UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington D.C. 20549
FORM 8-K/A
CURRENT REPORT
Pursuant to Section 13 OR 15(d) of The Securities Exchange Act of 1934
Date of Report (Date of Earliest Event Reported): March 18, 2011
(Amendment No. 1)
VIROLAB, INC.
(Exact name of registrant as specified in its charter)
|
Delaware
|
000-54059
|
27-2787170
|
||
|
(State or other jurisdiction
of Incorporation)
|
(Commission
File Number)
|
(IRS Employer
Identification No.)
|
|
1840 Gateway Drive, Suite 200, Foster City, CA
|
94404
|
|
|
(Address of Principal Executive Offices)
|
(Zip Code)
|
Registrant’s telephone number, including area code: (650) 283-2653
Accelerated Acquisitions X, Inc.
(Former name or former address, if changed since last report)
(Address of Principal Offices)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
|
r
|
Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
|
|
r
|
Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
|
|
r
|
Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
|
|
r
|
Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
|
Item 1.01 Entry into a Material Definitive Agreement
On March 8, 2011, Virolab, Inc. (the “Company”) entered into a Licensing Agreement (“Licensing Agreement”) with Virolab Nevada, LLC (“Licensor”) pursuant to which the Company was granted an exclusive, non-transferrable worldwide license for certain intellectual property, principally comprising technology, patents, intellectual property, know-how, trade secret information, clinical trial protocols and data, and commercial rights to an investigational therapeutic vaccine for the human papillomavirus, or HPV, and HPV-related cancers and a specific blood test for HPV (the “Technology”).
Except for the rights granted under the License Agreement, Licensor retains all rights, title and interest to the Technology and any improvements thereto—although the License includes the Company’s right to utilize such improvements.
The term of the License commences on the date of the Licensing Agreement and continues for thirty (30) years, provided that the Licensee is not in breach or default of any of the terms or conditions contained in this Agreement. In addition to other requirements, the continuation of the License is conditioned on the Company generating net revenues in the normal course of operations or the funding by the Company of specified amounts for qualifying research, development and commercialization expenses related to the Technology. In addition, the Company is required to fund certain specified expenses related to the commercialization of the Technology as specified in the License Agreement. The license is terminated upon the occurrence of events of default specified in the License Agreement.
Item 2.01 Completion of Acquisition or Disposition of Assets.
On March 8, 2011, the Company entered into the Licensing Agreement with Licensor pursuant to which the Company was granted an exclusive, non-transferrable worldwide license for certain intellectual property, principally comprising technology, patents, intellectual property, know-how, trade secret information, clinical trial protocols and data, and commercial rights to an investigational therapeutic vaccine for HPV and HPV-related cancers and a specific blood test for HPV.
The Licensor acquired the Technology from Virolab S de RL de CV (“Virolab Mexico”), a company based in Mexico City, Mexico. Ricardo Rosales, Ph.D. is the founder and President of Virolab Mexico and is a director of the Company. Dr. Rosales is also the sole shareholder of the Licensor. Dr. Rosales was Chief Executive Officer of the Company from February 27, 2011 through April 15, 2011. Virolab Mexico owns 22.350,000 shares of the Company’s outstanding common stock, representing an 88.2% ownership interest in the Company. Virolab Mexico purchased its shares in the Company on February 27, 2011 as disclosed in a Form 8-K filed on March 2, 2011. There were no other agreements between the Company and Virolab Mexico prior to the Share Purchase Agreement entered into on February 27, 2011.
Mr. Timothy Neher, the Company’s Chief Executive Officer prior to February 27, 2011, controls Accelerated Venture Partners, LLC (“AVP”), an entity which has agreed to provide financial advisory services to the Company. AVP owns 3,000,000 shares of the Company’s outstanding common stock, representing an 11.8% ownership interest in the Company (collectively, Virolab Mexico and AVP own 100% of the Company as there are no other stockholders). Up to 1,500,000 of AVP’s shares can be repurchased by the Company for $0.0001 per share under certain circumstances. AVP is entitled to receive specified cash compensation if the Company achieves certain financial milestones as outlined in the “Our Business” section below and as described in Exhibit 10.4 to the Current Report on Form 8-K filed on March 2, 2011.
Aside from the Licensor, Virolab Mexico, Dr. Rosales, AVP and Mr. Neher, no other parties have an interest related to the Share Purchase Agreement or the Licensing Transaction. The parties were introduced by an acquaintance with which both Dr. Rosales and Mr. Neher have previously conducted different business transactions, but who does not have a business relationship with the Company.
Under the terms of the Licensing Agreement, the Company has agreed to pay the Licensor two percent (2%) of any royalties received if the Company grants any third parties royalty-bearing licenses to the Technology. In addition, the Company has agreed to pay Licensor a royalty of one quarter of one percent (0.25%) of all gross revenue resulting from use of the Technology by the Company. In order to retain its rights, the Company must receive revenues or fund a minimum of $2 million in qualified research, development and commercialization expenses before the third anniversary of the Licensing Agreement (at least $0.5 million of which must be before the first anniversary of the Licensing Agreement and at least $1 million of which must be before the second anniversary of the Licensing Agreement). There are additional customary commercialization requirements in the Licensing Agreement, and this description is qualified by the terms of the Licensing Agreement attached to the Current Report on Form 8-K filed on March 18, 2011 as Exhibit 10.1.
- 2 -
Item 5.06 Change in Shell Company Status.
Prior to the Company’s entry into the business of research, development and commercialization of therapeutic vaccines and specific blood tests for diseases, such as HPV-related cancers, through the execution of the License Agreement as described in Item 1.01 above, the Company was a “shell company” (as such term is defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended). As a result of entering into this agreement and undertaking efforts into the research and development of therapeutic vaccines and diagnostic tests for human diseases, we ceased to be a shell company.
None of the Company’s securities are registered for resale with the Securities and Exchange Commission. The outstanding shares of common stock may only be resold through registration under the Securities Act of 1933, or under an applicable exemption from registration
OTHER PERTINENT INFORMATION
Unless specifically set forth to the contrary, when used herein, the terms “Virolab”, “Accelerated Acquisitions X, Inc.”, "we", "our", the "Company" and similar terms refer to Virolab, Inc., a Delaware corporation, which was formerly named Accelerated Acquisitions X, Inc. The name change was filed with the Delaware Secretary of State on April 5, 2011.
FORM 10 DISCLOSURE
Item 2.01(f) of Form 8-K states that if the registrant was a shell company, like our company, the registrant must disclose the information that would be required if the registrant were filing a general form for registration of securities on Form 10. Accordingly, we are providing below the information that would be included in a Form 10 if we were to file a Form 10.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING INFORMATION
This report contains forward-looking statements. These forward-looking statements are subject to known and unknown risks, uncertainties and other factors which may cause actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. These forward-looking statements were based on various factors and were derived utilizing numerous assumptions and other factors that could cause our actual results to differ materially from those in the forward-looking statements. These factors include, but are not limited to, our ability to develop our operations, our ability to satisfy our obligations, our ability to consummate the acquisition of additional assets, our ability to generate revenues and pay our operating expenses, our ability to raise capital as necessary, economic, political and market conditions and fluctuations, government and industry regulation, interest rate risk, U.S. and global competition, and other factors. Most of these factors are difficult to predict accurately and are generally beyond our control. You should consider the areas of risk described in connection with any forward-looking statements that may be made herein. Readers are cautioned not to place undue reliance on these forward-looking statements and readers should carefully review this report in its entirety, including the risks described in "Risk Factors" and the risk factors described in our other filings with the Securities and Exchange Commission. Except for our ongoing obligations to disclose material information under the Federal securities laws, we undertake no obligation to update any of our forward-looking statements. These forward-looking statements speak only as of the date of this report, and you should not rely on these statements without also considering the risks and uncertainties associated with these statements and our business.
None of the Company’s securities are registered for resale with the Securities and Exchange Commission. The outstanding shares of common stock may only be resold through registration under the Securities Act of 1933, or under an applicable exemption from registration.
OUR BUSINESS
From inception on May 4, 2010, Accelerated Acquisitions X, Inc. was organized as a vehicle to investigate and, if such investigation warranted, acquire a target company or business seeking the perceived advantages of being a publicly held corporation. Our principal business objectives were to achieve long-term growth potential through a combination with a business rather than immediate, short-term earnings.
On May 4, 2010, the Company sold 5,000,000 shares of Common Stock to Accelerated Venture Partners, LLC, or AVP, for an aggregate investment of $2,000. The Company sold these shares of Common Stock under the exemption from registration provided by Section 4(2) of the Securities Act.
- 3 -
On February 27, 2011, Virolab S de RL de CV (the “Purchaser” or “Virolab Mexico”) acquired 22,350,000 shares of the Company’s common stock par value $0.0001 for a price of $0.0001 per share. At the same time, AVP tendered 3,500,000 of their 5,000,000 shares of the Company’s common stock for cancellation. In addition, AVP received an option to purchase 1,500,000 shares of the Company’s common stock for a price of $0.0001 per share, which AVP immediately exercised. The 1,500,000 shares purchased under option by AVP are subject to repurchase by the Company as described in the February 27, 2011 Consulting Services Agreement disclosure below. Following these transactions, Virolab S de RL de CV., owned 88.2% of the Company’s 25,350,000 issued and outstanding shares of common stock and AVP owned the remaining 11.8%. Simultaneously with the share purchase, Ricardo Rosales, PhD was appointed to the Company’s Board of Directors. Such action represents a change of control of the Company.
Prior to the purchase of the shares, the Purchaser was not affiliated with the Company. However, the Purchaser will be deemed an affiliate of the Company after the share purchase as a result of its stock ownership interest in the Company and position on the Board of Directors. The purchase of the shares by the Purchaser was completed pursuant to a written Subscription Agreement with the Company. The purchase was not subject to any other terms and conditions other than the sale of the shares in exchange for the cash payment. Following the sale of common stock to the Purchaser, the Company was still seeking to achieve its objective of acquiring a target company or business.
On February 27, 2011, the Company entered into a Consulting Services Agreement with AVP. The agreement requires AVP to provide the Company with certain advisory services that include reviewing the Company’s business plan, identifying and introducing prospective financial and business partners, and providing general business advice regarding the Company’s operations and business strategy in consideration of (a) an option granted by the Company to AVP to purchase 1,500,000 shares of the Company’s common stock at a price of $0.0001 per share (the “AVP Option”) (which was immediately exercised by the holder) subject to a repurchase option granted to the Company to repurchase the shares at a price of $0.0001 per share in the event the Company fails to complete funding as detailed in the agreement subject to the following milestones:
|
|
·
|
Milestone 1 – Company’s right of repurchase will lapse with respect to 60% of the shares upon securing $5 million in available cash from funding;
|
|
|
·
|
Milestone 2 – Company’s right of repurchase will lapse with respect to 40% of the Shares upon securing $10 million in available cash (inclusive of any amounts attributable to Milestone 1);
|
and (b) cash compensation at a rate of $66,667 per month. The payment of the cash compensation is subject to the Company’s achievement of certain designated milestones, specifically, cash compensation of $400,000 is due consultant upon the achievement of Milestone 1, and an additional $400,000 is due upon the achievement of Milestone 2. Upon achieving each Milestone, the cash compensation is to be paid to consultant in the amount then due at the rate of $66,667 per month. The total cash compensation to be received by the consultant is not to exceed $800,000 unless Virolab receives an amount of funding in excess of the amount specified in Milestone 2. If the Company receives equity or debt financing that is an amount less than Milestone 1, in between any of the above Milestones or greater than the above Milestones, the cash compensation earned by the Consultant under this Agreement will be prorated according to the above Milestones. The Company also has the option to make a lump sum payment to AVP in lieu of the monthly cash payments.
On March 8, 2011, the Company entered into a Licensing Agreement (“Licensing Agreement”) with Virolab Nevada LLC (“Licensor”) pursuant to which the Company was granted an exclusive, non-transferrable worldwide license for the commercial rights to an investigational therapeutic vaccine for the human papillomavirus, or HPV, and HPV-related cancers and a specific blood test for HPV (the “Technology”) (see Item 1.01, above). Rather than acquiring an operating company, the Company acquired an exclusive license to trade secrets, patents, know-how and other intellectual property that had been developed in Mexico. We will lose our rights to the Technology that we licensed if we do not raise at least $0.5 million for its future development before March 8, 2012, if we do not raise at least $1.0 million for its future development before March 8, 2013, and if we do not raise at least $2.0 million for its future development before March 8, 2014.
Prior to the licensing transaction entered into on March 8, 2011, the Company was a “blank check company”. Shares of the Company’s common stock are not registered under the securities laws of any state or other jurisdiction, and accordingly there is no public trading market for our common stock. Therefore, outstanding shares of our common stock cannot be offered, sold, pledged or otherwise transferred unless subsequently registered pursuant to, or exempt from registration under, the Securities Act and any other applicable federal or state securities laws or regulations. Shares of our common stock including shares issued to AVP cannot be sold before or after an acquisition under the exemptions from registration provided by Rule 144 under or Section 4(1) of the Securities Act (“Rule 144”) if the Company is designated a “shell company,” and for 12 months after it ceases to be a “shell company,” provided the Company otherwise is in compliance with the applicable rules and regulations. Compliance with the criteria for securing exemptions under federal securities laws and the securities laws of the various states is extremely complex, especially in respect of those exemptions affording flexibility and the elimination of trading restrictions in respect of securities received in exempt transactions and subsequently disposed of without registration under the Securities Act or state securities laws.
- 4 -
AVP has indicated to the Company that it will pay the on-going administrative expenses of the Company until Virolab enters into a financing transaction. These on-going administrative expenses differ from the services covered in the Consulting Services Agreement. Through March 31, 2011, the Company has incurred approximately $64,000 of expenses related to its operations, and of this amount approximately $60,000 remained due to AVP. Virolab currently does not pay any cash compensation to its directors, officers or employees, but plans to do so if it succeeds in securing additional financing.
Throughout this document, we refer to our therapeutic vaccine as “investigational” or as a “candidate” because it has not received approval for commercial sale anywhere in the world, and we also refer to the investigational vaccine as “therapeutic” because it is designed to treat an existing viral infection and the resulting cervical lesions. Most of the currently approved vaccines for all diseases are not therapeutic, but instead are designed to prevent an infection from occurring before a patient is actually exposed to a virus or other infectious agent. We believe the intended therapeutic nature of our vaccine candidate differentiates it from other vaccines.
We believe that the HPV therapeutic vaccine technology that we have in development has the potential to be the first therapeutic to clear pre-cancerous and cancerous lesions of the Cervix along with the underlying HPV viral infection. This belief is based on the results of two phase 2 clinical trials and one phase 3 clinical conducted in Mexico in which cancerous and pre-cancerous cervical lesions were eliminated or regressed in 88% to 97% of the patients. In addition, detectable HPV virus was eliminated from approximately 50% of the 1,246 patients treated in the three trials. All studies showed the vaccine candidate to be well tolerated, although headaches and other flu-like symptoms appeared in some of the patients. More detailed results from the trials conducted by Virolab Mexico are contained in the section “Clinical Trials of Therapeutic Vaccine Candidate” below.
In addition to the therapeutic vaccine candidate, Virolab has licensed rights to a specific blood-based diagnostic test for HPV that is in development. Currently, diagnosis of an infection with HPV requires a gynecological examination and testing of cervical tissue samples. We believe that a diagnosis from a simple blood draw will be less expensive and more convenient for patients.
The Company is evaluating its options to commercialize its therapeutic vaccine candidate and diagnostic test. These product candidates have not been approved for commercial sale anywhere in the world, and each country has different requirements for approval, so the commercialization process is likely to be lengthy and complex. The Company may employ different strategies in different areas of the world, such as sublicensing development and commercialization rights for some territories while retaining rights for other territories. Based on the historical work of Virolab Mexico in the country of Mexico, the Company believes that the path to obtain government approval may be shorter in the Latin America countries.
The Company will not be able to commercialize either its vaccine candidate or its diagnostic test without additional capital. The Company is evaluating various means of raising this capital, including through the sale of equity securities, licensing agreements or other means. If the Company does not raise additional funds of at least $2 million for the advancement of its technology over the next three years it will lose its rights to the technology. The Company plans to seek at least $10 million of capital within the next 12 months, and plans to use these funds to determine the FDA’s requirements for approval of its vaccine candidate, begin working on these requirements, file patents on its technology, broaden its pipeline of investigational drugs, vaccines and/or diagnostics, establish a biotechnology laboratory, and continue the process of seeking approval of its vaccine candidate and diagnostic test outside of the United States. The Company is not presently able to allocate the specific costs for its plans because it does not yet know the requirements for approval of either its vaccine candidate or its diagnostic test.
HPV and Cervical Cancer Background
According to The World Health Organization, or WHO, cervical cancer is the second most frequent form of cancer among women in the world and second most frequent among women aged 15 – 44 years. WHO also reports that about 300 million women, about 10% of women worldwide, are estimated to be infected with HPV at a given time and that virtually 100% of cervical cancer cases are caused by HPV infection. HPV is transmitted through sexual contact and is extremely contagious. While the WHO estimates that an equal number of men are infected with HPV, in general HPV infected men do not show symptoms, although genital warts and codylomata may appear.
HPV-infected women can develop pre-cancerous lesions of increasing seriousness known as CIN 1, CIN 2, and CIN 3 which can develop into cancer and, if they do, can ultimately cause the death of the patient. Since the virus may foster the growth of cancer cells anytime from six months to 15 years after infection, women at risk currently have to undergo gynecological testing and pap-smears for early detection of potential cancer at least once a year.
Under current treatment practices, when a cervical lesion is detected, outpatient surgery is generally performed to remove the lesion. The underlying HPV infection, however, remains and new lesions can develop. Therefore, recommended check up frequency is increased to twice a year for these patients.
- 5 -
Virolab’s goal is to complete the clinical development of its therapeutic vaccine candidate, and seek regulatory approval to commercialize the therapeutic vaccine throughout the world. We believe that a vaccine which has potential to clear an underlying HPV infection from patients would be a cost effective and advantageous alternative to surgery that does not clear the underlying viral infection from patients. However, we must raise additional funds, conduct additional positive clinical trials and establish qualified manufacturing facilities in order for us to meet this goal of commercializing the vaccine candidate.
Virolab’s Therapeutic Vaccine Candidate
Below is an example of a cervical cancer before and after treatment with the investigational therapeutic vaccine candidate exclusively licensed to Virolab. This illustration was taken from one of the women that have been treated with the vaccine candidate in clinical trials.
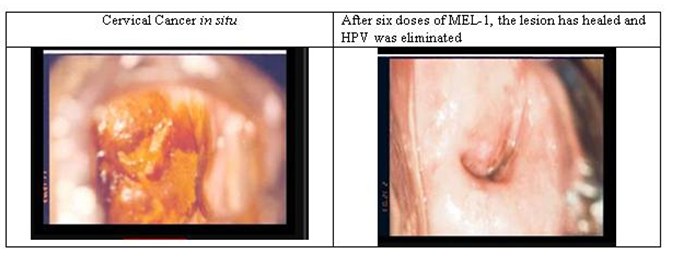
The technology licensed to Virolab involves the key process of developing production of the therapeutic vaccine candidate, which is referred to as MEL-1. MEL-1 is derived from the smallpox virus vaccine MVA, which was originally developed in Germany during the 1940s. The World Health Organization has approved MVA for use in other human vaccines in the early 1990s (note that this does not imply that the U.S. Food and Drug Administration, or FDA, or any other governmental regulatory agency will approve any vaccine based on the use of MVA). Scientists working for Virolab Mexico experimented by inserting certain genes from the Bovine Papillomavirus into MVA. One of the genes inserted was E2, an anti-tumor protein that self-regulates the activity of HVP’s tumor-generating proteins.
Under the protocols of completed phase 1, 2, and 3 clinical trials, the modified vaccine with the E2 gene inserted is injected directly into the cervix. This then produces an overdose of anti-tumor E2 protein immediately surrounding the HPV-infected tumor cells of the host. The immunological system of the patient reacts and destroys weakened HPV-infected cells. As a result, the patient develops antibodies against HPV and against additional foreign contents that may be contained in those cells.
The MEL-1 therapeutic vaccine has demonstrated in clinical trials conducted in Mexico that it can regress and heal pre-cancerous or cancerous lesions of the cervix in 88% to 97% of patients treated and eliminate detectable levels of HPV in approximately half of the treated patients. The standard treatments used in the clinical trials include weekly injections of MEL-1 for a period of six weeks. See more detailed information on the clinical trials in “Clinical Trials of Therapeutic Vaccine Candidate” below.
To our knowledge, no other treatment has demonstrated elimination of detectable levels of HPV from patients.
Virolab’s HPV Diagnostic Test
The HPV diagnostic test recently licensed to the Company, referred to as EDIVPH, uses a standard Enzyme-linked immunosorbent assay, or ELISA, procedure to test for HPV antibodies in the blood serum. ELISA tests are widely-used blood-based diagnostic method of detecting diseases. The advantage compared to other HPV diagnostics is the quick and easy access to blood samples requiring no gynecological examination, as the other marketed tests for HPV require. Virolab plans to investigate ways to commercialize its diagnostic test throughout the world. Virolab does not have approval to market the test in any countries, and government regulations vary from country to country.
Virolab Mexico has conducted a study of 172 HPV-infected women in which the HPV diagnostic test was compared to tissue samples using traditional HPV diagnostic methods. Results indicated that using different traditional diagnostic methods on the same patients did not consistently produce similar test results (approximately 20% could be diagnosed incorrectly as either positive or negative). The study was conducted in Mexico in patients with known current or previous HPV infections, and was published in the Journal of Medical Virology in 2001. Based on the results, Virolab believes that its blood-based test has potential to be more accurate. However, additional clinical comparisons will be necessary to verify this belief and additional clinical work will be necessary to seek approval to market the test.
- 6 -
Government Regulation
Regulations applicable to Therapeutic Vaccine Candidate
In the United States, drugs are subject to regulation under the Federal Food, Drug and Cosmetic Act, or the FDC Act. Biological products, in addition to being subject to provisions of the FDC Act, are regulated in the United States under the Public Health Service Act. Both statutes and related regulations govern, among other things, testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising, and other promotional practices.
Obtaining FDA approval or comparable approval from similar agencies in other countries is a costly and time-consuming process, and the results of seeking approval are uncertain. Generally, FDA approval requires that preclinical studies be conducted in the laboratory and in animal model systems to gain preliminary information on efficacy and to identify any major safety concerns. In the United States, the results of these studies are submitted as a part of an IND application which the FDA must review and allow before human clinical trials can start.
A company must submit an IND application or equivalent application in other countries for each proposed product and must conduct clinical studies to demonstrate the safety and efficacy of the product necessary to obtain FDA approval or comparable approval from similar agencies in other countries. For example, in the United States, the FDA receives reports on the progress of each phase of clinical testing and may require the modification, suspension, or termination of clinical trials if an unwarranted risk is presented to patients.
Obtaining FDA approval prior to marketing a pharmaceutical product typically requires several phases of clinical development to demonstrate the safety and efficacy of the product candidate. Clinical trials are the means by which experimental treatments are tested in humans, and are conducted following preclinical testing. Upon successful completion of clinical trials, approval to market the treatment for a particular patient population may be requested from the FDA and/or its counterparts in other countries.
Clinical trials are normally done in three phases. Phase 1 clinical trials are typically conducted with a small number of patients or healthy subjects to evaluate safety, determine a safe dosage range, identify side effects, and, if possible, gain early evidence of effectiveness. Phase 2 clinical trials are conducted with a larger group of patients to evaluate effectiveness of an investigational product for a defined patient population, and to determine common short-term side effects and risks associated with the drug. Phase 3 clinical trials involve large scale, multi-center, comparative trials that are conducted to evaluate the overall benefit-risk relationship of the investigational product and to provide an adequate basis for product labeling.
After completion of clinical trials of a new product, FDA marketing approval must be obtained, or equivalent approval by comparable agencies in other countries. For the FDA, if the product is regulated as a biologic, as we expect for our investigational therapeutic vaccine, a Biologics License Application, or BLA, is required. The BLA must include results of product development activities, preclinical studies, and clinical trials in addition to detailed chemistry, manufacturing and control information.
Applications submitted to the FDA are subject to an unpredictable and potentially prolonged approval process. Despite good-faith communication and collaboration between the applicant and the FDA during the development process, the FDA may ultimately decide, upon final review of the data, that the application does not satisfy its criteria for approval or requires additional product development or further preclinical or clinical studies. Even if FDA regulatory clearances are obtained, a marketed product is subject to continual review, and later discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market.
Before marketing clearance for a product can be secured, the facility in which the product is manufactured must be inspected by the FDA and must comply with current Good Manufacturing Practices, or cGMP, regulations. In addition, after marketing clearance is secured, the manufacturing facility may be inspected periodically for cGMP compliance by FDA inspectors.
Regulations Applicable to Diagnostic Test
In the United States, diagnostic tests are regulated by the FDA as medical devices. Medical devices are classified into one of three classes on the basis of the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices are subject to general controls, including labeling, premarket notification and adherence to FDA’s quality system regulations, which are device-specific good manufacturing practices. Class II devices are subject to general controls and special controls, including performance standards and postmarket surveillance. Class III devices are subject to most of the previously identified requirements as well as to premarket approval. Class I devices are exempt from premarket submissions to the FDA; most Class II devices require the submission of a 510(k) premarket notification to the FDA; and Class III devices require submission of a premarket approval application, or PMA. Most in vitro diagnostic tests are regulated as Class I or Class II devices and are either exempt from premarket notification or require a 510(k) submission. We presently do not know how the FDA will classify our HPV diagnostic test, but currently assume it is most likely to be a Class III device, since it would be the first blood-based diagnostic for HPV.
- 7 -
Class III devices require the submission and approval of a PMA prior to product sale. The PMA must be supported by detailed and comprehensive scientific evidence, including clinical data, to demonstrate the safety and efficacy of the medical device for its intended purpose. From start to finish the approval process may take several years, and the FDA may also request additional clinical data as a condition of approval or after the PMA is approved. Product changes after approval typically require a supplemental submission with FDA review cycles ranging from 30 to 180 days.
Any products manufactured or distributed pursuant to FDA clearances or approvals are subject to pervasive and continuing regulation by the FDA, including recordkeeping requirements, reporting of adverse experiences with the use of the device, and restrictions on advertising and promotion. Device manufacturers are required to register their establishments and list their devices with the FDA and are subject to periodic inspections by the FDA and certain state agencies. Noncompliance with applicable FDA requirements can result in, among other things, warning letters, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, refusal of the FDA to grant PMA approval for devices, withdrawal of PMA approvals, or criminal prosecution.
Clinical Trials of Therapeutic Vaccine Candidate
Medical protocols under which Virolab Mexico’s human clinical trials were conducted were reviewed in Mexico by the Health Ministry at the State and Governmental levels. Because the trials were not conducted in the United States, no protocols were submitted to or reviewed by the U.S. FDA, nor has an Investigational New Drug Application been submitted to the U.S. FDA. As is customary in human clinical trials, the proposed clinical trials of the therapeutic vaccine and subsequent results from its use were also reviewed by various Hospital Ethics Committees before each approved their institution’s participation in the trials. In addition, the clinical trials were conducted in accordance with international standards for Good Clinical Practice and all study patients met pre-defined enrollment criteria and provided signed informed consent after being fully advised of the risks and potential benefits of participating in the trials.
The therapeutic vaccine candidate licensed by Virolab is a recombinant live virus vaccine based on a highly attenuated vaccinia virus vector, known as MVA, or Modified Vaccinia Ankara. Viroloab’s licensors have spliced into MVA a gene encoding the E2 protein from the bovine papillomavirus, which is similar to HPV, the human papillomavirus, to create the Company’s therapeutic vaccine candidate. Virolab refers to its therapeutic vaccine candidate as the MEL-1 vaccine. MVA is a very well characterized vaccine that has been used commercially for decades and tested on more than 100,000 people as a vaccine to prevent smallpox. It is currently stockpiled by the U.S. Department of Health and Human Services and similar organizations in other countries in the event of future smallpox outbreaks.
Virolab’s MEL-1 therapeutic vaccine has been evaluated in multiple animal models (preclinical) and has completed phase 1, 2, and 3 clinical trials in Mexico. We expect additional clinical trials will be required before the vaccine candidate can be considered for approval in the United States.
The following table summarizes the results of the clinical trials conducted with Virolab’s MEL-1 HPV therapeutic vaccine candidate, all of which received requisite governmental approval in Mexico before initiation. Unless noted separately below, each of these trials evaluated the use of MEL-1 as a therapeutic vaccine for treating pre-cancerous or cancerous cervical lesions in women who have been infected with human papillomavirus. In each study MEL-1 was administered by injection directly into the cervix in a radial manner, at 12 o’clock, three o’clock, six o’clock, and nine o’clock positions, once per week for six weeks. Each weekly injection consisted of 107 MEL-1 virus units per dose. All women who enrolled in the trials completed the study. Efficacy was evaluated at study week 9, three weeks after completion of treatment and was determined by regression in lesion size and severity and reduction in detectable HPV viral DNA. According to the American Association of Cancer Research (AACR) task force on treatment and prevention of intra-epithelial neoplasia, obtaining a 50% regression rate in CIN2/3 with a new treatment is clinically meaningful. Regression Rate is defined by the AACR as both Complete Response (CR) and Partial Response (PR) where CR is regression to normal histology and PR is regression of CIN 2/3 to CIN 1, with no new CIN 2/3 lesions in at least 50% of treated patients (Clin. Cancer Res., (8):314-346, 2002).
- 8 -
|
Phase 1
|
1st Phase 2
|
2nd Phase 2
|
Phase 3
|
|
|
Primary endpoint
|
Human safety study
|
Safety and lesion reduction
|
Elimination of high-grade lesions
|
Reduction in lesions
|
|
Dates conducted
|
1999
|
2000-2002
|
2002-2004
|
2004-2008
|
|
Treatment protocol
|
Evaluate safety in volunteers
|
Evaluate vaccine candidate against cryosurgery
|
Evaluate vaccine candidate against conization
|
Evaluate reduction in viral load and reduction in lesions
|
|
Patient eligibility
|
Disease-free women with no cervical lesioins
|
Women with HPV and cervical lesions (CIN 1-3)
|
Women with HPV and high-grade lesions (CIN 2-3)
|
Women with HPV and pre-cancerous lesions (CIN 1-3) or carcinoma
|
|
Number of participants
|
120
|
78 total
36 MEL-1
42 Control (cryosurgery)
|
54 total
34 MEL-1
20 Control (conization)
|
1,176
All patients treated with MEL-1
|
|
Dosage and administration of study medication
|
MEL-1 vaccine injection into uterus once per week for 6 weeks at 107 MEL-1 virus units per dose.
|
MEL-1 vaccine injection into uterus once per week for 6 weeks at 107 MEL-1 virus units per dose.
|
MEL-1 vaccine injection into uterus once per week for 6 weeks at 107 MEL-1 virus units per dose.
|
MEL-1 vaccine injection into uterus once per week for 6 weeks at 107 MEL-1 virus units per dose.
|
|
Trial type
|
Open label safety study
|
Open-label vs. control
|
Open label vs. control
|
No control group
|
|
Trial outcome
|
Established initial safety database to allow commencement of trials to evaluate efficacy
|
Demonstrated lesion regression or elimination in 94% of MEL-1 treated patients and HPV virus elimination or significant reduction in 50% of MEL-1 treated patients
|
Elimination and regression of lesions in 88% of MEL-1 treated patients and HPV virus elimination or significant reduction in all MEL-1 patients
|
Elimination of lesions in 97% of participants. Elimination of HPV virus in 50% of patients
|
|
Trial location
|
Mexico City
|
Mexico City
|
Mexico City
|
Mexico City; Michoacan, Mexico; Barquisimento, Venezuela
|
|
Major adverse effects noted
|
Flu-like symptoms
|
Headaches, flu-like symptoms, chills, transient fever (>39º C), abdominal pain
|
Headaches, flu-like symptoms, chills, transient fever (>39º C), abdominal pain
|
Headaches, flu-like symptoms, chills, transient fever (>39º C), abdominal pain
|
|
Outcome publication
|
Not published
|
Human Gene Therapy, May 2004 (Peer Reviewed)
|
Cancer Gene Therapy, February 2006 (Peer Reviewed)
|
Publication submitted
|
In addition to the completed trials, in 2009 Virolab Mexico initiated an open-label phase 4 clinical trial at three different sites in Mexico. The Company refers to this trial as a phase 4 because this is how it is classified in Mexico, although the drug is not approved for commercial sale as is often the case with other phase 4 trials. To date, approximately 1,000 patients have been enrolled and treated in the trial, and efficacy results are consistent with the outcomes of the phase 2 and 3 clinical trials. Due to the Company’s limited funds, no on-going responsibility for the clinical trial has been assigned to Virolab, Inc., although it may at a future date if the Company is able to secure additional financing.
- 9 -
Patents
The company and its licensor have written a PCT patent application covering its therapeutic vaccine candidate MEL-1and the method of application directly to carcinomas and plans to submit the application shortly. The Company plans a similar filing in the United States. Virolab owns proprietary information and trade secrets relating to its HPV diagnostic technology and also plans patent applications. To date, no patents have issued.
Market and Competition for Therapeutic Vaccine
According to The World Health Organization, or WHO, cervical cancer is the second most frequent form of cancer among women in the world and second most frequent among women aged 15 – 44 years. WHO also reports that approximately 300 million women, about 10% of women worldwide, are estimated to be infected with HPV at a given time and that virtually 100% of cervical cancer cases are caused by HPV infection. Also according to WHO, of the women infected with HPV, over 500,000 will develop cervical cancer and over half of them will die from their HPV-caused cancer.
Based on extrapolations of CIN 1 lesions of 1.2 per 1,000 population, and CIN 2/3 lesions of 1.5 per 1,000 population (the Company’s own estimates), the Company estimates the candidate patients for its therapeutic vaccine as follows:
|
Region
|
Candidate Patients
|
||||
|
Americas
|
935,000 | ||||
|
Europe
|
880,000 | ||||
|
Asia
|
4,010,000 | ||||
|
Africa
|
840,000 | ||||
|
Oceania
|
36,000 | ||||
|
World Total
|
6,700,000 | ||||
The Company has not estimated prices yet for its therapeutic vaccine candidate, if it receives approval. Initial pricing studies are currently planned.
Two vaccines are currently available to prevent infection by some HPV types. These preventative vaccines are Gardasil, marketed by Merck, and Cervarix, marketed by GlaxoSmithKline. Both vaccines protect against initial infection with HPV types 16 and 18, which cause most of the HPV-associated cancer cases. Gardasil also protects against HPV types 6 and 11, which cause 90% of genital warts. However, the vaccines provide little benefit to women who have already been infected with HPV types 16 and 18. For this reason the vaccines are recommended primarily for those women who have not yet been exposed to HPV. Both preventative vaccines are delivered in three shots over six months. In most countries they are approved only for female use, but are approved for male use in relevant countries like USA and UK. These approved vaccines do not have any therapeutic effect on existing HPV infections or cervical lesions, the target market for Virolab’s therapeutic vaccine candidate.
We are also aware of several development-stage and established enterprises, including major pharmaceutical and biotechnology firms, which are actively engaged in infectious disease and cancer vaccine research and development. These include Crucell, Sanofi-Aventis, Novartis, GlaxoSmithKline plc, MedImmune, Inc., a wholly owned subsidiary of AstraZeneca, Merck and Pfizer Inc. The company may also experience competition from companies that have acquired or may acquire technologies from companies, universities and other research institutions. As these companies develop their technologies, they may develop proprietary technologies which may materially and adversely affect our business.
In addition, a number of companies are developing products to address the same diseases that we are targeting. For example, Merck and GlaxoSmithKline have products on the market for cervical cancer in the therapeutic setting. Transgene, Inc. has a cervical cancer product in Phase II trials. Inovio, Inc. is also working on a vaccine for cervical cancer.
- 10 -
There are seven main approaches that we are aware of under which companies are investigating HPV vaccine product candidates:
|
|
1.
|
Live vectors
|
|
|
2.
|
Peptides
|
|
|
3.
|
Proteins
|
|
|
4.
|
DNA
|
|
|
5.
|
RNA replicon
|
|
|
6.
|
Dendritic cell
|
|
|
7.
|
Tumor cell
|
Refer to Ma, et al., HPV and Therapeutic Vaccines: Where are we in 2010? Current Cancer Therapy Reviews, 2010, 6, 81-103, for a more thorough review of the potential approaches. Of the live vector approaches which are similar to the approach used to develop our therapeutic vaccine candidate, the Virolab investigational therapeutic vaccine is the only candidate using the E2 protein.
If any of our competitors develop products with efficacy or safety profiles significantly better than our products, we may not be able to commercialize our products. Some of our competitors and potential competitors have substantially greater product development capabilities and financial, scientific, marketing and human resources than we do. Competitors may develop products earlier, obtain FDA approvals for products more rapidly, or develop products that are more effective than the single vaccine candidate under development by us. We intend to seek to expand our technological capabilities to remain competitive, however, research and development by others may render our technologies or products obsolete or noncompetitive, or result in treatments superior to ours.
Our competitive position will be affected by the disease indications addressed by our product candidates and those of our competitors, the timing of market introduction for these products and the stage of development of other technologies to address these disease indications. For us and our competitors, proprietary technologies, the ability to complete clinical trials on a timely basis and with the desired results, and the ability to obtain timely regulatory approvals to market these product candidates are likely to be significant competitive factors. Other important competitive factors will include the efficacy, safety, ease of use, reliability, availability and price of products and the ability to fund operations during the period between technological conception and commercial sales.
The FDA and other regulatory agencies may expand current requirements applicable to virus-based products and product candidates, which may harm our competitive position relative to other companies developing virus-based products for similar indications.
Competition for the Blood Test
The Company’s EDIVPH blood test is the only method of detecting HPV of which we are aware that does not involve a gynecological examination or biopsies or smears from the cervix. Virolab’s HPV test is done as an immunological reaction in a sample of blood serum. Since a specialist does not need to examine the patient and technicians to take blood samples are widespread, we believe that our method will allow the examination of thousands of patients by a single lab-based technician familiar with the technology. We believe the diagnostic process can be readily automated and results can be obtained overnight.
Competitive HPV diagnostics methods include Hybrid Capture, Liquid Cytology, PCR and Thin Prep DNA determination, all of which require tissue samples from the cervix. Virolab believes that all of these diagnostic tests are readily used in practice, and each test’s usage depends on facilities available to diagnosing physicians and individual preferences.
Specific HPV diagnostic tests are increasingly being used in conjunction with routine annual gynecological examination and PAP smears as a means for identifying women at risk for developing cervical cancer. Currently, a portion of the tissue collected during a gynecological examination is utilized in a clinical laboratory for conducting an HPV test using one of the currently available test kits while the remainder of the tissue is examined microscopically for evidence of cancer cells or precancerous cellular changes. We believe a blood-based test will offer advantages for patients because they would not need a separate gynecological examination.
An experimental Light Sensitive Device method, involves the insertion of the device in the vagina and taking the sample to a lab with a spectrometer to measure light absorptions, but is not approved for use in most countries.
- 11 -
RISK FACTORS
Before you invest in our securities, you should be aware that there are various risks. You should consider carefully these risk factors, together with all of the other information included in this Current Report on Form 8-K, as well as elsewhere in our SEC filings before you decide to purchase our securities. Independent of the risk factors we list, you should further be aware that none of the Company’s securities are registered for resale with the Securities and Exchange Commission, so they can only be purchased under an applicable exemption from registration or if the Company registers the securities under the Securities Act of 1933. If any of the following risks and uncertainties develop into actual events, our business, financial condition or results of operations could be materially adversely affected.
Risks Related to Our Financial Condition
We need financing in the near term and may be unable to raise any funding needed, which could force us to abandon development of our investigational vaccine and diagnostic.
The Company has never previously raised money, and will need substantial funds in order to advance its investigational therapeutic HPV vaccine or its blood-based diagnostic test for HPV. Developing drugs, developing diagnostics, conducting clinical trials and commercializing medical products is expensive and neither of the Company’s potential products can be advanced without the Company obtaining significant additional capital. If additional capital is not obtained, we may have to cease operations and/or abandon rights to our products, and investors could realize no value for their investment in Virolab. The amount of our future funding requirements is currently uncertain, and will depend on many factors, including:
|
|
·
|
the requirements for approval of our investigational vaccine and diagnostic in different countries throughout the world;
|
|
|
·
|
progress and cost of our clinical trials and other research and development activities;
|
|
|
·
|
the costs of filing, prosecuting, defending and enforcing any patent applications, claims, patents and other intellectual property rights;
|
|
|
·
|
the cost and timing of securing manufacturing capabilities for our product candidates;
|
|
|
·
|
the terms and timing of any collaborative, licensing, acquisition or other arrangements that we may establish; and
|
|
|
·
|
the costs of establishing sales, marketing and distribution capabilities
|
Our cash may not be sufficient to support our currently limited operating activities through the end of the calendar year, even though we are not paying any salaries for our employees. Consequently, we will need to secure a source of capital and we presently do not have any arrangements in place to do so. In addition, we will lose our rights to the product candidates recently licensed if additional funding for use in the development of the product candidates is not obtained before March 8, 2012. Capital may not be available at all, in which case we will have to close operations and abandon rights to our product candidates.
To the extent we raise additional funds through collaboration or licensing arrangements, if we are able to do so, it will be necessary to relinquish some rights to our technologies or product candidates, and it may be necessary for us to grant licenses on terms that would otherwise be unfavorable to us. To the extent that we raise additional capital by issuing equity securities, our existing shareholders will experience dilution, which could be substantial. To the extent we raise additional capital by incurring debt, we would incur substantial interest obligations, may be required to pledge assets as security for the debt and may be constrained by restrictive financial and/or operational covenants that could limit our flexibility in conducting future business activities. Debt financing would also be senior to our stockholders’ interests in bankruptcy or liquidation. We may never succeed in raising additional capital of any form, and even if we do we may never achieve profitability.
We have a very limited operating history as we were formed as a shell company in May 2010 and, accordingly, you will not have any basis on which to evaluate our ability to achieve our business objectives.
We were formed as a shell company in May 2010 and did not have any operations until we licensed rights to an investigational therapeutic vaccine candidate and diagnostic test in March 2011. These product candidates have not been approved for sale anywhere in the world and we have not yet demonstrated our ability to successfully commercialize any product candidates. Successful commercialization will require us to, among other things, undertake human clinical trials; apply for and receive regulatory approvals once the clinical trials are completed; formulate and manufacture products; and conduct sales and marketing activities.
Our operations have been limited to organizing and staffing and acquiring our technology. These limited operations provide little or no basis for you to assess our ability to commercialize our product candidates and the advisability of investing in our securities.
Obtaining and maintaining the necessary U.S. and/or worldwide regulatory approvals for our product candidates will be time consuming, difficult and costly. If we fail to do so, we will be unable to commercialize our product candidates.
- 12 -
If we fail to obtain intellectual property rights, or otherwise secure exclusivity, for our investigational vaccine and diagnostic test, it will more difficult to obtain financing, enter into licensing or collaborative arrangements or sell the products at a reasonable profit, and our current funding is insufficient for us to seek our desired level of patent protection.
Our success, competitive position and future revenues will depend in part on our ability to obtain and then maintain patent protection for our product candidates, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of other third parties.
While we have plans to file patent applications, we have no issued patents. Our limited funds are not sufficient for us to currently secure patent protection or to pay fees required for translation or issuance of patents, if they were filed. We may never obtain sufficient exclusivity for our investigational therapeutic vaccine candidate or our diagnostic test to allow us an opportunity to sell them at a profit, in which case we may be forced to curtail or cease operations even if our development efforts are successful.
If we do ever successful obtain patent protection, in the future these patents may be challenged, invalidated or circumvented, which could limit our ability to prevent competitors from marketing related product candidates or could limit the length of the term of patent protection of our product candidates. In addition, the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may independently develop similar technologies. In addition, because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We have not registered any common stock for resale in the U.S. or elsewhere, and cannot assure you that our common stock will ever become liquid or trade on a securities exchange.
Our common stock has never traded. None of the Company’s securities are registered for resale with the Securities and Exchange Commission. The outstanding shares of common stock may only be resold through registration under the Securities Act of 1933, or under an applicable exemption from registration. We may never be able to register any shares with the Securities and Exchange Commission, and our stock may never actually trade. Although we intend to list our common stock on one of the Nasdaq markets or on the American Stock Exchange, we may never meet the listing requirements and may never be able to do so. Prior to a stock being listed on a stock exchange, its shares can only trade if a qualified market maker applies for and receives approval to make a market in the securities, companies cannot do so themselves. Our shares may never be quoted on a stock exchange or trade under alternative quotation system.
Risks Related to Our Business of Developing Investigational Therapeutic Vaccines and Diagnostics
Clinical trials are time-consuming and difficult and costly to design and implement
Human clinical trials are expensive to conduct and difficult to design and implement, in part because the science behind them is complex and they are therefore subject to rigorous regulatory requirements. Further, the medical, regulatory and commercial environment for pharmaceutical and vaccine products, such as our investigational therapeutic vaccine MEL-1, changes quickly and often in ways we may not be able to accurately predict. The clinical trial process is also very time-consuming. We estimate that clinical trials of our therapeutic vaccine candidate necessary to prepare regulatory submissions to seek approval to commercialize in the United States and Europe, and possibly other territories, will take at least several more years to complete. Furthermore, as failure can occur at any stage, we could encounter problems that cause us to abandon future clinical trials. The commencement and completion of clinical trials may be delayed by, among other things, changes in regulatory requirements, unforeseen safety issues, lack of effectiveness in clinical trials, slower than expected patient recruitment, inability to monitor patients adequately during or after treatment, inability or unwillingness of medical investigators to follow our clinical protocols, inability to maintain a sufficient supply of the investigational therapeutic vaccine to support the trials, suspension or termination of clinical trials for noncompliance with regulatory requirements and changes in clinical care protocols and standards of care within the institutions in which our trials take place.
In addition, we or the FDA may suspend our clinical trials at any time if it appears we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our Investigational New Drug Application, or IND, submissions or the conduct of these trials.
- 13 -
The results of our clinical trials may not support our product candidate claims.
Even if we are able to conduct clinical trials of our therapeutic vaccine candidate and diagnostic test, and the trials are completed as planned, we cannot be certain that the results of those trials will support our product candidate claims or that the FDA or foreign authorities will agree with our conclusions regarding the results. Success in preclinical testing or early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of any prior trials. The clinical trial process may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses, which could cause us to abandon a product candidate and may delay or cancel development of others. Any delay of our clinical trials will delay the filing of our BLAs and, ultimately, our ability to commercialize our product candidates and generate revenues. Any cancellation of our clinical trials will eliminate our ability to file a BLA for that product and eliminate our ability to generate any revenue from that product, unless we are later able to conduct a different trial that would satisfy the regulatory authorities. Negative results from a clinical trial, or positive results that are not robust enough to support approval, would be a major setback for us and could cause us to abandon development of one or more product candidates.
It is possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of our product candidate’s profile. Occurrence of any significant side effect could delay or terminate further development and hamper or prevent regulatory approval or marketing of our therapeutic vaccine candidate. Clinical trials of our diagnostic test may not support the efficacy of the test sufficiently to warrant its approval.
Obtaining and maintaining the necessary regulatory approvals in the U.S. and elsewhere for our product candidates will be time consuming, difficult and costly, and the results are uncertain. If we fail to obtain product approvals, we will be unable to commercialize our HPV product candidates.
Government regulations in the U.S. and other countries have a significant impact on our business and affect all aspects of drug and diagnostic research and development, manufacturing and marketing. We will require FDA approval to commercialize our product candidates in the U.S. and approvals from similar foreign regulatory authorities to commercialize our product candidates outside the U.S. In order to obtain FDA approval of a product candidate, we must submit to the FDA a BLA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research and development, including human clinical trials. These studies are time consuming, difficult and costly, and results are uncertain. We cannot predict whether our efforts will result in any vaccines or drugs that the FDA or other regulatory authorities consider safe and effective for humans. The results of our clinical studies in Mexico may not be replicated when we attempt to conduct similar trials in the Unites States or elsewhere. The FDA and other authorities have substantial discretion in the drug approval process, and may refuse to accept our application(s) or may deny approval. If the FDA does not accept or approve our applications, it may require us to conduct additional pre-clinical testing or manufacturing studies and submit that data before it will reconsider our application, or require us to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation, administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals will increase our operating expenses and delay the commercialization of our product candidates and our ability to derive product revenues from them.
Even if we comply with all FDA requests, the FDA may ultimately determine, under its statutory authority, to deny our requests for approval of our drug candidates. We cannot be certain that we will ever obtain regulatory clearance for any product candidate.
We have only one investigational therapeutic vaccine and only one diagnostic test that are currently being evaluated by us for potential commercial development, and even if our continued development of this product is successful, it will be several years before it can reach market.
Our current therapeutic vaccine product candidate MEL-1 is the only therapeutic product for which we have any rights, and our blood-based HPV diagnostic test is the only other product for which we have rights. Neither of these may ever be successfully marketed or manufactured, and our business would suffer substantial harm if they are not successfully commercialized. To date, these product candidates have only been tested in Mexico on a limited number of humans and the clinical trial database is not sufficient for approval in the United States, Europe and possibly other territories. The additional clinical trials required by the FDA and/or other regulatory authorities to obtain approval to market the product are likely to be long and complex and may take several years to complete. The FDA or other regulatory authorities may disagree with our clinical development plans, when formulated, and require us to conduct more extensive studies than we anticipate before considering our investigational drug for marketing approval. We cannot predict with any certainty when we might submit a BLA or PMA for regulatory approval for either of our product candidates, if at all.
- 14 -
Our development programs are likely to depend upon third-party researchers who are outside our control.
To develop our therapeutic vaccine candidate, we will be dependent upon clinical trial sites to administer the therapeutic vaccine to qualified patients that meet criteria specified in the clinical trial protocol. To develop our diagnostic product, we will be dependent on outside researchers to validate the accuracy of the test. Each of these trials is likely to require us to hire independent investigators and collaborators, such as medical institutions, clinical research organizations, and universities, to conduct our studies. We plan to contract with these collaborators, but they will not be our employees, and we cannot control the amount or timing of resources they devote to our programs. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking the programs ourselves. If outside collaborators fail to devote sufficient time and resources to our programs, or if their performance is substandard, the approval of our FDA applications and our introduction of new drugs, if any, will be delayed, which would harm our business, financial condition and results of operations. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators were to assist our competitors at our expense, our competitive position would be harmed.
We do not have current capability to manufacture our therapeutic vaccine candidate under FDA-required standards.
The manufacturing of biological agents such as our MEL-1 therapeutic vaccine candidate for HPV and cervical lesions is subject to a number of complex rules and regulations in the U.S. and elsewhere, often referred to as cGMP standards. We do not have any cGMP drug or vaccine manufacturing capability. Because our therapeutic vaccine candidate is manufactured using specialized types of live viruses, there are only a limited number of contract manufacturers with experience in this field. To commercialize our product candidate, we must build manufacturing facilities ourselves and/or rely on a limited number of vendors to supply usable product. The FDA may take the position that we and/or our chosen manufacturers do not have enough experience manufacturing the product, or that the quality standards are not sufficient for approval or commercialization. If we are unable to obtain sufficient supplies of product to complete clinical trials the product approval could be delayed or prevented. If we are unable to obtain sufficient supplies to commercialize the product we may never receive any revenues.
Risks Related to Management and our Company
We may not successfully manage our growth.
Our success will depend upon the effective management of the development of the MEL-1 therapeutic vaccine and the EDIVPH diagnostic test. We were formed as a shell company and have no operating history prior to March 2011, when we only began the process of commencing operations. We must build all capabilities ourselves, or acquire them, including operational, financial and management systems, the capability to conduct or oversee human clinical trials, and must hire and train additional qualified personnel. We also must put in place an adequate system of internal controls and do not have any system currently in place upon which to build. If we are unable to manage the development of our product candidates or our growth effectively, our business would be harmed.
There is currently no trading market for our common stock, and liquidity of shares of our common stock is limited.
Our shares of common stock are not registered under the securities laws of any state or other jurisdiction, and accordingly there is no public trading market for our common stock. Therefore, outstanding shares of our common stock cannot be offered, sold, pledged or otherwise transferred unless subsequently registered pursuant to, or exempt from registration under, the Securities Act and any other applicable federal or state securities laws or regulations. Shares of our common stock cannot be sold for at least 12 months after we cease to be a “shell company”, provided the Company otherwise is in compliance with the applicable rules and regulations. Compliance with the criteria for securing exemptions under federal securities laws and the securities laws of the various states is extremely complex, especially in respect of those exemptions affording flexibility and the elimination of trading restrictions in respect of securities received in exempt transactions and subsequently disposed of without registration under the Securities Act or state securities laws. We may never be able to register our shares, and a trading market for our common stock may never develop.
Our common stock is likely to be subject to the Penny Stock Regulations
Our stock is not publicly traded. None of our stock is registered for resale with the Securities and Exchange Commission, and it may not be sold absent such registration or appropriate exemptions from registration. We may never qualify for trading on a public exchange, and we may never be traded on an alternative listing service, such as the OTCBB or the Pink Sheets. If our stock ever commences trading, our common stock will likely be subject to the SEC's “penny stock” rules to the extent that the price remains less than $5.00. Those rules, which require delivery of a schedule explaining the penny stock market and the associated risks before any sale, may further limit your ability to sell your shares, or the price you receive for your shares upon the sale.
- 15 -
The SEC has adopted regulations which generally define “penny stock” to be an equity security that has a market price of less than $5.00 per share. Our common stock currently has no “market price” and when and if a trading market develops, may fall within the definition of penny stock and subject to rules that impose additional sales practice requirements on broker-dealers who sell such securities to persons other than established customers and accredited investors (generally those with assets in excess of $1,000,000, or annual incomes exceeding $200,000 or $300,000, together with their spouse).
For transactions covered by these rules, the broker-dealer must make a special suitability determination for the purchase of such securities and have received the purchaser's prior written consent to the transaction. Additionally, for any transaction, other than exempt transactions, involving a penny stock, the rules require the delivery, prior to the transaction, of a risk disclosure document mandated by the Commission relating to the penny stock market. The broker-dealer also must disclose the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and, if the broker-dealer is the sole market-maker, the broker-dealer must disclose this fact and the broker-dealer's presumed control over the market. Finally, monthly statements must be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks. Consequently, the `penny stock` rules may restrict the ability of broker-dealers to sell our common stock and may affect the ability of investors to sell their common stock in the secondary market.
The concentrated ownership of our common stock may have the effect of delaying or preventing mergers, acquisitions or other significant corporate transactions.
Virolab S de RL de CV, the company that developed our therapeutic vaccine and diagnostic candidates, owns approximately 88% of our outstanding common stock. One of the three members of our board of directors is the President and a majority shareholder of Virolab Mexico, and thus controls the voting discretion of these shares. Because of its concentrated ownership of our stock and position on our board, Virolab Mexico is currently able to control all matters requiring stockholder approval and is able to exercise significant influence over all matters requiring board approval, including the election of directors and approval of mergers, acquisitions and other significant corporate transactions. Virolab Mexico is also developing other biological therapeutics, and there may be conflicts between our drug development and commercialization interests and Virolab Mexico’s. In addition, if Virolab Mexico elects to sell any of our shares of common stock which they own, our stock price, if it is ever quoted, may decrease.
We have not voluntarily implemented various corporate governance measures, in the absence of which, shareholders may have more limited protections against interested director transactions, conflicts of interest and similar matters.
Recent Federal legislation, including the Sarbanes-Oxley Act of 2002, has resulted in the adoption of various corporate governance measures designed to promote the integrity of corporate management and the securities markets. Some of these measures have been adopted in response to legal requirements. Others have been adopted by companies in response to the requirements of national securities exchanges, such as the NYSE or the Nasdaq Stock Market, on which their securities are listed. Among the corporate governance measures that are required under the rules of national securities exchanges are those that address board of directors' independence, audit committee oversight, and the adoption of a code of ethics. We have not yet adopted any of these corporate governance measures and, since our securities are not listed on a national securities exchange, we are not required to do so. It is possible that if we were to adopt some or all of these corporate governance measures, stockholders would benefit from somewhat greater assurances that internal corporate decisions were being made by disinterested directors and that policies had been implemented to define responsible conduct. Prospective investors should bear in mind our current lack of corporate governance measures in formulating their investment decisions.
If we are unable to attract and retain key personnel and advisors, it may adversely affect our ability to obtain financing, pursue collaborations or develop or market our product candidates.
To pursue our business strategy, we will need to attract and retain qualified scientific personnel and managers, including personnel with expertise in clinical trials, government regulation, manufacturing, marketing and other areas. Competition for qualified personnel is intense among companies, academic institutions and other organizations. We presently have no cash to pay to any of our employees which will make it very difficult to attract and retain qualified personnel and advisors. If we are unable to attract and retain key personnel and advisors, it may negatively affect our ability to successfully develop, test, commercialize and market our products and product candidates.
- 16 -
We face intense and increasing competition and many of our competitors have significantly greater resources and experience.
Many other companies, as well as academic and governmental institutions, are pursuing other forms of treatment, prevention and detection of the diseases that we target. For example, several competitors are working on developing and testing HPV vaccines for the prevention or treatment of cervical and other cancers. Two HPV-preventative vaccines developed by our competitors have been approved for human use. Our competitors and potential competitors include large pharmaceutical companies. These companies have significantly greater financial and other resources and greater expertise than us in research and development, securing government contracts and grants to support research and development efforts, manufacturing, pre-clinical and clinical testing, obtaining regulatory approvals and marketing. This may make it easier for them to respond more quickly than us to new or changing opportunities, technologies or market needs. Many of these competitors operate large, well-funded research and development programs and have significant products approved or in development. Small companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical companies, government grants or through acquisition or development of intellectual property rights. Our potential competitors also include academic institutions, governmental agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for product and clinical development and marketing. Research and development by others may render our technologies or products obsolete or noncompetitive.
FINANCIAL INFORMATION
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Our discussion includes forward-looking statements within the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. All statements made which are not purely historical are forward-looking, and are based upon current expectations that involve risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated or implied in these forward-looking statements as a result of a number of factors, including those set forth under the “Risk Factors,” “Cautionary Notice Regarding Forward-Looking Statements” and “Our Business” sections in this Form 8-K. We use words such as “anticipate,” “estimate,” “plan,” “project,” “continuing,” “ongoing,” “expect,” “believe,” “intend,” “may,” “will,” “should,” “could,” and similar expressions to identify forward-looking statements.
Plan of Operation
On March 8, 2011, we entered into a Licensing Agreement (“Licensing Agreement”) with Virolab Nevada LLC (“Licensor”) pursuant to which we were granted an exclusive, non-transferrable worldwide license for certain intellectual property developed by Licensor, principally comprising the technology, intellectual property, know-how, trade secret information, clinical trial data, and all other commercial rights to an investigational therapeutic vaccine for HPV and HPV-related cancers and a blood test for HPV (the “Technology”). Before this transaction we were a shell company, as defined in Rule 12b-2 of the Securities Exchange Act of 1934
We believe that the HPV therapeutic vaccine technology that we have in development has the potential to be the first therapeutic to clear pre-cancerous and cancerous lesions of the Cervix along with the underlying HPV viral infection. This belief is based on the results of two phase 2 clinical trials and one phase 3 clinical conducted in Mexico in which cancerous and pre-cancerous cervical lesions were eliminated or regressed in 88% to 97% of the patients. In addition, detectable HPV virus was eliminated from approximately 50% of the 1,246 patients treated in the three trials. All studies showed the vaccine candidate to be well tolerated, although headaches and other flu-like symptoms appeared in some of the patients.
In addition to the therapeutic vaccine candidate, Virolab has licensed rights to a specific blood-based diagnostic test for HPV. Currently, diagnosis of an infection with HPV requires a gynecological examination as well as a cervical tissue sample. We believe that a diagnosis from a simple blood draw will be less expensive and more convenient for patients.
The Company is evaluating its options to commercialize its therapeutic vaccine candidate and diagnostic test. These product candidates have not been approved for commercial sale anywhere in the world, and each country has different requirements for approval, so the commercialization process is likely to be lengthy and complex. The Company may employ different strategies in different areas of the world, such as sublicensing development and commercialization rights for some territories while retaining rights for other territories. Based on the historical work of Virolab Mexico in the country of Mexico, the Company believes that the path to obtain government approval may be shorter in the Latin America countries.
- 17 -
The Company will not be able to commercialize either its therapeutic vaccine candidate or its diagnostic test without additional capital. The Company is evaluating various means of raising this capital, including through the sale of equity securities, licensing agreements or other means. If the Company does not raise additional funds of at least $2 million for the advancement of its technology over the next three years it will lose its rights to the technology (including at least $0.5 million of which must be raised before March 8, 2012, and $1 million of which must be raised before March 8, 2013). The Company plans to seek at least $10 million of capital within the next 12 months, and plans to use these funds to determine the FDA’s requirements for approval of its therapeutic vaccine candidate, begin working on these requirements, file patents on its technology, and continue the process of seeking approval of its therapeutic vaccine candidate and diagnostic test outside of the United States. The Company is not presently able to allocate the specific costs for its plans because it does not yet know the requirements for approval of either its therapeutic vaccine candidate or its diagnostic test. While the Company plans to raise additional capital, and may seek to do so by offering equity securities of the Company, none of the Company’s securities have been registered with the Securities and Exchange Commission, and they may only be sold under registration or an applicable exemption from registration.
If the Company or a sublicense is successful in commercializing its therapeutic vaccine candidate or diagnostic test, the Company will be obligated to pay the Licensor two percent (2%) of any royalties received if the Company grants any third parties royalty-bearing licenses to the Technology. In addition, the Company has agreed to pay Licensor a royalty of one quarter of one percent (0.25%) of all gross revenue resulting from use of the Technology by the Company.
Going Concern
Because we only had $200 in cash at June 30, 2010, which is insufficient to fund any operations, the report of our independent registered public accounting firm on our financial statements for the period ended June 30, 2010 contains an explanatory paragraph regarding their substantial doubt about our ability to continue as a going concern. Our auditors’ opinion is based upon our operating losses and our need to obtain additional financing to sustain operations. Our ability to continue as a going concern will be dependent upon our ability to obtain the necessary financing to meet our obligations and repay our liabilities when they become due, and to generate sufficient revenues from our operations to pay our operating expenses. We will need to raise substantial funds in order to develop the HPV products which we have recently licensed from Virolab Nevada, and if we cannot raise additional funds we may need to abandon development of these products and cease operations.
Results of Operations
Accelerated Acquisitions X, Inc. was incorporated on May 4, 2010, and has had minimal operations for all of its reporting periods. We have not had any revenue, and our cumulative expenses have aggregated $1,800 through December 31, 2010, which primarily comprised professional and legal fees and other costs related to the start-up and organization of our business.
Liquidity and Capital Resources
As of December 31, 2010, the Company had a cash balance of only $200. There were no liabilities.
We plan to measure our future liquidity primarily by the cash and working capital we will have available to fund our operations, if we are ever able to raise capital. To date we have not raised any capital and, accordingly, do not have any capital available to fund our operations. We will not be able to commercialize either our therapeutic vaccine candidate or our diagnostic test without additional capital. We are evaluating various means of raising our initial capital, including through the sale of equity securities, licensing agreements or other means. If we do not raise additional funds of at least $2 million for the advancement of our technology over the next three years, we will lose our rights to the technology; furthermore we must raise at least $0.5 million of these additional funds before March 8, 2012 or we will lose our rights to the technology.
Accelerated Venture Partners, LLC, or AVP, has agreed to fund certain administrative operating expenses of Virolab until the Company succeeds in raising additional funds, at which point the administrative operating expenses will be due. In addition, the Company is party to a consulting agreement with AVP under which AVP has agreed to provide the Company with certain advisory services that include reviewing the Company’s business plan, identifying and introducing prospective financial and business partners, and providing general business advice regarding the Company’s operations and business strategy. Under the consulting agreement, cash compensation of $400,000 is due to AVP upon the Company securing $5 million in available cash from funding, and an additional $400,000 is due upon the Company securing $10 million in available cash from funding (inclusive of the first $5 million). The cash compensation is to be paid to AVP at the rate of $66,667 per month. The total cash compensation to be received by AVP under the consulting agreement is not to exceed $800,000 unless the Company receives an amount of funding in excess of $10 million. If the Company receives equity or debt financing that is an amount less than $5 million, in between $5 million and $10 million, or greater than $10 million, the cash compensation earned by the AVP under its consulting services agreement will be prorated. The Company has the option to make a lump sum payment to AVP in lieu of the monthly cash payments.
- 18 -
We plan to seek to raise at least $10 million of capital through the sale of equity or other means that could dilute existing shareholders within the next 12 months, and plan to use these funds to determine the FDA’s requirements for approval of our therapeutic vaccine candidate, begin working on these requirements, file patents on our technology, broaden our pipeline of investigational drugs, vaccines and/or diagnostics, establish a biotechnology laboratory, and continue the process of seeking approval of our therapeutic vaccine candidate and diagnostic test outside of the United States, including Mexico and the Latin America countries. We are not presently able to determine the specific costs for our plans because we do not yet know the requirements for approval of either our therapeutic vaccine candidate or our diagnostic test.
We expect to incur losses for at least several years into the future as we develop our investigational therapeutic vaccine and diagnostic test, and we are unable to estimate when, if ever, we will receive revenue or have a positive cash flow.
Off-Balance Sheet Arrangements
The Company does not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on the Company’s financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Critical Accounting Policies
The Securities and Exchange Commission issued Financial Reporting Release No. 60, "Cautionary Advice Regarding Disclosure About Critical Accounting Policies" suggesting that companies provide additional disclosure and commentary on their most critical accounting policies. In Financial Reporting Release No. 60, the Securities and Exchange Commission has defined the most critical accounting policies as the ones that are most important to the portrayal of a company's financial condition and operating results, and require management to make its most difficult and subjective judgments, often as a result of the need to make estimates of matters that are inherently uncertain. Through the most recent financial reporting period presented, which ended on December 31, 2010, we did not have any operations and the nature of our business generally did not call for the preparation or use of estimates. Due to the fact that we did not have any operating business, we do not believe that we had any such critical accounting policies. We have not reported results since acquiring rights to an investigational therapeutic vaccine and diagnostic test for HPV. We plan to report the results of our operations for the fiscal year ended March 31, 2011 within the timeframe required for smaller reporting companies, and are evaluating critical accounting policies now that we have operations and are no longer a shell company.
PROPERTIES
Offices
At this time, the Company maintains its designated office at 1840 Gateway Drive, Suite 200, Foster City, CA 94404. The Company’s telephone number is 650-378-1232. The Company’s fax number is 650-378-1399. The Company does not presently have a website.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth certain information regarding beneficial ownership of the Company's Common Stock as of June 15, 2011, by: (i) each current director; each nominee for director, and executive officer of the Company; (ii) all directors and executive officers as a group; and (iii) each shareholder who owns more than five percent of the outstanding shares of the Company's Common Stock. Except as otherwise indicated, the Company believes each of the persons listed below possesses sole voting and investment power with respect to the shares indicated.
- 19 -
|
Name and Address
|
|
Number of Shares
|
|
Percentage Owned
|
|
|
Virolab S de RL de CV (3)
|
22,350,000
|
88.2%
|
|||
|
671 Montevideo Ave./ Lindavista
|
|||||
|
Mexico City, Mexico 07300
|
|||||
|
Ricardo Rosales (4)
|
22,350,000
|
88.2%
|
|||
|
Accelerated Venture Partners, LLC (5)
|
3,000,000
|
11.8%
|
|||
|
1840 Gateway Drive, Suite 200
|
|||||
|
Foster City CA, 94404
|
|||||
|
Timothy Neher (6)
|
3,000,000
|
11.8%
|
|||
|
James A. D. Smith (7)
|
104,167
|
*
|
|||
|
Matthew M. Loar (8)
|
0
|
*
|
|||
|
All officers and directors as a group
|
25,454,167
|
100.0%
|
____________
(1) This table is based upon 25,350,000 shares issued and outstanding as June 15, 2011, and options for 104,167 shares which vest within 60 days of June 15, 2011.
(2) Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission and includes voting and investment power with respect to the shares. Shares of Common Stock subject to options or warrants currently exercisable or exercisable within 60 days are deemed outstanding for computing the percentage of the person holding such options or warrants, but are not deemed outstanding for computing the percentage of any other person.
(3) Ricardo Rosales is majority shareholder and President of Virolab S de RL de CV and holds voting and dispositive power for these shares. Ricardo Rosales is a director of the Company.
(4) Shares are owned directly by Virolab S de RL de CV. Ricardo Rosales, a director of the Company, is President of Virolab S de RL de CV and holds voting and dispositive power for these shares.
(5) Timothy Neher is Managing Partner of Accelerated Venture Partners, LLC and holds voting and dispositive power for these shares. Timothy Neher is a director of the Company.
(6) Shares are owned directly by Accelerated Venture Partners, LLC. Timothy Neher, a director of the Company, is Managing Partner of Accelerated Venture Partners and holds voting and dispositive power for these shares.
(7) James A.D. Smith was appointed Chief Executive Officer on April 15, 2011. Pursuant to his employment agreement, shares represent shares to be granted under stock options which will vest prior to August 15, 2011.
(8) Matthew M. Loar was appointed Chief Financial Officer on May 23, 2011. Pursuant to his employment agreement, no shares to be granted under stock options will vest prior to August 15, 2011.
* represents less than 1%
- 20 -
DIRECTORS AND EXECUTIVE OFFICERS
The following individuals served as our executive officers and directors as of June 15, 2011:
|
Name
|
Age
|
Positions
|
|
Ricardo Rosales
|
50
|
Director
|
|
Timothy Neher
|
45
|
Director
|
|
James A. D. Smith
|
52
|
Chief Executive Officer
|
|
Matthew M. Loar
|
48
|
Chief Financial Officer
|
Ricardo Rosales was Chief Executive Officer, President, Secretary and Treasurer of the Company from February 27, 2011 through April 15, 2011. Dr. Rosales was also appointed to the board of directors on February 27, 2011, a position which he still holds. Dr. Rosales has over 15 years experience in vaccine research and is the founder and President of Virolab S de RL de CV, where he developed the investigational therapeutic vaccine and diagnostic test recently licensed to the Company. Dr. Rosales has a Ph.D. in Molecular Biology from the Luis Pasteur University in France, and did two years post-doctoral research at the National Institutes of Health in Bethesda, MD. He then spent numerous years at the Biomedical Research Institute of UNAM (National Autonomous University of Mexico).
Timothy Neher was the President, Secretary, Treasurer and sole director of the Company from its founding in May 2010 through February 27, 2011 when Mr. Neher resigned his positions as an officer of the company but not his position as a member of the board of directors. Mr. Neher is the founding partner of Accelerated Venture Partners, LLC, a private venture capital firm based in Foster City, California, and has over 15 years of experience in connection with the provision of debt and equity financing, mergers and public offering transactions. Timothy is the acting Chief Financial Officer, Treasurer and a Director of Mikojo, Inc. a public reporting company since 2009. Mr. Neher is also Director of Pinpointed Solutions Inc. a private company since 2008, Director of Ipaypod Inc., a private company since 2007 and Director of Internet Card Present, Inc., a private company since 2007. He is also the President, Secretary and sole director of following public reporting companies: Accelerated Acquisitions IV, Inc., Accelerated Acquisitions XI, Inc., Accelerated Acquisitions XII, Inc., Accelerated Acquisitions XIII, Inc. and Accelerated Acquisitions XIV, Inc. Prior to founding Accelerated Venture Partners, Internet Card Present Industries, Pinpointed Solutions and Ipaypod, Timothy was Chairman and Chief Executive Officer of Wherify Wireless, a private to public company from 1999 to 2007. Other past experience includes roles as VP of Marketing & Sales for CTH Consumer Plastics and VP of Operations for Windy City Product Development.
James A. D. Smith joined Virolab as Chief Executive Officer in April 2011. Before joining the Company, Mr. Smith was Chief Executive Officer of Carlsbad Pharmaceuticals from April 2009 through March 2011. Prior to Carlsbad, Mr. Smith was President and Chief Executive Officer at Genelabs Technologies, a company where he served in a variety of executive capacities over approximately 14 years, including five years as Chief Executive Officer. Genelabs was a Nasdaq-listed biopharmaceutical company focused on discovery and development of novel treatments for chronic hepatitis C virus infection; a product candidate for treatment of systemic lupus and an investigational vaccine for prevention of disease caused by infection with hepatitis E virus. The company was acquired by GlaxoSmithKline in 2008. Previously he had been with ICN Pharmaceuticals (now Valeant Pharmaceuticals International) and Viratek. Jim has almost 30 years of experience with vaccines and anti-viral products. He holds a B.A. in Biology from UC San Diego.
Matthew M. Loar joined Virolab as Chief Financial Officer in May 2011. Before joining the Company, Mr. Loar was Chief Financial Officer of Neurobiological Technologies, Inc., a Nasdaq-listed biopharmaceutical company, from April 2008 through September 2010. From 2006 through 2008 he was Chief Financial Officer of Osteologix, Inc. and from 2001 through 2006 Mr. Loar was Chief Financial Officer of Genelabs Technologies, Inc., a company which he joined in 1995. Earlier in his career, Mr. Loar held finance positions with an international manufacturing company and was an audit manager with an international public accounting firm. Mr. Loar is currently on the board of Neurobiological Technologies, Inc. and is a certified public accountant in California (inactive). Mr. Loar holds a B.A. degree from the University of California, Berkeley.
There are no family relationships between our officers and directors. Each director is elected at our annual meeting of stockholders and holds office until the next annual meeting of stockholders, or until his successor is elected and qualified.
- 21 -
EXECUTIVE COMPENSATION
The following table summarizes all compensation recorded by us in 2010 for our principal executive officers, each other executive officer serving as such whose annual compensation exceeded $100,000, and up to two additional individuals for whom disclosure would have been made in this table but for the fact that the individual was not serving as an executive officer of our Company at December 31, 2010:
None
Outstanding Equity Awards at Fiscal Year-End
The following table provides information concerning unexercised options, stock that has not vested and equity incentive plan awards for each named executive officer and director as of December 31, 2010 and March 31, 2011:
None
Compensation of Directors
We have not established standard compensation arrangements for our directors and the compensation, if any, payable to each individual for their service on our Board will be determined from time to time by our Board of Directors based upon the amount of time expended by each of the directors on our behalf and comparable director pay at other companies. To date, none of our directors have received any compensation for their services.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Related-Party Transactions
Licensing Agreement.
The Company is a party to the Licensing Agreement with Virolab Nevada, LLC which is owned by Ricardo Rosales, a director and majority shareholder of the Company. As a result, this may not be an arms-length agreement.
Consulting Services Agreement.
The Company is a party to the Consulting Services Agreement with Accelerated Venture Partners, LLC. The Company also pays Accelerated Venture Partners, LLC, for certain operating expenses which incurred by the Company but for which the Company does not have sufficient cash to pay. Accelerated Venture Partners, LLC is controlled by Timothy J. Neher, the founder of the Company and a director and shareholder of the Company. As a result, these may not be arms-length agreements.
- 22 -
Other.
The officers and directors for the Company are involved in other business activities and may, in the future, become involved in other business opportunities. If a specific business opportunity becomes available, such persons may face a conflict in selecting between the Company and their other business interest. The Company has not formulated a policy for the resolution of such conflicts.
Director Independence
The Company has no “independent” directors within the meaning of Nasdaq Listing Rule 5605(a)(2).
LEGAL PROCEEDINGS
None
MARKET PRICE OF AND DIVIDENDS ON THE REGISTRANT’S
COMMON EQUITY AND RELATED STOCKHOLDER MATTERS
Market Price of the Registrant’s Common Equity
Our stock does not trade on any established market. None of the Company’s securities are registered for resale with the Securities and Exchange Commission. The outstanding shares of common stock may only be resold through registration under the Securities Act of 1933, or under an applicable exemption from such registration.
Dividend Policy
We have never paid cash dividends on our common stock. Under Delaware law, we may declare and pay dividends on our capital stock either out of our surplus, as defined in the relevant Delaware statutes, or if there is no such surplus, out of our net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year. If, however, the capital of our company, computed in accordance with the relevant Delaware statutes, has been diminished by depreciation in the value of our property, or by losses, or otherwise, to an amount less than the aggregate amount of the capital represented by the issued and outstanding stock of all classes having a preference upon the distribution of assets, we are prohibited from declaring and paying out of such net profits any dividends upon any shares of our capital stock until the deficiency in the amount of capital represented by the issued and outstanding stock of all classes having a preference upon the distribution of assets shall have been repaired. We do not anticipate paying any dividends in the foreseeable future.
RECENT SALES OF UNREGISTERED SECURITIES
On May 4, 2010, the Registrant sold 5,000,000 shares of Common Stock to Accelerated Venture Partners, LLC for an aggregate investment of $2,000. The Registrant sold these shares of Common Stock under the exemption from registration provided by Section 4(2) of the Securities Act.
- 23 -
On February 27, 2011, the Registrant sold 22,350,000 shares of the Company’s common stock par value $0.0001 for a price of $0.0001 per share to Virolab S de RL de CV. At the same time, Accelerated Venture Partners, LLC agreed to tender 3,500,000 of their 5,000,000 shares of the Company’s common stock par value $0.0001 for cancellation. In addition, AVP received an option to purchase 1,500,000 shares of the Company’s common stock for a price of $0.0001 per share, which AVP immediately exercised. The 1,500,000 shares purchased under option by AVP was based upon 5% of the fully diluted shares then outstanding, including shares reserved for specific issuances such as future stock options. The AVP option shares are subject to repurchase by the Company if AVP does not materially contribute to specified financing milestones of the Company (60% of these shares may be repurchased by the Company if $5 million is not raised, and 40% may be repurchased if a total of $10 million is not raised). Following these transactions, Virolab S de RL de CV owned 88.2% of the Company’s 25,350,000 issued and outstanding shares of common stock and AVP owned the remaining 11.2% of the issued and outstanding shares. Simultaneously with the share purchase, Ricardo Rosales, PhD was appointed to the Company’s Board of Directors. Such action represents a change of control of the Company.
DESCRIPTION OF SECURITIES
Our authorized capital stock consists of 100,000,000 shares of common stock, par value $0.0001 per share, and 10,000,000 shares of preferred stock, par value $0.0001 per share, the rights and preferences of which may be established from time to time by our board. As of June 15, 2011, there were 25,350,000 shares of common stock and no shares of preferred stock issued and outstanding. There were no securities outstanding which were convertible into our common or preferred stock.
None of the Company’s securities are registered for resale with the Securities and Exchange Commission. The outstanding shares of common stock may only be resold through registration under the Securities Act of 1933, or under an applicable exemption from registration.
Common Stock
Holders of our common stock are entitled to one vote for each share on all matters voted upon by our stockholders, including the election of directors, and do not have cumulative voting rights. Subject to the rights of holders of any then outstanding shares of our preferred stock, our common stockholders are entitled to any dividends that may be declared by our board. Holders of our common stock are entitled to share ratably in our net assets upon our dissolution or liquidation after payment or provision for all liabilities and any preferential liquidation rights of our preferred stock then outstanding. Holders of our common stock have no preemptive rights to purchase shares of our stock. The shares of our common stock are not subject to any redemption provisions and are not convertible into any other shares of our capital stock. All outstanding shares of our common stock are fully paid and nonassessable. The rights, preferences and privileges of holders of our common stock will be subject to those of the holders of any shares of our preferred stock we may issue in the future.
Prior to the licensing transaction entered into on March 8, 2011, through which Virolab acquired its rights to the investigational HPV vaccine and diagnostic test, the Company was a “blank check company”. Virolab’s shares of common stock are not registered under the securities laws of any state or other jurisdiction, and accordingly there is no public trading market for our common stock. Therefore, outstanding shares of our common stock cannot be offered, sold, pledged or otherwise transferred unless subsequently registered pursuant to, or exempt from registration under, the Securities Act and any other applicable federal or state securities laws or regulations. Shares of our common stock including shares issued to AVP cannot be sold before or after an acquisition under the exemptions from registration provided by Rule 144 under or Section 4(1) of the Securities Act (“Rule 144”) if the Company is designated a “shell company,” and for 12 months after it ceases to be a “shell company,” provided the Company otherwise is in compliance with the applicable rules and regulations. Compliance with the criteria for securing exemptions under federal securities laws and the securities laws of the various states is extremely complex, especially in respect of those exemptions affording flexibility and the elimination of trading restrictions in respect of securities received in exempt transactions and subsequently disposed of without registration under the Securities Act or state securities laws.
Preferred Stock
Our board may, from time to time, authorize the issuance of one or more classes or series of preferred stock without stockholder approval. Subject to the provisions of our certificate of incorporation and limitations prescribed by law, our board is authorized to adopt resolutions to issue shares, establish the number of shares, change the number of shares constituting any series, and provide or change the voting powers, designations, preferences and relative rights, qualifications, limitations or restrictions on shares of our preferred stock, including dividend rights, terms of redemption, conversion rights and liquidation preferences, in each case without any action or vote by our stockholders. One of the effects of undesignated preferred stock may be to enable our board to discourage an attempt to obtain control of our company by means of a tender offer, proxy contest, merger or otherwise. The issuance of preferred stock may adversely affect the rights of our common stockholders by, among other things:
- 24 -
|
●
|
restricting dividends on the common stock;
|
|
●
|
diluting the voting power of the common stock;
|
|
●
|
impairing the liquidation rights of the common stock; or
|
|
●
|
delaying or preventing a change in control without further action by the stockholders.
|
Item 9.01 Financial Statements and Exhibits.
Financial statements and the licensing agreement are attached as exhibits to the Current Report on Form 8-K filed on March 18, 2011.
SIGNATURES
Pursuant to the requirements of the Securities and Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
VIROLAB, INC.
|
By:
|
/s/ James A.D. Smith
|
|
|
Name:
|
James A.D. Smith
|
|
|
Title:
|
Chef Executive Officer
|
Dated: June 24, 2011
- 25 -