Attached files
| file | filename |
|---|---|
| EX-2.1 - EXHIBIT 2.1 - Q Therapeutics, Inc. | v237419_ex2-1.htm |
| EX-16.1 - EXHIBIT 16.1 - Q Therapeutics, Inc. | v237419_ex16-1.htm |
| EX-99.2 - EXHIBIT 99.2 - Q Therapeutics, Inc. | v237419_ex99-2.htm |
| EX-10.1 - EXHIBIT 10.1 - Q Therapeutics, Inc. | v237419_ex10-1.htm |
| EX-99.1 - EXHIBIT 99.1 - Q Therapeutics, Inc. | v237419_ex99-1.htm |
| EX-10.3 - EXHIBIT 10.3 - Q Therapeutics, Inc. | v237419_ex10-3.htm |
| EX-99.3 - EXHIBIT 99.3 - Q Therapeutics, Inc. | v237419_ex99-3.htm |
| EX-10.2 - EXHIBIT 10.2 - Q Therapeutics, Inc. | v237419_ex10-2.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of
The Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): October 13, 2011
GRACE 2, INC.
(Exact name of registrant as specified in its charter)
|
Delaware
|
000-52062
|
20-3708500
|
||
|
(State or other jurisdiction of incorporation)
|
(Commission File Number)
|
(IRS Employer Identification No.)
|
||
|
615 Arapeen Drive, Suite 102
Salt Lake City, UT
|
84108
|
|||
|
(Address of principal executive offices)
|
(Zip Code)
|
Registrant’s telephone number, including area code:
Phone: 801-582-5400; Fax: 801-582-5401
N/A
(Former name or former address, if changed since last report.)
With a copy to:
Joseph M. Patricola, Esq.
The Sourlis Law Firm
The Courts of Red Bank
130 Maple Avenue, Suite 9B2
Red Bank, New Jersey 07701
Direct Dial: (732) 618-2843
T: (732) 530-9007
F: (732) 530-9008
JoePatricola@SourlisLaw.com
www.SourlisLaw.com
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
|
¨
|
Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
|
|
¨
|
Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
|
|
¨
|
Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
|
|
¨
|
Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
|
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Current Report on Form 8-K contains forward-looking statements that involve risks and uncertainties, principally in the sections entitled “Description of Business,” “Risk Factors,” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” All statements other than statements of historical fact contained in this Current Report on Form 8-K, including statements regarding future events, our future financial performance, business strategy and plans and objectives of management for future operations, are forward-looking statements. We have attempted to identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” or “will” or the negative of these terms or other comparable terminology. Although we do not make forward-looking statements unless we believe we have a reasonable basis for doing so, we cannot guarantee their accuracy. These statements are only predictions and involve known and unknown risks, uncertainties and other factors, including the risks outlined under “Risk Factors” or elsewhere in this Current Report on Form 8-K. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time and it is not possible for us to predict all risk factors, nor can we address the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause our actual results to differ materially from those contained in any forward-looking statements.
We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy, short-term and long-term business operations, and financial needs. These forward-looking statements are subject to certain risks and uncertainties that could cause our actual results to differ materially from those reflected in the forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in this Current Report on Form 8-K, and in particular, the risks discussed below and under the heading “Risk Factors” and those discussed in other documents we file with the Securities and Exchange Commission that are incorporated into this Current Report on Form 8-K by reference. The following discussion should be read in conjunction with our consolidated financial statements and notes thereto included in this Report. We undertake no obligation to revise or publicly release the results of any revision to these forward-looking statements. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this Current Report on Form 8-K may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
You should not place undue reliance on any forward-looking statement, each of which applies only as of the date of this Current Report on Form 8-K. Before you invest in our common stock, you should be aware that the occurrence of the events described in the section entitled “Risk Factors” and elsewhere in this Current Report on Form 8-K could negatively affect our business, operating results, financial condition and stock price. Except as required by law, we undertake no obligation to update or revise publicly any of the forward-looking statements after the date of this Current Report on Form 8-K to conform our statements to actual results or changed expectations.
2
ITEM 1.01. ENTRY INTO A MATERIAL DEFINITIVE AGREEMENT.
General
Grace 2, Inc. (hereinafter “Grace 2”, “Grace” or “the Company”), a corporation formed in the State of Delaware on October 27, 2005, which upon completion of the Company’s name change shall be known as Q Holdings, Inc. (throughout this Current Report on Form 8-K Grace 2 sometimes referred to as “Q Holdings” depending on context) is a holding company which, through its wholly owned subsidiary, Q Therapeutics, Inc., a corporation formed in the State of Delaware on March 28, 2002 (hereinafter “Q Therapeutics” or “Q”), is a biotechnology company headquartered in Salt Lake City, Utah. We believe that our company has taken a novel path to developing treatments for debilitating and sometimes fatal diseases of the central nervous system (“CNS”), utilizing and patenting ‘stem or progenitor cell’ therapeutics and their cellular progeny for the treatment of such devastating illnesses.
Merger Agreement
On October 13, 2011, Grace 2 entered into an Agreement and Plan of Merger (the “Agreement”) with Q Acquisition, Inc., a Delaware corporation and wholly owned subsidiary of the Company (“Merger Sub”), and Q Therapeutics. Pursuant to the Agreement, Merger Sub merged with and into Q Therapeutics with Q Therapeutics being the surviving corporation (the “Merger”). Upon the consummation of the Merger, the Company acquired 100% of Q Therapeutics. The Agreement is attached hereto as Exhibit 2.1 to this Current Report on Form 8-K.
As a result of the Merger:
|
|
·
|
Douglas Dyer, the sole director and officer of Grace 2, resigned.
|
|
|
·
|
Q Therapeutics shareholders received, on a pro rata basis, an aggregate of 22,671,923 shares of Grace 2 Common Stock (including shares underlying warrants and options), which immediately upon the consummation of the Merger, represented an 89.7% interest in the Company , thereby rendering Q Therapeutics shareholders the majority owners of the Company;
|
|
|
·
|
Grace 2 filed a Schedule 14C with the US Securities and Exchange Commission notifying the market of its immediate plans to change the Company’s corporate name to Q Holdings, Inc. by way of Amendment to the Company’s Certificate of Incorporation with the State of Delaware (Grace 2 thereafter referred to as “Q Holdings”);
|
|
|
·
|
Q Therapeutics’ existing senior management assumed the same positions with Q Holdings; and
|
|
|
·
|
Q Therapeutics became the wholly owned subsidiary of the Company with the Company conducting all of its operations through Q Therapeutics.
|
Securities Purchase Agreement
Immediately after the Merger, we entered into a securities purchase agreement (the “Purchase Agreement”) with certain accredited investors (the “Investors”) for the issuance and sale in a private placement of investment units (the “Units”), with each Unit consisting of (a) one share of the Company’s common stock, par value $0.0001 per share (the “Common Stock”), (b) a seven-year detachable warrant (“Warrant A”) to purchase one share of Common Stock with an exercise price of $1.00 per share, and (c) a seven-year detachable warrant (“Warrant B” and together with Warrant A, the “Warrants”) to purchase one share of Common Stock with an exercise price of $2.00 per share, for aggregate gross proceeds of $3,803,047 (the “Private Placement”). In the aggregate, we issued to the Investors a total of 3,803,047 shares of Common Stock and a seven-year Warrant A to purchase up to 3,803,047 shares of common stock and a seven-year Warrant B to purchase up to 3,803,047 shares of common stock.
3
Registration Rights Agreement
In connection with the Private Placement, we also entered into a registration rights agreement (the “Registration Rights Agreement”) with the Investors, pursuant to which the Company agreed to file a registration statement on Form S-1 (or any other applicable form exclusively for this offering) (the “Registration Statement”) with the Securities and Exchange Commission (the “SEC”) to register for resale (i) 100% of the shares of our common stock within the Units; and (ii) 100% of the shares of our common stock underlying the Warrants (collectively, the “Registrable Shares”), within 60 calendar days following the closing of the Private Placement, and use the Company’s best efforts to have the registration statement declared effective within 180 calendar days after closing of the Private Placement. If a Registration Statement covering the registration of the Registrable Shares is not filed with the SEC by the Required Filing Date or is not declared effective by the Required Effective Date, the Company shall pay to each Investor as liquidated damages, a cash payment equal to 3% of the aggregate amount invested by such Investor in the Private Placement for every 30-day period until the Registration Statement has been filed or declared effective, or a portion thereof. Such liquidated damages shall not exceed 18% per annum.
Series A Warrants
Each Series A Warrant:
|
(a)
|
entitles the holder or its registered assigns to purchase one share of common stock;
|
|
(b)
|
is exercisable any time after the Closing Date and expires on the date that is seven years following the original issuance date of the Warrants;
|
|
(c)
|
is exercisable, in whole or in part, at an exercise price of $1.00 per share (the “Exercise Price”);
|
|
(d)
|
is exercisable only for cash; and
|
|
(e)
|
is callable by us at a redemption price of $0.001 per warrant share by providing the holder ten day written notice (the “Redemption Period”); provided, however, that no redemption may occur unless (i) the Company’s Common Stock has had a per share closing sales price of at least $1.50 for ten consecutive trading days and (ii) at the date of the redemption notice and during the entire Redemption Period there is an effective registration statement covering the resale of the Warrant Stock.
|
Series B Warrants
Each Series B Warrant:
|
(a)
|
entitles the holder or its registered assigns to purchase one share of common stock;
|
4
|
(b)
|
is exercisable any time after the Closing Date and expires on the date that is seven years following the original issuance date of the Warrants;
|
|
(c)
|
is exercisable, in whole or in part, at an exercise price of $2.00 per share (the “Exercise Price”);
|
|
(d)
|
is exercisable only for cash; and
|
|
(e)
|
is callable by us at a redemption price of $0.001 per warrant share by providing the holder ten day written notice (the “Redemption Period”); provided, however, that no redemption may occur unless (i) the Company’s Common Stock has had a per share closing sales price of at least $3.00 for ten consecutive trading days and (ii) at the date of the redemption notice and during the entire Redemption Period there is an effective registration statement covering the resale of the Warrant Stock.
|
ITEM 2.01. COMPLETION OF ACQUISITION OR DISPOSITION OF ASSETS.
See “ITEM 1.01. ENTRY INTO A MATERIAL DEFINITIVE AGREEMENT.” above and incorporated by reference herein.
Description of Business
Company Overview
Q Holdings, Inc., through our wholly owned subsidiary Q Therapeutics, Inc. is a Salt Lake City, Utah based biopharmaceutical company that is developing human cell-based therapies intended to treat degenerative diseases of the brain and spinal cord, the primary components of the central nervous system (“CNS”). Q Therapeutics, Inc. was incorporated in the state of Delaware in March 2002.
The technology upon which these therapies are based was developed by Q Therapeutics’ co-founder Mahendra Rao, M.D., Ph.D., a global leader in glial stem cell biology, during Dr. Rao’s tenure as a Professor at the University of Utah and as Head of the Stem Cell Section at the National Institutes of Health. Q Therapeutics is managed by an experienced team of biotechnology executives with demonstrated start-up success and advised by leaders in the neurology and stem cell therapeutics fields.
Every year, hundreds of thousands of people suffer with debilitating neurodegenerative diseases such as Amyotrophic Lateral Sclerosis (ALS, or Lou Gehrig’s disease), Multiple Sclerosis, Transverse Myelitis and Spinal Cord Injuries. Traditional drugs tend to fail as long-term treatments of the damages to the CNS caused by these diseases due to the complex nature of these diseases and the nerve damage they inflict. Q Therapeutics is developing a new and nontraditional approach to improve the health of people suffering from neurodegenerative diseases: a human cell based product called Q-Cells®.
Q-Cells® are healthy human glial cells. The job of glial cells in the body is to support and protect neurons, which are the signal transmission lines of the nervous system, by forming an insulating “myelin sheath” around them and providing the necessary growth factors needed to maintain a healthy nervous system. Many neurodegenerative diseases arise when glial cells are damaged or destroyed, causing neurons to malfunction and eventually die. Q-Cells® technology aims to treat neurodegenerative diseases by supplementing the damaged or missing glia in the body with new, healthy cells that can help maintain and/or restore neuron function to a more robust state.
5
The diseases targeted by Q Therapeutics’ products are not well treated with current drug therapies. At best, patients suffering from these diseases can, in some cases, only hope to slow their inexorable progression and the associated disabilities. A handful of companies are exploring the possibility of harnessing the power of stem cells to treat these conditions, though no clear leader has emerged. In addition to utilizing its proprietary cellular products as therapeutic products, Q Therapeutics may evaluate novel ways to utilize these cells to screen for new drugs (small molecule compounds) that could also provide treatments for neurological diseases.
The Company believes that a worldwide market measured in the tens of billions of dollars exists for those companies whose cell-based treatments become commercial products. Q Therapeutics’ patent protected technology represents an opportunity to build on the recent advancements in the stem cell field and bring to market a therapeutic approach that will change the way medicine is practiced in treating many disabling and fatal conditions of the CNS.
Q Therapeutics is raising capital to complete preparations necessary to move into and initiate human clinical trials, initially treating patients that suffer from the always-fatal Amyotrophic Lateral Sclerosis (ALS, or Lou Gehrig’s Disease). Future trials are anticipated in other neurodegenerative diseases, such as Multiple Sclerosis and spinal cord injury.
Q Therapeutics’ Semi-Virtual Business Model
As described below, Q Therapeutics uses a semi-virtual business model to develop its products. Q believes this type of business model to be capital efficient while allowing it to augment its own expertise and capabilities with those of its development partners at the time that outside resources are needed. Q’s semi-virtual business model includes utilizing third parties for certain functions and outsourced services. For example, Q utilizes outside collaborators and contractors to:
|
|
·
|
Leverage its research and development resources by forming collaborations with outside scientists specialized in our areas of interest
|
|
|
·
|
Contract GLP and GMP manufacturing to facilities specialized in such production
|
|
|
·
|
Utilize experienced regulatory consultants to work with FDA
|
|
|
·
|
Contract safety/toxicology studies to qualified GLP labs
|
|
|
·
|
Conduct the clinical trials with physicians and institutions with relevant experience
|
|
|
·
|
Enter into pharmaceutical company collaborations to maximize product sales
|
Q Therapeutics – Taking a Cell-Based Therapeutic Approach to Treatment of Degenerative Conditions of the Central Nervous System
Q Therapeutics was founded in 2002 to further the development and commercialization of the pioneering work of Mahendra Rao, M.D., Ph.D., conducted at the University of Utah (Utah) and National Institutes of Health (NIH). Dr. Rao, a global leader in the development of stem cells as therapeutics, was one of the first to identify and seek patent coverage on stem cells and their progeny cells found in the CNS. After licensing Dr. Rao’s technology from Utah / NIH and raising capital, Q commenced operations in the spring of 2004 to develop cell based therapeutic products that can be sold as “off-the-shelf” pharmaceuticals. That work continues to advance Dr. Rao’s initial findings toward a commercial product.
6
Objectives of Company
Q Therapeutics aims to change the way medicine is practiced in the treatment of many debilitating and often fatal diseases of the brain and spinal cord. Q intends to bring its initial product, Q-Cells® to the market to treat Lou Gehrig’s disease, and eventually other indications, potentially including Multiple Sclerosis, spinal cord injury, Parkinson’s and Alzheimer’s diseases.
Initially, Q is targeting orphan diseases, where the Food and Drug Administration (FDA) allows applying for fast track approvals and market exclusivity, and for which smaller, less expensive clinical trials are often warranted. This approach can result in accelerated commercialization efforts while maintaining a financing approach focused on capital efficiency.
Q’s Approach to the Problem
Q Therapeutics believes it has taken a novel path to developing treatments for debilitating and sometimes fatal diseases of the CNS for which there are currently no long-term effective treatments. Traditional drugs have seen little success as long-term treatments for many neurodegenerative diseases, primarily due to the complex nature of those diseases and the difficulty of replacing damaged nerve cells, or neurons.
The majority of cells in the brain and spinal cord are glial cells that serve to support the health of the neurons. Without healthy and properly functioning glial cells, neurons cannot function properly and will eventually die. In many neurodegenerative diseases, damage to glial cells causes or accelerates loss of function and death of neurons. Q’s approach takes advantage of the normal support and repair mechanisms present in the healthy nervous system whereby supplementation of damaged glial cells with healthy glial cells in patients suffering from neurodegenerative diseases is intended to enable the return to healthy neuron function, thereby ameliorating the negative effects of the disease.
Q’s Initial Proprietary Product - Q-Cells®
Q is developing a new pharmaceutical product (Q-Cells®) intended to provide support to neurons in the patient’s CNS and ameliorate the deleterious effects of diseased cells. This product is comprised of healthy human glial cells that perform therapeutic tasks via multiple mechanisms:
(1) Serving as ‘mini-factories’ to produce the requisite growth factors and other nutrients as well as to remove toxic compounds, all needed for a healthy CNS; and
(2) Forming the insulating layer known as myelin around the axons of neurons, enabling their normal function for signal transmission.
Q-Cells® accomplish these complementary functions in a process that is unique to stem and progenitor cells. After Q-Cells® are injected into the CNS, they differentiate into the two mature glial cell subtypes found in the brain and spinal cord:
(1) Oligodendrocytes which provide the needed myelin insulation for proper signal transmission by the conducting neurons,
(2) Astrocytes, which provide support to neurons through production of growth factors and other trophic support mechanisms (Figure 1).
Q-Cells® technology aims to restore health and function to neurons prior to their death by using the natural support mechanisms in the CNS, rather than by replacing the neurons. No current drug can achieve this outcome.
7
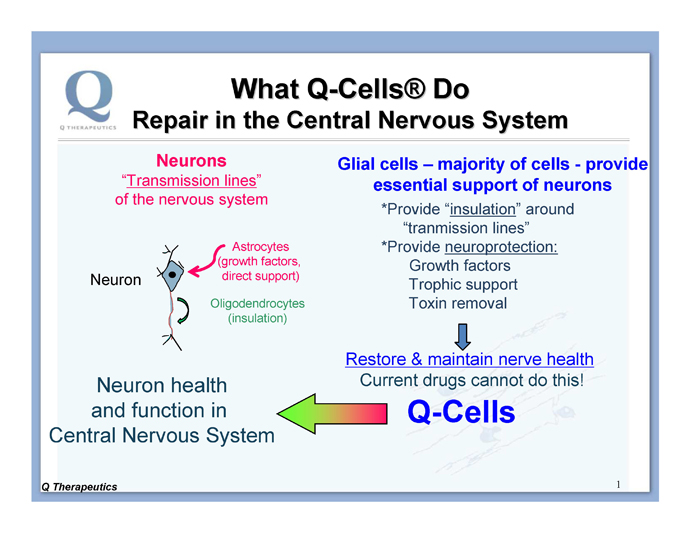
Figure 1. The majority of the cells in the brain and spinal cord are glia cells that provide support function for the neurons. Q-Cells® provide support for neurons, which Q believes is a more straightforward task than replacing neurons.
The Science behind Q-Cells®
During normal development, glia are formed by a progenitor cell called the glial-restricted precursor (“GRP”). Human GRPs, together with their progeny, have been trademarked as Q-Cells®. Q-Cells® naturally do three things in the CNS: replicate, migrate and differentiate into mature glial cells. The resulting differentiated cells (astrocytes and oligodendrocytes, shown in Figure 2) then can perform the support functions of these glial cells in vivo as described above. The Company is taking advantage of Q-Cells®’ natural abilities to follow the local molecular cues present in the brain or spinal cord, and intends to transplant Q-Cells® to treat certain diseases where glia are defective.
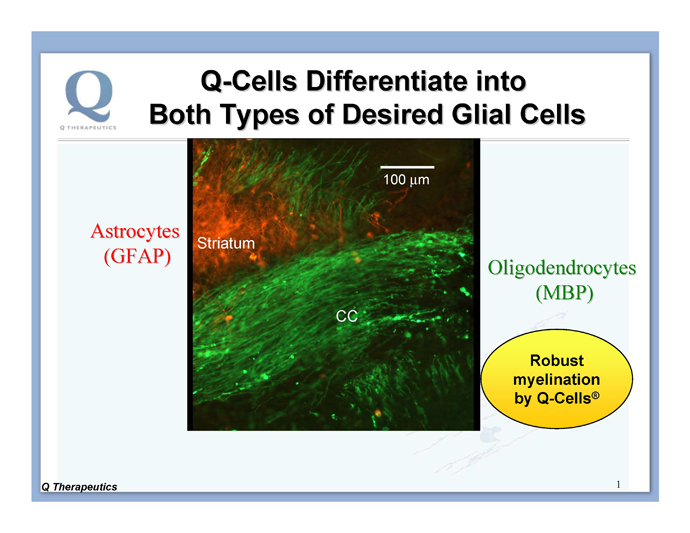
Figure 2. Q-Cells® differentiate into both types of glial cells: oligodendrocytes (green stain for myelin basic protein [MBP]) and astrocytes (red stain for human glial fibrillary acidic protein [GFAP]) in the shiverer mouse brain. Q-Cells® were implanted into the brains of newborn shiverer mice.
In demyelinating diseases such as Multiple Sclerosis, inflammatory attack damages the oligodendrocytes, resulting in demyelination, i.e., loss of the insulating myelin sheath; this is the primary pathology that causes loss of proper neuronal function and can lead to neuron death. Q-Cells® can follow endogenous cues to replace the missing myelin (which is part of the living oligodendrocyte cells produced by Q-Cells®) on the damaged neurons, which Q believes can provide restorative therapy for demyelinating diseases. Other drugs have not been able to achieve this.
In many other neurodegenerative diseases, the diseased astrocytes are dysfunctional or harmful, hastening neuronal death. Supplementing diseased astrocytes with healthy astrocytes that are produced by Q-Cells® may reduce or prevent neuronal death. Healthy astrocytes are an important part of normal homeostasis in the CNS.
Q-Cells® (whether isolated directly from somatic tissue or derived from CNS stem cells or pluripotent cells) are late-stage lineage-restricted progenitor cells and have a restricted differentiation path: they become astrocytes and oligodendrocytes. In contrast, transplantation of the earlier-stage CNS stem cells (NSCs or NEPs, see Figure 3) can produce neurons in vivo in addition to glial cells. The Company believes that this less-restricted differentiation potential of the stem cells (in contrast to the more differentiated lineage-restricted glial progenitor cells) of the CNS has the potential to produce neurons inappropriately, which may form aberrant synaptic connections leading to neurological complications. Q has confirmed that terminal differentiation of Q-Cells® in animal models is naturally restricted to the two desired cell types: astrocytes and oligodendrocytes.
8
Benefits and features of Q-Cells®
Advantages of Q-Cells® include:
|
|
(1)
|
Q-Cells® provide important growth factors and other trophic functions that support neuronal health, providing treatment options for diseases directly involving neuronal degeneration such as ALS and Parkinson’s disease.
|
|
|
(2)
|
Q-Cells® provide myelin repair functions for diseases involving demyelination such as Multiple Sclerosis, Transverse Myelitis, Cerebral Palsy, and Spinal Cord Injury.
|
|
|
(3)
|
When placed in the CNS, Q-Cells® predictably replicate, migrate, and terminally differentiate into physiologically relevant glial cells: oligodendrocytes and astrocytes.
|
|
|
(4)
|
Q-Cells® don’t give rise to appreciable numbers of neurons, reducing potential for unwanted effects due to aberrant neuronal connections.
|
|
|
(5)
|
The isolation process is readily scalable to GMP.
|
|
|
(6)
|
Tissue-sourced Q-Cells® spend little time in culture prior to freezing of the cells to produce the product for shipment to treatment sites, providing advantages in production.
|
For all these reasons, Q believes that Q-Cells® are a desirable product for CNS cell therapeutics.
Many normal CNS cells remain viable in the CNS through a person’s life; our clinical trials are intended to ascertain whether transplanted Q-Cells® function for significant periods of time, or if periodic (e.g., annual) treatments will be appropriate. This replacement therapy will build on the model of successful bone marrow transplants, where stem cells in donor bone marrow replace diseased blood cells. However, unlike bone marrow transplants, Q-Cells® will be banked in vials containing the cells and shipped frozen to hospitals and clinics. Thus, Q plans to sell products following the model of a traditional specialty pharmaceutical company in that Q-Cells® will be prepared and inventoried in advance, and will not be generated uniquely for each patient.
Although the CNS has a degree of immune privilege, Q plans to initially use marketed immunosuppressants for a transient period, to mitigate potential for immune rejection associated with transplants.
Q-Cells® Can Be Obtained from a Variety of Sources
Q-Cells® can be obtained from several sources (Figure 3): (i) from somatic tissue (termed adult stem cells, isolated from fetal or adult tissue), (ii) differentiated from CNS stem cells [neuroepithelial stem cells (NEP) or neural stem cells (NSC)] which can be isolated from somatic tissue or derived from pluripotent cells), or (iii) differentiated from pluripotent cells (e.g., embryonic stem cells (ESC) or other pluripotent cells such as induced pluripotent stem cells (iPSC)). The Company has intellectual property (IP) covering multiple sources, giving it a strong IP position. Because of this broad IP position, Q has the ability to follow a development path that enables cost and time-efficient development of its cellular therapeutics. For these reasons, Q has chosen to develop its initial Q-Cells® product by isolating the cells directly from somatic (fetal cadaver) tissue, followed by expansion in the production facility, as this provides cells that are at the desired cell type without the need for additional in vitro differentiation.
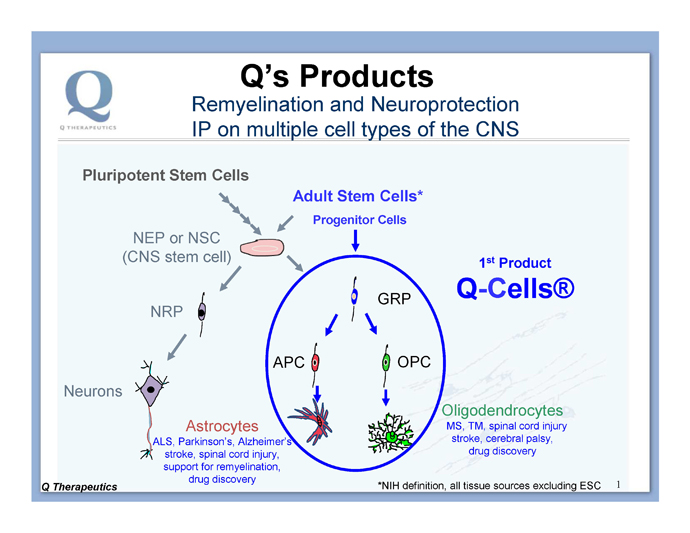
Figure 3. Stem and progenitor cells of the brain and spinal cord. Stem cells can be derived from pluripotent stem cells (e.g., ESCs or iPSCs) or isolated from somatic tissue sources (called Adult Stem Cells). The Company’s strategy is to start with the more straightforward path: isolate and use unmodified cells that are already, naturally, at the desired cell type. Q believes that the restricted fate of Q-Cells® lowers the chance of unwanted side effects, such as inappropriate production of neurons. NRP = neuronal restricted precursor, GRP = glial restricted precursor, APC = astrocyte precursor cell, OPC = oligodendrocyte precursor cell.
9
Manufacturing
The Company has developed manufacturing protocols to isolate Q-Cells® directly from somatic (fetal cadaver) tissue, without the need for additional differentiation in vitro. Q has developed proprietary methods to expand the cells at this stage to enable treatment of larger numbers of patients from each preparation. Q’s strategy is to start with the most straightforward path: isolate and characterize unmodified cells that are already, naturally, at the desired stage of differentiation, to achieve proof of activity in clinical trials. Q believes that this strategy enables development of Q-Cells® and other cellular therapeutics in the most cost and time-effective manner.
The Company will use a centralized laboratory (contract GMP cell production manufacturer) for cell isolation and expansion and to provide the necessary quality control. The clinical transplant sites will receive frozen cells, which they will subsequently thaw, wash and inject into the target site of the patient. Use of allogeneic cells (i.e., cells derived from a tissue not obtained from the patient), rather than autologous cells (cells derived from the patient’s own tissues) both enables use of what Q believes will be the most effective healthy cells as well as permits cell therapy to be an off-the-shelf treatment, promoting more widespread use. The manufacturing of the Q-Cells® is done via a proprietary process developed by Q. This process can be shared with Q’s contract manufacturer of choice, or with several manufacturers to mitigate the risk of loss of any one manufacturing subcontractor.
The Company is currently working with the GLP/GMP Cell Therapy Facility (CTF) operated by the University of Utah. Q may seek other manufacturers as its development programs progress, certainly once Q-Cells® have been approved for commercial sale.
Q Therapeutics’ Intellectual Property
Q Therapeutics has exclusive worldwide rights to its Q-Cells® product, either through an agreement with the University of Utah Research Foundation (“UURF”) or through owned, internally developed intellectual property. The Q patent portfolio encompasses five families of neural lineage progenitor or stem cell technologies. Currently, Q has rights to 14 (11 US) issued patents to which it has a license agreement with the University of Utah and the National Institutes of Health. This license provides for royalties on the Company’s and sublicensees’ sales and contains due diligence obligations and related provisions; a portion of the consideration to the University of Utah was equity in Q. In addition, Q has rights to 5 U.S., 2 Canadian, 1 Japanese, 1 South Korean, 1 PCT and 1 EPO pending patent applications in the field of neural lineage progenitor and stem cells.
Q also holds a U.S. Trademark to its first product name, Q-Cells® (serial number: 78869175; registration number: 3,385,490), and U.S. and international Trademarks/Service marks to: Q Therapeutics® (U.S. serial number: 78-415,125; U.S. registration number: 3,280,432; international registration number: 867,474).
Q’s issued/granted patents and patent applications that are licensed or owned by Q as of September 30, 2011, include the following:
Common Neural Progenitor for the CNS and PNS
Mahendra S. Rao, Tahmina Mujtaba
ISSUED U.S. Patent 6,830,927
Generalization, Characterization, and Isolation of Neuroepithelial Stem Cells and Lineage Intermediate Precursor
10
Mahendra S. Rao, Margot Mayer-Proschel, Tahmina Mujtaba
PCT/US98/ 09630, Filed 5/7/98, published PCT application
Isolation of Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel
ISSUED U.S. Patent 6,734,015
Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel and Anjali J. Kalyani
ISSUED U.S. Patent 6,787,353
Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel, Anjali J. Kalyani
PCT/US98/ 13875, filed, 7/3/98, published PCT application
Lineage Restricted Glial Precursors from the Central Nervous System
Mahendra S. Rao, Mark Noble, Margot Mayer-Proschel
ISSUED U.S. Patent 6,235,527
Lineage Restricted Glial Precursors from the Central Nervous System
Mahendra S. Rao, Mark Noble, Margot Mayer-Proschel
PCT/US98/ 24456, filed, 11/17/98, published PCT application
Generation, Characterization, and Isolation of Neuroepithelial Stem Cells and Lineage Restricted Intermediate Precursor
Mahendra S. Rao, Margot Mayer-Proschel, Tahmina Mujtaba
2,289,021, filed 5/7/98, published Canadian patent application
Generation, Characterization, and Isolation of Neuroepithelial Stem Cells and Lineage Restricted Intermediate Precursor
Mahendra S. Rao, Margot Mayer-Proschel, Tahmina Mujtaba
GRANTED Israeli Patent, 132584
[Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel, Anjali J. Kalyani
2,294,737, filed 7/3/98, published Canadian patent application
Note: the above Canadian patent application is abandoned with the right to revive until July 2012.]
Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel, Anjali J. Kalyani
GRANTED Israeli Patent, 133799
Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel, Anjali J. Kalyani
ISSUED Japanese Patent 4,371,179
Lineage-Restricted Precursor Cells Isolated From Mouse Neural Tube and Mouse Embryonic Stem Cells
Tahmina Mujtaba and Mahendra S. Rao
PCT/US00/ 12446, filed 5/5/00, published PCT application
11
Lineage Restricted Glial Precursors from the Central Nervous System
Mahendra S. Rao, Mark Noble and Margot Mayer-Proschel
ISSUED U.S. Patent 6,900,054
Method of Isolating Human Neuroepithelial Precursor Cells from Human Fetal Tissue
Margot Mayer-Proschel, Mahendra S. Rao, Patrick A. Tresco and Darin J. Messina
ISSUED U.S. Patent 6,852,532
Isolation of Mammalian CNS Glial Restricted Precursor Cells
Mahendra S. Rao and Margot Mayer-Proschel
ISSUED U.S. Patent 7,037,720
Isolation of Mammalian CNS Glial-Restricted Precursor Cells
Mahendra S. Rao and Margot Mayer-Proschel
ISSUED U.S. Patent 7,595,194
Generation, Characterization and Isolation of Neuroepithelial Stem Cells and Lineage Restricted Intermediate Precursor
Mahendra S. Rao and Margot Mayer-Proschel
12/568,419, filed 9/28/09, published U.S. patent application
Methods Using Lineage Restricted Glial Precursors from the Central Nervous System
Mahendra S. Rao, Mark Noble and Margot Mayer-Proschel
ISSUED U.S. Patent 7,214,372
Lineage Restricted Glial Precursors
Mahendra S. Rao and Tahmina Mujtaba
ISSUED U.S. Patent 7,795,021
Lineage Restricted Glial Precursors
Mahendra S. Rao and Tahmina Mujtaba
12/209,559, filed 9/12/08, published U.S. patent application
Pure Populations of Astrocyte Restricted Precursor Cells
Mahendra S. Rao, Tahmina Mujtaba and YuanYuan Wu
PCT/US2003 /002356, filed 1/23/03, published PCT application
Lineage-Restricted Neuronal Precursors
Mahendra S. Rao, Margot Mayer-Proschel and Anjali J. Kalyani
12/233,857, filed 9/19/08, published U.S. patent application
Pure Populations of Astrocyte Restricted Precursor Cells
Mahendra S. Rao, Tahmina Mujtaba, YuanYuan Wu and Ying Liu
2,473,749, filed 1/23/03, published Canadian patent application
Pure Populations of Astrocyte Restricted Precursor Cells
Mahendra S. Rao, Tahmina Mujtaba, YuanYuan Wu and Ying Liu
03732101.5, filed 1/23/03, published European patent application
Pure Populations of Astrocyte Restricted Precursor Cells
Mahendra S. Rao, Tahmina Mujtaba, YuanYuan Wu and Ying Liu
2008-139534, filed 1/23/03, published Japanese patent application
12
Pure Populations of Astrocyte Restricted Precursor Cells
Mahendra S. Rao, Tahmina Mujtaba, YuanYuan Wu and Ying Liu
2004-7011381, filed 1/23/03, published South Korean patent application
Pure Populations of Astrocyte Restricted Precursor Cells
Mahendra S. Rao, Tahmina Mujtaba, YuanYuan Wu and Ying Liu
12/433,060, filed 4/30/09, published U.S. patent application
Method of Isolating Human Neuroepithelial Precursor Cells from Human Fetal Tissue
Margot Mayer-Proschel, Mahendra S. Rao, Patrick A. Tresco and Darin J. Messina
ISSUED U.S. Patent 7,517,521
Method of Isolating Human Neuroepithelial Precursor Cells from Human Fetal Tissue
Margot Mayer-Proschel, Mahendra S. Rao, Patrick A. Tresco and Darin J. Messina
12/395,677, filed 3/1/09, published U.S. patent application
Methods and Compositions for Expanding, Identifying, Characterizing and Enhancing Potency of Mammalian-Derived Glial Restricted Progenitor Cells. Robert Sandrock, James Campanelli and Deborah A. Eppstein, PCT/US2010/055956, filed November 2010, published PCT application
In addition, Q has entered into out-license agreements with Life Technologies (LIFE), Molecular Transfer, Inc, (MTI), and xCell Science LLC, (xCell), under which the licensees may develop, manufacture and sell certain cell products covered by Q’s IP for research and drug discovery use only, for which Q (or its subsidiary NeuroQ) is anticipated to receive royalty payments, and in the case of xCell, Q may provide certain payments for research activities.
CNS Therapeutic Market Overview
The global CNS therapeutics market was valued at almost $100 billion in 2007. (Datamonitor Healthcare CNS Market Overview, 2008). This market is comprised of three main segments:
(1) Neurology (e.g. Parkinson’s and Alzheimer’s Diseases, Multiple Sclerosis, Spinal Cord Injury, ALS)
(2) Psychiatry (e.g. Depression, ADHD, Schizophrenia)
(3) Pain (e.g. Migraine).
While the psychiatry market represents nearly half of all dollars spent on CNS drug therapies, the neurology segment of the market is the fastest growing. Market growth overall is fueled by the increasing aging of the population as well as the increasing reported incidence of CNS disorders due to better diagnostic techniques.
The United States represents approximately half of this global market and is realizing the fastest growth rates. This is the result of three factors: Higher prices charged for the drugs themselves compared to other markets; the larger volume of patients seeking treatment; and the higher rates of pharmacotherapy compared to other countries.
13
The Unmet Need in the CNS Therapeutic Market
The CNS therapeutic market is based primarily on traditional drug therapies (small molecules and biologics). While this growing market is large and represents nearly 25% of all dollars spent on prescription pharmaceuticals worldwide, there remains a high unmet need in the treatment of many neurological diseases where current therapies are inadequate or yet to be developed. Two examples are Multiple Sclerosis (MS) and Lou Gehrig’s disease (Amyotrophic Lateral Sclerosis, or ALS).
The annual worldwide pharmacotherapy market for Multiple Sclerosis is approximately $10 billion. The vast majority of all approved MS therapies seek to slow disease progression by “knocking down” the autoimmune component of the disease, which causes the damage and destruction of the myelin sheath that surrounds the neurons. However there is no approved product that repairs this damage once it has occurred. Thus, while current therapies may slow or even halt the progression of the disease, once the damage is done, there is currently no means of repairing that damage. This represents a substantial unmet need in the MS disease space.
Similarly, there is only one approved drug for the treatment of ALS/Lou Gehrig’s disease. This drug, Rilutek ®, prolongs the life expectancy of ALS patients by approximately 60-90 days. ALS is always fatal, and there are no approved drugs that substantially slow or halt the progression of the disease, much less reverse its devastating effects. This too represents an unmet need in the CNS therapeutic market. ALS strikes approximately as many people (5,000-6,000) a year in the U.S. as does MS, but due to the rapid disease progression leading to death in 2-5 years after diagnosis, the estimated prevalence in the U.S. is only 30,000 patients, thus ALS is classified as an orphan disease.
Given these and many other shortcomings in the approved pharmacopeia for injuries and diseases of the CNS, there is an opportunity to substantially expand the CNS therapeutic market with new technologies that address the unmet needs. Stem cell-based therapy holds the promise of significantly expanding the CNS market and satisfying the unmet needs that exist.
The Potential of Stem Cell-Based Therapy
Stem cell based therapy is seen by many as the next great advance in the treatment of disease and injury. It holds the promise of better therapy for disorders which are currently not well treated and new therapies to meet currently unmet needs. Should this promise hold true, the currently available therapies could be displaced and the pharmacotherapuetic market substantially expanded as new stem and progenitor cell therapies are approved for human use.
The immense promise of stem cell biology has prompted the formation of several companies seeking to exploit the therapeutic potential of stem cells. Approaches to creating stem cell therapies fall into two broad categories:
|
|
(1)
|
Isolating and purifying new populations of stem or progenitor cells from various tissues, expanding them as necessary and transplanting the cells into various target organs. One challenge with this approach is ensuring that the exact desired population can be reproducibly purified and expanded. This affects safety and efficacy as well as the ease of scaled-up manufacturing.
|
|
|
(2)
|
Stimulating existing stem cells to “wake up,” expand and differentiate inside the body at an accelerated rate. Challenges associated with this approach include manipulating complex interactions among multiple growth factors and endogenous signals to generate the desired outcomes, as well as complications due to malfunctioning or mutant endogenous cells in certain diseases.
|
14
Q believes that the fastest route to a safe and effective product is through the first route – therapy with well characterized, unmodified cells that perform their tasks as nature intended, functioning as ‘mini-factories’ and playing roles appropriate to the specific disease state.
Several types of cells are under evaluation for use in cell therapy, including embryonic stem cells, human cord blood and placental stem cells, and fetal or adult tissue from various organ sites. Purified cell populations can be injected intravenously or transplanted directly into target organs such as brain, heart, pancreas, blood and bone. For most neurodegenerative diseases, Q believes that CNS cells, not hematopoietic or mesenchymal lineage cells, are needed.
Market Opportunity for Q Therapeutics
Q Therapeutics aims to change the way medicine is practiced in the treatment of many debilitating and often fatal diseases of the brain and spinal cord by bringing to market a patented cell-based therapeutic that addresses substantial unmet needs in the CNS therapeutic market today. The Company believes that its initial Q-Cells® product could meet a variety of these needs across a number of CNS diseases. By demonstrating that Q-Cells® are safe and effective first in smaller orphan indications and later in larger target markets, the Company believes it can both augment and/or displace current therapeutic approaches as well as expand the therapeutic market in currently untreatable CNS conditions. Should this prove true, Q-Cells® would address a multi-billion dollar market opportunity in treatment of neurodegenerative diseases for which there are no effective treatments.
Q intends to bring Q-Cells® to the market first to treat ALS (Lou Gehrig’s disease) to demonstrate the safety and efficacy of its cell based therapeutic. Thereafter, Q-Cells® will be brought to other indications potentially including Multiple Sclerosis, Spinal Cord Injury, Parkinson’s and Alzheimer’s diseases. Q estimates that the annual market opportunity for its initial orphan disease targets exceeds $1 billion worldwide. Application of its Q-Cells® product to other larger market diseases such as MS would substantially expand the market available to Q.
Orphan Disease Strategy for Initial Commercialization
Q’s first commercial targets are orphan diseases with billion-dollar market potential (Figure 4). The Orphan Drug Act of 1983 (in this paragraph, the “Act”) defined an “orphan drug” as a therapy intended to treat rare “orphan” diseases, those affecting fewer than 200,000 Americans. The benefits provided by the Act may include more rapid regulatory timelines, tax benefits, and seven-year market exclusivity for the first product approved for an indication. Despite the smaller numbers of affected patients, orphan diseases often have highly motivated patient advocacy groups that are eager to assist companies with patient recruitment and therapeutic development. Moreover, substantial annual per-patient treatment prices effectively offset the relatively small patient populations. Due to the focused nature of marketing permitted by targeting orphan indications, Q might be able to capture a significant portion of the U.S. market for certain orphan diseases without need for a major marketing partner, provided that it has adequate financial resources. This ability to target patients suffering from orphan diseases also provides an incentive to potential Q development partners with interest in funding development costs and providing developmental expertise in exchange for marketing rights.
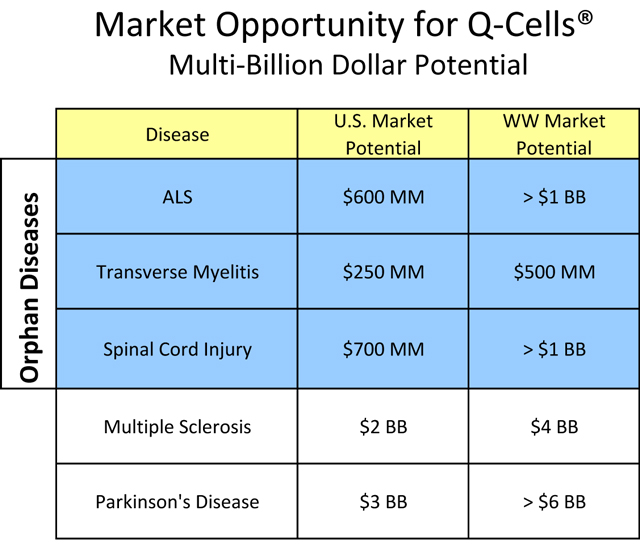
Figure 4. Multi-billion dollar market potential for Q-Cells®. The initial orphan diseases may allow Q or its partner to market the product with a focused specialty sales force. Market opportunity size was calculated using a lifetime treatment cost projection of $200,000, assuming 50%, 30% and 35% patient population penetration for the orphan diseases of ALS, Transverse Myelitis and Spinal Cord Injury, respectively, and 2% and 10% patient penetration for Multiple Sclerosis and Parkinson’s disease, respectively. Further market opportunities exist for additional neurodegenerative disease targets.
15
Q Market Development Strategy – Orphan Indications First, Larger Markets Follow
|
|
A.
|
Initial target indications
|
Q-Cells® offer multiple repair mechanisms from one product due to Q-Cells®’ ability to differentiate into both oligodendrocytes and astrocytes (discussed earlier in this Description of Business section). We have broadly characterized the disease targets into three general but overlapping groups: diseases primarily of myelination deficiencies, diseases where neuronal loss is the dominant mechanism, and diseases where both of the above fall into play. Evaluation of Q-Cells® in these various neurodegenerative diseases provides multiple shots on goal.
|
|
1.
|
Amyotrophic Lateral Sclerosis (ALS), or Lou Gehrig’s Disease
|
ALS is diagnosed in 5,000-6,000 new individuals per year in the U.S., comparable in annual incidence to Multiple Sclerosis. Patient death generally occurs 2-5 years after diagnosis, resulting in an estimated prevalence of 30,000 (ALS Association). ALS is a neurodegenerative disease causing progressive deterioration and loss of motor neurons, affecting both upper and lower motor neurons. The loss of nerve stimulus to specific muscles results in atrophy and progressive weakness that leads to paralysis. Death usually results from respiratory failure.
There is no curative treatment for ALS. The only drug approved for treatment for the disease—riluzole (Rilutek®) - is believed to reduce (but not reverse) damage to motor neurons by decreasing the release of glutamate, and prolongs survival by only 60-90 days.
Studies in mouse models of ALS suggest that abnormalities in astrocytes and microglia, in addition to motor neurons, play a role in disease onset as well as progression. Astrocytes play a prominent role in CNS homeostasis. In both ALS patients and animal models, astrocyte abnormalities and physiological dysfunction can be seen many months before motor neuron degeneration and precede clinical disease [literature references include Clement et al, Science 302, 113-117 (2003); Rothstein et al Neuron 18, 327-338 (1997); Lepore et al, Nature Neuroscience 11, 1294-1301 (2008); Yamanaka et al, Nature Neuroscience 11, 251-253 (2008); Wang et al, Human Molecular Genetics 20, 286-293 (2011)]. Further in support of the role of diseased astrocytes in causing motor neuron degeneration in ALS, the Maragakis lab recently demonstrated that transplanting rat astrocyte precursors carrying the SOD mutation (this mutation gives rise to ALS in patients carrying such mutation) into healthy rats results in degeneration of motor neurons in the previously healthy rats [Proc Natl Acad Sci on line edition Oct 2011 10.1073 (2011)]
This altered physiology of CNS astrocytes contributes to disease progression by resulting in further susceptibility to motor neuron loss. Supplementing these diseased astrocytes with healthy ones (such as occurs via transplantation of GRP cells) is a reasonable hypothesis for a promising therapeutic approach for slowing and/or halting the ALS disease course.
Maragakis and colleagues have demonstrated the validity of this hypothesis, showing therapeutic benefits following transplantation of rat GRPs into the SODG93A rat model (Lepore et al, Nature Neuroscience 11, 1294-1301 (2008)). Administration of rat GRPs (the rat cells homologous to human Q-Cells®) into the cervical spinal cord after onset of disease resulted in enhanced survival and motor function.
16
The results demonstrating that Q-Cells® exhibit characteristics of cell survival, migration and differentiation into mature glial phenotypes in animal models, in conjunction with safety data, the scientific literature and the above mentioned benefits obtained with rat GRPs implanted into the SODG93A rat, provide the rationale for testing Q-Cells® in clinical trials involving motor neuron degeneration in ALS patients. Implantation of Q-Cells®, which produce non-diseased astrocytes, may ultimately provide normal astrocytic function overcoming the dysfunction of the patient’s diseased astrocytes, thereby restoring homeostatic control and reducing or preventing further death of motor neurons.
|
|
2.
|
Demyelinating Diseases: Multiple Sclerosis and Transverse Myelitis
|
Q is also evaluating Q-Cells® in animal studies for Multiple Sclerosis (MS), a chronic autoimmune-triggered demyelinating neurodegenerative disease, and Transverse Myelitis (TM), a related but acute, localized inflammatory disease of the spinal cord. Multiple clinical and pathological studies suggest that there are many common features of the inflammation and neural injury between TM and MS, with a shared primary pathology of demyelination due to immune attack destroying the oligodendrocytes. TM may offer some technical advantages over MS for initial proof of concept clinical trials, as the inflammatory-induced lesion occurs in only one location in the spinal cord (often thoracic) and does not recur: i.e., TM is mono-focal and mono-phasic. Due to the similarity of the lesion pathology in TM and MS, demonstration of efficacy of remyelination in the orphan disease TM may be an indicator for efficacy in the more prevalent disease of MS. The rationale for using Q-Cells® in these indications is based on data obtained using both animal and human GRPs in models of disease. Demonstration of remyelination in lesions in TM and MS patients may also be relevant for developing treatments of other diseases in which demyelination is a significant factor such as cerebral palsy, white-matter stroke and certain traumatic spinal cord injuries.
Shiverer is an established myelin-deficiency model in which the entire CNS is defective in normal myelination. Q and its collaborators at Johns Hopkins have demonstrated that Q-Cells® engrafted into shiverer (a) effectively compete with host (defective) oligodendrocytes, (b) myelinate host axons resulting in normal myelin and (c) demonstrate benefits in a focal inflammatory spinal cord lesion model (Figure 5, Q data; Walczak et al, GLIA 59, 499-501 (2011)). Others have shown that human GRPs transplanted into shiverer/rag2 mice extended survival of a portion of the mice (Windrem et al, Cell Stem Cell 2, 553-565 (2008)). They also documented that the implanted human cells interacted effectively with the host proteins and did not leave the CNS. The efficacy of a localized treatment that does not produce toxicities in other organs is a significant finding. This is supported further by an extensive body of literature demonstrating remyelination by rodent glial precursors and related cells in multiple animal models [including publications from Blakemore (1999), Ben Hur (2006), Cummings (2005), Duncan (2004, 2005), Goldman (2008), Keirstead (2004, 2005), Whittemore (2010), Rao (1997-2010), Sandrock (2010), and Walczak (2011).] This myelination has occurred in both the presence and the absence of an ongoing inflammatory process.
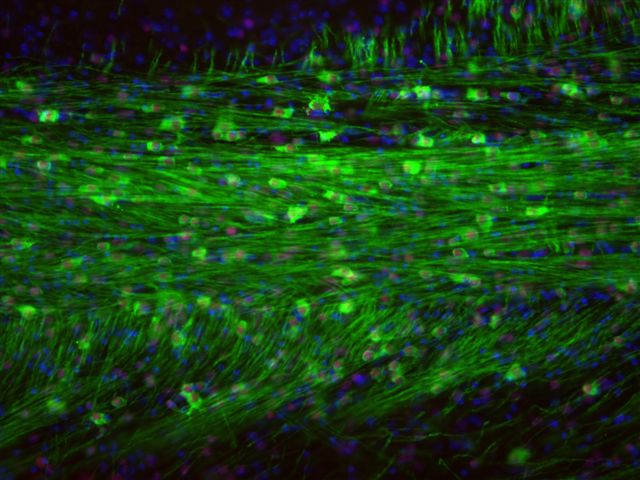
Figure 5. Myelination by implanted Q-Cells® that produced oligodendrocytes.
Shiverer is a mouse with a mutation that produces defective myelin basic protein (MBP) and hence defective myelin. Q-Cells® implanted in shiverer/rag2 brain mature into oligodendrocytes that produced normal MBP (green). All MBP seen here is produced by Q-Cells® that matured into oligodendrocytes (human cell-specific red nuclei stain).
17
|
|
3.
|
Traumatic Spinal Cord Injury
|
Spinal Cord Injury (“SCI”) due to trauma results in 10,000-12,000 people paralyzed each year in the U.S., and it is estimated that over 250,000 people are living with spinal cord injury in the U.S. This has tremendous costs both in terms of patient care, lost productivity as well as quality of life. It has been projected that the U.S. could save up to $400 billion in future healthcare costs if there were effective therapies to treat and prevent spinal cord injuries (Christopher and Dana Reeve Foundation, Centers for Disease Control, University of Alabama National Spinal Cord Injury Statistical Center). Q has collaborated with Itzhak Fischer, PhD, at Drexel University to test Q-Cells® in animal models of traumatic spinal cord injury. Recently published studies by Q and its collaborators at Drexel document the safety and statistically-significant, reproducible, disease modifying activity of Q-Cells transplanted into the injury site in an athymic rat model of thoracic contusion SCI (Jin et al., J Neurotrauma; 28(4):579-94; 2011).
|
4.
|
Parkinson’s Disease
|
Parkinson’s Disease (“PD”) affects more than one million patients in the U.S., and the numbers are expected to increase with the aging of the population. Symptoms of PD are caused by the gradual loss of neurons in a relatively small part of the brain, the substantia nigra. These neurons normally secrete dopamine, and lack of dopamine leads to symptoms such as shaking, rigidity, difficulty and slowness in movement, and postural instability. Although patients initially respond to treatment with L-dopa or dopaminergic agonists, over time this response decreases. Several early trials have found that administration of single growth factors failed to provide meaningful clinical benefits in PD patients.
Studies in animal Parkinson’s disease models have suggested non-cell autonomous killing of neurons, with diseased astrocytes playing a critical initiating role. Astrocytes in culture exert a protective effect on neuronal cells in a setting where both cell-types are co-cultured. Studies in a primate model of Parkinson’s disease showed functional benefits after implantation of neural cells. Upon autopsy, a large number of progeny astrocytes were found juxtaposed with the host nigrostriatal circuitry suggesting that the “homeostatic adjustments” to the microenvironment results in preservation of the remaining host nigrostriatal pathway (Redmond et al, Proceedings of National Academy of Sciences USA 104, 12175–12180 (2007)). As Q-Cells® produce healthy astrocytes, this provides potential for protective effects including those via production of multiple growth factors and other trophic support, rather than relying on a single factor. Q anticipates that it will pursue studies of Q-Cells® in Parkinson’s disease models upon availability of appropriate additional funding.
C. Other Disease Targets
Q-Cells® provide a platform technology that may be useful to treat not only demyelinating diseases, but other neurodegenerative diseases that can benefit from the neuronal support provided by growth factors and other trophic support by Q-Cells®. The patient populations and markets for Q’s follow-on therapeutic targets may be substantial, in addition to the opportunity in ALS, MS, TM, Spinal Cord Injury and Parkinson’s Disease discussed above:
|
|
·
|
Alzheimer’s Disease (“AD”) is a progressive, neurodegenerative disease characterized by abnormal clumps (amyloid plaques) and tangled bundles of fibers (neurofibrillary tangles) in the brain. Symptoms include memory loss, language deterioration and confusion and eventually lead to loss of cognition and personality. It is estimated that about 5.3 million people in the U.S. suffer from AD, with annual healthcare costs in excess of $170 billion. The number of patients is projected to reach 13.5 million by 2050 (Alzheimer’s Association, 2010).
|
18
Transplantation of neural cells that produce glial cells (both astrocytes and oligodendrocytes) as well as transplantation of astrocyte precursors, provided benefits in Alzheimer’s Disease mouse models, rescuing neurons and improving memory (Hampton et al, Journal of Neuroscience 30(3), 9973-9983 (2010); Blurton-Jones et al, PNAS 106(32), 13594-13599 (2009)). Since Q-Cells® mature into glial cells after transplantation, Q believes there may be an opportunity for ultimate use of Q-Cells® potentially to both slow decline and/or restore function for Alzheimer’s disease patients. Q is evaluating initiating a collaboration to further investigate the efficacy of Q-Cells® in animal models once additional financing has been obtained.
|
|
·
|
Traumatic Brain Injury (“TBI”) has been reported to occur in 1.4 million Americans each year, leaving 50,000 dead and more than 80,000/year with lifelong disabilities, and costing over $60 billion in 2000. The U.S. Centers for Disease Control estimates that at least 5.3 million Americans are living with the need for help in daily activities due to TBI. TBI is a leading injury for active military personnel in war zones. Other than acute phase treatments to prevent further injury (e.g., surgery, relief of pressure build-up), treatment of headaches, seizures and physical rehabilitation, there is no treatment for and nothing to reverse TBI. The multiple support functions of Q-Cells® may be beneficial in achieving recovery from TBI.
|
|
|
·
|
Cerebral Palsy (“CP”) has a U.S. patient population of about 750,000. An initial target may be for spastic diplegia, with involves injury to the cerebral cortex and represents approximately 70% of CP. As Q-Cells® can provide myelinating oligodendrocytes, they may provide benefits to CP patients.
|
|
|
·
|
Stroke afflicts almost 800,000 people and is the third leading cause of death in the U.S, with total annual costs over $40 billion. Market projections are significant even if only a small portion of these patients were treated. Q-Cells® may be beneficial in treating stroke both for restoring myelination, as well as the support provided by astrocytes to restoring neuron function.
|
|
|
·
|
Leukodystrophies and CNS storage diseases: These are several inherited pediatric diseases for which the molecular cause is known to involve a single gene defect. There are no treatment options for most of these diseases, often resulting in death at a young age. Although a few companies including Genzyme and Biomarin have developed enzyme replacement therapies to successfully treat systemic manifestations of a few of these diseases, the systemically-administered enzyme pharmaceuticals do not cross the blood-brain barrier to treat CNS targets. Such CNS diseases may provide future opportunity to Q as there are many storage disorders with severe CNS manifestations that involve destruction of oligodendrocytes with concomitant demyelination. Q-Cells®, as they are transplanted directly into the CNS, may be able to provide both the missing enzyme in the CNS as well as replace the destroyed oligodendrocytes and thus provide remyelination.
|
|
|
·
|
Peripheral Neuropathies have no effective treatments. In addition to causing severe pain, they are a leading cause of amputations in the developed world. Eight million diabetes patients have neuropathy in the U.S. alone. Q has patents on cells that support the peripheral nervous system.
|
Q-Cells® - Benefits of Localized Therapy
The ability of animal results to predict human results, and accordingly the drug development risk profile, depends on many factors including:
(1) The pharmacokinetic and metabolic profiles of a drug in animals vs. humans, which can greatly influence both efficacy and safety
19
(2) How the drug affects the target diseased and disease-causing tissues
(3) What the drug does to non-target cells.
Many drug candidates fail in development because they affect not only their intended target but have toxic off-target effects in other organ systems as well. For example, a drug intended to treat headache pain may fail to become a commercial product due to unacceptable toxic systemic effects in other organs, e.g., heart or kidneys. Q-Cells® are a localized treatment in the CNS. Studies conducted by Windrem et.al. (Cell Stem Cell 2, 553-565(2008)) indicate that human GRPs stop at the boundary where the central nervous system meets the peripheral nervous system. Q anticipates that this localized nature of the planned Q-Cell® therapy may reduce risk from systemic toxicity, which Q believes may reduce the risk of drug failure in early clinical trials due to systemic toxicities. Animal safety studies to be conducted are intended to evaluate safety both locally in the spinal cord and brain as well as systemically before initiating clinical trials.
In addition to the initial disease target of ALS, Q’s IP portfolio can be applied to additional follow-on indications as discussed above under Q Market Development Strategy. Q’s IP portfolio can also be applied to novel assays for drug screening, to identify new products for drug therapy with a focus on diseases of the central nervous system.
Competition
Q believes that Q-Cells® are different from other cell therapy products both in their novel mechanism of action / capabilities as well as the ease of production. The competitive landscape involves companies working on a range of alternative treatments for diseases of the CNS including traditional drug products, protein therapeutics, gene therapy and different types of cell therapy. Q believes that the cell therapy approach has many advantages over single drug therapeutics for many neurodegenerative diseases. Thus, the closest direct competition is other cell therapy companies working on the same disease targets.
Direct competition
A limited number of companies are working on the therapeutic neurological application of stem cells, including the public companies Geron (NASDAQ: GERN), StemCells Inc. (NASDAQ: STEM), ReNeuron (LSE: RENE.L), and Neuralstem (AMEX: CUR), and the private company California Stem Cells, Inc. Geron is conducting Phase 1 clinical trials in the U.S. with embryonic stem cell-derived oligodendrocyte precursor cells to treat traumatic spinal cord injury. Their approach involves isolating and expanding human embryonic stem cells, then differentiating them in culture to produce oligodendrocyte precursor cells. StemCells Inc. uses human fetal-derived neural stem cells and has completed an initial Phase 1 clinical trial in early infantile Batten disease, a rare congenital lysosomal storage disorder; this company also has filed an IND for Pelizaeus-Merzbacher disease, which is a very rare pediatric leukodystrophy. StemCells Inc. also recently initiated Phase 1 clinical trials in Switzerland in patients with traumatic spinal cord injury, and is conducting preclinical studies for retinal disorders. ReNeuron uses human fetal-derived neural stem cells that it has conditionally immortalized with a c-mycER gene, and it has commenced clinical trials in stroke patients in the United Kingdom. Neuralstem is conducting Phase 1 clinical trials in patients with ALS using a neural stem cell product. California Stem Cells is exploring use of embryonic stem cell-derived motor neurons in animal models of spinal muscular atrophy and ALS.
20
Why Q Therapeutics’ approach may prove superior
A noteworthy distinction between Q and other CNS cell therapy companies involves the specific properties of Q-Cells®. These cells produce mature glial cells (astrocytes and oligodendrocytes) that can both achieve myelination of neuronal axons as well as otherwise promote neuron health, while minimizing unwanted and potentially deleterious neuron formation. Q-Cells® are a more mature cell type - at a more advanced stage of differentiation - than are the cells used by StemCells Inc., ReNeuron, and Neuralstem, who all use the less-differentiated CNS stem cells that produce neurons as well as glial cells (see Figure 3). Unlike ReNeuron, Q does not use genetically immortalized cells, which Q believes could raise safety issues, e.g., concerns about deleterious effects such as tumor formation.
Truly pluripotent stem cells could also be a starting source. This approach requires the identification of proper signaling factors and establishing the sequence of use to correctly and safely induce differentiation into the desired CNS cell type and to ensure sufficient elimination of the undifferentiated ESCs which can form tumors, prior to transplantation. In contrast to Geron, Q-Cells® are not derived from undifferentiated (pluripotent) embryonic stem cells. Rather, Q-Cells® are isolated already at the desired final cell type, and require no in vitro differentiation protocols such as those used by Geron with its ESC-derived oligodendrocyte progenitor cells. Q believes that the progenitor cell therapies it is developing, generated from pre-formed cells that naturally occur, offer safety and efficacy benefits.
Other Potential Competitors
Another approach is the use of cells or virus-based vectors implanted in the brain as delivery vehicles for gene therapy, with the goal of achieving local production of desired proteins and growth factors. NsGene is studying the use of encapsulated, genetically-modified cells to secrete biological growth factors in the treatment of Alzheimer’s and Parkinson’s diseases. Ceregene is conducting clinical trials with virus-based vectors for gene therapy for Parkinson’s and Alzheimer’s diseases, with other disease targets in earlier research. Q’s approach differs significantly in that it does not rely on a single growth factor for success, and in that it does not use genetically-modified cells or viruses; rather, unmodified Q-Cells® produce the range of growth factors and trophic support that is typical of healthy glial cells.
Other companies working in the CNS space have focused on stimulating endogenous differentiation of nascent adult stem cells with administration of molecules to trigger their expansion and differentiation in vivo (e.g., NeuroNova, Braincells). Still other pharmaceutical and biotechnology companies (including, among others Genzyme, Biogen-Idec, Acorda, Merck-Serono, GlaxoSmithKline, Roche, and Sanofi-Aventis) have programs to identify therapeutics to treat neurodegenerative diseases such as MS, Parkinson’s disease and Alzheimer’s disease. Some of these programs might yield effective therapeutics that could compete with Q’s products, and which might prove less costly and easier to administer.
Description of Property.
Q Therapeutics utilizes leased office and laboratory space located at 615 Arapeen Drive, Suite 102, Salt Lake City, UT 84108, and owns the equipment it uses therein which is summarized in the Financial Statements. The cost of the lease is $12,192 per month for the year ending December 31, 2011.
Legal Proceedings.
None.
Submission of Matters to a Vote of Security Holders.
On October 12, 2011, the sole director of Grace 2, all of the directors of Q Acquisition, Inc., a majority of the shareholders of Q Acquisition, Inc., over two-thirds of the holders of Q Therapeutics Series A and Series B preferred stock, a majority of the common shareholders of Q Therapeutics and all of the directors of Q Therapeutics voted in favor of the transactions contemplated by the Merger Agreement.
21
Market for Common Equity and Related Stockholder Matters and Small Business Issuer Purchases of Equity Securities.
Market Information
Our common stock is not currently trading on any stock exchange and it is not quoted on any quotation system or traded in any other manner in the public markets. We are not aware of any market activity in our stock since inception through the date of this filing.
The Company, through a registered broker-dealer, plans to submit a Form 211 Application to FINRA, in an effort to have the Company’s Common Stock quoted on the OTC Bulletin Board. We cannot guarantee that we will obtain a listing. There is currently no trading activity in our securities, and there can be no assurance that a regular trading market for our common stock will ever be developed.
Holders
As of the date of this filing, there are 148 record holders of 24,557,632 shares of our common stock.
Dividends
We do not intend to pay cash dividends on our common stock and preferred stock for the foreseeable future, but currently intend to retain any future earnings to fund the development and growth of our business. The payment of dividends if any, on the common stock and the preferred stock will rest solely within the discretion of the Board of Directors and will depend, among other things, upon our earnings, capital requirements, financial condition, and other relevant factors. We have not paid or declared any dividends upon our common stock or preferred stock since inception.
Recent Sales of Unregistered Securities
In connection with the Merger Agreement, on October 13, 2011, we issued an aggregate of 22,671,923 shares of Common Stock (including shares underlying warrants and options), or 89.7% of the outstanding shares prior to the offering, to Q Therapeutics, Inc. Shareholders in exchange for 100% of the outstanding shares of Q Therapeutics, Inc., which merger resulted in Q Therapeutics, Inc. becoming our wholly owned subsidiary. These securities qualified for exemption under Section 4(2) of the Securities Act since the issuing of securities by us did not involve a public offering. The offering was not a “public offering” as defined in Section 4(2) due to the insubstantial number of persons involved in the deal, size of the offering, manner of the offering and number of securities offered. We did not undertake an offering in which we sold a high number of securities to a high number of investors. In addition, these shareholders had the necessary investment intent as required by Section 4(2) since they agreed to and received share certificates bearing a legend stating that such securities are restricted pursuant to Rule 144 of the Securities Act. This restriction ensures that these securities would not be immediately redistributed into the market and therefore not be part of a “public offering.” Based on an analysis of the above factors, we have met the requirements to qualify for exemption under Section 4(2) of the Securities Act for this transaction.
22
On August 30, 2011, Q Therapeutics, Inc. received $450,000 in bridge financing from investors, (including $90,000 in credit towards the advancement of consulting fees and out-of-pocket expenses) evidenced by secured convertible promissory notes (“Notes”) and a unit, consisting of an aggregate of (a) one, Seven-Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $1.00 per share of the Company’s common stock, (b) one Seven-Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $2.00 per share of the Company’s common stock and (c) one Seven-Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock with an exercise price equal to the lesser of (a) the price per share of the Company’s Shares issued in a financing or (b) the Fair Market Value (“FMV”) of the Company’s Shares. An automatic conversion feature is in place for each Note, whereby upon the attainment of the Company’s Private Placement Offering’s minimum raise amount of $3,000,000, each Note’s outstanding principal and interest balance will automatically be converted into common stock of Q Therapeutics, Inc. at the same purchase price and upon the same terms as investors in the Company’s Private Placement Offering. These securities qualified for exemption under Section 4(2) of the Securities Act since the issuance securities by us did not involve a public offering. The offering was not a “public offering” as defined in Section 4(2) due to the insubstantial number of persons involved in the deal, size of the offering, manner of the offering and number of securities offered. Q Therapeutics, Inc. did not undertake an offering in which we sold a high number of securities to a high number of investors. In addition, these shareholders had the necessary investment intent as required by Section 4(2) since they agreed to and received share certificates bearing a legend stating that such securities are restricted pursuant to Rule 144 of the Securities Act. This restriction ensures that these securities would not be immediately redistributed into the market and therefore not be part of a “public offering.” Based on an analysis of the above factors, Q Therapeutics, Inc. has met the requirements to qualify for exemption under Section 4(2) of the Securities Act for this transaction.
As more fully described in Item 1.01 above, on October 13, 2011, immediately following the Merger, pursuant to the Purchase Agreement, we consummated the Private Placement for the issuance and sale of Units, consisting of an aggregate of (a) one share of Common Stock, (b) one Seven-Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $1.00 per share, and (c) one Seven-Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $2.00 per share, for aggregate gross proceeds of $3,803,047. Such securities were not registered under the Securities Act. The issuance of these securities was exempt from registration under Regulation D, Regulation S and Section 4(2) of the Securities Act. We made this determination based on the representations of Investors, which included, in pertinent part, that such shareholders were either (a) “accredited investors” within the meaning of Rule 501 of Regulation D promulgated under the Securities Act, or (b) not a “U.S. person” as that term is defined in Rule 902(k) of Regulation S under the Act, and that such shareholders were acquiring our common stock, for investment purposes for their own respective accounts and not as nominees or agents, and not with a view to the resale or distribution thereof, and that the shareholders understood that the shares of our common stock may not be sold or otherwise disposed of without registration under the Securities Act or an applicable exemption therefrom.
In instances described above where we issued securities in reliance upon Regulation D, we relied upon Rule 506 of Regulation D of the Securities Act. These stockholders who received the securities in such instances made representations that (a) the stockholder is acquiring the securities for his, her or its own account for investment and not for the account of any other person and not with a view to or for distribution, assignment or resale in connection with any distribution within the meaning of the Securities Act, (b) the stockholder agrees not to sell or otherwise transfer the purchased shares unless they are registered under the Securities Act and any applicable state securities laws, or an exemption or exemptions from such registration are available, (c) the stockholder has knowledge and experience in financial and business matters such that he, she or it is capable of evaluating the merits and risks of an investment in us, (d) the stockholder had access to all of our documents, records, and books pertaining to the investment and was provided the opportunity to ask questions and receive answers regarding the terms and conditions of the offering and to obtain any additional information which we possessed or were able to acquire without unreasonable effort and expense, and (e) the stockholder has no need for the liquidity in its investment in us and could afford the complete loss of such investment. Management made the determination that the investors in instances where we relied on Regulation D are Accredited Investors (as defined in Regulation D) based upon management’s inquiry into their sophistication and net worth. In addition, there was no general solicitation or advertising for securities issued in reliance upon Regulation D.
23
As noted above, upon consummation of the Merger, all securities of Q Therapeutics issued and outstanding were converted into securities of the Company on a pro rata basis.
RISK FACTORS
An investment in our Company is extremely risky. You should carefully consider these risks, in addition to the other information presented in this Report, before deciding to our securities. If any of the following risks actually materialize, our business and prospects could be seriously harmed, the trading price and value of our securities could decline and you could lose all or part of your investment. The risks and uncertainties described below are not exclusive and are intended to reflect the material risks that are specific to us, material risks related to our industry and material risks related to companies that undertake a public offering or seek to maintain a class of securities that is registered or traded on an exchange or quoted on the over-the-counter market.
RISKS RELATED TO Q THERAPEUTICS, INC.’S BUSINESS
Our business is at an early stage of development.
Our business is at an early stage of development, in that we do not yet have product candidates in clinical trials or on the market. Our ability to develop product candidates that progress to and through clinical trials is subject to our ability to, among other things:
|
|
·
|
succeed in our research and development efforts;
|
|
|
·
|
select therapeutic compounds or cell therapies for development that have acceptable safety and efficacy profiles;
|
|
|
·
|
obtain required regulatory approvals;
|
|
|
·
|
finance, or obtain additional financing for, our clinical trials;
|
|
|
·
|
manufacture product candidates; and
|
|
|
·
|
collaborate successfully with clinical trial sites, academic institutions, physician investigators, clinical research organizations and other third parties, and achieve adequate enrollment of subjects.
|
We have a history of losses and anticipate future losses, and continued losses could impair our ability to sustain operations.
We have incurred operating losses every year since our incorporation in 2002. As of December 31 2010, our accumulated deficit was $14,835,100. Losses have resulted principally from costs incurred in connection with our research and development activities and from general and administrative costs associated with our operations. We expect to incur additional operating losses and, as our development efforts and clinical testing activities continue, our operating losses may increase in size.
Substantially all of our revenues to date have been from grants or license/research agreements. We may be unsuccessful in obtaining any new grants or entering into new license agreements that result in revenues. We do not expect that the revenues generated from any of these arrangements will be sufficient alone to continue or expand our research or development activities and otherwise sustain our operations.
24
Our ability to continue or expand our research and development activities and otherwise sustain our operations is dependent on our ability, alone or with others, to, among other things, manufacture and market therapeutic products. We also expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. This will result in decreases in our working capital, total assets and stockholders’ equity, which may not be offset by future financings. We will need to generate significant revenues to achieve profitability. We may not be able to generate these revenues, and we may never achieve profitability. Our failure to achieve profitability could negatively impact the market price of our common stock. Even if we do become profitable, we cannot assure you that we would be able to sustain or increase profitability on a quarterly or annual basis.
We will need additional capital to conduct our operations and develop our product candidates, and our ability to obtain the necessary funding is uncertain.
We will require substantial capital resources in order to conduct our operations and develop our product candidates, and we cannot assure you that our existing capital resources, grants, interest income and/or equipment financing arrangements will be sufficient to fund future planned operations. The timing and degree of any future capital requirements will depend on many factors, including:
|
|
·
|
the accuracy of the assumptions underlying our estimates for our capital needs for the remainder of the 2011 fiscal year and beyond;
|
|
|
·
|
the magnitude and scope of our research and development programs;
|
|
|
·
|
the progress we make in our research and development programs, preclinical development and clinical trials;
|
|
|
·
|
our ability to establish, enforce and maintain strategic arrangements for research, development, clinical testing, manufacturing and marketing;
|
|
|
·
|
the number and type of product candidates that we pursue;
|
|
|
·
|
the time and costs involved in obtaining regulatory approvals and clearances; and
|
|
|
·
|
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims.
|
We do not have any committed sources of capital. Additional financing through strategic collaborations, public or private equity financings, debt financing, capital lease transactions or other financing sources may not be available on acceptable terms, or at all. The receptivity of the public and private equity markets to proposed financings is substantially affected by the general economic, market and political climate and by other factors which are unpredictable and over which we have no control. Additional equity financings, if we obtain them, could result in significant dilution to our stockholders. Further, in the event that additional funds are obtained through arrangements with collaborative partners, these arrangements may require us to relinquish rights to some of our technologies, product candidates or proposed products that we would otherwise seek to develop and commercialize ourselves. If sufficient capital is not available, we may be required to delay, reduce the scope of or eliminate one or more of our programs, any of which could have a material adverse effect on our business.
25
Our business is dependent on limited product candidates.
At present, our ability to progress as a company is significantly dependent on our first product candidate for neurodegenerative diseases, with the initial clinical indication for ALS, which is in preclinical development. Any preclinical, regulatory or other development that significantly delays or prevents us from commencing or subsequently completing any of our trials, any material safety issue or adverse side effect to any study participant in these future trials, or the failure of trials to show the results expected would likely depress our stock price significantly and could prevent us from raising the additional capital we will need to further develop our cellular technologies. Moreover, any material adverse occurrence in our first clinical trials could substantially impair our ability to initiate clinical trials to test our stem cell therapies in other potential indications. Also, any material adverse event in clinical trials from other companies using cell therapy products could have a material adverse impact on our ability to carry out clinical trials. This, in turn, could adversely impact our ability to raise additional capital and pursue our planned research and development efforts.
Our business relies on cell therapy technologies that we may not be able to commercially develop.
We have concentrated the majority of our research on cell therapy technologies. Our ability to generate revenue and operate profitably will depend on being able to develop these technologies for human applications. These are emerging technologies and have limited human applications. We cannot guarantee that we will be able to develop our technologies or that such development will result in products with any commercial utility or value. We anticipate that the commercial sale of such products and royalty/licensing fees related to the technology will be our future primary sources of revenues. If we are unable to develop our technologies, we may never realize any revenue.
Our product development programs are based on novel technologies and are inherently risky.
We are subject to the risks of failure inherent in the development of products based on new technologies. The novel nature of these therapies creates significant challenges in regard to product development and optimization, manufacturing, government regulation, third-party reimbursement, and market acceptance. For example, the pathway to regulatory approval for cell-based therapies, including our product candidates, may be more complex and lengthy than the pathway for conventional drugs. These challenges may prevent us from developing and commercializing products on a timely or profitable basis or at all.
We intend to rely upon third-party manufacturers for our cells.
We currently have no internal manufacturing capability, and will rely extensively on licensees, strategic partners or third-party contract manufacturers or suppliers. Should we be forced to manufacture our cell therapy product, we cannot give you any assurance that we will be able to develop an internal manufacturing capability or procure alternative third-party suppliers. Moreover, we cannot give you any assurance that any contract manufacturers or suppliers we procure will be able to supply our product in a timely or cost effective manner or in accordance with applicable regulatory requirements or our specifications. We are presently working with the University of Utah’s Cell Therapy Facility in the production of Q-Cells®. We will need to change manufacturers at some point in the future in order to manufacture a product for commercial sale, among other things. When that change to a new manufacturer is made, it will likely entail additional expense to achieve technology transfer and validation, which could cause delays in manufacturing and product development activities.
We base our research and development on the use of human cells obtained from human tissue. The U.S. federal and state governments and other jurisdictions impose restrictions on the acquisition and use of human tissue, including those incorporated in federal Good Tissue Practice, or “GTP,” regulations. These regulatory and other constraints could prevent us from obtaining cells and other components of our products in the quantity or of the quality needed for their development or commercialization. These restrictions change from time to time and may become more onerous. Additionally, we may not be able to identify or develop reliable sources for the cells necessary for our potential products—that is, sources that follow all state and federal laws and guidelines for cell procurement. Certain components used to manufacture our stem and progenitor cell product candidates will need to be manufactured in compliance with the FDA’s Good Manufacturing Practices, or “GMP.” Accordingly, we will need to enter into supply agreements with companies that manufacture these components to GMP standards. There is no assurance that we will be able to enter into any such agreements.
26
Noncompliance with applicable requirements both before and after approval, if any, can subject us, our third-party suppliers and manufacturers and our other collaborators to administrative and judicial sanctions, such as, among other things, warning letters, fines and other monetary payments, recall or seizure of products, criminal proceedings, suspension or withdrawal of regulatory approvals, interruption or cessation of clinical trials, total or partial suspension of production or distribution, injunctions, limitations on or the elimination of claims we can make for our products, refusal of the government to enter into supply contracts or fund research, or government delay in approving or refusal to approve new drug applications.
Our inability to complete pre-clinical and clinical testing and receive regulatory approvals will impair our viability.
No assurances can be given that we will be able to commence the clinical trials or that any clinical trials will be completed or result in a successful outcome. If regulatory authorities do not approve our products or if we fail to maintain regulatory compliance, we would be unable to commercialize our therapeutic products, and our business and results of operations would be materially harmed.
Positive results from pre-clinical studies should not be relied upon as evidence that our clinical trials will succeed. Even if our product candidates achieve positive results in pre-clinical studies, we will be required to demonstrate through clinical trials that the product candidates are safe and effective for use in a diverse population before we can seek regulatory approvals for their commercial sale. There is typically an extremely high rate of attrition from the failure of product candidates as they proceed through clinical trials. If any product candidate fails to demonstrate sufficient safety and efficacy in any clinical trial, then we would experience potentially significant delays in, or be required to abandon, development of that product candidate. If we delay or abandon our development efforts of any of our product candidates, then we may not be able to generate sufficient revenues to become profitable, and our operations could be materially harmed.
Potential lead drug compounds or other product candidates and technologies require significant preclinical and clinical testing prior to regulatory approval in the United States and other countries. Our product candidates may prove to have undesirable and unintended side effects or other characteristics adversely affecting their safety, efficacy or cost-effectiveness that could prevent or limit their commercial use. In addition, our product candidates may not prove to be more effective for treating disease or injury than current therapies. Accordingly, we may have to delay or abandon efforts to research, develop or obtain regulatory approvals to market our product candidates. In addition, we will need to determine whether any of our potential products can be manufactured in commercial quantities at an acceptable cost. Our research and development efforts may not result in a product that can be or will be approved by regulators or marketed successfully. Competitors may have proprietary rights that prevent us from developing and marketing our products or they may sell similar, superior or lower cost products. Adverse results in research or clinical trials of others with cell therapy products may adversely affect regulatory or financial areas that could hinder our ability to continue with our clinical development. Because of the significant scientific, regulatory and commercial milestones that must be reached for any of our development programs or product candidates to be successful, any program or product candidate may be abandoned, even after we have expended significant resources, such as our investments or prospective investments in stem cell or cell therapy technologies, which could adversely affect our business and materially and adversely affect our stock price.
27
The science and technology of stem and progenitor cell biology are relatively new. There is no precedent for the successful commercialization of therapeutic product candidates being developed by Q Therapeutics based on these technologies. Therefore, our development programs are particularly risky and uncertain. In addition, we, and/or our collaborators must undertake significant research and development activities to develop product candidates based on these technologies, which will require additional funding and may take years to accomplish, if ever.
Our business is subject to ethical and social concerns and restrictions on use of stem or progenitor cells could prevent us from developing or gaining acceptance for commercially viable products based upon such technology and adversely affect the market price of our common stock.
The use of stem cells for research and therapy has been the subject of debate regarding ethical, legal and social issues. Negative public attitudes toward stem cell therapy could result in greater governmental regulation of stem cell therapies, which could harm our business. For example, concerns regarding such possible regulation could impact our ability to attract collaborators and investors. Existing and potential U.S. government regulation of human tissue may lead researchers to leave the field of stem and progenitor cell research or the country altogether, in order to assure that their careers will not be impeded by restrictions on their work. Similarly, these factors may induce graduate students to choose other fields less vulnerable to changes in regulatory oversight, thus exacerbating the risk that we may not be able to attract and retain the scientific personnel we need in the face of competition among pharmaceutical, biotechnology and health care companies, universities and research institutions for what may become a shrinking class of qualified individuals.
Some of our most important programs involve the use of progenitor cells that are derived from human fetal cadaver tissue. The use of these can give rise to ethical and social issues regarding elective termination of pregnancy and the appropriate use of tissue derived therefrom. Some political and religious groups have voiced opposition to these technologies and practices. We use progenitor cells, and may use stem cells, derived from fetal cadaver tissue. Our research related to these derivatives may become the subject of adverse commentary or publicity, which could significantly harm the market price of our common stock. Changes in federal or state laws regulating rights to elective terminations of pregnancy may adversely impact our ability to procure tissue for manufacturing our products.
We are not using cells derived from embryonic stem cells. Government imposed restrictions with respect to use of embryos in research and development still could have a negative material effect on our business, including reducing interest in the cell therapy field overall and reducing interest in financing our business, impairing our ability to establish critical partnerships and collaborations.
Changes in governmental regulations relating to funding of stem cell research may also materially impact our business and result in an increase to the volatility of the market price of our common stock. For example, in March 2009 President Obama issued Executive Order 13505, entitled “Removing Barriers to Responsible Scientific Research Involving Human Stem Cells.” As a result, the Secretary of Health and Human Services, through the Director of the National Institutes of Health (NIH), issued new guidelines relating to human stem cell research to allow federal funding for research using human embryonic stem cells (‘hESCs”) derived from embryos created by in vitro fertilization for reproductive purposes, but are no longer needed for that purpose. However, in August 2010, the Federal District Court for the District of Columbia issued a preliminary injunction prohibiting federal funding for hESC research. In September 2010, a federal appeals court lifted the injunction. Meanwhile, certain states are considering enacting, or already have enacted, legislation relating to stem cell research, including California, whose voters approved Proposition 71 to provide state funds for stem cell research in November 2004. In the United Kingdom and some other countries, the use of embryonic or fetal tissue in research (including the derivation of hESCs) is regulated by the government, whether or not the research involves government funding.
28
These potential effects and others may result in a decrease in the market price of our common stock.
We operate in leased facilities and cannot be assured that these facilities will be available to us upon expiration of our current lease.
We occupy laboratory and office space in facilities leased from a commercial landlord, and we have added improvements to these facilities at our own expense to meet our laboratory needs. We cannot be assured that we will be able to remain in the same space after our lease expires. If we need to move to a new facility, this will require additional expense to add the improvements needed for our operations as well as delay our development activities until we are fully operational in a new space.
We may engage in acquisitions or strategic transactions or make investments that could result in significant changes or management disruption and fail to enhance stockholder value.
We may engage in acquisitions or strategic transactions or make investments with the goal of maximizing stockholder value. We may acquire businesses, in-license new technologies for development, enter into joint ventures or other strategic transactions, and purchase equity and debt securities, including minority interests in publicly-traded and private companies, non-investment-grade debt securities, equity and debt mutual and exchange-traded funds, and corporate bonds/notes. Some of our strategic investments may entail a high degree of risk and will not become liquid until more than one year from the date of investment, if at all. Any or all of our acquisitions or strategic investments that we may undertake in the future may not generate financial returns or result in increased adoption or continued use of our technologies. In addition, our other investments may not generate financial returns or may result in losses due to market volatility, the general level of interest rates and inflation expectations. In some cases, we may be required to consolidate or record our share of the earnings or losses of those companies. Our share of any losses will adversely affect our financial results until we exit from or reduce our exposure to these investments. Achieving the anticipated benefits of acquisitions depends in part upon our ability to integrate the acquired businesses in an efficient and effective manner. The integration of companies that have previously operated independently may result in significant challenges, and we may be unable to accomplish the integration smoothly or successfully. We cannot assure you that the integration of acquired businesses with our business will result in the realization of the full benefits anticipated by us to result from the acquisition. We may not derive any commercial value from the acquired technology, products and intellectual property or from future technologies and products based on the acquired technology and/or intellectual property, and we may be subject to liabilities that are not covered by indemnification protection we may obtain.
Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses.
Changing laws, regulations and standards relating to corporate governance and public disclosure may create uncertainty regarding compliance matters. New or changed laws, regulations and standards are subject to varying interpretations in many cases. As a result, their application in practice may evolve over time. We are committed to maintaining high standards of corporate governance and public disclosure. Complying with evolving interpretations of new or changed legal requirements may cause us to incur higher costs as we revise current practices, policies and procedures, and may divert management time and attention from product development and revenue generation to compliance activities. If our efforts to comply with new or changed laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, our reputation might also be harmed.
29
Following the Merger as described herein, we are subject to the reporting requirements of federal securities laws, which can be expensive and may divert management’s time and Company resources from other projects, thus impairing our ability grow.
As a result of the Merger with Grace 2, we are now considered an SEC reporting company and, accordingly, subject to the information and reporting requirements of the Securities Exchange Act of 1934, as amended, and other federal securities laws, including compliance with the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”). The costs of preparing and filing annual, quarterly and current reports, proxy statements and other information with the SEC and furnishing audited reports to stockholders will cause our expenses to be higher than they would be if we remained privately held and did not consummate the Merger.
It may be time consuming, difficult and costly for us to develop and implement the internal controls and reporting procedures required by the Sarbanes-Oxley Act. We may need to hire additional financial reporting, internal controls and other finance personnel in order to develop and implement appropriate internal controls and reporting procedures. If we are unable to comply with the internal controls requirements of the Sarbanes-Oxley Act, then we may not be able to obtain the independent accountant certifications required by such Act, which may preclude us from keeping our filings with the SEC current.
If we fail to establish and maintain an effective system of internal control, we may not be able to report our financial results accurately or to prevent fraud. Any inability to report and file our financial results accurately and timely could harm our reputation and adversely impact the trading price of our Common Stock.
Effective internal control is necessary for us to provide reliable financial reports and prevent fraud. If we cannot provide reliable financial reports or prevent fraud, we may not be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed. As a result, our small size and any current internal control deficiencies may adversely affect our financial condition, results of operation and access to capital. We have not performed an in-depth analysis to determine if historical un-discovered failures of internal controls exist, and may in the future discover areas of our internal control that need improvement.
Public company compliance may make it more difficult to attract and retain officers and directors.
The Sarbanes-Oxley Act and new rules subsequently implemented by the SEC have required changes in corporate governance practices of public companies. As a public company, we expect these new rules and regulations to increase our compliance costs in 2011 and beyond and to make certain activities more time consuming and costly. As a public company, we also expect that these new rules and regulations may make it more difficult and expensive for us to obtain director and officer liability insurance in the future and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors or as executive officers.
30
RISKS RELATING TO OUR COMMON STOCK
There is currently no trading market for our Common Stock and we cannot ensure that one will ever develop or be sustained.
To date there has been no trading market for our Common Stock. We cannot predict how liquid the market for our Common Stock might become. Following the Merger as described herein, we anticipate having our Common Stock quoted for trading on the OTC Bulletin Board, however, we cannot be sure that such quotation will be obtained promptly, if at all. As soon as is practicable, we anticipate applying for listing of our Common Stock on either the NYSE, Amex, The NASDAQ Capital Market or other national securities exchange, assuming that we can satisfy the initial listing standards for such exchange.
We currently do not satisfy the initial listing standards, and cannot ensure that we will be able to satisfy such listing standards or that our Common Stock will be accepted for listing on any such exchange. Should we fail to satisfy the initial listing standards of such exchanges, or our Common Stock is otherwise rejected for listing and remain listed on the OTC Bulletin Board or suspended from the OTC Bulletin Board, the trading price of our Common Stock could suffer and the trading market for our Common Stock may be less liquid and our Common Stock price may be subject to increased volatility.
Furthermore, for companies whose securities are traded in the OTC Bulletin Board, it is more difficult (1) to obtain accurate quotations, (2) to obtain coverage for significant news events because major wire services generally do not publish press releases about such companies, and (3) to obtain needed capital.
We might not be successful in attaining a FINRA registered broker-dealer to apply to FINRA to have our Common Stock quoted on the OTCBB and if even if we are, the application might not be approved by FINRA.
Promptly after the Merger, we intend to solicit a FINRA registered broker-dealer to apply to FINRA to have our Common Stock quoted on the OTC Bulletin Board and to act as a market-maker in our Common Stock. We cannot give any assurances that we will be successful in soliciting a FINRA broker-dealer and that even if we are, that its application with FINRA will be approved. If our Common Stock is not quoted on the OTCBB, we intend to have our Common Stock listed on the OTC Pink Sheets which might make it more difficult for investors to trade their shares.
If and when our Common Stock becomes publicly traded, our Common Stock may be deemed a “penny stock,” which would make it more difficult for our investors to sell their shares.
If and when our Common Stock becomes publicly traded, our Common Stock may be subject to the “penny stock” rules adopted under Section 15(g) of the Exchange Act. The penny stock rules generally apply to companies whose common stock is not listed on The NASDAQ Stock Market or other national securities exchange and trades at less than $4.00 per share, other than companies that have had average revenue of at least $6,000,000 for the last three years or that have tangible net worth of at least $5,000,000 ($2,000,000 if the company has been operating for three or more years). These rules require, among other things, that brokers who trade penny stock to persons other than established customers complete certain documentation, make suitability inquiries of investors and provide investors with certain information concerning trading in the security, including a risk disclosure document and quote information under certain circumstances. Many brokers have decided not to trade penny stocks because of the requirements of the penny stock rules and, as a result, the number of broker-dealers willing to act as market makers in such securities is limited. If we remain subject to the penny stock rules for any significant period, it could have an adverse effect on the market, if any, for our securities. If our securities are subject to the penny stock rules, investors will find it more difficult to dispose of our securities.
31
Investor relations activities, nominal float and supply and demand factors may affect the price of our Common Stock.
We expect to utilize various techniques such as non-deal road shows and investor relations campaigns in order to create investor awareness for our Company. These campaigns may include personal, video and telephone conferences with investors and prospective investors in which our business practices are described. We may provide compensation to investor relations and financial advisory firms and pay for newsletters, websites, mailings and email campaigns that are produced by third-parties based upon publicly-available information concerning us. We will not be responsible for the content of analyst reports and other writings and communications by investor relations firms not authored by us or from publicly available information. We do not intend to review or approve the content of such analysts’ reports or other materials based upon analysts’ own research or methods. Investor relations firms should generally disclose when they are compensated for their efforts, but whether such disclosure is made or complete is not under our control. Our investors may be willing, from time to time, to encourage investor awareness through similar activities. Investor awareness activities may also be suspended or discontinued which may impact the trading market of our Common Stock.
The SEC and FINRA enforce various statutes and regulations intended to prevent manipulative or deceptive devices in connection with the purchase or sale of any security and carefully scrutinize trading patterns and company news and other communications for false or misleading information, particularly in cases where the hallmarks of pump and dump activities may exist, such as rapid share price increases or decreases. We and our shareholders may be subjected to enhanced regulatory scrutiny due to the small number of holders who initially will own the registered shares of our Common Stock publicly available for resale, and the limited trading markets in which such shares may be offered or sold which have often been associated with improper activities concerning penny-stocks, such as the OTC Bulletin Board or the OTCQB Marketplace (Pink OTC) or pink sheets. Until such time as the Common Stock sold in this Offering is registered and until such time as the restricted shares of the Company are registered or available for resale under Rule 144, there will continue to be a small percentage of shares held by a small number of investors, many of whom acquired such shares in privately negotiated purchase and sale transactions, that will constitute the entire available trading market. The Supreme Court has stated that manipulative action is a term of art connoting intentional or willful conduct designed to deceive or defraud investors by controlling or artificially affecting the price of securities. Often times, manipulation is associated by regulators with forces that upset the supply and demand factors that would normally determine trading prices. As described in this Memorandum, a small percentage of our outstanding Common Stock will initially be available for trading, held by a small number of individuals or entities. Accordingly, the supply of Common Stock for sale will be extremely limited for an indeterminate amount of time, which could result in higher bids, asks or sales prices than would otherwise exist. Securities regulators have often cited thinly-traded markets, small numbers of holders, and awareness campaigns as components of their claims of price manipulation and other violations of law when combined with manipulative trading, such as wash sales, matched orders or other manipulative trading timed to coincide with false or touting press releases. There can be no assurance that our or third-parties activities, or the small number of potential sellers or small percentage of stock in the float, or determinations by purchasers or holders as to when or under what circumstances or at what prices they may be willing to buy or sell stock, will not artificially impact (or would be claimed by regulators to have affected) the normal supply and demand factors that determine the price of stock.
Our stock price may be volatile.
The stock market in general, and the stock prices of technology-based and biotechnology / biopharmaceutical stocks in particular, have experienced volatility that often has been unrelated to the operating performance of any specific public company. The market price of our Common Stock is likely to be highly volatile and could fluctuate widely in price in response to various factors, many of which are beyond our control, including the following:
|
|
·
|
changes in our industry; competitive pricing pressures; our ability to obtain working capital financing; additions or departures of key personnel;
|
32
|
|
·
|
limited public float in the hands of a small number of persons whose sales or lack of sales could result in positive or negative pricing pressure on the market price for our Common Stock;
|
|
|
·
|
sales of our Common Stock;
|
|
|
·
|
our ability to execute our business plan;
|
|
|
·
|
operating results that fall below expectations;
|
|
|
·
|
loss of any strategic relationship;
|
|
|
·
|
regulatory developments;
|
|
|
·
|
changes in federal or state healthcare-related laws;
|
|
|
·
|
economic and other external factors;
|
|
|
·
|
period-to-period fluctuations in our financial results;
|
|
|
·
|
challenges and/or invalidation of key patents; and
|
|
|
·
|
inability to develop or acquire new or needed technology.
|
In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of our Common Stock.
Offers or availability for sale of a substantial number of shares of our Common Stock may cause the price of our Common Stock to decline.
If our stockholders sell substantial amounts of our Common Stock in the public market, including shares issued in past private offerings, upon the effectiveness of the registration statement required to be filed, or upon the expiration of any statutory holding period, under Rule 144, or upon expiration of lock-up periods applicable to outstanding shares, or issued upon the exercise of outstanding options or warrants, it could create a circumstance commonly referred to as an overhang and in anticipation of which the market price of our Common Stock could fall. The existence of an overhang, whether or not sales have occurred or are occurring, also could make more difficult our ability to raise additional financing through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate. The shares of Common Stock issued in the Merger to the current and former officers and directors, founders, and institutional shareholders of Q will be subject to a lock-up agreement prohibiting sales of such shares for a period of 12 months following the date on which the resale registration statement for the shares relating to our private offering is declared effective by the SEC. Following such date, all of those shares will become freely tradable, subject to securities laws and SEC regulations regarding sales by insiders. In addition, the shares of Common Stock sold in the private offering and the warrant shares will be freely tradable upon the earlier of: (i) effectiveness of a registration statement covering such shares and (ii) the date on which such shares may be sold without registration pursuant to Rule 144 (or other applicable exemption) under the Securities Act.
Our undesignated preferred stock may inhibit potential acquisition bids; this may adversely affect the market price for our common stock and the voting rights of holders of our common stock.
Our certificate of incorporation provides our Board of Directors with the authority to issue up 10,000,000 shares of undesignated preferred stock and to determine or alter the rights, preferences, privileges and restrictions granted to or imported upon these shares without further vote or action by our stockholders. As of the date of this Offering Memorandum, no shares of preferred stock have been designated and the Board of Directors still has authority to designate and issue up to 10,000,000 shares of preferred stock in one or more classes or series. The issuance of shares of preferred stock may delay or prevent a change in control transaction without further action by our stockholders. As a result, the market price of our common stock may be adversely affected. In addition, if we issue preferred stock in the future that has preference over our common stock with respect to the payment of dividends or upon our liquidation, dissolution or winding up, or if we issue preferred stock with voting rights that dilute the voting power of our common stock, the rights of holders of our common stock or the market price of our common stock could be adversely affected.
33
We have not paid dividends in the past and do not expect to pay dividends in the future. Any return on investment may be limited to the value of our Common Stock.
We have never paid cash dividends on our Common Stock and do not anticipate doing so in the foreseeable future. The payment of dividends on our Common Stock will depend on earnings, financial condition and other business and economic factors affecting us at such time as our board of directors may consider relevant. If we do not pay dividends, our Common Stock may be less valuable because a return on your investment will only occur if our stock price appreciates.
Because our directors and executive officers are among our largest stockholders, they can exert significant control over our business and affairs and have actual or potential interests that may depart from those of subscribers in our previous Private Offering.
Our directors and executive officers will own or control a significant percentage of the Common Stock following the Share Exchange and completion of our private placement offering. Additionally, the holdings of our directors and executive officers may increase in the future upon vesting or other maturation of exercise rights under any of the options or warrants they may hold or in the future be granted or if they otherwise acquire additional shares of our Common Stock. The interests of such persons may differ from the interests of our other stockholders, including purchasers of Units in our private placement offering. As a result, in addition to their board seats and offices, such persons will have significant influence over and control all corporate actions requiring stockholder approval, irrespective of how the Company’s other stockholders, including purchasers in our private placement offering, may vote, including the following actions: to elect or defeat the election of our directors; to amend or prevent amendment of our Certificate of Incorporation or Bylaws; to effect or prevent a merger, sale of assets or other corporate transaction; and to control the outcome of any other matter submitted to our stockholders for vote. Such person’s stock ownership may discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of the Company, which in turn could reduce our stock price or prevent our stockholders from realizing a premium over our stock price.
RISKS RELATED TO CLINICAL AND COMMERCIALIZATION ACTIVITIES
Delays in the commencement of clinical testing of our current and potential product candidates could result in increased costs to us and delay our ability to generate revenues.
The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
|
|
·
|
obtaining adequate financing;
|
|
|
·
|
demonstrating sufficient safety and efficacy to obtain regulatory clearance to commence a clinical trial;
|
|
|
·
|
manufacturing sufficient quantities or producing products meeting our quality standards of a product candidate;
|
|
|
·
|
obtaining an injection device or methodology meeting our quality standards;
|
|
|
·
|
obtaining approval of an IND application or proposed trial design from the FDA;
|
|
|
·
|
reaching agreement on acceptable terms with our collaborators on all aspects of the clinical trial, including the contract research organizations (“CROs”) and the trial sites; and
|
|
|
·
|
obtaining Institutional Review Board (“IRB”) approval to conduct a clinical trial at a prospective site.
|
34
In addition, clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size and nature of the patient population, the nature of the protocol, the proximity of patients to clinical sites, other competing clinical trials in the same disease indication, the availability of effective treatments for the relevant disease, and the eligibility criteria for the clinical trial. Delays in commencing clinical testing of our product candidates could prevent or delay us from obtaining approval for our product candidates.
We do not have experience as a company in conducting clinical trials, or in other areas required for the successful commercialization and marketing of our product candidates.
We have no experience as a company in conducting either early stage or large-scale, late stage clinical trials. We cannot be certain that planned clinical trials will begin or be completed on time, if at all. Large-scale trials would require either additional financial and management resources, or reliance on third-party clinical investigators, pharmaceutical partners, CROs and/or consultants. Relying on third-party clinical investigators, pharmaceutical partners or CROs may force us to encounter delays that are outside of our control. Any such delays could have a material adverse effect on our business.
We also do not currently have marketing and distribution capabilities for our product candidates. Developing an internal sales and distribution capability would be an expensive and time-consuming process. We may enter into agreements with third parties that would be responsible for marketing and distribution. However, these third parties may not be capable of successfully selling any of our product candidates. The inability to commercialize and market our product candidates could materially adversely affect our business.
Obtaining regulatory approvals to market our product candidates in the United States and other countries is a costly and lengthy process and we cannot predict whether or when we will be permitted to commercialize our product candidates.
Federal, state and local governments in the United States and governments in other countries have significant regulations in place that govern many of our activities and may prevent us from creating commercially viable products from our discoveries. The regulatory process, particularly for biopharmaceutical product candidates like ours, is uncertain, can take many years and requires the expenditure of substantial resources.
Our potential product candidates will require extensive preclinical and clinical testing prior to submission of any regulatory application to commence commercial sales. In particular, human pharmaceutical therapeutic product candidates are subject to rigorous requirements of the FDA in the United States and similar health authorities in other countries in order to demonstrate safety and efficacy. Data obtained from preclinical and clinical activities is susceptible to varying interpretations that could delay, limit or prevent regulatory agency approvals. In addition, delays or rejections may be encountered as a result of changes in regulatory agency policy during the period of product development and/or the period of review of any application for regulatory agency approval for a product candidate.
Any product candidate that we or our collaborators develop must receive all relevant regulatory agency approvals/clearance before it may be marketed in the United States or other countries. Obtaining regulatory approval/clearance is a lengthy, expensive and uncertain process. Because certain of our product candidates involve the application of new technologies or are based upon a new therapeutic approach, they may be subject to substantial additional review by various government regulatory authorities, and, as a result, the process of obtaining regulatory approvals/clearance for them may proceed more slowly than for product candidates based upon more conventional technologies.
35
Delays in obtaining regulatory agency approvals/clearance could:
|
|
·
|
significantly delay and harm the marketing of any products that we or our collaborators develop;
|
|
|
·
|
impose costly procedures upon our activities or the activities of our collaborators;
|
|
|
·
|
diminish any competitive advantages that we or our collaborators may attain; or
|
|
|
·
|
adversely affect our ability to receive royalties and generate revenues and profits.
|
Even if we commit the necessary time and resources, the required regulatory agency approvals/clearance may not be obtained for any product candidates developed by us or in collaboration with us. If we obtain regulatory agency approval/clearance for a new product, this approval/clearance may entail limitations on the indicated uses for which it can be marketed that could limit the potential commercial use of the product.
Failure to achieve continued compliance with government regulation over approved products could delay or halt commercialization of our products.
Approved products and their manufacturers are subject to continual review, and discovery of previously unknown problems with a product or its manufacturer may result in restrictions on the product or manufacturer, including withdrawal of the product from the market. The future sale by us or our collaborators of any commercially viable product will be subject to government regulation from several standpoints, including the processes of:
|
|
·
|
manufacturing;
|
|
|
·
|
adverse-event reporting;
|
|
|
·
|
advertising and promoting;
|
|
|
·
|
selling and marketing;
|
|
|
·
|
labeling; and
|
|
|
·
|
distribution.
|
If, and to the extent that, we are unable to comply with these regulations, our ability to earn revenues will be materially and negatively impacted.
Failure to comply with regulatory requirements can result in severe civil and criminal penalties, including but not limited to:
|
|
·
|
recall or seizure of products;
|
|
|
·
|
injunction against the manufacture, distribution and sales and marketing of products; and
|
|
|
·
|
criminal prosecution.
|
The imposition of any of these penalties or other commercial limitations could significantly impair our business, financial condition and results of operations.
RISKS RELATED TO PROTECTING OUR INTELLECTUAL PROPERTY
Impairment of our intellectual property rights may adversely affect the value of our technologies and product candidates and limit our ability to pursue their development.
Protection of our proprietary technology is critically important to our business. Our success will depend in part on our ability to obtain and enforce our patents and maintain trade secrets, both in the United States and in other countries. Further, our patents may be challenged, invalidated or circumvented, and our patent rights may not provide proprietary protection or competitive advantages to us. In the event that we are unsuccessful in obtaining and enforcing patents, we may not be able to further develop or commercialize our product candidates and our business would be negatively impacted.
36
The patent positions of pharmaceutical and biopharmaceutical companies, including ours, are highly uncertain and involve complex legal and technical questions. In particular, legal principles for biotechnology and pharmaceutical patents in the United States and in other countries are evolving, and the extent to which we will be able to obtain patent coverage to protect our technology, or enforce issued patents, is uncertain. In the United States, recent court decisions in patent cases as well as proposed legislative changes to the patent system only exacerbate this uncertainty. Furthermore, significant amendments to the regulations governing the process of obtaining patents were proposed in a new rule package by the United States Patent and Trademark Office (the Patent Office) in 2007. The proposed new rules were widely regarded as detrimental to the interests of biotechnology and pharmaceutical companies. The implementation of the rule package was blocked by a court injunction requested by a pharmaceutical company. The Patent Office challenged the court decision through an appeal to the U.S. Court of Appeals for the Federal Circuit (CAFC), but the appeal was dismissed in November 2009, after the Patent Office changed course and rescinded the proposed new rules. At this point we do not know whether the Patent Office will attempt to introduce new rules to replace those that were recently withdrawn or whether any such new rules would also be challenged.
We are uncertain of what patent coverage we will obtain for our products in Europe or Asia. At this time, we do not know whether or to what extent we will be able to obtain future patent protection for our technologies and products in Europe or Asia. If we are unable to protect our inventions related to cell therapy products in Europe or Asia, our future business opportunities could be negatively impacted.
Challenges to our patent rights can result in costly and time-consuming legal proceedings that may prevent or limit development of our product candidates.
Publication of discoveries in scientific or patent literature tends to lag behind actual discoveries by at least several months and sometimes several years. Therefore, the persons or entities that we or our licensors name as inventors in our patents and patent applications may not have been the first to invent the inventions disclosed in the patent applications or patents, or the first to file patent applications for these inventions. As a result, we may not be able to obtain patents for discoveries that we otherwise would consider patentable and that we consider to be extremely significant to our future success.
Where more than one party seeks U.S. patent protection for the same technology, the Patent Office may declare an interference proceeding in order to ascertain the party to which the patent should be issued. Patent interferences are typically complex, highly contested legal proceedings, subject to appeal. They are usually expensive and prolonged, and can cause significant delay in the issuance of patents. Moreover, parties that receive an adverse decision in an interference can lose important patent rights. Our pending patent applications, or our issued patents, may be drawn into interference proceedings which may delay or prevent the issuance of patents, or result in the loss of issued patent rights.
Outside of the United States, certain jurisdictions, such as Europe, New Zealand and Australia, permit oppositions to be filed against the granting of patents. Because our intent is to commercialize products internationally, securing both proprietary protection and freedom to operate outside of the United States is important to our business. We may be, in the future, involved in both opposing the grant of patents to others through such opposition proceedings and in defending our patent applications against oppositions filed by others.
European opposition and appeal proceedings can take several years to reach final decision. The potential oppositions discussed above reflect the complexity of the patent landscape in which we operate, and illustrate the risks and uncertainties.
37
Patent opposition proceedings are not currently available in the U.S. patent system, but legislation has been proposed to introduce them. However, issued U.S. patents can be reexamined by the Patent Office at the request of a third party. Patents owned or licensed by Q Therapeutics may therefore be subject to reexamination. As in any legal proceeding, the outcome of patent reexaminations is uncertain, and a decision adverse to our interests could result in the loss of valuable patent rights.
As more groups become engaged in scientific research and product development in the areas of stem cells, the risk of our patents being challenged through patent interferences, oppositions, reexaminations, litigation or other means will likely increase. Challenges to our patents through these procedures can be extremely expensive and time-consuming, even if the outcome is favorable to us. An adverse outcome in a patent dispute could severely harm our business by:
|
|
·
|
causing us to lose patent rights in the relevant jurisdiction(s);
|
|
|
·
|
subjecting us to litigation, or otherwise preventing us from commercializing potential products in the relevant jurisdiction(s);
|
|
|
·
|
requiring us to obtain licenses to the disputed patents;
|
|
|
·
|
forcing us to cease using the disputed technology; or
|
|
|
·
|
requiring us to develop or obtain alternative technologies.
|
Furthermore, if such challenges to our patent rights are not resolved promptly in our favor, our existing and future business relationships may be jeopardized and we could be delayed or prevented from entering into new collaborations or from commercializing certain products, which could materially harm our business.
If we fail to meet our obligations under license agreements, we may lose our rights to key technologies on which our business depends.
Our business depends on several critical technologies that are based in part on patents licensed from third parties. Those third-party license agreements impose obligations on us, such as payment obligations and obligations to diligently pursue development of commercial products under the licensed patents. If a licensor believes that we have failed to meet our obligations under a license agreement, the licensor could seek to limit or terminate our license rights, which could lead to costly and time-consuming litigation and, potentially, a loss of the licensed rights. During the period of any such litigation our ability to carry out the development and commercialization of potential products could be significantly and negatively affected. If our license rights were restricted or ultimately lost, our ability to continue our business based on the affected technology would be severely adversely affected.
We may be subject to litigation that will be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with our licensees, licensors, or others with whom we have contractual or other business relationships, or with our competitors or others whose interests differ from ours. If we are unable to resolve those conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against us. That litigation is likely to be expensive and may require a significant amount of management’s time and attention, at the expense of other aspects of our business. The outcome of litigation is always uncertain, and in some cases could include judgments against us that require us to pay damages, enjoin us from certain activities, or otherwise affect our legal or contractual rights, which could have a significant adverse effect on our business.
38
The development, manufacturing and commercialization of cell-based therapeutic products expose us to product liability claims, which could lead to substantial liability.
By developing and, ultimately, commercializing therapeutic products, we are exposed to the risk of product liability claims. Product liability claims against us could result in substantial litigation costs and damage awards against us. We intend to obtain liability insurance that covers our clinical trials, and we will need to increase our insurance coverage if and when we begin commercializing products. We may not be able to obtain insurance on acceptable terms, if at all, and the policy limits on our insurance policies may be insufficient to cover our liability.
We may be subject to infringement claims that are costly to defend, and which may limit our ability to use disputed technologies and prevents us from pursuing research and development or commercialization of potential products.
Our commercial success depends significantly on our ability to operate without infringing patents and the proprietary rights of others. Our technologies may infringe the patents or proprietary rights of others. In addition, we may become aware of discoveries and technology controlled by third parties that are advantageous to our programs. In the event our technologies infringe the rights of others or we require the use of discoveries and technology controlled by third parties, we may be prevented from pursuing research, development or commercialization of potential products or may be required to obtain licenses to those patents or other proprietary rights or develop or obtain alternative technologies. We have obtained or are negotiating licenses from universities and companies for technologies that we anticipate incorporating into our potential products, and we initiate negotiation for licenses to other technologies as the need or opportunity arises. We may not be able to obtain a license to patented technology on commercially favorable terms, or at all. If we do not obtain a necessary license, we may need to redesign our technologies or obtain rights to alternate technologies, the research and adoption of which could cause delays in product development. In cases where we are unable to license necessary technologies, we could be prevented from developing certain potential products. Our failure to obtain alternative technologies or a license to any technology that we may require to research, develop or commercialize our product candidates would significantly and negatively affect our business.
Much of the information and know-how that is critical to our business is not patentable and we may not be able to prevent others from obtaining this information and establishing competitive enterprises.
We sometimes rely on trade secrets to protect our proprietary technology, especially in circumstances in which we believe patent protection is not appropriate or available. We attempt to protect our proprietary technology in part by confidentiality agreements with our employees, consultants, collaborators and contractors. We cannot assure you that these agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets will not otherwise become known or be independently discovered by competitors, any of which would harm our business significantly.
39
RISKS RELATED TO OUR RELATIONSHIPS WITH THIRD PARTIES
We depend on other parties to help us develop, manufacture and test our product candidates, and our ability to develop and commercialize potential products may be impaired or delayed if collaborations are unsuccessful.
Our strategy for the development, clinical testing and commercialization of our product candidates requires that we enter into collaborations with corporate partners, licensors, licensees and others. We are dependent upon the subsequent success of these other parties in performing their respective responsibilities and the continued cooperation of our partners. By way of example, we have key agreements with Goodwin Biosciences, Inc. and University of Utah related to manufacture of our products, and are working with Johns Hopkins University and MPI Research, Inc. on preclinical studies for our first product. We have agreements with Life Technologies, Inc. concerning commercialization of certain of our products for the research market. Our collaborators, contractors and licensees may not cooperate with us or perform their obligations under our agreements with them. We cannot control the amount and timing of our collaborators’ resources that will be devoted to activities related to our collaborative agreements with them. Our collaborators may choose to pursue existing or alternative technologies in preference to those being developed in collaboration with us.
Under agreements with other parties, we may rely significantly on them to, among other activities:
|
|
·
|
conduct research and development activities and preclinical safety studies in conjunction with or for us;
|
|
|
·
|
manufacture product(s) or reagents used for such product manufacture for use by us, our contractors and our partners;
|
|
|
·
|
design and conduct advanced clinical trials in the event that we reach clinical trials;
|
|
|
·
|
fund research and development activities with us;
|
|
|
·
|
manage and license certain patent rights;
|
|
|
·
|
pay us fees upon the achievement of milestones; and
|
|
|
·
|
market with us any commercial products that result from our collaborations.
|
The development and commercialization of potential products will be delayed if collaborators or other partners fail to conduct these activities in a timely manner or at all. In addition, our collaborators could terminate their agreements with us and we may not receive any development or milestone payments. If we do not achieve milestones set forth in the agreements, or if our collaborators breach or terminate their collaborative agreements with us, our business may be materially harmed.
We also rely on other companies for certain reagent development, manufacturing or other technical scientific work, with respect to our cell products. We have contracts with these companies that specify the work to be done and results to be achieved, but we do not have direct control over their personnel or operations. If these companies do not perform the work that they were assigned, our ability to develop or manufacture our product candidates could be significantly harmed.
Our reliance on the activities of our consultants, research institutions, and scientific contractors, whose activities are not wholly within our control, may lead to delays in development of our product candidates.
We rely extensively upon and have relationships with scientific consultants at academic and other institutions, some of whom conduct research at our request, and other consultants who assist us in formulating our research and development and clinical strategy or other matters. These consultants are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. We have limited control over the activities of these consultants and, except as otherwise required by our collaboration and consulting agreements, can expect only limited amounts of their time to be dedicated to our activities.
In addition, we have formed research collaborations with academic and other research institutions in the U.S. These research facilities may have commitments to other commercial and noncommercial entities. We have limited control over the operations of these laboratories and can expect only limited amounts of their time to be dedicated to our research goals. If any of these third parties are unable or refuse to contribute to projects on which we need their help, our ability to generate advances in our technologies and develop our product candidates could be significantly harmed.
40
The loss of key personnel could slow our ability to conduct research and develop product candidates.
Our future success depends to a significant extent on the skills, experience and efforts of our executive officers and key members of our scientific staff. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. We may be unable to retain our current personnel or attract or assimilate other highly qualified management and scientific personnel in the future on acceptable terms. The loss of any or all of these individuals could harm our business and might significantly delay or prevent the achievement of research, development or business objectives.
Our product candidates are likely to be expensive to manufacture, and they may not be profitable if we are unable to significantly reduce the costs to manufacture them.
Our Q-Cells® and other cellular-based products could be more expensive to manufacture than other treatments currently on the market today or that will be on the market when our product is ready to be marketed. Our present manufacturing processes are conducted at a modest scale and we may be able to reduce manufacturing costs through process improvements, as well as through scale increases. If we are not able to do so, however, and, depending on the pricing of the potential product, the profit margin on our product may be significantly less than that of most drugs on the market today. If we are unable to scalably produce Q-Cells® at a commercially-acceptable manufacturing cost, it would impact the affordability of the therapy for patients and reduce our potential profitability.
The cell-based therapies we are developing may require large quantities of cells. We continue to develop processes to scale up production and increase yields of the cells in a cost-effective way. We may not be able to charge a high enough price for any cell therapy product we develop, even if it is safe and effective, to make a profit. If we are unable to realize significant profits from our potential product candidates, our business would be materially harmed.
Some of our competitors may develop technologies that are superior to or more cost-effective than ours, which may impact the commercial viability of our technologies and which may significantly damage our ability to sustain operations.
The pharmaceutical and biotechnology industries are intensely competitive. Other pharmaceutical and biotechnology companies and research organizations currently engage in or have in the past engaged in efforts related to the biological mechanisms that are the focus of our programs in glial cell therapies, including the study of neural stem cells as precursors for glial cells and use of embryonic stem cells to derive glial cells. In addition, other products and therapies that could compete directly with the product candidates that we are seeking to develop and market currently exist or are being developed by pharmaceutical and biopharmaceutical companies and by academic and other research organizations.
Other companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Academic institutions, government agencies and other public and private research organizations may also conduct research, seek patent protection and establish collaborative arrangements for research, clinical development and marketing of products similar to ours. These companies and institutions compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our programs.
41
In addition to the above factors, we expect to face competition in the following areas:
|
|
·
|
product efficacy and safety;
|
|
|
·
|
the timing and scope of regulatory consents;
|
|
|
·
|
availability of resources;
|
|
|
·
|
reimbursement coverage;
|
|
|
·
|
price; and
|
|
|
·
|
patent position, including potentially dominant patent positions of others.
|
As a result of the foregoing, our competitors may develop more effective or more affordable products, or achieve earlier patent protection or product commercialization than we do. Most significantly, competitive products may render any product candidates that we develop obsolete, which would negatively impact our business and ability to sustain operations. Although we believe that our approach holds the promise of achieving the desired safety and efficacy profiles for treatment of many neurodegenerative diseases, competitor companies may indeed demonstrate safety and efficacy of their competing products, as well as achieve a marketed product before we do.
To be successful, our product candidates must be accepted by the health care community, which can be very slow to adopt or unreceptive to new technologies and products.
Our product candidates and those developed by our collaborators, if approved for marketing, may not achieve market acceptance since hospitals, physicians, patients or the medical community in general may decide not to accept and utilize these products. The product candidates that we are attempting to develop represent substantial departures from established treatment methods and will compete with a number of conventional drugs and therapies manufactured and marketed by major pharmaceutical companies. The degree of market acceptance of any of our developed potential products will depend on a number of factors, including:
|
|
·
|
our establishment and demonstration to the medical community of the clinical efficacy and safety of our product candidates;
|
|
|
·
|
our ability to create products that are superior to alternatives currently on the market;
|
|
|
·
|
our ability to establish in the medical community the potential advantage of our treatments over alternative treatment methods; and
|
|
|
·
|
reimbursement policies of government and third-party payers.
|
If the health care community does not accept our potential products for any of the foregoing reasons, or for any other reason, our business would be materially harmed.
If we fail to obtain acceptable prices or adequate reimbursement for our product candidates, the use of our potential products could be severely limited.
Our ability to successfully commercialize our product candidates will depend significantly on our ability to obtain acceptable prices and the availability of reimbursement to the patient from third-party payers. In March 2010, President Obama signed the Patient Protection and Affordability Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the PPACA) into law. Focused on expanding healthcare coverage to millions of uninsured Americans and reducing the rate of increase in healthcare costs, the PPACA contains numerous initiatives that impact the pharmaceutical industry.
42
While the PPACA may increase the number of patients who have insurance coverage for our product candidates, its cost containment measures could also adversely affect reimbursement for our potential products. Cost control initiatives could decrease the price that we receive for any product candidate we may develop in the future. If our potential products are not considered cost-effective or if we fail to generate adequate third-party reimbursement for the users of our potential products and treatments, then we may be unable to maintain price levels sufficient to realize an appropriate return on our investment for potential products currently in development, which could have an adverse impact on our business. Furthermore, there have been challenges to the PPACA and if all or a portion of it were overturned this could have material adverse effects on our business.
Our competition has significantly greater experience and financial resources.
The biotechnology industry is characterized by intense competition. We compete against numerous companies, and many have substantially greater resources. Several such companies have initiated cell therapy research programs and/or efforts to treat the same diseases that we target. Companies such as Geron Corporation, Neuralstem, Inc., StemCells, Inc., and Genzyme Corporation as well as others, may have substantially greater resources and experience in our fields that put us at a competitive disadvantage.
RISKS RELATED TO ENVIRONMENTAL AND PRODUCT LIABILITY
Our activities involve hazardous materials, and improper handling of these materials by our employees or agents could expose us to significant legal and financial penalties.
Our research and development activities involve the controlled use of hazardous materials, chemicals and various compounds. As a consequence, we are subject to numerous environmental and safety laws and regulations, including those governing laboratory procedures, controlled substances, exposure to blood-borne pathogens and the handling of biohazardous materials. We may be required to incur significant costs to comply with current or future environmental laws and regulations and may be adversely affected by the cost of compliance with these laws and regulations. Although we believe that our safety procedures for using, handling, storing and disposing of hazardous materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. In the event of such an accident, state or federal authorities could curtail our use of these materials and we could be liable for any civil damages that result, the cost of which could be substantial. Further, any failure by us to control the use, disposal, removal or storage, or to adequately restrict the discharge, or assist in the cleanup, of hazardous chemicals or hazardous, infectious or toxic substances could subject us to significant liabilities, including joint and several liability under certain statutes. Any such liability could exceed our resources and could have a material adverse effect on our business, financial condition and results of operations. Additionally, an accident could damage our research and manufacturing facilities and operations.
Additional federal, state and local laws and regulations affecting us may be adopted in the future. We may incur substantial costs to comply with these laws and regulations and substantial fines or penalties if we violate any of these laws or regulations, which would adversely affect our business.
We may not be able to obtain or maintain sufficient insurance on commercially reasonable terms or with adequate coverage against potential liabilities in order to protect ourselves against product liability claims.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing and marketing of human therapeutic and research products. We may become subject to product liability claims if the use of our potential products is alleged to have injured subjects or patients. This risk exists for product candidates tested in human clinical trials as well as potential products that are sold commercially. We plan to obtain limited clinical trial liability insurance for our clinical trials and we may not be able to maintain this type of insurance for any of our clinical trials. In addition, product liability insurance is becoming increasingly expensive. Being unable to obtain or maintain product liability insurance in the future on acceptable terms or with adequate coverage against potential liabilities could have a material adverse effect on our business.
43
MANAGEMENT’S DISCUSSION AND ANALYSISOF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
The following discussion and analysis of the results of operations and financial condition of Q Therapeutics for the years ended December 31, 2010 and 2009 and for the six month period ended June 30, 2011 and 2010, should be read in conjunction with the Selected Consolidated Financial Data, Q Therapeutics’ financial statements, and the notes to those financial statements that are included elsewhere in this Current Report on Form 8-K. Our discussion includes forward-looking statements based upon current expectations that involve risks and uncertainties, such as our plans, objectives, expectations and intentions. Actual results and the timing of events could differ materially from those anticipated in these forward-looking statements as a result of a number of factors, including those set forth under the Risk Factors, Cautionary Notice Regarding Forward-Looking Statements and Business sections in this Form 8-K. We use words such as “anticipate,” “estimate,” “plan,” “project,” “continuing,” “ongoing,” “expect,” “believe,” “intend,” “may,” “will,” “should,” “could,” and similar expressions to identify forward-looking statements.
Company Overview
Q Therapeutics is a Salt Lake City, Utah based biopharmaceutical company that is developing human cell-based therapies intended to treat degenerative diseases of the brain and spinal cord, the primary components of the central nervous system (“CNS”). Q Therapeutics was incorporated in the state of Delaware in March, 2002.
The technology upon which these therapies are based was developed by Q’s co-founder Mahendra Rao, M.D., Ph.D., a global leader in glial stem cell biology, during Dr. Rao’s tenure as a Professor at the University of Utah and as Head of the Stem Cell Section at the National Institutes of Health. Q is managed by an experienced team of biotechnology executives with demonstrated start-up success and advised by leaders in the neurology and stem cell therapeutics fields.
Every year, hundreds of thousands of people suffer with debilitating neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease), Multiple Sclerosis, transverse myelitis and spinal cord injuries. Traditional drugs tend to fail as long-term treatments of the damages to the CNS caused by these diseases due to the complex nature of these diseases and the nerve damage they inflict. Q Therapeutics is developing a new and nontraditional approach to improve the health of people suffering from neurodegenerative diseases: a human cell based product called Q-Cells®.
Q-Cells® are healthy human glial cells. The job of glial cells in the body is to support and protect neurons, which are the signal transmission lines of the nervous system, by mechanisms including forming an insulating “myelin sheath” around them and providing the necessary growth factors needed to maintain a healthy nervous system. Many neurodegenerative diseases arise when glial cells are damaged or destroyed, causing neurons to malfunction and eventually die. Q-Cells® technology aims to treat neurodegenerative diseases by supplementing the damaged or missing glia in the body with new, healthy cells that can help maintain and/or restore neuron function to a more robust state.
The diseases targeted by Q’s products are not well treated with current drug therapies. At best, patients suffering from these diseases can, in some cases, only hope to slow their inexorable progression and the associated disabilities. A handful of companies are exploring the possibility of harnessing the power of stem cells to treat these conditions, though no clear leader has emerged. In addition to utilizing its proprietary cellular products as therapeutic products, Q may evaluate novel ways to utilize these cells to screen for new drugs (small molecule compounds) that could also provide treatments for neurological diseases.
44
The Company believes that a worldwide market measured in the tens of billions of dollars exists for those companies whose cell-based treatments become commercial products. Q Therapeutics’ patent protected technology represents an opportunity to build on the recent advancements in the stem cell field and bring to market a therapeutic approach that will change the way medicine is practiced in treating many disabling and fatal conditions of the CNS.
Q Therapeutics is raising capital to complete preparations necessary to move into and initiate human clinical trials, initially treating patients that suffer from the always-fatal amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease). Future trials are anticipated in other neurodegenerative diseases, such as Multiple Sclerosis and spinal cord injury.
Q’s Semi-Virtual Business Model
As described in this Report, the Company uses a semi-virtual business model to develop its products. Q believes this type of business model to be capital efficient while allowing it to augment its own expertise and capabilities with those of its development partners at the time that outside resources are needed. Q’s semi-virtual business model includes utilizing third parties for certain functions and outsourced services. For example, Q utilizes outside collaborators and contractors to:
|
|
·
|
Leverage its research and development resources by forming collaborations with outside scientists specialized in our areas of interest
|
|
|
·
|
Contract GLP and GMP manufacturing to facilities specialized in such production
|
|
|
·
|
Utilize experienced regulatory consultants to work with FDA
|
|
|
·
|
Contract safety/toxicology studies to qualified GLP labs
|
|
|
·
|
Conduct the clinical trials with physicians and institutions with relevant experience
|
|
|
·
|
Enter into pharmaceutical company collaborations to maximize product sales
|
Cell-Based Therapeutic Approach to Treatment of Degenerative Conditions of the Central Nervous System
Q Therapeutics was founded in 2002 to further the development and commercialization of the pioneering work of Mahendra Rao, M.D., Ph.D., conducted at the University of Utah (Utah) and National Institutes of Health (NIH). Dr. Rao, a global leader in the development of stem cells as therapeutics, was one of the first to identify and seek patent coverage on stem cells and their progeny cells found in the CNS. After licensing Dr. Rao’s technology from Utah / NIH and raising capital, Q commenced operations in the spring of 2004 to develop cell based therapeutic products that can be sold as “off-the-shelf” pharmaceuticals. That work continues to advance Dr. Rao’s initial findings toward a commercial product.
Objectives of Company
Q Therapeutics aims to change the way medicine is practiced in the treatment of many debilitating and often fatal diseases of the brain and spinal cord. Q intends to bring its initial product, Q-Cells® to the market to treat Lou Gehrig’s disease, and eventually other indications, potentially including Multiple Sclerosis, spinal cord injury, Parkinson’s and Alzheimer’s diseases.
45
Initially, Q is targeting orphan diseases, where the Food and Drug Administration (FDA) allows applying for fast track approvals and market exclusivity, and for which smaller, less expensive clinical trials are often warranted. This approach can result in accelerated commercialization efforts while maintaining a financing approach focused on capital efficiency.
Q’s Approach to the Problem
Q Therapeutics believes it has taken a novel path to developing treatments for debilitating and sometimes fatal diseases of the CNS for which there are currently no long-term effective treatments. Traditional drugs have seen little success as long-term treatments for many neurodegenerative diseases, primarily due to the complex nature of those diseases and the difficulty of replacing damaged nerve cells, or neurons.
The majority of cells in the brain and spinal cord are glial cells that serve to support the health of the neurons. Without healthy and properly functioning glial cells, neurons cannot function properly and will eventually die. In many neurodegenerative diseases, damage to glial cells causes or accelerates loss of function and death of neurons. Q’s approach takes advantage of the normal support and repair mechanisms present in the healthy nervous system whereby supplementation of damaged glial cells with healthy glial cells in patients suffering from neurodegenerative diseases is intended to enable the return to healthy neuron function and/or prevent additional degeneration, thereby ameliorating the negative effects of the disease.
2009 summary
Q Therapeutics focused its efforts on research and development of its proprietary neural cell products, including its human glial restricted progenitor Q-Cells® product, on maintaining and seeking additional protection for its intellectual property, and on fundraising. Q continued to work with its collaborators at Johns Hopkins University, University of Wisconsin, and Drexel University on development of Q-Cells for treatment of ALS, MS and spinal cord injury. Q also worked with the University of Utah Cell Therapy Facility to transfer manufacturing of Q-Cells® to GLP conditions. The translational costs to develop Q-Cells by Q and its collaborators/contractors was partially funded by grants from the Neilsen Foundation ($250,000 grant over a 2 year period commencing in 2008), the National Institutes of Health ($4 million grant in direct costs over a 4 year period commencing in 2009, milestone driven) and the Maryland Stem Cell Research Fund ($849,000 grant over a 3 year period commencing in 2008).
Q was issued three new patents as follows:
Japanese Patent 4371179 was granted 9/11/09 and related to neuronal restricted precursor cells;
U.S. Patent 7,595,194 issued 9/29/09 and has claims drawn to a method of isolating a pure population of mammalian CNS glial-restricted precursor cells from an isolated population of mammalian CNS neuroepithelial stems cells; and
U.S. Patent 7,517,521 issued 4/14/09 and has claims drawn to a method for transplanting an isolated population of human neuroepithelial precursor cells into an animal.
Q employed a semi-virtual business model as described in the Company Overview, which enabled it to utilize outside collaborators and contractors to augment product development activities without needing to build up internal infrastructure for many of these activities, such as manufacturing and GLP animal safety studies. Some of these activities were partially paid for by grant funding, reducing Q’s need for outside financing.
46
2010 summary
Q continued its work on development of Q-Cells® including with its collaborators, with the focus on development for ALS as the initial clinical indication in collaboration with Dr. Nicholas Maragakis’ laboratory at Johns Hopkins University. Q furthered manufacturing activities of Q-Cells® with increased production yields, resulting in filing of new patent applications. Q was awarded a grant from the federal government through the Patient Protection and Affordable Care Act for its ALS product development activities in 2009.
Q continued its work on defining and characterizing Q-Cells® and its ability to myelinate in animal models, including publication in peer reviewed journals (Sandrock et al, Regen Med 5, 381-394; 2010). Q also carried out research activities under an SBIR grant for development of an astrocyte cell product.
Q continued to prosecute and maintain its IP portfolio, resulting in issuance of one new patent in 2010:
U.S. Patent 7,795,021 issued 9/14/2010 and has claims drawn to an isolated population of mammalian CNS glial restricted precursor cells which generate oligodendrocytes and at least two distinct populations of astrocytes, but not neurons, said population of mammalian CNS glial restricted precursor cells being isolated from a mixture of cells by positive immunoselection with an A2B5 antibody.
Q employed a semi-virtual business model as described in the Company Overview, continuing to utilize outside collaborators and contractors to augment product development activities without needing to build up internal infrastructure for many of these activities, such as manufacturing and GLP animal safety studies. Some of these activities were partially paid for by grant funding, reducing Q’s need for outside financing. The Company continued fundraising activities.
2011 Summary through Q3
In 2011, Q focused on developing Q-Cells® for ALS and continued studies on safety and efficacy in animal models (collaboration with Dr. Maragakis lab, manuscript in press). In preparation for IND-enabling animal GLP safety studies, Q worked closely with its clinical collaborators on defining the clinical protocol, and with regulatory consultants and FDA on the design of preclinical studies to support the anticipated Phase I study after IND filing. Q also continued to work with its contractors on improvements on the manufacturing process in anticipation of GMP manufacturing for clinical trials.
To further define potential applications of Q-Cells®, Q furthered work with collaborators on use of Q-Cells® in animal model of spinal cord injury, both for inflammatory injury as well as mechanical injury. Recently published studies with Dr. Itzhak Fischer’s laboratory at Drexel University in an athymic rat model of thoracic contusion SCI document safety and statistically-significant, reproducible, disease modifying activity of Q-Cells transplanted into the injury site (Jin et al., J Neurotrauma 28(4), 579-94; 2011). Walczak et al (Glia 59, 499-510; 2011), in collaboration with Q, demonstrated safety and benefits of Q-Cells® in a model of focal inflammatory spinal cord injury.
Q continued to prosecute and maintain its IP portfolio.
Q established a subsidiary, NeuroQ Research, Inc, located in California, to carry out activities utilizing the Company’s intellectual property to develop products for the research market, and to carry out certain research and development activities of the Company’s therapeutic products.
47
Q continues to employ a semi-virtual business model as described in the Company Overview, utilizing outside collaborators and contractors to achieve product development activities without needing to build up internal infrastructure for many of these activities, especially such as manufacturing and GLP animal safety studies. Some of these activities are partially paid for by grant funding, reducing Q’s need for outside financing. Q works closely with the collaborators and contractors.
The Company anticipates that development activities and costs will increase going forward as we conduct the work necessary to include in our future IND submission. This includes GLP animal safety studies, injection device studies, manufacturing activities and working with clinical and regulatory consultants as well as other activities and costs. We may also increase research and development activities to evaluate use of our proprietary products in other disease indications, including working with outside collaborators. The Company’s general and administrative (“G&A”) expenses will increase going forward, including additional legal and administration expenses needed to comply with SEC and FDA, added costs such as insurance costs as we progress from a pre-clinical to a clinical company, as well as additional personnel costs.
Results of Operations
For the period from March 28, 2002 (date of inception) through June 30, 2011, the Company has not generated significant revenues and has been developing its products for commercial sale. Therefore, the Company is considered to be in the development stage.
Since entering the development stage, the Company has not generated revenues in excess of expenses and has been dependent on government grants and debt and equity raised from individual investors to sustain its operations. The Company’s products have not yet been approved by the U.S. Food and Drug Administration for commercial sale; therefore, the Company has not generated any revenues from product sales. The Company has incurred losses and used cash in operating activities since inception. As of June 30, 2011, the Company had an accumulated deficit of $15,428,733 and negative working capital. On August 30, 2011, the Company raised bridge financing totaling $450,000 and on October 13, 2011, the Company raised $3,803,047 in connection with a reverse merger with Grace 2, Inc. As a result, the negative factors above have been mitigated and there does not appear to be substantial doubt about the Company’s ability to continue as a going concern through at least June 30, 2012.
Results of operations for the six months ended June 30, 2011 as compared to the six months ended June 30, 2010:
|
|
For the Six Months
Ended
June 30,
|
|||||||||||||||
|
2011
|
2010
|
Change
|
%
|
|||||||||||||
|
Grant revenue
|
$ | 10,173 | $ | 50,907 | $ | (40,734 | ) | (80.0 | ) % | |||||||
|
License revenue
|
14,400 | 29,800 | (15,400 | ) | (51.7 | ) % | ||||||||||
|
Research and development expense
|
277,430 | 281,206 | (3,776 | ) | (1.3 | ) % | ||||||||||
|
General and administrative expense
|
340,673 | 395,298 | (54,625 | ) | (13.8 | ) % | ||||||||||
|
Loss from operations
|
(593,530 | ) | (595,797 | ) | (2,267 | ) | (0.4 | ) % | ||||||||
|
Other income (expense), net
|
(103 | ) | 8,966 | (9,069 | ) | (101.1 | ) % | |||||||||
|
Loss before provision for income taxes
|
(593,633 | ) | (586,831 | ) | 6,802 | 1.2 | % | |||||||||
|
Provision for income taxes
|
- | - | - | - | % | |||||||||||
|
Net loss
|
$ | (593,633 | ) | $ | (586,831 | ) | $ | (6,802 | ) | 1.2 | % | |||||
48
Grant Revenue
Grant revenue for the six months ended June 30, 2011 was $10,173, a decrease of $40,734 or (80.0)% from $50,907 for the six months ended June 30, 2010. The decrease was primarily attributable to fewer grants awarded to the Company.
License Revenue
License revenue for the six months ended June 30, 2011 was $14,400, a decrease of $15,400 or (51.7)% from $29,800 for the six months ended June 30, 2010. The decrease was primarily attributable to reduced sales of Q-cells to product development partners and collaborators.
Research and Development Expense
Total research and development expense decreased by $(3,776) or (1.3)% from $281,206 for the six months ended June 30, 2010 to $277,430 for the six months ended June 30, 2011. The decrease in research and development was primarily attributable to management’s efforts to contain costs and the resulting decrease in salaries and wage-related expenditures, offset by stock compensation related to the issuance of Series B Preferred Stock for research and development services valued at $44,750.
General and Administrative Expense
The following table breaks down the general and administrative expenses incurred by the Company:
|
|
For the Six Months
Ended
June 30,
|
|||||||||||||||
|
2011
|
2010
|
Change
|
%
|
|||||||||||||
|
Salaries and wage-related
|
$ | 149,671 | $ | 190,770 | $ | (41,099 | ) | (21.5 | ) % | |||||||
|
Professional fees
|
76,113 | 75,045 | 1,068 | 1.4 | % | |||||||||||
|
Stock-based compensation
|
14,541 | 53,590 | (39,049 | ) | (72.9 | ) % | ||||||||||
|
Rent
|
37,162 | 38,836 | (1,674 | ) | (4.3 | ) % | ||||||||||
|
Insurance
|
14,498 | 14,177 | 321 | 2.3 | % | |||||||||||
|
Other
|
48,688 | 22,880 | 25,808 | 112.8 | % | |||||||||||
|
Total
|
$ | 340,673 | $ | 395,298 | $ | (54,625 | ) | (13.8 | ) % | |||||||
The decrease in general and administrative expense was principally due to a decrease in salaries and wage-related expenses, stock-based compensation and professional fees partially offset by an increase in travel and conference expenses included in the other category.
49
Liquidity and Capital Resources
The following summarizes the key components of cash flows for the six months ended June 30:
|
2011
|
2010
|
|||||||
|
Net cash used in operating activities
|
$ | (365,244 | ) | $ | (649,902 | ) | ||
|
Net cash used in investing activities
|
(5,628 | ) | - | |||||
|
Net cash provided by financing activities
|
- | 975,000 | ||||||
|
Net change in cash
|
$ | (370,872 | ) | $ | 325,098 | |||
For the six months ended June 30, 2011, net cash used in operating activities totaled $(365,244) compared to $(649,902) for the six months ended June 30, 2010. The improvement of $284,658 resulted primarily from the collection of grants receivable and the timing of payments for accounts payable.
For the six months ended June 30, 2011, net cash used in investing activities was $(5,628). The change from the 2010 of $(5,628) was due to the fact that the Company did not expend any cash for additions to property and equipment during 2010.
Net cash provided by financing activities was $0 for the six months ended June 30, 2011, a decrease of $975,000 from net cash provided by financing activities for the six months ended June 30, 2010. The Company did not issue preferred stock for cash in 2011 as it did in 2010.
We believe that our current levels of cash, when combined with our expected cash flows from grants revenue and equity financing transactions, will be sufficient to meet our liquidity needs for at least the next 12 months. However, we will most likely require additional cash resources in the future, as we anticipate changed business conditions and other developments in the Company’s progress. We will need additional cash resources in the future if we pursue opportunities for investment, acquisition, strategic cooperation or other similar actions. To satisfy future cash requirements, we expect to seek funding through the issuance of debt or equity securities and the obtaining of a credit facility. Any future issuance of equity securities could cause dilution for our shareholders. Any incurrence of indebtedness will increase our debt service obligations and may cause us to be subject to restrictive operating and financial covenants. It is possible that financing will only be available to us in amounts or on terms that are not favorable to the Company, or not available at all.
Results of Operations for the Year Ended December 31, 2010 compared to the Year Ended December 31, 2009:
|
|
For the Years Ended
December 31,
|
|||||||||||||||
|
2010
|
2009
|
Change
|
%
|
|||||||||||||
|
Grant revenue
|
$ | 421,241 | $ | 47,251 | $ | 373,990 | 791.5 | % | ||||||||
|
License revenue
|
31,000 | 87,500 | (56,500 | ) | (64.6 | )% | ||||||||||
|
Research and development expense
|
501,900 | 406,499 | 95,401 | 23.5 | % | |||||||||||
|
General and administrative expense
|
826,330 | 485,235 | 341,095 | 70.3 | % | |||||||||||
|
Loss from operations
|
(875,989 | ) | (756,983 | ) | (119,006 | ) | (15.7 | )% | ||||||||
|
Other income, net
|
11,139 | 1,252 | 9,887 | 789.7 | % | |||||||||||
|
Loss before provision for income taxes
|
(864,850 | ) | (755,731 | ) | (109,119 | ) | (14.4 | )% | ||||||||
|
Provision for income taxes
|
- | - | - | - | % | |||||||||||
|
Net loss
|
$ | (864,850 | ) | $ | (755,731 | ) | $ | (109,119 | ) | (14.4 | )% | |||||
50
Grant Revenue
Grant revenue for the year ended December 31, 2010 was $421,241, an increase of $373,990, or 791.5%, from $47,251 for the year ended December 31, 2009. The increase was primarily attributable to new grants awarded to the Company.
License Revenue
License revenue for the year ended December 31, 2010 was $31,000, a decrease of $56,500, or (64.6)%, from $87,500 for the year ended December 31, 2009. The decrease was primarily attributable to the decreased sale of Q-cells to product development partners and collaborators.
Research and Development Expense
Total research and development expense increased by $95,401, or 23.5%, from $406,499 for the year ended December 31, 2009 to $501,900 for the year ended December 31, 2010. The increase in research and development expense was primarily attributable to increased salaries and wage-related expenditures as the Company moved forward on the development of its initial products.
General and Administrative Expense
The following table breaks down the general and administrative expenses of the Company:
|
|
For the Years Ended
December 31,
|
|||||||||||||||
|
2010
|
2009
|
Change
|
%
|
|||||||||||||
|
Salaries and wage-related
|
$ | 407,461 | $ | 184,874 | $ | 222,587 | 120.4 | % | ||||||||
|
Professional fees
|
181,615 | 93,651 | 87,964 | 93.9 | % | |||||||||||
|
Stock-based compensation
|
83,298 | 42,776 | 40,522 | 94.7 | % | |||||||||||
|
Rent
|
74,628 | 68,004 | 6,624 | 9.7 | % | |||||||||||
|
Insurance
|
32,178 | 33,888 | (1,710 | ) | (5.0 | )% | ||||||||||
|
Other
|
47,150 | 62,042 | (14,892 | ) | (24.0 | )% | ||||||||||
|
Total
|
$ | 826,330 | $ | 485,235 | $ | 341,095 | 70.3 | % | ||||||||
The increase in general and administrative expense was principally due to an increase in salaries and wage-related expenses, professional fees, stock compensation expense, and rent expense, offset by a decrease in insurance and bad debt expense, which is included in other, as quantified above. The increase in total general and administrative expenses was a direct result of the Company’s improved cash position due to the Company issuing preferred stock for cash during 2010.
51
Liquidity and Capital Resources
The following summarizes the key components of cash flows for the years ended December 31:
|
2010
|
2009
|
|||||||
|
Net cash used in operating activities
|
$ | (811,794 | ) | $ | (630,679 | ) | ||
|
Net cash used in investing activities
|
- | (2,541 | ) | |||||
|
Net cash provided by financing activities
|
1,221,845 | 55,000 | ||||||
|
Net change in cash
|
$ | 410,051 | $ | (578,220 | ) | |||
For the year ended December 31, 2010, net cash used in operating activities totaled $(811,794) compared to net cash used in operating activities for the year ended December 31, 2009 of $(630,679). The increase of $181,115 resulted mainly from an increase in the net loss of $109,119 to $(864,850) in 2010 from $(755,731) in 2009, as well as timing on collections of receivables and payment of payables.
For the year ended December 31, 2010, net cash provided by investing activities was $0. The change from 2009 of $(2,541) was the result of the Company making no purchases of property and equipment during 2010.
Net cash provided by financing activities was $1,221,845 for the year ended December 31, 2010, an increase of $1,166,845 from $55,000 of net cash provided by financing activities for the year ended December 31, 2009. The increase was due to the Company issuing preferred stock for cash.
We believe that our current levels of cash, in combination with cash flows from grants revenue and equity financing transactions will be sufficient to meet our anticipated cash needs for at least the next 12 months. However, we will most likely require additional cash resources in the future, as we anticipate changed business conditions and other developments in the Company’s progress. We will need additional cash resources in the future if we pursue opportunities for investment, acquisition, strategic cooperation or other similar actions. To satisfy future cash requirements, we expect to seek funding through the issuance of debt or equity securities and the obtaining of a credit facility. Any future issuance of equity securities could cause dilution for our shareholders. Any incurrence of indebtedness will increase our debt service obligations and may cause us to be subject to restrictive operating and financial covenants. It is possible that financing will only be available to us in amounts or on terms that are not favorable to the Company, or not available at all.
Recent Accounting Pronouncements
As of the date of this report, there are no accounting pronouncements that had not yet been adopted by the Company that we believe would have a material impact on our financial statements.
Quantitative and Qualitative Disclosures about Market Risks
None
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements, financings, or other relationships with unconsolidated entities or other persons, also known as “special purpose entities” (SPEs).
52
Our discussion and analysis of financial condition and results of operations is based upon the condensed financial statements included herein, which have been prepared in accordance with US generally accepted accounting principles and the requirements and regulations of the Securities and Exchange Commission. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of sales and expenses during the reporting period. Significant accounting policies and areas where substantial judgments are made by management include:
Estimates - Key management estimates include allowances for doubtful accounts, useful lives for property and equipment, valuation allowances for net deferred income tax assets, and valuation of stock-based compensation awards.
Stock-Based Compensation – Q Therapeutics calculates the estimated fair value of its stock options and warrants on the grant date using the Black-Scholes option-pricing model and recognizes the estimated fair value as compensation expense on a straight-line basis over the vesting period. The volatility assumption used in the Black-Scholes option-pricing model is based on the volatility of publicly traded companies in the same industry as Q Therapeutics. The expected term of the options and warrants granted represent the period of time that the options granted are expected to be outstanding. The risk free rate for periods within the contractual life of the option and warrant is based on the U.S. treasury securities constant maturity rate that corresponds to the expected term in effect at the time of grant.
Concentration of Credit Risk – Q Therapeutics’ cash is maintained in bank deposit accounts which occasionally may exceed federally insured limits. Cash equivalents consist of highly liquid securities with maturities of three months or less when purchased. Q Therapeutics has not experienced any losses with respect to these deposits.
Cash Equivalents – Q Therapeutics considers all highly liquid investments with an initial maturity of three months or less to be cash equivalents. During the periods presented, the Company had no cash equivalents.
Grants Receivable – Q Therapeutics applies for research grants generally as a sub-recipient to grants funded by government agencies through research universities. Q Therapeutics records grants receivable in accordance with the provisions of the grant agreement. The grants receivable are considered past due when payment has not been received within 30 days of the invoice date. The amounts of the specific reserves are estimated by management based on various assumptions including the age of the individual grant receivable, as well as changes in payment schedules and histories. Grants receivable balances are charged off against the allowance for doubtful accounts when the potential for recovery is remote. Recoveries of receivables previously charged off are recorded when payment is received.
Income Taxes – Q Therapeutics recognizes deferred income tax assets or liabilities for the expected future tax consequences of events that have been recognized in the financial statements or tax returns. Deferred income tax assets or liabilities are determined based upon the difference between the financial and income tax bases of assets and liabilities using enacted tax rates expected to apply when differences are expected to be settled or realized.
Net Loss Per Common Share – Basic earnings (loss) per share (“EPS”) is calculated by dividing income (loss) available to common stockholders by the weighted-average number of common shares outstanding during the period.
53
Diluted EPS is similar to Basic EPS except that the weighted-average number of common shares outstanding is increased using the treasury stock method to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. Such potentially dilutive common shares include convertible preferred stock, stock options, and warrants. Shares having an antidilutive effect on periods presented are not included in the computation of Diluted EPS.
54
FINANCIAL CONTROLS AND PROCEDURES.
Management’s Report on Internal Control over Financial Reporting
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Exchange Act. The Company’s internal control over financial reporting is a process designed under the supervision of the Company’s Chief Executive Officer and Chief Financial Officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the Company’s financial statements for external purposes in accordance with U.S. generally accepted accounting principles (US GAAP) and includes those policies and procedures that:
|
|
·
|
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
|
|
|
·
|
Provide reasonable assurance that the transactions are recorded as necessary to permit the preparation of financial statements in accordance with US GAAP, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
|
|
|
·
|
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
|
Management recognizes that there are inherent limitations in the effectiveness of any system of internal control. Accordingly, even effective internal control can provide only reasonable assurance with respect to financial statement preparation and may not prevent or detect misstatements. In addition, effective internal control at a point in time may become ineffective in future periods because of changes in conditions or due to deterioration in the degree of compliance with our established policies and procedures.
Effective as of June 30, 2011, Q Therapeutics, Inc.’s management assessed the effectiveness of the Company’s internal control over financial reporting based on criteria for effective internal control over financial reporting established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and SEC guidance on conducting such assessments.
Based on that evaluation, management concluded that, during the period covered by this report, such internal controls and procedures may not have been sufficient to detect the inappropriate application of US GAAP as more fully described below. This was due to deficiencies that existed in the design or operation of our internal control over financial reporting that adversely affected our internal controls and that taken together may be considered to be material weaknesses.
A material weakness is a deficiency, or combination of deficiencies, that results in more than a remote likelihood that a material misstatement of annual or interim financial statements will not be prevented or detected. In connection with the assessment described above, management identified the following control deficiencies that represent material weaknesses as of June 30, 2011:
|
|
1.
|
Inadequate segregation of incompatible duties due to our limited number of personnel, which could have resulted in a material misstatement that would otherwise have been prevented or detected;
|
55
|
|
2.
|
Lack of an established audit committee comprised of outside directors on the Company’s board of directors independent of management;
|
|
|
3.
|
Lack of sufficient resources to establish and operate an internal audit function; and
|
|
|
4.
|
Incomplete documentation of all proper accounting processes and procedures and the lack of a written code of ethics.
|
Management believes that the material weaknesses set forth above have not had an adverse effect on the Company’s financial reporting for fiscal 2011 and fiscal 2010. Management is also committed to improving the Company’s financial control capability. To the extent reasonably possible given our limited resources, we intend to address these weaknesses through measures that include, but are not limited to:
|
|
·
|
Considering the engagement of consultants to assist in ensuring that accounting policies and procedures are consistent across the organization and that we have adequate control over financial statement disclosures;
|
|
|
·
|
Adoption of a written code of ethics;
|
|
|
·
|
Appointing one or more outside directors to our board of directors who are willing to perform the audit oversight function and establishing a fully functioning audit committee;
|
|
|
·
|
Increasing our financial accounting staff, as resources allow, and segregate all key incompatible duties consistent with the control objectives.
|
We will continue to monitor and evaluate the effectiveness of our internal control, processes and procedures over financial reporting on an ongoing basis and are committed to taking further actions and implementing additional enhancements or improvements, as necessary and as funds allow.
Evaluation of disclosure controls and procedures
As of October 13, 2011, under the supervision and with the participation of management, including our Chief Executive Officer and Chief Financial Officer (the “Certifying Officers”), we conducted an evaluation of our Company’s disclosure controls and procedures. As defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act, the term “disclosure controls and procedures” means our controls and other procedures that are designed to ensure that information required to be disclosed in reports that we file or submit under the Securities Exchange Act of 1934 (the “Exchange Act”) is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is accumulated and communicated to our Company’s management, including the Certifying Officers, to allow timely decisions regarding required disclosure. Based on this evaluation, the Certifying Officers have concluded that our Company’s disclosure controls and procedures were not effective as of October 13, 2011. See Management’s Report on Internal Control over Financial Reporting for a discussion of the material weaknesses and its approach to address these matters.
56
Changes in Controls and Procedures
There were no significant changes made in our internal controls over financial reporting during the period ended June 30, 2011 that have materially affected or are reasonably likely to materially affect these controls.
Attestation Report of the Independent Registered Public Accounting Firm
This report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our independent registered public accounting firm pursuant to rules of the SEC that permit us to provide only management’s report in this report. We were not required to have, nor have we, engaged our independent registered public accounting firm to perform an audit of internal control over financial reporting pursuant to the rules of the SEC that permit us to provide only management’s report in this report.
In connection with the Merger Agreement, on October 13, 2011, we issued an aggregate of 22,671,923 shares of Common Stock (including shares underlying warrants and options), or 89.7% of the outstanding shares prior to the private placement, to Q Therapeutics, Inc. Shareholders in exchange for 100% of the outstanding shares of Q Therapeutics, Inc., which merger resulted in Q Therapeutic, Inc. becoming our wholly owned subsidiary. These securities qualified for exemption under Section 4(2) of the Securities Act since the issuance of securities by us did not involve a public offering. The offering was not a “public offering” as defined in Section 4(2) due to the insubstantial number of persons involved in the deal, size of the offering, manner of the offering and number of securities offered. We did not undertake an offering in which we sold a high number of securities to a high number of investors. In addition, these shareholders had the necessary investment intent as required by Section 4(2) since they agreed to and received share certificates bearing a legend stating that such securities are restricted pursuant to Rule 144 of the Securities Act. This restriction ensures that these securities would not be immediately redistributed into the market and therefore not be part of a “public offering.” Based on an analysis of the above factors, we have met the requirements to qualify for exemption under Section 4(2) of the Securities Act for this transaction.
On August 30, 2011, Q Therapeutics, Inc. received $450,000 in bridge financing from investors, (including $90,000 in credit towards the advancement of consulting fees and out-of-pocket expenses) evidenced by secured convertible promissory notes (“Notes”) and a unit, consisting of an aggregate of (a) one (1) Seven Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $1.00 per share of the Company’s common stock, (b) one (1) Seven Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $2.00 per share of the Company’s common stock and (c) one (1) Seven Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock with an exercise price equal to the lesser of (a) the price per share of the Company’s Shares issued in a financing or (b) the Fair Market Value (“FMV”) of the Company’s Shares [is (c) a derivative?]. An automatic conversion feature is in place for each Note, whereby upon the attainment of the Company’s Private Placement Offering’s minimum raise amount of $3,000,000, each Note’s outstanding principal and interest balance will automatically be converted into common stock of Q Therapeutics, Inc. at the same purchase price and upon the same terms as investors in the Company’s Private Placement Offering. These securities qualified for exemption under Section 4(2) of the Securities Act since the issuance securities by us did not involve a public offering. The offering was not a “public offering” as defined in Section 4(2) due to the insubstantial number of persons involved in the deal, size of the offering, manner of the offering and number of securities offered. Q Therapeutics, Inc. did not undertake an offering in which we sold a high number of securities to a high number of investors. In addition, these shareholders had the necessary investment intent as required by Section 4(2) since they agreed to and received share certificates bearing a legend stating that such securities are restricted pursuant to Rule 144 of the Securities Act. This restriction ensures that these securities would not be immediately redistributed into the market and therefore not be part of a “public offering.” Based on an analysis of the above factors, Q Therapeutics, Inc. has met the requirements to qualify for exemption under Section 4(2) of the Securities Act for this transaction.
57
As more fully described in Item 1.01 above, on October 13, 2011, immediately following the Merger, pursuant to the Purchase Agreement, we consummated the Private Placement for the issuance and sale of Units, consisting of an aggregate of (a) one share of Common Stock, (b) one Seven Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $1.00 per share, and (c) one (1) 7 Year Common Stock Purchase Warrant to purchase one share of the Company’s common stock exercisable at $2.00 per share, for aggregate gross proceeds of $3,803,047. Such securities were not registered under the Securities Act. The issuance of these securities was exempt from registration under Regulation D, Regulation S and Section 4(2) of the Securities Act. We made this determination based on the representations of Investors, which included, in pertinent part, that such shareholders were either (a) “accredited investors” within the meaning of Rule 501 of Regulation D promulgated under the Securities Act, or (b) not a “U.S. person” as that term is defined in Rule 902(k) of Regulation S under the Act, and that such shareholders were acquiring our common stock, for investment purposes for their own respective accounts and not as nominees or agents, and not with a view to the resale or distribution thereof, and that the shareholders understood that the shares of our common stock may not be sold or otherwise disposed of without registration under the Securities Act or an applicable exemption therefrom.
In instances described above where we issued securities in reliance upon Regulation D, we relied upon Rule 506 of Regulation D of the Securities Act. These stockholders who received the securities in such instances made representations that (a) the stockholder is acquiring the securities for his, her or its own account for investment and not for the account of any other person and not with a view to or for distribution, assignment or resale in connection with any distribution within the meaning of the Securities Act, (b) the stockholder agrees not to sell or otherwise transfer the purchased shares unless they are registered under the Securities Act and any applicable state securities laws, or an exemption or exemptions from such registration are available, (c) the stockholder has knowledge and experience in financial and business matters such that he, she or it is capable of evaluating the merits and risks of an investment in us, (d) the stockholder had access to all of our documents, records, and books pertaining to the investment and was provided the opportunity to ask questions and receive answers regarding the terms and conditions of the offering and to obtain any additional information which we possessed or were able to acquire without unreasonable effort and expense, and (e) the stockholder has no need for the liquidity in its investment in us and could afford the complete loss of such investment. Management made the determination that the investors in instances where we relied on Regulation D are Accredited Investors (as defined in Regulation D) based upon management’s inquiry into their sophistication and net worth. In addition, there was no general solicitation or advertising for securities issued in reliance upon Regulation D.
ITEM 4.01. CHANGES IN REGISTRANT’S CERTIFYING ACCOUNTANTS.
Effective as of October 12, 2011, the Company dismissed W. T. Uniack & Co., CPA’s P.C. (“Uniack”) as its independent registered public accounting firm. Uniack had previously been engaged as the principal accountant to audit Grace 2’s financial statements. The reason for the dismissal of Uniack is that, upon the consummation of the Merger, (i) the former shareholders of Q Therapeutics owned a majority of the outstanding shares of the Company’s common stock and (ii) Grace 2’s primary business unit became the business previously conducted by Q Therapeutics. It was more practical that Q Therapeutics’ independent auditors be engaged, going forward.
58
The report of Uniack on Grace 2’s financial statements for the period from October 27, 2005 (inception) to May 31, 2011 contained an adverse opinion or disclaimer of opinion and was qualified or modified as to uncertainty, audit scope or accounting principles. Uniack’s report contained a going concern opinion on the financial statements as of May 31, 2011 because Grace 2 was a “blank check” company with no assets and the sole shareholder of Grace 2 was not legally obligated to fund the activities of Grace 2.
The decision to change the Company’s independent registered public accounting firm was approved by the Company’s board of directors on October 12, 2011.
From October 27, 2005 through October 12, 2011, there were no disagreements between Grace 2 and Uniack on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure which, if not resolved to the satisfaction of Uniack, would have caused it to make reference to the matter in connection with Uniack’s reports.
The Company made the contents of this Current Report on Form 8-K available to Uniack and requested it to furnish a letter addressed to the SEC as to whether it agrees or disagrees with, or wishes to clarify our expression of our views, or wished to provide any additional information. A copy of Uniack’s letter to the SEC is included as Exhibit 16.1 to this Report.
On October 12, 2011, the Company engaged Tanner LLC (“Tanner”) as its new, independent registered public accounting firm. The appointment of Tanner was approved by our board of directors. During our two most recent fiscal years and the subsequent interim periods through October 12, 2011, Grace 2 did not consult Tanner regarding either: (i) the application of accounting principles to a specific completed or contemplated transaction, or the type of audit opinion that might be rendered on our financial statements; or (ii) any matter that was the subject of a disagreement as defined in Item 304(a)(1)(iv) of Regulation S-K.
Prior to engaging Tanner, the Company had not consulted Tanner regarding the application of accounting principles to any specified transaction, completed or proposed, or the type of audit opinion that might be rendered on the Company’s financial statements.
Principal Accountant Fees and Services.
Uniack & Associates, PC
Uniack served as Grace 2’s independent registered public accounting firm for the period from October 27, 2005 (inception) through October 12, 2011. Effective as of October 12, 2011, the Company dismissed Uniack as its independent registered public accounting firm. Uniack had previously been engaged as the principal independent registered public accounting firm to audit Grace 2’s financial statements. The reason for the dismissal of Uniack is that, upon the consummation of the transaction on October 13, 2011, (i) the former stockholders of Q Therapeutics owned a majority of the outstanding shares of Grace 2’s common stock and (ii) Grace 2’s primary business unit became the business previously conducted by Q Therapeutics. Therefore, in the discretion of management, it was deemed more practical that Q Therapeutics’ independent auditors be engaged, going forward.
Audit Fees
The aggregate fees billed by Uniack for professional services rendered for the audit of Grace 2’s annual financial statements included in Form 10-K and the review of financial statements included in Grace 2’s Quarterly Reports on Form 10-Q, and services that are normally provided in connection with statutory and regulatory filings were $5,000 and $2,000 for the fiscal years ended May 31, 2010 and 2009, respectively.
59
Audited-Related Fees
The aggregate fees billed by Uniack for assurance and related services related to the audit or review of Grace 2’s financial statements were $0 for each of the fiscal years ended May 31, 2010 and 2009, respectively.
Tax Fees
The aggregate fees billed by Uniack for professional services for tax compliance, tax advice, and tax planning were $0 for each of the fiscal years ended May 31, 2010 and 2009, respectively.
All Other Fees
The aggregate fees billed for other products and services were $0 for each of the fiscal years ended May 31, 2010 and 2009, respectively.
ITEM 5.01 CHANGES IN CONTROL OF REGISTRANT.
As described in Item 2.01, on October 13, 2011, Grace 2 entered into the Merger Agreement with Merger Sub and Q Therapeutics. Pursuant to the Agreement, Merger Sub merged with and into Q Therapeutics with Q Therapeutics being the surviving corporation. Upon the consummation of the Merger, the Company acquired 100% of Q Therapeutics. The Agreement is attached hereto as Exhibit 10.1 to this Form 8-K.
As a result of the Merger:
|
|
·
|
Douglas Dyer, the sole Director and Officer of Grace 2, resigned.
|
|
|
·
|
Q Therapeutics shareholders received, on a pro rata basis, an aggregate of 22,671,923 shares of Grace 2 Common Stock (including shares underlying warrants and options), which immediately upon the consummation of the Merger, represented an 89.7% interest in the Company , thereby rendering Q Therapeutics shareholders the majority owners of the Company;
|
|
|
·
|
Grace 2 filed a Schedule 14C with the US Securities and Exchange Commission notifying the market of its immediate plans to change the Company’s corporate name to Q Holdings, Inc. by way of Amendment to the Company Certificate of Incorporation with the State of Delaware (Grace 2 thereafter referred to as “Q Holdings”);
|
|
|
·
|
Q Therapeutics’ existing senior management assumed the same positions with Q Holdings; and
|
|
|
·
|
Q Therapeutics became the wholly owned subsidiary of the Company with the Company conducting all of its operations through Q Therapeutics.
|
Reference is made to the disclosures set forth under Item 2.01 of this Current Report on Form 8-K, which disclosure is incorporated herein by reference.
60
In connection with the Closing of the Merger, and as explained more fully in the above Item 2.01 and below in Item 5.02 of this Current Report on Form 8-K, Douglas Dyer resigned as the sole officer and director of the Company, and Messrs. Patel, Hutt, Goswami and Mss. Eppstein and Powers were appointed as members of the Company’s Board of Directors, with Mr. Patel serving as the Chairman. This change to the composition of the Board took effect upon expiration of 10 days following the Company’s filing of a Schedule 14F-1 with the SEC on October 3, 2011. The Board of Directors subsequently appointed Ms. Eppstein as the President and Chief Executive Officer of the Company, and Mr. Steven J. Borst as the Chief Financial Officer and Vice President of Corporate Development of the Company.
ITEM 5.02. DEPARTURE OF DIRECTORS OR CERTAIN OFFICERS; ELECTION OF DIRECTORS AND APPOINTMENT OF CERTAIN OFFICERS; COMPENSATORY ARRANGEMENTS OF CERTAIN OFFICERS.
Upon the consummation of the transactions contemplated by the Merger Agreement, Douglas Dyer, the sole officer and director of Grace 2, resigned effective immediately. The following section contains pertinent information related to the post-Merger composition of our Board of Directors and Officers.
Directors, Executive Officers, Promoters and Control Persons and Corporate Governance; Compliance with Section 16(a) of the Exchange Act.
As contemplated by the Agreement, upon consummation of the Merger, Messrs. Patel, Hutt, Goswami and Mss. Eppstein and Powers were appointed as members of the Company’s Board of Directors, with Mr. Patel serving as the Chairman. The Board of Directors subsequently appointed Ms. Eppstein as the President and Chief Executive Officer of the Company, and Mr. Borst as the Chief Financial Officer and Vice President of Corporate Development of the Company.
The new members of the Board of Directors of the Company will hold office for a period of one year or until his/her successor is elected and qualified. The officers of the Company are appointed by our Board of Directors and hold office until their death, resignation or removal from office. A summary of the composition of the Company’s directors and executive officers, their ages, positions held, are as follows:
|
Name
|
Age
|
Directors and Officers
|
||
|
Deborah A. Eppstein, Ph.D.
|
63
|
President, Chief Executive Officer and Director
|
||
|
Steven J. Borst, M.B.A.,
|
54
|
Chief Financial Officer and Vice President Corporate Development
|
||
|
Dinesh C. Patel, Ph.D.
|
61
|
Chairman of the Board of Directors
|
||
|
Peter Barton Hutt
|
76
|
Director
|
||
|
Joydeep Goswami, Ph.D., M.B.A.
|
40
|
Director
|
||
|
Linda F. Powers
|
56
|
Director
|
61
Business Experience of New Appointees
Deborah A. Eppstein, Ph.D., is President, CEO and a Member of the Board of Directors. Dr. Eppstein has more than 30 years of experience in the pharmaceutical and biotech industries, with the latter 18 on the entrepreneurial side. She was the founding CEO of Altea Therapeutics, a venture-backed company developing novel patch products for pain and diabetes, and for other macromolecular drugs. Previously, she was Vice President of Corporate Development at TheraTech, where she was involved with the IPO, partnering, achieving profitability and subsequent sale of the company. Earlier she was Director of Corporate Development and Department Head of biochemistry, virology and tumor biology at Syntex (now Roche). Dr. Eppstein received a B.A. from Grinnell, a Ph.D. in biochemistry from the University of Arkansas, and she conducted research in virology and cell biology as an NIH postdoctoral fellow at the University of California, Santa Barbara. Dr. Eppstein has received the Women’s Technology Leadership and Businesswoman of the Year awards in Salt Lake City.
Steven J. Borst, M.B.A., is Chief Financial Officer and Vice President of Corporate Development. Mr. Borst has both an operational and venture capital background, most recently as a founder and Managing Director of UpStart Ventures Management, an early-stage healthcare fund in Salt Lake City. He has also co-founded five Salt Lake biotech companies. Mr. Borst was previously associated with two Chicago-based venture funds and served in senior management positions with two venture-backed healthcare portfolio companies. Mr. Borst holds a B.S. degree in Industrial Engineering from the University of Michigan and an M.B.A. from the J.L. Kellogg Graduate School of Management at Northwestern University.
Board of Directors
In addition to Dr. Eppstein, the Q Board of Directors is composed of:
Dinesh C. Patel, Ph.D., serves as Chairman of the Company’s Board of Directors. Dr. Patel is a Managing Director and Founding Partner of vSpring Capital, an early-stage venture capital fund with over $400 million under management (2000 to present). From 1985-1999, Dr. Patel served as co-founder, Chairman, President and CEO of TheraTech, Inc., a Salt Lake City, Utah based company that has been a pioneer in the development and manufacturing of innovative drug delivery products. TheraTech went public in 1992, became profitable in 1997, and in 1999 was acquired for approximately $350 million by Watson Pharmaceuticals.
Dr. Patel has been the recipient of numerous awards including; Honorary Doctor of Business Degree, University of Utah (2008), the 2006 Utah Technology Council Hall of Fame, 2006 Pathfinder Award-Edison Showcase University of Utah, 2006 Ellis Island Medal of Honor, Utah Asian Chamber of Commerce Outstanding Business Award, US Small Business Administration’s Business Achiever Award, Scientific and Technology Award (State of Utah), Entrepreneur of the Year Award (Mountain West Capital Network) and Scientific and Technology Development Pioneer of Progress Award. Dr. Patel served as co-chair of Governor Huntsman’s transition team and is currently on the board of the Utah Policy Partnership (UPP), the board of the Utah Symphony & Opera, the Chairman of the USTAR Governing Authority board and sits on the Utah Technology Council executive committee. Dr. Patel received his undergraduate degree from India and his doctorate degree from the University of Michigan.
Peter Barton Hutt, Director, has been a partner of the Washington, D.C.-based law firm of Covington & Burling, specializing in food and drug law, since 1968, except for the period from 1971 to 1975 when he served as Chief Counsel of the FDA. He received a B.A., magna cum laude, from Yale University, a L.L.B. from Harvard Law School and a L.L.M. from New York University. Mr. Hutt has served on the boards of several publicly-traded biotechnology companies and is a member of the Institute of Medicine, National Academy of Sciences. Mr. Hutt has received numerous honors, including being named by the National Law Journal as one of the 40 best health care lawyers in the United States.
62
Joydeep Goswami, Ph.D., MBA, is currently the President of Life Technologies Japan, and leads the Japan business for all of Life Technologies products and services. Previously, Dr. Goswami was the Vice President/ General Manager of Primary and Stem Systems Group at Life Technologies. Before joining Invitrogen, Dr. Goswami spent 5 years with McKinsey & Co., during which he served major clients pharmaceutical, medical products, chemical, and technology industries in the U.S., Europe, Asia and Latin America on strategic and operational issues in the areas of Research & Development, Market Strategy, and Licensing strategy. Dr. Goswami holds a Ph.D. and M.S. in Chemical Engineering from the Massachusetts Institute of Technology and an MBA from MIT Sloan School of Management.
Linda F. Powers, Director, is a Managing Director and co-founder of Toucan Capital LLC, a Bethesda, MD venture fund that invested in Q’s Series A-2 round. She has more than 10 years of experience in seed and early-stage venture investing, and more than 18 years of experience in corporate finance and restructuring, mergers and acquisitions, joint ventures and intellectual property licensing. Ms. Powers holds an A.B. degree in economics from Princeton University, magna cum laude and Phi Beta Kappa, as well as a J.D. degree, magna cum laude, from Harvard Law School.
Scientific Advisors & Consultants
Ian D. Duncan, BVMS, Ph.D., is a Professor in the Department of Medical Sciences at the University of Wisconsin. He is a leading research authority on myelin repair by cell transplantation using stem cells, with Multiple Sclerosis as a main interest. Dr. Duncan has also done extensive studies of leukodystrophies with stem cells. He received degrees in veterinary medicine and a Ph.D. from Glasgow University in Scotland. Dr. Duncan collaborates with Q in running animal studies in inflammatory demyelinating diseases such as MS.
Douglas Kerr, M.D., Ph.D., is Medical Director, Neurology Research and Development at Biogen-Idec where he oversees late stage neurodegenerative programs, focusing on motor neuron disorders ALS and SMA. Dr. Kerr is an expert in autoimmune neurodegenerative diseases such as Multiple Sclerosis and Transverse Myelitis. Dr. Kerr previously established the world-leading Transverse Myelitis center at Johns Hopkins Medical School.
Nicholas J. Maragakis, M.D., is Associate Professor of Neurology at Johns Hopkins University. Dr. Maragakis treats patients with a variety of neuromuscular disorders, with an emphasis on patients with motor neuron diseases, such as Amyotrophic Lateral Sclerosis. This expertise is coordinated with the ALS clinic at Johns Hopkins, a multidisciplinary clinic. In addition to his clinical practice, Dr. Maragakis’ laboratory studies the role of astrocytes (the supporting cells of the brain and spinal cord) in causing and propagating neurological diseases, such as ALS. Q is conducting animal studies with Dr. Maragakis in models of ALS. Dr. Maragakis will be the PI for the Phase 1/2a clinical trial in ALS.
Thomas N. Parks, Ph.D., is Vice President of Research at the University of Utah School of Medicine, as well as Professor and Executive Director of the Brain Institute. He was a co-founder of NPS Pharmaceuticals, Inc. Dr. Parks received a B.S. in Biology from University of California, Irvine, and a Ph.D. in Psychobiology from Yale University.
Regulatory Consultants
The Company has engaged several experienced FDA consultants to advise on preclinical studies, clinical protocol development and assist in preparation of an IND application. They include:
63
|
|
·
|
Andra Miller, Ph.D. of The Biologics Consulting Group is a regulatory CMC specialist for cell therapy products since 2000. She previously spent 7 years at the CBER division of the FDA as an expert microbiologist for cell and gene therapy products.
|
|
|
Joy A. Cavagnaro, Ph.D. of Access Bio was Senior Pharmacologist at the CBER division of the FDA and was responsible for preclinical development and safety assessment of biological projects.
|
Involvement in Certain Legal Proceedings
None of our executive officers, directors or named consultants has, during the past five years:
|
|
(a)
|
Had any bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time;
|
|
|
(b)
|
Been convicted in a criminal proceeding or subject to a pending criminal proceeding;
|
|
|
(c)
|
Been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities, futures, commodities or banking activities; and
|
|
|
(d)
|
Been found by a court of competent jurisdiction (in a civil action), the Securities and Exchange Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated.
|
Family Relationships
None.
Section 16(A) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934, as amended, requires our executive officers and directors, and persons who beneficially own more than 10% of a registered class of our equity securities to file with the Securities and Exchange Commission initial statements of beneficial ownership, reports of changes in ownership and annual reports concerning their ownership of our common shares and other equity securities, on Forms 3, 4 and 5, respectively. Executive officers, directors and greater than 10% shareholders are required by the Securities and Exchange Commission regulations to furnish us with copies of all Section 16(a) reports they file. Based on our review of the copies of such forms received by us, and to the best of our knowledge, all executive officers, directors and greater than 10% shareholders filed the required reports in a timely manner.
Employment Agreements
The Company and Q Therapeutics have entered into employment agreements with Deborah A. Eppstein, President and Chief Executive Officer, and Steven J. Borst, Chief Financial Officer and Vice President of Corporate Development. The Q Therapeutics agreement with Ms. Eppstein provides for a base salary per year to be increased upon achievement of certain activities, and includes other financial benefits including a provision for a severance payment equal to one year’s salary in the case of her termination of employment without cause. In addition, both agreements include a one year non-compete provision, and a one year non-solicitation of the Company’s or Q Therapeutics’ employees in the event of termination of employment. Copies of the Q Holdings employment agreements are attached respectively as Exhibits 10.2 and 10.3 to this Current Report.
64
Our Board of Directors has not formally established separate audit, nominating or compensation committees though they perform many of the functions that would otherwise be delegated to such committees. Currently, our Board of Directors believes that the cost of establishing such committees, including the costs necessary to recruit and retain qualified independent directors to serve on our Board of Directors and such committees and the legal costs to properly form and document the authority, policies and procedures of such committees are not justified under our current circumstances. However, we anticipate that our Board of Directors will seek qualified independent directors to serve on the Board and ultimately form standing nominating and compensation committees and nominate other directors to serve on its audit committee.
Code of Ethics
Our Board of Directors has not established a Code of Ethics.
Executive Compensation.
Our Board of Directors has authorized the compensation of its officers with the following annual cash salaries:
|
Deborah A. Eppstein, President and Chief Executive Officer:
|
$ | 259,000 | ||
|
Steven J. Borst, CFO and Vice President of Corporate Development:
|
$ | 200,000 |
|
SUMMARY COMPENSATION TABLE
|
||||||||||||||||||||
|
Name
|
Title
|
Fiscal Year
|
Salary
|
Bonus
|
Other Comp.
|
Total
|
||||||||||||||
|
Deborah A.
|
President and
|
December 31, 2010
|
$ | 187,550 | $ | 0 | $ | 0 | $ | 187,550 | ||||||||||
| Eppstein, Ph.D. (1) | CEO |
December 31, 2009
|
$ | 89,830 | $ | 0 | $ | 0 | $ | 89,830 | ||||||||||
|
Steven J. Borst (2)
|
CFO and Vice President
|
December 31, 2010
|
$ | 122,500 | $ | 0 | $ | 0 | $ | 122,500 | ||||||||||
|
of Corporate Development
|
December 31, 2009
|
$ | 50,556 | $ | 0 | $ | 0 | $ | 50,556 | |||||||||||
|
(1)
|
As adjusted pursuant to the Merger, Dr. Eppstein has been granted 1,397,479 options to acquire the Company’s Common Stock comprising 448,956 options with a $0.08 strike price, 515,846 options with a $0.15 strike price, and 432,677 options with a $0.19 strike price.
|
|
(2)
|
As adjusted pursuant to the Merger, Mr. Borst has been granted 834,672 options to acquire the Company’s Common Stock comprising 209,995 options with a $0.08 strike price, 192,000 options with a $0.15 strike price, and 432,677 options with a $0.19 strike price.
|
Option Plan:
In 2002, Q adopted its current incentive option plan. As part of the option plan, the board of directors is given the authority to grant options to employees and directors at its sole discretion. As adjusted pursuant to the Merger, the entire option plan includes options to acquire 3,385,596 shares of Common Stock. All options under the plan have been granted except for options to acquire 228,648 shares of Common Stock. Upon the consummation of the Merger, options to purchase shares of Common Stock of Q were exchanged for options to purchase shares of Common Stock of the Company based on the same terms.
65
In connection with the Merger, a new option plan was authorized. The new option plan provides for up to 1,500,000 options to acquire Common Stock. There have been no options issued under this plan as of the date hereof.
Director Compensation:
Our directors are reimbursed for expenses incurred in connection with attending meetings. Other than a grant of options to Peter Barton Hutt to purchase 129,803 shares of Common Stock (as adjusted for the Share Exchange), our directors do not receive any compensation for serving on the board of directors.
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth certain information concerning the anticipated beneficial ownership of the Company’s Common Stock upon consummation of the Merger by (i) each person known by the Company to be the owner of more than 5% of the outstanding Common Stock, (ii) each director, (iii) each named executive officer, and (iv) all directors and executive officers as a group. In general, “beneficial ownership” includes those shares that a stockholder has the power to vote or the power to transfer, and stock options and other rights to acquire common stock that are exercisable currently or become exercisable within 60 days. Unless otherwise indicated, the address for each person is c/o Grace 2, Inc./Q Therapeutics, Inc., 615 Arapeen Drive, Suite 102, Salt Lake City, UT 84108. Upon consummation of the Merger and the initial closing of the Company’s Private Placement Offering, there were 24,557,632 shares of Common Stock issued and outstanding. In addition to the shares listed above, in connection with the Merger, previous warrant holders of Q Therapeutics exchanged all of their Q Therapeutics warrants for warrants to acquire 2,048,386 shares of Grace 2 Common Stock and previous option holders of Q Therapeutics exchanged all of their Q Therapeutics options for options to acquire 3,385,596 shares of Grace 2 Common Stock. In addition, the Company issued 7,606,094 warrants in connection with its Private Placement Offering.
|
Name of
Beneficial Owner
|
Class of Voting
Stock
|
Number of Shares of
Voting Stock
Beneficially Owned (1)
|
Percentage of Class
(1)
|
|||||||
|
Deborah A. Eppstein, Ph.D., President, CEO and Director
|
Common Stock
|
1,998,434 | (2) | 7.7 | % | |||||
|
Steven J. Borst, CFO and Vice President of Corporate Development`
|
Common Stock
|
1,337,326 | (3) | 5.3 | % | |||||
|
Dinesh C. Patel, Ph.D., Chairman of the Board of Directors, Managing Director, vSpring Capital, 2795 East Cottonwood Parkway, Salt Lake City, UT 84121
|
Common Stock
|
7,243,123 | (4) | 28.7 | % | |||||
|
Peter Barton Hutt, Director, Senior Counsel, Covington & Burling, 1201 Pennsylvania Ave., Washington, DC 20004
|
Common Stock
|
129,803 | (5) | 0.5 | % | |||||
|
Joydeep Goswami, Ph.D., M.B.A., President of Life Technologies –Japan, c/o Life Technologies 5791 Van Allen Way, Carlsbad, California 92008
|
Common Stock
|
2,445,564 | (6) | 10.0 | % | |||||
|
Linda F. Powers, Director, Managing Director, Toucan Capital, 4800 Montgomery Lane, Suite 801, Bethesda, MD 20814
|
Common Stock
|
1,497,496 | (7) | 6.1 | % | |||||
|
UpStart Life Sciences Capital, L.P., 417 Wakara Way, Suite 3510, Salt Lake City, UT 84108
|
Common Stock
|
2,433,288 | (8) | 9.2 | % | |||||
|
Cephalon, Inc., 41 Moores Road, Frazer, PA 19355
|
Common Stock
|
11,056,641 | (9) | 34.6 | % | |||||
|
Directors and Officers as a group (6 persons)
|
Common Stock
|
14,651,746 | 53.1 | % | ||||||
66
|
(1)
|
Based on 24,557,632 shares of Common Stock issued and outstanding as of October 13, 2011, post Merger, and any shares of Common Stock that the beneficial owner has the right to acquire within 60 days. Any securities not outstanding which are subject to such options, warrants, rights or conversion privileges shall be deemed to be outstanding for the purpose of computing the percentage of outstanding securities of the class owned by such person but shall not be deemed to be outstanding for the purpose of computing the percentage of the class by any other person.
|
|
(2)
|
Includes options to acquire 1,297,372 shares of Common Stock.
|
|
(3)
|
Includes options to acquire 743,986 shares of Common Stock and warrants to acquire 30,370 shares of Common Stock.
|
|
(4)
|
Includes 43,268 shares of Common Stock owned by Dr. Patel personally. The remaining shares, which include warrants to acquire 712,277 shares of Common Stock, are owned by vSpring Capital, Dr. Patel’s employer.
|
|
(5)
|
Includes options to acquire 129,803 shares of Common Stock.
|
|
(6)
|
Shares are owned by Life Technologies, Inc., Dr. Goswami’s employer.
|
|
(7)
|
Shares are owned by Toucan Capital, Ms. Powers’ employer, and include warrants to acquire 96,580 shares of Common Stock.
|
|
(8)
|
Includes warrants to acquire 1,822,192 shares of Common Stock.
|
|
(9)
|
Includes warrants to acquire 7,371,094 shares of Common Stock.
|
67
Certain Relationships and Related Transactions; and Director Independence.
Certain Relationships and Related Transactions
The Company utilizes the office space and equipment of its majority shareholder at no cost. Management believes the amounts are immaterial.
As of May 31, 2009, the majority shareholder advanced the Company $16,075 for working capital purposes. The advances are not documented and do not bear any specific repayment terms.
As of May 31, 2008, the former sole shareholder of the Company advanced the Company $9,694 for working capital purposes. These advances were classified as capital contributions and reflected as such in additional paid in capital.
Except as otherwise indicated herein, there have been no related party transactions, or any other transactions or relationships required to be disclosed pursuant to Item 404 of Regulation S-K.
Director Independence
Our determination of independence of our directors is made using the definition of “independent director” contained under NASDAQ Marketplace Rule 4200(a)(15), even though such definitions do not currently apply to us because we are not listed on NASDAQ. With the exception of Ms. Eppstein, who currently serves in the capacity of Officer and Director, we believe our Board Membership is composed of members who qualify as independent pursuant to this Rule.
|
ITEM 5.03
|
AMENDMENT TO THE ARTICLES OF INCORPORATION OR BYLAWS; CHANGE IN FISCAL YEAR.
|
On October 13, 2011, pursuant to the Merger, the Board adopted a resolution by unanimous written consent to effect a change of the Company’s fiscal year end from May 31st to December 31st. This change was made to be consistent with the fiscal year of Q Therapeutics, Inc., which is now our wholly owned subsidiary and operating company.
On October 13, 2011, pursuant to the Merger, the Board and Majority Shareholders adopted a resolution by joint unanimous written consent to effect a change of the Company’s name from Grace 2, Inc. to Q Holdings, Inc. This change will go into effect by the Company’s filing of a Certificate of Amendment to the Certificate of Incorporation with the Secretary of State of the State of Delaware, upon the expiration of 20 days following the Company’s filing of a Definitive Information Statement on Schedule 14C therein notifying Company shareholders of the pending name change.
ITEM 5.06. CHANGE IN SHELL COMPANY STATUS.
On October 13, 2011, the Company ceased to be a shell company as defined in Rule 12b-2 of the Exchange Act.
ITEM 9.01. FINANCIAL STATEMENTS AND EXHIBITS
(a) Financial Statements of Business Acquired
The Audited Consolidated Financial Statements of Q Therapeutics, Inc. as of December 31, 2010 and 2009 and for the years then ended are filed as Exhibit 99.1 to this current report and are incorporated herein by reference.
The Unaudited Consolidated Financial Statements of Q Therapeutics, Inc. as of June 30, 2011 and for the six months then ended are filed as Exhibit 99.2 to this current report and are incorporated herein by reference.
68
The unaudited pro forma condensed consolidated balance sheet as of June 30, 2011 and the unaudited pro forma condensed consolidated statements of operations for the six months ended June 30, 2011 and for the year ended December 31, 2010 to reflect the acquisition of the business operations of Q Therapeutics, Inc. are filed as Exhibit 99.3 to this current report and are incorporated herein by reference.
(c) Shell Company Transactions
Reference is made to Items 9.01(a) and 9.01(b) and the exhibits referred to therein which are incorporated herein by reference.
(d) Exhibits
|
Exhibit
No.
|
Description
|
|
|
2.1
|
Agreement and Plan of Merger by and between Grace 2, Inc., Q Merger Sub, Inc., and Q Therapeutics, Inc, dated October 13, 2011
|
|
|
3.1
|
Articles of Incorporation (1)
|
|
|
3.2
|
By Laws (1)
|
|
|
10.1
|
Exclusive License Agreement between Q Therapeutics, Inc. and the University of Utah Research Foundation dated October 5, 2005, as amended.
|
|
|
10.2
|
Employment Agreement between Q Holdings, Inc. and Deborah A. Eppstein PhD, dated October 13, 2011.
|
|
|
10.3
|
Employment Agreement between Q Holdings, Inc. and Steven J. Borst dated October 13, 2011.
|
|
|
16.1
|
Letter from W. T. Uniack & Co., CPA’s P.C. dated October 14, 2011.
|
|
|
99.1
|
The Audited Consolidated Financial Statements of Q Therapeutics as of December 31, 2010 and 2009
|
|
|
99.2
|
The Unaudited Consolidated Financial Statements of Q Therapeutics as of June 30, 2011
|
|
|
99.3
|
The Unaudited Pro Forma Financial Information and related notes thereto.
|
|
(1)
|
Incorporated by reference from the Company’s Registration Statement on Form 10SB (SEC File No. 000-52062) filed on June 19, 2006.
|
69
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
|
GRACE 2, INC.
|
|
|
|
||
|
Dated: October 18, 2011
|
By:
|
/s/ DEBORAH A. EPPSTEIN, PH.D.
|
|
|
Deborah A. Eppstein, Ph.D.
President and Chief Executive Officer, Director
(Principal Executive Officer)
|
|
|
Dated: October 18, 2011
|
By:
|
/s/ STEVEN J. BORST
|
|
|
Steven J. Borst, M.B.A.
Chief Financial Officer and Vice President of Corporate Development
(Principal Financial Officer,
Principal Accounting Officer)
|
70