Attached files
| file | filename |
|---|---|
| EX-99.1 - EX-99.1 - Radius Health, Inc. | a11-19031_1ex99d1.htm |
| EX-16.1 - EX-16.1 - Radius Health, Inc. | a11-19031_1ex16d1.htm |
| EX-99.2 - EX-99.2 - Radius Health, Inc. | a11-19031_1ex99d2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 8-K/A
(Amendment No. 1)
CURRENT REPORT
Pursuant to Section 13 or 15(d) of the
Securities Exchange Act of 1934
Date of Report (date of earliest event reported): May 17, 2011
RADIUS HEALTH, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
000-53173 |
|
80-0145732 |
|
(State of Incorporation) |
|
(Commission File Number) |
|
(IRS Employer Identification Number) |
201 Broadway, 6th Floor
Cambridge, MA 02139
(Address of principal executive offices) (Zip Code)
(617) 551-4700
(Registrant’s telephone number, including area code)
MPM ACQUISITION CORP.
c/o MPM Asset Management LLC, 200 Clarendon Street, 54th Floor, Boston, MA 02116
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
o Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
o Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
o Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
o Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
Item 1.01. Entry into a Material Definitive Agreement.
The disclosures set forth under Item 2.01 hereof are hereby incorporated by reference in this Item 1.01.
Item 2.01. Completion of Acquisition or Disposition of Assets.
Pursuant to an Agreement and Plan of Merger dated April 25, 2011 (the “Merger Agreement”), by and among MPM Acquisition Corp. (referred to herein as the “Company”, “Radius” or the “Registrant”), RHI Merger Corp., a Delaware corporation and wholly owned subsidiary of the Company (“MergerCo”), and Radius Health, Inc., a Delaware corporation (“Target”), MergerCo merged with and into Target, with Target remaining as the surviving entity and a wholly-owned operating subsidiary of the Company. This transaction is referred to throughout this report as the “Merger.” The Merger was effective as of May 17, 2011, upon the filing of a certificate of merger with the Delaware Secretary of State.
At the effective time of the Merger (the “Effective Time”), the legal existence of MergerCo ceased and all of the shares of Target’s common stock, par value $.01 per share (the “Target Common Stock”), and shares of Target’s preferred stock, par value $.01 per share (the “Target Preferred Stock”), that were outstanding immediately prior to the Merger were cancelled and each outstanding share of Target Common Stock outstanding immediately prior to the Effective Time was automatically converted into the right to receive one share of the Company’s Common Stock and each outstanding share of Target Preferred Stock outstanding immediately prior to the Effective Time was automatically converted into the right to receive one-tenth of one share of the Company’s Preferred Stock from the Company as consideration for the Merger. More specifically, each share of Series A-1 Convertible Preferred Stock of Target outstanding immediately prior to the Effective Time was converted into the right to receive 0.1 shares of Series A-1 Convertible Preferred stock of the Company; each share of Series A-2 Convertible Preferred Stock of Target outstanding immediately prior to the Effective Time was converted into the right to receive 0.1 shares of Series A-2 Convertible Preferred stock of the Company; each share of Series A-3 Convertible Preferred Stock of Target outstanding immediately prior to the Effective Time was converted into the right to receive 0.1 shares of Series A-3 Convertible Preferred stock of the Company; each share of Series A-4 Convertible Preferred Stock of Target outstanding immediately prior to the Effective Time was converted into the right to receive 0.1 shares of Series A-4 Convertible Preferred stock of the Company; each share of Series A-5 Convertible Preferred Stock of Target outstanding immediately prior to the Effective Time was converted into the right to receive 0.1 shares of Series A-5 Convertible Preferred stock of the Company; and each share of Series A-6 Convertible Preferred Stock of Target outstanding immediately prior to the Effective Time was converted into the right to receive 0.1 shares of Series A-6 Convertible Preferred stock of the Company. The Company assumed all options and warrants of Target outstanding immediately prior to the Effective Time, which shall become exercisable for shares of the Company’s Common Stock or Preferred Stock, as the case may be. See the description of the material terms of the options and warrants assumed in the merger in sections herein entitled “2003 Long-Term Incentive Plan” and “Description of Securities—Stock Purchase Warrants”, respectively. Target and the Company agreed to indemnify each of their officers and directors for their actions relating to the consideration, approval or consummation of the Merger Agreement, in accordance with an indemnity agreement (the “Indemnity Agreement”) entered into by and between Target, the Company and their respective officers before the closing of the merger. The Company’s entry into the Merger Agreement was disclosed on the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on April 29, 2011, which is hereby incorporated by reference, including the copy of the Merger Agreement filed as Exhibit 10.1 thereto.
Contemporaneously with the closing of the Merger, pursuant to the terms of a Redemption Agreement dated March 25, 2011 by and among the Company and its then-current stockholders, the Company completed the repurchase of 5,000,000 shares of Common Stock (the “Redemption”) from its former stockholders in consideration of an aggregate of $50,000. The 5,000,000 shares constituted all of the issued and outstanding shares of the Company’s capital stock, on a fully-diluted basis, immediately prior to the Merger. The Company’s entry into the Redemption Agreement was disclosed on the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on April 29, 2011, which is hereby incorporated by reference, including the copy of the Redemption Agreement filed as Exhibit 10.2 thereto. Also in connection with the Merger, the Company entered into Indemnification Agreements with each member of its board of directors, copies of which are filed here with as Exhibits 10.52 to and including Exhibit 10.62.
Upon completion of the Merger and the Redemption, the former stockholders of Target held 100% of the outstanding shares of capital stock of the Company. Accordingly, the Merger represents a change in control of the Company. As of the date of this report, there are 555,594 shares of our Common Stock and 1,549,130 shares of our Preferred Stock outstanding.
Pursuant to the Merger, we assumed all of the Target’s obligations under its existing contracts, including those filed herewith as material contracts. In particular, we have assumed the obligations of Target under that certain Series A-1 Convertible Preferred Stock Purchase Agreement (the “Original Purchase Agreement”) with certain investors listed therein (the “Investors”) pursuant to which, among other things, we are obligated to issue and sell to the Investors up to an aggregate of 789,553 shares of Series A-1 Convertible Preferred Stock, par value $.01 per share, to be completed in three closings (the initial closing, the “Stage I Closing”, the second closing, the “Stage II Closing” and the final closing, the “Stage III Closing”) (collectively, the “Series A-1 Financing”). The Original Purchase Agreement was subsequently amended by Amendment No. 1 thereto to eliminate all closing conditions previously provided for in the Original Purchase Agreement (as so amended, the “Purchase Agreement”). Upon notice from us, the Investors are obligated to purchase, and we are obligated to issue, 263,178 shares of our Series A-1 Convertible Preferred Closing at the Stage II Closing and 263,180 shares of our Series A-1 Convertible Preferred Stock at the Stage III Closing, each at a purchase price per share of $81.42. There are no conditions to funding if we notify the Investors of any such closing. A copy of the Purchase Agreement is attached hereto as Exhibit 10.26, and is incorporated herein by reference.
The Merger will be accounted for as a capital transaction. Upon effectiveness of the Merger, Target’s business plan became our business plan.
The foregoing description of the Merger Agreement, the Redemption Agreement, Purchase Agreement and the transactions contemplated thereby do not purport to be complete and are qualified in their entireties by reference to the Merger Agreement and the Redemption Agreement, respectively.
Following the Merger on May 17, 2011, our Board of Directors approved a transaction pursuant to which Target merged with and into the Company, leaving the Company as the surviving corporation (the “Short-Form Merger”). In connection with the Short-Form Merger, the Company relinquished its corporate name and assumed in its place the name “Radius Health, Inc.” The Short-Form and name change became effective on May 17, 2011, upon the filing of a Certificate of Ownership an Merger with the Delaware Secretary of State. Our certificate of incorporation, The Certificate of Ownership and Merger is filed as Exhibit 3.2 hereto.
On May 23, 2011, the Company entered into a Loan and Security Agreement with General Electric Capital Corporation (“GECC”) as agent and a lender, and Oxford Finance LLC (“Oxford” and together with GECC, the “Lenders”) as a lender, pursuant to which the lenders agreed to make available to the Company $25,000,000 in the aggregate over three term loans. The initial term loan was made on May 23, 2011 in an aggregate principal amount equal to $6,250,000 (the “Initial Term Loan”) and is repayable over a term of 42 months, including a six month interest only period. The Initial Term Loan bears interest at 10%. Pursuant to the Agreement, the Company may request two (2) additional term loans, the first, which must be funded not later than November 23, 2011, in an aggregate principal amount equal to $6,250,000 (the “Second Term Loan”) and the second, which must be funded not later than May 23, 2012, in an aggregate principal amount equal to $12,500,000 (the “Third Term Loan”). In the event the Second Term Loan is not funded on or before November 23, 2011, the Lenders’ commitment to make the Second Term Loan shall be terminated and the total commitment shall be reduced by $6,250,000. In the event the Third Term Loan is not funded on or before May 23, 2012, the Lenders’ commitment to make the Third Term Loan shall be terminated and the total commitment shall be further reduced by $12,500,000. Pursunt to the agreement, the Company agreed to issue to the Lenders (or their respective affiliates or designees) stock purchase warrants (collectively, the “Warrants”) to purchase in the aggregate a number of shares of the Company’s Series A-1 Preferred Stock equal to the quotient of (a) the product of (i) the amount of the applicable term loan multiplied by (ii) four percent (4%) divided by (b) the exercise price equal to $81.42 per share. The exercise period of each Warrant to be issued will expire ten (10) years from the date such Warrants are issued. On May 23, 2011, the Company issued a Warrant to each of GECC and Oxford for the purchase of 3,070 shares of Series A-1 Preferred stock.
DESCRIPTION OF THE BUSINESS OF RADIUS HEALTH, INC.
EXPLANATORY NOTE: Unless otherwise provided in this current report, all references in the balance of this current report to “we,” “us,” “our company,” “our,” or the “Company” refer to the combined Radius Health, Inc. entity after giving effect to the Merger and the Short-Form Merger.
Overview
Radius is a pharmaceutical company focused on acquiring and developing new therapeutics for the treatment of osteoporosis and other women’s health conditions. Our lead product candidate is BA058 Injection, a daily subcutaneous injection of our novel synthetic peptide analog of human parathyroid hormone-related protein (hPTHrP) for the treatment of osteoporosis. In April 2011, we began dosing of patients in a pivotal Phase 3 clinical study and expect to report top-line data from this study by late 2013. Based on our clinical and preclinical results to date, we believe that BA058 stimulates the rapid formation of new high quality bone in patients suffering from osteoporosis and may restore bone mineral density in these patients into the normal reference range. In addition to BA058 Injection, we are developing BA058 Microneedle Patch, a short wear time, transdermal form of BA058 that is delivered using a microneedle technology from 3M Drug Delivery Systems (3M). BA058 Microneedle Patch is being studied in a Phase 1b clinical study which began in December 2010. The BA058 Microneedle Patch may eliminate the need for daily injections and lead to better treatment compliance for patients. We believe that development costs for the BA058 Microneedle Patch will be lower than the development costs for BA058 Injection as it will not be necessary to conduct an additional fracture study for
this follow-on product. As a result of the compressed pathway for the BA058 Microneedle Patch, we expect that marketing approval of the BA058 Microneedle Patch can occur soon after the BA058 Injection.
While there are a number of drugs that help to reduce the rate of bone loss in patients suffering from osteoporosis, there are few that are able to build bone. The only approved therapy in the United States that increases bone mineral density (BMD) into the normal reference range in these patients is Forteo®, a daily subcutaneous injection of recombinant human parathyroid hormone (rhPTH(1-34)). The product is marketed by Eli Lilly and had reported worldwide sales of $830 million in 2010. We believe that BA058 may offer a number of important advantages over Forteo®, including greater efficacy, a faster benefit, a shorter course of therapy, an improved safety profile and no need to refrigerate in use BA058 Injection. We believe, if approved, the BA058 Injection and the BA058 Microneedle Patch will offer an attractive bone anabolic treatment option for prescribing physicians and women with compelling advantages in safety, efficacy and delivery over Forteo®.
Based upon guidance we have received from the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA), we believe that a single pivotal placebo-controlled, comparative Phase 3 study will be sufficient to support registration of BA058 Injection for the treatment of osteoporosis in both the United States and the European Union. Our planned study will enroll a total of 2,400 patients to be randomized equally to receive daily doses of one of the following: 80 micrograms (µg) of BA058, a matching placebo, or the approved dose of 20 µg of Forteo® for 18 months. The study will be powered to show that BA058 is superior to (i) placebo for fracture and (ii) Forteo® for greater BMD improvement at major skeletal sites and for a lower occurrence of hypercalcemia, a condition in which the calcium level in a patient’s blood is above normal. We believe that the study will also show that BMD gains for BA058 patients will be earlier than for Forteo® patients.
Market Opportunity for BA058
Osteoporosis is a disease characterized by low bone mass and structural deterioration of bone tissue, leading to an increase in fractures. The prevalence of osteoporosis is growing in developed nations with the aging of the populations. The National Osteoporosis Foundation (“NOF”) has estimated that (i) 10 million people in the United States, comprising eight million women and two million are men, already have osteoporosis and another 34 million have low bone mass placing them at increased risk for osteoporosis and (ii) osteoporosis was responsible for more than 2 million fractures in the United States in 2005 resulting in an estimated $19 billion in costs. The NOF expects that the number of fractures due to osteoporosis will rise to more than 3 million by 2025.
In 2011, Cowen and Company (“Cowen”), an investment banking firm, estimated that total worldwide sales of osteoporosis products was $7.6 billion in 2010. There are two main types of osteoporosis drugs now available in the United States: (i) anti-resorptive agents such as bisphosphonates including Actonel®, Boniva® or Reclast®, and Prolia® (a nuclear factor kB ligand (“RANKL”) inhibitor marketed by Amgen), as well as calcitonins and selective estrogen receptor modulators such as Evista® marketed by Lilly; and (ii) anabolic agents, with Forteo® being the only approved drug of this type. Anti-resorptive agents act to prevent further bone loss by inhibiting the breakdown of bone whereas anabolic agents stimulate bone formation to build high quality, new bone. The use of bisphosphonates have been associated with infrequent but serious adverse events such as osteonecrosis of the jaw, atrial fibrillation and anomalous fractures resulting from “frozen bone” that have created increasing concern with physicians and patients. Many physicians are seeking alternatives to current anti-resorptive therapies and we believe this will drive greater demand for bone anabolic agents in the future. We believe that there is a significant opportunity for a new anabolic agent such as BA058 that will increase bone mineral density to a greater degree and at a faster rate than Forteo® with added advantages in convenience and safety.
Our Strategy
We plan to build a pharmaceutical company focused on acquiring and developing new therapeutics for osteoporosis and women’s health by:
· Completing the single, pivotal Phase 3 clinical trial of BA058 Injection for the treatment of osteoporosis by the end of 2013;
· Pursuing the clinical development of BA058 Microneedle Patch as a follow-on product for the treatment of osteoporosis;
· Obtaining regulatory approval of BA058 Injection and BA058 Microneedle Patch for the treatment of osteoporosis, initially in the United States and subsequently in the European Union;
· Collaborating with third parties for the worldwide commercialization of BA058; and
· Collaborating with third parties for the further development and commercialization of RAD1901 and RAD140 on a worldwide basis.
To execute on our strategy, we have built a strong management team and Board of Directors with significant pharmaceutical development, regulatory and commercial experience.
Our Solution:
In addition to BA058 Injection and BA058 Microneedle Patch, we are currently conducting one other clinical and one preclinical program. Our second clinical stage product candidate is RAD1901, a selective estrogen receptor modulator, or SERM, licensed from Eisai Co (Eisai) in 2006 which has completed an initial Phase 2 clinical study for the treatment of vasomotor symptoms (hot flashes) in women entering menopause. Our third product candidate, RAD140, is a pre-investigational new drug, or IND, discovery. RAD140 is a selective androgen receptor modular, or SARM, that is an orally-active androgen agonist on muscle and bone and is a potential treatment for age-related muscle loss, frailty, weight loss associated with cancer cachexia and osteoporosis.
The following table summarizes the target indications, dosage forms, and stages of development for our product candidates.
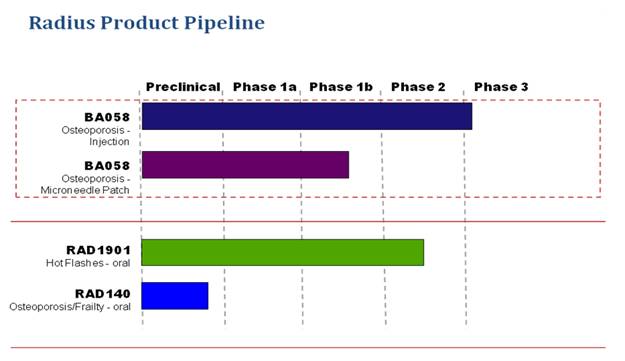
BA058
BA058 is a novel synthetic peptide analog of Parathyroid hormone-related peptide (hPTHrP) being developed by us as a bone anabolic treatment for osteoporosis. hPTHrP is a critical cytokine for the regulation of bone formation, able to rebuild bone with low associated risk of inducing hypercalcemia as a side-effect. We believe that BA058 is the most advanced hPTHrP analog in clinical development for the treatment of osteoporosis. We acquired and maintain exclusive worldwide rights (except Japan) to certain patents, data and technical information related to BA058 through a license agreement with Ipsen Pharma S.A.S (Ipsen) dated September 2005. Based on clinical and preclinical data to date, we believe that BA058 has the following important potential advantages over
Forteo® rhPTH(1-34), the only other approved anabolic agent for osteoporosis in the US:
· Greater efficacy,
· Faster benefit,
· Shorter treatment duration,
· Less hypercalcemia,
· No additional safety risks, and
· No refrigeration required in use.
BA058 Injection
In August 2009, we announced positive Phase 2 data that showed BA058 Injection produced faster and greater BMD increases at the spine and the hip after 6 months and 12 months of treatment than did Forteo®, which was a comparator in our study. Key findings were that the highest dose of BA058 tested of 80 µg increased mean lumbar spine BMD at 6 and 12 months by 6.7% and 12.9% compared to the increases seen with Forteo® trial arms of 5.5% and 8.6%, respectively. BA058 also produced increases in mean femoral neck BMD at the hip at 6 and 12 months of 3.1% and 4.1% compared to increases for Forteo® of 1.1% and 2.2%, respectively. We believe there to be a strong correlation between an increased level of BMD and a reduction in the risk of fracture for patients with osteoporosis. BA058 was generally safe and well tolerated in this study, with adverse events similar between the BA058, placebo and Forteo® groups. In addition, the occurrence of hypercalcemia as a side-effect was half that seen with Forteo® for the 80 µg dose of BA058.
In March 2011, we entered an agreement with Nordic Bioscience (Nordic) to manage the Phase 3 study of BA058 Injection. The study will be conducted in 8 countries at 11 centers operated by the Center for Clinical and Basic Research (CCBR). CCBR is a leading global clinical research organization (CRO) with extensive experience in global osteoporosis registration studies. We expect to report top-line data from the Phase 3 study of BA058 Injection by late 2013.
BA058 Microneedle Patch
In December 2010, we initiated a combined single and seven-day repeat-dose Phase 1 clinical study of the BA058 Microneedle Patch in healthy subjects with top-line data expected to be available by mid-2011. Following this Phase 1 study, we plan to select a dose range to conduct a Phase 2 clinical study comparing multiple daily doses of the BA058 Microneedle Patch to placebo and BA058 Injection using lumbar spine BMD at 6 months as the primary endpoint. We expect to begin the Phase 2 BA058 Microneedle Patch clinical study in mid 2012 with top-line data available in mid 2013. If the BA058 Injection product is already approved by the FDA, we believe that we will only need to conduct a single non-inferiority Phase 3 clinical study comparing the change in lumbar spine BMD at 12 months for patients dosed with the BA058 Microneedle Patch to patients dosed with the BA058 Injection. We believe that development costs for the BA058 Microneedle Patch will be lower than the injectable version as it will not be necessary to conduct an additional fracture study for this follow-on product. As a result of the compressed pathway, we expect that marketing approval of the BA058 Microneedle Patch can occur soon after the BA058 Injection.
Background on Osteoporosis
Osteoporosis is a disease characterized by low bone mass and structural deterioration of bone tissue, which can lead to an increase in fractures. On its website, www.nof.org, the National Osteoporosis Foundation (NOF) has estimated that 10 million people in the United States, comprising eight million women and two million men, already have osteoporosis and another 34 million have low bone mass placing them at increased risk for osteoporosis and broken bones. All bones become more fragile and susceptible to fracture as the disease progresses. People tend to be unaware that their bones are getting weaker, and a person with osteoporosis can fracture a bone from even a minor fall. Fractures due to osteoporosis are most likely to occur in the hip, spine and wrist. According to the NOF, osteoporosis was responsible for more than 2 million fractures in the United States in 2005 and is expected to rise to more than 3 million fractures by 2025. Vertebral (spinal) fractures may result in severe back pain, loss of height or spinal deformities. There were approximately 293,000 Americans age 45 and over admitted to hospitals in 2005 with a fracture of the femoral neck, a common type of hip fracture that is
associated with osteoporosis. A women’s lifetime risk of a hip fracture is equal to her combined risk of breast, uterine and ovarian cancer. An average of 24 percent of hip fracture patients aged 50 and over dies in the year following their fracture. An additional 20 percent of patients who were ambulatory before their hip fracture require long-term care.
The debilitating effects of osteoporosis have substantial costs. Loss of mobility, admission to nursing homes and dependence on caregivers are all common consequences of osteoporosis. The NOF has estimated that osteoporosis-related fractures were responsible for $19 billion in costs in 2005.
The prevalence of osteoporosis is growing and, according to the NOF, is significantly under-recognized and under-treated in the population. While the aging of the population is a primary driver of an increase in cases, osteoporosis is also increasing from the use of drugs that induce bone loss, such as chronic use of glucocorticoids for asthma, aromatase inhibitors that are increasingly used for breast cancer and the hormone therapies used for prostate cancer.
The range of treatment and prevention options for osteoporosis has expanded in recent years from anti-resorptive drugs that act to prevent bone loss by blocking bone resorption and now includes bisphosphonates, selective estrogen receptor modulators, calcitonins, and most recently in 2010, a RANKL inhibitor. Bisphosphonates remain the current standard of care with 2010 world-wide total sales of approximately $4.2 billion according to Cowen and Company’s report dated March 2011 and entitled Therapeutic Categories Outlook, led by Actonel®, Boniva®, and Fosamax®. Generic versions of Fosamax® (alendronate) became available in the US in 2008 and have now gained share from branded oral bisphosphonates.
The only anabolic (i.e., stimulating bone formation) drug approved in the U.S. for osteoporosis is Forteo®, which was approved by the FDA in December 2002. In 2011, the medical journal, Osteoporosis International, published results of a study indicating that patients’ preferences for osteoporosis medications are strongly influenced by the mode of administration. In particular, when given the choice of subcutaneously injected Forteo® versus other therapies, patients preferred the alternative drugs over Forteo, which requires once-daily, self-administered injections and must be refrigerated for storage in use. We believe that this research suggests that there is a substantial opportunity to optimize patient outcomes and expand the market by improved treatment compliance with a bone anabolic drug that offers an alternative to daily injection, is room-temperature-stable and requires a shorter treatment duration, such as the BA058 Microneedle Patch. Forteo® had world-wide sales of $615 million in 2008 and grew to $830 million in sales for 2010.
Clinical Development Program for BA058
Radius is developing BA058 for the prevention of fractures in postmenopausal women at risk of fracture from severe osteoporosis. Recognizing both the therapeutic potential of BA058 in this indication as well as the drawbacks inherent in self-injection therapies in this population, Radius is also developing the BA058 Microneedle Patch for transdermal administration of the product using a microneedle technology from 3M. We plan to develop and register BA058 Injection as our lead product, with the BA058 Microneedle Patch as a fast-following product that provides greater patient convenience. The ability of the Microneedle Patch to capitalize on the more extensive fracture study data of BA058 Injection will allow the patch product to be accelerated though later phase development without requiring its own fracture study.
Planned and Completed BA058 Studies
Planned Studies
BA058 Injection, Phase 3
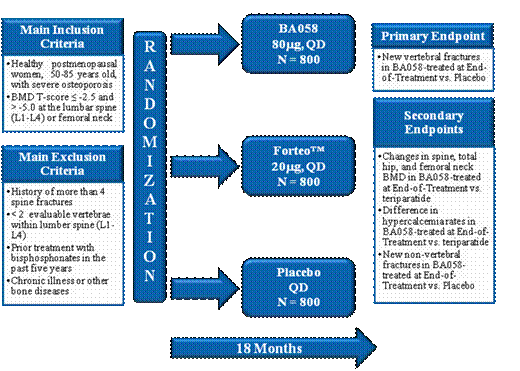
The Phase 3 study for BA058 Injection (Study BA058-05-003) was submitted as a draft protocol to IND 73,176 on December 18, 2009, and was the subject of a Type B End of Phase 2 Meeting conducted with the Agency on January 21, 2010. The protocol was subsequently revised and submitted to the FDA on December 17, 2010. The study is planned to enroll 2,400 patients at 11 medical centers in 8 countries in Europe, Latin America and Asia.
Study Objectives
The primary objective of this study is to determine the safety and efficacy of BA058 Injection 80 µg when compared to a matching placebo for prevention of vertebral fracture in otherwise healthy ambulatory postmenopausal women at risk of fracture from severe osteoporosis. Patients, investigators and independent assessors will be blinded as to treatment for that outcome. The secondary objectives of this study are to determine the safety and efficacy of BA058 80 µg when compared to placebo for prevention of non-vertebral fractures and for change in vertical height. Additional key secondary efficacy outcomes include BMD of spine, hip and femoral neck and hypercalcemia when compared to Forteo®.
Study Population
The study will enroll otherwise healthy ambulatory postmenopausal (> 5 years) women from 50 to 85 years of age (inclusive) who meet the study entry criteria and have provided written informed consent. The women will have a BMD T-score < -2.5 and > -5.0 at the lumbar spine or hip (femoral neck) by dual energy x-ray absorptiometry (DXA) and radiological evidence of two or more mild or one or more moderate lumbar or thoracic vertebral fractures, or history of low trauma forearm, humerus, sacral, pelvic, hip, femoral, or tibial fracture within the past 5 years. Postmenopausal women older than 65 who meet the above fracture criteria but have a T-score < -2.0 and > -5.0 may be enrolled. Women older than 65 who do not meet the fracture criteria may also be enrolled if their T-score is < -3.0 and > -5.0.
All patients are to be in good general health as determined by medical history, physical examination (including vital signs) and clinical laboratory testing.
Study Design
The planned 2,400 eligible patients will be randomized equally to receive one of the following: BA058 80 µg, a matching placebo, or Forteo® 20 µg for 18 months. Study drug will be blinded to patients and medical personnel until the randomization process is completed. Treatment with BA058 80 µg or placebo will remain blinded to all parties throughout the study. Forteo® comes as a proprietary prefilled drug and device combination that cannot be repackaged and therefore, its identity cannot be blinded to treating physicians and patients once use begins. Study medication will be self-administered daily by SC injection for a maximum of 18 months.
The dosages of study medications and the number of patients per group are shown in below.
Study BA058-05-003 — Medication Doses and Number of Patients per Group
|
Treatment Regimen |
|
Study Medication |
|
Daily Dose (SC) |
|
Duration |
|
Number of Patients |
|
|
1 |
|
BA058 |
|
80 µg |
|
18 months |
|
800 |
|
|
2 |
|
Placebo |
|
— |
|
18 months |
|
800 |
|
|
3 |
|
Forteo® |
|
20 µg |
|
18 months |
|
800 |
|
|
|
|
|
|
|
|
Total |
|
2,400 |
|
All enrolled patients will also receive Calcium and Vitamin D supplementation from the time of enrollment until the end of the Treatment Period; it will be recommended to patients that they also continue these supplements through the one month follow-up period.
Primary Efficacy Outcomes
The primary efficacy endpoint will be the number of BA058-treated patients showing new vertebral fractures at End-of-Treatment when compared to placebo as evaluated by a blinded assessor (radiologist) according to a standardized graded scale of severity of the vertebral deformity (Genant scale).
Secondary Efficacy Endpoints
Secondary efficacy parameters will also include reduction in the incidence of non-vertebral fractures (wrist, hip, rib, etc.) and reduction in moderate and severe vertebral fractures. Other secondary efficacy endpoints will include changes in BMD of the spine, hip, femoral neck and wrist from baseline to end-of-treatment as assessed by DXA.
Additional secondary endpoints will include change in standing height and changes in serum bone markers across treatment, such as N-terminal propeptide of type I procollagen PINP, osteocalcin and bone-specific alkaline phosphatase. The frequency of hypercalcemia across treatment groups will also be assessed.
Safety Outcomes
Safety evaluations to be performed will include physical examinations, vital signs, 12-lead ECGs, clinical laboratory tests and monitoring and recording of adverse events. Specific safety assessments will include post-dose (4 hours) determination of serum calcium, determination of creatinine clearance, post-dose ECG assessments at selected visits and assessments of postural hypotension (60 minutes post-dose) at selected clinic visits.
Bone biopsy of the iliac crest will be performed in a subset of patients receiving BA058 80 µg and Placebo (up to 100 per group) for assessment of quantitative bone histomorphometry and will be read blinded to treatment by an independent blinded assessor. Renal safety will be further evaluated in a subset of 100 patients in each treatment group by renal CT scan.
Overall study safety will be monitored by an independent Data Safety Monitoring Board.
BA058 Microneedle Patch Phase 2
We plan to conduct a Phase 2 randomized, placebo-controlled, parallel group dose-finding clinical trial in mid-2012. The study will evaluate the safety and efficacy of the daily BA058 Microneedle Patch in women with osteoporosis. We intend to enroll about 250 patients and the study will be similar in design to the Phase 2 study for BA058 Injection. The study will evaluate the effects of 3 doses of the BA058 Microneedle Patch, compared to placebo and BA058 Injection 80 µg on change in BMD and anabolic bone markers over 6 months of treatment. The study will be powered to detect clinically meaningful changes in these efficacy measures (BMD and bone biomarkers).
Safety will be assessed as changes in incidence of adverse events, changes in laboratory parameters - in particular serum calcium, change from baseline in vital signs and physical examination.
Study participation will be preceded by 4 weeks of pretreatment with Calcium and Vitamin D supplements and treatment conclusion will be followed by a one month period of safety observation.
Completed BA058 Studies
BA058 Injection, Phase 2
A Phase 2 dose-finding clinical trial (Study BA058-05-002) was conducted as a randomized, placebo-controlled, parallel group dose-finding study in the United States, Argentina, India and the United Kingdom. The purpose of the study was to evaluate the safety and efficacy of daily subcutaneous (SC) injections of BA058 Injection in women with osteoporosis. The study evaluated the effects of BA058 Injection at multiple doses (0, 20, 40 and 80 µg) on recovery of BMD, a marker of fracture risk, and on biomarkers of anabolic and resorptive activity in bone. The study also included a Forteo® treatment arm for reference. These efficacy measures (BMD and bone biomarkers) were powered for statistical significance. After the initial 24 weeks of treatment, eligible patients were
offered a second 24 weeks of their assigned treatment. Safety was assessed throughout the study and reported on at both 6 months and 12 months. BA058 Injection and BA058-placebo were self-administered using a prefilled cartridge in a pen-injector device. Forteo® was self-administered as the marketed product at the approved dose of 20 µg per day by SC injection. Four weeks prior to start of treatment, patients began taking Calcium and Vitamin D supplements that continued throughout the study.
A total of 270 patients (mean age: 65 years) entered the Pretreatment Period, 222 patients were randomized, and 221 patients received study treatment and were analyzed in the intent to-treat (ITT) population with 55 continuing into the Extension Period. A total of 155 patients were included in the Efficacy Population (Per Protocol) in the initial 24 weeks of treatment.
Initial 24 weeks of treatment
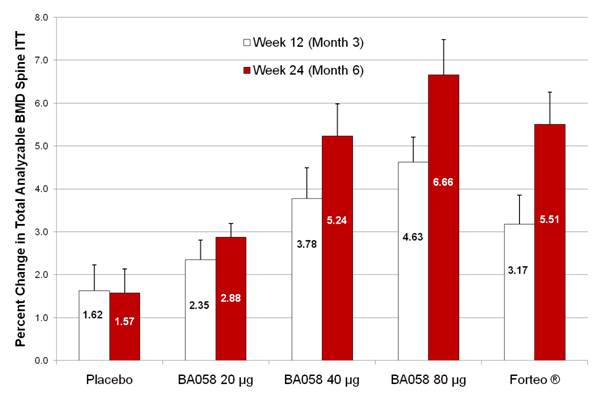
The efficacy results of Study BA058-05-002 confirm the preclinical and early clinical hypothesis that BA058 Injection induces a dose-dependent increase in BMD and in markers of bone remodeling measurable at both the 12-week and 24-week assessments.
At week 12, in the ITT population the mean percent change in total analyzable spine BMD increased with dose, Figure A. The mean gains in BMD (active treatment — placebo) for the BA058 Injection 40 µg and 80 µg groups were statistically significant (p = .0013 and p < 0.001, respectively). The difference was not statistically significant in the BA058 20 µg group and just missed significance in the Forteo® group (p = 0.055).
At week 24, the percent change from baseline continued to increase and was statistically significantly proportional to dose (p<0.001), Figure A. Again, the mean gain in total analyzable spine BMD was statistically significant for the BA058 Injection 40 µg (p = <0.001) and 80 µg ( p < 0.001) groups. The BMD gain at week 24 was also significant for the Forteo® group (p < 0.001), but not for the placebo group.
Figure A — Mean Standard Error of the Mean (SEM) Percent Change from Baseline at weeks 12 and 24 in Total Analyzable Spine BMD
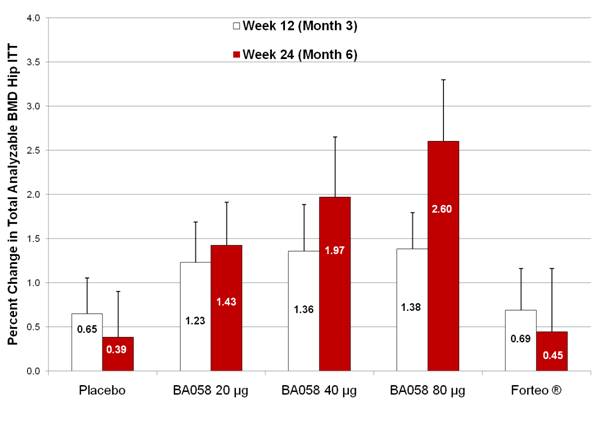
An even greater proportional response in BMD was elicited in the hip region. By week 24, mean percent changes in total analyzable hip BMD were 0.4%, 1.4%, 2.0% and 2.6% for the placebo, BA058 20 µg, BA058 40 µg, and BA058 80 µg groups, respectively; mean percent change in the Forteo® (0.5%) group was similar to placebo, Figure B. Total hip showed a clear dose response to BA058 and a more than five-fold benefit of BA058 80 µg over Forteo®. A similar relative benefit of BA058 80 µg over Forteo® was seen in all regions of the hip.
Figure B - Mean (SEM) Percent Change from Baseline at weeks 12 and 24 in Total Analyzable Hip BMD (ITT Population, N=221)
BA058 Injection also induced a dose-dependent rise in all markers of bone anabolic activity studied (N-terminal propeptide of type I procollagen PINP, bone specific alkaline phosphatase BSAP, and osteocalcin). The response to Forteo® was generally somewhat greater for all anabolic markers, but similar for bone resorption markers (C-telopeptides of type I collagen crosslinks [CTX] and N-telopeptides of type I collagen crosslinks [NTX]), consistent with published data on later attenuation of Forteo® BMD benefit.
BA058 Injection was well tolerated at all doses and safety events were consistent with usual medical events in a study population of this age and gender. The safety profile was also similar to that of Forteo® and there were no treatment-related SAEs; however, treatment-emergent adverse events (TEAEs) were reported by 74% of patients in the first 6 months of treatment, with a similar incidence across all treatment groups. The majority of TEAEs events were mild to moderate in severity and there were no deaths reported. Seven subjects discontinued due to adverse events,1in the BA058 20 µg group, 1 in the BA058 40 µg group, 3 in the BA058 80 µg group and 2 in the teriparatide group Eight patients (4%) experienced at least 1 severe AE and the incidence of such events was similar across treatment groups. Five TSAEs were reported in 3 patients, all unrelated to study drug. Local tolerance at the injection site was similar across treatment groups and fewer than 20% of subjects reported any one symptom, such as redness, at the injection site across the many months of injections.
Serum calcium levels were monitored throughout the study and clinically significant elevated levels (> 10.5 mg/dL) were observed in 40% of the Forteo® group while also observed in 4%, 12%, 19% and 18% of the placebo, BA058 Injection 20 µg, 40 µg and 80 µg groups. Most elevations were noted at the 4-hour post-injection time point. Clinically significant hypercalciuria (8%) and hypercalcemia (5%) were also more common in the Forteo® group.
Blood pressure was assessed throughout the study for postural change. Postural changes in blood pressure (predetermined level of change in systolic or diastolic from lying to standing) were reported in 7 patients, including 0%, 5%, 2%, 2% and 7% of patients in the placebo, BA058 Injection 20 µg, 40 µg, 80 µg and Forteo® groups, respectively. Pre-dose postural changes in blood pressure were similar across treatment groups. There were no clinically meaningful differences in ECG parameters between the placebo and active treatment groups.
Seventeen patients had low titer antibodies against BA058 after 6 months of treatment. Of these, 1 was in the placebo group, 2 were in the BA058 20 µg group, 8 were in the BA058 40 µg group and 6 were in the BA058 80 µg group. There were no associated safety events and no attenuation of treatment efficacy. One antibody-positive patient in the BA058 Injection 40 µg group was found to have evidence of neutralizing activity at 24 weeks without evidence of attenuation of drug efficacy, having a 9.3% gain in total analyzable spine BMD at the week 24 assessment.
Extended 24 weeks of treatment
Patients who completed the initial 24 weeks of treatment and continued to meet eligibility criteria were offered participation in the 24-week extension study in which they would continue their assigned treatment. On completion of the regulatory process to approve the study extension, 69 patients remained eligible and 55 participated, including 13, 10, 7, 11 and 14 patients in the BA058 Injection 20 µg, 40 µg, 80 µg, placebo and Forteo® groups, respectively. Forty-eight patients completed the extended treatment period.
BMD continued to increase during the extended 24 weeks of treatment, with the largest percent increases in total analyzable spine BMD, femoral neck BMD and total analyzable hip BMD observed in the BA058 Injection 80 µg group. By week 48, mean percent changes in spine BMD were 0.7%, 5.1%, 9.8% and 12.9% for the placebo, BA058 20 µg, BA058 40 µg and BA058 80 µg, groups, respectively, while mean percent change from baseline in the Forteo® group was 8.6%. At week 48, the mean femoral neck BMD in the BA058 Injection 80 µg group gained 4.1% compared to the mean of the Forteo® group at 2.2%. The respective results for total analyzable hip BMD were 0.7%, 2.2%, 2.1% and 2.7% for the placebo, BA058 20 µg, BA058 40 µg and BA058 80 µg groups, respectively; compared to 1.3% for the Forteo® group.
No treatment-related SAEs or deaths were reported during this time period. Two patients discontinued treatment, one for bilateral femoral hernias (BA058 Injection 80 µg) and one for moderate syncope (BA058 Injection 40 µg). TEAEs occurred in a similar proportion of patients in each treatment group across the 52-week study period and the majority of events were mild or moderate in severity. The profile of events was not different in the second 6 months of study treatment.
Local tolerance of study drug injections was also similar in the second 6 months of treatment. There were no safety signals observed in the evaluation of clinical laboratory parameters.
In conclusion, this study demonstrated that treatment with BA058 Injection induces a substantial positive change in BMD at both spine and hip in women with osteoporosis and achieves this benefit safely and with substantially less hypercalcemia effect than Forteo®.
BA058 Injection Phase 1 Trials
The First Phase 1 Trial
The first Phase 1 clinical trial was a single-dose study conducted as a randomized, double-blind, placebo-controlled, parallel-group dose escalation study of BA058 Injection in a vial formulation administered as a single SC dose to healthy male and female subjects (mean age: 61 years). The study administered single SC doses of 2, 5, 7.5, 10, 15, 20, 40, 60, 80, and 100 µg BA058 Injection or placebo. Sixteen subjects also received 2.5 µg BA058 Injection by the intravenous (IV) route and 15 µg SC in separate study periods. In total, 76 subjects received BA058 while 20 received a placebo. No elevation in serum calcium was observed at doses of 80 µg or lower and no clinically relevant effects of BA058 Injection on ECG or Holter monitor readings were observed. In summary, this study demonstrated that BA058 Injection is 100% bioavailable when administered by the SC route. BA058
Injection did not induce hypercalcemia and was well tolerated at doses up to 80 µg SC.
The Second Phase 1 Trial
The second Phase 1 clinical trial was a multi-dose study of BA058 Injection when administered as a single SC injection for 7 days. Thirty-nine healthy postmenopausal women (mean age: 60 years) received BA058 Injection (5, 20, 40 or 80 µg) or placebo administered SC. BA058 was well tolerated at all doses and there were no serious adverse events (AEs) and no discontinuations. All AEs were mild or moderate in intensity and did not appear to be dose-related.
BA058 was rapidly absorbed, reaching mean peak plasma concentration within 1 hour, had a rapid clearance and mean half-life values ranged from 1.05 to 2.59 hours. Following BA058 administration, serum PTH decreased and serum 1,25-dihydroxyvitamin D and serum P1NP rose in an apparent dose-dependent manner. Serum calcium showed a slight rise within the normal range following BA058 administration. Three BA058 and 2 placebo patients had isolated calcium values just above the normal range.
The Third Phase 1 Trial
The third Phase 1 clinical trial was a multi-dose study, with the same design as the Second Phase 1 Trial, but using a liquid prefilled multidose cartridge of BA058 and conducted at doses of 80, 100 and 120 µg. BA058 Injection or placebo was administered daily as a SC dose for 7 days to healthy postmenopausal women. Thirty healthy postmenopausal women (mean age: 61 years) were enrolled and 29 completed treatment.
BA058 Injection was well tolerated at doses of up to 100 µg but not at 120 µg which met criteria for termination of dose escalation. One patient in the 120 µg group was intolerant of study drug and was discontinued. All adverse events were mild or moderate in intensity. No study subject developed serum antibodies to BA058 following the 7 days of exposure. BA058 pharmacokinetics was again characterized by rapid absorption, reaching mean peak plasma concentration within approximately 0.5 hours; mean half-life values ranged from 1.13 to 1.65 hours. Similar responses in serum PTH, 1,25-dihydroxy Vitamin D and serum P1NP were observed. These higher doses of BA058 Injection were not associated with occurrence of hypercalcemia. In summary, BA058 Injection was well tolerated at up to 100 µg QD (or daily) for 7 days.
BA058 Microneedle Patch
First Phase 1 Trial
The objectives of the Phase 1 study were to determine the safety, pharmacokinetics and time course of delivery of BA058 Microneedle Patch in healthy postmenopausal women and to compare the PK profiles of BA058 Microneedle Patch delivered transdermally to BA058 Injection administered subcutaneously.
This study was a randomized, double-blind, placebo-controlled, ascending single-dose study and enrolled 38 healthy postmenopausal women (mean age 57.6). Subjects underwent up to 3 single dose exposures to BA058 Microneedle Patch, Placebo Microneedle Patch or BA058 Injection 80 µg over the course of 3 Study Periods.
Pharmacokinetic Results
BA058 Microneedle Patch was characterized by a rapid absorption and elimination. The Cmax and half-life times were shorter than for BA058 Injection administration.
Safety Results
The BA058 Microneedle Patch was well tolerated. Safety events were similar between the BA058 Microneedle Patch and BA058 Injection, with the majority of AEs being mild (99%) and, of these, most were reactions at the application site. There was no clinically notable difference in laboratory or cardiac safety parameters across doses of BA058 or routes of administration.
In conclusion, this Phase 1 study of the BA058 Microneedle Patch demonstrated that BA058 can safely be delivered by this route of administration.
Second Phase 1 Trial
A second Phase 1 single and multiple (7-day) application study of the BA058 Microneedle Patch is currently being conducted in the United States using an optimized Microneedle Patch system. The study is designed as a safety, dose-ranging and time-course pharmacokinetic and pharmacodynamic study. This Phase 1 study will investigate optimal dose, wear time and application site for transdermal delivery of BA058 using an optimized microneedle array.
The study will use a matrix design and will first establish optimal wear time before exploring the impact of application site in the range of doses chosen for evaluation. The results obtained using the BA058 Microneedle Patch will be referenced to those of BA058 Injection 80 µg.
Preclinical Pharmacology
In pharmacology studies conducted with BA058, the following has been shown:
· BA058 is a potent selective agonist of the human PTHR 1 receptor;
· In models of calcium mobilization, BA058 has significantly less calcium mobilizing activity at higher doses than the native hPTHrP(1-34), and less activity than hPTH(1-34);
· BA058 Injection stimulates the formation of normal, well-organized bone and restores BMD in ovariectomized, osteopenic rats and primates. BA058 exhibited the majority of its effects through the growth of trabecular bone without compromising cortical bone. Similar studies in rats with BA058 Microneedle Patch show comparable restoration of bone;
· BA058 Injection was well tolerated over a wide range of doses in two species, rats and primates, for up to 6 months and 9 months, respectively;
· Safety pharmacology studies demonstrated no respiratory, gastroenterologic, hematologic, renal or CNS effects. Tachycardia and hypotension were observed in dogs following both intravenous and subcutaneous administration, however such effects were not observed in other species;
· The No Observed Adverse Effect Level (NOAEL) was 15, 25 and 25 µg/kg/day in rats in the 4-, 13 and 26-week studies, respectively, and 100, 50 and < 10 µg/kg/day in monkeys in the 4-, 13- and 39-week studies, respectively; and
· Repeat SC dose studies in both rats and cynomolgus monkeys at doses up to 300 and 450 µg/kg/day, respectively, revealed a relatively fast absorption (Tmax from 0.083 to 1.0 hr); peak serum concentration (Cmax) and Area under the Curve (AUC), a measure of drug exposure, increased as the dose increased.
These preclinical studies suggest that compared to hPTH(1-34), BA058 Injection can potentially be used to restore lost BMD with a reduced risk of hypercalcemia and loss of cortical bone.
Planned and Active Preclinical Safety Studies
A two-year subcutaneous injection carcinogenicity study of BA058 in Fischer 344 albino rats is currently on-going and will assess the carcinogenic potential of BA058. The study is being conducted according to the provisions set forth in Guidance ICH-S1A, ICH-S1B, and ICH-S1C(R2), and the design was accepted by FDA on 15 July 2009. This study will evaluate 3 BA058 dose levels, and the doses were selected based upon findings and tolerance in completed long-term rat toxicology studies and the anticipated tolerance over a 2-year dosing period and, furthermore, represents a good exposure multiple over maximum clinical doses. An active comparator arm is also included; a cohort of rats will be dosed with hPTH (1-34), because it is anticipated that osteosarcoma will be observed over time. The active comparator will allow confirmation of the sensitivity of the model. This study will be
conducted in parallel to the Phase 3 clinical program.
Two preclinical bone quality studies will also be conducted, one in ovariectomized (OVX) rats for up to 12 months of daily BA058 subcutaneous injection, the second study in adult OVX monkeys for up to 18 months. The primary objective of these studies is to demonstrate that long-term treatment with BA058 Injection will not lead to deleterious effects on bone quality by determining BA058’s effect on the mass, architecture and strength of bones. These studies will be conducted in parallel to the Phase 3 clinical program and, in both studies, BA058 will be compared to placebo. The 12-month rat study is being performed in OVX skeletally mature Sprague-Dawley rats, an appropriate species for osteoporosis studies as a result of the cancellous bone changes and bone strength changes similarly noted in humans. In this study, a 13-week bone depletion period will occur after ovariectomy/sham surgery and prior to initiation of daily SC injection dosing with vehicle or three different dose levels of BA058.
The 16-month nonhuman primate study is being performed in OVX monkeys, a larger remodeling species whose bone depletion can be induced by estrogen deficiency, as in human menopause. In this study, an approximate 9-month bone depletion period will occur after OVX/sham surgery and prior to initiation of daily SC injection dosing with vehicle or three dose levels of BA058. The specific objectives and measured outcomes of both studies are to investigate the potential safety and efficacy of BA058 on prevention of bone loss. Retention of bone mass, both cortical bone - dominant in long bones, and cancellous bone - dominant in spinal bone, will be assess by BMD. Preservation of cortical and cancellous bone on strength will be determined by biomechanical testing. The mechanisms by which BA058 affects bone will be assessed by evaluation of biomarkers of bone turnover and histomorphometric indices of bone turnover. Pharmacokinetics of BA058 and development of antidrug antibodies will also be evaluated.
Manufacturing
BA058 API is manufactured on a contract basis by Lonza Group Ltd. (Lonza), under Good Manufacturing Practices (GMP) conditions using a solid phase peptide synthesis (SPPS) assembly process, and purification by high pressure liquid chromatography (HPLC). BA058 Injection is supplied to clinicians as a liquid in a multi-dose cartridge for use in a pen delivery device. The multi-dose cartridges are manufactured by VETTER Pharma Fertigung GmbH & Co (Vetter) for Ipsen. Ipsen in turn, is responsible for supplying Radius with quantities of BA058 Injection for use in certain clinical trials. The BA058 Microneedle Patch is manufactured by 3M based on their patented microneedle technology to administer drugs through the skin, as an alternative to subcutaneous injection.
Patents
Composition of matter of BA058 is claimed in issued patents in the United States (US 5,969,095), Europe, Australia, Canada, China, Hong Kong, Israel, South Korea, New Zealand, Poland, Russia, Singapore and Taiwan. These cases have a normal patent expiration date of 2016 absent the possibility of patent term extension. The phase 3 clinical dosage of BA058 by the subcutaneous route for use in treating osteoporosis is covered by US 7,803,770 until 2027 in the United States (absent extensions) and a related case is currently pending in Europe, China, Australia, Canada, Japan, Brazil, Mexico, Singapore, South Korea, India, Israel, New Zealand, Norway, Russia and Ukraine. A priority patent application covering various aspects of the BA058 for microneedle patch application has been filed in 2011 in the United States (US app. # 61/478,466). Any claims that might issue from app. # 61/478,466 will have a normal expiry date no earlier than 2031.
Competition
The development and commercialization of new products to treat osteoporosis and women’s health is highly competitive, and there will be considerable competition from major pharmaceutical, biotechnology and specialty pharmaceutical companies. Many of our competitors have substantially more resources than the Company, including both financial and technical. In addition, many of these companies have more experience than us in preclinical and clinical development, manufacturing, regulatory, and global commercialization. Competition for highly qualified employees is intense.
Potential competitors to Radius, in relation to BA058 include, but are not limited to, Amgen, Merck & Co, Novartis and Lilly and Zosano. Lilly launched Forteo® in December 2002 as the first-to-market anabolic or bone-building agent for the treatment of osteoporosis. Lilly has also announced that it is investigating a transdermal method of delivery of Forteo®. Zosano is also developing a transdermal form of rhPTH(1-34) that would compete with the BA058 Microneedle Patch.
Clinical Development Program for RAD1901
In June 2006, we exclusively licensed the worldwide rights (except Japan) to RAD1901 from Eisai. RAD1901 is a selective estrogen receptor modulator (SERM) being developed by us in an oral formulation as a treatment for vasomotor symptoms or hot flashes.
Background on Vasomotor Symptoms
Hot flashes and night sweats are a common symptom during menopause, and according to the Merck Manual, which can be found on the internet at www.merckmanuals.com/professional/print/sec19/ch260/ch260a.html, up to 85% of women experiencing them during the menopause transition, for a median duration of four years. In 2008, according to U.S. census data, more than 11.5 million women in the United States were in the 45 to 49 age range to enter menopause. In addition, most women receiving systemic therapy for breast cancer suffer hot flashes, often with more severe or prolonged symptoms than women experiencing menopause. These symptoms can disrupt sleep and interfere with quality of life. An estimated two million women undergo menopause every year in the U.S., with a total population of 50 million postmenopausal women.
Historically, hormone replacement therapy (HRT) with estrogen and/or progesterone was considered the most efficacious approach to relieving menopausal symptoms such as hot flashes. However, data from the Women’s Health Initiative (WHI) identified increased risks for malignancy and cardiovascular disease associated with estrogen therapy. Sales of HRT declined substantially after the release of the initial WHI data but have re-established growth, increasing more than 4% annually since 2004. HRT remains the current standard of care for many women suffering from hot flashes; however, due to concerns about the potential long-term risks and contraindications associated with HRT, we believe that there is a significant need for new therapeutic options to treat vasomotor symptoms. Pfizer’s Premarin family remains the market leader for drugs to manage menopausal symptoms with 2010 worldwide sales of $1 billion.
Pharmacologic Characteristics of RAD1901
RAD1901 has been shown to bind to the estrogen receptor alpha (ERa) and to have both estrogen-like and estrogen antagonist effects in different tissues. RAD1901 has also been shown to have both estrogen-like behavioral effects in animals and to reduce vasomotor signs in an animal model of menopausal hot flashes. In bone, RAD1901 protects against castration-induced bone loss while showing no unwanted stimulation of the endometrium. In cell culture, RAD1901 does not stimulate replication of breast cancer cells and antagonizes the stimulating effects of estrogen. Overall, therefore, RAD1901 exhibits a number of properties that would make it a suitable drug candidate for the management of menopausal symptoms, in particular the treatment of vasomotor symptoms.
Phase 1
A Phase 1 safety, pharmacokinetic and bioavailability study was conducted in 80 healthy postmenopausal women over a range of doses of RAD1901, including placebo. After single dosing with RAD1901 by mouth, the mean half-life ranged between 27.4 and 32.5 h. Bioavailability was determined to be approximately 10%. Food effect was also investigated and the presence of food was determined to increase absorption and delay clearance of RAD1901.
RAD1901 was generally well tolerated. All TEAEs were of mild intensity, with some increase in frequency at the higher doses in the multiple dose group, most commonly gastrointestinal symptoms and headache. There were no serious adverse events observed.
Phase 2
A Phase 2 proof of concept study was conducted in 100 healthy postmenopausal women using 4 doses of RAD1901 (10, 25, 50 and 100 mg) and placebo. The primary study outcome was reduction in the frequency and severity of moderate and severe hot flashes. While a classic dose-response effect was not demonstrated, efficacy was determined to occur at the 10 mg dose level which achieved a statistically significant reduction in the frequency of
moderate and severe hot flashes both by linear trend test and by comparison to placebo and in overall (mild-moderate-severe) hot flashes at either the 2-, 3- or 4-week time-points. A similar reduction in composite score (frequency x severity) was identified at all time-points, with a statistically significant difference from placebo achieved at the 2-, 3- or 4-week time-points. Numerical reductions in mean severity and mean daily severity were observed, but did not reach statistical significance.
No serious adverse events were reported during the course of the study. Overall, 69% of patients had an adverse event, generally mild or moderate in severity, with some evidence of dose dependency, and events were most commonly gastrointestinal symptoms and headache. Three severe adverse events occurred, one in a placebo patient, and were not considered treatment related. Two patients discontinued treatment due to an adverse event, neither in relation to the 10 mg dose.
Manufacturing
RAD1901 API is manufactured for Radius on a contract basis by Irix Pharmaceuticals, Inc. The present GMP manufacture of RAD1901 comprises 9 synthetic steps from a non-GMP starting material. The current process of manufacture requires no chromatographic separations. RAD1901 is a chiral material present as essentially one enantiomer.
Patents
RAD1901 as a composition of matter is covered by US patent 7,612,114 (normal expiry 2026 absent Hatch-Waxman extensions). A corresponding case has also been issued in Australia with related cases pending in Canada, India and Europe. A patent application covering methods of using RAD1901 for the treatment of hot flush has been filed in the US (published as US 2010/0105733A1), Europe and Canada and any claims issuing will have a normal expiry of 2027. In addition, a provisional dosage form application has been filed in the United States (US app# 61/334,095) and any claims that might issue from applications claiming priority to US app# 61/334,095 will have a normal expiry date no earlier than 2031.
Competition
Potential competitors to Radius in relation to RAD1901 include, but are not limited to, Pfizer and Depomed.
RAD140
Pharmacologic Characteristics of RAD140
RAD140 is a nonsteroidal, selective androgen receptor modulator that resulted from an internal drug discovery program that began in 2005. RAD140 has demonstrated potent anabolic activity on muscle and bone in preclinical studies and has completed 28-day preclinical toxicology studies in both rats and monkeys. Because of its high anabolic efficacy, receptor selectivity, potent oral activity and long duration half life, it is believed that RAD140 has clinical potential in a number of indications where the increase in lean muscle mass and/or bone density is beneficial such as treating the weight loss due to cancer cachexia, muscle frailty and osteoporosis.
Patents
RAD140 as a composition of matter and methods of using RAD140 is covered by pending patent applications in the US (e.g. US app#12/378,812)) and numerous additional countries worldwide. Any patents issued from these filings will have a normal expiry of 2029 absent any extensions.
Competition
Potential competitors to Radius in relation to RAD140 include, but are not limited to, GTx and Ligand.
Collaborations and License Agreements
Nordic Bioscience
We entered into a Letter of Intent with Nordic on September 3, 2010, pursuant to which we funded preparatory work by Nordic in respect of a Phase 3 clinical study of BA058 Injection. The Letter of Intent was extended on December 15, 2010 and on January 31, 2011. Pursuant to the Letter of Intent and the two extensions, we funded an aggregate $1,500,000 of preparatory work by Nordic during 2010 and funded and additional $750,000 of preparatory work by Nordic during 2011. On March 29, 2011, the Company and Nordic entered into a Clinical
Trial Services Agreement, a Work Statement NB-1 under such Clinical Trial Services Agreement and a related Stock Issuance Agreement. Pursuant to Work Statement NB-1, Nordic is managing the Phase 3 clinical study of BA058 Injection. Pursuant to the Stock Issuance Agreement, Nordic agreed to purchase the equivalent of €371,864 of our Series A-5 Convertible Preferred Stock at a price per share equal to $8.142. Nordic purchased 64,430 shares of Series A-5 Preferred Stock on May 17, 2011 for proceeds of $525,153.53 to the Company. The Stock Issuance Agreement provides that Nordic will receive additional shares of equity, which shall initially be the Series A-6 Convertible Preferred Stock, at certain times during the performance of the Phase 3 clinical study that is the subject of Work Statement NB-1.
3M
In December 2008, we entered into a Feasibility Agreement with 3M whereby 3M assessed the feasibility of developing a BA058 microneedle patch product and supplying the product for preclinical studies in an animal model. Upon completion of the feasibility study, during June 2009, we entered into a Development and Clinical Supplies Agreement with 3M under which 3M is responsible to develop a BA058 microneedle patch product and manufacture clinical and toxicology supplies of such patch product for preclinical, Phase 1 and Phase 2 studies on an exclusive basis. Radius pays 3M for services delivered pursuant to the Development and Clinical Supplies Agreement on a fee for service or a fee for deliverable basis as specified in the Development and Clinical Supplies Agreement. The Feasibility Agreement expired on or around September 2009, Radius has paid 3M approximately $4,003,000 in respect of services and deliverables delivered pursuant to the Feasibility Agreement and the Development and Clinical Supplies Agreement .
Ipsen Pharma
In September 2005, we entered into a License Agreement with Ipsen under which we exclusively licensed certain Ipsen compound technology and related patents covering BA058 to research, develop, manufacture and commercialize certain compounds and related products in all countries, except Japan (subject to certain co-marketing and co-promotion rights retained by Ipsen in France). Ipsen also granted us an exclusive right and license under the Ipsen compound technology and related patents to make and have made compounds or product in Japan. Ipsen also granted the Company an exclusive right and license under certain Ipsen formulation technology and related patents solely for purposes of enabling us to develop, manufacture and commercialize compounds and products covered by the compound technology license in all countries, except Japan and France. With respect to France, Ipsen retains co-marketing and co-promotion rights; if Ipsen exercises these rights then Ipsen may elect to receive a percentage of the aggregate revenue from the sale of products by both parties in France (subject to a cap) and Ipsen shall bear a corresponding percentage of the costs and expenses incurred by both parties with respect to such marketing and promotion efforts in France; Ipsen shall also pay Radius a royalty on Ipsen’s allocable portion of aggregate revenue from the sale of products by both parties in France. Specifically, we licensed US Patent No. 5,969,095, effective filing date (3/29/1996) entitled “Analogs of Parathyroid Hormone”, US Patent No. 6,544,949, effective filing date (3/29/1996) entitled “Analogs of Parathyroid Hormone” and the corresponding foreign patents and continuing patent applications. In addition, we have has rights to joint Ipsen/Company intellectual property including rights to US7803770, effective filing date (10/3/2007) and related patent applications both in the United States and worldwide (excluding Japan) that cover the method of treating osteoporosis using the phase 3 clinical dosage strength and form. As consideration for the rights to BA058 licensed to it by Ipsen, we paid Ipsen an initial license fee of $250,000. The license agreement requires us to make payments to Ipsen upon the achievement of certain development, regulatory and commercial milestones and we have at this time paid $750,000 in milestone payments and issued 17,326 shares of Series A-1 convertible preferred stock to Ipsen on May 17, 2011 in lieu of a cash payment due to Ipsen upon initiation of the first BA058 Phase 3 clinical study. If we commercialize a product that includes the compound licensed from Ipsen or any analog thereof, we will be obligated to pay to Ipsen fixed mid-single digit royalties based on net sales of the product on a country-by-country basis until the later of the last to expire of the licensed patents or ten years from the first commercial sale in such country. In the event that we sublicense the rights licensed from Ipsen to a third party, the Company is obligated to pay Ipsen a percentage of certain payments received from such sublicensee (in lieu of milestone payments not achieved at the time of such sublicense). The license agreement contains other customary clauses and terms as are common in similar agreements in the industry. The license agreement was amended on September 12, 2007 and May 11, 2011.
In January 2006, Radius and Ipsen, entered into a Pharmaceutical Development Agreement as contemplated by the License Agreement with Ipsen. The Pharmaceutical Development Agreement provides for the supply of quantities of licensed product for use in certain clinical trials. Beaufour Ipsen Industrie S.A.S is currently responsible for the supply of BA058 Injection in liquid form in a multi-dose cartridge for use in a pen delivery device. The multi-dose cartridges are manufactured for Beaufour Ipsen Industrie S.A.S by Vetter and the BA058 API manufactured by Lonza for Radius is delivered to Vetter for vialing. The Pharmaceutical Development Agreement was amended in July 2007, February 2009 and June 2010.
Eisai
In June 2006, we exclusively licensed the worldwide (except Japan) rights to research, develop, manufacture and commercialize RAD1901 and related products from Eisai. Specifically, we licensed the patent application that subsequently issued as US Patent No. 7612114, effective filing date (12/25/2003) entitled “Selective Estrogen Receptor Modulator”, the corresponding foreign patent applications and continuing patent applications. As consideration for the rights to RAD1901, we paid Eisai an initial license fee of $500,000. In connection with the License Agreement, we have agreed to pay Eisai certain fees payable upon the achievement of certain clinical and regulatory milestones. Should a product covered by the licensed technology be commercialized, we will be obligated to pay to Eisai royalties in a variable mid-single digit range based on net sales of the product on a country-by-country basis until the later of the last to expire of the licensed patents or the expiration of data protection clauses covering such product in such country; the royalty rate shall then be subject to reduction and the royalty obligation will expire at such time as sales of lawful generic version of such product account for more than a specified minimum percentage of the total sales of all products that contain the licensed compound. The Company also was granted the right to sublicense with prior written approval from Eisai, but subject to a right of first negotiation held by Eisai if we seek
to grant sublicenses limited to particular Asian countries. If we sublicense the licensed technology to a third party, we will be obligated to pay Eisai, in addition to the milestones referenced above, a percentage of certain fees it receives from such sublicensee and royalties based on net sales of the sublicense. The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Lonza
In October 2007, we entered into Development and Manufacturing Services Agreement with LONZA. Radius and Lonza have entered into a series of Work Orders pursuant to the Development and Manufacturing Services Agreement pursuant to which Lonza has performed pharmaceutical development and manufacturing services for the our BA058 product. Radius pays Lonza for services rendered and deliverables delivered pursuant to these work orders on a fee for service basis as specified in the applicable work statement.
Charles River Laboratories
In March 2004, we entered into a Laboratory Services and Confidentiality Agreement with Charles River Laboratories, Inc. (CRLI) and amended this agreement on November 7, 2008. Radius has entered into a series of letter agreements with CRLI pursuant to this Laboratory Services and Confidentiality Agreement, covering the performance of certain testing and analytical services concerning our product candidates. Radius pays CRLI for services rendered and deliverables delivered pursuant to these letter agreements on a fee for service basis as specified in the applicable letter agreement.
Copies of all of the foregoing material agreements are filed as exhibits hereto.
Government Regulation
U.S. —FDA Process The research, development, testing, manufacture, labeling, promotion, advertising, distribution, and marketing, among other things, of our products are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the “FDCA,” and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending New Drug Applications, or NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, and/or criminal prosecution. We expect that BA058, RAD1901 and RAD140 will each be subject to review by the FDA as a drug under NDA standards though we currently only have an active IND in relation to BA058 in the United States.
Drug Approval Process. None of our drugs may be marketed in the U.S. until the drug has received FDA approval. The steps required before a drug may be marketed in the U.S. include:
· preclinical laboratory tests, animal studies, and formulation studies;
· submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin;
· adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each indication;
· submission to the FDA of an NDA;
· satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practices, or “cGMPs”; and
· FDA review and approval of the NDA.
Preclinical tests include laboratory evaluation of product chemistry, toxicity, and formulation, as well as animal studies. The conduct of the preclinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in the IND. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. The Company cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND.
Clinical trials necessary for product approval are typically conducted in three sequential Phases, but the Phases may overlap. The study protocol and informed consent information for study subjects in clinical trials must also be approved by an Institutional Review Board for each institution where the trials will be conducted. Study subjects must sign an informed consent form before participating in a clinical trial. Phase 1 usually involves the initial introduction of the investigational drug into people to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness. Phase 2 usually involves trials in a limited patient population to (i) evaluate dosage tolerance and appropriate dosage; (ii) identify possible adverse effects and safety risks; and (iii) evaluate preliminarily the efficacy of the drug for specific indications. Phase 3 trials usually further evaluate clinical efficacy and test further for safety by using the drug in its final form in an expanded patient population. There can be no assurance that Phase 1, Phase 2, or Phase 3 testing will be completed successfully within any specified period of time, if at all. Furthermore, the Company or the FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
The FDCA permits FDA and the IND sponsor to agree in writing on the design and size of clinical studies intended to form the primary basis of an effectiveness claim in a New Drug Application (NDA). This process is known as Special Protocol Assessment or SPA. These agreements may not be changed after the clinical studies begin, except in limited circumstances.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. The testing and approval process requires substantial time, effort, and financial resources. The agencies review the application and may deem it to be inadequate to support the registration, and companies cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee.
The FDA has various programs, including fast track, priority review, and accelerated approval, that are intended to expedite or simplify the process for reviewing drugs, and/or provide for approval on the basis surrogate endpoints. Generally, drugs that may be eligible for one or more of these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that provide meaningful benefit over existing treatments. A company cannot be sure that any of its drugs will qualify for any of these programs, or that, if a drug does qualify, that the review time will be reduced.
Before approving an NDA, the FDA usually will inspect the facility or the facilities at which the drug is manufactured and will not approve the product unless the manufacturing is in compliance with current good manufacturing practice (cGMP). If the NDA and the manufacturing facilities are deemed acceptable by the Agency, the FDA may issue an approval letter, or in some cases, an approvable letter followed by an approval letter. Both letters usually contain a number of conditions that must be met in order to secure final approval of the NDA. When and if those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter. The approval letter authorizes commercial marketing of the drug for specific indications. As a condition of NDA approval, the FDA may require post-marketing testing and surveillance to monitor the drug’s safety or efficacy, or impose other conditions.
After approval, certain changes to the approved product, such as adding new indications, making certain manufacturing changes, or making certain additional labeling claims, are subject to further FDA review and approval. Before a company can market products for additional indications, it must obtain additional approvals from FDA. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. A company cannot be sure that any additional approval for new indications for any product candidate will be approved
on a timely basis, or at all.
Post-Approval Requirements. Often times, even after a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. If such post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA are required to: (i) report certain adverse reactions to the FDA, (ii) comply with certain requirements concerning advertising and promotional labeling for their products, and (iii) continue to have quality control and manufacturing procedures conform to cGMP after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of ongoing compliance with cGMP. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. We intend to use third party manufacturers to produce our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including withdrawal of the product from the market.
Hatch-Waxman Act: Under the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, Congress created an abbreviated FDA review process for generic versions of pioneer (brand name) drug products. In considering whether to approve such a generic drug product, the FDA requires that an Abbreviated New Drug Application, or ANDA, applicant demonstrate, among other things, that the proposed generic drug product’s active ingredient is the same as that of the reference product, that any impurities in the proposed product do not affect the product’s safety or effectiveness, and that its manufacturing processes and methods ensure the consistent potency and purity of its proposed product.
The Hatch-Waxman Act also provides three years of exclusivity for applications containing the results of new clinical investigations (other than bioavailability studies) essential to the FDA’s approval of new uses of approved products, such as new indications, dosage forms, strengths, or conditions of use. However, this exclusivity only protects against the approval of ANDAs and 505(b)(2) applications for the protected use and will not prohibit the FDA from accepting or approving ANDAs or 505(b)(2) applications for other products containing the same active ingredient. The Hatch-Waxman Act requires NDA applicants and NDA holders to provide certain information about patents related to the drug for listing in the FDA’s list of Approved Drug Products with Therapeutic Equivalence Evaluations (commonly known as the Orange Book). ANDA and 505(b)(2) applicants must then certify regarding each of the patents listed with the FDA for the reference product. A certification that a listed patent is invalid or will not be infringed by the marketing of the applicant’s product is called a “Paragraph IV certification.” If the ANDA or 505(b)(2) applicant provides such a notification of patent invalidity or noninfringement, then the FDA may accept the ANDA or 505(b)(2) application beginning four years after approval of the NDA. If an ANDA or 505(b)(2) application containing a Paragraph IV certification is submitted to the FDA and accepted as a reviewable filing by the agency, the ANDA or 505(b)(2) applicant then must provide, within 20 days, notice to the NDA holder and patent owner stating that the application has been submitted and providing the factual and legal basis for the applicant’s opinion that the patent is invalid or not infringed. The NDA holder or patent owner then may file suit against the ANDA or 505(b)(2) applicant for patent infringement. If this is done within 45 days of receiving notice of the Paragraph IV certification, a one-time 30-month stay of the FDA’s ability to approve the ANDA or 505(b)(2) application is triggered. The 30-month stay begins at the end of the NDA holder’s data exclusivity period, or, if data exclusivity has expired, on the date that the patent holder is notified of the submission of the ANDA. The FDA may approve the proposed product before the expiration of the 30-month stay if a court finds the patent invalid or not infringed or if the court shortens the period because the parties have failed to cooperate in expediting the litigation.
If we ever commercialize a product, the provisions of Hatch-Waxman may become significant to us.
EU—EMA Process
In the European Union, or the EU, medicinal products are authorized following a similar demanding process as that required in the U.S. Applications are based on the ICH Common Technical Document and must include a detailed plan for pediatric approval, if such approval is sought. Medicinal products must be authorized in one of two ways, either through the decentralized procedure or mutual recognition procedure by the competent authorities of the EU Member States, or through the centralized procedure by the European Commission following an opinion by the European Medicines Agency, or EMA. The authorization process is essentially the same irrespective of which route is used. In light of the fact that there is no policy at the EU level governing pricing and reimbursement, the 27 EU Member States each have developed their own, often varying, approaches. In many EU Member States, pricing negotiations must take place between the Marketing Authorization Holder and the competent national authorities before the product is sold in their market with Marketing Authorization Holders required to provide evidence demonstrating the pharmaco-economic superiority of their product in comparison with directly and indirectly competing products. We have reviewed our development program, proposed Phase 3 study design, and overall non-clinical and clinical data package to support future regulatory approval of the BA058 Injection with EMA but have not initiated any discussions with EMA with respect to seeking regulatory approval of our other products in Europe.
Good manufacturing practices. Like the FDA, the EMA, the competent authorities of the EU Member States and other regulatory agencies regulate and inspect equipment, facilities, and processes used in the manufacturing of pharmaceutical and biologic products prior to approving a product. If, after receiving clearance from regulatory agencies, a company makes a material change in manufacturing equipment, location, or process, additional regulatory review and approval may be required. Once we or our partners commercialize products, we will be required to comply with cGMP, and product-specific regulations enforced by, the European Commission, the EMA and the competent authorities of EU Member States following product approval. Also like the FDA, the EMA, the competent authorities of the EU Member States and other regulatory agencies also conduct regular, periodic visits to re-inspect equipment, facilities, and processes following the initial approval of a product. If, as a result of these inspections, it is determined that our or our partners’ equipment, facilities, or processes do not comply with applicable regulations and conditions of product approval, regulatory agencies may seek civil, criminal, or administrative sanctions and/or remedies against us, including the suspension of our manufacturing operations or the withdrawal of our product from the market.
Other International Markets—Drug approval process
In some international markets (e.g., China or Japan), although data generated in U.S. or EU trials may be submitted in support of a marketing authorization application, additional clinical trials conducted in the host territory, or studying people of the ethnicity of the host territory, may be required prior to the filing or approval of marketing applications within the country.
Pricing and Reimbursement
In the U.S. and internationally, sales of products that we market in the future, and our ability to generate revenues on such sales, are dependent, in significant part, on the availability and level of reimbursement from third-party payors such as state and federal governments, managed care providers, and private insurance plans. Private insurers, such as health maintenance organizations and managed care providers, have implemented cost-cutting and reimbursement initiatives and likely will continue to do so in the future. These include establishing formularies that govern the drugs and biologics that will be offered and also the out-of-pocket obligations of member patients for such products. In addition, particularly in the U.S. and increasingly in other countries, we are required to provide discounts and pay rebates to state and federal governments and agencies in connection with purchases of our products that are reimbursed by such entities. It is possible that future legislation in the U.S. and other jurisdictions could be enacted which could potentially impact the reimbursement rates for the products we are developing and may develop in the future and also could further impact the levels of discounts and rebates paid to federal and state government entities. Any legislation that impacts these areas could impact, in a significant way, our ability to generate revenues from sales of products that, if successfully developed, we bring to market.
There is no legislation at the EU level governing the pricing and reimbursement of medicinal products in the EU. As a result, the competent authorities of each of the 27 EU Member States have adopted individual strategies regulating the pricing and reimbursement of medicinal products in their territory. These strategies often vary widely in nature, scope and application. However, a major element that they have in common is an increased move towards reduction in the reimbursement price of medicinal products, a reduction in the number and type of products selected for reimbursement and an increased preference for generic products over innovative products. These efforts have mostly been executed through these countries’ existing price-control methodologies. The government of the UK, while continuing for now to utilize its established Pharmaceutical Pricing Reimbursement Scheme approach, has announced its intentions to phasing in, by 2014, a new value-based pricing approach, at least for new product introductions. Under this approach, in a complete departure from established methodologies, reimbursement levels of each drug will be explicitly based on an assessment of value, looking at the benefits for the patient, unmet need, therapeutic innovation, and benefit to society as a whole. It is increasingly common in many EU Member States for Marketing Authorization Holders to be required to demonstrate the pharmaco-economic superiority of their products as compared to products already subject to pricing and reimbursement in specific countries. In order for drugs to be evaluated positively under such criteria, pharmaceutical companies may need to re-examine, and consider altering, a number of traditional functions relating to the selection, study, and management of drugs, whether currently marketed, under development, or being evaluated as candidates for research and/or development.
Future legislation, including the current versions being considered at the federal level in the U.S. and at the national level in EU Member States, or regulatory actions implementing recent or future legislation may have a significant effect on our business. Our ability to successfully commercialize products depends in part on the extent to which reimbursement for the costs of our products and related treatments will be available in the U.S. and worldwide from government health administration authorities, private health insurers and other organizations. Substantial uncertainty exists as to the reimbursement status of newly approved health care products by third-party payors.
Sales and Marketing
The FDA regulates all advertising and promotion activities for products under its jurisdiction both prior to and after approval. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, and often reflect a physician’s belief that the off-label use is the best treatment for the patients. The FDA does not regulate the behavior of physicians in their choice of treatments, but FDA regulations do impose stringent restrictions on manufacturers’ communications regarding off-label uses. Failure to comply with applicable FDA requirements may subject a company to adverse publicity, enforcement action by the FDA, corrective advertising, consent decrees and the full range of civil and criminal penalties available to the FDA.
We may also be subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws make it illegal for a prescription drug manufacturer to solicit, offer, receive, or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Due to the breadth of the statutory provisions and the absence of guidance in the form of regulations and very few court decisions addressing industry practices, it is possible that our practices might be challenged under anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third-party payors (including Medicare and Medicaid) claims for reimbursed drugs or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal health care programs (including Medicare and Medicaid) and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties also can be imposed upon executive officers and employees, including criminal sanctions against executive officers under the so-called “responsible corporate officer” doctrine, even in situations where the executive officer did not intend to violate the law and was unaware of any wrongdoing. Given
the remedies that can be imposed on companies and individuals if convicted, allegations of such violations often result in settlements even if the company or individual being investigated admits no wrongdoing. Settlements often include significant civil sanctions, including fines and civil monetary penalties, and corporate integrity agreements. If the government were to allege or convict us or our executive officers of violating these laws, our business could be harmed. In addition, private individuals have the ability to bring similar actions. Our activities could be subject to challenge for the reasons discussed above and due to the broad scope of these laws and the increasing attention being given to them by law enforcement authorities. Further, there are an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state authorities.
Similar rigid restrictions are imposed on the promotion and marketing of medicinal products in the EU and other countries. Laws (including those governing promotion, marketing and anti-kickback provisions), industry regulations and professional codes of conduct often are strictly enforced. Even in those countries where we are not directly responsible for the promotion and marketing of our products, inappropriate activity by our international distribution partners can have implications for us.
Other Laws and Regulatory Processes
We are subject to a variety of financial disclosure and securities trading regulations as a public company in the U.S., including laws relating to the oversight activities of the Securities and Exchange Commission, or SEC, and, if any or our capital stock becomes listed on a national securities exchange, we will be subject to the regulations of such exchange on which our shares are traded. In addition, the Financial Accounting Standards Board, or FASB, the SEC, and other bodies that have jurisdiction over the form and content of our accounts, our financial statements and other public disclosure are constantly discussing and interpreting proposals and existing pronouncements designed to ensure that companies best display relevant and transparent information relating to their respective businesses.
Our international operations are subject to compliance with the Foreign Corrupt Practices Act, or the FCPA, which prohibits corporations and individuals from paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. We also may be implicated under the FCPA for activities by our partners, collaborators, contract research organizations, or CROs, vendors or other agents.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import and export and use and disposal of hazardous or potentially hazardous substances used in connection with our research work are or may be applicable to our activities. Certain agreements entered into by us involving exclusive license rights or acquisitions may be subject to national or supranational antitrust regulatory control, the effect of which cannot be predicted. The extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
Intellectual Property
As of July 20, 2011, we owned 1 issued U.S. patent, as well as 10 pending U.S. patent applications and 28 pending foreign patent applications in Europe and 16 other jurisdictions. As of July 20, 2011, we had licenses to 9 U.S. patents 1 U.S. patent applications, as well as numerous foreign counterparts to many of these patents and patent applications. We licensed these patents and patent applications on an exclusive basis for all countries except Japan though our rights in France with respect to BA058 are subject to certain co-promotion and co-marketing rights held by Ipsen and our rights to sublicense in certain Asia Pacific countries in respect of RAD1901 are subject to a right of first refusal held by Esai, all as described herein in our discussion of our license agreements with Ipsen and Esai.
Employees
As of the date of this Report, we employed 7 full-time employees and 3 part-time employees, 4 of whom held Ph.D. or M.D. degrees. Four of our employees were engaged in research and development activities and six were engaged in support administration, including business development, and finance. In addition, we intend to use clinical research organizations and third parties to perform our clinical studies and manufacturing.
Properties
Our executive offices are located at 201 Broadway, 6th Floor, Cambridge, MA 02139. Our telephone number is (617) 551-4700. The office space is approximately 5,700 square feet, and the lease expires on July 30, 2011.
Legal Proceedings
We are not currently involved in any material legal proceedings.
RISK FACTORS
In addition to the other information set forth in this Current Report on Form 8-K, you should carefully consider the factors discussed below when considering an investment in our capital stock. These are risks and uncertainties that could cause actual results to differ materially from the results contemplated by the forward-looking statements contained in this Current Report on Form 8-K. Because of the following factors, as well as other variables affecting our operating results, past financial performance should not be considered as a reliable indicator of future performance and investors should not use historical trends to anticipate results or trends in future periods. These risks are not the only ones facing the Company. Please also see “CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS” on page 35 of this Current Report on Form 8-K.
Risks Related to Owning our Capital Stock
There is not now and never has been any market for our securities and an active market may never develop. You may therefore be unable to re-sell shares of our securities at times and prices that you believe are appropriate. There is no market - active or otherwise - for our Common Stock or our Preferred Stock and neither is eligible for listing or quotation on any securities exchange, automated quotation system (e.g., Nasdaq) or any other over-the-counter market, such as the OTC Bulletin Board® (the “OTCBB”) or the Pink Sheets® (the “Pink Sheets”). Even if we are successful in obtaining approval to have our Common stock quoted on the OTCBB, it is unlikely that an active market for our Common Stock will develop any time soon thereafter. Accordingly, our Common Stock is highly illiquid. Because of this illiquidity, you will likely experience difficulty in re-selling such shares at times and prices that you may desire.
There is no assurance that our Common Stock will be listed on NASDAQ or any other securities exchange. We plan to seek listing of our Common Stock on Nasdaq or another national securities exchange or listed for quotation on the OTCBB, as soon as practicable. However, there is no assurance we will be able to meet the initial listing standards of either of those or any other stock exchange or automated quotation systems, or that we will be able to maintain a listing of our Common Stock on either of those or any other stock exchange or automated quotation system. We anticipate seeking a listing of our Common Stock on the OTCBB, the Pink Sheets or another over-the-counter quotation system, before our Common Stock is listed on the Nasdaq or a national securities exchange. An investor may find it more difficult to dispose of shares or obtain accurate quotations as to the market value of our Common Stock while our Common Stock is listed on the OTCBB. If our Common Stock is listed on the OTCBB, we would be subject to an SEC rule that, if it failed to meet the criteria set forth in such rule, imposes various practice requirements on broker-dealers who sell securities governed by the rule to persons other than established customers and accredited investors. Consequently, such rule may deter broker-dealers from recommending or selling our Common Stock, which may further limit its liquidity. This would also make it more difficult for us to raise additional capital.
Shares of our Capital Stock issued in the Merger will not be freely tradable under Securities Laws which will limit stockholders’ ability to sell such shares of our Capital Stock. Shares of our Preferred Stock and our Common Stock to be issued as consideration in the Merger pursuant the Merger Agreement will be deemed “Restricted Securities” under the federal securities laws, and consequently such shares may not be resold without registration under the Securities Act of 1933, as amended (the “Securities Act”), or without an exemption from the Securities Act. Further, Rule 144 covering resales of unregistered securities and promulgated under the Securities Act will not be available for resale of our capital stock unless or until one year following the date on which we file the information required by Form 10 as to the our business. In addition, all shares of our Preferred Stock issued in the Merger will be subject to a lock-up provision set forth in the applicable stockholders’ agreement. Each certificate evidencing shares of our capital stock to be issued pursuant to the Merger Agreement will bear a restrictive legend as to the nature of the restrictions on the transfer of such shares.
Because we became an operating company by means of a reverse merger, we may not be able to attract the attention of major brokerage firms. Additional risks may exist as a result of our becoming a public reporting operating company through a “reverse merger.” Security analysts of major brokerage firms may not provide coverage of our capital stock or business. Because we became a public reporting operating company through a reverse merger, there is no incentive to brokerage firms to recommend the purchase of our Common Stock. No
assurance can be given that brokerage firms will want to provide analyst coverage of our capital stock or business in the future.
The resale of shares covered by a registration statement could adversely affect the market price of our Common Stock in the public market, should one develop, which result would in turn negatively affect our ability to raise additional equity capital. The sale, or availability for sale, of our Common Stock in the public market pursuant to a registration statement may adversely affect the prevailing market price of our Common Stock and may impair our ability to raise additional capital by selling equity or equity-linked securities. Once effective, a registration statement will register the resale of a significant number of shares of our Common Stock. The resale of a substantial number of shares of our Common Stock in the public market could adversely affect the market price for our Common Stock and make it more difficult for you to sell shares of our Common Stock at times and prices that you feel are appropriate. Furthermore, we expect that, because there will be a large number of shares registered pursuant to a registration statement, selling stockholders will continue to offer shares covered by such registration statement for a significant period of time, the precise duration of which cannot be predicted. Accordingly, the adverse market and price pressures resulting from an offering pursuant to a registration statement may continue for an extended period of time and continued negative pressure on the market price of our Common Stock could have a material adverse effect on our ability to raise additional equity capital.
We are or will be subject to Sarbanes-Oxley and the reporting requirements of federal securities laws, which can be expensive. As a public reporting company, we are subject to the Sarbanes-Oxley Act of 2002, as well as the information and reporting requirements of the Securities Exchange Act of 1934, as amended, and other federal securities laws. The costs of compliance with the Sarbanes-Oxley Act and of preparing and filing annual and quarterly reports, proxy statements and other information with the SEC, and furnishing audited reports to stockholders, will cause our expenses to be higher than they would be if we were privately held.
For so long as shares of our Preferred Stock remain outstanding, if we are sold in a transaction yielding less than the liquidation preference payable in the aggregate to holders of our outstanding Preferred Stock, holders of our Common Stock may not receive any proceeds from such transaction and may lose their investment entirely. As of May 20, 2011, we have 555,594 shares of Common Stock; 413,254 shares of Series A-1 Stock; 983,208 shares of Series A-2 Stock; 142,227 shares of Series A-3 Stock; 3,998 shares of Series A-4 Stock; 6,443 shares of Series A-5 Stock; and assumed warrants to acquire 818 shares of Series A-1 Stock. As more fully described herein and in our certificate of incorporation, shares of our Preferred Stock outstanding at the time of a sale or liquidation of the Company will have a right to receive proceeds, if any, from any such transactions, before any payments are made to holders of our Common Stock. In the event that there are not enough proceeds to satisfy the entire liquidation preference of our Preferred Stock, holders of our Common Stock will receive nothing in respect of their equity holdings in the Company.
Risks Related to our Business
We currently have no product revenues and will need to raise additional capital to operate our business. To date, we have generated no product revenues. Until, and unless, we receive approval from the U.S. Food and Drug Administration, or FDA, and other regulatory authorities for its product candidates, we cannot sell our drugs and will not have product revenues. Currently, our only product candidates are BA058, RAD1901, and RAD140, and none of these products is approved by the FDA for sale. Therefore, for the foreseeable future, we will have to fund our operations and capital expenditures from cash on hand, licensing fees and grants and potentially, future offerings of our common or preferred stock. Currently, we believe that our cash on hand, which includes the $20.4 million in net proceeds received on May 17, 2011 from the first closing of the Series A-1 Financing, plus the proceeds of the two subsequent closings of the Series A-1 Financing which are available to us with no closing or other conditions, are sufficient to fund our operations through June 2012. However, changes may occur that would consume our available capital before that time, including changes in and progress of our development activities, acquisitions of additional candidates and changes in regulation.
We will need to seek additional sources of financing, which may not be available on favorable terms, if at all. Notwithstanding the expected completion of the subsequent two closings of the Series A-1 Financing, if we do not succeed in timely raising additional funds on acceptable terms, we may be unable to complete planned pre-
clinical and clinical trials or obtain approval of any product candidates from the FDA and other regulatory authorities. In addition, we could be forced to discontinue product development, reduce or forego sales and marketing efforts and forego attractive business opportunities. Any additional sources of financing will likely involve the issuance of additional equity securities, which will have a dilutive effect on stockholders.
We are not currently profitable and may never become profitable. We have a history of net losses and expect to incur substantial losses and negative operating cash flow for the foreseeable future, and may never achieve or maintain profitability. For the years ended December 31, 2010 and 2009, we had a net loss of $14.6 million and $15.1 million, respectively. As of March 31, 2011 we had an accumulated deficit of approximately $136.1 from the operations of target. Even if we succeed in developing and commercializing one or more product candidates, we expect to incur substantial losses for the foreseeable future and may never become profitable. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future as we:
· continue to undertake pre-clinical development and clinical trials for product candidates;
· seek regulatory approvals for product candidates;
· implement additional internal systems and infrastructure; and
· hire additional personnel.
We also expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability could negatively impact the value of our securities.
We have a limited operating history upon which to base an investment decision. We are a development-stage company and have not demonstrated an ability to perform the functions necessary for the successful commercialization of any product candidates. The successful commercialization of any product candidates will require us to perform a variety of functions, including:
· continuing to undertake pre-clinical development and clinical trials;
· participating in regulatory approval processes;
· formulating and manufacturing products; and
· conducting sales and marketing activities.
Our operations have been limited to organizing and staffing our company, acquiring, developing and securing its proprietary technology and undertaking pre-clinical and clinical trials of our product candidates. These operations provide a limited basis for you to assess our ability to commercialize our product candidates and the advisability of investing further in our securities.
We are heavily dependent on the success of the BA058 Injection, which is still under clinical development. We cannot be certain that BA058 Injection will receive regulatory approval or be successfully commercialized even if we receive regulatory approval. BA058 Injection is our only product candidate in late stage development, and our business currently depends heavily on its successful development, regulatory approval and commercialization. We have no drug products for sale currently and may never be able to develop marketable drug products. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, which regulations differ from country to country. We are not permitted to market BA058 Injection in the United States until it receives approval of a New Drug Application or NDA from the FDA, or in any foreign countries until it receives the requisite approval from such countries. In addition, the approval of BA058
Microneedle Patch as a follow-on product is dependent on an earlier approval of BA058 Injection. We have not submitted an NDA to the FDA or comparable applications to other regulatory authorities. Obtaining approval of an NDA is an extensive, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of BA058 Injection for many reasons, including:
· we may not be able to demonstrate that BA058 is safe and effective as a treatment for osteoporosis to the satisfaction of the FDA;
· the results of its clinical studies may not meet the level of statistical or clinical significance required by the FDA for marketing approval;
· the FDA may disagree with the number, design, size, conduct or implementation of our clinical studies;
· the clinical research organization, or CRO, that we retain to conduct clinical studies may take actions outside of our control that materially adversely impact our clinical studies;
· the FDA may not find the data from preclinical studies and clinical studies sufficient to demonstrate that BA058’s clinical and other benefits outweigh its safety risks;
· the FDA may disagree with our interpretation of data from our preclinical studies and clinical studies or may require that we conduct additional studies;
· the FDA may not accept data generated at its clinical study sites;
· if our NDA is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional preclinical studies or clinical studies, limitations on approved labeling or distribution and use restrictions;
· the FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval;
· the FDA may identify deficiencies in the manufacturing processes or facilities of our third-party manufacturers; or
· the FDA may change its approval policies or adopt new regulations.
Before we submit an NDA to the FDA for BA058 as a treatment for osteoporosis, we must initiate and complete our pivotal Phase 3 study, a thorough QT study, a renal safety study, an osteosarcoma study in rats, and bone quality studies in rats and monkey. We have not commenced all of these required studies and the results of these studies will have an important bearing on the approval of BA058. In addition to fracture and BMD, our pivotal Phase 3 study will measure a number of other potential safety indicators, including anti-BA058 antibodies which will have an important bearing on the approval of BA058. In addition, the results from the rat carcinogenicity study, which includes hPTH(1-34), as a comparator, may show that BA058 dosing results in more osteosarcomas than PTH which may have a material adverse bearing on approval of BA058.
If we do not obtain the necessary U.S. or worldwide regulatory approvals to commercialize any product candidate, we will not be able to sell our product candidates. We cannot assure you that we will receive the approvals necessary to commercialize any of our product candidates (BA058, RAD1901, and RAD140), or any product candidate we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any product candidate, we must submit to the FDA a New Drug Application, or NDA, demonstrating that the product candidate
is safe for humans and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as pre-clinical studies, as well as human tests, which are referred to as clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in drugs that the FDA considers safe for humans and effective for indicated uses. The FDA has substantial discretion in the drug approval process and may require us to conduct additional pre-clinical and clinical testing or to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during its regulatory review. Delays in obtaining regulatory approvals may:
· delay commercialization of, and our ability to derive product revenues from, our product candidate;
· impose costly procedures on us; and
· diminish any competitive advantages that we may otherwise enjoy.
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our NDAs. We may never obtain regulatory clearance for any of our product candidates (BA058, RAD1901, and RAD140). Failure to obtain FDA approval of any of our product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate can be developed. There is no guarantee that we will ever be able to develop or acquire another product candidate.
In foreign jurisdictions, we must receive approval from the appropriate regulatory authorities before we can commercialize any drugs. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. We cannot assure you that we will receive the approvals necessary to commercialize our product candidates for sale outside the United States.
Most of our product candidates are in early stages of clinical trials. Except for BA058, each of our other product candidates (RAD1901 and RAD140), are in early stages of development and requires extensive pre-clinical and clinical testing. We cannot predict with any certainty if or when we might submit an NDA for regulatory approval for any of our product candidates or whether any such NDA will be accepted.
Clinical trials are very expensive, time-consuming and difficult to design and implement. Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. A substantial portion of our BA058 development costs are denominated in euro and any adverse movement in the dollar/euro exchange rate will result in increased costs and require us to raise additional capital to complete the development of our products. The clinical trial process is also time consuming. We estimate that clinical trials of BA058 Injection will take at least three years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
· unforeseen safety issues;
· determination of dosing issues;
· lack of effectiveness during clinical trials;
· slower than expected rates of patient recruitment;
· inability to monitor patients adequately during or after treatment; and
· inability or unwillingness of medical investigators to follow our clinical protocols.
In addition, we or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our Investigational New Drug, or IND, submissions or the conduct of these trials. Therefore, we cannot predict with any certainty the schedule for future clinical trials.
The results of our clinical trials may not support its product candidate claims. Even if our clinical trials are completed as planned, we cannot be certain that the results will support our product candidate claims. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and pre-clinical testing. Our Phase 3 study of BA058 Injection for fracture prevention may not replicate the positive efficacy results for BMD from our Phase 2 study. The clinical trial process may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. This failure would cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. In addition, our clinical trials to date involve a small patient population. Because of the small sample size, the results of these clinical trials may not be indicative of future results.
Physicians and patients may not accept and use our drugs. Even if the FDA approves one or more of our product candidates, physicians and patients may not accept and use it. Acceptance and use of our product will depend upon a number of factors including:
· perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drug;
· cost-effectiveness of our product relative to competing products;
· availability of reimbursement for our product from government or other healthcare payers; and
· effectiveness of marketing and distribution efforts by us and its licensees and distributors, if any.
Because we expect sales of our current product candidates, if approved, to generate substantially all of its product revenues for the foreseeable future, the failure of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
Our drug-development program depends upon third-party researcher, investigators and collaborators who are outside our control. We depend upon independent researchers, investigators and collaborators, such as Nordic, to conduct our pre-clinical and clinical trials under agreements with us. These third parties are not our employees and we cannot control the amount or timing of resources that they devote to our programs. These third parties may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such programs ourselves. If outside collaborators fail to devote sufficient time and resources to our drug-development programs, or if their performance is substandard, the approval of our FDA applications, if any, and our introduction of new drugs, if any, will be delayed. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators assist competitors at our expense, our competitive position would be harmed.
We will rely exclusively on third parties to formulate and manufacture our product candidate. We have no experience in drug formulation or manufacturing and do not intend to establish our own manufacturing facilities. We lack the resources and expertise to formulate or manufacture our own product candidates. We have entered into agreements with contract manufacturers to manufacture BA058 Injection for use in clinical trial activities. These contract manufacturers are currently our only source for the production and formulation of BA058. We currently do not have sufficient clinical supplies of BA058 to complete the planned Phase 3 study for BA058 Injection but believe that our contract manufacturers will be able to produce sufficient supply of BA058 to complete all of the planned BA058 clinical studies. However, if our contract manufacturers are unable to produce, in a timely manner,
adequate clinical supplies to meet the needs of our clinical studies, we would be required to seek new contract manufacturers that may require us to modify our finished product formulation and modify or terminate our clinical studies for BA058. Any modification of our finished product or modification or termination of our Phase 3 clinical study could adversely affect our ability to obtain necessary regulatory approvals and significantly delay or prevent the commercial launch of the product, which would materially harm our business and impair our ability to raise capital.
We depend on a number of single source contract manufacturers to supply key components of BA058. For instance, we depend on Lonza, which produces supplies of bulk drug product of BA058 to support the BA058 Injection and BA058 Microneedle Patch clinical studies and potential commercial launch. We also depend on Vetter, and 3M for the production of finished supplies of BA058 Injection and BA058 Microneedle Patch, respectively. Because of our dependence on Vetter for the “fill and finish” part of the manufacturing process for BA058 Injection, we are subject to the risk that Vetter may not have the capacity from time to time to produce sufficient quantities of BA058 to meet the needs of our clinical studies or be able to scale to commercial production of BA058. Because the manufacturing process for BA058 Microneedle Patch requires the use of 3M’s proprietary technology, 3M is our sole source for finished supplies of BA058 Microneedle Patch.
While we are currently in discussions, to date, we have not entered into a long-term agreement with Lonza, Vetter or 3M, who each currently produces BA058 product on a purchase order basis for us. Accordingly, Lonza, Vetter and 3M could terminate our relationship at any time and for any reason. If our relationship with any of these contract manufacturers is terminated, or if they are unable to produce BA058 in required quantities, on a timely basis or at all, our business and financial condition would be materially harmed. If any of our current product candidates or any product candidates we may develop or acquire in the future receive FDA approval, we will rely on one or more third-party contractors to manufacture its drugs. Our anticipated future reliance on a limited number of third-party manufacturers exposes we to the following risks:
· we may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would require new testing and compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any.
· our third-party manufacturers might be unable to formulate and manufacture our drugs in the volume and of the quality required to meet our clinical needs and commercial needs, if any.
· our future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute its products.
· Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the Drug Enforcement Administration, and corresponding state agencies to ensure strict compliance with good manufacturing practice and other government regulations and corresponding foreign standards. We does not have control over third-party manufacturers’ compliance with these regulations and standards.
· If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to the innovation.
Each of these risks could delay our clinical trials, the approval, if any, of our product candidates by the FDA or the commercialization of our product candidates or result in higher costs or deprive us of potential product revenues.
We have no experience selling, marketing or distributing products and no internal capability to do so. We currently have no sales, marketing or distribution capabilities. We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our proposed products. Our future success depends, in
part, on our ability to enter into and maintain collaborative relationships for such capabilities, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We intend to pursue collaborative arrangements regarding the sales and marketing of our products, however, there can be no assurance that we will be able to establish or maintain such collaborative arrangements, or if able to do so, that they will have effective sales forces. To the extent that we decides not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with technical expertise. There can also be no assurance that we will be able to establish or maintain relationships with third party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receives will depend upon the efforts of such third parties, and there can be no assurance that such efforts will be successful. In addition, there can also be no assurance that we will be able to market and sell our products in the United States or overseas.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer. The market for our product candidates is characterized by intense competition and rapid technological advances. If any of our product candidates receives FDA approval, it will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. If we fail to develop BA058 Microneedle Patch, our commercial opportunity for BA058 will be limited. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We will compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have oncology compounds already approved or in development. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we does, as well as significantly greater experience in:
· developing drugs;
· undertaking pre-clinical testing and human clinical trials;
· obtaining FDA and other regulatory approvals of drugs;
· formulating and manufacturing drugs; and
· launching, marketing and selling drugs.
Developments by competitors may render our products or technologies obsolete or non-competitive. The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Some of the drugs that we are attempting to develop, such as BA058, RAD1901 and RAD140 will have to compete with existing therapies. In addition, a large number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we is targeting. We faces competition from pharmaceutical and biotechnology companies in the United States and abroad. In addition, companies pursuing different but related fields represent substantial competition. Many of these organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, longer drug development history in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to attract qualified personnel and parties for acquisitions, joint ventures or other collaborations, and therefore, we may not be able to hire or retain qualified personnel to run all facets of our business.
If our efforts to protect our intellectual property related to BA058, RAD1901 and/or RAD140 fail to adequately protect these assets, we may suffer the loss of the ability to license or successfully commercialize one
or more of these candidates. Our commercial success is significantly dependent on intellectual property related to that product portfolio. We are either the licensee or assignee of numerous issued and pending patent applications that cover various aspects of our assets including BA058, RAD1901 and RAD140.
Patents covering BA058 as a composition of matter have been issued in the United States (US patent No. 5,969,095), Europe and several additional countries. Because the BA058 composition of matter case was filed in 1996, it is expected to have a normal expiry of approximately 2016 in the United States (this date does not include the possibility of Hatch-Waxman extension) and additional countries where it has issued. Because of this, it is possible that the data exclusivity provisions as applied to new molecular entities may run longer than the issued composition of matter patents.
We and Ipsen are also coassignees to US patent No. 7,803,770 that we believe provides exclusivity until 2027 in the United States (absent any extensions) for the method of treating osteoporosis with the intended therapeutic dose. Because patents are both highly technical and legal documents that are frequently subject to intense litigation pressure, there is risk that one or more of the issued patents that are believed to cover BA058 when marketed will be found to be invalid, unenforceable and/or not infringed. In the absence of product exclusivity in the market, there is a high likelihood of multiple competitors selling the same product with a corresponding drop in pricing power and/or sales volume.
Currently, additional intellectual property covering the BA058 Microneedle Patch is the subject of a US provisional patent application with a priority date of 2011 and any issued claims resulting from this application will expire no earlier than 2031. However, pending patent applications in the United States and elsewhere may not issue since the interpretation of the legal requirements of patentability in view claimed inventions are not always predictable. Additional intellectual property covering the BA058 Microneedle Patch technology exists in the form of proprietary information contained by trade secrets. These can be accidentally disclosed to, independently derived by or misappropriated by competitors, possibly reducing or eliminating the exclusivity advantages of this form of intellectual property, thereby allowing those competitors more rapid entry into the market place with a competitive product thus reducing our marketing advantage of the BA058 Microneedle Patch. In addition, trade secrets may in some instances become publicly available required disclosures in regulatory files. Alternatively, competitors may sometimes reverse engineer a product once it becomes available on the market. Even where a competitor does not use an identical technology for the delivery of BA058, it is possible that they could achieve an equivalent or even superior result using another technology. Such occurrences could lead to either one or more alternative competitor products available on the market and/or one or more generic competitor products on the market with a corresponding decrease in market share and/or price for the BA058 Microneedle Patch.
Patents covering RAD1901 as a composition of matter have been issued in the United States, Australia and is pending in Europe and several additional countries. The RAD1901 composition of matter patent in the United States expires in 2025 (not including any Hatch-Waxman extension). Additional patent applications relating to methods of treating vasomotor symptoms, clinical dosage strengths and combination treatment modalities all covering RAD1901 have been filed. Since patents are both highly technical and legal documents that are frequently subject to intense litigation pressure, there is risk that one or more of the issued patents that are believed to cover RAD1901 when marketed will be found to be invalid, unenforceable and/or not infringed when subject to said litigation. In the absence of product exclusivity in the market, there is a high likelihood of multiple competitors selling the same product with a corresponding drop in pricing power and/or sales volume. Pending patent applications in the United States and elsewhere may not issue since the interpretation of the legal requirements of patentability in view of any claimed invention before that patent office are not always predictable. As a result, we Health could encounter challenges or difficulties in building, maintaining and/or defending its intellectual property rights protecting and defending our intellectual property both in the United States and abroad.
Patent applications covering RAD140 and other SARM compounds that are part of the we SARM portfolio have been filed in the United States and elsewhere. Since the RAD140 composition of matter case was effectively filed in 2009, if issued, it is expected to have a normal expiry of approximately 2029 in the United States (this does not include the possibility of USPTO patent term adjustment or Hatch-Waxman extension) and additional countries if/when it issues. Since patents are both highly technical and legal documents that are frequently subject to intense litigation pressure, there is risk that even if one or more RAD140 patents does issue and is asserted that the patent(s) will be found invalid, unenforceable and/or not infringed when subject to said litigation. Finally, the intellectual
property laws and practices can vary considerably from one country to another and also can change with time. As a result, we could encounter challenges or difficulties in building, maintaining and/or defending its intellectual property rights protecting and defending our intellectual property both in the United States and abroad.
Payments, fees, submissions and various additional requirements must be met in order for pending patent applications to advance in prosecution and issued patents to be maintained. Rigorous compliance with these requirements is essential to procurement and maintenance of patents integral to the we product portfolio. Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will come due for payment periodically throughout the lifecycle of patent applications and issued patents. In order to help ensure that we complies with any required fee payment, documentary and/or procedural requirements as they might relate to any patents for which we is an assignee or co-assignee, we employs competent legal help and related professionals as needed to comply with those requirements. Our outside patent counsel uses Computer Packages, Inc. for patent annuity payments. Failure to meet a required fee payment, document production of procedural requirement can result in the abandonment of a pending patent application or the lapse of an issued patent. In some instances the defect can be cured through late compliance but there are situations where the failure to meet the required event cannot be cured. Such an occurrence could compromise the intellectual property protection around a our preclinical or clinical candidate and possibly weaken or eliminate our ability to protect our eventual market share for that product.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages, and defend against litigation. If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and may have to:
· obtain licenses, which may not be available on commercially reasonable terms, if at all;
· abandon an infringing drug candidate;
· redesign its products or processes to avoid infringement;
· stop using the subject matter claimed in the patents held by others;
· pay damages; or
· defend litigation or administrative proceedings which may be costly whether we wins or loses, and which could result in a substantial diversion of its financial and management resources.
Our ability to generate product revenues will be diminished if our drugs sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement. Our ability to commercialize its drugs, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
· government and health administration authorities;
· private health maintenance organizations and health insurers; and
· other healthcare payers.
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payers increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs. Even if one of our product candidates is approved by the FDA, insurance coverage may not be available, and reimbursement levels may be inadequate, to cover such drug. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for one of our products, once approved, market acceptance of such product could be reduced.
We may not successfully manage our growth. Our success will depend upon the expansion of our
operations and the effective management of its growth, which will place a significant strain on our management and on administrative, operational and financial resources. To manage this growth, we may be required to expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage this growth effectively, our business would be harmed.
We may be exposed to liability claims associated with the use of hazardous materials and chemicals. Our research and development activities may involve the controlled use of hazardous materials and chemicals. Although we believe that our safety procedures for using, storing, handling and disposing of these materials comply with federal, state and local laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for any resulting damages and any liability could materially adversely affect its business, financial condition and results of operations. In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect its business, financial condition and results of operations.
We rely on key executive officers and scientific and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace. We are highly dependent on its principal scientific, regulatory and medical advisors. We do not have “key person” life policies for any of our officers. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, loss of customers and sales and diversion of management resources, which could adversely affect our operating results.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed. We will need to hire additional qualified personnel with expertise in pre-clinical testing, clinical research and testing, government regulation, formulation and manufacturing and sales and marketing. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits. The testing and marketing of medical products entail an inherent risk of product liability. If we cannot successfully defend our self against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
Certain statements contained in this prospectus that are forward-looking in nature are based on the current beliefs of our management as well as assumptions made by and information currently available to management, including statements related to the markets for our products, general trends in our operations or financial results, plans, expectations, estimates and beliefs. In addition, when used in this prospectus, the words “may,” “could,” “should,” “anticipate,” “believe,” “estimate,” “expect,” “intend,” “plan,” “predict” and similar expressions and their variants, as they relate to us or our management, may identify forward-looking statements. These statements reflect our judgment as of the date of this prospectus with respect to future events, the outcome of which is subject to risks, which may have a significant impact on our business, operating results or financial condition. You are cautioned that these forward-looking statements are inherently uncertain. Should one or more of these risks or uncertainties materialize, or should underlying assumptions prove incorrect, actual results or outcomes may vary materially from those described herein. We undertake no obligation to update forward-looking statements. The risks identified under the heading “Risk Factors” in this prospectus, among others, may impact forward-looking statements contained in this prospectus.
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
The following is a discussion of the financial condition and results of operations of the Target (Radius Health, Inc.) prior to the Merger and the Short-Form Merger and should be read in conjunction with the financial statements and the notes to those statements filed with, and hereby incorporated into, this Current Report on Form 8-K as Exhibit 99.1. This discussion includes forward-looking statements that involve risk and uncertainties. As a result of many factors, such as those set forth under “Risk Factors” in this Current Report on Form 8-K, actual results may differ materially from those anticipated in these forward-looking statements. The share numbers and per share numbers in this MD&A assume the completion of Merger and Short-Form Merger. The historical Target financial statements and related notes do not assume the completion of the Merger.
Overview
We are a pharmaceutical company focused on acquiring and developing new therapeutics for the treatment of osteoporosis and other women’s health conditions. We have three product candidates in development, the most advanced is BA058 Injection that has begun dosing of patients in a pivotal Phase 3 clinical study for the prevention of fractures in women suffering from osteoporsis. We are also developing the BA058 Microneedle Patch, a short wear time, transdermal form of BA058 that is based on a microneedle technology from 3M that is currently being studied in a Phase 1b clinical study. We believe that the BA058 Microneedle Patch may eliminate the need for injections and lead to better treatment compliance for patients. Our second clinical stage product candidate is RAD1901 which has completed an initial Phase 2 clinical study for the treatment of vasomotor symptoms (hot flashes) in women entering menopause. Our third product candidate, RAD140, in pre-IND discovery, is a potential treatment for age-related muscle loss, frailty, weight loss associated with cancer cachexia and osteoporosis.
In November 2005, we changed our name from Nuvios, Inc. to Radius Health, Inc. On May 17, 2011, the Merger and the Short-Form Merger were consummated thereby completing the combination of our historical business with the historical business of the Target. Since inception, our efforts and resources have been focused primarily on acquiring and developing BA058 and our other pharmaceutical product candidates, raising capital and recruiting personnel. We have no product sales to date and we will not receive any product sales until we receive approval for BA058 Injection from the FDA, or equivalent foreign regulatory bodies. However, developing pharmaceutical products is a lengthy and very expensive process. Assuming we do not encounter any unforeseen delays during the course of developing BA058, we do not expect to complete development and file for marketing approval in the United States for BA058 Injection and BA058 Microneedle Patch until approximately 2014 and 2016, respectively. Accordingly, our success depends not only on the safety and efficacy of BA058, but also on our ability to finance the development of these products, which will require substantial additional funding to complete development and file for marketing approval. In addition, we currently have no sales, marketing or distribution capabilities and thus our ability to market BA058 will depend in part on our ability to enter into and maintain collaborative relationships for such capabilities, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We are also evaluating strategic alternatives with respect to collaborating with third parties for the future development of RAD1901 and RAD140. Our ability to further develop these product candidates will be dependent upon the outcome of our collaboration strategy. Our major sources of working capital have been proceeds from various private financings, primarily the private sales of our preferred stock.
Financial Overview
Research and Development Expenses
Research and development expenses consist primarily of salaries and related personnel costs, fees paid to consultants and outside service providers for regulatory and quality assurance support, licensing of drug compounds, and other expenses relating to the manufacture, development, testing and enhancement of our product candidates. We expense our research and development cost as they are incurred.
Our lead product candidate is BA058 and it represents the largest portion of our research and development expenses for our product candidates. BA058 is a novel synthetic peptide analog of hPTHrP being developed by as a treatment for osteoporosis in both injection and transdermal routes of administration. BA058 Injection is currently in a Phase 3 study and BA058 Microneedle Patch is in a Phase 1b study. Our other clinical stage program is RAD1901, a selective estrogen receptor modulator, or SERM, which has completed an initial Phase 2 clinical study for the treatment of vasomotor symptoms (hot flashes) in women entering menopause. Our third product candidate, RAD140 is a selective androgen receptor modular, or SARM, is in pre-IND development.
The following table sets forth our research and development expenses related to BA058 injection, BA058 Microneedle Patch, RAD1901 and RAD140 for the years ended December 31, 2009 and 2010 and the three months ended March 31, 2010 and 2011. We began tracking program expenses for BA058 Injection in 2005, and program expenses from inception to March 31, 2011 were approximately $29.3 million. We began tracking program expenses for BA058 Microneedle Patch in 2007, and program expenses from inception to March 31, 2011 were approximately $6.0 million. We began tracking program expenses for RAD1901 in 2006, and program expenses from inception to March 31, 2011 were approximately $15.3 million. We began tracking program expenses for RAD140 in 2008, and program expenses from inception to March 31, 2011 were approximately $5.1 million. These expenses relate primarily to external costs associated with manufacturing, preclinical studies and clinical trial costs.
Costs related to facilities, depreciation, share-based compensation and research and development support services are not directly charged to programs as they benefit multiple research programs that share resources.
|
|
|
Year ended December 31, |
|
Three Months ended March 31, |
| ||||||||
|
|
|
2009 |
|
2010 |
|
2010 |
|
2011 |
| ||||
|
|
|
(in thousands) |
| ||||||||||
|
BA058 Injection |
|
$ |
3,671 |
|
$ |
4,664 |
|
$ |
676 |
|
$ |
2,977 |
|
|
BA058 Microneedle Patch |
|
2,819 |
|
1,863 |
|
462 |
|
577 |
| ||||
|
RAD1901 |
|
2,185 |
|
1,654 |
|
251 |
|
— |
| ||||
|
RAD140 |
|
2,031 |
|
313 |
|
157 |
|
20 |
| ||||
The majority of our external costs are spent on BA058, as costs associated with later stage clinical trials are, in most cases, more significant than those incurred in earlier stages of our pipeline. In April 2011, we began dosing of patients in a pivotal Phase 3 clinical study of BA058 Injection for the treatment of osteoporosis. In addition, in December 2010, we initiated a Phase 1b clinical study for BA058 Microneedle Patch. We expect that future development costs related to the BA058 Injection and BA058 Microneedle Patch programs will increase significantly through possible marketing approval in the United States in 2015 and 2017 and may exceed $160 million and $50 million, respectively. The successful development of the BA058 Injection and BA058 Microneedle Patch is subject to numerous risks and uncertainties associated with developing drugs, including the variables listed below. A change in the outcome of any of these variables with respect to the development of any of our product candidates could mean a significant change in the costs and timing associated with the development of that product candidate.
BA058 Injection is our only product candidate in late stage development, and our business currently depends heavily on its successful development, regulatory approval and commercialization. We have no drug products for sale currently and may never be able to develop marketable drug products. We have not submitted an NDA to the FDA or comparable applications to other regulatory authorities. Obtaining approval of an NDA is an extensive, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of BA058 Injection for many reasons, including:
· we may not be able to demonstrate that BA058 is safe and effective as a treatment for osteoporosis to the satisfaction of the FDA;
· the results of its clinical studies may not meet the level of statistical or clinical significance required by the FDA for marketing approval;
· the FDA may disagree with the number, design, size, conduct or implementation of our clinical studies;
· the clinical research organization, or CRO, that we retain to conduct clinical studies may take actions outside of our control that materially adversely impact our clinical studies or we could experience significant delays in enrollment in any of our clinical trials;
· the FDA may not find the data from preclinical studies and clinical studies sufficient to demonstrate that BA058’s clinical and other benefits outweigh its safety risks;
· the FDA may disagree with our interpretation of data from our preclinical studies and clinical studies or may require that we conduct additional studies;
· the FDA may not accept data generated at its clinical study sites;
· if our NDA is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional preclinical studies or clinical studies, limitations on approved labeling or distribution and use restrictions;
· the FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval;
· the FDA may identify deficiencies in the manufacturing processes or facilities of our
third-party manufacturers;
· the FDA may change its approval policies or adopt new regulations.
As a result of these uncertainties, other than as discussed above, we are unable to determine the duration and costs to complete current or future clinical stages of our other product candidates or when, or to what extent, we will generate revenues from the commercialization and sale of any of our product candidates. Development timelines, probability of success and development costs vary widely. We anticipate that we will make determinations as to which additional programs to pursue and how much funding to direct to each program on an ongoing basis in response to the scientific and clinical data of each product candidate, as well as ongoing assessments of such product candidate’s commercial potential and our ability to fund such product development. The lengthy process of securing FDA approvals for new drugs requires the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, regulatory approvals would materially adversely affect our product development efforts and our business overall.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related expense for executive, finance and other administrative personnel, professional fees, business insurance, rent, general legal activities, and other corporate expenses. We expect our general and administrative expenses to increase as a result of higher costs associated with being a public company.
Our results include non-cash compensation expense as a result of the issuance of stock and stock option grants. Compensation expense for options granted to employees and directors (excluding directors who are also scientific advisory board member or consultants) represent the difference between the fair value of our common stock and the exercise price of the options at the date of grant. Compensation for options granted to consultants has been determined based upon the fair value of the equity instruments issued and the unvested portion of such option grants is re-measured at each reporting period. The stock-based compensation expense is included in the respective categories of expense in the statement of operations (research and development and general and administrative expenses). We expect to record additional non-cash compensation expense in the future, which may be significant.
Interest Income and Interest Expense
Interest income reflects interest earned on our cash, cash equivalents and marketable securities.
Interest expense reflects interest due on a Loan and Security Agreement under which we made the final payment in 2009.
Accretion of Preferred Stock
Accretion of preferred stock reflects the periodic accretions of issuance costs, dividends and the investor rights/obligations on Target’s Series B and C redeemable convertible preferred stock to adjust the carrying value to its redemption value.
Critical Accounting Policies and Estimates
The preparation of our financial statement requires us to make certain estimates and assumptions that affect the reported amounts of assets and liabilities and expenses during the reported periods. We believe the following accounting policies are “critical” because they require us to make judgments and estimates about matters that are uncertain at the time we make the estimate, and different estimates, which would have been reasonable could have been used, which would have resulted in different financial results.
Accrued Clinical Expenses
As part of the process of preparing our financial statements, we are required to estimate our accrued
expenses. This process involves reviewing open contracts and purchase orders, communicating with our personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. Payments under some of the contracts we have with parties depend on factors, such as the milestones accomplished, successful enrollment of certain numbers of patients, site initiation and the completion of clinical trial milestones. Examples of estimated accrued clinical expenses include:
· fees paid to investigative sites and laboratories in connection with clinical studies;
· fees paid to CROs in connection with clinical studies, if CROs are used; and
· fees paid to contract manufacturers in connection with the production of clinical study materials.
In accruing clinical expenses, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If possible, we obtain information regarding unbilled services directly from these service providers. However, we may be required to estimate the cost of these services based on information available to us. If we underestimate or overestimate the cost associated with a trial or service at a given point in time, adjustments to research and development expenses may be necessary in future periods. Historically, our estimated accrued clinical expenses have approximated actual expense incurred. Subsequent changes in estimates may result in a material change in our accruals.
Research and Development Expenses
We account for research and development costs by expensing such costs to operations as incurred. Research and development costs primarily consist of personnel costs, outsourced research activities, laboratory supplies, and license fees.
Nonrefundable advance payments for goods or services to be received in the future for use in research and development activities are deferred and capitalized. The capitalized amounts will be expensed as the related goods are delivered or the services are performed. If expectations change such that we do not expect we will need the goods to be delivered or the services to be rendered, capitalized nonrefundable advance payments would be charged to expense.
Stock-based Compensation
We recognize the compensation cost of employee stock-based awards using the straight-line method over the requisite service period of the award, which is typically the vesting period. During both the years ended December 31, 2009 and 2010 and the three months ended March 31, 2011 and 2010, we recorded approximately $100,000, $100,000, $22,000 and $33,000 of employee stock-based compensation expense. We estimate the fair value of each option award using the Black-Scholes-Merton option-pricing model.
In calculating the estimated fair value of our stock options, the Black-Scholes-Merton option-pricing model requires the consideration of the following six variables for purposes of estimating fair value:
· The stock option exercise price,
· The expected term of the option,
· The grant date price of the Company’s common stock, which is issuable upon exercise of the option,
· The expected volatility of the Company’s common stock,
· The expected dividends on the Company’s common stock, and
· The risk-free rate for the expected option term.
The expected term of the stock options granted represents the period of time that options granted are expected to be outstanding. For options granted prior to January 1, 2008, the expected term was calculated using the “simplified” method as prescribed by the SEC’s Staff Accounting Bulletin No. 107, Share-Based Payment. For options granted after January 1, 2008, we calculated the expected term using similar assumptions. The expected volatility is a measure of the amount by our stock price is expected to fluctuate during the term of the options
granted. We determine the expected volatility based on a review of the historical volatility of similar publicly held companies in the biotechnology field over a period commensurate with the option’s expected term. We have never declared or paid any cash dividends on our common stock and we do not expect to do so in the foreseeable future. Accordingly, we use an expected dividend yield of zero. The risk-free interest rate is the implied yield available on U.S. Treasury issues with a remaining life consistent with the option’s expected term on the date of grant. We apply an estimated forfeiture rate to current period expense to recognize compensation expense only for those awards expected to vest. We estimate forfeitures based upon historical data, adjusted for known trends, and will adjust the estimate of forfeitures if actual forfeitures differ or are expected to differ from such estimates. Subsequent changes in estimated forfeitures are recognized through a cumulative adjustment in the period of change and also will impact the amount of stock-based compensation expense in future periods. The forfeiture rate was estimated to be 2.8% and 2.4% for the years ended December 31, 2010 and 2009, respectively.
The following table presents the grant dates and related exercise prices of stock options granted from January 1, 2009 to May 11, 2011:
|
Date of Issuance |
|
Nature of |
|
Number of |
|
Exercise |
|
Per Share |
|
Per Share |
| |||
|
April 9, 2009 |
|
Option grant |
|
9,666 |
|
$ |
1.20 |
|
$ |
1.20 |
|
$ |
0.70 |
|
|
December 2, 2009 |
|
Option grant |
|
5,000 |
|
$ |
1.20 |
|
$ |
1.20 |
|
$ |
0.68 |
|
|
October 12, 2010 |
|
Option grant |
|
256,666 |
|
$ |
1.35 |
|
$ |
1.35 |
|
$ |
0.76 |
|
|
November 30, 2010 |
|
Option grant |
|
1,666 |
|
$ |
1.35 |
|
$ |
1.35 |
|
$ |
0.76 |
|
(1) The per share estimated fair value of common stock represents the determination by our board of directors of the fair value of our common stock as of the date of grant, taking into account various objective and subjective factors and including the results, if applicable, of valuations of our common stock as discussed in the pages that follow.
(2) Our estimate of the per share weighted average fair value for stock option grants was computed based upon the Black-Scholes option-pricing model with the assumptions through December 31,2010 as disclosed in our financial statements.
We have historically granted stock options at exercise prices not less than the fair value of our common stock as determined by our board of directors, with input from management. Our board of directors has historically determined, with input from management, the estimated fair value of our common stock on the date of grant based on a number of objective and subjective factors, including:
· the prices at which we sold shares of convertible preferred stock;
· the superior rights and preferences of securities senior to our common stock at the time of each grant;
· the likelihood of achieving a liquidity event such as an initial public offering or sale of our company;
· our historical operating and financial performance and the status of our research and product development efforts; and
· achievement of enterprise milestones, including our entering into collaboration and license agreements;
Our board of directors also considered valuations provided by management in determining the fair value of our common stock. Such valuations were prepared as of December 3, 2008, December 2, 2009 and October 1, 2010, and valued our common stock at $1.05, $1.20 and $1.35 per share, respectively. The valuations have been used to estimate the fair value of our common stock as of each option grant date listed and in calculating stock-based compensation expense. Our board of directors has consistently used the most recent valuation provided by management for determining the fair value of our common stock unless a specific event occurs that necessitates an interim valuation.
The valuations were based on the guidance from the Valuation of Privately-Held-Company Equity Securities Issued as Compensation that was developed by staff of the American Institute of Certified Public
Accountants and a task force comprising representatives from the appraisal, preparer, public accounting, venture capital, and academic communities. The Option-pricing method was selected to value Radius’ common stock-based on the Company’s stage of development and the degree of uncertainty surrounding the future success of clinical trials for our Company’s product candidates. The option-pricing method treats common stock and preferred stock as call options on the enterprise’s value, with exercise prices based on the liquidation preference of the preferred stock. Under this method, the common stock has value only if the funds available for distribution to shareholders exceed the value of the liquidation preference at the time of a liquidity event (for example, merger of sale), assuming the enterprise has funds available to make a liquidation preference meaningful and collectible by the shareholders.
In the model, the exercise price is based on a comparison with the enterprise value rather than, as in the case of a “regular” call option, a comparison with a per-share stock price. Thus, common stock is considered to be a call option with a claim on the enterprise at an exercise price equal to the remaining value immediately after the preferred stock is liquidated. We used the Black-Scholes model to price the call option. Under the option-pricing method we had to consider the various terms of the stockholder agreements -including the level of seniority among the securities, dividend policy, conversion ratios, and cash allocations -upon liquidation of the enterprise
Results of Operations
The following discussion summarizes the key factors our management believes are necessary for an understanding of our financial statements.
|
|
|
Years ended December 31, |
|
Three Months ended March 31, |
| ||||||||
|
|
|
2009 |
|
2010 |
|
2010 |
|
2011 |
| ||||
|
|
|
(in thousands) |
| ||||||||||
|
Revenue: |
|
|
|
|
|
|
|
|
| ||||
|
Option Fee |
|
$ |
1,616 |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
| ||||
|
Research and development |
|
14,519 |
|
11,692 |
|
2,491 |
|
4,137 |
| ||||
|
General and administrative |
|
2,668 |
|
3,630 |
|
644 |
|
897 |
| ||||
|
Restructuring |
|
— |
|
217 |
|
— |
|
— |
| ||||
|
Loss from operations |
|
(15,571 |
) |
(15,539 |
) |
(3,135 |
) |
(5,034 |
) | ||||
|
Other income (expense): |
|
|
|
|
|
|
|
|
| ||||
|
Other Income |
|
— |
|
883 |
|
— |
|
10 |
| ||||
|
Other (expense) |
|
(7 |
) |
(59 |
) |
— |
|
— |
| ||||
|
Interest income, net |
|
489 |
|
85 |
|
27 |
|
14 |
| ||||
|
Net loss |
|
(15,089 |
) |
(14,630 |
) |
(3,108 |
) |
(5,010 |
) | ||||
|
Accretion of preferred stock |
|
(11,405 |
) |
(12,143 |
) |
(3,046 |
) |
(2,876 |
) | ||||
|
Net loss attributable to common stockholders |
|
$ |
(29,494 |
) |
$ |
(26,773 |
) |
$ |
(6,154 |
) |
$ |
(7,886 |
) |
Three months Ended March 31, 2011 and 2010
Revenue: There was no revenue for the three months ended March 31, 2011 or March 31, 2010.
|
|
|
Three months Ended |
|
Change |
| |||||||
|
|
|
2010 |
|
2011 |
|
$ |
|
% |
| |||
|
|
|
(dollars in thousands) |
| |||||||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
| |||
|
Research and development |
|
$ |
2,491 |
|
$ |
4,137 |
|
$ |
1,646 |
|
66 |
% |
|
General and administrative |
|
644 |
|
897 |
|
253 |
|
39 |
% | |||
|
Restructuring |
|
— |
|
— |
|
— |
|
|
| |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Total operating expenses |
|
$ |
3,135 |
|
5,034 |
|
1,899 |
|
61 |
| ||
Research and development expenses: For the three months ended March 31, 2011, research and development expense was $ 4,137,000 compared to $2,491,000 for the three months ended March 31, 2010, an increase of $ 1,646,000 and 66%. For the three months ended March 31, 2011, we incurred professional contract services associated with the development of BA058 Injection of $ 2,977,000 compared to $251,000 for the three months ended March 31, 2010. The increase was primarily the result of expenses incurred to initiate our Phase 3 study. These expenses included payments of approximately $2,503,000 to Nordic for upfront Phase 3 study expenses, as well as a $600,000 payment for materials required for the study. Additionally, we incurred $113,000 more in contract services associated with the development of BA058 Microneedle Patch. Offsetting these increases, we spent $ 131,000 less on miscellaneous supplies and services related to RAD140, and $676,000 less for professional contract services associated with the development of RAD1901 in the three months ended March 31, 2011 compared to the three months ended March 31, 2010. We also had reductions in facilities and outside services expensed of approximately $378,000 for the three months ended March 31, 2011 compared to the three months ended March 31, 2010. These reductions were attributable to the closure of our lab in September of 2010.
General and administrative expenses: For the three months ended March 31, 2011, general and administrative expense was $897,000 compared to $644,000 for the three months ended March 31, 2010, an increase of $253,000 and 39%. The increase is primarily the result of increased legal and accounting costs.
Restructuring
There were no restructuring charges for the three months ended March 31, 2011 and March 31, 2010.
Years ended December 31, 2010 and 2009
Revenue: For the year ended December 31, 2010, revenue was $0 compared to $1,616,000 for the year ended December 31, 2009. The revenue in 2009 relates solely to an option agreement signed with Novartis in 2007 pursuant to which Novartis obtained an option to license the exclusive worldwide rights (except Japan) to all formulations of BA058. Revenue was recognized ratably over the option period based on criteria specified in the agreement. The period of option exclusivity expired in 2009 without exercise by Novartis.
|
|
|
Years Ended |
|
Change |
| |||||||
|
|
|
2009 |
|
2010 |
|
$ |
|
% |
| |||
|
|
|
(dollars in thousands) |
| |||||||||
|
Operating expenses: |
|
|
|
|
|
|
|
|
| |||
|
Research and development |
|
$ |
14,519 |
|
$ |
11,692 |
|
$ |
(2,827 |
) |
(24 |
)% |
|
General and administrative |
|
2,668 |
|
3,630 |
|
962 |
|
36 |
% | |||
|
Restructuring |
|
0 |
|
217 |
|
217 |
|
|
| |||
|
|
|
|
|
|
|
|
|
|
| |||
|
Total operating expenses |
|
$ |
17,187 |
|
$ |
15,539 |
|
$ |
(1,548 |
) |
(9 |
)% |
Research and development expenses: For the year ended December 31, 2010, research and development expense was $ 11,692,000 compared to $14,519,000 for the year ended December 31, 2009, a decrease of $ 2,827,000 and 24%. For the year ended December 31, 2010, we incurred professional contract services associated with the development of BA058 Injection of approximately $ 4,664,000 compared to approximately $3,671,000 for the year ended December 31, 2009. The increase is attributable to a $1,000,000 up-front payment to Nordic for Phase 3 study expenses. Offsetting these increases, we incurred $956,000 less in contract services associated with the development of BA058 Microneedle Patch. The decrease was mainly the result of completion of the feasibility agreement with 3M for the Microneedle Patch in 2009. Additionally, we spent $1,717,000 less on RAD140 and $531,000 less on RAD1901 for professional contract services in the year ended December 31, 2010 compared to the year ended December 31, 2009 as we evaluate strategic options of the further development of these programs. Lastly, we experienced reductions in stock-based and other compensation of approximately $125,000, professional fees of approximately $234,000, and facility
and other miscellaneous costs of approximately $ 256,000, for the year ended December 30, 2010 compared to the year ended December 31, 2009. The reduction in compensation was the result of the achievement of certain milestones that generated higher stock-based compensation in 2009. The reduction in professional fees, facilities, and miscellaneous other costs was related to the curtailment in costs for the RAD140 and RAD1901 programs.
General and administrative expenses: For the year ended December 31, 2010, general and administrative expense was $3,630,000 compared to $2,668,000 for the year ended December 31, 2009, an increase of approximately $962,000 and 36%. The increase was attributable to an increase in compensation of approximately $279,000 and professional fees of approximately $715,000. The increase in compensation consisted mainly of management bonuses which were higher in 2010 than in 2009. The increase in professional fees included legal and accounting fees. These increases were offset by reductions in other individually insignificant accounts.
Restructuring
We incurred restructuring costs of approximately $217,000 in the year ended December 31, 2010 related to lease termination costs associated with vacating our laboratory space. No similar costs were incurred in 2009.
Interest Income
Interest income decreased approximately $404,000 from $489,000 in the year ended December 31, 2009 to $85,000 in the year ended December 31, 2010. The decrease is attributable to a lower average cash equivalents and marketable securities balance in 2010.
Other Income
Other income of $883,000 at December 31, 2010 included approximately $733,000 of grant proceeds from the Internal Revenue Service pursuant to the qualifying therapeutic discovery grant program and approximately $149,000 in proceeds from the sale of equipment.
Liquidity and Capital resources
From inception to March 31, 2011, we have incurred an accumulated deficit of $136.1 million, primarily as a result of expenses incurred through a combination of research and development activities related to our various product candidates and expenses supporting those activities.
We have financed our operations since inception primarily through the private sale of preferred stock as well as the receipt of $5,000,000 in fees associated with an option agreement. Total cash, cash equivalents and marketable securities as of March 31, 2011 were $12,074,293.
The following table sets forth the major sources and uses of cash for each of the periods set forth below:
|
|
|
Years ended December |
|
Change |
|
Three months ended |
|
Change |
| ||||||||||||||
|
|
|
2009 |
|
2010 |
|
$ |
|
% |
|
2010 |
|
2011 |
|
$ |
|
% |
| ||||||
|
|
|
(in thousands) |
|
|
|
|
|
(in thousands) |
|
|
|
|
| ||||||||||
|
Net cash provided by (used in): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Operating activities |
|
$ |
(18,293 |
) |
$ |
(12,986 |
) |
$ |
5,307 |
|
-29 |
% |
$ |
(4,230 |
) |
$ |
(6,454 |
) |
$ |
(2,224 |
) |
53 |
% |
|
Investing activities |
|
17,623 |
|
15,669 |
|
(1,954 |
) |
-11 |
% |
2,852 |
|
6,097 |
|
3,245 |
|
114 |
% | ||||||
|
Financing activities |
|
(8 |
) |
2 |
|
10 |
|
-125 |
% |
— |
|
— |
|
— |
|
0 |
% | ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
Net increase (decrease) in cash and cash equivalents |
|
(678 |
) |
2,685 |
|
3,363 |
|
-496 |
% |
(1,378 |
) |
(357 |
) |
1,021 |
|
-74 |
% | ||||||
Cash Flows From Operating Activities
The increase of $2,224,000 in net cash used in operations for the three months ended March 31, 2011 compared to the three months ended March 31, 2010 was primarily associated with an increase in net loss of $1,902,000 and net changes in working capital related to expenses incurred to initiate the Phase 3 clinical study for BA058 Injection, the most significant of which was a $311,000 change in accrued expenses.
The decrease of $5,307,000 in net cash used in operations for the year ended December 31, 2010 compared to the year ended December 31, 2009 was primarily associated with a $459,000 decrease in net loss and net changes in working capital, including a $2,413,000 million increase in accrued expenses related to preparations to initiate the Phase 3 clinical study for BA058 Injection, a $1,027,000 decrease in accounts payable and a decrease of $1,616,000 million in deferred revenue due to the expiration of the Novartis option agreement in 2009.
Cash Flows From Investing Activities
Net cash provided by investing activities increased by $3,245,000 for the three months ended March 31, 2011 compared to the three months ended March 31, 2010. The increase was primarily a result of a $3,234,000 increase in net cash proceeds from the sales and maturities of investments, net of purchases, in the three months ended March 31, 2011; offset by $11,000 used for the purchase of equipment.
Net cash provided by investing activities decreased by $1,954,000 for the year ended December 31, 2010 compared to the year ended December 31, 2009. The decrease was primarily a result of a $2,120,000 decrease in net cash proceeds from the sales and maturities of investments, net of purchases, in the year ended December 31, 2010; offset by $149,000 in proceeds from the sale of equipment.
Cash Flows From Financing Activities
There were no significant cash flows from financing activities for the three months ended March 31, 2011 and March 31, 2010 or the years ended December 31, 2010 and December 31, 2009.
Our continued operations will depend on whether we are able to raise additional funds through various potential sources, such as equity and debt financing and potential collaboration agreements. Through March 31, 2011, a significant portion of our financing has been through private placements of preferred stock. We will seek to continue to fund operations from cash on hand and through additional equity and/or debt financing and potential collaboration agreements. We can give no assurances that any additional capital that we are able to obtain will be sufficient to meet our needs. Based on our existing resources, which include the $21,428,000 of proceeds from the first closing of the Series A-1 convertible preferred stock (Series A-1 Preferred Stock) financing on May 17, 2011 and an irrevocable legally binding commitment effective May 11, 2011, for additional proceeds of $42,857,000 from the issuance of Series A-1 convertible preferred stock in two additional closings which are expected to take place in 2011, we believe that we have sufficient capital to fund our operations into May 2012, but will need additional financing thereafter until we can achieve profitability, if ever.
Financings
Through March 31, 2011, we received aggregate net cash proceeds of $105.9 million from the sale of shares of our preferred stock as follows:
|
Issue |
|
Year |
|
No. Shares |
|
Net Proceeds (in |
| |
|
Series B redeemable convertible preferred stock |
|
2003, 2004, 2005 |
|
1,599,997 |
|
23,775 |
| |
|
Series C redeemable convertible preferred stock |
|
2006, 2007, 2008 |
|
10,146,629 |
|
82,096 |
| |
|
|
|
|
|
11,746,626 |
|
$ |
105,871 |
|
On May 11, 2011 accredited investors in a Series A-1 convertible preferred stock financing (“Series A-1 Private Placement”) entered into an irrevocable legally binding commitment to purchase $64.3 million of Series A-1 convertible preferred stock in three closings. The first closing of the Series A-1 Private Placement occurred on May 17, 2011 and we received gross proceeds of approximately $21,428,000 through the sale of 2,631,845 shares of Series A-1 convertible preferred stock. Those shares were exchanged in the Merger for an aggregate of
263,177 Shares of Series A-1 Stock Preferred Shares of the Series A-1 Preferred Stock are convertible, in whole or in part, at the option of the holder at any time into shares of our common stock initially on a one-for-ten basis at an initial conversion price of $8.142 per share.
The Series A-1 Private Placement provides for additional Stage II and Stage III closings upon notice by us to the same accredited investors for an additional 526,358 shares of Series A-1 convertible preferred stock in consideration of gross proceeds of an additional $42,857,000. We expect to affect the Stage II and Stage III closings in 2011. Concurrently with the Stage I Closing of the Series A-1 Private Placement, we issued 64,430 shares of Series A-5 Preferred Stock to Nordic for gross proceeds of approximately $525,000.
On May 23, 2011, the Company entered into a Loan and Security Agreement with General Electric Capital Corporation (“GECC”) as agent and a lender, and Oxford Finance LLC (“Oxford” and together with GECC, the “Lenders”) as a lender, pursuant to which the lenders agreed to make available to the Company $25,000,000 in the aggregate over three term loans. The initial term loan was made on May 23, 2011 in an aggregate principal amount equal to $6,250,000 (the “Initial Term Loan”) and is repayable over a term of 42 months, including a six month interest only period. The initial term loan bears interest at 10%. Pursuant to the Agreement, the Company may request two (2) additional term loans, the first, which must be funded not later than November 23, 2011, in an aggregate principal amount equal to $6,250,000 (the “Second Term Loan”) and the second, which must be funded not later than May 23, 2012, in an aggregate principal amount equal to $12,500,000 (the “Third Term Loan”). In the event the Second Term Loan is not funded on or before November 23, 2011, the Lenders’ commitment to make the Second Term Loan shall be terminated and the total commitment shall be reduced by $6,250,000. In the event the Third Term Loan is not funded on or before May 23, 2012, the Lenders’ commitment to make the Third Term Loan shall be terminated and the total commitment shall be further reduced by $12,500,000. Pursuant to the agreement, the Company agreed to issue to the Lenders (or their respective affiliates or designees) stock purchase warrants (collectively, the “Warrants”) to purchase in the aggregate a number of shares of the Company’s Series A-1 Preferred Stock equal to the quotient of (a) the product of (i) the amount of the applicable term loan multiplied by (ii) four percent (4%) divided by (b) the exercise price equal to $81.42 per share. The exercise period of each Warrant to be issued will expire ten (10) years from the date such Warrants are issued. On May 23, 2011, the Company issued a Warrant to each of GECC and Oxford for the purchase of 3,203 shares of Series A-1 Preferred Stock.
License Agreement Obligations
BA058
In September, 2005, we exclusively licensed the worldwide rights (except Japan) to BA058 and analogs from Ipsen. Of particular relevance, Radius licensed US Patent No. 5,969,095, effective filing date (3/29/1996) entitled “Analogs of Parathyroid Hormone” that claims BA058 and US Patent No. 6,544,949, effective filing date (3/29/1996) entitled “Analogs of Parathyroid Hormone” that claims methods of treating osteoporosis using BA058 and pharmaceutical compositions comprising BA058, and the corresponding foreign patents and continuing patent applications. In addition, Radius has rights to joint Ipsen/Radius intellectual property related to BA058 including rights to the joint Radius/Ipsen derived intellectual property contained in US7803770, effective filing date (10/3/2007) and related patent applications both in the United States and worldwide (excluding Japan) that cover the method of treating osteoporosis using the phase 3 clinical dosage strength and form. In consideration for the rights to BA058 and in recognition of certain milestones having been met to date, Radius has paid to Ipsen an aggregate amount of $1,000,000 US dollars. The license agreement further requires Radius to make payments upon the achievement of certain future clinical and regulatory milestones. Should BA058 become commercialized, Radius will be obligated to pay to Ipsen an annual royalty based on net sales of the product. In the event that Radius sublicenses BA058 to a third party, Radius is obligated to pay royalties to Ipsen based on a fixed rate of fees or royalties received from the sublicense. Effective May, 11, 2011, Ipsen agreed to accept shares of Series A-1 Preferred Stock in lieu of a cash milestone payment. We issued 173,263 shares of Series A-1 Preferred Stock to Ipsen on May 17, 2011 to settle the liability. The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
RAD1901
In June, 2006, we exclusively licensed the worldwide rights (except Japan) to RAD1901 from Eisai. In consideration for the rights to BA058 and in recognition of certain milestones having been met to date, Radius has paid to Eisai an aggregate amount of $1,500,000 US dollars. The license agreement further requires Radius to make payments upon the achievement of certain future clinical and regulatory milestones. Should RAD1901 become commercialized, Radius will be obligated to pay to Eisai an annual royalty based on net sales of the product. In the event that Radius sublicenses RAD1901 to a third party, Radius is obligated to pay royalties to Eisai based on a fixed rate of fees or royalties received from the sublicense. The license agreement contains other customary clauses and terms as are common in similar agreements in the industry.
Net Operating Loss Carryforwards
As of December 31, 2010, we had federal and state net operating loss carryforwards of approximately $85,000,000 and $75,000,000, respectively. If not utilized, the net operating loss carryforwards will begin expiring in 2024 and 2016 for federal and state purposes, respectively.
Under Section 382 of the Code, substantial changes in our ownership may limit the amount of net operating loss carryforwards that could be utilized annually in the future to offset taxable income. Specifically, this limitation may arise in the event of a cumulative change in ownership of our company of more than 50% within a three-year period. Any such annual limitation may significantly reduce the utilization of the net operating loss carryforwards before they expire. The closing of this offering, together with private placements and other transactions that have occurred since our inception, may trigger an ownership change pursuant to Section 382, which could limit the
amount of net operating loss carryforwards that could be utilized annually in the future to offset taxable income, if any. Any such limitation, whether as the result of this offering, prior private placements, sales of common stock by our existing stockholders or additional sales of common stock by us after this offering, could have a material adverse effect on our results of operations in future years. We have not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since our inception, due to the significant costs and complexities associated with such study. In each period since our inception, we have recorded a valuation allowance for the full amount of our deferred tax asset, as the realization of the deferred tax asset is uncertain. As a result, we have not recorded any federal or state income tax benefit in our statement of operations.
Internal Control Over Financial Reporting
We are not currently required to comply with Section 404 of the Sarbanes-Oxley Act and are therefore not required to make an assessment of the effectiveness of our internal control over financial reporting. Further, our independent registered public accounting firm has not been engaged to express, nor have they expressed, an opinion on the effectiveness of our internal control over financial reporting. In connection with our becoming a public company, we intend to hire additional accounting personnel with public company and SEC reporting experience and to focus on implementing appropriate internal controls and other procedures.
Beginning with the year ending December 31, 2011, pursuant to Section 404 of the Sarbanes-Oxley Act, management will be required to deliver a report that assesses the effectiveness of our internal control over financial reporting. Under current SEC rules, our independent registered public accounting firm will also be required to deliver an attestation report on the effectiveness of our internal control over financial reporting beginning with the year ending December 31, 2011, unless we qualify for an exemption as a non-accelerated filer under the Dodd-Frank Wall Street Reform and Consumer Protection Act, enacted on July 21, 2010.
Changes in Registrant’s Certifying Accountants
In connection with the closing of the Merger on May 17, 2011, Ernst & Young LLP (“E&Y”), who was the independent registered public accounting firm for Target prior to the Merger, became the independent registered public accounting firm for the Company and Raich Ende Malter & Co. LLP (“REMC”) was dismissed as the independent registered public accountant for the Company. The decision to appoint E&Y and dismiss REMC was recommended, and subsequently approved, by the Board of Directors of the Company.
The reports of REMC on the Company’s financial statements for the period ended December 31, 2010 did not contain an adverse opinion or a disclaimer of opinion, and were not qualified or modified as to uncertainty, audit scope, or accounting principles.
In connection with the audits of the Company’s financial statements for each of the two years ended December 31, 2010, and in the subsequent interim period through REMC’s dismissal, there were no disagreements with REMC on any matters of accounting principles or practices, financial statement disclosures, or auditing scope or procedures, which if not resolved to REMC’s satisfaction would have caused REMC to make reference to the matter in their report.
In connection with the audited financial statements of the Company through REMC’s dismissal, there have been no reportable events with the Company as set forth in Item 304(a)(1)(v) of Regulation S-K.
The Company has requested that REMC furnish it with a letter addressed to the Securities & Exchange Commission stating whether it agrees with the above statements. A copy of the letter, dated July 12, is filed herewith as Exhibit 16.1.
Quantitative and Qualitative Disclosures about Market Risk
Our primary exposure to market risk is foreign currency exposure. A substantial portion of our BA058 development costs are denominated in euro and an immediate 10 percent adverse change in the dollar/euro exchange rate will result in increased costs and would have a material adverse impact on our financial statements and require us to raise additional capital to complete the development of our products. We do not hedge our foreign currency exchange rate risk.
We are also exposed to market risk related to changes in interest rates. As of December 31, 2010 and December 31, 2009, we had cash, cash equivalents and short-term investments of $18,551,000 and $31,722,000 million, respectively, consisting of money market funds, U.S. Treasuries, Certificates of Deposit and cash equivalents. This exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our investments are in short-term marketable securities. Our short-term investments are subject to interest rate risk and will fall in value if market interest rates increase. Due to the short-term duration of our investment portfolio and the low risk profile of our investments, an immediate 10 percent change in interest rates would not have a material effect on the fair market value of our portfolio. We have the ability to hold our short-investments until maturity, and therefore we would not expect our operations results or cash flows to be affected by any significant degree by the effect of a change in market interest rates on our investments. We carry our investments based on publicly available information. We do not currently have any hard to value investment securities or securities for which a market is not readily available or active.
We are not subject to significant credit risk as this risk does not have the potential to materially impact the value of assets and liabilities.
MANAGEMENT
Each executive officer and each member of our board of directors shall serve until his successor is elected and qualified.
|
Name |
|
Age |
|
Position |
|
|
|
|
|
|
|
C. Richard Lyttle, Ph.D |
|
66 |
|
Director, President and Chief Executive Officer |
|
Nick Harvey |
|
51 |
|
Senior Vice President, Chief Financial Officer, Treasurer and Secretary |
|
Louis O’Dea, MB |
|
60 |
|
Senior Vice President, Chief Medical Officer |
|
Gary Hattersley, PhD |
|
44 |
|
Vice President, Biology |
|
|
|
|
|
|
|
Alan Auerbach |
|
41 |
|
Director |
|
Jonathan Fleming |
|
53 |
|
Director |
|
Ansbert K. Gadicke, M.D. |
|
53 |
|
Director |
|
Kurt Graves |
|
43 |
|
Director |
|
Martin Muenchbach, Ph.D. |
|
40 |
|
Director |
|
Elizabeth Stoner, M.D. |
|
60 |
|
Director |
C. Richard Lyttle, PhD, Director, President and Chief Executive Officer, 66, has served as a member of our board of directors and as our President and Chief Executive Officer since November 2010. Prior to the Merger Short-Form Merger, Dr. Lyttle had been President and Chief Executive Officer and a Director of Target since August 2004. Dr. Lyttle is the former Vice President of Discovery for Women’s Health and Bone from 1998 to 2004, and the Women’s Health Research Institute at Wyeth from 1993 to 2004. Prior to joining Wyeth, Dr. Lyttle was Research Professor of Obstetrics, Gynecology, and Pharmacology at the University of Pennsylvania from 1979 to 1993. He received a PhD in Biochemistry from Queen’s University, Kingston, Ontario in 1972, followed by postdoctoral research at the Population Council at the Rockefeller University from 1973 to 1974, the Department of Biology at Queen’s University from 1974 to 1976, and at the University of Chicago from 1976 to 1979. Dr. Lyttle was selected as a director because of his business and professional experience.
Nick Harvey, Senior Vice President, Chief Financial Officer, Treasurer and Secretary, 51, has served as our Chief Financial Officer, Treasurer and Secretary since November 2010, and served as a member of our board of directors from November 2010 until the consummation of the Merger in May 2011. Prior to the Merger and Short-Form Merger, Mr. Harvey had served as Chief Financial Officer and Senior Vice President of Target since December 2006. Prior to joining Target, Mr. Harvey served as Managing Director of Shiprock Capital, LLC, a venture capital firm, from 2003 to 2006 and remains a member of the Board of that firm. Prior to Shiprock Capital, Mr. Harvey served as Chief Financial Officer of a number of venture-backed companies over a 10-year period, including LifetecNet from 2001 to 2002, Transfusion Technologies from 1999 to 2000, and Transcend Therapeutics from 1993 to 1999. Mr. Harvey received a Bachelor of Economics degree in 1980 and a Bachelor of Laws degree with first-class honors in 1983 from the Australian National University, and an MBA from the Harvard Business School in 1991. Mr. Harvey was selected as a director because of his business and professional experience.
Louis O’Dea, M.B., Senior Vice President, Chief Medical Officer, 60, has been Senior Vice President and Chief Medical Officer since the closing of the Merger in May 2011 and, prior to the Merger served in such capacity at Target since March 2006. Prior to joining Target, Dr O’Dea was Vice President and Head of Clinical Development for Reproductive Endocrinology and Metabolism at Serono, Inc. from 2004 to 2006. Joining Serono as a Medical Director in 1993 (1993-95), he was appointed Executive Medical Director in 1995 (1995-1998) and Vice President for Clinical Development in 1999 (1999-2006). From 2000 to 2002, Dr O’Dea served as Regional Medical Officer in Japan. Dr. O’Dea received his Medical Degree from University College Dublin, Ireland, in 1976. He received his postgraduate medical education in Internal Medicine and Endocrinology at McGill University, Canada from 1976-84 and is board certified in both specialties in Canada and USA. From 1984-88, Dr O’Dea undertook a Research and Clinical Fellowship in Reproductive Endocrinology at Massachusetts General Hospital, Harvard University and from 1988-95 was a member of the Medical Faculty of McGill University, Montreal, Canada, in the Departments of Medicine and Obstetrics-Gynecology. Dr O’Dea has also served on the Board of Directors of the Eliassen Group from 2007 to 2010 and is an advisor to Lineage Capital.
Gary Hattersley, PhD, Vice President, Biology, 44, has served as our Vice President of Biology since the closing of the Merger in May 2011 and had served in the same capacity at Target since April 2008. He also served as Target’s Director, Disease Biology & Pharmacology from 2003 to 2008. Prior to joining Target, Dr. Hattersley was a Senior Scientist at Millennium Pharmaceuticals from 2000 to 2003 with responsibility for the discovery and development of novel small-molecule agents for the treatment of osteoporosis and other metabolic bone diseases. Dr. Hattersley also held positions at Genetics Institute/Wyeth Research from 1992 to 2000 investigating the application of the bone morphogenetic proteins in bone and connective tissue repair and regeneration. Dr. Hattersley received a PhD in Experimental Pathology from St. George’s Hospital Medical School in London in 1991.
Alan H. Auerbach, 41, has been a director since the closing of the Merger in May 2011 and, prior to the Merger, was a director of Target since October 2010. Mr. Auerbach is currently the Founder, Chief Executive Officer and President of Puma Biotechnology, Inc., a company dedicated to in-licensing and developing drugs for the treatment of cancer and founded in 2010. Mr. Auerbach founded Cougar Biotechnology in May 2003 and served as the company’s Chief Executive Officer, President and a Member of its Board of Directors until July 2009 when Cougar was acquired by Johnson & Johnson for approximately $1 billion. From July 2009 until January 2010, Mr. Auerbach served as the Co-Chairman of the Integration Steering Committee at Cougar (as part of Johnson & Johnson) that provided leadership and oversight for the development and global commercialization of Cougar’s lead product candidate, abiraterone acetate, for the treatment of advanced prostate cancer. Prior to founding Cougar, from June 1998 to April 2003 Mr. Auerbach was a Vice President, Senior Research Analyst at Wells Fargo Securities, where he was responsible for research coverage of small- and middle- capitalization biotechnology companies, with a focus on companies in the field of oncology. He had primary responsibility for technical, scientific and clinical due diligence, as well as selection of biotechnology companies followed by the company. During 2002, Mr. Auerbach ranked second in the NASDAQ/Starmine survey of analyst performance for stock picking in biotechnology. From August 1997 to May 1998, Mr. Auerbach was a Vice President, Research Analyst at the Seidler Companies, Inc., where he was responsible for research coverage of small capitalization biotechnology companies. Prior to his work as a biotechnology analyst, Mr. Auerbach worked for Diagnostic Products Corporation, where he designed and implemented clinical trials in the field of oncology. Mr. Auerbach received a B.S. in Biomedical Engineering from Boston University and an M.S. in Biomedical Engineering from the University of Southern California. Mr. Auerbach was selected as a director because of his business and professional experience, including but not limited his leadership of Cougar Biotechnology in drug development, private and public financings and a successful sale of the business.
Jonathan Fleming, 53, has been a director since the closing of the Merger in May 2011 and prior to the Merger, was as director of Target since March 2009. Mr. Fleming is, the Managing General Partner of Oxford Bioscience Partners (OBP), an international venture capital firm specializing in life science technology-based investments, which he joined in August 1996 as a General Partner, with offices in Boston and Connecticut. Mr. Fleming has been in the investment business for more than 20 years and has launched and financed growth companies in the United States, Europe, and Israel. Prior to joining OBP in 1996, Mr. Fleming was a Founding General Partner of MVP Ventures in Boston from 1988 to 1996. He began his investment career with TVM Techno Venture Management in Munich, Germany in 1985. Mr. Fleming is also a co-founder of Medica Venture Partners, a venture capital investment firm specializing in early-stage healthcare and biotechnology companies in Israel. Mr. Fleming has been on the Board of Asterand plc (LSE: ATD) since September 2008 and is a director of several private companies including Leerink Swann, a Boston-based investment bank specializing in healthcare companies since June 1998, Laboratory Partners, a clinical diagnostic testing company, since June 2006, and Railrunner, a rail products and services company, since June 1999. Mr. Fleming is a Trustee of the Museum of Science in Boston, a Member of the Board of the New England Healthcare Institute, and a Senior Lecturer at the MIT Sloan School of Business. He holds an MPA from Princeton University and a BA from the University of California, Berkeley. Mr. Fleming brings to our board of directors strategic insight and experience with his long career in venture capital and investing in life sciences technology-based firms for over 20 years.
Ansbert K. Gadicke, MD, 53, has been a director since the closing of the Merger and, prior to the Merger, served a director of Target since November 2003. Dr. Gadicke is a Co-Founder and Managing Director of MPM Capital since August 1996 to date. He led MPM’s effort to build its Advisory and Investment Banking business from 1992 to 1996 and started its Asset Management business in 1996. Prior to founding MPM, Dr. Gadicke was employed by The Boston Consulting Group from 1989 to 1992. Dr. Gadicke received an M.D. from J.W. Goethe University in Frankfurt in 1983. He subsequently held research positions in biochemistry and molecular biology at the German Cancer Research Center from 1984 to 1986, Harvard University from 1987 to 1988, and the Whitehead Institute at MIT from 1988 to 1989. He has published in leading scientific publications including Nature and Cell. Dr. Gadicke is also a director of Cerimon Pharmaceuticals; Dragonfly Sciences; Solasia Pharma K.K., Tokyo; and Verastem, Inc.
He previously served as a director of Arriva Pharmaceuticals, BioMarin, Biovitrum, Chiasma, Coelacanth, Idenix, Kourion, MediGene, Omrix Biopharmaceuticals, Pharmasset, Inc.; PharmAthene, Transform, Xanodyne Pharmaceuticals, and ViaCell. He is a member of the Board of Fellows of Harvard Medical School. Dr. Gadicke was selected as a director because of his business and professional experience.
Kurt Graves, 43, has been a director since the closing of the Merger in May 2011. Mr. Graves is a global industry leader with more than twenty years of US and global general management experience in top-tier U.S. and Europe-based pharmaceutical and biotechnology companies. He served as an Executive Vice President, Chief Commercial Officer and Head of Strategic Development at Vertex Pharmaceuticals Inc. from July 2007 to October 2009 where he led the development of the company’s HCV and CF programs as well as the acquisition of Virochem Pharmaceuticals. Prior to joining Vertex, Mr. Graves held various leadership positions at Novartis Pharmaceuticals from 1999 to June 2007 including a member of the Executive Committee and the Global Head of the General Medicines Business, a $15 billion dollar business with 8 therapeutic area franchises. He was also the first Chief Marketing Officer for the Pharmaceuticals division from September 2003 June 2007. Prior to that, Mr. Graves served as Senior Vice President & General Manager—US Pharma & Commercial Operations; Vice President, Head of US Marketing & Primary Care Franchises; and Vice President & Business Unit Head: Respiratory, GI, Dermatology and Bone Franchises at Novartis.. Prior to joining Novartis in 1999, Mr. Graves held various commercial and general management positions since 1990 at Merck and Astra/Merck Pharmaceuticals including US GI Business Unit Head where he was responsible for developing and commercializing for Prilosec(R) and Nexium(R) and the Prilosec OTC alliance with P&G. Mr. Graves has been Executive Chairman and director of Intarcia Therapeutics, Inc. since August 2010. He has been Executive Chairman of Biolex Therapeutics, Inc. since November 2010. He has also been a director of Pulmatrix Therapeutics, Alevium Pharmaceuticals, and Springleaf Therapeutics since 2010 as well. Mr. Graves earned his B.S. in Biology from Hillsdale College and has attended numerous executive leadership programs at Harvard, Wharton, and University of Michigan. Mr. Graves was selected as a director because of his business and professional experience.
Martin Muenchbach, Ph.D., 40, has been a director since the closing of the Merger in May 2011 and prior to the Merger, he served as an observer to the Board of Directors of Target since February 2007. Mr. Muenchbach launched BB BIOTECH VENTURES II in, and has managed it since, 2004. Previously, he was Partner at BioMedinvest and Investment Advisor at HBM Partners. At BioMedinvest, from 2003 to 2004, he led several of the firm’s investments, and served on the board of portfolio companies. Before the merger of HBM Bioventures and NMT New Medical Technologies, he was Investment Manager at NMT from 1999 to 2003, where he was focusing on international private equity investments in biopharmaceutical and biotechnological companies. Before becoming a venture capitalist, from Dr. Münchbach gained experience in strategic marketing at Sanofi-Synthelabo. Dr. Münchbach holds a Ph.D. in Protein Chemistry, a MSc in Biochemistry and a Master in Industrial Engineering and Management from the Swiss Federal Institute of Technology (ETH), Zurich. Dr. Münchbach’s current board assignments include BioVascular Inc, Molecular Partners AG, Optimer Pharmaceuticals Inc, and Tioga Pharmaceuticals Inc. Dr. Münchbach was selected as a director because of his business and professional experience.
Elizabeth Stoner, M.D., 60, has been a director since the closing of the Merger in May 2011. Dr. Stoner has been a Managing Director at MPM Capital from October 2007 to date and is based at the Boston office. Dr. Stoner is also the Chief Development Officer of Rhythm Pharmaceuticals. She is an industry veteran with broad expertise in clinical research and pharmaceutical product development. Dr. Stoner joined MPM Capital following a 22 year career at Merck Research Laboratories where she started in 1985. At the time of her retirement from Merck in 2007, she served as a Senior Vice President of Global Clinical Development Operations with responsibility for the Merck’s clinical development activities in more than 40 countries. Dr. Stoner also oversaw the clinical development activities of Merck’s Japanese partner, Banyu, led the clinical development for the Merck/Schering-Plough Joint Venture for Zetia/Vytorin, and played a critical leadership role in Merck’s efforts to transform its worldwide clinical development operations. Earlier in her career at Merck, she had led the Proscar clinical development program from inception to establishing Merck as a leader in the field of prostate disease. As the Endocrine Therapeutic Head, Dr. Stoner’s responsibilities included all steroid and lipid metabolism, as well as the growth hormone secretagogue clinical research programs. Prior to her position at Merck, she served as an Assistant Professor of Pediatrics at Cornell University Medical College from 1982 to 1985. She has been a director of Momenta Pharmaceuticals Inc. since October 25, 2007. She also served as director for Metabasis Pharmaceuticals. Dr. Stoner is a Member of the Scientific Advisory Board at Solasia, Inc. She received an M.D. from Albert Einstein College of Medicine, an M.S. in Chemistry from the State University of New York at Stony Brook, and a B.S. in Chemistry from Ottawa University, Kansas. Dr. Stoner was selected as a director because of her knowledge and expertise in the development of pharmaceutical products.
Terms of Office; Voting Arrangements as to Directors
Our directors and officers have been appointed for a one-year term or until their respective successors are duly elected and qualified or until their earlier resignation or removal in accordance with our By-Laws.
Pursuant to a stockholders’ agreement and our certificate of incorporation:
(i) for so long as any shares of Series A-1 Stock are outstanding, the holders of a majority of the shares of Series A-1 Stock outstanding, voting as a separate class, shall have the right to elect two (2) members of the Board of Directors; and
(ii) Oxford Bioscience Partners IV L.P. (together with Saints Capital VI, L.P. and their respective affiliates and certain transferees), HealthCare Ventures VII, L.P. (together with its affiliates and certain transferees) and The Wellcome Trust Limited as trustee of the Wellcome Trust (together with its affiliates and certain transferees) (collectively, the “G3 Holders” and individually, each a “Group”) voting as a separate class shall have the right to elect one (1) member of the Board of Directors of the Corporation by majority vote of the shares of Series A-1 Stock held by them; provided, however, that in order to be eligible to vote or consent with respect to the election of such member of the Board of Directors, a G3 Holder together with members of such G3 Holders’ Group must hold greater than twenty percent (20%) of the shares of Series A-1 Stock purchased under the Series A-1 Stock Purchase Agreement by such G3 Holder and the members of such G3 Holders’ Group; and
(iii) MPM Capital L.P., voting as a separate class, shall have the right to elect one (1) member of the Board of Directors of the Corporation by majority vote of the shares of Series A-1 Stock held by MPM Capital L.P.; provided that such member of the Board of Directors shall be an individual with particular expertise in the development of pharmaceutical products; and, provided, further, that in order to be eligible to vote or consent with respect to the election of such member of the Board of Directors, MPM Capital L.P. together with members of the MPM Group (as defined in the Stockholders’ Agreement) must hold greater than twenty percent (20%) of the shares of Series A-1 Stock purchased under the Series A-1 Stock Purchase Agreement by MPM Capital L.P. and the members of the MPM Group.
As part of the stockholders’ agreement, certain of the stockholders will agree to vote in favor of all nominations pursuant to the foregoing.
Certain Relationships and Transactions
Significant Employees
As of the date hereof, we have no significant employees, other than the Company’s named executive officers.
Family Relationships
There are no family relationships among our directors or executive officers.
Involvement in Certain Legal Proceedings
To our knowledge, there have been no events under any bankruptcy act, no criminal proceedings and no Federal or State judicial or administrative orders, judgments, decrees or findings, no violations of any Federal or State securities law, and no violations of any Federal commodities law material to the evaluation of the ability and integrity of any director (existing or proposed), executive officer (existing or proposed), promoter or control person of the Company during the past ten (10) years.
Transactions with Related Persons
Since October 2010 until the closing of the Merger and Short-Form Merger, the Target funded our ongoing Exchange Act filing requirements and other costs associated with investigating and analyzing an acquisition. Management estimates such amounts to be de minimus. We have utilized the office space and equipment of MPM Asset Management LLC, our sole stockholder prior to the Redemption completed inconnection with the Merger and those of Target from time to time, in all cases, at no cost to us.
As described above, Dr. Lyttle, a current director and executive officer was the President and Chief Executive Officer of Target prior to the Merger, and each of Dr. Lyttle, Mr. Auerbach, Dr. Gadicke, and Mr. Fleming, each directors, served as directors of the Target prior to the Merger. In addition, certain investment funds affiliated with MPM Asset Management LLC (the sole stockholder of the Company prior to the Merger), MPM BioVentures III Fund, was an investor in Target prior to the Merger. Dr. Gadicke, the Managing Director of MPM Capital was a control person of the Company prior to the Merger and affiliated with major stockholders of the Target prior to the Merger. The shares held by MPM Asset Management LLC were repurchased by the Company for any aggregate purchase price of $50,000 plus reimbursement of certain costs contemporaneously with the closing of the Merger.
Policies and Procedures for Review, Approval or Ratification of Transactions with Related Persons
We do not have any special committee, policy or procedure related to the review, approval or ratification of related party transactions, other than as required by the Delaware General Corporation Law.
Director Independence
Our securities are not listed on a national securities exchange or on any inter-dealer quotation system which has a requirement that directors be independent. As both of our present directors are also executive officers of the Company, we do not presently have any independent directors. We evaluate independence by the standards for director independence established by applicable laws, rules, and listing standards, including, without limitation, the standards for independent directors established by the New York Stock Exchange, Inc., the NASDAQ National Market, and the Securities and Exchange Commission.
Subject to some exceptions, these standards generally provide that a director will not be independent if (a) the director is, or in the past three years has been, an employee of ours; (b) a member of the director’s immediate family is, or in the past three years has been, an executive officer of ours; (c) the director or a member of the director’s immediate family has received more than $120,000 per year in direct compensation from us other than for service as a director (or for a family member, as a non-executive employee); (d) the director or a member of the director’s immediate family is, or in the past three years has been, employed in a professional capacity by our independent public accountants, or has worked for such firm in any capacity on our audit; (e) the director or a member of the director’s immediate family is, or in the past three years has been, employed as an executive officer of a company where one of our executive officers serves on the compensation committee; or (f) the director or a member of the director’s immediate family is an executive officer of a company that makes payments to, or receives payments from, us in an amount which, in any twelve-month period during the past three years, exceeds the greater of $1,000,000 or two percent of that other company’s consolidated gross revenues.
Each of Alan Auerbach, Jonathan Fleming, Ansbert K. Gadicke, M.D., Kurt Graves, Martin Muenchbach, Ph.D. and Elizabeth Stoner, M.D. qualify as independent directors under the foregoing standard.
Board of Directors’ Meetings
During the fiscal year ended December 31, 2010, our board of directors did not meet. We did not hold an annual meeting in 2010. Our board of directors conducted all of its business and approved all corporate action during the fiscal year ended December 31, 2010 by the unanimous written consent of its members, in the absence of formal board meetings.
Committees of the Board of Directors
As our Common Stock is not presently listed for trading or quotation on a national securities exchange or NASDAQ, we are not presently required to have board committees. In addition, due to our small size and limited operations to date, we do not presently have an audit committee, compensation committee or nominating committee or other committees performing similar functions. Our entire board presently performs the functions that would otherwise be performed by such committees. We have not adopted any procedures by which security holders may
recommend nominees to our board of directors. We do not have a diversity policy. We do not have a qualified financial expert at this time because it has not been able to hire a qualified candidate. Further, we believe that it has inadequate financial resources at this time to hire such an expert. We intend to continue to search for a qualified individual for hire. The new board of directors will consider forming an audit committee, compensation committee and nominating and corporate governance committee.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our directors and officers, and persons who beneficially own more than ten percent (10%) of the Company’s Common Stock (collectively, the “Reporting Persons”), to file reports with the SEC of beneficial ownership and reports of changes in beneficial ownership of Common Stock on Forms 3, 4 and 5. Reporting Persons are required by applicable SEC rules to furnish us with copies of all such forms filed with the SEC pursuant to Section 16(a) of the Exchange Act. To our knowledge, based solely on our review of the copies of the Forms 3, 4 and 5 received by it during the fiscal year ended December 31, 2010, the Company believes that all reports required to be filed by such persons with respect to the Company’s fiscal year ended December 31, 2010 were timely filed, except the following reports: (a) Form 3 filed by C. Richard Lyttle on April 29, 2011; and (b) Form 3 filed by Nicholas Harvey on April 29, 2011.
Code of Ethics
We have not adopted a Code of Business Conduct and Ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions in that our officers and directors serve in these capacities.
Board Leadership Structure and Role on Risk Oversight
Dr. C. Richard Lyttle currently serves as our Chief Executive Officer, President and Director, and Nick Harvey currently serves as our Chief Financial Officer, Treasurer, and Secretary. At present, we have determined this leadership structure, together with the rest of our board of directors, is appropriate due to our small size and limited operations and resources. Our current directors are exclusively involved in the general oversight of risks that could affect our business. The proposed directors will continue to evaluate our leadership structure and modify such structure as appropriate based on our size, resources, and operations.
Indemnification of Directors and Officers
Section 145 of the DGCL permits indemnification of officers, directors and other corporate agents under certain circumstances and subject to certain limitations. Articles 7 and 8 of our certificate of incorporation provides that we will indemnify, to the fullest extent permitted by Section 145 of the DGCL, as amended from time to time, each person that such section grants the Corporation power to indemnify, including in circumstances in which indemnification is otherwise discretionary under Delaware law. In addition, we have entered into separate indemnification agreements with our directors and executive officers which would require us, among other things, to indemnify them against certain liabilities which may arise by reason of their status or service (other than liabilities arising from willful misconduct of a culpable nature). The indemnification provisions in our certificate of incorporation and the indemnification agreements to be entered into between us and our directors and executive officers may be sufficiently broad to permit indemnification of our directors and executive officers for liabilities (including reimbursement of expenses incurred) arising under the Securities Act. We also intend to maintain director and officer liability insurance, if available on reasonable terms, to insure our directors and officers against the cost of defense, settlement or payment of a judgment under certain circumstances.
Legal Proceedings
We are not aware of any legal proceedings in which any director, officer, or record or beneficial owner of 5% or more of our outstanding capital stock is a party adverse to us or has a material interest adverse to us, or an affiliate of such persons.
Stockholder Communication with the Board of Directors
Stockholders may send communications to our board of directors by writing to the Company, c/o Radius Health, Inc., 201 Broadway, 6th Fl., Cambridge, MA, 02116 Attention: Board of Directors.
Executive and Director Compensation
The following tables summarize all compensation earned by or paid to our Chief Executive and Financial Officer (Principal Executive and Financial Officer) and other named executive officers during the two fiscal years ended December 31, 2010 and 2009. We have not had any formal policy for determining the compensation of executive officers.
SUMMARY COMPENSATION TABLE
By Us Prior to the Merger
Prior to the Merger, we had not issued any stock options or maintained any stock option or other equity incentive plans. Prior to the Merger, we have no plans in place and never maintained any plans that provided for the payment of retirement benefits or benefits that will be paid primarily following retirement including, but not limited to, tax qualified deferred benefit plans, supplemental executive retirement plans, tax-qualified deferred contribution plans and nonqualified deferred contribution plans. Similarly, we had no contracts, agreements, plans or arrangements, whether written or unwritten, that provide for payments to any named executive officers or any other persons following, or in connection with the resignation, retirement or other termination of a named executive officer, or a change in control of us or a change in a named executive officer’s responsibilities following a change in control. The following table summarizes all compensation earned by or paid to Chief Executive Officer and Financial Officer during two fiscal years ended December 31, 2010 and 2009.
|
Name and |
|
Year |
|
Salary |
|
Bonus |
|
Stock |
|
Option |
|
Non- |
|
Non- |
|
All |
|
Total ($) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Steven St. Peter, |
|
2010(1) |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
President, Director, Principal Executive Officer and Principal Financial Officer |
|
2009 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
John Vander Vort, |
|
2010(1) |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
Secretary and Director |
|
2009 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Richard Lyttle, |
|
2010(2) |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
President, Director, Principal Executive Officer |
|
2009 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nick Harvey, |
|
2010(2) |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
|
Senior Vice President, Director Principal Financial Officer, Treasurer and Secretary |
|
2009 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
Notes:
(1) Resigned in November 2010.
(2) Appointed in November 2010.
By Target Prior to the Merger
The following tables summarize all compensation earned by or paid to Target’ Chief Executive and Financial Officer (Principal Executive and Financial Officer) and other named executive officers during the two fiscal years ended December 31, 2010 and 2009, as of May 23, 2011. Target did not have any formal policy for determining the compensation of executive officers. Instead, base salaries for our named executive officers typically are established through arm’s length negotiation at the time the executive is hired. On an annual basis, our board of directors reviews and evaluates, with input from our President and Chief Executive Officer, the need for adjustment of the base salaries of our executives based on changes and expected changes in the scope of an executive’s responsibilities, including promotions, the individual contributions made by and performance of the executive during the prior fiscal year, the executive’s performance over a period of years, overall labor market conditions, the relative ease or difficulty of replacing the executive with a well-qualified person, our overall growth and development as a company and general salary trends in our industry.
Each executive officer is eligible to receive an annual performance-based cash bonus, in an amount up to a fixed percentage of his base salary. At the beginning of each year the board develops with input from our President and Chief Executive Officer a list of corporate goals for the year that would be used as a guideline to assess the annual performance of the executive officers. As soon as practical after the year is completed, the board would review actual performance against the stated goals and determine subjectively what it believes to be the appropriate level of cash bonus. Whether or not a cash bonus is paid is entirely at the discretion of the board of directors.
As a public operating company, we intend to create a compensation committee that will be charged with developing a formal policy for determining and reviewing the compensation of executive officers on a regular basis.
|
Name and |
|
Year |
|
Salary |
|
Bonus |
|
Stock |
|
Option |
|
Non- |
|
Non- |
|
All |
|
Total ($) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Richard Lyttle, |
|
2010 |
|
378,622 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
1714.50 |
|
380,336.50 |
|
|
President, Director, and Chief Executive Officer |
|
2009 |
|
369,387 |
|
73,877 |
|
0 |
|
0 |
|
0 |
|
0 |
|
1,584.00 |
|
444,848.00 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Nick Harvey, |
|
2010 |
|
278,492 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
1,305.48 |
|
279,797.48 |
|
|
Treasurer, Secretary and Chief Fianncial Officer and Director |
|
2009 |
|
271,700 |
|
40,755 |
|
0 |
|
0 |
|
0 |
|
0 |
|
851.40 |
|
313,306.40 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Louis O’Dea, |
|
2010 |
|
319,363 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
1,032.00 |
|
320,395.00 |
|
|
Sr. Vice-President and Chief Medical Officer |
|
2009 |
|
311,574 |
|
51,410 |
|
0 |
|
0 |
|
0 |
|
0 |
|
104.64 |
|
363,088.64 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gary Hattersley, |
|
2010 |
|
223,860 |
|
0 |
|
0 |
|
0 |
|
0 |
|
0 |
|
240.00 |
|
224,100.00 |
|
|
Vice President, Biology & Pharmacology |
|
2009 |
|
218,400 |
|
27,300 |
|
0 |
|
0 |
|
0 |
|
0 |
|
240.00 |
|
245,940.00 |
|
Notes:
(1) Bonuses related to prior years are shown in same year, even if paid subsequent to year end. The amount of bonuses for the fiscal year ended in 2010 have not been determined yet.
(2) All amounts are attributable to life insurance premiums paid by Radius.
Each of the named executive officers are at-will employees eligible for discretionary bonus and equity incentive awards with certain severance rights discussed further below.
If his employment is terminated without cause or he resigns for good reason, Dr. Lyttle will receive 12 months salary in severance payments, payable in accordance with the payroll practice then in effect, and for a period of 12 months, the continued payment or subsidy of health insurance benefits to the same extent as being paid or subsidized at the time of termination. Upon a change of control, if not offered a position or terminated within 12 months following the closing, Dr. Lyttle shall be entitled to 18 months salary in severance payments and 50% of his then unvested options will become immediately vested and exercisable. Under certain circumstances additional options may vest.
If his employment is terminated without cause or he resigns with good reason, Mr. Harvey will receive 6 months salary in severance payments, payable in accordance with the payroll practice then in effect, and for a period of 6 months, the continuation of health insurance at no cost to him for 6 months and all options which would have vested in the 6 months following such termination but for such termination shall become immediately exercisable. If the Company is acquired, 50% of his then unvested options will become immediately vested and exercisable.
Following termination of employment without cause, and subject to signing a general release, Mr. Hattersley and Mr. O’Dea are entitled to severance payments equal to 6 months of their then current base salary (minus required withholdings). In addition, they elect and remain eligible for COBRA coverage during the such six month period, they are entitled to be reimbursed for the portion of COBRA premium would have been paid by the Company had such person remained employed by Company during such period.
Compensation of Directors
No member of our board of directors received any compensation for services as a director of the Company during the fiscal year ended December 31, 2010. Neither we nor Target has had any formal policy governing the compensation of directors.
During Target’s fiscal year ended December 31, 2010, Target granted to Alan Auerbach options to acquire 256,666 shares of Radius Common Stock at an exercise price of $1.35 per share. The following table summarizes all compensation earned by or paid to Mr. Auerbach in the fiscal year ended December 31, 2010. Mr. Auerbach was not a director of Target in 2009. Mr. Auerbach’s options were assumed by us in the Merger.
|
Name and |
|
Year |
|
Salary |
|
Bonus |
|
Stock |
|
Option |
|
Non- |
|
Non- |
|
All |
|
Total ($)(1) |
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||
|
Alan H. Auerbach (Director) |
|
2010 |
|
0 |
|
0 |
|
0 |
|
$ |
194,040 |
|
0 |
|
0 |
|
0 |
|
$ |
194,040 |
|
(1) The value of the option award was calculated based on 256,666 options outstanding at December 31, 2010 multiplied by the fair value per share of $0.756 that was derived using the Black-Scholes-Merton option pricing model. This model used the following assumptions in valuing the options; expected dividend yield of 0, risk-free interest rate of 1.92%, expected term of 6.25 years, and volatility of 58%.
Grants of Plan-Based Awards and Equity Awards
No plan-based awards or equity awards were granted to any of our or Target’s named executive officers or directors during the fiscal year ended December 31, 2010.
Option Exercises and Stock Vested; Outstanding Equity Awards at Fiscal Year End
No unexercised options or warrants were held by any of our named executive officers at December 31, 2010. No options to purchase our capital stock were exercised by any of our named executive officers, nor was any restricted stock held by such executive officers vested during the fiscal year ended December 31, 2010.
During the fiscal year ended December 31, 2010, no Target options held by named executive officers, director or proposed director were exercised. At the end of such period, the following options (which will be assumed in the Merger by the Company) were held by our named executive officers, directors and proposed Company directors. We assumed all Target options in the Merger:
|
|
|
Number of Unexercised |
|
|
|
|
| |||
|
Name |
|
Exercisable |
|
Unexercisable |
|
Exercise Price |
|
Expiration |
| |
|
C. Richard Lyttle |
|
101,562 |
|
6,770 |
|
$ |
1.50 |
|
10/28/14 |
|
|
|
|
148,605 |
|
9,906 |
|
$ |
0.90 |
|
7/12/17 |
|
|
|
|
190,005 |
|
12,667 |
|
$ |
1.20 |
|
5/8/18 |
|
|
|
|
80,977 |
|
5,398 |
|
$ |
1.50 |
|
12/3/18 |
|
|
Nicholas Harvey |
|
66,711 |
|
16,677 |
|
$ |
0.90 |
|
7/12/17 |
|
|
|
|
51,459 |
|
11,875 |
|
$ |
1.20 |
|
5/8/18 |
|
|
|
|
13,496 |
|
13,496 |
|
$ |
1.20 |
|
12/3/18 |
|
|
Louis O’Dea |
|
13,141 |
|
9,500 |
|
$ |
1.50 |
|
2/15/16 |
|
|
|
|
34,622 |
|
6,924 |
|
$ |
0.90 |
|
7/12/17 |
|
|
|
|
57,634 |
|
13,300 |
|
$ |
1.20 |
|
5/8/18 |
|
|
|
|
15,115 |
|
15,115 |
|
$ |
1.20 |
|
12/3/18 |
|
|
Gary Hattersley |
|
10,833 |
|
0 |
|
$ |
1.50 |
|
12/16/13 |
|
|
|
|
5,416 |
|
0 |
|
$ |
1.50 |
|
2/15/16 |
|
|
|
|
20,804 |
|
2,972 |
|
$ |
0.90 |
|
7/12/17 |
|
|
|
|
24,700 |
|
5,700 |
|
$ |
1.20 |
|
5/8/18 |
|
|
|
|
6,478 |
|
6,478 |
|
$ |
1.20 |
|
12/3/18 |
|
|
Alan Auerbach |
|
0 |
|
138,973 |
|
$ |
1.35 |
|
10/12/20 |
|
|
|
|
0 |
|
117,693 |
|
$ |
1.35 |
|
10/12/20 |
|
2003 Long-Term Incentive Plan
Generally, In the Merger, we assumed the Target’s 2003 Long-Term Incentive Plan (the “Incentive Plan”) and all options to acquire common stock of Target issued thereunder. The plan is intended to assist us and our affiliates in attracting and retaining employees and consultants of outstanding ability and to promote the identification of their interests with those of our stockholders and our affiliates. Under the plan, we are authorized to issue incentive stock options, nonstatutory stock options, rights, incentive stock grants, performance stock grants and restricted stock. Only incentive stock options and non-statutory stock options have been granted under the plan. We have 1,319,682 options issued and unexercised, 998,254 of which are vested. The Company has the right to issue additional awards under the plan for an aggregate of 315,194 shares of Common Stock. If an option or right expires or terminates for any reason (other than termination by virtue of the exercise of a related option or related right, as the case may be) without having been fully exercised, if shares of restricted stock are forfeited, or if shares covered by an incentive share award or performance award are not issued or are forfeited, the unissued or forfeited shares that had been subject to the award become available for the grant of additional awards
Administration. The compensation committee of the board of directors administers the incentive plan. In the event that there is no compensation committee, the board of directors administers the plan. The committee or the board may delegate authority to administer the incentive plan to any other committee. Subject to the terms of the incentive plan, the plan administrator (the board or its authorized committee) selects the recipients of awards and determine the:
· number of shares of common stock covered by the awards and the dates upon which such awards become exercisable or any restrictions lapse, as applicable;
· type of award and the price and method of payment for each such award;
· vesting period for options and restricted stock, and any acceleration;
· exercise price or purchase price of awards; and
· duration of options.
Incentive Stock Options. Incentive stock options are intended to qualify as incentive stock options under Section 422 of the Internal Revenue Code and are granted pursuant to incentive stock option agreements. The plan administrator determines the exercise price for an incentive stock option, which may not be less than 100% of the fair market value of the stock underlying the option determined on the date of grant. Notwithstanding the foregoing, incentive options granted to employees who own, or are deemed to own, more than 10% of our voting stock, must have an exercise price not less than 110% of the fair market value of the stock underlying the option determined on the date of grant.
Nonstatutory Stock Options. Nonstatutory stock options are granted pursuant to nonstatutory stock option agreements. The plan administrator determines the exercise price for a nonstatutory stock option.
Vesting. Options granted under the plan generally vest in sixteen (16) quarterly installments, each quarterly installment being equal in number of shares as possible (as determined by the Company in its reasonable discretion), with the first quarterly installment vesting one quarter after the date of the grant, and an additional quarterly installment vesting on the first day of each calendar quarter thereafter, until all of the shares subject to the option are fully vested and the option may be exercised as to 100% of the shares issuable upon exercise thereof.
Rights. Stock appreciation rights are granted pursuant to award agreements. Rights may be granted under the plan (a) in connection with, an at the same time as, the grant of an option under the plan, (b) by amendment of an outstanding option granted under the plan, or (c) independent of any option granted under the plan. A rights granted under the plan in relation to an option granted under the plan are referred to as a related right. Upon exercise, in whole or in part, the holder is entitled to receive either cash or that umber of shares, or a combination thereof, in an amount having an aggregate fair market value, as determined under the plan, as of the exercise date not to exceed the number of shares subject to the portion of the right exercised multiplied by an amount equal to the excess of (x) the fair market value of the right as of the exercise date over (b) either (i) the fair market value on the grant date if the right is not a related right or (ii) the exercise price of the related option if the right is a related right. The exercise, in whole or in part, of a related right reduces the number of shares subject to the related option and an exercise of a related options will reduce the number of shares subject to the related right.
Restricted Stock Awards. Restricted stock awards are granted pursuant to restricted stock award agreements. The purchase price for restricted stock awards are determined by the plan administrator. Restricted stock awards may be subject to a repurchase right in accordance with a vesting schedule determined by the plan administrator.
Performance Awards. Performance awards are granted pursuant award agreements that provide for the payment of cash and/or issuance of shares on terms and conditions determined by the plan administrator.
Other Equity Awards. The plan administrator may grant other awards based in whole or in part by reference to our common stock.
Changes to Capital Structure. In the event of certain types of changes in our capital structure, such as a share split, the number of shares reserved under the plan and the number of shares and exercise price or strike price, if applicable, of all outstanding awards will be appropriately adjusted.
Dividends. Any award under the plan may confer upon the recipient the right to receive dividend payments or dividend equivalent payments with respect to the shares subject to the award. Such dividend payments may be paid currently or credited to an account in favor of the recipient. Such dividends may be settled in cash or shares, as determined by the plan administrator.
Employment Agreements
Each of the named executive officers are at-will employees eligible for discretionary bonus and equity incentive awards with certain severance rights discussed further below.
Potential Payments upon Termination or Change in Control
If his employment is terminated without cause or he resigns for good reason, Dr. Lyttle will receive 12 months salary in severance payments, payable in accordance with the payroll practice then in effect, and for a period of 12 months, the continued payment or subsidy of health insurance benefits to the same extent as being paid or subsidized at the time of termination. Upon a change of control, if not offered a position or terminated within 12 months following the closing, Dr. Lyttle shall be entitled to 18 months salary in severance payments and 50% of his then unvested options will become immediately vested and exercisable. Under certain circumstances additional options may vest.
If his employment is terminated without cause or he resigns with good reason, Mr. Harvey will receive 6 months salary in severance payments, payable in accordance with the payroll practice then in effect, and for a period of 6 months, the continuation of health insurance at no cost to him for 6 months and all options which would have
vested in the 6 months following such termination but for such termination shall become immediately exercisable. If the Company is acquired, 50% of his then unvested options will become immediately vested and exercisable.
Following termination of employment without cause, and subject to signing a general release, Mr. Hattersley and Mr. O’Dea are entitled to severance payments equal to 6 months of their then current base salary (minus required withholdings). In addition, they elect and remain eligible for COBRA coverage during the such six month period, they are entitled to be reimbursed for the portion of COBRA premium would have been paid by the Company had such person remained employed by Company during such period.
Estimated Benefits and Payments Upon Termination of Employment
The following table describes the potential payments and benefits upon termination of our named executive officers’ employment before or after a change in control of our company as described above, as if each officer’s employment terminated as of December 31, 2010, the last business day of the 2010 fiscal year.
|
Name |
|
Benefit |
|
Termination |
| |
|
Rich Lyttle |
|
Severance |
|
$ |
567,933 |
|
|
|
|
Option Acceleration |
|
$ |
3,584 |
|
|
|
|
COBRA Premiums |
|
$ |
0 |
|
|
|
|
Vacation Payout |
|
$ |
14,562 |
|
|
|
|
Total Value |
|
$ |
586,079 |
|
|
|
|
|
|
|
| |
|
Nick Harvey |
|
Severance |
|
$ |
139,246 |
|
|
|
|
Option Acceleration |
|
$ |
5,655 |
|
|
|
|
COBRA Premiums |
|
$ |
10,382 |
|
|
|
|
Vacation Payout |
|
$ |
10,711 |
|
|
|
|
Total Value |
|
$ |
165,994 |
|
|
|
|
|
|
|
| |
|
Louis O’Dea |
|
Severance |
|
$ |
159,682 |
|
|
|
|
Option Acceleration |
|
$ |
0 |
|
|
|
|
COBRA Premiums |
|
$ |
10,382 |
|
|
|
|
Vacation Payout |
|
$ |
12,283 |
|
|
|
|
Total Value |
|
$ |
182,347 |
|
|
|
|
|
|
|
| |
|
Gary Hattersley |
|
Severance |
|
$ |
111,930 |
|
|
|
|
Option Acceleration |
|
$ |
0 |
|
|
|
|
COBRA Premiums |
|
$ |
0 |
|
|
|
|
Vacation Payout |
|
$ |
8,527 |
|
|
|
|
Total Value |
|
$ |
120,457 |
|
For purposes of valuing the severance and vacation payments in the table above, we used each executive officer’s base salary in effect at the end of 2010 and the number of accrued but unused vacation days at the end of 2010.
The value of option acceleration shown in the table above was calculated based on the assumption that the officer’s employment was terminated and the change in control (if applicable) occurred on December 31, 2010 and that the fair market value of our common stock on that date was $1.35. The value of the vesting acceleration was calculated by multiplying the number of unvested shares subject to each option by the difference between the fair market value of our common stock as of December 31, 2010 and the exercise price of the option.
Pension Benefits
No named executive officers received or held pension benefits during the fiscal year ended December 31, 2010.
Nonqualified Deferred Compensation
No nonqualified deferred compensation was offered or issued to any named executive officer during the fiscal year ended December 31, 2010.
Compensation Committee Interlocks and Insider Participation
We do not have a Compensation Committee. All compensation matters are approved by the full Board.
Security Ownership of Certain Beneficial Owners and Management
The following table sets forth (i) each person known by the Company to be the beneficial owner calculated in accordance with Rule 13d-3(d)(1) promulgated under the Exchange Act of more than 5% of the outstanding shares of Common Stock, (ii) each director and executive officer of the Company and (iii) all officers and directors as a group. Unless otherwise stated in the table or its footnotes, the person and entities listed below have the sole voting power and investment power with respect to the shares set forth next to one’s name. Unless otherwise noted, the address of each stockholder below is c/o Radius Health, Inc., 201 Broadway, 6th Floor Cambridge, MA 02139.
|
Name, (Title) and Address |
|
Shares |
|
Title of Class |
|
Percentage of |
|
|
C. Richard Lyttle, Ph.D. (Chief Executive Officer, President and Director) |
|
454,484(2) |
|
Common Stock |
|
51.60 |
% |
|
|
|
|
|
|
|
|
|
|
Nick Harvey (Chief Financial Officer, Treasurer, and Secretary) |
|
118,603(3) |
|
Common Stock |
|
22.04 |
% |
|
|
|
|
|
|
|
|
|
|
Louis O’Dea |
|
155,908(4) |
|
Common Stock |
|
26.02 |
% |
|
|
|
|
|
|
|
|
|
|
Gary Hattersley |
|
79,335(5) |
|
Common Stock |
|
12.50 |
% |
|
|
|
|
|
|
|
|
|
|
Dr. Ansbert Gadicke (Director) |
|
6,563,550(6) |
|
Common Stock |
|
92.20 |
% |
|
|
|
200,909(7) |
|
Series A-1 Preferred Stock |
|
48.62 |
% |
|
|
|
402,115(8) |
|
Series A-2 Preferred Stock |
|
40.90 |
% |
|
|
|
53,331(9) |
|
Series A-3 Preferred Stock |
|
37.50 |
% |
|
|
|
|
|
|
|
|
|
|
Alan H. Auerbach (Director) |
|
64,166(10) |
|
Common Stock |
|
10.35 |
% |
|
|
|
|
|
|
|
|
|
|
Jonathan Fleming (Director) |
|
1,349,510(11) |
|
Common Stock |
|
71.64 |
% |
|
|
|
109,718(12) |
|
Series A-2 Preferred Stock |
|
11.16 |
% |
|
|
|
25,233(13) |
|
Series A-3 Preferred Stock |
|
17.74 |
% |
|
|
|
|
|
|
|
|
|
|
Kurt Graves (Director) |
|
0 |
|
|
|
0 |
% |
|
|
|
|
|
|
|
|
|
|
Dr. Martin Muenchbach (Director) |
|
1,487,580(14) |
|
Common Stock |
|
72.81 |
% |
|
|
|
43,596(15) |
|
Series A-1 Preferred |
|
10.55 |
% |
|
|
|
105,162(16) |
|
Series A-2 Preferred |
|
10.70 |
% |
|
|
|
|
|
|
|
|
|
|
Elizabeth Stoner (Director) |
|
0 |
|
|
|
0 |
% |
|
Entities Affiliated with: |
|
|
|
|
|
|
|
|
MPM Bioventures III, L.P. |
|
|
|
|
|
|
|
|
c/o MPM Capital |
|
6,563,550(17) |
|
Common Stock |
|
92.20 |
% |
|
200 Carendon St |
|
200,909(18) |
|
Series A-1 Preferred |
|
48.62 |
% |
|
54th Fl. |
|
402,115(19) |
|
Series A-2 Preferred |
|
40.90 |
% |
|
Boston, MA 02116 |
|
53,331(20) |
|
Series A-3 Preferred |
|
37.50 |
% |
|
|
|
|
|
|
|
|
|
|
The Wellcome Trust Limited as trustee of |
|
|
|
|
|
|
|
|
The Wellcome Trust |
|
|
|
|
|
|
|
|
215 Euston Road |
|
2,358,470(21) |
|
Common Stock |
|
80.93 |
% |
|
London NW1 2BE |
|
25,522(22) |
|
Series A-1 Preferred Stock |
|
6.18 |
% |
|
England |
|
210,325 |
|
Series A-2 Preferred Stock |
|
21.39 |
% |
|
|
|
|
|
|
|
|
|
|
HealthCare Ventures VII, L.P. |
|
1,815,920(23) |
|
Common Stock |
|
80.08 |
% |
|
55 Cambridge Parkway, Suite 102 |
|
19,651(24) |
|
Series A-1 Preferred Stock |
|
4.76 |
% |
|
Cambridge, Massachusetts 02142-1234 |
|
98,278 |
|
Series A-2 Preferred Stock |
|
10.00 |
% |
|
|
|
63,663 |
|
Series A-3 Preferred Stock |
|
44.76 |
% |
|
|
|
|
|
|
|
|
|
|
Entities Affiliated with: |
|
|
|
|
|
|
|
|
Saints Captial VI, L.P. |
|
1,528,584(25) |
|
Common Stock |
|
73.89 |
% |
|
475 Sansome Street |
|
16,375(26) |
|
Series A-1 Preferred Stock |
|
3.96 |
% |
|
Suite 1850 |
|
109,718(27) |
|
Series A-2 Preferred Stock |
|
11.16 |
% |
|
San Francisco, CA 94111 |
|
25,233(28) |
|
Series A-3 Preferred Stock |
|
17.74 |
% |
|
|
|
|
|
|
|
|
|
|
BB Biotech Ventures II, L.P. |
|
|
|
|
|
|
|
|
Traflagar Court |
|
|
|
|
|
|
|
|
Les Banques |
|
|
|
|
|
|
|
|
St. Peter Port |
|
|
|
|
|
|
|
|
Guernsey |
|
1,487,580(29) |
|
Common Stock |
|
72.81 |
% |
|
Channel Islands |
|
43,596(30) |
|
Series A-1 Preferred Stock |
|
10.55 |
% |
|
GY1 3QL |
|
105,162 |
|
Series A-2 Preferred Stock |
|
10.70 |
% |
|
|
|
|
|
|
|
|
|
|
Entities Affiliated with: |
|
|
|
|
|
|
|
|
Oxford Bioscience Partners |
|
|
|
|
|
|
|
|
222 Berkley Street |
|
1,364,824(31) |
|
Common Stock |
|
71.64 |
% |
|
Suite 1650 |
|
109,718(32) |
|
Series A-2 Preferred Stock |
|
11.16 |
% |
|
Boston, MA 02116 |
|
25,233(33) |
|
Series A-3 Preferred Stock |
|
17.74 |
% |
|
|
|
|
|
|
|
|
|
|
Healthcare Private Equity Limited |
|
|
|
|
|
|
|
|
Partnership |
|
|
|
|
|
|
|
|
Edinburgh One |
|
|
|
|
|
|
|
|
Morrison Street |
|
628,910(34) |
|
Common Stock |
|
53.09 |
% |
|
Edinburgh EH3 8BE |
|
6,805 |
|
Series A-1 Preferred Stock |
|
1.65 |
% |
|
U.K |
|
56,086 |
|
Series A-2 Preferred Stock |
|
5.70 |
% |
|
|
|
|
|
|
|
|
|
|
Brookside Capital Partners Fund, L.P. |
|
|
|
|
|
|
|
|
Bain Capital, LLC |
|
|
|
|
|
|
|
|
111 Huntington Avenue |
|
409,400(35) |
|
Common Stock |
|
42.43 |
% |
|
Boston, MA 02199 |
|
40,940 |
|
Series A-1 Preferred Stock |
|
9.91 |
% |
|
|
|
|
|
|
|
|
|
|
BB Biotech Growth N.V. |
|
|
|
|
|
|
|
|
Asset Management BAB N.V. |
|
|
|
|
|
|
|
|
Ara Hill Top Building, Unit A-5 |
|
|
|
|
|
|
|
|
Pletterijweg Oost 1 |
|
409,400(36) |
|
Common Stock |
|
42.43 |
% |
|
Curaçao, Dutch Caribbean |
|
40,940 |
|
Series A-1 Preferred Stock |
|
9.91 |
% |
|
|
|
|
|
|
|
|
|
|
Ipsen Pharma S.A.S |
|
|
|
|
|
|
|
|
65, quai Georges Gorse |
|
|
|
|
|
|
|
|
92100 Boulogne Billancourt |
|
173,260(37) |
|
Common Stock |
|
23.77 |
% |
|
France |
|
17,326 |
|
Series A-1 Preferred Stock |
|
4.19 |
% |
|
|
|
|
|
|
|
|
|
|
Stavros C. Manolagas |
|
|
|
|
|
|
|
|
35 River Ridge Circle |
|
|
|
|
|
|
|
|
Little Rock, AR 72227 |
|
91,040(38) |
|
Common Stock |
|
16.39 |
% |
|
|
|
|
|
|
|
|
|
|
Nordic Bioscience |
|
|
|
|
|
|
|
|
Herlev Hovedgade 207 |
|
|
|
|
|
|
|
|
2730 Herlev |
|
64,430(39) |
|
|
|
10.39 |
% |
|
Denmark |
|
6,443 |
|
Series A-5 Preferred Stock |
|
100 |
% |
|
|
|
|
|
|
|
|
|
|
Michael Rosenblatt |
|
|
|
|
|
|
|
|
130 Lake Ave |
|
|
|
|
|
|
|
|
Newton, MA 02459 |
|
46,664(40) |
|
Common Stock |
|
8.40 |
% |
|
|
|
|
|
|
|
|
|
|
John Katzenellenbogen Trust |
|
|
|
|
|
|
|
|
704 West Pennsylvania Avenue |
|
|
|
|
|
|
|
|
Urbana, Illinois 61801 |
|
49,399(41) |
|
Common Stock |
|
8.89 |
% |
|
|
|
|
|
|
|
|
|
|
Chris Miller |
|
|
|
|
|
|
|
|
11Edgar Walker Court |
|
|
|
|
|
|
|
|
Hingham, MA 02043 |
|
33,355(42) |
|
Common Stock |
|
6.00 |
% |
|
|
|
|
|
|
|
|
|
|
Bart Henderson |
|
|
|
|
|
|
|
|
48 Prentiss Lane |
|
|
|
|
|
|
|
|
Belmont, MA 02478 |
|
30,468(43) |
|
Common Stock |
|
5.48 |
% |
|
|
|
|
|
|
|
|
|
|
All Officers and Directors as a group (10 individuals)(44) |
|
10,414,333 |
|
Common Stock |
|
96.17 |
% |
(1) Because shares of Preferred Stock vote together with Common Stock on an as-converted basis the percentages of beneficial ownership reported in this column do not reflect the beneficial owner’s voting percentage of our outstanding capital stock. A more accurate reflection of the each beneficial owner’s voting percentage is their percentage of the fully-diluted shares of Common Stock which equals 16,086,040 shares (the “Fully-Diluted Shares”), assuming the conversion of all issued and outstanding shares of Preferred Stock, the exercise of all issued and outstanding warrants to purchase Common Stock or Preferred Stock, as well as the conversion of all shares of Preferred Stock issuable upon exercise of such warrants. In order to provide accurate disclosure of the relevant voting percentages of each beneficial owner included in this table we have set forth each such beneficial owner’s ownership percentage of the Fully-Diluted Shares in each applicable footnote below.
(2) Includes 454,484 options to purchase our Common Stock anticipated to be exercisable within 60 days after May 23, 2011. Dr. Lyttle beneficially owns 3.24% of the Fully-Diluted Shares.
(3) Includes 118,603 options to purchase our Common Stock anticipated to be exercisable within 60 days after May 23, 2011. Mr. Harvey beneficially owns 0.92% of the Fully-Diluted Shares.
(4) Includes 155,908 options to purchase our Common Stock anticipated to be exercisable within 60 days after May 23, 2011. Mr. O’Deal beneficially owns 1.15% of the Fully-Diluted Shares.
(5) Includes 79,335 options to purchase our Common Stock anticipated to be exercisable within 60 days after May 23, 2011. Mr. Hattersley beneficially owns 0.49% of the Fully-Diluted Shares.
(6) Includes 82,220 shares of Common Stock issuable upon conversion of 8,222 shares of Company Series A-1 Preferred Stock, 121,940 shares of Common Stock issuable upon conversion of 12,194 shares of Company Series A-2 Preferred Stock, 29,850 shares of Common Stock issuable upon conversion of 2,985 shares of Company Series A-3 Preferred Stock issued to MPM BioVentures III, L.P. (“BV III”), in the Merger; 1,222,900 shares of Common Stock issuable upon conversion of 122,290 shares of Company Series A-1 Preferred Stock, 1,813,640 shares of Common Stock issuable upon conversion of 181,364 shares of Company Series A-2 Preferred Stock, and 443,950 shares of Common Stock issuable upon conversion of 44,395 shares of Company Series A-3 Preferred Stock issued to MPM BioVentures III-QP, L.P. (“BV III QP”), in the Merger; 103,350 shares of Common Stock issuable upon conversion of 10,335 shares of Company Series A-1 Preferred Stock, 153,270 shares of Common Stock issuable upon conversion of 15,327 shares of Company Series A-2 Preferred Stock, and 37,520 shares of Common Stock issuable upon conversion of 3,752 shares of Company Series A-3 Preferred Stock issued to MPM BioVentures III GmbH & Co. Beteiligungs K.G. (“BV III KG”), in the Merger; 36,930 shares of Common Stock issuable upon conversion of 3,693 shares of Company Series A-1 Preferred Stock, 54,770 shares of Common Stock issuable upon conversion of 5,477 shares of Company Series A-2 Preferred Stock, and 13,400 shares of Common Stock issuable upon conversion of 1,340 shares of Company Series A-3 Preferred Stock issued to MPM BiVentures III Parallel Fund, L.P. (“BV III PF”), in the Merger; 23,680 shares of Common Stock issuable upon conversion of 2,368 shares of Company Series A-1 Preferred Stock, 35,110 shares of Common Stock issuable upon conversion of 3,511 shares of Company Series A-2 Preferred Stock, and 8,590 shares of Common Stock issuable upon conversion of 859 shares of Company Series A-3 Preferred Stock issued to MPM Asset Management Investors 2003 BVIII LLC (“AM LLC”) in the Merger; 540,010 shares of Common Stock issuable upon conversion of 54,001 shares of Company Series A-1 Preferred Stock, and 1,842,420 shares of Common Stock issuable upon conversion of 184,242 shares of Company Series A-2 Preferred Stock issued to MPM Bio IV NVS Strategic Fund, L.P. (“MPM NVS”) in the Merger. Additionally, BV III has committed to purchase an aggregate of 6,874 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III QP has committed to purchase an aggregate of 102,238 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III KG has committed to purchase an aggregate of 8,640 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III PF has committed to purchase an aggregate of 3,087 shares of Company Series A-1 Preferred Stock at two subsequent closings, AM LLC has committed to purchase an aggregate of 1,979 shares of Company
Series A-1 Preferred Stock at two subsequent closings, and MPM NVS has committed to purchase an aggregate of 60,536 shares of Company Series A-1 Preferred Stock at two subsequent closings. MPM BioVentures III GP, L.P. and MPM BioVentures III LLC (“BV3LLC”), are the direct and indirect general partners of BV III, BV III QP, BV III KG, and BV III PF. MPM BioVentures IV GP LLC and MPM BioVentures IV LLC (“BV4LLC”) are the direct and indirect general partners of MPM NVS. Dr. Gadicke is a Series A Member of BV3LLC, Member of BV4LLC and Manager of AM LLC and shares voting and investment power with respect to these shares. Each entity mentioned above and Dr. Gadicke disclaim beneficial ownership of all shares not held by it or him of record. Dr. Gadicke may be deemed to beneficially own 40.80% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(7) Includes of 8,222 shares of Company Series A-1 Preferred Stock issued to BV III in the Merger; 122,290 shares of Company Series A-1 Preferred Stock issued to BV III QP in the Merger; 10,335 shares of Company Series A-1 Preferred Stock, issued to BV III KG in the Merger; 3,693 shares of Company Series A-1 Preferred Stock issued to BV III PF in the Merger; 2,368 shares of Company Series A-1 Preferred Stock issued to AM LLC in the Merger; and 54,001 shares of Company Series A-1 Preferred Stock issued to MPM NVS in the Merger. Dr. Gadicke is a Series A Member of BV3LLC, Member of BV4LLC and Manager of AM LLC and shares voting and investment power with respect to these shares. Each entity mentioned above and Dr. Gadicke disclaim beneficial ownership of all shares not held by it or him of record. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(8) Includes 12,194 shares of Company Series A-2 Preferred Stock issued BV III in the Merger; 181,364 shares of Company Series A-2 Preferred Stock issued to BV III QP in the Merger, 15,327 shares of Company Series A-2 Preferred Stock issued BV III KG in the Merger; 5,477 shares of Company Series A-2 Preferred Stock issued to BV III PF in the Merger; 3,511 shares of Company Series A-2 Preferred Stock issued to AM LLC in the Merger; and 184,242 shares of Company Series A-2 Preferred Stock issued to MPM NVS in the Merger. Dr. Gadicke is a Series A Member of BV3LLC, Member of BV4LLC and Manager of AM LLC and shares voting and investment power with respect to these shares. Each entity mentioned above and Dr. Gadicke disclaim beneficial ownership of all shares not held by it or him of record. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(9) Includes 2,985 shares of Company Series A-3 Preferred Stock issued to BV III in the Merger; 44,395 shares of Company Series A-3 Preferred Stock issued to BV III QP, in the Merger; 3,752 shares of Company Series A-3 Preferred Stock issued to BV III KG, in the Merger; 1,340 shares of Company Series A-3 Preferred Stock issued to BV III PF, in the Merger; and 859 shares of Company Series A-3 Preferred Stock issued to AM LLC in the Merger. Dr. Gadicke is a Series A Member of BV3LLC, Member of BV4LLC and Manager of AM LLC and shares voting and investment power with respect to these shares. Each entity mentioned above and Dr. Gadicke disclaim beneficial ownership of all shares not held by it or him of record. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(10) Includes 64,166 options to purchase our Common Stock anticipated to be exercisable within 60 days after May 23, 2011. Mr. Auerbach beneficially owns 0.40% of the Fully-Diluted Shares.
(11) Includes 15,173 shares of Common Stock and 1,086,280 shares of Common Stock issuable upon conversion of 108,628 shares of Company Series A-2 Preferred Stock, 249,830 shares of Common Stock issuable upon conversion of 24,983 shares of Company Series A-3 Preferred Stock (the “OBP IV Shares”) held directly by Oxford Bioscience Partners IV L.P. (“OBP IV”); and 151 shares of Common Stock and 10,900 shares of Common Stock issuable upon conversion of 1,090 shares of Company Series A-2 Preferred Stock, 2,500 shares of Common Stock issuable upon conversion of 250 shares of Company Series A-3 Preferred Stock (the “mRNA Fund II Shares”) held directly by mRNA Fund II L.P. (“mRNA Fund II”). The Oxford Shares are indirectly held by OBP Management IV L.P. (“OBP Management IV”), the sole general partner of each of OBP IV and mRNA Fund II, and the individual general partners of OBP Management IV. The individual general partners of OBP Management IV are Jonathan Fleming and Alan Walton. Each entity mentioned above and Messrs. Fleming and Walton disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Oxford Shares in which such entity or individual has no actual pecuniary interest therein. Mr. Fleming may be deemed to beneficially own 8.48% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(12) Includes 108,628 shares of Company Series A-2 Preferred Stock held directly by OBP IV (the “OBP IV A-2 Shares”) and 1,090 shares of Company Series A-2 Preferred Stock held directly by mRNA Fund II (the “mRNA Fund II A-2 Shares”). The OBP IV A-2 Shares and the mRNA Fund II A-2 Shares are indirectly held by Oxford Bioscience Partners IV L.P. (“OBP L.P.”), a member of OBP IV, and OBP Management IV, the sole general partner of OBP LP and mRNA II, and the individual managers of OBP Management IV. The individual managers of OBP Management IV are Jonathan Fleming and Alan Walton. Each entity mentioned above and Messrs. Fleming and Walton disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-2 Shares in which such entity or individual has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(13) Includes 24,983 shares of Company Series A-3 Preferred Stock held directly by OBP IV (the “OBP IV A-3 Shares”) and 250 shares of Company Series A-3 Preferred Stock held directly by mRNA Fund II (the “mRNA Fund II A-3 Shares”). The OBP IV A-3 Shares and the mRNA Fund II A-3 Shares are indirectly held by OBP LP, a member of OBP IV, and OBP Management IV, the sole general partner of OBP LP and mRNA II, and the individual managers of OBP Management IV. The individual managers of OBP Management IV are Jonathan Fleming and Alan Walton. Each entity mentioned above and Messrs. Fleming and Walton disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-3 Shares in which such entity or individual has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(14) Includes 435,960 shares of Common Stock issuable upon conversion of 43,596 shares of Company Series A-1 Preferred Stock (the “BBBV LP A-1 Preferred Stock”), and 1,051,620 shares of Common Stock issuable upon conversion of 105,162 shares of Company Series A-2 Preferred Stock (together with the BBBV LP A-1 Preferred Stock the “BBBV LP Shares”) issued to BB Biotech Ventures II L.P. (“BBBV LP”) in the Merger. Additionally, BBBV LP has committed to purchase an aggregate of 40,940 shares of Company Series A-1 Preferred Stock at two subsequent closings. BB Biotech Ventures GP (Guernsey) Limited (“BBBV Limited”) is the General Partner of BBBV LP. The directors of BBBV Limited are Jan Bootsma; Pascal Mahieux, and Ben Morgan. Dr. Muenchbach, the Senior Investment Advisor Private Equity at Bellevue Asset Management AG, advises Asset Management BAB N.V. (“AMB NV”) who,
pursuant to a services agreement with BAM AG, advises the directors of BBBV Limited. Each of the foregoing, except BBBV LP in the case of the BBBV LP Shares, disclaims beneficial ownership of the BBBV LP Shares except to the extent of their pecuniary interest therein, if any. Dr. Muenchbach may be deemed to beneficially own 9.25% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BBBV LP, BBBV Limited, Jan Bootsma, Pascal Mahieux, Ben Morgan, and Martin Muenchbach.
(15) Includes 43,596 shares of Company Series A-1 Preferred Stock issued to BB Biotech Ventures II L.P. in the Merger. BB Biotech Ventures II L.P. has committed to purchase an aggregate of 40,940 shares of Company Series A-1 Preferred Stock at two subsequent closings. Voting and investment power with respect to these shares is shared by the general partners of this fund.
(16) Includes 105,162 shares of Company Series A-2 Preferred Stock issued to BB Biotech Ventures II L.P. in the Merger. Voting and investment power with respect to these shares is shared by the general partners of this fund.
(17) Includes 82,220 shares of Common Stock issuable upon conversion of 8,222 shares of Company Series A-1 Preferred Stock, 121,940 shares of Common Stock issuable upon conversion of 12,194 shares of Company Series A-2 Preferred Stock, 29,850 shares of Common Stock issuable upon conversion of 2,985 shares of Company Series A-3 Preferred Stock issued to BV III in the Merger; 1,222,900 shares of Common Stock issuable upon conversion of 122,290 shares of Company Series A-1 Preferred Stock, 1,813,640 shares of Common Stock issuable upon conversion of 181,364 shares of Company Series A-2 Preferred Stock, and 443,950 shares of Common Stock issuable upon conversion of 44,395 shares of Company Series A-3 Preferred Stock issued to BV III QP, in the Merger; 103,350 shares of Common Stock issuable upon conversion of 10,335 shares of Company Series A-1 Preferred Stock, 153,270 shares of Common Stock issuable upon conversion of 15,327 shares of Company Series A-2 Preferred Stock, and 37,520 shares of Common Stock issuable upon conversion of 3,752 shares of Company Series A-3 Preferred Stock issued to BV III KG, in the Merger; 36,930 shares of Common Stock issuable upon conversion of 3,693 shares of Company Series A-1 Preferred Stock, 54,770 shares of Common Stock issuable upon conversion of 5,477 shares of Company Series A-2 Preferred Stock, and 13,400 shares of Common Stock issuable upon conversion of 1,340 shares of Company Series A-3 Preferred Stock issued to BV III PF, in the Merger; 23,680 shares of Common Stock issuable upon conversion of 2,368 shares of Company Series A-1 Preferred Stock, 35,110 shares of Common Stock issuable upon conversion of 3,511 shares of Company Series A-2 Preferred Stock, and 8,590 shares of Common Stock issuable upon conversion of 859 shares of Company Series A-3 Preferred Stock issued to MPM Asset Management Investors AM LLC in the Merger; 540,010 shares of Common Stock issuable upon conversion of 54,001 shares of Company Series A-1 Preferred Stock, and 1,842,420 shares of Common Stock issuable upon conversion of 184,242 shares of Company Series A-2 Preferred Stock issued to MPM NVS in the Merger. Additionally BV III has committed to purchase an aggregate of 6,874 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III QP has committed to purchase an aggregate of 102,238 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III KG has committed to purchase an aggregate of 8,640 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III PF has committed to purchase an aggregate of 3,087 shares of Company Series A-1 Preferred Stock at two subsequent closings, AM LLC has committed to purchase an aggregate of 1,979 shares of Company Series A-1 Preferred Stock at two subsequent closings, and MPM NVS has committed to purchase an aggregate of 60,536 shares of Company Series A-1 Preferred Stock at two subsequent closings. Voting and investment power is shared with Dr. Gadicke and the other general partners of these funds. MPM BioVentures and MPM BioVentures III LLC (“BV3LLC”), are the direct and indirect general partners of BV III, BV III QP, BV III KG, and BV III PF. Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III and Dennis Henner are Series A members of BV III LLC and managers of AM LLC. MPM BioVentures III Parallel Fund, L.P. MPM BioVentures IV GP LLC and MPM BioVentures IV LLC are the direct and indirect general partners of MPM. Luke Evnin, Ansbert Gadicke, Ashley L. Dombkowski, William Greene, Vaughn M. Kailian, James Paul Scopa, Steven St. Peter and John Vander Vort are members of BV IV LLC. Each fund mentioned above disclaims
beneficial ownership of all shares not held by it of record. Each fund mentioned above may be deemed to beneficially owns 40.80% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(18) Includes of 8,222 shares of Company Series A-1 Preferred Stock issued to BV III in the Merger; 122,290 shares of Company Series A-1 Preferred Stock issued to BV III QP, in the Merger; 10,335 shares of Company Series A-1 Preferred Stock, issued to BV III KG, in the Merger; 3,693 shares of Company Series A-1 Preferred Stock issued to BV III PF in the Merger; 2,368 shares of Company Series A-1 Preferred Stock, issued to AM LLC in the Merger; and 54,001 shares of Company Series A-1 Preferred Stock issued to MPM NVS in the Merger. BV III has committed to purchase an aggregate of 6,874 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III QP has committed to purchase an aggregate of 102,238 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III KG has committed to purchase an aggregate of 8,640 shares of Company Series A-1 Preferred Stock at two subsequent closings, BV III PF has committed to purchase an aggregate of 3,087 shares of Company Series A-1 Preferred Stock at two subsequent closings, AM LLC has committed to purchase an aggregate of 1,979 shares of Company Series A-1 Preferred Stock at two subsequent closings, and MPM NVS has committed to purchase an aggregate of 60,536 shares of Company Series A-1 Preferred Stock at two subsequent closings. Voting and investment power is shared with Dr. Gadicke and the other general partners of these funds. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(19) Includes 12,194 shares of Company Series A-2 Preferred Stock issued to BV III in the Merger; 181,364 shares of Company Series A-2 Preferred Stock issued to BV III QP, in the Merger 15,327 shares of Company Series A-2 Preferred Stock issued to BV III KG in the Merger; 5,477 shares of Company Series A-2 Preferred Stock issued to BV III PF, in the Merger; 3,511 shares of Company Series A-2 Preferred Stock issued to AM LLC in the Merger; and 184,242 shares of Company Series A-2 Preferred Stock issued to MPM NVS in the Merger. Voting and investment power is shared with Dr. Gadicke and the other general partners of these funds. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(20) Includes 2,985 shares of Company Series A-3 Preferred Stock issued to BV III in the Merger; 44,395 shares of Company Series A-3 Preferred Stock issued to BV III QP in the Merger; 3,752 shares of Company Series A-3 Preferred Stock issued to BV III KG, in the Merger; 1,340 shares of Company Series A-3 Preferred Stock issued BV III PF, in the Merger; and 859 shares of Company Series A-3 Preferred Stock issued to AM LLC in the Merger. Voting and investment power is shared with Dr. Gadicke and the other general partners of these funds. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BV III, BV III QP, BV III KG, BV III PF, AM LLC, MPM NVS, Luke Evnin, Ansbert Gadicke, Nicholas Galakatos, Michael Steinmetz, Kurt Wheeler, Nicholas Simon III, Dennis Henner, Ashley L. Dombkowski, William Greene, Vaughn Kailian, James Paul Scopa, Steven St. Peter, and John Vander Vort.
(21) Includes 255,220 shares of Common Stock issuable upon conversion of 25,522 shares of Company Series A-1 Preferred Stock, and 2,103,250 shares of Common Stock issuable upon conversion of 210,325 shares of Company Series A-2 Preferred Stock issued to The Wellcome Trust Limited as trustee of The Wellcome Trust in the Merger. Additionally, The Wellcome Trust Limited as trustee of The Wellcome
Trust has committed to purchase an aggregate of 51,044 shares of Company Series A-1 Preferred Stock at two subsequent closings. The Wellcome Trust Limited as trustee of The Wellcome Trust beneficially owns 14.66% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 25, 2011 by The Wellcome Trust Limited as trustee of The Wellcome Trust.
(22) The Wellcome Trust Limited as trustee of The Wellcome Trust has committed to purchase an aggregate of 51,044 shares of Company Series A-1 Preferred Stock at two subsequent closings.
(23) Includes 83,113 shares of Common Stock and 196,510 shares of Common Stock issuable upon conversion of 19,651 shares of Company Series A-1 Preferred Stock, 982,780 shares of Common Stock issuable upon conversion of 98,278 shares of Company Series A-2 Preferred Stock, 636,630 shares of Common Stock issuable upon conversion of 63,663 shares of Company Series A-3 Preferred Stock issued to HealthCare Ventures VII, L.P. (“HCVVII”) in the Merger. Additionally, HCVVII has committed to purchase an aggregate of 39,302 shares of Company Series A-1 Preferred Stock at two subsequent closings. HealthCare Partners VII, L.P. (“HCPVII”) is the General Partner of HCVVII. The General Partners of HCPVII are James H. Cavanaugh, Ph.D., Harold R. Werner, John W. Littlechild, Christopher Mirabelli, Ph.D., and Augustine Lawlor. HCVVII beneficially owns 11.81% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by HCVVII, HCPVII, James H. Cavanaugh, Ph.D., Harold R. Werner, John W. Littlechild, Christopher Mirabelli, Ph.D., and Augustine Lawlor.
(24) HCVVII has committed to purchase an aggregate of 39,302 shares of Company Series A-1 Preferred Stock at two subsequent closings.
(25) Includes: (i) 15,173 shares of Common Stock (the “OBP IV Common Shares”) held directly by OBP IV—Holdings LLC (“OBP IV”); (ii) 1,498,240 shares of Common Stock (the “OBP IV Conversion Shares” and, together with the OBP IV Common Shares, the “OBP IV Saints Shares”) issuable to OBP IV upon the conversion of 16,213 shares of Company Series A-1 Preferred Stock held directly by OBP IV, 108,628 shares of Company Series A-2 Preferred Stock held directly by OBP IV and 24,983 shares of Company Series A-3 Preferred Stock held directly by OBP IV; (iii) 151 shares of Common Stock (the “mRNA II Common Shares”) held directly by mRNA II—Holdings LLC (“mRNA Saints II”); (iv) 15,020 shares of Common Stock (the “mRNA II Conversion Shares” and, together with the mRNA II Common Shares, the “mRNA II Shares”) issuable to mRNA II upon the conversion of 162 shares of Company Series A-1 Preferred Stock held directly by mRNA II, 1,090 shares of Company Series A-2 Preferred Stock held directly by mRNA II and 250 shares of Company Series A-3 Preferred Stock held directly by mRNA II. The OBP Saints IV Shares and the mRNA Saints II Shares are referred to herein as the “Saints Shares.” The Saints Shares are indirectly held by Saints Capital Granite, L.P. (“Saints LP”), a member of OBP IV and mRNA II, Saints Capital Granite, LLC (“Saints LLC”), the sole general partner of Saints LP, and the individual managers of Saints LLC. The individual managers of Saints LLC are Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer. Each entity mentioned above and Messrs. Halsted, Quinlivan and Sawyer disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints Shares in which such entity or individual has no actual pecuniary interest therein. The entities affiliated with Saints Capital VI, L.P. may be deemed to beneficially own 9.5% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(26) Includes: 16,213 shares of Company Series A-1 Preferred Stock held directly by OBP IV, and 162 shares of Company Series A-1 Preferred Stock held directly by mRNA II. The Saints A-1 Shares are indirectly held by Saints LP, a member of OBP IV and mRNA II, Saints LLC, the sole general partner of Saints LP, and the individual managers of Saints LLC. The individual managers of Saints LLC are Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer . Each entity mentioned above and Messrs. Halsted, Quinlivan and Sawyer disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-1 Shares in which such entity or individual
has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(27) Includes: 108,628 shares of Company Series A-2 Preferred Stock held directly by OBP IV (the “OBP IV A-2 Shares”) and 1,090 shares of Company Series A-2 Preferred Stock held directly by mRNA II. (together with the OBP IV A-2 Shares, the “Saints A-2 Shares”). The Saints A-2 Shares are indirectly held by Saints LP, a member of OBP IV and mRNA II, Saints LLC, the sole general partner of Saints LP, and the individual managers of Saints LLC. The individual managers of Saints LLC are Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer . Each entity mentioned above and Messrs. Halsted, Quinlivan and Sawyer disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-2 Shares in which such entity or individual has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(28) Includes: 24,983 shares of Company Series A-3 Preferred Stock held directly by OBP IV (the “OBP IV A-3 Shares”); and 250 shares of Company Series A-3 Preferred Stock held directly by mRNA II. (together with the OBP IV A-3 Shares, the “Saints A-3 Shares”). The Saints A-3 Shares are indirectly held by Saints LP, a member of OBP IV and mRNA II, Saints LLC, the sole general partner of Saints LP, and the individual managers of Saints LLC. The individual managers of Saints LLC are Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer . Each entity mentioned above and Messrs. Halsted, Quinlivan and Sawyer disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-3 Shares in which such entity or individual has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(29) Includes BBBV LP Shares BBBV LP in the Merger. Additionally, BBBV LP has committed to purchase an aggregate of 40,940 shares of Company Series A-1 Preferred Stock at two subsequent closings. BBBV Limited is the General Partner of BBBV LP. The directors of BBBV Limited are Jan Bootsma; Pascal Mahieux, and Ben Morgan. Additionally, Martin Muenchbach, the Senior Investment Advisor Private Equity at Bellevue Asset Management AG, advises AMB NV who, pursuant to a services agreement with BAM AG, advises the directors of BBBV Limited. Each of the foregoing, except BBBV LP in the case of the BBBV LP Shares, disclaims beneficial ownership of the BBBV LP Shares except to the extent of their pecuniary interest therein, if any. BBBV LP beneficially owns 9.25% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by BBBV LP, BBBV Limited, Jan Bootsma, Pascal Mahieux, Ben Morgan, and Martin Muenchbach.
(30) BBBV LP has committed to purchase an aggregate of 40,940 shares of Company Series A-1 Preferred Stock at two subsequent closings.
(31) Includes the OBP IV Shares and the mRNA Fund II Shares. The OBP IV Shares are indirectly held by Oxford Bioscience
Partners IV L.P. (“OBP LP”), a member of OBP IV. The mRNA II Shares are indirectly held by mRNA Fund II L.P. (“mRNA LP”), a member of mRNA II. The Oxford Shares are indirectly held by OBP Management IV, the sole general partner of each of OBP LP and mRNA LP, and the individual general partners of OBP Management IV. The individual general partners of OBP Management IV are Jonathan Fleming and Alan Walton. Each of the entities and individuals mentioned above disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the OBP IV Shares and mRNA Fund II Shares in which such entity or individual has no actual pecuniary interest therein. The entities affiliated with Oxford Bioscience Partners may be deemed to beneficially own 8.48% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(32) Includes: 108,628 shares of Company Series A-2 Preferred Stock held directly by OBP IV and 1,090 shares of Company Series A-2 Preferred Stock held directly by mRNA II. The Saints A-2 Shares are indirectly held by OBP LP, a member of OBP IV, and OBP Management IV, the sole general partner of OBP LP and mRNA II, and the individual managers of OBP Management IV. The individual managers of OBP Management IV are Jonathan Fleming and Alan Walton. Each entity mentioned above and Messrs. Fleming and Walton disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-2 Shares in which such entity or individual has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(33) Includes: 24,983 shares of Company Series A-3 Preferred Stock held directly by OBP IV; and 250 shares of Company Series A-3 Preferred Stock held directly by mRNA II. The Saints A-3 Shares are indirectly held by OBP LP, a member of OBP IV, and OBP Management IV, the sole general partner of OBP LP and mRNA II, and the individual managers of OBP Management IV. The individual managers of OBP Management IV are Jonathan Fleming and Alan Walton. Each entity mentioned above and Messrs. Fleming and Walton disclaim beneficial ownership within the meaning of Section 16 of the Securities Exchange Act of 1934, as amended, or otherwise of such portion of the Saints A-2 Shares in which such entity or individual has no actual pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on May 27, 2011 by OBP IV, mRNA II, mRNA Fund II, OBP Management IV, Saints LP, Saints LLC, Jonathan Fleming, Alan Walton, Scott Halsted, David P. Quinlivan, and Kenneth B. Sawyer.
(34) Includes 68,050 shares of Common Stock issuable upon conversion of 6,805 shares of Company Series A-1 Preferred Stock, and 560,860 shares of Common Stock issuable upon conversion of 56,086 shares of Company Series A-2 Preferred Stock. Healthcare Private Equity Limited Partnership (“HPELP”) beneficially owns 3.91% of the Fully-Diluted Shares. The general partner or HPELP is Wavesly Healthcare Private Equity Limited Partnership (“WHPELP”). The controlling stockholders of WHPELP is SWIP Group Limited. Beneficial ownership information is based on information known to the Company.
(35) Includes 409,400 shares of Common Stock issuable upon conversion of 40,940 shares of Company Series A-1 Preferred Stock. Brookside Capital Investors, L.P. (“Brookside Investors”) is the sole general partner of Brookside Capital Partners Fund, L.P. (“Partners Fund”). Brookside Capital Management, LLC is the sole general partner of Brookside Investors. Partners Fund beneficially owns 2.55% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company.
(36) Includes 409,400 shares of Common Stock issuable upon conversion of 40,940 shares of Company Series A-1 Preferred Stock. BB Biotech Growth N.V. (“BB Growth”) is a wholly-owned subsidiary of BB
Biotech AG (“BB Biotech”). The directors and executive officers of BB Biotech are Dr. Thomas D. Szucs, Chairman and Director; Dr. Clive Meanwell, Vice Chairman and Director; and Dr. Erich Hunziker, Director. The directors and executive officers of BB Growth are Dr. Thomas D. Szucs, Statutory Director; Deanna Chemaly, Statutory Director; and Hugo Jan van Neutegem, Statutory Director. BB Growth beneficially owns 2.55% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on June 3, 2011 by BB Biotech and BB Growth.
(37) Includes 173,260 shares of Common Stock issuable upon conversion of 17,326 shares of Company Series A-1 Preferred Stock. Ipsen Pharma S.A.S (“Ipsen Pharma”) is a société par actions simplifiée organized under the laws of France and is a wholly-owned subsidiary of Ipsen S.A. (“Ipsen”), a société anonyme organized under the laws of France. Ipsen’s majority shareholder is Mayroy, a société anonyme organized under the laws of Luxembourg. The directors and executive officers of Ipsen Pharma are Christophe Jean, Director; Claude Bertrand, Director; Etienne De Blois, Director; Philippe Robert-Gorsse, Director; Eric Drape, Director; Claire Giraut, Director; Jean Fabre, Director; Jean-Pierre Dubuc, Director; Didier Veron, Director; and Marc De Garidel, President. The directors of Ipsen are Marc De Garidel, Director and Chief Executive Officer; Anne Beaufour, Director; Henri Beaufour, Director; Hervé Couffin, Director; Antoine Flochel, Director; Gérard Hauser, Director; Pierre Martinet, Director; René Merkt, Director; Yves Rambaud, Director; Klaus-Peter Schwabe, Director and Christophe Vérot, Director. The executive officers of Ipsen are Claire Giraut, Etienne de Blois, Christophe Jean, Claude Bertrand, and Eric Drape. The directors of Mayroy are Anne Beaufour, Antoine Flochel, Beech Tree SA, Bee Master B.V. Holding BV, Henri Beaufour, Klaus Peter Schwabe, and Jean-Pierre Diehl. Ipsen Pharma beneficially owns 1.08% of the Fully-Diluted Shares. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on June 23, 2011 by Ipsen Pharma and Ipsen.
(38) Stavros C. Manolagas beneficially owns 0.57% of the Fully-Diluted Shares.
(39) Includes 64,430 shares of Common Stock issuable upon conversion of 6,443 shares of Company Series A-5 Preferred Stock. Nordic Bioscience beneficially owns 0.40% of the Fully-Diluted Shares. Clinical Development VII A/S (“Nordic VII”). Nordic VII is a wholly-owned subsidiary of Nordic Bioscience Clinical Development A/S (“Nordic A/S”). Nordic A/S is wholly-owned subsidiary of Nordic Bioscience Holding A/S (“Nordic Holding”). Nordic Holding is majority owned by C.C. Consulting A/S (“C.C. Consulting”). Claus Chrstiansen, MD, and Bente Riis Chrstiansen each own 50% of C.C. Consulting. The entities and individuals mentioned above disclaim beneficial ownership of the share except to the extent of their pecuniary interest therein. Beneficial ownership information is based on information known to the Company and a Schedule 13D filed with the SEC on June 17, 2011 by Nordic VII.
(40) Michael Rosenblatt beneficially owns 0.29% of the Fully-Diluted Shares.
(41) Includes 8,961 shares of Common Stock held by Mr. Katzenellenbogen. Mr. Katzenellenbogen is the trustee of the Katzenellenbogen Trust. The Katzenellenbogen Trust may be deemed to beneficially own the shares held by Mr. Katzenellenbogen. The John Katzenellenbogen Trust beneficially owns 0.31% of the Fully-Diluted Shares.
(42) Chris Miller beneficially owns 0.21% of the Fully-Diluted Shares.
(43) Bart Henderson beneficially owns 0.19% of the Fully-Diluted Shares.
(44) The Directors and Officers as a group may be deemed to beneficially own 64.74% of the Fully-Diluted Shares.
Other Information
We file periodic reports with the SEC. You may obtain a copy of these reports by accessing the SEC’s website at http://www.sec.gov. You may also send communications to the Board of Directors at: Radius Health Inc., 201 Broadway, 6th Fl., Cambridge, MA, 02116 Attention: Board of Directors.
Market Price of and Dividends on Radius’ Common Equity and Related Stockholder Matters
There is not currently, and there has never been, any market for any of our securities. Our securities are not eligible for trading on any national securities exchange, the Nasdaq or other over-the-counter markets, including the Over-the-Counter Bulletin Board®.
As of May 20, 2011, we had outstanding 555,594 shares of common stock, 413,254 shares of Series A-1 Preferred Stock, 983,208 shares of Series A-2 Preferred Stock, 142,227 shares of Series A-3 Preferred Stock, 3,998 shares of Series A-4 Preferred Stock, 6,443 shares of Series A-5 Preferred Stock. The shares of Preferred Stock are convertible into shares of common stock as set forth in more detail under the caption “Description of Capital Stock” below. Additionally, we have outstanding options to purchase an aggregate of 1,461,865 of which 1,134,806 are vested shares of common stock and warrants to purchase an aggregate of 818 shares of Series A-1 Preferred Stock, and warrants to purchase 266 shares of common stock.
Additionally, 1,549,596 shares of our common stock, of which 1,549,130 shares are issuable upon conversion of preferred stock, and 818 shares are issuable upon exercise of warrants, are entitled to certain registration rights we have provided to the holders thereof.
Recent Sales of Unregistered Securities
The following summarizes all sales of unregistered securities by Target and the Company, as applicable, since November 2003, adjusted to reflect the Post-Reverse Split numbers, where applicable:
· On February 4, 2008, we sold 5,000,000 shares of Common Stock to MPM Asset Management for an aggregate purchase price equal to $50,000.
· On March 28, 2008, Target issued 3,471,849 shares of Series C Preferred Stock to existing accredited investors for an aggregate purchase price equal to $28,265,844.
· On November 14, 2008, Target issued 1,846,067 shares of Series C Preferred Stock to existing accredited investors for an aggregate purchase price equal to $15,029,639.
· On May 17, 2011, Target issued 9,832,133 shares of Series A-2 Convertible Preferred Stock, 1,422,300 shares of Series A-3 Convertible Preferred Stock, 40,003 Shares of Series A-4 Convertible Preferred stock and 555,549 shares of Common Stock to former stockholder of the Target pursuant to a recapitalization, including a reverse split, and completed a private placement of 2,631,845 shares of Series A-1 Preferred Stock to a group of accredited investors, including former stockholders of Target, all for an aggregate purchase price of $21,428,482. In addition, we issued 173,263 shares of Series A-1 Preferred Stock to Ipsen in lieu of a cash milestone payment on May 17, 2011.
· On May 17, 2011, Target issued 64,430 shares Series A-5 Preferred Stock to Nordic for an aggregate purchase price of $525,153.53.
· On May 17, 2011, as part of the recapitalization, the Company issued warrants to purchase an aggregate of 266 shares of Common Stock to SVB Financial Group in exchange for a previously issued warrant to purchase shares of the Target.
· On May 17, 2011, the Company issued warrants to purchase an aggregate of 8,188 shares of Series A-1 Preferred Stock to Leerink Swann LLC as consideration for services rendered in connection with the Series A-1 Financing.
· On May 17, 2011, the Company issued 413,254 shares of A-1 Convertible Preferred Stock, 983,208 shares of Series A-2 Convertible Preferred Stock, 142,227 shares of Series A-3 Convertible Preferred Stock, 3,998 shares of Series A-4 Convertible Preferred stock, 6,443 shares of Series A-5 Convertible Preferred Stock and 555,549 shares of Common Stock in the Merger to the former stockholders of target in exchange for shares held in the Target pursuant to Section 4(2) of the Securities Act and Rule 506.
· On May 23, 2011, the Company issued warrants to purchase an aggregate of 3,070 shares of Series A-1 Convertible Preferred Stock to lenders in connection with a debt financing pursuant to Section 4(2) and Rule 506.
The offers, sales, and issuances of the securities described above were deemed to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act or Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions acquired the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions had adequate access, through employment, business, stockholder, other relationships or the delivery of an information statement, to information about us.
In addition to the foregoing, from November 2003 until May 23, 2011, the Company issued options pursuant to the Target’s 2003 Long Term Incentive Plan to purchase an aggregate of 1,755,197 shares of Common Stock to employees, consultants and directors with a range of exercise prices from $0.15 to $1.50 per share. The issuance of options described above were deemed to be exempt from registration under the Securities Act in reliance upon Rule 701 promulgated under Section 3(b) of the Securities Act, as transactions pursuant to a written compensation benefit plan and contracts relating to compensation as provided under Rule 701.
Description of Securities
The following description of our capital stock being registered herein is a summary only and is qualified in its entirety by reference to our Certificate of Designations, Certificate of Incorporation, By-Laws, and the Stockholders’ Agreement which are included as Exhibits 3.1, 3.4, 3.5 and 4.1, respectively, of this Form 8-K.
General
As of the date of this prospectus we have authorized capital of 110,000,000 shares, of which 100,000,000 are designated as Common Stock, and 10,000,000 shares are Preferred Stock, of which 1,000,000 are currently designated as Series A-1 Preferred Stock, 983,213 are currently designated as Series A-2 Preferred Stock, 142,230 are currently designated as Series A-3 Preferred Stock, 4,000 are currently designated as Series A-4 Preferred Stock, 7,000 are currently designated as Series A-5 Preferred Stock, and 800,000 are currently designated as Series A-6 Preferred Stock. As of the date of this prospectus we have outstanding 555,594 shares of Common Stock and 1,549,130 shares of Preferred Stock, of which 413,254 shares are Series A-1 Stock; 983,208 shares are Series A-2 Stock; 142,227 shares are Series A-3 Stock; 3,998 shares are Series A-4 Stock; and 6,443 shares are Series A-5 Stock, such outstanding Common Stock and Preferred Stock are held by 59 holders of record. Additionally, we have outstanding three preferred stock purchase warrants (the “Preferred Stock Purchase Warrants”) that entitle the holders to purchase an aggregate of 3,888 shares of Series A-1 Stock at $81.42 per share and one common stock purchase warrant (the “Common Stock Purchase Warrant”) that entitles the holder to purchase 266 shares of Common Stock at $15.00 per share.
Additionally, there are 1,319,682 options issued pursuant to the Inventive Plan that are exercisable into an aggregate of 1,319,682 shares of Common Stock. A total of 2,015,666 shares of Common Stock are available under the Incentive Plan. We plan to file a Form S-8 with the SEC in order to register the 2,015,666 available under the Incentive Plan.
Dividend Rights
Other than with respect to the payment of the Series A-5 Accruing Dividend (as defined in the Certificate of Designations) which ranks senior in payment to any other dividends payable on any and all series of Preferred Stock and upon Liquidation (as defined in Section 4(b) of the Certificate of Designations) or an Event of Sale (as defined in Section 5 of Certificate of Designations), each share of Series A-1 Stock ranks equally with each other share of Series A-1 Stock and senior to all shares of Series A-2 Stock, Series A-3 Stock, Series A-4 Stock, Series A-5 Stock and Series A-6 Stock and senior to all shares of Common Stock and all other classes or series of stock not currently authorized by the Certificate of Designations, except as otherwise approved by the affirmative vote or consent of the holders of shares of Series A-1 Stock, Series A-2 Stock and/or Series A-3 Stock representing at least
70% of the voting power of the shares of Series A-1 Stock, Series A-2 Stock and Series A-3 Stock then outstanding (the “Senior Majority”); each share of Series A-2 Stock ranks equally with each other share of Series A-2 Stock and senior to all shares of Series A-3 Stock, Series A-4 Stock, Series A-5 Stock and Series A-6 Stock and senior to all shares of Common Stock and all other classes or series of stock not currently authorized by the Certificate of Designations, except as otherwise approved by the affirmative vote or consent of the Senior Majority; each share of Series A-3 Stock, Series A-5 Stock and Series A-6 Stock ranks equally with each other share of Series A-3 Stock, Series A-5 and Series A-6 Stock and senior to all shares of Series A-4 Stock and shares of Common Stock and all other classes or series of stock not currently authorized by the Certificate of Designations, except as otherwise approved by the affirmative vote or consent of the Senior Majority. Each share of Series A-4 Stock, ranks equally with each other share of Series A-4 Stock and senior to all shares of Common Stock and all other classes or series of stock not currently authorized by the Certificate of Designations, except as otherwise approved by the affirmative vote or consent of the Senior Majority.
Terms of Conversion
Each share of Preferred Stock is convertible into shares of Common Stock at any time at the election of the holder thereof at the then prevailing conversion price as determined in our Certificate of Incorporation. Each share of Preferred Stock is initially convertible into ten shares of Common Stock. In addition, pursuant to our Certificate of Incorporation, the Preferred Stock shall convert into Common Stock upon (i) an election to convert made by the Senior Majority or (ii) the listing of the Common Stock on a national securities exchange.
Redemption and Liquidation Rights
Shares of our Preferred Stock outstanding at the time of a sale or liquidation of the Company will have a right to receive proceeds, if any, from any such transactions, before any payments are made to holders of our Common Stock, except as otherwise approved by the affirmative vote or consent of the Senior Majority. In the event that there are not enough proceeds to satisfy the entire liquidation preference of our Preferred Stock, holders of our Common Stock will receive nothing in respect of their equity holdings in the Company.
Voting Rights
The Preferred Stock entitles each holder thereof to one vote for each share of Common Stock into which such share of Preferred Stock is convertible, voting together with the Common Stock as a single class, except with respect to certain amendments to the Certificate of Designations, authorization or issuance of any new class or series of senior or parity capital stock (subject to certain exceptions), any liquidation, dissolution or winding up of the Company which require the consent of the Senior Majority. Initially each share of Preferred Stock is convertible into ten shares of Common Stock, and, therefore, each holder of Preferred Stock is entitled to ten votes for every share of Preferred Stock held by them.
Board of Directors
Pursuant to the Stockholders’ Agreement and our Certificate of Incorporation:
(i) for so long as any shares of Series A-1 Stock are outstanding, the holders of a majority of the shares of Series A-1 Stock outstanding, voting as a separate class, shall have the right to elect two members of the Board of Directors;
(ii) The G3 Holders voting as a separate class shall have the right to elect one member of the Board of Directors of the Corporation by majority vote of the shares of Series A-1 Stock held by them; provided, however, that in order to be eligible to vote or consent with respect to the election of such member of the Board of Directors, a G3 Holder together with members of such G3 Holders’ Group must hold greater than (20%) of the shares of Series A-1 Stock purchased under the Series A-1 Stock Purchase Agreement by such G3 Holder and the members of such G3 Holders’ Group; and
(iii) MPM Capital L.P., voting as a separate class, shall have the right to elect one member of the Board of Directors of the Corporation by majority vote of the shares of Series A-1 Stock held by MPM Capital L.P.; provided that such member of the Board of Directors shall be an individual with particular expertise in the development of pharmaceutical products; and, provided, further, that in order to be eligible to vote or consent with respect to the election of such member of the Board of Directors, MPM Capital L.P. together with members of the MPM Group (as defined in the Stockholders’ Agreement) must hold greater than twenty percent (20%) of the shares of Series A-1 Stock purchased under the Series A-1 Stock Purchase Agreement by MPM Capital L.P. and the members of the MPM Group.
The balance of the board is elected by all of the stockholders acting as a single class and voting on an as converted basis.
Anti-dilution Protection
All shares of Preferred Stock shall have weighted-average anti-dilution protection (based on the initial conversion purchase price of $8.142 per share) and anti-dilution protection upon the occurrence of any subdivision or combination of the Common Stock, stock dividend and other distribution, reorganization, reclassification or similar event affecting the Common Stock, subject to certain exceptions.
Preemption Rights
Under the terms of the Stockholders’ Agreement, we are obligated to offer the holders of the Series A-1 Stock, the Series A-2 Stock and the Series A-3 Stock a right of first refusal with respect future equity issuances, subject to customary exceptions.
Indemnification
The Stockholders’ Agreement contains customary cross-indemnification provisions, pursuant to which we are obligated to indemnify the selling stockholders in the event of material misstatements or omissions in the registration statement attributable to us, and the selling stockholders are obligated to indemnify the us for material misstatements or omissions attributable to them.
Restrictions on Alienability
Pursuant to the Stockholders’ Agreement, for a period of 180 days from May 17, 2011, all other parties subject to the Stockholders’ Agreement have agreed, subject to certain exceptions, not to sell, assign, transfer, make a short sale of, loan, grant any option for the purchase of, or exercise registration rights with respect to any shares of Common Stock or other securities convertible into or exercisable for Common Stock, other than to a member of such Stockholders’ Group (as defined in the Stockholders’ Agreement). Notwithstanding the foregoing, on the 30th, 60th, 90th and 180th day after May 17, 2011, these restrictions shall cease to apply to 5%, 15%, 30% and 50%, respectively, of shares of Common Stock issued or issuable upon conversion of the Series A-1 Stock held or issuable to such stockholder at such time. These restrictions do not apply to block trades of 10,000 shares or more of Common Stock issued upon the conversion of Series A-1 Stock or, if, on or at any time after any days indicated in the previous sentence, the average of the closing bid and ask price of the our Common Stock if quoted on any electronic quotation system, including but not limited to the OTC:BB for the five trading days ending on such date, or the average last-sale price of our Common Stock if listed on a national securities exchange for the five trading days ending on such date, is greater than $16.29 per share the percentages set forth in the previous sentence shall be doubled.
Registration Rights
Pursuant to the terms of the Stockholders’ Agreement, on or before the 60th calendar day following the closing of the Merger, we are required to file a resale shelf registration statement (the “Resale Registration Statement”)
pursuant to Rule 415 of the Securities Act, covering the offering and resale of all of the Registrable Shares (meaning all of the Common Stock issued or issuable upon the conversion or exchange of the Preferred Stock). We are required to use reasonable best efforts to have the Resale Registration Statement declared effective by the 90th calendar day following the closing of the Merger unless we receive comments on the Resale Registration Statement from the SEC, in which case we shall use reasonable best efforts to have the Resale Registration Statement declared effective by the 180th calendar day following the closing of the Merger. In the event that the we are notified by the SEC that the Resale Registration Statement will not be reviewed or is no longer subject to further review and comments, we are required to use reasonable best efforts to have the Resale Registration Statement declared effective by the 5th trading day following such notification. We are required to use reasonable best efforts to keep the Resale Registration Statement continuously effective until all Registrable Shares have been sold or may be sold without volume limitations under Rule 144 promulgated under the Securities Act. In the event that any of these provisions are not complied with, as described in greater detail in the Stockholders’ Agreement, on each such Event Date (as defined in the Stockholders’ Agreement) and on each monthly anniversary of each such Event Date until such event is cured, the we are required to pay to each holder an amount in cash equal to one percent of the aggregate purchase price paid by such holder pursuant to the Purchase Agreement for any Registrable Securities (meaning the Preferred Stock) then held by such holder. The maximum aggregate liquidated damages payable to a holder under the Stockholders’ Agreement is 16 percent of the aggregate purchase price paid by such holder pursuant to the Purchase Agreement. Interest accrues at the rate of 10 percent per annum on any unpaid liquidated damages.
Demand Registration Rights. At such time as the Resale Registration Statement is no longer effective, subject to specified exceptions, any party to the Stockholders’ Agreement has the right to demand that we file an unlimited number of registration statements on Form S-3 covering the offering and sale of all or at least $1,000,000 worth of its Registrable Securities.
Piggyback Registration Rights. All parties to the Stockholders’ Agreement have piggyback registration rights. Under these provisions, if we register any securities for public sale, other than pursuant to a registration statement on Form S-4, Form S-8 or Form S-1 (only in connection with an initial public offering), these stockholders will have the right to include their shares in the registration statement, subject to customary exceptions. The underwriters of any underwritten offering will have the right to limit the number of shares having registration rights to be included in the registration statement.
Expenses of Registration. We are responsible for paying all registration expenses, other than underwriting discounts and commissions, related to any demand or piggyback registration.
Share Purchase Warrants
The exercise price and number of shares of Common Stock and Preferred Stock issuable on exercise of the Common Stock Purchase Warrant and the Preferred Stock Purchase Warrants, respectively, may be adjusted in upon certain events that result in a change of the number and/or class of the securities issuable upon exercise or conversion of the applicable Warrants. The exercise period of each warrant expires ten years from the date such warrant was issued. The Preferred Stock Purchase Warrants may be exercised for cash or pursuant to standard cashless exercise provisions. To the extent not previously exercised, the Preferred Stock Purchase Warrants issued to GE and Oxford Finance, referred to herein as the Lender Warrants, shall be deemed to have been automatically exercised pursuant to the cashless exercise provision of such Lender Warrant as of immediately before its expiration, involuntary termination or cancellation if the then fair market value of a share issuable upon exercise of such Lender Warrant exceeds the warrant’s exercise price, unless such automatic exercise is waived in writing prior the Lender to any such automatic exercise. To the extent not previously exercised the closing of any acquisition in which the consideration paid to the Company or stockholders is cash or cash equivalents, at the election of the holder of the Preferred Stock Purchase Warrants issued to Leerink and the Common Stock Purchase Warrants (all of which are issued to SVB Financial Group), the Company shall purchase the unexercised portion of any such warrant for cash for an amount equal to (a) the fair market value of any consideration that would have been received had such Leerink Warrant been exercised immediately before the record date for determining the shareholders entitled to participate in the proceeds of the acquisition, less (b) the aggregate exercise price of such warrant, but in no event less than zero.
Delaware Anti-Takeover Statute
We are subject to Section 203 of the Delaware General Corporation Law. This statute regulating corporate takeovers prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for three years following the date that the stockholder became an interested stockholder, unless:
· prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder;
· upon completion of the transaction that resulted in the interested stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers, and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
· on or subsequent to the date of the transaction, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock which is not owned by the interested stockholder.
Generally, a business combination includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. An interested stockholder is any person who, together with such person’s affiliates and associates (i) owns 15% or more of a corporation’s voting securities or (ii) is an affiliate or associate of a corporation and was the owner of 15% or more of the corporation’s voting securities at any time within the three year period immediately preceding a business combination of the corporation governed by Section 203. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage takeover attempts that might result in a premium over the market price, once a market exists, for the shares of common stock held by our stockholders.
Item 3.02. Unregistered Sales of Equity Securities
As disclosed under Item 2.01 above, in connection with the Merger, the Company issued an aggregate of 1,549,130 shares of its newly-designated Preferred Stock to the former holders of Radius Preferred Stock and 555,594 shares of Common Stock to the former holders of Radius Common Stock, all of which were unregistered. For these issuances, the Company relied on the exemptions from the registration requirements of the
Securities Act provided by Section 4(2) and Rule 506.
Item 4.01 Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
In connection with the closing of the Merger on May 17, 2011, Ernst & Young LLP (“E&Y”), who was the independent registered public accounting firm for Target prior to the Merger, became the independent registered public accounting firm for the Company and Raich Ende Malter & Co. LLP (“REMC”) was dismissed as the independent registered public accountant for the Company. The decision to appoint E&Y and dismiss REMC was recommended, and subsequently approved, by the Board of Directors of the Company.
The reports of REMC on the Company’s financial statements for the period ended December 31, 2010 did not contain an adverse opinion or a disclaimer of opinion, and were not qualified or modified as to uncertainty, audit scope, or accounting principles.
In connection with the audits of the Company’s financial statements for each of the two years ended December 31, 2010 through REMC’s dismissal, there were no disagreements with REMC on any matters of accounting principles or practices, financial statement disclosures, or auditing scope or procedures, which if not resolved to REMC’s satisfaction would have caused REMC to make reference to the matter in their report.
In connection with the audited financial statements of the Company through REMC’s dismissal, there have been no reportable events with the Company as set forth in Item 304(a)(1)(v) of Regulation S-K.
The Company has requested that REMC furnish it with a letter addressed to the Securities & Exchange Commission stating whether it agrees with the above statements. A copy of the letter, dated July 12, is filed herewith as Exhibit 16.1.
Item 5.01. Change in Control of Registrant
The disclosures set forth in Item 2.01 above are hereby incorporated by reference into this Item 5.01.
Item 5.02 Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers.
At the effective time of the Merger, the Company’s board of directors was reconstituted by the appointment of Ansbert Gadicke, MD, Jonathan Fleming, Alan Auerbach as directors (all of whom were directors of Radius immediately prior to the Merger), along with the new appointment of Martin Muenchbach, Ph.D, Kurt Graves, and Elizabeth Stoner, MD as directors and the resignation of Nick Harvey from his role as director of the Company.
At the effective time of the Merger, the following individuals (all of whom were officers of Target prior to the Merger) took the positions set after their names: Dr. Richard Lyttle (President and Chief Executive Officer); Dr. Louis O’Dea (Senior Vice President and Chief Medical Officer); Dr. Gary Hattersley (Vice President, Biology); and Nick Harvey (Senior Vice President and Chief Financial Officer, Secretary and Treasurer). Biographical and other information regarding these individuals is provided under the caption “Management” in Item 2.01 above, which is incorporated by reference into this Item 5.02.
Item 5.06. Change in Shell Company Status
As described in Item 1.01 and 2.01 above, which is incorporated by reference into this Item 5.06, the Company ceased being a shell company (as defined in Rule 12b-2 under the Exchange Act of 1934, as amended) upon completion of the Merger.
Item 9.01. Financial Statements and Exhibits.
(a) As a result of its acquisition of Target as described in Item 2.01, the registrant is filing herewith Target’s audited financial statements for the fiscal years ended December 31, 2010 and 2009 as Exhibit 99.1 to this current report and its unaudited financial statements for the three months ended March 31, 2011 and 2010 as Exhibit 99.1 to this current report.
(b) Pro forma combined financial information for the fiscal year ended December 31, 2010 is attached as Exhibit 99.2 to this current report and Pro forma combined financial information for the three months ended March 31, 2011 is attached as Exhibit 99.2.
(c) Exhibits.
|
Exhibit |
|
Description |
|
|
|
|
|
2.1 |
|
Agreement and Plan of Merger, dated April 25, 2011(1) |
|
|
|
|
|
3.1 |
|
Certificate of Designations of the Company, filed with the Secretary of State of Delaware on May 17, 2011(3) |
|
|
|
|
|
3.2 |
|
Certificate of Merger, filed with the Secretary of State of Delaware on May 17, 2011, by and between Radius Health, Inc. and RHI Merger Corp. (3) |
|
|
|
|
|
3.3 |
|
Certificate of Ownership relating to the merger of Radius Health, Inc. with and into MPM Acquisition Corp. (3) |
|
|
|
|
|
3.4 |
|
Certificate of Incorporation, as filed with the Delware Secretary of State on February 4, 2008.(2) |
|
|
|
|
|
3.5 |
|
By-Laws(2) |
|
|
|
|
|
4.1 |
|
Amended and Restated Stockholders’ Agreement, dated May 17, 2011, by and among the Company, as successor to Radius Health, Inc., and the Stockholders listed therein(3) |
|
10.1* |
|
Clinical Trial Services Agreement and Work Statement NB-1, dated March 29, 2011, by and between the Company, as successor to Radius Health, Inc., and Nordic BioScience Clinical Development VII A/S(3) |
|
|
|
|
|
10.2* |
|
Stock Issuance Agreement, dated March 29, 2011, by and between the Company, as successor to Radius Health, Inc., and Nordic BioScience Clinical Development VII A/S(3) |
|
|
|
|
|
10.3* |
|
Side Letter, dated March 29, 2011, by and between the Company, as successor to Radius Health, Inc., and Nordic BioScience Clinical Development VII A/S(3) |
|
|
|
|
|
10.4* |
|
License Agreement, dated September 27, 2005, by and between the Company, as successor to Radius Health, Inc., and SCRAS S.A.S., on behalf of itself and its Affiliates(3) |
|
|
|
|
|
10.5* |
|
Pharmaceutical Development Agreement, dated January 2, 2006, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
|
|
|
|
10.6* |
|
Amendment No. 1 to Pharmaceutical Development Agreement, dated January 1, 2007, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
|
|
|
|
10.7* |
|
License Agreement Amendment No. 1, dated September 12, 2007, by and between the Company, as successor to Radius Health, Inc., and SCRAS S.A.S. (3) |
|
|
|
|
|
10.8* |
|
Amendment No. 2 to Pharmaceutical Development Agreement, dated January 1, 2009, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
|
|
|
|
10.9* |
|
Amendment No. 3 to Pharmaceutical Development Agreement, dated June 16, 2010, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
|
|
|
|
10.10* |
|
License Agreement Amendment No. 2, dated May 11, 2011, by and between the Company, as successor to Radius Health, Inc., and Ipsen Pharma S.A.S. (3) |
|
|
|
|
|
10.11* |
|
Series A-1 Convertible Preferred Stock Issuance Agreement, dated May 11, 2011, by and between the Company, as successor to Radius Health, Inc., and Ipsen Pharma S.A.S. (3) |
|
|
|
|
|
10.12* |
|
Development and Manufacturing Services Agreement, dated October 16, 2007, by and between the Company, as successor to Radius Health, Inc., and LONZA Sales Ltd. (3) |
|
|
|
|
|
10.13* |
|
Work Order No. 2, dated January 15, 2010, by and between the Company, as successor to Radius Health, Inc., and LONZA Sales Ltd. (3) |
|
|
|
|
|
10.14* |
|
Amendment No. 3 to Work Order No.2, dated December 15, 2010, by and between the Company, as successor to Radius Health, Inc., and LONZA Sales Ltd. (3) |
|
|
|
|
|
10.15* |
|
Development and Clinical Supplies Agreement, dated June 19, 2009, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
10.16* |
|
Amendment No. 1, dated December 31, 2009, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.17* |
|
Amendment No. 2, dated September 16, 2010, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.18* |
|
Amendment No. 3, dated September 29, 2010, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.19* |
|
Change Order Form - Amendment No. 5, dated February 4, 2011, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.20* |
|
Amendment No. 4, dated March 2, 2011, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.21* |
|
Laboratory Services and Confidentiality Agreement, dated March 31, 2004, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories, Inc. (3) |
|
|
|
|
|
10.22* |
|
First Amendment to Laboratory Services and Confidentiality Agreement, dated November 7, 2008, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories, Inc. (3) |
|
|
|
|
|
10.23* |
|
Letter of Payment Authorization, dated November 20, 2010, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories Preclinical Services Montréal Inc. (3) |
|
|
|
|
|
10.24* |
|
Letter of Payment Authorization, dated February 7, 2011, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories Preclinical Services Montréal Inc. (3) |
|
|
|
|
|
10.25* |
|
License Agreement, dated June 29, 2006, by and between the Company, as successor to Radius Health, Inc., and Eisai Co., Ltd. (3) |
|
|
|
|
|
10.26 |
|
Series A-1 Purchase Agreement, dated April 25, 2011, by and among the Company, as successor to Radius Health, Inc., and the Investors listed therein(3) |
|
|
|
|
|
10.26.1 |
|
Amendment No. 1 to Series A-1 Convertible Preferred Stock Purchase Agreement, dated May 11, 2011(5) |
|
|
|
|
|
10.27** |
|
Radius Health, Inc. (f/k/a Nuvios, Inc.) 2003 Long-Term Incentive Plan, assumed in the Merger(3) |
|
|
|
|
|
10.28** |
|
Radius Health, Inc. First Amendment to 2003 Long-Term Incentive Plan effective as of December 15, 2006, assumed in the Merger(3) |
|
|
|
|
|
10.29** |
|
Radius Health, Inc. Second Amendment to 2003 Long-Term Incentive Plan effective as of March 28, 2008, assumed in the Merger(3) |
|
|
|
|
|
10.30** |
|
Radius Health, Inc. Third Amendment to 2003 Long-Term Incentive Plan effective as of November 14, 2008, assumed in the Merger(3) |
|
10.31 |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Form of Stock Option Agreement(3) |
|
|
|
|
|
10.32** |
|
Radius Health, Inc. (f/k/a Nuvios, Inc.) 2003 Long-Term Incentive Plan Stock Option Agreement, dated October 8, 2004, by and between the Company, as successor to Nuvios, Inc., and Richard Lyttle for Option No. 04-103(3) |
|
|
|
|
|
10.33** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Richard Lyttle for Option No. 07-08(3) |
|
|
|
|
|
10.34** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Richard Lyttle for Option No. 08-09(3) |
|
|
|
|
|
10.35** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Richard Lyttle for Option No. 08-14(3) |
|
|
|
|
|
10.36** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated February 15, 2006, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 06-07(3) |
|
|
|
|
|
10.37** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 07-07(3) |
|
|
|
|
|
10.38** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 08-05(3) |
|
|
|
|
|
10.39** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 08-10(3) |
|
|
|
|
|
10.40** |
|
Radius Health, Inc. (f/k/a Nuvios, Inc.) 2003 Long-Term Incentive Plan Stock Option Agreement, dated December 16, 2003, by and between the Company, as successor to Nuvios, Inc., and Gary Hattersley for Option No. 03-001(3) |
|
|
|
|
|
10.41** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated February 15, 2006, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 06-02(3) |
|
|
|
|
|
10.42** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 07-06(3) |
|
|
|
|
|
10.43** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 08-08(3) |
|
|
|
|
|
10.44** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 08-13(3) |
|
|
|
|
|
10.45** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey for Option No. 07-09(3) |
|
10.46** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey for Option No. 08-06(3) |
|
|
|
|
|
10.47** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey for Option No. 08-11(3) |
|
|
|
|
|
10.48 |
|
Employment Letter Agreement, dated July 2, 2004, by and between the Company, as successor to Nuvios, Inc., and C. Richard Edmund Lyttle(3) |
|
|
|
|
|
10.49 |
|
Employment Letter Agreement, November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Gary Hattersley(3) |
|
|
|
|
|
10.50 |
|
Employment Letter Agreement, dated January 30, 2006, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea(3) |
|
|
|
|
|
10.51 |
|
Employment Letter Agreement, dated November 15, 2006, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey(3) |
|
|
|
|
|
10.52 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Ansbert K. Gadicke(3) |
|
|
|
|
|
10.53 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and C. Richard Edmund Lyttle(3) |
|
|
|
|
|
10.54 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Martin Muenchbach(3) |
|
|
|
|
|
10.55 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Jonathan Fleming(3) |
|
|
|
|
|
10.56 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Kurt Graves(3) |
|
|
|
|
|
10.57 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Elizabeth Stoner(3) |
|
|
|
|
|
10.58 |
|
Indemnification Agreement, dated October 12, 2010, by and between the Company, as successor to Radius Health, Inc., and Alan Auerbach(3) |
|
|
|
|
|
10.59 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Michael Rosenblatt, M.D. (3) |
|
|
|
|
|
10.60 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Christopher Mirabelli(3) |
|
|
|
|
|
10.61 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Augustine Lawlor(3) |
|
|
|
|
|
10.62 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Edward Mascioli, M.D. (3) |
|
|
|
|
|
10.63 |
|
Consent to Sublease, dated January 14, 2011, by and among the Company, as successor to Radius Health, Inc., Sonos, Inc., and Broadway/Hampshire Associates Limited Partnership(3) |
|
10.64 |
|
Sublease, dated January 14, 2011, by and between the Company, as successor to Radius Health, Inc., and Sonos, Inc. (3) |
|
|
|
|
|
10.65 |
|
Amended and Restated Warrant to Purchase Common Stock, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and SVB Financial Group(3) |
|
|
|
|
|
10.66** |
|
Warrant to Purchase Series A-1 Convertible Preferred Stock, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Leerink Swann LLC(3) |
|
|
|
|
|
10.67 |
|
Redemption Agreement, by and between MPM Acquisition Corp. and MPM Asset Management LLC, dated April 25, 2011(1) |
|
|
|
|
|
10.68 |
|
Loan and Security Agreement, dated May 23, 2011, with General Electric Capital Corporation as agent and a lender, and Oxford Finance LLC as a lender(4) |
|
|
|
|
|
10.69 |
|
Promissory Note, dated May 23, 2011, issued by the Company to General Electric Capital Corporation in the principal amount of $12,500,000(4) |
|
|
|
|
|
10.70 |
|
Promissory Note, dated May 23, 2011, issued by the Company to Oxford Finance LLC in the principal amount of $3,125,000(4) |
|
|
|
|
|
10.71 |
|
Promissory Note, dated May 23, 2011, issued by the Company to Oxford Finance LLC in the principal amount of $9,375,000(4) |
|
|
|
|
|
10.72 |
|
Warrant to Purchase Shares of Series A-1 Convertible Preferred Stock, dated May 23, 2011, issued by the Company to GE Capital Equity Investments(4) |
|
|
|
|
|
10.73 |
|
Warrant to Purchase Shares of Series A-1 Convertible Preferred Stock, dated May 23, 2011, issued by the Company to Oxford Finance LLC(4) |
|
|
|
|
|
16.1 |
|
Letter from Raich Ende Malter & Co. LLP as to the change in certifying accountant, dated as of July 12, 2011 |
|
|
|
|
|
99.1 |
|
Audited financial statements of Target for the fiscal years ended December 31, 2010 and 2009 and Unaudited financial statements of Target for the three months ended March 31, 2011 and 2010 |
|
|
|
|
|
99.2 |
|
Unaudited Pro Forma Condensed Combined Financial Statements for the year ended December 31, 2010 and the three months ended March 31, 2011 |
*Confidential Treatment Requested by the Registrant. Redacted Portion Filed Separately with the Commission.
**Share numbers and per share prices are presented pre-Reverse Split completed by Radius Health, Inc. on May 17, 2011.
(1) Incorporated by reference to the Company’s Current Report on Form 8-K filed on April 29, 2011.
(2) Incorporated by reference to the Company’s Registration Statement on Form 10 Filed on April 16, 2008.
(3) Incorporated by reference to the Company’s Current Report on Form 8-K filed on May 23, 2011.
(4) Incorporated by reference to the Company’s Current Report on Form 8-K filed on May 27, 2011.
(5) Incorporated by reference to the Company’s Registration Statement on Form S-1 filed on June 23, 2011
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
|
|
RADIUS HEALTH, INC. | ||
|
|
| ||
|
|
| ||
|
Date: July 20, 2011 |
By: |
/s/ C. Richard Edmund Lyttle | |
|
|
|
Name: |
C. Richard Edmund Lyttle |
|
|
|
Title: |
President and Chief Executive Officer |
EXHIBIT INDEX
|
Exhibit |
|
Description |
|
|
|
|
|
2.1 |
|
Agreement and Plan of Merger, dated April 25, 2011(1) |
|
|
|
|
|
3.1 |
|
Certificate of Designations of the Company, filed with the Secretary of State of Delaware on May 17, 2011(3) |
|
|
|
|
|
3.2 |
|
Certificate of Merger, filed with the Secretary of State of Delaware on May 17, 2011, by and between Radius Health, Inc. and RHI Merger Corp. (3) |
|
|
|
|
|
3.3 |
|
Certificate of Ownership relating to the merger of Radius Health, Inc. with and into MPM Acquisition Corp. (3) |
|
|
|
|
|
3.4 |
|
Certificate of Incorporation, as filed with the Delaware Secretary of State on February 4, 2008(2)_ |
|
|
|
|
|
3.5 |
|
By-Laws(2) |
|
|
|
|
|
4.1 |
|
Amended and Restated Stockholders’ Agreement, dated May 17, 2011, by and among the Company, as successor to Radius Health, Inc., and the Stockholders listed therein(3) |
|
|
|
|
|
10.1* |
|
Clinical Trial Services Agreement and Work Statement NB-1, dated March 29, 2011, by and between the Company, as successor to Radius Health, Inc., and Nordic BioScience Clinical Development VII A/S(3) |
|
|
|
|
|
10.2* |
|
Stock Issuance Agreement, dated March 29, 2011, by and between the Company, as successor to Radius Health, Inc., and Nordic BioScience Clinical Development VII A/S(3) |
|
|
|
|
|
10.3* |
|
Side Letter, dated March 29, 2011, by and between the Company, as successor to Radius Health, Inc., and Nordic BioScience Clinical Development VII A/S(3) |
|
|
|
|
|
10.4* |
|
License Agreement, dated September 27, 2005, by and between the Company, as successor to Radius Health, Inc., and SCRAS S.A.S., on behalf of itself and its Affiliates(3) |
|
|
|
|
|
10.5* |
|
Pharmaceutical Development Agreement, dated January 2, 2006, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S(3) |
|
|
|
|
|
10.6* |
|
Amendment No. 1 to Pharmaceutical Development Agreement, dated January 1, 2007, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
|
|
|
|
10.7* |
|
License Agreement Amendment No. 1, dated September 12, 2007, by and between the Company, as successor to Radius Health, Inc., and SCRAS S.A.S. (3) |
|
|
|
|
|
10.8* |
|
Amendment No. 2 to Pharmaceutical Development Agreement, dated January 1, 2009, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
|
|
|
|
10.9* |
|
Amendment No. 3 to Pharmaceutical Development Agreement, dated June 16, 2010, by and between the Company, as successor to Radius Health, Inc., and Beaufour Ipsen Industrie S.A.S. (3) |
|
10.10* |
|
License Agreement Amendment No. 2, dated May 11, 2011, by and between the Company, as successor to Radius Health, Inc., and Ipsen Pharma S.A.S. (3) |
|
|
|
|
|
10.11* |
|
Series A-1 Convertible Preferred Stock Issuance Agreement, dated May 11, 2011, by and between the Company, as successor to Radius Health, Inc., and Ipsen Pharma S.A.S. (3) |
|
|
|
|
|
10.12* |
|
Development and Manufacturing Services Agreement, dated October 16, 2007, by and between the Company, as successor to Radius Health, Inc., and LONZA Sales Ltd. (3) |
|
|
|
|
|
10.13* |
|
Work Order No. 2, dated January 15, 2010, by and between the Company, as successor to Radius Health, Inc., and LONZA Sales Ltd. (3) |
|
|
|
|
|
10.14* |
|
Amendment No. 3 to Work Order No.2, dated December 15, 2010, by and between the Company, as successor to Radius Health, Inc., and LONZA Sales Ltd. (3) |
|
|
|
|
|
10.15* |
|
Development and Clinical Supplies Agreement, dated June 19, 2009, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.16* |
|
Amendment No. 1, dated December 31, 2009, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.17* |
|
Amendment No. 2, dated September 16, 2010, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.18* |
|
Amendment No. 3, dated September 29, 2010, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.19* |
|
Change Order Form - Amendment No. 5, dated February 4, 2011, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.20* |
|
Amendment No. 4, dated March 2, 2011, to the 3M Development Agreement, by and among the Company, as successor to Radius Health, Inc., and 3M Co. and 3M Innovative Properties Co. (3) |
|
|
|
|
|
10.21* |
|
Laboratory Services and Confidentiality Agreement, dated March 31, 2004, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories, Inc. (3) |
|
|
|
|
|
10.22* |
|
First Amendment to Laboratory Services and Confidentiality Agreement, dated November 7, 2008, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories, Inc. (3) |
|
|
|
|
|
10.23* |
|
Letter of Payment Authorization, dated November 20, 2010, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories Preclinical Services Montréal Inc. (3) |
|
10.24* |
|
Letter of Payment Authorization, dated February 7, 2011, by and between the Company, as successor to Radius Health, Inc., and Charles River Laboratories Preclinical Services Montréal Inc. (3) |
|
|
|
|
|
10.25* |
|
License Agreement, dated June 29, 2006, by and between the Company, as successor to Radius Health, Inc., and Eisai Co., Ltd. (3) |
|
|
|
|
|
10.26 |
|
Series A-1 Purchase Agreement, dated April 25, 2011, by and among the Company, as successor to Radius Health, Inc., and the Investors listed therein(3) |
|
|
|
|
|
10.26.1 |
|
Amendment No. 1 to Series A-1 Convertible Preferred Stock Purchase Agreement, dated May 11, 2011(5) |
|
|
|
|
|
10.27** |
|
Radius Health, Inc. (f/k/a Nuvios, Inc.) 2003 Long-Term Incentive Plan, assumed in the Merger(3) |
|
|
|
|
|
10.28** |
|
Radius Health, Inc. First Amendment to 2003 Long-Term Incentive Plan effective as of December 15, 2006, assumed in the Merger(3) |
|
|
|
|
|
10.29** |
|
Radius Health, Inc. Second Amendment to 2003 Long-Term Incentive Plan effective as of March 28, 2008, assumed in the Merger(3) |
|
|
|
|
|
10.30** |
|
Radius Health, Inc. Third Amendment to 2003 Long-Term Incentive Plan effective as of November 14, 2008, assumed in the Merger(3) |
|
|
|
|
|
10.31 |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Form of Stock Option Agreement(3) |
|
|
|
|
|
10.32** |
|
Radius Health, Inc. (f/k/a Nuvios, Inc.) 2003 Long-Term Incentive Plan Stock Option Agreement, dated October 8, 2004, by and between the Company, as successor to Nuvios, Inc., and Richard Lyttle for Option No. 04-103(3) |
|
|
|
|
|
10.33** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Richard Lyttle for Option No. 07-08(3) |
|
|
|
|
|
10.34** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Richard Lyttle for Option No. 08-09(3) |
|
|
|
|
|
10.35** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Richard Lyttle for Option No. 08-14 |
|
|
|
|
|
10.36** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated February 15, 2006, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 06-07(3) |
|
|
|
|
|
10.37** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 07-07(3) |
|
|
|
|
|
10.38** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 08-05(3) |
|
|
|
|
|
10.39** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea for Option No. 08-10(3) |
|
10.40** |
|
Radius Health, Inc. (f/k/a Nuvios, Inc.) 2003 Long-Term Incentive Plan Stock Option Agreement, dated December 16, 2003, by and between the Company, as successor to Nuvios, Inc., and Gary Hattersley for Option No. 03-001(3) |
|
|
|
|
|
10.41** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated February 15, 2006, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 06-02(3) |
|
|
|
|
|
10.42** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 07-06(3) |
|
|
|
|
|
10.43** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 08-08(3) |
|
|
|
|
|
10.44** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Gary Hattersley for Option No. 08-13(3) |
|
|
|
|
|
10.45** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated July 12, 2007, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey for Option No. 07-09(3) |
|
|
|
|
|
10.46** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated May 8, 2008, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey for Option No. 08-06(3) |
|
|
|
|
|
10.47** |
|
Radius Health, Inc. 2003 Long-Term Incentive Plan Incentive Stock Option Agreement, dated December 3, 2008, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey for Option No. 08-11(3) |
|
|
|
|
|
10.48 |
|
Employment Letter Agreement, dated July 2, 2004, by and between the Company, as successor to Nuvios, Inc., and C. Richard Edmund Lyttle(3) |
|
|
|
|
|
10.49 |
|
Employment Letter Agreement, November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Gary Hattersley(3) |
|
|
|
|
|
10.50 |
|
Employment Letter Agreement, dated January 30, 2006, by and between the Company, as successor to Radius Health, Inc., and Louis O’Dea(3) |
|
|
|
|
|
10.51 |
|
Employment Letter Agreement, dated November 15, 2006, by and between the Company, as successor to Radius Health, Inc., and Nick Harvey(3) |
|
|
|
|
|
10.52 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Ansbert K. Gadicke(3) |
|
|
|
|
|
10.53 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and C. Richard Edmund Lyttle(3) |
|
|
|
|
|
10.54 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Martin Muenchbach(3) |
|
|
|
|
|
10.55 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Jonathan Fleming(3) |
|
10.56 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Kurt Graves(3) |
|
|
|
|
|
10.57 |
|
Indemnification Agreement, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Elizabeth Stoner(3) |
|
|
|
|
|
10.58 |
|
Indemnification Agreement, dated October 12, 2010, by and between the Company, as successor to Radius Health, Inc., and Alan Auerbach(3) |
|
|
|
|
|
10.59 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Michael Rosenblatt, M.D. (3) |
|
|
|
|
|
10.60 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Christopher Mirabelli(3) |
|
|
|
|
|
10.61 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Augustine Lawlor(3) |
|
|
|
|
|
10.62 |
|
Indemnification Agreement, dated November 14, 2003, by and between the Company, as successor to Nuvios, Inc., and Edward Mascioli, M.D. (3) |
|
|
|
|
|
10.63 |
|
Consent to Sublease, dated January 14, 2011, by and among the Company, as successor to Radius Health, Inc., Sonos, Inc., and Broadway/Hampshire Associates Limited Partnership(3) |
|
|
|
|
|
10.64 |
|
Sublease, dated January 14, 2011, by and between the Company, as successor to Radius Health, Inc., and Sonos, Inc. (3) |
|
|
|
|
|
10.65 |
|
Amended and Restated Warrant to Purchase Common Stock, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and SVB Financial Group(3) |
|
|
|
|
|
10.66** |
|
Warrant to Purchase Series A-1 Convertible Preferred Stock, dated May 17, 2011, by and between the Company, as successor to Radius Health, Inc., and Leerink Swann LLC(3) |
|
|
|
|
|
10.67 |
|
Redemption Agreement, by and between MPM Acquisition Corp. and MPM Asset Management LLC, dated April 25, 2011(1) |
|
|
|
|
|
10.68 |
|
Loan and Security Agreement, dated May 23, 2011, with General Electric Capital Corporation as agent and a lender, and Oxford Finance LLC as a lender(4) |
|
|
|
|
|
10.69 |
|
Promissory Note, dated May 23, 2011, issued by the Company to General Electric Capital Corporation in the principal amount of $12,500,000(4) |
|
|
|
|
|
10.70 |
|
Promissory Note, dated May 23, 2011, issued by the Company to Oxford Finance LLC in the principal amount of $3,125,000(4) |
|
|
|
|
|
10.71 |
|
Promissory Note, dated May 23, 2011, issued by the Company to Oxford Finance LLC in the principal amount of $9,375,000(4) |
|
|
|
|
|
10.72 |
|
Warrant to Purchase Shares of Series A-1 Convertible Preferred Stock, dated May 23, 2011, issued by the Company to GE Capital Equity Investments(4) |
|
|
|
|
|
10.73 |
|
Warrant to Purchase Shares of Series A-1 Convertible Preferred Stock, dated May 23, 2011, issued by the Company to Oxford Finance LLC(4) |
|
|
|
|
|
16.1 |
|
Letter from Raich Ende Malter & Co. LLP as to the change in certifying accountant, dated as of July 12, 2011 |
|
|
|
|
|
99.1 |
|
Audited financial statements of Target for the fiscal years ended December 31, 2010 and 2009 and Unaudited financial statements of Target for the three months ended March 31, 2011 and 2010 |
|
|
|
|
|
99.2 |
|
Unaudited Pro Forma Condensed Combined Financial Statements for the year ended December 31, 2010 and the three months ended March 31, 2011 |
*Confidential Treatment Requested by the Registrant. Redacted Portion Filed Separately with the Commission.
**Share numbers and per share prices are presented pre-Reverse Split completed by Radius Health, Inc. on May 17, 2011.
(1) Incorporated by reference to the Company’s Current Report on Form 8-K filed on April 29, 2011.
(2) Incorporated by reference to the Company’s Registration Statement on Form 10 Filed on April 16, 2008.
(3) Incorporated by reference to the Company’s Current Report on Form 8-K filed on May 23, 2011.
(4) Incorporated by reference to the Company’s Current Report on Form 8-K filed on May 27, 2011.
(5) Incorporated by reference to the Company’s Registration Statement on Form S-1 filed on June 23, 2011.