreement upon a specified number of days’ prior notice. Upon the termination of the agreement, Takeda is obliged to assign to the Company the patents covering ITI-214 assigned to Takeda upon the execution of the agreement, to grant the Company a license to develop and commercialize licensed compounds developed by Takeda and to transfer to the Company certain materials, information and regulatory materials reasonably necessary for us to continue the development and commercialization of those compounds.
The Company evaluates all deliverables within an arrangement to determine whether or not they provide value on a stand-alone basis. Based on this evaluation, the deliverables were separated into units of accounting. The arrangement consideration that is fixed or determinable at the inception of the arrangement was allocated to the separate units of accounting based on their relative selling prices. The Company may exercise significant judgment in determining whether a deliverable is a separate unit of accounting, as well as in estimating the selling prices of such unit of accounting.
To determine the selling price of a separate deliverable, the Company uses the hierarchy as prescribed in ASC Topic 605-25 Revenue Recognition based on vendor-specific objective evidence (VSOE), third-party evidence (TPE) or best estimate of selling price (BESP). VSOE is based on the price charged when the element is sold separately and is the price actually charged for that deliverable. TPE is determined based on third-party evidence for a similar deliverable when sold separately and BESP is the price at which the Company would transact a sale if the elements of collaboration and license arrangements were sold on a stand-alone basis. The Company was not able to establish VSOE or TPE for the deliverables within collaboration and license arrangements, as the Company does not have a history of entering into such arrangements or selling the individual deliverables within such arrangements separately. In addition, there may be significant differentiation in these arrangements, which indicates that comparable third-party pricing may not be available. The Company determined that the selling price for the deliverables within collaboration and license arrangements should be determined using BESP. The process for determining BESP involved significant judgment on our part and included consideration of multiple factors such as estimated direct expenses and other costs, and available data.
During the three- and nine-months ended September 30, 2013, the Company recognized revenue of $0.7 million, and $1.9 million under this agreement, respectively. At September 30, 2013 and December 31, 2012, $0.4 million and $1.7 million of revenue, respectively, was deferred under this agreement.
Other License Agreement
In May 2002, the Company entered into a license agreement (the License) and research agreement with a university. Under the provisions of the License, the Company is entitled to use this organization’s patented technology and other intellectual property relating to diagnosis and treatment of central nervous system disorders.
The License expires upon expiration of the patent rights or 15 years subsequent to the first sale of products developed through this License. The Company is required to make future milestone payments for initiation of clinical trials and approval of a New Drug Application (NDA). Should the Company commercialize the technology related to this License, the Company would be required to make royalty payments, and would also be required to pay fees under any sublicense agreements with third parties.
In connection with the License, the Company issued 400,000 shares of common stock to the organization. Upon issuance of the shares, the Company recorded the estimated fair value of the shares issued, approximately $120,000, as research and development expense.
In addition, the Company is required to use at least $1.0 million annually of its resources for the development and commercialization of the technology until the Company submits an NDA. The Company met its spending requirements in 2012. There were no other payments made or required for the three- and nine-months ended September 30, 2013 and 2012 and the year ended December 31, 2012.
F-34
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholder of
Oneida Resources Corp.
Great Neck, NY
We have audited the accompanying balance sheets of Oneida Resources Corp. (a development stage company) (the “Company”) as of March 31, 2013 and the related statements of operations, stockholder’s deficiency, and cash flows for the period from August 29, 2012 (inception) through March 31, 2013. The Company’s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Oneida Resources Corp. as of March 31, 2013, and the results of its operations and its cash flows for the period from August 29, 2012 (inception) through March 31, 2013 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 3 to the financial statements, the Company is in the development stage and has incurred net losses since inception. This raises substantial doubt about its ability to continue as a going concern. Management’s plans in regards to these matters are also described in Note 3. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Raich Ende Malter & Co. LLP
Raich Ende Malter & Co. LLP
New York, New York
July 16, 2013
F-35
Table of Contents
(A Development Stage Company)
BALANCE SHEET
March 31, 2013
| ASSETS | ||||
| CURRENT ASSETS: |
||||
| Cash |
$ | 4,070 | ||
|
|
|
|||
| Total Current Assets |
4,070 | |||
|
|
|
|||
| TOTAL ASSETS |
$ | 4,070 | ||
|
|
|
|||
| LIABILITIES AND STOCKHOLDER’S DEFICIENCY | ||||
| CURRENT LIABILITIES: |
||||
| Accounts payable and accrued expenses |
$ | 26,170 | ||
|
|
|
|||
| Total Current Liabilities |
$ | 26,170 | ||
|
|
|
|||
| COMMITMENTS AND CONTINGENCIES |
— | |||
|
|
|
|||
| STOCKHOLDER’S DEFICIENCY: |
||||
| Preferred stock, $.0001 par value; 10,000,000 shares authorized; none issued and outstanding |
— | |||
| Common stock, $.0001 par value; 100,000,000 shares authorized; 5,000,000 shares issued and outstanding |
500 | |||
| Additional paid-in capital |
9,500 | |||
| Accumulated deficit during the development stage |
(32,100 | ) | ||
|
|
|
|||
| Total Stockholder’s Deficiency |
(22,100 | ) | ||
|
|
|
|||
| TOTAL LIABILITIES AND STOCKHOLDER’S DEFICIENCY |
$ | 4,070 | ||
|
|
|
|||
See accompanying notes to the financial statements.
F-36
Table of Contents
(A Development Stage Company)
STATEMENT OF OPERATIONS
FOR THE PERIOD AUGUST 29, 2012 (INCEPTION) TO MARCH 31, 2013
| REVENUES |
$ | — | ||
| GENERAL AND ADMINISTRATIVE EXPENSES |
32,100 | |||
|
|
|
|||
| (LOSS) BEFORE BENEFIT FROM INCOME TAXES |
(32,100 | ) | ||
| BENEFIT FROM INCOME TAXES |
— | |||
|
|
|
|||
| NET (LOSS) |
$ | (32,100 | ) | |
|
|
|
|||
| BASIC AND DILUTED LOSS PER SHARE |
— | |||
|
|
|
|||
| WEIGHTED AVERAGE NUMBER OF COMMON SHARES OUTSTANDING - BASIC AND DILUTED |
3,720,930 | |||
|
|
|
See accompanying notes to the financial statements.
F-37
Table of Contents
(A Development Stage Company)
STATEMENT OF STOCKHOLDER’S DEFICIENCY
FOR THE PERIOD FROM AUGUST 29, 2012 (INCEPTION) TO MARCH 31, 2013
| Preferred Stock | Common Stock | Additional Paid-in Capital |
Accumulated Deficit During the Development Stage |
Total Stockholder’s Deficiency |
||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
| Balance at August 29, 2012 (Inception) |
— | $ | — | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||||||
| Common stock issuance |
— | — | 5,000,000 | 500 | 9,500 | — | 10,000 | |||||||||||||||||||||
| Net (loss) |
— | — | — | — | — | (32,100 | ) | (32,100 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance at March 31, 2013 |
— | $ | — | 5,000,000 | $ | 500 | $ | 9,500 | $ | (32,100 | ) | $ | (22,100 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
See accompanying notes to the financial statements.
F-38
Table of Contents
(A Development Stage Company)
STATEMENT OF CASH FLOWS
FOR THE PERIOD AUGUST 29, 2012 (INCEPTION) TO MARCH 31, 2013
| CASH FLOWS FROM OPERATING ACTIVITIES: |
||||
| NET (LOSS) |
$ | (32,100 | ) | |
| ADJUSTMENT TO RECONCILE NET LOSS TO NET CASH USED IN OPERATING ACTIVITIES: |
||||
| Increase in accounts payable |
26,170 | |||
|
|
|
|||
| NET CASH USED IN OPERATING ACTIVITIES |
(5,930 | ) | ||
|
|
|
|||
| CASH FLOWS FROM FINANCING ACTIVITIES: |
||||
| Proceeds from issuance of common stock |
10,000 | |||
|
|
|
|||
| NET CASH PROVIDED BY FINANCING ACTIVITIES |
10,000 | |||
|
|
|
|||
| NET INCREASE IN CASH |
4,070 | |||
| CASH, BEGINNING OF PERIOD |
— | |||
|
|
|
|||
| CASH, END OF PERIOD |
$ | 4,070 | ||
|
|
|
See accompanying notes to the financial statements.
F-39
Table of Contents
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Note 1 - Organization and Business
Business Activity
Oneida Resources Corp., a Development Stage Company, (“the Company”) was incorporated in the state of Delaware on August 29, 2012 with the objective to acquire, or merge with, an operating business.
The Company was organized as a vehicle to investigate and, if such investigation warrants, acquire a target company or business seeking the perceived advantages of being a publicly traded corporation. The Company’s principal business objective over the next twelve months and beyond will be to achieve long-term growth potential through a combination with a business rather than immediate short-term earnings. The Company will not restrict its potential target companies to any specific business, industry or geographical location. The analysis of business opportunities will be undertaken by, or under the supervision of, the officers and directors of the Company.
Note 2 - Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the balance sheet and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash Equivalents
The Company considers highly liquid financial instruments purchased with a maturity of three months or less to be cash equivalents. There are no cash equivalents at the balance sheet date.
Income Taxes
The Company utilizes the accrual method of accounting for income taxes. Under the accrual method, deferred tax assets and liabilities are determined based on the differences between the financial reporting basis and the tax basis of the assets and liabilities and are measured using enacted tax rates and laws that will be in effect, when the differences are expected to reverse. An allowance against deferred tax assets is recognized, when it is more likely than not, that such tax benefits will not be realized.
The Company recognizes the financial statement benefit of an uncertain tax position only after considering the probability that a tax authority would sustain the position in an examination.
For tax positions meeting a “more-likely than-not” threshold, the amount recognized in the financial statements is the benefit expected to be realized upon settlement with the tax authority. For tax positions not meeting the threshold, no financial statement benefit is recognized. The Company recognizes interest and penalties, if any, related to uncertain tax positions in income tax expense. As of March 31, 2013, the Company has no accrued interest or penalties related to uncertain tax positions.
Loss Per Common Share
Basic loss per share is calculated using the weighted-average number of common shares outstanding during each reporting period. Diluted loss per share includes potentially dilutive securities such as
F-40
Table of Contents
ONEIDA RESOURCES CORP.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Note 2 - Summary of Significant Accounting Policies (cont’d.)
Loss Per Common Share (cont’d.)
outstanding options and warrants, using various methods such as the treasury stock or modified treasury stock method in the determination of dilutive shares outstanding during each reporting period. The Company does not have any potentially dilutive instruments for the period presented.
Emerging Growth Company
The Company is an “emerging growth company” and has elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies.
Recent Accounting Pronouncements
Management does not believe that any recently issued, but not yet effective accounting pronouncements, if adopted, would have a material effect on the accompanying financial statements.
Note 3 - Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern, which contemplates the recoverability of assets and the satisfaction of liabilities in the normal course of business.
The Company has incurred losses from inception of approximately $32,000, and has negative working capital of approximately $22,000 at March 31, 2013, which among other factors, raises substantial doubt about the Company’s ability to continue as a going concern. The ability of the Company to continue as a going concern is dependent upon management’s plan to find a suitable acquisition or merger candidate, raise additional capital from the sales of stock, and receive loans from related parties. The accompanying financial statements do not include any adjustments that might be required should the Company be unable to continue as a going concern.
Note 4 - Income Taxes
As of March 31, 2013, the Company has net operating loss carryforwards of approximately $32,000 to reduce future federal and state taxable income through 2033.
The Company currently has no federal or state tax examinations in progress nor has it had any federal or state examinations since its inception. All of the Company’s tax years are subject to federal and state tax examination.
F-41
Table of Contents
ONEIDA RESOURCES CORP.
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Note 4 - Income Taxes (cont’d.)
The benefit from income taxes consists of the following:
| For the Period August 29, 2012 (Inception) to March 31, 2013 |
||||
| Current Expense: |
||||
| Federal and State |
$ | — | ||
| Deferred tax benefit: |
||||
| Federal and State |
11,000 | |||
| Valuation allowance |
(11,000 | ) | ||
|
|
|
|||
| Total |
$ | — | ||
|
|
|
|||
The income tax benefit differs from the amount computed by applying the federal statutory income tax rate to the loss before income taxes due to the following:
| For the Period August 29, 2012 (Inception) to March 31, 2013 |
||||
| Statutory federal income tax rate |
(34)% | |||
| Valuation allowance |
34% | |||
|
|
|
|||
| Effective income tax rate |
0% | |||
|
|
|
|||
Note 5 - Common Stock
On August 29, 2012, the Company authorized one hundred million (100,000,000) shares of common stock. On October 15, 2012, the Company received a subscription for five million (5,000,000) shares of common stock. On October 23, 2012, the Company received payment of $10,000 for the subscription.
Note 6 - Preferred Stock
The Company is authorized to issue (10,000,000) shares of $.0001 par value preferred stock with designations, voting and other rights and preferences as may be determined from time to time by the Board of Directors of the Company.
Note 7 - Related Party Transactions
The Company utilizes the office space and equipment of its management at no cost.
On October 15, 2012, the Company issued a Promissory Note payable (the “Note”) to NLBDIT 2010 Enterprises, LLC. The Note bears interest at 6% and is payable upon completion of a business combination with a private company in a reverse merger or other transaction after which the Company would cease to be a shell company. At March 31, 2013, there is no outstanding balance.
Note 8 - Subsequent Events
Subsequent to March 31, 2013, professional fees of $3,400 were paid on behalf of the Company by Sunrise Financial Group Inc. (“SFG”).
Subsequent to March 31, 2013, the Company received approximately $6,000 relating to the promissory note with NLBDIT 2010 Enterprises, LLC for professional fees.
F-42
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution |
The following table sets forth the fees and expenses to be incurred in connection with the registration of the securities being registered hereby, all of which will be borne by us. Except for the SEC registration fee, all amounts are estimates.
| Description |
Amount | |||
| SEC registration fee |
$ | 19,031 | ||
| Printing expense |
7,823 | |||
| Accounting fees and expenses |
86,250 | |||
| Legal fees and expenses |
150,000 | |||
|
|
|
|||
| Total expenses |
$ | 263,104 | ||
|
|
|
|||
| Item 14. | Indemnification of Directors and Officers |
Delaware Law
Section 102 of the General Corporation Law of the State of Delaware permits a corporation to eliminate the personal liability of directors of a corporation to the corporation or its stockholders for monetary damages for a breach of fiduciary duty as a director, except where the director breached his duty of loyalty, failed to act in good faith, engaged in intentional misconduct or knowingly violated a law, authorized the payment of a dividend or approved a stock repurchase in violation of Delaware corporate law or obtained an improper personal benefit.
Section 145 of the General Corporation Law of the State of Delaware provides that a corporation has the power to indemnify a director, officer, employee, or agent of the corporation, or a person serving at the request of the corporation for another corporation, partnership, joint venture, trust or other enterprise in related capacities against expenses, including attorneys’ fees, judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with an action, suit or proceeding to which he was or is a party or is threatened to be made a party to any threatened, ending or completed action, suit or proceeding by reason of such position, if such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, in any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful, except that, in the case of actions brought by or in the right of the corporation, no indemnification shall be made with respect to any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or other adjudicating court determines that, despite the adjudication of liability but in view of all of the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
Amended and Restated Certificate of Incorporation and Restated Bylaws
Our restated certificate of incorporation and restated bylaws provide that we shall indemnify, to the fullest extent authorized by the Delaware General Corporation Law, each person who is involved in any litigation or other proceeding because such person is or was a director or officer of Intra-Cellular Therapies, Inc. or is or was serving as an officer or director of another entity at our request, against all expense, loss or liability reasonably incurred or suffered in connection therewith. Our restated certificate of incorporation and restated bylaws also provide that the right to indemnification includes the right to be paid expenses incurred in defending any proceeding in advance of its final disposition, provided, however, that such advance payment will only be made
II-1
Table of Contents
upon delivery to us of an undertaking, by or on behalf of the director or officer, to repay all amounts so advanced if it is ultimately determined that such director is not entitled to indemnification. If we do not pay a proper claim for indemnification in full within 60 days after we receive a written claim for such indemnification, except in the case of a claim for an advancement of expenses, in which case such period is 20 days, our restated certificate of incorporation and our restated bylaws authorize the claimant to bring an action against us and prescribe what constitutes a defense to such action.
Our restated certification of incorporation eliminates the liability of a director to us or our stockholders for monetary damages for such a breach of fiduciary duty as a director, except for liabilities arising:
| • | from any breach of the director’s duty of loyalty to us or our stockholders; |
| • | from acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; |
| • | under Section 174 of the Delaware General Corporation Law; and |
| • | from any transaction from which the director derived an improper personal benefit. |
Indemnification Agreements
We have entered into indemnification agreements with our directors and certain officers, in addition to the indemnification provided in our restated certificate of incorporation and our restated bylaws, and intend to enter into indemnification agreements with any new directors and executive officers in the future. These indemnification agreements may require us, among other things, to indemnify our directors and officers for some expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or officer in any action or proceeding arising out of his or her service as one of our directors or officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request.
In addition, we entered into an indemnity agreement with our former officer and director of the Company pursuant to which we agreed to indemnify such former officer and director for actions taken by him in his official capacity relating to the consideration, approval and consummation of the Merger and certain related transactions.
We have purchased and intend to maintain insurance on behalf of any person who is or was a director or officer against any loss arising from any claim asserted against him or her and incurred by him or her in any such capacity, subject to certain exclusions.
The foregoing discussion of our restated certificate of incorporation, restated bylaws, indemnification agreements, indemnity agreement, and Delaware law is not intended to be exhaustive and is qualified in its entirety by such restated certificate of incorporation, restated bylaws, indemnification agreements, indemnity agreement, or law.
| Item 15. | Recent Sales of Unregistered Securities |
Set forth below is information regarding shares of common stock, convertible preferred stock, convertible notes and warrants issued, and options granted, by us and by ITI within the past three years that were not registered under the Securities Act. Also included is the consideration, if any, received by us or ITI for such shares, notes, warrants and options and information relating to the section of the Securities Act, or rule of the Securities and Exchange Commission, under which exemption from registration was claimed. The number of shares and the warrant issued prior to the Merger described below in paragraphs C and D below do not reflect the exchange of shares in the Merger, which is further described in paragraph E below.
II-2
Table of Contents
Original Issuances of Stock, Convertible Notes and Warrants
A. On October 15, 2012, the Company issued an aggregate of 5,000,000 shares of its common stock to its then sole stockholder in exchange for $10,000, which represented all of the Company’s outstanding common stock immediately prior to the Merger.
B. On October 25, 2012, ITI issued convertible promissory notes having an aggregate principal amount of $14,343,795 to seven accredited investors. On November 14, 2012, ITI issued additional convertible promissory notes having an aggregate principal amount of $846,098 to two additional accredited investors. On March 20, 2013, ITI issued additional convertible promissory notes having an aggregate principal amount of $100,000 to two additional accredited investors. In connection with the private placement described in paragraph D below, the principal amount of all outstanding convertible promissory notes, plus accrued interest, converted into ITI common stock on August 29, 2013.
C. On April 19, 2013, ITI issued 36,450 shares of ITI common stock and a warrant to purchase 3,645 shares of ITI common stock to one accredited investor in exchange for biotechnology grant funding received from the investor.
D. On August 29, 2013, immediately prior to the Effective Time, ITI issued 18,889,307 shares of its common stock at a price of $3.1764 per share, or an aggregate purchase price of approximately $60.0 million, which included approximately $15.3 million in principal and $0.8 million in accrued interest from the conversion of ITI’s then outstanding convertible promissory notes (such that ITI received approximately $40.0 million in net proceeds, after expenses), to 206 accredited investors in a private placement. As part of the private placement, all outstanding convertible promissory notes, plus accrued interest thereon, described in paragraph B above, were converted into shares of ITI common stock at a price of $3.1764 per share. Leerink Swann LLC acted as sole lead placement agent and National Securities Corporation and Livingston Securities LLC acted as co-agents for purposes of the sale of ITI common stock in the private placement. Entities and individuals affiliated with Leerink Swann LLC purchased an aggregate of 346,302 shares of ITI common stock in the private placement on the same terms as the other investors.
E. On August 29, 2013, at the Effective Time, each share of ITI common stock and preferred stock that was issued and outstanding immediately prior to the Effective Time was automatically exchanged for 0.5 shares of our common stock. We issued an aggregate of 22,134,647 shares of our common stock upon such exchange of the outstanding shares of ITI common stock and preferred stock to ITI’s stockholders immediately prior to the Effective Time, which included no more than 35 non-accredited investors. In addition, at the Effective Time, we assumed the ITI 2003 Equity Incentive Plan, and all options to purchase ITI common stock then outstanding under the 2003 Equity Incentive Plan, and such options became exercisable for an aggregate of 1,462,380 shares of our common stock, subject to the vesting and other terms of such options. At the Effective Time, we also assumed the outstanding warrant to purchase ITI common stock described in paragraph C above, and such warrant became exercisable for 1,822 shares of our common stock.
F. From August 1, 2010 through the Effective Time, which occurred on August 29, 2013, ITI issued an aggregate of 519,936 shares of its common stock upon the exercise of stock options issued under the 2003 Equity Incentive Plan.
Stock Option Grants
From August 1, 2010 through the consummation of the Merger on August 29, 2013, ITI granted stock options under the 2003 Equity Incentive Plan to purchase an aggregate of 579,300 shares of common stock, net of forfeitures, at a weighted-average exercise price of $2.99 per share, to certain of its employees, consultants and directors.
II-3
Table of Contents
Securities Act Exemptions
We deemed the offers, sales and issuances of the securities described above under “—Original Issuances of Stock, Convertible Notes and Warrants” to be exempt from registration under the Securities Act in reliance on Section 4(a)(2) of the Securities Act, including Regulation D and Rule 506(b) promulgated thereunder, relative to transactions by an issuer not involving a public offering.
We deemed the grants of stock options described above under “—Stock Option Grants” to be exempt from registration under the Securities Act in reliance on Rule 701 of the Securities Act as offers and sales of securities under compensatory benefit plans and contracts relating to compensation in compliance with Rule 701. Each of the recipients of securities in any transaction exempt from registration either received or had adequate access, through employment, business or other relationships, to information about us.
All certificates representing the securities issued in the transactions described under “Recent Sales of Unregistered Securities” included appropriate legends setting forth that the securities had not been offered or sold pursuant to a registration statement and describing the applicable restrictions on transfer of the securities. There were no underwriters employed in connection with any of the transactions set forth in “Recent Sales of Unregistered Securities.”
| Item 16. | Exhibits and Financial Statement Schedules |
(a) See the Exhibit Index on the page immediately preceding the exhibits for a list of exhibits filed as part of this registration statement on Form S-1, which Exhibit Index is incorporated herein by reference.
(b) Financial Statement Schedules
Financial Statement Schedules are omitted because the information is included in our financial statements or notes to those financial statements.
| Item 17. | Undertakings |
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this Registration Statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the Registration Statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the Registration Statement. Notwithstanding the foregoing, any increase or any decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low end or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective Registration Statement;
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the Registration Statement or any material change to such information in the Registration Statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new Registration Statement relating to the securities to be offered therein, and the offering of such securities at that time shall be deemed to be an initial bona fide offering thereof.
II-4
Table of Contents
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which shall remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser: each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness; provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
II-5
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act, the registrant has duly caused this Amendment No. 2 to this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of New York, State of New York, on December 9, 2013.
| INTRA-CELLULAR THERAPIES, INC. | ||
| By: |
/s/ Sharon Mates, Ph.D. | |
| Sharon Mates, Ph.D. | ||
| Chairman, President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act, this Registration Statement has been signed below by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Sharon Mates, Ph.D. Sharon Mates, Ph.D. |
Chairman, President and Chief Executive Officer (principal executive officer) | December 9, 2013 | ||
| /s/ Lawrence J. Hineline Lawrence J. Hineline |
Vice President of Finance, Chief Financial Officer and Secretary (principal financial officer and principal accounting officer) | December 9, 2013 | ||
| * |
Director |
December 9, 2013 | ||
| Christopher Alafi, Ph.D. | ||||
| * |
Director |
December 9, 2013 | ||
| Richard Lerner, M.D. | ||||
| * |
Director |
December 9, 2013 | ||
| Joel S. Marcus | ||||
| * |
Director |
December 9, 2013 | ||
| Sir Michael Rawlins, M.D., FRCP, FMedSci |
||||
| *By: | /s/ Sharon Mates, Ph.D. |
December 9, 2013 | ||||
| Sharon Mates, Ph.D. Attorney-in-fact |
||||||
II-6
Table of Contents
EXHIBIT INDEX
| Exhibit |
Exhibit Description |
Filed Herewith |
Incorporated by Reference herein from Form or Schedule |
Filing Date |
SEC File/ | |||||||
| 2.1 | Agreement and Plan of Merger, dated as of August 23, 2013, by and among the Registrant, ITI, Inc. and Intra-Cellular Therapies, Inc. | 8-K (Exhibit 2.1) |
8/29/2013 | 000-54896 | ||||||||
| 2.2 | Agreement and Plan of Merger, dated as of August 29, 2013, by and between the Registrant and Intra-Cellular Therapies, Inc., relating to the name change of the Registrant. | 8-K (Exhibit 2.2) |
9/5/2013 | 000-54896 | ||||||||
| 3.1 | Restated Certificate of Incorporation of the Registrant, as filed with the Secretary of State of the State of Delaware on November 7, 2013. | S-1/A (Exhibit 3.1) |
11/26/13 |
333-191238 | ||||||||
| 3.2 | Certificate of Merger relating to the Merger of ITI, Inc. with and into Intra-Cellular Therapies, Inc., filed with the Secretary of State of the State of Delaware on August 29, 2013. | 8-K (Exhibit 3.3) |
9/5/2013 | 000-54896 | ||||||||
| 3.3 | Certificate of Ownership and Merger relating to the Merger of Intra-Cellular Therapies, Inc. with and into the Registrant, filed with the Secretary of State of the State of Delaware on August 29, 2013, relating to the name change of the Registrant. | 8-K (Exhibit 3.4) |
9/5/2013 | 000-54896 | ||||||||
| 3.4 | Restated Bylaws of the Registrant. | 8-K (Exhibit 3.5) |
9/5/2013 | 000-54896 | ||||||||
| 4.1 | Form of common stock certificate. | 8-K (Exhibit 4.1) |
9/5/2013 | 000-54896 | ||||||||
| 4.2 | .1 | Warrant to Purchase Common Stock dated April 19, 2013 issued to Alzheimer Drug Discovery Foundation, Inc. | 8-K (Exhibit 4.2.1) |
9/5/2013 | 000-54896 | |||||||
| .2 | Amendment dated August 29, 2013 to Warrant to Purchase Common Stock dated April 19, 2013 issued to Alzheimer Drug Discovery Foundation, Inc. | 8-K (Exhibit 4.2.2) |
9/5/2013 | 000-54896 | ||||||||
II-7
Table of Contents
| Exhibit |
Exhibit Description |
Filed Herewith |
Incorporated by Reference herein from Form or Schedule |
Filing Date |
SEC File/ | |||||||
| 5.1 | Opinion of Mintz, Levin, Cohn, Glovsky and Popeo, P.C. | S-1 (Exhibit 5.1) |
9/18/13 | 333-191238 | ||||||||
| 10.1 | .1 | License Agreement dated as of May 31, 2005 by and between Bristol-Meyers Squibb Company and Intra-Cellular Therapies, Inc.** | 8-K/A (Exhibit 10.1.1) |
10/31/2013 | 000-54896 | |||||||
| .2 | Amendment No. 1 to License Agreement dated as of November 3, 2010 by and between Bristol-Meyers Squibb Company and Intra-Cellular Therapies, Inc. | 8-K (Exhibit 10.1.2) |
9/5/2013 |
000-54896 | ||||||||
| 10.2 | License and Collaboration Agreement dated as of February 25, 2011 by and between Takeda Pharmaceutical Company Limited and Intra-Cellular Therapies, Inc.** | 8-K/A (Exhibit 10.2) |
10/31/2013 |
000-54896 | ||||||||
| 10.3 | Employment Agreement effective as of February 26, 2008 by and between Sharon Mates, Ph.D. and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.3) |
9/5/2013 | 000-54896 | ||||||||
| 10.4 | Employment Agreement effective as of February 26, 2008 by and between Lawrence J. Hineline and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.4) |
9/5/2013 | 000-54896 | ||||||||
| 10.5 | Employment Agreement effective as of February 26, 2008 by and between Allen Fienberg, Ph.D. and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.5) |
9/5/2013 | 000-54896 | ||||||||
| 10.6 | Employment Agreement effective as of February 26, 2008 by and between Lawrence Wennogle, Ph.D. and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.6) |
9/5/2013 | 000-54896 | ||||||||
| 10.7 | Offer Letter dated February 2, 2007 by Intra-Cellular Therapies, Inc. to Kimberly Vanover.* | 8-K (Exhibit 10.7) |
9/5/2013 | 000-54896 | ||||||||
| 10.8 | Employee Proprietary Information, Inventions, and Non-Competition Agreement effective as of September 1, 2003 by and between Sharon Mates, Ph.D. and Intra-Cellular Therapies, Inc.* |
8-K (Exhibit 10.8) |
9/5/2013 | 000-54896 | ||||||||
| 10.9 | Employee Proprietary Information, Inventions, and Non-Competition Agreement effective as of December 1, 2003 by and between Lawrence J. Hineline and Intra-Cellular Therapies, Inc.* |
8-K (Exhibit 10.9) |
9/5/2013 | 000-54896 | ||||||||
II-8
Table of Contents
| Exhibit |
Exhibit Description |
Filed Herewith |
Incorporated by Reference herein from Form or Schedule |
Filing Date |
SEC File/ | |||||||
| 10.10 | Employee Proprietary Information, Inventions, and Non-Competition Agreement effective as of June 3, 2002 by and between Allen Fienberg, Ph.D. and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.10) |
9/5/2013 | 000-54896 | ||||||||
| 10.11 | Employee Proprietary Information, Inventions, and Non-Competition Agreement effective as of January 1, 2003 by and between Lawrence Wennogle, Ph.D. and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.11) |
9/5/2013 | 000-54896 | ||||||||
| 10.12 | Employee Proprietary Information, Inventions, and Non-Competition Agreement effective as of March 5, 2007 by and between Kimberly E. Vanover, Ph.D. and Intra-Cellular Therapies, Inc.* | 8-K (Exhibit 10.12) |
9/5/2013 | 000-54896 | ||||||||
| 10.13 | Form of Indemnification Agreement by and between the Company and its directors and executive officers.* | 8-K (Exhibit 10.13) |
9/5/2013 | 000-54896 | ||||||||
| 10.14 | 2003 Equity Incentive Plan, as amended.* | 8-K (Exhibit 10.14) |
9/5/2013 | 000-54896 | ||||||||
| 10.15 | Form of Stock Option Agreement under the 2003 Equity Incentive Plan, as amended.* | 8-K (Exhibit 10.15) |
9/5/2013 | 000-54896 | ||||||||
| 10.16 | 2013 Equity Incentive Plan.* | 8-K (Exhibit 10.16) |
9/5/2013 | 000-54896 | ||||||||
| 10.17 | Redemption Agreement dated as of August 29, 2013 by and between the Registrant and NLBDIT 2010 Services, LLC. | 8-K (Exhibit 10.17) |
9/5/2013 | 000-54896 | ||||||||
| 10.18 | Indemnity Agreement dated as of August 29, 2013 by and among the Registrant, Intra-Cellular Therapies, Inc. and Samir N. Masri. | 8-K (Exhibit 10.18) |
9/5/2013 | 000-54896 | ||||||||
| 10.19 | Registration Rights Agreement dated as of August 29, 2013 by and among Intra-Cellular Therapies, Inc., the stockholders named therein and the Registrant. | 8-K (Exhibit 10.19) |
9/5/2013 | 000-54896 | ||||||||
| 16.1 | Letter from Raich Ende Malter & Co. LLP to the Securities and Exchange Commission, dated September 18, 2013. | S-1 (Exhibit 16.1) |
9/18/2013 | 333-191238 | ||||||||
II-9
Table of Contents
| Exhibit |
Exhibit Description |
Filed Herewith |
Incorporated by Reference herein from Form or Schedule |
Filing Date |
SEC File/ | |||||||
| 21.1 | Subsidiaries | S-1 (Exhibit 21.1) |
9/18/13 | 333-191238 | ||||||||
| 23.1 | Consent of Ernst & Young LLP | X | ||||||||||
| 23.2 | Consent of Raich Ende Malter & Co. LLP | X | ||||||||||
| 23.3 | Consent of Mintz, Levin, Cohn, Glovsky and Popeo, P.C. (included in Exhibit 5.1) | |||||||||||
| 24.1 | Power of Attorney (included on signature page to initial filing) | |||||||||||
| 101 | .INS | XBRL Instance Document.*** | S-1/A (Exhibit 101) |
11/26/13 | 333-191238 | |||||||
| .SCH | XBRL Taxonomy Extension Schema Document.*** | S-1/A (Exhibit 101) |
11/26/13 | 333-191238 | ||||||||
| .CAL | XBRL Taxonomy Extension Calculation Linkbase Document.*** | S-1/A (Exhibit 101) |
11/26/13 | 333-191238 | ||||||||
| .DEF | XBRL Taxonomy Extension Definition.*** | S-1/A (Exhibit 101) |
11/26/13 | 333-191238 | ||||||||
| .LAB | XBRL Taxonomy Extension Label Linkbase Document.*** | S-1/A (Exhibit 101) |
11/26/13 | 333-191238 | ||||||||
| .PRE | XBRL Taxonomy Presentation Linkbase Document*** | S-1/A (Exhibit 101) |
11/26/13 | 333-191238 | ||||||||
| * | Management contract or compensatory plan or arrangement. |
| ** | Confidential treatment has been granted for portions of this Exhibit. Redacted portions filed separately with the Securities and Exchange Commission. |
| *** | Submitted electronically herewith. In accordance with Rule 406T of Regulation S-T, the XBRL related information in Exhibit 101 of this Registration Statement is deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act, is deemed not filed for purposes of Section 18 of the Exchange Act, and otherwise is not subject to liability under these sections. |
II-10