Attached files
| file | filename |
|---|---|
| EX-1.1 - EX-1.1 - Regulus Therapeutics Inc. | d553765dex11.htm |
| EX-23.1 - EX-23.1 - Regulus Therapeutics Inc. | d553765dex231.htm |
| EX-10.48 - EX-10.48 - Regulus Therapeutics Inc. | d553765dex1048.htm |
| EX-10.50 - EX-10.50 - Regulus Therapeutics Inc. | d553765dex1050.htm |
| EX-10.46 - EX-10.46 - Regulus Therapeutics Inc. | d553765dex1046.htm |
| EX-10.47 - EX-10.47 - Regulus Therapeutics Inc. | d553765dex1047.htm |
| EX-10.49 - EX-10.49 - Regulus Therapeutics Inc. | d553765dex1049.htm |
Table of Contents
As filed with the Securities and Exchange Commission on June 26, 2013
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Regulus Therapeutics Inc.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 2834 | 26-4738379 | ||
| (State or Other Jurisdiction of Incorporation or Organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
3545 John Hopkins Ct.
Suite 210
San Diego, CA 92121
(858) 202-6300
(Address, Including Zip Code, and Telephone Number, Including Area Code, of Registrant’s Principal Executive Offices)
Kleanthis G. Xanthopoulos, Ph.D.
President and Chief Executive Officer
Regulus Therapeutics Inc.
3545 John Hopkins Court
Suite 210
San Diego, CA 92121
(858) 202-6300
(Name, Address, Including Zip Code, and Telephone Number, Including Area Code, of Agent for Service)
Copies to:
| Thomas A. Coll, Esq. Kenneth J. Rollins, Esq. Cooley LLP 4401 Eastgate Mall San Diego, CA 92121 (858) 550-6000 |
Mitchell S. Bloom, Esq. Maggie L. Wong, Esq. Goodwin Procter LLP 53 State Street Boston, MA 02109 (617) 570-1000 |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended (the “Securities Act”), check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer |
¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer |
¨ (Do not check if a smaller reporting company) | Smaller reporting company | x |
CALCULATION OF REGISTRATION FEE
|
| ||||
| Title of each class of securities to be registered | Proposed maximum aggregate offering price (1) |
Amount of registration fee | ||
| Common Stock, $0.001 par value per share |
$57,500,000 | $7,843.00 | ||
|
| ||||
| (1) | Estimated solely for the purpose of calculating the amount of the registration fee in accordance with Rule 457(o) under the Securities Act. Includes the offering price of shares that the underwriters have the option to purchase to cover over-allotments, if any. |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment that specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and we are not soliciting offers to buy these securities in any jurisdiction where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED JUNE 26, 2013
Shares

Common Stock
Regulus Therapeutics Inc. is offering shares of its common stock. Our common stock is listed on The NASDAQ Global Market under the symbol “RGLS.” On , 2013, the last reported sale price of our common stock on The NASDAQ Global Market was $ per share.
We are an emerging growth company as that term is used in the Jumpstart Our Business Startups Act of 2012 and, as such, have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings.
Investing in our common stock involves substantial risks. See “Risk factors” beginning on page 12.
PRICE $ A SHARE
| Per Share | Total | |||
| Public offering price | $ | $ | ||
| Underwriting discounts and commissions | $ | $ | ||
| Proceeds, before expenses, to us | $ | $ |
We have granted the underwriters an option for 30 days from the date of this prospectus to purchase up to of additional shares of our common stock to cover over-allotments. If the underwriters exercise the option in full, the total underwriting discounts and commissions payable by us will be $ , and the total proceeds to us, before expenses, will be $ .
The Securities and Exchange Commission and state securities regulators have not approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the shares of common stock to purchasers on , 2013.
| Lazard Capital Markets | Cowen and Company |
BMO Capital Markets |
| Needham & Company | Wedbush PacGrow Life Sciences |
The date of this prospectus is , 2013
Table of Contents
| Page | ||||
| 1 | ||||
| 12 | ||||
| 18 | ||||
| 20 | ||||
| 21 | ||||
| 22 | ||||
| 23 | ||||
| 24 | ||||
| 26 | ||||
| 28 | ||||
| 30 | ||||
| 68 | ||||
| 71 | ||||
| 74 | ||||
| 78 | ||||
| Material U.S. federal income tax consequences to non-U.S. holders of our common stock |
80 | |||
| 84 | ||||
| 88 | ||||
| 88 | ||||
| 88 | ||||
| 89 | ||||
You should rely only on the information contained in this prospectus and in any free writing prospectus that we may have provided to you in connection with this offering. Neither we nor any of the underwriters has authorized anyone to provide you with information different from, or in addition to, that contained in this prospectus or any such free writing prospectus. If anyone provides you with different or inconsistent information, you should not rely on it. Neither we nor any of the underwriters is making an offer to sell or seeking offers to buy shares of our common stock in any jurisdiction where or to any person to whom the offer or sale is not permitted. The information in this prospectus is accurate only as of the date on the front cover of this prospectus and the information in any free writing prospectus that we may have provided to you in connection with this offering is accurate only as of the date of that free writing prospectus. Our business, financial condition, results of operations and future growth prospects may have changed since those dates.
For investors outside the United States: neither we nor any of the underwriters has done anything that would permit this offering or possession or distribution of this prospectus or any free writing prospectus we may provide to you in connection with this offering in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus and any such free writing prospectus outside of the United States.
i
Table of Contents
This summary provides an overview of selected information contained elsewhere in this prospectus or incorporated by reference into this prospectus from our Annual Report on Form 10-K for the year ended December 31, 2012 and our other filings with the Securities and Exchange Commission listed in the section of this prospectus entitled “Incorporation of certain information by reference” and does not contain all of the information you should consider before investing in our common stock. You should carefully read this prospectus, the registration statement of which this prospectus is a part and the information incorporated by reference herein in their entirety before investing in our common stock, including the information discussed under “Risk factors” in this prospectus and in our Annual Report on Form 10-K for the year ended December 31, 2012 and our Quarterly Report on Form 10-Q for the quarter ended March 31, 2013 incorporated by reference herein, along with our financial statements and notes thereto that are incorporated by reference herein. Unless otherwise indicated herein, the terms “Regulus,” “we,” “our,” “us,” or “the Company” refer to Regulus Therapeutics Inc.
OVERVIEW
We are a biopharmaceutical company focused on discovering and developing first-in-class drugs that target microRNAs to treat a broad range of diseases. We were formed in 2007 when Alnylam Pharmaceuticals, Inc., or Alnylam, and Isis Pharmaceuticals, Inc., or Isis, contributed significant intellectual property, know-how and financial and human capital to pursue the development of drugs targeting microRNAs pursuant to a license and collaboration agreement. microRNAs are recently discovered, naturally occurring ribonucleic acid, or RNA, molecules that play a critical role in regulating key biological pathways. Scientific research has shown that the improper balance, or dysregulation, of microRNAs is directly linked to many diseases. We believe we have assembled the leading position in the microRNA field, including expertise in microRNA biology and oligonucleotide chemistry, a broad intellectual property estate, key opinion leaders and disciplined drug discovery and development processes. We refer to these assets as our microRNA product platform. We are using our microRNA product platform to develop chemically modified, single-stranded oligonucleotides that we call anti-miRs. We use these anti-miRs to modulate microRNAs and by doing so return diseased cells to their healthy state. We believe microRNAs may be transformative in the field of drug discovery and that anti-miRs may become a new and major class of drugs with broad therapeutic application much like small molecules, biologics and monoclonal antibodies.
We are currently optimizing anti-miRs in several distinct programs, both independently and with our strategic alliance partners, AstraZeneca AB, or AstraZeneca, GlaxoSmithKline plc, or GSK, and Sanofi. We also have a collaboration agreement with Biogen Idec MA Inc., or Biogen Idec, to evaluate the potential use of microRNA signatures as a biomarker for human patients with multiple sclerosis. Under these strategic alliances, we are eligible to receive up to approximately $1.3 billion in milestone payments upon successful commercialization of microRNA therapeutics for the programs contemplated by our agreements. These payments include up to $42.0 million upon achievement of preclinical and IND milestones, up to $272.0 million upon achievement of clinical development milestones, up to $305.0 million upon achievement of regulatory milestones and up to $670.0 million upon achievement of commercialization milestones.
We are currently executing on our ‘Road to the Clinic’ strategy which sets forth certain corporate goals that seek to advance our microRNA therapeutic pipeline towards the clinic. Specifically, we set the goal of nominating two microRNA candidates for clinical development in 2013. In May 2013, we announced our first clinical candidate, RG-101, for which we have full ownership and commercial rights. RG-101 is a GalNAc-conjugated microRNA anti-miR, which targets microRNA-122, for the treatment of patients with chronic hepatitis C virus infection, or HCV. We expect to submit our first investigational new drug
| 1 |
Table of Contents
application, or IND, to the U.S. Food and Drug Administration, or FDA, or equivalent foreign regulatory filing with foreign regulatory authorities, as applicable, for RG-101 in the first half of 2014. We anticipate that we will nominate a second clinical development candidate by the end of 2013.
POTENTIAL OF microRNA BIOLOGY
RNA plays an essential role in the process used by cells to encode and translate genetic information from DNA to proteins. RNA is comprised of subunits called nucleotides and is synthesized from a DNA template by a process known as transcription. Transcription generates different types of RNA, including messenger RNAs that carry the information for proteins in the sequence of their nucleotides. In contrast, microRNAs are small RNAs that do not code for proteins but rather are responsible for regulating gene expression by affecting the translation of target messenger RNAs. By interacting with many messenger RNAs, a single microRNA can regulate several genes that are instrumental for the normal function of a biological pathway. More than 500 microRNAs have been identified to date in humans, each of which is believed to interact with a specific set of genes that control key aspects of cell biology. Since most diseases are multi-factorial and involve multiple targets in a pathway, the ability to modulate gene networks by targeting a single microRNA provides a new therapeutic approach for treating complex diseases.
We believe that microRNA therapeutics have the potential to become a new and major class of drugs with broad therapeutic application for the following reasons:
| Ø | microRNAs are a recent discovery in biology and, up until now, have not been a focus of pharmaceutical research; |
| Ø | microRNAs play a critical role in regulating biological pathways by controlling the translation of many target genes; |
| Ø | microRNA therapeutics target entire disease pathways which may result in more effective treatment of complex multi-factorial diseases; |
| Ø | microRNA therapeutics can be produced with a more efficient rational drug design process; and |
| Ø | microRNA therapeutics may be synergistic with other therapies because of their different mechanism of action. |
OUR microRNA PRODUCT PLATFORM
We are the leading company in the field of microRNA therapeutics. Backed by our founding companies, Alnylam and Isis, we are uniquely positioned to leverage oligonucleotide technologies that have been proven in clinical trials. Central to achieving our goals is the know-how that we have accumulated in oligonucleotide design and how the specific chemistries behave in the clinical setting. We refer to this collective know-how, proprietary technology base, and its systematic application as our microRNA product platform.
We view the following as providing a competitive advantage for our microRNA product platform:
| Ø | a mature platform selectively producing multiple development candidates advancing to the clinic; |
| Ø | scientific advisors who are pioneers in the microRNA field; |
| Ø | access to proven RNA therapeutic technologies through our founding companies, as well as approximately 900 patents and patent applications relating to oligonucleotide technologies; |
| Ø | a leading microRNA intellectual property estate with access to over 170 patents and patent applications covering compositions and therapeutic uses; |
| 2 |
Table of Contents
| Ø | development expertise and financial resources provided by our three major strategic alliances with AstraZeneca, GSK and Sanofi; and |
| Ø | over 30 academic collaborations that help us identify new microRNA targets and support our early stage discovery efforts. |
The disciplined approach we take for the discovery and development of microRNA therapeutics is as important as the assets assembled to execute our plans and is based on the following four steps:
Step 1 - Evaluation of microRNA therapeutic opportunities
The initiation of our microRNA discovery and development efforts is based on rigorous scientific and business criteria, including:
| Ø | existence of significant scientific evidence to support the role of a specific microRNA in a disease; |
| Ø | availability of predictive preclinical disease models to test our microRNA development candidates; |
| Ø | ability to effectively deliver anti-miRs to the diseased cells or tissues; and |
| Ø | existence of a reasonable unmet medical need and commercial opportunity. |
Step 2 - Identification of microRNA targets
We identify microRNA targets through bioinformatic analysis of public and proprietary microRNA expression profiling data sets from samples of diseased human tissues. The analysis of such data sets can immediately highlight a potential role for specific microRNAs in the disease being studied. Further investigation of animal models that are predictive of human diseases in which those same microRNAs are also dysregulated provides additional data to support a new program. We have applied this strategy successfully in our existing programs and we believe that this approach will continue to help us identify clinically relevant microRNA targets.
Step 3 - Validation of microRNA targets
Our validation strategy is based on two distinct steps. First, using genetic tools, we determine whether up-regulation, or overproduction, of the microRNA in healthy animals can create the specific disease state and inhibition of the microRNA can lead to a therapeutic benefit. Second, using animal models predictive of human diseases, we determine whether pharmacological modulation of the up-regulated microRNA target with our anti-miRs can also lead to a therapeutic benefit. This validation process enables us to prioritize the best microRNA targets that appear to be key drivers of disease and not simply correlating markers.
Step 4 - Optimization of microRNA development candidates
We have developed a proprietary process that allows us to rapidly generate an optimized development candidate. Unlike traditional drug classes, such as small molecules, in which thousands of compounds must be screened to identify prospective leads, the fact that anti-miRs are mirror images of their target microRNAs allows for a more efficient rational design process. The optimization process incorporates our extensive knowledge base around oligonucleotide chemistry and anti-miR design to efficiently synthesize a starting pool of rationally designed anti-miRs to be evaluated in a series of proven assays and models. We also enhance our anti-miRs for distribution to the tissues where the specific microRNA target is causing disease.
OUR INITIAL DEVELOPMENT CANDIDATES
We are developing single-stranded oligonucleotides, which are chemically synthesized chains of nucleotides that are mirror images of specific target microRNAs. We incorporate proprietary chemical modifications to enhance drug properties such as potency, stability and tissue distribution. We refer to
| 3 |
Table of Contents
these chemically modified oligonucleotides as anti-miRs. Each anti-miR is designed to bind with and inhibit a specific microRNA target that is up-regulated in a cell and that is involved in the disease state. In binding to the microRNA, anti-miRs correct the dysregulation and return diseased cells to their healthy state. We have demonstrated therapeutic benefits of our anti-miRs in at least 20 different preclinical models of human diseases.
We have identified and validated several microRNA targets across a number of disease categories and are working independently and with our strategic alliance partners to optimize anti-miR development candidates. We expect that anti-miR development candidates will be easily formulated in saline solution and administered systemically or locally depending on the therapeutic indication. Our distinct therapeutic development programs are shown in the table below:
| microRNA target | anti-miR program | Commercial rights | ||
| miR-122 | RG-101 for HCV | Regulus* | ||
| miR-221 | Hepatocellular carcinoma | Regulus | ||
| miR-10b | Glioblastoma | Regulus | ||
| miR-21 | Hepatocellular carcinoma | Sanofi | ||
| miR-21 | Kidney fibrosis | Sanofi | ||
| miR-33 | Atherosclerosis | AstraZeneca |
| * | With the exception of RG-101, commercial rights for miR-122 target licensed to GSK. |
One aspect of our strategy is to pursue a balanced approach between product candidates that we develop ourselves and those that we develop with partners. We intend to focus our own resources on proprietary product opportunities in therapeutic areas where development and commercialization activities are appropriate for our size and financial resources, which we anticipate will include niche indications and orphan diseases. In therapeutic areas where costs are more significant, development timelines are longer or markets are too large for our capabilities, we will seek to secure partners with requisite expertise and resources.
Our approach has been validated to date by the following strategic alliances and collaborations with large pharmaceutical companies:
| Ø | In April 2008, we formed a strategic alliance with GSK to discover and develop microRNA therapeutics for immuno-inflammatory diseases. In February 2010, we and GSK expanded the alliance to include potential microRNA therapeutics for the treatment of HCV. In June 2013, we amended our agreement with GSK and agreed that RG-101 is fully-owned by us and that miR-122 remains a collaboration target under the agreement. |
| Ø | In June 2010, we formed a strategic alliance with Sanofi to discover and develop microRNA therapeutics for fibrotic diseases. In July 2012, we expanded the alliance to include potential microRNA therapeutics in oncology. The original research term for this strategic alliance expired in June 2013, upon which we and Sanofi entered into an option agreement pursuant to which we granted Sanofi an exclusive right to negotiate the co-development and commercialization of certain of our unencumbered microRNA programs through December 2013, for which Sanofi has agreed to pay us an upfront option fee of $2.5 million, $1.25 million of which is creditable against future amounts payable by Sanofi to us. In addition, Sanofi granted us an exclusive option, which also expires in December 2013 to negotiate the co-development and commercialization of miR-21. |
| Ø | In August 2012, we formed a strategic alliance with AstraZeneca to discover and develop microRNA therapeutics for cardiovascular diseases, metabolic diseases and oncology. |
| Ø | In August 2012, we entered into a collaboration agreement with Biogen Idec to evaluate the potential use of microRNA signatures as a biomarker for human patients with multiple sclerosis. In June 2013, we and Biogen Idec amended the collaboration agreement to update the research plan and criteria for success. |
| 4 |
Table of Contents
OUR STRATEGY
We are building the leading biopharmaceutical company focused on the discovery and development of first-in-class, targeted drugs based on our proprietary microRNA product platform. The key elements of our strategy are to:
| Ø | Rapidly advance our initial programs into clinical development. We are currently optimizing our proprietary and partnered anti-miRs for development candidate selection. Under our ‘Road to the Clinic’ strategy, we have nominated our fully-owned compound, RG-101, for the treatment of HCV as our first clinical candidate and expect to submit our first IND, or equivalent foreign regulatory filing, for RG-101 in the first half of 2014. We anticipate that we will nominate a second clinical candidate by the end of 2013. |
| Ø | Focus our resources on developing drugs for niche indications or orphan diseases. We believe that microRNA therapeutics have utility in almost every disease state as they regulate pathways, not single targets. We intend to focus on proprietary product opportunities in niche therapeutic areas where the development and commercialization activities are appropriate for our size and financial resources. |
| Ø | Selectively form strategic alliances to augment our expertise and accelerate development and commercialization. We have established strategic alliances with AstraZeneca, GSK and Sanofi and we will continue to seek partners who can bring therapeutic expertise, development and commercialization capabilities and funding to allow us to maximize the potential of our microRNA product platform. |
| Ø | Selectively use our microRNA product platform to develop additional targets. We have identified several other microRNA targets with potential for therapeutic modulation and will apply our rigorous scientific and business criteria to develop them. |
| Ø | Develop microRNA biomarkers to support therapeutic product candidates. We believe that microRNA biomarkers may be used to select optimal patient segments in clinical trials, to develop companion diagnostics, and to monitor disease progression or relapse. We believe these microRNA biomarkers can be applied toward drugs that we develop and drugs developed by other companies, including small molecules and monoclonal antibodies. |
| Ø | Maintain scientific and intellectual leadership in the microRNA field. We will continue to conduct research in the microRNA field to better understand this new biology and characterize the specific mechanism of action for our future drugs. This includes building on our strong network of key opinion leaders and securing additional intellectual property rights to broaden our existing proprietary asset estate. |
OUR LEADERSHIP
Our management has more than 50 years of collective experience leading the discovery and development of innovative therapeutics, including significant operational and financial experience with emerging biotechnology companies, which we believe is the ideal combination of talent to execute our strategy. In addition, our experienced board of directors, which includes representatives of our founding companies, Alnylam and Isis, provides significant support and guidance in all aspects of our business.
Our executive officers are:
| Ø | Kleanthis G. Xanthopoulos, Ph.D., our President and Chief Executive Officer, is an entrepreneur who has been involved in founding several companies, including Anadys Pharmaceuticals, Inc. (acquired by F. Hoffmann-La Roche Inc. in 2011), which he started as President and Chief Executive Officer. |
| 5 |
Table of Contents
| Ø | Neil W. Gibson, Ph.D., our Chief Scientific Officer, is a leading scientist focused on cancer research and drug development who previously served as Chief Scientific Officer of the Oncology Research Unit at Pfizer Inc. and as Chief Scientific Officer of OSI Pharmaceuticals, Inc. He was involved in the development of several commercial cancer drugs including Xalkori® (crizotinib), Nexavar® (sorafenib) and Tarceva® (erlotinib). |
Our executive team is supported by the following key personnel:
| Ø | Mary Glanville, our Senior Vice President of Human Capital, is an accomplished human resources executive in the life sciences industry who previously served in management roles at Anadys Pharmaceuticals, Inc. (acquired by F. Hoffman-La Roche Inc. in 2011), Inflazyme Inc. and Inex Pharmaceuticals Corp. |
| Ø | Victor Knopov, Ph.D., our Vice President, Pharmaceutical Development, is a leader in oligonucleotide drug delivery and pharmaceutical development who has held positions at Nitto Denko Technical Corporation, Bio-Medics, Inc., EnGene, Inc., Marina Biotech, Inc. and Inex Pharmaceuticals Corporation. Dr. Knopov has extensive knowledge of Chemistry, Manufacturing and Control, or CMC, development for various technology platforms including commercial production of enzymes, anticancer liposomal products as well as advanced delivery systems for antisense, plasmids and siRNA based on lipids, polymer nanoparticles and conjugated systems. |
| Ø | Daniel R. Chevallard, CPA, our Vice President, Finance and Accounting, is a corporate finance leader with public accounting expertise who previously held senior roles in corporate finance, accounting and financial reporting as a corporate controller and Senior Director, Finance at Prometheus Laboratories Inc. and who was a senior financial auditor at Ernst & Young LLP. |
Our executive team, key personnel and board of directors are supported by our scientific advisory board members, who are renowned pioneers in the microRNA field:
| Ø | David Baltimore, Ph.D., Chairman of our scientific advisory board and Professor of Biology at the California Institute of Technology, received the Nobel Prize in 1975 and is highly regarded as a pioneer in virology and immunology, with his current research investigating the role of microRNAs in immunity. Dr. Baltimore is also a member of our board of directors. |
| Ø | David Bartel, Ph.D., Professor of Biology at the Massachusetts Institute of Technology and the Whitehead Institute for Biomedical Research and an investigator at the Howard Hughes Medical Institute, studies microRNA genomics, target recognition and regulatory functions. |
| Ø | Gregory Hannon, Ph.D., Professor at the Cold Spring Harbor Laboratory and an investigator at the Howard Hughes Medical Institute, has identified and characterized many of the major biogenesis and effector complexes for microRNA biology. |
| Ø | Markus Stoffel, M.D., Ph.D., Professor of Metabolic Diseases at the Swiss Federal Institute of Technology, is focused on microRNA research and the regulation of glucose and lipid metabolism. |
| Ø | Thomas Tuschl, Ph.D., Professor and Head of the Laboratory for RNA Molecular Biology at the Rockefeller University and an investigator at the Howard Hughes Medical Institute, discovered many of the mammalian microRNA genes and has developed methods for characterization of small RNAs. |
| 6 |
Table of Contents
RISKS ASSOCIATED WITH OUR BUSINESS
Our business and ability to execute our business strategy are subject to a number of risks of which you should be aware before you decide to buy our common stock. In particular, you should consider the following risks, which are discussed more fully in the section entitled “Risk factors” in this prospectus and in our Annual Report on Form 10-K for the year ended December 31, 2012 and our Quarterly Report on Form 10-Q for the quarter ended March 31, 2013, incorporated by reference herein.
| Ø | We have never generated any revenue from product sales and may never become profitable. Even if this offering is successful, we may need to raise additional funds to support our operations and such funding may not be available to us on acceptable terms, or at all. |
| Ø | The approach we are taking to discover and develop drugs is novel and may never lead to marketable products. |
| Ø | All of our programs are still in preclinical development. Preclinical testing and clinical trials of our future product candidates may not be successful. If we are unable to successfully complete preclinical testing and clinical trials of our product candidates or experience significant delays in doing so, our business will be materially harmed. |
| Ø | We will depend on our strategic alliances for the development and eventual commercialization of certain future microRNA product candidates. If these strategic alliances are unsuccessful or are terminated, we may be unable to commercialize certain product candidates or generate future revenue from our development programs. |
| Ø | If we are unable to obtain or protect intellectual property rights related to our future products and product candidates, we may not be able to effectively develop or commercialize any of our product candidates or otherwise compete effectively in our markets. |
| Ø | We will need to expand our organization and we may experience difficulties in managing this growth, which could disrupt our operations. |
| Ø | Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel and our failure to do so might impede the progress of our research, development and commercialization objectives. |
CORPORATE INFORMATION
We were originally formed as a limited liability company under the name Regulus Therapeutics LLC in the State of Delaware in September 2007. In January 2009, we converted Regulus Therapeutics LLC to a Delaware corporation and changed our name to Regulus Therapeutics Inc. Our principal executive offices are located at 3545 John Hopkins Court, Suite 210, San Diego, California 92121, and our telephone number is (858) 202-6300. Our corporate website address is www.regulusrx.com. Information contained on or accessible through our website is not a part of this prospectus, and the inclusion of our website address in this prospectus is an inactive textual reference only.
We use “Regulus Therapeutics” as a trademark in the United States and other countries. We have filed for registration of this trademark in the United States and have registered it in the European Union and Switzerland. This prospectus contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this prospectus, including logos, artwork and other visual displays, may appear without the ® or ™ symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
| 7 |
Table of Contents
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of (a) December 31, 2017, (b) the last day of the fiscal year in which we have total annual gross revenue of at least $1.0 billion, or (c) the last day of the fiscal year in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th and (d) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. We refer to the Jumpstart Our Business Startups Act of 2012 herein as the “JOBS Act,” and references herein to “emerging growth company” shall have the meaning associated with it in the JOBS Act.
| 8 |
Table of Contents
The offering
| Common stock offered by us |
shares |
| Common stock to be outstanding after this offering |
shares |
| Over-allotment option |
The underwriters have an option for a period of 30 days to purchase up to additional shares of our common stock to cover over-allotments. |
| Use of proceeds |
We intend to use the net proceeds of this offering for preclinical and clinical development of our proprietary compound, RG-101, and our other initial microRNA development candidates, for the identification and validation of additional microRNA targets and for other general corporate purposes. See “Use of proceeds.” |
| Risk factors |
You should read the “Risk factors” section of this prospectus for a discussion of certain factors to consider carefully before deciding to purchase any shares of our common stock. |
| NASDAQ Global Market symbol |
RGLS |
The number of shares of our common stock to be outstanding after this offering is based on 35,965,371 shares of common stock outstanding as of March 31, 2013, and excludes:
| Ø | 4,742,780 shares of common stock issuable upon the exercise of outstanding stock options as of March 31, 2013, at a weighted average exercise price of $2.24 per share; |
| Ø | 2,191,925 shares of common stock reserved for future issuance under our 2012 equity incentive plan, or the 2012 Plan, plus any future increases in the number of shares of common stock reserved for issuance under the 2012 Plan pursuant to the evergreen provision; and |
| Ø | 481,274 shares of common stock reserved for future issuance under our 2012 employee stock purchase plan, or the ESPP, plus any future increases in the number of shares of common stock reserved for issuance under the ESPP pursuant to the evergreen provision. |
Unless otherwise indicated, all information contained in this prospectus, and the number of shares of common stock outstanding as of March 31, 2013 assumes no exercise by the underwriters of their over-allotment option to purchase up to an additional shares of our common stock.
| 9 |
Table of Contents
Summary financial data
The following table summarizes our financial data. We derived the summary statement of operations data for the years ended December 31, 2010, 2011 and 2012 from our audited financial statements and related notes incorporated by reference in this prospectus. We derived the summary statement of operations data for the three months ended March 31, 2012 and 2013 and balance sheet data as of March 31, 2013 from our unaudited financial statements and related notes incorporated by reference in this prospectus. Our historical results are not necessarily indicative of the results that may be expected in the future and results of interim periods are not necessarily indicative of the results for the entire year. The summary financial data should be read together with our financial statements and related notes incorporated by reference in this prospectus, “Selected financial data” and “Management’s discussion and analysis of financial condition and results of operations” appearing elsewhere or incorporated by reference in this prospectus.
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||
| Statement of operations data | 2010 | 2011 | 2012 | 2012 | 2013 | |||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||
| (unaudited) | ||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Revenue under strategic alliances and collaborations |
$ | 8,112 | $ | 13,767 | $ | 12,700 | $ | 3,344 | $ | 3,238 | ||||||||||
| Grant revenue |
489 | 22 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total revenues |
8,601 | 13,789 | 12,700 | 3,344 | 3,238 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
20,178 | 17,289 | 20,342 | 4,603 | 6,883 | |||||||||||||||
| General and administrative |
3,921 | 3,637 | 4,932 | 921 | 1,905 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
24,099 | 20,926 | 25,274 | 5,524 | 8,788 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(15,498 | ) | (7,137 | ) | (12,574 | ) | (2,180 | ) | (5,550 | ) | ||||||||||
| Other expense, net |
(91 | ) | (259 | ) | (4,844 | ) | (66 | ) | (1,689 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss before income taxes |
(15,589 | ) | (7,396 | ) | (17,418 | ) | (2,246 | ) | (7,239 | ) | ||||||||||
| Income tax (benefit) expense |
(30 | ) | 206 | (10 | ) | 1 | (10 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (15,559 | ) | $ | (7,602 | ) | $ | (17,408 | ) | $ | (2,247 | ) | $ | (7,229 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss per share, basic and diluted(1) |
$ | (85.82 | ) | $ | (2.12 | ) | $ | (13.06 | ) | $ | (0.20 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Shares used to compute basic and diluted net loss per share(1) |
88,582 | 8,212,538 | 171,998 | 35,872,606 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| (1) | See Note 2 of our Notes to Financial Statements incorporated by reference in this prospectus for an explanation of the method used to calculate the basic and diluted net loss per common share and the number of shares used in the computation of the share and per share data. No share or per share data have been presented for 2010 since we had no common shares outstanding during that year. |
| 10 |
Table of Contents
The unaudited as adjusted balance sheet data set forth below give effect to our issuance and sale of shares of our common stock in this offering at an assumed public offering price of $ per share, which is the last reported sale price of our common stock as reported on The NASDAQ Global Market on , 2013, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
| As of March 31, 2013 | ||||||||
| Balance sheet data | Actual | Pro forma as adjusted(1) |
||||||
| (unaudited, in thousands) | ||||||||
| Cash, cash equivalents and short-term investments |
$ | 90,715 | $ | |||||
| Working capital |
79,076 | |||||||
| Total assets |
97,027 | |||||||
| Convertible notes payable |
11,895 | 11,895 | ||||||
| Accumulated deficit |
(67,648 | ) | ||||||
| Total stockholders’ equity |
55,867 | |||||||
| (1) | Each $1.00 increase (decrease) in the assumed public offering price of $ per share, the last reported sale price of our common stock on The NASDAQ Global Market on , 2013, would increase (decrease) the as adjusted amount of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase of shares in the number of shares offered by us would increase the as adjusted amount of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $ million, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, each decrease of shares in the number of shares offered by us would decrease the as adjusted amount of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $ million, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The as adjusted information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
| 11 |
Table of Contents
A purchase of shares of our common stock is an investment in our securities and involves a high degree of risk. You should carefully consider the risks and uncertainties and all other information contained in or incorporated by reference in this prospectus, including the risks and uncertainties discussed under “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2012 and our Quarterly Report on Form 10-Q for the quarter ended March 31, 2013. All of these risk factors are incorporated by reference herein in their entirety. If any of these risks actually occur, our business, financial condition and results of operations would likely suffer. In that case, the market price of our common stock could decline, and you may lose part or all of your investment in our company. Additional risks of which we are not presently aware or that we currently believe are immaterial may also harm our business and results of operations.
RISKS RELATED TO THIS OFFERING AND OWNERSHIP OF OUR COMMON STOCK
The market price of our common stock may be highly volatile, and you may not be able to resell your shares at or above the public offering price.
The trading price of our common stock is likely to continue to be volatile. Since shares of our common stock were sold in our initial public offering in October 2012 at a price of $4.00 per share, our closing stock price as reported on The NASDAQ Global Market has ranged from $4.02 to $ , through , 2013.
Our stock price could be subject to wide fluctuations in response to a variety of factors, including the following:
| Ø | adverse results or delays in preclinical testing or clinical trials; |
| Ø | inability to obtain additional funding; |
| Ø | any delay in filing an IND or NDA for any of our future product candidates and any adverse development or perceived adverse development with respect to the FDA’s review of that IND or NDA; |
| Ø | failure to maintain our existing strategic alliances or enter into new alliances; |
| Ø | failure of our strategic alliance partners to elect to develop and commercialize product candidates under our alliance agreements or the termination of any programs under our alliance agreements; |
| Ø | failure by us or our licensors and strategic alliance partners to prosecute, maintain or enforce our intellectual property rights; |
| Ø | failure to successfully develop and commercialize our future product candidates; |
| Ø | changes in laws or regulations applicable to our preclinical and clinical development activities, product candidates or future products; |
| Ø | inability to obtain adequate product supply for our future product candidates or the inability to do so at acceptable prices; |
| Ø | adverse regulatory decisions; |
| Ø | introduction of new products, services or technologies by our competitors; |
| Ø | failure to meet or exceed financial projections we may provide to the public; |
| Ø | failure to meet or exceed the estimates and projections of the investment community; |
12
Table of Contents
Risk factors
| Ø | the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community; |
| Ø | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us, our strategic alliance partners or our competitors; |
| Ø | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| Ø | additions or departures of key scientific or management personnel; |
| Ø | significant lawsuits, including patent or stockholder litigation; |
| Ø | changes in the market valuations of similar companies; |
| Ø | sales of our common stock by us or our stockholders in the future; and |
| Ø | trading volume of our common stock. |
In addition, companies trading in the stock market in general, and The NASDAQ Global Market in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Our executive officers, directors, 5% stockholders and their affiliates beneficially own approximately 86% of our voting stock before this offering and, upon closing of this offering, that same group will beneficially own approximately % of our outstanding voting stock. Therefore, even after this offering, these stockholders will have the ability to influence us through this ownership position. These stockholders may be able to determine all matters requiring stockholder approval. For example, these stockholders, acting together, may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may believe are in your best interest as one of our stockholders.
Even if this offering is successful, we may need to raise additional funding, which may not be available on acceptable terms, or at all.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We expect our research and development expenses to substantially increase in connection with our ongoing activities, particularly as we advance our product candidates toward clinical programs. If we are unable to successfully complete this offering, we will need to seek alternative financing or change our operational plans to continue as a going concern. Even if this offering is successful, we may need to raise additional funds to support our operations and such funding may not be available to us on acceptable terms, or at all.
We estimate that the net proceeds from this offering will be approximately $ million. We expect that the net proceeds from this offering and our existing cash and cash equivalents, together with interest, will be sufficient to fund our current operations through at least the end of 2016. However, changing circumstances may cause us to consume capital more rapidly than we currently anticipate. For example, as we move our lead compounds through toxicology and other preclinical studies, also referred to as
13
Table of Contents
Risk factors
nonclinical studies, required to file an investigational new drug application, or IND, and as we conduct clinical development of RG-101 and any other future product candidates, we may have adverse results requiring that we find new product candidates. Additionally, our strategic alliance partners may not elect to pursue the development and commercialization of any of our microRNA product candidates that are subject to their respective strategic alliance agreements with us. Any of these events may increase our development costs more than we expect. We may need to raise additional funds or otherwise obtain funding through strategic alliances if we choose to initiate clinical trials for new product candidates other than programs currently partnered. In any event, we will require additional capital to obtain regulatory approval for, and to commercialize, future product candidates. Raising funds in the current economic environment, when the capital markets have been affected by the global recession, may present additional challenges.
If we are required to secure additional financing, such additional fundraising efforts may divert our management from our day-to-day activities, which may adversely affect our ability to develop and commercialize future product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we are unable to raise additional capital when required or on acceptable terms, we may be required to:
| Ø | significantly delay, scale back or discontinue the development or commercialization of any future product candidates; |
| Ø | seek strategic alliances for research and development programs at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; or |
| Ø | relinquish or license on unfavorable terms, our rights to technologies or any future product candidates that we otherwise would seek to develop or commercialize ourselves. |
If we are required to conduct additional fundraising activities and we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will be prevented from pursuing development and commercialization efforts, which will have a material adverse effect on our business, operating results and prospects.
We are an “emerging growth company,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an emerging growth company through December 31, 2017, although circumstances could cause us to lose that status earlier, including if the market value of our common stock held by non-affiliates exceeds $700.0 million as of any June 30 before that time or if we have total annual gross revenue of $1.0 billion or more during any fiscal year before that time, in which cases we would no longer be an emerging growth company as of the following December 31 or, if we issue more than $1.0 billion in non-convertible debt during any three year period before that time, we would cease to be an emerging growth company immediately. Even after we no longer qualify as an emerging growth company, we may still qualify as a “smaller reporting company” which would allow us to take
14
Table of Contents
Risk factors
advantage of many of the same exemptions from disclosure requirements including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in this prospectus and our periodic reports and proxy statements. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Sales of a substantial number of shares of our common stock in the public market by our existing stockholders could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our common stock.
We, along with our directors and executive management team have agreed that for a period of 90 days after the date of this prospectus, subject to specified exceptions, we or they will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock. Subject to certain limitations, approximately 140,327 shares will become eligible for sale upon expiration of such lock-up period, as calculated and described in more detail in the section entitled “Shares eligible for future sale.” In addition, shares issued or issuable upon exercise of options vested as of the expiration of the lock-up period will be eligible for sale at that time. Sales of stock by these stockholders could have a material adverse effect on the trading price of our common stock.
An additional 26,515,110 shares of our common stock are subject to lock-up agreements that were entered into in connection with our initial public offering, which lock-up agreements expire on October 4, 2013.
Certain holders of our securities are entitled to rights with respect to the registration of their shares under the Securities Act of 1933, as amended, or the Securities Act, subject to the applicable lock-up arrangement described above. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares held by our affiliates as defined in Rule 144 under the Securities Act. Any sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. These sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders.
15
Table of Contents
Risk factors
Pursuant to our 2012 equity incentive plan, or the 2012 Plan, our management is authorized to grant stock options and other equity-based awards to our employees, directors and consultants. The number of shares available for future grant under the 2012 Plan will automatically increase each year by up to 4% of all shares of our capital stock outstanding as of December 31 of the prior calendar year, subject to the ability of our board of directors to take action to reduce the size of the increase in any given year. Any such increase, of the maximum amount or a lesser amount, will cause our stockholders to experience additional dilution, which could cause our stock price to fall. Currently, we plan to register the increased number of shares available for issuance under the 2012 Plan each year.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds from this offering, including for any of the purposes described in the section entitled “Use of proceeds,” and you will not have the opportunity as part of your investment decision to assess whether the net proceeds are being used appropriately. Because of the number and variability of factors that will determine our use of the net proceeds from this offering , their ultimate use may vary substantially from their currently intended use. The failure by our management to apply these funds effectively could harm our business. Pending their use, we may invest the net proceeds from this offering in short-term, investment-grade, interest-bearing securities. These investments may not yield a favorable return to our stockholders.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three year period, the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited. We believe that, with our initial public offering and other transactions that have occurred over the past three years, we may have triggered an “ownership change” limitation. We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will therefore be limited to the appreciation of their stock.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States,
16
Table of Contents
Risk factors
involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions and inter partes reexamination proceedings before the U.S. Patent and Trademark Office and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our strategic alliance partners are pursuing development candidates. For example, we are aware that Santaris Pharma A/S, or Santaris, has patents and patent applications in the microRNA therapeutics space, including patents and patent applications related to targeting microRNAs, such as miR-122, for the treatment of disease. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our future product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our future product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our future product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our future product candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire. Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, including combination therapy, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product candidate unless we obtained a license or until such patent expires. In either case, such a license may not be available on commercially reasonable terms or at all.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our future product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
17
Table of Contents
Special note regarding forward-looking statements
This prospectus contains forward-looking statements. The forward-looking statements are contained principally in the sections entitled “Prospectus summary,” “Risk factors,” “Management’s discussion and analysis of financial condition and results of operations” and “Business” in this prospectus or incorporated by reference herein. These statements relate to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
| Ø | the initiation, cost, timing, progress and results of our research and development activities, preclinical studies and future clinical trials; |
| Ø | our ability to obtain and maintain regulatory approval of our future product candidates, and any related restrictions, limitations, and/or warnings in the label of an approved product candidate; |
| Ø | our ability to obtain funding for our operations; |
| Ø | our plans to research, develop and commercialize our future product candidates; |
| Ø | our strategic alliance partners’ election to pursue development and commercialization; |
| Ø | our ability to attract collaborators with development, regulatory and commercialization expertise; |
| Ø | our ability to obtain and maintain intellectual property protection for our future product candidates; |
| Ø | the size and growth potential of the markets for our future product candidates, and our ability to serve those markets; |
| Ø | our ability to successfully commercialize our future product candidates; |
| Ø | the rate and degree of market acceptance of our future product candidates; |
| Ø | our ability to develop sales and marketing capabilities, whether alone or with potential future collaborators; |
| Ø | regulatory developments in the United States and foreign countries; |
| Ø | the performance of our third-party suppliers and manufacturers; |
| Ø | the success of competing therapies that are or become available; |
| Ø | the loss of key scientific or management personnel; |
| Ø | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act; |
| Ø | our use of the proceeds from this offering; and |
| Ø | the accuracy of our estimates regarding expenses, future revenues, capital requirements and need for additional financing. |
In some cases, you can identify these statements by terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” or the negative of those terms, and similar expressions. These forward-looking statements reflect our management’s beliefs and views with respect to future events and are based on estimates and assumptions as of the date of this prospectus and are subject to risks and uncertainties. We discuss many of these risks in greater detail under the heading “Risk factors.” Moreover, we operate in a very
18
Table of Contents
Special note regarding forward-looking statements
competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Given these uncertainties, you should not place undue reliance on these forward-looking statements. The Private Securities Litigation Reform Act of 1995 and Section 27A of the Securities Act of 1933, as amended, do not protect any forward-looking statements that we make in connection with this offering.
You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements in this prospectus by these cautionary statements.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
19
Table of Contents
We estimate that we will receive net proceeds of approximately $ million (or approximately $ million if the underwriters’ over-allotment option is exercised in full) from the sale of the shares of common stock offered by us in this offering, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us.
We intend to use the net proceeds of this offering for preclinical and clinical development of our proprietary compound, RG-101, and our other initial microRNA development candidates, for the identification and validation of additional microRNA targets, and for capital expenditures, working capital and other general corporate purposes. We may also use a portion of the net proceeds to in-license, acquire or invest in complementary microRNA businesses, technologies, products or assets. However, we have no current commitments or obligations to do so. We cannot currently allocate specific percentages of the net proceeds that we may use for the purposes specified above. Accordingly, we will have broad discretion in the use of the net proceeds from this offering and could spend the proceeds in ways that do not improve our results of operations or enhance the value of our stock. Pending their use, we plan to invest the net proceeds from this offering in short- and intermediate-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the U.S. government.
20
Table of Contents
Price range of our common stock
Our common stock has been listed on the NASDAQ Global Market since October 4, 2012 under the symbol “RGLS”. Prior to that date, there was no public market for our common stock. Shares sold in our initial public offering on October 4, 2012 were priced at $4.00 per share.
On , 2013, the closing price for our common stock as reported on the NASDAQ Global Market was $ per share. The following table sets forth the ranges of high and low sales prices per share of our common stock as reported on the NASDAQ Global Market for the period indicated. Such quotations represent inter-dealer prices without retail markup, markdown or commission and may not necessarily represent actual transactions.
| Year Ended December 31, 2012 |
High | Low | ||
| Fourth Quarter (from October 4, 2012) |
$6.49 | $4.02 | ||
| Year Ended December 31, 2013 |
High | Low | ||
| First Quarter |
$7.89 | $4.67 | ||
| Second Quarter |
$ | $ | ||
| Third Quarter (through , 2013) |
$ | $ |
As of , 2013, there were stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers. The actual number of common stockholders is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
21
Table of Contents
Equity compensation plan information
The following table provides certain information with respect to all of our equity compensation plans in effect as of December 31, 2012:
| Plan Category | Number of securities to be issued upon exercise of outstanding restricted stock units and rights (a) |
Weighted-average exercise price of outstanding options (b) |
Number of securities remaining available for issuance under equity compensation plans (excluding securities reflected in column (a)) (c) |
|||||||||
| Equity compensation plans approved by security holders(1) |
4,719,799 | $ | 2.11 | 1,485,711 | ||||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
| Total |
4,719,799 | $ | 2.11 | 1,485,711 | ||||||||
| (1) | Available for the grant of future rights under our 2012 equity incentive plan, or 2012 Plan, and 2012 employee stock purchase plan, or ESPP, as of December 31, 2012, excluding future increases in the number of shares of common stock reserved for issuance under the 2012 Plan and ESPP pursuant to the evergreen provisions therein |
22
Table of Contents
We have never declared or paid any cash dividends on our common stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our board of directors may deem relevant.
23
Table of Contents
The following table sets forth our cash, cash equivalents and short-term investments, and our capitalization as of March 31, 2013:
| Ø | on an actual basis; |
| Ø | on a pro forma as adjusted basis to reflect the sale by us of shares of our common stock in the offering at an assumed public offering price of $ per share (the last reported sale price of our common stock, as reported on The NASDAQ Global Market on , 2013), after deducting the underwriting discounts and commissions and estimated offering costs payable by us. |
You should read this table together with “Management’s discussion and analysis of financial condition and results of operations” and our financial statements and the related notes appearing elsewhere in this prospectus or incorporated by reference herein.
| As of March 31, 2013 | ||||||||
| Actual | Pro forma as adjusted(1) |
|||||||
| (unaudited, in thousands, except share and per share data) |
||||||||
| Cash, cash equivalents and short-term investments |
$ | 90,715 | ||||||
|
|
|
|
|
|||||
| Convertible notes payable(2) |
11,895 | 11,895 | ||||||
| Convertible preferred stock; $0.001 par value: 10,000,000 shares authorized, no shares issued or outstanding, actual; 10,000,000 shares authorized, no shares issued or outstanding, pro forma as adjusted |
||||||||
| Stockholders’ equity: |
||||||||
| Common stock; $0.001 par value: 200,000,000 shares authorized, 35,965,371 shares issued and outstanding, actual; 200,000,000 shares authorized and shares issued and outstanding, pro forma as adjusted |
36 | |||||||
| Additional paid-in capital |
123,576 | |||||||
| Accumulated other comprehensive loss |
(37 | ) | (37 | ) | ||||
| Accumulated deficit |
(67,648 | ) | (67,648 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
55,867 | |||||||
|
|
|
|
|
|||||
| Total capitalization |
$ | 67,762 | ||||||
|
|
|
|
|
|||||
| (1) | Each $1.00 increase (decrease) in the assumed public offering price of $ per share, the last reported sale price of our common stock on The NASDAQ Global Market on , 2013, would increase (decrease) the as adjusted amount of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $ million, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. Each increase of shares in the number of shares offered by us would increase the as adjusted amount of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $ million, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, each decrease of shares in the number of shares offered by us would decrease the as adjusted amount of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $ million, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The as adjusted information discussed above is illustrative only and will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
24
Table of Contents
Capitalization
| (2) | In October 2012, in conjunction with our initial public offering, the promissory note we issued to Glaxo Group Limited, or GSK, in 2010 was amended and restated with a principal amount of $5.4 million. This amended note had a maturity date of three years from the anniversary of the agreement, or October 2015. At GSK’s option, this note shall be convertible into shares of our common stock at any time prior to the maturity date with a conversion equal to the quotient of all outstanding principal and interest divided by the initial public offering price of $4.00 per share. The value attributed to the convertible note payable in the table above reflects the fair value of the note, including the conversion option. The outstanding principal of the convertible note payable as of March 31, 2013 was $5.4 million, which would result in total capitalization of $74.2 million, on a pro forma basis. |
The number of shares of common stock shown as issued and outstanding on a pro forma as adjusted basis in the table is based on the number of shares of our common stock outstanding as of March 31, 2013, and excludes:
| Ø | 4,742,780 shares of common stock issuable upon the exercise of outstanding stock options as of March 31, 2013, at a weighted average exercise price of $2.24 per share; |
| Ø | 2,191,925 shares of common stock reserved for future issuance under our 2012 equity incentive plan, or the 2012 Plan, plus any future increases in the number of shares of common stock reserved for issuance under the 2012 Plan pursuant to the evergreen provision; and |
| Ø | 481,274 shares of common stock reserved for future issuance under our 2012 employee stock purchase plan, or the ESPP, plus any future increases in the number of shares of common stock reserved for issuance under the ESPP pursuant to the evergreen provision. |
25
Table of Contents
If you invest in our common stock in this offering, your ownership interest will be immediately diluted to the extent of the difference between the public offering price per share and the pro forma as adjusted net tangible book value per share of our common stock after this offering.
Our historical net tangible book value per share is determined by dividing our total tangible assets less total liabilities by the actual number of outstanding shares of our common stock. The historical net tangible book value of our common stock as of March 31, 2013 was $54.7 million, or $1.52 per share.
After giving effect to the sale of shares of our common stock offered by us at an assumed public offering price of $ per share (the last reported sale price of our common stock on The NASDAQ Global Market on , 2013), after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, our net tangible book value as of March 31, 2013, would have been approximately $ million, or $ per share of common stock. This represents an immediate increase in net tangible book value of $ per share to existing stockholders and an immediate dilution of $ per share to new investors purchasing shares of common stock in this offering at the assumed public offering price.
The following table illustrates this dilution on a per share basis:
| Assumed offering price per share |
$ | |||
| Historical net tangible book value per share as of March 31, 2013 |
$ | 1.52 | ||
| Increase in net tangible book value per share attributable to new investors |
||||
| Pro forma net tangible book value per share after the offering |
||||
|
|
|
|||
| Dilution per share to new investors |
$ | |||
|
|
|
Each $1.00 increase (decrease) in the assumed public offering price of $ per share would increase (decrease) our as adjusted net tangible book value after this offering by approximately $ million, or approximately $ per share, and the dilution per share to new investors by approximately $ per share, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. We may also increase or decrease the number of shares we are offering. An increase of shares in the number of shares offered by us would increase our as adjusted net tangible book value after this offering by approximately $ million, or $ per share, and the dilution per share to new investors would be $ per share, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, a decrease of shares in the number of shares offered by us would decrease our as adjusted net tangible book value after this offering by approximately $ million, or $ per share, and the dilution per share to new investors would be $ per share, assuming that the assumed public offering price remains the same, and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The information discussed above is illustrative only and will adjust based on the actual public offering price and other terms of this offering determined at pricing.
If the underwriters exercise in full their option to purchase up to additional shares of common stock at the assumed public offering price of $ per share, the as adjusted net tangible book value after this offering would be $ per share, representing an increase in net tangible book value of $ per share to existing stockholders and immediate dilution in net tangible book value of $ per share to investors purchasing our common stock in this offering at the assumed public offering price.
26
Table of Contents
Dilution
The number of shares of our common stock to be outstanding after this offering is based on 35,965,371 shares of common stock outstanding as of March 31, 2013, and excludes:
| Ø | 4,742,780 shares of common stock issuable upon the exercise of outstanding stock options as of March 31, 2013, at a weighted average exercise price of $2.24 per share; |
| Ø | 2,191,925 shares of common stock reserved for future issuance under our 2012 equity incentive plan, or the 2012 Plan, plus any future increases in the number of shares of common stock reserved for issuance under the 2012 Plan pursuant to the evergreen provision; and |
| Ø | 481,274 shares of common stock reserved for future issuance under our 2012 employee stock purchase plan, or the ESPP, plus any future increases in the number of shares of common stock reserved for issuance under the ESPP pursuant to the evergreen provision. |
27
Table of Contents
The following selected financial data should be read together with our financial statements and accompanying notes and “Management’s discussion and analysis of financial condition and results of operations” appearing elsewhere in this prospectus or incorporated by reference herein. The selected financial data in this section are not intended to replace our financial statements and the related notes. Our historical results are not necessarily indicative of our future results.
The selected statement of operations data for the years ended December 31, 2010, 2011 and 2012 and the selected balance sheet data as of December 31, 2011 and 2012 are derived from our audited financial statements incorporated by reference in this prospectus. The selected statement of operations data for the three months ended March 31, 2012 and 2013 and the selected balance sheet data as of March 31, 2013 are derived from our unaudited financial statements incorporated by reference in this prospectus. The unaudited financial statements have been prepared on a basis consistent with our audited financial statements included in this prospectus and include, in our opinion, all adjustments, consisting of normal recurring adjustments necessary for the fair presentation of the financial information in those statements.
| Year ended December 31, | Three months ended March 31, | |||||||||||||||||||
| Statement of operations data | 2010 | 2011 | 2012 | 2012 | 2013 | |||||||||||||||
| (in thousands, except share and per share data) | ||||||||||||||||||||
| (unaudited) | ||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Revenue under strategic alliances and collaborations |
$ | 8,112 | $ | 13,767 | $ | 12,700 | $ | 3,344 | $ | 3,238 | ||||||||||
| Grant revenue |
489 | 22 | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total revenues |
8,601 | 13,789 | 12,700 | 3,344 | 3,238 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
20,178 | 17,289 | 20,342 | 4,603 | 6,883 | |||||||||||||||
| General and administrative |
3,921 | 3,637 | 4,932 | 921 | 1,905 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
24,099 | 20,926 | 25,274 | 5,524 | 8,788 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(15,498 | ) | (7,137 | ) | (12,574 | ) | (2,180 | ) | (5,550 | ) | ||||||||||
| Other expense, net |
(91 | ) | (259 | ) | (4,844 | ) | (66 | ) | (1,689 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss before income taxes |
(15,589 | ) | (7,396 | ) | (17,418 | ) | (2,246 | ) | (7,239 | ) | ||||||||||
| Income tax (benefit) expense |
(30 | ) | 206 | (10 | ) | 1 | (10 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (15,559 | ) | $ | (7,602 | ) | $ | (17,408 | ) | $ | (2,247 | ) | $ | (7,229 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss per share, basic and diluted(1) |
$ | (85.82 | ) | $ | (2.12 | ) | $ | (13.06 | ) | $ | (0.20 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Shares used to compute basic and diluted net loss per share(1) |
88,582 | 8,212,538 | 171,998 | 35,872,606 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| (1) | See Note 2 of our Notes to Financial Statements incorporated by reference in this prospectus for an explanation of the method used to calculate the basic and diluted net loss per common share and the number of shares used in the computation of the share and per share data. No share or per share data have been presented for 2010 since we had no common shares outstanding during that year. |
28
Table of Contents
Selected financial data
| As of December 31, | As of March 31, | |||||||||||
| Balance sheet data | 2011 | 2012 | 2013 | |||||||||
| (unaudited) | ||||||||||||
| (in thousands) | ||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 38,144 | $ | 98,100 | $ | 90,715 | ||||||
| Working capital |
25,816 | 86,161 | 79,076 | |||||||||
| Total assets |
42,881 | 103,518 | 97,027 | |||||||||
| Convertible notes payable |
10,815 | 10,134 | 11,895 | |||||||||
| Accumulated deficit |
(43,011 | ) | (60,419 | ) | (67,648 | ) | ||||||
| Total stockholders’ (deficit) equity |
(41,494 | ) | 62,093 | 55,867 | ||||||||
29
Table of Contents
OVERVIEW
Our business
We are a biopharmaceutical company focused on discovering and developing first-in-class drugs that target microRNAs to treat a broad range of diseases. microRNAs are recently discovered, naturally occurring ribonucleic acid, or RNA, molecules that play a critical role in regulating key biological pathways. Scientific research has shown that the improper balance, or dysregulation, of microRNAs is directly linked to many diseases. We believe we have assembled the leading position in the microRNA field, including expertise in microRNA biology and oligonucleotide chemistry, a broad intellectual property estate, key opinion leaders and disciplined drug discovery and development processes. We refer to these assets as our microRNA product platform. We are using our microRNA product platform to develop chemically modified, single-stranded oligonucleotides that we call anti-miRs. We use these anti-miRs to modulate microRNAs and by doing so return diseased cells to their healthy state. We believe microRNAs may be transformative in the field of drug discovery and that anti-miRs may become a new and major class of drugs with broad therapeutic application much like small molecules, biologics and monoclonal antibodies. We are currently optimizing anti-miRs in several distinct programs, both independently and with our strategic alliance partners, AstraZeneca AB, or AstraZeneca, GlaxoSmithKline plc, or GSK, and Sanofi. We also have a collaboration agreement with Biogen Idec MA lnc. to evaluate the potential use of microRNA signatures as a biomarker for human patients with multiple sclerosis.
We are currently executing on our ‘Road to the Clinic’ strategy which sets forth certain corporate goals that seek to advance our microRNA therapeutic pipeline toward the clinic. Specifically, we set the goal of nominating two microRNA candidates for clinical development in 2013. In May 2013, we announced our first clinical candidate as RG-101, for which we have full ownership and commercial rights. RG-101 is a GalNAc-conjugated microRNA anti-miR, which targets microRNA-122 for the treatment of patients with chronic hepatitis C virus infection, or HCV. We expect to submit our first investigation new drug application, or IND, to the U.S. Food and Drug Administration, or FDA, or equivalent foreign regulatory filing with foreign regulatory authorities, as applicable, for RG-101 in the first half of 2014. We anticipate that we will nominate a second clinical candidate by the end of 2013.
RNA plays an essential role in the process used by cells to encode and translate genetic information from DNA to proteins. RNA is comprised of subunits called nucleotides and is synthesized from a DNA template by a process known as transcription. Transcription generates different types of RNA, including messenger RNAs that carry the information for proteins in the sequence of their nucleotides. In contrast, microRNAs are small RNAs that do not code for proteins, but rather are responsible for regulating gene expression by affecting the translation of target messenger RNAs. By interacting with many messenger RNAs, a single microRNA can regulate several genes that are instrumental for the normal function of a biological pathway. More than 500 microRNAs have been identified to date in humans, each of which is believed to interact with a specific set of genes that control key aspects of cell biology. Since most diseases are multi-factorial and involve multiple targets in a pathway, the ability to modulate gene networks by targeting a single microRNA provides a new therapeutic approach for treating complex diseases.
We were formed in 2007 when Alnylam Pharmaceuticals, Inc., or Alnylam, and Isis Pharmaceuticals, Inc., or Isis, contributed significant intellectual property, know-how and financial and human capital to pursue the development of drugs targeting microRNAs. This provided the foundation for our leadership position in the microRNA field and, since then, we have leveraged their RNA-based discovery and development expertise, established over more than 20 years, to build our own proprietary microRNA product platform that combines a deep understanding of biology with innovative chemistries and disciplined processes.
30
Table of Contents
Business
We are developing single-stranded oligonucleotides, which are chemically synthesized chains of nucleotides, that are mirror images of specific target microRNAs. We incorporate proprietary chemical modifications to enhance drug properties such as potency, stability and tissue distribution. We refer to these chemically modified oligonucleotides as anti-miRs. Each anti-miR is designed to bind with and inhibit a specific microRNA target that is up-regulated, or overproduced, in a cell and that is involved in the disease state. In binding to the microRNA, anti-miRs correct the dysregulation and return diseased cells to their healthy state. We have demonstrated therapeutic benefits of our anti-miRs in at least 20 different preclinical models of human diseases.
We have identified and validated several microRNA targets across a number of disease categories and are working independently and with our strategic alliance partners to optimize anti-miR development candidates. We expect that anti-miR development candidates will be easily formulated in saline solution and administered systemically or locally depending on the therapeutic indication. Our therapeutic development programs are shown in the table below:
| microRNA target | anti-miR program | Commercial rights | ||
| miR-122 | RG-101 for HCV | Regulus* | ||
| miR-221 | Hepatocellular carcinoma | Regulus | ||
| miR-10b | Glioblastoma | Regulus | ||
| miR-21 | Hepatocellular carcinoma | Sanofi | ||
| miR-21 | Kidney fibrosis | Sanofi | ||
| miR-33 | Atherosclerosis | AstraZeneca |
| * | With the exception of RG-101, commercial rights for miR-122 target licensed to GSK. |
One aspect of our strategy is to pursue a balanced approach between product candidates that we develop ourselves and those that we develop with partners. We intend to focus our own resources on proprietary product opportunities in therapeutic areas where development and commercialization are appropriate for our size and financial resources, which we anticipate will include niche indications and orphan diseases. In therapeutic areas where costs are more significant, development timelines are longer or markets are too large for our capabilities, we will seek to secure partners with requisite expertise and resources.
Our approach has been validated to date by the following strategic alliances and collaborations with large pharmaceutical companies:
| Ø | In April 2008, we formed a strategic alliance with GSK to discover and develop microRNA therapeutics for immuno-inflammatory diseases. In February 2010, we and GSK expanded the alliance to include potential microRNA therapeutics for the treatment of HCV. In June 2013, we amended our agreement with GSK and agreed that RG-101 is fully-owned by us and that miR-122 remains a collaboration target under the agreement. |
| Ø | In June 2010, we formed a strategic alliance with Sanofi to discover and develop microRNA therapeutics for fibrotic diseases. In July 2012, we expanded the alliance to include potential microRNA therapeutics in oncology. The original research term for this strategic alliance expired in June 2013, upon which we and Sanofi entered into a letter agreement pursuant to which we granted Sanofi an exclusive right to negotiate for the co-development and commercialization of certain of our unencumbered microRNA programs through December 2013, for which Sanofi has agreed to pay us an upfront option fee of $2.5 million, $1.25 million of which is creditable against future amounts payable by Sanofi to us. In addition, Sanofi granted us an exclusive option, which also expires in December 2013, to negotiate the co-development and commercialization of miR-21. |
| Ø | In August 2012, we formed a strategic alliance with AstraZeneca to discover and develop microRNA therapeutics for cardiovascular diseases, metabolic diseases and oncology. |
| Ø | In August 2012, we entered into a collaboration agreement with Biogen Idec to evaluate the potential use of microRNA signatures as a biomarker for human patients with multiple sclerosis. In June 2013, we and Biogen Idec amended the collaboration agreement to update the research plan and criteria for success. |
31
Table of Contents
Business
Under our existing strategic alliances with AstraZeneca, GSK and Sanofi, we are eligible to receive up to approximately $1.3 billion in milestone payments upon successful commercialization of microRNA therapeutics for the eight programs contemplated by our agreements. These payments include up to $42.0 million upon achievement of preclinical and IND milestones, up to $272.0 million upon achievement of clinical development milestones, up to $305.0 million upon achievement of regulatory milestones and up to $670.0 million upon achievement of commercialization milestones.
Our leadership
Our management has more than 50 years of collective experience leading the discovery and development of innovative therapeutics, including significant operational and financial experience with emerging biotechnology companies, which we believe is the ideal combination of talent to execute our strategy. In addition, our experienced board of directors, which includes representatives of our founding companies, Alnylam and Isis, provides significant support and guidance in all aspects of our business.
Our executive officers are:
| Ø | Kleanthis G. Xanthopoulos, Ph.D., our President and Chief Executive Officer, is an entrepreneur who has been involved in founding several companies, including Anadys Pharmaceuticals, Inc. (acquired by F. Hoffman-La Roche Inc. in 2011), which he started as President and Chief Executive Officer. |
| Ø | Neil W. Gibson, Ph.D., our Chief Scientific Officer, is a leading scientist focused on cancer research and drug development who previously served as Chief Scientific Officer of the Oncology Research Unit at Pfizer Inc. and as Chief Scientific Officer of OSI Pharmaceuticals, Inc. He was involved in the development of several commercial cancer drugs including Xalkori® (crizotinib), Nexavar® (sorafenib) and Tarceva® (erlotinib). |
Our executive team is supported by the following key personnel:
| Ø | Mary Glanville, our Senior Vice President of Human Capital, is an accomplished human resources executive in the life sciences industry who previously served in management roles at Anadys Pharmaceuticals, Inc. (acquired by F. Hoffman-La Roche Inc. in 2011), Inflazyme Inc. and Inex Pharmaceuticals Corp. |
| Ø | Victor Knopov, Ph.D., our Vice President, Pharmaceutical Development, is a leader in oligonucleotide drug delivery and pharmaceutical development who has held positions at Nitto Denko Technical Corporation, Bio-Medics, Inc., EnGene, Inc., Marina Biotech, Inc. and Inex Pharmaceuticals Corporation. Dr. Knopov has extensive knowledge of Chemistry, Manufacturing and Control, or CMC, development for various technology platforms including commercial production of enzymes, anticancer liposomal products as well as advanced delivery systems for antisense, plasmids and siRNA based on lipids, polymer nanoparticles and conjugated systems. |
| Ø | Daniel R. Chevallard, CPA, our Vice President, Finance and Accounting, is a corporate finance leader with public accounting expertise who previously held senior roles in corporate finance, accounting and financial reporting as a corporate controller and Senior Director, Finance at Prometheus Laboratories Inc. and who was a senior financial auditor at Ernst & Young LLP. |
Our executive team, key personnel and board of directors are supported by our scientific advisory board members, who are renowned pioneers in the microRNA field:
| Ø | David Baltimore, Ph.D., Chairman of our scientific advisory board and Professor of Biology at the California Institute of Technology, received the Nobel Prize in 1975 and is highly regarded as a pioneer in virology and immunology, with his current research investigating the role of microRNAs in immunity. Dr. Baltimore is also a member of our board of directors. |
32
Table of Contents
Business
| Ø | David Bartel, Ph.D., Professor of Biology at the Massachusetts Institute of Technology and the Whitehead Institute for Biomedical Research and an investigator at the Howard Hughes Medical Institute, studies microRNA genomics, target recognition and regulatory functions. |
| Ø | Gregory Hannon, Ph.D., Professor at the Cold Spring Harbor Laboratory and an investigator at the Howard Hughes Medical Institute, has identified and characterized many of the major biogenesis and effector complexes for microRNA biology. |
| Ø | Markus Stoffel, M.D., Ph.D., Professor of Metabolic Diseases at the Swiss Federal Institute of Technology, is focused on microRNA research and the regulation of glucose and lipid metabolism. |
| Ø | Thomas Tuschl, Ph.D., Professor and Head of the Laboratory for RNA Molecular Biology at the Rockefeller University and an investigator at the Howard Hughes Medical Institute, discovered many of the mammalian microRNA genes and has developed methods for characterization of small RNAs. |
THE POTENTIAL OF microRNA BIOLOGY
RNA plays an essential role in the process used by cells to encode and translate genetic information from DNA to proteins. RNA is comprised of subunits called nucleotides and is synthesized from a DNA template by a process known as transcription. Transcription generates different types of RNA, including messenger RNAs that carry the information for proteins in the sequence of their nucleotides. In contrast, microRNAs are small RNAs that do not code for proteins, but rather are responsible for regulating gene expression by affecting the translation of target messenger RNAs. This is achieved when the microRNA binds with its messenger RNA targets and blocks cell machinery, called ribosomes, from translating them into proteins, as shown below.
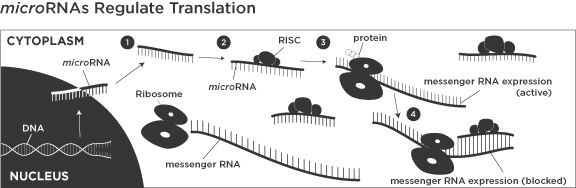
Step 1. microRNAs are transcribed from DNA in the nucleus and exported to the cytoplasm.
Step 2. In the cytoplasm, microRNAs associate with the RNA-induced silencing complex, or RISC.
Step 3. The microRNA in RISC targets specific messenger RNAs.
Step 4. The microRNA interaction with its target messenger RNAs blocks translation into proteins.
RNA therapeutics are drugs designed to specifically target RNA. The field of RNA therapeutics consists of various technologies including antisense therapeutics, RNAi therapeutics and microRNA therapeutics:
Antisense therapeutics—Antisense therapeutics are small oligonucleotides that target RNA through hybridization, a specific type of binding, and modulate the function of the targeted RNA. There are at least 12 known antisense mechanisms that can be exploited once an antisense drug binds to its target RNA. One of our founding companies, Isis, is leading the discovery and development of antisense therapeutics with over 25 drugs currently in development. Isis’ most advanced drug, KYNAMRO™,
33
Table of Contents
Business
which is partnered with Genzyme Corporation, a subsidiary of Sanofi, was approved by the FDA in January 2013 for the treatment of homozygous familial hypercholesterolemia. The majority of Isis’ antisense drugs in development bind to the specific RNAs of a particular gene, and ultimately inhibit or alter the expression of the protein encoded in the target gene.
RNAi therapeutics—RNAi therapeutics are RNA-like oligonucleotides that harness RNAi, a powerful and natural biologic mechanism that was awarded the 2006 Nobel Prize for Physiology or Medicine. RNAi therapeutics are rationally designed to silence disease causing genes. The molecule that mediates RNAi, small interfering RNA, or siRNA, binds with a cellular complex known as the RNA-induced silencing complex, or RISC. The siRNA within RISC is processed into single-stranded RNA that targets a specific messenger RNA and promotes its degradation through cleavage. In this way, RNAi therapeutics can be used to target specific disease causing genes. One of our founding companies, Alnylam, has shown human proof-of-concept in clinical trials with multiple RNAi drug candidates.
microRNA therapeutics—microRNA therapeutics are single- or double-stranded RNA-like oligonucleotides that are chemically modified and target specific microRNAs. Single-stranded microRNA therapeutics, or anti-miRs, are designed to bind and inhibit specific microRNAs that have been up-regulated in diseases as shown in the figure below. Double-stranded microRNA therapeutics, or miR-mimics, are designed to replace the activity of specific microRNAs that have been down-regulated in disease. In this way, microRNA therapeutics can be used to modulate specific microRNA targets and regulate entire biological pathways.
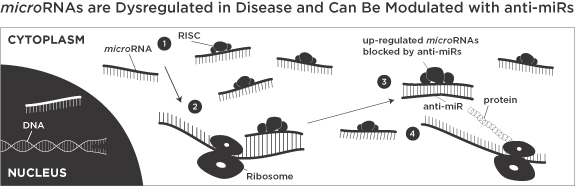
Step 1. microRNA expression is up-regulated in disease such that a specific microRNA is produced in excess amounts.
Step 2. The up-regulated microRNA targets messenger RNAs, resulting in lower levels of key proteins.
Step 3. The anti-miR therapeutic is delivered to the diseased cell and binds to the up-regulated microRNA, resulting in the elimination of excess microRNA.
Step 4. Use of the anti-miR therapeutic therefore restores the normal function of microRNA biology in the cell and corrects the disease.
34
Table of Contents
Business
microRNA THERAPEUTICS AS A NEW CLASS OF DRUGS
We believe that microRNA therapeutics have the potential to become a new and major class of drugs with broad therapeutic application. There are several reasons why microRNA therapeutics have transformative potential, some of which are listed below.
microRNAs represent a new drug target space—microRNAs are a recent discovery in biology and, up until now, have not been a focus of pharmaceutical research. Traditional drug classes cannot be used to target microRNAs because they are typically designed to bind and inhibit proteins, not RNA molecules. microRNA therapeutics provide the capability to very specifically modulate microRNAs and allow access to this new target space.
microRNAs are dysregulated in a broad range of diseases—microRNAs play a critical role in regulating biological pathways by controlling the translation of many target genes. More than 500 microRNAs have been identified to date in humans, each of which are believed to interact with a specific set of genes that control key aspects of cell biology. Thus the improper balance, or dysregulation, of microRNAs has been directly linked to numerous diseases, including cancer, diabetes, congestive heart failure, viral infections and macular degeneration.
microRNA therapeutics target entire disease pathways—microRNAs are naturally occurring molecules that have evolved to regulate gene networks responsible for entire biological pathways. Because of this unique attribute, the use of microRNA therapeutics may allow for more effective treatment of complex multi-factorial diseases in which the entire disease pathway can be addressed.
microRNA therapeutics can be produced with efficient rational design—Traditional drug classes, like small molecules, usually require screening of thousands of potential compounds to identify prospective leads. Given that microRNAs are a short sequence of nucleotides and that the corresponding sequence of the mirror image anti-miR is also known, microRNA therapeutics allow for a more efficient rational drug design process.
microRNA therapeutics may be synergistic with other therapies—Because of their completely different mechanisms of action, microRNA therapeutics and traditional drugs can be synergistic. In certain fields, such as cancer and infectious diseases, physicians typically treat patients with combinations of drugs that have different mechanisms in the hope that there will be complementary activity.
OUR microRNA PRODUCT PLATFORM
We are the leading company in the field of microRNA therapeutics dedicated to pioneering a new paradigm in treating serious diseases. We believe we have assembled the leading position in the microRNA field, including expertise in microRNA biology and oligonucleotide chemistry, broad intellectual property estate, key opinion leaders and disciplined drug discovery and development processes. We refer to these assets as our microRNA product platform. Backed by our founding companies, Alnylam and Isis, we are uniquely positioned to leverage oligonucleotide technologies that have been proven in clinical trials. Central to achieving our goals is the know-how that we have accumulated in oligonucleotide design and how the specific chemistries behave in the clinical setting.
We view the following as providing a competitive advantage for our microRNA product platform:
| Ø | a mature platform selectively producing multiple development candidates advancing to the clinic; |
| Ø | scientific advisors who are pioneers in the microRNA field, including the first researcher to discover microRNAs in humans; |
| Ø | access to proven RNA therapeutic technologies through our founding companies, as well as approximately 900 patents and patent applications relating to oligonucleotide technologies; |
35
Table of Contents
Business
| Ø | a leading microRNA intellectual property estate with access to over 170 microRNA patents and patent applications covering compositions and therapeutic uses; |
| Ø | development expertise and financial resources provided by our three major strategic alliances with AstraZeneca, GSK and Sanofi; and |
| Ø | over 30 academic collaborations that help us identify new microRNA targets and support our early stage discovery efforts. |
The disciplined approach we take to the discovery and development of microRNA therapeutics is as important as the assets assembled to execute on our plans. Beginning with how we evaluate a therapeutic opportunity and followed by the identification of a specific microRNA target, its validation and optimization of the development candidates that will go into clinical trials, each is the subject of proprietary standards and rules that increase our probability of technical success. Our disciplined approach is based on the following four steps:
Step 1 - Evaluation of microRNA therapeutic opportunities
The initiation of our microRNA discovery and development efforts is based on rigorous scientific and business criteria, including:
| Ø | existence of significant scientific evidence to support the role of a specific microRNA in a disease; |
| Ø | availability of predictive preclinical disease models to test our microRNA development candidates; |
| Ø | ability to effectively deliver anti-miRs to the diseased cells or tissues; and |
| Ø | existence of a reasonable unmet medical need and commercial opportunity. |
The advantage of our evaluation criteria is that they can be applied to a broad range of microRNA targets, allowing us to generate a focused portfolio of discovery programs that we believe have a high probability of clinical and commercial success.
Once we have decided to initiate a new program, we use a disciplined approach to identify novel microRNA targets, validate such novel microRNA targets and use our proprietary methodologies to optimize lead microRNA development candidates for IND-enabling studies and subsequent clinical development.
Step 2 - Identification of microRNA targets
We have developed a significant understanding and know-how of human microRNA biology and the biological pathways that are regulated by microRNAs. We identify microRNA targets through bioinformatic analysis of public and proprietary microRNA expression profiling data sets from samples of diseased human tissues. The analysis of such data sets can immediately highlight a potential role for specific microRNAs in the diseases being studied. Further investigation of animal models that are predictive of human diseases in which those same microRNAs are also dysregulated provides additional data to support a new program. We have applied this strategy successfully in our existing programs and we believe that this approach will continue to help us identify clinically relevant microRNA targets.
Step 3 - Validation of microRNA targets
Our validation strategy is based on two distinct steps. First, using genetic tools, we determine whether up-regulation of the microRNA in healthy animals can create the specific disease state and inhibition of the microRNA can lead to a therapeutic benefit. Second, using animal models predictive of human diseases, we determine whether pharmacological modulation of the up-regulated microRNA target with our anti-miRs can also lead to a therapeutic benefit. This validation process enables us to prioritize the best microRNA targets that appear to be key drivers of diseases and not simply correlating markers.
36
Table of Contents
Business
Step 4 - Optimization of microRNA development candidates
We have developed a proprietary process that allows us to rapidly generate an optimized development candidate following the identification and validation of a microRNA target. Unlike traditional drug classes, such as small molecules, in which thousands of compounds must be screened to identify prospective leads, the fact that anti-miRs are mirror images of their target microRNAs allows for a more efficient rational design process. The optimization process incorporates our extensive knowledge base around oligonucleotide chemistry and anti-miR design to efficiently synthesize a starting pool of rationally designed anti-miRs to be evaluated in a series of proven assays and models that quickly allow us to:
| Ø | identify our most effective anti-miRs in mechanistic cell-based assays; |
| Ø | evaluate the activity of our anti-miRs in preclinical animal models that are predictive of human diseases; and |
| Ø | eliminate anti-miRs that may trigger undesirable effects. |
We also enhance our anti-miRs for distribution to the tissues where the specific microRNA target is causing disease. For example, we employ a conjugate approach to successfully deliver anti-miRs to target tissues and achieve optimized cellular uptake. Our conjugate platform includes several receptor-mediated chemistries such as triantennary N-acetylgalactosamine, or GalNAc, conjugates. We have demonstrated the ability to achieve optimized delivery to cells in the liver with both intravenous and subcutaneous administration of GalNAc conjugates in multiple species and with multiple targets.
We believe that our optimization process provides us with a competitive advantage in the discovery and development of microRNA therapeutics. The lessons we learn from one program can be applied to other programs at an earlier stage in our portfolio, leading to potential acceleration of the advancement of those programs towards IND-enabling activities and future clinical development.
microRNA BIOMARKERS
Through our microRNA target identification and validation efforts we have developed proprietary technologies for microRNA profiling and analysis of human clinical samples such as tissue biopsies. More recently, microRNAs have been detected in bodily fluids such as blood, and emerging data generated by us and others have demonstrated that microRNA signatures in blood can mimic the expression profile observed in disease tissues.
The identification of dysregulated microRNAs from various human tissues and blood helps us identify and validate potential microRNA targets for therapeutic development. Equally important, such microRNAs may become biomarkers that can be used to select optimal patient segments for our clinical trials. We expect this personalized approach to clinical development to result in a significantly improved risk-benefit ratio in the appropriate patient populations and plan to implement the strategy in our programs.
We believe that microRNA biomarkers in the blood are of significant value and provide opportunities to develop companion diagnostics for any drugs that we may successfully develop and drugs developed by other companies. In August 2012, we entered into an agreement with Biogen Idec MA Inc. to collaborate on microRNA biomarkers for multiple sclerosis.
OUR INITIAL DEVELOPMENT CANDIDATES
We have demonstrated in at least 20 preclinical animal models that are predictive of human diseases that the inhibition of up-regulated microRNA targets using anti-miRs can modulate entire biological pathways, returning diseased cells to their healthy state. Based on the extensive preclinical data we have
37
Table of Contents
Business
generated to date, we believe that our microRNA product platform has the potential to provide significant therapeutic benefit in a broad range of human diseases. We have chosen to focus our initial efforts on select therapeutic areas with unmet medical needs including HCV, oncology and metabolic diseases. We have identified and validated several microRNA targets that play a role in these areas.
Our microRNA development candidates target miR-122 in HCV, miR-21 and miR-221 in hepatocellular carcinoma, miR-21 in kidney fibrosis, miR-33 in atherosclerosis and miR-10b in GBM.
RG-101 targeting miR-122 in hepatitis C virus infection
Market opportunity:
Hepatitis C is a result of a hepatocyte specific infection induced by the virus known as HCV. Chronic HCV may lead to significant liver disease, including chronic active hepatitis, cirrhosis, and hepatocellular carcinoma. According to the World Health Organization, up to 170 million people are chronically infected with HCV worldwide, and more than 350,000 people die from HCV annually. The CDC estimates that there are currently approximately 3.2 million persons infected with HCV in the United States.
Current treatments:
The current standard of care for HCV is a combination of injectable pegylated interferon-alfa, oral ribavirin and an oral protease inhibitor. Two protease inhibitors were approved for such combination treatment in 2011: telaprevir (marketed as Incivek® in North America by Vertex Pharmaceuticals Incorporated, as Incivo® in Europe by Johnson & Johnson and as TELAVIC® in Japan by Mitsubishi Tanabe Pharma Corporation) and boceprevir (marketed as Victrelis® by Merck & Co, Inc.). All-oral combination therapies that include new direct-acting antivirals are being developed and in clinical trials appear to achieve significant improvements in efficacy, tolerability and treatment duration. However, unmet needs will likely remain for certain segments of the HCV patient population, including those who have not responded at all to previous therapies and those who have relapsed following previous therapies.
Our program:
We have nominated RG-101 as our first microRNA clinical candidate for the treatment of HCV. RG-101, which is wholly-owned by us, is a hepatocyte-targeted GalNAc-conjugated anti-miR targeting microRNA-122, or miR-122. miR-122 is the most abundant microRNA in liver hepatocytes and HCV has evolved to utilize it as a viral replication factor such that the stability and propagation of HCV is dependent on the interaction of miR-122 and the HCV genome. Clinical trials have shown that inhibiting the miR-122 target with an oligonucleotide administered weekly can result in a three-fold logarithmic viral load reduction observed after five weekly doses in HCV patients.
We believe that RG-101 has several attractive properties that make our approach to treating HCV unique and differentiated among the current therapies:
| Ø | RG-101 employs a unique mechanism of action by targeting a host factor. We believe by targeting a host factor there is a lower likelihood for the virus to develop resistance, which has been observed with direct-acting antivirals. In addition, because of its unique mechanism of action, we believe that RG-101 will have minimal to potentially no negative drug-drug interactions with current and future direct-acting antiviral treatment regimens. |
| Ø | RG-101 has the potential to be a pan-genotypic agent. The miR-122 binding site of the HCV genome is one of the most highly conserved regions across all HCV genotypes. Therefore, RG-101 is expected to be active against all viral genotypes. In preclinical studies, RG-101 has demonstrated activity across a broad spectrum of HCV genotypes and against the most commonly identified HCV mutations detected in patients being treated with direct-acting antiviral therapy. |
38
Table of Contents
Business
| Ø | RG-101 has a convenient dosing regimen. The pharmacological activity of RG-101 has been shown in animal models to be sustained for more than 28 days after a single dose administration. These data suggest the feasibility of a convenient subcutaneous dosing regimen, to be administered during a patient’s regularly scheduled physician visit. |
| Ø | RG-101 is potent and has a favorable safety profile to date. In pharmacology studies using a human liver chimeric mouse model to assess potency against HCV, RG-101 demonstrated significant lowering of viral titer load. These experiments are helping us refine the initial starting dose for clinical trials in humans and assess the therapeutic index. Further, we did not identify any adverse effects that would preclude the development of RG-101 for the treatment of HCV. |
Given its attractive properties and preclinical data demonstrated to date, we believe that RG-101 may be an attractive agent to add to existing therapeutic regimens. We currently plan to develop RG-101 as a key component of an HCV combination regimen for patients who have failed, or are intolerant of, the current standard of care and specific patient populations such as HCV/HIV co-infection. In the near term, we plan to study RG-101 as monotherapy in healthy subjects and in HCV patients in Phase 1 clinical trials. We anticipate being able to evaluate the ability of a single dose of RG-101 to reduce viral load in HCV patients within the first year of the development program. If these clinical trials are successful, we expect to study RG-101 in Phase 2 clinical trials, in which we will focus primarily on demonstrating the efficacy of RG-101 in combination with other HCV therapeutics in patients who have failed other HCV treatments and in specific patient populations.
miR-21 and miR-221 for hepatocellular carcinoma
Market opportunity:
Hepatocellular carcinoma, or HCC, is the most prevalent form of liver cancer and is the most common cancer in some parts of the world, with more than 1 million new cases diagnosed each year worldwide according to the National Cancer Institute. According to the World Health Organization, liver cancer is the third leading cause of cancer deaths worldwide. According to recent reports from the Centers for Disease Control, or the CDC, HCC rates in the United States are increasing with common risk factors including alcohol consumption, metabolic syndrome and type 2 diabetes.
Current treatments:
Patients diagnosed with HCC have poor prognosis with a relatively low five-year survival rate of approximately 10%. Treatment options include surgical resection, transplantation and chemoembolization (delivery of drug directly to the tumor through a catheter). The only systemic drug therapy approved for the treatment of unresectable HCC is the multi-kinase inhibitor sorafenib (co-marketed by Bayer AG and Onyx Pharmaceuticals, Inc. as Nexavar®), which has been shown to be poorly tolerated and provides a 2.8-month overall survival benefit.
Our programs:
miR-21 is one of the most validated microRNA targets in oncology, with numerous scientific publications suggesting that miR-21 plays an important role in the initiation and progression of cancers including liver, kidney, breast, prostate, lung and brain. Similarly, miR-221 has been identified to be upregulated in multiple cancers including liver, kidney, prostate, brain, thyroid, ovarian, and breast cancer. In addition there is genetic evidence that links miR-21 and miR-221 to HCC. We are developing oncology anti-miRs targeting miR-21 and miR-221, which we refer to as anti-miR-21 and anti-miR-221, for HCC because our analysis of liver biopsies from HCC patients has shown that miR-21 and miR-221 are up-regulated relative to surrounding normal liver tissues. We have also shown that miR-21 and miR-221 levels are increased in a
39
Table of Contents
Business
genetically engineered mouse that develops HCC, thereby enabling us to test anti-miR-21 and anti-miR-221 in a preclinical model that mimics the human disease. Testing anti-miR-21 in this mouse model of HCC showed delayed tumor progression resulting in a survival rate of 80% at the study endpoint (compared to a 0% survival rate for the control group).
As part of our strategic alliance with Sanofi, we anticipate entering into an additional agreement under which we plan to nominate an anti-miR-21 development candidate for the treatment of HCC and file an IND and to pursue clinical development of such candidate together with Sanofi. We intend to independently file an IND, develop and commercialize an anti-miR-221 development candidate for the treatment of HCC.
miR-21 in kidney fibrosis
Market opportunity:
Fibrosis is the harmful build-up of excessive fibrous tissue leading to scarring and ultimately the loss of organ function. Fibrosis can affect any tissue and organ system, and is most common in the heart, liver, lung, peritoneum, and kidney. The fibrotic scar tissue is made up of extracellular matrix proteins. Fibrosis is widespread in industrialized nations and regularly leads to organ failure, contributing significantly to morbidity and mortality. Kidney fibrosis is the principal process underlying the progression of chronic kidney disease, or CKD, and ultimately leads to end-stage renal disease, a devastating disorder that requires dialysis or kidney transplantation. According to the CDC, more than 20 million people in the United States have CKD with over 100,000 patients starting treatment for end-stage renal disease annually. The National Kidney Foundation estimates that the annual cost of treating kidney failure in the U.S. is approximately $23.0 billion.
Current treatments:
Currently there are no approved drugs for fibrosis in the United States. In Europe, Asia and Japan there is only one approved therapy, pirfenidone (marketed as Esbriet® in Europe by InterMune, Inc. and as Pirespa™ in Japan by Shionogi & Co.), for lung fibrosis termed idiopathic pulmonary fibrosis, or IPF (scarring of the lung). The clinical results for pirfenidone concluded that it was able to improve progression-free survival and, to a lesser extent, improve pulmonary function allowing the approval for the treatment of mild-to-moderate IPF.
Our program:
Our lead program for fibrosis targets miR-21, which has been found in human tissue and animal models to be up-regulated in multiple fibrotic conditions. We and our academic collaborators have shown that either the absence of miR-21 or the inhibition of miR-21 reduces fibrosis in multiple preclinical models of organ fibrosis, including kidney and heart. We have also shown that anti-miR-21 treatment administered to preclinical animal models that are predictive of human kidney fibrosis can reduce fibrosis by up to 50%. In addition, the effects of our anti-miR-21 have been associated with improved kidney function and decreased mortality associated with injury to the kidney. Based on these data, we believe that anti-miR-21 could have therapeutic benefit in patients with CKD and kidney fibrosis.
As part of our strategic alliance with Sanofi, we anticipate entering into an additional agreement under which we will nominate an anti-miR-21 clinical candidate and file an IND initially for the treatment of Alport’s syndrome, an orphan genetic disorder caused by mutations in collagen synthesizing genes, that leads to kidney fibrosis.
40
Table of Contents
Business
miR-10b in glioblastoma
Market opportunity:
GBM, also known as glioblastoma or grade IV astrocytoma, is an aggressive tumor that forms from abnormal growth of glial (supportive) tissue of the brain. According to the New England Journal of Medicine, GBM is the most prevalent form of primary brain tumor and accounts for approximately 50% of the 22,500 new cases of brain cancer diagnosed in the United States each year. Treatment options are limited and expected survival is little over one year. GBM is considered a rare, or orphan, disease by the FDA and EMA.
Current treatments:
The standard of care for GBM involves surgical removal of the tumor followed by radiotherapy and chemotherapy with temozolomide (marketed as Temodar® and Temodal® by Merck & Co., Inc.), a non-specific cytotoxic agent approved for newly diagnosed GBM. Temozolomide has been shown to be poorly tolerated and provides a 2.5-month overall survival benefit. In addition, bevacizumab (marketed as Avastin® by Genentech Inc. and F. Hoffman-La Roche Ltd.) was granted accelerated approval in 2009 for the treatment of GBM with progressive disease in adult patients following prior therapy.
Our program:
Through proprietary bioinformatic analysis of academic laboratory profiling studies of GBM tumors, we have identified specific dysregulated microRNAs in distinct subtypes of the disease. Our analysis found that miR-10b is highly overexpressed, up to eight-fold, in a particular GBM patient population called the proneural subtype. Our findings show that treatment of GBM cell lines with anti-miRs targeting miR-10b, which we refer to as anti-miR-10b, reduces proliferation by blocking cell cycle progression and triggering cell death. In addition, we have shown in preclinical animal models of GBM, that direct injection into the tumor and spinal fluid achieves appropriate tissue delivery of anti-miRs for potential therapeutic effects.
We have a research collaboration with the Samsung Biomedical Research Institute to assist us in testing our anti-miR-10b development candidates in specialized preclinical models that mimic human brain cancer. In addition, we have funding support from Accelerate Brain Cancer Cure, or ABC2, a non-profit organization dedicated to accelerating therapies for brain cancer patients.
We intend to independently file an IND, develop and commercialize our anti-miR-10b development candidate for the treatment of GBM. Following the initiation of a Phase 1 clinical trial in patients with recurrent GBM to assess the safety and tolerability of our anti-miR-10b development candidate under the IND, we anticipate filing for orphan drug status for this development candidate. Upon identification of the maximum tolerated dose, we plan to enroll an expanded cohort using our microRNA biomarker strategy to identify patients with up-regulated miR-10b to further assess safety and evaluate efficacy on a preliminary basis in accordance with Response Criteria in Solid Tumors, or RECIST, measurement guidelines.
miR-33 in atherosclerosis
Market opportunity:
Atherosclerosis is the build up of plaque that occurs when cholesterol and inflammatory cells accumulate in blood vessels. These plaques can rupture, leading to slowing or blockage of blood flow and ultimately resulting in a heart attack or stroke. Scientific research has shown a strong correlation between high cholesterol levels and cardiovascular disease which, according to the CDC, is the leading cause of death in the United States.
41
Table of Contents
Business
Current treatments:
Most patients with atherosclerosis have high levels of a particular type of cholesterol particle known as LDL-C. The current standard of care is treatment with a class of drugs called statins that inhibit the production of cholesterol in the liver and therefore reduce the amount of LDL-C in circulation that might end up in plaques. However, such an approach has been shown to reduce the risk of a future heart attack or stroke by only approximately 30-40%. Recently, the scientific community has focused on another cholesterol particle known as HDL-C because it has been shown to remove cholesterol from plaques and transport it to the liver for excretion from the body.
Our program:
Our lead program for atherosclerosis targets miR-33, which has a unique mechanism of action for the management of cholesterol levels. The inhibition of miR-33 with our anti-miRs promotes reverse cholesterol transport, or RCT, which is the efflux of cholesterol from specific cholesterol-laden inflammatory cells called macrophages in atherosclerotic plaques. A natural consequence of enhancing RCT is an increase in the number of HDL-C particles that can remove cholesterol to the liver for excretion from the body. We are developing anti-miRs targeting miR-33, which we refer to as anti-miR-33. Treatment with anti-miR-33 in an atherosclerotic mouse model led to reduction in arterial plaque size by 35% and treatment in non-human primates increased circulating levels of HDL-C by 50%. By enhancing RCT, anti-miR-33 differs from other emerging therapeutic strategies that focus only on raising HDL-C in circulation.
In addition to direct benefits on atherosclerosis, treatment with anti-miR-33 in a preclinical study increased the breakdown of lipids, such as fatty acids, and enhanced signaling through the insulin receptor. These findings suggest that the inhibition of miR-33 could have additional benefits in other aspects of the metabolic syndrome, such as non-alcoholic steatohepatitis (fatty liver disease) and type-2 diabetes. We expect anti-miR-33 to be developed as a treatment for atherosclerosis, initially for patients at high-risk of recurrent cardiovascular events, such as heart attack.
As part of our strategic alliance with AstraZeneca, we, in consultation with AstraZeneca, plan to nominate an anti-miR-33 development candidate for the treatment of atherosclerosis and file an IND. We expect that AstraZeneca will initiate and fund Phase 1 clinical trials under the IND, according to a clinical development plan designed in consultation with us.
OUR STRATEGY
We are building the leading biopharmaceutical company focused on the discovery and development of first-in-class, targeted drugs based on our proprietary microRNA product platform. The key elements of our strategy are:
| Ø | Rapidly advance our initial programs into clinical development. We are currently optimizing our proprietary and partnered anti-miRs for development candidate selection. Under our “Road to the Clinic” strategy, we have nominated our proprietary compound, RG-101, in HCV as our first clinical candidate and expect to submit our first IND, or equivalent foreign regulatory filing, for RG-101 in the first half of 2014. We anticipate that we will nominate a second clinical candidate by the end of 2013. |
| Ø | Focus our resources on developing drugs for niche indications or orphan diseases. We believe that microRNA therapeutics have utility in almost every disease state as they regulate pathways, not single targets. We intend to focus on proprietary product opportunities in niche therapeutic areas where the development and commercialization activities are appropriate for our size and financial resources. |
42
Table of Contents
Business
| Ø | Selectively form strategic alliances to augment our expertise and accelerate development and commercialization. We have established strategic alliances with AstraZeneca, GSK and Sanofi and we will continue to seek partners who can bring therapeutic expertise, development and commercialization capabilities and funding to allow us to maximize the potential of our microRNA product platform. |
| Ø | Selectively use our microRNA product platform to develop additional targets. We have identified several other microRNA targets with potential for therapeutic modulation and will apply our rigorous scientific and business criteria to develop them. |
| Ø | Develop microRNA biomarkers to support therapeutic product candidates. We believe that microRNA biomarkers may be used to select optimal patient segments in clinical trials, to develop companion diagnostics, and to monitor disease progression or relapse. We believe these microRNA biomarkers can be applied toward drugs that we develop and drugs developed by other companies, including small molecules and monoclonal antibodies. |
| Ø | Maintain scientific and intellectual leadership in the microRNA field. We will continue to conduct research in the microRNA field to better understand this new biology and characterize the specific mechanism of action for our future drugs. This includes building on our strong network of key opinion leaders and securing additional intellectual property rights to broaden our existing proprietary asset estate. |
OUR STRATEGIC ALLIANCES AND COLLABORATIONS
Our goal is to discover and develop microRNA therapeutics. To access the substantial funding and expertise required to develop and commercialize microRNA therapeutics, we have formed and intend to seek other opportunities to form strategic alliances with pharmaceutical companies who can augment our industry leading microRNA expertise. To date, we have focused on forging a limited number of significant strategic alliances with leading pharmaceutical partners and academic laboratories where both parties contribute expertise to enable the discovery and development of potential microRNA therapeutics.
Under our existing strategic alliances with AstraZeneca, GSK and Sanofi, we are eligible to receive up to approximately $1.3 billion in milestone payments upon successful commercialization of microRNA therapeutics for the eleven programs contemplated by our agreements. These payments include up to $42.0 million upon achievement of preclinical and IND milestones, up to $272.0 million upon achievement of clinical development milestones, up to $305.0 million upon achievement of regulatory milestones and up to $670.0 million upon achievement of commercialization milestones.
Our strategic alliance with GlaxoSmithKline
In April 2008, we entered into a product development and commercialization agreement with Glaxo Group Limited, an affiliate of GlaxoSmithKline plc, or GSK. Under the terms of the agreement, we agreed to develop four programs of interest to GSK in the areas of inflammation and immunology and granted to GSK an option to obtain an exclusive worldwide license to develop, manufacture and commercialize products in each program. We are responsible for the discovery, optimization and development of anti-miR product candidates in each program through proof-of-concept, defined as the achievement of relevant efficacy and safety endpoints in the first clinical trial designed to show efficacy, safety and tolerability, unless GSK chooses to exercise its option at an earlier stage. Upon GSK exercising its option with respect to a particular program and paying an option exercise fee, we will grant GSK an exclusive worldwide license to develop drugs under the selected program, and GSK will thereafter be responsible for all development, manufacturing and commercialization activities and costs. As of the date of the agreement, GSK had pre-selected two microRNA alliance targets. In February 2010, we and GSK
43
Table of Contents
Business
expanded the alliance to include miR-122 for the treatment of HCV. In June 2013, we amended our agreement with GSK to specify that RG-101 is fully owned by us and that miR-122 will remain a collaboration target under the agreement. We will be solely responsible for the development, manufacture and commercialization of RG-101 and GSK has no rights to this compound.
Upon entering into the agreement, we received an upfront payment of $15.0 million as an option fee, and GSK loaned $5.0 million to us under a convertible note. In connection with the expansion of the alliance to include miR-122 for the treatment of HCV, in February 2010, GSK made an upfront payment to us of $3.0 million and loaned an additional $5.0 million to us pursuant to a second convertible note. The notes were amended and restated in July 2012 and upon closing of our initial public offering, the principal and accrued interest of one note was converted to 1,447,037 shares of our common stock. The note that did not automatically convert upon the initial public offering was restated in October 2012 and accrues interest at 3.297% with an adjusted face amount equal to the principal and accrued interest as of the completion of our initial public offering, which totaled approximately $5.4 million, and will mature in October 2015. At GSK’s option, this note is convertible into shares of our common stock at any time prior to the maturity date with a conversion equal to the quotient of all outstanding principal and interest divided by the initial public offering price of $4.00 per share. Under our strategic alliance with GSK, we earned a $500,000 milestone payment in each of May 2009 and July 2011. We are eligible to receive up to $144.5 million in preclinical, clinical, regulatory and commercialization milestone payments for each of the four microRNA programs under our alliance with GSK. We are also eligible to receive tiered royalties as a percentage of annual sales which can increase up to the low end of the 10 to 20% range. These royalties are subject to reduction upon the expiration of certain patents or introduction of generic competition into the market, or if GSK is required to obtain licenses from third parties to develop, manufacture or commercialize products under the alliance.
For each microRNA alliance target selected by GSK under the agreement, we commence a research program directed against such target under a research plan adopted by a joint committee to discover and optimize compounds that meet candidate selection criteria. On a program-by-program basis, GSK may exercise its option at any time on or before completion of the proof-of-concept trial. To exercise its option, GSK must pay us an option exercise fee, which varies depending on the stage of the program at which the option is exercised. Milestone payments payable by GSK are higher if GSK exercises its option upon completion of the proof-of-concept trial. Once a microRNA alliance target has been selected by GSK, neither party may work independently or with a third party on a microRNA compound designed to modulate an alliance target. In addition, during the research term, neither we nor our affiliates may work independently or with a third party on any compound that is designed to modulate an alliance target.
If GSK does not exercise its option, or ceases development after exercising its option with respect to a particular program, we will have all rights to develop or commercialize product candidates under the program (including the right to sublicense these rights to a third party) at our sole expense. In the event the product is eventually commercialized, GSK will be entitled to “reverse royalties” as a percentage of net sales, subject to certain caps.
Either party may terminate the agreement upon written notice in the event of the other party’s material breach, including the failure to comply with such party’s diligence obligations, that remains uncured for 90 days. GSK has the unilateral right to terminate the agreement in its entirety or on an alliance target basis upon 90 days’ prior written notice to us.
44
Table of Contents
Business
Our strategic alliance with Sanofi
Sanofi collaboration and license agreement
In June 2010, we entered into a collaboration and license agreement with Sanofi, which we subsequently amended, restated and superseded in July 2012. Under the terms of the agreement, we have agreed to collaborate with Sanofi to develop and commercialize licensed compounds targeting four microRNA alliance targets initially focused in the field of fibrosis. The agreement specified that miR-21 would be the first alliance target in the field of fibrosis and we granted Sanofi an exclusive worldwide license to develop and commercialize products under the alliance. The July 2012 amended and restated agreement expanded the alliance to include potential microRNA therapeutics in oncology.
Under the terms of the agreement, we have agreed to use commercially reasonable efforts to provide Sanofi with validated microRNA targets and are responsible for conducting all research and compound manufacturing activities until acceptance of an IND. After acceptance of the IND, Sanofi will assume all costs, responsibilities and obligations for further development and commercialization.
Under the terms of the agreement, we received an upfront payment of $25.0 million which was allocated to the research programs. In addition, Sanofi made a $10.0 million equity investment in the company. We also received $5.0 million for one year of research and development funding. Subsequently, we received $5.0 million for research and development funding following each of the first and second anniversaries of our entry into the agreement in June 2010. The research term ended in June 2013 per the terms of the agreement and therefore we have no further research obligations under the agreement. Under the terms of the agreement, Sanofi may continue development of any licensed compounds. We are also entitled to receive preclinical, clinical, regulatory and commercialization milestone payments of up to $213.0 million in the aggregate for all alliance product candidates. In addition, we are entitled to receive royalties based on a percentage of net sales which will range from the mid-single digits to the low end of the 10 to 20% range, depending upon the target and the volume of sales.
Sanofi may terminate the agreement in full or on a product-by-product basis by giving 30 days’ prior written notice to us. Either party may also terminate the agreement for a material breach by the other party which remains uncured after 120 days’ notice of such breach, except that we may not exercise this termination right until after the expiration of the research term if Sanofi is in breach of its obligations to use commercially reasonable efforts. In the event a program or the agreement is terminated by Sanofi, the rights to develop and commercialize product candidates in the terminated programs (including the right to sublicense these rights to a third party) returns to us. If we sublicense the rights to a third party, we will be required to pay a percentage of sublicense revenues to Sanofi in the low end of the 10 to 20% range, and if we commercialize a product on our own, we will be required to pay royalties in the low single digits to Sanofi as a percentage of net sales.
In April 2013, we entered into a letter agreement with Sanofi to extend the time period available to Sanofi to extend its option to extend the research term under the agreement. The original research term for our strategic alliance with Sanofi expired in June 2013, upon which we and Sanofi entered into an option agreement pursuant to which we have the exclusive right to negotiate with Sanofi to enter into a co-development and commercialization agreement for an anti-miR-21 for the treatment of oncology indications and Alport’s disease through December 2013 and pursuant to which we granted Sanofi an exclusive right to negotiate with us to enter into a development and license agreement for the co-development and commercialization of certain of our unencumbered microRNA programs through December 2013. In connection with the option agreement, Sanofi has agreed to pay us an upfront option fee of $2.5 million, $1.25 million of which is creditable against future payments payable by Sanofi to us.
45
Table of Contents
Business
Sanofi non-exclusive technology alliance and option agreement
Concurrently with the collaboration and license agreement, we also entered into a non-exclusive technology alliance and option agreement with Sanofi. Under this agreement, Sanofi received an option for a broader microRNA technology license. Sanofi may exercise the option at any time until the date 30 days after the third anniversary of the agreement, subject to a one-time extension and payment of an extension fee.
If Sanofi exercises its option under this agreement, we will receive a payment of up to $50.0 million, payable in installments. In return, Sanofi will receive a license to our microRNA product platform technology for research of microRNA compounds. The option also provides us with certain rights to participate in the development and commercialization of products. We are also entitled to receive a product-by-product milestone payment and royalties as a percentage of net sales in the low single digits for products commercialized by Sanofi.
Our strategic alliance with AstraZeneca
In August 2012, we entered into a collaboration and license agreement with AstraZeneca. Under the terms of the agreement, we have agreed to collaborate with AstraZeneca to identify, research and develop compounds targeting three microRNA alliance targets primarily in the fields of cardiovascular diseases, metabolic diseases and oncology and granted to AstraZeneca an exclusive, worldwide license to thereafter develop, manufacture and commercialize lead compounds designated by AstraZeneca in the course of the collaboration activities against the alliance targets for all human therapeutic uses. Under the terms of the agreement we are required to use commercially reasonable efforts to perform all research, development and manufacturing activities described in the research plan, at our cost, until the acceptance of an IND or the end of the research term, which extends until the fourth anniversary of the date of the agreement, and may be extended only by mutual written agreement of us and AstraZeneca. Following the earlier to occur of the acceptance of an IND in a major market or the end of the research term, AstraZeneca will assume all costs, responsibilities and obligations for further development, manufacture and commercialization of alliance product candidates. In April 2013, we and AstraZeneca amended the collaboration and license agreement to provide that we would undertake microRNA analysis of certain samples controlled by AstraZeneca in connection with miR-33 and another microRNA target. All results and inventions relating to the microRNA analysis of such clinical samples will be owned solely by AstraZeneca, but AstraZeneca granted us a perpetual, non-exclusive license to such results and inventions for our internal research and development purposes.
Under the terms of the agreement, we are entitled to receive an upfront payment of $3.0 million. If all three targets are successfully developed and commercialized through pre-agreed sales targets we could receive milestone payments up to $498.0 million, including up to $5.0 million for preclinical milestones, up to $123.0 million for clinical milestones, and up to $370.0 million for commercialization milestones. In addition, we are entitled to receive royalties based on a percentage of net sales which will range from the mid-single digits to the low end of the 10 to 20% range, depending upon the product and the volume of sales, which royalties may be reduced in certain, limited circumstances.
Either party may terminate the agreement upon written notice to the other party provided within 20 business days after the end of the research term if no lead compound has been designated at that time. Either party may also terminate the agreement in the event of the other party’s material breach which remains uncured for 40 business days following notice thereof (or 30 business days in the case of nonpayment). During the research term, AstraZeneca may terminate the agreement in its entirety or on a collaboration target-by-collaboration target basis at any time within 20 business days of our notice to AstraZeneca of the closing of a transaction that would result in a change of control. During the research term, AstraZeneca may also terminate the agreement in its entirety or on a collaboration target-by-
46
Table of Contents
Business
collaboration target basis for any reason upon 60 business days’ prior written notice to us. Following the research term, AstraZeneca may terminate the agreement in its entirety or on a collaboration target-by-collaboration target basis for any reason upon 12 months’ prior written notice to us.
Concurrently with the collaboration and license agreement, we entered into a Common Stock Purchase Agreement, or CSPA, with AstraZeneca, pursuant to which we agreed to sell to AstraZeneca an aggregate of $25.0 million of our common stock in a private placement concurrently with our initial public offering, at a price per share equal to the price at which we sold our common stock to the public in such initial public offering. In October 2012, in accordance with the CSPA, we sold AstraZeneca 6,250,000 shares of our common stock at a price per share of $4.00. Further, the CSPA stipulated that AstraZeneca could not sell, transfer, make any short sale of, or grant any option for the sale of any common stock for a 365-day period following the effective date of our initial public offering.
Our collaboration with Biogen Idec
In August 2012, we entered into a collaboration and license agreement with Biogen Idec pursuant to which we and Biogen Idec have agreed to collaborate on microRNA biomarkers for multiple sclerosis, or MS. Under the terms of the agreement, we granted Biogen Idec an exclusive, royalty free, worldwide license to our interest in the collaboration intellectual property for the purpose of commercializing non-microRNA products for the treatment, diagnosis and prevention of MS and non-MS diseases and disorders. We also granted Biogen Idec an exclusive, royalty-free, worldwide license, with the right to sublicense, to our interest in the collaboration intellectual property (and a non-exclusive license to our background intellectual property) for the purpose of commercializing products for the diagnosis of MS. We also granted Biogen Idec a right of first negotiation on certain commercial transactions relating to microRNA products which utilize intellectual property developed during the collaboration. Pursuant to the terms of the agreement, in August 2012 we received an upfront payment of $0.8 million. We can earn the following research milestones: $0.25 million for identification of a microRNA biomarker; $0.5 million for validation of the microRNA biomarker in a second independent sample set; and $0.5 million upon the refinement of the microRNA biomarker signature from a longitudinal study of patient samples on MS therapy. In June 2013, we and Biogen amended the agreement to update the research plan and criteria for success. In June 2013 we and Biogen Idec entered into an amendment agreement which revised the terms with respect to the Phase 1 research plan and Phase 1 development milestones under the collaboration and license agreement.
Concurrently with the collaboration and license agreement, we entered into a note purchase agreement with Biogen Idec, pursuant to which we issued Biogen Idec a convertible promissory note in the principal amount of $5.0 million. The $5.0 million note plus accrued interest converted into 1,256,232 shares of our common stock upon the closing of our initial public offering in October 2012 at a conversion price of $4.00 per share.
Either party may terminate the agreement upon the other party’s material breach which remains uncured for 60 days following notice thereof. If the agreement is terminated in connection with a party’s material breach, the non-breaching party will retain the license rights granted by the breaching party under the agreement, but all license rights granted by the non-breaching party to the breaching party will terminate. Each party may also terminate the agreement for convenience upon 30 days’ prior written notice to the other party, but in such case the terminating party will forfeit the license rights granted to it by the non-terminating party pursuant to the agreement, and the non-terminating party will retain the license rights granted by the terminating party.
Our strategic alliance with Alnylam and Isis
In September 2007, we entered into a license and collaboration agreement with Alnylam and Isis, which we subsequently amended, restated and superseded in January 2009, and further amended in June 2010 and
47
Table of Contents
Business
October 2011. Under the agreement, we acquired an exclusive, royalty-bearing, worldwide license, with rights to sublicense, to patent rights owned or licensed by Alnylam and Isis to develop, manufacture and commercialize products covered by the licensed patent rights for use in microRNA compounds which are microRNA antagonists and microRNA therapeutics containing these compounds. In addition, we have certain rights to miR-mimics. Under the agreement, we granted to both Alnylam and Isis a license to practice our intellectual property developed by us to the extent that it is useful specifically to Alnylam’s RNAi programs or Isis’ single-stranded oligonucleotide programs, but not including microRNA compounds or therapeutics that are the subject of our exclusive licenses from Alnylam and Isis.
We are required to use commercially reasonable efforts to develop and commercialize licensed products under the agreement. We are required to notify Alnylam and Isis when a program reaches development stage (defined as initiation of good laboratory practices, or GLP, toxicology studies) and whether or not we intend to pursue the program. Under the agreement, both Alnylam and Isis have an option to assume the development and commercialization of product candidates in a program that we do not pursue. If neither Alnylam nor Isis exercises this option, we are required to use our best efforts to finalize a term sheet with a third party with respect to such program. In the event we are unable to complete a transaction with a third party, both Alnylam and Isis have a second opt-in option.
If an election is made by either Alnylam or Isis (but not both) to opt-in, such party will pay us a one-time fixed payment, the amount of which will depend on whether the first or the second opt-in option was exercised, with a higher amount due if the first opt-in option was exercised. Clinical and regulatory milestones are also payable to us in the event the opt-in election is exercised. Such milestones total $64.0 million in the aggregate if the election is made during the first opt-in period or $15.7 million in the aggregate if the election is made at the second opt-in period. Tiered royalties are payable to us as a percentage of net sales on all products commercialized by the opt-in party. These royalties range from the low to middle single digits depending upon the volume of sales. The opt-in party is also entitled to sublicense the development program to a third party. In such a case, we are also entitled to receive a percentage of the sublicense income received by the opt-in party. The percentage payable depends upon the point at which the opt-in party sublicenses the program and ranges from the low end of the 10 to 20% range to the high end of the 40 to 50% range. The opt-in party is only required to pay the higher of the clinical and regulatory milestones or the sublicense income received in any calendar quarter. The opt-in party is also responsible for all third party payments due under other agreements as a result of the development. In the event both Alnylam and Isis elect to opt-in during either opt-in period, the parties have agreed to work together to amend the development plan to continue development of the project, including funding of such project and assignment of roles and responsibilities.
In the event we or one of our strategic alliance partners continues with the development of a program, each of Alnylam and Isis are entitled to royalties as a percentage of net sales. For products that we independently commercialize, these royalties will be in the low single digits. For products commercialized by a third-party collaborator, the royalties will be either the same percentage of net sales as described above or, if the sublicense does not provide a specified level of royalties to us or upon our election, a percentage of the sublicense income received by us from the strategic alliance partner and a modified royalty. The modified royalty would be based upon the lower of the single digit percentage discussed above or one third of the royalty received by us after payments made by us to third parties for development, manufacture and commercialization activities under other agreements. In addition, if we sublicense rights to a collaborator, we will be required to pay to each of Alnylam and Isis a percentage of our sublicense income in the mid-single digits. We are also responsible for payments due to third parties under other agreements as a result of our development activities, including payments owed by Alnylam and/or Isis under their agreements.
Under the October 2011 amendment, Alnylam and Isis granted us the right to research microRNA mimics under the licensed intellectual property of Alnylam and Isis. In the event we develop a miR-mimic, we must
48
Table of Contents
Business
first obtain approval from Alnylam and/or Isis, as applicable, and such approval is subject to the consent of applicable third parties, if any. No additional consideration will be owed by us to Alnylam or Isis for granting approval. We have the right to sublicense our research rights. We granted to both Alnylam and Isis a fully paid up, worldwide and exclusive license to any intellectual property developed by us and useful to their research programs and which are not microRNA antagonists or approved miR-mimics.
The agreement expires on the earlier of the cessation of development of the potential royalty-bearing products prior to the commercial sale of any such products anywhere in the world or following the first commercial sale of such products, the expiration of royalty obligations determined on a country-by-country and product-by-product basis.
OUR INTELLECTUAL PROPERTY AND TECHNOLOGY LICENSES
Intellectual property
We strive to protect and enhance the proprietary technologies that we believe are important to our business, including seeking and maintaining patents intended to cover our products and compositions, their methods of use and any other inventions that are important to the development of our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain our proprietary position in the field of microRNA therapeutics.
We believe that we have a strong intellectual property position and substantial know-how relating to the development and commercialization of microRNA therapeutics, consisting of:
| Ø | over 170 patents or patent applications that we own or have in-licensed from academic institutions and third parties including our founding companies, Alnylam and Isis, related to microRNA and microRNA drug products; and |
| Ø | approximately 900 patents or patent applications exclusively licensed from our founding companies, Alnylam and Isis, related to RNA technologies, including patent and patent applications relating to chemical modification of oligonucleotides that are useful for microRNA therapeutics. |
Our objective is to continue to expand our intellectual property estate through our multiple layer approach in order to protect our microRNA therapeutics and to maintain our leading position in the microRNA therapeutics field. Examples of the technologies covered by our patent portfolio are described below.
We have exclusively licensed patent rights from Julius-Maximilians-Universität Würzburg and Bayerische Patent Allianz GmBH, which we collectively refer to herein as the University of Würzburg, which rights encompass the use of anti-miR therapeutics targeting miR-21 for the treatment of fibrosis, including kidney, liver, lung and cardiac fibrosis. In collaboration with us, investigators at the University of Würzburg demonstrated that targeting miR-21 in a disease model resulted in beneficial phenotypic effects, including the inhibition of the development of fibrosis. The Würzburg-licensed patent portfolio includes more than 20 U.S. and foreign patents and patent applications. Any patents within this portfolio that have issued or may yet issue would have a statutory expiration date in 2029.
We and Alnylam have a co-exclusive license from Stanford University, or Stanford, to patent rights concerning the use of anti-miR therapeutics targeting miR-122 for the treatment of HCV. This patent
49
Table of Contents
Business
portfolio is based upon research conducted by Peter Sarnow, Ph.D. and colleagues at Stanford, demonstrating that miR-122 is required for HCV replication in mammalian cells. The Stanford-licensed portfolio includes more than 12 U.S and foreign patents and patent applications. Any patents within this portfolio that have issued or may yet issue would have a statutory expiration date in 2025.
In support of our program targeting miR-33, we have exclusively licensed from New York University, or NYU, patent rights encompassing the use of an anti-miR therapeutic targeting miR-33 for the treatment of atherosclerosis, metabolic syndrome and elevated triglycerides. In collaboration with us, Kathryn Moore, Ph.D. and colleagues at NYU demonstrated that inhibiting miR-33 has several therapeutic benefits, including reduction of atherosclerotic plaque size in an experimental model of atherosclerosis, in addition to reduction of serum triglycerides in non-human primates. The NYU-licensed patent rights include five US and foreign patent applications. Any patents that may issue from these applications would have a statutory expiration date in 2031.
Our portfolio of exclusively and jointly owned patent and patent applications is currently composed of over 55 U.S. and foreign patents and patent applications. We are the sole owner of over 50 of the patents and pending applications. We jointly own at least five of the patents and pending applications including those claiming methods for treating liver cancer, including HCC, using anti-miRs targeting miR-21. The patents have statutory expiration dates in through 2029. Any patents that may issue from the pending applications would have statutory expiration dates through 2034.
Our founding companies, Alnylam and Isis, each own or otherwise have rights to numerous patents and patent applications concerning oligonucleotide technologies and a substantial number of these patents and applications have been exclusively licensed to us for use in the microRNA field. The technologies covered in these patents and applications include various chemical modifications that are applicable to microRNA therapeutics. Among the licensed patents or patent applications, those covering key chemical modifications for use in microRNA drug products have statutory expiration dates through 2028.
We have a co-exclusive license to the patent portfolio owned by Max-Planck-Gesellschaft, or MPG, which has been granted to us by Max-Planck-Innovation GmbH, or MI, a wholly-owned subsidiary of MPG acting as MPG’s technology transfer agency. MPG and MI are collectively referred to herein as Max-Planck. This patent portfolio is based on the pioneering microRNA research conducted by Thomas Tuschl, Ph.D. and colleagues at the Max-Planck Institute of Biophysical Chemistry, which led to the discovery of over 100 human microRNA sequences, including microRNAs that are the focus of several of our programs. The patent rights encompass both microRNA mimic and anti-miR products. Our license is co-exclusive with our founding companies, Alnylam and Isis, for the exploitation of the Max-Planck patent rights for therapeutic uses. In addition, we also have a co-exclusive license to develop and commercialize diagnostics based upon the Max-Planck patent rights contained in these applications. The Max-Planck licensed patent portfolio, referred to herein as the Tuschl 3 patents, includes at least 25 U.S. and foreign patents and patent applications. Any patents within this portfolio that have issued or may yet issue would have a statutory expiration date in 2022.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing the non-provisional application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent.
The term of a patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration of a U.S. patent as compensation for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the
50
Table of Contents
Business
length of time the drug is under regulatory review. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of a new drug application, or NDA, we expect to apply for patent term extensions for patents covering our microRNA product candidates and their methods of use.
We may rely, in some circumstances, on trade secrets to protect our technology. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants, contractors or collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
OUR TECHNOLOGY LICENSES
Max-Planck
Therapeutic license
Prior to 2011, our access to the Tuschl 3 patents was derived from agreements between Max-Planck and our founding companies, Alnylam and Isis, for exclusive use in microRNA therapeutics. In April 2011, we entered into a direct, co-exclusive license with Max-Planck. The license provides to us, Alnylam and Isis, co-exclusively, access to the Tuschl 3 patents for therapeutic use. Max-Planck retains the right to practice the intellectual property licensed under the agreement for non-commercial purposes.
Under the terms of the license, we are permitted to sublicense our rights outright or as part of an alliance. The license requires that we use commercially reasonable diligence in developing and commercializing a product. In order to secure the license, we made an upfront payment of $400,000 to Max-Planck. We will be required to make payments based upon the initiation of clinical trials and/or product approval milestones totaling up to $1.6 million for each licensed product reaching such clinical stage. In addition to milestone payments, we will be required to pay royalties of a percentage of cumulative annual net sales of a licensed product commercialized by us or one of our strategic alliance partners. The percentage is in the low single digits, with the exact percentage depending upon whether the licensed product incorporates intellectual property covered by a Tuschl 3 patent that is still a pending application or, alternatively, an issued patent, and also upon the volume of annual sales. The royalties payable to Max-Planck are subject to reduction for any third party payments required to be made, with a minimum floor in the low single digits.
We may unilaterally terminate the license agreement upon three months’ notice and payment of all accrued amounts owing to Max-Planck. Max-Planck may terminate the agreement upon 30 days’ prior written notice if we challenge the validity of its patents, or in the event of our material breach which remains uncured after 60 days of receiving written notice of such breach (30 days in the case of nonpayment). Absent early termination, the agreement will automatically terminate upon the expiration or abandonment of all issued patents and filed patent applications with the patent rights covered by the agreement. The longest lived patent rights licensed to us under the agreement are currently expected to expire in September 2022.
51
Table of Contents
Business
Diagnostic license
In addition, in June 2009, we entered into a co-exclusive license with Max-Planck for use of the Tuschl 3 patents for diagnostic purposes. Under the terms of the license, we made an aggregate initial payment to Max-Planck of €175,000 in three installments, with €75,000 paid in June 2009 and €50,000 paid in each of June 2010 and June 2011. In addition, we made annual maintenance payments of €10,000 in 2011 and €20,000 in 2012 and will make an increased annual maintenance payment commencing in 2013 and thereafter during the term of the agreement. In addition to maintenance payments, we will be required to pay royalties of a percentage of net sales of licensed products. The percentage is in the mid-single digits in the event we market the product and low end of the 10 to 20% range in the event we sell the product through a distributor. The royalties payable to Max-Planck are reduced by the royalties payable to third parties but only if aggregate royalties payable to Max-Planck and third parties exceed a percentage in the mid-10 to 20% range.
We are required to use commercially reasonable efforts to develop and commercialize products under the agreement. Under the terms of the agreement, Max-Planck is permitted to provide up to three additional co-exclusive licenses to its diagnostic patent rights. We may unilaterally terminate the license agreement upon three months’ notice and payment of all accrued amounts owing to Max-Planck. Max-Planck may terminate the agreement upon 30 days’ prior written notice if we challenge the validity of its patent rights, or in the event of our material breach which remains uncured after 60 days of receiving written notice of such breach (30 days in the case of nonpayment). Absent early termination, the agreement will automatically terminate upon the expiration or abandonment of all issued patents and filed patent applications with the patent rights covered by the agreement. The longest lived patent rights licensed to us under the agreement are currently expected to expire in September 2022.
Max-Planck retains the right to practice the intellectual property licensed under the agreement for non-commercial purposes.
University of Würzburg
In May 2010, we exclusively licensed patent rights from the University of Würzburg which encompass the use of anti-miR therapeutics targeting miR-21 for the treatment of fibrosis, including kidney, liver, lung and cardiac fibrosis.
The University of Würzburg has reserved the right to use the licensed intellectual property for academic and non-commercial purposes. We have the right to grant sublicenses to third parties under the agreement provided such sublicense is for the purpose of developing or commercializing a product. We must obtain the University of Würzburg’s written consent to any such sublicense, which may not be unreasonably withheld. We must use commercially reasonable diligence in our efforts to develop, manufacture and commercialize a licensed product. We have assumed certain development milestone obligations and must report on our progress in achieving these milestones on an annual basis.
As a license issuance fee, we paid the University of Würzburg €300,000. In addition, upon commercialization of a product, we will pay to the University of Würzburg a percentage of net sales as a royalty. This royalty is in the low single digits and is reduced upon expiration of all patent claims covering the product. We also paid the University of Würzburg a partnership bonus of €200,000 upon entering into our strategic alliance agreement with Sanofi. Under the agreement, beginning January 1, 2020 and ending on the date we receive NDA approval for a licensed product, we will accrue a minimum royalty obligation of €150,000 per year, which will become payable upon approval of an NDA for a licensed product. After approval of an NDA for a licensed product, we will be required to pay the University of Würzburg an annual minimum royalty, which increases in the five years following approval up to a maximum of €3.0 million per year. The minimum royalties are creditable against actual royalties due and payable for the same calendar year.
52
Table of Contents
Business
In addition, we will be required to pay the University of Würzburg milestone payments of up to an aggregate of €1.75 million, based upon achievement of specified clinical and regulatory events. In the event we initiate a Phase 2 clinical trial for another indication with the same licensed product, we will be required to pay 50% of the milestone payments applicable to such milestone events. These milestone events are also tied to the due dates set forth in the commercialization plan but may be extended by delays caused by scientific challenges, regulatory requirements or other circumstances outside of our control. We must request an extension in writing explaining the cause for the delay and proposing new due dates. The University of Würzburg may accept the revised dates or reject them, in which case an arbitrator will set the revised dates.
We may terminate the agreement upon 30 days’ notice to the University of Würzburg. The University of Würzburg may terminate the agreement if we challenge the validity of its patent rights, or in the event of our nonpayment which remains uncured after 60 days of receiving written notice of such nonpayment. Absent early termination, the agreement will terminate upon the later of the expiration of the last to expire patent licensed to us under the agreement (which is currently expected to be in February 2029) or 10 years following the date of the most recent first commercial sale in a new country of a licensed product.
Stanford University
In August 2005, Alnylam and Isis entered into a co-exclusive license agreement with Stanford, relating to its patent applications claiming the use of miR-122 to reduce the replication of HCV. Upon our formation, we received access to the Stanford technology as an affiliate of Alnylam and Isis. In July 2009, Isis assigned its rights and obligations under the license agreement to us.
Under the license agreement, we are permitted to research, develop, manufacture and commercialize therapeutics for the treatment and prevention of HCV and related conditions. Diagnostics and reagents are specifically excluded from the license. In addition, the license provides a non-exclusive right to research, develop, manufacture and commercialize therapeutics for all conditions or diseases other than HCV. Stanford retained the right, on behalf of itself and all other non-profit academic institutions, to practice the licensed patents for non-profit purposes.
We are permitted to sublicense our rights under the agreement in connection with a bona fide partnership seeking to research and/or develop products under a jointly prepared research plan and which also includes a license to our intellectual property or in association with providing services to a sublicensee. In the event we receive an upfront payment in connection with a sublicense, we are obligated to pay to Stanford a one-time fixed payment amount, which amount will vary depending upon the size of upfront payment we receive. We must also make an annual license maintenance payment during the term of the agreement. The maintenance payments are creditable against royalty payments made in the same year. We will be required to pay milestones for an exclusively licensed product which will be payable upon achievement of specified regulatory and clinical milestones in an aggregate amount of up to $400,000. Milestones for a non-exclusively licensed product will be payable upon achievement of the same milestones in an aggregate amount of up to $300,000 for the first such product and up to $200,000 for the second such product. Upon commercialization of a product, we will be required to pay to Stanford a percentage of net sales as a royalty. This percentage is in the low single digits. The payment will be reduced by other payments we are required to make to third parties until a minimum royalty has been reached.
The agreement requires that we use commercially reasonable efforts to develop, manufacture and commercialize a licensed product and we have agreed to meet certain development and commercialization milestones.
We may terminate the agreement upon 30 days’ notice. Stanford may terminate the agreement in the event of our nonpayment or material breach which remains uncured after 60 days of receiving written
53
Table of Contents
Business
notice of such nonpayment or breach. Absent early termination, the agreement will automatically terminate upon the expiration of the last to expire patent licensed to us under the agreement, which is currently expected to be in May 2025.
New York University
In March 2011, we entered into an exclusive license with NYU related to our miR-33 program. The license provides us the right to develop, manufacture and commercialize therapeutics for the treatment or prevention of atherosclerotic plaque and/or other metabolic disorders under NYU’s patents. We are entitled to grant sublicenses under the agreement. NYU retains the right to practice the intellectual property licensed under the agreement for non-commercial purposes.
Under the terms of the agreement, we paid to NYU an upfront payment of $25,000. An equal additional payment will be required upon issuance of a patent containing a claim of treating or preventing disease. We will be required to make payments to NYU upon achievement of specified clinical and regulatory milestones of up to an aggregate of $925,000. These milestone payments will only be made after issuance of a therapeutic claim under the NYU patent applications. We are also required to pay royalties of a percentage of net sales for any product sold by us or a strategic alliance partner. The royalty rate is in the low single digits and is subject to reduction to a minimum amount in the event we are required to pay royalties to a third party. In the event we sublicense the NYU patents, NYU is also entitled to receive a percentage of the sublicense income received by us. The percentage payable depends upon the development stage of the program when the sublicense is completed with the highest percentage paid with submission of the first IND. The percentage thereafter declines until completion of the first Phase 2 clinical trial.
We are required, under the terms of the agreement, to use reasonable diligence to develop and commercialize a product and are required to provide NYU with annual reports detailing our progress in this regard. In particular, we are required to fulfill specific development and regulatory milestones by particular dates. The agreement may be terminated by either party upon written notice to the other party of its material breach of the agreement which has remained uncured for 60 days following written notice thereof (30 days in the case of nonpayment). We may also terminate the license upon 60 days’ notice in the event development of a product is not scientifically or commercially feasible. Absent early termination, the agreement will automatically terminate upon the expiration of the longest-lived patent rights covered by the agreement, which is currently expected to be in August 2031.
COMPETITION
The biotechnology and pharmaceutical industries are characterized by intense and rapidly changing competition to develop new technologies and proprietary products. While we believe that our proprietary asset estate and scientific expertise in the microRNA field provide us with competitive advantages, we face potential competition from many different sources, including larger and better-funded pharmaceutical companies. Not only must we compete with other companies that are focused on microRNA therapeutics but any products that we may commercialize will have to compete with existing therapies and new therapies that may become available in the future.
We are aware of several companies that are working specifically to develop microRNA therapeutics. These include the biotechnology companies Groove Biopharma, Inc., miRagen Therapeutics, Inc., Mirna Therapeutics, Inc., and Santaris Pharma A/S. These competitors also compete with us in recruiting human capital and securing licenses to complementary technologies or specific microRNAs that may be critical to the success of our business. They also compete with us for potential funding from the pharmaceutical industry.
In addition, we expect that for each disease category for which we determine to develop and apply our microRNA therapeutics there are other biotechnology companies that will compete against us by
54
Table of Contents
Business
applying marketed products and development programs using technology other than microRNA therapeutics. The key competitive factors that will affect the success of any of our development candidates, if commercialized, are likely to be their efficacy relative to such competing technologies, safety, convenience, price and the availability of reimbursement from government and other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors have products which are better in one or more of these categories.
MANUFACTURING
We contract with third parties to manufacture our compounds and intend to do so in the future. We do not own or operate and we do not expect to own or operate, facilities for product manufacturing, storage and distribution, or testing. We have personnel with extensive technical, manufacturing, analytical and quality experience and strong project management discipline to oversee contract manufacturing and testing activities, and to compile manufacturing and quality information for our regulatory submissions.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Our systems and contractors are required to be in compliance with these regulations, and this is assessed regularly through monitoring of performance and a formal audit program.
Drug substance
Our current drug substance supply chain involves various contractors that supply the raw materials and others that manufacture the anti-miR drug substance. We believe our current drug substance contractors have the scale, the systems and the experience to supply all planned IND-enabling studies, early clinical supplies and may be considered for later clinical trials and commercial manufacturing. To ensure continuity in our supply chain, we plan to establish supply arrangements with alternative suppliers for certain portions of our supply chain, as appropriate.
Our process uses common synthetic chemistry and readily available materials. We have established an ongoing program to identify possible process changes to improve purity, yield, manufacturability, and process changes will be implemented as warranted and appropriate. Based upon our knowledge of anti-sense compounds and conjugate chemistry, we do not anticipate any stability issues with our anti-miR product candidates.
Drug product
Our drug product is expected to consist of the anti-miR drug substance in a powdered form formulated in water or a saline solution for injection. Drug product manufacturing uses common processes and readily available materials. When a potential product is ready to commence IND-enabling studies, we will be required to commence drug product stability studies.
GOVERNMENT REGULATION AND PRODUCT APPROVAL
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of products such as those we are developing. Any product candidate that we develop must be approved by the FDA before it may be legally marketed in the United States and by the appropriate foreign regulatory agency before it may be legally marketed in foreign countries.
55
Table of Contents
Business
U.S. drug development process
In the United States, the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act, or FDCA, and implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial civil or criminal sanctions. FDA sanctions could include refusal to approve pending applications, withdrawal of an approval, clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, debarment, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| Ø | completion of nonclinical laboratory tests, animal studies and formulation studies according to good laboratory practices, or GLP, or other applicable regulations; |
| Ø | submission to the FDA of an application for an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| Ø | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as current good clinical practices, or GCPs, which are ethical and scientific quality standards and FDA requirements for conducting, recording and reporting clinical trials to assure that the rights, safety and well-being of trial participants are protected and to establish the safety and efficacy of the proposed drug for its intended use; |
| Ø | submission to the FDA of an NDA for a new drug; |
| Ø | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the drug is produced to assess compliance with the FDA’s current good manufacturing practice standards, or cGMP, to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| Ø | potential FDA audit of the nonclinical and clinical trial sites that generated the data in support of the NDA; and |
| Ø | FDA review and approval of the NDA. |
The lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources and approvals are inherently uncertain.
Before testing any compounds with potential therapeutic value in humans, the drug candidate enters the preclinical testing stage. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the drug candidate. The conduct of the preclinical tests must comply with federal regulations and requirements including GLP. The sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a drug candidate at any time before or during clinical trials due to safety concerns or non-compliance.
56
Table of Contents
Business
Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such trial.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and FDA to reach agreement on the next phase of development. Sponsors typically use the end of Phase 2 meeting to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support approval of the new drug. If this type of discussion occurred, a sponsor may be able to request a Special Protocol Assessment, or SPA, the purpose of which is to reach agreement with the FDA on the design of the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
According to a FDA guidance for industry on the SPA process, a sponsor which meets the prerequisites may make a specific request for a special protocol assessment and provide information regarding the design and size of the proposed clinical trial. The FDA is supposed to evaluate the protocol within 45 days of the request to assess whether the proposed trial is adequate, and that evaluation may result in discussions and a request for additional information. A SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the testing began.
Clinical trials involve the administration of the drug candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety. Each protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted in accordance with the FDA’s regulations comprising the good clinical practices requirements. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| Ø | Phase 1. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| Ø | Phase 2. The drug is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| Ø | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. Generally, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA. |
57
Table of Contents
Business
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication.
Annual progress reports detailing the results of the clinical trials must be submitted to the FDA and written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk for human subjects. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
U.S. review and approval processes
The results of product development, nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees; a waiver of such fees may be obtained under certain limited circumstances.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any drug for an indication for which orphan designation has been granted.
The FDA reviews all NDAs submitted to determine if they are substantially complete before it accepts them for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has 10 months in which to complete its initial review of a standard NDA and respond to the applicant, and six months for a priority NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the NDA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug or biological products or drug or biological products which present difficult questions of safety or efficacy to an advisory committee,
58
Table of Contents
Business
typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. During the drug approval process, the FDA also will determine whether a risk evaluation and mitigation strategy, or REMS, is necessary to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without a REMS, if required.
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND study requirements. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable it will outline the deficiencies in the submission and often will request additional testing or information.
The NDA review and approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA. The complete response letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials testing, which involves clinical trials designed to further assess a drug safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Orphan drug designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug or biological product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan product exclusivity, which
59
Table of Contents
Business
means that the FDA may not approve any other applications to market the same drug or biological product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors, however, may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity. Orphan product exclusivity also could block the approval of one of our products for seven years if a competitor obtains approval of the same drug or biological product as defined by the FDA or if our drug candidate is determined to be contained within the competitor’s product for the same indication or disease. If a drug or biological product designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan product exclusivity. Orphan drug status in the European Union has similar but not identical benefits in the European Union.
Expedited development and review programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. Unique to a Fast Track product, the FDA may consider for review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
Any product submitted to the FDA for marketing, including a Fast Track program, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Post-approval requirements
Any drug products for which we or our strategic alliance partners receive FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements, which
60
Table of Contents
Business
include, among others, standards for direct-to-consumer advertising, promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities, and promotional activities involving the internet. Failure to comply with FDA requirements can have negative consequences, including adverse publicity, enforcement letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses.
We will rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of any products that we may commercialize. Our strategic alliance partners may also utilize third parties for some or all of a product we are developing with such strategic alliance partner. Manufacturers of our products are required to comply with applicable FDA manufacturing requirements contained in the FDA’s cGMP regulations. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including withdrawal of the product from the market. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
U.S. patent term restoration and marketing exclusivity
Depending upon the timing, duration and specifics of the FDA approval of the use of our drug candidates, some of our United States patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The United States Patent and Trademark Office, or U.S. PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may intend to apply for restoration of patent term for one of our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA. We cannot be certain that the U.S. PTO and FDA would grant any patent term extension.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications of other companies seeking to reference another company’s NDA. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has
61
Table of Contents
Business
not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness. Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
U.S. Foreign Corrupt Practices Act
The U.S. Foreign Corrupt Practices Act, or FCPA, prohibits certain individuals and entities, including us, from promising, paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, directly or indirectly, to obtain or retain business or an improper advantage. The U.S. Department of Justice and the U.S. Securities and Exchange Commission, or SEC, have increased their enforcement efforts with respect to the FCPA. Violations of the FCPA may result in large civil and criminal penalties and could result in an adverse effect on a company’s reputation, operations, and financial condition. A company may also face collateral consequences such as debarment and the loss of export privileges.
Federal and state fraud and abuse laws
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain business practices in the biopharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for purchasing, leasing, ordering, or arranging for the purchase, lease, or order of any healthcare item or service reimbursable under Medicare, Medicaid, or other federally financed healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value, including for example, gifts, discounts, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of payment, ownership interests and providing anything at less than its fair market value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and our practices may not in all cases meet all of the criteria for statutory exemptions or safe harbor protection. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases, or recommendations may be subject to scrutiny if they
62
Table of Contents
Business
do not qualify for an exemption or safe harbor. . Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The reach of the Anti-Kickback Statute was also broadened by the Patient Protection and Affordable Health Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the PPACA, which, among other things, amends the intent requirement of the federal Anti-Kickback Statute. Pursuant to the statutory amendment, a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. In addition, the PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act (discussed below) or the civil monetary penalties statute, which imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
The federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, and thus non-reimbursable, uses. Many states also have statutes or regulations similar to the federal Anti-Kickback Statute and False Claims Act, which state laws apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. Also, the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created new federal criminal statutes that prohibit knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payers and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
Because of the breadth of these laws and the narrowness of the federal Anti-Kickback Statute’s safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business, financial condition and results of operations. If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations may be directly, or indirectly through our customers, distributors, or other business partners, subject to various federal and state fraud and abuse laws, including, without limitation, anti-kickback statutes and false claims statutes. These laws may impact, among other things, our proposed sales, marketing and education programs.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates”—independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
63
Table of Contents
Business
If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government healthcare programs, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our product candidates are ultimately sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
In the United States and foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the United States federal and state levels that seek to reduce healthcare costs. The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from Medicare Part D may result in a similar reduction in payments from non-governmental payors.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
In March 2010 the PPACA was enacted, which includes measures to significantly change the way healthcare is financed by both governmental and private insurers. Among the provisions of the PPACA of importance to the pharmaceutical and biotechnology industry are the following:
| Ø | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs, that began in 2011; |
64
Table of Contents
Business
| Ø | an increase in the rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13% of the average manufacturer price for branded and generic drugs, respectively; |
| Ø | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts to negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| Ø | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| Ø | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the Federal Poverty Level beginning in 2014, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| Ø | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| Ø | new requirements under the federal Open Payments program, created under Section 6002 of the PPACA and its implementing regulations, that manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) report annually to the U.S. Department of Health and Human Services, or HHS, information related to “payments or other transfers of value” made or distributed to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and that applicable manufacturers and applicable group purchasing organizations report annually to HHS ownership and investment interests held by physicians (as defined above) and their immediate family members, with data collection required beginning August 1, 2013 and reporting to the Centers for Medicare & Medicaid Services, or CMS, required by March 31, 2014 and by the 90th day of each subsequent calendar year; |
| Ø | a new requirement to annually report drug samples that manufacturers and distributors provide to physicians, effective April 1, 2012; |
| Ø | expansion of health care fraud and abuse laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers, and enhanced penalties for noncompliance; |
| Ø | a licensure framework for follow-on biologic products; |
| Ø | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| Ø | creation of the Independent Payment Advisory Board which, beginning in 2014, will have authority to recommend certain changes to the Medicare program that could result in reduced payments for prescription drugs and those recommendations could have the effect of law even if Congress does not act on the recommendations; and |
| Ø | establishment of a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending that began on January 1, 2011. |
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. In August 2011, the president signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction, or joint committee, to recommend proposals in spending reductions to Congress. The joint committee did not achieve its targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering automatic reductions to several
65
Table of Contents
Business
government programs. These reductions include aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. In January 2013, the president signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our financial operations.
Europe / rest of world government regulation
In addition to regulations in the United States, we and our strategic alliance partners will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we or our collaborators obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational drug or biological product under European Union regulatory systems, we or our strategic alliance partners must submit a marketing authorization application. The application used to file the NDA or BLA in the United States is similar to that required in the European Union, with the exception of, among other things, country-specific document requirements. In the European Economic Area, or EEA (which is comprised of the 27 Member States of the European Union plus Norway, Iceland and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations: the Community MA, which is issued by the European Commission through the Centralized Procedure based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, a body of the EMA, and which is valid throughout the entire territory of the EEA; and the National MA, which is issued by the competent authorities of the Member States of the EEA and only authorized marketing in that Member State’s national territory and not the EEA as a whole.
The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products containing a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union. The National MA is for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized
66
Table of Contents
Business
Procedure an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member state, or RMS. If the RMS proposes to authorize the product, and the other Member States do not raise objections, the product is granted a national MA in all the Member States where the authorization was sought. Before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we or our strategic alliance partners fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
EMPLOYEES
As of March 31, 2013, we had 71 full-time employees, 31 of whom have Ph.D. degrees. Of these full-time employees, 57 employees are engaged in research and development activities and 14 employees are engaged in finance, legal, human resources, facilities and general management. We have no collective bargaining agreements with our employees and we have not experienced any work stoppages. We consider our relations with our employees to be good.
FACILITIES
Our corporate headquarters are located in La Jolla, California. The facility we lease encompasses approximately 28,881 square feet of office and laboratory space. The lease for this facility expires in June 2017, subject to our option to renew for up to two additional three-year terms. We believe that our facility is sufficient to meet our needs and that suitable additional space will be available as and when needed.
LEGAL PROCEEDINGS
From time to time, we are subject to various legal proceedings and claims that arise in the ordinary course of our business activities. Although the results of litigation and claims cannot be predicted with certainty, as of the date of this prospectus, we do not believe we are party to any claim or litigation the outcome of which, if determined adversely to us, would individually or in the aggregate be reasonably expected to have a material adverse effect on our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
67
Table of Contents
Certain relationships and related party transactions
The following includes a summary of transactions since January 1, 2010 to which we have been a party, in which the amount involved in the transaction exceeded $120,000, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described under “Executive and director compensation.”
DIRECTOR AFFILIATIONS WITH OUR PRINCIPAL STOCKHOLDERS & STRATEGIC PARTNERS
Some of our directors or former directors, are affiliated with our principal stockholders and strategic partners as indicated in the table below:
| Director | Affiliation | |
| John M. Maraganore, Ph.D. |
Alnylam Pharmaceuticals, Inc. | |
| Stelios Papadopoulos, Ph.D. |
Biogen Idec | |
| B. Lynne Parshall |
Isis Pharmaceuticals, Inc. | |
| Douglas E. Williams, Ph.D. |
Biogen Idec | |
| Barry E. Greene (former director) |
Alnylam Pharmaceuticals, Inc. | |
| Stanley T. Crooke, M.D., Ph.D. (former director) |
Isis Pharmaceuticals, Inc. |
PREFERRED STOCK FINANCINGS
Series B Convertible Preferred Stock Financing. In October 2010, we issued and sold to Aventis Holdings, Inc. an aggregate of 2,499,999 shares of series B convertible preferred stock, at a purchase price of $4.00 per share, for aggregate consideration of $10.0 million. In connection with the October 2010 financing, we entered into an investor rights agreement with Aventis Holdings, Inc., containing information rights, rights of first refusal and registration rights, among other things. This investor rights agreement will terminate in October 2015, except for certain of the registration rights granted thereunder, as more fully described below in “Description of capital stock—Registration Rights.”
STRATEGIC ALLIANCES AND COLLABORATIONS
In April 2008, we entered into a product development and commercialization agreement with GSK, which was amended in February 2010, June 2010, July 2011, June 2012 and June 2013. Upon entering into the agreement, GSK loaned $5.0 million to us under a convertible promissory note, which was amended in January 2011, amended and restated in July 2012 and amended and restated in October 2012. This agreement and the associated convertible promissory note are described under “Business—Our Strategic Alliances.”
In January 2009, we entered into an amended and restated license and collaboration agreement with Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., which the parties amended in June 2010, October 2011. This agreement is described under “Business—Our Strategic Alliances.”
In February 2010, we entered into an exclusive license and nonexclusive option agreement with GSK. Upon entering into the agreement, GSK loaned $5.0 million to us under a convertible promissory note, which was amended and restated in July 2012. This agreement and the associated convertible promissory note are described under “Business—Our Strategic Alliances.”
In June 2010, we entered into a collaboration and license agreement and a non-exclusive technology alliance and option agreement with Sanofi, an affiliate of Aventis Holdings, Inc. In July 2012, we
68
Table of Contents
Certain relationships and related party transactions
amended and restated the 2010 license and collaboration agreement and further amended it by letter agreement in April 2013. In June 2013, we and Sanofi entered into an option agreement pursuant to which we granted Sanofi an exclusive right to negotiate with us to enter into a development and license agreement for the co-development and commercialization of certain of our unencumbered microRNA programs through December 2013, for which Sanofi has agreed to pay us an upfront option fee of $2.5 million, $1.25 million of which is creditable against future payments payable by Sanofi to us. In addition, Sanofi granted us an exclusive option, which also expires December 2013, to negotiate the co-development and commercialization of miR-21. These agreements are described under “Business—Our Strategic Alliances.”
In August 2012, we entered into a collaboration and license agreement with AstraZeneca, which the parties amended in April 2013. Upon entering into the agreement, AstraZeneca entered into a common stock purchase agreement pursuant to which we agreed to sell to AstraZeneca an aggregate of $25.0 million of our common stock in a separate private placement concurrent with the completion of this offering.These agreements are described under “Business—Our Strategic Alliances.”
In August 2012, we entered into a collaboration and license agreement with Biogen Idec, which the parties amended in June 2013. Upon entering into the agreement, Biogen Idec loaned $5.0 million to us under a convertible promissory note. These agreements are described under “Business—Our Strategic Alliances.”
In December 2012, we entered into a material transfer agreement with Alnylam Pharmaceuticals, Inc. for the purchase of GalNAc conjugate, a component of our drug product. We paid to Alnylam $150,000 for time and materials. In March 2013, we agreed to purchase additional GalNAc conjugate for our GLP studies and for Alnylam’s time to support the transfer of the GalNAc technologies to us and to our contract manufacturers. We expect to pay approximately $500,000 during 2013 for these activities consisting of pass through costs for the manufacture of the GalNAc conjugate and the time of Alnylam’s employees required in the manufacture of the conjugate and the technology transfer.
EMPLOYMENT ARRANGEMENTS
We have entered into written employment agreements with our executive officers, including our President and Chief Executive Officer, Kleanthis G. Xanthopoulos, Ph.D., our former Chief Operating Officer and Executive Vice President, Finance, Garry E. Menzel, Ph.D., and our Chief Scientific Officer, Neil W. Gibson, Ph.D. These agreements are described under “Executive and director compensation” in our Proxy Statement on Schedule 14A incorporated by reference herein.
STOCK OPTIONS GRANTED TO EXECUTIVE OFFICERS AND DIRECTORS
We have granted stock options to our executive officers and directors, as more fully described under “Executive and director compensation” in our Proxy Statement on Schedule 14A incorporated by reference herein.
INDEMNIFICATION AGREEMENTS
We have entered into separate indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated bylaws. These agreements, among other things, require us to indemnify our directors and executive officers for certain expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or executive officer in any action or proceeding arising out of his or her services as one of our directors or executive officers or any other company or enterprise to which the person provides services at our request. We believe that these indemnification agreements, together with the provisions in our bylaws, are necessary to attract and retain qualified persons as directors and officers.
69
Table of Contents
Certain relationships and related party transactions
POLICIES AND PROCEDURES FOR TRANSACTIONS WITH RELATED PERSONS
We have adopted a written related-person transactions policy that sets forth our policies and procedures regarding the identification, review, consideration, approval and oversight of “related-person transactions.” For purposes of our policy only, a “related-person transaction” is a past, present or future transaction, arrangement or relationship (or any series of similar transactions, arrangements or relationships) in which we and any “related person” are participants involving an amount that exceeds $120,000.
Transactions involving compensation for services provided to us by an employee, consultant or director will not be considered related-person transactions under this policy. A “related person,” as determined since the beginning of our last fiscal year, is any executive officer, director or a holder of more than five percent of our common stock, including any of their immediate family members and any entity owned or controlled by such persons.
The policy imposes an affirmative duty upon each director and executive officer to identify, and we will request that significant stockholders identify, any transaction involving them, their affiliates or immediate family members that may be considered a related party transaction before such person engages in the transaction. Under the policy, where a transaction has been identified as a related-person transaction, management must present information regarding the proposed related-person transaction to our audit committee (or, where review by our audit committee would be inappropriate, to another independent body of our board of directors) for review. The presentation must include a description of, among other things, the material facts, the direct and indirect interests of the related persons, the benefits of the transaction to us and whether any alternative transactions are available. In considering related-person transactions, our audit committee or other independent body of our board of directors takes into account the relevant available facts and circumstances including, but not limited to:
| Ø | the risks, costs and benefits to us of the transaction; |
| Ø | the impact on a director’s independence in the event the related person is a director, immediate family member of a director or an entity with which a director is affiliated; |
| Ø | the terms of the transaction; |
| Ø | the availability of other sources for comparable services or products; and |
| Ø | the terms available to or from, as the case may be, unrelated third parties or to or from our employees generally. |
In the event a director has an interest in the proposed transaction, the director must recuse himself or herself from the deliberations and approval. Our policy requires that, in reviewing a related party transaction, our audit committee must consider, in light of known circumstances, and determine in the good faith exercise of its discretion whether the transaction is in, or is not inconsistent with, the best interests of us and our stockholders.
COMPENSATION COMMITTEE INTERLOCKS AND INSIDER PARTICIPATION
None of our current or former executive officers serves as a member of the compensation committee. None of our officers serves, or has served during the last completed fiscal year on the board of directors or compensation committee, or other committee serving an equivalent function, of any other entity that has one or more of its executive officers serving as a member of our board of directors or our compensation committee. Prior to establishing the compensation committee, our full board of directors made decisions relating to compensation of our officers.
70
Table of Contents
The following table sets forth information regarding beneficial ownership of our capital stock as of May 31, 2013 by:
| Ø | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our common stock; |
| Ø | each of our directors; |
| Ø | each of our named executive officers; and |
| Ø | all of our directors and current executive officers as a group. |
The numbers of shares and percentage ownership information before the offering is based on 36,052,587 shares of common stock outstanding as of May 31, 2013. The numbers of shares and percentage ownership information after the offering is based on the sale of shares of common stock in this offering.
Information with respect to beneficial ownership has been furnished by each director, officer or beneficial owner of more than 5% of our common stock. We have determined beneficial ownership in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options that are either immediately exercisable or exercisable on or before July 30, 2013 which is 60 days after May 31, 2013. These shares are deemed to be outstanding and beneficially owned by the person holding those options for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
71
Table of Contents
Principal stockholders
Except as otherwise noted below, the address for each person or entity listed in the table is c/o Regulus Therapeutics Inc., 3545 John Hopkins Court, Suite 210, San Diego, California 92121.
| Name and address of beneficial owner |
Number of shares beneficially owned before offering(1) |
Number of shares beneficially owned after offering |
Percentage of shares beneficially owned before offering |
Percentage of shares beneficially owned after offering | ||||||||||
| 5% or greater stockholders |
||||||||||||||
| Isis Pharmaceuticals, Inc.(2). |
7,074,500 | 7,074,500 | 19.6 | % | ||||||||||
| 2855 Gazelle Court |
||||||||||||||
| Carlsbad, CA 92010 |
||||||||||||||
| AstraZeneca AB |
6,250,000 | 6,250,000 | 17.3 | % | ||||||||||
| SE-431 83 Molndal |
||||||||||||||
| Sweden |
||||||||||||||
| Alnylam Pharmaceuticals, Inc. |
6,150,500 | 6,150,500 | 17.1 | % | ||||||||||
| 300 Third Street, 3rd Floor |
||||||||||||||
| Cambridge, MA 02142 |
||||||||||||||
| Aventis Holdings, Inc. |
3,749,999 | 3,749,999 | 10.4 | % | ||||||||||
| c/o Sanofi |
||||||||||||||
| 54, rue La Boétie |
||||||||||||||
| 75414 Paris – France |
||||||||||||||
| Glaxo Group Limited(3) |
3,340,182 | 3,340,182 | 8.9 | % | ||||||||||
| 980 Great West Road |
||||||||||||||
| Brentford, Middlesex TW8 9GS |
||||||||||||||
| United Kingdom |
||||||||||||||
| Biotechnology Value Fund, L.P. and its affiliates(4) |
2,718,200 | 2,718,200 | 7.5 | % | ||||||||||
| 900 North Michigan Avenue, |
||||||||||||||
| Suite 1100 Chicago, Illinois 60611 |
||||||||||||||
| Directors and named executive officers |
||||||||||||||
| David Baltimore, Ph.D.(5) |
197,393 | 197,393 | * | |||||||||||
| Bruce L.A. Carter, Ph.D. |
— | — | * | |||||||||||
| Mark G. Foletta |
— | — | * | |||||||||||
| John M. Maraganore, Ph.D.(6) |
6,150,500 | 6,150,500 | 17.1 | % | ||||||||||
| Stelios Papadopoulos, Ph.D.(7) |
183,176 | 183,176 | * | |||||||||||
| B. Lynne Parshall(2)(8) |
7,074,500 | 7,074,500 | 19.6 | % | ||||||||||
| William H. Rastetter, Ph.D. |
— | — | * | |||||||||||
| Douglas Williams, Ph.D. |
— | — | * | |||||||||||
| Kleanthis G. Xanthopoulos, Ph.D.(9) |
964,671 | 964,671 | 2.6 | |||||||||||
| Garry E. Menzel, Ph.D.(10) |
466,508 | 466,508 | 1.3 | % | ||||||||||
| Neil W. Gibson, Ph.D.(11) |
121,192 | 121,192 | * | |||||||||||
| All current executive officers and directors as a group (11 persons)(12) |
15,157,940 | 15,157,940 | 40.1 | % | ||||||||||
| * | Represents beneficial ownership of less than one percent. |
| (1) | This table is based upon information supplied by officers, directors and principal stockholders and Schedules 13D and 13G filed with the SEC. Unless otherwise indicated in the footnotes to this table and subject to community property laws where applicable, the Company believes that each of the stockholders named in this table has sole voting and investment power with respect to the shares indicated as beneficially owned. |
| (2) | Includes 25,000 shares that Stanley T. Crooke, M.D., Ph.D, one of our former directors, has the right to acquire from us within 60 days of May 31, 2013 pursuant to the exercise of stock options. |
72
Table of Contents
Principal stockholders
| (3) | Includes 1,393,145 shares of our common stock issuable upon the conversion of $5,572,578 of outstanding principal plus accrued interest underlying an amended and restated convertible note at a price of $4.00 per share, based on interest accrued through July 30, 2013. |
| (4) | Based solely upon a schedule 13G filed with the SEC on February 14, 2013 by Biotechnology Value Fund, L.P. (“BVF”) reporting beneficial ownership as of December 31, 2012. According to the schedule 13G BVF has shared voting and dispositive power over 1,725,153 shares of our common stock, Biotechnology Value Fund II, L.P. (“BVF II”) has shared voting and dispositive power over 993,047 shares of our common stock, BVF Partners L.P. (“Partners”) has shared voting and dispositive power over 2,718,200 shares of our common stock, BVF Inc. has shared voting and dispositive power over 2,718,200 shares of our common stock, and Mark N. Lampert has shared voting and dispositive power over 2,718,200 shares of our common stock. Partners is the general partner of BVF and BVF II. BVF, Inc. is the general partner of Partners and Mr. Lampert is a director and officer of BVF Inc. Each of Partners, BVF Inc. and Mr. Lampert disclaims beneficial ownership of the shares beneficially owned by BVF and BVF II. |
| (5) | Includes 197,393 shares that Dr. Baltimore has the right to acquire from us within 60 days of May 31 2013 pursuant to the exercise of stock options. |
| (6) | Represents 6,150,500 shares held by Alnylam. Dr. Maraganore is an officer and director of Alnylam and therefore may be deemed to have voting or investment power over the shares held by Alnylam. Dr. Maraganore disclaims beneficial ownership over the shares held by Alnylam, except to the extent of his proportionate pecuniary interests therein as a stockholder of Alnylam. |
| (7) | Includes 183,176 shares that Dr. Papadopoulos has the right to acquire from us within 60 days of May 31, 2013 pursuant to the exercise of stock options. |
| (8) | Represents 7,074,500 shares beneficially owned by Isis Pharmaceuticals, Inc. Ms. Parshall is an officer and director of Isis and therefore may be deemed to have voting or investment power over the shares beneficially owned by Isis. Ms. Parshall disclaims beneficial ownership over the shares beneficially owned by Isis, except to the extent of her proportionate pecuniary interests therein as a stockholder of Isis. |
| (9) | Includes 82,216 shares held by The Xanthopoulos Family Trust dated September 30, 2011, of which entity Dr. Xanthopoulos is the sole trustee. Also includes 881,844 shares that Dr. Xanthopoulos has the right to acquire from us within 60 days of May 31, 2013 pursuant to the exercise of stock options. Effective as of June 21, 2013, Dr. Xanthopoulos was appointed as our principal financial and accounting officer, in addition to his other roles and responsibilities. |
| (10) | Includes 111,842 shares held by the Dr. Menzel and Mary E. Henshall Family Trust dated July 29, 2010, of which trust Dr. Menzel is a trustee. Also includes 354,666 shares that Dr. Menzel has the right to acquire from us within 60 days of May 31, 2013 pursuant to the exercise of stock options which were vested on the date of his resignation. Dr. Menzel resigned from all positions with the Company on June 21, 2013. |
| (11) | Includes 71,192 shares that Dr. Gibson has the right to acquire from us within 60 days of May 31, 2013 pursuant to the exercise of stock options. |
| (12) | Includes the shares described in notes (4) through (10). |
73
Table of Contents
Our amended and restated certificate of incorporation authorizes us to issue up to 200,000,000 shares of common stock, $0.001 par value, and 10,000,000 shares of preferred stock, $0.001 par value.
As of March 31, 2013, there were outstanding:
| Ø | 35,965,371 shares of common stock outstanding; and |
| Ø | 4,742,780 shares of common stock subject to outstanding options. |
As of March 31, 2013, we had 32 holders of record of our common stock. The actual number of stockholders is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
The following description of our capital stock is not complete and is subject to and qualified in its entirety by our amended and restated certificate of incorporation and amended and restated bylaws and by the provisions of applicable Delaware law.
COMMON STOCK
Voting
Our common stock is entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, including the election of directors, and does not have cumulative voting rights. Accordingly, the holders of a majority of the shares of our common stock entitled to vote in any election of directors can elect all of the directors standing for election.
Dividends
Subject to preferences that may be applicable to any then outstanding preferred stock, the holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds.
Liquidation
In the event of our liquidation, dissolution or winding up, holders of our common stock will be entitled to share ratably in the net assets legally available for distribution to stockholders after the payment of all of our debts and other liabilities, subject to the satisfaction of any liquidation preference granted to the holders of any outstanding shares of preferred stock.
Rights and Preferences
Holders of our common stock have no preemptive, conversion or subscription rights, and there are no redemption or sinking fund provisions applicable to our common stock. The rights, preferences and privileges of the holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of our preferred stock that we may designate and issue in the future.
Fully Paid and Nonassessable
All of our outstanding shares of common stock are, and the shares of common stock to be issued in this offering will be, fully paid and nonassessable.
74
Table of Contents
Description of capital stock
PREFERRED STOCK
Our board of directors has the authority, without further action by the stockholders, to issue up to 10,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each such series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any qualifications, limitations or restrictions thereon and to increase or decrease the number of shares of any such series, but not below the number of shares of such series then outstanding.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could, among other things, have the effect of delaying, deferring or preventing a change in our control that may otherwise benefit holders of our common stock and may adversely affect the market price of the common stock and the voting and other rights of the holders of common stock. As of March 31, 2013, there are no shares of preferred stock outstanding and we have no current plans to issue any shares of preferred stock.
REGISTRATION RIGHTS
Under our investor rights agreements entered into in connection with the issuances of our convertible preferred stock and the private placement that occurred concurrently with the closing of our initial public offering, the holders of 13,235,000 shares of common stock, or their transferees, have the right to require us to register their shares with the SEC so that those shares may be publicly resold, or to include their shares in any registration statement we file.
Form S-3 Registration Rights
If we are eligible to file a registration statement on Form S-3, one or more holders of registration rights have the right to demand that we file a registration statement on Form S-3 so long as the aggregate value of the securities to be sold under the registration statement on Form S-3 is at least $15.0 million subject to specified exceptions.
“Piggyback” Registration Rights
If we register any securities for public sale, holders of registration rights will have the right to include their shares in the registration statement. The underwriters of any underwritten offering will have the right to limit the number of shares having registration rights to be included in the registration statement, subject to specified limitations. We have obtained waivers of any and all rights to have shares included in this offering from all holders of such registration rights.
Expenses of Registration
Generally, we are required to bear all registration and selling expenses incurred in connection with the piggyback and Form S-3 registrations described above, other than underwriting discounts and commissions.
Expiration of Registration Rights
The piggyback and Form S-3 registration rights discussed above will terminate in October 2015, three years following the closing of our initial public offering or, as to a given holder of registrable securities, when such holder is able to sell, following the initial offering, all of their registrable securities in a single 90-day period under Rule 144 of the Securities Act.
75
Table of Contents
Description of capital stock
ANTI-TAKEOVER EFFECTS OF PROVISIONS OF OUR AMENDED AND RESTATED CERTIFICATE OF INCORPORATION, OUR BYLAWS AND DELAWARE LAW
Delaware Anti-Takeover Law
We are subject to Section 203 of the Delaware General Corporation Law, or Section 203. Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
| Ø | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; |
| Ø | the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| Ø | on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Section 203 defines a business combination to include:
| Ø | any merger or consolidation involving the corporation and the interested stockholder; |
| Ø | any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation; |
| Ø | subject to exceptions, any transaction involving the corporation that has the effect of increasing the proportionate share of the stock of any class or series of the corporation beneficially owned by the interested stockholder; |
| Ø | subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; and |
| Ø | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
76
Table of Contents
Description of capital stock
Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
Provisions of our amended and restated certificate of incorporation and amended and restated bylaws, may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our amended and restated certificate of incorporation and amended and restated bylaws:
| Ø | permit our board of directors to issue up to 10,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change in our control); |
| Ø | provide that the authorized number of directors may be changed only by resolution of the board of directors; |
| Ø | provide that all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of directors then in office, even if less than a quorum; |
| Ø | require that any action to be taken by our stockholders must be effected at a duly called annual or special meeting of stockholders and not be taken by written consent; |
| Ø | provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide notice in writing in a timely manner and also specify requirements as to the form and content of a stockholder’s notice; |
| Ø | do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); and |
| Ø | provide that special meetings of our stockholders may be called only by the Chairman of the Board, our Chief Executive Officer or by the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors. |
The amendment of any of these provisions, with the exception of the ability of our board of directors to issue shares of preferred stock and designate any rights, preferences and privileges thereto, would require approval by the holders of at least 66 2/3% of our then outstanding common stock.
NASDAQ GLOBAL MARKET LISTING
Our common stock is listed on The NASDAQ Global Market under the “RGLS” symbol.
TRANSFER AGENT AND REGISTRAR
The transfer agent and registrar for our common stock is Computershare Trust Company, N.A. The transfer agent and registrar’s address is 250 Royall Street, Canton, MA, 02021.
77
Table of Contents
Shares eligible for future sale
Prior to our initial public offering in October 2012, there was no public market for our common stock, and a liquid trading market for our common stock may not develop or be sustained after this offering. Future sales of substantial amounts of our common stock in the public market, including shares issued upon exercise of outstanding options and warrants, or the anticipation of these sales, could adversely affect prevailing market prices from time to time and could impair our ability to raise equity capital in the future.
Upon completion of this offering, we will have shares of common stock outstanding, assuming (1) no exercise of any options outstanding as of March 31, 2013 and (2) no exercise of the underwriters’ option to purchase additional shares from us. Of those shares, all of the shares sold in this offering and all 12,730,982 shares sold in our initial public offering will be freely tradable, except that any shares held by our “affiliates,” as that term is defined in Rule 144 under the Securities Act, or Rule 144, may only be sold in compliance with the limitations described below.
RULE 144
In general, under Rule 144, any person who is not our affiliate and has held their shares for at least six months, including the holding period of any prior owner other than one of our affiliates, may sell shares without restriction, subject to the availability of current public information about us. In addition, under Rule 144, any person who is not an affiliate of ours and has held their shares for at least one year, including the holding period of any prior owner other than one of our affiliates, would be entitled to sell an unlimited number of shares immediately upon the closing of this offering without regard to whether current public information about us is available. A person who is our affiliate or who was our affiliate at any time during the preceding three months and who has beneficially owned restricted securities for at least six months, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of shares within any three-month period that does not exceed the greater of:
| Ø | 1% of the number of shares of our common stock then outstanding, which will equal approximately shares immediately after this offering; and |
| Ø | the average weekly trading volume of our common stock on The NASDAQ Global Market during the four calendar weeks preceding the filing of a Notice of Proposed Sale of Securities Pursuant to Rule 144 with respect to the sale. |
Sales under Rule 144 by our affiliates are also subject to manner of sale provisions and notice requirements and to the availability of current public information about us.
Subject to the lock-up agreements described herein and under “Underwriting” included elsewhere in this prospectus, 23,321,605 shares of our common stock qualify for resale under Rule 144.
RULE 701
In general, under Rule 701 of the Securities Act, any of our stockholders who purchased shares from us in connection with a qualified compensatory stock plan or other written agreement before we became subject to the reporting requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, or the Exchange Act, is eligible to resell those shares in reliance on Rule 144. An affiliate of the issuer can resell shares in reliance on Rule 144 without having to comply with the holding period requirements of Rule 144, and a non-affiliate of the issuer can resell shares in reliance on Rule 144 without having to comply with the holding period requirements of Rule 144 and without regard to the volume of such sales or the availability of public information about the issuer.
78
Table of Contents
Shares eligible for future sale
As of March 31, 2013, options to purchase a total of 4,742,780 shares of common stock were outstanding, of which 2,396,165 were vested. Of the total number of shares of our common stock issuable under these options, substantially all are subject to contractual lock-up agreements with us or the underwriters from our initial public offering which lock-up agreements expire on October 4, 2013, and will become eligible for sale at the expiration of those agreements unless held by an affiliate of ours.
LOCK-UP AGREEMENTS
We, along with our directors and executive management team have agreed that for a period of 90 days after the date of this prospectus, subject to specified exceptions, we or they will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock. In addition, 26,515,110 shares of our common stock are subject to lock-up agreements that were entered into in connection with our initial public offering, which lock-up agreements expire on October 4, 2013. Upon expiration of the respective “lock-up” periods, certain of our stockholders will have the right to require us to register their shares under the Securities Act. See “Registration Rights” below.
REGISTRATION RIGHTS
The holders of 13,235,000 shares of our common stock are entitled to rights with respect to the registration of their shares under the Securities Act, subject to the lock-up arrangement described above. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates, immediately upon the effectiveness of the registration statement of which this prospectus is a part. Any sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock. See “Description of capital stock—Registration Rights.”
EQUITY INCENTIVE PLANS
Shares of our common stock issued under the 2012 Plan and the ESPP are available for sale in the open market, subject to Rule 144 volume limitations and the lock-up agreements described above, if applicable.
79
Table of Contents
Material U.S. federal income tax consequences to non-U.S. holders of our common stock
The following summary describes the material U.S. federal income and estate tax consequences of the acquisition, ownership and disposition of our common stock acquired in this offering by Non-U.S. Holders (as defined below). This discussion does not address all aspects of U.S. federal income and estate taxes that may be relevant to Non-U.S. Holders in light of their particular circumstances, does not deal with foreign, state and local tax consequences and does not address U.S. federal tax consequences other than income and estate taxes. Special rules different from those described below may apply to certain Non-U.S. Holders that are subject to special treatment under the Code, such as financial institutions, insurance companies, tax-exempt organizations, broker-dealers and traders in securities, U.S. expatriates, “controlled foreign corporations,” “passive foreign investment companies,” corporations that accumulate earnings to avoid U.S. federal income tax, persons that hold our common stock as part of a “straddle,” “hedge,” “conversion transaction,” “synthetic security” or integrated investment or other risk reduction strategy, partnerships and other pass-through entities, and investors in such pass-through entities or an entity that is treated as a disregarded entity for U.S. federal income tax purposes (regardless of its place of organization or formation). Such Non-U.S. Holders are urged to consult their own tax advisors to determine the U.S. federal, state, local and other tax consequences that may be relevant to them. Furthermore, the discussion below is based upon the provisions of the Code, and Treasury regulations, rulings and judicial decisions thereunder as of the date hereof, and such authorities may be repealed, revoked or modified, perhaps retroactively, so as to result in U.S. federal income and estate tax consequences different from those discussed below. We have not requested a ruling from the U.S. Internal Revenue Service, or IRS, with respect to the statements made and the conclusions reached in the following summary, and there can be no assurance that the IRS will agree with such statements and conclusions. This discussion assumes that the Non-U.S. Holder holds our common stock as a “capital asset” within the meaning of Section 1221 of the Code (generally, property held for investment).
The following discussion is for general information only and is not tax advice. Persons considering the purchase of our common stock pursuant to this offering should consult their own tax advisors concerning the U.S. federal income and estate tax consequences of acquiring, owning and disposing of our common stock in light of their particular situations as well as any consequences arising under the laws of any other taxing jurisdiction, including any state, local or foreign tax consequences.
For the purposes of this discussion, a “Non-U.S. Holder” is, for U.S. federal income tax purposes, a beneficial owner of common stock that has not been excluded from this discussion and is not a U.S. Holder. A “U.S. Holder” means a beneficial owner of our common stock that is for U.S. federal income tax purposes (a) an individual who is a citizen or resident of the United States, (b) a corporation or other entity treated as a corporation created or organized in or under the laws of the United States, any state thereof or the District of Columbia, (c) an estate the income of which is subject to U.S. federal income taxation regardless of its source or (d) a trust if it (1) is subject to the primary supervision of a court within the United States and one or more U.S. persons have the authority to control all substantial decisions of the trust or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person.
DISTRIBUTIONS
Subject to the discussion below, distributions, if any, made on our common stock to a Non-U.S. Holder of our common stock to the extent made out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles) generally will constitute dividends for U.S. tax purposes and will be subject to withholding tax at a 30% rate or such lower rate as may be specified by
80
Table of Contents
Material U.S. federal income tax consequences to non-U.S. holders of our common stock
an applicable income tax treaty. To obtain a reduced rate of withholding under a treaty, a Non-U.S. Holder generally will be required to provide us with a properly executed IRS Form W-8BEN, or other appropriate form, certifying the Non-U.S. Holder’s entitlement to benefits under that treaty. In the case of a Non-U.S. Holder that is an entity, Treasury Regulations and the relevant tax treaty provide rules to determine whether, for purposes of determining the applicability of a tax treaty, dividends will be treated as paid to the entity or to those holding an interest in that entity. If a Non-U.S. Holder holds stock through a financial institution or other agent acting on the holder’s behalf, the holder will be required to provide appropriate documentation to such agent. The holder’s agent will then be required to provide certification to us or our paying agent, either directly or through other intermediaries. If you are eligible for a reduced rate of U.S. federal withholding tax under an income tax treaty, you should consult with your own tax advisor to determine if you are able to obtain a refund or credit of any excess amounts withheld by timely filing an appropriate claim for a refund with the IRS.
We generally are not required to withhold tax on dividends paid to a Non-U.S. Holder that are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States (and, if required by an applicable income tax treaty, are attributable to a permanent establishment that such holder maintains in the United States) if a properly executed IRS Form W-8ECI, stating that the dividends are so connected, is furnished to us (or, if stock is held through a financial institution or other agent, to such agent). In general, such effectively connected dividends will be subject to U.S. federal income tax, on a net income basis at the regular graduated rates, unless a specific treaty exemption applies. A corporate Non- U.S. Holder receiving effectively connected dividends may also be subject to an additional “branch profits tax,” which is imposed, under certain circumstances, at a rate of 30% (or such lower rate as may be specified by an applicable treaty) on the corporate Non-U.S. Holder’s effectively connected earnings and profits, subject to certain adjustments.
To the extent distributions on our common stock, if any, exceed our current and accumulated earnings and profits, they will constitute a non-taxable return of capital and will first reduce your adjusted basis in our common stock, but not below zero, and then will be treated as gain and taxed in the same manner as gain realized from a sale or other disposition of common stock as described in the next section.
GAIN ON DISPOSITION OF OUR COMMON STOCK
Subject to the discussion below regarding backup withholding and foreign accounts, a Non-U.S. Holder generally will not be subject to U.S. federal income tax with respect to gain realized on a sale or other disposition of our common stock unless (a) the gain is effectively connected with a trade or business of such holder in the United States (and, if required by an applicable income tax treaty, is attributable to a permanent establishment that such holder maintains in the United States), (b) the Non-U.S. Holder is a nonresident alien individual and is present in the United States for 183 or more days in the taxable year of the disposition and certain other conditions are met, or (c) we are or have been a “United States real property holding corporation” within the meaning of Code Section 897(c)(2) at any time within the shorter of the five-year period preceding such disposition or such holder’s holding period. In general, we would be a United States real property holding corporation if interests in U.S. real estate comprised (by fair market value) at least half of our business assets. We believe that we are not, and do not anticipate becoming, a United States real property holding corporation. Even if we are treated as a United States real property holding corporation, gain realized by a Non-U.S. Holder on a disposition of our common stock will not be subject to U.S. federal income tax so long as (1) the Non-U.S. Holder owned, directly, indirectly and constructively, no more than 5% of our common stock at all times within the shorter of (i) the five-year period preceding the disposition or (ii) the holder’s holding period and (2) our common stock is regularly traded on an established securities market. There can be no assurance that our common stock will qualify or continue to qualify as regularly traded on an established securities market.
81
Table of Contents
Material U.S. federal income tax consequences to non-U.S. holders of our common stock
If you are a Non-U.S. Holder described in (a) above, you will be required to pay tax on the net gain derived from the sale at regular graduated U.S. federal income tax rates, unless a specific treaty exemption applies, and corporate Non-U.S. Holders described in (a) above may be subject to the additional branch profits tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty. If you are an individual Non-U.S. Holder described in (b) above, you will be required to pay a flat 30% tax on the gain derived from the sale, which gain may be offset by U.S. source capital losses (even though you are not considered a resident of the United States).
INFORMATION REPORTING REQUIREMENTS AND BACKUP WITHHOLDING
Generally, we or certain financial middlemen must report information to the IRS with respect to any dividends we pay on our common stock including the amount of any such dividends, the name and address of the recipient, and the amount, if any, of tax withheld. A similar report is sent to the holder to whom any such dividends are paid. Pursuant to tax treaties or certain other agreements, the IRS may make its reports available to tax authorities in the recipient’s country of residence.
Dividends paid by us (or our paying agents) to a Non-U.S. Holder may also be subject to U.S. backup withholding. U.S. backup withholding generally will not apply to a Non-U.S. Holder who provides a properly executed IRS Form W-8BEN or otherwise establishes an exemption. The current backup withholding rate is 28%.
Under current U.S. federal income tax law, U.S. information reporting and backup withholding requirements generally will apply to the proceeds from a disposition of our common stock effected by or through a U.S. office of any broker, U.S. or foreign, except that information reporting and such requirements may be avoided if the holder provides a properly executed IRS Form W-8BEN or otherwise meets documentary evidence requirements for establishing Non-U.S. Holder status or otherwise establishes an exemption. Generally, U.S. information reporting and backup withholding requirements will not apply to a payment of disposition proceeds to a Non-U.S. Holder where the transaction is effected outside the United States through a non-U.S. office of a non-U.S. broker. Information reporting and backup withholding requirements may, however, apply to a payment of disposition proceeds if the broker has actual knowledge, or reason to know, that the holder is, in fact, a U.S. person. For information reporting purposes, certain brokers with substantial U.S. ownership or operations will generally be treated in a manner similar to U.S. brokers.
If backup withholding is applied to you, you should consult with your own tax advisor to determine if you are able to obtain a tax benefit or credit with respect to such backup withholding.
FOREIGN ACCOUNTS
A U.S. federal withholding tax of 30% may apply to dividends paid after December 31, 2013 and the gross proceeds from a disposition of our common stock paid after December 31, 2016 to a foreign financial institution (as specifically defined for this purpose), including when the foreign financial institution holds our common stock on behalf of a non-U.S. Holder, unless such institution enters into an agreement with the U.S. government to withhold on certain payments and to collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which includes certain equity and debt holders of such institution, as well as certain account holders that are foreign entities with U.S. owners). This U.S. federal withholding tax of 30% will also apply to dividends paid after December 31, 2013 and the gross proceeds from a disposition of our common stock paid after December 31, 2016 to a non-financial foreign entity unless such entity provides the withholding agent with either a certification that it does not have any substantial direct or indirect U.S. owners or provides information regarding direct and indirect U.S. owners of the entity. Under certain circumstances, a Non-
82
Table of Contents
Material U.S. federal income tax consequences to non-U.S. holders of our common stock
U.S. Holder might be eligible for refunds or credits of such taxes. An intergovernmental agreement between the United States and an applicable foreign country may modify the requirements described in this paragraph. Holders are encouraged to consult with their own tax advisors regarding the possible implications of the legislation on their investment in our common stock.
FEDERAL ESTATE TAX
An individual Non-U.S. Holder who is treated as the owner of, or has made certain lifetime transfers of, an interest in our common stock will be required to include the value thereof in his or her gross estate for U.S. federal estate tax purposes, and may be subject to U.S. federal estate tax unless an applicable estate tax treaty provides otherwise, even though such individual was not a citizen or resident of the United States at the time of his or her death.
THE PRECEDING DISCUSSION OF U.S. FEDERAL INCOME AND ESTATE TAX CONSIDERATIONS IS FOR GENERAL INFORMATION ONLY. IT IS NOT TAX ADVICE. EACH PROSPECTIVE INVESTOR SHOULD CONSULT ITS OWN TAX ADVISOR REGARDING THE TAX CONSEQUENCES OF PURCHASING, HOLDING AND DISPOSING OF OUR COMMON STOCK, INCLUDING THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAW.
83
Table of Contents
Subject to the terms and conditions set forth in an underwriting agreement dated the date of this prospectus among us and the underwriters named below, we have agreed to sell to the underwriters, and each underwriter has severally agreed to purchase from us, the number of shares of common stock listed next to its name in the following table. Lazard Capital Markets LLC is acting as book-running manager for the offering and as representative of the underwriters.
| Underwriters | Number of shares | |
| Lazard Capital Markets LLC |
||
| Cowen and Company, LLC |
||
| BMO Capital Markets Corp. |
||
| Needham & Company, LLC |
||
| Wedbush Securities Inc. |
||
|
| ||
| Total |
||
|
|
The underwriters are committed to purchase all the shares of common stock offered by us if they purchase any shares. The underwriting agreement also provides that if an underwriter defaults, the purchase commitments of nondefaulting underwriters may be increased or the offering may be terminated. The underwriters are not obligated to purchase the shares of common stock covered by the underwriters’ over-allotment option described below. The underwriters are offering the shares, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel, and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officer’s certificates and legal opinions. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
DISCOUNTS AND COMMISSIONS
The underwriters propose initially to offer the shares to the public at the public offering price set forth on the cover page of this prospectus and to dealers at that price less a concession not in excess of $ per share. After the initial offering of the shares, the public offering price and other selling terms may be changed by the representative.
The following table shows the public offering price, underwriting discounts and commissions and proceeds before expenses to us. The information assumes either no exercise or full exercise by the underwriters of their over-allotment option.
| Per share | Without option | With option | ||||
| Public offering price |
||||||
| Underwriting discounts and commissions |
||||||
| Proceeds, before expenses, to us |
The total estimated expenses of the offering, including registration, filing and listing fees, printing fees and legal and accounting expenses, but excluding underwriting discounts and commissions, are approximately $ million and are payable by us.
Lazard Frères & Co. LLC referred this transaction to Lazard Capital Markets LLC and will receive a referral fee from Lazard Capital Markets LLC in connection therewith.
84
Table of Contents
Underwriting
OVER-ALLOTMENT OPTION
We have granted the underwriters an option to purchase up to additional shares of common stock at the public offering price, less underwriting discounts and commissions. The underwriters may exercise this option for 30 days from the date of this prospectus. If any shares are purchased pursuant to this over-allotment option, the underwriters will purchase the additional shares in approximately the same proportions as shown in the table above. If any of these additional shares are purchased, the underwriters will offer the additional shares on the same terms as those on which the shares are being offered. We will pay the expenses associated with the exercise of the over-allotment option.
LOCK-UP AGREEMENTS
We, our executive management team and directors have entered into lock-up agreements with the underwriters in connection with this offering. Under these agreements, we, our executive management team and directors have agreed, subject to specified exceptions, not to sell or transfer any common stock or securities convertible into, or exchangeable or exercisable for, common stock, during a period ending 90 days after the date of this prospectus without first obtaining the written consent of Lazard Capital Markets LLC. Specifically, we and these other individuals and entities have agreed not to:
| Ø | offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, any shares of common stock or any securities convertible into or exercisable or exchangeable for common stock or publicly disclose the intention to do any of the foregoing; |
| Ø | enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of our common stock or publicly disclose the intention to do any of the foregoing; or |
| Ø | make any demand for or exercise any right with respect to, the registration of any shares of common stock or any security convertible into or exercisable or exchangeable for common stock. |
The restrictions described above do not apply to:
| Ø | the sale of shares of common stock to the underwriters pursuant to the underwriting agreement; |
| Ø | the issuance by us of shares of common stock upon the exercise of an option or warrant or the conversion of a security outstanding on the date of this prospectus of which the underwriters have been advised in writing or that is described in this prospectus; |
| Ø | transactions by security holders relating to any shares of common stock or other securities acquired in open market transactions after the closing of this offering; |
| Ø | the establishment of a 10b5-1 trading plan under the Exchange Act by a security holder for the sale of shares of common stock, provided that such plan does not provide for the transfer of common stock during the restricted period; |
| Ø | exercises of options or warrants to purchase shares of common stock or other securities; |
| Ø | transfers of shares of common stock or other securities to us in connection with the exercise of any stock options held by the security holder to the extent, but only to the extent, as may be necessary to satisfy tax withholding obligations pursuant to our equity incentive or other plans; |
| Ø | transfers to us in connection with the repurchase of shares of common stock or other securities issued pursuant to equity incentive plans or pursuant to agreements disclosed herein, in each case only in connection with a termination of the security holder’s employment with us; |
| Ø | transfers by security holders of shares of common stock or other securities as a bona fide gift by will or intestate succession, or to a trust for a direct or indirect benefit of the security holder or a member of the immediate family of the security holder; or |
85
Table of Contents
Underwriting
| Ø | transfers by distribution by security holders of shares of common stock or other securities to general or limited partners, members, or stockholders of the security holder or to any investment fund or other entity controlled or managed by the security holder. |
provided that in the case of each of the preceding two types of transactions, the transfer does not involve a disposition for value and each transferee or distributee signs and delivers a lock-up agreement agreeing to be subject to the restrictions on transfer described above.
In addition, 26,665,437 shares of our common stock are subject to lock-up agreements that were entered into in connection with our initial public offering, which lock-up agreements expire on October 4, 2013.
INDEMNIFICATION
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, and to contribute to payments that the underwriters may be required to make for these liabilities.
NASDAQ GLOBAL MARKET LISTING
Our common stock is listed on The NASDAQ Global Market under the “RGLS” symbol.
PRICE STABILIZATION, SHORT POSITIONS AND PENALTY BIDS
In order to facilitate the offering of our common stock, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the price of our common stock. In connection with the offering, the underwriters may purchase and sell our common stock in the open market. These transactions may include short sales, purchases on the open market to cover positions created by short sales and stabilizing transactions. Short sales involve the sale by the underwriters of a greater number of shares of common stock than they are required to purchase in the offering. “Covered” short sales are sales made in an amount not greater than the underwriters’ option to purchase additional shares of common stock in the offering. The underwriters may close out any covered short position by either exercising their over-allotment option or purchasing shares of common stock in the open market. In determining the source of shares of common stock to close out the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the over-allotment option. “Naked” short sales are sales in excess of the over-allotment option. The underwriters must close out any naked short position by purchasing shares of common stock in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of our common stock in the open market after pricing that could adversely affect investors who purchase in the offering. Stabilizing transactions consist of various bids for or purchases of shares of common stock made by the underwriters in the open market prior to the completion of the offering.
Similar to other purchase transactions, the underwriters’ purchases to cover the syndicate short sales may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market.
The underwriters have advised us that, pursuant to Regulation M of the Securities Act, they may also engage in other activities that stabilize, maintain or otherwise affect the price of our common stock, including the imposition of penalty bids. This means that if the representative of the underwriters purchase common stock in the open market in stabilizing transactions or to cover short sales, the representative can require the underwriters that sold those shares as part of this offering to repay the underwriting discount received by them.
86
Table of Contents
Underwriting
The underwriters make no representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common stock. In addition, neither we nor the underwriters make any representation that the underwriters will engage in these transactions or that these transactions, once commenced, will not be discontinued without notice.
ELECTRONIC OFFER, SALE AND DISTRIBUTION OF SHARES
A prospectus in electronic format may be made available on the websites maintained by one or more underwriters or selling group members, if any, participating in the offering. The underwriters may agree to allocate a number of shares of common stock to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the representative to underwriters and selling group members that may make Internet distributions on the same basis as other allocations. Other than the prospectus in electronic format, the information on the underwriters’ websites and any information contained in any other website maintained by the underwriters is not part of this prospectus or the registration statement of which this prospectus forms a part.
NOTICE TO NON-U.S. INVESTORS
In relation to each member state of the European Economic Area that has implemented the Prospectus Directive, each of which we refer to as a relevant member state, with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state, or the relevant implementation date, an offer of securities described in this prospectus may not be made to the public in that relevant member state other than:
| Ø | to legal entities that are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
| Ø | to any legal entity that has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43.0 million and (3) an annual net turnover of more than €50.0 million, as shown in its last annual or consolidated accounts; |
| Ø | to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive) subject to obtaining the prior consent of representative for any such offer; or |
| Ø | in any other circumstances that do not require the publication of a prospectus pursuant to Article 3 of the Prospectus Directive; |
provided that no such offer of securities shall require us or any underwriter to publish a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an “offer of shares to the public” in relation to any shares of common stock in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the shares to be offered so as to enable an investor to decide to purchase or subscribe for the shares, as the same may be varied in that member state by any measure implementing the Prospectus Directive in that member state and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each relevant member state.
OTHER RELATIONSHIPS
From time to time, certain of the underwriters and their affiliates have provided, and may provide in the future, various advisory, investment and commercial banking and other services to us in the ordinary course of business, for which they have received and may continue to receive customary fees and commissions.
87
Table of Contents
The validity of the shares of common stock being offered by this prospectus will be passed upon for us by Cooley LLP, San Diego, California. The underwriters are being represented by Goodwin Procter LLP, Boston, Massachusetts.
Ernst & Young LLP, independent registered public accounting firm, has audited our financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2012, as set forth in their report, which is incorporated by reference in this prospectus and elsewhere in the registration statement. Our financial statements are incorporated by reference in reliance on Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
Where you can find additional information
We have filed with the SEC a registration statement on Form S-1 under the Securities Act, with respect to the shares of common stock being offered by this prospectus. This prospectus does not contain all of the information in the registration statement and its exhibits. For further information with respect to us and the common stock offered by this prospectus, we refer you to the registration statement and its exhibits. Statements contained in this prospectus as to the contents of any contract or any other document referred to are not necessarily complete, and in each instance, we refer you to the copy of the contract or other document filed as an exhibit to the registration statement. Each of these statements is qualified in all respects by this reference.
You can read our SEC filings, including the registration statement, over the Internet at the SEC’s website at www.sec.gov. You may also read and copy any document we file with the SEC at its public reference facilities at 100 F Street NE, Washington, D.C. 20549. You may also obtain copies of these documents at prescribed rates by writing to the Public Reference Section of the SEC at 100 F Street NE, Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference facilities. You may also request a copy of these filings, at no cost, by writing us at 3545 John Hopkins Court, Suite 210, San Diego, California 92121 or telephoning us at (858) 202-6300.
We are subject to the information and periodic reporting requirements of the Exchange Act, and we will file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information are available for inspection and copying at the public reference room and website of the SEC referred to above. We maintain a website at http://www.hyperiontx.com. You may access our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act with the SEC free of charge at our website as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC. The information contained in, or that can be accessed through, our website is not part of this prospectus.
88
Table of Contents
Incorporation of certain information by reference
The SEC allows us to “incorporate by reference” information from other documents that we file with it, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus. Information in this prospectus supersedes information incorporated by reference that we filed with the SEC prior to the date of this prospectus.
We incorporate by reference into this prospectus and the registration statement of which this prospectus is a part the information or documents listed below that we have filed with the SEC (Commission File No. 001-35670):
| Ø | our annual report on Form 10-K for the year ended December 31, 2012, filed with the SEC on February 19, 2013; |
| Ø | the portions of our Definitive Proxy Statement on Schedule 14A filed on April 30, 2013 that are deemed “filed” with the SEC under the Exchange Act; |
| Ø | our quarterly report on Form 10-Q for the quarter ended March 31, 2013, filed with the SEC on May 15, 2013; |
| Ø | the description of our common stock contained in our Registration Statement on Form 8-A filed on September 27, 2012, including any amendment or report filed for the purpose of updating such description; and |
| Ø | our current reports on Form 8-K filed with the SEC on June 10, 2013, June 12, 2013 and June 26, 2013 (other than portions of those documents not deemed to be filed). |
We will furnish without charge to you, on written or oral request, a copy of any or all of the documents incorporated by reference, including exhibits to these documents. You should direct any requests for documents by writing us at 3545 John Hopkins Court, Suite 210, San Diego, California 92121 or telephoning us at (858) 202-6300.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus will be deemed modified, superseded or replaced for purposes of this prospectus to the extent that a statement contained in this prospectus modifies, supersedes or replaces such statement.
89
Table of Contents
Shares

Common Stock
Lazard Capital Markets
Cowen and Company
BMO Capital Markets
Needham & Company
Wedbush PacGrow Life Sciences
, 2013
Table of Contents
Part II
Information not required in prospectus
ITEM 13. OTHER EXPENSES OF ISSUANCE AND DISTRIBUTION.
The following table sets forth all costs and expenses, other than underwriting discounts and commissions, payable by Regulus Therapeutics Inc., or the Registrant, in connection with the sale of the common stock being registered. All amounts shown are estimates except for the Securities and Exchange Commission, or the SEC, registration fee, the FINRA filing fee and The NASDAQ Global Market filing fee.
| Amount to be paid | ||||
| SEC registration fee |
$ | 7,843 | ||
| FINRA filing fee |
9,125 | |||
| Blue sky qualification fees and expenses |
* | |||
| Printing and engraving expenses |
* | |||
| Legal fees and expenses |
* | |||
| Accounting fees and expenses |
* | |||
| Transfer agent and registrar fees and expenses |
* | |||
| Miscellaneous expenses |
* | |||
|
|
|
|||
| Total |
$* | * | ||
|
|
|
|||
| * | To be filed by amendment. |
ITEM 14. INDEMNIFICATION OF DIRECTORS AND OFFICERS.
The Registrant is incorporated under the laws of the State of Delaware. Section 145 of the Delaware General Corporation Law provides that a Delaware corporation may indemnify any persons who were, are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was an officer, director, employee or agent of such corporation, or is or was serving at the request of such corporation as an officer, director, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal. A Delaware corporation may indemnify any persons who were, are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses (including attorneys’ fees) actually and reasonably incurred.
II-1
Table of Contents
Part II
Information not required in prospectus
The Registrant’s amended and restated certificate of incorporation and amended and restated bylaws, provide for the indemnification of its directors and officers to the fullest extent permitted under the Delaware General Corporation Law.
Section 102(b)(7) of the Delaware General Corporation Law permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability for any:
| Ø | transaction from which the director derives an improper personal benefit; |
| Ø | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| Ø | unlawful payment of dividends or redemption of shares; or |
| Ø | breach of a director’s duty of loyalty to the corporation or its stockholders. |
The Registrant’s amended and restated certificate of incorporation includes such a provision. Expenses incurred by any officer or director in defending any such action, suit or proceeding in advance of its final disposition shall be paid by the Registrant upon delivery to it of an undertaking, by or on behalf of such director or officer, to repay all amounts so advanced if it shall ultimately be determined that such director or officer is not entitled to be indemnified by the Registrant.
Section 174 of the Delaware General Corporation Law provides, among other things, that a director who willfully or negligently approves of an unlawful payment of dividends or an unlawful stock purchase or redemption, may be held liable for such actions. A director who was either absent when the unlawful actions were approved or dissented at the time may avoid liability by causing his or her dissent to such actions to be entered in the books containing minutes of the meetings of the board of directors at the time such action occurred or immediately after such absent director receives notice of the unlawful acts.
As permitted by the Delaware General Corporation Law, the Registrant has entered into separate indemnification agreements with each of its directors and executive officers, that require the Registrant to indemnify such persons for certain expenses (including attorneys’, witness or other professional fees) actually and reasonably incurred by such persons in connection with any action, suit or proceeding (including derivative actions), whether actual or threatened, to which any such person may be made a party by reason of the fact that such person is or was a director or officer or is or was acting or serving as an officer, director, employee or agent of the Registrant or any of its affiliated enterprises. Under these agreements, the Registrant is not required to provided indemnification for certain matters, including:
| Ø | indemnification beyond that permitted by the Delaware General Corporation Law; |
| Ø | indemnification for any proceeding with respect to the unlawful payment of remuneration to the director or officer; |
| Ø | indemnification for certain proceedings involving a final judgment that the director or officer is required to disgorge profits from the purchase or sale of the Registrant’s stock |
| Ø | indemnification for proceedings involving a final judgment that the director’s or officer’s conduct was in bad faith, knowingly fraudulent or deliberately dishonest or constituted willful misconduct or a breach of his or her duty of loyalty, but only to the extent of such specific determination; |
II-2
Table of Contents
Part II
Information not required in prospectus
| Ø | indemnification for proceedings or claims brought by an officer or director against us or any of the Registrant’s directors, officers, employees or agents, except for claims to establish a right of indemnification or proceedings or claims approved by the Registrant’s board of directors or required by law; |
| Ø | indemnification for settlements the director or officer enters into without the Registrant’s consent; or |
| Ø | indemnification in violation of any undertaking required by the Securities Act or in any registration statement filed by the Registrant. |
The indemnification agreements also set forth certain procedures that will apply in the event of a claim for indemnification thereunder.
Except as otherwise disclosed under the heading “Business—Legal Proceedings” in this registration statement, there is at present no pending litigation or proceeding involving any of the Registrant’s directors or executive officers as to which indemnification is required or permitted, and the Registrant is not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
The Registrant has an insurance policy in place that covers its officers and directors with respect to certain liabilities, including liabilities arising under the Securities Act of 1933, as amended, or the Securities Act, or otherwise.
The Registrant plans to enter into an underwriting agreement which provides that the underwriters are obligated, under some circumstances, to indemnify the Registrant’s directors, officers and controlling persons against specified liabilities, including liabilities under the Securities Act.
ITEM 15. RECENT SALES OF UNREGISTERED SECURITIES.
The following sets forth information regarding all unregistered securities sold by the Registrant since January 1, 2010:
| (1) | In October 2010, we issued and sold an aggregate of 2,499,999 shares of series B convertible preferred stock to one accredited investor at a purchase price of $4.00 per share, for aggregate gross proceeds of $10.0 million. Upon completion of our initial public offering, these shares converted into 1,249,999 shares of common stock. |
| (2) | In July 2012, we issued two convertible notes in an aggregate principal amount of $5.0 million each with a maturity date of February 25, 2013 to Glaxo Group Limited. These notes amend, restate and supersede notes originally issued in April 2008 and February 2010. The principal amount plus accrued interest under the note originally issued in April 2008 automatically converted into 1,447,037 shares of our common stock in connection with our initial public offering in October 2012. In accordance with the terms of the note originally issued in February 2010, in October 2012 we issued a convertible promissory note in the principal amount of approximately $5.4 million to GSK, the New GSK Note. The New GSK Note is convertible into shares of our common stock at the conversion price of $4.00 per share for until the maturity date of October 9, 2015. |
| (3) | In August 2012, we issued a convertible note in an aggregate principal amount of $5.0 million with a maturity date of February 15, 2013 to Biogen Idec. The principal plus accrued interest under the note converted into 1,256,232 shares of our common stock in connection with our initial public offering in October 2012. |
II-3
Table of Contents
Part II
Information not required in prospectus
| (4) | In October 2012, concurrently with the closing of our initial public offering, we issued and sold 6,250,000 shares of common stock in a private placement to AstraZeneca at the initial public offering price of $4.00 per share. |
| (5) | From January 2010 to September 2012, we granted stock options under our 2009 equity incentive plan to purchase 1,469,719 shares of common stock (net of expirations, exercises and cancellations) to our employees, directors and consultants, having exercise prices ranging from $0.38 to $2.66 per share. |
No underwriters were involved in the foregoing sales of securities. The offers, sales and issuances of the securities described in paragraphs (1), (2), (3) and (4) were deemed to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act and Rule 506 promulgated under Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions was an accredited investor within the meaning of Rule 501 of Regulation D under the Securities Act and had adequate access, through employment, business or other relationships, to information about the Registrant.
The offers, sales and issuances of the securities described in paragraph (5) were deemed to be exempt from registration under the Securities Act in reliance on Rule 701 in that the transactions were under compensatory benefit plans and contracts relating to compensation as provided under Rule 701. The recipients of such securities were the Registrant’s employees, directors or bona fide consultants and received the securities under the 2009 equity incentive plan. Appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions had adequate access, through employment, business or other relationships, to information about the Registrant.
ITEM 16. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES.
(a) Exhibits
See the Exhibit Index on the page immediately preceding the exhibits for a list of exhibits filed as part of this registration statement on Form S-1, which Exhibit Index is incorporated herein by reference.
(b) Financial Statement Schedules
Not applicable.
ITEM 17. UNDERTAKINGS.
The undersigned Registrant hereby undertakes to provide to the underwriters at the closing specified in the Underwriting Agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or
II-4
Table of Contents
Part II
Information not required in prospectus
paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The Registrant hereby undertakes that:
| (a) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (b) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-5
Table of Contents
Signatures
Pursuant to the requirements of the Securities Act, the Registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of San Diego, State of California, on the 26th day of June, 2013.
| REGULUS THERAPEUTICS INC. | ||
| By: |
/s/ Kleanthis G. Xanthopoulos, Ph.D. | |
| Kleanthis G. Xanthopoulos, Ph.D. President and Chief Executive Officer | ||
Each person whose individual signature appears below hereby authorizes and appoints Kleanthis G. Xanthopoulos, Ph.D., with full power of substitution and resubstitution and full power to act, as his or her true and lawful attorney-in-fact and agent to act in his or her name, place and stead and to execute in the name and on behalf of each person, individually and in each capacity stated below, and to file any and all amendments to this registration statement, including any and all post-effective amendments and amendments thereto, and any subsequent registration statement relating to the same offering as this registration statement that is to be effective upon filing pursuant to Rule 462(b) under the Securities Act and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing, ratifying and confirming all that said attorney-in-fact and agent or his substitute may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Act, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Kleanthis G. Xanthopoulos, Ph.D. Kleanthis G. Xanthopoulos, Ph.D. |
President, Chief Executive Officer and Member of the Board of Directors (Principal Executive Officer and Principal Financial and Accounting Officer) |
June 26, 2013 | ||
| /s/ Stelios Papadopoulos, Ph.D. Stelios Papadopoulos, Ph.D. |
Chairman of the Board and Member of the Board of Directors |
June 26, 2013 | ||
| David Baltimore, Ph.D. |
Member of the Board of Directors |
|||
| /s/ Bruce L.A. Carter, Ph.D. Bruce L.A. Carter, Ph.D. |
Member of the Board of Directors | June 26, 2013 | ||
| /s/ Mark G. Foletta Mark G. Foletta |
Member of the Board of Directors | June 26, 2013 | ||
II-6
Table of Contents
Signatures
| Signature | Title | Date | ||
| /s/ John M. Maraganore, Ph.D. John M. Maraganore, Ph.D. |
Member of the Board of Directors | June 26, 2013 | ||
| /s/ B. Lynne Parshall B. Lynne Parshall |
Member of the Board of Directors |
June 26, 2013 | ||
| /s/ William H. Rastetter, Ph.D. William H. Rastetter, Ph.D. |
Member of the Board of Directors |
June 26, 2013 | ||
| /s/ Douglas E. Williams, Ph.D. Douglas E. Williams, Ph.D. |
Member of the Board of Directors |
June 26, 2013 | ||
II-7
Table of Contents
Exhibit index
| Exhibit number |
Description of document | |
| 1.1 | Form of Underwriting Agreement. | |
| 3.1 | Amended and Restated Certificate of Incorporation of the Registrant (incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K, filed with the SEC on October 11, 2012). | |
| 3.3 | Amended and Restated Bylaws of the Registrant (incorporated by reference to Exhibit 3.2 to the Registrant’s Current Report on Form 8-K, filed with the SEC on October 11, 2012). | |
| 4.1 | Form of Common Stock Certificate of the Registrant (incorporated by reference to Exhibit 4.1 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 5.1(1) | Opinion of Cooley LLP. | |
| 10.1+ | Form of Indemnity Agreement between the Registrant and its directors and officers (incorporated by reference to Exhibit 10.1 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.2+ | 2009 Equity Incentive Plan, as amended, and Form of Stock Option Grant Notice, Option Agreement and Form of Notice of Exercise (incorporated by reference to Exhibit 10.2 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.3+ | 2012 Equity Incentive Plan and Form of Stock Option Agreement and Form of Stock Option Grant Notice thereunder (incorporated by reference to Exhibit 10.3 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.4+ | Non-Employee Director Compensation Policy (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.5+ | 2012 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.5 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.6+ | Amended and Restated Employment Agreement between the Registrant and Kleanthis G. Xanthopoulos, Ph.D., dated June 15, 2012 (incorporated by reference to Exhibit 10.6 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.7+ | Amended and Restated Employment Agreement between the Registrant and Garry E. Menzel, Ph.D., dated June 15, 2012 (incorporated by reference to Exhibit 10.7 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.8+ | Employment Agreement between the Registrant and Neil W. Gibson, Ph.D., dated June 15, 2012 (incorporated by reference to Exhibit 10.8 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
Table of Contents
Exhibit index
| Exhibit number |
Description of document | |||
| 10.9 | Lease between the Registrant and BMR-John Hopkins Court LLC, a Delaware limited liability company, dated March 19, 2010 (incorporated by reference to Exhibit 10.9 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.10 | First Amendment to Lease between the Registrant and BMR-John Hopkins Court LLC, a Delaware limited liability company, dated April 26, 2010 (incorporated by reference to Exhibit 10.10 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.11 | Second Amendment to Lease between the Registrant and BMR-John Hopkins Court LLC, a Delaware limited liability company, dated January 26, 2011 (incorporated by reference to Exhibit 10.11 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.12 | Third Amendment to Lease between the Registrant and BMR-3545-3575 John Hopkins LP, a Delaware limited partnership (formerly known as BMR-John Hopkins Court LLC), dated February 27, 2012 (incorporated by reference to Exhibit 10.12 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.13 | Fourth Amendment to Lease between the Registrant and BMR-3545-3575 John Hopkins LP, a Delaware limited partnership (formerly known as BMR-John Hopkins Court LLC), dated November 19, 2012 (incorporated by reference to Exhibit 10.13 to the Registrant’s Annual Report on Form 10-K (File No. 001-35670), originally filed with the SEC on February 19, 2013). | |||
| 10.14 | Fifth Amendment to Lease between the Registrant and BMR-3545-3575 John Hopkins LP, a Delaware limited partnership (formerly known as BMR-John Hopkins Court LLC), dated February 21, 2013 (incorporated by reference to Exhibit 99.1 to the Registrant’s Current Report on Form 8-K, filed with the SEC on June 26, 2013). | |||
| 10.15 | * | Founding Investor Rights Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated January 1, 2009 (incorporated by reference to Exhibit 10.13 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.16 | Amendment Number One to the Founding Investor Rights Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated June 7, 2010 (incorporated by reference to Exhibit 10.14 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.17 | Amendment Number Two to the Founding Investor Rights Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated October 27, 2010 (incorporated by reference to Exhibit 10.15 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.18 | Investor Rights Agreement between the Registrant and Aventis Holdings, Inc., dated October 27, 2010 (incorporated by reference to Exhibit 10.16 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
Table of Contents
Exhibit index
| Exhibit number |
Description of document | |
| 10.19* | Amended and Restated License and Collaboration Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated January 1, 2009 (incorporated by reference to Exhibit 10.17 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.20* | Amendment Number One to the Amended and Restated License and Collaboration Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated June 10, 2010 (incorporated by reference to Exhibit 10.18 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.21* | Amendment Number Two to the Amended and Restated License and Collaboration Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated October 25, 2011 (incorporated by reference to Exhibit 10.19 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.22* | Product Development and Commercialization Agreement between the Registrant and Glaxo Group Limited, dated April 17, 2008 (incorporated by reference to Exhibit 10.20 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.23* | Amendment #1 to the Product Development and Commercialization Agreement between the Registrant and Glaxo Group Limited, dated February 24, 2010 (incorporated by reference to Exhibit 10.21 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.24* | Amendment #2 to the Product Development and Commercialization Agreement between the Registrant and Glaxo Group Limited, dated June 16, 2010 (incorporated by reference to Exhibit 10.22 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.25* | Amendment #3 to the Product Development and Commercialization Agreement between the Registrant and Glaxo Group Limited, dated June 30, 2011 (incorporated by reference to Exhibit 10.23 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.26* | Exclusive License and Nonexclusive Option Agreement between the Registrant and Glaxo Group Limited, dated February 24, 2010 (incorporated by reference to Exhibit 10.24 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.27* | Co-Exclusive License Agreement among the Board of Trustees of the Leland Stanford Junior University, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated August 31, 2005 (incorporated by reference to Exhibit 10.25 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.28 | Assignment Agreement between the Registrant and Isis Pharmaceuticals, Inc., dated July 13, 2009 (incorporated by reference to Exhibit 10.26 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
Table of Contents
Exhibit index
| Exhibit number |
Description of document | |||
| 10.29 | * | License Agreement between the Registrant and Max-Planck-Innovation GmbH, dated June 5, 2009 (incorporated by reference to Exhibit 10.27 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.20 | * | Amended and Restated License Agreement among Max-Planck-Innovation GmbH, the Registrant, Isis Pharmaceuticals, Inc. and Alnylam Pharmaceuticals, Inc., dated April 18, 2011 (incorporated by reference to Exhibit 10.28 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.31 | * | NYU-Regulus License Agreement by and between the Registrant and New York University, dated March 28, 2011 (incorporated by reference to Exhibit 10.29 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.32 | * | Exclusive Patent License Agreement between the Registrant and Bayerische Patent Allianz GmbH, dated May 18, 2010 (incorporated by reference to Exhibit 10.30 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.33 | * | Amended and Restated Collaboration and License Agreement between the Registrant and Sanofi, dated July 16, 2012 (incorporated by reference to Exhibit 10.31 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.34 | * | Non-Exclusive Technology Alliance and Option Agreement between the Registrant and Sanofi, dated June 21, 2010 (incorporated by reference to Exhibit 10.32 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.35 | Amended and Restated Convertible Promissory Note No. 1 made by the Registrant in favor of Glaxo Group Limited, dated July 27, 2012 (incorporated by reference to Exhibit 10.33 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.36 | Amended and Restated Convertible Promissory Note No. 2 made by the Registrant in favor of Glaxo Group Limited, dated July 27, 2012 (incorporated by reference to Exhibit 10.34 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.37 | * | Amendment #4 to the Product Development and Commercialization Agreement between the Registrant and Glaxo Group Limited, dated June 29, 2012 (incorporated by reference to Exhibit 10.35 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
| 10.38 | Amendment Number Three to the Founding Investor Rights Agreement among the Registrant, Alnylam Pharmaceuticals, Inc. and Isis Pharmaceuticals, Inc., dated July 24, 2012 (incorporated by reference to Exhibit 10.36 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |||
| 10.39 | * | Collaboration and License Agreement between the Registrant and AstraZeneca AB, dated August 14, 2012 (incorporated by reference to Exhibit 10.37 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | ||
Table of Contents
Exhibit index
| Exhibit number |
Description of document | |
| 10.40 | Common Stock Purchase Agreement between the Registrant and AstraZeneca AB, dated August 14, 2012 (incorporated by reference to Exhibit 10.38 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.41* | Collaboration and License Agreement between the Registrant and Biogen Idec MA Inc., dated August 15, 2012 (incorporated by reference to Exhibit 10.39 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.42 | Note Purchase Agreement between the Registrant and Biogen Idec MA Inc., dated August 15, 2012 (incorporated by reference to Exhibit 10.40 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.43 | Convertible Promissory Note made by the Registrant in favor of Biogen Idec MA Inc., dated August 15, 2012 (incorporated by reference to Exhibit 10.41 to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-183384), originally filed with the SEC on August 17, 2012). | |
| 10.44 | Investor Rights Agreement, dated October 10, 2012, between AstraZeneca AB and the Registrant (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed with the SEC on October 11, 2012). | |
| 10.45 | Convertible Promissory Note, dated October 10, 2012, made by the Registrant in favor of Glaxo Group Limited (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K, filed with the SEC on October 11, 2012). | |
| 10.46† | Amendment #5 to the Product Development and Commercialization Agreement between the Registrant and Glaxo Group Limited, dated June 6, 2013. | |
| 10.47 | Letter Agreement dated April 26, 2013 between the Registrant and Sanofi. | |
| 10.48† | Option Letter Agreement dated June 21, 2013 between the Registrant and Sanofi. | |
| 10.49† | Amendment No. 1 (to Collaboration and License Agreement) between the Registrant and AstraZeneca AB, dated April 30, 2013. | |
| 10.50† | Amendment No. 1 to Collaboration and License Agreement between the Registrant and Biogen Idec MA Inc., dated June 24, 2013. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 23.2(1) | Consent of Cooley LLP. Reference is made to Exhibit 5.1. | |
| 24.1 | Power of Attorney. Reference is made to the signature page hereto. | |
| (1) | To be filed by amendment. |
| + | Indicates management contract or compensatory plan. |
| * | We have received confidential treatment for certain portions of this agreement, which have been omitted and filed separately with the SEC pursuant to Rule 406 under the Securities Act of 1933, as amended. |
| † | We have requested confidential treatment for certain portions of this agreement. |