Attached files
| file | filename |
|---|---|
| EX-2.2 - EX-2.2 - Ignyta, Inc. | d608873dex22.htm |
| EX-4.1 - EX-4.1 - Ignyta, Inc. | d608873dex41.htm |
| EX-2.1 - EX-2.1 - Ignyta, Inc. | d608873dex21.htm |
| EX-3.1 - EX-3.1 - Ignyta, Inc. | d608873dex31.htm |
| EX-10.1 - EX-10.1 - Ignyta, Inc. | d608873dex101.htm |
| EX-10.6 - EX-10.6 - Ignyta, Inc. | d608873dex106.htm |
| EX-10.4 - EX-10.4 - Ignyta, Inc. | d608873dex104.htm |
| EX-10.8 - EX-10.8 - Ignyta, Inc. | d608873dex108.htm |
| EX-99.2 - EX-99.2 - Ignyta, Inc. | d608873dex992.htm |
| EX-10.3 - EX-10.3 - Ignyta, Inc. | d608873dex103.htm |
| EX-10.5 - EX-10.5 - Ignyta, Inc. | d608873dex105.htm |
| EX-16.1 - EX-16.1 - Ignyta, Inc. | d608873dex161.htm |
| EX-21.1 - EX-21.1 - Ignyta, Inc. | d608873dex211.htm |
| EX-10.9 - EX-10.9 - Ignyta, Inc. | d608873dex109.htm |
| EX-10.7 - EX-10.7 - Ignyta, Inc. | d608873dex107.htm |
| EX-10.2 - EX-10.2 - Ignyta, Inc. | d608873dex102.htm |
| EX-99.1 - EX-99.1 - Ignyta, Inc. | d608873dex991.htm |
| EX-99.3 - EX-99.3 - Ignyta, Inc. | d608873dex993.htm |
| EX-10.10 - EX-10.10 - Ignyta, Inc. | d608873dex1010.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): October 31, 2013
IGNYTA, INC.
(Exact Name of Registrant as Specified in its Charter)
| Nevada | 333-183886 | 59-3564984 | ||
| (State of Incorporation) | (Commission File Number) |
(IRS Employer Identification No.) |
11095 Flintkote Avenue, Suite D
San Diego, California 92121
(Address of principal executive offices, including zip code)
Registrant’s telephone number, including area code: (858) 255-5959
Infinity Oil & Gas Company
750 Broadway
Woodmere, NY 11598
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Table of Contents
Ignyta, Inc., a Nevada corporation formerly known as Infinity Oil & Gas Company, is providing the disclosure contained in this Current Report on Form 8-K in connection with the closing of the Merger (as defined in Item 2.01 of this Current Report on Form 8-K) on October 31, 2013, under the following items of Form 8-K: Item 1.01, Item 2.01, Item 3.02, Item 3.03, Item 4.01, Item 5.01, Item 5.02, Item 5.03, Item 5.06, Item 5.07, and Item 9.01. A table of contents of this Current Report on Form 8-K is as follows:
As used in this Current Report on Form 8-K, unless the context indicates or otherwise requires, all references to “Ignyta” refer to Ignyta, Inc., a Nevada corporation formerly known as Infinity Oil & Gas Company; all references to “Ignyta Operating” refer to Ignyta Operating, Inc., a Delaware corporation formerly known as Ignyta, Inc. that became the wholly owned subsidiary of Ignyta following the completion of the Merger, as described in this report; all references to the “Combined Company” refer to Ignyta and its subsidiaries, including Ignyta Operating; and all references to “we,” “our” and “us” refer to the Combined Company from and after the closing of the Merger.
Ignyta and Ignyta Operating effected reverse stock splits of their capital stock, at the ratios of 100-to-one and three-to-one, respectively, shortly prior to and in connection with the transactions described in this report. Unless the context indicates or otherwise requires, all share numbers and share price data included in this Current Report on Form 8-K relating to the common stock of Ignyta and the capital stock of Ignyta Operating have been adjusted to give effect to those reverse stock splits.
We have registered trademarks for Ignyta®, Methylome®, and Trailblaze®, and pending trademark applications for Oncolome™ and Actagene™. All other trademarks, trade names and service marks included in this Current Report on Form 8-K are the property of their respective owners.
1
Table of Contents
Statements in this Current Report on Form 8-K that are not descriptions of historical facts are forward-looking statements that are based on management’s current expectations and assumptions and are subject to risks and uncertainties. If such risks or uncertainties materialize or such assumptions prove incorrect, our business, operating results, financial condition and stock price could be materially negatively affected. In some cases, you can identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” “will,” “would” or the negative of these terms or other comparable terminology. Factors that could cause actual results to differ materially from those currently anticipated include those set forth in the section titled “Risk Factors” including, without limitation, risks relating to:
| • | the results of our research and development activities, including uncertainties relating to the discovery of potential product candidates and the preclinical and clinical testing of our product candidates; |
| • | the early stage of our product candidates presently under development; |
| • | our ability to obtain and, if obtained, maintain regulatory approval of our current product candidates, and any of our other future product candidates, and any related restrictions, limitations, and/or warnings in the label of any approved product candidate; |
| • | our need for substantial additional funds in order to continue our operations, and the uncertainty of whether we will be able to obtain the funding we need; |
| • | our ability to retain or hire key scientific or management personnel; |
| • | our ability, with partners, to validate, develop and obtain regulatory approval of companion diagnostics for our product candidates; |
| • | our ability to protect our intellectual property rights that are valuable to our business, including patent and other intellectual property rights; |
| • | our dependence on third-party manufacturers, suppliers, research organizations, testing laboratories and other potential collaborators; |
| • | our ability to develop successful sales and marketing capabilities in the future as needed; |
| • | the size and growth of the potential markets for any of our approved product candidates, and the rate and degree of market acceptance of any of our approved product candidates; |
| • | competition in our industry; and |
| • | regulatory developments in the United States and foreign countries |
We operate in a very competitive and rapidly-changing environment and new risks emerge from time to time. As a result, it is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Moreover, neither we nor any other person assumes responsibility for the accuracy and completeness of the forward-looking statements. The forward-looking statements included in this report speak only as of the date hereof, and except as required by law, we undertake no obligation to update publicly any forward-looking statements for any reason after the date of this report to conform these statements to actual results or to changes in our expectations.
2
Table of Contents
| Item 1.01 | Entry into a Material Definitive Agreement. |
Merger Agreement
On October 31, 2013, Ignyta, IGAS Acquisition Corp., a Delaware corporation and a wholly-owned subsidiary of Ignyta (Merger Sub), and Ignyta Operating entered into an Agreement and Plan of Merger and Reorganization (the Merger Agreement). The Merger Agreement provides for the merger of Merger Sub with and into Ignyta Operating (the Merger), with Ignyta Operating surviving the transaction as a wholly owned subsidiary of Ignyta. The Merger closed on October 31, 2013 concurrently with the execution and delivery of the Merger Agreement. Reference is made to the description of the Merger and the Merger Agreement included in Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference. The description of the Merger Agreement set forth in this report is qualified in its entirety by reference to the full text of that document, which is attached hereto as Exhibit 2.2 and is incorporated herein by reference.
Indemnification Agreements
On October 31, 2013, our Board of Directors approved a form of indemnification agreement to be entered into between us and our directors and certain executive officers. The indemnification agreement requires that we, under the circumstances and to the extent provided for therein, indemnify such persons to the fullest extent permitted by applicable law against certain expenses and other amounts incurred by any such person as a result of such person being made a party to certain actions, suits and proceedings by reason of the fact that such person is or was a director, officer, employee or agent of Ignyta, any entity that was a predecessor corporation of Ignyta or any of its affiliates. The rights of each person who is a party to an indemnification agreement are in addition to any other rights such person may have under applicable law, our Amended and Restated Articles of Incorporation, our Bylaws, any other agreement, a vote of our stockholders, a resolution adopted by our Board of Directors or otherwise. Immediately following the closing of the Merger on October 31, 2013, we entered into indemnification agreements in the form approved by our Board of Directors with each of our newly appointed executive officers and directors, consisting of Jonathan E. Lim, M.D., Zachary Hornby, Patrick O’Connor, Ph.D., Alexander Casdin and Heinrich Dreismann, Ph.D. The foregoing is only a brief description of the indemnification agreement, does not purport to be a complete description of the rights and obligations of the parties thereunder and is qualified in its entirety by reference to the form of indemnification agreement filed as Exhibit 10.10 to this Current Report on Form 8-K and incorporated herein by reference.
| Item 2.01 | Completion of Acquisition of Disposition of Assets. |
The Merger closed on October 31, 2013 concurrently with the execution and delivery of the Merger Agreement.
Effective as of October 31, 2013, prior to the execution and delivery of the Merger Agreement and the concurrent closing of the Merger, (i) Ignyta amended and restated its Articles of Incorporation to, among other things, change its name from Infinity Oil & Gas Company to “Ignyta, Inc.” and to effect a 100-to-one reverse stock split, resulting in 87,336 outstanding shares of Ignyta’s common stock, (ii) the Board of Directors of Ignyta declared a $3.50 per share cash dividend to its stockholders of record, and (iii) Ignyta repurchased 80,000 shares of its common stock (on a post reverse stock split basis) at a price of $0.99 per share from its principal stockholder, Betty Sytner.
Also on October 31, 2013, prior to the execution and delivery of the Merger Agreement and the concurrent closing of the Merger, (i) the holders of all series of outstanding preferred stock of Ignyta Operating, consisting of series A preferred stock and series B preferred stock, voluntarily converted such shares into shares of Ignyta Operating’s common stock in accordance with the certificate of incorporation of Ignyta Operating and at the then-effective conversion rates therefor, which were one-to-one in all cases, and (ii) Ignyta Operating amended its certificate of incorporation to change its name to “Ignyta Operating, Inc.” and to effect a three-to-one reverse stock split of its capital stock, resulting in 4,916,469 outstanding shares of Ignyta Operating’s common stock, outstanding warrants to acquire up to an aggregate of 25,001 shares of Ignyta Operating’s common stock, and outstanding options granted under Ignyta Operating’s Amended and Restated 2011 Stock Incentive Plan (the Ignyta Plan) to purchase up to an aggregate of 358,986 shares of Ignyta Operating’s common stock.
At the closing of the Merger and pursuant to the terms of the Merger Agreement, Ignyta issued an aggregate of 4,916,469 shares of its common stock to the former stockholders of Ignyta Operating in exchange for all of the outstanding shares of Ignyta Operating’s capital stock. That number of shares was negotiated and agreed to by Ignyta and Ignyta Operating prior to entering into the Merger Agreement. As a result, the equity holders of Ignyta Operating became entitled to receive one
3
Table of Contents
share of Ignyta’s common stock, or the right to acquire one share of Ignyta’s common stock, in exchange for each one share of Ignyta Operating’s common stock, or right to acquire one share of Ignyta Operating’s common stock, held by them prior to the closing of the Merger. As of immediately following the closing of the Merger, Ignyta Operating has become a wholly-owned subsidiary of Ignyta, and the former stockholders of Ignyta Operating collectively own approximately 99.85% of the outstanding shares of Ignyta’s common stock. In addition, pursuant to the terms of the Merger Agreement, Ignyta has assumed (i) the Ignyta Plan, under which an aggregate of 342,209 shares are reserved for issuance pursuant to future equity grants, (ii) the obligation to issue up to an aggregate of 358,986 shares of its common stock upon the exercise of all options granted under the Ignyta Plan that were outstanding as of immediately prior to the closing of the Merger, and (iii) the obligation to issue up to an aggregate of 25,001 shares of its common stock upon the exercise of two warrants previously issued by Ignyta Operating and outstanding as of immediately prior to the closing of the Merger.
In connection with the closing of the Merger, Ignyta Operating changed its name from “Ignyta, Inc.” to “Ignyta Operating, Inc.”
The Merger Agreement includes customary representations, warranties and covenants made by Ignyta and Ignyta Operating as of specific dates. The assertions embodied in those representations and warranties were made solely for purposes of the Merger Agreement and are not intended to provide factual, business, or financial information about Ignyta, Ignyta Operating or the Combined Company. Moreover, those representations and warranties generally were made solely for the benefit of the parties to the Merger Agreement, and some or all of them (i) may not be accurate or complete as of any specified date, (ii) may be subject to a contractual standard of materiality different from those generally applicable to stockholders or different from what a stockholder might view as material, and/or (iii) may have been qualified by certain disclosures of Ignyta or Ignyta Operating not reflected in the Merger Agreement. The description of the Merger Agreement set forth in this report does not purport to be complete and is qualified in its entirety by reference to the full text of that document. A copy of the Merger Agreement is attached to this Current Report on Form 8-K as Exhibit 2.2 and is incorporated herein by reference.
Post-Merger Ownership of Ignyta
As of immediately after the closing of the Merger, our securities (on a fully diluted basis) are owned as follows:
| • | Former holders of Ignyta Operating’s common stock hold an aggregate of 4,916,469 shares of our common stock, or approximately 87.02% on a fully diluted basis; |
| • | Holders of our common stock prior to the closing of the Merger hold an aggregate of 7,336 shares of our common stock, or approximately 0.13% on a fully diluted basis; |
| • | Holders of Ignyta Operating’s outstanding warrants, which we have assumed, have the right to acquire up to an aggregate of 25,001 shares of our common stock, or approximately 0.44% on a fully diluted basis; |
| • | Holders of outstanding options granted under the Ignyta Plan, which we have assumed, have the right to purchase up to an aggregate of 358,986 shares of our common stock, or approximately 6.35% on a fully diluted basis; and |
| • | 342,209 shares of our common stock are reserved for issuance pursuant to future equity grants to employees, directors and consultants under the Ignyta Plan, representing approximately 6.06% on a fully diluted basis. |
Accounting Treatment of the Merger
The Merger is being accounted for as a reverse-merger and recapitalization. Ignyta Operating is the acquirer for financial reporting purposes and Ignyta is the acquired company. Consequently, the assets and liabilities and the operations that will be reflected in the historical financial statements prior to the Merger will be those of Ignyta Operating and will be recorded at the historical cost basis of Ignyta Operating, and the consolidated financial statements after completion of the Merger will include the assets and liabilities of Ignyta and Ignyta Operating, the historical operations of Ignyta Operating and the operations of the Combined Company from and after the closing date of the Merger.
Tax Treatment; Smaller Reporting Company
The Merger is intended to constitute a tax-free reorganization within the meaning of the Internal Revenue Code of 1986. Following the Merger, the Combined Company continues to be a “smaller reporting company,” as defined in Item 10(f)(1) of Regulation S-K, as promulgated by the Securities and Exchange Commission (the SEC).
4
Table of Contents
Background of Ignyta; Form 10 Information
Ignyta was incorporated on August 21, 2012 in the State of Nevada with the name “Infinity Oil & Gas Company.” Ignyta filed a registration statement on Form S-1 (File No. 333-183886) that was declared effective by the SEC on December 13, 2012, and sold an aggregate of 7,336 shares of its common stock (on a post reverse stock split basis) under that registration statement. Prior to the Merger, Ignyta intended to pursue a business in buying property for oil and gas drilling, buying oil and gas leases and acquiring and managing oil and gas royalties. Upon the closing of the Merger, Ignyta has abandoned those business plans and is now pursuing the business of Ignyta Operating.
Prior to the closing of the Merger, Ignyta was a “shell company,” as such term is defined in Rule 12b-2 under the Securities Exchange Act of 1934 (the Exchange Act). Accordingly, pursuant to the requirements of Item 2.01(f) and Item 5.01(a)(8) of Form 8-K, this Item 2.01 sets forth the information that would be required if the Combined Company were filing a general form for registration of a class of securities on Form 10 under the Exchange Act, with such information reflecting the Combined Company and its securities upon completion of the Merger. The Combined Company intends to carry on the business of Ignyta Operating. Upon closing the Merger, our executive office is the San Diego, California office of Ignyta Operating.
Corporate Overview
Ignyta was incorporated under the laws of the State of Nevada on August 21, 2012 with the name “Infinity Oil & Gas Company.” Ignyta changed its name to “Ignyta, Inc.” concurrently with the closing of the Merger. On October 31, 2013, Ignyta effected a 100-to-one reverse stock split of its issued and outstanding shares of common stock, and all share information in this report with respect to Ignyta gives retroactive effect to that reverse stock split.
Ignyta Operating was incorporated under the laws of the State of Delaware on August 29, 2011 with the name “NexDx, Inc.” Ignyta Operating changed its name to “Ignyta, Inc.” on October 8, 2012, and changed its name to “Ignyta Operating, Inc.” in connection with the closing of the Merger. On May 20, 2013, Ignyta Operating completed its acquisition of Actagene Oncology, Inc. (Actagene), which merged with and into Ignyta Operating on that date. On October 31, 2013, in connection with the closing of the Merger, (i) all then-outstanding shares of each series of Ignyta Operating’s preferred stock were voluntarily converted into shares of Ignyta Operating’s common stock in accordance with Ignyta Operating’s certificate of incorporation, and (ii) Ignyta Operating effected a three-to-one reverse stock split of its issued and outstanding shares of capital stock. All share information in this report with respect to Ignyta Operating’s capital stock gives retroactive effect to that reverse stock split.
On October 30, 2013, Ignyta formed IGAS Acquisition Corp., a wholly owned subsidiary formed for the purpose of the Merger, and on October 31, 2013, that wholly owned subsidiary merged with and into Ignyta Operating, and Ignyta Operating became our wholly owned subsidiary. Concurrent with the closing of the Merger, Ignyta has abandoned it pre-Merger business plan in the oil and gas industry, and we are now solely pursuing the business of Ignyta Operating in the oncology drug development industry. The following discussion describes the business now being collectively pursued by the Combined Company.
Business Overview
We are a precision medicine biotechnology company dedicated to discovering or acquiring, then developing and commercializing, precisely targeted new drugs for cancer patients whose tumors harbor specific molecular alterations. We pursue an integrated drug and diagnostic, or Rx/Dx, strategy, where we anticipate pairing each of our drug candidates with biomarker-based companion diagnostics, developed by us or by third parties with which we may partner, that are designed to identify the patients that are most likely to benefit from the use of the drugs we may develop. Our current development plans focus on two product candidates: RXDX-101, a tyrosine kinase inhibitor directed to the TrkA, ROS1 and ALK proteins, which is in a Phase I/II clinical study in molecularly defined patient populations for the treatment of solid tumors; and RXDX-102, a tyrosine kinase inhibitor directed to the Trk family tyrosine kinase receptors, TrkA, TrkB and TrkC, which is currently in preclinical development for the treatment of multiple cancers. We have entered into a license agreement granting us exclusive global development and marketing rights to RXDX-101 and RXDX-102, which will become effective upon our satisfaction of certain financing conditions set forth in that license agreement. We also have three discovery stage programs, Spark-1, Spark-2, and Spark-3, directed to emerging oncology targets identified through mining our database of information from proprietary and publicly available tumor samples, called Oncolome™. We
5
Table of Contents
currently have no products that have obtained marketing approval in any jurisdiction, have not generated revenues since inception and do not expect to do so in the foreseeable future due to the early stage nature of our current product candidates, and we have an accumulated deficit as of June 30, 2013 of approximately $3.5 million.
From our inception, we have focused on discovering novel biomarkers that define diseases based on our belief that such biomarkers could provide rich biological insight into the underlying pathophysiology that drives the clinical symptomatology of those diseases. One of our core platforms for revealing multivariate biomarkers that characterize diseases of interest is epigenetic analysis, particularly assessment of DNA methylation signatures. Our initial business strategy was to use epigenetic biomarkers to develop new biomarker-based molecular diagnostic assays to help physicians differentially diagnose clinically confounding diseases, particularly chronic autoimmune and rheumatic diseases. However, in part due to macroeconomic challenges facing the molecular diagnostics industry, we determined that a more valuable deployment of our biomarker discovery engine would be to seek biomarkers that can serve as novel disease targets for therapeutic intervention. As a consequence, in May 2013, we acquired Actagene, a discovery stage precision medicine company applying genomic insights to discover new biomarkers and targets for cancer therapeutics. With the acquisition of Actagene we added important members to our management and drug discovery team, which is utilizing genetic and epigenetic analysis to discover and understand genes that are inappropriately activated in tumors. Our current focus is to identify genes and pathways that are altered in tumors of interest and to then acquire or develop drugs that target the proteins encoded by those genes and test those drugs in precise patient populations who have the underlying molecular alteration that our drug candidates seek to address.
To identify molecular alterations that drive cancers, we mine both publicly available, as well as proprietary, tumor repositories to seek genetic (e.g., sequence mutations, fusions, inversions, translocations, copy number variants) and epigenetic (e.g., differential DNA methylation) changes that are common across cancers. We aggregate these tumor data along with detailed de-identified patient phenotypic information into our proprietary in-house OncolomeTM database. Our Oncolome database currently consists of data from hundreds of proprietary tumor samples, as well as publicly available data from tens of thousands of tumor samples.
We currently pursue a two-pronged strategy to leverage the biomarker insights that we have gained through our genetic and epigenetic mining of Oncolome, as well as the knowledge of cancer biology of our management and drug discovery team.
In the first case, when we identify a molecular alteration that is driving the growth of tumors in cancers of interest and if there is already a company(ies) developing a drug candidate(s) that targets that specific molecular alteration, we plan to seek to in-license what we believe to be the most promising or most advanced drug candidate(s) available for licensing. This approach is exemplified by our in-license of RXDX-101 and RXDX-102 from Nerviano Medical Sciences S.r.l. (NMS), an Italian state-owned biopharmaceutical company based in Nerviano, Italy, pursuant to a license agreement entered in October 2013 that will become effective upon the completion of a financing pursuant to which we or our affiliates receive gross proceeds of at least $20 million. We believe that RXDX-101 is the most clinically advanced inhibitor of TrkA, a target that we believe is an activating alteration in several cancers with high unmet need. RXDX-101 also has been observed to have potent activity against ROS1, a second cancer target against which there are no approved products, and ALK, a clinically and commercially validated oncology target. RXDX-102 has potential to be one of the first pan-Trk (TrkA, TrkB and TrkC) inhibitors in active clinical development. We believe each of these agents has a potential opportunity to be a first-in-class drug against important molecular targets that are driving alterations in various cancers.
In the second case, when we identify an activating molecular alteration that drives the growth of tumors in cancers of interest and there is no known company(ies) developing a drug candidate(s) that targets that specific molecular alteration, we plan to seek to initiate target validation and drug discovery activities against such molecular target. This approach is exemplified by our Spark programs. To date, we have identified six molecular targets, denoted Spark-1 through Spark-6, that appear to be commonly altered in different cancer tissues. To our knowledge, no other commercial entity is currently developing clinical stage drug candidates that are specifically directed to these molecular targets. We have prioritized three of these six targets, denoted Spark-1, Spark-2 and Spark-3, and have initiated target validation and drug discovery activities against some of these molecular targets.
Our ability to identify innovative cancer targets and develop drugs against them is enabled by, and dependent on, a set of essential capabilities and the experience of our drug discovery and management team. Key aspects of our core drug discovery capabilities include the ability to perform x-ray crystallography on protein targets, conduct in silico structure based drug design and run virtual chemistry screens. Once compounds with activity against our target have been identified by those or other tests and procedures, our drug discovery and scientific team further pursues the drug development process. The members of our team have significant experience in medicinal chemistry, lead optimization, ADME & PK
6
Table of Contents
(the study of absorption, distribution, metabolism, excretion, and pharmacokinetics), preclinical development and clinical development, and have collectively led or contributed to the development of more than a dozen drugs approved by the U.S. Food and Drug Administration (FDA), including several cancer therapeutics.
Cancer Background
Cancer is a heterogeneous group of diseases characterized by uncontrolled cell division and growth. Cancerous cells that arise in the lymphatic system and bone marrow are referred to as hematological tumors. Cancer cells that arise in other tissues or organs are referred to as solid tumors. Researchers believe that exposure to chemical agents, viruses and various forms of radiation can cause genetic alterations that cause cancer. Genetic predisposition also can increase the risk of cancer in some people. Epigenetic factors are also increasingly believed to contribute to development of cancer.
Cancer is the second leading cause of death in the United States, exceeded only by heart disease. The American Cancer Society (ACS) estimates that in 2013, there will be approximately 1.6 million new cases of cancer and approximately 580,000 deaths from cancer in the United States. The World Health Organization estimated that 7.6 million people worldwide died of cancer in 2008. According to ACS data, lung, colon and rectal, breast, and prostate cancer are the most prevalent cancers in the United States.
The most common methods of treating patients with cancer are surgery, radiation and drug therapy. A cancer patient often receives treatment with a combination of these methods. Surgery and radiation therapy are particularly effective when the disease is localized. Physicians generally use systemic drug therapies when the cancer has spread beyond the primary site or cannot otherwise be treated through surgery. The goal of drug therapy is to damage and kill cancer cells or to interfere with the molecular and cellular processes that control the development, growth and survival of cancer cells. In many cases, drug therapy entails the administration of several different drugs in combination. Over the past several decades, drug therapy has been evolving from non-specific drugs that kill both healthy and cancerous cells, to drugs that target specific molecular pathways involved in cancer and, more recently, to therapeutics that target specific activating alterations that are the “drivers” of cancer.
Cytotoxic Chemotherapies. The earliest approach to pharmacological cancer treatment was to develop drugs referred to as cytotoxic drugs that kill rapidly proliferating cancer cells through non-specific mechanisms, such as deterring cell metabolism or causing damage to cellular components required for survival and rapid growth. While these drugs have been effective in the treatment of some cancers, many unmet medical needs for the treatment of cancer remain. Also, cytotoxic drug therapies act in an indiscriminate manner, killing healthy, as well as cancerous, cells. Due to their mechanism of action, many cytotoxic drugs have a narrow dose range above which the toxicity causes unacceptable or even fatal levels of damage to healthy cells and below which the drugs are not effective in eradicating cancer cells.
Targeted Therapies. The next approach to pharmacological cancer treatment was to develop drugs, referred to as targeted therapeutics, that target specific biological molecules in the human body that play a role in rapid cell growth and the spread of cancer. Targeted therapeutics are designed to preferentially kill cancer cells and spare normal cells, to improve efficacy and minimize side effects. The drugs are designed to attack either a target that causes uncontrolled growth of cancer cells because of either a specific genetic alteration primarily found in cancer cells but not in normal cells, or a target that cancer cells are more dependent on for their growth than normal cells. These drugs focus on eradicating processes that help the cancer cell survive, but not on the oncogenes, which are the drivers or cause of the cancer itself.
Oncogene-Targeted Therapies. A more recent approach to pharmacological cancer treatment is to develop drugs that affect the drivers that cause uncontrolled growth of cancer cells because of a specific activating molecular alteration. In some cases these agents may be initially identified as targeted therapeutics without knowledge, at the time of development, of the underlying genetic change causing the disease. One primary shortcoming of this approach is that historically it has not been pursued systematically, but rather has tended to follow a conventional trial and error approach to drug discovery. Clinical development of oncogene-targeted therapies has involved the treatment of large populations from which a defined subpopulation that responds to treatment is identified through post-hoc analysis, after the trial has been completed. As a result, this approach can be time-consuming and costly, with success often uncertain.
Strategy
Our goal is to become a preeminent precision medicine oncology company by developing the next generation of therapeutics that treat cancer by targeting specific oncogenic activating molecular alterations and the corresponding patient populations. We believe our competitive advantage lies at the nexus of our two fundamental approaches: (1) a bottom up,
7
Table of Contents
data driven, unbiased, genome-wide multi-omics (e.g., DNA sequence, DNA methylation, DNA expression, protein expression) approach to mining extensive tumor data to identify activating alterations and their key biomarkers; and (2) a top down “drug hunter” approach of applying our senior scientific leadership team’s many decades of successful cancer drug discovery and development experience. Key elements of our strategy are to:
| • | Utilize public and proprietary sources of tumor samples and cancer data so that we are informed by a rich knowledge base. We have assembled a proprietary database of hundreds of tumor samples from primary human tumors from multiple solid tissues and hematological cancers. We supplement our proprietary database of tumor data by electronically integrating publicly available databases of tumor data. The combined database, with data from tens of thousands of tumor samples, is called OncolomeTM. Oncolome consists of elements such as DNA sequences, gene copy number variants, and RNA transcript levels. This database also contains information on patient characteristics (such as age, gender, diagnosis, and treatments) and, in some cases, analysis from such patients of ex-vivo chemosensitivity of their tumor cells to approved anticancer agents. We apply disciplined bioinformatic mining strategies and complex biostatisical algorithms to the data available in Oncolome, with the goal of identifying non-obvious trends and biomarkers that indicate activating alterations that drive cancer biology. |
| • | Apply a multi-omics approach to discover activating molecular alterations that drive cancer biology. We believe that genetic insight can be very valuable in understanding cancer biology, but that the exploration of biological factors in addition to genetics can provide a more comprehensive understanding of the precise activating molecular alterations that drive oncogenicity. Thus, when we mine Oncolome to seek new cancer biomarkers and potential drug targets, we often explore epigenetic phenomena, such as DNA methylation patterns, in addition to DNA sequencing and transcript counting. Our team has identified potential cancer targets that are marked by epigenetic alterations that we may not have identified had we applied a genetic approach alone. |
| • | Leverage deep cancer biology expertise and systems biology understanding to identify the specific role of activating alterations. Our senior scientific leadership team has been involved with the discovery or development of eight approved cancer drugs and has a vast knowledge of the pathways involved with tumorigenicity. We aim to apply this knowledge, along with gene pathway mapping software, to gain insight into the biomarkers that are revealed from our unbiased genome-wide mining of Oncolome. We believe that this approach could expose unique druggable targets that are actually distinct from the specific biomarkers or activating alterations that characterize the cancer of interest. |
| • | Deploy drug design tools to develop small molecule inhibitors of activating targets. Our team has extensive experience with x-ray crystallography, structure based drug design and virtual screening, in addition to more traditional chemistry screening methods and medicinal chemistry. We believe that by using these tools, we can more efficiently discover novel chemical series that bind to and inhibit our protein targets without incurring the expense of developing and maintaining a large chemical library and automated high throughput screening infrastructure. |
| • | Employ a capital-efficient drug development team. The members of our development leadership team have served in positions at global pharmaceutical organizations, and importantly, each has also worked productively in resource-constrained environments, such as at start-up biotechnology companies. Key members of our team have also led critical disciplines such as oncology discovery biology, chemistry, ADME & PK, and clinical development of approved products. This set of diverse experiences provides our team with the knowledge of how to develop novel drug candidates, but the ability to do so in a capital-efficient fashion. |
| • | Test our drug candidates only in the patients that we believe are most likely to derive benefit. We plan to use biomarkers both to identify the activating molecular alterations that represent the drug targets that we wish to pursue, and to precisely define the patient populations in which we would test those drug candidates based on the presence of the biomarkers associated with those specific alterations. If our product candidates demonstrate a therapeutic benefit in those specific molecularly defined patients, then, provided that we are able to complete appropriate clinical trials and obtain regulatory approvals for those product candidates, we intend to use biomarkers to inform physicians which patients are strong candidates to receive commercial access to the applicable drugs. |
| • | Develop, or pursue relationships with third parties to develop, companion diagnostics to assist in identifying appropriate patients for any product candidates we are able to successfully commercialize. We believe that the availability of high quality companion diagnostics is essential to formalize biomarker discovery and utilization |
8
Table of Contents
| into a platform that can be used by regulators, physicians, payors and, most importantly, patients themselves, to facilitate administration of the applicable therapeutics to the most appropriate patients. A companion diagnostic is a test or measurement that evaluates the presence of biomarkers in a patient, which information can then assist physicians in selecting the specific drugs or treatments that may be most effective for that patient. With respect to our proposed and potential future product candidates, we believe that any high quality companion diagnostics that we or a third parties are able to successfully develop, could be used to select patients for late stage clinical testing, to inform regulators precisely which patients should be indicated for access to the therapeutics, to advise physicians and patients which individuals are good candidates for treatment with the therapeutics, and to guide payors as to the value the therapeutics provide to well-defined patients and the circumstances under which the therapeutics should be reimbursed. |
| • | In-license development candidates that meet our strict criteria. In some instances, the most promising oncogenic activating gene alteration targets that we identify through our analyses may be the subject of a compound already in development with potent activity against such target. In these cases, we may attempt to in-license such compounds if they meet our strict scientific and development criteria, particularly if we believe that their therapeutic potential could be better realized by us. This approach is exemplified by our recent in-license of RXDX-101 and RXDX-102, two investigational agents with first-in-class potential against the TrkA receptor, a target that we prioritized for development based on our analyses using Oncolome. |
| • | Seek and maintain commercial rights and, when and if appropriate, establish commercialization and marketing capabilities. We currently have exclusive worldwide commercialization rights to all of our programs in development. We intend, when and if it makes strategic and operational sense, to retain these commercial rights and those for any future product candidates we may pursue on a territory-by-territory basis and establish internal commercialization and marketing capabilities. |
Pipeline
Consistent with our strategy, each of our initial two in-licensed product candidates and each of our three internal discovery programs, for all of which we hold or have entered into agreements granting us exclusive global marketing rights, is being developed for precise biomarker-defined precise patient groups. Each of our product candidates is in the early stage of development, and we anticipate that it will likely be several years before any of our product candidates could be commercialized. The following table summarizes the status of our current product candidates and programs:
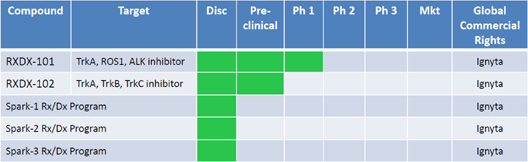
We have only recently entered into a license agreement to obtain the rights to develop our two lead product candidates, RXDX-101 and RXDX-102. That license agreement, entered in October 2013 with NMS, will not become effective until the completion of a financing of equity or debt securities pursuant to which we or our affiliates receive gross proceeds of at least $20 million. As a result, all discovery-stage, preclinical and clinical studies and other development activities relating to those product candidates that have been conducted to date have been performed by NMS and any third parties with which it has contracted. We have had no involvement or input in, nor have we had any control over, any of those activities. All of the descriptions of those product candidates in this report have been generated based on information provided by NMS or, in some cases, such as the graphic disclosure of preclinical study results for RXDX-101 and RXDX-102, are included in the form provided to us by NMS. As discussed below under the heading “Collaborations and License Agreements—NMS,” we will begin the process of obtaining all data relating to these product candidates from NMS following the effective date of the license agreement, which data transfer process we expect will continue for a number of months. We have not validated any of the information included in this report that has been provided by, or based on
9
Table of Contents
information provided by, NMS, and will not be fully able to do so until that data transfer process is complete. NMS has consented to our use of the data it has provided in this report.
RXDX-101: Lead Oncology Clinical Asset
RXDX-101 is a new chemical entity that we are in-licensing upon the effectiveness of our license agreement with NMS entered in October 2013. RXDX-101 is an orally available, selective tyrosine kinase inhibitor of the TrkA, ROS1 and ALK proteins. RXDX-101 is designed as a targeted therapeutic candidate to treat patients with cancers that harbor activating alterations to TrkA, ROS1 and ALK. Candidate alterations include gene rearrangements or mutations, splice variants, increased gene copy number and increased gene expression.
Rationale for Targeting TrkA, ROS1 and ALK
About TrkA (Tropomyosin Receptor Kinase A). The Trk (tropomyosin receptor kinase)/NTRK (neurotrophin tyrosine receptor kinase) family tyrosine kinase receptors, which include TrkA/NTRK1, TrkB/NTRK2 and TrkC/NTRK3, are activated by neurotrophins, a family of nerve growth factors. The Trk family members play a key role in normal central and peripheral neuronal cell development and differentiation. They regulate the survival (or prevention of programmed cell death) and maintain the function of neuronal cells throughout the body. Trk receptors are found on a number of different cell types, and many non-neuronal cells also produce neurotrophins. Deregulated kinase activities of Trk family members occur due to gene rearrangements and translocations, mutations, overexpression and alternative splicing and are associated with a number of human neuronal and non-neuronal cancers. Oncogenic TrkA translocations (fusion proteins with tropomycin-3) have been reported in colorectal, non small cell lung (NSCLC), papillary thyroid, pancreatic and certain prostate cancers. The TrkA fusion protein has a constitutively active kinase that provides the driving force for transformation and tumor progression, via the relay of growth and survival signals within cancer cells. In addition, TrkA overexpression and activation of kinase driven signal transduction pathways can be activated by its neural growth factor (NGF) ligand, produced by tumors or non-tumor cells. The growth and survival of cancers such as ovarian, breast, and oral squamous cancers are maintained by TrkA/NGF auto-stimulation and often occur early in the process of tumor formation. Further, in neuroblastomas, a type of extracranial solid cancer, a TrkA splice variant (TrkAIII) can be produced that switches TrkA to an oncogene, which promotes tumor progression often with a more aggressive character. TrkAIII containing tumors are resistant to chemotherapy-induced cell death, and they induce the formation of new blood vessels (angiogenesis) to allow the tumors to grow larger and metastasize.
About ROS1. ROS1 belongs to the insulin-receptor superfamily. Like other tyrosine kinase receptor molecules, it plays a role in relaying growth signals from the environment outside the cell into the cell’s nucleus. ROS1 is one of two orphan receptor tyrosine kinase family members with no known binding ligand. Genetic changes in ROS1 such as fusions, rearrangements, mutations, or copy number increases, create oncogenes, which can lead to cancer. Molecular rearrangements of ROS1 create fusion proteins with constitutively active kinase domains that activate downstream signaling pathways, which lead to oncogenic properties in cells, including uncontrolled proliferation and resistance to cell death with increased tumor cell survival. ROS1 was first discovered in NSCLC patients in the form of a ROS fusion protein (six different partners for ROS1). Two other genetic rearrangements of ROS1 have been detected in a variety of other cancers, including glioblastoma multiforme, cholangiocarcinoma, ovarian cancer, gastric adenocarcinoma, colorectal cancer, inflammatory myofibroblastic tumor, angiosarcoma and epitheloid hemangioendothelioma.
About ALK (Anaplastic lymphoma kinase). ALK also belongs to the insulin-receptor superfamily and is related to ROS1. ALK was first identified in anaplastic lymphomas, a distinct subset of non-Hodgkin’s lymphoma. Molecular changes in ALK through gene rearrangements, mutations, and overexpression lead to the formation of at least 14 ALK oncogenes. Aberrant ALK fusion proteins spontaneously form molecular structures that lead to self-activation and constitutive activity within cancer cells, via activation of signal transduction pathways and intracellular kinases that drive uncontrolled tumor cell growth, metabolism, and survival. In addition to anaplastic lymphomas, ALK oncogenes are found in a number of cancers such as NSCLC, diffuse large B-cell lymphoma, neuroblastomas, inflammatory myofibroblastic tumors and possibly subsets of esophageal/gastric and renal cell cancers. A currently available ALK inhibitor drug, crizotinib, has demonstrated potent in vitro, in vivo and human anti-tumor activity, validating the utility of ALK inhibitors. However, the rapid emergence of crizotinib-resistant tumors (especially in NSCLC) and the poor penetration of crizotinib into the brain for treating brain metastases support the need for the development of improved ALK inhibitors with better penetration of the blood brain barrier, a separation of circulating blood from the brain, and activity against crizotinib-resistant ALK mutations.
10
Table of Contents
Incidence of TrkA, ROS1 and ALK Mutations; Opportunity for RXDX-101
Research to date indicates that TrkA, ROS1 and ALK gene rearrangements and fusion proteins are most prevalent in solid tumors. Each of these genes also appears to be overexpressed in a portion of certain tumor types, though the importance of overexpression of these genes in cancer biology is not currently well understood.
| • | TrkA appears to be rearranged across a range of tumor types with a frequency usually in the low single digit percentages. Studies suggest that TrkA is rearranged in ALK mutation negative and epidermal growth factor receptor (EGFR) mutation negative non small cell lung adenocarcinoma patients, as well as in colorectal adenocarcinoma patients and in papillary thyroid cancer patients. |
| • | ROS1 appears to be rearranged across a range of tumor types with a frequency usually in the low single digit percentages. Studies suggest that ROS1 is rearranged in non small cell lung adenocarcinoma cancer patients, stomach cancer patients, glioblastoma patients, and cholangiocarcinoma patients. |
| • | ALK appears to be rearranged across a range of tumor types with a frequency usually in the single digit percentages. Studies suggest that ALK is rearranged in non small cell lung adenocarcinoma cancer patients, neuroblastoma patients and anaplastic large cell lymphoma patients. |
The potential ability of RXDX-101 to act as a potent inhibitor of the TrkA, ROS1, and ALK proteins, as well as its observed ability to be administered orally and reach systemic circulation (oral bioavailability) and its observed ability to cross the blood brain barrier in preclinical studies, attracted us to the profile of this drug candidate and support the market opportunity for the product.
RXDX-101 Preclinical Data
RXDX-101 is an orally available potent inhibitor of the TrkA, ROS1 and ALK tyrosine kinases. In vitro, RXDX-101 achieves low nanomolar inhibition of TrkA, ROS1 and ALK. RXDX-101 has been preclinically tested in vivo in three species to date, the mouse, rat and dog. It has demonstrated in vivo antitumor activity against various TrkA, ROS1 or ALK-driven mouse xenograft models of different human cancers, has also demonstrated oral bioavailability in all three species tested, and has been observed to efficiently cross the blood brain barrier in all three species tested.
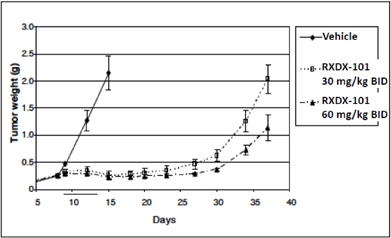
The graphs below depict the results of some of the preclinical studies of RXDX-101 conducted to date. Each of the studies whose results are shown below involved the administration of RXDX-101 orally twice daily for 10 days in mouse xenograft models of various cancers driven by one of the molecular targets of RXDX-101, TrkA, ROS1 or ALK. All of those studies were conducted by NMS or its third party contractors, we had no involvement in the conduct of such studies and the graphs below were provided by NMS.
| • | The following graph demonstrates the in vivo anti-tumor activity observed with the use of RXDX-101 against a TrkA-driven mouse xenograft model of human colorectal cancer: |
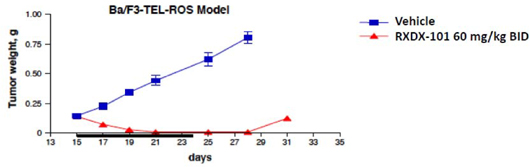
| • | The following graph demonstrates the in vivo anti-tumor activity observed with the use of RXDX-101 against a ROS1-driven Ba/F3 mouse xenograft model: |
11
Table of Contents
| • | The following graph demonstrates the in vivo anti-tumor activity observed with the use of RXDX-101 against an ALK-driven mouse xenograft model of human NSCLC: |
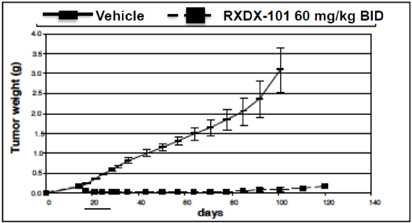
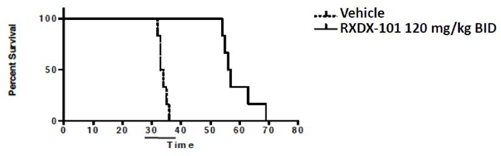
| • | The following graph demonstrates the survival benefit observed with the use of RXDX-101 against an ALK-driven mouse xenograft model of brain metastases associated with human NSCLC, which provides support for the product candidate’s potential ability to cross the blood brain barrier: |
Phase I/II Clinical Trial
NMS has filed a Clinical Trial Application (CTA) under the European Directive to the Italian Competent Authority that enabled NMS to commence a Phase I/II clinical study in patients with solid tumors that are positive for alterations in TrkA, ROS1 or ALK. This study, which is currently ongoing at two clinical sites in Italy, is an open label trial that has two phases. The first phase is a Phase I dose escalation phase that will include 20 to 30 patients, depending on when the maximum tolerated dose is achieved, with solid tumors with genetic mutations of TrkA, ROS1 or ALK. The second phase is an expansion phase utilizing the recommended Phase II dose identified in the first phase and is expected to include several cohorts of patients that have alterations to TrkA, ROS1 or ALK. Although we have not yet determined the types of cancer we may study in the second phase of this trial, we currently anticipate that the cohorts will consist of colorectal
12
Table of Contents
cancer and NSCLC, among other cancer types. To date, 11 patients have been dosed in the first, dose escalation phase of this trial, and the two trial sites in Italy are currently enrolling additional patients. We intend to file an investigational new drug application (IND) and add additional sites in the United States, as well as additional sites in Europe. Based on currently available data and certain assumptions regarding the ongoing clinical trial and FDA requirements, we project that such an IND application could be submitted as early as 2014.
The Phase I/II trial is not powered to show results with statistical significance. Statistical significance means that an effect is unlikely to have occurred by chance. Clinical trial results are considered statistically significant when the probability of the results occurring by chance, rather than from the efficacy of the drug candidate, is sufficiently low. Since this trial is not powered to show results with statistical significance, the results from the trial may be attributable to chance and not the clinical efficacy of RXDX-101. This trial design is customary for a Phase I and some Phase II clinical trials, the principal purpose of which is to provide the basis for the design of larger, definitive trials that are powered by the addition of more patients to potentially show statistical significance. Pending guidance from regulatory agencies such as the FDA, we would likely design any later stage trials that are intended to support marketing approval applications to show statistical significance. We would do so by enrolling a larger number of patients based on the clinical data observed in earlier trials.
The primary objectives of the trial are to evaluate the safety and tolerability of RXDX-101 and to determine its maximum tolerated dose when administered to patients with TrkA-, ROS1- or ALK-positive solid tumors.
Secondary objectives of this trial are to:
| • | determine the process by which RXDX-101 is distributed and metabolized in the body, which is referred to as pharmacokinetics; |
| • | assess the biochemical and physiological effects of RXDX-101 on the human body, which is referred to as pharmacodynamics; and |
| • | evaluate any early evidence of anti-tumor activity in patients with TrkA-, ROS1- or ALK-positive tumors |
Patients treated with RXDX-101 have experienced some adverse events, which have been predominantly gastrointestinal or constitutional in nature, but there have been no dose limiting toxicities or Grade 3 or Grade 4 treatment-related adverse events experienced by any of the patients treated with RXDX-101 in this trial to date.
If we observe an anti-tumor effect in one or more of the proposed expansion cohorts, such results, pending discussions with regulatory agencies such as the FDA, could serve as the basis to either initiate randomized Phase II studies or to potentially enter directly into registration-enabling clinical studies under an accelerated registration path.
RXDX-101 Companion Diagnostic
Several companion diagnostic technologies are available for measuring alterations in TrkA, ROS1 and ALK. There is an FDA-approved FISH (fluorescence in situ hybridization) test for measuring ALK translocations (Vysis manufactured by Abbott Molecular). There is also a commercially available FISH test for measuring ROS1 fusion proteins, and we are aware of at least one group that has developed a FISH test for measuring TrkA fusion proteins. TrkA fusion proteins can also be measured by IHC (immunohistochemistry) using commercially available antibodies. In addition, NMS has developed and is expected to transition to us PCR (polymerase chain reaction) assays for measuring fusion proteins for each of TrkA, ROS1 and ALK. Finally, several commercial, as well as academic, groups evaluate sequence mutations and translocations of TrkA, ROS1 and ALK by next generation sequencing. It is our intent to evaluate each of these candidate diagnostic approaches for measuring alterations to TrkA, ROS1 and ALK and select a technology to be pursued by us or, most likely for later stage development and commercialization, a third party collaborator, after taking into consideration scientific, as well as commercial, factors.
RXDX-102: Preclinical Asset
RXDX-102 is a second new chemical entity that we are in-licensing upon the effectiveness of our license agreement with NMS entered in October 2013. RXDX-102 is an orally available, selective pan-TRK tyrosine kinase inhibitor, or inhibitor of the TrkA, TrkB and TrkC proteins. RXDX-102 is designed as an oncogene-targeted therapeutic candidate to treat patients with cancers that harbor activating alterations to TrkA, TrkB or TrkC. Candidate alterations include gene rearrangements or mutations, increased gene copy number and increased gene expression. RXDX-102 is a preclinical product candidate, and we anticipate that we will next pursue repeat dose toxicology studies of this product candidate in a rodent and non-rodent species in compliance with the FDA’s good laboratory practice (GLP) regulations. Based on
13
Table of Contents
currently available data and estimates of our scientific team, we believe an IND application could potentially be submitted for RXDX-102 as early as 2014.
Rationale for Targeting TrkA, TrkB and TrkC
About TrkA. See the description of TrkA set forth above under the same heading in the discussion about RXDX-101.
About TrkB. TrkB acts as an oncogene when overexpressed in neuroblastomas and ovarian cancer. TrkB expression can respond to its growth factor ligand, BDNF, produced by tumor cells or non-tumor cells around the tumor, including immune cells such as macrophages. Activated TrkB receptors relay growth and survival signals into the cancer cells and amplify the expression of additional oncogenes such as mycN. Tumors expressing TrkB oncogenes are more aggressive, drug resistant, highly angiogenic, and more invasive for establishing metastatic tumors. Studies have shown that patients with TrkB driven tumors have poor survival.
About TrkC. Neurotrophin-3 is the normal growth factor for TrkC. Oncogenic translocations involving TrkC kinase domain generate fusion proteins that have been identified in acute myeloid leukemia, salivary gland carcinoma, adult secretory breast cancer, congenital fibrosarcoma, and pediatric nephroma and neuroblastoma. Depending on the tumor type, TrkC expression can accelerate angiogenesis and can be associated with perineural skin invasion (basal cell and cutaneous squamous cell carcinomas) via expression of proteases to break barriers and migration molecules to establish metastatic tumors.
RXDX-102 Preclinical Data
RXDX-102 is an orally available selective inhibitor of TrkA, TrkB and TrkC. In in vitro studies performed to date, RXDX-102 achieves single digit nanomolar inhibition of TrkA, TrkB and TrkC enzymatic assays. RXDX-102 has been tested in vivo in four species to date, the mouse, rat, dog and primate. It has demonstrated in vivo antitumor activity against various TrkA, TrkB or TrkC-driven mouse xenograft models of cancer, and has also demonstrated oral bioavailability in all four species tested to date.
The graphs below depict the results of some of the preclinical studies of RXDX-102 conducted to date. Each of the studies whose results are shown below involved the administration of RXDX-102 orally twice daily for 10 days in mouse xenograft models of various cancers driven by one of the molecular targets of RXDX-102, TrkA, TrkB or TrkC. All of those studies were conducted by NMS or its third party contractors, we had no involvement in the conduct of such studies and the graphs below were provided by NMS.
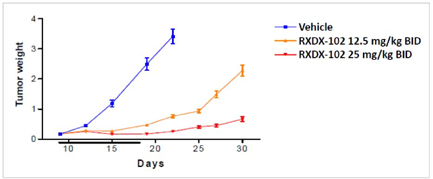
| • | The following graph demonstrates the in vivo anti-tumor activity observed with the use of RXDX-102 against a TrkA-driven mouse xenograft model of human colorectal cancer: |
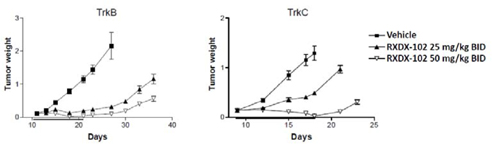
| • | The following graph demonstrates the in vivo anti-tumor activity observed with the use of RXDX-102 against a TrkB-driven Ba/F3 mouse xenograft model and a TrkC-driven Ba/F3 mouse xenograft model: |
14
Table of Contents
RXDX-102 Companion Diagnostic
We intend to pursue a companion diagnostic strategy for RXDX-102 similar to that described above for RXDX-101 under the heading “RXDX 101 Companion Diagnostic.” We plan to compare multiple possible diagnostic methods, such as IHC, rtPCR and next generation sequencing, to determine the most appropriate diagnostic method for patient selection for RXDX-102.
Spark-1 through Spark-6
In our mining of the Oncolome database for molecular alterations that frequently occur in tumor tissue samples to date, we have identified six molecular targets, which, when altered, we believe to drive tumorigenesis. We denote these six targets as Spark-1 through Spark-6, respectively. We have prioritized three of these targets (Spark-1 through Spark-3) and have initiated target validation and small molecule drug discovery activities against some of these targets. Such discovery activities include, or may in the future include, but are not limited to: x-ray crystallography, structure based drug design, virtual screening, in vitro screening, in vivo screening, medicinal chemistry and lead optimization. We aim to have developed our first IND candidate against one of the Spark-1, Spark-2 or Spark-3 targets as early as 2015.
Collaborations and License Agreements
NMS
We entered into a license agreement with NMS on October 10, 2013, which was amended on October 25, 2013, and which grants us exclusive global rights to develop and commercialize RXDX-101 and RXDX-102. The license agreement will become effective upon the completion of a financing of equity or debt securities pursuant to which we or our affiliates receive gross proceeds of at least $20 million. Our development rights under the license agreement are exclusive for the term of the agreement with respect to RXDX-101 and RXDX-102 and also, as to NMS, are exclusive for a five-year period with respect to any product candidate with activity against the target proteins of RXDX-101 and RXDX-102, and include the right to grant sublicenses. The license agreement provides that we will assume control of financial and all other responsibility for the ongoing Phase I/II clinical trial of RXDX-101 that is currently being conducted by NMS and for continued preclinical development of RXDX-102. We are obligated under the license agreement to use commercially reasonable efforts to develop and commercialize RXDX-101 and RXDX-102, and, with the exception of transfer to us without cost of NMS’ existing inventory of RXDX-101 and RXDX-102 material, we are responsible for all remaining development and commercialization costs for RXDX-101 and RXDX-102.
Under the terms of the license agreement, we have agreed to issue to NMS a warrant to acquire up to 16,667 shares of our common stock upon the effective date of the license agreement, which will have an exercise price per share equal to that used in the financing that causes the license agreement to become effective, and will be exercisable at any time at the option of the holder from the effective date of the license agreement until the five-year anniversary thereof. We are also obligated to make an up-front payment to NMS of $7.0 million on the earlier of 10 days following the effective date of the license agreement and December 31, 2013, $1.0 million of which NMS may elect to receive in the form of shares of our common stock at a price per share equal to that used in the financing that causes the license agreement to become effective, and the remainder of which shall be paid in cash. If we fail to make this upfront payment, the license agreement terminates. When and if commercial sales of RXDX-101 or RXDX-102 begin, we are obligated to pay NMS tiered royalties ranging from a mid-single digit percentage to a low double digit percentage of our net sales, depending on the amount of our net sales, with standard provisions for royalty offsets to the extent we need to obtain any rights from third parties to commercialize either RXDX-101 or RXDX-102. We are obligated under the terms of the license agreement to engage NMS to perform services valued at $1 million or more between the effective date of the license agreement and December 31, 2014, which services could include, among others at our election, manufacture and supply services, technology transfer activities, preclinical
15
Table of Contents
activities, process development activities and assay development activities. We are also required to make development and regulatory milestone payments to NMS of up to $105.0 million in the aggregate if specified clinical study initiations and regulatory approvals are achieved across multiple products or indications. The first such milestone payment is not due until we elect to initiate the first randomized Phase II clinical study, which, based on our current estimates and certain assumptions, we anticipate could occur as early as 2015.
The license agreement with NMS provides that NMS will transfer to us all data, technology and know-how related to RXDX-101 and RXDX-102 and necessary for their continued development. That data and technology transfer will commence upon the effectiveness of the license agreement, and we anticipate that the transfer will be complete in 2014. Our ability to continue all ongoing studies, design and commence any new studies or trials and solidify our development plans for RXDX-101 and RXDX-102 is dependent on that transfer process being completed successfully and in a timely manner.
The license agreement will not become effective if we do not satisfy the financing conditions to its effectiveness by December 31, 2013. Additionally, following its effectiveness, the license agreement with NMS will remain in effect until the expiration of all of our royalty and sublicense revenue obligations to NMS, determined on a product-by-product and country-by-country basis, unless we elect to terminate the license agreement earlier. If we fail to meet our obligations under the license agreement and are unable to cure such failure within specified time periods, NMS can terminate the license agreement, resulting in a loss of our rights to RXDX-101 and RXDX-102.
Competition
The pharmaceutical and biotechnology industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our technology, development experience, scientific knowledge and strategies provide us with competitive advantages, we face competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions. Any product candidates that we are able to successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future. Our competitors may develop or market products or other novel technologies that are more effective, safer, more convenient or less costly than any that may be commercialized by us, or may obtain regulatory approval for their products more rapidly than we may obtain approval for ours.
The acquisition or licensing of pharmaceutical products is also very competitive, and a number of more established companies, some of which have acknowledged strategies to license or acquire products and many of which are bigger than us and have more institutional experience and greater cash flows than we have, may have competitive advantages over us, as may other emerging companies taking similar or different approaches to product licenses and/or acquisitions. In addition, a number of established research-based pharmaceutical and biotechnology companies may acquire products in late stages of development to augment their internal product lines, which may provide those companies with an even greater competitive advantage.
Many of our competitors will have substantially greater financial, technical and human resources than we have. Additional mergers and acquisitions in the pharmaceutical industry may result in even more resources being concentrated in some of our competitors. Competition may increase further as a result of advances made in the commercial applicability of technologies and greater availability of capital for investment in these fields. Our success will be based in part on our ability to build, obtain regulatory approval for and market acceptance of, and actively manage a portfolio of drugs that addresses unmet medical needs and creates value in patient therapy.
We compete in the segments of the pharmaceutical, biotechnology and other related markets that pursue precision medicine approaches to combatting activating molecular alterations in cancer. There are a number of other companies presently working to develop therapies for cancer in the field of precision medicines, including divisions of large pharmaceutical companies and biotechnology companies of various sizes.
The most common methods of treating patients with cancer are surgery, radiation and drug therapy, including chemotherapy, hormone therapy and targeted drug therapy or a combination of such methods. There are a variety of available drug therapies marketed for cancer. In many cases, these drugs are administered in combination to enhance efficacy. While our product candidates, if any are approved, may compete with these existing drug and other therapies, to the extent they are ultimately used in combination with or as an adjunct to these therapies, our product candidates may not be competitive with them. Some of the currently approved drug therapies are branded and subject to patent protection, and
16
Table of Contents
others are available on a generic basis. Many of these approved drugs are well established therapies and are widely accepted by physicians, patients and third-party payors. As a result, market acceptance of, and a significant share of the market for, any of our product candidates that we successfully introduce to the market will pose challenges.
In addition to currently marketed therapies, there are also a number of medicines in late stage clinical development to treat cancer. These medicines in development may provide efficacy, safety, convenience and other benefits that are not provided by currently marketed therapies and may not be provided by any of our current or future product candidates. As a result, they may provide significant competition for any of our product candidates.
RXDX-101
RXDX-101 has demonstrated potent activity in testing to date against the three molecular targets, TrkA, ROS1 and ALK. We may pursue indications in cancers where any one or more of these three genes are altered.
Based on publicly available information, we believe that other pharmaceutical companies may be seeking to develop TrkA selective inhibitors. Although we are unaware of other compounds that are specifically directed to TrkA activating alterations in cancer that are more advanced in clinical development than RXDX-101, any of the TrkA selective inhibitors being developed could progress through clinical development faster than or during a similar timeframe as RXDX-101.
We also believe that other pharmaceutical companies may be seeking to develop ROS1 selective inhibitors, and are aware of several such products currently in clinical development by other companies.
Xalkori® is the only drug currently approved in the U.S. to treat ALK-mutant NSCLC. In addition, we are aware of several products in clinical development targeting cancer-causing mutant forms of ALK for the treatment of NSCLC patients, some of which are more advanced in clinical development than RXDX-101. We believe RXDX-101 potentially offers several important advantages over Xalkori, including potentially superior efficacy due to activity against certain ALK resistant mutations, as well as potentially increased ability to cross the blood brain barrier, therefore offering an opportunity for clinical activity against brain metastases that are common in ALK mutant NSCLC.
RXDX-102
RXDX-102 has demonstrated potent activity against three molecular targets, TrkA, TrkB and TrkC. We may pursue indications in cancers where any one or more of these three genes are altered.
The only compound of which we are presently aware that is currently in clinical development and targets TrkA, TrkB or TrkC activating alterations is Daiichi Sankyo and its subsidiary Plexxikon’s PLX-7486, which has activity against TrkA, among other molecular targets, and we believe to be in a Phase I clinical study. We believe that other pharmaceutical companies may be seeking to develop TrkA, TrkB or TrkC selective inhibitors that may enter clinical development before or during a similar timeframe as RXDX-102.
Spark-1 through Spark-3
Spark-1, Spark-2 and Spark-3 represent activating gene alterations that we believe to drive cancer biology in certain tumors. To our knowledge, there are no commercial entities actively developing clinical stage drugs against any of these three targets. We believe that other pharmaceutical companies may seek to develop selective inhibitors against the Spark-1, Spark-2 or Spark-3 targets and that these potential inhibitors may enter clinical development before or during a similar timeframe as the compounds that we aim to develop against one or more of these three targets.
17
Table of Contents
Commercialization
We have not yet established a sales, marketing or product distribution infrastructure because our lead candidates are still in discovery, preclinical or early clinical development. We generally aim to retain commercial rights in the United States for any of our product candidates for which we receive marketing approvals. We currently anticipate that, when appropriate, we will seek to access the United States oncology market through a focused, specialized, internal sales force.
Subject to receiving marketing approvals, we expect to commence commercialization activities by building a focused internal sales and marketing team in the United States to sell our products. We believe that such an approach will enable us to address the community of oncologists who are the key specialists in treating the patient populations for which our current product candidates are being developed. Outside the United States, we may enter into distribution and other marketing arrangements with third parties for any of our product candidates that obtain marketing approval in foreign jurisdictions.
We also aim to build a marketing and sales management force to create and implement marketing strategies for any products that we market through our own sales teams and to oversee and support our sales force. We anticipate that our goals for any such marketing force include developing educational initiatives with respect to any approved products and establishing relationships with thought leaders in relevant fields of medicine.
We currently expect that any third parties with which we may collaborate in the future on the development of any commercial companion diagnostics for use with our therapeutic products will most likely hold the commercial rights to those diagnostic products. We expect that we would coordinate closely with any future diagnostic collaborators in connection with the marketing and sale of such diagnostic products and our related therapeutic products.
Manufacturing
We do not own or operate, and currently have no plans to establish, any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacture of our product candidates for preclinical and clinical testing, as well as for commercial manufacture of any products that we may commercialize. Our license agreement with NMS requires NMS to provide us with its existing inventory of clinical supply of RXDX-101, which can help support our planned expansion cohorts of the ongoing Phase I/II clinical study of that product candidate. We aim to engage, by entering into a supply agreement or through another arrangement, NMS and/or third party manufacturers to provide us with additional RXDX-101 clinical supply. We do not currently have any long-term supply commitments or other arrangements in place, and may obtain our supplies from NMS or any other manufacturer on a purchase order basis or through a formal supply agreement. We also do not currently have arrangements in place for redundant supply of bulk drug substance. For all of our product candidates, we aim to identify and qualify manufacturers to provide the active pharmaceutical ingredient and fill-and-finish services prior to submission of a new drug application (NDA) to the FDA.
RXDX-101 and RXDX-102 are organic compounds of low molecular weight, generally called small molecules. We believe that they can be manufactured in reliable and reproducible synthetic processes from readily available starting materials. We believe that the chemistry is amenable to scale-up and does not require unusual or expensive equipment in the manufacturing process. We expect to continue to develop drug candidates that can be produced cost-effectively at contract manufacturing facilities.
We generally expect to rely on third parties for the manufacture of any companion diagnostics we or our collaborators may develop.
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain proprietary or intellectual property protection for our product candidates and our core technologies, including novel biomarker and diagnostic discoveries and other know-how, to operate without infringing on the proprietary rights of others and to prevent others from infringing our proprietary or intellectual property rights. We expect that we will seek to protect our proprietary and intellectual property position by, among other methods, licensing or filing our own U.S., international and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business. We also rely on trade secrets, know-how and continuing technological innovation to develop and maintain our proprietary and intellectual property position, which we generally seek to protect through contractual obligations with third parties.
18
Table of Contents
We currently, and expect that we will continue to, file or license patent applications directed to our key product candidates in an effort to establish intellectual property positions regarding new chemical entities relating to these product candidates, as well as uses of new chemical entities in the treatment of various cancers. We also intend to seek patent protection, if available, with respect to biomarkers that may be useful in selecting the right patient population for use of any of our product candidates. Following the effective date of our license agreement with NMS, we will own or exclusively license a patent portfolio consisting of two issued U.S. patents and their respective counterparts in a number of foreign jurisdictions, nine pending U.S. patent applications, two pending applications under the Patent Cooperation Treaty and corresponding pending patent applications in a number of foreign jurisdictions. The two issued U.S. patents and one of the U.S. applications cover RXDX-101 and RXDX-102, and those two issued patents are expected to expire in 2028 and 2027 without patent term extension. The remaining pending U.S. patent applications and a significant portion of our pending patent applications in foreign jurisdictions pertain to our DNA methylation biomarkers and our platform for generating DNA methylation biomarkers, as well as the use of such biomarkers to diagnose, prognose and select treatments for certain autoimmune diseases, which activities we are not presently pursuing as a material aspect of our business and operations. We would expect that any patents that may issue from the pending U.S. patent applications would likely expire between 2031 and 2033; however, any and all of these patent applications may not result in issued patents.
In addition to the patent applications that we have filed as of the date of this report, we intend to file additional applications covering potential discoveries that we may make in relation to our drug discovery and biomarker activities directed to the Spark-1 through Spark-6 targets. We plan to continue to expand our intellectual property portfolio by filing patent applications directed to dosage forms, methods of treatment and additional inhibitor compounds of oncology molecular targets and their derivatives. Specifically, we anticipate that we will seek patent protection in the United States and internationally for novel compositions of matter covering the compounds, the chemistries and processes for manufacturing these compounds, the use of these compounds in a variety of therapies and the use of biomarkers for patient selection for these compounds. However, these or other patent applications that we may file or license from third parties may not result in the issuance of patents, and any issued patents may cover limited claims that reduce their value and/or may be challenged, invalidated or circumvented. See “Risk Factors—Rights Related to Our Intellectual Property.”
In addition to patents, we hold three trademarks in the United States, for Ignyta®, Methylome® and Trailblaze®, and have two trademark applications pending in the United States for Oncolome™ and Actagene™. We also rely upon unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our collaborators, scientific advisors, employees and consultants, and invention assignment agreements with our employees and selected consultants, scientific advisors and collaborators. The confidentiality agreements are designed to protect our proprietary information and, in the case of agreements or clauses requiring invention assignment, to grant us ownership of technologies that are developed through a relationship with a third-party.
With respect to our proprietary DNA methylation analysis platform, we consider trade secrets and know-how to be a critical component of our intellectual property. Trade secrets and know-how can be difficult to protect. In particular, with respect to this technology platform we anticipate that these trade secrets and know-how will over time be disseminated within the industry through independent development, the publication of journal articles describing the methodology and the movement of personnel skilled in the art from academic to industry scientific positions. As a result, those proprietary trade secrets and know-how may lose their value to us over a period of time, and we may lose any competitive advantage afforded by them as they become public knowledge.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, import and export of pharmaceutical products, such as those we are developing.
United States Drug Approval Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (FDCA), and implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applying company to a variety of administrative or judicial sanctions.
19
Table of Contents
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests, animal studies and formulation studies in compliance with GLP regulations; |
| • | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| • | approval by an independent institutional review board (IRB) at each clinical site before each trial may be initiated; |
| • | performance of adequate and well-controlled human clinical trials in accordance with good clinical practices (GCP) to establish the safety and efficacy of the proposed drug for each indication; |
| • | submission to the FDA of an NDA; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices (CGMP) requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
| • | FDA review and approval of the NDA. |
Preclinical Studies and IND
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as in vitro and animal studies to assess the potential for adverse events and, in some cases, to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including GLP regulations for safety/toxicology studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some long-term preclinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the trial on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include, among other things, the requirement that all research subjects provide their informed consent in writing before their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the safety and effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination.
Human clinical trials are typically conducted in the following three sequential phases, which may overlap or be combined:
| • | Phase I: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
| • | Phase II: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| • | Phase III: The drug is administered to an expanded patient population in adequate and well-controlled clinical trials to generate sufficient data to statistically confirm the efficacy and safety of the product for approval for specified indications, to establish the overall risk-benefit profile of the product and to provide adequate information for the labeling of the product. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and, more frequently, if serious adverse events occur. Phase I, Phase II and Phase III clinical trials may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any
20
Table of Contents
time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing Approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. Under federal law, the submission of most NDAs is additionally subject to a substantial application user fee.
The FDA generally conducts a preliminary review of all NDAs within the first 60 days after submission before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information in connection with this preliminary review rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is subject to further review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA has agreed to specified performance goals in the review of NDAs. Under these goals, the FDA has committed to review most such applications for non-priority products within 10 months, and most applications for priority review products, that is, drugs that the FDA determines represent a significant improvement over existing therapy, within six months. The review process may be extended by the FDA for three additional months to consider certain information or clarification regarding information already provided in the submission. The FDA may also refer applications for novel drugs or products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA is not required to adhere its review time goals, and its review could experience delays that cause those goals to not be met.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and are adequate to assure consistent production of the product within required specifications. In addition, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP and integrity of the clinical data submitted.
The testing and approval process for each product candidate requires substantial time, effort and financial resources, and each may take many years to complete. Data obtained from preclinical and clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval of an application for a product candidate on a timely basis, or at all. Further, applicants often encounter difficulties or unanticipated costs in their efforts to develop product candidates and secure necessary governmental approvals, which could delay or preclude the marketing of those products.
After the FDA’s evaluation of the NDA and inspection of the manufacturing facilities, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA may then issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval and refuse to approve the NDA.
Post-Market Drug Regulation
If the FDA approves a drug product for commercial marketing, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase IV clinical trials, be conducted to further assess a drug’s safety and/or other factors after approval, require testing and surveillance programs to monitor the product after commercialization and/or patients using the product for observation of the product’s long-term effects, or impose other conditions, including distribution restrictions or other risk management mechanisms, including Risk Evaluation and Mitigation Strategies (REMs), which can materially affect
21
Table of Contents
the potential market and profitability of the product. Any approved product is also subject to requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion, labeling, and reporting of adverse experiences with the product. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and re-approval.
In addition, drug manufacturers with which we partner and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon drug developers and their manufacturers. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, imposition of post-market studies or clinical trials to assess new safety risks or imposition of distribution or other restrictions under a REMs program. Other potential consequences of a failure to comply with regulatory requirements during or after the FDA approval process include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| • | fines, warning letters or holds on post-approval clinical trials; |
| • | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; or |
| • | consent decrees, injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off label uses, and a company that is found to have improperly promoted off label uses may be subject to significant liability.
Programs for Expedited Approval
The FDA has developed certain programs and designations that enable NDAs for product candidates meeting specified criteria to be eligible for certain expedited approval processes such as the fast track designation, priority review, accelerated approval, and breakthrough therapy designation. Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
Fast Track Designation. The FDA is required to facilitate the development and expedite the review of drugs that are intended for the treatment of a serious or life-threatening condition for which there is no effective treatment and that demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new drug candidate may request the FDA to designate the product for a specific indication as a fast track product concurrent with or after the filing of the IND for the product candidate. The FDA must determine if the product candidate qualifies for fast track designation within 60 days after receipt of the sponsor’s request.
In addition to other benefits, such as the ability to use surrogate endpoints (see the description of surrogate endpoints under “—Accelerated Approval” below) and have greater interactions with the FDA, the FDA may initiate review of sections of a fast track product’s NDA before the application is complete. This rolling review is available if the applicant provides and the FDA approves a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s review time goal for a fast track application does not begin until the last section of the NDA is submitted. In addition, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
22
Table of Contents
Priority Review. Under FDA policies, a product candidate may be eligible for priority review, or review generally within a six-month timeframe from the time a complete application is received. Products regulated by the FDA’s Center for Drug Evaluation and Research (CDER) are eligible for priority review if they provide a significant improvement compared to marketed products in the treatment, diagnosis or prevention of a disease. A fast track designated product candidate would ordinarily meet the FDA’s criteria for priority review.
Accelerated Approval. Under the FDA’s accelerated approval regulations, the FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit. In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase IV or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, would allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Breakthrough Therapy Designation. Under the provisions of the new Food and Drug Administration Safety and Innovation Act (FDASIA) enacted in 2012, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Alternative Approval Pathways
In addition to the expedited approval programs and designations, the FDA also recognizes certain other designations and alternative approval pathways that afford certain benefits over filing a traditional NDA, such as the orphan drug designation and alternative types of NDAs under the Hatch-Waxman Act.
Orphan Drugs. Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally defined as a disease or condition that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the drug and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the United States for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same orphan indication, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity in that it is shown to be safer, more effective or makes a major contribution to patient care. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
The Hatch-Waxman Act: Abbreviated New Drug Applications. In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s product or a method of using the product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an abbreviated new drug application (ANDA). Generally, an ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths, dosage form and route of administration as the listed drug and has been shown to be bioequivalent through in vitro or in vivo testing or otherwise to the listed drug. ANDA applicants are not required to conduct or submit results of preclinical or clinical tests to prove the safety or effectiveness of their drug product, other than the requirement for bioequivalence testing. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can be and are often substituted by pharmacists under prescriptions written for the original listed drug.
23
Table of Contents
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book, except for patents covering methods of use for which the ANDA applicant is not seeking approval. Specifically, the applicant must certify with respect to each patent that:
| • | the required patent information has not been filed; |
| • | the listed patent has expired; |
| • | the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or |
| • | the listed patent is invalid, unenforceable or will not be infringed by the new product. |
A certification that the new product will not infringe the already approved product’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the applicant does not challenge the listed patents or indicate that it is not seeking approval of a patented method of use, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days after the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA also will not be approved until any applicable non-patent exclusivity period, such as exclusivity for obtaining approval of a new chemical entity, for the referenced product has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active part of any molecule (moiety) during which ANDAs for generic versions of those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original product approval. Federal law provides for a period of three years of exclusivity during which the FDA cannot grant effective approval of an ANDA if a listed drug contains a previously approved active moiety, but FDA requires as a condition of approval new clinical trials conducted by or for the sponsor. This three-year exclusivity period often protects changes to a previously approved drug product, such as a new dosage form, route of administration, combination or indication. Under the Best Pharmaceuticals for Children Act, federal law also provides that periods of patent and non-patent marketing exclusivity listed in the Orange Book for a drug may be extended by six months if the NDA sponsor conducts pediatric studies identified by the FDA in a written request. If such a written request is issued by the FDA, the FDA must grant pediatric exclusivity no later than six months prior to the date of expiration of patent or non-patent exclusivity in order for the six-month pediatric extension to apply to that exclusivity period.
The Hatch-Waxman Act: Section 505(b)(2) New Drug Applications. Most drug products obtain FDA marketing approval pursuant to an NDA or an ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the FDA’s previous approval of a similar product, or published literature, in support of its application.
505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. If the 505(b)(2) applicant can establish that reliance on the FDA’s previous approval is scientifically appropriate, it may eliminate the need to conduct certain preclinical or clinical studies of the new product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. As a result, approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
24
Table of Contents
Combination Products
The FDA regulates combinations of products that cross FDA centers, such as drug, biologic or medical device components that are physically, chemically or otherwise combined into a single entity, as a combination product. The FDA center with primary jurisdiction for the combination product will take the lead in the premarket review of the product, with the other center consulting or collaborating with the lead center.
The FDA’s Office of Combination Products (OCP) determines which center will have primary jurisdiction for the combination product based on the combination product’s “primary mode of action.” A mode of action is the means by which a product achieves an intended therapeutic effect or action. The primary mode of action is the mode of action that provides the most important therapeutic action of the combination product, or the mode of action expected to make the greatest contribution to the overall intended therapeutic effects of the combination product.
Often it is difficult for the OCP to determine with reasonable certainty the most important therapeutic action of the combination product. In those difficult cases, the OCP will consider consistency with other combination products raising similar types of safety and effectiveness questions, or which center has the most expertise to evaluate the most significant safety and effectiveness questions raised by the combination product.
A sponsor may use a voluntary formal process, known as a Request for Designation, when the product classification is unclear or in dispute, to obtain a binding decision as to which center will regulate the combination product. If the sponsor objects to that decision, it may request that the agency reconsider the decision.
FDA Regulation of Companion Diagnostics
We may seek to develop, or seek to partner with third parties to develop in vitro and in vivo companion diagnostics for use in selecting the patients that we believe will respond to our drug therapeutics.
FDA officials have issued draft guidance that, when finalized, would address issues critical to developing in vitro companion diagnostics, such as biomarker qualification, establishing clinical validity, the use of retrospective data, the appropriate patient population and when the FDA will require that the device and the drug be approved simultaneously. The draft guidance issued in July 2011 states that if safe and effective use of a therapeutic product depends on an in vitro diagnostic, then the FDA generally will require approval or clearance of the diagnostic at the same time that the FDA approves the therapeutic product. The FDA has yet to issue further guidance regarding these matters, and it is unclear whether it will do so or what the scope of any additional guidance would be.
The FDA previously has required in vitro companion diagnostics intended to select the patients who will respond to a cancer treatment to obtain pre-market approval (PMA) simultaneously with approval of the drug.
PMA Approval Pathway
A medical device, including an in vitro diagnostic (IVD) to be commercially distributed in the United States must receive either 510(k) clearance or PMA approval from the FDA prior to marketing. There are three classes of medical devices recognized by the FDA, Class I (the least regulated), Class II, and Class III (the most regulated), and devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life supporting or implantable devices, or devices deemed not substantially equivalent to a previously 510(k) cleared device or a pre-amendment Class III device for which PMA applications have not been called, are placed in Class III requiring PMA approval. The PMA approval pathway requires proof of the safety and effectiveness of the device to the FDA’s satisfaction.
A PMA application for an IVD must provide extensive preclinical and clinical trial data. Preclinical data for an IVD includes many different tests, including how reproducible the results are when the same sample is tested multiple times by multiple users at multiple laboratories. The clinical data need to establish that the test is sufficiently safe, effective and reliable in the intended use population. In addition, the FDA must be convinced that a device has clinical utility, meaning that an IVD provides information that is clinically meaningful. A biomarker’s clinical significance may be obvious, or the applicant may be able to rely upon published literature or submit data to the FDA to show clinical utility.
A PMA application also must provide information about the device and its components regarding, among other things, device design, manufacturing and labeling. The sponsor must pay an application fee to the FDA upon submission of a PMA.
25
Table of Contents
As part of the PMA review, the FDA will typically inspect the manufacturer’s facilities for compliance with Quality System Regulation (QSR) requirements, which impose elaborate testing, control, documentation and other quality assurance procedures.
Upon submission, the FDA determines if the PMA application is sufficiently complete to permit a substantive review, and, if so, the FDA accepts the application for filing. The FDA then commences an in-depth review of the PMA application. The entire process typically takes one to three years from submission of the PMA, but may take longer. The review time is often significantly extended as a result of the FDA asking for more information or clarification of information already provided. The FDA also may respond with a not approvable determination based on deficiencies in the application and require additional clinical trials that are often expensive and time-consuming to conduct and can substantially delay approval.
During the review period, an FDA advisory committee, typically a panel of clinicians, may be convened to review the PMA application and recommend to the FDA whether, or upon what conditions, the device should be approved. Although the FDA is not bound by the advisory panel decision, the panel’s recommendation is important to the FDA’s overall decision-making process.
If the FDA’s evaluation of the PMA application is favorable, the FDA typically issues an approvable letter requiring the applicant’s agreement to specific conditions, such as changes in labeling, or specific additional information, such as submission of final labeling, in order to secure final approval of the PMA. If the FDA concludes that the applicable criteria have been met, the FDA will issue a PMA for the approved indications, which can be more limited than those originally sought by the applicant. The PMA can include post-approval conditions that the FDA believes necessary to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution. Failure to comply with the conditions of approval can result in an enforcement action, including the loss or withdrawal of the approval.
Even after approval of a PMA, a new PMA or PMA supplement may be required in the event of a modification to the device, its labeling or its manufacturing process. Supplements to a PMA often require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to the information needed to support the proposed change from the product covered by the original PMA.
Clinical Trials and IDEs
A clinical trial is almost always required to support a PMA application. In some cases, one or more smaller Investigational Device Exemption (IDE) studies may precede a pivotal clinical trial intended to demonstrate the safety and efficacy of the investigational device.
All clinical studies of investigational devices must be conducted in compliance with the FDA’s requirements. If an investigational device could pose a significant risk to patients pursuant to FDA regulations, the FDA must approve an IDE application prior to initiation of investigational use. IVD trials usually do not require an IDE, as the FDA does not judge them to be a significant risk because the results do not affect the patients in the study. However, for a trial where the IVD result directs the therapeutic care of patients with cancer, we believe that the FDA may consider the investigation to present significant risk and require an IDE application.
An IDE application must be supported by appropriate data, such as laboratory test results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The FDA typically grants IDE approval for a specified number of patients. A non-significant risk device does not require FDA approval of an IDE. Both significant risk and non-significant risk investigational devices require approval from IRBs at the study centers where the device will be used.
During the critical trial, the sponsor must comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting and record keeping. The investigators must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices and comply with all reporting and record keeping requirements. Prior to granting PMA approval, the FDA typically inspects the records relating to the conduct of the study and the clinical data supporting the PMA application for compliance with applicable requirements.
Although the QSR does not fully apply to investigational devices, the requirement for controls on design and development does apply. The sponsor also must manufacture the investigational device in conformity with the quality controls described in the IDE application and any conditions of IDE approval that the FDA may impose with respect to manufacturing.
26
Table of Contents
Post-Market Device Regulation
After a device obtains FDA approval and is on the market, numerous regulatory requirements apply. These requirements include the QSR, labeling regulations, the FDA’s general prohibition against promoting products for unapproved or “off label” uses, the Medical Device Reporting regulation, which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur, and the Reports of Corrections and Removals regulation, which requires manufacturers to report recalls and field actions to the FDA if initiated to reduce a risk to health posed by the device or to remedy a violation of the FDCA.
The FDA enforces these requirements by inspection and market surveillance. If the FDA finds a violation, it can institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions such as fines, injunctions and civil penalties; recall or seizure of products; operating restrictions, partial suspension or total shutdown of production; refusing requests for PMA approval of new products; withdrawing PMA approvals already granted; and criminal prosecution.
Foreign Regulation
To obtain marketing approval of a drug under European Union regulatory systems, we may submit marketing authorization applications (MAAs) either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The centralized procedure is compulsory for medicines produced by specified biotechnological processes, products designated as orphan medicinal products, and products with a new active substance indicated for the treatment of specified diseases, and optional for those products that are highly innovative or for which a centralized process is in the interest of patients. Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Scientific Advice Working Party of the Committee of Medicinal Products for Human Use (CHMP). Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, defined by three cumulative criteria of the seriousness of the disease, such as heavy disabling or life-threatening diseases, to be treated; the absence or insufficiency of an appropriate alternative therapeutic approach; and anticipation of high therapeutic benefit. In this circumstance, the European Medicines Agency (EMA) ensures that the opinion of the CHMP is given within 150 days.
The EMA grants orphan drug designation to promote the development of products that may offer therapeutic benefits for life-threatening or chronically debilitating conditions affecting not more than five in 10,000 people in the European Union. In addition, orphan drug designation can be granted if the drug is intended for a life threatening, seriously debilitating or serious and chronic condition in the European Union and without incentives it is unlikely that sales of the drug in the European Union would be sufficient to justify developing the drug. Orphan drug designation is only available if there is no other satisfactory method approved in the European Union of diagnosing, preventing or treating the condition, or if such a method exists, the proposed orphan drug will be of significant benefit to patients. Orphan drug designation provides opportunities for free protocol assistance, fee reductions for access to the centralized regulatory procedures before and during the first year after marketing authorization and between six and 10 years of market exclusivity following drug approval.
The decentralized procedure for submitting an MAA provides an assessment of an application performed by one member state, known as the reference member state, and the approval of that assessment by one or more other member states, known as concerned member states. Under this procedure, an applicant submits an application, or dossier, and related materials, including a draft summary of product characteristics, and draft labeling and package leaflet, to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state cannot approve the assessment report and related materials on the grounds of potential serious risk to public health, the disputed points may eventually be referred to the European Commission, whose decision is binding on all member states. Prior to submitting an MAA for use of drugs in pediatric populations the EMA requires submission of, or a request for waiver or deferral of, a Pediatric Investigation Plan.
In the European Union, new chemical entities qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. This data exclusivity, if granted, prevents regulatory authorities in the
27
Table of Contents
European Union from assessing a generic (abbreviated) application for eight years, after which generic marketing authorization can be submitted but not approved for two years. Even if a compound is considered to be a new chemical entity and the sponsor is able to gain the prescribed period of data exclusivity, another company nevertheless could also market another version of the drug if such company can complete a full MAA with a complete human clinical trial database and obtain marketing approval of its product.
Additional Regulations and Environmental Matters
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws, which generally will not be applicable to us or our product candidates unless and until we obtain FDA marketing approval for any of our product candidates, include anti-kickback statutes, false claims statutes and regulation regarding providing drug samples.
The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. Violations of the federal anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws.
The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Additionally, the Prescription Drug Marketing Act (PDMA) imposes requirements and limitations upon the provision of drug samples to physicians, as well as prohibits states from licensing distributors of prescription drugs unless the state licensing program meets certain federal guidelines that include minimum standards for storage, handling and record keeping. In addition, the PDMA sets forth civil and criminal penalties for violations. If we obtain approval from the FDA to market any of our drug product candidate, these product sampling restrictions may impact and curtail our marketing efforts to physicians.
Further, sales of any of our product candidates that may be approved will depend, in part, on the extent to which the cost of the product will be covered by third party payors. Third party payors may limit coverage to an approved list of products, or formulary, which might not include all drug products approved by the FDA for an indication. Any product candidates for which we obtain marketing approval may not be considered medically necessary or cost-effective by third party payors, and we may need to conduct expensive pharmacoeconomic studies in the future to demonstrate the medical necessity and/or cost effectiveness of any such product. The U.S. government, state legislatures and foreign governments have shown increased interest in implementing cost containment programs to limit government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Continued interest in and adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals such as the drug candidates we are developing.
In addition to regulatory schemes that apply, or may in the future apply, to our business, we are or may become subject to various environmental, health and safety laws and regulations governing, among other things, laboratory procedures and any use and disposal by us of hazardous or potentially hazardous substances in connection with our research and development activities. We do not presently expect such environmental, health and safety laws or regulations to materially impact our present or planned future activities.
28
Table of Contents
Our Scientific Advisors
We have assembled a world-class scientific advisory board that includes renowned experts in oncology, autoimmune disease, epigenetics, drug discovery and translational medicine. These advisors work in close collaboration with our scientific and drug discovery team to identify new research directions and accelerate our target validation and drug discovery programs.
| Name |
Primary Affiliation | |
| James Bristol, Ph.D. | — | |
| Sai-Hong Ignatius Ou, M.D., Ph.D. | University of California, Irvine | |
| Daniel D. Von Hoff, M.D., F.A.C.P. | Translational Genomics Research Institute | |
| Mary “Peggy” Crow, M.D. | The Hospital for Special Surgery | |
| Gary Firestein, M.D. | University of California, San Diego | |
| V. Michael Holers, M.D. | University of Colorado | |
| Tom Huizinga, M.D. | Leiden University Medical Center | |
| Bruce Richardson, M.D., Ph.D. | University of Michigan | |
| Wei Wang, Ph.D. | University of California, San Diego |
Employees
As of the date of this report, we have six full-time employees, including four employees with M.D. or Ph.D. degrees, and two part-time employees. Of these full-time and part-time employees, five employees are engaged in research and development activities. Patrick O’Connor, our Chief Scientific Officer, went on medical leave as of September 2013, and we expect him to return in January 2014. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
Facilities
We occupy approximately 3,945 rentable square feet of office and laboratory space in San Diego, California under a lease that expires in August 2016 and provides for our monthly rent payment of approximately $7,350, which amount will increase beginning in March 2014 to approximately $9,941 per month and by additional amounts in the years thereafter, and an additional 1,841 rentable square feet of laboratory space in San Diego, California under a lease that expires in November 2014 and provides for our monthly rent payment of approximately $3,774. We believe that our facilities are sufficient to meet our current needs and that suitable additional space will be available as and when needed.
Legal Proceedings
Neither we nor our subsidiaries are currently a party to, nor is our property the subject of, any material legal proceedings.
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, together with the other information contained in this report, including our financial statements and the related notes attached as exhibits, before making any decision to invest in shares of our common stock. If any of the events discussed in the risk factors below occurs, our business, operations, financial condition and cash flows could be materially harmed. If that were to happen, the trading price of our common stock could decline, and you could lose all or part of your investment.
Risks Related to Our Financial Position and Capital Requirements
We have incurred significant losses since our inception and anticipate that we will continue to incur losses for the foreseeable future. We are a clinical-stage company with no approved products, and have generated no revenue to date and may never generate revenue or achieve profitability.
We are a clinical-stage biopharmaceutical company with a limited operating history. We have not generated any revenue to date and are not profitable, and have incurred losses in each year since our inception in August 2011. Our net loss for the
29
Table of Contents
year ended December 31, 2012 and for the six months ended June 30, 2013 was $1.3 million and $2.2 million, respectively. As of June 30, 2013, we had an accumulated deficit of $3.5 million. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for, our product candidates, and begin to commercialize any approved products. We are currently focused primarily on the development of our in-licensed clinical and preclinical product candidates RXDX-101 and RXDX-102 and our discovery stage programs Spark-1, Spark-2 and Spark-3, which we believe will result in our continued incurrence of significant research and development and other expenses related to those programs. If the clinical trials for any of our products fail or produce unsuccessful results and those product candidates do not gain regulatory approval, or if any of our product candidates, if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our stockholders’ equity and working capital.
We will need substantial additional funding to continue our operations. We may not be able to raise capital when needed, if at all, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts and could cause our business to fail.
Our operations have consumed substantial amounts of cash since inception. We expect to need substantial additional funding to pursue the clinical development of our product candidates and launch and commercialize any product candidates for which we receive regulatory approval, which may include building internal sales and marketing forces to address certain markets.
We expect our existing cash and cash equivalents will be sufficient to fund our capital requirements for at least the next two months. We will require additional capital for the further development and commercialization of our product candidates and may need to raise additional funds sooner if we choose to expand more rapidly than we currently anticipate. Further, we expect our expenses to increase in connection with our ongoing activities, particularly as we continue the ongoing Phase I/II clinical trial of RXDX-101 and prepare for and initiate a Phase I clinical trial of RXDX-102, and continue research and development and initiate additional clinical trials of, and seek regulatory approval for, these and other product candidates. In addition, if we obtain regulatory approval for any of our product candidates, we expect to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution. Furthermore, upon the closing of the Merger, we expect to incur additional costs associated with operating as a public company. We may also encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may increase our capital needs and/or cause us to spend our cash resources faster than we expect. Accordingly, we will need to obtain substantial additional funding in order to continue our operations. As noted in our unaudited financial statement for the six months ended June 30, 2013, the uncertainties surrounding our ability to fund our operations raise substantial doubt about our ability to continue as a going concern.
To date, we have financed our operations entirely through investments by founders and other investors and the incurrence of debt, and we expect to continue to do so in the foreseeable future. We may also seek funding through collaborative arrangements. Additional funding from those or other sources may not be available when or in the amounts needed, on acceptable terms, or at all. If we raise capital through the sale of equity, or securities convertible into equity, it would result in dilution to our then existing stockholders, which could be significant depending on the price at which we may be able to sell our securities. If we raise additional capital through the incurrence of indebtedness, we would likely become subject to covenants restricting our business activities, and holders of debt instruments may have rights and privileges senior to those of our equity investors. In addition, servicing the interest and principal repayment obligations under debt facilities could divert funds that would otherwise be available to support research and development, clinical or commercialization activities. If we obtain capital through collaborative arrangements, these arrangements could require us to relinquish rights to our technology or product candidates and could result in our receipt of only a portion of the revenues associated with the partnered product.
If we are unable to raise capital when needed or on attractive terms, we could be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts. Any of these events could significantly harm our business, financial condition and prospects.
Our short operating history may hinder our ability to successfully meet our objectives, and may limit the amount of information about us upon which you can base an evaluation of our business and prospects.
Ignyta Operating’s operations commenced in August 2011. Our initial focus was on the discovery and development of biomarkers and molecular and companion diagnostic tests for certain autoimmune diseases. Only since May 2013 have we refocused our efforts on precision medicines for the treatment of cancers. Consequently, we have limited experience and
30
Table of Contents
have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. Further, the early stage nature of our business results in a limited operating history upon which you can evaluate our business and prospects. Our lead product candidates are in the earliest stages of development, have not obtained regulatory marketing approval, have never generated any sales and will require extensive testing before commercialization. Our limited operating history may adversely affect our ability to implement our business strategy and achieve our business goals, which include, among others, the following activities:
| • | develop our product candidates using unproven technologies; |
| • | obtain the human and financial resources necessary to develop, test, manufacture and market our product candidates; |
| • | engage corporate partners to assist in developing, testing, manufacturing and marketing our product candidates; |
| • | continue to build and maintain an intellectual property portfolio covering our technology and our product candidates; |
| • | satisfy the requirements of clinical trial protocols, including patient enrollment, establish and demonstrate the clinical efficacy and safety of our product candidates and obtain necessary regulatory approvals; |
| • | market our product candidates that receive regulatory approvals to achieve acceptance and use by the medical community in general; |
| • | maintain, grow and manage our internal teams as and to the extent we increase our operations and develop new segments of our business; |
| • | develop and maintain successful collaboration, strategic and other relationships for the development and commercialization of our product candidates and those of our partners that receive regulatory approvals; and |
| • | manage our cash flows and any growth we may experience in an environment where costs and expenses relating to clinical trials, regulatory approvals and commercialization continue to increase. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop product candidates, raise capital, expand our business or continue our operations.
Risks Related to our Employees
If we are not able to attract and retain highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and pharmaceuticals industries depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our management, scientific and medical personnel, especially Jonathan Lim, our President, Chief Executive Officer and Chairman of the Board, and Patrick O’Connor, our Senior Vice President, Research and Chief Scientific Officer, whose services are critical to the successful implementation of our product candidate development and regulatory strategies. As of the date of this report, Patrick O’Connor is on medical leave and is expected to return in early 2014. Further, as our approach is built in part upon the drug discovery and development experience of our scientific “drug hunter” team, which we believe is a significant contributor to our competitive advantage, we are dependent on the maintenance and growth of that team with qualified members containing high levels of expertise in specific scientific fields.
We are not aware of any present intention of any of our executive officers or other members of management to leave our company, but our industry tends to experience a high rate of turnover of management personnel and our personnel are generally able to terminate their relationships with us on short notice. All of our employment arrangements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. Additionally, several members of our scientific team are consultants rather than employees, and could terminate their consulting relationship with us at any time or with short notice, depending on the terms of their respective consulting agreements with us. The loss of the services of any of our executive officers or other key employees and our inability to find suitable replacements could potentially harm our business, financial condition and prospects. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior and mid-level managers as well as junior and mid-level scientific and medical personnel.
Moreover, there is intense competition for a limited number of qualified personnel among biopharmaceutical, biotechnology, pharmaceutical and other businesses. Many of the other pharmaceutical companies against which we compete for qualified personnel have greater financial and other resources, different risk profiles, longer histories in the
31
Table of Contents
industry and greater ability to provide valuable cash or stock incentives to potential recruits than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than what we are able to offer as an early stage company. If we are unable to continue to attract and retain high quality personnel, the rate and success at which we can develop and commercialize product candidates will be limited.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, with contractual provisions and other procedures, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employers. Litigation may be necessary to defend against any such claims.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact contributes to the development of intellectual property that we regard as our own. Further, the terms of such assignment agreements may be breached and we may not be able to successfully enforce their terms, which may force us to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of intellectual property rights we may regard and treat as our own.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause our business to suffer.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA or EMA regulations, provide accurate information to the FDA or EMA, comply with manufacturing standards we have established, comply with federal, state and international healthcare fraud and abuse laws and regulations as they become applicable to our operations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we currently take and the procedures we may establish in the future as our operations and employee base expand to detect and prevent this type of activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure by our employees to comply with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
Risks Related to the Discovery and Development of Our Product Candidates
We will not obtain development rights to our two lead product candidates until we satisfy certain financing conditions.
In October 2013, we entered into a license agreement with NMS that grants us exclusive rights to commercialize our two lead product candidates, RXDX-101 and RXDX-102. The effectiveness of the license agreement is conditioned upon the completion of a financing of equity or debt securities pursuant to which we or our affiliates receive gross proceeds of at least $20 million. If the required financing has not been completed by December 31, 2013, then the license agreement will not become effective and will automatically terminate. As of the date of this report, neither we nor any of our affiliates have contractual obligations or other firm commitments for any future funding toward the satisfaction of that financing condition. As is further described in the other risks set forth under the heading “Risk Factors,” funding may not be available to us when or in the amounts needed, on acceptable terms, or at all. As a result, the license agreement with NMS may never become effective and we may never have any rights to pursue either of our two proposed lead product candidates. If that were to occur, our business plans would require material modification and our business, prospects and operations would be materially harmed. Further, we cannot commence any development plans or activities relating to those
32
Table of Contents
lead product candidates unless and until the financing condition set forth in the license agreement is satisfied and the license agreement becomes effective. As a result, if we are not able to quickly obtain the required funding, then our development objectives would experience delays and our operations would suffer.
We are heavily dependent on the success of our two early-stage lead product candidates, both of which will require significant additional efforts to develop and may prove not to be viable for commercialization.
To date, we have invested significant efforts in the acquisition of our two lead product candidates. Our future success is substantially dependent on our ability to successfully develop, obtain regulatory approval for, and then successfully commercialize these two product candidates. One of our product candidates, RXDX-101, is in clinical trials, while our second product candidate, RXDX-102, is in preclinical development. Our business depends entirely on the successful development, clinical testing and commercialization of these and any other product candidates we may seek to develop in the future, which may never occur.
Before we could generate any revenues from sales of our lead product candidates, we must complete the following activities for each of them, any one of which we may not be able to successfully complete:
| • | conduct substantial additional clinical development; |
| • | manage clinical, preclinical and manufacturing activities; |
| • | achieve regulatory approval in multiple jurisdictions; |
| • | establish manufacturing relationships for the supply of the applicable product candidate; |
| • | build a commercial sales and marketing team, if we choose to market any such product ourselves; |
| • | develop and implement marketing strategies; |
| • | develop and/or work with third-party collaborators to develop companion diagnostics and conduct clinical testing and achieve regulatory approvals for those companion diagnostics; and |
| • | invest significant additional cash in each of the above activities. |
If the results of the ongoing RXDX-101 Phase I/II clinical trial are not successful, we may not be able to use those results as the basis for advancing the product candidate into further clinical development. In that case, we may not have the resources to conduct new clinical trials, and/or we may determine that further clinical development of this product candidate is not justified and may decide to discontinue the program. Clinical testing of RXDX-102 has not yet commenced, and the results of any future preclinical or clinical studies, if unsuccessful, could lead to our abandonment of the development of that product candidate as well. If studies of these product candidates produce unsuccessful results and we are forced or elect to cease their development, our business and prospects would be substantially harmed.
We may not be able to pursue further clinical development of our lead product candidates, or such development may be delayed, if NMS does not provide to us on a timely basis its data, technology and know-how related to those product candidates, or if we experience challenges assuming control of ongoing testing and studies.
All development to date of RXDX-101 and RXDX-102 has been conducted by their licensor, NMS. In connection with our license of the rights to develop those product candidates, NMS has agreed that it will, over a period of time, transfer to us all data, technology and know-how relating to those product candidates and that is reasonably necessary for their continued development, including detailed information regarding the performance and results of the preclinical and clinical studies conducted to date. However, NMS may not provide that material to us on a timely basis within the terms of the license agreement and/or the material NMS provides may be incomplete. We will not be able to assess the development work conducted to date and solidify our prospective development plans for these product candidates until we have obtained, processed and reviewed that material. Additionally, in connection with the transition of any ongoing studies and trials for these product candidates from NMS’s control to our control, we may seek to engage a third party CRO to pursue all further development efforts. However, we will not be able to engage a CRO to pursue those further development activities, including the continuation of the Phase I/II clinical trial for RXDX-101, until necessary data and technology have been provided by NMS. Further, NMS’s failure to provide complete information about its development activities to date could create challenges in connection with our preparation and submission of any future regulatory filings relating to these product candidates. These potential data and technology transfer and other transition challenges could result in increased costs to us, and could cause us to experience delays in, or be forced to discontinue, the further development of our lead product candidates, which would materially harm our business and operational results.
33
Table of Contents
Preclinical and clinical testing of our lead product candidates that has been conducted to date may not have been performed in compliance with applicable regulatory standards, which could lead to increased costs or material delays for their further development.
We have only recently entered a license agreement relating to our lead product candidates, and the development to date of those product candidates has been conducted wholly by NMS or any third parties with which it has contracted. As a result, we have not been involved with nor have we had any control over any of those development activities. Although NMS has provided certain information regarding those product candidates and the related preclinical and clinical studies conducted to date, including certain data that is included in this report, we have not received and do not yet have access to comprehensive information regarding those development activities, including the raw data from all studies that have been conducted, information regarding their design, procedural implementation and structure and information regarding the manufacture of the product candidate used in the studies. Because we have had no input on NMS’ development to date of these product candidates, we may discover that all or certain elements of the trials and studies it has performed have not been in compliance with applicable regulatory standards or have otherwise been deficient, and that advancement of the development of these product candidates on the basis of those trials and studies is not warranted. Further, the development of each of our lead product candidates to date has been conducted only in Europe. As a result, although those studies may meet the standards of applicable European regulatory bodies, the structure and design of those clinical and preclinical studies may not meet applicable FDA standards to allow immediate further development of those product candidates in the United States, and also may not meet the standards of any regulatory authority in any foreign country in which we desire to pursue marketing approval for these product candidates. As of the date of this report, although we are in the process of receiving data regarding, and assuming control of, those studies, we do not yet have access to sufficient information to fully assess the standard and design of prior clinical and preclinical work. If the studies conducted to date have not been in compliance with applicable regulatory standards or are otherwise not eligible for continued development in the United States, then we may be forced to conduct new studies in order to progress their development, which we may not have the funding or other resources to complete and which would severely delay our development plans for these product candidates. Any such deficiency in the prior development of these product candidates would significantly harm our business plans and prospects.
Our research and development is based on a rapidly evolving area of science, and our approach to drug discovery and development is novel and may never lead to marketable products.
Biopharmaceutical product development is generally a highly speculative undertaking and by its nature involves a substantial degree of risk. The specific line of our business, the discovery of personalized drug therapeutics for patients with molecularly defined cancers, is an emerging field, and the scientific discoveries that form the basis for our efforts to develop product candidates are relatively new. Further, the scientific evidence to support the feasibility of developing product candidates based on those discoveries is both preliminary and limited. Although epigenetic regulation of gene expression plays an essential role in biological function, very few drugs premised on epigenetics have been discovered. Moreover, drugs based on an epigenetic mechanism that have received marketing approval are not targeted to differentially methylated genes, which is the focus of our epigenetic research and development. Although research suggests that differential methylation of certain genes can cause them to drive particular human cancers, we are not aware of any company that has translated these biological observations into a drug that has received marketing approval. As a result, identifying drug targets based in part on differential gene methylation, which is a fundamental aspect of our business approach, may not lead to the discovery or development of any drugs that successfully treat patients with molecularly defined cancers. The failure of the scientific underpinnings of our business model to produce viable product candidates would substantially harm our operations and prospects.
We may not be successful in our efforts to build a pipeline of product candidates.
A key element of our strategy is to use and expand our product platform to build a pipeline of small molecule inhibitors of genetically and epigenetically altered targets, and progress those product candidates through clinical development for the treatment of a variety of different types of cancer. Although our research efforts to date have resulted in identification of a series of genetically or epigenetically altered cancer drug targets, we may not be able to develop product candidates that are safe and effective inhibitors of all or any of these targets. Even if we are successful in building a pipeline, the potential product candidates that we identify may not be suitable for clinical development for a number of reasons, including causing harmful side effects or demonstrating other characteristics that indicate a low likelihood of receiving marketing approval and achieving market acceptance. If our methods of identifying potential product candidates fail to produce a pipeline of potentially viable drug candidates, then our success as a business will be dependent on the success of fewer potential product candidates, which introduces risks to our business model and potential limitations to our success.
34
Table of Contents
Clinical drug development involves a lengthy and expensive process with uncertain outcomes, and any of our clinical trials or studies could produce unsuccessful results or fail at any stage in the testing process.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Additionally, any positive results of preclinical studies and early clinical trials of a product candidate may not be predictive of the results of later-stage clinical trials, such that product candidates may reach later stages of clinical trials and fail to show the desired safety and efficacy traits despite having shown indications of those traits in preclinical studies and initial clinical trials. For example, although the preclinical and early clinical results for our lead product candidates have been positive, those results and the results that may be generated in the ongoing Phase I/II clinical trial for RXDX-101 do not imply that later clinical trials will demonstrate similar results. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. The results of any future clinical trials we conduct may not be successful.
Although there is a clinical trial ongoing for RXDX-101, of which we will commence the process of assuming control upon the effectiveness of our license agreement with NMS, and although we are planning to initiate clinical trials for RXDX-102 as early as 2014, we may experience delays in pursuing those or any other clinical or preclinical studies, and any planned clinical trials may not begin on time, may require redesign, may not enroll sufficient patients in a timely manner, and may not be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays related to:
| • | obtaining regulatory approval to commence a trial; |
| • | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| • | obtaining IRB approval at each trial site; |
| • | recruiting suitable patients to participate in a trial; |
| • | developing and validating companion diagnostics on a timely basis; |
| • | changes in dosing or administration regimens; |
| • | having patients complete a trial or return for post-treatment follow-up; |
| • | clinical sites deviating from trial protocol or dropping out of a trial; |
| • | regulators instituting a clinical hold due to observed safety findings; |
| • | adding new clinical trial sites; or |
| • | manufacturing sufficient quantities of product candidate for use in clinical trials. |
We plan to rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials. Although we expect that we will have agreements in place with CROs governing their committed activities and conduct, we will have limited influence over their actual performance. As a result, we ultimately will not have control over a CRO’s compliance with the terms of any agreement it may have with us, its compliance with applicable regulatory requirements, or its adherence to agreed time schedules and deadlines, and a future CRO’s failure to perform those obligations could subject any of our clinical trials to delays or failure.
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by the Data Safety Monitoring Board (DSMB) for the trial, if applicable, or by the FDA, EMA or other regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, EMA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. If we experience delays in the completion of, or suspension or termination of, any clinical trial for our product candidates, the commercial prospects of the product candidate will be harmed, and our ability to generate product revenues from the product candidate will be delayed or eliminated. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. The occurrence of any of these events could harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of the applicable product candidate.
35
Table of Contents
If we experience delays or difficulties in the enrollment of patients in clinical trials, those clinical trials could take longer than expected to complete and our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials for our product candidates if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States. In particular, because we are focused on patients with molecularly defined cancers, our pool of suitable patients may be smaller and more selective and our ability to enroll a sufficient number of suitable patients may be limited or take longer than anticipated. For example, NMS’ enrollment for the Phase I/II clinical trial of RXDX-101 has been slow because of delays in recruiting suitable patients. In addition, some of our competitors have ongoing clinical trials for product candidates that treat the same indications as our product candidates, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ product candidates.
Patient enrollment for any of our clinical trials may also be affected by other factors, including without limitation:
| • | the severity of the disease under investigation; |
| • | the frequency of the molecular alteration we are seeking to target in the applicable trial; |
| • | the eligibility criteria for the study in question; |
| • | the perceived risks and benefits of the product candidate under study; |
| • | the extent of the efforts to facilitate timely enrollment in clinical trials; |
| • | the patient referral practices of physicians; |
| • | the ability to monitor patients adequately during and after treatment; and |
| • | the proximity and availability of clinical trial sites for prospective patients. |
Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs for our product candidates, and we may not have or be able to obtain sufficient cash to fund such increased costs when needed, which could result in the further delay or termination of the trial.
Consistent with our general product development strategy, we intend to design the Phase II aspect of the ongoing Phase I/II clinical trial of RXDX-101, the planned Phase I clinical trial of RXDX-102 and any future trials for those or other product candidates to include some patients with the applicable molecular alteration that causes the disease, with a view to assessing possible early evidence of potential therapeutic effect. If we are unable to locate and include such patients in those trials, then our ability to make those early assessments and to seek participation in FDA expedited review and approval programs, including breakthrough therapy and fast track designation, or otherwise to seek to accelerate clinical development and regulatory timelines, could be compromised.
The approval processes of regulatory authorities are lengthy, time consuming, expensive and inherently unpredictable. If we are unable to obtain approval for our product candidates from applicable regulatory authorities, we will not be able to market and sell those product candidates in those countries or regions and our business will be substantially harmed.
The time required to obtain approval by the FDA, EMA and comparable foreign authorities is unpredictable, but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not submitted an NDA or similar filing or obtained regulatory approval for any product candidate in any jurisdiction and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval for many reasons, including any one or more of the following:
| • | the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| • | we may be unable to demonstrate to the satisfaction of the FDA, EMA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication; |
36
Table of Contents
| • | the results of clinical trials may not meet the level of statistical significance required by the FDA, EMA or comparable foreign regulatory authorities for approval; |
| • | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| • | the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| • | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| • | the FDA, EMA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; |
| • | the FDA, EMA or comparable foreign regulatory authorities may fail to approve the companion diagnostics we contemplate developing internally or with partners; and |
| • | the approval policies or regulations of the FDA, EMA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
The time and expense of the approval process, as well as the unpredictability of future clinical trial results and other contributing factors, may result in our failure to obtain regulatory approval to market, in one or more jurisdictions, RXDX-101, RXDX-102, our discovery stage Spark-1 – Spark-6 programs, or any other product candidates we may seek to develop in the future, which would significantly harm our business, results of operations and prospects.
In order to market and sell our products in any jurisdiction, we or our third party collaborators must obtain separate marketing approvals in that jurisdiction and comply with its regulatory requirements. The approval procedure can vary drastically among countries, and each jurisdiction may impose different testing and other requirements to obtain and maintain marketing approval. Further, the time required to obtain those approvals may differ substantially among jurisdictions. In addition, in many countries outside the United States, it is required that the product be approved for reimbursement before the product can be approved for sale in that country. Moreover, approval by the FDA or an equivalent foreign authority does not ensure approval by regulatory authorities in any other countries or jurisdictions. As a result, the ability to market and sell a product candidate in more than one jurisdiction can involve the significant additional time, expense and effort to undertake separate approval processes, and would subject us and our collaborators to the numerous and varying post-approval requirements of each jurisdiction governing commercial sales, manufacturing, pricing and distribution of our product candidates. We or any third parties with whom we may collaborate may not have the resources to pursue those approvals, and we or they may not be able to obtain any approvals that are pursued. The failure to obtain marketing approval for our product candidates in foreign jurisdictions could severely limit their potential market and ability to generate revenue.
In addition, even if we were to obtain regulatory approval in one or more jurisdictions, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for our products, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing circumstances could materially harm the commercial prospects for our product candidates.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
To date, patients treated with RXDX-101 have experienced some adverse events, which have been predominantly gastrointestinal or constitutional in nature. While we have not yet initiated clinical trials for RXDX-102, as is the case with many oncology drugs, it is likely that there may be side effects associated with its use. Results of our trials for these or other product candidates could reveal a high and unacceptable severity and frequency of these or other side effects. In such an event, our trials could be suspended or terminated and the FDA, EMA or comparable foreign regulatory authorities could order us to cease further development of, or deny approval of, our product candidates for any or all targeted indications. Further, any observed drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial, or result in potential product liability claims. Any of these occurrences may materially harm our business, financial condition and prospects.
Additionally, if one or more of our product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
37
Table of Contents
| • | regulatory authorities may withdraw approvals of such product; |
| • | regulatory authorities may require additional warnings on the product’s label; |
| • | we may be required to create a medication guide for distribution to patients that outlines the risks of such side effects; |
| • | we could be sued and held liable for harm caused to patients; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Failure to successfully validate, develop and obtain regulatory approval for companion diagnostics could harm our drug development strategy and operational results.
As one of the central elements of our business strategy and clinical development approach, we seek to identify molecularly-defined subsets of patients within a disease category that may derive selective and meaningful benefit from the product candidates we are developing. We anticipate that the development of companion diagnostics concurrently with our drug candidates will help us more accurately identify the patients that belong to the target subset, both during our clinical trials and in connection with the commercialization of our product candidates. We do not plan to internally commercialize, or seek regulatory approval of, companion diagnostics and, as a result, we will likely rely on third party collaborators to successfully commercialize companion diagnostics. To date, we have not developed relationships with any such third-party collaborators to develop companion diagnostics for any of our product candidates. We may not be able to establish arrangements with any such third-party collaborators for the development and production of companion diagnostics when needed or on terms that are beneficial to us, or at all, which could negatively affect our development efforts with respect to our drug product candidates and materially harm our business, operations and prospects.
Companion diagnostics are subject to regulation by the FDA and comparable foreign regulatory authorities as medical devices and require separate regulatory approval prior to their commercialization. We are likely to be dependent on the sustained cooperation and effort of any third-party collaborators with whom we may partner in the future to develop and obtain approval for these companion diagnostics. We and our potential future collaborators may encounter difficulties in developing and obtaining approval for these companion diagnostics, including issues relating to the selectivity and/or specificity of the diagnostic, analytical validation, reproducibility, or clinical validation. Any delay or failure by us or our potential future collaborators to develop or obtain regulatory approval of any companion diagnostics could delay or prevent approval of our related product candidates. In addition, our potential future collaborators may encounter production difficulties that could constrain the supply of the companion diagnostics, and we or they may experience difficulties gaining acceptance of the use of the companion diagnostics in the clinical community. In addition, the third parties with whom we may contract to develop and produce companion diagnostics could decide to discontinue selling or manufacturing the companion diagnostic, and we may not be able to enter into arrangements with other parties to obtain supplies of alternative diagnostic tests on a timely basis or reasonable terms, or at all. The occurrence of any such event could adversely affect and/or delay the development or commercialization of our product candidates.
We may expend our limited resources to pursue a particular product candidate or indication that does not produce any commercially viable products and may fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must focus our efforts on particular research programs and product candidates for specific indications. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Further, our resource allocation decisions may result in our use of funds for research and development programs and product candidates for specific indications that may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. Any such failure to improperly assess potential product candidates could result in missed opportunities and/or our focus on product candidates with low market potential, which would harm our business and financial condition.
38
Table of Contents
We may not be able to obtain orphan drug exclusivity for the product candidates for which we seek it, which could limit the potential profitability of such product candidates.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals annually in the United States. Generally, if a product with an orphan drug designation subsequently receives the first marketing approval for the indication for which it receives the designation, then the product is entitled to a period of marketing exclusivity that precludes the applicable regulatory authority from approving another marketing application for the same drug for the exclusivity period.
We expect that we may in the future pursue an orphan drug designation for at least some of our product candidates. However, obtaining an orphan drug designation can be difficult and we may not be successful in doing so for any of our product candidates. Even if we were to obtain orphan drug exclusivity for a product candidate, that exclusivity may not effectively protect the product from the competition of different drugs for the same condition, which could be approved during the exclusivity period. Additionally, after an orphan drug is approved, the FDA could subsequently approve another application for the same drug for the same condition if the FDA concludes that the later drug is shown to be safer, more effective or makes a major contribution to patient care. The failure to obtain an orphan drug designation for any drug candidates we may develop for the treatment of rare cancers, and/or the inability to maintain that designation for the duration of the applicable exclusivity period, could reduce our ability to make sufficient sales of the applicable drug candidate to balance our expenses incurred to develop it, which would have a negative impact on our operational results and financial condition.
If we seek and obtain a fast track or breakthrough therapy designation by the FDA for any of our product candidates, such designations may not actually lead to a faster development or regulatory review or approval process or any other material benefits.
We may in the future seek fast track designation for some of our product candidates that reach the regulatory review process. If a drug candidate is intended for the treatment of a serious or life-threatening condition and the drug candidate demonstrates the potential to address unmet medical needs for this condition, the sponsor may apply to the FDA for a fast track designation for the drug candidate. The FDA has broad discretion over whether to grant a fast track designation and, as a result, even our product candidates that may be eligible for such a designation may not receive it. Even if we were to receive fast track designation for any of our product candidates, the designation may not result in a materially faster development process, review or approval compared to conventional FDA procedures. Additionally, the FDA could withdraw a fast track designation if it believes that the designation is no longer supported by data from our clinical development program.
Additionally, we may in the future seek a breakthrough therapy designation for some of our product candidates that reach the regulatory review process. A breakthrough therapy is a drug candidate that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and that, as indicated by preliminary clinical evidence, may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies by the FDA are eligible for accelerated approval and increased interaction and communication with the FDA designed to expedite the development and review process.
As with fast track designation, designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and may determine not to grant such a designation. Even if we receive a breakthrough therapy designation for any of our product candidates, the designation may not result in a materially faster development process, review or approval compared to conventional FDA procedures. Further, obtaining a breakthrough therapy designation does not assure or increase the likelihood of the FDA’s approval of the applicable product candidate. In addition, even if one or more of our product candidates qualifies as a breakthrough therapy, the FDA could later determine that those products no longer meet the conditions for the designation or determine not to shorten the time period for FDA review or approval.
As a result, even if a fast track or breakthrough therapy designation is granted for any product candidate for which we seek such designations, we may not experience any material expediting of or noticeable benefit relating to the FDA’s review and approval, which could result in delayed marketing approval of the applicable product candidate and our resulting inability to generate revenues from any sales of the product until later periods.
39
Table of Contents
Risks Related to Our Dependence on Third Parties
We plan to rely on third parties to conduct preclinical and clinical trials of our product candidates. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We plan to rely upon third-party CROs to execute our preclinical and clinical trials and to monitor and manage data produced by and relating to those trials. To date, we have not established relationships with any such third-party CROs for any preclinical or clinical studies, although we expect that we will pursue such relationships in the near future in connection with clinical and preclinical work for our lead product candidates, including continuation of the ongoing Phase I/II clinical trial for RXDX-101. We may not be able to establish arrangements with CROs when needed or on terms that are acceptable to us, or at all, which could negatively affect our development efforts with respect to our drug product candidates and materially harm our business, operations and prospects.
As a result of our planned use of CROs, we will have only limited control over certain aspects of their activities. Nevertheless, we will be responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on any CRO will not relieve us of our regulatory responsibilities. Based on our present expectations, we and our CROs will be required to comply with GCP for all of our product candidates in clinical development. Regulatory authorities enforce GCP through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCP, the clinical data generated in the applicable trial may be deemed unreliable and the FDA, EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving a product candidate for marketing, which we may not have sufficient cash or other resources to support and which would delay our ability to generate revenue from any sales of such product candidate. In addition, we expect that our clinical trials will be required to be conducted with product produced under cGMP regulations. Our failure to comply with those regulations may require us to repeat clinical trials, which would also require significant cash expenditures and delay the regulatory approval process.
Although we currently do not have any relationships in place with CROs, we anticipate that any agreements governing our relationships with CROs we may engage in the future may provide those CROs with certain rights to terminate a clinical trial under specified circumstances. If a CRO we engage in the future terminates its relationship with us during the performance of a clinical trial, we would be forced to seek an engagement with a substitute CRO, which we may not be able to do on a timely basis or on commercially reasonable terms, if at all, and the applicable trial would experience delays or may not be completed. In addition, CROs we engage will not be our employees, and except for remedies available to us under any agreements we enter with them, we will be unable to control whether or not they devote sufficient time and resources to our clinical, nonclinical and preclinical programs. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to a failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for, or successfully commercialize, the affected product candidates. As a result, our operations and the commercial prospects for the effected product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
We plan to rely completely on third parties to manufacture our preclinical and clinical drug supplies and any approved product candidates, and our operations could be harmed if those third parties fail to provide sufficient quantities of product in accordance with applicable regulatory and contractual obligations.
We do not currently have, nor do we plan to acquire, the infrastructure or capability internally to manufacture our preclinical and clinical drug supplies for use in the conduct of our clinical trials or commercial quantities of any product candidates that may obtain regulatory approval. As a result, we expect that we will need to rely completely on third party manufacturers for those services. Other than the limited supply of RXDX-101 and RXDX-102 materials that NMS has agreed to provide to us in connection with recent in-license of the rights to develop those product candidates, we have no commitments from any third-party manufacturer for the supply of any of our product candidates for use in preclinical and clinical trials. We expect that we will seek to establish contractual arrangements with third-party manufacturers, which could include NMS or any other third party, for that purpose in the near future, but we may not be able to establish those relationships when needed, on reasonable terms, or at all. Any failure to secure sufficient supply of our product candidates for clinical testing or, in the future, commercial purposes would materially harm our operations and financial results.
40
Table of Contents
We expect that the facilities to be used by any contract manufacturers we engage to manufacture our product candidates will be required to be approved by the FDA pursuant to inspections in connection with its regulatory approval process. We will not control the manufacturing process of, and will be dependent on, our contract manufacturing partners for compliance with cGMP for the manufacture of clinical and, if regulatory approval is obtained, commercial quantities of our product candidates. In addition, we expect to have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If any of our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, EMA or other comparable foreign authorities, we would be prevented from obtaining regulatory approval for our product candidates unless and until we engage a substitute contract manufacturer that can comply with such requirements, which we may not be able to do. Any such failure by any of our contract manufacturers would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We expect to rely on our manufacturers to purchase from third-party suppliers the materials necessary to produce our product candidates for our clinical trials. We do not have, nor do we expect to enter, any agreements for the commercial production of these raw materials, and we do not expect to have any control over the process or timing of our manufacturers’ acquisition of raw materials needed to produce our product candidates. Any significant delay in the supply of a product candidate or the raw material components thereof for an ongoing clinical trial due to a manufacturer’s need to replace a third-party supplier of raw materials could considerably delay completion of our clinical trials, product testing and potential regulatory approval of our product candidates. Additionally, if our future manufacturers or we are unable to purchase these raw materials to commercially produce any of our product candidates that gains regulatory approvals, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of our product candidates.
Risks Related to the Commercialization of Our Product Candidates
Even if we receive regulatory approval for any of our product candidates, we will be subject to ongoing regulatory obligations and review. Maintaining compliance with ongoing regulatory requirements may result in significant additional expense to us, and any failure to maintain such compliance could subject us to penalties and cause our business to suffer.
Any regulatory approvals that we receive for our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed, or contain requirements for potentially costly post-marketing testing, including Phase IV clinical trials, and surveillance to monitor the safety and efficacy of the product candidate. In addition, if the FDA, EMA or a comparable foreign regulatory authority approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP for all manufacture of the applicable product and GCP for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| • | fines, warning letters or holds on post-approval clinical trials; |
| • | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; and |
| • | consent decrees, injunctions or the imposition of civil or criminal penalties. |
The FDA’s or EMA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are otherwise not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
41
Table of Contents
We currently have no marketing and sales force. If we are unable to establish effective marketing and sales capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to effectively market and sell our product candidates, if approved, or generate product revenues.
We currently do not have a marketing or sales team for the marketing, sales and distribution of any of our product candidates that are able to obtain regulatory approval. In order to commercialize any product candidates, we must build on a territory-by-territory basis marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If our product candidates receive regulatory approval, we intend to establish an internal sales and marketing team with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time consuming and will require significant attention of our executive officers to manage. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of any of our products that we obtain approval to market. With respect to the commercialization of all or certain of our product candidates, we may choose to collaborate, either globally or on a territory-by-territory basis, with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements when needed on acceptable terms or at all, we may not be able to successfully commercialize any of our product candidates that receive regulatory approval or any such commercialization may experience delays or limitations. If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
Our commercial success depends upon attaining significant market acceptance of our product candidates, if approved, among physicians, patients, healthcare payors and major operators of cancer clinics.
Even if we obtain regulatory approval for our product candidates, the products may not gain market acceptance among physicians, health care payors, patients and the medical community, which is critical to commercial success. Market acceptance of any product candidate for which we receive approval depends on a number of factors, including:
| • | the efficacy and safety as demonstrated in clinical trials; |
| • | the timing of market introduction of the product candidate, any associated companion diagnostic, and/or competitive products; |
| • | the clinical indications for which the drug is approved; |
| • | the approval, availability, market acceptance and reimbursement for any companion diagnostic; |
| • | the ability of a companion diagnostic to successfully identify all tested patients that harbor the underlying molecular alteration that our product targets; |
| • | acceptance of the drug as a safe and effective treatment by physicians, major operators of cancer clinics and patients; |
| • | the size of the markets for the product candidate, based on the size of the patient subsets that we are targeting, in the territories for which we gain regulatory approval and have commercial rights; |
| • | the potential and perceived advantages of the product candidate over alternative treatments, especially with respect to patient subsets that we are targeting with the product candidate; |
| • | the safety of the product candidate as demonstrated through broad commercial use including, potentially, under conditions not tested in clinical trials; |
| • | the cost of treatment in relation to alternative treatments; |
| • | the availability of adequate reimbursement and pricing by third-party payors and government authorities; |
| • | relative convenience and ease of administration; |
| • | the prevalence and severity of adverse side effects; and |
| • | the effectiveness of our sales, marketing and distribution efforts. |
If our product candidates are approved but fail to achieve an adequate level of acceptance by key market participants, we will not be able to generate significant revenues, and we may not become or remain profitable.
We face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. In addition, the competition in the oncology market is intense. We have competitors both in the
42
Table of Contents
United States and internationally, including major multinational pharmaceutical companies, biotechnology companies and universities and other research institutions.
With respect to our two lead product candidates, we are aware of one agent that has been approved by the FDA for ALK-positive NSCLC, which is Pfizer’s Xalkori®/crizotinib, and we are aware of several other products in development targeting TrkA, ROS1 and/or ALK for the treatment of cancer, some of which may be in a more advanced stage of development than RXDX-101. There are also many other compounds directed to other molecular targets that are in clinical development by a variety of companies to treat cancer types that we may choose to pursue with RXDX-101 or RXDX-102.
Many of our competitors have substantially greater financial, technical and other resources than we do, such as larger research and development staff and experienced marketing and manufacturing organizations. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in certain of our competitors. As a result, these companies may be able to obtain regulatory approval more rapidly than we can and may be more effective in selling and marketing their products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing drug products that are more effective or less costly to produce or purchase on the market than any drug candidate we are currently developing or that we may seek to develop in the future. If approved, our product candidates will face competition from commercially available drugs as well as drugs that are in the development pipelines of our competitors.
Established pharmaceutical companies may invest heavily to accelerate discovery and development of or in-license novel compounds that could make our product candidates less competitive. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA, EMA or other regulatory approval, or discovering, developing and commercializing medicines before we do, which would have a material adverse impact on our business and ability to achieve profitability from future sales of our approved product candidates, if any.
Reimbursement may be limited or unavailable in certain market segments for our product candidates, which could make it difficult for us to sell on a profitable basis any products for which we obtain marketing approvals.
There is significant uncertainty related to the third-party coverage and reimbursement of newly approved drugs. Market acceptance and sales of any of our product candidates that obtain regulatory approval in domestic or international markets will depend significantly on the availability of adequate coverage and reimbursement from third-party payors for any of our product candidates, and may be affected by existing and future healthcare reform measures.
Pricing and reimbursement for any of our approved product candidates is uncertain. Government authorities and other third-party payors decide which drugs they will pay for and establish reimbursement levels for them, and obtaining coverage and reimbursement approval for a product from any such third-party payor is a time consuming and costly process. Adoption of our product candidates by the medical community may be limited if doctors, patients and other key market participants do not receive adequate partial or full reimbursement for our approved products, if any. As a result, any denial of private or government payor coverage or inadequate reimbursement for use of our product candidates, if any are commercialized, could harm our business and reduce our prospects for generating revenue.
Further, there have been, and may continue to be, legislative and regulatory proposals at the federal and state levels and in foreign jurisdictions directed at broadening the availability and containing or lowering the cost of healthcare. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare may adversely affect our ability to set prices for our products that would
43
Table of Contents
allow us to achieve or sustain profitability. In addition, governments may impose price controls on any of our products that obtain marketing approval, which may adversely affect our future profitability.
In some foreign countries, particularly in the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can be a long and expensive process after the receipt of marketing approval for a product candidate. To obtain reimbursement or pricing approval in some countries, we may be required to conduct additional clinical trials that compare the cost-effectiveness of our product candidates to other available therapies. If reimbursement of our product candidates is unavailable or limited in scope or amount in a particular country, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability for sales of any of our product candidates that are approved for marketing in that country.
We could be subject to product liability lawsuits based on the use of our product candidates in clinical testing or, if obtained, following marketing approval and commercialization. If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to cease clinical testing or limit commercialization of our product candidates.
We could be subject to product liability lawsuits if any product candidate we develop allegedly causes injury or is found to be otherwise unsuitable for human use during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates, if approved. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| • | decreased demand for our product candidates; |
| • | injury to our reputation; |
| • | withdrawal of clinical trial participants; |
| • | initiation of investigations by regulators; |
| • | costs to defend the related litigation; |
| • | a diversion of management’s time and our resources; |
| • | substantial monetary awards to trial participants or patients; |
| • | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| • | loss of revenues from product sales; and |
| • | the inability to commercialize our product candidates. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the clinical testing and commercialization of products we develop. We do not currently carry product liability insurance. We may seek to obtain such insurance after we assume control of clinical trials of RXDX-101 from NMS, but we may not be able to obtain the levels of coverage desired on acceptable terms, or at all. If we do secure product liability insurance, we may subsequently determine that additional amounts of coverage would be desirable at later stages of clinical development of our product candidates or upon commencing commercialization of any product candidate that obtains required approvals, but we may not be able to obtain such additional coverage amounts when needed on acceptable terms, or at all. Unless and until we obtain such insurance, we would be solely responsible for any product liability claims relating to our preclinical and clinical development activities. Further, even after any such insurance coverage is obtained, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by any insurance policies we may then have or that is in excess of the limits of our insurance coverage. We would be required to pay any amounts awarded by a court or negotiated in a settlement that exceed the coverage limitations or that are not covered by any product liability insurance we may obtain, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
44
Table of Contents
Risks Related to Our Intellectual Property
If we breach any of the agreements under which we license from third parties the commercialization rights to our product candidates, we could lose license rights that are important to our business and our operations could be materially harmed.
Upon the effectiveness of our license agreement with NMS, we will in-license from NMS the use, development and commercialization rights for our two lead product candidates, RXDX-101 and RXDX-102. As a result, our current business plans are dependent upon our satisfaction of certain conditions to the effectiveness of that agreement, and thereafter upon our maintenance of that agreement and the rights we license under it. Our license agreement with NMS provides that it will not become effective until the completion of a financing of equity or debt securities pursuant to which we or our affiliates receive gross proceeds of at least $20 million. If the required financing has not been completed by December 31, 2013, then the license agreement will not become effective and will automatically terminate. Additionally, the license agreement provides that, following its effectiveness, we are subject to diligence obligations relating to the commercialization and development of RXDX-101 and RXDX-102, milestone payments, royalty payments and other obligations. In addition to our license agreement with NMS, we may seek to enter into additional agreements with other third parties in the future granting similar license rights with respect to other potential product candidates. If we fail to comply with any of the conditions or obligations or otherwise breach the terms of our license agreement with NMS, or any future license agreement we may enter on which our business or product candidates are dependent, NMS or other licensors may have the right to terminate the applicable agreement in whole or in part and thereby extinguish our rights to the licensed technology and intellectual property and/or any rights we have acquired to develop and commercialize certain product candidates, including, with respect to our license agreement with NMS, RXDX-101 and RXDX-102. The loss of the rights licensed to us under our license agreement with NMS, or any future license agreement that we may enter granting rights on which our business or product candidates are dependent, would eliminate our ability to further develop the applicable product candidates and would materially harm our business, prospects, financial condition and results of operations.
If our efforts to protect the proprietary nature of the intellectual property related to our technologies are not adequate, we may not be able to compete effectively in our market and our business would be harmed.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our technologies. Any disclosure to or misappropriation by third parties of our proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding any competitive advantage we may derive from the proprietary information.
The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. The patent applications we own or license may fail to result in issued patents in the United States or in foreign countries. Third parties may challenge the validity, enforceability or scope of any issued patents we own or license or any applications that may successfully issue in the future, which may result in those patents being narrowed, invalidated or held unenforceable. Even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from developing similar products that do not infringe the claims made in our patents. If the breadth or strength of protection provided by the patents we hold or pursue is threatened, our ability to commercialize any product candidates with technology protected by those patents could be threatened. Further, if we encounter delays in our clinical trials, the period of time during which we would have patent protection for any covered product candidates that obtain regulatory approval would be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, we cannot be certain at the time of filing that we are the first to file any patent application related to our product candidates.
The license agreement with NMS will grant us an exclusive, worldwide license under a portfolio of patents and patent applications directed to the RXDX-101 and RXDX-102 composition of matter, which begin to expire in 2028 for the patents and applications relating to RXDX-101 and in 2027 for the patents and applications relating to RXDX-102. While patent term extensions under the Hatch-Waxman Act in the United States and under supplementary protection certificates in Europe may be available to extend our patent exclusivity for either RXDX-101 or RXDX-102, the applicable patents may not meet the specified conditions for eligibility for any such term extension and, even if eligible, we may not be able to obtain any such term extension. Further, because filing, prosecuting and defending patents in multiple jurisdictions can be expensive, we may elect to pursue patent protection relating to RXDX-101 and RXDX-102 or any other product candidates we may pursue in only certain jurisdictions. As a result, competitors would be permitted to use our technologies in jurisdictions where we have not obtained patent protection to develop their own products, any of which could compete with our product candidates.
45
Table of Contents
In addition to the protection afforded by patents, we seek to rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our discovery platform and drug development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees and certain consultants and advisors to assign inventions to us, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, our trade secrets and other proprietary information may be disclosed or competitors may otherwise gain access to such information or independently develop substantially equivalent information. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant difficulty in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain the competitive advantage that we believe is provided by such intellectual property, which could materially adversely affect our market position and business and operational results.
Claims that we infringe the intellectual property rights of others may prevent or delay our drug discovery and development efforts.
Our research, development and commercialization activities, as well as any product candidates or products resulting from those activities, may infringe or be accused of infringing a patent or other form of intellectual property under which we do not hold a license or other rights. Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents of which we are currently unaware with claims that cover the use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If our activities or product candidates infringe the patents or other intellectual property rights of third parties, the holders of such intellectual property rights may be able to block our ability to commercialize such product candidates unless we obtain a license under the intellectual property rights or until any applicable patents expire or are determined to be invalid or unenforceable.
Defense of any intellectual property infringement claims against us, regardless of their merit, would involve substantial litigation expense and would be a significant diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, obtain one or more licenses from third parties, limit our business to avoid the infringing activities, pay royalties and/or redesign our infringing product candidates, any or all of which may be impossible or require substantial time and monetary expenditure. Further, if we were to seek a license from the third party holder of any applicable intellectual property rights, we may not be able to obtain the applicable license rights when needed or on reasonable terms, or at all. The occurrence of any of the above events could prevent us from continuing to develop and commercialize one or more of our product candidates and our business could materially suffer.
We may desire to, or be forced to, seek additional licenses to use intellectual property owned by third parties, and such licenses may not be available on commercially reasonable terms or at all.
A third party may hold intellectual property, including patent rights, that are important or necessary to the development of our product candidates, in which case we would need to obtain a license from that third party or develop a different formulation of the product that does not infringe upon the applicable intellectual property, which may not be possible. Additionally, we may identify product candidates that we believe are promising and whose development and other intellectual property rights are held by third parties. In such a case, we may desire to seek a license to pursue the development of those product candidates, as we have done with RXDX-101 and RXDX-102. Any license that we may desire to obtain or that we may be forced to pursue may not be available when needed on commercially reasonable terms or at all. Any inability to secure a license that we need or desire could have a material adverse effect on our business, financial condition and prospects.
The patent protection covering some of our product candidates may be dependent on third parties, who may not effectively maintain that protection.
While we intend to, and expect that we will, seek and gain the right to fully prosecute any patents covering product candidates we may in-license from third-party owners, there may in the future be instances when platform technology patents that cover our product candidates remain controlled by our licensors. If any of our future licensing partners retain
46
Table of Contents
the right to prosecute patents covering the product candidates we license from them and fail to appropriately maintain that patent protection, we may not be able to prevent competitors from developing and selling competing products and our ability to generate revenue from any commercialization of the affected product candidates may suffer.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our potential licensors. To attempt to stop infringement or unauthorized use, we may need to file infringement claims, which can be expensive and time-consuming and distract management. If we pursue any infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the relevant technology on the grounds that our patents do not cover the technology in question. Further, the legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, which could reduce the likelihood of success of any infringement proceeding we pursue in any such jurisdiction. An adverse result in any infringement litigation or defense proceedings could put one or more of our patents at risk of being invalidated, held unenforceable, or interpreted narrowly and could put our patent applications at risk of not issuing, which could limit the ability of our product candidates to compete in those jurisdictions.
Interference proceedings provoked by third parties or brought by the United States Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to use it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms, or at all. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We currently, and expect in the future to continue to, seek to protect these trade secrets, in part, by entering into confidentiality agreements with parties who have access to them, such as our employees, collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for any such disclosure. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they disclose the trade secrets, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
Risks Related to Managing Any Growth We May Experience
We will need to grow the size of our organization, and we may experience difficulties in managing any growth we may achieve.
As of the date of this report, we have six full-time employees. As our development and commercialization plans and strategies develop, we expect to need additional research, development, managerial, operational, sales, marketing, financial, accounting, legal and other resources. Future growth would impose significant added responsibilities on members of management, including:
| • | effectively managing our clinical trials and submissions to regulatory authorities for marketing approvals; |
| • | effectively managing our discovery research and preclinical development; |
| • | identifying, recruiting, maintaining, motivating and integrating additional employees; |
| • | effectively managing our internal development efforts; |
| • | establishing relationships with third parties essential to our business and ensuring compliance with our contractual obligations to such third parties; |
47
Table of Contents
| • | developing and managing new divisions of our internal business, including any sales and marketing segment we elect to establish; |
| • | maintaining our compliance with public company reporting and other obligations, including establishing and maintaining effective internal control over financial reporting and disclosure controls and procedures; and |
| • | improving our managerial, development, operational and finance systems. |
We may not be able to accomplish any of those tasks, and our failure to do so could prevent us from effectively managing future growth, if any, and successfully growing our company.
We may in the future be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If we are unable to comply with any such laws, we could face substantial penalties.
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations may be directly, or indirectly through our customers, subject to various federal and state fraud and abuse laws, including, without limitation, anti-kickback and false claims statutes. These laws may impact, among other things, any sales, marketing and education programs we may develop in the future and the manner in which we implement any of those programs. In addition, we may be subject to federal and state patient privacy regulations, such as the federal Health Insurance Portability and Accountability Act of 1996 (HIPAA). If our operations are found to be in violation of any of those laws or any other governmental regulations that may apply to us in connection with marketing and sales of any product candidates that may gain regulatory approval, we may be subject to penalties, including civil and criminal penalties, damages, fines and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial condition.
If we fail to comply with environmental, health and safety laws and regulations that apply to us, we could become subject to fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of any hazardous materials we use and wastes we produce. The use of these materials in our business could result in contamination or injury, which could cause damage for which we may be responsible but may not have sufficient resources to pay. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with these laws and regulations, which we may not be able to afford.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts or impact the research activities we pursue, particularly with respect to research involving human subjects or animal testing. Our failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions, which could cause our financial condition to suffer.
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be limited or eliminated as a result of the Merger.
We have incurred substantial losses during our history and do not expect to become profitable in the foreseeable future and may never achieve profitability. To the extent we continue to generate taxable losses, unused losses will carry forward to offset future taxable income, if any, until such unused losses expire. Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-ownership change net operating loss carryforwards and other pre-ownership change tax attributes to offset its post-ownership change income may be limited. We expect that we will experience an ownership change as a result of the Merger, and as a result may lose some or all of the benefit of our net operating loss carryforwards. As of December 31, 2012, we had federal and state net
48
Table of Contents
operating loss carryforwards of approximately $1.3 million that could be limited or eliminated if the Merger is an ownership change, or if we experience any other ownership change, which could have an adverse effect on our results of operations.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and we may incur substantial costs to attempt to recover or reproduce the data. If any disruption or security breach resulted in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and/or the further development of our product candidates could be delayed.
Our operations are vulnerable to interruption by natural disasters, power loss, terrorist activity and other events beyond our control, the occurrence of which could materially harm our business.
Businesses located in California have, in the past, been subject to electrical blackouts as a result of a shortage of available electrical power, and any future blackouts could disrupt our operations. We are vulnerable to a major earthquake, wildfire and other natural disasters, and we have not undertaken a systematic analysis of the potential consequences to our business as a result of any such natural disaster and do not have an applicable recovery plan in place. We do not carry any business interruption insurance that would compensate us for actual losses from interruption of our business that may occur, and any losses or damages incurred by us could cause our business to materially suffer.
Risks Related to the Merger and Ownership of our Common Stock
There is not now, and there may never be, an active, liquid and orderly trading market for our common stock, which may make it difficult for you to sell your shares of our common stock.
There is not now, nor has there been since our inception, any trading activity in our common stock or a market for shares of our common stock, and an active trading market for our shares may never develop or be sustained. As a result, investors in our common stock must bear the economic risk of holding those shares for an indefinite period of time. Although our common stock is quoted on the OTC Bulletin Board (OTCBB), an over-the-counter quotation system, trading of our common stock is extremely limited and sporadic and at very low volumes. We do not now, and may not in the future, meet the initial listing standards of any national securities exchange. We presently anticipate that our common stock will continue to be quoted on the OTCBB or another over-the-counter quotation system in the foreseeable future. In those venues, our stockholders may find it difficult to obtain accurate quotations as to the market value of their shares of our common stock, and may find few buyers to purchase their stock and few market makers to support its price. As a result of these and other factors, you may be unable to resell your shares of our common stock at or above the price for which you purchased them, or at all. Further, an inactive market may also impair our ability to raise capital by selling additional equity in the future, and may impair our ability to enter into strategic partnerships or acquire companies or products by using our shares of common stock as consideration.
Our share price is volatile and may be influenced by numerous factors, some of which may be beyond our control.
The trading price of our common stock is likely to be highly volatile, and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this report, these factors include:
| • | The product candidates we seek to pursue, and our ability to obtain rights to develop, commercialize and market those product candidates; |
| • | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
| • | actual or anticipated adverse results or delays in our clinical trials; |
| • | our failure to commercialize our product candidates, if approved; |
| • | unanticipated serious safety concerns related to the use of any of our product candidates; |
| • | adverse regulatory decisions; |
49
Table of Contents
| • | additions or departures of key scientific or management personnel; |
| • | changes in laws or regulations applicable to our product candidates, including without limitation clinical trial requirements for approvals; |
| • | disputes or other developments relating to patents and other proprietary rights and our ability to obtain patent protection for our product candidates; |
| • | our dependence on third parties, including CROs as well as our potential partners that provide us with companion diagnostic products; |
| • | failure to meet or exceed any financial guidance or expectations regarding development milestones that we may provide to the public; |
| • | actual or anticipated variations in quarterly operating results; |
| • | failure to meet or exceed the estimates and projections of the investment community; |
| • | overall performance of the equity markets and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; |
| • | conditions or trends in the biotechnology and biopharmaceutical industries; |
| • | introduction of new products offered by us or our competitors; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| • | our ability to maintain an adequate rate of growth and manage such growth; |
| • | issuances of debt or equity securities; |
| • | sales of our common stock by us or our stockholders in the future, or the perception that such sales could occur; |
| • | trading volume of our common stock; |
| • | ineffectiveness of our internal control over financial reporting or disclosure controls and procedures; |
| • | general political and economic conditions; |
| • | effects of natural or man-made catastrophic events; and |
| • | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the stocks of small-cap biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our common stock.
Our common stock may be a “penny stock.”
Generally, a “penny stock” is an equity security that is not listed on a national securities exchange and has a market price of less than $5.00 per share, subject to specific exceptions. Our common stock presently has, and since our inception has had, no trading activity to support a market price, but historical sales of our common stock have all been at a price per share less than $5.00. As a result, our common stock may be considered to be a penny stock. Regulations imposed by the SEC and other regulatory authorities requiring, among other things, that broker-dealers effecting transactions in a penny stock make certain disclosures to and obtain a written suitability statement from potential purchasers, could restrict the ability of broker-dealers to sell our common stock if it were to be considered a penny stock, which could affect the ability of our stockholders to sell their shares of our stock. In addition, if our common stock continues to be quoted on the OTCBB, then our stockholders may find it difficult to obtain accurate quotations for our stock, and may find few buyers to purchase our stock and few market makers to support its price.
FINRA sales practice requirements may limit a stockholder’s ability to buy and sell our stock.
In addition to rules applicable to “penny stock,” the Financial Industry Regulatory Authority (FINRA) has adopted rules requiring that, in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA has indicated its belief that there is a high probability that speculative low-priced securities will not be suitable for at least some customers. These FINRA requirements make it more difficult for broker-dealers to recommend that at least some of their customers buy our common stock, which may limit the ability of our stockholders to buy and sell our common stock and could have an adverse effect on the market for and price of our shares.
50
Table of Contents
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
Any trading market for our common stock that may develop will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on us or our business. If no securities or industry analysts commence coverage of our company, the trading price for our stock would be negatively affected. If securities or industry analysts initiate coverage, and one or more of those analysts downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.
We may have material liabilities that are not discovered until after the closing of the Merger.
As a result of the Merger, the former business plan and management of Ignyta, previously known as Infinity Oil & Gas Company, have been replaced with the business and management team of Ignyta Operating. Prior to the Merger, there were no relationships or other connections among the businesses or individuals associated with those two entities. As a result, Ignyta may have material liabilities that are not discovered until after the Merger is completed. The Combined Company could experience losses as a result of any such undisclosed liabilities that are discovered following the Merger, which could materially harm our business and financial condition. Although the Merger Agreement contains customary representations and warranties from Ignyta concerning its assets, liabilities, financial condition and affairs, there may be limited or no recourse against Ignyta’s pre-Merger stockholders or principals in the event those representations prove to be untrue. As a result, the stockholders of the Combined Company following the closing of the Merger will bear some, or all, of the risks relating to any such unknown or undisclosed liabilities.
We may be exposed to additional risks as a result of “going public” by means of a reverse merger transaction.
We may be exposed to additional risks because the business of Ignyta Operating has become a public company through a “reverse merger” transaction. There has been increased focus by government agencies on transactions such as the Merger in recent years, and we may be subject to increased scrutiny by the SEC and other government agencies and holders of our securities as a result of the completion of that transaction. Further, since we existed as a “shell company” under applicable rules of the SEC prior to the closing of the Merger on October 31, 2013, we are subject to certain restrictions and limitations for certain specified periods of time relating to potential future issuances of our securities and compliance with applicable SEC rules and regulations. Additionally, our “going public” by means of a reverse merger transaction may make it more difficult for us to obtain coverage from securities analysts of major brokerage firms following the Merger because there may be little incentive to those brokerage firms to recommend the purchase of our common stock. The occurrence of any such event could cause our business or stock price to suffer.
We will incur increased costs associated with, and our management will need to devote substantial time and effort to, compliance with public company reporting and other requirements.
As a public company, and particularly if and after we cease to be an “emerging growth company” or a “smaller reporting company,” we will incur significant legal, accounting and other expenses that Ignyta Operating did not incur as a private company. In addition, the rules and regulations of the SEC and national securities exchanges impose numerous requirements on public companies, including requirements relating to our corporate governance practices, with which we will now need to comply. Further, upon becoming subject to the Exchange Act, we will be required to, among other things, file annual, quarterly and current reports with respect to our business and operating results. Our management and other personnel will need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations, and our efforts and initiatives to comply with those requirements could be expensive.
Ignyta Operating was not subject to requirements to establish, and did not establish, internal control over financial reporting and disclosure controls and procedures prior to the Merger. Our management team and Board of Directors will need to devote significant efforts to maintaining adequate and effective disclosure controls and procedures and internal control over financial reporting in order to comply with applicable regulations, which may include hiring additional legal, financial reporting and other finance staff. Additionally, any of our efforts to improve our internal controls and design, implement and maintain an adequate system of disclosure controls may not be successful and will require that we expend significant cash and other resources.
51
Table of Contents
We have elected under the JOBS Act to delay the adoption of new or revised accounting pronouncements applicable to public companies until such pronouncements are made applicable to private companies.
Under the Jumpstart Our Business Startups Act of 2012 (JOBS Act), an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected to take advantage of this extended transition period. Since we will not be required to comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies, our financial statements may not be comparable to the financial statements of companies that comply with the effective dates of those accounting standards.
Until we register a class of our securities under Section 12 or become subject to Section 15(d) of the Exchange Act, we will be a “voluntary filer.”
We are not currently required under Section 13 or 15(d) of the Exchange Act to file periodic reports with the SEC. We have in the past voluntarily elected to file some or all of these reports to ensure that sufficient information about us and our operations is publicly available to our stockholders and potential investors. Because we are a voluntary filer, we are considered a non-reporting issuer under the Exchange Act. Until we become subject to the reporting rules under the Exchange Act, we are not required to file annual, quarterly or current reports and could cease doing so at any time. Additionally, until we register a class of our securities under Section 12 of the Exchange Act, we are not be subject to the SEC’s proxy rules, and large holders of our capital stock will not be subject to beneficial ownership reporting requirements under Sections 13 or 16 of the Exchange Act and their related rules. As a result, our stockholders and potential investors may not have available to them as much or as robust information as they may have if and when we become subject to those requirements.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Certain of our executive officers, directors and large stockholders own a significant percentage of our outstanding capital stock. Immediately after the closing of the Merger, our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates beneficially own approximately 72.40% of our outstanding voting stock. Accordingly, even after giving effect to the Merger, our directors and executive officers have significant influence over our affairs due to their substantial ownership coupled with their positions on our management team, and have substantial voting power to approve matters requiring the approval of our stockholders. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This concentration of ownership in our Board of Directors and management team and certain other large stockholders may prevent or discourage unsolicited acquisition proposals or offers for our common stock that some of our stockholders may believe is in their best interest.
Shares of our common stock that have not been registered under federal securities laws are subject to resale restrictions imposed by Rule 144, including those set forth in Rule 144(i) which apply to a former “shell company.”
Prior to the closing of the Merger, we were deemed a “shell company” under applicable SEC rules and regulations, because we had no or nominal operations and either no or nominal assets, assets consisting solely of cash and cash equivalents, or assets consisting of any amount of cash and cash equivalents and nominal other assets. Pursuant to Rule 144 (Rule 144), promulgated under the Securities Act of 1933, as amended (the Securities Act), sales of the securities of a former shell company, such as us, under that rule are not permitted until at least 12 months have elapsed from the date on which this report, reflecting our status as a non-shell company, is filed with the SEC. As a result, most of our stockholders will be forced to hold their shares of our common stock for at least that 12-month period before they are eligible to sell those shares, and even after that 12-month period, sales may not be made under Rule 144 unless we and the selling stockholders are in compliance with other requirements of Rule 144. Further, it will be more difficult for us to raise funding to support our operations through the sale of debt or equity securities unless we agree to register such securities under the Securities Act, which could cause us to expend additional time and cash resources. Additionally, our previous status as a shell company could also limit our use of our securities to pay for any acquisitions we may seek to pursue in the future (although none are currently planned). The lack of liquidity of our securities as a result of the inability to sell under Rule 144 for a longer period of time could cause the market price of our securities to decline.
52
Table of Contents
If we issue additional shares of our capital stock in the future, our existing stockholders will be diluted.
Our Amended and Restated Articles of Incorporation authorize the issuance of up to 100,000,000 shares of our common stock and up to 10,000,000 shares of preferred stock with the rights, preferences and privileges that our Board of Directors may determine from time to time. In addition to capital raising activities, which we expect to pursue in order to raise the funding we will need in order to continue our operations, other possible business and financial uses for our authorized capital stock include, without limitation, future stock splits, acquiring other companies, businesses or products in exchange for shares of our capital stock, issuing shares of our capital stock to partners or other collaborators in connection with strategic alliances, attracting and retaining employees by the issuance of additional securities under our equity compensation plans, or other transactions and corporate purposes that our Board of Directors deems are in the best interest of our company. Additionally, shares of our capital stock could be used for anti-takeover purposes or to delay or prevent changes in control or our management. Any future issuances of shares of our capital stock may not be made on favorable terms or at all, they may not enhance stockholder value, they may have rights, preferences and privileges that are superior to those of our common stock, and they may have an adverse effect on our business or the trading price of our common stock. The issuance of any additional shares of our common stock will reduce the book value per share and may contribute to a reduction in the market price of the outstanding shares of our common stock. Additionally, any such issuance will reduce the proportionate ownership and voting power of all of our current stockholders.
Sales of a substantial number of shares of our common stock in the public market, or the perception that such sales could occur, could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after the legal restrictions on resale discussed in this report lapse or after those shares become registered for resale pursuant to an effective registration statement, the trading price of our common stock could decline. Upon the closing of the Merger, a total of 4,923,805 shares of our common stock are outstanding. Of those shares, only approximately 7,336 are currently freely tradable, without restriction, in the public market. Although we have no present intent to file a registration statement for the resale of any of the shares of our common stock that are outstanding as of the closing of the Merger and no holders of such shares presently hold registration rights with respect to those shares, if we elect to pursue the registration of any of those shares under the Securities Act in the future, those shares that become registered would be freely tradable without restriction, except for shares held by our affiliates, and any sales of those shares or any perception in the market that such sales may occur could cause the trading price of our common stock to decline.
In addition, shares of common stock that are either subject to outstanding options or reserved for future issuance under our equity incentive plans will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, Rule 144 and Rule 701 under the Securities Act, and any future registration of such shares under the Securities Act. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans or otherwise, could result in dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors in a prior transaction may be materially diluted by subsequent sales. Additionally, any such sales may result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to those of holders of our common stock.
Pursuant to the Ignyta Plan, which we assumed upon the closing of the Merger from Ignyta Operating, we are authorized to grant future equity awards to our employees, directors and consultants for up to an aggregate of 342,209 shares of our common stock. Additionally, we have assumed upon the closing of the Merger all options previously granted under the Ignyta Plan and outstanding as of the closing of the Merger, which, following the closing of the Merger and giving effect to our and Ignyta Operating’s reverse stock splits, are exercisable for up to 358,986 shares of our common stock. Further, upon the completion of the Merger, warrants to acquire shares of Ignyta Operating’s common stock have converted by their terms into warrants to acquire up to 25,001 shares of Ignyta’s common stock. Any future grants of options, warrants or other securities exercisable or convertible into our common stock, or the exercise or conversion of such shares, and any sales of such shares in the market, could have an adverse effect on the market price of our common stock.
53
Table of Contents
Some provisions of our charter documents and Nevada law may discourage an acquisition of us by others, even if the acquisition may be beneficial to some of our stockholders.
Provisions in our Amended and Restated Articles of Incorporation and Bylaws as in effect upon the closing of the Merger, as well as certain provisions of Nevada law, could make it more difficult for a third-party to acquire us, even if doing so may benefit some of our stockholders. These provisions include the authorization of 10,000,000 shares of “blank check” preferred stock, the rights, preferences and privileges of which may be established and shares of which may be issued by our Board of Directors at its discretion from time to time and without stockholder approval.
Because we are incorporated in Nevada, we may be governed by Nevada’s statutes governing combinations with interested stockholders and control share acquisitions, which may discourage, delay or prevent someone from acquiring us or merging with us, whether or not it is desired by or beneficial to our stockholders. Pursuant to our Amended and Restated Articles of Incorporation and our Bylaws, we have elected not to be governed by Nevada’s laws governing combinations with interest stockholders, and as a result will only be subject to those laws upon a future amendment to the applicable provisions of the Amended and Restated Articles of Incorporation. Under Nevada’s laws governing combinations with interested stockholders, a corporation may not, in general, engage in certain types of business combinations with any beneficial owner of 10% or more of the corporation’s voting shares or an affiliate of the corporation who at any time within two years immediately prior to the date in question was the beneficial owner of 10% or more of the corporation’s voting shares, unless the holder has held the stock for two years or the board of directors approved the beneficial owner’s acquisition of its shares, the board of directors approved the transaction before the beneficial owner acquired its shares, or holders of at least a majority of the outstanding voting power approve the transaction after the beneficial owner acquired its shares. In addition, Nevada’s control share acquisition laws prohibit a purchaser of the shares of an “issuing corporation” from voting those shares, under certain circumstances and subject to certain limitations, after crossing specified threshold ownership percentages, unless the purchaser obtains the approval of the issuing corporation’s disinterested stockholders. As the control share acquisition law only applies to an “issuing corporation,” which is a corporation with 200 or more stockholders of record and at least 100 stockholders of record with addresses in Nevada appearing on the stock ledger of the corporation, we do not presently believe that the control share acquisition laws are applicable to us. However, such control share acquisition laws could become applicable to us in the future, and could have an anti-takeover effect.
Any provision of our Amended and Restated Articles of Incorporation or Bylaws or of Nevada law that is applicable to us that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock in the event that a potentially beneficial acquisition is discouraged, and could also affect the price that some investors are willing to pay for our common stock.
The elimination of personal liability against our directors and officers under Nevada law and the existence of indemnification rights held by our directors, officers and employees may result in substantial expenses.
Our Amended and Restated Articles of Incorporation and our Bylaws eliminate the personal liability of our directors and officers to us and our stockholders for damages for breach of fiduciary duty as a director or officer to the extent permissible under Nevada law. Further, our Amended and Restated Articles of Incorporation and our Bylaws and individual indemnification agreements we have entered with each of our directors and executive officers provide that we are obligated to indemnify each of our directors or officers to the fullest extent authorized by the Nevada law and, subject to certain conditions, advance the expenses incurred by any director or officer in defending any action, suit or proceeding prior to its final disposition. Those indemnification obligations could expose us to substantial expenditures to cover the cost of settlement or damage awards against our directors or officers, which we may be unable to afford. Further, those provisions and resulting costs may discourage us or our stockholders from bringing a lawsuit against any of our current or former directors or officers for breaches of their fiduciary duties, even if such actions might otherwise benefit our stockholders.
We do not intend to pay cash dividends on our capital stock in the foreseeable future.
Other than the cash dividend paid in connection with the Merger, we have never declared or paid any dividends on our common stock and do not anticipate paying any dividends in the foreseeable future. Any future payment of cash dividends in the future would depend on our financial condition, contractual restrictions, solvency tests imposed by applicable corporate laws, results of operations, anticipated cash requirements and other factors and will be at the discretion of the our Board of Directors. Our stockholders should not expect that we will ever pay cash or other dividends on our outstanding capital stock.
54
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes and other financial information attached as exhibits to this report. Some of the information contained in this discussion and analysis or set forth elsewhere in this report, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties as described under the heading “Forward-Looking Statements” elsewhere in this report. You should review the “Risk Factors” section of this report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
All references to “we,” “us,” “our” and “Ignyta Operating” in this discussion and analysis refer solely to Ignyta Operating, Inc., a Delaware corporation formerly known as “Ignyta, Inc.” Ignyta Operating become the wholly owned subsidiary of Ignyta, Inc., a Nevada corporation formerly known as “Infinity Oil & Gas Company” (Ignyta), upon the closing of a merger (the Merger) pursuant to which a wholly owned subsidiary of Ignyta formed solely for the purpose of the Merger merged with and into Ignyta Operating. The Merger is accounted for as a reverse merger and recapitalization, with Ignyta Operating as the acquirer and Ignyta as the acquired company for financial reporting purposes. As a result, the assets and liabilities and the operations that will be reflected in the historical financial statements prior to the Merger will be those of Ignyta Operating and will be recorded at the historical cost basis of Ignyta Operating, and the consolidated financial statements after completion of the Merger will include the assets and liabilities of Ignyta and Ignyta Operating, the historical operations of Ignyta Operating and the operations of the combined enterprise of Ignyta and Ignyta Operating from and after the closing date of the Merger.
Overview
We were incorporated under the laws of the State of Delaware on August 29, 2011 with the name “NexDx, Inc.” We changed our name to “Ignyta, Inc.” on October 8, 2012, and changed our name to “Ignyta Operating, Inc.” in connection with the closing of the Merger. On May 20, 2013, we completed our acquisition of Actagene Oncology, Inc. (Actagene), which merged with and into our company on that date. On October 31, 2013, prior to the closing of the Merger, (i) all then-outstanding shares of each series of our preferred stock were voluntarily converted by the holders thereof into shares of our common stock in accordance with our certificate of incorporation, and (ii) we effected a three-to-one reverse stock split of our issued and outstanding shares of capital stock. All share information in this discussion and analysis relating to our capital stock gives retroactive effect to that reverse stock split. On October 31, 2013, a wholly owned subsidiary of Ignyta merged with and into our company, pursuant to which we have become the wholly owned subsidiary of Ignyta.
We are a precision medicine biotechnology company dedicated to discovering or acquiring, then developing and commercializing, precisely targeted new drugs for cancer patients whose tumors harbor specific molecular alterations. We pursue an integrated drug and diagnostic, or Rx/Dx, strategy, where we anticipate pairing each of our drug candidates with biomarker-based companion diagnostics, developed by us or by third parties with which we may partner, that are designed to identify the patients that are most likely to benefit from the use of the drugs we may develop. Our current development plans focus on two product candidates: RXDX-101, a tyrosine kinase inhibitor directed to the TrkA, ROS1 and ALK proteins, which is in a Phase I/II clinical study in molecularly defined patient populations for the treatment of solid tumors; and RXDX-102, a tyrosine kinase inhibitor directed to the Trk family tyrosine kinase receptors, TrkA, TrkB and TrkC, which is currently in preclinical development for the treatment of multiple cancers. We have entered into a license agreement granting us exclusive global development and marketing rights to RXDX-101 and RXDX-102, which will become effective upon our satisfaction of certain financing conditions set forth in that license agreement. We also have three discovery stage programs, Spark-1, Spark-2, and Spark-3, directed to emerging oncology targets identified through mining of our database of information from proprietary and publicly available tumor samples, called Oncolome™. Our strategy is to leverage the biomarker insights that we gain through our genetic and epigenetic mining of Oncolome and the knowledge of cancer biology of our management and drug discovery team, with the goal of discovering, validating, developing and commercializing a pipeline of novel drug candidates for the treatment of cancer.
Since inception, our operations have focused on organizing and staffing our company, business planning, raising capital, assembling our core capabilities in genetic and epigenetic based biomarker and drug target discovery, and identifying potential product candidates. In the future, as we discover or acquire product candidates for development, we expect that our operations will also include preparing, managing and conducting preclinical and clinical studies and trials, preparing
55
Table of Contents
regulatory submissions relating to those product candidates, if regulatory approval is obtained, pursuing the commercialization of our product candidates, and establishing and managing relationships with third parties in connection with all of those activities. To date, we have financed our operations primarily through funding received from private placement offerings of our preferred stock and under a loan agreement. We have had no revenue to date. Since our inception, and through June 2013, we have raised an aggregate of approximately $7.5 million to fund our operations, of which approximately $6.0 million has been received from our issuance and sale of our preferred stock and approximately $1.5 million has been received under our loan and security agreement with Silicon Valley Bank (SVB).
Since inception, we have incurred significant operating losses. Our net losses were $2.2 million, $1.3 million and $0.1 million for the six months ended June 30, 2013 and for the years ended December 31, 2012 and 2011, respectively. As of June 30, 2013, we had an accumulated deficit of approximately $3.5 million. We expect to continue to incur significant expenses and operating losses over the next several years. Our net losses may fluctuate significantly from quarter to quarter and from year to year. We anticipate that our expenses will increase significantly as we assume control of the ongoing studies and trials of RXDX-101 and RXDX-102; plan for the commencement of potential Phase II clinical development activities for RXDX-101; advance the preclinical and potential clinical development of RXDX-102; pursue the initial stages of development of our Spark-1 through Spark-3 programs; continue to discover, validate and develop additional novel product candidates; expand and protect our intellectual property portfolio; and hire additional scientific, business, accounting and financial personnel. In addition, we expect to incur additional costs associated with operating as a public company.
Recent Developments
Merger with Ignyta
We completed the Merger on October 31, 2013, pursuant to which a wholly owned subsidiary of Ignyta that was formed solely for the purpose of the Merger merged with and into us. Upon the closing of the Merger, we have become the wholly owned subsidiary of Ignyta and have changed our name to Ignyta Operating, Inc. In connection with the Merger, the holders of shares of our preferred stock elected, in accordance with the terms of our certificate of incorporation then in effect, to convert all issued and outstanding shares of all classes of our preferred stock into shares of our common stock at the applicable conversion rate therefor, which in each case was one-to-one, with such conversion taking effect immediately prior to the closing of the Merger. Upon completion of the Merger, all holders of shares of our common stock and outstanding options and warrants to purchase shares of our common stock became entitled to receive or became entitled to the right to acquire one share of the common stock of Ignyta, for each one share of our common stock held by them or which they had the right to acquire.
License Agreement with NMS
We entered into a license agreement with NMS on October 10, 2013, which was amended on October 25, 2013, and which grants us exclusive global rights to develop and commercialize RXDX-101 and RXDX-102. The license agreement will become effective upon the completion of a financing of equity or debt securities pursuant to which we or our affiliates receive gross proceeds of at least $20 million. Our development rights under the license agreement are exclusive for the term of the agreement with respect to RXDX-101 and RXDX-102 and also, as to NMS, are exclusive for a five-year period with respect to any product candidate with activity against the target proteins of RXDX-101 and RXDX-102, and include the right to grant sublicenses. The license agreement provides that we will assume control of and financial and all other responsibility for the ongoing Phase I/II clinical trial of RXDX-101 that is currently being conducted by NMS and for continued preclinical development of RXDX-102. We are obligated under the license agreement to use commercially reasonable efforts to develop and commercialize RXDX-101 and RXDX-102, and, with the exception of transfer to us without cost of NMS’ existing inventory of RXDX-101 and RXDX-102 material, we are responsible for all remaining development and commercialization costs for RXDX-101 and RXDX-102.
Under the terms of the license agreement, we have agreed to issue to NMS a warrant to acquire up to 16,667 shares of our common stock upon the effective date of the license agreement, which will have an exercise price per share equal to that used in the financing that causes the license agreement to become effective, and will be exercisable at any time at the option of the holder from the effective date of the license agreement until the five-year anniversary thereof. We are also obligated to make an up-front payment to NMS of $7.0 million on the earlier of 10 days following the effective date of the license agreement and December 31, 2013, $1.0 million of which NMS may elect to receive in the form of shares of our common stock at a price per share equal to that used in the financing that causes the license agreement to become effective, and the remainder of which shall be paid in cash. If we fail to make this upfront payment, the license agreement terminates. When and if commercial sales of RXDX-101 or RXDX-102 begin, we are obligated to pay NMS tiered royalties ranging from a
56
Table of Contents
mid-single digit percentage to a low double digit percentage of our net sales, depending on the amount of our net sales, with standard provisions for royalty offsets to the extent we need to obtain any rights from third parties to commercialize either RXDX-101 or RXDX-102. We are obligated under the terms of the license agreement to engage NMS to perform services valued at $1 million or more between the effective date of the license agreement and December 31, 2014, which services could include, among others at our election, manufacture and supply services, technology transfer activities, preclinical activities, process development activities and assay development activities. We are also required to make development and regulatory milestone payments to NMS of up to $105.0 million in the aggregate if specified clinical study initiations and regulatory approvals are achieved across multiple products or indications. The first such milestone payment is not due until we elect to initiate the first randomized Phase II clinical study, which, based on our current estimates and certain assumptions, we anticipate could occur as early as 2015.
The license agreement with NMS provides that NMS will transfer to us all data, technology and know-how related to RXDX-101 and RXDX-102 and necessary for their continued development. That data and technology transfer will commence upon the effectiveness of the license agreement, and we anticipate that the transfer will be complete in 2014. Our ability to continue all ongoing studies, design and commence any new studies or trials, and solidify our development plans for RXDX-101 and RXDX-102 is dependent on that transfer process being completed successfully and in a timely manner.
The license agreement will not become effective if we do not satisfy the financing conditions to its effectiveness by December 31, 2013. Additionally, following its effectiveness, the license agreement with NMS will remain in effect until the expiration of all of our royalty and sublicense revenue obligations to NMS, determined on a product-by-product and country-by-country basis, unless we elect to terminate the license agreement earlier. If we fail to meet our obligations under the license agreement and are unable to cure such failure within specified time periods, NMS can terminate the license agreement, resulting in a loss of our rights to RXDX-101 and RXDX-102.
Acquisition of Actagene
In May 2013, we acquired Actagene, a discovery stage precision medicine company applying genomic insights to discover new biomarkers and targets for cancer therapeutics, by way of its merger with and into us on May 20, 2013. Prior to our acquisition of Actagene, our business focus was on the development of new biomarker-based molecular diagnostic assays to facilitate the diagnosis of certain clinically confounding diseases, such as chronic autoimmune and rheumatic diseases. With the acquisition of Actagene, we shifted the focus of our business to the use of biomarkers to discover and develop drug candidates for the treatment of cancer. Several members of our drug discovery and scientific team, which we consider to be a valuable asset and an important element of our strategy, joined our company in connection with the acquisition of Actagene. All consideration paid by us in connection with our acquisition of Actagene was paid with shares of our common stock, totaling an aggregate of 1,583,336 shares. The merger with Actagene was accounted for as a combination of entities under common control, and as a result the shares issued as merger consideration were valued for accounting purposes at $0.003 per share (see footnote 2 of our unaudited financial statements for the six months ended June 30, 2013 attached as Exhibit 99.1 to this Current Report on Form 8-K).
Financial Operations Overview
Revenue
To date, we have not generated any revenue from product sales or otherwise, and do not expect to generate any revenue from the sale of products in the near future.
In the future, we expect that we will seek to generate revenue primarily from product sales, but may also seek to generate revenue from research funding, milestone payments and royalties on future product sales in connection with any out-license or other strategic relationships we may establish.
Research and Development Expenses
Research and development expenses consist primarily of costs incurred for our research activities, including our drug and biomarker discovery efforts and the development of our product candidates, which include:
| • | employee-related expenses, including salaries, benefits and stock-based compensation expense; |
57
Table of Contents
| • | expenses incurred under agreements with third parties, including consultants and advisors we engage for research-related services and, in the future, any contract research organizations (CROs) that we may engage in connection with conducting preclinical and clinical activities on our behalf; |
| • | the cost of laboratory supplies; and |
| • | facilities, depreciation, and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance and other operating costs. |
Research and development costs are expensed as incurred. Nonrefundable advance payments for goods or services to be received in the future for use in research and development activities are deferred and capitalized. The capitalized amounts are expensed as the related goods are delivered or the services are performed.
We have not yet begun tracking our internal and external research and development costs on a program-by-program basis. As such, we do not have historical research and development expenditures by program and we use our employee and infrastructure resources across multiple research and development programs. The following table sets forth our research and development expenses for the periods presented:
| Years ended December 31, | Six Months ended June 30, | |||||||||||||||
| 2012 | 2011 | 2013 | 2012 | |||||||||||||
| Total research and development expenses |
$ | 708,043 | $ | 39,870 | $ | 1,220,664 | $ | 236,224 | ||||||||
Research and development activities are central to our business model. Our research and development programs that we expect will be our focus in the immediate future consist of the development of RXDX-101 and RXDX-102, for which we will acquire exclusive development rights upon the effectiveness of our license agreement with NMS, and drug discovery activities for the development of our Spark-1, Spark-2 and Spark-3 programs. All of those research and development programs are in the early stage, and since product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials, we expect research and development costs relating to each of those programs to increase significantly for the foreseeable future. However, the successful development of any of those product candidates, or any others we may seek to pursue, is highly uncertain. As such, at this time, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts that will be necessary to complete the remainder of the development of these product candidates, or whether any of these product candidates will reach successful commercialization. We are also unable to predict when, if ever, any net cash inflows will commence from any of the product candidates we currently or may in the future pursue. This lack of predictability is due to the numerous risks and uncertainties associated with developing medicines, many of which, such as our ability to obtain approvals to market and sell those medicines from the United Stated Food and Drug Administration (FDA) and other applicable regulatory authorities, are beyond our control, including the uncertainty of:
| • | establishing an appropriate safety profile with toxicology studies adequate to submit to the FDA in an investigational new drug application (IND) or comparable applications to foreign regulatory authorities; |
| • | successful enrollment in and adequate design and completion of clinical trials; |
| • | receipt of marketing approvals from applicable regulatory authorities, including the FDA and comparable foreign authorities; |
| • | establishing commercial manufacturing capabilities or, more likely, seeking to establish arrangements with third-party manufacturers; |
| • | obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidates; |
| • | launching commercial sales of the products, if and when approved, including establishing an internal sales and marketing force or establishing relationships with third parties for such purpose; |
| • | developing and commercializing, individually or with third-party collaborators, companion diagnostics; and |
| • | a continued acceptable safety profile of the products following approval, if any. |
A change in the outcome of any of these variables with respect to the development of any of our product candidates would significantly change the costs, timing and likelihood of success associated with the development of that product candidate.
58
Table of Contents
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and other related costs, including stock-based compensation, for personnel in executive, finance, accounting, business development, legal and human resources functions. Other significant costs include facility costs not otherwise included in research and development expenses, legal fees relating to patent and corporate matters and fees for accounting and consulting services.
We anticipate that our general and administrative expenses will increase in the future to support continued research and development activities, potential commercialization of our product candidates and increased costs of operating as a public company. These increases will likely include increased costs related to the hiring of additional personnel and increased fees to outside consultants, lawyers and accountants, among other expenses. Additionally, we anticipate increased costs associated with operating as a public company, including expenses related to services associated with maintaining compliance with requirements of the Securities and Exchange Commission (SEC), insurance and investor relations costs.
Critical Accounting Policies and Estimates
This discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which we have prepared in accordance with United States generally accepted accounting principles. The preparation of these consolidated financial statements requires us to make estimates, judgments and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported amounts of expenses during the reporting periods. We base our estimates on historical experience and on various other factors and assumptions that we believe are reasonable under the circumstances at the time the estimates are made, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We periodically evaluate our estimates and judgments, including those described in greater detail below, in light of changes in circumstances, facts and experience.
Our critical accounting policies are those accounting principles generally accepted in the United States of America that require us to make subjective estimates and judgments about matters that are uncertain and are likely to have a material impact on our financial condition and results of operations, as well as the specific manner in which we apply those principles. Our significant accounting policies are described in more detail in the notes to our financial statements included as exhibits to this report. We believe the critical accounting policies used in the preparation of our financial statements that require significant estimates and judgments are as follows:
Revenue Recognition
To date, we have not generated any revenue.
Income Taxes
Deferred income taxes are recognized for the tax consequences in future years of differences between the tax basis of assets and liabilities and their financial reporting amounts at each year end based on enacted tax laws and statutory tax rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense is the combination of the tax payable for the year and the change during the year in deferred tax assets and liabilities.
Cash and Cash Equivalents
We consider all highly liquid investments with an original maturity of 90 days or less when purchased to be cash equivalents. Cash equivalents primarily represent amounts invested in money market funds whose cost equals market value.
Stock-Based Compensation
We account for stock-based compensation in accordance with Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Topic 718, Compensation – Stock Compensation, which establishes accounting for equity instruments exchanged for employee services. Under such provisions, stock-based compensation cost is measured at the grant date, based on the calculated fair value of the award, and is recognized as an expense, under the straight-line method, over the employee’s requisite service period (generally the vesting period of the equity grant).
59
Table of Contents
We account for equity instruments, including restricted stock or stock options, issued to non-employees in accordance with authoritative guidance for equity based payments to non-employees. Stock options issued to non-employees are accounted for at their estimated fair value determined using the Black-Scholes option-pricing model. The fair value of options granted to non-employees is re-measured as they vest, and the resulting increase in value, if any, is recognized as expense during the period the related services are rendered. Restricted stock issued to non-employees is accounted for at their estimated fair value as they vest.
Recently Issued Accounting Pronouncements
There are no recent accounting pronouncements likely to have a material impact on the financial statements.
JOBS Act
In April 2012, the Jumpstart Our Business Startups Act of 2012 (JOBS Act) was enacted. Section 107(b) of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended (Securities Act), for complying with new or revised accounting standards. Thus, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have irrevocably elected to avail ourselves of this extended transition period and, as a result, we will not be required to adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies.
We are in the process of evaluating the benefits of relying on other exemptions and reduced reporting requirements under the JOBS Act afforded to us for so long as we are an emerging growth company. Subject to certain conditions, we intend to rely on certain of these exemptions, including without limitation (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act and (ii) complying with any requirement that may be adopted by the PCAOB regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an emerging growth company until the earlier of (i) the last day of the fiscal year in which we have total annual gross revenues of $1 billion or more; (ii) the last day of the fiscal year following the fifth anniversary of the date of the first sale of the common stock of Ignyta pursuant to an effective registration statement under the Securities Act, which was on February 15, 2013; (iii) the date on which we have issued more than $1 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
Results of Operations
Comparison of Six Months Ended June 30, 2013 and 2012
The following table summarizes our results of operations for the six months ended June 30, 2013 and 2012, together with the changes in those items in dollars and as a percentage:
| Six months ended June 30, |
Dollar change |
|||||||||||||||
| (in thousands) |
2013 | 2012 | % change | |||||||||||||
| Revenue |
$ | — | $ | $ | — | — | % | |||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
1,220 | 236 | 984 | 417 | ||||||||||||
| General and administrative |
904 | 161 | 743 | 461 | ||||||||||||
| Loss from operations |
(2,124 | ) | (397 | ) | (1,727 | ) | 435 | |||||||||
| Other income (expense) |
(30 | ) | — | (30 | ) | N/A | ||||||||||
| Provision for income taxes |
2 | 1 | 1 | 100 | ||||||||||||
| Net loss |
$ | (2,156 | ) | $ | (398 | ) | $ | (1,758 | ) | 442 | % | |||||
Revenue. We did not record any revenue for the six months ended June 30, 2013 and June 30, 2012.
Research and development expense. Research and development expense increased by approximately $984,000 to approximately $1,220,000 for the six months ended June 30, 2013 from approximately $236,000 for the six months ended
60
Table of Contents
June 30, 2012, an increase of 417%. The increase in research and development expenses was primarily attributable to an increase in activities related to our biomarker discovery programs and platform technologies, on which we incurred an increase of expenses between periods of approximately $480,000. Many of those activities were associated with our prior business focus on developing biomarker-based molecular assays for chronic autoimmune and rheumatic diseases. In addition, we incurred an increase between periods of approximately $114,000 for facilities related expenses, and approximately $390,000 for personnel expenses related to hiring and engaging additional employees and consultants.
General and administrative expense. General and administrative expenses increased by approximately $743,000 to approximately $904,000 for the six months ended June 30, 2013 from approximately $161,000 for the six months ended June 30, 2012, an increase of 461%. The increase in general and administrative expenses was primarily attributable to increased personnel costs of approximately $380,000, increased facilities related expenses of approximately $61,000 and increased audit, legal and intellectual property costs of approximately $300,000.
Other income (expense). Interest expense increased by approximately $30,000 to approximately $30,000 for the six months ended June 30, 2013, from approximately $0 for the six months ended June 30, 2012. The increase in interest expense was primarily attributable to increased interest owed under our loan agreement with SVB after the initial funding of the loan in late June 2012 offset by the change in the fair value of the warrant liability.
Provision for income tax. The provision for income taxes increased by approximately $1,000 to approximately $2,000 for the six months ended June 30, 2013, from approximately $1,000 for the six months ended June 30, 2012, an increase of 100%. The increase in the provision for income taxes for the six months ended June 30, 2013 was primarily attributable to an increase in corporate filing fees to the State of Delaware.
Comparison of Years Ended December 31, 2012 and 2011
The following table summarizes our results of operations for the years ended December 31, 2012 and 2011, together with the changes in those items in dollars and as a percentage:
| Years ended December 31, | Dollar change |
|||||||||||||||
| (in thousands) |
2012 | 2011 | % change | |||||||||||||
| Revenue |
$ | — | $ | — | $ | — | — | % | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
708 | 40 | 668 | 1,670 | ||||||||||||
| General and administrative |
548 | 39 | 509 | 1,305 | ||||||||||||
| Loss from operations |
(1,256 | ) | (79 | ) | (1,177 | ) | 1,490 | |||||||||
| Other income (expense) |
(23 | ) | — | (23 | ) | N/A | ||||||||||
| Provision for income taxes |
1 | — | 1 | N/A | ||||||||||||
| Net loss |
$ | (1,280 | ) | $ | (79 | ) | $ | (1,201 | ) | 1,520 | % | |||||
Revenue. We did not record any revenue for the years ended December 31, 2012 and December 31, 2011.
Research and development expense. Research and development expense increased by approximately $668,000 to approximately $708,000 in 2012 from approximately $40,000 in 2011, an increase of 1,670%. The increase in research and development expense was primarily attributable to hiring research and development staff, establishing our laboratory and other facilities and conducting activities to establish our epigenetic platform for identifying biomarkers of disease, particularly relating to our prior business focus on developing biomarker-based molecular assays for chronic autoimmune and rheumatic diseases. Negligible expenses were incurred in 2011 on such activities due to our company’s very early stage of research.
General and administrative expense. General and administrative expense increased by approximately $509,000 to $548,000 in 2012 from approximately $39,000 in 2011, an increase of 1,305%. The increase in general and administrative expense was primarily attributable to hiring general and administrative staff, for which we incurred approximately $272,000 in 2012 and approximately $6,000 in 2011, and establishing our offices and legal and intellectual property costs, for which we incurred approximately $276,000 in 2012 and approximately $34,000 in 2011.
61
Table of Contents
Other income (expense). Interest expense increased by approximately $23,000 to $23,000 in 2012, from approximately $0 in 2011. The increase in interest expense was primarily attributable to interest owed under our loan agreement with SVB, which we entered in June 2012.
Provision for income taxes. During 2011, we incurred no income taxes. During 2012 we incurred an income tax provision of approximately $1,000.
Liquidity and Capital Resources
Sources of Liquidity
Since our inception, and through June 30, 2013, we have raised an aggregate of approximately $7,005,000 to fund our operations, of which approximately $6,005,000 was received from our issuance and sale of our preferred stock and approximately $1,000,000 was received from the incurrence of indebtedness under our loan agreement with SVB. We have also received a small amount of funding from our issuance of common stock, through the exercise of stock options and upon issuance to our founders in August and September 2011. As of June 30, 2013, we had approximately $3,235,000 in cash and cash equivalents. Following June 30, 2013, in July 2013, we obtained an additional $500,000 under our loan agreement with SVB.
Preferred stock financings. We have received approximately $6,005,000 from the issuance and sale of our series A preferred stock and our series B preferred stock. We received approximately $500,000 from our issuance and sale of an aggregate of 833,334 shares of our series A preferred stock at a price per share of $0.60 to one investor in October 2011 and March 2012. We received approximately $5,505,000 from our issuance and sale of an aggregate of 1,835,000 shares of our series B preferred stock at a price per share of $3.00 to a number of investors in June 2012 and December 2012.
SVB loan and security agreement. We entered into a loan and security agreement with SVB in June 2012, which was amended in February 2013. Pursuant to the terms of the loan agreement, SVB granted us a loan in principal amount of $500,000 in June 2012, made an additional loan advance to us in principal amount of $500,000 in February 2013 and made a further loan advance to us in principal amount of $500,000 in July 2013. The amounts loaned to us under the loan agreement bear interest at a rate of 4.77% for the first loan advance, 4.00% for the second loan advance and 4.04% for the third loan advance, and are payable in monthly installments through June 1, 2015 for the first loan advance and September 1, 2015 for the second and third loan advances. Pursuant to the loan agreement, we are bound by certain affirmative and negative covenants setting forth actions that we must and must not take during the term of the loan agreement, and all amounts owed under the loan agreement may be declared due and payable by SVB upon the occurrence of an event of default under the loan agreement, which include, among other things, the occurrence of certain bankruptcy events, our failure to make payments under the loan agreement when due, and our breach of any representation or covenant in the loan agreement. We have granted SVB a security interest in substantially all of our personal property, rights and assets, other than our intellectual property, to secure our payment of all amounts owed by us to SVB under the loan agreement.
Cash Flows
The following table provides information regarding our cash flows for the years ended December 31, 2012 and 2011, and the six months ended June 30, 2013 and 2012:
| Years ended, December 31, |
Six months ended, June 30, |
|||||||||||||||
| (in thousands) |
2012 | 2011 | 2013 | 2012 | ||||||||||||
| Net cash (used in) operating activities |
$ | (991 | ) | $ | (65 | ) | $ | (2,056 | ) | $ | (333 | ) | ||||
| Net cash (used in) investing activities |
(306 | ) | (2 | ) | (247 | ) | (24 | ) | ||||||||
| Net cash provided by financing activities |
6,173 | 223 | 506 | 2,829 | ||||||||||||
| Net increase (decrease) in cash and cash equivalents |
$ | 4,876 | $ | 156 | $ | (1,797 | ) | $ | 2,472 | |||||||
Net cash used in operating activities. The use of cash in all periods resulted primarily from our net losses adjusted for non-cash charges and changes in components of working capital. Net cash used in operating activities was approximately $333,000 during the six months ended June 30, 2012 compared to approximately $2,056,000 during the six months ended
62
Table of Contents
June 30, 2013. The increase in cash used in operating activities in the first six months of 2013 was driven primarily by an increase in net loss during the six months ended June 30, 2013 as compared to the six months ended June 30, 2012.
Net cash used in operating activities was approximately $65,000 for the year ended December 31, 2011 compared to approximately $991,000 for the year ended December 31, 2012. The increase in cash used in operating activities during 2012 was driven primarily by an increase in net loss. This loss was partially offset by a decrease in working capital, including an increase in accounts payable and accrued expenses and other liabilities minus an increase in prepaid expenses and other current assets.
Net cash used in investing activities. Net cash used in investing activities was approximately $24,000 during the six months ended June 30, 2012 compared to approximately $247,000 during the six months ended June 30, 2013. The cash used in investing activities for the six months ended June 30, 2013 and 2012 was primarily the result of purchases of equipment.
Net cash used in investing activities was approximately $2,000 during the year ended December 31, 2011 compared to approximately $306,000 during the year ended December 31, 2012. The cash used in investing activities for the year ended December 31, 2012 was the result of increased purchases of equipment of approximately $304,000. The minimal cash used in investing activities for the year ended December 31, 2011 was also the result of purchases of equipment.
Net cash provided by financing activities. Net cash provided by financing activities was approximately $2,829,000 during the six months ended June 30, 2012 compared to approximately $506,000 during the six months ended June 30, 2013. The cash provided by financing activities for the six months ended June 30, 2013 was primarily the result of the incurrence of indebtedness under our loan agreement with SVB in February 2013, resulting in gross proceeds of $500,000. The cash provided by financing activities for the six months ended June 30, 2012 was the result of the issuance and sale of our series A preferred stock in March 2012, resulting in gross proceeds of $250,000, the issuance and sale of our series B preferred stock in June 2012, resulting in gross proceeds of $2,100,000, and the incurrence of indebtedness under our loan agreement with SVB in June 2012, resulting in gross proceeds of $500,000.
Net cash provided by financing activities was approximately $223,000 during the year ended December 31, 2011 compared to approximately $6,173,000 during the year ended December 31, 2012. The cash provided by financing activities during the year ended December 31, 2012 was the result of the issuance and sale of our series A preferred stock in March 2012, resulting in gross proceeds of $250,000, and the issuance and sale of our series B preferred stock in June 2012 and December 2012, collectively resulting in gross proceeds of approximately $5,505,000, and the incurrence of indebtedness under our loan agreement with SVB in June 2012, resulting in gross proceeds of $500,000. The cash provided by financing activities for the year ended December 31, 2011 was the result of the issuance and sale of our series A preferred stock in October 2011, resulting in gross proceeds of $250,000.
Funding Requirements
We expect our expenses to increase in connection with our ongoing activities, particularly as we assume rights to, and operational and financial responsibility for, the clinical development and manufacturing of RXDX-101 and RXDX-102 and seek to continue the research and development of, initiate or continue, as applicable, clinical trials of, and seek marketing approval for, those product candidates and our Spark-1 through Spark-3 programs. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to product sales, marketing, manufacturing and distribution to the extent that such sales, marketing and distribution are not the responsibility of any collaborators with whom we may engage. Further, we expect to incur additional costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or future commercialization efforts. As noted in our unaudited financial statements for the six months ended June 30, 2013, the uncertainties surrounding our ability to fund our operations raise substantial doubt about our ability to continue as a going concern.
We expect that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements for at least the next two months. Our future capital requirements will depend on many factors, including:
| • | the scope, progress, results and costs of drug discovery, preclinical development, laboratory testing and clinical trials for our product candidates; |
| • | the scope, progress, results and costs of companion diagnostic development for our product candidates; |
| • | the extent to which we acquire or in-license other medicines, biomarkers and/or technologies; |
| • | the costs, timing and outcome of regulatory review of our product candidates; |
63
Table of Contents
| • | the achievement of development milestones that trigger payments due to our licensing partners; |
| • | the costs of future commercialization activities, including product sales, marketing, manufacturing and distribution, for any of our product candidates for which we receive marketing approval (to the extent that such sales, marketing, manufacturing and distribution are not the responsibility of collaborators with whom we may engage); |
| • | revenue, if any, received from commercial sales of our product candidates, should any of our product candidates receive marketing approval; |
| • | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; and |
| • | our ability to establish and maintain development, manufacturing or commercial collaborations on favorable terms, if at all. |
Identifying potential product candidates and conducting preclinical testing and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our commercial revenues, if any, will be derived from sales of medicines that we do not expect to be commercially available for many years, if at all. Accordingly, we will need to continue to rely on additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances and licensing arrangements. Any or all of those sources of funding may not be available when needed on acceptable terms or at all. We do not have any committed external source of funds. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise funds through collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings or relationships with third parties when needed or on acceptable terms, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
Contractual Obligations
The following table summarizes our significant contractual obligations as of their payment due dates by period as of June 30, 2013:
| Payments due by period | ||||||||||||||||||||
| (in thousands) |
Total | July 1, 2013 – December 31, 2013 |
January 1, 2014 – December 31, 2016 |
January 1, 2017 – December 31, 2018 |
After December 31, 2018 |
|||||||||||||||
| Operating lease obligations(1) |
$ | 557 | $ | 81 | $ | 476 | $ | — | $ | — | ||||||||||
| Other(2) |
90 | 15 | 45 | 30 | — | |||||||||||||||
| Total contractual cash obligations |
$ | 647 | $ | 96 | $ | 521 | $ | 30 | $ | — | ||||||||||
| (1) | Represents future minimum lease payments under our two non-cancelable operating leases for our facilities and under one equipment lease, one of which expires by its terms on October 31, 2013. The minimum lease payments reflected do not include any related common area maintenance charges, utilities or real estate taxes. |
| (2) | Consists of an annual maintenance payment of $15,000 that we are required to pay under an in-license agreement for certain intellectual property and technology relating to biomarker-based molecular diagnostic assays. Annual maintenance payment obligations extend through the term of the license agreement, which is tied to the life of the patents subject to the agreement. |
64
Table of Contents
In addition to our contractual obligations set forth in the table above, we are bound by the following additional contractual obligations under agreements we have entered subsequent to June 30, 2013: (i) pursuant to the terms of our license agreement with NMS, which we entered in October 2013 and which will become effective upon our satisfaction of certain financing conditions, we are obligated to engage NMS to perform services for us valued at $1 million or more between the effective date of the license agreement and December 31, 2014, which services could include, among others at our election, manufacture and supply services, technology transfer activities, preclinical activities, process development activities and assay development activities, and we are obligated to make an up-front payment to NMS of $7.0 million on the earlier of 10 days following the effective date of the license agreement and December 31, 2013, $1.0 million of which NMS may elect to receive in the form of shares of our common stock at a price per share equal to that used in the financing that causes the license agreement to become effective, and the remainder of which shall be paid in cash, and (ii) pursuant to the terms of an operating lease agreement for a new facility, which replaces an older lease agreement expiring on October 31, 2013, and the term of which commences in November 2013, we are obligated to make rent payments totaling approximately $45,000 for the 12-month term of the lease agreement.
We enter into contracts in the normal course of business with vendors for research studies and other services and products for operating purposes, which generally provide for termination within 30 days of notice, and therefore are cancelable contracts and not included in the table of contractual obligations above.
We have obligations to make future payments to third parties that become due and payable on the achievement of certain development, regulatory and commercial milestones. Since the achievement and timing of these milestones is not fixed and determinable, and we typically have the ability to terminate the agreements upon 60-90 days’ notice, such commitments have not been included in our consolidated balance sheets or in the table of contractual obligations above.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under applicable SEC rules.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information regarding the beneficial ownership of our common stock by (i) each person who, to our knowledge, owns more than 5% of our common stock, (ii) each of our current directors and the named executive officers of Ignyta Operating identified under the heading “Executive Compensation” below, (iii) certain of our other executive officers that have been appointed as such upon the closing of the Merger and that may be named executive officers of Ignyta for the fiscal year ending December 31, 2013, and (iv) all of those directors and executive officers as a group. We have determined beneficial ownership in accordance with applicable rules of the SEC, which generally provide that beneficial ownership includes voting or investment power with respect to securities. Except as indicated by the footnotes to the table below, we believe, based on the information furnished to us, that the persons named in the table have sole voting and investment power with respect to all shares of common stock that they beneficially own, subject to applicable community property laws.
The information set forth in the table below is based on 4,923,805 shares of our common stock issued and outstanding on October 31, 2013 immediately after to giving effect to the closing of the Merger. In computing the number of shares of common stock beneficially owned by a person and the percentage ownership of that person, we deemed to be outstanding all shares of common stock subject to options, warrants or other convertible securities held by that person that are currently exercisable or will be exercisable within 60 days after October 31, 2013. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Except as otherwise noted, the address for each person listed in the table below is c/o Ignyta, Inc., 11095 Flintkote Avenue, Suite D, San Diego, California 92121.
65
Table of Contents
| Name and Address of Beneficial Owner | Number of Shares Beneficially Owned |
Percentage Beneficially Owned |
||||||
| 5%+ Stockholders: |
||||||||
| City Hill Venture Partners I, LLC (1) |
2,983,334 | 60.59 | % | |||||
| Directors and Executive Officers: |
||||||||
| Jonathan E. Lim, M.D. (2) |
2,990,972 | 60.65 | % | |||||
| Patrick O’Connor, Ph.D. |
500,000 | 10.15 | % | |||||
| Zachary Hornby (3) |
42,985 | * | ||||||
| Alex Casdin (4) |
53,055 | 1.08 | % | |||||
| Heinrich Dreismann, Ph.D. (5) |
4,027 | * | ||||||
| All Current Directors and Executive Officers as a Group (5 persons) |
3,591,039 | 72.55 | % |
| * | Less than 1%. |
| (1) | Dr. Lim is the Managing Partner of City Hill Ventures Partners I, LLC and has sole voting and investment control with respect to the securities that it holds of record. Dr. Lim disclaims beneficial ownership of these securities except to the extent of his pecuniary interest therein. |
| (2) | Represents (a) 2,983,334 shares of our common stock held by City Hill Venture Partners I, LLC, with respect to which Dr. Lim has sole voting and investment control, and (b) 7,638 shares underlying an option held by Dr. Lim and exercisable within 60 days following October 31, 2013. |
| (3) | Represents (a) 31,667 shares of our common stock held of record by Mr. Hornby, and (b) 11,318 shares underlying an option held by Mr. Hornby and exercisable within 60 days following October 31, 2013. |
| (4) | Represents (a) 50,000 shares of our common stock held of record by Mr. Casdin, and (b) 3,055 shares underlying an option held by Mr. Casdin and exercisable within 60 days following October 31, 2013. |
| (5) | Represents 4,027 shares underlying an option held by Dr. Dreismann and exercisable within 60 days following October 31, 2013. |
Directors, Executive Officers and Other Non-Executive Officers
The table below sets forth the name, age and position of each of our directors and executive officers and certain other non-executive officer members of our scientific and drug development team. Each of the directors and executive officers listed below joined Ignyta upon the closing of the Merger on October 31, 2013, and each of the members of our non-executive officer management team listed below hold their respective positions with Ignyta Operating.
| Name | Age | Position | ||
| Directors and Executive Officers: | ||||
| Jonathan E. Lim, M.D. | 41 | President, Chief Executive Officer and Chairman of the Board (Principal Executive Officer) | ||
| Patrick O’Connor, Ph.D. | 53 | Chief Scientific Officer, Sr. Vice President, Research | ||
| Zachary Hornby | 34 | Chief Financial Officer, and Vice President, Corporate Development (Principal Financial and Accounting Officer) | ||
| Alexander Casdin | 46 | Director | ||
| Heinrich Dreismann, Ph.D. | 60 | Director | ||
| Non-Executive Officer Management Team: | ||||
| James Freddo, M.D. | 58 | Consulting Chief Medical Officer | ||
| Jean-Michel Vernier, Ph.D. | 52 | Vice President, Chemistry | ||
| Paul Pearson, Ph.D. | 53 | Consulting Vice President of PK, Drug Metabolism & Safety | ||
| Dave Matthews, Ph.D. | 70 | Consulting Vice President, Crystallography | ||
| Robert Shoemaker, Ph.D. | 32 | Director of Bioinformatics | ||
Business Experience
The following is a brief account of the education and business experience of our current directors and executive officers:
66
Table of Contents
Jonathan E. Lim, M.D. Dr. Lim is a co-founder of Ignyta Operating and joined that company as Chairman, President and Chief Executive Officer at its inception in August 2011. Prior to joining Ignyta Operating, Dr. Lim most recently served as Chairman and Chief Executive Officer of Eclipse Therapeutics, Inc., a private biotechnology company discovering and developing monoclonal antibody therapeutics targeting cancer stem cells, that he co-founded in March 2011 as a spinout from Biogen Idec and that was sold to Bionomics Ltd., an Australian public biotechnology company discovering and developing drugs targeting oncology and central nervous system disorders, in September 2012. Dr. Lim currently serves as a member of the Board of Directors of Bionomics Ltd. Prior to founding Eclipse Therapeutics, Dr. Lim served as the President, Chief Executive Officer and a Director of Halozyme Therapeutics, Inc., a public biotechnology company, from May 2003 to December 2010. Prior to that, Dr. Lim’s experience included management consulting at McKinsey & Company, a National Institutes of Health Postdoctoral Fellowship at Harvard Medical School and two years of general surgery residency at New York Hospital-Cornell. Dr. Lim has B.S. and M.S. degrees from Stanford, an M.D. from McGill University and an M.P.H. from Harvard University. We believe that Dr. Lim adds value to our Board of Directors based on his intimate knowledge of our business plans and strategies as a co-founder of our business and his extensive experience as an executive officer and director of multiple public and private biotechnology companies.
Patrick O’Connor, Ph.D. Dr. O’Connor joined Ignyta Operating in May 2013 as Chief Scientific Officer and Senior Vice President, Research, after Ignyta Operating acquired Actagene, a discovery stage precision medicine company that Dr. O’Connor founded in February 2013 and for which he was serving as Chief Executive Officer. Dr. O’Connor is currently on medical leave, and we expect him to return in January 2014. Prior to founding Actagene, Dr. O’Connor was a Scientific Advisory Board member and the Head of Oncology at Ruga Corporation, a private oncology biopharmaceutical company that he joined in 2012 when Ruga acquired Selexagen Therapeutics, a private cancer therapeutics company that Dr. O’Connor co-founded in early 2009 and where he had been serving as Chief Scientific Officer. Prior to Selexagen, Dr. O’Connor was Vice President of Research at Halozyme Therapeutics, a public biotechnology company, from 2008 to 2009. Prior to Halozyme Therapeutics, Dr. O’Connor was SVP and Head of Research at Ardea Biosciences, a private biotechnology small-molecule therapeutics company, from 2007 to 2008. Prior to Ardea Biosciences, Dr. O’Connor served as the Global Research Therapeutic Area Head for Oncology at Pfizer, a global research-based pharmaceutical company, from 1998 to 2007, following Pfizer’s acquisition of Agouron/Warner-Lambert, a company Dr. O’Connor joined as Head of the Oncology Research Division in 1998. Prior to joining the pharmaceutical industry, Dr. O’Connor spent 10 years at the National Cancer Institute in Bethesda, Maryland. Dr. O’Connor is a Senior Editor of Cancer Research, and serves on the Scientific Advisory Boards of Molecular Response and Deciphera Pharmaceuticals. Dr. O’Connor is also currently on the board of directors of Selexagen Therapeutics. He gained his Ph.D. from the University of Manchester in England where he was a Venborough Scholar.
Zachary Hornby. Mr. Hornby joined Ignyta Operating in August 2012 as Vice President, Corporate Development and was appointed as its Chief Financial Officer in August 2013. Prior to joining Ignyta Operating, Mr. Hornby served as senior director of business development at Fate Therapeutics, a public biopharmaceutical stem cell discovery and development company, from August 2010 to August 2012. Prior to Fate Therapeutics, Mr. Hornby was director of business development at Halozyme Therapeutics, a public biotechnology company, from January 2008 to August 2010. Prior to Halozyme Therapeutics, Mr. Hornby was senior product manager at Neurocrine Biosciences, a public biopharmaceutical company, from June 2006 to January 2008. Prior to Neurocrine Biosciences, Mr. Hornby served as a life sciences consultant at L.E.K. Consulting and in regulatory affairs and business development roles at Transkaryotic Therapies (acquired by Shire Pharmaceuticals in 2005), an orphan drug discovery and development company. Mr. Hornby holds B.S. and M.S. degrees in biology from Stanford University and an M.B.A. from Harvard Business School.
Alexander Casdin. Mr. Casdin joined the Board of Directors of Ignyta upon the closing of the Merger on October 31, 2013. Alex Casdin is a private investor focused on the healthcare sector. From October 2011 through September 2012, Mr. Casdin was the Chief Financial Officer of Sophiris Bio, Corp., a Canadian public urology company. Prior to Sophiris Bio, Mr. Casdin served as the Vice President, Finance of Amylin Pharmaceuticals, a biopharmaceutical company that was acquired by Bristol-Myers Squibb in 2012, a position he held from October 2009 to October 2011. Prior to his position at Amylin Pharmaceuticals, Mr. Casdin founded and operated Casdin Advisors LLC, where he served as a strategic advisor to companies in the life sciences industry. Before founding Casdin Advisors, Mr. Casdin was the Chief Executive Officer and Portfolio Manager of Cooper Hill Partners, LLC, a healthcare investment fund. Mr. Casdin has also held previous positions at Pequot Capital Management and Dreyfus Corporation. Mr. Casdin currently serves on the board of directors of DiaVacs, a private clinical stage biotechnology company focused on a treatment for Type 1 Diabetes, and as a member of the advisory boards of the Hassenfeld Center For Cancer & Blood Disorders and the Social Enterprise Program of the Columbia Business School, each of which are non-profit entities, and served on the board of directors of DUSA Pharmaceuticals, a specialty pharmaceutical company in the field of dermatology that was previously listed on the NASDAQ Stock Market and was acquired by Sun Pharmaceutical Industries Limited in December 2012, from January
67
Table of Contents
2009 until December 2012. Mr. Casdin earned his B.A. degree from Brown University and his M.B.A., Beta Gamma Sigma, from Columbia Business School. We believe that Mr. Casdin adds value to our Board of Directors based on his experience with the financing and other aspects of company-building for enterprises in our industry.
Heinrich Dreismann, Ph.D. Dr. Dreismann joined the Board of Directors of Ignyta upon the closing of the Merger on October 31, 2013. Dr. Dreismann currently serves on the boards of directors of several public and private diagnostic companies, including Myriad Genetics, Inc., a public molecular diagnostic company, GeneNews, a Canadian public molecular diagnostics company, and Med BioGene, Inc., a Canadian public life sciences company focused on genomic-based clinical laboratory diagnostic tests. Dr. Dreismann also served on the board of directors of Shrink Nanotechnologies, Inc., a nanotechnology company, from June 2009 until November 2011. Dr. Dreismann completed a career at Roche Molecular Systems in 2006, where he served since 1985 and held several senior positions, including President and Chief Executive Officer of Roche Molecular Systems, Head of Global Business Development at Roche Diagnostics and Member of Roche’s Global Diagnostic Executive Committee. Dr. Dreismann earned a Master of Science in biology and a Doctor of Philosophy in microbiology/molecular biology from Westfaelische Wilhelms University in Muenster, Germany. He conducted his Post-Doctoral studies in microbial genetics at the Centre d’Etudes Nucleaires de Saclay, France. We believe that Dr. Dreismann adds value to our Board of Directors based on his experience as a member of boards of directors and senior management of public companies and his expertise in the molecular diagnostics field.
The following is a brief account of the education and business experience of the current non-executive officer members of our scientific and drug development team:
James Freddo, M.D. Dr. Freddo joined Ignyta Operating July 2013 as a consultant holding the position of Chief Medical Officer. Prior to joining Ignyta Operating, he served as a consultant from April 2012 until May 2012 and as the Executive Vice President, Clinical Development and Chief Medical Officer from June 2012 until May 2013, in each case for Ruga Corporation, a private oncology biopharmaceutical company. Prior to that, he was the Chief Medical Officer and Senior Vice President, Drug Development at Anadys Pharmaceuticals, a drug development company focused on small molecule therapeutics that was previously listed on the NASDAQ Stock Market and was acquired by Roche in 2011, from July 2006 until March 2012, where he also served as a member of the Board of Directors from January 2011 until November 2011. Prior to joining Anadys Pharmaceuticals, Dr. Freddo served at Pfizer, a global research-based pharmaceutical company, in La Jolla, California from June 2002 until July 2006, holding the positions of Vice President, Clinical Site Head and Development Site Head and, prior to that, Executive Director and leader of Oncology Clinical Development. Prior to joining Pfizer, Dr. Freddo held a variety of senior management positions at Wyeth-Ayerst Research from 1996 to 2002, in the Oncology, Infectious Diseases and Transplantation Immunology therapeutic areas. He has also served as a member of the Board of Directors for InfuSystems, Inc., a public healthcare products and services company, from 2008 until 2011. Dr. Freddo received an M.D. degree from the University of North Carolina, Chapel Hill, completed his residency training at University of California, San Diego and returned to Chapel Hill for his fellowship training in gynecologic oncology.
Jean-Michel Vernier, Ph.D. Dr. Vernier joined Ignyta Operating in June 2013 as Vice President, Chemistry, after Ignyta Operating acquired Actagene, a discovery stage precision medicine company for which he was serving as a consultant holding the position of Vice President, Chemistry at the time of the acquisition. Prior to joining Actagene, Dr. Vernier served as Head of Chemistry of Ruga Corporation, a private oncology biopharmaceutical company, from February 2012 until April 2013. Prior to Ruga, Dr. Vernier was a Co-Founder and Vice President of Chemistry at Selexagen Therapeutics, a private cancer therapeutics company, from October 2010 until January 2012. Prior to co-founding Selexagen Therapeutics, Dr. Vernier served as Vice President of Discovery Chemistry at Ardea Biosciences, a private biotechnology small-molecule therapeutics company, from 2007 until 2010. Dr. Vernier has also led chemistry at Valeant Pharmaceuticals, Merck Research Laboratories and SIBIA Neurosciences. Dr. Vernier received a Ph.D. in synthetic organic chemistry from the University Louis Pasteur, Strasbourg, France and was a postdoctoral fellow at Colorado State University.
Paul Pearson, Ph.D. Dr. Pearson joined Ignyta Operating in July 2013 as a consultant holding the position of Vice President of Pharmacokinetics, Drug Metabolism & Safety, after Ignyta Operating acquired Actagene, a discovery stage precision medicine company for which he was serving as a consultant holding the position of Vice President of Pharmacokinetics, Drug Metabolism & Safety at the time of the acquisition. Dr. Pearson also currently serves as the President of Pearson Pharma Partners, a private drug development consulting company, and has served in such position since May 2008. Prior to joining Actagene, Dr. Pearson served as a consultant from August 2010 until June 2012 and as the Vice President, Preclinical Development from July 2012 until March 2013, in each case for Ruga Corporation, a private oncology biopharmaceutical company. Prior to that, Dr. Pearson served as Global Head and Vice President, Pharmacokinetics and Drug Metabolism (PKDM) at Amgen, Inc., a public biotechnology company focused on developing
68
Table of Contents
human therapeutics, from October 2003 to April 2008. Prior to Amgen, Dr. Pearson was Executive Director of Preclinical Drug Metabolism at Merck Research Laboratories, a global healthcare therapeutics company, from January 1998 until September 2003. Prior to Merck, Dr. Pearson held positions of increasing importance in drug metabolism at the Upjohn Company, a pharmaceutical manufacturing company. Dr. Pearson is the Editor of the Handbook of Drug Metabolism (2009).
Dave Matthews, Ph.D. Dr. Matthew joined Ignyta Operating in June 2013 as a consultant holding the position of Vice President of Crystallography, after Ignyta Operating acquired Actagene, a discovery stage precision medicine company for which he was serving as a consultant holding the position of Vice President of Crystallography at the time of the acquisition. Dr. Matthews is also currently Chairman of the Scientific Advisory Board for a broad-based consortium of partners participating in the Bill and Melinda Gates Foundation-funded initiative for “Structure Guided Drug Discovery for Tuberculosis and Malaria.” Prior to joining Actagene, Dr. Matthews served as a consultant for Ruga Corporation, a private oncology biopharmaceutical company, from May 2012 until November 2012. Prior to that, Dr. Matthews was a scientific founder of Selexagen Therapeutics, Inc., a private cancer therapeutics company, in March 2008. Prior to Selexagen Therapeutics, Dr. Matthews served on the Medicines for Malaria Venture’s Expert Scientific Advisory Committee from November 2005 to November 2010. Prior to that, Dr. Matthews was Distinguished Research Fellow, Head of Structural Biology, Computational Chemistry, and Bioinformatics at Pfizer, a global research-based pharmaceutical company, from Pfizer’s acquisition of Warner Lambert and Agouron Pharmaceuticals in 2000 until 2005. Prior to that, Dr. Matthews was the scientific founder of Agouron Pharmaceuticals in 1985. Prior to Agouron Pharmaceuticals, Dr. Matthews was a postdoctoral fellow and later a faculty member in the Department of Chemistry at the University of California, San Diego from 1971 until 1985. Dr. Matthews received his Ph.D. in physical chemistry from the University of Illinois, Urbana-Champaign.
Robert Shoemaker, Ph.D. Dr. Shoemaker joined Ignyta Operating in January 2012 as Director of Bioinformatics. Prior to joining Ignyta Operating, Dr. Shoemaker was a Scientist at Illumina, Inc., a public life science company focused on the analysis of genetic variation and function, from March 2011 to January 2012. Prior to Illumina, Dr. Shoemaker was a Graduate Student and Postdoctoral Researcher at the University of California, San Diego from 2005 until 2011. Dr. Shoemaker has a Ph.D. and M.S. in chemistry, B.S. in biochemistry, and B.A. in German literature from the University of California, San Diego.
Term of Office of Directors
Our directors are elected at each annual meeting of stockholders and serve until the next annual meeting of stockholders or until their successor has been duly elected and qualified, or until their earlier death, resignation or removal.
Family Relationships
There are no family relationships among any of our current or former directors or executive officers.
Involvement in Certain Legal Proceedings
None of our directors, executive officers, significant employees, promoters or control persons has been involved in any legal proceeding in the past 10 years that would require disclosure under Item 401(f) of Regulation S-K promulgated under the Securities Act.
Committees of the Board of Directors
Our Board of Directors has not established a separate standing audit committee within the meaning of Section 3(a)(58)(A) of the Exchange Act or separate standing nominating or compensation committees, or committees performing similar functions, nor has it adopted charters for any such committee. Due to the present and prior size of our Board of Directors, our Board of Directors believes that it is not necessary to have separate standing audit, nominating or compensation committees at this time because the functions of each such committee are adequately performed by our full Board of Directors. However, it is anticipated that our Board of Directors will form separate standing audit, nominating and compensation committees, with the audit committee including an audit committee financial expert and the audit and compensation committees consisting solely of independent directors, if and when our Board of Directors determines that the establishment of such committees is advisable as we seek to further develop our business and operations and potentially expand the size of our Board of Directors.
69
Table of Contents
Nominations to the Board of Directors
Director candidates are considered based upon various criteria, including without limitation their broad-based business and professional skills and experiences, knowledge of the industry in which we operate and ability to add perspectives relating to that industry, expertise in the precision medicine biotechnology field, concern for the long-term interests of our stockholders, diversity, and personal integrity and judgment. Our Board of Directors has a critical role in guiding our strategic direction and overseeing the management of our business, and accordingly, we seek to attract and retain highly qualified directors who have sufficient time to engage in the activities of our Board of Directors and to understand and enhance their knowledge of our industry and business plans.
Stockholder Communications
Although we do not have a formal policy regarding stockholder communications with our Board of Directors, stockholders may communicate with our Board of Directors, or any individual director on our Board of Directors, by writing to us at the address of our principal executive offices, addressing the communication to the attention of our Chief Executive Officer, and specifying the Board of Directors or, if applicable, the individual member thereof as the intended recipient of the communication.
Board Leadership Structure and Role in Risk Oversight
Jonathan E. Lim, M.D. currently serves as our principal executive officer and the Chairman of the Board of Directors. Although the Board of Directors does not have a formal policy regarding whether the same person should serve as both the principal executive officer and the Chairman of the Board, the Board of Directors has determined that appointing Dr. Lim to both such positions is presently in the best interests of Ignyta and its stockholders. Dr. Lim’s founding role with Ignyta Operating provides him with an in-depth knowledge of the industry and strategic priorities of the Combined Company, and his positions as the principal executive officer and Chairman of the Board enable him to facilitate effective communication among management and the Board of Directors, providing an effective, aligned leadership structure for our present operations. The Board of Directors will continue to evaluate our leadership structure and modify it as appropriate based on the size, resources and operations of the Combined Company.
The role of our Board of Directors is to oversee our risk management function. Members of our management team report to our Board of Directors on areas of material risk to us, including operational, financial, legal and regulatory, and strategic and other risks, and provide it with all information necessary to enable our directors to develop a fulsome understanding of the applicable risk and conduct an evaluation of the risk and management’s manner of addressing it. If an identified area of risk poses an actual or potential conflict with management, our non-employee directors may conduct the evaluation. It is anticipated that our Board of Directors will in the future establish more formal procedures regarding the scope and administration of its risk oversight role.
Compensation Committee Interlocks and Insider Participation
Our Board of Directors has not established a separate standing compensation committee. None of our current or former executive officers serves, or during our last completed fiscal year has served, as a member of the board of directors or compensation committee, or other committee serving an equivalent function, of any other entity that has one or more of its executive officers serving as a member of our Board of Directors.
Code of Ethics
We have not adopted a formal code of ethics within the meaning of Item 406 of Regulation S-K promulgated under the Securities Act that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and that that establishes, among other things, procedures for handling actual or apparent conflicts of interest. Our Board of Directors intends to adopt such a formal code of ethics when it deems appropriate based on the size of our operations and personnel.
70
Table of Contents
From the inception of Ignyta to the date of this report, no compensation has been earned by or paid to any of Ignyta’s named executive officers, which have consisted of (i) its principal executive officer, and (ii) its next most highly compensated executive officer other than its principal executive officer serving as an executive officer as of the end of its most recently completed fiscal year and whose total compensation exceeded $100,000 during that fiscal year (of which there were none).
Ignyta Operating became our wholly owned subsidiary upon the closing of the Merger on October 31, 2013. The following table summarizes the compensation earned in each of Ignyta Operating’s fiscal years ended December 31, 2012 and 2011 by its named executive officers, which consist of (i) its principal executive officer, and (ii) its next most highly compensated executive officer other than its principal executive officer serving as an executive officer as of December 31, 2012 and whose total compensation exceeded $100,000 in during the year ended December 31, 2012 (of which there were none).
Summary Compensation Table
| Name and Principal Position |
Year ended December 31, |
Salary | Stock awards ($) |
Option awards ($) |
All other compensation ($) |
Total | ||||||||||||||||||
| Jonathan E. Lim, M.D., |
2012 | $ | 45,000 | — | — | — | $ | 45,000 | ||||||||||||||||
| President and CEO (1) |
2011 | $ | — | — | — | — | $ | — | ||||||||||||||||
| (1) | Dr. Lim co-founded Ignyta Operating in August 2011, but did not become an employee of Ignyta Operating and did not begin earning compensation for the services he performed for Ignyta Operating, as an employee or otherwise, until July 1, 2012. During the period from July 1, 2012 through December 31, 2012, Dr. Lim’s annual base salary for his service as an employee of Ignyta Operating was $100,000, of which he earned a pro-rated amount in the 2012 fiscal year as reflected in the table above based on the term of his service as an employee of Ignyta Operating during that fiscal year. Effective as of January 1, 2013, Dr. Lim’s annual base salary was increased to $250,000. |
Upon Dr. Lim’s appointment as President and Chief Executive Officer of Ignyta immediately following the closing of the Merger on October 31, 2013, the members of our Board of Directors, excluding Dr. Lim, approved his receipt of an annual base salary of $250,000, which maintains the compensation earned by Dr. Lim immediately prior to the closing of the Merger as an executive officer of Ignyta Operating. The amount of Dr. Lim’s annual base salary or any other compensation he may receive as an executive officer of Ignyta may be modified at any time at the discretion of our Board of Directors.
Employment Agreements
Ignyta Operating does not have, and has not in the past had, formal employment agreements with its named executive officer or any of its other employees, who all have served as “at will” employees.
Immediately following the closing of the Merger on October 31, 2013, Dr. Lim was appointed as the President, Chief Executive Officer and Chairman of the Board of Ignyta. He will serve in those positions as an “at will” employee of Ignyta, and will not have a formal employment agreement with Ignyta unless and until Dr. Lim and our Board of Directors, or a committee thereof, approve the terms of any such agreement.
Outstanding Equity Awards at Fiscal Year-End
There were no outstanding equity compensation awards held by Ignyta Operating’s named executive officer as of the end of its last completed fiscal year on December 31, 2012.
Director Compensation
Dr. Lim has been the sole director of Ignyta Operating since its inception. Dr. Lim received no compensation for his service as a director of Ignyta Operating during the fiscal year ended December 31, 2012 that is not reflected under the heading “Summary Compensation Table” above.
71
Table of Contents
Potential Payments upon Termination or Change in Control
Except as described below, neither Ignyta nor Ignyta Operating has, and neither such entity had immediately prior to the closing of the Merger, any agreements, plans or arrangements that provide for payments or benefits to their respective named executive officers in connection with the resignation, retirement or other termination of a named executive officer, a change in control of the applicable entity, or a change in a named executive officer’s responsibilities following a change in control of the applicable entity.
The Ignyta Plan, which was assumed by Ignyta upon the closing of the Merger, provides that the administrator of the plan has the authority to provide for the full or partial automatic vesting and exercisability of outstanding unvested awards under the Ignyta Plan in connection with certain corporate events and change in control transactions. The named executive officer of Ignyta Operating for its fiscal year ended December 31, 2012 and the executive officers of Ignyta as of immediately following the closing of the Merger hold outstanding option awards granted under the Ignyta Plan and may be granted option or other equity awards in the future under the Ignyta Plan. There has been no acceleration of vesting or exercisability for any outstanding options under the Ignyta Plan, in connection with the Merger or any other corporate event or change in control transaction of Ignyta Operating.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Related Party Transactions
Ignyta
Except as described below and except for employment compensation, since our inception in August 2012, there has not been, nor is there currently proposed, any transaction to which we are or were a party in which the amount involved exceeds the lesser of $120,000 and 1% of the average of our total assets at year-end for the last two completed fiscal years, and in which any of our current directors, executive officers, holders of more than 5% of any class of our voting securities or any of their respective affiliates or immediate family members, had, or will have, a direct or indirect material interest.
We have entered into indemnification agreements with each of our directors and executive officers. Each of those indemnification agreements is in the form approved by our Board of Directors. Reference is made to the description of the indemnification agreements included under the heading “Indemnification of Directors and Officers”, which description is incorporated herein by reference. The description of the indemnification agreements set forth in this report is qualified in its entirety by reference to the full text of the form indemnification agreement, which is attached hereto as Exhibit 10.10 and is incorporated herein by reference.
Each of our directors and officers that was an investor in or held a director, officer or other position with Ignyta Operating prior to the closing of the Merger, which includes all of our current directors and executive officers, were issued shares of our common stock as consideration for the cancellation of their equity holdings in Ignyta Operating upon the closing of the Merger. See the information under the heading “Security Ownership of Certain Beneficial Owners and Management” for information about each such party’s current beneficial ownership in our common stock.
Ignyta Operating
Except as described below and except for employment compensation, since the inception of Ignyta Operating in August 2011, there has not been, nor is there currently proposed, any transaction to which it was or is a party in which the amount involved exceeds the lesser of $120,000 and 1% of the average of Ignyta Operating’s total assets at year-end for the last two completed fiscal years, and in which any of its directors, executive officers, holders of more than 5% of any class of our voting securities or any of their respective affiliates or immediate family members, had, or will have, a direct or indirect material interest.
On July 26, 2011 and March 21, 2012, City Hill Venture Partners I, LLC (City Hill) purchased an aggregate of 833,334 shares of the series A preferred stock of Ignyta Operating, for a per share purchase price of $0.60 and an aggregate purchase price of $500,000. In addition, (i) on June 22, 2012, City Hill and Alexander Casdin, among other investors, purchased shares of the series B preferred stock of Ignyta Operating for a per share purchase price of $3.00, with City Hill purchasing 500,000 shares for an aggregate purchase price of $1,500,000, and Mr. Casdin purchasing 33,334 shares for an aggregate
72
Table of Contents
purchase price of $100,000, and (ii) on December 21, 2012, City Hill, Mr. Casdin and Zachary Hornby, among other investors, purchased shares of the series B preferred stock of Ignyta Operating for a per share purchase price of $3.00, with City Hill purchasing an additional 83,334 shares for an aggregate purchase price of $250,000, Mr. Casdin purchasing an additional 16,667 shares for an aggregate purchase price of $50,000, and Mr. Hornby purchasing 8,334 shares for an aggregate purchase price of $25,000. Jonathan E. Lim, the current President, Chief Executive Officer and Chairman of the Board of Ignyta and the sole director of Ignyta Operating, is the Managing Partner of City Hill, and City Hill was a holder of more than 5% of the outstanding capital stock of Ignyta Operating prior to the closing of the Merger and is a holder of more than 5% of the outstanding capital stock of Ignyta following the closing of the Merger and on the date of this report. Mr. Casdin is currently a member of the Board of Directors of Ignyta. Mr. Hornby currently serves as the Chief Financial Officer and Vice President, Corporate Development of Ignyta and in the same roles for Ignyta Operating.
On May 20, 2013, Ignyta Operating closed its acquisition of Actagene, by way of Actagene’s merger with and into Ignyta Operating. As consideration for the cancellation of the shares of Actagene held by its stockholders upon the closing of that merger, Ignyta Operating issued to each such stockholder a number of shares of Ignyta Operating common stock based on a specified ratio. Jonathan E. Lim was a director, and City Hill was the controlling stockholder, of both Ignyta Operating and Actagene immediately prior to the closing of the merger. As a result of its equity ownership of Actagene, City Hill was issued an aggregate of 1,000,000 shares of Ignyta Operating common stock as consideration upon the closing of the merger. In addition, Dr. Patrick O’Connor, our current Chief Scientific Officer and Senior Vice President, Research, was a director, executive officer and stockholder of Actagene prior to the closing of the merger. As a result of his equity ownership of Actagene, Dr. O’Connor was issued an aggregate of 500,000 shares of Ignyta Operating common stock as consideration upon the closing of the merger. Pursuant to a valuation completed shortly following the merger with Actagene, Ignyta Operating’s common stock had a fair market value of $1.02 per share as of the date of such valuation, resulting in an aggregate fair market value of approximately $1,020,000 and $510,000 of the shares of Ignyta Operating’s common stock issued to City Hill (and thereby controlled by Dr. Lim) and Dr. O’Connor, respectively, in connection with the merger with Actagene. Ignyta Operating has accounted for the merger with Actagene as a combination of entities under common control, and as a result the shares issued as merger consideration were valued for accounting purposes at $0.003 per share (see footnote 2 of our unaudited financial statements for the six months ended June 30, 2013 attached as Exhibit 99.1 to this Current Report on Form 8-K).
Review, Approval or Ratification of Transactions with Related Persons
Due to the small size of our company, we do not at this time have a formal written policy regarding the review of related party transactions, and rely on our full Board of Directors to review, approve or ratify such transactions and identify and prevent conflicts of interest. Our Board of Directors reviews any such transaction in light of the particular affiliation and interest of any involved director, officer or other employee or stockholder and, if applicable, any such person’s affiliates or immediate family members. Management aims to present transactions to our Board of Directors for approval before they are entered into or, if that is not possible, for ratification after the transaction has occurred. If our Board of Directors finds that a conflict of interest exists, then it will determine the appropriate action or remedial action, if any. Our Board of Directors approves or ratifies a transaction if it determines that the transaction is consistent with our best interests and the best interest of our stockholders.
Director Independence
In connection with the closing of the Merger, our Board of Directors undertook a review of the composition of our Board of Directors and independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our Board of Directors has determined that Mr. Alexander Casdin and Dr. Heinrich Dreismann would qualify as “independent” as that term is defined by NASDAQ Listing Rule 5605(a)(2). Further, although we do not presently have separately standing audit, nominating or compensation committees of our Board of Directors, our Board of Directors has determined that each of Mr. Casdin and Dr. Dreismann would qualify as “independent” under NASDAQ Listing Rules applicable to such board committees. Dr. Jonathan Lim would not qualify as “independent” under applicable NASDAQ Listing Rules applicable to the Board of Directors generally or to separately designated board committees because he currently serves as our President and Chief Executive Officer. In making such determinations, our Board of Directors considered the relationships that each of our non-employee directors has with the Combined Company and all other facts and circumstances deemed relevant in determining independence, including the beneficial ownership of our capital stock by each non-employee director.
Subject to some exceptions, NASDAQ Listing Rule 5605(a)(2) provides that a director will only qualify as an “independent director” if, in the opinion of our Board of Directors, that person does not have a relationship that would interfere with the
73
Table of Contents
exercise of independent judgment in carrying out the responsibilities of a director, and that a director cannot be an “independent director” if (a) the director is, or in the past three years has been, an employee of ours; (b) a member of the director’s immediate family is, or in the past three years has been, an executive officer of ours; (c) the director or a member of the director’s immediate family has received more than $120,000 per year in direct compensation from us within the preceding three years, other than for service as a director or benefits under a tax-qualified retirement plan or non-discretionary compensation (or, for a family member, as a non-executive employee); (d) the director or a member of the director’s immediate family is a current partner of our independent public accounting firm, or has worked for such firm in any capacity on our audit at any time during the past three years; (e) the director or a member of the director’s immediate family is, or in the past three years has been, employed as an executive officer of a company where one of our executive officers serves on the compensation committee; or (f) the director or a member of the director’s immediate family is an executive officer, partner or controlling shareholder of a company that makes payments to, or receives payments from, us in an amount which, in any twelve-month period during our past three fiscal years, exceeds the greater of 5% of the recipient’s consolidated gross revenues for that year or $200,000 (except for payments arising solely from investments in our securities or payments under non-discretionary charitable contribution matching programs). Additionally, in order to be considered an independent member of an audit committee under Rule 10A-3 of the Exchange Act, a member of an audit committee may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other committee of the board of directors, accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the applicable company or any of its subsidiaries or otherwise be an affiliated person of the applicable company or any of its subsidiaries.
MARKET PRICE OF AND DIVIDENDS ON THE REGISTRANT’S COMMON EQUITY
AND RELATED STOCKHOLDER MATTERS
Market Information
Our common stock is presently not traded on any market or securities exchange. Our common stock is currently quoted on the OTCBB over-the-counter quotation system under the ticker symbol “IGASD”, which will change to the ticker symbol “RXDX” on the 20th business day following the effect of our 100-to-one reverse stock split. There is not currently, and there has not been since our inception, any trading of our shares of common stock on the OTCBB or any other over-the-counter market, and as a result there is no established trading market for our common stock. As of the date of this Current Report on Form 8-K and after giving effect to the Merger, there are: (i) outstanding options to purchase up to 358,986 shares of our common stock; (ii) outstanding warrants to purchase up to 25,001 shares of our common stock; and (iii) 7,336 outstanding shares of our common stock that have been registered under the Securities Act and are freely tradeable.
Holders
As of October 31, 2013 immediately following the closing of the Merger, there were 65 holders of record of our common stock.
Dividends
As of the date of this Current Report on Form 8-K, other than the dividend declared in connection with the Merger, we have never declared nor paid any cash dividends to stockholders. We do not intend to pay cash dividends on our common stock for the foreseeable future, and currently intend to retain any future earnings to fund our operations and the development and growth of our business. The declaration of any future cash dividend, if any, would be at the discretion of our Board of Directors and would depend upon our earnings, if any, our capital requirements and financial position, our general economic conditions, and other pertinent conditions.
Shares Eligible for Future Sale
Upon the completion of the Merger, we had 4,923,805 shares of common stock outstanding, of which our directors and executive officers beneficially own an aggregate of 3,591,039 shares. Of those outstanding shares, 7,336 shares of our common stock are freely tradeable, without restriction, as of the date of this Current Report on Form 8-K. No shares issued in connection with the Merger can be publicly sold under Rule 144 promulgated under the Securities Act until 12 months after the date of filing this Current Report on Form 8-K.
74
Table of Contents
In general, Rule 144 provides that (i) any of our non-affiliates that has held restricted common stock for at least 12 months is thereafter entitled to sell its restricted stock freely and without restriction, provided that we remain compliant and current with our SEC reporting obligations, and (ii) any of our affiliates, which includes our directors, executive officers and other person in control of us, that has held restricted common stock for at least 12 months is thereafter entitled to sell its restricted stock subject to the following restrictions: (a) we are compliant and current with our SEC reporting obligations, (b) certain manner of sale provisions are satisfied, (c) a Form 144 is filed with the SEC, and (d) certain volume limitations are satisfied, which limit the sale of shares within any three-month period to a number of shares that does not exceed the greater of 1% of the total number of outstanding shares. A person who has ceased to be an affiliate at least three months immediately preceding the sale and who has owned such shares of common stock for at least one year is entitled to sell the shares under Rule 144 without regard to any of the limitations described above.
Securities Authorized for Issuance under Equity Compensation Plans
Equity Compensation Plan Information
In October 2011, Ignyta Operating’s Board of Directors and the holders of at least a majority of its then-outstanding capital stock approved and adopted the Ignyta Plan. Ignyta Operating has not adopted any other stockholder-approved or non-stockholder approved equity compensation plans.
The following table provides information with respect to the Ignyta Plan as of December 31, 2012:
| Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) |
Weighted-average exercise price of outstanding options, warrants and rights (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) |
||||||||||
| Plan Category |
||||||||||||
| Equity compensation plans approved by security holders |
156,659 | $ | 0.36 | 10,007 | ||||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
| Total |
156,659 | $ | 0.36 | 10,007 | ||||||||
In February 2013, Ignyta Operating’s Board of Directors and the holders of at least a majority of its then-outstanding capital stock approved an amendment to the Ignyta Plan to, among other things, increase the number of shares of its common stock available for issuance thereunder from 166,666 shares to 666,666 shares. Effective as of immediately prior to the closing of the Merger on October 31, 2013, Ignyta Operating’s Board of Directors and the holders of at least a majority of its then-outstanding capital stock approved a further amendment to the Ignyta Plan to, among other things, increase the number of shares of its common stock available for issuance thereunder from 666,666 shares to 712,652 shares. Effective upon the closing of the Merger, Ignyta assumed the Ignyta Plan and the obligation to issue all outstanding options and other awards outstanding thereunder as of the closing of the Merger.
The following table provides information with respect to the Ignyta Plan as of immediately prior to the closing of the Merger on October 31, 2013:
| Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) |
Weighted-average exercise price of outstanding options, warrants and rights (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) |
||||||||||
| Plan Category |
||||||||||||
| Equity compensation plans approved by security holders |
358,986 | $ | 0.71 | 296,223 | ||||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
| Total |
358,986 | $ | 0.71 | 296,223 | ||||||||
75
Table of Contents
Description of the Ignyta Plan
The purpose of the Ignyta Plan is to attract and retain the best available personnel and provide additional incentives to such personnel through the grant of equity awards. The Ignyta Plan permits the grant of a variety of forms of awards to employees, directors and/or consultants of Ignyta or its affiliated companies. The total number of shares reserved for issuance under the Ignyta Plan is 712,652 shares and, after accounting for outstanding awards granted under the Ignyta Plan prior to the closing of the Merger, the number of shares of our common stock reserved for issuance pursuant to future grants under the Ignyta Plan is 342,209 shares. Our Board of Directors currently serves as the administrator of the Ignyta Plan. As of the date of this Current Report on Form 8-K, the only equity awards that have been granted under the Ignyta Plan are those that were granted by Ignyta Operating prior to the closing of the Merger and have been assumed by us upon the closing of the Merger.
The following is a summary of the principal provisions of the Ignyta Plan, as presently in effect following the assumption thereof by Ignyta upon the closing of the Merger. The below summary is not a complete description of the Ignyta Plan and is qualified in its entirety by reference to the Ignyta Plan, which is attached to this Current Report on Form 8-K as Exhibit 10.1.
Types and Terms of Awards. The Ignyta Plan provides for the grant of stock options, restricted stock, restricted stock units, dividend equivalent rights, and stock appreciation rights. Stock options granted under the Ignyta Plan may be either incentive stock options, or non-qualified stock options. Incentive stock options may be granted only to employees. Awards other than incentive stock options may be granted to employees, consultants and directors of us or our related entities. To the extent that the aggregate fair market value of the shares subject to stock options designated as incentive stock options that become exercisable for the first time by a participant during any calendar year exceeds $100,000, such excess stock options will be treated as non-qualified stock options. The terms of each award granted under the Ignyta Plan will be designated in an award agreement.
Awards may be granted subject to vesting schedules and restrictions on transfer and repurchase or forfeiture rights in favor of Ignyta as determined by the administrator and as specified in the applicable award agreement under the Ignyta Plan. The administrator generally has the authority, in its discretion, to select employees, consultants and directors to whom awards may be granted from time to time, to determine whether and to what extent awards are granted, to determine the number of shares or the amount of other consideration to be covered by each award (subject to certain limitations), to approve award agreements for use under the Ignyta Plan, to determine the terms and conditions of any award (including the vesting schedule applicable to the award), to amend the terms of any outstanding award granted under the Ignyta Plan (subject to certain limitations), to construe and interpret the terms of the Ignyta Plan and awards granted thereunder, to establish additional terms, conditions, rules or procedures to accommodate the rules or laws of applicable non-U.S. jurisdictions, and to take such other action not inconsistent with the terms of the Ignyta Plan, as the administrator deems appropriate.
The term of any award granted under the Ignyta Plan will be stated in the applicable award agreement, provided that the term may not exceed 10 years (or five years in the case of an incentive stock option granted to any participant who owns stock representing more than 10% of our combined voting power or any parent or subsidiary of us).
The Ignyta Plan authorizes the administrator to grant stock options at an exercise price not less than 100% of the fair market value of our common stock on the date the stock option is granted (or 110%, in the case of an incentive stock option granted to any employee who owns stock representing more than 10% of our combined voting power or any parent or subsidiary of us). In the case of stock appreciation rights, and awards intended to qualify as performance-based compensation, the base appreciation amount or purchase price, if any, shall be not less than 100% of the fair market value per share on the date of grant. In the case of all other awards granted under the Ignyta Plan, the exercise or purchase price shall be determined by the administrator.
Section 162(m) of the Internal Revenue Code. The maximum number of shares with respect to which options and stock appreciation rights may be granted to a participant during a calendar year is 666,666 shares, provided that a participant may
76
Table of Contents
be granted options and stock appreciation rights for an additional 333,333 shares in connection with the commencement of service for Ignyta. For awards of restricted stock and restricted stock units that are intended to be performance-based compensation under Section 162(m) of the Internal Revenue Code (the Code), the maximum number of shares subject to those awards that may be granted to a participant during a calendar year is 333,333 shares.
In order for restricted stock and restricted stock units to qualify as performance-based compensation, the administrator must establish a performance goal with respect to such award in writing not later than 90 days after the commencement of the services to which it relates (or, if earlier, the date after which 25% of the period of service to which the performance goal relates has elapsed) and while the outcome is substantially uncertain. In addition, the performance goal must be stated in terms of an objective formula or standard.
The Ignyta Plan includes the following performance criteria that may be considered by the administrator when granting performance-based awards: increase in share price, earnings per share, total stockholder return, return on equity, return on assets, return on investment, net operating income, cash flow, revenue, economic value added, personal management objectives, or other measure of performance selected by the administrator of the Ignyta Plan.
Certain Adjustments. Subject to any required action by our stockholders, the number of shares covered by outstanding awards, the number of shares that have been authorized for issuance under the Ignyta Plan, the exercise or purchase price of each outstanding award, the maximum number of shares or amount that may be granted subject to awards to any participant, and the like, shall be proportionally adjusted by the administrator of the Ignyta Plan in the event of (i) any increase or decrease in the number of issued shares resulting from a stock split, reverse stock split, stock dividend, combination or reclassification or similar event affecting the shares, (ii) any other increase or decrease in the number of issued shares effected without receipt of consideration by Ignyta, or (iii) any other transaction with respect to our shares including a corporate merger, consolidation, acquisition of property or stock, separation (including a spin-off or other distribution of stock or property), reorganization, liquidation (whether partial or complete), distribution of cash or other assets to stockholders other than a normal cash dividend, or any similar transaction; provided, however, that conversion of any convertible securities of Ignyta shall not be deemed to have been “effected without receipt of consideration.”
Corporate Transaction and Change in Control. The administrator of the Ignyta Plan shall have the authority, exercisable either in advance of any actual or anticipated Corporate Transaction or Change in Control, as those terms are defined in the Ignyta Plan, or at the time of an actual Corporate Transaction or Change in Control and exercisable at the time of the grant of an award under the Ignyta Plan or any time while an award remains outstanding, to provide for the full or partial automatic vesting and exercisability of one or more outstanding unvested awards under the Ignyta Plan and the full or partial release from restrictions on transfer and repurchase or forfeiture rights of such awards in connection with a Corporate Transaction or Change in Control, on such terms and conditions as the administrator may specify. The Administrator also shall have the authority to condition any such award vesting and exercisability or release from such limitations upon the subsequent termination of the continuous service of the participant within a specified period following the effective date of the Corporate Transaction or Change in Control. The administrator may provide that any awards so vested or released from such limitations in connection with a Change in Control shall remain fully exercisable until the expiration or earlier termination of the award. Upon the consummation of a Corporate Transaction, all outstanding awards shall terminate unless assumed by the successor corporation or its parent.
Amendment, Suspension or Termination of the Plan. The Ignyta Board of Directors may at any time amend, suspend or terminate the Ignyta Plan. The Ignyta Plan will terminate on October 7, 2021, unless earlier terminated by the Ignyta Board of Directors. To the extent necessary to comply with applicable provisions of federal securities laws, state corporate and securities laws, the Code, applicable rules of any stock exchange or national market system, and the rules of any foreign jurisdiction applicable to awards granted to residents of the jurisdiction, we shall obtain stockholder approval of any such amendment to the Ignyta Plan in such a manner and to such a degree as so required.
RECENT SALES OF UNREGISTERED SECURITIES
Ignyta
In August 2012, Ignyta issued 160,000 shares of its common stock to Betty Sytner, one of its officers and sole director, pursuant to the exemption from registration contained in Section 4(a)(2) of the Securities Act. Ms. Sytner was
77
Table of Contents
supplied with the same information that could be found in a registration statement on Form S-1 and is a sophisticated investor.
Reference is made to the disclosure set forth under Item 3.02 of this Current Report on Form 8-K, which is incorporated herein by reference.
Ignyta Operating
Upon its inception in August 2011, Ignyta Operating issued to four individuals that founded the company an aggregate of 666,668 shares of its common stock at a purchase price per share of $0.003 and for an aggregate purchase price of $2,000. The issuance and sale of such securities was not registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act. In determining that the issuance of such securities qualified for an exemption under Section 4(a)(2) of the Securities Act, Ignyta Operating relied on the following facts: the securities were issued to recipients that each represented that it was an “accredited investor” as defined in Rule 501 promulgated under the Securities Act, it was acquiring the securities for investment purposes and without a view toward disposition thereof and it had sufficient investment experience to evaluate the risks of the investment; Ignyta Operating used no advertising or general solicitation in connection with the issuance and sale of the securities; and the securities were issued as restricted securities.
In October 2011, Ignyta Operating issued and sold to one investor 416,667 shares of its series A preferred stock at a purchase price per share of $0.60 and for an aggregate purchase price of $250,000. In March 2012, Ignyta Operating issued and sold to the same investor an additional 416,667 share of its series A preferred stock at a purchase price per share of $0.60 and for an aggregate purchase price of $250,000. The issuance and sale of such securities was not registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. In determining that the issuance of such securities qualified for an exemption under Section 4(a)(2) of the Securities Act, Ignyta Operating relied on the following facts: the securities were issued to one investor that represented that it was an “accredited investor” as defined in Rule 501 promulgated under the Securities Act, it was acquiring the securities for investment purposes and without a view toward disposition thereof, and it had sufficient investment experience to evaluate the risks of the investment; Ignyta Operating used no advertising or general solicitation in connection with the issuance and sale of the securities; and the securities were issued as restricted securities.
In June 2012, Ignyta Operating issued and sold to six investors an aggregate of 700,000 shares of its series B preferred stock at a purchase price per share of $3.00 and for an aggregate purchase price of $2,100,000. In December 2012, Ignyta Operating issued and sold to fifteen investors, including three investors from the June 2012 sales, an aggregate of 1,135,000 shares of its series B preferred stock at a purchase price per share of $3.00 and for an aggregate purchase price of $3,405,000. The issuance and sale of such securities was not registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. In determining that the issuance of such securities qualified for an exemption under Section 4(a)(2) of the Securities Act, Ignyta Operating relied on the following facts: the securities were issued to investors that each represented that it was an “accredited investor” as defined in Rule 501 promulgated under the Securities Act, it was acquiring the securities for investment purposes and without a view toward disposition thereof, and it had sufficient investment experience to evaluate the risks of the investment; Ignyta Operating used no advertising or general solicitation in connection with the issuance and sale of the securities; and the securities were issued as restricted securities.
In June 2012, upon entry into and as a condition of a loan and security agreement with Silicon Valley Bank (SVB), Ignyta Operating issued to SVB a warrant to acquire up to 8,334 shares of its series B preferred stock at an exercise price of $3.00 per share. In February 2013, upon entry into and as a condition of an amendment to the loan agreement relating to an increase in the amount funded thereunder, Ignyta Operating issued to SVB an additional warrant to acquire up to a number of shares of its series B preferred stock equal to 5% of the amount funded by SVB under the loan agreement after the date of issuance of the warrant, resulting in an aggregate of 16,667 shares of its series B preferred stock following SVB’s two advances under the loan agreement after such date of issuance, at an exercise price of $3.00 per share. Each such warrant is exercisable at any time at the option of the holder until the seven-year anniversary of its date of issuance. Pursuant to and in accordance with its terms, each such warrant, (i) upon the conversion of Ignyta Operating’s preferred stock into common stock prior to the closing of the Merger, was converted into a warrant to acquire the same number of shares of Ignyta Operating’s common stock, and (ii) upon the closing of the Merger, was assumed by Ignyta and became a warrant to acquire the number of shares of Ignyta’s common stock that would have been issued to the warrantholder on the closing of the Merger had the warrant been fully exercised at such time. The issuance and sale of those warrants and any shares issuable upon their exercise has not
78
Table of Contents
been registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act. In determining that the issuance of such securities qualified for an exemption under Section 4(a)(2) of the Securities Act, Ignyta Operating relied on the following facts: the securities were issued to one recipient that represented that it was an “accredited investor” as defined in Rule 501 promulgated under the Securities Act, it was acquiring the securities for investment purposes and without a view toward disposition thereof, and it had sufficient investment experience to evaluate the risks of the investment; Ignyta Operating used no advertising or general solicitation in connection with the issuance and sale of the securities; and the securities were issued as restricted securities.
In May 2013, Ignyta Operating acquired Actagene by way of Actagene’s merger with and into Ignyta Operating on May 20, 2013. All consideration paid by Ignyta Operating in connection with its acquisition of Actagene was paid with shares of its common stock, totaling an aggregate of 1,583,336 shares. The issuance and sale of such securities was not registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. In determining that the issuance of such securities qualified for an exemption under Section 4(a)(2) of the Securities Act, Ignyta Operating relied on the following facts: the securities were issued to parties that each represented that it was an “accredited investor” as defined in Rule 501 promulgated under the Securities Act, it was acquiring the securities for investment purposes and without a view toward disposition thereof, and it had sufficient investment experience to evaluate the risks of the investment; Ignyta Operating used no advertising or general solicitation in connection with the issuance and sale of the securities; and the securities were issued as restricted securities.
Since its inception in August 2011, Ignyta Operating has granted, under the Ignyta Operating Plan, options to purchase up to an aggregate of 442,983 shares of its common stock to 31 different employees, consultants or other service providers as compensation for services rendered to Ignyta Operating. The issuance of those options and any shares issuable upon their exercise has not been registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration under Rule 701 promulgated under the Securities Act. In determining that the issuance of such securities qualified for an exemption under Rule 701 promulgated under the Securities Act, Ignyta Operating relied on the following facts: the securities were issued under the Ignyta Operating Plan, a written compensatory benefit plan intended to comply with Rule 701; the recipients of the securities were bona fide service providers to Ignyta Operating; and the securities were issued as restricted securities.
The following describes the material terms of the capital stock of Ignyta. The following description does not purport to be complete and is subject to, and qualified in its entirety by reference to, Ignyta’s Amended and Restated Articles of Incorporation and Bylaws, which are attached as Exhibits 3.1 and 3.2, respectively, to this Current Report on Form 8-K. All Ignyta stockholders are urged to read our Amended and Restated Articles of Incorporation and Bylaws carefully and in their entirety.
Authorized Capital Stock; Issued and Outstanding Capital Stock
Effective October 31, 2013, we amended and restated our Articles of Incorporation to increase our authorized common stock, par value $0.00001 per share from 25,000,000 shares to 100,000,000 shares of common stock and authorize 10,000,000 shares of preferred stock, par value $0.00001 per share.
Also on October 31, 2013, in connection with the Merger and pursuant to our Amended and Restated Articles of Incorporation, we effected a reverse stock split at a ratio of 100-to-one, such that each 100 shares of our common stock issued and outstanding immediately prior to the effective time of the reverse stock split was automatically combined and converted, without any action on the part of the stockholder thereof, into one fully paid and nonassessable share of our common stock. All share information in this Current Report on Form 8-K with respect to our common stock gives retroactive effect to that reverse stock split. Also on October 31, 2013 and immediately following the effect of the reverse stock split, we declared a $3.50 per share cash dividend to our stockholders of record and repurchased 80,000 shares of our common stock at a price of $0.99 per share from our principal stockholder, each on a post reverse stock split basis.
As of October 31, after giving effect to the closing of the Merger, there were a total of 4,923,805 shares of our common stock issued and outstanding and no shares of preferred stock issued and outstanding.
79
Table of Contents
Common Stock
The holders of our common stock are entitled to one vote per share on all matters submitted to vote of our stockholders, including the election of directors. Holders of our common stock are not entitled to cumulate their votes for the election of directors. Except as otherwise required by law, or as otherwise fixed by resolution or resolutions of our Board of Directors with respect to one or more series of our preferred stock, the entire voting power and all voting rights shall be vested exclusively in our common stock.
Holders of our common stock will not be entitled to receive dividends except if declared by our Board of Directors and will not be entitled to a liquidation preference in respect of their shares of common stock. Upon liquidation, dissolution or winding up of our company, the holders of our common stock will be entitled to receive pro rata all assets remaining for distribution to stockholders after the payment of all of our liabilities and of all preferential amounts to which any series of our preferred stock may be entitled.
Holders of our common stock will have no preemptive or subscription rights, and will have no rights to convert their common stock into any other securities. The common stock will not be subject to call or redemption.
Preferred Stock
Our Amended and Restated Articles of Incorporation authorize our Board of Directors to fix or alter the dividend rights, dividend rate, conversion rights, voting rights, rights and terms of redemption (including sinking fund provisions), redemption price or prices, and dissolution preferences or any wholly unissued series of our preferred stock, and the number of shares constituting any such series and the designation thereof, or any of them. Our Amended and Restated Articles of Incorporation also provide that our Board of Directors is expressly authorized to increase or decrease (but not below the number of shares of such series of preferred stock then outstanding) the number of shares of any series of preferred stock subsequent to the issue of shares of that series.
Anti-Takeover Provisions
Nevada law contains provisions governing the combination of a Nevada corporation with two hundred (200) or more stockholders of record with an “interested shareholder” (a person who is the direct or indirect beneficial owner of ten percent (10%) or more of the voting power of the outstanding voting shares of a corporation), which generally prohibit such transaction for three (3) years following the time that such person becomes an “interested shareholder,” subject to certain exceptions. Generally, such provisions may have the effect of delaying or making it more difficult to effect a change in control of our company. Pursuant to our Amended and Restated Articles of Incorporation, we have elected to opt out of these provisions, which will permit us to engage in such transactions with “interested shareholders.”
Nevada law also contains provisions governing acquisition of a controlling interest of a Nevada corporation. These provisions provide generally that any person or entity that acquires a certain percentage of the outstanding voting shares of a Nevada corporation may be denied voting rights with respect to the acquired shares, unless the holders of a majority of the voting power of the corporation, excluding shares as to which any of such acquiring person or entity, an officer or a director of the corporation, and an employee of the corporation exercises voting rights, elect to restore such voting rights in whole or in part. These provisions apply whenever a person or entity acquires shares that, but for the operation of these provisions, would bring voting power of such person or entity in the election of directors within any of the following three ranges: (i) 20% or more but less than 33 1⁄3%; (ii) 33 1⁄3% or more but less than or equal to 50%; or (iii) more than 50%. The stockholders or board of directors of a corporation may elect to exempt the stock of the corporation from these provisions through adoption of a provision to that effect in its articles of incorporation or bylaws. Our articles of incorporation and bylaws do not exempt our common stock from these provisions. These provisions are applicable only to a Nevada corporation that: (i) has 200 or more stockholders of record, at least 100 of whom have addresses in Nevada appearing on the stock ledger of the corporation; and (ii) does business in Nevada directly or through an affiliated corporation. As a result, we do not presently believe that Nevada’s controlling interest acquisition provisions are currently applicable to us.
Our Amended and Restated Articles of Incorporation and Bylaws may delay or discourage transactions involving an actual or potential change of control of our company or change in our Board of Directors, including transactions in which our stockholders might otherwise receive a premium for their shares of our common stock, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our
80
Table of Contents
common stock. Among other things, our Amended and Restated Articles of Incorporation and Bylaws and applicable Nevada law:
| • | permit our Board of Directors to issue up to 10,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate (including dividend rights, dividend rate, conversion rights, voting rights, rights and terms of redemption (including sinking fund provisions), redemption price or prices, and dissolution preferences); |
| • | provide that, subject to the rights of any series of preferred stock to elect directors, directors may only be removed, subject to any limitation imposed by law, by the holders of at least 2⁄3 of the voting power of all of our then-outstanding shares of the capital stock entitled to vote generally at an election of directors; |
| • | provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by a vote of a majority of directors then in office; and |
| • | do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose). |
The amendment of any of these provisions would require approval by the majority of our Board of Directors or by holders of at least a majority of the voting power of all of our then-outstanding common stock entitled to vote, voting together as a single class.
Warrants
SVB Warrants
In connection with the loan and security agreement between Ignyta Operating and SVB, Ignyta Operating issued to SVB two warrants to acquire shares of its capital stock. The first such warrant was issued on June 25, 2012, has a term of exercise of seven years, and, as of immediately prior to the closing of the Merger, provided for the purchase by SVB of up to 8,334 fully paid and nonassessable shares of Ignyta Operating’s series B preferred stock at an exercise price of $3.00 per share. The second such warrant was issued on February 27, 2013 and the number of shares issuable thereunder increased on July 19, 2013, has a term of exercise of seven years, and, as of immediately prior to the closing of the Merger, provided for the purchase by SVB of up to 16,667 fully paid and nonassessable shares of Ignyta Operating’s series B preferred stock at an exercise price of $3.00 per share. The terms of each such warrant provide for anti-dilution and other adjustments in the event of certain stock dividends, stock splits, recapitalizations, reclassifications and consolidations.
In connection with the conversion of all issued and outstanding shares of all series of Ignyta Operating’s preferred stock and, immediately thereafter, the reverse stock split of all issued and outstanding shares of Ignyta Operating’s common stock, immediately prior to the closing of the Merger the warrants issued to SVB converted into, collectively, a right to acquire up to 25,001 shares of Ignyta Operating’s common stock at an exercise price per share equal to $3.00. Upon the closing of the Merger, pursuant to and in accordance with the terms and provisions of the warrants and the Merger Agreement, such warrants were assumed by us and have become exercisable for up to 25,001 shares of our common stock at an exercise price of $3.00 per share. The aggregate purchase price of each of the warrants issued to SVB may be paid by either cash or, at the option of SVB, through a customary cashless exercise process. The warrants issued to SVB are attached to this Current Report on Form 8-K as Exhibits 10.5 and 10.6, respectively, and are incorporated herein by reference.
NMS Warrant
Pursuant to the terms of the license agreement between Ignyta Operating and NMS, we have agreed to issue to NMS, upon the completion of a financing of equity or debt securities pursuant to which we or our affiliates receive gross proceeds of at least $20 million, a warrant to acquire up to 16,667 shares of our common stock. That warrant is to have an exercise price per share equal to that used in the financing that causes the license agreement with NMS to become effective, and is to be exercisable at the option of NMS, in whole or in part, at any time after the effective date of the license agreement until the five-year anniversary thereof. The license agreement between Ignyta Operating and NMS is attached to this Current Report on Form 8-K as Exhibit 10.4 and is incorporated herein by reference.
81
Table of Contents
NMS Equity Rights
Pursuant to the terms of the license agreement between Ignyta Operating and NMS, upon the earlier of 10 days following the effective date of the license agreement and December 31, 2013, Ignyta Operating will owe to NMS an upfront license fee totaling $7.0 million. NMS may elect to receive up to $1.0 million of that upfront license fee in the form of shares of our common stock, which would be issued at a price per share equal to that used in the financing that causes the license agreement to become effective and subject to substantially the same other terms as that financing and all applicable law, including without limitation customary representations and warranties by NMS. The license agreement between Ignyta Operating and NMS is attached to this Current Report on Form 8-K as Exhibit 10.4 and is incorporated herein by reference.
Stock Transfer Agent
Our stock transfer agent for our securities will be Olde Monmouth Stock Transfer Co., Inc., 200 Memorial Parkway, Atlantic Highlands, New Jersey 07716, telephone (732) 872-2727.
INDEMNIFICATION OF DIRECTORS AND OFFICERS
Subsection 1 of Section 78.7502 of the Nevada Revised Statutes (the Nevada Law) empowers a corporation to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the fact that he or she is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by him or her in connection with such action, suit or proceeding if he or she (i) is not liable pursuant to Section 78.138 of the Nevada Law or (ii) acted in good faith and in a manner which he or she reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful. Section 78.138 of the Nevada Law provides that, with certain exceptions, a director or officer is not individually liable to the corporation or its stockholders or creditors for any damages as a result of any act or failure to act in his or her capacity as a director or officer unless it is proven that (i) his or her act or failure to act constituted a breach of his or her fiduciary duties as a director or officer, and (ii) his or her breach of those duties involved intentional misconduct, fraud or a knowing violation of law.
Subsection 2 of Section 78.7502 empowers a corporation to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that such person acted in any of the capacities set forth above against expenses, including amounts paid in settlement and attorneys’ fees actually and reasonably incurred by him or her in connection with the defense or settlement of such action or suit if he or she (i) is not liable pursuant to Section 78.138 of the Nevada Law, or (ii) acted in good faith and in a manner which he or she reasonably believes to be in or not opposed to the best interests of the corporation, except that no indemnification may be made in respect of any claim, issue or matter as to which such person shall have been adjudged by a court of competent jurisdiction to be liable to the corporation or for amounts paid in settlement to the corporation, unless and only to the extent that the court in which such action or suit was brought or other court of competent jurisdiction determines that, in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses as the court deems proper.
Section 78.7502 further provides that to the extent that a director, officer, employee or agent of a corporation has been successful in the defense of any action, suit or proceeding referred to in subsections (1) and (2), or in the defense of any claim, issue or matter therein, he or she shall be indemnified against expenses (including attorneys’ fees) actually and reasonably incurred by him or her in connection with the defense. Subsection 3 of Section 78.751 of the Nevada Law provides that the indemnification provided for by Section 78.7502 does not exclude any other rights to which the indemnified party may be entitled (except that indemnification will generally not be available to a director or officer if a final adjudication establishes that his or her acts or omissions involved intentional misconduct, fraud or a knowing violation of the law and was material to the cause of action) and that the indemnification shall continue for directors, officers, employees or agents who have ceased to hold such positions, and inures to the benefit of their heirs, executors and administrators.
82
Table of Contents
Section 78.752 of the Nevada Law empowers the corporation to purchase and maintain insurance or make other financial arrangements on behalf of a person who is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation or other enterprise, for any liability asserted against him or her and expenses incurred by him or her in any such capacity or arising out of his or her status as such whether or not the corporation has the power to indemnify him or her against such liabilities or expenses. As of the date of this report, we have obtained a customary directors’ and officers’ liability insurance policy.
Our Amended and Restated Articles of Incorporation provide for indemnification of our directors and officers, substantially identical in scope to that permitted under the Nevada Law. Our Amended and Restated Articles of Incorporation provide, pursuant to Subsection 2 of Section 78.751, that the expenses of our directors and officers incurred in defending any action, suit or proceeding, whether civil, criminal, administrative or investigative, must be paid by us as they are incurred and in advance of the final disposition of the action, suit or proceeding, upon delivery, if required by Nevada Law, of an undertaking by or on behalf of the director or officer to repay all amounts so advanced if it is ultimately determined that the director or officer is not entitled to be indemnified by us.
We have also entered into separate indemnification agreements consistent with Nevada law and the form approved by our Board of Directors with each of our current directors and executive officers, and we contemplate entering into such indemnification agreements with directors and certain executive officers that may be elected or appointed in the future, as the case may be. The information set forth under the heading “Indemnification Agreements” in Item 1.01 of this Current Report on Form 8-K is incorporated herein by reference.
Reference is made to the financial statements and pro forma financial information relating to Ignyta Operating contained in Item 9.01 of this Current Report on Form 8-K, which is incorporated herein by reference.
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS
ON ACCOUNTING AND FINANCIAL DISCLOSURE
Reference is made to the disclosure set forth in Item 4.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
83
Table of Contents
| Item 3.02 | Unregistered Sales of Equity Securities. |
Reference is made to the disclosure set forth under Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference. Upon the closing of the Merger, we issued 4,916,469 shares of our common stock to 27 former stockholders of Ignyta Operating in exchange for all of the outstanding shares of Ignyta Operating’s capital stock. The issuance and sale of such securities was not registered under the Securities Act, and such securities were issued in reliance upon an exemption from registration afforded by Section 4(a)(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. In determining that the issuance of such securities qualified for an exemption under Section 4(a)(2) of the Securities Act, we relied on the following facts: the securities were issued to recipients that each represented that it was an “accredited investor” as defined in Rule 501 promulgated under the Securities Act, it was acquiring the securities for investment purposes and without a view toward disposition thereof, and it had sufficient investment experience to evaluate the risks of the investment; we used no advertising or general solicitation in connection with the issuance and sale of the securities; and the securities were issued as restricted securities.
| Item 3.03 | Material Modification of Rights of Security Holders. |
Reference is made to the disclosure set forth under Item 5.03 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
| Item 4.01 | Changes in Registrant’s Certifying Accountant. |
(a) Effective on October 31, 2013 and with the approval of our Board of Directors, we dismissed Dov Weinstein & Co. C.P.A (“Weinstein”) as our independent registered public accounting firm engaged to audit our financial statements.
The reports issued by Weinstein dated September 30, 2013 and September 5, 2012 relating to its audits of our balance sheets as of August 31, 2013 and 2012, and the related statements of operations, changes in stockholders’ equity and cash flows for each of the fiscal years then ended and for the period from inception (August 21, 2012) through August 31, 2013, contained an explanatory paragraph stating that there was substantial doubt about our ability to continue as a going concern. Other than as disclosed above, such reports did not contain an adverse opinion or disclaimer of opinion and were not qualified as to uncertainty, audit scope or accounting principles.
Our decision to dismiss Weinstein is not the result of any disagreement between us and Weinstein on matters of accounting principles or practices, financial statement disclosure or auditing scope or procedures. During our two most recent fiscal years, there were no disagreements with Weinstein on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedures, which disagreements, if not resolved to the satisfaction of Weinstein, would have caused Weinstein to make a reference to the subject matter of the disagreement in connection with its reports. Pursuant to the rules of the SEC applicable to smaller reporting companies, Weinstein was not required to provide an attestation as to the effectiveness of our internal control over financial reporting for any period since our inception.
Other than as disclosed above, there were no reportable events (as that term is defined in Item 304(a)(1)(v) of Regulation S-K) during our two most recent fiscal years. Our Board of Directors discussed the subject matter referred to above with Weinstein. We authorized Weinstein to respond fully and without limitation to all requests of our successor accountant concerning all matters related to the annual and interim periods audited and reviewed by Weinstein, including with respect to the subject matter of any reportable event.
We provided Weinstein with a copy of the above disclosures it is making in response to Item 4.01 of this Current Report on Form 8-K and requested that Weinstein furnish a letter addressed to the SEC stating whether or not it agrees with the above statements, and, if not, stating the respects in which it does not agree. A copy of the letter dated October 28, 2013, is filed as Exhibit 16.1 to this Current Report on Form 8-K.
(b) Effective on October 31, 2013 and with the approval of our Board of Directors, we have engaged Mayer Hoffman McCann P.C. (“MHM”) as our new independent registered public accounting firm. MHM was engaged by Ignyta Operating before it became our wholly owned subsidiary to audit its financial statements for the years ended December 31, 2012 and 2011 and the related statements of operations, changes in stockholders’ deficit and cash flows for each of the years then ended and for the period from inception (August 29, 2011) through June 30, 2013, which are filed as Exhibits 99.1 and 99.2 to this Current Report on Form 8-K.
84
Table of Contents
During our two most recent fiscal years and through the date of our engagement of MHM, neither we nor anyone on our behalf consulted with MHM regarding either (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered with respect to our financial statements, and no written report or oral advice was provided to us by MHM that was an important factor considered by us in reaching a decision as to any accounting, auditing or financial reporting issue; or (ii) any matter that was the subject of a disagreement (as that term is defined in Item 304(a)(1)(iv) of Regulation S-K promulgated under the Securities Act and the related instructions) or a reportable event (as that term is defined in Item 304(a)(1)(v) of Regulation S-K) relating to our company.
| Item 5.01 | Changes in Control of Registrant. |
Reference is made to the disclosure set forth under Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
| Item 5.02 | Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers. |
(b) — (c): Effective upon the closing of the Merger on October 31, 2013, our executive officers prior to the Merger, Betty Sytner (former President, Treasurer, Chief Executive Officer and Chief Financial Officer) and Eileen Friedman (former Secretary), each tendered her resignation from all positions then held with our company. Following such resignation, the members of our Board of Directors that were elected in connection with the closing of the Merger, as described in part (d) of this Item 5.02 below, appointed as the executive officers of Ignyta the individuals to the executive officer positions set forth under the heading “Management—Directors, Executive Officers and Other Non-Executive Officers” in Item 2.01 of this Current Report on Form 8-K.
Each of our newly appointed executive officers will serve in his positions as an “at will” employee of our company, and will not have a formal employment agreement with us unless and until our Board of Directors, or a committee thereof, and the applicable executive officer have approved the terms of any such agreement. Upon the appointment of our new executive officers immediately following the closing of the Merger, our Board of Directors, excluding Dr. Lim with respect to his compensation, approved (i) an annual base salary of $250,000 for Dr. Lim, (ii) an annual base salary of $200,000 for Zachary Hornby, and (iii) an annual base salary of $250,000 for Patrick O’Connor. Patrick O’Connor is presently on medical leave, and is expected to return in January 2014. The amount of each of our executive officer’s annual base salary or any other form of compensation to be received by him may be modified at any time at the discretion of our Board of Directors.
In connection with the closing of the Merger, we assumed the Ignyta Plan and the obligation to issue shares of our common stock upon the exercise of all options granted thereunder that were outstanding as of the closing of the Merger. At the time of our assumption of the Ignyta Plan, there were 342,209 shares available for issuance pursuant to future grants thereunder. Our principal executive officer, president, principal financial officer, principal accounting officer, principal operating officer and persons performing similar functions are eligible to receive additional awards under the Ignyta Plan. Additionally, as of the closing of the Merger, we assumed (i) the obligation to issue to Jonathan E. Lim, our newly appointed President, Chief Executive Officer and principal executive officer, up to an aggregate of 33,333 shares of our common stock upon the exercise of options granted under the Ignyta Plan that were outstanding as of the closing of the Merger, (ii) the obligation to issue to Zachary Hornby, our newly appointed Chief Financial Officer, Vice President, Research and Development, and principal financial and accounting officer, up to an aggregate of 76,666 shares of our common stock upon the exercise of options granted under the Ignyta Plan that were outstanding as of the closing of the Merger, and (iii) the obligation to issue to Patrick O’Connor, our Chief Scientific Officer and Sr. Vice President, Research, up to an aggregate of 20,000 shares of our common stock upon the exercise of options granted under the Ignyta Plan that were outstanding as of the closing of the Merger.
For certain biographical, related party and other information regarding our newly appointed executive officers, see the disclosure under the heading “Management” and “Certain Relationships and Related Transactions, and Director Independence—Related Party Transactions” in Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
(d) Effective upon the closing of the Merger, our sole director prior to the Merger, Betty Sytner, (i) resigned as a director, and (ii) appointed as our new directors the three individuals identified as directors under the heading “Management—Directors, Executive Officers and Other Non-Executive Officers” in Item 2.01 of this Current Report on Form 8-K. Following the closing of the Merger, our newly elected directors appointed Jonathan E. Lim as the Chairman of the Board.
85
Table of Contents
We presently have no formal plan for compensating our directors for their service as our directors, and none of our newly appointed directors has received compensation for his service as a director of our company to date. Dr. Dreismann and Mr. Casdin have each provided advisory services relating to product development efforts and commercial opportunities for Ignyta Operating in the past, which advisory relationships were terminated effective upon Ignyta Operating becoming our wholly owned subsidiary and the appointment of each such individual to our Board of Directors. In connection with those advisory services, Dr. Dreismann was paid cash compensation totaling $27,500 in 2012 and $22,500 in 2013 through the date of termination of his advisory relationship, and each of Dr. Dreismann and Mr. Casdin were issued options to acquire Ignyta Operating’s common stock, which we have assumed upon the closing of the Merger. See the disclosure under the heading “Security Ownership of Certain Beneficial Owners and Management” for information about each such party’s current beneficial ownership in our common stock.
No standing committees of our Board of Directors have been established and, as a result, none of our current directors is a member of any such committee. Further, there are no arrangements or understandings pursuant to which any of our current directors was appointed as a director.
For certain biographical, related party and other information regarding our newly appointed directors, see the disclosure under the heading “Management” and “Certain Relationships and Related Transactions, and Director Independence—Related Party Transactions” in Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
(e) Effective upon the closing of the Merger, we assumed the Ignyta Plan. As of the date of this report, there are outstanding options to purchase up to an aggregate of 358,986 shares of our common stock, all of which were outstanding and assumed by us as of the closing of the Merger, and 342,209 shares of common stock reserved for issuance pursuant to future equity grants under the Ignyta Plan.
Reference is made to the description of the Ignyta Plan set forth under the heading “Market Price of and Dividends on Registrant’s Common Equity and Related Stockholder Matters—Securities Authorized for Issuance under Equity Compensation Plans” in Item 2.01 of this Current Report on Form 8-K, which description is incorporated herein by reference. The description of the Ignyta Plan contained in this report does not purport to be complete, and is qualified in its entirety by reference to the full text of the Ignyta Plan, which is attached hereto as Exhibit 10.1 and is incorporated herein by reference.
| Item 5.03 | Amendments to Articles of Incorporation or Bylaws; Change in Fiscal Year. |
Amendments to Articles of Incorporation
Prior to the closing of the Merger, we amended and restated our Articles of Incorporation in their entirety, for the purpose of, among other things, (i) changing our name from “Infinity Oil & Gas Company” to “Ignyta Inc.”, (ii) increasing the number of authorized shares of our common stock from 25,000,000 to 100,000,000; (iii) authorizing the issuance of up to 10,000,000 shares of preferred stock, in one or more series and with such rights, preferences and privileges as our Board of Directors may determine, and (iv) effecting a 100-to-one reverse stock split of our common stock. Our Board of Directors approved the amendment and restatement of our Articles of Incorporation on October 21, 2013, and as described under Item 5.07 of this Current Report on Form 8-K, stockholders holding 91.6% of the then outstanding shares of our common stock approved the amendment to our Articles of Incorporation on October 21, 2013. Our Amended and Restated Articles of Incorporation became effective on October 31, 2013 and are filed as Exhibit 3.1 to this Current Report on Form 8-K.
As a result of the reverse stock split effected by our Amended and Restated Articles of Incorporation, every 100 shares of our outstanding common stock prior to the effect of that amendment have been combined and reclassified into one share of our common stock, and the number of outstanding shares of our common stock has been reduced from 8,733,600 to 87,336. No fractional shares will be issued in connection with the reverse stock split, and any of our stockholders that would have been entitled to receive a fractional share as a result of the reverse stock split will instead receive one whole share of our common stock in lieu of such fractional share. The reverse stock split will not in itself affect any stockholder’s ownership percentage of our common stock, except to the extent that any fractional share is rounded up to the nearest whole share. Beginning with the opening of trading on November 1, 2013, our common stock is expected to commence trading on the OTCBB on a post reverse stock split basis.
In accordance with rules and regulations promulgated by FINRA, the amendments to our Articles of Incorporation to change our name, increase the number of authorized shares of our common stock, authorize preferred stock, and effect the
86
Table of Contents
100-to-one reverse stock split are expected to become effective upon receipt of FINRA’s approval of those changes on the morning of November 1, 2013. In connection with the change of our name to “Ignyta, Inc.”, FINRA has assigned us a new stock symbol, “RXDX”, which is expected to take effect on or about 20 business days following the effect of the reverse stock split on November 1, 2013.
Change in Fiscal Year
Pursuant to the approval of our Board of Directors, our fiscal year end has been changed from August 31 to December 31, which is the fiscal year end of Ignyta Operating. The Merger is being accounted for as a reverse acquisition, with Ignyta Operating regarded as the accounting acquirer. Commencing with the periodic report for the quarter ended September 30, 2013 we intend to file annual and quarterly reports based on the December 31 fiscal year end of Ignyta Operating. Such financial statements will depict the operating results of Ignyta Operating, including the acquisition of Infinity Oil & Gas Company, from Ignyta Operating’s inception on August 29, 2011. In reliance on Section III.F of the SEC’s Division of Corporate Finance: Frequently Requested Accounting and Financial Reporting Interpretations and Guidance dated March 31, 2001, we do not intend to file a transition report.
| Item 5.06 | Change in Shell Company Status. |
Upon the closing of the Merger on October 31, 2013, we ceased to be a “shell company” as defined in Rule 12b-2 of the Exchange Act. Reference is made to the disclosure under Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
| Item 5.07 | Submission of Matters to a Vote of Security Holders. |
On October 21, 2013, stockholders holding 91.6% of the then outstanding shares of our common stock executed a written consent in lieu of meeting to approve the amendment and restatement of our Articles of Incorporation to, among other things:
| • | change the name of Ignyta from “Infinity Oil & Gas Company” to “Ignyta, Inc.”; |
| • | increase the number of authorized shares of our common stock from 25,000,000 to 100,000,000; |
| • | authorize the issuance of up to 10,000,000 shares of preferred stock, in one or more series and with such rights, preferences and privileges as our Board of Directors may determine; and |
| • | effect a 100-to-one reverse stock split; |
On October 31, 2013, stockholders holding 91.6% of our then issued and outstanding shares of our common stock executed a written consent in lieu of meeting the approve the Merger Agreement and all transactions and agreements contemplated thereby, including the consummation of the Merger; the issuance of 4,916,469 shares of Ignyta’s common stock to the former stockholder of Ignyta Operating as consideration for the Merger; the assumption of the Ignyta Plan and all outstanding options thereunder; the assumption of Ignyta Operating’s outstanding warrants; and the execution and filing of the Certificate of Merger with the Secretary of State of the State of Delaware to effect the Merger.
| Item 9.01 | Financial Statements and Exhibits. |
(a) Financial Statements of Businesses Acquired. In accordance with Item 9.01(a), the following are filed as exhibits to this Current Report on Form 8-K:
| • | Unaudited financial statements of Ignyta Operating for the six months ended June 30, 2013 and 2012 are filed as Exhibit 99.1. |
| • | Audited financial statements of Ignyta Operating for the years ended December 31, 2012 and 2011 are filed as Exhibit 99.2. |
(b) Pro Forma Financial Information. In accordance with Item 9.01(b), the unaudited pro forma financial information of Ignyta and its wholly owned subsidiary Ignyta Operating as of the fiscal year ended December 31, 2012 and the six months ended June 30, 2013 are filed as Exhibit 99.3 to this Current Report on Form 8-K.
(c) Shell Company Transactions. Reference is made to Items 9.01(a) and 9.01(b) and the exhibits referred to therein, which are incorporated herein by reference.
87
Table of Contents
(d) Exhibits. Reference is made to the Exhibit Index following the signature page of this Current Report on Form 8-K, which is incorporated herein by reference.
88
Table of Contents
SIGNATURE
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| Dated: October 31, 2013 | IGNYTA, INC. | |||||
| By: | /s/ Jonathan Lim, M.D. | |||||
| Name: | Jonathan Lim, M.D. | |||||
| Title: | President and Chief Executive Officer | |||||
89
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 2.1 | Agreement and Plan of Reorganization, dated May 7, 2013, by and between Ignyta Operating, Inc. (then known as Ignyta, Inc.) and Actagene Oncology, Inc. | |
| 2.2 | Agreement and Plan of Merger and Reorganization, dated October 31, 2013, by and among Ignyta, Inc. (then known as Infinity Oil & Gas Company), IGAS Acquisition Corp., and Ignyta Operating, Inc. (then known as Ignyta, Inc.). | |
| 3.1 | Amended and Restated Articles of Incorporation of Ignyta, Inc., as amended by Certificate of Amendment to Articles of Incorporation of Ignyta, Inc. | |
| 3.2 | Bylaws of Ignyta, Inc. (incorporated by reference to Exhibit 3.2 to the Company’s Registration Statement on Form S-1 filed with the SEC on September 13, 2013). | |
| 4.1 | Form of Common Stock certificate. | |
| 10.1 | Ignyta, Inc. Amended and Restated 2011 Stock Incentive Plan. | |
| 10.2 | Form of Stock Option Award Agreement under the Ignyta, Inc. Amended and Restated 2011 Stock Incentive Plan. | |
| 10.3 | Form of Restricted Stock Award Agreement under the Ignyta, Inc. Amended and Restated 2011 Stock Incentive Plan. | |
| 10.4 | License Agreement, dated October 10, 2013, by and between Ignyta Operating, Inc. (then known as Ignyta, Inc.) and Nerviano Medical Sciences, S.r.l., as amended by that certain Amendment No. 1 to License Agreement, dated October 25, 2013, by and between Ignyta Operating, Inc. (then known as Ignyta, Inc.) and Nerviano Medical Sciences, S.r.l. (Confidential treatment has been requested with respect to portions of this exhibit pursuant to Rule 24b-2 of the Exchange Act and these confidential portions have been redacted from the document filed as an exhibit to this report. A complete copy of this agreement, including the redacted terms, has been separately filed with the SEC). | |
| 10.5 | Warrant to Purchase Stock, issued by Ignyta Operating, Inc. (then known as NexDx, Inc.) to Silicon Valley Bank on June 25, 2012 and assumed by Ignyta, Inc. (formerly known as Infinity Oil & Gas Company). | |
| 10.6 | Warrant to Purchase Stock, issued by Ignyta Operating, Inc. (then known as Ignyta, Inc.) to Silicon Valley Bank on February 27, 2013 and assumed by Ignyta, Inc. (formerly known as Infinity Oil & Gas Company). | |
| 10.7 | Loan and Security Agreement, dated June 25, 2012, by and between Ignyta Operating, Inc. (then known as NexDx, Inc.) and Silicon Valley Bank, as amended by the First Amendment to Loan and Security Agreement, dated February 27, 2013, by and between Ignyta Operating, Inc. (then known as Ignyta, Inc.) and Silicon Valley Bank. | |
| 10.8 | Standard Industrial/Commercial Multi-Tenant Lease – Gross, dated August 7, 2013, by and between Ignyta Operating, Inc. (then known as Ignyta, Inc.) and Robert C. Kyle as Trustee of the Robert C. Kyle 1979 Insurance Trust and Barbara Ann Battey as the Trustee of the Barbara Ann Battey Trust dated January 27, 2000. | |
| 10.9 | Lease, date February 19, 2013, by and between Ignyta Operating, Inc. (then known as Ignyta, Inc.) and BMR-Coast 9 LP. | |
| 10.10 | Form of Indemnification Agreement by and between Ignyta, Inc. and each of its current directors and executive officers. | |
| 16.1 | Letter from Dov Weinstein & Co. C.P.A, dated October 30, 2013. | |
| 21.1 | List of Subsidiaries. | |
| 99.1 | Unaudited condensed financial statements of Ignyta Operating, Inc. (then known as Ignyta, Inc.) for the six months ended June 30, 2013 and 2012. | |
| 99.2 | Audited financial statements of Ignyta Operating, Inc. (now known as Ignyta, Inc.) for the years ended December 31, 2012 and 2011. | |
| 99.3 | Pro forma financial information of Ignyta, Inc. and its wholly owned subsidiary Ignyta Operating, Inc. |
90