Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) | |
| OF THE SECURITIES EXCHANGE ACT OF 1934 | ||
| For the fiscal year ended December 31, 2010 |
or
| ¨ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) | |
| OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission file number 0-19825
SciClone Pharmaceuticals, Inc.
(Exact name of Registrant as specified in its charter)
| Delaware | 94-3116852 | |
| (State or other jurisdiction of Incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 950 Tower Lane, Suite 900 Foster City, California |
94404 | |
| (Address of principal executive offices) | (Zip Code) | |
(650) 358-3456
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Common Stock, $0.001 par value | The NASDAQ Global Market of the NASDAQ Stock Market Inc. | |
| (Title of Class) | (Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ¨ No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act (Check one):
Large accelerated filer ¨ Accelerated filer x Non-accelerated filer ¨ Smaller Reporting Company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting stock held by non-affiliates of SciClone Pharmaceuticals, Inc. was approximately $126,519,000 as of June 30, 2010, based upon the closing price of SciClone Pharmaceuticals Inc.’s Common Stock on The NASDAQ Global Market of the NASDAQ Stock Market Inc. on such date. Shares of Common Stock held by each executive officer and director have been excluded from the calculation because such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of March 29, 2011, there were 48,040,125 shares of the Registrant’s Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Part III incorporates by reference from the definitive proxy statement for the Company’s 2011 Annual Meeting of Stockholders to be filed with the Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K (the “Proxy Statement”).
Table of Contents
As used in this Annual Report, the terms “we,” “us,” “our,” the “Company” and “SciClone” mean SciClone Pharmaceuticals, Inc. and its subsidiaries (unless the context indicates a different meaning). SciClone, the SciClone logo and ZADAXIN are registered U.S. trademarks and SCV-07 is a trademark of SciClone Pharmaceuticals, Inc. All other Company names and trademarks included in this Annual Report are trademarks, registered trademarks or trade names of their respective owners.
2
Table of Contents
NOTE REGARDING FORWARD-LOOKING STATEMENTS:
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. These forward-looking statements are based on our current expectations, estimates and projections about our business, industry, management’s beliefs and certain assumptions made by us. Words such as “anticipate,” “expect,” “intend,” “plan,” “believe” or similar expressions are intended to identify forward-looking statements including those statements we make regarding our future financial results; anticipated product sales; the sufficiency of our resources to complete clinical trials and other new product development initiatives; the timing and outcome of clinical trials; the timing of completion of therapy and observation of our clinical trials; ZADAXIN®’s ability to complement existing therapies; prospects for ZADAXIN and our plans for its enhancement and commercialization; future size of the worldwide hepatitis B virus (“HBV”) and hepatitis C virus (“HCV”) and other markets; research and development and other expense levels, the ability of our suppliers to continue financially viable production of our products; cash and other asset levels; and the allocation of financial resources to certain trials and programs. These statements are not guarantees of future performance and are subject to certain risks, uncertainties and assumptions that are difficult to predict. Therefore, our actual results could differ materially and adversely from those expressed in any forward-looking statements as a result of various factors including, but not limited to, those described under the caption “Risk Factors” in this Annual Report on Form 10-K. We undertake no obligation to revise or update publicly any forward-looking statements for any reason.
PART I
| Item 1. | Business |
OVERVIEW
SciClone Pharmaceuticals (NASDAQ: SCLN) is a revenue-generating, China-centric, specialty pharmaceutical company with a substantial international business and a product portfolio of novel therapies for cancer and infectious diseases. We are focused on continuing international sales growth, while containing clinical development and other costs.
Our international business and corporate strategy is focused primarily on the People’s Republic of China (“China”) where we believe we have built a solid reputation and established a strong brand through our many years of experience marketing ZADAXIN. We believe these strengths position us to benefit from expanding pharmaceutical markets in China and elsewhere. We believe China will rank second among global pharmaceutical markets by 2015, with projected annual growth rates of more than 20% annually over the next several years. We seek to grow our sales in this region while we leverage our strong balance sheet for mergers and acquisitions (“M&A”), in-licensing and product acquisitions.
ZADAXIN, our brand of thymalfasin or thymosin alpha 1, has regulatory approval in over 30 countries for the treatment of HBV, as a vaccine adjuvant, for the treatment of HCV, and certain cancers. All of our revenues are derived from sales of ZADAXIN, substantially most of which is sold to China. To continue to grow ZADAXIN sales to China, our sales force of over 200 people is focused on increasing sales to the country’s largest hospitals (class 3 with over 500 beds) as well as smaller hospitals (class 2). These hospitals serve Tier 1 and Tier 2 cities which are the largest, and generally have the most affluent populations. In China, they are mainly located in the eastern portion of the nation.
We continue to look for M&A or in-licensing opportunities of late-stage or approved branded, well-differentiated products. To date, we have in-licensed two products that are part of this international commercial growth strategy, DC Bead® and ondansetron RapidFilm®. We are working towards regulatory approval in China for these in-licensed candidates.
DC Bead, an in-licensed product for which we have commercialization rights in China, is a novel treatment for advanced liver cancer which is currently approved in approximately 40 countries worldwide, including
3
Table of Contents
countries in the European Union (“EU”) and the U.S. We commenced a small local registration trial in 2010 which has completed enrollment of approximately 40 advanced liver cancer patients at several liver cancer treatment centers. The primary objective is safety and the secondary objective is efficacy, as measured by tumor response. We believe we may receive regulatory approval for DC Bead in late 2011.
Our second in-licensed product as part of this international commercial growth strategy is ondansetron RapidFilm. This product is an oral thin film formulation of ondansetron, a serotonin 5-HT3 receptor antagonist commonly used to treat and prevent nausea and vomiting caused by chemotherapy, radiotherapy and surgery. In March 2010, we announced that approval was granted for ondansetron RapidFilmTM under the EU decentralized procedure in 16 major EU countries. We have commercialization rights for this product in China including Hong Kong and Macau, and Vietnam, and we are working towards filing a clinical trial application in 2011 as part of the product registration process which will begin the regulatory approval process in China.
Outside of China, SciClone’s clinical development strategy is focused on driving cost-efficient phase 1 and 2 development of promising compounds while seeking development partners for more costly phase 3 trials. We are currently developing SCV-07, a small molecule synthetic peptide with immunomodulating properties, in a phase 2b clinical trial for the prevention of oral mucositis (“OM”).
OM is a common, painful, debilitating complication of cancer treatment, and we estimate that medical costs for the treatment of oral mucositis were approximately $4.2 billion in the U.S. and $10 billion worldwide in 2010. Approximately 450,000 patients per year in the U.S. suffer from OM during cancer therapy. In May 2010, we announced top-line results from our phase 2a proof of concept clinical trial of SCV-07 to modify the course of oral mucositis in patients with head and neck cancer receiving chemoradiotherapy regimens.
Based on the findings from the phase 2a study and discussions with the U.S. Food and Drug Administration, we announced the enrollment of the first patient in the Company’s phase 2b clinical trial of SCV-07 for the prevention of OM in January of 2011. The 2b study is examining three doses of SCV-07, including two higher doses than those used in the Company’s recent phase 2a study, to assess the drug’s impact on modifying the course of OM in patients with head and neck cancer.
The United States Securities and Exchange Commission (“SEC”) and the United States Department of Justice (“DOJ”) are each conducting formal investigations of us regarding a range of matters including the possibility of violations of the Foreign Corrupt Practices Act (“FCPA”). In response to these matters, our board of directors has appointed a special committee of independent directors to oversee the Company’s response to the government inquiries. Based on an initial review, the special committee has decided to undertake an independent investigation as to matters reflected in and arising from the SEC and DOJ investigations including, but not limited to, certain sales and marketing matters in China, in order to evaluate whether any violation of the FCPA or other laws occurred. The special committee has not concluded its investigation or reached any conclusions, to date, nor has the special committee or management made a final determination regarding whether any violation of the FCPA or any other law or regulation has occurred. However, we anticipate that the special committee will finalize its findings in the near future, which may include findings regarding potential FCPA issues and recommendations regarding additional remediation measures and possible suggestions regarding personnel issues. In the course of its review, the special committee has instructed management to (i) evaluate and to expand the Company’s training of employees regarding understanding and compliance with laws including the FCPA and other anti-bribery laws and regulations, (ii) evaluate our existing compliance and anti-bribery guidelines and to prepare a new, more detailed, guideline for implementation after review by our Board and/or committees of the Board, and (iii) implement a pre-approval policy for certain expenses including payments for, or reimbursement of, third-party gifts, travel expenses, honoraria and sponsorships of certain third party events.
We have consistently maintained a policy that our employees comply with all applicable laws. However, in the course of evaluating our existing training, policies and internal controls over financial reporting and expense approval and expense reimbursement practices, and of the matters brought to the attention of management by the special committee, we determined that our guidelines, employee training and expense approval practices and our
4
Table of Contents
internal controls with respect to certain matters, including the payment for, or reimbursement of, third party gifts, travel expenses, honoraria and sponsorship of certain third party events required improvements in order to provide reasonable assurance that our policy on compliance with applicable laws was effective. In addition, management determined that we had a material weakness in our internal control over financial reporting as of December 31, 2010 relating to our implementation of our policy on compliance with laws. During the first quarter of fiscal 2011, we initiated or commenced various remediation efforts to provide reasonable assurance of our implementation of our policy on compliance with laws, including the FCPA and other anti-bribery laws. See “Management’s Annual Report on, and Changes in, Internal Control over Financial Reporting” for further information regarding this material weakness and our remediation efforts.
Since our special committee has not reached any conclusions and has not reported its findings to the Board, none of the disclosures in this Form 10-K are intended to, or do, represent the special committee’s finding or conclusions. We anticipate that the special committee will conclude its investigation soon. We anticipate that the special committee may make more specific or different findings in addition to the disclosures herein. We also anticipate that the special committee may recommend additional remediation measures to those we have already taken or commenced.
Following our announcement of the SEC and DOJ investigations, various shareholder litigations were filed and further litigation may be filed. We cannot predict the outcome of these matters, but we have incurred and anticipate that we will continue to incur substantial expenses in the course of the investigations, and in connection with current or future litigation. If it is determined that sales or marketing activities have occurred that violate, or may have violated, the FCPA or other laws, our future operating results could be adversely affected. In addition, we could incur additional expenses related to fines, or to remediative measures including changes in our compliance programs, internal controls or disclosure controls.
SciClone Pharmaceuticals, Inc. was organized in 1990 as a California corporation and reincorporated in Delaware in 2003. Our corporate headquarters are located in Foster City, California. For information about our revenues from external customers, measures of our profit and loss, our total assets and other financial matters, you should read our Consolidated Financial Statements provided in Part II, Item 8 of this Form 10-K.
BUILDING A LEADING INTERNATIONAL PHARMACEUTICAL BUSINESS
Our Established Business in China
In China, we have an established business with growing product revenue and positive cash flow. We are committed to building on this base and introducing additional pharmaceutical products to meet the country’s evolving healthcare needs. China state leaders have agreed about a new health care reform plan which, among other things, is seeking to expand patient access to pharmaceuticals. We believe the China pharmaceutical market may grow more than 20% annually over the next several years.
We launched ZADAXIN in China in 1996 and by 2010 our worldwide sales of this product reached more than $85 million, 96% of which was sales to China. Today, ZADAXIN is one of the largest imported pharmaceutical products in China, measured by revenue. Our volume market share of thymalfasin is approximately 5%. Over the last decade, SciClone China has established a sales and marketing organization and extensive importation and distribution channels which have resulted in strong growth in sales and substantial cash flow. Through our sales organization of more than 200 medical representatives, we believe we have developed a good reputation and relationships with physicians and administrators in over 500 hospitals in the major cities in China. We have built a strong commercial presence in liver disease, cancer and the intensive care setting. We are expanding geographically in China to position the Company for further growth. ZADAXIN has strong brand recognition and is positioned as a high-quality, imported, premium-priced product. ZADAXIN is approved in China for the treatment of HBV and for use as a vaccine adjuvant. In China, orders for ZADAXIN
5
Table of Contents
are filled largely by distributors and sub distributors who purchase ZADAXIN from our selected, established, government-licensed importing agents.
China accounted for approximately 96%, 96%, and 94%, respectively, of our product sales for the years ended December 31, 2010, 2009 and 2008. In 2010, Shanghai Lingyun Pharmaceutical Company Ltd. and Guangdong South Pharmaceutical Foreign Trade Company Ltd. accounted for 74% and 14% of our product sales, respectively. In 2009, Shanghai Lingyun Pharmaceutical Company Ltd. and China National Pharmaceutical Foreign Trade Corporation accounted for 66% and 27% of our product sales, respectively. In 2008, Shanghai Lingyun Pharmaceutical Company Ltd. and China National Pharmaceutical Foreign Trade Corporation accounted for 68% and 22% of our product sales, respectively. No other customers accounted for more than 10% of product sales in those periods. In recent months, Sinopharm Group Co. Limited acquired a majority interest in two of our large importers, Shanghai Lingyun Pharmaceutical Company Ltd. and Guangdong South Pharmaceutical Foreign Trade Company Ltd. We do not believe these acquisitions will impact our sales. As of December 31, 2010, approximately $30.1 million, or 98% of our accounts receivable were attributable to two customers in China.
The Chinese government is increasing its efforts to reduce overall health care costs, including pricing controls on pharmaceutical products. Individual provinces in China and, in some cases, individual hospitals can and have established pricing requirements for a product to be included on formulary lists. In some cases, these prices have been significantly lower than our distributors have been selling ZADAXIN, in which case we have been removed from formulary lists, which consequently has reduced sales to certain hospitals and could adversely affect our future sales. The process and timing for any price restrictions is unpredictable. In addition, we are aware that ZADAXIN may be used on an off-label basis, and the Chinese government’s pricing, reimbursement or other actions might reduce such uses. We are working on these regulatory processes as well as on potential changes in our business model depending on potential outcomes. We believe we will be able to successfully manage our business in China through this process, however if a substantial reduction in the sale price to hospitals occurred, our gross margins would be substantially reduced.
International Sales and Marketing
ZADAXIN is approved in over 30 countries, primarily in China, Pacific Rim, Latin America, Eastern Europe, and the Middle East and where our distributors are largely responsible for the marketing and sales of the product. ZADAXIN’s approvals are principally for the treatment of HBV and as a vaccine adjuvant, with additional approvals in certain countries for the treatment of HCV, or as a chemotherapy adjuvant for cancer patients with weakened immune systems. We sell ZADAXIN in various international markets through our wholly owned subsidiary, SciClone Pharmaceuticals International Ltd. (“SPIL”).
SPIL is registered in the Cayman Islands and has its principal office in Hong Kong. SPIL orders ZADAXIN from our European manufacturer and contracts with a third party for the storage of our finished goods inventory at warehousing facilities in Hong Kong. SPIL then distributes our product worldwide from these warehousing facilities based on purchase orders from our customers. Under our established distribution arrangements, local importers and distributors are responsible for the importation, inventory, distribution and invoicing of ZADAXIN after importation.
Product sales were $85.1 million, $72.4 million, and $54.1 million for the years ended December 31, 2010, 2009 and 2008, respectively, and all were from sales of ZADAXIN.
Broadening SciClone’s International Product Portfolio
Currently, we are evaluating opportunities to acquire or in-license the marketing rights to other proprietary products in China that can be effectively and efficiently marketed to physicians by our team of medical representatives. To maintain maximum efficiency in our existing operations, we plan to continue to focus on
6
Table of Contents
product opportunities in the areas of oncology, including supportive care, infectious disease and intensive care medicine; the areas that we have an established sales presence.
We acquired the exclusive China marketing rights to DC Bead, an embolizing and chemotherapy releasing device for liver cancer, for which we filed a regulatory application with the SFDA. We plan to obtain regulatory approval for this product in late 2011. DC Bead is currently approved in Europe for use in the treatment of malignant hypervascularized tumors such as hepatocellular carcinoma (“HCC”), or primary liver cancer. China accounts for approximately one-half of the world’s liver cancer cases and more than 300,000 people die annually from this disease in China, making this an important unmet medical need.
In addition, in July 2009, we entered into a licensing agreement with APR Applied Pharma Research S.A. (“APR”) for the commercialization rights of APR’s anti-nausea drug ondansetron RapidFilm in China including Hong Kong and Macau, and Vietnam. Ondansetron RapidFilm is an oral thin film formulation of ondansetron, a serotonin 5-HT3 receptor antagonist commonly used to treat and prevent nausea and vomiting caused by chemotherapy, radiotherapy or surgery. We plan to file a clinical trial application as part of the product registration process in 2011. We have ten years of marketing and sales exclusivity from APR for China including Hong Kong and Macau, and Vietnam following regulatory approval.
DEVELOPING PRODUCTS FOR THE U.S. AND EUROPEAN MARKETS
SCV-07 Indications
Our proprietary drug candidate SCV-07 (gamma-D-glutamyl-L-tryptophan) is a small molecule which stimulates the immune system, possibly through inhibition of STAT3 signaling and the resulting effects on T-helper 1 cells. SCV-07 has been shown to be efficacious in animal models of immune-sensitive diseases, including viral infections and cancers, and in the enhancement of response to vaccines. SCV-07 has been shown to be safe and well tolerated in clinical trials at varied single and multi-dose levels and is orally bioavailable, differentiating it from other immunomodulators.
Phase 2b Oral Mucositis Trial Protocol
Oral mucositis (“OM”) is a common painful, debilitating, and costly toxicity caused by chemoradiotherapy regimens used to treat cancer.
Based on the findings from our phase 2a OM study and discussions with the U.S. Food and Drug Administration, we announced the enrollment of the first patient in the Company’s phase 2b clinical trial of SCV-07 for the prevention of OM in January of 2011. The 2b study is examining three doses of SCV-07, including two higher doses than those used in the Company’s recent phase 2a study, to assess the drug’s impact on modifying the course of OM in patients with head and neck cancer. The multicenter, randomized, double-blind, placebo-controlled study is designed to enroll approximately 160 patients who are receiving standard chemoradiation therapy for treatment of cancers of the head and neck. Patients are being randomly assigned to one of the trial’s four treatment arms: SCV-07 at doses of 0.1 mg/kg, 0.3 mg/kg or 1 mg/kg, or placebo. The study’s primary efficacy endpoint is the reduction in proportion of patients with clinically assessed ulcerative OM (WHO Grade ³ 2) at the time that they have received a cumulative radiation dose of 45 Gy. The study’s secondary endpoints include, incidence and duration of ulcerative and severe (WHO Grade ³ 3) OM, analgesic use and pain assessments, quality of life measurements, gastrostomy tube placement and use, breaks in radiation or chemotherapy treatment, and unscheduled office or hospital visits. The trial is further evaluating SCV-07’s safety and tolerability in the patient population, as well as the role played by specific genetic profiles on patient response to the treatment.
Phase 2a Oral Mucositis Trial Protocol
We conducted a multi-center, randomized, double-blind, placebo-controlled dose-ranging phase 2a clinical trial to assess the safety and efficacy of SCV-07 for the delay to onset and severity of severe OM in 60 subjects
7
Table of Contents
receiving chemoradiation therapy for the treatment of cancers of the head and neck. This study was conducted at 21 centers in the United States and included approximately 20 patients in each of the three treatment cohorts. Each cohort received placebo, SCV-07 at a dose of 0.02 mg/kg, or SCV-07 at a dose of 0.10 mg/kg. The treatment period was approximately seven weeks depending on the patient’s prescribed radiation plan, with a follow-up visit approximately 30 days following the last day of radiation therapy. All the patients were treated with a standard cisplatin regimen in addition to radiation therapy. The primary efficacy endpoint was delay of onset and severity of severe OM.
This phase 2a study showed a signal towards delay to onset of severe OM, the study’s primary endpoint, in the higher dose group. The incidence of severe OM after 5 weeks of chemoradiation was 30% lower in the patients in the high dose treatment arm (0.1 mg/kg; 17 patients) compared to the 20 patients who received placebo (29% vs. 42%). A post hoc data analysis of ulcerative mucositis (WHO grades 2-4), a major cause of morbidity associated with OM, was also conducted. In the patients treated with the high dose of SCV-07 the onset of any ulcerative mucositis was delayed in 24% of patients at doses of radiation up to 50Gy (after approximately 5 weeks of treatment) while 100% of patients in the placebo group suffered from ulcerative mucositis at 35Gy (after approximately 3.5 weeks of treatment). Supportive of these findings, the high dose SCV-07 group also showed improvement over placebo for exploratory endpoints, including the use of gastric tube feedings, unplanned office or emergency room visits, and breaks in planned course of radiation therapy of one week or longer.
The incidence of severe OM after 5 weeks of chemoradiation was 50% higher in the patients in the low-dose treatment arm (0.02 mg/kg; 20 patients) compared to the 20 patients who received placebo (63% vs. 42%). However, the post hoc data analysis of ulcerative mucositis showed that the onset of any ulcerative mucositis was delayed in 5% of patients treated with doses of radiation up to 50Gy compared to 100% of patients in the placebo group who suffered from ulcerative mucositis at 35Gy. Regarding exploratory endpoints, the low dose group showed similar results to the placebo group. Importantly, both doses of SCV-07 were shown to be safe and well tolerated with no drug-related serious adverse events reported in either arm.
Researchers also identified two unique gene clusters that were associated with subjects who responded to treatment in our phase 2a study. We believe that the discovery of these gene clusters may assist in providing the framework for effectively identifying those patients most likely to respond to SCV-07 in future clinical trials based on their individual genomic profile or gene signature.
Phase 2 HCV Trial Protocol
We conducted a multicenter, multidose, open-label 2b study designed to evaluate the safety and immunomodulatory effects of SCV-07 as a monotherapy or in combination with ribavirin in non–cirrhotic patients with genotype 1 chronic HCV who had relapsed after at least 44 weeks of treatment with pegylated interferon and ribavirin. The study, which monitored biomarkers of immune activation and HCV viral load dynamics, included two treatment cohorts of 20 patients each, who received SCV-07 at a dose of either 0.1 mg/kg or 1.0 mg/kg. The treatment period was approximately eight weeks long, including four weeks of SCV-07 monotherapy followed by four weeks of SCV-07 in combination with ribavirin. Although the data showed an interesting biological signal, due to the rapidly changing landscape of effective treatments which increase the complexity and risks of developing drugs for chronic HCV, we have decided not to continue development in this indication.
Preclinical Data
SCV-07 has been shown to alter certain intracellular signaling pathways in certain subsets of white blood cells which affects their differentiation into CD-4 helper cells, specifically towards differentiation into the Th1 subset of CD-4 helper cells (Th1 cells secrete cytokines such as interleukin-2 (IL-2) and gamma interferon that can help the immune response). This shift to the Th1 subset can explain how SCV-07 helps protect against
8
Table of Contents
damage to cells from radiation treatment, as well as interfere with infectious diseases and cancer. Publications outline the efficacy of SCV-07 treatment in various animal models, including prevention of oral mucositis in response to radiation therapy, treatment of several immune-sensitive cancers such as melanoma, and treatment of several infectious diseases such as tuberculosis, herpes simplex virus, papilloma virus, and Pichinde virus.
SciClone’s Lead Product ZADAXIN (Thymalfasin)
ZADAXIN is SciClone’s synthetic preparation of thymalfasin, scientifically referred to as thymosin alpha 1, a thymic peptide which circulates in the blood naturally and is instrumental in the immune response to certain cancers and viral infections. Published scientific and clinical studies have shown that thymalfasin helps enhance response to vaccines, and to stimulate and direct the body’s immune response to eradicate infectious diseases like HBV and HCV, as well as certain cancers. Thymalfasin appears to be well tolerated with few reports of significant side effects or toxicities associated with its use.
Thymalfasin elicits a variety of immune system responses. Acting on intracellular signaling pathways, thymalfasin increases the Th1 subset of T-helper cells, which leads to a boost in production of antibodies in response to vaccines, and assists with fighting invading viruses and cancers. Thymalfasin also results in decreased CD-4 cell differentiation into the Th2 subset of CD-4 helper cells that produce cytokines, such as IL-4, which are associated with persistence of viral infection, and stimulates several other components of the immune response that help the body attack and kill virally-infected or tumor cells.
Thymalfasin for Enhancement of Response to Vaccines
Clinical trials have demonstrated that thymalfasin increases response to influenza and hepatitis B vaccines in the elderly and in hemodialysis patients. In elderly subjects, thymalfasin was also shown to decrease the incidence of influenza from 19% in subjects given an influenza vaccine alone, to 6% in subjects receiving thymalfasin treatment in addition to the influenza vaccine. For these clinical trials, the treatment regimen involved 8 to 10 injections of 1.6 mg doses of thymalfasin. A pilot clinical study conducted in 2009/2010 by our partner Sigma-Tau in Italy showed that higher doses of thymalfasin (3.2 or 6.4 mg) given only twice (seven days prior to vaccination and on the day of vaccination) led to a statistically significant increase in percent of subjects who seroconverted to the H1N1 vaccine (MF59 adjuvanted monovalent vaccine, Focetria™ from Novartis), and an increase in total titers, when measured at 21 or 42 days after vaccination. When evaluated at 84 and 168 days after vaccination, the seroconversion rates were similar for patients receiving ZADAXIN and those receiving vaccine alone. These data indicate that the enhancement effect of ZADAXIN, while significantly higher in the critical first six weeks following vaccination, was reduced at later time points and no longer significantly different compared to the vaccine alone. These promising data support the utility of thymalfasin for use in vaccine enhancement.
INTELLECTUAL PROPERTY AND PROPRIETARY RIGHTS
Patents
We seek regulatory approval for our products in disease areas with high unmet medical need, significant market potential and where we have a proprietary position through patents covering use, manufacturing process, or composition of matter for our products. For our lead product ZADAXIN®, we are the licensee or owner of patents and patent applications relating to thymalfasin and its use for a number of diseases. In particular, we are the licensee or owner of patents and applications in the United States and internationally including China that are directed to thymalfasin therapy for the treatment of hepatitis B and hepatitis C as a monotherapy or in combination with other therapeutics including drugs with or without regulatory approval for marketing. In addition, we are the licensee or owner of several pending patent applications in the United States and internationally that are directed to thymalfasin therapy for vaccine enhancement. If issued, these patents should have expiring patent terms from 2025 to 2030. We are also the licensee or owner of several pending patent
9
Table of Contents
applications in the United States and internationally that are directed to thymalfasin therapy for the treatment of melanoma. If issued, these patents should have expiring patent terms from 2026 to 2028. We are also the licensee or owner of patents and pending patent applications in the United States and internationally that are directed to thymalfasin therapy for reducing side effects of chemotherapy. The expiring patent terms for issued or to be issued patents are from 2020 to 2021. We have also applied for patents in the United States and internationally that are directed to thymalfasin therapy for the treatment of other infectious diseases such as Acute infection, Aspergillus infection and Severe Acute Respiratory Syndrome (“SARS”), as well as other indications such as treatment of septic shock. In addition, we have patents or patent applications directed to thymalfasin conjugates and the use of thymalfasin to stimulate the immune system in general.
We own patents directed to the composition of matter of SCV-07 and related products as well as their use as immunomodulators in general in the United States, China and a number of international markets, excluding Russia. These patents have expiring patent terms ranging from 2016 in the United States to 2018 outside of the United States. In addition, we own issued patents and have pending patent applications that are directed to SCV-07 therapy for the treatment of oral mucositis. The expiring patent terms for issued or to be issued patents are from 2028 to 2031. We have also filed for various applications pending in the United States and internationally that are directed to SCV-07 therapy for the treatment of a number of diseases, including the treatment of hepatitis C and various other infectious diseases. If issued, these patents should have expiring patent terms from 2025 to 2031.
With respect to our issued patents in the U.S. and Europe, we are also entitled to obtain a patent term extension to extend the patent expiration date. For example, in the US, we can apply for a patent term extension of up to 5 years for one of the patents covering ZADAXIN or SCV-07 once ZADAXIN or SCV-07 is approved by the Federal Drug Administration (“FDA”). The exact duration of the extension depends on the time we spend in clinical trials as well as getting a New Drug Application Approval from the FDA.
ZADAXIN in China
| Granted Patents Relevant to Approved Indication (not yet expired or abandoned) |
Approved Indication |
Year of Expiration |
||||
| ZL 93120725 [China] |
Chronic Hepatitis B | 2013 | ||||
| ZL 99811382.4 [China] |
Chronic Hepatitis B | 2019 | ||||
SCV-07 in Europe, U.S. and China
| Granted Patents Relevant to Compositions or Indications Currently in Clinical trial in the U.S. |
Year of Expiration |
|||
| 5,744,452 [U.S.] |
2016 | |||
| 5,916,878 [U.S.] |
2016 | |||
| 7,906,486 [U.S.] |
2028 | |||
| 1042286 [E.P.] |
2018 | |||
| ZL 98813799.2 [China] |
2018 | |||
| Pending Applications Relevant to Compositions or Indications Currently in Clinical trial in the U.S. |
Year of Expiration |
|||
| 13/013,340 [U.S.] |
2028 | |||
| 61/319,050 [U.S.] |
2031 | |||
| 61/366,655 [U.S.] |
2031 | |||
| 61/386,442 [U.S.] |
2031 | |||
| 61/418/830 [U.S.] |
2031 | |||
| 200880011935.2 [China] |
2028 | |||
| 08725404.1 [E.P.] |
2028 | |||
10
Table of Contents
Proprietary Rights
In addition to patent protection, we intend to use other means to protect our proprietary rights. We may pursue marketing exclusivity periods that are available under regulatory provisions in certain countries including the United States, Europe, Japan, and China. For example, if we are the first to obtain market approval of a product, e.g., thymalfasin or SCV-07 in the United States, we would expect to receive at least 5 years of market exclusivity.
Furthermore, orphan drug exclusivity has been or may be sought where available. Such exclusivity has a term of 7 years in the United States and 10 years in Europe. We have obtained orphan drug designation for thymalfasin for the treatment of malignant melanoma and chronic hepatitis B in the United States and for the treatment of hepatocellular carcinoma in the United States and in Europe. Orphan drug protection has been or may be sought where available if such protection also grants seven years of market exclusivity. We have filed trademark applications worldwide for ZADAXIN and other trademarks that appear on our commercial packaging and promotional literature. Copyrights for the commercial packaging may prevent counterfeit products or genuine but unauthorized products from entering a particular country by parallel importation. Brand and trademark protection are particularly important to us in China. We are implementing anti-counterfeiting measures on commercial packaging and we are registering the packaging with customs departments in countries where such procedures exist. We rely upon trade secrets, which we seek to protect in part by entering into confidentiality agreements with our employees, consultants, corporate partners, suppliers, and licensees.
MANUFACTURING
ZADAXIN is manufactured for us in Europe and the U.S. by third parties under exclusive contract manufacturing and supply agreements. We closely monitor production runs of ZADAXIN and conduct our own quality assurance audit programs. We believe the manufacturing facilities of our contract suppliers are in compliance with the FDA’s current Good Manufacturing Practices (“GMP”), and European equivalents of such standards. In order to sell ZADAXIN to the licensed importers in China, our manufacturers must 1) be approved by the Italian Ministry of Health (“AIFA”) and 2) be accepted by the SFDA, the Chinese regulatory agency, and we must obtain an Imported Drug License from the SFDA permitting the importation of ZADAXIN into China. The license must be renewed every five years, and our next renewal will be required in 2013. If we change manufacturers, these changes must 1) be approved by AIFA in Italy and 2) be accepted by the SFDA, and we must obtain a new Imported Drug License from the SFDA.
In the event of the termination of an agreement with any single supplier, we believe that we would be able to enter into arrangements with other suppliers with similar terms. We do not intend at this time to acquire or establish our own dedicated manufacturing facilities for any of our products. We believe that our current manufacturing partners for ZADAXIN have enough manufacturing capacity to meet potential market demand. We also believe that our current manufacturing partners in the U.S. and Europe for our other drug candidates will be able to meet our clinical trial needs.
COMPETITION
Our competition for sales of ZADAXIN in China is primarily from generic drug manufacturers located in China who sell their product at lower prices. We compete with them based upon our reputation as a provider of a high quality product, including the fact that our product is produced at European GMP facilities.
Our competitors for future products include pharmaceutical companies, biotechnology firms, universities and other research institutions, both in the United States and abroad, that are actively engaged in research and development or marketing of products in the therapeutic areas we are pursuing. Currently, competitors are marketing drugs for HBV, HCV and cancer, or have products in clinical trials. We believe that the principal
11
Table of Contents
competitive factors in this industry for a marketed drug include the efficacy, safety, price, therapeutic regimen, manufacturing, quality assurance and associated patents and the capabilities of its marketer.
In addition, most of our competitors, particularly large pharmaceutical companies, have substantially greater financial, technical, regulatory, manufacturing, marketing and human resource capabilities than us. Most of them also have extensive experience in undertaking the preclinical and clinical testing and in obtaining the regulatory approvals necessary to market drugs. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated with our competitors.
We believe continuing advancements in and increasing awareness of the use of therapeutics which boost the immune system to fight cancer and infectious diseases may create new competitors.
For the treatment of HBV, current therapies being marketed by competitors include interferon alpha, in standard and pegylated forms, nucleoside analogues such as lamivudine, nucleotide analogue adefovir, and nucleoside analogue, entecavir. In addition to these products, in our largest market, China, ZADAXIN faces competition from other synthetic and generic biological extracts, which are locally manufactured and significantly lower priced.
Future clinical trials may or may not show ZADAXIN, SCV-07, DC Bead, or ondansetron RapidFilm, to have advantages or value over such existing or future competitive products.
RESEARCH AND DEVELOPMENT
A substantial portion of our operating expenses to date are related to research and development (“R&D”). R&D expenses consist of independent R&D costs and costs associated with collaborative R&D and in-licensing arrangements. A substantial portion of our development expense is third party cost relating to the conduct of clinical trials. R&D expenses were $12.4 million, $16.5 million and $22.9 million, for the years ended December 31, 2010, 2009, and 2008, respectively. Our research and development strategy is focused on driving cost-efficient phase 1 and 2 development of promising compounds while seeking development partners for more costly phase 3 trials. Licensed technology as well as products developed by outside parties are an additional source of potential products.
EMPLOYEES
As of December 31, 2010, we had 261 employees: 34 in the United States, 216 in China, and 11 in other countries. From time to time, we engage the services of consultants worldwide with pharmaceutical and business backgrounds to assist in our product development and ZADAXIN commercialization activities. We plan to leverage our key personnel by continuing to make extensive use of clinical research organizations, contract laboratories, development consultants and collaborations with pharmaceutical companies to develop and market our products.
GOVERNMENT REGULATION
Regulation by governmental authorities in the United States, China and other foreign countries is a significant factor in the manufacturing and marketing of our products, as well as in ongoing research and development activities and in pre-clinical and clinical trials and testing related to our products. Our products in clinical development in the United States, China and other foreign countries are subject to approval by the FDA, the SFDA and similar regulatory authorities. Manufacturing establishments are subject to inspections by regulatory authorities at the federal, state and local level and must comply with current GMP as established in various jurisdictions. In complying with GMP standards, manufacturers must continue to expend time, money and effort in the area of production and quality assurance to ensure ongoing full technical compliance. As we do not manufacture our products, we depend on third parties to meet requisite GMP standards.
12
Table of Contents
China
In China, the pharmaceutical industry is subject to extensive government regulation and supervision. The regulatory framework addresses all aspects of operating in the pharmaceutical industry, including approval, pricing, re-imbursement, production, licensing and certification requirements and procedures, periodic renewal and reassessment processes, registration of new drugs and environmental protection.
The SFDA is the authority that monitors and supervises the administration of pharmaceutical products and medical appliances and equipment as well as food, health food and cosmetics in China. The primary responsibilities of the SFDA include:
| • | monitoring and supervising the administration of pharmaceutical products, medical appliances and equipment as well as food, health food and cosmetics in China; |
| • | formulating administrative rules and policies concerning the supervision and administration of food, health food, cosmetics and the pharmaceutical industry; |
| • | evaluating, registering and approving of new drugs, generic drugs, imported drugs and traditional Chinese medicine; |
| • | approving and issuing permits for the manufacture and export/import of pharmaceutical products and medical appliances and equipment and approving the establishment of enterprises to be engaged in the manufacture and distribution of pharmaceutical products; and |
| • | examining and evaluating the safety of food, health food and cosmetics and handling significant accidents involving these products. |
The Ministry of Health (“MOH”) is an authority at the ministerial level under the State Council and is primarily responsible for national public health in China and has administrative responsibility for the SFDA. The MOH performs a variety of tasks in relation to the health industry such as establishing social medical institutes, promulgating national regulations, and producing professional codes of ethics for public medical personnel. The MOH is also responsible for international issues, such as that pertinent to foreign companies and governments.
Drug Administration Laws and Regulations
The China Drug Administration Law and related regulations provide the legal framework for the establishment of pharmaceutical manufacturing enterprises, pharmaceutical trading enterprises and for the administration of pharmaceutical products including the development and manufacturing of new drugs, the import of pharmaceuticals, and the regulation of packaging, trademarking and advertising of pharmaceutical products in China.
Permits and Licenses for Importation, Manufacturing and Registration of Drugs
Imported Drug License. Our strategy in China to date has been to seek approval for the import into China of drugs approved in other markets. We must obtain an Imported Drug License from the SFDA to import a pharmaceutical product into China.
To qualify to receive an Imported Drug License from the SFDA, each manufacturing establishment must be registered with the FDA or European (“EMEA”) regulatory authorities where the product is registered for sale and listed on the Certificate of Pharmaceutical Product (“CPP” or Country of Origin Approval). In general, the SFDA also requires that an imported drug must also have country of origin approval for the same indication that an Imported Drug License is applied for.
13
Table of Contents
As a result, in order to obtain and maintain an Imported Drug License in China, we or our partners must also meet the regulatory requirements for the country of origin of the pharmaceutical products we import, or are seeking to import, into China.
The process for applying for and obtaining an Imported Drug License can be protracted and uncertain. In addition to the submission of clinical data from trials outside China, the SFDA may require additional clinical data, including studies in China, and it may conduct its own inspection and testing of manufacturing facilities and of finished product. An Imported Drug License needs to be renewed every five years. Further, if the manufacturer of the pharmaceutical product changes, an additional approval is required from the SFDA, and approval will also have to be obtained in the country from which the product is imported.
For ZADAXIN, the CPP is in Italy, and was issued by the AIFA and the named manufacturer is Patheon Italia S.p.A. We and our partners need to maintain these approvals.
China requires that products with an Imported Drug License be imported through approved importing agents. At each port of entry, prior to moving the product forward to the distributors, government-licensed importing agents must process and evaluate each shipment to determine whether such shipment satisfies China’s quality control requirements.
GMP Certificates. Our current products and our clinical candidates in China are all manufactured outside China and are subject to GMP standards in the country in which they are manufactured. Our manufacturers are subject to site inspections by the regulatory authorities in the jurisdictions in which they are located. The issuance and renewal of an Imported Drug License is dependent, among other things, upon maintaining manufacturing standards that comply with the GMP standards of a widely recognized regulatory authority, such as the FDA or EMEA.
If we were to manufacture product in China, or obtain product from Chinese contract manufacturers, such manufacture would be subject to similar GMP standards established in China and administered by local authorities.
Distribution of Pharmaceutical Products
According to the China Drug Administration Law and related regulations a manufacturer of pharmaceutical products in China can only engage in the trading of the pharmaceutical products that the manufacturer has produced itself. In addition, such manufacturer can only sell its products to:
| • | wholesalers and retailers holding pharmaceutical trading permits; |
| • | other holders of pharmaceutical manufacturing permits; or |
| • | medical practitioners holding medical practice permits. |
A pharmaceutical manufacturer in China is prohibited from selling its products to end-users, or individuals or entities other than holders of Pharmaceutical Trading Permits, the pharmaceutical manufacturing permits or the medical practice permits.
A pharmaceutical distributor (including wholesalers and retailers) must satisfy requirements as to personnel with pharmaceutical expertise, appropriate warehousing and sanitary environment compatible to the distributed pharmaceutical products; quality management and compliance with regulations to ensure the quality of the distributed pharmaceutical products. Operations of pharmaceutical distributors must be conducted in accordance with the Pharmaceutical Operation Quality Management Rules and require a certificate from the SFDA. Pharmaceutical distributors must comply with record keeping requirements regarding the products sold.
14
Table of Contents
Price Control and Competitive Bidding
The administration of price control over pharmaceutical products is vested in the national and provincial price administration authorities. Depending on the categories of pharmaceutical products in question, the prices of pharmaceutical products listed in the State Basic Medical Insurance and Work Injury Insurance Drug Catalogue, drugs with patents and other drugs whose production or trading may constitute monopolies are subject to the control of the National Development and Reform Commission (“NDRC”) of China and the relevant provincial or local price administration authorities. For pharmaceutical products manufactured or imported into China, the national price administration authority from time to time publishes price control lists specifying pricing ceilings for specific pharmaceuticals. The Ministry of Labor (“MOL”) and Social Security, together with other government authorities, determines which medicines are to be included in or removed from the Catalogue for the national medical insurance program, and under which category a medicine should fall, both of which affect the amounts reimbursable to program participants for their purchases of those medicines. These determinations are based on a number of factors, including price and efficacy. A national medical insurance program participant can be reimbursed for the full cost of a Category A medicine and 60 to 90% of the cost of a Category B medicine. In November 2009, thymalfasin, the generic chemical name for our pharmaceutical product ZADAXIN, was included as a Category B product in the National Reimbursed Drug List (“NRDL”). The main purpose of the NDRC and the NRDL price control policy is to establish new maximum allowable prices for listed pharmaceutical products, which in many cases will be below previously established prices, thus lowering the prices for many approved pharmaceutical products. The provincial price administration authorities also publish price control lists for pharmaceutical products. Pursuant to the NDRC and Measures for Medicine Pricing by the Government, the price ceiling is determined whether drugs are deemed essential drugs and included on the National Essential Drug List (“NEDL”) or non-essential drugs, which could be included in the NRDL and subject to price control and include reference to the quality of the product, whether the products are newly developed products, whether the products have patent protection in China, and the status of implementing the GMP Guidelines by the manufacturer of the relevant product.
The prices of pharmaceutical products included in the price control lists are subject to adjustment upon approval by the price administration authorities from time to time. Pharmaceutical enterprises in China are required to submit cost related information such as raw material prices regularly to the relevant authorities so that the authorities may take into account the prevailing market conditions when setting the prices. The price administration authorities may approve adjustments to the price of pharmaceutical products upon the pharmaceutical manufacturer’s request if material changes in the cost structure in producing the pharmaceutical products or significant changes in demand for these pharmaceutical products are recognized.
In each province where we market our products, distributors participate in a government-sponsored competitive bidding process every year or every few years for procurement by state-owned hospitals of a medicine included in the provincial medicine catalogs. A government-appointed committee reviews bids submitted by pharmaceutical companies and selects one or more medicines for treatment of a particular medical condition. The selection is based on a number of factors, including whether the product is on the NEDL or the NRDL, bid price, quality and manufacturer’s reputation and service. The bid price of the selected medicine will become the price required for purchases of that medicine by all state-owned hospitals in the relevant province or local district.
Health Insurance System
The MOL and Social Security are responsible for the reform of the medical insurance system. As part of the reform of the state basic medical insurance system for employees in the urban areas, the MOL and Social Security, the MOH, the SFDA and various other governmental departments jointly issued the State Basic Medical Insurance and Work Injury Insurance Drug Catalogue, as amended, or Catalogue, with a view to enhancing the management of the use of drugs under the medical insurance system. The drugs listed in the Catalogue are covered by the national medical insurance program.
15
Table of Contents
Reimbursement under the National Medical Insurance Program
Although it is designated as a national program, the implementation of the national medical insurance program is delegated to various provincial governments, each of which has established its own medicine catalog. A provincial government must include all Category A medicines listed in the Catalogue in its provincial medicine catalog, but may use its discretion based on its own selection criteria to add other medicines to, or exclude Category B medicines listed in the Catalogue from, its provincial medicine catalog, so long as the combined numbers of the medicines added and excluded do not exceed 15% of the number of the Category B medicines listed in the Catalogue. In addition, provincial governments may not downgrade a nationally classified Category A medicine to Category B. The total amount of reimbursement for the cost of prescription and over-the-counter medicines, in addition to other medical expenses, for an individual program participant in a calendar year is capped at the amount in that participant’s individual account. The amount in a participant’s account varies, depending upon the amount of contributions from the participant and his or her employer. Generally, program participants who are from relatively wealthier eastern parts of China and relatively wealthier metropolitan centers have greater amounts in their individual accounts than those from less developed provinces.
Prescription Regulations
As announced by MOH, the Prescription Administrative Measures, regulating prescription of drugs, took effect on May 1, 2007, which stipulates that doctors may only use the generic names of drugs in their prescriptions instead of brand names and that medical institutions offer patients the same type of drug from no more than two separate pharmaceutical companies. The purpose of this legislation is to minimize the practice of doctors receiving kickbacks from pharmaceutical companies for prescribing higher priced, or even unneeded, drugs to patients.
United States, Europe and Other Countries
We have several products in development for markets in the United States, Europe and elsewhere. The regulatory regime in the United States and Europe is similar in many respects to the regulatory system in China, although the regulatory system has been developed over many decades and, we believe, is subject to greater and more certain regulatory oversight than the Chinese regulatory system. The steps required before a new drug may be distributed commercially generally include:
| • | conducting appropriate pre-clinical laboratory evaluations, including animal studies, in compliance with the FDA’s Good Laboratory Practice (“GLP”) requirements, to assess the potential safety and efficacy of the product, and to characterize and document the product’s chemistry, manufacturing controls, formulation and stability; |
| • | submitting the results of these evaluations and tests to the FDA, along with manufacturing information, analytical data, and protocols for clinical studies, in an Investigational New Drug Application (“IND”), and receiving approval from the FDA that the clinical studies proposed under the IND are allowed to proceed; |
| • | obtaining approval of Institutional Review Boards (“IRBs”) to administer the product to humans in clinical studies; |
| • | conducting adequate and well-controlled human clinical trials in compliance with the FDA’s Good Clinical Practice (“GCP”) requirements that establish the safety and efficacy of the product candidate for the intended use, typically in the same Phase 1, Phase 2 and Phase 3 steps described above for China; |
| • | development of manufacturing processes which conform to FDA current Good Manufacturing Practices, or cGMPs, as confirmed by FDA inspection; |
| • | submitting to the FDA the results of pre-clinical studies, clinical studies, and adequate data on chemistry, manufacturing and control information to ensure reproducible product quality batch after batch, in a New Drug Application (“NDA”) or Biologics License Application (“BLA”); |
16
Table of Contents
| • | obtaining FDA approval of the NDA, including inspection and approval of the product manufacturing facility as compliant with cGMP requirements, prior to any commercial sale or shipment of the pharmaceutical agent. |
When used in connection with trials and filings in other countries, terms such as “phase 1,” “phase 2,” “phase 3,” “phase 4,” “new drug application” and “marketing application” refer to what we believe are comparable trials and filings in these other countries.
The process in the United States of obtaining regulatory approval is lengthy, uncertain, and requires the expenditure of substantial resources. Each NDA must be accompanied by a user fee, pursuant to the requirements of the Prescription Drug User Fee Act, or PDUFA, and its amendments.
After FDA approval has been obtained, the FDA requires post-marketing reporting to monitor the side effects of the drug. This may include phase 4 studies in which the drug is studied in an expanded patient population in a post-approval setting for continued monitoring of safety and sometimes continued efficacy. Further studies may be required to provide additional data on the product’s risks, benefits, and optimal use, and will be required to gain approval for the use of the product as a treatment for clinical indications other than those for which the product was initially tested. Results of post-marketing programs may limit or expand the further marketing of the product. Further, if there are any modifications to the drug, including changes in indication, labeling, or a change in the manufacturing process or manufacturing facility, an NDA or BLA supplement may be required to be submitted to the FDA. We market or intend to market products in other countries and each country has regulations for the regulation of approval, marketing and sale of pharmaceutical products. We must comply with the regulations of each country in which we seek approval of and intend to market and sell any product.
AGREEMENTS WITH THIRD PARTIES
Sigma-Tau Agreement. Our collaboration with Sigma-Tau Finanziaria S.p.A. (“Sigma-Tau”) is governed by an agreement entered into in 2000 with a term expiring in March 2012 unless renewed, as well as by amendments to the agreement regarding particular development efforts. Under the agreement, we licensed to Sigma-Tau exclusive ZADAXIN development and marketing rights that cover all countries in the European Union as defined on January 1, 1995, in addition to Iceland, Norway and Switzerland. In addition, the agreement governed our joint collaboration on the development of thymalfasin in certain indications, and for the sharing of intellectual property in our respective territories. The agreement also provides that if Sigma-Tau sells ZADAXIN in the licensed territory, it will purchase the product from us at a specified price, subject to certain adjustments. We do not currently anticipate that Sigma-Tau will sell any ZADAXIN other than nominal amounts in Italy.
There are no on-going development or reimbursement obligations under the agreement, and there are no milestones or similar terms currently in effect. However, we continue to cooperate with Sigma-Tau on exploring the development of thymalfasin in other indications, including as a vaccine adjuvant, and in aspergillosis.
Biocompatibles Agreement. In June 2006 we entered into a licensing and distribution arrangement with Biocompatibles UK LTD (“Biocompatibles”) for the distribution of DC BeadTM in China. The agreement provides us exclusive marketing rights in China. Under the agreement, if and when product approval is obtained in China, we would purchase product from Biocompatibles for sale in China at a price specified in the agreement. The purchase prices are subject to adjustment in certain circumstances. To maintain our exclusive rights, we must meet certain unit volume requirements.
Applied Pharma Research s.a. Agreement. In July 2009 we entered into a licensing and distribution arrangement with Applied Pharma Research s.a. (“APR”) for ondansetron RapidFilmTM and paid an upfront fee of $1 million. The agreement grants us exclusive marketing rights in China including Hong Kong and Macau, and Vietnam. If product approval is obtained in China, we would purchase product from APR at a price specified
17
Table of Contents
in the agreement for sale in our territory. In addition to the purchase price of the product, the agreement provides for additional payments by us to APR upon achievement of first product approval and of specified sales levels.
SCV-07 Agreement. In December 2010, we entered into an assignment and purchase of intellectual property rights to purchase the rights, titles and interest in and to the worldwide (except as to Russia) technology and patent rights for SCV-07 for an initial fee of $125,000 and a milestone payment of $1,750,000 due upon our receiving government approval to commercially market and sell SCV-07 product in specified territories.
THIRD-PARTY REIMBURSEMENT
Our ability to successfully commercialize our products may depend in part on the extent to which coverage and reimbursement to patients for our products will be available from government health care programs, private health insurers and other third-party payors or organizations. Significant uncertainty exists as to the reimbursement status of new therapeutic products, such as ZADAXIN. In most of the markets in which we are currently approved to sell ZADAXIN, reimbursement for ZADAXIN under government or private health insurance programs is not yet widely available, and in many of these countries government resources and per capita income may be so low that our products will be prohibitively expensive. We believe that many of the sales of ZADAXIN in China are made with some third party reimbursement. We anticipate reimbursement to be approved when a new price is approved for ZADAXIN. In the United States, Europe and Japan, proposed health care reforms could limit the amount of governmental or third-party reimbursement available for our products should they be approved for sale in these markets. Various governments and third-party payors are trying to contain or reduce the costs of health care through various means. We believe that there will continue to be legislative efforts and proposals to implement such government controls.
AVAILABLE INFORMATION
We file electronically with the Securities and Exchange Commission (“the Commission” or the “SEC”) our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended. The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is http://www.sec.gov.
You may obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, on the day of filing with the SEC on our website on the World Wide Web at http://www.sciclone.com, by contacting the Investor Relations Department at our corporate offices by calling 800-724-2566 or by sending an e-mail message to investorrelations@sciclone.com.
| Item 1A. | Risk Factors |
You should carefully consider the risks described below, in addition to the other information in this report on Form 10-K, before making an investment decision. Each of these risk factors could adversely affect our business, financial condition, and operating results as well as adversely affect the value of an investment in our common stock.
Our stock price may be volatile, and an investment in our stock could suffer a decline in value.
We have a history of operating losses and an accumulated deficit. Although we reported net income of $21.1 million and $11.9 million for the years ended December 31, 2010 and 2009, respectively, we have experienced significant operating losses in the past, and as of December 31, 2010, we had an accumulated deficit of
18
Table of Contents
approximately $143.8 million. If our operating expenses were to increase or if we were not able to increase or sustain revenue, we may not achieve profitability over the next 12 months.
The market price of our common stock has experienced, and may continue to experience, substantial volatility due to many factors, some of which we have no control over, including:
| • | developments related to the pending United States Securities and Exchange Commission (“SEC”) and United States Department of Justice (“DOJ”) investigations, our efforts to cooperate with the investigations and events related to pending litigations; |
| • | government regulatory action affecting our Company or our drug products or our competitors’ drug products in China, the United States and other foreign countries, including the effect of any change in the governmentally permitted maximum listed price for ZADAXIN (“thymosin alpha 1 or thymalfasin”); |
| • | actual or anticipated fluctuations in our quarterly operating results; |
| • | progress and results of clinical trials; |
| • | progress of thymalfasin and SCV-07 through the regulatory process, especially regulatory actions and the adequacy of clinical data and documentation for regulatory purposes; |
| • | finding a partner for late-stage trials of our clinical development candidates; |
| • | progress of DC Bead® and ondansetron RapidFilm® through required clinical studies and the regulatory process, especially regulatory actions and the adequacy of clinical data and documentation for regulatory purposes in China including Hong Kong and Macau, and Vietnam; |
| • | timing and achievement of our corporate milestones; |
| • | changes in our agreements or relationships with collaborative partners; |
| • | announcements of technological innovations or new products by us or our competitors; |
| • | announcement and completion of corporate acquisition, merger, licensing or marketing arrangements, or sales of assets; |
| • | developments or disputes concerning patent or proprietary rights; |
| • | changes in the composition of our management team or board of directors; |
| • | changes in company assessments or financial estimates by securities analysts; |
| • | changes in assessments of our internal controls over financial reporting; |
| • | general stock market conditions and fluctuations for the emerging growth and pharmaceutical market sectors; |
| • | economic and political conditions in the United States or abroad; and |
| • | broad financial market fluctuations in the United States, Europe or Asia. |
Our revenue is dependent on our sale of ZADAXIN in China, and if we experience difficulties in our foreign sales efforts, our operating results and financial condition will be harmed.
Our product revenue is highly dependent on the sale of ZADAXIN in China. If we experience difficulties in our foreign sales efforts in China, our business will suffer and our operating results and financial condition will be harmed. For the years ended December 31, 2010, 2009 and 2008, approximately 96%, 96% and 94%, respectively, of our ZADAXIN sales were to customers in China. Sales of ZADAXIN in China may be limited due to the low average personal income, lack of patient cost reimbursement, poorly developed infrastructure and competition from other products, including generics. ZADAXIN sales growth in recent years has benefited from
19
Table of Contents
the rapidly growing Chinese economy and growing personal disposable income. Sales of ZADAXIN in China could be adversely affected by a slowing or downturn of the Chinese economy and from the decisions of the National Development and Reform Commission (“NDRC”) pricing reform anticipated to be made in 2011.
In China, ZADAXIN is approved for the treatment of hepatitis B virus (“HBV”) and as a vaccine adjuvant. We face competition from pharmaceutical companies who are aggressively marketing competing products for the treatment of HBV and for other indications where we believe ZADAXIN may be used on an off-label basis. In addition, several local companies are selling lower-priced, locally manufactured generic thymalfasin, which is a competitive product and is selling in substantial and increasing quantities. While generic products outsell ZADAXIN in unit volumes, we have been able to maintain a pricing advantage through the reputation of our imported, branded product. We believe such competition to continue with added new local manufacturers of generic thymalfasin and there could be a negative impact on the price and the volume of ZADAXIN sold in China, which would harm our business. Our efforts to in-license or acquire other pharmaceutical products for marketing in China and other markets may be unsuccessful or even if successful may not have a meaningful effect on our dependence on ZADAXIN sales in those markets.
In November 2009, thymalfasin, the generic chemical name for our pharmaceutical product ZADAXIN, was included as a Category B product in the National Reimbursed Drug List (“NRDL”) and pricing for ZADAXIN on the NRDL is still being reviewed by the authorities. The price for pharmaceutical products is regulated in China both at the national and at the provincial level. These regulations, as well as regulation of the importation of pharmaceutical products may reduce prices for ZADAXIN to levels significantly below those that would prevail in an unregulated market, limit the volume of product which may be imported and sold or place high import duties on the product, any of which may limit the growth of our revenues or cause them to decline. The Chinese government is increasing its efforts to reduce overall health care costs, including pricing controls on pharmaceutical products. Individual provinces in China and, in some cases, individual hospitals can and have established pricing requirements for a product to be included on formulary lists. In some cases, these prices have been significantly lower than our distributors have been selling ZADAXIN, in which case we have been removed from formulary lists, which consequently has reduced sales to certain hospitals and could adversely affect our future sales. The process and timing for any price restrictions is unpredictable. In addition, we are aware that ZADAXIN may be used on an off-label basis, and the Chinese government’s pricing, reimbursement or other actions might reduce such uses. We are working on these regulatory processes as well as on potential changes in our business model depending on potential outcomes. We believe we will be able to successfully manage our business in China through this process, however maximum prices could be set at some time in the future which could adversely affect our results or require substantial changes in our business model which may be difficult to implement.
Importers and distributors of ZADAXIN avail themselves of funds in China from banks to purchase, hold and distribute ZADAXIN. Substantial increases in restrictions on fund availability and/or increases in borrowing costs could limit the ability of our importers and distributors to finance their import and distribution process.
We have received regulatory approvals to import and market ZADAXIN in China and to manufacture ZADAXIN and export the product from Italy. In order to continue our sales to China, we need to maintain these approvals. Our license to import ZADAXIN into China needs to be renewed every five years and the next renewal is required in 2013. Although we were successful in obtaining a renewal in 2008, there is no assurance that we will receive renewals in the future when applied for or that the renewals will not be conditioned or limited in ways that limit our ability to sell ZADAXIN to China. Further, our licenses to manufacture and export ZADAXIN from Italy are dependent upon our continuing compliance with regulations in Italy. Our business would be adversely affected if we are not able to maintain these approvals. In order to sell ZADAXIN to the licensed importers in China, our manufacturers must 1) be approved by the Italian Ministry of Health (“AIFA”) and 2) be accepted by the State Food and Drug Administration of China (“SFDA”), the Chinese equivalent to the United States Food and Drug Administration (“FDA”), and we must obtain an Imported Drug License from the SFDA permitting the importation of ZADAXIN into China. The license must be renewed every five years, and if
20
Table of Contents
we change manufacturers, these changes must 1) be approved by the AIFA in Italy and 2) be accepted by the SFDA. When we change manufacturers we must obtain a new approval. The SFDA, the FDA, AIFA and other regulatory agencies may, and have, changed their internal administrative rules in ways that may delay or complicate the regulatory process. Those changes are not always disclosed or known to us and we may experience unexpected delays or additional costs as a result of such changes.
Our ZADAXIN sales and operations in other parts of China and the world are subject to a number of risks and increasing regulations, including difficulties and delays in obtaining registrations, renewals of registrations, permits, pricing approvals and reimbursement, increasing regulation of product promotion and selling practices, unexpected changes in regulatory requirements and political instability. In addition, during the second quarter of 2009 we experienced a strong upsurge in ZADAXIN sales which we believe was attributable both to the increasing penetration of ZADAXIN within the Chinese market, as well as concerns in China from the H1N1 flu virus. Although we believe that ZADAXIN sales have returned to levels more consistent with our established business, if distributors and hospitals that purchase ZADAXIN stockpile more ZADAXIN than needed for current use, our sales of ZADAXIN may suffer as distributors and hospitals use ZADAXIN already in their inventory before purchasing additional product from us. This could lead to uneven future revenue results for ZADAXIN and in turn materially impact our cash flow and business condition.
We experience other issues with managing foreign sales operations including long payment cycles, potential difficulties in accounts receivable collection and, especially from significant customers, fluctuations in the timing and amount of orders and the adverse effect of any of these issues on our business could be increased due to the concentration of our business with a small number of distributors. Problems with collections from, or sales to, any one of those distributors could materially adversely affect our results. Operations in foreign countries including China also expose us to risks relating to difficulties in enforcing our proprietary rights, currency fluctuations and adverse or deteriorating economic conditions. If we experience problems with obtaining registrations, complying with reimbursement rules or compliance with foreign country or applicable U.S. laws, adverse or mixed outcome of clinical studies anywhere in the world, or if we experience difficulties in payments or intellectual property matters in foreign jurisdictions, or if significant political, economic or regulatory changes occur, our results could be adversely affected.
Our operations throughout the world including China are potentially subject to the laws and regulations of the United States including the Foreign Corrupt Practices Act (“FCPA”), in addition to the laws and regulations of the local countries. Regulation in China of the activities in the pharmaceutical industry has increased and may continue to undergo significant and unanticipated changes. A number of companies have faced significant expenses or fines as a result of the increasing regulation of, and enforcement activity regarding, the pharmaceutical industry.
The Chinese government has been engaged in an extensive economic stimulus program in China over the past year. We believe that the Chinese government is reducing these stimulus programs. We believe these programs have had broad effects across the Chinese economy, and the reduction or withdrawal of these programs could adversely affect spending in numerous ways, including reduction of spending on pharmaceutical products including ZADAXIN.
We face risks related to the potential outcomes of the SEC investigation regarding FCPA compliance and other matters and DOJ investigation regarding the FCPA and our own investigation into such matters, including potential penalties, substantial expenses and the use of significant management time and attention, and changes in our marketing and sales practices that could affect our ability to generate revenue, any of which could adversely affect our business.
In August 2010, we received notices of investigations by U.S. government agencies that relate to our operations in China including compliance with the FCPA and we subsequently initiated an internal investigation regarding these matters. In connection with the formal, non public SEC investigation, the SEC issued a subpoena
21
Table of Contents
to us requesting documents regarding a range of matters including but not limited to documents relating to potential payments or transfer of anything of value to regulators and government-owned entities in China; documents relating to bids or contracts with state or government-owned entities in China; documents relating to intermediary or local agent of the Company in China; documents regarding the Company’s ethics and anti-corruption policies, training, and audits; and documents relating to certain Company financial and other disclosures made by the Company. The DOJ is currently conducting an investigation of us in connection with compliance with the FCPA, as to which they have advised us that the DOJ has information about the Company’s practices suggesting possible violations. We have been cooperating with, and will continue to cooperate with, the investigations by and inquiries from the SEC and DOJ. In response to these matters, our board of directors has appointed a special committee of independent directors to oversee our response to the government inquiry. Based on an initial review, the special committee is conducting an independent investigation as to matters reflected in and arising from the SEC and DOJ investigations including, but not limited to, certain sales and marketing matters in China, in order to evaluate whether any violation of the FCPA or other laws occurred. We are unable to predict what consequences, if any, that any investigation by any regulatory agency or by our special committee may have on us. These and any other regulatory investigations and our cooperation with them will result in substantial legal and accounting expenses, and could divert management’s attention from other business concerns and harm our business. If we fail to comply with regulations or to carry out controls on our Chinese or other foreign operations in a manner that satisfies all applicable laws, our business would be harmed. Any civil or criminal action commenced against us by a regulatory agency could result in administrative orders against us, the imposition of significant penalties and/or fines against us, and/or the imposition of civil or criminal sanctions against certain of our officers, directors and/or employees. The investigations, results of the investigations, or remedial actions we may take, if any, as a result of such investigations, may adversely affect our business in China. If we are subject to an adverse finding resulting from the SEC and DOJ investigations, or from our own independent investigation, we could be required to pay damages or penalties or have other remedies imposed upon us. In addition, we could incur additional expenses related to fines, or to remediative measures including if we determine that our compliance programs, internal controls or disclosure controls are not effective. The period of time necessary to resolve our independent investigation, and the investigation by the DOJ and the SEC is uncertain, and these matters could require significant management and financial resources which could otherwise be devoted to the operation of our business.
We are taking actions to improve our practices and policies regarding compliance with law, and have identified a material weakness in our internal control over financial reporting relating to our implementation of our policy on compliance with laws.
We have consistently maintained a policy that our employees comply with all applicable laws. However, in the course of evaluating our existing training, policies and internal controls over financial reporting and expense approval and expense reimbursement practices, and of the matters brought to the attention of management by the special committee, we determined that our guidelines, employee training and expense approval practices and our internal controls with respect to certain matters, including the payment for, or reimbursement of, third party gifts, travel expenses, honoraria and sponsorship of certain third party events required improvements in order to provide reasonable assurance that our policy on compliance with applicable laws was effective. In addition, management determined that we had a material weakness in our internal control over financial reporting as of December 31, 2010 relating to our implementation of our policy on compliance with laws.
During the first quarter of fiscal 2011, we initiated or commenced various remediation efforts to provide reasonable assurance of our implementation of our policy on compliance with laws, including the FCPA and other anti-bribery laws as described in “Management’s Annual Report on, and Changes in, Internal Control over Financial Reporting”. As a result of adopting new practices, guidelines and other measures, certain of our sales and marketing practices will change. In particular these actions will impose restrictions and limitations on certain expenses including payments for, or reimbursement of, third-party gifts, travel expenses, honoraria and sponsorships of medical, scientific or other third party events. Our efforts to implement these changes will
22
Table of Contents
require substantial effort and expense, and changes in types or amount of expenses we’ll pay or reimburse could adversely affect our ability to generate revenue.
We have been named as a party to purported stockholder class actions and purported derivative lawsuits, and we may be named in additional litigation, all of which will require significant management time and attention and result in significant legal expenses and may result in an unfavorable outcome which could have a material adverse effect on our business, financial condition, results of operations and cash flows.
Certain of our officers and directors have been named as nominal defendants in three derivative lawsuits purportedly filed on behalf of the Company. The lawsuits which have been consolidated into one action assert claims for breach of fiduciary duty, abuse of control, unjust enrichment and corporate waste based on alleged violations of the Foreign Corrupt Practices Act. We and certain of our officers have also been named as defendants in two purported class action lawsuits which alleged violations of the Securities Exchange Act of 1934, as amended, in connection with allegedly false, misleading and incomplete statements issued by the defendants related to potential violations of the Foreign Corrupt Practices Act, our reported revenues, income and sales growth, and marketing and sales activities. Although the class action lawsuits have been dismissed, additional class action lawsuits could be filed. In addition, our board of directors has received a demand from a purported stockholder demanding that the board of directors take actions to remedy breaches of fiduciary duties by the directors and certain officers relating to alleged violations of the FCPA and securities laws. We cannot predict whether these or other lawsuits or demands are likely to result in any material recovery by, or expense to, SciClone. We believe we will continue to incur legal fees in responding to these lawsuits and to the demand. The expense of defending such litigation may be significant. The amount of time to resolve this and any additional lawsuit is unpredictable and these actions may divert management’s attention from the day-to-day operations of our business, which could adversely affect our business, results of operations and cash flows.
We are at risk of additional securities class action and derivative lawsuits.
Securities class action and derivative lawsuits are often filed against public companies following a decline in the market price of their securities. We were sued recently after our announcement regarding SEC and DOJ investigations and we and certain of our officers and directors have been named as parties in purported stockholder class actions and derivative lawsuits. The class action lawsuits have been dismissed and we are continuing to litigate the derivative lawsuits. We may be named in additional litigation, all of which will require significant management time and attention and result in significant legal expenses and may result in an unfavorable outcome which could have a material adverse effect on our business, financial condition, results of operations and cash flows. We may experience stock price volatility in the future, either related to announcements regarding the SEC and DOJ investigation, our own investigations related thereto or other matters. This risk is especially relevant for us because biotechnology companies have experienced greater than average stock price volatility in recent years. Such litigation could result in additional substantial costs and a diversion of management’s attention and resources, which could harm our business.
Our sales of ZADAXIN may fluctuate due to seasonality of product orders and sales in any quarter may not be indicative of future sales.
Our sales for the quarter ended June 30, 2009 were greatly affected by the demand in China for ZADAXIN which we believe was in connection with Influenza A (H1N1). Similarly, our sales for the quarter ended September 30, 2003 were greatly affected by the demand in China for ZADAXIN in connection with the treatment of Severe Acute Respiratory Syndrome (“SARS”). To date, SARS has not re-emerged, like influenza, as a seasonal public health problem. However, it is not possible to predict what effect, if any, H1N1, SARS, or similar epidemics would have on future sales of ZADAXIN, if they were to emerge. Although we do not market ZADAXIN for use in treating epidemic diseases such as Influenza A (H1N1) or SARS, if ZADAXIN is purchased in connection with outbreaks of seasonal viral contagions, product sales could become more
23
Table of Contents
concentrated in certain quarters of the calendar year, quarterly sales levels could fluctuate and sales in any quarter may not be indicative of sales in future periods.
We may lose market share or otherwise fail to compete effectively in the intensely competitive pharmaceutical industry.
Competition in the pharmaceutical industry in China is intense, and we believe that competition will increase. Our success depends on our ability to compete in this industry, but we cannot assure you that we will be able to successfully compete with our competitors. Increased competitive pressure could lead to intensified price-based competition resulting in lower prices and margins, which would hurt our operating results. We cannot assure you that we will compete successfully against our competitors or that our competitors, or potential competitors, will not develop drugs or other treatments for our targeted indications that will be superior to ours.
We depend on sales to China, and global conditions could negatively affect our operating results or limit our ability to expand our operations outside of China. Changes in China’s political, social, regulatory and economic environment may affect our financial performance.
A large majority of our sales are to China. Heightened tensions resulting from the current geopolitical conditions in the Middle East, North Korea and elsewhere could worsen, causing disruptions in foreign trade, which would harm our sales. In particular, our commercial product is manufactured in Europe and distributed by us from our operations in Hong Kong. Any disruption of our supply and distribution activities due to geopolitical conditions could decrease our sales and harm our operating results.
With respect to China, our financial performance may be affected by changes in China’s political, social, regulatory and economic environment. The role of the Chinese central and local governments in the Chinese economy is significant. Chinese policies toward economic liberalization, and laws and policies affecting foreign companies, currency exchange rates and other matters could change, resulting in greater restrictions on our ability to do business in China. Any imposition of surcharges or any increase in Chinese tax rates could hurt our operating results. The Chinese government could revoke, terminate or suspend our license for national security and similar reasons without compensation to us. If the government of China were to take any of these actions, we would be prevented from conducting all or part of our business. Any failure on our part to comply with governmental regulations could result in the loss of our ability to market our products in China.
Because of China’s tiered method of importing and distributing finished pharmaceutical products, our quarterly results may vary substantially from one period to the next.
China uses a tiered method to import and distribute finished pharmaceutical products. At each port of entry, and prior to moving the product forward to the distributors, government-licensed importing agents must process and evaluate each shipment to determine whether such shipment satisfies China’s quality assurance requirements. In order to efficiently manage this process, the importing agents typically place large, and therefore relatively few, orders within an annual period. Therefore, our sales to an importing agent can vary substantially from quarter to quarter depending on the size and timing of the orders, which has in the past and may in the future cause our quarterly results to fluctuate. We rely on a limited number of importers, in any given quarter, to supply most of our product in China. The Chinese government has been encouraging a consolidation of the government-licensed importing agents and in recent months Sinopharm Group Co. Limited acquired a majority interest in two of our large importers in China. Because we use a small number of importing agents in China, our receivables from any one importing agent are material, and if we were unable to collect receivables from any importer, our business and cash-flow would be adversely affected. These importers are not obligated to place purchase orders for our product, and if they determined for any reason not to place purchase orders, we would need to seek alternative licensed importers, which could cause short term fluctuations in our business.
24
Table of Contents
The existence of counterfeit pharmaceutical products in China’s pharmaceutical retail market may damage our brand and reputation and have a material adverse effect on our business, financial condition, results of operations and prospects.
Certain medicine products distributed or sold in China’s pharmaceutical retail market, including those appearing to be our products, may be counterfeit. Counterfeit products are products sold under the same or very similar brand names and/or having a similar appearance to genuine products. Counterfeit products, including counterfeit pharmaceutical products, are a significant problem in China. Such products divert sales from genuine products, often are of lower cost, often are of lower quality (having different ingredients or formulations, for example), and have the potential to damage the reputation for quality and effectiveness of the genuine product. The counterfeit pharmaceutical product regulation control and enforcement system in China is not able to completely eliminate production and sale of counterfeit pharmaceutical products. Any sale of counterfeit products resulting in adverse side effects to consumers may subject us to negative publicity and expenses. It could have a material adverse effect on our business, financial condition, results of operations and prospects.
We may be subject to currency exchange rate fluctuations, which could adversely affect our financial performance.
Most of our product sales are denominated in U.S. dollars. Fluctuation in the U.S. dollar exchange rate with local currency directly affects the customer’s cost for our product. In particular, a stronger U.S. dollar vis-à-vis the local currency would tend to have an adverse effect on sales and potentially on collection of accounts receivable. China currently maintains the value of the renminbi in a narrow currency trading band that may or may not fluctuate based upon government policy. Depending on market conditions and the state of the Chinese economy, China has intervened in the foreign exchange market in the past to prevent significant short-term fluctuations in the renminbi exchange rate, and it could make future adjustments, including moving to a managed float system, with opportunistic interventions. This reserve diversification may negatively impact the United States dollar and U.S. interest rates. A trend to a stronger U.S. dollar would erode margins earned by our Chinese importers and prompt them to ask us to lower our prices. We are subject to currency exchange rate fluctuations as a result of expenses incurred by our foreign operations. In particular, one of our supply arrangements under which we purchase finished products is denominated in euros and costs of our operations in China are paid in local currency. Consequently, changes in exchange rates could unpredictably and adversely affect our operating results and could result in exchange losses. To date, we have not hedged against the risks associated with fluctuations in exchange rates and, therefore, exchange rate fluctuations could have a material adverse impact on our future operating results and stock price.
Our business strategy is dependent on our ability to in-license or otherwise acquire the rights to develop and commercialize products, particularly in China. If we fail to acquire such rights or are unsuccessful in our efforts to develop such products and obtain regulatory approval to market and successfully commercialize them, our business will suffer.
All of our products, including ZADAXIN, the only one for which we have sales revenue, have been in-licensed by us. We do not conduct product discovery and typically have in-licensed our products from third parties who have discovered the products and conducted at least some pre-clinical research on them. We are particularly focused on in-licensing products for the China market and the competition for attractive products to in-license is intense, and we cannot assure you that we will be able to in-license products in the future on acceptable terms, if at all. In addition, we face the risks of developing the in-licensed products and the risks and uncertainties of conducting clinical trials and seeking regulatory approval to market the in-licensed products, all of which require years of effort and the commitment of significant financial resources. Our ability to grow our business requires the development and commercialization of additional products. If we are unable to in-license or acquire products on acceptable terms and successfully develop and commercialize them, our business could be harmed.
25
Table of Contents
We may engage in acquisitions and must successfully integrate any acquired products or companies in order to avoid a material disruption to our business and material adverse effects to our financial results.
We may engage in one or more acquisitions of products or companies. Acquisitions involve numerous risks, including the following:
| • | difficulties and costs in integrating the operations, technologies, products and personnel of the acquired businesses; |
| • | inadequate internal control procedures and disclosure controls to comply with the requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or poor integration of a target company’s or businesses’ procedures and controls; |
| • | potential difficulties in complying with the Foreign Corrupt Practices Act; |
| • | diversion of management’s attention from normal daily operations of the business; |
| • | potential difficulties in completing projects associated with in-process research and development; |
| • | difficulties in entering markets in which we have no or limited direct prior experience and where competitors in such markets have stronger market positions; |
| • | insufficient net revenue to offset increased expenses associated with acquisitions; |
| • | potential failure to achieve commercial expectations; |
| • | potential loss of key employees of the acquired companies; and |
| • | difficulty in forecasting revenues and margins. |
Acquisitions may also cause us to:
| • | issue common stock that would dilute our current shareholders’ percentage ownership; |
| • | assume liabilities, some of which may be unknown at the time of such acquisitions; |
| • | record goodwill and intangible assets that will be subject to impairment testing and potential periodic impairment charges; |
| • | incur amortization expenses related to certain intangible assets; |
| • | incur large and immediate write-offs of in-process research and development costs; or |
| • | become subject to litigation. |
Mergers and acquisitions of pharmaceutical companies inherently entail risk, and no assurance can be given that any future acquisitions will be successful or will not adversely affect our business, operating results, or financial condition. Failure to manage and successfully integrate acquisitions could harm our business and operating results in a material way. Even when an acquired company has already developed and marketed products, there can be no assurance of the continued prospects or success of such products or that pre-acquisition due diligence will have identified all possible issues that might arise with respect to such products.
We may not be able to successfully develop or commercialize our products.
While we have sales of ZADAXIN in certain markets, regulatory approval does not exist at this time for ZADAXIN in other key markets. In 2006, we acquired the rights to distribute DC Bead in China, but we must receive regulatory approval before we can commercialize this product. We previously believed that regulatory approval for DC Bead in China would be forthcoming in 2009. However, the SFDA required us to conduct a local trial in China to supplement data already obtained from a previous DC Bead study. This study is underway and if the trial results are positive, we believe we may receive regulatory approval for DC Bead in late 2011,
26
Table of Contents
although timing may be delayed if there are additional requirements requested by the SFDA. Our other potential products presently are SCV-07 and ondansetron RapidFilm and each of them is in an earlier stage of development than ZADAXIN. Clinical trials outcomes are uncertain. For example, in October 2009, we halted our phase 2 trial of RP101 and we have no plans to proceed with further development of RP101 at this time. We may consider and undertake various strategies to expand our portfolio of potential products, including acquiring product candidate rights through licenses or other relationships, or through other strategic relationships including acquisitions of other companies that may have proprietary rights to other development candidates or the capability to discover new drug candidates. Such transactions could require a substantial amount of our financial resources, or, if equity is involved, may result in substantial dilution to current stockholders. Strategic transactions also require substantial management time and effort and are subject to various risks that could adversely affect us or our financial results.
To fully develop our products, substantial resources are required for extensive research, development, pre-clinical testing, clinical trials, and manufacturing scale-up and regulatory approval prior to the potential products being ready for sale. We cannot assure that our efforts will produce commercially viable products. We face significant technological risks inherent in developing these products. We may also abandon some or all of our proposed products before they become commercially viable. For ondansetron RapidFilm, we are obligated to make a milestone payment upon regulatory approval. If any of our products, even if developed and approved, cannot be successfully commercialized in a timely manner, our business will be harmed and the price of our stock may decline.
We have not yet sold any product other than ZADAXIN and our sales have been primarily to a single country, China. Our future revenue growth depends to a great extent on increased sales of ZADAXIN to China. If we fail to successfully market ZADAXIN, our revenue and operating results will be limited. If unexpected and serious adverse events are reported, or if expected efficacy results are not achieved, it would have a material adverse effect on our business. Our future revenue will also depend in part on our ability to develop other commercially viable and accepted products, such as DC Bead, ondansetron RapidFilm, and SCV-07. Market acceptance of our products will depend on many factors, including our ability to convince prospective customers to use our products as an alternative to other treatments and therapies and to convince prospective strategic partners to market our products effectively and to manufacture our products in sufficient quantities with acceptable quality and at an acceptable cost. In addition, doctors must opt to use treatments involving our products. If doctors elect to use a different course of treatment, demand for our drug products would be reduced. Failure to do any of the above will lead to an unfavorable outcome on the results of our operations.
We cannot predict the safety profile of the use of thymalfasin, DC Bead, ondansetron RapidFilm or SCV-07 when used in combination with other drugs.
Many of our trials involve the use of thymalfasin in combination with other drugs. SCV-07 may be developed to be used in combination with other drugs. Some of these drugs, particularly pegylated interferon alpha, ribavirin, non-pegylated interferon alpha, and dacarbazine are known to cause adverse patient reactions. We cannot predict how thymalfasin, DC Bead, ondansetron RapidFilm or SCV-07 will work with other drugs, including causing possible adverse side effects not directly attributable to the other drugs that could compromise the safety profile of thymalfasin, DC Bead, ondansetron RapidFilm or SCV-07 when used in certain combination therapies.
Final results from our proposed or ongoing clinical trials for thymalfasin, SCV-07, ondansetron RapidFilm, and DC Bead may differ materially from interim or pre-clinical trial results. These clinical trials could be affected by the future actions of our partners, unexpected delays, unanticipated patient dropout rates or adverse side effects, future actions by the SFDA or the FDA or equivalent regulatory authorities in Europe or other countries or additional expenses.
Our ability to sustain operating profitability depends in part on our ability to commence, execute and complete clinical programs and obtain additional regulatory approvals for ZADAXIN and other drug candidates.
27
Table of Contents
Clinical trials are inherently risky and may reveal that our product candidates are ineffective or have unanticipated side effects and/or drug interactions that may significantly decrease the likelihood of regulatory approval. In December 2010, we announced topline results from our phase 2b clinical trial of SCV-07 for treatment of chronic hepatitis C. Although the data showed an interesting biological signal, due to the rapidly changing landscape of effective treatments which increase the complexity and risks of developing drugs for chronic HCV, we decided not to continue development in this indication. In October 2009, we announced the discontinuation of our phase 2 clinical trial evaluating RP101, a nucleoside analog known as BVdU, for the treatment of late-stage pancreatic cancer. This decision followed the recommendation of the trial’s Data Safety Monitoring Committee (“DSMC”) based upon the data reviewed and we have no plans to proceed with further development of RP101 at this time. In addition, on November 5, 2008, we announced the top-line results from a large, randomized, phase 3 clinical trial evaluating thymalfasin in combination with pegylated interferon alpha-2a (“peg-IFN-2a”) and ribavirin (“RBV”) as a treatment for patients with hepatitis C virus (“HCV”) who have not responded to prior therapy consisting of peg-IFN and RBV alone (current standard of care). The thymalfasin treated group did not achieve statistical significance for the primary end point of sustained virological response (“SVR”) as assessed in the primary analysis population, i.e. the intent-to-treat population. In the prospectively defined secondary population of patients who completed the full course of 48 weeks of treatment with thymalfasin in addition to peg-IFN-2a and RBV (“Completer Population”), the primary endpoint was achieved with statistical significance. These results did not meet our expectations based upon prior clinical trials. Similarly, the results of our thymalfasin phase 2 melanoma clinical trial do not necessarily predict future clinical or commercial success. Finally, the results of studies in DC Bead, ondansetron RapidFilm, SCV-07, including a phase 2a for oral mucositis, do not predict clinical or commercial success.
We may face numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent commercialization of our product candidates including: our product candidate clinical trials may not prove statistical significance; negative or inconclusive clinical trial results may require us to conduct further testing or we may choose to abandon projects that we had been expecting to complete; clinical trials may be halted due to safety reasons; patient dropout rates in our clinical trial may be higher than anticipated; the FDA, its European equivalent EMEA or the SFDA may not approve our products for commercialization or may require additional clinical trial data prior to approving our products; and/or our future expenses and ability to proceed with a trial may be dependent in part on finding a partner. Moreover, if the outcome of any of these trials is unsuccessful, or even if successful, does not achieve commercially meaningful results, our business could be harmed.
If third-party reimbursement is not available or patients cannot otherwise pay for ZADAXIN, DC Bead, ondansetron RapidFilm, or SCV-07, we may not be able to successfully market them.
Significant uncertainty exists as to the reimbursement status of new therapeutic products, such as ZADAXIN, and once commercialized, DC Bead, ondansetron RapidFilm and SCV-07. We cannot assure you that third-party insurance coverage and reimbursement will be available for therapeutic products we might develop. Although ZADAXIN receives some limited reimbursement in certain provinces in China, we cannot assure you that we will be able to maintain existing reimbursements or increase third-party payments for ZADAXIN or obtain third-party payments for DC Bead in China or ondansetron RapidFilm in China including Hong Kong and Macau, or Vietnam. The failure to obtain or maintain third-party reimbursement for our products would harm our business. Further, we cannot assure you that additional limitations will not be imposed in the future in the United States or elsewhere on drug coverage and reimbursement due to proposed health care reforms. In many emerging markets where we have marketing rights to ZADAXIN, but where government resources and per capita income may be so low that our products will be prohibitively expensive, we may not be able to market our products on economically favorable terms, if at all.
Efforts by governmental and third-party payers to contain or reduce health care costs or the announcement of legislative proposals or reforms to implement government controls could cause us to reduce the prices at which we market our drugs, which would reduce our gross margins and may harm our business.
28
Table of Contents
If we do not maintain regulatory approval for thymalfasin or obtain regulatory approval for SCV-07, ondansetron RapidFilm or DC Bead for the intended indications that we are evaluating, our revenues will be limited and our operating results may be materially affected.
Our ability to execute on our business strategy is largely dependent on our ability to maintain regulatory approval for the use of thymalfasin, and obtain regulatory approval for the use of SCV-07 in major markets, excluding Russia, for the use of ondansetron RapidFilm in China including Hong Kong and Macau, and Vietnam, and for the use of DC Bead in China. The regulatory approval processes in the United States, Europe and China are demanding and typically require 12 months or more in the United States and China and 18 months or more in Europe from the date of submission of an NDA. We have committed significant resources, including capital and time, to develop these products, and intend to continue to do so, including the initiation and execution of additional clinical trials, with the goal of obtaining such approvals. If we do not obtain these approvals, we will be unable to achieve any revenue from these products in these major markets and our thymalfasin sales in other jurisdictions could decline.
We believed that regulatory approval for DC Bead would be forthcoming in 2009. However, the SFDA required us to conduct a local trial in China to supplement data already obtained from a previous DC Bead study. The study is underway and if the trial results are positive, we believe we may receive regulatory approval for DC Bead in late 2011. However, we cannot give assurance that such submission will be approved by the regulatory authorities. If additional clinical trials in China are required as part of the regulatory process, the regulatory submission for marketing approval could be delayed for a considerable period of time, and there can be no assurance that the results of clinical trials would support a regulatory submission or that the regulatory authorities would approve the product to be commercialized and sold in China. To the extent that additional information or clinical trials are required by the regulatory authorities, or we do not receive regulatory approval in China, our future sales potential in China could be harmed.
All new drugs, including our products, which have been developed or are under development, are subject to extensive and rigorous regulation by the FDA, SFDA and similar regulatory agencies. These regulations govern, among other things, the development, testing, manufacturing, labeling, storage, pre-market approval, importation, advertising, promotion, sale and distribution of our products. These regulations may change from time to time and new regulations may be adopted.
Satisfaction of government regulations may take several years and the time needed to satisfy them varies substantially based on the type, complexity and novelty of the pharmaceutical product. As a result, government regulation may cause us to delay the introduction of, or prevent us from marketing, our existing or potential products for a considerable period of time and impose costly procedures upon our activities. Even if we obtain regulatory approval for our products, such approval may impose limitations on the indicated uses for which our products may be marketed. Further unsatisfactory data resulting from clinical trials may also adversely affect our ability to market and sell thymalfasin in markets where it is approved for sale.
We rely on third parties who are our sole source suppliers for our clinical trial and commercial products and their inability to deliver products that meet our quality-control standards could delay or harm one or more important areas of our business including our sales, clinical trials or the regulatory approval process.
We rely on third parties, who are subject to regulatory oversight, to supply our commercial products. Any deficiencies or shortages in supply of our commercial products would adversely affect our ability to realize our sales plans. For example, the manufacturing of the raw material and the processing to finished product of ZADAXIN is done in few batches in any given three-month period and any manufacturing errors have the potential to require a product recall. We currently have only one approved finished vial manufacturer and two approved active pharmaceutical ingredient (“API”) suppliers. If we experience a problem with the manufacturer or our suppliers our sales may suffer. During 2006, we experienced failures and lower yields on production runs from our sole supplier of bulk API product for the manufacture of ZADAXIN for our current markets, including
29
Table of Contents
China. During 2009, we experienced quality-control problems with a component of our ZADAXIN kit. During 2010, we experienced difficulties validating upgrades to equipment with one of our API manufacturers. Although we are taking steps to ensure that such problems do not continue, there is no assurance that we will either be successful in doing so with our current supplier or be able to timely and cost-effectively qualify new suppliers for this component. Manufacturing interruptions or failure or delay of product to meet quality assurance specifications could adversely affect shipments and recognition of sales of ZADAXIN in any period and impair our relationships with customers and our competitive position and may increase the cost of material produced.
The process of registering new suppliers for ZADAXIN with regulatory agencies in markets where thymalfasin is approved for sale, including China quality assurance and other steps could cause delays or interruptions of supply in certain other markets. In some countries, a manufacturing change may require additional regulatory approvals that may delay thymalfasin marketing approvals in new markets. In addition, if sales of ZADAXIN were to increase dramatically, our third-party suppliers may not be able to supply ZADAXIN either quickly enough or at a commercially reasonable cost, which could limit our ability to satisfy increased demand or could adversely affect the ability of these suppliers to provide products for our clinical trials. If any of our suppliers are unable to match our need for supply, either because of product defects, inability to increase supply in the face of increased demand, or maintain financially viable businesses, our ability to affect our sales and protect our brand reputation would be materially impaired, thereby materially and adversely affecting our sales and results of operations. From time-to-time, we have to apply for approval from SFDA for a revised Import Drug List due to changes to the ZADAXIN packaging, label and registration resulting from the additions or replacement of primary and secondary manufacturers and suppliers which could cause delays or interruptions in the supply of ZADAXIN.
We also rely on third parties, who are subject to regulatory oversight, to supply drug product for our clinical trials. For example, Biocompatibles is the sole supplier of DC Bead, Applied Pharma Research S.A. (“APR”) is the sole supplier of ondansetron RapidFilm, and Solvay Peptides S.A. is our sole supplier of SCV-07. Any unanticipated deficiencies in these suppliers, or the suppliers of our raw materials, and/or recall of the manufacturing lots or similar action regarding the pegylated interferon alpha or ribavirin used in our clinical trials could delay the trials or detract from the integrity of the trial data and its potential use in future regulatory filings. In addition, manufacturing interruptions or failure to comply with regulatory requirements by any of these suppliers could significantly delay clinical development of potential products and reduce third-party or clinical researcher interest and support of proposed trials. These interruptions or failures could also impede commercialization of our products and impair our competitive position.
We rely on third parties for development of our products and the inability of any of these parties to reliably, timely or cost-effectively provide us with their obligated services could materially harm the timing of bringing our products to market and accordingly adversely affect our business.
We rely on third parties, such as contract research organizations, medical institutions, clinical investigators, contract laboratories, and collaborative partners in the conduct of clinical trials for our product candidates. If these parties, whom we do not control, do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines or choose not to continue their relationship with us, if the third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical or clinical activities may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize our product candidates.
Commercialization of some of our products depends on collaborations with others. If our collaborators are not successful, or if we are unable to find future collaborators, we may not be able to properly develop and commercialize our products.
We depend in part on our distributors and business partners to develop or promote our drugs, and if they are not successful in their efforts or fail to do so, our business will suffer. For example, Sigma-Tau is responsible for
30
Table of Contents
the development and marketing of ZADAXIN in most of Europe. Biocompatibles is providing SciClone with product samples and the necessary supporting documents to obtain regulatory approval in China for DC Bead, APR’s manufacturing partner Labtec GmbH is providing SciClone with the product samples, and APR is providing SciClone with the necessary supporting documents to obtain regulatory approval in China including Hong Kong and Macau, and Vietnam for ondansetron RapidFilm. We generally do not have control over the amount and timing of resources that our business partners devote to our collaborative efforts, and some have not always performed as or when expected. If they do not perform their obligations as we expect, particularly obligations regarding clinical trials, our development expenses would increase and the development or sale of our products could be limited or delayed, which could hurt our business and cause our stock price to decline. In addition, our relationships with these companies may not be successful. Disputes may arise with our collaborators, including disputes over ownership rights to intellectual property, know-how or technologies developed with our collaborators. We may not be able to negotiate similar additional arrangements in the future to develop and commercialize ZADAXIN or other products.
If we are unable to retain our key personnel, or are unable to attract and retain additional, highly skilled and experienced personnel, including the ability to expand our sales staff, our business will suffer.
We are highly dependent upon our ability to attract and retain qualified personnel because of the specialized, scientific and worldwide nature of our business. Further, we are also dependent on our ability to appropriately staff these personnel in appropriate positions as our business fluctuates. There is intense competition for qualified management, scientific, clinical, regulatory, and sales and marketing personnel in the pharmaceutical industry. There is significant turnover in the industry in China in particular, and we have also experienced turnover in our sales personnel. We may not be able to attract and retain the qualified personnel we need to grow and develop our business globally. Conversely, in the event that we need to reduce the size of a particular aspect of our business, we are also dependent on our ability to make such adjustments while retaining suitably skilled personnel. Further, our efforts to in-license or acquire, develop and commercialize product candidates for China require the addition of clinical and regulatory personnel and the capabilities to expand our sales and marketing operation. In addition, we assign numerous key responsibilities to a limited number of individuals, and we would experience difficulty in finding immediate replacements for any of them were any one of them to choose to leave employment with us.
Depending upon the results of the investigation into matters including compliance with the FCPA we could determine to undertake corrective measures which could have adverse effects on our business, including the loss of personnel, and changes in marketing, sales and educational practices or programs. If we were unable to attract and retain qualified personnel as needed or promptly replace those employees who are critical to our product development and commercialization, the development and commercialization of our products would be adversely affected. At this time, we do not maintain “key person” life insurance for any of our personnel.
We may need to obtain additional funding to support our long-term product development and commercialization programs.
We believe our existing cash and investments and ongoing revenue generating business operations will be sufficient to support our current operating plan for at least the next 12 months. However, our ability to achieve and sustain operating profitability is dependent on numerous factors including our ability to achieve our goal of increasing sales of ZADAXIN, securing regulatory approval for DC Bead in China, and for ondansetron RapidFilm in China including Hong Kong and Macau, and Vietnam, the execution and successful completion of thymalfasin, SCV-07, and DC Bead clinical trials and securing partnerships for those programs that lead to regulatory approvals in major pharmaceutical markets. We cannot assure you that such funds from operating activities will be sufficient, or that we will attain profitable operations in future periods. In addition, we intend to develop other products and we may need additional funds in the future to support such development and to support future growth and achieve profitability. If we need to raise additional funds in the future and such funds
31
Table of Contents
are not available on reasonable terms, if at all, our commercialization efforts may be impeded, our revenues may be limited and our operating results may suffer.
If we fail to protect our products, technologies and trade secrets, we may not be able to successfully use, manufacture, market or sell our products, or we may fail to advance or maintain our competitive position, and we have limited intellectual property protection in China.
Our success depends significantly on our ability to obtain and maintain meaningful patent protection for our products and technologies and to preserve our trade secrets. Our pending patent applications may not result in the issuance of patents in the future. Our patents or patent applications may not have priority over others’ applications. Our existing patents and additional patents that may be issued, if any, may not provide a competitive advantage to us or may be invalidated or circumvented by our competitors. Others may independently develop similar products or design around patents issued or licensed to us. Patents issued to, or patent applications filed by, other companies could harm our ability to use, manufacture, market or sell our products or maintain our competitive position with respect to our products. Although many of our patents relating to thymalfasin have expired, including composition of matter patents, we have rights to other patents and patent applications relating to thymalfasin and thymalfasin analogues, including method of use patents with respect to the use of thymalfasin for certain indications. Additionally, thymosin alpha 1 (“thymalfasin”), the chemical composition of thymalfasin, has received Orphan Drug designation in the United States for the treatment of stage IIb through stage IV melanoma. If other parties develop generic forms of thymalfasin for other indications, including conducting clinical trials for such indications, our patents and other rights might not be sufficient to prohibit them from marketing and selling such generic forms of thymalfasin. If other parties develop analogues or derivatives of thymalfasin, our patents and other rights might not be sufficient to prohibit them from marketing these analogues or derivatives.
Pharmaceutical products are either not patentable or have only recently become patentable in some of the countries in which we market or may market thymalfasin. We do not have composition patent claims directed to the same form of thymalfasin currently marketed in China, our largest market, although we do have other type of patent claims, pending or issued, directed to other aspects of thymalfasin therapy. Other companies market generic thymalfasin in China, sometimes in violation of our patent, trademark or other rights which, to date, we have defended by informing physicians and hospitals of the practice. Past enforcement of intellectual property rights in many of these countries, including China in particular, has been limited or non-existent. Future enforcement of patents and proprietary rights in many other countries will likely be problematic or unpredictable. Moreover, the issuance of a patent in one country does not assure the issuance of a similar patent in another country. Claim interpretation and infringement laws vary by nation, so the extent of any patent protection is uncertain and may vary in different jurisdictions.
If we are involved in intellectual property claims and litigation, the proceedings may divert our resources and subject us to significant liability for damages, substantial litigation expense and the loss of our proprietary rights.
Our commercial success depends in part on our not infringing valid, enforceable patents or proprietary rights of third parties, and not breaching any licenses that may relate to our technologies and products. In addition, we may not be aware of all patents or patent applications that may impact our ability to make, use or sell any of our potential products. For example, U.S. patent applications may be kept confidential for 12 or more months while pending in the Patent and Trademark Office, and patent applications filed in foreign countries are often first published nine months or more after filing. It is possible that we may unintentionally infringe these patents or other patents or proprietary rights of third parties. We may in the future receive notices claiming infringement from third parties as well as invitations to take licenses under third-party patents. Any legal action against us or our collaborative partners claiming damages and seeking to enjoin commercial activities relating to our products and processes affected by third-party rights may require us or our collaborative partners to obtain licenses in order to continue to manufacture or market the affected products and processes. Our efforts to defend
32
Table of Contents
against any of these claims, regardless of merit, would require us to devote resources and attention that could have been directed to our operations and growth plans. In addition, these actions may subject us to potential liability for damages. We or our collaborative partners may not prevail in a patent action and any license required under a patent may not be made available on commercially acceptable terms, or at all. Any conflicts resulting from the patent rights of others could significantly reduce the coverage of our patents and limit our ability to obtain meaningful patent protection.
If other companies obtain patents with conflicting claims, we may be required to obtain licenses to those patents or develop or obtain alternative technology to manufacture or market the affected products and processes. We may not be able to obtain any such licenses on acceptable terms or at all. Any failure to obtain such licenses could delay or prevent us from pursuing the development or commercialization of our potential products. Our efforts to defend against any of these claims would require us to devote resources and attention that could have been directed to our operations and growth plans.
We may need to initiate litigation, which could be time-consuming and expensive, to enforce our proprietary rights or to determine the scope and validity of others’ rights. If litigation results, a court may find our patents or those of our licensors invalid or may find that we have infringed on a competitor’s rights. If any of our competitors have filed patent applications in the United States which claim technology we also have invented, the Patent and Trademark Office may require us to participate in expensive interference proceedings to determine who has the right to a patent for the technology. These actions may subject us to potential liability for damages. We or our collaborative partners may not prevail in a patent action and any license required under a patent may not be made available on commercially acceptable terms, or at all.
Substantial sales of our stock or equity in our subsidiaries or the exercise or conversion of options or convertible securities may impact the market price of our common stock.
Our collaborative partner Sigma-Tau and affiliates hold a substantial amount of our stock. The stock is freely tradable and Sigma-Tau is not under any obligation to SciClone which would prevent it from selling some or all of the stock it holds except for applicable U.S. insider trading regulations with respect to possession of material non-public information by Sigma-Tau or its officers and directors.
In May 2009, we filed a Form S-3 Shelf registration with the SEC which was later declared effective by the SEC and will allow us to sell securities in one or more offerings. Future issuances of substantial amounts of our common stock could adversely affect the market price of our common stock. Similarly, if we raise additional funds through the issuance of common stock or securities convertible into or exercisable for common stock or sell equity in a subsidiary, the percentage ownership of our present stockholders of the respective entities will be reduced and the price of our common stock may fall.
Our cash and investments are subject to certain risks which could materially adversely affect our overall financial position.
We invest our cash in accordance with an established internal policy and customarily in instruments which historically have been highly liquid and carried relatively low risk. However, with turmoil in the credit markets, similar types of investments have experienced losses in value or liquidity issues which differ from their historical pattern. For example, we routinely have invested in money market funds with large financial institutions. One or more of these funds could experience losses or liquidity problems and, although to date some of the largest financial institutions who sponsor such funds have offset similar losses, there is no assurance that our financial institutions would either not incur losses or would offset any losses were they to occur.
Should any of our cash investments permanently lose value or have their liquidity impaired, it would have a material and adverse effect on our overall financial position by imperiling our ability to fund our operations and
33
Table of Contents
forcing us to seek additional financing sooner than we would otherwise and such financing may not be available on commercially attractive terms.
In addition, financial instruments may subject us to a concentration of credit risk. Most of our cash, and cash equivalents are held by a limited number of financial institutions including all of our term deposits held by financial institutions in Hong Kong and offshore. Such term deposits do not exceed federally insured limits and to date, we have not experienced any losses on our deposits of cash and cash equivalents. However, if any of these instruments permanently lost value or had their liquidity impaired, it would also have a material and adverse effect on our overall financial position by imperiling our ability to fund our operations and forcing us to seek additional financing sooner than we would otherwise and such financing may not be available on commercially attractive terms.
New accounting pronouncements may impact our financial position or results of operations.
Future changes in financial accounting standards may cause adverse, unexpected fluctuations in the timing of the recognition of revenues or expenses and may affect our financial position or results of operations. New pronouncements and varying interpretations of pronouncements have occurred with frequency and may occur in the future and this may lead to changes in our accounting policies in the future.
We have material weaknesses in our internal control over financial reporting and if we are not able to remediate these weaknesses, we may not be able to accurately report our financial results. As a result, current and potential stockholders could lose confidence in our financial reporting, which would harm our business and the trading price of our stock.
Effective internal controls are necessary for us to provide reliable financial reports and to protect from fraudulent, illegal or unauthorized transactions. If we cannot provide effective controls and reliable financial reports, our business and operating results could be harmed. Moreover, as a United States-based corporation doing business in China, these controls often need to satisfy the requirements of Chinese law as well as the requirements of United States law which frequently differ in certain aspects. Our management has determined that as of December 31, 2010, we had two material weaknesses in our internal control over financial reporting that have not been remediated. The material weaknesses relate to our controls over (i) our implementation of our policy on compliance with laws and (ii) our accounting for income taxes, as discussed in Item 9A of this report. In addition, we determined that our disclosure controls were not working effectively as of December 31, 2010. Although we are taking steps to remediate these material weaknesses, there can be no assurance that we will be successful in this regard. Any failure to implement and maintain controls over our financial reporting, or difficulties encountered in the implementation of improvements in our controls, could cause us to fail to meet our reporting obligations. Any failure to improve our internal controls or to address identified weaknesses in the future, including with respect to our clinical research expenses, if they were to occur, could also cause investors to lose confidence in our reported financial information, which could have a negative impact on the trading price of our stock.
New legislation may impact our financial position or results of operations.
Compliance with changing regulations concerning corporate governance and public disclosure has resulted in and may continue to result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new SEC regulations and The NASDAQ Stock Market rules, are creating uncertainty for companies such as ours and costs are increasing as a result of this uncertainty and other factors. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment has and may continue to result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
34
Table of Contents
Currently a majority of our revenue is generated from customers located outside the United States, and a substantial portion of our assets, including employees, are located outside the United States. United States income taxes and foreign withholding taxes have not been provided on undistributed earnings of non-United States subsidiaries, because such earnings are intended to be indefinitely reinvested in the operations of those subsidiaries. The United States government may propose initiatives that would substantially reduce our ability to defer U.S. taxes including: repealing deferral of U.S. taxation of foreign earnings, eliminating utilization or substantially reducing our ability to claim foreign tax credits, and eliminating various tax deductions until foreign earnings are repatriated to the United States. If any of these proposals are constituted into legislation, they could increase our U.S. income tax liability and as a result have a negative impact on our financial position and results of operations.
Anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us, which may be beneficial to our stockholders, more difficult.
Certain anti-takeover provisions of Delaware law and our charter documents as currently in effect may make a change in control of our company more difficult, even if a change in control would be beneficial to our stockholders. Our charter documents contain certain anti-takeover provisions, including provisions in our certificate of incorporation providing that stockholders may not cumulate votes, stockholders’ meetings may be called by stockholders only if they hold 25% or more of our common stock and provisions in our bylaws providing that the stockholders may not take action by written consent. Additionally, our board of directors has the authority to issue 10 million shares of preferred stock and to determine the terms of those shares of stock without any further action by the stockholders. The rights of holders of our common stock are subject to the rights of the holders of any preferred stock that may be issued. The issuance of preferred stock could make it more difficult for a third-party to acquire a majority of our outstanding voting stock. Delaware law also prohibits corporations from engaging in a business combination with any holders of 15% or more of their capital stock until the holder has held the stock for three years unless, among other possibilities, the board of directors approves the transaction. Our board of directors may use these provisions to prevent changes in the management and control of our company. Also, on December 18, 2006, our board of directors declared a dividend distribution of one Preferred Stock Purchase Right (collectively, the “Rights”) for each outstanding share of our Common Stock, each Right which entitles the registered holder to purchase from the Company one one-thousandth of a share of the Company’s Series D Preferred Stock, $0.001 par value, at a price of $25.00 pursuant to a Rights Agreement dated as of December 19, 2006, between the Company and Mellon Investor Services LLC. The Rights have certain anti-takeover effects. Under certain circumstances the Rights could cause substantial dilution to a person or group who attempts to acquire the Company on terms not approved by our board of directors. Although the Rights should not interfere with an acquisition of the Company approved by the board, the Rights may have the effect of delaying and perhaps improving the terms of an acquisition for our stockholders, or deterring an acquisition of the Company. Also, under applicable Delaware law, our board of directors may adopt additional anti-takeover measures in the future.
We may be subject to product liability lawsuits, and our insurance may be inadequate to cover damages.
Clinical trials of any of our current and potential products or the actual commercial sales of our product may expose us to liability claims from the use of these products. We currently carry product liability insurance. However, we cannot be certain that we will be able to maintain insurance on acceptable terms, if at all, for clinical and commercial activities or that the insurance would be sufficient to cover any potential product liability claim or recall. If we fail to have sufficient coverage, our business, results of operations and cash flows could be adversely affected.
If we are unable to comply with environmental and other laws and regulations, our business may be harmed.
We are subject to various federal, state and local laws, regulations and recommendations relating to the use, manufacture, storage, handling and disposal of hazardous materials and waste products (including radioactive
35
Table of Contents
compounds and infectious disease agents), as well as safe working conditions, laboratory and manufacturing practices and the experimental use of animals. The extent of government regulation that might result from future legislation or administrative action in these areas cannot be accurately predicted.
We do not currently maintain hazardous materials at our facilities. While we outsource our research and development programs involving the controlled use of biohazardous materials, if in the future we conduct these programs ourselves, we might be required to incur significant cost to comply with environmental laws and regulations. Further, in the event of an accident, we would be liable for any damages that result, and the liability could exceed our resources.
Our business and operations are subject to the risks of being based in particular locations known for earthquakes, other natural catastrophic disasters and service interruptions.
Our corporate headquarters are located in the Silicon Valley area of Northern California, a region known for seismic activity. Although we maintain a disaster recovery policy that includes storage of important corporate data in a different geographic region of the United States, all of our significant corporate data is stored in our headquarters facility and accordingly, a significant natural disaster, such as an earthquake, could have a material adverse impact on our business, operating results, and financial condition. Most of our sales are into China for which we maintain a sole warehouse for finished goods in Hong Kong, which can experience severe typhoon storms, earthquakes or other natural catastrophic disasters. Although our distributors in China may maintain several months supply of our product, were our warehouse capability to be interrupted, either through a natural disaster such as flooding or through a service interruption, such as a lack of electricity to power required air conditioning, our ability to timely deliver finished product to China could be adversely affected which in turn would materially adversely affect our sales and ensuing operating results.
We May Be Affected by Climate Change and Market or Regulatory Responses to Climate Change
Climate change, including the impact of global warming, could have a material adverse effect on our results of operations, financial condition, and liquidity if it were to disrupt the demand, supply or delivery of product, management of our business, or result in cost increases as a result of government regulation.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We currently lease approximately 22,000 square feet of office space for our corporate headquarters in Foster City, California and lease approximately 12,000 square feet of office space in Beijing, Hong Kong, Shanghai, Singapore, Guangzhou, and Vietnam. We believe that our existing facilities will be adequate for our current needs and that additional space will be available as needed.
| Item 3. | Legal Proceedings |
In August 2010, a purported securities class action lawsuit was filed in the United States District Court for the Northern District of California, naming us and certain of our officers as defendants. In September 2010, a second purported securities class action lawsuit was filed in the same court. The lawsuits alleged violations of the Securities Exchange Act of 1934, as amended, in connection with allegedly false, misleading and incomplete statements issued by the defendants related to potential violations of the Foreign Corrupt Practices Act, our reported revenues, income and sales growth, and marketing and sales activities. Plaintiffs sought damages, an award of their costs and attorney’s fees, and injunctive and/or equitable relief on behalf of a purported class of stockholders who purchased our common stock during the period between May 11, 2009 and August 10, 2010.
36
Table of Contents
On October 27, 2010, the securities class action lawsuits were consolidated under the caption In re SciClone Pharmaceuticals, Inc. Securities Litigation, Case No. CV 10-03584-JW, and the court appointed lead plaintiffs. Plaintiffs were ordered by the court to file an amended consolidated complaint on or before November 29, 2010. On November 24, 2010, before filing an amended complaint, the parties stipulated to the voluntary dismissal of the case without prejudice. Plaintiffs may re-file the complaint at a later date.
In September 2010, three derivative lawsuits were filed purportedly on behalf of the Company in California Superior Court for the County of San Mateo naming certain of our officers and directors as defendants. The lawsuits assert claims for breach of fiduciary duty, abuse of control, unjust enrichment and corporate waste based on alleged violations of the Foreign Corrupt Practices Act. Plaintiffs seek damages, restitution and injunctive and/or equitable relief purportedly on behalf of the Company, and an award of their costs and attorney’s fees. On January 13, 2011, the derivative lawsuits were consolidated into a single action under the caption In re SciClone Pharmaceuticals, Inc. Shareholder Derivative Litigation, Case No. CIV 499030, and the court appointed lead plaintiffs’ counsel. The court ordered plaintiffs to file a consolidated complaint on or before March 18, 2011. On February 28, 2011, the parties agreed to a temporary stay of the consolidated action until and including April 15, 2011, subject to further applications of the parties to extend the stay, pending completion of the Company’s internal investigation.
While Company management believes that we have meritorious defenses and intend to defend these lawsuits vigorously, these lawsuits and any other related lawsuits are subject to inherent uncertainties, and the actual cost will depend upon many unknown factors. The outcome of the litigation is necessarily uncertain. We could be forced to expend significant resources in the defense of this lawsuit and we may not prevail.
On November 3, 2010, the board of directors received a letter from a purported stockholder demanding that the board of directors take actions to remedy alleged breaches of fiduciary duties by the directors and certain officers relating to alleged violations of the FCPA and other securities laws. The board responded to the demand letter and informed the shareholder that the board would consider the demand after its internal investigation is completed.
| Item 4. | Removed and Reserved |
37
Table of Contents
PART II
| Item 5. | Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock trades on The NASDAQ Global Market of the NASDAQ Stock Market under the symbol “SCLN.”
The following table sets forth the high and low sales prices per share for the quarterly periods indicated, as reported by The NASDAQ Stock Market. The quotations shown represent inter-dealer prices without adjustment for retail markups, markdowns, or commissions, and may not necessarily reflect actual transactions.
| Price Range Common Stock |
||||||||
| High | Low | |||||||
| 2010 |
||||||||
| 4th quarter |
$ | 4.34 | $ | 2.55 | ||||
| 3rd quarter |
3.67 | 2.08 | ||||||
| 2nd quarter |
4.50 | 2.65 | ||||||
| 1st quarter |
4.30 | 2.35 | ||||||
| 2009 |
||||||||
| 4th quarter |
$ | 4.41 | $ | 2.09 | ||||
| 3rd quarter |
5.33 | 2.50 | ||||||
| 2nd quarter |
2.77 | 1.21 | ||||||
| 1st quarter |
1.41 | 0.75 | ||||||
Stockholders
As of March 11, 2011, there were approximately 348 holders of record of our common stock and 48,040,125 shares of common stock issued and outstanding.
Dividends
We have not paid any dividends on our common stock during the fiscal years ended December 31, 2010, 2009, and 2008 and currently intend to retain any future earnings for use in our business.
Securities Authorized for Issuance Under Equity Compensation Plans
The information required by Item 201(d) of Regulation S-K is incorporated by reference from the section entitled “Securities Authorized for Issuance under Equity Compensation Plans” in Part III, Item 12 of this Form 10-K.
38
Table of Contents
Performance Graph
The following line graph compares the annual percentage change in (i) the cumulative total stockholder return on the Company’s Common Stock since December 31, 2005, with (ii) the cumulative total return on (a) The NASDAQ Composite Index and (b) the NASDAQ Biotechnology Index. The comparison assumes (i) an investment of $100 on December 31, 2005 in each of the foregoing indices and (ii) reinvestment of dividends, if any. The stock price performance shown on the graph below is not necessarily indicative of future stock price performance.
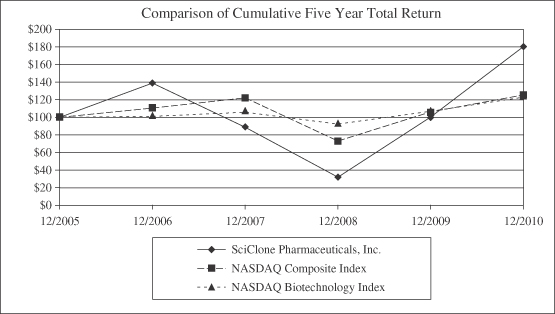
39
Table of Contents
| Item 6. | Selected Financial Data |
This section presents selected historical financial data for each of the last five fiscal years and is qualified by reference to and should be read in conjunction with the consolidated financial statements and notes thereto included elsewhere in this Annual Report on Form 10-K. The selected balance sheets data at December 31, 2010 and 2009 and the selected statement of operations data for each year ended December 31, 2010, 2009, and 2008 have been derived from our audited financial statements that are included elsewhere in this report. The selected balance sheets data at December 31, 2008, 2007, and 2006 and the statements of operations for each year ended December 31, 2007 and 2006 have been derived from our audited financial statements not included in this report. Historical results are not necessarily indicative of the results to be expected in the future.
| Year Ended December 31, | ||||||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| Statement of Operations data: |
||||||||||||||||||||
| Product sales |
$ | 85,112 | $ | 72,411 | $ | 54,108 | $ | 37,038 | $ | 32,433 | ||||||||||
| Contract revenue |
— | — | 5 | 20 | 229 | |||||||||||||||
| Total revenues |
85,112 | 72,411 | 54,113 | 37,058 | 32,662 | |||||||||||||||
| Cost of product sales |
12,691 | 11,960 | 10,299 | 7,088 | 7,235 | |||||||||||||||
| Gross margin |
72,421 | 60,451 | 43,814 | 29,970 | 25,427 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
12,415 | 16,531 | 22,857 | 16,552 | 13,399 | |||||||||||||||
| Sales and marketing |
22,006 | 18,805 | 16,853 | 13,575 | 11,223 | |||||||||||||||
| General and administrative |
15,606 | 12,521 | 12,538 | 11,139 | 9,729 | |||||||||||||||
| Total operating expenses |
50,027 | 47,857 | 52,248 | 41,266 | 34,351 | |||||||||||||||
| Income (loss) from operations |
22,394 | 12,594 | (8,434 | ) | (11,296 | ) | (8,924 | ) | ||||||||||||
| Interest and investment income |
105 | 153 | 608 | 1,629 | 1,764 | |||||||||||||||
| Interest expense |
(195 | ) | (179 | ) | (31 | ) | (20 | ) | (94 | ) | ||||||||||
| Other income (expense), net |
953 | 18 | (16 | ) | 40 | 7,981 | ||||||||||||||
| Income (loss) before provision for income tax |
23,257 | 12,586 | (7,873 | ) | (9,647 | ) | 727 | |||||||||||||
| Provision for income tax |
2,176 | 641 | 475 | 301 | — | |||||||||||||||
| Net income (loss) |
$ | 21,081 | $ | 11,945 | $ | (8,348 | ) | $ | (9,948 | ) | $ | 727 | ||||||||
| Earnings (loss) per share: |
||||||||||||||||||||
| Basic net income (loss) per share |
$ | 0.44 | $ | 0.26 | $ | (0.18 | ) | $ | (0.22 | ) | $ | 0.02 | ||||||||
| Diluted net income (loss) per share |
$ | 0.43 | $ | 0.25 | $ | (0.18 | ) | $ | (0.22 | ) | $ | 0.02 | ||||||||
| Weighted average shares used in computing: |
||||||||||||||||||||
| Basic net income (loss) per share |
47,624 | 46,574 | 46,212 | 46,100 | 45,901 | |||||||||||||||
| Diluted net income (loss) per share |
49,414 | 47,135 | 46,212 | 46,100 | 46,072 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Balance Sheet data: |
||||||||||||||||||||
| Cash, cash equivalents, and short-term investments |
$ | 56,142 | $ | 31,333 | $ | 27,773 | $ | 35,209 | $ | 41,894 | ||||||||||
| Accounts receivable |
30,671 | 21,394 | 11,927 | 12,650 | 13,277 | |||||||||||||||
| Inventories |
7,078 | 10,149 | 6,056 | 5,579 | 3,232 | |||||||||||||||
| Long-term investments |
— | — | 1,485 | — | — | |||||||||||||||
| Restricted investments |
380 | 486 | 457 | 72 | 698 | |||||||||||||||
| Working capital |
83,819 | 55,937 | 37,793 | 45,400 | 53,079 | |||||||||||||||
| Total assets |
97,807 | 66,900 | 51,905 | 58,659 | 62,584 | |||||||||||||||
| Long-term borrowing on line of credit |
2,500 | — | — | — | — | |||||||||||||||
| Long-term liabilities |
990 | 979 | 779 | 341 | 68 | |||||||||||||||
| Total stockholders’ equity |
82,188 | 57,393 | 40,903 | 47,259 | 54,634 | |||||||||||||||
40
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
This Management’s Discussion and Analysis of Financial Condition and Results of Operations should be read in conjunction with the “Selected Financial Data” and our consolidated financial statements and related notes thereto included elsewhere in this Annual Report on Form 10-K. This Management’s Discussion and Analysis of Financial Condition and Results of Operations and other parts of this Annual Report on Form 10-K contain forward-looking statements which involve risks and uncertainties. See “Note Regarding Forward-Looking Statements” and “Risk Factors” contained in this Annual Report on Form 10-K.
Overview
SciClone Pharmaceuticals (NASDAQ: SCLN) is a revenue-generating, China-centric, specialty pharmaceutical company with a substantial international business and a product portfolio of novel therapies for cancer and infectious diseases. We are focused on continuing international sales growth, while containing clinical development and other costs.
Our international business and corporate strategy is focused primarily on the People’s Republic of China (“China”) where we believe we have built a solid reputation and established a strong brand through our many years of experience marketing ZADAXIN. We believe these strengths position us to benefit from expanding pharmaceutical markets in China and elsewhere. We believe China will rank second among global pharmaceutical markets by 2015, with projected annual growth rates of more than 20% annually over the next several years. We seek to grow our sales in the region while we leverage our strong balance sheet for mergers and acquisitions (“M&A”), in-licensing and product acquisitions.
ZADAXIN, our brand of thymalfasin or thymosin alpha 1, has regulatory approval in over 30 countries for the treatment of HBV, as a vaccine adjuvant, for the treatment of HCV, and certain cancers. All of our revenues are derived from sales of ZADAXIN, substantially most of which is sold to China. To continue to grow ZADAXIN sales to China, our sales force of over 200 people is focused on increasing sales to the country’s largest hospitals (class 3 with over 500 beds) as well as smaller hospitals (class 2). These hospitals serve Tier 1 and Tier 2 cities which are the largest, and generally have the most affluent populations. In China, they are mainly located in the eastern portion of the nation.
As our approval in China is an Imported Drug License, all sales are made to licensed importers. Most of our sales are made to two such importers and in recent months, Sinopharm Group Co. Limited acquired a majority interest in these two large importers. To date, we have been able to maintain our gross margin in part due to relatively stable or even decreasing costs of sales, and in part due to maintaining a relatively stable sales price. We anticipate that gross margins will remain in the historical range, however prices in China are subject to regulation, and are currently under review by regulatory authorities, and if a substantial reduction in the sale price to hospitals occurred, our gross margins would be substantially reduced. We believe our current cash and investment position and the gross profit from our product sales provide the financial resources to execute on our strategy for continued growth in our international business and for development of our pipeline of late-stage product candidates.
We continue to look for M&A or in-licensing opportunities of late-stage or approved branded, well-differentiated products that are approved or have a clear regulatory pathway. To date, we have in-licensed two products that are part of this international commercial growth strategy, DC Bead® and ondansetron RapidFilm®. We are working towards regulatory approval in China for these in-licensed candidates.
DC Bead, an in-licensed product for which we have commercialization rights in China, is a novel treatment for advanced liver cancer and is currently approved in approximately 40 countries worldwide, including countries in the European Union (“EU”) and the U.S. We commenced a small local registration trial in 2010 which has completed enrollment of approximately 40 advanced liver cancer patients at several liver cancer treatment centers. The primary objective is safety and the secondary objective is efficacy, as measured by tumor response. We believe we may receive regulatory approval for DC Bead in late 2011.
41
Table of Contents
Our second in-licensed product as part of this international commercial growth strategy is ondansetron RapidFilm. This product is an oral thin film formulation of ondansetron, a serotonin 5-HT3 receptor antagonist commonly used to treat and prevent nausea and vomiting caused by chemotherapy, radiotherapy and surgery. In March 2010, we announced that approval was granted for ondansetron RapidFilmTM under the EU decentralized procedure in 16 major EU countries. We have commercialization rights for this product in China including Hong Kong and Macau, and Vietnam, and we are working towards filing a clinical trial application in 2011 as part of the product registration process which will begin the regulatory approval process in China.
Outside of China, SciClone’s clinical development strategy is focused on driving cost-efficient phase 1 and 2 development of promising compounds while seeking development partners for more costly phase 3 trials. We are currently developing SCV-07, a small molecule synthetic peptide with immunomodulating properties, in a phase 2b clinical trial for the prevention of oral mucositis (“OM”).
OM is a common, painful, debilitating complication of cancer treatment, and we estimate that medical costs for the treatment of oral mucositis were approximately $4.2 billion in the U.S. and $10 billion worldwide in 2010. Approximately 450,000 patients per year in the U.S. suffer from OM during cancer therapy. In May 2010, we announced top-line results from our phase 2a proof of concept clinical trial of SCV-07 to modify the course of oral mucositis in patients with head and neck cancer receiving chemoradiotherapy regimens.
Based on the findings from the phase 2a study and discussions with the U.S. Food and Drug Administration, we announced the enrollment of the first patient in the Company’s phase 2b clinical trial of SCV-07 for the prevention of OM in January of 2011. The 2b study is examining three doses of SCV-07, including two higher doses than those used in the Company’s recent phase 2a study, to assess the drug’s impact on modifying the course of OM in patients with head and neck cancer. In addition, the trial is further evaluating SCV-07’s safety and tolerability in this patient population, as well as the role played by specific genetic profiles on patient response to the treatment. The multicenter, randomized, double-blind, placebo-controlled study is designed to enroll approximately 160 patients who are receiving standard chemoradiation therapy for treatment of cancers of the head and neck. Patients are being randomly assigned to one of the trial’s four treatment arms: SCV-07 at doses of 0.1 mg/kg, 0.3 mg/kg or 1 mg/kg, or placebo. The study’s primary efficacy endpoint is the reduction in proportion of patients with clinically assessed ulcerative OM (WHO Grade ³ 2) at the time that they have received a cumulative radiation dose of 45 Gy. The study’s secondary endpoints include, incidence and duration of ulcerative and severe (WHO Grade ³ 3) OM, analgesic use and pain assessments, quality of life measurements, gastrostomy tube placement and use, breaks in radiation or chemotherapy treatment, and unscheduled office or hospital visits.
The United States Securities and Exchange Commission (“SEC”) and the United States Department of Justice (“DOJ”) are each conducting formal investigations of us regarding a range of matters including the possibility of violations of the Foreign Corrupt Practices Act (“FCPA”). In response to these matters, our board of directors has appointed a special committee of independent directors to oversee the Company’s response to the government inquiries. Based on an initial review, the special committee has decided to undertake an independent investigation as to matters reflected in and arising from the SEC and DOJ investigations including, but not limited to, certain sales and marketing matters in China, in order to evaluate whether any violation of the FCPA or other laws occurred. The special committee has not concluded its investigation or reached any conclusions, to date, nor has the special committee or management made a final determination regarding whether any violation of the FCPA or any other law or regulation has occurred. However, we anticipate that the special committee will finalize its findings in the near future, which may include findings regarding potential FCPA issues and recommendations regarding additional remediation measures and possible suggestions regarding personnel issues. In the course of its review, the special committee has instructed management to (i) evaluate and to expand the Company’s training of employees regarding understanding and compliance with laws including the FCPA and other anti-bribery laws and regulations, (ii) evaluate our existing compliance and anti-bribery guidelines and to prepare a new, more detailed, guideline for implementation after review by our Board and/or committees of the Board, and (iii) implement a pre-approval policy for certain expenses including payments for, or reimbursement of, third-party gifts, travel expenses, honoraria and sponsorships of certain third party events.
42
Table of Contents
We have consistently maintained a policy that our employees comply with all applicable laws. However, in the course of evaluating our existing training, policies and internal controls over financial reporting and expense approval and expense reimbursement practices, and of the matters brought to the attention of management by the special committee, we determined that our guidelines, employee training and expense approval practices and our internal controls with respect to certain matters, including the payment for, or reimbursement of, third party gifts, travel expenses, honoraria and sponsorship of certain third party events required improvements in order to provide reasonable assurance that our policy on compliance with applicable laws was effective. In addition, management determined that we had a material weakness in our internal control over financial reporting as of December 31, 2010 relating to our implementation of our policy on compliance with laws. During the first quarter of fiscal 2011, we initiated or commenced various remediation efforts to provide reasonable assurance of our implementation of our policy on compliance with laws, including the FCPA and other anti-bribery laws. See “Management’s Annual Report on, and Changes in, Internal Control over Financial Reporting” for further information regarding this material weakness and our remediation efforts.
Since our special committee has not reached any conclusions and has not reported its findings to the Board, none of the disclosures in this Form 10-K are intended to, or do, represent the special committee’s finding or conclusions. We anticipate that the special committee will conclude its investigation soon. We anticipate that the special committee may make more specific or different findings in addition to the disclosures herein. We also anticipate that the special committee may recommend additional remediation measures to those we have already taken or commenced.
Following our announcement of the SEC and DOJ investigations, various shareholder litigations were filed and further litigation may be filed. We cannot predict the outcome of these matters, but we have incurred and anticipate that we will continue to incur substantial expenses in the course of the investigations, and in connection with current or future litigation. If it is determined that sales or marketing activities have occurred that violate, or may have violated, the FCPA or other laws, our future operating results could be adversely affected. In addition, we could incur additional expenses related to fines, or to remediative measures including changes in our compliance programs, internal controls or disclosure controls.
We believe our cash and investments as of December 31, 2010 and ongoing revenue generating business operations will be sufficient to support our current operating plan for at least the next 12 months. Our results may fluctuate from quarter to quarter and we may report quarterly losses in the future.
Critical Accounting Policies and Significant Judgments and Estimates
General
We have identified the policies below as critical to our business operations and the understanding of our results of operations. The impact and any associated risks related to these policies on our business operations is discussed throughout “Management’s Discussion and Analysis of Financial Condition and Results of Operations” where such policies affect our reported and expected financial results. For a detailed discussion on the application of these and other accounting policies, see Note 1 in the “Notes to our Consolidated Financial Statements” in Part II, Item 8 of this Annual Report on Form 10-K. Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United Sates, which require us to make estimates and assumptions that affect the reported amount of assets and liabilities, disclosure of contingent assets and liabilities at the date of our financial statements, and the reported amounts of revenue and expenses during the reporting period. On an on-going basis, we evaluate the relevance of our estimates and judgments. We base our estimates on historical experience and on various other market specific assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. There can be no assurance that actual results will not differ from those estimates.
43
Table of Contents
Revenue Recognition
We recognize revenue from product sales at the time of delivery. There are no significant customer acceptance requirements or post-shipment obligations on our part, except for sales to a new market where acceptance requirements have to be met. Sales to importing agents or distributors are recognized at time of shipment when title to the product is transferred to them. Importing agents or distributors do not have contractual rights of return except under limited terms regarding product quality. However, we are expected to replace products that have expired or are deemed to be damaged or defective when delivered. We estimate expected returns primarily on historical patterns. Historically, we have had no product returns of damaged, defective or expired product. As such, no amount was accrued for product returns as of December 31, 2010 and 2009 in the respective consolidated balance sheets. Payments by the importing agents and distributors are not contingent upon sale to the end user by the importing agents or distributors. Amounts invoiced relating to arrangements where collectability is uncertain and revenue cannot be recognized are reflected on our balance sheet as deferred revenue and recognized as the applicable revenue recognition criteria are satisfied.
Cash Equivalents and Investments
We record investments at fair value, as determined by available information on the consolidated balance sheet date. An investment is considered impaired if the fair value of the investment is less than its cost. If, after consideration of all available evidence to evaluate the realizable value of its investment, impairment is determined to be other-than-temporary, then an impairment loss is recognized in the Consolidated Statement of Operations.
Fair Value of Financial Instruments
We record our financial assets including cash equivalents and accrued liabilities at cost, which approximates fair value due to their short-term nature. Investments in marketable securities are recorded at their estimated fair value. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The three levels of input are:
Level 1—Quoted prices in active markets for identical assets or liabilities.
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
Where quoted prices are available in an active market, we determine fair value based upon quoted market prices, and classify these values in level 1 of the valuation hierarchy. If quoted market prices are not available, fair values are based upon observable inputs such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities and are classified in level 2 of the valuation hierarchy. When quoted prices and observable inputs are unavailable, fair values are based on internally developed cash flow models and are classified in level 3 of the valuation hierarchy. The internally developed cash flow models primarily use, as inputs, estimates for interest rates and discount rates including yields of comparable traded instruments adjusted for illiquidity and other risk factors, amount of cash flows and expected holding periods of the assets. These inputs reflect our assumptions about the assumptions market participants would use in pricing
44
Table of Contents
the assets including assumptions about risk developed based on the best information available in the circumstances. Our assessment of the significance of a particular input to the fair value measurements requires judgment, and may affect the valuation of the assets being measured and their placement within the fair value hierarchy.
Accounts Receivable
Accounts receivable are recorded net of the allowance for doubtful accounts. We maintain an allowance for doubtful accounts for estimated losses resulting from the inability of customers to make required payments, when appropriate. We record our allowance for doubtful accounts based on our assessment of various factors. When estimating the need for an allowance for doubtful accounts, we consider historical payment patterns of our customers, the circumstances of each individual customer and their geographic region including a review of the local economic environment, the age of the accounts receivable balances, credit quality of our customers, and other factors that may affect customers’ ability to pay. At December 31, 2010, no allowance for doubtful accounts was considered necessary.
Inventories
Our inventories are stated at the lower of cost or market, with cost determined on a first-in, first-out basis and include amounts related to materials, labor and overhead. In assessing the ultimate realization of inventories, we are required to make judgments as to future demand requirements and compare that with the current inventory levels. If our current assumptions about future production or inventory levels and demand were to change or if actual market conditions are less favorable than those projected by management, inventory write-downs may be required which could negatively impact our gross margins and results of operations. If obsolete items are observed and there are no alternate uses for the inventory, we will record a write-down to net realizable value in the period that the impairment is first recognized.
Accrued Expenses
We make estimates of our accrued expenses as of each balance sheet date in our consolidated financial statements based on facts and circumstances known to us. Examples of estimated accrued expenses include fees paid to contract research organizations and investigative sites in connection with clinical trials, fees paid to contract manufacturers in connection with the production of clinical trial materials, and professional services. We periodically confirm the accuracy of our estimates with selected service providers and make adjustments, if necessary. Expenses related to clinical trials generally are accrued based on estimates of work performed or the level of patient enrollment and activities according to the protocols and agreements. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Payments under certain contracts depend on factors such as the achievement of certain events, the successful enrollment of patients, and the completion of portions of the clinical trial or similar conditions. The objective of our accrual policy is to match the recording of expenses to the actual services received and efforts expended. We monitor planned protocols, work performed, patient enrollment levels and related activities to the extent possible and adjust estimates accordingly.
We record as liabilities estimated amounts for litigation, claims or other legal actions that are probable and can be reasonably estimated. The likelihood of a material change in these estimated reserves is dependent on the possible outcome of settlement negotiations, regulatory or judicial review and the development of facts and circumstances in extended litigation which could change claims or assessments when both the amount and range of loss on some outstanding litigation is uncertain. We disclose in the footnotes of the financial statements when we are unable to make a reasonable estimate of a material liability that could result from unfavorable outcomes. As events occur, we will assess the potential liability related to any pending litigation, claims or other legal actions and adjust our estimates accordingly. Such adjustments could materially impact our financial statements.
45
Table of Contents
Stock-Based Compensation
Stock-based compensation is estimated at the date of grant based on the fair value of the award using the Black-Scholes option-pricing model and the portion that is ultimately expected to vest is recognized as expense ratably (as the awards vest) over the requisite service period, which is generally one or four years, or over the period of time we estimate that it is probable that the performance goal of a performance-based option will be achieved. We estimate pre-vesting forfeitures at the time of grant by analyzing historical data and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. The total expense recognized over the vesting period will only be for those awards that ultimately vest. Certain target-stock-price-based options were valued using the Monte Carlo simulation options pricing model and recognized to expense over the service periods for each of the vesting portions of these awards which were six or eight years. The option-pricing models require the use of certain subjective assumptions, including the expected volatility of the market price of the underlying stock and the expected term of the award. Expected volatility is based on the historical volatility of our stock. Expected term is derived from historical data on employee exercises and terminations, or the contractual life of the award for target-stock-price-based options. We review our valuation assumptions at each grant date, and, as a result, valuation assumptions used to value stock-based compensation of awards granted in future periods may change.
Income Taxes
Income taxes are accounted for under the liability method. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities as measured by the enacted tax rates that will be in effect when these differences reverse. We provide a valuation allowance against net deferred tax assets if, based upon the available evidence, it is more-likely-than-not that the deferred tax assets will not be realized. Realization of our deferred tax assets is dependent upon the generation of future taxable income, the timing and amount of which are uncertain. Accordingly, our deferred tax assets have been fully offset by a valuation allowance. The tax years 1995-2010 remain open to examination by the major taxing jurisdictions to which we are subject. Furthermore, we have been notified by the Internal Revenue Service that our 2008 U.S. federal tax return has been selected for examination, but the examination process has not yet commenced.
We record liabilities related to uncertain tax positions in accordance with the guidance that clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. We do not believe any such uncertain tax positions currently pending will have a material adverse effect on our Consolidated Financial Statements, although an adverse resolution of one or more of these uncertain tax positions in any period could have a material impact on the results of operations for that period.
Results of Operations
Product Sales and Gross Margin:
The following tables summarize the year over year changes in our product sales (in thousands), in our cost of product sales (in thousands) and our product gross margin:
| Years Ended December 31, | ||||||||||||||||||||
| 2010 | Change | 2009 | Change | 2008 | ||||||||||||||||
| Product Sales |
$ | 85,112 | 18 | % | $ | 72,411 | 34 | % | $ | 54,108 | ||||||||||
| Cost of Product Sales |
12,691 | 6 | % | 11,960 | 16 | % | 10,299 | |||||||||||||
| Product Gross Margin |
85.1 | % | 83.5 | % | 81.0 | % | ||||||||||||||
Product sales were $85.1 million, $72.4 million, and $54.1 million for the years ended December 31, 2010, 2009, and 2008, respectively. Product sales to China are denominated in U.S. dollars and accounted for
46
Table of Contents
approximately $82.0 million, $69.7 million and $50.7 million, or 96%, 96% and 94% of sales, respectively, for the years ended December 31, 2010, 2009, and 2008. Our revenue growth was attributable to a higher volume of ZADAXIN sales due to further market penetration in China and modest price increases from the 2008 to 2010 periods. All product sales in each period were derived from sales of ZADAXIN. For the year ended December 31, 2009, we believe there was also increased demand for ZADAXIN as a result of the H1N1 flu virus. In 2010, Shanghai Lingyun Pharmaceutical Company Ltd. and Guangdong South Pharmaceutical Foreign Trade Company Ltd. accounted for 74% and 14% of our sales, respectively. In 2009, Shanghai Lingyun Pharmaceutical Company Ltd. and China National Pharmaceutical Foreign Trade Corporation accounted for 66% and 27% of our sales, respectively. In 2008, Shanghai Lingyun Pharmaceutical Company Ltd. and China National Pharmaceutical Foreign Trade Corporation accounted for 68% and 22% of our sales, respectively. No other customers accounted for more than 10% of sales in those periods. In recent months, Sinopharm Group Co. Limited acquired a majority interest in two of our large importers, Shanghai Lingyun Pharmaceutical Company Ltd. and Guangdong South Pharmaceutical Foreign Trade Company Ltd. We do not believe these acquisitions will impact our sales. Our experience with our largest importers has been good and we anticipate that we will continue to sell a majority of our product to them. We expect our unit sales for the year ending December 31, 2011 will increase, compared to sales for the year ended December 31, 2010, related to further market penetration in China, however there are discussions that government reimbursement levels in China for listed drugs could be reviewed in 2011 and that ZADAXIN’s list price could be included in such a review.
Gross margin was 85.1%, 83.5%, and 81.0% for the years ended December 31, 2010, 2009, and 2008, respectively. The increase in gross margin for the year ended December 31, 2010, compared to the years ended December 31, 2009 and 2008, was attributable to higher sales of ZADAXIN resulting in volume discounts and lower per vial production costs, and was also attributable to lower royalty expense in the 2010 and 2009 periods. We also recorded a charge of $630,000 associated with our finished goods inventory related to vials made obsolete due to labeling requirements arising from our renewed import license in 2008. No significant charges were recorded by us associated with our product inventory during the years ended December 31, 2010 or 2009.
We expect cost of product sales and gross margins to fluctuate from period to period depending upon the level of sales and price of ZADAXIN, the absorption of product-related fixed costs, currency exchange fluctuations, any charges associated with excess or expiring finished product inventory and the timing of other inventory period costs.
Research and Development (“R&D”):
The following tables summarize the year over year changes in our R&D expenses (in thousands):
| Years Ended December 31, | ||||||||||||||||||||
| 2010 | Change | 2009 | Change | 2008 | ||||||||||||||||
| Research and Development |
$ | 12,415 | -25 | % | $ | 16,531 | -28 | % | $ | 22,857 | ||||||||||
Research and development (“R&D”) expenses were $12.4 million, $16.5 million, and $22.9 million for the years ended December 31, 2010, 2009, and 2008, respectively. The decrease of $4.1 million, or 25%, in R&D expenses for the year ended December 31, 2010, compared to the year ended December 31, 2009, was primarily related to the timing of clinical trial-related expenses, including the completion of enrollment of patients in our phase 2a clinical trial of SCV-07 for the delay to onset of severe oral mucositis in the first quarter of 2010 and the discontinuation of the RP101 clinical trial for the treatment of pancreatic cancer last year. These decreases were partially offset by increased expenses related to our SCV-07 phase 2b clinical trials for the treatment of hepatitis c virus (“HCV”) and oral mucositis (“OM”) in the 2010 period.
The decrease of $6.3 million, or 28%, in R&D expenses for the year ended December 31, 2009, compared to the year ended December 31, 2008, was primarily related to lower expenses in the year ended December 31, 2009 for our RP101 clinical trial which was discontinued, offset partially by increased expense related to our
47
Table of Contents
SCV-07 phase 2 clinical trials for the treatment of OM and HCV. Further, we made a $1.3 million milestone payment upon first patient dosing in the RP101 phase 2 clinical trial that occurred during the year ended December 31, 2008, and there was no corresponding payment in 2009. These decreases were partially offset by a $1.0 million license fee payment related to our licensing agreement with APR Applied Pharma Research S.A. (“APR”) that occurred during the year ended December 31, 2009.
The major components of R&D expenses include salaries and other personnel-related expenses, including associated stock-based compensation, facility-related expenses, depreciation of facilities and equipment, license-related fees, services performed by clinical research organizations and research institutions and other outside service providers, and the sharing of certain costs for the development of ZADAXIN by our partner, Sigma-Tau.
The initiation, continuation, and completion of our current clinical development programs has had and is expected to continue to have a significant effect on our research and development expenses. Actual costs incurred in future periods will vary depending in particular upon timeline and design of further clinical trials and final decisions regarding the timing and expense-sharing arrangements for these trials, though we expect our research and development expenses to increase for the year ending December 31, 2011, compared to those incurred for the year ended December 31, 2010, primarily related to our SCV-07 phase 2b trial in oral mucositis for which we began enrolling patients in January 2011. We are evaluating opportunities to acquire or in-license the marketing rights to proprietary products primarily in China, which may result in increased research and development expenses due to license fee payments or other expenses related to in-licensing and development of new products in the future.
Sales and Marketing:
The following tables summarize the year over year changes in our sales and marketing expenses (in thousands):
| Years Ended December 31, | ||||||||||||||||||||
| 2010 | Change | 2009 | Change | 2008 | ||||||||||||||||
| Sales and Marketing |
$ | 22,006 | 17 | % | $ | 18,805 | 12 | % | $ | 16,853 | ||||||||||
Sales and marketing expenses were $22.0 million, $18.8 million, and $16.9 million for the years ended December 31, 2010, 2009, and 2008, respectively. The increase in sales and marketing expenses of $3.2 million or 17% for the year ended December 31, 2010, compared to the year ended December 31, 2009, was primarily related to increased conferences and local training seminars and an increase in employee-related costs associated with growth in our sales force and our sales efforts for ZADAXIN in the 2010 period. The increase in sales and marketing expenses of $2.0 million or 12% for the year ended December 31, 2009, compared to the year ended December 31, 2008, was primarily due to an increase in conference and seminar expenses, an increase in compensation and benefits expense as a result of increased bonus and sales incentives and an increase in the number of sales personnel, and an increase in rent and business tax expense, all related to our expanding sales efforts. We expect sales and marketing expenses for the year ending December 31, 2011 to be higher than those incurred for the year ended December 31, 2010 due to increased sales efforts, primarily in China.
General and Administrative:
The following tables summarize the year over year changes in our general and administrative expenses (in thousands):
| Years Ended December 31, | ||||||||||||||||||||
| 2010 | Change | 2009 | Change | 2008 | ||||||||||||||||
| General and Administrative |
$ | 15,606 | 25 | % | $ | 12,521 | 0 | % | $ | 12,538 | ||||||||||
General and administrative expenses were $15.6 million, $12.5 million, and $12.5 million for the years ended December 31, 2010, 2009, and 2008, respectively. During the year ended December 31, 2010, general and
48
Table of Contents
administrative expenses increased $3.1 million or 25% compared to the year ended December 31, 2009, primarily as a result of higher corporate and legal expenses in connection with responding to the SEC and DOJ investigations announced in August 2010, shareholder litigations that have been filed following the announcement of those investigations, and the conduct of an independent investigation by a special committee of our board of directors in order to evaluate whether any violation of the FCPA or other laws occurred, as well as our business development efforts for China. During the year ended December 31, 2009, general and administrative expenses decreased $17,000 or 0% compared to the year ended December 31, 2008. Although we incurred increased expenses related to business development efforts in China during 2009, the increases were offset by decreased accounting and legal fees. We expect our general and administrative expenses will increase in 2011, related to corporate and legal expenses in connection with responding to the SEC and DOJ investigations announced in August 2010, shareholder litigations that have been filed following the announcement of those investigations, and the conduct of an independent investigation by a special committee of our board of directors in order to evaluate whether any violation of the FCPA or other laws occurred, as well as our expanding operations and ongoing business development efforts for China.
Other Interest and Investment Income:
Interest and investment income was approximately $0.1 million, $0.2 million, and $0.6 million for the years ended December 31, 2010, 2009, and 2008, respectively. Although our cash balances increased during 2010, interest income decreased for the year ended December 31, 2010 compared to the year ended December 31, 2009 as a result of lower returns due to a declining interest rate environment in the 2010 period. The decrease for the year ended December 31, 2009 compared to the year ended December 31, 2008, resulted from lower cash and investments balances and lower returns due to a declining interest rate environment in the 2009 period.
Interest and Investment Expense:
Interest and investment expense was $0.2 million, $0.2 million, and $31,000 for the years ended December 31, 2010, 2009, and 2008, respectively. For the year ended December 31, 2010, interest and investment expense of $0.2 million included the amortization and write-off of the remaining loan origination fees related to our $6.0 million terminated Silicon Valley Bank line of credit, and the interest and amortization of the loan origination fees related to our new $15.0 million Silicon Valley Bank line of credit established in October 2010. The increase in interest expense for the year ended December 31, 2009, compared to the year ended December 31, 2008 resulted from the amortization of loan origination fees and a credit insurance policy related to our $6.0 million Silicon Valley Bank line of credit that was established in November 2008. We expect that interest and investment expense for the year ending December 31, 2011 will remain at a comparable level to those incurred for the year ended December 31, 2010.
Other Income:
In November 2010, we were awarded approximately $1.0 million in non-taxable grants as part of the U.S. Department of Treasury’s Therapeutic Discovery Project Program related to our research and development activities in SCV-07 and ZADAXIN contributing to an increase in other income of $0.9 million for the year ended December 31, 2010 compared to December 31, 2009. We do not expect any income related to non-taxable grants for the year ending December 31, 2011.
Provision for Income Tax:
Provision for income tax of $2.2 million, $0.6 million and $0.5 million for the years ended December 31, 2010, 2009 and 2008, respectively, relates to our foreign operations in China. The increases resulted from an increase in operating activities in China and an increase in the statutory tax rate in China. In addition, for the year ended December 31, 2010 we recorded an additional tax expense of $0.8 million related to our uncertain tax position in China. The statutory tax rate in China was 22%, 20% and 18% for the years ended December 31,
49
Table of Contents
2010, 2009 and 2008, respectively. Our statutory tax rate in China is 24% in 2011. We expect the provision for income tax to decrease for the year ending December 31, 2011, compared to the year ended December 31, 2010, as we do not expect a comparable tax expense for uncertain tax positions in 2011.
We have not recorded any U.S. federal or state income tax expense for the years ended December 31, 2010, 2009, and 2008. Undistributed earnings of our foreign subsidiaries amounted to approximately $12.4 million at December 31, 2010. These earnings are considered to be permanently reinvested and accordingly, no deferred U.S. income taxes have been provided thereon.
At December 31, 2010, we had net operating loss carryforwards for federal income tax purposes of approximately $122.7 million that expire in the years 2011 through 2028.
At December 31, 2010, we had federal research and development, orphan drug and investment tax credit carryforwards of approximately $12.3 million that expire in the years 2011 through 2030.
Because of the “change in ownership” provisions of the Internal Revenue Code, a portion of our net operating loss carryforwards and tax credit carryforwards may be subject to an annual limitation regarding their utilization against taxable income in future periods. As a result of the annual limitation, a portion of these carryforwards may expire before ultimately becoming available to reduce future income tax liabilities.
Liquidity and Capital Resources
Days’ sales outstanding in accounts receivable, using the average receivables method, were 98, 101, and 103 days in 2010, 2009, and 2008, respectively. The majority of our sales are to customers in China where our accounts receivable collections have standard credit terms ranging from 90 to 180 days.
The following tables summarize our cash and investments and our cash flow activities as of the end of, and for each of, the periods presented (in thousands):
| As of December 31, | ||||||||
| 2010 | 2009 | |||||||
| Cash and investments |
$ | 56,522 | $ | 31,819 | ||||
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cash provided by (used in): |
||||||||||||
| Operating activities |
$ | 20,660 | $ | (452 | ) | $ | (4,945 | ) | ||||
| Investing activities |
$ | (1,316 | ) | $ | (148 | ) | $ | 572 | ||||
| Financing activities |
$ | 3,901 | $ | 2,625 | $ | 149 | ||||||
At December 31, 2010 and 2009, we had $56.5 million and $31.8 million, respectively, in cash and investments of which $56.1 million was invested at December 31, 2010 in unrestricted cash, short-term foreign U.S. term deposits, and money market accounts. Net cash provided by (used in) operating activities was $20.7 million, ($0.5) million, and ($4.9) million, for the years ended December 31, 2010, 2009, and 2008, respectively. Net cash used in operating activities for the years ended December 31, 2010, 2009 and 2008 primarily reflected the net income (loss) for the period, adjusted for non-cash items such as stock-based compensation expense, depreciation and amortization expense and changes in operating assets and liabilities. For the year ended December 31, 2010, such changes included an increase in accounts receivable of $9.3 million, compared to the year ended December 31, 2009, related to increased sales and fluctuations in the timing of cash receipts from customers. All of our accounts receivable were current as of December 31, 2010. In addition, inventory levels decreased $3.1 million for the year ended December 31, 2010, compared to the year ended December 31, 2009,
50
Table of Contents
primarily related to an increase in sales and a decrease in production during fiscal 2010, and accounts payable and other accrued liabilities increased $3.7 million mainly related to an increase in legal and other costs associated with the SEC and DOJ investigations, shareholder litigations that have been filed following the announcement of those investigations, and the conduct of an independent investigation by a special committee of our board of directors, and an increase in accrued taxes and tax reserves.
For the year ended December 31, 2009, such changes included a $9.5 million increase in accounts receivable, compared to the year ended December 31, 2008 related to increased sales and a reduction in payment terms that occurred at the end of fiscal 2008 with our largest customer in China reducing their payment terms from 180 days to 90 days. In addition, inventory levels increased by $4.1 million for the year ended December 31, 2009 compared to the year ended December 31, 2008 to meet expected sales demands.
Net cash (used in) provided by investing activities was ($1.3) million, ($0.1) million and $0.6 million for the years ended December 31, 2010, 2009 and 2008, respectively. Cash (used in) provided by investing activities was primarily related to purchases of investments, net of proceeds from sale or maturities of investments, and purchases of property and equipment.
Net cash provided by financing activities was $3.9 million, $2.6 million, and $0.1 million for the years ended December 31, 2010, 2009 and 2008, respectively, and consisted of proceeds from the exercise of stock options made under our stock award plans and the issuance of stock under our employee stock purchase plan. During the year ended December 31, 2010, we also received proceeds of $2.5 million from borrowing on our line of credit.
The following table summarizes our future contractual obligations as of December 31, 2010 (in thousands).
| Payments Due by Period | ||||||||||||||||
| Contractual Obligations |
Total | Less than 1 | 2-3 years | 4 years | ||||||||||||
| Operating leases(1) |
$ | 5,506 | $ | 1,806 | $ | 2,956 | $ | 744 | ||||||||
| Purchase obligations(2) |
4,999 | 4,999 | — | — | ||||||||||||
| Other liabilities(3) |
300 | — | 300 | — | ||||||||||||
| Total |
$ | 10,805 | $ | 6,805 | $ | 3,256 | $ | 744 | ||||||||
| (1) | These are future minimum rental commitments for office space and copiers leased under non-cancelable operating lease arrangements. |
| (2) | These consist of purchase obligations with manufacturers. |
| (3) | This amount represents a discretionary accrued bonus payable to our chief executive officer as of December 31, 2010 based on the achievement of performance targets over the years 2009-2011. |
We have also entered into a purchase obligation with our primary manufacturer and supplier of bulk drug substance to purchase an aggregate amount of 70% of our total annual forecasted requirements of bulk drug substance from them, subject to certain conditions.
On October 1, 2010, our subsidiaries, SciClone Pharmaceuticals International Ltd. and SciClone Pharmaceuticals International China Holding Ltd. as borrowers, terminated the existing $6 million line-of-credit facility with Silicon Valley Bank (“SVB”) and entered into a new loan and security agreement with SVB (“the Debt Financing Facility”) for a debt financing facility up to $15 million for a term of 24 months. SciClone Pharmaceuticals, Inc. is the guarantor of the facility. The Debt Financing Facility bears interest on borrowed funds at the bank’s prime rate plus 1.25% (5.25% at December 31, 2010) on outstanding balances and is secured by a first priority secured interest in all of our assets, including intellectual property in an event of default. We are required to meet certain financial covenants, including minimum liquidity, as defined, and are subject to
51
Table of Contents
certain minimum fees and interest payments. We are also required to meet certain operating covenants that limit our ability to incur liabilities, create liens, make capital expenditures, pay dividends or distributions, make investments, and dispose of assets. As of December 31, 2010, we had borrowed $2.5 million on the Debt Financing Facility and we were required to maintain a debt coverage ratio of 1.35 to 1 equal to $3.4 million of cash in SVB cash accounts to meet our financial liquidity covenant. We were in compliance with all covenants as of December 31, 2010. The Debt Financing Facility expires October 1, 2012, and upon termination all amounts borrowed must be repaid in full.
In May 2009, we filed a shelf registration statement on Form S-3 with the SEC under which we may offer and sell up to $50.0 million of our securities, assuming we continue to meet the SEC’s eligibility requirements for primary offerings on Form S-3.
We believe that our existing cash and investments and ongoing revenue generating business operations will be sufficient to support our current operating plan for at least the next 12 months. We have no current commitments to offer and sell any securities that may be offered or sold pursuant to our registration statement. To the extent that we raise additional capital by issuing equity securities, our stockholders may experience dilution. Debt financing, if available, may subject us to restrictive covenants and significant interest costs. To the extent that we raise additional funds through collaboration and licensing arrangements, we would be required to relinquish some rights to our technologies, product candidates or marketing territories. Additional financing or collaboration and licensing arrangements may not be available when needed either at all or, on favorable terms.
We intend to continue to explore alternatives for financing to provide additional flexibility in managing our operations, in-licensing or acquiring new products, particularly in China, and funding our clinical trials. The unavailability or the inopportune timing of any financing could prevent or delay our long-term product development and commercialization programs, either of which could hurt our business. We cannot assure you that funds from financings, if any, will be sufficient to conduct and complete further clinical trials or to acquire or in-license additional products. The need, timing and amount of any such financing would depend upon numerous factors, including the status of the pending regulatory investigations and pending litigations, the level and price of ZADAXIN sales, the timing and amount of manufacturing costs related to ZADAXIN, the availability of complementary products, technologies and businesses, the initiation and continuation of preclinical and clinical trials and testing, the timing of regulatory approvals, developments in relationships with existing or future collaborative parties, the status of competitive products, and various alternatives for financing. We have not determined the timing or structure of any transaction.
Related Party Transactions
We have licensed to our largest shareholder, Sigma-Tau Finanziaria S.p.A. (“Sigma-Tau”), exclusive ZADAXIN (“thymalfasin or thymosin alpha 1”) development and marketing rights that cover all countries in the European Union as defined on January 1, 1995, in addition to Iceland, Norway and Switzerland. Our collaboration with Sigma-Tau is governed by an agreement entered into in 2000 with a term expiring in March 2012 unless renewed, as well as by amendments to the agreement regarding particular development efforts. In addition, the agreement governed our joint collaboration on the development of thymalfasin in certain indications, and for the sharing of intellectual property in the respective territories. The agreement also provides that if Sigma-Tau sells ZADAXIN in the licensed territory, it will purchase the product from us at a specified price, subject to certain adjustments. We do not currently anticipate that Sigma-Tau will sell any ZADAXIN other than nominal amounts in Italy.
There are no on-going development or reimbursement obligations under the agreement, and there are no milestones or similar terms currently in effect. However, we continue to cooperate with Sigma-Tau on exploring the development of thymalfasin in other indications, including as a vaccine adjuvant and in aspergillosis.
52
Table of Contents
Off-Balance Sheet Arrangements
There were no off-balance sheet arrangements in 2010, 2009, or 2008.
Recent Accounting Guidance
In January 2010, the Financial Accounting Standards Board (“FASB”) issued FASB Accounting Standards Update (“ASU”) No. 2010-06, Fair Value Measurements and Disclosures (Topic 820)—Improving Disclosures about Fair Value Measurements. The ASU requires new disclosures about significant transfers in and out of Levels 1 and 2 fair value measurements and separate disclosures about purchases, sales, issuances and settlements relating to Level 3 fair value measurements. The ASU also clarifies existing disclosure requirements regarding inputs and valuation techniques, as well as the level of disaggregation for each class of assets and liabilities for which separate fair value measurements should be disclosed. We adopted ASU 2010-06 at the beginning of fiscal 2010. The adoption of this ASU did not have a material impact on our condensed consolidated financial statements.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we invest in money market funds, term deposits, U.S. Treasury, or government agency notes. All of our investments mature within one year from date of purchase except for our Italian state bonds which mature in 2013. Our investment securities may be subject to interest rate risk and could decrease in value if market interest rates rise. To minimize this risk, we primarily hold securities that are short-term in duration and maintain an average maturity of less than one year. We believe that our exposure to interest rate risk is not significant and a 1% movement in market interest rates would not have a significant impact to the total value of our investment portfolio at December 31, 2010.
We do not hold any derivative financial instruments for speculation or trading purposes. Most of our sales have been in U.S. dollars. Our purchases with contract manufacturers are denominated in U.S. dollars and euros and costs of our sales and marketing efforts in China are paid in local currency. In addition, we have certain cash balances, other assets and liabilities denominated in euros and renminbi. As a result, we are exposed to foreign currency rate fluctuations, and we do not hedge against the risk associated with such fluctuations. Consequently, changes in exchange rates could result in material exchange losses and could unpredictably, materially and adversely affect our operating results and stock price. Such losses have not been significant to date.
53
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
SCICLONE PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||||
| 55 | ||||
| 56 | ||||
| 57 | ||||
| 58 | ||||
| 59 | ||||
| 60 | ||||
54
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of SciClone Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of SciClone Pharmaceuticals, Inc. as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2010. Our audits also included the financial statement schedule listed at Item 15(a). These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of SciClone Pharmaceuticals, Inc. at December 31, 2010 and 2009, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), SciClone Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 31, 2011 expressed an adverse opinion on the effectiveness of internal control over financial reporting.
/s/ Ernst & Young LLP
Palo Alto, California
March 31, 2011
55
Table of Contents
SCICLONE PHARMACEUTICALS, INC.
(In thousands, except share and per share amounts)
| December 31, 2010 |
December 31, 2009 |
|||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 53,017 | $ | 29,687 | ||||
| Short-term investments |
3,125 | 1,646 | ||||||
| Accounts receivable |
30,671 | 21,394 | ||||||
| Inventories |
7,078 | 10,149 | ||||||
| Prepaid expenses and other current assets |
2,057 | 1,518 | ||||||
| Restricted short-term investments |
— | 71 | ||||||
| Total current assets |
95,948 | 64,465 | ||||||
| Property and equipment, net |
588 | 771 | ||||||
| Restricted investments |
380 | 415 | ||||||
| Other assets |
891 | 1,249 | ||||||
| Total assets |
$ | 97,807 | $ | 66,900 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 3,882 | $ | 2,339 | ||||
| Accrued liabilities |
8,247 | 6,048 | ||||||
| Deferred revenue |
— | 141 | ||||||
| Total current liabilities |
12,129 | 8,528 | ||||||
| Long-term borrowing on line of credit |
2,500 | — | ||||||
| Other long-term liabilities |
990 | 979 | ||||||
| Commitments and contingencies (see Note 9 and 16) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock; $0.001 par value; 10,000,000 shares authorized; no shares issued and outstanding |
— | — | ||||||
| Common stock; $0.001 par value; 75,000,000 shares authorized; 48,011,235 and 47,217,944 shares issued and outstanding at December 31, 2010 and 2009, respectively |
48 | 47 | ||||||
| Additional paid-in capital |
225,897 | 222,229 | ||||||
| Accumulated other comprehensive income |
67 | 22 | ||||||
| Accumulated deficit |
(143,824 | ) | (164,905 | ) | ||||
| Total stockholders’ equity |
82,188 | 57,393 | ||||||
| Total liabilities and stockholders’ equity |
$ | 97,807 | $ | 66,900 | ||||
See notes to consolidated financial statements.
56
Table of Contents
SCICLONE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenues: |
||||||||||||
| Product sales |
$ | 85,112 | $ | 72,411 | $ | 54,108 | ||||||
| Contract revenue |
— | — | 5 | |||||||||
| Total revenues |
85,112 | 72,411 | 54,113 | |||||||||
| Cost of product sales |
12,691 | 11,960 | 10,299 | |||||||||
| Gross margin |
72,421 | 60,451 | 43,814 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development (including $0, $58 and $322 of related party research and development in 2010, 2009 and 2008, respectively) |
12,415 | 16,531 | 22,857 | |||||||||
| Sales and marketing |
22,006 | 18,805 | 16,853 | |||||||||
| General and administrative |
15,606 | 12,521 | 12,538 | |||||||||
| Total operating expenses |
50,027 | 47,857 | 52,248 | |||||||||
| Non-operating income (expense): |
||||||||||||
| Income (loss) from operations |
22,394 | 12,594 | (8,434 | ) | ||||||||
| Interest and investment income |
105 | 153 | 608 | |||||||||
| Interest and investment expense |
(195 | ) | (179 | ) | (31 | ) | ||||||
| Other income (expense), net |
953 | 18 | (16 | ) | ||||||||
| Income (loss) before provision for income tax |
23,257 | 12,586 | (7,873 | ) | ||||||||
| Provision for income tax |
2,176 | 641 | 475 | |||||||||
| Net income (loss) |
$ | 21,081 | $ | 11,945 | $ | (8,348 | ) | |||||
| Basic net income (loss) per share |
$ | 0.44 | $ | 0.26 | $ | (0.18 | ) | |||||
| Diluted net income (loss) per share |
$ | 0.43 | $ | 0.25 | $ | (0.18 | ) | |||||
See notes to consolidated financial statements.
57
Table of Contents
SCICLONE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands)
| Common stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| Balance at December 31, 2007 |
46,121 | $ | 46 | $ | 215,633 | $ | 82 | $ | (168,502 | ) | $ | 47,259 | ||||||||||||
| Issuance of common stock from exercise of stock options |
98 | — | 146 | — | — | 146 | ||||||||||||||||||
| Stock short-swing profit |
— | — | 3 | — | — | 3 | ||||||||||||||||||
| Issuance of warrants related to line of credit |
— | — | 30 | — | — | 30 | ||||||||||||||||||
| Compensation related to stock option awards |
— | — | 1,892 | — | — | 1,892 | ||||||||||||||||||
| Net loss |
— | — | — | — | (8,348 | ) | (8,348 | ) | ||||||||||||||||
| Net change in unrealized losses and foreign currency translation on foreign-denominated available-for-sale securities |
— | — | — | (60 | ) | — | (60 | ) | ||||||||||||||||
| Net change in unrealized gains/losses on available-for-sale securities |
— | — | — | (102 | ) | — | (102 | ) | ||||||||||||||||
| Foreign currency translation |
— | — | — | 83 | — | 83 | ||||||||||||||||||
| Total comprehensive loss |
(8,427 | ) | ||||||||||||||||||||||
| Balance at December 31, 2008 |
46,219 | 46 | 217,704 | 3 | (176,850 | ) | 40,903 | |||||||||||||||||
| Issuance of common stock from exercise of stock options |
999 | 1 | 2,624 | — | — | 2,625 | ||||||||||||||||||
| Compensation related to stock option awards |
— | — | 1,901 | — | — | 1,901 | ||||||||||||||||||
| Net income |
— | — | — | — | 11,945 | 11,945 | ||||||||||||||||||
| Net change in unrealized losses and foreign currency translation on foreign-denominated available-for-sale securities |
— | — | — | 26 | — | 26 | ||||||||||||||||||
| Net change in unrealized gains/losses on available-for-sale securities |
— | — | — | 4 | — | 4 | ||||||||||||||||||
| Foreign currency translation |
— | — | — | (11 | ) | — | (11 | ) | ||||||||||||||||
| Total comprehensive income |
11,964 | |||||||||||||||||||||||
| Balance at December 31, 2009 |
47,218 | 47 | 222,229 | 22 | (164,905 | ) | 57,393 | |||||||||||||||||
| Issuance of common stock from exercise of stock options and employee stock purchase plan |
746 | 1 | 1,401 | — | — | 1,402 | ||||||||||||||||||
| Issuance of common stock from exercise of warrants, net of repurchases |
47 | — | — | — | — | — | ||||||||||||||||||
| Compensation related to stock option awards |
— | — | 2,267 | — | — | 2,267 | ||||||||||||||||||
| Net income |
— | — | — | — | 21,081 | 21,081 | ||||||||||||||||||
| Net change in unrealized losses and foreign currency translation on foreign-denominated available-for-sale securities |
— | — | — | (36 | ) | — | (36 | ) | ||||||||||||||||
| Net change in unrealized gains/losses on available-for-sale securities |
— | — | — | (2 | ) | — | (2 | ) | ||||||||||||||||
| Foreign currency translation |
— | — | — | 83 | — | 83 | ||||||||||||||||||
| Total comprehensive income |
21,126 | |||||||||||||||||||||||
| Balance at December 31, 2010 |
48,011 | $ | 48 | $ | 225,897 | $ | 67 | $ | (143,824 | ) | $ | 82,188 | ||||||||||||
See notes to consolidated financial statements.
58
Table of Contents
SCICLONE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Operating activities: |
||||||||||||
| Net income (loss) |
$ | 21,081 | $ | 11,945 | $ | (8,348 | ) | |||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: |
||||||||||||
| Non-cash expense related to stock-based compensation |
2,224 | 1,867 | 1,843 | |||||||||
| Depreciation and amortization |
556 | 530 | 353 | |||||||||
| Realized (gain) loss on investments |
(226 | ) | (103 | ) | 315 | |||||||
| Other non-cash expense (income) |
249 | 83 | (248 | ) | ||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(9,277 | ) | (9,467 | ) | 723 | |||||||
| Inventories |
3,115 | (4,059 | ) | (428 | ) | |||||||
| Prepaid expenses and other assets |
(674 | ) | 247 | 1,243 | ||||||||
| Accounts payable |
1,543 | 144 | 258 | |||||||||
| Accrued liabilities (including $0, ($1.4) million and ($0.2) million due to related party in 2010, 2009 and 2008, respectively) |
2,199 | (1,980 | ) | (1,057 | ) | |||||||
| Deferred revenue |
(141 | ) | 141 | (37 | ) | |||||||
| Long-term liabilities |
11 | 200 | 438 | |||||||||
| Net cash provided by (used in) operating activities |
20,660 | (452 | ) | (4,945 | ) | |||||||
| Investing activities: |
||||||||||||
| Purchases of property and equipment |
(133 | ) | (192 | ) | (373 | ) | ||||||
| Proceeds from sale or maturities of available-for-sale investments |
7,283 | 44 | 2,593 | |||||||||
| Proceeds from the sale of trading securities |
1,800 | — | — | |||||||||
| Purchases of investments |
(10,266 | ) | — | (1,648 | ) | |||||||
| Net cash (used in) provided by investing activities |
(1,316 | ) | (148 | ) | 572 | |||||||
| Financing activities: |
||||||||||||
| Proceeds from borrowing on line of credit |
2,500 | — | — | |||||||||
| Proceeds from issuances of common stock |
1,401 | 2,625 | 149 | |||||||||
| Net cash provided by financing activities |
3,901 | 2,625 | 149 | |||||||||
| Effect of exchange rate changes on cash and cash equivalents |
85 | (11 | ) | 80 | ||||||||
| Net increase (decrease) in cash and cash equivalents |
23,330 | 2,014 | (4,144 | ) | ||||||||
| Cash and cash equivalents, beginning of year |
29,687 | 27,673 | 31,817 | |||||||||
| Cash and cash equivalents, end of year |
$ | 53,017 | $ | 29,687 | $ | 27,673 | ||||||
| Supplemental disclosures of cash flow information: |
||||||||||||
| Income taxes paid related to foreign operations |
$ | 829 | $ | 608 | $ | 559 | ||||||
| Interest and unused line fees paid related to line of credit |
$ | 37 | $ | — | $ | — | ||||||
See notes to consolidated financial statements.
59
Table of Contents
SCICLONE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 — The Company and Summary of Significant Accounting Policies
The Company
SciClone Pharmaceuticals, Inc. (“SciClone” or the “Company”) is a revenue-generating, global specialty pharmaceutical company with a substantial international business, based primarily in the People’s Republic of China (“China”), and with a product portfolio of novel therapies for cancer and infectious diseases. ZADAXIN®, the Company’s brand of thymalfasin or thymosin alpha 1 and its primary product, has regulatory approval in over 30 countries. All of the Company’s revenues are derived from sales of ZADAXIN, substantially most of which is sold to China, through the Company’s wholly-owned subsidiary SciClone Pharmaceuticals International Ltd. The Company has in-licensed the commercialization rights of DC BeadTM in China, a novel treatment for advanced liver cancer which is currently approved in 40 countries worldwide, including Europe and the U.S., and commenced a small local registration trial in China in 2010 as required by the Chinese State Food and Drug Administration for regulatory approval. The Company has also in-licensed the commercialization rights to an anti-nausea drug ondansetron RapidFilmTM in China including Hong Kong and Macau, and Vietnam, for which the Company intends to seek regulatory approval. The Company’s proprietary in-licensed compound SCV-07 is in phase 2 clinical development for the treatment of oral mucositis.
Presentation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries, SciClone Pharmaceuticals International Ltd. (“SPIL”), SciClone Pharmaceuticals International China Holding Ltd. (“SPIL China”), SciClone Pharmaceuticals (China) Ltd., SciClone Italy S.R.L., and SciClone Pharmaceuticals Hong Kong Limited. SPIL is registered in the Cayman Islands with its principal office located in Hong Kong. SPIL China is registered in Cayman Islands with its principal office located in Hong Kong. SciClone Pharmaceuticals (China) Ltd. is registered in China with its principal office located in Shanghai. SciClone Italy S.R.L. is registered in Italy with its principal office located in Rome. SciClone Pharmaceuticals Hong Kong Limited is registered in Hong Kong with its principal office located in Hong Kong. All significant intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ materially from those estimates.
Cash Equivalents and Investments
Cash equivalents consist of highly liquid investments with maturities of three months or less on the date of purchase. The Company records its investments at fair value, as determined by available information on the consolidated balance sheet date. The Company’s available-for-sale portfolio at December 31, 2010 primarily consisted of foreign U.S. dollar term deposits and restricted long-term Italian state bonds. At December 31, 2009, the Company’s trading portfolio consisted of auction-rate securities (“ARS”) and its available-for-sale portfolio primarily consisted of corporate equity securities, a certificate of deposit and restricted long-term Italian state bonds.
Unrealized gains or losses on available-for-sale securities are included in accumulated other comprehensive income on the consolidated balance sheet. Realized gains and losses and declines in value judged to be other-than-temporary on available-for-sale securities are included in earnings. Gains or losses and declines in value judged to be other-than-temporary on trading securities are included in earnings. The amortized cost of securities
60
Table of Contents
is adjusted for amortization of premiums and accretion of discounts to maturity and is included in earnings. The cost of securities sold is based on the specific identification method.
Available-for-sale investments are evaluated for impairment each reporting period. An investment is considered impaired if the fair value of the investment is less than its cost. If, after consideration of all available evidence to evaluate the realizable value of its investment, impairment is determined to be other-than-temporary, then an impairment loss is recognized in the Consolidated Statement of Operations.
For the years ended December 31, 2010, 2009 and 2008, net change in unrealized (loss) gains, including foreign currency translation on foreign-denominated securities, of approximately ($38,000), $30,000, and ($0.2) million, on available-for-sale securities, respectively, were included in accumulated other comprehensive income.
For the years ended December 31, 2010, 2009 and 2008, the Company realized gains (losses) of $0.2 million, $0.1 million and ($0.3) million, respectively, substantially related to its ARS trading securities.
Fair Value of Financial Instruments
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The three levels of input are:
Level 1—Quoted prices in active markets for identical assets or liabilities.
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
Where quoted prices are available in an active market, the Company determines fair value based upon quoted market prices, and classifies these values in level 1 of the valuation hierarchy. If quoted market prices are not available, fair values are based upon observable inputs such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities and are classified in level 2 of the valuation hierarchy. When quoted prices and observable inputs are unavailable, fair values are based on internally developed cash flow models and are classified in level 3 of the valuation hierarchy. The internally developed cash flow models primarily use, as inputs, estimates for interest rates and discount rates including yields of comparable traded instruments adjusted for illiquidity and other risk factors, amount of cash flows and expected holding periods of the assets. These inputs reflect the Company’s assumptions about the assumptions market participants would use in pricing the assets including assumptions about risk developed based on the best information available in the circumstances. The Company’s assessment of the significance of a particular input to the fair value measurements requires judgment, and may affect the valuation of the assets being measured and their placement within the fair value hierarchy.
Other financial instruments, including accrued liabilities, are carried at cost, which the Company believes approximates fair value because of the short-term maturity of these instruments.
61
Table of Contents
Concentration of Risk
Financial instruments that potentially subject the Company to a concentration of credit risk consist of cash, cash equivalents and investments. The Company is exposed to credit risk in the event of default by the institutions holding the cash, cash equivalents and investments to the extent of the amounts recorded on the consolidated balance sheet. Most of the Company’s cash and cash equivalents are held by financial institutions that the Company believes are of high credit quality. At times, deposits may exceed government insured limits. The Company has not experienced any losses on its deposits of cash and cash equivalents.
China uses a tiered method to import and distribute products. The distributors make the sales in the country, but the product is imported for them by licensed importers. For the years ended December 31, 2010, 2009 and 2008, sales to four importing agents in China accounted for 96%, 96% and 94%, respectively, of the Company’s product sales. In 2010, the two largest importers accounted for 74% and 14% of sales, respectively. In 2009, the two largest importers accounted for 66% and 27% of sales, respectively. In 2008, the two largest importers accounted for 68% and 22% of sales, respectively. No other importer accounted for more than 10% of sales in 2010, 2009 or 2008. The Company’s two largest importers were the same for the years ending December 31, 2010, 2009, and 2008 and in recent months, a third party acquired a majority interest in these two largest importers. As of December 31, 2010, approximately $30.1 million, or 98%, of the Company’s accounts receivable were attributable to these two importing agents in China. The Company performs on-going credit evaluations of its customers’ financial condition, and generally does not require collateral from its customers.
The Company currently relies on two suppliers to provide key components to its ZADAXIN manufacturing supply. Although there are a limited number of manufacturers who would be able to meet the requirements to manufacture these components, management believes that other suppliers could provide similar components on comparable terms. A change in suppliers, however, could cause a delay in manufacturing and a possible loss of sales, which would affect operating results adversely.
Accounts Receivable
Accounts receivable are recorded net of an allowance for doubtful accounts. The Company maintains allowances for doubtful accounts for estimated losses resulting from the inability of customers to make required payments, when appropriate. The Company records its allowance for doubtful accounts based on its assessment of various factors. When estimating the need for an allowance for doubtful accounts, the Company considers historical payment patterns of its customers, the circumstances of each individual customer and their geographic region including a review of the local economic environment, the age of the accounts receivable balances, credit quality of its customers, and other factors that may affect customers’ ability to pay. At December 31, 2010 and 2009, no allowance for doubtful accounts was considered necessary.
Inventories
Inventories consist of raw materials, work in progress and finished goods products. Inventories are stated at the lower of cost or market, with cost determined on a first-in, first-out basis, and include amounts related to materials, labor and overhead. The Company periodically reviews the inventory in order to identify obsolete items. If obsolete items are observed and there are no alternate uses for the inventory, the Company will record a write-down to net realizable value in the period that the impairment is first recognized.
Property and Equipment
Property and equipment is stated at cost, less accumulated depreciation. Depreciation is recorded over the estimated useful lives of the respective assets (generally three to five years) on the straight-line basis. Leasehold improvements are amortized over the shorter of the estimated useful life or lease term on the straight-line basis. The Company’s policy is to identify and record impairment losses, if necessary, on property and equipment when events and circumstances indicate that the assets might be impaired and the undiscounted cash flows estimated to be generated by those assets are less than the carrying amounts of those assets.
62
Table of Contents
Intangible Assets
Intangible assets with definite lives are amortized over their estimated useful lives and are reviewed for impairment when facts or circumstances suggest that the carrying value of these assets may not be recoverable. The Company’s policy is to identify and record impairment losses, if necessary, on intangible product rights when events and circumstances indicate that the assets might be impaired and the undiscounted cash flows estimated to be generated by those assets are less than the carrying amounts of those assets.
Accrued Expenses
The Company’s management makes estimates of its accrued expenses as of each balance sheet date in its consolidated financial statements based on facts and circumstances known to them. Examples of estimated accrued expenses include fees paid to contract research organizations and investigative sites in connection with clinical trials, fees paid to contract manufacturers in connection with the production of clinical trial materials, and professional services. The Company periodically confirms the accuracy of its estimates with selected service providers and makes adjustments, if necessary. Expenses related to clinical trials generally are accrued based on estimates of work performed or the level of patient enrollment and activities according to the protocols and agreements. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Payments under certain contracts depend on factors such as the achievement of certain events, the successful enrollment of patients, and the completion of portions of the clinical trial or similar conditions. The objective of the Company’s accrual policy is to match the recording of expenses to the actual services received and efforts expended. The Company’s management monitors planned protocols, work performed, patient enrollment levels and related activities to the extent possible and adjust estimates accordingly.
The Company records as liabilities estimated amounts for litigation, claims or other legal actions that are probable and can be reasonably estimated. The likelihood of a material change in these estimated reserves is dependent on the possible outcome of settlement negotiations, regulatory or judicial review and the development of facts and circumstances in extended litigation which could change claims or assessments when both the amount and range of loss on some outstanding litigation is uncertain. The Company discloses in the footnotes of the financial statements when it is unable to make a reasonable estimate of a material liability that could result from unfavorable outcomes. As events occur, the Company assesses the potential liability related to any pending litigation, claims or other legal actions and adjusts its estimates accordingly. Such adjustments could materially impact its financial statements.
Foreign Currency Translation
The Company translates the assets and liabilities of its foreign subsidiaries stated in local functional currencies to U.S. dollars at the rates of exchange in effect at the end of the period. Revenues and expenses are translated using rates of exchange in effect during the period. Gains and losses from the translation of financial statements denominated in foreign currencies are included as a separate component of accumulated other comprehensive income in the statement of stockholders’ equity.
The Company records foreign currency transactions at the exchange rate prevailing at the date of the transaction with resultant gains and losses being included in results of operations. Foreign currency transaction gains and losses have not been significant for any period presented.
Revenue Recognition
The Company recognizes revenue from product sales at the time of delivery. There are no significant customer acceptance requirements or post-shipment obligations on the part of the Company, except for sales to a new market where acceptance requirements have to be met. Sales to importing agents or distributors are recognized at time of shipment when title to the product is transferred to them. Importing agents or distributors
63
Table of Contents
do not have contractual rights of return except under limited terms regarding product quality. However, the Company is expected to replace products that have expired or are deemed to be damaged or defective when delivered. The Company estimates expected returns primarily on historical patterns. Historically, the Company has had no product returns of damaged, defective or expired product. As such, no amount was accrued for product returns as of December 31, 2010 and 2009 in the respective consolidated balance sheets. Payments by the importing agents and distributors are not contingent upon sale to the end user by the importing agents or distributors. Amounts invoiced relating to arrangements where collectability is uncertain and revenue cannot be recognized are reflected on the Company’s balance sheet as deferred revenue and recognized as the applicable revenue recognition criteria are satisfied.
Contract revenue for research and development is recorded as earned based on performance requirements of the contract. Nonrefundable contract fees for which no further performance obligations exist, and for which there is no continuing involvement by the Company, are recognized as contract revenue on the earlier of when the payments are received or when collection is reasonably assured.
Revenue associated with substantive performance milestones is recognized based on the achievement of the milestones, as defined in the respective agreements and provided that (i) the milestone event is substantive and its achievement is not reasonably assured at the inception of the agreement and (ii) there are no future performance obligations associated with the milestone payment.
Research and Development Expenses
Research and development costs are expensed as incurred. These costs consist primarily of salaries and other personnel-related expenses, including associated stock-based compensation, facility-related expenses, depreciation of facilities and equipment, license-related fees, services performed by clinical research organizations and research institutions and other outside service providers, and certain costs shared for the development of ZADAXIN by the Company’s partner, Sigma-Tau Finanziaria S.p.A (“Sigma-Tau”).
Expenses related to clinical trials generally are accrued based on estimates of work performed or the level of patient enrollment and activities according to the protocols and agreements. The Company monitors planned protocols, work performed, patient enrollment levels and related activities to the extent possible and adjusts estimates accordingly. Nonrefundable advance payments for research and development goods or services are recognized as expense as the related goods are delivered or the related services are provided.
Shipping and Handling Costs
Shipping and handling costs incurred for inventory purchases and product shipments are included in cost of product sales for all periods presented.
Advertising Expenses
Advertising costs are expensed as incurred and are included in sales and marketing expenses for all periods presented. Advertising expenses for the years ended December 31, 2010, 2009 and 2008 were $0.2 million, $0.3 million, and $0.2 million, respectively.
Stock-Based Compensation
The Company records stock-based compensation costs relating to share-based payment transactions, including stock options and employee stock purchase plans. Stock-based compensation expense is estimated at the date of grant based on the fair value of the award using the Black-Scholes option-pricing model. The stock-based compensation costs that are ultimately expected to vest are recognized as expense ratably (as the awards vest) over the requisite service period, which is generally one or four years for stock options and three months for the employee stock purchase plan. The Company estimates pre-vesting forfeitures at the time of grant by
64
Table of Contents
analyzing historical data and revises those estimates in subsequent periods if actual forfeitures differ from those estimates. The total expense recognized over the vesting period will only be for those awards that ultimately vest. Certain target-stock-price-based options were valued using the Monte Carlo simulation options pricing model and recognized to expense over the service periods for each of the vesting portions of these awards which were six or eight years. Refer also to Note 13, “Stockholders’ Equity,” in the Notes to Consolidated Financial Statements for further information regarding stock-based compensation.
Income Taxes
Income taxes are accounted for under the liability method. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities as measured by the enacted tax rates that will be in effect when these differences reverse. The Company provides a valuation allowance against net deferred tax assets if, based upon the available evidence, it is more-likely-than-not that the deferred tax assets will not be realized. The Company’s policy is to recognize interest and penalties related to the estimated obligations for tax positions as a component of income tax expense. The amount of accrued interest related to tax positions taken on our tax returns and included in accrued liabilities was $0.2 million and $0 at December 31, 2010 and 2009, respectively.
The Company records liabilities related to uncertain tax positions in accordance with the guidance that clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The Company does not believe any such uncertain tax positions currently pending will have a material adverse effect on its Consolidated Financial Statements, although an adverse resolution of one or more of these uncertain tax positions in any period could have a material impact on the results of operations for that period.
Comprehensive Income
Comprehensive income is comprised of net income (loss) and other comprehensive income (loss). The following table summarizes the components of accumulated other comprehensive income (in thousands):
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Net unrealized gain (loss) on available-for-sale securities |
$ | — | $ | 2 | $ | (2 | ) | |||||
| Net unrealized loss and foreign currency translation adjustment on |
(70 | ) | (34 | ) | (60 | ) | ||||||
| Cumulative foreign currency translation adjustment |
137 | 54 | 65 | |||||||||
| Accumulated other comprehensive income |
$ | 67 | $ | 22 | $ | 3 | ||||||
Net Income (Loss) Per Share
Basic net income (loss) per share has been computed by dividing net income (loss) by the weighted-average number of shares of common stock outstanding for the period. Diluted net income (loss) per share is computed by dividing net income (loss) by the weighted-average number of common equivalent shares outstanding for the period. Diluted net income (loss) per share includes any dilutive impact from outstanding stock options, warrants and the employee stock purchase plan using the treasury stock method.
65
Table of Contents
The following is a reconciliation of the numerator and denominators of the basic and diluted net income (loss) per share computations for the year ended December 31 (in thousands, except per share amounts):
| 2010 | 2009 | 2008 | ||||||||||
| Numerator: |
||||||||||||
| Net income (loss). |
$ | 21,081 | $ | 11,945 | $ | (8,348 | ) | |||||
| Denominator: |
||||||||||||
| Weighted-average shares outstanding used to compute basic net income (loss) |
47,624 | 46,574 | 46,212 | |||||||||
| Effect of dilutive securities |
1,790 | 561 | — | |||||||||
| Weighted-average shares outstanding used to compute diluted net income (loss) per share |
49,414 | 47,135 | 46,212 | |||||||||
| Basic net income (loss) per share |
$ | 0.44 | $ | 0.26 | $ | (0.18 | ) | |||||
| Diluted net income (loss) per share |
$ | 0.43 | $ | 0.25 | $ | (0.18 | ) | |||||
For the years ended December 31, 2010, 2009 and 2008, approximately 2,872,513, 6,107,498 and 7,968,269 shares, respectively, related to outstanding stock options and warrants were excluded from the calculation of diluted net income (loss) per share because their inclusion would have been anti-dilutive. In addition, for the years ended December 31, 2010 and 2009, 155,000 and 710,959 shares, respectively, subject to market or performance conditions were excluded from the calculation of diluted net income per share because the performance or market criteria had not been met.
Segment Information
The Company operates in one segment (refer to Note 17).
Recent Accounting Guidance
In January 2010, the Financial Accounting Standards Board (“FASB”) issued FASB Accounting Standards Update (“ASU”) No. 2010-06, Fair Value Measurements and Disclosures (Topic 820)—Improving Disclosures about Fair Value Measurements. The ASU requires new disclosures about significant transfers in and out of Levels 1 and 2 fair value measurements and separate disclosures about purchases, sales, issuances and settlements relating to Level 3 fair value measurements. The ASU also clarifies existing disclosure requirements regarding inputs and valuation techniques, as well as the level of disaggregation for each class of assets and liabilities for which separate fair value measurements should be disclosed. The Company adopted ASU 2010-06 at the beginning of fiscal 2010. The adoption of this ASU did not have a material impact on the Company’s condensed consolidated financial statements.
66
Table of Contents
Note 2 — Cash, Cash Equivalents and Investments
The following is a summary of cash, cash equivalents, and investments (in thousands):
| December 31, 2010 | ||||||||||||||||
| Amortized Cost |
Unrealized Gains |
Unrealized Losses for More Than 12 Months |
Estimated Fair Value |
|||||||||||||
| Cash and cash equivalents: |
||||||||||||||||
| Cash |
$ | 28,112 | $ | — | $ | — | $ | 28,112 | ||||||||
| Money market funds |
12,900 | — | — | 12,900 | ||||||||||||
| Foreign U.S. dollar term deposits |
12,005 | — | — | 12,005 | ||||||||||||
| Total cash and cash equivalents |
$ | 53,017 | $ | — | $ | — | $ | 53,017 | ||||||||
| Available-for-sale investments: |
||||||||||||||||
| Foreign U.S. dollar term deposits maturing within 6 months |
$ | 3,125 | $ | — | $ | — | $ | 3,125 | ||||||||
| Restricted long-term Italian state bonds maturing in 2013 |
450 | — | (70 | ) | 380 | |||||||||||
| Total available-for-sale investments |
$ | 3,575 | $ | — | $ | (70 | ) | $ | 3,505 | |||||||
| December 31, 2009 | ||||||||||||||||
| Amortized Cost |
Unrealized Gains |
Unrealized Losses for More Than 12 Months |
Estimated Fair Value |
|||||||||||||
| Cash and cash equivalents: |
||||||||||||||||
| Cash |
$ | 17,541 | $ | — | $ | — | $ | 17,541 | ||||||||
| Money market funds |
16 | — | — | 16 | ||||||||||||
| Foreign U.S. dollar term deposits |
12,130 | — | — | 12,130 | ||||||||||||
| Total cash and cash equivalents |
$ | 29,687 | $ | — | $ | — | $ | 29,687 | ||||||||
| Available-for-sale investments: |
||||||||||||||||
| Certificates of deposit maturing within 1 year |
$ | 76 | $ | — | $ | — | $ | 76 | ||||||||
| Restricted long-term Italian state bonds maturing in 2013 |
449 | — | (34 | ) | 415 | |||||||||||
| Corporate equity securities |
51 | 2 | — | 53 | ||||||||||||
| Total available-for-sale investments |
$ | 576 | $ | 2 | $ | (34 | ) | $ | 544 | |||||||
| Trading security investments: |
||||||||||||||||
| Auction rate securities maturing after 20 years |
$ | 1,588 | $ | — | $ | — | $ | 1,588 | ||||||||
| Total trading security investments |
$ | 1,588 | $ | — | $ | — | $ | 1,588 | ||||||||
The Company’s restricted long-term Italian state bonds secure its Italian value added tax filing arrangements. The unrealized losses on the bonds mainly relate to loss on foreign currency translation. The Company intends and has the ability to hold its restricted Italian state bond investments until recovery and is not likely to sell the investments before maturity.
The Company’s certificate of deposit for $0.1 million at December 31, 2009 secured the Company’s letter of credit required under its European value added tax filing arrangements.
67
Table of Contents
Note 3 — Fair Value Measurements
The following table represents the Company’s fair value hierarchy for its financial assets (cash equivalents, investments and other current assets) measured at fair value on a recurring basis (in thousands):
| Fair Value Measurements at December 31, 2010 Using | ||||||||||||||||
| Description |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
Balance as of December 31, 2010 |
||||||||||||
| Money market funds |
$ | 12,900 | $ | — | $ | — | $ | 12,900 | ||||||||
| Foreign U.S. dollar term deposits |
— | 15,130 | — | 15,130 | ||||||||||||
| Restricted long-term Italian state bonds |
380 | — | — | 380 | ||||||||||||
| Total |
$ | 13,280 | $ | 15,130 | $ | — | $ | 28,410 | ||||||||
| Fair Value Measurements at December 31, 2009 Using | ||||||||||||||||
| Description |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
Balance as of December 31, 2009 |
||||||||||||
| Money market funds |
$ | 16 | $ | — | $ | — | $ | 16 | ||||||||
| Foreign U.S. dollar term deposits |
— | 12,130 | — | 12,130 | ||||||||||||
| Certificates of deposit |
— | 76 | — | 76 | ||||||||||||
| Corporate equity securities |
53 | — | — | 53 | ||||||||||||
| Restricted long-term Italian state bonds |
415 | — | — | 415 | ||||||||||||
| Auction rate securities |
— | — | 1,588 | 1,588 | ||||||||||||
| Put option |
— | — | 202 | 202 | ||||||||||||
| Total |
$ | 484 | $ | 12,206 | $ | 1,790 | $ | 14,480 | ||||||||
The following table provides a summary of changes in fair value of the Company’s level 3 financial assets during fiscal 2008, 2009 and 2010 (in thousands):
| Auction Rate Securities |
Put Option |
|||||||
| Balance at December 31, 2007 |
$ | 2,825 | $ | — | ||||
| Total realized gain (loss) included in other income (expense) |
(315 | ) | 285 | |||||
| Proceeds from sales, net of purchases |
(1,025 | ) | — | |||||
| Balance at December 31, 2008 |
1,485 | 285 | ||||||
| Total realized gain (loss) included in other income (expense) |
103 | (83 | ) | |||||
| Balance at December 31, 2009 |
1,588 | 202 | ||||||
| Proceeds from sales |
(1,800 | ) | — | |||||
| Total realized gain (loss) included in other income (expense) |
212 | (202 | ) | |||||
| Balance at December 31, 2010 |
$ | — | $ | — | ||||
In November 2008, the Company accepted an Auction Rate Securities Rights Offer (the “Settlement Agreement”) from UBS AG under which, in return for a general release of claims and the grant of a right to UBS AG to purchase the Company’s ARS at any time for full par value, the Company received the right to require UBS AG to purchase the Company’s ARS beginning in June 2010 (the “Rights”). By entering into the Settlement Agreement, the Company (1) received the right (the “Put Option”) to sell these auction rate securities back to the investment firm at par, at its sole discretion, anytime during the period from June 30, 2010 through July 2, 2012,
68
Table of Contents
and (2) gave the investment firm the right to purchase these auction rate securities or sell them on the Company’s behalf at par anytime after the execution of the Settlement Agreement through July 2, 2012. On June 30, 2010, the Company exercised the Put Option and sold its ARS back to the investment firm.
Note 4 — Inventories
Inventories consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Raw materials |
$ | 590 | $ | 2,446 | ||||
| Work in progress |
355 | 1,048 | ||||||
| Finished goods |
6,133 | 6,655 | ||||||
| $ | 7,078 | $ | 10,149 | |||||
Note 5 — Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Prepaid insurance |
$ | 499 | $ | 406 | ||||
| Prepaid clinical trial expense |
828 | 397 | ||||||
| Grant receivable |
268 | — | ||||||
| Other prepaid expenses |
462 | 715 | ||||||
| $ | 2,057 | $ | 1,518 | |||||
Note 6 — Property and Equipment
Property and equipment consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Office equipment |
$ | 868 | $ | 793 | ||||
| Leasehold improvements |
735 | 732 | ||||||
| Office furniture and fixtures |
619 | 616 | ||||||
| Software |
108 | 68 | ||||||
| Vehicle |
— | 57 | ||||||
| 2,330 | 2,266 | |||||||
| Less accumulated depreciation |
(1,742 | ) | (1,495 | ) | ||||
| Net property and equipment |
$ | 588 | $ | 771 | ||||
Depreciation expense was $0.3 million for each of the years ended December 31, 2010, 2009 and 2008.
Note 7 — Other Assets
Other assets consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Value added tax receivable |
$ | 441 | $ | 732 | ||||
| Lease deposits |
249 | 247 | ||||||
| Intangible assets, net |
53 | 192 | ||||||
| Other assets |
148 | 78 | ||||||
| $ | 891 | $ | 1,249 | |||||
69
Table of Contents
Note 8 — Accrued Liabilities
The following is a summary of accrued liabilities (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accrued compensation |
$ | 2,277 | $ | 1,913 | ||||
| Accrued taxes, tax reserves and interest |
1,683 | 309 | ||||||
| Accrued sales and marketing expenses |
1,558 | 1,394 | ||||||
| Accrued professional fees |
1,184 | 394 | ||||||
| Accrued clinical trial expense |
444 | 452 | ||||||
| Accrued manufacturing costs |
361 | 808 | ||||||
| Other |
740 | 778 | ||||||
| $ | 8,247 | $ | 6,048 | |||||
Note 9 — Commitments
Leases
In May 2007, the Company entered into a non-cancelable operating lease agreement for its corporate headquarters (“the Lease”) effective from July 1, 2007 through June 30, 2014, with an option to renew for an additional five year period. In September 2008, the Company entered into an amendment to the Lease for additional office space (“the Expansion Agreement”) that expires on June 30, 2014, with an option to renew for an additional five year period. Both the Lease and Expansion Agreements contain rent escalations of approximately 4% and 6% per year, respectively. The Company is recognizing the rental expense on a straight-line basis over the lease terms. Under the terms of the Lease and the Expansion Agreements, the Company was provided allowances in the amounts of approximately $0.2 million and $0.5 million, respectively, towards the cost of its leasehold improvements and as an incentive to rent, respectively. The Company has recorded these allowances as deferred rent which is being amortized over the lease terms as a reduction of rent expense. The leases require the Company to pay insurance and taxes and its pro-rata share of operating expenses.
The Company also leases office facilities and equipment outside the U.S. under non-cancelable operating lease agreements and subleases certain office facilities to a third party. Rent expense for the years ended December 31, 2010, 2009, and 2008 was $1.6 million, $1.6 million and $1.1 million, respectively. Future minimum lease payments and sublease rental income under non-cancelable facility and equipment operating lease agreements as of December 31, 2010, were as follows (in thousands):
| Year ended: |
Minimum Lease Payments |
Sublease Rental Income |
Net Minimum Lease Payments |
|||||||||
| 2011 |
$ | 1,876 | $ | 70 | $ | 1,806 | ||||||
| 2012 |
1,524 | — | 1,524 | |||||||||
| 2013 |
1,432 | — | 1,432 | |||||||||
| 2014 |
744 | — | 744 | |||||||||
| $ | 5,576 | $ | 70 | $ | 5,506 | |||||||
Note 10 — Silicon Valley Bank Line-of-Credit
On October 1, 2010, the Company’s subsidiaries, SciClone Pharmaceuticals International Ltd. and SciClone Pharmaceuticals International China Holding Ltd. as borrowers, terminated the existing $6 million Credit Facility with Silicon Valley Bank (“SVB”) and entered into a $15 million loan and security agreement with SVB (“the Debt Financing Facility”). SciClone Pharmaceuticals, Inc. is the guarantor of the Debt Financing Facility. The Debt Financing Facility bears interest on borrowed funds at SVB’s prime rate plus 1.25% (5.25% at December 31, 2010) on outstanding balances and is secured by a first priority secured interest in all of the
70
Table of Contents
Company’s assets, including intellectual property in an event of default. The Company is required to meet certain financial covenants, including minimum liquidity, as defined, and is subject to certain minimum fees and interest payments. The Company is also required to meet certain operating covenants that limit its ability to incur liabilities, create liens, make capital expenditures, pay dividends or distributions, make investments, and dispose of assets. The Debt Financing Facility expires October 1, 2012, and upon termination all amounts borrowed must be repaid in full. As of December 31, 2010, the Company had borrowed $2.5 million on the Debt Financing Facility and was required to maintain debt coverage ratio of 1.35 to 1 equal to $3.4 million of cash in SVB cash accounts to meet its financial liquidity covenant. The Company was in compliance with all covenants as of December 31, 2010. The Company capitalized $0.1 million in costs associated with the origination of the $15 million facility, which are being amortized to interest and investment expense over the term of the Debt Financing Facility. The Company recognized to expense approximately $0.1 of unamortized loan origination fees associated with the initial $6 million Credit Facility upon termination.
In connection with the initial Credit Facility entered into in November 2008, the Company issued Silicon Valley Bank a five-year warrant to purchase 60,000 shares of the Company’s common stock at an exercise price of $0.86 per share. Silicon Valley Bank exercised the warrant in full on December 8, 2010 in a net share settlement resulting in the issuance of 47,132 shares of the Company’s common stock.
Note 11 — Related Party Transactions
The Company has licensed to its largest shareholder, Sigma-Tau, exclusive ZADAXIN (thymalfasin or thymosin alpha 1) development and marketing rights that cover all countries in the European Union as defined on January 1, 1995, in addition to Iceland, Norway and Switzerland. The Company’s collaboration with Sigma-Tau is governed by an agreement entered into in 2000 with a term expiring in March 2012 unless renewed, as well as by amendments to the agreement regarding particular development efforts. In addition, the agreement governed the Company’s joint collaboration on the development of thymalfasin in certain indications, and for the sharing of intellectual property in the respective territories. The agreement also provides that if Sigma-Tau sells ZADAXIN in the licensed territory, it will purchase the product from SciClone at a specified price, subject to certain adjustments. The Company does not currently anticipate that Sigma-Tau will sell any ZADAXIN other than nominal amounts in Italy.
Pursuant to the agreement, Sigma-Tau conducted trials in Europe for the treatment of malignant melanoma and hepatitis C and the Company conducted certain trials in the U.S. for the treatment of hepatitis C, and each party agreed to provide certain funding and support for the development efforts of the other. The development obligations to each other were completed when Sigma-Tau completed the phase 3 hepatitis C triple therapy clinical trial in Europe and delivered a final report on the trial in October 2009. The Company paid Sigma-Tau an aggregate of $4.0 million of funding support during the course of patient enrollment and trial period including the period of completion of the final report. The Company’s accounting policy for recording funding amounts due to Sigma-Tau was to record the amounts to research and development expense over the trial period including the period of completion of the final report. Based on the level of activity in this trial, the Company recorded $0.1 million and $0.3 million of research and development expense related to this trial in the years ended December 31, 2009 and 2008, respectively.
There are no on-going development or reimbursement obligations under the agreement, and there are no milestones or similar terms currently in effect. However, the Company continues to cooperate with Sigma-Tau on exploring the development of thymalfasin in other indications, including as a vaccine adjuvant and in aspergillosis.
Note 12 — Income Taxes
The Company recorded income tax expense of $2.2 million, $0.6 million, and $0.5 million for the years ended December 31, 2010, 2009 and 2008, respectively, related to its operations in China. The Company’s
71
Table of Contents
statutory tax rate in China was 22%, 20% and 18% for the years ended December 31, 2010, 2009, and 2008, respectively. The Company has not recorded any U.S. federal or state income tax expense for the years ended December 31, 2010, 2009 and 2008. Undistributed earnings of the Company’s foreign subsidiaries that are considered to be permanently invested outside the U.S. and for which no U.S. taxes have been provided amounted to approximately $12.4 million at December 31, 2010.
The domestic and foreign components of income (loss) before provision for tax for the years ended December 31 are as follows (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Domestic |
$ | (19,315 | ) | $ | (19,554 | ) | $ | (25,922 | ) | |||
| Foreign |
42,572 | 32,140 | 18,049 | |||||||||
| Pre-tax income (loss) |
$ | 23,257 | $ | 12,586 | $ | (7,873 | ) | |||||
A reconciliation of the statutory federal income tax rate of 34% to the actual tax rate for the years ended December 31 is as follows (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Tax at federal statutory rate |
$ | 7,907 | $ | 4,279 | $ | (2,677 | ) | |||||
| Foreign income tax at different rates |
(13,085 | ) | (10,279 | ) | (5,661 | ) | ||||||
| Taxable dividend from foreign subsidiary |
6,724 | 5,440 | 4,177 | |||||||||
| Tax reserve accrual |
795 | — | — | |||||||||
| Net operating losses not benefited |
— | 916 | 4,239 | |||||||||
| Change in valuation adjustment |
(318 | ) | — | — | ||||||||
| Stock-based compensation |
53 | 122 | 91 | |||||||||
| Non deductible expenses |
350 | 281 | 308 | |||||||||
| Other |
(250 | ) | (118 | ) | (2 | ) | ||||||
| Income tax expense |
$ | 2,176 | $ | 641 | $ | 475 | ||||||
Significant components of the Company’s deferred tax assets at December 31 are as follows (in thousands):
| 2010 | 2009 | |||||||
| Net operating loss carryforwards |
$ | 40,324 | $ | 43,439 | ||||
| Research and development credit carryforwards |
10,284 | 8,010 | ||||||
| Other |
4,855 | 4,731 | ||||||
| Gross deferred tax assets |
55,463 | 56,180 | ||||||
| Valuation allowance |
(55,463 | ) | (56,180 | ) | ||||
| Total deferred tax assets |
$ | — | $ | — | ||||
Realization of deferred tax assets is dependent upon the Company generating future taxable income, the timing and amount of which are uncertain. Accordingly, the deferred tax assets have been fully offset by a valuation allowance. The valuation allowance (decreased) increased by approximately $(0.7) million, $(6.7) million, and $13.0 million, in the years ended December 31, 2010, 2009 and 2008, respectively.
At December 31, 2010, the Company had federal net operating loss carryforwards of approximately $122.7 million that expire in the years 2011 through 2028, and federal research and development, orphan drug and investment tax credit carryforwards of approximately $12.3 million that expire in the years 2011 through 2020. At December 31, 2010, the Company has state net operating loss carryforwards of approximately $33.7 million that begin to expire in the year 2012, if not utilized, and state research and development tax credit carryforwards
72
Table of Contents
of approximately $2.1 million that do not expire. Approximately $9.1 million of the operating loss carryforwards relate to benefits associated with stock option deductions that, when recognized, $3.6 million will be credited directly to stockholders’ equity.
Utilization of the Company’s net operating loss and credit carryforwards may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such annual limitation could result in the expiration of the net operating loss and credit carryforwards before utilization.
As of December 31, 2010, the unrecognized tax benefit was $4.2 million, of which $0.6 million, if recognized would affect the effective tax rate, and $3.6 million was offset by a valuation allowance. A reconciliation of the beginning and ending amount of unrecognized tax benefit is as follows (in thousands):
| Balance as of January 1, 2010 |
$ | 2,818 | ||
| Tax positions related to current year: |
||||
| Additions for current year items |
313 | |||
| Additions for prior year items |
1,127 | |||
| Reductions for prior year items |
(33 | ) | ||
| Balance as of December 31, 2010 |
$ | 4,225 | ||
The tax years 1995-2010 remain open to examination by the major taxing jurisdictions to which the Company is subject. Furthermore, the Company has been notified by the Internal Revenue Service that its 2008 U.S. federal tax return has been selected for examination, but the examination process has not yet commenced. Although potential resolution of uncertain tax positions involve multiple tax periods and jurisdictions, it is reasonably possible that a reduction of up to $0.6 million of unrecognized tax benefits may occur within the next 12 months.
Note 13 — Stockholders’ Equity
Stock Award Plans
The Company’s 1995 Equity Incentive Plan has reserved 6,100,000 shares of common stock for issuance and permits the grant of incentive stock options, nonstatutory stock options and other forms of equity compensation. Although the 1995 Plan has expired, the outstanding stock options relating to it are fully valid.
The Company’s 2005 Equity Incentive Plan (the “2005 Plan”) has reserved 10,600,000 shares of common stock for issuance. The 2005 Plan permits the grant of incentive stock options, nonstatutory stock options, restricted stock units, performance shares and other forms of equity compensation. As of December 31, 2010, approximately 3,679,000 shares of common stock were available for future issuance under the 2005 Plan.
Under the 1995 and 2005 Plans, options are exercisable upon conditions determined by the board of directors and expire ten years from the date of grant. Options are generally granted at fair market value on the date of grant and vest over time, generally four years, or upon achievement of certain market and service conditions. See Stock-Based Compensation.
The Company’s 1995 Nonemployee Director Stock Option Plan (the “1995 Director Plan”) had reserved 750,000 shares of common stock for issuance. The 1995 Director Plan permits the grant of nonqualified stock options to nonemployee directors. Although the 1995 Directors Plan has expired, the outstanding stock options relating to it are fully valid.
The Company’s 2004 Outside Directors Stock Option Plan (the “2004 Director Plan”) has reserved 1,765,000 shares of common stock for issuance. The 2004 Director Plan automatically grants nonqualified stock options to nonemployee directors upon their appointment or first election to the Company’s board of directors
73
Table of Contents
(“Initial Grant”) and annually upon their reelection to the board of directors at the Company’s Annual Meeting of Stockholders (“Annual Grant”). As of December 31, 2010, approximately 425,000 shares of common stock were available for future issuance under the 2004 Director Plan.
Under the 1995 and 2004 Director Plans, options are granted at fair market value on the date of grant and expire ten years from the date of grant. Initial Grants become exercisable in three equal annual installments beginning on the first anniversary of the date of grant, and Annual Grants become exercisable in twelve equal monthly installments from the date of grant, subject in each case to the director’s continuous service on the Company’s board of directors.
Certain stock option awards are subject to accelerated vesting if there is a change in control.
Stock-Based Compensation
The compensation cost that has been charged against income for the stock award plans was $2.2 million, $1.9 million and $1.8 million, respectively, for the years ended December 31, 2010, 2009, and 2008. Compensation cost capitalized in inventory was $44,000, $35,000 and $49,000, respectively, for the years ended December 31, 2010, 2009, and 2008. There has been no income tax benefit recognized in the income statement for share-based compensation arrangements.
The fair value of each non performance-based and non target stock-price-based option granted under the Company’s stock award plans is estimated on the date of grant using the Black-Scholes option valuation model and the single option approach with the following weighted-average assumptions for the years ended December 31:
| Stock Option Plan |
2010 |
2009 |
2008 |
|||||||||
| Risk-free interest rate |
2.39 | % | 1.79 | % | 2.76 | % | ||||||
| Dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | ||||||
| Volatility factor of the expected market price of our common stock |
72.50 | % | 70.38 | % | 66.57 | % | ||||||
| Weighted-average expected life of option (years) |
5.03 | 4.83 | 4.83 | |||||||||
The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The expected dividend yield is based on the Company’s history and expectations of no dividend payouts. Expected volatility is based on the historical volatility of the Company’s stock. The expected term of options granted is derived from historical data on employee exercises and terminations.
The following table summarizes stock option activity as of December 31, 2010, and changes during the year then ended is presented below (in thousands, except per share amounts):
| Options Outstanding | ||||||||||||||||||||
| Shares Available For Grant |
Number of Shares |
Weighted- Average Exercise Price Per Share |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value |
||||||||||||||||
| Balance at December 31, 2009 |
2,283 | 7,939 | $ | 2.93 | ||||||||||||||||
| 2005 Plan shares reserved |
2,800 | — | — | |||||||||||||||||
| Options cancelled |
1,223 | (1,223 | ) | $ | 5.35 | |||||||||||||||
| Options granted |
(1,475 | ) | 1,475 | $ | 3.46 | |||||||||||||||
| Options exercised |
— | (730 | ) | $ | 1.87 | |||||||||||||||
| Plan shares expired |
(727 | ) | — | — | ||||||||||||||||
| Balance at December 31, 2010 |
4,104 | 7,461 | $ | 2.74 | 6.22 | $ | 12,034 | |||||||||||||
| Vested and expected to vest after |
7,209 | $ | 2.74 | 6.16 | $ | 11,638 | ||||||||||||||
| Exercisable at December 31, 2010 |
4,128 | $ | 3.07 | 4.96 | $ | 5,819 | ||||||||||||||
74
Table of Contents
The Company granted to its chief executive officer target-stock-price-based options to purchase 600,000 and 300,000 shares of the Company’s common stock at an exercise price per share of $2.49 and $1.81, respectively. The options have a term of 10 years and shares of such option were to vest upon the Company’s common stock trading for at least 30 consecutive calendar days at or greater than a target closing stock price as reported on The NASDAQ Stock Market, of (a) $4.50 on or before June 2, 2009 for 150,000 shares, (b) $6.00 on or before June 2, 2010 for 150,000 shares, (c) $8.00 on or before June 2, 2011 for 150,000 shares, (d) $10.00 on or before June 2, 2012 for 150,000 shares, (e) $12.00 on or before June 2, 2013 for 150,000 shares, and (f) $14.00 on or before June 2, 2014 for 150,000 shares, each price as adjusted for stock dividends, stock splits or similar changes in the Company’s capital structure. These grants were considered awards with market and service conditions and compensation expense was recognized for these options as long as the service requirements were met, even if the market conditions were not reached. Because of the market conditions of the grants, the Monte Carlo simulation option pricing model was used to calculate the grant date fair value per share of $1.70 and $0.88, respectively, related to each of the six vesting portions of these awards with the assumptions of a risk-free interest rate of 5.10% and 3.88% respectively, a volatility factor of 94% and 89.14%, respectively, dividend yield of 0%, and an expected life of 10 years. The related compensation costs were being expensed over the service periods for each of the six vesting portions of the awards.
In May 2010, the Company amended the target-stock-price-based options to purchase an aggregate of 750,000 shares of the Company’s common stock that were still subject to vesting that had been granted to its chief executive officer. The amendment modified the vesting provisions of the options such that 1/36th of the unvested stock options vest monthly over a three-year period, with initial vesting occurring on June 1, 2010. The fair value of the modified award was estimated using the Black-Scholes option valuation model with the assumptions of a risk-free rate of 2.28%, a volatility factor of 71.83%, a dividend yield of 0%, and an expected life of 5.07 years. The incremental compensation cost resulting from the modification and the remaining unrecognized compensation expense from the original award are being recognized ratably over the three-year vesting period. The Company recorded expense of $0.3 million, $0.2 million and $0.3 million for the years ended December 31, 2010, 2009 and 2008, respectively, related to these options.
The Company has granted certain performance-based options to purchase shares of the Company’s common stock at an exercise price equal to the closing price of a share of the Company’s common stock as of the grant date. The options will fully vest upon meeting a performance goal within an established time frame. If the performance goal is met for the option within the established time frame, the option generally has a ten-year term measured from the date of grant. If the performance goal is not met within the established time frame, the option expires in its entirety. The grant date fair value per share of the awards has been calculated using the Black-Scholes option pricing model using the following assumptions: expected terms ranging from 4.83-5.02 years, volatility factor ranging from 69.06-72.66%, and risk free interest rates ranging from 1.72-2.43%. The Company recognizes expense related to a performance-based option over the period of time the Company determines that it is probable that the performance goal will be achieved.
During 2009, the Company extended the period in which two of the Company’s departed board members may exercise their outstanding vested stock options following the cessation of their service to the Company from ninety days to the second anniversary of the date of cessation of service which ceased on June 9, 2009. The Company recorded expense of $0.1 million for the year ended December 31, 2009 related to these modifications.
The weighted-average fair value of stock options granted for the years ended December 31, 2010, 2009 and 2008 was $2.10, $0.76, and $0.98, respectively. The intrinsic value of options at time of exercise was $1.3 million, $1.7 million, and $0.1 million for the years ended December 31, 2010, 2009, and 2008, respectively. The estimated fair value of shares vested for the years ended December 31, 2010, 2009, and 2008 was $1.9 million, $1.5 million, and $1.6 million, respectively. As of December 31, 2010, unamortized compensation expense related to unvested options was approximately $3.9 million. The weighted average period over which compensation expense related to these options will be recognized is approximately 2.27 years. Cash received from stock option exercises was $1.4 million, $2.6 million, and $0.1 million for the years ended December 31, 2010, 2009 and 2008, respectively.
75
Table of Contents
Employee Stock Purchase Plan
As of December 31, 2010, 1,300,000 shares of our common stock are reserved for issuance under the Company’s Employee Stock Purchase Plan (“ESPP”). Under the terms of the ESPP, eligible employees may choose to have up to 15% of their salary withheld to purchase the Company’s common stock and may purchase up to 1,000 shares per offering period. Each offering under the ESPP is for a three-month period. Commencing with the offering period beginning June 1, 2010, the purchase price of the stock issued under the ESPP will be equal to 85% of the lower of the fair market value of a share of common stock on the first day of the offering or on the final day of the offering period. For the year ended December 31, 2010, the Company recorded $16,000 of expense related to the ESPP and issued 16,426 shares. There was no expense recorded or shares issued related to the ESPP during the years ended December 31, 2009 and 2008. As of December 31, 2010, approximately 566,000 shares of common stock were available for issuance under the ESPP.
Stockholder Rights Agreement
On December 18, 2006, the Company’s board of directors declared a dividend distribution of one Preferred Stock Purchase Right (collectively, the “Rights”) for each outstanding share of the Company’s Common Stock, each Right which entitles the registered holder to purchase from the Company one one-thousandth of a share of the Company’s Series D Preferred Stock, $0.001 par value, at a price of $25.00 pursuant to a Rights Agreement dated as of December 19, 2006, between the Company and Mellon Investor Services LLC (the “Rights Agreement”). The Rights, which will initially trade with the Common Stock, become exercisable when a person or group acquires 15% or more of the Company’s Common Stock without prior board approval. In that event, the Rights permit the Company’s stockholders, other than the acquiror, to purchase the Company’s Common Stock having a market value of twice the exercise price of the Rights, in lieu of the Preferred Stock. Alternatively, when the Rights become exercisable, the Company’s board of directors may authorize the issuance of one share of the Company’s Common Stock in exchange for each Right that is then exercisable. In addition, in the event of certain business combinations, the Rights permit the purchase of the Common Stock of an acquiror at a 50% discount. Rights held by the acquiror will become null and void in each case. Prior to a person or group acquiring 15%, the Rights can be redeemed for $0.001 each by action of the board. The Rights Agreement contains an exception to the 15% ownership threshold for shares currently beneficially owned by Sigma-Tau Finanziaria S.p.A. The Rights expire on December 19, 2016. The Rights Agreement includes a requirement that a committee of independent directors evaluate the Rights Agreement at least every three years.
Note 14 — 401k Plan
The Company has a pre-tax savings plan covering most U.S. employees, which qualifies under Section 401(k) of the Internal Revenue Code. Under the plan, eligible employees may contribute a portion of their pre-tax salary, subject to certain limitations. The Company contributes and matches 50% of the employee contributions. Company contributions, which can be terminated at the Company’s discretion, were approximately $0.2 million for each of the years ended December 31, 2010, 2009, and 2008.
Note 15 — Other Income
Other income for the year ended December 31, 2010 included approximately $1.0 million in non-taxable grants awarded to the Company in November 2010 related to its research and development activities in SCV-07 and ZADAXIN as part of the U.S. Department of Treasury’s Therapeutic Discovery Project Program.
Note 16 — Other Corporate Matters
On August 5, 2010, SciClone was contacted by the United States Securities and Exchange Commission (“SEC”) and advised that the SEC has initiated a formal, non-public investigation of SciClone, and the SEC issued a subpoena to SciClone requesting a variety of documents and other information. The subpoena requests documents relating to a range of matters including, but not limited to, potential payments or transfers of anything
76
Table of Contents
of value to regulators and government-owned entities in China, bids or contracts with state or government-owned entities in China, any joint venture partner, intermediary or local agent of the Company in China, the Company’s ethics and anti-corruption policies, training, and audits, and certain company financial and other disclosures. On August 6, 2010, the Company received a letter from the United States Department of Justice (“DOJ”) indicating that the DOJ was investigating Foreign Corrupt Practices Act (“FCPA”) issues in the pharmaceutical industry generally, and that the DOJ had information about the Company’s practices suggesting possible violations. The Company intends to cooperate fully with the SEC and DOJ in the conduct of their investigations.
In response to these matters, the Company’s board of directors has appointed a special committee of independent directors to oversee the Company’s response to the government inquiry. Based on an initial review, the special committee has decided to undertake an independent investigation as to matters reflected in and arising from the SEC and DOJ investigations including, but not limited to, certain sales and marketing matters in China, in order to evaluate whether any violation of the FCPA or other laws occurred. The special committee has not concluded its investigation or reached any conclusions, to date, nor has the special committee or management made a final determination regarding whether any violation of the FCPA or any other law or regulation has occurred. However, we anticipate that the special committee will finalize its findings in the near future, which may include findings regarding potential FCPA issues and recommendations regarding additional remediation measures and possible suggestions regarding personnel issues. In the course of its review, the special committee has instructed management to (i) evaluate and to expand the Company’s training of employees regarding understanding and compliance with laws including the FCPA and other anti-bribery laws and regulations, (ii) evaluate our existing compliance and anti-bribery guidelines and to prepare a new, more detailed, guidelines for implementation after review by our Board and/or committees of the Board, and (iii) implement a pre-approval policy requiring our compliance officer to pre-approve certain expenses including payments for, or reimbursement of, third party gifts, travel expenses, honoraria and sponsorships of certain third party events.
We have consistently maintained a policy that our employees comply with all applicable laws. However, we have reached certain conclusions regarding the effectiveness of our implementation of that policy. In the course of the evaluation of our existing training, policies and internal controls over financial reporting and expense approval and expense reimbursement practices, and of the matters brought to the attention of management by the special committee, we determined that our guidelines, employee training and expense approval practices and our internal controls with respect to certain matters, including the payment for, or reimbursement of, third party gifts, travel expenses, honoraria and sponsorship of certain third party events required improvements in order for management to provide reasonable assurance of our implementation of our policy on compliance with applicable laws. In addition, management determined that we had a material weakness in our internal control over financial reporting as of December 31, 2010 relating to our implementation of our policy on compliance with laws described in Item 9A of this report.
During the first quarter of fiscal 2011, we initiated or commenced various remediation efforts to provide reasonable assurance of our implementation of our policy on compliance with laws, including the FCPA and other anti-bribery laws, as described in Item 9A of this report. We also determined that our internal controls have not provided adequate information for us to monitor our policy on compliance with laws.
Following the Company’s announcement of these investigations, purported class actions naming SciClone and certain of its officers as defendants were filed and derivate lawsuits purportedly on behalf of the Company were filed naming certain of its officers and directors as defendants. See Item 3, “Legal Proceedings.”
Based on the information obtained to date, the Company has determined that any potential liability that may result is not probable or cannot be reasonably estimated and therefore no accrual was made related to these matters in its consolidated financial statements as of December 31, 2010. As events occur, the Company will assess the potential liability related to its pending investigations and class action and derivative lawsuits and adjust its estimates accordingly. Such adjustments could materially impact the Company’s financial statements.
77
Table of Contents
Note 17 — Significant Geographic Information
The Company operates in one business segment: the development and commercialization of novel therapeutics to treat chronic and life threatening diseases. Through December 31, 2010, all of the Company’s product revenues were generated from the sale of ZADAXIN.
The President and CEO has been identified as the Chief Operating Decision Maker (“CODM”) because he has final authority over resource allocation decisions and performance assessment. The CODM does not receive discrete financial information at a level below that of the business segment.
The Company’s domestic operations primarily consist of research and development. The Company’s wholly owned international subsidiary, SciClone Pharmaceuticals International Ltd., is engaged in sales and marketing and product distribution worldwide.
Information regarding geographic areas is as follows (in thousands):
| Revenue for the Year Ended December 31, |
Long Lived Assets December 31, |
|||||||
| 2010 |
||||||||
| U.S |
$ | — | $ | 695 | ||||
| China |
82,012 | 202 | ||||||
| Rest of world |
3,100 | 582 | ||||||
| Total |
$ | 85,112 | $ | 1,479 | ||||
| 2009 |
||||||||
| U.S |
$ | — | $ | 877 | ||||
| China |
69,696 | 217 | ||||||
| Rest of world |
2,715 | 926 | ||||||
| Total |
$ | 72,411 | $ | 2,020 | ||||
| 2008 |
||||||||
| U.S |
$ | — | $ | 1,549 | ||||
| China |
50,724 | 287 | ||||||
| Rest of world |
3,389 | 180 | ||||||
| Total |
$ | 54,113 | $ | 2,016 | ||||
78
Table of Contents
Note 18 — Selected Quarterly Financial Data (unaudited)
| Three Months Ended | ||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| (in thousands, except per share amounts) | ||||||||||||||||
| 2010 |
||||||||||||||||
| Product sales |
$ | 17,962 | $ | 20,694 | $ | 22,840 | $ | 23,616 | ||||||||
| Cost of product sales |
2,759 | 3,540 | 3,146 | 3,246 | ||||||||||||
| Gross margin |
15,203 | 17,154 | 19,694 | 20,370 | ||||||||||||
| Net income |
4,193 | 5,480 | 7,628 | 3,780 | (1) | |||||||||||
| Basic net income per share |
$ | 0.09 | $ | 0.12 | $ | 0.16 | $ | 0.08 | ||||||||
| Diluted net income per share |
$ | 0.09 | $ | 0.11 | $ | 0.16 | $ | 0.08 | ||||||||
| 2009 |
||||||||||||||||
| Product sales |
$ | 15,069 | $ | 21,971 | $ | 17,240 | $ | 18,131 | ||||||||
| Cost of product sales |
2,665 | 3,422 | 3,098 | 2,775 | ||||||||||||
| Gross margin |
12,404 | 18,549 | 14,142 | 15,356 | ||||||||||||
| Net income |
$ | 96 | $ | 7,339 | (2) | $ | 2,064 | $ | 2,446 | |||||||
| Basic and diluted net income per share |
$ | 0.00 | $ | 0.16 | $ | 0.04 | $ | 0.05 | ||||||||
| (1) | During the three months ended December 31, 2010, the Company recorded $1.5 million of income tax expense related to its foreign operations in China, including $1.3 million of additional tax expense for the fourth quarter of 2010, of which $0.8 million was to establish a reserve for the Company’s uncertain tax position in China. |
| (2) | During the three months ended June 30, 2009, product sales increased attributable to higher volume of ZADAXIN which the Company believes was primarily due to increased demand as a result of the H1N1 flu virus and further market penetration in China. |
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Securities Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed under the Exchange Act is accumulated and communicated to management, including our principal executive officer and our principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. As of the end of the period covered by this report, the Company carried out an evaluation under the supervision and with the participation of its management, including the Company’s President and Chief Executive Officer (“CEO”) and its Senior Vice President and Chief Financial Officer (“CFO”), of the effectiveness of the design and operation of the Company’s disclosure controls and procedures in ensuring that material information required to be disclosed in the Company’s reports filed or submitted under the Exchange Act, has been made known to them in a timely fashion. Based on this evaluation, the CEO and CFO concluded that our disclosure controls and procedures were not effective in timely alerting them to material information relating to us and our consolidated subsidiaries as required to be disclosed in the reports we file and submit under the Exchange Act as of December 31, 2010, due to the material weaknesses disclosed below. In particular, the Company’s policies and procedures were not sufficient to give reasonable assurance that matters involving potential violations of the FCPA or other laws were appropriately identified, reported and evaluated, nor that tax exposures and valuation allowances related to foreign uncertain tax positions were correctly calculated and recorded.
79
Table of Contents
Management’s Annual Report on Internal Control over Financial Reporting
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. The Company’s internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. The Company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. All control systems have inherent limitations so that no evaluation of controls can provide absolute assurance that all control issues are detected. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our management, we assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2010, based on the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control-Integrated Framework.
Based on that evaluation and assessment, our management concluded that our internal control over financial reporting was not effective as of December 31, 2010 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with accounting principles generally accepted in the United States of America, to the extent and for the reasons set forth below. Our management reviewed the results of its evaluation and assessment with our Audit Committee.
Our management identified two material weaknesses in internal control over financial reporting as of December 31, 2010. A material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
Implementation of Our Policy on Compliance with Laws
In connection with our special committee’s investigation, our management identified a material weakness in our internal control over financial reporting regarding the implementation of our policy on compliance with applicable laws as of December 31, 2010. Our conclusion that we have this material weakness in our internal control over financial reporting is not based on quantified misstatements in our historical financial statements or our financial statements as of and for the period ended December 31, 2010, but instead on the determination that we did not design or maintain sufficient policies, procedures, controls, communications or training to mitigate the risk of violations of law, including the FCPA.
We do not presently anticipate that the material weakness in our internal control over financial reporting as of December 31, 2010, identified in this section, will have any material effect on our prior years previously reported financial results or our financial results for our fiscal year ended December 31, 2010.
80
Table of Contents
The deficiencies contributing to this material weakness in our internal control over financial reporting as of December 31, 2010, are as follows:
Inadequate Entity-Level Controls
As of December 31, 2010, we identified a number of entity-level control deficiencies that relate both to the design and to the operating effectiveness of our internal controls and include:
| • | While our procedures for approval of expenses or expense reimbursement required documentation supporting such reimbursements, this documentation in certain instances was inadequate and the review of that supporting documentation was inadequate to ensure consistent application of policies on reimbursement; |
| • | Our lack of sufficiently detailed written guidelines regarding reimbursement to physicians for attendance at medical and scientific conferences or other events, gifts and on compliance with laws, including a detailed training program; |
| • | Our lack of an entity level pre-approval policy administered by appropriate personnel, in particular for the reimbursement of third party gifts, travel expenses, honoraria and sponsorship including expenses for attendance at medical and scientific conferences or other events; and |
| • | Our failure to adequately monitor and test our training, approval and other controls over compliance with laws. |
Remediation
During the first quarter of fiscal 2011, we undertook or initiated several measures to address the above deficiencies, including the following measures, which we believe will remediate the material weakness:
| • | Our initiation of more in-depth, Company-wide, comprehensive training of our personnel in the requirements of the FCPA and other laws, including training with respect to those areas of our operations that are most likely to raise FCPA compliance concerns; |
| • | Our adoption of a policy regarding the pre-approval of expenses for, or reimbursement of third parties for, certain travel expenses and sponsorship of attendance at medical, scientific or other events including review by our Compliance Officer or designee of these expenses; |
| • | Our determination to adopt and begin the process of implementing more detailed guidelines on gifts, reimbursement to doctors for attendance at medical and scientific or other events and on compliance with laws; |
| • | Our identification of the need for more detail and back-up documentation for expense approvals or reimbursement requests relating to various expenses including third-party gifts, travel expenses, honoraria and sponsorship of attendance at medical, scientific or other events and our decision to implement at least two additional policies regarding these matters; |
| • | Our determination to implement regular evaluation by management of the effectiveness and sufficiency of our controls, guidelines and procedures relating to our policy on compliance with laws and regulations. |
Accounting for Income Taxes
In our assessment of the effectiveness of internal control over financial reporting as of December 31, 2010, we have also identified a material weakness in the Company’s internal controls over financial reporting for income taxes. The Company’s processes, procedures and controls related to financial reporting were not effective to ensure that tax exposures were correctly calculated and recorded. Specifically, our process of identifying and
81
Table of Contents
evaluating our foreign uncertain tax positions and related reserves were not designed effectively to provide for adequate and timely identification and recognition of income tax expense in accordance with U.S. GAAP, and there was a reasonable possibility that a material misstatement would not be prevented or detected in the consolidated financial statements. This material weakness resulted in errors in the provision for income tax, current liabilities and footnotes of our condensed consolidated financial statements.
The principal deficiencies contributing to the material weakness in internal controls over financial reporting for income taxes as of December 31, 2010, are as follows:
| • | Our requirements for supporting documentation related to expenses deducted on foreign tax returns were not sufficiently clear to ensure the deductibility of all such expenses; and |
| • | We do not have a process for evaluating changes that may be impacted by the international tax environment, where such changes may result in a significant or material increase of our foreign uncertain tax positions and related reserves. |
Remediation
We plan to initiate several measures to address the above deficiencies, including the following measures, which we believe will remediate the material weakness:
| • | Revision of existing policies and procedures related to documentation of expenses to ensure that documentation is sufficiently clear to ensure that all such expenses will meet the more likely than not threshold of the foreign tax authorities; and |
| • | Establishing a process for evaluating changes that may be impacted by the international tax environment, where such changes may result in a significant or material increase of our foreign uncertain tax positions and related reserves. |
Ernst & Young LLP, an independent registered public accounting firm, has audited and issued an attestation report on the effectiveness of the Company’s internal control over financial reporting as of December 31, 2010 and, based on that audit, issued an adverse opinion, as stated in their report which is included below.
82
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of SciClone Pharmaceuticals, Inc.
We have audited SciClone Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). SciClone Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. In its assessment, management has identified material weaknesses in controls related to its process for ensuring compliance with laws and regulations and in its process for income taxes. These material weaknesses were considered in determining the nature, timing, and extent of audit tests applied in our audit of the 2010 financial statements, and this report does not affect our report dated March 31, 2011 on those financial statements.
In our opinion, because of the effect of the material weaknesses described above on the achievement of the objectives of the control criteria, Sciclone Pharmaceuticals, Inc. has not maintained effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
/s/ Ernst & Young LLP
Palo Alto, California
March 31, 2011
83
Table of Contents
Changes in Internal Controls
Except as discussed above, there has been no change in the Company’s internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Securities Act of 1934, as amended) that was identified in connection with the evaluation thereof that occurred during the fourth quarter of 2010 that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting. Please see the discussion above in “Management’s Annual Report on, and Changes in, Internal Control over Financial Reporting” with respect to remediation efforts we implemented subsequent to December 31, 2010.
Limitations of the Effectiveness of Internal Controls
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the internal control system are met. Because of inherent limitations in any control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected. We are continuously seeking to improve the efficiency and effectiveness of our operations and of our internal controls. This results in refinements to processes throughout our organization.
| Item 9B. | Other Information |
None.
PART III
Certain information required by Part III is incorporated by reference from our definitive Proxy Statement to be filed with the Securities and Exchange Commission in connection with the solicitation of proxies for our 2011 Annual Meeting of Stockholders (the “Proxy Statement”) not later than 120 days after the end of the fiscal year covered by this report, and certain information therein is incorporated in this report by reference.
| Item 10. | Directors, Executive Officers, and Corporate Governance |
The information required by Item 401 of Regulation S-K is incorporated by reference from the Proxy Statement under the caption “Proposal No. 1 Election of Directors — Nominees,” and “Proposal No. 1 Election of Directors — Board Meetings and Committees — Audit Committee.” Information relating to the executive officers of the Company is incorporated by reference from the Proxy Statement under the caption “Executive Compensation and Other Matters — Executive Officers”.
The information required by Item 405 of Regulation S-K is incorporated by reference from the Proxy Statement under the caption “Compensation Committee Report — Section 16(a) Beneficial Ownership Reporting Compliance”.
Code of Ethics
We have adopted a code of business conduct and ethics, and a policy providing for the reporting of potential violations of the code, for directors, officers (including our principal executive officer, principal financial officer, principal accounting officer or controller) and employees, known as the Corporate Code of Conduct and Ethics (including “Whistle blowing” in the case of Violations of the Company Policies) (the “Code of Conduct”). The Code of Conduct is available on our website at www.sciclone.com under the section “Investor Relations — Corporate Governance”. Additionally, stockholders may request a free copy of the Code of Conduct by contacting the Investor Relations Department at our corporate offices by calling 800-724-2566 or by sending an e-mail message to investorrelations@sciclone.com.
84
Table of Contents
| Item 11. | Executive Compensation |
The information required by this Item 11 is incorporated by reference from the Proxy Statement under the captions “Executive Compensation and Other Matters” and “Corporate Governance — Compensation of Directors.”
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Securities Authorized for Issuance under Equity Compensation Plans
The following table provides certain information regarding our compensation plans in effect as of December 31, 2010 (in thousands, except per share amounts):
| Plan Category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) |
Weighted- average exercise price of outstanding options, warrants and rights (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) |
|||||||||
| Equity compensation plans approved by security holders: |
||||||||||||
| 1995 Equity Incentive Plan(1) |
626 | $ | 5.17 | — | ||||||||
| 1995 Nonemployee Director Stock Option Plan(1) |
200 | 6.15 | — | |||||||||
| SciClone Pharmaceuticals, Inc. Employee Stock Purchase Plan |
— | — | 566 | |||||||||
| 2004 Outside Directors Stock Option Plan |
1,340 | 2.72 | 425 | |||||||||
| 2005 Equity Incentive Plan |
5,295 | 2.32 | 3,679 | |||||||||
| Equity compensation plans not approved by security holders: |
— | — | — | |||||||||
| Total |
7,461 | $ | 2.93 | 4,670 | ||||||||
| (1) | Although the 1995 Equity Incentive Plan and 1995 Nonemployee Director Stock Option Plan have expired, the outstanding stock options relating to them are fully valid. |
The information required by Item 403 of Regulation S-K is incorporated by reference from the Proxy Statement under the caption “Security Ownership of Certain Beneficial Owners and Management.”
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this Item is incorporated by reference from the Proxy Statement under the captions “Compensation Committee Report — Transactions with Related Persons” and “Corporate Governance — Director Independence.”
| Item 14. | Principal Accounting Fees and Services |
The information required by this Item 14 is incorporated by reference from the Proxy Statement under the caption “Proposal No. 2 Ratification of Appointment of Independent Registered Public Accounting Firm — Principal Accountant Fees.”
85
Table of Contents
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
Item 15 (a). The following documents are filed as part of this Annual Report on Form 10-K:
(1) Financial Statements. The following financial statements of the Company are contained in Item 8, Part II on pages 55-79 of this Annual Report on Form 10-K:
Report of Independent Registered Public Accounting Firm.
Consolidated Balance Sheets at December 31, 2010 and 2009.
Consolidated Statements of Operations for each of the three years in the period ended December 31, 2010.
Consolidated Statements of Stockholders’ Equity for the three years ended December 31, 2010.
Consolidated Statements of Cash Flows for each of the three years in the period ended December 31, 2010.
Notes to Consolidated Financial Statements.
(2) Financial Statement Schedules
The following schedule required to be filed by Item 8 of this form and Item 15(d) is contained on page 92 of this Report:
Schedule II — Valuation and Qualifying Accounts for each of the three years in the period ended December 31, 2010.
All other schedules have been omitted because they are either inapplicable or the required information has been given in the consolidated financial statements or the notes thereto.
(3) Exhibits.
Refer to Item 15(b) below.
Item 15 (b). Exhibits.
Exhibits (numbered in accordance with Item 601 of Regulation S-K):
| Exhibit Number |
Description | |
| 3(i).1(1) | Amended and Restated Certificate of Incorporation. | |
| 3(i).2(15) | Certificate of Designation, Preferences and Rights of the Terms of the Series D Preferred Stock, as filed by SciClone Pharmaceuticals, Inc. with the Secretary of State of the State of Delaware as of December 22, 2006. | |
| 3(ii).1(1) | Bylaws. | |
| 4.9(14) | Rights Agreement, effective as of December 19, 2006, between the Company and Mellon Investor Services LLC, as rights agent (including as Exhibit A the form of Certificate of Designation, Preferences and Rights of the Series D Preferred Stock, as Exhibit B the form of Right Certificate, and as Exhibit C the Summary of Terms of Rights Agreement). | |
| 10.1(2)** | Registrant’s 1995 Equity Incentive Plan, together with forms of agreement thereunder. | |
86
Table of Contents
| Exhibit Number |
Description | |
| 10.2(2)** | Registrant’s 1995 Nonemployee Director Stock Option Plan, together with forms of agreement thereunder. | |
| 10.3(13)** | Registrant’s 2005 Equity Incentive Plan, effective as of June 7, 2005, as amended on February 22, 2007. | |
| 10.4(13)** | Registrant’s 2004 Outside Directors Stock Option Plan, as amended on June 7, 2005 and as further amended on February 22, 2007. | |
| 10.5(7)** | Change in Control Agreement between the Company and Hans P. Schmid dated as of April 22, 2003. | |
| 10.6(8)** | Form of Indemnity Agreement by and between the Registrant and each director and executive officer of SciClone Pharmaceuticals, Inc. | |
| 10.7(3) | Alpha Rights Acquisition Agreement by and between the Registrant and Alpha 1 Biomedicals, Inc., dated December 17, 1997. | |
| 10.8(4)* | Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement by and between the Company and Sigma-Tau Industrie Farmadeutiche Riunite S.p.A. dated as of March 3, 2000. | |
| 10.9(6)* | Amendment No. 1 to the Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement by and between the Company and Sigma-Tau Farmadeutiche Riunite S.p.A. dated as of December 19, 2001. | |
| 10.10(10)* | Amendment No. 2 to the Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement between the Registrant and Sigma-Tau Industrie Farmacetiche Riunite S.p.A., dated May 20, 2004. | |
| 10.11(10)* | Amendment No. 3 to the Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement between the Registrant and Sigma-Tau Industrie Farmacetiche Riunite S.p.A., dated May 21, 2004. | |
| 10.12(5) | Acquisition Agreement between the Company and Sclavo S.p.A. dated April 20, 1998. | |
| 10.13(5) | First Amendment to Acquisition Agreement between the Company and Sclavo S.p.A., dated April 20, 1998. | |
| 10.14(9)* | Manufacturing and Supply Agreement between SciClone Pharmaceuticals International Ltd. and Patheon Italia S.p.A. dated as of November 1, 2002. | |
| 10.15(11)** | Amendment to Offer Letter dated as of February 24, 2006 by and between SciClone Pharmaceuticals International, Ltd. and Hans P. Schmid including offer letter dated May 21, 2001 between SciClone Pharmaceuticals International, Ltd. and Hans P. Schmid as Exhibit A thereto. | |
| 10.16(12)** | Employment Agreement between SciClone Pharmaceuticals, Inc. and Friedhelm Blobel, Ph.D. dated as of April 23, 2006 and effective as of June 2, 2006. | |
| 10.17(12)** | Change in Control Agreement between SciClone Pharmaceuticals, Inc. and Friedhelm Blobel, Ph.D. dated as of April 23, 2006 and effective as of June 2, 2006. | |
| 10.18(16)* | Manufacturing Supply Agreement Between SciClone Pharmaceuticals International Ltd. and Lonza Sales Ltd., executed as of May 23, 2008. | |
| 10.19(17)** | Employment Agreement between SciClone Pharmaceuticals, Inc. and Gary Titus, dated as of November 21, 2008 and effective as of December 8, 2008. | |
87
Table of Contents
| Exhibit Number |
Description | |
| 10.20(18) | Settlement Agreement between Sigma-Tau Finanziaria, S.p.A. and SciClone Pharmaceuticals, Inc. dated March 30, 2009. | |
| 10.21(18) | Letter Agreement related to Thymosin Alpha 1 between SciClone Pharmaceuticals, Inc. and Sigma-Tau Industrie Farmaceutiche Riunite S.p.A., dated March 30, 2009. | |
| 10.22(19)** | Amendment No. 1 to Change of Control Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. dated April 7, 2009. | |
| 10.23(19)** | Amendment No. 1 to Employment Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. dated April 7, 2009. | |
| 10.24(20)** | Change of Control Agreement between Mr. Gary Titus and SciClone Pharmaceuticals, Inc. dated December 8, 2008. | |
| 10.25(23)** | Description of Executive Incentive Plan. | |
| 10.26(22) | Loan and Security Agreement by and among SciClone Pharmaceuticals International Ltd., Sciclone Pharmaceuticals International China Holding Ltd., and Silicon Valley Bank, dated as of October 1, 2010. | |
| 10.27(21)** | The SciClone Pharmaceuticals, Inc. Employee Stock Purchase Plan, as amended. | |
| 10.28(24) | Assignment and Purchase of Intellectual Property Rights Agreement by and among Edward T. Wei, Cragmont Pharmaceuticals, LLC, and certain Russian Inventors and Russian Companies and SciClone Pharmaceuticals, Inc., dated as of December 28, 2010. | |
| 10.29(24)* | Amendment No. 1 to Manufacturing and Supply Agreement between SciClone Pharmaceuticals International Ltd. and Lonza Sales Ltd., effective December 31, 2010. | |
| 10.30(24)** | Executive Severance Agreement between Gary Titus and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |
| 10.31(24)** | Executive Severance Agreement between Hans P. Schmid and SciClone Pharmaceuticals, Inc. effective June 14, 2010. | |
| 10.32(24)** | Executive Severance Agreement between Israel Rios, M.D. and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |
| 10.33(24)** | Amendment No. 2 to Change of Control Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |
| 10.34(24)** | Amendment No. 2 to Employment Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |
| 10.35(24)** | Amendment No. 1 to Change of Control Agreement between Gary S. Titus and SciClone Pharmaceuticals, Inc. effective July 9, 2010. | |
| 10.36(24)** | Amendment No. 1 to Change of Control Agreement between Hans P. Schmid and SciClone Pharmaceuticals, Inc. effective July 9, 2010. | |
| 10.37(24)** | Amendment No. 1 to Change of Control Agreement between Israel Rios, M.D. and SciClone Pharmaceuticals, Inc. effective July 9, 2010. | |
| 14 | See “Code of Ethics” in Item 10: Executive Officers and Directors, of this Annual Report on Form 10-K. | |
| 21.1(24) | Subsidiaries of Registrant. | |
| 23.1(24) | Consent of Independent Registered Public Accounting Firm. | |
88
Table of Contents
| Exhibit Number |
Description | |
| 24.1(24) | Power of Attorney. See page 91. | |
| 31.1(24) | Rule 13a-14(a) Certification of Principal Executive Officer. | |
| 31.2(24) | Rule 13a-14(a) Certification of Principal Financial Officer. | |
| 32.1(24) | Section 1350 Certification of Principal Executive Officer. | |
| 32.2(24) | Section 1350 Certification of Principal Financial Officer. | |
| * | Certain information in this exhibit has been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request under 17 C.F.R. Sections 200.80(b)(4), 200.83 and 230.46. |
| ** | Management compensatory plan or arrangement. |
| (1) | Incorporated by reference from the Company’s Current Report on 8-K filed on July 28, 2003. |
| (2) | Incorporated by reference from the Company’s Registration Statement on Form S-8 (No. 33-80911) filed with the Commission on December 28, 1995. |
| (3) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on January 26, 1998. |
| (4) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 20, 2000. |
| (5) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 1998. |
| (6) | Incorporated by reference from the Company’s Annual Report on Form 10-K for the year ended December 31, 2002. |
| (7) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2003. |
| (8) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2003. |
| (9) | Incorporated by reference from the Company’s Annual Report on Form 10-K for the year ended December 31, 2003. |
| (10) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2004. |
| (11) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on February 28, 2006. |
| (12) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 25, 2006. |
| (13) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2007 filed on August 8, 2007. |
| (14) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on December 22, 2006. |
| (15) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on December 28, 2006. |
| (16) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2008 filed on August 1, 2008. |
| (17) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on December 4, 2008. |
| (18) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 1, 2009. |
| (19) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 8, 2009. |
89
Table of Contents
| (20) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q filed on May 11, 2009. |
| (21) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q filed on August 9, 2010. |
| (22) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q filed on November 8, 2010. |
| (23) | Incorporated by reference from the Company’s Annual Report on Form 10-K filed on March 16, 2010. |
| (24) | Filed herewith. |
Item 15 (c). See Item 15(a) above.
90
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| SCICLONE PHARMACEUTICALS, INC. | ||
| By: | /S/ FRIEDHELM BLOBEL, PH.D. | |
| Friedhelm Blobel, Ph.D. President and Chief Executive Officer (Principal Executive Officer) | ||
Date: March 31, 2011
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Friedhelm Blobel, Ph.D. and Gary Titus, and each of them, his attorneys-in-fact and agents, each with the power of substitution and resubstitution, for him in any and all capacities, to sign this Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting to said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary, to be done in connection therewith, as fully as to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either of them, or their or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ FRIEDHELM BLOBEL, PH.D. (Friedhelm Blobel, Ph.D.) |
President and Chief Executive Officer, Director (Principal Executive Officer) |
March 31, 2011 | ||
| /S/ GARY S. TITUS (Gary S. Titus) |
Senior Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) |
March 31, 2011 | ||
| /S/ ROBERTO CAMERINI, M.D. (Roberto Camerini, M.D.) |
Director |
March 31, 2011 | ||
| /S/ RICHARD J. HAWKINS (Richard J. Hawkins) |
Director |
March 31, 2011 | ||
| /S/ TREVOR M. JONES, PH.D. (Trevor M. Jones, Ph.D.) |
Director |
March 31, 2011 | ||
| /S/ IRA D. LAWRENCE, M.D. (Ira D. Lawrence. M.D.) |
Director |
March 31, 2011 | ||
| /S/ GREGG A. LAPOINTE (Gregg A. Lapointe) |
Director |
March 31, 2011 | ||
| /S/ JON S. SAXE (Jon S. Saxe) |
Chairman of the Board of Directors |
March 31, 2011 | ||
| /S/ DEAN S. WOODMAN (Dean S. Woodman) |
Director |
March 31, 2011 | ||
91
Table of Contents
SCHEDULE II — VALUATION AND QUALIFYING ACCOUNTS
SCICLONE PHARMACEUTICALS, INC.
Accounts Receivable Allowances for Uncollectible Accounts (in thousands):
| Balance at Beginning of Period |
Charged to Costs and Expenses |
Deductions for Amounts Recovered |
Deductions for Amounts Written Off |
Balance at End of Period |
||||||||||||||||
| Year Ended December 31, 2010 |
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Year Ended December 31, 2009 |
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Year Ended December 31, 2008 |
$ | 15 | $ | — | $ | (15 | ) | $ | — | $ | — | |||||||||
92
Table of Contents
INDEX TO EXHIBITS
| Exhibit Number |
Description | |||
| 3(i).1(1) | Amended and Restated Certificate of Incorporation. | |||
| 3(i).2(15) | Certificate of Designation, Preferences and Rights of the Terms of the Series D Preferred Stock, as filed by SciClone Pharmaceuticals, Inc. with the Secretary of State of the State of Delaware as of December 22, 2006. | |||
| 3(ii).1(1) | Bylaws. | |||
| 4.9(14) | Rights Agreement, effective as of December 19, 2006, between the Company and Mellon Investor Services LLC, as rights agent (including as Exhibit A the form of Certificate of Designation, Preferences and Rights of the Series D Preferred Stock, as Exhibit B the form of Right Certificate, and as Exhibit C the Summary of Terms of Rights Agreement). | |||
| 10.1(2)** | Registrant’s 1995 Equity Incentive Plan, together with forms of agreement thereunder. | |||
| 10.2(2)** | Registrant’s 1995 Nonemployee Director Stock Option Plan, together with forms of agreement thereunder. | |||
| 10.3(13)** | Registrant’s 2005 Equity Incentive Plan, effective as of June 7, 2005, as amended on February 22, 2007. | |||
| 10.4(13)** | Registrant’s 2004 Outside Directors Stock Option Plan, as amended on June 7, 2005 and as further amended on February 22, 2007. | |||
| 10.5(7)** | Change in Control Agreement between the Company and Hans P. Schmid dated as of April 22, 2003. | |||
| 10.6(8) ** | Form of Indemnity Agreement by and between the Registrant and each director and executive officer of SciClone Pharmaceuticals, Inc. | |||
| 10.7(3) | Alpha Rights Acquisition Agreement by and between the Registrant and Alpha 1 Biomedicals, Inc., dated December 17, 1997. | |||
| 10.8(4)* | Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement by and between the Company and Sigma-Tau Industrie Farmadeutiche Riunite S.p.A. dated as of March 3, 2000. | |||
| 10.9(6)* | Amendment No. 1 to the Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement by and between the Company and Sigma-Tau Farmadeutiche Riunite S.p.A. dated as of December 19, 2001. | |||
| 10.10(10)* | Amendment No. 2 to the Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement between the Registrant and Sigma-Tau Industrie Farmacetiche Riunite S.p.A., dated May 20, 2004. | |||
| 10.11(10)* | Amendment No. 3 to the Expanded and Amended Thymosin Alpha 1 License, Distributorship and Supply Agreement between the Registrant and Sigma-Tau Industrie Farmacetiche Riunite S.p.A., dated May 21, 2004. | |||
| 10.12(5) | Acquisition Agreement between the Company and Sclavo S.p.A. dated April 20, 1998. | |||
| 10.13(5) | First Amendment to Acquisition Agreement between the Company and Sclavo S.p.A., dated April 20, 1998. | |||
| 10.14(9)* | Manufacturing and Supply Agreement between SciClone Pharmaceuticals International Ltd. and Patheon Italia S.p.A. dated as of November 1, 2002. | |||
93
Table of Contents
| Exhibit Number |
Description | |||
| 10.15(11)** | Amendment to Offer Letter dated as of February 24, 2006 by and between SciClone Pharmaceuticals International, Ltd. and Hans P. Schmid including offer letter dated May 21, 2001 between SciClone Pharmaceuticals International, Ltd. and Hans P. Schmid as Exhibit A thereto. | |||
| 10.16(12)** | Employment Agreement between SciClone Pharmaceuticals, Inc. and Friedhelm Blobel, Ph.D. dated as of April 23, 2006 and effective as of June 2, 2006. | |||
| 10.17(12)** | Change in Control Agreement between SciClone Pharmaceuticals, Inc. and Friedhelm Blobel, Ph.D. dated as of April 23, 2006 and effective as of June 2, 2006. | |||
| 10.18(16)* | Manufacturing Supply Agreement Between SciClone Pharmaceuticals International Ltd. and Lonza Sales Ltd., executed as of May 23, 2008. | |||
| 10.19(17)** | Employment Agreement between SciClone Pharmaceuticals, Inc. and Gary Titus, dated as of November 21, 2008 and effective as of December 8, 2008. | |||
| 10.20(18) | Settlement Agreement between Sigma-Tau Finanziaria, S.p.A. and SciClone Pharmaceuticals, Inc. dated March 30, 2009. | |||
| 10.21(18) | Letter Agreement related to Thymosin Alpha 1 between SciClone Pharmaceuticals, Inc. and Sigma-Tau Industrie Farmaceutiche Riunite S.p.A., dated March 30, 2009. | |||
| 10.22(19)** | Amendment No. 1 to Change of Control Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. dated April 7, 2009. | |||
| 10.23(19)** | Amendment No. 1 to Employment Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. dated April 7, 2009. | |||
| 10.24(20)** | Change in Control Agreement between Mr. Gary Titus and SciClone Pharmaceuticals, Inc. dated December 8, 2008. | |||
| 10.25(23)** | Description of Executive Incentive Plan. | |||
| 10.26(22) | Loan and Security Agreement by and among SciClone Pharmaceuticals International Ltd., Sciclone Pharmaceuticals International China Holding Ltd., and Silicon Valley Bank, dated as of October 1, 2010. | |||
| 10.27(21)** | The SciClone Pharmaceuticals, Inc. Employee Stock Purchase Plan, as amended. | |||
| 10.28(24) | Assignment and Purchase of Intellectual Property Rights Agreement by and among Edward T. Wei, Cragmont Pharmaceuticals, LLC, and certain Russian Inventors and Russian Companies and SciClone Pharmaceuticals, Inc., dated as of December 28, 2010. | |||
| 10.29(24)* | Amendment No. 1 to Manufacturing and Supply Agreement between SciClone Pharmaceuticals International Ltd. and Lonza Sales Ltd., effective December 31, 2010. | |||
| 10.30(24)** | Executive Severance Agreement between Gary Titus and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |||
| 10.31(24)** | Executive Severance Agreement between Hans P. Schmid and SciClone Pharmaceuticals, Inc. effective June 14, 2010. | |||
| 10.32(24)** | Executive Severance Agreement between Israel Rios, M.D. and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |||
| 10.33(24)** | Amendment No. 2 to Change of Control Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |||
94
Table of Contents
| Exhibit Number |
Description | |
| 10.34(24)** | Amendment No. 2 to Employment Agreement between Dr. Friedhelm Blobel and SciClone Pharmaceuticals, Inc. effective May 4, 2010. | |
| 10.35(24)** | Amendment No. 1 to Change of Control Agreement between Gary S. Titus and SciClone Pharmaceuticals, Inc. effective July 9, 2010. | |
| 10.36(24)** | Amendment No. 1 to Change of Control Agreement between Hans P. Schmid and SciClone Pharmaceuticals, Inc. effective July 9, 2010. | |
| 10.37(24)** | Amendment No. 1 to Change of Control Agreement between Israel Rios, M.D. and SciClone Pharmaceuticals, Inc. effective July 9, 2010. | |
| 14 | See “Code of Ethics” in Item 10: Executive Officers and Directors, of this Annual Report on Form 10-K. | |
| 21.1(24) | Subsidiaries of Registrant. | |
| 23.1(24) | Consent of Independent Registered Public Accounting Firm. | |
| 24.1(24) | Power of Attorney. See page 91. | |
| 31.1(24) | Rule 13a-14(a) Certification of Principal Executive Officer. | |
| 31.2(24) | Rule 13a-14(a) Certification of Principal Financial Officer. | |
| 32.1(24) | Section 1350 Certification of Principal Executive Officer. | |
| 32.2(24) | Section 1350 Certification of Principal Financial Officer. | |
| * | Certain information in this exhibit has been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request under 17 C.F.R. Sections 200.80(b)(4), 200.83 and 230.46. |
| ** | Management compensatory plan or arrangement. |
| (1) | Incorporated by reference from the Company’s Current Report on 8-K filed on July 28, 2003. |
| (2) | Incorporated by reference from the Company’s Registration Statement on Form S-8 (No. 33-80911) filed with the Commission on December 28, 1995. |
| (3) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on January 26, 1998. |
| (4) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 20, 2000. |
| (5) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 1998. |
| (6) | Incorporated by reference from the Company’s Annual Report on Form 10-K for the year ended December 31, 2002. |
| (7) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2003. |
| (8) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2003. |
| (9) | Incorporated by reference from the Company’s Annual Report on Form 10-K for the year ended December 31, 2003. |
| (10) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2004. |
| (11) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on February 28, 2006. |
| (12) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 25, 2006. |
| (13) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2007 filed on August 8, 2007. |
| (14) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on December 22, 2006. |
95
Table of Contents
| (15) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on December 28, 2006. |
| (16) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2008 filed on August 1, 2008. |
| (17) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on December 4, 2008. |
| (18) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 1, 2009. |
| (19) | Incorporated by reference from the Company’s Current Report on Form 8-K filed on April 8, 2009. |
| (20) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q filed on May 11, 2009. |
| (21) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q filed on August 9, 2010. |
| (22) | Incorporated by reference from the Company’s Quarterly Report on Form 10-Q filed on November 8, 2010. |
| (23) | Incorporated by reference from the Company’s Annual Report on Form 10-K filed on March 16, 2010. |
| (24) | Filed herewith. |
96