Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - CURAXIS PHARMACEUTICAL Corp | Financial_Report.xls |
| EX-31.2 - CERTIFICATION - CURAXIS PHARMACEUTICAL Corp | curx_ex312.htm |
| EX-32.1 - CERTIFICATION - CURAXIS PHARMACEUTICAL Corp | curx_ex321.htm |
| EX-32.2 - CERTIFICATION - CURAXIS PHARMACEUTICAL Corp | curx_ex322.htm |
| EX-31.1 - CERTIFICATION - CURAXIS PHARMACEUTICAL Corp | curx_ex311.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington D.C. 20549
FORM 10-K
þ ANNUAL REPORT UNDER SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended: December 31, 2011
¨ TRANSITION REPORT UNDER SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission file number: 333-150937
Curaxis Pharmaceutical Corporation
(Exact name of registrant as specified in its charter)
|
Nevada
|
26-1919261
|
|
|
(State or other jurisdiction of
incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
1004 Chagford Way
Raleigh, NC 27614
(Address of principal executive offices)
(888) 919-2873
(Registrant’s telephone number, including area code)
Securities registered under Section 12(b) of the Exchange Act: None
Securities registered under Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.:
|
Large accelerated filer
|
¨
|
Non-accelerated filer
|
¨
|
|
|
Accelerated filer
|
¨
|
Smaller reporting company
|
þ
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of registrant’s voting and non-voting common equity held by non-affiliates (as defined by Rule 12b-2 of the Exchange Act) computed by reference to the closing price of such common equity on June 30, 2011, was $28,562,164. As of March 28, 2012, the issuer has one class of common equity, and the number of shares outstanding of such common equity was 83,554,940.
INFORMATION REGARDING FORWARD-LOOKING STATEMENTS
Statements contained in this Annual Report on Form 10-K which are not historical facts are forward-looking statements within the meaning of Section 21E of the U.S. Securities Exchange Act of 1934, as amended. A forward-looking statement may contain words such as “anticipate that,” “believes,” “continue to,” “estimates,” “expects to,” “hopes,” “intends,” “plans,” “to be,” “will be,” “will continue to be,” or similar words. These forward-looking statements include the statements in this Report regarding: our expected financial position and operating results; our business strategy; future developments in our markets and the markets in which we expect to compete; our future ability to fund our operations; our development of new products and relationships; our ability to increase our customer base; the impact of entering new markets; our future cost of revenue, gross margins and net losses; our future restructuring, research and development, sales and marketing, general and administrative, and depreciation and amortization expenses; our future interest expenses; the value of our goodwill and other intangible assets; our future capital expenditures and capital requirements; our financing plans; the outcome of any contingencies and the anticipated impact of changes in applicable accounting rules.
The accuracy of these forward-looking statements may be impacted by a number of business risks and uncertainties that could cause actual results to differ materially from those projected or anticipated. These risks include the risks described in “Item 1A — Risk Factors” below. We do not undertake any obligation to update this forward-looking information, except as required under applicable law.
2
TABLE OF CONTENTS
|
PART I
|
PAGE
|
||||
|
Item 1.
|
Business.
|
4 | |||
|
Item 1A.
|
Risk Factors.
|
25 | |||
|
Item 1B.
|
Unresolved Staff Comments.
|
44 | |||
|
Item 2.
|
Properties.
|
45 | |||
|
Item 3.
|
Legal Proceedings.
|
45 | |||
|
Item 4.
|
Mine Safety Disclosures.
|
45 | |||
|
PART II
|
|||||
|
Item 5.
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
|
46 | |||
|
Item 6.
|
Selected Financial Data.
|
47 | |||
|
Item 7.
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations.
|
47 | |||
|
Item 7A.
|
Quantitative and Qualitative Disclosures about Market Risk.
|
57 | |||
|
Item 8.
|
Financial Statements and Supplementary Data.
|
57 | |||
|
Item 9.
|
Changes In and Disagreements with Accountants on Accounting and Financial Disclosure.
|
58 | |||
|
Item 9A.
|
Controls and Procedures.
|
58 | |||
|
Item 9B.
|
Other Information.
|
58 | |||
|
PART III
|
|||||
|
Item 10.
|
Directors, Executive Officers and Corporate Governance.
|
59 | |||
|
Item 11.
|
Executive Compensation.
|
64 | |||
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
|
68 | |||
|
Item 13.
|
Certain Relationships and Related Transactions, and Director Independence.
|
69 | |||
|
Item 14.
|
Exhibits, Financial Statement Schedules.
|
69 | |||
|
PART IV
|
|||||
|
Item14.
|
Principal Accountant Fees and Services.
|
69 | |||
|
Item15.
|
Exhibits, Financial Statement Schedules.
|
70 | |||
|
SIGNATURES
|
72 | ||||
|
CONSOLIDATED FINANCIAL STATEMENTS
|
73 | ||||
3
PART I
ITEM 1. BUSINESS.
Corporate History
We were incorporated on February 1, 2008, under the laws of the State of Nevada under the name Auto Search Cars, Inc. On February 8, 2010, we entered into a merger agreement (as amended on July 29, 2010, the “Merger Agreement”) by and among Auto Search Cars, Inc., Auto Search Cars Acquisition Corp., a Delaware Corporation (“Acquisition Corp.”), and Curaxis Pharmaceutical Corporation, a corporation incorporated in Delaware on February 27, 2001 (“Curaxis”) (the “Merger”).
On July 29, 2010 (the “Closing Date”), pursuant to the Merger Agreement, (i) the common stock of Curaxis (the “Curaxis Common Stock”) became no longer outstanding and existing, and was cancelled and retired, and (ii) each holder of Curaxis Common Stock ceased to have any rights with respect to their shares of Curaxis Common Stock, except the right to receive shares of our common stock. Each holder of Curaxis Common Stock received one (1) share of Auto Search common stock for each share of Curaxis Common Stock they own immediately prior to completion of the Merger. Each share of the Auto Search Common Stock issued to the Curaxis stockholders pursuant to the Merger Agreement was restricted from trading or resale for a period of one (1) year commencing at the effective date of the Merger. Such restriction did not apply to Auto Search Common Stock issued in exchange for shares of Curaxis Common Stock issued in Curaxis' Bridge Financing (as defined in the Merger Agreement). Auto Search stockholders continued to own their existing shares, which were not affected by the Merger.
On the Closing Date, Curaxis and Acquisition Corp. merged with and into one another, with the surviving corporation being Curaxis. Simultaneously with the filing of a Certificate of Merger with the State of Delaware, Curaxis amended its Certificate of Incorporation in order to change its name to Curaxis Pharma Corp. On the Closing Date, Curaxis Pharma Corp. became our subsidiary.
On July 30, 2010, we entered into an agreement and plan of merger with Curaxis Pharmaceutical Corporation, a Nevada corporation formed solely for the purpose of a name change (the “Short-Form Merger”). Pursuant to the Short-Form Merger, we changed our name to Curaxis Pharmaceutical Corporation.
As the result of the Merger, we acquired Curaxis Pharmaceutical Corporation. Curaxis was incorporated on February 27, 2001, under the laws of the State of Delaware.
In connection with the Merger dated as of July 29, 2010 and described in our Form 8-K filed on June 30, 2010, the Company, on August 11, 2011, entered into an Agreement and Plan of Merger (the “Curaxis Merger Agreement”) by and between the Company and Curaxis Pharma Corporation, a Delaware corporation and a wholly owned subsidiary of the Company (“Pharma”), pursuant to which Pharma merged with and into the Company and the Company is the surviving corporation.
Curaxis is an emerging specialty pharmaceutical company with a hormone drug product candidate for the treatment of Alzheimer’s disease and multiple cancers. Its therapeutic platform is based on the hypothesis that many diseases of aging may be caused by age-related changes in the function of the hypothalamic-pituitary-gonadal (HPG) axis. The HPG axis is a hormonal endocrine feedback loop that controls development, reproduction and aging in animals. This drug development platform is built on the premise that hormones associated with this feedback loop are beneficial early in life, when they promote growth and development, but are harmful later in life when the mechanism for feedback is compromised, thereby leading to disease processes, including pathologies associated with Alzheimer’s disease and various cancers. Curaxis believed that its discovery of similar hormonal signaling mechanisms at the cellular level in brain tissue from Alzheimer’s patients and in multiple tumors would enable it to develop significant new treatments for Alzheimer’s disease as well as many cancers.
Curaxis is a development stage company and has not yet (i) received FDA approval for any of its products; and (ii) generated any commercial revenue through the sale of its products.
4
Curaxis’ most advanced product is VP4896, a proprietary, small, biodegradable implant that is comprised of leuprolide acetate and a polymer to be used for the treatment of Alzheimer’s disease. The Company had previously conducted several clinical trials for this indication, the most recent of which it were forced to terminate in 2006 due to financial constraints. Since terminating those trials, it had been unable to advance the clinical development of its Alzheimer’s disease candidate due to a lack of financial resources. The Board and the executive management team is focused on attracting bridge financing for ongoing operational support and has a strategy in place to identify and attract a strategic partner to absorb a portion of the clinical trial cost and processes to advance the Company’s lead drug candidate into a Phase IIb or registration clinical trial. There is no guarantee that the Company will be successful in its efforts.
Our Alzheimer’s Disease Program
Leuprolide acetate, a hormone analogue of naturally occurring gonadotropin-releasing hormone (GnRH), has been widely used over the past twenty years for the treatment of a number of hormone-related disorders, most notably prostate cancer and endometriosis and precocious puberty, and has a well-established safety profile. Curaxis has conducted extensive pre-clinical and clinical studies exploring the use of leuprolide acetate for the treatment of Alzheimer’s disease in mild-to-moderate patients. The results to date are encouraging and point especially to a potentially significant new treatment for women with Alzheimer’s disease. Women represent approximately two-thirds of Alzheimer’s patients. However, our ability to conduct further clinical development of Curaxis’ Alzheimer’s disease candidate will be dependent on our ability to raise the funds required to pay for such further clinical development activities.
Men present greater challenges in the use of leuprolide to treat Alzheimer’s disease because leuprolide suppresses their production of testosterone, which could necessitate patient self-administration of supplemental testosterone and which can lead to wide swings in testosterone blood serum levels. Therefore, in the near-term, we plan to concentrate our development efforts on the use of leuprolide acetate to treat women, although we will continue our efforts to better understand mechanisms that might lead to optimum outcomes in men.
Alzheimer’s Disease
Background and Demographics
Alzheimer’s disease is named after Dr. Alois Alzheimer, a German physician, who first described the disease in 1906. Alzheimer’s disease (“AD”) is a progressive, degenerative, and ultimately terminal brain disorder that gradually destroys a person’s memory and ability to learn, reason, make judgments, communicate and carry out daily activities. Commercially marketed AD drug medications work to modestly delay the onset of symptoms and slightly improve cognition. There is currently no treatment that stops or materially slows the progression of AD. As a result, AD is one of the world’s largest unmet medical needs.
In 2009, the global market (consisting of US, France, Germany, Italy, Spain, UK and Japan) for AD drug medications generated revenues of over $8.0 billion with the US market as the single-largest market, accounting for over half of all global revenues. According to BCC Research, AD market value is expected to increase to $9.4 billion by 2014. It is expected that the AD drug market will continue to experience strong growth for the foreseeable future driven by increasing AD prevalence, physician and patient awareness, improved diagnostic tools, increasing prices, the introduction of new products, and expanded indications for existing products. In addition to all of these drivers, the aging of the world’s population is the single greatest contributor to the need for more and better medications.
AD Severity
As a progressive, degenerating disease, the AD population has been segmented into three categories of severity of illness - mild, moderate or severe. However, there is no consensus as to distinct cut-off points from one category to the next as symptoms can be seen at any point along the disease progression. The two general categories used for regulatory approval of AD treatments are Mild to Moderate and Moderate to Severe. These categories correspond with neuropsychological assessments such as the Mini Mental Status Examination (MMSE) and the Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-Cog), both standard measures in clinical trials of potential AD treatments. Curaxis has focused development of VP4896 on patients with mild to moderate AD.
5
AD Prevalence
According to the Alzheimer’s Association, an estimated 5.4 million Americans have AD in 2011. This number includes 5.2 million people age 65 and older and at least 200,000 individuals younger than 65 with early-onset Alzheimer’s. The Alzheimer’s Association estimates there are approximately 500,000 Americans younger than 65 years of age that have Alzheimer’s or some other form of dementia, and has suggested that at least 40 to 50 percent of them are likely to have AD.
By age group, the proportion and number of the 5.4 million Americans age 65 and over with AD breaks down as follows:
|
●
|
Age 65-74: 324,000 (6%)
|
|
●
|
Age 75-84: 2,430,000 (45%)
|
|
●
|
Age 85+: 2,430,000 (45%)
|
|
●
|
13 percent, or one in eight, persons age 65 and over have AD.
|
|
●
|
Nearly half of persons over age 85 have AD.
|
|
●
|
65% of persons with AD are female
|
|
●
|
Every 69 seconds, someone in America develops AD; by mid-century, someone will develop Alzheimer’s every 33 seconds.
|
AD is the sixth leading cause of death across all ages in the United States. It is the fifth leading cause of death for those aged ≥ 65 years.
These figures reflect the total number of Americans estimated to have Alzheimer’s, whether or not they have ever been diagnosed with the disease. Many people with AD and other dementias have not been diagnosed, and while some may have been diagnosed, their diagnosis may have been omitted from their medical record. In one study of patients aged 65, assessing seven urban, racially diverse, primary care practices in Indianapolis, less than one fifth of those with Alzheimer’s or another dementia had a diagnosis of the condition in the patient’s medical record. It is clear that a new therapy with enhanced activity would increase the number of diagnoses and those who are eligible for treatment.
The Cost of Alzheimer's Disease in the United States
According to revised figures by the Alzheimer's Association:
|
●
|
By 2050, the number of individuals in this country 65 and older with Alzheimer's could range from 11 million to 16 million, without new and more effective therapies.
|
|
●
|
Medicare payments for services to beneficiaries aged ≥ 65 with AD and other dementias are almost 3 times higher than for beneficiaries without these conditions. Total payments in 2011 for healthcare care, long-term care, and hospice services for people aged ≥ 65 with AD and other dementias are expected to be $183 billion (not including the contributions of unpaid caregivers).
|
|
●
|
In 2010, nearly 15 million family and other unpaid caregivers provided an estimated 17 billion hours of care to people with AD and other dementias, a contribution valued at more than $202 billion.
|
6
|
●
|
Families who hire in-home caregivers face annual costs of up to $200,000, while families that decide to provide care themselves spend as many as 70 hours a week, severely compromising their own earning potential and costing employers an estimated $61 billion annually in lost productivity.
|
|
●
|
Caring for a person with AD or other dementia is very difficult, and therefore many family and other unpaid caregivers experience high levels of emotional stress and depression. The physical and emotional impact of caregiving on AD and other dementia caregivers is estimated to result in $7.9 billion in increased healthcare cost in the United States.
|
Current Scientific Theories
Curaxis’ LH Hypothesis
Based upon Curaxis’ pre-clinical and clinical studies, we believe that inappropriately elevated levels of pituitary hormones produced in response to normal aging may be important in promoting the development of Alzheimer’s disease. At the time of menopause in females, when estrogen production by the ovaries falls precipitously, gonadotropins (luteinizing hormone (LH) and follicle-stimulating hormone (FSH)) become elevated. In aging males, testosterone production by the testes drops about one percent per year, leading to a more gradual increase in gonadotropins over time. Our preclinical research suggests that LH, which is released by the pituitary gland, is associated with many of the pathologies currently being researched in Alzheimer’s disease, including amyloid beta deposition which leads to formation of plaques in the brain tissue, tau phosphorylation (addition of phosphate groups to tau protein) which leads to formation of neurofibrillary tangles inside neurons, abnormal cell division, oxidative stress, and inflammation. We believe that elevated levels of LH that are known to occur with aging contribute to many of these pathological changes, ultimately resulting in cognitive decline and Alzheimer’s disease. We refer to the hypothesis that links LH to the development of Alzheimer’s disease as the LH Hypothesis.
Our lead product candidate, VP4896, is designed to dramatically reduce levels of LH in the bloodstream and in brain tissue through administration of a long-term, controlled release dose of leuprolide acetate, which suppresses LH production. We hold an issued United States patent with claims directed to the treatment of Alzheimer’s disease by administering any agent, including leuprolide acetate that reduces or eliminates serum levels of gonadotropins. In Curaxis’ Phase I trial, trial 105, its proprietary dosage form of leuprolide acetate, VP4896, has been demonstrated to dramatically suppress or eliminate LH levels in the serum of healthy male and female subjects.
The following summarizes the prominent hypotheses regarding the cause of Alzheimer’s disease and provides an overview of evidence we have that links the LH Hypothesis to each of these hypotheses.
Beta Amyloid Hypothesis
There are several hypotheses regarding the cause of Alzheimer’s disease, the predominant one being the beta amyloid hypothesis. The assumption behind this hypothesis is that amyloid beta protein, which makes up the plaques present in the brains of Alzheimer’s disease patients, is toxic and is the causative agent of the disease. The generally accepted view is that inhibiting the production of, and enhancing the clearance of, amyloid beta protein would reduce the formation of and possibly eliminate amyloid plaques that might be toxic to neurons, therefore preventing or treating Alzheimer’s disease. Based on this hypothesis, many companies have designed therapies to suppress or eliminate amyloid beta protein in order to affect the rate of progression of Alzheimer’s disease. Research based on the beta amyloid hypothesis has been ongoing for two decades without yielding any approved therapies to date.
While we do not specifically target amyloid beta elimination, we have evidence that links leuprolide acetate treatment in animal models to significant reductions in amyloid beta concentrations, and more importantly to a decreased rate of cognitive decline.
7
In a paper published in the Journal of Biological Chemistry in May 2004, Curaxis showed that leuprolide treatment of normal mice significantly reduced the concentrations of brain amyloid beta (1-42) by 71% after four weeks, and amyloid beta (1-40) by 40% after eight weeks. Amyloid beta (1-42) and (1-40) are different segments of amyloid present in the brain, with amyloid beta (1-42) representing a more toxic form of the protein (Luteinizing hormone, a reproductive regulator that modulates the processing of amyloid-beta precursor protein and amyloid-beta deposition).
In a paper published in Biochemica Biphysica Acta in April 2006, Curaxis showed that leuprolide, when given to mice with Alzheimer’s-like disease, significantly reduced concentrations of brain amyloid beta, and resulted in stabilized, and in some cases, improved cognition when leuprolide treated mice were compared to those which received placebo (Luteinizing hormone modulates cognition and amyloid-ß deposition in Alzheimer APP transgenic mice).
Tau Hypothesis
Another prominent theory is the Tau Hypothesis. Tau proteins contribute to the internal structural architecture of neurons. In Alzheimer’s disease, tau proteins gain unusually high levels of phosphate groups (become phosphorylated) and a portion of the protein is apparently sliced off to create protein fragments. These altered tau protein fragments lose their ability to function normally and stick together in structures called neurofibrillary tangles which form within the neuron and are thought to be neurotoxic. These tangles, together with plaques formed by amyloid beta, are the primary hallmarks of Alzheimer’s disease found in the brains of Alzheimer’s disease patients when autopsied.
While we do not target the reduction of neurofibrillary tangles, we have evidence that LH, which is suppressed by VP4896, actively and rapidly induces the phosphorylation of tau proteins and may therefore be one of the causative agents for tangle formation. We have evidence from laboratory experiments that LH treatment of neuroblastoma cell lines results in increased phosphorylation of tau protein, whereas leuprolide acetate reduces the levels and slows the kinetics of tau phosphorylation.
Abnormal Cell Division Hypothesis
Another hypothesis, called the cell cycle hypothesis, proposes that neurological and biochemical changes associated with Alzheimer’s disease are caused by the abnormal re-entry of brain cells into the cell division cycle, a process by which one cell replicates itself and divides into two cells. In general, it is thought that adult brain cells have lost the ability to successfully divide. Thus, this hypothesis suggests that when adult brain cells are stimulated to divide, the neurological changes seen in Alzheimer’s disease result. The proponents of this hypothesis believe that instead of successfully completing cell division, these brain cells die, and this brain cell death is believed to produce the clinical deficits observed in Alzheimer’s disease patients. We have evidence from laboratory experiments that LH treatment of glioblastoma and neuroblastoma cells in culture causes increased cell division that can subsequently be prevented by concomitant treatment with leuprolide acetate.
Oxidative Stress
Oxidative stress results from the production of toxic chemical by-products that arise from normal cellular metabolism and has also been implicated and studied as a potential cause of Alzheimer’s disease. These toxic by-products may cause direct damage to cells or may induce genetic mutations or DNA damage. With respect to the LH hypothesis, treatment of a neuronal cell line with LH in vitro has produced preliminary data that suggests that LH may inhibit enzymes that are essential to the body’s management of oxidative stress. In other words, LH may enhance oxidative stress and the elimination of LH might reduce the impact of oxidative stress on cells.
Inflammation
Inflammation, which is a normal physiological reaction to infection, can sometimes produce damage. Autoimmune diseases such as rheumatoid arthritis and multiple sclerosis are examples of undesirable and uncontrolled inflammatory events. Inflammation has been implicated as a possible cause of Alzheimer’s disease and it has been suggested that reducing chronic inflammation might provide benefit. Several anti-inflammatory therapies, including drugs as common as Ibuprofen, have been clinically tested for their ability to benefit Alzheimer’s patients. To date, anti-inflammatory therapies have not proven successful. With respect to the LH hypothesis, laboratory data suggests that LH promotes the production of growth factors (cytokines) that enhance or magnify the inflammatory response and LH may, therefore, contribute to Alzheimer’s disease by exacerbating inflammatory processes in the brain.
8
LH Hypothesis – The Human Reproductive Hormone Feedback Loop
Most biochemical processes in the body are tightly regulated and are subject to both positive and negative feedback. Positive feedback promotes a certain reaction, such as the synthesis of a hormone, while negative feedback inhibits a reaction or event. The concentration of certain hormones secreted by a region of the brain called the hypothalamus, the pituitary gland and the gonads is regulated by a feedback loop that has both positive and negative feedback components. The loop is initiated when the hypothalamus releases gonadotropin-releasing hormone, or GnRH. GnRH then stimulates the pituitary to secrete the two gonadotropins—LH and FSH. The gonadotropins bind to receptors on the gonads, the ovaries in females and the testicles in males, and stimulate the gonads to produce the sex steroid hormones, estrogen and testosterone.
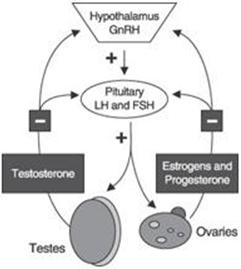
Once the hypothalamus senses that the sex steroid hormones are at an acceptable level, it reduces the release of GnRH. The reduced level of GnRH provides feedback to the pituitary gland to reduce the secretion of gonadotropins, resulting in reduced gonadotropin levels. Reduced gonadotropin levels then provide feedback to the gonads to reduce the production of the sex steroid hormones. Once the hypothalamus senses the sex steroid hormones dropping below a particular level, the hypothalamus increases the release of GnRH, which re-initiates the hormonal feedback loop and the production of the two gonadotropins.
Our Scientific Approach
Our scientific approach is based on the observation that many diseases of aging may be caused by the age-related changes in levels of reproductive hormones that are secreted by the hypothalamus, the pituitary gland and the gonads. This approach is built on the premise that these hormones are beneficial early in life, because they regulate and promote development and growth through cell division and differentiation in order to achieve reproduction, but are harmful later in life because they become unregulated and cause abnormal processes including pathologies associated with AD. We believe that this change in hormone levels is a primary cause of many age-related diseases, including Alzheimer’s disease and various cancers.
We believe that the gonadotropin LH is an important and pivotal cause of Alzheimer’s disease. Our research suggests that LH serves as the catalyst that potentially leads to increased production of amyloid beta protein, drives changes in tau protein that may lead to formation of neurofibrillary tangles, and possibly impacts other factors such as oxidative stress and inflammation which, in total, lead to the cognitive decline associated with Alzheimer’s disease. We base these beliefs on both experimental evidence and scientific observations, principally resulting from Curaxis’ work and the work of our consultants.
9
Preclinical Support for the Combined Treatment of Leuprolide Acetate and Acetylcholinesterase Inhibitors in Alzheimer’s Disease
Curaxis’ Phase II proof of concept study in female patients with mild-to-moderate Alzheimer’s disease demonstrated efficacy of 22.5 mg Lupron Depot™ when administered together with acetylcholinesterase inhibitors (AChEIs), as assessed by the ADAS-cog, ADCS-CGIC and ADCS-ADL outcome measures, when compared to patients receiving AChEIs alone. Administration of AChEIs is the current standard of care for Alzheimer’s patients. AChEIs inhibit the enzymatic activity of acetylcholinesterase, thereby increasing levels of the neurotransmitter acetylcholine. As the positive effect of leuprolide acetate plus AChEIs was an unexpected clinical result, subsequent pre-clinical work has focused on understanding the potential interaction between the LH and acetylcholine biological signaling pathways in neurons. In an attempt to better understand the mechanism of action or potential functional overlap between GnRH analogues and AChEIs, cell culture studies were performed to explore the role of AChEIs in modulation of the hypothalamic-pituitary-gonadal (HPG) axis of hormones and receptors, using brain cancer cells. Curaxis’ findings in these studies demonstrate that an AChEI can modulate HPG axis genes and may support the clinical findings that the two drugs together provide more benefit than either drug alone. A patent application detailing the use of GnRH analogues in combination with AChEIs or NMDA receptor antagonists to treat Alzheimer’s disease and mild cognitive impairment is pending in the United States, Europe and other countries.
Current Treatments for Alzheimer’s Disease
There are three marketed acetylcholinesterase inhibitors (Aricept, Razadyne, and Exelon) administered as oral medications or by absorption through the skin in a patch. This class of drugs accounted for approximately $5 billion in US sales in 2010. Namenda is an NMDA receptor antagonist that had estimated US sales of $1.5 billion in 2010. While many drugs are in development currently, the majority of Phase II/III AD clinical trials have failed to demonstrate consistent and sustainable efficacy in AD patients. According to www.clinicaltrials.gov, there are 238 open interventional studies in AD as of October 2011 (representing all phases of development).
Our Drug Candidate: Memryte
Memryte (generic name: VP4896) is a proprietary, small, biodegradable implant comprised of leuprolide acetate and a polymer (synthetic compound). Each implant is approximately 1.5 millimeters in diameter and 3.0 centimeters in length. Memryte is designed to provide controlled, long-term, sustained release of leuprolide acetate. The polymer in Memryte is similar to the material in resorbable surgical stitches and degrades in the body. As a result, and in contrast to some other types of implantable drug delivery devices, surgical removal is not required. Memryte implants are inserted subcutaneously through a needle in the fleshy region on the side of the patient’s abdomen. The procedure is conducted in a physician’s office using a local anesthetic and takes approximately ten minutes. Administration of leuprolide acetate using the Memryte implant has been demonstrated in our Phase I ALADDIN 105 trial to minimize an initial dosage peak and provide a steady leuprolide acetate level throughout the two-month dosing period.
Curaxis administered an injectable formulation of leuprolide acetate in its two Phase II dose-ranging clinical trials for Alzheimer’s disease while it was developing Memryte. Leuprolide acetate has been marketed for almost two decades, primarily as a treatment for advanced prostate cancer. It is also approved for use in other hormone-related conditions, such as endometriosis and anemia caused by uterine fibroids in women, and precocious puberty in children. It is administered for these indications by intramuscular injection. The safety profile of leuprolide acetate has been well established over many years. The most common side effects are similar to those seen with menopause and surgical castration, such as hot flashes and osteoporosis. Since all of the women who enter our trials are post-menopausal, these side effects should be nominal. As a raw material, leuprolide acetate is available from a number of manufacturers.
10
Clinical Trials
Curaxis has conducted several trials in our clinical program to evaluate leuprolide acetate for the treatment of mild to moderate AD. We call our clinical trial program “Antigonadotropin-Leuprolide in Alzheimer’s Disease Drug INvestigation,” for which we use the acronym ALADDIN. Our clinical program includes:
|
●
|
A completed Phase I safety and pharmacokinetic study of VP4896, a proprietary, sustained-release implant formulation.
|
|
●
|
Two completed Phase II clinical trials, which used an injectable formulation of leuprolide acetate.
|
|
●
|
The first of two planned Phase III trials was fully enrolled with 612 subjects in August 2006. Due to funding requirements and constraints, the trial was discontinued early, in late 2006.
|
These clinical trials are summarized in the following table:
|
Trial Name / Clinical Phase
|
Enrollment Criteria
|
Number of Clinical Sites
|
Number of Participants
|
Trial Duration
|
Status
|
|
ALADDIN 103/ Phase II
|
Women aged 65 years or older with mild to moderate Alzheimer’s disease; patients were allowed, but not required, to receive AChEIs during and prior to trial
|
5 in the United States
|
108
|
48 weeks
|
Completed
|
|
ALADDIN 104 / Phase II
|
Men aged 65 years or older with mild to moderate Alzheimer’s disease; patients were allowed, but not required, to receive AChEIs during and prior to trial
|
17 in the United States
|
119
|
48 weeks
|
Completed
|
|
ALADDIN 105 / Phase I
|
Healthy women aged 45 to 70 years and men aged 50 to 70 years
|
1 in the United States
|
50
|
24 weeks
|
Completed
|
|
ALADDIN 301 / Phase III truncated
|
Men and women aged 60 years or older with mild to moderate Alzheimer’s disease; patients were required to receive AChEIs during and for at least 120 days prior to trial
|
62 in the United States and Canada
|
612 recruited
|
56 weeks
|
Discontinued on October 18, 2006 as a result of financial constraints
|
We employed a variety of clinically validated and FDA accepted efficacy measurements in our AD clinical trials. These include:
|
●
|
The Alzheimer’s Disease Assessment Scale-Cognitive Subscale, or ADAS-cog. ADAS-cog is a standard test of memory and cognition that is designed to measure changes in AD subjects that might occur in response to a pharmacological intervention, or the action of a given drug. An increasing score on this measure indicates cognitive decline, while a decreasing score indicates cognitive improvement. This is the most widely used and accepted test in clinical trials of AD subjects, and is the most widely used primary clinical endpoint.
|
|
●
|
The Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change, or ADCS-CGIC. ADCS-CGIC is a global measure of a subject’s change in condition from baseline in AD. The score is based on information gained from interviews with the subject and the subject’s caregiver. This test is usually included in clinical trials as a secondary endpoint.
|
|
●
|
Alzheimer’s Disease Cooperative Study-Activities of Daily Living Inventory, or ADCS-ADL. ADCS-ADL is a comprehensive rating scale designed to measure a subject’s capacity to perform activities of daily living, including ability to eat, dress, bathe, telephone, travel, shop and perform other household chores. The score is based on interviews of the subject’s caregiver. An increasing score on this measure indicates additional capacity to perform activities of daily living, while a decreasing score indicates less capacity. This test is usually included as a secondary endpoint.
|
11
The ADAS-cog, the ADCS-CGIC and the ADCS-ADL are recognized by the FDA and regulatory agencies outside the United States as the most commonly used assessments for endpoints in Alzheimer’s disease clinical trials. To minimize the side effects associated with the loss of testosterone due to leuprolide acetate therapy, male subjects who received leuprolide acetate in these clinical trials also received testosterone on a daily basis in the form of a gel applied to the skin. To prevent the patients or the study personnel from learning of the treatment assignment, male subjects randomly assigned to placebo received a placebo gel on a daily basis. Since women with Alzheimer’s disease are post-menopausal and are no longer producing much estrogen, side effects associated with the loss of estrogen are minimal and do not require hormonal supplementation.
The intent-to-treat population in these trials consists of participants who received at least one dose of randomized drug and who had at least one assessment beyond baseline of at least one primary efficacy variable. For data analysis purposes, an intent-to-treat analysis is based on the initial treatment intent, not on the treatment eventually administered, and is intended to reduce data artifacts. For patients who do not complete the trial, the last assessment taken is carried forward to each of the subsequent assessment time points and is used in the final analysis data set. These data points are referred to as last observations carried forward (LOCF).
In general, the results of Curaxis’ Phase II dose-ranging studies in women were encouraging and pointed to a potentially significant new treatment for women with Alzheimer’s disease. The results in men have been less encouraging, in part because a number of men on placebo unexpectedly performed at or above baseline during the trials. Therefore, for the short-and medium-term, we plan to concentrate our development efforts on the use of leuprolide acetate to treat women, although we will continue our efforts to better understand mechanisms that might lead to improved outcomes in men.
Phase II / ALADDIN I
Curaxis has completed a randomized, double-blind, placebo-controlled, dose-ranging, 48-week, Phase II clinical trial to assess the efficacy and safety of an injectable formulation of leuprolide acetate on cognitive and global function in women with mild to moderate Alzheimer’s disease. We call this clinical trial ALADDIN I or trial 103. The trial was conducted at five investigative study sites in the United States. Women aged 65 or older with mild to moderate Alzheimer’s disease were eligible to participate in the trial. Patients were allowed to receive AChEIs during the trial if they began taking this medication at least 90 days prior to the trial and continued a stable dose throughout the trial.
A total of 109 women were enrolled in this study, 108 of which were included in the intent-to-treat population and assigned to one of three groups comprised of 36 participants each:
|
●
|
a low dose leuprolide acetate group; 11.25 mg of leuprolide every 12 weeks;
|
|
●
|
a high dose leuprolide acetate group; 22.5 mg of leuprolide every 12 weeks; and
|
|
●
|
a placebo group.
|
Each participant was administered an injection of leuprolide acetate or placebo (saline) once every 12 weeks during the trial. The primary efficacy endpoints of the trial were patient scores on the ADAS-cog and the ADCS-CGIC at 48 weeks compared to baseline. There were various secondary efficacy endpoints, including patient scores on the ADCS-ADL at 48 weeks compared to baseline. There was a trend at week 48 in favor of the high dose leuprolide acetate group in this Phase II trial, indicating a relative stabilization of the disease compared to the placebo group. However, Curaxis did not achieve the primary efficacy endpoints or any of the secondary efficacy endpoints in this trial with statistical significance.
12
In accordance with the written statistical analysis plan Curaxis had previously adopted for ALADDIN I, it also performed an analysis of 78 patients in the intent-to-treat population who were taking AChEIs, comparing results for the group of 24 patients treated with AChEIs plus the high dose of leuprolide acetate used in the study and the group of 28 patients treated with AChEIs plus the 11.25 mg dose of leuprolide acetate against the results for a group of 26 patients who were treated with AChEIs and received placebo in the study. The results for the group that received an AChEI plus the 11.25 mg dose of leuprolide acetate were not statistically significantly different from the results for the group that received an AChEI plus placebo.
As described below, the group that received the 22.5 mg dose of leuprolide acetate plus an AChEI demonstrated a benefit in comparison to the group that received an AChEI plus placebo. In addition, on each of the seven occasions during the 48-week study at which Curaxis assessed these two groups, the mean score of the 22.5 mg dose of leuprolide acetate plus AChEI group was more favorable than the mean score of the placebo plus AChEI group on each of the ADAS-cog, ADCS-CGIC and ADCS-ADL measures.
Statistical significance is measured by a p-value, which is a mathematical calculation used to determine the likelihood that the measured result was obtained by chance. A p-value of 0.05 means that the probability that the result occurred by chance is one in twenty. A low p-value indicates a greater likelihood that the observed result did not occur by chance, and therefore implies greater statistical significance.
For purposes of this subgroup analysis of the results of our ALADDIN I trial, Curaxis calculated p-values in two different ways. First it calculated unadjusted p-values, which indicate statistical significance as if this subgroup analysis had been a primary efficacy endpoint. However, because it performed several comparisons, it was required to adjust the p-values of the results to account for multiple statistical comparisons. This was done by using what is referred to as the Bonferroni correction, which applies an estimated statistical penalty to account for the number of comparisons made.
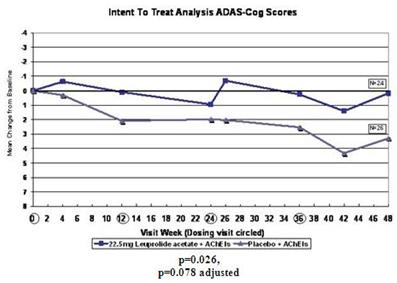
In this subgroup analysis, the mean ADAS-cog score in the group receiving the 22.5 mg dose of leuprolide acetate and an AChEI worsened by 0.18 points at week 48 from baseline compared to a mean worsening of 3.30 points in the group receiving placebo and an AChEI. The p-value for this difference was 0.026 on an unadjusted basis and 0.078 on an adjusted basis.
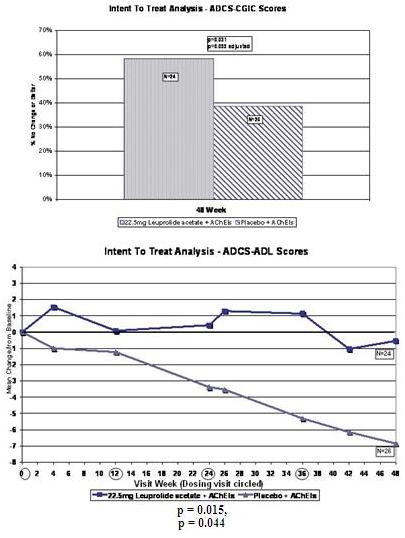
In the ADCS-CGIC analysis, 58% of the subgroup receiving the 22.5 mg dose of leuprolide acetate and an AChEI scored no change or better at week 48 in comparison with baseline versus 38% of the subgroup receiving placebo and an AChEI. The p-value for this difference was 0.031 on an unadjusted basis and 0.093 on an adjusted basis.
The mean ADCS-ADL score in the subgroup receiving the 22.5 mg dose of leuprolide acetate and an AChEI declined 0.54 points at week 48 from baseline compared to a mean decline of 6.85 points in the subgroup receiving placebo and an AChEI. The p-value for this difference was 0.015 on an unadjusted basis and 0.044 on an adjusted basis.
13
The following graphs summarize the results of this subgroup analysis.
14
ALADDIN 301
In August 2006, Curaxis completed enrollment in ALADDIN 301, the first of its two randomized, double-blind, placebo-controlled, 56-week, Phase III clinical trials of the VP4896 implant for the treatment of mild to moderate Alzheimer’s disease as adjunctive (combination) therapy to AChEIs. Men and women aged 60 or older who had been diagnosed with mild to moderate Alzheimer’s disease were eligible to participate in the trials. One of the trial inclusion criteria was that all subjects shall have been on an AChEI for at least 120 days prior to baseline and continue to receive that AChEI throughout the trial.
The protocol provided for study participants to be randomly assigned to VP4896 or placebo in a ratio of three to two (3:2). VP4896 or placebo was administered every eight weeks in the form of a subcutaneous implant. Each participant received two implants on each occasion. The dosage level from the two implants was approximately equal to the high dose of leuprolide acetate being administered in ALADDIN II and 150% of the high dose in ALADDIN I. Curaxis method of administration in these trials was different from its ALADDIN I and ALADDIN II clinical trials, in which leuprolide acetate was administered by injection at 12-week intervals over a 48-week period.
The primary evaluation of the efficacy of VP4896 in the treatment of mild to moderate Alzheimer’s disease was measured by the difference from placebo in the scores on ADAS-cog and ADCS-CGIC compared to baseline. Secondary efficacy endpoints in each trial were to assess the efficacy of VP4896 compared to placebo as measured by change from baseline in a variety of other commonly used Alzheimer’s disease measurements, including ADCS-ADL, at 50 weeks.
In October 2006, Curaxis discontinued this trial due to financial constraints and converted it to a Phase II trial. At the time this trial was terminated, approximately 625 patients had been enrolled at 62 sites in the United States and Canada. After this discontinuation, Curaxis had approximately 369 patients who had received their 24 week assessment, approximately 193 patients who had received their 32/34 week assessment, and approximately 89 patients who had received their 40 week assessment.
In May 2007, in accordance with the written statistical analysis plan Curaxis had previously adopted for the trial, it completed an analysis of the results of ALADDIN 301 for (1) men and women as a single group and (2) for women separately, for participants who reached their 24, 32/34 or 40 week assessments. The analysis of the combined group of men and women showed no significant differences in favor of the group that received VP4896, in part because a number of men on placebo unexpectedly performed at or above baseline during the trials. However, the results in women who received VP4896 supported the efficacy signal in women that was demonstrated in ALADDIN 103. The following graphs reflect the results of the analysis for women in ALADDIN 301.
15
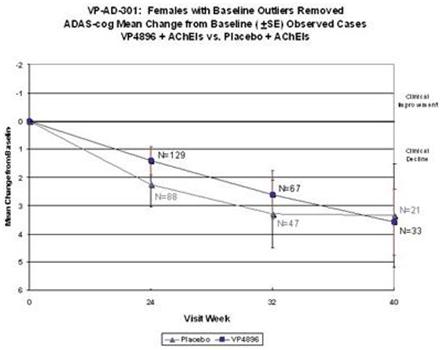
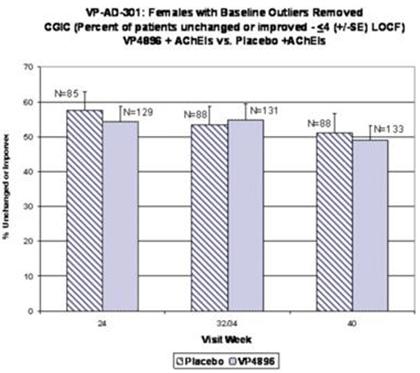
Female patients with ADAS-cog baseline scores that were considered to be extreme observations (too low or too high) were removed from the analysis (10 patients were removed from both groups; placebo and VP4896). Average (mean) ADAS-cog scores (changes from baseline scores) were plotted and the numbers of patients included at each time point are given. The graph above includes the observed cases (patients that actually had an assessment at the specified visits) at 24, 32/34 and 40 weeks. VP4896-treated females demonstrated better ADAS-cog scores at 24 and 32/34 weeks, compared to placebo females.
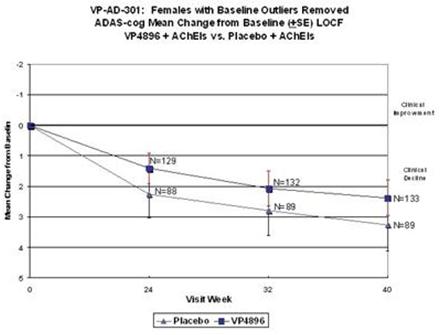
Female patients with ADAS-cog baseline scores that were considered to be extreme observations (too low or too high) were removed from the analysis (10 patients were removed from both groups; placebo and VP4896). Average (mean) ADAS-cog scores (changes from baseline scores) were plotted and the numbers of patients included at each time point are given. The graph above includes the last-observation-carried-forward (LOCF) cases at 24, 32/34 and 40 weeks, i.e., all of the females completed through week 24 and if they discontinued after weeks 24 or 32/34, their scores are included in the 40 week data point. VP4896-treated females demonstrated better ADAS-cog scores at 24, 32/34 and 40 weeks, compared to placebo females.
16
Female patients with ADAS-cog baseline scores that were considered to be extreme observations (too low or too high) were removed from the analysis (10 patients were removed from both groups; placebo and VP4896).
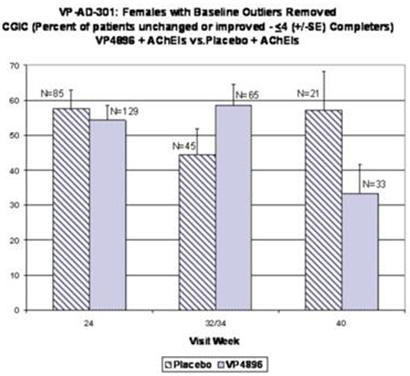
Female patients with ADAS-cog baseline scores that were considered to be extreme observations (too low or too high) were removed from the analysis (10 patients were removed from both groups; placebo and VP4896). Average (mean) clinical global impression of change (CGIC) scores were plotted and the numbers of patients included at each time point are given. The bars represent the percent of females that were scored as unchanged or better at each time point. The graph above includes the last-observation-carried-forward (LOCF) cases at 24, 32/34 and 40 weeks, i.e., all of the females completed through week 24 and if they discontinued after weeks 24 or 32/34, their scores are included in the 40 week data point. VP4896-treated females demonstrated better CGIC scores at 32/34 weeks, compared to placebo females.
17
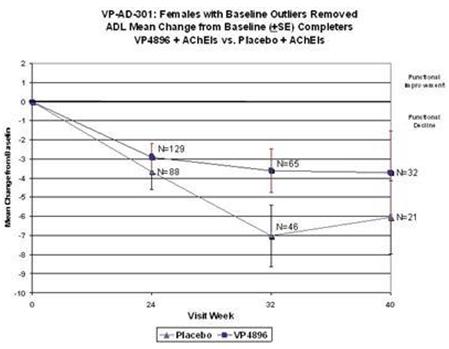
Female patients with ADAS-cog baseline scores that were considered to be extreme observations (too low or too high) were removed from the analysis (10 patients were removed from both groups; placebo and VP4896). Average (mean) ADCS-ADL scores (changes from baseline scores) were plotted and the numbers of patients included at each time point are given. The graph above includes the observed cases (patients that actually had assessments at the specified visits) at 24, 32/34 and 40 weeks. VP4896-treated females demonstrated better ADL scores at 24, 32/34 and 40 weeks, compared to placebo females.

Female patients with ADAS-cog baseline scores that were considered to be extreme observations (too low or too high) were removed from the analysis (10 patients were removed from both groups, placebo and VP4896). Average (mean) ADCS-ADL change from baseline scores were plotted and the numbers of patients included at each time point are given. The graph above includes the last-observation-carried-forward (LOCF) cases at 24, 32/34 and 40 weeks, i.e., all of the females completed through week 24 and if they discontinued after weeks 24 or 32/34, their scores are included in the 40 week data point. VP4896-treated females demonstrated better ADL scores at 24, 32/34 and 40 weeks, compared to placebo females.
ALADDIN II
Curaxis had completed a randomized, double-blind, placebo controlled, dose-ranging, 48-week, Phase II clinical trial, ALADDIN II, to assess the efficacy and safety of an injectable formulation of leuprolide acetate on cognitive and global function in men with mild to moderate Alzheimer’s disease. It called this clinical trial ALADDIN II or trial 104. The trial was conducted at 17 investigative study sites in the United States. Men aged 65 or older with mild to moderate Alzheimer’s disease were eligible to participate in the trial. Patients were allowed to receive AChEIs during the trial if they began taking this medication at least 60 days prior to the trial and continued a stable dose throughout the trial.
A total of 119 men were enrolled in this study and were included in the intent-to-treat population and assigned to one of three groups comprised of:
|
●
|
a low dose leuprolide acetate group; 22.5 mg of leuprolide per 12 weeks; 39 men
|
|
●
|
a high dose leuprolide acetate group; 33.75 mg of leuprolide per 12 weeks; 42 men and
|
|
●
|
a placebo group; 38 men.
|
18
Each participant was administered an injection of leuprolide acetate or placebo (saline) once every 12 weeks during the trial. The primary efficacy endpoints of the trial were a patient’s score on the ADAS-cog and the ADCS-CGIC at 48 weeks compared to baseline. There were various secondary efficacy endpoints, including a patient’s score on the ADCS-ADL at 48 weeks compared to baseline. The low and high doses of leuprolide acetate produced very similar results. Curaxis did not achieve the primary efficacy endpoints or any of the secondary efficacy endpoints in this trial with statistical significance. The p values for the endpoints in this trial were 0.729 for ADAS-cog, 0.530 for ADCS-CGIC and 0.232 for ADCS-ADL.
Curaxis analyzed data from patients receiving either 22.5 mg or 33.75 mg doses of leuprolide acetate in conjunction with AChEIs and compared those to placebo (saline) patients also receiving AChEIs. There was a small but not statistically significant signal on the ADAS-cog in favor of both leuprolide treatment groups at week 48. Curaxis did not achieve its primary or secondary statistical endpoints for either group.Results on the ADCS-CGIC rating demonstrated that 44% of the men who received 22.5 mg of leuprolide acetate plus AChEIs, and 47% of the men who received 33.75 mg of leuprolide acetate plus AChEIs, were either improved or showed no change compared to 31% of the men who received placebo plus AChEIs. The p-value for this difference was 0.18 on an adjusted basis.
In the ALADDIN II study, leuprolide acetate administered as an injection was well tolerated at both dose levels without any evidence of a dose-related increase in adverse events. The safety profile in ALADDIN II was similar to that of ALADDIN I.
Phase I / ALADDIN 105
Curaxis had completed a Phase I single center, randomized, double-blind, placebo-controlled, multiple-dose and formulation comparison study of VP4896. It enrolled 50 healthy volunteers in this study consisting of post-menopausal women aged 45 to 70 years and men between the ages of 50 and 70 years. The primary objective of this trial was to determine the safety and tolerability of VP4896 implants at various dose combinations monitored over a period of 24 weeks. Secondary objectives were to study the pharmacokinetics (the processes by which a drug is absorbed, distributed, metabolized and eliminated by the body) of VP4896 implants over a period of up to 48 weeks and to compare the pharmacokinetic profile of a single administration of a VP4896 implant with the pharmacokinetic profile of a single administration of one-month leuprolide acetate by injection over a period of eight weeks.
The implants provided a relatively constant systemic release of leuprolide acetate over an eight-week dosing interval. Following the first 24 hours of dosing, steady concentrations of the drug were established, rising slowly to a peak that occurred at approximately four weeks and, thereafter, declining slowly to the end of the eight-week dosing period to a level similar to the level at the end of 24 hours.
There were no serious adverse events reported in this trial. All safety events in this trial believed to be related to leuprolide acetate were consistent with those listed in the package insert for leuprolide acetate’s approved indications. Some patients experienced minor bruising or irritation as a result of the implant procedure that resolved within two weeks.
Development Plan for Alzheimer’s Program
Curaxis has conducted four randomized, double-blind, placebo-controlled, studies exploring the use of leuprolide acetate for the treatment of mild to moderate AD. In the first two of those studies, ALADDIN I AND ALADDIN II, the Company utilized an injectable formulation of leuprolide acetate in 108 women and 119 men aged 65 or older, respectively, as a treatment for mild to moderate AD. Also, the Company has completed an analysis of data from ALADDIN 301, which was originally designed to serve as a Phase III trial of VP4896 in mild to moderate AD, which supported results the Company achieved in women in the ALADDIN I trial. The Phase II efficacy results in females provide sufficient clinical evidence to continue development of VP4896 in the treatment of mild to moderate AD in women. The Company anticipates that the next step in the clinical development plan is a Phase IIb study in 200-250 females with trial duration of 12 – 18 months. The study likely will include two treatment groups: placebo implant (acetylcholinesterase inhibitors only) and VP4896 implants plus acetylcholinesterase inhibitors (45.2 mg implants every 8 weeks). The approximate cost of the proposed trial is $12 to $15 million. The Company, however, is working with outside consultants to better define the indication of treatment and will seek regulatory agreement prior to finalizing the study protocol. If by design, the proposed trial meets the requirements of a registration trial, additional costs may be incurred. However, the timeline to potential commercialization will be accelerated.
19
In order to initiate our clinical development plan, the Company we will need to raise approximately $20 million during the next year to cover the costs associated with the proposed study and related administrative and overhead costs. Curaxis had been unable to advance the clinical development of Memryte since 2006 due to a lack of financial resources and there can be no assurance that we will be successful in raising the funds needed to commence and complete the proposed study. Furthermore, even if we are successful in raising the funds necessary to restart the clinical development of our Alzheimer’s disease candidate, there can be no assurance that we will be able to successfully complete all phases of such clinical development. Drug development is extremely costly and complex and requires multiple clinical trials and we may be unable to obtain suitable financing for all such trials and, even if we are successful in obtaining suitable financing, we may be unable to complete all required clinical trials, due to an inability to recruit an adequate number of clinical trial sites or an adequate number of patients to participate in those trials or other reasons.
Our Oncology Program
Curaxis had conducted an extensive preclinical research program in the use of leuprolide acetate to treat a number of cancers, including hormone refractory prostate cancer, brain cancers, kidney cancer, pancreatic cancer and non-small-cell lung cancer.
Its work in oncology is based on new insights into the growth of cancer cells that have been discovered by its scientists. In particular, its scientific findings relating to autocrine (cell produces factors that regulate its own functions)-paracrine (cell produces factors that regulate the functions of adjacent cells) signaling and the replication of the hypothalamic-pituitary-gonadal hormonal feedback loop (the HPG axis) inside of cancer cells point to a previously unknown mechanism that drives the growth of cancer cells and an entirely new method of attacking those cancer cells; i.e., using high doses of leuprolide acetate to eliminate the gonadotropins that may be driving the growth of those cells.
Manufacturing
We do not own or intend to own manufacturing facilities for the production of clinical or commercial quantities of leuprolide acetate or any of the compounds that Curaxis had been testing in its pre-clinical programs. We currently rely, and our strategy is to continue to rely, on third parties for the manufacture of our product candidates and any products for which we may obtain regulatory approval to commercially manufacture and market.
The implants for VP4896 are manufactured by Durect Corporation using a melt extrusion process in which leuprolide acetate is mixed with a polymer. The mixture is then formed and cut into small rod-shaped implants that are approximately 1.5 millimeters in diameter and 3.0 centimeters in length.
Durect Agreement
In July 2002, Curaxis entered into a Feasibility, Development and Commercialization Agreement with Southern Biosystems, Inc., a company which subsequently merged into its parent corporation, Durect. Under this agreement, Durect produces VP4896 for us using Durect’s biodegradable polymeric implant technology to provide a sustained release formulation of leuprolide acetate to treat Alzheimer’s disease. Durect has the right to subcontract its responsibilities under the agreement, provided that Durect remains responsible for its obligations under the agreement. Under the agreement, Durect has granted Curaxis a worldwide, exclusive license to manufacture, market and sell VP4896.
We must pay all of Durect’s costs associated with development and regulatory approval activities and make payments to Durect upon the achievement of specified development and regulatory milestones for VP4896. Curaxis has made milestone payments to Durect totaling $500,000 and we are obligated to make additional payments, up to an aggregate maximum amount of $2.5 million, if specified additional milestones are met. We have agreed to purchase quantities of VP4896 from Durect at transfer prices equal to specified percentages of Durect’s fully allocated costs to produce it. We are also obligated to pay royalties to Durect on commercial sales of VP4896 based on the following schedule: (i) 10% on annual revenues through $250 million, (ii) 12% on annual revenues in excess of $250 million up through $500 million, and (iii) 14% on annual revenues in excess of $500 million. These royalties are payable to Durect for the maximum time permitted by law in each country, which may be longer than the life of any patents that Durect licensed to us. The term of the agreement is open-ended and the rights and responsibilities of the parties to the agreement continue until the agreement is terminated pursuant to its terms. Either party to the agreement may terminate the agreement in the event of a default by the other party in any of its material obligations under the agreement if the default is not cured within thirty days of receipt of written notice from the other party of the agreement. In addition, either party to the agreement may immediately terminate the agreement if the other party to the agreement files a petition in bankruptcy or insolvency and such petition is not dismissed within sixty days.
20
Under the agreement, Durect granted Curaxis an exclusive license under its intellectual property rights to use its polymeric implant technology to develop and commercialize VP4896 on a worldwide basis for the treatment of Alzheimer’s disease. The Durect intellectual property licensed by Curaxis includes patent applications covering the VP4896 formulation and the method of making VP4896. Curaxis’ license from Durect of its technology does not extend to the use of VP4896 for the treatment of any other indications. It is required under the agreement to diligently, and in accordance with the timelines for clinical development set by Curaxis, conduct all required clinical trials, obtain all necessary regulatory approvals and commercialize VP4896 in the United States for Alzheimer’s disease.
Covidien/Mallinckrodt Agreement
On July 28, 2008, Curaxis entered into a supply agreement with Mallinckrodt, Inc. (“Mallinkrodt”), a subsidiary of Covidien, Ltd. (“Covidien”), under which Curaxis had agreed to purchase 75% of its annual requirements of leuprolide acetate from Mallinckrodt for a period of five years beginning January 1, 2009, and ending December 31, 2013, at an initial price of $550 per gram (the “Covidien Agreement”). The initial price is subject to annual adjustment upward and downward based on changes in Mallinckrodt’s costs and production methods. The Covidien Agreement is automatically renewable for a second five-year term beginning January 1, 2014, unless Mallinckrodt elects not to renew the Covidien Agreement for the second five-year term. In connection with the execution of the Covidien Agreement, Mallinckrodt paid Curaxis the sum of $500,000. The Company has been in default of the Agreement since January 1, 2009, as it was unable to accept product. Covidien has informed the Company that they will supply product on a prospective basis, as needed, under the terms of the original agreement.
Southridge Agreement
On May 28, 2009, Curaxis entered into a transaction management agreement with Southridge Business Solutions Group, LLC, of Ridgefield, Connecticut (“Southridge”), to assist Curaxis in restructuring its balance sheet, principally through negotiations with several large trade creditors to reduce their claims, and to assist Curaxis in effectuating a merger with a suitable public corporation (the “Transaction Management Agreement”). Pursuant to the Transaction Management Agreement, Curaxis had worked with Southridge to reduce its trade debt by approximately $6,000,000 and entered into the Merger Agreement with Auto Search Cars, Inc. Under the terms of the Transaction Management Agreement, Curaxis is obligated to pay Southridge a management fee of $10,000 per month, from June 1, 2009 through the first anniversary of the closing of the Merger with Auto Search, or July 2011. Curaxis may terminate the Transaction Management Agreement at any time by giving 90 days’ advance notice to Southridge. In addition, Curaxis was obligated to issue to Southridge warrants equal to 3% of Curaxis’ outstanding stock as of the date of the Merger with Auto Search at an exercise price of $0.001 per share. A total of 2,149,148 warrants have been issued to Southridge under the agreement. The agreement with Southridge was terminated effective May 31, 2011.
21
Canterbury Agreement
On June 12, 2009, Curaxis entered into a letter agreement with Canterbury Investment Partners, LLC of Hingham, Massachusetts (“Canterbury”), to assist Curaxis in a range of issues in connection with its contemplated merger with a public shell corporation, including its negotiations with Southridge, in developing a strategy to negotiate reductions, deferrals and/or equity exchanges with its trade creditors and in raising funds from current or prospective investors and in negotiating the terms of the acquisition of a public corporation (the “Letter Agreement”). Under the terms of the Letter Agreement, Curaxis paid Canterbury a retainer of $50,000 and is also obligated to pay Canterbury a monthly fee of $5,833 for a period of twelve (12) months from September 2009 through August 2010. In addition, under the Letter Agreement, Curaxis has issued a warrant to Canterbury to purchase 854,358 shares of Curaxis Common Stock at a price of $0.22 per share. Following the closing of the Merger with Auto Search, Curaxis is obligated to pay Canterbury the sum of $8,000 per month for a period of six (6) months for Canterbury’s assistance in developing and implementing a strategy to support Curaxis in its dealings with the financial community. Mark Pompeo, who is the manager of Canterbury, is the brother of Ronald Pompeo, who was a director of Curaxis from July 3, 2009 to June 24, 2011. The agreement with Canterbury was terminated effective January 31, 2011.
Equity Credit Agreement
On September 16, 2010, the Company entered into a Private Equity Credit Agreement (the “Equity Credit Agreement”) which was amended and restated on December 6, 2010, with Southridge Partners II, LP (the “Investor”), a limited partnership organized and existing under the laws of the State of Delaware and an affiliate of Southridge. Pursuant to this Equity Credit Agreement, the Investor committed to purchase up to $25 million of the Company’s common stock over the course of thirty six months, commencing the effective date of the initial registration statement covering the registrable securities pursuant to the Equity Credit Agreement. The put option price is set at 95% of the average of three lowest closing bid price (the “Bid Price”) of any two applicable trading days, consecutive or inconsecutive, during the five trading day period (the “Valuation Period”), commencing the date a put notice (the “Put Notice”) is delivered to the Investor (the “Put Date”) in a manner provided by the Equity Credit Agreement.
In addition, pursuant to the Equity Credit Agreement, in each Put Notice, the Company is required to specify a minimum stock price which in no event shall be less than 80% of the average of the Bid Price during the three trading day period commencing the Put Date (the “Floor Price”). In the event the Bid Price decreases below the Floor Price during the Valuation Period, the Investor’s obligation to fund one-fifth of the Put Amount for each such trading day shall terminate and the Put amount shall be adjusted accordingly.
On December 7, 2011, the Company filed a registration statement on Form S-1, which was subsequently amended on January 13, 2011, to register 13,000,000 shares of the Company’s common stock to be issued under the terms of the Equity Credit Agreement (the “Registration Statement”). The Registration Statement was declared effective by the SEC on January 26, 2011. The “Registrable Securities” include the Put Shares, any Blackout Shares (as defined in the Equity Credit Agreement) and any securities issued or issuable with respect to any of the foregoing by way of exchange, stock dividend or stock split or in connection with a combination of shares, recapitalization, merger, consolidation or other reorganization or otherwise. To date, the Company has issued 5,563,689 shares of common stock under the Equity Credit Agreement.
Upon execution of the Equity Credit Agreement, the Company issued 820,856 warrants to the Investor in accordance with the Transaction Management Agreement with Southridge. The exercise price is equal to 120% of the average closing price of the Company's stock for the previous 20 days or $1.52.
Sales and Marketing
We intend to maximize the commercial potential of our product candidates by selectively entering into collaboration agreements with leading pharmaceutical and biotechnology companies to assist us in furthering the development of our product candidates. In particular, we intend to enter into these third-party arrangements for target indications in which our potential collaborator has particular expertise or that involve a large, primary care market that must be served by large sales and marketing organizations. In entering into these collaboration agreements, our goal will be to maintain co-promotion or co-commercialization rights in the United States and, in some cases, other markets.
22
We expect to contract with third parties to warehouse and distribute our products and to provide administrative functions, such as accounts receivable management and other similar activities.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions.
Many of our competitors have significantly greater financial resources and experience in research and development, manufacturing, pre-clinical testing, clinical trials, regulatory approvals and marketing approved products than we do. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunity will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects, are easier to administer or are less expensive than any products that we may develop. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
If approved, our Alzheimer’s product will compete against the four drugs currently approved for the treatment of Alzheimer’s disease. There are three marketed acetylcholinesterase inhibitors (Aricept, Razadyne, and Exelon) administered as oral medications or by absorption through the skin in a patch. This class of drugs accounted for approximately $5 billion in US sales in 2010. Namenda is an NMDA receptor antagonist that had estimated US sales of $1.5 billion in 2010. These products have well-known brand names, are distributed by large pharmaceutical companies and have achieved widespread acceptance among physicians and patients. In addition, our product could face competition from other leuprolide acetate products that are already on the market or may later be approved for other indications, if they are used or prescribed off label for Alzheimer’s disease.
In addition, there are many companies and academic and research institutions researching and developing potential treatments for Alzheimer’s disease. Some of these companies are large pharmaceutical companies, while others are smaller companies that may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and biotechnology companies.
Patents and Proprietary Rights
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing United States and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
Our patent portfolio includes one issued U.S. patent and patent applications in the United States, foreign countries and regions under the Patent Cooperation Treaty. Our U.S. patent covers the use of leuprolide acetate in therapeutically effective amounts to treat Alzheimer’s disease through a reduction or elimination of blood serum levels of FSH and LH. This U.S. patent expires in 2018. Should the FDA approve the use of our leuprolide acetate formulation (VP4896) for the treatment of Alzheimer’s disease, this patent may be eligible for patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. A foreign counterpart application to this patent is pending only in Canada. We are aware of one issued third party U.S. patent, filed after our issued U.S. patent was filed, which may nonetheless be prior art to our patent and which might form the basis of an interference proceeding or invalidity challenge. See “Risk Factors—If we infringe or are alleged to infringe intellectual property rights of third parties, it will adversely affect our business.”
23
Our portfolio also includes patent applications directed to the treatment of Alzheimer’s disease with a combination of leuprolide acetate and AChEIs and/or NMDA receptor antagonists, to the treatment of various diseases and disorders associated with senescence, including Parkinson’s disease and prostate cancer, to the treatment of brain cancers, and to the treatment of cancers that express the genes of the HPG axis. Counterpart filings to these patent applications have been made in other jurisdictions, including Europe and Japan. As described above, Curaxis also holds an exclusive license from Durect for the sustained release technology for VP4896 for the treatment of Alzheimer’s disease. Leuprolide acetate, the active pharmaceutical ingredient in VP4896, is off-patent; therefore we expect to rely on method of use and formulation patents to protect our market position.
We may rely, in some circumstances, on trade secrets to protect our technology. However, trade secrets are difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, scientific advisors and other contractors, as well as physical security of our premises and our information technology systems. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Regulatory Matters
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing and export and import of pharmaceutical products, such as those we are developing. The process of obtaining regulatory approvals and the subsequent substantial compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Government Regulation
In the United States, the information that must be submitted to the FDA in order to obtain approval to market a new drug varies depending on whether the drug is a new product whose safety and effectiveness has not previously been demonstrated in humans or a drug whose active ingredient(s) and certain other properties are the same as those of a previously approved drug. A new drug will follow the New Drug Application, or NDA, route.
NDA Approval Processes
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, and implements regulations. Failure to comply with the applicable regulatory requirements at any time may result in administrative or judicial sanctions. These sanctions could include the FDA’s imposition of a clinical hold on trials, refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The testing and approval process requires substantial time, effort, and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval.
Post-Approval Requirements
After regulatory approval of a product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of an NDA, the FDA may require post marketing testing and surveillance to monitor the product’s safety or efficacy. In addition, holders of an approved NDA are required to report certain adverse reactions and production problems to the FDA to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes certain procedural, substantive, and record keeping requirements. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
24
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from country to country.
Under European Union regulatory systems, we may submit marketing authorizations either under a decentralized procedure or, if our product is eligible, under a centralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval.
Reimbursement
In both domestic and foreign markets, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third party payors to those who use our products. Third party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. These third party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the coverage status and reimbursement amount of newly approved healthcare product candidates. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Depending on the results of such studies, our product candidates may not be considered cost-effective. Adequate third party reimbursement may not be available to health care providers to cover the costs of using any products we may develop, which may not allow us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Number of Total Employees and Number of Full Time Employees
We believe that our success will depend greatly on our ability to identify, attract, and retain capable employees. As of December 31, 2011, we had two full time employees. Our employees are not represented by any collective bargaining unit, and we believe our relations with our employees are good.
Employment Agreements
We do not have any employment agreements in place and do not anticipate entering into any employment agreements in the foreseeable future.
ITEM 1A. RISK FACTORS.
RISKS RELATED TO OUR BUSINESS
WE EXPECT TO INCUR LOSSES AT LEAST FOR THE FORESEEABLE FUTURE AND MAY NEVER ACHIEVE OR MAINTAIN PROFITABILITY.
We expect to incur operating losses at least for the foreseeable future. If we are unable to raise the funds necessary to pay our expenses, including expenses accumulated in our clinical trial activities to date, we may be unable to continue operations.
25
To become and remain profitable, we must succeed in developing and commercializing drugs with significant market potential. This will require us to be successful in a range of challenging activities for which we are only in the preliminary stages: developing drugs, obtaining regulatory approval for them, and manufacturing, marketing and selling them. We may never succeed in these activities and may never generate revenues that are significant or large enough to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations.
DUE TO A LACK OF FINANCIAL RESOURCES, WE HAVE BEEN UNABLE TO ADVANCE THE CLINICAL DEVELOPMENT OF OUR LEAD PRODUCT CANDIDATE SINCE 2006.
Our lead product candidate is a hormone for the treatment of Alzheimer’s disease. We have previously conducted several clinical trials for this indication, the most recent of which we were forced to terminate in 2006 due to financial constraints. Since terminating those trials, we have been unable to advance the clinical development of our Alzheimer’s disease candidate due to a lack of financial resources. We anticipate that we will be able to raise the funds necessary to restart the clinical development of our Alzheimer’s disease candidate, but there can be no assurance that we will be able to do so. Furthermore, even if we are successful in raising the funds necessary to restart the clinical development of our Alzheimer’s disease candidate, there can be no assurance that we will be able to successfully complete all phases of such clinical development. Drug development is extremely costly and complex and requires multiple clinical trials and we may be unable to obtain suitable financing for all such trials and, even if we are successful in obtaining suitable financing, we may be unable to complete all required clinical trials due to an inability to recruit an adequate number of clinical trial sites or an adequate number of patients to participate in those trials or other reasons.
WE WILL NEED SUBSTANTIAL ADDITIONAL FUNDING AND MAY BE UNABLE TO RAISE CAPITAL WHEN NEEDED, WHICH WOULD FORCE US TO DELAY, REDUCE OR ELIMINATE OUR PRODUCT DEVELOPMENT PROGRAMS OR COMMERCIALIZATION EFFORTS.
We expect our research and development expenses to increase in connection with our ongoing activities, particularly as we conduct clinical trials for our candidate for the treatment of mild to moderate Alzheimer’s disease. In addition, subject to regulatory approval of any of our product candidates, we expect to incur significant commercialization expenses for product sales, marketing, securing commercial quantities of product from our manufacturers and distribution. We will need substantial additional funding to complete clinical trials and fund initial commercialization costs of our therapeutic candidates and to advance our earlier stage preclinical programs, and may be unable to raise capital when needed or on attractive terms, which would force us to delay, reduce or eliminate our research and development programs or commercialization efforts. At present, sufficient funds are not available to continue operations for an extended period.
Our future capital requirements will depend on many factors, including:
|
●
|
the progress and results of clinical trials of our lead candidate, VP4896, for mild to moderate Alzheimer’s disease;
|
|
●
|
the costs of establishing sales and marketing functions, outsourcing manufacturing needs and obtaining pre-launch inventory of VP4896;
|
|
●
|
the scope, progress, results and cost of pre-clinical development and laboratory testing and clinical trials for our other product candidates;
|
|
●
|
the costs, timing and outcome of regulatory review of VP4896 for mild to moderate Alzheimer’s disease and of our other product candidates;
|
26
|
●
|
the number and development requirements of other product candidates;
|
|
●
|
the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims;
|
|
●
|
our success in entering into collaboration agreements with leading pharmaceutical and biotechnology companies to assist in furthering the development of product candidates; and
|
|
●
|
the timing, receipt and amount of sales or royalties, if any, from VP4896 and other potential products.
|
Until such time, if ever, when we can generate substantial product revenues, we expect to finance our cash needs through public or private equity offerings, debt financings and possibly corporate collaboration and licensing arrangements. If we raise additional funds by issuing equity securities, our stockholders may experience dilution. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any debt financing or additional equity that we raise may contain terms, such as liquidation and other preferences that are not favorable to our Company or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
WE ARE STILL IN THE INITIAL STAGES OF OUR BUSINESS PLAN, WHICH MAY MAKE IT DIFFICULT FOR YOU TO EVALUATE THE SUCCESS OF OUR BUSINESS TO DATE AND TO ASSESS OUR FUTURE VIABILITY.
Our operations have been limited to organizing and staffing our Company, acquiring, developing and securing our technology and undertaking pre-clinical studies and limited clinical trials of our most advanced product candidate, VP4896. We have not yet demonstrated our ability to successfully complete large-scale, pivotal clinical trials, obtain regulatory approval, manufacture a commercial scale product or arrange for a third party to do so on our behalf or conduct sales and marketing activities necessary for successful product commercialization.
In addition, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. We will need to transition from our research focus to supporting commercial activities, and we may not be successful in such transition.
WE DEPEND HEAVILY ON THE SUCCESS OF OUR MOST ADVANCED PRODUCT CANDIDATE, VP4896 FOR MILD TO MODERATE ALZHEIMER’S DISEASE, WHICH IS STILL IN CLINICAL DEVELOPMENT. IF WE ARE UNABLE TO COMMERCIALIZE VP4896 FOR THIS INDICATION, OR EXPERIENCES SIGNIFICANT DELAYS IN DOING SO, OUR BUSINESS WILL BE MATERIALLY HARMED.
We have invested a significant portion of our efforts and financial resources in the development of our most advanced product candidate, VP4896, for mild to moderate Alzheimer’s disease. The success of our VP4896 Alzheimer’s disease program will depend on several factors, including the following:
|
●
|
successful patient enrollment in, and completion of, clinical trials;
|
|
●
|
receipt of marketing approvals from the FDA and similar foreign regulatory authorities;
|
|
●
|
obtaining commercial quantities of leuprolide acetate, the active ingredient of VP4896;
|
|
●
|
obtaining commercial quantities of VP4896 from our supplier, Durect Corporation;
|
|
●
|
establishing a sales and marketing infrastructure to market and sell VP4896 in the United States and internationally, whether alone or in collaboration with others; and
|
|
●
|
acceptance of the product by patients, the medical community and third party payors.
|
27
Our ability to generate product revenues, which we do not expect will occur for at least the next several years, if ever, will depend heavily on the successful development and commercialization of VP4896. If we are unable to commercialize VP4896 for mild to moderate Alzheimer’s disease, or experience significant delays in doing so, our business will be materially harmed.
IF WE ARE UNABLE TO ACHIEVE STATISTICAL SIGNIFICANCE ON THE PRIMARY EFFICACY ENDPOINTS IN CLINICAL TRIALS OF VP4896 FOR THE TREATMENT OF MILD TO MODERATE ALZHEIMER’S DISEASE, WE MAY NOT BE SUCCESSFUL IN OBTAINING FDA APPROVAL FOR VP4896, WHICH WOULD MATERIALLY HARM OUR BUSINESS.
In ALADDIN I, our Phase II clinical trial for women with mild to moderate Alzheimer’s disease involving an injectable formulation of leuprolide acetate, we identified a trend in favor of the high dose leuprolide acetate group versus placebo, but we did not achieve statistical significance on the primary efficacy endpoints or any of the secondary efficacy endpoints in this clinical trial. The primary efficacy endpoints of the trial were a patient’s score on each of the ADAS-cog, a test of memory and cognition, and the ADCS-CGIC, a global measure of a subject’s change in condition, at 48 weeks compared to baseline. There were various secondary efficacy endpoints, including a patient’s score on the ADCS-ADL, a measurement of a patient’s capacity to perform activities of daily living, at 48 weeks compared to baseline. Analysis of the same primary efficacy endpoints at the completion of the 48-week men’s Phase II study, ALADDIN II, also did not demonstrate statistical significance.
We performed a preplanned subgroup analysis of 78 women out of the 108 women who participated in ALADDIN I, the Phase II clinical trial in women. Of this subgroup of 78 women, which included all of the patients who were taking an acetylcholinesterase inhibitor (AChEI) and excluded the 30 patients who were not taking an AChEI, 28 patients were taking an AChEI plus 11.25 mg of leuprolide acetate, 24 patients were taking an AChEI plus 22.5 mg of leuprolide acetate and 26 patients were taking an AChEI plus placebo. The results for the group of 28 patients that received an AChEI plus the 11.25 mg dose of leuprolide acetate were not statistically significantly different from the results for the group that received an AChEI plus placebo. The results for the group of 24 patients that received an AChEI plus the 22.5 mg of leuprolide acetate compared to the group of 26 patients that received an AChEI plus placebo were statistically significant with respect to the patients’ scores on both primary endpoints, the ADAS-cog and ADCS-CGIC as well as on the ADCS-ADL, a secondary endpoint, prior to applying any statistical adjustment. However, because we performed several comparisons, we were required to adjust the p-values (a mathematical calculation used to determine the likelihood that the measured result was obtained by chance) of the results to account for multiple comparisons. This was done by using what is referred to as the Bonferroni correction, which applies an estimated statistical penalty to account for the number of comparisons made. Specifically, the unadjusted p-values for this subgroup were 0.026 for the ADAS-cog, 0.031 for the ADCS-CGIC and 0.015 for ADCS-ADL. The adjusted p-values for this subgroup were 0.078 for the ADAS-cog, 0.093 for the ADCS-CGIC and 0.044 for ADCS-ADL. Therefore, after applying the Bonferroni correction, we were only able to demonstrate statistical significance with respect to the ADCS-ADL.
We likewise performed an analysis of 90 men out of 119 men who participated in ALADDIN II, the Phase II clinical trial in men. Of this subgroup of 90 men, which included all of the patients who were taking an AChEI and excluded the 29 patients who were not taking an AChEI, 27 patients were taking an AChEI plus a 22.5 mg dose of leuprolide acetate, 34 patients were taking an AChEI plus a 33.75 mg dose of leuprolide acetate and 29 patients were taking an AChEI plus placebo. The results for both leuprolide-treated groups were not significantly different from the results for the group that received an AChEI plus placebo. The treated groups of 27 and 34 patients given 22.5 mg and 33.75 mg of leuprolide acetate, respectively, demonstrated a slight positive signal on the ADAS-cog assessment compared to placebo at 48 weeks. Results on the ADCS-CGIC rating demonstrated that 44% of the men who received 22.5 mg of leuprolide acetate plus AChEIs, and 47% of the men who received 33.75 mg of leuprolide acetate plus AChEIs, were either improved or showed no change compared to 31% of the men who received placebo plus AChEIs.
28
In addition, we used a commercially available formulation of leuprolide acetate administered by injection in its Phase II trials for the treatment of mild to moderate Alzheimer’s disease, while our planned clinical trials will test VP4896, a biodegradable polymeric implant formulation of leuprolide acetate, for this indication. Leuprolide acetate administered through an implant may not have the same effect as leuprolide acetate injections had in our earlier Phase II clinical trials and may cause side effects not seen in those Phase II clinical trials. ALADDIN 105 was a Phase I trial performed by us to test the safety and tolerability of the VP4896 implant and the implantation procedure as well as the pharmacokinetics (absorption, distribution, metabolism, and elimination), of leuprolide delivery from the implant. The results of that study suggest that the VP4896 implant is safe and well tolerated. ALADDIN 105 also demonstrated that the VP4896 implant releases leuprolide acetate in a steady and sustained way over the 8-week dosing period. In addition, our planned clinical trials may be at dosing levels that are higher than the doses of leuprolide acetate used in ALADDIN I and ALADDIN II.
The differences in dosing levels and methods of administration, as well as other factors, may have unexpected effects on the outcome of our clinical trials. If we are unable to achieve statistical significance on the primary efficacy endpoints in pivotal Phase III clinical trials, we may not be successful in obtaining FDA approval for VP4896, which would materially harm our business.
OUR PROGRAM FOR VP4896 IS BASED ON THE LH HYPOTHESIS, WHICH IS NOT VIEWED AS THE PREDOMINANT HYPOTHESIS REGARDING THE POSSIBLE CAUSES OF ALZHEIMER’S DISEASE. THIS ADDS TO THE RISK OF OUR DEVELOPMENT EFFORT AND MAY AFFECT PHYSICIAN AND PATIENT ACCEPTANCE AND USE OF VP4896 AND THE WILLINGNESS OF THIRD PARTIES TO PROVIDE REIMBURSEMENT.
Our program for VP4896 is based on the LH hypothesis, which is not the predominant view regarding the possible causes of Alzheimer’s disease. The LH hypothesis proposes that the known neurological and biochemical changes associated with Alzheimer’s disease are caused by elevated levels of the pituitary hormone, leuteinizing hormone (LH). This hypothesis suggests that when LH levels increase as the body ages, the neurological changes seen in Alzheimer’s disease result. The beta amyloid hypothesis, however, proposes that amyloid beta protein, which makes up the plaques present in the brains of Alzheimer’s disease patients, is toxic, and is the causative agent of the disease. Under this hypothesis, inhibiting the production of, and enhancing the clearance of, amyloid beta protein plaques may prevent or treat Alzheimer’s disease. The beta amyloid hypothesis is the predominant view. While preclinical data now support the notion that the LH hypothesis may be interrelated with the amyloid hypothesis and other hypotheses of Alzheimer’s disease pathology, the unproven nature of the LH hypothesis adds to the risk that our development effort may not be successful. In particular, no drugs for the treatment of Alzheimer’s disease have been successfully developed based on the LH hypothesis. Moreover, the predominant status of the beta amyloid hypothesis may impede our development and commercialization efforts. For example, the predominant status of the beta amyloid hypothesis in the medical community may adversely affect our ability to recruit patients for our clinical trials or, if we successfully complete clinical development of this product candidate and obtain regulatory approval, may adversely affect our ability to recruit sales and marketing personnel, may affect physician and patient acceptance and use of the product and the willingness of third parties to provide reimbursement, any of which could materially harm our business.
IF CURAXIS’ PRECLINICAL STUDIES DO NOT PRODUCE SUCCESSFUL RESULTS OR ITS CLINICAL TRIALS DO NOT DEMONSTRATE SAFETY AND EFFICACY IN HUMANS, CURAXIS WILL NOT BE ABLE TO COMMERCIALIZE ITS PRODUCT CANDIDATES.
Before obtaining regulatory approval for the sale of its product candidates, Curaxis must conduct, at its own expense, extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of its product candidates. Preclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. Specifically, the results of Curaxis’ Phase II trials were collected from a limited number of subjects and may not be indicative of results that it will obtain in later studies which are expected to be of greater size and scope.
29
A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize VP4896 or our other product candidates, including:
|
●
|
regulatory authorities, institutional review boards, or ethics committees may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site;
|
|
●
|
our clinical trials may produce negative or inconclusive results, and we may decide, or regulatory authorities may require us, to conduct additional clinical trials or we may abandon projects that we expect to be promising;
|
|
●
|
enrollment in our clinical trials may be slower than we currently anticipate, resulting in significant delays. Additionally, participants may drop out of our clinical trials at a higher rate than we anticipate;
|
|
●
|
our third party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner;
|
|
●
|
we might have to suspend or terminate our clinical trials if the participants are being exposed to unacceptable health risks;
|
|
●
|
regulatory authorities, institutional review boards, or ethics committees may require that we hold, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements;
|
|
●
|
the cost of our clinical trials may be greater than we anticipate;
|
|
●
|
the supply or quality of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate; and
|
|
●
|
the effects of our product candidates may not achieve the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics.
|
If we are required to conduct additional clinical trials or other testing of VP4896 or our other product candidates beyond those that we currently contemplate, if we are unable to successfully complete our clinical trials or other testing, or if the results of these trials or tests are not positive or are only modestly positive, we may:
|
●
|
be delayed in obtaining marketing approval for our product candidates;
|
|
●
|
not be able to obtain marketing approval; or
|
|
●
|
obtain approval for indications that are not as broad as we intended.
|
Our product development costs will also increase if we experience delays in testing or approvals. We do not know whether any clinical trials will begin as planned, need to be restructured or will be completed on schedule, if at all. Significant clinical trial delays could also shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to commercialize our products or product candidates.
USE OF THIRD PARTIES TO MANUFACTURE OUR PRODUCT CANDIDATES, INCLUDING VP4896, MAY INCREASE THE RISK THAT WE WILL NOT HAVE SUFFICIENT QUANTITIES OF OUR PRODUCT CANDIDATES OR SUCH QUANTITIES AT AN ACCEPTABLE COST, AND CLINICAL DEVELOPMENT AND COMMERCIALIZATION OF OUR PRODUCT CANDIDATES COULD BE DELAYED, PREVENTED OR IMPAIRED.
We do not own or operate manufacturing facilities for clinical or commercial production of our product candidates. We have limited personnel with experience in drug manufacturing and we lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. Our strategy is to outsource all manufacturing of our product candidates and products, including VP4896, to third parties.
30
Reliance on third party manufacturers entails risks to which we would not be subject if we manufactured product candidates or products ourselves, including:
|
●
|
reliance on the third party for regulatory compliance and quality assurance;
|
|
●
|
the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and
|
|
●
|
the possible termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
|
Our manufacturers may not be able to comply with current Good Manufacturing Practice, or cGMP, regulations or other regulatory requirements or similar regulatory requirements outside the United States. Our failure, or the failure of our third party manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates.
Our product candidates and any products that we may develop may compete with other product candidates with respect to the availability of manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations that are both capable of manufacturing for us and willing to do so. If the third parties that we engage to manufacture product for our clinical trials should cease to continue to do so for any reason, we likely would experience delays in advancing these trials while we identify and qualify replacement suppliers and we may be unable to obtain replacement supplies on terms that are favorable to us. In addition, if we are not able to obtain adequate supplies of our product candidates or the drug substances used to manufacture them, it will be more difficult for us to develop our product candidates and compete effectively.
Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins and our ability to develop product candidates and commercialize any products that receive regulatory approval on a timely and competitive basis.
WE RELY ON DURECT CORPORATION, OUR SOLE SOURCE PROVIDER OF VP4896, TO PRODUCE VP4896 FOR OUR CLINICAL TRIALS AND WILL RELY ON DURECT TO PRODUCE COMMERCIAL DRUG SUPPLIES OF VP4896. DURECT HAS LIMITED EXPERIENCE PRODUCING BIODEGRADABLE IMPLANTS AND MAY NOT BE ABLE TO PROVIDE VP4896 TO SATISFY OUR REQUIREMENTS.
We rely on Durect Corporation, or Durect, as our sole source provider of VP4896 for use in our clinical trials and, if we receive marketing approval, will rely on Durect as our sole source provider for commercial supply of VP4896. VP4896 requires precise, high quality manufacturing. Durect’s failure to achieve and maintain satisfactory standards could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could materially harm our business. Durect may encounter manufacturing difficulties involving production yields, quality control and quality assurance. Durect is subject to ongoing periodic unannounced inspection by the FDA and corresponding state and foreign agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. We do not control Durect’s compliance with these regulations and standards.
To date, Durect has not produced commercial supply of a biodegradable implant for any drug approved for human use. If we receive marketing approval for and commercially launch VP4896, we anticipate that Durect will need to expand its manufacturing capacity for this drug, possibly materially. Durect may not be able to increase its manufacturing capacity for VP4896 in a timely or economic manner, or at all. Moreover, significant scale up of manufacturing may require additional validation studies, which the FDA must review and approve.
31
If Durect is unable to successfully increase the manufacturing capacity for VP4896 and we are unable to establish alternative manufacturing capabilities, the commercial launch of VP4896 may be delayed or there may be a shortage in supply. If Durect is unable to successfully increase manufacturing capacity for VP4896 at an acceptable cost, our commercialization of VP4896 could be delayed, prevented or impaired, including through a reduction of our gross margins on product sales.
We could lose supply of VP4896 from Durect if Durect elects to discontinue supply to us pursuant to Durect’s right to do so beginning two years after the first commercial sale or use of VP4896 or if Durect simply fails to or is unable to supply us at the levels required under the agreement. If we lose supply of VP4896 from Durect, we will need to establish an alternative source of supply. The loss of supply and need to establish alternative supply could result in lengthy delays in clinical trials, significant interruption of commercial supplies and substantial additional costs. Switching manufacturers may be difficult because, among other reasons, the number of potential manufacturers is limited, and the FDA must approve any replacement manufacturer. Such approval would require new clinical trials and compliance inspections. If we are able to transfer Durect’s manufacturing technology and process to a new manufacturer, which we have the right to do in circumstances specified in our agreement with Durect, to receive FDA approval would require bioequivalence or similar clinical trials, which are generally simpler and less costly than pivotal clinical trials. If we are not able to obtain such transfer, including as a result of Durect’s refusal to adhere to its contractual obligations, such approval would require new pivotal clinical trials. Accordingly, it may be difficult or impossible for us to find a replacement manufacturer for Durect on acceptable terms, in a manner that avoids supply interruption or possibly at all.
We could also lose our supply of VP4896 from Durect if our agreement with Durect terminates for any reason. In that case, in addition to the risks and consequences associated with having to secure alternative supply arrangements for VP4896 described above, we would be subject to the risks and consequences associated with losing our license to the Durect technology that is included in VP4896, and having to find and test alternative sustained release technology, as described in detail in the risk factor below entitled “If we fail to comply with our obligations in our intellectual property agreements with Durect or other third parties, we could lose license rights that are important to our business.”
WE RELY ON THIRD PARTIES TO CONDUCT OUR CLINICAL TRIALS AND THOSE THIRD PARTIES MAY NOT PERFORM SATISFACTORILY, INCLUDING FAILING TO MEET ESTABLISHED DEADLINES FOR THE COMPLETION OF SUCH TRIALS.
We do not have the ability to independently conduct clinical trials for our product candidates and we rely on third parties, such as contract research organizations, clinical data management organizations, medical institutions, and clinical investigators, to perform this function. Our reliance on these third parties for clinical development activities reduces our control over these activities. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we will not be able to obtain, or may be delayed in obtaining, regulatory approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
We also rely on other third parties to store and distribute drug supplies for our clinical trials for VP4896. Any performance failure on the part of our existing or future distributors could delay clinical development or regulatory approval of our product candidates or commercialization of our products, producing additional losses and depriving us of potential product revenue.
32
WE MAY NOT BE SUCCESSFUL IN ESTABLISHING COLLABORATIONS, WHICH COULD ADVERSELY AFFECT OUR ABILITY TO DISCOVER, DEVELOP AND, PARTICULARLY IN INTERNATIONAL MARKETS, COMMERCIALIZE PRODUCTS.
We intend to selectively enter into collaboration agreements with leading pharmaceutical and biotechnology companies to assist us in furthering the development of our product candidates. In particular, we intend to enter into these third-party arrangements for target indications in which our potential collaborator has particular expertise or that involve a large, primary care market that must be served by large sales and marketing organizations. In entering into these collaboration agreements, our goal will be to maintain co-promotion or co-commercialization rights in the United States and, in some cases, other markets. If we are unable to reach agreements with suitable collaborators, we may fail to meet our business objectives for the affected product or program. We face, and will continue to face, significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements. The terms of any additional collaborations or other arrangements that we establish may not be favorable to us. Moreover, these collaborations or other arrangements may not be successful.
IF WE ARE UNABLE TO OBTAIN AND ENFORCE PATENT PROTECTION FOR OUR DISCOVERIES, OUR ABILITY TO SUCCESSFULLY COMMERCIALIZE OUR PRODUCT CANDIDATES WILL BE HARMED AND WE MAY NOT BE ABLE TO OPERATE OUR BUSINESS PROFITABLY.
Our success depends, in part, on our ability to protect proprietary methods and technologies that we develop under the patent and other intellectual property laws of the United States and other countries, so that we can prevent others from using our inventions and proprietary information. However, we may not hold proprietary rights to some patents related to our current and/or future products and technologies. Because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing, or in some cases not at all, and because publications of discoveries in scientific literature lag behind actual discoveries, we cannot be certain that we were the first to make the inventions claimed in our issued patent or pending patent applications, or that we were the first to file for protection of the inventions set forth in our patent applications. As a result, we may be required to obtain licenses under third-party patents to market our proposed products. If licenses are not available to us on acceptable terms, or at all, we will not be able to market the affected products.
Our strategy depends on our ability to rapidly identify and seek patent protection for our discoveries. This process is expensive and time consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. Despite our efforts to protect our proprietary rights, unauthorized parties may be able to obtain and use information that we regard as proprietary. The issuance of a patent does not guarantee that it is valid or enforceable, so even if we obtain patents, they may not be valid or enforceable against third parties. In addition, the issuance of a patent does not guarantee that we have the right to practice the patented invention. Third parties may have blocking patents that could be used to prevent us from marketing our own patented-product or method and practicing our own patented technology.
Our pending patent applications and the pending patent applications of Durect that we have licensed may not result in issued patents. If patents do not issue with respect to such patent applications or if the claims allowed are too narrow, our business may be harmed. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the United States Patent and Trademark Office and its foreign counterparts use to grant patents are not always applied predictably or uniformly and can change. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents. The laws of some foreign countries do not protect proprietary information to the same extent as the laws of the United States, and many companies have encountered significant problems and costs in protecting their proprietary information in those foreign countries. Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims allowed in any patents issued to us or to others. The allowance of broader claims may increase the incidence and cost of patent interference proceedings and/or opposition proceedings, and the risk of infringement litigation. On the other hand, the allowance of narrower claims may limit the value of our proprietary rights.
Our issued patent and any future patents that we may own or license may not contain claims sufficiently broad to protect us against third parties with similar technologies or products, or provide us with any competitive advantage. Moreover, our patents and any patent for which we have licensed rights may be challenged, narrowed, invalidated or circumvented. For example, we are aware of one issued third party U.S. patent, filed after our issued U.S. patent was filed, which may nonetheless be prior art to our patent and which might form the basis of an interference proceeding or invalidity challenge. We believe that our claimed methods of treatment for Alzheimer’s disease are patentably distinct over the third party patent, that the third party patent does not enable our methods of treatment and, therefore, that our patent claims should be found valid and enforceable. However, the patent office or a court might disagree with us. An interference or invalidity challenge to our issued patent could result in the narrowing or loss of our patent claims.
If the patents that we own or license are invalidated or otherwise limited, other companies will be better able to develop products and technologies that compete with ours, which could adversely affect our competitive business position, business prospects and financial condition.
33
THERE ARE LIMITATIONS ON OUR PATENT RIGHTS RELATING TO OUR PRODUCT CANDIDATES AND TECHNOLOGIES, INCLUDING WITH RESPECT TO THE USE OF VP4896 TO TREAT ALZHEIMER’S DISEASE, THAT MAY AFFECT OUR ABILITY TO EXCLUDE THIRD PARTIES FROM COMPETING AGAINST US IF WE RECEIVE APPROVAL TO MARKET THESE PRODUCT CANDIDATES OR TECHNOLOGIES.
Our proprietary rights relating to our product candidates and technologies, including with respect to the use of VP4896 to treat Alzheimer’s disease, are limited in ways that may affect our ability to exclude third parties from competing against us if we receive regulatory approval to market these product candidates and technologies. In particular:
|
●
|
We do not own or license composition of matter patents covering leuprolide acetate, the active pharmaceutical ingredient of VP4896, or methods of making leuprolide acetate. Moreover, the active ingredient that we are testing in our most advanced preclinical programs is also leuprolide acetate. Leuprolide acetate is currently marketed for a number of other indications. As a result, competitors can offer and sell leuprolide acetate products so long as they do not infringe, or contribute to the infringement of, or actively induce infringement of, any patents or other proprietary rights that we or others may have the rights to enforce;
|
|
●
|
The principal patent protection that covers, or that we expect will cover, the use of VP4896 to treat Alzheimer’s disease and leuprolide acetate to treat other indications stems from a method of use patent and patent applications. This type of patent only protects specified methods of using specified products according to the patent claims. This type of patent does not limit a competitor from making and marketing a product that is identical to our product for an indication that is outside of the patented method. Moreover, physicians may prescribe such a competitive identical product for off label indications that are covered by the applicable patents. In particular, leuprolide acetate, the active pharmaceutical ingredient in VP4896 and the drug being tested by us in our most advanced preclinical programs, is currently marketed for a number of other indications. Although off label prescriptions may infringe or contribute to or actively induce infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute. Consequently, VP4896, if approved, could face competition from leuprolide acetate products that are already on the market, or may later be approved, through off label usage of these products to treat Alzheimer’s disease that may be difficult or impossible for us to prevent because our primary patent protection is limited to method of use claims; and
|
|
●
|
We have not applied for method of use patent protection outside of the United States and Canada for use of VP4896 to treat Alzheimer’s disease. As a result, there is a significant risk that we will face early generic competition for VP4896 for the treatment of Alzheimer’s disease in international markets. This risk may be increased if competitors are able to develop formulations of leuprolide acetate, such as other polymeric implants, that are as effective as VP4896 may prove to be or if competitors can demonstrate that commercially available leuprolide acetate can be administered by injection in a manner that is as effective as VP4896 may prove to be.
|
These limitations on our patent rights may result in competitors taking product sales away from us, which would reduce our revenues and harm our business.
34
IF WE FAIL TO COMPLY WITH OUR OBLIGATIONS IN OUR INTELLECTUAL PROPERTY AGREEMENTS WITH DURECT OR OTHER THIRD PARTIES, WE COULD LOSE LICENSE RIGHTS THAT ARE IMPORTANT TO OUR BUSINESS.
We are a party to a license agreement with Durect pursuant to which Durect grants to us a worldwide, royalty-bearing exclusive license relating to Durect’s polymeric implant technology to develop and commercialize VP4896 for the treatment of Alzheimer’s disease. This technology includes patent applications covering the VP4896 formulation and the method of making VP4896. If we fail to perform our obligations under our agreement with Durect, Durect may terminate the agreement and our license to its patents and its manufacturing know-how. If this happened, we would be required to develop or license an alternative sustained release technology for VP4896. This would likely involve reformulating or otherwise changing VP4896. As a result, the FDA would likely require us to perform new pivotal clinical trials of the reformulated or otherwise changed version of VP4896. This could result in lengthy delays in clinical trials, significant interruption of commercial supplies and substantial additional costs. This would substantially harm our business. Furthermore, acceptable alternative technology may not exist, may not be something we are able to develop and may not be available to us on reasonable terms or possibly at all.
In addition to clinically developing VP4896 for mild to moderate Alzheimer’s disease, we also are working on the pre-clinical development of leuprolide acetate for the treatment of various cancers. We may determine to develop an implantable formulation of leuprolide acetate for these additional indications. However, because our license from Durect is limited to the field of Alzheimer’s disease, we will not be able to develop a leuprolide acetate implant based on Durect’s technology for these additional indications without obtaining an appropriate amendment of our license from Durect. If Durect will not agree to such an amendment, we could seek to develop other polymeric implants of leuprolide acetate that are outside of Durect’s patents and know-how. However, it might be time consuming and expensive for us to do so and might deprive us of the clinical and regulatory benefits that we believe will be available to us from developing a leuprolide acetate implant based on Durect’s technology for additional indications.
We may also enter into additional licenses in the future. Our existing license with Durect imposes, and we expect future licenses with third parties will impose, various diligence, milestone payment, royalty, insurance and other obligations on us. If we fail to comply with these obligations, the licensor may have the right to terminate the license, in which event we might not be able to market any product that is covered by the licensed patents.
IF WE ARE UNABLE TO PROTECT THE CONFIDENTIALITY OF OUR PROPRIETARY INFORMATION AND KNOW-HOW, THE VALUE OF OUR TECHNOLOGY AND PRODUCTS COULD BE ADVERSELY AFFECTED.
In addition to patented technology, we rely upon unpatented proprietary technology, processes and know-how, including particularly Durect’s manufacturing know-how relating to the production of VP4896. We seek to protect our unpatented proprietary information in part by confidentiality agreements with our employees, consultants and third parties. These agreements may be breached and we may not have adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently developed by competitors. If we are unable to protect the confidentiality of our proprietary information and know-how, competitors may be able to use this information to develop products that compete with our products, which could adversely impact our business. We could be similarly harmed if Durect’s confidential know-how relating to the production of VP4896 becomes known to our competitors.
IF WE INFRINGE OR ARE ALLEGED TO INFRINGE INTELLECTUAL PROPERTY RIGHTS OF THIRD PARTIES, IT WILL ADVERSELY AFFECT OUR BUSINESS.
Our research, development and commercialization activities, as well as any product candidates or products resulting from these activities, may infringe or be claimed to infringe patents or patent applications under which we do not hold licenses or other rights. Third parties may own or control these patents and patent applications in the United States and abroad. These third parties could bring claims against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
As a result of patent infringement claims, or in order to avoid potential claims, we or our current or future collaborators may choose or be required to seek a license from the third party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. This could harm our business significantly.
35
For example, we are aware of one issued third party U.S. patent, filed after our issued patent was filed, which includes a claim directed to a method for the treatment of a neurodegenerative disorder which could form the basis of an infringement claim. We believe that if this patent were asserted against us in an infringement action relating to VP4896 for the treatment of mild to moderate Alzheimer’s disease, the claims should be held invalid. An issued U.S. patent, however, is entitled to a presumption of validity as a matter of law. If the claims of this third party patent were found valid and interpreted to cover the use of VP4896 for the treatment for Alzheimer’s disease, we would be liable for past damages for infringement and might be required to obtain a license under the patent to manufacture and market VP4896. If a license were not available to us on acceptable terms, or at all, we might not be able to market VP4896.
Moreover, the field of polymeric implants for sustained drug release is highly competitive, and we are aware of third party patents directed to technologies similar to that of Durect. Although we are not aware of any third party patents that would be literally infringed by the commercial development of the VP4896 implant, we are aware of third party patents which include claims directed to implants containing a copolymer of lactic acid and glycolic acid, that could be asserted against our product in the United States under the doctrine of equivalents. Even if a U.S. patent claim is not literally infringed, the doctrine of equivalents permits a court to find infringement if the differences between the accused product or process and the patent claims are insubstantial. If a court were to find a third party patent infringed under the doctrine of equivalents, the consequences could be the same as those of a finding of literal infringement, including liability for past damages and the possibility of being enjoined from commercializing the affected product or process. In the event of an injunction, we would be required to cease marketing VP4896, but could develop an alternative sustained release formulation which would not infringe any third party patents. Development of an alternative sustained release formulation would require substantial additional time and expense, including additional regulatory review, all of which could harm our business significantly.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including interference proceedings declared by the United States Patent and Trademark Office and opposition proceedings in the European Patent Office, regarding intellectual property rights with respect to our products and technology. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time.
IF WE ARE NOT ABLE TO OBTAIN REQUIRED REGULATORY APPROVALS, WE WILL NOT BE ABLE TO COMMERCIALIZE OUR PRODUCT CANDIDATES, AND OUR ABILITY TO GENERATE REVENUE WILL BE MATERIALLY IMPAIRED.
Our product candidates and the activities associated with their development and commercialization, including their testing, manufacture, safety, efficacy, record keeping, labeling, storage, approval, advertising, promotion, sale and distribution, are subject to comprehensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in other countries. Failure to obtain regulatory approval for a product candidate will prevent us from commercializing the product candidate.
We have not received regulatory approval to market any of our product candidates in any jurisdiction. We have only limited experience in filing the applications necessary to gain regulatory approvals and expect to rely on third party contract research organizations to assist us in this process. Securing FDA approval requires the submission of extensive pre-clinical and clinical data, information about product manufacturing processes and inspection of facilities and supporting information to the FDA for each therapeutic indication to establish the product candidate’s safety and efficacy. Our future products may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use.
36
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon, among other things, the type, complexity and novelty of the product candidates involved. Changes in the regulatory approval policy during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. The FDA has substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional pre-clinical, clinical or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the product not commercially viable.
OUR PRODUCTS COULD BE SUBJECT TO RESTRICTIONS OR WITHDRAWAL FROM THE MARKET AND WE MAY BE SUBJECT TO PENALTIES IF WE FAIL TO COMPLY WITH REGULATORY REQUIREMENTS OR IF WE EXPERIENCE UNANTICIPATED PROBLEMS WITH OUR PRODUCTS, WHEN AND IF ANY OF THEM ARE APPROVED.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and comparable regulatory bodies. These requirements include submissions of safety and other post-marketing information and reports, registration requirements, cGMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents and requirements regarding the distribution of samples to physicians and record keeping. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with our products, manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in:
|
●
|
restrictions on such products, manufacturers or manufacturing processes;
|
|
●
|
warning letters;
|
|
●
|
withdrawal of the products from the market;
|
|
●
|
refusal to approve pending applications or supplements to approved applications that we submit;
|
|
●
|
recall;
|
|
●
|
fines;
|
|
●
|
suspension or withdrawal of regulatory approvals;
|
|
●
|
refusal to permit the import or export of our products;
|
|
●
|
product seizure; and
|
|
●
|
injunctions or the imposition of civil or criminal penalties.
|
37
FAILURE TO OBTAIN REGULATORY APPROVAL IN INTERNATIONAL JURISDICTIONS WOULD PREVENT US FROM MARKETING OUR PRODUCTS ABROAD.
We intend to have our products marketed outside the United States. In order to market our products in the European Union and many other foreign jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market.
THE COMMERCIAL SUCCESS OF ANY PRODUCTS THAT WE MAY DEVELOP WILL DEPEND UPON THE DEGREE OF MARKET ACCEPTANCE BY PHYSICIANS, PATIENTS, HEALTHCARE PAYORS AND OTHERS IN THE MEDICAL COMMUNITY.
Any products that we bring to the market may not gain market acceptance by physicians, patients, healthcare payors and others in the medical community. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenues and we may not become profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, including:
|
●
|
the prevalence and severity of any side effects;
|
|
●
|
the efficacy and potential advantages over alternative treatments;
|
|
●
|
the ability to offer our product candidates for sale at competitive prices;
|
|
●
|
relative convenience and ease of administration;
|
|
●
|
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
|
|
●
|
the strength of marketing and distribution support; and
|
|
●
|
sufficient third party coverage or reimbursement.
|
IF WE ARE UNABLE TO ESTABLISH SALES AND MARKETING CAPABILITIES OR ENTER INTO AGREEMENTS WITH THIRD PARTIES TO MARKET AND SELL OUR PRODUCT CANDIDATES, WE MAY BE UNABLE TO GENERATE PRODUCT REVENUES.
We do not currently have a sales or marketing organization and have limited experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product, we must either develop a sales and marketing organization or outsource these functions to third parties. We intend to selectively enter into collaboration agreements with leading pharmaceutical and biotechnology companies to assist us in furthering the development of our product candidates. In particular, we intend to enter into these third-party arrangements for target indications in which our potential collaborator has particular expertise or that involve a large, primary care market that must be served by large sales and marketing organizations. Our goal in these collaborations will be to maintain co-promotion or co-commercialization rights in the United States and, in some cases, other markets. There are risks involved with establishing our own sales and marketing capabilities, as well as in entering into arrangements with third parties to perform these services. For example, developing a sales force is expensive and time consuming and could delay any product launch. We will begin to incur many sales and marketing costs prior to receiving marketing approval for our product candidates. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or prohibited as a result of FDA requirements or other reasons, our investment in these functions would be lost. If we enter into arrangements with third parties to perform sales and marketing services, our product revenues could be lower than if we directly sold and marketed our products. In addition, any revenues received under such arrangements will depend on the skills and efforts of others, and we do not know whether these efforts will be successful.
38
IF THIRD PARTY PAYORS DO NOT PROVIDE COVERAGE FOR PRODUCTS WE MAY DEVELOP, OR IF THE AMOUNT THAT A THIRD PARTY PAYOR MAY REIMBURSE HEALTH CARE PROVIDERS WHO USE ANY PRODUCTS THAT WE MAY DEVELOP IS NOT ADEQUATE TO COMPENSATE THE HEALTH CARE PROVIDER FOR THE USE OF ANY SUCH PRODUCT, OUR REVENUES AND PROSPECTS FOR PROFITABILITY WILL SUFFER.
Our revenues and profits will depend heavily on purchases of any products we may develop by health care providers who receive reimbursement from third party payors, including governmental and private payors, both in the United States and abroad. Whether health care providers will purchase any products we develop will depend in part upon the coverage policies and availability of adequate reimbursement from such third party payors. Obtaining reimbursement from a third party payor will depend on whether a product is covered under the third party payor’s benefit plan. In general, to be covered, a product must fall within a designated benefit category and be medically reasonable and necessary for treatment. However, even if covered, the amount and method of reimbursement will vary based on factors such as the particular site of service in which the product is used.
Obtaining a determination that a product is covered is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payor. We may not be able to provide data sufficient to gain coverage. Even when a payor determines that a product is covered, the payor may impose limitations that preclude payment for some uses that are approved by the FDA or comparable authorities but are determined by the payor to not be medically reasonable and necessary. Moreover, eligibility for coverage does not imply that any product will be covered in all cases or that reimbursement will be available at a rate that permits the health care provider to cover its costs of using the product.
Most persons suffering from Alzheimer’s disease are elderly, and therefore we expect that coverage and reimbursement for VP4896 for this indication in the United States will be primarily through the Medicare program. Our business would be materially adversely affected if the Medicare program were to determine that it will not: (1) cover VP4896 for the treatment of Alzheimer’s disease, or any other products we may develop; or (2) adequately reimburse health care providers for the use of VP4896 or any other products we may develop.
Beginning in 2005, pursuant to the Medicare Prescription Drug, Improvement and Modernization Act of 2003, Medicare changed the formula used to calculate the reimbursement for physician-administered drugs. The price established for VP4896 under the Medicare program under the new formula will probably be less than under the previous formula. In addition, alternative methods for calculating reimbursement are used under the Medicare program depending on the benefit category and site of service, which could lower reimbursement still further. In addition, starting in January 2006, physicians now have the option of being reimbursed directly by Medicare or participating in the Competitive Acquisition Program, or CAP, whereby the physician has the option to obtain drugs from a CAP vendor who negotiates prices with drug companies and obtains reimbursement from Medicare for the drug. If substantial numbers of physicians participate in the CAP, it will likely decrease the price we can obtain for VP4896 from the CAP vendor, if it is approved for marketing.
Any products we may develop may also receive reimbursement under Medicaid. If the state-specific Medicaid programs do not provide adequate coverage and reimbursement for any products we may develop, it may have a negative impact on our operations.
The scope of coverage and payment policies varies among third-party private payors, including indemnity insurers, employer group health insurance programs and managed care plans. Such third party carriers may base their coverage and reimbursement on the coverage and reimbursement rate paid by carriers for Medicare beneficiaries. Furthermore, many such payors are investigating or implementing methods for reducing health care costs, such as the establishment of prospective payment systems. Cost containment pressures have led to an increased emphasis on the use of cost-effective products by health care providers. If private payors do not provide adequate coverage or reimbursement for any products we may develop, it could have a negative effect on revenues and results of operations.
39
FOREIGN GOVERNMENTS TEND TO IMPOSE STRICT PRICE CONTROLS, WHICH MAY ADVERSELY AFFECT OUR REVENUES, IF ANY.
In some foreign countries, particularly the countries of the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
IF PRODUCT LIABILITY LAWSUITS ARE BROUGHT AGAINST US, WE MAY INCUR SUBSTANTIAL LIABILITIES AND MAY BE REQUIRED TO LIMIT COMMERCIALIZATION OF ANY PRODUCTS THAT WE MAY DEVELOP.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and will face an even greater risk if we commercially sell any products that we may develop. Because VP4896 is being developed as an implant that will contain eight weeks’ supply of drug, we may face higher product liability risk than if our product was administered in another form, such as a daily pill. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
|
●
|
decreased demand for any product candidates or products that we may develop;
|
|
●
|
injury to our reputation;
|
|
●
|
withdrawal of clinical trial participants;
|
|
●
|
costs to defend the related litigation;
|
|
●
|
substantial monetary awards to trial participants or patients;
|
|
●
|
loss of revenue; and
|
|
●
|
the inability to commercialize any products that we may develop.
|
We have product liability insurance that covers our clinical trials up to a $10 million annual aggregate limit and subject to a per claim deductible. The amount of insurance that we currently hold may not be adequate to cover all liabilities that may incur. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for any products. Insurance coverage is increasingly expensive. We may not be able to maintain insurance coverage at a reasonable cost and we may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise.
WE FACE SUBSTANTIAL COMPETITION, WHICH MAY RESULT IN OTHERS DISCOVERING, DEVELOPING OR COMMERCIALIZING PRODUCTS BEFORE OR MORE SUCCESSFULLY THAN WE DO.
The development and commercialization of new drugs is highly competitive. We face competition with respect to our current product candidates and any products we may seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. Our competitors may develop products that are more effective, safer, more convenient or less costly than any that we are developing or that would render our product candidates obsolete or non-competitive. Our competitors may also obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours.
40
We believe that many competitors are attempting to develop therapeutics for Alzheimer’s disease, including academic institutions, government agencies, public and private research organizations, large pharmaceutical companies and smaller more focused companies.
There are currently four drugs approved for the treatment of Alzheimer’s disease in the United States. In many cases, these products have well-known brand names, are distributed by large pharmaceutical companies with substantial resources and experience and have achieved widespread acceptance among physicians and patients. See “Business—Alzheimer’s disease.” In addition, we are aware of product candidates of third parties that are in development, which, if approved, would compete against our product candidates, if approved.
Durect has made a commitment to us not to develop, commercialize or license to someone else formulations of leuprolide acetate that release a similar dose per unit of time as does VP4896, but has otherwise not committed to us that it will not develop other leuprolide acetate products, including sustained release leuprolide acetate products, using its technologies. As a result, Durect could collaborate with a third party to develop a product having a different dose that competes with VP4896, in some cases using the same Durect sustained release technology as is included in VP4896. Finally, as described under “Risk Factors—There are limitations on our patent rights relating to our product candidates and technologies, including with respect to the use of VP4896 to treat Alzheimer’s disease, that may affect our ability to exclude third parties from competing against us if we receive approval to market these product candidates or technologies,” to the extent that we market products with active ingredients that do not have composition of matter patent protection and are approved for other indications, such as leuprolide acetate, it is possible that we will experience competition from off label sales of products containing these active ingredients.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to or necessary for our programs or advantageous to our business.
VP4896, our lead product candidate for the treatment of mild to moderate Alzheimer’s disease, is a small biodegradable implant that is administered once every eight weeks by a physician in the physician’s office through a minor surgical procedure. All drugs currently marketed for the treatment of Alzheimer’s disease, and many of the drugs under development for this indication, are orally self-administered pills. If VP4896’s administration method is not accepted by patients or physicians, we may not generate significant product revenues and our business may be materially harmed.
OUR BUSINESS ACTIVITIES INVOLVE THE USE OF HAZARDOUS MATERIALS, WHICH REQUIRE COMPLIANCE WITH ENVIRONMENTAL AND OCCUPATIONAL SAFETY LAWS REGULATING THE USE OF SUCH MATERIALS. IF WE VIOLATE THESE LAWS, WE COULD BE SUBJECT TO SIGNIFICANT FINES, LIABILITIES OR OTHER ADVERSE CONSEQUENCES.
Our research and development programs involve the controlled use of hazardous materials. Accordingly, we are subject to federal, state and local laws governing the use, handling and disposal of these materials. Although we believe that our safety procedures for handling and disposing of these materials comply in all material respects with the standards prescribed by state and federal regulations, we cannot completely eliminate the risk of accidental contamination or injury from these materials. In addition, our collaborators may not comply with these laws. In the event of an accident or failure to comply with environmental laws, we could be held liable for damages that result, and any such liability could exceed our assets and resources.
41
WE MUST COMPLY WITH FEDERAL, STATE AND FOREIGN LAWS, REGULATIONS, AND OTHER RULES RELATING TO THE HEALTH CARE BUSINESS AND, IF WE ARE UNABLE TO FULLY COMPLY WITH SUCH LAWS, REGULATIONS AND OTHER RULES, WE COULD FACE SUBSTANTIAL PENALTIES.
We are or will be, directly or indirectly through our customers, subject to extensive regulation by the federal government, the states and foreign countries in which we may conduct our business. The laws that directly or indirectly affect our ability to operate our business include the following:
|
●
|
the federal Medicare and Medicaid Anti-Kickback law, which prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce either the referral of an individual, or furnishing or arranging for a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid Programs;
|
|
●
|
other Medicare laws, regulations, rules, manual provisions and policies that prescribe the requirements for coverage and payment for services performed by our customers, including the amount of such payment;
|
|
●
|
the federal False Claims Act, which imposes civil and criminal liability on individuals and entities who submit, or cause to be submitted, false or fraudulent claims for payment to the government;
|
|
●
|
the federal False Statements Act, which prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; and
|
|
●
|
state and foreign law equivalents of the foregoing.
|
If our operations are found to be in violation of any of the laws, regulations, rules or policies described above or any other law or governmental regulation to which we or our customers are or will be subject, or if the interpretation of the foregoing changes, we may be subject to civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs and the curtailment or restructuring of our operations. Similarly, if our customers are found non-compliant with applicable laws, they may be subject to sanctions, which could also have a negative impact on us. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations, and additional legal or regulatory change. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
WE HAVE SUBSTANTIAL INDEBTEDNESS TO TRADE CREDITORS AND SEVERAL TRADE CREDITORS HAVE OBTAINED JUDGMENTS AGAINST US.
We have substantial indebtedness to trade creditors relating to goods and services provided to us in connection with our truncated Phase III trials. In general, our relationship with our trade creditors have remained cordial despite this indebtedness and the vast majority of our trade creditors have cooperated with us in our efforts to defer payments to them until we can obtain further financing. However, there can be no assurance that our trade creditors will continue to be cooperative and one or more of them may choose to bring suit to enforce their claims. One trade creditor won a judgment of approximately $1,100,000 against us and we subsequently entered into a negotiated payment arrangement with this creditor, which reduced the ultimate liability to $900,000. In addition, several other trade creditors have obtained default judgments against us, although to our knowledge none of such creditors have initiated any court proceedings to enforce those judgments. To date, the total amount of judgments rendered against us is $1.4 million.
42
OUR FUTURE SUCCESS DEPENDS ON OUR ABILITY TO RETAIN OUR CHIEF EXECUTIVE OFFICER AND OTHER KEY EXECUTIVES AND TO ATTRACT, RETAIN AND MOTIVATE QUALIFIED PERSONNEL.
We are highly dependent on Timothy R. Wright, our Chairman and Chief Executive Officer, Judith S. T. Geaslen, our Vice President of Finance and members of our current Board of Directors. The loss of the services of any of these persons might impede the achievement of our research, development, and commercialization objectives. At this time, we do not have employment agreements with Mr. Wright and Ms. Geaslen. The Company does not expect to enter into any employment contacts with its officers in the near term as due to the Company’s current financial condition it is unlikely that the Company would be able to reasonably meet established financial commitments of such contracts.
Recruiting and retaining qualified scientific personnel and sales and marketing personnel will also be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
RISKS RELATED TO OUR COMMON STOCK
ANY ADDITIONAL FUNDING WE ARRANGE THROUGH THE SALE OF OUR COMMON STOCK WILL RESULT IN DILUTION TO EXISTING SECURITY HOLDERS.
We will have to raise additional capital in order for our business plan to succeed. Wherever possible, our board of directors will attempt to use non-cash consideration to satisfy obligations. Therefore, our most likely source of additional capital will be through the sale of additional shares of our common stock. Our board of directors has authority, without action or vote of the shareholders, to issue all or part of the authorized 480,000,000 shares. In addition, if a trading market develops for our common stock, we may attempt to raise capital by selling shares of our common stock, possibly at a discount to market. Such issuances will cause our security holders’ interests in our Company to be diluted, which may negatively affect the value of their shares.
CURRENTLY, THERE IS NO PUBLIC MARKET FOR OUR SHARES OF COMMON STOCK, AND THERE CAN BE NO ASSURANCE THAT ANY PUBLIC MARKET WILL EVER DEVELOP OR THAT OUR COMMON STOCK WILL BE QUOTED FOR TRADING.
Our common stock is listed on the OTC Markets OTCQB under the symbol “CURX. QB.” However, there is a limited trading market for our shares of common stock. Even if we are successful in developing a public market, there may not be enough liquidity in such market to enable our security holders to sell their stock. If a public market for our common stock does not develop, investors may not be able to re-sell the shares of its common stock, rendering their shares effectively worthless and resulting in a complete loss of their investment. We cannot predict the extent to which investor interest in our Company will lead to the development of an active, liquid trading market. Active trading markets generally result in lower price volatility and more efficient execution of buy and sell orders for investors.
IN THE EVENT THAT A PUBLIC MARKET DOES DEVELOP, THE PRICE OF OUR COMMON STOCK WILL BE SUBJECT TO SIGNIFICANT PRICE FLUCTUATIONS.
Until an orderly market develops in our common stock, if ever, the price at which our common stock trades is likely to fluctuate significantly. Prices for our common stock will be determined in the marketplace and may be influenced by many factors, including the depth and liquidity of the market for shares of our common stock, developments affecting our business, including the impact of the factors referred to elsewhere in these Risk Factors, investor perception of our Company and general economic and market conditions. No assurances can be given that an orderly or liquid market will ever develop for the shares of our stock.
In addition, our common stock may not be followed by any market analysts, and there may be few institutions acting as market makers for our common stock. Currently, there are five institutions acting as market makers, but there can be no guarantee that they will continue to do so or that any other institutions in the future will act as a market make for our common stock. Either of these factors could (i) adversely affect the trading price of our common stock and (ii) severely limit the liquidity of our common stock.
43
ANY TRADING MARKET THAT MAY DEVELOP MAY BE RESTRICTED BY VIRTUE OF STATE SECURITIES “BLUE SKY” LAWS, MAKING IT DIFFICULT OR IMPOSSIBLE FOR OUR SECURITY HOLDERS TO SELL SHARES OF ITS COMMON STOCK IN THOSE STATES.
There is a limited public market for our shares of common stock, and there can be no assurance that any public market will develop in the foreseeable future. Transfer of our common stock may also be restricted under the securities regulations and laws promulgated by various states and foreign jurisdictions, commonly referred to as “Blue Sky” laws, which prohibit trading absent compliance with individual state laws. Absent compliance with such individual state laws, our common stock may not be traded in such jurisdictions.
BECAUSE WE ARE SUBJECT TO THE “PENNY STOCK” RULES, THE LEVEL OF TRADING ACTIVITY IN OUR COMMON STOCK MAY BE REDUCED.
Broker-dealer practices in connection with transactions in “penny stocks” are regulated by penny stock rules adopted by the Securities and Exchange Commission. Penny stocks generally are equity securities with a price of less than $5.00 (other than securities registered on some national securities exchanges). The penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document that provides information about penny stocks and the nature and level of risks in the penny stock market. The broker-dealer also must provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction, and, if the broker-dealer is the sole market maker, the broker-dealer must disclose this fact and the broker-dealer’s presumed control over the market, and monthly account statements showing the market value of each penny stock held in the customer’s account. In addition, broker-dealers who sell these securities to persons other than established customers and “accredited investors” must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. Consequently, these requirements may have the effect of reducing the level of trading activity, if any, in the secondary market for a security subject to the penny stock rules, and investors in our common stock may find it difficult to sell their shares.
WE DO NOT EXPECT TO PAY DIVIDENDS TO HOLDERS OF OUR COMMON STOCK IN THE FORESEEABLE FUTURE. AS A RESULT, HOLDERS OF OUR COMMON STOCK MUST RELY ON STOCK APPRECIATION FOR ANY RETURN ON THEIR INVESTMENT.
We have not declared any dividends since our inception, and we do not plan to declare any dividends in the foreseeable future. If we were to become profitable, it would be expected that all of such earnings would be retained to support our business. Accordingly, holders of our common stock will have to rely on capital appreciation, if any, to earn a return on their investment in our common stock.
CERTAIN PROVISIONS OF OUR CHARTER COULD DISCOURAGE POTENTIAL ACQUISITION PROPOSALS OR CHANGE IN CONTROL.
Our board of directors, without further stockholder approval, may issue preferred stock that would contain provisions that could have the effect of delaying or preventing a change in control or which may prevent or frustrate any attempt by stockholders to replace or remove the current management. The issuance of additional shares of preferred stock could also adversely affect the voting power of the holders of our common stock, including the loss of voting control to others.
WE HAVE RECENTLY CHANGED OUR SENIOR MANAGEMENT TEAM
During 2011, we experienced the resignation of a majority of our executive officers and directors, and currently have only two executive officers, one of whom is serving as Chief Executive Officer on an interim basis. While our new executive officer and directors have prior experience in the biotechnology field, there are no assurance that they will be able to secure necessary financing or be able to properly implement the Company business plan.
ITEM 1B. UNRESOLVED STAFF COMMENTS.
Not applicable.
44
ITEM 2. PROPERTIES.
Our executive office is currently leased to us by Judith Geaslen, Vice President - Finance, who provides the space to us at no charge. The address of our executive office is 1004 Chagford Way, Raleigh, NC 27614. We are confident that as the Company’s need for space increases, suitable space will be available at commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS.
In 2008, Aptuit, Inc. obtained a judgment for approximately $1.1M in the Circuit Court of Jackson County, Missouri at Kansas City, relating to work it performed for Curaxis in connection with our truncated ALADDIN 301 trial. Aptuit’s claims were based on invoices previously rendered to Curaxis in connection with that work and on a promissory note that Curaxis had delivered to Aptuit in connection with those invoices. We subsequently entered into a settlement agreement with Aptuit that requires payment of $900,000 over a two year period to satisfy this judgment. Payments to date under the settlement agreement total $400,000.
In 2009, Genzyme Corporation obtained a judgment for $425,000 in the United States District Court for the District of Massachusetts, relating to leuprolide supplied to Curaxis in 2006 for our truncated ALADDIN 301 trial. Curaxis did not oppose the entry of this judgment, as it had previously acknowledged the validity of this debt to Genzyme. There have been no actions initiated by, or on behalf of, the creditor in the last six months with respect to settlement and/or payment of this judgment.
In 2009, Margolin Brain Institute obtained a default judgment for $128,469, including interest and attorneys fees, in the Superior Court of the State of California, Fresno County - Civil Division, relating to work it performed in our truncated ALADDIN 301 trial. There have been no actions initiated by the creditor in the last 12 months with respect to settlement and/or payment of this judgment.
Other than listed above, we are currently not involved in any litigation that we believe could have a material adverse effect on our financial condition or results of operations. There is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of the executive officers of our company or any of our subsidiaries, threatened against or affecting our company, our common stock, any of our subsidiaries or of our companies or our subsidiaries’ officers or directors in their capacities as such, in which an adverse decision could have a material adverse effect.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable.
45
PART II
ITEM 5. MARKET FOR COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
(a) Market Information
Our common stock is quoted on the OTCQB under the trading symbol “CURX.QB.” To date, our common stock has traded with limited volume.
The following table sets forth the range of the high and low bid quotations of the Common Stock for the past two years in the over-the-counter market, as reported by the OTC Bulletin Board and in the Pink Sheets. The quotations reflect inter-dealer prices without retail mark-up, mark-down or commission, and may not represent actual transactions.
Calendar Quarter Ended:
|
High
|
Low
|
|||||||
|
2011
|
||||||||
|
December 31
|
$
|
0.15
|
0.04
|
|||||
|
September 30
|
$
|
0.45
|
0.08
|
|||||
|
June 30
|
$
|
0.57
|
0.26
|
|||||
|
March 31
|
$
|
0.90
|
0.46
|
|||||
|
2010
|
||||||||
|
December 31
|
$
|
1.47
|
0.28
|
|||||
|
September 30
|
$
|
1.90
|
0.90
|
|||||
|
June 30
|
$
|
1.30
|
0.40
|
|||||
|
March 31
|
$
|
1.00
|
0.20
|
|||||
(b) Holders
As of March 28, 2012, the Company had 1,254 stockholders of record of our common stock. This figure does not take into account those shareholders whose certificates are held in the name of broker-dealers, “street name,” or other nominees.
(c) Dividends
We have not declared any dividends in the past, and we do not plan to declare dividends in the future.
Recent Sales of Unregistered Securities
During 2011, we issued 190,000 shares of common stock in exchange for consulting services rendered and 329,800 shares of common stock for services rendered by members of the Board of Directors.
During 2011, we issued 2,482,632 shares of common stock as a result of warrant conversions. At December 31, 2011, warrants outstanding for the purchase or the Company’s common stock were as follows:
|
# of Shares
|
Exercise Price
|
Expiration Date
|
|||||
| 844,400 | $ | 0.22 |
August 31, 2016
|
||||
| 3,589,500 | $ | 0.001 |
July 29, 2017
|
||||
| 820,800 | $ | 1.52 |
September 15, 2015
|
||||
| 100,000 | $ | 0.30 |
August 31, 2015
|
||||
| 5,354,700 | |||||||
46
During 2010, the Company issued 500 shares of Series A and 500 shares of Series B convertible preferred stock. Each share of Series A and Series B preferred stock has a par value of $.0001 and an initial stated value equal to $1,000. Each share of preferred stock shall be convertible at any time at the option of the Holder into common stock. The number of shares of common stock to be issued upon conversion of each share of preferred stock will be determined by dividing the initial stated value of the preferred stock plus dividends added to the stated value upon such election by the Holder, by a pre-determined conversion rate. The predetermined conversion rates for the Series A and Series B preferred stock is $0.35 and $0.50, respectively. Assuming conversion of all shares of Series A and B preferred stock issued, 2,549,398 shares of the Company’s common stock would be issued.
During 2011, the Company issued 636 shares of Series C convertible preferred stock. Each share of Series C stock has a par value of $.0001 and an initial stated value equal to $1,000. Each share of preferred stock shall be convertible at any time at the option of the Holder into common stock. The number of shares of common stock to be issued upon conversion of each share of preferred stock will be determined by dividing the initial stated value of the preferred stock plus dividends added to the stated value upon such election by the Holder, by a pre-determined conversion rate. The predetermined conversion rates for the Series C preferred stock is $0.50. During 2011, the company issued 1,200,000 shares of commons stock upon conversion of 600 shares of the Series C convertible preferred stock. Assuming conversion of the remaining shares of Series C preferred stock issued, 77,740 shares of the Company’s common stock would be issued.
During 2011, we issued 372,330 shares of common stock upon the exercise of outstanding stock options. At December 31, 2011, 3,736,000 options were outstanding for the purchase of shares of the company’s stock. The weighted average exercise price and the weighted average contractual term of the outstanding options were $1.64 and 5.24 years, respectively.
ITEM 6. SELECTED FINANCIAL DATA.
Not applicable for smaller reporting companies.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following discussion and analysis of our financial condition and results of our operations should be read in conjunction with the financial statements and the notes thereto of Curaxis Pharmaceutical Corporation which appear elsewhere in this Annual Report. The results shown herein are not necessarily indicative of the results to be expected for any future periods.
Forward Looking Statements
The Company is including the following cautionary statement in this Annual Report on Form 10-K for any forward-looking statements made by, or on behalf of, the Company. Forward-looking statements include statements concerning plans, objectives, goals, strategies, future events or performance and underlying assumptions and other statements which are other than statements of historical facts. Certain statements contained herein are forward-looking statements and accordingly involve risks and uncertainties which could cause actual results or outcomes to differ materially from those expressed in the forward-looking statements. Our expectations, beliefs and projections are expressed in good faith and are believed by us to have a reasonable basis, including without limitation, management’s examination of historical operating trends, data contained in our records and other data available from third parties, but there can be no assurance that management’s expectation, beliefs or projections will result or be achieved or accomplished. In addition to other factors and matters discussed elsewhere herein, the following are important factors that, in our view, could cause actual results to differ materially from those discussed in the forward-looking statements: technological advances by our competitors, changes in health care reform, including reimbursement programs, capital needs to fund any delays or extensions of development programs, delays in the manufacture of new and existing products by us or third party contractors, market acceptance of our products, the loss of any key employees, delays in obtaining federal, state or local regulatory clearance for new installations and operations, changes in governmental regulations, and the availability of capital on terms satisfactory to us. We are also subject to numerous Risk Factors relating to manufacturing, regulatory, financial resources and personnel as described in this Annual Report. We disclaim any obligation to update any forward-looking statements to reflect events or circumstances after the date hereof.
47
Overview
We are an emerging specialty pharmaceutical company with a pipeline of product candidates in Alzheimer’s disease and oncology. Curaxis’ most advanced product candidate is Memryte (generic name VP4896), a proprietary, small, biodegradable implant that is comprised of leuprolide acetate and a polymer. Curaxis’ therapeutic platform is based on the hypothesis that many diseases of aging may be caused by age-related changes in the function of the hypothalamic-pituitary-gonadal (HPG) axis. The HPG axis is a hormonal endocrine feedback loop that controls development, reproduction and aging in animals. We believe that Curaxis’ discovery of similar hormonal signaling mechanisms at the cellular level in brain tissue from Alzheimer’s patients and in multiple tumors may enable us to develop significant new treatments for Alzheimer’s disease as well as many tumors.
From inception through early 2006, Curaxis completed two Phase II clinical trials, Aladdin I and Aladdin II, to support the efficacy of its drug candidate in women and men, respectively. In addition, a Phase I trial was completed to support safety and the drug release profile of the proprietary subcutaneous implant. In August 2006, Curaxis completed enrollment in Aladdin 301, the first of its two Phase III clinical trials of the VP4896 implant for the treatment of mild to moderate Alzheimer’ s disease. In October 2006, this trial was discontinued due to financial constraints and converted to a Phase II trial. Curaxis has not conducted material research and development activities for Memryte since terminating its clinical trials and closing its research facility in 2007.
Since Curaxis’ inception, it has not received approval to market any product, has had no revenues from product sales and has devoted substantially all of its efforts to the development of its treatment for mild to moderate Alzheimer’s disease. Curaxis funded its operations primarily through the sale of common stock, preferred stock and warrants to private investors, which have provided net cash proceeds of approximately $76.5 million as of December 31, 2011. Curaxis has never been profitable and, as December 31, 2011, it had an accumulated deficit of $88.5 million. Curaxis had net income of $1.3 million for the year ended December 31, 2011 and a net loss of $88.4 million for the cumulative period from inception through December 31, 2011. Net income for 2011 was the result of a gain of $2.2 million recognized on the reduction of liabilities at December 31, 2011; the obligations principally relate to goods acquired or services rendered while the Company was actively engaged in the terminated Phase III clinical trial. The balances subject to reduction include accounts payable represented by vendor invoices submitted to the company for payment, retained billings and accruals for estimated services rendered by vendors through the termination of the Phase III clinical trial but not billed to date. Very little, if any, collection activity has occurred. In the opinion of Company’s counsel as a result of the passage of time these liabilities cannot be legally enforced against the Company. A net loss of $2.2 million was recorded for the ended December 31, 2010. At December 31, 2011 the Company had $22 thousand in cash and cash equivalents.
We are continuing our business plan of developing our drug candidate, Memrtye, for the treatment of mild to moderate Alzheimer’s disease. We expect our minimum cash needs to fund our operating expenses and capital expenditures, excluding direct costs of a potential Phase IIb or registration trial, for the twelve-month period ending December 31, 2012 to be approximately $2.2 million. We expect to incur significant and increasing operating losses for at least the foreseeable future as we advance our drug candidate through clinical trials and seek regulatory approval and the eventual commercialization if and when, we may receive regulatory approval. We will need to generate significant revenues to achieve profitability and we may never do so.
We do not expect to generate product revenue for at least the next several years, if at all. If any of our programs experience delays or do not result in a commercial product, we would not generate revenue from that program in a timely manner or at all. We expect that our operating expenses will continue to increase and may vary substantially from quarter to quarter and year-to-year based on the timing of clinical trial patient enrollment and our other research and development activities. In particular, as we initiate a potential Phase IIb and/or pivotal Phase III trials of Memryte, we expect that our research and development expenses will increase significantly. We expect general and administrative costs also to increase as we add personnel. In connection with our preparation for the commercial launch of Memryte, we expect that our capital expenditures may increase as a result of increased manufacturing equipment costs. We believe that period-to-period comparisons of our results of operations are not meaningful and should not be relied on as indicative of our future performance.
48
The successful development of pharmaceuticals, including our lead product candidate, Memryte, is uncertain. We estimate that we will incur substantial expenses over the next several years in order to complete the clinical development of Memryte in accordance with our current development plan and seek FDA approval to market Memryte. If we or the FDA cause a change or delay in our development plan for Memryte, our costs could increase substantially.
It is our intent to seek an alliance with a strategic partner to assist in the funding and execution of drug development activities. We cannot reasonably estimate or know the nature, timing and estimated expenses of the efforts necessary to complete the remainder of the development of, or the period, if ever, in which material net cash inflows will commence from Memryte for this indication or other product candidates due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
|
●
|
the scope, rate of progress and expense of our clinical trials and other research and development activities;
|
|
●
|
future clinical trial results;
|
|
●
|
the expense, timing and outcome of seeking regulatory approvals;
|
|
●
|
the expense of establishing clinical and commercial supplies of our product candidates and any products that we may develop;
|
|
●
|
the effect of competing technological and market developments; and
|
|
●
|
the expense of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights.
|
Critical Accounting Policies and Estimates
The discussion and analysis of Curaxis’ financial condition and results of operations set forth below are based on its financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America, or “GAAP.” The preparation of these financial statements requires Curaxis to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. Curaxis based its estimates on historical experience and on various other assumptions that it believed to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe the following accounting policies are the most critical to Curaxis’ financial statements. We believe they are important to the presentation of its financial condition and require the greatest degree of management judgment to make the estimates necessary to ensure their fair presentation.
Accrued Expenses
As part of the process of preparing financial statements, Curaxis is required to estimate accrued expenses. This process involves identifying services which have been performed on its behalf and estimating the level of service performed and the associated cost incurred for such service as of each balance sheet date in its financial statements. Services for which it must estimate accrued expenses include:
|
●
|
costs of services performed by contract research organizations and clinical sites in conjunction with clinical trials;
|
|
●
|
costs of services performed by contract manufacturers in conjunction with the production of clinical trial materials; and
|
|
●
|
professional service fees and expenses.
|
49
In accruing service fees, Curaxis estimates the time period over which services will be provided and the level of effort in each period. If the actual timing of the provision of services or the level of effort varies from the estimate, it will adjust the accrual accordingly. The majority of its service providers invoice Curaxis monthly in arrears for services performed. In the event that Curaxis does not identify costs that have begun to be incurred or it underestimates or overestimates the level of services performed or the costs of such services, its actual expenses could differ from such estimates. The date on which some services commence, the level of services performed on or before a given date and the cost of such services are often subjective determinations. Curaxis made judgments based upon the facts and circumstances known to it in accordance with GAAP.
Research and Development
Curaxis expenses all research and development costs as incurred. Research and development expense includes, among other things, clinical trial costs. It accounts for its clinical trial costs by estimating the total cost to treat a patient in each clinical trial and recognizing this cost, based on a variety of factors, beginning with the preparation for the clinical trial. This estimated cost includes payments to its contract research organizations for trial site and patient-related costs, including laboratory costs related to the conduct of the trial, and other costs. Its cost per patient varies based on the type of clinical trial, the site of the clinical trial and the length of the treatment period for each patient. As actual costs become known to it, it adjusts its accrual. These changes in estimates may result in a material change in its clinical study accrual, which could materially affect its results of operations. Research and development expenses includes those costs described under “Financial Operations Overview—Research and Development” below.
Stock-Based Compensation
As a normal practice, Curaxis compensates employees and non-employee directors through stock-based compensation. It applies ASC 718 Compensation – Stock Compensation, to stock-based compensation awards. ASC 718 requires the measurement and recognition of non-cash compensation expense for all share-based payment awards made to employees and directors, including employee stock options related to its 2001 Stock Option Plan and 2005 Stock Option Plan, based on fair values. Curaxis adopted this Statement on January 1, 2006 using the modified prospective application.
Stock compensation arrangements with non-employee service providers are accounted for in accordance ASC 505-50 Equity-Based Payments to Non-Employees, using a fair value approach. The compensation costs of these arrangements are subject to re-measurement over the vesting terms as earned.
The measurement of stock-based compensation requires the use of complex option pricing models and application of judgment in selecting the appropriate valuation assumptions, such as volatility and expected term. Curaxis values its stock-based compensation using the Black-Scholes option pricing model, in accordance with the requirements of ASC 718.
Curaxis does not have sufficient history of exercise behavior to develop estimates of expected terms since as of December 31, 2011, only two grants had been exercised since inception of the plans. The expected remaining terms are based on the estimated time period to elapse prior to commercialization of Curaxis’ initial product. For options to outside consultants, Curaxis uses a term which is equal to the average period required for the options to fully vest and the contractual life of the option. As Curaxis gathers more history regarding its option exercise pattern, and as information regarding the option pattern of comparable companies in its industry becomes publicly available, it will review these estimates and revise them as appropriate. Different estimates of volatility and expected term could materially change the value of an option and the resulting expense.
In addition, Curaxis monitors equity instruments with non-standard provisions, such as performance-based vesting conditions, accelerated vesting based on achievement of performance milestones and features that require instruments to be accounted for as liabilities. Curaxis accounts for transactions in which services are received from non-employees in exchange for equity instruments based on the fair value of such services received or of the equity instruments issued, whichever is more reliably measured, in accordance with ASC 718 and ASC 505-50.
50
Accounting for equity instruments granted or issued by us under ASC 718 and ASC 505-50 requires fair value estimates of the equity instrument granted or sold. If Curaxis’ estimates of the fair value of these equity instruments are too high or too low, its expenses may be over or understated. Prior to the Merger transaction, the fair value of its common stock was determined by its board of directors. In the absence of a public trading market for its common stock, its board of directors considered objective and subjective factors in determining the fair value of its common stock. In all periods, the board of directors evaluated events that provided indicators of the fair value of its common stock. These events included, depending on the period, the purchase price of its common stock that was issued in private placements through July 29, 2010 and the results of its clinical trials. Based on these factors, its board of directors determined that the value of the common stock underlying the stock options granted to employees through the Merger date of July 29, 2010 had a fair value that was equal to the exercise price.
Recently Issued Accounting Pronouncements
Curaxis’ management does not believe that any recently issued, but not yet effective, accounting standards if currently adopted would have a material effect on the accompanying financial statements.
Financial Operations Overview
Revenue
Curaxis had not yet generated any product revenue and does not expect to generate any product revenue for at least the next several years. It plans to seek to generate revenue from product sales if any of its drug candidates receive marketing approval and it begins commercial activities. It expected that any revenue it generates would fluctuate from quarter-to-quarter as a result of the timing, nature and amount of its commercialization activities, whether through direct sales by it or by payments under distribution or other collaboration agreements.
Research and Development Expense
Research and development expense consists primarily of:
|
●
|
salaries and related expenses for personnel engaged in research and development activities;
|
|
●
|
fees paid to contract research organizations and clinical sites in conjunction with clinical trials;
|
|
●
|
fees paid to research organizations in conjunction with preclinical studies;
|
|
●
|
costs of materials used in research and development;
|
|
●
|
consulting and sponsored research fees paid to third parties;
|
|
●
|
fees paid to external legal counsel and third parties filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and
|
|
●
|
depreciation of capital assets used to discover and develop our product candidates.
|
Curaxis expensed both internal and external research and development costs as incurred. Research and development expenses have diminished over the last several years due to termination of its clinical trials and closure of its research facility.
Substantially all research and development expenses incurred by Curaxis to date relate to its efforts to develop Memryte for the treatment of mild to moderate Alzheimer’s disease. Curaxis expects that a substantial percentage of its research and development expense in the future will be incurred in support of its current and future pre-clinical and clinical development programs. These expenditures are subject to numerous uncertainties in timing and cost to completion.
51
General and Administrative
General and administrative expense consists primarily of salaries and related expenses for personnel in administrative, finance, operations and human resource functions. Other costs include fees for general legal and other professional services, accounting fees, and directors’ and officers’ liability insurance premiums.
Marketing
Marketing expense consists primarily of salaries and related expenses for personnel responsible for clinical trial recruitment and costs of attendance at various scientific and industry meetings. Other costs include advertising for patient enrollment of clinical trials, promotion materials relating to scientific publications and fees for professional services related to public relations. Curaxis expects these expenses to increase significantly as it begins to develop its own commercialization capabilities.
Net Interest Income (expense)
Interest income consists of interest earned on Curaxis’ cash and cash equivalents. Interest expense consists of interest incurred on notes and mortgage debt payable and equipment and insurance premium debt financing.
Results of Operations
The results of operations of Curaxis for each of the years ended December 31, 2011, and 2010 are as follows:
|
Year Ended December 31,
|
||||||||
|
2011
|
2010
|
|||||||
|
(Dollars in thousands)
|
||||||||
|
Operating expenses:
|
||||||||
|
Research and development
|
$
|
33
|
78
|
|||||
|
General and administrative
|
1,889
|
1,859
|
||||||
|
Total operating expenses
|
1,922
|
1,937
|
||||||
|
Loss from operations
|
(1,922
|
)
|
(1,937
|
)
|
||||
|
Gain on debt restructuring
|
-
|
204
|
||||||
|
Gain on reduction of liablities
|
2,233
|
-
|
||||||
|
Interest income (expense), net
|
(263
|
)
|
(175
|
)
|
||||
|
Interest expense from derivative liability
|
-
|
(2,586
|
)
|
|||||
|
Change in fair value of derivative liability
|
1,286
|
2,250
|
|
|||||
|
Net income (loss)
|
$
|
1,334
|
(2,244
|
|||||
Curaxis accounts for equity awards to employees and non-employee directors and service providers under ASC 718 Compensation – Stock Compensation and ASC 505-50 Equity-based Payments to Non-Employees. As of December 31, 2011, there was $0.3 million of total unrecognized compensation costs related to unvested stock options. That cost is expected to be amortized on a straight-line basis over a weighted average service period of 1.6 years. Stock-based compensation expense recorded in the years ended December 31, 2011 and 2010 was $0.2 million and $0.5 million, respectively. During the year ended December 31, 2011, 130 thousand unvested stock options were cancelled upon the resignation of certain executive officers and directors. In addition, the Company extended the contractual life of 1.5 million vested options held by the former employees as of their respective resignation date. No additional expense was recognized as a result of the modifications.
52
Year Ended December 31, 2011 Compared to Year Ended December 31, 2010
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Research and development costs
|
$
|
33
|
|
$
|
78
|
$
|
(45)
|
(57.7)
|
%
|
|||||||
|
Percent of total operating expenses
|
1.7
|
%
|
4.0
|
%
|
||||||||||||
During 2007, Curaxis terminated all clinical trial activities and closed its’ research facility. Further, the Company did not employ any personnel devoted to research and development activities in 2011 and 2010. As a result, research and development expenses in 2011 and 2010 were limited to legal fees associated with the pursuit and maintenance of various patents and pending patent applications related to our Alzheimer’s disease and oncology drug candidates and depreciation of remaining capital assets used in the manufacturing of our proprietary implant. All capital assets used in the manufacturing of our propriety implant were fully depreciated at December 31, 2009.
The decrease in research and development expenses for the year ended December 31, 2011, as compared to the same period of 2010, reflects monies received under a government grant program in July of 2011. Specifically, the Company received $39 thousand in grants proceeds awarded by the US Department of Health and Human Services under the Qualifying Therapeutic Discovery Project Program. The application for the grant was filed by the Company in 2010 based on estimated research and development expenses and subsequently amended in 2011 to reflect actual 2010 research and development expenses incurred. Due to the contingent nature of the award, no recognition of the potential award was made until actual receipt of the proceeds.
Legal expense fees for the year ended December 31, 2011, as compared to the same period of 2010, decreased $6 thousand as very limited work was completed on our patent positions in the earlier part of 2011.
General and Administrative Expenses
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
General and administrative expenses
|
$
|
1,889
|
$
|
1,859
|
$
|
30
|
1.6
|
%
|
||||||||
|
Percent of total operating expenses
|
98.3
|
%
|
96.0
|
%
|
||||||||||||
The increase in general and administrative expenses for the year ended December 31, 2011, as compared to the same period for 2010, is related to a increase in consulting and advisory fees of $49 thousand, an increase in Directors’ and Officers’ (“D&O”) liability insurance of $75 thousand, an increase in Board of Directors fees of $112 thousand and an increase in expense associated with investor relations activities of $33 thousand.
The increase in consulting and advisory fees recognized reflects expenses associated with the recruitment of the new Board of Directors and executive officers; fees paid to a European advisor upon commencement of Company’s European capital market initiatives in 2011; and fees paid to advisory groups engaged to assist the Company with identification of financing alternatives and overall domestic market exposure. These fees recognized in 2011 were offset by a reduction in fees recognized under the Company’s agreements with Southridge and Canterbury and a reduction in payments to miscellaneous consultants..
Effective June 24, 2011, two of the executive officers of the Company resigned and five of the existing Board members resigned, six new members were appointed to the Board of Directors and one of the new Board members was appointed as Interim Chief Executive Officer. Expenses totaling $119 were recognized by the Company for professional recruitment and legal services rendered to effect the transition.
On February 8, 2011, the Company’s stock was approved for listing on the Frankfurt Exchange under the symbol “8CX.” In anticipation of the listing, the Company engaged a European advisor, Continental Advisors, to complement its European capital markets initiatives as well as explore further business development or European strategic partnerships. Fees paid to Continental Advisors in 2011 totaled $202 thousand. Along with the advisory services rendered, the fees also reflect work contracted by Continental Advisors on the Company’s behalf for preparation and submission of the foreign exchange application, public relations activities, analyst research, and escrow agent and stock placement fees.
53
During 2011 the Company engaged two advisory groups to assist in the identification of financing alternatives and to increase the Company’s domestic market exposure. Fees paid to the advisory groups totaled $84 thousand; in lieu of cash payments, the company issued restricted stock to cover a significant portion of the billings. Specifically, during 2011, the company issued a total of 190 thousand shares of common stock to the identified groups with an estimated fair value of $84 thousand.
During the year ended December 31, 2011, total expenses recognized under the under agreements executed between the Company and Southridge and Canterbury totaled $58 thousand compared to $415 thousand for the corresponding period of 2010. Pursuant to the executed agreements, Southridge and Canterbury were to assist Curaxis in negotiating settlements with its creditors to eliminate or substantially reduce and defer its trade date and assist Curaxis in merging with a publicly traded corporation to establish a public trading market for its stock. Compensation for the services rendered includes monthly fees plus warrants issued for the purchase of the Company’s stock at agreed upon terms upon consummation of the proposed merger transaction. The agreement with Canterbury expired during the first quarter of 2011 and the agreement with Southridge was terminated in May 2011. In addition, the expense reported for 2010 includes warrant compensation recognized under the Southridge agreement and totaling $181 thousand. No warrant compensation was recognized in 2011 under the agreement.
The premium expense for D&O liability insurance totaled $126 thousand for the year ended December 31, 2011 compared to $51 thousand for the year ended December 31, 2010. Board of Directors fees for the year ended December 31, 2011 totaled $136 thousand compared to $24 thousand for the year ended December 31, 2010. The Company did not recognize expenses for D&O insurance premiums and Board of Directors fees prior to the merger transaction effective July 29, 2010.
During 2011, the Company filed its first Annual Report and Proxy statement and completed its first annual Shareholders meeting; completed the initial conversion of its corporate financial filings to the XRBL platform as required by the SEC and experienced increased stock issuance activity related to warrant exercises and puts under the Equity Credit Agreement. These activities resulted in an overall increase in expenses associated with investor relations of $33 thousand.
The increases noted above were offset by a decrease in payroll and related benefit expense of $73 thousand; a decrease in website development and public relations fees of $84 thousand; a decrease in occupancy expense of $60 thousand; and a decrease in accounting and auditing fees of $19 thousand.
Payroll and related benefit expense for the year ended December 31, 2011, totaled $763 thousand compared to $836 thousand for the corresponding period of 2010. No compensation was paid or accrued for any employees for the two month period ended December 31, 2010 which reduced the 2010 expense from a projected $946 thousand expense to the reported $836 thousand. Effective June 24, 2011, two of the executive officers of the Company resigned and five of the existing Board members resigned, six new members were appointed to the Board of Directors and one of the new Board members was appointed as Interim Chief Executive Officer. The decrease in headcount resulted in a decrease of $101 thousand in the 2011 expense recorded as compared to the projected 2010 expense. The reduced expense for 2011 also reflects a reduction is stock based compensation of $58 thousand, a reduction in bonus expense of $15 thousand and a reduction in health insurance and other benefits expense of $9 thousand.
During the year ended December 31, 2011, expenses recognized for website development and maintenance and public relations fees totaled $14 thousand compared to $98 thousand for the corresponding period of 2010. Significant work was completed during 2010 with respect to the Company’s website in preparation of the merger transaction; expenses recognized during 2010 for development and maintenance of the website totaled $41 thousand compared to $8 thousand recognized for the corresponding period of 2011. In addition, fees paid for the promotion of the listing of the Company’s stock and public communication of the merger transaction during the 2010 totaled $57 thousand compared to fees totaling $6 thousand paid for similar services in 2011.
For the year ended December 31, 2011, occupancy expense totaled $27 thousand compared to $91 thousand for the corresponding period of 2010, a decrease of $63 thousand. The 2010 expense includes monthly rent recognized under the lease for the corporate office location vacated in 2008. The original lease term expired in April of 2011. Further, the 2010 expense includes rent of the corporate office locations in Atlanta and Durham. The master lease for these managed office spaces expired in February of 2011.
54
Accounting and auditing fees totaled $54 thousand for the year ended December 31, 2011 compared to $76 thousand for the comparable period of 2010; a decrease of $22 thousand. The 2010 expense reflects billings of $28 thousand related to work completed in conjunction with the merger transaction. These fees were offset by additional fees in 2011 for services rendered in conjunction with the Company’s quarterly filings with the SEC.
Gain on Debt Restructuring
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Gain on debt restructuring
|
$ | - | $ | 204 | $ | (204 | ) | (100.0 | ) % | |||||||
On January 22, 2010 the Company and a Creditor reached a settlement agreement whereby the Company will continue to make payments totaling $900 thousand over the next two years to satisfy, in full, its obligation. Further, the agreement restricts the Company from transferring or pledging its intellectual property without prior consent of the Creditor. Immediately preceding the settlement balances recorded on the Company’s financial records with respect to this vendor account included accrued interest and notes payable of $135 thousand and $924 thousand, respectively. The company evaluated the settlement under ASC 470-60, Troubled Debt Restructuring by Debtors. A gain totaling $159 thousand was realized on the vendor settlement.
During 2009, one of the Company’s creditors filed a law suit against the Company relating to a claim for $45 thousand for services rendered in connection with the Company’s terminated Phase III clinical trial, VP-AD-301. On March 3, 2010, the suit was dismissed with prejudice. A gain of $45 thousand was realized on the vendor settlement.
Gain on Reduction of Liabilities
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2009
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Gain on reduction of liabilities
|
$
|
2,233
|
$
|
-
|
$
|
2,233
|
100.0
|
%
|
||||||||
The Company recorded a gain from the reduction in liabilities of $2.2 million in 2011. The obligations principally relate to goods acquired or services rendered while the Company was actively engaged in the terminated Phase III clinical trial. The balances reported include accounts payable represented by vendor invoices submitted to the company for payment, retained billings and accruals for services rendered by vendors but not billed to date. Very little, if any, collection activity has occurred. In the opinion of Company’s counsel as a result of the passage of time these liabilities cannot be legally enforced against the Company.
Interest expense, Net
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Interest expense, net
|
$
|
263
|
$
|
175
|
$
|
88
|
50.3
|
%
|
||||||||
Interest expense, net for the year ended December 31, 2011 and 2010 mainly includes interest accrued on Curaxis’ outstanding promissory notes payable to certain vendors and clinical sites. The majority of the notes outstanding were issued during 2007 and 2008 to satisfy, in part, liabilities generated for services performed in conjunction with the terminated Phase III clinical trial.
55
The increase in expense for the year ended December 31, 2011, as compared to the respective period of 2010, can be can be attributed to interest expense accrued on delayed payments on the restructured debt totaling $84 thousand and interest accrued on the promissory note executed in conjunction with the merger transaction totaling $4 thousand.
Interest from derivative liability
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Interest from derivative liability
|
$
|
-
|
$
|
2,586
|
$
|
(2,586
|
)
|
(100.0
|
%)
|
|||||||
Interest from derivative liability for the year ended December 31, 2010 represents the fair value adjustment recorded upon the initial sale of the preferred securities. We have an obligation to make cash payments to the holders of the preferred stock for any loss that could be realized if the holder converts the preferred stock and we subsequently fail to deliver the certificates representing the shares to be issued upon the conversion by the third trading day of such conversion. Accordingly, the preferred stock has been accounted for as a derivative liability. The fair value of the derivative liability is based on the number of potential common shares issuable upon conversion multiplied by the quoted market price of the Company’s stock as of the date each preferred stock is issued and revalued at each reporting period thereafter.
Change in fair value of derivative liability
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Change in fair value of derivative liability
|
$
|
1,286
|
$
|
2,250
|
$
|
(964
|
)
|
(42.9
|
%)
|
|||||||
During the year ended December 31, 2010, the Company issued Series A and Series B Convertible preferred stock. Because the Series A and Series B stock provide for a potential cash settlement in certain circumstances, the preferred stock has been classified as a derivative liability at December 31, 2011 and 2010. The liability has been recorded at fair value based on potential shares of common stock issuable upon conversion multiplied by the current quoted market price of the company’s common stock and is subject to re measurement.
The change in fair value recognized for the year ended December 31, 2011, reflects the decrease in the quoted market price of the Company’s stock from January 1, 2011 through December 31, 2011, coupled with the capitalization of unpaid dividends at December 15, 2011. The quoted market prices of the Company’s stock at January 1, 2011 and December 31, 2011, were $0.55 and $0.04 per share, respectively. Dividends capitalized at December 15, 2011, totaled $52 thousand representing an increase in the number of shares subject to conversion of 120,800.
The change in fair value recognized for the year ended December 31, 2010 reflects the decrease in the quoted market price of the Company’s stock from the date of the initial issuance of the stock through December 31, 2010. The quoted market price of the Company’s stock at the date of issuance for the Series A and Series B convertible preferred stock was $ 1.60 and $1.30 per share, respectively. The quoted market price of the Company’s stock at the December 31, 2010 was $0.55 per share.
Net Income (Loss)
|
Year Ended
|
||||||||||||||||
|
December 31,
|
||||||||||||||||
|
2011
|
2010
|
$ Change
|
% Change
|
|||||||||||||
|
(Dollars in thousands)
|
||||||||||||||||
|
Net income (loss)
|
$
|
1,334
|
$
|
(2,244
|
)
|
$
|
3,578
|
159.5
|
%
|
|||||||
Net income for the year ended December 31, 2011, was $1.3 million, including a $2.2 million gain relating to the reduction of liabilities, compared to a net loss of $2.2 million, including a $0.2 million gain relating to the restructuring of trade debt, for the same period in 2010. The increase in net income reported is primarily due to the gain recognized on the reduction of liabilities and the elimination of interest recognized on the convertible preferred stock September of 2010 offset by a reduction in the fair value adjustment recognized on preferred stock.
56
Liquidity and Capital Resources
Curaxis has been a developmental stage pharmaceutical company, has had no FDA approved products and has generated no commercial revenue. Since inception, Curaxis has incurred losses from operations and has reported negative cash flows.
As of December 31, 2011, Curaxis had an accumulated deficit of $88.5 million and cash and cash equivalents of $22 thousand. The cash position of Curaxis continues to be insufficient to satisfy its obligations with many vendors. Curaxis will continue to incur operating losses and will not recognize revenues until it is successful in developing and commercializing its lead product candidate, VP4986, for the treatment of Alzheimer’s disease. We expect our minimum cash needs to fund our operating expenses and capital expenditures, excluding direct costs of a potential Phase IIb or registration trial, for the twelve-month period ending December 31, 2012 to be approximately $2.2 million.
Historically, Curaxis had relied on private placements of its common stock and the issuance of preferred stock to fund operations.In 2009, Curaxis entered into a series of agreements with Southridge and Canterbury, in order to restructure its balance sheet and establish a trading market for its common stock. Under the terms of those agreements, Southridge and Canterbury have assisted Curaxis in negotiating settlements with its creditors to eliminate or substantially reduce and defer its trade debt. To date, Curaxis had realized reductions in excess of $6 million in liabilities as the result of the combined efforts. In addition, Southridge assisted Curaxis in securing a $25 million equity facility under which Curaxis can periodically, over a period of three years, sell up to $25 million of its common stock to a third party or affiliate of Southridge. The registration statement which registered 13,000,000 shares of the Company’s common stock to be issued under the terms of the Equity Credit Agreement became effective January 26, 2011. The proceeds of the equity facility will be used to fund the next step in Curaxis’ clinical development plan, a Phase IIb study in approximately 200-250 women. To date, cash proceeds received for draws under the equity line have totaled $526 thousand which have been used to meet current operating needs. Common stock issued as a result of such draws total 5.6 million shares. Because our ability to draw down amounts under the Equity Credit Agreement is subject to a number of conditions, and due to the fact that the number of remaining registered shares approximates 7.4 million, there is no guarantee that we will be able to draw down any significant portion of the remaining $25 million commitment available to us under the Equity Credit Agreement.
On June 27, 2011, the Company announced changes to its Board of Directors (the “Board”) and its executive management team to better position the Company for its next phase of growth and development. The new Board has a wealth of experience and comprehensive knowledge of life sciences, operations and strategic management in the area of pharmaceuticals. In addition, the newly appointed Board members have significant experience in the necessary industry arenas to execute the Company’s business plan on an accelerated basis. The Board and the executive management team is focused on attracting bridge financing for ongoing operational support and has a strategy in place to identify and attract a strategic partner to absorb a portion of the clinical trial cost and processes to advance the Company’s lead drug candidate into a Phase IIb or registration clinical trial. Currently, the Company is doing its best to secure such financing.
Off-Balance Sheet Arrangements
Curaxis had no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on its financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to stockholders.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Not applicable to smaller reporting companies.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
Our financial statements are contained in pages F-1 through F-25 which appear at the end of this Annual Report.
57
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES.
(a) Evaluation of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our Chief Executive Officer and Principle Accounting Officer, we conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (“Exchange Act”), as of December 31, 2011. Disclosure controls and procedures are those controls and procedures designed to provide reasonable assurance that the information required to be disclosed in our Exchange Act filings is (1) recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission’s rules and forms, and (2) accumulated and communicated to management, including our Chief Executive Officer and Principle Accounting Officer, as appropriate, to allow timely decisions regarding required disclosure.
Based on that evaluation, the Chief Executive Officer and Principle Accounting Officer concluded that, as of December 31 2011, the Company had a material weakness with respect to its disclosure controls and procedures because it did not have a sufficient number of personnel with an appropriate level of knowledge and experience of generally accepted accounting principles in the United States of America (U.S. GAAP) that are commensurate with the Company’s financial reporting requirements. As a result of the material weakness, the Chief Executive Officer and Principle Accounting Officer concluded that as of December 31, 2011, the Company’s disclosure controls and procedures were ineffective.
(b) Management’s Annual Report on Internal Control over Financial Reporting
Management, including our Chief Executive Officer and Principle Accounting Officer,, is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a – 15(f). Management conducted an assessment as of December 31, 2011 of the effectiveness of our internal control over financial reporting based on the framework in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on that evaluation, the Chief Executive Officer and Chief Financial Officer concluded that, as of December 31 2011, the Company had a material weakness with respect to its internal controls over financial reporting because it did not have a sufficient number of personnel to provide for adequate segregation of duties and independent review and approval of specific transactions. In addition, the Company lacked a sufficient number of personnel with an appropriate level of knowledge and experience of generally accepted accounting principles in the United States of America (U.S. GAAP).
This Annual Report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit us to provide only management’s report in this Annual Report.
(c) Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting during the most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION.
Not applicable.
58
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICES AND CORPORATE GOVERNANCE.
Directors and Executive Officers
The following table and text sets forth the names and ages of all our directors and executive officers and our key management personnel as of March 28, 2012. All of our directors serve until the next annual meeting of stockholders and until their successors are elected and qualified, or until their earlier death, retirement, resignation or removal. Executive officers serve at the discretion of the Board of Directors, and are elected or appointed to serve until the next Board of Directors meeting following the annual meeting of stockholders. Also provided is a brief description of the business experience of each director and executive officer and the key management personnel during the past five years and an indication of directorships held by each director in other companies subject to the reporting requirements under the Federal securities laws.
|
Name
|
Age
|
Title
|
||
|
Timothy R. Wright
|
54
|
Chairman of the Board and Interim Chief Executive Officer
|
||
|
Judith S. T. Geaslen
|
50
|
Vice President of Finance (Principle Accounting Officer)
|
||
|
Michel W. George
|
63
|
Independent Director
|
||
|
K. Ivan F. Gothner
|
53
|
Independent Director
|
||
|
Stephen J. Leary
|
63
|
Independent Director
|
||
|
Michael R. Miller
|
61
|
Independent Director
|
||
|
Terence S. Novak
|
55
|
Independent Director
|
||
|
Bert A. Spilker
|
69
|
Independent Director
|
Timothy R. Wright was appointed to the Board of Directors effective June 24, 2011, and was named Interim Chief Executive Officer effective June 27, 2011. Mr. Wright served as President of Pharmaceuticals and Medical Imaging for Covidien, a large global healthcare company specializing in the manufacture of medical devices and supplies, diagnostic imaging agents and pharmaceuticals, from 2007 to 2010. Mr. Wright also served as President of Global Operations for Elan Biopharmaceuticals, a neuroscience-based biotechnology company which includes research, development and commercial activities for neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease and autoimmune diseases, from 2000 to 2004. Additionally, Mr. Wright served as Senior Vice President of Strategy and Corporate and Development for DuPont Merck Pharmaceuticals Company, a DuPont Merck joint venture focused on the research, development and delivery of pharmaceuticals to treat unmet medical needs, from 1984 to 1999. Currently, Mr. Wright serves on the board of directors of Agenus, a late stage immunology based biotechnology company developing novel technologies to treat cancer and infectious diseases. He has held this directorship since 2006. Mr. Wright has served on several boards in the past, including AAI Pharma, from 2004 through 2006, CeNes from 2002 through 2004, Shanghai Harvestor from 2002 through 2004 and Dupont Sankyo from 1998 through 1999. Mr. Wright received his Bachelor of Science from The Ohio State University in 1981.
Judith S. T. Geaslen has served as our Vice President of Finance since the effective date of the Merger. Ms. Geaslen had been Curaxis’ Vice President of Finance and Chief Accounting Officer since January 2010 after serving as Corporate Controller and Accountant since July 2004. From March 1999 to April, 2000, Ms. Geaslen served as Vice President and Corporate Controller at Wilmington Trust Corporation, a financial holding company engaged in providing a range of banking and other financial services through its banking and other subsidiaries, and from1994 to 1999, served as Vice President and Manager of the Asset Review Division. From September 1984 to September 1994, Ms. Geaslen was employed by Ernst & Young, a registered public accounting firm, mostly recently serving as Audit Senior Manager. Ms. Geaslen is a graduate of Saint Mary’s College, Notre Dame, Indiana.
59
Michael W. George was appointed to the Board of Directors effective June 24, 2011. Mr. George is currently the Chief Executive Officer of Michael George & Associates, a consulting firm specializing in assisting pharmaceutical and diagnostic firms with developing infrastructure, a position he has held since 2006. Mr. George served as the Executive Vice President of aaiPharma Inc., a pharmaceutical company and clinical research organization, from 2004 to 2006. From 1998 to 2001, Mr. George served as Chief Executive Officer of Urocor, Inc., a publicly traded company specializing in diagnostic services for Urologists (detecting prostate and bladder cancers). From 1989 to 1998, Mr. George held several positions at DuPont Merck Pharmaceutical Company, a DuPont Merck joint venture focused on the research, development and delivery of pharmaceuticals to treat unmet medical needs, including President North America, President International and finally Corporate Executive Vice President. Mr. George served on the Board of Directors of Avanir Pharmaceuticals, a publically traded product development company that has successfully received FDA approval for two commercial products, from 1998 to 2004 and served as the Chair of the board designated Governance Committee for two years. In addition, Mr. George served on the Board of Directors for FeRx, a privately held product development company, from 2002 to 2004. Mr. George received his Bachelor of Science in Business Administration from Central Missouri State University in 1970 and his M.B.A. from New Hampshire College in 1984.
K. Ivan F. Gothner was appointed to the Board of Directors effective June 24, 2011. Mr. Gothner is currently the Managing Director and Founder of Adirondack Partners, LLC, a private merchant banking firm which focuses on serving small and mid-sized growth companies, a position he has held since 1992. Mr. Gothner currently serves on the Board of Directors of AgFeed Industries, Inc., an international agribusiness with operations in the U.S. and China, a position he has held since 2009. Recently, Mr. Gothner was appointed Chairman and Interim CEO of AgFeed Industries, Inc. Mr. Gothner served on the Board of Directors of Covenant Group of China, Inc., US based holding company that focuses on making majority investments in high growth companies based in and operating in the People's Republic of China from 2000 to 2011. Mr. Gothner has also served on the Board of Directors of ArtID, LLC, a private company providing online exhibition space to artists. Mr. Gothner also served as a director for Global Matrechs, Inc. from 2006 through 2008. Additionally, Mr. Gothner served as Senior Vice President for Barclays Bank, where he was responsible for establishing an investment banking unit to serve small and mid-sized companies, from 1990 to 1992. From 1986 through 1990, Mr. Gothner was with Kleinwort Benson Limited where he was the general manager for a specialized fund that invested in small and mid-sized companies. Mr. Gothner received a Bachelor's of Art from Columbia College in 1980, and an MIA from Columbia University's School of International Affairs in 1982.
Stephen J. Leary was appointed to the Board of Directors effective June 24, 2011. Mr. Leary currently serves as Principal of Professional Management Healthcare Consultants, Inc., a practice management and financial consulting firm providing services to medical, dental and veterinary practices located throughout the Carolinas and Virginia, a position he has held since 1979. Mr. Leary is the President Elect and Chairman of the Executive Board of the National Society of Certified Health Care Business Consultants (“NSCHCBC”), a national organization dedicated to serving the needs of consultants who provide ethical, confidential and professional advice to the healthcare industry. Mr. Leary has served as a member of the Board of Directors of NSCHCBC since 2007. Additionally, Mr. Leary was a member of the Board of Directors for the National Association of Health Care Consultants, an association of consultants specializing in the business of medical, dental and other health care practices from 2000 to 2007. Mr. Leary received his Bachelor of Science in Business Administration from Saint Leo University, Tampa, Florida in 1971. Mr. Leary has been a shareholder, individually and as a member and Co-Founder of a large investment group, in the Company since 2004.
Michael R. Miller was appointed to the Board of Directors effective June 24, 2011. Mr. Miller has been a member of the Board of Directors of Christiana Care Health System, one of the largest health care providers in the mid Atlantic region since 2008 and is also the Chair of the Audit Committee, a position he has held since 2009. Mr. Miller is also currently a part time Chief Financial Officer of the Delaware Art Museum, a position he has held since 2007. Mr. Miller was Chief Financial Officer and Senior Vice President for DuPont Merck Pharmaceutical Company, a DuPont Merck joint venture focused on the research, development and delivery of pharmaceuticals to treat unmet medical needs from 1995 until his retirement in 1999. Mr. Miller served as Controller at DuPont Merck, from 1991 to 1995. Prior to joining DuPont Merck, Mr. Miller was employed by E.I. DuPont de Nemours, where, over the course of his tenure, he held numerous finance staff and management positions, including, financial manager for operations in Latin America and a five year assignment in Europe handling merger and acquisition activities. Mr. Miller received his Bachelor of Science degree from Indiana University in 1973.
60
Terence Novak was appointed to the Board of Directors effective June 24, 2011. Mr. Novak is currently President of Norwich Pharmaceuticals, a comprehensive global provider of contract development and manufacturing services, a position he has held since January 2011. Mr. Novak also currently serves on the Board of Directors of Frontline Pharmaceuticals, a start-up specialty pharmaceutical company, a position he has held since January 2010. Mr. Novak has sat on the Editorial Advisory Board of Contract Pharma magazine, which provides a mix of industry news, technical features and association event coverage with respect to the pharmaceutical industry, since 2005 through the present. Mr. Novak served as President of North America and Chief Commercial Officer at Patheon, Inc., a leading global provider of contract dosage form development and manufacturing services to the pharmaceutical and biotechnology industries, from 2008 to 2010. Mr. Novak also served as Director of Patheon Puerto Rico from 2008 to 2009. Additionally, Mr. Novak served as President of DSM Pharmaceuticals, Inc., the contract manufacturing business arm of Netherlands-based Royal DSM, from 2007 to 2008. Mr. Novak has over 30 years of experience in the pharmaceutical and biotech industries, including 20 years in business development, sales, marketing and manufacturing operations. Mr. Novak received a Bachelor of Science degree in Biology from Muhlenberg College in 1978.
Bert A. Spilker was appointed to the Board of Directors effective August 12, 2010. Dr. Spilker is founder and President of Bert A. Spilker & Associates, LLC, a leader in drug development, clinical trials and FDA advisory services. Prior to his current position, Dr. Spilker served as the Senior Vice President of Scientific and Regulatory Affairs for PhRMA (Pharmaceutical Research and Manufacturers of America) based in Washington, D.C. Formerly Dr. Spilker was President and co-founder of Orphan Medical, Inc., a public pharmaceutical company acquired by Jazz Pharmaceuticals in 2005. He is well known as the author of 16 books on clinical trial methods and the processes of drug discovery and development. These books are considered by many as the standard references on clinical trials and drug development.
There are no family relationships among our officers, directors and significant employees.
Board of Directors
Members of our board of directors are elected for one-year terms serving until the next annual stockholders’ meeting or until their death, resignation, retirement, removal, disqualification, or until a successor has been elected and qualified. All officers are appointed annually by our board of directors and serve at the pleasure of our board of directors. Currently, our directors are entitled to receive compensation of $6,000 per fiscal quarter for their service on our board of directors and $2,000 per fiscal quarter for their service as a Chairman of one of the standing committees of the Board.
Director Independence
On an annual basis, each director and executive officer will be obligated to disclose any transactions with our Company and any of its subsidiaries in which a director or executive officer, or any member of his or her immediate family, have a direct or indirect material interest. Following completion of these disclosures, our board of directors will make an annual determination as to the independence of each director using the current standards for “independence” that satisfy both the criteria for the Nasdaq and the NYSE Amex Equities.
As of March 28, 2012, the Curaxis board of directors determined that the following directors are independent under these standards: (i) Mr. Michael George, (ii) Mr. K. Ivan F. Gothner (iii) Stephen Leary, (iv) Mr. Michael Miller (v) Mr. Terrance Novak and (vi) Dr. Bert Spilker.
Board Meetings and Annual Meeting Attendance
The Board of Directors met 14 times during the year ended December 31, 2011. During their tenure, all board members attended more than 75% of the scheduled meetings.
61
The 2010 Annual Meeting of the Company’s shareholders was held on Tuesday, November 8, 2011 in Raleigh, North Carolina. Although not required, all members were in attendance with the exception of Mr. Michael Miller and Dr. Bert Spilker.
Committees of the Board of Directors
Audit Committee
The members of the Audit Committee are K. Ivan F. Gothner, Stephen J. Leary and Michael R. Miller, who serves as chairperson. The Audit Committee has a written charter, which is available at our website at www.curaxispharma.com. The Audit Committee charter requires that the Audit Committee consist of three or more members of the board of directors, each of whom are independent as defined by the corporate governance listing standards of the NYSE Amex. The board of directors has determined that each of the members of the Audit Committee is independent, as defined by Rule 10A-3 of the Securities Exchange Act of the 1934 (the “Exchange Act”), and the corporate governance listing standards. The board of directors also has determined that Mr. Michael Miller is an “audit committee financial expert” as defined in Item 401(h) of Regulation S-K.
The primary functions of the audit committee are to oversee: (i) the audit of our consolidated financial statements provided to the SEC and our stockholders; (ii) our internal financial and accounting processes; and (iii) the independent audit process. Additionally, the audit committee has responsibilities and authority necessary to comply with Rule 10A-3(b) (2), (3), (4), and (5) of the Exchange Act, concerning the responsibilities relating to: (a) registered public accounting, (b) complaints relating to accounting, internal accounting controls or auditing matters, (c) authority to engage advisors and (d) funding. These and other aspects of the audit committee’s authority are more particularly described in the audit committee charter.
The Audit Committee has oversight responsibility for quality and integrity of our consolidated financial statements. The committee meets privately with members of our independent registered public accounting firm, has the sole authority to retain and dismiss the independent registered public accounting firm and reviews their performance and independence from management. The independent registered public accounting firm has unrestricted access and reports directly to the committee. The Audit Committee has met four (4) times since it was established in June of 2011.
The Audit Committee is responsible for review and approval of audit and non-audit services to be provided to us by our independent registered public accounting firm, RRBB, LLP. The Audit Committee has approved all audit services to be provided by RRBB through December 31, 2011.
Compensation Committee
The members of the Compensation Committee are Michael W. George, as chairperson, Michael R. Miller and Terrance Novak. All members of the Compensation Committee are independent within the corporate governance listing standards of the NYSE Amex. The Compensation Committee has a written charter, which is available at our website at www.curaxispharma.com.
The functions performed by the Compensation Committee include reviewing and approving all compensation arrangements for our executive officers and administering our equity incentive plans. The Compensation Committee has met two (2) times since its establishment in July 2011. At the first meeting, the committee approved recommendations for compensation plans effective July 1, 2011 for the current officers and directors.
Nominating and Governance Committee
The members of the Nominating and Governance Committee are Michael W. George and Stephen J. Leary, who serves as chairperson. All members of the Nominating and Governance Committee are independent within the corporate governance listing standards of the NYSE Amex. The Nominating and Governance Committee has a written charter, which is available at our website at www.curaxispharma.com. Subsequent to its establishment in August 2011, the Nominating and Governance Committee met one (1) time.
62
The functions of the Nominating and Governance Committee include determining and recommending to the board of directors the slate of director nominees for election to the board of directors at each annual stockholders’ meeting and identifying and recommending director candidates to fill vacancies occurring between annual stockholders’ meetings. In addition, the Nominating and Governance Committee reviews, evaluates and recommends changes to our corporate governance guidelines and policies, and monitors our compliance with these corporate governance guidelines and policies.
Research and Development Committee
The Research and Development Committee was established in August 2011. The Committee is chaired by Dr. Bert Spilker who is independent within the corporate governance listing standards of the NYSE Amex. The research and development committee has a written charter, which is available at our website at www.curaxispharma.com.
The purpose of the Research and Development Committee is to provide advice, clarity, understanding and guidance to the Board of Directors on scientific and medical matters involving the Company’s discovery and development programs and projects, relationships with academic and other external research and development organizations, intellectual property issues and acquisitions of new technology or products.
Board’s Role in Risk Oversight
Our Board, through its four standing committees, has an advisory role in risk oversight for the Company. The Company’s management maintains primary responsibility for the risk management of the Company. The current trends of increased regulation, litigation and political and economic volatility make it extremely difficult to predict the type and magnitude of risks the Company faces. The Board relies on the representations of management, the external audit of our financial and operating results, the Company’s systems of internal controls and the historically conservative practices of the Company to provide comfort on the Company’s ability to manage its risks.
Code of Ethics
Effective August 11, 2011, the Board adopted a Code of Business Conduct and Ethics (the “Code”) that applies to all of our employees, officers and directors. The Code is available at our website at www.curaxispharma.com.
Involvement in Certain Legal Proceedings
To the best of our knowledge, during the past five years, none of the following occurred with respect to our present or former director, executive officer, or employee: (1) any bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; (2) any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); (3) being subject to any order, judgment or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his or her involvement in any type of business, securities or banking activities; and (4) being found by a court of competent jurisdiction (in a civil action), the SEC or the Commodities Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended or vacated.
63
ITEM 11. EXECUTIVE COMPENSATION.
Summary Compensation Table
The following summary compensation table sets forth all compensation awarded to, earned by, or paid to the named executive officers by us during the years ended December 31, 2011 and 2010.
| Name and Principal Position |
Year
|
Salary
($)
|
Bonus
($)
|
Stock
Awards
($)
|
Option
Awards
($)
|
Non-Equity
Incentive
Plan
Compensation
($)
|
All Other
Compensation
($)
|
Total
($)
|
||||||||||||||
|
(a)
|
(b)
|
(c)
|
(d)
|
(e)
|
(f)
|
(g)
|
(h)
|
(i)
|
||||||||||||||
|
Timothy R. Wright,
|
2011
|
$ | 140,000 | (4) |
None
|
None
|
$ | 37,765 | (6) |
None
|
None
|
$ | 177,765 | |||||||||
|
Interim Chief Executive Officer and Chairman (1)
|
2010
|
None
|
None
|
None
|
None
|
None
|
None
|
None
|
||||||||||||||
|
Judith S. T. Geaslen
|
2011
|
$ | 153,600 | (5) |
None
|
None
|
$ | 16,955 | (6) |
None
|
None
|
$ | 170,555 | |||||||||
|
Vice President, Finance
|
2010
|
$ | 113,833 |
None
|
None
|
$ | 34,240 | (7) |
None
|
$ | 5,700 | (8) | $ | 153,773 | ||||||||
|
Patrick S. Smith,
|
2011
|
$ | 143,500 | (2) |
None
|
None
|
$ | 1,277 | (6) |
None
|
None
|
$ | 144,777 | |||||||||
|
Former President, Former Chief Executive Officer
|
2010
|
$ | 224,708 |
None
|
None
|
$ | 32,490 | (7) |
None
|
$ | 24,934 | (9) | $ | 282,132 | ||||||||
|
and Former Chairman of the Board (2)
|
||||||||||||||||||||||
|
David J. Corcoran,
|
2011
|
$ | 119,500 | (3) |
None
|
None
|
$ | 958 | (6) |
None
|
None
|
$ | 120,458 | |||||||||
|
Former Executive Vice President, Former Chief
|
2010
|
$ | 185,083 |
None
|
None
|
$ | 24,369 | (7) |
None
|
$ | 7,125 | (10) | $ | 216,577 | ||||||||
|
Financial Officer and Former Director (3)
|
||||||||||||||||||||||
|
(1)
|
Mr. Wright was elected to the Board and appointed Interim Chief Executive Officer effective June 27, 2011.
|
|
(2)
|
Mr. Smith resigned from his position effective June 24, 2011; at the date of resignation, accrued and unpaid compensation totaled $48,183. The earned compensation is to be paid to the former officer in quarterly installments commencing September 15, 2011. No payments have been made to date. In addition, the Company extended the contractual life of all outstanding vested stock options held by the respective officer at the date of resignation.
|
|
(3)
|
Mr. Corcoran resigned from his position effective June 24, 2011; at the date of resignation, accrued and unpaid compensation totaled $72,583. The earned compensation is to be paid to the former officer in quarterly installments commencing September 15, 2011. No payments have been made to date. In addition, the Company extended the contractual life of all outstanding vested stock options held by the respective officer at the date of resignation.
|
|
(4)
|
Includes $140,000 in accrued and unpaid wages at December 31, 2011.
|
|
(5)
|
Includes $76,651 in accrued and unpaid wages at December 31, 2011.
|
|
(6)
|
Represents the aggregate expense under ASC 718 recognized by Curaxis for 2011 as a result of option awards held by the applicable executive officer, disregarding estimated forfeitures. Assumptions made in the valuation were consistent with those applied in years 2010. For a description of these assumptions, see Notes 2 and 11 to the financial statements for the year ended December 31, 2011.
|
|
(7)
|
Represents the aggregate expense under ASC 718 recognized by Curaxis for 2010 as a result of option awards held by the applicable executive officer, disregarding estimated forfeitures. For a description of these assumptions made in the evaluation, see Notes 2 and 11 to the financial statements for the year ended December 31, 2011.
|
64
|
(8)
|
Represents car allowance of $5,700.
|
|
(9)
|
Represents car allowance of $9,500 plus payment for certain travel expenses incurred not directly associated with the Company.
|
|
(10)
|
Represents car allowance of $7,125.
|
Explanatory Information Relating to 2011 Summary Compensation Table
Please note the following points in connection with the information in the 2011 Summary Compensation Table:
|
●
|
The Compensation Committee of the Board of Directors is charged with reviewing and approving all compensation arrangements for our executive officers and administering our equity incentive plans. The Compensation Committee approved recommendations for compensation plans effective July 1, 2011 for the current officers and directors. In developing the approved recommendations, the Committee reviewed individual performance, as appropriate, professional attributes and proven capabilities and competitive considerations. The Committee also identified performance goals for the officers with respect to funding objectives and clinical initiatives. Measurement of the performance goals will be considered in developing prospective compensation recommendations.
|
OUTSTANDING EQUITY AWARDS AT 2011 FISCAL YEAR-END
|
Option Awards
|
Stock Awards
|
||||||||||||||||||||||||||||
|
Equity
|
|||||||||||||||||||||||||||||
|
Incentive
|
|||||||||||||||||||||||||||||
|
Equity
|
Plan
|
||||||||||||||||||||||||||||
|
Incentive
|
Awards:
|
||||||||||||||||||||||||||||
|
Plan
|
Market
|
||||||||||||||||||||||||||||
|
Awards:
|
or Payout
|
||||||||||||||||||||||||||||
|
Number
|
Value of
|
||||||||||||||||||||||||||||
|
Number
|
Market
|
of Unearned
|
Unearned
|
||||||||||||||||||||||||||
|
Number of
|
Number of
|
of
|
Value of
|
Shares,
|
Shares,
|
||||||||||||||||||||||||
|
Securities
|
Securities
|
Shares or
|
Shares or
|
Units or
|
Units or
|
||||||||||||||||||||||||
|
Underlying
|
Underlying
|
Units of
|
Units of
|
Other
|
Other
|
||||||||||||||||||||||||
|
Unexercised
|
Unexercised
|
Option
|
Stock That
|
Stock That
|
Rights
|
Rights That
|
|||||||||||||||||||||||
|
Options
|
Options
|
Exercise
|
Option
|
Have Not
|
Have Not
|
That Have
|
Have
|
||||||||||||||||||||||
| (#) | (#) |
Price
|
Expiration
|
Vested
|
Vested
|
Not Vested
|
Not Vested
|
||||||||||||||||||||||
|
Name
|
Exercisable
|
Unexercisable
|
($)
|
Date
|
(#) |
($)
|
(#) |
($)
|
|||||||||||||||||||||
|
(a)
|
(b)
|
(c)
|
(d)
|
(e)
|
(f)
|
(g)
|
(h)
|
(i)
|
|||||||||||||||||||||
|
Timothy R. Wright (1)
|
150,000
|
150,000
|
$
|
0.20
|
08/11/21
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(2)
|
3,500
|
—
|
$
|
4.00
|
12/01/14
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(3)
|
5 ,000
|
—
|
$
|
8.00
|
01/17/15
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(4)
|
1,000
|
—
|
$
|
10.00
|
05/04/15
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(5)
|
25,000
|
—
|
$
|
6.00
|
05/18/16
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(6)
|
30,000
|
—
|
$
|
1.51
|
02/05/17
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(7)
|
100,000
|
—
|
$
|
0.20
|
02/19/19
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen(8)
|
20,000
|
30,000
|
$
|
0.20
|
01/18/20
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Judith S. T. Geaslen (1)
|
25,000
|
25,000
|
$
|
0.20
|
08/11/21
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Patrick S. Smith(9)
|
240,000
|
—
|
$
|
0.83
|
06/20/12
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Patrick S. Smith(10)
|
80,000
|
—
|
$
|
2.00
|
09/15/13
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Patrick S. Smith(11)
|
40 ,000
|
—
|
$
|
8.00
|
01/17/15
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
Patrick S. Smith(12)
|
96,000
|
—
|
$
|
1.51
|
02/05/17
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
David J. Corcoran(9)
|
180,000
|
—
|
$
|
0.83
|
06/20/12
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
David J. Corcoran(10)
|
60,000
|
—
|
$
|
2.00
|
09/15/13
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
David J. Corcoran(11)
|
30 ,000
|
—
|
$
|
8.00
|
01/17/15
|
—
|
—
|
—
|
—
|
||||||||||||||||||||
|
David J. Corcoran(12)
|
72,000
|
—
|
$
|
1.51
|
02/05/17
|
||||||||||||||||||||||||
65
|
(1)
|
These stock options vested as to 50% of the shares on the grant date of August 11, 2011 and 50% vest on August 11, 2012 based on continued employment through August 11, 2012.
|
|
(2)
|
These stock options granted to Ms. Geaslen upon employment vested as to 20% of the shares on December 1, 2005 and 20% vested on each December 1st thereafter based on continued employment through December 1, 2009.
|
|
(3)
|
These stock options vested as to 20% of the shares on January 17, 2006 and 20% vested on each January 17th thereafter based on continued employment through January 17, 2010.
|
|
(4)
|
These stock options vested as to 20% of the shares on May 4, 2006 and 20% vested on each May 4th thereafter based on continued employment through May 4, 2010.
|
|
(5)
|
These stock options vest as to 20% on May 18, 2007 and 20% vested on each May 18th thereafter based on continued employment through May 18, 2011.
|
|
(6)
|
These stock options vested as to 20% of the shares on January 1, 2008 and 20% vested on each January 1st thereafter based on continued employment through January 1, 2012.
|
|
(7)
|
These stock options vested as to 100% of the shares on the grant date of February 19, 2009.
|
|
(8)
|
These stock options vest as to 20% of the shares on January 18, 2011and 20% vest on each January 18th thereafter based on continued employment through January 18, 2015.
|
|
(9)
|
These stock options vested as to 20% of the shares on June 20, 2003 and 20% vested on each June 20th thereafter based on continued employment through June 20, 2007. Named officer resigned effective June 24, 2011; the Company extended the exercise period of the vested options to the original expiration date.
|
|
(10)
|
These stock options vested as to 20% of the shares on September 15, 2004 and 20% vested on each September 15th thereafter based on continued employment through September 15, 2008. Named officer resigned effective June 24, 2011; the Company extended the exercise period of the vested options to the original expiration date.
|
|
(11)
|
These stock options vested as to 20% of the shares on January 17, 2006 and 20% vested on each January 17th thereafter based on continued employment through January 17, 2010. Named officer resigned effective June 24, 2011; the Company extended the exercise period of the vested options to the original expiration date.
|
|
(12)
|
These stock options vested as to 20% of the shares on January 1, 2008 and 20% vested on each January 1st thereafter based on continued employment through January 1, 2012. Named officer resigned effective June 24, 2011. Upon resignation, the unvested options were cancelled. The Company extended the exercise period of the vested options to the original expiration date.
|
66
DIRECTOR COMPENSATION
2011 SUMMARY COMPENSATION TABLE
The following summary compensation table sets forth all compensation awarded to, earned by, or paid to the named directors by us during the years ended December 31, 2011.
|
Name and Principal Position
|
Fees earned or paid in cash
($)
|
Stock
Awards
($)
|
Option
Awards
($)
|
Non-Equity
Incentive Plan
Compensation
($)
|
All Other
Compensation
($)
|
Total
($)
|
||||||||||||
|
(a)
|
(c) (4)
|
(e)
|
(f)
|
(g)
|
(g)
|
(h)
|
||||||||||||
|
Michael W. George, Director (1)
|
$ | 16,000 | (5) |
None
|
$ | 9,441 | (9) |
None
|
None
|
$ | 25,441 | |||||||
|
K. Ivan F. Gothner, Director (1)
|
$ | 12,000 | (6) |
None
|
$ | 9,441 | (9) |
None
|
None
|
$ | 21,441 | |||||||
|
Stephen J. Leary, Director (1)
|
$ | 16,000 | (5) |
None
|
$ | 9,441 | (9) |
None
|
None
|
$ | 25,441 | |||||||
|
Michael R. Miller, Director (1)
|
$ | 16,000 | (7) |
None
|
$ | 9,441 | (9) |
None
|
None
|
$ | 25,441 | |||||||
|
Terence S. Novak, Director (1)
|
$ | 12,000 | (6) |
None
|
$ | 9,441 | (9) |
None
|
None
|
$ | 21,441 | |||||||
|
Bert A. Spilker, Director (2)
|
$ | 28,000 | (8) |
None
|
$ | 35,770 | (9) |
None
|
None
|
$ | 63,770 | |||||||
|
Sheldon L. Goldberg, Director (3)
|
$ | 12,000 |
None
|
$ | 4,062 | (9) |
None
|
None
|
$ | 16,062 | ||||||||
|
William E. McConville, Director(3)
|
$ | 12,000 |
None
|
$ | 2,981 | (9) |
None
|
None
|
$ | 14,981 | ||||||||
|
Ronald V. Pompeo, Director(3)
|
$ | 12,000 |
None
|
-0- | (9) |
None
|
None
|
$ | 12,000 | |||||||||
|
(1)
|
Director elected to the Board of Directors effective June 27, 2011.
|
|
(2)
|
Dr. Spilker was elected to the Board effective August 2010.
|
|
(3)
|
Director resigned from his position effective June 24, 2011; at the date of resignation, accrued and unpaid directors’ fees totaled $18,000. Fees are to be paid to the former director in quarterly installments commencing September 15, 2011. No payments have been made to date. In addition, the Company extended the contractual life of all outstanding vested stock options held by the respective director at the date of resignation.
|
|
(4)
|
Directors are entitled to receive compensation of $6,000 per fiscal quarter for their service on our board of directors and $2,000 per fiscal quarter for their service as a Chairman of one of the standing committees of the Board.
|
|
(5)
|
Includes $8,000 in fees accrued and unpaid at December 31, 2011.
|
|
(6)
|
Includes $6,000 in fees accrued and unpaid at December 31, 2011.
|
|
(7)
|
Includes $16,000 in fees accrued and unpaid at December 31, 2011.
|
|
(8)
|
Includes $28,000 in fees accrued and unpaid at December 31, 2011. In addition, fees totaling $6,000 were accrued for the year ended December 31, 2010 which remain unpaid at December 31, 2011.
|
|
(9)
|
Represents the aggregate expense under ASC 718 recognized by Curaxis for 2011 as a result of option awards held by the applicable director, disregarding estimated forfeitures. Assumptions made in the valuation were consistent with those applied in years 2010 and 2009. For a description of these assumptions, see Notes 2 and 11 to the financial statements for the year ended December 31, 2011.
|
67
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
As of March 28, 2012 our authorized capitalization was 500,000,000 shares, consisting of 480,000,000 shares of common stock, $0.0001 par value per share and 20,000,000 shares of preferred stock, $0.0001 par value per share. As of March 28, 2012, there were 83,554,940 shares of our common stock outstanding. The following table sets forth, as of March 28, 2012, the number of shares of our common stock owned by (i) each person who is known by us to own of record or beneficially five percent (5%) or more of our outstanding shares, (ii) each of our directors, (iii) each of our executive officers and (iv) all of our directors and executive officers as a group. Unless otherwise indicated, each of the persons listed below has sole voting and investment power with respect to the shares of our common stock beneficially owned.
Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or dispositive power with respect to securities. Ordinary shares relating to options or warrants currently exercisable, or exercisable within 60 days of March 28, 2012 and convertible preferred stock are deemed outstanding for computing the percentage of the person holding such securities but are not deemed outstanding for computing the percentage of any other person. Except as indicated by footnote, and subject to the community property laws where applicable, the persons or entities named in the tables have sole voting and dispositive power with respect to all shares shown as beneficially owned by them. Except as otherwise noted in the tables below, the address of each person or entity listed in the table is c/o Curaxis Pharmaceutical Corporation, 1004 Chagford Way, Raleigh, NC 27614.
|
Name and Address
|
Amount and
Nature of
Beneficial
Ownership
of Ordinary
Shares(1)
|
Percentage
of Total
|
||||||
|
DIRECTORS AND EXECUTIVE OFFICERS
|
||||||||
|
Timothy R. Wright
|
150,000
|
(2)
|
—
|
%
|
||||
|
Chairman of the Board and Interim Chief Executive Officer
|
||||||||
|
Judith S. T. Geaslen,
|
454,300
|
(3)
|
—
|
%
|
||||
|
Vice President of Finance
|
||||||||
|
Michael George
|
446,729
|
(4)
|
—
|
%
|
||||
|
Director
|
||||||||
| K. Ivan F. Gothner |
258,171
|
(4)
|
—
|
%
|
||||
|
Director
|
|
|||||||
|
Stephen Leary
|
926,099
|
(5)
|
1.0
|
%
|
||||
|
Director
|
||||||||
|
Michael Miller
|
37,500
|
(4)
|
—
|
%
|
||||
|
Director
|
||||||||
|
Terence Novak
|
258,171
|
(4)
|
—
|
%
|
||||
|
Director
|
|
|
|
|||||
|
Bert A. Spilker
|
60,500
|
(6)
|
—
|
%
|
||||
|
Director
|
||||||||
|
All directors and executive officers as a group (8 persons)
|
2,591,470
|
(8)
|
2.90
|
%
|
||||
|
SHAREHOLDERS OWNING MORE THAN 5%
|
||||||||
|
Richard L. Bowen
|
10,162,560
|
11.4
|
%
|
|||||
| Patrick S. Smith | 5,351,000 | (7) | 6.0 | % | ||||
|
CP Acquisition Partners LP
|
4,508,827
|
(7)
|
5.1
|
%
|
||||
|
—
|
Less than 1%
|
|
(1)
|
Based on 88,909,614 outstanding shares assuming the exercise of 5,354,674 warrants issued and outstanding, but assuming no exercise of outstanding options. In addition, shares outstanding do not assume conversion of Series A, B and C preferred stock.
|
68
|
(2)
|
Includes 150,000 shares of common stock issuable upon exercise of stock options exercisable within 60 days of March 28, 2012.
|
|
(3)
|
Includes 209,500 shares of common stock issuable upon exercise of stock options exercisable within 60 days of March 28, 2012, and 4,000 shares of common stock held by Wachovia Securities as custodian fbo Judith S. T. Geaslen. Also includes 200,000 shares held by Donald. P Geaslen and 10,000 shares of common stock held by Wachovia Securities as custodian fbo Donald P. Geaslen, spouse of Ms. Geaslen.
|
|
(4)
|
Includes 37,500 shares of common stock issuable upon exercise of stock options exercisable within 60 days of March 28, 2012.
|
|
(5)
|
Includes 37,500 shares of common stock issuable upon exercise of stock options exercisable within 60 days of March 28, 2012. Also includes 6,090 shares of common stock held in the name of Patricia C. Leary, spouse of Mr. Leary and 265,000 shares of common stock held in Professional Mgmt of Raleigh Inc. PSP U/A DTD 07/01/85 FBO Stephen J. Leary of which Mr. Leary is the trustee.
|
|
(6)
|
Includes 57,500 shares of common stock issuable upon the exercise of stock options exercisable within 60 days of September 12, 2011.
|
|
(7)
|
Includes 456,000 shares of common stock issuable upon exercise of stock options exercisable within 60 days of September 12, 2011. Also includes 4,345,000 shares of common stock owned by the Patrick S. Smith Charitable Remainder Trust, of which Mr. Smith is the trustee, and 300,000 shares of common stock held by RBC Dain Rauscher Inc., as custodian for Patrick S. Smith IRA.
|
|
(8)
|
Includes 4,508,827 shares of common stock held by CP Acquisition Partners and its affiliate, Southridge Partners II LP. Reported amount does not include 4,410,316 warrants held by those affiliates. In addition, CP Acquisition Partners LP holds 500 shares of convertible Series A, 500 shares of convertible Series B, and 36 shares of convertible Series C preferred stock, which are convertible into a total of 2,653,409 shares of common stock, and a Promissory note, which is convertible into 567,225 shares of common stock. All warrants, preferred stock and the note are subject to certain conversion restrictions.
|
|
(9)
|
See note (1)-(6) above.
|
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Any future transactions or loans between us and our officers, directors, principal stockholders or affiliates will be on terms no less favorable to us than could be obtained from an unaffiliated third party, and will be approved by a majority of disinterested directors.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES.
Summary of Principal Accountant Fees for Professional Services Rendered
The following table presents the aggregate fees for professional audit services and other services rendered by Rosenberg Rich Baker Berman & Company, our independent registered public accountants in 2011 and 2010.
|
|
Fiscal Years Ended
December 31,
|
|||||||
|
|
2011
|
2010
|
||||||
|
Audit and Audit Related Fees
|
$
|
49,000 (1)
|
$
|
50,000 (3)
|
||||
|
Tax Fees
|
$
|
-
|
$
|
-
|
||||
|
All Other Fees
|
$
|
5,195 (2)
|
$
|
26,220 (4)
|
||||
|
(1)
|
For 2011, audit and audit related fees were for professional services rendered for the audit of the Company’s financial statements for the fiscal year and review of the Company’s quarterly financial statements included in its Form 10Q filings for the three quarters ended September 30, 2011.
|
|
(2)
|
For 2011, all other fees are for services rendered related to our Post-Effective Amendment to our Registration Statement filed on Form S-1; our Definitive Proxy Statement filed on Schedule 14A, our application for a dual listing of our stock on the Frankfurt Exchange; and tax advisory services related to the procedural filing requirements for federal and state taxes subsequent to the merger.
|
|
(3)
|
For 2010, audit and audit related fees were for professional services rendered for the audit of the Company’s financial statements for the fiscal year and review of the Company’s quarterly financial statements included in its Registration Statements and in its Form 10Q filing for the quarter ended September 30, 2010.
|
|
(4)
|
For 2010, all other fees are for services rendered related to our Registrations Statements filed on Form S-4 relating to the Merger Transaction and Form S-1 related to our Equity Credit Agreement.
|
69
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
|
Exhibit No.
|
Description
|
|
|
2.1
|
Agreement and Plan of Merger and Plan of Reorganization by and Among Auto Search Cars, Inc., Auto Search Cars Acquisition Corp., and Curaxis Pharmaceutical Corporation
|
|
|
3.1
|
Articles of Incorporation (incorporated by reference to the Company’s Registration Statement on Form S-1 (as filed with the SEC on May 15, 2008 (Registration No. 333-150937))
|
|
|
3.2
|
Certificate of Designation of Preferences, Rights and Limitations of the Curaxis Pharmaceutical Corporation Series A Convertible Preferred Stock, as filed with the Nevada Secretary of State on September 23, 2010 (as filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K, filed on September 29, 2010)
|
|
|
3.3
|
Certificate of Designation of Preferences, Rights and Limitations of the Curaxis Pharmaceutical Corporation Series B Convertible Preferred Stock, as filed with the Nevada Secretary of State on September 29, 2010 (as filed as Exhibit 3.1 to the Company’s Current Report on Form 8-K, filed on October 1, 2010)
|
|
|
3.4
|
Certificate of Designation of Preferences, Rights and Limitations of the Curaxis Pharmaceutical Corporation Series C Convertible Preferred Stock, as filed with the Nevada Secretary of State on March 29, 2011 (as filed on Form 10-K with the SEC on March 30, 2011).
|
|
|
3.5
|
By-laws of the Company (incorporated by reference to the Company’s Registration Statement on Form S-1, filed with the SEC on May 15, 2008 (Registration No. 333-150937))
|
|
|
10.1
|
Feasibility, Development and Commercialization Agreement, dated July 22, 2002, by and between Voyager Pharmaceutical Corporation, a Delaware corporation, and Southern Biosystems, Inc., an Alabama corporation (Durect) (as filed on Form S-4/A with the SEC on June 21, 2010)
|
|
|
10.2
|
Amendment No.1 to Feasibility, Development and Commercialization Agreement, effective January 23, 2007, by and between Durect Corporation and Voyager Pharmaceutical Corporation (as filed on Form S-4/A with the SEC on June 21, 2010)
|
|
|
10.3
|
Supply Agreement, dated January 1, 2009, by and between Voyager, Inc. and Mallinckrodt Inc. (as filed on Form S-4/A with the SEC on June 21, 2010)
|
|
|
10.4
|
Consulting Agreement, dated June 12, 2009, by and between Voyager Pharmaceutical Corporation and Canterbury Investment Partners LLC (as filed on Form S-4/A with the SEC on June 21, 2010)
|
|
|
10.5
|
Transaction Management Agreement, dated May 28, 2009, by and between Voyager Pharmaceutical Corporation and Southridge Business Solutions Group LLC (as filed on Form S-4/A with SEC on June 21, 2010)
|
|
|
10.6
|
Amended and Restated Equity Credit Agreement by and between Curaxis Pharmaceutical Corp. and Southridge Partners II, LP, dated December 6, 2010 (as filed as Exhibit 10.8 on Form S-1 with the SEC on December 8, 2010)
|
|
|
10.7
|
Amended and Restated Registration Rights Agreement by and between Curaxis Pharmaceutical Corp. and Southridge Partners II, LP, dated December 6, 2010 (as filed as Exhibit 10.9 on Form S-1 with the SEC on December 8, 2010)
|
70
|
10.8
|
Series A Convertible Stock Purchase Agreement, by and between Curaxis Pharmaceutical Corporation and C P Acquisition Partners LP, dated September 30, 2010 (as filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K, as filed with the SEC on October 1, 2010)
|
|
|
10.9
|
Series B Convertible Stock Purchase Agreement, by and between Curaxis Pharmaceutical Corporation and C P Acquisition Partners LP, dated September 30, 2010 (as filed as Exhibit 10.2 to the Company’s Current Report on Form 8-K, as filed with the SEC on October 1, 2010)
|
|
|
10.10
|
Series C Convertible Stock Purchase Agreement, by and between Curaxis Pharmaceutical Corporation and C P Acquisition Partners LP, dated March 30, 2011 (as filed on Form 10-K with the SEC on March 30, 2011)
|
|
|
31.1
|
Certification by the Principal Executive Officer of Registrant pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Rule 13a-14(a) or Rule 15d-14(a)).*
|
|
|
31.2
|
Certification by the Principal Financial Officer of Registrant pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Rule 13a-14(a) or Rule 15d-14(a)).*
|
|
|
32.1
|
Certification by the Principal Executive Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.*
|
|
|
32.2
|
Certification by the Principal Financial Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.*
|
|
|
99.1
|
Curaxis Pharmaceutical Corporation 2011 Incentive Stock Plan
|
71
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
CURAXIS PHARMACEUTICAL CORPORATION
|
||
|
Dated: March 29, 2012
|
By:
|
/s/ Timothy R. Wright
|
|
Timothy R. Wright
|
||
|
Chief Executive Officer
(Principal Executive Officer)
|
||
|
Dated: March 29, 2012
|
By:
|
/s/ Judith S.T. Geaslen
|
|
Judith S.T. Geaslen
|
||
|
Vice President of Finance
(Principal Financial Officer)
(Principal Accounting Officer)
|
||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ Timothy R. Wright
|
Chairman of the Board and Interim Chief Executive Officer
|
March 29, 2012
|
||
|
Timothy R. Wright
|
(Principal Executive Officer)
|
|||
|
/s/ Judith S. T. Geaslen
|
Vice President of Finance (Principal Financial Officer)
|
March 29, 2012
|
||
|
Judith S. T. Geaslen
|
(Principal Accounting Officer)
|
|||
|
/s/ Michel W. George
|
Independent Director
|
March 29, 2012
|
||
|
Michel W. George
|
||||
|
/s/ K. Ivan F. Gothner
|
Independent Director
|
March 29, 2012
|
||
|
K. Ivan F. Gothner
|
||||
|
/s/ Stephen J. Leary
|
Independent Director
|
March 29, 2012
|
||
|
Stephen J. Leary
|
||||
|
/s/ Michael R. Miller
|
Independent Director
|
March 29, 2012
|
||
|
Michael R. Miller
|
||||
|
/s/ Terence S. Novak
|
Independent Director
|
March 29, 2012
|
||
|
Terence S. Novak
|
||||
|
/s/ Bert A. Spilker
|
Independent Director
|
March 29, 2012
|
||
|
Bert A. Spilker
|
72
Curaxis Pharmaceutical Corporation
(A Development Stage Company)
Financial Statements
December 31, 2010 and 2011
73
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Index to Financial Statements
|
Report of Independent Registered Public Accounting Firm
|
F-1
|
|
Balance Sheets
|
F-2
|
|
Statements of Operations
|
F-3
|
|
Statements of Changes in Stockholders’ Equity (Deficit)
|
F-4 – F-5
|
|
Statements of Cash Flows
|
F-6 – F-7
|
|
Notes to Financial Statements
|
F-8 – F-25
|
74
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Curaxis Pharmaceutical Corporation
(A Development Stage Company):
We have audited the accompanying balance sheets of Curaxis Pharmaceutical Corporation (a Development Stage Company) as of December 31, 2010 and 2011, and the related statements of operations and cash flows for each of the two years in the period ended December 31, 2011 and the changes in stockholders' equity (deficit), for the period from January 1, 2006 to December 31, 2011. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We did not audit the financial statements of Curaxis Pharmaceutical Corporation for the period from inception to December 31, 2005.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, based on our audits, the financial statements referred to above present fairly, in all material respects, the financial position of Curaxis Pharmaceutical Corporation as of December 31, 2010 and 2011, and the consolidated results of its operations and its cash flows for each of the two years in the period ended December 31, 2011, and the changes in stockholders' equity (deficit), for the period from January 1, 2006 to December 31, 2011 in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company is in the development stage, has incurred significant losses from operations since its inception, has a net stockholders’ deficit and, is currently in default of several credit agreements. These factors raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Rosenberg Rich Baker Berman & Company
Somerset, New Jersey
March 28, 2012
F-1
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Balance Sheets
(in thousands)
|
December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Assets
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 2 | $ | 22 | ||||
|
Prepaid assets
|
301 | 321 | ||||||
|
Security deposit
|
6 | - | ||||||
|
Total current assets
|
309 | 343 | ||||||
|
Property and equipment, net
|
2 | 1 | ||||||
|
Other assets
|
612 | 314 | ||||||
|
Total assets
|
923 | 658 | ||||||
|
Liabilities and Stockholders’ Equity (Deficit)
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
2,793 | 1,746 | ||||||
|
Accrued expenses
|
1,231 | 1,068 | ||||||
|
Current portion of capital lease obligation
|
4 | 4 | ||||||
|
Notes payable
|
2,525 | 3,774 | ||||||
|
Total current liabilities
|
6,553 | 6,592 | ||||||
|
Derivative instrument
|
1,336 | 99 | ||||||
|
Deferred purchase credit
|
500 | 500 | ||||||
|
Deferred revenue
|
1,727 | 1,727 | ||||||
|
Long-term notes payable
|
1,250 | - | ||||||
|
Total liabilities
|
11,366 | 8,918 | ||||||
|
Commitments and contingencies
|
― | ― | ||||||
|
Stockholders’ equity (deficit):
|
||||||||
|
Preferred Stock, $0.0001 par value; 20,000 authorized; 1 and 1 shares issued and outstanding at December 31, 2010 and 2011, respectively.
|
― | ― | ||||||
|
Common stock, $.0001 par value; 480,000 authorized; 72,216 and 80,120 shares issued and outstanding at December 31, 2010 and 2011, respectively
|
7 | 8 | ||||||
|
Additional paid-in capital
|
79,458 | 80,247 | ||||||
|
Subscription receivable
|
(104 | ) | - | |||||
|
Deficit accumulated during the development stage
|
(89,804 | ) | (88,515 | ) | ||||
|
Total stockholders’ equity (deficit)
|
(10,443 | ) | (8,260 | ) | ||||
|
Total liabilities and stockholders’ equity (deficit)
|
$ | 923 | $ | 658 | ||||
The accompanying notes are an integral part of these financial statements.
F-2
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Statements of Operations
(in thousands, except per share amounts)
|
For the Years Ended
December 31,
|
Cumulative from Inception (February 27, 2001) to December 31,
|
|||||||||||
|
2010
|
2011
|
2011
|
||||||||||
|
(unaudited)
|
||||||||||||
|
Operating expenses
|
||||||||||||
|
Research and development
|
$ | 78 | $ | 33 | $ | 61,466 | ||||||
|
General and administrative
|
1,859 | 1,889 | 28,566 | |||||||||
|
Marketing
|
- | - | 5,557 | |||||||||
|
Loss on investment in real estate
|
- | - | 1,091 | |||||||||
|
Loss on lease termination
|
- | - | 374 | |||||||||
|
Total operating (income) expenses
|
1,937 | 1,922 | 97,054 | |||||||||
|
Operating (loss) income
|
(1,937 | ) | (1,922 | ) | (97,054 | ) | ||||||
|
Gain on debt restructuring
|
204 | - | 6,766 | |||||||||
|
Gain on reduction of liabilities
|
- | 2,233 | 2,233 | |||||||||
|
Interest income (expense), net
|
(175 | ) | (263 | ) | (1,355 | ) | ||||||
|
Interest from derivative liability
|
(2,586 | ) | - | (2,586 | ) | |||||||
|
Change in fair value of derivative liability
|
2,250 | 1,286 | 3,536 | |||||||||
|
Net income (loss)
|
$ | (2,244 | ) | $ | 1,334 | $ | (88,460 | ) | ||||
|
Basic net income (loss) per share
|
$ | (0.03 | ) | $ | 0.02 | |||||||
|
Diluted net income (loss) per share
|
$ | (0.03 | ) | $ | 0.02 | |||||||
|
Weighted average common shares outstanding – basic
|
67,323 | 75,859 | ||||||||||
|
Weighted average common shares outstanding – diluted
|
67,323 | 80,082 | ||||||||||
The accompanying notes are an integral part of these financial statements.
F-3
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Statements of Changes in Stockholders’ Equity (Deficit)
(in thousands)
|
Preferred stock
|
Common stock
|
Additional
Paid-in-
|
Subscription | Deficit Accumulated during the Development | |||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
receivable
|
Stage
|
Total
|
||||||||||||||||||||||||||
|
Balances at Inception (February 27, 2001)
|
― | $ | ― | ― | $ | ― | $ | ― | $ | ― | $ | ― | $ | ― | |||||||||||||||||||
|
Sale of common stock
|
25,657 | 26 | 1,073 | ― | 1,099 | ||||||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 100 | ― | ― | 100 | |||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (1,219 | ) | (1,219 | ) | |||||||||||||||||||||||
|
Balances at December 31, 2001
|
― | ― | 25,657 | 26 | 1,173 | ― | (1,219 | ) | (20 | ) | |||||||||||||||||||||||
|
Sale of common stock
|
― | ― | 4,054 | 4 | 4,105 | ― | ― | 4,109 | |||||||||||||||||||||||||
|
Issuance of common stock in exchange for consulting services
|
― | ― | 12 | ― | 10 | ― | ― | 10 | |||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 272 | ― | ― | 272 | |||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (3,190 | ) | (3,190 | ) | |||||||||||||||||||||||
|
Balances at December 31, 2002
|
― | ― | 29,723 | 30 | 5,560 | ― | (4,409 | ) | 1,181 | ||||||||||||||||||||||||
|
Sale of common stock
|
― | ― | 4,794 | 5 | 8,239 | ― | ― | 8,244 | |||||||||||||||||||||||||
|
Issuance of common stock in exchange for consulting services
|
― | ― | 36 | ― | 55 | ― | ― | 55 | |||||||||||||||||||||||||
|
Repurchase of common stock
|
― | ― | (330 | ) | ― | (379 | ) | ― | ― | (379 | ) | ||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 78 | ― | ― | 78 | |||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (6,633 | ) | (6,633 | ) | |||||||||||||||||||||||
|
Balances at December 31, 2003
|
― | ― | 34,223 | 35 | 13,553 | ― | (11,042 | ) | 2,546 | ||||||||||||||||||||||||
|
Sale of common stock
|
― | ― | 4,388 | 4 | 12,491 | ― | ― | 12,495 | |||||||||||||||||||||||||
|
Issuance of common stock in exchange for consulting services
|
― | ― | 20 | ― | 66 | ― | ― | 66 | |||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (11,626 | ) | (11,626 | ) | |||||||||||||||||||||||
|
Balances at December 31, 2004
|
― | ― | 38,631 | 39 | 26,110 | ― | (22,668 | ) | 3,481 | ||||||||||||||||||||||||
|
Sale of common stock
|
― | ― | 3,305 | 3 | 28,839 | ― | ― | 28,842 | |||||||||||||||||||||||||
|
Issuance of common stock in exchange for consulting services
|
― | ― | 4 | ― | 33 | ― | ― | 33 | |||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 10 | ― | ― | 10 | |||||||||||||||||||||||||
|
Repurchase of common stock
|
― | ― | (1 | ) | ― | (9 | ) | ― | ― | (9 | ) | ||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (32,904 | ) | (32,904 | ) | |||||||||||||||||||||||
|
Balances at December 31, 2005
|
― | ― | 41,939 | 42 | 54,983 | ― | (55,572 | ) | (547 | ) | |||||||||||||||||||||||
|
Sale of common stock
|
― | ― | 1,513 | 1 | 15,127 | ― | ― | 15,128 | |||||||||||||||||||||||||
|
Sale of common stock under
warrants
|
― | ― | 2,886 | 3 | 286 | ― | ― | 289 | |||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 1,155 | ― | ― | 1,155 | |||||||||||||||||||||||||
|
Repurchase of common
stock
|
― | ― | (150 | ) | ― | ― | ― | ― | ― | ||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (28,235 | ) | (28,235 | ) | |||||||||||||||||||||||
|
Balances at December 31, 2006
|
― | ― | 46,188 | $ | 46 | $ | 71,551 | $ | ― | $ | (83,807 | ) | $ | (12,210 | ) | ||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-4
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Statements of Changes in Stockholders’ Equity (Deficit), continued
(in thousands)
|
Preferred stock
|
Common stock
|
Additional
Paid-in-
|
Subscription | Deficit Accumulated during the Development | ||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
receivable
|
Stage
|
Total
|
|||||||||||||||||||||||||
|
Sale of common stock
|
― | $ | ― | 1,822 | $ | 2 | $ | 2,336 | $ | ― | $ | ― | $ | 2,338 | ||||||||||||||||||
|
Sale of common stock
under warrants
|
1,971 | 2 | 171 | ― | 173 | |||||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 889 | ― | ― | 889 | ||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (6,744 | ) | (6,744 | ) | ||||||||||||||||||||||
|
Balances at December 31, 2007
|
― | ― | 49,981 | 50 | 74,947 | ― | (90,551 | ) | (15,554 | ) | ||||||||||||||||||||||
|
Sale of common stock
|
― | ― | 5,120 | 5 | 518 | ― | ― | 523 | ||||||||||||||||||||||||
|
Sale of common stock
under warrants
|
― | ― | 41 | ― | 4 | ― | ― | 4 | ||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 469 | ― | ― | 469 | ||||||||||||||||||||||||
|
Net loss
|
― | ― | ― | ― | ― | ― | (1,986 | ) | (1,986 | ) | ||||||||||||||||||||||
|
Balances at December 31, 2008
|
― | ― | 55,142 | 55 | 75,938 | ― | (92,537 | ) | (16,544 | ) | ||||||||||||||||||||||
|
Sale of common stock,
net
|
― | ― | 6,071 | 6 | 1,096 | ― | ― | 1,102 | ||||||||||||||||||||||||
|
Sale of common stock under warrants
|
― | ― | 1,448 | 1 | 143 | ― | ― | 144 | ||||||||||||||||||||||||
|
Issuance of warrants
|
― | ― | ― | ― | 330 | ― | ― | 330 | ||||||||||||||||||||||||
|
Stock grant
|
― | ― | 150 | ― | 30 | ― | ― | 30 | ||||||||||||||||||||||||
|
Stock-based
compensation
|
― | ― | ― | ― | 361 | ― | ― | 361 | ||||||||||||||||||||||||
|
Net income
|
― | ― | ― | ― | ― | ― | 4,987 | 4,987 | ||||||||||||||||||||||||
|
Balances at December 31, 2009
|
― | ― | 62,811 | 62 | 77,898 | ― | (87,550 | ) | (9,590 | ) | ||||||||||||||||||||||
|
Sale of common stock,
net
|
― | ― | 1,182 | 1 | 219 | ― | ― | 220 | ||||||||||||||||||||||||
|
Sale of common stock
under warrants
|
― | ― | 528 | 1 | 50 | 51 | ||||||||||||||||||||||||||
|
Issuance of warrants
|
― | ― | ― | ― | 1,111 | ― | ― | 1,111 | ||||||||||||||||||||||||
|
Recapitalization of Company upon Merger effective July 29, 2010
|
― | ― | 7,695 | (57 | ) | (51 | ) | ― | ― | (108 | ) | |||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 231 | ― | 231 | |||||||||||||||||||||||||
|
Sale of Preferred stock
|
1 | ― | ― | ― | ― | (104 | ) | ― | (104 | ) | ||||||||||||||||||||||
|
Dividends on convertible preferred
stock 4%
|
― | ― | ― | ― | ― | ― | (10 | ) | (10 | ) | ||||||||||||||||||||||
|
Net loss
|
(2,244 | ) | (2,244 | ) | ||||||||||||||||||||||||||||
|
Balances at December 31, 2010
|
1 | ― | 72,216 | 7 | 79,458 | (104 | ) | (89,804 | ) | (10,443 | ) | |||||||||||||||||||||
|
Issuance of common stock under Equity Credit Agreement, net
|
― | ― | 3,329 | ― | 200 | ― | ― | 200 | ||||||||||||||||||||||||
|
Sale of common stock under warrants
|
― | ― | 2,483 | ― | 35 | 35 | ||||||||||||||||||||||||||
|
Sale of common stock under options
|
― | ― | 372 | ― | 1 | ― | ― | 1 | ||||||||||||||||||||||||
|
Issuance of common stock in exchange for consulting services
|
520 | 1 | 111 | 112 | ||||||||||||||||||||||||||||
|
Stock-based compensation
|
― | ― | ― | ― | 173 | ― | 173 | |||||||||||||||||||||||||
|
Sale of Preferred stock
|
1 | ― | ― | ― | 266 | 104 | ― | 370 | ||||||||||||||||||||||||
|
Conversion of Preferred stock to common stock
|
(1 | ) | ― | 1,200 | ― | ― | ― | ― | ― | |||||||||||||||||||||||
|
Dividends on convertible preferred
stock 4%
|
― | ― | ― | ― | 3 | ― | (45 | ) | (42 | ) | ||||||||||||||||||||||
|
Net income
|
― | 1,334 | 1,334 | |||||||||||||||||||||||||||||
|
Balances at December 31, 2011
|
1 | $ | ― | 80,120 | $ | 8 | $ | 80,247 | $ | ― | $ | (88,515 | ) | $ | (8,260 | ) | ||||||||||||||||
The accompanying notes are an integral part of these financial statements.
F-5
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Statements of Cash Flows
(in thousands)
|
For the Years Ended December 31,
|
Cumulative from Inception
(February 27, 2001) to
December 31,
|
|||||||||||
|
2010
|
2011
|
2011
|
||||||||||
|
(unaudited)
|
||||||||||||
|
Cash flows from operating activities
|
||||||||||||
|
Net income (loss)
|
$ | (2,244 | ) | $ | 1,334 | $ | (88,460 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash used in operating activities: | ||||||||||||
|
Depreciation
|
- | 1 | 995 | |||||||||
|
Stock-based compensation expense
|
460 | 173 | 4,243 | |||||||||
|
Common stock issued in exchange for consulting services
|
― | 112 | 276 | |||||||||
|
Loss (gain) on disposal of property and equipment estate
|
― | ― | 169 | |||||||||
|
Loss on investment in real estate
|
― | ― | 1,091 | |||||||||
|
Gain on the reduction of liabilities
|
― | (2,233 | ) | (2,233 | ) | |||||||
|
Gain on restructuring of trade debt
|
(204 | ) | ― | (6,766 | ) | |||||||
|
Change in fair value of derivative liability
|
(2,250 | ) | (1,286 | ) | (3,536 | ) | ||||||
|
Interest from derivative liability
|
2,586 | ― | 2,586 | |||||||||
|
Changes in operating assets and liabilities:
|
||||||||||||
|
Prepaid expenses
|
103 | 6 | 57 | |||||||||
|
Accounts receivable
|
11 | ― | ― | |||||||||
|
Security deposits
|
(2 | ) | 6 | ― | ||||||||
|
Accounts payable
|
211 | 413 | 8,150 | |||||||||
|
Accrued expenses
|
11 | 617 | 3,070 | |||||||||
|
Deferred purchase credit
|
― | ― | 500 | |||||||||
|
Deferred revenue
|
― | ― | 1,727 | |||||||||
|
Accrued compensation
|
― | ― |
―
|
|||||||||
|
Net cash used in operating activities
|
(1,318 | ) | (857 | ) | (78,131 | ) | ||||||
|
Cash flows from investing activities
|
||||||||||||
|
Purchase of property and equipment
|
(2 | ) | ― | (1,195 | ) | |||||||
|
Purchase of real estate investment
|
― | ― | (3,155 | ) | ||||||||
|
Proceeds from the sale of property and equipment
|
― | ― | 50 | |||||||||
|
Proceeds from the sale of real estate
|
― | ― | 2,064 | |||||||||
|
Net cash provided by (used in) investing activities
|
$ | (2 | ) | $ | ― | $ | (2,236 | ) | ||||
The accompanying notes are an integral part of these financial statements.
F-6
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Statements of Cash Flows, continued
(in thousands)
|
For the Years Ended December 31,
|
Cumulative from Inception (February 27, 2001) to December 31,
|
|||||||||||
|
2010
|
2011
|
2011
|
||||||||||
|
(unaudited)
|
||||||||||||
|
Cash flows from financing activities
|
||||||||||||
|
Issuance of common stock
|
$ | 272 | $ | 508 | $ | 75,270 | ||||||
|
Issuance of preferred stock
|
896 | 266 | 1,162 | |||||||||
|
Proceeds from subscription receivable on preferred stock
|
― | 104 | 104 | |||||||||
|
Issuance of notes payable
|
― | ― | 5,011 | |||||||||
|
Issuance of long-term debt
|
― | ― | 592 | |||||||||
|
Payments on notes payable
|
(477 | ) | (1 | ) | (1,343 | ) | ||||||
|
Payments on capital lease obligation
|
― | ― | (16 | ) | ||||||||
|
Repurchase of common stock
|
― | ― | (388 | ) | ||||||||
|
Repayments of notes payable to related parties
|
― | ― | (202 | ) | ||||||||
|
Payments on long-term debt
|
(3 | ) | ||||||||||
|
Proceeds from notes payable to related parties
|
― | ― | 202 | |||||||||
|
Repayment of mortgage note payable
|
― | ― | (3,100 | ) | ||||||||
|
Proceeds from the issuance of mortgage note
|
― | ― | 3,100 | |||||||||
|
Net cash provided by financing activities
|
691 | 877 | 80,389 | |||||||||
|
Net increase (decrease) in cash and cash equivalents
|
(629 | ) | 20 | 22 | ||||||||
|
Cash and cash equivalents, beginning of period
|
631 | 2 | ― | |||||||||
|
Cash and cash equivalents, end of period
|
$ | 2 | $ | 22 | $ | 22 | ||||||
|
Supplemental disclosure of cash flow information:
|
||||||||||||
|
Cash paid for interest
|
$ | 15 | $ | 3 | $ | 260 | ||||||
|
Non-cash financing activities:
|
||||||||||||
|
Conversion of trade payables to notes
|
$ | ― | $ | ― | $ | 5,603 | ||||||
|
Restructuring of trade debt
|
$ | 204 | $ | ― | $ | 6,766 | ||||||
|
Issuance of warrant for accrued liability
|
$ | 129 | $ | ― | $ | 129 | ||||||
|
Issuance of warrants for prepaid asset
|
$ | (881 | ) | $ | ― | $ | (881 | ) | ||||
|
Net liabilities acquired in Merger Transaction
|
$ | (92 | ) | $ | ― | $ | (92 | ) | ||||
|
Subscription receivable for issuance of preferred stock
|
$ | 104 | $ | ― | $ | 104 | ||||||
The accompanying notes are an integral part of these financial statements.
F-7
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(1) Description of Business and Basis of Presentation
The Company is an emerging specialty pharmaceutical company with a hormone drug product candidate for the treatment of Alzheimer’s disease and multiple cancers. Our therapeutic platform is based on the hypothesis that many diseases of aging may be caused by age-related changes in the function of the hypothalamic-pituitary-gonadal (HPG) axis. The HPG axis is a hormonal endocrine feedback loop that controls development, reproduction and aging in animals. This drug development platform is built on the premise that hormones associated with this feedback loop are beneficial early in life, when they promote growth and development, but are harmful later in life when the mechanism for feedback is compromised, thereby leading to disease processes, including pathologies associated with Alzheimer’s disease and various cancers. We believe our discovery of similar hormonal signaling mechanisms at the cellular level in brain tissue from Alzheimer’s patients and in multiple tumors will enable us to develop significant new treatments for Alzheimer’s disease as well as many cancers.
Corporate History
The Company incorporated in Nevada on February 1, 2008, under the name Auto Search Cars, Inc. The Company initially was engaged in the development of a web-based e-commerce site platform to provide information for the sale of vehicles and vehicle financing and warranties. However, no significant activities related to the website development occurred. On February 8, 2010, the Company entered into an Agreement and Plan of Merger and Plan of Reorganization (the “Merger Agreement”) with Auto Search Cars Acquisition Corp. (“Acquisition Sub”), a Delaware corporation wholly owned by the Company, and Curaxis Pharmaceutical Corporation, a Delaware corporation (“Curaxis Delaware”), pursuant to which Acquisition Sub would be merged with and into the Curaxis Delaware with Curaxis Delaware continuing as the surviving wholly-owned subsidiary of the Company. Upon effectiveness of the Merger Agreement, the Company ceased its e-commerce business and initiated the business plan of Curaxis Delaware. On July 29, 2010, the parties executed an amendment to the Merger Agreement in order to amend the consideration paid for the cancellation of certain shares of the Corporation’s common stock.
On July 29, 2010, the merger transaction was closed. Pursuant to the amended Merger Agreement, the Company issued 64.2 million shares of its common stock to the holders of common stock of the Curaxis Delaware (the “Merger”). Under the terms of the Merger, all but 8.7 million of these shares were restricted from trading for a period of one year from the closing of the Merger. In addition, each issued Curaxis Delaware Warrant (as defined in the Merger Agreement) were converted into warrants to purchase an equal number of shares of Auto Search’s common stock at the exercise price defined in the Company’s warrants. Further the sole officer of Auto Search agreed to cancel 181,285,000 shares of Auto Search common stock in exchange for payment of $100 thousand dollars in the form of a promissory note and the issuance of 3,589,460 warrants to purchase common stock of the surviving corporation.
On July 30, 2010, the Company entered into an agreement and plan of merger with Curaxis Pharmaceutical Corporation, a Nevada corporation formed solely for the purpose of a name change. Pursuant to the short-form merger, the Company changed its name to Curaxis Pharmaceutical Corporation (“Curaxis” or the “Company”).
In connection with the Merger dated as of July 29, 2010 and described in our Form 8-K filed on June 30, 2010 , the Company on August 11, 2011 entered into an Agreement and Plan of Merger (the “Curaxis Merger Agreement”) by and between the Company and Curaxis Pharma Corporation, a Delaware corporation and a wholly owned subsidiary of the Company (“Pharma”), pursuant to which Pharma merged with and into the Company and the Company is the surviving corporation.
Basis of Presentation
The Company is a going concern development stage company and has focused its efforts to date on raising capital and research and development. The accompanying financial statements have been prepared on a basis which assumes that the Company will continue as a going concern and which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has a limited operating history and has incurred significant losses from operations since its inception. The cash position of the Company has deteriorated significantly over the last three years and the Company has been unable to satisfy its outstanding liabilities with many vendors and is currently in default of several credit agreements. In late 2006, the Company terminated its Phase III clinical trial VP-AD-301 and in 2007 closed its research facility. These factors raise substantial doubt as to the Company’s ability to continue as a going concern. The accompanying financial statements do not include adjustments that might result from this uncertainty.
The accompanying notes are an integral part of these financial statements.
F-8
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
In 2009, the Company entered into a series of agreements with Southridge Business Solutions Group (“Southridge”) and Canterbury Investment Partners LLC (“Canterbury”), in order to restructure its balance sheet and establish a trading market for its common stock. Under the terms of those agreements, Southridge and Canterbury have assisted Curaxis in negotiating settlements with its creditors to eliminate or substantially reduce and defer its trade debt. To date, the Company has realized reductions in excess of $6 million in liabilities as the result of the combined efforts. In addition, Southridge assisted Curaxis in securing a $25 million equity facility under which the Company can periodically, over a period of three years, sell up to $25 million of its common stock to an affiliate of Southridge. On December 8, 2010, the Company filed a registration statement on Form S-1 to register 13,000,000 shares of common stock to be reserved for issuance under the equity line facility. The proceeds of the equity facility will be used to meet current operating requirements and to fund the next step in Curaxis’ clinical development plan.
On June 27, 2011, the Company announced changes to its Board of Directors (the “Board”) and its executive management team to better position the Company for its next phase of growth and development. The new Board has a wealth of experience and comprehensive knowledge of life sciences, operations and strategic management in the area of pharmaceuticals. In addition, the newly appointed Board members have significant experience in the necessary industry arenas to execute the Company’s business plan on an accelerated basis. The Board and the executive management team is focused on attracting bridge financing for ongoing operational support and has a strategy in place to identify and attract a strategic partner to absorb a portion of the clinical trial cost and processes to advance the Company’s lead drug candidate into a Phase IIb or Phase III clinical trial. Currently, the Company is doing its best to secure such financing.
Assuming favorable results from the proposed trial are achieved, Curaxis’ management expects to fund the remaining development of its product candidate through public equity offerings, debt financings and possibly corporate collaborations and licensing arrangements. However, the ability to secure such funding on a prospective basis is uncertain. If Curaxis is unable to secure funding as intended, Curaxis will be forced to delay, reduce or eliminate its research and development programs or commercialization efforts. There can be no assurance that the Company will be successful in achieving its goals.
(2) Summary of Significant Accounting Policies
Use of Estimates - The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Fair Value of Financial Instruments - The carrying amounts for cash, cash equivalents, accounts receivables, prepaid assets, accounts payable, and accrued expenses approximate fair value because of their short-term nature. We have determined that it is not practical to estimate the fair value of our notes payable because of their unique nature and the costs that would be incurred to obtain an independent valuation. We do not have comparable outstanding debt on which to base an estimated current borrowing rate or other discount rate for purposes of estimating the fair value of the notes payable and we have not yet obtained or developed a valuation model. Additionally, we are engaged in research and development activities and have not yet developed products for sale. Accordingly, at this stage of our development, a credit risk assessment is highly judgmental. These factors all contribute to the impracticability of estimating the fair value of the notes payable. At December 31, 2011, the carrying value of the notes payable and accrued interest was $3.8 million and $710 thousand, respectively.
Fair Value Measurements - The authoritative guidance for fair value measurements defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or the most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Market participants are buyers and sellers in the principal market that are (i) independent, (ii) knowledgeable, (iii) able to transact, and (iv) willing to transact. The guidance describes a fair value hierarchy based on the levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
|
●
|
Level 1 — Quoted prices in active markets for identical assets or liabilities
|
|
●
|
Level 2 — Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or corroborated by observable market data or substantially the full term of the assets or liabilities
|
|
●
|
Level 3 — Unobservable inputs that are supported by little or no market activity and that are significant to the value of the assets or liabilities
|
The accompanying notes are an integral part of these financial statements.
F-9
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(2) Summary of Significant Accounting Policies (continued)
Derivative Instrument - The Derivative instrument consists of the Series A and B convertible preferred stock, which has certain cash settlement provisions. This financial instrument is recorded in the balance sheet at fair value as a liability. Changes in fair value are recognized in earnings in the period of change.
Cash and Cash Equivalents - The Company considers all highly liquid investments with a maturity of three months or less at the date of purchase to be cash equivalents. Cash and cash equivalents are stated at cost and consist of bank deposits and a money market fund that invests in short-term debt securities. The carrying amount of cash and cash equivalents approximates fair value.
Property and Equipment - Property and equipment is stated at cost. Equipment acquired under capital leases is initially recorded at the present value of the minimum lease payments at inception of the lease.
Depreciation is computed using the straight-line method over the estimated useful lives of the assets which range from three to five years. Leasehold improvements and equipment held under capital leases are amortized over the related lease terms, which is shorter than the estimated useful lives of the assets.
Impairment of Long-Lived Assets - Long-lived assets are reviewed for impairment when events or changes in circumstances indicate the book value of the assets may not be recoverable. In accordance with ASC 360-10-35-15 Impairment or Disposal of Long-Lived Assets, recoverability is measured by comparing the book value of the asset to the future net undiscounted cash flows expected to be generated by the asset.
No events or changes in circumstances have been identified which would impact the recoverability of the Company’s long-lived assets reported at December 31, 2010 or 2011.
Stock-Based Compensation - The Company applies ASC 718 Compensation – Stock Compensation, to stock-based compensation awards. ASC 718 requires the measurement and recognition of non-cash compensation expense for all share-based payment awards made to employees and directors, including employee stock options related to our 2001 Stock Option Plan and 2005 Stock Option Plan, based on fair values. The Company adopted this Statement on January 1, 2006 using the modified prospective application.
Stock compensation arrangements with non-employee service providers are accounted for in accordance ASC 505-50 Equity-Based Payments to Non-Employees, using a fair value approach. The compensation costs of these arrangements are subject to re measurement over the vesting terms as earned.
Research and Development - Research and development costs include all direct costs related to the development of the Company’s products, including clinical trial costs, salaries and related benefits of personnel, costs of producing drug material for clinical trials and fees paid to consultants. Research and development costs are expensed as incurred.
Income Taxes - Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities, and their respective tax bases and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. The Company has established a valuation allowance against its deferred tax assets due to the uncertainty surrounding the realization of such assets.
On January 1, 2007, the Company adopted ASC 740-10-25, Income Taxes- Overall-Recognition. The Company did not have any unrecognized tax benefits and there was no effect on our financial condition or results of operations as a result of adopting this standard. The Company’s practice is to recognize interest and /or penalties related to income tax matters in income tax expense as incurred.
The accompanying notes are an integral part of these financial statements.
F-10
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(2) Summary of Significant Accounting Policies (continued)
Certain Risks and Uncertainties - The Company’s product candidates under development require approval from the United States Food and Drug Administration (“FDA”) or other international regulatory agencies prior to commercial sales. There can be no assurance our products will receive the necessary clearance. If the Company is denied clearance or clearance is delayed, it may have a material adverse impact on the Company.
The Company’s product is concentrated in rapidly changing, highly competitive markets, which are characterized by rapid technological advances, changes in customer requirements and evolving regulatory requirements and industry standards. Any failure by the Company to anticipate or to respond adequately to technological developments in our industry, changes in customer requirements or changes in regulatory requirements or industry standards, or any significant delays in the development or introduction of products or services, could have a material adverse effect on the Company’s business, operating results and future cash flows. The Company relies on one corporation as a sole source provider of Memryte for use in its clinical trials.
Recently Issued Accounting Pronouncements - Management does not believe that any recently issued, but not yet effective, accounting standards if currently adopted would have a material effect on the accompanying financial statements
(3) Prepaid Expenses and Other Assets
Prepaid expenses and other assets consist of the following at (in thousands):
|
December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Prepaid insurance premiums
|
$ | 28 | $ | 24 | ||||
|
Deposits for professional services
|
4 | 3 | ||||||
|
Prepaid financing costs represented by warrants issued under Equity Credit Agreement (Note 7)
|
881 | 608 | ||||||
| 913 | 635 | |||||||
|
Less: Non-current portion
|
(612 | ) | (314 | ) | ||||
| $ | 301 | $ | 321 | |||||
(4) Property and Equipment, Net
Property and equipment consists of the following at (in thousands):
|
December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Furniture and office equipment
|
$ | 86 | $ | 82 | ||||
|
Laboratory and manufacturing equipment
|
241 | 241 | ||||||
| 327 | 323 | |||||||
|
Accumulated depreciation and amortization
|
(325 | ) | (322 | ) | ||||
| $ | 2 | $ | 1 | |||||
Depreciation expense totaled $-0- thousand and $1thousand for the years ended December 31, 2010 and 2011, respectively.
The accompanying notes are an integral part of these financial statements.
F-11
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(5) Accrued Expenses
Accrued expenses consist of the following (in thousands):
|
December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Development and clinical trial expenses
|
$ | 773 | $ | - | ||||
|
Interest
|
448 | 710 | ||||||
|
Dividends payable (Note 9)
|
10 | 4 | ||||||
|
Accrued compensation
|
- | 354 | ||||||
| $ | 1,231 | $ | 1,068 | |||||
(6) Notes payable
Notes payable consist of the following (in thousands):
|
December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Notes payable to clinical research sites
|
$ | 405 | $ | 404 | ||||
|
Promissory Note dated September 15, 2006
|
1,925 | 1,925 | ||||||
|
Promissory Installment Note dated September 26, 2006
|
500 | 500 | ||||||
|
Promissory Note dated July 1, 2007
|
300 | 300 | ||||||
|
Promissory Note dated August 21, 2007
|
420 | 420 | ||||||
|
Promissory Note and Security Agreement dated September 22, 2008
|
26 | 26 | ||||||
|
Default judgment dated October 29, 2009
|
99 | 99 | ||||||
|
Promissory note dated July 29, 2010
|
100 | 100 | ||||||
| 3,775 | 3,774 | |||||||
|
Less: Current portion of notes payable
|
2,525 | 3,774 | ||||||
| $ | 1,250 | $ | - | |||||
Weighted average interest rates on current notes payable at December 31, 2010 and 2011 were 8.5% and 9.8%, respectively.
Notes payable to clinical sites represent unsecured promissory notes executed during 2007 and 2008 to repay clinical sites for services performed in conjunction with the Company’s terminated Phase III clinical trial, VP-AD-301. The notes were issued for six-month to one year terms and bear interest at an annual rate of 12% to 18%. At December 31, 2011, all notes payable to clinical sites had matured and were in default for lack of payment.
The accompanying notes are an integral part of these financial statements.
F-12
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(6) Notes payable (continued)
Promissory note dated September 16, 2006 in the amount of $4 million was executed in conjunction with a Repayment agreement between the Company and a significant vendor. The note was issued to satisfy, in part, liabilities generated for service performed in conjunction with our terminated Phase II clinical trial. The unsecured note bears interest at 12.0% and required installment payments commencing October 1, 2006. At the request of the Company, the Repayment Agreement has been amended to cure all events of default for non-payment. However, payment of principal in full and accrued interest to date was due on May 31, 2008. On September 23, 2009, the Company reached an agreement with this vendor to satisfy amounts owed related to notes payable and trade debts outstanding. The agreement requires the Company to make payments totaling $2 million commencing September 1, 2010 and ending March 31, 2012. The Company has the right to prepay any balances due without penalty. In addition, the company agreed to prepay the entire balance of such payments in the event of an acquisition of all or substantially all of the Company’s assets or stock; in the event the company’s lead product candidate is licensed to a third party; in the event any patent related to the Company’s lead product candidate is sold to any third party; or in the event the Company obtains financing in excess of $20 million. Any payment not made in accordance with the agreement shall bear interest at 18% per annum. At December 31, 2011, the Company was in default of payments totaling $675 thousand for which interest accrued to date totaled $95 thousand. Immediately preceding the settlement balances recorded on the Company’s financial records with respect to this vendor account included accounts payable, accrued interest and notes payable of $4.7 million, $1.1 million and $2.8 million, respectively. The company evaluated the settlement under ASC 470-60, Troubled Debt Restructuring by Debtors. As a result, a gain totaling $6.5 million was realized on the settlement. The basic and diluted EPS on the aggregate gain for the year ended December 31, 2009 were $0.11.
Promissory Installment Note dated September 26, 2006 in the amount of $1.1 million was executed in conjunction with Letter of Agreement between the Company and a significant vendor to satisfy liabilities generated for the receiving, distribution and return processing of clinical trial material. The note is secured against inventory held by the vendor and required monthly payments commencing September 26, 2006 of principal plus a 1% fee on the outstanding balance. The Company was in default for nonpayment as of December 31, 2006. On April 3, 2008, a Final and Amended Judgment was awarded on behalf of the creditor against the Company in the amount $924 thousand principal and $135 thousand in accrued interest and late fees, for a total of $1.059 million. On January 22, 2010 the Company and Creditor reached a settlement agreement whereby the Company will make payments totaling $900,000 over a two year period to satisfy, in full, its obligation. Further, the agreement restricts the Company from transferring or pledging its intellectual property without prior consent of the Creditor. As a result of the settlement, the Company recognized a gain of $159 thousand. The basic and diluted EPS on the aggregate gain for the year ended December 31, 2010 were $0.00.
Promissory Note dated July 1, 2007 in the amount of $300 thousand was executed in conjunction with a Termination of Sublease Agreement between the Company and its’ landlord as consideration for early termination of the sublease agreement. The original sublease agreement for corporate office space was executed on April 20, 2005 and was to expire on March 31, 2011. The promissory note bears interest at 10%. Payment of principal and all accrued interest was due June 30, 2009. The Promissory note was in default for non-payment as of July 1, 2009.
Promissory note dated August 21, 2007 in the amount of $274 thousand was executed in conjunction with a Structured Settlement Agreement between the Company and a significant vendor to satisfy, in part, payments due to the vendor for the purchase of clinical trial material. The unsecured note bears interest at an annual rate of 8% and required payment in full not later than July 15, 2012. In addition, the Company was required to make graduated monthly payments commencing June 25, 2007 to satisfy amounts outstanding at the date of the agreement not reflected in the Promissory Note. At December 31, 2008, the Company was in default of the Structured Settlement Agreement for non-payment of the monthly amounts set forth by the agreement. On April 6, 2009, an Order of Default Judgment was awarded on behalf of the creditor against the Company in the amount of $420 thousand principal and $5 thousand in accrued interest and fees, for a total of $425 thousand. The principal balance of $420 reflects the balance outstanding on the original Promissory note of $274 thousand and amounts recorded as trade payables immediately preceding the issuance of the Default Judgment of $146 thousand. The principal balance due on the Default Judgment has been reclassified as a current note payable at December 31, 2010 and 2011.
Promissory Note and Security Agreement dated September 22, 2008 in the amount of $27 thousand was executed pursuant to the terms of the Fifth Amendment to Lease between the Company and it’s landlord as consideration for the Company to relocate and reduce the size of its’ leased office space. The Promissory note bears interest at an annual rate of 9% and requires equal monthly installment payments to be made commencing November 1, 2008 through April 11, 2011. The Promissory note balance of $26 thousand at December 31, 2010 was in default for non-payment. As a result, the note was reclassified as a current liability at December 31, 2010. No payments were made on the note in 2011.
A default judgment was issued on October 29, 2009 on behalf of a creditor seeking payment of amounts due for services rendered with respect to the Company’s terminated Phase III clinical trial, VP-AD-301. The judgment requires payment of the principal amount of $99 thousand plus interest accrued at an annual rate of 10%.
Promissory note dated July 29, 2010 was executed in conjunction with the Merger transaction consummated on July 29, 2010 for $100 thousand. The note bears interest at an annual rate of 8% and was payable on or before July 29, 2011.
The accompanying notes are an integral part of these financial statements.
F-13
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(7) Equity Credit Agreement
On September 16, 2010, the Company entered into a Private Equity Credit Agreement (the “Equity Credit Agreement”) with Southridge Partners II, LP (the “Investor”), a limited partnership organized and existing under the laws of the State of Delaware and an affiliate of Southridge. Pursuant to this Equity Credit Agreement, the Investor shall commit to purchase up to $25 million of the Company’s common stock over the course of thirty-six months commencing the effective date of the initial registration statement covering the registrable securities pursuant to the Equity Credit Agreement. The put option price is 95% of the average of three lowest closing bid price (the “Bid Price”) of any two applicable trading days, consecutive or inconsecutive, during the five trading day period (the “Valuation Period”) commencing the date a put notice (the “Put Notice”) is delivered to the Investor (the “Put Date”) in a manner provided by the Equity Credit Agreement.
In addition, pursuant to the Equity Credit Agreement, in each Put Notice, the Company is required to specify a minimum stock price which in no event shall be less than 80% of the average of the Bid Price during the five trading day period commencing the Put Date (the “Floor Price”). In the event the Bid Price decreases below the Floor Price during the Valuation Period, the Investors obligation to fund one-fifth of the put amount for each such trading day shall terminate and the put amount shall be adjusted accordingly.
On December 7, 2010, the Company filed a Registration on Form S-1 which was subsequently amended on January 13, 2011 to register 13,000,000 shares of the Company’s common stock to be issued under the terms of the Equity Credit Agreement. The Registration Statement was declared effective January 26, 2011. The “Registrable Securities” include the Put Shares, any Blackout Shares (as defined in the Equity Credit Agreement) and any securities issued or issuable with respect to any of the foregoing by way of exchange, stock dividend or stock split or in connection with a combination of shares, recapitalization, merger, consolidation or other reorganization or otherwise. From January 26, 2011, to December 31, 2011, the Investor purchased 3.3 million shares of common stock pursuant to the terms of the Equity Credit Agreement, for which the proceeds to the Company totaled $200 thousand. These proceeds are net of expenses of $272 thousand associated with the warrants issued to the Investor upon execution of the Equity Credit Agreement.
Upon execution of the Equity Credit Agreement, the Company issued 820,856 warrants to the Investor in accordance with the Transaction Management Agreement with Southridge. The exercise price is equal to 120% of the average closing price of the Company's stock for the previous 20 days or $1.52. The fair value of the warrants totaling $881 thousand has been recognized as prepaid financing costs as of the agreement date and I being amortized over the life of the agreement or 36 months commencing January 26, 2011. The fair value of the warrants issued under the Equity Credit Agreements was determined using a Black-Scholes valuation model in accordance with ASC 505-50 Equity Based Payments to Non- Employees. Assumptions used in the underlying calculation included a risk-free interest rate of 1.48%, expected term of 5 years, expected volatility of 100% and expected dividends of 0%.
(8) Common Stock and Stock Purchase Warrants
At December 31, 2010 and 2011, the Company was authorized to issue 480 million shares of common stock, par value $0.0001 per share. The holders of common stock are entitled to receive dividends whenever funds are legally available and when declared by the Board of Directors. Each share of common stock is entitled to one vote.
The accompanying notes are an integral part of these financial statements.
F-14
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(8) Common Stock and Stock Purchase Warrants (continued)
In May 2003, the Company declared a 3 for 1 stock split of its common stock in the form of a stock dividend. All common share and per share information in the financial statements and related notes have been retroactively restated to reflect the stock split.
Upon inception, the Company issued 5,728 million shares of common stock to its founders. Issuances of common stock through private placements are as follows (in thousands except for per share amounts):
|
Shares
|
Proceeds
|
Price per share
|
||||||||||
|
(unaudited)
|
(unaudited)
|
(unaudited)
|
||||||||||
|
March through August 2001
|
1,268 | $ | 846 | $ | 0.66 | |||||||
|
November 2001
|
4 | 3 | 0.83 | |||||||||
|
January through July 2002
|
2,591 | 2,158 | 0.83 | |||||||||
|
August through December 2002
|
1,463 | 1,951 | 1.33 | |||||||||
|
January through May 2003
|
1,719 | 2,292 | 1.33 | |||||||||
|
June through October 2003
|
2,581 | 5,162 | 2.00 | |||||||||
|
December 2003
|
164 | 410 | 2.50 | |||||||||
|
January through April 2004
|
3,371 | 8,429 | 2.50 | |||||||||
|
February through July 2004
|
1,017 | 4,066 | 4.00 | |||||||||
|
January through March 2005
|
2,102 | 16,814 | 8.00 | |||||||||
|
June through July 2005
|
1,203 | 12,028 | 10.00 | |||||||||
In November 2001, the Company issued 18,657,000 shares of common stock in exchange for 3,007,000 shares of Old Voyager common stock which were originally issued in March 2001 for $250 thousand.
In May 2003, the Company repurchased and reissued 120,000 shares of common stock for $100 thousand, or $0.83 per share. During the same period, the Company repurchased and reissued 210 thousand shares of common stock for $280 thousand, or $1.33 per share.
From January 2006 through May 2007, the Company raised $16.6 million through the sale of 1,661,000 units at $10 per unit. A unit consisted of one share of common stock and warrants to purchase two additional shares of common stock at $12 per share. The Company amended this offering on August 21, 2006. Under the amendment each unit consists of one share of common stock and warrants to purchase six additional shares of common stock at $0.10 per share. At December 31, 2011, the remaining outstanding warrants to purchase 2.8 million shares of common stock expired.
From June 2007 to December 2008, the Company raised $1.4 million through the sale of 6,792,000 shares of common stock at $1.00 per share. This offering was amended on August 22, 2007 and March 6, 2008 to reduce the pricing to $.50 and $.20 per share, respectively and 3,669,000 additional shares were issued on a retroactive basis. The accounting for the issuance of these additional shares followed the character of the initial offering and no expense was recorded.
During 2008, the Company issued stock purchase warrants to select vendors to satisfy, in part, liabilities generated from services performed in conjunction with our terminated Phase III clinical trial, VP-AD-301. The warrants provide for the purchase 54,800 shares of common stock at $.50 per share and expired on December 31, 2011.
From January 2009 through February 5, 2010, the Company raised $1.4 million through the sale of 7.1 million shares of common stock at $0.20 per share and the issuance of 1.6 million shares of common stock upon exercise of outstanding warrants at $0.10 per share. These proceeds are net of expenses associated with warrants to be issued to outside consultants for services rendered in connection with the bridge financing round totaling $129 thousand. The Company raised an additional $39 thousand subsequent to the closing of the bridge financing round through December 31, 2010 from the issuance of 389 thousand shares of common stock upon exercise of outstanding warrants at $0.10 per share and the cashless issuance of 10 thousand shares of common stock upon exercise of outstanding warrants at $0.22 per share.
The accompanying notes are an integral part of these financial statements.
F-15
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(8) Common Stock and Stock Purchase Warrants (continued)
On May 28, 2009, the company entered into a Transaction Management Agreement with Southridge Business Solutions Group, LLC (“Southridge”). Under the terms of the agreement, the Company retained Southridge, on a non-exclusive basis, to provide services to enable the Company to restructure its balance sheet and execute a potential public offering or other public transaction. As part of the consideration for the investment advisory services to be provided, the Company issued a total of 2,149,148 warrants to Southridge to purchase the Company’s stock at $.001 per share. The warrants provide for a cashless exercise and expire seven years after the date of issuance. The estimated fair value of the warrants issued totaled $428 thousand as determined using a Black-Scholes valuation model in accordance with ASC 505-50 Equity-Based Payments to Non-Employees. Assumptions used in the underlying calculation included a risk-free interest rate of 2.34%, expected term of 7 years, expected volatility of 100% and expected dividends of 0%. At December 31, 2009, the Company had amortized $246 thousand of the expense associated with the warrants. In addition, a prepaid balance of $83 was recorded. During the year ended December 31, 2010, the remaining $182 thousand in expense was amortized by the Company.
On November 25, 2009, the Company executed an engagement letter for legal services to be provided in conjunction with a proposed Merger transaction. Fees for the work to be performed include the commitment of the Company to issue shares of common stock of the surviving corporation in an amount equivalent to .25% of the anticipated outstanding common shares post Merger. Upon closing of the Merger transaction 179 thousand shares were issued for legal services rendered.
On December 2, 2009, the Board of Directors granted 150 thousand shares of common stock, valued at $30 thousand, to a former executive officer to satisfy, in full, amounts due under an original five year employment contract.
During the year ended December 31, 2011, the Company issued 190 thousand shares of common stock in exchange for consulting services and 330 thousand shares of common stock for services rendered by members of the Board of Directors, which had a total fair value of $84 thousand and $28 thousand, respectively.
At December 31, 2010 and 2011 outstanding warrants to purchase the Company’s common stock are as follows:
|
December 31,
|
|||||||||||||
|
2010
|
2011
|
Exercise Price
|
Expiration Date | ||||||||||
|
Warrants issued January 2006 through May 2007 in conjunction with private placement of common stock.
|
3,095,800 | - | $ | 0.10 |
December 31, 2011
|
||||||||
|
Warrants issued to select vendors to satisfy, in part, liabilities generated from services performed in conjunction with our terminated Phase III clinical trial, VP-AD-301.
|
54,800 | - | $ | 0.50 |
December 31, 2011
|
||||||||
|
Warrants issued under Transaction Management Agreement executed with Southridge.
|
2,149,100 | - | $ | 0.001 |
August 31, 2016
|
||||||||
|
Warrants issued to outside consultants for services rendered in connection with the financing round completed on February 5, 2010.
|
844,400 | 844,400 | $ | 0.22 |
August 31, 2016
|
||||||||
|
Warrants issued as consideration for the cancellation of 181,285,000 shares of common stock upon consummation of the Merger transaction effective July 29, 2010.
|
3,589,500 | 3,589,500 | $ | 0.001 |
July 29, 2017
|
||||||||
|
Warrants issued to Southridge under the Transaction Management Agreement upon execution of Equity Credit Agreement dated September 16, 2010.
|
820,800 | 820,800 | $ | 1.52 |
September 15, 2015
|
||||||||
|
Warrants issued August 31, 2010 to outside vendor for development of the Company’s website.
|
100,000 | 100,000 | $ | 0.30 |
August 31, 2015
|
||||||||
| 10,654,400 | 5,354,700 | ||||||||||||
The accompanying notes are an integral part of these financial statements.
F-16
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(8) Common Stock and Stock Purchase Warrants (continued)
During the year ended December 31, 2010, 520 thousand warrants were exercised at $0.10 per share and 10,000 warrants were exercised at $0.22 per share.
During the year ended December 31, 2011, the Company issued 2.1 million shares and 334 thousand shares of common stock upon exercise of outstanding warrants at $0.001 per share and $0.10 per share, respectively. In addition, the Company issued 372 thousand shares of common stock upon the exercise of outstanding stock options at $.003 per share.
(9) Convertible Preferred Stock
On September 23, 2010 and September 29, 2010, the Company amended its certificate of incorporation by filing certificates of designation with Secretary of State of Nevada that designated two series of convertible preferred stock (the “Series A Convertible Preferred Stock” and the “Series B Convertible Preferred Stock”), respectively. Each share of Series A and Series B preferred stock has a par value of $.0001 and an initial stated value equal to $1,000. The Company is to pay cumulative dividends at the rate of 4% per annum payable quarterly in arrears beginning December 15, 2010. Dividends may be paid in cash or at the Company’s irrevocable option, in shares at the lesser of (A) the pre-determined conversion price or (B) 80% of the ten previous days Volume Weighted Average Price (“VWAP”). If dividend payments are not made by the third trading date following the specified Dividend Payment Date, the Holder may treat the amount of such dividend as added to the Stated value of the preferred stock issued. Each share of preferred stock shall be convertible at any time at the option of the Holder into common stock. The number of shares of common stock to be issued upon conversion of each share of preferred stock will be determined by dividing the initial stated value of the preferred stock by a pre-determined conversion rate. The predetermined conversion rates for the Series A and Series B preferred stock is $0.35 and $0.50, respectively.
On September 30, 2010, the Company entered into a Series A Convertible Preferred Stock Purchase Agreement (the “Series A Agreement”), by and between the Company and C P Acquisition Partners LP, an affiliate of Southridge, (the “Purchaser”) Pursuant to the terms of the Series A Agreement, the Company sold 500 shares of the Company’s Series A Convertible Preferred Stock for a total purchase price of $500,000.
On September 30, 2010, the Company entered into a Series B Convertible Preferred Stock Purchase Agreement (the “Series B Agreement”), by and between the Company and the Purchaser. Pursuant to the terms of the Series B Agreement, the Company sold 500 shares of the Company’s Series B Convertible Preferred Stock for a total purchase price of $500,000. Cash proceeds of $396 thousand have been received by the Company as of December 31, 2010. A subscription receivable in the amount of $104 thousand is recognized as of December 31, 2010. During the year ended December 31, 2011, the Company received $104 thousand due on the subscription receivable.
We have an obligation to make a cash payment to the holders of the Series A and Series B preferred stock for any loss that could be realized if the holders convert the preferred stock and we subsequently fail to deliver a certificate representing the shares to be issued upon such conversion by the third trading day after such conversion and the holders purchase, in an arm’s-length open market transaction or otherwise, shares of Common Stock (the “Covering Shares”) in order to make delivery in satisfaction of a sale of Common Stock by the converting Holder (the “Sold Shares”), which delivery such converting Holder anticipated to make using the shares to be issued upon such conversion (a “Buy-In”). “Buy-In Adjustment Amount” means the amount equal to the excess, if any, of (i) the converting Holder’s total purchase price (including brokerage commissions, if any) for the Covering Shares associated with a Buy-In, over (ii) the net proceeds (after brokerage commissions, if any) received by the converting Holder from the sale of the Sold Shares. Accordingly, the Series A and Series B preferred shares have been accounted for as a liability. Their fair value is estimated, at the end of each quarterly reporting period, based on the potential shares of common stock issuable upon conversion multiplied by the current quoted market price of the Company’s common stock. The Company estimated the fair value of the Series A and Series B preferred shares on the grant date to be $3.6 million. The excess of the estimated fair value as on the grant date over the stated par value totaling $2.6 million was recorded as interest expense. The fair value of the Series A and Series B preferred shares has decreased by $3.5 million subsequent to the grant date through December 31, 2011. The fluctuation has been recorded in the statements of operations.
The accompanying notes are an integral part of these financial statements.
F-17
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(9) Convertible Preferred Stock (continued)
On March 29, 2011, the Company amended its certificate of incorporation by filing certificates of designation with the Secretary of State of Nevada that designated a third series of convertible preferred stock (the “Series C Convertible Preferred Stock”). Each share of Series C Convertible Preferred Stock has a par value of $0.0001 and an initial stated value equal to $1,000. The Company is to pay cumulative dividends at the rate of 4% per annum, payable quarterly in arrears beginning June 15, 2011. Dividends may be paid in cash or at the Company’s irrevocable option, in shares at the lesser of (A) the pre-determined conversion price or (B) 80% of the ten previous days’ Volume Weighted Average Price (VWAP). If dividend payments are not made by the third trading date following the specified Dividend Payment Date, the Holder may treat the amount of such dividend as added to the Stated value of the preferred stock issued. Each share of preferred stock shall be convertible at any time at the option of the holder into common stock. The number of shares of common stock to be issued upon conversion of each share of Series C Convertible Preferred Stock will be determined by dividing the initial stated value of the Series C Convertible Preferred Stock by a pre-determined conversion rate of $0.50.
On March 30, 2011 the Company entered into a Series C Convertible Preferred Stock Purchase Agreement (the “Series C Agreement”), by and between the Company and C P Acquisition Partners LP, an affiliate of Southridge, (the “Purchaser”). Pursuant to the terms of the Series C Agreement, the Company sold 636 shares of the Company’s Series C Convertible Preferred Stock for a total purchase price of $265,900. On April 26, 2011, the Company issued 1,200,000 shares of common stock to the Purchaser upon conversion of 600 shares of the Company’s Series C Convertible Preferred Stock at the pre-determined conversion rate.
Dividends payable in arrears on all issued preferred stock at December 31, 2011 totaled $56 thousand of which $52 thousand has been added to the Stated Value of the Series A, B and C preferred stock issued at the election of the respective Holders.
(10) Fair Value
In accordance with FASB ASC 820, “Fair Value Measurements and Disclosures”, the following table represents the Company’s fair value hierarchy for its financial assets and liabilities measured at fair value on a recurring basis as December 31, 2010 and 2011 (in thousands):
|
Level 2 - At December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Derivative instruments (long term)
|
$
|
1,336
|
$
|
99
|
||||
|
Total
|
$
|
1,336
|
$
|
99
|
||||
(11) Stock Option Plan
During 2001, the Company adopted the 2001 Stock Option Plan (the “2001 Plan”) under which 1.5 million shares of the Company’s common stock was reserved for issuance to employees, directors and consultants. Options granted under the Plan may be incentive stock options or non-qualified stock options. On January 31, 2005, the Company adopted the 2005 Stock Option Plan (the “2005 Plan”) with terms and conditions similar to the 2001 Plan under which 3.5 million shares of the Company’s common stock are reserved for issuance to employees, directors and consultants. The Board of Directors determines the exercise price, term and the period over which options become exercisable at each grant date. The Board of Directors determines what it deems to be the fair market value of the stock based on the price per share of the most recent private placement of common stock and other relevant information. Since inception, all options were granted to employees with an exercise price equal to the deemed fair value of the common stock on the date of grant. Under the 2001 Plan and the 2005 Plan, options vest over various periods, generally annually over five years, and expire not more than ten years after the date of grant.
The accompanying notes are an integral part of these financial statements.
F-18
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(11) Stock Option Plan (continued)
The Company has granted 684 thousand stock options outside of the approved 2001 Plan and 2005 Plan to several consultants that vested upon completion of certain services. In addition, the Company has granted 53 thousand options under the 2005 Plan to other non-employees. Upon adoption of ASC 718, the Company continues to account for the resulting non-employee stock-based compensation for all options granted to non-employees based on the estimated fair value of the options, determined using the Black-Scholes option valuation model, in accordance with ASC 505-50, and amortizes such expense on a straight –line basis. The measurement date for the calculation of compensation expense is considered to be the date when all services have been rendered or the date that options are fully vested.
During the years ended December 31, 2010 and 2011, the Company recognized stock-based compensation expense related to its stock option plans in the accompanying statements of operations as follows (in thousands):
|
Year ended December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Employee-related stock-based compensation
|
$ | 231 | $ | 173 | ||||
|
Non-employee related stock based compensation
|
229 | - | ||||||
| $ | 460 | $ | 173 | |||||
Total stock-based compensation expense/ (benefit) recognized in the accompanying statement of operations is included in the General and administrative expense category.
The Company estimates the fair value of stock options at the date of grant using the Black-Scholes option valuation model. The Company calculated the fair value of employee stock options for the years ended December 31, 2010 and 2011, using the following assumptions:
|
Year ended December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Risk-free interest rate
|
2.15 to 3.20
|
% | 1.65 | % | ||||
|
Expected term (in years)
|
7.5 | 7.5 | ||||||
|
Expected volatility
|
100 | % | 100 | % | ||||
|
Expected dividends
|
0 | % | 0 | % | ||||
The risk-free rate is estimated to equal U.S. Treasury security rates for applicable terms. Estimated volatility is based on the historical stock price volatility of comparable companies’ common stock, as our stock does not have sufficient historical trading activity as we became a public reporting company during 2010. In addition, the Company does not have sufficient history of exercise behavior to develop estimates of expected term. To date, only two blocks of options have been exercised; the exercise of these options occurred subsequent to the granting of options in 2011. The expected term is based on the estimated time period to elapse prior to commercialization of the Company’s initial product. For options to outside consultants, the Company uses a term which is equal to the average the period required for the options to fully vest and the contractual life of the option. As the Company gathers more history regarding its option exercise pattern, and as information regarding the option pattern of comparable companies in its industry becomes publically available, it will review these estimates and revise them as appropriate. Different estimates of volatility and expected term could materially change the value of an option and the resulting expense.
The accompanying notes are an integral part of these financial statements.
F-19
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(11) Stock Option Plan (continued)
The following table summarizes the Company’s stock option activity under the 2001 Plan, the 2005 Plan, as well as 674,000 options granted to consultants outside of the 2001 and 2005 Plans, including number of shares (“Number”) and weighted average exercise price (“WAEP”) for the years ended December 31, 2010 and 2011:
|
Number of Shares
|
Weighted Average Exercise Price
|
Weighted-Average Contractual Term
|
Weighted Average Grant Date Fair Value per Share
|
|||||||||||||
|
Outstanding at December 31, 2009
|
3,277,000 | $ | 1.87 | 5.15 | $ | 1.07 | ||||||||||
|
Options granted
|
260,000 | $ | 0.20 | |||||||||||||
|
Options cancelled
|
(9,000 | ) | $ | 0.20 | ||||||||||||
|
Outstanding at December 31, 2010
|
3,528,000 | $ | 1.75 | 4.54 | $ | 1.07 | ||||||||||
|
Options granted
|
800,000 | $ | 0.20 | |||||||||||||
|
Options exercised
|
(372,000 | ) | $ | 0.003 | ||||||||||||
|
Options cancelled
|
(220,000 | ) | $ | 0.87 | ||||||||||||
|
Outstanding at December 31, 2011
|
3,736,000 | $ | 1.64 | 5.24 | $ | 0.94 1.07 | ||||||||||
|
Exercisable at December 31, 2011
|
3,136,000 | $ | 1.92 | 4.48 | $ | 1.03 | ||||||||||
The weighted-average grant-date fair value of options granted during the years ended December 31, 2010 and 2011 were $0.17and $1.06. No options were exercised during the two years ended December 31, 2011.
A summary of the status of the Company’s non-vested shares as of December 31, 2011:
|
Weighted-
|
||||||||
|
Average Grant
|
||||||||
|
Shares
|
Date Fair Value
|
|||||||
|
Non-vested at January 1, 2011
|
406,000 | $ | 1.05 | |||||
|
Options Granted
|
800,000 | 0.17 | ||||||
|
Options vested
|
(386,000 | ) | (0.24 | ) | ||||
|
Options cancelled
|
(220,000 | ) | (0.77 | ) | ||||
|
Non-vested at December 31, 2011
|
600,000 | $ | 0.50 | |||||
As of December 31, 2011, there was $270 thousand of total unrecognized compensation cost related to non-vested share based compensation arrangements granted under the Plans. That cost is expected to be recognized over a weighted–average period of 1.6 years. The total fair value of shares vested during the years ended December 31, 2010, and 2011 was $271 thousand, and $94 thousand, respectively.
During the years end December 31, 2009 and 2011, the Company extended the contractual life of 397 thousand and 1.5 million vested options, respectively, held by several former employees. No additional expense was recognized as a result of the modifications.
The accompanying notes are an integral part of these financial statements.
F-20
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(12) Stock Incentive Plan
On December 9, 2011, the Company adopted the Curaxis Pharmaceutical Corporation 2011 Incentive Stock Plan (the “Incentive Plan”) designed to retain directors, executives and selected employees and consultants and reward them for making major contributions to the success of the Company. Under the provisions of the Plan, the Company can grant common stock to eligible persons awarding them with a proprietary interest in the growth and performance of the Company. Persons eligible to receive grants under the plan include directors, officers, employees or consultants to the Company. The term consultant shall mean any person, other than an employee, who is engaged by the Company to render services and is compensated for such services, including, but not limited to, outside legal counsel. On December 9, 2011, the Company filed a Registration on Form S-8 to register 7.5 million shares of the Company’s common stock to be issued under the terms of the Incentive Plan. The Registration Statement was declared effective December 9, 2011. From December 9 to December 31, 2011, no stock awards were granted under the Incentive Plan.
(13) Gain on the Reduction of Liabilities
The Company recorded a gain from the reduction in liabilities of $2.2 million in 2011. The obligations principally relate to goods acquired or services rendered while the Company was actively engaged in the terminated Phase III clinical trial. The balances reported include accounts payable represented by vendor invoices submitted to the company for payment, retained billings and accruals for estimated services rendered by vendors through the termination of the Phase III clinical trial but not billed to date. Very little, if any, collection activity has occurred. In the opinion of Company’s counsel as a result of the passage of time these liabilities cannot be legally enforced against the Company. The basic and diluted EPS on the aggregate gain for the year ended December 31, 2011 were $0.03.
(14) Earnings Per Share
In accordance with ASC 260, basic net loss per common share is computed by dividing net loss by the weighted-average number of common shares outstanding. Diluted net loss per common share is computed similarly to basic net loss per share, except that the denominator is increased to include all potential dilutive common shares, including outstanding options and warrants. Potentially dilutive common shares have been excluded from the diluted loss per common share computation for the year ended December 31, 2010 because such securities have an anti-dilutive effect on loss per share due to the Company’s net loss.
The following table sets forth a reconciliation of the number of shares used in the basic and diluted EPS computation for the years ended December 31, 2010 and 2011:
|
|
at December 31,
|
|||||||
|
2010
|
2011
|
|||||||
|
Weighted average common shares outstanding
|
67,323 | 75,859 | ||||||
|
Dilutive securities - Options
|
- | 388 | ||||||
|
Dilutive securities - Warrants
|
- | 3,835 | ||||||
|
Diluted average common stock equivalents outstanding
|
67,323 | 80,082 | ||||||
The following table sets forth the number of potential shares of common stock that have been excluded from diluted earnings per share because their effect was anti-dilutive (in thousands):
|
|
at December 31,
|
|||||||
|
2010
|
2011
|
|||||||
|
Outstanding stock options
|
3,528 | 3,348 | ||||||
|
Outstanding warrants
|
10,654 | 1,520 | ||||||
|
Outstanding preferred stock at conversion
|
2,429 | 2,627 | ||||||
| 16,611 | 7,495 | |||||||
The accompanying notes are an integral part of these financial statements.
F-21
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(15) Income Taxes
No provision for Federal or state income taxes has been recorded as the Company has incurred net operating losses since inception.
Significant components of the Company’s deferred tax assets at December 31, 2010 and 2011 consist of the following (in thousands):
|
December 31,
|
||||||||
|
2010
|
2011
|
|||||||
|
Deferred tax assets
|
||||||||
|
Domestic and operating loss carryforwards
|
$ | 32,476 | $ | 31,585 | ||||
|
Research and development credits
|
2,756 | 2,756 | ||||||
|
Contributions
|
28 | - | ||||||
|
Fixed assets
|
4 | 5 | ||||||
|
Accrued compensation
|
- | 134 | ||||||
|
Stock based compensation
|
1,201 | 1.268 | ||||||
|
Derivative liabilities
|
129 | 496 | ||||||
|
Total deferred tax asset
|
36,594 | 36,244 | ||||||
|
Valuation allowance for deferred assets
|
(36,594 | ) | (36,244 | ) | ||||
|
Net deferred tax asset
|
$ | ― | $ | ― | ||||
At December 31, 2010 and 2011, the Company provided a full valuation allowance against its net deferred tax assets since realization of these benefits is not considered to be more likely than not. The decrease in the valuation allowance of $350 thousand for 2011 resulted from the reduction in the net operating loss carryforward of $2.2 million. The reduction represents the gain recognized on the reduction in liabilities which was excluded from taxable income due to the insolvent status of the Company immediately prior to the release of the obligations. The increase in the valuation allowance of $867 for 2010 resulted primarily from the additional net operating loss carryforward generated.
At December 31, 2011, the Company had Federal net operating loss carryforwards of $83.6 million that expire in varying amounts between 2023 and 2031, and state net operating loss carryforwards of $83.6 million that expire in varying amounts between 2023 and 2026. Additionally, the Company has research and development credit carryforwards of $2.7 million as of December 31, 2011 for Federal tax purposes that expire in varying amounts between 2022 and 2027.
The Tax Reform Act of 1986 contains provisions which limit the ability to utilize net operating loss carryforwards and tax credit carryforwards in the case of certain events including significant changes in ownership interests. If the Company’s NOLs and/or tax credits are limited, and the Company has taxable income which exceeds the permissible yearly NOL, the Company would incur a Federal income and/or state tax liability even though NOLs would be available in future years.
Taxes computed at the statutory Federal income tax rate of 34 percent are reconciled to the provision for income taxes for the years ended December 31 as follows (in thousands):
|
2010
|
% of
pretax
loss
|
2011
|
% of
pretax
loss
|
|||||||||||||
|
United States Federal tax (benefit) at
statutory rate
|
$ | (763 | ) | (34.0 | ) | $ | 454 | 34.0 | ||||||||
|
State taxes (benefit) (net of federal benefit)
|
(35 | ) | (1.6 | ) | 20 | 1.5 | ||||||||||
|
Change in valuation reserve
|
867 | 38.6 | (350 | ) | (26.2 | ) | ||||||||||
|
Other non-deductible expenses
|
(69 | ) | (3.0 | ) | (124 | ) | (9.3 | ) | ||||||||
|
Provision for income taxes
|
$ | ― | ― | $ | ― | ― | ||||||||||
The accompanying notes are an integral part of these financial statements.
F-22
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(16) Commitments and Contingencies
Leases
The Company is obligated under a capital lease that expired in 2009 for office equipment. Included in property and equipment are assets recorded under this capital lease obligation with a cost of $20,200 at December 31, 2011. The assets were fully depreciated at December 31, 2007. No payments were made under the agreement in 2010 and 2011.
The Company had several non-cancelable operating leases, primarily for facilities and office equipment. Rental expense, including maintenance charges, for operating leases during 2010 and 2011 was $85 thousand and $22 thousand, respectively.
Effective September 22, 2008 the Company agreed to a Fifth Amendment to Lease agreement with its landlord. Pursuant to the terms of the amendment, the Company reduced the size of its’ leased space and relocated its’ office within the building. The term of the amended lease expires on April 30, 2011. In consideration for the reduced lease obligation, the Company agreed to pay the landlord a relocation fee of $27 thousand pursuant to the terms of a Promissory note. No payments have been made under the amended agreement during the three years ended December 31, 2011. Payments in arrears total $74 thousand at December 31, 2011.
Management Advisory and Consulting Agreements
Southridge Agreement
On May 28, 2009, Curaxis entered into a transaction management agreement with Southridge Business Solutions Group, LLC, of Ridgefield, Connecticut (“Southridge” or “Selling Security Holder”), to assist Curaxis in restructuring its balance sheet, principally through negotiations with several large trade creditors to reduce their claims, and to assist Curaxis in effectuating a merger with a suitable public corporation (the “Transaction Management Agreement”). Pursuant to the Transaction Management Agreement, Curaxis had worked with Southridge to reduce its trade debt by approximately $6,000,000 and entered into the Merger Agreement with Auto Search Cars, Inc. Under the terms of the Transaction Management Agreement, Curaxis is obligated to pay Southridge a management fee of $10,000 per month, from June 1, 2009 through the first anniversary of the closing of the Merger with Auto Search, or July 29, 2011. Curaxis may terminate the Transaction Management Agreement at any time by giving 90 days’ advance notice to Southridge. In addition, Curaxis was obligated to issue to Southridge warrants equal to 3% of Curaxis’ outstanding stock as of the date of the Merger with Auto Search at an exercise price of $0.001 per share. A total of 2,149,148 warrants have been issued to Southridge under the agreement. On May 27, 2011, the Transaction Management Agreement was terminated by Southridge.
Fees recognized by the Company under the terms of the Agreement totaled $301 thousand and $50 thousand for the years ended December 31, 2010 and 2011, respectively. The fees recognized include the amortization of the fair value of the warrants issued under the agreement totaling $181 thousand and $-0- thousand, respectively. The fair value of the warrants issued by Curaxis was determined using a Black-Scholes valuation model in accordance with ASC 505-50 Equity-Based payments to Non-Employees. Assumptions used in the underlying calculation included a risk-free interest rate of 2.34%, expected term of 7 years, expected volatility of 100% and expected dividends of 0%.
Canterbury Agreement
On June 12, 2009, Curaxis entered into a letter agreement with Canterbury Investment Partners, LLC of Hingham, Massachusetts (“Canterbury”), to assist Curaxis in a range of issues in connection with its contemplated merger with a public shell corporation, including its negotiations with Southridge, in developing a strategy to negotiate reductions, deferrals and/or equity exchanges with its trade creditors and in raising funds from current or prospective investors and in negotiating the terms of the acquisition of a public corporation (the “Letter Agreement”). Under the terms of the Letter Agreement, Curaxis paid Canterbury a retainer of $50,000 and is also obligated to pay Canterbury a monthly fee of $5,833 for a period of twelve (12) months from September 2009 through August 2010. In addition, under the Letter Agreement, Curaxis has issued a warrant to Canterbury to purchase 854,358 shares of Curaxis Common Stock at a price of $0.22 per share. Following the closing of the Merger with Auto Search, Curaxis is obligated to pay Canterbury the sum of $8,000 per month for a period of six (6) months for Canterbury’s assistance in developing and implementing a strategy to support Curaxis in its dealings with the financial community.
The accompanying notes are an integral part of these financial statements.
F-23
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(16) Commitments and Contingencies (continued)
Fees recognized by the Company under the terms of the Agreement, including the initial retainer, totaled $159 thousand and $8 thousand for the years ended December 31, 2010 and 2011, respectively. The fees recognized include the amortization of the fair value of the warrants issued under the agreement totaling $113 thousand and $16 thousand, respectively. The fair value of the warrants issued by Curaxis was determined using a Black-Scholes valuation model in accordance with ASC 505-50 Equity-Based payments to Non-Employees. Assumptions used in the underlying calculation included a risk-free interest rate of 2.98%, expected term of 5 years, expected volatility of 100% and expected dividends of 0%.
GroupMark Agreement
On September 15, 2009, Curaxis entered into an agreement with GroupMark Financial Services Ltd. (“GroupMark”) to perform investor relation services and develop Curaxis’ new website. The Company is to pay GroupMark an Investor Development fee of $35,000 of which $25,000 had been paid as of December 31, 2009. In addition, the Company paid an affiliate of GroupMark, $15,000 during the year ended December 31, 2009 for the development of its new website. Further, as part for the consideration for the services rendered, upon completion of the website in 2010, Curaxis issued 100,000 warrants to GroupMark to purchase shares of the Company’s common stock at $.030 per share. The warrants provide for a cashless exercise and expire five years from the date of issuance or August 31, 2015.
Supplier Arrangement
The Company has entered into a non-exclusive worldwide agreement with Durect Corporation to develop and manufacture clinical and commercial supplies of the Company’s unique formulation for Alzheimer’s’ treatment. The Company has an exclusive worldwide license to Durect’s sustained release technology used in the production of this formulation. During the years ended December 31, 2004 and 2005, the Company made payments of $250 thousand and $250 thousand, respectively, upon initiation of the first Phase I trial for the product and initiation of the first Phase III trial for the product. These milestone payments were recorded as research and development expense. The Company will make additional payments of up to $2.5 million to Durect upon the achievement of certain development and regulatory milestones, as well as payments for research and development expenditures, and royalties based on product sales. The agreement is non-cancelable by Durect, except for material default by the Company. Research and development expenses recognized under the contract totaled $1.7 million for the year ended December 31, 2006. As of December 31, 2006, the Company was in default of the agreement due to non –payment.
On January 23, 2007 the agreement with Durect Corporation was amended to provide financial assistance to the Company. Pursuant to the Amendment, Durect permanently waived its right to receive payments for development costs incurred totaling $727 thousand and its’ right to terminate the agreement as allowed for non-payment for outstanding development costs. Further, Durect remitted $1 million to the Company to assist in funding work required to compile and analyze data relating to the terminated Phase III clinical trial. In consideration for the above, the Company agreed to increase future royalty payments due Durect on product sales from a range of 5% to 7% to a range of 10% to 14% dependent upon volumes and to make a minimum royalty payment of $250 thousand annually commencing the year in which the first commercial sale is made. The liabilities forgiven by Durect and the financial payment made totaling $1.8 million have been recorded as deferred credits by the Company. The liability will be amortized over the projected royalty stream upon commercialization of the respective product.
On July 28, 2008, the Company entered into a Supply Agreement with Mallinckrodt Inc. to provide the Company with Lueprolide Acetate for a period of five years commencing January 1, 2009. Under the terms of the Agreement, the company is required to purchase 75% of its annual requirements for the product from Mallinckrodt at an agreed upon price of $550 per gram. The price will be adjusted annually to reflect cost increases of the supplier on a dollar-for dollar basis. The price to be paid by the Company will be reduced by $50 per gram from the price in effect once the first volume of 75% of its annual requirements is accepted under the “FMOC” manufacturing process as defined by the agreement. The agreement will be automatically renewed for an additional 5 year term unless Mallinkrodt provides written notice at least six months prior to the end of the initial term. The Agreement may be terminated by written agreement of the parties. Upon execution of the Agreement, Mallinkckrodt paid $500 thousand to the Company with the understanding that the Company would be in a position to accept product as of January 1, 2009. The Company has recorded the payment as a deferred purchase credit at December 31, 2008. The Company was in default of the Agreement at January 1, 2009, as it was unable to accept product.
The accompanying notes are an integral part of these financial statements.
F-24
Curaxis Pharmaceutical Corporation
(A Development Stage Corporation)
Notes to Financial Statements
(16) Commitments and Contingencies (continued)
Contingencies
The Company recorded a gain from the reduction in liabilities of $2.2 million in 2011. The obligations principally relate to goods acquired or services rendered while the Company was actively engaged in the terminated Phase III clinical trial. Very little, if any, collection activity has occurred. The balances reported include accounts payable represented by vendor invoices submitted to the Company for payment, retained billings, and accruals for estimated services rendered by vendors through the termination of the Phase III clinical trial but not billed to dale. While it is unlikely, however that vendors possibly may will pursue the collection of a portion of the liabilities released, if such action is taken, the Company based on the opinion of the Company’s counsel, believes that the final outcome of any such proceedings in a court of law will not have an adverse impact on the operations of the Company and its financial position.
(17) Related Party Transactions
During the two years ended December 2008, the Company funded travel expenses of an executive officer not directly related to the Company’s business. Travel expenses paid by the Company on behalf of the officer totaled $14 thousand. In addition, during the year ended December 31, 2009, the Company advanced funds to the executive officer for travel and office expenses totaling $16,000. Payments made to the Company by the employee and actual expenses incurred on the Company’s behalf totaled $10 thousand and $9 thousand for the years ended December 31, 2008 and 2009, respectively. A receivable in the amount of $11 thousand was recorded at December 31, 2009. During the year ended December 31, 2010, the executive officer satisfied the liability to the Company in full.
(18) Subsequent Events
From January 1 to March 28, 2012, the Investor purchased 2.2 million shares of common stock pursuant to the terms of the Equity Credit Agreement for which the proceeds to the Company totaled $54 thousand.
On February 20, 2012, the Company executed a Promissory Note payable to Southridge Partners II, LP. The $111 thousand unsecured note bears interest at 6% per annum and is payable on or before June 30, 2011. The proceeds of the note were used to meet current operating needs of the Company.
On March 6, 2012, the Company issued 500,000 shares of registered common stock to outside counsel under the terms of the Stock Incentive Plan for legal services rendered.
The accompanying notes are an integral part of these financial statements.
F-25