Attached files
| file | filename |
|---|---|
| EX-10.7 - EXHIBIT 10.7 - ENDOCYTE INC | d316117dex107.htm |
| EX-32.1 - EXHIBIT 32.1 - ENDOCYTE INC | d316117dex321.htm |
| EX-10.9 - EXHIBIT 10.9 - ENDOCYTE INC | d316117dex109.htm |
| EX-23.1 - EXHIBIT 23.1 - ENDOCYTE INC | d316117dex231.htm |
| EX-31.1 - EXHIBIT 31.1 - ENDOCYTE INC | d316117dex311.htm |
| EX-31.2 - EXHIBIT 31.2 - ENDOCYTE INC | d316117dex312.htm |
| EXCEL - IDEA: XBRL DOCUMENT - ENDOCYTE INC | Financial_Report.xls |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) |
| OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the fiscal year ended December 31, 2011 |
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) |
| OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the transition period from to |
Commission file number 001-35050
ENDOCYTE, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 35-1969-140 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification Number) | |
| 3000 Kent Avenue, Suite A1-100 West Lafayette, IN 47906 | ||
| (Address of Registrant’s principal executive offices) | ||
Registrant’s telephone number, including area code: (765) 463-7175
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $.001 par value | NASDAQ Global Market | |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer þ | Smaller reporting company ¨ | |||
| (Do not check if a smaller reporting company) | ||||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ¨ Yes þ No
The aggregate market value of the registrant’s common stock, $0.001 par value per share, held by non-affiliates of the registrant, based upon the closing price of the Common Stock on the Nasdaq Global Market on June 30, 2011, was approximately $323.5 million (excludes shares of the registrant’s common stock held as of such date by officers, directors and stockholders that the registrant has concluded are or were affiliates of the registrant, exclusion of such shares should not be construed to indicate that the holder of any such shares possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.)
Number of shares of the registrant’s Common Stock, $0.001 par value, outstanding on March 1, 2012: 35,793,078
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive Proxy Statement to be delivered to stockholders in connection with the 2012 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
ENDOCYTE, INC
ANNUAL REPORT ON FORM 10-K FOR THE YEAR ENDED DECEMBER 31, 2011
INDEX
This annual report contains certain statements that are forward-looking statements within the meaning of federal securities laws. When used in this report, the words “may,” “will,” “should,” “could,” “would,” “anticipate,” “estimate,” “expect,” “plan,” “believe,” “predict,” “potential,” “project,” “target,” “forecast,” “intend” and similar expressions are intended to identify forward-looking statements. Forward-looking statements are subject to risks and uncertainties that could cause actual results to differ materially from those projected. These risks and uncertainties include the important risks and uncertainties that may affect our future operations that we describe in Part I, Item 1A — Risk Factors of this report, including, but not limited to, statements regarding the progress and timing of clinical trials, the safety and efficacy of our product candidates, the goals of our development activities, estimates of the potential markets for our product candidates, estimates of the capacity of manufacturing and other facilities to support our product candidates, projected cash needs and our expected future revenues, operations and expenditures. Readers of this report are cautioned not to place undue reliance on these forward-looking statements. While we believe the assumptions on which the forward-looking statements are based are reasonable, there can be no assurance that these forward-looking statements will prove to be accurate. This cautionary statement is applicable to all forward-looking statements contained in this report.
| Page | ||||||
| PART I | ||||||
| ITEM 1. | 1 | |||||
| ITEM 1A. | 39 | |||||
| ITEM 1B. | 59 | |||||
| ITEM 2. | 59 | |||||
| ITEM 3. | 60 | |||||
| ITEM 4. | 60 | |||||
| PART II | ||||||
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES | 60 | ||||
| ITEM 6. | 62 | |||||
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS | 64 | ||||
| ITEM 8. | 78 | |||||
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE | 102 | ||||
| ITEM 9A. | 102 | |||||
| ITEM 9B. | 102 | |||||
| PART III | ||||||
| ITEM 10. | 103 | |||||
| ITEM 11. | 103 | |||||
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS | 103 | ||||
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE | 104 | ||||
| ITEM 14. | 104 | |||||
| PART IV | ||||||
| ITEM 15. | 105 | |||||
| SIGNATURES | 106 | |||||
| EXHIBIT INDEX | 107 | |||||
| Item | 1. Business |
Overview
We are a biopharmaceutical company developing targeted therapies for the treatment of cancer and inflammatory diseases. We use our proprietary technology to create novel small molecule drug conjugates, or SMDCs, and companion imaging diagnostics. Our SMDCs actively target receptors that are over-expressed on diseased cells, relative to healthy cells. This targeted approach is designed to enable the treatment of patients with highly active drugs at greater doses, delivered more frequently, and over longer periods of time than would be possible with the untargeted drug alone. We are also developing companion imaging diagnostics for each of our SMDCs that are designed to identify the patients whose disease over-expresses the target of the therapy and who are therefore more likely to benefit from treatment. This combination of an SMDC with its companion imaging diagnostic is designed to personalize the treatment of patients by delivering effective therapy, selectively to diseased cells, in the patients most likely to benefit.
Our lead SMDC, EC145, targets the folate receptor, which is frequently over-expressed in some of the most prevalent, and difficult to treat solid tumor indications. We identify the presence of the folate receptor in cancer patients by using EC20, our proprietary companion imaging diagnostic for EC145. We have chosen platinum-resistant ovarian cancer, or PROC, a highly treatment-resistant disease, as our lead indication for development of EC145 because of the high unmet need in treating this patient population and the high percentage of ovarian cancer patients whose tumors over-express the targeted folate receptor. In the final progression free survival, or PFS, analysis of PRECEDENT, our randomized phase 2 clinical trial in women with PROC, EC145 increased PFS from a median of 2.7 months to a median of 5.0 months, representing an 85 percent improvement over standard therapy (p=0.031). We studied a subset of patients in which 100 percent of their target lesions over-expressed the folate receptor as determined by an EC20 scan, patients which we refer to as FR(++). We treated these FR(++) patients with a combination of EC145 and pegylated liposomal doxorubicin, or PLD (marketed in the U.S. under the brand name Doxil), and observed a median PFS of 5.5 months compared to a median of 1.5 months for patients receiving PLD alone, an improvement of over 260 percent. The hazard ratio was 0.381 (p=0.018), or a reduction in the risk of progression of 61.9 percent.
In December 2011, we announced the results of supplemental analyses we had conducted of the PRECEDENT trial data. We believe these findings continue to support the robustness of the PRECEDENT trial results, particularity in the group of FR (++) patients. The primary endpoint of the open-label PRECEDENT trial was PFS based on investigator assessment. Sensitivity analyses demonstrated these results were robust when adjusted for a variety of potential imbalances (e.g., demographic, prognostic, etc.). The assessments made by the blinded independent review committee, or IRC, shared a high level of agreement with investigator assessments, confirming investigator-assessed progression for 74 percent of all patients. In addition, the metrics designed to identify an investigator's potential bias to delay assessed progressions for patients in the EC145 study arm or to accelerate assessed progressions in the PLD control arm did not reveal such a bias. The IRC results also correlate with the mechanism of action, reflecting greater reduction in the risk of progression with increasing presence of folate receptor. In FR(++) patients, the IRC reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.465 suggests a reduction in risk of progression in the EC145 study arm of 53 percent (p=0.0498). In the FR(+) patient population, which includes the FR(++) subgroup, the IRC also reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.652 was not statistically significant in this broader group.
The results of the supplemental analyses also confirmed the reliability of our companion imaging diagnostic EC20 to select targeted patients. In order to confirm the reliability of EC20 to select these patients, independent readers evaluated EC20 images from the PRECEDENT trial to measure the agreement rate between readers. The evaluation resulted in an 87 percent agreement rate in selecting patients with at least one tumor positive for the
folate-receptor, FR(+), and an 85 percent agreement rate for patients with all folate-receptor positive tumors, FR(++). These rates of agreement compare favorably to internal standards established for this evaluation. In addition, intra-reader agreement was measured. This is an assessment of the consistency of the same reader to select patients when reviewing images at different times. Intra-reader rates were 90 percent and 95 percent for the independent readers in selecting FR(+) patients and 95 percent and 100 percent in selecting FR(++) patients. These rates of agreement also compare favorably to internal standards established for this evaluation.
The PRECEDENT trial was not statistically powered to show a survival advantage and the results did not indicate a trend toward benefit in either arm. The hazard ratio was 1.010 for the intent-to-treat population and 1.097 for the FR(++) population. On an adjusted basis, which accounts for potential differences in demographics and prognostic factors, the overall survival hazard ratio was 0.864 in the intent-to-treat population and 0.481 in the FR(++) group, although these result were not statistically significant.
We are preparing applications for conditional marketing authorization to be filed with the European Medicines Agency, or EMA, for our lead drug candidate EC145 for the treatment of PROC and its companion imaging diagnostic EC20 for patient selection. The marketing authorization applications will be supported by four clinical studies: a Phase 1 study in solid tumors, two single-agent, single-arm Phase 2 studies in ovarian cancer and non small cell lung cancer, and the PRECEDENT trial, a randomized study in PROC. This plan was based on consultation with the EMA, including a meeting with the Scientific Advice Working Party and written advice from the Committee for Medicinal Products for Human Use, or CHMP. Discussions with European Health Authorities about the contents of the marketing applications occurred both before and after the supplemental analyses were finalized. We plan to seek conditional marketing authorization for treatment of patients with folate receptor positive PROC as selected by EC20. EC145 and EC20 have also both been granted orphan drug status for ovarian cancer by the European Commission, which provides ten years of marketing exclusivity of the product candidate if approved for marketing for the designated orphan indication in the European Union, or EU, and makes us eligible for protocol assistance and other benefits in connection with the application process. We expect to file both applications in the third quarter of 2012.
We began enrolling patients in PROCEED, our phase 3 registration trial for EC145 and EC20, in May 2011 but enrollment stopped later in the year due to global shortages of PLD. In March 2012, we announced that the U.S. Food and Drug Administration, or FDA, approved our plans to import our supply of PLD from Europe into the U.S. for use in this trial and we plan to resume enrollment in the U.S. PROCEED is a randomized, double-blinded trial of EC145 in combination with PLD compared to PLD plus placebo. The patient population, those with PROC, will be the same as in the PRECEDENT trial, and the primary endpoint will be PFS in patients selected by EC20 as folate receptor positive in all tumors, FR(++). The secondary endpoint will be overall survival, or OS, in this same population. Projected enrollment of FR(++) patients is more than 200. Based on feedback from EU health authorities and the FDA, we are evaluating the inclusion of additional patients for exploratory analysis in order to assess potential benefit in patients with less than all of their target tumors positive for the folate receptor. While those plans are being finalized, patients will be enrolled regardless of the FR status, although the primary endpoint will include only FR(++) patients. The trial will be conducted in approximately 150 sites in the United States, Canada, Europe and Asia. We expect final primary PFS data from this study in the first half of 2014.
We are also developing EC145 for use in non-small cell lung cancer, or NSCLC, where we have completed a phase 2 single-arm clinical trial in heavily pre-treated patients and observed a disease control rate, or DCR, of 57 percent at the eight week assessment in patients whose target tumors were all identified as over-expressing the folate receptor. This compares to historical DCRs ranging from 21 to 30 percent reported in other trials of approved therapies in less heavily pre-treated patients. In a subset of FR(++) patients who had received three or fewer prior therapies, the DCR was 70 percent. We also evaluated OS in FR(++) patients (n=14) compared to patients in which at least one of the target lesions, but not all, over-expressed the folate receptor, which patients we refer to as FR(+) (n=14). Median OS improved from 3.4 months for FR(+) patients to 10.9 months for FR(++) patients. The hazard ratio was 0.539, meaning FR(++) patients were 46.1 percent less likely to die when compared to FR(+) patients when receiving EC145 (p=0.209).
2
Based on results of our single-arm, single agent phase 2 clinical trial of EC145 in patients with heavily pre-treated NSCLC, we plan to begin enrollment in a randomized phase 2 trial in the second quarter of 2012. The trial is designed to enroll up to 200 patients with adenocarcinoma of the lung who have failed one prior line of therapy. Patients will be selected based on EC20 scan results and only FR(++) patients will be included. The trial design is intended to evaluate the safety and efficacy of EC145 in second line NSCLC as a single agent and in combination with docetaxel, an FDA approved and commonly used second line chemotherapy. The study will have three arms: docetaxel alone; EC145 alone; and EC145 plus docetaxel. The primary outcome measure will be PFS with secondary measures of OS, tumor response and duration of response. Final PFS data is expected to be available by early 2014.
Our imaging studies with our lead companion imaging diagnostic, EC20, and the analysis of tumor biopsies, have shown that the folate receptor is also over-expressed in a broad range of other solid tumors, including non-small cell lung, breast, colorectal, kidney, endometrial and other cancers. We are using companion imaging diagnostics that target the folate receptor and other target receptors, including prostate-specific membrane antigen, or PSMA, to guide future development of our SMDCs in other oncology indications and inflammatory diseases.
We currently have no commercial products and we have not received regulatory approval for, nor have we generated commercial revenue from, any of our product candidates.
Our Strategy
Our strategy is to develop and commercialize SMDCs to treat patients who suffer from a variety of cancers and inflammatory diseases that are not well addressed by currently available therapies. The critical components of our business strategy are to:
| • | Obtain marketing approval of our lead SMDC, EC145, for use in women with PROC. We plan to submit applications to the EMA for conditional marketing authorization of EC145 and EC20 in Europe in the third quarter of 2012. In addition, in May 2011 we initiated a randomized, controlled, double-blinded phase 3 registration trial, PROCEED, for the use of EC145 to treat women with PROC. Enrollment stopped later in the year due to global shortages of PLD, however, we announced in March 2012 that the FDA approved our plans to import our supply of PLD from Europe into the U.S. for use in this trial and we plan to resume enrollment in the U.S. If successful, we plan to submit the results of the PROCEED trial, supported by the results of a Phase 1 study in solid tumors, two single-agent, single-arm Phase 2 studies in ovarian cancer and non small cell lung cancer, and the PRECEDENT trial to the FDA as the basis for our application for marketing approval in the U.S. |
| • | Expand use of EC145 to other indications. We are also developing EC145 for the treatment of NSCLC, and plan to conduct a controlled, randomized, phase 2 trial of EC145 in patients with folate receptor positive second line NSCLC. In this trial, we will use EC20 to select and enroll patients whose cancer over-expresses the folate receptor and who we believe are therefore more likely to respond to EC145. Folate receptors are over-expressed on a wide variety of tumors, including breast, colorectal, kidney, endometrial and other cancers. We estimate, based on worldwide cancer incidence rates, our own imaging studies and analysis of tumor biopsies that there are over one million newly diagnosed cancer patients per year in the United States, Europe and Japan whose tumors over-express the folate receptor. We intend to use EC20 to identify additional cancer indications for EC145 and to identify individual patients within each cancer indication who may be most suitable for treatment. |
| • | Build a pipeline of SMDCs by leveraging our technology platform. We believe that the modular approach of our technology platform will allow us to quickly and efficiently expand our pipeline of SMDC candidates featuring various combinations of our targeting ligands, linker systems and drug payloads. We currently have four SMDCs and two companion imaging diagnostics in clinical development. |
3
| • | Develop companion imaging diagnostics for each of our therapies. We believe there is a significant opportunity to create targeted therapies where individual patients are selected based upon the use of non-invasive imaging diagnostic tools. Our companion imaging diagnostics may lower the risk of development of our SMDCs by allowing us to select for our clinical trials only those patients whose disease over-expresses the receptor targeted by our SMDCs. This benefit may, upon regulatory approval, extend to clinical practice by giving physicians the information they need to prescribe our SMDCs to patients who are most likely to respond to our therapy. |
| • | Build commercial capabilities and partner to maximize the value of our SMDCs. To date, we have retained all worldwide commercial rights to our SMDCs. We intend to commercialize our oncology SMDCs in the United States through our own focused sales force that we would build in connection with such commercialization efforts, or by co-promoting these SMDCs in collaboration with one or more larger pharmaceutical companies that have established capabilities in commercializing cancer therapies. Outside of the United States, we will consider partnering with established international pharmaceutical companies to maximize the value of our pipeline. In the large inflammatory disease markets, we currently expect to out-license our SMDCs in order to mitigate their higher costs of development and commercialization. |
Our Technology Platform
Our technology platform has enabled us to develop multiple new SMDCs for a range of disease indications. Each SMDC is comprised of three modules: a targeting ligand, a linker and a drug payload. Our companion imaging diagnostics employ the same modular structure as our SMDCs replacing the drug payload with an imaging agent.
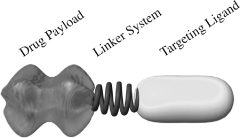
Targeting Ligand. Our technology is founded on our high-affinity small molecule ligands that bind to over-expressed receptors on target cells, while largely avoiding healthy cells. We are developing a number of targeting ligands to address a broad range of cancers and inflammatory diseases.
Linker System. Our linker system attaches the targeting ligand to the drug payload or imaging agent. It is designed to be stable in the bloodstream, and to release the active drug from the targeting ligand when the SMDC is taken up by the diseased cell. The linker system can be customized for each SMDC and each companion imaging diagnostic to improve its pharmacologic properties.
Drug Payload. This module is the biologically active component of our SMDCs. The majority of our drug payloads are highly active molecules that are too toxic to be administered in their untargeted forms at therapeutic dose levels. We are using drug payloads in our SMDCs that were shown in our in vitro preclinical studies to be between 10,000 and 100,000 times more potent than traditional cancer cell-killing drugs such as cisplatin.
With our modular approach, we use a variety of different targeting ligands, linker systems and drug payloads to create a pipeline of novel SMDC candidates for clinical development. For example, our PSMA targeting technology uses a targeting ligand that specifically binds to a receptor over-expressed on the surface of prostate cancer cells. We have developed alternative linker systems that modulate the pharmacologic and biodistribution properties of our SMDCs. In addition, we have developed a linker system that allows us to
4
conjugate multiple drug payloads to a single targeting ligand, thus offering the potential to simultaneously disrupt multiple pathways within cancer cells, forming a novel strategy for addressing drug resistance. We can also attach a wide variety of different drug payloads to our targeting ligands to address different disease indications. For example, we have SMDCs in preclinical development which incorporate proven anti-cancer and anti-inflammatory drug classes, such as microtubule destabilizers, DNA alkylators, proteasome inhibitors and mTOR inhibitors.
We own or have rights to 78 issued patents and 188 patent applications worldwide covering our core technology, SMDCs and companion imaging diagnostics. Our U.S. patent covering our core technology and our lead SMDC, EC145, expires in 2026, and our U.S. patents covering the EC145 companion imaging diagnostic, EC20, expire in 2024. In 2012, the United States Patent and Trademark Office awarded us a patent with claims specifically directed to EC145 entitled “Vitamin Receptor Binding Drug Delivery Conjugates”.
Companion Imaging Diagnostics
Our technology allows us to create companion imaging diagnostics intended for use with each of our SMDCs. To create our companion imaging diagnostics, we replace the drug payload of the SMDC with an imaging agent that is easily seen with widely available nuclear imaging equipment. Because the targeting ligand found on the companion imaging diagnostic is identical to that found on the therapeutic SMDC, our companion imaging diagnostics allow us to obtain full-body real-time images of tumors that over-express the target for that particular SMDC. This is accomplished without requiring an invasive tissue biopsy or reliance on archived tissue samples.
The information provided by our companion imaging diagnostics is used throughout the development of every new SMDC. In both preclinical and clinical trials, a companion imaging diagnostic is used to validate targeting of our SMDC to specific tissues and cells. These companion imaging diagnostics also allow for the screening of large patient populations to select diseases where a high percentage of the patient population have tumors or diseased cells that over-express the molecular target. These companion imaging diagnostics may also enable us to expand the use of our SMDCs to cancer indications where the percentage of patients who over-express a given receptor target of interest may be relatively low. Upon regulatory approval, we believe companion imaging diagnostics, such as EC20, will help to identify patients who will most likely benefit from treatment with our SMDCs. As a result, use of our companion imaging diagnostics may broaden the commercial use of our SMDCs. In our phase 2 single-arm and phase 2 randomized PRECEDENT clinical trials with EC145, we have seen correlations between favorable therapeutic outcomes and uptake of our companion imaging diagnostic, and, through supplemental analyses of the data, were able to validate with high rates of agreement through independent readers.
Lead SMDC Candidate (EC145)
Our lead SMDC candidate, EC145, consists of a highly cytotoxic anti-cancer drug, DAVLBH, joined by a linker system to the targeting ligand, folate. DAVLBH is a member of a class of proven anti-cancer drugs that destabilize microtubules within the cell, leading to cell death. As folate is required for cell division, many rapidly dividing cancer cell types have been found to over-express high-affinity folate receptors. In clinical trials using our companion imaging diagnostic, EC20, we found that ovarian, non-small cell lung, breast, colorectal, kidney, endometrial and other cancers over-express folate receptors. EC145 binds to these folate receptors on cancer cells with high-affinity and is internalized through a process known as endocytosis. Once EC145 is inside the cell, the linker system is cleaved, releasing the active drug payload within the cancer cell.
Advanced Clinical Trials
We have completed final PFS analysis for PRECEDENT, our randomized phase 2 clinical trial of 149 women with PROC. PRECEDENT is a randomized, controlled clinical trial in which patients received EC145 in combination with PLD, versus PLD alone. PLD is a current standard of care for PROC. The primary endpoint of
5
the trial is PFS which refers to the period of time that begins when a patient enters the clinical trial and ends when either the patient dies, or the patient’s cancer has grown by a RECIST-specified percentage or has spread to a new location in the body. RECIST refers to the response evaluation criteria in solid tumors, a set of published rules that define when the patients’ disease shrinks, remains stable, or progresses. Historically, PROC has proven difficult to treat, and no approved therapy has extended either PFS or OS in a randomized clinical trial. OS refers to the period of time that begins when a patient enters the clinical trial and ends when the patient dies.
At PRECEDENT’s final PFS analysis of 149 patients and 95 PFS events, combination therapy with EC145 and PLD increased median PFS by 85 percent over therapy with PLD alone. PFS increased from a median of 2.7 months in the PLD control arm to a median of 5.0 months in the EC145 and PLD combination therapy arm (p=0.031). The hazard ratio was 0.626, meaning patients receiving EC145 were 37.4 percent less likely to have died or have their cancer progress compared to patients receiving only PLD.
Kaplan-Meier curve for PFS in PRECEDENT
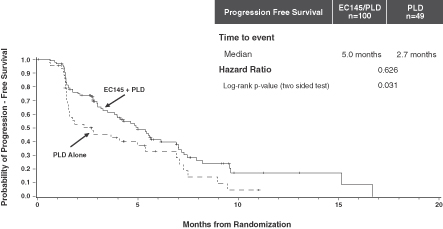
This observed improvement in PFS was provided in the context of low additional toxicity over that seen in patients receiving PLD alone. There was no statistically significant difference in adverse events in the combination arm of the PRECEDENT trial compared with the control arm (p=0.210).
In December 2011, we announced the results of supplemental analyses of PRECEDENT data. We believe these findings continue to support the robustness of the PRECEDENT trial results, particularity in the group of FR (++) patients. The primary endpoint of the open-label PRECEDENT trial was PFS based on investigator assessment. Sensitivity analyses demonstrated these results were robust when adjusted for a variety of potential imbalances (e.g., demographic, prognostic, etc.). The IRC assessments shared a high level of agreement with investigator assessments, confirming investigator-assessed progression for 74 percent of all patients. In addition, the metrics designed to identify an investigator's potential bias to delay assessed progressions for patients in the EC145 study arm or to accelerate assessed progressions in the PLD control arm did not reveal such a bias. The IRC results also correlate with the mechanism of action, reflecting greater reduction in the risk of progression with increasing presence of folate receptor. In FR(++) patients, the IRC reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.465 suggests a reduction in risk of progression in the EC145 study arm of 53 percent (p=0.0498). In the FR(+) patient population, which includes the FR(++) subgroup, the IRC also reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.652 was not statistically significant in this broader group.
The predictive power of our EC20 companion imaging diagnostic was also evaluated in the PRECEDENT trial. In an analysis of FR(++) patients, an increased improvement in PFS was observed. In this subgroup of
6
38 patients, PFS improved from a median of 1.5 months for patients receiving PLD alone to a median of 5.5 months for patients receiving the combination of EC145 and PLD, an improvement of over 260 percent. The hazard ratio was 0.381 (p=0.018), or a reduction in the risk of progression of 61.9 percent.
The results of the supplemental analyses also confirmed the reliability of our companion imaging diagnostic EC20 to select targeted patients. In order to confirm the reliability of EC20 to select these patients, independent readers evaluated EC20 images from the PRECEDENT trial to measure the agreement rate between readers. The evaluation resulted in an 87 percent agreement rate in selecting patients with at least one tumor positive for the folate-receptor, FR(+), and an 85 percent agreement rate for patients with all folate-receptor positive tumors, FR(++). These rates of agreement compare favorably to internal standards established for this evaluation. In addition, intra-reader agreement was measured. This is an assessment of the consistency of the same reader to select patients when reviewing images at different times. Intra-reader rates were 90 percent and 95 percent for the independent readers in selecting FR(+) patients and 95 percent and 100 percent in selecting FR(++) patients. These rates of agreement also compare favorably to internal standards established for this evaluation.
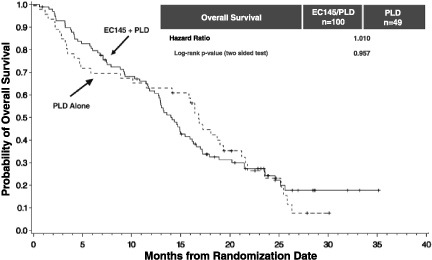
The PRECEDENT trial was not statistically powered to show a survival advantage and the results did not indicate a trend toward benefit in either arm. The hazard ratio was 1.010 for the intent-to-treat population and 1.097 for the FR(++) population. On an adjusted basis, which accounts for potential differences in demographics and prognostic factors, the overall survival hazard ratio was 0.864 in the intent-to-treat population and 0.481 in the FR(++) group, although these result were not statistically significant.
PROCEED is our phase 3 registration trial for U.S. approval of EC145 for the treatment of women with PROC and U.S. approval of EC20 for patient selection. If EC145 and EC20 receive Conditional Marketing Authorisation in the EU, this phase 3 trial serves to provide comprehensive data for conversion to regular Marketing Authorisation. The study design was discussed with Health Authorities in EU and with FDA. Basic agreement on the trial design has been reached; however, final details of the statistical analyses are being discussed with the FDA’s imaging and oncology divisions. PROCEED shares the same fundamental design characteristics of the PRECEDENT trial, except that it is a double-blinded trial, it measures PFS based on radiological progression alone without including clinical progression, and the primary efficacy analysis will be on FR(++) patients. The endpoint, patient population, dose and schedule are the same as those used in the PRECEDENT trial. In contrast to PRECEDENT, the PLD control arm in PROCEED will include a placebo in order to blind the study, which will be dosed on the same schedule as EC145. As was the case with the PRECEDENT trial, PROCEED’s primary endpoint is PFS.
All patients in the PROCEED trial are being imaged with EC20 prior to treatment. In our clinical trials that incorporated EC20 to date, we saw that approximately 80 percent of ovarian cancer patients over-express the folate receptor, and approximately 40 percent of patients have tumors that are FR(++). The primary endpoint of the phase 3 PROCEED trial is PFS in the FR(++) patient population. Based on feedback from EU health authorities and the FDA, we are evaluating the inclusion of additional patients for exploratory analysis in order to assess potential benefit in patients with less than all of their target tumors positive for the folate receptor. While those plans are being finalized, patients will be enrolled regardless of the FR status, although the primary endpoint will include only FR(++) patients.
PROCEED is powered to demonstrate a minimum 67 percent improvement in median PFS and a hazard ratio of 0.60 in the FR(++) patient population, which compares to a 265 percent improvement in median PFS and a hazard ratio 0.381 observed in PRECEDENT. PROCEED is also powered to demonstrate a minimum 56 percent improvement in the secondary endpoint of median OS.
7
The table below compares the design characteristics of the PROCEED and PRECEDENT trials.
| PRECEDENT | PROCEED | |||
| Number of Patients |
149 | More than 200 FR(++) patients* | ||
| Clinical Sites |
65 | Approximately 150 | ||
| Patient Population |
Platinum-resistant ovarian cancer | |||
| Blinding |
Open-label | Double-blinded | ||
| Treatment Arm |
EC145 and PLD | |||
| Control Arm |
PLD | PLD and Placebo | ||
| Primary Endpoint |
PFS (radiologic and clinical) | PFS (radiologic only) | ||
| Powered for OS |
No | Yes | ||
| EC145 Dose |
2.5 mg intravenous, 3 times per week in weeks 1 and 3, on a 28 day cycle | |||
| PLD Dose |
50 mg/m(2) intravenous on day 1, on a 28 day cycle | |||
| FR(++) Hazard Ratio |
0.381 (actual) | 0.600 | ||
| Cross-Over |
Not allowed | |||
| EC20 Scan |
Enrolled regardless of scan results | Only FR(++) included in primary
end point* | ||
| * | Based on feedback from the EU health authorities and the FDA, we are evaluating the optimal process to assess potential benefit in patients with less than all of their target tumors positive for the folate receptor. This would be an exploratory analysis, not included in the primary efficacy endpoint of the trial. As this plan is being finalized, all patients, regardless of their FR status, may enroll. |
Regulatory Strategy
During the second quarter of 2011, we announced our plan to file applications with the EMA for conditional marketing authorization for our lead drug candidate EC145 for the treatment of PROC and its companion imaging diagnostic EC20 for patient selection. We confirmed this plan after meeting with EU health authorities following the supplemental analyses of the PRECEDENT trial, announced in December 2011. The filings will be supported by four clinical studies: a Phase 1 study in solid tumors, two single-agent, single-arm Phase 2 studies in ovarian cancer and non small cell lung cancer, and the PRECEDENT trial, a randomized study in PROC. This plan reflects consultation with the EMA, including the Scientific Advice Working Party, and written advice from the Committee for Medicinal Products for Human Use, or CHMP, as well as discussions with our assigned MAA rapporteures and co-rapporteures. We plan to seek conditional marketing authorization for treatment of patients with folate receptor positive PROC as selected by EC20. We expect to file both applications in the third quarter of 2012.
The therapeutic, EC145, and the diagnostic imaging agent, EC20, have also both been granted orphan drug status for ovarian cancer by the European Commission. EMA's Orphan Medicinal Product Designation is designed to promote the development of drugs that may provide significant benefit to patients suffering from rare, life-threatening diseases. This designation will provide ten years of marketing exclusivity if the product candidate is approved for marketing for the designated orphan indication in the European Union. It also provides special incentives for sponsors, including eligibility for protocol assistance and possible exemptions or reductions in certain regulatory fees during development or at the time of application for marketing approval.
In addition to data already available from the phase 2 trials, we conducted supplemental analyses of the PRECEDENT trial. These included the following data:
| • | EC20 validation: We conducted an inter-reader analysis to determine the reliability of EC20. Two independent radiologists read images from the PRECEDENT study and the evaluation resulted in an 87 percent agreement rate in selecting patients with at least one tumor positive for the folate-receptor, FR (+), and an 85 percent agreement rate in selecting patients with all folate-receptor positive tumors, FR (++). This provides evidence supporting the reliability of the EC20 scan process to consistently classify patients. |
8
| • | Blinded assessment of CT scans: Results from the PRECEDENT trial published to date have been based on analyses of CT scans by investigators who were not blinded to the treatment arm or EC20 scan status. Sensitivity analyses demonstrated these results were robust when adjusted for a variety of potential imbalances (e.g., demographic, prognostic, etc.). Radiologists blinded to the patients’ EC20 scan results and their treatment arm performed an independent assessment of the CT scans used to measure progression of disease in the PRECEDENT trial. The IRC assessments shared a high level of agreement with investigator assessments, confirming investigator-assessed progression for 74 percent of all patients. In addition, the metrics designed to identify an investigator’s potential bias to delay assessed progressions for patients in the EC145 study arm or to accelerate assessed progressions in the PLD control arm did not reveal such a bias. Also consistent with the site results, the IRC results also correlate with the mechanism of action, reflecting greater reduction in the risk of progression with increasing presence of folate receptor. In FR(++) patients, the IRC reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.465 suggests a reduction in risk of progression in the EC145 study arm of 53 percent (p=0.0498). In the FR(+) patient population, which includes the FR(++) subgroup, the IRC also reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.652 was not statistically significant in this broader group. |
| • | Overall survival: The PRECEDENT trial was not statistically powered to show a survival advantage and the results did not indicate a trend toward benefit in either arm. The hazard ratio was 1.010 for the intent-to-treat population and 1.097 for the FR(++) population. On an adjusted basis, which accounts for potential differences in demographics and prognostic factors, the overall survival hazard ratio was 0.864 in the intent-to-treat population and 0.481 in the FR(++) group, although these result were not statistically significant. |
In the U.S., if PROCEED meets the primary endpoint with limited additional toxicity over the PLD control arm, we intend to file new drug applications, or NDAs, for EC145 and EC20 with the FDA, for use of EC145 in combination with PLD for the treatment of positive patients with PROC. If final analyses indicate a broader FR definition also shows benefit, those patients will we included in the NDA. The PROCEED trial will also serve as the confirmatory trial for the potential conditional marketing authorization in Europe.
The FDA has stated that PROCEED must provide evidence of robust statistically significant and clinically meaningful benefit. If we fail to demonstrate a benefit of this magnitude, we would expect that the FDA would require us to conduct a second phase 3 clinical trial in order to file an NDA and receive marketing approval of EC145 for the treatment of PROC. In addition, the results of PROCEED may not yield safety and efficacy results sufficient to be approved by the FDA for commercial sale.
EC145 in Lung Cancer
Our second indication with EC145 is second line NSCLC. Lung cancer is the leading cause of cancer-related death worldwide and an area of high unmet medical need. Although several therapies are commercially available for the treatment of first and second line NSCLC, ultimately, in most patients the therapy fails and their cancer grows. In our clinical trials that incorporated EC20, approximately 80 percent of NSCLC patients over-express the folate receptor. As a result, we believe NSCLC is also an attractive indication for EC145 development. In a phase 2 single-arm trial in NSCLC patients who had at least one tumor that over-expressed the folate receptor, EC145 met the primary endpoint by demonstrating clinical benefit. At the eight week assessment of the patients, the DCR was 57 percent in the patients whose target tumors were all identified as over-expressing the folate receptor. This compares to historical DCRs ranging from 21 to 30 percent reported in other trials of approved therapies in less heavily pre-treated patients. In a subset of patients who had received three or fewer prior therapies and whose target tumors were all positive for the folate receptor, the DCR was 70 percent. DCR is the percentage of patients with complete response, partial response or stable disease, which has been shown to correlate with OS in NSCLC.
9
We also evaluated OS in FR(++) patients (n=14) compared to patients in which at least one of the target lesions, but not all, over-expressed the folate receptor, such patients we refer to as FR(+) (n=14). Median OS improved from 3.4 months for FR(+) patients to 10.9 months for FR(++) patients. The hazard ratio was 0.539, meaning FR(++) patients were 46.1 percent less likely to die when compared to FR(+) patients when receiving EC145 (p=0.209).
Based on results of our single-arm, single agent phase 2 clinical trial of EC145 in patients with heavily pre-treated NSCLC, we plan to begin enrollment in a randomized phase 2 trial in the second quarter of 2012. The trial is designed to enroll up to 200 patients with adenocarcinoma of the lung who have failed one prior line of therapy. Patients will be selected based on EC20 scan results and only FR(++) patients will be included. The trial design is intended to evaluate the safety and efficacy of EC145 in second line NSCLC as a single agent and in combination with docetaxel, an FDA approved and commonly used second line chemotherapy. The study will have three arms: docetaxel alone; EC145 alone; and EC145 plus docetaxel. The primary outcome measure will be PFS with secondary measures of OS, tumor response and duration of response. Final PFS data is expected to be available by early 2014.
Inflammatory Diseases
Beyond cancer, we have discovered that activated macrophages, a type of white blood cell found at sites of acute and chronic inflammation, also over-express the folate receptor. Activated macrophages release a variety of mediators of inflammation that contribute to a broad range of diseases, such as rheumatoid arthritis, osteoarthritis, inflammatory bowel disease and psoriasis. We have a number of SMDCs in preclinical development for autoimmune diseases that are designed to inhibit the production of pro-inflammatory cytokines by activated macrophages.
Our Small Molecule Drug Conjugate (SMDC) Technology
Traditional cytotoxic cancer chemotherapies kill rapidly dividing cancer and normal cells in an indiscriminate manner, leading to significant toxicity in patients. The need for patients to recover from this toxicity can limit the ability to deliver effectively-dosed cancer therapy. In addition, cancer therapies for a given tumor type are generally selected based on observations of efficacy and toxicity in that patient population and not, in most cases, based on an understanding of the differences between tumors on a molecular level. In response to these limitations, a number of targeted therapies were developed to be more selective, including monoclonal antibody-based therapies. Due to their selectivity against certain cancers, antibody therapies have achieved tremendous therapeutic and financial success in recent years. According to Roche Holdings’ publicly available information, the three largest cancer drugs in the world, Avastin, Herceptin and Rituxan, are monoclonal antibodies, with collective sales of $9.9 billion in 2010.
For certain cancers, antibodies alone are not sufficiently effective to achieve meaningful clinical benefit. This limitation has led to the development of a new class of agents called antibody drug conjugates, or ADCs. ADCs are comprised of a monoclonal antibody, which is used to target the specific cancer, attached via a linker system to a cell-killing drug. In clinical trials ADCs have enabled the targeted delivery of highly active anti-cancer drugs, improving response rates in several cancer indications, with generally less toxicity than standard chemotherapy. However, ADCs also have limitations. First, larger molecules, like ADCs, do not penetrate dense solid tumors as efficiently as small molecules, and as a result, ADC efficacy may be compromised due to limited accessibility to the target cells. Second, the slow clearance of antibodies from a patient’s bloodstream may lead to increased toxicity. The longer half-life of ADCs has also limited the development of antibody-based imaging diagnostics due to the poor image quality associated with the high background noise caused by ADCs remaining in a patient’s bloodstream. Third, ADCs are biologic molecules that are costly and often complex to manufacture.
10
We believe our SMDC platform represents a novel approach, comparable to ADCs in its ability to deliver highly active drug payloads in a targeted manner, but also with a number of potential advantages:
| • | Small size to better penetrate solid tumors. We believe a key characteristic of our SMDCs is their ability to penetrate deeply into dense solid tumors. The targeting ligands for our SMDCs are approximately 300 times smaller in molecular weight than a typical antibody incorporated in ADCs. This may result in greater uptake and higher concentrations of these molecules within solid tumors. |
| • | Rapid clearance for reduced toxicity. The circulating half-life of ADCs currently in development generally range from several hours to several days. In contrast, our SMDCs are engineered to provide rapid uptake in targeted cells and rapid clearance from the bloodstream with a half-life of approximately 20 minutes. As a result of this shorter half-life, we believe there is reduced risk that our SMDCs will release the unconjugated drug payload into the blood stream. In our phase 1 trial of EC145, only four of 410, or less than one percent, of the blood samples analyzed had quantifiable levels of the unconjugated drug payload, and all four of these positive samples had concentrations near the lowest level of detection, and at a level where no significant toxicity was found. |
| • | Companion imaging diagnostics for targeted therapy. A companion imaging diagnostic can be created for each of our SMDCs. Because of the modular nature of our SMDC technology, the drug payload can be replaced with a radioisotope imaging agent, such as technetium-99m, or Tc-99m, that we employ in EC20, to create a companion imaging diagnostic designed to target the same diseased cells as the SMDC. The companion imaging diagnostic is intended to allow for real-time, full-body assessment of the receptor target without requiring an invasive tissue biopsy. Using full-body imaging, the receptor expression can be measured in every tumor and monitored throughout treatment. In our clinical trials that combined EC145 with EC20, we have seen correlations between favorable therapeutic outcomes and increased uptake of EC20. |
| • | Cost-effective and simple to manufacture. Given the increasing pressure on drug pricing posed by payors, costs of development and manufacturing are increasingly important. Our SMDCs are relatively simple to manufacture and do not have the complexity and expense of biological molecules, like antibodies and ADCs. |
11
SMDC Pipeline
We have a pipeline of multiple SMDCs and companion imaging diagnostics that are in varying stages of clinical and preclinical development, all of which use our platform SMDC targeting technology. A summary of our most advanced development pipeline MDCs and companion imaging diagnostics are as follows:
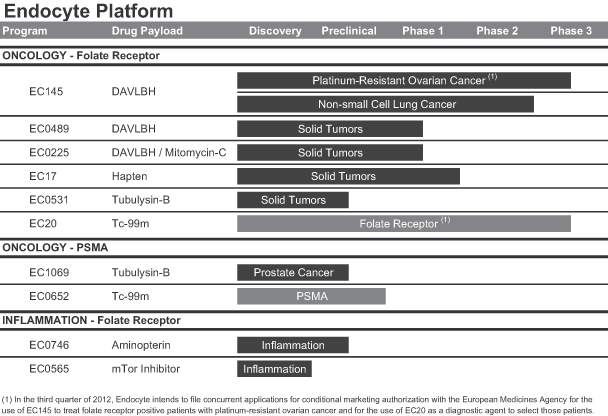
EC145: Folate Receptor Targeted Therapy
EC145 is designed to deliver a highly cytotoxic drug payload directly to folate receptors that are over-expressed on cancer cells, with low toxicity to healthy cells. EC145 consists of a targeting ligand, folate, conjugated via a linker system to an anti-cancer drug payload, DAVLBH. DAVLBH is derived from a proven class of anti-cancer drugs and is a potent destabilizer of microtubules. Since microtubules are critical for the separation of chromosomes during cell division, disruption of this microtubule system with DAVLBH promotes cell death.
Folate Receptors in Cancer Cells
Folate is a nutrient required by all living cells, and it is essential for cellular division. As depicted in the image below, folate enters human cells via two distinct transport systems, the reduced folate carrier pathway, or RFC, which has low affinity for folate and the folate receptor pathway, which has high-affinity for folate. The RFC is the predominant route by which normal cells access folate circulating in the body. The RFC is a transport protein that is expressed on virtually all cells in the body. In contrast, rapidly dividing cancer cells over-express the high-affinity folate receptor. The folate receptor captures folate from outside the cell and transports it inside by engulfing it within a vesicle called an endosome. Once internalized, the folate receptor releases the folate and is then recycled back to the cell surface where it resumes its function of capturing circulating folates.
12
Cellular Uptake of Folate
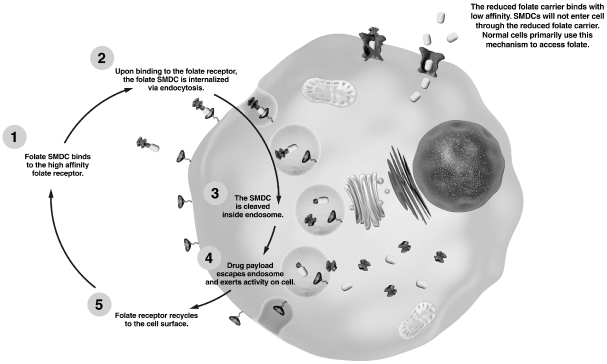
The folate receptor is not significantly expressed on most normal tissues. Lung, brain, small intestine, kidney and activated macrophages are the normal tissues known to express the folate receptor. In the lung, brain and small intestine, the folate receptor does not face the bloodstream, thus these folate receptors are not accessible to our folate-targeted SMDCs. In the kidney, the folate receptor functions as a salvage receptor that captures folates and transports them back into the blood stream to prevent folate deficiency. Although our SMDCs are also shuttled from the urine back into the blood using this folate receptor-based system, the linker system remains stable during this re-absorptive process to prevent release of the drug payload within the kidney. As a result, we have not observed any SMDC-related kidney toxicities throughout our preclinical or clinical trials. Activated macrophages also express the folate receptor. SMDCs like EC145 target folate receptor-expressing activated macrophages; however, these types of cells are not rapidly dividing, and as a result, anti-cancer drug payloads that disrupt cellular division processes are inactive against this cell type.
Elevated expression of the folate receptor occurs in several cancer types, and may be associated with the more aggressive growth characteristics of cancer cells as compared to normal cells. We believe that cancer cells over-express the folate receptor as a mechanism to capture additional folate to support rapid cell growth. The graph below shows the percentage of patients with different cancer types that are known to over-express the folate receptor. We estimate, based on worldwide cancer incidences combined with our own imaging studies that there are over one million newly diagnosed cancer patients per year in the United States, Europe and Japan whose tumors over-express the folate receptor.
13
Folate Receptor Positive Cancers by Cancer Type
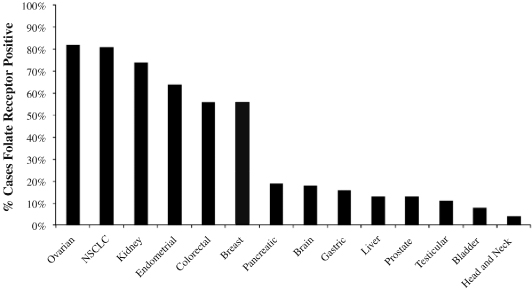
Source: American Cancer Society and Endocyte estimates based upon imaging studies and tissue biopsies.
EC145 takes advantage of the natural process of enhanced uptake of folate by cancer cells via the folate receptor by linking an active drug to a folate targeting ligand to create a tumor-targeted SMDC. Following transit through the bloodstream and entry into tumor tissue, EC145 binds to the externally-oriented folate receptor with high-affinity. Endocytosis of EC145 entraps it within a vesicle. As shown in the image on the previous page, drug payload release occurs within that vesicular compartment. The other pathway for folate to be taken up into cells, the RFC, is highly specific to folate but will not readily take up EC145 into the cell due to its molecular structure. As a result, EC145 is highly specific to cancer cells that over-express the folate receptor compared with normal cells which express the RFC.
When tested preclinically as a single agent, EC145 therapy has been observed to eliminate human tumors in mice across multiple folate receptor positive tumor models, using regimens that caused little to no observable toxicity. In the same models, treatment at the maximum tolerated dose, or MTD, with the free drug, DAVLBH, generated only modest or temporary tumor responses, and always in association with substantial toxicity. In addition, EC145 therapy caused no anti-tumor responses in preclinical tumor models that did not express the folate receptor, thus confirming the SMDC’s specificity to the target receptor.
EC145 has also been evaluated in preclinical models for activity in combination with several approved chemotherapeutic agents. For example, EC145 has shown significant anti-tumor responses in animals when dosed in combination with approved drugs, such as PLD, cisplatin, topotecan, bevacizumab, docetaxel, carboplatin, erlotinib and paclitaxel protein-bound. The toxicity profile of EC145, particularly its lack of hematologic toxicity, makes this SMDC a good potential candidate for combination therapies.
If we are successful in obtaining regulatory approval for EC145 in second or third line therapies, we intend to explore combinations with other drugs, several of which are frequently used as first line therapies in a variety
14
of tumors that over-express the folate receptor. First line therapy refers to initial cancer treatments, while second or third line therapies refer to subsequent treatments following disease progression.
Ovarian Cancer
Market Opportunity
Ovarian cancer is a significant cause of patient morbidity, and is the leading cause of gynecologic cancer mortality in the United States. According to the American Cancer Society, approximately 22,280 new cases of ovarian cancer are projected in the United States in 2012. Of those ovarian cancer cases, approximately 50 percent of patients will eventually develop PROC. Mortality rates remain high, with nearly 15,000 deaths from ovarian cancer each year in the United States alone.
While the treatment of ovarian cancer depends on the stage of the disease, the initial therapy almost always involves surgical removal of the cancer from as many sites as possible followed by platinum-based chemotherapy. For women with advanced ovarian cancer who respond to initial platinum-based chemotherapy, most will eventually experience recurrence or progression of their cancer. Patients whose cancer recurs or progresses after initially responding to surgery and primary chemotherapy can be placed into one of two groups based on the time from completion of platinum therapy to disease recurrence or progression, referred to as the platinum-free interval:
| • | Platinum-sensitive. Women with platinum-sensitive ovarian cancer have a platinum-free interval of greater than six months. Upon disease recurrence or progression, these patients are believed to benefit from additional exposure to platinum-based chemotherapy. |
| • | Platinum-resistant. Women with platinum-resistant ovarian cancer have a platinum-free interval of six months or less. These patients are much more resistant to standard chemotherapy and will typically receive PLD or topotecan or participate in a clinical trial. Overall response rate measured by RECIST, or ORR, for these subsequent therapies is in the range of 10 to 20 percent with median OS of approximately 11 to 12 months. |
There are currently only two approved therapies for women with PROC, PLD and topotecan. In clinical trials, neither of these drugs has demonstrated a statistically significant increase in PFS or OS in this indication. The last drug approved in this patient population was PLD, which was granted accelerated approval in 1999 based on an ORR of 13.8 percent (n = 145). The “n” represents the total patient population utilized in the reported data. ORR refers to the sum of complete and partial tumor responses seen, divided by the total number of evaluated patients. More recently, phase 3 trials of gemcitabine, trabectedin, patupilone and phenoxodiol have shown no statistically significant benefit over PLD in terms of either PFS or OS in women with PROC.
We have chosen PROC as our lead indication for EC145 because of the large unmet need in treating this patient population, the high levels of over-expression of the folate receptor in this tumor type, the enhanced therapeutic effect EC145 had with PLD in preclinical studies, and the acceptable clinical safety profile seen to date with EC145, which may avoid increasing the toxicities seen with PLD. We chose to develop EC145 in conjunction with PLD because it is commonly used as a second line therapy and has a better safety profile than topotecan, the other approved second line therapy.
Phase 1 Clinical Trial
We completed a phase 1 safety and dose-finding trial in 32 patients designed to determine the MTD of EC145 in patients with a variety of different solid tumors. In the trial, we established a dose regimen of three times per week, every other week, at 2.5 mg per day, which was the MTD. This dose regimen showed preliminary signs of efficacy as a monotherapy in heavily pre-treated late-stage cancer patients with a variety of tumor types, including a partial tumor response and long-term disease stabilization in ovarian cancer (n=2) and long-term stabilization in other cancer types. This dose regimen was well tolerated. Toxicities most commonly
15
seen in the trial were constipation, fatigue, nausea, vomiting, abdominal pain and anemia, many of which are observed in late-stage cancer patients. The primary dose-limiting toxicity for EC145 was a significant but spontaneously reversible constipation/ileus observed at doses above the MTD.
Phase 2 Single-Arm Clinical Trial
We have completed a phase 2 single-arm clinical trial designed to evaluate the safety and efficacy of EC145 in women with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer. The primary objectives were to collect data on the clinical benefit of EC145 therapy, defined as the number of patients who received six or more cycles of therapy, to explore the safety of EC145 in this drug-resistant patient population, and to assess the degree to which patients who had at least one tumor that over-expressed the targeted folate receptor responded to therapy, thereby enabling us to better identify a target population in which to conduct a randomized clinical trial of EC145 in the future.
Prior to treatment with EC145, patients were scanned with EC20 to determine whether their tumors over-expressed the folate receptor. In addition to standard eligibility criteria, patients were required to have PROC or refractory ovarian cancer, meaning disease that did not respond or progressed during the most recent platinum-based chemotherapy, and at least a single RECIST-measurable tumor. EC145 was administered as a bolus dose three days per week, on weeks one and three of a four-week cycle. Forty-nine women, with a median age of 62 years, were enrolled into the trial. Participants had been heavily pre-treated prior to participation in the trial, having received a median of four prior chemotherapeutic regimens. All of these patients had advanced disease and most had a heavy tumor burden. Although the trial did not achieve the threshold for efficacy, the effectiveness of EC145 was also evaluated based on DCR and ORR. These analyses indicated that a subset of patients exhibited evidence of anti-tumor effect such as tumor shrinkage and disease stabilization, with low toxicity.
In the final analysis of the trial data, the DCR for the 45 eligible patients was 42.2 percent with two women achieving a partial response, and the ORR for the eligible patients was 5 percent. An additional analysis established that EC145 was more active in patients previously treated with less than four therapies resulting in a DCR of 60.0 percent and an ORR of 13.3 percent. Safety data indicated that EC145 was very well tolerated, with no Grade 4 drug-related toxicities. The most frequent Grade 3 drug-related toxicities were fatigue in 8.2 percent of the patients and constipation in 8.2 percent of the patients. In addition, the safety data indicated that EC145 did not produce overlapping toxicity with the existing second line therapeutic agents used, such as topotecan and PLD.
Each patient who was scanned with EC20 was given a score, computed by dividing the number of positive tumors by the total number of target tumors. For example, a patient with a total of four target tumors, two of which over-expressed the folate receptor, would have a patient score of 50 percent. Patients were divided into three groups based on their EC20 scores as follows:
| • | the FR(++) group was characterized as having 100 percent of their tumors tested positive for the folate receptor; |
| • | the FR(+) group score was 1 to 99 percent positive or at least one, but not all of their tumors tested positive for the folate receptor; and |
| • | the FR(-) group score was 0 percent positive because none of their tumors tested positive for the folate receptor. |
Separate analyses were performed to assess the degree to which FR(++) and FR(+) patients responded to therapy with EC145. Of the 145 evaluable tumors, only tumors in FR(++) and FR(+) patients treated with EC145 showed tumor shrinkage of greater than 20 percent. No tumors of FR(-) patients showed a decrease of greater than 20 percent. These results were statistically significant, indicating that EC20 uptake by tumors correlates with response to EC145 treatment (p=0.0022).
16
The ORR and DCR were calculated for each of the three groups. FR(++) patients had the highest DCR, 57 percent, followed by FR(+) at 36 percent and FR(-) patients at 33 percent. In a subgroup analysis of less heavily pre-treated patients, those treated with three or fewer prior regimens, the DCR for the FR(++) group was 86 percent versus 50 percent and zero percent in the FR(+) and FR(-) groups, respectively. Similarly, the ORR in the FR(++) subgroup was the highest at 14 percent, while the ORR for the FR(+) subgroup and for the FR(-) subgroup was 13 percent and zero percent, respectively. Results from this trial indicate a potential correlation between EC20 binding levels and anti-tumor response at the level of the individual patient and the individual tumor.
Platinum REsistant Ovarian Cancer Evaluation of Doxil and EC145 CombiNation Therapy (PRECEDENT): Randomized Phase 2 Clinical Trial in PROC
PRECEDENT is a multicenter, open-label, randomized phase 2 clinical trial of 149 patients comparing EC145 and PLD in combination, versus PLD alone, in women with PROC. The trial completed enrollment in June 2010 and final PFS analysis of the data has been conducted and was presented at the annual meeting of the American Society of Clinical Oncology in June 2011.
We chose PFS as the primary endpoint of the trial as PFS is a clinically meaningful endpoint in this patient population and allows for a more rapid assessment of results than OS. PFS was measured based upon investigator assessment using both radiological measurements based on RECIST, as well as assessment of clinical progression. Secondary endpoints include OS, ORR and safety and tolerability of EC145 in combination with PLD. To minimize the potential for bias and to ensure the integrity of the OS measurement, cross-over was not allowed. In addition, the trial explored the correlation between therapeutic response and EC20 imaging results.
Eligible patients were randomized in a 2 to 1 ratio to either the EC145 and PLD arm or to the PLD alone arm. PLD was selected as the comparator because it is approved and widely used in PROC and EC145 in combination with PLD was more effective than PLD alone in our preclinical studies. Patients are dosed with EC145 three times per week every other week and PLD is administered once every 28 days in both population arms, consistent with the standard of care. All patients enrolled at clinical centers with nuclear imaging capabilities were scanned with our EC20 companion imaging diagnostic within 28 days prior to the initiation of treatment (113 patients).
PRECEDENT is a multicenter trial involving 65 sites in the United States, Canada and Poland. Patients were stratified based upon their geographic location, primary versus secondary platinum resistance and level of the tumor marker (CA-125). Most of the demographics and disease characteristics were well-balanced between the arms. There were two characteristics slightly imbalanced in the arms of the trial, both of which should have contributed to a poorer prognosis for patients receiving the combination of EC145 and PLD versus patients receiving PLD alone. Specifically, the number of patients with hepatic and pulmonary metastases at enrollment were greater in the combination arm (38.0 percent) compared to the PLD alone arm (22.4 percent). Also, the median cumulative length of tumor at enrollment in the combination arm was 9.3 cm compared to 5.6 cm in the PLD alone arm.
A Data Safety Monitoring Board, or DSMB, monitored the trial and conducted multiple safety reviews and a pre-specified interim analysis. This interim analysis was prepared by an independent biostatistician and reviewed by the DSMB on February 26, 2010. The DSMB recommendation was to continue the trial to full accrual with no protocol modifications.
At PRECEDENT’s final PFS analysis of 149 patients and 95 PFS events, we reported an 85 percent increase of median PFS from 2.7 months in the PLD arm to 5.0 months in the EC145 and PLD treatment arm (p=0.031). The hazard ratio was 0.626, meaning patients receiving EC145 were 37.4 percent less likely to have died or have their cancer progress compared to patients receiving only PLD. The median PFS seen in the control arm was consistent with historical data in this disease setting. This benefit in PFS was provided in the context of low additional toxicity over the current standard of care. We believe that EC145 and PLD is the first combination to show a meaningful improvement in PFS over standard therapy for the treatment of PROC.
17
In December 2011, we announced the results of supplemental analyses we had conducted of the PRECEDENT trial data. We believe these findings continue to support the robustness of the PRECEDENT trial results, particularity in the group of FR (++) patients. The primary endpoint of the open-label PRECEDENT trial was PFS based on investigator assessment. Sensitivity analyses demonstrated these results were robust when adjusted for a variety of potential imbalances (e.g., demographic, prognostic, etc.). The assessments made by the blinded independent review committee, or IRC, shared a high level of agreement with investigator assessments, confirming investigator-assessed progression for 74 percent of all patients. In addition, the metrics designed to identify an investigator's potential bias to delay assessed progressions for patients in the EC145 study arm or to accelerate assessed progressions in the PLD control arm did not reveal such a bias. The IRC results also correlate with the mechanism of action, reflecting greater reduction in the risk of progression with increasing presence of folate receptor. In FR(++) patients, the IRC reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.465 suggests a reduction in risk of progression in the EC145 study arm of 53 percent (p=0.0498). In the FR(+) patient population, which includes the FR(++) subgroup, the IRC also reflected a 2.5 months improvement in median PFS from 1.5 months in the PLD control arm to 4.0 months in the EC145 study arm. The hazard ratio of 0.652 was not statistically significant in this broader group.
Kaplan-Meier curve for PFS in PRECEDENT
EC145’s companion imaging diagnostic, EC20, was used to correlate folate receptor over-expression with EC145 efficacy in PROC. Patients in the PRECEDENT trial were imaged with EC20 prior to enrollment and target lesions were read to determine whether patients were FR(++), FR(+) or FR(-). In the FR(++) subgroup of 38 patients, patients whose target lesions all over-expressed the folate receptor, PFS improved from a median of 1.5 months for patients receiving PLD alone to a median of 5.5 months for patients receiving the combination of EC145 and PLD, an improvement of over 260 percent. The hazard ratio was 0.381 (p=0.018) or a reduction in the risk of progression of 61.9 percent.
The PRECEDENT trial was not statistically powered to show a survival advantage and the results did not indicate a trend toward benefit in either arm. The hazard ratio was 1.010 for the intent-to-treat population and 1.097 for the FR(++) population. On an adjusted basis, which accounts for potential differences in demographics and prognostic factors such as platinum free intervals, the overall survival hazard ratio was 0.864 in the intent-to-treat population and 0.481 in the FR(++) group, although these result were not statistically significant.
18
Kaplan-Meier curve for OS in PRECEDENT
We also examined other secondary endpoints, including ORR. The ORR at the initial scan was 28.0 percent in the treatment arm as compared to 16.3 percent in the control arm. Consistent with RECIST, the protocol required follow-up scans at least four weeks later to confirm responses. The ORR at the confirmatory scan was 18.0 percent in the treatment arm as compared to 12.2 percent in the control arm.
At the final PFS analysis, the combination therapy was generally well tolerated. The EC145 and PLD combination arm received a 62 percent greater cumulative dose of PLD because these patients remained in the trial for a longer duration due to improved PFS. Despite this higher cumulative dose of PLD in the combination arm, total drug-related adverse events and serious adverse events were similar between arms. Review of toxicity data indicate that the number of patients reporting at least one treatment-emergent drug-related serious adverse event resulting in discontinuation from the trial was 2.8 percent (n=3) for the EC145 and PLD combination arm versus 4.0 percent (n=2) for the PLD single agent arm of the trial. No patient in either arm was known to have died from drug-related adverse events while receiving treatment or within 30 days of receiving treatment. Toxicity levels in the combination arm are similar to historical levels of toxicity experienced in patients receiving PLD as a single agent.
19
PRECEDENT Trial Grade 3-4 Toxicities(1)
(At final PFS Analysis)
| Hematological Toxicities |
EC145 and PLD (n=107) |
PLD (n=50) |
||||||
| Neutropenia < 1,000/mm3 |
13 (12.1%) | 2 (4.0%) | ||||||
| Febrile neutropenia |
1 (0.9%) | 1 (2.0%) | ||||||
| Anemia < 8 g/dL |
7 (6.5%) | 2 (4.0%) | ||||||
| Thrombocytopenia < 50,000/mm3 |
2 (1.9%) | 1 (2.0%) | ||||||
| Leukopenia < 2,000/mm3 |
18 (16.8%) | 2 (4.0%) | ||||||
| Lymphopenia < 500/mm3 |
19 (17.8%) | 9 (18.0%) | ||||||
| Non-Hematological Toxicities |
EC145 and PLD (n=107) |
PLD (n=50) |
||||||
| Stomatitis |
6 (5.6%) | 2 (4.0%) | ||||||
| PPE syndrome |
12 (11.2%) | 1 (2.0%) | ||||||
| (1) | Hematological toxicities are based on lab values regardless of causality. Non-hematological toxicities were drug-related toxicities occurring in five percent or more of patients in at least one arm of the trial. |
Trial for Women With Platinum Resistant Ovarian Cancer Evaluating EC145 in Combination with Doxil (PROCEED): Phase 3 Clinical Trial for Approval of EC145 in PROC
The PROCEED trial is designed to support the approval of EC145 in combination with PLD for the treatment of women with PROC. PROCEED is a double-blinded, multicenter, international, randomized phase 3 clinical trial in more than 200 folate receptor positive patients at approximately 150 sites comparing EC145 and PLD to placebo and PLD in women with PROC. We initiated enrollment of the PROCEED trial in May 2011 but enrollment stopped later in the year due to global shortages of PLD. In March 2012, we announced that the FDA approved our plans to import our supply of PLD from Europe into the U.S. for use in this trial and we plan to resume enrollment in the U.S.
We designed our phase 3 registration trial following our end of phase 2 meeting with the FDA held on May 24, 2010 and an additional meeting on March 9, 2012. Fundamental design characteristics of the PROCEED trial closely match those of the PRECEDENT trial. The endpoint, patient population, dose and schedule, are the same as those used in the PRECEDENT trial. The only significant design changes are that the phase 3 trial will utilize a placebo in order to double-blind the investigators and patients to which treatment arm they are on, the PFS endpoint will be based only on radiologic assessments with no clinical progression allowed, FR(-) and FR(+) patients will be excluded from the primary endpoint, and the trial will be powered for OS as a secondary endpoint. In addition, we expect to add additional countries to those used in the PRECEDENT trial, which included the United States, Canada and Poland.
In the PRECEDENT trial, the FR(++) subgroup received the most benefit from EC145 therapy, with a hazard ratio of 0.381 (p=0.018). Because this data indicates a strong correlation between FR(++) scans and EC145 efficacy, the primary endpoint in the phase 3 PROCEED clinical trial will be PFS. The secondary endpoint will be overall survival, or OS, in this same population. Projected enrollment of FR(++) patients is more than 200. Based on feedback from EU health authorities and the FDA, we are evaluating the inclusion of additional patients for exploratory analysis in order to assess potential benefit in patients with less than all of their target tumors positive for the folate receptor. While those plans are being finalized, patients will be enrolled regardless of the FR status, although the primary endpoint will include only FR(++) patients. We expect final primary PFS data from this study in the first half of 2014.
20
The PROCEED trial is designed to meet the key elements that we believe, based on meetings, will be required by the FDA for approval of new drugs based on PFS in this patient population:
| • | Clinically meaningful improvement in PFS. The trial is designed to show approximately a 67 percent improvement in median PFS, which we believe is clinically meaningful in this patient population, where there is a high unmet medical need and no approved drug has demonstrated a statistically significant improvement in median PFS or OS. Our PRECEDENT randomized phase 2 clinical trial showed a statistically significant PFS benefit that corresponds to approximately an 85 percent improvement in median PFS. |
| • | Powered to evaluate OS. The trial is powered to detect a 56 percent improvement in median OS. To ensure the integrity of the OS measurement, we do not allow cross-over between the trial arms. |
| • | Blinded radiologic assessment of PFS. The trial is double-blinded to minimize any occurrence of bias, and the PFS analysis will be based only on radiologic assessment. The trial includes a supportive PFS analysis based on a centralized blinded independent review confirmed by a centralized blinded independent review. |
| • | Favorable risk/benefit analysis. The final results of the phase 2 trial indicated a significant and clinically meaningful benefit in PFS in the context of manageable toxicity. No statistically significant safety differences were seen between EC145 and PLD versus PLD alone either in overall adverse events or serious adverse events. We believe that in this area of high unmet medical need, this is a favorable risk/benefit assessment, which we believe can be repeated in PROCEED. |
Non-Small Cell Lung Cancer
Market Opportunity
Lung cancer is the number one cause of cancer deaths worldwide. According to the American Cancer Society, it is projected that there will be approximately 192,000 patients in the United States diagnosed with NSCLC and approximately 136,000 will die of the disease. Although numerous drugs are available for the treatment of NSCLC patients, according to a study published in Lung Cancer in 2003, the disease of more than 75 percent of patients progresses in less than eight weeks following second line or third line therapy. These findings underscore the need for continued development of new therapeutics, especially for refractory and progressive disease. In our clinical trials that incorporated EC20 in approximately 60 patients, the tumors in approximately 80 percent of heavily pre-treated NSCLC patients over-expressed the folate receptor. As a result, we have investigated the use of EC145 for the treatment of NSCLC patients in our phase 2 single-arm trial in this indication.
Clinical Trials in NSCLC: Phase 2 Single-Arm Clinical Trial
We conducted a phase 2 single-arm trial of EC145 in 43 patients with NSCLC. Prior to treatment with EC145, patients were scanned with EC20 to determine whether their tumors over-expressed the folate receptor. Only FR(++) and FR(+) patients were eligible for the trial. In addition to standard eligibility criteria, patients were required to have a diagnosis of adenocarcinoma of the lung and to have at least a single RECIST-measurable tumor. There were no limits to the maximum number of prior therapies allowed in this patient population. EC145 was administered as a bolus dose of 1 mg per day five days per week, during weeks one through three and five through seven of the eight week induction period, followed by 2.5 mg per day, three days per week every other week thereafter as maintenance. CT scans were performed every eight weeks and adverse events were assessed using standard criteria.
The trial gathered data on efficacy, including DCR and ORR, to characterize the toxicity profile of EC145 in the target population. The trial was also designed to assess whether EC20 tumor scans that were positive for the over-expression of the folate receptor correlated with anti-tumor response. All participants in the trial were
21
FR(++) and FR(+). The trial was conducted in a heavily pre-treated patient population, with a median age of 62 and a median of three prior chemotherapeutic regimens with a range of two to nine treatments. Additional analysis confirmed that the group had large-volume disease with a median cumulative tumor length of 7.9 cm. The trial met the primary endpoint of clinical benefit, which was defined as more than 20.0 percent of the patients completing four months of therapy. Safety data for all 43 participants indicated that EC145 was well tolerated, with no Grade 4 drug-related toxicities. The most frequently observed drug-related Grade 3 toxicity was fatigue (4.7 percent).
At the eight week patient assessment, the disease control rate, or DCR, was 57 percent in the patients who had all of their target tumors identified as over-expressing the folate receptor. This compares to DCRs for approved therapies of 21 to 30 percent reported in trials of less heavily pre-treated patients. In a subset of only FR(++) patients who had received three or fewer prior therapies, the DCR was 70 percent. DCR has been shown to correlate with OS in NSCLC.
| All Patients | FR(++) | FR(+) | ||||||||||
| EC145 (all patients) |
34.5% | 57.1% | 14.3% | |||||||||
| EC145 (three or fewer prior therapies) |
42.9% | 70.0% | 20.0% | |||||||||
| Historical benchmark (two or three prior therapies) |
21% to 30% | — | — | |||||||||
We also evaluated OS in FR(++) patients (n=14) compared to FR(+) patients (n=14). Median OS improved from 3.4 months for FR(+) patients to 10.9 months for FR(++) patients. The hazard ratio was 0.539, meaning FR(++) patients were 46.1 percent less likely to die when compared to FR(+) patients when receiving EC145 (p=0.209).
NSCLC Development Plan
Based on results of our single-arm, single agent phase 2 clinical trial of EC145 in patients with heavily pre-treated NSCLC, we plan to begin enrollment in a randomized phase 2 trial in the second quarter of 2012. The trial is designed to enroll up to 200 patients with adenocarcinoma of the lung who have failed one prior line of therapy. Patients will be selected based on EC20 scan results and only FR(++) patients will be included. The trial design is intended to evaluate the safety and efficacy of EC145 in second line NSCLC as a single agent and in combination with docetaxel, an FDA approved and commonly used second line chemotherapy. The study will have three arms: docetaxel alone; EC145 alone; and EC145 plus docetaxel. The primary outcome measure will be PFS with secondary measures of OS, tumor response and duration of response. Final PFS data is expected to be available by early 2014.
EC20: Lead Companion Imaging Diagnostic for Folate-Targeted Therapies
We believe the future of medicine includes not only safer and more effective drugs, but also the ability to identify the appropriate therapy for a particular patient. We are committed to this approach, which is commonly referred to as personalized medicine or predictive medicine.
Because of the modular nature of our SMDC technology, the drug payload can be replaced with a radioisotope imaging agent, such as Tc-99m, which we employ in EC20, to create a companion imaging diagnostic designed to target the same diseased cells as the SMDC. The companion imaging diagnostic allows for real-time, full-body assessment of the receptor target without requiring an invasive tissue biopsy. Using full-body imaging, the receptor expression can be measured in every tumor and monitored throughout treatment. As described earlier, our PROC and NSCLC clinical trials combining EC145 with its companion imaging diagnostic have shown correlations between favorable therapeutic outcomes and increased uptake of EC20.
EC20 is the companion imaging diagnostic for all of our SMDCs that target the folate receptor. EC20 is a conjugate of the targeting ligand folate and the radioisotope imaging agent Tc-99m. Following intravenous administration, EC20 rapidly binds to tumors that over-express the folate receptor, allowing the treating
22
physician to distinguish between patients who are FR(++), FR(+) or FR(-). EC20 enables high quality diagnostic scans one to two hours following its administration as a result of the quick clearance from the blood of EC20 not taken up by cells which over-express the folate receptor.
Potential key advantages of EC20 over tissue-based samples include:
| • | minimally invasive (not requiring biopsy); |
| • | real-time assessment of tumor receptor expression (as opposed to analysis based on archived tissue); |
| • | greater sensitivity because EC20 binds to all forms of the folate receptor (tissue sample analysis may understate folate receptor expression by not recognizing all forms of the folate receptor); |
| • | greater specificity because EC20 distinguishes between those receptors accessible to folate in the blood and those not accessible (tissue sample analysis may overstate folate receptor expression); and |
| • | full-body evaluation (as opposed to samples of tumor which may or may not be indicative of all areas of disease). |
EC20 Development Plan
In the U.S., EC20 is being developed under the FDA’s published guidance regarding usage of imaging agents for therapeutic management of patients. Consistent with this guidance, our development strategy for EC20 will be to show that the presence of EC20 uptake in tumors is predictive of improved therapeutic outcomes resulting from treatment with EC145.
With an estimated incidence of over one million newly diagnosed cancer patients in the United States, Europe and Japan who have tumors that over-express the folate receptor, EC20 could, if approved, represent a substantial commercial opportunity for us as a screening diagnostic. Companion imaging diagnostics are an important part of our development and commercial plan, which may allow us not only to provide a targeted therapy, but also a truly personalized and more effective therapy. This could benefit patients, doctors and payors by allowing doctors to select only patients who are likely to benefit from our therapies.
We plan to file an NDA with the FDA for the separate approval of EC20 for use in women with PROC. We intend to use the data obtained from PROCEED for the basis of the filing of this NDA. However, there can be no assurance the FDA will not require additional clinical trials of EC20 prior to approval.
In Europe, we intend to file applications for conditional marketing authorization with the EMA in the third quarter of 2012 for the use of EC20 as a diagnostic agent to select patients most likely to respond to EC145. We will file EC20 and EC145 concurrently for the selection and treatment of folate receptor positive patients with platinum-resistant ovarian cancer. We conducted an inter-reader analysis to determine the reliability of EC20. Two independent radiologists read images from the PRECEDENT study and the evaluation resulted in an 87 percent agreement rate in selecting patients with at least one tumor positive for the folate-receptor, FR (+), and an 85 percent agreement rate in selecting patients with all folate-receptor positive tumors, FR (++). This provides evidence supporting the reliability of the EC20 scan process to consistently classify patients.
Other Pipeline Programs
Folate Receptor Targeted Programs
EC0489 is a conjugate that utilizes the same targeting ligand, folate, and the same drug payload, DAVLBH, as EC145, but it includes an alternative linker system. This alternative linker system yields a different biodistribution of the drug compared to EC145, which may allow for higher dosage of drug payload. EC0489 is currently in a phase 1 dose escalation trial evaluating the safety of the drug in patients with solid cancer tumors. To date, 51 patients have been treated with EC0489. This trial will also allow us to evaluate the utility of our alternative linker system technology and its potential for use in the construction of future SMDCs. Following the conclusion of the phase 1 trial, we will evaluate future development options for EC0489.
23
EC0225 is a conjugate of folate and two distinct and highly active drugs, DAVLBH and mitomycin-C. DAVLBH is a microtubule destabilizer and mitomycin-C is a DNA alkylator. These two drugs are attached to a single targeting ligand and are brought into the targeted cancer cell via endocytosis, at which point both drugs are concurrently released. The anticipated advantage of using two drugs is that the payload is doubled, which may increase the overall anti-cancer activity of the SMDC. Attaching drugs with different mechanisms of action may allow them to overcome drug resistance. The drug payloads within EC0225 utilize distinct mechanisms of action to kill target cancer cells, thus making this SMDC a targeted combination therapy within a single drug. EC0225 is currently being evaluated in an open-label phase 1 dose escalation trial to assess its safety in patients with solid tumors, and to determine the dose for future trials. To date, 66 patients have been treated with EC0225.
EC17 is a conjugate of folate and a hapten molecule. This SMDC also utilizes our folate targeting ligand, but instead of DAVLBH, EC17 incorporates hapten as the drug payload, and delivers this drug payload to the tumor surface. When bound to cancer cells, EC17 is designed to elicit an immunologic response from the host immune system in order to facilitate tumor-cell killing. We completed a phase 1 trial with EC17 during which the drug was administered safely with a confirmed anti-hapten antibody response and evidence of anti-tumor activity. We are currently evaluating future development options for EC17.
EC0531 is a conjugate of folate and a drug payload of tubulysin, a microtubule destabilizer that, in our in vitro models, showed approximately 100,000 times more potency than cisplatin. Tubulysin alone is too toxic to be used in patients rendering it impractical for therapeutic use. In contrast, our folate receptor-targeted tubulysin SMDC, EC0531, is curative in multiple xenograft models under conditions that produce no observable toxicities. We plan to file an IND for EC0531 or a similar agent by early 2013.
Prostate Specific Membrane Antigen Targeting Programs
Leveraging our ligand and linker system expertise that we have obtained from our folate SMDC programs, we recently introduced into clinical trials EC0652, a companion imaging diagnostic for a new class of SMDCs whose targeting ligand binds to PSMA. PSMA is a receptor target that is predominantly over-expressed on prostate cancer cells.
EC0652 is a conjugate of a proprietary PSMA targeting ligand and the imaging agent Tc-99m. EC0652 is used to support SMDCs that target PSMA. In preclinical studies, EC0652 was found to specifically bind to PSMA-expressing tumor cells and it allowed for the non-invasive, real-time assessment of functionally active and drug-accessible PSMA-expressing tumor cells. EC0652 is currently in early clinical trials to validate specificity and biodistribution of the targeting ligand. In addition, we are currently evaluating EC1069, an SMDC for prostate cancer therapy based on the PSMA targeting ligand.
Inflammatory Disease Programs
During the clinical development of EC20, it was discovered that patients with active inflammatory conditions, such as arthritic knees, displayed areas of EC20 uptake in non-cancerous regions of the body. Based on this observation, we began to test preclinical models of rheumatoid arthritis and discovered that activated, but not resting, macrophages present within the inflamed joints over-expressed the folate receptor. Activated macrophages are a type of white blood cell involved in a variety of inflammatory diseases, and they are responsible for the release of pro-inflammatory molecules, such as tumor necrosis factor alpha, or TNF-alpha as well as other cytokines, into the body. Commercially available drugs such as etanercept and adalimumab, both designed to neutralize TNF-alpha biological activity, have proven to be effective for the treatment of a variety of inflammatory diseases. In 2011, these products generated $11.6 billion in annual sales according to Amgen’s and Abbott Laboratories’ publicly available filings. Although effective in some patients, other patients may fail to respond to these costly biological products because the activated macrophages secrete a variety of pro-inflammatory agents, in addition to TNF-alpha, into the systemic circulation, which results in continuing inflammation, causing a decreased therapeutic effect.
24
Our strategy for treating chronic inflammatory disorders differs from these available drugs that neutralize specific secreted factors such as TNF-alpha because our technology targets the folate receptor over-expressed on activated macrophages, which we believe are the source of pro-inflammatory agents, like TNF-alpha. We are developing a new class of SMDCs designed to neutralize, or de-activate, the activated macrophage itself. Preclinical data suggests that our SMDC candidates may suppress the secretion of all mediators of inflammation. Our oncology class of SMDCs, such as EC145, will target folate receptor-expressing activated macrophages; however, these types of cells are not rapidly dividing, and as a result, anti-cancer drug payloads that would otherwise disrupt cellular division processes are inactive against this cell type.
Utilizing an identical strategy to that we applied in the development of our oncology programs, SMDCs designed for treating inflammation may be constructed with more potent forms of known, active drugs, and then used along with companion imaging diagnostics to provide personalized therapy. Using our folate receptor-targeted EC20 companion imaging diagnostic in both preclinical and clinical trials, we have identified a number of diseases involving activated macrophages that over-express the folate receptor, including rheumatoid arthritis, osteoarthritis, inflammatory bowel disease and psoriasis.
In our preclinical models of rheumatoid arthritis, SMDCs targeted to activated macrophages result in significant reduction in inflammation and prevention of bone destruction that often accompanies these diseases. For example, EC0746 is an SMDC constructed with the targeting ligand, folate, and an inhibitor of cellular metabolism, called aminopterin. We observed in preclinical models that EC0746 was safe and reduced inflammation more than the most commonly prescribed anti-inflammatory agent, methotrexate, and the anti-TNF-alpha agent, etanercept.
Competition
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to our SMDCs. A number of multinational pharmaceutical companies and large biotechnology companies are pursuing the development of or are currently marketing pharmaceuticals that target ovarian cancer and NSCLC, or other oncology pathways on which we are focusing. It is possible that the number of companies seeking to develop products and therapies for the treatment of unmet needs in oncology will increase.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of drugs, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than we may be in obtaining approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any drug we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our product candidates. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available.
Recent successes with targeted therapies in solid tumors, such as those found in colon, breast and lung cancers, have led to a marked increase in research and development in targeted treatments for cancer, including cancer of the ovary. We are aware of a number of companies that have ongoing programs to develop both small molecules and biologics to treat patients with ovarian cancer. Roche Holdings is currently testing bevacizumab in women with ovarian cancer, including a phase 3 clinical trial in women with PROC. Nektar Therapeutics, using its conjugate technology, is conducting a phase 2 clinical trial of NKTR-102 for use in patients with solid tumor malignancies, including PROC, colorectal, and breast cancers. Sunesis Pharmaceuticals conducted a phase 2 single-arm trial of voreloxin, an anti-cancer quinolone derivative, in a number of patient populations, including PROC. Sanofi-Aventis is conducting a phase 2 single-arm trial of its drug BSI-201, a PARP inhibitor currently in a number of patient populations, including ovarian cancer. Eli Lilly is conducting a phase 2 single-arm trial of its drug LY573636.
25
We believe that our ability to successfully compete will depend on, among other things:
| • | our ability to design and successfully execute appropriate clinical trials; |
| • | our ability to recruit and enroll patients for our clinical trials; |
| • | the results of our clinical trials and the efficacy and safety of our SMDCs and companion imaging diagnostics; |
| • | the cost of treatment in relation to alternative therapies; |
| • | the speed at which we develop our SMDCs and companion imaging diagnostics; |
| • | achieving and maintaining compliance with regulatory requirements applicable to our business; |
| • | the timing and scope of regulatory approvals, including labeling; |
| • | adequate levels of reimbursement under private and governmental health insurance plans, including Medicare; |
| • | our ability to protect intellectual property rights related to our SMDCs and companion imaging diagnostics; |
| • | our ability to commercialize and market any of our products that may receive regulatory approval; |
| • | our ability to have our partners manufacture and sell commercial quantities of any approved SMDCs and companion imaging diagnostics to the market; and |
| • | acceptance of SMDCs and companion imaging diagnostics by physicians, other healthcare providers and patients. |
In addition, the biopharmaceutical industry is characterized by rapid technological change. Our future will depend in large part on our ability to maintain a competitive position with respect to these technologies. Our competitors may render our technologies obsolete by advances in existing technological approaches or the development of new or different approaches, potentially eliminating the advantages in our drug discovery process that we believe we derive from our research approach and proprietary technologies. Also, because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our SMDCs or companion imaging diagnostics obsolete or less competitive.
Manufacturing
To date, our SMDCs and companion imaging diagnostics have been manufactured in small and medium size quantities for preclinical studies, clinical trials and validation activities by third-party manufacturers and we intend to continue to do so in the future. We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of our SMDC candidates. We currently have no plans to build our own clinical or commercial scale manufacturing capabilities. To meet our projected needs for commercial manufacturing, third parties with whom we currently work have sufficient capacity, but will need to increase their scale of production or we will need to secure alternate suppliers. While individual contract manufacturers have demonstrated the capability to produce quantities of our SMDCs sufficient to support our ongoing clinical trials for EC145 and other SMDCs, we continue to engage alternative suppliers to provide supply in the event of a failure to meet demand on the part of a single contract manufacturer. In addition, the linker system for EC145 is obtained from a single source supplier, AmbioPharm, and we are currently assessing alternate suppliers to prevent a possible disruption of manufacturing of EC145. We currently have, or have access to, sufficient supplies of all of the key components of EC145 to manufacture EC145 to conduct and complete our ongoing phase 3 clinical trial for EC145. We have identified several manufacturers that have the capacity to manufacture
26
EC145 in the quantities that our development and future commercialization efforts, if any, may require. We have utilized two such suppliers to date. We expect to continue to depend on third-party contract manufacturers for the foreseeable future.
Our SMDCs and companion imaging diagnostics require precise, high quality manufacturing. Failure by our contract manufacturers to achieve and maintain high manufacturing standards could result in patient injury or death, product recalls or withdrawals, delays or failures in testing or delivery, cost overruns, or other problems that could seriously harm our business.
As a result of the potency of our compounds, we do not expect the quantities required at full commercial scale to present a challenge to our third-party manufacturing partners. Although we rely on contract manufacturers, we have personnel with extensive manufacturing experience to oversee the relationships with our contract manufacturers.
Sales and Marketing
Our operations to date have been limited to organizing and staffing our company, acquiring, developing and securing our technology, undertaking preclinical testing and clinical studies of our SMDCs and companion imaging diagnostics and engaging in research and development under collaboration agreements. We have not yet demonstrated an ability to obtain regulatory approval, formulate and manufacture commercial-scale products, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, it is difficult to predict our future success and the viability of any commercial programs that we may choose to take forward.
Subject to successful completion of our PROCEED clinical trial for EC145 and FDA approval of EC145 for women with PROC, it is our objective to establish EC145 in combination with PLD as the therapy of choice for PROC patients. If EC145 is approved by the FDA, we intend to build the commercial infrastructure sufficient to market and sell EC145 and EC20 in the United States. This infrastructure is expected to include a specialty sales force of approximately 50 to 75 representatives, sales management, sales and distribution support staff and internal marketing staff. Following approval, but in advance of distribution, we expect to make a significant investment in marketing efforts to support the successful launch of EC145.
Outside of the United States, we may choose to utilize strategic partners or contract sales organizations to support the sales, marketing and distribution of EC145 and other approved SMDCs.
Employees
As of December 31, 2011, we had a total of 61 full-time employees, of whom 51 were engaged in research and development activities. None of our employees is represented by a labor union or subject to a collective bargaining agreement. We have not experienced a work stoppage and consider our relations with our employees to be good.
Government Regulation
Government authorities in the United States (including federal, state and local authorities) and in other countries, extensively regulate, among other things, the manufacturing, research and clinical development, marketing, labeling and packaging, distribution, post-approval monitoring and reporting, advertising and promotion, and export and import of pharmaceutical products, such as those we are developing. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and related regulations. Drugs are also subject to other federal, state and local statutes and regulations. Failure to comply with the applicable U.S. regulatory requirements at any time during the product development process,
27
approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the imposition by the FDA or an Institutional Review Board, or IRB, of a clinical hold on trials, the FDA’s refusal to approve pending applications or supplements, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The Investigational New Drug Process
An Investigational New Drug application, or an IND, is a request for authorization from the FDA to administer an investigational drug to humans. Such authorization must be secured before commencing clinical trials of any new drug candidate in humans.
The central focus of the initial IND submission is on the general investigational plan and the protocol(s) for human studies. The IND also includes results of animal studies or other human studies, as appropriate, as well as manufacturing information, analytical data and any available clinical data or literature to support the use of the investigational new drug. An IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical trials as outlined in the IND. In such a case, the IND may be placed on clinical hold until any outstanding concerns or questions are resolved.
Clinical trials involve the administration of the investigational drug to patients under the supervision of qualified investigators in accordance with Good Clinical Practices, or GCPs. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the efficacy criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical site’s IRB before the trials may be initiated. All participants in clinical trials must provide their informed consent in writing prior to their enrollment in the trial.
The clinical investigation of an investigational drug is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined. The three phases of an investigation are as follows:
| • | Phase 1. Phase 1 involves the initial introduction of an investigational new drug into humans. Phase 1 clinical trials are typically closely monitored and may be conducted in patients with the target disease or condition or healthy volunteers. These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. During phase 1 clinical trials, sufficient information about the investigational drug’s pharmacokinetics and pharmacologic effects may be obtained to permit the design of well-controlled and scientifically valid phase 2 clinical trials. The total number of participants included in phase 1 clinical trials varies, but generally ranges from 20 to 80. |
| • | Phase 2. Phase 2 includes the controlled clinical trials conducted to preliminarily or further evaluate the effectiveness of the investigational drug for a particular indication in patients with the disease or condition under study, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks associated with the drug. Phase 2 clinical trials are typically well controlled, closely monitored and conducted in a limited patient population, usually involving no more than several hundred participants. |
| • | Phase 3. Phase 3 clinical trials are generally controlled clinical trials conducted in an expanded patient population generally at geographically dispersed clinical trial sites. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained, and are intended to further evaluate dosage, effectiveness and safety, to establish the overall benefit-risk relationship of the investigational drug, and to provide an adequate basis for product approval by the FDA. Phase 3 clinical trials usually involve several hundred to several thousand participants. |
28
The decision to terminate development of an investigational drug may be made by either a health authority body such as the FDA, or by us for various reasons. Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known as a data safety monitoring board or committee. This group provides a recommendation of whether or not a trial may move forward at pre-specified check points based on access that only the group maintains to available data from the trial. Suspension or termination of development during any phase of clinical trials can occur if it is determined that the participants or patients are being exposed to an unacceptable health risk. We may decide to suspend or terminate development based on evolving business objectives and/or competitive climate.
In addition, there are requirements and industry guidelines to require the posting of ongoing clinical trials on public registries and the disclosure of designated clinical trial results and related payments to healthcare professionals.
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational drug information is submitted to the FDA in the form of a New Drug Application, or NDA, except under limited circumstances, requesting approval to market the product for one or more indications.
The NDA Approval Process
In order to obtain approval to market a drug in the United States, a marketing application must be submitted to the FDA that provides data establishing the safety and effectiveness of the investigational drug for the proposed indication to the FDA’s satisfaction. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators.
The FDA will initially review the NDA for completeness before it accepts the NDA for filing. The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Based on phase 3 clinical trial results submitted in an NDA, upon the request of an applicant a priority review designation may be granted to a product by the FDA, which sets the target date for FDA action on the application at six months. Priority review is given where preliminary estimates indicate that a product, if approved, has the potential to provide a safe and effective therapy where no satisfactory alternative therapy exists, or a significant improvement compared to marketed products is possible. If criteria are not met for priority review, the standard FDA review period is ten months. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Before approving an NDA, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. If the FDA determines the application, manufacturing process or manufacturing
29
facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process for a drug requires substantial time, effort and financial resources and this process may take several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to develop our SMDCs or companion imaging diagnostics and secure necessary governmental approvals, which could delay or preclude us from marketing our products. Even if the FDA approves an SMDC or companion imaging diagnostic, it may limit the approved indications for use or place other conditions on approval that could restrict commercial application, such as a requirement that we implement special risk management measures through a Risk Evaluation and Mitigation Strategy. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Regulation
After regulatory approval of a drug product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of an NDA, the FDA may require post-marketing testing, including phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. Regulatory approval of oncology products often requires that patients in clinical trials be followed for long periods to determine the OS benefit of the drug. The FDA has indicated to us that if we receive approval of EC145 for treatment of PROC based on PFS data from the PROCEED trial, we will be required to continue to follow patients in that trial to determine the OS benefit of the drug. In addition, as a holder of an approved NDA, we would be required to report, among other things, certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for any of our products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval to assure and preserve the long-term stability of the drug product. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes extensive procedural, substantive and record-keeping requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our SMDCs. Future FDA and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
30
Europe/Rest of World Government Regulation
In addition to regulations in the United States, we will be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In Europe, for example, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of an investigational drug under European Union regulatory systems, we must submit a marketing authorization application. This application is similar to the NDA in the United States, with the exception of, among other things, country-specific document requirements. Medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures, and our SMDCs and companion imaging diagnostics would fall under the centralized authorization procedure.
The EMA may grant conditional marketing authorization for a product that addresses an unmet medical need while additional development work is being completed. Conditional marketing authorization is subject to annual review by the EMA, and such authorization may be limited or even withdrawn by the EMA is subsequent development work is not satisfactory.
The EMA implemented the centralized procedure for the approval of human medicines to facilitate marketing authorizations that are valid throughout the European Union. This procedure results in a single marketing authorization issued by the EMA that is valid across the European Union, as well as Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for human medicines that are: derived from biotechnology processes, such as genetic engineering; contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions; and officially designated orphan medicines.
Under the Centralized Procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Committee for Medicinal Products for Human Use, or CHMP). Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, defined by three cumulative criteria: the seriousness of the disease to be treated; the absence or insufficiency of an appropriate alternative therapeutic approach; and anticipation of high therapeutic benefit. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days.
For other countries outside of the European Union, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the other applicable regulatory requirements.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
31
Compliance
During all phases of development (pre- and post-marketing), failure to comply with the applicable regulatory requirements may result in administrative or judicial sanctions. These sanctions could include the following actions by the FDA or other regulatory authorities: imposition of a clinical hold on trials; refusal to approve pending applications; withdrawal of an approval; warning letters; product recalls; product seizures; total or partial suspension of production or distribution; product detention or refusal to permit the import or export of products; injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain FDA approvals. Our SMDCs may not be considered medically necessary or cost-effective. A payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In 2003, the United States government enacted legislation providing a partial prescription drug benefit for Medicare recipients, which became effective at the beginning of 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through prescription drug plans operating pursuant to this legislation. These plans will likely negotiate discounted prices for our products. Federal, state and local governments in the United States continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the European Community, governments influence the price of pharmaceutical products through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular SMDC to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert a commercial pressure on pricing within a country.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time.
32
Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Under the Veterans Health Care Act, or VHCA, drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including U.S. Department of Veterans Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Recent legislative changes purport to require that discounted prices be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities or register their sales representatives, as well as prohibiting pharmacies and other healthcare entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing, and prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
Patents and Proprietary Rights
Because of the length of time and expense associated with bringing new products to market, biopharmaceutical companies have traditionally placed considerable importance on obtaining and maintaining patent protection for significant new technologies, products and processes.
Our success depends in part on our ability to protect the proprietary nature of our SMDC candidates, technology, and know-how, to operate without infringing on the proprietary rights of others, and to prevent others from infringing our proprietary rights. As a matter of policy, we seek to protect our proprietary position by, among other methods, filing United States and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation, and in-licensing opportunities to develop and maintain our proprietary position.
33
We have applied, and are applying, for patents directed to our three main areas of focus: anti-tumor therapeutics and diagnostics, anti-inflammation therapeutics and diagnostics and immunotherapy therapeutics and diagnostics, both in the United States and, when appropriate, in other countries. We own or have rights to 78 issued patents and 188 applications worldwide covering our core technology, SMDCs and companion imaging diagnostics.
Currently, we own eight issued U.S. patents in whole. Four of these patents relate to EC145 or EC20 compositions of matter — patent number 7,601,332, or the ‘332 patent, entitled “Vitamin Receptor Binding Drug Delivery Conjugates”, patent number 8,105,568 or the ‘568 patent, with the same title, patent number 7,128,893, or the ‘893 patent, entitled “Vitamin-Targeted Imaging Agents” and patent number 7,862,798, or the ‘798 patent, also entitled “Vitamin-Targeted Imaging Agents”. The ‘332 patent issued on October 13, 2009 in the United States and is scheduled to expire in 2026. The ‘332 patent includes claims covering EC145, among other compounds. The ‘568 patent is a continuation to the ‘332 patent and claims the EC145 structure specifically. In 2012, the United States Patent and Trademark Office awarded us a patent with claims specifically directed to EC145 entitled “Vitamin Receptor Binding Drug Delivery Conjugates”, patent number 8,105,658, which is a patent continuation to ‘332. Additionally, we have filed a continuation patent applications to the ‘568 patent and prosecution is ongoing. With respect to EC145 coverage, we have patents issued in the United States, Japan, Australia, China, India, New Zealand and South Africa with continuations filed in the United States, Japan and China, and pending patent applications in Canada, Europe, Japan, Israel, Taiwan, Argentina and Venezuela, and the patents also claim related chemical structures, pharmaceutical compositions, and methods for linking vitamins to drugs through our linker system. We also have filed several patent applications related to EC145 specifically, such as using EC20 to predict patients’ response to EC145 and the combination of EC145 with PLD.
The ‘893 patent and ‘798 patent issued on October 31, 2006 and on January 4, 2011, respectively, in the United States and are both scheduled to expire in 2024. The ‘893 patent and ‘798 patent include claims covering EC20, among other compounds. Additionally, we have filed continuation patent applications to the ‘893 patent and prosecution is ongoing. The ‘798 patent was one such continuation application issued as a patent. Notably, the ‘893 patent has foreign patent counterparts and to date, the ‘893 patent equivalent has been issued in several countries worldwide. In Europe, drug product claims covering some EC20 formulations have been issued. We continue to develop strategies to increase the breadth of EC20 coverage in Europe.
We have filed additional patent applications worldwide to protect our innovations such as multidrug ligand conjugates including EC0225, spacer conjugates including EC0489, tubulysin conjugates including EC0531 and conjugates directed to the PSMA, including EC0651. EC0651 is owned by the Purdue Research Foundation, a non-profit organization, which manages the intellectual property of Purdue University, and exclusively licensed to us.
We entered into two exclusive, worldwide licenses for a number of patents and patent applications, owned by the Purdue Research Foundation, for select folate-targeted technology and for select technology related to PSMA. The folate-technology license was originally entered into on July 17, 1998 with an effective date as of December 21, 1995, and was restated on October 21, 1998. The PSMA license was entered into on March 1, 2010.
Under the two Purdue Research Foundation licenses, there are 35 issued and 70 pending patent applications worldwide which have yet to issue or grant. Most of our licensed intellectual property is under the folate-targeted license. In the United States, we license nine issued patents under the folate-targeted license and none under the PSMA license. Each of the folate-targeted license and PSMA license expire on the expiration date of the last to expire of the patents licensed thereunder, respectively, including those that are issued on patents currently pending and on matters not yet filed. As a result, the final termination date of the Purdue Research Foundation licenses is indeterminable until the last such patents issue and results of potential patent extensions are known. We may terminate the Purdue Research Foundation licenses without cause with 60 days notice. Purdue Research Foundation may terminate the licenses for material default by us which is not cured within 90 days notice by Purdue Research Foundation or upon 60 days notice in the event we fail to meet public demand for approved products covered by the licensed patents after a six month cure period following commercial introduction. The Purdue Research Foundation licenses also contain standard provisions allowing Purdue Research Foundation to terminate upon our bankruptcy. We have royalty obligations to Purdue Research Foundation based on sales of
34
products that are designed, developed or tested using the licensed technology as well as annual minimum royalty obligations. Pursuant to our exclusive license agreement with Purdue Research Foundation relating to folate, we are obligated to pay an annual minimum royalty of $12,500 until commercial sales commence, following which time the payment of single digit royalty rates will commence. Pursuant to our exclusive license agreement with Purdue Research Foundation relating to PSMA, we are obligated to make annual minimum payments of $15,000 until commercial sales commence, following which time the payment of single digit royalty rates will commence, along with an annual milestone payment of $100,000. In addition, certain clinical and regulatory milestone payments of $500,000 along with sales-based milestones related to third-party sales are also payable. We are also subject to penalties totaling $300,000 if certain diligence milestones are not met. Future milestone payments in excess of $500,000 may be waived by Purdue Research Foundation. We do not anticipate incurring liabilities for EC145 royalty payments based on the estimated timing of when we may have commercial sales and the expiration of the applicable patents.
Most of our portfolio consists of intellectual property we exclusively license from Purdue Research Foundation or which we own ourselves. Generally, the intellectual property licensed from Purdue Research Foundation is early stage and relates to methods that were invented in the laboratory of Professor Philip Low. Internally, we typically develop these methods further and refine them to determine the commercial applicability. Additionally, these early-stage patents often provide us protection from competitors while we evaluate commercial possibilities of a specific program. For example, some of the very early patents we licensed from Purdue Research Foundation covered methods of delivering folate attached to targeting ligand across a cell membrane. We were able to use the patent protection afforded by such early patents to develop folate conjugates, including the invention and clinical development, and in the future, the commercialization of our linker system incorporated in EC145.
Due to the use of federal funds in the development of some of the folate-related technology at Purdue Research Foundation, the U.S. government has the irrevocable, royalty-free, paid-up right to practice and have practiced certain patents throughout the world, should it choose to exercise such rights, namely to three early-stage United States patents issued to Purdue Research Foundation and to two pending jointly owned Purdue Research Foundation patent applications.
Our development strategy also employs lifecycle management positions. For example, the final results of our PRECEDENT trial indicated a PFS benefit of PLD and EC145 over PLD alone. As noted above, we filed a patent application directed to this combination. In evaluating our current plans for development, it is likely that we will apply for regulatory approval for this combination. If we are able to obtain patent approval for this indication, then it will provide several years of additional coverage above and beyond the expiration of certain patents relating to EC145 alone. As a result, our general guiding strategy is to obtain patent coverage for innovations that show a clinical benefit to patients and an economic opportunity for us.
Pursuant to a license agreement with BMS we co-own several patent applications directed to epothilone with BMS. BMS notified us of their intent to terminate our license agreement in June 2010 and in July 2010 also notified us of their intent to abandon certain of the patent applications subject to the license related to folate conjugates with epothilone.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in granting a patent, or may be shortened if a patent is terminally disclaimed over another patent.
The patent term of a patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension
35
cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other non-U.S. jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products.
While we pursue patent protection and enforcement of our SMDC candidates and aspects of our technologies when appropriate, we also rely on trade secrets, know-how and continuing technological advancement to develop and maintain our competitive position. To protect this competitive position, we regularly enter into confidentiality and proprietary information agreements with third parties, including employees, independent contractors, suppliers and collaborators. Our employment policy requires each new employee to enter into an agreement containing provisions generally prohibiting the disclosure of confidential information to anyone outside of us and providing that any invention conceived by an employee within the scope of his or her employment duties is our exclusive property. We have a similar policy with respect to independent contractors, generally requiring independent contractors to enter into an agreement containing provisions generally prohibiting the disclosure of confidential information to anyone outside of us and providing that any invention conceived by an independent contractor within the scope of his or her services is our exclusive property with the exception of contracts with universities and colleges that may be unable to make such assignments. Furthermore, our know-how that is accessed by third parties through collaborations and research and development contracts and through our relationships with scientific consultants is generally protected through confidentiality agreements with the appropriate parties. We cannot, however, assure you that these protective arrangements will be honored by third parties, including employees, independent contractors, suppliers and collaborators, or that these arrangements will effectively protect our rights relating to unpatented proprietary information, trade secrets and know-how. In addition, we cannot assure you that other parties will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our proprietary information and technologies.
Additionally, there can be no assurance that our patents will provide significant protection, competitive advantage or commercial benefit. The validity and enforceability of patents issued to biopharmaceutical companies has proven highly uncertain. For example, legal considerations surrounding the validity of patents in the fields of biopharmaceuticals are in transition, and we cannot assure you that the historical legal standards surrounding questions of validity will continue to be applied or that current defenses relating to issued patents in these fields will be sufficient in the future. In addition, we cannot assure you as to the degree and range of protections any of our patents, if issued, may afford us or whether patents will be issued. For example, patents which may issue to us may be subjected to further governmental review that may ultimately result in the reduction of their scope of protection, and pending patent applications may have their requested breadth of protection significantly limited before being issued, if issued at all. Further, since publication of discoveries in scientific or patent literature often lags behind actual discoveries, we cannot assure you that we were the first creator of inventions covered by our pending patent applications, or that we were the first to file patent applications for these inventions.
Many biopharmaceutical companies and university and research institutions have filed patent applications or have received patents in our areas of product development. Many of these entities’ applications, patents and other intellectual property rights could prevent us from obtaining patents or could call into question the validity of any of our patents, if issued, or could otherwise adversely affect the ability to develop, manufacture or commercialize SMDC candidates. In addition, certain parts of our technology originated from third-party sources. These third-party sources include academic, government and other research laboratories, as well as the public domain. If use of technology incorporated into or used to produce our SMDCs is challenged, or if a conflicting patent issued to others is upheld in the courts or if a conflicting patent application filed by others is issued as a patent and is upheld, we may be unable to market one or more of our SMDCs, or we may be required to obtain a license to market those SMDCs. To contend with these possibilities, we may have to enter into license agreements in the future with third parties for technologies that may be useful or necessary for the manufacture or commercialization of some of our SMDCs. In addition, we are routinely in discussions with academic and
36
commercial entities that hold patents on technology or processes that we may find necessary in order to engage in some of our activities. However, we cannot assure you that these licenses, or any others that we may be required to obtain to market our SMDCs, will be available on commercially reasonable terms, if at all, or that we will be able to develop alternative technologies if we cannot obtain required licenses.
To protect our rights to any of our issued patents and proprietary information, we may need to litigate against infringing third parties, or avail ourselves of the courts or participate in hearings to determine the scope and validity of those patents or other proprietary rights. These types of proceedings are often costly and could be very time-consuming to us, and we cannot assure you that the deciding authorities will rule in our favor. An unfavorable decision could allow third parties to use our technology without being required to pay us licensing fees or may compel us to license needed technologies to avoid infringing third-party patent and proprietary rights; or even could result in the invalidation or a limitation in the scope of our patents or forfeiture of the rights associated with our patents or pending patent applications. Although we believe that we would have valid defenses to allegations that our current SMDCs, production methods and other activities infringe the valid and enforceable intellectual property rights of any third parties, we cannot be certain that a third party will not challenge our position in the future. Even if some of these activities were found to infringe a third party’s patent rights, we may be found to be exempt from infringement under 35 U.S.C. § 271(e) to the extent that these are found to be pre-commercialization activities related to our seeking regulatory approval for a SMDC. However, the scope of protection under 35 U.S.C. § 271(e) is uncertain and we cannot assure you that any defense under 35 U.S.C. § 271(e) would be successful. Further, the defense under 35 U.S.C. § 271(e) is only available for pre-commercialization activities, and could not be used as a defense for sale and marketing of any of our SMDCs. There has been, and we believe that there will continue to be, significant litigation in the biopharmaceutical and pharmaceutical industries regarding patent and other intellectual property rights.
Corporate Information
We were incorporated in the State of Indiana in 1995 and we were reincorporated in the State of Delaware in 2001. Our principal executive offices are located at 3000 Kent Avenue, Suite A1-100, West Lafayette, Indiana 47906, and our telephone number is (765) 463-7175.
Available Information
Our website address is www.endocyte.com. Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act are available or may be accessed free of charge through www.sec.gov and the Investor Relations section of our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission, or SEC. Our website and the information contained therein or connected thereto are not intended to be incorporated by reference into this Annual Report on Form 10-K or any other report we file with the SEC.
The name “Endocyte” and our logo are our trademarks. All other trademarks and trade names appearing in this annual report are the property of their respective owners.
37
Executive Officers of the Registrant
The following table sets forth certain information concerning our executive officers as of March 1, 2012:
| Name |
Age |
Position | ||
| P. Ron Ellis |
50 | President and Chief Executive Officer | ||
| Michael A. Sherman |
45 | Chief Financial Officer | ||
| Philip S. Low, Ph.D. |
64 | Chief Science Officer | ||
| Christopher P. Leamon, Ph.D. |
45 | Vice President of Research | ||
| Chandra D. Lovejoy |
40 | Vice President of Regulatory Affairs | ||
| Binh Nguyen, M.D., Ph.D. |
53 | Vice President of Clinical Affairs | ||
| Allen R. Ritter, Ph.D. |
50 | Vice President of Manufacturing and Chemistry Manufacturing Control | ||
| Beth A. Taylor |
46 | Corporate Controller |
P. Ron Ellis is one of our founders and has served as our President and Chief Executive Officer since January 1996 and as a member of our Board of Directors since December 1995. From May 1987 to December 1995, Mr. Ellis served in various positions at Hill-Rom Company, but most recently as Vice President of Strategy and Corporate Development of the specialty care division. Mr. Ellis holds a B.S. in computer science and an M.B.A. from Brigham Young University and a certification in regulatory affairs from Purdue University.
Michael A. Sherman has served as our Chief Financial Officer since October 2006. From December 1994 to October 2006, Mr. Sherman served in various executive roles, but most recently as Vice President of Finance and Strategic Planning from May 2004 to October 2006, of Guidant Corporation, or Guidant, a cardiovascular device manufacturer acquired by Boston Scientific Corporation, a medical device company, in April 2006. Mr. Sherman holds a B.A. in economics from DePauw University and an M.B.A. from the Amos Tuck School, Dartmouth College.
Philip S. Low, Ph.D. is one of our founders and has served as our Chief Science Officer since April 1998 and as a member of our Board of Directors since December 1995. Dr. Low has served on the faculty at Purdue University since August 1976, where he is currently the Ralph C. Corley Distinguished Professor of Chemistry. Dr. Low holds a B.S. in chemistry from Brigham Young University and a Ph.D. in biochemistry from the University of California, San Diego.
Christopher P. Leamon, Ph.D. has served as our Vice President of Research since April 2000. From February 1999 to April 2000, Dr. Leamon served as our Director of Biology and Biochemistry. Prior to joining us, Dr. Leamon was employed in the pharmaceutical industry where he conducted discovery research in the field of peptide, oligonucleotide, liposome and DNA drug delivery for GlaxoWellcome, a healthcare company, in December 2000, and Isis Pharmaceuticals, a biomedical pharmaceutical company. Dr. Leamon holds a B.S. in chemistry from Baldwin Wallace College and a Ph.D. in biochemistry from Purdue University.
Chandra D. Lovejoy has served as our Vice President of Regulatory Affairs since May 2010. From December 2007 to May 2010, Ms. Lovejoy served as our Director of Regulatory Affairs. Ms. Lovejoy served in various positions at Genentech, a biotechnology company and an indirectly wholly owned subsidiary of Roche Holdings, a healthcare company, including Manager of Regulatory Affairs. Ms. Lovejoy holds an M.S. in regulatory affairs from San Diego State University and a B.S. in organizational behavior from the University of San Francisco.
Binh Nguyen, M.D., Ph.D. has served as our Vice President of Clinical Affairs since June 2011. From August 2005 to June 2011, he served as Chief Medical Officer at Tigris Pharmaceuticals, Inc. and prior to August 2005, he worked at Eli Lilly & Company for 10 years, most recently as Executive Director, Global Oncology Platform Team, for which he was responsible for the global development of Gemzar ® , Alimta ® , and Enzastaurin. Dr. Nguyen received his Ph.D. in organic chemistry from Georgetown University and his M.D. from the University of Maryland. He completed fellowships at the National Cancer Institute and worked at the FDA’s Division of Oncology as a medical reviewer.
38
Allen R. Ritter, Ph.D. has served as our Vice President of Manufacturing and Chemistry Manufacturing Control, or CMC, since December 2005. From May 2004 to December 2005, Dr. Ritter served as our Director of Development, CMC and Manufacturing. Dr. Ritter holds a B.S. in chemistry from St. Olaf College, an M.S. in organic chemistry from the University of Pittsburgh, and a Ph.D. in synthetic organic chemistry from the University of Notre Dame.
Beth A. Taylor has served as our Corporate Controller since January 2011. She served as Corporate Controller of Author Solutions, Inc. from December 2009 to December 2010, as Vice President of Accounting, Financial Reporting and Control at Harlan Laboratories, Inc. from July 2007 to May 2009 and in various roles at Republic Airways Holdings, Inc. from May 1999 through June 2007, including Vice President and Corporate Controller from August 2004 through June 2007. Ms. Taylor holds a B.S. in Accounting from Indiana University.
| Item | 1A. Risk Factors |
Risk factors which could cause actual results to differ from our expectations and which could negatively impact our financial condition and results of operations are discussed below and elsewhere in this report. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that are currently not believed to be significant to our business may also affect our actual results and could harm our business, financial condition and results of operations. If any of the risks or uncertainties described below or any additional risks and uncertainties actually occur, our business, results of operations and financial condition could be materially and adversely affected.
Risks Related to Our Business and Industry
We have incurred significant losses since our inception and anticipate that we will continue to incur losses for the foreseeable future. We may never achieve or sustain profitability.
We are a clinical-stage biopharmaceutical company with a limited operating history. Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We are not profitable and have incurred losses in each year since our inception in December 1995. We have not generated any revenue from product sales to date. We continue to incur significant research and development and other expenses related to our ongoing operations. Our net loss for the year ended December 31, 2011 was $40.5 million. As of December 31, 2011, we had a retained deficit of $138.6 million. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our development of, and seek regulatory approvals for, our small molecule drug conjugates, or SMDCs, and companion imaging diagnostics, and begin to commercialize any approved products. As such, we are subject to all the risks incident to the creation of new SMDCs and companion imaging diagnostics, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. If our product candidates fail in clinical trials, or do not gain regulatory approval, or fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
We are a clinical-stage company with no approved products, which makes it difficult to assess our future viability.
We were incorporated in December 1995, are a clinical-stage company and, as of December 31, 2011, have not derived any revenue from the sales of our products. Our operations to date have been limited to organizing and staffing our company, acquiring, developing and securing our technology, undertaking preclinical studies and clinical trials of our product candidates and engaging in research and development under collaboration agreements. We have not yet demonstrated an ability to obtain regulatory approval, formulate and manufacture commercial-scale products, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, it is difficult to predict our future success and the viability of any commercial programs that we may choose to take forward. From our inception through December 31, 2011, we have derived
39
non-grant related revenues of $12.1 million from payments under collaborative agreements with Bristol-Myers Squibb, or BMS, Sanofi-Aventis and On Target Laboratories L.L.C., or On Target. The agreement with On Target is the only agreement that is currently in force. We will be entitled to receive minimum royalty payments from On Target annually based on net sales, but there is no guarantee that a commercial product will be developed and approved for commercial sales.
We are highly dependent on the success of our lead SMDC, EC145, and we cannot give any assurance that we will successfully complete its clinical development, or that it will receive regulatory approval or be successfully commercialized.
Our lead SMDC, EC145, has been evaluated in a randomized phase 2 clinical trial for the treatment of women with platinum-resistant ovarian cancer, or PROC, and is currently being evaluated in a randomized phase 3 clinical trial in the same indication. In addition, we recently completed a phase 2 single-arm clinical trial for heavily pre-treated non-small cell lung cancer, or NSCLC, and are planning an additional randomized phase 2 clinical trial in the same indication. Our future trials may not be successful, and EC145 may never receive regulatory approval or be successfully commercialized. We may fail to obtain necessary marketing approvals for EC145 from the U.S. Food and Drug Administration, or FDA, the European Medicines Authority, or EMA, or other non-U.S. regulatory authorities if our clinical development program for EC145 fails to demonstrate that it is safe and effective to the satisfaction of such authorities, or if we have inadequate financial or other resources to advance EC145 through the necessary development activities. Even if EC145 receives regulatory approval, we may not be successful in marketing it for a number of reasons, including the introduction by our competitors of more clinically-effective or cost-effective alternatives or failure in our sales and marketing efforts. Any failure to obtain approval of EC145 and successfully commercialize it would have a material and adverse impact on our business.
The results of previous clinical trials may not be predictive of future results, and our current and planned clinical trials may not satisfy the requirements of the FDA or non-U.S. regulatory authorities.
The clinical trials of our product candidates are, and the manufacturing and marketing of any approved products will be, subject to extensive and rigorous review and regulation by numerous government authorities in the United States, Europe and in other countries where we intend to test and market our product candidates. Before obtaining regulatory approvals for the commercial sale of any product candidate, we must demonstrate through preclinical testing and clinical trials that the product candidate is safe and effective for use in each indication for which we intend to market such product candidate. This process can take many years and requires the expenditure of substantial financial and human resources and may include post-marketing trials and surveillance. To date, we have not completed any randomized phase 3 clinical trials. We have completed two phase 2 single-arm and one phase 2 randomized clinical trials with EC145 for the treatment of patients with PROC and NSCLC. In May 2011, we began evaluating EC145 in a phase 3 clinical trial, known as PROCEED, in PROC. We had to stop patient enrollment later in the year due to global shortages of pegylated liposomal doxorubicin, or PLD (marketed in the U.S. under the brand name Doxil), but we have plans to resume enrollment in the U.S. We have three other product candidates in phase 1 clinical trials. In addition, we have other product candidates in the discovery and preclinical testing stages.
Positive results from preclinical studies and early clinical trials should not be relied upon as evidence that later-stage or large-scale clinical trials will succeed. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, even after promising results in earlier trials. We will be required to demonstrate with substantial evidence through adequate and well-controlled clinical trials, including PROCEED, our phase 3 trial of EC145 for the treatment of women with PROC, that our product candidates are safe and effective for use in the target population before we can seek regulatory approvals for their commercial sale in the United States.
In our end of phase 2 meeting with the FDA related to EC145, the FDA stated that, because of the difficulty in reliably determining cancer progression based on imaging studies in ovarian cancer, its office policy is to require overall survival, or OS, to be the primary endpoint for an ovarian cancer registration trial. However, the
40
FDA stated that we may choose, at our own risk, to conduct a phase 3 trial in which progression free survival, or PFS, is the primary endpoint; provided that for such a trial to be the basis for approval, the PFS results must be very robust statistically and clinically meaningful, and the trial must be powered to demonstrate a statistically significant OS benefit. In a follow-up meeting in March, 2012, the FDA reiterated this position and added that the design of the Phase 3 PROCEED trial could lead to an accelerated approval depending on the results. The primary endpoint of the phase 3 PROCEED trial is PFS in the FR(++) patient population. Based on feedback from EU health authorities and the FDA, we are evaluating the inclusion of additional patients for exploratory analysis in order to assess potential benefit in patients with less than all of their target tumors positive for the folate receptor. While those plans are being finalized, patients will be enrolled regardless of the FR status, although the primary endpoint will include only FR(++) patients. Even if our phase 3 trial meets its PFS primary endpoint, a positive trend in OS at the time of filing our new drug application, or NDA, may be required for approval or the FDA may delay consideration of approval until final OS data becomes available, which would result in significant additional costs and delay our ability to market EC145 for this indication. The FDA also noted that the final OS analysis from our phase 3 trial would be required as a post-marketing commitment should approval be granted based upon PFS. In addition, if the FDA approves EC145 based upon meeting our PFS primary endpoint, in certain circumstances the approval could be withdrawn if any required post-marketing trials or analyses do not meet FDA requirements. Furthermore, as is typical for cancer drug approvals, the FDA stated that for the initial approval of EC145 to be based on a single phase 3 clinical trial, the trial must provide evidence of persuasive and robust statistically significant clinical benefit such that it would be considered unethical to conduct another trial. If we fail to demonstrate a benefit of this magnitude in PROCEED, we would expect that the FDA would require us to conduct a second phase 3 trial in order to receive marketing approval of EC145 for the treatment of PROC. Such a requirement would result in significant additional cost and would delay our ability to market EC145 for this indication.
Patients in PROCEED are being imaged with our companion imaging diagnostic, EC20, prior to treatment with EC145. Although EC20 is part of our phase 3 trial design, there can be no assurance that this trial will provide a sufficient basis for approval of an NDA for EC20. Similarly, we can provide no assurance to you that EC145 will be approved without EC20 approval.
The FDA and other regulatory authorities may change requirements for the approval of our product candidates even after reviewing and providing non-binding comment on a protocol for a pivotal phase 3 clinical trial that has the potential to result in FDA approval. In addition, regulatory authorities may also approve any of our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
Our efforts to obtain conditional marketing authorization for EC145 and EC20 from the European Medicines Agency may be unsuccessful.
We expect to file applications with the EMA in the third quarter of 2012 for conditional marketing authorization for EC145 for the treatment of PROC and for EC20 for patient selection. These filings will be based on the results of our randomized phase 2 clinical trial, which we refer to as the PRECEDENT trial, which investigated EC145 in combination with standard chemotherapy PLD for treatment of women with PROC and which also evaluated the utility of EC20 for patient selection. Our filings will be supported by four clinical studies: a Phase 1 study in solid tumors, two single-agent, single-arm Phase 2 studies in ovarian cancer and non small cell lung cancer, and the PRECEDENT trial, a randomized study in platinum-resistant ovarian cancer.
We cannot predict with any certainty whether the EMA will grant the marketing authorizations that we intend to seek in these applications. Marketing authorizations based on phase 2 randomized studies are unusual and, if granted, are subject to significant conditions, which likely would include requirements to:
| • | complete the phase 3 study; |
41
| • | confirm that the patient risk-benefit is positive; and |
| • | complete an annual renewal process. |
If the EMA were to grant conditional marketing authorization of EC145 and EC20 based on our phase 2 studies, that authorization could be further limited or even withdrawn if our required phase 3 studies fail to demonstrate evidence of persuasive and robust statistically significant clinical benefit or result in unexpected safety concerns with the study drugs. We cannot give any assurance that the EMA will approve our applications for conditional marketing authorization or that, if approved, that the labeling restrictions and other approval conditions will enable us to profitably commercialize these drug candidates in the European Union. Conditional approval by the EMA would not authorize us to commercialize EC145 or EC20 in any country outside the European Union and would not be expected to have any beneficial effect on our ability to obtain regulatory approval from the FDA or other regulatory agencies.
There is a high risk that our development and clinical activities will not result in commercial products, and we will have invested in our current development and clinical programs, to the exclusion of others, for several more years before it is known whether one or more of our product candidates will receive regulatory approval or be commercially introduced.
Our product candidates are in various stages of development and are prone to the risks of failure inherent in biopharmaceutical development. We will need to complete significant additional clinical trials before we can demonstrate that our product candidates are safe and effective to the satisfaction of the FDA, the EMA or other non-U.S. regulatory authorities. Clinical trials are expensive and uncertain processes that take years to complete. Failure can occur at any stage of the process. Further, even if our product candidates receive required regulatory approvals, we cannot assure you that they will be successful commercially. In addition, we have a large number of product candidates in our development pipeline, and while we invest in the technology and indications that we believe are most promising, financial and resource constraints may require us to forego or delay opportunities that may ultimately have greater commercial potential than those programs we are currently actively developing.
The coverage and reimbursement status of newly approved biopharmaceuticals is uncertain, and failure to obtain adequate coverage and adequate reimbursement of EC145 or other product candidates could limit our ability to generate revenue.
There is significant uncertainty related to the third-party coverage and reimbursement of newly approved drugs. The commercial success of our product candidates, including EC145, in both domestic and international markets will depend in part on the availability of coverage and adequate reimbursement from third-party payors, including government payors, such as the Medicare and Medicaid programs, and managed care organizations. Government and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for new drugs and, as a result, they may not cover or provide adequate payment for our product candidates. These payors may conclude that our product candidates are less safe, less effective or less cost-effective than existing or later introduced products, and third-party payors may not approve our product candidates for coverage and reimbursement or may cease providing coverage and reimbursement for these product candidates. Because each country has one or more payment systems, obtaining reimbursement in the United States and internationally may take significant time and cause us to spend significant resources. The failure to obtain coverage and adequate reimbursement for our product candidates or healthcare cost containment initiatives that limit or deny reimbursement for our product candidates may significantly reduce any future product revenue.
In the United States and in other countries, there have been and we expect there will continue to be a number of legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. International, federal and state lawmakers regularly propose and, at times, enact legislation that would result in significant changes to the healthcare system, some of which are intended to contain or reduce the costs of medical products and services. The U.S. government and other governments have shown significant
42
interest in pursuing healthcare reform, as evidenced by the recent passing of the Patient Protection and Affordable Care Act and its amendment, the Health Care and Education Reconciliation Act. Such government-adopted reform measures may adversely impact the pricing of healthcare products and services in the United States or internationally and the amount of reimbursement available from governmental agencies or other third-party payors. In addition, in some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. The continuing efforts of U.S. and other governments, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce healthcare costs may adversely affect our ability to set satisfactory prices for our products, to generate revenues, and to achieve and maintain profitability.
In some countries, particularly in the European Union, prescription drug pricing is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product candidate. To obtain reimbursement or pricing approval in some countries, we may be required to conduct additional clinical trials that compare the cost-effectiveness of our product candidates to other available therapies. If reimbursement of our product candidates is unavailable or limited in scope or amount in a particular country, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability of our products in such country.
Our development activities could be delayed or stopped for a number of reasons, many of which are outside our control, including failure to recruit and enroll patients for clinical trials.
Each of our clinical trials requires the investment of substantial expense and time and the timing of the commencement, continuation and completion of these clinical trials may be subject to significant delays relating to various causes. We do not know whether our current clinical trials will be completed on schedule, or at all, and we cannot guarantee that our future planned clinical trials will begin on time, or at all. Clinical trials must be conducted in accordance with FDA or other applicable foreign government guidelines and are subject to oversight by the FDA, other foreign governmental agencies and independent institutional review boards, or IRBs, at the medical institutions where the clinical trials are conducted. In addition, clinical trials must be conducted with supplies of our product candidates produced under current Good Manufacturing Practice, or cGMP, and other requirements in foreign countries, and may require large numbers of test patients. Our current and planned clinical trials could be substantially delayed or prevented by several factors, including:
| • | limited number of, and competition for, suitable sites to conduct our clinical trials; |
| • | government or regulatory delays and changes in regulatory requirements, policy and guidelines; |
| • | delay or failure to obtain sufficient supplies of the product candidate for, or other drugs used in, our clinical trials as a result of our suppliers’ non-compliance with cGMP, or for other reasons; |
| • | delay or failure to reach agreement on acceptable clinical trial agreement terms with prospective sites or investigators; and |
| • | delay or failure to obtain IRB approval to conduct a clinical trial at a prospective site. |
The completion of our clinical trials could also be substantially delayed or prevented by several factors, including:
| • | slower than expected rates of patient recruitment and enrollment; |
| • | unforeseen safety issues; |
| • | lack of efficacy evidenced during clinical trials, which risk may be heightened given the advanced state of disease and lack of response to prior therapies of patients in our clinical trial for EC145 in PROC; |
| • | termination of our clinical trials by an IRB at one or more clinical trial sites; |
43
| • | inability or unwillingness of patients or medical investigators to follow our clinical trial protocols; and |
| • | inability to monitor patients adequately during or after treatment or high patient dropout rates. |
For example, we experienced slower than expected rates of patient recruitment and enrollment with our PRECEDENT trial due to a number of reasons, including slower than expected clinical trial site activations due to prolonged contract negotiations and delays in scheduling or approval by IRBs, lack of qualified patients at a particular site, competition with other clinical trials for patients, and clinical investigator scheduling and availability due to vacations or absences. We also have experienced a delay in enrollment of the PROCEED trial due to the lack of supply of PLD.
Our clinical trials may be suspended or terminated at any time by the FDA, other regulatory authorities or us. For example, a Data Safety Monitoring Board will monitor PROCEED and could recommend closing the trial based on the results of a pre-specified interim futility analysis or any observed unexpected safety concern that may occur during the trial. Failure or significant delay in completing clinical trials for our product candidates could materially harm our financial results and the commercial prospects for our product candidates.
Even if we are able to obtain regulatory approval of EC145 based on our initial phase 3 clinical trial, marketing will be limited to our intended indication of PROC and not ovarian cancer generally, or any other type of cancer.
Even if we are able to obtain regulatory approval of EC145 based on our initial phase 3 clinical trial, PROCEED, and formulate and manufacture a commercial-scale product, our marketing of EC145 will be limited to our initial intended indication of PROC and not ovarian cancer generally, or any other type of cancer. According to the American Cancer Society, approximately 22,280 new cases of ovarian cancer are projected in the United States in 2012. Of those ovarian cancer cases, approximately 50 percent of patients will eventually develop PROC. Marketing of EC145, if approved for our intended indication, will be limited to those women with ovarian cancer who demonstrate a resistance to platinum-based therapies and who are FR(+) or FR(++). The intended indication for use may be further limited to only patients who are FR(++). Marketing efforts for EC145 outside of our approved indication of PROC will require additional regulatory approvals, which we may never pursue or receive.
Our product candidates may cause undesirable side effects that could delay or prevent their regulatory approval or commercialization.
Common side effects of EC145 include abdominal pain, vomiting, constipation, nausea, fatigue, loss of appetite and peripheral sensory neuropathy. Because our products have been tested in relatively small patient populations and for limited durations to date, additional side effects may be observed as their development progresses.
Undesirable side effects caused by any of our product candidates could cause us or regulatory authorities to interrupt, delay or discontinue clinical trials and could result in the denial of regulatory approval by the FDA, the EMA or other non-U.S. regulatory authorities for any or all targeted indications. This, in turn, could prevent us from commercializing our product candidates and generating revenues from their sale. In addition, if one of our products receives marketing approval and we or others later identify undesirable side effects caused by this product:
| • | regulatory authorities may withdraw their approval of this product; |
| • | we may be required to recall this product, change the way this product is administered, conduct additional clinical trials or change the labeling of this product; |
| • | this product may be rendered less competitive and sales may decrease; or |
| • | our reputation may suffer generally both among clinicians and patients. |
44
Any one or a combination of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent us from generating significant revenues from the sale of the product.
We may not obtain government regulatory approval to market our product candidates or negotiate satisfactory pricing for our product candidates which could adversely impact our future profitability.
We intend to seek approval to market certain of our product candidates in both the United States and in non-U.S. jurisdictions. Prior to commercialization, each product candidate will be subject to an extensive and lengthy governmental regulatory approval process in the United States and in other countries. In order to market our products in the European Union and many other non-U.S. jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. Although we intend to file applications with the EMA in the third quarter 2012 seeking conditional marketing authorization for EC145 and EC20 based on the results of our randomized phase 2 PRECEDENT trial and supplemental analyses of that trial’s results, we have not yet filed for marketing approval for any of our product candidates in the U.S. or in any other jurisdictions and may not receive the approvals necessary to commercialize our product candidates in any market. We may not be able to obtain regulatory approval for any product candidates, or even if approval is obtained, the labeling for such products may place restrictions on their use that could materially impact the marketability and profitability of the product subject to such restrictions. Satisfaction of these regulatory requirements, which includes satisfying the FDA and foreign regulatory authorities that the product is both safe and effective for its intended uses, typically takes several years or more depending upon the type, complexity, novelty and safety profile of the product and requires the expenditure of substantial resources. Uncertainty with respect to meeting the regulatory requirements governing our product candidates may result in excessive costs or extensive delays in the regulatory approval process, adding to the already lengthy review process.
Also, the approval procedure varies among countries and can involve additional testing and data review. The time and safety and efficacy data required to obtain foreign regulatory approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory agencies in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory agencies in other countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in other jurisdictions, including approval by the FDA. The failure to obtain regulatory approval in any jurisdiction could materially harm our business.
We may require substantial additional funding which may not be available to us on acceptable terms, or at all.
We are advancing multiple product candidates through clinical development. We believe that our current cash position, including cash equivalents and short term investments is sufficient to fund the operations, including PROCEED, our phase 3 clinical trial of EC145 and EC20, through the availability of final primary PFS data from that study, which we, assuming the availability of PLD for the trial, anticipate will be in the first half of 2014 If we were to receive conditional marketing authorization in Europe of EC145 and EC20 prior to the completion of PROCEED, this could impact the enrollment timeline as European sites would transition from clinical trials to commercial use. This could delay the availability of final data from the PROCEED trial. We may be able to mitigate this potential delay by adding clinical trial sites in locations where conditional marketing authorization has not been granted. We would require additional funding through public or private equity or debt financings or other sources, such as strategic partnerships or licensing arrangements, to commercialize any of our SMDCs or companion imaging diagnostics. In addition, if the FDA requires us to undertake a second phase 3 clinical trial or obtain final OS data from PROCEED we will also require additional funding.
45
Our future funding requirements will depend on many factors, including but not limited to:
| • | our need to expand our research and development activities; |
| • | the rate of progress and cost of our clinical trials and the need to conduct clinical trials beyond those planned; |
| • | the outcome of the applications we intend to file with the EMA for conditional marketing authorization for EC145 and EC20; |
| • | the costs associated with establishing a sales force and commercialization capabilities; |
| • | the costs of acquiring, licensing or investing in businesses, product candidates and technologies; |
| • | the costs and timing of seeking and obtaining approval from the FDA and non-U.S. regulatory authorities; |
| • | our ability to maintain, defend and expand the scope of our intellectual property portfolio; |
| • | our need and ability to hire additional management and scientific and medical personnel; |
| • | the effect of competing technological and market developments; |
| • | our need to implement additional internal systems and infrastructure, including financial and reporting systems necessary to comply with section 404 of the Sarbanes Oxley Act; and |
| • | the economic and other terms and timing of collaboration, licensing or other arrangements into which we may enter. |
Until we can generate a sufficient amount of revenue to finance our cash requirements, which we may never do, we expect to finance future cash needs primarily through public or private equity or debt financings or other sources, such as strategic partnerships or licensing arrangements. We do not know whether additional funding will be available on acceptable terms, or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs, or enter into collaboration or other arrangements with other companies to provide such funding for one or more of such clinical trials or programs in exchange for our affording such partner commercialization or other rights to the product candidates that are the subject of such clinical trials or programs.
In addition, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned. Also, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Raising additional capital may cause dilution to existing stockholders, restrict our operations or require us to relinquish rights.
We may seek the additional capital necessary to fund our operations through public or private equity or debt financings or other sources, such as strategic partnerships or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of the current stockholders will be diluted and the terms may include liquidation or other preferences that adversely affect their rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures, or declaring dividends, or which impose financial covenants on us that limit our operating flexibility to achieve our business objectives. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. In addition, we cannot assure you that additional funds will be available to us on favorable terms or at all.
46
If our competitors develop and market products that are more effective, safer or less expensive than our product candidates, our commercial opportunities will be negatively impacted.
The life sciences industry is highly competitive, and we face significant competition from many pharmaceutical, biopharmaceutical and biotechnology companies that are researching and marketing products designed to address various types of cancer and other indications we treat or may treat in the future. We are currently developing cancer therapeutics that will compete with other drugs and therapies that currently exist or are being developed. Also, our lead SMDC, EC145, is being clinically developed not as a primary therapy but as a therapy for patients whose tumors have developed resistance to chemotherapy, which limits its potential addressable market. Products we may develop in the future are also likely to face competition from other drugs and therapies. Many of our competitors have significantly greater financial, manufacturing, marketing and drug development resources than we do. Large biopharmaceutical companies, in particular, have extensive experience in clinical testing and in obtaining regulatory approvals for drugs. Additional mergers and acquisitions in the biopharmaceutical industry may result in even more resources being concentrated by our competition. Competition may increase further as a result of advances in the commercial applicability of technologies currently being developed and a greater availability of capital investment in those fields. These companies also have significantly greater research and marketing capabilities than we do. Some of the companies developing products which may compete with EC145 include Roche Holdings, Nektar Therapeutics, Sunesis Pharmaceuticals, Eli Lilly, Sanofi, and Amgen. In addition, many universities and U.S. private and public research institutes are active in cancer research, the results of which may result in direct competition with EC145 or other of our product candidates.
In certain instances, the drugs which will compete with our product candidates are widely available or established, existing standards of care. To compete effectively with these drugs, our product candidates will need to demonstrate advantages that lead to improved clinical safety or efficacy compared to these competitive products. We cannot assure you that we will be able to achieve competitive advantages versus alternative drugs or therapies. If our competitors market products that are more effective, safer or less expensive than our product candidates, if any, or that reach the market sooner than our product candidates, if any, we may not achieve commercial success.
We believe that our ability to successfully compete will depend on, among other things:
| • | our ability to design and successfully execute appropriate clinical trials; |
| • | our ability to recruit and enroll patients for our clinical trials; |
| • | the results of our clinical trials and the efficacy and safety of our product candidates; |
| • | the speed at which we develop our product candidates; |
| • | achieving and maintaining compliance with regulatory requirements applicable to our business; |
| • | the timing and scope of regulatory approvals, including labeling; |
| • | adequate levels of reimbursement under private and governmental health insurance plans, including Medicare; |
| • | our ability to protect intellectual property rights related to our product candidates; |
| • | our ability to commercialize and market any of our product candidates that may receive regulatory approval; |
| • | our ability to have our partners manufacture and sell commercial quantities of any approved product candidates to the market; |
| • | acceptance of our product candidates by physicians, other healthcare providers and patients; and |
| • | the cost of treatment in relation to alternative therapies. |
47
In addition, the biopharmaceutical industry is characterized by rapid technological change. Our future success will depend in large part on our ability to maintain a competitive position with respect to these technologies. Our competitors may render our technologies obsolete by advances in existing technological approaches or the development of new or different approaches, potentially eliminating the advantages in our drug discovery process that we believe we derive from our research approach and proprietary technologies. Also, because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively.
If we fail to attract and retain key management and scientific personnel, we may be unable to successfully develop or commercialize our product candidates.
Our success as a specialized scientific business depends on our continued ability to attract, retain and motivate highly qualified management and scientific and clinical personnel. The loss of the services of any of our senior management could delay or prevent the commercialization of our product candidates.
We may not be able to attract or retain qualified management and scientific personnel in the future due to the intense competition for a limited number of qualified personnel among biopharmaceutical, biotechnology, pharmaceutical and other businesses, particularly in Indiana. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will impede the achievement of our research and development objectives, our ability to raise additional capital and our ability to implement our business strategy.
As we evolve from a company primarily involved in clinical development to a company also involved in commercialization, we may encounter difficulties in managing our growth and expanding our operations successfully.
As we advance our product candidates through clinical trials, we will need to expand our development, regulatory, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to manage additional relationships with such third parties, as well as additional collaborators and suppliers.
Maintaining these relationships and managing our future growth will impose significant added responsibilities on members of our management and other personnel. We must be able to: manage our development efforts effectively; manage our clinical trials effectively; hire, train and integrate additional management, development, administrative and sales and marketing personnel; improve our managerial, development, operational and finance systems; and expand our facilities, all of which may impose a strain on our administrative and operational infrastructure.
If we are unable to establish sales, marketing and distribution capabilities or to enter into agreements with third parties to do so, we may be unable to successfully market and sell any products, even if we are able to obtain regulatory approval.
We currently have no marketing, sales or distribution capabilities. If our product candidates receive regulatory approval, we intend to establish our sales and marketing organization with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time consuming. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of these products. With respect to our product candidates, we may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. To the extent that we enter into co-promotion or other licensing arrangements, our product revenue is likely to be lower than if we directly marketed or sold our products. In addition, any revenue we receive will depend in whole or in part upon the efforts of such third parties, which may not be successful and are generally not within our control. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to
48
successfully commercialize any of our product candidates that receive regulatory approval. If we are not successful in commercializing our product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
If we do not establish development or commercialization collaborations, we may have to alter our development and marketing plans.
Our development programs and potential commercialization of our product candidates will require substantial additional cash to fund expenses. Our strategy includes potentially selectively collaborating with leading biopharmaceutical, pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of some of our product candidates in the United States or internationally. Although we are not currently party to any collaboration agreements, we may enter into collaborations in the future, especially for target indications in which the potential collaborator has particular therapeutic expertise or for markets outside of the United States. We face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on acceptable terms, or at all. If that were to occur, we may have to curtail the development of a product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization, reduce the scope of our sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all. If we do not have sufficient funds, we will not be able to bring our product candidates to market and generate product revenue. In addition, even if we do enter into one or more development or commercialization arrangements, we cannot assure you that the objectives of such arrangements will be realized or that the arrangement will not be terminated or expire. For example, we previously entered into an exclusive license agreement with BMS in a collaboration to develop and commercialize folate conjugates, which was terminated by BMS in June 2010, we believe as a result of a change in its strategic focus.
We rely on third parties to conduct clinical trials for our product candidates and plan to rely on third parties to conduct future clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, it may cause delays in commencing and completing clinical trials of our product candidates or we may be unable to obtain regulatory approval for or commercialize our product candidates.
Clinical trials must meet applicable FDA and foreign regulatory requirements. We do not have the ability to independently conduct phase 2 or phase 3 clinical trials for any of our product candidates. We rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories, to conduct all of our clinical trials of our product candidates; however, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA, the EMA and other non-U.S. regulatory authorities require us to comply with regulations and standards, commonly referred to as Good Clinical Practices for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements.
We or the third parties we rely on may encounter problems in clinical trials that may cause us or the FDA or foreign regulatory agencies to delay, suspend or terminate our clinical trials at any phase. These problems could include the possibility that we may not be able to manufacture sufficient quantities of materials for use in our clinical trials, conduct clinical trials at our preferred sites, enroll a sufficient number of patients for our clinical trials at one or more sites, or begin or successfully complete clinical trials in a timely fashion, if at all. Furthermore, we, the FDA or foreign regulatory agencies may suspend clinical trials of our product candidates at any time if we or they believe the subjects participating in the trials are being exposed to unacceptable health risks, whether as a result of adverse events occurring in our trials or otherwise, or if we or they find deficiencies in the clinical trial process or conduct of the investigation.
49
The FDA and foreign regulatory agencies could also require additional clinical trials before or after granting of marketing approval for any products, which would result in increased costs and significant delays in the development and commercialization of such products and could result in the withdrawal of such products from the market after obtaining marketing approval. Our failure to adequately demonstrate the safety and efficacy of a product candidate in clinical development could delay or prevent obtaining marketing approval of the product candidate and, after obtaining marketing approval, data from post-approval studies could result in the product being withdrawn from the market, either of which would likely have a material adverse effect on our business.
We rely on third parties to manufacture and supply our product candidates.
We do not currently own or operate manufacturing facilities for clinical or commercial production of our product candidates. We lack the resources and the capability to manufacture any of our product candidates on a clinical or commercial scale. We rely on four third-party suppliers to make the key components of EC145. The linker system for EC145 is currently obtained from a single source supplier and we have identified two alternate suppliers to prevent a possible disruption of manufacturing EC145. We believe that we currently have, or have the ability to access, sufficient supplies of all of the key components of EC145 in sufficient quantities to conduct and complete our PROCEED clinical trial and any other clinical trials related to EC145. We also believe that the suppliers we have identified will have the capacity to manufacture EC145 in the quantities that our development and future commercialization efforts may require. We do not have any long-term supply arrangements with any of these third parties and obtain our raw materials on a purchase order-basis. We expect to continue to depend on third-party contract manufacturers for the foreseeable future.
If for some reason our contract manufacturers cannot perform as agreed, we may be unable to replace them in a timely manner and the production of our product candidates would be interrupted, resulting in delays in clinical trials and additional costs. For example, we are currently obtaining clinical trial quantities of EC145 and our other product candidates from our contract manufacturers. We have no experience with managing the manufacturing of commercial quantities of any of our product candidates and scaling-up production to commercial quantities could take us significant time and result in significant costs, both of which could delay commercialization of EC145 for PROC, NSCLC or any other indication or of any of our other SMDCs. Switching manufacturers may be difficult because the number of potential manufacturers is limited and the FDA must approve any replacement manufacturer prior to manufacturing our product candidates. Such approval would require new testing and compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our product candidates after receipt of FDA approval to manufacture any of our product candidates. It may be difficult or impossible for us to find a replacement manufacturer on acceptable terms quickly, or at all.
To date, our product candidates have been manufactured in small quantities for preclinical studies and clinical trials by third-party manufacturers. If the FDA or other regulatory agencies approve any of our product candidates for commercial sale, we expect that we would continue to rely, at least initially, on third-party manufacturers to produce commercial quantities of our approved product candidates, as is the case with EC145. These manufacturers may not be able to successfully increase the manufacturing capacity for any of our approved product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. Additionally, any third-party manufacturer we retain to manufacture our product candidates on a commercial scale must pass an FDA pre-approval inspection for conformance to the cGMPs before we can obtain approval of our product candidates. If we are unable to successfully increase the manufacturing capacity for a product candidate in conformance with cGMPs, the regulatory approval or commercial launch of such products may be delayed or there may be a shortage in supply.
Our product candidates require precise, high quality manufacturing. Failure by our contract manufacturers to achieve and maintain high manufacturing standards could result in patient injury or death, product recalls or withdrawals, delays or failures in testing or delivery, cost overruns, or other problems that could seriously harm our business. Contract manufacturers may encounter difficulties involving production yields, quality control and
50
quality assurance. These manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state and non-U.S. authorities to ensure strict compliance with cGMP and other applicable government regulations and corresponding foreign standards; however, we do not have control over third-party manufacturers’ compliance with these regulations and standards.
We are subject to risks associated with the availability of key raw materials, such as technetium-99m, as well as drugs used in our clinical trials, such as Doxil.
Our EC20 companion imaging diagnostic requires the use of the radioisotope technetium-99m, or Tc-99m, and there have been historical periods in which supply was not able to satisfy demand. Tc-99m for nuclear medicine purposes is usually extracted from Tc-99m generators, which contain molybdenum-99, or Mo-99, as the usual parent nuclide for Tc-99m. The majority of Mo-99 produced for Tc-99m medical use comes from fission of highly enriched uranium from only five reactors around the world located in Canada, Belgium, South Africa, the Netherlands and France. Although Tc-99m is used in various nuclear medicine diagnostics utilized by healthcare providers, Tc-99m has a very short half-life (6 hours). As a result, healthcare providers extract Tc-99m from generators which use Mo-99. Mo-99 itself has a short half-life (2.75 days) and is sent to the nuclear medicine pharmacy directly from one of the five reactors. Accordingly, Tc-99m diagnostics are made on-site at the clinic, and neither Tc-99m nor Mo-99 can be inventoried. Sources of Tc-99m may be insufficient for our clinical trial site needs due to its limited supply globally. For example, global shortages of Tc-99m emerged in the past few years because aging nuclear reactors in the Netherlands and Canada that provided about two-thirds of the world’s supply of Mo-99 were shut down repeatedly for extended maintenance periods and two replacement Canadian reactors constructed in the 1990s were closed before beginning operation for safety reasons.
We use, and plan to continue to use, EC20 or other companion imaging diagnostics that employ Tc-99m in our clinical trials. For example, EC20 is a component of PROCEED and, in the future, if our clinical trial sites are not able to obtain sufficient quantities of Tc-99m for use in EC20, we may not be able to gather sufficient data on EC20 during PROCEED and as a result, the approval of EC20 may be delayed. In addition, to the extent the approval of our product candidates depends on the screening and monitoring of the patient population with a companion imaging diagnostic such as EC20 in our clinical trials, we would experience a corresponding delay in approval and commercialization of these SMDCs if we are not able to obtain sufficient Tc-99m.
In July 2011, the manufacturer of Doxil announced that supplies of the drug are limited, and later in the year we stopped enrollment in the PROCEED trial due to global shortages. We have purchased a sufficient quantity of Doxil to begin enrolling patients and have received approval from the FDA to import the drug into the U.S. for use in the PROCEED trial. Our current supply of Doxil is not sufficient to complete the PROCEED trial. Additional disruptions in the availability of Doxil could delay our PROCEED trial.
If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, we could be forced to pay substantial damage awards.
The use of any of our product candidates in clinical trials, and the sale of any approved products, may expose us to product liability claims. We currently maintain product liability insurance coverage in an amount which we believe is adequate for our clinical trials currently in progress and those recently completed. We monitor the amount of coverage we maintain as the size and design of our clinical trials evolve and intend to adjust the amount of coverage we maintain accordingly. However, we cannot assure you that such insurance coverage will fully protect us against some or all of the claims to which we might become subject. We might not be able to maintain adequate insurance coverage at a reasonable cost or in sufficient amounts or scope to protect us against potential losses. In the event a claim is brought against us, we might be required to pay legal and other expenses to defend the claim, as well as uncovered damages awards resulting from a claim brought successfully against us. Furthermore, whether or not we are ultimately successful in defending any such claims, we might be required to direct financial and managerial resources to such defense and adverse publicity could result, all of which could harm our business.
51
Each of our product candidates will remain subject to ongoing regulatory review even if it receives marketing approval. If we or our contractors fail to comply with continuing regulations, we or they may be subject to enforcement action that could adversely affect us.
Even if our product candidates become approved products, we and our contractors will continue to be subject to pervasive regulation by the FDA and other regulatory authorities. We and our contractors will continue to be subject to FDA requirements governing among other things the manufacture, packaging, sale, promotion adverse event reporting, storage and recordkeeping of our approved products. Although we have not received any notice that we are the subject of any FDA enforcement action, it is possible that we may be in the future and that could have a material adverse effect on our business. We may be slow to adapt, or may never adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements. If we or any of our contractors fail to comply with the requirements of the FDA and other applicable U.S. or foreign governmental or regulatory authorities or previously unknown problems with our products, manufacturers or manufacturing processes are discovered, we or the contractor could be subject to administrative or judicially imposed sanctions, including: restrictions on the products, the manufacturers or manufacturing processes we use, warning letters, civil or criminal penalties, fines, injunctions, product seizures or detentions, import bans, voluntary or mandatory product recalls and publicity requirements, suspension or withdrawal of regulatory approvals, total or partial suspension of production, and refusal to approve pending applications for marketing approval of new products to approved applications.
We deal with hazardous materials and must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our activities involve the controlled storage, use, and disposal of hazardous materials, including corrosive, explosive and flammable chemicals, biologic waste and various radioactive compounds. We are subject to federal, state, and local laws and regulations governing the use, manufacture, storage, handling, and disposal of these hazardous materials. Although we believe that our safety procedures for the handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials.
In the event of an accident, state or federal authorities may curtail our use of these materials, and we could be liable for any civil damages, which may exceed our financial resources and may seriously harm our business. We currently maintain insurance covering hazardous waste clean up costs in an amount we believe to be sufficient for typical risks regarding our handling of these materials, however, this amount of coverage may not be sufficient to cover extraordinary or unanticipated events. Additionally, an accident could damage, or force us to temporarily shut down, our operations.
Our ability to use net operating losses to offset future taxable income is subject to certain limitations.
Under Section 382 of the U.S. Internal Revenue Code, or Code, a corporation that experiences a more-than 50 percent ownership change over a three-year testing period is subject to limitations on its ability to utilize its pre-change NOLs to offset future taxable income. We have determined that we experienced a change in ownership as defined under Section 382 of the Code as a result of the public offering in August 2011. As a result, the future use of our net operating losses and credit equivalents will be limited to approximately $17.7 million per year. At December 31, 2011 we recorded a full valuation allowance against our net operating loss carryforwards of approximately $58.7 million, as we believe it is more likely than not they will not be fully realized.
Risks Related to Intellectual Property
Our proprietary rights may not adequately protect our technologies and product candidates.
Our commercial success will depend in part on our ability to obtain additional patents and protect our existing patent position as well as our ability to maintain adequate protection of other intellectual property for our
52
technologies, product candidates, and any future products in the United States and other countries. If we do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate any competitive advantage we may have, which could harm our business and ability to achieve profitability. The laws of some foreign countries do not protect our proprietary rights to the same extent or in the same manner as U.S. laws, and we may encounter significant problems in protecting and defending our proprietary rights in these countries. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies, product candidates and any future products are covered by valid and enforceable patents or are effectively maintained as trade secrets.
We apply for patents covering both our technologies and product candidates, as we deem appropriate. However, we may fail to apply for patents on important technologies or product candidates in a timely fashion, or at all. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products and technologies. We cannot be certain that our patent applications will be approved or that any patents issued will adequately protect our intellectual property. For example, our issued patents do not claim composition of matter protection for the drug payloads connected to the linker system and targeting ligand modules of our SMDCs. In addition, we generally do not control the patent prosecution of subject matter that we license from others, including those licensed from Purdue Research Foundation, a non-profit organization which manages the intellectual property of Purdue University. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we would over our own and would need to involve Purdue Research Foundation in legal proceedings to enforce these intellectual property rights. Moreover, the patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions for which important legal principles are often evolving and remain unresolved. As a result, the validity and enforceability of patents cannot be predicted with certainty. In addition, we do not know whether:
| • | we or our licensors were the first to make the inventions covered by each of our issued patents and pending patent applications; |
| • | we or our licensors were the first to file patent applications for these inventions; |
| • | any of our product candidates will be Orange Book eligible; |
| • | others will independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | any of our or our licensors’ pending patent applications will result in issued patents; |
| • | any of our or our licensors’ patents will be valid or enforceable; |
| • | any patents issued to us or our licensors and collaboration partners will provide us with any competitive advantages, or will be challenged by third parties; |
| • | we will develop additional proprietary technologies that are patentable; |
| • | the U.S. government will exercise any of its statutory rights to our intellectual property that was developed with government funding; or |
| • | our business may infringe the patents or other proprietary rights of others. |
The actual protection afforded by a patent varies based on products or processes, from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country, the validity and enforceability of the patents and our financial ability to enforce our patents and other intellectual property. Our ability to maintain and solidify our proprietary position for our products will depend on our success in obtaining effective claims and enforcing those claims once granted. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, narrowed, invalidated or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against
53
competitors with similar products. Due to the extensive amount of time required for the development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We also rely on trade secrets to protect some of our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to maintain. While we use reasonable efforts to protect our trade secrets, our or any of our collaboration partners’ employees, consultants, contractors or scientific and other advisors may unintentionally or willfully disclose our proprietary information to competitors and we may not have adequate remedies in respect of that disclosure. Enforcement of claims that a third party has illegally obtained and is using trade secrets is expensive, time consuming and uncertain. In addition, non-U.S. courts are sometimes less willing than U.S. courts to protect trade secrets. If our competitors independently develop equivalent knowledge, methods and know-how, we would not be able to assert our trade secrets against them and our business could be harmed.
The intellectual property protection for our product candidates is dependent on third parties.
With respect to patent applications relating to our product candidates that incorporate patents licensed from Purdue Research Foundation, the right and obligation to prosecute and maintain the patents and patent applications covered by these license agreements are retained by Purdue Research Foundation. Generally, we do not have the right to prosecute and maintain such patents in our territories, unless Purdue Research Foundation elects not to file, prosecute or maintain any or all of such patents. We would need to determine, with our other potential partners, who would be responsible for the prosecution of patents relating to any joint inventions. If any of our licensing partners fail to appropriately prosecute and maintain patent protection for any of our product candidates, our ability to develop and commercialize those product candidates may be adversely affected and we may not be able to prevent competitors from making, using and selling competing products.
If we breach any of the agreements under which we license commercialization rights to our product candidates or technology from third parties, we could lose license rights that are important to our business.
We license the use, development and commercialization rights for some of our product candidates, and we expect to enter into similar licenses in the future. For example, we licensed exclusive worldwide rights from Purdue Research Foundation, pursuant to a license agreement, which enables us to use and administer EC145 in the treatment of cancer. Under this license we are subject to commercialization and development, diligence obligations, sublicense revenue sharing requirements, royalty payments and other obligations. If we fail to comply with any of these obligations or otherwise breach this license agreement or any other current or future licenses, our licensing partners may have the right to terminate the license in whole or in part or to terminate the exclusive nature of the license. Generally, the loss of any of current or future licenses or the exclusivity rights provided therein could materially harm our financial condition and operating results.
The patent protection for our product candidates may expire before we are able to maximize their commercial value, which may subject us to increased competition and reduce or eliminate our opportunity to generate product revenue.
The patents for our product candidates have varying expiration dates and, if these patents expire, we may be subject to increased competition and we may not be able to recover our development costs or market any of our approved products profitably. For example, one of our U.S. patents claims compounds encompassing EC145 and is due to expire in 2026, and two of our other U.S. patents claim compounds encompassing EC20 and are due to expire in 2024. In some of the larger potential market territories, such as the United States and Europe, patent term extension or restoration may be available to compensate for time taken during aspects of the product’s development and regulatory review. However, we cannot be certain that such an extension will be granted, or if granted, what the applicable time period or the scope of patent protection afforded during any extension period will be. In addition, even though some regulatory authorities may provide some other exclusivity for a product
54
under their own laws and regulations, we may not be able to qualify the product or obtain the exclusive time period. If we are unable to obtain patent term extension/restoration or some other exclusivity, we could be subject to increased competition and our opportunity to establish or maintain product revenue could be substantially reduced or eliminated. Furthermore, we may not have sufficient time to recover our development costs prior to the expiration of our U.S. and non-U.S. patents.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
If we are sued for infringing intellectual property rights of third parties, litigation will be costly and time-consuming and could prevent us from developing or commercializing our product candidates.
Our commercial success depends, in part, on our not infringing the patents and proprietary rights of other parties and not breaching any collaboration or other agreements we have entered into with regard to our technologies and product candidates. Numerous third-party U.S. and non-U.S. issued patents and pending applications exist in the areas of targeted therapy and targeted diagnostics, including cytotoxic agents and other active compounds and formulations comprising such compounds.
Because patent applications can take several years to issue, if they are issued at all, there may currently be pending applications, unknown to us, that may result in issued patents that cover our technologies or product candidates. It is uncertain whether the issuance of any third-party patent would require us to alter our products or processes, obtain licenses or cease activities related to the development or commercialization of our product candidates. If we wish to use the technology or compound claimed in issued and unexpired patents owned by others, we may need to obtain a license from the owner, enter into litigation to challenge the validity of the patents or incur the risk of litigation in the event that the owner asserts that any of our product candidates infringe its patents. The failure to obtain a license to technology or the failure to challenge an issued patent that we may require to discover, develop or commercialize our products may have a material adverse impact on us.
There is a substantial amount of litigation involving intellectual property in the biopharmaceutical industry generally. If a third party asserts that our products or technologies infringe its patents or other proprietary rights, we could face a number of risks that could seriously harm our results of operations, financial condition and competitive position, including:
| • | infringement and other intellectual property claims, which would be costly and time-consuming to defend, whether or not the claims have merit, and which could delay the regulatory approval process and divert management’s attention from our business; |
| • | substantial damages for past infringement, which we may have to pay if a court determines that our product candidates or technologies infringe a competitor’s patent or other proprietary rights; |
55
| • | a court prohibiting us from selling or licensing our technologies or our product candidates unless the third-party licenses its patents or other proprietary rights to us on commercially reasonable terms, which it is not required to do; |
| • | if a license is available from a third party, we may have to pay substantial royalties or lump sum payments or grant cross-licenses to our patents or other proprietary rights to obtain that license; and |
| • | redesigning our products so they do not infringe, which may not be possible or may require substantial monetary expenditure and time. |
Although we are not currently a party to any legal proceedings relating to our intellectual property, in the future, third parties may file claims asserting that our technologies or products infringe on their intellectual property. We cannot predict whether third parties will assert these claims against us or against the current or future licensors of technology licensed to us, or whether those claims will harm our business. If we are forced to defend against these claims, whether they are with or without any merit, whether they are resolved in favor of or against us or our licensors, we may face costly litigation and diversion of management’s attention and resources. As a result of these disputes, we may have to develop costly non-infringing technology, or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, if at all, which could seriously harm our business or financial condition.
One or more third-party patents or patent applications may conflict with patent applications to which we have rights. Any such conflict may substantially reduce the coverage of any rights that may issue from the patent applications to which we have rights. If third parties file patent applications in technologies that also claim technology to which we have rights, we may have to participate in interference proceedings with the U.S. Patent and Trademark Office, or USPTO, or non-U.S. patent regulatory authorities, as applicable, to determine priority of invention.
We may become involved in lawsuits to enforce our patents or other intellectual property rights, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe our patents or other intellectual property rights. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming. To the extent such claims relate to patents held by the Purdue Research Foundation, it would have to file such an infringement lawsuit since we do not have the independent right to enforce the Purdue Research Foundation’s intellectual property. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings brought by the USPTO may be necessary to determine the priority of inventions with respect to our patents and patent applications or those of our current or future collaborators. An unfavorable outcome could require us to cease using the technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if a prevailing party does not offer us a license on terms that are acceptable to us. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distraction of our management and other employees. We may not be able to prevent, alone or with our collaborators, misappropriation of our proprietary rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential and proprietary information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
56
Risks Related to Ownership of Our Common Stock
The price of our common stock has been volatile and our shares may suffer a decline in value.
Since becoming a public company in February 2011, we have experienced volatility in the trading price of our common stock. Factors that could cause volatility in the market price of our common stock include, but are not limited to the risk factors identified above as well as:
| • | results from, supplemental analyses of and any delays in, our current or planned clinical trials, including PRECEDENT and PROCEED; |
| • | announcements of FDA non-approval of our product candidates, including EC145, or delays in FDA or other non-U.S. regulatory authority review processes; |
| • | FDA or other U.S. or non-U.S. regulatory actions affecting us or our industry; |
| • | litigation or public concern about the safety of our product candidates; |
| • | failure or discontinuation of any of our research or clinical trial programs; |
| • | delays in the commercialization of our product candidates; |
| • | our ability to effectively partner with collaborators to develop or sell our products; |
| • | market conditions in the pharmaceutical, biopharmaceutical and biotechnology sectors and issuance of new or changed securities analysts’ reports or recommendations; |
| • | actual and anticipated fluctuations in our quarterly operating results; |
| • | developments or disputes concerning our intellectual property or other proprietary rights; |
| • | introduction of technological innovations or new products by us or our competitors; |
| • | issues in manufacturing our product candidates; |
| • | market acceptance of our product candidates; |
| • | deviations in our operating results from the estimates of securities analysts; |
| • | coverage and reimbursement policies of governments and other third-party payors; |
| • | sales of our common stock by our officers, directors or significant stockholders; |
| • | price and volume fluctuations in the overall stock market from time to time; |
| • | general economic conditions and trends; |
| • | major catastrophic events; |
| • | our ability to expand our operations, domestically and internationally, and the amount and timing of expenditures related to this expansion; and |
| • | additions or departures of key personnel. |
In addition, the stock markets in general, and the markets for biopharmaceutical, pharmaceutical and biotechnology stocks in particular, have experienced extreme volatility that has been often unrelated to the operating performance of the issuer. These broad market fluctuations may adversely affect the trading price or liquidity of our common stock. In the past, when the market price of a stock has been volatile, holders of that stock have sometimes instituted securities class action litigation against the issuer. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the attention of our management would be diverted from the operation of our business, which could result in the delays of our clinical trials or commercialization efforts.
57
Sales of substantial amounts of our shares could adversely affect the market price of our common stock.
Sales of substantial amounts of our common stock in the public market, or the perception that these sales could occur, could cause the market price of our common stock to decline. These sales could also make it more difficult for us to raise capital by selling equity or equity-related securities in the future at a time and price that we deem appropriate.
As of December 31, 2011, we had 35,784,485 of our common stock outstanding. All of the outstanding shares are freely transferable without restriction under the Securities Act 1933, as amended, or the Securities Act, unless held by our “affiliates” as that term is used in Rule 144 promulgated under the Securities Act. Shares held by our affiliates may be sold in the public market pursuant to Rule 144 or another exemption from registration.
In addition, in the event that we propose to register any of our securities under the Securities Act, either for our own account or for the account of other securityholders, certain holders of more than one percent of the outstanding shares of our common stock and other registrable securities are entitled to notice of such registration and are entitled to include their common stock in such registration, subject to certain marketing and other limitations. The holders of at least 50 percent of these registrable securities have the right to require us, on not more than two occasions, to file a registration statement on Form S-1 under the Securities Act in order to register the resale of shares of their common stock, subject to our right, in certain circumstances, to defer such registrations. Further, these holders may require us to register the resale of all or a portion of their shares on a registration statement on Form S-3, subject to certain conditions and limitations. Finally, these holders have certain “piggyback” registration rights. If we propose to register any of our equity securities under the Securities Act, other than pursuant to the registration rights noted above or specified excluded registrations, which include the registration of the shares issued and issuable under our equity incentive plans, holders may require us to include all or a portion of their registrable securities in the registration and in any related underwritten offering. The market price of our common stock could decline if these securityholders exercise their registration rights or are otherwise perceived by the market as intending to sell their shares.
Our existing stockholders have substantial control of our management and affairs, which could limit your ability to influence the outcome of key transactions, including a change of control.
Our directors, executive officers and each of the three stockholders who own greater than five percent of our outstanding common stock and their affiliates, in the aggregate, beneficially own approximately 46.3 percent of the outstanding shares of our common stock. As a result, these stockholders, if acting together, could influence or control matters requiring approval by our stockholders, including the election of directors and the approval of mergers, acquisitions or other extraordinary transactions. This concentration of ownership may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company and might affect the market price of our common stock.
Provisions in our certificate of incorporation and bylaws and under Delaware law might discourage, delay or prevent a change of control of our company or changes in our management and, therefore, depress the trading price of our common stock.
Our certificate of incorporation and bylaws contain provisions that could depress the trading price of our common stock by acting to discourage, delay or prevent a change of control of our company or changes in our management that our stockholders may deem advantageous. These provisions include:
| • | establishing a classified board so that not all members of our Board of Directors are elected at one time; |
| • | authorizing “blank check” preferred stock that our Board of Directors could issue to increase the number of outstanding shares to discourage a takeover attempt; |
| • | eliminating the ability of stockholders to call a special stockholder meeting; |
58
| • | eliminating the ability of stockholders to act by written consent; |
| • | being subject to provisions of Section 203 of the Delaware General Corporate Law regulating corporate takeovers; |
| • | providing that our Board of Directors is expressly authorized to make, alter or repeal our bylaws; and |
| • | establishing advance notice requirements for nominations for elections to our Board of Directors or for proposing other matters that can be acted upon by stockholders at stockholder meetings. |
If we fail to establish and maintain proper internal controls, our ability to produce accurate financial statements or comply with applicable regulations could be impaired.
We will become subject to Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, beginning with our annual report for the year ending December 31, 2011. Section 404 requires management to assess and report annually on the effectiveness of internal control over financial reporting and identify any material weaknesses in internal control over financial reporting. Beginning with the year ending December 31, 2012, Section 404 of the Sarbanes-Oxley Act requires our independent registered public accounting firm to issue an attestation report as to management’s assessment of the effectiveness of internal control over financial reporting. We anticipate that compliance with Section 404 of the Sarbanes-Oxley Act will increase our legal, accounting and financial compliance costs, may make related activities more difficult, time-consuming and costly and may also place undue strain on our personnel, systems and resources.
If we conclude that our internal control over financial reporting is not effective, we cannot be certain as to the timing of completion of our evaluation, testing and remediation actions or their effect on our operations because there is presently no precedent available by which to measure compliance adequacy. As a consequence, we may not be able to remediate in time to meet our deadline for compliance with Section 404 of the Sarbanes-Oxley Act. Also, there can be no assurance that we will not identify one or more material weaknesses in our internal controls in connection with evaluating our compliance with Section 404 of the Sarbanes-Oxley Act. The presence of material weaknesses could result in financial statement errors which, in turn, could require us to restate our operating results.
If we are unable to conclude that we have effective internal control over financial reporting or if our independent auditors are unwilling or unable to provide us with an attestation report on the effectiveness of internal control over financial reporting as required by Section 404 of the Sarbanes-Oxley Act, investors may lose confidence in our operating results, our stock price could decline and we may be subject to litigation or regulatory enforcement actions. In addition, if we are unable to meet the requirements of Section 404 of the Sarbanes-Oxley Act, we may not be able to remain listed on The NASDAQ Global Market.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
Our offices are located in two primary leased facilities, a 15,700 square foot facility in West Lafayette, Indiana in the Purdue University Research Park and a 4,400 square foot corporate office space in Indianapolis, Indiana. The West Lafayette facility includes both administrative and research laboratory space. The lease for this facility expires December 31, 2012, and we do not foresee risk in obtaining an extension to this lease agreement for similar terms or having a disruption to our operations. The Indianapolis offices are used exclusively for corporate and administrative functions. The lease for this facility expires in November 2015. We believe the existing facilities are sufficient to meet our current and near-term needs.
59
| Item 3. | Legal Proceedings |
We are not currently a party to any material legal proceedings.
| Item 4. | Mine Safety Disclosures |
Not applicable.
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information and Stockholders
Shares of our common stock began trading on the The Nasdaq Global Market (symbol ECYT) on February 4, 2011. Prior to that time, there was no established public trading market for our common stock. The following table sets forth the high and low sale prices per share for our common stock on The Nasdaq Global Market for the periods indicated:
| High | Low | |||||||
| 2011 |
||||||||
| First Quarter (beginning February 4, 2011) |
$ | 9.33 | $ | 6.15 | ||||
| Second Quarter |
$ | 14.50 | $ | 8.25 | ||||
| Third Quarter |
$ | 14.80 | $ | 9.35 | ||||
| Fourth Quarter |
$ | 11.63 | $ | 3.02 | ||||
As of March 1, 2012, there were 64 stockholders of record of our common stock. The number of record holders is based upon the actual number of holders registered on the books of the company at such date and does not include holders of shares in “street name” or persons, partnerships, associations, corporations or other entities identified in security position listings maintained by depositories.
We have never declared or paid any dividends on our capital stock. We currently intend to retain all future earnings for the operation and expansion of our business and, therefore, we do not anticipate declaring or paying cash dividends for the foreseeable future. The payment of dividends will be at the discretion of our board of directors and will depend on our results of operations, capital requirements, financial condition, prospects, contractual arrangements, any limitations on payment of dividends present in our current and future debt agreements, and other factors that our Board of Directors may deem relevant. In addition, provisions contained in our current credit facility restrict our ability to pay dividends.
Unregistered Sales of Securities
During the period covered by this report, we did not offer or sell any securities in transactions except from registration under the Securities Act of 1933, as amended, or the Securities Act.
Issuer Purchases of Equity Securities
We did not purchase any of our equity securities during the period covered by this report.
Use of Proceeds from Registered Securities
Our initial public offering of common stock was effected through a Registration Statement on Form S-1 (File No. 333-166904) that was declared effective by the SEC on February 4, 2011. We registered an aggregate
60
of 14,375,000 shares of our common stock at an aggregate offering price of approximately $86.3 million. On February 9, 2011, we completed the sale of all 14,375,000 shares of our common stock at a price to the public of $6.00 per share, including the sale of 1,875,000 shares of common stock in connection with the underwriters’ exercise of their overallotment option, for aggregate gross proceeds of $86.3 million. The offering commenced on February 4, 2011 and terminated upon the sale of all of the registered securities in the offering. RBC Capital Markets, LLC and Leerink Swann LLC acted as joint book-running managers for the offering. Wedbush Securities, Inc. and Robert W. Baird & Co. acted as co-managers. There were no selling stockholders in the offering.
We paid $5.5 million in underwriting discounts and commissions to the underwriters in connection with the offering and incurred additional costs of approximately $2.6 million in connection with the offering, which when added to the underwriting discounts and commissions paid, amounts to total expenses of approximately $8.1 million. Thus, the net offering proceeds to us, after deducting underwriting discounts and offering expenses, were approximately $78.2 million. No offering expenses were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning ten percent or more of any class of our equity securities or to any other affiliates.
As of December 31, 2011, we have used approximately $40.1 million of the net proceeds from the initial public offering to fund the phase 3 clinical trial of EC145 and EC20, preparing the applications for conditional marketing authorization from the EMA, preparing for the randomized phase 2 trial of EC145 and EC20 in NSCLC and for working capital expenditures and other general corporate purposes. We have invested the unused proceeds from the offering in short-term interest-bearing, investment grade securities. There has been no material change in our planned use of proceeds from the initial offering from that described in the final prospectus filed with the SEC pursuant to Rule 424(b) on February 9, 2011.
Comparative Stock Performance Graph
The information included under the heading “Comparative Stock Performance Graph” in this Item 5 of Part II of this Annual Report on Form 10-K shall not be deemed to be “soliciting material” or subject to Regulation 14A or 14C, shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, or otherwise subject to the liabilities of that section, nor shall it be deemed incorporated by reference in any filing under the Securities Act or the Exchange Act.
61
Set forth below is a graph comparing the total cumulative returns of ECYT, the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph assumes $100 was invested on February 4, 2011 in our common stock and each of the indices and that all dividends, if any, are reinvested.
| 2/4/11 | 12/31/11 | |||||||
| Endocyte, Inc. |
$ | 100.00 | $ | 48.64 | ||||
| NASDAQ Composite Index |
$ | 100.00 | $ | 94.08 | ||||
| NASDAQ Biotechnology Index |
$ | 100.00 | $ | 110.71 | ||||
| Item 6. | Selected Financial Data |
We have derived the selected statement of operations data for the year ended December 31, 2009, 2010 and 2011 and selected balance sheet data as of December 31, 2010 and 2011 from our audited financial statements and related notes included in this annual report. We have derived the statement of operations data for the year ended December 31, 2007 and 2008 and the balance sheet data as of December 31, 2007, 2008 and 2009 from our audited financial statements not included in this annual report. Our historical results are not necessarily indicative of the results to be expected for any future period. The following selected financial data should be read
62
in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and related notes included elsewhere in this annual report.
| Year Ended December 31, | ||||||||||||||||||||
| 2007 | 2008 | 2009 | 2010 | 2011 | ||||||||||||||||
| (In thousands, Except Per Share Amounts) | ||||||||||||||||||||
| Statement of operations data: |
||||||||||||||||||||
| Total revenue |
$ | 1,082 | $ | 500 | $ | 3,000 | $ | — | $ | 191 | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
11,305 | 13,323 | 14,804 | 14,561 | 28,828 | |||||||||||||||
| General and administrative |
4,401 | 4,786 | 3,934 | 6,039 | 10,000 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
15,706 | 18,109 | 18,738 | 20,600 | 38,828 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(14,624 | ) | (17,609 | ) | (15,738 | ) | (20,600 | ) | (38,637 | ) | ||||||||||
| Other income (expense): |
||||||||||||||||||||
| Interest income |
1,297 | 682 | 49 | 8 | 129 | |||||||||||||||
| Interest expense |
(25 | ) | (1,579 | ) | (1,436 | ) | (1,065 | ) | (1,988 | ) | ||||||||||
| Other income (expense) |
(306 | ) | 13 | 119 | 1,564 | (36 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total other income (expense) |
966 | (884 | ) | (1,268 | ) | 507 | (1,895 | ) | ||||||||||||
| Loss before income taxes |
(13,658 | ) | (18,493 | ) | (17,006 | ) | (20,093 | ) | (40,532 | ) | ||||||||||
| Income tax (benefit) expense |
— | — | — | — | — | |||||||||||||||
| Net loss |
$ | (13,658 | ) | $ | (18,493 | ) | $ | (17,006 | ) | $ | (20,093 | ) | $ | (40,532 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted loss per share(1) |
$ | (15.76 | ) | $ | (20.54 | ) | $ | (18.67 | ) | $ | (21.77 | ) | $ | (1.40 | ) | |||||
| Shares used in computation of basic and diluted loss per share(1)(2) |
866,517 | 900,127 | 911,066 | 923,007 | 29,003,991 | |||||||||||||||
| (1) | On January 10, 2011, we effected a 1.00 for 1.91 reverse stock split. All historical common stock and per share information has been changed to reflect the stock split. |
| (2) | Diluted weighted average common shares outstanding are identical to basic weighted average common shares outstanding and diluted earnings per share is identical to basic earnings per share because common share equivalents are excluded from the calculations of diluted weighted average common shares outstanding for those quarters, as their effect is anti-dilutive. |
| As of December 31, | ||||||||||||||||||||
| 2007 | 2008 | 2009 | 2010 | 2011 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Balance sheet data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 31,735 | $ | 18,383 | $ | 23,909 | $ | 16,873 | $ | 128,085 | ||||||||||
| Working capital |
29,737 | 11,486 | 15,476 | 12,377 | 124,286 | |||||||||||||||
| Total assets |
34,354 | 20,188 | 25,268 | 21,214 | 131,675 | |||||||||||||||
| Total debt |
9,952 | 14,384 | 8,977 | 14,804 | 12,833 | |||||||||||||||
| Subordinated notes(1) |
— | — | — | 9,529 | — | |||||||||||||||
| Total stockholders’ equity (deficit) |
(41,318 | ) | (59,412 | ) | (76,058 | ) | (96,911 | ) | 113,372 | |||||||||||
| (1) | As of December 31, 2010, our subordinated notes were treated as share-settled debt under ASC 480-10-25-14 and were recorded at fair value. The subordinated notes accrued interest in kind at an annual rate of 10.0 percent. Subsequent to December 31, 2010, we issued an addition $3.7 million of subordinated notes. All of the subordinated notes plus accrued and unpaid interest thereon were automatically converted into 2,335,823 shares of common stock upon the completion of our initial public offering. |
63
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes included in this annual report. Please note that the Company effected a 1.00 for 1.91 stock split on January 10, 2011, and all historical common stock and per share information has been changed to reflect the stock split.
Overview
We are a biopharmaceutical company developing targeted therapies for the treatment of cancer and inflammatory diseases. We use our proprietary technology to create novel small molecule drug conjugates, or SMDCs, and companion imaging diagnostics. Our SMDCs actively target receptors that are over-expressed on diseased cells, relative to healthy cells. This targeted approach is designed to enable the treatment of patients with highly active drugs at greater doses, delivered more frequently, and over longer periods of time than would be possible with the untargeted drug alone. We are also developing companion imaging diagnostics for each of our SMDCs that are designed to identify the patients whose disease over-expresses the target of the therapy and who are therefore more likely to benefit from treatment. This combination of an SMDC with its companion imaging diagnostic is designed to personalize the treatment of patients by delivering effective therapy, selectively to diseased cells, in the patients most likely to benefit.
Our lead SMDC candidate, EC145, targets the folate receptor, which is frequently over-expressed on cancer cells. We have chosen platinum-resistant ovarian cancer, or PROC, a highly treatment-resistant disease, as our lead indication for development of EC145 because of the high unmet need in treating this patient population and the high percentage of ovarian cancer patients whose tumors over-express the targeted folate receptor. We conducted a multicenter, open-label randomized phase 2 clinical trial of EC145 in 149 women with PROC, referred to as the PRECEDENT trial. Based upon our findings from the PRECEDENT trial, we initiated enrollment of our PROCEED trial, a phase 3 registration trial in women with PROC, in the first half of 2011 but enrollment stopped later in the year due to global shortages of pegylated liposomal doxorubicin, or PLD (marketed in the U.S. under the brand name Doxil). In March 2012, we announced that the U.S. Food and Drug Administration, or FDA approved our plans to import our supply of PLD from Europe into the U.S. so that we can resume PROCEED enrollment in the U.S. In 2012, we will be increasing the amount of time and resources, both financial and personnel, devoted to our EC145 program in PROC.
We are currently preparing to file marketing authorization applications to the European Medicines Agency, or EMA, for EC145 for the treatment of PROC and for EC20 for patient selection in the third quarter of 2012. These filings will be based on the results and supplemental analyses of our PRECEDENT trial. Our filings will be supported by four clinical studies: a Phase 1 study in solid tumors, two single agent, single-arm Phase 2 studies in ovarian cancer and non-small cell lung cancer (NSCLC), and the PRECEDENT trial, a randomized study in PROC. The results of the PRECEDENT trial demonstrated a statistically significant delay in disease progression or death in the overall population, with the largest improvement observed in the FR(++) patient population, those with all tumors positive for the folate receptor. Women with FR(++) platinum resistant ovarian cancer who received EC145-based therapy experienced a 62 percent decrease in their risk of progression [HR 0.381, p= 0.018] compared to women receiving chemotherapy alone. Median progression free survival (PFS; the time without disease progressing) in the EC145 based treatment arm was 5.5 months compared to 1.5 months of women who received chemotherapy alone. Tumor shrinkage (overall response rate) was observed in 17.3 percent of women receiving the EC145-based therapy compared to 6.7 percent in patients treated with chemotherapy alone.
In addition to PROC, we are pursuing clinical trials of EC145 in other indications and we also plan to advance other SMDCs and companion imaging diagnostics through development as preclinical and clinical trial results merit and funding permits. We plan to begin enrollment in a randomized phase 2 trial of EC145 and EC20 for the treatment of second line NSCLC in the second quarter of 2012.
On February 9, 2011, we sold 14,375,000 shares of common stock, including 1,875,000 shares of common stock pursuant to the exercise of the over-allotment option by the underwriters in our initial public offering.
64
Proceeds, net of underwriting discounts, commissions and other transaction costs, were approximately $78.2 million. Upon the completion of the offering, all of our outstanding Subordinated Convertible Promissory Notes, or Subordinated Notes, plus accrued and unpaid interest thereon were automatically converted into 2,335,823 shares of common stock using a conversion price of $5.10 per share (85% of the original issue price of the shares sold in the initial public offering), all of our outstanding preferred stock was converted into an aggregate 11,747,563 shares of common stock, and all of the outstanding warrants to purchase our preferred stock were converted into warrants to purchase common stock.
On August 2, 2011, we sold 4,968,321 shares of common stock in a public offering, including 871,489 shares of common stock pursuant to the exercise of the underwriters’ over-allotment. Proceeds, net of underwriting discounts, commissions and other transaction costs, were approximately $66.7 million.
Financial Operations Overview
We have never been profitable and have incurred significant net losses since our inception. As of December 31, 2011, we had a retained deficit of $138.6 million. We expect to continue to incur significant and increasing operating losses for the next several years as we pursue the advancement of our SMDCs and companion imaging diagnostics through the research, development, regulatory and commercialization processes. We expect that our current cash position, including cash equivalents and short term investments are sufficient to fund our operations through the end of the first quarter of 2014. Our current operating plan includes completion of PROCEED, the phase 3 clinical trial of EC145 and EC20, through the availability of final primary PFS data from that study which is anticipated to be in the first half of 2014, assuming the availability of PLD, the preparation of applications to the EMA for conditional marketing authorization for EC145 and EC20, pre-commercial activities in Europe, a randomized phase 2 trial of EC145 and EC20 for the treatment of second line NSCLC and to advance our earlier stage phase 1 and preclinical pipeline. If we were to receive conditional marketing approval in Europe of EC145 and EC20 prior to the completion of the PROCEED study, this could impact the enrollment timeline as European sites would transition from clinical trials to commercial use. This could delay the availability of final data from the PROCEED trial. We may be able to mitigate this potential delay by adding clinical trial sites in locations where conditional marketing approval has not been granted. If the FDA requires us to undertake a second phase 3 clinical trial or obtain final OS data from PROCEED or we initiate significant investments in commercial capabilities, we will require additional financing through public or private equity or debt financings or other sources, such as strategic partnerships or licensing arrangements, to fund the additional activities. Such funding may not be available on favorable terms, or at all. Our failure to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue our business strategies.
The PROCEED trial was initiated in May 2011 but enrollment stopped later in the year due to global shortages of Doxil. We have purchased a sufficient quantity of Doxil and have received approvel from the FDA to import the drug into the U.S. for use in the PROCEED trial. We have plans to resume enrollment in the U.S. If the supplies of Doxil remain limited for an extended period of time, our PROCEED trial may be significantly delayed.
Revenue
To date, we have generated no revenue from sales of our SMDCs or companion imaging diagnostics. All of our revenue has been derived from license fees, milestone payments and government grants.
In the future, we may generate revenue from a combination of direct sales of our SMDCs and companion imaging diagnostics, license fees, milestone payments and royalties in connection with strategic collaborations. We expect that any revenue we generate will fluctuate from quarter to quarter as a result of the timing and amount of license fees, achievement of performance-based milestones and other payments received under such collaborations, and the amount and timing of payments that we receive upon the sale of our SMDCs and companion imaging diagnostics, to the extent any are successfully commercialized. Based upon our SMDCs and companion imaging diagnostics currently in development and the stage of development, we do not expect to
65
generate revenue from product sales until 2013 at the earliest. If we or our strategic partners fail to complete the development of our SMDCs and companion imaging diagnostics in a timely manner or obtain regulatory approval for them, our ability to generate future revenue, and our results of operations and financial position, would be materially adversely affected.
Research and Development Expenses
Research and development expenses consist of expenses incurred in connection with the discovery and development of our SMDCs and companion imaging diagnostics, including:
| • | employee-related expenses, which include salaries and stock-based compensation expense; |
| • | expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a portion of our preclinical studies; |
| • | the cost of acquiring and manufacturing clinical trial materials; |
| • | license fees for and milestone payments related to in-licensed products and technology; |
| • | costs associated with non-clinical activities and regulatory approvals; and |
| • | research supplies. |
We expense research and development costs as incurred. License fees and milestone payments related to in-licensed products and technology and research supplies are expensed if it is determined that they have no alternative future use.
Conducting a significant amount of research and development is central to our business model. Our SMDCs and companion imaging diagnostics in later stages of clinical development generally have higher development costs than those in earlier stages of development, primarily due to the increased size and duration of late stage clinical trials. We plan to increase our research and development expenses for the foreseeable future due to the PROCEED trial and the Phase 2 trial for second line NSCLC for our most advanced SMDC, EC145, and to further advance our earlier-stage research and development projects.
Our internal resources, employees and infrastructure are not directly tied to any individual research project and are typically deployed across multiple projects. Through our clinical development programs, we are advancing our SMDCs and companion imaging diagnostics in parallel for multiple therapeutic indications and through our preclinical development programs we are seeking to develop potential SMDCs and companion imaging diagnostics for additional disease indications. The following table sets forth costs incurred on a program-specific basis for identified lead candidate SMDCs and companion imaging diagnostics, excluding personnel-related costs. Discovery Research includes such costs for projects where no lead candidate has yet been identified. All employee-related expenses for those employees working in research and development functions are included in Research and Development Payroll.
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| EC145 |
$ | 6,495 | $ | 6,844 | $ | 17,786 | ||||||
| EC20 |
298 | 322 | 2,259 | |||||||||
| EC0489 |
753 | 883 | 439 | |||||||||
| EC0225 |
643 | 418 | — | |||||||||
| EC17 |
19 | — | — | |||||||||
| Discovery Research |
1,285 | 1,318 | 1,723 | |||||||||
| Research and Development Payroll |
5,311 | 4,776 | 6,621 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total Research and Development Expenses |
$ | 14,804 | $ | 14,561 | $ | 28,828 | ||||||
|
|
|
|
|
|
|
|||||||
66
The following table identifies the current status of our major research and development projects and our currently expected near-term milestone timing:
| Project |
Status | Expected Near-Term Milestones | ||
| EC145 |
Phase 2 | Initiating re-start of phase 3 trial | ||
| EC20 |
Phase 2 | Initiating re-start of phase 3 trial | ||
| EC0489 |
Phase 1 | Completed phase 1 trial in 2011 | ||
| EC0225 |
Phase 1 | Evaluating future development options | ||
| EC17 |
Phase 2 | Evaluating future development options |
General and Administrative Expenses
General and administrative expenses consist principally of salaries and stock-based compensation for personnel in executive, finance, business development, legal and human resources functions. Other general and administrative expenses include employee benefits, facility, patent filing and prosecution costs, and professional service fees.
We anticipate that our general and administrative expenses will increase in the future primarily for the following reasons:
| • | increased payroll and expanded infrastructure as a result of more advanced development activity and potential preparation for commercial operations; |
| • | increased expenses related to becoming a public company, including increased legal, accounting and investor relations fees, higher director compensation and increased insurance premiums; and |
| • | expenses related to the sales and marketing of our SMDCs and companion imaging diagnostics in anticipation of commercial launch before we receive regulatory approval. |
Other Income
Other income consists primarily of gains or losses on sales of equipment, amounts related to the change in the fair value of our preferred stock warrant liability and income for a tax credit award.
Interest Income and Interest Expense
Interest income consists of interest earned on our cash, cash equivalents and short-term investments. The primary objective of our investment policy is capital preservation. Interest expense consists primarily of interest, amortization of debt discount and amortization of deferred financing costs associated with our previous credit facility with General Electric Capital Corporation, or GECC, and Oxford Finance Corporation, or Oxford and our current facility with Mid Cap Financial, or Mid-Cap and Silicon Valley Bank, or SVB.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to accrued expenses and stock-based compensation. We base our estimates on historical experience, known trends and events and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
67
Our significant accounting policies are described in more detail in Note 2 of the notes to our financial statements appearing elsewhere in this annual report. We believe the following accounting policies to be most critical to the judgments and estimates used in preparation of our financial statements and have been reviewed and discussed with our audit committee.
Revenue Recognition
To date, our revenues have been generated primarily through collaborative research, development and commercialization agreements. The terms of these collaborative agreements typically include payments to us of one or more of the following: nonrefundable, upfront license fees, milestone payments and royalties on future product sales.
When evaluating multiple element arrangements, we consider whether the components of the arrangement represent separate units of accounting. This evaluation requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
We typically have received upfront, nonrefundable payments when licensing our intellectual property in conjunction with a research and development agreement. Upfront payments, if they are nonrefundable and not contingent on further performance by us, are recognized when due pursuant to the terms of the underlying contract or when cash is received, if collectability is uncertain.
Our licensing agreements may also contain milestone payments. Revenues from milestones are recognized upon the achievement of the milestone event if the event is substantive, objectively determinable and represents an important point in the development life cycle of the SMDC. If not considered substantive, milestones are initially deferred and recognized over the remaining performance obligation.
We have not received any royalty revenues related to SMDC or companion imaging diagnostic sales to date.
Grant revenue is recognized when earned, which is in the period in which qualifying expenditures are incurred.
Accrued Clinical Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued expenses. This process involves reviewing open contracts, identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We estimate our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Expense accruals related to clinical trial activity typically comprise the majority of these accruals. Examples of estimated expenses related to clinical trial activity include:
| • | fees paid to contract research organizations in connection with clinical trials; |
| • | fees paid to investigative sites in connection with clinical trials; and |
| • | fees paid to vendors in connection with preclinical development activities. |
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and contract research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under certain contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services are performed and the level of effort to be expended in
68
each period. If the actual timing of the performance of services or the level of the effort varies from our estimate, we adjust the accrual accordingly. Although our estimates in the past have not been materially different from amounts actually incurred, and we do not expect our estimates to be materially different from amounts actually incurred in the future, if our estimate of the status and timing of services performed differs from the actual status and timing of services performed we may report amounts that are too high or too low in any particular period. Based on our level of clinical trial expenses for the twelve months ended December 31, 2011, a five percent change in our estimate could result in an adjustment to our accrued clinical trial expense in future periods of approximately $10,000.
Stock-Based Compensation
Effective January 1, 2006, we adopted the recognition provisions of Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Compensation — Stock Compensation, which we refer to as ASC 718, using the prospective transition method. Generally, options fully vest over four years from the grant date and have a term of ten years. Options granted in connection with an employee’s commencement of employment generally vest over a four-year period with half of the shares subject to the grant vesting after two years of employment and remaining options vesting monthly over the remainder of the four-year period. Options granted for performance or promotions vest monthly over a four-year period. Unexercised stock options terminate on the tenth anniversary date after the date of grant. The Company recognizes the stock-based compensation expense over the requisite service period of the individual grantees, which generally equals the vesting period. In connection with the adoption of ASC 718, the Company reassessed the valuation methodology for stock options and the related input assumptions. As a result, beginning with the 2006 stock option grant, the Company utilizes a Black-Scholes option-pricing model to estimate the value of stock options. The Black-Scholes model allows the use of a range of assumptions related to volatility, risk-free interest rate, and employee exercise behavior. Prior to the initial public offering, the expected volatility was derived from the historical volatility of the NASDAQ Biotechnology Index, which the Company determined was a proxy for its volatility. Since the Company has elected to use an index to determine the value of its stock options, it is applying the calculated value approach for a private company instead of using fair value. Since August 2010, all grants have been valued under the fair value provisions of ASC 718. Since the Company completed its initial public offering in February 2011, it does not have sufficient history as a publicly traded company to evaluate volatility. As a result, the Company has used an average of several peer companies’ volatilities to determine a reasonable estimate of volatility. For purposes of identifying similar entities, the Company considered characteristics such as industry, length of trading history, market capitalization and similar product pipelines. The risk-free interest rate is derived from the weighted-average yield of a Treasury security with the same term as the expected life of the options and the dividend yield is based on historical experience and the Company’s estimate of future dividend yields. Prior to February 2011, the expected life of the options was based on the weighted-average life of the Company’s historical option grants, and the dividend yield is based on historical experience and the Company’s estimate of future dividend yields. Subsequent to February 2011 the initial public offering, due to the lack of available quarterly data for the peer companies and insufficient history as a public company, the Company elected to use the “simplified” method for “plain vanilla” options to estimate the expected term of the stock options grants. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option.
The value of our stock options was estimated at the grant date using the following assumptions:
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Volatility |
35.59 | % | 36.26 | % | 82.77 | % | ||||||
| Expected Term (in years) |
10.0 | 9.8 | 6.2 | |||||||||
| Risk-Free Interest Rates |
3.67 | % | 3.52 | % | 2.27 | % | ||||||
| Dividend Yield |
0.00 | % | 0.00 | % | 0.00 | % | ||||||
69
In accordance with ASC 718, we recognized stock-based compensation expense of approximately $382,000, $524,000 and $1,960,000 for the year ended December 31, 2009, 2010 and 2011, respectively. As of December 31, 2011, we had $5.2 million in total unrecognized compensation expense, net of related forfeiture estimates, which we expect to recognize over a weighted-average period of approximately 1.6 years.
In May 2011, the Company adopted and granted awards under a new performance-based restricted stock unit (“RSU”) program (the “2011 RSU Program”) under the Company’s 2010 Equity Incentive Plan. Each unit represents an amount equal to one share of the Company’s common stock. The RSUs will be earned, in whole or in part, based on performance and service conditions. The performance condition is based upon whether the Company receives regulatory approval to sell a therapeutic product, and the awards include a target number of RSUs that will vest upon a First Commercial Approval, and a maximum number of RSUs that will vest upon a Second Commercial Approval. The RSUs will vest fifty percent based on the performance condition of commercial approval and fifty percent one year thereafter to fulfill the service condition, which requires the employee to remain employed by the company.
As of December 31, 2011, the Company has 271,213 RSUs outstanding. The unrecorded stock compensation expense is based on number of units granted, less estimated forfeitures based on the Company’s historical forfeiture rate of 6.49%, and the closing market price of the Company’s common stock at the grant date. As of December 31, 2011, the performance condition of obtaining regulatory approval has not been achieved, therefore, no vesting has occurred. The awards are being accounted for under ASC 718, and compensation expense is to be recorded if the Company has determined that it is probable that the performance conditions will be achieved. As of December 31, 2011, it was not probable that the performance conditions will be achieved, therefore, no compensation expense was recorded for the year ended December 31, 2011. Unrecorded compensation expense for the 2011 RSU program as of December 31, 2011 was $2.7 million.
Results of Operations
Comparison of Year Ended December 31, 2010 and 2011
| Year Ended December 31, |
Increase/ (Decrease) |
% | ||||||||||||||
| 2010 | 2011 | |||||||||||||||
| (In thousands) | ||||||||||||||||
| Statement of operations data: |
||||||||||||||||
| Revenue |
$ | — | $ | 191 | $ | 191 | 100 | % | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
14,561 | 28,828 | 14,267 | 98 | % | |||||||||||
| General and administrative |
6,039 | 10,000 | 3,961 | 66 | % | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
20,600 | 38,828 | 18,228 | 88 | % | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(20,600 | ) | (38,637 | ) | 18,037 | 88 | % | |||||||||
| Interest income |
8 | 129 | 121 | 1,513 | % | |||||||||||
| Interest expense |
(1,065 | ) | (1,988 | ) | 923 | 87 | % | |||||||||
| Other income (expense), net |
1,564 | (36 | ) | (1,600 | ) | (102 | )% | |||||||||
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (20,093 | ) | $ | (40,532 | ) | $ | 20,439 | 102 | % | ||||||
|
|
|
|
|
|
|
|||||||||||
Revenue
For the year ended December 31, 2011, we earned revenue from a licensing agreement with On Target Laboratories, L.L.C., or On Target, which was entered into in September 2011. Under such license agreement, we granted On Target exclusive worldwide licenses to develop and commercialize products relating to the
70
compound comprising Endocyte’s Folate and DUPA ligands and other certain Licensed Patents. On Target is solely responsible for conducting research and development, seeking regulatory approval and commercialization of products. The Company received nonrefundable upfront license fees totaling $191,000 from On Target in December 2011 and the amount was recognized as collaboration revenue in the same period. On Target is considered a related party as the company was founded by a relative of the Company’s Chief Scientific Officer. The Company believes that the terms of the agreement are no less favorable than terms available in an arm’s length transaction.
Research and Development
The increase in research and development expense in 2011 compared to 2010 was attributable to a $11.9 million increase in clinical trial and product expenses principally due to the PROCEED trial, and a $4.0 million increase in manufacturing and regulatory expenses related to the anticipated regulatory filing with the EMA for EC145 and EC20, including process and method validations for EC145 and EC20. The increase also included a $1.8 million increase in compensation expense due to $347,000 of severance compensation charges and increased bonus accrual and stock-based compensation expense in 2011.
Included in research and development expense were stock-based compensation charges of $402,000 and $1,077,000 for the years ended December 31, 2010 and 2011, respectively. The compensation charge for year ended December 31, 2011 included a $133,000 charge relating to severance stock-based compensation.
Research and development expenses include expense of $391,000 and $518,000 for the years ended 2010 and 2011, respectively, for company-funded research at Purdue University, the primary employer of our Chief Science Officer.
General and Administrative
The increase in general and administrative expenses in 2011 compared to 2010 was primarily attributable to an increase in professional fees and insurance associated with being a public company and compensation expenses.
Included in general administrative expenses were stock-based compensation charges of $122,000 and $883,000 for the years ended 2010 and 2011, respectively.
Interest Income
The increase in interest income in 2011 compared to 2010 resulted from an increase in short-term investments due to the proceeds from the public stock offerings completed in 2011.
Interest Expense
The increase in interest expense in 2011 compared to 2010 was due to increased borrowings under the loan agreement with Mid-Cap and SVB and the interest payable on outstanding subordinated notes which were converted to common shares at the initial public offering.
Other Income, Net
Other income decreased in 2011 compared to 2010 due to receiving an award of $1.5 million under section 48D of the Code for Qualifying Therapeutic Discovery Projects in 2010. The other expense in 2011 related primarily to charitable contributions and foreign currency losses.
71
Comparison of Year Ended December 31, 2009 and 2010
| Year
Ended December 31, |
Increase/ (Decrease) |
|||||||||||||||
| 2009 | 2010 | % | ||||||||||||||
| (In thousands) | ||||||||||||||||
| Statement of operations data: |
||||||||||||||||
| Revenue |
$ | 3,000 | $ | — | $ | (3,000 | ) | (100 | )% | |||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
14,804 | 14,561 | (243 | ) | (2 | )% | ||||||||||
| General and administrative |
3,934 | 6,039 | 2,105 | 54 | % | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
18,738 | 20,600 | 1,862 | 9 | % | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(15,738 | ) | (20,600 | ) | 4,862 | 30 | % | |||||||||
| Interest income |
49 | 8 | (41 | ) | (84 | )% | ||||||||||
| Interest expense |
(1,436 | ) | (1,065 | ) | (371 | ) | (26 | )% | ||||||||
| Other income, net |
119 | 1,564 | 1,445 | (1,214 | )% | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (17,006 | ) | $ | (20,093 | ) | $ | 3,087 | 18 | % | ||||||
|
|
|
|
|
|
|
|||||||||||
Revenue
For the year ended December 31, 2009, we earned revenue from a licensing agreement with Bristol-Myers Squibb, or BMS, which was entered into on December 22, 2005. Under such license agreement, we granted BMS an exclusive worldwide license to develop and commercialize SMDCs using folate as the targeting ligand and epothilone as the drug payload. In 2009, BMS advanced this folate-epothilone SMDC, which BMS called BMS-753453, into a phase 2 clinical trial, which triggered a $3.0 million milestone payment under the license. On June 3, 2010, BMS notified us of their intent to terminate the license. We believe BMS elected to terminate the license as a result of a change in its strategic focus. We received an aggregate of $8.1 million from BMS from the fourth quarter 2005 to the third quarter of 2009 pursuant to this license.
Research and Development
The decrease in research and development expense in 2010 compared to 2009 was primarily the result of a decrease in payroll expense due to a shift of management focus from research and development activities to general and administrative functions during 2010. These decreases were partially offset by increased manufacturing costs incurred as preparation for the EC 145 Phase 3 clinical trial.
Included in research and development expense were stock-based compensation charges of $332,000 and $402,000 for the years ended December 31, 2009 and 2010, respectively.
Research and development expenses include expense of $352,000 and $391,000 for the years ended 2009 and 2010, respectively, for company-funded research at Purdue University, the primary employer of our Chief Science Officer.
General and Administrative
The increase in general and administrative expenses in 2010 compared to 2009 was primarily attributable to increases in patent prosecution expenses associated with the growing portfolio and professional fees associated with various partnering and financing activities, including the preparation for our initial public offering.
Included in general administrative expenses were stock-based compensation charges of $50,000 and $122,000 for the years ended 2009 and 2010, respectively.
72
Interest Income
The decrease in interest income in 2010 compared to 2009 resulted from a decrease in balances available for investment due to the use of these funds for operations.
Interest Expense
The decrease in interest expense in 2010 compared to 2009 was due to the decrease in the outstanding and unpaid principal under our previous credit facility with GECC and Oxford, which was repaid in August 2010, partially offset by the outstanding and unpaid principal and interest incurred under our new credit facility.
Other Income, Net
Other income increased in 2010 compared to 2009 primarily due to receiving an award of $1.5 million under section 48D of the Code for Qualifying Therapeutic Discovery Projects. Other income, net in 2010 also includes a loss on extinguishment of debt of $144,000, which was offset by an increase in income of $219,000 related to the change in the fair value of our preferred stock warrant liability.
Liquidity and Capital Resources
We have funded our operations principally through private placements of equity and debt securities, revenue from strategic collaborations, revenue from grants, loans, private equity and debt financings and, most recently, the two public offerings of common stock we completed in 2011. As of December 31, 2011, we had cash, cash equivalents and short-term investments of $128.1 million.
In August 2010, we entered into a $15.0 million loan agreement with Mid-Cap and SVB to pay-off an existing loan that was set to mature in 2011. Upon execution of the agreement, we drew $10.0 million in principal. In December 2010, we amended the loan agreement in order to access the remaining tranche of $5.0 million. In September 2011, we modified the loan agreement to revise the repayment terms. As revised, we are required to make interest-only monthly payments through December 2012 followed by 36 months of interest and principal payments. This modification will provide us with additional working capital of $7.4 million over the 15 month period of interest-only payments.
On October 29, 2010, we were notified we had been awarded a total of $1.5 million under section 48D of the Code for Qualifying Therapeutic Discovery Projects. In November 2010, we received $1.4 million of this award, and the remaining $0.1 million was received in February 2011. This was accounted for as other income in 2010.
In December 2010, we issued $8.1 million of Subordinated Notes, and as of December 31, 2010, the Subordinated Notes were treated as share-settled debt under ASC 480-10-25-14 and were recorded at fair value. The Subordinated Notes accrued interest in kind at an annual rate of 10.0 percent. In January 2011, we issued an additional $3.7 million of Subordinated Notes to certain accredited investors.
On February 9, 2011, we sold 14,375,000 shares of common stock in our initial public offering. Proceeds, net of underwriting discounts, commissions and other transaction costs, were $78.2 million. Upon the completion of the offering, all outstanding Subordinated Notes, plus accrued and unpaid interest thereon were automatically converted into 2,335,823 shares of common stock using a conversion price of $5.10 per share (85% of the original issue price of the shares sold in the initial public offering), all outstanding shares of preferred stock were converted into an aggregate 11,747,563 shares of common stock, and all outstanding warrants to purchase preferred stock were converted into warrants to purchase common stock.
On August 2, 2011, we sold 4,968,321 shares of common stock in a public offering. Proceeds, net of underwriting discounts, commissions and other transaction costs, were approximately $66.7 million.
73
Currently, our primary uses of working capital are for conducting PROCEED, the phase 3 clinical trial of EC145 and EC20, preparing the applications for conditional marketing authorization from the EMA for EC145 and EC20 and preparing for the randomized phase 2 trial of EC145 and EC20 in NSCLC.
The following table sets forth the primary sources and uses of cash for each of the periods set forth below:
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Net cash used in operating activities |
$ | (15,396 | ) | $ | (20,745 | ) | $ | (34,585 | ) | |||
| Net cash provided by (used in) investing activities |
(1,866 | ) | 15,010 | (67,311 | ) | |||||||
| Net cash provided by financing activities |
21,083 | 13,908 | 146,381 | |||||||||
| Effect of exchange rate |
— | — | (5 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net increase in cash and cash equivalents |
$ | 3,821 | $ | 8,173 | $ | 44,480 | ||||||
|
|
|
|
|
|
|
|||||||
Operating Activities
The use of cash in all periods primarily resulted from our net losses adjusted for non-cash items and changes in operating assets and liabilities. The decrease in cash used for the year ended 2010 compared to 2009 resulted from an increase in collaboration revenue received from BMS offset by a decrease in interest income. The increase in cash used in 2011 compared to 2010 was due to increases in clinical trial expenses, manufacturing and regulatory expenses related to the anticipated regulatory filing with the EMA for EC145 and EC20 and general and administrative expenses relating to being a public company.
Investing Activities
The cash provided by (used in) investing activities for each of the three years was due primarily to the net result of maturities and sales of short-term investments, which was partially offset by capital expenditures for equipment of $155,000 in 2009, $201,000 in 2010 and $528,000 in 2011.
Financing Activities
The cash provided by financing activities in 2009 was from the sale and issuance of 3,282,456 shares of Series C-3 convertible preferred stock for total net proceeds of $26.6 million, which was partially offset by principal payments of $5.5 million on our prior credit facility with GECC and Oxford. For 2010, our financing activities consisted of repaying our credit facility with GECC and Oxford of $9.0 million, receiving proceeds from our new credit facility with Mid-Cap and SVB of $15.0 million and receiving gross proceeds of $8.1 million for the issuance of subordinated notes. The cash provided by financing activities in 2011 primarily consisted of the $3.6 million in proceeds from the issuance of Subordinated Notes and $144.9 million in proceeds from the public offerings in February and August and $596,000 in payments from the exercise of stock options. These receipts were partially offset by $2.7 million of principal payments on the Mid-Cap and SVB loan facility.
Credit Facilities
In August 2010, we entered into a $15.0 million credit facility with Mid-Cap and SVB to pay-off a prior credit facility to fund research and development and for general corporate purposes. We drew down $10.0 million in principal amount upon signing the agreement at a fixed 9.75 percent interest rate and drew down the remaining $5.0 million in December 2010. Repayment of the principal began in April 2011 following a seven-month interest-only period. In September 2011, we modified the loan agreement to provide for interest-only monthly payments from October 2011 to December 2012, followed by 36 months of interest and principal payments. This modification will provide the Company with additional working capital of $7.4 million over the 15 month interest-only period. The loan is collateralized by a security interest in all of our assets, excluding
74
intellectual property. The loan agreement includes customary covenants and may be accelerated upon the occurrence of any event of default in the loan agreement, including defects in collateral securing the loan, judgment defaults or any event or development that has a material adverse effect, as defined in the agreement. The failure of any preclinical study or clinical trial will not, in and of itself, constitute a material adverse effect. We are in compliance with all material covenants and other obligations in the loan agreement.
Operating Capital Requirements
If we obtain conditional marketing approval in Europe, we anticipate commercializing our first product in 2013 at the earliest. Therefore, we anticipate we will continue to generate significant losses for the next several years as we incur expenses to complete our clinical trial programs for EC145 and EC20 in PROC and NSCLC, build commercial capabilities, develop our pipeline, initiate clinical trials of earlier stage products and expand our corporate infrastructure.
If our available cash, cash equivalents and short-term investments are insufficient to satisfy our liquidity requirements, or if we develop additional opportunities to do so, we may seek to sell additional equity or debt securities, obtain additional credit facilities or refinance our current credit facility with Mid-Cap and SVB. The sale of additional equity and debt securities may result in additional dilution to our stockholders. If we raise additional funds through the issuance of debt securities or convertible preferred stock, these securities may have rights senior to those of our common stock and could contain covenants that would restrict our operations. We may require additional capital beyond our currently forecasted amounts. Any such required additional capital may not be available on reasonable terms, if at all. If we were unable to obtain additional financing, we may be required to reduce the scope of, delay or eliminate some or all of our planned research, development and commercialization activities, which could harm our business.
Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amounts of our working capital requirements. Our future funding requirements will depend on many factors, including but not limited to:
| • | the number and characteristics of the SMDCs and companion imaging diagnostics we pursue; |
| • | the scope, progress, results and costs of researching and developing our SMDCs and companion imaging diagnostics and conducting preclinical and clinical trials; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for our SMDCs and companion imaging diagnostics; |
| • | the cost of commercialization activities if any of our SMDCs and companion imaging diagnostics are approved for sale, including marketing sales and distribution costs; |
| • | the cost of manufacturing any SMDCs and companion imaging diagnostics we successfully commercialize; |
| • | our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; and |
| • | the timing, receipt and amount of sales of, or royalties on, our SMDCs and companion imaging diagnostics, if any. |
75
Contractual Obligations and Commitments
The following table summarizes our contractual obligations at December 31, 2011:
| Total | Less than 1 Year |
1 to 3 Years |
4 to 5 Years |
|||||||||||||
| (In thousands) | ||||||||||||||||
| Short-term and long-term debt (including interest) |
$ | 16,021 | $ | 1,218 | $ | 9,848 | $ | 4,955 | ||||||||
| Operating lease obligations |
712 | 465 | 167 | 80 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total contractual cash obligations |
$ | 16,733 | $ | 1,683 | $ | 10,015 | $ | 5,035 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
In addition, we have certain obligations under licensing agreements with third parties. In October 2007, we entered into an exclusive worldwide license with R&D Biopharmaceuticals to research, develop, and commercialize products containing conjugates of folate receptor targeting compounds and tubulysin compounds. In February 2011, this licensing agreement was assigned by R&D Biopharmaceuticals to Trientlgasse. Under this license agreement, we may be required to make $6.3 million in additional contingent payments upon the achievement of specific scientific, clinical and regulatory milestones, in addition to royalties upon commercial sales. Pursuant to our exclusive license agreement with Purdue Research Foundation relating to folate, we are obligated to pay an annual minimum royalty of $12,500 until commercial sales commence, following which time the payment of royalties with market rates will commence. Pursuant to our exclusive license agreement with Purdue Research Foundation relating to PSMA, we are obligated to pay annual minimum payments of $15,000 until commercial sales commence, following which time the payment of royalties with market rates will commence, along with an annual milestone payment of $100,000. In addition, certain clinical and regulatory milestone payments of $500,000 along with sales-based milestones related to third-party sales are also payable. We are also subject to penalties totaling $300,000 if certain diligence milestones are not met. Future milestone payments in excess of $500,000 may be waived by Purdue Research Foundation. There are no material obligations greater than five years. We do not anticipate incurring liabilities for EC145 royalty payments based on the estimated timing of when we may have commercial sales and the expiration of the applicable patents.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under rules promulgated by the Securities and Exchange Commission.
Tax Loss Carryforwards
As of December 31, 2011, we have net operating loss carryforwards of approximately $132.5 million and $131.1 million to offset future federal and state income taxes, respectively. These federal and state loss carryforwards expire at various times beginning in 2022. We also have research and development tax credit carryforwards of approximately $5.0 million to offset future federal income taxes and approximately $1.7 million to offset future state income taxes. The federal and state tax credits expire at various times through 2029. We have determined that we experienced a change in ownership as defined under Section 382 of the U.S. Internal Revenue Code as a result of the public offering in August 2011. As a result, the future use of our net operating losses and credit equivalents will be limited to approximately $17.7 million per year. At December 31, 2011 we recorded a full valuation allowance against our net operating loss carryforwards of approximately $58.7 million, as we believe it is more likely than not they will not be fully realized. If we determine in the future that we will be able to realize all or a portion of our net operating loss carryforwards, an adjustment to our net operating loss carryforwards would increase net income in the period in which we make such a determination.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risks |
We are exposed to market risk related to changes in interest rates. As of December 31, 2010 and December 31, 2011 we had cash, cash equivalents and short-term investments of $16.9 million and
76
$128.1 million, respectively. The short-term investments consisted of U.S. government money market funds, U.S. Treasuries, U.S. Government agency obligations and cash equivalents. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our investments are in short-term marketable securities. Our short-term investments are subject to interest rate risk and will fall in value if market interest rates increase. Due to the short-term duration of our investment portfolio and the low risk profile of our investments, an immediate 10 percent change in interest rates would not have a material effect on the fair market value of our portfolio. We have the ability to hold our short-investments until maturity, and therefore we do not expect that our results of operations or cash flows would be adversely affected by any change in market interest rates on our investments. We carry our investments based on publicly available information. We do not currently have any investment securities for which a market is not readily available or active. In addition, our loan agreement with Mid-Cap and SVB requires us to pay interest at fixed rates.
We do not believe that any credit risk is likely to have a material impact on the value of our assets and liabilities.
We contract with contract research organizations and investigational sites globally. We may be subject to fluctuations in foreign currency rates in connection with these agreements. A ten percent fluctuation in foreign currency rates would not have a material impact on our financial statements. We currently do not hedge our foreign currency exchange rate risk, but as our operations in foreign countries expand, we may consider the use of hedges.
77
| Item 8. | Financial Statements and Supplementary Data |
ENDOCYTE, INC.
INDEX TO FINANCIAL STATEMENTS
78
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Endocyte, Inc.
We have audited the accompanying consolidated balance sheets of Endocyte, Inc. as of December 31, 2011 and 2010, and the related consolidated statements of operations, convertible preferred stock, stockholders’ equity (deficit) and comprehensive loss, and cash flows for each of the three years in the period ended December 31, 2011. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Endocyte, Inc. at December 31, 2011 and 2010, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles.
Indianapolis, Indiana
March 28, 2012
79
ENDOCYTE, INC.
| December 31, | ||||||||
| 2010 | 2011 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 16,872,783 | $ | 61,352,483 | ||||
| Short-term investments |
— | 66,732,242 | ||||||
| Prepaid expenses |
2,637,783 | 1,122,979 | ||||||
| Other assets |
539,659 | 548,320 | ||||||
|
|
|
|
|
|||||
| Total current assets |
20,050,225 | 129,756,024 | ||||||
| Property and equipment, net |
863,008 | 1,107,851 | ||||||
| Other noncurrent assets |
300,985 | 811,229 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 21,214,218 | $ | 131,675,104 | ||||
|
|
|
|
|
|||||
| Liabilities, Convertible Preferred Stock, and Stockholders’ Equity (Deficit) |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 755,208 | $ | 2,125,500 | ||||
| Accrued wages and benefits |
155,102 | 1,032,334 | ||||||
| Accrued clinical trial expenses |
1,305,496 | 665,477 | ||||||
| Accrued interest payable |
140,427 | 105,942 | ||||||
| Other current liabilities |
775,691 | 1,540,749 | ||||||
| Current portion of long-term debt |
4,318,078 | — | ||||||
| Preferred stock warrants |
223,032 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
7,673,034 | 5,470,002 | ||||||
| Long-term debt, net of current portion |
11,123,311 | 12,833,179 | ||||||
| Subordinated notes |
9,529,413 | — | ||||||
|
|
|
|
|
|||||
| Total liabilities |
28,325,758 | 18,303,181 | ||||||
| Convertible preferred stock, $0.001 par value 14,310,992 shares authorized; 11,747,563 shares issued and outstanding at December 31, 2010 |
89,799,483 | — | ||||||
| Stockholders’ equity (deficit): |
||||||||
| Common stock: $0.001 par value, 100,000,000 shares authorized; 1,203,228 and 35,784,485 shares issued and outstanding at December 31, 2010 and 2011 |
1,203 | 35,785 | ||||||
| Additional paid-in capital |
1,156,681 | 251,942,922 | ||||||
| Accumulated other comprehensive income |
— | (6,264 | ) | |||||
| Retained deficit |
(98,068,907 | ) | (138,600,520 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity (deficit) |
(96,911,023 | ) | 113,371,923 | |||||
|
|
|
|
|
|||||
| Total liabilities, convertible preferred stock, and stockholders’ equity (deficit) |
$ | 21,214,218 | $ | 131,675,104 | ||||
|
|
|
|
|
|||||
See accompanying notes.
80
ENDOCYTE, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| 2009 | 2010 | 2011 | ||||||||||
| Revenue: |
||||||||||||
| Grant revenue |
$ | — | $ | — | $ | — | ||||||
| Collaboration revenue |
3,000,000 | — | 191,023 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenue |
3,000,000 | — | 191,023 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
14,803,741 | 14,560,821 | 28,827,746 | |||||||||
| General and administrative |
3,934,259 | 6,039,145 | 10,000,000 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
18,738,000 | 20,599,966 | 38,827,746 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(15,738,000 | ) | (20,599,966 | ) | (38,636,723 | ) | ||||||
| Other income (expense): |
||||||||||||
| Interest income |
49,315 | 7,683 | 129,449 | |||||||||
| Interest expense |
(1,435,988 | ) | (1,064,556 | ) | (1,987,873 | ) | ||||||
| Other income (expense) |
118,988 | 1,564,117 | (36,466 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
(17,005,685 | ) | (20,092,722 | ) | (40,531,613 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss per share — basic and diluted |
$ | (18.67 | ) | $ | (21.77 | ) | $ | (1.40 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted-average number of common shares used in net loss per share calculation — basic and diluted |
911,066 | 923,007 | 29,003,991 | |||||||||
See accompanying notes.
81
ENDOCYTE, INC.
CONSOLIDATED STATEMENTS OF CONVERTIBLE PREFERRED STOCK, STOCKHOLDERS’ EQUITY (DEFICIT) AND COMPREHENSIVE LOSS
| Convertible Preferred Stock |
Common Stock | Additional Paid-In Capital |
Accumulated Other Comprehensive Income (Loss) |
Retained Deficit |
Comprehensive Loss |
|||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Total | ||||||||||||||||||||||||||||||||
| Balances, December 31, 2008 |
8,465,107 | $ | 63,197,046 | 1,133,579 | $ | 1,133 | $ | 1,533,435 | $ | 23,563 | $ | (60,970,500 | ) | $ | (59,412,369 | ) | $ | (18,397,674 | ) | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Issuance of Series C-3 convertible preferred stock, net |
3,282,456 | 26,602,437 | — | — | — | — | — | |||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 381,877 | — | — | 381,877 | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | (17,005,685 | ) | (17,005,685 | ) | (17,005,685 | ) | |||||||||||||||||||||||||
| Unrealized loss on securities |
— | — | — | — | — | (21,386 | ) | — | (21,386 | ) | (21,386 | ) | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balances, December 31, 2009 |
11,747,563 | 89,799,483 | 1,133,579 | 1,133 | 1,915,312 | 2,177 | (77,976,185 | ) | (76,057,563 | ) | (17,027,071 | ) | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Fair value adjustment of the subordinated notes |
— | — | — | — | (1,429,412 | ) | — | — | (1,429,412 | ) | ||||||||||||||||||||||||||
| Exercise of stock options |
— | — | 69,649 | 70 | 147,219 | — | — | 147,289 | ||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 523,562 | — | — | 523,562 | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (20,092,722 | ) | (20,092,722 | ) | (20,092,722 | ) | ||||||||||||||||||||||||
| Unrealized loss on securities |
— | — | — | — | — | (2,177 | ) | — | (2,177 | ) | (2,177 | ) | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balances, December 31, 2010 |
11,747,563 | $ | 89,799,483 | 1,203,228 | $ | 1,203 | $ | 1,156,681 | $ | — | $ | (98,068,907 | ) | $ | (96,911,023 | ) | $ | (20,094,899 | ) | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Fair value adjustment of the subordinated notes |
— | — | — | — | (644,118 | ) | — | — | (644,118 | ) | ||||||||||||||||||||||||||
| Net proceeds from public offerings |
— | — | 20,214,810 | 20,215 | 144,882,518 | — | — | 144,902,733 | ||||||||||||||||||||||||||||
| Conversion of preferred stock to common stock |
(11,747,563 | ) | (89,799,483 | ) | 11,747,563 | 11,747 | 89,787,736 | — | — | 89,799,483 | ||||||||||||||||||||||||||
| Reclassify warrants to equity |
— | — | — | — | 223,031 | — | — | 223,031 | ||||||||||||||||||||||||||||
| Conversion of subordinated notes to common stock |
— | — | 2,335,823 | 2,336 | 13,981,441 | — | — | 13,983,777 | ||||||||||||||||||||||||||||
| Exercise of stock options |
— | — | 283,061 | 284 | 595,288 | — | — | 595,572 | ||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | 1,960,345 | — | — | 1,960,345 | ||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (40,531,613 | ) | (40,531,613 | ) | (40,531,613 | ) | ||||||||||||||||||||||||
| Unrealized loss on foreign exchange translation |
— | — | — | — | — | (5,718 | ) | — | (5,718 | ) | (5,718 | ) | ||||||||||||||||||||||||
| Unrealized gain on securities |
— | — | — | — | — | (546 | ) | — | (546 | ) | (546 | ) | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balances December 31, 2011 |
— | $ | — | 35,784,485 | $ | 35,785 | $ | 251,942,922 | $ | (6,264 | ) | $ | (138,600,520 | ) | $ | 113,371,923 | $ | (40,537,877 | ) | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
See accompanying notes.
82
ENDOCYTE, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (17,005,685 | ) | $ | (20,092,722 | ) | $ | (40,531,613 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation |
231,890 | 260,571 | 262,518 | |||||||||
| Stock-based compensation |
381,877 | 523,562 | 1,960,345 | |||||||||
| Loss on disposal of property and equipment |
3,588 | — | 20,876 | |||||||||
| Loss on extinguishment of debt |
— | 144,284 | — | |||||||||
| Accretion of bond discount |
(17,250 | ) | (3,075 | ) | 343,342 | |||||||
| Non cash interest expense |
210,809 | 160,844 | 646,069 | |||||||||
| Decrease in fair value on preferred stock warrants |
(74,118 | ) | (237,457 | ) | — | |||||||
| Extinguishment of liability |
— | 18,389 | — | |||||||||
| Change in operating assets and liabilities: |
||||||||||||
| Accounts and accrued interest receivable |
— | — | (548,320 | ) | ||||||||
| Prepaid expenses and other assets |
269,326 | (2,323,788 | ) | 1,480,653 | ||||||||
| Accounts payable |
493,263 | (118,140 | ) | 943,024 | ||||||||
| Accrued interest, wages, benefits and other liabilities |
109,959 | 922,386 | 838,490 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(15,396,341 | ) | (20,745,146 | ) | (34,584,616 | ) | ||||||
| Investing activities |
||||||||||||
| Purchases of property and equipment |
(155,441 | ) | (200,527 | ) | (234,969 | ) | ||||||
| Purchases of investments |
(35,496,785 | ) | (12,295,754 | ) | (236,930,028 | ) | ||||||
| Proceeds from sale of investments |
33,785,889 | 27,507,030 | 169,853,899 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
(1,866,337 | ) | 15,010,749 | (67,311,098 | ) | |||||||
| Financing activities |
||||||||||||
| Proceeds from issuance of convertible preferred stock, net of issuance costs |
26,602,437 | — | — | |||||||||
| Proceeds from borrowings, net of issuance costs |
— | 14,857,292 | — | |||||||||
| Proceeds from issuance of subordinated convertible notes, net of issuance costs |
— | 8,040,837 | 3,590,837 | |||||||||
| Principal payments on borrowings |
(5,518,941 | ) | (9,035,548 | ) | (2,708,011 | ) | ||||||
| Proceeds from initial public offering, net of issuance costs |
— | — | 78,167,843 | |||||||||
| Proceeds from second public offering, net of issuance costs |
— | — | 66,734,890 | |||||||||
| Loss on Extinguishment of debt |
— | (102,062 | ) | — | ||||||||
| Proceeds from the exercise of stock options |
— | 147,289 | 595,573 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
21,083,496 | 13,907,808 | 146,381,132 | |||||||||
|
|
|
|
|
|
|
|||||||
| Effect of exchange rate |
— | — | (5,718 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net increase in cash and cash equivalents |
3,820,818 | 8,173,411 | 44,479,700 | |||||||||
| Cash and cash equivalents at beginning of period |
4,878,554 | 8,699,372 | 16,872,783 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of period |
$ | 8,699,372 | $ | 16,872,783 | $ | 61,352,483 | ||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes.
83
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | Nature of Business and Organization |
Endocyte, Inc. (the “Company”) was incorporated on December 6, 1995. The Company is a biopharmaceutical company developing targeted therapies for the treatment of cancer and inflammatory diseases. The Company uses its proprietary technology to create novel small molecule drug conjugates (“SMDCs”), and companion imaging diagnostics. The SMDCs actively target receptors that are over-expressed on diseased cells, relative to healthy cells. This targeted approach is designed to enable the treatment of patients with a highly active drug at greater doses, delivered more frequently, and over longer periods of time than would be possible with the untargeted drug alone. The Company is also developing companion imaging diagnostics for each of its SMDCs that are designed to identify the patients whose disease over-expresses the target of the therapy and who are therefore more likely to benefit from treatment.
On July 21, 2011, the Company formed Endocyte Europe B.V., a limited liability company in The Netherlands. This wholly-owned subsidiary has been formed to assist with the administration of the filing of applications with the European Medicines Authority (“EMA”) and pre-commercial planning activities.
Public Offerings
On February 9, 2011, the Company completed its initial public offering of 14,375,000 shares of common stock, including 1,875,000 shares of common stock pursuant to the exercise of the over-allotment option by the underwriters. Proceeds, net of underwriting discounts, commissions and other transaction costs were approximately $78.2 million. Upon the closing of the offering, the Subordinated Notes (see footnote 9) automatically converted into 2,335,823 shares of common stock using a conversion price of $5.10 per share (85% of the original issue price of the shares sold in the initial public offering), all of the outstanding shares of preferred stock were converted to common stock and the outstanding warrants to purchase Series C-3 preferred stock were converted to warrants to purchase common stock and reclassified from a liability to equity.
On August 2, 2011, the Company completed a public offering of 4,968,321 shares of common stock, including 871,489 shares of common stock pursuant to the exercise of the over-allotment by the underwriters. Proceeds, net of underwriting discounts, commissions and other transaction costs were approximately $66.7 million.
| 2. | Significant Accounting Policies |
Stock Split
On January 10, 2011, the Company effected a 1.00 for 1.91 reverse stock split. All historical common stock and per share information has been changed to reflect the stock split.
Basis of Presentation
The accompanying consolidated financial statements include the accounts of Endocyte, Inc. and Endocyte Europe B.V., and all intercompany amounts have been eliminated. The consolidated financial statements are prepared in conformity with U.S. generally accepted accounting principles (“GAAP”). Subsequent events have been evaluated through the date of issuance, which is the same as the date the Form 10-K is filed with the Securities Exchange Commission.
Reclassifications
Certain amounts in the 2010 consolidated financial statements have been reclassified to be consistent with the 2011 presentation.
84
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Cash and Cash Equivalents
The Company considers cash and all highly liquid investments with an original maturity of three months or less at the date of purchase to be cash equivalents, except for those funds managed by the Company’s investment manager, which are classified as short-term investments. Cash equivalents consist primarily of money market instruments.
Short-Term Investments
Short-term investments consist primarily of investments with original maturities greater than three months and less than one year when purchased. Management determines the appropriate classification of marketable securities at the time of purchase and reevaluates such designation as of each balance sheet date. All securities held at December 31, 2011, were classified as available-for-sale as defined by the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 320, Investments — Debt and Equity Securities (“ASC 320”). Available-for-sale securities are carried at fair value, with the unrealized gains and losses reported in other comprehensive income. Realized gains and losses and declines in value judged to be other-than-temporary on available-for-sale securities are included in other income. The Company considers and accounts for other-than-temporary impairments according to ASC 320. The cost of securities sold is based on the specific-identification method. Discounts and premiums on debt securities are amortized to interest income and expense over the term of the security.
Prepaid Offering Expenses
Prepaid expenses as of December 31, 2010 include $1.9 million of deferred costs for legal, accounting and other direct costs related to the Company’s initial public offering. These costs were reclassified to additional paid-in capital in 2011 as a reduction of the initial public offering proceeds.
Property and Equipment
Property and equipment are stated at cost and are being depreciated using the straight-line method over estimated useful lives, which range from three to seven years.
Licenses and Patents
Licenses and patent costs are expensed as incurred as the Company does not believe there is an alternate future use for the costs. Licenses are classified as research and development and patents are classified as general and administrative expenses in the consolidated statements of operations.
Long-Lived Assets
The Company reviews long-lived assets, including property and equipment, for impairment when events or changes in business conditions indicate that their full carrying value may not be fully recoverable.
Leases
The Company evaluates all leases to determine whether they should be accounted for as operating or capital leases.
85
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Preferred Stock Warrants
The Company accounted for its preferred stock warrants under ASC Topic 480, Distinguishing Liabilities from Equity. The preferred stock warrants were classified as current liabilities at December 31, 2010 and were recorded at fair value. In connection with the initial public offering, the preferred stock warrants converted to warrants to purchase our common stock and were reclassified to stockholders’ equity. Any changes to fair value were recorded as other income (expense) through the date of the initial public offering. Subsequent to the initial public offering, the warrants no longer meet the definition of a liability and are not subject to a fair value adjustment.
Revenue Recognition
The Company recognizes revenues from license agreements when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the fee is fixed or determinable, and there is reasonable assurance that the related amounts are collectible in accordance with ASC Topic 605, Revenue Recognition (“ASC 605”). When evaluating multiple element arrangements, we consider whether the components of the arrangement represent separate units of accounting. This evaluation requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
The Company has generated revenues as a result of out-license agreements. The Company immediately recognizes the full amount of milestone payments due upon the achievement of the milestone event if the event is substantive, objectively determinable, and represents an important point in the development life cycle of the product. Milestone payments earned are recorded in collaboration revenue. The Company recognizes nonrefundable up-front fees when performance conditions by the Company have been achieved and there is reasonable assurance that the related amounts are collectible.
Research and Development Expenses
Research and development expenses represent costs associated with the ongoing development of SMDCs and companion imaging diagnostics and include salaries, supplies, and expenses for clinical trials. The Company records accruals for clinical trial expenses based on the estimated amount of work completed. The Company monitors patient enrollment levels and related activities to the extent possible through internal reviews, correspondence, and discussions with research organizations.
Upfront payments made in connection with business collaborations and research and development arrangements are evaluated under ASC Subtopic 730-20, Research and Development Arrangements. Upfront payments made in connection with business development collaborations are expensed as research and development costs, as the assets acquired do not have alternative future use. Amounts related to future research and development are capitalized as prepaid research and development and are expensed over the service period based upon the level of services provided. As of December 31, 2011, the Company had $1,065,792 of capitalized research and development costs included in prepaid expense and noncurrent assets.
Stock-Based Compensation
The Company has accounted for all options granted subsequent to January 1, 2006, pursuant to ASC Topic 718, Compensation — Stock Compensation (“ASC 718”), which requires the recognition of the fair value or calculated value for nonpublic entities, of stock-based compensation in net income. Stock-based compensation consists of stock options, which are granted to employees at exercise prices at or above the fair market value of the Company’s common stock on the dates of grant. The Company used the calculated value to measure its stock-based compensation.
86
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Prior to January 1, 2006, in accordance with ASC 718, the Company used the intrinsic value method to account for stock options. The Company issued its stock options at exercise prices at or above the estimated fair market value of the Company’s common stock on the dates of grant. Accordingly, no compensation expense was recognized for options granted to employees in the prior periods.
The Company elected the prospective transition method for the adoption of ASC 718, which requires that nonpublic companies that had previously measured compensation cost using the minimum value method continue to account for equity awards outstanding at the date of adoption in the same manner as they had been accounted for prior to adoption. For all awards granted, modified, or settled after the date of adoption, the Company recognizes compensation cost based on the grant-date value estimated in accordance with the provisions of ASC 718.
Net Loss Per Share
The Company calculates basic net loss per share based on the weighted-average number of outstanding common shares. The Company calculates diluted net loss per share based on the weighted-average number of outstanding common shares plus the effect of dilutive securities.
Income Taxes
The Company accounts for income taxes under the liability method in accordance with the provision of ASC Topic 740, Income Taxes (“ASC 740”). ASC 740 requires recognition of deferred taxes to provide for temporary differences between financial reporting and tax basis of assets and liabilities. Deferred taxes are measured using enacted tax rates expected to be in effect in a year in which the basis difference is expected to reverse. The Company continues to record a valuation allowance for the full amount of deferred tax assets, which would otherwise be recorded for tax benefits relating to operating loss and tax credit carryforwards, as realization of such deferred tax assets cannot be determined to be more likely than not.
In June 2006, the FASB issued authoritative guidance on accounting for uncertainty in income taxes, amended by Accounting Standards Update No. (“ASU”) 2009-06 in September 2009 on accounting for uncertain tax positions, which clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements. This guidance prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return and provides guidance on derecognition of tax benefits, classification on the balance sheet, interest and penalties, accounting in interim periods, disclosure, and transition.
Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. Accumulated other comprehensive loss consists of unrealized losses on foreign exchange translation and changes in unrealized gains and losses on available-for-sale securities. Comprehensive loss from operations was calculated as follows:
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Net loss |
$ | (17,005,685 | ) | $ | (20,092,722 | ) | $ | (40,531,613 | ) | |||
| Unrealized loss on foreign exchange translation |
— | — | (5,718 | ) | ||||||||
| Unrealized loss on available-for-sale securities |
(21,386 | ) | (2,177 | ) | (546 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive loss |
$ | (17,027,071 | ) | $ | (20,094,899 | ) | $ | (40,537,877 | ) | |||
|
|
|
|
|
|
|
|||||||
87
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Segment Information
Operating segments are defined as components of an enterprise engaging in business activities for which discrete financial information is available and regularly reviewed by the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating segment and the Company operates in only two geographic segments.
| 3. | New Accounting Pronouncements |
Recently Adopted Accounting Standards
As of January 1, 2011, we adopted on a prospective basis the accounting updates to guidance ASC 605 “Revenue Recognition”, subtopic ASC 605-25 “Revenue with Multiple Element Arrangements” and subtopic ASC 605-28 “Revenue Recognition-Milestone Method”, which provides accounting guidance for revenue recognition for arrangements with multiple deliverables and guidance on defining the milestone and determining when the use of the milestone method of revenue recognition for research and development transactions is appropriate, respectively. These updates will result in different accounting treatment for future new collaboration arrangements. The adoption of ASC 605-25 “Revenue with Multiple Element Arrangements” and the election of the milestone method under subtopic ASC 605-28 “Revenue Recognition-Milestone Method” did not have a material impact on our consolidated financial statements.
In April 2010, the FASB ratified ASU No. 2010-17 guidance related to the milestone method of revenue recognition. The ASU provides guidance on defining a milestone under ASC 605. This guidance states that an entity can make an accounting policy election to recognize a payment that is contingent upon the achievement of a substantive milestone in its entirety in the period in which the milestone is achieved. This guidance was effective for the Company as of January 1, 2011, and did not have a material impact to the Company’s financial position or results of operations.
Recently Issued Accounting Standards
In June 2011, the FASB issued ASU No. 2011-05, “Presentation of Comprehensive Income” an update to ASC Topic 220, “Comprehensive Income”. This update requires that all nonowner changes in stockholders’ equity be presented either in a single continuous statement of comprehensive income or in two separate but consecutive statements. This update is to be applied retrospectively and is effective for financial statements issued for fiscal years, and interim periods within those years, beginning after December 15, 2011, and interim and annual periods thereafter. This update will be effective for the Company beginning January 1, 2012. The Company does not expect the adoption of this guidance to have a material impact on its consolidated financial statements.
| 4. | Net Loss Per Share |
Basic net loss per share is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration for common stock equivalents. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method and the if-converted method. For purposes of this calculation, convertible preferred stock, stock options and warrants are considered to be common stock equivalents and are only included in the calculation of diluted net loss per share when their effect is dilutive.
88
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following tables and discussion provide a reconciliation of the numerator and denominator of the basic and diluted net loss per share computations. The calculation below provides net loss, weighted-average common shares outstanding, and the resultant net loss per share on both a basic and diluted basis for the year ended December 31, 2009, 2010 and 2011.
Historical net loss per share
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Numerator: |
||||||||||||
| Net loss |
$ | (17,005,685 | ) | $ | (20,092,722 | ) | $ | (40,531,613 | ) | |||
| Denominator: |
||||||||||||
| Weighted-average common shares outstanding |
911,066 | 923,007 | 29,003,991 | |||||||||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (18.67 | ) | $ | (21.77 | ) | $ | (1.40 | ) | |||
|
|
|
|
|
|
|
|||||||
As of December 31, 2009, 2010 and 2011 the following number of potential common stock equivalents were outstanding:
Common stock equivalents
| Year Ended December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Outstanding common stock options |
1,588,209 | 2,019,333 | 2,592,009 | |||||||||
| Outstanding restricted stock |
222,510 | 222,510 | 271,213 | |||||||||
| Outstanding warrants |
69,294 | 133,968 | 133,968 | |||||||||
| Shares issuable upon conversion of preferred shares |
11,747,563 | 11,747,563 | — | |||||||||
| Shares issuable upon conversion of subordinated notes |
— | 1,596,066 | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
13,627,576 | 15,719,440 | 2,997,190 | |||||||||
|
|
|
|
|
|
|
|||||||
These common stock equivalents were excluded from the determination of diluted net loss per share due to their anti-dilutive effect on earnings.
| 5. | Short-Term Investments |
Effective January 1, 2008, the Company adopted ASC Topic 820, Fair Value Measurements and Disclosures (“ASC 820”). ASC 820, which defines fair value, establishes a framework for measuring fair value in GAAP, and expands disclosures about fair value measurements.
ASC 820 establishes a three-level valuation hierarchy for fair value measurements. These valuation techniques are based upon the transparency of inputs (observable and unobservable) to the valuation of an asset or liability as of the measurement date. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect the Company’s market assumptions. These two types of inputs create the following fair value hierarchy:
Level 1 — Valuation is based on quoted prices for identical assets or liabilities in active markets.
89
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Level 2 — Valuation is based on quoted prices for similar assets or liabilities in active markets, or other inputs that are observable for the asset or liability, either directly or indirectly, for the full term of the financial instrument.
Level 3 — Valuation is based upon other unobservable inputs that are significant to the fair value measurement.
The fair value of the Company’s fixed income securities is based on a market approach using quoted market values.
The following tables summarize the fair value of cash and cash equivalents and short-term investments as of December 31, 2011:
| Description |
Cost | Level 1 | Fair Value (Carrying Value) |
|||||||||
| Cash and cash equivalents |
$ | 61,352,483 | $ | 61,352,483 | $ | 61,352,483 | ||||||
|
|
|
|
|
|
|
|||||||
| Short-term investments |
||||||||||||
| U. S. treasury obligations |
$ | 23,013,799 | $ | 23,016,965 | $ | 23,016,965 | ||||||
| U.S. government agency obligations |
43,718,989 | 43,715,277 | 43,715,277 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total Short-term investments |
$ | 66,732,788 | $ | 66,732,242 | $ | 66,732,242 | ||||||
|
|
|
|
|
|
|
|||||||
Total unrealized gross gains were $0 and $5,741 for the year ended December 31, 2010 and 2011, respectively. Total unrealized gross losses were $0 and $6,287 for the year ended December 31, 2010 and 2011, respectively. The Company does not consider any of the unrealized losses to be other-than-temporary impairments. The Company had no investment securities as of December 31, 2010.
| 6. | Property and Equipment |
Property and equipment consisted of the following:
| Estimated Useful Lives |
December 31, | |||||||||||
| 2010 | 2011 | |||||||||||
| Laboratory equipment |
7 | $ | 2,453,788 | $ | 2,213,774 | |||||||
| Office equipment and software |
3-5 | 582,072 | 506,246 | |||||||||
| Leasehold improvements |
7 | 140,101 | 144,409 | |||||||||
| Assets not in service |
— | 293,269 | ||||||||||
|
|
|
|
|
|||||||||
| 3,175,961 | 3,157,698 | |||||||||||
| Less accumulated depreciation |
(2,312,953 | ) | (2,049,847 | ) | ||||||||
|
|
|
|
|
|||||||||
| $ | 863,008 | $ | 1,107,851 | |||||||||
|
|
|
|
|
|||||||||
Assets not in service represents new laboratory equipment that was not installed and ready to use at December 31, 2011. The total amount of depreciation expense for the year ended December 31, 2009, 2010 and 2011 were $231,890, $260,571 and $262,518 respectively.
90
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 7. | Long-Term Debt |
Long-term debt consisted of the following:
| December 31, | ||||||||
| 2010 | 2011 | |||||||
| Notes payable to Mid-Cap and SVB, with fixed interest rate of 9.75%, monthly payments through December 1, 2015 |
15,637,500 | 12,929,489 | ||||||
|
|
|
|
|
|||||
| Less unamortized discount |
(196,111 | ) | (96,310 | ) | ||||
|
|
|
|
|
|||||
| 15,441,389 | 12,833,179 | |||||||
| Less current portion, including unamortized discount |
(4,318,078 | ) | — | |||||
|
|
|
|
|
|||||
| $ | 11,123,311 | $ | 12,833,179 | |||||
|
|
|
|
|
|||||
In August, 2010 the Company obtained a $15.0 million loan commitment from Mid Cap Financial (“Mid-Cap”) and Silicon Valley Bank (“SVB”) to pay-off the existing loan commitment from General Electric Capital Corporate (“GECC”) and Oxford Finance Corporation (“Oxford”) that was set to mature in March and July 2011, and to fund research and development and for corporate purposes. Upon execution of the agreement in August 2010, the Company drew $10.0 million in principal. In December 2010, the Company amended their term loan arrangement with Mid-Cap and SVB in order to access the remaining tranche of $5.0 million. Prior to September 30, 2011, the Company was required to make interest-only monthly payments on the notes payable to Mid-Cap and SVB through March 2011, and thereafter monthly principal and interest payments of $566,612. The loan with Mid-Cap and SVB was modified on September 30, 2011. Under this modification, the Company is required to make interest-only monthly payments from October 2011 through December 2012. Starting January 2013, the Company is required to make principal payments of $341,448 and interest payments at a rate of 9.75% per annum. The loan is collateralized by a security interest in all of the Company’s assets, excluding intellectual property. The loan agreement includes customary covenants, including those that require prior written consent of the lenders before the Company can incur or prepay indebtedness, create additional liens, sell, or transfer any material portion of its assets. The loan agreement also contains customary events of defaults, and also includes a material adverse effect clause. The Company is in compliance with all material covenants and other obligations in the loan agreement. In connection with the loan during 2010, the Company issued warrants to lenders to purchase an aggregate of 64,674 shares of Series C-3 convertible preferred stock. The fair value of the preferred stock warrants issued was $219,322 on the dates of issuance and was based on observable inputs using quoted market values and the income approach as derived by the Black-Scholes model. The terms of these preferred stock warrants and the fair value assumptions and inputs are detailed in Note 8.
Interest paid was $1,272,942, $842,141 and $1,334,507 for the year ended December 31, 2009, 2010 and 2011, respectively.
Aggregate maturities of debt due after December 31, 2011, are as follows:
| 2012 |
$ | — | ||
| 2013 |
4,097,371 | |||
| 2014 |
4,097,371 | |||
| 2015 |
4,734,747 | |||
|
|
|
|||
| 12,929,489 | ||||
| Less unamortized discount |
(96,310 | ) | ||
|
|
|
|||
| $ | 12,833,179 | |||
|
|
|
91
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 8. | Preferred Stock Warrants |
In 2007, the Company issued warrants as consideration in connection with its loan commitment from GECC and Oxford to purchase 69,294 of Series C-3 convertible preferred stock, maturing in 2017. These preferred stock warrants were exercisable upon issuance, and no contingent conversion feature exists. Upon closing of the initial public offering, these warrants were converted to warrants to purchase common stock and were reclassified to stockholders’ equity.
In August 2010 and December 2010, the Company issued warrants as consideration in connection with its loan commitment from Mid-Cap and SVB to purchase either Series C-3 preferred stock or preferred stock offered in a subsequent offering. The number of preferred stock warrants issued was based on the Series C-3 convertible preferred stock and a $8.12 per share exercise price. The preferred stock warrants will mature in 2020, are exercisable upon issuance and do not contain contingent conversion features. Upon closing of the initial public offering, these warrants were converted to warrants to purchase common stock and were reclassified to stockholders’ equity.
At December 31, 2010, the liability was measured at fair value, and any changes to fair value were recorded in the statement of operations as other income (expense). The fair value of the GECC and Oxford and Mid-Cap and SVB preferred stock warrants was determined using the Black-Scholes valuation model as of December 31, 2010 based upon the following assumptions:
| 2010 | ||||
| Exercise price |
$ | 8.12 | ||
| Convertible preferred stock price |
$ | 6.00 | ||
| Risk-free interest rate |
2.9-3.7 | % | ||
| Expected life |
7-10 years | |||
| Dividend yield |
0.00 | % | ||
| Expected volatility |
22.69-29.55 | % | ||
The fair value of the preferred stock warrants was based upon observable inputs using quoted market values and the income approach as derived by the Black-Scholes model. Volatility is derived from the historical volatility of the NASDAQ Biotechnology Index over the remaining expected life of the warrant. The warrants contain anti-dilution and change in control provisions that can impact conversion.
The carrying and fair values of the preferred stock warrants are $223,032 as of December 31, 2010. The preferred stock warrants were classified as Level 2 within the ASC 820 fair value hierarchy. The Company recorded other income in 2009 and 2010 of $74,118 and $219,068 respectively, due to the change in fair value on the warrants.
| 9. | Subordinated Notes |
In December 2010, the Company issued $8.1 million of Subordinated Convertible Promissory Notes (the “Subordinated Notes”) and in January 2011 the Company issued an additional $3.7 million of Subordinated Notes. The Subordinated Notes were converted into shares of our common stock upon the closing of our initial public stock offering on February 9, 2011. The Subordinated Notes, plus accrued and unpaid interest thereon, were converted into 2,335,823 shares of common stock using a conversion price of $5.10 per share (85% of offering price). As of December 31, 2010, the Subordinated Notes were treated as share-settled debt under ASC 480-10-25-14 and were recorded at fair value. The Subordinated Notes accrued interest in kind at an annual rate of 10.0 percent and were not due until maturity.
92
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 10. | Leases |
Future minimum lease payments for noncancellable operating leases as of December 31, 2011, are as follows:
| 2012 |
$ | 465,090 | ||
| 2013 |
82,627 | |||
| 2014 |
84,826 | |||
| 2015 |
79,696 | |||
| 2016 |
— | |||
|
|
|
|||
| Total minimum lease payments |
$ | 712,239 | ||
|
|
|
Rent expense for operating leases was $355,601, $367,144 and $421,297 for the year ended December 31, 2009, 2010 and 2011, respectively. In 2010, the Company entered into a noncancelable operating lease, with minimum lease payments that total $406,009 and are payable through 2015.
| 11. | Convertible Preferred Stock |
On July 29, 2009, the Company increased the number of Series C-3 convertible preferred stock authorized for issuance to 5,235,602 shares. On October 8, 2009, the Company increased the number of Series C-3 convertible preferred stock authorized for issuance to 5,549,738 shares. Between August 3, 2009 and October 13, 2009, the Company sold 3,282,456 shares of Series C-3 convertible preferred stock for $8.12 per share for proceeds totaling $26,645,524, net of offering costs of $43,097. In December 2010, the Company increased the number of Series C-3 convertible preferred stock authorized for issuance to 7,643,979 shares to allow for the conversion of the Subordinated Notes and the issuance of Series C-3 warrants associated with the Company’s credit facility.
All of the Company’s outstanding shares of preferred stock converted into 11,747,563 shares of common stock upon the closing of the Company’s initial public offering in 2011. The Company’s total outstanding convertible preferred stock, with a par value of $0.001 per share, consisted of the following as of December 31, 2010:
| Convertible |
Shares Authorized |
Shares Issued |
Price per Share |
Total Proceeds (Net of Offering Costs) |
||||||||||||
| Series A-1 |
750,261 | 750,167 | $ | 1.91 | $ | 1,470,370 | ||||||||||
| Series A-2 |
241,884 | 241,634 | 5.73 | 1,430,749 | ||||||||||||
| Series B |
936,649 | 936,241 | 8.12 | 7,530,180 | ||||||||||||
| Series C-1 |
1,937,172 | 1,895,765 | 8.12 | 15,320,640 | ||||||||||||
| Series C-2 |
2,801,047 | 2,787,791 | 8.12 | 22,447,353 | ||||||||||||
| Series C-3 |
7,643,979 | 5,135,965 | 8.12 | 41,600,191 | ||||||||||||
|
|
|
|||||||||||||||
| $ | 89,799,483 | |||||||||||||||
|
|
|
|||||||||||||||
93
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Company’s total proceeds, net of offering costs, for its outstanding convertible preferred stock, with a par value of $0.001 per share, consisted of the following:
| Convertible Preferred Stock |
2010 | |||
| Series A-1 |
$ | 1,470,370 | ||
| Series A-2 |
1,430,749 | |||
| Series B |
7,530,180 | |||
| Series C-1 |
15,320,640 | |||
| Series C-2 |
22,447,353 | |||
| Series C-3 |
41,600,191 | |||
|
|
|
|||
| $ | 89,799,483 | |||
|
|
|
|||
| 12. | Stockholders’ Equity (Deficit) |
Common Stock
In conjunction with the issuance of Series B convertible preferred stock, the Company issued common stock to several Series B investors. The Company had the right, but not the obligation, to repurchase the common stock, if certain conditions were not met by the Series B investors. Based on the post-money valuation of the Company on February 9, 2011, 196,335 of the shares of common stock subject to certain repurchase conditions were released to several Series B investors. Additionally, based on the market capitalization of the Company reaching a certain threshold later in 2011, the remaining 26,175 shares of common stock subject to certain repurchase conditions were released.
Stock-Based Compensation Plans
The Company has had stock-based compensation plans since 1997. The plans adopted in 1997 and 2007 provided only for awards of stock options. The 2010 Equity Incentive Plan (the “2010 Plan”), which is the only plan under which awards may currently be made, authorizes awards in the form of stock options, stock appreciation rights, restricted stock, restricted stock units (“RSUs”), performance units and performance shares. Awards under the 2010 Plan may be made to employees, directors and certain contractors as determined by the compensation committee of the board of directors. There were 2,486,663and 3,795,563 shares of common stock authorized and reserved at December 31, 2010 and 2011 under these plans.
Stock Options
Under the various plans, employees have been granted incentive stock options, while directors and contractors have been granted non-qualified options. The plans allow the holder of an option to purchase common stock at the exercise price, which was at or above the fair value of the Company’s common stock on the date of grant. At December 31, 2011, there were 1,159,964 shares of common stock reserved for issuance pursuant to outstanding awards made under the 2010 Plan and 331,465 additional shares of common stock reserved for issuance under future awards. In addition, there were 2,019,333 and 1,703,258 shares of common stock reserved at December 31, 2010 and 2011, respectively, for issuance pursuant to outstanding stock options granted under the 1997 and 2007 plans.
Generally, options granted under the 1997 and 2007 plans fully vest four years from the grant date and have a term of ten years. Options granted under the 1997 and 2007 plans in connection with an employee’s commencement of employment generally vest over a four-year period with one-half of the shares subject to the grant vesting after two years of employment and remaining options vesting monthly over the remainder of the
94
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
four-year period. Options granted for performance or promotions vest monthly over a four-year period. Generally, options granted under the 2010 Plan vest annually over a four year period. Unexercised stock options terminate on the tenth anniversary date after the date of grant. The Company recognizes the stock-based compensation expense over the requisite service period of the individual grantees, which generally equals the vesting period. The Company utilizes a Black-Scholes option-pricing model to estimate the value of stock options. The Black-Scholes model allows the use of a range of assumptions related to volatility, risk-free interest rate, and employee exercise behavior. Since the Company does not yet have sufficient history as a publicly traded company to evaluate volatility, the Company has used an average of several peer companies’ volatilities to determine a reasonable estimate of volatility. For purposes of identifying similar entities, the Company considered characteristics such as industry, length of trading history, market capitalization and similar product pipelines.
Due the lack of available quarterly data for these peer companies and insufficient history as a public company, the Company elected to use the “simplified” method for “plain vanilla” options to estimate the expected term of the stock options grants. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option. The risk-free interest rate is derived from the weighted-average yield of a Treasury security with the same term as the expected life of the options and the dividend yield is based on historical experience and the Company’s estimate of future dividend yields.
The weighted-average value of the individual options granted during 2009, 2010 and 2011 were determined using the following assumptions:
| Year Ended December 31, | ||||||
| 2009 | 2010 | 2011 | ||||
| Weighted-average volatility |
35.59% | 36.26% | 82.77% | |||
| Risk-free interest rate |
3.67% | 3.52% | 2.27% | |||
| Weighted-average expected life (in years) |
10.0 | 9.8 | 6.2 | |||
| Dividend yield |
0.00% | 0.00% | 0.00% | |||
The resulting value of options granted was $1,226,783, and $6,328,869 for the year ended December 31, 2010 and 2011, respectively, which will be amortized into income over the remaining requisite service period. The Company recognized stock-based compensation cost, net of forfeitures, in the amount of $381,877, $523,562
95
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
and $1,960,345 for the year ended December 31, 2009, 2010 and 2011, respectively. The Company’s stock option activity and related information are summarized as follows:
| Options | Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (In Years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at January 1, 2009 |
1,354,321 | $ | 2.72 | |||||||||||||
| Granted during year |
374,227 | 2.54 | ||||||||||||||
| Exercised during year |
— | — | ||||||||||||||
| Expired during year |
(119,607 | ) | 6.58 | |||||||||||||
| Forfeited during year |
(20,732 | ) | 2.34 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2009 |
1,588,209 | $ | 2.39 | 7.23 | $ | 1,113,770 | ||||||||||
|
|
|
|||||||||||||||
| Exercisable at December 31, 2009 |
980,812 | 2.23 | 6.44 | 442,029 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at January 1, 2010 |
1,588,209 | $ | 2.39 | |||||||||||||
| Granted during year |
555,987 | 4.22 | ||||||||||||||
| Exercised during year |
(69,649 | ) | 2.11 | |||||||||||||
| Expired during year |
(8,224 | ) | 7.64 | |||||||||||||
| Forfeited during year |
(46,990 | ) | 5.08 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2010 |
2,019,333 | $ | 2.82 | 7.01 | $ | 8,887,814 | ||||||||||
|
|
|
|||||||||||||||
| Exercisable at December 31, 2010 |
1,271,226 | 2.41 | 6.10 | $ | 6,108,168 | |||||||||||
|
|
|
|||||||||||||||
| Outstanding at January 1, 2011 |
2,019,333 | $ | 2.82 | |||||||||||||
| Granted during year |
926,956 | 9.39 | ||||||||||||||
| Exercised during year |
(283,061 | ) | 2.10 | |||||||||||||
| Expired during year |
(23,039 | ) | 7.64 | |||||||||||||
| Forfeited during year |
(48,180 | ) | 7.07 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2011 |
2,592,009 | 5.12 | 7.38 | 1,652,483 | ||||||||||||
| Exercisable at December 31, 2011 |
1,339,341 | 2.69 | 5.97 | 1,523,957 | ||||||||||||
96
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following is a rollforward of the Company’s nonvested stock options from December 31, 2008 to December 31, 2011.
| Options | Weighted- Average Grant Date Value |
|||||||
| Nonvested stock options at January 1, 2009 |
554,335 | $ | 1.05 | |||||
| Granted during year |
374,227 | 0.92 | ||||||
| Vested during year |
(180,826 | ) | 1.15 | |||||
| Expired during year |
(119,607 | ) | 6.59 | |||||
| Forfeited during year |
(20,732 | ) | 1.24 | |||||
|
|
|
|||||||
| Nonvested at December 31, 2009 |
607,397 | $ | 1.01 | |||||
|
|
|
|||||||
| Nonvested stock options at January 1, 2010 |
607,397 | $ | 1.01 | |||||
| Granted during year |
555,987 | 2.21 | ||||||
| Vested during year |
(360,063 | ) | 1.31 | |||||
| Expired during year |
(8,224 | ) | 4.41 | |||||
| Forfeited during year |
(46,990 | ) | 1.43 | |||||
|
|
|
|||||||
| Nonvested at December 31, 2010 |
748,107 | $ | 1.97 | |||||
| Nonvested stock options at January 1, 2011 |
748,107 | $ | 1.97 | |||||
| Granted during year |
926,956 | 6.83 | ||||||
| Vested during year |
(330,132 | ) | 1.97 | |||||
| Expired during year |
(44,083 | ) | 5.22 | |||||
| Forfeited during year |
(48,180 | ) | 4.81 | |||||
|
|
|
|||||||
| Nonvested at December 31, 2011 |
1,252,668 | 5.34 | ||||||
The total grant date value of options vested during 2009, 2010 and 2011 was $351,664, $521,196 and $650,068, respectively. As of December 31, 2010 and December 31, 2011, the total remaining unrecognized compensation cost related to nonvested stock options granted, was $1,204,746 and $5,225,153, respectively, which will be amortized over the remaining requisite service period. The intrinsic value of options exercised was $302,160 and $1,965,031 for the year ended December 31, 2010 and December 31, 2011, respectively.
Restricted Stock Units
In May 2011, the Company adopted and granted awards under a new performance-based restricted stock unit (“RSU”) program (the “2011 RSU Program”) under the Company’s 2010 Equity Incentive Plan. Each unit represents an amount equal to one share of the Company’s common stock. The RSUs will be earned, in whole or in part, based on performance and service conditions. The performance condition is based upon whether the Company receives regulatory approval to sell a therapeutic product, and the awards include a target number of RSUs that will vest upon a First Commercial Approval, and a maximum number of RSUs that will vest upon a Second Commercial Approval. The RSUs will vest fifty percent based on the performance condition of commercial approval and fifty percent one year thereafter to fulfill the service condition, which requires the employee to remain employed by the company.
As of December 31, 2011, the Company has 271,213 RSU awards outstanding. The unrecorded stock compensation expense is based on number of units granted, less estimated forfeitures based on the Company’s
97
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
historical forfeiture rate of 6.49%, and the closing market price of the Company’s common stock at the grant date. As of December 31, 2011, the performance condition of obtaining regulatory approval has not been achieved, therefore, no vesting has occurred. The awards are being accounted for under ASC 718, and compensation expense is to be recorded if the Company has determined that it is probable that the performance conditions will be achieved. As of December 31, 2011, it was not probable that the performance conditions will be achieved, therefore, no compensation expense was recorded for the year ended December 31, 2011. Unrecorded compensation expense for the 2011 RSU program as of December 31, 2011 was $2.7 million.
| 13. | Income Taxes |
A reconciliation of the expected income tax benefit (expense) computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows for year ended December 31, 2009, 2010, and 2011:
| December 31, | ||||||||||||
| 2009 | 2010 | 2011 | ||||||||||
| Income tax computed at federal statutory tax rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State taxes |
8.5 | % | 8.4 | % | 6.5 | % | ||||||
| Research and development credits |
5.7 | % | 4.8 | % | 3.9 | % | ||||||
| Permanent differences |
(3.6 | )% | (0.7 | )% | 0.1 | % | ||||||
| Other |
0.2 | % | (0.1 | )% | (6.3 | )% | ||||||
| Change in valuation allowance |
(44.8 | )% | (46.4 | )% | (38.2 | )% | ||||||
|
|
|
|
|
|
|
|||||||
| Total |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
|
|
|
|
|
|
|
|||||||
At December 31, 2011, the Company has net operating loss carryforwards totaling approximately $132,500,000 and $131,100,000 for federal and state income taxes, respectively, that may be used to offset future taxable income. If not used, the carryforwards will begin expiring in the year 2022. As of December 31, 2009 and December 31, 2010, the Company had not experienced a change in ownership, as defined under Section 382 of the U.S. Internal Revenue Code (the “Code”). The Company has determined that it experienced such a change in ownership as a result of the public offering in August 2011. As a result, the future use of its net operating losses and credit equivalents will be limited to approximately $17,700,000 per year. Due to uncertainty surrounding the realization of its deferred tax assets, the Company has recorded an equal and offsetting valuation allowance against its net deferred tax assets.
Net deferred tax assets and liabilities are comprised of the following:
| December 31, | ||||||||
| 2010 | 2011 | |||||||
| Net operating loss carryforwards |
$ | 37,987,000 | $ | 51,224,000 | ||||
| Research and development credit carryforwards |
5,169,000 | 6,716,000 | ||||||
| Stock Options |
— | 438,000 | ||||||
| Accrued Wages |
— | 315,000 | ||||||
| Accrued vacation |
49,000 | 52,000 | ||||||
| Other |
(1,000 | ) | (54,000 | ) | ||||
|
|
|
|
|
|||||
| 43,204,000 | 58,691,000 | |||||||
| Less valuation allowance |
(43,204,000 | ) | (58,691,000 | ) | ||||
|
|
|
|
|
|||||
| $ | — | $ | — | |||||
|
|
|
|
|
|||||
98
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 14. | Collaborative Arrangements |
In December 2005, the Company entered into a collaborative agreement with Bristol-Myers Squibb (“BMS”) granting BMS an exclusive worldwide license to its proprietary patent rights and know-how related to methods and compositions useful in making folate conjugates to develop and commercialize folate conjugates of epothilone compounds. To date, the Company received upfront payments, milestone payments, and maintenance payments totaling $8.1 million from BMS. As all payments are not contingent on specific performance or ongoing responsibilities of the Company, the Company recognized the upfront revenue upon contract signing, milestone revenue when milestones were achieved, and annual maintenance fees when they were due. BMS terminated this agreement during 2010. As a result, the Company does not expect to receive additional milestones or royalties associated with this program.
In October 2007, the Company entered into an exclusive worldwide license with R&D Biopharmaceuticals to research, develop, and commercialize products containing conjugates of folate receptor targeting compounds and tubulysin compounds. The Company paid an upfront fee of $300,000 as research and development and has since paid $50,000 in annual maintenance fees. In February 2011, this licensing agreement was assigned by R&D Biopharmaceuticals to Trientlgasse. The Company could pay $6,300,000 in additional contingent payments upon the achievement of specific scientific, clinical, and regulatory milestones, in addition to royalties upon commercial sales. All payments have been expensed as research and development as incurred, as there is no alternate future use for this technology.
In December 1995, as amended in October 1998, the Company entered into an exclusive license agreement with Purdue Research Foundation, which licenses the right under certain patents to the Company. The Company is obligated to pay an annual minimum royalty of $12,500 until commercial sales commence, following which time the payment of single digit royalty rates will commence. All payments have been expensed as incurred, as the Company does not believe there is an alternate future use for this technology.
In December 2009, the Company entered into a financial term sheet to be incorporated into a written license agreement with Purdue Research Foundation for a patent related to prostate cancer. The agreement was signed and became effective on March 1, 2010. Pursuant to the exclusive license agreement, the Company is subject to annual payments of $15,000, payable until first commercial sale, following which time the payment of single digit royalty rates will commence. In addition, certain clinical and regulatory milestone payments of $500,000 along with sales-based milestones related to third-party sales are also payable. The Company is also subject to penalties totaling $300,000 if certain diligence milestones are not met. Subsequent to the first commercial sales, the annual milestone is $100,000. Future milestone payments in excess of $500,000 may be waived by Purdue Research Foundation.
| 15. | Related-Party Transactions |
The Company funds research at the employer of one of its founders and current Chief Science Officer. Amounts included in research and development expenses were $352,165, $391,325 and $517,500, for the year ended December 31, 2009, 2010 and 2011, respectively.
During the past three years, the Company has completed various financing transactions which included the issuance of our Preferred Stock and Subordinated Notes. In each case, certain of our existing stockholders participated by purchasing the securities.
In September 2011, the Company entered into exclusive worldwide licenses with On Target Laboratories, L.L.C. (“On Target”) to develop and commercialize products relating to the compound comprising Endocyte’s Folate and DUPA ligands and other certain Licensed Patents. On Target was founded by a relative of the
99
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Company’s Chief Scientific Officer. The Company believes that the terms of the agreement are no less favorable than terms that would have been available in an arm’s length transaction. On Target is solely responsible for conducting research and development, seeking regulatory approval and commercialization of products. The Company received nonrefundable upfront license fees totaling $191,000 from On Target in December 2011. The Company has determined that the deliverables under this agreement do not meet the criteria required for separate accounting units for the purposes of revenue recognition, and as a result, the Company recognizes revenue from non-refundable, upfront fees when the performance condition has been achieved and there is reasonable assurance of collectability. If On Target fails to meet minimum spend requirements in 2011 and 2012 on research and development the Company has the right to terminate the agreement. The Company will be entitled to receive minimum royalty payments annually based on net sales, but there is no guarantee that a commercial product will be developed and approved for commercial sales. The Company will also be entitled to reimbursement of expenses relating to patent expenses and payments to the inventors and Purdue University if certain milestones are met. During the fourth quarter of 2011, the Company received a $50,000 reimbursement from On Target for payments made to inventors for issued patents.
| 16. | Retirement Plans |
The Company maintains a 401(k) retirement savings plan to provide retirement benefits for substantially all of its employees. Participants in the plan may elect to contribute a portion of their annual compensation to the plan, limited to the maximum allowed by the Code. The Company does not currently match 401(k) contributions.
| 17. | Selected Quarterly Financial Data (unaudited) |
The following table summarizes the unaudited statements of operations for each quarter of 2011 and 2010 (in thousands except per share amounts):
| Quarters Ended | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| 2011 |
||||||||||||||||
| Revenue |
$ | — | $ | — | $ | — | $ | 191 | ||||||||
| Cost and Expenses: |
||||||||||||||||
| Research and development |
4,439 | 7,721 | 8,915 | 7,753 | ||||||||||||
| General and administrative |
2,074 | 2,341 | 2,723 | 2,862 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(6,513 | ) | (10,062 | ) | (11,638 | ) | (10,424 | ) | ||||||||
| Interest, net |
(679 | ) | (453 | ) | (414 | ) | (313 | ) | ||||||||
| Other |
3 | (4 | ) | (18 | ) | (17 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (7,189 | ) | $ | (10,519 | ) | $ | (12,070 | ) | $ | (10,754 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted net loss per share(1) |
$ | (0.43 | ) | $ | (0.35 | ) | $ | (0.36 | ) | $ | (0.30 | ) | ||||
| Weighted average common shares outstanding — basic and diluted(2) |
16,907,823 | 29,693,004 | 33,414,303 | 35,745,364 | ||||||||||||
100
ENDOCYTE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| Quarters Ended | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| 2010 |
||||||||||||||||
| Revenue |
$ | — | $ | — | $ | — | $ | — | ||||||||
| Cost and Expenses: |
||||||||||||||||
| Research and development |
3,899 | 3,701 | 3,671 | 3,290 | ||||||||||||
| General and administrative |
1,476 | 1,546 | 1,700 | 1,317 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(5,375 | ) | (5,247 | ) | (5,371 | ) | (4,607 | ) | ||||||||
| Interest, net |
(246 | ) | (201 | ) | (217 | ) | (393 | ) | ||||||||
| Other |
12 | 26 | (126 | ) | 1,652 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (5,609 | ) | $ | (5,422 | ) | $ | (5,714 | ) | $ | (3,348 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Basic and diluted net loss per share(1) |
$ | (6.16 | ) | $ | (5.93 | ) | $ | (6.16 | ) | $ | (3.57 | ) | ||||
| Weighted average common shares outstanding — basic and diluted(2) |
911,066 | 914,580 | 927,703 | 937,088 | ||||||||||||
| (1) | Per common share amounts for the quarters and full years have been calculated separately. Accordingly, the sum of quarterly amounts may not equal the annual amount because of differences in the weighted average common shares outstanding during each period, principally due to the effect of share issuances by the Company during the year. |
| (2) | Diluted weighted average common shares outstanding are identical to basic weighted average common shares outstanding and Diluted EPS is identical to Basic EPS for all quarters of 2011 and 2010 because common share equivalents are excluded from the calculations of diluted weighted average common shares outstanding for those quarters, as their effect is anti-dilutive. |
101
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
As of December 31, 2011, management, with the participation of our Chief Executive Officer and Chief Financial Officer, performed an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act. Our disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and that such information is accumulated and communicated to our management, including the Chief Executive Officer and the Chief Financial Officer, to allow timely decisions regarding required disclosures. Because of inherent limitations, any controls and procedures, no matter how well designed and operated, can provide only reasonable, and not absolute, assurance of achieving the desired control objective. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2011, the design and operation of our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting during the quarter ended December 31, 2011, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) of the Exchange Act. Internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in the Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management’s assessment included evaluation of such elements as the design and operating effectiveness of key financial reporting controls, process documentation, accounting policies, and our overall control environment. Based on this evaluation, our management concluded that, as of December 31, 2011, our internal control over financial reporting was effective based on such criteria.
| Item 9B. | Other Information |
None.
102
| Item 10. | Directors, Executive Officers and Corporate Governance |
Directors
The information required by this item set forth under the caption “Proposal No. 1 — Election of Directors” in our definitive Proxy Statement to be used in connection with the 2012 Annual Meeting of Stockholders (the “2012 Proxy Statement”) and incorporated herein by reference.
Executive Officers
Information regarding our executive officers is set forth in Item 1 of Part I of this annual report under the caption “Executive Officers of the Registrant.”
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that applies to all of our directors and officers and other employees, including our principal executive officer, principal financial officer and principal accounting officer. This code is publicly available through the Investor Relations section of our website at www.endocyte.com. To the extent permissible under applicable law, the rules of the SEC or NASDAQ listing standards, we intend to post on our website any amendment to the code of business conduct and ethics, or any grant of a waiver from a provision of the code of business conduct and ethics, that requires disclosure under applicable law, the rules of the SEC or NASDAQ listing standards.
Audit Committee
The information required by this item relating to our audit committee is set forth under the caption “Corporate Governance” and “Meetings and Committees of the Board” in the 2012 Proxy Statement and incorporated herein by reference.
Section 16(a) Beneficial Ownership Reporting Compliance
The information required by this item relating to our audit committee is set forth under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in the 2012 Proxy Statement and incorporated herein by reference.
Corporate Governance
The information required by this item relating to the procedures by which stockholders may recommend nominees to the board of directors is set forth under the caption “Corporate Governance Matters” in the 2012 Proxy Statement and incorporated herein by reference.
| Item 11. | Executive Compensation |
The information required by this item is set forth under the captions “Executive Compensation” and “Meetings and Committees of the Board” in the 2012 Proxy Statement and incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item with respect to security ownership of certain beneficial owners and management is set forth under the caption “Beneficial Ownership of Common Stock by Certain Stockholders and Management” in the 2012 Proxy Statement and incorporated herein by reference.
103
Equity Compensation Plan Information
The following table provides information regarding our equity compensation plans as of December 31, 2011:
| Equity Compensation Plan Information | ||||||||||||
| Plan Category |
Number
of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights (a) |
Weighted Average Exercise Price of Outstanding Options, Warrants and Rights (b) |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plan (Excluding Securities Referenced in Column (a)) (c) |
|||||||||
| Equity compensation plans approved by security holders:(1) |
2,592,009 | $ | 5.12 | 331,465(2) | ||||||||
| Equity compensation plans not approved by security holders: |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
2,592,009 | $ | 5.12 | 331,465 | ||||||||
|
|
|
|
|
|
|
|||||||
| (1) | Consists of the 1997 Stock Plan, the 2007 Stock Plan and the 2010 Equity Incentive Plan. |
| (2) | The 1997 Stock Plan provides for the grant of incentive stock options, nonqualified stock options and stock purchase rights. The original number of shares available for awards under the 1997 Stock Plan was 1,047,120. On December 31, 2011 there were no shares available for awards under the 1997 Stock Plan. The 2007 Stock Plan provides for the grant of incentive stock options, nonqualified stock options and stock purchase rights. The original number of shares available for awards under the 2007 Stock Plan was 1,730,237. On December 31, 2011 there were no shares available for awards under the 2007 Stock Plan. The 2010 Equity Incentive Plan provides for incentive stock options, nonqualified stock options, stock appreciation rights, restricted stock, restricted stock units, performance units and performance shares. The original number of shares available for awards under the 2010 Equity Incentive Plan was 1,498,929, and on December 31, 2011, there were 331,465 shares available for awards under the 2010 Equity Incentive Plan. Effective January 1, 2012, the Board of Directors increased the number of shares available for awards under the 2010 Equity Incentive Plan by 1,400,000 shares. |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this item is set forth under the captions “Corporate Governance” and “Transactions with Related Persons” in the 2012 Proxy Statement and incorporated herein by reference.
| Item 14. | Principal Accounting Fees and Services |
The information required by this item is set forth under “Proposal 2 — Ratification of Independent Registered Public Accounting Firm” in the 2012 Proxy Statement and incorporated herein by reference.
104
| Item 15. | Exhibits, Financial Statement Schedules. |
(a) 1. Financial Statements
The following financial statements of Endocyte, Inc. and its subsidiaries are set forth in Part II, Item 8.
2. Financial Statement Schedules
Financial statement schedules are omitted because they are not applicable or the required information is shown in the financial statements or the notes thereto.
3. Exhibits
A list of exhibits required to be filed as part of this report is set forth in the Index to Exhibits, which immediately precedes such exhibits and is incorporated herein by reference.
105
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ENDOCYTE, INC. | ||
| By: |
/s/ P. Ron Ellis | |
| P. Ron Ellis President and Chief Executive Officer | ||
Date: March 28, 2012
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ P. Ron Ellis Ron Ellis |
Director, President and Chief Executive Officer (Principal Executive Officer) |
March 28, 2012 | ||
| /s/ Michael A. Sherman Michael A. Sherman |
Chief Financial Officer (Principal Financial Officer) |
March 28, 2012 | ||
| /s/ Beth A. Taylor Beth A. Taylor |
Corporate Controller (Principal Accounting Officer) |
March 28, 2012 | ||
| /s/ John C. Aplin John C. Aplin |
Chairman of the Board of Directors | March 28, 2012 | ||
| /s/ Philip S. Low Philip S. Low |
Director and Chief Science Officer | March 28, 2012 | ||
| /s/ Douglas G. Bailey Douglas G. Bailey |
Director | March 28, 2012 | ||
| /s/ Keith E. Brauer Keith E. Brauer |
Director | March 28, 2012 | ||
| /s/ John G. Clawson John G. Clawson |
Director | March 28, 2012 | ||
| /s/ Ann F. Hanham Ann F. Hanham |
Director | March 28, 2012 | ||
| /s/ Fred A. Middleton Fred A. Middleton |
Director | March 28, 2012 | ||
|
James S. Shannon |
Director | March 28, 2012 |
106
| Exhibit Number |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation of Endocyte, Inc. (incorporated by reference to Exhibit 3.1 to Annual Report on Form 10-K for the year ended December 31, 2010 filed March 18, 2011). | |
| 3.2 | Amended and Restated Bylaws of Endocyte, Inc. (incorporated by reference to Exhibit 3.2 to Annual Report on Form 10-K for the year ended December 31, 2010 filed March 18, 2011). | |
| 4.1 | Specimen Common Stock Certificate (incorporated by reference to Exhibit 4.1 of Amendment No. 3 to Form S-1 (Registration No. 333-168904) filed January 12, 2011). | |
| 4.2 | Third Amended and Restated Investor Rights Agreement dated March 9, 2007, among Endocyte, Inc. and the parties set forth therein, as amended (incorporated by reference to Exhibit 4.2 of Amendment No. 1 to Form S-1 (Registration No. 333-168904) filed September 28, 2010). | |
| 4.3 | Warrant to Purchase Shares of Series C-3 Preferred Stock issued by Endocyte, Inc. to General Electric Capital Corporation on December 31, 2007 (incorporated by reference to Exhibit 4.3 to Form S-1 (Registration No. 333-168904) filed August 17, 2010). | |
| 4.4 | Warrant to Purchase Shares of Series C-3 Preferred Stock issued by Endocyte, Inc. to Oxford Finance Corporation on December 31, 2007 (incorporated by reference to Exhibit 4.4 to Form S-1 (Registration No. 333- 168904) filed August 17, 2010). | |
| 4.5 | Warrant to Purchase Stock issued by Endocyte, Inc. to Silicon Valley Bank on August 27, 2010 (incorporated by reference to Exhibit 4.5 of Amendment No. 1 to Form S-1 (Registration No. 333- 168904) filed September 28, 2010). | |
| 4.6 | Warrant to Purchase Stock issued by Endocyte, Inc. to Midcap Funding III, LLC on August 27, 2010 (incorporated by reference to Exhibit 4.6 of Amendment No. 1 to Form S-1 (Registration No. 333-168904) filed September 28, 2010). | |
| 10.1* | Form of Director and Executive Officer Indemnification Agreement (incorporated by reference to Exhibit 10.1 to Form S-1 (Registration No. 333-168904) filed August 17, 2010). | |
| 10.2* | 1997 Stock Plan and forms of option agreements thereunder (incorporated by reference to Exhibit 10.2 to Form S-1 (Registration No. 333-168904) filed August 17, 2010). | |
| 10.3* | 2007 Stock Plan and forms of option agreements thereunder (incorporated by reference to Exhibit 10.3 to Form S-1 (Registration No. 333-168904) filed August 17, 2010). | |
| 10.4* | 2010 Equity Incentive Plan (incorporated by reference to Exhibit 10.4 to Form S-1 (Registration No. 333- 168904) filed August 17, 2010). | |
| 10.5* | Form of Option Agreement under the 2010 Equity Incentive Plan (incorporated by reference to Exhibit 10.5 to Annual Report on Form 10-K for the year ended December 31, 2010 filed March 18, 2011). | |
| 10.6* | 2010 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.5 to Form S-1 (Registration No. 333-168904) filed August 17, 2010). | |
| 10.7** | Lease dated January 1, 2012 between Endocyte, Inc. and American Yorkshire Club. | |
| 10.8 | Lease dated May 30, 2008 between Endocyte, Inc. and Zeller Management Corporation, amended (incorporated by reference to Exhibit 10.7 to Form S-1 (Registration No. 333-168904) filed August 17, 2010). | |
| 10.9 | Lease dated January 3, 2012 between Endocyte, Inc. and Purdue Research Foundation. | |
| 10.10 | Amended and Restated Exclusive License Agreement dated October 21, 1998 between Endocyte, Inc. and Purdue Research Foundation, as amended (incorporated by reference to Exhibit 10.11 of Amendment No. 1 to Form S-1 (Registration No. 333-168904) filed September 28, 2010). | |
| 10.11 | Exclusive License Agreement effective March 1, 2010 between Endocyte, Inc. and Purdue Research Foundation (incorporated by reference to Exhibit 10.12 of Amendment No. 1 to Form S-1 (Registration No. 333- 168904) filed September 28, 2010). |
| Exhibit Number |
Description | |
| 10.12* | Form of Change in Control Agreements each dated August 25, 2010 between Endocyte, Inc. and certain of its named executive officers (incorporated by reference to Exhibit 10.13 of Amendment No. 1 to Form S-1 (Registration No. 333-168904) filed September 28, 2010). | |
| 10.13* | Change in Control Agreement dated August 25, 2010 between Endocyte, Inc. and P. Ron Ellis (incorporated by reference to Exhibit 10.14 of Amendment No. 1 to Form S-1 (Registration No. 333-168904) filed September 28, 2010). | |
| 10.14* | Change in Control Agreement dated August 25, 2010 between Endocyte, Inc. and Michael A. Sherman (incorporated by reference to Exhibit 10.15 of Amendment No. 1 to Form S-1 (Registration No. 333- 168904) filed September 28, 2010). | |
| 10.15 | Patent Assignment Agreement dated November 1, 2007 among Endocyte, Inc. and the parties set forth therein (incorporated by reference to Exhibit 10.16 of Amendment No. 1 to Form S-1 (Registration No. 333- 168904) filed September 28, 2010). | |
| 10.16 | Loan and Security Agreement dated August 27, 2010 among Endocyte, Inc., Midcap Funding III, LLC and Silicon Valley Bank, as amended on December 14, 2010 (incorporated by reference to Exhibit 10.17 of Amendment No. 2 to Form S-1 (Registration No. 333-168904) filed December 15, 2010). | |
| 10.17 | Note Purchase Agreement, dated December 14, 2010 among Endocyte, Inc. and the parties set forth therein (incorporated by reference to Exhibit 10.18 of Amendment No. 2 to Form S-1 (Registration No. 333- 168904) filed December 15, 2010). | |
| 10.18 | Form of Subordination Agreement between Endocyte, Inc. and the parties set forth in the Note Purchase Agreement dated December 14, 2010 (incorporated by reference to Exhibit 10.19 of Amendment No. 2 to Form S-1 (Registration No. 333-168904) filed December 15, 2010). | |
| 10.19 | Form of Endocyte, Inc. 2010 Equity Incentive Plan Performance-Based Restricted Stock Unit Award Agreement (2011 RSU Program) (incorporated by reference to Exhibit 10.1 to Current Report on Form 8-K filed June 2, 2011). | |
| 10.20 | Second Loan Modification Agreement dated July 19, 2011 among Endocyte, Inc. and Midcap Funding V, LLC (incorporated by reference to Exhibit 10.23 to Amendment No. 1 for Form S-1 (Registration No. 333-175573) filed July 21, 2011). | |
| 23.1 | Consent of Ernst & Young, LLP, Independent Registered Public Accounting Firm. | |
| 31.1 | Certification pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934 of the Chief Executive Officer, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934 of the Chief Financial Officer, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101^ | The following materials from Registrant’s Annual Report on Form 10-K for the year ended December 31, 2011, formatted in Extensible Business Reporting Language (XBRL) includes: (i) Consolidated Balance Sheets at December 31, 2010 and 2011, (ii) Consolidated Statements of Operations for the years ended December 31, 2009, 2010 and 2011, (iii) Consolidated Statements of Convertible Preferred Stock, Stockholders’ Equity (Deficit) and Comprehensive Loss for the years ended December 31, 2009, 2010 and 2011, (iv) Consolidated Statements of Cash Flows for years ended December 31, 2010 and 2011 and (v) Notes to Consolidated Financial Statements. |
| * | Indicates management contracts or compensatory plans or arrangements. |
| ** | Application has been made to the Securities and Exchange Commission to seek confidential treatment of certain provisions of this exhibit. Omitted material for which confidential treatment has been requested has been filed separately with the Securities Exchange Commission. |
| ^ | XBRL information is furnished and not filed or a part of a registration statement or prospectus for purposes of sections 11 or 12 of the Securities Exchange Act of 1933, as amended, is deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934, as amended, and otherwise is not subject to liability under these sections. |