Attached files
| file | filename |
|---|---|
| EX-31.2 - ASSEMBLY BIOSCIENCES, INC. | v216864_ex31-2.htm |
| EX-32.2 - ASSEMBLY BIOSCIENCES, INC. | v216864_ex32-2.htm |
| EX-32.1 - ASSEMBLY BIOSCIENCES, INC. | v216864_ex32-1.htm |
| EX-31.1 - ASSEMBLY BIOSCIENCES, INC. | v216864_ex31-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the fiscal year ended December 31, 2010
|
¨
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the Transition Period from to
Commission File Number 001-35005
VENTRUS BIOSCIENCES, INC.
(Exact name of registrant specified in its charter)
|
Delaware
|
2834
|
20-8729264
|
|
(State or Other Jurisdiction of
Incorporation or Organization)
|
(Primary Standard Industrial
Classification Code Number)
|
(I.R.S. Employer
Identification No.)
|
99 Hudson Street, 5th Floor
New York, New York 10013
(Address of Principal Executive Offices)
(212) 554-4506
(Telephone Number, Including Area Code)
Securities Registered Pursuant to Section 12(b) of the Exchange Act:
|
Title of Each Class
|
Name of Exchange on which Registered
|
|
|
Common Stock, $0.001 Par Value
|
Nasdaq Capital Market
|
Securities Registered Pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act.
Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer ¨
|
Accelerated filer ¨
|
Non-accelerated filer ¨
|
Smaller reporting company x
|
||
|
|
|
|
|
(Do not check if a smaller reporting company)
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting stock held by non-affiliates of the registrant, as of December 31, 2010, was approximately $37,348,210. Such aggregate market value was computed by reference to the closing price of the common stock as reported on the Nasdaq Capital Market on December 31, 2010. For purposes of making this calculation only, the registrant has defined affiliates as including only directors and executive officers and shareholders holding greater than 10% of the voting stock of the registrant as of December 31, 2010. The registrant used December 31, 2010 as the measurement date because it completed its initial public offering on December 22, 2010 and prior to that time no market existed for its voting stock.
As of April 8, 2011 there were 7,189,706 shares of the registrant’s common stock, $0.001 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The Company’s definitive Proxy Statement for its 2011 Annual Meeting of Stockholders (certain parts, as indicated in Part III).
VENTRUS BIOSCIENCES, INC.
TABLE OF CONTENTS
|
Page
|
||
|
PART I
|
1
|
|
|
Item 1.
|
Business
|
1
|
|
Item 2.
|
Properties
|
54
|
|
Item 3.
|
Legal Proceedings
|
54
|
|
Item 4.
|
[Removed and Reserved]
|
54
|
|
PART II
|
54
|
|
|
Item 5.
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
|
54
|
|
Item 6.
|
Selected Financial Data
|
54
|
|
Item 7.
|
Management’s Discussion and Analysis of Financial Condition and Results of Operation
|
55
|
|
Item 8.
|
Financial Statements and Supplementary Data
|
63
|
|
Item 9.
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
|
63
|
|
Item 9A.
|
Controls and Procedures
|
64
|
|
Item 9B.
|
Other Information
|
64
|
|
PART III
|
65
|
|
|
Item 10.
|
Directors and Executive Officers of the Registrant
|
65
|
|
Item 11.
|
Executive Compensation
|
65
|
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
|
65
|
|
Item 13.
|
Certain Relationships and Related Transactions, and Director Independence
|
66
|
|
Item 14.
|
Principal Accounting Fees and Services
|
66
|
|
Item 15.
|
Exhibits and Financial Statement Schedules
|
66
|
PART I
Item 1. Business
Overview
We are a development stage specialty pharmaceutical company focused on the development of late-stage prescription drugs for gastrointestinal disorders for which there are no approved prescription drugs in the U.S., specifically hemorrhoids, anal fissures and fecal incontinence. We are not aware of any prescription treatments for these conditions that have been approved by the U.S. Food and Drug Administration, or FDA, yet there are approximately 12.5 million Americans suffering from hemorrhoids, 7 million from fecal incontinence and over 4 million from anal fissures. Our lead product, Inferanserin (VEN 309) is a new chemical entity, or NCE, for the topical treatment of hemorrhoids. In multiple clinical studies in 359 patients, VEN 309 demonstrated good tolerability and no severe adverse events, and statistically significant improvements in bleeding, itchiness and pain. We have filed a special protocol assessment, or SPA, with the FDA to allow us to begin the first of two Phase III clinical trials for VEN 309.
Our additional product candidate portfolio consists of two in-licensed late-stage drugs intended to treat anal fissures (VEN 307) and fecal incontinence (VEN 308). These candidates are two molecules that were previously approved and marketed for other indications and that have been formulated into our proprietary topical treatments for these new gastrointestinal indications. In August 2007, we had a pre-investigational new drug, or IND, meeting with the FDA concerning VEN 307 (diltiazem cream for the treatment of pain from anal fissures) where it was established that next clinical studies needed for approval were two pivotal Phase III trials, preceded (if conducted in the U.S.) by three short-term dermal toxicology studies. In June 2007, we had a pre-IND meeting with the FDA concerning VEN 308 (phenylephrine gel for the treatment of fecal incontinence associated with ileal pouch anal anastomosis) where it was established that the next clinical study in the program should be a Phase II(b) study where multiple doses will be assessed and that existing toxicology data are sufficient to support this study. We have not had further meetings with the FDA on either VEN 307 or VEN 308 since the meetings in 2007. The development of the three products, VEN 307, VEN 308 and VEN 309, was delayed subsequent to the FDA meetings due to a lack of financial resources prior to the completion of our initial public offering in December 2010. We intend to use the proceeds from that offering to advance VEN 309 and VEN 307 through the next stage of development.
Major pharmaceutical progress has been made in the gastrointestinal therapeutic areas of gastroesophageal reflux, peptic ulcer disease and inflammatory bowel disease. However, many major gastrointestinal disorders still lack medical treatments. We are pursuing treatments for three of the ten most prevalent gastrointestinal disorders in the U.S. We estimate that the patient population of these three disorders exceeds 23 million adults in the U.S., based on the data we cite for each indication in this report.
Our Products and Development Strategy
Our three late-stage product candidates are:
Iferanserin ointment (VEN 309) for the topical treatment of hemorrhoids. Hemorrhoids, which are characterized by the inflammation and swelling of veins around the anus or lower rectum, can cause bleeding, itching, pain and difficulty defecating. Iferanserin (VEN 309), a NCE formulated as an ointment for intra-anal application, has highly selective, antagonistic activity against peripheral 5-HT 2 A receptors involved in clotting and the contraction of arteries and veins, two events believed to be associated with hemorrhoid formation. By limiting 5-HT2 A receptor activity, VEN 309 improves the flow of blood out of the dilated veins that comprise the hemorrhoid, thereby reducing bleeding, itchiness and pain. As reported by the National Institute of Diabetes and Digestive Kidney Diseases, hemorrhoids affect approximately 12.5 million adults in the U.S. Despite such a high prevalence, we are not aware of any FDA-approved prescription drugs for the treatment of hemorrhoids in the U.S. While there are commonly used prescription drugs in the U.S. for hemorrhoids in the U.S., such as Anusol, none have been approved by the FDA because they entered the market prior to 1962 and have not been designated by the FDA as safe and effective. Various combination products (such as the Preparation H line of products) are available in the U.S. over-the-counter, or OTC, under the FDA’s OTC monograph rule. The great majority of these treatments provide only temporary relief from the symptoms of hemorrhoids and do not address the cause of hemorrhoids. These treatments’ mechanism of action is either general, such as steroids, or acting as a protective coating on the hemorrhoid, in the case of most of the Preparation H products, or unknown, in the case of herbal remedies, and we are not aware of any reports published in medical journals on the efficacy or safety of any product currently marketed in the U.S. for the treatment of hemorrhoids. We believe VEN 309 to be more effective than the currently available conventional hemorrhoid topical therapies and more attractive than surgical procedures, which are the only other currently validated treatment options.
1
We have licensed Iferanserin ointment (VEN 309) from Sam Amer & Co., Inc., or Amer, who had developed VEN 309 through Phase II studies and up to readiness for Phase III studies in the U.S. and Europe. Our license includes rights to all existing intellectual property and any further improvements on VEN 309 owned by Amer for the topical treatment of anorectal disorders.
Diltiazem cream (VEN 307), a topical treatment for the relief of pain associated with anal fissures. Anal fissures are small tears or cuts in the skin that lines the anus. They can be extremely painful, cause bleeding and often require surgery, which itself can have unsatisfactory outcomes. At present, we are not aware of any FDA-approved drugs for the treatment of anal fissures. Diltiazem cream, however, is currently used as the preferred treatment by many gastroenterologists across the U.S. in a version that must be specially mixed for each patient in the pharmacy. Topical nitroglycerine has also been used in this way but has a higher rate of side effects than topical diltiazem, notably headaches. Custom-mixed diltiazem, however, is not an FDA-approved use nor is the cost reimbursed by Medicare or health insurance plans. When applied topically for the treatment of anal fissures, diltiazem, which has been used for decades for hypertension and angina, dilates the blood vessels supplying the region, reduces anal sphincter tone, and thereby substantially decreases pain. In the majority of multiple clinical trials conducted against placebo or topical nitroglycerine conducted between 1999 and 2002 by various researchers, diltiazem cream significantly reduced the pain associated with anal fissures. Our product VEN 307 is a proprietary formulation of diltiazem that when applied topically is only minimally absorbed, at one-tenth the amount of the lowest dose used for cardiovascular treatment. We believe this low absorption improves VEN 307’s safety profile and lowers the risk of side effects. We expect to capture immediate market share if VEN 307 is approved due to its known efficacy among gastroenterologists, its ease of prescription as a pre-formulated FDA-approved product with no need for custom mixing necessary at the pharmacy, and the ability for patients to be reimbursed through their health plan or Medicare. We have licensed the exclusive North American rights to VEN 307 for the topical treatment of anal fissures from S.L.A. Pharma who has completed early-stage clinical trials, toxicology studies and manufacturing for VEN 307 up to the end of Phase II.
Phenylephrine gel (VEN 308) for the treatment of fecal incontinence associated with ileal pouch anal anastomosis, an FDA orphan indication. Ileal pouch anal anastomosis, or IPAA, is a surgical procedure used as part of a colectomy, which is a treatment for patients with ulcerative colitis. Fecal incontinence resulting from dysfunctional sphincter tone is a common consequence of this procedure. According to a U.S. community based epidemiology study (Nelson et al., JAMA, 1995), 2.2% of U.S. adults suffer from fecal incontinence, which we estimate to be approximately 7 million people, based on 2009 Census Bureau adult population estimates. Currently, there are few options available to treat this problem, consisting of bulk laxatives, fiber diets, Imodium, which is a treatment for diarrhea, and invasive surgical procedures. In addition, Oceana Therapeutics is developing Solesta™, an injectable inert bulking agent product approved in the European Union for the treatment of fecal incontinence in adult patients who have failed conservative therapy. Solesta is injected submucosally around the anal sphincter and consequently has to be administered in an outpatient setting by qualified physicians. Oceana Therapeutics is currently pursuing approval of Solesta by the FDA. Also, Norgine plans to conduct a Phase I trial with NRL001, a suppository formulation of an alpha adrenergic stimulating agent for the treatment of fecal incontinence, which is anticipated to start in Europe in early 2011. We are not aware of any FDA-approved drugs for fecal incontinence. In multiple clinical trials with patients suffering from IPAA-associated fecal incontinence, topical phenylephrine significantly (and in some patients, dramatically) improved patient bowel control. In clinical trials with other forms of incontinence, improvements were also observed following application of topical phenylephrine, depending on the cause of the incontinence.
2
Our product VEN 308 is a gel formulation of phenylephrine. Applied topically, VEN 308 increases anal sphincter tone, thereby improving fecal incontinence in patients where sphincter tone is the major cause of their symptoms, such as post-IPAA surgery. We believe VEN 308 has significant advantages over the limited treatment options currently available for fecal incontinence associated with IPAA including, but not limited to, increased efficacy and/or reduced invasiveness. We have licensed the exclusive North American rights to VEN 308 from S.L.A. Pharma who developed the specific formulation of phenylephrine for the topical use in fecal incontinence and developed the manufacturing method. S.L.A. Pharma’s previous partner, Solvay, conducted important pharmacokinetic studies. We do not expect to continue developing VEN 308 in the short term.
Our Development Efforts
We do not own and did not develop any of our product candidates. We have licensed our three product candidates from third parties. All clinical trials to date have been conducted either by the licensor, the licensor’s previous partners or by independent investigators, as have the preclinical studies and product formulation activities. Since the time we licensed these products, we have focused our efforts on establishing and clarifying the regulatory pathway for late phase clinical trials and regulatory approval, and on establishing the contract manufacturing capacity and methods necessary to allow late phase clinical trials to proceed, all of which will be conducted by contracted third parties under our direction. These development efforts have not required many employees and we have historically operated with only a handful of employees with the scientific expertise necessary to progress our product candidates down the development path outlined above. This has helped us contain our operating costs. Subsequent to the completion of our initial public offering in late December 2010, we hired five employees and contracted with three individuals or entities to complete our staffing needs for our planned initial Phase III trial of VEN 309. However, we remain dependent on the availability and competency of these third parties for the continued development of our product candidates.
Our Management
Although incorporated in 2005, we began active operations in the spring of 2007 upon the licensing of VEN 307 and VEN 308 by Paramount BioSciences from S.L.A. Pharma. Shortly thereafter, we hired Thomas Rowland as our chief executive officer (who was then and remains one of our directors), Dr. Terrance Coyne as our chief medical officer, and Dr. John Dietrich as our vice president of clinical operations, as well as other employees. Due to our lack of capital, Drs. Coyne and Dietrich resigned in February 2009. Mr. Rowland resigned as our chief executive officer in February 2009, but he continued to act as our president from the date of his resignation in February 2009 until May 2010. Simultaneously with the resignation of Dr. Dietrich, we entered into a consulting agreement with him whereby he provides consultation on manufacturing, preclinical and clinical aspects of our drug programs on an as-needed basis. These arrangements with Mr. Rowland and Dr. Dietrich allowed us to continue minimal operations following their resignations until June 2010. Between February 2009 and June 2010, our only business activities consisted of maintaining our licenses with S.L.A. Pharma and Amer and financing and business development activities. Upon the successful completion of our convertible promissory note offering in May 2010, our Board of Directors determined to proceed with our initial public offering to raise capital to finance the partial development of VEN 309 and VEN 307. To conserve our resources, and recognizing that permanent employment would be dependent on our raising capital in our initial public offering, in June 2010, we entered into consulting agreements with Dr. Russell Ellison, our Chief Executive Officer and Chief Medical Officer, and David Barrett, our Chief Financial Officer. Between June 2010 and December 2010, our only business activities consisted of maintaining our licenses with S.L.A. Pharma and Amer and activities connected with our initial public offering. Effective on the completion of our initial public offering, we entered into employment agreements with Dr. Ellison and Mr. Barrett. From late December 2010 through February 2011, we completed the staffing for our planned development of VEN 309, including the extension of the consulting agreement with Dr. Dietrich through February 2012. We also added a clinician, two clinical project managers, a head of manufacturing, and an executive assistant, on a contract or permanent employment basis.
3
IFERANSERIN OINTMENT (VEN 309)
Background on hemorrhoids
Incidence and prevalence
Hemorrhoids are a common anal disorder, characterized by bleeding, itching, pain, swelling, tenderness and difficulty defecating. Based on information from an article entitled The Prevalence of Hemorrhoids and Chronic Constipation by J. Johanson and A. Sonnenberg published in Gastroenterology (1990; 98: 380-386), the point prevalence of symptomatic hemorrhoids in the U.S. adult population is approximately 5.7%, representing approximately 12.5 million cases based on 2009 population data published by the U.S. Census Bureau. The prevalence of hemorrhoids peaks in adults aged 45 to 65 years.
Patho-physiology of hemorrhoids
Hemorrhoids are symptomatic abnormalities of normal vascular structures in the anal canal that are manifested by dilation of the local arteries and veins due to constriction and partial obstruction of the exiting colonic veins. Although the exact mechanism for hemorrhoid formation is not clear, the progressive occlusion of venous exit vessels (e.g., as seen in straining during defecation, heavy lifting and pregnancy) is thought to produce stretching of the vessels in the hemorrhoidal plexus combined with vascular stasis. This stasis causes exposure of the blood to collagen, which in turn causes platelet clumping with the release of the platelet’s artery and vein constricting contents, including serotonin, which via stimulation of the 5HT2 receptor causes localized constriction of the exit arteries and veins, where most of the vascular smooth muscles are, and, in combination with other factors, causes a cascade effect producing clot formation. These events result in additional stasis of the blood, perpetuating and further worsening the situation. As hemorrhoids worsen, the trapped blood forms piles (protruding skin folds filled with static and thrombosed blood), initially above the pectinate line (internal hemorrhoids) and then below the pectinate line (external hemorrhoids). The classification of internal hemorrhoid grades by Banov is accepted by most specialists. This system consists of four grades and symptoms: first degree (grade I): hemorrhoids bleed but do not protrude; second degree (grade II): hemorrhoids protrude but reduce on their own; third degree (grade III): hemorrhoids protrude and require manual re-insertion; and fourth degree (grade IV): hemorrhoids protrude and cannot be manually re-inserted.
The cardinal symptom and most common manifestation of internal hemorrhoids is bleeding. Bleeding is often the only sign in grade I hemorrhoids, but it can also be accompanied by other symptoms as the hemorrhoids further enlarge, such as discomfort, itching, prolapse, and fecal soilage.
Current treatments
Despite the high prevalence of hemorrhoids, we are not aware of any FDA-approved prescription drugs for the treatment of hemorrhoids in the U.S. While there are commonly used prescription drugs for hemorrhoids in the U.S., such as Anusol, none have been approved by the FDA because they entered the market prior to 1962 and have not been designated by the FDA as safe and effective. Various combination products (such as the Preparation H line of products) are available in the U.S. under the FDA’s OTC monograph rule. The great majority of these treatments provide only temporary relief from the symptoms of hemorrhoids and do not address the cause of hemorrhoids. These treatments’ mechanism of action is either general, such as steroids, or acting as a protective coating on the hemorrhoid, in the case of most of the Preparation H products, or unknown, in the case of herbal remedies, and we are not aware of any reports published in medical journals on the efficacy or safety of any product currently marketed in the U.S. for the treatment of hemorrhoids. By contrast, our product, iferanserin ointment (VEN 309), has highly selective, antagonistic activity against peripheral 5-HT2 A receptors (5HT2 A >5HT2C>>5HT2B) involved in clotting and the contraction of arteries and veins, two events believed to be associated with hemorrhoid formation. By limiting 5-HT2 A receptor activity, VEN 309 improves the flow of blood out of the dilated veins that comprise the hemorrhoid, thereby reducing bleeding, itchiness and pain. We believe that the potential for side effects is likely to be limited because iferanserin is topically applied and iferanserin does not enter the brain to affect 5HT2 CNS receptors, at the exposures seen with topical application. In multiple clinical trials, iferanserin ointment significantly reduced bleeding, pain and itchiness compared to placebo with minimal adverse effects. As a result, we believe VEN 309 to be more effective and/or less invasive than the currently available conventional hemorrhoid topical therapies and more attractive than surgical procedures, which are the only other currently validated treatment options. Patients with persistent symptoms, especially bleeding, usually require an invasive procedure. The most common is rubber band ligation, which involves banding the internal hemorrhoid for four to seven days. Other procedures are the injection of a sclerosing agent, electrocoagulation, light therapy and hemorrhoidectomy. Most physicians treating hemorrhoids start with conservative therapy consisting of diet modification, fiber, sitz baths and stool softeners. In addition to this conservative therapy, physicians might prescribe topical steroids. The only other alternatives are invasive procedures and/or surgery. Because of the lack of effective prescription products, most hemorrhoid patients will use over-the-counter preparations or the prescription drugs available, which are similar to the over-the-counter treatment, but formulated with a higher dose of topical steroid. According to IMS Health (2006), 4.0 million prescriptions are written per year in the U.S. for unapproved hemorrhoid prescription products and 22 million units per year are sold in the U.S. for the unapproved OTC hemorrhoid products. If VEN 309 receives FDA approval in the U.S., we expect our competition for patient use and physician prescribing will be these drugs which have not been approved by the FDA and lack any medical study dated supporting their efficacy and safety. In Europe it appears that, from our discussions with experts and staff from other companies, many products exist, differently from country to country, and are mostly herbal extracts and mixtures in topical and systemic forms which are either prescribed or available over-the-counter. We do not have market data concerning these products in Europe, other than product acceptance market research, nor is their precise regulatory status clear to us.
4
INFERANSERIN OINTMENT (VEN 309) DEVELOPMENT
Background on Iferanserin
The early proof of concept for the utilization of a 5-HT2 A antagonist for the treatment of hemorrhoid was developed by Sam Amer PhD, a former director of research and development at Bristol Myer. Dr. Amer explored the potential application of serotonin drugs, which would not enter the brain at therapeutic concentrations, for use in various venous conditions. After successful pre-clinical and clinical experiments, Dr. Amer filed a method of use patent covering this molecule in 1992. Dr. Amer subsequently separated the S-isomer from this racemic mixture and filed new composition of matter patents for the S-isomer in 1998. Also in 1998, the early stage product was licensed to Tsumura, a Japanese company. Tsumura conducted over 350 pre-clinical and six clinical studies, but was not able to continue development due to financial difficulty and returned the product to Dr. Amer. Upon the return, Dr. Amer’s company, Sam Amer & Co., Inc., or Amer, conducted a double-blind, placebo controlled, multi-center confirmatory non-pivotal phase III study in Europe. After the successful completion of that study in 2003, Novartis Pharmaceuticals licensed iferanserin from Amer to be part of its gastroenterology portfolio strategy. Novartis improved the iferanserin manufacturing processes and completed important toxicology and metabolite studies. In 2005, Novartis’ lead gastroenterology product, Zelnorm™ was experiencing increased FDA scrutiny on the safety of that product, which would ultimately lead to its eventual withdrawal from the market. We believe that with the impending loss of their lead gastroenterology product, Novartis decided to dissolve the gastrointestinal franchise. In 2005, Novartis returned iferanserin to Amer. According to Amer, no safety or clinical issues were ever communicated as reasons for the return.
On February 5, 2008, in conjunction with Amer, we held an End of Phase II meeting with the FDA, to confirm the U.S. regulatory status and pathway to a new drug application, or NDA, for iferanserin ointment where it was agreed that the product may enter late-stage Phase III development. In March 2008, we licensed exclusive worldwide rights to develop and market iferanserin ointment for the treatment of anorectal disorders from Amer.
Mechanism of action on iferanserin
Iferanserin has selective antagonistic activity against 5-HT2 receptors, especially against those involved in contraction of vascular smooth muscle and platelet aggregation (clotting), the 5HT2A receptors. It is a particularly potent high-affinity antagonist of 5HT2A, has less affinity for and is a moderate antagonist of 5HT2C and has considerably less affinity for 5HT2B receptors. In a specific validated model, iferanserin did not demonstrate any agonism activity at 5HT2B receptors, but did demonstrate moderate antagonistic activity. Unlike other 5HT2 receptor antagonists, iferanserin’s 5HT2 receptor antagonism, clinically, is entirely peripheral, meaning it occurs outside the central nervous system because iferanserin does not cross the bloodbrain barrier except in extremely high exposures far above those seen with topical application. Studies conducted in 1997 and 1998 by Amer in rats addressed the potential effects of iferanserin on impaired rectal mucosal blood flow and increased peripheral vascular resistance after administration of serotonin or thrombin. At doses of 3 mg/kg and above administered intrarectally, iferanserin improved rectal mucosal blood flow and normalized the peripheral vascular resistance. Iferanserin had minimal effects on arterial blood pressure.
5
Preclinical safety
Iferanserin has been extensively tested in multiple preclinical models. The iferanserin exposure from dosing in humans topically using 0.5% applied twice daily (the dose to be used in our planned studies) ranges from 1/17th to 1/88th of the exposure that produces toxicity and from 1/45th to 1/85th of the exposure that produces cardiovascular effects in animal toxicology studies and 1/60th − 1/100th of the exposure that produces these effects in vitro. In addition, iferanserin exhibits low systemic exposure, with less than 10% bioavailability, based on a pre-clinical rat study.
Clinical trials and patent status
A total of seven clinical trials with iferanserin have been completed by Amer (excluding Japan) and Tsumura in Japan between 1993 and 2003. One Phase I study and one Phase II study were completed using the racemic mixture of iferanserin. After the successful Phase II proof-of-principle study, the licensor, Amer, separated the R- and S-isomers (the two active components of most small molecule pharmaceuticals), determined that the primary activity was focused in the S-isomer and filed a patent claiming this isomer. The patent issued in the U.S. and other countries and expires in 2015. In the U.S., the patent was filed with Dr. Amer as the inventor and in all foreign countries with Amer as the assignee. After the development of the S-isomer in the mid 1990s and the patent filing in 1998, the remaining studies — two Phase I studies, two Phase II studies, and one Phase III study — were all conducted with the S-isomer product. This development progression (racemic to S-isomer) is a common pharmaceutical practice, enabling companies to use the purest form of the molecule in late-stage clinical trials.
Our license agreement with Amer includes the rights to all intellectual property owned by or assigned to Amer as well as to any new improvements owned by or assigned to Amer. Different concentrations of a drug are separately patentable. Because of unexpected differences between concentrations of the product that were observed in the clinical program (i.e. that 0.5% concentration is superior to a 0.25% and a higher 1.0% concentration in the comprehensive reduction in hemorrhoid symptoms), which data have not been previously published, we filed in August 2010 a patent claiming our specific concentration range (among other claims) which, we believe, if issued, would be considered new art and provide patent protection for 20 additional years. Dr. Amer is the inventor in this U.S. application and the assignee in the patent application. However the original S-isomer patent could be challenged by a third party and invalidated, and the concentration patent may never issue and even if issued could be challenged by a third party, in which case we would have five years of U.S. market exclusivity under the Hatch-Waxman Act.
An investigator IND for iferanserin was filed with the FDA in November 1991 and transferred to Amer as the sponsor in January 1994 and remains open.
Trial Results
Overall safety
In the seven clinical studies of iferanserin conducted by Amer and Tsumura in 359 individuals, of whom 220 were exposed to iferanserin, the adverse effects, at least possibly related to the iferanserin administration, were mostly gastrointestinal (diarrhea, lower abdominal discomfort, residual stools, and anal irritation). These events were considered mild by the investigators and required no medical treatment. There were no serious adverse events judged by the investigator as related to iferanserin and no mortality in these studies. There was one report of exacerbation of atopic dermatitis requiring observation in hospital with an uncertain relationship to iferanserin.
6
Clinical Pharmacology in Normal Volunteers (Phase I)
Two clinical pharmacology studies were conducted in Japan by Tsumura in 1998 and 1999 in 18 healthy volunteers exposed to a single dose and in six healthy volunteers exposed to six days of dosing with the 1% preparation. Three mild adverse events where the drug could not be ruled out were observed in three patients in the single dose group and four mild adverse events were observed in three patients in the multi-dose group. There is no accumulation of the drug on twice daily dosing and the half life at one and six days is 1.6 hours. Peak concentrations are similar at one and six days and well below the lowest exposure where toxicity was observed in toxicology experiments in animals.
One patient was identified as having a very compromised activity of an enzyme, CYP2D6, and the maximum concentration of the drug in this patient was three times the maximum observed in the other patients and the total exposure (AUC) was 17 times that observed in the other patients. However, these exposures to the drug were still well below the lowest exposures where toxicity was observed in animal toxicology experiments, and this patient did not experience any adverse events.
As is typical of several modern drugs for depression such as Fluoxetine and older drugs such as tri-cyclic anti-depression agents and other drugs extensively prescribed, iferanserin is an inhibitor of the enzyme CYP2D6 and is at least partially dependant on this enzyme for its metabolism. Therefore kinetic interactions with other drugs that are potent inhibitors of CYP2D6 and or are highly dependent on CYP2D6 for their metabolism are possible. There are several of these drugs and most are psychiatric medications, and one is tamoxifen. We will exclude patients from the clinical trials who are taking such drugs, and will be conducting extensive drug-drug interaction studies as part of our clinical pharmacology program to clarify which drugs could be affected by or could affect iferanserin. We intend to conduct these studies contingent on having sufficient resources after the completion of the first planned Phase III trial.
Proof-of-concept study (U.S.)
A double-blind, placebo-controlled study of 26 patients conducted by Amer that was completed in August 1992 and published in August 1994 was the first clinical trial to test the activity of the racemic mixture of iferanserin. Topical 1% iferanserin ointment was applied three times daily for five days to calculate the effect on bleeding and other symptoms in patients with grade I to III external hemorrhoids. Treatment produced statistically significant improvements in ease of defecation, throbbing, fullness, bleeding and tenderness. Itchiness and pain were also reduced following treatment. These positive treatment effects started immediately after treatment and were maintained throughout the study.
Early Phase II dose-ranging study (Japan)
Topical iferanserin ointment, in twice-a-day doses of 0.25%, 0.5%, and 1.0%, was provided to 72 patients for 14 days to treat symptomatic internal and mixed internal/external hemorrhoids. A total of 68 patients were evaluable for analysis: 23 patients in the 0.25% dose group, 24 patients in the 0.5% dose group, and 21 patients in the 1.0% dose group.
There was a significant change in ease of defecation between dose groups by day 7 but no other differences in improvements of symptoms among the three dose groups. Anal discomfort and pain persistence improved with increasing dose on a visual analog scale, or VAS, of pain. For the symptom of bleeding, a significant difference between dose levels (P = 0.016) and a paired comparison statistical analysis showed that the 0.5% dose was more effective than either the 0.25% dose or the 1.0% dose. By day 14, hemorrhoid swelling was reduced in the 0.5% dose group (41%) and the 1.0% dose group (43%). A review of patient diaries revealed that all symptoms started improvement on day 1, with improvement peaking at day 7 and being maintained to day 14. Comparison of all doses showed, unexpectedly, that the 0.5% dose provided the most consistent improvements.
7
There were 45 adverse events, but only five (11%) were judged as related to iferanserin ointment. These iferanserin-related adverse events were mostly mild diarrhea or lower abdominal discomfort, which required no medical treatment. Laboratory tests were generally normal, with the exception of one case of mild elevation of total bilirubin one month after trial completion, which required no therapy. Further evaluation of metabolites revealed no relationship to adverse events. The unexpected and novel finding that 0.5% concentration is superior to both a lower (0.2%) and higher (1%) concentration supports our patent claiming a specific concentration range that we filed in August 2010, which, if issued will expire in 2030.
Late Phase II study (Japan)
A double-blind, placebo-controlled trial was conducted by Tsumura Company with three different concentrations of iferanserin ointment (0.25%, 0.5% and 1%) administered twice daily for four weeks for treatment of 104 patients with grade I to III internal hemorrhoids. The study was completed in July 2002 and published in February 2003. Inclusion criteria required a minimal degree of either bleeding or prolapse. The primary endpoint was physician-rated size reduction of the hemorrhoids; secondary endpoints included subjective symptoms as assessed by patient diaries and VAS. By day 28, compared with placebo, the concentrations of 0.5% and 1% of iferanserin showed the most consistent improvements across groups for secondary symptoms, such as bleeding, pain severity and duration, and ease of defecation.
PhaseIIB/III study (E.U.)
Based on the results of the two Tsumura Phase II trials, a double-blind, randomized, placebo-controlled study was conducted by Amer to compare 0.5% iferanserin ointment with placebo when administered twice daily for 14 days for treatment of 121 patients with symptomatic grade I to III internal hemorrhoids. The disease specific inclusion criterion was diagnosis of grade I − III hemorrhoids with bleeding episodes of at least every other day during the last two weeks before enrollment in the study. Exclusion criteria included patients with protruding or irreducible hemorrhoids (grade IV), and patients with anal fistulas, periproctitis or hemorrhagic diathesis. Daily patient diaries for bleeding, itching and pain/discomfort were recorded for 14 days, and patient assessments were recorded at days 7 and 14 based on a 10-point scale. A physician evaluation for prolapse and size occurred at baseline and day 14. Fifty-six patients, each in the active and placebo treatment groups, were evaluable for the primary endpoint, which was bleeding. Not all patients had each of the other symptoms, but sufficient numbers were available for statistical analyses to be performed for bleeding, pain, itching and dyschezia, which is extensive straining with stools.
Compared with placebo, iferanserin ointment significantly reduced bleeding (P < 0.05) by day 3, a reduction maintained to day 14 (Figure 1). Total cessation of bleeding occurred in 89% of the iferanserin-treated patients versus 68% of the placebo-treated patients. Compared with placebo, iferanserin ointment also significantly reduced itching (P < 0.05) by day 3. Total elimination of itching by day 14 was achieved in 90% of the iferanserin-treated patients versus 62% of the placebo-treated patients. Finally, compared with placebo, iferanserin ointment significantly reduced pain (P < 0.05) by day 3, effecting a total elimination of pain in 78% of patients versus 46% of patients in the placebo group. There were also no clinically significant adverse findings for either iferanserin or placebo.
8
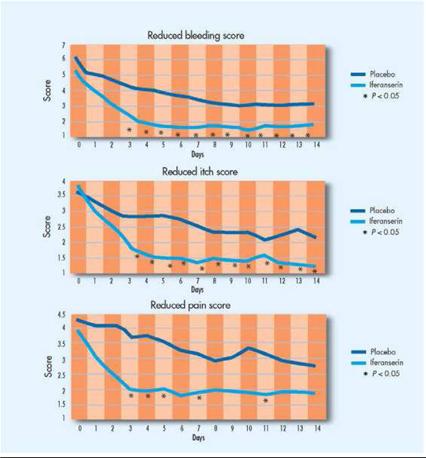
Figure 1. In a Phase III double-blind, randomized, placebo-controlled study of 121 patients with grades I to III internal hemorrhoids, iferanserin ointment significantly improved bleeding, itching, and pain.
After the end of Phase II meeting with the FDA and as part of the SPA review process, we commissioned a post hoc analysis of the study for the end point that the FDA agreed would be the primary efficacy endpoint for the pivotal trials. This endpoint is defined as time to cessation of bleeding that lasts for three days or more for which iferanserin 0.5% twice daily will be compared with placebo. In this analysis, the median time to cessation of bleeding in this 14 day study was 10.5 days in the placebo group and 4.5 days in the treatment group which was statistically significant (P < 0.01) (Figure 2).
9

Figure 2. In a Phase III double-blind, randomized, placebo-controlled study of 121 patients with grades I to III internal hemorrhoids, the median time to complete cessation of bleeding was 4.5 days for iferanserin ointment versus 10.5 days for placebo (p 0.01).
In this Phase III double-blind, randomized, placebo-controlled study of 121 patients with grades I to III internal hemorrhoids, iferanserin provided rapid and sustained improvements of the main symptoms of this disorder: bleeding, itching and pain. Maximal improvements of symptoms occurred by day 7 and were maintained to day 14 at the end of the trial.
Iferanserin ointment (VEN 309) development plan
At the end-of Phase II meeting held in February 2008, the FDA advised us that, as is common for chronic or repeated use drugs, it would require for submission of the NDA:
|
|
·
|
a total safety database of 1,500 patients exposed to iferanserin, a proportion of which need to be followed for repeat use for six months and 12 months (standard International Conference on Harmonization recommendation);
|
|
|
·
|
included in this safety database, two placebo controlled studies would be required with the primary endpoint being time to cessation of bleeding for a minimum of three days;
|
|
|
·
|
also included in the safety database a clinical pharmacology program consisting of a thorough QT study (standard for most drugs), drug-drug interaction studies, and pharmacology in special populations will be required; and
|
|
|
·
|
as is usual for chronic or repeated use drugs, carcinogenicity studies in two species exposed for 104 weeks, preceded by dose ranging studies and 6 months toxicology in rats and 9 months in dogs.
|
As the carcinogenicity study (including the prior dose ranging study) will take up to 40 months to complete, we intend to conduct the Phase III studies sequentially as this will not delay the program, will conserve funds and allow adjustments (for example, increased sample size) to the second Phase III study to optimize its potential. We anticipate that we will initiate the first patient randomized into the first Phase III trial on or about the end of the second quarter of 2011, and that data will be available in the first quarter of 2012. We also intend to initiate the dose ranging part of the carcinogenicity studies in 2011, and to initiate the carcinogenicity studies themselves in the second half of 2011. We originally filed in June 2008 an SPA with the FDA to ensure their explicit agreement with our Phase III protocol for VEN 309. Due to lack of funds we could not follow up or complete the process but were able to resume with another filing in March 2010 and received comments in May 2010. We filed another submission in July 2010 which could not be processed because the FDA required us to reformulate the questions set forth in the filing. In August and September 2010, we had a series of emails and telephone calls with the FDA in which we believed that agreement has been reached on the precise definition of the endpoints and how to assess recurrence of hemorrhoids in the study and on October 28, 2010 we filed another submission reflecting these discussions.
10
In that SPA, we originally proposed for the pivotal trial design:
|
|
·
|
400 patients randomized, double blind, to either placebo ointment or iferanserin 0.5%, both applied twice daily (to be conducted in 60 community sites in the U.S. and Canada);
|
|
|
·
|
14 days treatment with follow up at 28 days;
|
|
|
·
|
Rolling over all patients to active treatment after 28 days double blind follow up visit, to be followed for 12 months, with retreatment for recurrence monitored;
|
|
|
·
|
Inclusion criteria to include patients with symptomatic Grade I to III internal hemorrhoids, bleeding from hemorrhoids every day for the two days immediately preceding the day that they are randomized and study medication applied, with pain or itching accompanying the bleeding for the two days;
|
|
|
·
|
Exclusion criteria to exclude patients with grade IV hemorrhoids; thrombosed internal or external hemorrhoids; laxatives, anticoagulants, over-the-counter anti-hemorrhoidal agents, topical steroids, suppositories of any kind, non-steroidal anti-inflammatory drugs (NSAIDs), Cox-2 inhibitors, and other drugs and conditions including potent inhibitors of CYP2D6 such as fluoxitene; and
|
|
|
·
|
The primary endpoint will be time to cessation of bleeding for a minimum of 3 days and secondary endpoints will be cessation of pain and cessation of itching for three days.
|
In March 2011, we had a formal meeting with the FDA to discuss the feedback received on the SPA that we submitted to the FDA in October 2010, in order to resolve remaining issues that would allow an agreement of the protocol between us and the FDA. The primary focus of the meeting was the FDA’s recommendations for changes to the definitions of the primary and key secondary efficacy endpoints of the protocol submitted in the SPA. We viewed the suggestions as improvements to the endpoints as well as enhancing their clinical meaningfulness and readily agreed to the changes. We submitted a new SPA on March 16, 2011, that includes a revised protocol, including the newly defined endpoints, and expect a response within 45 days, which is the customary FDA review period.
For the double-blind part of the study, where patients are treated twice daily for two weeks and then followed up on Day 28, the improved, FDA-recommended definitions for the endpoints, which remain subject to FDA agreement with the protocol for the SPA, are:
|
|
·
|
Primary: Proportion of patients with cessation of bleeding by the end of Day 7 that persists for the remainder of the treatment period (through Day 14) (replaced cessation of bleeding for a minimum of 3 days); and
|
|
|
·
|
Key Secondary: Proportion of patients with cessation of pain and/or itching by the end of Day 7 that persists for the remainder of the treatment period (through Day 14) (replaced cessation of pain and cessation of itching for three days).
|
We have modeled the potential performance of these new endpoints for the Phase III study using data from a prior double-blind Phase IIb study conducted in Germany which randomized 121 patients to iferanserin or placebo ointment. In the German study, using the statistical methodology proposed for the analyses of the primary endpoint in our planned Phase III study, the difference between the proportion of patients responding to treatment under the new endpoint definition for cessation of bleeding in the iferanserin arm (57% responders) and the placebo arm (20% responders) was considerable with a p <.0001 (Figure 3).This is an improvement over the prior endpoints, which were time-to-bleeding cessation (defined as three consecutive days of no bleeding) as the primary, and proportion of patients who had three days cessation of pain and/or itching as the secondary, due to a more rigorous definition of the endpoint in terms of the duration of effect required for a response. In fact, the difference in proportion of responders between treatment arms in this analysis of the proposed revised primary endpoint is almost twice that seen in an analysis of the previously defined primary endpoint, mostly due to the much lower response in the placebo group as would be expected with a more rigorous definition. Similarly, analyses of the key secondary endpoints of pain and/or itching also showed considerable differences between iferanserin and placebo.
11
We believe the new endpoint definitions confirm the projected power of > 90% for the primary endpoint and > 90% for the key secondary endpoints for the proposed Phase III study design of 400 patients randomized 1:1 to iferanserin or placebo ointment. Since the study size and power appear to be re-affirmed by this change, and since all of our clinical study sites will be using central Institutional Review Boards (IRBs) with rapid review times, and contracting with sites is already underway, we believe that our estimated timelines for study start (mid-summer 2011), completion of enrollment (year-end 2012), and availability of data (first quarter 2012), remain unaffected by the proposed new endpoints.

Figure 3. Analysis of new endpoints in the Phase IIB study: In a Phase III double-blind, randomized, placebo-controlled study of 121 patients with grades I to III internal hemorrhoids, 57% of iferanserin treated patients had cessation of bleeding by day 7 that continued through day 14 versus 20% of placebo treated patients (p < 0.0001).
After the results of the Phase III study are available, and if we raise additional capital, we intend to continue the carcinogenicity study, conduct the 6 and 9 month chronic toxicology studies and launch either an identical Phase III trial and a safety study, or a larger Phase III trial to provide adequate numbers of patients exposed, and to complete the clinical pharmacology program which, will include extensive drug-drug interaction studies to clarify the CYP2D6 interactions and a ‘‘thorough QT study’’ to test the arrythmogenic potential, which studies are routinely required by the FDA. We will also explore at that time the feasibility of lifecycle options for follow-on products such as combinations with steroids and other agents or different formulations such as suppositories, which could be developed for launch after approval of the original VEN 309 product.
We expect that the earliest we will be able to file a NDA with the FDA will be mid 2014, and the earliest the product could be approved in the U.S. would be in 2015. However, the Phase III trial may not meet the primary endpoint, or unexpected safety problems could arise, or even if the study is successful we may not be able to obtain more capital for other reasons, in which case we may not be able to complete the development of the product and we may not be able to effect the payments due to Amer on a timely basis, which could result in the loss of our rights to the product.
Commercial summary for iferanserin (VEN 309)
Market research regarding hemorrhoids
Market research conducted in 2001 by Amer with both patients and physicians shows a significant dissatisfaction with current treatment options and the need for a product that relieves multiple hemorrhoidal symptoms. In a survey conducted with 57 hemorrhoid patients, average satisfaction with current prescription treatment was rated at 6.0 on a 10-point scale. The most desired treatment effects of a new hemorrhoidal medication that patients described would be ‘‘fast onset,’’ and ‘‘bleeding cessation.’’ The most frequent hemorrhoidal symptoms these patients reported experiencing were itching (79%), bleeding (77%) and pain (68%).
12
A research study conducted by Amer of 40 physicians (30 primary-care physicians, five proctologists, and five colon and rectal surgeons) evaluated their satisfaction with current treatment for hemorrhoidal treatment on a 10-point scale. The level of satisfaction with current treatment for reducing bleeding was 6.4; for relieving itch, 7.1; and for reducing pain, 6.8. The physicians indicated that the most desirable treatment effects of new hemorrhoidal medication would be ‘‘fast onset (2 to 3 days)’’ and ‘‘multi-symptom relief.’’ Another research study of 98 physicians showed that most physicians would replace their current first line therapy with iferanserin ointment, if it is approved.
DILTIAZEM CREAM (VEN 307)
Background on anal fissure
Incidence and prevalence
Anal fissure, which is a crack in the skin of the anal canal that results from reduced blood supply to the area and/or from increased sphincter tone, is a common anal disorder characterized by severe anal pain and bleeding with or after bowel movements. Because there are no approved pharmacological treatments for anal fissure, many cases progress to surgery because of the severe pain. There are no formal epidemiology studies for anal fissure, but its prevalence has been estimated indirectly. When 1,500 unselected neurological inpatients were screened in studies between 1990 and 1998 conducted in the U.S. by Dr. Wolfgang Jost, the prevalence of anal fissure was estimated at 1.6% in males and 2.2% in females. By extrapolation to the 2009 U.S. population of 227 million adults, we estimate that the general prevalence rate is 1.9%, with approximately 4.3 million current cases.
Physiology of anal fissure
Although hypertonia, or an increase in tightness of muscle tone, of the internal anal sphincter, or IAS, is associated with anal fissure, its contribution to the cause of anal fissure remains unclear. Hypertonia of the IAS does, however, contribute to chronic anal fissure. Anatomical, angiographic, and blood-flow studies have shown that the vascular supply of the anal epithelium, or tissue lining the anus, is very poor in the posterior midline, the anal area most commonly affected by fissures. Thus, it is possible that decreased anodermal blood supply to this area contributes to the pain and ischemia, or decrease in the blood supply, of traumatized anal epithelium, perpetuating ulceration and preventing healing. Whether the primary event for anal fissure is hypertonia of the IAS or decreased blood supply, hypertonia itself reduces vascular perfusion in the anal area. This reduction of vascular perfusion has been compared with that associated with ischemic pain in the lower limbs.
Current treatments
Presently, there are no FDA-approved drugs of which we are aware for the treatment of anal fissure in the U.S. The clinical goal in treating anal fissures is to reduce the pain associated with the fissure long enough for it to heal naturally and prevent the patient from having to resort to surgery. Currently, most physicians start treatment with diet modification, fiber, sitz baths and stool softeners. If these conservative treatments fail, physicians proceed to pharmacologic therapy, prescribing topical steroids or by directing special pharmacies to create compound topical formulations by mixing raw diltiazem, and in some cases nitroglycerin, into a cream for topical use by fissure patients. If these pharmacologic treatments fail to manage the pain, physicians consider, and often perform, surgery. In some instances, physicians initially prescribe pharmacologic therapy in addition to conservative treatments; in other instances because of the severe pain, they initially perform surgery.
13
The purpose of surgery is to reduce hypertonia of the IAS by either manual dilatation or lateral sphincterotomy. Both procedures are highly successful in relieving the pain and promoting healing of fissures. Although a relatively simple and effective surgical procedure, lateral sphincterotomy is also associated with short-term mild-to-moderate fecal incontinence. This is not an insignificant adverse effect and can become permanent or at least chronic in a fairly high percentage of patients. Studies have shown 6-8% of patients had incontinence to flatus or minor fecal soiling at a time greater than five years after surgery. In another study, at a mean follow-up time of 66.6 months (range 30-84 months), 10% of patients who had a lateral internal sphincterotomy were incontinent.
Over the last decades, a drug developer attempted to gain FDA approval for the topical treatment of anal fissures with nitroglycerin, an agent that reduces IAS and anal fissure pain. Early attempts to develop nitroglycerin utilizing a healing endpoint failed as it was discovered most fissures will heal naturally if the patient can endure the pain for the first several weeks of the disorder. However, it was discovered during development that lowering IAS hypertonia did have a significant benefit in reducing the pain associated with anal fissures. The subsequent pivotal studies with pain as a primary endpoint demonstrated a 33% reduction in pain scores in patients with baseline pain score >48 (1-100 mm on the visual analog scale, or VAS). However because the developer did not use minimum pain scores as an inclusion criteria, the overall effect was diluted to 22%. In addition, 64% of subjects reported headaches, which is a known systemic side effect of nitroglycerin. The FDA denied its approval, concluding that the risk benefit ratio for nitroglycerin as topical treatment for anal fissure pain was not favorable due to the modest overall effect and high incidence of systemic side effects. We have planned a clinical program that focuses on pain as the primary endpoint and includes only patients who have adequate pain scores on entry into the studies, which we believe will avoid the modest effects seen in the earlier study. In addition, based on results of previously published trials (such as Kocher et al. 2002; see Table 1 below), we believe that the side effects of diltiazem cream are likely to be less than those observed with topical nitroglycerin, which primarily were headaches.
DILTIAZEM CREAM (VEN 307) DEVELOPMENT
Background on diltiazem
Diltiazem was first approved in 1982 in oral form for the treatment of angina and high blood pressure. It has been prescribed in the U.S. for millions of patients in oral dosages typically from 240 mg to 360 mg per day. In contrast, daily doses of VEN 307 for treatment of anal fissures will range from 15 to 45 mg. Because of the extensive patient exposure to diltiazem as a cardiovascular agent and the wide safety margin as a low dose topical therapy, we intend to develop the topical formulation as a Section 505(b)(2) NDA, as agreed with the FDA at our pre- IND meeting in August 2007. A special NDA procedure, known as a ‘‘section 505(b)(2) application’’ or a ‘‘paper NDA,’’ allows an applicant to seek approval on the basis of a combination of a prior approval of a similar product or published literature, and some new clinical studies conducted or sponsored by the applicant. Section 505(b)(2) applications are often used for changes in a drug that require clinical investigations and thus cannot be handled through the generic drug process, such as a new indication or change in dosage.
Compounded diltiazem (prepared by the pharmacist, for each patient, using a general cream base and diltiazem from oral formulations) is currently listed in the U.S. and E.U. anal fissure treatment guidelines as a preferred agent prior to attempting surgery. According to advice we have received from members of our scientific advisory board, who are experts in gastroenterology and gastrointestinal surgery, compounded diltiazem is utilized by many colorectal and gastroenterology specialists each year for the treatment of anal fissures and, according to these experts, has also reduced the number of surgeries required. As a result, awareness and utilization of diltiazem as an effective treatment for anal fissures is high among physicians that treat this disorder. However, compounded diltiazem for anal fissure is not an FDA-approved use nor is it an FDA-approved product, and as such, the cost is not reimbursed by Medicare or health insurance plans. Data on unit and dollar volumes of compounded preparations are not routinely collected and not available to us. We expect to capture immediate market share if VEN 307 is approved due to its known efficacy and the current use of the compounded version. We expect that VEN 307 will be highly competitive with the compounded version because of the ease of prescription (already formulated, and approved by the FDA), with no need for custom mixing at the pharmacy, and because VEN 307 will be eligible for reimbursement under Medicare and other health plans, which the compounded version is not. For these reasons, we believe that the use of the compounded form of diltiazem will greatly decrease if VEN 307 is approved. The use of diltiazem for the treatment of anal fissures was first discovered at St. Mark gastroenterology teaching hospital in London. Professors Kamm and Robins filed the original method of use patent application in 1996. In 1997, diltiazem patent application and rights were assigned to S.L.A. Pharma, who filed the current patent application in 1998 (the original 1996 patent had lapsed). In 2001, North American rights were licensed to Solvay Pharmaceuticals, SA. During the time that Solvay held the rights, it improved the manufacturing processes and formulation and conducted important pharmacokinetic studies. In 2004, the new CEO of Solvay Pharmaceuticals refocused the R&D strategy on CNS and cardio-metabolic programs, discontinuing gastroenterology and women’s health projects. Consequently, in 2005, the license rights to diltiazem cream were returned to S.L.A. Pharma. From 2005 to the March 2007 licensing by Paramount BioSciences, S.L.A. Pharma focused on regulatory and manufacturing priorities, preparing diltiazem for further development.
14
In August 2007, we acquired North American rights to diltiazem from Paramount BioSciences, which previously acquired rights from S.L.A. Pharma in the United Kingdom for developing and marketing a proprietary diltiazem cream for relief of pain associated with anal fissures. We incurred a liability to Paramount BioSciences in the amount of $1,087,876, which represented the fees Paramount BioSciences had paid through August 2007 for both VEN 307 and VEN 308. Paramount BioSciences had acquired the S.L.A. rights in March 2007 and began working with Ventrus immediately to advance the development of these assets while an asset transfer agreement was finalized. S.L.A. Pharma is developing diltiazem cream for the European market and S.L.A. Pharma began a Phase III clinical trial in the E.U. in November 2010. We are financially supporting the E.U. trial and are obligated to make the following payments to S.L.A. Pharma for VEN 307 development milestones.
|
Amount Due
|
Date Due
|
Fee Description
|
||
|
$41,500
|
Monthly beginning October 1, 2010, and continuing until S.L.A. Pharma is no longer managing the development program for VEN 307.
|
Project management fees for VEN 307.
|
||
|
$600,000
|
December 31, 2010
|
Development costs for VEN 307.
|
||
|
$800,000
|
Upon the completion of enrollment into the Phase III clinical trial that S.L.A. Pharma is conducting in Europe, anticipated at the end of 2011.
|
Development costs for VEN 307.
|
||
|
Up to $400,000
|
If contingencies are met, payable monthly as invoiced by S.L.A. Pharma.
|
Development expenses for VEN 307. Contingent upon (i) receipt of a final study report from the S.L.A. Pharma Phase III VEN 307 trial in Europe (anticipated in the third quarter of 2012), and (ii) if we have raised net proceeds of at least $20.0 million from sales of securities and/or licensing of rights to our products by that time.
|
In August 2007, we concluded a pre-IND meeting with the FDA in anticipation of our IND submission for permission to initiate Phase III trials in the U.S. This meeting also afforded us an opportunity to gain agreement on the key design issues of the studies (including the one which S.L.A. Pharma is implementing) and additional information required for an approval of an NDA. We anticipate the availability of data from the S.L.A Phase III study in the second quarter of 2012 and, if the E.U. trial is successful, we plan to initiate the U.S. pivotal program by the second half of 2012, contingent on the availability of additional capital. We expect to collaborate closely with S.L.A. Pharma in order to leverage clinical data for different regulatory agencies and to rationalize manufacturing capacity.
Mechanism of action
The mechanism of action for topical diltiazem cream was demonstrated in human pharmacodynamic studies that showed an anal maximal resting pressure, or MRP, reduction of 28% that was sustained for 3−5 hours. This MRP reduction is believed to decrease the pain associated with anal fissures by normalizing internal anal sphincter pressure, which improves vascular blood supply and reduces ischemic pain.
15
Preclinical safety
Studies have been conducted in rabbits and guinea pigs to assess the topical safety of diltiazem cream. Clinicians treated rabbits in and around the anus with 2% diltiazem or placebo cream twice daily for 90 days to evaluate the chronic safety of the product. Although exterior anal tissue showed an increase in erythema, or redness of the skin, and edema, or accumulation of fluid beneath the skin, the clinicians concluded that these effects were due to the application procedure, to a possible reaction to latex gloves or to both. There were no histological findings. In this study, topical 2% diltiazem cream had no other adverse effects. Clinicians used guinea pigs to assess the potential for 2% diltiazem cream to elicit contact sensitization, or skin reaction to the application. This study did not demonstrate any sensitization potential of the diltiazem cream in guinea pigs.
Investigator-initiated clinical studies (studies sponsored by individual clinicians)
The investigator studies conducted with diltiazem cream applied topically in the perianal area in normal subjects and in patients with anal fissures are summarized in Table 1. These studies were conducted by independent investigators and not by us or any partner of ours. The year the study was published is given in the column headed ‘‘Study.’’
Table 1. Summary of Investigator-initiated clinical studies.
|
Study
|
Condition,
treatment, dosage
|
Study design,
endpoints
|
Efficacy
|
Adverse events
|
||||
|
Carapeti, E.A., et al, Gut, 45:719 – 722, 1999
|
10 normal subjects; placebo (PBO) or diltiazem (DTZ) gel (0.1%, 0.5%, 1%, 2%, 5%, and 10%)
|
DTZ or PBO gel applied once to anal margin; maximum resting anal pressure (MRP) and anodermal blood flow measured starting 1 hour after treatment
|
DTZ decreased MRP at concentrations of 1% and higher, maximum decrease of 28% at 2% gel, no further effect of 5% or 10%; effect at 2% lasted 3 – 5 hours; no change in blood flow
|
No local or systemic adverse events (AEs) reported
|
||||
|
Carapeti, E.A., et al, Dis Colon rectum, 43:1359 – 1362, 2000
|
15 patients with chronic anal fissures (CAF); 2% DTZ gel, three times-per-day (TID) for 8 weeks
|
DTZ gel applied to anal margin; MRP, anodermal blood flow and healing rate monitored every 2 weeks, daily diary cards for worst pain (scale of 0 – 10) of the day
|
Fissures healed in 67% of subjects; significant decrease in MRP and pain (decreased from 5.5 pretreatment to 1 post-treatment); no effect on blood flow
|
No AEs
|
||||
|
Bhardwaj, R., et al, Annual Meeting of British Association of Colon proctologists, Brighton, United Kingdom, 2000
|
44 patients with CAF, 2% DTZ gel, TID for 8 weeks
|
27 patients assessed at 2 months, 15 patients evaluated at 4 months (included 9 who had healed at 2 months and remained healed); assessed for healing, pain, rectal bleeding, MRP
|
Fissures healed in 56% of subjects at 2 months, 73% at 4 months; pain abolished in 88%, bleeding in 92%; MRP decreased by 24% at 2 months
|
1 patient had minor incontinence to flatus
|
||||
|
Jonas, M., et al, Dis Colon rectum, 44:1074 – 1078, 2001
|
50 patients with CAF, 24 treated with oral DTZ (60 mg), 26 with topical DTZ (2% gel), twice per day (BID) for 8 weeks
|
DTZ gel applied 1cm inside anus and to anal margin; pain, bleeding, perianal irritation (all 3 measured on a scale of 1 – 100 mm), MRP, healing monitored every 2 weeks
|
Fissures healed in 38% of subjects (oral) vs. 65% (topical) (9 in each group had previously failed on glyceryl trinitrate (GTN); 7 of these healed on topical vs. 1 on oral DTZ); both oral and topical DTZ decreased MRP; pain, bleeding and irritation reduced by both formulations (pain went from 70 to 7 after 8 weeks on oral, from 68 to 3 on topical)
|
No AEs in topical group; AEs reported in 8 patients on oral DTZ (headaches, nausea and/or vomiting, rash, decreased sense of taste and smell)
|
||||
|
Knight, J,S., et al, Br J Surg, 88:553 – 556, 2001
|
71 patients with CAF, 2% DTZ gel, BID, additional 8-12 weeks for subjects who did not heal on original regimen
|
DTZ applied perianally; healing monitored;
|
75% healed after 2-3 months, a total of 89% healed after a median duration of 9 weeks (range of 2-16 weeks); after a median of 32 weeks follow-up (range 14 – 67 weeks) 66% symptom-free, 17% had mild symptoms, and 7% had reoccurrence
|
4 patients reported perianal dermatitis, 1 reported headache
|
16
|
Study
|
Condition,
treatment, dosage
|
Study design,
endpoints
|
Efficacy
|
Adverse events
|
||||
|
Griffin, N., et al, Colorectal Dir, 4:430 – 435, 2002
|
47 patients with CAF who failed topical GTN, 2% DTZ cream, BID for 8 weeks
|
Treatment administered in anal verge; daily diary for pain, bleeding and itching (scale of 0 – 100); healing monitored
|
Fissures healed in 48% of subjects; pain and bleeding decreased after 8 weeks, no effect on itching; 2 patients relapsed after median duration of follow-up 45 weeks (range 23 – 54)
|
1 patient developed a local perianal rash; up to 25% reported increased perianal itch
|
||||
|
DasGupta, R., et al, Colorectal Dir, 4:20 – 22, 2002
|
23 patients with CAF, 2% DTZ gel, TID for up to 12 weeks
|
DTZ applied to lower half of anal canal, healing monitored
|
Fissures healed in 48% of subjects, in a median of 8 weeks (range 1 – 12 weeks); of 8 who had previously failed GTN, 6 (75%) healed; no recurrences at 3 months
|
No AEs
|
||||
|
Kocher, H.M., et al, Br J Surg, 89:413 – 417, 2002
|
60 patients with CAF, 0.2% GTN ointment (29 patients) or 2% DTZ cream (31 patients), BID for 6 – 8 weeks
|
DTZ or GTN applied to anal verge, monitored every 3 weeks for healing; pain recorded on VAS (0 – 100) scale
|
At 8 weeks fissures healed or improved in 12 and 13 patients, respectively, after GTN (86%) vs. 8 (healed) and 16 (improved) after DTZ (77%); both decreased pain to approximately same extent; at 12 weeks 2 GTN patients had recurred vs. none in the DTZ group
|
21/29 GTN subjects (72%) reported AEs vs. 13/31 (42%) in DTZ group; 17 /29 in GTN group had headaches, vs. 8/31 of DTZ patients
|
||||
|
Bielecki, K., et al, Colorectal Dir, 5:256 – 257, 2003
|
43 patients with CAF, 0.5% GTN ointment (21 patients) or 2% DTZ ointment (22 patients), BID for 8 weeks
|
Patients monitored 3 times during treatment
|
Fissures healed in 86% of GTN, 86% of subjects with DTZ, 3 failures in each group
|
Mainly headache in 7 GTN patients (33%), no AEs reported in DTZ patients
|
||||
|
Shrivastava, U.K., et al, Surg Today, 37:482 – 485, 2007
|
90 patients with CAF; 2% DTZ ointment (30 patients), 0.2% GTN ointment (30 patients), BID; no treatment (30 patients)
|
Treatments applied BID to anus, patients monitored for healing and pain (VAS) twice 2 per week then every 2 weeks
|
Fissures healed in 80%, 73% and 33% for DTZ, GTN and control subjects, respectively; mean time for healing 6.6 weeks, 7.0 weeks and 7.6 weeks for DTZ, GTN and controls, respectively; pain decreased by 75% for DTZ, 59% for GTN and 29% for controls at 6 weeks; recurrence rate 12.5%, 32% and 50% for DTZ, GTN and controls, respectively
|
No AEs in DTZ patients, 67% of GTN patients had headaches
|
DTZ = diltiazem; GTN = glyceryltrinitrate (nitroglycerin)
Clinical trials of diltiazem cream sponsored by S.L.A. Pharma
In 2004 and 2005, S.L.A. Pharma assessed the pharmacokinetic profile of topical diltiazem cream over a four-day period in subjects with anal fissure. Clinical dosing was completed in November 2005 and published in January 2007. Clinicians treated patients with eight doses of either 2%, 4%, or 8% diltiazem cream. Clinicians administered a single dose perianally on day 1, followed by doses three times a day on days 2 and 3, followed by another single dose on day 4. The clinicians collected blood over 24 hours on days 1 and 4. Maximum blood levels and area under the curve increased with the dose, and there appeared to be accumulation of diltiazem in blood on day 4 after multiple dosing. The time to maximum blood levels was five to seven hours, and the plasma half-life was less than 12 hours. However, the maximum amount of diltiazem that was absorbed was much less (at least five-fold less) than observed after oral dosing. Side effects, such as anal irritation, headache, and nausea, were mild.
Blood pressure was measured at the following times after the single dose on days 1 and 4: predose, 15, 30 and 45 minutes and 1, 1.5, 2, 4 and 8 hours after dosing. The relatively small maximum mean decreases (mmHg) in blood pressure in patients receiving 2%, 4% and 8% cream (3-4 patients per group) by day 4 ranged from 4 to 8mmHg systolic blood pressure, or SBP, and 4 to 6 mmHg diastolic blood pressure, or DBP. The changes were, in general, transient and asymptomatic and blood pressure had returned to at or near baseline by the next reading. There was no clear dose-related effect among the 2%, 4% and 8% creams with respect to decreases in blood pressure. In clinical trials with oral diltiazem for hypertension, the patients receiving placebo had mean decreases of blood pressure from 2 to 4 mmHg.
17
S.L.A. Pharma compared the effect of 2% diltiazem cream with 0.2% glyceryltrinitrate cream in subjects with chronic anal fissure. This study was completed in January 2001 and published in October 2001. Clinicians applied the preparations in and around the anus twice daily for six weeks. Nine of the 31 patients treated with diltiazem and three of the 29 patients treated with glyceryltrinitrate withdrew from the study by eight weeks. In the diltiazem group, 26% of the patients experienced healed fissures and 52% of patients experienced improved fissures. In the glyceryltrinitrate group, 41% of patients experienced healed fissures and 45% of patients experienced improved fissures. There was no significant difference in the healing rates between the groups. Both treatments resulted in a significant decrease in pain. Four weeks after the end of treatment, no fissures recurred in patients treated with diltiazem, but fissures recurred in two patients treated with glyceryltrinitrate. Compared with 18 treatment-emergent adverse events reported by 13 patients (42%) receiving diltiazem, there were 33 adverse events reported by 21 patients (72%) receiving glyceryltrinitrate. Eight patients receiving diltiazem complained of nine headaches, 17 patients receiving glyceryltrinitrate complained of 20 headaches.
Similar to the early glyceryltrinitrate, or GTN, development program that found healing to be a difficult and inappropriate endpoint for registration trials, S.L.A. Pharma also pursued a healing endpoint strategy in early development. In an exploratory trial sponsored by S.L.A. Pharma that was completed in February 2002 and published in February 2003, the effects of 2% diltiazem cream on healing rates were compared with placebo cream in patients with severe chronic anal fissure. Thirty-one patients were randomized to each treatment group. Creams were applied twice daily for eight weeks. At the end of eight weeks, there was no difference in the healing rates between patients receiving diltiazem (10%) and patients receiving placebo (19%). No difference was observed in the secondary endpoints, which is likely due to the assessment being made only at the end of the study, not daily as in the other trials, which showed a positive outcome in these endpoints. Fifteen patients receiving diltiazem reported 28 adverse events and 12 patients receiving placebo received 18 adverse events. Seven patients receiving diltiazem and three patients receiving placebo reported a rash or pruritus, or itchiness. Headaches were reported in the same number of patients in both treatment groups.
Summary of studies to date
The topical application of diltiazem cream provides pain relief associated with anal fissure and has also been found to be associated with healing. The effects of diltiazem cream are comparable to those observed for treatment of anal fissure with topical application of GTN, but diltiazem cream is much better tolerated. Based on currently available data and discussion with the FDA, we think it is clear that relief of pain associated with anal fissures is the preferred clinical endpoint. Our belief is supported by the study by U.K. Shrivastava, et al., published in Surgery Today, 37:482 − 485, 2007 (see Table 1 above), which compared GTN and diltiazem perianally compared with standard care alone. In this trial, pain decreased by 75% for diltiazem compared with 29% for controls at six weeks. In almost all studies with either GTN or diltiazem where pain was measured, results are consistent whereas with healing as an endpoint results are variable.
Our belief that relief of pain associated with anal fissures is the preferred clinical endpoint is further supported by market research that identified clinicians’ primary treatment goal as pain relief. Importantly, the diltiazem mechanism of action for pain relief is to reduce IAS pressure which addresses the underlying cause of anal fissure pain.
18
Diltiazem cream (VEN 307) development plan
In August 2007, we had a pre-IND meeting with the FDA concerning VEN 307 (diltiazem cream for the treatment of pain from anal fissures) where it was established that next clinical studies needed for approval were two pivotal Phase III trials, preceded (if conducted in the U.S.) by three short-term dermal toxicology studies. Prior to conducting clinical Phase III studies in the U.S., we must complete three short-term dermal toxicology studies and file an IND for FDA approval. We plan to employ a two-pronged development strategy for VEN 307. While S.L.A. Pharma is conducting the first Phase III clinical trial in the E.U. which is anticipated to be complete in the second quarter of 2012, we intend to initiate development of a superior formulation with new intellectual property in the form of an extended release formulation. There are several proven methodologies for extended release topical formulations, and we believe that diltiazem is readily drugable in this regard. We intend to assess three to four alternatives preclinically with multiple contractors, and then assess absorption and effect on IAS pressure with the most promising, while we will file PCP applications for the specific technology combined with diltiazem for all formulations that are technically feasible.
The patent for the existing formulation that S.L.A. Pharma is using has not yet been issued for reasons of lack of novelty and inventiveness (prior art) and S.L.A. Pharma filed an appeal to the USPTO Board of Appeal. The Appeal of US Patent Application No. 09/355,928 was addressed on August 31, 2010 wherein the Appeal Board affirmed the novelty of the topical use of diltiazem while maintaining the lack of inventive step rejection over the prior art, with the proviso that additional data were necessary to show unexpected results. As such, on October 25, 2010, S.L.A. Pharma filed a Request for Continued Examination, or RCE, to continue prosecution of the pending application and to provide the additional data requested by the Appeal Board. S.L.A Pharma also introduced additional arguments to the PTO Examiner to address recent legal decisions by the Federal Circuit Courts, relevant to the inventive step for the topical use of diltiazem, which have not been previously considered by the PTO examiner. We expect the first PTO action to be made within 12 months.
If there is successful completion of the E.U. trial, we will make the final decision on which formulation to pursue depending on several factors, including whether the new formulation is clinically superior, our access to capital, clinical and regulatory considerations, and our assessment of the then-current state of our intellectual property estate. If the new U.S. developed formulation is superior and the other factors are met, we plan to initiate two pivotal trials in parallel in order to complete the NDA for an estimated FDA submission in 2013. If the new formulation is not superior, and/or we judge the existing formulation to be patentable, we plan to finish clinical development utilizing the current formulation which would require one additional pivotal Phase III study in the U.S., and expect to continue to pursue other lifecycle options such as combination with other drugs. Both development pathways could result in a NDA submission in 2013.
Commercial summary for diltiazem cream (VEN 307)
Market research regarding anal fissure
Eidetics, a Boston-based research company, conducted quantitative market research in 2003 and reported that on average primary-care physicians see 23 anal fissure patients per month, gastroenterologists see 17 anal fissure patients per month, and colon and rectal surgeons see 31 anal fissure patients per month. Physicians in all three medical specialties indicated that there is a significant unmet need for therapeutic choices for anal fissure. Only 8% of primary-care physicians, 5% of gastroenterologists, and 27% of colon and rectal surgeons reported being ‘‘very satisfied’’ with current treatment options. All three medical specialties reported failure rates exceeding 50% for current first-line therapy in patients with anal fissure. Given this unmet medical need and the absence of other approved drugs in the U.S., we believe that up to 2.0 million patients per year could benefit from treatment with VEN 307.
PHENYLEPHRINE GEL (VEN 308)
Background on fecal incontinence
Incidence and prevalence
According to a U.S. community based epidemiology study (Nelson et al., JAMA, 1995), 2.2% of U.S. adults suffer from fecal incontinence, which we estimate to be approximately 7 million people, based on 2009 Census Bureau adult population estimates.
19
The IPAA orphan population
Patients with an ileal pouch anal anastomosis, or IPAA, secondary to a total colectomy, tend to have a high incidence of fecal incontinence, up to 30%, according to a 1987 study conducted by Dr. John Pemberton and others at the Mayo Medical School. The surgery associated with IPAA can weaken sphincters and muscles necessary for continence and therefore can result in incontinence. About 30% of patients with ulcerative colitis, a form of inflammatory bowel disease which has a prevalence of 700,000 patients in the U.S. (according to Datamonitor 2008) will have had a colectomy, almost always an IPAA procedure (according to McGlauchlin and Clark, Practical Gastroenterology, 8/2008). IPAA-related fecal incontinence is considered an orphan indication by the FDA and the European Medicines Agency, or EMEA. Patients who undergo ileal pouch anal anastomosis are prone to fecal incontinence. In 2006, the total population of patients with IPAA-related fecal incontinence in the U.S. was estimated to be 50,000 to 100,000, according to IMS Health, Inc.
Physiology of fecal incontinence
Continence is a complex physiological action that requires the presence of a series of anatomical barriers preventing the movement of feces through the anus. The puborectalis muscle works with the internal and external anal sphincters to control continence. If any of these three barriers are dysfunctional, incontinence can occur in a wide range of severity. Specifically, anal sphincter weakness has long been associated with fecal incontinence. Abnormal fibrosis, reduced elasticity, insensitivity to norepinephrine and spontaneous relaxation are associated with anal sphincter weakness.
Current treatments
To our knowledge, there are no FDA approved drugs for the treatment of fecal incontinence. Most physicians start with conservative therapy, which consists of diet modification, sitz baths and over-the-counter antidiarrheal medication. In addition to conservative therapy, physicians might prescribe antidiarrheal medication or recommend surgery.
The most common surgical procedure is sphincteroplasty for patients with physical injury to the anal sphincter. Success rates for this type of surgery are low and most of the benefit decreases with time. Solesta™, which is being developed by Oceana Therapeutics, is an injectable inert bulking agent product approved in the European Union for the treatment of fecal incontinence in adult patients who have failed conservative therapy. Solesta is injected submucosally around the anal sphincter and consequently has to be administered in an outpatient setting by qualified physicians. Oceana Therapeutics is currently pursuing approval of Solesta by the FDA and in December 2010 an FDA Advisory Committee recommended Solesta for approval. In addition, Norgine plans to conduct a Phase I trial with NRL001, a suppository formulation of an alpha adrenergic stimulating agent for the treatment of fecal incontinence, which is anticipated to start in Europe in early 2011.
Background on phenylephrine
Phenylephrine has been available since the early 1940s in oral and nasal form for the treatment of nasal congestion. It has also been used as a topical ophthalmic agent since 1936. Phenylephrine is prescribed more than 17 million times per year in the United States, with 99% of the prescriptions being for cough/cold oral preparations. The typical oral dosing is 40 mg to 60 mg per day. Because of the extensive patient exposure to phenylephrine, we intend to develop the topical formulation as a Section 505(b)(2) NDA. The use of phenylephrine for the treatment of fecal incontinence was first discovered at St. Mark gastroenterology teaching hospital in London. Professors Kamm and Robins filed the original method of use patents in 1996. In 1997, phenylephrine patent application and rights were assigned to S.L.A. Pharma. In 2001, S.L.A. Pharma licensed North American rights to Solvay Pharmaceuticals, SA. During the time that Solvay held the rights, it improved the manufacturing processes and formulation and conducted important pharmacokinetic studies. In 2004, the new CEO of Solvay Pharmaceuticals refocused its R&D strategy on CNS and cardio-metabolic programs, discontinuing gastroenterology and women’s health projects. Consequently, in 2005, the licensed rights to phenylephrine gel were returned to S.L.A. Pharma. From 2005 to the March 2007 licensing by Paramount BioSciences, S.L.A. Pharma focused on regulatory and manufacturing priorities, preparing the asset for further development.
20
In August 2007, we acquired North American rights to phenylephrine gel from Paramount BioSciences, which previously acquired rights from S.L.A. Pharma in the United Kingdom in March 2001 for developing and marketing a proprietary phenylephrine gel for the treatment of fecal incontinence. We incurred a liability to Paramount BioSciences of $1,087,876, which represented the fees Paramount BioSciences had paid through August 2007 for both VEN 307 and VEN 308. Paramount BioSciences had acquired the S.L.A. rights in March 2007 and began working with Ventrus immediately to advance the development of these assets while an asset transfer agreement was finalized.
We expect to collaborate closely with S.L.A. Pharma to leverage clinical data for different regulatory agencies and to rationalize manufacturing capacity.
Our total payment obligation for VEN 308 will not exceed $1,200,000. S.L.A. Pharma has billed us for, and we have paid, $973,500 of services through December 31, 2010. This leaves $226,500 in possible additional payments. However, we currently have no further payment obligations for VEN 308 unless we agree with S.L.A. Pharma to additional services outside the scope of the agreement.
Mechanism of action (MOA)
The MOA for topical phenylephrine gel is to increase resting anal sphincter pressure, thus increasing patient bowel control. Phenylephrine gel’s MOA makes it an attractive candidate for any patient population that suffers from incontinence characterized as leaking/seeping fecal incontinence.
Preclinical safety
A mouse lymph node assay conducted by S.L.A. Pharma did not show phenylephrine hydrochloride to be a sensitizer (meaning a chemical that induces an allergic reaction after repeated exposure) because the drug was not associated with any type of delayed hypersensitization. In another S.L.A. Pharma study, contact sensitization potential, as measured in guinea pigs, under the conditions of the study, a 20% gel was considered to be a strong sensitizer to guinea pig skin. A 28-day study by S.L.A. Pharma in rabbits, in which 10% and 20% phenylephrine gel (900 mg) was applied three times each day to the dorsum, demonstrated mild inflammation which may have been exacerbated by animals biting the site of application. These studies were primarily conducted at St. Mark’s Hospital in the U.K. in the 1990s.
Investigator-initiated clinical studies
A number of investigator studies have been conducted with phenylephrine applied topically for the treatment of fecal incontinence and are summarized in Table 2. These studies were conducted by independent investigators and not by us or any partner of ours. The year the study was published is given under the column headed ‘‘Study.’’ One of these studies was conducted in patients with IPAA-related fecal incontinence. In one specific randomized controlled trial, phenylephrine significantly reduced the incontinence score (P = 0.015) and improved subjective measures (P = 0.04) compared with placebo. For some patients in this study, phenylephrine totally eliminated nocturnal episodes of fecal incontinence. No patient discontinued treatment during the study due to side effects. Studies in patients whose incontinence was more related to factors other than anal sphincter tone (many patients in the passive fecal incontinence studies) showed less response. As a result, our development plan will initially focus on the orphan IPAA indication.
21
Table 2. Investigator-initiated studies of topical phenylephrine gel for treatment of fecal incontinence.
|
Study
|
Condition, treatment, dosage
|
Summary of results
|
||
|
Carapeti, E.A., et al, Br J Surg. 86:267 – 270, 1999
|
Normal subjects, phenylephrine gel (5%, 10%, 20%, 30%) applied once to anal verge
|
Resting anal pressure increased by 8% to 33%, effect lasted for median of 7 hours, no change in pulse
|
||
|
Carapeti, E.A., et al, Dis Colon rectum, 43:1059 – 1063, 2000
|
IPAA-related FI, 10% phenylephrine or placebo gel, 2 times/day for 4 weeks
|
50% (6/12) of phenylephrine subjects improved vs 8% (1/12) placebo, 33% had cessation of FI on phenylephrine, 0% on placebo, phenylephrine increased anal pressure. No reported side effects.
|
||
|
Carapeti, E.A., et al, Br J Surg, 87:35 – 42, 2000
|
Passive FI, 10% phenylephrine vs placebo cream, 2 times/day for 4 weeks
|
No effect of phenylephrine or placebo on incontinence or anal pressure, 17% of phenylephrine and 6% of placebo patients had > 75% improvement
|
||
|
Cheetham, M.J., et al, 2000
|
Passive FI, 20% phenylephrine or placebo gel, 2 times/day for 4 weeks
|
No effect of phenylephrine or placebo on incontinence, anal pressure, blood pressure, or pulse rate
|
||
|
Sasse, K.L., et al, Dis Colon rectum, 43:A2, 2000
|
FI, 10% phenylephrine cream, 24 weeks
|
Increased anal pressure, improved incontinence
|
||
|
Cheetham, M.J., et al, Gut, 43:356 – 359, 2001
|
Passive FI, placebo or phenylephrine gel (10%, 20%, 30%, or 40%) as single application
|
Anal pressure increased in dose-related manner after phenylephrine, no effect on pulse, transient perianal burning
|
||
|
Mutch, M.G., et al, 2002
|
Passive FI, 10% phenylephrine cream, 3 times/day for 30 days
|
Phenylephrine improved incontinence score, anal pressure, and anal sphincter length
|
FI = fecal incontinence; IPAA = ileal pouch anal anastomosis
Clinical trials
Solvay Pharmaceuticals assessed the safety and pharmacokinetic profile of intra-anal and perianal application of phenylephrine gel in healthy volunteers in 2004 in a study completed in March 2004 and published in May 2004. The phenylephrine gel was applied as a single dose either intra-anally at doses of 5, 10, 25, 50, or 100 mg, or perianally at doses of 100, 200, or 400 mg. Blood samples were collected out to 24 hours after dosing.
Perianal application of phenylephrine gel resulted in much less absorption than intra-anal application: at a perianal dose of 400 mg, blood levels were comparable to what was seen after intra-anal treatment with 10 mg to 25 mg.
Intra-anal application of phenylephrine was associated with increased blood pressure that lasted for approximately three hours, whereas these effects were not seen with perianal treatment. The most frequent side effects were headache and goosebumps after intra-anal application of phenylephrine gel which were not seen with perianal application, and anal/rectal pain after perianal application of phenylephrine gel.
Summary of studies to date
Topical pheny lephrine gel has demonstrated efficacy for the treatment of fecal incontinence associated with IPAA. Pharmacokinetic studies have shown a superiority of perianal dosing which yielded low systemic absorption while still providing the desired local therapeutic effect. No hemodynamic effects where observed when phenylephrine gel was administered perianally at up to 8 times the therapeutic dose. Therefore, further development of the drug will focus solely on perianal application.
Phenylephrine gel (VEN 308) development plan
Based on pre-IND meetings with the FDA in 2007, we are planning to initiate the U.S. Phase IIb dose ranging study in support of the orphan indication of IPAA-related fecal incontinence. Assuming we raise sufficient capital in the future, we plan to start this study in 2012 to finalize the dose and clinical endpoints. We expect to conclude VEN 308 development and submit the orphan NDA in 2015. Orphan status provides seven years exclusivity from the date of approval during which time we will pursue several potential lifecycle opportunities.
22
Commercial summary for phenylephrine gel (VEN 308)
Market research regarding fecal incontinence
Quantitative market research conducted in 2003 by Eidetics reported that, on average, primary-care physicians see 23 fecal incontinence patients per month, gastroenterologists see 20 fecal incontinence patients per month, and colon and rectal surgeons see 14 fecal incontinence patients per month. Physicians categorize fecal incontinence according to its underlying cause. This market research was not designed to eliminate double counting of referred patients and has not been used in calculating commercial potential. However, these data do indicate the volume of patients seen at each type of practice irrespective of whether the same patient has been seen by another physician, and any one of these physicians can initiate a prescription for the product.
Physicians in all three medical specialties indicated that there is a significant unmet need for therapeutic choices for fecal incontinence. Only 4% of primary-care physicians, 3% of gastroenterologists, and 7% of colon and rectal surgeons reported being ‘‘very satisfied’’ with current treatment options.
Government Regulation
General
The production, distribution, and marketing of products employing our technology, and its development activities, are subject to extensive governmental regulation in the U.S. and in other countries. In the U.S., our products are regulated as drugs and are subject to the Federal Food, Drug, and Cosmetic Act, as amended, and the regulations of the FDA, as well as to other federal, state, and local statutes and regulations. These laws, and similar laws outside the U.S., govern the clinical and preclinical testing, manufacture, safety, effectiveness, approval, labeling, distribution, sale, import, export, storage, recordkeeping, reporting, advertising, and promotion of our products. Product development and approval within this regulatory framework, if successful, will take many years and involve the expenditure of substantial resources. Violations of regulatory requirements at any stage may result in various adverse consequences, including the FDA’s and other health authorities’ delay in approving or refusal to approve a product. Violations of regulatory requirements also may result in enforcement actions.
The following provides further information on legal and regulatory matters that have the potential to affect our operations or future marketing of products.
Research, Development and Product Approval Process
The research, development, and approval process in the U.S. and elsewhere is intensive and rigorous and generally takes many years to complete. The typical process the FDA requires before a therapeutic drug may be marketed in the U.S. includes:
• preclinical laboratory and animal tests performed under the FDA’s Good Laboratory Practices regulations, or GLPs;
• submission to the FDA of an application for an IND, which must become effective before human clinical trials may commence;
• preliminary human clinical studies to evaluate the drug and its manner of use;
• adequate and well-controlled human clinical trials to establish whether the drug is safe and effective for its intended uses;
• FDA review of whether the facility in which the drug is manufactured, processed, packed, or held meets standards designed to assure the product’s continued quality; and
23
• submission of a marketing application to the FDA, and review and approval of the application by the FDA.
During preclinical testing, studies are performed with respect to the chemical and physical properties of candidate formulations. These studies are subject to GLP requirements. Biological testing is typically done in animal models to demonstrate the activity of the compound against the targeted disease or condition and to assess the apparent effects of the new product candidate on various organ systems, as well as its relative therapeutic effectiveness and safety. An IND must be submitted to the FDA and become effective before studies in humans may commence.
Clinical trial programs in humans generally follow a three-phase process. Typically, Phase I studies are conducted in small numbers of healthy volunteers or, on occasion, in patients afflicted with the target disease. Phase I studies are conducted to determine the metabolic and pharmacological action of the product candidate in humans and the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. In Phase II, studies are generally conducted in larger groups of patients having the target disease or condition in order to validate clinical endpoints, and to obtain preliminary data on the effectiveness of the product candidate and optimal dosing. This phase also helps determine further the safety profile of the product candidate. In Phase III, large-scale clinical trials are generally conducted in patients having the target disease or condition to provide sufficient data for the statistical proof of effectiveness and safety of the product candidate as required by U.S. and foreign regulatory agencies.
Before proceeding with a study, sponsors may seek a written agreement from the FDA regarding the design, size, and conduct of a clinical trial. This is known as a Special Protocol Assessment, or SPA. Among other things, SPAs can cover clinical studies for pivotal trials whose data will form the primary basis to establish a product’s efficacy. An SPA helps establish up-front agreement with the FDA about the adequacy of a clinical trial design to support a regulatory approval, but the agreement is not binding if new circumstances arise. There is no guarantee that a study will ultimately be adequate to support an approval even if the study is subject to an SPA.
United States law requires that studies conducted to support approval for product marketing be ‘‘adequate and well controlled.’’ In general, this means that either a placebo or a product already approved for the treatment of the disease or condition under study must be used as a reference control. Studies must also be conducted in compliance with good clinical practice requirements, and informed consent must be obtained from all study subjects.
The clinical trial process for a new compound can take 10 years or more to complete. The FDA may prevent clinical trials from beginning or may place clinical trials on hold at any point in this process if, among other reasons, it concludes that study subjects are being exposed to an unacceptable health risk. Trials may also be prevented from the beginning or may be terminated by institutional review boards, which must review and approve all research involving human subjects. Side effects or adverse events that are reported during clinical trials can delay, impede, or prevent marketing authorization. Similarly, adverse events that are reported after marketing authorization can result in additional limitations being placed on a product’s use and, potentially, withdrawal of the product from the market.
Following the completion of clinical trials, the data are analyzed to determine whether the trials successfully demonstrated safety and effectiveness and whether a product approval application may be submitted. In the U.S., if the product is regulated as a drug, a new drug application, or NDA, must be submitted and approved before commercial marketing may begin. The NDA must include a substantial amount of data and other information concerning the safety and effectiveness of the compound from laboratory, animal, and human clinical testing, as well as data and information on manufacturing, product quality and stability, and proposed product labeling. A special NDA procedure, known as a ‘‘section 505(b)(2) application’’ or a ‘‘paper NDA,’’ allows an applicant to seek approval on the basis of a combination of a prior approval of a similar product or published literature, and some new clinical studies conducted or sponsored by the applicant. Section 505(b)(2) applications are often used for changes in a drug that require clinical investigations and thus cannot be handled through the generic drug process, such as a new indication or change in dosage form.
24
Each domestic and foreign manufacturing establishment, including any contract manufacturers we may decide to use, must be listed in the NDA and must be registered with the FDA. The application generally will not be approved until the FDA conducts a manufacturing inspection, approves the applicable manufacturing process for the drug product, and determines that the facility is in compliance with cGMP requirements. Each NDA submitted for FDA approval is usually reviewed for administrative completeness and reviewability within 45 to 60 days following submission of the application. If deemed complete, the FDA will ‘‘file’’ the NDA, thereby triggering substantive review of the application. The FDA can refuse to file any NDA that it deems incomplete or not properly reviewable. The FDA has established performance goals for the review of NDAs — 6 months for priority applications and 10 months for standard applications. However, the FDA is not legally required to complete its review within these periods, and these performance goals may change over time. Moreover, the outcome of the review, even if generally favorable, typically is not an actual approval but an ‘‘action letter’’ that describes additional work that must be done before the application can be approved. The FDA’s review of an application may involve review and recommendations by an independent FDA advisory committee. Even if the FDA approves a product, it may limit the approved therapeutic uses for the product as described in the product labeling, require that warning statements be included in the product labeling, require that additional studies be conducted following approval as a condition of the approval, impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval.
Significant legal and regulatory requirements also apply after FDA approval to market under an NDA. These include, among other things, requirements related to adverse event and other reporting, product advertising and promotion and ongoing adherence to cGMPs, as well as the need to submit appropriate new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. The FDA also enforces the requirements of the Prescription Drug Marketing Act which, among other things, imposes various requirements in connection with the distribution of product samples to physicians.
The regulatory framework applicable to the production, distribution, marketing, and/or sale, of our products may change significantly from the current descriptions provided herein in the time that it may take for any of our products to reach a point at which an NDA is approved.
Overall research, development, and approval times depend on a number of factors, including the period of review at FDA, the number of questions posed by the FDA during review, how long it takes to respond to the FDA’s questions, the severity or life-threatening nature of the disease in question, the availability of alternative treatments, the availability of clinical investigators and eligible patients, the rate of enrollment of patients in clinical trials, and the risks and benefits demonstrated in the clinical trials.
Orphan Drugs
Under the Orphan Drug Act, special incentives exist for companies to develop products for rare diseases or conditions, which are defined to include those diseases or conditions that affect fewer than 200,000 people in the U.S. Companies may request that the FDA grant a drug orphan designation prior to approval. Products designated as orphan drugs are eligible for special grant funding for research and development, FDA assistance with the review of clinical trial protocols, potential tax credits for research, reduced filing fees for marketing applications, and a special seven-year period of market exclusivity after marketing approval. Orphan drug exclusivity prevents FDA approval of applications by others for the same drug and the designated orphan disease or condition. The FDA may approve a subsequent application from another entity if the FDA determines that the application is for a different drug or different use, or if the FDA determines that the subsequent product is clinically superior, or that the holder of the initial orphan drug approval cannot assure the availability of sufficient quantities of the drug to meet the public’s need. A grant of an orphan designation is not a guarantee that a product will be approved. If a sponsor receives orphan drug exclusivity upon approval, there can be no assurance that the exclusivity will prevent another entity or a similar drug from receiving approval for the same or other uses.
25
Other United States Regulatory Requirements
In the U.S., the research, manufacturing, distribution, sale, and promotion of drug and biological products are potentially subject to regulation by various federal, state, and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services, other divisions of the U.S. Department of Health and Human Services (for example, the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing, and scientific/ educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provision of the Health Insurance Portability and Accountability Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection, unfair competition, and other laws. Moreover, we are now, and in the future may become subject to, additional federal, state, and local laws, regulations, and policies relating to safe working conditions, laboratory practices, the experimental use of animals, and/or the use, storage, handling, transportation, and disposal of human tissue, waste, and hazardous substances, including radioactive and toxic materials and infectious disease agents used in conjunction with our research work.
Foreign Regulatory Requirements
We and our collaborative partners may be subject to widely varying foreign regulations, which may be quite different from those of the FDA, governing clinical trials, manufacture, product registration and approval, and pharmaceutical sales. Whether or not FDA approval has been obtained, we or our collaboration partners must obtain a separate approval for a product by the comparable regulatory authorities of foreign countries prior to the commencement of product marketing in these countries. In certain countries, regulatory authorities also establish pricing and reimbursement criteria. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. In addition, under current U.S. law, there are restrictions on the export of products not approved by the FDA, depending on the country involved and the status of the product in that country.
Reimbursement and Pricing Controls
In many of the markets where we or our collaborative partners might commercialize a product following regulatory approval, the prices of pharmaceutical products are subject, by law, to direct price controls and to drug reimbursement programs with varying price control mechanisms. Public and private health care payors control costs and influence drug pricing through a variety of mechanisms, including through negotiating discounts with the manufacturers and through the use of tiered formularies and other mechanisms that provide preferential access to certain drugs over others within a therapeutic class. Payors also set other criteria to govern the uses of a drug that will be deemed medically appropriate and therefore reimbursed or otherwise covered. In particular, many public and private health care payors limit reimbursement and coverage to the uses of a drug that are either approved by the FDA or that are supported by other appropriate evidence (for example, published medical literature) and appear in a recognized drug compendium. Drug compendia are publications that summarize the available medical evidence for particular drug products and identify which uses of a drug are supported or not supported by the available evidence and whether or not such uses have been approved by the FDA. For example, in the case of Medicare coverage for physician-administered oncology drugs, the Omnibus Budget Reconciliation Act of 1993, with certain exceptions, prohibits Medicare carriers from refusing to cover unapproved uses of an FDA-approved drug if the unapproved use is supported by one or more citations in the American Hospital Formulary Service Drug Information, the American Medical Association Drug Evaluations, or the U.S. Pharmacopoeia Drug Information. Another commonly cited compendium, for example under Medicaid, is the DRUGDEX Information System.
26
License Agreements & Intellectual Property
General
Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets and operate without infringing on the proprietary rights of other parties, both in the U.S. and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the U.S. and abroad. However, patent protection may not afford us with complete protection against competitors who seek to circumvent our patents. We also depend upon the skills, knowledge, experience and know-how of our management and research and development personnel, as well as that of our advisors, consultants and other contractors. To help protect our proprietary know-how, which is not patentable, and for inventions for which patents may be difficult to enforce, we currently rely and will in the future rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
We do not own and did not develop any of our product candidates. We have licensed our three product candidates from third parties. All clinical trials to date have been conducted either by the licensor, the licensor’s previous partners or by independent investigators, as have the preclinical studies and product formulation activities. Since we licensed these products, we have focused our efforts on establishing and clarifying the regulatory pathway for late phase clinical trials and regulatory approval, and on establishing the contract manufacturing capacity and methods necessary to allow late phase clinical trials to proceed, all of which will be conducted by contracted third parties under our direction. We are dependent on the availability and competence of these third parties for the continued development of our product candidates.
License Agreements
In March 2007, pursuant to an Exclusive License Agreement, S.L.A. Pharma granted Paramount BioSciences an exclusive, royalty-bearing license to sell, make and use diltiazem for treatment, through topical administration, of anal fissures and phenylepherine for treatment, through topical administration, of fecal incontinence in the U.S., Canada and Mexico. Pursuant to the Exclusive License Agreement, Paramount BioSciences was obligated to form a company to develop the technologies referenced in the Exclusive License Agreement and issue to S.L.A. Pharma that number of shares equal to 5% of such company’s outstanding common stock as of the effective date of the Exclusive License Agreement. To satisfy this obligation, Paramount BioSciences formed our company and we issued 18,401 shares of our common stock to S.L.A. Pharma in August 2007. In the event we closed an equity financing with gross proceeds of not less than $5,000,000 and the 18,401 shares issued to S.L.A. Pharma did not have a fair market value at least equal to $500,000 (calculated by multiplying the number of shares by the price per share paid in the financing), we were required to issue to S.L.A. Pharma that number of additional shares of our common stock so that, when added to the 18,401 shares initially issued, the new and old shares would have a fair market value equal to $500,000 (based on the price per share paid in the financing). As a result, upon the closing of our initial public offering on December 22, 2010, based on the initial offering price of $6.00, we issued S.L.A. Pharma 64,933 shares of our common stock.
In August 2007, pursuant to an Assignment and Assumption Agreement, Paramount BioSciences sold all of its rights in and arising out of the Exclusive License Agreement with S.L.A. Pharma to us for $1,087,876, which represented the fees Paramount BioSciences had paid through August 2007 for both VEN 307 and VEN 308. The corresponding U.S. and foreign patents and applications for the two compounds have been licensed to us under the Assignment and Assumption Agreement (the technology referred to collectively as the Compound Technology). As consideration in part for the rights to the Compound Technology, an initial licensing fee of $250,000 was paid to S.L.A. Pharma (this amount was paid by Paramount BioSciences and was included in the consideration paid by us to Paramount BioSciences in connection with the Assignment and Assumption Agreement). In the event that the Compound Technology is commercialized, we are obligated to pay to S.L.A. Pharma annual royalties ranging from the mid to upper single digit percentages, based upon net sales of the product. In addition, we are required to make payments to S.L.A. Pharma up to an aggregate amount of $20 million upon the achievement of various milestones related to regulatory events. Should we make any improvements regarding the Compound Technology, we are required to grant S.L.A. Pharma licenses to use such improvements.
27
We also are required to reimburse S.L.A. Pharma for clinical development costs associated with the technology development of both VEN 307 and VEN 308. Our total payment obligation for these development costs for VEN 307 will not exceed $4,000,000. As of December 31, 2010, we had made $3,200,000 of such payments, including $600,000 paid on December 31, 2010, which represented past development costs we had accrued. Upon completion of enrollment into the Phase III trial in Europe, which we anticipate to occur at the end of 2011, we will be obligated to pay S.L.A. Pharma $800,000. S.L.A. Pharma has not completed recruitment of patients into the Phase III trial and therefore we had not recorded the $800,000 expense at December 31, 2010. In addition, both we and S.L.A. Pharma have agreed to add additional services outside the scope of the agreement in which case we are obligated to pay an additional $400,000 above the $4,000,000 cap. The additional amount will begin to be due only if and when we receive a final study report from S.L.A. Pharma from the Phase III VEN 307 trial in Europe (anticipated in the third quarter of 2012) and if we have raised net proceeds of at least $20.0 million from sales of securities and or the licensing of rights to the products. S.L.A. Pharma has not provided the services for this additional work and therefore we have not recorded any additional expenses.
As of December 31, 2010, we had made $973,500 in payments to S.L.A. Pharma relating to developing
VEN 308, including a payment of $373,500 on September 29, 2010 for services related to managing the project from January 2010 through September 2010. These project management fees were terminated effective
October 1, 2010. We do not expect to continue developing VEN 308 in the short term and therefore do not expect to make any additional payments.
Our future known payment obligations to S.L.A. Pharma are as follows.
|
Amount Due
|
Date Due
|
Fee Description
|
||
|
$41,500
|
Monthly beginning October 1, 2010, and continuing until S.L.A. Pharma is no longer managing the development program for VEN 307.
|
Project management fees for VEN 307.
|
||
|
$800,000
|
Upon the completion of enrollment into the Phase III clinical trial that S.L.A. Pharma is conducting in Europe, anticipated at the end of 2011.
|
Development costs for VEN 307.
|
||
|
Up to $400,000
|
If contingencies are met, payable monthly as invoiced by S.L.A. Pharma.
|
Development expenses for VEN 307. Contingent upon (i) receipt of a final study report from the S.L.A. Pharma Phase III VEN 307 trial in Europe (anticipated in the third quarter of 2012), and (ii) if we have raised net proceeds of at least $20.0 million from sales of securities and/or licensing of rights to our products by that time.
|
We issued an additional 2,016 shares of our common stock to S.L.A. Pharma pursuant to the terms of the fourth amendment to the license agreement entered into in December 2009 and issued a warrant to purchase 13,605 shares of our common stock at an exercise price of $1.24 per share pursuant to the terms of the sixth amendment entered into on August 30, 2010. The sixth amendment benefited us by providing for an extension of the next $600,000 development fee, due September 30, 2010 to December 31 2010, an extension of the next $800,000 payment for diltiazem (VEN 307) development costs, due February 28, 2011 to within 14 days of S.L.A. Pharma providing us with written notification of the completion of enrollment in its Phase III trial for VEN 307 in Europe, which we expect to occur in the third quarter of 2011, and the cancellation of all future phenylephrine (VEN 308) monthly project management fees of $41,500 per month beginning after September 30, 2010, resulting in significant short term savings.
28
The Exclusive License Agreement with S.L.A. Pharma is terminable by us for any reason upon 90 days’ written notice, and by either party in the event of a material breach or default of the Exclusive License Agreement or either party becomes bankrupt or insolvent. In addition, S.L.A. Pharma may terminate the Exclusive License Agreement at any time, with one month’s notice in the event that a third party wishes to enter into a license agreement for VEN 307 and VEN 308 and has entered into an agreement to that effect, provided that within that the termination will not be effective if within that one-month period we pay all then required payments under the agreement. If the license is terminated in any of these situations, we would have no further payment obligations to S.L.A. Pharma. In the event we have a ‘‘change in control’’ prior to the completion of the Phase III study for VEN 307 and we terminate the license within 30 days of the change in control, we must pay the balance of all payments (including the contingent $400,000) owed for VEN 307 even if S.L.A. Pharma has not actually incurred those costs. In the event we have a ‘‘change in control’’ after the completion of the Phase III study for VEN 307 and we terminate the license within 30 days of the change in control, we must pay the balance of all payments (including the contingent $400,000) owed for VEN 307 even if S.L.A. Pharma has not actually incurred those costs plus any other development expenses mutually agreed upon, but excluding the $41,500 monthly payments for VEN 307 and any monthly payments that might have been agreed to and initiated for VEN 308. A ‘‘change in control’’ is defined as a merger or other reorganization of our company in which our stockholders prior to the transaction do not own a majority of the voting stock of the surviving or successor entity, the sale by one or more of our stockholders of a majority of our voting securities, or the sale of all or substantially all of our assets related to VEN 307 and VEN 308. A ‘‘change in control’’ does not include a bona fide financing transaction in which voting control transfers to one or more persons or entities who acquire our securities in the transaction.
The patents for the intellectual property that we have licensed from S.L.A. Pharma are summarized below.
VEN 307: Diltiazem Cream for Anal Fissure
United States Patent Application No. 09/335,928
|
|
·
|
Title: ‘‘Topical Pharmaceutical Composition comprising a Cholinergic Agent or a Calcium Channel Blocker.’’ This application is still pending and currently under appeal in the United States Patent and Trademark Office, or USPTO.
|
|
|
·
|
Filed under Patent Treaty Cooperation: February 23, 1998
|
|
|
·
|
Filed in the United States: August 12, 1999
|
|
|
·
|
The patent for the existing formulation that S.L.A. Pharma is using has not yet been issued for reasons of lack of novelty and inventiveness (prior art) and S.L.A. Pharma filed an appeal to the PTO Board of Appeal. The Appeal of U.S. Patent Application No. 09/355,928 was addressed on August 31, 2010 wherein the Appeal Board affirmed the novelty of the topical use of diltiazem while maintaining the lack of inventive step rejection over the prior art, with the proviso that additional data were necessary to show unexpected results. As such, on October 25, 2010, S.L.A. Pharma filed a Request for Continued Examination (RCE) to continue prosecution of the pending application and to provide the additional data requested by the Appeal Board to show the unexpected results of using topical diltiazem. S.L.A. Pharma introduced additional arguments to the PTO examiner to address recent legal decisions by the Federal Circuit Courts, relevant to the inventive step for the topical use of diltiazem, which decisions have not been previously considered by the PTO examiner. We expect the first PTO action to be made by the end of 2011.
|
|
|
·
|
The inventors are: Michael A. Kamm and Robin K.S. Phillips
|
|
|
·
|
The original assignee of the patent was: S.L.A. Pharma AG
|
|
|
·
|
S.L.A. Pharma AG is responsible for the prosecution of this patent and we are responsible for the costs of such prosecution:
|
|
|
·
|
Expiration Date: If the patent application is found allowable by the Appeal Board at the USPTO the patent will expire on February 23, 2018, if all maintenance fees are paid.
|
Canadian Patent No. 2,281,755
|
|
·
|
Title: ‘‘Topical Pharmaceutical Composition comprising a Cholinergic Agent or a Calcium Channel Blocker’’
|
|
|
·
|
Filed under Patent Treaty Cooperation: February 23, 1998
|
|
|
·
|
Entered National Stage in Canada: August 23, 1999
|
29
|
|
·
|
The Patent was granted on: November 11, 2006
|
|
|
·
|
The inventors of the patent are: Michael A. Kamm and Robin K.S. Phillips
|
|
|
·
|
The original assignee of the patent was: S.L.A. Pharma AG
|
|
|
·
|
S.L.A. Pharma AG is responsible for the prosecution of this patent and Ventrus Biosciences is responsible for the costs of such prosecution.
|
|
|
·
|
Expiration date of the patent: February 23, 2018, assuming all maintenance fees are paid.
|
VEN 308: Phenylephrine gel for fecal incontinence
U.S. Patent No. 6,635,678
|
|
·
|
Title: ‘‘Pharmaceutical composition for treating fecal incontinence’’
|
|
|
·
|
Filed under Patent Treaty Cooperation: December 23, 1997
|
|
|
·
|
Entered National Stage in U.S.: August 24, 1999
|
|
|
·
|
The Patent was granted on: October 21, 2003
|
|
|
·
|
The inventors of the patent are: Michael A. Kamm and Robin K.S. Phillips
|
|
|
·
|
The original assignee of the patent was: S.L.A. Pharma AG
|
|
|
·
|
S.L.A. Pharma AG is responsible for the prosecution of this patent and Ventrus Biosciences is responsible for the costs of such prosecution
|
|
|
·
|
Expiration date of the patent: December 23, 2017, assuming all maintenance fees are paid.
|
Canadian Patent No. 2,275,663
|
|
·
|
Title: ‘‘Pharmaceutical compositions comprising alpha-adrenergic agonists for the treatment of fecal incontinence’’
|
|
|
·
|
Filed under Patent Treaty Cooperation: December 23, 1997
|
|
|
·
|
Entered National Stage in Canada: June 18, 1999
|
|
|
·
|
The Patent was granted on: March 18, 2008
|
|
|
·
|
The inventors of the patent are: Michael A. Kamm and Robin K.S. Phillips
|
|
|
·
|
The original assignee of the patent was: S.L.A. Pharma AG
|
|
|
·
|
S.L.A. Pharma AG is responsible for the prosecution of this patent and Ventrus BioSciences is responsible for the costs of such prosecution
|
|
|
·
|
Expiration date of the patent: December 23, 2017, assuming all maintenance fees are paid.
|
In March 2008, we entered into a license agreement with Amer whereby we acquired patent rights to iferanserin (VEN 309) for the topical treatment of any anorectal disorders. At this time we are only pursuing VEN 309 as a treatment for hemorrhoids. However, Amer has rights to the use of VEN for wound healing to which we also would have rights to develop VEN 309 as a topical treatment for anorectal wounds. We have no current plans to pursue this indication. Amer also has a patent application pending for iferanserin as a treatment for pulmonary arterial hypertension, or PAH, which is not an anorectal disorder and to which we have no rights. The administration of iferanserin for PAH would not be a topical formulation, but instead would be systemic (taken orally or by injection) or used in a pulmonary formulation, such as an inhaler, and therefore would not compete with VEN 309.
We may be required to make future milestone payments to Amer totaling up to $20 million upon the achievement of various milestones related to regulatory or commercial events. In the event that VEN 309 is commercialized, we are obligated to pay to Amer annual royalties ranging from the upper single to lower double digit percentages for sales in the U.S. and ranging from the low to mid single digit percentages for sales outside of the U.S. The license agreement is terminable by either party for cause, upon 30 days notice and subject to a 60-day cure period, upon notice if either party becomes bankrupt or insolvent or at any time after the expiration of the Royalty Period for any Licensed Product (as such terms are defined in the Exclusive License Agreement) upon 90 days’ written notice. We may terminate the license agreement upon 30 days’ written notice in the event any safety, efficacy or regulatory issues prevent development or commercialization of the VEN 309.
30
The patents for the intellectual property that we have licensed from Amer are summarized below.
US Patent No. 5,266,571 ‘‘Treatment of Hemorrhoids with 5-HT2 Antagonists’’
|
|
·
|
Filed in United States, Europe (EP 0684816) (Germany, Great Britain, Austria, Greece, France, Portugal,
|
Luxemburg, Ireland, Spain, Denmark, Switzerland, Belgium, Sweden, and Netherland), Japan (2807092), and Korea (0278522).
|
|
·
|
All patents have been granted.
|
|
|
·
|
In the U.S. the patent was filed with the Sam Amer as the inventor and in all foreign countries, Sam
|
Amer & Co., Inc. as the assignee.
|
|
·
|
Ventrus is responsible for further prosecution.
|
|
|
·
|
U.S. patent will expire on January 9, 2012, all other foreign patents will expire on February 19, 2013, if all maintenance fees are timely paid.
|
US Patent No. 5,605,902 ‘‘5-HT2 Receptor Antagonist Compositions Useful in Treating Venous Conditions’’
|
|
·
|
Filed in the United States only.
|
|
|
·
|
Patent has been granted.
|
|
|
·
|
In the U.S. the patent was filed with the Sam Amer as the inventor.
|
|
|
·
|
Ventrus is responsible for further prosecution.
|
|
|
·
|
U.S. patent will expire on January 9, 2012, if all maintenance fees are timely paid.
|
US Patent No. 5,780,487 ‘‘S-2’-[2-(1-Methyl-2-Piperidyl) ethyl] Cinnamanilide’’ (this is the VEN 309 compound being developed by Ventrus)
|
|
·
|
Filed in United States, Europe (EP 0973741) (Germany, Great Britain, France, Switzerland, Spain), Japan (520835/98), Norway (19994181) and Korea (10-997007763).
|
|
|
·
|
U.S. and Europe granted patent, Norway, Japan and Korea are pending.
|
|
|
·
|
In the U.S. the patent was filed with the Sam Amer as the inventor and in all foreign countries, Sam Amer & Co., Inc. as the assignee.
|
|
|
·
|
Ventrus is responsible for further prosecution.
|
|
|
·
|
U.S. patent will expire on August 7, 2015, all other foreign patents will expire on January 23, 2018, if all maintenance fees are timely paid.
|
Newly Filed Applications ‘‘Methods and Compositions for Treating Internal and External Hemorrhoids’’
US Patent Application No. 12/860,974 filed on August 23, 2010
PCT International Application No. PCT/US2010/046260, filed on August 23, 2010
|
|
·
|
Both patents are still pending.
|
|
|
·
|
In the U.S. the patent application was filed with the Sam Amer as the inventor and in the PCT International application, Sam Amer & Co., Inc. as the assignee.
|
|
|
·
|
Ventrus is responsible for further prosecution.
|
|
|
·
|
Any patent that is granted from these applications will expire on August 23, 2030.
|
Under both the S.L.A. Pharma and the Amer license agreements, we are responsible for the costs of prosecution of the patent, as well as any new patent filings for the licensed products. While we will pay these costs, S.L.A. Pharma and Amer will retain ownership of the respective patents although we will have the rights to license the technology underlying the patents for the duration of the respective license agreement.
31
Employees
Our activities to date have consisted of establishing and clarifying the regulatory pathway for the late phase clinical trials and regulatory approval of our product candidates, primarily VEN 309 and VEN 307, and on establishing the contract manufacturing capacity and methods, and study start up procedures necessary to allow the first Phase III clinical trial of VEN 309 to proceed. All of these planned trials will be conducted by third parties. All clinical trials to date have been conducted either by the licensor, the licensor’s previous partners or by independent investigators, as have the preclinical studies and product formulation activities. Consequently, we have needed only a few employees with medical expertise and drug development experience and a limited number of administrative employees.
As of February 28, 2011, we had five employees and had contracted with three consultants on manufacturing, preclinical and clinical aspects of our drug programs. We use these consulting agreements to avoid the costs customarily associated with employees until our financial resources will allow us to hire additional employees.
Where you can find additional information
Our website address is www.ventrusbio.com. We make available free of charge through our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC.
Risk Factors
Certain statements in this Annual Report on Form 10-K, including certain statements contained in “Description of Business” and “Management’s Discussion and Analysis,” constitute “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. The words or phrases “can be,” “may,” “could,” “would,” “expects,” “believes,” “seeks,” “estimates,” “projects” and similar words and phrases are intended to identify such forward-looking statements. Such forward-looking statements are subject to various known and unknown risks and uncertainties, including those described on the following pages, and we caution you that any forward-looking information provided by us is not a guarantee of future performance. Our actual results could differ materially from those anticipated by such forward-looking statements due to a number of factors, some of which are beyond our control. All such forward-looking statements are current only as of the date on which such statements were made. We do not undertake any obligation to publicly update any forward-looking statement to reflect events or circumstances after the date on which any such statement is made or to reflect the occurrence of unanticipated events.
Risks Related to Our Financial Condition
We have had negative cash flows from operations and might not be able to generate sufficient cash to service our existing indebtedness and any additional indebtedness could have a material adverse effect on our financial health.
Our ability to make payments on our indebtedness depends on our ability to generate cash in the future. We expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. As of December 31, 2010, we were obligated to pay the following indebtedness:
|
|
·
|
an aggregate of $1,573,000 in principal under promissory notes issued to Paramount Credit Partners, an affiliate of Paramount BioSciences, of which Dr. Lindsay A. Rosenwald, our largest stockholder, is the sole member, which are due on the earlier of December 31, 2013 or the completion by us of a transaction, or series of related transactions, subsequent to our initial public offering, including an equity offering, sale of assets, licensing or strategic partnership, in which we raise at least $5,000,000 in gross cash proceeds;
|
32
|
|
·
|
$800,000 in principal under a promissory note issued to Israel Discount Bank of New York, which is guaranteed by Lindsay A. Rosenwald, the sole member of Paramount BioSciences, which is due on September 22, 2011, but which we agreed with Dr. Rosenwald to repay upon the completion of our initial public offering (we repaid this in January 2011 with proceeds from the sale of shares of our common stock pursuant to the exercise of the underwriters’ over-allotment option on January 7, 2011); and
|
|
|
·
|
$420,000 in principal under a promissory note issued to Israel Discount Bank of New York, which is guaranteed by Lindsay A. Rosenwald, which is due on demand or on November 4, 2011, but which we agreed with Dr. Rosenwald to pay upon the completion of our initial public offering (we repaid this in January 2011 with proceeds from the sale of shares of our common stock pursuant to the exercise of the underwriters’ over-allotment option on January 7, 2011).
|
There can be no assurance that we will be in a position to pay any of this indebtedness when due or obtain extensions of the maturity dates. We anticipate that we will need to secure additional funding in order for us to be able to pay our obligations when due.
Moreover, this level of debt could have important consequences to our stockholders. For example, it could:
|
|
·
|
make it more difficult for us to satisfy our obligations with respect to any other indebtedness and obligations, including payments owed to our licensors;
|
|
|
·
|
limit our flexibility in planning for the development, clinical testing, approval and marketing of our products;
|
|
|
·
|
place us at a competitive disadvantage compared to any of our competitors that are less leveraged than we are;
|
|
|
·
|
increase our vulnerability to both general and industry-specific adverse economic conditions; and
|
|
|
·
|
limit our ability to obtain additional funds.
|
The addition of further debt to our current debt levels could make it more difficult for us to repay our indebtedness and meet our other obligations and would intensify the leverage-related risks that we now face.
Our failure to meet our substantial obligations to our licensors could result in the termination of our licenses or put substantial burdens on our financial position.
We have in-licensed all of our product candidates. Our licenses require us to make substantial up-front, milestone, and royalty payments. If we fail to make these payments, the licensors might terminate the licenses on relatively short notice, in which event we would not be able to commercialize drug candidates that were covered by the licenses.
We license two of our product candidates, VEN 307 and VEN 308, from S.L.A. Pharma, a Swiss corporation, and have obligations related to VEN 308 and to fund S.L.A. Pharma’s development efforts for VEN 307 in the E.U., all of which are set forth in the chart below.
|
Amount Due
|
Date Due
|
Fee Description
|
||
|
$41,500
|
Monthly beginning October 1, 2010, and continuing until S.L.A. Pharma is no longer managing the development program for VEN 307.
|
Project management fees for VEN 307.
|
||
|
$800,000
|
Upon the completion of enrollment into the Phase III clinical trial that S.L.A. Pharma is conducting in Europe, anticipated at the end of 2011.
|
Development costs for VEN 307.
|
||
|
Up to $400,000
|
If contingencies are met, payable monthly as invoiced by S.L.A. Pharma.
|
Development expenses for VEN 307. Contingent upon (i) receipt of a final study report from the S.L.A. Pharma Phase III VEN 307 trial in Europe (anticipated in the third quarter of 2012), and (ii) if we have raised net proceeds of at least $20.0 million from sales of securities and/or licensing of rights to our products by that time.
|
33
In addition, if we commercialize a product candidate, we must pay S.L.A. Pharma annual royalties ranging from the mid to upper single digit percentages, based upon net sales of the product. We also are required to make future milestone payments totaling up to $20 million upon the achievement of various milestones related to regulatory events for both VEN 307 and VEN 308, the earliest of which is not anticipated until 2015. In the event we breach these obligations, we could lose our rights to VEN 307 or VEN 308, or both, depending on the breach, which would have a material adverse effect on our business and prospects.
We have also entered into a license agreement with Amer, a California company, whereby we acquired patent rights to iferanserin, VEN 309, for the topical treatment of anorectal disorders. We are required to make future milestone payments totaling up to $20 million upon the achievement of various milestones related to regulatory or commercial events. If we commercialize iferanserin, we will be obligated to pay to Amer annual royalties ranging from the upper single to lower double digit percentages for sales in the U.S. and ranging from the low to mid single digit percentages for sales outside of the U.S., based upon sales of the product. In the event we breach these obligations, we could lose rights to VEN 309, which would have a material adverse effect on our business and prospects.
Risks Related to Our Business
We have a limited operating history and a history of operating losses, and expect to incur significant additional operating losses.
We were established in October 2005, began active operations in the spring of 2007 and have only a limited operating history. Therefore, there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We have generated losses since we began operations and, as of December 31, 2010, we had a deficit accumulated during the development stage of $33,184,354. We expect to incur substantial additional losses over the next several years as our research, development, pre-clinical testing, and clinical trial activities increase. The amount of future losses and when, if ever, we will achieve profitability are uncertain. We have no products that have generated any commercial revenue, do not expect to generate revenues from the commercial sale of products unless and until our product candidates are approved by the FDA for sale, and might never generate revenues from the sale of products.
We are not currently profitable and might never become profitable.
We have a history of losses and expect to incur substantial losses and negative operating cash flow for the foreseeable future, and we might never achieve or maintain profitability. Even if we succeed in developing and commercializing one or more product candidates, we expect to incur substantial losses for the foreseeable future. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future as we:
34
|
|
·
|
continue to undertake development and clinical trials for product candidates;
|
|
|
·
|
seek regulatory approvals for product candidates;
|
|
|
·
|
implement additional internal systems and infrastructure; and
|
|
|
·
|
hire additional personnel.
|
Our ability to generate revenue and achieve profitability will depend on, among other things:
|
|
·
|
successful completion of animal tests, which are referred to as pre-clinical studies, as well as human tests, which are referred to as clinical trials, for our product candidates;
|
|
|
·
|
obtaining necessary regulatory approvals from the FDA and international regulatory agencies;
|
|
|
·
|
establishing manufacturing, sales, and marketing arrangements with third parties; and
|
|
|
·
|
raising sufficient funds to finance our activities.
|
We might not succeed at any of these undertakings. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations might be materially adversely affected.
We expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We might not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability would negatively impact the value of our common stock.
We have no approved products.
To date, we have no approved product on the market and have generated no product revenues. Unless and until we receive approval from the FDA and other regulatory authorities for our product candidates, we cannot sell our drugs and will not have product revenues. Therefore, for the foreseeable future, we will have to fund all of our operations and capital expenditures from the net proceeds of this offering, cash on hand, licensing fees and grants and future securities offerings.
We are a development-stage company and might not be able to commercialize any product candidates.
We are a development-stage company and have not demonstrated our ability to perform the functions necessary for the successful commercialization of any product candidates. The successful commercialization of any product candidates will require us to perform a variety of functions, including:
|
|
·
|
continuing to undertake preclinical development and clinical trials;
|
|
|
·
|
participating in regulatory approval processes;
|
|
|
·
|
formulating and manufacturing products; and
|
|
|
·
|
conducting sales, marketing and distribution activities.
|
Our development of current and future product candidates is subject to the risks of failure and delay inherent in the development of new pharmaceutical products and products based on new technologies, including:
|
|
·
|
delays in product development, clinical testing, or manufacturing;
|
|
|
·
|
unplanned expenditures in product development, clinical testing, or manufacturing;
|
|
|
·
|
failure of a product candidate to demonstrate acceptable safety and efficacy;
|
|
|
·
|
failure to receive regulatory approvals;
|
|
|
·
|
emergence of superior or equivalent products;
|
|
|
·
|
inability to manufacture on our own, or through any others, product candidates on a commercial scale or at a financially viable cost; and
|
|
|
·
|
failure to achieve market acceptance.
|
35
Because of these risks, our research and development efforts might not result in any commercially viable products. If we do not successfully complete a significant portion of these development efforts, obtain required regulatory approvals, or have commercial success with any approved products, our business, financial condition and results of operations will be materially harmed.
We will need additional financing to fund our activities in the future.
We anticipate that we will incur operating losses for the foreseeable future. We expect that our resources on hand as of the date of this report will provide us with sufficient capital to fund our operations into the third quarter of 2012. However, we might consume our available capital before that time if, for example, we are not efficient in developing our product candidates and conducting clinical trials or if regulatory requirements change.
Moreover, we believe we will require substantial funds in the future to support our operations. We anticipate that to complete the clinical trial process to obtain the approval of our product candidates will cost approximately $15 million for VEN 307, $20 million for VEN 308 and $20 million for VEN 309. We might seek equity or debt financings in the future to fund our operations. However, there is no assurance that we will be successful in raising the additional capital we need to fund our business plan on terms that are acceptable to us, or at all. If we do not succeed in raising additional funds on acceptable terms, we might be unable to initiate or complete clinical trials or obtain approval of any product candidate from the FDA and other regulatory authorities. In addition, we could be forced to discontinue product development, forego sales and marketing efforts, sacrifice attractive business opportunities, cease operations entirely and sell or otherwise transfer all or substantially all of our remaining assets.
We are dependent on license relationships.
We have acquired, by license, technology that is critical to our business, and we might enter into additional licenses in the future. Licenses to which we are a party contain, and we expect that any future licenses will contain, provisions requiring up-front, milestone, and royalty payments to licensors. If we fail to comply with these obligations to a licensor, that licensor might have the right to terminate the license on relatively short notice, in which event we would not be able to commercialize drug candidates or technologies that were covered by the license. Also, the milestone and other payments associated with these licenses will make it less profitable for us to develop our drug candidates.
We have identified material weaknesses in our financial reporting process.
We have identified material weaknesses in our financial reporting process with respect to lack of accounting expertise, segregation of duties and lack of independent review over financial reporting. We have also identified numerous errors in the accounting for routine transactions and non-routine, complex transactions, including with respect to the valuation of common stock and derivative securities, the recording of debt discount and related amortization for warrants issued in connection with debt financings and calculation of deferred tax assets. The material weaknesses identified with respect to lack of accounting expertise and segregation of duties relate to the policies and procedures that:
|
|
·
|
pertain to the procedures to ensure that information required to be disclosed is properly gathered and reported;
|
|
|
·
|
pertain to the maintenance of records that accurately and fairly reflect our transactions and dispositions of our assets;
|
|
|
·
|
provide reasonable assurance that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
|
36
|
|
·
|
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
|
We have taken the following measures to address the material weaknesses identified by us and improve our periodic financial statement reporting process:
|
|
·
|
hired a permanent Chief Financial Officer to strengthen our internal staffing and technical expertise in financial accounting and reporting;
|
|
|
·
|
upgraded our accounting software system in the first quarter of 2011;
|
|
|
·
|
limited access to the accounting and information systems and related data to strengthen segregation of duties; and
|
|
|
·
|
implemented in the fourth quarter of 2010 procedures and controls in the financial statement close process to improve the accuracy and timeliness of the preparation of quarterly and annual financial statements.
|
There can be no assurance that we will be able to successfully implement our plans to remediate the material weaknesses in our financial reporting process. Our failure to successfully implement our plans to remediate these material weaknesses could cause us to fail to meet our reporting obligations, to produce timely and reliable financial information, and to effectively prevent fraud. Additionally, such failure could cause investors to lose confidence in our reported financial information, which could have a negative impact on our financial condition and stock price.
Corporate and academic collaborators might take actions to delay, prevent, or undermine the success of our products.
Our operating and financial strategy for the development, clinical testing, manufacture, and commercialization of drug candidates heavily depends on collaborating with corporations, academic institutions, licensors, licensees, and other parties. However, there can be no assurance that we will successfully establish these collaborations. In addition, should a collaboration be terminated, replacement collaborators might not be available on attractive terms, or at all. The activities of any collaborator will not be within our control and might not be within our power to influence. There can be no assurance that any collaborator will perform its obligations to our satisfaction or at all, that we will derive any revenue or profits from these collaborations, or that any collaborator will not compete with us. If any collaboration is not successful, we might require substantially greater capital to undertake development and marketing of our proposed products and might not be able to develop and market these products effectively, if at all. In addition, a lack of development and marketing collaborations might lead to significant delays in introducing proposed products into certain markets and/or reduced sales of proposed products in such markets.
We rely on data provided by our collaborators and others that has not been independently verified and could prove to be false, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, clinical trials, and our business. If these third parties provide inaccurate, misleading, or incomplete data, our business, prospects, and results of operations could be materially adversely affected.
37
We rely exclusively on third parties to formulate and manufacture our product candidates.
We have no experience in drug formulation or manufacturing and do not intend to establish our own manufacturing facilities. We lack the resources and expertise to formulate or manufacture our own product candidates, which are currently being manufactured entirely by commercial third parties. If any product candidate we might develop or acquire in the future receives FDA approval, we will rely on one or more third-party contractors to manufacture our products. Our anticipated future reliance on a limited number of third-party manufacturers exposes us to the following risks:
|
|
·
|
We might be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would generally require new testing and compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any.
|
|
|
·
|
Our third-party manufacturers might be unable to formulate and manufacture our drugs in the volume and of the quality required to meet our clinical and commercial needs, if any.
|
|
|
·
|
Our future contract manufacturers might not perform as agreed or might not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products.
|
|
|
·
|
Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with the FDA’s Current Good Manufacturing Practices, or cGMP, and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards.
|
|
|
·
|
If any third-party manufacturer makes improvements in the manufacturing process for our products, we might not own, or might have to share, the intellectual property rights to the innovation with our licensors.
|
|
|
·
|
We might compete with other companies for access to these manufacturers’ facilities and might be subject to manufacturing delays if the manufacturers give other clients higher priority than us.
|
Each of these risks could delay our clinical trials or the approval, if any, of our product candidates by the FDA or the commercialization of our product candidates and could result in higher costs or deprive us of potential product revenues. As a result, our business, financial condition, and results of operations might be materially harmed.
If we or our collaborators are unable to manufacture our products in sufficient quantities or are unable to obtain regulatory approvals for the manufacturing facility, we might be unable to meet demand for our product, and we might lose potential revenues.
We require access to, or development of, facilities to manufacture a sufficient supply of our product candidates in order to complete our clinical trials and commercialize our product candidates. We currently contract with an outside source to manufacture our compounds for our clinical needs. If, for any reason, we become unable to rely on our current source or any future source to manufacture our product candidates, either for clinical trials or, at some future date, for commercial quantities, then we would need to identify and contract with additional or replacement third-party manufacturers to manufacture compounds for preclinical, clinical and commercial purposes. We might not be successful in identifying additional or replacement third-party manufacturers, or in negotiating acceptable terms with any that we do identify. Such third-party manufacturers must receive FDA approval before they can produce clinical material or commercial product, and any that we identify might not receive such approval. If we are unable to secure and maintain third-party manufacturing capacity, the development and sales of our products and our financial performance might be materially affected.
38
Before we can begin to commercially manufacture our product candidates, we must obtain regulatory approval of the manufacturing facility and process. Manufacturing of drugs for clinical and commercial purposes must comply with cGMPs and applicable non-U.S. regulatory requirements. The cGMP requirements govern quality control and documentation policies and procedures. Complying with cGMP and non-U.S. regulatory requirements will require that we expend time, money, and effort in production, recordkeeping, and quality control to assure that the product meets applicable specifications and other requirements. We, or our contracted manufacturing facility, must also pass a pre-approval inspection prior to FDA approval. Failure to pass a pre-approval inspection might significantly delay FDA approval of our products. If we fail to comply with these requirements, we would be subject to possible regulatory action and might be limited in the jurisdictions in which we are permitted to sell our products. As a result, our business, financial condition, and results of operations might be materially harmed.
Preclinical and clinical trials required for our product candidates are expensive and time-consuming, and their outcome is uncertain.
In order to obtain FDA approval to market a new drug product, we must demonstrate safety and effectiveness in humans. To meet these requirements, we must conduct extensive preclinical testing and adequate and well-controlled clinical trials. Conducting clinical trials is a lengthy, time consuming, and expensive process. The length of time might vary substantially according to the type, complexity, novelty, and intended use of the product candidate, and often can be several years or more per trial. Delays associated with products for which we are directly conducting preclinical or clinical trials might cause us to incur additional operating expenses. The commencement and rate of completion of clinical trials might be delayed by many factors, including, for example:
|
|
·
|
inability to manufacture sufficient quantities of qualified materials under cGMP for use in clinical trials;
|
|
|
·
|
slower than expected rates of patient recruitment;
|
|
|
·
|
failure to recruit a sufficient number of patients;
|
|
|
·
|
modification of clinical trial protocols;
|
|
|
·
|
changes in regulatory requirements for clinical trials;
|
|
|
·
|
the lack of effectiveness during clinical trials;
|
|
|
·
|
the emergence of unforeseen safety issues;
|
|
|
·
|
delays, suspension, or termination of clinical trials by the institutional review board responsible for overseeing the study at a particular study site; and
|
|
|
·
|
government, institutional review board or other regulatory delays or clinical holds requiring suspension or termination of the trials.
|
The results from preclinical testing and early clinical trials are not necessarily predictive of results obtained in later clinical trials. Accordingly, even if we obtain or have obtained positive results from preclinical or early clinical trials, we might not achieve the same success in future clinical trials. Clinical trials might not provide statistically significant data supporting a product candidate’s safety and effectiveness to meet the requisite regulatory approvals.
The failure of clinical trials to demonstrate safety and effectiveness for the desired indications could harm the development of that product candidate and other product candidates. This failure could cause us to abandon a product candidate and could delay development of other product candidates. Any delay in, or termination of, our clinical trials would delay the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. Any change in, or termination of, our clinical trials could materially harm our business, financial condition, and results of operation.
39
Existing and unforeseen safety issues could hinder the development of our product candidates and their adoption, if approved.
Iferanserin (VEN 309), like numerous other drugs, is dependent on the CYP2D6 enzyme for its metabolism. An important property of CYP2D6 is that its activity is affected by genetic variability in individuals, including individuals who are CYP2D6 deficient and that its activity can be reduced by certain drugs. If this enzyme is inhibited by other medications being taken by a patient or the patient has a genetically reduced amount or a deficiency of the enzyme, and the patient takes iferanserin, the patient might have a higher level of iferanserin in his or her blood and might experience side effects although we are unaware of what the side effects might be. One patient in one of our Phase I studies had a genetic reduction of this enzyme and did experience substantially higher levels of iferanserin in his blood. However, no side effects were observed in this patient. There are several well known drugs that also are dependent on CYP2D6, including several antidepressants as well as tamoxifen. We might restrict the use of iferaserin in patients taking medications that inhibit or are dependent on the CYP2D6 enzyme, depending on the outcome of clinical drug-drug interaction clinical studies that we intend to undertake subsequent to the completion of the first Phase III clinical trial with this product. Iferanserin (VEN 309) has demonstrated arrythmogenic potential in in vitro (hERG channel) studies at exposures 60-100 times the dose expected in humans when using the 0.5% concentration applied topically twice daily. We expect to conduct an arrhythmia clinical study (“thorough QT study”) as part of our Phase III clinical pharmacology program, which studies are routinely required by the FDA. Even though VEN 309 has a wide safety margin in this area, we cannot be certain of the outcome of this study, and demonstration of clinically meaningful arrhythmia risks could compromise or prevent the approvability of the product in major markets.
Both diltiazem and phenylephrine have been safely used extensively for decades when given orally at much higher exposures (blood levels) than currently under study in the topical application of VEN 307 and VEN 308.
Despite these safety records, other safety issues could arise during testing of our products, which might delay testing or prevent further development entirely. If a product is approved, any limitation on use that might be necessary could hinder its adoption in the marketplace. In addition, if any product is approved, it could be used against any instructions that we publish that limit its use, which could subject us to litigation.
If we cannot compete successfully for market share against other drug companies, we might not achieve sufficient product revenues and our business will suffer.
If our product candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing drugs might provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or might offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we might not achieve sufficient product revenues and our business will suffer.
We might compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we do, as well as significantly greater experience in:
|
|
·
|
developing drugs;
|
|
|
·
|
undertaking pre-clinical testing and human clinical trials;
|
|
|
·
|
obtaining FDA and other regulatory approvals of drugs;
|
|
|
·
|
formulating and manufacturing drugs; and
|
|
|
·
|
launching, marketing and selling drugs.
|
We might not obtain the same resources and experience as our competitors. If we are unable to perform these tasks effectively and efficiently, our results of operations might be materially adversely affected.
40
Developments by competitors might render our products or technologies obsolete or non-competitive.
The pharmaceutical and biotechnology industries are intensely competitive. We might compete with organizations that are developing treatments for the indications that our products target.
To our knowledge, there are currently no approved drugs for treatment of anal fissures, though ProStrakan Group plc is currently developing a topical nitroglycerin for such treatment. For the treatment of fecal incontinence, to our knowledge there are currently no products approved or in development although there are two non-drug products in development. For the treatment of hemorrhoids, some physicians are known to prescribe topical steroids, although such treatment has not been approved by the FDA for this indication. Further, many hemorrhoid sufferers use Wyeth’s Preparation H® or similar products for symptomatic relief (active ingredients can vary by country but generally include glycerin, phenylephrine HCl, pramoxine HCl, and white petrolatum). No data is publicly available regarding the clinical efficacy of this or other over-the-counter symptomatic treatments for hemorrhoids. Finally, there are surgical devices being studied for the treatment of hemorrhoids. If our competitors develop effective treatments for anal fissure, fecal incontinence or hemorrhoids and successfully commercialize those treatments, our business and prospects might be materially harmed.
If we are not able to develop collaborative marketing relationships with licensees or partners, or create an effective internal sales, marketing, and distribution capability, we might be unable to market our products successfully.
To market our products, we will have to establish our own marketing and sales force or out-license our product candidates to, or collaborate with, larger firms with experience in marketing and selling pharmaceutical products. There can be no assurance that we will be able to successfully establish our own marketing capabilities or establish marketing, sales, or distribution relationships with third parties; that such relationships, if established, will be successful; or that we will be successful in gaining market acceptance for our products. To the extent that we enter into any marketing, sales, or distribution arrangements with third parties, our product revenues will be lower than if we marketed and sold our products directly, and any revenues we receive will depend upon the efforts of such third parties. If we are unable to establish such third-party sales and marketing relationships, or choose not to do so, we will have to establish our own in-house capabilities. We, as a company, have no experience in marketing or selling pharmaceutical products and currently have no sales, marketing, or distribution infrastructure. To market any of our products directly, we would need to develop a marketing, sales, and distribution force that both has technical expertise and the ability to support a distribution capability. To establish our own marketing, sales, and distribution capacity would significantly increase our costs, and require substantial additional capital. In addition, there is intense competition for proficient sales and marketing personnel, and we might not be able to attract individuals who have the qualifications necessary to market, sell, and distribute our products. There can be no assurance that we will be able to establish internal marketing, sales, or distribution capabilities.
Physicians and patients might not accept and use our drugs.
Even if the FDA approves one of our product candidates, physicians and patients might not accept and use it. Acceptance and use of our products will depend upon a number of factors, including:
|
|
·
|
perceptions by members of the health care community, including physicians, about the safety and effectiveness of our product;
|
|
|
·
|
cost-effectiveness of our product relative to competing product or therapies;
|
|
|
·
|
availability of reimbursement for our product from government or other healthcare payors; and
|
|
|
·
|
effective marketing and distribution efforts by us and our licensees and distributors, if any.
|
41
If our current product candidates are approved, we expect sales to generate substantially all of our revenues for the foreseeable future, and as a result, the failure of these products to find market acceptance would harm our business and would require us to seek additional financing.
Our ability to generate product revenues will be diminished if our products sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our products, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
|
|
·
|
government and health administration authorities;
|
|
|
·
|
private health maintenance organizations and health insurers; and
|
|
|
·
|
other healthcare payors.
|
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payors, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payors increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs. Even if our product candidates are approved by the FDA, insurance coverage might not be available, and reimbursement levels might be inadequate, to cover our products. If government and other healthcare payors do not provide adequate coverage and reimbursement levels for our products, once approved, market acceptance of such products could be reduced.
Proposals to modify the current health care system in the United States to improve access to health care and control its costs are continually being considered by the federal and state governments. In March 2010, the U.S. Congress passed landmark healthcare legislation. We cannot predict what impact on federal reimbursement policies this legislation will have in general or on our business specifically. Members of the U.S. Congress and some state legislatures are seeking to overturn at least portions of the legislation and we expect they will continue to review and assess this legislation and possibly alternative health care reform proposals. We cannot predict whether new proposals will be made or adopted, when they may be adopted or what impact they may have on us if they are adopted.
Health administration authorities in countries other than the United States may not provide reimbursement for our products at rates sufficient for us to achieve profitability, or at all. Like the United States, these countries have considered health care reform proposals and could materially alter their government-sponsored health care programs by reducing reimbursement rates.
Any reduction in reimbursement rates under Medicare or foreign health care programs could negatively affect the pricing of our products. If we are not able to charge a sufficient amount for our products, then our margins and our profitability will be adversely affected.
If we lose key management or scientific personnel, cannot recruit qualified employees, directors, officers, or other significant personnel or experience increases in our compensation costs, our business might materially suffer.
We are highly dependent on the services of our Chief Executive Officer and Chief Medical Officer, Dr. Russell H. Ellison. The employment agreement with Dr. Ellison does not ensure the retention of Dr. Ellison. This is also true for our other management team members, both present and future. Furthermore, our future success also depends, in part, on our ability to identify, hire, and retain additional management team members as our operations glow. We expect to experience intense competition for qualified personnel and might be unable to attract and retain the personnel necessary for the development of our business. Finally, we do not currently maintain, nor do we intend to obtain in the future, “key man” life insurance that would compensate us in the event of the death or disability of any of the members of our management team.
42
If we cannot enforce non-compete and confidentiality provisions applicable to our employees and consultants, our business might materially suffer.
We include a non-compete provision in any employment agreement we enter into with an employee including Dr. Ellison, that runs during the term of the agreement and for six months after termination. This non-compete provision was also included in employment agreements with our former chief executive officer, chief medical officer and chief scientific officer which have lapsed.
We include a confidentiality provision in any employment or consulting agreement we enter into with an employee or a consultant. The confidentiality provision runs during the term of the agreement and thereafter without limit. As a result, the confidentiality provisions contained in the employment agreements with our former chief executive officer, chief medical officer and chief scientific officer remain in effect and are in effect under all of our current consulting agreements.
All of our employees or consultants to date have been subject to an employment or a consulting agreement. For future employees with whom we do not enter into an employment agreement, we will enter into a confidentiality agreement with the same provisions described above.
To be able to enforce these non-compete and confidentiality provisions we would need to know of any breach and have sufficient funds to enforce the provisions. We cannot assure you that we would know of or be able to afford enforcement of any breach. In addition, such provisions are subject to state law and interpretation by courts, which could limit the scope and duration of these provisions. Any limitation on or non-enforcement of these non-compete and confidentiality provisions could have an adverse effect on our business.
If we are unable to hire additional qualified personnel, our ability to grow our business might be harmed.
At February 28, 2011, we had only five employees and three consultants to carry out our business plan. While we believe this will provide us with sufficient staffing to develop VEN 309 through the second quarter of 2012, we will need to hire or contract with additional qualified personnel with expertise in preclinical testing, clinical research and testing, government regulation, formulation and manufacturing and sales and marketing to commercialize VEN 309 and to develop VEN 307 and VEN 308. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for these individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
We might not successfully manage our growth.
Our success will depend upon the expansion of our operations and the effective management of our growth, which will place a significant strain on our current and future management and other administrative and operational resources. To manage this growth, we must expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage our growth effectively, our business would be harmed.
43
We might seek to develop our business through acquisitions of or investment in new or complementary businesses, products or technologies, and the failure to manage these acquisitions or investments, or the failure to integrate them with our existing business, could have a material adverse effect on us.
We might consider opportunities to acquire or invest in other technologies, products and businesses that might enhance our capabilities or complement our current product candidates. Potential and completed acquisitions and strategic investments involve numerous risks, including potential problems or issues associated with the following:
|
|
·
|
assimilating the purchased technologies, products or business operations;
|
|
|
·
|
maintaining uniform standards, procedures, controls and policies;
|
|
|
·
|
unanticipated costs associated with the acquisition or investment;
|
|
|
·
|
diversion of our management’s attention from our preexisting business;
|
|
|
·
|
maintaining or obtaining the necessary regulatory approvals or complying with regulatory standards; and
|
|
|
·
|
adverse effects on existing business operations.
|
We have no current commitments with respect to any acquisition or investment. We do not know if we will identify suitable acquisitions, whether we will be able to successfully complete any acquisitions, or whether we will be able to successfully integrate any acquired product, technology or business into our business or retain key personnel, suppliers or collaborators.
Our ability to successfully develop our business through acquisitions would depend on our ability to identify, negotiate, complete and integrate suitable target businesses or technologies and obtain any necessary financing. These efforts could be expensive and time consuming and might disrupt our ongoing operations. If we are unable to integrate efficiently any acquired business, technology or product into our business, our business and financial condition might be adversely affected.
Risks Related to Our Regulatory and Legal Environment
We are subject to extensive and costly government regulation.
Product candidates employing our technology are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of the United States Department of Health and Human Services, the United States Department of Justice, state and local governments, and their respective foreign equivalents. The FDA regulates the research, development, preclinical and clinical testing, manufacture, safety, effectiveness, record-keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of pharmaceutical products. The FDA regulates small molecule chemical entities, whether administered orally, topically or by injection, as drugs, subject to an NDA, under the Federal Food, Drug, and Cosmetic Act. If products employing our technologies are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not they have obtained FDA approval for a given product and its uses. Such foreign regulation might be equally or more demanding than corresponding United States regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling our products. The regulatory review and approval process, which includes preclinical testing and clinical trials of each product candidate, is lengthy, expensive, and uncertain. We or our collaborators must obtain and maintain regulatory authorization to conduct clinical trials and approval for each product we intend to market, and the manufacturing facilities used for the products must be inspected and meet legal requirements. Securing regulatory approval requires submitting extensive preclinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish the product’s safety and efficacy for each intended use. The development and approval process might take many years, requires substantial resources, and might never lead to the approval of a product.
44
Even if we are able to obtain regulatory approval for a particular product, the approval might limit the indicated medical uses for the product, limit our ability to promote, sell, and distribute the product, require that we conduct costly post-marketing surveillance, and/or require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as, for example, manufacturing changes or revised labeling, might require further regulatory review and approval. Once obtained, any approvals might be withdrawn, including, for example, if there is a later discovery of previously unknown problems with the product, such as a previously unknown safety issue.
If we, our collaborators, or our contract manufacturers fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in, among other things, delays in the approval of applications or supplements to approved applications; refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; warning letters; fines; import and export restrictions; product recalls or seizures; injunctions; total or partial suspension of production; civil penalties; withdrawals of previously approved marketing applications or licenses; recommendations by the FDA or other regulatory authorities against governmental contracts; and/or criminal prosecutions.
We might not obtain the necessary U.S. or worldwide regulatory approvals to commercialize any product candidate.
We cannot assure you that we will receive the approvals necessary to commercialize for sale any of our product candidates, or any product candidate we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any product candidate, we must submit to the FDA an NDA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research, pre-clinical studies, and clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in drugs that the FDA considers safe for humans and effective for their indicated uses. The FDA has substantial discretion in the drug approval process and might require us to conduct additional pre-clinical and clinical testing, perform post-marketing studies, or otherwise limit or impose conditions on any approval we obtain. The approval process might also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals might:
|
·
|
delay commercialization of, and our ability to derive product revenues from, our product candidates;
|
|
·
|
impose costly procedures on us; and
|
|
·
|
diminish any competitive advantages that we might otherwise enjoy.
|
Even if we comply with all FDA requests, the FDA might ultimately reject one or more of our NDAs. We cannot be sure that we will ever obtain regulatory clearance for our product candidates. Failure to obtain FDA approval of our product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate can be developed or obtained. There is no guarantee that we will ever be able to develop or acquire another product candidate.
45
In foreign jurisdictions, we must receive approval from the appropriate regulatory authorities before we can commercialize any drugs. The risks associated with foreign regulatory approval processes are similar to the risks associated with the FDA approval procedures described above. We cannot assure you that we will receive the approvals necessary to commercialize our product candidates for sale outside the U.S.
Even if approved, our products will be subject to extensive post-approval regulation.
Once a product is approved, numerous post-approval requirements apply. Among other things, the holder of an approved NDA is subject to periodic and other FDA monitoring and reporting obligations, including obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for changes to the approved product, product labeling, or manufacturing process. Application holders also must submit advertising and other promotional material to the FDA and report on ongoing clinical trials. The FDA also has the authority to require changes in the labeling of approved drug products and to require post-marketing studies.
Advertising and promotional materials must comply with FDA rules in addition to other applicable federal and state laws. The distribution of product samples to physicians must comply with the requirements of the Prescription Drug Marketing Act. Manufacturing facilities remain subject to FDA inspection and must continue to adhere to the FDA’s cGMP requirements. Application holders must obtain FDA approval for product, manufacturing, and labeling changes, depending on the nature of the change. Sales, marketing, and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veteran’s Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws.
Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. In addition, even if we comply with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval.
We face the risk of product liability claims and might not be able to obtain insurance.
Our business exposes us to the risk of product liability claims that are inherent in the development of drugs. If the use of one or more of our or our collaborators’ drugs harms people, we might be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, pharmaceutical companies or others selling our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop. We currently do not carry clinical trial insurance or product liability insurance. We intend to obtain such insurance in the future. We cannot predict all of the possible harms or side effects that might result and, therefore, the amount of insurance coverage we hold now or in the future might not be adequate to cover all liabilities we might incur. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for our drug candidates in development, but we might be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which might materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused by our or our collaborators’ products, our liability could exceed our total assets and our ability to pay the liability. A successful product liability claim or series of claims brought against us would decrease our cash and could cause the value of our common stock to decrease.
46
We might be exposed to liability claims associated with the use of hazardous materials and chemicals.
Our research, development and manufacturing activities and/or those of our third-party contractors might involve the controlled use of hazardous materials and chemicals. Although we will strive to have our safety procedures for using, storing, handling and disposing of these materials comply with federal, state and local laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for any resulting damages, and any liability could materially adversely affect our business, financial condition and results of operations. In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products might require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations. We currently do not carry hazardous materials liability insurance. We intend to obtain such insurance in the future.
Risks Related to Our Intellectual Property
Our license agreement with S.L.A. Pharma is subject to termination if a third party wishes to license VEN 307 and VEN 308 in certain events.
We have in-licensed the rights to VEN 307 and VEN 308 from S.L.A. Pharma. S.L.A. Pharma may terminate the license agreement at any time with one month’s notice in the event that a third party wishes to enter into a license agreement for VEN 307 and VEN 308 and has entered into an agreement to that effect, provided that the termination will not be effective if within that one-month period we pay all then required development payments related to VEN 307 under the agreement, which could total an aggregate of $1.4 million depending on when this event occurs. If the license agreement were terminated, our business would be materially harmed.
A patent has not been issued for VEN 307 and might never be issued.
No patent has been issued for VEN 307. S.L.A. Pharma has a pending application in the United States that includes claims that cover topical treatment for the relief of pain associated with anal fissures. This patent has not yet issued for reasons related to the U.S Patent and Trademark Office, or PTO, examiner’s concerns about novelty and lack of inventiveness over prior art. S.L.A. Pharma filed an appeal of the examiner’s findings to the PTO Board of Appeal. The appeal of US Patent Application No. 09/355,928 was addressed on August 31, 2010 wherein the Appeal Board affirmed the novelty of the topical use of diltiazem while maintaining the lack of inventive step rejection over the prior art, with the proviso that additional data were necessary to show unexpected results. As a result, on October 25, 2010, S.L.A. Pharma filed a Request for Continued Examination, or RCE, with the PTO to continue prosecution of the pending application and to provide the additional data requested by the Appeal Board. S.L.A. Pharma also introduced additional arguments to the PTO to address recent legal decisions by the U.S. Federal Circuit Courts, relevant to the inventive step for the topical use of diltiazem, which decisions have not been previously considered by the PTO examiner. We expect the first PTO action to be made within 12 months. However these additional arguments may not be persuasive and the patent may never issue, or, if issued, may be invalidated.
If the patent does not issue, we will not have the exclusive rights to the intellectual property behind VEN 307, although we will have market exclusivity in the U.S. for three years after FDA approval, if granted. After that, competitors would be able to use and sell the technology without having to pay us to do so, which would greatly diminish the value of the technology to us. In such event, our business will be materially harmed.
47
Our business depends on protecting our intellectual property.
If we and our licensors do not obtain protection for our respective intellectual property rights, our competitors might be able to take advantage of our research and development efforts to develop competing drugs.
Our success, competitive position and future revenues, if any, depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. To date, we hold some exclusive patent rights, including rights under U.S. patents and patent applications as well as rights under foreign patents and patent applications. We anticipate filing additional patent applications both in the U.S. and in other countries, as appropriate. However, the patent process is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents. These risks and uncertainties include the following:
|
·
|
Our patent rights might be challenged, invalidated, or circumvented, or otherwise might not provide any competitive advantage;
|
|
·
|
Our competitors, many of which have substantially greater resources than we do and many of which might make significant investments in competing technologies, might seek, or might already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our potential products either in the United States or in international markets;
|
|
·
|
As a matter of public policy regarding worldwide health concerns, there might be significant pressure on the U.S. government and other international governmental bodies to limit the scope of patent protection both inside and outside the U.S. for disease treatments that prove successful; and
|
|
·
|
Countries other than the U.S. might have less restrictive patent laws than those upheld by U.S. courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products.
|
In addition, the U.S. Patent and Trademark Office and patent offices in other jurisdictions have often required that patent applications concerning pharmaceutical and/or biotechnology-related inventions be limited or narrowed substantially to cover only the specific innovations exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents might be substantially narrower than anticipated.
In addition to patents, we also rely on trade secrets and proprietary know-how. Although we take measures to protect this information by entering into confidentiality and inventions agreements with our employees, scientific advisors, consultants, and collaborators, we cannot provide any assurances that these agreements will not be breached, that we will be able to protect ourselves from the harmful effects of disclosure if they are breached, or that our trade secrets will not otherwise become known or be independently discovered by competitors. If any of these events occurs, or we otherwise lose protection for our trade secrets or proprietary know-how, the value of this information may be greatly reduced.
Patent and other intellectual property protection is crucial to the success of our business and prospects, and there is a substantial risk that such protections will prove inadequate. Our business and prospects will be harmed if these protections prove insufficient.
48
We rely on trade secret protections through confidentiality agreements with our employees, customers and other parties, and the breach of these agreements could adversely affect our business and prospects.
We rely on trade secrets, which we seek to protect, in part, through confidentiality and non-disclosure agreements with our employees, collaborators, supplies, and other parties. There can be no assurance that these agreements will not be breached, that we would have adequate remedies for any such breach or that our trade secrets will not otherwise become known to or independently developed by our competitors. We might be involved from time to time in litigation to determine the enforceability, scope and validity of our proprietary rights. Any such litigation could result in substantial cost and divert management’s attention from our operations.
If we infringe the rights of third parties we might have to forgo selling our future products, pay damages, or defend against litigation.
If our product candidates, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we might have to:
|
·
|
obtain licenses, which might not be available on commercially reasonable terms, if at all;
|
|
·
|
abandon an infringing product candidate;
|
|
·
|
redesign our products or processes to avoid infringement;
|
|
·
|
stop using the subject matter claimed in the patents held by others;
|
|
·
|
pay damages; and/or
|
|
·
|
defend litigation or administrative proceedings which might be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources.
|
Any of these events could substantially harm our earnings, financial condition and operations.
Risks Related to our Common Stock
There are interlocking relationships among us and certain affiliates of Paramount BioSciences, LLC, which might present potential conflicts of interest.
Dr. Lindsay Rosenwald is the Chairman, Chief Executive Officer and sole stockholder of Paramount BioCapital, Inc., or Paramount, and is the sole member of Paramount BioSciences, LLC. We acquired the rights to VEN 307 and VEN 308 from Paramount BioSciences who had licensed them from S.L.A. Pharma. Dr. Rosenwald individually and through entities he controls and trusts established for his family beneficially owned as of February 28, 2011 approximately 13% of our issued and outstanding common stock, excluding any shares issuable upon the exercise of warrants. Dr. Rosenwald’s beneficial ownership includes shares issued upon the automatic conversion of promissory notes held by affiliates of Paramount BioSciences and Capretti Grandi, LLC and shares issuable upon the conversion of warrants held by affiliates of Paramount Credit Partners, LLC. Moreover, Dr. Rosenwald has the right to purchase additional shares of our common stock pursuant to purchase right agreements with certain employees of Paramount BioSciences or its affiliates. All share amounts and ownership percentages give effect to the 1-for-12.4 reverse stock split that we effected on November 10, 2010.
In consideration of his guaranteeing the $800,000 promissory note we issued to Israel Discount Bank of New York in September 2010, we entered into a letter agreement with Dr. Rosenwald whereby Dr. Rosenwald has the right to attend our Board meetings and to appoint two directors to our Board. Dr. Rosenwald has not exercised his right to appoint those directors. If and when appointed, these directors would be subject to stockholder approval at the expiration of their terms. This board representation, coupled with his beneficial ownership of approximately 13% of the common stock of our company, increases Dr. Rosenwald’s ability to influence our board of directors and the management of our company. Dr. Rosenwald’s rights will terminate upon the earlier to occur of (a) August 30, 2015, (b) the merger, consolidation or sale of all or substantially all of our stock or assets in a transaction or series of transactions immediately after which our stockholders as of immediately prior to the transaction hold less than 50% of the outstanding voting securities of the surviving, acquiring or parent corporation, or (c) Dr. Rosenwald’s ownership of our company is less than 5.0% of the outstanding shares of our capital stock.
49
At December 31, 2010, we had borrowed from Paramount Credit Partners, an entity whose managing member is Dr. Rosenwald, an aggregate principal amount of $1,573,000.
As of December 31, 2010, we owe Paramount Corporate Development, LLC, an affiliate of Dr. Rosenwald’s, $100,000 for services previously rendered and for which there is no due date.
Generally, Delaware corporate law, under which we are governed, requires that any transactions between us and any of our affiliates be on terms that, when taken as a whole, are substantially as favorable to us as those then reasonably obtainable from a person who is not an affiliate in an arms-length transaction. We believe that the terms of our relationships with Paramount BioSciences and its affiliates satisfy the requirement of Delaware law, but in the event that one or more parties challenges the fairness of such terms, we might have to expend substantial resources in resolving the challenge, and we can make no guarantees as to the result.
None of our affiliates, Paramount BioSciences, its affiliates or Dr. Rosenwald is obligated pursuant to any agreement or understanding with us to make any additional products or technologies available to us, nor can there be any assurance, and we do not expect and purchasers of our common stock should not expect, that any biomedical or pharmaceutical product or technology identified by such affiliates, Paramount BioSciences, its affiliates or Dr. Rosenwald in the future will be made available to us. In addition, certain of our current officers and directors or certain of any officers or directors hereafter appointed or elected might from time to time serve as officers or directors of other biopharmaceutical or biotechnology companies. There can be no assurance that such other companies will not have interests in conflict with our own.
We are controlled by our current officers, directors and principal stockholders.
As of February 28, 2011, our directors, officers, principal stockholders and their affiliates beneficially owned approximately 13.5% of our issued and outstanding capital stock, excluding any shares issuable upon the exercise of options or warrants. Accordingly, our officers, directors, principal stockholders and their affiliates control the election of our Board of Directors and the outcome of issues submitted to our stockholders, including any merger, consolidation, or sale of all or substantially all of our assets. The interests of our executive officers, directors and principal stockholders and their affiliates might not coincide with the interests of other holders of our capital stock. This concentration of ownership may harm the value of our common stock by, among other things:
|
·
|
delaying, deferring or preventing a change in control of our company;
|
|
·
|
impeding a merger, consolidation, takeover or other business combination involving our company; or
|
|
·
|
causing us to enter into transaction or agreements that are not in the best interests of all stockholders.
|
As of February 28, 2011, Dr. Lindsay Rosenwald, our largest stockholder and the sole member of Paramount BioSciences LLC, beneficially owned approximately 13% of our issued and outstanding capital stock, excluding any shares issuable upon the exercise of warrants.
In consideration of his guaranteeing the $800,000 promissory note we issued to Israel Discount Bank of New York in September 2010, we entered into a letter agreement with Dr. Rosenwald whereby Dr. Rosenwald has the right to attend our Board meetings and to appoint two directors to our Board. Dr. Rosenwald has not exercised his right to appoint a director. If and when appointed, these directors would be subject to stockholder approval at the expiration of their terms. This board representation, coupled with his beneficial ownership of approximately 13% of the common stock of our company, increases Dr. Rosenwald’s ability to influence our board of directors and the management of our company.
50
All share amounts and ownership percentages give effect to the 1-for-12.4 reverse stock split that we effected on November 10, 2010.
We might not be able to maintain the listing of our common stock on the Nasdaq Capital Market.
Our common stock is listed on the Nasdaq Capital Market under the symbol “VTUS.” We might not be able to maintain the listing standards of that exchange. If we fail to maintain the listing requirements, our common stock might trade on the OTC Bulletin Board or in the “pink sheets” maintained by Pink OTC Markets, Inc. These alternative markets are generally considered to be markets that are less efficient and less broad than the Nasdaq Capital Market.
The price of our common stock might fluctuate significantly, and you could lose all or part of your investment.
The trading price of our common stock might be volatile and subject to wide price fluctuations in response to various factors, including:
|
·
|
success or failure of our product candidates;
|
|
·
|
results of our clinical trials and other studies;
|
|
·
|
actual or anticipated fluctuations in our quarterly financial and operating results;
|
|
·
|
the overall performance of the equity markets;
|
|
·
|
changes in interest rates;
|
|
·
|
introduction of new products or announcements of significant contracts, acquisitions or capital commitments by us or our competitors;
|
|
·
|
legislative, political or regulatory developments;
|
|
·
|
issuance of new or changed securities analysts’ reports or recommendations, or the announcement of any changes to our credit rating;
|
|
·
|
additions or departures of key personnel;
|
|
·
|
availability of capital;
|
|
·
|
changes in accounting standards, policies, guidance, interpretations or principles;
|
|
·
|
threatened or actual litigation and government investigations;
|
|
·
|
future sales of our common stock;
|
|
·
|
investor perceptions of us and the pharmaceutical industry;
|
|
·
|
sale of shares of our common stock by our significant stockholders or members of our management; and
|
|
·
|
general economic conditions.
|
These and other factors might cause the market price of our common stock to fluctuate substantially, which might limit or prevent investors from readily selling their shares of our common stock and might otherwise negatively affect the liquidity of our common stock. In addition, in recent years, the stock market has experienced significant price and volume fluctuations. This volatility has had a significant impact on the market price of securities issued by many companies across many industries. The changes frequently appear to occur without regard to the operating performance of the affected companies. Accordingly, the price of our common stock could fluctuate based upon factors that have little or nothing to do with our company, and these fluctuations could materially reduce our share price.
51
We will need additional financing to fund our activities in the future, which likely will dilute our stockholders.
We anticipate that we will incur operating losses for the foreseeable future. Moreover, we believe we will require substantial funds in the future to support our operations. We expect to seek equity or debt financings in the future to fund our operations. The issuance of additional equity securities, or convertible debt or other derivative securities, likely will dilute some if not all of our then existing stockholders, depending on the financing terms.
The requirements of being a public company adds to our operating costs and might strain our resources and distract our management.
As a public company, we face increased legal, accounting, administrative and other costs and expenses not faced by private companies. We are subject to the reporting requirements of the Securities Exchange Act of 1934, which requires that we file annual, quarterly and current reports with respect to our business and financial condition, and the rules and regulations implemented by the SEC, the Sarbanes-Oxley Act of 2002, and the Nasdaq Capital Market, each of which imposes additional reporting and other obligations on public companies. These rules and regulations increase our legal and financial compliance costs and make some activities more time-consuming and costly, although we are currently unable to estimate these costs with any degree of certainty. Complying with these requirements might divert management’s attention from other business concerns, which could have a material adverse effect on our prospects, business, and financial condition.
Additionally, the expenses incurred by public companies generally for reporting and corporate governance purposes have been increasing. These increased costs will require us to divert a significant amount of money that we could otherwise use to develop our product candidates or otherwise expand our business. If we are unable to satisfy our obligations as a public company, we could be subject to delisting of our common stock, fines, sanctions and other regulatory action and potentially civil litigation.
Our internal control over financial reporting currently has material weaknesses, and failure to achieve and maintain effective internal control over financial reporting could have a material adverse effect on our business and stock price.
As a public company, we will have to maintain internal control over financial reporting in a manner that meets the standards of publicly traded companies. We anticipate being required to meet these standards in the course of preparing our financial statements as of and for the year ending December 31, 2011, and our management will be required to report on the effectiveness of our internal control over financial reporting as of December 31, 2011. The rules governing the standards that must be met for our management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation. We are in the process of reviewing, documenting and testing our internal control over financial reporting. We might encounter problems or delays in completing the implementation of any changes necessary to make a favorable assessment of our internal control over financial reporting. If we cannot favorably assess the effectiveness of our internal control over financial reporting, investors could lose confidence in our financial information and the price of our common stock could decline.
We do not intend to pay dividends for the foreseeable future and our stock may not appreciate in value.
We currently intend to retain our future earnings, if any, to finance the operation and growth of our business and do not expect to pay any cash dividends in the foreseeable future. As a result, the success of an investment in shares of our common stock will depend upon any future appreciation in its value. There is no guarantee that shares of our common stock will appreciate in value or that the price at which our stockholders have purchased their shares will be able to be maintained.
52
Shares currently eligible for sale and shares that will become eligible for sale in the near future may adversely affect the market price of our common stock, as the future market sale of a substantial amount of outstanding stock in the public marketplace could reduce the price of our common stock.
Holders of a significant number of our shares and/or their designees are eligible to sell our shares of common stock by means of ordinary brokerage transactions in the open market without any restrictions, such as limitation imposed by Rule 144, promulgated under the Securities Act. In addition, lock-up agreements that our executive officers and directors and certain of our stockholders entered into in connection with our initial public offering will expire 180 days after December 22, 2010, which will allow those shares to be sold. Any substantial sale of common stock, whether individually or in the aggregate, may have an adverse effect on the market price of our common stock.
Several provisions of the Delaware General Corporation Law and our amended and restated certificate of incorporation and bylaws could discourage, delay or prevent a merger or acquisition, which could adversely affect the market price of our common stock.
Several provisions of the Delaware General Corporation Law and our amended and restated certificate of incorporation and bylaws could discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, and the market price of our common stock could be reduced as a result. These provisions include:
|
·
|
“blank check” preferred stock;
|
|
·
|
prohibiting us from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder unless certain provisions are met;
|
|
·
|
prohibiting cumulative voting in the election of directors;
|
|
·
|
limiting the persons who may call special meetings of stockholders; and
|
|
·
|
establishing advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted on by stockholders at stockholder meetings.
|
53
Item 2. Properties
We occupy space on the 5th floor at 99 Hudson Street, New York, New York 10013. We rent this space pursuant to a lease that runs until June 2012. We believe our current facilities are suitable and adequate for our activities until such time as we hire a significant number of additional employees or consultants.
Item 3. Legal Proceedings
We are not a party to any legal proceedings and we are not aware of any claims or actions pending or threatened against us. In the future, we might from time to time become involved in litigation relating to claims arising from our ordinary course of business.
Item 4. [Removed and Reserved]
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is traded under the symbol “VTUS” and is quoted on the Nasdaq Capital Market. The following table sets forth the high and low sales prices for shares of our common stock, as reported by Nasdaq for the periods indicated.
|
2010
|
||||||||
|
High
|
Low
|
|||||||
|
First Quarter*
|
$ | - | $ | - | ||||
|
Second Quarter*
|
$ | $ | ||||||
|
Third Quarter*
|
$ | $ | ||||||
|
Fourth Quarter
|
$ | 7.71 | $ | 6.00 | ||||
* Our common stock began trading on the Nasdaq Capital Market on December 17, 2010, on a “when-issued” basis. On December 23, 2010, the first trading day after the distribution, “when-issued” trading with respect to our common stock ended and “regular way” trading began. As a result, our stock was not listed in the first three quarters of 2010 and only listed for 10 trading days in the fourth quarter of 2010.
On March 28, 2011 the closing price for the common stock as reported on the Nasdaq Capital Market was $10.42.
As of March 28, 2011 there were 193 stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers. We believe that, when our record holders and stockholders whose shares were held in nominee or street name by brokers are combined, we have an aggregate of 850 stockholders.
Equity Compensation Plans
The information required by Item 4 of Form 10-K regarding equity compensation plans is incorporated herein by reference to “Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” in this report.
Item 6. Selected Financial Data
Not applicable.
54
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operation
Business Overview
You should read the following discussion and analysis together with our financial statements and the notes to those statements included elsewhere in this report. This discussion contains forward-looking statements that involve risks and uncertainties. As a result of many factors, such as those set forth under ‘‘Business - Risk Factors’’ and elsewhere in this report, our actual results may differ materially from those anticipated in these forward-looking statements.
Unless otherwise indicated, all share amounts and prices take into account the 1-for-12.4 reverse stock split effected on November 10, 2010.
Overview
We are a pharmaceutical company that seeks to develop therapeutic products for the treatment of gastrointestinal disorders, specifically hemorrhoids, anal fissures and fecal incontinence. We have in-licensed all of the products in our current pipeline.
We have several proprietary product candidates that we have licensed that are in clinical development that address large market opportunities, including our most advanced product candidates, VEN 309 (iferanserin) and VEN 307 (diltiazem cream). VEN 309, a topical form of iferanserin which blocks a peripheral (outside the central nervous system) serotonin receptors, is being developed for the topical treatment of hemorrhoids, where it can reduce the bleeding, itchiness, and pain associated with the condition. Approximately 12.5 million people in the U.S. suffer from hemorrhoids and we are not aware of any FDA-approved prescription drugs for this condition. VEN 307 is a proprietary topical formulation of the drug diltiazem which we are developing for the treatment of anal fissures. Over 4 million people in the U.S. suffer from anal fissures and even though gastroenterology specialists will prescribe a pharmacy-prepared cream (made for each patient) of diltiazem or glyceryl trinitrate (a heart drug), to our knowledge, there are no drugs with FDA approval for this condition. Diltiazem is a drug that has been used in millions of patients orally for hypertension and angina, and our formulation, applied peri-anally, reduces the pain associated with the reduced blood supply in this disease, at a dose substantially below its usual oral dosage in hypertension and angina patients.
We have met with the FDA regarding our plans for the development of VEN 309, VEN 307 and VEN 308. We intend to initiate and conduct a Phase III clinical trial in the U.S. with VEN 309 beginning approximately mid - 2011 and initiate a long term carcinogenicity study. Depending on our assessment of the data generated by the Phase III trial, which is expected in the first quarter of 2012, as well as on other factors, including our access to capital, clinical and regulatory considerations, and our assessment of the then-current state of our intellectual property estate, we intend to initiate and conduct the second Phase III trial, which, together with the first study, other small pharmacology studies, and the carcinogenicity study (which we plan to complete after the second trial) will comprise the data needed to be able to submit a NDA to the FDA, which we anticipate could occur as early as 2014.
Our partner for VEN 307, S.L.A. Pharma, began conducting the first Phase III clinical trial with VEN 307 in Europe in November 2010 and expects to continue it in 2011. At the same time we intend to conduct a formulation program with contract manufacturers to create a new, improved formulation of topical diltiazem, with new intellectual property protections. We expect to receive the data from the first Phase III trial in the second quarter of 2012 and aim to have completed our formulation program by that time. Depending on our assessment of the data generated by this study and on whether the new formulation is superior to the existing version, as well as on other factors, including our access to capital, clinical and regulatory considerations, and our assessment of the then-current state of our intellectual property estate, we intend to initiate either one additional Phase III study in the U.S. with the existing formulation or two additional Phase III clinical trials in the U.S. with the new formulation, to be run in parallel. We anticipate that both program options could provide sufficient data for a NDA submission to the FDA in 2013.
55
Since our inception, we have had no revenue from product sales, and have funded our operations principally through debt financings. Our operations to date have been primarily limited to organizing and staffing our company, licensing our product candidates, developing clinical trials for our product candidates, establishing manufacturing for our product candidates and maintaining and improving our patent portfolio. We have generated significant losses to date, and we expect to continue to generate losses as we progress towards the commercialization of our product candidates, including VEN 307 and VEN 309. As of December 31, 2010, we had a deficit accumulated during the development stage of $33,184,354. Because we do not generate revenue from any of our product candidates, our losses will continue as we advance our product candidates towards regulatory approval and eventual commercialization. As a result, our operating losses are likely to be substantial over the next several years. We are unable to predict the extent of any future losses or when we will become profitable, if at all.
We believe that our existing cash will be sufficient to fund our projected operating requirements until into the third quarter of 2012, while we anticipate receiving data from the key clinical trials with VEN 309 in the first quarter of 2012 and VEN 307 in the second quarter of 2012. Until we can generate a sufficient amount of product revenue, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaborations and licensing arrangements.
Financial Operations Overview
Critical Accounting Policies
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or GAAP. Our significant accounting policies are more fully described in Note 1 to the financial statements included in this report. The following accounting policies are critical to fully understanding and evaluating our financial results.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires our management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent liabilities at the date of the financial statements as well as the reported revenue, if any, and expenses during the reporting periods. On an ongoing basis, our management evaluates their estimates and judgments. Management bases estimates on historical experience and on various other factors that they believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results might differ from these estimates under different assumptions or conditions.
Stock-Based Compensation
We account for stock options granted to employees, which are measured at grant date, based on the estimated fair value of the award, and are recognized as expense over the employee’s requisite service period on a straight-line basis. We account for stock options and warrants granted to non-employees on a fair value basis using the Black-Scholes option pricing model. The initial non-cash charge to operations for nonemployee options and warrants with vesting are revalued at the end of each reporting period based upon the change in the fair value of the options and recognized as consulting expense over the related service period. For the purpose of valuing options and warrants granted to employees and non-employees, we use the Black-Scholes option pricing model. To determine the risk-free interest rate, we utilize the U.S. Treasury yield curve in effect at the time of grant with a term consistent with the expected term of the awards. We estimated the expected life of the options granted based on anticipated exercises in the future periods assuming the success of our business model as currently forecasted. For warrants and non-employee options, we use the contractual term of the warrant, the length of the note or option as the expected term. The expected dividend yield reflects our current and expected future policy for dividends on our common stock. The expected stock price volatility for our stock options will be calculated by examining historical volatilities for publicly traded industry peers as we do not now and for the near future will not have any significant trading history for our common stock. Forfeiture rates will be calculated based on the expected service period for our employees.
56
Accounting for Convertible Debt and Debt Issued with Stock Purchase Warrants
The proceeds from any financing in which we issue warrants to purchase our common stock are first allocated to the warrants based upon their estimated relative fair values as of the closing date.
Warrants, or any other detachable instruments issued in connection with debt financing agreements, are accounted for using the relative fair value method and allocated to additional paid-in capital and recorded as a reduction in the carrying value of the related debt. This discount is amortized to interest expense from the issuance date through the maturity date of the debt using the straight-line method.
When the convertible feature of conventional convertible debt provides for a rate of conversion that is below market value, this feature is characterized as a beneficial conversion feature, or BCF. Prior to the determination of the BCF, the proceeds from the debt instrument are first allocated between the convertible debt and any detachable free-standing instruments that are included, such as common stock warrants. We have disclosed the contingent nature of our BCFs and have recorded them as such in our financial statements since the conversion took place in the current year.
Off-Balance Sheet Arrangements
Since our inception, we have not engaged in any off-balance sheet arrangements, including the use of structured finance, special purpose entities or variable interest entities.
Research and Development Expense
Research and development expenses consist primarily of costs associated with (i) internal costs associated with our development activities; (ii) payments we make to third party contract research organizations, contract manufacturers, and consultants; (iii) technology and intellectual property license costs; (iv) patent reimbursements. All research and development is expensed as incurred. License fees and pre-approved milestone payments due under each research and development arrangement that are paid prior to regulatory approval are expensed when the license is entered into or the milestone is achieved.
Conducting a significant amount of research and development is central to our business model. Through December 31, 2010, we incurred $14,251,561 in research and development expenses since our inception in October 2005. Product candidates in later-stage clinical development generally have higher development costs than those in earlier stages of development, primarily due to the significantly increased size and duration of the clinical trials. We plan to increase our research and development expenses for the foreseeable future in order to complete development of our two most advanced product candidates, VEN 309 and VEN 307. The following table summarizes the research and development expenses related to our two most advanced product candidates and other projects. The table reflects expenses directly attributable to each development candidate, which are tracked on a project basis.
|
YE 2009
|
YE 2010
|
Period from
October 7, 2005
(inception) to
Dec. 31, 2010
|
||||||||||
|
VEN 307
|
$ | 155,000 | $ | 1,309,501 | $ | 3,802,001 | ||||||
|
VEN 309
|
$ | 2,734,147 | $ | 379,237 | $ | 8,439,983 | ||||||
|
Other
|
$ | 53,845 | $ | 161,928 | $ | 2,009,577 | ||||||
57
The process of conducting pre-clinical studies and clinical trials necessary to obtain FDA approval is costly and time consuming. The probability of success for each product candidate and clinical trial may be affected by a variety of factors, including, among others, the quality of the product candidate’s early clinical data, investment in the program, competition, manufacturing capabilities and commercial viability. As a result of the uncertainties discussed above, the uncertainty associated with clinical trial enrollments and the risks inherent in the development process, we are unable to determine with certainty the duration and completion costs of current or future clinical stages of our product candidates or when, or to what extent, we will generate revenues from the commercialization and sale of any of our product candidates. Development timelines, probability of success and development costs vary widely. Based on their current status, we anticipate that to complete the clinical trial process and commercialize our product candidates will cost approximately $15 million for VEN 307, $20 million for VEN 308 and $20 million for VEN 309. These estimates could change significantly depending on the progress, timing and results of non-clinical and clinical trials. We will need to raise additional funds in order to fully complete the development of VEN 307 and VEN 309.
General and Administrative Expense
General and administrative expense consists primarily of salaries, consulting fees and other related costs, professional fees for legal services and accounting services, insurance and travel expenses, as well as the option expense associated with the grants of options to our employees and directors in 2010. We expect that our general and administrative expenses will increase due to our recent staffing and as we add additional personnel and comply with the reporting obligations applicable to public companies. From our inception in October 2005 through December 30, 2010, we spent $5,520,678 on general and administrative expense.
Interest Expense
Interest expense consists of interest incurred on the 5% related parties’ promissory notes from October 2005 to June 2008, the 8% related parties’ promissory notes from July 2008 to December 2010, the 10% Paramount Credit Partners notes from January 2009 to June 2010, the 8% senior convertible notes from December 2007 to December 2008, the 10% senior convertible notes from December 2008 to December 2010, the 8% 2010 senior convertible notes from February 2010 to December 2010, our letter of credit borrowings and interest due on our license fee payments. Additionally, interest expense includes the beneficial conversion feature of conventional convertible debt that was converted below market value as well as amortization of debt discount and deferred financing costs, as well as the debt discount for warrants issued in connection with debt financings.
Results of Operations
Comparison of the Years Ended December 31, 2010 and December 31, 2009
Research and Development Expense
Research and development expense was $1,850,667 for the year ended December 31, 2010, a decrease of $1,092,325, or 37%, from $2,942,992 for the year ended December 31, 2009. The primary reason for the decrease was the contractual payment of approximately $1,600,000 that was expensed by the Company in 2009. Also contributing to the increase in research and development was an increase in payments to Sam Amer and higher patent fees. The decrease was offset by the expense from the issuance of a warrant to purchase shares of our common stock issued to S.L.A. Pharma in August 2010 and additional shares of common stock issued to S.L.A. Pharma in December 2010 as a result of our initial public offering share price. These issuances were at a discount to the market price and therefore the warrants had a significant value. We expect to incur higher development costs in the future due to initiation of the Phase III clinical trial as well as product development and manufacturing costs to support the clinical study.
General and Administrative Expense
General and administrative expense was $2,915,590 for the year ended December 31, 2010, an increase of $2,518,352, or 634%, from $397,238 for the year ended December 31, 2009. The increase was primarily due to $2,298,782 of compensation expense related to options granted to our employees and directors in 2010. In addition, professional fees increased by approximately $210,000, or 98% over 2009 due to the use of consultants to oversee our operations and prepare us for being a public company.
58
Interest Expense
Interest expense was $10,530,099 for the year ended December 31, 2010, an increase of $9,409,288, or 84%, from $1,120,811 for the year ended December 31, 2009. The increase was primarily due to the $6,001,496 beneficial conversion feature associated with the conversion of the 2007 convertible notes and 2010 convertible notes in December 2010 in connection with our initial public offering as well as $2,484,927 we expensed as amortization of debt discount associated with warrants issued with the 2010 notes. Interest expense paid or payable in cash was $1,329,925 for the year ended December 31, 2010, an increase of $595,773, or 81%, from the $734,152 for the year ended December 31, 2009. In addition, we had higher interest expense in 2010 due to the issuance of the 2010 convertible notes in February, April and May of 2010 and the interest charges from one of our licensors.
Liquidity and Capital Resources
Sources of Liquidity
As a result of our significant research and development expenditures and the lack of any approved products to generate product sales revenue, we have not been profitable and have generated operating losses since we were incorporated in October 2005. We have funded our operations through December 31, 2010, and prior to our initial public offering, which closed on December 22, 2010, principally with the sale of $11,923,586 in convertible notes, $1,573,000 in non-convertible notes and $934,141 in equity financing. On December 22, 2010 we closed on our initial public offering and received net proceeds of approximately $15.7 million. In connection with the offering, the Paramount/Capretti notes, 2010 senior convertible notes and the 2007 senior convertible notes converted into an aggregate of 3,334,085 shares of common stock.
Notes Payable
On October 7, 2005, we issued a 5% promissory note payable to Paramount BioSciences, LLC, an affiliate of Lindsay A. Rosenwald, a significant stockholder of our company. This note and all accrued interest were to mature on October 7, 2008, or earlier if certain events occurred. The note was amended to extend the maturity date to October 7, 2009. On June 16, 2008, this note was voluntarily converted into shares of our common stock and a warrant to purchase shares of our common stock (together, a ‘‘unit’’) at a price of $60.39 per unit, the price of a concurrent financing. At the time of the conversion, the outstanding balance due under this note was $1,396,672 which was converted into 23,128 shares of our common stock and a warrant to purchase 4,805 shares of our common stock for which we recorded a charge of $266,243. Upon conversion, the note was automatically cancelled.
On July 12, 2007, we issued an 8% promissory note payable to Paramount BioSciences. This note and all accrued interest mature on July 12, 2010, or earlier if certain events occur. On June 16, 2008, this note was voluntarily converted into shares of our common stock and a warrant to purchase shares of our common stock at a price of $60.39 per unit, the price of a concurrent financing. At the time of the conversion, the outstanding balance due under this note was $406,562 which was converted into 6,733 shares of our common stock and a warrant to purchase 1,347 shares of our common stock for which we recorded a charge of $74,617. Upon conversion, the note was automatically cancelled.
On July 23, 2008, we issued an 8% promissory note payable to Paramount BioSciences and on April 24, 2008, we issued an 8% promissory note payable to Capretti Grandi, LLC, an entity affiliated with Lindsay A. Rosenwald. Other than the maturity date, these notes have identical terms. All amounts outstanding under these notes originally were to mature and be payable on September 10, 2010 and April 24, 2012, respectively. Pursuant to an amendment dated December 21, 2009, all unpaid principal and accrued interest on these loans immediately and automatically converted into shares of our common stock at the close of our initial public offering on December 22, 2010. At the time of conversion, the outstanding balance, including accrued interest, on these notes was $1,131,656.
59
During 2009, we issued four separate 10% promissory notes (collectively, the ‘‘PCP Notes’’) to Paramount Credit Partners, LLC, an entity whose managing member is Lindsay A. Rosenwald. Specifically, the PCP Notes consist of a note in the principal amount of $1,100,000 issued on January 23, 2009, a note in the principal amount of $100,000 issued on March 25, 2009, a note in the principal amount of $250,000 issued on June 1, 2009 and a note in the principal amount of $123,000 issued on June 24, 2009. Interest on the PCP Notes is payable quarterly, in arrears, and the principal matures on the earlier of (i) December 31, 2013 or (ii) the completion by us of a transaction, subsequent to our initial public offering, involving the sale of equity securities, sale of assets, licensing, strategic partnership or otherwise, in which we raise at least $5,000,000 in gross cash proceeds. In addition, Paramount Credit Partners received five-year warrants (‘‘PCP Warrants’’) to purchase, at an exercise price of 110% of the lowest price paid for securities in a Qualified Financing (as defined below), a number of shares of our common stock equal to 40% of the principal amount of each PCP Note purchased divided by the lowest price paid for securities in a Qualified Financing prior to the two-year anniversary of such PCP Note. As a result of our initial public offering, the PCP warrants are exercisable for an aggregate of 104,867 shares, at a per share exercise price of $6.60. As of December 31, 2010, the principal amount outstanding under these notes is $1,573,000. The PCP Notes are not convertible. We intend to pay the PCP Notes when due with the proceeds from a future financing.
We paid interest owed to Paramount Credit Partners for the first and second quarters of 2010 and the first quarter of 2009. For the second, third and fourth quarters of 2009 and the third and fourth quarters of 2010, we had insufficient funds to pay the quarterly interest amount owed to Paramount Credit Partners, and carried these as accounts payable on our balance sheet. Interest amounts for these three quarterly periods in 2009 and two quarterly periods in 2010 were paid directly by Lindsay A. Rosenwald to Paramount Credit Partners, pursuant to certain guarantee obligations owed by Dr. Rosenwald under Paramount Credit Partners’ operating agreement. In January 2011, we repaid Dr. Rosenwald for all of these quarterly payments with the proceeds from our IPO.
2007 Senior Convertible Notes
During 2007 and 2008, we issued 8% senior convertible notes in connection with a private placement in the aggregate principal amount of $5,305,000 (the ‘‘Bridge Notes’’). The Bridge Notes were originally scheduled to mature on December 20, 2008, but we exercised our option to extend the maturity date to December 20, 2009, at an increased interest rate of 10%. We subsequently solicited the consent of the noteholders to an additional extension of the maturity date of the Bridge Notes to September 10, 2010, and in September 2010, obtained the consent for an additional extension of the maturity date to December 31, 2010. The Bridge Notes, plus all accrued interest thereon, automatically converted into an aggregate of 1,642,802 shares of our common stock upon the close of our initial public offering, at a conversion price of $4.20 , which was equal to 70% of the $6.00 purchase price paid for our stock in the initial public offering.
In connection with the offering of the Bridge Notes, Paramount BioCapital and third party agents received warrants (the ‘‘Placement Warrants’’) to purchase, at an exercise price of 110% of the lowest price paid for securities in a Qualified Financing (as defined in the Placement Warrants), a number of shares of our common stock equal to 10% of the principal amount of the notes purchased, less any amount used to repay the related party notes, or amounts due to Paramount BioSciences or its affiliates or employees as finder’s fees, payments under the consulting services agreement with Paramount Corporate Development LLC, an affiliate of Dr. Rosenwald or other similar payments, divided by the lowest price paid for securities in a Qualified Financing prior to December 21, 2009. If the Qualified Financing did not occur on or before December 21, 2009, the Placement Warrants will be exercisable for a number of shares of our common stock equal to 10% of the principal amount of the Notes purchased, less any amount used to repay the related party notes, or amounts due to Paramount BioSciences or its affiliates or employees as finder’s fees, payments under the services agreement or other similar payments, divided by $12.40, at a per share exercise price of $12.40 and are exercisable for seven years. Since the Qualified Financing did not occur by such date, the Placement Warrants are now exercisable into 42,782 shares of our common stock, at a per share exercise price of $12.40.
60
2010 Senior Convertible Notes
In February, March, April and May 2010, we issued 8% senior convertible notes in connection with a private placement in the aggregate principal amount of $3,425,000. These notes originally matured on September 10, 2010, but in September 2010, we obtained the consent of the noteholders to extend the maturity date to December 31, 2010. Upon the closing of our initial public offering on December 22, 2010, the 2010 Notes plus all accrued but unpaid interest thereon converted automatically into shares of our common stock at a per share price of $4.20, which is 70% of the $6.00 price at which shares of common stock were sold in the initial public offering (the “IPO Price”). Each noteholder also holds a warrant to purchase a number of shares of our common stock equal to 50% of the principal amount of the notes purchased by it divided by the IPO Price at a per share exercise price equal to $6.60, which is 110% of the IPO Price, subject to adjustment. Each of these warrants will expire and no longer be exercisable after February 26, 2015.
On February 26, 2010, a note similar to those discussed above in the aggregate principal amount of $2,192,433 (which maturity date also was extended to December 31, 2010) and related warrant were issued to Paramount BioSciences for the cancellation of a portion of the debt outstanding under the 8% promissory note issued to Paramount BioSciences on July 23, 2008, which is not included in the $3,425,000 of aggregate principal amount of notes issued in the 2010 senior convertible note private placement. Including such converted debt, the total aggregate principal amount of 2010 senior convertible notes is $5,617,433.
Net Cash Used in Operating Activities
Net cash used in operations was $5,252,826 for the year ended December 31, 2010. The net loss for the year ended December 31, 2010 was higher than cash used in operating activities by $10,037,801. The primary reasons for the difference are the $6,001,496 we expensed in beneficial conversion feature associated with the conversion of the notes that converted on December 22, 2010 in connection with our initial public offering, the option expense of $2,298,782 related to the grant of options to our employees and directors in 2010, the expense of $915,118 related to the warrants issued in connection with the conversion of related party notes in 2010 as part of our 2010 convertible note financing, and the $389,532 expense related to the issuance of a warrant in August 2010 and common stock in December 2010 to S.L.A. Pharma, as well as amortization of deferred financing costs and debt discount and interest payable on the 2010 notes.
Net Cash Used in Investing Activities
No significant cash was used in investing activities for the years ended December 31, 2010 and 2009.
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $19,704,194 for the year ended December 31, 2010. Net cash provided by financing activities consisted primarily of proceeds of approximately $15.2 from our initial public offering that closed in December 22, 2010, proceeds of approximately $3.4 million from the issuance of the 2010 convertible notes in 2010, proceeds of approximately $2.1 million from the conversion of related party debt into 2010 convertible notes, and proceeds of approximately $1.2 million from the notes issued to Israel Discount bank of New York.
Funding Requirements
We expect to incur losses for the foreseeable future. We expect to incur increasing research and development expenses, including expenses related to our recently hired personnel and planned additional clinical trials. We expect that our general and administrative expenses will also increase as we expand our finance and administrative staff, add infrastructure, and incur additional costs related to being a public company, including directors’ and officers’ insurance, investor relations programs, and increased professional fees. Our future capital requirements will depend on a number of factors, including the timing and outcome of clinical trials and regulatory approvals, the costs involved in preparing, filing, prosecuting, maintaining, defending, and enforcing patent claims and other intellectual property rights, the acquisition of licenses to new products or compounds, the status of competitive products, the availability of financing, and our success in developing markets for our product candidates.
61
Our expected future expenditures related to product development, based on our capital position at December 31, 2010, are as follows:
|
·
|
conduct a Phase III clinical trial of iferanserin (VEN 309) in the treatment of hemorrhoids, carcinogenicity testing and developing new intellectual property: $8,000,000;
|
|
·
|
payment to S.L.A. Pharma of our licensing obligations for diltiazem cream (VEN 307) and development of an improved formulation for use in Phase III studies in the U.S. on completion of S.L.A. Pharma’s European study, payment to S.L.A. Pharma of our licensing obligations for phenylephrine gel and preparation of a Phase II clinical trial: $4,200,000 (includes the repayment of the $800,000 note issued to Israel Discount Bank); and
|
|
·
|
general and administrative expenses: approximately $2,800,000.
|
We believe that our existing cash will be sufficient to enable us to fund our operating expenses and capital expenditure requirements into the third quarter of 2012. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect, which would cause us to require additional capital earlier. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials.
We do not anticipate that we will generate product revenue for at least the next several years. In the absence of additional funding, we expect our continuing operating losses to result in increases in our cash used in operations over the next several quarters and years.
We may need to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. We do not currently have any commitments for future external funding. We may need to raise additional funds more quickly if one or more of our assumptions prove to be incorrect or if we choose to expand our product development efforts more rapidly than we presently anticipate, and we may decide to raise additional funds even before we need them if the conditions for raising capital are favorable. We may seek to sell additional equity or debt securities or obtain a bank credit facility. The sale of additional equity or debt securities, if convertible, could result in dilution to our stockholders. The incurrence of indebtedness would result in increased fixed obligations and could also result in covenants that would restrict our operations.
Additional equity or debt financing, grants, or corporate collaboration and licensing arrangements may not be available on acceptable terms, if at all. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our planned commercialization efforts or obtain funds through arrangements with collaborators or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
Material Weaknesses in Internal Control Over Financial Reporting
We have identified material weaknesses in our financial reporting process with respect to lack of accounting expertise, segregation of duties and lack of independent review over financial reporting. We have also identified numerous errors in the accounting for routine transactions and non-routine, complex transactions, including with respect to the valuation of common stock and derivative securities, the recording of debt discount and related amortization for warrants issued in connection with debt financings and calculation of deferred tax assets. The material weaknesses identified with respect to lack of accounting expertise and segregation of duties relate to the policies and procedures that:
|
·
|
pertain to the procedures to ensure that information required to be disclosed is properly gathered and reported;
|
62
|
·
|
pertain to the maintenance of records that accurately and fairly reflect our transactions and dispositions of our assets;
|
|
·
|
provide reasonable assurance that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
|
|
·
|
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
|
We took the following measures to address the material weaknesses identified by us and improve our periodic financial statement reporting process:
|
·
|
hired a permanent Chief Financial Officer to strengthen our internal staffing and technical expertise in financial accounting and reporting;
|
|
·
|
upgraded our accounting software system in the first quarter of 2011;
|
|
·
|
limited access to the accounting and information systems and related data to strengthen segregation of duties; and
|
|
·
|
implemented in the fourth quarter of 2010 procedures and controls in the financial statement close process to improve the accuracy and timeliness of the preparation of quarterly and annual financial statements.
|
There can be no assurance that we will be able to successfully implement our plans to remediate the material weaknesses in our financial reporting process. Our failure to successfully implement our plans to remediate these material weaknesses could cause us to fail to meet our reporting obligations, to produce timely and reliable financial information, and to effectively prevent fraud.
Cautionary Note Regarding Forward-Looking Statements
This report contains estimates and forward-looking statements, principally in “Business-Risk Factors,” and this “Management’s Discussion and Analysis of Financial Condition and Results of Operation.” These constitute forward-looking statements under the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Our estimates and forward-looking statements are mainly based on our current expectations and estimates of future events and trends, which affect or might affect our businesses and operations. Although we believe that these estimates and forward-looking statements are based upon reasonable assumptions, they are subject to several risks and uncertainties and are made in light of information currently available to us. Many important factors, in addition to the factors described in this report, might adversely affect our results as indicated in the forward-looking statements. Our estimates and forward-looking statements may be influenced by the following factors, among others: our ability to obtain FDA approval of our product candidates; our anticipated capital expenditures and our estimates regarding our capital requirements; our liquidity and working capital requirements; our need to obtain additional funding and our ability to obtain future funding on acceptable terms; and our ability to retain and hire necessary employees and to staff our operations appropriately.
Item 8. Financial Statements and Supplementary Data
The financial statements required to be filed pursuant to this Item 8 are appended to this report. An index of those financial statements is found on page F-1.
Not applicable.
63
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain a system of disclosure controls and procedures, as defined in Exchange Act Rule 13a-15(e),which is designed to provide reasonable assurance that information, which is required to be disclosed in our reports filed pursuant to the Securities and Exchange Act of 1934, as amended (the “Exchange Act”), is accumulated and communicated to management in a timely manner. At the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer of the effectiveness of the design and operation of our disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(b). Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective.
Changes in Internal Control Over Financial Reporting
During 2010, we took the following measures to address the material weaknesses that we have identified and improve our periodic financial statement reporting process:
|
·
|
hired a permanent Chief Financial Officer to strengthen our internal staffing and technical expertise in financial accounting and reporting;
|
|
·
|
upgraded our accounting software system in the first quarter of 2011;
|
|
·
|
limited access to the accounting and information systems and related data to strengthen segregation of duties; and
|
|
·
|
implemented in the fourth quarter of 2010 procedures and controls in the financial statement close process to improve the accuracy and timeliness of the preparation of quarterly and annual financial statements.
|
Other than the matters discussed above, there were no other significant changes in our internal control over financial reporting or in other factors that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Internal Control over Financial Reporting
This annual report does not include a report of our management’s assessment regarding internal control over financial reporting or an attestation report by our registered public accounting firm regarding internal control over financial reporting due to a transition period established by rules of the Securities and Exchange Commission for newly public companies.
Item 9B. Other Information
64
PART III
Item 10. Directors and Executive Officers of the Registrant
We have adopted a written code of ethics and business conduct that applies to our directors, executive officers and all employees. We intend to disclose any amendments to, or waivers from, our code of ethics and business conduct that are required to be publicly disclosed pursuant to rules of the SEC by filing such amendment or waiver with the SEC. This code of ethics and business conduct can be found in the corporate governance section of our website, www.ventrusbio.com .
The other information required by this Item is incorporated by reference to the information under the sections captioned “Proposal No. 1 – Election of Directors,” “Executive Compensation,” “Section 16(A) Beneficial Ownership Reporting Compliance,” and “Corporate Governance and Board Matters.”
Item 11. Executive Compensation
The information required by this Item is incorporated by reference to the information under the sections captioned “Director Compensation,” “Executive Compensation – Summary Compensation Table,” and “Executive Compensation – Outstanding Equity Awards at Fiscal Year-End 2010” contained in the proxy statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Equity Compensation Plans
The following table sets forth the indicated information as of December 31, 2010 with respect to our equity compensation plans:
|
Plan Category
|
Number of securities
to be issued
upon exercise of
outstanding options,
warrants and rights
|
Weighted-average
exercise price of
outstanding options,
warrants and rights
|
Number of securities
remaining available for
future issuance under
equity compensation plans
|
|||||||||
|
Equity compensation plans approved by our shareholders:
|
||||||||||||
|
2007 Stock Plan
|
2,016 | $ | 6.00 | -0- | ||||||||
|
2010 Stock Plan
|
1,077,759 | $ | 6.00 | 1,389,441 | ||||||||
|
Equity compensation plans not approved by our shareholders:
|
||||||||||||
|
2008 Placement Agent Warrants
|
42,782 | $ | 12.40 | -0- | ||||||||
|
Licensor Warrants
|
13,605 | $ | 1.24 | -0- | ||||||||
|
Consultant Warrants
|
87,770 | $ | 6.22 | -0- | ||||||||
|
2010 Placement Agent Warrants
|
89,000 | $ | 7.50 | -0- | ||||||||
|
Underwriter Warrants
|
197,200 | $ | 7.50 | -0- | ||||||||
|
Total
|
1,510,132 | $ | 6.44 | 1,389,441 | ||||||||
Our equity compensation plan consists of the 2007 Stock Plan and the 2010 Stock Plan, both of which were approved by our stockholders. Our equity compensation arrangements that have not been approved by our stockholders consist of warrants to purchase shares of our common stock issued to: Paramount BioCapital as placement agent in our 2008 common stock offering; S.L.A. Pharma to whom we issued a warrant for 13,605 shares as part of an amendment to the license agreement between us and S.L.A. Pharma for VEN 307 and VEN 308; three consultants; National Securities Corporation as placement agent in our 2010 convertible note offering; and the underwriters of our IPO.
65
The other information required by this Item is incorporated by reference to the information under the section captioned “Security Ownership of Certain Beneficial Owners and Management” contained in the proxy statement.
The information required by this Item is incorporated by reference to the information under the section captioned “Certain Relationships and Related Transactions” and “Corporate Governance and Board Matters – Independence of Directors” contained in the proxy statement.
Item 14. Principal Accounting Fees and Services
The information required by this Item is incorporated by reference to the information under the section captioned “Auditor and Audit Committee Matters” contained in the proxy statement.
(a) Exhibits. The following exhibits are filed as part of this registration statement:
|
Exhibit No.
|
Description
|
|
|
1.1
|
Form of Underwriting Agreement.(1)
|
|
|
3.1
|
Amended and Restated Certificate of Incorporation dated November 11, 2010.(2)
|
|
|
3.2
|
Amended and Restated Bylaws dated July 12, 2010.(5)
|
|
|
4.1
|
Specimen of Common Stock Certificate.(3)
|
|
|
4.2
|
Form of Convertible Promissory Note issued to investors between December 2007 and March 2008, as amended in December 14, 2009.(5)
|
|
|
4.3
|
Form of Warrant issued to investors between June and September 2008.(5)
|
|
|
4.4
|
Form of Convertible Promissory Note issued to Paramount BioSciences, LLC and Capretti Grandi, LLC in 2008 and 2009, as amended on December 21, 2009.(4)
|
|
|
4.5
|
Warrants issued to Paramount Credit Partners, LLC on January 23, March 25, June 1 and June 24, 2009.(4)
|
|
|
4.6
|
Form of Convertible Promissory Note issued to investors and Paramount BioCapital, Inc. in February, March and April 2010.(5)
|
|
|
4.7
|
Form of Convertible Promissory Note issued to investors in May 2010.(4)
|
|
|
4.8
|
Form of Warrant issued to investors in February and March, 2010.(4)
|
|
|
4.9
|
Form of Warrant issued to investors in May 2010.(4)
|
|
|
4.10
|
Form of Placement Agent Warrant issued to Paramount BioCapital, Inc. on March 11, 2008.(5)
|
|
|
4.11
|
Placement Agent Warrants issued to National Securities Corporation on February 26, March 31 and May 6, 2010, as amended October 28, 2010 and November 30, 2010.(1)
|
|
|
4.12
|
Warrant issued to S.L.A. Pharma AG on August 30, 2010.(4)
|
|
|
4.13
|
Form of underwriters warrant.(1)
|
|
|
10.1*
|
Exclusive License Agreement dated March 23, 2007 by and between S.L.A. Pharma AG, and Paramount BioSciences, LLC, as amended on July 24, 2008, November 20, 2008, June 1, 2009, December 18, 2009 and June 24, 2010 and letter agreements dated October 27, 2008, November 20, 2008 and January 22, 2009.(2)
|
|
|
10.2
|
Assignment and Assumption Agreement dated August 2, 2007, by and between Paramount BioSciences LLC and Ventrus Biosciences, Inc.(5)
|
66
|
10.3*
|
License Agreement dated March 10, 2008 by and between Sam Amer & Co., Inc. and Ventrus Biosciences, Inc., as amended on July 31, 2008, September 29, 2008, November 17, 2008, and letter agreements dated March 13, 2009, August 18, 2009, May 13, 2009 and December 15, 2009.(5)
|
|
|
10.4
|
Amended and Restated Consulting Agreement dated July 19, 2010 between Russell H. Ellison and Ventrus Biosciences, Inc.(5)
|
|
|
10.5
|
Amended and Restated Employment Agreement dated July 19, 2010 between Russell H. Ellison and Ventrus Biosciences, Inc.(5)
|
|
|
10.6
|
Amended and Restated Consulting Agreement dated July 19, 2010 between David J. Barrett and Ventrus Biosciences, Inc.(5)
|
|
|
10.7
|
2007 Stock Incentive Plan.(5)
|
|
|
10.8
|
Consulting Agreement dated March 1, 2009 between John Dietrich and Ventrus Biosciences, Inc.(4)
|
|
|
10.9
|
Consulting Agreement dated May 11, 2010 between Timothy Hofer and Ventrus Biosciences, Inc.(4)
|
|
|
10.10
|
Amendment No. 6, dated August 30, 2010, to Exclusive License Agreement between S.L.A. Pharma AG and Paramount BioSciences, LLC (assigned to Ventrus Biosciences, Inc.)(4)
|
|
|
10.11
|
Senior promissory notes issued by Ventrus Biosciences, Inc. to Paramount Credit Partners, LLC on January 23, March 25, June 1 and June 24, 2010 and Waiver Agreement and Amendment dated as of August 30, 2010.(3)
|
|
|
10.12
|
Employment Agreement dated November 11, 2010 between David J. Barrett and Ventrus Biosciences, Inc.(2)
|
|
|
10.14
|
2010 Equity Incentive Plan.(4)
|
|
|
31.1
|
Certification of the Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
31.2
|
Certification of the Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
32.1
|
Certification of the Chief Executive Officer Pursuant to 18 U.S. C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
32.2
|
Certification of the Chief Financial Officer Pursuant to 18 U.S. C. Section 1350 as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
*
|
Certain information in this exhibit has been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request.
|
|
|
(1)
|
Incorporated by reference to the exhibit filed in the Registrant’s Amendment No. 4 to Registration Statement on Form S-1 filed on December 6, 2010.
|
|
|
(2)
|
Incorporated by reference to the exhibit filed in the Registrant’s Amendment No. 3 to Registration Statement on Form S-1 filed on November 16, 2010.
|
|
|
(3)
|
Incorporated by reference to the exhibit filed in the Registrant’s Amendment No. 2 to Registration Statement on Form S-1 filed on October 29, 2010.
|
|
|
(4)
|
Incorporated by reference to the exhibit filed in the Registrant’s Amendment No. 1 to Registration Statement on Form S-1 filed on October 4, 2010.
|
|
|
(5)
|
Incorporated by reference to the exhibit filed in the Registrant’s Registration Statement on Form S-1 filed on July 20, 2010.
|
67
SIGNATURES
In accordance with Section 13 or 15(d) of the Exchange Act, the Registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| VENTRUS BIOSCIENCES, INC. | ||
|
Date: April 13, 2011
|
By:
|
/s/ Russell H. Ellison
|
| Name: Russell H. Ellison | ||
| Title: Chief Executive Officer | ||
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, this report has been signed by the following persons on behalf of the registrant in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ Russell H. Ellison
|
Chief Executive Officer
|
|||
|
Russell H. Ellison
|
(Principal Executive Officer) and Director
|
April 13, 2011
|
||
|
/s/ David J. Barrett
|
Chief Financial Officer
|
|||
|
David J. Barrett
|
(Principal Financial and Accounting Officer)
|
April 13, 2011
|
||
|
/s/ Mark Auerbach
|
||||
|
Mark Auerbach
|
Director
|
April 13, 2011
|
||
|
/s/ Joseph Felder
|
||||
|
Joseph Felder
|
Director
|
April 13, 2011
|
||
|
/s/ Myron Z. Holubiak
|
||||
|
Myron Z. Holubiak
|
Director
|
April 13, 2011
|
||
|
/s/ Thomas A. Rowland
|
||||
|
Thomas A. Rowland
|
Director
|
April 13, 2011
|
S-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Ventrus BioSciences, Inc.
We have audited the accompanying balance sheets of Ventrus BioSciences, Inc. (a development stage company) (the “Company”) as of December 31, 2010 and 2009, and the related statements of operations, stockholders’ equity (deficiency) and cash flows for each of the years then ended and for the period October 7, 2005 (inception) to December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting as of December 31, 2010. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Ventrus BioSciences, Inc. as of December 31, 2010 and 2009 and the results of its operations and cash flows for each the years then ended and for the period October 7, 2005 (inception) to December 31, 2010, in conformity with accounting principles generally accepted in the United States of America.
/s/ EisnerAmper LLP
New York, New York
April 12, 2011
F-1
Ventrus Biosciences, Inc.
(A Development Stage Company)
|
Balance Sheets
|
||||||||
|
December 31, 2010
|
December 31, 2009
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash
|
$ | 14,571,055 | $ | 81,288 | ||||
|
Other current assets
|
18,915 | 2,519 | ||||||
|
Total current assets
|
14,589,970 | 83,807 | ||||||
|
Computer equipment, net of accumulated depreciation of $27,260 and $14,734
|
- | 12,525 | ||||||
|
Deferred financing costs, net
|
26,631 | 69,922 | ||||||
|
Total assets
|
$ | 14,616,601 | $ | 166,254 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIENCY)
|
||||||||
|
Current liabilities:
|
||||||||
|
Borrowings under short-term note and line of credit
|
$ | 419,380 | $ | 320,000 | ||||
|
Accounts payable
|
312,896 | 398,055 | ||||||
|
Accrued expenses
|
||||||||
|
License fees
|
- | 2,919,423 | ||||||
|
Other
|
- | $ | 44,982 | |||||
|
2007 Senior convertible notes
|
- | 5,305,000 | ||||||
|
Interest payable – 2007 senior convertible notes
|
- | 986,838 | ||||||
|
Notes payable – Paramount BioSciences, LLC
|
- | 2,215,591 | ||||||
|
Interest payable – Paramount BioSciences, LLC
|
- | 59,719 | ||||||
|
Term Note - bank
|
800,000 | - | ||||||
|
Interest payable – related party
|
187,536 | 107,840 | ||||||
|
Total current liabilities
|
1,719,812 | 12,357,448 | ||||||
|
Notes payable - Paramount Credit Partners, LLC (net of discount of $302,327 in 2010 and $401,546 in 2009)
|
1,270,673 | 1,171,454 | ||||||
|
Total liabilities
|
2,990,485 | 13,528,902 | ||||||
|
Commitments (see Notes 5 and 7)
|
||||||||
|
Stockholders’ equity:
|
||||||||
|
Preferred stock, $.001 par value; 5,000,000 shares authorized, none issued
|
- | - | ||||||
|
Common stock, $.001 par value; 25,000,000 shares authorized; 6,746,365 and 447,347 issued and outstanding at December 31, 2010 and 2009 respectively
|
6,746 | 447 | ||||||
|
Additional paid-in capital
|
44,803,724 | 4,530,634 | ||||||
|
Deficit accumulated during the development stage
|
(33,184,354 | ) | (17,893,729 | ) | ||||
|
Total stockholders’ equity (deficiency)
|
11,626,116 | (13,362,648 | ) | |||||
|
Total liabilities and stockholders’ equity (deficiency)
|
$ | 14,616,601 | $ | 166,254 | ||||
F-2
Ventrus Biosciences, Inc.
(A Development Stage Company)
|
Statements of Operations
|
||||||||||||
|
Year Ended
December 31, 2010
|
Year Ended
December 31,
2009
|
Period from October 7,
2005 (Inception) to
December 31, 2010
|
||||||||||
|
Operating expenses:
|
||||||||||||
|
Research and development
|
$ | 1,850,667 | $ | 2,942,992 | $ | 14,251,561 | ||||||
|
General and administrative
|
2,915,590 | 397,238 | 5,520,678 | |||||||||
|
Loss from operations
|
(4,766,257 | ) | (3,340,230 | ) | (19,772,239 | ) | ||||||
|
Interest income
|
5,730 | 140 | 19,719 | |||||||||
|
Interest expense:
|
||||||||||||
|
Beneficial conversion feature
|
(6,001,496 | ) | - | (6,001,496 | ) | |||||||
|
Amortization of debt discount and warrants
|
(2,484,927 | ) | (78,504 | ) | (2,563,431 | ) | ||||||
|
Interest expense
|
(2,043,676 | ) | (1,120,811 | ) | (4,866,908 | ) | ||||||
| (10,530,099 | ) | (1,199,315 | ) | (13,431,835 | ) | |||||||
|
Net loss
|
(15,290,625 | ) | (4,539,405 | ) | $ | (33,184,354 | ) | |||||
|
Basic and diluted net loss per common share
|
$ | (24.67 | ) | $ | (10.02 | ) | ||||||
|
Weighted average common shares outstanding -basic and diluted
|
619,923 | 445,040 | ||||||||||
F-3
Ventrus Biosciences, Inc.
(A Development Stage Company)
Statement of Cash Flows
|
Year
ended December
31, 2010
|
Year
ended December
31, 2009
|
Period from
October 7, 2005
(Inception) to
December 31, 2010
|
||||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net loss
|
(15,290,625 | ) | (4,539,405 | ) | (33,184,354 | ) | ||||||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Stock-based compensation
|
2,356,087 | 123,758 | 2,957,322 | |||||||||
|
Stock issued in connection with license agreement
|
389,597 | 25,000 | 414,825 | |||||||||
|
Charge resulting from beneficial conversion feature
|
6,001,496 | - | 6,001,496 | |||||||||
|
Stock issued to vendor
|
- | 5,000 | 5,000 | |||||||||
|
Warrants issued in connection with related party note conversion
|
915,118 | - | 1,255,978 | |||||||||
|
Amortization of deferred financing costs and debt discount
|
2,327,193 | 116,952 | 3,137,052 | |||||||||
|
Non-cash research and development
|
- | - | 1,087,876 | |||||||||
|
Interest payable - 2007 Senior convertible notes
|
611,266 | 573,708 | 1,598,104 | |||||||||
|
Interest payable - 2010 Senior convertible notes
|
354,269 | - | 354,269 | |||||||||
|
Expenses paid on behalf of the Company satisfied through the issuance of notes
|
- | - | 227,910 | |||||||||
|
Interest payable - related parties
|
94,912 | 55,841 | 266,279 | |||||||||
|
Interest payable - Paramount Credit Partners, LLC
|
79,696 | 107,840 | 187,536 | |||||||||
|
Depreciation
|
12,525 | 7,511 | 27,260 | |||||||||
|
Changes in operating assets and liabilities:
|
||||||||||||
|
Prepaid research and development
|
- | 800,000 | - | |||||||||
|
Other current assets
|
(16,396 | ) | 2,649 | (18,915 | ) | |||||||
|
Accounts payable and accrued expenses
|
(3,049,564 | ) | (762,773 | ) | 312,896 | |||||||
|
Net cash used in operating activities
|
(5,214,427 | ) | (3,483,919 | ) | (15,369,467 | ) | ||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Purchase of office and computer equipment
|
- | (2,573 | ) | (27,260 | ) | |||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Net Proceeds from IPO
|
15,184,344 | - | 15,184,344 | |||||||||
|
Proceeds from 2010 Senior convertible notes
|
3,425,000 | - | 3,425,000 | |||||||||
|
Proceeds from notes payable to Paramount Credit Partners, LLC
|
- | 1,573,000 | 1,573,000 | |||||||||
|
Proceeds from notes payable to related parties
|
950,562 | 1,905,390 | 5,041,953 | |||||||||
|
Proceeds from 2007 Senior convertible notes
|
- | - | 5,305,000 | |||||||||
|
Proceeds from private placement
|
- | - | 1,146,024 | |||||||||
|
Payment for deferred financing costs
|
(755,092 | ) | (76,461 | ) | (1,431,603 | ) | ||||||
|
Proceeds from utilization of short-term note and line of credit
|
99,380 | 150,000 | 419,380 | |||||||||
|
Proceeds from term note payable
|
800,000 | - | 800,000 | |||||||||
|
Repayment of notes payable - related party
|
- | - | (1,500,000 | ) | ||||||||
|
Proceeds from receipt of subscriptions
|
- | - | 4,684 | |||||||||
|
Net cash provided by financing activities
|
19,704,194 | 3,551,929 | 29,967,782 | |||||||||
|
Net increase in cash
|
14,489,767 | 65,437 | 14,571,055 | |||||||||
|
Beginning of period
|
81,288 | 15,851 | - | |||||||||
|
End of period
|
14,571,055 | 81,288 | $ | 14,571,055 | ||||||||
|
Supplemental schedule of non-cash financing activities:
|
||||||||||||
|
Warrants issued to placement agent
|
$ | - | - | $ | 341,334 | |||||||
|
Warrants issued to investors in connection with convertible notes
|
$ | 1,166,989 | - | $ | 1,166,989 | |||||||
|
Debt discount on Paramount Credit Partners, LLC notes
|
$ | - | - | $ | 480,049 | |||||||
|
Related party notes and accrued interest converted to 2010 Senior convertible notes
|
$ | 2,192,433 | - | $ | 3,995,667 | |||||||
|
Notes and accrued interest converted to common stock
|
$ | 14,003,158 | - | $ | 14,003,158 | |||||||
|
Supplemental disclosure – cash paid for interest
|
$ | 76,899 | $ | 344,974 | $ | 408,073 | ||||||
F-4
Ventrus Biosciences, Inc.
(A Development Stage Company)
Statements of Changes in Stockholders' Equity (Deficiency)
Period from October 7, 2005 (Inception) to December 31, 2010
|
Common Stock
|
Additional Paid-
|
Deficit
Accumulated
During the
Development
|
||||||||||||||||||
|
Shares
|
Amount
|
in Capital
|
Stage
|
Total
|
||||||||||||||||
|
Issuance of common stock to founders and employees at $0.0124 per share in March and April 2007
|
368,012 | $ | 368 | $ | 4,196 | - | $ | 4,564 | ||||||||||||
|
Issuance of common stock to founders and employees at $0.0124 per share in May and June 2007
|
9,677 | 10 | 110 | - | 120 | |||||||||||||||
|
Issuance of common stock to licensor at $0.0124 per share in August 2007
|
18,401 | 18 | 210 | - | 228 | |||||||||||||||
|
Stock-based compensation for the period from January to December 2007
|
- | - | 16,655 | - | 16,655 | |||||||||||||||
|
Warrants issued in connection with senior convertible notes in 2007
|
- | - | 164,284 | - | 164,284 | |||||||||||||||
|
Net loss
|
- | - | - | (4,567,894 | ) | (4,567,894 | ) | |||||||||||||
|
Balance at December 31, 2007
|
396,090 | 396 | 185,455 | (4,567,894 | ) | (4,382,043 | ) | |||||||||||||
|
Warrants issued in connection with senior convertible notes in January, February and March 2008
|
- | - | 177,050 | - | 177,050 | |||||||||||||||
|
Issuance of common stock in financing at $60.39 per share in June and September 2008 (net of expenses of $216,567)
|
18,977 | 19 | 929,438 | - | 929,457 | |||||||||||||||
|
Conversion of related party notes and interest payable at $60.39 per share in June 2008
|
29,861 | 30 | 1,803,204 | - | 1,803,234 | |||||||||||||||
|
Warrants issued in connection with related party note conversion in June 2008
|
- | - | 340,860 | - | 340,860 | |||||||||||||||
|
Stock-based compensation for the period from January to December 2008
|
- | - | 460,822 | - | 460,822 | |||||||||||||||
|
Net loss
|
- | - | - | (8,786,430 | ) | (8,786,430 | ) | |||||||||||||
|
Balance at December 31, 2008
|
444,928 | 445 | 3,896,829 | (13,354,324 | ) | (9,457,050 | ) | |||||||||||||
|
Stock-based compensation for the period from January to December 2009
|
- | - | 123,758 | - | 123,758 | |||||||||||||||
|
Warrants issued in connection with Paramount Credit Partner LLC notes in January, March and June 2009
|
- | - | 480,049 | - | 480,049 | |||||||||||||||
|
Common Stock issued to licensor in December 2009 at $12.40 per share
|
2,016 | 2 | 24,998 | - | 25,000 | |||||||||||||||
|
Common Stock issued to vendor in December 2009 at $12.40 per share
|
403 | - | 5,000 | - | 5,000 | |||||||||||||||
|
Net loss
|
- | - | - | (4,539,405 | ) | (4,539,405 | ) | |||||||||||||
|
Balance at December 31, 2009
|
447,347 | 447 | 4,530,634 | (17,893,729 | ) | (13,362,648 | ) | |||||||||||||
|
Warrant issued to licensor in connection with amendment to the agreement in August 2010
|
- | - | 161,552 | - | 161,552 | |||||||||||||||
|
Stock based compensation for the period from January to December 2010
|
- | - | 2,194,535 | - | 2,194,535 | |||||||||||||||
|
Conversion of notes and accrued interest to common stock in December 2010
|
3,334,085 | 3,334 | 13,999,824 | - | 14,003,158 | |||||||||||||||
|
Beneficial conversion charge recorded on notes and interest converted to common stock in December 2010
|
- | - | 6,001,496 | - | 6,001,496 | |||||||||||||||
|
Common stock issued in IPO in December 2010, net of related costs
|
2,900,000 | 2,900 | 15,181,444 | - | 15,184,344 | |||||||||||||||
|
Fair value of warrants issued with Senior convertible notes in December 2010
|
- | - | 2,344,708 | - | 2,344,708 | |||||||||||||||
|
Common Stock issued to Licensor for amendment in December 2010
|
64,933 | 65 | 389,532 | - | 389,597 | |||||||||||||||
|
Net loss
|
- | - | - | (15,290,625 | ) | (15,290,625 | ) | |||||||||||||
|
Balance at December 31, 2010
|
6,746,365 | 6,746 | 44,803,725 | (33,184,354 | ) | 11,626,116 | ||||||||||||||
F-5
VENTRUS BIOSCIENCES, INC.
(A Development Stage Company)
December 31, 2010
Notes to Financial Statements
Note 1 — Organization, Business and Basis of Presentation:
Organization and business:
Ventrus BioSciences, Inc., formerly known as South Island BioSciences, Inc. (“Ventrus” or the “Company”) was incorporated in the State of Delaware on October 7, 2005 and commenced operations in April 2007. Ventrus is a specialty pharmaceutical company focused on the late-stage development and commercialization of gastrointestinal products.
On December 22, 2010, the Company issued 2,900,000 shares of its common stock in an initial public offering (the “IPO”) and raised net proceeds of $15,184,344. On January 7, 2011, the Company issued an additional 435,000 shares of its common stock to fulfill the over-allotment option that it granted to the underwriters as part of the IPO and raised net proceeds of $2,420,775. On December 22, 2010, in connection with the consummation of the IPO, the Company converted $14,003,158 of convertible notes and accrued interest by issuing an aggregate of 3,334,085 shares to holders of the convertible notes. In addition, the Company issued 64,933 shares of its common stock, valued at $389,597 to S.L.A. Pharma, AG, a Swiss corporation (“S.L.A. Pharma”) on December 22, 2010 pursuant to the terms of an amendment to a license agreement.
Basis of presentation:
The accompanying audited financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America.
The Company’s primary activities since incorporation have been organizational activities, including recruiting personnel, acquiring licenses for its pharmaceutical compound pipeline, performing business and financial planning, performing research and development, and raising funds through the issuance of debt and common stock.
The Company is in the development stage and has funded its operations primarily through the issuance of equity and debt. The Company expects to continue to expend substantial amounts for continued product research, development, and commercialization activities for the foreseeable future. Management believes the Company’s funds are sufficient to continue operations through the second quarter of 2012. Management's plans with respect to funding this development are to secure additional equity, if possible, and to secure additional strategic alliances that will provide available cash funding for operations. Continuation of the Company is dependent on its ability to obtain additional financing and, ultimately, on its ability to achieve profitable operations. There is no assurance, however, that such financing will be available or that the Company's efforts ultimately will be successful.
On November 10, 2010, the Company effected a 1-for-12.4 reverse stock split of its common stock. All share and per share information in these financial statements have been adjusted to give effect to the reverse stock split.
Note 2 — Summary of Significant Accounting Policies:
Cash:
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash. The Company maintains its cash in bank deposit and other accounts, the balances of which, at times and at December 31, 2010, exceed Federally insured limits.
F-6
Use of estimates:
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Significant estimates inherent in the preparation of the accompanying financial statements include the fair value of stock options and warrants granted to employees, consultants , directors, investors, licensors, placement agents and underwriters.
Computer equipment:
Computer equipment is stated at cost and depreciated using the straight-line method over the estimated useful life of the related assets of three years. At December 31, 2010, computer equipment was fully depreciated.
Stock based compensation:
The Company’s share-based compensation cost is measured at grant date, based on the estimated fair value of the award, and is recognized as expense over the employee’s requisite service period on a straight-line basis. The Company accounts for stock options and warrants granted to non-employees on a fair value basis using the Black-Scholes option pricing model. The initial non-cash charge to operations for non-employee options and warrants with vesting are revalued at the end of each reporting period based upon the change in the fair value of the options and recognized as consulting expense over the related vesting period.
Warrants issued with convertible notes:
For the purpose of valuing the warrants issued with convertible notes (See Notes 3, 8 and 9), the Company used the Black-Scholes option pricing model utilizing the assumptions noted in those Notes. To determine the risk-free interest rate, the Company utilized the U.S. Treasury yield curve in effect at the time of grant with a term consistent with the expected term of the Company’s awards. The Company estimated the expected life of the options granted based on anticipated exercises in the future periods assuming the success of its business model as currently forecasted. The expected dividend yield reflects the Company’s current and expected future policy for dividends on its common stock. The expected stock price volatility for the Company’s stock options was calculated by examining historical volatilities for publicly traded industry peers as the Company did not have any trading history for its common stock at the time the grants were issued. The Company will continue to analyze the expected stock price volatility and expected term assumptions as more historical data for the Company’s common stock becomes available.
In accordance with ASC Topic 470-20, “Debt with Conversion and Other Options,” warrants, or any other detachable instruments issued in connection with debt financing agreements, are accounted for using the relative fair value method and allocated to additional paid-in capital and recorded as a reduction in the carrying value of the related debt. This discount is amortized to interest expense from the issuance date through the maturity date of the debt using the effective interest method.
Beneficial Conversion Feature:
When the conversion feature of conventional convertible debt provides for a rate of conversion that is below market value, this feature is characterized as a beneficial conversion feature (“BCF”). Prior to the determination of the BCF, the proceeds from the debt instrument are first allocated between the convertible debt and any detachable free standing instruments that are included, such as common stock warrants. The Company has disclosed the contingent nature of its BCFs (See Notes 3, 8 and 9) and has recorded their effects.
F-7
Research and development:
Research and development expenses include personnel and facility-related expenses, third party contract services including clinical trial costs, manufacturing and process development costs, research costs and other consulting services. Research and development costs are expensed as incurred. In instances where the Company enters into agreements with third parties for clinical trials, manufacturing and process development, research and other consulting activities, costs are expensed as services are performed. Amounts due under such arrangements may be either fixed fee or fee for service, and may include upfront payments, monthly payments, and payments upon the completion of milestones or receipt of deliverables.
The Company’s accruals for clinical trials are based on estimates of the services received and pursuant to contracts with the respective clinical trial centers and clinical research organizations. In the normal course of business, the Company contracts with third parties to perform various clinical trial activities in the ongoing development of potential products. The financial terms of these agreements are subject to negotiation and variation from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the achievement of certain events, the successful enrollment of patients, and the completion of portions of the clinical trial or similar conditions. The objective of the Company’s accrual policy is to match the recording of expenses in its financial statements to the actual services received. As such, expense accruals related to clinical trials are recognized based on the estimate of the degree of completion of the event or events specified in the specific clinical study or trial contract.
Income taxes:
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when it is more likely than not that some or all of the deferred tax assets will not be realized.
Loss per common share:
Basic loss per common share excludes dilution and is computed by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted loss per common share reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised or converted into common stock or resulted in the issuance of common stock that then shared in the earnings of the entity unless inclusion of such shares would be anti-dilutive. Since the Company has only incurred losses, basic and diluted loss per share is the same. The number of potentially dilutive securities excluded at December 31, 2010 and 2009 was 2,093,064 and 168,885, respectively.
Fair value measurements:
The carrying value of the senior convertible notes, related party notes, and Paramount Credit Partners, LLC notes approximate fair value due to the short-term nature of these notes and the related interest rates approximate market rates.
Note 3 — Related Party Transactions:
Consulting services:
Effective April 2007, the Company began accruing monthly fees for consulting services at a rate of $25,000 per month to Paramount Corporate Development, LLC (“Paramount”), which is an affiliate of Dr. Lindsay A. Rosenwald, M.D., a significant investor in and stockholder of the Company. This agreement was terminated as of August 31, 2008. For the period from October 7, 2005 (inception) through August 31, 2008, $425,000 was incurred under this arrangement. As of December 31, 2010 and 2009, the Company had $100,000 outstanding under this arrangement, which is included in accounts payable.
F-8
Notes payable:
On October 7, 2005, the Company issued a 5% promissory note payable to Paramount BioSciences, LLC (“PBS”), an affiliate of Dr. Rosenwald, to borrow funds as needed. This note and all accrued interest were to mature on October 7, 2008, or earlier if certain events occurred. The note was amended to extend the maturity date to October 7, 2009. On June 16, 2008, this note was voluntarily converted into units consisting of one share of common stock and one warrant to purchase common stock of the Company at a price of $60.39 per unit, the price of a concurrent financing (see Note 6). At the time of the conversion, the outstanding balance due under this note was $1,396,672, which was converted into 23,128 shares of the Company’s common stock and a warrant to purchase 4,805 shares of the Company’s common stock for which the Company recorded a BCF charge of $266,243. Upon conversion, the note was automatically cancelled. Each warrant has a seven-year term and an exercise price of $66.46.
On July 12, 2007, the Company issued an 8% promissory note payable to an entity related to Dr. Rosenwald, to borrow funds as needed. This note and all accrued interest were to mature on July 12, 2010, or earlier if certain events occurred. On June 16, 2008, this note was voluntarily converted into units consisting of one share of common stock and one warrant to purchase common stock of the Company at a price of $60.39 per unit, the price of a concurrent financing. At the time of the conversion, the outstanding balance due under this note was $406,562, which was converted into 6,733 shares of the Company’s common stock and a warrant to purchase 1,346 shares of the Company’s common stock for which the Company recorded a BCF charge of $74,617. Upon conversion, the note was automatically cancelled. Each warrant has a seven-year term and an exercise price of $66.46.
The fair value of the warrants granted was based on the following assumptions:
|
Risk-free interest rate
|
3.89 | % | ||
|
Expected volatility
|
128.18 | % | ||
|
Expected life of warrants
|
7 years
|
|||
|
Expected dividend yield
|
0 | % | ||
On July 23, 2008 and April 24, 2009, the Company issued an 8% promissory note to PBS and an 8% promissory note to Capretti Grandi, LLC, another entity related to Dr. Rosenwald, to borrow funds as needed. Originally, all amounts outstanding under these notes matured and were payable on July 23, 2010 and April 24, 2012. On December 21, 2009, these notes were amended to provide that all loans (including principal and accrued interest thereon) made by PBS and Capretti Grandi, LLC after September 30, 2009 shall immediately and automatically be converted into the same equity or derivative securities as are issued in any equity or derivative equity financing consummated by the Company on or after September 30, 2009 (that does not otherwise constitute a Qualified Financing, as defined below), on the same terms and conditions that such equity securities are offered in such non-Qualified Financing. A “Qualified Financing” means the closing of an equity financing or series of related equity financings by the Company resulting in aggregate gross cash proceeds (before brokers’ fees or other transaction related expenses) of at least $10,000,000. These notes were further amended to provide that all remaining amounts outstanding under the notes will automatically convert into the Company’s equity securities issued in the Company’s next equity financing at a conversion price equal to 70% of the lowest per unit price paid for such securities in cash by investors in such Qualified Financing. As of December 31, 2009, the aggregate principal amount outstanding under these notes was $2,215,591 and the accrued interest due was $59,719. On February 1, 2010, the Company received an additional $950,000 in loan proceeds from PBS. On February 26, 2010, a portion of the PBS notes outstanding ($2,192,433) was converted into 2010 convertible notes (the “2010 Notes”) . The Company valued the beneficial conversion feature of the 2010 Notes at $939,614, which was recorded as interest expense after the Qualified Financing was completed. The Company computed the conversion feature to be $939,614 by dividing the amount of debt and interest ($2,192,433), which is convertible into common stock by the conversion rate (70%). From this amount ($3,132,047) the amount of debt and interest ($2,192,433) was subtracted to determine the amount by which the instrument if-converted exceeds the conversion amount ($939,614). On December 22, 2010, in connection with the IPO, the remaining principal amount of $1,001,153 and accrued interest of $130,533 were converted into 269,449 shares of common stock at a price of $4.20, a conversion price equal to 70% of the lowest per unit price paid for such securities in cash by investors. The Company valued the beneficial conversion feature of the remaining notes at $485,008, which was recorded as interest expense. The Company computed the conversion feature to be $485,008 by dividing the amount of debt and interest ($1,131,686), which is convertible into common stock by the conversion rate (70%). From this amount ($1,616,694) the amount of debt and interest ($1,131,686) was subtracted to determine the amount by which the instrument if-converted exceeds the conversion amount ($485,008). Upon conversion, these notes were automatically cancelled.
F-9
During 2009, the Company issued four separate 10% promissory notes (collectively, the “PCP Notes”) to Paramount Credit Partners, LLC (“PCP”), an entity whose managing member is Dr. Rosenwald. Specifically, the PCP Notes consist of a note in the principal amount of $1,100,000 issued on January 23, 2009, a note in the principal amount of $100,000 issued on March 25, 2009, a note in the principal amount of $250,000 issued on June 1, 2009 and a note in the principal amount of $123,000 issued on June 24, 2009. Interest on the PCP Notes are payable quarterly, in arrears, and the principal matures on the earlier of (i) December 31, 2013 or (ii) the completion by the Company of a transaction, subsequent to the Company’s IPO, including an equity offering, sale of assets, licensing or strategic partnership, in which the Company raises at least $5,000,000 in gross cash proceeds. In addition, PCP received five-year warrants (“PCP Warrants”) to purchase, at an exercise price of 110% of the lowest price paid for securities in a Qualified Financing, a number of shares of the Company’s common stock equal to 40% of the principal amount of each PCP Note purchased divided by the lowest price paid for securities in a Qualified Financing prior to the two-year anniversary of such PCP Note. The Company allocated proceeds of $480,049 from the sale of the PCP Notes to the warrants at the time of issuance, which are recorded as a debt discount and reduced the carrying values of the PCP Notes. Such discount is being amortized to interest expense over the term of the PCP Notes. As of December 31, 2010, the principal amount outstanding under these notes is $1,573,000. The fair value of the warrants granted was based on the following assumptions:
|
Risk-free interest rate
|
1.64% – 2.58 | % | ||
|
Expected volatility
|
104.11% – 110.89 | % | ||
|
Expected life of warrants
|
5 years
|
|||
|
Expected dividend yield
|
0 | % | ||
On December 22, 2010, in connection with the completion of the IPO, the amount and exercise price of the PCP Warrants was fixed at 104,867 shares of the Company’s common stock with an exercise price of $6.60 per share. During 2010, PCP transferred the rights to an aggregate of $1,147,000 in principal amount of the PCP Notes and an aggregate of 76,553 of PCP Warrants to various employees of affiliates of PCP, none of whom are related parties. As a result, at December 31, 2010, PCP owned only $426,000 in principal amount of the PCP Notes and only 28,314 PCP Warrants.
On December 22, 2010, in connection with the completion of the IPO and pursuant to the terms of the warrants held by the purchasers of the 2010 Notes, the above related party holders of 2010 Notes were issued 182,703 warrants with a per share exercise price of $6.60. Each of these warrants will expire and no longer be exercisable after February 26, 2015. The Company valued these warrants at $915,118 using the Black-Scholes option pricing model, and the Company expensed the entire amount as interest expense in 2010. The fair value of the warrants granted was based on the following assumptions:
F-10
|
Risk-free interest rate
|
2.30% – 2.55 | % | ||
|
Expected volatility
|
124.46% – 129.05 | % | ||
|
Expected life of warrants
|
5 years
|
|||
|
Expected dividend yield
|
0 | % | ||
Line of Credit and Term Note:
On December 3, 2008, the Company, PBS and various other private pharmaceutical companies in which Dr. Rosenwald is a significant investor and stockholder, entered into a loan agreement with Bank of America, N.A. for a line of credit of $2,000,000. PBS pledged collateral consisting of personal assets securing the Company’s and the other borrowers’ obligations to Bank of America, N.A. under the loan agreement. Interest on amounts borrowed under the line of credit was accrued and was payable on a monthly basis at an annual rate equal to the London Interbank Offered Rate (LIBOR) plus 1%. On November 10, 2009, the parties entered into Amendment No. 1 to the Loan Agreement, which extended the initial one-year term for an additional year, such that it was to mature on November 5, 2010, and reduced the aggregate amount available under the line of credit to $1,000,000. Under the loan agreement, the Company’s liability under the line of credit was several, not joint, with respect to the payment of all obligations thereunder. As of December 31, 2009, the amount borrowed by the Company under the Bank of America, N.A. line of credit was $320,000. In November 2010, the Company paid off the Bank of America, N.A. line of credit with proceeds from a promissory note issued to Israel Discount Bank.
On September 23, 2010, the Company borrowed $800,000 from Israel Discount Bank of New York (“Israel Discount Bank”). The promissory note the Company issued to Israel Discount Bank to evidence the loan is guaranteed by Dr. Rosenwald. The interest rate on the note is equal to the interest rate that Israel Discount Bank will pay on the cash accounts at Israel Discount Bank maintained by Dr. Rosenwald and pledged to secure the note, plus 1%. At December 31, 2010 the interest rate was 1.45%. The note is due on September 22, 2011. As of December 31, 2010, the amount borrowed by the Company that was outstanding under the Israel Discount Bank promissory note was $800,000. In consideration of his guaranteeing the $800,000 promissory note the Company issued to Israel Discount Bank, the Company entered into a letter agreement with Dr. Rosenwald whereby Dr. Rosenwald has the right to attend meetings of the Company’s board of directors and to appoint two directors to the board. Dr. Rosenwald has not exercised his right to appoint these directors. If and when appointed, these directors would be subject to stockholder approval at the expiration of their terms. The rights granted to Dr. Rosenwald in connection with his guarantee continue until specified termination conditions.
On November 5, 2010, the Company borrowed an additional $420,000 from Israel Discount Bank of New York. The promissory note issued to Israel Discount Bank to evidence the loan is guaranteed by Dr. Rosenwald. The interest rate on the note is equal to the interest rate that Israel Discount bank will pay on the cash accounts at Israel Discount Bank maintained by Dr. Rosenwald and pledged to secure the note, plus 1%. At December 31, 2010 the interest rate was 1.45%. The note is due on demand or on November 4, 2011. The Company used the proceeds from the note to pay off the Bank of America, N.A. line of credit.
The Company repaid the Israel Discount Bank promissory notes in full in January 2011, using proceeds received upon the exercise by the underwriters of the Company’s IPO of their over-allotment option, which closed on January 7, 2011.
F-11
Placement Agent:
In connection with the offering of the 2010 Notes and related warrants, National Securities Corporation (“National”) and the Company entered into a placement agency agreement dated January 5, 2010, as amended on January 29, 2010, and a placement agency agreement dated April 14, 2010, as amended on April 30, 2010. Pursuant to these agreements, the Company paid National cash fees of $671,592, which consisted of placement agent commissions of $561,743 and non-accountable expense reimbursements of $109,849. In addition, the Company issued National warrants to purchase an aggregate of 89,000 shares of common stock, with an exercise price of $7.50. In addition, the Company paid National’s outside counsel $32,500 for its services as placement agent counsel. Dr. Lindsay A. Rosenwald beneficially owns, indirectly, a controlling interest in the parent holding company of National.
In connection with the Company’s IPO, National, Rodman & Renshaw (“Rodman”) and the Company entered into an underwriting agreement dated December 16, 2010, pursuant to which at closing on December 22, 2010, the Company paid National and Rodman cash fees of $1,662,400, which consisted of underwriting discounts of $1,261,500 and non-accountable expense reimbursements of $261,000. In addition, the Company issued to each of National and Rodman warrants to purchase an aggregate of 98,600 shares of common stock with an exercise price of $7.50.
The Company also granted National the exclusive right until May 6, 2011 to act as lead placement agent on the next private placement of the Company’s securities, or as lead managing underwriter on the initial public offering of the Company’s securities, with the compensation being paid to National with respect to such financing to be mutually agreed to by the parties in good faith with respect to such financing. The IPO satisfied this obligation.
Note 4 — Income Taxes:
There was no current or deferred income tax provision for the years ended December 31, 2010 and 2009.
The Company’s deferred tax assets as of December 31, consist of the following:
|
2010
|
2009
|
|||||||
|
Net operating loss
|
$ | 8,255,000 | $ | 6,248,000 | ||||
|
Stock-based compensation
|
1,291,000 | 267,000 | ||||||
| Research and development credits | 245,000 | 229,000 | ||||||
|
Totals
|
9,791,000 | 6,744,000 | ||||||
|
Less: valuation allowance
|
(9,791,000 | ) | (6,744,000 | ) | ||||
|
|
$ | — | $ | — | ||||
A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. The net increase in the total valuation allowance for the years ended December 31, 2010 and 2009 was $3,047,000 and $2,215,000, respectively. The tax benefit assumed the Federal statutory tax rate of 34% and a state and local tax rate of 11% and has been fully offset by the aforementioned valuation allowance.
At December 31, 2010, the Company had potentially utilizable Federal and state net operating loss tax carryforwards of approximately $18,500,000, expiring through 2030.
An ownership change under Internal Revenue Code (“IRC”) Section 382 is likely to have occurred due to common stock issued in the IPO and debt conversions in December 2010. Due to the change in ownership provisions of the IRC, the availability of the Company’s net operating loss carry forwards may be subject to annual limitations against taxable income in future periods, which could substantially limit the eventual utilization of such carry forwards. The Company has not analyzed the historical or potential impact of its equity financings on beneficial ownership and therefore no determination has been made whether the net operating loss carry forward is subject to any IRC Section 382 limitation. To the extent there is a limitation, there would be a reduction in the deferred tax asset with an offsetting reduction in the valuation allowance.
F-12
|
2010
|
2009
|
|||||||
|
Statutory Federal tax rate
|
(34 | )% | (34 | )% | ||||
|
Statutory income tax rate (net of Federal)
|
(7 | )% | (7 | )% | ||||
|
Warrant amortization and beneficial conversion charges
|
19 | % | 9 | % | ||||
|
Effect of valuation allowance
|
22 | % | 32 | % | ||||
|
Effective tax rate
|
— | % | — | % | ||||
Management believes that the Company does not have any uncertain tax positions that will result in a material impact on the Company’s financial statements. The Company files income tax returns in the U.S. federal and applicable state jurisdictions. There are currently no federal or state income tax examinations and therefore all years are statutorally open and subject to examination. If and when applicable, the Company will recognize interest and penalties as income tax expense.
Note 5 — Commitments:
Employment agreements:
Dr. Ellison serves as the Company’s Chief Executive Officer pursuant to an employment agreement entered into in June 2010, and amended and restated in July 2010, that became effective on December 22, 2010, upon the closing of the IPO. The employment agreement provides for a base salary of $375,000 per year, a guaranteed bonus of $75,000 per year and an annual performance-based bonus of up to 50% of his base salary. The agreement also provides incentive bonuses of $250,000 and $500,000 in the event that the Company’s market capitalization exceeds specified levels, which has not yet occurred. On December 22, 2010, Dr. Ellison received a grant of options to purchase 573,599 shares of the Company’s common stock at $6.00 per share, which amount was equal to 7.5% of the Company’s fully diluted capitalization at that date. One-third of these options vested at grant and the remaining options vest in equal amounts on the first and second anniversaries of the grant. Consequently, 382,398 options were unvested at December 31, 2010. The options expire on December 22, 2020. The Company recognized $951,355 as compensation expense related to these options as of December 31, 2010.
Mr. Barrett currently serves as our Chief Financial Officer pursuant to an employment agreement entered into in June 2010, and amended and restated in July 2010, that became effective upon December 22, 2010, upon the closing of the IPO. The employment agreement provides for a base salary of $250,000 per year. The agreement also provides incentive bonuses of $250,000 and $500,000 in the event that the Company’s market capitalization exceeds specified levels, which has not yet occurred. On December 22, 2010, Mr. Barrett received a grant of options to purchase 305,920 shares of the Company’s common stock at $6.00 per share, which amount was equal to 4.0% of the Company’s fully diluted capitalization at that date. One-third of these options vested at grant and the remaining options vest in equal amounts on the first and second anniversaries of the grant. Consequently, 203,947 options were unvested at December 31, 2010. The options expire on December 22, 2020. The Company recognized $507,390 as compensation expense related to these options as of December 31, 2010.
Debt repayments:
The scheduled payments based on maturities of current and long-term debt at December 31, 2010 are as follows:
|
Year:
|
||||
|
2011
|
$ | 1,219,380 | ||
|
2012
|
— | |||
|
2013
|
1,573,000 | |||
|
Total debt outstanding
|
$ | 2,792,380 | ||
F-13
Consulting Agreements
Effective May 11, 2010, the Company entered into a consulting agreement with Timothy Hofer, pursuant to which Mr. Hofer provides the Company with consulting services focused on general business and company development. Mr. Hofer is also a former employee of PBS, a related party. This consulting agreement is for a period of one year, subject to renewal for such longer period as the Company may agree in writing with Mr. Hofer, and may be terminated by either party upon 30 days’ prior written notice.
Under the terms of the consulting agreement with Mr. Hofer and as compensation for his services thereunder, the Company granted Mr. Hofer a fully vested ten-year warrant to purchase 76,480 shares of the Company’s common stock, at an exercise price of $6.00 per share. The Company recognized $372,103 of consulting expense as of December 31, 2010 because the warrants were fully vested on that date.
Note 6 — Stockholders’ Transactions:
Common Stock Transactions:
On November 10, 2010, the Company amended and restated its certificate of incorporation which, among other things, increased the authorized shares of common stock from 25,000,000 to 50,000,000 and effected a 1-for-12.4 reverse stock split. All shares and per share amounts reflect the effects of the reverse split.
During March and April 2007, the Company issued 368,012 shares of common stock to its founders for $4,564, or $0.0124 per share.
During May and June 2007, the Company issued 9,677 shares of common stock to its employees for $120, or $0.0124 per share. During August 2007, the Company issued 18,401 shares of common stock at $0.0124 per share in accordance with the license agreement between the Company and S.L.A. Pharma (see note 7). During 2007, the Company recorded $228 of stock-based research and development expense in connection with this license.
During June through September 2008, the Company issued 18,977 shares of common stock and 3,796 warrants at $60.39 per unit (consisting of a share of common stock with 20% warrant coverage) in connection with a private placement financing at $60.39 per unit. Each warrant has a seven-year term and an exercise price of $66.46. The Company raised $929,457 of net proceeds.
During July 2008, the Company issued 29,861 shares of common stock and 6,151 warrants at $60.39 per unit (consisting of a share of common stock with 20% warrant coverage) to related parties in connection with the conversion of amounts outstanding under certain promissory notes (see Note 3). Each warrant has a seven-year term and an exercise price of $66.46.
The fair value of the warrants granted, mentioned in the two preceding paragraphs, was based on the following assumptions:
|
Risk-free interest rate
|
3.89 | % | ||
|
Expected volatility
|
128.18 | % | ||
|
Expected life of warrants
|
5 years
|
|||
|
Expected dividend yield
|
0 | % | ||
During December 2009, the Company issued 2,016 shares of common stock to S.L.A. Pharma pursuant to an amendment to the license agreement between the Company and S.L.A. Pharma, and 403 shares of common stock to a vendor, each at a value of $12.40 per share, recording an expense of $25,000 and $5,000 to research and development expense, respectively.
F-14
In connection with the Company’s IPO, all of the issued and outstanding convertible notes issued in 2007 and 2010 converted into shares of common stock pursuant to the terms of those notes. All principal and accrued interest on the 2007 and 2010 convertible notes converted at per share price of $4.20, which was 70% of the public offering price of $6.00 per share in the IPO, resulting in an aggregate of 1,642,802 shares of common stock issued upon conversion of the 2007 convertible notes and an aggregate of 1,421,834 shares of common stock issued upon conversion of the 2010 convertible notes. Also in connection with the IPO, and pursuant to their terms, the promissory notes issued to PBS and Capretti Grandi LLC, were converted at a per share price of $4.20, which was 70% of the public offering price of $6.00 per share in the IPO, resulting in an aggregate of 269,449 shares of common stock issued upon conversion of these notes.
On December 22, 2010, the Company issued 2,900,000 shares of its common stock in an IPO at $6.00 per share and received net proceeds of $15,184,344, after deduction of underwriting discounts, commissions and other expenses related to the IPO.
Pursuant to the terms of the license agreement between the Company and S.L.A Pharma, the Company was obligated to issue to S.L.A. Pharma that number of additional shares of common stock so that, when added to the 18,401 shares initially issued, the new and old shares had an estimated fair market value equal to $500,000 (based on the price per share paid in the financing). The closing of the Company’s IPO triggered this obligation. As a result, the Company issued 64,933 shares of its common stock to S.L.A. Pharma on December 22, 2010. The Company valued the stock issuance to S.L.A. Parma at $389,597 and expensed the full amount to research and development expense as of December 31, 2010.
On January 7, 2011, the Company issued 435,000 shares of its common stock to fulfill the over-allotment option that it granted to the underwriters as part of its IPO at a price of $6.00 per share and received net proceeds of $2,420,775, after deduction of underwriting discounts and commissions. Including the over-allotment shares, a total of 3,335,000 shares were sold in the IPO, resulting in gross proceeds of approximately $20 million.
Common stock options and warrants:
In 2007, the Company established a stock incentive plan (the “2007 Plan”) under which incentive stock and/or options could be granted to officers, directors, consultants and key employees of the Company for the purchase of up to 483,871 shares of the Company’s common stock. The options could have a maximum term of ten years, vest over a period to be determined by the Company’s Board of Directors and have an exercise price at or above fair market value on the date of grant.
There were no options issued under the 2007 Plan in 2008 or 2009.
On May 11, 2010, the Company granted options to purchase 2,016 shares of its common stock to a director under the 2007 Plan with an exercise price of $6.00. The Company valued these options at $9,714 and expensed the full amount on the grant date since the options were fully vested.
All outstanding options under the 2007 Plan have fully vested by the end of 2010. The Company terminated the 2007 Plan in July 2010, but the 2,016 options granted under the 2007 Plan remain outstanding.
During 2007, the Company granted 12,903 warrants to various consultants with an exercise price of $7.69 per share. Each warrant granted during 2007 vests equally over a three-year period and has a seven-year term. During 2008, 1,613 of these warrants were forfeited due to the consultant’s relationship with the Company ending prior to the vesting period. All of the warrants that remain outstanding were fully vested at December 31, 2010.
F-15
On August 30, 2010, the Company issued a warrant to purchase 13,605 shares of its common stock with an exercise price of $1.24 per share to S.L.A. Pharma (see Note 7) pursuant to an amendment to the license agreement between the Company and S.L.A. Pharma. The warrant was fully vested at issuance and the Company recognized the full amount of $161,552 of stock-based research and development expense as of December 31, 2010. The fair value of the warrants granted and the related fair value adjustments at the end of each reporting period were based on the following assumptions:
|
2007
|
2008
|
2009
|
2010
|
|||||||||||||
|
Risk-free interest rate
|
4.00 | % | 1.55% – 3.61 | % | 1.67% – 2.69 | % | 0.75 | % | ||||||||
|
Expected volatility
|
65.55 | % | 104.78% – 219.91 | % | 128.96% – 163.74 | % | 113.31 | % | ||||||||
|
Expected life of warrants (in years)
|
7 years
|
7 years
|
7 years
|
3 years
|
||||||||||||
|
Expected dividend yield
|
0 | % | 0 | % | 0 | % | 0 | % | ||||||||
In August 2010, the Company’s stockholders approved the 2010 Equity Incentive Plan (the “2010 Plan”). The 2010 Plan authorizes the Company to issue equity incentive awards in the form of shares, options or other awards based on our common stock as part of an overall compensation package to provide performance-based compensation to attract and retain qualified personnel. The 2010 Plan reserves up to 2,467,200 shares of the Company’s common stock. In November 2010, the Company granted options to non-employee directors to purchase an aggregate of 160,000 shares under the 2010 Plan. In addition, under Dr. Ellison’s and Mr. Barrett’s respective employment agreements, in connection with the closing of the Company’s IPO, the Company granted to Dr. Ellison and Mr. Barrett options to purchase shares of the Company’s common stock with an exercise price of $6.00, which was equal to the initial public offering price per share, in an amount equal to 7.5% (573,599 shares) and 4.0% (305,920 shares), respectively, of the Company’s fully diluted capitalization on that date.
In addition to the options and warrants discussed above, in connection with the Company’s financings in 2007, 2008, 2009 and 2010, the Company issued warrants to investors and/or placement agents to purchase shares of common stock as well as certain consulting warrants (See Notes 3, 8 and 9).
A summary of the Company’s warrant activity and related information is as follows:
|
Year Ended
|
Year Ended
|
|||||||||||||||
|
December 31, 2009
|
December 31, 2010
|
|||||||||||||||
|
Shares
|
Weighted Average
Exercise Price
|
Shares
|
Weighted Average
Exercise Price
|
|||||||||||||
|
Outstanding at beginning of period
|
64,018 | $ | 33.86 | 168,885 | $ | 11.67 | ||||||||||
|
Granted
|
104,867 | $ | 6.60 | 767,924 | $ | 6.84 | ||||||||||
|
Outstanding at end of year
|
168,885 | $ | 11.43 | 936,809 | $ | 7.71 | ||||||||||
|
Warrants exercisable at end of period
|
168,885 | $ | 11.43 | 936,809 | $ | 7.71 | ||||||||||
All outstanding warrants have vested and no additional expense is expected to be recorded in future years.
A summary of the Company’s option activity and related information is as follows:
|
Year Ended
December 31, 2010
|
||||||||||||
|
Shares
|
Weighted Average Exercise Price
|
Aggregate Intrinsic Value
|
||||||||||
|
Outstanding at beginning of period
|
0 | - | ||||||||||
|
Granted
|
1,156,255 | $ | 6.01 | $ | 705,406 | |||||||
|
Outstanding at end of year
|
1,156,255 | $ | 6.01 | $ | 705,406 | |||||||
|
Options exercisable at end of period
|
473,991 | $ | 6.00 | $ | 293,874 | |||||||
|
Vested or expected to vest at December 31
|
1,156,255 | |||||||||||
|
Shares available on December 31 for options that may be granted
|
1,389,441 | |||||||||||
F-16
Estimated future stock-based compensation expense relating to stock options is as follows:
|
Calendar Years Ending December 31,
|
Future
Stock Option
Compensation
Expense
|
|||
|
2011
|
$ | 1,573,110 | ||
|
2012
|
1,491,158 | |||
|
2013
|
60,680 | |||
|
Total estimated future stock-based compensation expense – stock options
|
$ | 3,124,947 | ||
The weighted average remaining contractual life of options outstanding at December 31, 2010 is approximately 18 months.
Note 7 — License Agreements:
In March 2007, pursuant to an Exclusive License Agreement, S.L.A. Pharma granted PBS a royalty-bearing license to sell, make and use diltiazem for treatment, through topical administration, of anal fissures and phenylepherine for treatment, through topical administration, of fecal incontinence in the United States, Canada and Mexico. Pursuant to the Exclusive License Agreement, PBS was obligated to form a company to develop the technologies referenced in the Exclusive License Agreement and issue a number of shares equal to 5% of such company’s outstanding common stock as of the effective date of the Exclusive License Agreement. On August 2, 2007, the Company issued 18,401 shares to S.L.A. Pharma to satisfy this obligation. In addition, the Company was obligated to issue to S.L.A. Pharma that number of additional shares of common stock so that the number of shares following specific transactions would have a fair market value equal to $500,000. On December 22, 2010, the Company issued S.L.A Pharma an additional 64,933 shares valued at $389,597 to satisfy this obligation. See Note 6.
In August 2007, pursuant to an Assignment and Assumption Agreement, PBS sold all of its rights in and arising out of the Exclusive License Agreement with S.L.A. Pharma to Ventrus for $1,087,876. The corresponding U.S. and foreign patents and applications for the two compounds have been licensed to Ventrus under the Assignment and Assumption Agreement (the technology referred to collectively as the “Compound Technology”). As consideration in part for the rights to the Compound Technology, an initial licensing fee of $250,000 was paid to S.L.A. Pharma and $50,000 for reimbursement of clinical development costs incurred by S.L.A. Pharma (these amounts were paid by PBS and were included in the consideration paid by the Company to PBS in connection with the Assignment and Assumption Agreement). In the event that the Compound Technology is commercialized, the Company is obligated to pay to S.L.A. Pharma annual royalties, based upon net sales of the product. In addition, the Company is required to make payments to S.L.A. Pharma up to an aggregate amount of $20 million upon the achievement of various milestones related to regulatory events. Should the Company make any improvements regarding the Compound Technology, the Company is required to grant S.L.A. Pharma licenses to use such improvements.
As compensation for S.L.A. Pharma’s participation in the management and the development of the technologies, Ventrus is required to make two separate payments to S.L.A. Pharma of $41,500 per month each (aggregate $83,000 per month) (“Monthly Payments”) for diltiazem and phenylephrine. Per the agreement, Ventrus’ obligation to make these monthly payments was to terminate upon a new drug application (“NDA”) filing. Pursuant to certain amendments to the Exclusive License Agreement, the Company had been accruing the monthly payments ($41,500) for phenylephrine under the Exclusive License Agreement from January 31, 2010 until September 30, 2010 the date of ending the development efforts on phenylephrine. The Company continued to pay S.L.A. Pharma the monthly payments of $41,500 per month for diltiazem and has been current in such payments. At December 31, 2010, the Company has paid all contractual payments relating to the license agreement.
F-17
Ventrus is also required to reimburse S.L.A. Pharma for clinical development costs associated with the technology development of both diltiazem and phenylephrine. Ventrus’ total payment obligation for the diltiazem project shall not exceed $4,000,000. Ventrus made $3,200,000 and $1,650,000 of payments to S.L.A. Pharma through December 31, 2010 and December 31, 2009 and expects to make payments upon completion of recruitment into the Phase III trial in Europe, of $800,000. S.L.A. Pharma has not completed the recruitment of patients into the Phase III trial and therefore Ventrus has not accrued the $800,000 expense at December 31, 2010. In addition, both Ventrus and S.L.A. Pharma have agreed to add additional services outside the scope of the agreement for $400,000. The services have not yet been provided by S.L.A. Pharma. Ventrus’ total payment obligation for the phenylephrine project shall not exceed $1,200,000. S.L.A. Pharma has provided and billed Ventrus for $600,000 of services for the phenylephrine project through December 31, 2009. S.L.A. Pharma did not provide or bill the Company for any services for the phenylephrine project in 2010 and management does not expect to be billed for any services for the phenylephrine project in the foreseeable future.
In March 2008, Ventrus entered into an exclusive worldwide license agreement with Sam Amer & Co., Inc., a California company (“Amer”), whereby Ventrus acquired certain patent rights to iferanserin (the “Technology”) for the topical treatment of any anorectal disorders. Ventrus is obligated to pay Amer (i) a monthly consulting fee of $7,500 through May 2010, (ii) a license fee of $2,050,000, (iii) late fees of $7,500 per month starting July 2009 until the successful completion of the Phase III trials (iv) interest payments totaling $595,000 and (v) additional late fees of $7,500 per month if an NDA is not submitted by September 2010. In addition, Ventrus may be required to make future milestone and royalty payments totaling up to $20 million upon the achievement of various milestones related to regulatory or commercial events. The license agreement is terminable by either party for cause and, upon 30 days notice in the event any safety, efficacy or regulatory issues prevent development or commercialization of the technology. At December 31, 2010, the Company had made all contractual payments relating to the license agreement.
In December 2009, the Company and Amer supplemented the license agreement and added an additional licensing fee of $20,000 for six months. After the fourth month, the Company and Amer agreed that the additional license would not be needed and, therefore, the Company did not pay the last two months.
Note 8 — Private Placements:
2007 Senior convertible notes:
During 2007 and 2008, the Company issued 8% senior convertible notes in connection with a private placement in the aggregate principal amount of $5,305,000 (the “Bridge Notes”). The Bridge Notes were originally scheduled to mature on December 20, 2008, but the Company exercised its option to extend the maturity date to December 20, 2009, at an increased interest rate of 10%. The Company subsequently obtained the consent of the noteholders to an additional extension of the maturity date of the Bridge Notes to September 10, 2010 and again to December 31, 2010. The Bridge Notes, plus all accrued interest thereon, would automatically convert into the same securities issued in the Company’s next Qualified Financing (as defined below), at a conversion price equal to 70% of the lowest per unit price paid for such securities in cash. The completion of the Company’s IPO triggered the automatic conversion of the Bridge Notes. Upon conversion, the Bridge Notes were automatically cancelled. The Company valued the beneficial conversion feature of the 2007 Notes at $2,957,187, which was recorded as interest expense after the Qualified Financing was completed. The Company computed the conversion feature to be $2,957,187 by dividing the amount of debt and interest ($6,899,770), which is convertible into common stock by the conversion rate (70%). From this amount ($9,856,957) the amount of debt and interest ($6,899,770) was subtracted to determine the amount by which the instrument if-converted exceeds the conversion amount ($2,957,187).
F-18
In connection with the offering of the Bridge Notes, Paramount Biocapital, Inc. (“PCI”) and the Company entered into a placement agency agreement dated October 9, 2007, pursuant to which the Company paid PCI and third party agents cash commissions of $243,600 and $19,250, respectively, for its services. The Company agreed to additional services by PCI during the 18-month period subsequent to March 11, 2008 which expired without any further amounts being paid. PCI is a related party to the Company since it is wholly-owned by Dr. Rosenwald.
In addition, PCI and third party agents received seven-year warrants (the “Placement Warrants”). The amount of shares and the exercise price were to be determined based on whether a qualified financing occurred on or before December 21, 2009. The qualified financing did not occur by such date and as a result the number of shares subject to the Placement Warrants is 42,782 shares, an amount equal to 10% of the principal amount of the Bridge Notes purchased, divided by $12.40, with an exercise price equal to $12.40.PCI subsequently transferred the Placement Warrants among various of its employees. The Company estimated the value of the Placement Warrants using the Black-Scholes option pricing model at approximately $341,000 and recorded them as deferred financing costs, which were amortized to interest expense over the term of the Bridge Notes. The fair value of the Placement Warrants granted was based on the following assumptions:
|
Risk-free interest rate
|
3.01% – 3.84 | % | ||
|
Expected volatility
|
63.69% – 123.73 | % | ||
|
Expected life of warrants
|
7 years
|
|||
|
Expected dividend yield
|
0 | % | ||
Note 9 — 2010 Senior convertible notes:
In February, March, April and May 2010, the Company issued 8% senior convertible notes in connection with a private placement in the aggregate principal amount of $3,425,000 (the “2010 Notes”). The 2010 Notes mature on September 10, 2010. Upon the closing of a Qualified Financing (as defined below), the 2010 Notes plus any accrued but unpaid interest thereon would convert automatically into shares of the Company’s common stock at 70% of the price at which shares of common stock are sold in the Qualified Financing (the “IPO Price”). The completion of the Company’s IPO triggered the automatic conversion of the 2010 Notes. Upon conversion, the 2010 Notes were automatically cancelled. The Company valued the beneficial conversion feature of the 2010 Notes at $1,619,687, which was recorded as interest expense after the Qualified Financing was completed. The Company computed the conversion feature to be $1,619,687 by dividing the amount of debt and interest ($3,779,269), which is convertible into common stock by the conversion rate (70%). From this amount ($5,398,956) the amount of debt and interest ($3,779,269) was subtracted to determine the amount by which the instrument if-converted exceeds the conversion amount ($1,619,687).
Each 2010 Noteholder holds a warrant to purchase that number of shares of the Company’s common stock equal to 50% of the principal amount of the 2010 Notes purchased by it divided by the IPO Price at a per share exercise price equal to 110% of the IPO Price, subject to adjustment. Each of these warrants will expire and no longer be exercisable after February 26, 2015. In connection with the Company’s IPO, the number of shares of common stock issuable pursuant to these warrants is an aggregate of 285,417 shares with an exercise price of $6.60 per share. The Company valued these warrants at $1,429,590 using the Black-Scholes option pricing model and has expensed such amount as of December 31, 2010. The fair value of the warrants granted was based on the following assumptions:
F-19
|
Risk-free interest rate
|
2.02 | % | ||
|
Expected volatility
|
124 | % | ||
|
Expected life of warrants
|
5 years
|
|||
|
Expected dividend yield
|
0 | % | ||
On February 26, 2010, a 2010 Note in the aggregate principal amount of $2,192,433 and related warrant were issued to PBS for the cancellation of certain debt (as discussed in Note 3 above), which is not included in the $3,425,000 of aggregate principal amount of 2010 Notes issued in the private placement. Including such converted debt, the total aggregate principal amount of 2010 Notes was $5,617,433. In connection with the Company’s IPO, these 2010 Notes converted into an aggregate of 1,421,834 shares of common stock. Upon conversion, these 2010 Notes were automatically cancelled.
Note 10 — Subsequent Events:
Common Stock Options:
In January and February 2011, the Company granted options to purchase 250,000 shares to one of its directors, options to purchase an aggregate of 286,800 shares to three employees and options to purchase an aggregate of 364,240 shares to five consultants, all pursuant to the 2010 Plan with exercise prices at or greater than the then market value of our common stock ($6.24 - $14.63).
F-20