Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - VACCINOGEN INC | v349778_ex23-1.htm |
| EX-10.23 - EXHIBIT 10.23 - VACCINOGEN INC | v349778_ex10-23.htm |
As filed with the Securities and Exchange Commission on July 12, 2013
Registration No. _____________
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
VACCINOGEN, INC.
(Exact name of registrant as specified in its charter)
| Maryland | 5090 | 14-1997223 | ||
| (State or other Jurisdiction of Incorporation) | (Primary Standard Classification Code) | (IRS Employer Identification No.) |
5300 Westview Drive, Suite 406
Frederick, MD 21703
Tel.: (301) 668-8400
(Address and Telephone Number of Registrant’s Principal
Executive Offices and Principal Place of Business)
Michael G. Hanna, Jr., Ph.D
Chief Executive Officer
VACCINOGEN, INC.
5300 Westview Drive, Suite 406
Frederick, MD 21703
Tel.: (301) 668-8400
þ(Name, Address and Telephone Number of Agent for Service)
Copies of communications to:
Marc A. Indeglia, Esq.
Gregory R. Carney, Esq.
Indeglia& Carney, P.C.
1900 Main Street, Suite 300
Irvine, CA 92614
Tel No.: (949) 861-3321
Fax No.: (949) 861-3324
Approximate date of commencement of proposed sale to the public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. þ
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act of 1933, please check the following box and list the Securities Act registration Statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If delivery of the prospectus is expected to be made pursuant to Rule 434, please check the following box. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | o | Accelerated filer | o |
| Non-accelerated filer | o | Smaller reporting company | x |
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to be Registered | Amount to be Registered (1) | Proposed Maximum Offering Price Per Share (2) | Proposed Maximum Aggregate Offering Price (2) | Amount of Registration Fee | ||||||||||||
| Common Stock, par value $0.0001 | 6,000,000 | $ | 5.50 | $ | 33,000,000 | $ | 4501.20 | |||||||||
| (1) | This Registration Statement covers the Offering of common stock of the Company according to an Investment Agreement and for the resale by the selling stockholder named in this Prospectus. The Company is making a good faith estimate on the number of shares that it may issue under the Investment Agreement. |
| (2) | The proposed maximum offering price per share and the proposed maximum aggregate offering price have been estimated solely for the purpose of calculating the amount of the registration fee in accordance with Rule 457(c) under the Securities Act of 1933 on the basis of the average of the high and low prices of the Common Stock on the OTC Markets on July 9, 2013, a date within 5 trading days prior to the date of the filing of this registration statement. |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with section 8(a) of the securities act of 1933 or until the registration statement shall become effective on such date as the commission, acting pursuant to said section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
| PREPRELIMINARY PROSPECTUS | SUBJECT TO COMPLETION | DATED July 12, 2013 |
6,000,000 Common Shares
VACCINOGEN, INC.
This prospectus relates to the resale of up to 6,000,000 shares of the common stock of Vaccinogen, Inc., a Maryland corporation, by Kodiak Capital Group, LLC, a Delaware limited liability company (“Kodiak ” or “Selling Shareholder”), a selling shareholder pursuant to a Put Notice(s) under an Investment Agreement, as amended (the “Investment Agreement”) that we have entered into with Kodiak. The Investment Agreement permits us to sell shares of our common stock to Kodiak enabling us to put up to $26 million of common stock to Kodiak. The registration statement covers the offer and possible sale of approximately $33 million in common stock based on our July 9, 2013 closing market price of $5.50 per share before the discount offered to Kodiak. We will not receive any proceeds from the sale of these shares of common stock offered by Kodiak. However, we will receive proceeds from the sale of securities pursuant to each Put Notice we send to Kodiak. We will bear all costs associated with this registration.
The total amount of shares of common stock which may be sold pursuant to this Prospectus would constitute 16.1% of our issued and outstanding common stock as of July 9, 2013, if all of the shares had been sold by that date. As of July 9, 2013, the closing market price of the Company’s common stock was $5.50. Based on that price, and disregarding limitations on the number of shares Kodiak may hold at any given time and the maximum advance provisions of the Investment Agreement, the maximum amount of shares of common stock which may be sold would be 5,909,091, representing 15.9% of the outstanding common stock as of July 9, 2013 if all shares were sold by that date.
Kodiak is an “underwriter” within the meaning of the Securities Act of 1933, as amended (the “Securities Act”) in connection with the resale of our common stock under the Investment Agreement. Kodiak will pay us 80% of the lowest daily VWAP of the common stock during the five consecutive trading days immediately following the date of our notice to Kodiak of our election to put shares pursuant to the Investment Agreement. There are no underwriting agreements in place.
Our shares of common stock are currently traded on the OTC Markets Group (OTC Pink Tier) under the symbol "VGEN." We have filed a general registration statement on Form 10 and we intend to immediately upgrade our tier on the OTC Link to OTC.QB upon effectiveness of such registration statement.
Our Independent Registered Public Accounting Firm has raised substantial doubts about our ability to continue as a going concern.
Investing in our common stock involves a high degree of risk. Before buying any shares, you should carefully read the discussion of material risks of investing in our common stock in “Risk Factors” beginning on page 4 of this prospectus.
Neither the U.S. Securities and Exchange Commission nor any state securities commission has approved or disapproved of anyone’s investment in these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The Date of This Prospectus Is: _____________, 2013
TABLE OF CONTENTS
| PAGE | |
| Forward-looking Statements | 1 |
| Prospectus Summary | 2 |
| Risk Factors | 4 |
| Use of Proceeds | 17 |
| Selling Shareholders | 17 |
| Plan of Distribution | 18 |
| Determination of Offering Price | 19 |
| Dilution | 19 |
| Penny Stock Considerations | 19 |
| Description of Business | 21 |
| Description of Property | 47 |
| Legal Proceedings | 47 |
| Security Ownership of Certain Beneficial Owners and Management | 48 |
| Directors, and Executive Officers | 49 |
| Executive Compensation | 53 |
| Certain Relationships and Related Transactions | 55 |
| Market For Common Equity and Related Stockholder Matters | 56 |
| Description of Securities | 57 |
| Legal Matters | 60 |
| Experts | 60 |
| Available Information | 61 |
| Index to Financial Statements | 62 |
| Management’s Discussion and Analysis of Financial Condition and Results of Operations | 63 |
| Selected Financial Data | 74 |
| Supplementary Financial Information | 74 |
| Changes in and Disagreements with Accounts on Accounting and Financial Disclosure | 74 |
| Quantitative and Qualitative Disclosures about Market Risk | 74 |
FORWARD-LOOKING STATEMENTS
Some of the statements contained in this Offering Circular are forward-looking statements within the meaning of the Securities Act of 1933 (the “Securities Act”) and the Securities Exchange Act of 1934 (the “Exchange Act”) and are subject to the safe harbor created by the Private Securities Litigation Reform Act of 1995. We have based these forward-looking statements largely on our expectations and projections about future events and financial trends affecting the financial condition and/or operating results of our business. Forward-looking statements involve risks and uncertainties; particularly those risks and uncertainties inherent in the process of discovering, developing and commercializing drugs that are safe and effective for use as human therapeutics. There are important factors that could cause actual results to be substantially different from the results expressed or implied by these forward-looking statements, including, among other things:
| · | whether we have adequate financial resources and access to capital to fund commercialization of OncoVAX®, the Human Monoclonal Antibodies (HuMabs) Program and that of other potential product candidates we may develop; |
| · | our ability to successfully manufacture OncoVAX® and other product candidates in necessary quantities with required quality; |
| · | our ability to successfully obtain regulatory approvals and commercialize our products that are under development and develop the infrastructure necessary to support commercialization if regulatory approvals are received; |
| · | our ability to complete and achieve positive results in ongoing and new clinical trials; |
| · | our dependence on single-source vendors for some of the components used in our product candidates; |
| · | the extent to which the costs of any products that we are able to commercialize will be reimbursable by third-party payers; |
| · | the extent to which any products that we are able to commercialize will be accepted by the market; |
| · | our dependence on our intellectual property and ability to protect our proprietary rights and operate our business without conflicting with the rights of others; |
| · | the effect that any intellectual property litigation, or product liability claims may have on our business and operating and financial performance; |
| · | our expectations and estimates concerning our future operating and financial performance; |
| · | the impact of competition and regulatory requirements and technological change on our business; |
| · | our ability to recruit and retain key personnel; |
| · | our ability to enter into future collaboration agreements; |
| · | anticipated trends in our business and the biotechnology industry generally; and |
| · | other factors set forth under the heading “Risk Factors” herein. |
In addition, in this Offering Circular, the words “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “plan,” “expect,” “potential,” or “opportunity,” the negative of these words or similar expressions, as they relate to us, our business, future financial or operating performance or our management, are intended to identify forward-looking statements. We do not intend to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise. Past financial or operating performance is not necessarily a reliable indicator of future performance and you should not use our historical performance to anticipate results or future period trends.
The terms "Vaccinogen," "our" and "we," and the “Company” as used in this prospectus, refer to Vaccinogen, Inc.
| 1 |
You should rely only on the information contained in this prospectus. We have not authorized any other person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate as of the date on the front cover of this prospectus only. Our business, financial condition, results of operations and prospects may have changed since that date.
We intend to furnish our shareholders with annual reports containing consolidated financial statements audited by an independent accounting firm.
PROSPECTUS SUMMARY
This summary highlights selected information contained elsewhere in this prospectus. This summary does not contain all the information that you should consider before investing in the common stock. You should carefully read the entire prospectus, including “Risk Factors”, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the Consolidated Financial Statements, before making an investment decision.
Vaccinogen is a biotechnology company with more than three decades of research into combating cancer by using the body’s own immune system. It is the developer of OncoVAX®, the only patient specific immunotherapy for Stage II colon cancer and has the following mission:
| 1. | Complete the pivotal Phase III clinical trial of this autologous vaccine, OncoVAX®, which is a new paradigm to prevent the recurrence of stage II colon cancer. The planned trial will closely replicate a successful prior Phase IIIa study showing 50% improvement in recurrence free survival and overall survival in the Stage II target patient population. |
| 2. | Develop near term revenues by establishing licensing arrangements for distribution of OncoVAX® in certain territories as well as for additional carcinoma indications. |
| 3. | Develop diagnostic and therapeutic drug products as well as near-term revenues by exploiting its distinctive Human Monoclonal Antibodies (“HuMabs”) technology. |
Our executive office is located at 5300 Westview Drive, Suite 406, Frederick, MD 21703. Our telephone number is 301-668-8400. Our Internet address is www.vaccinogeninc.com.
Kodiak Investment Agreement
This prospectus relates to the resale of up to 6,000,000 shares of our common stock by Kodiak. Kodiak will obtain our common stock pursuant to the Investment Agreement entered into by Kodiak and us, dated July 18, 2012, as amended on July 8, 2013.
Although the Company is not mandated to sell shares under the Investment Agreement, the Investment Agreement gives the Company the option to sell to Kodiak, up to $26,000,000 worth of our common stock (“Shares”), par value $0.0001 per share over an eighteen (18) month period. At the date of filing, we may not obtain the full $26,000,000 in funding if the price of our common stock decreases (and remains below) 2% of the current market price. The $26,000,000 was stated as the total amount of available funding in the Agreement because this was the maximum amount that Kodiak agreed to offer us in funding. There is no assurance that the market price of our common stock will remain at its current price or increase substantially in the future. The number of common shares that remains issuable may not be sufficient, dependent upon the share price, to allow us to access the full amount contemplated under the Agreement. Therefore, we may not have access to the remaining commitment under the Investment Agreement unless the market price of our common stock remains at its current price or increases from it current level. Based on our stock price as of July 9, 2013, the registration statement covers the offer and possible sale of only approximately $26.4 million worth of our shares at current discounted market price of $4.40 or approximately 80% of $5.50 (our market price at July 9, 2013)
The purchase price of the common stock shall be set at eighty percent (80%) of the lowest daily VWAP of the common stock during the pricing period. The pricing period shall be the five (5) consecutive trading days immediately after the Put Notice Date.
In addition, there is an ownership limit for Kodiak of 9.99%.
| 2 |
On any Closing Date, we shall deliver to Kodiak the number of shares of the Common Stock registered in the name of Kodiak as specified in the Put Notice. In addition, we must deliver the other required documents, instruments and writings required. Kodiak is not required to purchase the shares unless:
| · | Our Registration Statement with respect to the resale of the shares of Common Stock delivered in connection with the applicable put shall have been declared effective. |
| · | We shall have obtained all material permits and qualifications required by any applicable state for the offer and sale of the Registrable Securities. |
| · | We shall have filed with the SEC in a timely manner all reports, notices and other documents required. |
We believe that we will be able to meet all of the above obligations mandated in Investment Agreement set forth above. We are aware that if we fail to perform our obligations and we fail to deliver to Kodiak on the Put Date the shares of Common Stock corresponding to the applicable Put, Kodiak shall suffer financial hardship and therefore we acknowledge that we will be liable for any and all losses, commission, fees, interest, legal fees or any other financial hardships caused to Kodiak. Fees and penalties for such losses (liquidated damages) to Kodiak shall be paid by the Company in accordance with the following schedule:
| Payments for Each Number of Days Overdue | For each $10,000 Worth of Common Stock | |||
| 1 | $ | 100 | ||
| 2 | $ | 200 | ||
| 3 | $ | 300 | ||
| 4 | $ | 400 | ||
| 5 | $ | 500 | ||
| 6 | $ | 600 | ||
| 7 | $ | 700 | ||
| 8 | $ | 800 | ||
| 9 | $ | 900 | ||
| 10 | $ | 1000 | ||
| Over 10 | $1000 + $200 for each Business Day beyond the tenth day | |||
| 3 |
RISK FACTORS
The following is a summary of the risk factors that we believe are most relevant to our business. You should understand that it is not possible to predict or identify all such factors. Consequently, you should not consider the following to be a complete discussion of all potential risks or uncertainties. We undertake no obligation to publicly update forward-looking statements, whether as a result of new information, future events, or otherwise.
RISK FACTORS RELATED TO THE OFFERING
Existing stockholders may experience significant dilution from the sale of our common stock pursuant to the Kodiak Capital Group Investment Agreement.
The sale of our common stock to Kodiak Capital Group, LLC in accordance with the Investment Agreement may have a dilutive impact on our shareholders. As a result, our net income per share could decrease in future periods and the market price of our common stock could decline. In addition, the lower our stock price is at the time we exercise our put options, the more shares of our common stock we will have to issue to Kodiak Capital Group LLC in order to exercise a put under the Investment Agreement. If our stock price decreases, then our existing shareholders would experience greater dilution for any given dollar amount raised through the Offering.
The perceived risk of dilution may cause our stockholders to sell their shares, which may cause a decline in the price of our common stock. Moreover, the perceived risk of dilution and the resulting downward pressure on our stock price could encourage investors to engage in short sales of our common stock. By increasing the number of shares offered for sale, material amounts of short selling could further contribute to progressive price declines in our common stock.
The issuance of shares pursuant to the Kodiak Investment Agreement may have a significant dilutive effect.
Depending on the number of shares we issue pursuant to the Kodiak Investment Agreement, it could have a significant dilutive effect upon our existing shareholders. Although the number of shares that we may issue pursuant to the Investment Agreement will vary based on our stock price (the higher our stock price, the less shares we have to issue) the information set out below indicates the potential dilutive effect to our shareholders, based on different potential future stock prices, if the full amount of the Investment Agreement is realized.
Dilution based upon common stock put to Kodiak Capital Group and the stock price discounted to Kodiak Capital Group’s purchase price of 80% of the lowest daily volume weighted average price (VWAP) during the pricing period. The example below illustrates dilution based upon a $5.50 market price/$4.40 purchase price and other increased/decreased prices:
$26,000,000 Put
| Stock Price(Kodiak Purchase Price) | Shares Issued | Percentage of Outstanding Shares (1) | ||||||
| $6.875 ($5.50) +25% | 4,727,273 | 13.15 | % | |||||
| $5.50 ($4.40) | 5,909,091 | 15.91 | % | |||||
| $4.125 ($3.30) -25% | 7,878,788 | 20.15 | % | |||||
| $2.75 ($2.20) -50% | 11,918,182 | 27.45 | % | |||||
| $1.375 ($1.10) -75% | 23,636,364 | 43.08 | % | |||||
| (1) | Based on 31,227,756 shares outstanding as of June 26, 2013. |
| 4 |
Kodiak Capital Group, LLC will pay less than the then-prevailing market price of our common stock which could cause the price of our common stock to decline.
Our common stock to be issued under the Kodiak Investment Agreement will be purchased at a twenty (20%) discount or 80% of the lowest daily VWAP during the five trading days immediately following our notice to Kodiak Capital Group, LLC of our election to exercise our "put" right.
Kodiak Capital Group, LLC has a financial incentive to sell our shares immediately upon receiving the shares to realize the profit between the discounted price and the market price. If Kodiak Capital Group, LLC sells our shares, the price of our common stock may decrease. If our stock price decreases, Kodiak may have a further incentive to sell such shares. Accordingly, the discounted sales price in the Investment Agreements may cause the price of our common stock to decline.
Kodiak Capital Group, LLC has entered into similar agreements with other public companies and may not have sufficient capital to meet our put notices.
Kodiak Capital Group LLC has entered into similar investment agreements with other public companies, and some of those companies have filed registration statements with the intent of registering shares to be sold to Kodiak pursuant to investment agreements. We do not know if management at any of the companies who have or will have effective registration statements intend to raise funds now or in the future, what the size or frequency of each put request would be, if floors will be used to restrict the amount of shares sold, or if the investment agreement will ultimately be cancelled or expire before the entire amount of shares are put to Kodiak. Since we do not have any control over the requests of these other companies, if Kodiak Capital Group LLC receives significant requests, it may not have the financial ability to meet our requests. If so, the amount of available funds may be significantly less than we anticipate.
We are registering an aggregate of 6,000,000 shares of common stock to be issued under the Kodiak Investment Agreement. The sale of such shares could depress the market price of our common stock.
We are registering an aggregate of 6,000,000 shares of common stock under the registration statement of which this Prospectus forms a part for issuance pursuant to the Kodiak Investment Agreement. The sale of these shares into the public market by Kodiak could depress the market price of our common stock.
We May Not Have Access to the Full Amount under the Investment Agreement.
During the period ended July 9, 2013, the closing price of our common stock was $5.50 based on very little volume. There is no assurance that the market price of our common stock will remain at its current price or increase from its current level. The entire commitment under the Investment Agreement is for $26,000,000. Therefore, we may not have access to the remaining commitment under the Investment Agreement unless the share price of our common stock remains at or increases from its current level.
Unless an active trading market develops for our securities, investors may not be able to sell their shares.
We are a reporting company and our common shares are quoted on the OTC Link (OTC Pink Tier under the symbol “VGEN” (and we will immediately upgrade our tier to OTC.QB upon effectiveness of our recently filed Form 10 registration statement). However, there is not currently an active trading market for our common stock and an active trading market may never develop or, if it does develop, may not be maintained. Failure to develop or maintain an active trading market will have a generally negative effect on the price of our common stock, and you may be unable to sell your common stock or any attempted sale of such common stock may have the effect of lowering the market price and therefore your investment could be a partial or complete loss.
Since our common stock is thinly traded it is more susceptible to extreme rises or declines in price, and you may not be able to sell your shares at or above the price paid.
Since our common stock is thinly traded its trading price is likely to be highly volatile and could be subject to extreme fluctuations in response to various factors, many of which are beyond our control, including (but not necessarily limited to):
| 5 |
| · | the trading volume of our shares; | |
| · | the number of securities analysts, market-makers and brokers following our common stock; | |
| · | changes in, or failure to achieve, financial estimates by securities analysts; | |
| · | new products or services introduced or announced by us or our competitors; | |
| · | actual or anticipated variations in quarterly operating results; | |
| · | conditions or trends in our business industries; | |
| · | announcements by us of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments; | |
| · | additions or departures of key personnel; | |
| · | sales of our common stock and | |
| · | general stock market price and volume fluctuations of publicly-traded, and particularly microcap, companies. |
Investors may have difficulty reselling shares of our common stock, either at or above the price they paid for our stock, or even at fair market value. The stock markets often experience significant price and volume changes that are not related to the operating performance of individual companies, and because our common stock is thinly traded it is particularly susceptible to such changes. These broad market changes may cause the market price of our common stock to decline regardless of how well we perform as a company. In addition, there is a history of securities class action litigation following periods of volatility in the market price of a company’s securities. Although there is no such litigation currently pending or threatened against the Company, such a suit against us could result in the incursion of substantial legal fees, potential liabilities and the diversion of management’s attention and resources from our business. Moreover, and as noted below, our shares are currently traded on the OTC Bulletin Board and, further, are subject to the penny stock regulations. Price fluctuations in such shares are particularly volatile and subject to manipulation by market-makers, short-sellers and option traders.
RISKS RELATED TO OUR BUSINESS
Our success is dependent upon OncoVAX® achieving regulatory approval and acceptance in the marketplace, the failure of either will have a negative effect on our business, financial condition and results of operations.
Our future growth and profitability will depend on its ability to introduce and market OncoVAX®. There can be no assurance that we will be able to obtain necessary regulatory approvals for OncoVAX® in a timely manner, if at all. Our failure to introduce and market OncoVAX® in a timely manner would have a material adverse effect on our business, financial condition and results of operations.
Even if OncoVAX® is approved for marketing by the FDA and other regulatory authorities, there can be no assurance that it will be commercially successful or that we will be successful producing OncoVAX® on a commercial scale at a cost that will enable us to realize a profit. Despite the results of the clinical trials of OncoVAX®, there can be no assurance that oncologists and other physicians will refer patients for treatment with OncoVAX®. Market acceptance also could be affected by the availability of third-party reimbursement. Failure of OncoVAX® to achieve significant market acceptance could have a material adverse effect on our business, financial condition and results of operations.
We have a history of significant losses from continuing operations and expect to continue such losses for the foreseeable future.
Since our inception, our expenses have substantially exceeded our revenues, resulting in continuing losses and an accumulated deficit. Because we presently have no product revenues and we are committed to continuing our product research, development and commercialization programs, we will continue to experience significant operating losses unless and until we complete the development of OncoVAX® and other new products and these products have been clinically tested, approved by the FDA and successfully marketed.
| 6 |
We have devoted our resources to developing a new generation of products but will not be able to market these products until we have completed clinical testing and obtain all necessary governmental approvals. In addition, our products are still in development and testing and cannot be marketed until we have completed clinical testing and obtained necessary governmental approval. Accordingly, our revenue sources are, and will remain, extremely limited until our products are clinically tested, approved by the FDA and successfully marketed. We cannot guarantee that our product will be successfully tested, approved by the FDA or marketed, successfully or otherwise, at any time in the foreseeable future or at all.
We have no internal sales or marketing capability and must enter into alliances with others possessing such capabilities to commercialize our products successfully.
We intend to market our products, if and when such products are approved for commercialization by the FDA, either directly or through other strategic alliances and distribution arrangements with third parties. There can be no assurance that we will be able to enter into third-party marketing or distribution arrangements on advantageous terms or at all. To the extent that we do enter into such arrangements, we will be dependent on our marketing and distribution partners. In entering into third-party marketing or distribution arrangements, we expect to incur significant additional expense. There can be no assurance that, to the extent that we sell products directly or we enter into any commercialization arrangements with third parties, such third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance for our products and services.
If we do not raise additional capital, we may not be able to complete the development, testing and commercialization of our treatment systems or continue as a going concern.
To complete the development and commercialization of our product, we will need to raise substantial amounts of additional capital. We do not have any committed sources of financing and we cannot offer any assurances that additional funding will be available in a timely manner, on acceptable terms or at all.
In the event we cannot raise sufficient capital, we may be required to delay, scale back or eliminate certain aspects of our operations or attempt to obtain funds through unfavorable arrangements with partners or others that may force us to relinquish rights to certain of our technologies, products or potential markets or that could impose onerous financial or other terms. Furthermore, if we cannot fund our ongoing development and other operating requirements we may no longer be able to continue as a going concern.
We rely on third parties to conduct all of our clinical trials. If these third parties do not successfully carry out their contractual duties, comply with budgets and other financial obligations or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our product candidates in a timely or cost-effective manner.
We rely, and expect to continue to rely, on third-party Clinical Research Organizations to conduct all of our clinical trials. Because we do not conduct our own clinical trials, we must rely on the efforts of others and cannot always control or accurately predict the timing of such trials, the costs associated with such trials or the procedures that are followed for such trials. We do not anticipate significantly increasing our personnel in the foreseeable future and therefore, expect to continue to rely on third parties to conduct all of our future clinical trials. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they do not carry out the trials in accordance with budgeted amounts, if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or for other reasons, or if they fail to maintain compliance with applicable government regulations and standards, our clinical trials may be extended, delayed or terminated or may become prohibitively expensive, and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates.
| 7 |
We depend on key personnel for our continued operations and future success and a loss of certain key personnel could significantly hinder our ability to move forward with our business plan.
Because of the specialized nature of our business, we are highly dependent on our ability to identify, hire, train and retain highly qualified scientific and technical personnel for the research and development activities we conduct or sponsor. The loss of one or more certain key executive officers, or scientific officers, including Michael G. Hanna, Jr, Ph.D., would be significantly detrimental to us. In addition, recruiting and retaining qualified scientific personnel to perform research and development work is critical to our success. There is intense competition for qualified personnel in the areas of our present and planned activities, and there can be no assurance that we will be able to continue to attract and retain the qualified personnel necessary for the development of our business. The failure to attract and retain such personnel or to develop such expertise would adversely affect our business.
If an event of default is declared under agreements with Organon Teknika, including the security agreement, we could lose possession of the OncoVAX® intellectual property.
In connection with certain prior agreements between Intracel Holdings Corporation and Organon Teknika (now Merck), Intracel entered into a security agreement granting Organon Teknika a first priority security interest in all of intellectual property intellectual property and patents related to OncoVAX®. The security agreement states that if an event of default occurs, the secured party has the right to take possession of the OncoVAX® intellectual property to satisfy the obligations under these agreements. We assumed these obligations under our License and subsequent Asset Transfer Agreement. Certain required payments have not been made to Organon Teknika. Although no formal event of default has been issued and discussions and negotiations to resolve the matter is continuing, loss of our OncoVAX® intellectual property would adversely affect our business.
If an event of default is declared under our security agreement with The Abell Foundation, we could lose possession of the OncoVAX® intellectual property.
In connection with our promissory note issued to The Abell Foundation, we granted The Abell Foundation a security interest in our patents related to OncoVAX®. The security agreement states that if an event of default occurs, the secured party has the right to take possession of the OncoVAX® patents to satisfy the obligations under these agreements. Although we are not currently in default under these agreements, loss of our OncoVAX® patents would adversely affect our business.
Our business is subject to numerous and evolving state, federal and foreign regulations and we may not be able to secure the government approvals needed to develop and market our products.
Our research and development activities, pre-clinical tests and clinical trials, and ultimately the manufacturing, marketing and labeling of our products, all are subject to extensive regulation by the FDA and foreign regulatory agencies. Pre-clinical testing and clinical trial requirements and the regulatory approval process typically take years and require the expenditure of substantial resources. Additional government regulation may be established that could prevent or delay regulatory approval of our product candidates. Delays or rejections in obtaining regulatory approvals would adversely affect our ability to commercialize any product candidates and our ability to generate product revenues or royalties.
The FDA and foreign regulatory agencies require that the safety and efficacy of product candidates be supported through adequate and well-controlled clinical trials. If the results of pivotal clinical trials do not establish the safety and efficacy of our product candidates to the satisfaction of the FDA and other foreign regulatory agencies, we will not receive the approvals necessary to market such product candidates. Even if regulatory approval of a product candidate is granted, the approval may include significant limitations on the indicated uses for which the product may be marketed.
Many states in which we do, or in the future may do, business, or in which our products may be sold, impose licensing, labeling or certification requirements that are in addition to those imposed by the FDA. There can be no assurance that one or more states will not impose regulations or requirements that have a material adverse effect on our ability to sell our products.
In many of the foreign countries in which we may do business or in which our products may be sold, we will be subject to regulation by national governments and supranational agencies as well as by local agencies affecting, among other things, product standards, packaging requirements, labeling requirements, import restrictions, tariff regulations, duties and tax requirements. There can be no assurance that one or more countries or agencies will not impose regulations or requirements that could have a material adverse effect on our ability to sell our products.
| 8 |
Legislative and regulatory changes affecting the health care industry could adversely affect our business.
Political, economic and regulatory influences are subjecting the healthcare industry to potential fundamental changes that could substantially affect our results of operations. There have been a number of government and private sector initiatives during the last few years to limit the growth of healthcare costs, including price regulation, competitive pricing, coverage and payment policies, comparative effectiveness of therapies, technology assessments and managed-care arrangements. It is uncertain which legislative proposals, if any, will be adopted (or when) or what actions federal, state, or private payors for health care treatment and services may take in response to any health care reform proposals or legislation. We cannot predict the effect health care reforms may have on our business and we can offer no assurances that any of these reforms will not have a material adverse effect on our business. These actual and potential changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective treatments. In addition, uncertainty remains regarding proposed significant reforms to the U.S. healthcare system.
The success of our products may be harmed if the government, private health insurers and other third-party payors do not provide sufficient coverage or reimbursement.
Our ability to commercialize our products successfully will depend in part on the extent to which reimbursement for the costs of such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payors. The reimbursement status of newly approved medical products is subject to significant uncertainty. We cannot guarantee that adequate third-party insurance coverage will be available for us to establish and maintain price levels sufficient for us to realize an appropriate return on our investment in developing new therapies. Government, private health insurers and other third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement for new therapeutic products approved for marketing by the FDA. Accordingly, even if coverage and reimbursement are provided by government, private health insurers and third-party payors for uses of our products, market acceptance of these products would be adversely affected if the reimbursement available proves to be unprofitable for health care providers.
We face intense competition and the failure to compete effectively could adversely affect our ability to develop and market our products.
There are many companies and other institutions engaged in research and development of various technologies for cancer treatment products. We believe that the level of interest by others in investigating the potential of possible competitive treatments and alternative technologies will continue and may increase. Potential competitors engaged in all areas of cancer treatment research in the United States and other countries include, among others, major pharmaceutical companies, specialized technology companies, and universities and other research institutions. Most of our current and potential competitors have substantially greater financial, technical, human and other resources, and may also have far greater experience than do we, both in pre-clinical testing and human clinical trials of new products and in obtaining FDA and other regulatory approvals. One or more of these companies or institutions could succeed in developing products or other technologies that are more effective than the products and technologies that we have been or are developing, or which would render our technology and products obsolete and non-competitive. Furthermore, if we are permitted to commence commercial sales of any of our products, we will also be competing, with respect to manufacturing efficiency and marketing, with companies having substantially greater resources and experience in these areas.
OncoVAX® reg; is based on novel technologies, which may raise new regulatory issues that could delay or make FDA approval more difficult.
The process of obtaining required FDA and other regulatory approvals, including foreign approvals, is expensive, often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. OncoVAX® and our immunotherapies are novel; therefore, regulatory agencies may lack experience with them, which may lengthen the regulatory review process, increase our development costs and delay or prevent commercialization of OncoVAX® and our other active immunotherapy products under development.
| 9 |
Our products may not be accepted in the marketplace; therefore we may not be able to generate significant revenue, if any.
Even if OncoVAX® or any of our other potential products is approved and sold, physicians and the medical community may not ultimately use them or may use them only in applications more restricted than we expect. Our products, if successfully developed, will compete with a number of traditional products and immunotherapies manufactured and marketed by major pharmaceutical and other biotechnology companies. Our products will also compete with new products currently under development by such companies and others. Physicians will only prescribe a product if they determine, based on experience, clinical data, side effect profiles and other factors, that it is beneficial and preferable to other products currently in use. Many other factors influence the adoption of new products, including marketing and distribution restrictions, course of treatment, adverse publicity, product pricing, the views of thought leaders in the medical community, and reimbursement by government and private third party payers.
Technologies for the treatment of cancer are subject to rapid change, and the development of treatment strategies that are more effective than our technologies could render our technologies obsolete.
Various methods for treating cancer currently are, and in the future are expected to be, the subject of extensive research and development. Many possible treatments that are being researched, if successfully developed, may not require, or may supplant, the use of our technologies. The successful development and acceptance of any one or more of these alternative forms of treatment could render our technology obsolete as a cancer treatment method.
We may not be able to hire or retain key officers or employees that we need to implement our business strategy and develop our products and business.
Our success depends significantly on the continued contributions of our executive officers, scientific and technical personnel and consultants, and on our ability to attract additional personnel as we seek to implement our business strategy and develop our products and businesses. During our operating history, we have assigned many essential responsibilities to a relatively small number of individuals. However, as our business and the demands on our key employees expand, we have been, and will continue to be, required to recruit additional qualified employees. The competition for such qualified personnel is intense, and the loss of services of certain key personnel or our inability to attract additional personnel to fill critical positions could adversely affect our business. Further, we do not carry "key man" insurance on any of our personnel. Therefore, loss of the services of key personnel would not be ameliorated by the receipt of the proceeds from such insurance.
Products we may potentially commercialize and market may be subject to promotional limitations.
The FDA has the authority to impose significant restrictions on an approved product through the product label and allowed advertising, promotional and distribution activities. The FDA also may require us to undertake post-marketing clinical trials. If the results of such post-marketing studies are not satisfactory, the FDA may withdraw marketing authorization or may condition continued marketing on commitments from us that may be expensive and/or time consuming to fulfill. Even if we receive FDA and other regulatory approvals, if we or others identify adverse side effects after any of our products are on the market, or if manufacturing problems occur, regulatory approval may be suspended or withdrawn and reformulation of our products, additional clinical trials, changes in labeling of our products and additional marketing applications may be required, any of which could harm our business and cause our stock price to decline.
RISKS RELATING TO MANUFACTURING ACTIVITIES
We and our contract manufacturers are subject to significant regulation with respect to manufacturing of our products.
All of those involved in the preparation of a therapeutic drug for clinical trials or commercial sale, including our existing (and any future) supply contract manufacturers and clinical trial investigators, are subject to extensive regulation. Components of a finished therapeutic product approved for commercial sale or used in late-stage clinical trials must be manufactured in accordance with the FDA’s current Good Manufacturing Practices, or equivalent cGMP regulations developed by authorities in other countries. These regulations govern manufacturing processes and procedures and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved for sale. Our facilities and quality systems and the facilities and quality systems of some or all of our third-party contractors must be inspected and audited routinely for compliance with applicable United States and other country regulations. Our current Emmen facility is operating under cGMP protocols with an expected cGMP recertification inspection in 3rd quarter 2013. We expect to receive recertificated cGMP status in 4th quarter 2013. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulation occurs independent of such an inspection or audit, we, the FDA, or governmental authorities in other countries may require remedial measures that may be costly and/or time consuming for us or a third-party to implement and that may include the temporary or permanent suspension or change of a clinical trial or commercial sales, recalls, market withdrawals, seizures or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business. In addition, we will be required to have a cGMP facility in the United States prior to receipt of FDA approval for our OncoVAX® and other products. Failure to procure or construct a U.S. cGMP facility will delay our ability to secure FDA approval and would have a material adverse effect on our operations.
| 10 |
RISKS FROM COMPETITIVE FACTORS
Our competitors may develop and market products that are less expensive, more effective, safer or reach the market sooner, which may diminish or eliminate the commercial success of any products we may commercialize.
Competition in the cancer therapeutics field is intense and is accentuated by the rapid pace of advancements in product development. In addition, we compete with other clinical-stage companies and institutions for clinical trial participants, which could reduce our ability to recruit participants for our clinical trials. Delay in recruiting clinical trial participants could adversely affect our ability to bring a product to market prior to our competitors. Further, research and discoveries by others may result in breakthroughs that render potential products obsolete before they generate revenue.
Some of our competitors in the cancer therapeutics field have substantially greater research and development capabilities and manufacturing, marketing, financial and managerial resources than we do. Acquisitions of competing companies by large pharmaceutical and biotechnology companies could enhance our competitors’ resources. In addition, our competitors may obtain patent protection or FDA approval and commercialize products more rapidly than we do, which may impact future sales of our products. We expect that competition among products approved for sale will be based, among other things, on product efficacy, price, safety, reliability, availability, patent protection, and sales, marketing and distribution capabilities. Our profitability and financial position will suffer if our products receive regulatory approval, but cannot compete effectively in the marketplace.
RISKS RELATING TO OUR CLINICAL TRIAL AND PRODUCT DEVELOPMENT INITIATIVES
Clinical trials for product candidates must satisfy stringent regulatory requirements or we may be unable to utilize the results.
The clinical trials for OncoVAX® and of any product candidates that we develop must comply with regulations by numerous federal, state and local government authorities in the United States, principally the FDA, and by similar governmental authorities in other countries. Clinical trials are subject to continuing oversight by governmental regulatory authorities and institutional review boards and must meet the requirements of these authorities in the United States and other countries, including those for informed consent and good clinical practices. We may not be able to comply with these requirements, which could disqualify completed or ongoing clinical trials. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of our product candidates, including the following:
| • | safety and efficacy results from human clinical trials may show the product candidate to be less effective or safe than desired or earlier results may not be replicated in later clinical trials; | ||
| • | the results of pre-clinical studies may be inconclusive or they may not be indicative of results that will be obtained in human clinical trials; | ||
| • | after reviewing relevant information, including pre-clinical testing or human clinical trial results, we may abandon or substantially restructure programs that we might previously have believed to be promising; | ||
| • | we, the FDA, an IRB, an EC, or similar regulatory authorities in other countries may suspend or terminate clinical trials if the participating patients are being exposed to unacceptable health risks or for other reasons; and | ||
| • | the effects of our product candidates may not be the desired effects or may include undesirable side effects or other characteristics that interrupt, delay or cause us or the FDA, or equivalent governmental authorities in other countries, to halt clinical trials or cause the FDA or non-United States regulatory authorities to deny approval of the product candidate for any or all target indications. |
| 11 |
Each phase of clinical testing is highly regulated, and during each phase there is risk that we will encounter serious obstacles or will not achieve our goals, and accordingly we may abandon a product in which we have invested substantial amounts of time and money. In addition, we must provide the FDA and foreign regulatory authorities with pre-clinical and clinical data that demonstrate that our product candidates are safe and effective for each target indication before they can be approved for commercial distribution. We cannot state with certainty when or whether any of our products now under development will be approved or launched; or whether any products, once approved and launched, will be commercially successful.
The FDA, other non-United States regulatory authorities, or an Advisory Committee may determine our clinical trials data regarding safety or efficacy are insufficient for regulatory approval.
Although we obtain guidance from regulatory authorities on certain aspects of our clinical development activities, these discussions are not binding obligations on regulatory authorities. Regulatory authorities may revise or retract previous guidance or may disqualify a clinical trial in whole or in part from consideration in support of approval of a potential product for commercial sale or otherwise deny approval of that product. Even if we obtain successful clinical safety and efficacy data, we may be required to conduct additional, expensive trials to obtain regulatory approval. FDA, or equivalent other country authorities, may elect to obtain advice from outside experts regarding scientific issues and/or marketing applications under FDA or other country authority review through the FDA’s Advisory Committee process or other country procedures. Views of the Advisory Committee or other experts may differ from those of the FDA, or equivalent other country authority, and may impact our ability to commercialize a product candidate.
If we encounter difficulties enrolling patients in our clinical trials, our trials could be delayed or otherwise adversely affected.
Clinical trials for our product candidates may require that we identify and enroll a large number of patients with the disease under investigation. We may not be able to enroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a study, to complete our clinical trials in a timely manner.
Patient enrollment is affected by factors including:
| • | design of the trial protocol; | ||
| • | the size of the patient population; | ||
| • | eligibility criteria for the study in question; | ||
| • | perceived risks and benefits of the product candidate under study; | ||
| • | availability of competing therapies and clinical trials; | ||
| • | efforts to facilitate timely enrollment in clinical trials; | ||
| • | patient referral practices of physicians; | ||
| • | the ability to monitor patients adequately during and after treatment and | ||
| • | geographic proximity and availability of clinical trial sites for prospective patients. |
Additionally, even if we are able to identify an appropriate patient population for a clinical trial, there can be no assurance that the patients will continue in the clinical trial through completion.
If we have difficulty enrolling or maintaining a sufficient number of patients with sufficient diversity to conduct our clinical trials as planned, we may need to delay or terminate ongoing or planned clinical trials, either of which would have a negative effect on our business.
| 12 |
RISKS RELATED TO REGULATION OF THE PHARMACEUTICAL INDUSTRY
OncoVAX® and our other products in development cannot be sold if we do not maintain or gain required regulatory approvals.
Our business is subject to extensive regulation by numerous state and federal governmental authorities in the United States, including the FDA, and potentially by foreign regulatory authorities, with regulations differing from country to country. In the United States, the FDA regulates, among other things, the pre-clinical testing, clinical trials, manufacturing, safety, efficacy, potency, labeling, storage, record keeping, quality systems, advertising, promotion, sale and distribution of therapeutic products. Other applicable non-United States regulatory authorities have equivalent powers. Failure to comply with applicable requirements could result in, among other things, one or more of the following actions: withdrawal of product approval, notices of violation, untitled letters, warning letters, fines and other monetary penalties, unanticipated expenditures, delays in approval or refusal to approve a product candidate; product recall or seizure; interruption of manufacturing or clinical trials; operating restrictions; injunctions; and criminal prosecution. We are required in the United States and in foreign countries to obtain approval from regulatory authorities before we can manufacture, market and sell our products.
Obtaining regulatory approval for marketing of a product candidate in one country does not assure we will be able to obtain regulatory approval in other countries. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries. Once approved, the FDA and other United States and non-United States regulatory authorities have substantial authority to limit the uses or indications for which a product may be marketed, restrict distribution of the product, require additional testing, change product labeling or mandate withdrawal of our products. The marketing of our approved products will be subject to extensive regulatory requirements administered by the FDA and other regulatory bodies, including: the manufacturing, testing, distribution, labeling, packaging, storage, reporting and record-keeping related to the product, advertising, promotion, and adverse event reporting requirements. In addition, incidents of adverse drug reactions, unintended side effects or misuse relating to our products could result in required post-marketing studies, additional regulatory controls or restrictions, or even lead to withdrawal of a product from the market.
In general, the FDA and equivalent other country authorities require labeling, advertising and promotional materials to be truthful and not misleading, and marketed only for the approved indications and in accordance with the provisions of the approved label. If the FDA or other regulatory authorities were to challenge our promotional materials or activities, they may bring enforcement action.
Our failure to obtain approval, significant delays in the approval process, or our failure to maintain approval in any jurisdiction will prevent us from selling a product in that jurisdiction. Any product and its manufacturer will continue to be subject to strict regulations after approval, including but not limited to, manufacturing, quality control, labeling, packaging, adverse event reporting, advertising, promotion and record-keeping requirements. Any problems with an approved product, including the later exhibition of adverse effects or any violation of regulations could result in restrictions on the product, including its withdrawal from the market, which could materially harm our business. The process of obtaining approvals in foreign countries is subject to delay and failure for many of the same reasons.
Failure to comply with foreign regulatory requirements governing human clinical trials and failure to obtain marketing approval for product candidates could prevent us from selling our products in foreign markets, which may adversely affect our operating results and financial condition.
The requirements governing the conduct of clinical trials, manufacturing, testing, product approvals, pricing and reimbursement outside the United States vary greatly from country to country. In addition, the time required to obtain approvals outside the United States may differ significantly from that required to obtain FDA approval. We may not obtain foreign regulatory approvals in the timeframe we may desire, if at all. Approval by the FDA does not assure approval by regulatory authorities in other countries, and foreign regulatory authorities could require additional testing. Failure to comply with these regulatory requirements or obtain required approvals could impair our ability to develop foreign markets for our products and may have a material adverse effect on our business and future prospects.
RISKS IN PROTECTING OUR INTELLECTUAL PROPERTY
If we are unable to protect our proprietary rights or to defend against infringement claims, we may not be able to compete effectively or operate profitably.
We invent and develop technologies that are the basis for or incorporated in our potential products. We protect our technology through United States and foreign patent filings, trademarks and trade secrets. We have issued patents, and applications for United States and foreign patents in various stages of prosecution. We expect that we will continue to file and prosecute patent applications and that our success depends in part on our ability to establish and defend our proprietary rights in the technologies that are the subject of issued patents and patent applications.
| 13 |
The fact that we have filed a patent application or that a patent has issued, however, does not ensure that we will have meaningful protection from competition with regard to the underlying technology or product. Patents, if issued, may be challenged, invalidated, declared unenforceable or circumvented or may not cover all applications we may desire. Our pending patent applications as well as those we may file in the future may not result in issued patents. Patents may not provide us with adequate proprietary protection or advantages against competitors with, or who could develop, similar or competing technologies or who could design around our patents. Patent law relating to the scope of claims in the pharmaceutical field in which we operate is continually evolving and can be the subject of some uncertainty. The laws providing patent protection may change in a way that would limit protection.
We also rely on trade secrets and know-how that we seek to protect, in part, through confidentiality agreements. Our policy is to require our officers, employees, consultants, contractors, manufacturers, outside scientific collaborators and sponsored researchers and other advisors to execute confidentiality agreements. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us be kept confidential and not disclosed to third parties except in specific limited circumstances. We also require signed confidentiality agreements from companies that receive our confidential data. For employees, consultants and contractors, we require confidentiality agreements providing that all inventions conceived while rendering services to us shall be assigned to us as our exclusive property. It is possible, however, that these parties may breach those agreements, and we may not have adequate remedies for any breach. It is also possible that our trade secrets or know-how will otherwise become known to or be independently developed by competitors.
We are also subject to the risk of claims, whether meritorious or not, that our products or immunotherapy candidates infringe or misappropriate third-party intellectual property rights. Defending against such claims can be quite expensive even if the claims lack merit. And if we are found to have infringed or misappropriated a third-party’s intellectual property, we could be required to seek a license or discontinue our products or cease using certain technologies or delay commercialization of the affected product or products, and we could be required to pay substantial damages, which could materially harm our business.
We may be subject to litigation with respect to the ownership and use of intellectual property that will be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us. That litigation is likely to be expensive and may require a significant amount of management’s time and attention, at the expense of other aspects of our business.
Litigation relating to the ownership and use of intellectual property is expensive, and our position as a relatively small company in an industry dominated by very large companies may cause us to be at a disadvantage in defending our intellectual property rights and in defending against claims that our immunotherapy candidates infringe or misappropriate third-party intellectual property rights. Even if we are able to defend our position, the cost of doing so may adversely affect our profitability. We may in the future be subject to patent litigation and may not be able to protect our intellectual property at a reasonable cost if such litigation is initiated. The outcome of litigation is always uncertain, and in some cases could include judgments against us that require us to pay damages, enjoin us from certain activities or otherwise affect our legal or contractual rights, which could have a significant adverse effect on our business.
Our Success Will Depend In Part On Our Ability To Grow And Diversify, Which In Turn Will Require That We Manage And Control Our Growth Effectively.
Our business strategy contemplates growth and diversification. Our ability to manage growth effectively will require that we continue to expend funds to improve our operational, financial and management controls, reporting systems and procedures. In addition, we must effectively expand, train and manage our employees. We will be unable to manage our businesses effectively if we are unable to alleviate the strain on resources caused by growth in a timely and successful manner. There can be no assurance that we will be able to manage our growth and a failure to do so could have a material adverse effect on our business.
| 14 |
A Significant Delay In Procuring Recertification of the cGMP Status Of Our Emmen-Based Manufacturing Facility Could Cause a Delay in the Commencement of Our Clinical Trials, Which Delay Could Have A Material Adverse Effect On Our Business.
Our Emmen-based manufacturing facility previously enjoyed a cGMP rating, which permits its use in the manufacturing the vaccines in the upcoming phase III trial. Certain on-going maintenance and re-qualification of the facility and its equipment are essential to maintain its cGMP rating. We expect to have a recertification inspection in 3rd quarter 2013 and expect to receive recertified cGMP status in the 4th quarter of 2013. Any delay in receiving recertified cGMP status will inhibit our ability to commence our Phase IIIb clinical trials which could ultimately have an adverse effect on our business.
RISKS RELATED TO OUR COMMON STOCK
Transfers of our securities may be restricted by virtue of state securities “blue sky” laws which prohibit trading absent compliance with individual state laws. These restrictions may make it difficult or impossible to sell shares in those states.
Transfers of our securities may be restricted under the securities regulations laws promulgated by various states and foreign jurisdictions, commonly referred to as “blue sky” laws. Absent compliance with such individual state laws, our common stock may not be traded in such jurisdictions. Because the securities registered hereunder have not been registered for resale under the blue sky laws of any state, the holders of such shares and persons who desire to purchase them should be aware that there may be significant state blue sky law restrictions upon the ability of investors to sell the securities and of purchasers to purchase the securities. These restrictions may prohibit the trading of our securities. Investors should consider the secondary market for our securities to be a limited one.
We have no immediate plans to pay dividends.
We have not paid any cash dividends to date and do not expect to pay dividends for the foreseeable future. We intend to retain earnings, if any, as necessary to finance the operation and expansion of our business.
Our officers, directors and principal stockholders collectively own a substantial portion of our outstanding common stock, and as long as they do, they may be able to control the outcome of stockholder voting.
Our officers and directors are collectively the beneficial owners of approximately 43% of the outstanding shares of our common stock as of the date of this Registration Statement. In addition, our principal stockholder is the beneficial owner of approximately 43% of the outstanding shares of our common stock as of the date of this Offering Circular. Accordingly, these stockholders, individually and as a group, may be able to control us and direct our affairs and business, including any determination with respect to a change in control, future issuances of common stock or other securities, declaration of dividends on the common stock and the election of directors.
We have the ability to issue additional shares of our common stock and shares of preferred stock without asking for stockholder approval, which could cause your investment to be diluted.
Our Articles of Incorporation authorizes the Board of Directors to issue up to 200,000,000 shares of common stock and up to 50,000,000 shares of preferred stock. The power of the Board of Directors to issue shares of common stock, preferred stock or warrants or options to purchase shares of common stock or preferred stock is generally not subject to stockholder approval. Accordingly, any additional issuance of our common stock, or preferred stock that may be convertible into common stock, may have the effect of diluting your investment.
| 15 |
By issuing preferred stock, we may be able to delay, defer, or prevent a change of control.
Our Articles of Incorporation permits us to issue, without approval from our stockholders, a total of 50,000,000 shares of preferred stock. Our Board of Directors can determine the rights, preferences, privileges and restrictions granted to, or imposed upon, the shares of preferred stock and to fix the number of shares constituting any series and the designation of such series. It is possible that our Board of Directors, in determining the rights, preferences and privileges to be granted when the preferred stock is issued, may include provisions that have the effect of delaying, deferring or preventing a change in control, discouraging bids for our common stock at a premium over the market price, or that adversely affect the market price of and the voting and other rights of the holders of our common stock.
Once publicly trading, the application of the “penny stock” rules could adversely affect the market price of our common shares and increase your transaction costs to sell those shares.
The Securities and Exchange Commission has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require:
| • | that a broker or dealer approve a person's account for transactions in penny stocks and |
| • | the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. |
In order to approve a person's account for transactions in penny stocks, the broker or dealer must:
| • | obtain financial information and investment experience objectives of the person and |
| • | make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. |
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the Commission relating to the penny stock market, which, in highlight form:
| • | sets forth the basis on which the broker or dealer made the suitability determination and |
| • | that the broker or dealer received a signed, written agreement from the investor prior to the transaction. |
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks
Should our stock become quoted on the OTC bulletin board, if we fail to remain current on our reporting requirements, we could be removed from the OTC bulletin board which would limit the ability of broker-dealers to sell our securities and the ability of stockholders to sell their securities in the secondary market.
Companies quoted on the Over-The-Counter Bulletin Board, such as we are seeking to become, must be reporting issuers under Section 12 of the Securities Exchange Act of 1934, as amended, and must be current in their reports under Section 13, in order to maintain price quotation privileges on the OTC Bulletin Board. If we fail to remain current on our reporting requirements, we could be removed from the OTC Bulletin Board. As a result, the market liquidity for our securities could be severely adversely affected by limiting the ability of broker-dealers to sell our securities and the ability of stockholders to sell their securities in the secondary market. In addition, we may be unable to get re-quoted on the OTC Bulletin Board, which may have an adverse material effect on our Company.
| 16 |
We face risks related to compliance with corporate governance laws and financial reporting standard.
The Sarbanes-Oxley Act of 2002, as well as related new rules and regulations implemented by the SEC and the Public Company Accounting Oversight Board, require changes in the corporate governance practices and financial reporting standards for public companies. These new laws, rules and regulations, including compliance with Section 404 of the Sarbanes-Oxley Act of 2002 relating to internal control over financial reporting (“Section 404”), will materially increase the Company's legal and financial compliance costs and make some activities more time-consuming, burdensome and expensive. Any failure to comply with the requirements of the Sarbanes-Oxley Act of 2002, our ability to remediate any material weaknesses that we may identify during our compliance program, or difficulties encountered in their implementation, could harm our operating results, cause us to fail to meet our reporting obligations or result in material misstatements in our financial statements. Any such failure could also adversely affect the results of the periodic management evaluations of our internal controls and, in the case of a failure to remediate any material weaknesses that we may identify, would adversely affect the annual auditor attestation reports regarding the effectiveness of our internal control over financial reporting that are required under Section 404 of the Sarbanes-Oxley Act. Inadequate internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain qualified board members.
As a public company, we incur significant legal, accounting and other expenses that we would not incur as a private company, including costs associated with public company reporting requirements. We also incur costs associated with the Sarbanes-Oxley Act of 2002, as amended, the Dodd-Frank Wall Street Reform and Consumer Protection Act and related rules implemented or to be implemented by the SEC and the NYSE AMEX. The expenses incurred by public companies generally for reporting and corporate governance purposes have been increasing. We expect these rules and regulations to increase our legal and financial compliance costs and to make some activities more time-consuming and costly, although we are currently unable to estimate these costs with any degree of certainty. These laws and regulations could also make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. These laws and regulations could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as our executive officers and may divert management’s attention. Furthermore, if we are unable to satisfy our obligations as a public company, we could be subject to delisting of our common stock, fines, sanctions and other regulatory action and potentially civil litigation.
Use of Proceeds
We will not receive any proceeds from the sale of the shares of our common stock by the selling stockholders. All proceeds from the sale of such shares will be for the account of the selling stockholders. We will pay for expenses of this offering, except that the selling stockholders will pay any broker discounts or commissions or equivalent expenses applicable to the sale of their shares.
Selling Stockholders
We agreed to register for resale shares of common stock of the selling security holder. The selling security holder may from time to time offer and sell any or all of their shares that are registered under this prospectus. The selling security holder and any participating broker-dealers are “underwriters” within the meaning of the Securities Act of 1933, as amended. All expenses incurred with respect to the registration of the common stock will be borne by us, but we will not be obligated to pay any underwriting fees, discounts, commissions or other expenses incurred by the selling security holder in connection with the sale of such shares.
The following table provides information regarding the beneficial ownership of our common stock held by each of the selling shareholders as of the date of this prospectus, including:
| 17 |
1. the number of shares owned by each prior to this offering;
2. the total number of shares that are to be offered by each;
3. the total number of shares that will be owned by each upon completion of the offering;
4. the percentage owned by each upon completion of the offering and
5. the identity of the beneficial holder of any entity that owns the shares.
The named party beneficially owns and has sole voting and investment power over all shares or rights to the shares, unless otherwise shown in the table. The numbers in this table assume that none of the selling shareholders sells shares of common stock not being offered in this prospectus or purchases additional shares of common stock, and assumes that all shares offered are sold. The percentages are based on 31,227,756 shares of common stock outstanding on June 26, 2013.
| Name of Selling Shareholder | Shares Owned Prior to this Offering(1) | Total Number of Shares to be Offered for Selling Shareholder Account | Total Shares to be Owned Upon Completion of this Offering | Percent Owned Upon Completion of this Offering | ||||||||||||
| Kodiak Capital Group, LLC(2) | 236,364 | 6,000,000 | 236,364 | <1 | %* | |||||||||||
*None of the selling shareholders: (i) has had a material relationship with us other than as a shareholder at any time within the past three years or (ii) has ever been one of our officers or directors.
(1) The actual number of shares of common stock offered in this prospectus, and included in the registration statement of which this prospectus is a part, includes such additional number of shares of common stock as may be issued or issuable upon a put notice under the Agreement with Kodiak.
(2) Ryan Hodson is the Managing Director of Kodiak Capital Group, LLC and, in that capacity, has the authority to direct voting and investment decisions regarding the securities.
PLAN OF DISTRIBUTION
Investment Agreement / Registration Rights Agreement
On July 18, 2012, we entered into the Investment Agreement (as amended on July 8, 2013) and a Registration Rights Agreement with Kodiak Capital Group, LLC in order to establish a possible source of funding for us.
Under the Investment Agreement, Kodiak has agreed to provide us with up to $26,000,000 of funding over an eighteen (18) month period from the effective date of this prospectus; 6,000,000 shares of our common stock are being registered pursuant to this prospectus. During this period, we can deliver a put under the Investment Agreement by selling shares of our common stock to Kodiak and Kodiak will be obligated to purchase the shares. A put transaction must close before we can deliver another put notice to Kodiak.
We may request a put by sending a put notice to Kodiak, stating the amount of the put. During the five trading days following a put notice, we will calculate the amount of shares we will sell to Kodiak and the purchase price per share. The number of shares of Common Stock that Kodiak shall purchase pursuant to each put notice shall be determined by dividing the amount of the put by the purchase price.
The purchase price per share of common stock will be set at eighty-percent (80%) of the lowest daily VWAP common stock during the pricing period, which the five consecutive days following a put notice.
| 18 |
There is no minimum amount we can put to kodiak at any one time. Upon effectiveness of the Registration Statement, the Company shall deliver instructions to its transfer agent to issue shares of Common Stock to Kodiak free of restrictive legends on or before each closing date.
Pursuant to the Investment Agreement, Kodiak and its affiliates shall not be issued shares of our common stock that would result in its beneficial ownership equaling more than 9.99% of our outstanding common stock.
Kodiak will not enter into any short selling or any other hedging activities during the pricing period. On July 18, 2012, we entered into a Registration Rights Agreement with Kodiak requiring, among other things, that we prepare and file with the SEC a Registration Statement on Form S-1 covering the shares issuable to Kodiak under the Investment Agreement.
As per the Investment Agreement, none of Kodiak’s obligations thereunder are transferrable and may not be assigned to a third party.
DETERMINATION OF OFFERING PRICE
The Selling Stockholder may sell its shares in the over-the-counter market or otherwise, at market prices prevailing at the time of sale, at prices related to the prevailing market price, or at negotiated prices. We will not receive any proceeds from the sale of shares by the Selling Stockholder.
DILUTION
“Dilution” represents the difference between the Offering price of the shares of common stock and the net tangible book value per share of common stock immediately after completion of the Offering. “Net Tangible Book Value” is the amount that results from subtracting total liabilities from total tangible assets. Please refer to the following table presenting the number of shares issued and the corresponding price per share paid before this Offering for more information. Following is a table detailing dilution as of March 31, 2012, to investors if 100%, 75%, 50%, or 10% of the Offering is sold.
| Percent of Offering sold | 100 | % | 75 | % | 50 | % | 25 | % | ||||||||
| Net Tangible Book Value Per Share Prior to Stock Sale | $ | (0.55 | ) | $ | (0.55 | ) | $ | (0.55 | ) | $ | (0.55 | ) | ||||
| Pro Forma Net Tangible Book Value Per Share After Stock Sale | 0.24 | 0.07 | (0.12 | ) | (0.33 | ) | ||||||||||
| Increase in net book value per share due to stock sale | 0.80 | 0.62 | 0.43 | 0.23 | ||||||||||||
| Net Dilution (Purchase price of $4.40less Pro Forma Net Tangible Book Value per share) | $ | 4.16 | $ | 4.33 | $ | 4.52 | $ | 4.73 |
PENNY STOCK CONSIDERATIONS
While our current stock price is $5.50 per share, our common stock could become a penny stock if our stock price decreases; therefore, trading in our securities is subject to penny stock considerations. Broker-dealer practices in connection with transactions in “penny stocks” are regulated by certain penny stock rules adopted by the Securities and Exchange Commission.
Penny stocks generally are equity securities with a price of less than $5.00 (other than securities registered on certain national securities exchanges or quoted on the NASDAQ system). Penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document that provides information about penny stocks and the risks in the penny stock market. The broker-dealer also must provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction, and monthly account statements showing the market value of each penny stock held in the customer’s account. The broker-dealer must also make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. These requirements may have the effect of reducing the level of trading activity, if any, in the secondary market for a security that becomes subject to the penny stock rules. The additional burdens imposed upon broker-dealers by such requirements may discourage broker-dealers from effecting transactions in our securities, which could severely limit their market price and liquidity of our securities. These requirements may restrict the ability of broker-dealers to sell our common stock and may affect your ability to resell our common stock.
| 19 |
State Securities - Blue Sky Laws
There is no public market for our common stock, and there can be no assurance that any market will develop in the foreseeable future. Transfer of our common stock may also be restricted under the securities regulations or laws promulgated by various states and foreign jurisdictions, commonly referred to as “Blue Sky” laws. Absent compliance with such individual state laws, our common stock may not be traded in such jurisdictions. Because the securities registered hereunder have not been registered for resale under the Blue Sky laws of any state, the holders of such shares and persons who desire to purchase them in any trading market that might develop in the future, should be aware that there may be significant state Blue-Sky law restrictions upon the ability of investors to sell the securities and of purchasers to purchase the securities. Accordingly, investors may not be able to liquidate their investments and should be prepared to hold the shares of our common stock for an indefinite period of time.
Selling security holders may contact us directly to ascertain procedures necessary for compliance with Blue Sky Laws in the applicable states relating to sellers and/or purchasers of our shares of common stock.
We intend to apply for listing in Mergent, Inc. Securities Manual which, once published, will provide us with “manual” exemptions (as described below) in approximately 33 states as indicated in CCH Blue Sky Law Desk Reference at Section 6301 entitled “Standard Manuals Exemptions.”
Thirty-three states have what is commonly referred to as a “manual exemption” for secondary trading of securities such as those to be resold by selling security holders under this Prospectus. In these states, so long as we obtain and maintain a listing in Mergent, Inc. or Standard and Poor’s Corporate Manual, secondary trading of our common stock can occur without any filing, review or approval by state regulatory authorities in these states. These states are Alaska, Arizona, Arkansas, Colorado, Connecticut, District of Columbia, Florida, Hawaii, Idaho, Indiana, Iowa, Kansas, Maine, Maryland, Massachusetts, Michigan, Mississippi, Missouri, Nebraska, New Jersey, New Mexico, North Carolina, North Dakota, Ohio, Oklahoma, Oregon, Rhode Island, South Carolina, Texas, Utah, Washington, West Virginia and Wyoming. We cannot secure this listing, and thus this qualification, until after the Registration Statement, of which this Prospectus forms a part, is declared effective. Once we secure this listing, secondary trading can occur in these states without further action.
We currently do not intend to and may not be able to qualify our common stock for resale in other states which require shares to be qualified before they can be resold by our security holders.
Limitations Imposed by Regulation M
Under applicable rules and regulations under the Securities Exchange Act of 1934, as amended, any person engaged in the distribution of the shares may not simultaneously engage in market making activities with respect to our common stock for a period of two business days prior to the commencement of such distribution. In addition and without limiting the foregoing, each selling security holder will be subject to applicable provisions of the Securities Exchange Act of 1934, as amended, and the associated rules and regulations thereunder, including, without limitation, Regulation M, which may limit the timing of purchases and sales of shares of our common stock by the selling security holders. We will make copies of this Prospectus available to the selling security holders and will inform them of the need for delivery of copies of this Prospectus to purchasers at or prior to the time of any sale of the shares offered hereby. We assume no obligation to so deliver copies of this Prospectus or any related Prospectus supplement.
| 20 |
DESCRIPTION OF BUSINESS
Overview
Vaccinogen, Inc. (referred to as “Vaccinogen”, “the Company”, “we”, “us”, or “our”), a Maryland corporation, is a biotechnology company focused on the development and commercialization cancer vaccines and immunotherapeutic products for cancers and infectious diseases. Our primary product is OncoVAX®, a cancer vaccine for the post-surgical treatment of Stage II colon cancer. We believe that OncoVAX® is the first immunotherapy for Stage II colon cancer.
We currently employ 10 persons. Our executive offices are located at 5300 Westview Drive, Suite 406, Frederick, MD 21703. Our telephone number is (301) 668-8400 and our Internet address is www.vaccinogen.com.
Company History
We are a Maryland corporation that was originally formed as a Delaware corporation on May 2, 2007. On November 23, 2010, the Company changed its domicile from Delaware to Maryland.
Agreements with Intracel Holdings Corporation
License Agreement
On October 10, 2007, we entered into a License Agreement with Intracel (the “License Agreement”), for exclusive rights to use the OncoVAX® technology platform. OncoVAX® is an active specific immunotherapy (“ASI”) that uses the patient’s own cancer cells to block the return of colon cancer following surgery. In consideration of the license, we (i) agreed to issue equity equal to 10% of the fully diluted capitalization of the Company (ii) assumed liabilities of Intracel to Organon Teknika Corporation (“Organon”) under an October 30, 2007 Letter Agreement between Intacel and Organon; (iii) $450,000 on cash for settling trade payable related to the OncoVAX® intellectual property and (iv) royalty payments based on future sales of OncoVAX®. The License Agreement provided Intracel with antidilution rights with respect to its 10% equity issuance. The licensing arrangement also contained a provision such that if we obtained certain levels of financing in a specified time period, all rights to OncoVAX® would transfer to us.
Asset Transfer Agreement and Stock Exchange Agreement
On June 24, 2010, we entered into an Asset Transfer Agreement with Intracel pursuant to which we acquired all of the intellectual property associated with OncoVAX®. Upon execution of the transfer agreement, all significant terms of the license agreement were terminated except the provision for royalties on future sales. In consideration of the asset transfer, we agreed to assume all of Intracel’s obligations to Organon and agreed to enter into a stock exchange agreement with Intracel. Under the terms of the June 24, 2010 Stock Exchange Agreement, we issued 3,471,766 shares of its Series B Preferred Stock in exchange for the assets transferred under the Asset Transfer Agreement and all of our common stock and Series AA Preferred Stock held by Intracel. The Series B Preferred Stock issued to Intracel (and its designees) constituted 20% of the issued and outstanding stock of the Company on a fully-diluted basis. Intracel (and the other investors under the Stock Exchange Agreement) were granted anti-dilution rights with respect to its series B preferred stock. In addition, we agreed that Series B Preferred holders’ ownership position (and corresponding anti-dilution rights) would increase to 50% upon failure of us to meet certain defined milestones, which included but were not limited to, us attaining certain levels of financing. We did not meet these criteria and consequently, during December 2010, were required to increase the Series B Preferred holders total ownership interest to 50% through the issuance of additional Series B Preferred Stock to Intracel and related parties to Intracel. Currently, Intracel’s ownership percentage is approximately 40% and the remaining 10% resides with related parties to Intracel.
| 21 |
Acquisition of Manufacturing Assets
On October 23, 2007, Vaccinogen acquired out of bankruptcy court in the Netherlands, certain other manufacturing assets that had been previously owned and operated by Intracel’s wholly owned Netherlands based subsidiary. In connection with this asset acquisition, the Company formed its wholly owned subsidiary, Vaccinogen BV, for the purposes of continuing development of OncoVAX®.
Operating Strategy
We will maintain a cGMP facility and outsource clinical activities to contract research organizations (“CRO) in an effort to achieve the following:
| 1. | Complete the pivotal Phase IIIb clinical trial of this autologous vaccine, OncoVax®, which is a new paradigm to prevent the recurrence of stage II colon cancer. The planned trial will closely replicate a successful prior Phase IIIa. |
| 2. | Develop near term revenues by establishing licensing arrangements for distribution of OncoVax® in certain territories as well as for additional carcinoma indications. |
| 3. | Develop diagnostic and therapeutic drug products as well as near-term revenues by exploiting its distinctive Human Monoclonal Antibodies (“HuMabs”) technology. |
Vaccines in Immunotherapy of Cancer
In 2007 a major review of active immunotherapy’s, so called "Cancer Vaccines", (as distinguished from passive immunotherapy with immune stimulators or monoclonal antibodies) and the various scientific and business factors that have contributed to the disappointing results in this field was presented by Finke et al1. Over the past decade many Phase III clinical trials with cancer vaccines have failed to achieve significant clinical results2. It is interesting that one of the first general considerations of the review was "Select the most informative targets". They point out that ideally the target should be tumor-specific and that "it is important to use the intended study population to assess the proportion of tumors that express the target of choice and the proportion of cells within each tumor that express it." We believe this indicates that the review focused on antigen discovery and was emphasizing the use of common antigens and presumably based on the assumption of inter-and intra-tumor homogeneity.
Since 2007, cancer genome DNA sequencing data for several carcinomas have provided what we believe is indisputable evidence of extreme genetic diversity or heterogeneity both within tumors or among tumors. Thus, the concept of tumor homogeneity was in our opinion a mistaken assumption and as such we believe this probably was the weakest underlying biologic premise of the past active specific immunotherapy efforts of the past two decades. Cancer is a genetic disease; the genetic sequencing data of tumor cells over the last few years, based on second-generation DNA sequencing technology, clearly reveals that there has been an underestimation of the degree of tumor heterogeneity and the consequences of this in the antigen discovery process of cancer vaccine development. In erroneously treating cancer as a homogeneous disease, the industry has failed in attempts to adequately train the immune system to recognize the abundant foreign or “non-self” components of an individual tumor and defend the body against it. It is understood that you cannot treat a heterogeneous disease with a homogeneous treatment. It is well recognized that the surgical resection is the only treatment for reducing bulky tumors. In addition, by using the patients’ primary tumor resected at surgery as the source of the antigens, embraces the heterogeneity of cancer cells. It does this by using a cancer vaccine as a preventative measure against metastasis. Thus, cancer vaccines hold tremendous promise.
1Finke LH, Wentworth K, Blumenstein B, Rudolph NS, Levitsky H, HoosA.Lessons from randomized phase III studies with active cancer immununotherapies—outcomes from the 2006 meeting of the Cancer Vaccine Consortium (CVC). Vaccine 2007;25 (Suppl 2):B97-109; PMID:17916465
2Kudrin A and Hanna MG Jr. (Guest Editors) Special Focus Cancer Commentaries Series, Human Vaccines &Immunotherapeutics, Vol 8 Issue 8, August 2012.
| 22 |
To further expand on this topic of heterogeneity, Wood et al3 asked the question "how many genes are mutated in a human tumor." They analyzed this question in breast and colorectal cancer; they reported that there are ~80 DNA mutations that alter amino acids in a typical cancer. These altered proteins are all candidates for unique markers or tumor-specific antigens. The study included an analysis of the sequences of 20,857 transcripts from 18,191 human genes including the great majority that encode for proteins. Examining the overall distribution of these mutations in different cancers of the same type leads to a new view of cancer genome landscapes. Although the numbers of mutant genes in breast and colorectal cancers were similar, the particular genes that were mutated were quite different, as were the type of mutations found. The surprising discovery that has an enormous impact on cancer vaccines or active specific immunotherapy, is that of the ~80 mutations in an individual tumor only ~3 of these ~80 mutations were common and thus would be shared antigens in two different tumors. Consequently, with polyvalent cancer vaccines, a robust and therapeutic immune response cannot be provided by allogeneic cells or even a relatively minor component of "off the shelf" common antigens. We believe these results demonstrate that the antigen discovery aspect of cancer vaccines takes on a whole new level of complexity and is fraught with new hurdles. Autologous tumor cell vaccines become a much more technologically and immunologically sound approach to cancer vaccines, in our opinion. This, for active immunotherapy cancer treatment advocates, is not all bad. The findings confirm our belief that the genetic lesions that are unique to the original tumor cells, "the trunk of the evolutionary tree" are consistently expressed.
Mainstream pharma continues to advocate for more targeted therapies for smaller patient populations. The hundreds and thousands of cancer mutations identified over the past few years probably makes this approach impractical. There is one evolutionary approach that already exists to address the magnitude of cancer diversity. The immune system is designed by nature to protect against diversity of disease. The immune system constantly protects humans from an array of deadly foreign pathogens, viruses and proteins. With the exception of safe drinking water, no other modality, not even antibiotics, has, in our opinion, had such a major effect on mortality reduction and population survival and growth.
OncoVAX® Overview
We believe that OncoVAX® is the first colon cancer vaccine to demonstrate effectiveness in both preventing cancer recurrence after surgical resection of the primary tumor and addressing the diversity of cancer cells. Currently in a pivotal Phase III trial under the auspices of an FDA granted Special Protocol Assessment (SPA) and a Fast Track designation, OncoVAX® uses the patients live, metabolically active, sterile and nontumorigenic tumor cells to mobilize the body's immune system to prevent the recurrence of colon cancer following surgery. Embracing the now widely recognized heterogeneity of cancer, OncoVAX® uses a patient's own tumor to stimulate a broad and effective immune response against the diversity and uniqueness of that patient's cancer cells. The OncoVAX® technology platform is being tested in Stage II colon cancer after standard of care surgical resection. The global incidence of Stage I-IV colon cancer is 900,000 patients per year of which 269,000 are Stage II. The prevalence of Stage II, an unmet medical need, has grown with the emergence of more rigorous screening practices and endoscopic examinations. Stage II colon cancer is forecasted to be about 46% of the US and EU colon cancer at diagnosis by 2020.
3Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal and colorectal cancers. Science 2007; 318:1108-13; PMID:17932254
| 23 |
OncoVAX® Clinical Trials in Colon Cancer Patients
Since 1980 investigations by Hanna and colleagues4, 5, 6, 7 have translated the principles and procedures of active specific immunotherapy as observed in the L10, inbred and syngeneic guinea pigs into phase I, II, and III adjuvant therapy clinical trials in patients with colon cancer. These clinical trials were designed based on the evidence in the animal models that the immune system can be educated to control a limited systemic tumor burden remaining after surgical excision of solid tumors. In the translation of the animal model studies to the clinic, the antigen discovery selection of the human vaccine was an important decision. In 1977 Fidler and Kripke published in Science8 the discovery of phenotypic heterogeniety in transplanted tumors. Clones derived in vitro from a parent culture of muring malignant melanoma cells varied greatly in their ability to produce metastatic colonies in the lungs upon intravenous inoculation into syngeneic mice. This suggested that the parent tumor is heterogeneous and that highly metastatic tumor cell variants pre-exist in the parental population. Having been present at the NCI laboratories while this work was bering performed and published, Dr. M. G. Hanna Jr. recognized that if intratumoral phenotypic heterogeniety for metastasis exists it would be probable that intertumoral and intratumoral antigenic heterogeneity might also exist. Therefore, it was theorized that the use of autologous tumor cell vaccines from the primary tumor would obviate the antigenic diversity in any immunotherapeutic approach involving tumor cell vaccines. Over 30 years later, we believe this strategy has been validated. Vaccinogen's technology tailors a specific vaccine for each patient from their own tumor, remaining agnostic as to which antigens will prove to be the most functional in individual cases.
Clinical testing of OncoVAX® as an adjuvant to surgical resection began in 1980 with a single center pilot study of 5 subjects with colon cancer. To date 757 subjects with colorectal cancer, of which 720 had colon cancer, have been enrolled in trials of OncoVAX® (see Table 1 for summary). In addition, a bioequivalency study (2002-01) enrolled 15 patients with colon cancer. This study was conducted to compare the immunogenicity, as determined by the magnitude of delayed type hypersensitivity (DTH) responses to tumor cells alone, of vaccines manufactured by the current, sterile process with historical data from the phase III clinical study (8701). We believe that the results from this study unequivocally support the premise that the immunogenicity of vaccines produced by either process are comparable.
In these trials, excluding the bioequivalency study, 385 subjects were randomized to receive OncoVAX®, of which 353 received at least one vaccination; 372 subjects were treated with surgery alone. In addition, subjects with colorectal cancer were enrolled in three separate trials that assessed the effects of ASI with OncoVAX® in combination with chemotherapy as adjuvant therapy to surgical resection. Lastly, two trials have been performed using autologous tumor cells/BCG in melanoma (n=86) and renal cell carcinoma (n=14).
Table 1 OncoVAX® Clinical Trials for Colon Cancer
| Protocol Number |
Trial Phase |
Number of Patients |
Number of Vaccine Doses |
Disease and Stage |
Manufacturing* |
| 8101 | I | 5 Treated | 3 | Colon Cancer Stage III/IV | Deentralized, Non Validated, Non cGMP |
| 8102 | II/III |
47 Control
50 Treated |
3 | Colorectal Cancer Stage I-IV | Decentralized, Non Validated, Non cGMP |
| 5283 | III |
207 Control
205 Treated |
3 | Colon Cancer Stage II/III | Decentralized, Non Validated, Non cGMP |
| 8701 | III |
128 Control
126 Treated |
4 | Colon Cancer Stage I-III | Centralized, Validated, cGMP |
| ASI-2002-01 | I/II | 15 Treated | 4 | Colon Cancer Stage II/III | Centralized, Validated, cGMP |
4Hanna MG Jr, Peters LC.Immunotherapy of established micrometastases with Bacillus Calmette-Guerin tumor cell vaccine.Cancer Res 38:204, 1978.
5Hanna MG Jr, Peters LC. BCG immunotherapy: Efficacy of BCTG-induced tumor immunity in guinea pigs with regional tumor and/or visceral micrometastases. In Immunotherapy of Human Cancer. Raven Press, 1978, pp 11-129.
6Hanna MG Jr, Active specific immunotherapy of residual micrometastases: A comparison of postoperative treatment with BCG-tumor cell vaccine to preoperative intratumoral BCG injection. In Immunobiology and Immunotherapy of Cancer. WD Terry, Y Yamamura, eds, Elsevier/North Holland, 1979, pp 331-350
7Hanna MG Jr, Brandhorst JS, Peters LC. Active specific immunotherapy of residual micrometastasis: An evaluation of sources, doses and ratios of BCG with tumor cells. Cancer ImmunolImmunother 7:165, 1979
8Fidler IJ, Kripke ML. Metastasis results from pre-existing variant cells within a malignant tumor. Science 197:4306 893-895, August 1977.
| 24 |
*Centralized = Manufacture at a single, centralized location; Decentralized = manufacture at each investigational site.
Study 8102 used a three-vaccine regimen as did 5283. In Study 8102, manufacture was conducted at the institution of the principal investigator, Dr. Herbert Hoover9, although this institution changed twice during the study, while in 5283, manufacture was decentralized. Each clinical site manufactured the vaccine, without Quality Assurance or adequate Quality control. While both of these studies provided encouraging clinical results, with respect to tumor immunity as measured by delayed-type hypersensitivity (DTH) and induction of T-cell immunity, neither of these studies demonstrated statistically significant clinical benefit in an Intent to Treat analysis of recurrence free survival or overall survival. In the ECOG study 528310, an analysis by the ECOG statisticians of the correlation of clinical benefit to the quality of the vaccine showed a significant correlation. Further, in study 8102, a subset of subjects underwent DTH testing that revealed a deterioration of the immune response to the standard vaccine at 6 months. It was concluded that a fourth booster immunization at 6 months after surgical resection would enhance the waning immune response to autologous tumor cells. Hence a four vaccine regimen was tested and manufacturing was centralized at the Free University of Amsterdam11. In this study (8701), subjects with Stage II disease demonstrated what we believe were both clinically meaningful and statistically significant outcomes in both recurrence-free interval (p=0.008) and recurrence-free survival (p=0.015). The 5-year event-free rates also demonstrated a clinically and statistically meaningful outcome in overall survival.
Phase IIIa Trial 8701
The 8701 Phase IIIa trial was conducted in 254 patients with Stage II or III colon cancer. In this study a planned, prospective stratification by tumor stage, was conducted in order to legitimately analyze the clinical benefit in each stage of tumor. Thus, this was not a retrospective subgroup analysis. Patients were randomized 1:1 to receive surgery and then observation or immunotherapy according to the protocol that Vaccinogen plans to use in the upcoming confirmatory Phase IIIb trial that will be a key use of proceeds from this offering. The treatment schedule is depicted in the figure below. After approximately one month to recover from surgery and regain their full immune function, patients receive isolated irradiated tumor cells in combination with BCG in two injections a week apart. A week later, a third injection of irradiated tumor cells alone was given. A fourth injection was given as a booster six months after the surgery. This was the first clinical trial where the manufacturing process was optimized in a central processing facility to provide for the three induction vaccinations and the boost at 6 months. This turned out to be the optimum regimen for OncoVAX®.
9Hoover HC Jr, Brandhorst J, Peters C, Surdyke MG, et al. Adjuvant active specific immunotherapy for human colorectal cancer – 6.5-year median follow-up of a phase III prospectively randomized trial. J Clin Oncology, 11:3 390-399, 1993
10Harris JE, Ryan L, Hoover HC Jr, Stuart RK, Oken MN, Benson AB, et al. Adjuvant active specific immunotherapy of stage II and III colon cancer with an autologous tumor cell vaccine: ECOG study E5283. Journal of Clinical Oncology Vol 18 148-157, January 2000
11Vermorken JB, Claessen AME, van Tinteren H, Gall HE, et al. Active specific immunotherapy for stage II and III human colon cancer. A randomized Trial. The Lancet 353 345-350, January 1999
| 25 |
VACCINATION PROTOCOL FOR OncoVAX® IN 8701 AND PLANNED PHASE IIIb TRIAL
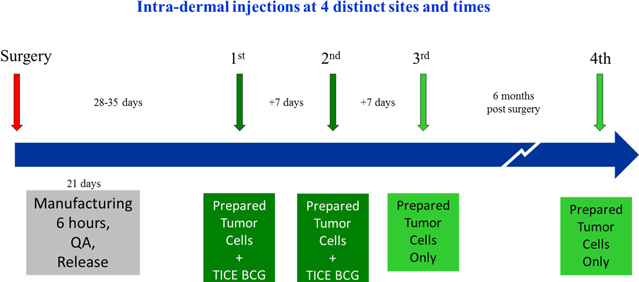
In Study 8701, all patients underwent surgery, and their resected tumors were sent to Vaccinogen’s central processing laboratory. Eight vials of isolated, irradiated tumor cells were sent back to the patient’s clinic along with two vials of BCG.
Recurrence-free survival is the most appropriate endpoint for studies of Stage II colon cancer since patients with recurrent disease will receive treatment with other modalities and their overall survival (OS) will consequently depend in part on the efficacy of other therapies. The outcome of the pre-specified analysis of the stratified Stage II patients from trial 8701 has been particularly informative in planning the upcoming confirmatory Phase IIIb trial in Stage II disease.
| 26 |
OVERALL SURVIVAL IN STUDY 8701
OncoVAX® - Clinical Results
8701 Study - Overall Survival in Stage II Patients
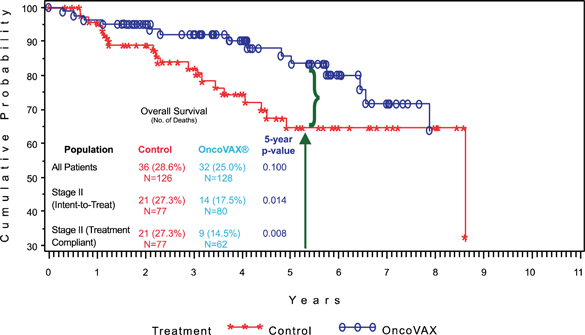
Trends towards efficacy in OS and RFS were not statistically significant in the full intent-to-treat population. A prespecified stratification of the trial to analyze by tumor stage demonstrated that Stage II patients separately reached statistical significance with a p value of 0.015 on a log rank, and 0.008 on a five year event free analysis.
| 27 |
RECURRENCE-FREE SURVIVAL IN STUDY 8701
OncoVAX® - Clinical Results
8701 Study - Recurrence-Free Survival in Stage II Patients
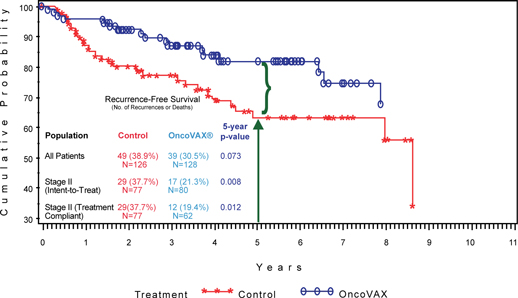
This published, randomized Phase IIIa clinical trial was stratified by tumor stage so that a legitimate analysis by tumor stage could be calculated. These results are for Stage II colon cancer. Some benefit was seen in Stage III colon cancer however the results were not statistically significant. This study was accepted by the FDA as supportive data for the next Phase IIIb clinical trial to be conducted under an FDA granted SPA with fast tract designation. The disease-free survival clinical endpoint will be used for the interim analysis, which is an accepted basis for FDA approval.
The results were published in the British Medical Journal, The Lancet January 30, 1999; 353: 345-350.
| 28 |
RECURRENCE-FREE INTERVAL IN STUDY 8701
OncoVAX® - Clinical Results
8701 Study - Recurrence-Free Interval (RFI) in Stage II Patients
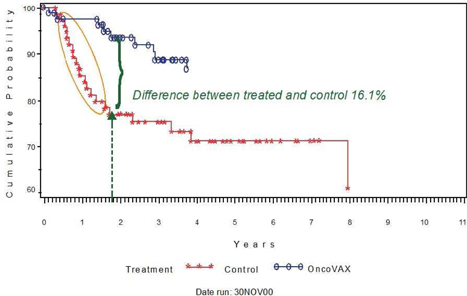
Greatest difference between treated and control in RFI at 18 months post Vx
Patients with Stage II disease who received all four inoculations had superior clinical outcomes to those who received fewer than four inoculations. Relative risk of death or recurrence (recurrence-free survival) for all patients receiving four inoculations was 0.61 (p = 0.034) and relative risk of death or recurrence for patients with Stage II disease receiving four inoculations was 0.40 (p = 0.007). Relative risk of death from any cause in Stage II patients receiving four inoculations was 0.46 (p = 0.046). See the Kaplan-Meier curves in the figure above. Recurrence free survival is the agreed endpoint for Vaccinogen’s Phase IIIb trial. In summary in the surgery only group one can estimate that approximately one out of three patients will reccur with cancer, in the OncoVAX® treated group one can estimate that one out of ten patients will reccur with cancer.
To emphasize the robustness of the clinical benefit of OncoVAX® in these colon cancer patients a recent review of the patient follow-up was conducted by Vermorken and colleagues12. The remarkable result was that the observed delta and significant benefit in recurrence-free survival measured at 5 years was reevaluated at 15 years. The results showed that OncoVAX® treated patients versus control at 15 year follow-up was HR=0.62 (95%, CI:34-0.96) log rank p-value 0.03). This type of robust benefit is what is requisite in effective therapies.
Planned Trials - Stage II Colon Cancer, Phase IIIb Trial
The FDA has requested a second, confirmatory, randomized controlled Phase III trial of OncoVAX® in Stage II colon cancer. The principal objective of Vaccinogen is to organize a "best of breed" team to conduct a confirmatory Phase IIIb trial in Stage II colon cancer. The protocol of this trial, including endpoints and the statistical analysis plan is the subject of an SPA agreement granted by the FDA.
12Weger de VA, Turksma AW, Voorham, QJM, Euler Z, et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite stable and microsatellite instable colon cancer. Clin Cancer Res Feb 1, 2012, 18;882
| 29 |
This study will be carried out under a Special Protocol Assessment (SPA) that was negotiated with the FDA in 2010. An SPA granted by the FDA provides a mechanism for the sponsors and the FDA to reach agreement on size, execution and analysis of a clinical trial that is intended to form the primary basis for regulatory approval. The primary endpoint of this pivotal Phase IIIb trial is recurrence-free survival (RFS) with an interim analysis after 70 “events,” and a final primary analysis three years following completed enrollment. “Events” are incidents of either recurrent disease or death. The study is powered at 90% to detect a 50% improvement in RFS versus resection only for final analysis with an adjustment for interim analysis. The power calculations in the study assume an event rate and distribution that match those observed in the 8701 Phase IIIa study. If a robust p value is achieved at the interim analysis, the Biologics License Application (BLA) can be filed with the FDA’s center for Biologics Evaluation and Research (CBER) at that point. Past clinical trials using the optimal regimen with four immunizations will be accepted as supportive studies during the FDA review of the BLA.
The Phase IIIb study will enroll 550 patients, randomized 1:1 to receive surgery alone, or surgery plus OncoVAX®. Patients will be followed on a regular schedule to see whether and when their cancer might reappear. The experience with OncoVAX® in the 8701 study showed that in the relevant Stage II group, disease recurrence happened more quickly and more frequently in those patients who received surgery alone. If the Phase IIIb trial replicates the experience of the 8701 patients, the interim analysis after 2/3 of the expected events should give a clear and statistically significant separation between the Kaplan-Meier curves at that point, and would form the basis for a BLA seeking marketing approval from the FDA. We expect to begin Phase IIIb trials in the 4th quarter of 2013.
It is important to emphasize that the FDA has agreed that RFS is the most appropriate endpoint to evaluate a trial in Stage II colon cancer. The alternative in cancer studies is often OS. An OS endpoint has several drawbacks in this case. The average age of patients presenting with colon cancer is 65 years, so deaths from causes other than the tumor will dilute the outcome in both arms. Patients in either arm whose disease recurs will receive chemotherapy and other treatments for their metastatic disease, and the success of these treatments will dilute the impact of earlier therapies such as OncoVAX® on OS. From a patient perspective, being disease free is clearly a meaningful endpoint.
Planned Trial - Phase I/II Trial in Stage III Colon Cancer
When patients present for colon cancer surgery, it is not clear in advance whether they have Stage II or Stage III disease. Vaccinogen’s planned Phase IIIb trial will enroll only patients with Stage II tumors, but the clinical sites will perform surgery and capture tumors on many Stage III patients. Stage III tumors may hypothetically be more difficult to treat since micrometastases may be more common, more aggressive, or larger. Any vaccination protocol also needs to accommodate the schedule of adjuvant chemotherapy that is given to Stage III colon cancer patients in the period after their surgery. There is a risk that the immunosuppression that accompanies chemotherapy could interfere with the effect of the fourth, booster vaccination. Nonetheless, there is a considerable unmet need in Stage III colon cancer as well as a significant market opportunity.
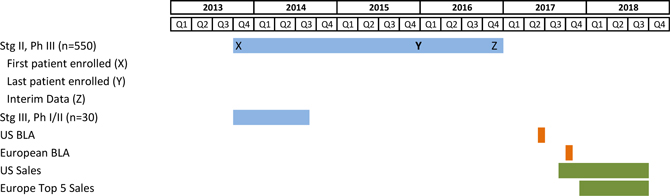
The expense of conducting a parallel registration program in Stage III disease is likely beyond our current means, but with relatively modest additional funds, we could conduct a small pilot program in parallel to the pivotal Stage II trial to demonstrate the ability to generate vaccine from Stage III patients and to accommodate their adjuvant chemotherapy schedule. This pilot trial will have DTH and safety endpoints to show whether chemotherapy interferes with the expected immunogenicity of the vaccine. This trial should enroll 30 patients and is planned to start one month after the Stage II, Phase IIIb pivotal trial. DTH data will become available for each patient when they receive their third immunization, which is contemplated to be before their chemotherapy begins. Full data from this trial could be available as early as 4Q 2013. A successful outcome of this study could pave the way for a future program in Stage III disease.
| 30 |
Additional Applications of Technology
Our technology may also have application in other tumor types, notably melanoma and renal cell carcinoma (RCC). Clinical development in both these tumor types is more challenging than in Stage II colon cancer because there are approved chemotherapies and numerous investigational agents crowding the landscape in each.
Human Monoclonal Antibodies (“HuMabs”) Program
Monoclonal antibody (Mab) technologies evolved rapidly over the last 20 years. Antibody based therapeutics now make up a significant and growing portion of total pharmaceutical sales. Data Monitor’s Monoclonal Antibody: Update 2009 reports global sales of monoclonal antibodies (Mabs) reached $32.2 billion in 2008 and forecast an increase to nearly $60 billion by 2014. KirhamMaggon of the Knol-Beta Publishing Group reports that global Mab sales reached $48 billion in 2010 and that the top five selling Mab (Remicade, Avastin, Rituxan, Humira and Herceptin) each had sales gains of $1 billion over 2009 levels. An additional $10 billion in sales of Mabs for diagnostics and as reagents for R&D results in a total market approaching $60 billion.
We believe that the ascendancy of Mab in the industry is based on the fundamental role that antibodies play in our natural defense system. Mabs seek out, recognize and bind to a particular site on cells, viruses and other organisms in a highly specific manner. We believe this makes them an effectively targeted approach to fight or detect many diseases. Mabs are a part of the body's own natural disease fighting system. Consequently, they have lower side effects compared to small molecule based treatments. In addition, antibody products generally have a shorter development time and in our opinion attractive IP benefits compared with small molecules. We expect interest in Mab technology will continue to grow particularly in the oncology field as the cancer genome is being revealed. Last year alone over 400 random mutational events were discovered yielding 400 new markers. This number is expected to rise into the thousands (MR Stratton, Science, 331 pp 1553-1558). Most major pharmaceutical companies now have Mab projects in their R&D plans. Hence, M&A and Mab deals in R&D and marketing have increased again last year. We believe that we are well positioned to exploit these developments by building novel fully human monoclonal antibody libraries. We intend to implement a strategy to leverage our planned phase III trial to participate in this burgeoning market.
We believe that our previous Phase III OncoVAX® trials, demonstrated that the immunized patients, while mounting a robust and tumoricidal cytotoxic T-cell response, also produced circulating B-cells, which have the potential to differentiate and produce tumor specific, fully human monoclonal antibodies (HuMabs). B-cells are a specialized type of blood cell that retains a memory of the interaction, an immunological history of the protective response. We plan to collect this rare set of B-cells generated from the treated portion of the enrolled patient population. We plan to use the new B-cells and perpetuate them into libraries that can be screened by potential partners to create new therapeutics, vaccines, diagnostics, imaging agents and as research tools.
We believe this novel asset, the B-cell collection, and potentially materials developed during the 8701 trial and predecessor company efforts, represent a valuable asset that can generate significant upfront fees, development milestones and royalties via collaborations and licensing activities that will complement the OncoVAX® investment opportunity.
History
The team of scientists led by Michael G. Hanna, Jr., Ph.D., Vaccinogen’s Founder, Chairman and Chief Executive Officer discovered that the autologous colon tumor immunized patients, while mounting a robust and tumoricidal cytotoxic T-cell response, also during a restricted period of time produced circulating B-cells which were capable of being perpetuated into tumor specific, human monoclonal antibody producing cells (HuMabs). A research program was created that over the course of several years assembled a large and most unique array of fully human monoclonal antibodies. These HuMabs were products of the antibody forming B-cells in circulation in the colon tumor immunized patients. HuMabs may be effective in both, diagnosis and treatment of disease, and overcome many of the limitations of cytotoxic drugs. Biopharmaceutical companies that exploit monoclonal antibodies as the source for developing new commercial products have made great strides in new product efficacy and generally experience shorter development cycles. The powerful combination of those two factors has made monoclonal antibodies a most attractive source for the development activities in the biopharmaceutical marketplace.
| 31 |
Competitive Advantage
During the course of the first OncoVAX® Phase III trial (8701), Dr. Hanna’s team recognized that the tumor cell destruction was mediated by cytotoxic, specifically immunized T-cells, and also for evidence of a humeral (antibody) mediated immune response. It was discovered that following the second OncoVAX® dose involving the patients’ own tumor cells and BCG, the patients produced tumor specific antibody producing circulating B-cells.
We believe our competitive advantage is the unique access to and ownership of a valuable educated B-cell repertoire to be collected from immunized patients in the upcoming Phase III trial as well as the insight and experience from legacy efforts to build a HuMab based business unit. From this institutional knowledge, we should be able to leverage a plethora of new development and production strategies as well as technologies to efficiently and effectively exploit this opportunity. Selected contractors and collaborators will collect blood from patients over the first year of the trial and develop libraries from these unique HuMabs.
We believe that we two other competitive advantages – these Mabs are fully human and have high avidity binding potential.
The most important point to understand is that no company in our opinion (other than us) can generate antibodies in a set of patients based on a clinical response to a sterile, colon cancer vaccine.
Fully Human Monoclonal Antibodies
The industry’s original Mab products were mouse or chimeric or humanized resulting in significant drug development challenges as they:
• Do not interact efficiently with the human immune system,
• Have rapid clearance, potency issues,
• Can cause allergic reactions,
• Often trigger a human anti-mouse antibody (HAMA) response, and
• Often require royalty payments to third party owners of the tools and methods to optimize the drug product.
By contrast, Vaccinogen’s HuMabs are fully human (not humanized) and carcinoma tumor specific. Their properties allow them to be administered safely to humans in large quantities with potential application in chronic as well as acute disease settings. They are also expected to be of interest in the vaccine, imaging and diagnostics markets. Furthermore, they may have applications in indications beyond the cancer market.
Immune Libraries
We believe that our repertoire of educated B-cells from OncoVAX® immunized patients will allow the creation of a unique library comprised solely of human “immune” responses from immunized patients. This means that the antibodies were created naturally in the human body (matured in-vivo). These antibodies are far more “potent” and vastly superior to those developed in-vitro (outside the body) in their ability to bind with and neutralized the target disease.
These two characteristics, “fully human” and “immune”, provide us with what we believe is valuable competitive advantage and an asset that can complement the OncoVAX® investment opportunity.
| 32 |
We intend to launch a pivotal Phase III trial of autologous colon tumor vaccines. The 275 treated patients will yield immune B-cells from which multiple phage expression libraries can be produced. Our intended approach to exploit this market opportunity is to execute strategic partnerships with a select group of qualified pharma and genomics companies, allowing them access to its monoclonal library. Our partners will “mine” the libraries using antigens or target molecules they own, to identify potential antibodies for further development. Ultimately, we expect these antibodies will be developed and commercialized into products for use in the detection, treatment, and prevention of malignant diseases. This is very timely since based on the explosion of genomic research into cancer cells, discoveries of the random mutational events have in 2011 alone yielded 400 new markers and possibly 1,000 in 2012. The utility of safe and effective high avidity HuMab to these new markers would be a powerful application.
The business model may provide us with multiple potential funding events as our partners explore, discover, qualify, and ultimately commercialize a product using one of our antibodies. In general terms, the potential funding events for each single antibody targeted for a specific indication will follow a pattern similar to the following.
| Development Stage | Agreement Stage | Timing |
| Exploration | Technology Access | Lump-sum on signing, and annual renewal fees |
| Discovery | Technology Development | Lump-sum on identification of target antibody |
| Qualification | Developmental Collaboration | Payments at various stages throughout the FDA approval process |
| Commercialization | Royalty | Quarterly payments |
Technology access and collaboration agreements can generate $2 million to $5 million per antibody target and provide for enrollment of more than one target per Agreement, as well as royalties ranging from 5% to 15% of sales.
| 33 |
HuMabs FORECAST OF LICENSING REVENUE
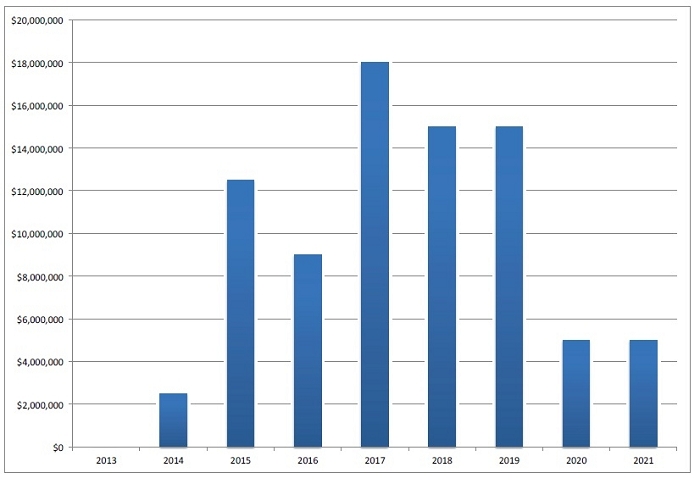
The landscape of the biopharmaceutical industry is undergoing transformation. The patents for some of the most successful commercial pharmaceutical products are expiring. Pharmaceutical companies are discovering that the scientific breakthroughs in the area of human genetics can easily render a successful product obsolete at a very early stage in its product life cycle. Over 400 known cancer genes were discovered in 2011 and thousands of complete cancer genome sequences will be available by 2018. So, we expect that the drug makers are searching for ways to respond to the need for developing new products quickly, while reducing development costs and lowering their exposure to the risk of unplanned obsolescence.
We believe that the HuMab derived from phage libraries of our immune B-cells can be used to diagnose, treat or prevent tumor progression. The safety of the fully HuMab has been declared by the European Medicines Agency which stated that as a tumor imaging agent, the treatments could not be given sooner than once a month. One treatment program that Vaccinogen holds several patents on and has investigated with HuMab is pretargeting. This approach consisted of injecting maximum tolerable dose of HuMab conjugated to a proprietary linker. Once the tumor is saturated, inject a radioisotope, toxin or chemotherapeutic dose, which will bind to the tumor bound antibody linker and spare the normal tissue. We did several patients with a variety of late stage carcinomas and the approach showed promise in preventing progression of tumors.
The pharmaceutical industry appears to have settled on monoclonal antibodies as one answer to these challenges. Human monoclonal antibodies offer the industry a safer and less expensive path to a more expeditious product development cycle, shortening the time to market for each new drug and allowing the pharmaceuticals to strength their product portfolios. We believe that our fully human immune repertoire is unique and extensive therefore attractive to strategic partners.
| 34 |
Prostate vs. Colon Cancer Vaccines
The global incidence of Stage I-IV colon cancer is about 900,000 cases per year, which is larger than the 780,000 cases of prostate cancer supporting market forecasts for Dendreon, Bavarian Nordic and other, less advanced, competitors. With respect to the Stage II (and potentially Stage III) colon cancer market addressed by OncoVAX®, Vaccinogen estimates the collective incidence is about 269,000 cases of Stage II and about 420,000 cases of Stage III colon cancers, a collective market that is only slightly smaller than the targeted patient populations for therapeutic prostate cancer vaccines.
There are important differences between hormone refractory prostate and Stage II colon cancers. All patients with hormone refractory prostate cancer are likely to die of their disease and have an urgent need for therapy. No current therapy, including the vaccines under development, cures these patients. Months of extended life represent the best outcome to date. The majority of patients with Stage II colon cancer are cured by surgery. Unfortunately the minority that is not surgically cured is significant (circa 30%) and the patients who relapse will likely die of their disease. Post-surgical Stage II colon cancer patients typically have a very low burden of tumor cells, giving the best opportunity for an immunologic cure as well as deferral of relapse. There is no way to tell in advance which Stage II patients are at most risk of relapse and so Vaccinogen’s market models assume that all Stage II patients are candidates for immunotherapy.
| 35 |
Colon Cancer Commercial Market Opportunity
COLON CANCER WORLD MARKET VALUE
$Amounts in Thousands
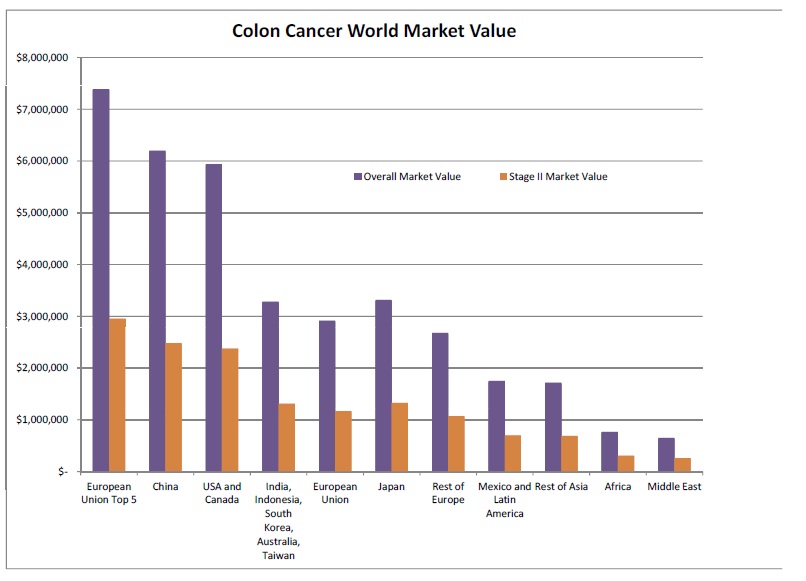
Colon cancer represents the third most common form of cancer in both the US and Europe. American Cancer Society statistics suggest there will be about 102,000 new cases of colon cancer diagnosed within the US. The European Cancer Observatory estimates that there were about 245,000 new cases of colon cancer across Europe.
Colon cancer is segmented at diagnosis into four disease stages. In Stage I, the cancer is confined to the mucosa and sub-mucosa of the colon. Stage II tumors have penetrated farther into the muscles around the colon, but the tumor has not visibly spread to lymph nodes of more distant sites. Stage III tumors have either advanced local spread or have spread to lymph nodes. Stage IV tumors have distant metastases at diagnosis.
| 36 |
Statistical information on the relative staging of colon cancer is collected and analyzed by a variety of government and industry groups. One of the most comprehensive cancer tumor registries for the US market is the Surveillance, Epidemiology and End Results Data (SEER) database. The Journal of the National Cancer Institute also publishes a variety of analytical studies largely relying on the SEER database for evaluation of cancer incidence, staging and survival.
Vaccinogen’s analysis of available colon cancer data suggests that the proportion of Stage I and IV patients has remained relatively constant over the past decade. However, there has been an on-going increase in the proportion of patients being diagnosed with earlier stage localized disease (Stage II), largely resulting from more consistent screening practices in the US and improving diagnostic technologies (fecal occult blood tests, sigmoidoscopy, colonoscopy, CEA screening). Available data suggests that Stage III is still a significant, but declining, percentage of reported colon cancers.
The table below highlights some the shifts in historical and anticipated incidence of Stage I-IV colon cancer in the US.
COLON CANCER INCIDENCE BY STAGE
| 1990s1 | 20042 | 2010E3 | 2020E3 | |||||||||||||
| Stage I | 15 | % | 8 | % | 10 | % | 10 | % | ||||||||
| Stage II | 36 | % | 39 | % | 40 | % | 46 | % | ||||||||
| Stage III | 28 | % | 39 | % | 37 | % | 31 | % | ||||||||
| Stage IV | 22 | % | 14 | % | 13 | % | 13 | % | ||||||||
| 1 Journal of the National Cancer Institute, Vol. 96, No. 19 | ||||||||||||||||
| 2 SEER Data | ||||||||||||||||
| 3 Company Estimates | ||||||||||||||||
Epidemiology to Market Characteristics
In Stage I colon cancer, the disease is treated completely through surgical intervention. There is an unmet medical need with respect to more effective Stage II colon cancer therapies, since recommended surgical intervention does not appear to fully eradicate micrometastases and leads to recurrence rates of approximately 30%. Chemotherapy is not recommended in Stage II colon cancer, with numerous studies suggesting no benefit. More serious Stage III colon cancer is treated with a combination of surgery and chemotherapy as a standard of care. Advanced Stage IV cases involve treatment with chemotherapeutics and/or surgical intervention.
Stage II Tumor Colon Cancer Market
While the Stage II colon cancer market is considerable, Vaccinogen’s OncoVAX® production methodology requires a total of about 3 grams of tumor to provide enough material for the production of a full complement of vaccine for a course of therapy. Based on prior clinical trial experience, Vaccinogen estimates that about 30% of surgically resected Stage II tumors will not meet minimum size requirements for vaccine production. Vaccinogen’s colon cancer market models and forecasts reduce the targeted Stage II total market to reflect this minimum tumor size constraint. Vaccinogen further adjusted down targeted Stage II colon cancer figures by an additional 10% to account for potential logistical problems inherent with its centralized manufacturing requirements, damages, etc.
A review of available data on estimated annual colon cancer incidence is produced by the American Cancer Society, European Cancer organizations, SEER and the Journal of the National Cancer Institute. This statistical work and related assumptions provides the foundation for determining the scope and magnitude of colon cancer across major global regions, and was further segmented to evaluate distinct Stage II colon cancer market opportunities for OncoVAX®.
| 37 |
United States Trend
The overall incidence of colorectal cancer has actually been declining modestly in the US, from a total of about 112,000 cases in 2007 to 102,000 cases in 2011. The reasons for this decline are not completely understood, but may include factors such as reduced smoking, higher aspirin use, changes in diet, etc. For purposes of forecasting, the Company has assumed a modest decline in the net number of new colon cancers over the next ten years. This reduction in overall colon cancer cases in the US is largely offset by a greater proportion of disease detected at Stage II.
More rigorous screening for colon cancer has led to improvement in earlier detection, giving a significant redistribution of the TNM (tumor, node, metastases) staging of the disease. Over the past decade, there has been a gradual shift toward earlier detection and therefore more localized disease as well as relative declines in advanced disease. Vaccinogen estimates that Stage IV disease has declined significantly over the past 20 years and now account for only about 13% of cases in the US. Stage III has also been gradually declining, with a corresponding increase in earlier disease increasing the proportion of patients with Stage II disease. Vaccinogen projects that the proportion of overall cancer assigned to Stage II should gradually increase to 46% of colon cancer patients in 2020 and beyond.
European Trends
The European market for colon cancer is considerably larger than the US and still growing, owing to sheer population demographics. Vaccinogen estimates that many of the more wealthy major European markets (France, Germany, Italy, Spain, UK) have characteristics closely mirroring US colon cancer staging, but with a slightly higher proportion of Stage III and Stage IV disease. This trend is even more pronounced toward Stages III and IV in other Eastern European geographies with less comprehensive screening. For Eastern Europe (and other less developed healthcare economies), Stage II is estimated to represent about 20%-25% of total colon cancer diagnosis. Similar to the shift in the US, Vaccinogen projects that Stage II colon cancer should gradually build to a level of about 45% of all cases in major EU economies and to about 35% of all cases in emerging Europe by 2020.
Rest of World (ROW)
Rates of colon cancer are actually rising in Japan, and changes in diet and higher rates of obesity are seen as key drivers for increasing colon cancer incidence through major emerging economies such as China. While sheer population demographics suggest a significant incidence of colon cancer in the region, the lack of rigorous screening protocols suggest that disease is usually detected at even later stages than in the US or Europe. Vaccinogen forecasts assume that only about 25%-26% of colon cancer is Stage II across the collective rest of world geographies. Vaccinogen forecasts that this proportion of Stage II cases will gradually increase, but only reach about the 35%-36% level by 2020.
Global Totals
These factors point to a considerable market for OncoVAX® in a growing and sizeable global market for Stage II colon cancer. In the US, Vaccinogen estimates that the overall number of Stage II colon cancer cases will range between 41,000 to 46,000 over the next decade. For the top five major European countries, Vaccinogen forecasts growth in the Stage II colon cancer market will increase from about 61,000 in 2010, to a level of 74,000 by 2020. Other rest of world geographies are collectively estimated to grow from a level of about 201,000 in 2010 to about 252,000 by 2020, albeit largely confined with less developed healthcare economies.
OncoVAX® Market Penetration Assumptions
Geographies with high insurance coverage as well as comprehensive government-controlled healthcare are major market opportunities for OncoVAX®. The enormous populations in numerous developing and emerging economies are also attractive, although the high price anticipated for OncoVAX® will be a barrier in many poorer economies.
The uptake in new cancer medications is a function of relative levels of effectiveness, physician education as well as reimbursement. Since OncoVAX® is expected to provide a significant improvement in survival benefit for Stage II cancer patients, Vaccinogen anticipates rapid and healthy market penetration within the colon cancer treatment community following approval. These market penetration trajectory assumptions are applied against the total addressable market forecasts for each year to derive a net OncoVAX® vaccine volume forecast.
The following chart highlights our forecast for relative share uptake in addressable Stage II colon cancer populations across the US, Europe Top 5, Other Geographies. The fundamental assumption in the analysis is strong uptake in traditional US oncology markets, somewhat lower net penetration across Europe and lower relative penetration across the collective Rest of World (ROW) economies.
| 38 |
Our market penetration assumptions anticipate relatively solid uptake in US Stage II colon cancer markets, with utilization rising steadily to over 50% of the adjusted Stage II markets by 2020. Vaccinogen is forecasting a similarly robust uptake, but lower net share uptake, for OncoVAX® in major European economies. Owing to more strict healthcare utilization pressures in Eastern Europe, the Company is anticipating that European uptake will run at a level of about one-half that of the US. This also accounts for divergent healthcare reimbursement trends across various Western and Eastern European geographies.
The analysis takes a somewhat conservative view of the opportunity for OncoVAX® in major Pacific Rim and other ROW geographies. Japan and the growing China market represent significant market opportunities, although much of Asia is still too poor to afford expensive new oncology products. While the sheer size of populations within the region is large, We anticipate that the market penetrations will run at a rate lower than those seen in the major markets. Market penetration rates are decidedly lower for the rest of the world, where the year six penetration is forecast at about 11%. The richest economy in the ROW bucket is Japan, where considerable unmet medical need and ability to pay is balanced against very slow regulatory and reimbursement processes.
OncoVAX® MARKET PENETRATION ASSUMPTIONS BY GEOGRAPHY
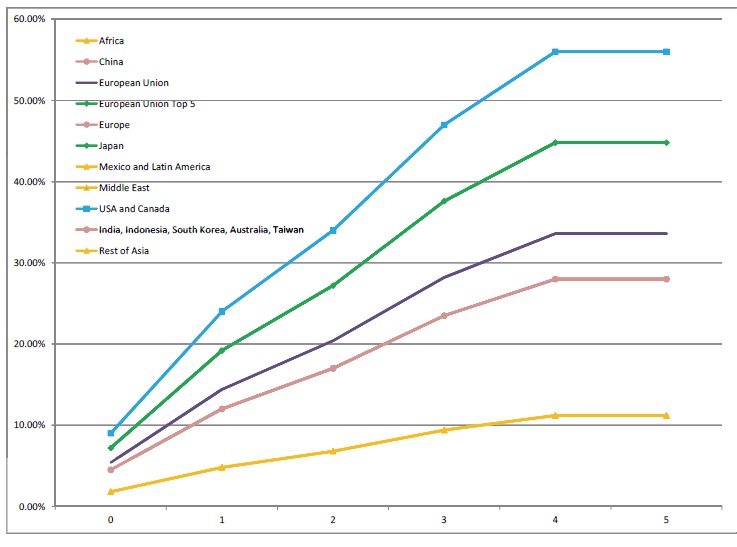
| 39 |
OncoVAX® Pricing
We anticipate pricing for OncoVAX® in the US at about $54,000. For benchmarking purposes, it is worth highlighting that the announced US price for Dendreon’s Provenge is $93,000. The model contemplates a $45,000 price point for OncoVAX® in the top five European geographies and $38,000/treatment for other European as well as Pacific Rim/ROW geographies. Naturally, the efficacy demonstrated in the contemplated Phase IIIb trial will be important for OncoVAX® pricing.
If successful, OncoVAX® could provide significant economic benefits to the healthcare system in addition to direct benefits to individual patients. The elimination or lengthy deferral of expensive chemotherapy, surgery and radiotherapy, and general end-of-life care should provide a significant benefit to the paying systems. Of course, extra years of life and economic productivity are to be expected based on the results of OncoVAX®’s previous Phase III trials. If as many as half the patients who would otherwise have suffered a near-term recurrence of their cancer are spared that fate by treatment with OncoVAX®, the savings to the healthcare system will be large.
A recently published January 2011 study on projections on the cost of cancer care in the US in the Journal of the National Cancer Institute suggest that the average male (>65 years old) colorectal patient has initial costs of about $52,000 for the first 12 months of care, annual continuing costs of about $4,500, and final year of life costs of about $86,000. The clear need to reduce colorectal cancer recurrence is further reinforced by a June 2007 study entitled “Projecting the National Economic Burden of Colorectal Cancer for the U.S. through the Year 2020”. This study estimates that by the year 2020, projected costs of colorectal cancer care in the U.S. for initial, continuing and last year of life phases will grow by about 50% to levels of $5.1 billion, $3.8 billion and $4.3 billion, respectively.
Stage III Colon Cancer Opportunity
The shift in colon cancer diagnosis to earlier Stage II disease is expected to lead to a gradual reduction in the proportion of patients diagnosed with later Stage III disease. We believe that OncoVAX® has significant potential in Stage III disease, and plans to conduct an inexpensive pilot study with a surrogate endpoint to test the feasibility of giving the vaccine in conjunction with chemotherapy. We do not plan to conduct registrational studies for Stage III disease as a use of proceeds from this offering. Larger studies in Stage III disease could begin after successful data from Stage II trials. Such a timeline could introduce a label indication for Stage III disease in a 2019 timeframe. Although Stage III is modestly declining, it still represents a potentially attractive commercial target. Our modeling suggests a US market for Stage III of about 35,000 cases in 2020. Europe Top 5 Stage III colon cancer collectively should be over 57,000 cases, while ROW geographies will account for over 241,000 cases in 2020. Our current revenue forecast contains no revenues from Stage III.
2013-2015: Costs to Pivotal Data & Approval
Manufacturing Costs
We have a manufacturing facility operating under cCMP standards and protocols in Emmen, The Netherlands that will process all tumors for the planned clinical trials. We expect cGMP recertification inspection in the 3rd quarter of 2013 with re-certified cGMP status in the 3rd/4th quarter of 2013. The facility is expected to cost $2.1 million to operate in 2013, rising to $4.9 and $7.4 million for 2014 and 2015. The Emmen facility’s six employees are anticipated to increase to eighteen by year-end 2014. Each tumor has a projected variable cost to process of approximately $3,500. The company has projected the hiring of a US based VP of Manufacturing in 2013.
Research and Development
We anticipate spending $39 million in R&D in the period 2013 through 2015. Third party costs associated with the 550 patient pivotal trial are estimated at $45,000 per patient, and total $23.8 million over 2013-2015. The third party trial costs for the 30 patient, Stage III, Phase I/II trial are estimated at approximately $31,000 per patient, and total $1.0 million over the clinical trial period. We estimate spending $5.0 million on the HuMabs project between 2013 and 2015.
To support the R&D effort we anticipate increasing R&D headcount from one to thirteen by year-end 2014. Additional hires are expected to include a Vice Presidents of Regulatory and Clinical Affairs, a Director of Regulatory, Director of HuMabs and additional clinical support staff. Costs for these staff and associated expenses are projected to be approximately $2.6 million a year.
| 40 |
Sales & Marketing
We have a Business Development Managing Director based in The Netherlands. We plan to hire a Director of Marketing and Director of BD - HuMabs and a US Vice President of Business Development in 2014. The company projects two additional hires in 2015. The Sales and Marketing expense reflect these personnel costs.
General & Administrative
We currently have three employees allocated to G&A with plans to add three additional employees by year end 2013 and eight additional employees by year end 2014 and six more employees in 2015, bringing the total to twenty by the end of 2015.
Preparatory Equipment
In 2013, we anticipate spending on average $87,500 at each of approximately 47 clinical trial sites on preparatory equipment (including nitrogen freezers, glove boxes, centrifuges, etc.).
Manufacturing Build Out
We plan to explore the feasibility and cost of upgrading its Emmen facility to qualify as a commercial vaccine-manufacturing site for European based treatments and sales. Emmen currently has the projected capacity to manufacture 2,000 vaccines, the equivalent of over $100 million in annual sales at current pricing. The financial forecast also includes $23 million by 2016 towards the cost of building an additional manufacturing facility as our primary manufacturing facility servicing US and international treatments and sales.
2015 – 2016: Costs to Commercialize
Revenues
OncoVAX® sales are projected to start in Q3 2017 in the US and Q4 2018 in Europe. In 2017, Vaccinogen anticipates selling approximately 188 vaccines in the US at approximately $54,000 per vaccine treatment.
We anticipate a rest of world partnership in which its partner both manufactures and distributes the vaccine and pays upfront fees, milestones and a royalty modeled at 20% to Vaccinogen. The model estimates $217 million in OncoVAX® upfront milestones for one region. There is upside potential in the licensing of additional regions, indications and partners sharing in the development costs of the OncoVAX® Project.
We estimate12 HuMabs licensing deals from 2013 through 2017. The projected revenue for this period is approximately $42.0 Million.
Royalties Owed
We have agreed to pay Organon (now part of Merck) a royalty of 10% of the net sales of OncoVAX® (and all other TICE BCG related products) until payment of $3.5 million (and accrued interest) and 3% for five years thereafter.
We are also required to pay Intracel a royalty in conjunction with the Asset Transfer Agreement tiered between 3% and 5% of sales.
Supplies
Organon Teknika produces a key product (TICE BCG) used in processing our OncoVAX® vaccines. We previously acquired from Organon Teknika (now part of Merck) all TICE BCG necessary for conducting our Phase III trials. We believe we will be able to procure addition quantities of TICE BCG from Organon as necessary in the future.
We believe that we have exclusive use of TICE BCG with autologous cancer cells through our current patent portfolio.
| 41 |
Manufacturing Costs
Our forecast assumes spending $10.3 and $23.51 million on Manufacturing in 2016 and 2017. Within the 2016 budget is a $15 million expenses in preparation of the commercialization of OncoVAX® in Q3 2017.
Research and Development
Our forecast assumes spending $8.7 and $9.3 million on R&D in 2016 and 2017.
Sales and Marketing
We anticipate spending approximately $10.7 and $26.7 million on Sales and Marketing in 2016 and 2017. The increase in 2017 is to support the pre launch and launch commercialization of OncoVAX®.
General & Administrative
We anticipate General and Administrative spend of $8.2 and $14.0 million in 2016 and 2017.
Intellectual Property
Intellectual property protection is important to our ability to successfully commercialize its innovative technology. We have broad patents covering the OncoVAX® technology in the U.S. and eight other countries, Australia, Canada, Switzerland, Germany, France, Great Britain, Ireland and Italy. These patents and applications provide broad coverage for the production of autologous cancer vaccines. The key protection of OncoVAX®, in addition to considerable expertise protected as trade secrets, is the broad, issued patent protection around the production of autologous, sterile, metabolically active cancer vaccines developed by us. This patent, entitled “Sterile Immunogenic Non-Tumorigenic Tumor Cell Composition and Methods” was issued in 2009 and expires no sooner than 2025. We believe that sterility will be required for any product to reach the US market and likely in any other market with an approved sterile vaccine like the one we have developed. This could result in a regulatory barrier to entry to competitors. Our intellectual property is pledged as collateral under certain financing arrangements. See “Description of Securities—The Abell Foundation Financings, —Agreements with Organon Teknika Corporation.”
We hold 1 U.S. patent to related technologies and including the sterility patent referred to above, 2 U.S. patents total.
Outside of the United States, we have, in certain territories, corresponding issued patents related to OncoVAX®. Patent expiration dates may be subject to patent term extension depending on certain factors. In addition, following expiration of a basic product patent or loss of patent protection resulting from a legal challenge, it may be possible to continue to obtain commercial benefits from other characteristics such as clinical trial data, product manufacturing trade secrets, uses for products, and special formulations of the product or delivery mechanisms.
We intend to continue using our scientific experience to pursue and patent new developments to enhance our position in the cancer field. Patents, if issued, may be challenged, invalidated, declared unenforceable, circumvented or may not cover all applications we desire. Thus, any patent that we own or license from third parties may not provide adequate protection against competitors. Our pending patent applications, those we may file in the future, or those we may license from third parties may not result in issued patents. Also, patents may not provide us with adequate proprietary protection or advantages against competitors with, or who could develop, similar or competing technologies, or who could design around our patents. In addition, future legislation may impact our competitive position in the event brand-name and follow-on biologics do not receive adequate patent protection. From time to time, we have received invitations to license third-party patents.
We also rely upon unpatented proprietary know-how and continuing technological innovation and other trade secrets to develop and maintain our competitive position, in part by using confidentiality agreements. Our policy is to require our officers, employees, consultants, contractors, manufacturers, outside scientific collaborators and sponsored researchers and other advisors to execute confidentiality agreements. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us be kept confidential and not disclosed to third parties except in specific limited circumstances. We also require signed confidentiality agreements from companies that receive our confidential data. For employees, consultants and contractors, we require agreements providing that all inventions conceived while rendering services to us shall be assigned to us as our exclusive property. We hold considerable proprietary expertise related to the OncoVAX® technology, including the production of autologous cancer vaccines.
| 42 |
We have brand names for our OncoVAX® products and related technologies, and anticipate filing 5 trademark applications for these and related marks.
Competition
The biotechnology and biopharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. Pharmaceutical and biotechnology companies, academic institutions and other research organizations are actively engaged in the discovery, research and development of products designed to address prostate cancer and other indications. There are products currently under development by other companies and organizations that could compete with OncoVAX® or other products that we are developing.
Our competitors include major pharmaceutical companies. These companies may have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing. In addition, smaller competitors may collaborate with these large established companies to obtain access to their resources.
Competition among products approved for sale will be based upon, among other things, efficacy, reliability, product safety, price-value analysis, and patent position. Our competitiveness will also depend on our ability to advance our product candidates, license additional technology, maintain a proprietary position in our technologies and products, obtain required government and other approvals on a timely basis, attract and retain key personnel and enter into corporate relationships that enable us and our collaborators to develop effective products that can be manufactured cost-effectively and marketed successfully.
Regulatory
General
Government authorities in the United States and other countries extensively regulate, among other things, the pre-clinical and clinical testing, manufacturing, labeling, storage, record-keeping, advertising, promotion, export, marketing and distribution of biologic products. In the United States, the FDA subjects pharmaceutical and biologic products to rigorous review under the Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal statutes and regulations.
FDA Approval Process
To obtain approval of our product candidates from the FDA, we must, among other requirements, submit data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product candidate. In most cases, this entails extensive laboratory tests and pre-clinical and clinical trials. The collection of these data, as well as the preparation of applications for review by the FDA, are costly in time and effort, and may require significant capital investment. We may encounter significant difficulties or costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing any products we may develop.
A company typically conducts human clinical trials in three sequential phases, but the phases may overlap. Phase 1 trials consist of testing of the product in a small number of patients or healthy volunteers, primarily for safety at one or more doses. Phase 1 trials in cancer are often conducted with patients who are not healthy and who have end-stage or metastatic cancer. Phase 2 trials, in addition to safety, evaluate the efficacy of the product in a patient population somewhat larger than Phase 1 trials. Phase 3 trials typically involve additional testing for safety and clinical efficacy in an expanded population at geographically dispersed test sites. Prior to commencement of each clinical trial, a company must submit to the FDA a clinical plan, or “protocol,” which must also be approved by the Institutional Review Boards at the institutions participating in the trials. The trials must be conducted in accordance with the FDA’s good clinical practices. The FDA may order the temporary or permanent discontinuation of a clinical trial at any time.
To obtain marketing authorization, a company must submit to the FDA the results of the pre-clinical and clinical testing, together with, and among other things, detailed information on the manufacture and composition of the product, in the form of a new drug application or, in the case of a biologic such as OncoVAX®, a biologics license application.
| 43 |
We are also subject to a variety of regulations governing clinical trials and sales of our products outside the United States. Whether or not FDA approval has been obtained, approval of conduct of a clinical trial or authorization of a product by the comparable regulatory authorities of foreign countries and regions must be obtained prior to the commencement of marketing the product in those countries. The approval process varies from one regulatory authority to another and the time may be longer or shorter than that required for FDA approval. In the E.U., Canada and Australia, regulatory requirements and approval processes are similar, in principle, to those in the United States.
Fast Track Designation/Priority Review
Congress enacted the Food and Drug Administration Modernization Act of 1997 (the “Modernization Act”) in part to ensure the availability of safe and effective drugs, biologics and medical devices by expediting the development and review for certain new products. The Modernization Act establishes a statutory program for the review of Fast Track products, including biologics. A Fast Track product is defined as a new drug or biologic intended for the treatment of a serious or life-threatening condition that demonstrates the potential to address unmet medical needs for this condition. Under the Fast Track program, the sponsor of a new drug or biologic may request that the FDA designate the drug or biologic as a Fast Track product at any time during the development of the product, prior to a new drug application submission.
Post-Marketing Obligations
The Food and Drug Administration Amendments Act of 2007 expanded FDA authority over drug products after approval. All approved drug products are subject to continuing regulation by the FDA, including record-keeping requirements, reporting of adverse experiences with the product, sampling and distribution requirements, notifying the FDA and gaining its approval of certain manufacturing or labeling changes, complying with certain electronic records and signature requirements, submitting periodic reports to the FDA, maintaining and providing updated safety and efficacy information to the FDA, and complying with FDA promotion and advertising requirements. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action, criminal prosecution, or civil penalties.
The FDA may require post-marketing studies or clinical trials to develop additional information regarding the safety of a product. These studies or trials may involve continued testing of a product and development of data, including clinical data, about the product’s effects in various populations and any side effects associated with long-term use. The FDA may require post-marketing studies or trials to investigate known serious risks or signals of serious risks or identify unexpected serious risks and may require periodic status reports if new safety information develops. Failure to conduct these studies in a timely manner may result in substantial civil fines.
Drug and biologics manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and to list their products with the FDA. The FDA periodically inspects manufacturing facilities in the United States and abroad in order to assure compliance with the applicable cGMP regulations and other requirements. Facilities also are subject to inspections by other federal, foreign, state or local agencies. In complying with the cGMP regulations, manufacturers must continue to assure that the product meets applicable specifications, regulations and other post-marketing requirements. We must ensure that any third-party manufacturers continue to ensure full compliance with all applicable regulations and requirements. Failure to comply with these requirements subjects the manufacturer to possible legal or regulatory action, such as suspension of manufacturing or recall or seizure of product.
Also, newly discovered or developed safety or efficacy data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, additional pre-clinical or clinical studies, or even in some instances, revocation or withdrawal of the approval. Violations of regulatory requirements at any stage, including after approval, may result in various adverse consequences, including the FDA’s withdrawal of an approved product from the market, other voluntary or FDA-initiated action that could delay or restrict further marketing, and the imposition of civil fines and criminal penalties against the manufacturer and Biologics License Applications (“BLA”) holder. In addition, later discovery of previously unknown problems may result in restrictions on the product, manufacturer or BLA holder, including withdrawal of the product from the market. Furthermore, new government requirements may be established that could delay or prevent regulatory approval of our products under development, or affect the conditions under which approved products are marketed.
| 44 |
Federal Anti-Kickback, False Claims Laws & The Federal Physician Payment Sunshine Act
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws are relevant to certain marketing practices in the pharmaceutical industry. These laws include anti-kickback statutes, false claims statutes, and the federal Physician Payment Sunshine Act. The federal healthcare program anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, lease, order or recommendation of, any good or service for which payment may be made under federal health care programs such as the Medicare and Medicaid programs. For example, this statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the federal anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. There are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, however, the exceptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exception or safe harbor may be subject to scrutiny.
Federal false claims laws prohibit, among other things, any person from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment, or knowingly making, or causing to be made, a false record or statement material to a false or fraudulent claim. For example, several pharmaceutical and other healthcare companies have faced enforcement actions under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, anti-kickback statute violations and certain marketing practices, including off-label promotion, may also implicate false claims laws. Federal false claims laws violations may result in imprisonment, criminal fines, civil monetary damages and penalties and exclusion from participation in federal healthcare programs. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs. A number of states have anti-kickback laws that apply regardless of the payor.
In addition, the federal Physician Payment Sunshine Act, when implemented will require the reporting by drug manufacturers of “payments or transfers of value” made or distributed to physicians and teaching hospitals, with limited exceptions. Failure to comply with the reporting obligations may result in civil monetary penalties.
State Laws
Marketing Restrictions and Disclosure Requirements. A number of states, such as Minnesota, Massachusetts and Vermont, have requirements that restrict pharmaceutical marketing activities that go beyond commitments made related to adhering to the Pharmaceutical Research and Manufacturers of America Code for Interactions with Healthcare Professionals. These state requirements limit the types of interactions we may have with healthcare providers licensed in these jurisdictions. In addition, a number of states have laws that require pharmaceutical companies to track and report payments, gifts and other benefits provided to physicians and other health care professionals and entities. Still other state laws mandate implementation of specific compliance policies to regulate interactions with health care professionals.
Healthcare Reform. Certain states, such as Massachusetts, are pursuing their own programs for health reform. These programs may include cost containment measures that could affect state healthcare benefits, particularly for higher priced drugs. Under the federal Patient Protection and Affordable Care Act, states will have authority to define packages of “essential health benefits” that health plans in the individual and small group markets must offer beginning in 2014. The definition of these packages could affect coverage of our products by those plans.
Sale of Pharmaceutical Products. In addition, in the United States, many states have enacted their own laws and statutes applicable to the sale of pharmaceutical products within the state, with which we must comply. We are also subject to certain state privacy and data protection laws and regulations.
Data Privacy
Numerous federal and state laws, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use and disclosure of personal information. Other countries also have, or are developing, laws governing the collection, use and transmission of personal information. In addition, most healthcare providers who prescribe our product and from whom we obtain patient health information are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”). We are not a HIPAA covered entity and therefore, these privacy and security requirements do not apply to us. However, we could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a covered entity in a manner that is not authorized or permitted by HIPAA or for aiding and abetting the violation of HIPAA. We are unable to predict whether our actions could be subject to prosecution in the event of an impermissible disclosure of health information to us. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues with the potential to affect our business, including recently enacted laws in a majority of states requiring security breach notification. These laws could create liability for us or increase our cost of doing business.
| 45 |
Price Controls
In many of the markets in which we may do business in the future, the prices of pharmaceutical products are subject to direct price controls (by law) and to drug reimbursement programs with varying price control mechanisms. In the United States, the Medicare program is administered by CMS. Coverage and reimbursement for products and services under Medicare are determined in accordance with the Social Security Act and pursuant to regulations promulgated by CMS as well as the agency’s subregulatory coverage and reimbursement determinations. The methodology under which CMS makes coverage and reimbursement determinations is subject to change, particularly because of budgetary pressures facing the Medicare program. For example, the Medicare Modernization Act of 2003 made changes in reimbursement methodology that reduced the Medicare reimbursement rates for many drugs, including oncology therapeutics. In the past year, Congress has considered additional reductions in Medicare reimbursement for drugs as part of legislation to reduce the budget deficit. Similar legislation could be enacted in the future. The Medicare regulations and interpretive determinations that determine how drugs and services are covered and reimbursed also are subject to change.
We intend to make OncoVAX® available to patients that are eligible for Medicaid benefits. A condition of federal funds being made available to cover our products under Medicaid and Medicare Part B will be Vaccinogen’s participation in the Medicaid drug rebate program, established by the Omnibus Budget Reconciliation Act of 1990, Pub. L. 101-508, and as amended by subsequent legislation, including the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010 and subsequent legislation (collectively, “PPACA” ).
The availability of federal funds under Medicaid and Medicare Part B to pay for OncoVAX® and any other products that are approved for marketing also is conditioned on our participation in the Public Health Service 340B drug pricing program. The 340B drug pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. These covered entities include hospitals that serve a disproportionate share of poor Medicare beneficiaries, as well as a variety of community health clinics and other recipients of health services grant funding. PPACA expanded the 340B program to include certain free standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by the Act. The 340B ceiling price for a drug is calculated using a statutory formula that is based on the AMP and Medicaid rebate amount for the drug. Any revisions to previously reported Medicaid pricing data also may require revisions to the 340B ceiling prices that were based on those data and could require the issuance of refunds.
European Regulatory Authorities
In the European Union, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of such products to consumers. The approach taken varies from member state to member state. Some jurisdictions operate positive and/or negative list systems under which products may be marketed only once a reimbursement price has been agreed. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products, as exemplified by the role of the National Institute for Health and Clinical Excellence in the United Kingdom, which evaluates the data supporting new medicines and passes reimbursement recommendations to the government. In addition, in some countries cross-border imports from low-priced markets (parallel imports) exert commercial pressure on pricing within a country.
| 46 |
Environmental and Safety Laws
We are subject to a variety of federal, state and local regulations relating to the use, handling, storage and disposal of hazardous materials, including chemicals and radioactive and biological materials. Our operations produce such hazardous waste products. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by federal, state and local regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. We generally contract with third parties for the disposal of such substances, and store our low level radioactive waste at our facilities until the materials are no longer considered radioactive. We are also subject to various laws and regulations governing laboratory practices and the experimental use of animals.
We are also subject to regulation by the Occupational Safety and Health Administration (“OSHA”), and the Environmental Protection Agency (the “EPA”), and to regulation under the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other regulatory statutes, and may in the future be subject to other federal, state or local regulations. OSHA and/or the EPA may promulgate regulations that may affect our research and development programs.
Taxes
We have not filed any U.S. tax returns since 2007. We anticipate filing all outstanding U.S. tax returns in fiscal 2013.
There is an issue with penalties regarding the U.S. information reporting Form 5471 for both the Netherlands and Switzerland. We failed to file all applicable foreign information returns for the 2007-2011 annual periods. The IRS has been imposing penalties on a regular basis for the failure of U.S. taxpayers to file Form 5471. The penalty is $10,000 per entity for each year. A U.S. income tax return is not considered complete without the filing of the report. Accordingly, management has concluded that a $20,000 income tax (ASC 740-10) penalty has been incurred for 2011. None has been incurred for 2012 because a corporate tax return extension has been filed. The details of these amounts are as follow: The Netherlands was formed in 2007 and Switzerland was formed in 2008 and dissolved in 2011. Therefore, $70,000, $90,000 and $90,000 are the penalty liabilities as of December 31, 2010, 2011 and 2012. Current tax expense was $20,000 for the year ended December 31, 2011 and none for 2012.
Employees
As of December 31, 2012, we had 10 full time employees. None of our employees are subject to a collective bargaining agreement or represented by a labor or trade union, and we believe that our relations with our employees are good. In addition, we also have 8 technical and administrative consultants that we may hire as additional full time employees as our financial condition improves.
DESCRIPTION OF PROPERTY
Our executive offices are located at 5300 Westview Drive, Suite 406, Frederick, Maryland 21703 and comprise approximately 4,064 square feet. We lease our executive offices at a rate of $5,089 per month, which lease term ends on October 31, 2013, with an optional additional six month extension period until April 30, 2014.
We also maintain offices, laboratory and scientific manufacturing space at Marco Polostraat 27. 7825 VM, Emmen, The Netherlands which is 9,600 square feet. We lease this space at a price of $5,804 per month, which lease term ends on November 30, 2013. We have the right to extend the term for successive one year periods.
LEGAL PROCEEDINGS
We are currently in a dispute with a vendor regarding amounts owed for services performed. A demand for payment under a written agreement has been made in the amount of approximately $150,000. Management believes the vendor did not perform under the terms of the contract and contends that no amounts are due. Settlement discussions between the parties are continuing. We have reserved $75,000 under our financial statements for resolution of this matter.
We are party to claims and litigation that arise in the normal course of business. Management believes that the ultimate outcome of these claims and litigation will not have a material impact on our financial position or results of operations.
| 47 |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following tables sets forth information as of June 26, 2013 as to each person or group who is known to us to be the beneficial owner of more than 5% of our outstanding voting securities and as to the security and percentage ownership of each of our executive officers and directors and of all of our officers and directors as a group. As of June 26, 2013 we had 31,227,756 shares of common stock outstanding.
Beneficial ownership is determined under the rules of the Securities and Exchange Commission and generally includes voting or investment power over securities. Except in cases where community property laws apply or as indicated in the footnotes to this table, we believe that each stockholder identified in the table possesses sole voting and investment power over all shares of common stock shown as beneficially owned by the stockholder.
Shares of common stock subject to options or warrants that are currently exercisable or exercisable within 60 days of the date of this Offering Circular are considered outstanding and beneficially owned by the person holding the options for the purpose of computing the percentage ownership of that person but are not treated as outstanding for the purpose of computing the percentage ownership of any other person.
| Name And Address (1) | Number Of Shares Beneficially Owned | Percentage Owned | ||||||
| 5% Stockholders: | ||||||||
| Intracel Holdings Corporation (2) | 13,324,864 | 42.67 | % | |||||
| Executive Officers: | ||||||||
| Michael G. Hanna, Jr. Ph.D | 3,949,289 | (3) | 12.57 | % | ||||
| Andrew Tussing | 421,002 | (4) | 1.35 | % | ||||
Directors: | ||||||||
| John Nicolis | 3,947,160 | (5) | 12.16 | % | ||||
| Daniel Fitzgerald | 606,212 | (6) | 1.94 | % | ||||
| Alan Cohen | 2,511,362 | (7) | 8.04 | % | ||||
| Daniel Kane | 2,511,362 | (7) | 8.04 | % | ||||
| All directors and officers as a group (6 persons) | 13,946,387 | 42.64 | % | |||||
| (1) | Unless otherwise indicated the address is c/o Vaccinogen, Inc., 5300 Westview Drive, Suite 406, Frederick, MD 21701. |
| (2) | The address is 550 Highland Street, Suite 417, Frederick, MD 21701. |
| (3) | Includes 200,000 shares under presently exercisable warrants and 23,188 shares in respect of his indirect ownership interest in Intracel Holdings Corporation. |
| (4) | Includes 20,575 shares under presently exercisable warrants. |
| (5) | Includes 1,194,748 shares under presently exercisable warrants, 179,216 shares held under trusts under which Mr. Nicolis is the trustee and 42,336 shares under restricted stock grants. |
| (6) | Includes 9,090 shares under restricted stock grants and 39,142 shares in respect of his indirect ownership interest in Intracel Holdings Corporation. |
| (7) | Includes 7,272 shares under restricted stock grants and 1,729,581 shares in respect of his indirect ownership interest of Intracel Holdings Corporation. |
| 48 |
DIRECTORS AND EXECUTIVE OFFICERS
Set forth below is certain information concerning our directors and executive officers:
| Name | Age | Position | ||
| Michael G. Hanna, Jr., Ph.D. | 76 | Chairman and Chief Executive Officer | ||
| Andrew Tussing | 48 | President, Chief Operating Officer and acting Chief Financial Officer | ||
| John Nicolis | 60 | Director | ||
| Daniel Fitzgerald | 60 | Director | ||
| Alan Cohen | 76 | Director | ||
| Daniel Kane | 55 | Director |
Michael G. Hanna, Jr., Ph.D., is our Chief Executive Officer and Chairman of our Board of Directors. Dr. Hanna has been with Vaccinogen since its founding in 2007 and developer of OncoVAX®, Vaccinogen’s lead project. Prior to Vaccinogen, Dr. Hanna served as Chairman (Emeritus) and Chief Scientific Officer of Intracel Resources, an integrated biopharmaceutical company that developed cancer vaccines and immunotherapeutic and diagnostic products for both cancers and infectious diseases. Dr. Hanna also served as President and Chief Executive Officer of PerImmune, Inc. before it and Intracel Corp. merged in 1998. From 1985 to 1994, he was the Chief Operating Officer of Organon Teknika Biotechnology Research Institute and Senior Vice President of Organon Teknika Corporation, a subsidiary of Akzo Nobel, N.V., The Netherlands. Prior to that, he was Director of the National Cancer Institute, Frederick Cancer Research Center.
Dr. Hanna received a doctoral degree in experimental pathology and immunology from the University of Tennessee.
Andrew Tussing is our President, Chief Operating Officer and acting Chief Financial Officer. Mr. Tussing has served as an officer of Vaccinogen since its founding in 2007 (of which he is a co-founder). Previously, Mr. Tussing was active in investment banking activities at 1st BridgeHouse Securities and as Managing Director of Growth5 – a venture capital marketing strategy firm. Prior to that, Mr. Tussing was a co-founder of Milestone International Asset Management, which developed hedge-fund-related solutions for financial advisors. He also served in senior roles at Pershing, a division of the Bank of New York, and at Deutsche Bank’s Alex. Brown division where he was Director of Strategic Development, Marketing & Sales for its Correspondent Services business.Mr. Tussing has a BS in Business Administration from the University of Baltimore.
John Nicolis, Co-Chairman, has served as one of our directors since inception in 2007. Mr.Nicolis is Managing Director of Optimal Fund Management, a Sydney, Australia-based investment management company with over US $2.4 Billion in assets; he specializes in Asia Pacific equities.
Mr. Nicolis has almost 20 years' experience in equity sales with Salomon Brothers (Citigroup) and has spent a considerable portion of this time working in Tokyo. More recently, he was based in Melbourne as Head of Equities for Japan, Asia and Australia with overall responsibility for marketing of the company's brokering services as well as the company’s operational activities.
Mr. Nicolis holds a Bachelor of Arts majoring in Japanese and Chinese.
Daniel Fitzgerald has served as a director since 2009. Mr. Fitzgerald worked in fixed income on Wall Street for over thirty years. He is a founding partner of Pinewood Capital Partners in Greenwich, CT, specializing in high yield debt; a market neutral fund. Prior to that, he co-created the Gleacher Nat West High Yield department, specializing in underwriting research and trading in high yield securities. Before that, Mr. Fitzgerald was Managing Director in high yield for 15 years at Donaldson, Lufkin and Jenrette. He has a BS in accounting from Manhattan College. Mr. Fitzgerald is a former member of the Pathmark Board of Directors (2000-2007) and was instrumental in the sale of Pathmark to GAP (Great Atlantic and Pacific) for US$1.35 billion dollars. Mr. Fitzgerald currently manages a family office.
Alan Cohen has served as a director since January 2011.Mr. Cohen, Chairman of Abacus Advisors, has more than 25 years of experience working with businesses in all aspects of management and operations. He has been an active participant in seminars on turnaround management, has lectured extensively on restructuring and asset-based lending, and has served as a consultant and advisor to numerous Fortune 500 companies and many of the country’s leading banks and financial institutions.
| 49 |
Daniel Kane has served as a director since January 2011. Mr. Kane is Managing Member of Tiger Capital Group, LLC, and The Nassi Group, LLC. He has over twenty years’ experience managing appraisals, real estate, intellectual property, various acquisitions and liquidations. Prior to joining these companies, Mr. Kane practiced for over ten years as a CPA.
Mr. Kane is currently on the board of Intracel Holdings Corp. and a former board member of Official Pillowtex, LLC (which was sold to Iconix Brand Group), and a former board member of Unwired Technology (which was sold to American Capital, Ltd.).
Mr. Kane received his MS degree from the Stern School of Business at New York University.
Significant Employees and Advisors
Debra R. Hoopes, CPA, MBA, Chief Financial Advisor.
Debra R. Hoopes is our Chief Financial Adviser, a position she has held since August, 2012. Ms. Hoopes has over 20 years of corporate finance and management experience across various industries and includes both private and public companies in the CFO role. Ms. Hoopes has held CFO and COO positions for various companies through her consulting company Hoopes Management and Advisory Services dba CFO Link, including RO12, Inc., Hatteras Networks, OpenQ and Certification Partners. In addition, Ms. Hoopes served as CFO for Catuity, Inc. and Intersections, Inc., both Nasdaq listed companies. Ms. Hoopes earned her B.S. in accounting at Virginia Tech, her MBA at George Washington University and obtained her CPA in the Commonwealth of Virginia in 1987, license 32837.
John Powers, Ph.D., Logistics and Manufacturing Expert/Advisor
Dr. Powers has thirty-two years of experience in the life sciences, and spent twenty-three years successfully starting, growing then selling three small biotech companies. Dr. Powers founded and ownsKsD Scientific, a biotechnology-consulting firm and is currently helping start Baltimore BioWorks (Baltimore, MD) a minority-owned vocational biotech training company funded by The Abell Foundation. Dr. Powers was involved in Phase III clinical trial work with Hoffman LaRoche; then worked with the Centers for Disease Control (Atlanta, GA) after which he went to the National Institutes of Health (Bethesda, MD). Dr. Powers is the author of numerous papers, abstracts and manuals, holds four biotech product patents and has filed and received over a dozen FDA 510(k) approvals.
Over the past few years The Abell Foundation has engaged KsD Scientific to investigate several companies to analyze investment opportunities, perform scientific and business due diligence which include; cancer vaccines, nanoparticle fluorophores and bacteriophage applications.
John Powers, Ph.D., MBA did his undergraduate and graduate work at the University of Maryland (College Park, MD) and the Maryland Medical School (Baltimore, MD).
Sanda Milder, BSc, Site Director
Ms. Milder is the Site Director as well as the Operations Manager. At the Emmen, The Netherlands, manufacturing facility she is responsible for the overall plant and oversees and coordinates the manufacturing functions to process the vaccines as well as being responsible for all administrative functions. Ms. Milder maintains the Standard Operating Procedures for the manufacture of vaccines and interfaces with other plant management to ensure a safe, clean manufacturing environment and product.
Scientific, business and personal key elements:
| · | GLP and GMP compliance |
| · | Regulatory agencies interactions |
| · | Nontraditional manager, inventive and optimistic |
| · | Able to work accurately and pay attention to detail |
| 50 |
| · | Excellent communication skills, multilingual |
Ms. Milder has a Bachelor of Science in Biochemistry, Cum Laude; and several vocational certifications in quality control and quality assurance.
Herbert C. Hoover, Jr., MD, Medical Advisor
Dr. Hoover serves as a Consulting Medical Advisor for Vaccinogen and has been the one clinician involved in all of the clinical trials of OncoVAX®. Dr. Hoover initiated the original pilot trial of active specific immunotherapy in patients with colon cancer at Johns Hopkins Hospital in 1981. He served as a medical advisory role with Organon Teknika/Biotechnology Research, PerImmune and Intracel.
Dr. Hoover has had numerous honors and awards throughout his career including the Charles B. Thornton Advanced Technology Achievement Award (for the development of tumor-specific human monoclonal antbodies) as well as the Best Doctors in America recognition. He shares multiple patents for the development of monoclonal antibodies and active-specific immunotherapy and has published nearly 100 scientific articles.
Dr. Hoover received his MD from the University of Kansas School of Medicine in 1970. His surgical residency was served at the Massachusetts General Hospital and Harvard Medical School from 1970 to 1977 with a Chief Residency in 1978. He was a Clinical Associate in the Surgery Branch of the NCI, NIH in Bethesda from 1972 to 1974.
Director Independence and Qualifications
Our Board of Directors has determined that Messrs. Nicolas, Fitzgerald, Cohen and Kane are “independent” as defined under the standards set forth in Section 121A of the American Stock Exchange Company Guide. In making this determination, the Board of Directors considered all transactions set forth under “Certain Relationships and Related Transactions” below.
We considered Dr. Hanna’s prior experience in the biotechnology industry, including his development of the OncoVAX® vaccine as well as his position as a director of the National Cancer Institute were important factors in concluding that he was qualified to serve as one of our directors. Regarding Mr. Nicolis, we considered his investment and financial experience as important factors in concluding that he was qualified to serve as one of our directors.. Regarding Mr. Fitzgerald, we considered his financial experience as well as his role as a former director of Pathmark as important factors in concluding that he was qualified to serve on our board of directors. Regarding Mr. Cohen, we considered his prior experience working with portfolio companies and his role as consultant to Fortune 500 companies as important factors in concluding that he was qualified to serve on our board of directors. Regarding Mr. Kane, we considered his prior experience as a CPA as well as his prior experience serving as a director for other companies as important factors in concluding that he was qualified to serve as one of our directors.
Involvement in Certain Legal Proceedings
None of our directors or executive officers has, during the past five years:
| · | been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offences); |
| · | been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities, futures, commodities or banking activities or |
| · | been found by a court of competent jurisdiction (in a civil action), the Securities and Exchange Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated. |
| 51 |
Communications with the Board of Directors
Stockholders can send communications to the Board of Directors by sending a certified or registered letter to the Chairman of the Board, care of the Secretary, at our main business address set forth above. Communications that are threatening, illegal, or similarly inappropriate, and advertisements, solicitations for periodical or other subscriptions, and other similar communications will generally not be forwarded to the Chairman.
The Medical Advisory Board
Our Medical Advisory Board is comprised of internationally recognized individuals in the fields of oncology and cancer surgery. Our Medical Advisory Board advises and assists our management by reviewing and evaluating our research strategy and research, development and clinical programs. To accomplish this purpose, the Medical Advisory Board will review and monitor the processes and procedures, infrastructure underlying our major discovery and clinical development programs and consists of the following individuals:
Michael G. Hanna, Jr., Ph.D., our Chairman of the Board and Chief Executive Officer.
John S. Macdonald, MD, professor of medicine, New York Medical College. Previously, Dr. Macdonald has served on the medical staffs at Georgetown University Medical School, University of Kentucky, Temple University New York Medical College and Saint Vincent's Hospital’s Comprehensive Cancer Center where he was chief of medical oncology. Dr. Macdonald graduated from Harvard University medical school and is board certified in internal medicine and medical oncology as well as a specialist in gastrointestinal cancer and cancer immunotherapy. Dr. Macdonald is the recipient of the distinguished American Cancer Society Junior Faculty Fellowship as well as having received numerous awards and distinctions including being named in the Best Doctors in America listing and the Good Housekeeping "Best 300 Doctors in America."
Benjamin S. Carson, MD, Director of Pediatric Neurosurgery at Johns Hopkins Hospital and professor of neurosurgery, oncology, plastic surgery and pediatrics at John Hopkins University and co-director of The Johns Hopkins Craniofacial Center. Dr. Carson's other surgical innovations have included the first intrauterine procedure to relieve pressure on the brain of a hydrocephalic fetal twin, and a hemispherectomy, in which a young girl suffering from uncontrollable seizures had one half of her brain removed. In 1987, Dr. Carson made medical history by being the first surgeon to successfully separate conjoined twins (the Binder twins) who had been joined at the back of the head (craniopagus twins). The 70-member surgical team, led by Dr. Carson, worked for 22 hours. At the end, the twins were successfully separated and can now survive independently. Dr. Carson is a member of the American Academy of Achievement, and the Horatio Alger Association of Distinguished Americans. In 2008, the White House awarded Benjamin Carson the Presidential Medal of Freedom, the nation's highest civilian honor. As an internationally renowned physician, Dr. Carson has authored over 100 neurosurgical publications, along with four best-selling books, and has been awarded 60 honorary doctorate degrees and dozens of national merit citations.
Herbert C. Hoover, MD, Ph.D., our Medical Advisor
Jan Vermorken, MD, Ph.D., Editor-in-Chief of Annals of Oncology Dr. Vermorken served on the medical staff of oncology, at the University Hospital VrijeUniversiteit , The Netherlands and is Chairman of the Netherlands Society of Oncology,Emeritus Professor of Oncology at the University of Antwerp and past head of the Department of Oncology at University Hospital Antwerp. Dr. Vermorken was principal investigator of the OncoVAX® 8701 phase IIIa clinical trial Jan Vermorken is Emeritus Professor of Oncology at the University of Antwerp and past head of the Department of Oncology at University Hospital Antwerp. Dr. Vermorken’s main field of interest is in gynecological oncology, and head & neck oncology. He coordinated large trials in breast and colon cancer including OncoVAX®’s phase III trial (8701). His main research areas concern early clinical and pharmacological studies with new drugs, studies on the interaction of chemotherapy and radiation therapy, HPV in various malignancies and immunological approaches. Dr. Vermorken is a member of the European Organization for Research and Treatment of Cancer (EORTC) Gynecological Cancer Group (GCG) and the ROTC Head and Neck Cancer Group (HNCG). He chaired the EORTC-GCG from 1983 to 1989 and the EORTC-HNCG from 2006 to 2009. He is still an active member with both groups. He is an officer for ESMO, was member of the ESMO executive committee during the time that he chaired the ESMO National Representative Committee (1991 – 1996) and the ESMO Education Committee (1996 – 2002) and is a prominent member of the ESMO faculty. He is also member of the International Gynecological Cancer Society (IGCS), was founding chair of the Gynecological Cancer Intergroup (GCIG) and was Chairman of the Belgian Association of Cancer Research (BACR) from 2003 to 2011, now still being part of its board.
| 52 |
EXECUTIVE COMPENSATION
The following table sets forth the compensation paid to the Chief Executive Officer and our other executive officers for services rendered during the fiscal years ended December 31, 2012, and 2011.
SUMMARY COMPENSATION TABLE
| Change in | ||||||||||||||||||||||||||||||||||||
| pension value | ||||||||||||||||||||||||||||||||||||
| and non- | ||||||||||||||||||||||||||||||||||||
| Non-equity | qualified deferred | |||||||||||||||||||||||||||||||||||
| Stock | Option | incentive plan | compensation | All other | ||||||||||||||||||||||||||||||||
| Name and principal position | Year | Salary | Bonus | Awards | Awards | compensation | earnings | compensation | Total | |||||||||||||||||||||||||||
| ($) | ($) | ($) | ($) | ($) | ($) | ($) | ($) | |||||||||||||||||||||||||||||
| Michael G. Hanna, Jr., Ph.D. | 2012 | 350,000 | — | — | — | — | — | — | 350,000 | |||||||||||||||||||||||||||
| Chairman and Chief Executive
Officer (since July 2012) | 2011 | 350,000 | — | — | — | — | — | — | 350,000 | |||||||||||||||||||||||||||
| Andrew Tussing | 2012 | 250,000 | — | — | — | — | — | — | 250,000 | |||||||||||||||||||||||||||
| President (since July 2012)
and Chief Operating Officer | 2011 | 250,000 | — | — | — | — | — | — | 250,000 | |||||||||||||||||||||||||||
| Michael Kranda | 2012 | 200,000 | — | — | — | — | — | — | 200,000 | |||||||||||||||||||||||||||
| Former President and Chief
Executive Officer (from September 2010 - June 2012) | 2011 | 400,000 | — | — | — | — | — | — | 400,000 | |||||||||||||||||||||||||||
Outstanding Equity Awards
There were no outstanding equity awards, unexercised options, unvested stock, or equity incentive plan awards as of December 31, 2012 for any of the executive officers named in the Summary Compensation Table above.
| 53 |
Employment Agreements
Employment Agreement with Michael G. Hanna, Jr., Ph.D.
We entered into a written employment agreement with Michael G. Hanna, Jr., Ph.D, our Chairman of the Board and Chief Executive Officer on February 1, 2010 and amended on December 21, 2010. Pursuant to the agreement Dr. Hanna is entitled to an annual base salary of $350,000 paid semi-monthly. In addition, the agreement provides for an Annual Bonus of up to seventy-five (75%) of Dr. Hanna’s annual base salary, depending on the satisfaction of performance criteria for each calendar year. No later than March 1st of each calendar year, the Board of Directors shall approve performance goals for the calendar year (either as presented by Dr. Hanna, or with reasonable modifications desired by the Board). Such approved performance goals shall indicate the manner in which the Executive's Annual Bonus (if any) will be determined upon partial satisfaction of one or more of the goals. Any Annual Bonus due (less all amounts required to be withheld under applicable law) shall be paid to Dr. Hanna by the March 15th of the calendar year following the calendar year for which it is payable.
Dr. Hanna’s employment agreement also provides for reimbursement of reasonable business expenses and entitlement to reimbursement by us for expenses incurred in a calendar year with respect to tax, financial, family office and legal planning services (including tax return preparation), up to a maximum of $10,000 per calendar month during the Term.
The term of Dr. Hanna’s employment agreement currently ends on December 31, 2014 but will be extended for additional two (2) year periods unless either party, in its or his sole discretion, provides written notice of an intention to not renew the Agreement at the end of the Term at least one hundred and twenty (120) days prior to the end of the Term.
Employment Agreement with Andrew L. Tussing
We entered into a written employment agreement with Andrew L. Tussing, our Chief Operating Officer on February 1, 2010 and President effective July, 2012 and amended on December 21, 2010. Pursuant to the agreement Mr. Tussing is entitled to an annual base salary of $250,000 paid semi-monthly. In addition, the agreement provides for an Annual Bonus of up to seventy-five (75%) of the annual base salary, depending on the satisfaction of performance criteria for each calendar year. No later than February 1st of each calendar year, Mr. Tussing shall submit to our Chief Executive Officer performance goals for the calendar year No later than March 1st of each calendar year, the Chief Executive Officer shall approve performance goals for the calendar year (either as presented by the Mr. Tussing, or with reasonable modifications desired by the Chief Executive Officer). Fifty percent of the bonus will be paid on personal achievements. Such approved performance goals shall indicate the manner in which the Executive's Annual Bonus (if any) will be determined upon partial satisfaction of one or more of the goals. The other fifty percent of the bonus will be a function of our performance and meeting plan for the Calendar year. Any Annual Bonus due (less all amounts required to be withheld under applicable law) shall be paid to Mr. Tussing by the March 15th of the calendar year following the calendar year for which it is payable.
Mr. Tussing’s employment agreement also provides for reimbursement of reasonable business expenses and entitlement to reimbursement by us for expenses incurred in a calendar year with respect to tax, financial, family office and legal planning services (including tax return preparation), up to a maximum of $10,000 per calendar month during the Term.
The term of Mr. Tussing’s employment agreement currently ends on December 31, 2014 but will be extended for additional two (2) year periods unless either party, in its or his sole discretion, provides written notice of an intention to not renew the Agreement at the end of the Term at least one hundred and twenty (120) days prior to the end of the Term.
Potential Payments upon Termination
Both Michael G. Hanna, Jr. Ph.D. and Andrew L. Tussing have entered into an employment agreements. Under the terms of their respective Employment Agreement, if (i) the executive resigns with “good reason” or if the we terminates executive’s employment without cause such executive will be paid its base salary for the greater of (A) the remainder of the Term or (B) one year with payments due during the first 90 day period following separation to be paid in a lump sun on the final regularly scheduled paydate preceding the end of such 90-day period and the remaining payment to be made when due on our regular payroll schedule or (ii) such executive’s employment is terminated due to death or disability, such executive (or their estate) will be paid a lump sum cash payment equal to one year of the executive’s base salary. No payments are required to be made to executive upon a change of control (as defined in the employment agreements).
The following table sets forth quantitative information with respect to potential payments to be made to each of Dr. Hanna and Mr. Tussing upon termination in various circumstances. The potential payments are based on each of the executive officer’s Employment Agreement discussed above. For a more detailed description of the Employment Agreement, see the “Employment Agreements” section above.
| 54 |
| Name | Payments upon Death Disability (1) | Payments upon Termination without Cause and Resignation for Good Reason (1)(2) |
|||||
| Michael G. Hanna, Jr., Ph.D | $ | 350,000 | $350,000-$700,000 | ||||
| Andrew L. Tussing | $ | 250,000 | $250,000-$500,000 | ||||
| (1) | Based on current annual base salary of $350,000 for Dr. Hanna and $250,000 for Mr. Tussing. |
| (2) | Each executive entitled to be paid its base salary for the greater of the remainder of its employment term or one-year. |
Benefits
Continued medical and dental benefits as provided by the Company from time to time for its employees, at the Company’s expense, for the period of time equal to the shorter of the Severance Period or the maximum period of COBRA continuation coverage provided under Section 4980B(f) of the Internal Revenue Code (with such coverage to be treated as COBRA coverage); provided, however, that the Company shall not subsidize COBRA continuation coverage to the extent that the Executive is eligible for the COBRA subsidy under the American Recovery and Reinvestment Act of 2009.
Compensation of Directors
| Fees Earned or | ||||||||||||
| Paid in | Stock | Option | All Other | |||||||||
| Name | Cash | Awards ($) | Awards ($) | Compensation | Total ($) | |||||||
| Alan Cohen | — | $ | 20,000 | (1) | — | — | — | |||||
| Daniel Fitzgerald | 20,000 | (1) | ||||||||||
| Dan Kane | 20,000 | (1) | ||||||||||
| John Nicolis | 20,000 | (1) | ||||||||||
| (1) | We granted each of our non-employee directors 3,636 shares of restricted common stock on December 31, 2012 for their services as a director for which they will become vested if, and only if, the Corporation consummates a "Public Offering" or a "Deemed Liquidation Event" (as defined in the Third Amended and Restated Certificate of Incorporation of the Corporation) within ten (10) years after the Grant Date thereof. |
From time to time we may engage certain members of the Board of Directors to perform services on our behalf. In such cases, we compensate the members for their services at rates no more favorable than could be obtained from unaffiliated parties. Other than as set forth above, we did not engage any members of the Board of Directors to perform services our behalf in 2012.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Intracel Holdings Corporation
Prior to our formation, our Chief Executive Officer was an employee and minority shareholder of Intracel and currently, he holds less than 1% interest in Intracel. In addition, three of our directors (Daniel Fitzgerald, Daniel Kane and Alan Cohen) are shareholders of Intracel and both Mr. Kane and Mr. Cohen are directors of Intracel. Intracel is our largest stockholder holding approximately 43% of our outstanding stock.
| 55 |
Pursuant to the October 2007 License Agreement, the Company agreed to pay Intracel the following royalties on the Net Sales of Colon Cancer Products (as defined): (i) 3% of net sales on the first $350.0 million of Net Sales of Colon Cancer Products occurring in the calendar year; (ii) 4% of net sales of Net Sales of Colon Cancer Products occurring in the calendar year in excess of $350.0 million and up to and including $700.0 million and (iii) 5%of net sales of Net Sales of Colon Cancer Products occurring in the calendar year in excess of $750.0 million
On April 15, 2012, Intracel loaned us $30,000 under a promissory note. The note is unsecured, non-interest bearing, and becomes due on the date on which a minimum equity raise of $1.0 million occurs. Currently the note is in default.
Due to Officers/Directors
As of December 31, 2011, our CEO agreed to convert $462,500 compensation owed to him into a payable that may be satisfied with issuance of restricted Common Stock, subject to restrictions yet to be defined. The restricted Common Stock is issuable if and only if we complete a financing round that provides us bona fide equity capital of at least $35.0 million.
On February 14, 2012, John Nicolis, one of our directors, paid one of our vendors an outstanding amount of $169,729. This money was used to pay for travel expenses associated with raising additional equity. In consideration of this payment, we issued Mr. Nicolis 30,860 units from our 2012 unit offering consisting of 30,860 shares of common stock and 9,258 warrants.
In April 2012, our Chief Executive Officer, Dr. Hanna, loaned us $10,000. The loan is evidenced by a non-interest bearing unsecured promissory note which is due and payable upon the date on which we receive gross proceeds of equity or debt securities of at least $20 million. As of the date of this registration statement, $4,099 remains outstanding under this note.
We believe that the foregoing transactions were in our best interests. Consistent with Section 2-419 of the Maryland General Corporation Law, it is our current policy that all transactions between us and our officers, directors and their affiliates will be entered into only if such transactions are approved by a majority of the disinterested directors, are approved by vote of the stockholders, or are fair to us as a corporation as of the time it is us at is authorized, approved or ratified by the board. We will conduct an appropriate review of all related party transactions on an ongoing basis, and, where appropriate, we will utilize our audit committee for the review of potential conflicts of interest.
Director Independence
Our Board of Directors has determined that each of Messrs. Nicolas, Fitzgerald, Cohen and Kane is “independent” as defined under the standards set forth in Section 121A of the American Stock Exchange Company Guide. In making this determination, the Board of Directors considered all transactions set forth under “Certain Relationships and Related Transactions” above.
MARKET FOR COMMON EQUITY AND RELATED STOCKHOLDER MATTERS.
Market information
On March 8, 2013, FINRA assigned our common stock the trading symbol “VACC.” Effective March 14, 2013, the trading symbol for our common stock was changed to “VGEN”. Our stock has been approved for quotation on the OTC Link (OTC Pink Tier). We plan to immediately upgrade our tier on the OTC Link to OTC.QB upon effectiveness of our recently filed General Registration Statement on Form 10. The table below sets forth the high and low bid prices for our common stock for each quarter since March 8, 2013, as reported on the OTC Link
| 56 |
We consider our stock to be “thinly traded” and any reported sale prices may not be a true market-based valuation of our stock. Some of the bid quotations from the OTC Link set forth below may reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions.
| 2013 (OTC Link) | High Bid | Low Bid | ||||||
| First quarter | $ | - | $ | - | ||||
| Second quarter | $ | 7.00 | $ | 5.50 | ||||
As of June 26, 2013, there were 31,227,756 shares of common stock outstanding, which were held by approximately 230 record stockholders. In addition, we have reserved 2,915,710 shares of common stock for issuance upon exercise of outstanding warrants.
Transfer Restrictions under our Charter
Pursuant to the our Articles of Amendment and Restatement filed on August 1, 2012 (the “Initial Date”), from and until the date that is one hundred eighty (180) days following the effectiveness of the first Form S-1 filed by us with the SEC, no shareholder may transfer more than 5% of the shares held by it on the Initial Date, unless approved by the board of directors. Any transfer in violation of this provision will be void. The restrictions on transfer do not apply certain limited exceptions such as dispositions by gift, will or the laws of descent and distribution and for any transfer of shares acquired from the Company after the Initial Date.
Dividend Policy
We have not paid cash dividends since our inception and we do not contemplate paying dividends in the foreseeable future.
Securities Authorized for Issuance Under Equity Compensation Plans. We did not have any equity compensation plans as of the year ended December 31, 2012.
DESCRIPTION OF SECURITIES
Common Stock
We are authorized by our Certificate of Incorporation to issue 200,000,000 shares of common stock, $0.0001 par value.
As of June 6, 2013 we had issued and outstanding approximately 31,122,755 shares of common stock. Holders of our common stock are entitled to one vote per share on all matters subject to stockholder vote. If the Board of Directors were to declare a dividend out of funds legally available therefore, all of the outstanding shares of common stock would be entitled to receive such dividend ratably. We have never declared dividends and we do not intend to declare dividends in the foreseeable future. If our business was liquidated or dissolved, holders of shares of common stock would be entitled to share ratably in assets remaining after satisfaction of our liabilities.
Pursuant to the our Articles of Amendment and Restatement filed on August 1, 2012 (the “Initial Date”), from and until the date that is one hundred eighty (180) days following the effectiveness of the first Form S-1 filed by us with the SEC, no shareholder may transfer more than 5% of the shares held by it on the Initial Date, unless approved by the board of directors. Any transfer in violation of this provision will be void. The restrictions on transfer do not apply certain limited exceptions such as dispositions by gift, will or the laws of descent and distribution and for any transfer of shares acquired from the Company after the Initial Date.
Holders of common stock do not have cumulative voting rights.
| 57 |
Preferred Stock
Our Certificate of Incorporation permits us to issue up to 50,000,000 shares of preferred stock, par value $0.0001 per share. The preferred stock may be issued in any number of series, as determined by the Board of Directors, the Board may by resolution fix the designation and number of shares of any such series of preferred stock and may determine, alter or revoke the rights, preferences, privileges and restrictions pertaining to any wholly unissued series and the Board may increase or decrease the number of shares of any such series (but not below the number of shares of that series then outstanding.)
We have no preferred stock issued or outstanding.
Description of the Warrants issued in our 2012-2013 Unit Offering
2012-2013 Unit Offering Warrants
The Warrants issued in our 2012-2013 Unit Offering (with each Unit consisting of one share of common stock and 0.3 shares of common stock) enable the holder to purchase the shares of Common Stock underlying the Warrants at $6.05 per whole share, during a five year term commencing on the final closing of the Offering. In the event a Warrant is partially exercised, a new Warrant for the remaining number of such shares will be issued upon surrender of the Warrant at the principal office of the Company. There will be no fractional share exercise.
The Warrant is transferable by the registered holder thereof in person or in writing, but only in the manner and subject to the limitations provided in the Warrant. Warrant transfers will be effected upon surrender of the Warrant at the Company’s principal executive offices (or such other office or agency of the Company as may be designated by notice to the holder thereof). Upon any such transfer, a new Warrant of different denominations, of like tenor and representing in the aggregate the right to purchase a like number of shares of Common Stock will be issued to the transferee in exchange for the original Warrant.
Warrant holders are not entitled to vote, receive dividends, or exercise any of the rights of a stockholder of the Company for any purpose until the Warrants have been duly exercised and payment of the purchase price has been made. The Warrants are issued in unregistered form and may be presented for transfer, exchange, or exercise at the corporate office of the Company.
The Warrants may be exercised, in whole or in part, upon surrender of the Warrant on or prior to the expiration date at the offices of the Company, together with the “Form of Exercise Notice” attached to the Warrant filled outand executed as indicated, accompanied by payment of the full exercise price for thenumber of Warrants being exercised. Payment must be either (i) in the form of a certified or official bank check made payable to the Company or by wire transfer for the account of the Company or (ii) delivery to the Company of a written notice of an election to effect a “Cashless Exercise” (as defined in Section 1(b) of the Warrant) for the Warrant Shares specified in the Exercise Agreement.
The Warrants contain “piggyback” registration rights for the resale of the shares issuable upon exercise of the Warrants.
The Warrants a contain standard anti-dilution provision in the event the Company sells any of its shares of common stock, or securities convertible into common stock at less than the applicable exercise price of the Warrants. In such event, the exercise price of the Warrants will be adjusted as set forth in the Warrant.
Placement Agents Warrants for 2012-2013 Unit Offering
We have agreed to issue First Liberties Financial (FLF) a warrant to acquire such number of shares of common stock as is equal to 5% of the number of Units sold through FLF in the 2012-2013 Unit Offering. These warrants will be issued on terms identical to the Investor Warrants, except that the FLF Warrants have an exercise price of $5.50. To date no warrants have been issued to FLF.
| 58 |
The Abell Foundation Financings
Between October 26, 2011 and February 16, 2012, we sold $1,800,000 of principal amount of secured promissory notes to The Abell Foundation. The notes had an initial 6-month term bearing interest at eight percent (8%) per annum; payable at maturity. Upon an event of default the interest rate increases to 10% per annum. The notes were secured by all accounts, chattel paper, deposit accounts, equipment, general intangibles, instruments, inventory, investment property and letter of credit rights. In April 2013, the maturity date on the note was extended to May 31, 2013. In consideration of the extension, we also granted Abell a security interest in our patents related to OncoVax® under a Patent Security Agreement.
On May 31, 2013, the maturity date on the note was extended to July 31, 2013. In consideration of the extension, we agreed that the Notes would be paid concurrently with the closing of each issuance or sale of additional shares of capital stock, or securities directly or indirectly convertible or exchangeable for capital stock (each an “Equity Issuance”) occurring after May 28, 2013 in an amount equal to (a) twenty percent (20%) of the nest $3,778,771 of gross proceeds of such Equity Issuance(s), (b) twenty-five percent (25%) of the next $6,000,000 of gross proceeds of such Equity Issuance(s), and (c) one hundred percent (100%) of the net proceeds of all Equity Issuance(s) thereafter to pay the remaining amount due under the Note, if any.
Warrants
In connection with the secured promissory notes referenced above, we issued Abell a warrant to purchase the number of shares equal to $800,000 divided by 85% of the purchase price per share of stock sold in our first venture capital financing resulting in proceeds of not less than $20,000,000. In connection with the January 2013 extension of maturity date, we increased the amount of shares issuable under the warrant to $1.1 million divided by 85% of the purchase price per share of stock sold in our first venture capital financing resulting in proceeds of not less than $25,000,000. The exercise price equals 85% of the purchase price per share in such venture capital financing, during a 10-year term. In the event a Warrant is partially exercised, a new Warrant for the remaining number of such shares will be issued upon surrender of the Warrant at the principal office of the Company. There will be no fractional share exercise.
The warrant holders are not entitled to vote, receive dividends, or exercise any of the rights of a stockholder of the Company for any purpose until the warrants have been duly exercised and payment of the purchase price has been made. The warrants are issued in unregistered form and may be presented for transfer, exchange, or exercise at the corporate office of the Company.
The Warrants may be exercised, in whole or in part, upon surrender of the Warrant on or prior to the expiration date at the offices of the Company, together with the “Form of Exercise Notice” attached to the Warrant filled out and executed as indicated, accompanied by payment of the full exercise price for the number of Warrants being exercised. Payment must be in the form of a certified or official bank check made payable to the Company or by wire transfer for the account of the Company.
Investment Agreement
On January 16, 2013, we entered into an investment agreement with The Abell Foundation, Inc. under which at the request of the investor we will issue and sell to the investor up to $5.0 million of common stock. The number of shares issued will be based on the lowest price paid by any purchaser of shares in the any Venture Capital Financing (as defined in the agreement). The investor will not exercise this above option unless the Company has received $25.0 million in gross proceeds under its financing arrangements with Kodiak Capital and an additional $10.0 million from investors other than Kodiak.
Description of Other Warrants
We have issued 5-year warrants to certain officer and other providers to purchase 2,433,165 shares of our common stock, with exercise prices beginning at $1.00 per share. In the event a Warrant is partially exercised, a new Warrant for the remaining number of such shares will be issued upon surrender of the Warrant at the principal office of the Company. There will be no fractional share exercise.
| 59 |
Warrant holders are not entitled to vote, receive dividends, or exercise any of the rights of a stockholder of the Company for any purpose until the Warrants have been duly exercised and payment of the purchase price has been made. The Warrants are issued in unregistered form and may be presented for transfer, exchange, or exercise at the corporate office of the Company.
The Warrants may be exercised, in whole or in part, upon surrender of the Warrant on or prior to the expiration date at the offices of the Company, together with the “Form of Exercise Notice” attached to the Warrant filled out and executed as indicated, accompanied by payment of the full exercise price for the number of Warrants being exercised. Payment must be in the form of a certified or official bank check made payable to the Company or by wire transfer for the account of the Company. In addition, these warrants contain a “cashless exercise” provision.
Bridge Loan 2012
Between April 2012 and September 2012, we issued unsecured promissory notes to several investors in the aggregate principal amount of $1,019,000. These notes are unsecured, and the interest payable on this balance is equal to the principal amount borrowed, or $1,019,000 for total principal and interest of $2,038,000 due at repayment. The notes mature after a date on which a transaction occurs involving the issuance or sale of additional equity of Vaccinogen that results in at least $20.0 million in gross proceeds. Although management anticipates that the maturity will occur by July 2013, there is no guarantee that the maturity date will occur and if the maturity date does not occur we will have no obligation to repay principal amount to investors or make the interest payment to investors. In May 2013, noteholders with an aggregate principal amount of $419,000 in unsecured notes (with $419,000 in accrued interest) cancelled their notes in consideration of their subscription for 152,359 Units.
Agreements with Organon Teknika Corporation
On October 31, 2007, Intracel Holdings Corporation entered into a letter agreement with Organon Teknika Corporation (which is now part of Merck Corporation) to settle all liabilities owed to OTC under that certain Product Supply Agreement with OTC and related agreements (the “Letter Agreement”). Under the Letter Agreement, Intracel settled all liabilities with OTC for an amount equal to $4,000,000, of which $500,000 was paid concurrently with execution of the Letter Agreement and $500,000 was to be paid within 1 year of the Letter Agreement. In connection with our License Agreement and subsequent Asset Transfer Agreement with Intracel, we assumed Intracel’s obligations under the Letter Agreement. At OTC’s option, the second $500,000 installment was payable in shares of our common stock. The remaining monies are to be paid upon receipt of marketing approval of OncoVAX® by the United States Food and Drug Administration. Payments of these amounts were secured by the OncoVAX® intellectual property and patents. Payment of the second $500,000 has not been made, although no formal notice or assertion of default has been issued. We are currently in discussions with the secured party to resolve this matter. If any notice of default is declared, we would have 45-days to cure prior to an actual event of default.
Transfer Agent
The transfer agent for our common stock is Globex Transfer, LLC, 780 Deltona Blvd., Suite 202, Deltona, FL 32725, telephone: (813) 344-4490.
LEGAL MATTERS
The validity of the common stock offered by this prospectus will be passed upon for us by Indeglia& Carney, P.C., Irvine, California.
EXPERTS
The financial statements of our company included in this prospectus and in the registration statement have been audited by BDO USA, LLP., independent registered public accounting firm, to the extent and for the periods set forth in their report appearing elsewhere herein and in the registration statement, and are included in reliance on such report, given the authority of said firm as an expert in auditing and accounting.
| 60 |
AVAILABLE INFORMATION
We are filing with the SEC this registration statement on Form S-1 under the Securities Act with respect to the common stock offered hereby. This prospectus, which constitutes part of the registration statement, does not contain all of the information set forth in the registration statement and the exhibits and schedule thereto, certain parts of which are omitted in accordance with the rules and regulations of the SEC. For further information regarding our common stock and our company, please review the registration statement, including exhibits, schedules and reports filed as a part thereof. Statements in this prospectus as to the contents of any contract or other document filed as an exhibit to the registration statement, set forth the material terms of such contract or other document but are not necessarily complete, and in each instance reference is made to the copy of such document filed as an exhibit to the registration statement, each such statement being qualified in all respects by such reference.
We are also subject to the informational requirements of the Exchange Act which requires us to file reports, proxy statements and other information with the SEC. Such reports, proxy statements and other information along with the registration statement, including the exhibits and schedules thereto, may be inspected at public reference facilities of the SEC at 100 F Street N.E., Washington D.C. 20549. Copies of such material can be obtained from the Public Reference Section of the SEC at prescribed rates. You may call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference room. Because we file documents electronically with the SEC, you may also obtain this information by visiting the SEC’s Internet website at http://www.sec.gov.
| 61 |
INDEX TO FINANCIAL STATEMENTS
Audited Statements;
| Page | ||
| Report of Independent Registered Public Accounting Firm | F-3 | |
| Consolidated Balance Sheets as of December 31, 2012 and 2011 | F-5 | |
| Consolidated Statements of Operations for the years ended December 31, 2012 and 2011 | F-6 | |
| Consolidated Statements of Comprehensive Loss for the years ended December 31, 2012 and 2011 | F-7 | |
| Consolidated Statements of Changes in Stockholders’ Equity (Deficit) as of December 31, 2012 and 2011 | F-8 | |
| Consolidated Statement of Cash Flows for the years ended December 31, 2012 and 2011 | F-9 | |
| Notes to Consolidated Financial Statements | F-10 - F-35 |
Unaudited Statements:
| Condensed Consolidated Balance Sheets as of March 31, 2013 and December 31, 2012 (audited) | F-40 | |
| Unaudited Condensed Consolidated Statements of Operations for the three months ended March 31, 2013 and 2012 | F-41 | |
| Unaudited Condensed Consolidated Statements of Comprehensive Loss for the three months ended March 31, 2013 and 2012 | F-42 | |
| Unaudited Condensed Consolidated Statement of Changes in Stockholders’ Equity as of March 31, 2013 | F-43 | |
| Unaudited Condensed Consolidated Statements of Cash Flows for the three months ended March 31, 2013 and 2012 | F-44 | |
| Notes to Unaudited Condensed Consolidated Financial Statements | F-45- F-69 |
| 62 |
Vaccinogen Inc. and Subsidiaries
Consolidated Financial Statements
Years Ended December 31, 2012 and 2011
Vaccinogen Inc. and Subsidiaries
Consolidated Financial Statements
Years Ended December 31, 2012 and 2011
VACCINOGEN INC. AND SUBSIDIARIES
Contents
| Report of Independent Registered Public Accounting Firm | F-3 |
| Consolidated Financial Statements | |
| Consolidated Balance Sheets | F-5 |
| Consolidated Statements of Operations | F-6 |
| Consolidated Statements of Comprehensive Loss | F-7 |
| Consolidated Statements of Changes in Stockholders’ Equity (Deficit) | F-8 |
| Consolidated Statements of Cash Flows | F-9 |
| Notes to Consolidated Financial Statements | F-10 - F-35 |
| F-2 |
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
Vaccinogen, Inc.
Frederick, Maryland
We have audited the accompanying consolidated balance sheets of Vaccinogen Inc. and subsidiaries as of December 31, 2012 and 2011 and the related consolidated statements of operations, comprehensive loss, changes in stockholders’ equity (deficit), and cash flows for years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Vaccinogen Inc. and subsidiaries at December 31, 2012 and 2011, and the results of their operations and their cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has suffered recurring losses from operations, has a negative working capital, and losses are expected to continue in the future. These factors raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Bethesda, Maryland
April 22, 2013
BDO USA, LLP, a Delaware limited liability partnership, is the U.S. member of BDO International Limited, a UK company limited by guarantee, and forms part of the international BDO network of independent member firms.
BDO is the brand name for the BDO network and for each of the BDO Member Firms.
| F-3 |
Consolidated
Financial Statements
| F-4 |
VACCINOGEN INC. AND SUBSIDIARIES
Consolidated Balance Sheets
| December 31, | 2012 | 2011 | ||||||
| Assets | ||||||||
| Current Assets | ||||||||
| Cash and cash equivalents | $ | 113,840 | $ | 236,681 | ||||
| Restricted cash | 42,044 | 40,481 | ||||||
| Inventory | 102,174 | 105,621 | ||||||
| Prepaid expenses and other current assets | 140,343 | 75,411 | ||||||
| Total Current Assets | 398,401 | 458,194 | ||||||
| Prepaid expenses | 1,301,463 | - | ||||||
| Property and equipment, net | 107,455 | 162,616 | ||||||
| Intangible assets, net | 69,428,831 | 76,132,103 | ||||||
| Total Assets | $ | 71,236,150 | $ | 76,752,913 | ||||
| Liabilities, Redeemable Preferred Stock, and Stockholders’ Equity (Deficit) | ||||||||
| Current Liabilities | ||||||||
| Notes payable | $ | 5,300,000 | $ | 4,975,373 | ||||
| Accounts payable | 2,021,039 | 1,058,006 | ||||||
| Financial instruments | 2,590,655 | 36,940 | ||||||
| Accrued interest | 929,915 | 672,899 | ||||||
| Accrued compensation | 620,212 | 170,379 | ||||||
| Related party payable | 34,099 | - | ||||||
| Accrued expenses and other liabilities | 400,678 | 233,734 | ||||||
| Total Current Liabilities | 11,896,598 | 7,147,331 | ||||||
| Commitments and Contingencies | ||||||||
| Redeemable Preferred Stock | ||||||||
| Series AA Preferred Stock, redeemable, convertible, $0.0001 par value; 0 and 15,000,000 shares authorized; 0 and 913,361 share issued and outstanding at December 31, 2012 and 2011, respectively. | - | 8,993,418 | ||||||
| Series B Preferred Stock, redeemable, convertible, $0.0001 par value; 0 and 35,000,000 shares authorized; 0 and 14,425,377 shares issued and outstanding at December 31, 2012 and 2011, respectively. | - | 110,135,112 | ||||||
| Stockholders’ Equity (Deficit) | ||||||||
| Preferred stock, $0.0001 par value; 50,000,000 shares authorized; 0 shares issued and outstanding | - | - | ||||||
| Common stock, $0.0001 par value; 200,000,000 and 75,000,000 share authorized; 30,601,700 and 12,003,455 share issued and outstanding at December 31, 2012 and 2011, respectively. | 3,060 | 1,200 | ||||||
| Additional paid-in capital | 138,118,424 | - | ||||||
| Accumulated other comprehensive loss | (83,152 | ) | (37,669 | ) | ||||
| Accumulated deficit | (78,698,780 | ) | (49,486,479 | ) | ||||
| Total Stockholders’ Equity (Deficit) | 59,339,552 | (49,522,948 | ) | |||||
| Total Liabilities, Redeemable Preferred Stock, and Stockholders’ Equity (Deficit) | $ | 71,236,150 | $ | 76,752,913 | ||||
See accompanying notes to consolidated financial statements.
| F-5 |
VACCINOGEN INC. AND SUBSIDIARIES
Consolidated Statements of Operations
| Years ending December 31, | 2012 | 2011 | ||||||
| Revenues | $ | - | $ | - | ||||
| Operation Expenses: | ||||||||
| Research and development | 8,139,038 | 8,564,519 | ||||||
| General and administrative | 2,960,939 | 2,783,460 | ||||||
| Total Operating Expenses | 11,099,977 | 11,347,979 | ||||||
| Loss From Operations | (11,099,977 | ) | (11,347,979 | ) | ||||
| Loss on Financial Instruments | (1,281,916 | ) | - | |||||
| Interest and Other Expenses | (312,257 | ) | (176,489 | ) | ||||
| Net Loss | $ | (12,694,150 | ) | $ | (11,524,468 | ) | ||
| Less: Accretion of preferred stock | (16,518,151 | ) | (27,667,590 | ) | ||||
| Net loss available to common stockholders | $ | (29,212,301 | ) | $ | (39,192,058 | ) | ||
| Basic and diluted weighted average share outstanding | 19,686,792 | 12,003,455 | ||||||
| Basic and diluted loss per common share | $ | (1.48 | ) | $ | (3.27 | ) | ||
See accompanying notes to consolidated financial statements.
| F-6 |
VACCINOGEN INC. AND SUBSIDIARIES
Consolidated Statements of Comprehensive Loss
| Years ended December 31, | 2012 | 2011 | ||||||
| Comprehensive Loss | ||||||||
| Net loss | $ | (12,694,150 | ) | $ | (11,524,468 | ) | ||
| Foreign currency translation adjustments | (45,483 | ) | 34,839 | |||||
| $ | (12,739,633 | ) | $ | (11,489,629 | ) | |||
See accompanying notes to consolidated financial statements.
| F-7 |
VACCINOGEN INC. AND SUBSIDIARIES
Consolidated Statements of Changes in Stockholders’ (Deficit)/Equity
| Common Stock | ||||||||||||||||||||||||
| Shares | Amount | Additional Paid- In Capital | Accumulated Deficit | Accumulated Other Comprehensive Loss | Total Stockholders’ Equity (Deficit) | |||||||||||||||||||
| Balance, January 1, 2011 | 11,893,455 | $ | 1,189 | $ | 7,594,709 | $ | (18,458,493 | ) | $ | (72,508 | ) | $ | (10,935,103 | ) | ||||||||||
| Accretion on preferred stock | - | - | (8,164,072 | ) | (19,503,518 | ) | - | (27,667,590 | ) | |||||||||||||||
| Stock-based compensation | 110,000 | 11 | 569,447 | - | - | 569,458 | ||||||||||||||||||
| Stock dividends | - | - | (84 | ) | - | - | (84 | ) | ||||||||||||||||
| Other comprehensive income | - | - | - | - | 34,839 | 34,839 | ||||||||||||||||||
| Net loss | - | - | - | (11,524,468 | ) | - | (11,524,468 | ) | ||||||||||||||||
| Balance, December 31, 2011 | 12,003,455 | $ | 1,200 | $ | - | $ | (49,486,479 | ) | $ | (37,669 | ) | $ | (49,522,948 | ) | ||||||||||
| Accretion on preferred stock | - | - | - | (16,518,151 | ) | - | (16,518,151 | ) | ||||||||||||||||
| Conversion of preferredstock to common stock | 18,163,748 | 1,816 | 135,644,865 | - | - | 135,646,681 | ||||||||||||||||||
| Issuance of common stock to Kodiak | 236,364 | 24 | 1,301,463 | - | - | 1,301,487 | ||||||||||||||||||
| Issuance of common stock for cash | 167,273 | 17 | 725,740 | - | - | 725,757 | ||||||||||||||||||
| Conversion of payable to common stock | 30,860 | 3 | 133,621 | - | - | 133,624 | ||||||||||||||||||
| Warrants issued for services | - | - | 312,735 | - | - | 312,735 | ||||||||||||||||||
| Other comprehensive loss | - | - | - | - | (45,483 | ) | (45,483 | ) | ||||||||||||||||
| Net loss | - | - | - | (12,694,150 | ) | - | (12,694,150 | ) | ||||||||||||||||
| Balance, December 31, 2012 | 30,601,700 | $ | 3,060 | $ | 138,118,424 | $ | (78,698,780 | ) | $ | (83,152 | ) | $ | 59,339,552 | |||||||||||
See accompanying notes to consolidated financial statements.
| F-8 |
VACCINOGEN INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
| Years ending December 31, | 2012 | 2011 | ||||||
| Cash Flows from Operating Activities | ||||||||
| Net loss | $ | (12,694,150 | ) | $ | (11,524,468 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 109,817 | 95,656 | ||||||
| Amortization of intangible assets | 6,703,273 | 6,706,552 | ||||||
| Loss on financial instruments | 1,281,916 | - | ||||||
| Stock based compensation | - | 569,458 | ||||||
| Stock based compensation – non employees | 312,735 | - | ||||||
| Non-cash interest | 47,077 | 12,313 | ||||||
| Changes in operating assets and liabilities, net: | ||||||||
| Restricted cash | (711 | ) | (707 | ) | ||||
| Prepaid expenses and other assets | (65,420 | ) | 86,032 | |||||
| Accrued interest | 257,017 | 137,691 | ||||||
| Accounts payable and accrued expenses and other liabilities | 1,775,543 | 405,884 | ||||||
| Net cash use In operating activities | (2,272,903 | ) | (3,511,589 | ) | ||||
| Cash flow from investing activities | ||||||||
| Purchase of property and equipment | (48,282 | ) | (80,230 | ) | ||||
| Net cash used in investing activities | (48,282 | ) | (80,230 | ) | ||||
| Cash flows from financing activities | ||||||||
| Proceeds from 2012 bridge loan | 1,019,000 | - | ||||||
| Proceeds from notes payable | 300,000 | 1,500,000 | ||||||
| Proceeds from issuance of preferred stock | - | 1,102,448 | ||||||
| Proceeds from issuance of common stock and warrants | 920,002 | - | ||||||
| Dividends on common stock | - | (84 | ) | |||||
| Net cash provided by financial activities | 2,239,002 | 2,602,364 | ||||||
| Change in foreign currency translation | (40,658 | ) | 25,378 | |||||
| Net decrease in cash and cash equivalents | (122,841 | ) | (964,077 | ) | ||||
| Cash and cash equivalents, beginning of year | 236,681 | 1,200,758 | ||||||
| Cash and cash equivalents, end of year | $ | 113,840 | $ | 236,681 | ||||
See accompanying notes to consolidated financial statements.
| F-9 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
1. Organization
The Business
Vaccinogen, Inc. (the “Company” or “Vaccinogen”), a biotechnology Company headquartered in Frederick, Maryland, was incorporated in the State of Delaware during 2007 for the purpose of developing therapies and vaccines to combat cancer by using the body’s own immune system. On November 23, 2010, the Company changed its domicile from Delaware to Maryland by means of a merger of the Company with and into its wholly owned subsidiary Vaccinogen I, Inc., a Maryland corporation.
On October 10, 2007, the Company entered into a license agreement with Intracel Holdings Corporation (“Intracel”), a related party, for the exclusive and indefinite rights to use the OncoVAX® technology platform. OncoVAX® is an active specific immunotherapy (“ASI”) that uses the patient’s own cancer cells to create a vaccine that in turn is used to block the return of cancer following surgery. In June 2010, the Company entered into an agreement with Intracel (the “Asset Transfer Agreement”) whereby the Company acquired title to the patents associated with the OncoVAX® (See Note 4). On October 23, 2007, Vaccinogen acquired out of bankruptcy, certain tangible assets that had been previously owned and used by Intracel’s wholly owned subsidiary in the Netherlands. These assets will be used to conduct research and development and in the commercialization of OncoVAX® to produce vaccines. In connection with the acquisition of these assets, the Company formed a wholly owned subsidiary, Vaccinogen BV, for the purposes of continuing development of OncoVAX®.
2. Going Concern
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As of December 31, 2012, the Company had an accumulated deficit of approximately $78.7 million and negative working capital of approximately $11.5 million. Since inception, the Company has financed its activities principally from the proceeds from the issuance of equity and debt securities and loans from officers.
The Company’s ability to continue as a going concern is dependent upon the Company’s ability to raise additional debt and equity capital. As discussed in Note 5 to these consolidated financial statements, the Company has entered into a series of agreements with the Kodiak Capital Group, LLC (“Kodiak”) to provide up to $26 million of additional equity capital. The proceeds from the agreements with Kodiak would primarily be used to continue the Company’s research and development activities including the furtherance of clinical trials using OncoVAX® to develop cancer related vaccines. However, Kodiak is not required to provide funding until certain conditions are met, including the registration and trading of the Company’s equity securities as defined in those agreements. There can be no assurance that the Company will meet the conditions under which Kodiak will be required to provide the equity capital or that the capital available under such agreements will be sufficient to allow the Company to funds its continuing research and development activities. If the Company is unable to raise the additional equity capital from Kodiak, the Company will need to seek alternative sources of debt or equity capital. There can be no assurance that such capital will be available in sufficient amounts or on terms acceptable to the Company. These factors raise substantial doubt about the Company's ability to continue as a going concern. The accompanying financial statements do not include any adjustments relating to the recoverability of the recorded assets or the classification of liabilities that may be necessary should the Company be unable to continue as a going concern.
| F-10 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
3. Summary of Significant Accounting Policies
Principles of Consolidation
The consolidated financial statements include accounts of Vaccinogen and its wholly owned subsidiary, Vaccinogen BV (a company incorporated in the Netherlands). All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in its financial statements. On an ongoing basis, the Company evaluates the estimates used in recording common stock warrant related liabilities, derivative financial instruments, stock based compensation, and where applicable, the fair value of assets. The Company may base such estimates on various assumptions which it believes to be reasonable under the circumstances. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid securities with a maturity of three months or less at acquisition to be cash equivalents. Cash and cash equivalents include demand deposits with financial institutions and at times the amounts may exceed federally insured deposit limits. The Company has not experienced any losses and does not believe it is exposed to any significant credit risk related to demand deposits.
Restricted Cash
Restricted cash represents monies pledged by the Company’s foreign subsidiary for a lease obligation related to the manufacturing facility and to the Dutch government as required for companies with irradiator equipment.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents. The Company maintains its cash and cash equivalents with high-credit-quality financial institutions in the United States.
Cash and cash equivalents are maintained at financial institutions and, at times, balances may exceed federally insured limits. All non-interest bearing cash balances were fully insured at December 31, 2012 due to a temporary federal program in effect from December 31, 2010 through December 31, 2012. Under the program, there is no limit to the amount of insurance for eligible accounts. Beginning 2013, insurance coverage will revert to $250,000 per depositor at each financial institution, and noninterest bearing cash balances may again exceed federally insured limits.
| F-11 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Inventory
Inventory is reported at the lower of cost or market value. The Company analyzes its inventory and writes down inventory that has become obsolete, or has a cost basis in excess of its expected net realizable value and inventory quantities in excess of expected requirements. Inventory primarily consists of a product used in creating vaccines using the OncoVAX® technology platform.
Property and Equipment
Property and equipment are recorded at cost and are depreciated or amortized over their estimated useful lives using the straight-line method. Estimated useful lives are as follows:
| Machinery and equipment | 3 – 5 years | |||
| Automobile | 3 – 5 years | |||
| Furniture and fixtures | 3 years | |||
| Computers and software | 3 years |
Maintenance and repairs are charged to expense as incurred. Major betterments and improvements, which extend the useful life of the underlying assets, are capitalized and depreciated.
Intangible Assets
Intangible assets consist primarily of the cost of acquired patents associated with OncoVAX® to be used in research and development and the commercialization of cancer related vaccines. The Company has capitalized the cost of the acquired patents because the Company has identified alternative future research and development efforts for numerous forms of cancer which it intends to pursue and for which management believes will result in commercialization of related vaccines. Acquired patents are carried at cost less accumulated amortization. Amortization is calculated on a straight-line basis, over the estimated useful economic life of the patent, which is 15 years for OncoVAX®.
Impairment of Long-Lived Assets
Long-lived assets, including identifiable intangible assets with finite lives, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. The Company has determined that no impairment has occurred as of December 31, 2012.
Redeemable Preferred Stock
Conditionally redeemable preferred stock whose redemption is outside the control of the Company is recorded as temporary equity in accordance with Accounting Standards Codification (“ASC”) Topic 480 Distinguishing Liabilities from Equity (“Topic 480”) and Securities and Exchange Commission (“SEC”) guidance contained in SEC Accounting Release Series No. 268.
| F-12 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Foreign Currency Translation
The financial statements of foreign subsidiaries are maintained in their functional currency, which generally is the local currency. The assets and liabilities are translated to U.S. dollars using the exchange rate in effect at the balance sheet date. Revenues, expenses and cash flows of these operations are translated using average exchange rates during the reporting period which they occur. The resulting translation adjustments are reflected in other comprehensive loss. As of December 31, 2012, the assets and net deficit of Vaccinogen BV, excluding intercompany balances, were approximately $177,000 and $204,000, respectively. As of December 31, 2011, the net assets and net deficit of Vaccinogen BV, excluding intercompany balances, were approximately $196,000 and 280,000, respectively. The net loss of Vaccinogen BV was approximately $1.2 million and $1.3 million for the years ended December 31, 2012 and 2011 respectively.
Revenue Recognition
To date, the Company has not earned any revenues as the use of OncoVAX® to create cancer related vaccines is still undergoing clinical trials and has not received regulatory approval for commercialization and sale.
Research and Development Expense
Research and development costs are expensed as incurred. Research and development expenses primarily include the amortization of intangible assets, cost of conducting clinical trials, compensation and related overhead for employees, consultants, facilities costs and the cost of materials purchased for research and development.
Stock-Based Compensation
The Company measures the cost of employee services received in exchange for stock options or restricted stock awards based upon the fair value of the award on the date of the grant. The Company recognizes the estimated grant date fair value of the award as stock-based compensation expense on a straight-line basis over the requisite service period, which is generally the vesting period.
The Company initially measures the cost of awards granted to non-employees based on the fair value of the award on the date of grant however such cost is re-measured at the end of each reporting period until performance is fully satisfied or services are rendered by the non-employee.
The fair value of stock options granted is calculated using the Black-Scholes option-pricing model, which requires the use of subjective assumptions including volatility, expected term, risk-free rate, and the fair value of the underlying common stock. The fair value of non-vested stock awards is determined based upon the estimated fair value of the Company's common stock.
Income Taxes
Deferred income tax assets and liabilities are determined based on differences between the financial statements and tax basis of assets and liabilities, as measured using the enacted tax rates, which are expected to be in effect when the differences reverse. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
| F-13 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The tax effects of uncertain tax positions are recognized in the financial statements only if the position is more likely than not to be sustained on audit, based on the technical merits of the position. For tax positions meeting the more likely than not threshold, the amount recognized in the consolidated financial statements is the largest benefit that had greater than 50% likelihood of being realized. Management has not identified any uncertain tax positions with the exception of income tax return filing penalties and accordingly has established a liability under ASC 740-10 (“FIN 48”). It is the Company’s accounting policy to account for ASC 740-10 related penalties and interest in other liabilities/expenses and not include it in the income tax provision of consolidated statement of operations. The Company has identified its U.S. Federal income tax return and its state return in Maryland as its major tax jurisdictions. Tax returns for fiscal years 2007 and forward are still open for examination.
Financial Instruments
Warrants Accounted for as Liabilities
Abell Warrants
In October 2011, the Company entered into a borrowing arrangement with The Abell Foundation (“Abell”). In connection with that arrangement, the Company also issued warrants (the “Abell Warrants”) exercisable into common stock of the Company. In February 2012, the Company and Abell amended the agreement to provide for additional borrowings. In 2013, the borrowing arrangement was amended on two occasions, first to extend the maturity to March 31, 2013 and a second time to extend the maturity date to May 31, 2013.
The number of shares issuable pursuant to the Abell Warrants was originally determined based upon a fixed amount of $500,000 divided by 85% of the per share price of stock sold in the next qualifying round of venture capital financing (defined as a round that raised at least $20 million). In connection with February 2012 amendment to the borrowing arrangement, the fixed amount used to determine the ultimate number of shares into which the Abell Warrants are exercisable was increased to $800,000. In connection with the 2013 amendments to the borrowing arrangement, the fixed amount used to determine the ultimate number of shares into which the Abell Warrants are exercisable was increased to $1.1 million and the total proceeds of the next qualifying round of venture capital financing was increased to $35 million. The Abell Warrants have a contractual term of 10 years and were fully vested upon issuance.
The Abell Warrants represent a fixed obligation that is to be settled through the issuance of a variable number of shares of the Company’s common stock. Consistent with the provisions of Topic 480, the Company has concluded that the Abell Warrants should be accounted for as a liability. The Company is required to record the Abell Warrants at their estimated fair value at the end of each reporting period, with changes in the estimated fair value recorded in the consolidated statements of operations as a component of other income (expense). As of December 31, 2012 and 2011, the estimated fair value of the Abell Warrants was $831,806 and $36,940, respectively. The change in the estimated fair value of the liability of $772,416 and zero was recorded as an expense and classified in Loss on Financial Instruments in the accompanying consolidated statements of operations for the years ended December 31, 2012 and 2011, respectively.
| F-14 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Derivative Financial Instruments
The Company may enter into transactions that represent free-standing or embedded derivative financial instruments as those terms are defined in ASC Topic 815 Derivatives and Hedging (“Topic 815”). The Company records the estimated fair value of derivative financial instruments in its consolidated balance sheets and records changes in the estimated fair value of derivative financial instruments as income or expense in its consolidated statements of operations.
Round C Warrants
From October 2012 through December 2012, the Company issued warrants to certain investors in the common stock of the Company (the “Round C Warrants”). A total of 59,439 shares of common stock are issuable under the Round C Warrants. The Round C Warrants have an exercise price of $6.05, a contractual term of 5 years and were fully vested upon issuance.
The terms of the Round C Warrants provide for "down-round" anti-dilution adjustments in certain situations whereby the Company sells or issues (a) stock at a price per share less than the exercise price of the Round C Warrants or (b) equity linked financial instruments with an exercise price less than the exercise price of the Round C Warrants. Consistent with the provisions of ASC Topic 815-40, the Round C Warrants are classified as derivative financial instruments. The Company is required to record the estimated fair value of derivative financial instruments at the end of each reporting period, with changes in the estimated fair value of such derivatives recorded in the consolidated statements of operations as a component of Loss on Financial Instruments. As of December 31, 2012, the estimated fair value of the liability associated with the Round C Warrants was $230,349 and is included in derivative financial instruments in the accompanying consolidated balance sheets. There was no change in the fair value of the Round C Warrants during 2012, accordingly no gain or loss was recorded in the accompanying consolidated statements of operations.
2012 Bridge Loan
Between April 2012 and October 2012, the Company entered into transactions with various investors which resulted in the Company raising $1,019,000 from the issuance of unsecured notes payable (collectively the “Bridge Loan”). The Bridge Loan has no contractual maturity date, and is repayable only in the event that the Company closes on a future round of equity financing which results in gross proceeds of at least $20 million. If the Company fails to raise sufficient additional capital, there is no obligation to pay interest or repay any amount borrowed under the Bridge Loan. Should the Company be successful in raising sufficient equity capital, the Company must repay an amount to the investors equal to 2 times the amount originally raised.
The Company has classified the Bridge Loan as a derivative financial instrument. As of December 31, 2012, the estimated fair value of liability associated with the Bridge Loan was $1,528,500 which has been recorded and included in derivative financial instruments in the accompanying consolidated balance sheet. The change in the estimated fair value of the Bridge Loan from the dates of issuance through December 31, 2012 of approximately $509,500 has been recorded as an expense and classified in Loss on Financial Instruments in the accompanying consolidated statements of operations for the year ending December 31, 2012.
| F-15 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Net Loss Per Share
Basic loss per share is determined by dividing loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted loss per share is computed by dividing the loss attributable to common stockholders by the weighted-average number of common share equivalents outstanding for the period. The treasury stock method is used to determine the dilutive effect of the Company's outstanding stock warrants, unvested restricted stock and the if-converted method is used to determine the dilutive effect of convertible preferred stock and convertible debt. The following common stock equivalents were excluded in the calculation of diluted loss per share because their effect would be anti-dilutive:
| 2012 | 2011 | |||||||
| Series AA preferred stock | - | 1,507,666 | ||||||
| Series B preferred stock | - | 16,656,082 | ||||||
| Convertible debt | 136,799 | 281,690 | ||||||
| Restricted stock awards | 200,732 | 1,274,253 | ||||||
| Warrants | 662,212 | 547,505 | ||||||
Recent Accounting Pronouncements
In May 2011, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2011-04, Fair Value Measurement (“ASU 2011-04”), which contains amendments to achieve common fair value measurement and disclosures in U.S. GAAP and International Financial Reporting Standards. ASU 2011-04 explains how to measure fair value for financial reporting and requires certain disclosures related to fair value measurements. The guidance does not require fair value measurements in addition to those already required or permitted by other Topics. The provisions of ASU 2011-04 became effective January 1, 2012. The adoption of ASU 2011-04 did not have a material effect on the Company's consolidated results of operations, financial position or liquidity.
4. Agreements with Intracel
License Agreement
On October 10, 2007, the Company entered into an agreement (the “License Agreement”) with Intracel Holdings Corporation (“Intracel”), for the exclusive and indefinite rights to license and use the OncoVAX® technology platform. OncoVAX® is an active specific immunotherapy (“ASI”) that uses the patient’s own cancer cells to block the return of cancer following surgery. In exchange for the rights to OncoVAX®, the Company (i) agreed to issue equity securities equal to 10% of the fully diluted capitalization of the Company, (ii) assumed liabilities of Intracel to Organon Teknika Corporation (“Organon”) totaling $4.0 million under an October 31, 2007 Letter Agreement between Intracel and Organon, (iii) agreed to pay $450,000 in cash for settling trade payable related to the OncoVAX® intellectual property, and (iv) agreed to make royalty payments to Intracel based on future sales of OncoVAX®. The terms of the securities issued to Intracel provided Intracel with anti-dilution rights with respect to its 10% ownership interest (See Note 10). The License Agreement also contained a provision such that if the Company obtained specified levels of financing in a specified time period, that title to OncoVAX® would transfer to the Company without further consideration. If the Company did not reach the specified levels of financing in the specified period of time, Intracel could cancel the License Agreement and could re-purchase the rights to OncoVAX®. The Company did not obtain the necessary financing in the period specified.
| F-16 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Asset Transfer Agreement and Stock Exchange Agreement
As a result of the Company’s inability to raise the necessary capital under the License Agreement, the Company and Intracel negotiated amended terms to the License Agreement. On June 24, 2010, the Company and Intracel entered into the Asset Transfer Agreement pursuant to which the title to all of the intellectual property associated with OncoVAX® was transferred to the Company. Under the Asset Transfer Agreement, the Company also agreed to exchange the previously issued common stock and Series AA preferred stock representing a 10% interest in the Company for shares of its Series B preferred stock equal to a 20% interest in the Company on a fully diluted basis. The terms and conditions of the Series B preferred stock provided Intracel with anti-dilution rights with respect to its 20% ownership interest (See Note 10). In addition, the Company agreed that Intracel’s ownership position (and corresponding anti-dilution rights) would increase to 50% upon failure of the Company to meet certain defined milestones, which included but were not limited to, the Company attaining specified levels of additional financing. The Company did not meet these milestones and consequently, in December 2010, was required to increase Intracel’s total ownership interest in the Company to 50% through the issuance of additional shares of Series B preferred stock. As of December 31, 2012, Intracel directly owns approximately 40% of the Company on a fully diluted basis, and certain stockholders of Intracel own approximately 10% of the Company on a fully diluted basis. Intracel also continues to hold certain royalty rights associated with future commercial sales of vaccines developed using OncoVAX®.
5. Contingent Equity Lines of Credit
Initial Equity Line of Credit
On July 18, 2012, the Company entered into an Investment Agreement (“Initial Investment Agreement”) with Kodiak Capital Group, LLC (“Kodiak”). The Investment Agreement provides the Company an equity line whereby the Company can issue and sell to Kodiak, from time to time, shares of the Company’s common stock up to an aggregate purchase price of $1.0 million (the “Initial Kodiak Shares”) during the Initial Open Period (as defined below). Under the terms of the Investment Agreement, the Company has the right to deliver from time to time a written notice (the “Notice”) to Kodiak stating the dollar amount of Initial Kodiak Shares the Company intends to sell to Kodiak with the price per share based on the following formula: eighty percent (80%) of the lowest daily volume-weighted average price of the Company’s common stock during the period beginning on the date of the Notice and ending five (5) days thereafter. Under the Initial Investment Agreement, the Company may not deliver the Notice until the Company becomes quoted or listed on a Principal Market (as defined in the Initial Investment Agreement, which includes the Over-the-Counter Bulletin Board and the OTC Market Group’s OTC Link quotation system) (the “Effective Date”). Additionally, provided that the Investment Agreement does not terminate earlier, the Company has a twelve (12) month period, beginning on the trading day immediately following the Effective Date, during which it may deliver the Notice or Notices to Kodiak (the “Initial Open Period”). In addition, the Company cannot submit a new Notice until the closing of the previous Notice, and in no event shall Kodiak be entitled to purchase that number of Initial Kodiak Shares which when added to the sum of the number of shares of common stock already beneficially owned by Kodiak would exceed 9.99% of the number of shares of common stock outstanding on the applicable closing date.
| F-17 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The Initial Investment Agreement also provides that the Company shall not be entitled to deliver a Notice and Kodiak shall not be obligated to purchase any Initial Kodiak Shares unless each of the following conditions are satisfied: (i) at all times during the period beginning on the date of the Notice and ending on the date of the related closing, the Company’s common stock has been listed on the Principal Market and shall not have been suspended from trading thereon for a period of two (2) consecutive trading days during the Open Period; (ii) the Company has complied with its obligations and is otherwise not in breach of or in default under the Initial Investment Agreement, or any other agreement executed in connection therewith; (iii) no injunction has been issued and remains in force, and no action has been commenced by a governmental authority which has not been stayed or abandoned, prohibiting the purchase or the issuance of the Initial Kodiak Shares; and (iv) the issuance of the Shares will not violate any shareholder approval requirements of the market or exchange on which the Company’s common stock are principally listed.
The Investment Agreement will terminate when any of the following events occur: (i) Kodiak has purchased an aggregate of $1.0 million of the Company’s common stock, (ii) on the date which is twelve months (12) months following the effectiveness of the registration statement, or (iii) upon written notice from the Company to Kodiak. Similarly, the Initial Investment Agreement, may, at the option of the non-breaching party, terminate if Kodiak or the Company commits a material breach, or becomes insolvent or enters bankruptcy proceedings.
Subsequent Equity Line of Credit
On July 18, 2012, the Company entered into an Investment Agreement (“Subsequent Investment Agreement”) with Kodiak. The Subsequent Investment Agreement provides the Company an equity line (the “Subsequent Financing”) whereby the Company can issue and sell to Kodiak, from time to time, shares of the Company’s common stock up to an aggregate purchase price of $25.0 million (the “Subsequent Kodiak Shares”) during the Subsequent Open Period (as defined below). Under the terms of the Subsequent Investment Agreement, the Company has the right to deliver from time to time a Notice to Kodiak stating the dollar amount of Subsequent Kodiak Shares the Company intends to sell to Kodiak with the price per share based on the following formula: eighty percent (80%) of the lowest daily volume-weighted average price of the Company’s common stock during the period beginning on the date of the Notice and ending five (5) days thereafter. Under the Subsequent Investment Agreement, the Company may not deliver the Notice until after the resale of the Subsequent Kodiak Shares has been registered pursuant to a registration statement filed with the Securities and Exchange Commission. Additionally, provided that the Subsequent Investment Agreement does not terminate earlier, the Company has an eighteen (18) month period, beginning on the trading day immediately following the effectiveness of the registration statement, during which it may deliver the Notice or Notices to Kodiak (the “Subsequent Open Period”). In addition, the Company cannot submit a new Notice until the closing of the previous Notice, and in no event shall Kodiak be entitled to purchase that number of Subsequent Kodiak Shares which when added to the sum of the number of shares of common stock already beneficially owned by Kodiak would exceed 9.99% of the number of shares of common stock outstanding on the applicable closing date.
| F-18 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The Subsequent Investment Agreement also provides that the Company shall not be entitled to deliver a Notice and Kodiak shall not be obligated to purchase any Subsequent Kodiak Shares unless each of the following conditions are satisfied: (i) a registration statement has been declared effective and remains effective for the resale of the Subsequent Kodiak Shares until the closing with respect to the subject Notice; (ii) at all times during the period beginning on the date of the Notice and ending on the date of the related closing, the Company’s common stock has been listed on the Principal Market as defined in the Subsequent Investment Agreement (which includes, among others, the Over-the-Counter Bulletin Board and the OTC Market Group’s OTC Link quotation system) and shall not have been suspended from trading thereon for a period of two (2) consecutive trading days during the Open Period; (iii) the Company has complied with its obligations and is otherwise not in breach of or in default under the Subsequent Investment Agreement, the Registration Rights Agreement or any other agreement executed in connection therewith; (iv) no injunction has been issued and remains in force, and no action has been commenced by a governmental authority which has not been stayed or abandoned, prohibiting the purchase or the issuance of the Subsequent Kodiak Shares; and (v) the issuance of the Shares will not violate any shareholder approval requirements of the market or exchange on which the Company’s common stock are principally listed.
The Subsequent Investment Agreement will terminate when any of the following events occur: (i) Kodiak has purchased an aggregate of $25.0 million of the Company’s common stock, (ii) on the date which is eighteen months (18) months following the effectiveness of the registration statement, or (iii) upon written notice from the Company to Kodiak. Similarly, this Subsequent Investment Agreement, may, at the option of the non-breaching party, terminate if Kodiak or the Company commits a material breach, or becomes insolvent or enters bankruptcy proceedings.
6. Property and Equipment
Property and equipment consisted of the following:
| December 31, | 2012 | 2011 | ||||||
| Machinery and equipment | $ | 753,769 | $ | 724,757 | ||||
| Automobile | 3,965 | 10,877 | ||||||
| Furniture and fixtures | 6,690 | 8,283 | ||||||
| Computers and software | 1,921 | 5,076 | ||||||
| 766,345 | 748,993 | |||||||
| Less accumulated depreciation | (658,890 | ) | (586,377 | ) | ||||
| $ | 107,455 | $ | 162,616 | |||||
Depreciation expense was $109,817 and $95,656 for the years ended December 31, 2012 and 2011, respectively.
7. Intangible Assets
Intangible assets consist of the capitalized costs associated with the acquisition of patents related to OncoVAX® (the “Intellectual Property”) and the costs associated with website development and domain names.
As discussed in Note 4 to these consolidated financial statements, the total purchase price for the Intellectual Property was ultimately determined based upon the estimated fair value of the Series B preferred stock representing a 50% stock ownership in the Company, the value of cash payments made of $450,000 and, obligations of Intracel assumed of $4 million.
| F-19 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Intangible assets by major asset class were as follows at December 31, 2012:
| Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | ||||||||||
| Intellectual Property | $ | 84,481,856 | $ | 15,093,909 | $ | 69,387,947 | ||||||
| Other Intangible Assets | 121,944 | 81,060 | 40,884 | |||||||||
| $ | 84,603,800 | $ | 15,174,969 | $ | 69,428,831 | |||||||
Intangible assets by major asset class were as follows at December 31, 2011:
| Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | ||||||||||
| Intellectual Property | $ | 84,481,856 | $ | 8,395,379 | $ | 76,086,477 | ||||||
| Other Intangible Assets | 121,944 | 76,318 | 45,626 | |||||||||
| $ | 84,603,800 | $ | 8,471,697 | $ | 76,132,103 | |||||||
The amortization expense for intangible assets was approximately $6.7 million and $6.7 million for the years ended December 31, 2012 and 2011, respectively. The weighted average amortization period for intangible assets was 12.6 years.
The estimated future amortization relating to all intangible assets that are recorded in the consolidated balance sheets as of December 31, 2012 is as follows:
| Years ending December 31, | ||||
| 2013 | $ | 6,698,530 | ||
| 2014 | 6,698,530 | |||
| 2015 | 6,698,530 | |||
| 2016 | 6,698,530 | |||
| 2017 | 6,698,530 | |||
| Thereafter | 35,936,181 | |||
| $ | 69,428,831 | |||
8. Notes Payable
Notes payable are as follows:
| December 31, | 2012 | 2011 | ||||||
| Organon Obligation | $ | 3,500,000 | $ | 3,500,000 | ||||
| Abell Loan | 1,800,000 | 1,475,373 | ||||||
| $ | 5,300,000 | $ | 4,975,373 | |||||
| F-20 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Organon Obligation
Organon, currently owned by Merck & Co, Inc., manufactures a key component used with the OncoVAX® technology. In 2007, in conjunction with the Agreement with Intracel, the Company assumed $4.0 million of related liabilities from Intracel due to Organon (“Organon Obligation”). Of the $4.0 million due to Organon, $500,000 was paid at the time of the Agreement. The remaining $3.5 million was due in installments with an additional $500,000 (plus accrued interest) payable the first year but no later than one year after the agreement date of October 31, 2007. Organon may elect to receive this first $500,000 installment in stock. Commencing one year after the earlier of the first marketing approval of OncoVAX® by the United States Food and Drug Administration or the European Medicines Agency or October 31, 2007, Vaccinogen would make an annual payment of $1.0 million to Organon until repayment of the entire liability amount. The obligation accrued interest based on a simple annual interest rate based on the US prime lending rate, which was 3.25% as of December 31, 2012. Interest expense under this agreement was approximately $114,000 for the years ended December 31, 2012 and December 31, 2011, respectively. This obligation was secured by the OncoVAX® Intellectual Property. While the Company has not paid the installment due one year after the Agreement, no event of default has been declared by Organon or its successors including Merck & Co, Inc. Due to the right to declare an event of default and to accelerate all amounts owed on this obligation, all amounts owed under the Agreement have been classified as current in the accompanying consolidated balance sheets. If an event of default were declared, the Company would need to pay the principal payment of $500,000 plus accrued interest within 45 days in order to cure such default.
Abell Loan
On October 26, 2011, the Company obtained a $1.5 million working capital loan from The Abell Foundation Inc. (“Abell”). The loan (the “Abell Loan”) was originally due on April 26, 2012, with 8% simple interest accruing and payable on the maturity date. On February 16, 2012, Vaccinogen received an additional $300,000, thereby increasing the amount outstanding to $1.8 million. In January 2013, the maturity of the Abell Loan was extended to March 31, 2013. In April 2013, the borrowing arrangement was again amended to extend the maturity date to May 31, 2013, at which time all principal plus accrued interest is due in full. Payments of amounts due are required prior to the maturity date based on a percentage of proceeds received from the Company’s subsequent equity financing transactions, as outlined in the agreement. The Abell Loans are secured by all accounts, chattel paper, deposit accounts, equipment, general intangibles, instruments, inventory, investment property and letter of credit rights. The amendments to the Abell Loan were accounted for as modifications as defined under ASC Topic 470-50, Debt,
Modifications and Extinguishments
Under the terms of the loan, in the event of default, the interest rate would increase to 10% per annum. Interest expense under the Abell Loan was approximately $143,000 and $20,000 for the years ended December 31, 2012 and December 31, 2011, respectively.
As described in Note 3 to these consolidated financial statements, in connection with the Abell Loan and the various amendments, the Company issued the Abell Warrants which are exercisable into common stock of the Company. The number of shares into which the Abell Warrants are exercisable was revised with each amendment to the Abell Loan and is ultimately equal $1.1 million divided by 85% of the purchase price per share of stock sold in the Company’s next venture capital financing resulting in proceeds of not less than $35.0 million.
| F-21 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The fair value of the Abell Warrants issued in connection with the Abell Loan and subsequent amendments were recorded upon issuance as a debt discount based upon the estimated fair value and are being amortized as additional interest expense using the effective interest method.
9. Fair Value Measurements
Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction occurring in the most advantageous market. The Company determines fair value based on a hierarchy that priorities valuation techniques used to measure fair value based on observable and unobservable inputs. Observable inputs reflect market data obtained from independent sources. Unobservable inputs reflect assumptions based on the best information available.
The three levels of the fair value hierarchy are:
| Level 1 — | Inputs are quoted prices for identical assets or liabilities in an active market |
| Level 2 — | Inputs include quoted prices in active markets for similar assets and liabilities, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (interest rates and yield curves), and inputs that are derived principally from or corroborated by observable market data correlation or other means |
| Level 3 — | Inputs that are unobservable and significant to the fair value measurement |
The Company is required to record or disclose the fair value of certain assets and liabilities. The fair value guidance described above is used in measuring and recording the fair value of the liability associated with the Abell Warrants, and the fair value of the financial derivatives including the Round C Warrants and the Bridge Financing. This fair value guidance also applies to the disclosure of the fair value of financial instruments not otherwise recorded in the Company’s consolidated balance sheet at fair value.
The Company’s financial instruments measured on a recurring basis using fair value estimates are as follows:
| December 31, 2012 | ||||||||||||||||
| Description | Total | Level 1 | Level 2 | Level 3 | ||||||||||||
| Abell Warrants | $ | 831,806 | $ | - | $ | - | $ | 831,806 | ||||||||
| Round C Warrants | 230,349 | - | - | 230,349 | ||||||||||||
| Bridge Loan | 1,528,500 | - | - | 1,528,500 | ||||||||||||
| $ | 2,590,655 | $ | - | $ | - | $ | 2,590,655 | |||||||||
| December 31, 2011 | ||||||||||||||||
| Description | Total | Level 1 | Level 2 | Level 3 | ||||||||||||
| Abell Warrants | $ | 36,940 | $ | - | $ | - | $ | 36,940 | ||||||||
| $ | 36,940 | $ | - | $ | - | $ | 36,940 | |||||||||
| F-22 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The following is a reconciliation of level 3 fair value measurements from January 1, 2011 to December 31, 2012.
| Abell Warrants | Round C Warrants | Bridge Loan | ||||||||||
| Balance, January 1, 2011 | $ | - | $ | - | $ | - | ||||||
| Issuance of securities | 36,940 | - | - | |||||||||
| Balance, December 31, 2011 | 36,940 | - | - | |||||||||
| Issuance of securities | 22,450 | 230,349 | 1,019,000 | |||||||||
| Fair value change included in earnings | 772,416 | 509,500 | ||||||||||
| Balance, December 31, 2012 | $ | 831,806 | $ | 230,349 | $ | 1,528,500 | ||||||
Abell Warrants and Round C Warrants
The fair value of the Abell Warrants and the Round C Warrants are estimated at the end of each reporting period using an option pricing model. More specifically, the Black-Scholes option pricing model was utilized in the valuation of the Abell Warrants and a Monte Carlo simulation methodology was utilized in the valuation of the Round C Warrants. The following assumptions were used:
| Abell Warrants | Round C Warrants | |||||||||||||||
| 2012 | 2011 | 2012 | 2011 | |||||||||||||
| Volatility | 90 | % | 90 | % | 90% | - | ||||||||||
| Exercise price | $ | 4.68 | $ | 4.68 | $3.05-$6.96 | - | ||||||||||
| Stock price at December 31 | $ | 5.71 | $ | 1.60 | $5.71 | - | ||||||||||
| Risk free interest rate | 0.27 | % | 0.25 | % | 0.68- 0.72% | - | ||||||||||
| Dividend yield | 0 | % | 0 | % | 0% | - | ||||||||||
| Expected life | 9 | 10 | 4.8 - 5.0 | - | ||||||||||||
As described in Note 3 to the consolidated financial statements, the exercise price of the Abell Warrants is ultimately dependent upon the per share price and size of future rounds of equity financing. The Black-Scholes option pricing model was used to value the Abell Warrants as management believes that it can reasonably estimate the terms and conditions of future equity offerings that would impact the valuation of the Abell Warrants. Management’s ability to estimate these terms is based in part upon the terms and conditions of binding agreements to raise future equity capital in place at the time of each valuation.
As described in Note 3 to the consolidated financial statements, the Round C Warrants include a form of anti-dilution protection that may result in future adjustments to the terms of the warrants. A Monte Carlo simulation approach was used to value the Round C Warrants since the terms are subject to adjustment based on future issuances of the Company’s stock. This approach incorporates a range of simulated future stock prices to derive the range of potential exercise prices used as inputs to the model.
Because of the inherent subjectivity in the assumptions used to estimate the fair value of the Abell Warrants and the Round C Warrants, the Company considers the derived fair value to have been determined using Level 3 inputs.
| F-23 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Significant changes to the assumptions used in the Company’s model would result in changes in the fair value of the Abell Warrants and the Round C Warrants.
Bridge Loan
The estimated fair value of the Bridge Loan was determined based upon the present value of probability weighted cash flows, using assumptions about the timing and amount of future cash flows and discount rates that management considers to be appropriate in the circumstances. Because of the inherent subjectivity in management’s assumptions, the Company considers the derived fair value to have been determined using Level 3 inputs.
Significant changes to the assumptions used in the Company’s model would result in changes in the fair value of Bridge Loan.
Disclosure of the Fair Value of Financial Instruments
Cash and cash equivalents, accounts receivable, and accounts payable, are carried at amounts that approximate their fair values due to the short term nature of these financial instruments. The fair value of the Abell Loan approximates its carrying value due to the short term nature of the Abell Loan’s maturity. The fair value of the Organon Obligation approximates its carrying value as the note is due on demand.
10. Redeemable Preferred Stock and Stockholders’ Equity (Deficit)
As of January 1, 2011, the aggregate number of shares which the Company was authorized to issue was 75,000,000 shares of common stock with par value of $.0001 per share and 50,000,000 shares of preferred stock. The Company designated 15,000,000 shares of preferred stock as Series AA Convertible Redeemable Preferred Stock with a par value of $.0001 per share (“Series AA”) and 35,000,000 shares of preferred stock as Series B Convertible Redeemable Preferred Stock with a par value of $.0001 per share (“Series B”).
In August 2012, the Company amended and restated its Certificate of Incorporation to increase the number of shares authorized for issuance to 200,000,000 shares of common stock with a par value $.0001 and 50,000,000 shares of preferred stock with a par value of $.0001 per share.
Common Stock
On August 1, 2012, the Company issued 1,507,666 shares of common stock to the holders of the Series AA preferred stock in consideration for the conversion of all outstanding shares of Series AA preferred stock.
On August 1, 2012, the Company issued 16,656,082 shares of common stock to the former holders of our Series B Preferred Stock in consideration for the conversion of all outstanding shares of Series B preferred stock.
| F-24 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
During 2012, the Company issued 236,364 shares of common stock to Kodiak in exchange for their commitment to enter into the equity financing agreements described in Note 5 to these consolidated financial statements. The Company estimated the fair value of the common stock issued to be approximately $1.3 million and has recorded that value as a deferred cost. These costs will be offset against the anticipated proceeds of the equity financing agreements with Kodiak. The carrying value associated with these shares of common stock is included in long-term prepaid expenses in the accompanying consolidated balance sheet as of December 31, 2012. The amount will be written off if no proceeds are received or it is determined that financing will not be probable.
During 2012, the Company raised additional capital of $920,002 through the issuance of 167,273 shares of common stock and common stock warrants (Round C Warrants) to purchase 50,181 shares of common stock. $725,757 was allocated to the common stock and $194,245 was allocated to the common stock warrants. The common stock warrants were determined to be a derivative financial instrument. In addition, the Company satisfied a payable to a board member of the Company in the amount of $169,729 by issuing 30,860 shares of common stock and common stock warrants (Round C Warrants) exercisable into 9,258 shares of common stock. $133,624 was allocated to the common stock and $36,105 was allocated to the common stock warrants. The common stock warrants were determined to be a derivative financial instrument. The terms and conditions of the Round C Warrants are described in Note 3 to the consolidated financial statements.
Series AA Preferred Stock
In 2010, the Company created a class of preferred stock known as Series AA preferred stock and authorized 15,000,000 shares for issuance as Series AA preferred stock. All shares of Series AA were issued with an original purchase price of $9.0797 per share (“Series AA Original Issuance Price”). As noted above, all shares of Series AA were converted into common stock of the Company in 2012.
Dividends - Dividends on Series AA were cumulative and accrue at a rate of 7% of the Series AA Original Issuance Price, payable in cash or through the issuance of additional shares of Series AA when and if declared. If paid in stock, the shares to be issued are calculated by dividing the accrued dividend amount by the Series AA Original Issuance Price.
Conversion - Each share of Series AA was convertible at any time at the option of the holder or automatically upon a qualified public offering. The conversion ratio was one to one, subject to adjustment for specific dilutive events. In addition, the conversion ratio would be adjusted in the event the Company issues additional Series B shares, under the Mandatory Series B issuance described below, such that the Series AA in the aggregate could convert shares of Series AA into the same percentage of the outstanding common stock, on a fully-diluted and as-converted basis, as such holders of Series AA were entitled to convert immediately prior to the issuance of the Mandatory Series B Issuance. The conversion feature was determined to be clearly and closely related to the Series AA as that term is defined under Topic 815 and, as a result, is not required to be accounted for as a free standing derivative financial instrument. Management has also determined that the conversion feature does not represent a beneficial conversion feature as defined in ASC Topic 740-20, Debt, Beneficial Conversion (“Topic 470-20”).
| F-25 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Voting and Board Representation - The holders of Series AA generally were entitled to vote, together with the holders of Series B and common stock. Each preferred stockholder was entitled to the number of votes equal the number of shares of common stock into which each share of Series A were convertible at the time of such vote. The holders of Series AA were entitled to elect one member to the board of directors of the Company.
Redemption – Prior to the conversion into common stock in 2012, the Series AA were subject to redemption at the option of the holder beginning February 5, 2015 for the Series AA Original Issuance price plus accrued and unpaid dividends.
Liquidation Preference - The holders of Series AA had liquidation preference over the holders of Series B and Common Stock. In the event of liquidation, holders of Series AA would be entitled to receive, prior to any distributions to the holders of Series B or common stock, an amount per share equal to the greater of (i) the Series AA Original Issuance Price plus accrued and unpaid dividends or (ii) such amount per share payable had the shares of Series AA been converted into common stock prior to liquidation.
From January 13, 2011 to October 24, 2011, the Company issued 123,015 shares of Series AA Preferred Stock to 15 third party investors in a private offering at a price of $9.0797 per share resulting in net proceeds of approximately $1.1 million after approximately $14,000 in related stock issuance costs.
The activity related to Series AA Preferred Stock from January 1, 2011 through December 31, 2012 is as follows:
| Series AA Preferred Stock | ||||||||
| Shares | Amount | |||||||
| Balance, January 1, 2011 | $ | 790,346 | $ | 7,299,388 | ||||
| Issuance of Series AA preferred stock for cash, net of issuance costs | 123,015 | 1,102,448 | ||||||
| Accretion on preferred stock | - | 591,582 | ||||||
| Balance, December 31, 2011 | 913,361 | 8,993,418 | ||||||
| Accretion on preferred stock | - | 415,074 | ||||||
| Conversion of preferred stock to common stock | (913,361 | ) | (9,408,492 | ) | ||||
| Balance, December 31, 2012 | $ | - | $ | - | ||||
Series B Preferred Stock
In 2010, the Company created a class of preferred stock known as Series B preferred stock and authorized 35,000,000 shares for issuance as Series B Preferred Stock. All shares of Series B were issued pursuant to a Stock Exchange Agreement entered into concurrently with the June 2010 Asset Transfer Agreement with Intracel. As noted above, all shares of Series B were converted into common stock in 2012.
| F-26 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Dividends - Dividends on Series B were cumulative and accrue at a rate of 7% of $9.0797 per share, payable in cash or through the issuance of additional shares of Series B when and if declared. If paid in stock, the shares to be issued were calculated by dividing the accrued dividend amount by $9.0797.
Conversion - Each share of Series B was convertible at any time at the option of the holder or automatically upon a qualified public offering at a conversion ratio of one to one, subject to adjustment for specific dilutive events. The conversion feature was determined to be clearly and closely related to the Series B as that term is defined by Topic 815 and, as a result, is not required to be accounted for as a free standing derivative financial instrument. Management has also determined that the conversion feature does not represent a beneficial conversion feature as defined in Topic 470-20.
Issuance of Additional Series B - The holders of Series B had the right to receive additional shares of Series B in an amount necessary to cause the holders of Series B holders to maintain their equity ownership of the Company on a fully-diluted and as-converted following any anti-dilutive event. These anti-dilution rights are further explained below.
Voting and Board Representation - The holders of Series B generally were entitled to vote, together with the holders of Series B and common stock. Each preferred stockholder was entitled to the number of votes equal the number of shares of Common Stock into which each share of Series B were convertible at the time of such vote. The holders of Series B were entitled to elect 20% of the members to the board of directors of the Company, but not less than one member.
Redemption - Prior to conversion into common stock in 2012, the Series B were subject to redemption at the option of the holder beginning February 5, 2015 for the $9.0797 per share plus accrued and unpaid dividends.
Liquidation Preference - The holders of Series B had liquidation preference over the holders of common stock. In the event of liquidation, holders of Series B would be entitled to receive, after payment of the Series A liquidation preference but prior to any distributions to the holders of Common Stock, an amount per share equal to the greater of (i) the Series AA Original Issuance Price plus accrued and unpaid dividends or (ii) such amount per share payable had the shares of Series B been converted into common prior to liquidation.
Anti-dilution Rights - As discussed in Note 4 to these consolidated financial statements the Series B preferred stock issued to Intracel in 2010 requires the Company to maintain Intracel’s ownership interest in the Company at not less than 50% of the total outstanding equity ownership of the Company on a fully-diluted and as-converted basis. Pursuant to these provisions, rights to additional shares of Series B preferred stock in the amount of 1,972,919 shares in 2010, 233,620 shares in 2011; and 24,166 shares in 2012 accumulated in those periods, respectively. During 2012, all outstanding shares of Series B were converted to common shares. The conversion ratio was adjusted as a result of these provisions increasing the number of common shares issued by 2,230,705 shares. These anti-dilution rights expired upon conversion of the Series B preferred stock to common stock in 2012. The anti-dilution rights afforded to holders of the Series B were determined to be clearly and closely related to the Series B as that term is defined in Topic 815 and as a result these rights are not required to be accounted for as a free standing derivative financial instrument.
| F-27 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The subscription agreement for the Company’s most recent financing through the issuance of common stock (“Round C”) provides a form of anti-dilution protection to subscribers. Pursuant to the subscription agreement, if the market price of the Company’s common stock on the effective date of the Company’s initial public offering (“IPO Price”) is less than $5.50 per share, then the Company will issue to each subscriber additional shares of common stock. The number of shares issued would equal the difference between a) the number of shares that would have been issued if the price per unit was equal to the greater of the IPO Price or $5.00 and b) the number of shares originally issued to the subscriber. The anti-dilution rights are determined to be clearly and closely related to the Round C common stock as that term is defined in Topic 815 and as a result these rights are not required to be accounted for as a free standing derivative financial instrument.
The activity related to Series B Preferred Stock from January 1, 2011 through December 31, 2012 is as follows:
| Series B Preferred Stock | ||||||||
| Shares | Amount | |||||||
| Balance, January 1, 2011 | $ | 14,425,377 | $ | 83,059,104 | ||||
| Accretion on preferred stock | - | 27,076,008 | ||||||
| Balance, December 31, 2011 | 14,425,377 | 110,135,112 | ||||||
| Accretion on preferred stock | - | 16,103,077 | ||||||
| Conversion of preferred stock to common stock | (14,425,377 | ) | (126,238,189 | ) | ||||
| Balance, December 31, 2012 | $ | - | $ | - | ||||
11. Stock-Based Compensation
Restricted Stock
The Company from time to time has issued shares of restricted common shares to employees. From August 2010 through December 31, 2012, the Company issued 119,734 shares of restricted common stock to employees whose vesting is contingent upon a successful initial public stock offering with a 10 year contractual life. In 2007 and 2010 the Company issued 180,000 and 1.1 million shares, respectively of restricted common stock to employees whose vesting is dependent upon future employment. Those shares generally vest over periods of 4 to 5 years.
The Company records compensation expense for the award of restricted stock based upon the awards fair value determined as the based difference in the estimated fair value of the Company’s common stock and the price paid by the employee, if any, generally on the date of grant. The fair value of restricted stock awards is recognized as compensation expense over the service period which is generally the same as the vesting period. No compensation cost has been or will be recognized for the restricted stock awards whose vesting is contingent upon a successful initial public offering until such event is probable of occurring. As of December 31, 2012, management has determined that such an event is not yet probable of occurring. Compensation expense related to awards with service based vesting was zero and $569,458 during the years ended December 31, 2012 and December 31, 2011, respectively.
| F-28 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The following table summarizes activity related to restricted-stock awards for 2011 and 2012. The weighted average fair values for the awards below are based on the fair value at the grant date of the respective awards, which is equal to the value of the Company’s common stock on such date.
| 2012 | 2011 | |||||||||||||||
| Shares | Weighted Average Fair Value | Shares | Weighted Average Fair Value | |||||||||||||
| Balance, beginning of year | 985,190 | $ | 3.59 | 1,080,646 | $ | 6.62 | ||||||||||
| Granted | 14,544 | $ | 5.71 | 14,544 | $ | 1.60 | ||||||||||
| Vested | - | - | (110,000 | ) | $ | 3.65 | ||||||||||
| Forfeitures | (880,000 | ) | $ | 3.65 | - | - | ||||||||||
| Balance, end of year | 119,734 | $ | 3.42 | 985,190 | $ | 3.59 | ||||||||||
As of December 31, 2012, total unrecognized compensation costs related to nonvested restricted stock awards to purchase 119,734 shares of common stock was approximately $410,000, which will be recognized upon a successful initial public offering of the Company’s stock. The nonvested restricted stock awards have a weighted average remaining contractual term of 8.0 years. The total fair value of awards vested during the years ended December 31, 2012 and 2011 was approximately zero and $569,458, respectively.
In December 2010, the Company has committed to issue $462,500 of restricted stock, subject to restrictions yet to be defined, to an officer of the Company if and only if the Company completes a financing round that provides bona fide equity capital to the Company of at least $35.0 million.
On July 17, 2012, the Company’s board of directors approved the grant of 200,000 options to acquire Vaccinogen common stock to be awarded to existing employees. No options have been issued through December 31, 2012. On October 23, 2012, the Company committed to grant an option to purchase 20,000 shares of common stock to an employee if the Company’s stock begins trading on the “Over the Counter Bulletin Board”. The exercise price of the award will be equal to the Company’s stock closing price on the first day of trading on the “Over the Counter Bulletin Board” and the awards will vest over four years from that date.
Stock Purchase Warrants
From time to time the Company has issued stock purchase warrants to non-employees in exchange for services rendered. As of December 31, 2012, approximately 785,575 warrants were issued and outstanding with exercise prices ranging from $1.00 to $5.50. To date, all warrants have been issued for past services, with the exercise prices at least equal to the then estimated fair value of the underlying security, were fully vested upon issuance, and had contractual terms ranging from 2.5 to 7.5 years. During 2012, 80,000 fully vested warrants were issued and $312,735 of expense was recognized based on the fair value of the warrant measured on the date of grant. The fair value of the warrants was estimated on the date of grant using the Black-Scholes model assuming volatility of 90%, dividend yield of 0%, and a risk free interest rate of 0.27%.
| F-29 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The following table summarizes the warrant activity for 2012 and 2011.
| 2012 | 2011 | |||||||||||||||
| Shares | Weighted Average Fair Value | Shares | Weighted Average Fair Value | |||||||||||||
| Balance, beginning of year | 705,575 | $ | 1.33 | 705,575 | $ | 1.33 | ||||||||||
| Granted | 80,000 | $ | 2.87 | - | - | |||||||||||
| Balance, end of year | 785,575 | $ | 1.48 | 705,575 | $ | 1.33 | ||||||||||
The following table summarizes information on warrants outstanding as of December 31, 2012:
| Exercise price | Shares | Weighted Average Remaining Life | Weighted Average Exercise Price | |||||||||||
| $ | 1.00 | 705,575 | 2.06 | $ | 1.00 | |||||||||
| $ | 5.50 | 80,000 | 4.75 | $ | 5.50 | |||||||||
| Total | 785,575 | |||||||||||||
12. Commitments and Contingencies
Leases
The Company leases office space, a manufacturing facility, and equipment under operating leases expiring in 2013. In addition, the Company leases storage facilities on a month to month basis. Rent expense was approximately $147,000 and $134,000 for the years ended December 31, 2012 and December 31, 2011, respectively.
Minimum future rental payments under non-cancelable operating leases, including amendments to leases entered through the date the financial statements were available to be issued, total $123,247 for 2013.
Royalty Agreement with Intracel
Pursuant to the Agreement, the Company agreed to pay Intracel the following royalties on the Net Sales of Colon Cancer Products (as defined): (i) 3% of net sales on the first $350.0 million of Net Sales of Colon Cancer Products occurring in the calendar year; (ii) 4% of net sales of Net Sales of Colon Cancer Products occurring in the calendar year in excess of $350.0 million and up to and including $750.0 million and (iii) 5% of net sales of Net Sales of Colon Cancer Products occurring in the calendar year in excess of $750.0 million.
Royalty Agreement with Organon
The Company has agreed to pay Organon a royalty of 10% of the Net Sales of OncoVAX® (and all other TICE BCG related products, if any) until the Organon Obligation is paid in full, including interest, and 3% for 5 years thereafter.
| F-30 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
Litigation
The Company may be subject to certain claims arising in the ordinary course of business. The Company and a vendor are in dispute over amounts owed for services performed. A claim has been filed against the Company in the amount of approximately $150,000. Management believes the vendor did not perform under the terms of the contract and contends that no amounts are due to the vendor. Management has offered to settle the matter for $75,000 to be paid upon the Company’s successful initial public offering and has accrued $75,000 in the accompanying financial statements.
13. Related Party Transactions
Vaccinogen’s chief executive officer is a minority shareholder of Intracel and currently, holds less than 1% interest in Intracel.
On April 15, 2012, Intracel provided an unsecured note payable in the amount of $30,000. The note is unsecured, non-interest bearing, and becomes due on the date on which a minimum equity raise of $1.0 million occurs. Currently the note is in default. The carrying value of the note payable to Intracel is included in related party payable in the accompanying consolidated financial statements as of December 31, 2012.
In 2012, an executive of the Company loaned the Company $10,000. As of December 31, 2012, the balance due of $4,099 is past due and in default. The carrying value of this amount due to the Company executive is included in related party payable in the accompanying consolidated financial statements as of December 31, 2012.
As discussed in Note 10, on February 14, 2012, a member of the board of directors of the Company, agreed to loan the Company money to pay one of the Company’s vendors an outstanding amount of $169,729. The Company subsequently issued to this board member 30,860 shares of common stock and a warrant exercisable into 9,258 warrants (Round C Warrant).
| F-31 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
14. Income Taxes
The Company is subject to U.S. Federal, state and foreign income taxes. The provision/(benefit) for income taxes for the tax years ended December 31, 2012 and December 31, 2011 are as follows:
| December 31, | ||||||||
| 2012 | 2011 | |||||||
| Current: | ||||||||
| Federal | $ | - | $ | - | ||||
| State | - | - | ||||||
| Foreign | - | - | ||||||
| Total Current | - | - | ||||||
| Deferred: | ||||||||
| Federal | (3,567,528 | ) | (3,388,520 | ) | ||||
| State | (571,329 | ) | (542,662 | ) | ||||
| Foreign | (231,982 | ) | (307,371 | ) | ||||
| Total Deferred | (4,370,839 | ) | (4,238,553 | ) | ||||
| Valuation Allowance | 4,370,839 | 4,238,553 | ||||||
| Total Tax Provision/(Benefit) | $ | - | $ | - | ||||
The benefit for income taxes differed from the amount computed by applying the federal statutory income tax rate to loss before income taxes due to the following items for the years ended December 31, 2012 and 2011:
| December 31, | 2012 | 2011 | ||||||
| Tax benefit at statutory rates | $ | (4,370,839 | ) | $ | (4,238,553 | ) | ||
| Change in valuation allowance | 4,370,839 | 4,238,553 | ||||||
| Total Tax Provision/(Benefit) | $ | - | $ | - | ||||
The Company has approximately $3.5 million of net operating losses (“NOLs”) available for federal and state purposes as of December 31, 2012, which will expire between 2027 and 2032.
The Company has approximately $8.2 million of net operating loss (“NOL”) available for foreign purposes as of December 31, 2012, which will expire between 2016 and 2021.
Pursuant to Section 382 of the Internal Revenue Code (“IRC Sec. 382”), the Company believes that it may have undergone ownership changes in the past. As a result, the use of the Company’s net operating losses may be subject to an annual limitation under IRC Sec. 382.
| F-32 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
The temporary differences that arise between financial statement reporting and income tax basis carrying amounts of assets and liabilities give rise to deferred income taxes. The Company’s deferred income tax assets and liabilities as of December 31, 2012 and 2011 were as follows:
| December 31, | 2012 | 2011 | ||||||
| Total current deferred tax assets | $ | - | $ | - | ||||
| Long-term deferred tax assets: | ||||||||
| Start-up expenditures and amortization | 11,595,539 | 7,682,365 | ||||||
| U.S. net operating loss | 1,388,768 | 1,163,084 | ||||||
| Foreign net operating loss | 1,630,858 | 1,398,877 | ||||||
| Total long-term deferred income taxes: | 14,615,165 | 10,244,326 | ||||||
| Valuation allowance | (14,615,165 | ) | (10,244,326 | ) | ||||
| Total Current and Deferred Income Tax Assets/(Liabilities) | $ | - | $ | - | ||||
Management assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to utilize the existing deferred tax assets. A significant piece of the objective negative evidence evaluated was the cumulative loss incurred over the three year period ended December 31, 2012. Such objective evidence limits the ability to consider other subjective evidence such as our projection for future growth.
On the basis of this evaluation, as of December 31, 2012 and 2011, a valuation allowance of $14.6 million and $10.2 million has been recorded to record the portion of the deferred tax asset that is more likely than not to be realized. The amount of the deferred tax asset considered realizable, however, could be adjusted if estimates of future taxable income during the carryforward period are reduced or increased or if objective negative evidence in the form of cumulative losses is no longer present and additional weight may be given to subjective evidence such as our projections for growth.
The Company is subject to income taxes in the U.S. as well as certain state and foreign jurisdictions. Tax regulations within each jurisdiction are subject to interpretation and require significant judgment to apply.
Management has not identified any uncertain tax positions with the exception of income tax return filing penalties and accordingly has established a liability under ASC 740-10 (“FIN 48”). It is the Company’s accounting policy to account for ASC 740-10 related penalties and interest in other liabilities/expenses and not include it in the income tax provision of consolidated statement of operations. The Company has net operating loss carryovers therefore, the statutes of limitation, for U.S. purposes, remain open until the net operating losses are utilized. The Company is not currently under examination by any taxing authority.
The total unrecognized tax benefits as of December 31, 2012 and 2011 were $0, with penalties and interest accrued of $90,000 for both years. The Company does not expect its unrecognized tax benefits to significantly increase or decrease over the next 12 months.
| F-33 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
| December 31, | 2012 | 2011 | ||||||
| Balance at January 1, | $ | 90,000 | $ | 70,000 | ||||
| Penalties and interest | - | 20,000 | ||||||
| Balance at December 31, | $ | 90,000 | $ | 90,000 | ||||
The following table summarizes the open tax years for each major jurisdiction:
| Jurisdiction | Open Tax Years | |
| United States Federal | 2007-2012 | |
| Switzerland | 2008-2012 | |
| Netherlands | 2007-2012 |
The Company’s various tax positions could be challenged by the Internal Revenue Service (“IRS”) at such time the Company becomes profitable. The Company has treated the acquisition of the OncoVAX® technology as the acquisition of a portfolio of assets and not as a trade or business. The Company has taken the position that the purchase price paid in stock represents a taxable acquisition under the Internal Revenue Code (“IRC”) with a taxable basis in those assets equal to the fair market value of the stock paid. In the event the Company is challenged upon receiving a tax benefit, the Company could be subject to income tax understatement penalties.
15. Supplemental Disclosure of Cash Flow Information
For the years ended December 31, 2012 and December 31, 2011 the Company paid interest costs of $55,241 and $7,045, respectively.
During 2012, the Company issued 236,634 shares of common stock, valued at approximately $1.3 million, to Kodiak in satisfaction of the commitment fund for the equity line of credit.
During 2012, the Company issued 18,163,748 shares of common stock to former holders of preferred stock in consideration for the conversion of all outstanding shares of preferred stock.
During 2012, the Company satisfied a payable to a board member of the Company in the amount of $169,729 by issuing 30,860 shares of common stock and common stock warrants (Round C Warrants) exercisable into 9,258 shares of common stock.
16. Subsequent Events
Subsequent events were evaluated through April 22, 2013, the date the consolidated financial statements were available to be issued. In addition to events occurring after December 31, 2012 previously described herein, the following additional transactions occurred subsequent to December 31, 2012.
January 1, 2013 through April 22, 2013, the Company raised additional capital totaling approximately $0.97 million (net of issuance costs) through the issuance of 176,697 shares of Common Stock and warrants to purchase 53,008 shares of Common Stock.
| F-34 |
VACCINOGEN INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
On January 16, 2013, the Company entered into an investment agreement whereby at the request of the investor the Company shall issue and sell to the investor up to $5.0 million of common stock. The number of shares issued will be based on the lowest price paid by any purchaser of shares in the Company’s Venture Capital Financing (as defined in the agreement). The investor will not exercise this above option unless the Company has received $25.0 million dollars pursuant to the equity lines with Kodiak described in Note 5 and an additional $10.0 million from investors other than Kodiak.
On March 20, 2013 the Company entered into an Investment Banking Agreement with John Thomas Financial (JTF) for the purpose of introducing potential investors, acting as agent with respect to private placement of securities, assist in presentations to the Company’s Board of Directors and assist in the Company’s efforts to have its securities listed on a national stock exchange. Upon execution of the agreement an initial non-refundable retainer of twenty-five thousand dollars ($25,000) was paid. JTF will be compensated in cash in the amount of eight percent ( 8%) of gross cash proceeds and warrants to purchase a number of shares of the Company’s common stock equal to five percent (5%) of the shares of common stock issued under the agreement unless the gross proceeds exceed $7,500,000 when JTF will receive warrants to purchase a number of shares of the Company’s common stock equal to ten percent (10%) of the shares of common stock issued under the agreement.
In April 2013, the board of directors authorized the Company to offer the investors in the 2012 Bridge Loan, the option to convert the amount otherwise due and payable to them in the event of a successful qualified offering, or $2,038,000, into common stock of the Company, at a per share price equal to that provided in the Round C common stock offering, or $5.50 per share plus 30% warrant coverage. The bridge loan holder will have until May 3, 2013 or the close of the C round, whichever comes first. In order to accommodate all bridge loan holders, the total dollar value of common stock issuable in the Round C offering was increased from $11 million to $13 million.
| F-35 |
Vaccinogen, Inc. and Subsidiaries
Unaudited Condensed Consolidated Financial Statements
For the Three Months Ended March 31, 2013 and 2012
| F-36 |
| Vaccinogen, Inc. and Subsidiaries |
| Unaudited Condensed Consolidated Financial Statements |
| For the Three Months Ended March 31, 2013 and 2012 |
| F-37 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Contents |
| Unaudited Condensed Consolidated Financial Statements | ||
| Condensed Consolidated Balance Sheets as of March 31, 2013 (unaudited) and December 31, 2012 | F-40 | |
| Unaudited Condensed Consolidated Statements of Operations for the Three Months Ended March 31, 2013 and March 31, 2012 | F-41 | |
| Unaudited Condensed Consolidated Statements of Comprehensive Loss for the Three Months Ended March 31, 2013 and March 31, 2012 | F-42 | |
| Unaudited Condensed Consolidated Statement of Changes in Stockholders’ Equity for the Three Months Ended March 31, 2013 | F-43 | |
| Unaudited Condensed Consolidated Statements of Cash Flows for the Three Months Ended March 31, 2013 and March 31, 2012 | F-44 | |
| Notes to Unaudited Condensed Consolidated Financial Statements | F-45 - F-69 |
| F-38 |
| Unaudited Condensed Consolidated |
| Financial Statements |
| F-39 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Condensed Consolidated Balance Sheets |
| March 31, 2013 | December 31, 2012 | |||||||
| (Unaudited) | ||||||||
| Assets | ||||||||
| Current Assets | ||||||||
| Cash and cash equivalents | $ | 124,169 | $ | 113,840 | ||||
| Restricted cash | 40,933 | 42,044 | ||||||
| Inventory | 101,367 | 102,174 | ||||||
| Prepaid expenses and other current assets | 167,615 | 140,343 | ||||||
| Total Current Assets | 434,084 | 398,401 | ||||||
| Prepaid expenses | 1,301,463 | 1,301,463 | ||||||
| Property and equipment, net | 96,777 | 107,455 | ||||||
| Intangible assets, net | 67,753,014 | 69,428,831 | ||||||
| Total Assets | $ | 69,585,338 | $ | 71,236,150 | ||||
| Liabilities and Stockholders' Equity | ||||||||
| Current Liabilities | ||||||||
| Notes payable | $ | 5,300,000 | $ | 5,300,000 | ||||
| Accounts payable | 2,315,387 | 2,021,039 | ||||||
| Financial instruments | 9,128,119 | 2,590,655 | ||||||
| Accrued interest | 955,803 | 929,915 | ||||||
| Accrued compensation | 765,040 | 620,212 | ||||||
| Related party payable | 34,099 | 34,099 | ||||||
| Accrued expenses and other liabilities | 391,132 | 400,678 | ||||||
| Total Current Liabilities | 18,889,580 | 11,896,598 | ||||||
| Total Liabilities | 18,889,580 | 11,896,598 | ||||||
| Commitments and Contigencies | ||||||||
| Stockholders' Equity | ||||||||
| Preferred stock, $0.0001 par value: 50,000,000 shares authorized; 0 shares issued and outstanding | - | - | ||||||
| Common stock, $0.0001 par value; 200,000,000 and 200,000,000 shares authorized; 30,778,397 and 30,601,700 shares issued and outstanding at March 31, 2013 and December 31, 2012, respectively. | 3,078 | 3,060 | ||||||
| Additional paid-in capital | 138,921,218 | 138,118,424 | ||||||
| Accumulated other comprehensive loss | (47,689 | ) | (83,152 | ) | ||||
| Accumulated deficit | (88,180,849 | ) | (78,698,780 | ) | ||||
| Total Stockholders' Equity | 50,695,758 | 59,339,552 | ||||||
| Total Liabilities and Stockholders' Equity | $ | 69,585,338 | $ | 71,236,150 | ||||
See accompanying notes to unaudited condensed consolidated financial statements.
| F-40 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Unaudited Condensed Consolidated Statements of Operations |
| For the Three Months Ended March 31, | ||||||||
| 2013 | 2012 | |||||||
| Revenues | $ | - | $ | - | ||||
| Operating Expenses: | ||||||||
| Research and Development | 2,073,184 | 2,025,004 | ||||||
| General and administrative expenses | 6,929,760 | 653,572 | ||||||
| Total Operating Expenses | 9,002,944 | 2,678,576 | ||||||
| Loss From Operations | (9,002,944 | ) | (2,678,576 | ) | ||||
| Loss on Financial Instruments | (413,895 | ) | (92,368 | ) | ||||
| Interest Expense and Other Expenses | (65,230 | ) | (131,938 | ) | ||||
| Net Loss | $ | (9,482,069 | ) | $ | (2,902,882 | ) | ||
| Less: Accretion of preferred stock | - | (7,023,167 | ) | |||||
| Net loss available to common stockholders | $ | (9,482,069 | ) | $ | (9,926,049 | ) | ||
| Basic and diluted weighted average shares outstanding | 19,794,127 | 12,003,455 | ||||||
| Basic and diluted loss per common share | $ | (0.48 | ) | $ | (0.83 | ) | ||
See accompanying notes to unaudited condensed consolidated financial statements.
| F-41 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Unaudited Condensed Consolidated Statements of Comprehensive Loss |
| For the Three Months Ended March 31, | ||||||||
| 2013 | 2012 | |||||||
| Comprehensive Loss | ||||||||
| Net loss | $ | (9,482,069 | ) | $ | (2,902,882 | ) | ||
| Foreign currency translation adjustments | 35,463 | 28,198 | ||||||
| Total Comprehensive Loss | $ | (9,446,606 | ) | $ | (2,874,684 | ) | ||
See accompanying notes to unaudited condensed consolidated financial statements.
| F-42 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Unaudited Condensed Consolidated Statement of Changes in Stockholders’ Equity |
| For the Three Months Ended March 31, 2013 |
| Stockholders' Equity | ||||||||||||||||||||||||
| Accumulated Other | Total | |||||||||||||||||||||||
| Common Stock | Additional | Accumulated | Comprehensive | Stockholders' | ||||||||||||||||||||
| Shares | Amount | Paid-In Capital | Deficit | Loss | Equity | |||||||||||||||||||
| Balance, Janaury 1, 2013 | 30,601,700 | $ | 3,060 | $ | 138,118,424 | $ | (78,698,780 | ) | $ | (83,152 | ) | $ | 59,339,552 | |||||||||||
| Issuance of common stock for cash | 176,697 | 18 | 802,794 | - | - | 802,812 | ||||||||||||||||||
| Other comprehensive income | - | - | - | - | 35,463 | 35,463 | ||||||||||||||||||
| Net Loss | - | - | - | (9,482,069 | ) | - | (9,482,069 | ) | ||||||||||||||||
| Balance, March 31, 2013 | 30,778,397 | $ | 3,078 | $ | 138,921,218 | $ | (88,180,849 | ) | $ | (47,689 | ) | $ | 50,695,758 | |||||||||||
See accompanying notes to unaudited condensed consolidated financial statements.
| F-43 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Unaudited Condensed Consolidated Statements of Cash Flows |
| Three Months Ended March 31, | ||||||||
| 2013 | 2012 | |||||||
| Cash Flows From Operating Activities | ||||||||
| Net loss | $ | (9,482,069 | ) | $ | (2,902,882 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 7,356 | 10,763 | ||||||
| Amortization of intangible assets | 1,675,817 | 1,674,839 | ||||||
| Loss on financial instruments | 413,895 | 92,368 | ||||||
| Stock based compensation - non-employees | 5,954,545 | - | ||||||
| Non-cash interest expense | - | 68,825 | ||||||
| Changes in operating assets and liabilities, net: | ||||||||
| Changes in restricted cash | (165 | ) | (263 | ) | ||||
| Prepaid expenses and other assets | (28,294 | ) | 48,147 | |||||
| Accrued interest | 25,888 | 75,342 | ||||||
| Accounts payable and accrued expenses and other liabilities | 431,490 | 447,177 | ||||||
| Net Cash Used In Operating Activities | (1,001,537 | ) | (485,684 | ) | ||||
| Cash Flows From Investing Activities | ||||||||
| Purchases of property and equipment | (722 | ) | (57,823 | ) | ||||
| Net Cash Used In Investing Activities | (722 | ) | (57,823 | ) | ||||
| Cash Flows From Financing Activities | ||||||||
| Proceeds from notes payable | - | 300,000 | ||||||
| Proceeds from issuance of common stock and warrants | 971,836 | - | ||||||
| Net Cash Provided by Financing Activities | 971,836 | 300,000 | ||||||
| Impact of foreign currency translation on cash and cash equivalents | 40,752 | 7,648 | ||||||
| Net Increase (Decrease) in Cash and Cash Equivalents | 10,329 | (235,859 | ) | |||||
| Cash and Cash Equivalents, beginning of period | 113,840 | 236,681 | ||||||
| Cash and Cash Equivalents, end of period | $ | 124,169 | $ | 822 | ||||
See accompanying notes to unaudited condensed consolidated financial statements.
| F-44 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
1. Organization
The Business
Vaccinogen, Inc. (the “Company” or “Vaccinogen”), a biotechnology Company headquartered in Frederick, Maryland, was incorporated in the State of Delaware during 2007 for the purpose of developing therapies and vaccines to combat cancer by using the body’s own immune system. On November 23, 2010, the Company changed its domicile from Delaware to Maryland by means of a merger of the Company with and into its wholly owned subsidiary Vaccinogen I, Inc., a Maryland corporation.
On October 10, 2007, the Company entered into a license agreement with Intracel Holdings Corporation (“Intracel”), a related party, for the exclusive and indefinite rights to use the OncoVAX® technology platform. OncoVAX® is an active specific immunotherapy (“ASI”) that uses the patient’s own cancer cells to create a vaccine that in turn is used to block the return of cancer following surgery. In June 2010, the Company entered into an agreement with Intracel (the “Asset Transfer Agreement”) whereby the Company acquired title to the patents associated with the OncoVAX® (See Note 4). On October 23, 2007, Vaccinogen acquired out of bankruptcy, certain tangible assets that had been previously owned and used by Intracel’s wholly owned subsidiary in the Netherlands. These assets will be used to conduct research and development and in the commercialization of OncoVAX® to produce vaccines. In connection with the acquisition of these assets, the Company formed a wholly owned subsidiary, Vaccinogen BV, for the purposes of continuing development of OncoVAX®.
2. Going Concern
The accompanying unaudited condensed consolidated financial statements have been prepared assuming the Company will continue as a going concern. As of March 31, 2013, the Company had an accumulated deficit of approximately $88.2 million and negative working capital of approximately $18.5 million. Since inception, the Company has financed its activities principally from the proceeds from the issuance of equity and debt securities and loans from officers.
The Company’s ability to continue as a going concern is dependent upon the Company’s ability to raise additional debt and equity capital. As discussed in Note 5 to these unaudited condensed consolidated financial statements, the Company has entered into a series of agreements with the Kodiak Capital Group, LLC (“Kodiak”) to provide up to $26 million of additional equity capital. The proceeds from the agreements with Kodiak would primarily be used to continue the Company’s research and development activities including the furtherance of clinical trials using OncoVAX® to develop cancer related vaccines. However, Kodiak is not required to provide funding until certain conditions are met, including the registration and trading of the Company’s equity securities as defined in those agreements. There can be no assurance that the Company will meet the conditions under which Kodiak will be required to provide the equity capital or that the capital available under such agreements will be sufficient to allow the Company to funds its continuing research and development activities. If the Company is unable to raise the additional equity capital from Kodiak, the Company will need to seek alternative sources of debt or equity capital. There can be no assurance that such capital will be available in sufficient amounts or on terms acceptable to the Company. These factors raise substantial doubt about the Company's ability to continue as a going concern. The accompanying unaudited condensed consolidated financial statements do not include any adjustments relating to the recoverability of the recorded assets or the classification of liabilities that may be necessary should the Company be unable to continue as a going concern.
| F-45 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
3. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited condensed consolidated financial statements as of March 31, 2013 and for the three months ended March 31, 2013 and 2012, respectively include the accounts of Vaccinogen, Inc. and its wholly owned subsidiary and have been prepared in accordance with the rules and regulations of the Securities and Exchange Commission (“SEC”) and, therefore, omit or condense certain disclosures and other information required under generally accepted accounting principles in the United States of America (US GAAP) for complete financial statements. These unaudited condensed consolidated financial statements should therefore be read in conjunction with the audited consolidated financial statements and the accompanying notes for the year ended December 31, 2012.
In the opinion of management, the accompanying unaudited condensed consolidated financial statements reflect all the adjustments and reclassifications necessary for a fair presentation for the periods presented in accordance with US GAAP. The results for the three months ended March 31, 2013 are not necessarily indicative of the results to be expected for the full year.
Principles of Consolidation
The unaudited condensed consolidated financial statements include accounts of Vaccinogen and its wholly owned subsidiary, Vaccinogen BV (a company incorporated in the Netherlands). All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in its financial statements. On an ongoing basis, the Company evaluates the estimates used in recording common stock warrant related liabilities, derivative financial instruments, stock based compensation, and where applicable, the fair value of assets. The Company may base such estimates on various assumptions which it believes to be reasonable under the circumstances. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid securities with a maturity of three months or less at acquisition to be cash equivalents. Cash and cash equivalents include demand deposits with financial institutions and at times the amounts may exceed federally insured deposit limits. The Company has not experienced any losses and does not believe it is exposed to any significant credit risk related to demand deposits.
| F-46 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Restricted Cash
Restricted cash represents monies pledged by the Company’s foreign subsidiary for a lease obligation related to the manufacturing facility and to the Dutch government as required for companies with irradiator equipment.
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents. The Company maintains its cash and cash equivalents with high-credit-quality financial institutions in the United States.
Cash and cash equivalents are maintained at financial institutions and, at times, balances may exceed federally insured limits. The temporary federal program in effect from December 31, 2010 through December 31, 2012 that fully insured all non-interest bearing cash balances expired on December 31, 2012. Beginning 2013, insurance coverage will revert to $250,000 per depositor at each financial institution, and noninterest bearing cash balances may again exceed federally insured limits.
Inventory
Inventory is reported at the lower of cost or market value. The Company analyzes its inventory and writes down inventory that has become obsolete, or has a cost basis in excess of its expected net realizable value and inventory quantities in excess of expected requirements. Inventory primarily consists of a product used in creating vaccines using the OncoVAX® technology platform.
Property and Equipment
Property and equipment are recorded at cost and are depreciated or amortized over their estimated useful lives using the straight-line method. Estimated useful lives are as follows:
| Machinery and equipment | 3 – 5 years |
| Automobile | 3 – 5 years |
| Furniture and fixtures | 3 years |
| Computers and software | 3 years |
Maintenance and repairs are charged to expense as incurred. Major betterments and improvements, which extend the useful life of the underlying assets, are capitalized and depreciated.
Intangible Assets
Intangible assets consist primarily of the cost of acquired patents associated with OncoVAX® to be used in research and development and the commercialization of cancer related vaccines. The Company has capitalized the cost of the acquired patents because the Company has identified alternative future research and development efforts for numerous forms of cancer which it intends to pursue and which management believes will result in commercialization of related vaccines. Acquired patents are carried at cost less accumulated amortization. Amortization is calculated on a straight-line basis, over the estimated useful economic life of the patent, which is 15 years for OncoVAX®.
| F-47 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Impairment of Long-Lived Assets
Long-lived assets, including identifiable intangible assets with finite lives, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. The Company has determined that no impairment has occurred as of March 31, 2013.
Foreign Currency Translation
The financial statements of foreign subsidiaries are maintained in their functional currency, which generally is the local currency. The assets and liabilities are translated to U.S. dollars using the exchange rate in effect at the balance sheet date. Revenues, expenses and cash flows of these operations are translated using average exchange rates during the reporting period which they occur. The resulting translation adjustments are reflected in other comprehensive loss. As of March 31, 2013, the assets and net deficit of Vaccinogen BV, excluding intercompany balances, were approximately $252,000 and $314,000, respectively. As of December 31, 2012, the net assets and net deficit of Vaccinogen BV, excluding intercompany balances, were approximately $177,000 and 204,000, respectively. The net loss of Vaccinogen BV was approximately $387,000 and $273,000 for the quarters ended March 31, 2013 and 2012, respectively.
Revenue Recognition
To date, the Company has not earned any revenues as the use of OncoVAX® to create cancer related vaccines is still undergoing clinical trials and has not received regulatory approval for commercialization and sale.
Research and Development Expense
Research and development costs are expensed as incurred. Research and development expenses primarily include the amortization of intangible assets, cost of conducting clinical trials, compensation and related overhead for employees, consultants, facilities costs and the cost of materials purchased for research and development.
Stock-Based Compensation
The Company measures the cost of employee services received in exchange for stock options or restricted stock awards based upon the fair value of the award on the date of the grant. The Company recognizes the estimated grant date fair value of the award as stock-based compensation expense on a straight-line basis over the requisite service period, which is generally the vesting period.
The Company initially measures the cost of awards granted to non-employees based on the fair value of the award on the date of grant however such cost is re-measured at the end of each reporting period until performance is fully satisfied or services are rendered by the non-employee.
The fair value of stock options granted is calculated using the Black-Scholes option-pricing model, which requires the use of subjective assumptions including volatility, expected term, risk-free rate, and the fair value of the underlying common stock. The fair value of non-vested stock awards is determined based upon the estimated fair value of the Company's common stock.
| F-48 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Income Taxes
Deferred income tax assets and liabilities are determined based on differences between the financial statements and tax basis of assets and liabilities, as measured using the enacted tax rates, which are expected to be in effect when the differences reverse. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
The tax effects of uncertain tax positions are recognized in the financial statements only if the position is more likely than not to be sustained on audit, based on the technical merits of the position. For tax positions meeting the more likely than not threshold, the amount recognized in the consolidated financial statements is the largest benefit that had greater than 50% likelihood of being realized. Management has not identified any uncertain tax positions with the exception of income tax return filing penalties and accordingly has established a liability under Accounting Standards Codification (“ASC”) Topic 740-10 (“FIN 48”). It is the Company’s accounting policy to account for Topic 740-10 related penalties and interest in other liabilities/expenses and not include it in the income tax provision of consolidated statement of operations. The Company has identified its U.S. Federal income tax return and its state return in Maryland as its major tax jurisdictions. Tax returns for fiscal years 2007 and forward are still open for examination.
Financial Instruments
Stock Awards Accounted for as Liabilities
Abell Warrants
In October 2011, the Company entered into a borrowing arrangement with The Abell Foundation (“Abell”). In connection with that arrangement, the Company also issued warrants (the “Abell Warrants”) exercisable into common stock of the Company. In February 2012, the Company and Abell amended the agreement to provide for additional borrowings. In January 2013, the maturity of the Abell Loan was extended to March 31, 2013. In April 2013, the borrowing arrangement was again amended to extend the maturity date to May 31, 2013. On May 31, 2013, the borrowing arrangement was amended to extend the maturity date to July 31, 2013, at which time all principal plus accrued interest is due in full.
The number of shares issuable pursuant to the Abell Warrants was originally determined based upon a fixed amount of $500,000 divided by 85% of the per share price of stock sold in the next qualifying round of venture capital financing (defined as a round that raised at least $20 million). In connection with February 2012 amendment to the borrowing arrangement, the fixed amount used to determine the ultimate number of shares into which the Abell Warrants are exercisable was increased to $800,000. In connection with the January 2013 amendment to the borrowing arrangement, the fixed amount used to determine the ultimate number of shares into which the Abell Warrants are exercisable was increased to $1.1 million and the total proceeds of the next qualifying round of venture capital financing was increased to $35 million. The Abell Warrants have a contractual term of 10 years and were fully vested upon issuance.
| F-49 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
The Abell Warrants represent a fixed obligation that is to be settled through the issuance of a variable number of shares of the Company’s common stock. Consistent with the provisions of ASC Topic 480, Distinguishing Liabilities from Equity, the Company has concluded that the Abell Warrants should be accounted for as a liability. The Company is required to record the Abell Warrants at their estimated fair value at the end of each reporting period, with changes in the estimated fair value recorded in the unaudited condensed consolidated statements of operations as a component of other income (expense). As of March 31, 2013 and December 31, 2012, the estimated fair value of the Abell Warrants was $984,618 and $831,806, respectively. The change in the carrying value of the liability includes an increase of $275,813 related to the increase in the fixed dollar amount used to determine the number of warrants issuable associated with the January 2013 amendment, and a decrease in the estimated fair value of the liability of $123,001 from its carrying value as of December 31, 2012. The increase in fair value attributable to the amendment to the warrant agreement has been included in the determination of the loss resulting from the deemed extinguishment of the Abell Loan in January 2013 and has been included in Loss on Financial Instruments in the accompanying unaudited condensed consolidated statement of operations for the three months ended March 31, 2013. The decrease attributable to the change in the fair value was recorded as a gain and classified in Loss on Financial Instruments in the accompanying unaudited condensed consolidated statements of operations for the three months ended March 31, 2013. The change in fair value of the Abell Warrants from December 31, 2011 to March 31, 2012, was a loss of $92,368.
Abell Investment Option
On January 16, 2013, the Company entered into an investment agreement with the Abell under which Abell was granted an option to acquire up to $5.0 million of common stock of the Company (the “Abell Option”). The number of shares to be issued will be based on the lowest price paid by any purchaser of shares in a subsequent round of equity financing meeting certain conditions defined in the agreement. Abell cannot exercise its rights to purchase any stock unless the Company has received $25.0 million dollars pursuant to the equity lines with Kodiak described in Note 5 and an additional $10.0 million from investors other than Kodiak. The term of the agreement and Abell’s right to exercise is perpetual.
The Abell Option represents a fixed obligation that is to be settled through the issuance of a variable number of shares of the Company’s common stock. Consistent with the provisions of ASC Topic 480, the Company has concluded that the Abell Option should be accounted for as a liability and should be recorded as the conditions necessary to trigger the holders rights to exercise are considered by management to be probable of occurring as of March 31, 2013. The Company is required to record the Abell Option at its estimated fair value at the end of each reporting period, with changes in the estimated fair value recorded in the unaudited condensed consolidated statements of operations as a component of general and administrative expenses. As of March 31, 2013, the estimated value of the Abell Option is $5,954,545, which the Company has recorded as a liability. The Company has classified the carrying value of the Abell Option in Financial Instruments in the accompanying unaudited condensed consolidated balance sheet.
Derivative Financial Instruments
The Company may enter into transactions that represent free-standing or embedded derivative financial instruments as those terms are defined in ASC Topic 815 Derivatives and Hedging (“Topic 815”). The Company records the estimated fair value of derivative financial instruments in its consolidated balance sheets and records changes in the estimated fair value of derivative financial instruments as income or expense in its consolidated statements of operations.
| F-50 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Round C Warrants
From October 2012 through December 2012, and then again from January 2013 through March 2013, the Company issued warrants to certain investors in the common stock of the Company (the “Round C Warrants”). Round C Warrants to acquire 59,439 shares of common stock were issued in 2012 and Round C Warrants to acquire 53,008 shares of common stock were issued in the three month period ended March 31, 2013. The Round C Warrants have an exercise price of $6.05, a contractual term of 5 years and were fully vested upon issuance.
The terms of the Round C Warrants provide for "down-round" anti-dilution adjustments in certain situations whereby the Company sells or issues (a) stock at a price per share less than the exercise price of the Round C Warrants or (b) equity linked financial instruments with an exercise price less than the exercise price of the Round C Warrants. Consistent with the provisions of ASC Topic 815-40, the Round C Warrants are classified as derivative financial instruments. The Company is required to record the estimated fair value of derivative financial instruments at the end of each reporting period, with changes in the estimated fair value of such derivatives recorded in the consolidated statements of operations as a component of Loss on Financial Instruments. As of March 31, 2013 and December 31, 2012, the estimated fair value of the liability associated with the Round C Warrants was $354,756 and $230,349, respectively, and is included in derivative financial instruments in the accompanying unaudited condensed consolidated balance sheets. The change in the estimated fair value of the Round C Warrants liability for the three month period ended March 31, 2013 of $44,617 has been included as a gain and classified in Loss on Financial Instruments in the accompanying unaudited condensed consolidated statements of operations.
2012 Bridge Loan
Between April 2012 and October 2012, the Company entered into transactions with various investors which resulted in the Company raising $1,019,000 from the issuance of unsecured notes payable (collectively the “Bridge Loan”). The Bridge Loan has no contractual maturity date, and is repayable only in the event that the Company closes on a future round of equity financing which results in gross proceeds of at least $20 million. If the Company fails to raise sufficient additional capital, there is no obligation to pay interest or repay any amount borrowed under the Bridge Loan. Should the Company be successful in raising sufficient equity capital, the Company must repay an amount to the investors equal to 2 times the amount originally raised.
In April 2013, the board of directors authorized the Company to offer the investors in the 2012 Bridge Loan, the option to convert the amount otherwise due and payable to them in the event of a successful qualified offering, or $2,038,000, into common stock of the Company, at a per share price equal to that provided in the Round C common stock offering, or $5.50 per share plus 30% warrant coverage. The bridge loan holder will have until May 3, 2013 or the close of the C round, whichever comes first. In order to accommodate all bridge loan holders, the total dollar value of common stock issuable in the Round C offering was increased from $11 million to $13 million.
The Company has classified the Bridge Loan as a derivative financial instrument. As of March 31, 2013, and December 31, 2012, the estimated fair value of the liability associated with the Bridge Loan was $1,834,200 and $1,528,500 respectively, which has been recorded and included in Financial Instruments in the accompanying unaudited condensed consolidated balance sheets. The change in the estimated fair value of the Bridge Loan for the three months ended March 31, 2013 of $305,700 has been recorded as an expense and classified in Loss on Financial Instruments in the accompanying unaudited condensed consolidated statements of operations for the three months ended March 31, 2013.
| F-51 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Net Loss Per Share
Basic loss per share is determined by dividing loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted loss per share is computed by dividing the loss attributable to common stockholders by the weighted-average number of common share equivalents outstanding for the period. The treasury stock method is used to determine the dilutive effect of the Company's outstanding stock warrants, unvested restricted stock and the if-converted method is used to determine the dilutive effect of convertible preferred stock and convertible debt. The following common stock equivalents were excluded in the calculation of diluted loss per share because their effect would be anti-dilutive:
| For the three months ended March 31, | 2013 | 2012 | ||||||
| Series AA preferred stock | - | 1,507,666 | ||||||
| Series B preferred stock | - | 16,656,082 | ||||||
| Convertible debt | 91,324 | 312,500 | ||||||
| Restricted stock awards | 207,997 | 1,274,253 | ||||||
| Warrants | 610,467 | 264,591 | ||||||
| Abell Option | 223,122 | - | ||||||
4. Agreements with Intracel
License Agreement
On October 10, 2007, the Company entered into an agreement (the “License Agreement”) with Intracel Holdings Corporation (“Intracel”), for the exclusive and indefinite rights to license and use the OncoVAX® technology platform. OncoVAX® is an active specific immunotherapy (“ASI”) that uses the patient’s own cancer cells to block the return of cancer following surgery. In exchange for the rights to OncoVAX®, the Company (i) agreed to issue equity securities equal to 10% of the fully diluted capitalization of the Company, (ii) assumed liabilities of Intracel to Organon Teknika Corporation (“Organon”) totaling $4.0 million under an October 31, 2007 Letter Agreement between Intracel and Organon, (iii) agreed to pay $450,000 in cash for settling trade payable related to the OncoVAX® intellectual property, and (iv) agreed to make royalty payments to Intracel based on future sales of OncoVAX®. The terms of the securities issued to Intracel provided Intracel with anti-dilution rights with respect to its 10% ownership interest (See Note 10). The License Agreement also contained a provision such that if the Company obtained specified levels of financing in a specified time period, that title to OncoVAX® would transfer to the Company without further consideration. If the Company did not reach the specified levels of financing in the specified period of time, Intracel could cancel the License Agreement and could re-purchase the rights to OncoVAX®. The Company did not obtain the necessary financing in the period specified.
| F-52 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Asset Transfer Agreement and Stock Exchange Agreement
As a result of the Company’s inability to raise the necessary capital under the License Agreement, the Company and Intracel negotiated amended terms to the License Agreement. On June 24, 2010, the Company and Intracel entered into the Asset Transfer Agreement pursuant to which the title to all of the intellectual property associated with OncoVAX® was transferred to the Company. Under the Asset Transfer Agreement, the Company also agreed to exchange the previously issued common stock and Series AA preferred stock representing a 10% interest in the Company for shares of its Series B preferred stock equal to a 20% interest in the Company on a fully diluted basis. The terms and conditions of the Series B preferred stock provided Intracel with anti-dilution rights with respect to its 20% ownership interest (See Note 10). In addition, the Company agreed that Intracel’s ownership position (and corresponding anti-dilution rights) would increase to 50% upon failure of the Company to meet certain defined milestones, which included but were not limited to, the Company attaining specified levels of additional financing. The Company did not meet these milestones and consequently, in December 2010, was required to increase Intracel’s total ownership interest in the Company to 50% through the issuance of additional shares of Series B preferred stock. As of March 31, 2013, Intracel directly owns approximately 40% of the Company on a fully diluted basis, and certain stockholders of Intracel own approximately 10% of the Company on a fully diluted basis. Intracel also continues to hold certain royalty rights associated with future commercial sales of vaccines developed using OncoVAX®.
5. Contingent Equity Lines of Credit
Initial Equity Line of Credit
On July 18, 2012, the Company entered into an Investment Agreement (“Initial Investment Agreement”) with Kodiak Capital Group, LLC (“Kodiak”). The Investment Agreement provides the Company an equity line whereby the Company can issue and sell to Kodiak, from time to time, shares of the Company’s common stock up to an aggregate purchase price of $1.0 million (the “Initial Kodiak Shares”) during the Initial Open Period (as defined below). Under the terms of the Investment Agreement, the Company has the right to deliver from time to time a written notice (the “Notice”) to Kodiak stating the dollar amount of Initial Kodiak Shares the Company intends to sell to Kodiak with the price per share based on the following formula: eighty percent (80%) of the lowest daily volume-weighted average price of the Company’s common stock during the period beginning on the date of the Notice and ending five (5) days thereafter. Under the Initial Investment Agreement, the Company may not deliver the Notice until the Company becomes quoted or listed on a Principal Market (as defined in the Initial Investment Agreement, which includes the Over-the-Counter (“OTC”) Bulletin Board and the OTC Market Group’s OTC Link quotation system) (the “Effective Date”). Additionally, provided that the Investment Agreement does not terminate earlier, the Company has a twelve (12) month period, beginning on the trading day immediately following the Effective Date, during which it may deliver the Notice or Notices to Kodiak (the “Initial Open Period”). In addition, the Company cannot submit a new Notice until the closing of the previous Notice, and in no event shall Kodiak be entitled to purchase that number of Initial Kodiak Shares which when added to the sum of the number of shares of common stock already beneficially owned by Kodiak would exceed 9.99% of the number of shares of common stock outstanding on the applicable closing date.
| F-53 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
The Initial Investment Agreement also provides that the Company shall not be entitled to deliver a Notice and Kodiak shall not be obligated to purchase any Initial Kodiak Shares unless each of the following conditions are satisfied: (i) at all times during the period beginning on the date of the Notice and ending on the date of the related closing, the Company’s common stock has been listed on the Principal Market and shall not have been suspended from trading thereon for a period of two (2) consecutive trading days during the Open Period; (ii) the Company has complied with its obligations and is otherwise not in breach of or in default under the Initial Investment Agreement, or any other agreement executed in connection therewith; (iii) no injunction has been issued and remains in force, and no action has been commenced by a governmental authority which has not been stayed or abandoned, prohibiting the purchase or the issuance of the Initial Kodiak Shares; and (iv) the issuance of the Shares will not violate any shareholder approval requirements of the market or exchange on which the Company’s common stock are principally listed.
The Investment Agreement will terminate when any of the following events occur: (i) Kodiak has purchased an aggregate of $1.0 million of the Company’s common stock, (ii) on the date which is twelve months (12) months following the effectiveness of the registration statement, or (iii) upon written notice from the Company to Kodiak. Similarly, the Initial Investment Agreement, may, at the option of the non-breaching party, terminate if Kodiak or the Company commits a material breach, or becomes insolvent or enters bankruptcy proceedings.
Subsequent Equity Line of Credit
On July 18, 2012, the Company entered into an Investment Agreement (“Subsequent Investment Agreement”) with Kodiak. The Subsequent Investment Agreement provides the Company an equity line (the “Subsequent Financing”) whereby the Company can issue and sell to Kodiak, from time to time, shares of the Company’s common stock up to an aggregate purchase price of $25.0 million (the “Subsequent Kodiak Shares”) during the Subsequent Open Period (as defined below). Under the terms of the Subsequent Investment Agreement, the Company has the right to deliver from time to time a Notice to Kodiak stating the dollar amount of Subsequent Kodiak Shares the Company intends to sell to Kodiak with the price per share based on the following formula: eighty percent (80%) of the lowest daily volume-weighted average price of the Company’s common stock during the period beginning on the date of the Notice and ending five (5) days thereafter. Under the Subsequent Investment Agreement, the Company may not deliver the Notice until after the resale of the Subsequent Kodiak Shares has been registered pursuant to a registration statement filed with the Securities and Exchange Commission. Additionally, provided that the Subsequent Investment Agreement does not terminate earlier, the Company has an eighteen (18) month period, beginning on the trading day immediately following the effectiveness of the registration statement, during which it may deliver the Notice or Notices to Kodiak (the “Subsequent Open Period”). In addition, the Company cannot submit a new Notice until the closing of the previous Notice, and in no event shall Kodiak be entitled to purchase that number of Subsequent Kodiak Shares which when added to the sum of the number of shares of common stock already beneficially owned by Kodiak would exceed 9.99% of the number of shares of common stock outstanding on the applicable closing date.
| F-54 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
The Subsequent Investment Agreement also provides that the Company shall not be entitled to deliver a Notice and Kodiak shall not be obligated to purchase any Subsequent Kodiak Shares unless each of the following conditions are satisfied: (i) a registration statement has been declared effective and remains effective for the resale of the Subsequent Kodiak Shares until the closing with respect to the subject Notice; (ii) at all times during the period beginning on the date of the Notice and ending on the date of the related closing, the Company’s common stock has been listed on the Principal Market as defined in the Subsequent Investment Agreement (which includes, among others, the Over-the-Counter Bulletin Board and the OTC Market Group’s OTC Link quotation system) and shall not have been suspended from trading thereon for a period of two (2) consecutive trading days during the Open Period; (iii) the Company has complied with its obligations and is otherwise not in breach of or in default under the Subsequent Investment Agreement, the Registration Rights Agreement or any other agreement executed in connection therewith; (iv) no injunction has been issued and remains in force, and no action has been commenced by a governmental authority which has not been stayed or abandoned, prohibiting the purchase or the issuance of the Subsequent Kodiak Shares; and (v) the issuance of the Shares will not violate any shareholder approval requirements of the market or exchange on which the Company’s common stock are principally listed.
The Subsequent Investment Agreement will terminate when any of the following events occur: (i) Kodiak has purchased an aggregate of $25.0 million of the Company’s common stock, (ii) on the date which is eighteen months (18) months following the effectiveness of the registration statement, or (iii) upon written notice from the Company to Kodiak. Similarly, this Subsequent Investment Agreement, may, at the option of the non-breaching party, terminate if Kodiak or the Company commits a material breach, or becomes insolvent or enters bankruptcy proceedings.
6. Property and Equipment
Property and equipment consisted of the following:
| March 31, 2013 | December 31, 2012 | |||||||
| Machinery and equipment | $ | 742,728 | $ | 753,769 | ||||
| Automobile | 3,845 | 3,965 | ||||||
| Furniture and fixtures | 6,690 | 6,690 | ||||||
| Computers and software | 1,386 | 1,921 | ||||||
| 754,649 | 766,345 | |||||||
| Less accumulated depreciation | (657,872 | ) | (658,890 | ) | ||||
| $ | 96,777 | $ | 107,455 | |||||
Depreciation expense was $7,356 and $10,763 for the three month ended March 31, 2013 and 2012, respectively.
7. Intangible Assets
Intangible assets consist of the capitalized costs associated with the acquisition of patents related to OncoVAX® (the “Intellectual Property”) and the costs associated with website development and domain names.
As discussed in Note 4 to these unaudited condensed consolidated financial statements, the total purchase price for the Intellectual Property was ultimately determined based upon the estimated fair value of the Series B preferred stock representing a 50% stock ownership in the Company, the value of cash payments made of $450,000 and, obligations of Intracel assumed of $4 million. Intangible assets by major asset class were as follows at March 31, 2013:
| F-55 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
| Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | ||||||||||
| Intellectual Property | $ | 84,481,856 | $ | 16,768,540 | $ | 67,713,316 | ||||||
| Other Intangible Assets | 121,944 | 82,246 | 39,698 | |||||||||
| $ | 84,603,800 | $ | 16,850,786 | $ | 67,753,014 | |||||||
Intangible assets by major asset class were as follows at December 31, 2012:
| Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | ||||||||||
| Intellectual Property | $ | 84,481,856 | $ | 15,093,909 | $ | 69,387,947 | ||||||
| Other Intangible Assets | 121,944 | 81,060 | 40,884 | |||||||||
| $ | 84,603,800 | $ | 15,174,969 | $ | 69,428,831 | |||||||
The amortization expense for intangible assets was approximately $1.7 million for both the three months ended March 31, 2013 and 2012, respectively. The weighted average amortization period for intangible assets was 12.3 years.
The estimated future amortization relating to all intangible assets that are recorded in the unaudited condensed consolidated balance sheets as of March 31, 2013 is as follows:
| Years ending December 31, | ||||
| 2013 | $ | 5,022,713 | ||
| 2014 | 6,698,530 | |||
| 2015 | 6,698,530 | |||
| 2016 | 6,698,530 | |||
| 2017 | 6,698,530 | |||
| Thereafter | 35,936,181 | |||
| $ | 67,753,014 | |||
| F-56 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
8. Notes Payable
Notes payable are as follows:
| March 31, 2013 | December 31, 2012 | |||||||
| Organon Obligation | $ | 3,500,000 | $ | 3,500,000 | ||||
| Abell Loan | 1,800,000 | 1,800,000 | ||||||
| $ | 5,300,000 | $ | 5,300,000 | |||||
Organon Obligation
Organon, currently owned by Merck & Co, Inc., manufactures a key component used with the OncoVAX® technology. In 2007, in conjunction with the Agreement with Intracel, the Company assumed $4.0 million of related liabilities from Intracel due to Organon (“Organon Obligation”). Of the $4.0 million due to Organon, $500,000 was paid at the time of the Agreement. The remaining $3.5 million was due in installments with an additional $500,000 (plus accrued interest) payable the first year but no later than one year after the agreement date of October 31, 2007. Organon may elect to receive this first $500,000 installment in stock. Commencing one year after the earlier of the first marketing approval of OncoVAX® by the United States Food and Drug Administration or the European Medicines Agency or October 31, 2007, Vaccinogen would make an annual payment of $1.0 million to Organon until repayment of the entire liability amount. The obligation accrued interest based on a simple annual interest rate based on the US prime lending rate, which was 3.25% as of March 31, 2013. Interest expense related to this agreement was $28,438 and $28,438 for the three months ended March 31, 2013 and 2012, respectively. This obligation was secured by the OncoVAX® Intellectual Property. While the Company has not paid the installment due one year after the Agreement, no event of default has been declared by Organon or its successors including Merck & Co, Inc. Due to the right to declare an event of default and to accelerate all amounts owed on this obligation, all amounts owed under the Agreement have been classified as current in the accompanying unaudited condensed consolidated balance sheets. If an event of default were declared, the Company would need to pay the principal payment of $500,000 plus accrued interest within 45 days in order to cure such default.
Abell Loan
On October 26, 2011, the Company obtained a $1.5 million working capital loan from The Abell Foundation Inc. (“Abell”). The loan (the “Abell Loan”) was originally due on April 26, 2012, with 8% simple interest accruing and payable on the maturity date. On February 16, 2012, Vaccinogen received an additional $300,000, thereby increasing the amount outstanding to $1.8 million. In January 2013, the maturity of the Abell Loan was extended to March 31, 2013. In April 2013, the borrowing arrangement was again amended to extend the maturity date to May 31, 2013. On May 31, 2013, the borrowing arrangement was amended to extend the maturity date to July 31, 2013, at which time all principal plus accrued interest is due in full. . The 2012 amendment to the Abell Loan was accounted for as a modification and the January 2013 amendment was accounted for as an extinguishment as those terms are defined under ASC Topic 470-50, Debt, Modifications and Extinguishments.
| F-57 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Payments of amounts due are required prior to the maturity date based on a percentage of proceeds received from the Company’s subsequent equity financing transactions, as outlined in the agreement. The Abell Loans are secured by all accounts, chattel paper, deposit accounts, equipment, general intangibles, instruments, inventory, investment property and letter of credit rights.
Under the terms of the loan, in the event of default, the interest rate increases to 10% per annum. Interest expense under the Abell Loan was $36,000 and $35,308 for the three months ended March 31, 2013 and 2012, respectively.
As described in Note 3 to these unaudited condensed consolidated financial statements, in connection with the Abell Loan and the various amendments, the Company issued the Abell Warrants which are exercisable into common stock of the Company. The number of shares into which the Abell Warrants are exercisable was revised with each amendment to the Abell Loan and is ultimately equal $1.1 million divided by 85% of the purchase price per share of stock sold in the Company’s next venture capital financing resulting in proceeds of not less than $35.0 million.
The fair value of the Abell Warrants issued in connection with the Abell Loan in 2011 and subsequent amendment in February 2012 were recorded upon issuance as a debt discount based upon the estimated fair value and were amortized as additional interest expense through the original maturity date. The Company recorded $68,825 of additional interest expense related to the amortization of debt discount for the three months ended March 31, 2012. The fair value of the Abell Warrants of $275,813 issued in connection with the January 2013 amendment were included in the determination of the loss associated with the deemed extinguishment of the Abell Loan at that time. That loss has been classified within Loss on Financial instruments in the accompanying unaudited condensed consolidated statements of operations for the three months ended March 31, 2013.
9. Fair Value Measurements
Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction occurring in the most advantageous market. The Company determines fair value based on a hierarchy that priorities valuation techniques used to measure fair value based on observable and unobservable inputs. Observable inputs reflect market data obtained from independent sources. Unobservable inputs reflect assumptions based on the best information available.
The three levels of the fair value hierarchy are:
| Level 1 — | Inputs are quoted prices for identical assets or liabilities in an active market |
| Level 2 — | Inputs include quoted prices in active markets for similar assets and liabilities, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (interest rates and yield curves), and inputs that are derived principally from or corroborated by observable market data correlation or other means |
| Level 3 — | Inputs that are unobservable and significant to the fair value measurement |
| F-58 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
The Company is required to record or disclose the fair value of certain assets and liabilities. The fair value guidance described above is used in measuring and recording the fair value of the liability associated with the Abell Warrants, and the fair value of the financial derivatives including the Round C Warrants and the Bridge Financing. This fair value guidance also applies to the disclosure of the fair value of financial instruments not otherwise recorded in the Company’s consolidated balance sheet at fair value.
The Company’s financial instruments measured on a recurring basis using fair value estimates are as follows:
| March 31, 2013 | ||||||||||||||||
| Description | Total | Level 1 | Level 2 | Level 3 | ||||||||||||
| Abell Warrants | $ | 984,618 | $ | - | $ | - | $ | 984,618 | ||||||||
| Abell Option | 5,954,545 | 5,954,545 | ||||||||||||||
| Round C Warrants | 354,756 | - | - | 354,756 | ||||||||||||
| Bridge Loan | 1,834,200 | - | - | 1,834,200 | ||||||||||||
| $ | 9,128,119 | $ | - | $ | - | $ | 9,128,119 | |||||||||
| December 31, 2012 | ||||||||||||||||
| Description | Total | Level 1 | Level 2 | Level 3 | ||||||||||||
| Abell Warrants | $ | 831,806 | $ | - | $ | - | $ | 831,806 | ||||||||
| Round C Warrants | 230,349 | - | - | 230,349 | ||||||||||||
| Bridge Loan | 1,528,500 | - | - | 1,528,500 | ||||||||||||
| $ | 2,590,655 | $ | - | $ | - | $ | 2,590,655 | |||||||||
The following is a reconciliation of level 3 fair value measurements for the three months ended March 31, 2013 and 2012, respectively.
| Abell Warrants | Abell Options | Round C Warrants | Bridge Loan | |||||||||||||
| Balance, December 31, 2012 | $ | 831,806 | $ | - | $ | 230,349 | $ | 1,528,500 | ||||||||
| Issuance of securities | 275,813 | 5,954,545 | 169,024 | - | ||||||||||||
| Fair value change included in earnings | (123,001 | ) | - | (44,617 | ) | 305,700 | ||||||||||
| Balance, March 31, 2013 | $ | 984,618 | $ | 5,954,545 | $ | 354,756 | $ | 1,834,200 | ||||||||
| Abell Warrants | Round C Warrants | Bridge Loan | ||||||||||
| Balance, December 31, 2011 | $ | 36,940 | $ | - | $ | - | ||||||
| Issuance of securities | 79,022 | - | - | |||||||||
| Fair value change included in earnings | 92,368 | - | - | |||||||||
| Balance, March 31, 2012 | $ | 208,330 | $ | - | $ | - | ||||||
| F-59 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Abell Warrants and Round C Warrants
The fair value of the Abell Warrants and the Round C Warrants are estimated at the end of each reporting period using an option pricing model. More specifically, the Black-Scholes option pricing model was utilized in the valuation of the Abell Warrants and a Monte Carlo simulation methodology was utilized in the valuation of the Round C Warrants. The following assumptions were used to estimate the fair value of the warrants as of March 31, 2013 and December 31, 2012:
| Abell Warrants | Round C Warrants | |||||||
| March 31, 2013 | December 31, 2012 | March 31, 2013 | December 31, 2012 | |||||
| Volatility | 80% | 90% | 90% | 90% | ||||
| Exercise price | $4.68 | $4.68 | $6.05 | $3.05-$6.96 | ||||
| Stock price | $5.42 | $5.71 | $5.42 | 5.71 | ||||
| Risk free interest rate | 1.58-1.83% | 0.27% | 0.66-0.73% | 0.68-.072% | ||||
| Dividend yield | 0% | 0% | 0% | 0% | ||||
| Expected life | 8.6-9.8 | 9.0 | 4.6-5.0 | 4.8-5.0 | ||||
As described in Note 3 to these unaudited condensed consolidated financial statements, the exercise price of the Abell Warrants is ultimately dependent upon the per share price and size of future rounds of equity financing. The Black-Scholes option pricing model was used to value the Abell Warrants as management believes that it can reasonably estimate the terms and conditions of future equity offerings that would impact the valuation of the Abell Warrants. Management’s ability to estimate these terms is based in part upon the terms and conditions of binding agreements to raise future equity capital in place at the time of each valuation.
As described in Note 3 to these unaudited condensed consolidated financial statements, the Round C Warrants include a form of anti-dilution protection that may result in future adjustments to the terms of the warrants. A Monte Carlo simulation approach was used to value the Round C Warrants since the terms are subject to adjustment based on future issuances of the Company’s stock. This approach incorporates a range of simulated future stock prices to derive the range of potential exercise prices used as inputs to the model.
Because of the inherent subjectivity in the assumptions used to estimate the fair value of the Abell Warrants and the Round C Warrants, the Company considers the derived fair value to have been determined using Level 3 inputs.
Significant changes to the assumptions used in the Company’s model would result in changes in the fair value of the Abell Warrants and the Round C Warrants.
Abell Option
As described in Note 3 to these unaudited condensed consolidated financial statements, the number of shares issuable under the Abell Option is dependent upon the lowest price paid for shares in a future qualified round of equity financing. Management has valued the Abell Option based upon an estimate of the fair value of the Company’s underlying stock because management believes it can reasonably estimate the occurrence and the terms of the future equity offering necessary to trigger Abell’s right to exercise the option and establish an exercise price, and because the right to exercise the option has no expiration.
| F-60 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Because of the inherent subjectivity in the assumptions used to estimate the fair value of the Company’s common stock, the Company considers the derived fair value to have been determined using Level 3 inputs.
Significant changes to the assumptions used in the Company’s model would result in changes in the fair value of the Abell Option.
Bridge Loan
The estimated fair value of the Bridge Loan was determined based upon the present value of probability weighted cash flows, using assumptions about the timing and amount of future cash flows and discount rates that management considers to be appropriate in the circumstances. Because of the inherent subjectivity in management’s assumptions, the Company considers the derived fair value to have been determined using Level 3 inputs.
Significant changes to the assumptions used in the Company’s model would result in changes in the fair value of Bridge Loan.
Disclosure of the Fair Value of Financial Instruments
Cash and cash equivalents, accounts receivable, and accounts payable, are carried at amounts that approximate their fair values due to the short term nature of these financial instruments. The fair value of the Abell Loan approximates its carrying value due to the short term nature of the Abell Loan’s maturity. The fair value of the Organon Obligation approximates its carrying value as the note is due on demand.
10. Redeemable Preferred Stock and Stockholders’ Equity
As of January 1, 2011, the aggregate number of shares which the Company was authorized to issue was 75,000,000 shares of common stock with par value of $.0001 per share and 50,000,000 shares of preferred stock. The Company designated 15,000,000 shares of preferred stock as Series AA Convertible Redeemable Preferred Stock with a par value of $.0001 per share (“Series AA”) and 35,000,000 shares of preferred stock as Series B Convertible Redeemable Preferred Stock with a par value of $.0001 per share (“Series B”).
In August 2012, the Company amended and restated its Certificate of Incorporation to increase the number of shares authorized for issuance to 200,000,000 shares of common stock with a par value $.0001 and 50,000,000 shares of preferred stock with a par value of $.0001 per share.
Common Stock
On August 1, 2012, the Company issued 1,507,666 shares of common stock to the holders of the Series AA preferred stock in consideration for the conversion of all outstanding shares of Series AA preferred stock.
On August 1, 2012, the Company issued 16,656,082 shares of common stock to the former holders of our Series B Preferred Stock in consideration for the conversion of all outstanding shares of Series B preferred stock.
| F-61 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
In August 2012, the Company issued 236,364 shares of common stock to Kodiak in exchange for their commitment to enter into the equity financing agreements described in Note 5 to these unaudited condensed consolidated financial statements. The Company estimated the fair value of the common stock issued to be approximately $1.3 million and has recorded that value as a deferred cost. These costs will be offset against the anticipated proceeds of the equity financing agreements with Kodiak. The carrying value associated with these shares of common stock is included in long-term prepaid expenses in the accompanying unaudited condensed consolidated balance sheet as of December 31, 2012. The amount will be written off if no proceeds are received or it is determined that financing will not be probable.
From October 2012 through December 2012, the Company raised additional capital totaling approximately $920,002 through the issuance of 167,273 shares of common stock (Round C Stock) and common stock warrants (Round C Warrants) to purchase 50,181 shares of common stock. The Company allocated $725,757 of the total proceeds to the common stock and $194,245 of the total proceeds to the common stock warrants.
From January 1, 2013 through March 31, 2013, the Company raised additional capital totaling approximately $972,000 (net of issuance costs) from the issuance of 176,697 shares of Round C Stock and additional Round C Warrants to purchase 53,008 shares of common stock. The Company allocated $802,812 of the total 2013 proceeds to the common stock and $169,024 of the total proceeds to the common stock warrants.
In addition, in December of 2012, the Company satisfied a payable to a board member of the Company in the amount of $169,729 by issuing 30,860 shares of Round C Stock and Round C Warrants exercisable into 9,258 shares of common stock. The Company allocated $133,624 to the common stock and $36,105 of the total proceeds to the common stock warrants.
The Round C Warrants described above are derivative financial instruments. The terms and conditions of the Round C Warrants are described in Note 3 to the consolidated financial statements.
On March 20, 2013 the Company entered into an Investment Banking Agreement with John Thomas Financial (“JTF”) for the purpose of introducing potential investors, acting as agent with respect to private placement of securities, assist in presentations to the Company’s Board of Directors and assist in the Company’s efforts to have its securities listed on a national stock exchange. Upon execution of the agreement an initial non-refundable retainer of twenty-five thousand dollars ($25,000) was paid. JTF will be compensated in cash in the amount of eight percent (8%) of gross cash proceeds and warrants to purchase a number of shares of the Company’s common stock equal to five percent (5%) of the shares of common stock issued under the agreement unless the gross proceeds exceed $7,500,000 when JTF will receive warrants to purchase a number of shares of the Company’s common stock equal to ten percent (10%) of the shares of common stock issued under the agreement.
Series AA Preferred Stock
In 2010, the Company created a class of preferred stock known as Series AA preferred stock and authorized 15,000,000 shares for issuance as Series AA preferred stock. All shares of Series AA were issued with an original purchase price of $9.0797 per share (“Series AA Original Issuance Price”). As noted above, all shares of Series AA were converted into common stock of the Company in 2012.
| F-62 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Dividends - Dividends on Series AA were cumulative and accrue at a rate of 7% of the Series AA Original Issuance Price, payable in cash or through the issuance of additional shares of Series AA when and if declared. If paid in stock, the shares to be issued are calculated by dividing the accrued dividend amount by the Series AA Original Issuance Price.
Conversion - Each share of Series AA was convertible at any time at the option of the holder or automatically upon a qualified public offering. The conversion ratio was one to one, subject to adjustment for specific dilutive events. In addition, the conversion ratio would be adjusted in the event the Company issues additional Series B shares, under the Mandatory Series B issuance described below, such that the Series AA in the aggregate could convert shares of Series AA into the same percentage of the outstanding common stock, on a fully-diluted and as-converted basis, as such holders of Series AA were entitled to convert immediately prior to the issuance of the Mandatory Series B Issuance. The conversion feature was determined to be clearly and closely related to the Series AA as that term is defined under Topic 815 and, as a result, is not required to be accounted for as a free standing derivative financial instrument. Management has also determined that the conversion feature does not represent a beneficial conversion feature as defined in ASC Topic 740-20, Debt, Beneficial Conversion (“Topic 470-20”).
Voting and Board Representation - The holders of Series AA generally were entitled to vote, together with the holders of Series B and common stock. Each preferred stockholder was entitled to the number of votes equal the number of shares of common stock into which each share of Series A were convertible at the time of such vote. The holders of Series AA were entitled to elect one member to the board of directors of the Company.
Redemption – Prior to the conversion into common stock in 2012, the Series AA were subject to redemption at the option of the holder beginning February 5, 2015 for the Series AA Original Issuance price plus accrued and unpaid dividends.
Liquidation Preference - The holders of Series AA had liquidation preference over the holders of Series B and Common Stock. In the event of liquidation, holders of Series AA would be entitled to receive, prior to any distributions to the holders of Series B or common stock, an amount per share equal to the greater of (i) the Series AA Original Issuance Price plus accrued and unpaid dividends or (ii) such amount per share payable had the shares of Series AA been converted into common stock prior to liquidation.
From January 13, 2011 to October 24, 2011, the Company issued 123,015 shares of Series AA Preferred Stock to 15 third party investors in a private offering at a price of $9.0797 per share resulting in net proceeds of approximately $1.1 million after approximately $14,000 in related stock issuance costs.
| F-63 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
The activity related to Series AA Preferred Stock for the three month period ended through March 31, 2012 is as follows:
| Series AA Preferred Stock | ||||||||
| Shares | Amount | |||||||
| Balance, December 31, 2011 | 913,361 | $ | 8,993,418 | |||||
| Accretion on preferred stock | - | 145,636 | ||||||
| Balance, March 31, 2012 | 913,361 | $ | 9,139,054 | |||||
Series B Preferred Stock
In 2010, the Company created a class of preferred stock known as Series B preferred stock and authorized 35,000,000 shares for issuance as Series B Preferred Stock. All shares of Series B were issued pursuant to a Stock Exchange Agreement entered into concurrently with the June 2010 Asset Transfer Agreement with Intracel. As noted above, all shares of Series B were converted into common stock in 2012.
Dividends - Dividends on Series B were cumulative and accrue at a rate of 7% of $9.0797 per share, payable in cash or through the issuance of additional shares of Series B when and if declared. If paid in stock, the shares to be issued were calculated by dividing the accrued dividend amount by $9.0797.
Conversion - Each share of Series B was convertible at any time at the option of the holder or automatically upon a qualified public offering at a conversion ratio of one to one, subject to adjustment for specific dilutive events. The conversion feature was determined to be clearly and closely related to the Series B as that term is defined by Topic 815 and, as a result, is not required to be accounted for as a free standing derivative financial instrument. Management has also determined that the conversion feature does not represent a beneficial conversion feature as defined in Topic 470-20.
Issuance of Additional Series B - The holders of Series B had the right to receive additional shares of Series B in an amount necessary to cause the holders of Series B holders to maintain their equity ownership of the Company on a fully-diluted and as-converted following any anti-dilutive event. These anti-dilution rights are further explained below.
Voting and Board Representation - The holders of Series B generally were entitled to vote, together with the holders of Series B and common stock. Each preferred stockholder was entitled to the number of votes equal the number of shares of Common Stock into which each share of Series B were convertible at the time of such vote. The holders of Series B were entitled to elect 20% of the members to the board of directors of the Company, but not less than one member.
Redemption - Prior to conversion into common stock in 2012, the Series B were subject to redemption at the option of the holder beginning February 5, 2015 for the $9.0797 per share plus accrued and unpaid dividends.
| F-64 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
Liquidation Preference - The holders of Series B had liquidation preference over the holders of common stock. In the event of liquidation, holders of Series B would be entitled to receive, after payment of the Series A liquidation preference but prior to any distributions to the holders of Common Stock, an amount per share equal to the greater of (i) the Series AA Original Issuance Price plus accrued and unpaid dividends or (ii) such amount per share payable had the shares of Series B been converted into common prior to liquidation.
Anti-dilution Rights - As discussed in Note 4 to these unaudited condensed consolidated financial statements the Series B preferred stock issued to Intracel in 2010 requires the Company to maintain Intracel’s ownership interest in the Company at not less than 50% of the total outstanding equity ownership of the Company on a fully-diluted and as-converted basis. Pursuant to these provisions, rights to additional shares of Series B preferred stock in the amount of 1,972,919 shares in 2010, 233,620 shares in 2011; and 24,166 shares in 2012 accumulated in those periods, respectively. During 2012, all outstanding shares of Series B were converted to common shares. The conversion ratio was adjusted as a result of these provisions increasing the number of common shares issued by 2,230,705 shares. These anti-dilution rights expired upon conversion of the Series B preferred stock to common stock in 2012. The anti-dilution rights afforded to holders of the Series B were determined to be clearly and closely related to the Series B as that term is defined in Topic 815 and as a result these rights are not required to be accounted for as a free standing derivative financial instrument.
The subscription agreement for the Company’s most recent financing through the issuance of common stock (“Round C”) provides a form of anti-dilution protection to subscribers. Pursuant to the subscription agreement, if the market price of the Company’s common stock on the effective date of the Company’s initial public offering (“IPO Price”) is less than $5.50 per share, then the Company will issue to each subscriber additional shares of common stock. The number of shares issued would equal the difference between a) the number of shares that would have been issued if the price per unit was equal to the greater of the IPO Price or $5.00 and b) the number of shares originally issued to the subscriber. The anti-dilution rights are determined to be clearly and closely related to the Round C common stock as that term is defined in Topic 815 and as a result these rights are not required to be accounted for as a free standing derivative financial instrument.
The activity related to Series B Preferred Stock for the three month period ended March 31, 2012 is as follows:
| Series B Preferred Stock | ||||||||
| Shares | Amount | |||||||
| Balance, December 31, 2011 | 14,425,377 | $ | 110,135,112 | |||||
| Accretion on preferred stock | - | 6,877,531 | ||||||
| Balance, March 31, 2012 | 14,425,377 | $ | 117,012,643 | |||||
| F-65 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
11. Stock-Based Compensation
Restricted Stock
The Company from time to time has issued shares of restricted common shares to employees. From August 2010 through December 31, 2012, the Company issued 119,734 shares of restricted common stock to employees whose vesting is contingent upon a successful initial public stock offering with a 10 year contractual life. In 2007 and 2010 the Company issued 180,000 and 1.1 million shares, respectively of restricted common stock to employees whose vesting is dependent upon future employment. Those shares generally vest over periods of 4 to 5 years.
The Company records compensation expense for the award of restricted stock based upon the awards fair value determined as the based difference in the estimated fair value of the Company’s common stock and the price paid by the employee, if any, generally on the date of grant. The fair value of restricted stock awards is recognized as compensation expense over the service period which is generally the same as the vesting period. No compensation cost has been or will be recognized for the restricted stock awards whose vesting is contingent upon a successful initial public offering until such event is probable of occurring. As of March 31, 2013, management has determined that such an event is not yet probable of occurring.
No restricted stock was awarded and no shares of outstanding restricted stock vested in the three month periods ended March 31, 2013 and 2012. As a result, no compensation expense has been recorded in the three month periods ended March 31, 2013 and 2012, respectively. As of March 31, 2013, total unrecognized compensation costs related to nonvested restricted stock awards to purchase 119,734 shares of common stock was approximately $410,000, which will be recognized upon a successful initial public offering of the Company’s stock. The nonvested restricted stock awards have a weighted average remaining contractual term of approximately 8.0 years.
In December 2010, the Company committed to issue $462,500 of restricted stock, subject to restrictions yet to be defined, to an officer of the Company if and only if the Company completes a financing round that provides bona fide equity capital to the Company of at least $35.0 million.
On July 17, 2012, the Company’s board of directors approved the grant of 200,000 options to acquire Vaccinogen common stock to be awarded to existing employees. No options have been issued through December 31, 2012. On October 23, 2012, the Company committed to grant an option to purchase 20,000 shares of common stock to an employee if the Company’s stock begins trading on the “Over the Counter Bulletin Board”. The exercise price of the award will be equal to the Company’s stock closing price on the first day of trading on the “Over the Counter Bulletin Board” and the awards will vest over four years from that date. No compensation cost has been or will be recognized for stock options whose vesting is contingent upon a successful securities registration until such event is probable of occurring. As of March 31, 2013, management has determined that such an event is not yet probable of occurring.
Stock Purchase Warrants
From time to time the Company has issued stock purchase warrants to non-employees in exchange for services rendered. As of March 31, 2013, and December 31, 2012, 785,575 warrants were issued and outstanding with exercise prices ranging from $1.00 to $5.50. To date, all warrants have been issued for past services, with the exercise prices at least equal to the then estimated fair value of the underlying security, were fully vested upon issuance, and had contractual terms ranging from 2.5 to 7.5 years. No stock warrants were issued to non-employees for services in the three month periods ended March 31, 2013 and 2012, respectively.
| F-66 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
The following table summarizes information related to warrants outstanding issued to non-employees in exchange for services as of March 31, 2013:
| Exercise price | Shares | Weighted Average Remaining Life | Weighted Average Exercise Price | |||||||||||
| $ | 1.00 | 705,575 | 2.06 | $ | 1.00 | |||||||||
| $ | 5.50 | 80,000 | 4.75 | $ | 5.50 | |||||||||
| Total | 785,575 | |||||||||||||
12. Commitments and Contingencies
Leases
The Company leases office space, a manufacturing facility, and equipment under operating leases expiring in 2013. In addition, the Company leases storage facilities on a month to month basis. Rent expense was approximately $18,627 and $20,435 for the quarters ended March 31, 2013 and 2012, respectively.
Minimum future rental payments under non-cancelable operating leases, including amendments to leases entered through the date the financial statements were available to be issued, total $91,388 for 2013.
Royalty Agreement with Intracel
Pursuant to the Agreement, the Company agreed to pay Intracel the following royalties on the Net Sales of Colon Cancer Products (as defined): (i) 3% of net sales on the first $350.0 million of Net Sales of Colon Cancer Products occurring in the calendar year; (ii) 4% of net sales of Net Sales of Colon Cancer Products occurring in the calendar year in excess of $350.0 million and up to and including $750.0 million and (iii) 5% of net sales of Net Sales of Colon Cancer Products occurring in the calendar year in excess of $750.0 million.
Royalty Agreement with Organon
The Company has agreed to pay Organon a royalty of 10% of the Net Sales of OncoVAX® (and all other TICE BCG related products, if any) until the Organon Obligation is paid in full, including interest, and 3% for 5 years thereafter.
Litigation
The Company may be subject to certain claims arising in the ordinary course of business. The Company and a vendor are in dispute over amounts owed for services performed. A claim has been filed against the Company in the amount of approximately $150,000. Management believes the vendor did not perform under the terms of the contract and contends that no amounts are due to the vendor. Management has offered to settle the matter for $75,000 to be paid upon the Company’s successful initial public offering and has accrued $75,000 in the accompanying financial statements.
| F-67 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
13. Related Party Transactions
Vaccinogen’s chief executive officer is a minority shareholder of Intracel and currently, holds less than 1% interest in Intracel.
On April 15, 2012, Intracel provided an unsecured note payable in the amount of $30,000. The note is unsecured, non-interest bearing, and becomes due on the date on which a minimum equity raise of $1.0 million occurs. Currently the note is in default. The carrying value of the note payable to Intracel is included in related party payable in the accompanying unaudited condensed consolidated financial statements as of March 31, 2013.
In 2012, an executive of the Company loaned the Company $10,000. As of March 31, 2013, the balance due of $4,099 is past due and in default. The carrying value of this amount due to the Company executive is included in related party payable in the accompanying unaudited condensed consolidated financial statements as of March 31, 2013.
As discussed in Note 10, on February 14, 2012, a member of the board of directors of the Company, agreed to loan the Company money to pay one of the Company’s vendors an outstanding amount of $169,729. The Company subsequently issued to this board member 30,860 shares of common stock and a Round C Warrant exercisable into 9,258 shares of common stock of the Company.
14. Supplemental Disclosure of Cash Flow Information
For the quarters ended March 31, 2013 and 2012 and the Company paid interest costs of $38,550 and $1,421, respectively.
15. Subsequent Events
Subsequent events were evaluated through June 11, 2013, the date the unaudited condensed consolidated financial statements were available to be issued. In addition to events occurring after December 31, 2012 previously described herein, the following additional transactions occurred subsequent to March 31, 2013.
In April 2013, the board of directors authorized the Company to offer the investors in the 2012 Bridge loan, the option to convert the amount otherwise due and payable to them in the event of a successful qualified offering, or $2,038,000, into common stock of the Company, at a per share price equal to that of previously issued Round C Stock offering, or $5.50 per share, plus Round C Warrants equivalent to 30% of the shares of Round C Stock issued in the conversion. The holders of the 2012 Bridge Loan were given until May 3, 2013 or the close of the offering period for the Round C Stock, whichever comes first. Investors owning $838,000 of the $2,038,000 of the 2012 Bridge Loan have indicated their intent to convert their interests in exchange for 152,359 shares of Round C Stock and the issuance of Round C Warrants exercisable into 45,705 shares of common stock.
For the period of April 1, 2013 to June 11, 2013, the Company raised $1,056,000 of capital from the issuance of 192,000 shares of common stock and Round C Warrants exercisable into 57,599 shares of common stock.
| F-68 |
| VACCINOGEN, INC. AND SUBSIDIARIES |
| Notes to Unaudited Condensed Consolidated Financial Statements |
On May 13, 2013 Vaccinogen and Kodiak Capital Group, LLC terminated the Initial Equity Line of Credit as discussed in Note 5.
| F-69 |
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS OR PLAN OF OPERATION
This section of the Registration Statement includes a number of forward-looking statements that reflect our current views with respect to future events and financial performance. Forward-looking statements are often identified by words like believe, expect, estimate, anticipate, intend, project and similar expressions, or words which, by their nature, refer to future events. You should not place undue certainty on these forward-looking statements. These forward-looking statements are subject to certain risks and uncertainties that could cause actual results to differ materially from our predictions.
Our Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A) is provided in addition to the accompanying financial statements and notes to assist readers in understanding our results of operations, financial condition and cash flows. Our MD&A is organized as follows:
| • | Overview — Discussion of our business and overall analysis of financial and other highlights affecting the Company in order to provide context for the remainder of MD&A. | |
| • | Trends & Outlook — Discussion of what we view as the overall trends affecting our business and the strategy for 2013 and beyond. | |
| • | Critical Accounting Policies— Accounting policies that we believe are important to understanding the assumptions and judgments incorporated in our reported financial results and forecasts. | |
| • | Results of Operations— Analysis of our financial results comparing 2012 and 2011. | |
| • | Liquidity and Capital Resources— An analysis of changes in our balance sheet and cash flows and discussion of our financial condition and future liquidity needs. |
The various sections of this MD&A contain a number of forward-looking statements. Words such as “expects,” “goals,” “plans,” “believes,” “continues,” “may,” and variations of such words and similar expressions are intended to identify such forward-looking statements. In addition, any statements that refer to projections of our future financial performance, our anticipated growth and trends in our businesses, and other characterizations of future events or circumstances are forward-looking statements. Such statements are based on our current expectations and could be affected by the uncertainties and risk factors described throughout this filing and particularly in the “Overview” and “Trends & Outlook” section. Our actual results may differ materially.
Overview
We are a biotechnology company with more than three decades of research into combating cancer by using the body’s own immune system. Vaccinogen is the developer of OncoVax®, which we believe is the only patient specific immunotherapy for Stage II colon cancer and our technology may also have application in other tumor types, notably melanoma and renal cell carcinoma.
Colon cancer represents the third most common form of cancer in both the US and Europe. American Cancer Society statistics suggest there will be about 102,000 new cases of colon cancer diagnosed within the US. The European Cancer Observatory estimates that there were about 245,000 new cases of colon cancer across Europe.
We have broad patents covering the OncoVax® technology in the U.S. and eight other countries, Australia, Canada, Switzerland, Germany, France, Great Britain, Ireland and Italy. These patents and applications provide broad coverage for the production of autologous cancer vaccines. The key protection of OncoVax®, in addition to considerable expertise protected as trade secrets, is the broad, issued patent protection around the production of autologous, sterile, metabolically active cancer vaccines developed by us. This patent, entitled “Sterile Immunogenic Non-Tumorigenic Tumor Cell Composition and Methods” was issued in 2009 and expires no sooner than 2025. We believe that sterility will be required for any product to reach the US market and likely in any other market with an approved sterile vaccine like the one we have developed. This could result in a regulatory barrier to entry to competitors.
| 63 |
We hold 1 U.S. patent to related technologies and including the sterility patent referred to above, 2 U.S. patents total.
We also rely upon unpatented proprietary know-how and continuing technological innovation and other trade secrets to develop and maintain our competitive position. We hold considerable proprietary expertise related to the OncoVax® technology, including the production of autologous cancer vaccines. We have brand names for our OncoVax® products and related technologies, and anticipate filing 5 trademark applications for these and related marks.
All of our research efforts to date are at the pre-clinical or clinical stage of development. We are focused on leveraging our key assets, including our intellectual property, our scientific team and our facilities, to advance our technologies.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our common stock held by non-affiliates exceeds $700 million as of any June 30, in which case we would no longer be an emerging growth company as of the following December 31.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies.
Operating Strategy
We will maintain a cGMP facility and outsource clinical activities to contract research organizations (“CRO) in an effort to achieve the following:
| 1. | Complete the pivotal Phase IIIb clinical trial of this autologous vaccine, OncoVax®, which is a new paradigm to prevent the recurrence of stage II colon cancer. The planned trial will closely replicate a successful prior Phase IIIa. |
| 2. | Develop near term revenues by establishing licensing arrangements for distribution of OncoVax® in certain territories as well as for additional carcinoma indications. |
| 3. | Develop diagnostic and therapeutic drug products as well as near-term revenues by exploiting its distinctive Human Monoclonal Antibodies (“HuMabs”) technology. |
Trends & Outlook
Revenue
To date, the Company has not earned any revenues as the use of OncoVax® to create cancer related vaccines is still undergoing clinical trials and has not yet received regulatory approval for commercialization and sale.
We are mainly focused on: preparing for the initiation of Phase IIIb clinical trials relating to OncoVax® and our research regarding human monoclonal antibodies (HuMabs).
| 64 |
On a long-term basis, we anticipate that our revenue will be derived primarily from global sales of our OncoVax® product and licensing fees .Commercialization of the OncoVax® product is dependent on successful completion of a Phase IIIb clinical trial and subsequent approval by the FDA.
Research & Development Expenses
Our research and development costs consist of expenses incurred in activities connected with the development of a stage II colon cancer vaccine. These expenses consist primarily of the amortization of intangible assets, cost of conducting clinical trials, salaries and related expenses for personnel, fees paid to professional service providers and, costs of a manufacturing facility in The Netherlands. We expect that research and development expenses will increase in the near term with the onset of the Phase IIIb clinical trial.
Clinical Trials
The FDA has requested a second, confirmatory, randomized controlled Phase III trial of OncoVax® in Stage II colon cancer. The principal objective of Vaccinogen is to organize a "best of breed" team to conduct a confirmatory Phase IIIb trial in Stage II colon cancer. The protocol of this trial, including endpoints and the statistical analysis plan is the subject of an SPA agreement granted by the FDA.
This study will be carried out under a Special Protocol Assessment (SPA) that was negotiated with the FDA in 2010. An SPA granted by the FDA provides a mechanism for the sponsors and the FDA to reach agreement on size, execution and analysis of a clinical trial that is intended to form the primary basis for regulatory approval. The primary endpoint of this pivotal Phase IIIb trial is recurrence-free survival (RFS) with an interim analysis after 70 “events,” and a final primary analysis three years following completed enrollment. “Events” are incidents of either recurrent disease or death. The study is powered at 90% to detect a 50% improvement in RFS versus resection only for final analysis with an adjustment for interim analysis. The power calculations in the study assume an event rate and distribution that match those observed in the 8701 Phase IIIa study. If a robust p value is achieved at the interim analysis, the Biologics License Application (BLA) can be filed with the FDA’s center for Biologics Evaluation and Research (CBER) at that point. Past clinical trials using the optimal regimen with four immunizations will be accepted as supportive studies during the FDA review of the BLA.
The Phase IIIb study will enroll 550 patients, randomized 1:1 to receive surgery alone, or surgery plus OncoVax®. Patients will be followed on a regular schedule to see whether and when their cancer might reappear. The experience with OncoVax® in the 8701 study showed that in the relevant Stage II group, disease recurrence happened more quickly and more frequently in those patients who received surgery alone. If the Phase IIIb trial replicates the experience of the 8701 patients, the interim analysis after 2/3 of the expected events should give a clear and statistically significant separation between the Kaplan-Meier curves at that point, and would form the basis for a BLA seeking marketing approval from the FDA.
It is important to emphasize that the FDA has agreed that RFS is the most appropriate endpoint to evaluate a trial in Stage II colon cancer. The alternative in cancer studies is often overall survival (OS). An OS endpoint has several drawbacks in this case. The average age of patients presenting with colon cancer is 65 years, so deaths from causes other than the tumor will dilute the outcome in both arms. Patients in either arm whose disease recurs will receive chemotherapy and other treatments for their metastatic disease, and the success of these treatments will dilute the impact of earlier therapies such as OncoVax® on OS. From a patient perspective, being disease free is clearly a meaningful endpoint.
General and Administrative Expenses
Our general and administrative (“G&A”) expenses consist of the general costs, expenses and salaries for the operation and maintenance of our business. We anticipate that general and administrative expenses will increase as we progress through the clinical phase of development into commercialization.
| 65 |
Critical Accounting Policies
Our financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses. Note 3 of the Notes to Financial Statements describes the significant accounting policies used in the preparation of the financial statements. Certain of these significant accounting policies are considered to be critical accounting policies, as defined below.
A critical accounting policy is defined as one that is both material to the presentation of our financial statements and requires management to make difficult, subjective or complex judgments that could have a material effect on our financial condition and results of operations. Specifically, critical accounting estimates have the following attributes: (1) we are required to make assumptions about matters that are highly uncertain at the time of the estimate; and (2) different estimates we could reasonably have used, or changes in the estimate that are reasonably likely to occur, would have a material effect on our financial condition or results of operations.
Estimates and assumptions about future events and their effects cannot be determined with certainty. We base our estimates on historical experience and on various other assumptions believed to be applicable and reasonable under the circumstances. These estimates may change as new events occur, as additional information is obtained and as our operating environment changes. These changes have historically been minor and have been included in the financial statements as soon as they became known. Based on a critical assessment of our accounting policies and the underlying judgments and uncertainties affecting the application of those policies, management believes that our financial statements are fairly stated in accordance with accounting principles generally accepted in the United States, and present a meaningful presentation of our financial condition and results of operations. We believe the following critical accounting policies reflect our more significant estimates and assumptions used in the preparation of our financial statements:
Use of Estimates—Our financial statements have been prepared in accordance with accounting principles generally accepted in the United States and, accordingly, require management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in the financial statements. On an ongoing basis, the Company evaluates the estimates used in recording common stock warrant related liabilities, derivative financial instruments, stock based compensation, and where applicable, the fair value of assets. The Company may base such estimates on various assumptions including information received from valuation consultants which it believes to be reasonable under the circumstances.. Actual results could differ from those estimates.
Intangible Assets— Intangible assets consist primarily of the cost of acquired patents associated with OncoVax® to be used in research and development and the commercialization cancer related vaccines. The Company has capitalized the cost of the acquired patents because the Company has identified alternative future research and development efforts for numerous forms of cancer which it intends to pursue and for which management believes will result in commercialization of related vaccines. Acquired patents are carried at cost less accumulated amortization. Amortization is calculated on a straight-line basis, over the estimated useful economic life of the patent, which is 15 years for OncoVax®.
Long-lived assets, including identifiable intangible assets with finite lives, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. The Company has determined that no impairment has occurred as of December 31, 2011 and December 31, 2012.
Fair Value of Financial Instruments - Fair value is defined as the price at which an asset could be exchanged or a liability transferred (an exit price) in an orderly transaction between knowledgeable, willing parties in the principal or most advantageous market for the asset or liability. Where available, fair value is based on observable market prices or parameters or derived from such prices or parameters. Where observable prices or inputs are not available, valuation models are applied.
| 66 |
Financial assets recorded at fair value in the accompanying financial statements are categorized based upon the level of judgment associated with the inputs used to measure their fair value. Hierarchical levels, as defined by the new guidance related to fair value measurements and disclosures, and directly related to the amount of subjectivity associated with the inputs to fair valuation of these assets and liabilities, are as follows:
| Level 1- | Inputs are unadjusted, quoted prices in active markets for identical assets at the reporting date. Active markets are those in which transactions for the asset or liability occur in sufficient frequency and volume to provide pricing information on an ongoing basis. | |
| We carry no investments classified as Level 1. | ||
| Level 2 - | Inputs are other than quoted prices included in Level 1, which are either directly or indirectly observable for the asset or liability through correlation with market data at the reporting date and for the duration of the instrument's anticipated life. | |
| We carry no investments classified as Level 2. | ||
| Level 3- |
Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities and which reflect management's best estimate of what market participants would use in pricing the asset or liability at the reporting date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model.
Our warranty and Bridge Loan obligations are considered Level 3. |
For a further discussion regarding fair value measurements, see Note 9 on Fair Value in the Notes to Financial Statements.
Accounting for Warrants – We have adopted FASB guidance related to determining whether an instrument or embedded feature is indexed to an entity’s own stock. This guidance applies to any freestanding financial instruments or embedded features that have the characteristics of a derivative, as defined in ASC Topic 815, Derivatives and hedging, and to any freestanding financial instruments that are potentially settled in an entity’s own common stock. As a result, certain of our warrants were considered to be derivatives and were valued using various assumptions as they are recorded as liabilities.
Research and Development Costs – Research and development costs that do not have alternative future use are expensed as incurred. Research and development expenses primarily include the amortization of intangible assets, cost of conducting clinical trials, compensation and related overhead for employees, consultants, facilities costs and the cost of materials purchased for research and development.
Stock Based Compensation – The Company measures the cost of employee services received in exchange for stock options or restricted stock awards based upon the fair value of the award on the date of the grant. The Company recognizes the estimated grant date fair value of the award as stock-based compensation expense on a straight-line basis over the requisite service period, which is generally the vesting period.
The Company initially measures the cost of awards granted to non-employees based on the fair value of the award on the date of grant however such cost is re-measured at the end of each reporting period until performance is fully satisfied or services are rendered by the non-employee.
The fair value of stock options granted is calculated using the Black-Scholes option-pricing model, which requires the use of subjective assumptions including volatility, expected term, risk-free rate, and the fair value of the underlying common stock. The fair value of non-vested stock awards is determined based upon the estimated fair value of the Company's common stock.
| 67 |
Results of Operations
Consolidated Statement of Operations Data
| Three Months | ||||||||||||||||
| Years Ended December 31, | Ended March 31, | |||||||||||||||
| 2012 | 2011 | 2013 | 2012 | |||||||||||||
| Revenues | $ | - | $ | - | $ | - | $ | - | ||||||||
| Operating Expenses: | ||||||||||||||||
| Research and development | 8,139,038 | 8,564,519 | 2,073,184 | 2,025,004 | ||||||||||||
| General and administrative expenses | 2,960,939 | 2,783,460 | 6,929,760 | 653,572 | ||||||||||||
| Total Operating Expenses | 11,099,977 | 11,347,979 | 9,002,944 | 2,678,576 | ||||||||||||
| Loss from Operations | (11,099,977 | ) | (11,347,979 | ) | (9,002,944 | ) | (2,678,576 | ) | ||||||||
| Loss on Financial Instruments | (1,281,916 | ) | - | (413,895 | ) | (92,368 | ) | |||||||||
| Interest and Other Expenses | (312,257 | ) | (176,489 | ) | (65,230 | ) | (131,938 | ) | ||||||||
| Net Loss | $ | (12,694,150 | ) | $ | (11,524,468 | ) | $ | (9,482,069 | ) | $ | (2,902,882 | ) | ||||
Revenue
Since inception, we have not earned any revenues as the use of OncoVax® to create cancer related vaccines is still undergoing clinical trials and has not yet received regulatory approval for commercialization and sale.
Operating Expenses – Three Months Ended March 31, 2013 and 2012
Our operating expenses totaled $9,002,944 and $2,678,576 in the three months ended March 31, 2013 and March 31, 2012 respectively. The operating expenses are discussed in further detail below.
Research and Development Expenses
| Three Months Ended March 31 | Change in 2013 v. 2012 | |||||||||||||
| 2013 | 2012 | $ | % | |||||||||||
| $ | 2,073,184 | $ | 2,025,004 | $ | 48,180 | 2.38 | % | |||||||
Research and development expenses increased to $2,073,184 in the three months ended March 31, 2013, or approximately 2.38%, from $2,025,004 in the three months ended March 31, 2012. These expenses include $1,675,817 and $1,674,839 for amortization of intangibles, related to OncoVax® intellectual property, for the three months ended March 31, 2013 and 2012 respectively. The increase was primarily attributable to increased activity in preparation of the upcoming Phase IIIb clinical trial.
| 68 |
General and Administrative Expenses
| Three Months Ended March 31 | Change in 2013 v. 2012 | |||||||||||||
| 2013 | 2012 | $ | % | |||||||||||
| $ | 6,929,760 | $ | 653,572 | $ | 6,276,188 | * | ||||||||
| * Not meaningful | ||||||||||||||
General and administrative expenses increased to $6,929,760 in the three months ended March 31, 2013 from $653,272 in the comparable period in 2012. The increase of $6,276,188, from 2012 to 2013 was attributable to the estimated value of the Abell Option issued with respect to the Abell Investment Agreement in January 2013 ($5,954,545) and increases in accounting fees ($475,000) offset by a decrease in salaries ($130,000). The Abell Investment agreement is discussed in Note 3 of the unaudited condensed consolidated financial statements.
Operating Expenses – Years Ended December 31, 2012 and 2011
Our operating expenses totaled $11,099,977 in the year ended December 31, 2012 and $11,347,979 in the year ended December 31, 2011. The operating expenses are discussed in further detail below.
Research and Development Expenses
| Year Ended December 31 | Change in 2012 v. 2011 | |||||||||||||
| 2012 | 2011 | $ | % | |||||||||||
| $ | 8,139,038 | $ | 8,564,519 | $ | (425,481 | ) | -4.97 | % | ||||||
Research and development expenses decreased to $8,139,038 in the year ended December 31, 2012, or approximately 4.97%, from $8,564,519 in the year ended December 31, 2011. These expenses include $6,703,273 and $6,706,552 for amortization of intangibles, related to OncoVax® intellectual property, for the years ended December 31, 2012 and 2011 respectively. The decrease of $425,481, or 4.97% was primarily attributable to a decrease in salaries ($201,000), Phase IIIb clinical trial expenses ($52,000), travel and entertainment ($27,000) and stock based compensation ($34,000).
General and Administrative Expenses
| Year Ended December 31 | Change in 2012 v. 2011 | |||||||||||||
| 2012 | 2011 | $ | % | |||||||||||
| $ | 2,960,939 | $ | 2,783,460 | 177,479 | 6.38 | % | ||||||||
General and administrative expenses increased to $2,960,939 in the year ended December 31, 2012, or approximately 6.38%, from $2,783,460 in the comparable period in 2011. The increase of $177,479, from 2011 to 2012 was primarily attributable to increases in accounting and consulting fees.
NonoperatingExpense – Three Months Ended March 31, 2013 and 2012
Nonoperating expenses were $479,125 and $224,306 in the three months ended March 31, 2013 and 2012 respectively. The nonoperating expenses are discussed below.
| 69 |
Loss on Financial Instruments
| Three Months Ended March 31 | Change in 2013 v. 2012 | |||||||||||||
| 2013 | 2012 | $ | % | |||||||||||
| $ | 413,895 | $ | 92,368 | $ | 321,527 | 348.09 | % | |||||||
Loss on Financial Instruments was generated due to changes in the estimated fair value at March 31, 2013 for the Bridge Loan in the amount of approximately $306,000 and offset by gains in the estimated fair value of Abell Warrants of approximately $123,000 and the Round C Warrants of approximately $45,000.
Additionally, during the period ended March 31, 2013 the Company issued additional warrants associated with the January 2013 amendment as a result of the deemed extinguishment of the Abell Loan. The fair value of the warrants ($275,813) is included in the Loss on Financial Instruments. Further discussion of this transaction can be found in Note 3 and Note 9 to the unaudited condensed consolidated financial statements.
The March 31, 2012 Loss on Financial Instruments was solely attributable to the change in the estimated fair value of the Abell Warrants.
Interest and Other Expenses
| Three Months Ended March 31 | Change in 2013 v. 2012 | |||||||||||||
| 2013 | 2012 | $ | % | |||||||||||
| $ | 65,230 | $ | 131,938 | $ | (66,708 | ) | -50.56 | % | ||||||
Interest Expense decreased to $65,230 in the three months ended March 31, 2013, or approximately 51% from $131,938 in the comparable period in 2012.
NonoperatingExpense - Years Ended December 31, 2012 and 2011
Nonoperating expenses were $1,594,173 and $176,489 in the years ended December 31, 2012 and 2011, respectively. The nonoperating expenses are discussed below.
Loss on Financial Instruments
| Year Ended December 31 | Change in 2012 v. 2011 | |||||||||||||
| 2012 | 2011 | $ | % | |||||||||||
| $ | 1,281,916 | $ | - | 1,281,916 | * | |||||||||
* not meaningful
Loss on Financial Instruments was generated due to changes in the estimated fair value at December 31, 2012 for the Abell Warrants in the amount of approximately $772,000 and the 2012 Bridge Loan in the amount of approximately $510,000, both of which are discussed in Note 3 of the financial statements.
Interest and Other Expenses
| Year Ended December 31 | Change in 2012 v. 2011 | |||||||||||||
| 2012 | 2011 | $ | % | |||||||||||
| $ | 312,257 | $ | 176,489 | 135,768 | 76.93 | % | ||||||||
| 70 |
Interest and Other Expense increased to $312,257 in the year ended December 31, 2012, or approximately 76.9% from $176,489 in the comparable period in 2011. The increase of $135,768 is attributable to primarily additional short-term financing of continuing operations. Additional funding was provided by an increase in the Note Payable to the Abell Foundation ($300,000) as discussed in Note 8 of the financial statements and the 2012 Bridge Loan financing ($1,019,000), a derivative financial instrument as discussed in Note 3 of the financial statements.
Liquidity and Capital Resources
Since our inception, we have financed our operations through the private placement of our securities.
| Three Months | ||||||||||||||||
| Years Ended December 31, | Ended March 31, | |||||||||||||||
| 2012 | 2011 | 2013 | 2012 | |||||||||||||
| Net cash provided by (used in) operating activities | $ | (2,272,903 | ) | $ | (3,511,589 | ) | $ | (1,001,537 | ) | $ | (485,684 | ) | ||||
| Net cash used in investing activities | (48,282 | ) | (80,230 | ) | (722 | ) | (57,823 | ) | ||||||||
| Net cash provided by (used in) financing activities | 2,239,002 | 2,602,364 | 971,836 | 300,000 | ||||||||||||
| Effect of foreign exchange rate changes on cash and equivalents | (40,658 | ) | 25,378 | 40,752 | 7,648 | |||||||||||
| Net increase (decrease) in cash and equivalents | $ | (122,841 | ) | $ | (964,077 | ) | $ | 10,329 | $ | (235,859 | ) | |||||
Cash & Cash Equivalents
Total cash and cash equivalents was $124,169 at March 31, 2013, compared with $113,840 at December 31, 2012, for an increase of $10,329 in the three months ended March 31, 2013. The increase in cash and cash equivalents was is primarily attributable to cash of $971,836 received from our private placement of stock and warrants and $40,752 from impact of foreign currency on cash and cash equivalents, which amounts were offset by $1,001,537 of net cash used in operating activities and $722 of net cash used in investing activities in the three months ended March 31, 2013.
Total cash and cash equivalents was $113,840 at December 31, 2012, compared with $236,681 at December 31, 2011. The decrease in cash and cash equivalents of $122,841 or 51.9%, from 2011 to 2012, was primarily attributable to funding operations without adequate cash flows from financing activities in 2012.
Net Cash Used in Operating Activities
Operating activities required $1,001,537 and $485,684 for the three months ended March 31, 2013 and 2012 respectively. The increase of $515,853 or 106.21%, from 2012 to 2013 was primarily attributable to an increase in our accounts payable balance of approximately $294,000 as we experienced increased accounting, legal and consulting expenses.
Operating activities required $2,272,903 and $3,511,589 for the years ended December 31, 2012, and 2011 respectively. The decrease of $1,238,686 or 35.27%, from 2011 to 2012 was primarily attributable to an increase in our accounts payable balance of $1,000,000 as we experienced increased accounting, legal and consulting expenses.
Net Cash Used in Investing Activities
We invested $722 and $57,823 in the three months ended March 31, 2013 and 2012 respectively in purchases of property and equipment.
| 71 |
We invested $48,282 and $80,230 in the years ended December 31, 2012 and 2011 respectively in purchases of property and equipment.
Net Cash Provided by Financing Activities.
Listed below are key financing transactions entered into by us during 2013:
| · | During the three months ended March 31, 2013 we raised additional capital of approximately $972,000 through the issuance of 176,697 Units (each Unit consisting of 1 share of common stock and a common stock purchase warrant to purchase 0.3 shares of common stock). |
Listed below are key financing transactions entered into by us during 2012:
| · | We raised additional capital of approximately $920,000 through the issuance of 167,273 Units (each Unit consisting of 1 share of common stock and a common stock warrant to purchase 0.3 shares of common stock). |
| · | Between April 2012 and October 2012, we issued $1,019,000 in unsecured notes payable to various investors. |
| · | On February 16, 2012, we received an additional $300,000, from The Abell Foundation, on the working capital loan originally taken in 2011 in the amount of $1.5 million, thereby increasing the amount outstanding to $1.8 million. |
Listed below are key financing transactions entered into by us during 2011
| · | On October 26, 2011, we obtained a $1.5 million working capital loan from The Abell Foundation Inc. |
| · | From January 13, 2011 to October 24, 2011, we issued 123,015 shares of Series AA Preferred Stock resulting in net proceeds of approximately $1.1 million. |
Future Liquidity & Needs
We have incurred significant operating losses and negative cash flows since inception. We do not expect to be profitable in the next several years, but rather expect to incur additional operating losses. We have limited liquidity and capital resources and must obtain significant additional capital resources in order to sustain our product development efforts, for our Phase IIIb clinical trial, pursuit of regulatory approvals, acquisition of capital equipment, costs to maintain office and manufacturing facilities, for general and administrative expenses and other working capital requirements. We rely on cash balances and the proceeds from the offering of our securities to fund our operations.
Because we have recurring losses and negative cash flows from operating activities and have significant future commitments, substantial doubt exists regarding our ability to remain in operation at the same level we are currently performing. Further, the report of BDO USA, LLP, our independent registered accounting firm, with respect to our financial statements at December 31, 2012 and December 31, 2011 contains an explanatory paragraph as to our potential inability to continue as a going concern. Additionally, this may adversely affect our ability to obtain new financing on reasonable terms or at all.
As discussed in Note 5 of the consolidated financial statements, the Company has entered into a series of agreements with the Kodiak Capital Group, LLC (“Kodiak”) to provide up to $26 million of additional equity capital, although subsequent to the date of the financial statements we and Kodiak mutually agreed to terminate the Investment Agreement for Kodiak’s investment of up to $1 million. On July 8, 2013, Kodiak agreed to increase its commitment under its remaining Investment Agreement to $26 million. The proceeds from the agreements with Kodiak would primarily be used to continue the Company’s research and development activities including the furtherance of clinical trials using OncoVax® to develop cancer related vaccines. However, Kodiak is not required to provide funding until certain conditions are met, including the registration and trading of the Company’s equity securities as defined in those agreements. There can be no assurance that the Company will meet the conditions under which Kodiak will be required to provide the equity capital of that the capital available under such agreements will be sufficient to allow the Company to funds its continuing research and development activities. If the Company is unable to raise the additional equity capital from Kodiak, the Company will need to seek alternative sources of debt or equity capital. There can be no assurance that such capital will be available in sufficient amounts or on terms acceptable to the Company.
| 72 |
In addition, we are continuing our Unit offering of common stock and warrants. The total offering is for $13,038,000.
Contractual Obligations
As of March 31, 2013 and December 31, 2012, our contractual obligations for the next five years, and thereafter were as follows:
Future minimum operating lease payments:
| March 31, 2013 | December 31, 2012 | |||||||
| 2013 | $ | 91,388 | $ | 123,247 | ||||
| 2014 | - | |||||||
| 2015 | - | |||||||
| 2016 | - | |||||||
| 2017 | - | |||||||
| 2018 and thereafter | - | |||||||
| Total minimum payments | $ | $ | 123,247 | |||||
Off-Balance Sheet Arrangements
We do not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities that would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. As such, we are not exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in those types of relationships.
| 73 |
SELECTED FINANCIAL DATA
Not applicable because we are a smaller reporting company.
SUPPLEMENTARY FINANCIAL INFORMATION
Not applicable because we are a smaller reporting company.
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
We have not had changes in or disagreements with accountants on accounting and financial disclosure. BDO USA, LLP. has served as our registered independent public accounting firm since 2012. There have been no changes in or disagreements on accounting or financial disclosure matters.
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable because we are a smaller reporting company.
| 74 |
6,000,000 Common Shares
VACCINOGEN, INC.
PROSPECTUS
YOU SHOULD RELY ONLY ON THE INFORMATION CONTAINED IN THIS DOCUMENT OR THAT WE HAVE REFERRED YOU TO. WE HAVE NOT AUTHORIZED ANYONE TO PROVIDE YOU WITH INFORMATION THAT IS DIFFERENT. THIS PROSPECTUS IS NOT AN OFFER TO SELL COMMON STOCK AND IS NOT SOLICITING AN OFFER TO BUY COMMON STOCK IN ANY STATE WHERE THE OFFER OR SALE IS NOT PERMITTED.
Until ___________, 2013, all dealers that effect transactions in these securities whether or not participating in this offering may be required to deliver a prospectus. This is in addition to the dealer’s obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
The Date of This Prospectus Is: _____ __, 2013
| 75 |
PART II — INFORMATION NOT REQUIRED IN THE PROSPECTUS
Item. 13 Other Expenses Of Issuance And Distribution.
The estimated costs of this offering are as follows:
| Securities and Exchange Commission registration fee | $ | 4,501.20 | ||
| Transfer Agent Fees* | $ | 80.00 | ||
| Accounting fees and expenses* | $ | 420,000.00 | ||
| Legal fees and expenses* | $ | 127,000.00 | ||
| Edgar filing, printing and engraving fees* | $ | 17,000.00 | ||
| TOTAL | $ | 568.581.20 |
*Indicates expenses that have been estimated for filing purposes.
All amounts are estimates other than the Securities and Exchange Commission’s registration fee.
All amounts are estimates other than the Commission’s registration fee. We are paying all expenses of the offering listed above. No portion of these expenses will be borne by the selling shareholders. The selling shareholders, however, will pay any other expenses incurred in selling their common stock, including any brokerage commissions or costs of sale.
Item.14 Indemnification Of Directors And Officers.
As permitted by Section 2-405.2 of the Maryland General Corporation Law, our Articles of Incorporation includes a provision that provides to the maximum extend that Maryland law permits limitation of liability, no present or former director or officer shall be liable to the to the corporation or its stockholders for money damages.
In addition, as permitted by Section 2-418 of the Maryland General Corporation Law, our bylaws provide that to the maximum extent permitted by Maryland law in effect from time to time, the corporation shall indemnify and, without requiring a preliminary determination of the ultimate entitlement to indemnification, shall pay or reimburse reasonable expenses in advance of final disposition of a proceeding to (a) any individual who is a present or former director or officer of the corporation and who is made or threatened to be made a party to the proceeding by reason of his or her service in that capacity or (b) any individual who, while a director or officer of the corporation and at the request of the corporation, serves or has served as a director, officer, partner, trustee, member or manager of another corporation, real estate investment trust, limited liability company, partnership, joint venture, trust, employee benefit plan or other enterprise and who is made or threatened to be made a party to the proceeding by reason of his or her service in that capacity. The rights to indemnification and advance of expenses provided by the charter of the corporation and these bylaws shall vest immediately upon election of a director or officer. The corporation may, with the approval of its board of directors, provide such indemnification and advance for expenses to an individual who served a predecessor of the corporation in any of the capacities described in (a) or (b) above and to any employee or agent of the corporation or a predecessor of the corporation. The indemnification and payment or reimbursement of expenses provided in these bylaws shall not be deemed exclusive of or limit in any way other rights to which any person seeking indemnification or payment or reimbursement of expenses may be or may become entitled under any bylaw, resolution, insurance, agreement or otherwise. Neither the amendment nor repeal of this Section, nor the adoption or amendment of any other provision of the bylaws or charter of the corporation inconsistent with this Section, shall apply to or affect in any respect the applicability of the preceding paragraph with respect to any act or failure to act which occurred prior to that amendment, repeal or adoption.
| II-1 |
We may enter into indemnification agreements with each of our directors and officers that provide the maximum indemnity allowed to directors and officers by Section 2-418 of the Maryland General Corporation Law and the bylaws as well as certain additional procedural protections.
The indemnification provisions in the bylaws and the indemnification agreements which we may enter into with our directors and officers may be sufficiently broad to permit indemnification of our directors and officers for liabilities arising under the Securities Act. However, insofar as indemnification for liabilities arising under the Securities Act may be permitted for our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by us for expenses incurred or paid by a director, officer or controlling person in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, we will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether our indemnification is against public policy as expressed in the Securities Act and will be governed by the final adjudication of the issue by the court.
Disclosure of Commission Position of Indemnification for Securities Act Liabilities
Insofar as indemnification for liabilities arising under the Act, may be permitted to our directors, officers and controlling persons pursuant to the foregoing provisions, or otherwise, we have been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable.
Item 15. Recent Sales of Unregistered Securities
Common Stock
During, 2010, we issued 685,866 shares of common stock to Intracel in respect of its anti-dilution rights under the License Agreement with Intracel Holdings Corporation. The issuance was exempt under Rule 506 of the Securities Act of 1933, as amended.
During 2010, we issued 2,859,098 shares of common stock to the former holders of its Series A Preferred Stock in consideration of the full conversion of the Series A Preferred Stock to common stock. The issuances were exempt under Section 3(a)(9) of the Securities Act of 1933, as amended.
On December 31, 2011, we issued 3,636 shares to each of our non-employee directors (John Nicolis, Dan Fitzgerald, Daniel Kane and Alan Cohen) in consideration of their services as a director in 2012. The share were valued at $5.50, or $20,000 per issuance. These issuances were exempt under Rule 506 of the Securities Act of 1933, as amended. The shares of common stock were issued as restricted securities and all certificates issues contained a legend stating the shares have not been registered under the Securities Act and setting forth or referring to the restrictions on transferability and the sale of the shares under the Securities Act
| II-2 |
On August 1, 2012, we issued 1,507,666 shares of common stock to the former holders of our Series AA Preferred Stock in consideration of the full conversion of the Series AA Preferred Stock to common stock. The issuances were exempt under Section 3(a)(9) of the Securities Act of 1933, as amended.
On August 1, 2012, we issued 16,656,082 shares of common stock to the former holders of our Series B Preferred Stock in consideration of the full conversion of the Series B Preferred Stock to common stock. The issuances were exempt under Section 3(a)(9) of the Securities Act of 1933, as amended.
On August 1, 2012, we issued 236,634 shares of common stock to Kodiak Capital Group, LLC as a commitment fee under an investment agreement. The issuance was exempt under Section 4(2) of the Securities Act of 1933, as amended.
From September 1, 2012 through June 26, 2013, we issued 824,189 units (“Units”), of which 671,830 units were purchased for cash and 152,359 units were purchased in consideration of cancellation of outstanding indebtedness. Each Unit consists of 1 share of common stock and a warrant to purchase 0.3 shares of common stock. The purchase price for each unit was $5.50 per unit and the exercise price for each warrant was $6.05 per shares. We received gross proceeds of $3,695,065 from the sale of the units for cash. The proceeds were used for working capital and general corporate purposes. These issuances were exempt under Rule 506 of the Securities Act of 1933, as amended. All investors in Unit offering were accredited investors and were solicited by officers (Michael Hanna and Andrew Tussing) of Vaccinogen and all investors had a substantial pre-existing relationship with such officers of Vaccinogen. The shares of common stock and warrants received were issued as restricted securities and all certificates issues contained a legend stating the shares have not been registered under the Securities Act and setting forth or referring to the restrictions on transferability and the sale of the shares under the Securities Act.
The Units referred to in the paragraph above contained anti-dilutions rights such that f the market price of our Common Stock on the effective date of our first S-1 registration statement to filed with the SEC (the “Effectiveness Date Market Price”) is less than $5.50 per share, then we will issue to each subscriber of the Units, for no additional consideration, such number of shares of Common Stock equal to the difference between (x) the number of shares of our Common Stock that would have been issued to purchaser if the per-Unit purchase price for such shares had been equal to either (1) the Effectiveness Date Market Price or (2) $5.00, whichever is greater and (y) the number of shares of the Common Stock originally issued to purchaser upon the closing of the sale of Units.
On December 26, 2012, we issued 110,000 shares of our common stock to our former President, Michael Kranda, in consideration of his Separation Agreement of August 2012. The shares were valued at $110,000 or $1.00 per share. These issuances were exempt under Rule 506 of the Securities Act of 1933, as amended. The shares of common stock were issued as restricted securities and all certificates issues contained a legend stating the shares have not been registered under the Securities Act and setting forth or referring to the restrictions on transferability and the sale of the shares under the Securities Act
On December 31, 2012, we issued 3,636 shares to each of our non-employee directors (John Nicolis, Dan Fitzgerald, Daniel Kane and Alan Cohen in consideration of their services as a director in 2012. The shares were valued at $5.50, or $20,000 per issuance. These issuances were exempt under Rule 506 of the Securities Act of 1933, as amended. The shares of common stock were issued as restricted securities and all certificates issues contained a legend stating the shares have not been registered under the Securities Act and setting forth or referring to the restrictions on transferability and the sale of the shares under the Securities Act.
| II-3 |
Series A Preferred Stock
On January 30, 2010, we issued 9,022 shares of our Series A Preferred Stock to Intracel in respect of its anti-dilution rights under the License Agreement. The issuance was exempt under Rule 506 of the Securities Act of 1933, as amended.
On January 30, 2010, we issued 81,205 shares of its Series A Preferred Stock to the holders of our Series A preferred stock as a stock dividend. The issuance was exempt under Rule 506 of the Securities Act of 1933, as amended
Series AA Preferred Stock
From February 8, 2010 to November 8, 2010 we issued 790,346 shares of Series AA Preferred Stock to 19 third party investors in a private offering at a price of $9.0797 per share resulting in approximately $7 million of net proceeds. The proceeds were used for general corporate and working capital. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
From January 13, 2011 to October 24, 2011, the Company issued 123,015 shares of Series AA Preferred Stock to 15 third party investors in a private offering at a price of $9.0797 per share resulting in net proceeds of approximately $1.1 million. The proceeds were used for general corporate and working capital. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
Series B Preferred Stock
On June 24, 2010, we issued 3,451,7655 shares of Series B Preferred Stock to Intracel and its nominees in consideration of the asset transfer and other shares of Company stock held by Intracel pursuant to the June 24, 2010 Asset Transfer Agreement and Stock Exchange Agreements equaling a 20% interest in Vaccinogen. In December 2010, we issued an additional 10,973,612 shares of Series B Preferred Stock to all holders of our Series B Preferred Stock to increase their ownership percentage to 50% pursuant to the terms of the Investor Rights Agreement dated June 24, 2010, entered into concurrently with the Asset Transfer Agreement. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
During 2011, we issued 2,206,539 shares of Series B Preferred Stock to Intracel in consideration of its anti-dilution rights and due to our failure to meet certain milestones as set forth in its Agreements with Intracel. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
During 2012, we issued 24,166 shares of Series B Preferred Stock to Intracel in consideration of its anti-dilution rights. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
The Abell Foundation Financings
Secured Promissory Notes
| II-4 |
Between October 26, 2011 and February 16, 2012, we sold $1,800,000 of principal amount of secured promissory notes to The Abell Foundation. The notes had an initial 6-month term year term and bear interest at eight percent (8%) per annum; payable at maturity. The note was amended in January 2013 to extend the maturity date to March 31, 2013. Upon an event of default the interest rate increases to 10% per annum. The notes were secured by all accounts, chattel paper, deposit accounts, equipment, general intangibles, instruments, inventory, investment property and letter of credit rights. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended. In April 2013, the maturity date on the note was extended to May 31, 2013. In consideration of the extension, we also granted Abell a security interest in our patents related to OncoVAX® under a Patent Security Agreement.
On May 31, 2013, the maturity date on the note was extended to July 31, 2013. In consideration of the extension, we agreed that the Notes would be paid concurrently with the closing of each issuance or sale of additional shares of capital stock, or securities directly or indirectly convertible or exchangeable for capital stock (each an “Equity Issuance”) occurring after May 28, 2013 in an amount equal to (a) twenty percent (20%) of the nest $3,778,771 of gross proceeds of such Equity Issuance(s), (b) twenty-five percent (25%) of the next $6,000,000 of gross proceeds of such Equity Issuance(s), and (c) one hundred percent (100%) of the net proceeds of all Equity Issuance(s) thereafter to pay the remaining amount due under the Note, if any.
Warrants
In connection with the secured promissory notes referenced above, we issued Abell a warrant to purchase the number of shares equal to $800,000 divided by 85% of the purchase price per share of stock sold in our first venture capital financing resulting in proceeds of not less than $20,000,000. In connection with the January 2013 extension of maturity date, we increased the amount of shares issuable under the warrant to $1.1 million divided by 85% of the purchase price per share of stock sold in our first venture capital financing resulting in proceeds of not less than $25,000,000. The exercise price equals 85% of the purchase price per share in such venture capital financing, during a 10-year term. The issuance was exempt under Rule 506 of the Securities Act of 1933, as amended.
Investment Agreement
On January 16, 2013, we entered into an investment agreement with The Abell Foundation, Inc. under which at the request of the investor we will issue and sell to the investor up to $5.0 million of common stock. The number of shares issued will be based on the lowest price paid by any purchaser of shares in the any Venture Capital Financing (as defined in the agreement). The investor will not exercise this above option unless the Company has received $25.0 million in gross proceeds under its financing arrangements with Kodiak Capital and an additional $10.0 million from investors other than Kodiak.
Warrants
From September 1, 2012 through June 26, 2013, we issued warrants to purchase 247,251 shares of common stock as part of a unit offering consisting of common stock and warrants. These issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
| II-5 |
Promissory Notes
Between April 2012 and September 2012, we issued unsecured promissory notes to several investors in the aggregate principal amount of $1,019,000. These notes are unsecured, and the interest payable on this balance is equal to the principal amount borrowed, or $1,019,000 in total interest due. The notes mature after a date on which a transaction occurs involving the issuance or sale of additional equity of Vaccinogen that results in at least $20.0 million in gross proceeds. These note holders have the option to convert all principal and accrued interest ($2,038,000 total) into units of common stock and warrants on the same terms as the other investors subscribing for such units. No fees are payable in connection with any Units issued in consideration of cancellation of indebtedness. In May 2013, noteholders with an aggregate principal amount of $419,000 in unsecured notes (with $419,000 in accrued interest) cancelled their notes in consideration of their subscription for 152,359 Units.
The proceeds were used for working capital and general corporate purposes. The issuances were exempt under Rule 506 of the Securities Act of 1933, as amended.
| II-6 |
Item 16. Exhibits.
| Exhibit No. | Description | |
| 2.1 | Asset Transfer Agreement dated June 24, 2012 by and among Vaccinogen, Inc., Intracel Holdings Corporation and the other parties set forth therein.* | |
| 3.1 | Articles of Incorporation of Vaccinogen 1, Inc. dated November 23, 2012. * | |
| 3.2 | Articles of Merger of Vaccinogen, Inc., Delaware corporation with and into Vaccinogen 1, Inc., a Maryland corporation (including name change to “Vaccinogen Inc.”) dated November 23, 2012.* | |
| 3.3 | Articles of Amendment and Restated of Vaccinogen, Inc. dated August 1, 2012.* | |
| 3.4 | Bylaws.* | |
| 4.1 | Investor Rights Agreement dated June 24, 2010 by and among Vaccinogen, Inc., Intracel Holdings Corporation and the other parties set forth therein.* | |
| 4.2 | Fourth Amended and Restated Promissory Note dated May 31, 2013 in the amount of $1,800,000 made by Vaccinogen, Inc. in favor of The Abell Foundation, Inc.* | |
| 4.3 | Common Stock Purchase Warrant issued to The Abell Foundation.* | |
| 4.4 | Form of Promissory Note (2012 bridge note offering).* | |
| 4.5 | Form of Warrant (2012 Unit Offering).* | |
| 5.1 | Opinion of Indeglia & Carney, P.C.** | |
| 10.1 | License Agreement dated October 10, 2007 between Vaccinogen, Inc. and Intracel Holdings Corporation.* | |
| 10.2 | Letter Agreement dated October 31, 2007 by and among Intracel Holdings Corporation, Intracel Acquisition Holding Company LLC, Organon Teknika Corp. and Organon Biosciences International BV.* | |
| 10.3 | New Security Agreement dated October 31, 2007 by and among Intracel Holdings Corporation, Organon Teknika Corp. and Organon Biosciences International BV.* | |
| 10.4 | Extension and Second Amendment to Lease dated October 23, 2012 by and between Martens Properties, LLLP and Vaccinogen.* | |
| 10.5 | Security Agreement dated October 26, 2011 by and between Vaccinogen, Inc. and The Abell Foundation, Inc.* |
| II-7 |
| 10.6 | Stock Exchange Agreement dated as of June 24, 2010 by and between Vaccinogen, Inc. Intracel Holdings Corporation and the other parties thereto.* | |
| 10.7 | Agreement dated April 16, 2012 between the Company and Oncology Trials Insight, Inc.* | |
| 10.8 | Investment Agreement dated July 18, 2012 between Kodiak Capital Group, LLC and Vaccinogen, Inc..* | |
| 10.9 | Investment Agreement dated January 16, 2013 between The Abell Foundation and Vaccinogen, Inc.* | |
| 10.10 | Registration Rights Agreement dated July 18, 2012 between Kodiak Capital Group, LLC and Vaccinogen.* | |
| 10.11 | Registration Rights Agreement dated June 24, 2010 by and between Vaccinogen, Inc. Intracel Holdings Corporation and the other parties thereto (series B preferred stockholders). * | |
| 10.12 | Amended and Restated Registration Rights Agreement by and between Vaccinogen, Inc., and the other parties thereto (series AA preferred stockholders).* | |
| 10.13 | Advisory Agreement dated May 1, 2009 between Alms and Associates, Inc. and Vaccinogen, Inc.* | |
| 10.14 | Agreement dated October 10, 2012 between Marquant Partners and Vaccinogen, Inc.* | |
| 10.15 | Employment Agreement between Vaccinogen and Michael G. Hanna, Jr., Ph.D.* | |
| 10.16 | Employment Agreement between Vaccinogen and Andrew Tussing.* | |
| 10.17 | Amendment No. 2 to Note and Warrant Purchase Agreement dated January 16, 2013 between Vaccinogen and The Abell Foundation. | |
| 10.18 | Patent Security Agreement dated April 2013 by and between Vaccinogen and The Abell Foundation, Inc.* | |
| 10.19 | Amendment No. 3 to Note and Warrant Purchase Agreement dated April 18, 2013 between Vaccinogen and The Abell Foundation.* | |
| 10.20 | Placement Agent Agreement with First Liberties Financial.* | |
| 10.21 | Extension and Third Amendment to Lease dated April 2013 by and between Martens Properties LLLP and Vaccinogen.* | |
| 10.22 | Form of Subscription Agreement (2012-2013 Unit Offering)* | |
| 10.23 | Amendment to Investment Agreement dated July 8, 2013. |
| II-8 |
| 21.1 | Subsidiaries.* | |
| 23.1 | Consent of Independent Public Accountants, BDO USA, LLP. | |
| 23.2 | Consent of Indeglia & Carney, P.C. (part of Exhibit 5.1)** |
*Filed as an exhibit to our Registration on Form 10 filed with the SEC on July 5, 2013 (File No. 000-54997) and incorporated herein by reference.
**To be filed by amendment.
Item 17. Undertakings.
The undersigned registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
i. To include any prospectus required by section 10(a)(3) of the Securities Act of 1933;
ii. To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the "Calculation of Registration Fee" table in the effective registration statement.
iii. To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
| II-9 |
(5) Each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(6) That, for the purpose of determining liability of the registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities: The undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
i. Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
ii. Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
iii. The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
iv. Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
| II-10 |
SIGNATURES
In accordance with the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-1 and authorized this registration statement to be signed on its behalf by the undersigned on July 12, 2013.
| VACCINOGEN, INC. | ||
| By: | /s/ Michael G. Hanna, Jr., Ph.D. | |
| Michael G. Hanna Jr., Ph.D. | ||
Chief Executive Officer,
Principal (Principal Executive Officer ) | ||
Pursuant to the requirements of the Securities Act of 1933, as amended, this Registration Statement has been signed by the following person in the capacities and on the date indicated.
| Signatures | Title | Date | ||
| /s/ Michael G. Hanna, Jr., Ph.D | Chairman and Chief Executive Officer | July 12, 2013 | ||
| Michael G. Hanna, Jr., Ph.D | (Principal Executive Officer) | |||
| /s/ Andrew Tussing | President, Chief Operating Officer and | July 12, 2013 | ||
| Andrew Tussing | acting Chief Financial Officer | |||
| (Principal Financial and Accounting Officer) | ||||
| /s/ John Nicolis | Director | July 12, 2013 | ||
| John Nicolis | ||||
| /s/ Daniel Fitzgerald | Director | July 12, 2013 | ||
| Daniel Fitzgerald | ||||
| /s/ Alan Cohen | Director | July 12, 2013 | ||
| Alan Cohen | ||||
| /s/ Daniel Kane | Director | July 12, 2013 | ||
| Daniel Kane |