SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
Annual Report Pursuant to Section 13 or 15(d) of
the Securities Exchange Act of 1934
For the Fiscal Year Ended December 31, 2010
Commission File No. 000-51290
EpiCept Corporation
(Exact name of registrant as specified in its charter)
| |
|
|
|
| Delaware
|
|
52-1841431 |
| (State or other jurisdiction of
|
|
(IRS Employer Identification No.) |
| incorporation or organization) |
|
|
777 Old Saw Mill River Road
Tarrytown, NY 10591
(Address of principal executive offices) (zip code)
Registrant’s telephone number, including area code: (914) 606-3500
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, par value $.0001 per share
(Title of Class)
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark whether the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes o No þ.
Indicate by check mark whether the registrant is not required to file reports pursuant to
Section 13 or 15(d) of the Exchange Act. Yes o No þ.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed
by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. Yes þ No o.
Indicate by check mark whether the registrant has submitted electronically and posted on its
corporate website, if any, every Interactive Data File required to be submitted and posted pursuant
to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the
registrant was required to submit and post such files).
Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation
S-K is not contained herein and will not be contained, to the best of registrant’s knowledge, in
definitive proxy or information statements incorporated by reference in Part III of this Form 10-K
or any amendments to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated
filer, a non-accelerated filer or a smaller reporting company. See definitions of “large
accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the
Exchange Act.
| |
|
|
|
|
|
|
|
| Large accelerated filer o
|
|
Accelerated filer o
|
|
Non-accelerated filer o
|
|
Smaller reporting company þ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Exchange Act). Yes o No þ.
As of June 30, 2010, the last business day of the registrant’s most recently completed second
fiscal quarter, the aggregate market value of shares of common stock held by non-affiliates was
$43,244,458.
As of March 30, 2011, the registrant had 70,989,292 shares of its common stock, par value
$.0001 per share, outstanding.
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements that are subject to risks and
uncertainties. All statements other than statements of historical fact included in this Form 10-K
are forward-looking statements. Forward-looking statements give our current expectations and
projections relating to our financial condition, results of operations, plans, objectives, future
performance and business. You can identify forward-looking statements by the fact that they do not
relate strictly to historical or current facts. These statements may include words such as
“anticipate,” “estimate,” “expect,” “project,” “plan,” “intend,” “believe,” “may,” “should,” “can
have,” “likely” and other words and terms of similar meaning in connection with any discussion of
the timing or nature of future operating or financial performance or other events.
These forward-looking statements are based on assumptions that we have made in light of our
industry experience and on our perceptions of historical trends, current conditions, expected
future developments and other factors we believe are appropriate under the circumstances. As you
read and consider this Form 10-K, you should understand that these statements are not guarantees of
performance or results. They involve risks, uncertainties (some of which are beyond our control)
and assumptions. Although we believe that these forward-looking statements are based on reasonable
assumptions, you should be aware that many factors could affect our actual financial results and
cause them to differ materially from those anticipated in the forward-looking statements. These
factors include, among others:
| |
• |
|
the risk that our securities may be delisted from Nasdaq; |
| |
| |
• |
|
the risk that Ceplene® will not receive regulatory approval or marketing
authorization in the United States or Canada; |
| |
| |
• |
|
the risk that Ceplene® will not achieve significant commercial success; |
| |
| |
• |
|
the risk that Ceplene® will not be reimbursed or receive an acceptable price
for reimbursement in certain countries; |
| |
| |
• |
|
the risk that any required post-approval clinical studies for Ceplene® will
not be successful; |
| |
| |
• |
|
the risk that we will not be able to maintain our final regulatory approval or marketing
authorization for Ceplene®; |
| |
| |
• |
|
the risks associated with the adequacy of our existing cash resources and our ability to
continue as a going concern; |
| |
| |
• |
|
the risks associated with our ability to continue to meet our obligations under our
existing debt agreements; |
| |
| |
• |
|
the risk that Myrexis’s development of Azixa™ will not be successful; |
| |
| |
• |
|
the risk that Azixa™ will not receive regulatory approval or achieve significant
commercial success; |
| |
| |
• |
|
the risk that we will not receive any significant payments under our agreement with
Myrexis; |
| |
| |
• |
|
the risk that the development of our other apoptosis product candidates will not be
successful; |
| |
| |
• |
|
the risk that clinical trials for EpiCeptTM NP-1 or crolibulinTM
will not be successful; |
| |
| |
• |
|
the risk that EpiCeptTM NP-1 or crolibulinTM will not receive
regulatory approval or achieve significant commercial success; |
| |
| |
• |
|
the risk that we will not be able to find a partner to help conduct the Phase III trials
for EpiCeptTM NP-1 on attractive terms, a timely basis or at all; |
| |
| |
• |
|
the risk that our other product candidates that appeared promising in early research and
clinical trials do not demonstrate safety and/or efficacy in larger-scale or later stage
clinical trials; |
| |
| |
• |
|
the risk that we will not obtain approval to market any of our product candidates; |
| |
| |
• |
|
the risks associated with dependence upon key personnel; |
| |
| |
• |
|
the risks associated with reliance on collaborative partners and others for further
clinical trials, development, manufacturing and commercialization of our product candidates; |
| |
| |
• |
|
the cost, delays and uncertainties associated with our scientific research, product
development, clinical trials and regulatory approval process; |
| |
| |
• |
|
our history of operating losses since our inception; |
| |
| |
• |
|
the highly competitive nature of our business; |
| |
| |
• |
|
risks associated with litigation; |
| |
| |
• |
|
risks associated with our ability to protect our intellectual property; and |
| |
| |
• |
|
the other factors described under “Risk Factors” and “Management’s Discussion and
Analysis of Financial Condition and Results of Operations.” |
There may be other factors that may cause our actual results to differ materially from the
forward-looking statements. Because of these factors, we caution that you should not place undue
reliance on any of our forward-looking statements. Further, any forward-looking statement speaks
only as of the date on which it is made. New risks and uncertainties arise from time to time, and
it is impossible for us to predict those events or how they may affect us. Except as required by
law, we have no duty to, and do not intend to, update or revise the forward-looking statements in
this Form 10-K after the date of this Form 10-K. This Form 10-K also contains market data related
to our business and industry. This market data includes projections that are based on a number of
assumptions. If these assumptions turn out to be incorrect, actual results may differ from the
projections based on these assumptions. As a result, our markets may not grow at the rates
projected by these data, or at all. The failure of these markets to grow at these projected rates
may have a material adverse effect on our business, financial condition, results of operations and
the
market price of our common stock. We do not undertake to discuss matters relating to our ongoing
clinical trials or our regulatory strategies beyond those which have already been made public or
discussed herein. As used herein, references to “we,” “us,” “our,” “EpiCept” or the “Company” refer
to EpiCept Corporation and its subsidiaries. References in this Form 10-K to the “FDA” means the
U.S. Food and Drug Administration.
3
We are a specialty pharmaceutical company focused on the clinical development and
commercialization of pharmaceutical products for the treatment of cancer and pain. We focus our
clinical development efforts on innovative cancer therapies and topically delivered analgesics
targeting peripheral nerve receptors. Our lead product is Ceplene®, which when used
concomitantly with low-dose interleukin-2, or IL-2, is intended as remission maintenance therapy in
the treatment of acute myeloid leukemia, or AML, for adult patients who are in their first complete
remission. Ceplene® is approved in the Europe Union, or EU, and Israel.
In addition to Ceplene®, we have two other oncology compounds including
crolibulinTM, a novel small molecule vascular disruption agent, or VDA, and apoptosis
inducer for the treatment of patients with solid tumors and AzixaTM, an apoptosis
inducer with VDA activity that is currently in Phase II clinical trials in patients with primary
glioblastoma and cancer that has metastasized to the brain. We also have a late-stage pain product
candidate, EpiCeptTM NP-1 cream, which we refer to as NP-1, for the treatment of
peripheral neuropathies. We recently concluded a Phase IIb clinical trial of NP-1 in which we
studied its safety and efficacy in patients suffering from chemotherapy-induced neuropathy, or CPN,
compared with placebo. We believe this portfolio of oncology and pain management product
candidates lessens our reliance on the success of any single product or product candidate.
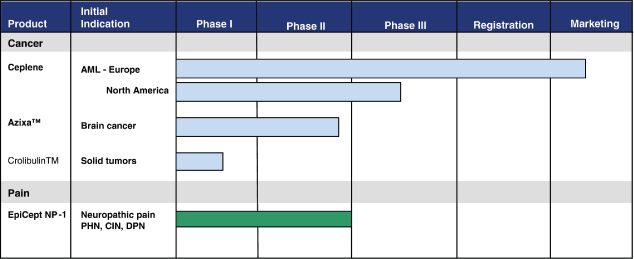
Product Portfolio
The following chart illustrates the depth of our product pipeline at December 31, 2010:
Cancer
Cancer is the second leading cause of death in the United States. Half of all men and one
third of all women in the United States will develop cancer during their lifetimes. Today, millions
of people are living with cancer or have had cancer. Although there are many kinds of cancer, they
are all caused by the out-of-control growth of abnormal cells. Normal body cells grow, divide, and
die in an orderly fashion. During the early years of a person’s life, normal cells divide more
rapidly until the person becomes an adult. After that, cells in most parts of the body divide only
to replace worn-out or dying cells and to repair injuries. Because cancer cells continue to grow
and divide, they are different from normal cells. Instead of dying, they outlive normal cells and
continue to form new abnormal cells.
Cancer usually forms as a tumor. However, some cancers, like leukemia, do not form tumors.
Instead, these cancer cells involve the blood and blood-forming organs and circulate through other
tissues where they grow. Often, cancer cells travel to other parts of the body where they begin to
grow and replace normal tissue. Different types of cancer can behave very differently. For example,
lung cancer and breast cancer are very different diseases. They grow at different rates and respond
to different treatments. That is
why people with cancer need treatment that is aimed at their particular kind of cancer. The
risk of developing most types of cancer can be reduced by changes in a person’s lifestyle, for
example, by quitting smoking and following a better diet. The sooner a cancer is found and
treatment begins, the better are the chances for long term survival.
4
Ceplene®
AML is the most deadly and most common type of acute leukemia in adults. There are
approximately 40,000 AML patients in the EU, with 16,000 new cases occurring each year.
Additionally, there are approximately 13,000 new cases of AML and 9,000 deaths caused by this
cancer each year in the United States. Once diagnosed with AML, patients typically receive
induction and consolidation chemotherapy, with the majority achieving complete remission. However,
about 70-80% of patients who achieve first complete remission will relapse, with the median time in
remission before relapse with current treatments being only 12 months. Less than 15% of relapsed
patients survive long-term.
Ceplene® (histamine dihydrochloride) is our proprietary product for the remission
maintenance and prevention of relapse in adult patients with AML in first remission. Ceplene®
is to be administered in conjunction with low-dose IL-2 and is designed to protect
lymphocytes responsible for immune-mediated destruction of residual leukemic cells.
Ceplene®reduces the formation of oxygen radicals from phagocytes, inhibiting
nicotinamide adenine dinucleotide phosphate-oxidase, or NADPH oxidase, and protecting
IL-2-activated Natural Killer cells, or NK-cells, and Thymus cells, or T-cells. These two kinds of
cells, NK-cells and T-cells, possess an ability to kill and support the killing of cancer cells and
virally infected cells.
In October 2008, we received a full marketing authorization from the European Commission, or
EU, for Ceplene®. The approval allows Ceplene® to be marketed in the 27
member states of the EU, as well as in Iceland, Liechtenstein and Norway. The approval by the
European Commission is based, in part, on the results of the single pivotal 320-patient Phase III
trial for Ceplene® in conjunction with low dose IL-2. The primary result of this trial
was that treatment with Ceplene/IL-2 significantly reduced the occurrence of relapse among AML
patients in complete remission. The improvement of long-term leukemia-free survival in patients
receiving Ceplene/IL-2 exceeded 50%. Moreover, Ceplene® was well tolerated in this
patient population and conferred an acceptable risk benefit profile for AML patients.
Ceplene is an Orphan Medicinal Product in the EU for the treatment of AML. As a result of its
designation, we have been granted market exclusivity in the EU until October 2018. As part of
receiving marketing authorization under Exceptional Circumstances for Ceplene®, we are
required to perform two post-approval clinical studies that have now been combined into a single
clinical study. The first part of the study will seek to further elucidate the clinical
pharmacology of Ceplene® by assessing certain immune biomarkers in AML patients in first
remission. The second part of the study will assess the effect of Ceplene/IL-2 on the development
of minimal residual disease in the same patient population.
In January 2010, we entered into an exclusive commercialization agreement for
Ceplene® with Meda AB, a leading international specialty pharmaceutical company based in
Stockholm, Sweden. Under the terms of the agreement, we granted Meda the right to market
Ceplene® in Europe and several other countries including Japan, China, and Australia.
We received a $3 million fee on signing and received an additional $2 million upon the first
commercial launch of Ceplene in a major European market. The commercial launch of
Ceplene® commenced in the United Kingdom in April 2010 and launches continue to be
initiated in other European countries. We will also receive other milestone payments and an
escalating percentage royalty on net sales in the covered territories. We are responsible for
Ceplene’s commercial supply.
In November 2009, Health Canada screened and accepted for review a New Drug Submission (“NDS”)
for Ceplene® for remission maintenance treatment of AML in Canada. During the third
quarter of 2010, our appeal of a decision by Health Canada not to provide data protection for
Ceplene® was denied by a federal court in Canada. Lack of data protection in Canada for an
innovative drug such as Ceplene® eliminates any right to sales exclusivity and enables competition
to seek approval to sell generic equivalents immediately. In November 2010, we appealed the
court’s decision and withdrew our New Drug Submission until the appeals process is completed. We
retain the right to re-file the application at any time over the next five years without prejudice.
In June 2010, we filed a New Drug Application, or NDA, with the United States Food and Drug
Administration, or FDA for Ceplene® in the same indication. In August 2010, we received
a refusal to file letter from the FDA on the NDA. In the FDA’s preliminary review of the NDA, the
FDA concluded that the application did not establish Ceplene®’s therapeutic contribution in its
combination with IL-2, and recommended that an additional pivotal trial assessing Ceplene®’s
contribution and using overall survival as a primary endpoint be conducted. In October 2010, we
reached an agreement with the FDA on a two-arm, randomized, open-label trial that will compare the
efficacy of Ceplene® plus low-dose IL-2 to standard of care in this indication. Based on FDA
guidance, the primary endpoint of the trial will be overall patient survival. Our previous Phase
III trial demonstrated a statistically significant prolongation of the primary endpoint of
leukemia-free survival and extended overall survival by more than one year in
patients in their first complete remission. As a next step, we will submit to the FDA a
detailed Phase III protocol. The FDA, via the Special Protocol Assessment (SPA) procedure, will
provide guidance on specific sections of the protocol including the adaptive design with
prospectively defined rules for sample size selection for both futility and expected success. We
also recently filed an application with the FDA to grant Ceplene® fast track status. In addition to
other benefits, if granted, this should permit an expedited review of the Ceplene® NDA once it is
filed.
5
In December 2010, Ceplene® was approved for marketing in Israel. Megapharm Ltd. is
EpiCept’s partner in Israel and upon Ministry of Health approval of labeling and other technical
matters the company is expected to launch Ceplene® in the first half of 2011.
CrolibulinTM
CrolibulinTM is a novel small molecule vascular disruption agent, or VDA, and
apoptosis inducer for the treatment of patients with solid tumors. CrolibulinTM has
shown promising vascular targeting activity with potent anti-tumor activity in pre-clinical in
vitro and in vivo studies. The molecule has been shown to induce tumor cell apoptosis and
selectively inhibit growth of proliferating cell lines, including multi-drug resistant cell lines.
Murine models of human tumor xenografts demonstrated crolibulinTM inhibits growth of
established tumors of a number of different cancer types. In preclinical tumored animal models,
combination therapy has demonstrated synergistic activity.
In November 2004, two publications appeared in Molecular Cancer Therapeutics discussing
crolibulinTM, a journal of the American Association of Cancer Research ( “Discovery and
mechanism of action of a novel series of apoptosis inducers with potential vascular targeting
activity”, Kasibhatla, S., Gourdeau, H., Meerovitch, K., Drewe, J., Reddy, S., Qiu, L., Zhang, H.,
Bergeron, F., Bouffard, D., Yang, Q., Herich, J., Lamothe, S., Cai, S. X., Tseng, B., Mol. Cancer
Ther. 2004 vol. 3 pp. 1365-1374; and “Antivascular and antitumor evaluation of
2-amino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4H-chromenes, a novel series of anticancer
agents”, Henriette Gourdeau, Lorraine Leblond, Bettina Hamelin, Clemence Desputeau, Kelly Dong,
Irenej Kianicka, Dominique Custeau, Chantal Boudreau, Lilianne Geerts, Sui-Xiong Cai, John Drewe,
Denis Labrecque, Shailaja Kasibhatla, and Ben Tseng, Mol. Cancer Ther. 2004 vol. 3 pp.1375-1384).
The manuscripts characterize crolibulinTM as a potent caspase activator demonstrating
vascular targeting activity and potent antitumor activity in pre-clinical in vitro and in vivo
studies. CrolibulinTM appeared highly effective in mouse tumor models, producing tumor
necrosis at doses that correspond to only 25% of the maximum tolerated dose, or MTD. Moreover, in
combination treatment, crolibulinTM significantly enhanced the antitumor activity of
cisplatin/taxane, resulting in tumor-free animals.
In October 2007, we completed a Phase I clinical trial for crolibulinTM. We
successfully identified the maximum tolerated dose, or MTD, of crolibulinTM in the Phase
I study. The MTD was below the dose which produced the expected toxicity based on preclinical
studies at higher doses. CrolibulinTM was administered as a single agent in increasing
doses to small cohorts of patients with advanced solid tumors. A total of seventeen patients were
enrolled in the study. The drug was tested in a variety of cancer types including melanoma,
prostate, lung, breast, colon, and pancreatic cancers. The study, which was initiated in December
2006, was conducted at three cancer centers in the United States. In addition to determining
crolibulin’s MTD, the primary objective of the study was to determine the pharmacokinetic profile
of the drug. In 2008, we enrolled an additional sixteen patients into the study, lengthening the
infusion period of the drug in order to increase the MTD. The studies helped characterize the
pharmacodynamic effects on tumor blood flow and identified early signs of objective anti-tumor
response as measured by computed axial tomography, or CT scans, magnetic resonance imaging, or MRI,
or positron emission tomography, or PET scan, in advanced cancer patients with well vascularized
solid tumors.
In December 2010, the National Cancer Institute, or NCI, initiated a Phase II trial with
crolibulinTM. The trial will assess the drug’s efficacy and safety in combination with
cisplatin in patients with anaplastic thyroid cancer, or ATC. The Phase II trial consists of two
stages. The primary objective of the first stage is to assess the safety and tolerability of
cisplatin and crolibulinTM given in 21-day treatment cycles. The study will assess the
toxicities of crolibulinTM co-administered with cisplatin, evaluate dose-limiting
toxicities, or DLTs, and determine the MTD for the combination. The primary objective in the second
stage of the trial will be to compare the combination of crolibulinTM and cisplatin
against cisplatin alone in adult ATC patients by assessing the duration of progression-free
survival, or PFS. An important secondary objective is the comparison of the response rates
evaluated by Response Evaluation Criteria in Solid Tumors, or RECIST. Up to 70 patients are planned
to be enrolled in the trial.
6
Azixaä
AzixaTM is a compound discovered internally and licensed to Myrexis Inc. for
clinical development. AzixaTM demonstrated a broad range of anti-tumor activities
against many tumor types in various animal models as well as activity against different types of
multi-drug resistant cell lines. The Phase I clinical testing was conducted by Myrexis on patients
with solid tumors with a particular
focus on brain cancers or brain metastases due to the pharmacologic properties of
AzixaTM in pre-clinical animal studies that indicated higher drug levels in the brain
than in the blood. In March 2007, Myrexis initiated two Phase II clinical trials for
AzixaTM in patients with primary brain cancer and in patients with melanoma that has
spread to the brain. The trials are designed to assess the safety profile of AzixaTM
and the extent to which it can improve the overall survival of these patients. In June 2010,
Myrexis reported results from two of the Phase II trials, in metastatic melanoma in combination
with temozolomide and in recurrent glioblastoma in combination with carboplatin, at the annual
meeting of the American Society for Clinical Oncology
(ASCO) that resulted in
durable responses with no additive toxicity in patients. Updated results from an ongoing,
open-label Phase 2a monotherapy study of Azixa™ in treatment-experienced patients with glioblastoma
multiforme (GBM), an aggressive brain tumor, were presented in November 2010 demonstrating durable
responses in patients who had failed both first and second line therapy. Data from a sub-group of
patients with recurrent GBM who are naïve to Avastin is expected to be reported in the first half
of 2011.
In December 2010, Myrexis began a Phase IIb clinical study of AzixaTM to treat GBM.
The study will enroll as many as 120 patients in the U.S. and India in order to evaluate
AzixaTM combination therapy as a first-line GBM treatment. AzixaTM has
received orphan drug status in the United States for the treatment of glioblastoma.
Peripheral Neuropathy
Peripheral neuropathy is a medical condition caused by damage to the nerves in the peripheral
nervous system. The peripheral nervous system includes nerves that run from the brain and spinal
cord to the rest of the body. A 2006 study conducted by Business Insight stated that peripheral
neuropathy affects over 15 million people in the United States and is associated with conditions
that injure peripheral nerves, including herpes zoster, or shingles, diabetes, HIV/AIDS and other
diseases. It can also be caused by trauma or may result from surgical procedures. Peripheral
neuropathy is usually first felt as tingling and numbness in the hands and feet. Symptoms can be
experienced in many ways, including burning, shooting pain, throbbing or aching. Peripheral
neuropathy can cause intense chronic pain that, in many instances, is debilitating.
Post-herpetic neuralgia, or PHN, is one type of peripheral neuropathic pain associated with
herpes zoster, or shingles, which exists after the rash has healed. According to Datamonitor, PHN
affects over 100,000 people in the United States each year. PHN causes pain on and around the area
of skin that was affected by the shingles rash. Most people with PHN describe their pain as “mild”
or “moderate.” However, the pain can be severe in some cases. PHN pain is usually a constant,
burning or gnawing pain but can be an intermittent sharp or stabbing pain. Current treatments for
PHN have limited effectiveness, particularly in severe cases and can cause significant adverse side
effects. One of the initial indications for our NP-1 product candidate is for the treatment of
peripheral neuropathy in PHN patients.
Painful diabetic peripheral neuropathy, or DPN, is common in patients with long-standing Type
1(juvenile) and Type 2 (adult onset) diabetes mellitus. An estimated 18.2 million people have
diabetes mellitus in the United States. The prevalence of neuropathy approaches 50% in those with
diabetes mellitus for greater than 25 years. Specifically, the lifetime incidence of DPN is 11.6%
and 32.1% for Type 1 and Type 2 diabetes, respectively. Common symptoms of DPN are sharp, stabbing,
burning pain, or allodynia, which is pain to light touch, with numbness and tingling of the feet
and sometimes the hands.
Various drugs are currently used in the treatment of DPN. These include tricyclic
antidepressants, or TCA’s, such as amitriptyline, anticonvulsants such as gabapentin, serotonin and
norepinephrine re-uptake inhibitors (e.g., duloxetine), and opioids (e.g., oxycodone).
Unfortunately, the use of these drugs is often limited by the extent of the pain relief provided
and the occurrence of significant central nervous system, or CNS, side effects such as dizziness,
somnolence, and confusion. Because of its limited systemic absorption into the blood, NP-1 topical
cream potentially fulfills the unmet need for a safe, better tolerated, and effective agent for
painful DPN.
Cancer neuropathies represent a large unmet market. Neuropathic pain is caused by injury to,
or compression of, the structures of the peripheral and central nervous system. Chemotherapy or
radiotherapy treatments that patients receive for their cancer often results in neuropathic pain
and is considered a side effect of the cancer treatment. Chemotherapeutic agents, including vinca
alkaloids, cisplatin and paclitaxel, are associated with peripheral neuropathies. Neuropathic pain
is often described as sharp, tingling, burning or shooting. The National Cancer Institute estimates
that 30-40% of cancer patients treated with chemotherapy experience symptoms of CPN, which impairs
their quality of life and ability to function. The debilitating, chronic pain of CPN is one of the
most common reasons why cancer patients stop their treatment early. More than one million breast
cancer survivors in the United States alone suffer from this disease, for which there is no
effective therapy.
7
EpiCeptTM NP-1
EpiCeptTM NP-1, or NP-1, is a prescription topical analgesic cream containing a
patented formulation of two FDA-approved drugs; amitriptyline, which is a widely-used
antidepressant, and ketamine, an NMDA antagonist that is used as an intravenous anesthetic. NP-1 is
designed to provide effective, long-term relief from the pain caused by peripheral neuropathies.
Since each of these ingredients has been shown to have significant analgesic effects and because
NMDA (N-methyl-D-aspartic acid) antagonists, such as ketamine, have demonstrated the ability to
enhance the analgesic effects of amitriptyline, we believe the combination is a good candidate for
the development of a new class of analgesics. We believe that NP-1 can be used in conjunction with
orally delivered analgesics, such as gabapentin.
NP-1 is an odorless, white vanishing cream that is applied twice daily and is quickly absorbed
into the applied area. We believe the topical delivery of its patented combination represents a
fundamentally new approach for the treatment of pain associated with peripheral neuropathy. In
addition, we believe that the topical delivery of our product candidate will significantly reduce
the risk of adverse side effects and drug to drug interactions associated with the systemic
delivery of the active ingredients. The results of our clinical trials to date have demonstrated
the safety of the cream for use for up to one year and a potent analgesic effect in subjects with
both post-herpetic neuralgia and other types of peripheral neuropathy, such as those with diabetic,
traumatic and surgical causes.
In February 2011, we presented data from our Phase IIb trial for EpiCept™ NP-1 in
chemotherapy-induced peripheral neuropathy, or CPN, which is being conducted by National Cancer
Institute, or NCI. CPN may affect 50% of women undergoing treatment for breast cancer. A common
therapeutic agent for the treatment of advanced breast cancer is paclitaxel, and as many as 80% of
the patients with advanced breast cancer experience some signs and symptoms of CPN, such as
burning, tingling pain associated sometimes with mild muscular weakness, after high dose paclitaxel
administration. The multi-center, double-blind, randomized, placebo-controlled study was conducted
within a network of approximately 25 sites under the direction of the NCI funded Community Clinical
Oncology Program, or CCOP. More than 460 cancer survivors suffering from painful CPN were enrolled
in the six-week study. The results of the trial in the intent to treat, or ITT, population
demonstrated that the change in average daily neuropathy intensity scores in the NP-1 group
achieved a statistically significant reduction in CPN intensity versus placebo (p<0.001), which
was the trial’s primary endpoint. Additionally, a pre-specified subgroup of the ITT population,
those patients who previously received taxane chemotherapy, also showed a statistically significant
reduction in average daily neuropathy intensity scores (p=0.034). This subgroup constituted more
than 50% of the ITT population. Secondary efficacy endpoints confirmed the superiority of NP-1 vs.
placebo. Furthermore, the safety profile of NP-1 was comparable to placebo.
In January 2009, we completed a Phase IIb, multi-center, randomized, placebo controlled trial
in approximately 360 patients evaluating the analgesic properties and safety of NP-1 cream in
patients with PHN. This trial compared the efficacy and safety of NP-1 against both gabapentin, the
leading drug prescribed for this indication, and placebo. The first primary endpoint was the
change in pain intensity over the four week duration of the trial. The data demonstrated that NP-1
achieved statistically significant superior efficacy compared with placebo (p=0.024). An additional
primary endpoint, to demonstrate that NP-1 was not inferior to gabapentin in reducing pain, was
also met. A key secondary endpoint measured in the trial from a responder analysis indicated that
63% of patients in the NP-1 treatment arm achieved a reduction in pain scores of at least 30%,
significantly higher than that of patients in the placebo arm (p=0.033). Top-line results further
indicate that NP-1 achieved a superior safety profile when compared with gabapentin, especially
with regard to dizziness and somnolence, as evaluated by the reporting of adverse events. NP-1 has
received orphan drug protection for the treatment of PHN.
In February 2008, we completed a Phase II clinical trial in 215 patients suffering from DPN.
The results of this double-blind, placebo-controlled study demonstrated that the primary endpoint,
the difference in changes in pain intensity between NP-1 and placebo over the four week duration of
the trial, nearly reached statistical significance (p=0.0715). The analgesic benefits of NP-1
continued to build over time during the course of the study. Key secondary endpoints measured in
the trial from a responder analysis indicate that 60% of patients in the NP-1 treatment arm
achieved a reduction of pain scores of at least 30% compared with 48% of patients in the placebo
arm (p=0.076). In addition, 33% of patients in the NP-1 treatment arm achieved a reduction in pain
scores of at least 50% compared with 21% of patients in the placebo arm (p=0.078). All pain
scores measured trended in favor of the NP-1 treated patients over the placebo group, indicative of
an analgesic effect in this type of peripheral neuropathic pain. We concluded that data derived
from the trial support the continued study of NP-1 in late-stage pivotal clinical trials.
8
Our Strategic Alliances
Meda AB
In January 2010, we entered into an exclusive commercialization agreement for
Ceplene® with Meda AB, a leading international specialty pharmaceutical company based in
Stockholm, Sweden. Under the terms of the agreement, we granted Meda the right to market Ceplene
in Europe and several other countries including Japan, China, and Australia. Pursuant to the terms
of the agreement, we received a $3 million fee on signing and an additional $2 million milestone
payment in May 2010 upon the first commercial sale of Ceplene® in a major European
market. Additional potential payments include a $5 million payment upon achievement of a
regulatory milestone and up to $30 million in sales-based milestones that commence upon attainment
of at least $50 million in annual sales. We will receive an escalating percentage royalty on net
sales in the covered territories ranging from the low teens to the low twenties and will be
responsible for Ceplene’s commercial supply. The initial term of this agreement is ten years and
is subject to automatic two year extensions at Meda’s option. The agreement can be terminated at
any time by Meda upon six months’ prior written notice. The agreement can also be terminated by
mutual agreement or for cause.
Myrexis, Inc.
We licensed the MX90745 series of caspase-inducer anti-cancer compounds to Myrexis in 2003.
Under the terms of the agreement, we granted to Myrexis a research license to develop and
commercialize any drug candidates from the series of compounds with a non-exclusive, worldwide,
royalty-free license, without the right to sublicense the technology. Myrexis is responsible for
the worldwide development and commercialization of any drug candidates from the series of
compounds. We also granted to Myrexis a worldwide royalty bearing development and commercialization
license with the right to sublicense the technology. The agreement requires that Myrexis make
licensing, research and milestone payments to the Company totaling up to $27 million, of which $3.0
million was paid and recognized as revenue, assuming the successful commercialization of the
compound for the treatment of cancer, as well as pay an escalating mid to high single digit
percentage royalty on product sales. In March 2007, Myrexis initiated Phase II clinical trials
for AzixaTM, a MX90745 series compound. In March 2008, we received a milestone payment
of $1.0 million following dosing of the first patient in these trials. In December 2010, Myrexis
began a Phase IIb clinical study of AzixaTM to treat glioblastoma multiforme, an
aggressive brain tumor in order to evaluate AzixaTM combination therapy as a first-line
GBM treatment.
DURECT Corporation
In December 2006, we entered into a license agreement with DURECT Corporation, pursuant to
which we granted DURECT the exclusive worldwide rights to certain of our intellectual property for
a transdermal patch containing bupivacaine for the treatment of back pain. Under the terms of the
agreement, we received a $1.0 million upfront payment. In September 2008, we amended our license
agreement with DURECT. Under the terms of the amended agreement, we granted DURECT royalty-free,
fully paid up, perpetual and irrevocable rights to the intellectual property licensed as part of
the original agreement in exchange for a cash payment of $2.25 million from DURECT.
Endo Pharmaceuticals, Inc.
In December 2003, we entered into a license agreement with Endo under which we granted Endo
(and its affiliates) the exclusive (including as to us and our affiliates) worldwide right to
commercialize LidoPAIN BP, a lidocaine patch developed by EpiCept for the treatment of acute back
pain. We also granted Endo worldwide rights to use certain of our patents for the development of
certain other non-sterile, topical lidocaine patches, including Lidoderm®, Endo’s
non-sterile topical lidocaine-containing patch for the treatment of chronic lower back pain. Upon
the execution of the Endo agreement, we received a non-refundable payment of $7.5 million. Other
potential compensation under this agreement includes additional milestone payments of up to $82.5
million related to both our LidoPAIN BP product and Lidoderm® and a royalty on net sales
of LidoPAIN BP. We do not expect to receive further compensation under this agreement.
The last published clinical study of Lidoderm® in back pain occurred in 2005. We
believe that no measurable progress has occurred with Lidoderm® for back pain and do not
expect further revenue from this license at this time.
Manufacturing
We currently intend to outsource all of our manufacturing activities. We believe that this
strategy will enable us to direct operational and financial resources to the development of our
product candidates rather than diverting resources to establishing a
manufacturing infrastructure. Under the terms of a supply agreement with Meda, we are
responsible for supplying commercial quantities of Ceplene® based on Meda’s estimate of
sales in the licensed territories.
9
We have entered into arrangements with qualified third parties for the manufacture of
Ceplene® and for the formulation and manufacture of our clinical supplies. We may enter
into additional written supply agreements in the future. We generally purchase our supplies from
current suppliers pursuant to purchase orders. We plan to use a single, separate third party
manufacturer for Ceplene® and each of our product candidates for which we are
responsible for manufacturing. In some cases, the responsibility to manufacture product, or to
identify suitable third party manufacturers, may be assumed by our licensees. We cannot assure you
that our current manufacturers can successfully increase their production to meet full commercial
demand. We believe that in most cases there are several manufacturing sources available to us,
including our current manufacturers, which can meet our commercial supply requirements on
commercially reasonable terms. We will continue to look for and secure the appropriate
manufacturing capabilities and capacity to ensure commercial supply at the appropriate time.
Sales and Marketing
We hired a small marketing staff in 2009 and 2010 in anticipation of the start of commercial
activities for Ceplene® in the United States. After receiving a refusal to file letter
from the FDA on the NDA for Ceplene®, we eliminated our marketing staff and scaled back our
commercial activities in the United States. In order to commercially market Ceplene® or
any of our product candidates in the United States upon receipt of marketing approval, we must
either develop an internal sales and marketing infrastructure or collaborate with third parties
with sales and marketing expertise. We have retained the rights to commercialize NP-1 and
crolibulinTM worldwide and to market Ceplene globally except for Europe and certain
Pacific rim countries. In addition, we have granted Myrexis exclusive worldwide commercialization
rights, with rights to sublicense, for AzixaTM. We will likely market our products in
international markets outside of North America through collaborations with third parties. We intend
to make decisions regarding internal sales and marketing of our product candidates on a
product-by-product and country-by-country basis.
Intellectual Property
Our commercial success will depend in part on obtaining and maintaining patent protection and
trade secret protection of our technologies and drug candidates as well as successfully defending
these patents against third-party challenges. We have various compositions of matter and use
patents, which have claims directed to our product candidates or their methods of use. Our patent
policy is to retain and secure patents for the technology, inventions and improvements related to
our core portfolio of product candidates. We also rely on trade secrets, technical know-how and
continuing innovation to develop and maintain our competitive position.
The following is a summary of the patent position relating to our three in-house product
candidates:
Ceplene® — The intellectual property protection surrounding our histamine
technology includes 24 U.S. patents issued or allowed, with the term for the latest one expiring in
February 2023. Patents issued or pending in the international markets concern specific therapeutic
areas or manufacturing. Claims include the therapeutic administration of histamine or any H2
receptor agonist in the treatment of cancer, infectious diseases and other diseases, either alone
or in combination therapies, the novel synthetic method for the production of pharmaceutical-grade
histamine dihydrochloride, the mechanism of action including the binding receptor and pathway, and
the rate and route of administration.
CrolibulinTM — The intellectual property protection regarding this compound is
covered by two issued U.S. patents, with the latest one expiring in May 2022 and one application
pending covering the composition and uses of this compound and structurally related analogs.
Additional foreign patent applications are pending in major pharmaceutical markets outside the
United States.
EpiCeptTM NP-1 — We own a U.S. patent with claims directed to a formulation
containing a combination of amitriptyline and ketamine, which can be used as a treatment for the
topical relief of pain, including neuropathic pain, that expires in August 2021. We also have a
license to additional patents, which expire in September 2015 and May 2018, and which have claims
directed to topical uses of tricyclic antidepressants, such as amitriptyline, and NMDA antagonists,
such as ketamine, as treatments for relieving pain, including neuropathic pain.
We may seek to protect our proprietary information by requiring our employees, consultants,
contractors, outside partners and other advisers to execute, as appropriate, nondisclosure and
assignment of invention agreements upon commencement of their employment or engagement. We also
require confidentiality or material transfer agreements from third parties that receive our
confidential data or materials.
10
We also rely on trade secrets to protect our technology, especially where we do not believe
patent protection is appropriate or obtainable. However, trade secrets are difficult to protect.
While we use reasonable efforts to protect our trade secrets, our employees, consultants,
contractors, partners and other advisors may unintentionally or willfully disclose information to
competitors. Enforcing a claim that a third party illegally obtained and is using trade secrets is
expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the
United States are sometimes less willing to protect trade secrets. Moreover, our competitors may
independently develop equivalent knowledge, methods and know-how.
The pharmaceutical, biotechnology and other life sciences industries are characterized by the
existence of a large number of patents and frequent litigation based upon allegations of patent
infringement. While our drug candidates are in clinical trials, and prior to commercialization, we
believe our current activities fall within the scope of the exemptions provided by 35 U.S.C.
Section 271(e) in the United States and Section 55.2(1) of the Canadian Patent Act, each of which
covers activities related to developing information for submission to the FDA and its counterpart
agency in Canada. As our drug candidates progress toward commercialization, the possibility of an
infringement claim against us increases. While we attempt to ensure that our drug candidates and
the methods we employ to manufacture them do not infringe other parties’ patents and other
proprietary rights, competitors or other parties may assert that we infringe on their proprietary
rights.
For a discussion of the risks associated with our intellectual property, see Item 1A. “Risk
Factors — Risks Relating to Intellectual Property.”
License Agreements
We have in the past licensed and will continue to license patents from collaborating research
groups and individual inventors.
Epitome/Dalhousie
In August 1999, we entered into a sublicense agreement with Epitome Pharmaceuticals Limited
under which we were granted an exclusive license to certain patents for the topical use of
tricyclic anti-depressants and NMDA antagonists as topical analgesics for neuralgia that were
licensed to Epitome by Dalhousie University. These and other patents cover the combination
treatment consisting of amitriptyline and ketamine in NP-1. This technology has been incorporated
into NP-1. In July 2007, we converted the sublicense agreement previously established with Epitome
Pharmaceuticals Limited, related to NP-1, into a direct license with Dalhousie University. Under
this new arrangement, we gained more favorable terms, including a lower maintenance fee obligation
and reduced royalty rate on future product sales.
We have been granted worldwide rights to make, use, develop, sell and market products utilizing the
licensed technology in connection with passive dermal applications. We are obligated to make
payments to Dalhousie totaling $0.9 million, of which $0.2 million has been paid, upon achievement
of specified milestones and to pay single-digit royalties based on annual net sales derived from
the products incorporating the licensed technology. We are obligated to pay Dalhousie an annual
maintenance fee of $0.5 million until the license agreement expires or is terminated, or an NDA for
NP-1 is filed with the FDA, otherwise Dalhousie will have the option to terminate the contract. The
license agreement with Dalhousie terminates upon the expiration of the last to expire licensed
patent. The sublicense agreement with Epitome terminated in July 2007. During 2010, 2009 and
2008, we paid Epitome a fee of $0.0, $0.3 million and $0.3 million, respectively. During 2010, 2009
and 2008, we paid Dalhousie a maintenance fee of $0.5 million, $0.5 million and $0.4 million,
respectively. These payments were expensed to research and development.
Hellstrand
In October 1999, we entered into a royalty agreement with Dr. Kristoffer Hellstrand under
which we have an exclusive license to certain patents for Ceplene® configured for the
systemic treatment of cancer, infectious diseases, autoimmune diseases and other medical
conditions. Under this agreement, we paid Dr. Hellstrand $1 million in 1999 and will owe a royalty
of 1% of net sales. In November 2009 we expanded our collaboration with Dr. Hellstrand by
concluding a new agreement in which we acquired rights to develop certain new compounds. As
compensation for this agreement and for providing certain ongoing consulting services, we awarded
Dr. Hellstrand options to purchase 50,000 shares of our common stock and agreed to pay a monthly
consulting fee. During 2010 and 2009, Dr. Hellstrand earned $0.1 and $0 million in royalties based
on net sales of Ceplene®.
Shire Biochem
In March 2004 and as amended in January 2005, we entered into a license agreement reacquiring
the rights to the MX2105 series of apoptosis inducer anti-cancer compounds from Shire BioChem,
Inc., formerly known as BioChem Pharma, Inc., which had previously announced that oncology would no
longer be a therapeutic focus of the company’s research and development efforts.
Under the agreements, Shire BioChem agreed to assign and/or license to us rights it owned
under or shared under its oncology research program. The agreement requires that we provide Shire
BioChem a portion of any sublicensing payments we receive if we relicense the series of compounds,
and make milestone payments to Shire BioChem totaling up to $26 million, assuming the successful
commercialization of a compound for the treatment of a cancer indication, as well as pay a royalty
on product sales. At December 31, 2010, we accrued a license fee of $0.6 million payable as a
result of the commencement of a Phase I clinical trial for crolibulinTM in 2006.
11
Government Regulation
United States
The FDA and comparable state and local regulatory agencies impose substantial requirements
upon the clinical development, manufacture, marketing and distribution of drugs. These agencies and
other federal, state and local entities regulate research and development activities and the
testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping,
approval, advertising and promotion of our product candidates. In the United States, the FDA
regulates drugs under the Federal Food, Drug, and Cosmetic Act, and implementing regulations. The
process required by the FDA before our product candidates may be marketed in the United States
generally involves the following:
| |
• |
|
completion of extensive pre-clinical laboratory tests, pre-clinical animal studies and
formulation studies all performed in accordance with the FDA’s good laboratory practice, or
GLP, regulations; |
| |
| |
• |
|
submission to the FDA of an Investigational New Drug, or IND, application that must
become effective before clinical trials may begin; |
| |
| |
• |
|
performance of adequate and well-controlled clinical trials to establish the safety and
efficacy of the product candidate for each proposed indication; |
| |
| |
• |
|
submission of an NDA to the FDA; |
| |
| |
• |
|
satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities
at which the product is produced to assess compliance with current GMP, or cGMP,
regulations; and |
| |
| |
• |
|
FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of
the drug. |
The testing and approval process requires substantial time, effort and financial resources,
and we cannot be certain that any approvals for our product candidates will be granted on a timely
basis, if at all.
Pre-clinical Activities. Pre-clinical activities include laboratory evaluation of product
chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The
results of pre-clinical tests, together with manufacturing information and analytical data, are
submitted as part of an IND application to the FDA. The IND automatically becomes effective 30 days
after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or
questions about the conduct of the clinical trial, including concerns that human research subjects
will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must
resolve any outstanding concerns before the clinical trial can begin. Our submission of an IND, or
those of our collaborators, may not result in FDA authorization to commence a clinical trial. A
separate submission to an existing IND must also be made for each successive clinical trial
conducted during product development, and the FDA must grant permission before each clinical trial
can begin. Further, an independent institutional review board, or IRB, for each medical center
proposing to conduct the clinical trial must review and approve the plan for any clinical trial
before it commences at that center, and it must monitor the study until completed. The FDA, the IRB
or the sponsor may suspend a clinical trial at any time on various grounds, including a finding
that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing
also must satisfy extensive Good Clinical Practice, or GCP, regulations and regulations for
informed consent of subjects.
Clinical Trials. For purposes of NDA submission and approval, clinical trials are typically
conducted in the following three sequential phases, which may overlap:
| |
• |
|
Phase I: Studies are initially conducted in a limited population to test the drug
candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion
in healthy humans or, on occasion, in subjects. In some cases, a sponsor may decide to
run what is referred to as a “Phase Ib” evaluation, which is a second safety-focused
Phase I
clinical trial typically designed to evaluate the impact of the drug candidate in
combination with currently approved drugs. |
12
| |
• |
|
Phase II: Studies are generally conducted in a limited patient population to identify
possible adverse effects and safety risks, to determine the efficacy of the drug
candidate for specific targeted indications and to determine dose tolerance and optimal
dosage. Multiple Phase II clinical trials may be conducted by the sponsor to obtain
information prior to beginning larger and more expensive Phase III clinical trials. In
some instances, a sponsor may decide to run what is referred to as a “Phase IIa”
clinical trial, which is designed to provide dose-ranging and additional safety and
pharmaceutical data. In other cases, a sponsor may decide to run what is referred to as
a “Phase IIb” evaluation, which is a second, confirmatory Phase II clinical trial that
could, if positive and accepted by the FDA, serve as a pivotal clinical trial in the
approval of a drug candidate. |
| |
• |
|
Phase III: These are commonly referred to as pivotal studies. When Phase II clinical
trials demonstrate that a dose range of the drug candidate is effective and has an
acceptable safety profile, Phase III clinical trials are undertaken in large patient
populations to further evaluate dosage, to provide substantial evidence of clinical
efficacy and to further test for safety in an expanded and diverse patient population at
multiple, geographically dispersed clinical trial sites. |
In some cases, the FDA may give conditional approval of an NDA for a drug candidate on the
sponsor’s agreement to conduct additional clinical trials to further assess the drug’s safety and
effectiveness after NDA approval. Such post-approval trials are typically referred to as Phase IV
clinical trials.
New Drug Application. The results of drug candidate development, pre-clinical testing,
chemistry and manufacturing controls and clinical trials are submitted to the FDA as part of an
NDA. The NDA also must contain extensive manufacturing information. The FDA has 60 days to decide
whether to file the NDA so that it can be reviewed. The FDA can refuse to file an application that
is incomplete. In accordance with the Prescription Drug User Fee Act (PDUFA), the FDA’s Center for
Drug Evaluation and Research (CDER) expects to review and act on at least 90 percent of NDAs for
standard drugs no later than 10 months after the applications are received. The review goal is six
months for priority drugs. The review process is often significantly extended by FDA
requests for additional information or clarification. The FDA may refer the NDA to an advisory
committee for review, evaluation and recommendation as to whether the application should be
approved. The FDA is not bound by the recommendation of an advisory committee, but it generally
follows such recommendations. The FDA may deny approval of an NDA if the applicable regulatory
criteria are not satisfied, or it may require additional clinical data or an additional pivotal
Phase III clinical trial. Even if such data is submitted, the FDA may ultimately decide that the
NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive
and the FDA may interpret data differently than we do. Once issued, the FDA may withdraw drug
approval if ongoing regulatory requirements are not met or if safety problems occur after the drug
reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials,
and surveillance programs to monitor the effect of approved products that have been commercialized,
and the FDA has the power to prevent or limit further marketing of a drug based on the results of
these post-marketing programs. Drugs may be marketed only for the approved indications and in
accordance with the provisions of the approved label. Further, if there are any modifications to
the drug, including changes in indications, labeling or manufacturing processes or facilities, we
may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require
us to develop additional data or conduct additional pre-clinical studies and clinical trials.
Satisfaction of FDA regulations and requirements or similar requirements of state, local and
foreign regulatory agencies typically takes several years, and the actual time required may vary
substantially based upon the type, complexity and novelty of the product or disease. Government
regulation may delay or prevent marketing of drug candidates for a considerable period of time and
impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant
approvals for new indications for our drug candidates on a timely basis, if at all. Even if a drug
candidate receives regulatory approval, the approval may be significantly limited to specific
usages, patient populations and dosages. Further, even after regulatory approval is obtained, later
discovery of previously unknown problems with a drug may result in restrictions on the drug or even
complete withdrawal of the drug from the market. Delays in obtaining, or failures to obtain,
regulatory approvals for any of our drug candidates would harm its business. In addition, we cannot
predict what additional governmental regulations may arise from future U.S. governmental action.
Any drugs manufactured or distributed by us or our collaborators pursuant to FDA approvals are
subject to continuing regulation by the FDA, including record keeping requirements and reporting of
adverse experiences associated with the drug. Drug manufacturers and their subcontractors are
required to register their establishments with the FDA and certain state agencies and are subject
to periodic unannounced inspections by the FDA and certain state agencies for compliance with
ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation
requirements upon us and our third-party manufacturers. Failure to comply with the statutory and
regulatory requirements can subject a manufacturer to potential legal or regulatory action, such as
warning letters, suspension of manufacturing, seizure of product, injunctive action or civil
penalties. We
cannot be certain that we or our present or future third-party manufacturers or suppliers will
be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If our
present or future third-party manufacturers or suppliers are not able to comply with these
requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution,
or withdraw approval of the NDA for that drug.
13
The FDA closely regulates the post-approval marketing and promotion of drugs, including
standards and regulations for direct-to-consumer advertising, off-label promotion,
industry-sponsored scientific and educational activities and promotional activities involving the
Internet. A company can make only those claims relating to safety and efficacy that are approved by
the FDA. Failure to comply with these requirements can result in adverse publicity, warning
letters, corrective advertising and potential civil and criminal penalties. Physicians may
prescribe legally available drugs for uses that are not described in the drug’s labeling and that
differ from those tested by us and approved by the FDA. Such off-label uses are common across
medical specialties. Physicians may believe that such off-label uses are the best treatment for
many patients in varied circumstances. The FDA does not regulate the behavior of physicians in
their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’
communications regarding off-label use.
Section 505(b)(2) Drug Applications. Once an FDA-approved new drug is no longer
patent-protected, another company may sponsor a new indication, a new use or put the drug in a new
dosage form. Each new indication from a different company requires an NDA filing. As an alternate
path to FDA approval for new or improved formulations of previously approved products, a company
may file a Section 505(b)(2) NDA. Section 505(b)(2) permits the filing of an NDA where at least
some of the information required for approval comes from studies not conducted by or for the
applicant and for which the applicant has not obtained a right of reference. However, this NDA does
not have to contain all of the information or data that was submitted with the original NDA because
of the FDA’s prior experience with the drug product. An original NDA for an FDA-approved new drug
would have required numerous animal toxicology studies that have been reviewed by the FDA. These
can be referenced in the 505(b)(2) NDA submitted by the new applicant. Many studies in humans that
support the safety of the drug product may be in the published literature. The FDA allows the new
sponsor company to submit these publications to support its 505(b)(2) NDA. By allowing the new
sponsor company to use this information, the time and cost required to obtain approval for a drug
product for the new indication can be greatly reduced. The FDA may also require companies to
perform additional studies or measurements to support the change from the approved product. The FDA
may then approve the new product candidate for all or some of the label indications for which the
referenced product has been approved, as well as for any new indication sought by the Section
505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an
already approved product, the applicant is required to certify to the FDA concerning any patents
listed for the approved product in the FDA’s Orange Book publication. Specifically, the applicant
must certify that: (i) the required patent information has not been filed; (ii) the listed patent
has expired; (iii) the listed patent has not expired, but will expire on a particular date and
approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be
infringed by the new product. If the applicant does not challenge the listed patents, the Section
505(b)(2) application will not be approved until all the listed patents claiming the referenced
product have expired. The Section 505(b)(2) application also will not be approved until any
non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed
in the Orange Book for the referenced product has expired.
Foreign Regulation
Whether or not we obtain FDA approval for a product, we must obtain approval of a product by
the comparable regulatory authorities of foreign countries before we can commence clinical trials
or marketing of the product in those countries. The approval process varies from country to
country, and the time may be longer or shorter than that required for FDA approval. The
requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement
also vary greatly from country to country. Although governed by the applicable country, clinical
trials conducted outside of the United States typically are administered with the three-phase
sequential process that is discussed above under “Government Regulation — United States.” However,
the foreign equivalent of an IND is not a prerequisite to performing pilot studies or Phase I
clinical trials.
Under European Union regulatory systems, we may submit marketing authorization applications
either under a centralized or decentralized procedure. The centralized procedure, which is required
for oncology products and is available for medicines produced by biotechnology or which are highly
innovative, provides for the grant of a single marketing authorization that is valid for all member
states. This authorization is a marketing authorization application, or MAA. The decentralized
procedure provides for mutual recognition of national approval decisions. Under this procedure, the
holder of a national marketing authorization may submit an application to the remaining member
states. Within 90 days of receiving the applications and assessment report, each member state must
decide whether to recognize approval. This procedure is referred to as the mutual recognition
procedure.
14
In addition, regulatory approval of prices is required in most countries other than the United
States. We face the risk that the resulting prices would be insufficient to generate an acceptable
return to us or our collaborators.
Corporate Information
We were incorporated in Delaware in March 1993. We have two wholly-owned subsidiaries, EpiCept
GmbH, based in Munich, Germany, which is engaged in commercial activities on our behalf, and Maxim
Pharmaceuticals, Inc., which is currently inactive. Our principal executive offices are located at
777 Old Saw Mill River Road, Tarrytown, NY, and our telephone number is (914) 606-3500. Our website
address is www.epicept.com. Our website, and the information contained in our website, is not a
part of this annual report.
Employees
As of March 30, 2011, our workforce consists of 14 full-time employees, three of whom hold a
Ph.D. or M.D., and one of whom holds another advanced degree. We have no collective bargaining
agreements with our employees and have not experienced any work stoppages. We believe that our
relations with our employees are good.
Research and Development
Since our inception, we have made substantial investments in research and development. In the
years ended December 31, 2010, 2009 and 2008, we incurred research and development expenses of $8.1
million, $11.6 million and $12.6 million, respectively.
Availability of SEC Filings
We have filed reports, proxy statements and other information with the SEC. Copies of our
reports, proxy statements and other information may be inspected and copied at the public reference
facilities maintained by the SEC at SEC Headquarters, Public Reference Section, 100 F Street, N.E.,
Washington D.C. 20549. The public may obtain information on the operation of the SEC’s public
reference facilities by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that
contains reports, proxy statements and other information regarding us. The address of the SEC
website is http://www.sec.gov. We will also provide copies of our Forms 8-K, 10-K, 10-Q,
Proxy and Annual Report at no charge available through our website at www.epicept.com as soon as
reasonably practicable after filing electronically such material with the SEC. Copies are also
available, without charge, from EpiCept Corporation, 777 Old Saw Mill River Road, Tarrytown, NY,
10591.
15
An investment in our common stock involves a high degree of risk. You should carefully
consider the risk factors described below as well as the other information contained in this Annual
Report before buying shares of our common stock. If any of the following risks or uncertainties
occurs, our business, financial conditions and operating results could be materially and adversely
affected. As a result, the trading price of our common stock could decline and you may lose all or
a part of your investment in our common stock.
Risks Relating to our Financial Condition
We have limited liquidity and, as a result, may not be able to meet our obligations.
As of December 31, 2010 we had approximately $2.4 million in cash and cash equivalents. In
March 2011, we received $4.3 million cash, net of $0.3 million in transaction costs, from the
issuance of approximately 7.1 million shares of our common stock and warrants to purchase 5.3
million shares of our common stock. In February 2011, we received $6.6 million cash, net of $0.5
million in transaction costs, from the issuance of approximately 8.9 million shares of our common
stock and warrants to purchase 3.6 million shares of our common stock. Our anticipated average
monthly cash operating expenses in 2011 is approximately $1.1 million per month. In addition, we
are required to make an interest and principal payment to our lender tbg on June 30, 2011 in the
amount of approximately $0.5 million. We believe that our cash is sufficient to fund operations
into 2012. We may receive cash from certain of our licensing partners during 2011 for achievement
of clinical milestones, and we may raise additional funds through the issuance of debt and/or
equity to meet our cash needs. To conserve cash, we may delay or cancel some of our planned
development activities that are not related to Ceplene® during 2011.
In November 2010 we provided an update with respect to our financing plans. The key element
of the plan is a non-equity financing transaction that, if fully completed, will support our
operations for an additional year. We anticipate that the transaction, which is subject to
completion of due diligence and execution of mutually-satisfactory documentation, will close in
2011, but should the transaction not close or the proceeds be less than anticipated we may
determine to seek additional or alternative sources or types of financing, including equity
financing.
We plan to out license our NP-1 compound to a third party who will agree to complete clinical
development and commercialize the product upon receipt of necessary regulatory approvals.
Discussions with prospective partners are continuing, however at this time we are unable to
determine whether or when such an agreement might be concluded or the amount of any fees that may
be paid to us in 2011 in connection with the agreement.
If additional funds are raised by issuing equity, substantial dilution to existing
shareholders may result. If we fail to obtain capital when required, we may be forced to delay,
scale back, or eliminate some or all of our commercialization and clinical efforts for
Ceplene® and our research and development programs or to cease operations entirely.
We have a history of losses and have never generated significant revenue from product sales and we
expect to incur substantial losses in the future.
We have incurred significant losses since our inception, and we expect that we will experience
net losses and negative cash flow for the foreseeable future. Since our inception in 1993, we have
incurred significant net losses in each year. Our losses have resulted principally from expenses
incurred in connection with our development activities and from general and administrative expenses
associated with our operations. Our net loss for the years ended December 31, 2010, 2009 and 2008
was $15.5 million, $38.8 million and $25.4 million, respectively. As of December 31, 2010 and 2009,
our accumulated deficit was $250.6 million and $235.0 million, respectively. We may never generate
sufficient net revenue to achieve or sustain profitability.
We expect to continue to incur significant expenses over the next several years as we:
| |
• |
|
apply for marketing approval of Ceplene® in the U.S., Canada, and other
countries |
| |
| |
• |
|
continue to conduct clinical trials for Ceplene® and our product
candidates; |
| |
| |
• |
|
seek regulatory approvals for Ceplene® and our product candidates; |
| |
| |
• |
|
develop, formulate and commercialize our product candidates; |
| |
| |
• |
|
implement additional internal controls and reporting systems and further develop our
corporate infrastructure; |
16
| |
• |
|
acquire or in-license additional products or technologies or expand the use of our
technologies; and |
| |
• |
|
maintain, defend and expand the scope of our intellectual property. |
| |
• |
|
hire additional personnel. |
We expect that we will have large fixed expenses in the future, including significant expenses
for research and development and selling, general and administrative expenses. We will need to
generate significant revenues to achieve and maintain profitability. If we cannot successfully
develop, obtain regulatory approvals for and commercialize our product candidates, we will not be
able to generate significant revenue from product sales or achieve profitability in the future. As
a result, our ability to achieve and sustain profitability will depend on our ability to generate
and sustain substantially higher revenue while maintaining reasonable cost and expense levels.
We may not be able to continue as a going concern.
Our recurring losses from operations and our stockholders’ deficit raise substantial doubt
about our ability to continue as a going concern and as a result our independent registered public
accounting firm included an explanatory paragraph in its report on our consolidated financial
statements for the year ended December 31, 2010, which is included herein, with respect to this
uncertainty. We will need to generate significant revenue from the sale of Ceplene® or
raise additional capital to continue to operate as a going concern. In addition, the perception
that we may not be able to continue as a going concern may cause others to choose not to deal with
us due to concerns about our ability to meet our contractual obligations and may adversely affect
our ability to raise additional capital.
Our quarterly financial results are likely to fluctuate significantly, which could have an adverse
effect on our stock price.
Our quarterly operating results will be difficult to predict and may fluctuate significantly
from period to period, particularly because we are a relatively small company and we have not
generated any meaningful revenue to date. The level of our revenues and expenses and our results of
operations at any given time could fluctuate as a result of any of the following factors:
| |
• |
|
research and development expenses incurred and other operating expenses; |
| |
• |
|
results of our clinical trials; |
| |
• |
|
our ability to obtain regulatory approval for our product candidates; |
| |
• |
|
our ability to achieve milestones under our strategic relationships on a timely basis
or at all; |
| |
• |
|
timing of new product offerings, acquisitions, licenses or other significant events
by us or our competitors; |
| |
• |
|
regulatory approvals and legislative changes affecting the products we may offer or
those of our competitors; |
| |
• |
|
our ability to establish and maintain a productive sales force; |
| |
• |
|
demand and pricing of any of our products; |
| |
• |
|
physician and patient acceptance of our products; |
| |
• |
|
levels of third-party reimbursement for our products; |
| |
• |
|
interruption in the manufacturing or distribution of our products; |
| |
• |
|
the effect of competing technological and market developments; |
| |
• |
|
litigation involving patents, licenses or other intellectual property rights; and |
| |
• |
|
product failures or product liability lawsuits. |
With the exception of Ceplene®, we have not yet obtained regulatory approval for
any of our product candidates. In addition, we do not manufacture products ourselves or conduct
significant sales and marketing activities. Consequently, it is difficult to make any predictions
about our future success, viability or profitability based on our historical operations. It is also
difficult to predict the timing of the achievement of various milestones under our strategic
relationships. In addition, our operating expenses may continue to increase as we develop product
candidates and build commercial capabilities. Accordingly, we may experience significant quarterly
losses.
17
Because of these factors, our operating results in one or more future quarters may fail to
meet the expectations of securities analysts or investors, which could cause our stock price to
decline significantly.
We have had limited operating activities, which may make it difficult for you to evaluate the
success of our business to date and to assess our future viability.
Our activities to date have been limited to organizing and staffing our operations, acquiring,
developing and securing our technology, licensing product candidates, and undertaking preclinical
and clinical studies and clinical trials. With the exception of Ceplene®, we have not
yet demonstrated an ability to obtain regulatory approval, manufacture products or conduct sales
and marketing activities. Consequently, it is difficult to make any predictions about our future
success, viability or profitability based on our historical operations.
Clinical and Regulatory Risks
Other than the marketing authorization for Ceplene® in the European Union and
Israel, we currently have no products approved for sale and we cannot guarantee you that we will
ever obtain regulatory approval for such other product candidates, which could delay or prevent us
from being able to generate revenue from product sales.
All of our product candidates are subject to extensive government regulations related to
development, clinical trials, manufacturing and commercialization. The process of obtaining
approvals from the FDA, European Medicines Agency for the Evaluation of Medicinal Products, or EMA,
and other governmental and similar international regulatory agencies is costly, time consuming,
uncertain and subject to unanticipated delays. The FDA, EMA and similar international regulatory
authorities may not ultimately approve the candidate for commercial sale in any jurisdiction.
Despite the fact we received the marketing authorization for Ceplene® in the
European Union and Israel, we may not receive regulatory approval outside of the European Union and
Israel, including in the United States or Canada. The FDA, EMA and similar international regulators
may refuse to approve an application for approval of a drug candidate if they believe that
applicable regulatory criteria are not satisfied. The FDA, EMA or similar international regulators
may also require additional testing for safety and efficacy. Any failure or delay in obtaining
these approvals could prohibit or delay us from marketing product candidates. If our other product
candidates do not meet applicable regulatory requirements for approval, we may not have the
financial resources to continue research and development of these product candidates and we may not
generate significant revenues from the commercial sale of any of our products.
To obtain regulatory approval for our other product candidates, we or our partners must
conduct extensive human tests, which are referred to as clinical trials, as well as meet other
rigorous regulatory requirements. Satisfaction of all regulatory requirements typically takes many
years and requires the expenditure of substantial resources.
We currently have several product candidates in various stages of clinical testing. All of our
product candidates are prone to the risks of failure inherent in drug development and testing.
Product candidates in later-stage clinical trials may fail to show desired safety and efficacy
traits despite having progressed through initial clinical testing. In addition, the data collected
from clinical trials of our product candidates may not be sufficient to support regulatory
approval, or regulators could interpret the data differently than we do. The regulators may require
us or our partners to conduct additional clinical testing, in which case we would have to expend
additional time and resources. The approval process may also be delayed by changes in government
regulation, future legislation or administrative action or changes in regulatory policy that occur
prior to or during regulatory review.
We and other drug development companies have suffered set backs in late-stage clinical trials
even after achieving promising results in early stage development. Accordingly, the results from
completed preclinical studies and early stage clinical trials may not be predictive of results in
later stage trials and may not be predictive of the likelihood of regulatory approval. Any failure
or significant delay in completing clinical trials for our product candidates, or in receiving
regulatory approval for the sale of our product candidates, may severely harm our business and
delay or prevent us from being able to generate revenue from product sales, and our stock price
will likely decline.
We may not be able to obtain regulatory approval in the United States for Ceplene®, our
lead product candidate, which could delay or prevent us from being able to generate revenue from
sales of Ceplene®, and require additional expenditures.
None of our products has received regulatory approval in the United States. In August 2010,
we received a refusal to file letter from the U.S. Food and Drug Administration (FDA) on our New
Drug Application (NDA) for Ceplene®.
18
In October 2010, we reached an agreement on all outstanding issues with the FDA on a
regulatory path forward for Ceplene®. The FDA indicated that a new trial should be
conducted. As a next step, we will submit a detailed Phase III protocol. The FDA, via the Special
Protocol Assessment (SPA) procedure, will provide guidance on specific sections of the protocol.
However, it will take a considerable amount of time and money to conduct a new trial, and we may
not be able to obtain the necessary financing to see a new trial through to completion. Even if we
are able to complete a new trial, the trial may not be successful, and even if the new trial is
successful and we submit a new NDA for Ceplene®, the FDA may nevertheless again issue a
refusal to file, which means the FDA will not review our NDA, or even if it files our NDA it may
recommend that our NDA not be approved.
If the FDA accepts our submission, we may be unsuccessful in our efforts to obtain a marketing
approval from the FDA. In the event we do not obtain marketing approval, we may appeal, but such an
appeal may not be successful. A negative decision would delay or prevent us from generating revenue
from product sales of Ceplene® in the United States for the foreseeable future and may
require us to conduct additional costly and time-consuming clinical trials.
The FDA may also require additional testing for safety and efficacy. Any failure or delay in
obtaining a filing decision or an approval could prohibit or delay us from marketing product
candidates. If our product candidates do not meet applicable regulatory requirements for approval,
we may not have the financial resources to continue research and development of these product
candidates, and we may not generate significant revenues from the commercial sale of any of our
products in the U.S.
We may not be able to maintain data protection in Canada for Ceplene®, which could
limit our ability to generate revenue from sales of Ceplene® in Canada.
In November 2009, Health Canada accepted for review a New Drug Submission, or NDS, for
Ceplene® for the treatment of AML in Canada. We received a denial for data protection
for Ceplene® in Canada in the fourth quarter of 2009. We are currently appealing this
denial for data protection. In November 2010, we withdrew our application for approval of
Ceplene® in Canada. We retain the right to re-file the application at any time over the
next five years without prejudice. If we are unable to maintain data protection in Canada for
Ceplene®, we may never generate revenue from sales of Ceplene® in Canada.
Clinical trial designs that were discussed with regulatory authorities prior to their commencement
may subsequently be considered insufficient for approval at the time of application for regulatory
approval.
We or our partners discuss with and obtain guidance from regulatory authorities on clinical
trial protocols. Over the course of conducting clinical trials, circumstances may change, such as
standards of safety, efficacy or medical practice, which could affect regulatory authorities’
perception of the adequacy of any of our clinical trial designs or the data we develop from our
studies. Changes in circumstances could affect our ability to conduct clinical trials as planned.
Even with successful clinical safety and efficacy data, we may be required to conduct additional,
expensive trials to obtain regulatory approval.
We may not be able to maintain European Union regulatory approval for Ceplene®, our
lead product candidate, which could delay or prevent us from being able to generate revenue from
sales of Ceplene® and require additional expenditures.
Ceplene® is our lead product candidate and our only product candidate
currently under regulatory consideration. In October 2008, Ceplene® was
granted full marketing authorization by the European Commission, which allows
Ceplene® to be marketed in the 27 member states of the European Union, as well
as in Iceland, Liechtenstein and Norway. Ceplene® is to be administered in
conjunction with low-dose interleukin-2 (IL-2). As part of granting the marketing authorization
under Exceptional Circumstances, we have agreed to perform two post-approval clinical studies, that
have now been combined into a single clinical study. The first part of the study will seek to
further elucidate the clinical pharmacology of Ceplene® by assessing certain
biomarkers in AML patients in first remission. The second part of the study will assess the effect
of Ceplene®/IL-2 on the development of minimal residual disease in the same patient
population. We may not receive a positive outcome in this study, and our marketing authorization in
the European Union may be terminated. A negative outcome or terminated marketing authorization
would delay or prevent us from generating revenue from product sales of Ceplene®
and may require us to conduct additional costly and time-consuming clinical trials. There is
no assurance that we will be able to maintain governmental regulatory approvals to market
Ceplene® in Europe. If we are unable to maintain regulatory approval to market
Ceplene® in Europe, our business, financial condition and results of operations
would be materially and adversely affected.
19
If we receive regulatory approval, our marketed products will also be subject to ongoing FDA
and/or foreign regulatory agency obligations and continued regulatory review, and if we fail to
comply with these regulations, we could lose approvals to market any products, and our business
would be seriously harmed.
Following initial regulatory approval of any of our product candidates, we will be subject to
continuing regulatory review, including review of adverse experiences and clinical results that are
reported after our products become commercially available. This would include results from any
post-marketing tests or vigilance required as a condition of approval. The manufacturer and
manufacturing facilities we use to make any of our product candidates will also be subject to
periodic review and inspection by the FDA or foreign regulatory agencies. If a previously unknown
problem or problems with a product, manufacturing or laboratory facility used by us is discovered,
the FDA or foreign regulatory agency may impose restrictions on that product or on the
manufacturing facility, including requiring us to withdraw the product from the market. Any changes
to an approved product, including the way it is manufactured or promoted, often require FDA
approval before the product, as modified, can be marketed. We and our manufacturers will be subject
to ongoing FDA requirements for submission of safety and other post-market information. If we or
our manufacturers fail to comply with applicable regulatory requirements, a regulatory agency may:
| |
• |
|
impose civil or criminal penalties; |
| |
• |
|
suspend or withdraw regulatory approval; |
| |
• |
|
suspend any ongoing clinical trials; |
| |
• |
|
refuse to approve pending applications or supplements to approved applications; |
| |
• |
|
impose restrictions on operations; |
| |
• |
|
close the facilities of manufacturers; or |
| |
• |
|
seize or detain products or require a product recall. |
In addition, the policies of the FDA or other applicable regulatory agencies may change and
additional government regulations may be enacted that could prevent or delay regulatory approval of
our product candidates. We cannot predict the likelihood, nature, or extent of adverse government
regulation that may arise from future legislation or administrative action, either in the United
States or abroad.
Any regulatory approval we receive for our product candidates will be limited to those indications
and conditions for which we are able to show clinical safety and efficacy.
Any regulatory approval that we may receive for our current or future product candidates will
be limited to those diseases and indications for which such product candidates are clinically
demonstrated to be safe and effective. For example, in addition to the FDA approval required for
new formulations, any new indication to an approved product also requires FDA approval. If we are
not able to obtain regulatory approval for a broad range of indications for our product candidates,
our ability to effectively market and sell our product candidates may be greatly reduced and may
harm our ability to generate revenue.
Our lead product candidate, Ceplene®, which when used concomitantly with low-dose
interleukin-2, is intended only for remission maintenance therapy in the treatment of AML for adult
patients in their first complete remission. Any other indications or uses of
Ceplene® would require additional regulatory approval for us to market
Ceplene® for these indications.
While physicians may choose to prescribe drugs for uses that are not described in the
product’s labeling and for uses that differ from those tested in clinical studies and approved by
regulatory authorities, our regulatory approvals will be limited to those indications that are
specifically submitted to the regulatory agency for review. These “off-label” uses are common
across medical specialties and may constitute the best treatment for many patients in varied
circumstances. Regulatory authorities in the United States generally do not regulate the behavior
of physicians in their choice of treatments. Regulatory authorities do, however, restrict
communications by pharmaceutical companies on the subject of off-label use. If our promotional
activities fail to comply with these regulations or guidelines, we may be subject to warnings from,
or enforcement action by, these authorities. In addition, our failure
to follow regulatory rules and guidelines relating to promotion and advertising may cause the
regulatory agency to delay its approval or refuse to approve a product, the suspension or
withdrawal of an approved product from the market, recalls, fines, disgorgement of money, operating
restrictions, injunctions or criminal prosecutions, any of which could harm our business.
20
The results of our clinical trials are uncertain, which could substantially delay or prevent us
from bringing our product candidates to market.
Before we can obtain regulatory approval for a product candidate, we must undertake extensive
clinical testing in humans to demonstrate safety and efficacy to the satisfaction of the FDA or
other regulatory agencies. Clinical trials are very expensive and difficult to design and
implement. The clinical trial process is also time consuming. The commencement and completion of
our clinical trials could be delayed or prevented by several factors, including:
| |
• |
|
delays in obtaining regulatory approvals to commence or continue a study; |
| |
• |
|
delays in reaching agreement on acceptable clinical trial parameters; |
| |
• |
|
slower than expected rates of patient recruitment and enrollment; |
| |
• |
|
inability to demonstrate effectiveness or statistically significant results in our
clinical trials; |
| |
• |
|
unforeseen safety issues; |
| |
• |
|
uncertain dosing issues; |
| |
• |
|
inability to monitor patients adequately during or after treatment; and |
| |
• |
|
inability or unwillingness of medical investigators to follow our clinical protocols. |
We cannot assure you that our planned clinical trials will begin or be completed on time or at
all, or that they will not need to be restructured prior to completion. Significant delays in
clinical testing will impede our ability to commercialize our product candidates and generate
revenue from product sales and could materially increase our development costs. Completion of
clinical trials may take several years or more, but the length of time generally varies according
to the type, complexity, novelty and intended use of a drug candidate. The cost of clinical trials
may vary significantly over the life of a project as a result of differences arising during
clinical development, including:
| |
• |
|
the number of sites included in the trials; |
| |
• |
|
the length of time required to enroll suitable patient subjects; |
| |
• |
|
the number of patients that participate in the trials; |
| |
• |
|
the number of doses that patients receive; |
| |
• |
|
the duration of follow-up with the patient; |
| |
• |
|
the product candidate’s phase of development; and |
| |
• |
|
the efficacy and safety profile of the product. |
The use of FDA-approved therapeutics in certain of our pain product candidates could require us to
conduct additional preclinical studies and clinical trials, which could increase development costs
and lengthen the regulatory approval process.
Certain of our pain product candidates utilize proprietary formulations and topical delivery
technologies to administer FDA-approved pain management therapeutics. We may still be required to
conduct preclinical studies and clinical trials to determine if our product candidates are safe and
effective. In addition, we may also be required to conduct additional preclinical studies and Phase
I clinical trials to establish the safety of the topical delivery of these therapeutics and the
level of absorption of the therapeutics into the bloodstream. The FDA may also require us to
conduct clinical studies to establish that our delivery mechanisms are safer or more effective than
the existing methods for delivering these therapeutics. As a result, we may be required to conduct
complex clinical trials, which could be expensive and time-consuming and lengthen the anticipated
regulatory approval process.
21
In some instances, we rely on third parties, over which we have little or no control, to conduct
clinical trials for our products and their failure to perform their obligations in a timely or
competent manner may delay development and commercialization of our product candidates.
The nature of clinical trials and our business strategy requires us to rely on clinical
research centers and other third parties to assist us with clinical testing and certain research
and development activities, such as our agreement with Myrexis, Inc. related to the MX90745 series
of apoptosis-inducer anti-cancer compounds. As a result, our success is dependent upon the success
of these third parties in performing their responsibilities. We cannot directly control the
adequacy and timeliness of the resources and expertise applied to these activities by such third
parties. If such contractors do not perform their activities in an adequate or timely manner, the
development and commercialization of our product candidates could be delayed. In addition, we rely
on Myrexis for research and development related to the MX90745 series of apoptosis-inducer
anti-cancer compounds. We may enter into similar agreements from time to time with additional third
parties for our other product candidates whereby these third parties undertake significant
responsibility for research, clinical trials or other aspects of obtaining FDA approval. As a
result, we may face delays if Myrexis or these additional third parties do not conduct clinical
studies and trials, or prepare or file regulatory related documents, in a timely or competent
fashion. The conduct of the clinical studies by, and the regulatory strategies of, Myrexis or these
additional third parties, over which we have limited or no control, may delay or prevent regulatory
approval of our product candidates, which would delay or limit our ability to generate revenue from
product sales.
Risks Relating to Commercialization
We and our partner may not be able to successfully market and sell Ceplene®.
Even though Ceplene® was granted full marketing authorization by the European
Commission for the remission maintenance and prevention of relapse in adult patients with Acute
Myeloid Leukemia in first remission, our partner, Meda AB, may not be able to effectively market
and sell Ceplene®. We are reliant on Meda AB to generate revenue from sales of
Ceplene® in Europe and certain other countries on our behalf.
We expect to incur substantial net losses, in the aggregate and on a per share basis, for the
foreseeable future as Meda AB launches and commercializes Ceplene® and we seek FDA
approval to market Ceplene® in the United States. We are unable to predict the extent of
these future net losses, or when we may attain profitability, if at all. These net losses, among
other things, have had and will continue to have an adverse effect on our stockholders’ deficit. We
anticipate that for the foreseeable future our ability to generate revenues and achieve
profitability will be dependent on the successful commercialization of Ceplene®. If we
are unable to generate significant revenue from Ceplene®, or attain profitability, we
may not be able to sustain our operations.
Ceplene® may fail to achieve market acceptance, which could harm our business.
Even though Ceplene® was granted full marketing authorization by the European
Commission for the remission maintenance and prevention of relapse in adult patients with Acute
Myeloid Leukemia in first remission, physicians may choose not to prescribe this product, and
third-party payers may choose not to pay for it. Accordingly, we may be unable to generate
significant revenue or become profitable.
Acceptance of Ceplene® will depend on a number of factors including:
| |
• |
|
acceptance of Ceplene® by physicians and patients as a safe and effective
treatment; |
| |
• |
|
availability of reimbursement for our product from government or healthcare payors; |
| |
• |
|
cost effectiveness of Ceplene®; |
| |
• |
|
the effectiveness of our and Meda’s or collaboration partners’ sales and marketing
efforts; |
| |
• |
|
relative convenience and ease of administration; |
| |
• |
|
prevalence and severity of side effects; and |
| |
• |
|
availability of competitive products. |
If Ceplene® fails to achieve market acceptance, our business, financial condition
and results of operations would be materially and adversely affected.
22
We are dependent upon collaborative arrangements for the further development and
commercialization of Ceplene®. These collaborative arrangements may place the
development and commercialization of Ceplene® outside of our control, may require us
to relinquish important rights or may otherwise be on terms unfavorable to us.
We have entered into a collaborative arrangement with Meda to market and sell
Ceplene® in Europe and certain other countries and we may enter into other
collaborations with third parties to further develop and commercialize Ceplene®. We may
not be able to enter into collaborative arrangements on attractive terms, on a timely basis or at
all. Dependence on collaborators for the development and commercialization of Ceplene®
subjects us to a number of risks, including:
| |
• |
|
we may not be able to control the amount and timing of resources that our
collaborators devote to the development or commercialization of Ceplene® or to
their marketing and distribution, which could adversely affect our ability to obtain
milestone and royalty payments; |
| |
• |
|
disputes may arise between us and our collaborators that result in the delay or
termination of the commercialization of our product candidates or that result in costly
litigation or arbitration that diverts management’s attention and resources; |
| |
• |
|
our collaborators may experience financial difficulties; |
| |
• |
|
collaborators may not properly maintain or defend our intellectual property rights or
may use our proprietary information in such a way as to expose us to potential
litigation, jeopardize or lessen the value of our proprietary information, or weaken or
invalidate our intellectual property rights; |
| |
• |
|
business combinations or significant changes in a collaborator’s business strategy may
also adversely affect a collaborator’s willingness or ability to complete its obligations
under any arrangement; |
| |
• |
|
a collaborator could independently move forward with a competing product candidate
developed either independently or in collaboration with others, including our
competitors; and |
| |
• |
|
the collaborations may be terminated or allowed to expire, which would delay product
development and commercialization efforts. |
If our arrangement with Meda is not successful or we are not able to enter into other
collaborative arrangements on commercially attractive terms, on a timely basis or at all, or if any
of the risks occur and we are unable to successfully manage such risks, our business, financial
condition and results of operations would be materially and adversely affected.
If we fail to enter into and maintain successful strategic alliances for our product candidates,
we may have to reduce or delay our product commercialization or increase our expenditures.
Our strategy for developing, manufacturing and commercializing potential product candidates in
multiple therapeutic areas currently requires us to enter into and successfully maintain strategic
alliances with pharmaceutical companies that have product development resources and expertise,
established distribution systems and direct sales forces to advance our development programs and
reduce our expenditures on each development program and market any products that we may develop. We
have formed a strategic alliance with Myrexis with respect to the MX90745 series of
apoptosis-inducer anti-cancer compounds and with DURECT for our intellectual property for a
transdermal patch containing bupivacaine for the treatment of back pain. We may not be able to
negotiate additional strategic alliances on acceptable terms, or at all.
We have entered into collaborative arrangements with respect to marketing or selling
Ceplene® in Europe, certain Pacific rim countries and Israel. We may rely on
collaborative partners to market and sell Ceplene® in other international markets. We
cannot assure you that we will be able to enter into any more such arrangements on terms favorable
to us, or at all.
If we are unable to maintain our existing strategic alliances or establish and maintain
additional strategic alliances, we may have to limit the size or scope of, or delay, one or more of
our product development or commercialization programs, or undertake the various activities at our
own expense. In addition, our dependence on strategic alliances is subject to a number of risks,
including:
| |
• |
|
the inability to control the amount or timing of resources that our collaborators may
devote to developing the product candidates; |
| |
• |
|
the possibility that we may be required to relinquish important rights, including
intellectual property, marketing and distribution rights; |
23
| |
• |
|
the receipt of lower revenues than if we were to commercialize such products
ourselves; |
| |
• |
|
our failure to receive future milestone payments or royalties should a collaborator
fail to commercialize one of our product candidates successfully; |
| |
• |
|
the possibility that a collaborator could separately move forward with a competing
product candidate developed either independently or in collaboration with others,
including our competitors; |
| |
• |
|
the possibility that our collaborators may experience financial difficulties; |
| |
• |
|
business combinations or significant changes in a collaborator’s business strategy
that may adversely affect that collaborator’s willingness or ability to complete its
obligations under any arrangement; and |
| |
• |
|
the chance that our collaborators may operate in countries where their operations
could be negatively impacted by changes in the local regulatory environment or by
political unrest. |
If the market does not accept and use our product candidates, we will not achieve sufficient
product revenues and our business will suffer.
If we receive regulatory approval to market our product candidates, physicians, patients,
healthcare payors and the medical community may not accept and use them. The degree of market
acceptance and use of any approved products will depend on a number of factors, including:
| |
• |
|
perceptions by members of the healthcare community, including physicians, about the
safety and effectiveness of our products; |
| |
• |
|
cost effectiveness of our products relative to competing products; |
| |
• |
|
relative convenience and ease of administration; |
| |
• |
|
availability of reimbursement for our products from government or healthcare payors;
and |
| |
• |
|
effectiveness of marketing and distribution efforts by us and our licensees and
distributors. |
Because we expect to rely on sales and royalties generated by our current lead product,
Ceplene®, for a substantial portion of our product revenues for the foreseeable future,
the failure of any of these drugs to find market acceptance would harm our business and could
require us to seek additional funding to continue our other development programs.
Our product candidates could be rendered obsolete by technological change and medical
advances, which would adversely affect the performance of our business.
Our product candidates may be rendered obsolete or uneconomical by the development of medical
advances to treat the conditions that our product candidates are designed to address. Pain
management therapeutics are the subject of active research and development by many potential
competitors, including major pharmaceutical companies, specialized biotechnology firms,
universities and other research institutions. Research and development by others may render our
technology or product candidates obsolete or noncompetitive or result in treatments or cures
superior to any therapy we developed. Technological advances affecting costs of production could
also harm our ability to cost-effectively produce and sell products.
We have no manufacturing capacity and anticipate continued reliance on third parties for the
manufacture of our product candidates.
We do not currently operate manufacturing facilities for Ceplene® or any of our
product candidates. We lack the resources and the capabilities to manufacture Ceplene®
or any of our product candidates. We currently rely on one or more contract manufacturers for
Ceplene® and each product candidate to supply, store and distribute drug supplies for
commercial use and for our clinical trials. Any performance failure or delay on the part of our
existing manufacturers could result in lost sales or delay clinical development or regulatory
approval of our product candidates and their commercialization, producing additional losses and
depriving us of potential product revenues.
24
If the FDA or other regulatory agencies approve any of our product candidates for commercial
sale, the product will need to be manufactured in larger quantities. Some of our product candidates
have been manufactured in only small quantities for preclinical and clinical trials. In those
cases, our third party manufacturers may not be able to successfully increase their manufacturing
capacity in a timely or economical manner, or at all. We may be forced to identify alternative or
additional third party manufacturers, which may prove difficult because the number of potential
manufacturers is limited and the FDA must approve any replacement contractor prior to manufacturing
our products. Such approval would require new testing and compliance inspections. In addition, a
new manufacturer would have to be educated in, or develop substantially equivalent processes for,
production of our product candidates. It may be difficult or impossible for us to find a
replacement manufacturer on acceptable terms quickly, or at all. If we are unable to successfully
increase the manufacturing capacity for a drug candidate in a timely and economical manner, the
regulatory approval or commercial launch of any related products may be delayed or there may be a
shortage in supply, both of which may have an adverse effect on our business.
Our product candidates require precise, high quality manufacturing. A failure to achieve and
maintain high manufacturing standards, including the incidence of manufacturing errors, could
result in patient injury or death, product recalls or withdrawals, delays or failures in product
testing or delivery, cost overruns or other problems that could seriously hurt our business.
Manufacturers often encounter difficulties involving production yields, quality control and quality
assurance, as well as shortages of qualified personnel. These manufacturers are subject to ongoing
periodic unannounced inspection by the FDA, the U.S. Drug Enforcement Agency and corresponding
state and foreign agencies to ensure strict compliance with current Good Manufacturing Practice and
other applicable government regulations and corresponding foreign standards; however, we do not
have control over third party manufacturers’ compliance with these regulations and standards. If
one of our manufacturers fails to maintain compliance, the production of our product candidates
could be interrupted, resulting in delays, additional costs and potentially lost revenues.
Additionally, third-party manufacturers must pass a pre-approval inspection before we can obtain
marketing approval for any of our products in development.
Furthermore, our existing and future contract manufacturers may not perform as agreed or may
not remain in the contract manufacturing business for the time required to successfully produce,
store and distribute our product candidates. We may not own, or may have to share, the intellectual
property rights to such innovation. In the event of a natural disaster, equipment failure, power
failure, strike or other difficulty, we may be unable to replace our third party manufacturers in a
timely manner.
We may be the subject of costly product liability claims or product recalls, and we may be unable
to obtain or maintain insurance adequate to cover potential liabilities.
The risk of product liability is inherent in the development, manufacturing and marketing of
human therapeutic products. Regardless of merit or eventual outcome, product liability claims may
result in:
| |
• |
|
reduced revenues or a total withdrawal of a product from one or more markets; |
| |
• |
|
delays in, or failure to complete, our clinical trials; |
| |
• |
|
withdrawal of clinical trial participants; |
| |
• |
|
decreased demand for our product candidates; |
| |
• |
|
injury to our reputation; |
| |
• |
|
substantial monetary awards against us; and |
| |
• |
|
diversion of management or other resources from key aspects of our operations. |
Product liability claims could result in an FDA investigation of the safety or efficacy of our
products or our marketing programs. An FDA investigation could also potentially lead to a recall of
our products or more serious enforcement actions, or limitations on the indications for which our
products may be used, or suspension or withdrawal of approval.
We cannot be certain that the coverage limits of the insurance policies or those of our
strategic partners will be adequate. We further intend to expand our insurance coverage to include
the sale of commercial products if marketing approval is obtained for our product candidates. We
may not be able to obtain additional insurance or maintain our existing insurance coverage at a
reasonable cost or at all. If we are unable to obtain sufficient insurance at an acceptable cost or
if a claim is brought against us, whether fully covered by insurance or not, our business, results
of operations and financial condition could be materially adversely affected.
25
The coverage and reimbursement status of newly approved healthcare drugs is uncertain and failure
to obtain adequate coverage and reimbursement could limit our ability to market our products.
Our ability to commercialize any products successfully will depend in part on the extent to
which reimbursement will be available from governmental and other third-party payors, both in the
United States and in foreign markets. The amount reimbursed for our products may be insufficient to
allow them to compete effectively with products that are reimbursed at a higher level. If the price
we are able to charge for any product we develop is inadequate in light of our development costs,
our profitability would be reduced.
Reimbursement by a governmental and other third-party payor may depend upon a number of
factors, including the governmental and other third-party payor’s determination that the use of a
product is:
| |
• |
|
a covered benefit under its health plan; |
| |
• |
|
safe, effective and medically necessary; |
| |
• |
|
appropriate for the specific patient; |
| |
• |
|
neither experimental nor investigational. |
Obtaining reimbursement approval for a product from each third-party and governmental payor is
a time consuming and costly process that could require us to provide supporting scientific,
clinical and cost effectiveness data for the use of our products to each payor. We may not be able
to provide data sufficient to obtain reimbursement.
Eligibility for coverage does not imply that any drug product will be reimbursed in all cases
or at a rate that allows us to make a profit. Interim payments for new products, if applicable, may
also not be sufficient to cover our costs and may not become permanent. Reimbursement rates may
vary according to the use of the drug and the clinical setting in which it is used, may be based on
payments allowed for lower-cost drugs that are already reimbursed, may be incorporated into
existing payments for other products or services and may reflect budgetary constraints and/or
Medicare or Medicaid data used to calculate these rates. Net prices for products also may be
reduced by mandatory discounts or rebates required by government health care programs or by any
future relaxation of laws that restrict imports of certain medical products from countries where
they may be sold at lower prices than in the United States.
The health care industry is experiencing a trend toward containing or reducing costs through
various means, including lowering reimbursement rates, limiting therapeutic class coverage and
negotiating reduced payment schedules with service providers for drug products. There have been,
and we expect that there will continue to be, federal and state proposals to constrain expenditures
for medical products and services, which may affect reimbursement levels for our future products.
In addition, the Centers for Medicare and Medicaid Services frequently change product descriptors,
coverage policies, product and service codes, payment methodologies and reimbursement values.
Third-party payors often follow Medicare coverage policies and payment limitations in setting their
own reimbursement rates and may have sufficient market power to demand significant price
reductions.
Foreign governments tend to impose strict price controls, which may adversely affect our future
profitability.
In some foreign countries, particularly in the European Union, prescription drug pricing is
subject to governmental control. In these countries, pricing negotiations with governmental
authorities can take considerable time after the receipt of marketing approval for a product. To
obtain reimbursement or pricing approval in some countries, we may be required to conduct a
clinical trial that compares the cost-effectiveness of our product candidates to other available
therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if
pricing is set at unsatisfactory levels, our profitability would be reduced.
26
Risks Relating to Our Business and Industry
Our failure to attract and retain skilled personnel could impair our product development and
commercialization efforts.
Our success is substantially dependent on our continued ability to attract, retain and
motivate highly qualified management, scientific and technical personnel and our ability to develop
and maintain important relationships with leading institutions, clinicians and scientists. We are
highly dependent upon our key management personnel, particularly John V. Talley, our President and
Chief Executive Officer, Robert W. Cook, our Senior Vice President and Chief Financial Officer, Dr.
Stephane Allard, our
Chief Medical Officer and Dr. Dileep Bhagwat, our Senior Vice President, Pharmaceutical
Development. We are also dependent on certain scientific and technical personnel. The loss of the
services of any member of senior management, or scientific or technical staff may significantly
delay or prevent the achievement of product development, commercialization and other business
objectives. Messrs. Talley and Cook have entered into employment agreements with us. However,
either of them may decide to voluntarily terminate his employment with us. We do not maintain
key-man life insurance on any of our employees.
We believe that we will need to recruit additional management and technical personnel. There
is currently a shortage of, and intense competition for, skilled executives and employees with
relevant scientific and technical expertise, and this shortage may continue. The inability to
attract and retain sufficient scientific, technical and managerial personnel could limit or delay
our product development efforts, which would reduce our ability to successfully commercialize
product candidates and our business.
Our competitors may develop and market drugs that are less expensive, safer, or more effective,
which may diminish or eliminate the commercial success of any of our product candidates.
The biotechnology and pharmaceutical industries are highly competitive and characterized by
rapid technological change. Because we anticipate that our research approach will integrate many
technologies, it may be difficult for us to stay abreast of the rapid changes in technology. If we
fail to stay at the forefront of technological change, we will be unable to compete effectively.
Our competitors may render our technologies obsolete by advances in existing technological
approaches or the development of different approaches by one or more of our current or future
competitors.
We will compete with Pfizer and Endo in the treatment of neuropathic pain. There are also many
companies, both publicly and privately held, including well-known pharmaceutical companies and
academic and other research institutions, engaged in developing pharmaceutical products for the
treatment of life-threatening cancers and diseases.
Our competitors may:
| |
• |
|
develop and market product candidates that are less expensive and more effective than
our future product candidates; |
| |
• |
|
adapt more quickly to new technologies and scientific advances; |
| |
• |
|
commercialize competing product candidates before we or our partners can launch any
product candidates developed from our product candidates; |
| |
• |
|
initiate or withstand substantial price competition more successfully than we can; |
| |
• |
|
have greater success in recruiting skilled scientific workers from the limited pool
of available talent; |
| |
• |
|
more effectively negotiate third-party licenses and strategic alliances; and |
| |
• |
|
take advantage of acquisition or other opportunities more readily than we can. |
We will compete for market share against fully-integrated pharmaceutical companies and smaller
companies that are collaborating with larger pharmaceutical companies, new companies, academic
institutions, government agencies and other public and private research organizations. Many of
these competitors, either alone or together with their partners, may develop new product candidates
that will compete with our product candidates, as these competitors may operate larger research and
development programs or have substantially greater financial resources than us. Our competitors may
also have significantly greater experience in:
| |
• |
|
undertaking preclinical testing and human clinical trials; |
| |
• |
|
building relationships with key customers and opinion-leading physicians; |
| |
• |
|
obtaining and maintaining FDA and other regulatory approvals of drugs; |
| |
• |
|
formulating and manufacturing drugs; and |
| |
• |
|
launching, marketing and selling drugs. |
These and other competitive factors may negatively impact our financial performance.
27
EpiCept GmbH, our German subsidiary, is subject to various risks associated with its international
operations.
Our subsidiary, EpiCept GmbH, operates in Germany, and we face a number of risks associated
with its operations, including:
| |
• |
|
difficulties and costs associated in complying with German laws and regulations; |
| |
• |
|
changes in the German regulatory environment; |
| |
• |
|
increased costs associated with operating in Germany; |
| |
• |
|
increased costs and complexities associated with financial reporting; and |
| |
• |
|
difficulties in maintaining international operations. |
Expenses incurred by our German operations are typically denominated in euros. In addition,
EpiCept GmbH has incurred indebtedness that is denominated in euros and requires that interest be
paid in euros. As a result, our costs of maintaining and operating our German subsidiary, and the
interest payments and costs of repaying its indebtedness, increase if the value of the U.S. dollar
relative to the euro declines.
Risks Relating to Intellectual Property
If we are unable to protect our intellectual property, our competitors could develop and market
products with features similar to our products and demand for our products may decline.
Our commercial success will depend in part on obtaining and maintaining patent protection and
trade secret protection of our technologies and product candidates as well as successfully
defending these patents and trade secrets against third party challenges. We will only be able to
protect our intellectual property from unauthorized use by third parties to the extent that valid
and enforceable patents or trade secrets cover them.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and
involve complex legal and factual questions for which important legal principles remain unresolved.
In addition, changes in either the patent laws or in interpretations of patent laws in the United
States or other countries may diminish the value of our intellectual property. Accordingly, we
cannot predict the breadth of claims that may be allowed or enforced in our patents or in third
party patents.
The degree of future protection for our proprietary rights is uncertain because legal means
afford only limited protection and may not adequately protect our rights or permit us to gain or
keep our competitive advantage. For example:
| |
• |
|
we might not have been the first to make the inventions covered by each of its
pending patent applications and issued patents, and we could lose our patent rights as a
result; |
| |
• |
|
we might not have been the first to file patent applications for these inventions or
our patent applications may not have been timely filed, and we could lose our patent
rights as a result; |
| |
• |
|
others may independently develop similar or alternative technologies or duplicate any
of our technologies; |
| |
• |
|
it is possible that none of our pending patent applications will result in issued
patents; |
| |
• |
|
our issued patents may not provide a basis for commercially viable drugs or
therapies, may not provide us with any protection from unauthorized use of our
intellectual property by third parties, and may not provide us with any competitive
advantages; |
| |
• |
|
our patent applications or patents may be subject to interference, opposition or
similar administrative proceedings; |
| |
• |
|
we may not develop additional proprietary technologies that are patentable; or |
| |
• |
|
the patents of others may have an adverse effect on our business. |
Moreover, the issuance of a patent is not conclusive as to its validity or enforceability and
it is uncertain how much protection, if any, will be afforded by our patents if we attempt to
enforce them and they are challenged in court or in other proceedings, such as oppositions, which
may be brought in U.S. or foreign jurisdictions to challenge the validity of a patent. A third
party may challenge the validity or enforceability of a patent after its issuance by the U.S.
Patent and Trademark Office, or USPTO.
28
The defense and prosecution of intellectual property suits, interferences, oppositions and
related legal and administrative proceedings in the United States are costly, time consuming to
pursue and result in diversion of resources. The outcome of these proceedings is uncertain and
could significantly harm our business.
We will also rely on trade secrets to protect our technology, especially where we do not
believe patent protection is appropriate or obtainable. However, trade secrets are difficult to
protect. We will use reasonable efforts to protect our trade secrets, our employees, consultants,
contractors, outside scientific partners and other advisors may unintentionally or willfully
disclose its confidential information to competitors. Enforcing a claim that a third party
improperly obtained and is using our trade secrets is expensive and time consuming, and the outcome
is unpredictable. In addition, courts outside the United States are sometimes less willing to
protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge,
methods and know-how.
If we are not able to defend the patent protection position of our technologies and product
candidates, then we will not be able to exclude competitors from marketing product candidates that
directly compete with our product candidates, and we may not generate enough revenue from our
product candidates to justify the cost of their development and to achieve or maintain
profitability.
If we are sued for infringing intellectual property rights of third parties, such litigation will
be costly and time consuming, and an unfavorable outcome could increase our costs or have a
negative impact on our business.
Our ability to commercialize our products depends on our ability to sell our products without
infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and
pending applications, which are owned by third parties, exist with respect to the therapeutics
utilized in our product candidates and topical delivery mechanisms. Because we are utilizing
existing therapeutics, we will continue to need to ensure that we can utilize these therapeutics
without infringing existing patent rights. Accordingly, we have reviewed related patents known to
us and, in some instances, licensed related patented technologies. In addition, because patent
applications can take several years to issue, there may be currently pending applications, unknown
to us, which may later result in issued patents that the combined organization’s product candidates
may infringe. There could also be existing patents of which we are not aware that our product
candidates may inadvertently infringe.
We cannot assure you that any of our product candidates does not infringe the intellectual
property of others. There is a substantial amount of litigation involving patent and other
intellectual property rights in the biotechnology and biopharmaceutical industries generally. If a
third party claims that we infringe on their technology, we could face a number of issues that
could increase its costs or have a negative impact on its business, including:
| |
• |
|
infringement and other intellectual property claims which, with or without merit, can
be costly and time consuming to litigate and can delay the regulatory approval process
and divert management’s attention from our core business strategy; |
| |
• |
|
substantial damages for past infringement, which we may have to pay if a court
determines that our products infringes a competitor’s patent; |
| |
• |
|
an injunction prohibiting us from selling or licensing our product unless the patent
holder licenses the patent to us, which the holder is not required to do; and |
| |
• |
|
if a license is available from a patent holder, we may have to pay substantial
royalties or grant cross licenses to our patents. |
We may be subject to damages resulting from claims that our employees have wrongfully used or
disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at other biotechnology or pharmaceutical
companies, including competitors or potential competitors. We may be subject to claims that we or
these employees have inadvertently or otherwise used or disclosed trade secrets or other
proprietary information of their former employers. Litigation may be necessary to defend against
these claims. If we fail in defending such claims, in addition to paying monetary claims, we may
lose valuable intellectual property rights or personnel. A loss of key research personnel or their
work product could hamper or prevent our ability to commercialize certain product candidates, which
could severely harm our business. Litigation could result in substantial costs and be a distraction
to management.
29
Risks Relating to our Common Stock
Our common stock may be delisted from The Nasdaq Capital Market or the Nasdaq OMX Stockholm
Exchange, which may make it more difficult for you to sell your shares.
In October 2010, we received a letter from the Nasdaq Listing Qualifications Department
stating that we are not in compliance with the continued listing requirements of The Nasdaq Capital
Market because the bid price of our common stock had closed below the minimum $1.00 per share
requirement for 30 consecutive business days. We have been provided a period of 180 calendar days,
or until April 4, 2011, to regain compliance with the minimum bid price rule. If at any time
before April 4, 2011, the bid price of our common stock closes at $1.00 per share or higher for a
period determined by Nasdaq (which shall be a minimum of 10 consecutive business days), Nasdaq will
provide us with written notification that we are in compliance with the rule.
In the event that we do not regain compliance with the minimum bid price rule by April 4,
2011, Nasdaq will determine whether we meet the initial listing criteria, with the exception of bid
price, for The Nasdaq Capital Market and, if we do, we will be granted an additional compliance
period of 180 calendar days.
In the event that we are not eligible for the minimum bid price additional compliance period,
we will have the right to appeal a determination to delist our common stock. Our common stock
would remain listed on The Nasdaq Capital Market until the completion of the appeal process.
Delisting of our common stock by The Nasdaq Capital Market may result in the delisting of our
common stock on the Nasdaq OMX Stockholm Exchange in Sweden and the delisting of our common stock
on The Nasdaq Capital Market or the Nasdaq OMX Stockholm Exchange would adversely affect the market
price and liquidity of our common stock and warrants, your ability to sell your shares of our
common stock and our ability to raise capital.
We expect that our stock price will fluctuate significantly due to external factors, which could
cause the value of your investment to decline.
Securities markets worldwide have experienced, and are likely to continue to experience,
significant price and volume fluctuations. This market volatility, as well as general economic,
market or political conditions, could reduce the market price of our common stock regardless of our
operating performance.
Since January 30, 2007, our common stock trades on The Nasdaq Capital Market and on the Nasdaq
OMX Stockholm Exchange. From January 5, 2006 through January 29, 2007, our common stock traded on
The Nasdaq National Market. Prior to January 4, 2006, our common stock did not trade on an
exchange. Sales of substantial amounts of our common stock in the public market through equity
financing or otherwise could adversely affect the prevailing market prices of the common stock and
our ability to raise equity capital in the future. In particular, as of March 30, 2011 we have
outstanding warrants to purchase approximately 33.4 million shares of our common stock, and the
market price of our common stock could decline as a result of exercises or sales by our existing
warrant holders in the market or the perception that these exercises or sales could occur. These
exercises or sales might also make it more difficult for us to sell equity securities at a time and
price that we deem appropriate.
If securities or industry analysts do not publish research or reports about us, if they change
their recommendations regarding our stock adversely or if our operating results do not meet their
expectations, our stock price and trading volume could decline.
The trading market for our common stock is influenced by the research and reports that
industry or securities analysts publish about us. If one or more of these analysts cease coverage
of us or fail to regularly publish reports on us, we could lose visibility in the financial
markets, which in turn could cause our stock price or trading volume to decline. Moreover, if one
or more of the analysts who covers us downgrades our stock or if our operating results do not meet
their expectations, our stock price could decline.
Future sales of common stock may cause our stock price to fall.
As of March 30, 2011, we have outstanding and exercisable warrants to purchase approximately
17.0 million shares of our common stock with exercise prices ranging from $0.75 — $2.08. We also
have a significant number of warrants outstanding with exercise prices ranging from $2.58 — $5.64
that are currently exercisable. The market price of our common stock could decline as a result of
exercises or sales by our existing warrant holders and stockholders in the market or the perception
that these exercises or sales could occur. These sales might also make it more difficult for us to
sell equity securities or convertible debt securities at a time and price that we deem appropriate.
30
Provisions of our charter documents or Delaware law could delay or prevent an acquisition of us,
even if the acquisition would be beneficial to our stockholders, and could make it more difficult
for you to change management.
Provisions of our certificate of incorporation and bylaws may discourage, delay or prevent a
merger, acquisition or other change in control that stockholders may consider favorable, including
transactions in which stockholders might otherwise receive a premium for their shares. This is
because these provisions may prevent or frustrate attempts by stockholders to replace or remove our
management. These provisions include:
| |
• |
|
a classified board of directors; |
| |
• |
|
a prohibition on stockholder action through written consent; |
| |
• |
|
a requirement that special meetings of stockholders be called only by the board of
directors or a committee duly designated by the board of directors whose powers and
authorities include the power to call such special meetings; |
| |
• |
|
advance notice requirements for stockholder proposals and nominations; and |
| |
• |
|
the authority of the board of directors to issue preferred stock with such terms as
the board of directors may determine. |
In addition, Section 203 of the Delaware General Corporation Law prohibits a publicly held
Delaware corporation from engaging in a business combination with an interested stockholder,
generally a person that together with its affiliates owns or within the last three years has owned
15% of voting stock, for a period of three years after the date of the transaction in which the
person became an interested stockholder, unless the business combination is approved in a
prescribed manner. Accordingly, Section 203 may discourage, delay or prevent a change in control of
us.
As a result of these provisions in our charter documents and Delaware law, the price investors
may be willing to pay in the future for shares of our common stock may be limited.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash
dividends in the foreseeable future.
We have never paid cash dividends on any of our classes of capital stock to date, and we
intend to retain our future earnings, if any, to fund the development and growth of our business.
In addition, the terms of existing or any future debt may preclude us from paying these dividends.
As a result, capital appreciation, if any, of our common stock will be your sole source of gain for
the foreseeable future.
The requirements of being a public company may strain our resources and distract management.
As a public company, we are subject to the reporting requirements of the Exchange Act, the
Sarbanes-Oxley Act and the listing requirements of The Nasdaq Capital Market and the Nasdaq OMX
Stockholm Exchange. The obligations of being a public company require significant additional
expenditures and place additional demands on our management as we comply with the reporting
requirements of a public company. We may need to hire additional accounting and financial staff
with appropriate public company experience and technical accounting knowledge.
31
|
|
|
| ITEM 1B. |
|
UNRESOLVED STAFF COMMENTS |
We have no written comments regarding our periodic or current reports from the staff of the
Securities and Exchange Commission that were issued 180 days or more preceding the end of our 2010
fiscal year that remain unresolved.
We lease approximately 10,000 square feet located at 777 Old Saw Mill River Road, Tarrytown,
NY; the lease expires in February 2012. We also lease approximately 3,000 square feet in Munich,
Germany; the lease expires in July 2014. We currently lease approximately 23,500 rentable square
feet of laboratory and office space in San Diego, California that we are attempting to sublease. We
believe that our existing facilities will be adequate to accommodate our business needs for the
foreseeable future.
|
|
|
| ITEM 3. |
|
LEGAL PROCEEDINGS |
None.
|
|
|
| ITEM 4. |
|
REMOVED AND RESERVED |
32
PART II
|
|
|
| ITEM 5. |
|
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF
EQUITY SECURITIES |
Price Range of Common Stock
Our common stock is traded on both The Nasdaq Capital Market and the Nasdaq OMX Stockholm Exchange
under the symbol “EPCT.” The following table sets forth the range of high and low sales prices per
share for the common stock as reported on The Nasdaq Capital Market during the periods indicated.
For ease of comparison, all of the prices in the following table have been adjusted to reflect the
1:3 reverse split of our common stock, which was effective for the start of trading on January 15,
2010, as if such reverse split had been in effect during the periods indicated. Prior to January
4, 2006, all of our common stock, par value $.0001 per share, was privately held. We have never
declared or paid dividends on our common stock and we do not anticipate paying any cash dividends
on our capital stock in the foreseeable future. We currently intend to retain all available funds
and any future earnings to fund the development and growth of our business.
| |
|
|
|
|
|
|
|
|
| |
|
Price Range |
|
| |
|
High |
|
|
Low |
|
For Year Ended December 31, 2010 |
|
|
|
|
|
|
|
|
First Quarter |
|
$ |
2.73 |
|
|
$ |
1.74 |
|
Second Quarter |
|
|
2.12 |
|
|
|
0.95 |
|
Third Quarter |
|
|
1.15 |
|
|
|
0.52 |
|
Fourth Quarter |
|
|
1.09 |
|
|
|
0.33 |
|
| |
|
|
|
|
|
|
|
|
| |
|
Price Range |
|
| |
|
High |
|
|
Low |
|
For Year Ended December 31, 2009 |
|
|
|
|
|
|
|
|
First Quarter |
|
$ |
2.82 |
|
|
$ |
1.26 |
|
Second Quarter |
|
|
3.75 |
|
|
|
1.29 |
|
Third Quarter |
|
|
3.06 |
|
|
|
1.68 |
|
Fourth Quarter |
|
|
2.73 |
|
|
|
1.68 |
|
The high and low sales prices for the Common Stock during the first quarter of 2011 (through
March 28, 2011) were $0.93 and $0.57 respectively. The closing price on March 28, 2011 was $0.69.
33
Performance Graph
The following graph and table compare the cumulative total return of our common stock, the
Nasdaq Composite Index and the Nasdaq Biotechnology Index, as described below, for the period
beginning January 4, 2006 (the date we became a public company) and ending December 31, 2010,
assuming an initial investment of $100 and the reinvestment of any dividends. We obtained the
information reflected in the graph and table from independent sources we believe to be reliable,
but we have not independently verified the information.
COMPARISON
OF CUMULATIVE TOTAL RETURN
ASSUMES
$100 INVESTED ON JAN. 06, 2006
ASSUMES DIVIDEND REINVESTED
FISCAL YEAR ENDING DEC. 31, 2010
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Period Ending |
|
| Company/Market/Peer Group |
|
1/5/2006 |
|
|
12/31/2006 |
|
|
12/31/2007 |
|
|
12/31/2008 |
|
|
12/31/2009 |
|
|
12/31/2010 |
|
EpiCept Corporation |
|
$ |
100.00 |
|
|
$ |
16.82 |
|
|
$ |
14.94 |
|
|
$ |
7.53 |
|
|
$ |
6.82 |
|
|
$ |
3.41 |
|
NASDAQ Composite |
|
$ |
100.00 |
|
|
$ |
109.92 |
|
|
$ |
121.51 |
|
|
$ |
72.88 |
|
|
$ |
105.90 |
|
|
$ |
124.98 |
|
NASDAQ Biotechnology |
|
$ |
100.00 |
|
|
$ |
98.48 |
|
|
$ |
103.05 |
|
|
$ |
90.37 |
|
|
$ |
104.80 |
|
|
$ |
120.78 |
|
Performance Graph and related information shall not be deemed “soliciting material” or to be
“filed” with the Securities and Exchange Commission, nor shall such information be incorporated by
reference into any future filing under the Securities Act or Exchange Act, each as amended, except
to the extent that we specifically incorporate it by reference into such filing.
34
|
|
|
| ITEM 6. |
|
SELECTED FINANCIAL DATA |
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Years Ended December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006(1) |
|
| |
|
(Dollars in thousands, except share and per share data) |
|
Consolidated Statements of Operations Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue |
|
$ |
994 |
|
|
$ |
414 |
|
|
$ |
265 |
|
|
$ |
327 |
|
|
$ |
2,095 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Costs of goods sold |
|
|
997 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
General and administrative |
|
|
7,244 |
|
|
|
7,548 |
|
|
|
9,599 |
|
|
|
11,759 |
|
|
|
14,242 |
|
Research and development |
|
|
8,127 |
|
|
|
11,603 |
|
|
|
12,623 |
|
|
|
15,312 |
|
|
|
15,675 |
|
Acquired in-process research and development |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
33,362 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total costs and expenses |
|
|
16,368 |
|
|
|
19,151 |
|
|
|
22,222 |
|
|
|
27,071 |
|
|
|
63,279 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(15,374 |
) |
|
|
(18,737 |
) |
|
|
(21,957 |
) |
|
|
(26,744 |
) |
|
|
(61,184 |
) |
Other expense, net |
|
|
(159 |
) |
|
|
(20,073 |
)(5) |
|
|
(3,422 |
) |
|
|
(1,945 |
) |
|
|
(4,269 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss before (expense)/benefit for income taxes |
|
|
(15,533 |
) |
|
|
(38,810 |
) |
|
|
(25,379 |
) |
|
|
(28,689 |
) |
|
|
(65,453 |
) |
Income tax (expense)/benefit |
|
|
(4 |
) |
|
|
(4 |
) |
|
|
(3 |
) |
|
|
(4 |
) |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
(15,537 |
) |
|
|
(38,814 |
) |
|
|
(25,382 |
) |
|
|
(28,693 |
) |
|
|
(65,453 |
) |
Deemed dividend and redeemable convertible
preferred stock dividends |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(8,963 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss attributable to common stockholders |
|
$ |
(15,537 |
) |
|
$ |
(38,814 |
) |
|
$ |
(25,382 |
) |
|
$ |
(28,693 |
) |
|
$ |
(74,416 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted loss per common share(2) |
|
$ |
(0.32 |
) |
|
$ |
(0.97 |
) |
|
$ |
(1.23 |
) |
|
$ |
(2.37 |
) |
|
$ |
(9.21 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding(2) |
|
|
47,853,560 |
|
|
|
40,139,299 |
|
|
|
20,685,710 |
|
|
|
12,129,258 |
|
|
|
8,077,625 |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
As of December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
2007 |
|
|
2006(1) |
|
| |
|
(Dollars in thousands) |
|
Consolidated Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
2,435 |
|
|
$ |
5,142 |
|
|
$ |
790 |
|
|
$ |
4,943 |
|
|
$ |
14,097 |
|
Working capital (deficit) |
|
|
(1,887 |
) |
|
|
1,407 |
|
|
|
(8,535 |
) |
|
|
(8,208 |
)(3) |
|
|
(4,481 |
)(3) |
Total assets |
|
|
4,689 |
|
|
|
7,514 |
|
|
|
2,271 |
|
|
|
7,398 |
|
|
|
18,426 |
|
Long-term debt |
|
|
448 |
|
|
|
967 |
|
|
|
277 |
|
|
|
375 |
|
|
|
447 |
|
Accumulated deficit |
|
|
(250,582 |
) |
|
|
(235,045 |
) |
|
|
(196,231 |
) |
|
|
(170,849 |
) |
|
|
(142,156 |
)(4) |
Total stockholders’ deficit |
|
|
(14,135 |
) |
|
|
(9,079 |
) |
|
|
(17,730 |
) |
|
|
(14,177 |
) |
|
|
(9,373 |
) |
| |
|
|
| (1) |
|
On January 4, 2006, we completed our merger with Maxim Pharmaceuticals, Inc. |
| |
| (2) |
|
On January 4, 2006, there was a one-for-four reverse stock split and on January 14, 2010
there was a one-for-three reverse stock split. All prior periods have been retroactively
adjusted to reflect the reverse stock splits. |
| |
| (3) |
|
Our debt owed to Hercules of $7.3 million and $10.0 million at December 31, 2007 and 2006,
respectively, which matured on April 1, 2009 contained a subjective acceleration clause and
accordingly was classified as a current liability in accordance with Financial Accounting
Standard Board, or FASB, Accounting Standards Codification, or ASC, 470-10, “Debt — Overall”,
or ASC 470-10. |
| |
| (4) |
|
Includes the in-process research and development of $33.4 million acquired upon the
completion of our merger with Maxim Pharmaceuticals, Inc. on January 4, 2006 and the
beneficial conversion features of $8.6 million related to the conversion of certain of our
notes outstanding and preferred stock into our common stock and from certain anti-dilution
adjustments to our preferred stock as a result of the exercise of the bridge warrants. |
| |
| (5) |
|
Amortization of debt issuance costs and discount and interest expense were both accelerated
in 2009 as a result of the conversion of $24.5 million of our 7.5556% convertible subordinated
notes due 2014 into approximately 9.1 million shares of our common stock in 2009. Interest
expense for the year ended December 31, 2009 included $10.5 million in amortization of debt
issuance costs and discount and $9.3 million in interest expense paid from escrowed cash that
represented the make-whole interest payment that would have been due at maturity of such
notes. |
35
|
|
|
| ITEM 7. |
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion of our financial condition and results of operations
in conjunction with our consolidated financial statements and the related notes included elsewhere
in this report. This discussion contains forward-looking statements that involve risks and
uncertainties. As a result of many factors, including those set forth under the section entitled
“Risk Factors” and elsewhere in this report, our actual results may differ materially from those
anticipated in these forward-looking statements.
Overview
We are a specialty pharmaceutical company focused on the clinical development and
commercialization of pharmaceutical products for the treatment of cancer and pain. We focus our
clinical development efforts on innovative cancer therapies and topically delivered analgesics
targeting peripheral nerve receptors. Our lead product is Ceplene®, which when used
concomitantly with low-dose interleukin-2, or IL-2, is intended as remission maintenance therapy in
the treatment of acute myeloid leukemia, or AML, for adult patients who are in their first complete
remission. In addition to Ceplene®, we have two other oncology compounds and a
late-stage pain product candidate for the treatment of peripheral neuropathies in clinical
development. We believe this portfolio of oncology and pain management product candidates lessens
our reliance on the success of any single product or product candidate.
In October 2008, the European Commission issued a formal marketing authorization for
Ceplene® in the European Union, or EU. In January 2010 we completed an
agreement with Meda AB of Sweden to market and sell Ceplene® in Europe and certain Pacific Rim
countries. The commercial launch of Ceplene® commenced in the United Kingdom in April
2010 and launches continue in other European countries. In December 2010, Ceplene® was
approved for marketing in Israel. Megapharm Ltd. is our partner in Israel and upon Ministry of
Health approval of labeling and other technical matters we expect to launch Ceplene® in
the first half of 2011.
In November 2009, Health Canada screened and accepted for review a New Drug Submission (“NDS”)
for Ceplene® for remission maintenance treatment of AML in Canada. During the third
quarter of 2010, our appeal of a decision by Health Canada not to provide data protection for
Ceplene® was denied by a federal court in Canada. Lack of data protection in Canada for an
innovative drug such as Ceplene® eliminates any right to sales exclusivity by us and enables
competition to seek approval to sell generic equivalents immediately. In November 2010, we
appealed the court’s decision and withdrew our New Drug Submission until the appeals process is
completed. We retain the right to re-file the application at any time over the next five years
without prejudice.
In June 2010, we filed a New Drug Application, or NDA, with the United States Food and Drug
Administration, or FDA for Ceplene® in the same indication. In August 2010, we received
a refusal to file letter from the FDA. In the FDA’s preliminary review of the Ceplene®
NDA, the FDA concluded that the application did not establish Ceplene’s therapeutic contribution in
its combination with IL-2, and recommended that an additional pivotal trial assessing Ceplene’s
contribution and using overall survival as a primary endpoint be conducted. In October 2010, we
reached an agreement with the FDA for a two-arm, randomized, open-label trial that will compare the
efficacy of Ceplene® plus low-dose IL-2 to standard of care in this indication. Based on FDA
guidance, the primary endpoint of the trial will be overall patient survival. Our previous Phase
III trial demonstrated a statistically significant prolongation of the primary endpoint of
leukemia-free survival and extended overall survival by more than one year in patients in their
first complete remission. As a next step, we will submit a detailed Phase III protocol. The FDA,
via the Special Protocol Assessment (SPA) procedure, will provide guidance on specific sections of
the protocol including the adaptive design with prospectively defined rules for sample size
selection for both futility and expected success. We also recently filed an application with the
FDA to grant Ceplene® fast track status. In addition to other benefits, if granted, this should
permit an expedited review of the Ceplene® New Drug Application (NDA) once it is filed.
Our other oncology compounds include crolibulinTM, a novel small molecule vascular
disruption agent, or VDA, and apoptosis inducer for the treatment of patients with solid tumors.
In December 2010, the National Cancer Institute (NCI) initiated a Phase II trial with
crolibulinTM. The trial will assess the drug’s efficacy and safety in combination with
cisplatin in patients with anaplastic thyroid cancer (ATC). AzixaTM, an apoptosis
inducer with VDA activity licensed by us to Myrexis, Inc., or Myrexis, as part of an exclusive,
worldwide development and commercialization agreement, is currently in Phase II clinical trials in
patients with primary glioblastoma, and cancer that has metastasized to the brain. In
December 2010, Myrexis began a Phase IIb clinical study of AzixaTM to treat glioblastoma
multiforme, an aggressive brain tumor, in order to evaluate AzixaTM combination therapy
as a first-line GBM treatment. The study will enroll as many as 120 patients in the U.S. and
India. AzixaTM has received orphan drug status in the United States for the treatment
of glioblastoma.
36
Our late-stage pain product candidate, EpiCeptTM NP-1 cream, which we refer to as
NP-1, is a prescription topical analgesic cream designed to provide effective long-term relief of
pain associated with peripheral neuropathies. In February 2011, we presented data from our Phase
IIb trial for EpiCept™ NP-1 in chemotherapy-induced peripheral neuropathy (CPN) which is being
conducted by National Cancer Institute (NCI). CPN may affect 50% of women undergoing treatment for
breast cancer. A common therapeutic agent for the treatment of advanced breast cancer is
paclitaxel, and as many as 80% of the patients with advanced breast cancer experience some signs
and symptoms of CPN, such as burning, tingling pain associated sometimes with mild muscular
weakness, after high dose paclitaxel administration. The multi-center, double-blind, randomized,
placebo-controlled study was conducted within a network of approximately 25 sites under the
direction of the NCI funded Community Clinical Oncology Program (CCOP). More than 460 cancer
survivors suffering from painful CPN were enrolled in the six-week study. The results of the trial
in the intent to treat (ITT) population demonstrated that the change in average daily neuropathy
intensity scores in the NP-1 group achieved a statistically significant reduction in CPN intensity
versus placebo (p<0.001), which was the trial’s primary endpoint. Additionally, a pre-specified
subgroup of the ITT population, those patients who previously received taxane chemotherapy, also
showed a statistically significant reduction in average daily neuropathy intensity scores
(p=0.034). This subgroup constituted more than 50% of the ITT population. Secondary efficacy
endpoints confirmed the superiority of NP-1 vs. placebo. Furthermore, the safety profile of NP-1
was comparable to placebo.
Ceplene® has been granted full marketing authorization by the European Commission
for the remission maintenance and prevention of relapse in adult patients with Acute Myeloid
Leukemia in first remission. None of our other drug candidates has received FDA or foreign
regulatory marketing approval. In order to grant marketing approval, the FDA or foreign regulatory
agencies must conclude that our clinical data and that of our collaborators establish the safety
and efficacy of our drug candidates. Furthermore, our strategy includes entering into collaborative
arrangements with third parties to participate in the clinical development and commercialization of
our products. In the event that third parties have control over the preclinical development or
clinical trial process for a product candidate, the estimated completion date would largely be
under control of that third party rather than under our control. We cannot forecast with any degree
of certainty which of our drug candidates will be subject to future collaborations or how such
arrangements would affect our development plan or capital requirements.
We are subject to a number of risks associated with companies in the specialty pharmaceutical
industry. Principal among these are risks associated with our ability to obtain regulatory approval
for our product candidates, our ability to adequately fund our operations, dependence on
collaborative arrangements, the development by us or our competitors of new technological
innovations, dependence on key personnel, protection of proprietary technology and compliance with
the FDA and other governmental regulations. We have yet to generate significant product revenues
from any of our product candidates. We have financed our operations primarily through the proceeds
from the sale of common stock, warrants, debt instruments, cash proceeds from collaborative
relationships and investment income earned on cash balances and short-term investments.
We have prepared our consolidated financial statements under the assumption that we are a
going concern. We have devoted substantially all of our cash resources to research and development
programs and selling, general and administrative expenses, and to date we have not generated any
significant revenues from the sale of products. Since inception, we have incurred significant net
losses each year. As a result, we have an accumulated deficit of $250.6 million as of December 31,
2010. Our recurring losses from operations and the accumulated deficit raise substantial doubt
about our ability to continue as a going concern. The financial statements do not include any
adjustments that might result from the outcome of this uncertainty. Our losses have resulted
principally from costs incurred in connection with our development activities and from selling,
general and administrative expenses. Even if we succeed in developing and commercializing one or
more of our product candidates, we may never become profitable. We expect to continue to incur
significant expenses over the next several years as we:
| |
• |
|
apply for marketing approval of Ceplene® in the U.S., Canada, and other
countries; |
| |
• |
|
continue to conduct clinical trials for our product candidates; |
| |
• |
|
seek regulatory approvals for our product candidates; |
| |
• |
|
develop, formulate, and commercialize our product candidates; |
| |
• |
|
implement additional internal systems and develop new infrastructure; |
| |
• |
|
acquire or in-licenses additional products or technologies or expand the use of our
technologies; |
| |
• |
|
maintain, defend and expand the scope of our intellectual property; and |
| |
• |
|
hire additional personnel. |
Cash at December 31, 2010, plus $4.6 million of cash received in March 2011 from the issuance
of approximately 7.1 million shares of our common stock and warrants to purchase 5.3 million shares
of our common stock and $7.1 million of cash received in February 2011 from the issuance of
approximately 8.9 million shares of our common stock and warrants to purchase 3.6 million shares of
our common stock will be sufficient to fund our operations and meet our debt service requirements
into 2012. To conserve cash, we may delay or cancel some of our planned development activities
that are not related to Ceplene® during 2011.
37
We have two wholly-owned subsidiaries, Maxim Pharmaceuticals, Inc., which is currently
inactive, and EpiCept GmbH, based in Munich, Germany, which is engaged in commercial activities on
our behalf.
Reverse Split of Common Stock
On January 14, 2010, we effected a previously authorized 1-for-3 reverse stock split of our
common stock. The reverse stock split took effect at the start of trading on January 15, 2010 on a
1-for-3 split-adjusted basis. All prior periods have been retroactively adjusted to reflect the
reverse stock split.
The Nasdaq Capital Market Listing
On February 2, 2010, Nasdaq Listing Qualifications Department notified us that we have
regained compliance with the minimum bid price requirement in Listing Rule 5550(a)(2) and met the
requirements of the Nasdaq Listing Qualification Panel (the “Panel”) decision dated November 2,
2009. Accordingly, the Panel has determined to continue the listing of our common stock on The
Nasdaq Stock Market. The Panel had previously determined to continue our listing subject to the
condition that, on or before February 1, 2010, we evidence a closing bid price of $1.00 per share
or more for at least the ten prior consecutive trading days. On January 29, 2010, our closing bid
price was $2.37 per share, the tenth consecutive day it had exceeded the $1.00 per share threshold.
Accordingly, we satisfied the Panel’s condition and the delisting proceeding was closed.
On October 6, 2010, we received two letters from the Nasdaq Listing Qualifications Department.
One letter states that we are not in compliance with the continued listing requirements of The
Nasdaq Capital Market because the bid price of our common stock closed below the minimum $1.00 per
share requirement for 30 consecutive business days (pursuant to Listing Rule 5550(a)(2)). Pursuant
to Nasdaq Listing Rule 5810(c)(3)(A), we have been provided a period of 180 calendar days, or until
April 4, 2011, to regain compliance with the minimum bid price rule. If at any time before April 4,
2011, the bid price of our common stock closes at $1.00 per share or higher for a period determined
by Nasdaq (which shall be a minimum of 10 consecutive business days), Nasdaq will provide written
notification to us that we comply with the Rule.
The second letter from Nasdaq stated that we were not in compliance with the continued listing
requirements of The Nasdaq Capital Market because the market value of our listed securities fell
below $35 million for 30 consecutive business days (pursuant to Listing Rule 5550(b)(2)). Pursuant
to Nasdaq Listing Rule 5810(c)(3)(C), we were provided a period of 180 calendar days, or until
April 4, 2011, to regain compliance with the market value standard. If at any time before April 4,
2011, the market value of our listed securities closed at $35 million or more for a period
determined by Nasdaq (which shall be a minimum of 10 consecutive business days), Nasdaq was to
provide written notification to us that we comply with the Rule. On December 29, 2010, the Company
received notification from the Nasdaq Listing Qualifications Department that it had regained
compliance with the market value standard pursuant to Listing Rule 5550(b)(2) and this matter is
now closed.
We remain out of compliance with the continued listing requirements of The Nasdaq Capital
Market because the bid price of our common stock remains below the minimum $1.00 per share
requirement (pursuant to Listing Rule 5550(a)(2)). In the event that we do not regain compliance
with the minimum bid price rule by April 4, 2011, Nasdaq will determine whether we meet the initial
listing criteria, with the exception of bid price, for The Nasdaq Capital Market and, if we do, we
will be granted an additional compliance period of 180 calendar days. In the event that the
Company is not eligible for the minimum bid price additional compliance period the Company will
have the right to appeal a determination to delist the Company’s securities. The Company’s
securities would remain listed on The Nasdaq Capital Market until the completion of the appeal
process.
Recent Events
In March 2011, we sold approximately 7.1 million shares of our common stock and warrants to
purchase 5.3 million shares of our common stock for gross proceeds of $4.6 million, $4.3 million
net of $0.3 million in transaction costs.
In February 2011, we sold approximately 8.9 million shares of our common stock and warrants to
purchase 3.6 million shares of our common stock for gross proceeds of $7.1 million, $6.6 million
net of $0.5 million in transaction costs.
38
In February 2011, we presented data from our Phase IIb trial for EpiCept™ NP-1 in
chemotherapy-induced peripheral neuropathy, or CPN, which is being conducted by National Cancer
Institute, or NCI. CPN may affect 50% of women undergoing treatment for breast cancer. A common
therapeutic agent for the treatment of advanced breast cancer is paclitaxel, and as many as 80% of
the patients with advanced breast cancer experience some signs and symptoms of CPN, such as
burning, tingling pain associated sometimes with mild muscular weakness, after high dose paclitaxel
administration. The multi-center, double-blind, randomized, placebo-controlled study was conducted
within a network of approximately 25 sites under the direction of the NCI
funded Community Clinical Oncology Program, or CCOP. More than 460 cancer survivors suffering
from painful CPN were enrolled in the six-week study. The results of the trial in the intent to
treat, or ITT, population demonstrated that the change in average daily neuropathy intensity scores
in the NP-1 group achieved a statistically significant reduction in CPN intensity versus placebo
(p<0.001), which was the trial’s primary endpoint. Additionally, a pre-specified subgroup of the
ITT population, those patients who previously received taxane chemotherapy, also showed a
statistically significant reduction in average daily neuropathy intensity scores (p=0.034). This
subgroup constituted more than 50% of the ITT population. Secondary efficacy endpoints confirmed
the superiority of NP-1 vs. placebo. Furthermore, the safety profile of NP-1 was comparable to
placebo.
License Agreements
In January 2010, we entered into an exclusive commercialization agreement for
Ceplene® with Meda AB, a leading international specialty pharmaceutical company based in
Stockholm, Sweden. Under the terms of the agreement, we granted Meda the right to market
Ceplene® in Europe and several other countries including Japan, China, and Australia.
We received a $3 million fee on signing and received an additional $2 million upon the first
commercial launch of Ceplene in a major European market, which have been deferred and are being
recognized as revenue ratably over the life of the commercialization agreement with Meda.
Additional potential payments include a $5 million payment upon achievement of a regulatory
milestone and up to $30 million in sales-based milestones that commence upon attainment of at least
$50 million in annual sales. We will also receive an escalating percentage royalty on net sales in
the covered territories ranging from the low teens to the low twenties and we will be responsible
for Ceplene’s commercial supply. The initial term of this agreement is ten years and is subject to
automatic two year extensions at Meda’s option. The agreement can be terminated at any time by Meda
upon six months prior written notice. The agreement can also be terminated by mutual agreement or
for cause.
In connection with our merger with Maxim Pharmaceuticals on January 4, 2006, we acquired a
license agreement with Myrexis under which we licensed the MX90745 series of caspase-inducer
anti-cancer compounds to Myrexis. Myrexis has initiated clinical trials for AzixaTM, a
MX90745 series compound, for the treatment of brain cancer. Under the terms of the agreement, Maxim
granted to Myrexis a research license to develop and commercialize any drug candidates from the
series of compounds during the Research Term (as defined in the agreement) with a non-exclusive,
worldwide, royalty-free license, without the right to sublicense the technology. Myrexis is
responsible for the worldwide development and commercialization of any drug candidates from the
series of compounds. Maxim also granted to Myrexis a worldwide royalty bearing development and
commercialization license with the right to sublicense the technology. The agreement requires that
Myrexis make licensing, research and milestone payments to us totaling up to $27 million, of which
$3.0 million was paid and recognized as revenue by Maxim prior to the merger on January 4, 2006,
assuming the successful commercialization of the compound for the treatment of cancer, as well as
pay an escalating mid to high single digit percentage royalty on product sales. In March 2008, we
received a milestone payment of $1.0 million following dosing of the first patient in a Phase II
registration sized clinical trial, which has been deferred and is being recognized as revenue
ratably over the life of the last to expire patent that expires in July 2024. As of December 31,
2010, we recorded inception to date revenue of $0.2 million related to this license agreement.
In December 2006, we entered into a license agreement with DURECT, pursuant to which we
granted DURECT the exclusive worldwide rights to certain of our intellectual property for a
transdermal patch containing bupivacaine for the treatment of back pain. Under the terms of the
agreement, we received a $1.0 million payment. In September 2008, we amended our license agreement
with DURECT. Under the terms of the amended agreement, we granted DURECT royalty-free, fully paid
up, perpetual and irrevocable rights to the intellectual property licensed as part of the original
agreement in exchange for a cash payment of $2.25 million from DURECT. As of December 31, 2010, we
recorded inception to date revenue of $0.8 million related to this license agreement.
In December 2003, we entered into a license agreement with Endo under which we granted Endo
(and its affiliates) the exclusive (including as to us and our affiliates) worldwide right to
commercialize LidoPAIN BP. We also granted Endo worldwide rights to certain of our other patents
used by Endo in the development of certain Endo products, including Lidoderm®, Endo’s
topical lidocaine-containing patch, for the treatment of chronic lower back pain. Upon the
execution of the Endo agreement, we received a non-refundable payment of $7.5 million, which has
been deferred and is being recognized as revenue on the proportional performance method. We may
receive payments of up to $52.5 million upon the achievement of various milestones relating to
product development and regulatory approval for both our LidoPAIN BP product candidate and licensed
Endo product candidates, including Lidoderm®, so long as, in the case of Endo’s product
candidate, our patents provide protection thereof. We are also entitled to receive royalties from
Endo based on the net sales of LidoPAIN BP. These royalties are payable until generic equivalents
to the LidoPAIN BP product are available or until expiration of the patents covering LidoPAIN BP,
whichever is sooner. We are also eligible to receive milestone payments from Endo of up to
approximately $30.0 million upon the achievement of specified net sales milestones for licensed
Endo products, including Lidoderm®, so long as our patents provide protection thereof.
The future amount of milestone payments we are eligible to receive under the Endo agreement is
$82.5 million. As of December 31, 2010 we recorded
inception to date revenue related to this license agreement in the amount of $1.7 million of
which $14,000 was recorded as revenue during 2010.
39
Under the terms of the license agreement, we are responsible for continuing and completing the
development of LidoPAIN BP, including the conduct of all clinical trials and the supply of the
clinical products necessary for those trials and the preparation and submission of the NDA in order
to obtain regulatory approval for LidoPAIN BP. Endo remains responsible for continuing and
completing the development of Lidoderm® for the treatment of chronic lower back pain,
including the conduct of all clinical trials and the supply of the clinical products necessary for
those trials. No progress in the development of LidoPAIN BP or Lidoderm with respect to back pain
has been reported. Accordingly, we do not expect to receive any further cash compensation pursuant
to this license agreement.
Off Balance Sheet Arrangements
We do not have any relationships with unconsolidated entities or financial partnerships, such
as entities often referred to as structured finance or special purpose entities, which would have
been established for the purpose of facilitating off-balance sheet arrangements or other
contractually narrow or limited purposes. In addition, we do not engage in trading activities
involving non-exchange traded contracts. Therefore, we are not materially exposed to any financing,
liquidity, market or credit risk that could arise if we had engaged in these relationships. We do
not have relationships or transactions with persons or entities that derive benefits from their
non-independent relationship with us or our related parties.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on
our consolidated financial statements, which have been prepared in accordance with accounting
principles generally accepted in the United States. The preparation of these financial statements
requires that we make estimates and judgments affecting the reported amounts of assets, liabilities
and expenses and related disclosure of contingent assets and liabilities. We review our estimates
on an ongoing basis. We base our estimates on historical experience and on various other
assumptions that we believe to be reasonable under the circumstances. Actual results may differ
from these estimates under different assumptions or conditions. While our significant accounting
policies are described in more detail in the notes to our consolidated financial statements
included in this annual report, we believe the following accounting policies to be critical to the
judgments and estimates used in the preparation of our consolidated financial statements.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally
accepted in the United States of America requires management to make estimates and assumptions that
affect the reported amounts of assets and liabilities and disclosure of contingent assets and
liabilities at the date of the financial statements. Estimates also affect the reported amounts of
revenues and expenses during the reporting period and stock-based compensation. Actual results
could differ from those estimates.
Revenue Recognition
We recognize revenue relating to our collaboration agreements in accordance with the SEC Staff
Accounting Bulletin (“SAB”) 104, Revenue Recognition, and FASB Accounting Standards Codification
(“ASC”) 605-25, “Revenue Recognition — Multiple Element Arrangements” (“ASC 605-25”). Revenue
under collaborative arrangements may result from license fees, milestone payments, research and
development payments and royalties.
Our application of these standards involves subjective determinations and requires management
to make judgments about value of the individual elements and whether they are separable from the
other aspects of the contractual relationship. We evaluate our collaboration agreements to
determine units of accounting for revenue recognition purposes. For collaborations containing a
single unit of accounting, we recognize revenue when the fee is fixed or determinable,
collectibility is assured and the contractual obligations have occurred or been rendered. For
collaborations involving multiple elements, our application requires management to make judgments
about value of the individual elements and whether they are separable from the other aspects of the
contractual relationship. To date, we have determined that our upfront non-refundable license fees
cannot be separated from our ongoing collaborative research and development activities to the
extent such activities are required under the agreement and, accordingly, do not treat them as a
separate element. We recognize revenue from non-refundable, up-front licenses and related payments,
not specifically tied to a separate earnings process, either on the proportional performance method
with respect to our license with Endo, or ratably over either the development period or the later
of (1) the conclusion of the royalty term on a jurisdiction by jurisdiction basis; and (2) the
expiration of the last EpiCept licensed patent as we do with respect to our license with DURECT,
Myrexis and GNI, Ltd., or GNI.
40
Proportional performance is measured based on costs incurred compared to total estimated costs
over the development period which approximates the proportion of the value of the services provided
compared to the total estimated value over the development period. The proportional performance
method currently results in revenue recognition at a slower pace than the ratable method as many of
our costs are incurred in the latter stages of the development period. We periodically review our
estimates of cost and the length of the development period and, to the extent such estimates
change, the impact of the change is recorded at that time. During each of the years 2010, 2009 and
2008 we increased the estimated development period by an additional twelve months to reflect
additional time required to obtain clinical data from our partner.
We will recognize milestone payments as revenue upon achievement of the milestone only if (1)
it represents a separate unit of accounting as defined in ASC 605-25; (2) the milestone payments
are nonrefundable; (3) substantive effort is involved in achieving the milestone; and (4) the
amount of the milestone is reasonable in relation to the effort expended or the risk associated
with the achievement of the milestone. If any of these conditions are not met, we will recognize
milestones as revenue in accordance with our accounting policy in effect for the respective
contract. At the time of a milestone payment receipt, we will recognize revenue based upon the
portion of the development services that are completed to date and defer the remaining portion and
recognize it over the remainder of the development services on the proportional or ratable method,
whichever is applicable. When payments are specifically tied to a separate earnings process,
revenue will be recognized when the specific performance obligation associated with the payment has
been satisfied. Deferred revenue represents the excess of cash received compared to revenue
recognized to date under licensing agreements.
We have chosen early adoption of the milestone method of revenue recognition as defined in ASC
605-28, “Revenue Recognition — Milestone Method” on a prospective basis as of January 1, 2010.
Under this method of revenue recognition, we will recognize into revenue research-related milestone
payments for which there is substantial uncertainty at the date the arrangement is entered into
that the event will be achieved, when that event can only be achieved based in whole or in part on
our performance or a specific outcome resulting from our performance and, if achieved, would result
in additional payments being due to us. This accounting will be applicable to research milestones
under the license agreement entered into with Meda AB in 2010.
The Meda AB agreement entitles us to a $3.0 million upfront payment, a $2.0 million payment
upon launch of Ceplene® in a major country in the European Union, and milestone payments
and royalties on sales of Ceplene®. The $3.0 million upfront payment received in the
first quarter of 2010 and the $2.0 million payment upon launch of Ceplene® in a major
country in the European Union received in the second quarter of 2010 have been deferred and are
being recognized as revenue ratably over the life of the commercialization agreement with Meda AB.
Royalty revenue is recognized in the period the sales occur, provided that the royalty amounts
are fixed or determinable, collection of the related receivable is reasonably assured and we have
no remaining performance obligations under the arrangement providing for the royalty. If royalties
are received when we have remaining performance obligations, they would be attributed to the
services being provided under the arrangement and, therefore, recognized as such obligations are
performed under either the proportionate performance or straight-line methods, as applicable.
Royalty Expense
Upon receipt of marketing approval and commencement of commercial sales, some of which may not
occur for several years, we will owe royalties to licensors of certain patents generally based upon
net sales of the respective products. Under a license agreement with respect to NP-1, we are
obligated to pay royalties based on annual net sales derived from the products incorporating the
licensed technology. Under a license agreement with respect to crolibulinTM, we are
required to provide a portion of any sublicensing payments we receive if we relicense the series of
compounds or make milestone payments, assuming the successful commercialization of the compound by
us for the treatment of a cancer indication, as well as pay a royalty on product sales. Under a
royalty agreement with respect to Ceplene®, we are obligated to pay royalties based on
annual net sales derived from the products incorporating the licensed technology. In each case, our
royalty obligation ends the later of (1) the conclusion of the royalty term on a jurisdiction by
jurisdiction basis; and (2) the expiration of the last EpiCept licensed patent.
Stock-Based Compensation
We record stock-based compensation expense at fair value in accordance with the Financial
Accounting Standards Board, or FASB, issued ASC 718-10, “Compensation — Stock Compensation” (“ASC
718-10”). We utilize the Black-Scholes valuation method to recognize compensation expense over the
vesting period. Certain assumptions need to be made with respect to utilizing the Black-Scholes
valuation model, including the expected life, volatility, risk-free interest rate and anticipated
forfeiture of the stock options. The expected life of the stock options was calculated using the
method allowed by the provisions of ASC 718-10. In accordance with ASC 718-10, the simplified
method for “plain vanilla” options may be used where the expected term is equal to the
vesting term plus the original contract term divided by two. The risk-free interest rate is
based on the rates paid on securities issued by the U.S. Treasury with a term approximating the
expected life of the options. Estimates of pre-vesting option forfeitures are based on our
experience. We will adjust our estimate of forfeitures over the requisite service period based on
the extent to which actual forfeitures differ, or are expected to differ, from such estimates.
Changes in estimated forfeitures will be recognized through a cumulative catch-up adjustment in the
period of change and will also impact the amount of compensation expense to be recognized in future
periods.
41
We account for stock-based transactions with non-employees in which services are received in
exchange for the equity instruments based upon the fair value of the equity instruments issued, in
accordance with ASC 718-10 and ASC 505-50, “Equity-Based Payments to Non-Employees.” The two
factors that most affect charges or credits to operations related to stock-based compensation are
the estimated fair market value of the common stock underlying stock options for which stock-based
compensation is recorded and the estimated volatility of such fair market value. The value of such
options is periodically remeasured and income or expense is recognized during the vesting terms.
Accounting for stock-based compensation granted by us requires fair value estimates of the
equity instrument granted or sold. If our estimate of the fair value of stock-based compensation is
too high or too low, it will have the effect of overstating or understating expenses. When
stock-based grants are granted in exchange for the receipt of goods or services, we estimate the
value of the stock-based compensation based upon the value of our common stock.
During the years 2010 and 2009, we issued approximately 0.8 million and 0.7 million stock
options, respectively, with varying vesting provisions to certain of our employees and directors.
Based on the Black-Scholes valuation method (volatility — 108.0% -109.0%, risk free rate — 2.17%
- 2.39%, dividends — zero, weighted average life — 5 years; forfeiture — 10%), for the grants
issued in 2010, we estimated $1.3 million of share-based compensation will be recognized as
compensation expense over the vesting period, which will be amortized over the weighted average
remaining requisite service period of 3.25 years. During years 2010 and 2009, we recognized total
share-based compensation of approximately $0.9 million and $1.4 million, related to the options
granted during 2010, 2009, 2008, 2007, 2006 and the unvested outstanding Maxim options as of
January 4, 2006 that were converted into EpiCept options based on the vesting of those options
during 2006. Future grants of options will result in additional charges for stock-based
compensation that will be recognized over the vesting periods of the respective options.
Derivatives
As a result of certain financings, derivative instruments were created that we have measured
at fair value and mark to market at each reporting period. Fair value of the derivative instruments
will be affected by estimates of various factors that may affect the respective instrument,
including our cost of capital, the risk free rate of return, volatility in the fair value of our
stock price, future foreign exchange rates of the U.S. dollar to the euro and future profitability
of our German subsidiary. At each reporting date, we review applicable assumptions and estimates
relating to fair value and record any changes in the statement of operations.
Foreign Exchange Gains and Losses
We have a 100%-owned subsidiary in Germany, EpiCept GmbH, that performs certain
commercialization activities on our behalf. EpiCept GmbH has generally been unprofitable since its
inception. Its functional currency is the euro. The process by which EpiCept GmbH’s financial
results are translated into U.S. dollars is as follows: income statement accounts are translated at
average exchange rates for the period and balance sheet asset and liability accounts are translated
at end of period exchange rates. Translation of the balance sheet in this manner affects the
stockholders’ deficit account, referred to as the cumulative translation adjustment account. This
account exists only in EpiCept GmbH’s U.S. dollar balance sheet and is necessary to keep the
foreign balance sheet stated in U.S. dollars in balance.
Certain of our debt instruments, originally expressed in German deutsche marks, are now
denominated in euros. Changes in the value of the euro relative to the value of the U.S. dollar
will affect the U.S. dollar value of our indebtedness at each reporting date as substantially all
of our assets are held in U.S. dollars. These changes are recognized by us as a foreign currency
transaction gain or loss, as applicable, and are reported in other expense or income in our
consolidated statements of operations.
42
Research and Development Expenses
We expect that a large percentage of our future research and development expenses will be
incurred in support of current and future preclinical and clinical development programs. These
expenditures are subject to numerous uncertainties in timing and cost to completion. We test our
product candidates in numerous preclinical studies for toxicology, safety and efficacy. We then
conduct early stage clinical trials for each drug candidate. As we obtain results from clinical
trials, we may elect to discontinue or delay
clinical trials for certain product candidates or programs in order to focus resources on more
promising product candidates or programs. Completion of clinical trials may take several years but
the length of time generally varies according to the type, complexity, novelty and intended use of
a drug candidate. The cost of clinical trials may vary significantly over the life of a project as
a result of differences arising during clinical development, including:
| |
• |
|
the number of sites included in the trials; |
| |
• |
|
the length of time required to enroll suitable patients; |
| |
• |
|
the number of patients that participate in the trials; |
| |
• |
|
the number of doses that patients receive; |
| |
• |
|
the duration of follow-up with the patient; |
| |
• |
|
the product candidate’s phase of development; and |
| |
• |
|
the efficacy and safety profile of the product. |
Expenses related to clinical trials are based on estimates of the services received and
efforts expended pursuant to contracts with multiple research institutions and clinical research
organizations that conduct clinical trials on the our behalf. The financial terms of these
agreements are subject to negotiation and vary from contract to contract and may result in uneven
payment flows. If timelines or contracts are modified based upon changes in the clinical trial
protocol or scope of work to be performed, estimates of expenses are modified accordingly on a
prospective basis.
Ceplene® has been granted full marketing authorization by the European Commission
for the remission maintenance and prevention of relapse in adult patients with Acute Myeloid
Leukemia in first remission. None of our other drug candidates has received FDA or foreign
regulatory marketing approval. In order to grant marketing approval, the FDA or foreign regulatory
agencies must conclude that our clinical data and that of our collaborators establish the safety
and efficacy of our drug candidates. Furthermore, our strategy includes entering into
collaborations with third parties to participate in the development and commercialization of our
products. In the event that third parties have control over the preclinical development or clinical
trial process for a product candidate, the estimated completion date would largely be under control
of that third party rather than under our control. We cannot forecast with any degree of certainty
which of our drug candidates will be subject to future collaborations or how such arrangements
would affect our development plan or capital requirements.
Recent Accounting Pronouncements
In March 2010, the FASB issued ASU 2010-17 “Revenue Recognition — Milestone Method” (ASC
Topic 605). The guidance in this consensus allows the use of the milestone method as an acceptable
revenue recognition methodology when an arrangement includes substantive milestones. The guidance
provides a definition of substantive milestone and should be applied regardless of whether the
arrangement includes single or multiple deliverables or units of accounting. The scope of this
consensus is limited to the transactions involving milestones relating to research and development
deliverables. The guidance includes enhanced disclosure requirements about each arrangement,
individual milestones and related contingent consideration, information about substantive
milestones and factors considered in the determination. The consensus is effective prospectively to
milestones achieved in fiscal years, and interim periods within those years, after June 15, 2010.
Early application and retrospective application are permitted. We have chosen early adoption of
this pronouncement effective January 1, 2010 on a prospective basis. The early adoption did not
have a material effect on our financial statements.
On October 7, 2009, the FASB issued ASU 2009-13, “Multiple Revenue Arrangements — a Consensus
of the FASB Emerging Issues Task Force” which supersedes certain guidance in ASC 605-25, “Revenue
Recognition-Multiple Element Arrangements,” and requires an entity to allocate arrangement
consideration to all of its deliverables at the inception of an arrangement based on their relative
selling prices (i.e., the relative-selling-price method). The use of the residual method of
allocation will no longer be permitted in circumstances in which an entity recognized revenue for
an arrangement with multiple deliverables subject to ASC 605-25. ASU 2009-13 also requires
additional disclosures. We adopted the provisions of ASU 2009-13 beginning on January 1, 2011.
Based on our current revenue arrangements, the adoption of ASU 2009-13 is not expected to have a
material impact on our financial statements.
43
Results of Operations
Years Ended December 31, 2010 and 2009
Revenues. During the years 2010 and 2009, we recognized revenue of approximately $0.8 million
and $0.4 million, respectively, from prior upfront licensing fees and milestone payments received
from our strategic alliances and royalties with respect to certain technology; and $0.2 million and
$0, respectively, in product revenues from the sales of Ceplene® to Meda. We recognized revenue
from our agreement with Endo using the proportional performance method with respect to LidoPAIN BP,
from our agreement with Meda using the milestone method and on a straight line method over the life
of the last to expire patent with Myrexis, DURECT and GNI. During the years 2010 and 2009, we
recognized revenue of $33,000 and $40,000 from royalties with respect to acquired Maxim technology.
The current portion of deferred revenue as of December 31, 2010 of $0.9 million represents our
estimate of revenue to be recognized over the next twelve months primarily related to the upfront
payments from Meda, Myrexis, Endo, DURECT and GNI, Ltd.
Cost of goods sold. Cost of goods sold was $1.0 million and $0 for the years 2010 and 2009,
respectively. Cost of goods sold related solely to the costs of sales of Ceplene®, including
manufacturing costs and royalty expense related to sales of Ceplene®. During 2010, we expensed
$0.9 million of Ceplene® inventory as we believe such inventory will not be sold prior to reaching
its product expiration date.
Selling, general and administrative expense. General and administrative expense decreased by
4%, or $0.3 million, to $7.2 million for 2010 from $7.5 million in 2009. The overall decrease in
administrative expense can be attributed to continued cost reduction efforts in 2010. For 2010,
stock-based compensation charges amounted to $0.7 million, or a decrease of $0.2 million from 2009.
In addition, our accounting, investor relations and public reporting expense decreased $0.3
million for 2010 as compared to the same period in 2009, partially offset by higher legal and other
administrative expenses of $0.3 million related to exploring alternative financing opportunities.
Selling expenses have been significantly reduced, and we expect general and administrative expenses
to remain at approximately current levels over the next few quarters.
Research and development expense. Research and development expense decreased by 30%, or $3.5
million, to $8.1 million for 2010 from $11.6 million for 2009. The decrease was primarily
attributable to $2.0 million in lower salary and salary related expenses and facility costs related
to closing our research facility in San Diego in 2009, lower clinical trial expenses for Ceplene®
of $0.9 million, lower license fees of $0.5 million and lower stock-based compensation charges of
$0.2 million, partially offset by higher regulatory fees of $0.3 million. During 2010, our research
and development efforts were focused on our open label trial of Ceplene® that will meet our
post-approval requirements with the EMA and on preparation of the NDA for Ceplene® that was
submitted in June 2010. During 2009, our clinical efforts were focused on the completion of our
clinical trials of NP-1 and our Phase I clinical trial of crolibulinTM and the
commencement of our open label trial of Ceplene® that will meet our post-approval requirements with
the EMA.
For the years ended December 31, 2010 and 2009, we incurred the following research and development
expense:
| |
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Direct Expenses: |
|
|
|
|
|
|
|
|
Ceplene® |
|
$ |
4,081 |
|
|
$ |
3,896 |
|
NP-1 |
|
|
523 |
|
|
|
971 |
|
CrolibulinTM |
|
|
131 |
|
|
|
705 |
|
Other projects |
|
|
16 |
|
|
|
46 |
|
|
|
|
|
|
|
|
|
|
|
4,751 |
|
|
|
5,618 |
|
|
|
|
|
|
|
|
Indirect Expenses: |
|
|
|
|
|
|
|
|
Staffing |
|
|
1,492 |
|
|
|
2,591 |
|
Other indirect |
|
|
1,884 |
|
|
|
3,394 |
|
|
|
|
|
|
|
|
|
|
|
3,376 |
|
|
|
5,985 |
|
|
|
|
|
|
|
|
Total Research & Development |
|
$ |
8,127 |
|
|
$ |
11,603 |
|
|
|
|
|
|
|
|
Direct expenses consist primarily of fees paid to vendors and consultants for services related
to preclinical product development, clinical trials, and manufacturing of the respective products.
Generally, we have flexibility with respect to the timing and magnitude of a significant portion of
our direct expenses. Indirect expenses are those expenses we incur that are not allocated by
project, which consist primarily of the salaries and benefits of our research and development staff
and related premises.
44
Other income (expense), net. Our other income (expense), net consisted of the following for
the years ended December 31, 2010 and 2009:
| |
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Other income (expense), net consist of: |
|
|
|
|
|
|
|
|
Interest expense |
|
$ |
(361 |
) |
|
$ |
(20,021 |
) |
Change in value of warrants and derivatives |
|
|
— |
|
|
|
(305 |
) |
Interest income |
|
|
7 |
|
|
|
32 |
|
Other Income |
|
|
733 |
|
|
|
— |
|
Foreign exchange gain (loss) |
|
|
(538 |
) |
|
|
221 |
|
|
|
|
|
|
|
|
Other expense, net |
|
$ |
(159 |
) |
|
$ |
(20,073 |
) |
|
|
|
|
|
|
|
During 2010, we recorded other expense, net of $0.2 million as compared to other expense, net
of $20.1 million during 2009. The $19.9 million decrease in other expense, net was primarily
related to $10.5 million in amortization of debt issuance costs and discount and $9.3 million in
interest expense, which was paid from restricted cash, as a result of the conversion of $24.5
million of our 7.5556% convertible subordinated notes due 2014 into approximately 9.1 million
shares of our common stock in 2009. Amortization of debt issuance costs was accelerated in
December 2010 as a result of the termination of the Equity Distribution Agreement with Maxim Group
LLC, which established an at-the-market program through which we had the right to sell, from time
to time and at our sole discretion, shares of our common stock having an aggregate offering price
of up to $15 million. Other income (expense) was negatively impacted by a $0.8 million change in
foreign exchange gains though it was substantially offset by $0.7 million of other income from the
Qualifying Therapeutic Discovery Project Program, or QTDP.
In October 2010, we were notified by the Internal Revenue Service that our application for the
QTDP was certified and a grant in the amount of $0.7 million was approved. This grant was provided
under a new section of the Internal Revenue Code that was enacted as part of the Patient Protection
and Affordable Care Act of 2010. The program, which provides for grants and tax credits, is
intended to benefit discovery projects that show a reasonable potential to result in new therapies
to treat areas of unmet medical need, or detect or treat chronic or acute diseases and conditions;
reduce long-term growth of health care costs in the United States; or significantly advance the
goal of curing cancer within 30 years. We received the grant in November 2010.
Income Taxes. Income tax expense for each of the years ended December 31, 2010 and 2009 was
$4,000. As of December 31, 2010 and 2009, we had federal net operating loss carryforwards, or NOLs,
of $120.9 and $90.8 million, state NOLs of $115.6 and $95.5 million, and foreign NOLs of $15.6 and
$15.0 million respectively, available to reduce future taxable income. Our federal and state NOLs
will begin to expire after 2012 through 2030. In accordance with ASC 740, “Income Taxes,” we have
provided a valuation allowance for the full amount of our net deferred tax assets because it is not
more likely than not that we will realize future benefits associated with these deferred tax assets
at December 31, 2010 and 2009.
Years Ended December 31, 2009 and 2008
Revenues. During the years 2009 and 2008, we recognized revenue of approximately $0.4 million
and $0.3 million, respectively, from prior upfront licensing fees and milestone payments received
from Myrexis, Endo, DURECT and GNI, Ltd. and royalties with respect to certain technology; and
sales of Ceplene® via the named-patient program. We recognized revenue from our agreement with Endo
using the proportional performance method with respect to LidoPAIN BP and on a straight line method
over the life of the last to expire patent with Myrexis and DURECT. During the years 2009 and 2008,
we recognized revenue of $40,000 and $43,000 from royalties with respect to acquired Maxim
technology.
The current portion of deferred revenue as of December 31, 2009 of $0.4 million represents our
estimate of revenue to be recognized over the next twelve months primarily related to the upfront
payments from Myrexis, Endo, DURECT and GNI, Ltd.
Selling, general and administrative expense. Selling, general and administrative expense
decreased by 21%, or $2.1 million, to $7.5 million for 2009 from $9.6 million in 2008. The overall
decrease in administrative expense can be attributed to a cost reduction effort implemented in 2008
and continued into 2009. For 2009, stock-based compensation charges amounted to $0.9 million, or a
decrease of $0.9 million from 2008. In addition, our legal, accounting and public reporting
expense decreased $0.5 million and our facility, insurance and other administrative expenses
decreased $0.7 million for 2009 as compared to the same period in 2008.
Research and development expense. Research and development expense decreased by 8%, or $1.0
million, to $11.6 million for 2009 from $12.6 million for 2008. The decrease was primarily
attributable to lower clinical trial and consulting expenses of $0.4
million, lower salary and salary related expenses of $1.1 million and lower patent expenses of
$0.2 million in 2009 as compared to the same period in 2008, partially offset by a $0.8 million
facility expense and $0.2 million in severance expense related to closing our research facility in
San Diego.
45
During 2009, our clinical efforts were focused on the completion of our clinical trials of
NP-1 and our Phase I clinical trial of crinobulin and the commencement of our open label trial of
Ceplene® that will meet our post-approval requirements with the EMA. During 2008, our clinical
efforts were focused on the clinical trials of NP-1 and the ongoing Phase I clinical trial of
crinobulin, preparation for the Oral Explanation meeting with the CHMP, the scientific committee of
the EMA, regarding the remaining outstanding issues on the MAA for Ceplene® and
preparation for the reexamination of the negative determination issued by the CHMP regarding our
marketing application for Ceplene®.
For the years ended December 31, 2009 and 2008, we incurred the following research and development
expense:
| |
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
Direct Expenses: |
|
|
|
|
|
|
|
|
Ceplene® |
|
$ |
3,896 |
|
|
$ |
2,004 |
|
NP-1 |
|
|
971 |
|
|
|
2,584 |
|
Crinobulin |
|
|
705 |
|
|
|
908 |
|
Other projects |
|
|
46 |
|
|
|
357 |
|
|
|
|
|
|
|
|
|
|
|
5,618 |
|
|
|
5,853 |
|
|
|
|
|
|
|
|
Indirect Expenses: |
|
|
|
|
|
|
|
|
Staffing |
|
|
2,591 |
|
|
|
3,855 |
|
Other indirect |
|
|
3,394 |
|
|
|
2,915 |
|
|
|
|
|
|
|
|
|
|
|
5,985 |
|
|
|
6,770 |
|
|
|
|
|
|
|
|
Total Research & Development |
|
$ |
11,603 |
|
|
$ |
12,623 |
|
|
|
|
|
|
|
|
Direct expenses consist primarily of fees paid to vendors and consultants for services related
to preclinical product development, clinical trials, and manufacturing of the respective products.
Generally, we have flexibility with respect to the timing and magnitude of a significant portion of
our direct expenses. Indirect expenses are those expenses we incur that are not allocated by
project, which consist primarily of the salaries and benefits of our research and development staff
and related premises.
Other income (expense), net. Our other income (expense), net consisted of the following for
the years ended December 31, 2009 and 2008:
| |
|
|
|
|
|
|
|
|
| |
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
Other income (expense), net consist of: |
|
|
|
|
|
|
|
|
Interest expense |
|
$ |
(20,021 |
) |
|
$ |
(1,266 |
) |
Change in value of warrants and derivatives |
|
|
(305 |
) |
|
|
113 |
|
Interest income |
|
|
32 |
|
|
|
33 |
|
Loss on extinguishment of debt |
|
|
— |
|
|
|
(1,975 |
) |
Foreign exchange gain (loss) |
|
|
221 |
|
|
|
(327 |
) |
|
|
|
|
|
|
|
Other expense, net |
|
$ |
(20,073 |
) |
|
$ |
(3,422 |
) |
|
|
|
|
|
|
|
During 2009, we recorded other expense, net of $20.1 million as compared to other expense, net
of $3.4 million during 2008. The $16.7 million increase in other expense, net was primarily
related to $10.5 million in amortization of debt issuance costs and discount and $9.3 million in
interest expense, which was paid from restricted cash, as a result of the conversion of $24.5
million of our 7.5556% convertible subordinated notes due 2014 into approximately 9.1 million
shares of our common stock in 2009. Other income (expense) was positively impacted by a $0.5
million change in foreign exchange gains though it was substantially offset by a $0.4 million
decrease in the fair value of certain warrants and derivatives. The Company experienced a loss on
extinguishment of debt of $2.0 million in 2008.
Income Taxes. Income tax expense for the years ended December 31, 2009 and 2008 was $4,000 and
$3,000, respectively. As of December 31, 2009 and 2008, we had federal net operating loss
carryforwards, or NOLs, of $90.8 and $87.4 million, state NOLs of $95.5 and $95.6 million, and
foreign NOLs of $15.0 and $14.7 million respectively, available to reduce future taxable income.
Our federal and state NOLs will begin to expire after 2012 through 2029. In accordance with ASC
740, “Income Taxes,” we have provided a valuation allowance for the full amount of our net deferred
tax assets because it is not more likely than not that we will realize future benefits associated
with these deferred tax assets at December 31, 2009 and 2008.
46
Liquidity and Capital Resources
We have devoted substantially all of our cash resources to research and development programs
and general and administrative expenses. To date, we have not generated any significant revenues
from the sale of products and may not generate any such revenues for a number of years, if at all.
As a result, we have incurred an accumulated deficit of $250.6 million as of December 31, 2010, and
we anticipate that we will continue to incur operating losses in the future. Our recurring losses
from operations and our stockholders’ deficit raise substantial doubt about our ability to continue
as a going concern. Should we be unable to generate sufficient revenue from the sale of
Ceplene® or raise adequate financing in the future, operations will need to be scaled
back or discontinued. Since our inception, we have financed our operations primarily through the
proceeds from the sales of common and preferred securities, debt, revenue from collaborative
relationships, investment income earned on cash balances and short-term investments and the sales
of a portion of our New Jersey net operating loss carryforwards.
The following table describes our liquidity and financial position on December 31, 2010 and
2009.
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Working capital (deficit) |
|
$ |
(1,887 |
) |
|
$ |
1,407 |
|
Cash and cash equivalents |
|
|
2,435 |
|
|
|
5,142 |
|
Notes and loans payable, current portion |
|
|
524 |
|
|
|
985 |
|
Notes and loans payable, long term portion |
|
|
448 |
|
|
|
967 |
|
Working Capital (Deficit)
As of December 31, 2010, we had a working capital deficit of $1.9 million, consisting of
current assets of $2.9 million and current liabilities of $4.8 million. This represents an
unfavorable change in working capital of approximately $3.3 million from our working capital of
$1.4 million consisting of current assets of $6.9 million and current liabilities of $5.5 million
as of December 31, 2009. We funded our working capital deficit and the cash portion of our 2009
operating loss with $18.3 million in gross proceeds from our November 2010, June 2010 and June 2009
financings, $0.1 million in proceeds from the sale of our stock utilizing our at-the-market program
and the receipt of $5.0 million from Meda in connection with the Ceplene® European marketing and
distribution agreement.
Cash and Cash Equivalents
At December 31, 2010, our cash and cash equivalents totaled $2.4 million. At December 31,
2009, cash and cash equivalents totaled $5.1 million. Our cash and cash equivalents consist
primarily of an interest bearing money market account. In January 2010, we received $3.0 million
from Meda in connection with the signing of the Ceplene® European marketing and distribution
agreement and in May 2010 we received $2.0 million from Meda in connection with the launch of
Ceplene® in Europe. In June 2010, we sold approximately 6.1 million shares of common stock and
warrants to purchase 11.0 million shares of common stock for gross proceeds of $6.7 million, $6.2
million net of $0.5 million in transactions costs. In November 2010, we sold approximately 3.3
million shares of common stock and warrants to purchase 1.5 million shares of common stock for
gross proceeds of $2.0 million, $1.9 million net of $0.1 million in transactions costs. During
2010, we sold approximately 31,000 shares of common stock through our at-the-market program for
gross proceeds of $0.1 million and we received proceeds of $0.8 million from the exercise of
approximately 1.4 million warrants.
In June 2009, we sold approximately 4.0 million shares of common stock and warrants to
purchase 1.4 million shares of common stock for gross proceeds of $9.6 million, $8.9 million net of
$0.7 million in transaction costs. In February 2009, we received net proceeds of approximately
$14.0 million, after $1.6 million in transaction costs and establishing a restricted cash account
of $9.4 million for make-whole interest, from the issuance of $25.0 million principal aggregate
amount of 7.5556% convertible senior subordinated notes due February 2014 and five and one-half
year warrants to purchase approximately 4.2 million shares of the Company’s common stock at an
exercise price of $3.105 per share.
47
Current and Future Liquidity Position
During 2010, we raised gross proceeds of $8.7 million, $8.1 million net of $0.6 million in
transaction costs. In addition, we received $0.8 million upon exercise of certain warrants and a
$0.7 million grant from the Internal Revenue Service under the Qualifying Therapeutic Discovery
Project Program. As of December 31, 2010 we had approximately $2.4 million in cash and cash
equivalents. In March 2011, we sold approximately 7.1 million shares of our common stock and
warrants to purchase 5.3 million
shares of our common stock for gross proceeds of $4.6 million, $4.3 million net of $0.3
million in transaction costs. In February 2011, we sold approximately 8.9 million shares of our
common stock and warrants to purchase 3.6 million shares of our common stock for gross proceeds of
$7.1 million, $6.6 million net of $0.5 million in transaction costs. Our anticipated average
monthly cash operating expenses in 2011 is approximately $1.1 million per month. In addition, we
are required to make a final interest and principal payment to our lender tbg on June 30, 2011 in
the amount of approximately $0.5 million. We believe that our cash is sufficient to fund operations
into 2012. We may receive cash from certain of our licensing partners during 2011 for achievement
of clinical milestones, and we anticipate raising additional funds through the issuance of debt
and/or equity to meet our cash needs. To conserve cash, we may delay or cancel some of our planned
development activities that are not related to Ceplene during 2011.
In November 2010 we provided an update with respect to our financing plans. The key element
of the plan is a non-equity financing transaction that, if fully completed, will support our
operations for an additional year. We anticipate that the transaction, which is subject to
completion of due diligence and execution of mutually-satisfactory documentation, will close in
2011, but should the transaction not close or the proceeds be less than anticipated we may
determine to seek additional or alternative sources or types of financing, including equity
financing.
We plan to out license our NP-1 compound to a third party who will agree to complete clinical
development and commercialize the product upon receipt of necessary regulatory approvals.
Discussions with prospective partners are continuing, however at this time we are unable to
determine whether or when such an agreement might be concluded or the amount of any fees that may
be paid to us in 2011 in connection with the agreement.
If additional funds are raised by issuing equity, substantial dilution to existing
shareholders may result. If we fail to obtain capital when required, we may be forced to delay,
scale back, or eliminate some or all of our commercialization efforts for Ceplene and our research
and development programs or to cease operations entirely.
Our future capital uses and requirements depend on numerous forward-looking factors. These
factors include, but are not limited to, the following:
| |
• |
|
revenues generated from the sale of Ceplene® in Europe, including payments
from our marketing partner; |
| |
• |
|
manufacturing costs of Ceplene® |
| |
• |
|
the timing, receipt and amount of front-end fees and milestone payments that may
become payable through an NP-1 license; |
| |
• |
|
progress in our research and development programs, as well as the magnitude of these
programs; |
| |
• |
|
the timing, receipt and amount of milestone and other payments, if any, from present
and future collaborators, if any; |
| |
• |
|
the ability to establish and maintain additional collaborative arrangements; |
| |
• |
|
the resources, time and costs required to successfully initiate and complete our
preclinical and clinical trials, obtain regulatory approvals, protect our intellectual
property; |
| |
• |
|
the cost of preparing, filing, prosecuting, maintaining and enforcing patent claims;
and |
| |
• |
|
the timing, receipt and amount of sales and royalties, if any, from our potential
products. |
Our ability to raise additional capital will depend on financial, economic and market
conditions and other factors, many of which are beyond our control. We cannot be certain that such
additional funding will be available upon acceptable terms, or at all. To the extent that we raise
additional capital by issuing equity securities, our then-existing stockholders may experience
further dilution. Our sales of equity have generally included the issuance of warrants, and if
these warrants are exercised in the future, stockholders may experience significant additional
dilution. We may not be able to raise additional capital through the sale of our securities which
would severely limit our ability to fund our operations. Debt financing, if available, may subject
us to restrictive covenants that could limit our flexibility in conducting future business
activities. Given our available cash resources, existing indebtedness and results of operations,
obtaining debt financing may not be possible. To the extent that we raise additional capital
through collaboration and licensing arrangements, it may be necessary for us to relinquish valuable
rights to our product candidates that we might otherwise seek to develop or commercialize
independently.
48
Operating Activities
Net cash used in operating activities was $10.4 million in 2010, as compared to $29.5 million
in 2009 and $15.6 million in 2008. In 2010, cash was primarily used to fund our net loss for the
year for research and development, selling, general, administrative and other expenses. The 2010
net loss was partially offset by non-cash charges of $0.9 million in stock-based compensation, $0.2
million in amortization of deferred financing costs and discount on loans and $0.2 million of
depreciation and amortization expense. The 2010 net loss was also partially offset by a $5.0
million increase in deferred revenue in connection with the Ceplene® European marketing
and distribution agreement with Meda, a $0.9 million expense for excess inventory of
Ceplene® that we believe will not
be sold prior to reaching its product expiration date and $0.5 million in foreign exchange
loss. Inventory increased by $0.5 million as we prepared for the launch of Ceplene in Europe and
deferred revenue decreased by $0.8 million to account for the portion of the Meda, Myrexis, Endo
and DURECT deferred revenue recognized as revenue. Accounts payable and accrued expenses also
decreased by $0.9 million as a result of payments to our vendors.
In 2009, cash was primarily used to fund our net loss for the year for research and
development, general, administrative and interest expense. The net loss was partially offset by
non-cash charges of $10.5 million in amortization of deferred financing costs and discount on
loans, $1.4 million of stock-based compensation and $0.4 million of depreciation and amortization
expense. Inventory increased by $1.3 million as we prepared for the launch of Ceplene in Europe and
deferred revenue decreased by $0.4 million to account for the portion of the Myrexis, Endo and
DURECT deferred revenue recognized as revenue. Accounts payable and accrued expenses also decreased
by $1.8 million as a result of payments to our vendors.
Investing Activities
Net cash provided by investing activities was $0.1 million in 2010 compared with net cash used
in investing activities was $0.1 million in 2009 and net cash provided by investing activities of
$0.3 million in 2008. In 2010, cash was provided by the release of $40,000 from restricted cash to
pay for interest on the 7.5556% notes and $27,000 from the sale of equipment in our San Diego
research facility. In 2009, cash was used to establish restricted cash for a $9.4 million
make-whole interest payment resulting from the issuance of $25.0 million principal aggregate amount
of 7.5556% convertible senior subordinated notes, the release of $9.3 million from restricted cash
to pay for interest on the 7.5556% notes as the result of the conversion of $24.5 million in
aggregate principal amount of the 7.5556% notes, $0.1 million in proceeds from the sale of
equipment in our San Diego research facility and $27,000 for the purchase of equipment. We do not
anticipate significant capital expenditures in the near future.
Financing Activities
Net cash provided by financing activities was $7.6 million in 2010 compared to $33.9 million
in 2009 and $11.1 million in 2008. In June 2010, we raised $6.7 million in gross proceeds, $6.2
million net of $0.5 million in transaction costs, in connection with the issuance of common stock
and warrants. In November 2010, we raised $2.0 million gross proceeds, $1.9 million net of $0.1
million in transaction costs, in connection with the issuance of common stock and warrants.
During 2010, we sold approximately 31,000 shares of common stock through our at-the-market program
for gross proceeds of $0.1 million. We also received proceeds of $0.8 million related to the
exercise warrants in 2010 and paid $0.9 million towards the remaining balance of our loan with tbg.
In 2009, we issued $25.0 million principal aggregate amount of 7.5556% convertible senior
subordinated notes, netting us $14.0 million after $1.6 million in transaction costs and
establishing a restricted cash account of $9.4 million for make-whole interest. In June 2009, we
raised $9.6 million gross proceeds, $8.9 million net of $0.7 million in transaction costs, in
connection with the issuance of common stock and warrants. We also received proceeds of $3.9
million related to the exercise of approximately 3.4 million warrants in 2009. We repaid the
outstanding loan balance with Hercules of approximately $8,000 and deferred financing costs of $0.2
million, resulting in our having no further obligations under this loan agreement. We also repaid
$1.0 million of the subordinated convertible notes due April 10, 2009 and paid $0.9 million towards
the remaining balance of our loan with tbg.
Contractual Obligations
As of December 31, 2010, the annual amounts of future minimum payments under debt obligations,
interest, lease obligations and other long term liabilities consisting of research, development,
consulting and license agreements (including maintenance fees) are as follows (in thousands of U.S.
dollars, using exchange rates where applicable in effect as of December 31, 2010):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Less than |
|
|
|
|
|
|
|
|
|
|
More than |
|
|
|
|
| |
|
1 Year |
|
|
1 - 3 Years |
|
|
3 - 5 Years |
|
|
5 Years |
|
|
Total |
|
Notes and loans payable |
|
$ |
524 |
|
|
$ |
62 |
|
|
$ |
500 |
|
|
$ |
— |
|
|
$ |
1,086 |
|
Interest expense |
|
|
67 |
|
|
|
78 |
|
|
|
3 |
|
|
|
— |
|
|
|
148 |
|
Operating leases |
|
|
1,089 |
|
|
|
1,469 |
|
|
|
— |
|
|
|
— |
|
|
|
2,558 |
|
Other obligations |
|
|
2,386 |
|
|
|
2,318 |
|
|
|
159 |
|
|
|
— |
|
|
|
4,863 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
$ |
4,066 |
|
|
$ |
3,927 |
|
|
$ |
662 |
|
|
$ |
— |
|
|
$ |
8,655 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
49
€1.5 Million Due 2011. In August 1997, our subsidiary, EpiCept GmbH entered into a ten-year
non-amortizing loan in the amount of €1.5 million with Technologie-Beteiligungs Gesellschaft mbH
der Deutschen Ausgleichsbank, or tbg. The loan initially bore interest at 6% per annum. Tbg was
also entitled to receive additional compensation equal to 9% of the annual surplus (income
before taxes, as defined in the agreement) of EpiCept GmbH, reduced by any other compensation
received from EpiCept GmbH by virtue of other loans to or investments in EpiCept GmbH provided that
tbg is an equity investor in EpiCept GmbH during that time period. We considered the additional
compensation element based on the surplus of EpiCept GmbH to be a derivative. We assigned no value
to the derivative at each reporting period as no surplus of EpiCept GmbH was anticipated over the
term of the agreement. In addition, any additional compensation as a result of surplus would be
reduced by the additional interest noted below.
At the demand of tbg, additional amounts could have been due at the end of the loan term up to
30% of the loan amount, plus 6% of the principal balance of the loan for each year after the
expiration of the fifth complete year of the loan period, such payments to be offset by the
cumulative amount of all payments made to tbg from the annual surplus of EpiCept GmbH. We were
accruing these additional amounts as additional interest up to the maximum amount due over the term
of the loan.
On December 20, 2007, EpiCept GmbH entered into a repayment agreement with tbg, whereby
EpiCept GmbH paid tbg approximately €0.2 million ($0.2 million) in January 2008, representing all
interest payable to tbg as of December 31, 2007. The loan balance of €1.5 million ($2.0 million),
plus accrued interest at a rate of 7.38% per annum beginning January 1, 2008 was required to be
repaid to tbg no later than June 30, 2008. Tbg waived any additional interest payments of
approximately €0.5 million ($0.7 million). EpiCept GmbH considered this a substantial modification
to the original debt agreement and has recorded the new debt at its fair value in accordance with
ASC 470-50, “Modifications and Extinguishments” (“ASC 470-50”). As a result of the modification to
the original debt agreement, EpiCept GmbH recorded a gain on the extinguishment of debt of $0.5
million in December 2007. Accrued interest attributable to the additional interest payments
totaled $0 at December 31, 2009 and 2008.
On May 14, 2008, EpiCept GmbH entered into a prolongation of the repayment agreement with tbg,
whereby the loan balance of €1.5 million ($2.0 million) was required to be repaid to tbg no later
than December 31, 2008. Interest continued to accrue at a rate of 7.38% per annum and all the
provisions of the repayment agreement dated December 20, 2007 continued to apply without change.
On November 26, 2008, EpiCept GmbH entered into a second amendment to the repayment agreement
with tbg, whereby the loan balance of €1.5 million ($2.0 million) was required to be repaid to tbg
no later than June 30, 2009. Interest will continue to accrue at a rate of 7.38% per annum and all
the provisions of the repayment agreement dated December 20, 2007 will continue to apply without
change.
On June 25, 2009, EpiCept GmbH entered into a third amendment to the repayment agreement with
tbg, whereby €0.3 million of the loan balance of €1.5 million ($2.1 million) plus accrued interest
of €56,000 ($0.1 million) was repaid to tbg on June 30, 2009. The remaining loan balance of €1.2
million ($1.7 million) plus accrued interest will be paid in four semi-annual installments of €0.3
million ($0.4 million) beginning December 31, 2009. Interest will continue to accrue at a rate of
7.38% per annum and all the provisions of the repayment agreement dated December 20, 2007 will
continue to apply without change.
$25.0 million 7.5556% Convertible Notes Due 2014. On February 4, 2009, we issued $25.0 million
principal aggregate amount of 7.5556% convertible subordinated notes due February 2014 and five
and-a-half year warrants to purchase approximately 4.2 million shares of common stock at an
exercise price of $3.105 per share. Each $1,000 in principal aggregate amount of the notes is
initially convertible into approximately 371 shares of Common Stock, at the option of the holders
or upon specified events, including a change of control, and if the Company’s common stock trades
at or greater than $5.10 a share for 20 out of 30 days, beginning February 9, 2010. Upon any
conversion or redemption of the notes, the holders will receive a make-whole payment in an amount
equal to the interest payable through the scheduled maturity of the converted or redeemed notes,
less any interest paid before such conversion or redemption. Interest is due and payable on the
notes semi-annually in arrears on June 30 and December 31, beginning on June 30, 2009.
The Company allocated the $25.0 million in proceeds between the convertible subordinated notes
and the warrants based on their relative fair values. The Company calculated the fair value of the
warrants at the date of the transaction at approximately $8.8 million with a corresponding amount
recorded as a debt discount. The debt discount is being accreted over the life of the outstanding
convertible subordinated notes using the effective interest method. At the date of the transaction,
the fair value of the warrants of $8.8 million was determined utilizing the Black-Scholes option
pricing model under the following assumptions: dividend yield of 0%, risk free interest rate of
1.99%, volatility of 118% and an expected life of five years. During 2009, the Company recognized
approximately $8.6 million of non-cash interest expense related to the accretion of the debt
discount as a result of the conversion of $24.5 million of the convertible subordinated notes into
approximately 9.1 million shares of the Company’s common stock.
As of December 31, 2010, after giving effect to the conversions into common stock, the
remaining aggregate principal amount of the Notes outstanding is $0.5 million.
50
$0.8 million Due 2012. In July 2006, Maxim, our wholly-owned subsidiary, issued a six-year
non-interest bearing promissory note in the amount of $0.8 million to Pharmaceutical Research
Associates, Inc., or PRA, as compensation for PRA assuming the liability on a lease in San Diego,
CA. The note is payable in seventy-two equal installments of approximately $11,000 per month. We
terminated our lease of certain property in San Diego, CA as part of our exit plan upon the
completion of the merger with Maxim on January 4, 2006. Our loan balance at December 31, 2010 is
$0.2 million.
Other Commitments. Our long-term commitments under operating leases shown above consist of
payments relating to our facility lease in Tarrytown, New York, which expires in February 2012, and
Munich, Germany, which was due to expire in July 2011. In January 2011, our operating lease in
Munich, Germany was extended to July 2014. Long-term commitments under operating leases for
facilities leased by Maxim and retained by us relate primarily to the research and development site
at 6650 Nancy Ridge Drive in San Diego, which is leased through October 2013. In June 2008, we
defaulted on our lease agreement for the premises located in San Diego, California by failing to
make the monthly rent payment. As a result, the landlord exercised their right to draw down the
full letter of credit, amounting to approximately $0.3 million, and applied approximately $0.2
million to unpaid rent. The remaining balance of $0.1 million is classified as prepaid rent. At
December 31, 2010, we are current with our lease payments on this facility. We discontinued our
drug discovery activities at this location and are currently looking to sublease the premises
located in San Diego, California.
We have a number of research, consulting and license agreements that require us to make
payments to the other party to the agreement upon us attaining certain milestones as defined in the
agreements. As of December 31, 2010, we may be required to make future milestone payments, totaling
approximately $4.9 million, under these agreements, depending upon the success and timing of future
clinical trials and the attainment of other milestones as defined in the respective agreement. Our
current estimate as to the timing of other research, development and license payments, assuming all
related research and development work is successful, is listed in the table above in “Other
obligations.”
We are also obligated to make future royalty payments to three of our collaborators under
existing license agreements, based on net sales of Ceplene®, NP-1 and
crolibulinTM, to the extent revenues on such products are realized. We cannot reasonably
determine the amount and timing of such royalty payments and they are not included in the table
above.
|
|
|
| ITEM 7A. |
|
QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISKS |
The financial currency of our German subsidiary is the euro. As a result, we are exposed to
various foreign currency risks. First, our consolidated financial statements are in U.S. dollars,
but a portion of our consolidated assets and liabilities is denominated in euros. Accordingly,
changes in the exchange rate between the euro and the U.S. dollar will affect the translation of
our German subsidiary’s financial results into U.S. dollars for purposes of reporting consolidated
financial results. We also bear the risk that interest on our euro-denominated debt, when
translated from euros to U.S. dollars, will exceed our current estimates and that principal
payments we make on those loans may be greater than those amounts currently reflected on our
consolidated balance sheet. If the U.S. dollar depreciation to the euro had been 10% more
throughout 2010, we estimate that our interest expense and the fair value of our euro-denominated
debt would have increased by $8,000 and $41,000, respectively. Historically, fluctuations in
exchange rates resulting in transaction gains or losses have had a material effect on our
consolidated financial results. We have not engaged in any hedging activities to minimize this
exposure, although we may do so in the future.
Our exposure to interest rate risk is limited to interest income sensitivity, which is
affected by changes in the general level of U.S. interest rates, particularly because the majority
of our investments are in short-term debt securities and bank deposits. The primary objective of
our investment activities is to preserve principal while at the same time maximizing the income we
receive without significantly increasing risk. To minimize risk, we maintain our portfolio of cash
and cash equivalents in a variety of interest- bearing instruments, primarily bank deposits and
money market funds, which may also include U.S. government and agency securities, high-grade U.S.
corporate bonds and commercial paper. Due to the nature of our short-term and restricted
investments, we believe that we are not exposed to any material interest rate risk. We do not have
any relationships with unconsolidated entities or financial partnerships, such as entities often
referred to as structured finance or special purpose entities, which would have been established
for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or
limited purposes. In addition, we do not engage in trading activities involving non-exchange traded
contracts. Therefore, we are not materially exposed to any financing, liquidity, market or credit
risk that could arise if we had engaged in these relationships. We do not have relationships or
transactions with persons or entities that derive benefits from their non-independent relationship
with us or our related parties.
51
|
|
|
| ITEM 8. |
|
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
See our consolidated financial statements filed with this Annual Report on Form 10-K under
Item 15 below.
|
|
|
| ITEM 9. |
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
52
|
|
|
| ITEM 9A. |
|
CONTROLS AND PROCEDURES |
Disclosure Controls and Procedures
Our Chief Executive Officer and Chief Financial Officer, with the assistance of other members
of our management, have evaluated the effectiveness of our disclosure controls and procedures (as
defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period
covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer
and Chief Financial Officer have concluded that, as of December 31, 2010, our disclosure controls
and procedures were effective.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over
financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. The
Company’s internal control over financial reporting is a process designed to provide reasonable
assurance regarding the reliability of financial reporting and the preparation of financial
statements for external purposes in accordance with accounting principles generally accepted in the
United States of America.
Because of its inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Therefore, even those systems determined to be effective can provide only
reasonable assurance with respect to financial statement preparation and presentation. Also,
projections of any evaluation of effectiveness to future periods are subject to the risk that
controls may become inadequate because of changes in conditions, or that the degree of compliance
with the policies or procedures may deteriorate.
Management assessed the effectiveness of the Company’s internal control over financial
reporting as of December 31, 2010. Management based this assessment on criteria for effective
internal control over financial reporting described in “Internal Control-Integrated Framework”
issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management’s
assessment included an evaluation of the design of the Company’s internal control over financial
reporting and testing of the operational effectiveness of its internal control over financial
reporting. Management reviewed the results of its assessment with the Audit Committee.
There have not been any changes in our internal control over financial reporting (as defined
in Rule 13a-15(f)) during the fiscal quarter ended December 31, 2010 that have materially affected,
or are reasonably likely to materially affect, the Company’s internal control over financial
reporting.
Based on this assessment, management determined that, as of December 31, 2010, the Company’s
internal control over financial reporting was effective to provide reasonable assurance regarding
the reliability of financial reporting and the preparation of financial statements for external
purposes in accordance with accounting principles generally accepted in the United States of
America.
|
|
|
| ITEM 9B. |
|
OTHER INFORMATION |
None.
53
PART III
|
|
|
| ITEM 10. |
|
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Management and Board of Directors
The information concerning our directors, executive officers and corporate governance required
under this Item is incorporated herein by reference from our proxy statement, which will be filed
with the Securities and Exchange Commission, relating to our 2011 Annual Meeting of Stockholders
(referred to as our 2011 Proxy Statement).
|
|
|
| ITEM 11. |
|
EXECUTIVE COMPENSATION |
The information required under this Item is incorporated herein by reference from our 2011
Proxy Statement.
|
|
|
| ITEM 12. |
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED
STOCKHOLDER MATTERS |
The information required under this Item is incorporated herein by reference from our 2011
Proxy Statement.
|
|
|
| ITEM 13. |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE |
The information required under this Item is incorporated herein by reference from our 2011
Proxy Statement.
|
|
|
| ITEM 14. |
|
PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required under this Item is incorporated herein by reference from our 2011
Proxy Statement.
54
PART IV
|
|
|
| ITEM 15. |
|
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
| (a) |
|
(1) and (2) Financial Statements and Financial Statement Schedules |
The following consolidated financial statements of EpiCept Corporation and subsidiaries, the notes
thereto and the related report thereon of independent registered public accounting firm are filed
under Item 8 of this report.
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets as of December 31, 2010 and 2009
Consolidated Statements of Operations for the Years Ended December 31, 2010, 2009 and 2008
Consolidated Statements of Stockholders’ Deficit for the Years Ended December 31, 2010, 2009 and
2008
Consolidated Statements of Cash Flows for the Years Ended December 31, 2010, 2009 and 2008
Notes to Consolidated Financial Statements
Schedules for which provision is made in the applicable accounting regulations of the Securities
and Exchange Commission are omitted because they either are not required under the related
instructions, are inapplicable, or the required information is shown in the consolidated financial
statements or the notes thereto.
| (a) |
|
(3) See Exhibits Index. |
| |
| (b) |
|
Exhibits. See Item 15 (a) (3) above. |
| |
| (c) |
|
Financial Statement Schedules |
None.
55
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934,
Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto
duly authorized.
| |
|
|
|
|
| |
EPICEPT CORPORATION
|
|
| |
By: |
/s/ John V. Talley
|
|
| |
|
John V. Talley |
|
| |
|
President and Chief Executive Officer
March 31, 2011 |
|
| |
Pursuant to the requirements of the Securities Act of 1934, this report has been signed by the
following persons in the capacities indicated and on the dates indicated:
| |
|
|
|
|
| Signature |
|
Title |
|
Date |
/s/ John V. Talley
John V. Talley
|
|
Director, President and Chief Executive Officer
(Principal
Executive Officer)
|
|
March 31, 2011 |
|
|
|
|
|
/s/ Robert W. Cook
Robert W. Cook
|
|
Chief Financial Officer
(Principal
Financial and Accounting Officer)
|
|
March 31, 2011 |
|
|
|
|
|
/s/ Robert G. Savage
Robert G. Savage
|
|
Director
|
|
March 31, 2011 |
|
|
|
|
|
/s/ Guy C. Jackson
Guy C. Jackson
|
|
Director
|
|
March 31, 2011 |
|
|
|
|
|
/s/ Gerhard Waldheim
Gerhard Waldheim
|
|
Director
|
|
March 31, 2011 |
|
|
|
|
|
/s/ Wayne P. Yetter
Wayne P. Yetter
|
|
Director
|
|
March 31, 2011 |
|
|
|
|
|
/s/ A. Collier Smyth
A. Collier Smyth
|
|
Director
|
|
March 31, 2011 |
56
INDEX TO FINANCIAL STATEMENTS
EPICEPT CORPORATION AND SUBSIDIARIES CONSOLIDATED FINANCIAL STATEMENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
Epicept Corporation
Tarrytown, NY
We have audited the accompanying consolidated balance sheets of EpiCept Corporation and
subsidiaries (the “Company”) as of December 31, 2010 and 2009, and the related consolidated
statements of operations, stockholders’ deficit, and cash flows for each of the three years in the
period ended December 31, 2010. These consolidated financial statements are the responsibility of
the Company’s management. Our responsibility is to express an opinion on these consolidated
financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight
Board (United States). Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are free of material misstatement. The
Company is not required to have, nor were we engaged to perform, an audit of its internal control
over financial reporting. Our audits included consideration of internal control over financial
reporting as a basis for designing audit procedures that are appropriate in the circumstances, but
not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control
over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on
a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit
also includes assessing the accounting principles used and significant estimates made by
management, as well as evaluating the overall financial statement presentation. We believe that our
audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects,
the financial position of EpiCept Corporation and subsidiaries as of December 31, 2010, and 2009,
and the results of their operations and their cash flows for each of the three years in the period
ended December 31, 2010, in conformity with accounting principles generally accepted in the United
States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company
will continue as a going concern. As discussed in Note 2 to the consolidated financial statements,
the Company’s recurring losses from operations and stockholders’ deficit raise substantial doubt
about its ability to continue as a going concern. Management’s plans concerning these matters are
also described in Note 2 to the consolidated financial statements. The consolidated financial
statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Deloitte & Touche LLP
Parsippany, New Jersey
March 31, 2011
F-2
EpiCept Corporation and Subsidiaries
Consolidated Balance Sheets
(In thousands, except share and per share amounts)
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
ASSETS
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
2,435 |
|
|
$ |
5,142 |
|
Inventory |
|
|
179 |
|
|
|
1,315 |
|
Prepaid expenses and other current assets |
|
|
333 |
|
|
|
414 |
|
|
|
|
|
|
|
|
Total current assets |
|
|
2,947 |
|
|
|
6,871 |
|
Restricted cash |
|
|
189 |
|
|
|
227 |
|
Property and equipment, net |
|
|
222 |
|
|
|
360 |
|
Long-term inventory |
|
|
825 |
|
|
|
— |
|
Deferred financing costs |
|
|
506 |
|
|
|
28 |
|
Identifiable intangible asset, net |
|
|
— |
|
|
|
28 |
|
|
|
|
|
|
|
|
Total assets |
|
$ |
4,689 |
|
|
$ |
7,514 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ DEFICIT
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
1,244 |
|
|
$ |
1,808 |
|
Accrued research contract costs |
|
|
859 |
|
|
|
1,174 |
|
Other accrued liabilities |
|
|
1,286 |
|
|
|
1,072 |
|
Notes and loans payable, current portion |
|
|
524 |
|
|
|
985 |
|
Deferred revenue, current portion |
|
|
921 |
|
|
|
425 |
|
|
|
|
|
|
|
|
Total current liabilities |
|
|
4,834 |
|
|
|
5,464 |
|
|
|
|
|
|
|
|
Notes and loans payable |
|
|
62 |
|
|
|
618 |
|
7.5556% Convertible Subordinated Notes Due 2014 |
|
|
386 |
|
|
|
349 |
|
Deferred revenue |
|
|
12,905 |
|
|
|
9,197 |
|
Deferred rent and other noncurrent liabilities |
|
|
637 |
|
|
|
965 |
|
|
|
|
|
|
|
|
Total long term liabilities |
|
|
13,990 |
|
|
|
11,129 |
|
|
|
|
|
|
|
|
Total liabilities |
|
|
18,824 |
|
|
|
16,593 |
|
|
|
|
|
|
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
Stockholders’ Deficit: |
|
|
|
|
|
|
|
|
Common stock, $.0001 par value; authorized
225,000,000 shares; issued 54,991,535 shares
and 44,051,782 shares at December 31, 2010 and
2009, respectively |
|
|
5 |
|
|
|
4 |
|
Additional paid-in capital |
|
|
210,540 |
|
|
|
203,445 |
|
Warrants |
|
|
27,227 |
|
|
|
24,503 |
|
Accumulated deficit |
|
|
(250,582 |
) |
|
|
(235,045 |
) |
Accumulated other comprehensive loss |
|
|
(1,250 |
) |
|
|
(1,911 |
) |
Treasury stock, at cost (4,167 shares) |
|
|
(75 |
) |
|
|
(75 |
) |
|
|
|
|
|
|
|
Total stockholders’ deficit |
|
|
(14,135 |
) |
|
|
(9,079 |
) |
|
|
|
|
|
|
|
Total liabilities and stockholders’ deficit |
|
$ |
4,689 |
|
|
$ |
7,514 |
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these consolidated financial statements.
F-3
EpiCept Corporation and Subsidiaries
Consolidated Statements of Operations
(In thousands, except share and per share amounts)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net product sales |
|
$ |
158 |
|
|
$ |
— |
|
|
$ |
— |
|
Licensing and other revenue |
|
|
836 |
|
|
|
414 |
|
|
|
265 |
|
|
|
|
|
|
|
|
|
|
|
Revenue |
|
|
994 |
|
|
|
414 |
|
|
|
265 |
|
|
|
|
|
|
|
|
|
|
|
| |
Costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Costs of goods sold |
|
|
997 |
|
|
|
— |
|
|
|
— |
|
Selling, general and administrative |
|
|
7,244 |
|
|
|
7,548 |
|
|
|
9,599 |
|
Research and development |
|
|
8,127 |
|
|
|
11,603 |
|
|
|
12,623 |
|
|
|
|
|
|
|
|
|
|
|
Total costs and expenses |
|
|
16,368 |
|
|
|
19,151 |
|
|
|
22,222 |
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
|
|
(15,374 |
) |
|
|
(18,737 |
) |
|
|
(21,957 |
) |
|
|
|
|
|
|
|
|
|
|
Other income (expense), net: |
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
7 |
|
|
|
32 |
|
|
|
33 |
|
Foreign exchange gain (loss) |
|
|
(538 |
) |
|
|
221 |
|
|
|
(327 |
) |
Interest expense (See note 3) |
|
|
(361 |
) |
|
|
(20,021 |
) |
|
|
(1,266 |
) |
Other income (See note 3) |
|
|
733 |
|
|
|
— |
|
|
|
— |
|
Change in value of warrants and derivatives |
|
|
— |
|
|
|
(305 |
) |
|
|
113 |
|
Loss on extinguishment of debt |
|
|
— |
|
|
|
— |
|
|
|
(1,975 |
) |
|
|
|
|
|
|
|
|
|
|
Other expense, net |
|
|
(159 |
) |
|
|
(20,073 |
) |
|
|
(3,422 |
) |
|
|
|
|
|
|
|
|
|
|
Loss before income tax expense |
|
|
(15,533 |
) |
|
|
(38,810 |
) |
|
|
(25,379 |
) |
Income tax expense |
|
|
(4 |
) |
|
|
(4 |
) |
|
|
(3 |
) |
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(15,537 |
) |
|
$ |
(38,814 |
) |
|
$ |
(25,382 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted loss per common share |
|
$ |
(0.32 |
) |
|
$ |
(0.97 |
) |
|
$ |
(1.23 |
) |
Weighted average common shares outstanding |
|
|
47,853,560 |
|
|
|
40,139,299 |
|
|
|
20,685,711 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-4
EpiCept Corporation and Subsidiaries
Consolidated Statements of Stockholders’ Deficit
(In thousands, except share and per share amounts)
For the Years Ended December 31, 2008, 2009 and 2010
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
|
|
|
|
|
|
Other |
|
|
|
|
|
|
|
| |
|
Common Stock |
|
|
Paid-In |
|
|
|
|
|
|
Accumulated |
|
|
Comprehensive |
|
|
Treasury |
|
|
Stockholders’ |
|
| |
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Warrants |
|
|
Deficit |
|
|
(Loss) Income |
|
|
Stock |
|
|
Deficit |
|
| |
Balance at January 1, 2008 |
|
|
15,294,008 |
|
|
|
2 |
|
|
|
148,770 |
|
|
|
10,025 |
|
|
|
(170,849 |
) |
|
|
(2,050 |
) |
|
|
(75 |
) |
|
|
(14,177 |
) |
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(25,382 |
) |
|
|
— |
|
|
|
— |
|
|
|
(25,382 |
) |
Foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
432 |
|
|
|
— |
|
|
|
432 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(25,382 |
) |
|
|
432 |
|
|
|
— |
|
|
|
(24,950 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercise of warrants |
|
|
2,097,860 |
|
|
|
— |
|
|
|
4,159 |
|
|
|
(1,599 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,560 |
|
Issuance of common stock and warrants, net |
|
|
8,718,304 |
|
|
|
1 |
|
|
|
8,386 |
|
|
|
4,930 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
13,317 |
|
Issuance of common stock under ESPP |
|
|
166,667 |
|
|
|
— |
|
|
|
102 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
102 |
|
Issuance of common stock as payment of
loan |
|
|
1,197,411 |
|
|
|
— |
|
|
|
1,850 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,850 |
|
Issuance of restricted common stock, net |
|
|
11,468 |
|
|
|
— |
|
|
|
50 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
50 |
|
Issuance of warrants |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,288 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,288 |
|
Amortization of deferred stock
compensation |
|
|
— |
|
|
|
— |
|
|
|
2,230 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,230 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2008 |
|
|
27,485,718 |
|
|
|
3 |
|
|
|
165,547 |
|
|
|
14,644 |
|
|
|
(196,231 |
) |
|
|
(1,618 |
) |
|
|
(75 |
) |
|
|
(17,730 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(38,814 |
) |
|
|
— |
|
|
|
— |
|
|
|
(38,814 |
) |
Foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(293 |
) |
|
|
— |
|
|
|
(293 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(38,814 |
) |
|
|
(293 |
) |
|
|
— |
|
|
|
(39,107 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclass warrants from liability to equity |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,288 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,288 |
|
Issuance of common stock warrants in
connection with 7.5556% convertible
subordinated notes |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
8,777 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
8,777 |
|
Issuance of common stock and warrants, net |
|
|
4,000,000 |
|
|
|
— |
|
|
|
6,900 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
6,900 |
|
Issuance of restricted common stock and
restricted stock units, net |
|
|
22,670 |
|
|
|
— |
|
|
|
55 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
55 |
|
Issuance of common stock under ESPP |
|
|
49,065 |
|
|
|
— |
|
|
|
75 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
75 |
|
Conversion of 7.5556% convertible
subordinated notes into Common Stock |
|
|
9,074,065 |
|
|
|
1 |
|
|
|
24,499 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
24,500 |
|
Exercise of warrants |
|
|
3,361,375 |
|
|
|
— |
|
|
|
4,976 |
|
|
|
(1,206 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,770 |
|
Exercise of stock options |
|
|
58,889 |
|
|
|
— |
|
|
|
53 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
53 |
|
Amortization of deferred stock
compensation |
|
|
— |
|
|
|
— |
|
|
|
1,340 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,340 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2009 |
|
|
44,051,782 |
|
|
$ |
4 |
|
|
$ |
203,445 |
|
|
$ |
24,503 |
|
|
$ |
(235,045 |
) |
|
$ |
(1,911 |
) |
|
$ |
(75 |
) |
|
$ |
(9,079 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(15,537 |
) |
|
|
— |
|
|
|
— |
|
|
|
(15,537 |
) |
Foreign currency translation adjustment |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
661 |
|
|
|
— |
|
|
|
661 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(15,537 |
) |
|
|
661 |
|
|
|
— |
|
|
|
(14,876 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock and warrants,
net of issuance costs |
|
|
9,445,554 |
|
|
|
1 |
|
|
|
4,483 |
|
|
|
3,604 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
8,088 |
|
Issuance of common stock resulting from
reverse split |
|
|
3,828 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Issuance of restricted common stock and
restricted stock units, net |
|
|
22,349 |
|
|
|
— |
|
|
|
30 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
30 |
|
Issuance of common stock under ESPP |
|
|
32,821 |
|
|
|
— |
|
|
|
23 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
23 |
|
Exercise of warrants |
|
|
1,435,201 |
|
|
|
— |
|
|
|
1,653 |
|
|
|
(880 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
773 |
|
Amortization of deferred stock
compensation |
|
|
— |
|
|
|
— |
|
|
|
906 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
906 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2010 |
|
|
54,991,535 |
|
|
$ |
5 |
|
|
$ |
210,540 |
|
|
$ |
27,227 |
|
|
$ |
(250,582 |
) |
|
$ |
(1,250 |
) |
|
$ |
(75 |
) |
|
$ |
(14,135 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these consolidated financial statements.
F-5
EpiCept Corporation and Subsidiaries
Consolidated Statements of Cash Flows
(In thousands)
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(15,537 |
) |
|
$ |
(38,814 |
) |
|
$ |
(25,382 |
) |
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
170 |
|
|
|
368 |
|
|
|
237 |
|
Gain on disposal of assets, net |
|
|
(27 |
) |
|
|
(115 |
) |
|
|
(64 |
) |
Foreign exchange loss (gain) |
|
|
538 |
|
|
|
(221 |
) |
|
|
327 |
|
Stock-based compensation expense |
|
|
935 |
|
|
|
1,395 |
|
|
|
2,281 |
|
Amortization of deferred financing costs and discount on loans |
|
|
151 |
|
|
|
10,508 |
|
|
|
431 |
|
Change in value of warrants and derivatives |
|
|
— |
|
|
|
305 |
|
|
|
(113 |
) |
Excess inventory expense |
|
|
852 |
|
|
|
— |
|
|
|
— |
|
Loss on extinguishment of debt |
|
|
— |
|
|
|
— |
|
|
|
1,725 |
|
Change in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
Increase in inventory |
|
|
(541 |
) |
|
|
(1,315 |
) |
|
|
— |
|
Decrease (increase) in prepaid expenses and other current assets |
|
|
81 |
|
|
|
(19 |
) |
|
|
211 |
|
Decrease in other assets |
|
|
— |
|
|
|
26 |
|
|
|
1 |
|
(Decrease) increase in accounts payable |
|
|
(602 |
) |
|
|
(769 |
) |
|
|
1,474 |
|
(Decrease) increase in accrued research contract costs |
|
|
(314 |
) |
|
|
100 |
|
|
|
(103 |
) |
Increase (decrease) in other accrued liabilities |
|
|
19 |
|
|
|
(1,035 |
) |
|
|
353 |
|
Increase in deferred revenue |
|
|
5,000 |
|
|
|
— |
|
|
|
3,375 |
|
Recognition of deferred revenue |
|
|
(796 |
) |
|
|
(368 |
) |
|
|
(222 |
) |
(Decrease) increase in other liabilities |
|
|
(329 |
) |
|
|
501 |
|
|
|
(168 |
) |
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities |
|
|
(10,400 |
) |
|
|
(29,453 |
) |
|
|
(15,637 |
) |
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Restricted cash from issuance of 7.5556% convertible subordinated notes |
|
|
— |
|
|
|
(9,444 |
) |
|
|
— |
|
Release of restricted cash in connection with conversion of 7.5556%
convertible subordinated notes |
|
|
38 |
|
|
|
9,288 |
|
|
|
— |
|
Release of letter of credit from restricted cash |
|
|
— |
|
|
|
— |
|
|
|
265 |
|
Purchase of property and equipment |
|
|
(2 |
) |
|
|
(27 |
) |
|
|
(37 |
) |
Proceeds from sale of property and equipment |
|
|
27 |
|
|
|
131 |
|
|
|
64 |
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities |
|
|
63 |
|
|
|
(52 |
) |
|
|
292 |
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from exercise of stock options and warrants |
|
|
796 |
|
|
|
3,899 |
|
|
|
2,661 |
|
Proceeds from issuance of common stock and warrants, net |
|
|
8,088 |
|
|
|
8,759 |
|
|
|
13,277 |
|
Proceeds from loans and warrants |
|
|
— |
|
|
|
25,000 |
|
|
|
1,000 |
|
Repayment of loans |
|
|
(888 |
) |
|
|
(2,096 |
) |
|
|
(5,794 |
) |
Debt issuance costs |
|
|
(359 |
) |
|
|
(1,688 |
) |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities |
|
|
7,637 |
|
|
|
33,874 |
|
|
|
11,144 |
|
|
|
|
|
|
|
|
|
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
(7 |
) |
|
|
(17 |
) |
|
|
48 |
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents |
|
|
(2,707 |
) |
|
|
4,352 |
|
|
|
(4,153 |
) |
Cash and cash equivalents at beginning of year |
|
|
5,142 |
|
|
|
790 |
|
|
|
4,943 |
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of year |
|
$ |
2,435 |
|
|
$ |
5,142 |
|
|
$ |
790 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
137 |
|
|
$ |
9,477 |
|
|
$ |
872 |
|
Cash paid for income taxes |
|
|
4 |
|
|
|
4 |
|
|
|
3 |
|
Supplemental disclosure of non-cash financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Conversion of 7.5556% convertible subordinated notes into common stock |
|
|
— |
|
|
|
24,500 |
|
|
|
— |
|
Conversion of senior secured term loan into common stock |
|
|
— |
|
|
|
— |
|
|
|
1,850 |
|
Reclassification of warrants from equity to liability |
|
|
— |
|
|
|
— |
|
|
|
418 |
|
Reclassification of warrants from liability to equity |
|
|
— |
|
|
|
2,288 |
|
|
|
(305 |
) |
Unpaid costs associated with issuance of common stock |
|
|
— |
|
|
|
— |
|
|
|
124 |
|
Unpaid financing, initial public offering costs and acquisition costs |
|
|
233 |
|
|
|
— |
|
|
|
263 |
|
The accompanying notes are an integral part of these consolidated financial statements.
F-6
EPICEPT CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years Ended December 31, 2010, 2009 and 2008
1. Reverse Split of Common Stock
On January 14, 2010, EpiCept Corporation (“EpiCept” or the “Company”) effected a previously
authorized 1-for-3 reverse stock split of its common stock. The reverse stock split took effect at
the start of trading on January 15, 2010 on a 1-for-3 split-adjusted basis. All prior periods have
been retroactively adjusted to reflect the reverse stock split.
2. Organization and Description of Business
EpiCept is a specialty pharmaceutical company focused on the development and commercialization
of pharmaceutical products for the treatment of cancer and pain. The Company’s strategy is to focus
its development efforts on innovative cancer therapies and topically delivered analgesics targeting
peripheral nerve receptors. The Company’s lead product is Ceplene®, which when used
concomitantly with low dose interleukin-2 (“IL-2”) is intended as remission maintenance therapy in
the treatment of acute myeloid leukemia, or AML, for adult patients who are in their first complete
remission. In addition to Ceplene®, the Company has two other oncology compounds and a
late-stage pain product candidate for the treatment of peripheral neuropathies in clinical
development. The Company believes this portfolio of oncology and pain management product
candidates lessens its reliance on the success of any single product or product candidate.
In October 2008, the European Commission issued a formal marketing authorization for
Ceplene® in the European Union. In January 2010, EpiCept completed an agreement with
Meda AB of Sweden to market and sell Ceplene® in Europe and certain Pacific Rim countries. The
commercial launch of Ceplene® commenced in the United Kingdom in April 2010 and launches
are continuing in other European countries. In December 2010, Ceplene® was approved for
marketing in Israel. Megapharm Ltd. is EpiCept’s partner in Israel and upon Ministry of Health
approval of labeling and other technical matters the Company is expected to launch Ceplene®
in the first half of 2011.
In June 2010, the Company filed a New Drug Application (“NDA”) with the United States Food and
Drug Administration (“FDA”) for the same indication. In August 2010, the Company received a
refusal to file letter from the FDA on the NDA for Ceplene®. In the FDA’s preliminary review of
the Ceplene® NDA, the FDA concluded that the application did not establish Ceplene®’s
therapeutic contribution in its combination with IL-2, and recommended that an additional pivotal
trial assessing Ceplene®’s contribution and using overall survival as a primary endpoint be
conducted. In October 2010, the Company reached an agreement with the FDA for a two-arm,
randomized, open-label trial that will compare the efficacy of Ceplene® plus low-dose IL-2 to
standard of care in this indication. Based on FDA guidance, the primary endpoint of the trial will
be overall patient survival. The Company’s previous Phase III trial demonstrated a statistically
significant prolongation of the primary endpoint of leukemia-free survival and extended overall
survival by more than one year in patients in their first complete remission. As a next step,
EpiCept will submit to the FDA a detailed Phase III protocol. The FDA, via the Special Protocol
Assessment (SPA) procedure, will provide guidance on specific sections of the protocol including
the adaptive design with prospectively defined rules for sample size selection for both futility
and expected success. The Company also recently filed an application with the FDA to grant Ceplene®
fast track status. In addition to other benefits, if granted, fast track status permits an
expedited review of the Ceplene® New Drug Application (NDA) once it is filed.
The Company’s other oncology compounds include crolibulinTM, a novel small molecule
vascular disruption agent (“VDA”), and apoptosis inducer for the treatment of patients with solid
tumors and lymphomas. In December 2010, the National Cancer Institute (“NCI”) initiated a Phase II
clinical trial for crolibulinTM to assess the drug’s efficacy and safety in combination
with cisplatin in patients with anaplastic thyroid cancer (ATC). AzixaTM, an apoptosis
inducer with VDA activity licensed by the Company to Myrexis, Inc. (formerly Myriad
Pharmaceuticals, Inc.), as part of an exclusive, worldwide development and commercialization
agreement, is currently in Phase II clinical trials in patients with primary glioblastoma and
cancer that has metastasized to the brain.
F-7
The Company’s late-stage pain product candidate, EpiCeptTM NP-1 Cream,
(“NP-1”), is a prescription topical analgesic cream designed to provide effective long-term relief
of pain associated with peripheral neuropathies. In January 2009, the Company concluded a Phase II
clinical trial of NP-1 in which the Company studied its safety and efficacy in patients suffering
from post-herpetic neuralgia, or PHN, compared to gabapentin and placebo. This trial met its
primary endpoints. In February 2008, the Company concluded a Phase II clinical study of NP-1 in
patients suffering from diabetic peripheral neuropathy, or DPN. Both studies support the
advancement of NP-1 into a registration-sized trial. In February 2011, the Company presented data
from its Phase IIb trial for
EpiCept™ NP-1 in chemotherapy-induced peripheral neuropathy (“CPN”) which is being conducted
by NCI. CPN may affect 50% of women undergoing treatment for breast cancer. A common therapeutic
agent for the treatment of advanced breast cancer is paclitaxel, and as many as 80% of the patients
with advanced breast cancer experience some signs and symptoms of CPN, such as burning, tingling
pain associated sometimes with mild muscular weakness, after high dose paclitaxel administration.
The multi-center, double-blind, randomized, placebo-controlled study was conducted within a network
of approximately 25 sites under the direction of the NCI funded Community Clinical Oncology Program
(“CCOP”). More than 460 cancer survivors suffering from painful CPN were enrolled in the six-week
study. The results of the trial in the intent to treat (“ITT”) population demonstrated that the
change in average daily neuropathy intensity scores in the NP-1 group achieved a statistically
significant reduction in CPN intensity versus placebo (p<0.001), which was the trial’s primary
endpoint. Additionally, a pre-specified subgroup of the ITT population, those patients who
previously received taxane chemotherapy, also showed a statistically significant reduction in
average daily neuropathy intensity scores (p=0.034). This subgroup constituted more than 50% of the
ITT population. Secondary efficacy endpoints confirmed the superiority of NP-1 vs. placebo.
Furthermore, the safety profile of NP-1 was comparable to placebo.
Ceplene® has been granted full marketing authorization by the European
Commission for remission maintenance and prevention of relapse in adult patients with Acute Myeloid
Leukemia in first remission. None of the Company’s other drug candidates has received FDA or
foreign regulatory marketing approval. In order to grant marketing approval, the FDA or foreign
regulatory agencies must conclude that the Company’s clinical data and that of its collaborators
establish the safety and efficacy of its drug candidates. Furthermore, the Company’s strategy
includes entering into collaborative arrangements with third parties to participate in the clinical
development and commercialization of its products. In the event that third parties have control
over the preclinical development or clinical trial process for a product candidate, the estimated
completion date would largely be under control of that third party rather than under the Company’s
control. The Company cannot forecast with any degree of certainty which of its drug candidates will
be subject to future collaborations or how such arrangements would affect the Company’s development
plan or capital requirements.
The Company is subject to a number of risks associated with companies in the specialty
pharmaceutical industry. Principal among these are risks associated with the Company’s ability to
obtain regulatory approval for its product candidates, its ability to adequately fund its
operations, dependence on collaborative arrangements, the development by the Company or its
competitors of new technological innovations, the dependence on key personnel, the protection of
proprietary technology, the compliance with the FDA and other governmental regulations. The Company
has yet to generate significant product revenues from any of its product candidates. The Company
has financed its operations primarily through the proceeds from the sales of common stock,
warrants, debt instruments, cash proceeds from collaborative relationships and investment income
earned on cash balances and short-term investments.
The Company has prepared its consolidated financial statements under the assumption that it is
a going concern. The Company has devoted substantially all of its cash resources to research and
development programs and general and administrative expenses, and to date it has not generated any
significant revenues from the sale of products. Since inception, the Company has incurred
significant net losses each year. As a result, the Company has an accumulated deficit of $250.6
million as of December 31, 2010. The Company’s recurring losses from operations and the accumulated
deficit raise substantial doubt about its ability to continue as a going concern. The consolidated
financial statements do not include any adjustments that might result from the outcome of this
uncertainty. The Company’s losses have resulted principally from costs incurred in connection with
its development activities and from general and administrative expenses. Even if the Company
succeeds in developing and commercializing one or more of its product candidates, the Company may
never become profitable. The Company expects to continue to incur significant expenses over the
next several years as it:
| |
• |
|
applies for marketing approval of Ceplene® in the U.S., Canada, and other
countries |
| |
• |
|
continues to conduct clinical trials for its product candidates; |
| |
• |
|
seeks regulatory approvals for its product candidates; |
| |
• |
|
develops, formulates, and commercializes its product candidates; |
| |
• |
|
implements additional internal systems and develops new infrastructure; |
| |
• |
|
acquires or in-licenses additional products or technologies or expand the use of its
technologies; |
| |
• |
|
maintains, defends and expands the scope of its intellectual property; and |
| |
• |
|
hires additional personnel. |
F-8
Cash at December 31, 2010, plus $4.6 million of cash received in March 2011 from the issuance
of approximately 7.1 million shares of our common stock and warrants to purchase 5.3 million shares
of our common stock and $7.1 million of cash received in February 2011 from the issuance of
approximately 8.9 million shares of our common stock and warrants to purchase 3.6 million shares of
our common stock will be sufficient to fund the Company’s operations and meet our debt service
requirements into 2012.
In November 2010 the Company provided an update with respect to its financing plans. The key
element of the plan is a non-equity financing transaction that, if fully completed, will support
the Company’s operations for an additional year. The Company anticipates that the transaction,
which is subject to completion of due diligence and execution of mutually-satisfactory
documentation, will close in 2011, but should the transaction not close or the proceeds be less
than anticipated the Company may determine to seek additional or alternative sources or types of
financing, including equity financing.
3. Significant Accounting Policies
Consolidation
The accompanying consolidated financial statements include the accounts of EpiCept Corporation
and the Company’s 100%-owned subsidiaries. All inter-company transactions and balances have been
eliminated.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally
accepted in the United States of America requires management to make estimates and assumptions that
affect the reported amounts of assets and liabilities and disclosure of contingent assets and
liabilities at the date of the financial statements. Estimates also affect the reported amounts of
revenues and expenses during the reporting period. Actual results could differ from those
estimates.
Revenue Recognition
The Company recognizes revenue relating to its collaboration agreements in accordance with the
Securities and Exchange Commission’s Staff Accounting Bulletin No. 104, Revenue Recognition, and
FASB Accounting Standards Codification (“ASC”) 605-25, “Revenue Recognition — Multiple Element
Arrangements” (“ASC 605-25”). Revenue under collaborative arrangements may result from license
fees, milestone payments, research and development payments and royalty payments.
The Company’s application of these standards requires subjective determinations and requires
management to make judgments about value of the individual elements and whether they are separable
from the other aspects of the contractual relationship. The Company evaluates its collaboration
agreements to determine units of accounting for revenue recognition purposes. To date, the Company
has determined that its upfront non-refundable license fees cannot be separated from its ongoing
collaborative research and development activities and, accordingly, does not treat them as a
separate element. The Company recognizes revenue from non-refundable, upfront licenses and related
payments, not specifically tied to a separate earnings process, either on the proportional
performance method or ratably over either the development period in which the Company is obligated
to participate on a continuing and substantial basis in the research and development activities
outlined in the contract, or the later of 1) the conclusion of the royalty term on a jurisdiction
by jurisdiction basis or 2) the expiration of the last EpiCept licensed patent. Ratable revenue
recognition is only utilized if the research and development services are performed systematically
over the development period. Proportional performance is measured based on costs incurred compared
to total estimated costs to be incurred over the development period which approximates the
proportion of the value of the services provided compared to the total estimated value over the
development period. The Company periodically reviews its estimates of cost and the length of the
development period and, to the extent such estimates change, the impact of the change is recorded
at that time.
EpiCept recognizes milestone payments as revenue upon achievement of the milestone only if (1)
it represents a separate unit of accounting as defined in ASC 605-25; (2) the milestone payments
are nonrefundable; (3) substantive effort is involved in achieving the milestone; and (4) the
amount of the milestone is reasonable in relation to the effort expended or the risk associated
with the achievement of the milestone. If any of these conditions is not met, EpiCept will
recognize milestones as revenue in accordance with its accounting policy in effect for the
respective contract. For current agreements, EpiCept recognizes revenue for milestone payments
based upon the portion of the development services that are completed to date and defers the
remaining portion and recognizes it over the remainder of the development services on the
proportional or ratable method, whichever is applicable. Deferred revenue represents the excess of
cash received compared to revenue recognized to date under licensing agreements.
F-9
EpiCept has chosen early adoption of the milestone method of Revenue Recognition as defined in
ASC 605-28, “Revenue Recognition — Milestone Method” on a prospective basis as of January 1, 2010.
Under this method of revenue recognition, the Company will recognize into revenue research-related
milestone payments for which there is substantial uncertainty at the date the arrangement is
entered into that the event will be achieved, when that event can only be achieved based in whole
or in part on EpiCept’s performance or a specific outcome resulting from EpiCept’s performance and,
if achieved, would result in additional payments being due to EpiCept. This accounting will be
applicable to research milestones under the license agreement entered into with Meda AB in 2010 and
all future agreements.
Revenue from the sale of product is recognized when title and risk of loss of the product is
transferred to the customer. Provisions for discounts, early payments, rebates, sales returns and
distributor chargebacks under terms customary in the industry, if any, are provided for in the same
period the related sales are recorded.
Royalty revenue is recognized in the period in which the sales occur, provided that the
royalty amounts are fixed or determinable, collection of the related receivable is reasonably
assured and the Company has no remaining performance obligations under the arrangement providing
for the royalty. If royalties are received when the Company has remaining performance obligations,
they would be attributed to the services being provided under the arrangement and, therefore,
recognized as such obligations are performed under either the proportionate performance or ratable
methods, as applicable.
Foreign Currency
The financial statements of the Company’s foreign subsidiary are translated into U.S. dollars
using the period-end exchange rate for all balance sheet accounts and the average exchange rates
for income statement accounts. Adjustments resulting from translation have been reported in other
comprehensive loss.
Gains or losses from foreign currency transactions relating to inter-company debt are recorded
in the consolidated statements of operations in other income (expense).
Stock-Based Compensation
The Company has various stock-based compensation plans for employees and outside directors,
which are described more fully in Note 11 “Stock Options and Warrants.”
Income Taxes
The Company accounts for income taxes in accordance with ASC 740, “Income Taxes.” The Company
files income tax returns in the U.S. federal jurisdiction and various state and foreign
jurisdictions. The Company’s income tax returns for tax years after 2006 are still subject to
review. The Company does not believe there will be any material changes in its unrecognized tax
positions over the next 12 months.
The Company’s policy is to recognize interest and penalties accrued on any unrecognized tax
benefits as a component of operating expense. The Company did not have any accrued interest or
penalties associated with any unrecognized tax benefits, nor was any interest expense recognized
during the years ended December 31, 2010, 2009 and 2008. Income tax expense for the years ended
December 31, 2010, 2009 and 2008 is primarily due to minimum state and local income taxes.
The Company accounts for its income taxes under the asset and liability method. Under the
asset and liability method, deferred tax assets and liabilities are recognized based upon the
differences arising from carrying amounts of the Company’s assets and liabilities for tax and
financial reporting purposes using enacted tax rates in effect for the year in which the
differences are expected to reverse. The effect on the deferred tax assets and liabilities of a
change in tax rates is recognized in the period when the change in tax rates is enacted. A
valuation allowance is established when it is determined that it is more likely than not that some
portion or all of the deferred tax assets will not be realized. As of December 31, 2010 and 2009, a
full valuation allowance has been applied against the Company’s net deferred tax assets because it
is not more likely than not that the Company will realize future benefits associated with these
deferred tax assets (See Note 12).
F-10
Loss Per Share
Basic and diluted loss per share is computed by dividing loss attributable to common
stockholders by the weighted average number of shares of common stock outstanding during the
period. Diluted weighted average shares outstanding excludes shares underlying unvested restricted
stock, restricted stock units, stock options and warrants, since the effects would be
anti-dilutive. Accordingly, basic and diluted loss per share is the same. Such excluded shares are
summarized as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Common stock options |
|
|
3,044,073 |
|
|
|
2,412,780 |
|
|
|
1,986,689 |
|
Restricted stock (unvested) |
|
|
— |
|
|
|
9,956 |
|
|
|
22,936 |
|
Restricted stock units (unvested) |
|
|
75,441 |
|
|
|
92,999 |
|
|
|
101,341 |
|
Shares issuable upon conversion of convertible debt |
|
|
185,185 |
|
|
|
185,185 |
|
|
|
370,834 |
|
Warrants |
|
|
23,704,930 |
|
|
|
12,989,703 |
|
|
|
11,296,433 |
|
|
|
|
|
|
|
|
|
|
|
Total shares excluded from calculation |
|
|
27,009,629 |
|
|
|
15,690,623 |
|
|
|
13,778,233 |
|
|
|
|
|
|
|
|
|
|
|
Interest Expense:
Interest expense consisted of the following for the years ended December 31, 2010, 2009 and
2008:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
$ |
(210 |
) |
|
$ |
(9,513 |
) |
|
$ |
(835 |
) |
Amortization of debt issuance costs and discount |
|
|
(151 |
) |
|
|
(10,508 |
) |
|
|
(431 |
) |
|
|
|
|
|
|
|
|
|
|
Total interest expense |
|
$ |
(361 |
) |
|
$ |
(20,021 |
) |
|
$ |
(1,266 |
) |
|
|
|
|
|
|
|
|
|
|
Amortization of debt issuance costs was accelerated in December 2010 as a result of the
termination of the Equity Distribution Agreement with Maxim Group LLC, which established an
at-the-market program through which the Company had the right to sell, from time to time and at its
sole discretion, shares of its common stock having an aggregate offering price of up to $15
million. Amortization of debt issuance costs and discount and interest expense were both
accelerated in 2009 as a result of the conversion of $24.5 million of the Company’s 7.5556%
convertible subordinated notes due 2014 into approximately 9.1 million shares of the Company’s
common stock in 2009. Interest expense for the year ended December 31, 2009 included $10.5 million
in amortization of debt issuance costs and discount and $9.3 million in interest expense paid from
escrowed cash that represented the make-whole interest payment that would have been due at maturity
of such notes.
Other Income:
In October 2010, the Company was notified by the Internal Revenue Service that its application
for the Qualifying Therapeutic Discovery Project Program was certified and a grant in the amount of
$0.7 million was approved. This grant was provided under a new section of the Internal Revenue
Code that was enacted as part of the Patient Protection and Affordable Care Act of 2010. The
program, which provides for grants and tax credits, is intended to benefit discovery projects that
show a reasonable potential to result in new therapies to treat areas of unmet medical need, or
detect or treat chronic or acute diseases and conditions; reduce long-term growth of health care
costs in the United States; or significantly advance the goal of curing cancer within 30
years. The Company received the grant in November 2010 which was recorded as income in
other income (expense).
Cash Equivalents
The Company considers all highly liquid investments with a maturity of 90 days or less when
purchased to be cash equivalents.
Restricted Cash
The Company has lease agreements for the premises it occupies. Letters of credit in lieu of
lease deposits for leased facilities totaling $0.1 million are secured by restricted cash in the
same amount at December 31, 2010 and 2009. During 2008, the Company failed to make certain payments
on its lease agreement for the premises located in San Diego, California. As a result, the landlord
exercised their right to draw down the full letter of credit, amounting to approximately $0.3
million, and applied approximately $0.2 million to unpaid rent. The remaining balance of $0.1
million is classified as prepaid expense.
F-11
On February 4, 2009, the Company issued $25.0 million in principal aggregate amount of 7.5556%
convertible subordinated notes due 2014. In connection with the issuance of these notes, the
Company was required to deposit approximately $9.4 million in escrow for up to twenty-four months
for the purposes of paying the interest on the notes and the make-whole payments upon conversion or
redemption. As the result of the conversion of $24.5 million of these notes, approximately $9.3
million was released from restricted cash to fund the make-whole interest payment and $0.1 million
remained in escrow for payment of the interest on the remaining notes not converted and was
accordingly classified as restricted cash at December 31, 2010.
Inventory
Inventories are valued at the lower of cost (first-in, first-out) or market. The Company
periodically evaluates its inventories and will reduce inventory to its net realizable value
depending on certain factors, such as product demand, remaining shelf life, future marketing
plans, obsolescence and slow-moving inventories.
As of December 31, 2010 and 2009, inventory consists of the following:
| |
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Raw materials |
|
$ |
144 |
|
|
$ |
131 |
|
Work-in-process |
|
|
860 |
|
|
|
1,147 |
|
Finished goods |
|
|
— |
|
|
|
37 |
|
|
|
|
|
|
|
|
Total inventory |
|
$ |
1,004 |
|
|
$ |
1,315 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Recognized as: |
|
|
|
|
|
|
|
|
Inventory |
|
$ |
179 |
|
|
$ |
1,315 |
|
Long-term inventory |
|
|
825 |
|
|
|
— |
|
During 2010, the Company expensed $0.9 million of Ceplene® inventory as it believes such
inventory will not be sold prior to reaching its product expiration date. The portion of inventory
classified as long-term is not expected to be realized in cash or sold or consumed during the
normal operating cycle of the Company.
Prepaid Expenses and Other Current Assets
As of December 31, 2010 and 2009, prepaid expenses and other current assets include the
following:
| |
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Prepaid expenses |
|
$ |
151 |
|
|
$ |
167 |
|
Prepaid insurance |
|
|
158 |
|
|
|
209 |
|
Other |
|
|
24 |
|
|
|
38 |
|
|
|
|
|
|
|
|
Total prepaid expenses and other current assets |
|
$ |
333 |
|
|
$ |
414 |
|
|
|
|
|
|
|
|
Property and Equipment
Property and equipment consists of office furniture and equipment, laboratory equipment, and
leasehold improvements stated at cost. Furniture and equipment are depreciated on a straight-line
basis over their estimated useful lives ranging from five to seven years. Leasehold improvements
are amortized on a straight-line basis over the shorter of the lease term or the estimated useful
life of the asset. Maintenance and repairs are charged to expense as incurred.
Deferred Financing Costs
Deferred financing costs represent legal and other costs and fees incurred to negotiate and
obtain debt financing. Deferred financing costs are capitalized and amortized using the effective
interest method over the life of the applicable financing. During the first quarter of 2009, the
Company incurred approximately $1.6 million in deferred financing costs from the issuance of $25.0
million in principal aggregate amount of 7.5556% convertible subordinated notes due 2014. As a
result of the conversion of $24.5 million of these notes, amortization of the deferred financing
costs was accelerated and amortization expense of $1.6 million was recorded in 2009. As of
December 31, 2010 and 2009, deferred financing costs were approximately $0.5 million and $28,000,
respectively. Amortization expense was $0.2 million, $1.8 million and $0.4 million for 2010, 2009
and 2008, respectively.
F-12
Identifiable Intangible Asset
Identifiable intangible asset consisted of the assembled workforce acquired in the merger with
Maxim Pharmaceuticals Inc. (“Maxim”) in January 2006. The assembled workforce was being amortized
on the greater of the straight-line basis or actual assembled workforce turnover over six years.
The gross carrying amount of the assembled workforce was $0.5 million, which has been fully
amortized as of December 31, 2010. Approximately $0.2 million of amortization was recorded in 2009
due to the layoff of most of the Company’s employees at its facility in San Diego, California in
the first half of 2009. Assembled workforce amortization is recorded in research and development
expense. During each of the years 2010, 2009 and 2008, the Company recorded assembled workforce
amortization of $28,000, $0.2 million and $0.1 million, respectively.
Deferred Rent and Other Noncurrent Liabilities
Deferred rent and other noncurrent liabilities represents deferred rent expense on our
facilities in Tarrytown, NY and San Diego, CA. In accordance with accounting principles generally
accepted in the United States of America, the Company recognizes rental expense, including tenant
improvement allowances, on a straight-line basis over the life of the leases or useful life,
whichever is shorter, irrespective of the timing of payments to or from the lessor. During the
second quarter of 2009, the Company ceased use of its discovery research facility in San Diego, CA
as a result of the Company’s previously announced decision to discontinue its drug discovery
activities. In accordance with ASC 420-10, “Exit or Disposal Cost Activities” (“ASC 420-10”), the
Company recorded a liability of $0.8 million, included in research and development expense on the
consolidated statements of operations, on the cease-use date based on the fair value of the costs
that are expected to be incurred under the lease of the facility. The fair value of the liability
at the cease-use date was determined based on the remaining rental payments, reduced by estimated
sublease rental income that could be reasonably obtained for the property. As of December 31, 2010
and 2009, the Company had deferred rent of $0.6 million and $1.0 million, respectively, that is
being amortized through October 2013.
Impairment of Long-Lived Assets
The Company performs impairment tests on its long-lived assets when circumstances indicate
that their carrying amounts may not be recoverable. If required, recoverability is tested by
comparing the estimated future undiscounted cash flows of the asset or asset group to its carrying
value. If the carrying value is not recoverable, the asset or asset group is written down to fair
value. No such impairments have been identified with respect to the Company’s long-lived assets,
which consist primarily of property and equipment at December 31, 2010.
Derivatives
The Company accounts for its derivative instruments in accordance with ASC 815-10,
“Derivatives and Hedging” (“ASC 815-10”). ASC 815-10 establishes accounting and reporting
standards requiring that derivative instruments, including derivative instruments embedded in other
contracts, be recorded on the balance sheet as either an asset or liability measured at its fair
value. ASC 815-10 also requires that changes in the fair value of derivative instruments be
recognized currently in results of operations unless specific hedge accounting criteria are met.
The Company has not entered into hedging activities to date. As a result of certain financings (see
Note 7), derivative instruments were created that are measured at fair value and marked to market
at each reporting period. Changes in the derivative value are recorded as change in value of
warrants and derivatives on the consolidated statements of operations.
Other Comprehensive Loss
A summary of other comprehensive loss for the years ended December 31, 2010 and 2009 are as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
| |
Net loss |
|
|
(15,537 |
) |
|
|
(38,814 |
) |
|
|
(25,382 |
) |
Foreign currency translation gain (loss) |
|
|
661 |
|
|
|
(293 |
) |
|
|
432 |
|
|
|
|
|
|
|
|
|
|
|
Total other comprehensive loss |
|
$ |
(14,876 |
) |
|
$ |
(39,107 |
) |
|
$ |
(24,950 |
) |
|
|
|
|
|
|
|
|
|
|
At December 31,
2010 and 2009, the Company’s only element of accumulated other
comprehensive loss was foreign currency translation adjustments of ($1.3)
million and ($1.9) million, respectively.
F-13
Fair Value of Financial Instruments
The Company applies ASC Topic 820, Fair Value Measurements and Disclosures (“ASC 820”) to all
financial instruments that are being measured and reported on a fair value basis, non-financial
assets and liabilities measured and reported at fair value on a non-recurring basis, and
disclosures of fair value of certain financial assets and liabilities.
The following fair value hierarchy is used in selecting inputs for those instruments measured
at fair value that distinguishes between assumptions based on market data (observable inputs) and
the Company’s assumptions (unobservable inputs). The hierarchy consists of three levels:
| |
• |
|
Level 1 — Quoted prices in active markets for identical assets or liabilities. |
| |
• |
|
Level 2 — Inputs other than Level 1 that are observable for similar assets or
liabilities either directly or indirectly. |
| |
• |
|
Level 3 — Prices or valuation techniques that require inputs that are both
significant to the fair value measurement and unobservable. |
The financial instruments recorded in the Company’s Consolidated Balance Sheets consist
primarily of cash and cash equivalents, money market funds, accounts payable and the Company’s debt
obligations. The carrying amounts of the Company’s cash and cash equivalents and accounts payable
approximate fair value due to their short-term nature. The carrying amount of the Company’s money
market funds approximates fair value based on quoted market prices of the identical underlying
security in active markets, which approximate market rates and is considered a Level 1 asset. The
fair market value of the Company’s convertible and non-convertible loans is based on the present
value of their cash flows discounted at a rate that approximates current market returns for issues
of similar risk
The carrying amount and estimated fair values of the Company’s debt instruments are as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
At December 31, |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
Carrying |
|
|
Fair |
|
|
Carrying |
|
|
Fair |
|
| |
|
Amount |
|
|
Value |
|
|
Amount |
|
|
Value |
|
| |
|
(In millions) |
|
Non-convertible loans |
|
|
0.6 |
|
|
|
0.6 |
|
|
|
1.6 |
|
|
|
1.7 |
|
Convertible loans |
|
|
0.5 |
|
|
|
0.6 |
|
|
|
0.5 |
|
|
|
0.6 |
|
Recent Accounting Pronouncements
In March 2010, the FASB issued ASU 2010-17 “Revenue Recognition — Milestone Method” (ASC
Topic 605).” The guidance in this consensus allows the use of the milestone method as an acceptable
revenue recognition methodology when an arrangement includes substantive milestones. The guidance
provides a definition of substantive milestone and should be applied regardless of whether the
arrangement includes single or multiple deliverables or units of accounting. The scope of this
consensus is limited to the transactions involving milestones relating to research and development
deliverables. The guidance includes enhanced disclosure requirements about each arrangement,
individual milestones and related contingent consideration, information about substantive
milestones and factors considered in the determination. The consensus is effective prospectively to
milestones achieved in fiscal years, and interim periods within those years, after June 15, 2010.
Early application and retrospective application are permitted. The Company has chosen early
adoption of this pronouncement effective January 1, 2010 on a prospective basis. The early adoption
did not have a material effect on the Company’s consolidated financial statements.
On October 7, 2009, the FASB issued ASU 2009-13, “Multiple Revenue Arrangements — a Consensus
of the FASB Emerging Issues Task Force” which supersedes certain guidance in ASC 605-25, “Revenue
Recognition-Multiple Element Arrangements,” and requires an entity to allocate arrangement
consideration to all of its deliverables at the inception of an arrangement based on their relative
selling prices (i.e., the relative-selling-price method). The use of the residual method of
allocation will no longer be permitted in circumstances in which an entity recognized revenue for
an arrangement with multiple deliverables subject to ASC 605-25. ASU 2009-13 also requires
additional disclosures. The Company adopted the provisions of ASU 2009-13 beginning on January 1,
2011. Based on the Company’s current revenue arrangements, the adoption of ASU 2009-13 is not
expected to have a material impact on the Company’s consolidated financial statements.
F-14
4. License Agreements
Meda AB
In January 2010, the Company entered into an exclusive commercialization agreement for
Ceplene® with Meda AB (“Meda”), a leading international specialty pharmaceutical company
based in Stockholm, Sweden. Under the terms of the agreement, the Company granted Meda the right
to market Ceplene® in Europe and several other countries including Japan, China, and
Australia. The Company received a $3.0 million fee on signing and an additional $2.0 million
milestone payment in May 2010 upon the first commercial sale of Ceplene® in a major
European market, which have been deferred and are being recognized as revenue ratably over the life
of the commercialization agreement with Meda. Additional potential payments include a $5 million
payment upon achievement of a regulatory milestone and up to $30 million in sales-based milestones
that commence upon attainment of at least $50 million in annual sales. The Company will also
receive an escalating percentage royalty on net sales in the covered territories ranging from the
low teens to the low twenties and is responsible for Ceplene’s commercial supply. The initial term
of this agreement is ten years and is subject to automatic two year extensions at Meda’s option.
The agreement can be terminated at any time by Meda upon six months prior written notice. The
agreement can also be terminated by mutual agreement or for cause. In 2010, the Company recognized
revenue from Meda of approximately $0.4 million relating to the signing fee and milestone payment
and $0.2 million on commercial sales of Ceplene®. No revenue was recognized from Meda
prior to January 2010.
Hellstrand
In October 1999, the Company entered into a royalty agreement with Dr. Kristoffer Hellstrand
under which the Company has an exclusive license to certain patents for Ceplene®
configured for the systemic treatment of cancer, infectious diseases, autoimmune diseases and other
medical conditions. A predecessor company previously paid Dr. Hellstrand $1 million and agreed to
pay a royalty of 1% on net sales of Ceplene®. In November 2009 the Company expanded its
collaboration with Dr. Hellstrand by concluding a new agreement in which the Company acquired
rights to develop certain new compounds. As compensation for this agreement and for providing
certain ongoing consulting services, the Company awarded Dr. Hellstrand options to purchase 50,000
shares of its common stock having a fair value of $0.1 million and agreed to pay a monthly
consulting fee. In 2010, the Company paid $0.1 million in royalties to Dr. Hellstrand. No
royalties were paid to Dr. Hellstrand prior to 2010.
Epitome/Dalhousie
In July 2007, the Company entered into a direct license with Dalhousie University under which
the Company was granted an exclusive license to certain patents for the topical use of tricyclic
anti-depressants and NMDA antagonists as topical analgesics for neuralgia that were licensed to
Epitome by Dalhousie University. These, and other patents, cover the combination treatment
consisting of amitriptyline and ketamine in EpiCeptTM NP-1. This technology has been
incorporated into EpiCept NP-1.
The Company has been granted worldwide rights to make, use, develop, sell and market products
utilizing the licensed technology in connection with passive dermal applications. The Company is
obligated to make payments to Dalhousie upon achievement of specified milestones and to pay
royalties based on annual net sales derived from the products incorporating the licensed
technology. The Company is obligated to pay Dalhousie an annual maintenance fee until the license
agreement expires or is terminated, or an NDA for NP-1 is filed with the FDA, or Dalhousie will
have the option to terminate the contract. The license agreement with Dalhousie terminates upon the
expiration of the last to expire licensed patent. During 2010, 2009 and 2008, the Company paid
Dalhousie a maintenance fee of $0.5 million, $0.5 million and $0.4 million, respectively. These
payments were expensed to research and development in their respective years.
Myrexis, Inc.
In connection with its merger with Maxim Pharmaceuticals on January 4, 2006, EpiCept acquired
a license agreement with Myrexis Inc. (“Myrexis”) under which the Company licensed the MX90745
series of caspase-inducer anti-cancer compounds to Myrexis. Under the terms of the agreement, Maxim
granted to Myrexis a research license to develop and commercialize any drug candidates from the
series of compounds during the Research Term (as defined in the agreement) with a non-exclusive,
worldwide, royalty-free license, without the right to sublicense the technology. Myrexis is
responsible for the worldwide development and commercialization of any drug candidates from the
series of compounds. Maxim also granted to Myrexis a worldwide royalty bearing development and
commercialization license with the right to sublicense the technology. The agreement requires that
Myrexis make licensing, research and milestone payments to the Company totaling up to $27 million,
of which $3.0 million was paid and recognized as revenue by Maxim prior to the merger on January 4,
2006, assuming the successful commercialization of the compound for the
treatment of cancer, as well as pay an escalating mid to high single digit percentage royalty
on product sales. In March 2008, the Company received a milestone payment of $1.0 million
following dosing of the first patient in a Phase II registration sized clinical trial, which has
been deferred and is being recognized as revenue ratably over the life of the last to expire patent
that expires in July 2024. In each of the years 2010, 2009 and 2008, the Company recorded revenue
from Myrexis of approximately $0.1 million.
F-15
DURECT Corporation
In December 2006, the Company entered into a license agreement with DURECT Corporation
(“DURECT”), pursuant to which it granted DURECT the exclusive worldwide rights to certain of its
intellectual property for a transdermal patch containing bupivacaine for the treatment of back
pain. Under the terms of the agreement, EpiCept received a $1.0 million payment which has been
deferred and is being recognized as revenue ratably over the life of the last to expire patent that
expires in March 2020. In September 2008, the Company amended its license agreement with DURECT.
Under the terms of the amended agreement, the Company granted DURECT royalty-free, fully paid up,
perpetual and irrevocable rights to the intellectual property licensed as part of the original
agreement in exchange for a cash payment of $2.25 million from DURECT, which has also been deferred
and is being recognized as revenue ratably over the last patent life. In 2010, 2009 and 2008, the
Company recorded revenue from DURECT of approximately $0.3 million, $0.3 million and $0.1 million,
respectively.
Endo Pharmaceuticals Inc.
In December 2003, the Company entered into a license agreement with Endo Pharmaceuticals Inc.
(“Endo”) under which it granted Endo (and its affiliates) the exclusive (including as to the
Company and its affiliates) worldwide right to commercialize LidoPAIN BP. The Company also granted
Endo worldwide rights to use certain of its patents for the development of certain other
non-sterile, topical lidocaine containing patches, including Lidoderm®, Endo’s topical
lidocaine-containing patch for the treatment of chronic lower back pain. Upon the execution of the
Endo agreement, the Company received a non-refundable payment of $7.5 million, which has been
deferred and is being recognized as revenue on the proportional performance method. The Company is
eligible to receive payments of up to $52.5 million upon the achievement of various milestones
relating to product development and regulatory approval for both the Company’s LidoPAIN BP product
and licensed Endo products, including Lidoderm®, so long as, in the case of Endo’s
product candidate, the Company’s patents provide protection thereof. The Company is also entitled
to receive royalties from Endo based on the net sales of LidoPAIN BP. These royalties are payable
until generic equivalents to the LidoPAIN BP product are available or until expiration of the
patents covering LidoPAIN BP, whichever is sooner. The Company is also eligible to receive
milestone payments from Endo of up to approximately $30.0 million upon the achievement of specified
net sales milestones for licensed Endo products, including Lidoderm®, so long as the
Company’s patents provide protection thereof. The future amount of milestone payments the Company
is eligible to receive under the Endo agreement is $82.5 million. In 2010, 2009 and 2008, the
Company recorded revenue from Endo of approximately $14,000, $24,000 and $33,000, respectively.
Under the terms of the license agreement, the Company is responsible for continuing and
completing the development of LidoPAIN BP, including the conduct of all clinical trials and the
supply of the clinical products necessary for those trials and the preparation and submission of
the NDA in order to obtain regulatory approval for LidoPAIN BP. Endo remains responsible for
continuing and completing the development of Lidoderm® for the treatment of chronic
lower back pain, including the conduct of all clinical trials and the supply of the clinical
products necessary for those trials. No progress in the development of LidoPAIN BP or Lidoderm
with respect to back pain has been reported. Accordingly, the Company does not expect to receive
any further cash compensation pursuant to this license agreement.
The Company has the option to negotiate a co-promotion arrangement with Endo for LidoPAIN BP
or similar product in any country in which an NDA (or foreign equivalent) filing has been made
within thirty days of such filing. The Company also has the right to terminate its license to Endo
with respect to any territory in which Endo has failed to commercialize LidoPAIN BP within three
years of the receipt of regulatory approval permitting such commercialization.
Shire BioChem
In March 2004 and as amended in January 2005, Maxim entered into a license agreement
reacquiring the rights to the MX2105 series of apoptosis inducer anti-cancer compounds from Shire
Biochem, Inc (formerly known as BioChem Pharma, Inc.) which had previously announced that oncology
would no longer be a therapeutic focus of the company’s research and development efforts. Under the
agreement, all rights and obligations of the parties under the July 2000 agreement were terminated
and Shire BioChem agreed to assign and/or license to the Company rights it owned under or shared
under the prior research program. The agreement did not require any up-front payments, however, the
Company is required to provide Shire Biochem a portion of any sublicensing
payments the Company receives if the Company relicenses the series of compounds or make
milestone payments to Shire BioChem totaling up to $26.0 million, assuming the successful
commercialization of the compounds by the Company for the treatment of a cancer indication, as well
as pay a royalty on product sales. The Company accrued a license fee expense of $0.5 million upon
the commencement of a Phase I clinical trial for crolibulinTM in 2006. This amount, and
approximately $0.1 million in interest, remains accrued and unpaid as of December 31, 2010.
F-16
5. Property and Equipment
Property and equipment consist of the following:
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Furniture, office and laboratory equipment |
|
$ |
977 |
|
|
$ |
980 |
|
Leasehold improvements |
|
|
763 |
|
|
|
764 |
|
|
|
|
|
|
|
|
|
|
|
1,740 |
|
|
|
1,744 |
|
Less accumulated depreciation |
|
|
(1,518 |
) |
|
|
(1,384 |
) |
|
|
|
|
|
|
|
|
|
$ |
222 |
|
|
$ |
360 |
|
|
|
|
|
|
|
|
Depreciation expense was approximately $0.1 million, $0.2 million and $0.2 million for the
years ended December 31, 2010, 2009 and 2008, respectively. The Company sold excess equipment
during 2010, 2009 and 2008 resulting in a gain of $27,000, $0.1 million and $0.1 million,
respectively.
6. Other Accrued Liabilities
Other accrued liabilities consist of the following:
| |
|
|
|
|
|
|
|
|
| |
|
December 31 |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Accrued professional fees |
|
$ |
277 |
|
|
$ |
96 |
|
Accrued salaries and employee benefits |
|
|
554 |
|
|
|
709 |
|
Other accrued liabilities |
|
|
455 |
|
|
|
267 |
|
|
|
|
|
|
|
|
|
|
$ |
1,286 |
|
|
$ |
1,072 |
|
|
|
|
|
|
|
|
7. Notes, Loans and Financing
The Company is a party to several loan agreements in the following amounts, none of which had
covenant requirements at December 31, 2010:
| |
|
|
|
|
|
|
|
|
| |
|
December 31 |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Ten-year, non-amortizing loan due June 30, 2011(1) |
|
$ |
410 |
|
|
$ |
1,326 |
|
7.5556% convertible subordinated notes due February 9, 2014(2) |
|
|
500 |
|
|
|
500 |
|
July 2006 note payable due July 1, 2012(3) |
|
|
176 |
|
|
|
277 |
|
|
|
|
|
|
|
|
Total notes and loans payable and 7.5556% Convertible Subordinated Notes, before debt discount |
|
|
1,086 |
|
|
|
2,103 |
|
Less: Debt discount |
|
|
114 |
|
|
|
151 |
|
|
|
|
|
|
|
|
Total notes and loans payable and 7.5556% Convertible Subordinated Notes |
|
|
972 |
|
|
|
1,952 |
|
Less: Notes and loans payable, current portion |
|
|
524 |
|
|
|
985 |
|
|
|
|
|
|
|
|
Notes and loans payable and 7.5556% Convertible Subordinated Notes, long-term |
|
$ |
448 |
|
|
$ |
967 |
|
|
|
|
|
|
|
|
| |
|
|
| (1) |
|
In August 1997, EpiCept GmbH, a wholly-owned subsidiary of EpiCept, entered into a ten-year
non-amortizing loan in the amount of €1.5 million with Technologie-Beteiligungs Gesellschaft
mbH der Deutschen Ausgleichsbank (“tbg”). The loan initially bore interest at 6% per annum.
Tbg was also entitled to receive additional compensation equal to 9% of the annual surplus
(income before taxes, as defined in the agreement) of EpiCept GmbH, reduced by any other
compensation received from EpiCept GmbH by virtue of other loans to or investments in EpiCept
GmbH provided that tbg is an equity investor in EpiCept GmbH during that time period. The
Company considered the additional compensation element based on the surplus of EpiCept GmbH to
be a derivative. The Company assigned no value to the derivative at each reporting period as
no surplus of EpiCept
GmbH was anticipated over the term of the agreement. In addition, any additional compensation as
a result of surplus would be reduced by the additional interest noted below. |
F-17
| |
|
|
| |
|
At the demand of tbg, additional amounts could have been due at the end of the loan term
up to 30% of the loan amount, plus 6% of the principal balance of the note for each year
after the expiration of the fifth complete year of the loan period, such payments to be
offset by the cumulative amount of all payments made to the lender from the annual surplus
of EpiCept GmbH. The Company was accruing these additional amounts as additional interest
up to the maximum amount due over the term of the loan. |
| |
| |
|
On December 20, 2007, Epicept GmbH entered into a repayment agreement with tbg, whereby
Epicept GmbH paid tbg approximately €0.2 million ($0.2 million) in January 2008,
representing all interest payable to tbg as of December 31, 2007. The loan balance of €1.5
million ($2.0 million), plus accrued interest at a rate of 7.38% per annum beginning
January 1, 2008 was to be repaid to tbg no later than June 30, 2008. Tbg waived any
additional interest payments of approximately €0.5 million ($0.7 million). Epicept GmbH
considered this a substantial modification to the original debt agreement and has recorded
the new debt at its fair value in accordance with ASC 470-50, “Modifications and
Extinguishments” (“ASC 470-50”). As a result of the modification to the original debt
agreement, EpiCept GmbH recorded a gain on the extinguishment of debt of $0.5 million in
December 2007. |
| |
| |
|
On May 14, 2008, Epicept GmbH entered into a prolongation of the repayment agreement with
tbg, whereby the loan balance of €1.5 million ($2.0 million) was required to be repaid to
tbg no later than December 31, 2008. Interest continued to accrue at a rate of 7.38% per
annum and all the provisions of the repayment agreement dated December 20, 2007 continued
to apply without change. |
| |
| |
|
On November 26, 2008, EpiCept GmbH entered into a second amendment to the repayment
agreement with tbg, whereby the loan balance of €1.5 million ($2.1 million) was required to
be repaid to tbg no later than June 30, 2009. Interest continued to accrue at a rate of
7.38% per annum and all the provisions of the repayment agreement dated December 20, 2007
continued to apply without change. |
| |
| |
|
On June 25, 2009, EpiCept GmbH entered into a third amendment to the repayment agreement with
tbg, whereby €0.3 million of the loan balance of €1.5 million ($2.1 million) plus accrued
interest of €56,000 ($0.1 million) was repaid to tbg on June 30, 2009. The remaining loan
balance of €1.2 million ($1.7 million) at June 30, 2009 plus accrued interest will be paid in
four semi-annual installments of €0.3 million ($0.4 million) beginning December 31, 2009.
Interest will continue to accrue at a rate of 7.38% per annum and all the provisions of the
repayment agreement dated December 20, 2007 will continue to apply without change. |
| |
| (2) |
|
On February 4, 2009, the Company issued $25.0 million in principal aggregate amount of
7.5556% convertible subordinated notes due February 2014 and five and-a-half year warrants to
purchase approximately 4.2 million shares of common stock at an exercise price of $3.105 per
share. Each $1,000 in principal aggregate amount of the notes is initially convertible into
approximately 371 shares of Common Stock, at the option of the holders or upon specified
events, including a change of control, and if the Company’s common stock trades at or greater
than $5.10 a share for 20 out of 30 days, beginning February 9, 2010. Upon any conversion or
redemption of the notes, the holders will receive a make-whole payment in an amount equal to
the interest payable through the scheduled maturity of the converted or redeemed notes, less
any interest paid before such conversion or redemption. Interest is due and payable on the
notes semi-annually in arrears on June 30 and December 31, beginning on June 30, 2009. |
| |
| |
|
The Company allocated the $25.0 million in proceeds between the convertible subordinated
notes and the warrants based on their relative fair values. The Company calculated the fair
value of the warrants at the date of the transaction at approximately $8.8 million with a
corresponding amount recorded as a debt discount. The debt discount is being accreted over
the life of the outstanding convertible subordinated notes using the effective interest
method. At the date of the transaction, the fair value of the warrants of $8.8 million was
determined utilizing the Black-Scholes option pricing model under the following
assumptions: dividend yield of 0%, risk free interest rate of 1.99%, volatility of 118% and
an expected life of five years. During 2009, the Company recognized approximately $8.6
million of non-cash interest expense related to the accelerated accretion of the debt
discount as a result of the conversion of $24.5 million of the convertible subordinated
notes into approximately 9.1 million shares of the Company’s common stock. |
| |
| |
|
As of December 31, 2010, after giving effect to the conversions into common stock, the remaining
principal aggregate amount of the notes due 2014 outstanding is $0.5 million. |
| |
| (3) |
|
In July 2006, the Company entered into a six-year non-interest bearing promissory note in the
amount of $0.8 million with Pharmaceutical Research Associates, Inc., (“PRA”) as compensation
for PRA assuming liability on a lease of premises in San Diego, CA. The fair value of the note
(assuming an imputed 11.6% interest rate) was $0.6 million and broker fees amounted to $0.2
million at issuance. The note is payable in seventy-two equal installments of $11,000 per
month. The loan balance at December 31, 2010 was $0.2 million. |
F-18
At December 31, 2010, contractual principal payments due on loans and notes payable are as
follows:
| |
|
|
|
|
| |
|
As of December 31, |
|
| Year Ending |
|
2010 |
|
| |
|
(in thousands) |
|
| |
2011 |
|
$ |
524 |
|
2012 |
|
|
62 |
|
2013 |
|
|
— |
|
2014 |
|
|
500 |
|
|
|
|
|
Total |
|
$ |
1,086 |
|
|
|
|
|
8. Preferred Stock
The Company has authorized 5 million undesignated preferred shares. No such preferred stock
was issued and outstanding as of December 31, 2010 and 2009, respectively.
9. Common Stock and Common Stock Warrants
In July 2009, upon receipt of the approval of stockholders, the Company amended its
certificate of incorporation to increase the number of authorized shares of common stock from
175,000,000 to 225,000,000 shares.
In November 2010, the Company raised $2.0 million in gross proceeds, $1.9 million net of $0.1
million in transactions costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
3.3 million shares of the Company’s common stock were sold at a price of $0.61 per share. Five
year common stock purchase warrants were issued to investors granting them the right to purchase
approximately 1.3 million and 0.2 million shares of the Company’s common stock at an exercise price
of $0.56 and $0.7625 per share, respectively. The Company allocated the $2.0 million in gross
proceeds between the common stock and the warrants based on their relative fair values.
Approximately $0.5 million of this amount was allocated to the warrants. The warrants meet the
requirements of and are being accounted for as equity in accordance with ASC 815-40.
In June 2010, the Company raised $6.7 million in gross proceeds, $6.2 million net of $0.5
million in transactions costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
6.1 million shares of the Company’s common stock were sold at a price of $1.10 per share. Five
year common stock purchase warrants were issued to investors granting them the right to purchase
approximately 4.6 million and 0.3 million shares of the Company’s common stock at an exercise price
of $1.64 and $1.375 per share, respectively. In addition, one year common stock purchase warrants
were issued to investors granting them the right to purchase approximately 6.1 million shares of
the Company’s common stock at an exercise price of $1.64 per share. The Company allocated the $6.7
million in gross proceeds between the common stock and the warrants based on their relative fair
values. Approximately $3.2 million of this amount was allocated to the warrants. The warrants meet
the requirements of and are being accounted for as equity in accordance with ASC 815-40.
In June 2009, the Company raised $9.6 million in gross proceeds, $8.9 million net of $0.7
million in transaction costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
4.0 million shares of the Company’s common stock were sold at a price of $2.40 per share. Two and
one-half year common stock purchase warrants were issued to investors granting them the right to
purchase approximately 1.4 million shares of the Company’s common stock at an exercise price of
$2.70 per share. The Company allocated the $9.6 million in gross proceeds between the common stock
and the warrants based on their
relative fair values. Approximately $2.0 million of this amount was allocated to the
warrants. The warrants did not meet the requirements of being accounted for as equity in accordance
with ASC 815-40 since the Company did not have an adequate number of authorized shares to reserve
for the exercise of the warrants. Therefore, the value of the warrant shares were classified as a
liability and were marked to market at June 30, 2009 resulting in a loss of $0.3 million in the
statement of operations. On July 2, 2009, the warrants were reclassified from liability to equity
in accordance with ASC 815-40 as a result of stockholders approving an increase in the number of
authorized shares of the Company’s common stock.
F-19
On August 11, 2008, the Company raised $4.0 million in gross proceeds, $3.7 million net of
$0.3 million in transactions costs, through a public offering of common stock and common stock
purchase warrants registered pursuant to a shelf registration statement on Form S-3 registering the
issuance and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt
securities, convertible debt securities and/or warrants to purchase the Company’s securities.
Approximately 1.7 million shares of the Company’s common stock were sold at a price of $2.2767 per
share. Five year common stock purchase warrants were issued to investors granting them the right
to purchase approximately 0.9 million shares of the Company’s common stock at an exercise price of
$1.89 per share and approximately 0.1 million shares of the Company’s common stock at an exercise
price of $2.85 per share. In addition, in consideration of the receipt of $1.3 million in
connection with the exercise of all the warrants issued in connection with the Company’s public
offering on August 1, 2008 discussed below, the Company agreed to issue to the investors in that
offering new warrants to purchase up to approximately 0.9 million shares of Common Stock of the
Company with an exercise price of $2.079 per share. Such warrants are exercisable until August 11,
2013. The Company allocated the $3.7 million in gross proceeds between the common stock and the
warrants based on their relative fair values. $1.7 million of this amount was allocated to the
warrants. The warrants meet the requirements of and are being accounted for as equity in accordance
with ASC 815-40.
On August 1, 2008, the Company raised $3.0 million in gross proceeds, $2.8 million net of $0.2
million in transactions costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
1.8 million shares of the Company’s common stock were sold at a price of $1.62 per share. Five
year common stock purchase warrants were issued to investors granting them the right to purchase
approximately 0.9 million shares of the Company’s common stock at an exercise price of $1.44 per
share and approximately 0.1 million shares of the Company’s common stock at an exercise price of
$2.04 per share. The Company allocated the $3.0 million in gross proceeds between the common stock
and the warrants based on their relative fair values. $0.9 million of this amount was allocated to
the warrants. The warrants meet the requirements of and are being accounted for as equity in
accordance with ASC 815-40.
On July 15, 2008, the Company raised $0.5 million in gross proceeds, $0.5 million net of
$50,000 in transactions costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
0.67 million shares of the Company’s common stock were sold at a price of $0.75 per share. Five
year common stock purchase warrants were issued to investors granting them the right to purchase
approximately 0.7 million shares of the Company’s common stock at an exercise price of $1.17 per
share. The Company allocated the $0.5 million in gross proceeds between the common stock and the
warrants based on their relative fair values. $0.2 million of this amount was allocated to the
warrants. The warrants meet the requirements of and are being accounted for as equity in accordance
with ASC 815-40.
On June 23, 2008, the Company raised $2.0 million in gross proceeds, $1.8 million net of $0.2
million in transactions costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
2.67 million shares of the Company’s common stock were sold at a price of $0.75 per share. Five
year common stock purchase warrants were issued to investors granting them the right to purchase
approximately 2.8 million shares of the Company’s common stock at an exercise price of $1.17 per
share. The Company allocated the $2.0 million in gross proceeds between the common stock and the
warrants based on their relative fair values. $0.8 million of this amount was allocated to the
warrants. The warrants meet the requirements of and are being accounted for as equity in accordance
with ASC 815-40.
On June 23, 2008, the Company entered into the second amendment to a senior secured term loan.
Under this amendment, the Company issued the senior lender warrants to purchase an aggregate of 1.3
million shares of the Company’s common stock at an exercise price of $1.17 per share and an
aggregate of 0.3 million shares of the Company’s common stock at an exercise price of $1.23 per
share.
F-20
On May 7, 2008, in connection with a second amendment to the warrant agreement on a senior
secured term loan, the terms of the warrants issued to the senior lender were adjusted to grant the
senior lender the right to purchase an aggregate of 0.7 million shares of the Company’s common
stock at an exercise price of $0.90 per share.
On March 6, 2008, the Company raised $5.0 million in gross proceeds, $4.7 million net of $0.3
million in transaction costs, through a public offering of common stock and common stock purchase
warrants registered pursuant to a shelf registration statement on Form S-3 registering the issuance
and sale of up to $50.0 million of the Company’s common stock, preferred stock, debt securities,
convertible debt securities and/or warrants to purchase the Company’s securities. Approximately
1.8 million shares of the Company’s common stock were sold at a price of $2.7675 per share. Five
year common stock purchase warrants were issued to investors granting them the right to purchase
approximately 1.0 million shares of the Company’s common stock at an exercise price of $2.58 per
share. The Company allocated the $5.0 million in gross proceeds between the common stock and the
warrants based on their relative fair values. $1.3 million of this amount was allocated to the
warrants. The warrants meet the requirements of and are being accounted for as equity in accordance
with ASC 815-40.
10. Commitments and Contingencies
Leases
The Company leases facilities and certain equipment under agreements through 2013 accounted
for as operating leases. The leases generally contain renewal options and require the Company to
pay all executory costs such as maintenance and insurance. In January 2011, the Company renewed its
lease in Munich, Germany through July 2014. Rent expense approximated $1.1 million, $1.3 million
and $1.4 million for the years ended December 31, 2010, 2009, and 2008, respectively.
Future minimum rental payments under non-cancelable operating leases as of December 31, 2010
are as follows:
| |
|
|
|
|
| |
|
As of December 31, |
|
| Year Ending December 31, |
|
2010 |
|
| |
|
(in thousands) |
|
| |
2011 |
|
$ |
1,089 |
|
2012 |
|
|
808 |
|
2013 |
|
|
661 |
|
|
|
|
|
|
|
$ |
2,558 |
|
|
|
|
|
Consulting Contracts and Employment Agreements
The Company is a party to a number of research, consulting, and license agreements, which
require the Company to make payments to the other party to the agreement upon the other party
attaining certain milestones as defined in the agreements. As of December 31, 2010, the Company may
be required to make future milestone payments, totaling approximately $4.9 million, under these
agreements, of which approximately $2.4 million is payable during 2011 and approximately $2.5
million is payable from 2012 through 2015. The Company is obligated to make future royalty payments
to three of its collaborators under existing license agreements, including ones based on net sales
of NP-1 and the other based on net sales of crolibulinTM, to the extent revenues on such
products are realized. Under the Company’s agreement with Dalhousie University, the Company is
obligated to pay an annual maintenance fee so long as no commercial product sales have occurred and
the Company desires to maintain its rights under the license agreement. During each of the years
2010, 2009 and 2008, the Company paid Dalhousie a maintenance fee of $0.5 million.
The Company’s Board of Directors ratified the employment agreements between the Company and
its chief executive officer and chief financial officer dated as of October 28, 2004. The
employment agreements cover the term through December 31, 2010, and provide for base salary,
discretionary compensation, stock option awards, and reimbursement of reasonable expenses in
connection with services performed under the employment agreements. The agreements also compensate
such officers in the event of their death or disability, termination without cause, or termination
within one year of an initial public offering or a change of control, as defined in the respective
employment agreements. Both employment agreements were automatically renewed for another year,
ending December 31, 2011.
F-21
Litigation
There are no material legal proceedings pending against the Company as of December 31, 2010.
11. Share-Based Payments
The Company records stock-based compensation expense at fair value in accordance with ASC
718-10, “Compensation — Stock Compensation” (“ASC 718-10”). The Company utilizes the
Black-Scholes valuation method for determination of share-based compensation expense. Certain
assumptions need to be made with respect to utilizing the Black-Scholes valuation model, including
the expected life, volatility, risk-free interest rate and anticipated forfeiture of the stock
options. The expected life of the stock options was calculated using the method allowed by the
provisions of ASC 718-10. In accordance with ASC 718-10, the simplified method for “plain vanilla”
options may be used where the expected term is equal to the vesting term plus the original contract
term divided by two. The risk-free interest rate is based on the rates paid on securities issued by
the U.S. Treasury with a term approximating the expected life of the options. Estimates of
pre-vesting option forfeitures are based on our experience. The Company will adjust its estimate of
forfeitures over the requisite service period based on the extent to which actual forfeitures
differ, or are expected to differ, from such estimates. Changes in estimated forfeitures will be
recognized through a cumulative catch-up adjustment in the period of change and will also impact
the amount of compensation expense to be recognized in future periods.
The Company accounts for stock-based transactions with non-employees in which services are
received in exchange for the equity instruments based upon the fair value of the equity instruments
issued, in accordance with ASC 718-10 and ASC 505-50, “Equity-Based Payments to Non-Employees.” The
two factors that most affect charges or credits to operations related to stock-based compensation
are the estimated fair market value of the common stock underlying stock options for which
stock-based compensation is recorded and the estimated volatility of such fair market value. The
value of such options is periodically remeasured and income or expense is recognized during the
vesting terms.
2005 Equity Incentive Plan
The 2005 Equity Incentive Plan (the “2005 Plan”) was adopted on September 1, 2005, approved by
stockholders on September 5, 2005 and became effective on January 4, 2006. The 2005 Plan provides
for the grant of incentive stock options, within the meaning of Section 422 of the Internal Revenue
Code, to EpiCept’s employees and its parent and subsidiary corporations’ employees, and for the
grant of nonstatutory stock options, restricted stock, restricted stock units, performance-based
awards and cash awards to its employees, directors and consultants and its parent and subsidiary
corporations’ employees and consultants. Options are granted and vest as determined by the Board of
Directors. A total of 13,000,000 shares of EpiCept’s common stock are reserved for issuance
pursuant to the 2005 Plan. No optionee may be granted an option to purchase more than 1,500,000
shares in any fiscal year. Options issued pursuant to the 2005 Plan have a maximum maturity of 10
years and generally vest over 4 years from the date of grant. The Company records stock-based
compensation expense at fair value. For the year ended December 31, 2010, the Company issued
approximately 0.8 million options to purchase common stock to certain of its employees and members
of its board of directors and estimated $1.3 million of share-based compensation will be recognized
as compensation expense over the vesting periods.
The following table presents the 2010, 2009 and 2008 total employee, board of directors and
third party stock-based compensation expense resulting from the issuance of stock options,
restricted stock, restricted stock units and the Employee Stock Purchase Plan included in the
consolidated statement of operations:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
For the Years Ended December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
| |
General and administrative |
|
$ |
654 |
|
|
$ |
889 |
|
|
$ |
1,792 |
|
Research and development |
|
|
281 |
|
|
|
506 |
|
|
|
489 |
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation costs before income taxes |
|
|
935 |
|
|
|
1,395 |
|
|
|
2,281 |
|
Benefit for income taxes (1) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
Net compensation expense |
|
$ |
935 |
|
|
$ |
1,395 |
|
|
$ |
2,281 |
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
| (1) |
|
The stock-based compensation expense has not been tax-effected due to the recording of a full
valuation allowance against net deferred tax assets. |
F-22
Summarized information for stock option grants for the year ended December 31, 2010 is as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
Weighted Average |
|
|
|
|
| |
|
|
|
|
|
Weighted Average |
|
|
Remaining Contractual |
|
|
Aggregate Intrinsic |
|
| |
|
Options |
|
|
Exercise Price |
|
|
Term (years) |
|
|
Value |
|
Options outstanding at December 31, 2009 |
|
|
2,412,780 |
|
|
$ |
8.60 |
|
|
|
|
|
|
|
|
|
Granted |
|
|
823,750 |
|
|
|
1.93 |
|
|
|
|
|
|
|
|
|
Exercised |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
Forfeited |
|
|
(179,471 |
) |
|
|
3.22 |
|
|
|
|
|
|
|
|
|
Expired |
|
|
(12,986 |
) |
|
|
9.48 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options outstanding at December 31, 2010 |
|
|
3,044,073 |
|
|
$ |
7.11 |
|
|
|
7.13 |
|
|
$ |
34,358 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested or expected to vest at December 31, 2010 |
|
|
2,947,712 |
|
|
$ |
7.27 |
|
|
|
6.41 |
|
|
$ |
34,358 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options exercisable at December 31, 2010 |
|
|
2,080,461 |
|
|
$ |
9.45 |
|
|
|
6.41 |
|
|
$ |
34,358 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following table summarizes information about stock options outstanding at December 31,
2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Options Outstanding |
|
|
Options Exercisable |
|
| |
|
Options |
|
|
Weighted- |
|
|
Weighted- |
|
|
Shares |
|
|
Weighted- |
|
| |
|
Outstanding at |
|
|
Average |
|
|
Average |
|
|
Exercisable at |
|
|
Average |
|
| |
|
December 31, |
|
|
Remaining |
|
|
Exercise |
|
|
December 31, |
|
|
Exercise |
|
| Range of Exercise Price |
|
2010 |
|
|
Contractual Life |
|
|
Price |
|
|
2010 |
|
|
Price |
|
$0.72 – 1.89 |
|
|
1,076,790 |
|
|
8.1 years |
|
$ |
1.49 |
|
|
|
623,172 |
|
|
$ |
1.35 |
|
2.11 – 4.89 |
|
|
1,199,774 |
|
|
7.7 years |
|
$ |
3.00 |
|
|
|
689,780 |
|
|
$ |
3.48 |
|
6.00 – 10.20 |
|
|
72,716 |
|
|
5.6 years |
|
$ |
7.94 |
|
|
|
72,716 |
|
|
$ |
7.94 |
|
17.52 – 24.12 |
|
|
641,991 |
|
|
5.0 years |
|
$ |
17.58 |
|
|
|
641,991 |
|
|
$ |
17.58 |
|
32.07 – 141.21 |
|
|
52,802 |
|
|
1.7 years |
|
$ |
86.55 |
|
|
|
52,802 |
|
|
$ |
86.55 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,044,073 |
|
|
|
|
|
|
$ |
7.11 |
|
|
|
2,080,461 |
|
|
$ |
9.45 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The total intrinsic value of options exercised during 2010, 2009 and 2008 was approximately
$0, $0.1 million and $0, respectively. Intrinsic value is measured using the fair market value at
the date of exercise (for shares exercised) or at December 31, 2010 (for outstanding options), less
the applicable exercise price.
The weighted-average fair value of the stock option awards granted to employees was $1.93,
$1.89 and $1.80 for the years ended December 31, 2010, 2009, and 2008, respectively, and was
estimated at the date of grant using the Black-Scholes option-pricing model and the assumptions
noted in the following table:
| |
|
|
|
|
|
|
| |
|
2010 |
|
2009 |
|
2008 |
| |
Expected life |
|
5 years |
|
5 years |
|
5 years |
Expected volatility |
|
108.0% to 109.0% |
|
110.0% to 118.0% |
|
87.5% to 118.0% |
Risk-free interest rate |
|
2.17% to 2.39% |
|
1.67% to 2.52% |
|
2.96% to 3.34% |
Dividend yield |
|
0% |
|
0% |
|
0% |
The expected life of the stock options was calculated using the method allowed by the
provisions of ASC 718-10. In accordance with ASC 718-10, the simplified method for “plain vanilla”
options may be used where the expected term is equal to the vesting term plus the original contract
term divided by two. For the years 2010, 2009 and 2008, the Company used its historical volatility
rate as management believes that this rate will be representative of future volatility over the
expected term of the options. The risk-free interest rate is based on the rates paid on securities
issued by the U.S. Treasury with a term approximating the expected life of the options. The Company
has never paid cash dividends, and does not currently intend to pay cash dividends, and thus has
assumed a 0% dividend yield.
Pre-vesting forfeitures. Estimates of pre-vesting option forfeitures are based on the
Company’s experience. Currently, the Company uses a forfeiture rate of 10%. The Company will adjust
its estimate of forfeitures over the requisite service period based on the extent to which actual
forfeitures differ, or are expected to differ, from such estimates. Changes in estimated
forfeitures will be recognized through a cumulative catch-up adjustment in the period of change and
will also impact the amount of compensation expense to be recognized in future periods.
F-23
Following the departure of a former director, the Company agreed to extend the period during
which he would be entitled to exercise certain vested stock options to purchase the Company’s
common stock from three months following the effective date of his resignation, February 3, 2009,
to six months following such effective date. The Company recorded compensation expense related to
the modification of the exercise period of $3,000 in the first quarter of 2009.
As of December 31, 2010, the total remaining unrecognized compensation cost related to
non-vested stock options, restricted stock and restricted stock units amounted to $1.2 million,
which will be amortized over the weighted-average remaining requisite service period of 2.7 years.
Restricted Stock
Restricted stock was issued to certain employees in 2007, and entitled the holder to receive a
specified number of shares of the Company’s common stock over a four year, monthly vesting term.
This restricted stock grant is accounted for at fair value at the date of grant and an expense is
recognized during the vesting term. Summarized information for restricted stock grants for the year
ended December 31, 2010 is as follows:
| |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
Weighted Average |
|
| |
|
Restricted Stock |
|
|
Grant Date Value Per Share |
|
| |
Nonvested at December 31, 2009 |
|
|
9,956 |
|
|
$ |
4.38 |
|
Granted |
|
|
— |
|
|
|
— |
|
Vested |
|
|
(8,958 |
) |
|
|
4.38 |
|
Forfeited |
|
|
(998 |
) |
|
|
— |
|
|
|
|
|
|
|
|
|
Nonvested at December 31, 2010 |
|
|
— |
|
|
$ |
4.38 |
|
|
|
|
|
|
|
|
|
Restricted Stock Units
Restricted stock units were issued to certain employees and non-employee members of the
Company’s Board of Directors in 2010 and 2009, which entitle the holder to receive a specified
number of shares of the Company’s common stock at the end of the two year or four year vesting
term. This restricted stock unit grant is accounted for at fair value at the date of grant and an
expense is recognized during the vesting term. Summarized information for restricted stock unit
grants for the year ended December 31, 2010 is as follows:
| |
|
|
|
|
|
|
|
|
| |
|
Restricted Stock |
|
|
Weighted Average |
|
| |
|
Units |
|
|
Grant Date Value Per Share |
|
| |
Nonvested at December 31, 2009 |
|
|
92,999 |
|
|
$ |
3.45 |
|
Granted |
|
|
— |
|
|
|
— |
|
Vested |
|
|
(13,391 |
) |
|
|
1.35 |
|
Forfeited |
|
|
(4,167 |
) |
|
|
4.02 |
|
|
|
|
|
|
|
|
|
Nonvested at December 31, 2010 |
|
|
75,441 |
|
|
$ |
3.72 |
|
|
|
|
|
|
|
|
|
2009 Employee Stock Purchase Plan
The 2009 Employee Stock Purchase Plan (the “2009 ESPP”) was adopted by the Board of Directors
on December 19, 2008, subject to stockholder approval, and was approved by the stockholders at the
Company’s 2009 Annual Meeting held on June 2, 2009. The 2009 ESPP was effective on January 1, 2009
and a total of 1,000,000 shares of common stock have been reserved for sale. The 2009 ESPP is
implemented by offerings of rights to all eligible employees from time to time. Unless otherwise
determined by the Company’s Board of Directors, common stock is purchased for accounts of employees
participating in the 2009 ESPP at a price per share equal to the lower of (i) 85% of the fair
market value of a share of the Company’s common stock on the first day the offering or (ii) 85% of
the fair market value of a share of the Company’s common stock on the last trading day of the
purchase period. The initial period commenced January 1, 2009 and ended on June 30, 2009. Each
subsequent offering period will have a six-month duration.
2005 Employee Stock Purchase Plan
The 2005 Employee Stock Purchase Plan (the “2005 ESPP”) was adopted on September 1, 2005 and
approved by the stockholders on September 5, 2005. The Employee Stock Purchase Plan became
effective on January 4, 2006 and a total of 500,000 shares of common stock were reserved for sale.
The Company commenced the administration of the 2005 ESPP in November 2007. The 2005 ESPP is
implemented by offerings of rights to all eligible employees from time to time. Unless otherwise
determined by the
Company’s Board of Directors, common stock is purchased for accounts of employees
participating in the 2005 ESPP at a price per share equal to the lower of (i) 85% of the fair
market value of a share of the Company’s common stock on the first day the offering or (ii) 85% of
the fair market value of a share of the Company’s common stock on the last trading day of the
purchase period. The initial period commenced November 16, 2007 and ended June 30, 2008.
F-24
The number of shares to be purchased at each balance sheet date is estimated based on the
current amount of employee withholdings and the remaining purchase dates within the offering
period. The fair value of share options expected to vest is estimated using the Black-Scholes
option-pricing model. Share options for employees entering the 2009 ESPP and 2005 ESPP were
estimated using the Black-Scholes option-pricing model and the assumptions noted on the table
below.
| |
|
|
|
|
|
|
| |
|
2010 |
|
2009 |
|
2008 |
Expected life |
|
.50 years |
|
.50 years |
|
.50 years |
Expected volatility |
|
110.0% |
|
114.0%-118.0% |
|
97.0% |
Risk-free interest rate |
|
0.20% |
|
0.28%-0.33% |
|
2.13% |
Dividend yield |
|
0% |
|
0% |
|
0% |
As of December 31, 2010, 78,267 shares were issued under the 2009 ESPP and 166,667 shares were
issued under the 2005 ESPP. For the years ended December 31, 2010, 2009 and 2008, the Company
recorded an expense of $23,000, $38,000 and $45,000, respectively, based on the estimated number of
shares to be purchased.
Warrants
The following table summarizes information about warrants outstanding at December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Expiration |
|
|
Common Shares |
|
|
|
|
| Issued in Connection With |
|
Date |
|
|
Issuable |
|
|
Exercise Price |
|
Acquisition of Maxim January 2006 |
|
|
2011 |
|
|
|
595 |
|
|
$ |
111.93 |
|
February 2006 stock issuance |
|
|
2011 |
|
|
|
340,069 |
|
|
|
12.00 |
|
December 2006 stock issuance |
|
|
2011 |
|
|
|
1,284,933 |
|
|
|
4.41 |
|
June 2007 stock issuance |
|
|
2012 |
|
|
|
889,576 |
|
|
|
8.79 |
|
Termination of sublicense agreement |
|
|
2012 |
|
|
|
83,333 |
|
|
|
5.88 |
|
October 2007 stock issuance |
|
|
2013 |
|
|
|
709,745 |
|
|
|
5.64 |
|
December 2007 stock issuance |
|
|
2013 |
|
|
|
555,555 |
|
|
|
4.50 |
|
March 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
1,011,744 |
|
|
|
2.58 |
|
2006 Senior Secured Term Loan |
|
|
2013 |
|
|
|
27,968 |
|
|
|
1.17 |
|
2006 Senior Secured Term Loan |
|
|
2013 |
|
|
|
325,203 |
|
|
|
1.23 |
|
June 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
766,667 |
|
|
|
1.17 |
|
July 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
33,333 |
|
|
|
1.17 |
|
August 1, 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
92,166 |
|
|
|
2.04 |
|
August 11, 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
60,409 |
|
|
|
1.89 |
|
August 11, 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
460,830 |
|
|
|
2.08 |
|
August 11, 2008 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
86,748 |
|
|
|
2.85 |
|
February 2009 convertible debt (See Note 7) |
|
|
2014 |
|
|
|
3,703,704 |
|
|
|
3.11 |
|
February 2009 convertible debt (See Note 7) |
|
|
2013 |
|
|
|
462,963 |
|
|
|
3.87 |
|
June 2009 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
1,400,000 |
|
|
|
2.70 |
|
June 2009 stock issuance (See Note 9) |
|
|
2013 |
|
|
|
200,000 |
|
|
|
3.00 |
|
June 2010 stock issuance (See Note 9) |
|
|
2011 |
|
|
|
6,136,363 |
|
|
|
1.64 |
|
June 2010 stock issuance (See Note 9) |
|
|
2015 |
|
|
|
4,602,273 |
|
|
|
1.64 |
|
June 2010 stock issuance (See Note 9) |
|
|
2015 |
|
|
|
306,818 |
|
|
|
1.38 |
|
November 2010 stock issuance (See Note 9) |
|
|
2015 |
|
|
|
163,935 |
|
|
|
0.76 |
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
|
|
|
|
|
23,704,930 |
|
|
$ |
2.77 |
|
|
|
|
|
|
|
|
|
|
|
|
F-25
12. Income Taxes
Deferred income taxes reflect the net effects of temporary differences between the carrying
amounts of assets and liabilities for financial reporting purposes and the amounts used for income
tax purposes. The Company’s deferred tax assets relate primarily to its net operating loss
carryforwards and other balance sheet basis differences. In accordance with ASC 740, “Income
Taxes,” the Company recorded a valuation allowance to fully offset the net deferred tax asset,
because it is not more likely than not that the Company will realize future benefits associated
with these deferred tax assets at December 31, 2010 and 2009. The change in the valuation allowance
for the years ended December 31, 2010 and 2009 was an increase of approximately $11.9 million and
$0.4 million, respectively. The increases in the valuation allowance of $11.9 million and $0.4
million during the years ended December 31, 2010 and 2009 were net of an increase (decrease) in the
valuation allowance of $6.2 million and $(15.0) million, respectively, as a result of adjustments
to the gross deferred tax asset related to the net operating loss carryforward due to an ownership
change under Internal Revenue Code Section 382 in 2009, which was subsequently adjusted in 2010.
Significant components of the Company’s deferred tax assets at December 31, 2010 and 2009 are as
follows:
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in thousands) |
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
Patent costs |
|
$ |
731 |
|
|
$ |
887 |
|
Stock-based compensation |
|
|
5,318 |
|
|
|
5,093 |
|
Accrued liabilities — U.S. |
|
|
275 |
|
|
|
237 |
|
Accrued liabilities — foreign |
|
|
347 |
|
|
|
395 |
|
Deferred revenue |
|
|
4,882 |
|
|
|
3,220 |
|
Fixed assets |
|
|
695 |
|
|
|
724 |
|
Deferred rent |
|
|
273 |
|
|
|
322 |
|
Other |
|
|
(76 |
) |
|
|
(86 |
) |
Warrant |
|
|
45 |
|
|
|
554 |
|
Credits |
|
|
3,611 |
|
|
|
3,501 |
|
Net operating loss carryforwards — U.S. |
|
|
47,530 |
|
|
|
36,515 |
|
Net operating loss carryforwards — foreign |
|
|
4,116 |
|
|
|
4,514 |
|
|
|
|
|
|
|
|
Total deferred tax assets |
|
|
67,747 |
|
|
|
55,876 |
|
Valuation allowance |
|
|
(67,747 |
) |
|
|
(55,876 |
) |
|
|
|
|
|
|
|
Net deferred tax assets |
|
$ |
— |
|
|
$ |
— |
|
|
|
|
|
|
|
|
A reconciliation of the federal statutory tax rate and the effective tax rates for the years
ended December 31, 2010, 2009 and 2008 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
For the Year Ended |
|
| |
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
Statutory tax rate |
|
|
(34.0 |
)% |
|
|
(34.0 |
)% |
|
|
(34.0 |
)% |
State income taxes, net of federal benefit |
|
|
(5.5 |
) |
|
|
(5.5 |
) |
|
|
(5.5 |
) |
Equity-based compensation |
|
|
0.7 |
|
|
|
0.5 |
|
|
|
0.0 |
|
Foreign deferred tax adjustment |
|
|
3.8 |
|
|
|
0.0 |
|
|
|
0.0 |
|
Other |
|
|
(1.9 |
) |
|
|
0.0 |
|
|
|
0.2 |
|
Change in valuation allowance |
|
|
36.9 |
|
|
|
39.0 |
|
|
|
39.3 |
|
|
|
|
|
|
|
|
|
|
|
Effective tax rate |
|
|
(0 |
)% |
|
|
(0 |
)% |
|
|
(0 |
)% |
|
|
|
|
|
|
|
|
|
|
The principal differences between the U.S. statutory tax rate of 34% and the Company’s
effective tax rates of (0)% for the years ended December 31, 2010, 2009 and 2008 is primarily due
to the Company’s valuation allowance and the write-down of net operating loss carryforwards due to
an ownership change under Internal Revenue Code Section 382 in 2009.
F-26
The Company has approximately $252.1 million of net operating loss carryforwards (federal,
state and foreign) and tax credit carryforwards of $3.6 million as of December 31, 2010. The
Company reduced its tax attributes (NOL’s and tax credits) as a result of the Company’s ownership
changes in 2009 and 2007 and the limitation placed on the utilization of its tax attributes as a
substantial portion of the NOL’s and tax credits generated prior to the ownership change will
likely expire unused. Accordingly, the federal NOL’s were reduced by approximately $272.0 million
and the tax credit carryforwards were reduced by approximately $7.3 million.
| |
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
| |
|
(in millions) |
|
| |
Federal NOL’s |
|
$ |
120.9 |
|
|
$ |
90.8 |
|
State NOL’s |
|
|
115.6 |
|
|
|
95.5 |
|
Foreign NOL’s |
|
|
15.6 |
|
|
|
15.0 |
|
|
|
|
|
|
|
|
Total NOL’s |
|
$ |
252.1 |
|
|
$ |
201.3 |
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
| |
|
(in thousands) |
|
Federal Credits |
|
$ |
406 |
|
|
$ |
301 |
|
State Credits |
|
|
3,205 |
|
|
|
3,199 |
|
|
|
|
|
|
|
|
Total Credits |
|
$ |
3,611 |
|
|
$ |
3,500 |
|
|
|
|
|
|
|
|
The Company’s federal NOL’s of $120.9 million and state NOL’s of $115.6 million begin to
expire after 2012 up through 2030. The Company’s foreign NOL’s of $15.6 million do not expire. The
Company’s federal and state tax credits of $3.6 million begin to expire in 2024 through 2030.
A reconciliation of the beginning and ending amount of gross unrecognized tax benefits is as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
December 31, |
|
|
December 31, |
|
|
December 31, |
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
| |
Balance at January 1, |
|
$ |
— |
|
|
$ |
115 |
|
|
$ |
115 |
|
Additions (reductions) related to tax positions |
|
|
— |
|
|
|
(115 |
) |
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
115 |
|
|
|
|
|
|
|
|
|
|
|
F-27
13. Segment and Geographic Information
The Company operates as one reportable segment. The Company maintains development operations
in the United States and Germany. Geographic information for the years ended December 31, 2010,
2009 and 2008 are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
2010 |
|
|
2009 |
|
|
2008 |
|
| |
|
(in thousands) |
|
Revenue |
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
993 |
|
|
$ |
412 |
|
|
$ |
262 |
|
Germany |
|
|
1 |
|
|
|
2 |
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
994 |
|
|
$ |
414 |
|
|
$ |
265 |
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
14,187 |
|
|
$ |
38,505 |
|
|
$ |
24,218 |
|
Germany |
|
|
1,350 |
|
|
|
309 |
|
|
|
1,164 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
15,537 |
|
|
$ |
38,814 |
|
|
$ |
25,382 |
|
|
|
|
|
|
|
|
|
|
|
Total Assets |
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
4,564 |
|
|
$ |
7,417 |
|
|
$ |
2,219 |
|
Germany |
|
|
125 |
|
|
|
97 |
|
|
|
52 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
4,689 |
|
|
$ |
7,514 |
|
|
$ |
2,271 |
|
|
|
|
|
|
|
|
|
|
|
Long Lived Assets, net |
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
211 |
|
|
$ |
348 |
|
|
$ |
499 |
|
Germany |
|
|
11 |
|
|
|
12 |
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
222 |
|
|
$ |
360 |
|
|
$ |
502 |
|
|
|
|
|
|
|
|
|
|
|
14. Quarterly Results (Unaudited)
Summarized quarterly results of operations for the years ended December 31, 2010 and 2009 are
as follows (in thousands except per share and share amounts):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, 2010 |
|
| |
|
First |
|
|
Second |
|
|
Third |
|
|
Fourth |
|
| |
|
(in thousands, except for share and per share amounts) |
|
Revenue |
|
$ |
195 |
|
|
$ |
251 |
|
|
$ |
257 |
|
|
$ |
291 |
|
Total costs and expenses |
|
|
4,124 |
|
|
|
4,335 |
|
|
|
4,265 |
|
|
|
3,644 |
|
Net loss |
|
|
(4,508 |
) |
|
|
(4,889 |
) |
|
|
(3,168 |
) |
|
|
(2,972 |
) |
Basic and diluted loss per common share(1) |
|
|
(0.10 |
) |
|
|
(0.11 |
) |
|
|
(0.06 |
) |
|
|
(0.06 |
) |
Weighted average shares outstanding |
|
|
44,160,266 |
|
|
|
44,274,642 |
|
|
|
50,393,488 |
|
|
|
52,466,655 |
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, 2009 |
|
| |
|
First |
|
|
Second |
|
|
Third |
|
|
Fourth |
|
| |
|
(in thousands, except for share and per share amounts) |
|
Revenue |
|
$ |
115 |
|
|
$ |
91 |
|
|
$ |
116 |
|
|
$ |
92 |
|
Total costs and expenses |
|
|
4,197 |
|
|
|
5,541 |
|
|
|
5,136 |
|
|
|
4,278 |
|
Net loss |
|
|
(22,487 |
) |
|
|
(7,079 |
) |
|
|
(4,813 |
) |
|
|
(4,436 |
) |
Basic and diluted loss per common share(1) |
|
|
(0.68 |
) |
|
|
(0.18 |
) |
|
|
(0.11 |
) |
|
|
(0.10 |
) |
Weighted average shares outstanding |
|
|
32,894,822 |
|
|
|
39,727,916 |
|
|
|
43,740,961 |
|
|
|
44,031,387 |
|
| |
|
|
| (1) |
|
Basic and diluted loss per common share are computed independently for each of the periods
presented. Accordingly, the sum of the quarterly loss per common share amounts may not agree
to the total for the year. |
15. Subsequent Events (Unaudited)
In February 2011, the Company sold approximately 8.9 million shares of its common stock and
warrants to purchase 3.6 million shares of its common stock for gross proceeds of $7.1 million,
$6.6 million net of $0.5 million in transaction costs.
In March 2011, the Company sold approximately 7.1 million shares of its common stock and
warrants to purchase 5.3 million shares of its common stock for gross proceeds of $4.6 million,
$4.3 million net of $0.3 million in transaction costs.
F-28
Exhibit Index
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description of Exhibits |
| |
2.1 |
|
|
Agreement and Plan of Merger, dated as of September 6, 2005, among EpiCept Corporation,
Magazine Acquisition Corp. and Maxim Pharmaceuticals, Inc. (incorporated by reference to
Exhibit 2.1 to Maxim Pharmaceuticals Inc.’s Current Report on Form 8-K filed September 6,
2005). |
| |
|
|
|
|
| |
3.1 |
|
|
Third Amended and Restated Certificate of Incorporation of EpiCept Corporation (incorporated
by reference to Exhibit 3.1 to EpiCept Corporation’s Current Report on Form 8-K filed May 21,
2008). |
| |
|
|
|
|
| |
3.2 |
|
|
Amendment to the Third Amended and Restated Certificate of Incorporation (incorporated by
reference to Exhibit 3.1 to EpiCept Corporation’s Current Report on Form 8-K filed July 9,
2009). |
| |
|
|
|
|
| |
3.3 |
|
|
Certificate of Amendment to the Third Amended and Restated Certificate of Incorporation, filed
with the Secretary of State of the State of Delaware on January 14, 2010 (incorporated by
reference to Exhibit 3.1 to EpiCept Corporation’s Current Report on Form 8-K filed January 14,
2010). |
| |
|
|
|
|
| |
3.4 |
|
|
Amended and Restated Bylaws of EpiCept Corporation (incorporated by reference to Exhibit 3.1
to EpiCept Corporation’s Current Report on Form 8-K filed February 18, 2010). |
| |
|
|
|
|
| |
4.1 |
|
|
Specimen Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the EpiCept’s
Corporation’s Registration Statement on Form S-1 (File No. 333-121938) (the “EpiCept Form
S-1”)). |
| |
|
|
|
|
| |
4.2 |
|
|
Convertible Debenture due April 10, 2009 (incorporated by reference to Exhibit 4.1 to EpiCept
Corporation’s Current Report on Form 8-K, filed December 9, 2008). |
| |
|
|
|
|
| |
4.3 |
|
|
Indenture between EpiCept Corporation and Bank of New York Mellon, as Trustee, dated February
9, 2009 (incorporated by reference to Exhibit 4.1 to Epicept Corporation’s Current Report on
Form 8-K, filed February 10, 2009). |
| |
|
|
|
|
| |
4.4 |
|
|
Form of Series A Warrant (incorporated by reference to Exhibit 4.1 to EpiCept Corporatiion’s
Current Report on Form 8-k filed July 1, 2010). |
| |
|
|
|
|
| |
4.5 |
|
|
Form of Series B Warrant (incorporated by reference to Exhibit 4.2 to EpiCept Corporatiion’s
Current Report on Form 8-k filed July 15, 2010). |
| |
|
|
|
|
| |
4.6 |
|
|
Form of Warrant (incorporated by reference to Exhibit 4.1 to EpiCept Corporatiion’s Current
Report on Form 8-k filed November 12, 2010). |
| |
|
|
|
|
| |
10.1 |
|
|
Form of Indemnification Agreement between EpiCept Corporation and each of its directors and
executive officers (incorporated by reference to Exhibit 10.1 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
†10.2 |
|
|
1995 Stock Option Plan (incorporated by reference to Exhibit 10.2 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
†10.3 |
|
|
2005 Equity Incentive Plan (Amended and Restated May 23, 2007) (incorporated by reference to
Exhibit 10.1 to EpiCept’s Current Report on Form 8-K filed May 30, 2007). |
| |
|
|
|
|
| |
†10.4 |
|
|
2005 Employee Stock Purchase Plan (Incorporated by reference to Exhibit 10.4 of the Form S-4). |
| |
|
|
|
|
| |
†10.5 |
|
|
Employment Agreement, dated as of October 28, 2004, between EpiCept Corporation and John V.
Talley (incorporated by reference to Exhibit 10.5 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
†10.6 |
|
|
Employment Agreement, dated as of October 28, 2004, between EpiCept Corporation and Robert
Cook (incorporated by reference to Exhibit 10.6 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.7 |
|
|
License Agreement, dated as of December 18, 2003, between Endo Pharmaceuticals Inc. and
EpiCept Corporation (incorporated by reference to Exhibit 10.9 to the EpiCept Form S-1). |
F-29
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description of Exhibits |
| |
10.8 |
|
|
Royalty Agreement, dated as of July 16, 2003, between EpiCept Corporation and R. Douglas
Cassel, M.D. (incorporated by reference to Exhibit 10.10 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.9 |
|
|
Cooperation Agreement between APL American Pharmed Labs, Inc. and
Technologie-Beteiligungs-Gesellschaft mbH, dated August 1997 (incorporated by reference to
Exhibit 10.13 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.10 |
|
|
Investment Agreement between Pharmed Labs GmbH and Technologie-Beteiligungs-Gesellschaft mbH,
dated August 1997 (incorporated by reference to Exhibit 10.14 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.11 |
|
|
Investment Agreement among Pharmed Labs GmbH, American Pharmed Labs, Inc. and
Technologie-Beteiligungs-Gesellschaft mbH, dated February 17, 1998 (incorporated by reference
to Exhibit 10.15 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.12 |
|
|
Lease Agreement between BMR-Landmark at Eastview LLC, as Landlord, and EpiCept Corporation, as
Tenant, dated August 28, 2006 (incorporated by reference to Exhibit 10.12 to EpiCept
Corporation’s Annual Report on Form 10-K filed March 18, 2008). |
| |
|
|
|
|
| |
10.13 |
|
|
First Exchange Option Agreement, dated as of December 31, 1997, by and between American
Pharmed Labs, Inc. and tbg Technologie-Beteiligungs-Gesellschaft mbg der Deutschen
Ausgleichsbank (incorporated by reference to Exhibit 10.22 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.14 |
|
|
Second Exchange Option Agreement, dated as of February 17, 1998, by and between American
Pharmed Labs, Inc. and tbg Technologie-Beteiligungs-Gesellschaft mbh der Deutschen
Ausgleichsbank (incorporated by reference to Exhibit 10.23 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.15 |
|
|
Amendment to Second Exchange Option Agreement, dated as of August 26, 2005, by and between
EpiCept Corporation and tbg Technologie-Beteiligungs-Gesellschaft mbh der Deutschen
Ausgleichsbank (incorporated by reference to Exhibit 10.28 to the EpiCept Form S-4). |
| |
|
|
|
|
| |
†10.16 |
|
|
Amended and Restated Note Purchase Agreement (the “Note Purchase Agreement”), dated as of
March 3, 2005, by and among EpiCept Corporation and the Purchasers indentified therein
(incorporated by reference to Exhibit 10.18 to the EpiCept Form S-1). |
| |
|
|
|
|
| |
10.17 |
|
|
Amendment No. 1 to License Agreement between EpiCept Corporation and DURECT Corporation, dated
as of September 12, 2008 (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s
Current Report on Form 8-K filed September 17, 2008). |
| |
|
|
|
|
| |
10.18 |
|
|
Loan and Security Agreement with Hercules Technology Growth Capital, Inc., dated August 30,
2008 (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s Current Report on
Form 10-Q filed November 9, 2006). |
| |
|
|
|
|
| |
10.19 |
|
|
First Amendment to the Loan and Security Agreement with Hercules Technology Growth Capital,
Inc., dated May 7, 2008 (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s
Current Report on Form 8-K filed May 9, 2008). |
| |
|
|
|
|
| |
10.20 |
|
|
Second Amendment to Loan and Security Agreement, by and among EpiCept Corporation, Maxim
Pharmaceuticals Inc. and Hercules Technology Growth Capital, Inc., dated as of June 23, 2008
(incorporated by reference to Exhibit 10.4 to EpiCept Corporation’s Current Report on Form 8-K
filed June 23, 2008). |
| |
|
|
|
|
| |
10.21 |
|
|
Warrant Agreement with Hercules Technology Growth Capital, Inc., dated August 30, 2008
(incorporated by reference to Exhibit 10.2 to EpiCept Corporation’s Current Report on Form
10-Q filed November 9, 2006). |
| |
|
|
|
|
| |
10.22 |
|
|
Second Amendment to the Warrant Agreement with Hercules Technology Growth Capital, Inc., dated
May 7, 2008 (incorporated by reference to Exhibit 10.2 to EpiCept Corporation’s Current Report
on Form 8-K filed May 9, 2008). |
| |
|
|
|
|
| |
10.23 |
|
|
First Amendment to the Deposit Account Control Agreement with Hercules Technology Growth
Capital, Inc., dated May 7, 2008 (incorporated by reference to Exhibit 10.3 to EpiCept
Corporation’s Current Report on Form 8-K filed May 9, 2008). |
F-30
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description of Exhibits |
| |
10.24 |
|
|
Common Stock Warrant, by and between EpiCept Corporation and Hercules Technology Growth
Capital, Inc., dated as of June 23, 2008 (incorporated by reference to Exhibit 10.6 to EpiCept
Corporation’s Current Report on Form 8-K filed June 23, 2008). |
| |
|
|
|
|
| |
10.25 |
|
|
Amendment to the Repayment Agreement by and between EpiCept Corporation and
Technologie-Beteiligungs Gesellschaft mbH der Deutschen Ausgleichsbank, dated May 23, 2008
(incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s Current Report on Form 8-K
filed May 28, 2008). |
| |
|
|
|
|
| |
10.26 |
|
|
Securities Purchase Agreement, dated June 28, 2007 (incorporated by reference to Exhibit 10.1
to EpiCept’s Current Report on Form 8-K filed June 29, 2007). |
| |
|
|
|
|
| |
10.27 |
|
|
Form of Warrant, dated as of June 28, 2007 (incorporated by reference to Exhibit 10.3 to
EpiCept Corporation’s Current Report on Form 8-K filed June 29, 2007). |
| |
|
|
|
|
| |
10.28 |
|
|
Securities Purchase Agreement, dated October 10, 2007 (incorporated by reference to Exhibit
10.1 to EpiCept Corporation’s Current Report on Form 8-K filed October 11, 2007). |
| |
|
|
|
|
| |
10.29 |
|
|
Form of Warrant, dated as of October 10, 2007 (incorporated by reference to Exhibit 10.2 to
EpiCept Corporation’s Current Report on Form 8-K filed October 11, 2007). |
| |
|
|
|
|
| |
10.30 |
|
|
Securities Purchase Agreement, dated December 4, 2007 (incorporated by reference to Exhibit
10.1 to EpiCept Corporation’s Current Report on Form 8-K filed December 5, 2007). |
| |
|
|
|
|
| |
10.31 |
|
|
Form of Common Stock Purchase Warrant, dated as of December 4, 2007 (incorporated by reference
to Exhibit 10.2 to EpiCept Corporation’s Current Report on Form 8-K filed December 5, 2007). |
| |
|
|
|
|
| |
10.32 |
|
|
Securities Purchase Agreement, dated March 6, 2008 (incorporated by reference to Exhibit 10.1
to EpiCept Corporation’s Current Report on Form 8-K dated March 6, 2008). |
| |
|
|
|
|
| |
10.33 |
|
|
Securities Purchase Agreement, dated as of June 23, 2008 (incorporated by reference to Exhibit
10.1 to EpiCept Corporation’s Current Report on Form 8-K filed June 25, 2008). |
| |
|
|
|
|
| |
10.34 |
|
|
Form of Warrant, dated as of June 23, 2008 (incorporated by reference to Exhibit 10.6 to
EpiCept Corporation’s Current Report on Form 8-K filed June 23, 2008). |
| |
|
|
|
|
| |
10.35 |
|
|
Securities Purchase Agreement, dated July 15, 2008 (incorporated by reference to Exhibit 10.1
to EpiCept Corporation’s Current Report on Form 8-K filed July 16, 2008). |
| |
|
|
|
|
| |
10.36 |
|
|
Form of Warrant, dated as of July 15, 2008 (incorporated by reference to Exhibit 10.2 to
EpiCept Corporation’s Current Report on Form 8-K filed July 16, 2008). |
| |
|
|
|
|
| |
10.37 |
|
|
Securities Purchase Agreement, dated August 1, 2008 (incorporated by reference to Exhibit 10.1
to EpiCept Corporation’s Current Report on Form 8-K filed August 4, 2008). |
| |
|
|
|
|
| |
10.38 |
|
|
Form of Warrant, dated as of August 1, 2008 (incorporated by reference to Exhibit 10.2 to
EpiCept Corporation’s Current Report on Form 8-K filed August 4, 2008). |
| |
|
|
|
|
| |
10.39 |
|
|
Securities Purchase Agreement, dated August 11, 2008 (incorporated by reference to Exhibit
10.1 to EpiCept Corporation’s Current Report on Form 8-K filed August 12, 2008). |
| |
|
|
|
|
| |
10.40 |
|
|
Form of Warrant, dated as of August 11, 2008 (incorporated by reference to Exhibit 10.2 to
EpiCept Corporation’s Current Report on Form 8-K filed August 12, 2008). |
| |
|
|
|
|
| |
10.41 |
|
|
Securities Purchase Agreement, dated as of December 8, 2008 (incorporated by reference to
Exhibit 10.2 to EpiCept Corporation’s Current Report on Form 8-K filed December 9, 2008). |
F-31
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description of Exhibits |
| |
10.42 |
|
|
2009 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.1 to EpiCept
Corporation’s Registration Statement on Form S-8 filed December 23, 2008). |
| |
|
|
|
|
| |
10.43 |
|
|
Second Amendment to the Repayment Agreement by and between EpiCept Corporation and
Technologie-Beteiligungs Gesellschaft mbH der Deutschen Ausgleichsbank, dated November 26,
2008 (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s Current Report on
form 8-K filed December 3, 2008). |
| |
|
|
|
|
| |
10.44 |
|
|
Placement Agent Agreement (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s
Current Report on form 8-K filed December 9, 2008) |
| |
|
|
|
|
| |
10.45 |
|
|
Securities Purchase Agreement, dated as of February 4, 2009 (incorporated by reference to
Exhibit 10.1 to EpiCept Corporation’s Current Report on Form 8-K filed February 10, 2009). |
| |
|
|
|
|
| |
10.46 |
|
|
Form of Warrant (incorporated by reference to Exhibit 10.2 to EpiCept Corporation’s Current
Report on
Form 8-K filed February 10, 2009). |
| |
|
|
|
|
| |
10.47 |
|
|
Placement Agent Agreement, dated February 4, 2009 (incorporated by reference to Exhibit 10.3
to EpiCept Corporation’s Current Report on Form 8-K filed February 10, 2009). |
| |
|
|
|
|
| |
10.48 |
|
|
Securities Purchase Agreement, dated as of June 18, 2009 (incorporated by reference to Exhibit
10.1 to EpiCept Corporation’s Current Report on Form 8-K filed June 19, 2009). |
| |
|
|
|
|
| |
10.49 |
|
|
Form of Warrant (incorporated by reference to Exhibit 10.2 to EpiCept Corporation’s Current
Report on
Form 8-K filed June 19, 2009). |
| |
|
|
|
|
| |
10.50 |
|
|
Placement Agent Agreement, dated June 18, 2009 (incorporated by reference to Exhibit 10.3 to
EpiCept Corporation’s Current Report on Form 8-K filed June 19, 2009). |
| |
|
|
|
|
| |
10.51 |
|
|
Amendment to the Repayment Agreement, dated June 25, 2009 (English translation from the
original German) (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s Current
Report on Form 8-K filed June 30, 2009). |
| |
|
|
|
|
| |
10.52 |
|
|
License and Supply Agreement, dated as of January 8, 2010, between EpiCept Corporation,
EpiCept GmbH and Meda AB (incorporated by reference to Exhibit 10.1 to EpiCept Corporation’s
Quarterly Report on Form 10-Q for the quarter ended March 31, 2010). |
| |
|
|
|
|
| |
10.53 |
† |
|
Amended and Restated Employment Agreement, by and between EpiCept Corporation and John V.
Talley, dated as of June 9, 2010 (incorporated by reference to Exhibit 10.1 to EpiCept
Corporation’s Current Report on
Form 8-K filed June 15, 2010). |
| |
|
|
|
|
| |
10.54 |
|
|
Securities Purchase Agreement, dated June 25, 2010 (incorporated by reference to Exhibit 10.1
to EpiCept Corporation’s Current Report on Form 8-K filed July 1, 2010). |
| |
|
|
|
|
| |
10.55 |
|
|
Placement Agent Agreement, dated June 22, 2010 (incorporated by reference to Exhibit 10.2 to
EpiCept Corporation’s Current Report on Form 8-K filed July 1, 2010). |
| |
|
|
|
|
| |
10.56 |
|
|
Amendment No. 1 to Securities Purchase Agreement, dated July 8, 2010 (incorporated by
reference to Exhibit 10.1 to EpiCept Corporation’s Current Report on Form 8-K filed July 14,
2010). |
| |
|
|
|
|
| |
10.57 |
† |
|
Amended and Restated Employment Agreement, by and between EpiCept Corporation and Robert W.
Cook, dated as of July 21, 2010 (incorporated by reference to Exhibit 10.1 to EpiCept
Corporation’s Current Report on Form 8-K filed July 26, 2010). |
| |
|
|
|
|
| |
10.58 |
† |
|
Form of executive Severance Agreement, dated as of July 21, 2010 (incorporated by reference to
Exhibit 10.2 to EpiCept Corporation’s Current Report on Form 8-K filed July 26, 2010). |
F-32
| |
|
|
|
|
| Exhibit |
|
|
| Number |
|
Description of Exhibits |
| |
10.59 |
|
|
Securities Purchase Agreement, dated November 5, 2010 (incorporated by reference to Exhibit
10.2 to EpiCept Corporation’s Current Report on Form 8-K filed November 12, 2010). |
| |
|
|
|
|
| |
11.1 |
|
|
Statement Regarding Computation of Per Share Earnings (incorporated by reference to the Notes
to Consolidated Financial Statements included in Part II of this Report). |
| |
|
|
|
|
|
21.1 |
* |
|
List of Subsidiaries of EpiCept Corporation. |
| |
|
|
|
|
| |
23.1 |
* |
|
Consent of Deloitte & Touche LLP, Independent Registered Public Accounting Firm. |
| |
|
|
|
|
|
31.1 |
* |
|
Certification of Chief Executive Officer, pursuant to Exchange Act Rules
13a-14(a) and 15(d)-14(a), adopted pursuant to Section 302 of the Sarbanes-Oxley
Act of 2002. |
| |
|
|
|
|
|
31.2 |
* |
|
Certification of Chief Financial Officer, pursuant to Exchange Act Rules
13a-14(a) and 15(d)-14(a), adopted pursuant to Section 302 of the Sarbanes-Oxley
Act of 2002. |
| |
|
|
|
|
|
32.1 |
** |
|
Certification of Chief Executive Officer, pursuant to 18 U.S.C. 1350, adopted
pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| |
|
|
|
|
|
32.2 |
** |
|
Certification of Chief Financial Officer, pursuant to pursuant to 18 U.S.C.
1350, adopted Section 906 of the Sarbanes-Oxley Act of 2002. |
| |
|
|
| * |
|
Filed herewith. |
| |
| ** |
|
Furnished herewith. |
| |
| † |
|
Management contract or compensatory plan or arrangement |
| (c) |
|
Financial Statements Schedules. |
None.
F-33