Attached files
| file | filename |
|---|---|
| 8-K - FORM 8-K - PUMA BIOTECHNOLOGY, INC. | d429464d8k.htm |
| EX-1.1 - UNDERWRITING AGREEMENT - PUMA BIOTECHNOLOGY, INC. | d429464dex11.htm |
| EX-99.3 - PRESS RELEASE - PUMA BIOTECHNOLOGY, INC. | d429464dex993.htm |
| EX-99.1 - PRESS RELEASE - PUMA BIOTECHNOLOGY, INC. | d429464dex991.htm |
| EX-99.4 - RISK FACTORS - PUMA BIOTECHNOLOGY, INC. | d429464dex994.htm |
| EX-99.2 - PRESS RELEASE - PUMA BIOTECHNOLOGY, INC. | d429464dex992.htm |
Exhibit 99.5
BUSINESS
As used herein, unless the context requires otherwise, the terms “Company,” “we,” “our” and “us” refer to Puma Biotechnology, Inc., a Delaware corporation formed on April 27, 2007 and formerly known as Innovative Acquisitions Corp., and the term “Former Puma” refers to Puma Biotechnology, Inc., a private Delaware corporation that merged with and into us in October 2011.
Company Overview
We are a development-stage biopharmaceutical company that acquires and develops innovative products for the treatment of various forms of cancer. We focus on in-licensing drug candidates that are undergoing or have already completed initial clinical testing for the treatment of cancer and then seek to further develop those drug candidates for commercial use.
We currently license the rights to three drug candidates:
| • | PB272 (neratinib (oral)), which we are developing for the treatment of advanced breast cancer patients and non-small cell lung cancer patients; |
| • | PB272 (neratinib (intravenous)), which we are developing for the treatment of advanced cancer patients; and |
| • | PB357, which we believe can serve as a backup compound to PB272, and which we are evaluating for further development in 2013. |
We are initially focused on developing neratinib for the treatment of patients with human epidermal growth factor receptor type 2, or HER2, positive metastatic breast cancer. Studies show that approximately 20% to 25% of breast cancer tumors have an over-expression of the HER2 protein. Women with breast cancer that over-expresses HER2, referred to as HER2 positive breast cancer, are at greater risk for disease progression and death than women whose tumors do not over-express HER2. Therapeutic strategies, such as the use of Herceptin (trastuzumab) and Perjeta (pertuzumab), both produced by Genentech, and Tykerb (lapatinib), produced by GlaxoSmithKline, given in combination with chemotherapy have been developed to improve the treatment of this cancer by blocking HER2. Based on pre-clinical and clinical studies to date, we believe that neratinib may offer an advantage over existing treatments by more potently inhibiting HER2 at a site distinct from those targeted by pertuzumab, trastuzumab, and lapatinib and by acting via a mechanism different from those of other HER2 active drugs.
Currently, the FDA approved first-line therapy for treatment of HER2 positive metastatic breast cancer is the combination of Perjeta plus Herceptin and taxane chemotherapy. The current FDA-approved second-line therapy is Tykerb, given in combination with the chemotherapy drug capecitabine. As a single agent in patients who have failed first line treatment, Tykerb has demonstrated an objective response rate of approximately 5% to 7% and a progression free survival of between eight and nine weeks. In a Phase III clinical trial, patients with HER2 positive metastatic breast cancer who received the combination of Tykerb plus capecitabine demonstrated a median progression free survival, or PFS, of 27.1 weeks and a response rate of 23.7%. Another treatment regimen that is used in patients who have failed first line treatment is the combination of the chemotherapy drug vinorelbine given in combination with Herceptin, which has been shown to have an objective response rate of approximately 25% and a progression free survival of 22 weeks.
Data from a recently completed Phase II clinical trial of neratinib administered as a single agent to patients with HER2 positive metastatic breast cancer demonstrated an objective response rate of 24% and median PFS of 22.3 weeks for patients who had previously been treated with trastuzumab, and an objective response rate of 56% and median PFS of 39.6 weeks for patients who had not previously been treated with trastuzumab. Additionally, data from over 3,000 patients treated with neratinib, either as a single agent or in combination with other anti-cancer drugs, also suggests a manageable safety profile. Diarrhea has been the most common side effect, but appears to be manageable with antidiarrheal agents and dose modification.
We license the exclusive worldwide rights to our current drug candidates from Pfizer Inc., or Pfizer, which had previously been responsible for the clinical trials regarding neratinib. We have modified Pfizer’s clinical development strategy and during the next 12 to 18 months plan to:
| • | commence Phase III clinical trials to evaluate the use of neratinib in combination with chemotherapy and other anti-cancer drugs as a second or third-line treatment for HER2 positive breast cancer; |
| • | initiate Phase II clinical trials to evaluate the use of neratinib for the treatment of HER2 mutated non-small cell lung cancer and in patients with a newly identified breast cancer mutation in HER2 negative breast cancer; |
| • | continue the ongoing Phase II clinical trial of neratinib in the neoadjuvant treatment of HER2 positive breast cancer and the ongoing Phase II trial of neratinib in patients with HER2 positive metastatic breast cancer that has metastasized to the brain; and |
| • | continue to evaluate the application of neratinib in the treatment of other forms of HER resistant cancers where there may be unmet medical needs. |
Our President and Chief Executive Officer, Alan Auerbach, has extensive experience in identifying and developing drug candidates for use in the treatment of cancer. He was the founder, President and Chief Executive Officer of Cougar Biotechnology, Inc., or Cougar, where he was responsible for in-licensing and developing abiraterone acetate for the treatment of advanced prostate cancer. Mr. Auerbach progressed abiraterone acetate into two Phase III clinical trials before Cougar was purchased by Johnson & Johnson in 2009.
Our Strategy
Our strategy is to become a leading oncology-focused biopharmaceutical company. The key elements of our strategy are as follows:
| • | Advance PB272 (neratinib (oral)), our lead drug candidate, toward regulatory approval and commercialization. We are primarily focused on developing neratinib for the treatment of patients with HER2 positive metastatic breast cancer. We plan to modify the previous clinical development strategy that Pfizer employed by focusing our planned Phase II and Phase III clinical trials on the use of neratinib as a second- or third-line treatment option, which we believe may be underserved by current treatment alternatives and where clinical trials have shown substantial levels of activity. We are also focusing on the development of neratinib in the neoadjuvant treatment of patients with HER2 positive breast cancer and in patients with HER2 positive metastatic breast cancer that has metastasized to the brain. |
| • | Expand our product pipeline by pursuing additional applications of neratinib. We believe there are additional applications for neratinib in the treatment of HER2 mutated non-small cell lung cancer, which we also believe may be underserved by current treatment alternatives, in the treatment of patients with a newly identified breast cancer mutation in HER2 negative breast cancer and in the treatment of tumor types where HER2 is overexpressed, and we intend to further evaluate the safety and efficacy of neratinib for treating these cancers. |
| • | Focus on developing innovative cancer therapies. We focus on oncology drug candidates in order to capture efficiencies and economies of scale. We believe that drug development for cancer markets is particularly attractive because relatively small clinical trials can provide meaningful information regarding patient response and safety. Furthermore, we believe that our capabilities are well suited to the oncology market and represent distinct competitive advantages. |
| • | Build a sustainable pipeline by employing multiple therapeutic approaches and disciplined decision criteria based on clearly defined proof of principal goals. We seek to build a sustainable product pipeline by employing multiple therapeutic approaches and by acquiring drug candidates belonging to known drug classes. In addition, we employ disciplined decision criteria to assess drug candidates, favoring drug candidates that have undergone at least some clinical study. Our decision to license a drug candidate will also depend on the scientific merits of the technology; the costs of the transaction and other economic terms of the proposed license; the amount of capital required to |
| develop the technology; and the economic potential of the drug candidate, should it be commercialized. We believe this strategy minimizes our clinical development risk and allows us to accelerate the development and potential commercialization of current and future drug candidates. We intend to pursue regulatory approval for a majority of our drug candidates in multiple indications. |
| • | Evaluate the commercialization strategies on a product-by-product basis in order to maximize the value of each. As we move our drug candidates through development toward regulatory approval, we will evaluate several options for each drug candidate’s commercialization strategy. These options include building our own internal sales force; entering into a joint marketing partnership with another pharmaceutical company or biotechnology company, whereby we jointly sell and market the product; and out-licensing our product, whereby another pharmaceutical company or biotechnology company sells and markets our product and pays us a royalty on sales. Our decision will be made separately for each product and will be based on a number of factors including capital necessary to execute on each option, size of the market that needs to be addressed and terms of potential offers from other pharmaceutical and biotechnology companies. It is too early for us to know which of these options we will pursue for our drug candidates, assuming their successful development. |
Product Development Pipeline
Breast Cancer Overview
Breast cancer is the leading cause of cancer death among women worldwide, with approximately 1 million new cases reported each year and more than 400,000 deaths per year. Approximately 20% to 25% of breast cancer tumors show over-expression of the HER2 protein. Women with breast cancer that overexpresses HER2 are at greater risk for disease progression and death than women whose tumors do not over-express HER2. Therapeutic strategies have been developed to block HER2 in order to improve the treatment of this cancer.
Trastuzumab and pertuzumab are monoclonal antibodies that bind to the HER2 protein and thereby cause the cells to cease reproducing. Trastuzumab and pertuzumab given in combination with chemotherapy is the current first line standard of care for HER2 positive metastatic breast cancer. Lapatinib is a small molecule that also binds to the HER2 protein and causes the cell to cease reproducing. The current FDA-approved second-line therapy is lapatinib given in combination with the chemotherapy drug capecitabine. Unfortunately, most patients with HER2 positive breast cancer eventually develop resistance to these treatments, resulting in disease progression. For these reasons, there is a need for alternatives to block HER2 signaling in patients who fail pertuzumab, trastuzumab and lapatinib. PB272 is an orally active small molecule that inhibits HER2 at a different site and uses a different mechanism than trastuzumab. As a result, we believe that PB272 may have utility in patients with HER2 positive metastatic breast cancer who have failed treatment with trastuzumab.
The following chart shows each of our current drug candidates and their clinical development stage:
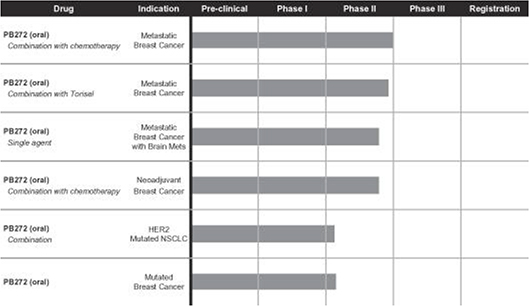
PB272 (neratinib (oral))—Breast Cancer
Neratinib is a potent irreversible tyrosine kinase inhibitor, or TKI, that blocks signal transduction through the epidermal growth factor receptors, or EGFRs, HER1, HER2 and HER4. We believe neratinib has clinical application in the treatment of several cancers, including breast cancer and non-small cell lung cancer and other tumor types that overexpress HER2. Our initial focus is on the development of neratinib as an oral treatment of patients with HER2 positive metastatic breast cancer.
Advantages of Neratinib
Based on pre-clinical and clinical studies to date, we believe that neratinib may offer an advantage over existing treatments that are used in the treatment of patients with HER2 positive metastatic breast cancer who have failed first-line therapy, including treatment with trastuzumab. Currently, the treatment of metastatic breast cancer patients who have failed first-line therapy with pertuzumab and trastuzumab involves continuing treatment with chemotherapy given in combination with either trastuzumab or lapatinib. We believe that by more potently inhibiting HER2 at a different site and acting via a mechanism different from those of pertuzumab, trastuzumab or lapatinib, neratinib may have potential advantages over these existing treatments, most notably due to its increased selectivity and stronger inhibition of the HER2 target enzyme.
Clinical Trials of Neratinib in Patients with Metastatic Breast Cancer
Trials of Neratinib as a Single Agent. In 2009, Pfizer presented data at the CTRC-AACR San Antonio Breast Cancer Symposium from a Phase II trial of neratinib administered as a single agent to patients with HER2 positive metastatic breast cancer. Final results from this trial were published in the Journal of Clinical Oncology in March 2010.
The trial involved a total of 136 patients, 66 of whom had received prior treatment with trastuzumab and 70 of whom had not received prior treatment with trastuzumab. The results of the study showed that neratinib was reasonably well tolerated among both the pretreated patients and the patients who had not received prior treatment with trastuzumab. Diarrhea was the most common side effect, but was manageable with antidiarrheal agents and dose modification. Efficacy results from the trial showed that the objective response rate was 24% for patients who had received prior trastuzumab treatment and 56% for patients with no prior trastuzumab treatment. Furthermore, the median PFS was 22.3 weeks for the patients who had received prior trastuzumab and 39.6 weeks for the patients who had not received prior trastuzumab.
Trials of Neratinib in Combination with Other Anti-Cancer Drugs. At the 2010 San Antonio Breast Cancer Symposium, Pfizer presented data from Phase II trials of neratinib when given in combination with other anti-cancer drugs that are currently used for the treatment of HER2 positive metastatic breast cancer. One Phase II trial evaluated the safety and efficacy of neratinib given in combination with the anti-cancer drug paclitaxel in patients with HER2 positive metastatic breast cancer. The results presented showed that for the 66 patients in the trial who had previously been treated with at least one prior line of therapy, the combination of neratinib with paclitaxel was shown to have a favorable safety profile that was similar to that of each drug when given alone. The efficacy results from the trial demonstrated an objective response rate of 74% and PFS of 63.1 weeks.
Pfizer also presented data from a second Phase II trial at the 2010 San Antonio Breast Cancer Symposium, which evaluated the safety and efficacy of neratinib when given in combination with the anti-cancer drug vinorelbine in patients with HER2 positive metastatic breast cancer. In the 56 patients who had not been previously treated with the anti-HER2 therapy lapatinib, treatment with the combination of vinorelbine plus neratinib resulted in an overall response rate of 57% and PFS was 44.1 weeks. For those patients who had received prior treatment with lapatinib, the overall response rate was 50%. The combination of vinorelbine and neratinib was generally well tolerated.
Data from a third Phase II study, in which patients with confirmed ErbB2 positive (HER2 positive) metastatic breast cancer who had failed treatment with trastuzumab and taxane chemotherapy were given PB272 in combination with capecitabine, was presented at the 2011 San Antonio Breast Cancer Symposium. The results of the study showed that the combination of PB272 and capecitabine had acceptable tolerability. The efficacy results from the trial showed that for the 61 patients in the trial who had not been previously treated with the HER2 targeted anticancer drug lapatinib, there was an overall response rate of 64% and a clinical benefit rate of 72%. In addition, for the seven patients in the trial who had previously been treated with lapatinib, there was an overall response rate of 57% and a clinical benefit rate of 71%. The median PFS for patients who had not received prior treatment with lapatinib was 40.3 weeks and the median PFS for the patients who had received prior lapatinib treatment was 35.9 weeks.
Puma anticipates initiating a Phase III trial of neratinib plus capecitabine in HER2 positive metastatic breast cancer patients who have failed first-line therapy in late 2012 or early 2013. We anticipate that this trial will be a randomized trial of neratinib plus capecitabine versus lapatinib plus capecitabine.
In 2010, Pfizer also initiated a Phase I/II trial of neratinib in combination with the anti-cancer drug temsirolimus, or Torisel, in patients with HER2 positive metastatic breast cancer who have failed multiple prior treatments. The study enrolled patients with either HER2 positive metastatic breast cancer and disease progression on trastuzumab or with triple negative breast cancer. The preliminary Phase II results of this trial were presented at the 2011 San Antonio Breast Cancer Symposium. The results of the study showed that the combination of PB272 and temsirolimus had acceptable tolerability. The efficacy results from the trial showed that for the 15 patients with HER2 positive disease, nine patients, or 60%, experienced a partial response and one patient, or 7%, experienced stable disease for greater than six months, which translates to a clinical benefit rate of 67%. Patients who experienced a partial response to the combination of neratinib plus temsirolimus demonstrated a maximum change in the size of their target lesions of between 33% and 83%. None of the five patients with triple-negative breast cancer demonstrated a partial response or stable disease for greater than six months. We anticipate that data from this trial will be presented in the fourth quarter of 2012 and, in the first quarter of 2013, we expect to commence a Phase III trial of neratinib in combination with temsirolimus in patients with HER2 positive metastatic breast cancer who have failed multiple prior treatments.
Approximately one-third of the patients with HER2 positive metastatic breast cancer develop metastases that spread to their brain. The current antibody based treatments, including Herceptin and Perjeta, do not enter the brain and therefore are not believed to be effective in treating these patients. In a Phase II trial with Tykerb given as a single agent, Tykerb demonstrated a 6% objective response rate in the patients with HER2 positive metastatic breast cancer whose disease spread to their brains. In January 2012, a Phase II trial of neratinib as a single agent in patients with HER2 positive metastatic breast cancer that has spread to their brains was initiated in conjunction with the Dana Farber Translational Breast Cancer Research Consortium. We anticipate that results from this trial will be presented in 2013.
At the 2010 San Antonio Breast Cancer Symposium, the results of the Neoadjuvant Lapatinib and/or Trastuzumab Treatment Optimisation) Study, or the Neo-ALTTO study, were presented. In this trial, patients with HER2 positive breast cancer were randomized to receive either the combination of paclitaxel plus trastuzumab, the combination of paclitaxel plus lapatinib or the combination of paclitaxel plus trastuzumab plus lapatinib, and neoadjuvant (preoperative) therapy. The results of the trial demonstrated that the patients who received the combination of paclitaxel plus trastuzumab demonstrated a pathological complete response rate of 29.5%, the patients who received paclitaxel plus lapatinib had a pathological complete response rate of 24.7% and the patients who received the combination of paclitaxel plus trastuzumab plus lapatinib had a pathological complete response rate of 51.3%.
In 2010, Pfizer, in collaboration with the National Surgical Adjuvant Breast and Bowel Project, or NSABP, a clinical trials cooperative group supported by the National Cancer Institute, or NCI, initiated a study to investigate the use of neratinib as a neoadjuvant (preoperative) therapy for newly diagnosed HER2 positive breast cancer. In this trial, a total of 129 patients are randomized to receive either neratinib plus the chemotherapy drug paclitaxel or trastuzumab plus paclitaxel prior to having surgery to remove their tumors. The purpose of this study is to test whether adding neratinib to paclitaxel chemotherapy is better than trastuzumab plus paclitaxel chemotherapy before having surgery. This trial has been modified to include a third treatment arm where patients will receive the combination of neratinib plus trastuzumab plus paclitaxel prior to having surgery to remove their tumors. We anticipate that enrollment in all three arms of this trial will continue through the end of 2012 and that results from this trial will be presented in 2013.
Also in 2010, the Foundation for the National Institutes of Health initiated the I-SPY 2 TRIAL (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging and Molecular Analysis 2). Patients with newly diagnosed HER2 positive breast cancer are randomized to receive either neratinib plus the chemotherapy drug paclitaxel or trastuzumab plus paclitaxel prior to having surgery to remove their tumors (neoadjuvant therapy). The purpose of this study is to test whether adding neratinib to paclitaxel chemotherapy is better than trastuzumab plus paclitaxel chemotherapy before having surgery. We anticipate that this trial will be modified in 2012 to include a third treatment arm where patients will receive the combination of neratinib plus trastuzumab plus paclitaxel prior to having surgery to remove their tumors. We anticipate that enrollment in all three arms of this trial will continue through the end of 2012.
Discontinued Studies. Pfizer had previously been sponsoring two additional clinical trials of neratinib. The first trial, referred to as the NEfERTT™ trial, was a Phase II randomized trial of neratinib in combination with the anti-cancer drug paclitaxel versus trastuzumab in combination with paclitaxel for the treatment of patients who have not received previous treatment for HER2 positive metastatic breast cancer. The second trial, referred to as the ExteNET™ trial, was a Phase III study investigating the effects of neratinib after adjuvant trastuzumab in patients with early stage breast cancer. On October 5, 2011, we announced that enrollment in the ExteNET trial was terminated and that both the NEfERTT and the ExteNET trials were going to be wound down. We anticipate that completion of these wind-down activities will continue in 2012. We are responsible for any activities associated with winding down these trials during 2012 and beyond.
PB272 (neratinib (oral))—Other Potential Applications
Approximately 2% to 4% of patients with non-small cell lung cancer have a HER2 mutation in the kinase domain. This mutation is believed to narrow the ATP binding cleft which results in increased tyrosine kinase activity. The mutation is also believed to result in increased PI3K activity and mTOR activation. Published data suggests that patients with HER2 mutated non-small cell lung cancer do not respond to platinum chemotherapy and do not respond to EGFR inhibitors. Pfizer previously conducted a Phase I trial of neratinib given in combination with the anticancer drug temsirolimus in patients with solid tumors. In this trial, seven patients with HER2 mutated non-small cell lung cancer were enrolled in the trial. These patients had received a median of three prior treatments for their disease. The results from the trial were presented at the 2011 American Society of Clinical Oncology (ASCO) Annual Meeting and at the 2012 International Association for the Study of Lung Cancer meeting and demonstrated that for the six evaluable patients, two (33%) patients demonstrated a partial radiological response and three patients had stable disease evidenced by tumor shrinkage of between approximately 5% and 28%. We anticipate initiating a Phase II randomized trial of neratinib plus temsirolimus in patients with HER2 mutated non-small cell lung cancer in the fourth quarter of 2012.
In September 2012, a new mutation in patients with HER2 negative breast cancer was identified as part of a study performed by the Cancer Genome Atlas Network and published in Nature. We believe this mutation may occur in an estimated 2% of patients with HER2 negative breast cancer. We are aware of results from third party preclinical studies that we believe suggest that neratinib is active in HER2 negative breast cancer cells that have this mutation and that neratinib has more anticancer activity than either trastuzumab or lapatinib in cells with this mutation. We anticipate that this preclinical data will be presented in the fourth quarter of 2012. In the fourth quarter of 2012 or the first quarter of 2013, we anticipate initiating a Phase II trial of neratinib in HER2 negative breast cancer patients who have this newly identified mutation.
PB272 (neratinib (intravenous))
We also plan to develop neratinib as an intravenously administered agent. In pre-clinical studies the intravenous version of neratinib resulted in higher exposure levels of neratinib in pre-clinical models. We believe that this may result in higher blood levels of neratinib in patients, and this may translate into enhanced efficacy. We plan to file the IND for the intravenous formulation of neratinib in 2013.
PB357
PB357 is an orally administered agent that is an irreversible TKI that blocks signal transduction through the epidermal growth factor receptors, HER1, HER2, and HER4. PB357 is structurally similar to PB272. Pfizer completed single dose Phase I trials of PB357. We are evaluating PB357 and considering options relative to its development in 2013.
Plan of Development
We plan to conduct additional clinical trials of neratinib in patients with HER2 positive metastatic breast cancer over the next 12 to 18 months. In one trial we plan to further investigate the efficacy of neratinib when given in combination with chemotherapy in patients with HER2 positive metastatic breast cancer who have previously been treated with at least one prior line of treatment. In another, we plan to investigate the efficacy of neratinib in patients with HER2 positive metastatic breast cancer with brain metastases. We will also continue the ongoing trial of neratinib in combination with the anti-cancer drug temsirolimus in patients with HER2 positive metastatic breast cancer. We are also continuing the development of neratinib in the neoadjuvant treatment of patients with HER2 positive breast cancer.
We also plan to conduct a Phase II clinical trial of neratinib in HER2 mutated non-small cell lung cancer patients and in HER2 negative breast cancer patients with a newly identified mutation during 2012.
Clinical Testing of Our Products in Development
Each of our products in development, and likely all future drug candidates we in-license, will require extensive pre-clinical and clinical testing to determine the safety and efficacy of the product applications prior to seeking and obtaining regulatory approval. This process is expensive and time consuming. In completing these trials, we are dependent upon third-party consultants, consisting mainly of investigators and collaborators, who will conduct such trials.
We and our third-party consultants conduct pre-clinical testing in accordance with Good Laboratory Practices, or GLP, and clinical testing in accordance with Good Clinical Practice standards, or GCP, which are international ethical and scientific quality standards utilized for pre-clinical and clinical testing, respectively. GCP is the standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials, and is required by the FDA to be followed in conducting clinical trials. Additionally, our pre-clinical and clinical testing completed in the European Union is conducted in accordance with applicable EU standards, such as the EU Clinical Trials Directive (Directive 2001/20/EC of April 4, 2001), or the EU Clinical Trials Directive, and the national laws of the Member Estates of the EU implementing its provisions.
Competition
The development and commercialization of new products to treat cancer is highly competitive, and we expect considerable competition from major pharmaceutical, biotechnology and specialty cancer companies. As a result, there are and will likely continue to be extensive research and substantial financial resources invested in the discovery and development of new cancer products. Our potential competitors include, but are not limited to,
Genentech, GlaxoSmithKline, Roche, Boehringer Ingelheim, Takeda, Array Biopharma and Ambit Biosciences. We are an early-stage company with no history of operations and we only recently acquired the rights to the drug candidates we expect to develop. Many of our competitors have substantially more resources than we do, including both financial and technical. In addition, many of our competitors have more experience than we have in pre-clinical and clinical development, manufacturing, regulatory and global commercialization. We are also competing with academic institutions, governmental agencies and private organizations that are conducting research in the field of cancer. We anticipate that we will face intense competition.
We expect that our products under development and in clinical trials will address major markets within the cancer sector. Our competition will be determined in part by the potential indications for which drugs are developed and ultimately approved by regulatory authorities. Additionally, the timing of market introduction of some of our potential products or of competitors’ products may be an important competitive factor. Accordingly, the speed with which we can develop products, complete pre-clinical testing, clinical trials and approval processes, and supply commercial quantities to market are expected to be important competitive factors. We expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price, reimbursement and patent position.
Intellectual Property and License Agreements
We hold a worldwide exclusive license under our license agreement with Pfizer to four granted U.S. patents and nine pending U.S. patent applications, as well as foreign counterparts thereof and other patent applications and patents claiming priority therefrom.
In the U.S., we have a license to an issued patent, which currently will expire in 2025, for the composition of matter of neratinib, our lead compound. We have a license to an issued U.S. patent covering a family of compounds including neratinib, as well as equivalent patents in the European Union and Japan, that currently expire in 2019. We also have a license to an issued U.S. patent for the use of neratinib in the treatment of breast cancer, which currently expires in 2025, and an issued U.S. polymorph patent for neratinib, which currently expires in 2028. In jurisdictions which permit such, we will seek patent term extensions where possible for certain of our patents. We plan to pursue additional patents in and outside the U.S. covering additional therapeutic uses and polymorphs of neratinib from these existing applications. In addition, we will pursue patent protection for any new discoveries or inventions made in the course of our development of neratinib.
If we obtain marketing approval for neratinib or other drug candidates in the U.S. or in certain jurisdictions outside the U.S., we may be eligible for regulatory protection, such as five years of new chemical entity exclusivity, and as mentioned above, up to five years of patent term extension potentially available in the United States under the Hatch-Waxman Act. In addition, eight to 11 years of data and marketing exclusivity potentially are available for new drugs in the European Union; up to five years of patent extension are potentially available in Europe (Supplemental Protection Certificate), and eight years of data exclusivity are potentially available in Japan. There can be no assurance that we will qualify for any such regulatory exclusivity, or that any such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies. See “Government Regulation” below.
Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. However, even patent protection may not always afford us with complete protection against competitors who seek to circumvent our patents. See “Risk Factors—Risks Related to Our Intellectual Property—Our proprietary rights may not adequately protect our intellectual property and potential products, and if we cannot obtain adequate protection of our intellectual property and potential products, we may not be able to successfully market our potential products.”
We depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors, none of which is patentable. To help protect our proprietary know-how, which is not patentable, and inventions for which patents may be difficult to obtain or enforce, we rely
on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
License Agreements
In August 2011, Former Puma entered into an agreement pursuant to which Pfizer agreed to grant to Former Puma a worldwide license for the development, manufacture and commercialization of neratinib (oral), neratinib (intravenous), PB357, and certain related compounds. Pursuant to the terms of the agreement, the license would not become effective until Former Puma closed a capital raising transaction in which it raised at least $25 million in aggregate net proceeds and had a net worth of at least $22.5 million. Upon the closing of the financing that preceded the Merger, this condition was satisfied.
We assumed the license agreement, in accordance with its terms, in the Merger. The license is exclusive with respect to certain patent rights owned or licensed by Pfizer. Under the license agreement, Pfizer is obligated to transfer to us certain information, records, regulatory filings, materials and inventory controlled by Pfizer and relating to or useful for developing these compounds and to continue to conduct certain ongoing clinical studies until a certain time. After that time, we are obligated to continue such studies pursuant to an approved development plan, including after the license agreement terminates for reasons unrelated to Pfizer’s breach of the license agreement, subject to certain specified exceptions. We are also obligated to commence a new clinical trial for a product containing one of these compounds within a specified period of time and use commercially reasonable efforts to complete such trial and achieve certain milestones as provided in a development plan. If certain of our out-of-pocket costs in completing such studies exceed a mutually agreed amount, Pfizer will pay for certain additional out-of-pocket costs to complete such studies. We must use commercially reasonable efforts to develop and commercialize products containing these compounds in specified major-market countries and other countries in which we believe it is commercially reasonable to develop and commercialize such products.
As consideration for the license, we are required to make payments totaling $187.5 million upon the achievement of certain milestones if all such milestones are achieved. Should we commercialize any of the compounds licensed from Pfizer or any products containing any of these compounds, we will be obligated to pay to Pfizer incremental annual royalties between approximately 10% and 20% of net sales of all such products, subject, in some circumstances, to certain reductions. Our royalty obligation continues, on a product-by-product and country-by-country basis, until the later of (i) the last to expire valid claim of a licensed patent covering the applicable licensed product in such country, or (ii) the earlier of generic competition for such licensed product reaching a certain level of sales in such country or expiration of a certain time period after first commercial sale of such licensed product in such country. In the event that we sublicense the rights granted to us under the license agreement with Pfizer to a third party, the same milestone and royalty payments are required.
We can terminate the license agreement at will at any time after April 4, 2013 or for safety concerns, in each case upon specified advance notice. Each party may terminate the license agreement if the other party fails to cure any breach of a material obligation by such other party within a specified time period. Pfizer may terminate the license agreement in the event of our bankruptcy, receivership, insolvency or similar proceeding. The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Government Regulation
United States—FDA Process
The research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing, among other things, of drug products are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, fines, civil penalties, product recalls, product seizures, total or partial suspension of production or distribution, injunctions and/or criminal prosecution.
Drug Approval Process. None of our drug product candidates may be marketed in the United States until the drug has received FDA approval. The steps required before a drug may be marketed in the United States generally include the following:
| • | completion of extensive pre-clinical laboratory tests, animal studies, and formulation studies in accordance with the FDA’s GLP regulations; |
| • | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each proposed indication; |
| • | submission to the FDA of an NDA after completion of all pivotal clinical trials; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished drug product are produced and tested to assess compliance with cGMPs; and |
| • | FDA review and approval of the NDA prior to any commercial marketing or sale of the drug in the United States. |
The development and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
Pre-clinical tests include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies. The conduct of the pre-clinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the pre-clinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about the conduct of the trial, such as whether human research subjects will be exposed to an unreasonable health risk. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. We cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin.
Clinical trials involve administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be provided to the FDA as part of a separate submission to the IND. Further, an IRB for each medical center proposing to conduct the clinical trial must review and approve the study protocol and informed consent information for study subjects for any clinical trial before it commences at that center, and the IRB must monitor the study until it is completed. There are also requirements governing reporting of ongoing clinical trials and clinical trial results to public registries. Study subjects must sign an informed consent form before participating in a clinical trial.
Clinical trials necessary for product approval typically are conducted in three sequential phases, but the phases may overlap. Phase I usually involves the initial introduction of the investigational drug into a limited population, typically healthy humans, to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness. Phase II usually involves trials in a limited patient population to (i) evaluate dosage tolerance and appropriate dosage; (ii) identify possible adverse effects and safety risks; and (iii) evaluate preliminarily the efficacy of the drug for specific targeted indications. Multiple Phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase III clinical trials. Phase III trials, commonly referred to as pivotal studies, are undertaken in an expanded patient population at multiple, geographically dispersed clinical trial centers to further evaluate clinical efficacy and test further for safety by using the drug in its final form. There can be no assurance that Phase I, Phase II or Phase III testing will be completed successfully within any specified period of time, if at all. Furthermore, we, the FDA or an IRB may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Moreover, the FDA may approve an NDA for a product candidate, but require that the sponsor conduct additional clinical trials to further assess the drug after NDA approval under a post-approval commitment. Post-approval trials are typically referred to as Phase IV clinical trials.
During the development of a new drug, sponsors are given an opportunity to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase II, and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and the FDA to reach an agreement on the next phase of development. Sponsors typically use the end of Phase II meeting to discuss their Phase II clinical results and present their plans for the pivotal Phase III clinical trial that they believe will support approval of the new drug. A sponsor may request a Special Protocol Assessment, or SPA, the purpose of which is to reach an agreement with the FDA that the protocol design, clinical endpoints, and statistical analyses are acceptable to support regulatory approval of the product candidate with respect to effectiveness in the indication studied. If such an agreement is reached, it will be documented and made part of the administrative record, and it will be binding on the FDA except in limited circumstances such as if the FDA identifies a substantial scientific issue essential to determining the safety or effectiveness of the product after clinical studies begin, or if the sponsor fails to follow the protocol that was agreed upon with the FDA. There is no guarantee that a study will ultimately be adequate to support an approval even if the study is subject to an SPA.
Concurrent with clinical trials, companies usually complete additional animal safety studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and the manufacturer must develop methods for testing the quality, purity and potency of the final drugs. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
Assuming successful completion of the required clinical testing, the results of pre-clinical studies and of clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. An NDA must be accompanied by a significant user fee, which is waived for the first NDA submitted by a qualifying small business. In July 2012, the Food and Drug Administration Safety and Innovation Act, or FDASIA, was signed into law. Among other things, FDASIA reauthorizes the FDA’s authority to collect user fees from industry participants to fund reviews of innovator drugs.
The testing and approval process requires substantial time, effort and financial resources. The FDA will review the NDA and may deem it to be inadequate to support approval, and we cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but it typically follows such recommendations.
Before approving an NDA, the FDA inspects the facility or the facilities at which the drug and/or its active pharmaceutical ingredient is manufactured and will not approve the product unless the manufacturing is in compliance with cGMPs. If the FDA evaluates the NDA and the manufacturing facilities are deemed acceptable, the FDA may issue an approval letter, or in some cases a Complete Response Letter. The approval letter authorizes commercial marketing of the drug for specific indications. As a condition of NDA approval, the FDA may require post-marketing testing and surveillance to monitor the drug’s safety or efficacy, or impose other conditions. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or additional pivotal Phase III clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, pre-clinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Alternatively, the FDA could also approve the NDA with a Risk Evaluation and Mitigation Strategy, or REMS, to mitigate risks of the drug, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. Once the FDA approves a drug, the FDA
may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, and surveillance programs to monitor the safety effects of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
Expedited Review and Approval. The FDA has various programs, including Fast Track, priority review and accelerated approval, which are intended to expedite or simplify the process for reviewing drugs, and/or provide for approval on the basis of surrogate endpoints. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will not be shortened. Generally, drugs that may be eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious or life-threatening diseases or conditions and which demonstrate the potential to address an unmet medical need. Priority review is designed to give drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of 10 months. Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track designated drug and expedite review of the application for a drug designated for priority review. Accelerated approval provides an earlier approval of drugs to treat serious or life-threatening diseases or conditions, including a fast track product, upon a determination that the product has an effect on a surrogate endpoint, which is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform post-marketing clinical trials. Pursuant to FDASIA, the FDA is required to issue draft guidance on expedited review and approval programs by July 9, 2013.
Post-Approval Requirements. After a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. In addition, certain changes to an approved product, such as adding new indications, making certain manufacturing changes, or making certain additional labeling claims, are subject to further FDA review and approval. Before a company can market products for additional indications, it must obtain additional approvals from the FDA, typically a new NDA. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. A company cannot be sure that any additional approval for new indications for any product candidate will be approved on a timely basis, or at all.
If post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA are required to (i) report certain adverse reactions to the FDA and maintain pharmacovigilance programs to proactively look for these adverse events; (ii) comply with certain requirements concerning advertising and promotional labeling for their products; and (iii) continue to have quality control and manufacturing procedures conform to cGMPs after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of ongoing compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. We intend to use third-party manufacturers to produce our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product after approval may result in restrictions on a product, manufacturer or holder of an approved NDA, including recall of the product from the market or withdrawal of approval of the NDA for that drug.
Patent Term Restoration and Marketing Exclusivity. Depending upon the timing, duration and specifics of FDA approval of the use of our drugs, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However,
patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the extension must be requested prior to expiration of the patent. The United States Patent and Trademark Office, or USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Data and market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent data exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct, or obtain a right of reference to all of the pre-clinical studies, adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials and approval of foreign countries or economic areas, such as the EU, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
In the European Economic Area, or EEA, which is comprised of the 27 member states of the EU, or Member States, plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of MAs:
| • | The Community MAs—These are issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and are valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, and medicinal products indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA; for products that constitute a significant therapeutic, scientific or technical innovation; or for products that are in the interest of public health in the EU. |
| • | National MAs—These are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, and are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National MA in |
| any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure, an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State. The competent authority of the Reference Member State prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling or packaging proposed by the Reference Member State, the product is subsequently granted a National MA in all the Member States (i.e., in the Reference Member State and the Member States Concerned). |
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA assess the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
As in the United States, it may be possible in foreign countries to obtain a period of market and/or data exclusivity that would have the effect of postponing the entry into the marketplace of a competitor’s generic product. For example, if any of our products receive marketing approval in the EEA, we expect they will benefit from eight years of data exclusivity and ten years of marketing exclusivity. An additional non-cumulative one-year period of marketing exclusivity is possible if during the data exclusivity period (the first eight years of the 10-year marketing exclusivity period), we obtain an authorization for one or more new therapeutic indications that are deemed to bring a significant clinical benefit compared to existing therapies. The data exclusivity period begins on the date of the product’s first marketing authorization in the EU and prevents generics from relying on the marketing authorization holder’s pharmacological, toxicological and clinical data for a period of eight years. After eight years, a generic product application may be submitted and generic companies may rely on the marketing authorization holder’s data. However, a generic cannot launch until two years later (or a total of 10 years after the first marketing authorization in the EU of the innovator product), or three years later (or a total of 11 years after the first marketing authorization in the EU of the innovator product) if the marketing authorization holder obtains marketing authorization for a new indication with significant clinical benefit within the eight-year data exclusivity period. In Japan our products may be eligible for eight years of data exclusivity. There can be no assurance that we will qualify for such regulatory exclusivity, or that such exclusivity will prevent competitors from seeking approval solely on the basis of their own studies.
When conducting clinical trials in the EU, we must adhere to the provisions of the EU Clinical Trials Directive and the laws and regulations of the EU Member States implementing them. These provisions require, among other things, that the prior authorization of an Ethics Committee and the competent Member State authority is obtained before commencing the clinical trial.
Pricing and Reimbursement
In the United States and internationally, sales of products that we market in the future, and our ability to generate revenues on such sales, are dependent, in significant part, on the availability of adequate coverage and reimbursement from third-party payors, such as state and federal governments, managed care providers and private insurance plans. Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and the out-of-pocket obligations of member patients for such products. We may need to conduct pharmacoeconomic studies to demonstrate the cost effectiveness of our products for formulary coverage and reimbursement. Even with such studies, our products may be considered less safe, less effective or less cost-effective than existing products, and third-party payors may not provide coverage and reimbursement for our product candidates, in whole or in part.
In addition, particularly in the U.S. and increasingly in other countries, we are required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our products that are reimbursed by such entities. It is possible that future legislation in the United States and other jurisdictions could be enacted to potentially impact reimbursement rates for the products we are developing and may develop in the future and could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market.
Political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our future business. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, the PPACA, enacted in March 2010, substantially changes the way healthcare is financed by both governmental and private insurers. Among other cost containment measures, PPACA establishes:
| • | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; |
| • | a new Medicare Part D coverage gap discount program, in which pharmaceutical manufacturers who wish to have their drugs covered under Part D must offer discounts to eligible beneficiaries during their coverage gap period, or the donut hole; and |
| • | a new formula that increases the rebates a manufacturer must pay under the Medicaid Drug Rebate Program. |
In the future, there may continue to be additional proposals relating to the reform of the U.S. healthcare system. Future legislation, including the current versions being considered at the federal level in the United States, or regulatory actions implementing recent or future legislation may have a significant effect on our business. Our ability to successfully commercialize products depends in part on the extent to which reimbursement for the costs of our products and related treatments will be available in the United States and worldwide from government health administration authorities, private health insurers and other organizations. The adoption of certain proposals could limit the prices we are able to charge for our products, the amounts of reimbursement available for our products, and limit the acceptance and availability of our products. Therefore, substantial uncertainty exists as to the reimbursement status of newly approved health care products by third-party payors.
Sales and Marketing
The FDA regulates all advertising and promotion activities for products under its jurisdiction prior to and after approval, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to collect additional data or conduct additional pre-clinical studies and clinical trials. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, and often reflect a physician’s belief that the off-label use is the best treatment for the patients. The FDA does not regulate the behavior of physicians in their choice of treatments, but FDA regulations do impose stringent restrictions on manufacturers’ communications regarding off-label uses. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
Outside the United States, our ability to market a product is contingent upon obtaining marketing authorization from the appropriate regulatory authorities. The requirements governing marketing authorization, pricing and reimbursement vary widely from country to country.
We may also be subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of
business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations and very few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented, for payment to third-party payors (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws.
Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal health care programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties also may be imposed upon executive officers and employees, including criminal sanctions against executive officers under the so-called “responsible corporate officer” doctrine, even in situations where the executive officer did not intend to violate the law and was unaware of any wrongdoing. Given the penalties that may be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the government was to allege or convict us or our executive officers of violating these laws, our business could be harmed. In addition, private individuals have the ability to bring similar actions. Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. Further, there are an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state authorities.
Manufacturing
We do not currently have our own manufacturing facilities. We intend to continue to use our financial resources to accelerate development of our drug candidates rather than diverting resources to establish our own manufacturing facilities. We intend to meet our pre-clinical and clinical trial manufacturing requirements by establishing relationships with third-party manufacturers and other service providers to perform these services for us. While our drug candidates were being developed by Pfizer, both the drug substance and drug product were manufactured by third-party contractors. We are currently using the same third-party contractors to manufacture, supply, store and distribute drug supplies for our clinical trials.
Should any of our drug candidates obtain marketing approval, we anticipate establishing relationships with third-party manufacturers and other service providers in connection with commercial production of our products. We have some flexibility in securing other manufacturers to produce our drug candidates; however, our alternatives may be limited due to proprietary technologies or methods used in the manufacture of some of our drug candidates.
Other Laws and Regulatory Processes
We are subject to a variety of financial disclosure and securities trading regulations as a public company in the United States, including laws relating to the oversight activities of the SEC, and we are also subject to the regulations of the New York Stock Exchange, the exchange on which our shares are traded. In addition, the FASB, the SEC, and other bodies that have jurisdiction over the form and content of our accounts, our financial statements and other public disclosure are constantly discussing and interpreting proposals and existing pronouncements designed to ensure that companies best display relevant and transparent information relating to their respective businesses.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, experimental use of animals, and the purchase, storage, movement, import and export, and use and disposal of hazardous or potentially hazardous substances used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights or acquisitions may be subject to national or supranational antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation that might result from future legislation or administrative action cannot accurately be predicted.
Research and Development Expenses
Research and development activities, which include personnel costs, research supplies, clinical and preclinical study costs, are the primary source of our overall expenses. Such expenses related to the research and development of our product candidates totaled $23.6 million for the six months ended June 30, 2012, $0.8 million for the year ended December 31, 2011, and $0.8 million from September 15, 2010 (Former Puma’s date of inception) through December 31, 2011.
Employees
As of June 30, 2012, we had 41 employees, all of whom are full-time employees. We believe our relations with our employees are good. Over the course of the next year, we anticipate hiring up to 10 additional full-time employees devoted to clinical activities, four additional full-time employees for the regulatory and quality assurance function, and two additional full-time employees for general and administrative activities. In addition, we intend to continue to use clinical research organizations and third parties to perform our clinical studies and manufacturing.
Properties
We lease approximately 13,254 square feet of office space in the building located at 10880 Wilshire Boulevard for use as our corporate headquarters. Our lease commenced in December 2011 and terminates in December 2018, with an option to extend for an additional five-year term. We have also signed a lease for additional office space in South San Francisco, California. This lease is for seven years and is expected to commence on or about October 1, 2012. We believe our office space will be adequate to meet current and anticipated future requirements and that additional or substitute space will be available as needed to accommodate any expansions that our operations require.
Legal Proceedings
We are not involved in any pending legal proceedings and are not aware of any threatened or contemplated legal proceedings against us.