Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - TENGION INC | ex23-1.htm |
| EX-32.2 - EXHIBIT 32.2 - TENGION INC | ex32-2.htm |
| EX-31.1 - EXHIBIT 31.1 - TENGION INC | ex31-1.htm |
| EX-23.2 - EXHIBIT 23.2 - TENGION INC | ex23-2.htm |
| EX-32.1 - EXHIBIT 32.1 - TENGION INC | ex32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - TENGION INC | ex31-2.htm |
| EX-10.29 - EXHIBIT 10.29 - TENGION INC | ex10-29.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| [X] |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the fiscal year ended December 31, 2010
OR
| [ ] |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from _____ to _____
Commission file number 001-34688
|
Tengion, Inc.
(Exact name of registrant as specified in its charter)
|
|
|
Delaware
(State or other jurisdiction of incorporation or organization)
|
20-0214813
(I.R.S. Employer Identification No.)
|
|
2900 Potshop Lane, Suite 100
East Norriton, PA 19403
(Address of principal executive offices)
(610) 292-8364
(Registrant’s telephone number, including area code)
|
|
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes [ ] No [X]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [ ] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer, as defined in Rule 12b-2 of the Exchange Act.
Large accelerated filer [ ] Accelerated filer [ ] Non-accelerated filer [X] Smaller reporting company [ ]
Indicate by check mark whether the registrant is a shell company, as defined in Rule 12b-2 of the Exchange Act. Yes [ ] No [X]
The aggregate market value of the registrant’s common stock held by non-affiliates of the registrant as of June 30, 2010 was $24,737,900. This calculation excludes 5,704,657 shares held on June 30, 2010 by directors, executive officers, and one holder of more than 10% of the registrant’s common stock. As of March 24, 2011, there were 23,471,249 shares of the registrant’s common stock outstanding.
TENGION, INC.
FORM 10-K
INDEX
|
Part I
|
||
|
Item 1.
|
Business
|
|
|
Item 1A.
|
Risk Factors
|
|
|
Item 1B.
|
Unresolved Staff Comments
|
|
|
Item 2.
|
Properties
|
|
|
Item 3.
|
Legal Proceedings
|
|
|
Item 4.
|
(Removed and Reserved)
|
|
|
Part II
|
||
|
Item 5.
|
Market for Registrant’s Common Equity, Related Stockholder Matters
|
|
|
and Issuer Purchases of Equity Securities
|
||
|
Item 6.
|
Selected Financial Data
|
|
|
Item 7.
|
Management’s Discussion and Analysis of Financial Condition and
|
|
|
Results of Operations.
|
||
|
Item 7A.
|
Quantitative and Qualitative Disclosures about Market Risk
|
|
|
Item 8.
|
Financial Statements and Supplementary Data
|
|
|
Item 9.
|
Changes in and Disagreements with Accountants on Accounting
|
|
|
and Financial Disclosure
|
||
|
Item 9A.
|
Controls and Procedures
|
|
|
Item 9B.
|
Other Information
|
|
|
Part III
|
||
|
Item 10.
|
Directors, Executive Officers and Corporate Governance
|
|
|
Item 11.
|
Executive Compensation
|
|
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management
|
|
|
and Related Stockholder Matters
|
||
|
Item 13.
|
Certain Relationships and Related Party Transactions, and Director
|
|
|
Independence
|
||
|
Item 14.
|
Principal Accounting Fees and Services
|
|
|
Part IV
|
||
|
Item 15.
|
Exhibits and Financial Statement Schedules
|
|
|
Signatures
|
||
Tengion® and the Tengion logo® are our registered trademarks and Tengion Neo-Urinary Conduit™, Tengion Neo-Kidney™, Tengion Neo-Kidney Augment™, Tengion Neo-Vessel™, Tengion Neo-Vessel Replacement™, Tengion Neo-Bladder Replacement™, Neo-Bladder Augment™, Tengion Organ Regeneration Platform™ and Organ Regeneration Platform™ are our trademarks. Other names are for informational purposes only and may be trademarks of their respective owners.
PART I
Item 1. Business
Overview
We believe we are the only regenerative medicine company focused on discovering, developing, manufacturing and commercializing a range of neo-organs, which we define as products composed of living cells, with or without synthetic or natural materials, implanted into the body to incorporate, replace or regenerate a damaged tissue or organ. We currently create these functional neo-organs using a patient’s own cells, or autologous cells, in conjunction with our Organ Regeneration Platform. We believe our proprietary product candidates harness the intrinsic regenerative pathways of the body to regenerate a range of native-like organs and tissues. Our neo-organs are designed to avoid the need to substitute other tissues of the body for a purpose to which they are poorly suited. In addition, our product candidates are intended to eliminate the need for donor organs and the administration of anti-rejection medications. We produce neo-organs in our scalable manufacturing facilities using efficient and repeatable proprietary processes, and have implanted neo-organs in our clinical trials. Building on our clinical and preclinical experience, we are initially leveraging our Organ Regeneration Platform to develop our Neo-Urinary Conduit for bladder cancer patients who are in need of a urinary diversion. We intend to develop our technology to address unmet medical needs in urologic, renal, and other diseases and disorders.
Regenerative medicine is an interdisciplinary field combining expertise in developmental biology, medicine and engineering with the goal of restoring native-like organ and tissue function. The premise of regenerative medicine is that the body has the intrinsic capacity to heal and regenerate. Liver tissue, for example, will naturally regenerate following partial resection. In many situations, however, the appropriate conditions that facilitate the regenerative process in the body may not be present for reasons such as the absence of appropriate gene expression or the presence of an underlying disease process. Regenerative medicine seeks to provide the necessary stimulus, environment or foundation to promote the body’s innate regenerative capacity. While the FDA has approved regenerative medicine products for skin applications and repair of selected cartilage defects, we believe that we are the only company using regenerative medicine to harness the body’s ability to regenerate a range of functional, native-like organs and tissues.
Our Organ Regeneration Platform has enabled us to develop our product candidates from proof of concept to first-in-man clinical trials in as short as 24 months. Our technology has been applied in two Phase II clinical trials for our Neo-Bladder Augment. Recent advances in our platform technology have enabled us to significantly simplify our process of manufacturing neo-organs, which we believe will allow us to address new and larger market opportunities. Our lead product candidate, our Neo-Urinary Conduit, is intended to replace the use of bowel tissue in bladder cancer patients requiring a non-continent urinary diversion after bladder removal surgery, or cystectomy. We are able to manufacture our Neo-Urinary Conduit using a proprietary process that takes four weeks or less and uses only smooth muscle cells derived from a routine fat biopsy. We are currently conducting a Phase I clinical trial for our Neo-Urinary Conduit in bladder cancer patients.
We were incorporated in Delaware in 2003. Our corporate headquarters are located at 2900 Potshop Lane, Suite 100, East Norriton, Pennsylvania and our telephone number is (267) 960-4800.
Our Organ Regeneration Platform
Our Organ Regeneration Platform is based on extensive work that began in the early 1990s at Children’s Hospital Boston and Massachusetts Institute of Technology, and continued at the Wake Forest Institute for Regenerative Medicine and our company. Our proprietary approach involves the use of a combinatorial method, testing different combinations of cell type and scaffold material. We believe this approach enables us to accurately identify the combination of various cell types and scaffold materials for a specific organ necessary to elicit a regenerative response and guides our decisions for subsequent rounds of testing. We own or license over 30 U.S. patents and patent applications and over 100 international patents and filings related to our Organ Regeneration Platform and product candidates.
The organ regeneration process enabled by our proprietary Organ Regeneration Platform involves the following steps:
|
|
·
|
Isolation and expansion of progenitor cells. Our autologous organ regeneration process begins with our receipt of a small tissue sample, generally obtained by a routine biopsy of the patient. This sample is then sent to our pilot manufacturing facility in North Carolina where our scientists engage in a specialized process to isolate the necessary committed progenitor cells, of one or more types as needed, that form the basis of the target organ’s or tissue’s essential function. Committed progenitor cells have been programmed by the body to become specific cell types, but are not yet developed into a single cell type, retaining the ability to promote regeneration. We obtain the requisite progenitor cells from various sources. For example, for our Neo-Urinary Conduit, we select smooth muscle progenitor cells from a small sample of adipose, or fat tissue. In the case of our Neo-Kidney Augment, we obtain a tissue sample through a small gauge needle biopsy of the kidney. We then use our proprietary cell growth process to grow, or expand, the specifically isolated progenitor cells ex vivo, or outside of the body, until an adequate number of cells are produced.
|
|
|
·
|
Seeding and growth. Once we have completed our cell expansion process, we separately place, or seed, these cell populations on a bioabsorbable scaffold. Depending upon the type of neo-organ being created, we use biomaterials for the scaffold that, when combined with the isolated and expanded cell populations, promote the desired regenerative outcome. We select the type of material based upon our knowledge of the organ or tissue we are seeking to regenerate and extensive testing to determine the optimal material characteristics, treatment and shape that will encourage cell growth and catalyze the body’s regenerative power. Composition and design of the biomaterials are important elements of our technology that help to ensure native-like tissue regeneration and ease of delivery through, where possible, minimally invasive surgical procedures.
|
2
We then place the bioabsorbable scaffold, seeded with the cell populations, in a bioreactor, or a closed container used for enhancing biological growth under controlled conditions. Further manipulation and handling of the neo-organ is done without removal from the bioreactor, which greatly reduces the risk of contamination. The expanded cell populations attach to the biomaterials and begin to form appropriate cellular and tissue layers for the neo-organ, until ready for implantation.
|
|
·
|
Implantation. The neo-organ is typically shipped from our manufacturing facility via a standard overnight courier service and delivered directly to the patient’s surgeon in as short as four weeks after we receive the patient’s biopsy. Each surgeon implanting our neo-organs will be trained and provided with specific procedures and protocols based on general surgical techniques to implant the neo-organ in the patient.
|
|
|
·
|
Regeneration. Based on data from preclinical and clinical studies, we believe that the neo-organ serves as a template for the body to regenerate native-like organs and tissues. Blood vessels and nerves grow into the implanted neo-organ and the scaffold is gradually absorbed by the body. In preclinical tests, we have observed that the newly grown tissue integrates with its surroundings and becomes substantially indistinguishable over time from the native organ. We have also observed that, in this regenerative process, the body regulates the growth and development of the organ to ensure that it is not under- or over-developed.
|
Our Strategy
Our goal is to become the leading regenerative medicine company focused on the development and commercialization of neo-organs for a variety of diseases and disorders. To achieve this objective, we intend to:
|
|
·
|
Advance the Tengion Neo-Urinary Conduit. We are devoting a significant portion of our resources and business efforts to completing the development of our lead product candidate, the Neo-Urinary Conduit. We have an active IND and are currently conducting a Phase I clinical trial in bladder cancer patients who require removal of their bladders and are in need of a urinary diversion.
|
|
|
·
|
Leverage our Organ Regeneration Platform to develop additional neo-organs. We believe our technology is broadly applicable to other indications including certain types of urologic, renal, gastrointestinal and vascular diseases. We have generated significant proprietary know-how and intellectual property in the development and manufacture of our various product candidates. We plan to continue to apply our technologies to other neo-organs as treatments for other conditions, such as our Neo-Kidney Augment for patients with advanced chronic kidney disease, or CKD.
|
3
|
|
·
|
Become a fully integrated company, developing, manufacturing and commercializing neo-organs. We believe our scalable manufacturing facilities will position us well for the commercialization of our product candidates. We believe a small direct sales force will be sufficient to market to the specialty surgeons for our targeted indications in North America and Europe.
|
|
|
·
|
Selectively pursue strategic partnerships to accelerate and maximize the potential of our product candidates and technology while preserving significant commercial rights. We intend to selectively pursue strategic partnership opportunities that we believe may allow us to accelerate the development or maximize the value of our product candidates and technology by leveraging the scientific, clinical development, manufacturing, commercialization and/or financial strengths of leading biotechnology and pharmaceutical companies while still preserving significant commercial rights.
|
Product Pipeline
Leveraging our Organ Regeneration Platform, we are currently developing the following product candidates with potential applications in urologic, renal, gastrointestinal and vascular diseases:

4
Urologic Product Candidates
We believe that there will be numerous opportunities for our technology across a variety of medical conditions. We are focusing on the development of product candidates to target applications where the current standard of care involves surgical procedures to replace diseased bladder tissue through the use of bowel tissue harvested from the patient. According to the Agency for Healthcare Research and Quality in the United States and similar government data sources in Europe, we estimate there are a total of approximately 28,000 such procedures performed every year in the United States and the European Union. We have developed a portfolio of product candidates in this area, with clinical experience in two product candidates.
We currently have two product candidates for the treatment of patients who require removal of their bladder in connection with the treatment of bladder, abdominal or pelvic cancer, or other severe bladder disease. Our Neo-Urinary Conduit, for which we have an active IND and are currently conducting a Phase I clinical trial, is a combination of bioabsorbable materials and autologous smooth muscle cells that are isolated and expanded ex-vivo by our scientists. We believe our Neo-Urinary Conduit will develop into physiologically functional tissue with a structure, diverting urine from the ureters to an ostomy bag. Similarly, our Neo-Bladder Replacement, for which we believe we have completed all preclinical development necessary to prepare an IND, is a combination of bioabsorbable materials and autologous smooth muscle cells cultured by our scientists that we believe will serve as a functioning bladder, eliminating the need for an ostomy bag, for patients who have their bladders removed due to cancer. We also have a product candidate, our Neo-Bladder Augment, for the treatment of neurogenic bladder, or dysfunctional bladder due to some form of neurologic disease or condition, for which treatment often requires an augmentation of the bladder in order to relieve high bladder pressure and incontinence. While we have conducted two Phase II clinical trials of this product candidate, we currently are not actively developing our Neo-Bladder Augment and are directing the majority of our resources toward the development of our Neo-Urinary Conduit and other development programs in our pipeline.
We believe that our urologic product candidates, which use autologous cells and develop into functional bladder tissue, will avoid many of the complications associated with using bowel tissue in urologic procedures.
5
Market Overview
The table below indicates the number of urologic surgical procedures performed per year in the United States and the European Union for our targeted indications.
|
Estimated Surgical Procedures Per Year
|
United States(1)
|
European Union(2)
|
||
|
Urinary Conduit
|
||||
|
Cancer
|
9,300
|
10,800
|
||
|
Other
|
1,700
|
2,300
|
||
|
Total
|
11,000
|
13,100
|
||
|
Bladder Replacement
|
700
|
900
|
||
|
Bladder Augment
|
||||
|
Spina Bifida
|
380
|
350
|
||
|
Spinal Cord Injury
|
140
|
130
|
||
|
Other
|
680
|
620
|
||
|
Total
|
1,200
|
1,100
|
||
|
Total Surgical Procedures
|
12,900
|
15,100
|
_______________________
Sources:
|
(1)
|
US: Agency for Healthcare Research and Quality.
|
|
(2)
|
EU: Government data sources for the United Kingdom, France and Germany; other EU countries estimated from population census.
|
Bladder Cancer
According to the National Cancer Institute, bladder cancer is the sixth most common form of cancer in the United States. The American Cancer Society estimates there were 71,000 new cases of bladder cancer diagnosed in the United States in 2009, and 14,000 deaths. The European Cancer Observatory estimates that there were over 100,000 new cases of bladder cancer in Europe in 2006. In the United States, there are approximately 10,000 cases per year of bladder cancer requiring bladder removal, according to data compiled from a 2005 National Inpatient Sample, or NIS, performed by the Healthcare Cost and Utilization Project, or HCUP, a family of healthcare databases and related software tools and projects developed through a federal-state-industry partnership and sponsored by the Agency for Healthcare Research and Quality.
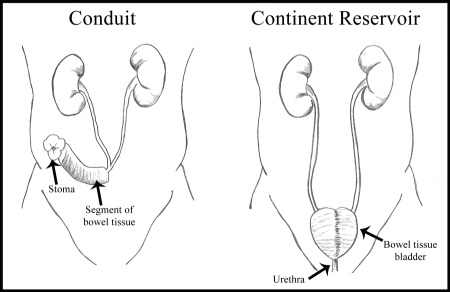
Following removal of a bladder, patients require some form of urinary diversion. Most patients are currently treated by using a segment of bowel tissue to construct a conduit for urine to exit from the body. In its simplest form, the reconstruction involves creating a tubular structure out of bowel tissue and then connecting it to the ureters at one end and the skin at the other in a procedure that was pioneered in the 1930s. Urine output is not controlled and the patient wears a collection device at all times.
The other diversionary option for patients is the creation of a continent reservoir which is most commonly a bladder-shaped pouch fashioned from bowel tissue, to which the ureters and urethra are connected. We believe that less than 10% of the urinary diversions performed after bladder removal are bladder replacements and the remaining diversions are urinary conduits, based upon data compiled from a 2005 NIS performed by HCUP. There are multiple factors that affect the decision concerning which form of post-bladder removal urinary diversion is optimal for a given patient, including stage of disease, health and mental status, manual dexterity, physique and, importantly, patient preference.
6
The graphic below illustrates two urinary diversion options for patients who require bladder removal.
Limitations of Current Therapies
There are many complications associated with surgery to remove or repair the bladder, such as adhesions and obstructions. In the case of traditional surgery to construct a conduit for urine to exit from the body into an ostomy bag, complications such as ureteral detachment, ureteral stricture and hydroureter / hydronephrosis, or swelling in the ureters or kidneys resulting from the backup of urine, are related to the attachment of the ureters to the conduit. Other complications, such as stomal stenosis, involve the attachment of the conduit to the abdominal wall.
While there is variation across procedures and patient types, there are risks and complications common to all procedures that rely on harvesting bowel tissue and placing it in the urinary tract. These complications may include:
|
|
·
|
Bowel complications. Bowel surgery required to harvest tissue for reconstructive use can result in complications, such as prolonged recovery of bowel function, ileus (temporary bowel paralysis), obstructions, leaks and fistulas. Because vitamin B12 is absorbed in the bowel tissue, the loss of tissue can result in anemia and neurologic abnormalities. Additionally, malabsorption of salts and lipids can lead to diarrhea. Patients with neurogenic bladder are prone to bowel movement problems even before surgery and the removal of bowel tissue may either exacerbate existing conditions or create new motility problems. These conditions further contribute to the substantial physical and psychological morbidity in these patients.
|
7
|
|
·
|
Absorption issues. Use of bowel tissue often leads to electrolyte and metabolic imbalances, which can cause bone loss. Certain drugs taken by the patient may be reabsorbed by the implanted bowel tissue, potentially leading to unintended toxic levels. The exposure of intestinal surface to urine also results in the inappropriate absorption of ammonium, chloride and hydrogen ions as well as potassium loss, leading to chronic metabolic imbalances or abnormalities.
|
|
|
·
|
Infection. Persistent and recurrent infections are common in patients with bowel tissue reconstruction. For example, based upon an article entitled “Long Term Outcome of Ideal Conduit Diversion” by S. Madersbacher, et. al., which appeared in the Journal of Urology in 2003, as many as 23% of patients with bowel tissue conduits have recurrent urinary tract infections, or UTIs, and according to “Use of intestinal segments in urinary diversion” by D. M. Dahl and W. S. McDougal in Campbell-Walsh Urology in 2007, approximately 10% to 17% of bowel tissue conduit patients have UTIs that reach the kidney. Bacteria normally found in bowel tissue can serve as a source of infection and septic complications when repositioned into the urinary tract. One of the consequences of persistent infection is the development of stones, hard masses which can cause pain, bleeding, obstruction of urine or infections.
|
|
|
·
|
Mucus. Bowel tissue repositioned in the urinary tract secretes mucus into the urine. Mucus increases the risk of stone formation and the viscosity of urine, and in the case of bladder augments, may require bladder irrigation and more frequent catheterization.
|
|
|
·
|
Cancer. Malignancy, although rare, is a well-recognized complication following enterocystoplasty and other reconstructive surgeries that incorporate bowel segments into the genitourinary tract. This is of greatest concern in the population of spina bifida patients receiving a bladder augment because of the young age of the patients at the time of surgery.
|
Patients requiring some form of urinary diversion with today’s standard of care are at risk of complications associated with the use of bowel tissue as well as those associated with the surgery to harvest the bowel tissue.
Our Solutions
Our Neo-Urinary Conduit and Neo-Bladder Replacement are being developed to address unmet needs in patients who require removal of their bladder in connection with treatment for bladder, abdominal or pelvic cancer or other severe bladder diseases. We believe our product candidates have the potential to be the clinical treatment of choice for the majority of these conditions.
8
The Tengion Neo-Urinary Conduit
Our Neo-Urinary Conduit is a combination of autologous cells and bioabsorbable scaffold that is intended to catalyze regeneration of a native bladder tissue conduit, passively transporting urine from the ureters, through a stoma, or hole in the abdomen, into a standard ostomy bag. We expect that it will be a safe and effective alternative to the creation of a urinary diversion from bowel tissue after bladder removal. Our Neo-Urinary Conduit is intended to avoid complications such as bowel obstruction, urine absorption, infection and mucus secretion associated with the use of bowel tissue in the urinary tract, as well as the potential surgical issues that arise from the procedure involved in harvesting bowel tissue. We produce our Neo-Urinary Conduit using smooth muscle cells from a routine fat biopsy and not cells from the diseased bladder, eliminating the risk of reintroducing cancerous cells from the bladder into the patient.
We produce our Neo-Urinary Conduit for our Phase I clinical trial at our cGMP qualified clinical production facility using our Organ Regeneration Platform. We are able to deliver our Neo-Urinary Conduit in four weeks or less after we receive the patient’s fat biopsy. This timing is consistent with the clinical practice of bladder removal in these patients. To create our Neo-Urinary Conduit, we isolate the smooth muscle cells from the biopsy, expand the cells ex vivo and then seed them onto a bioabsorbable scaffold, which is composed of biomaterials similar to those used in bioabsorbable sutures. Each surgeon implanting one of our Neo-Urinary Conduits will be trained in and receive specific procedures and protocols based on routine surgical techniques to implant our Neo-Urinary Conduit in the patient. We have an active IND and are currently conducting a Phase I clinical trial for the treatment of bladder cancer patients who require bladder removal.
Preclinical History and Background of the Tengion Neo-Urinary Conduit
Our preclinical studies utilized various large animal models for bladder removal and implantation of our product candidate. In these studies, animals receiving our product candidate underwent bladder removal and the animals’ ureters were attached to one end of our Neo-Urinary Conduit using bioabsorbable sutures and the other end was connected to the skin in the abdominal wall, providing an outlet for urine to flow out of the body.
Principal observations of our Neo-Urinary Conduit in preclinical animal models have shown that:
|
|
·
|
Implantation of our Neo-Urinary Conduit created from fat-derived smooth muscle cells results in the formation of a functional conduit.
|
|
|
·
|
By three months post-implantation the scaffold is no longer present and is replaced by a tri-layered native-like bladder tissue consisting of urothelium, submucosa and smooth muscle cells.
|
|
|
·
|
There is no evidence of abnormal cell growth, tissue development or adverse immune response, or adverse systemic effects in response to the biomaterials, autologous cells, or our Neo-Urinary Conduit.
|
9
|
|
·
|
Our Neo-Urinary Conduit regenerates native-like bladder tissue with complete mucosal lining at the ureteral and skin junctions. The function of the regenerated bladder tissue is similar to native bladder tissue in that it allows elimination of urine in a water-tight fashion and does not absorb urine or secrete mucus or electrolytes.
|
These preclinical animal studies suggest that by using current standards of clinical care and specific surgical and post-operative procedures associated with urinary conduits, our Neo-Urinary Conduit may be safe and effective for treating patients who have had their bladders removed.
Clinical Development Plan
We have an active IND for our Neo-Urinary Conduit and commenced a Phase I study in March 2010. The program is designed to provide data to support the use of our Neo-Urinary Conduit in patients who are undergoing bladder removal due to bladder cancer.
This trial is an open-label, single-arm study which is currently expected to enroll up to ten patients. This trial is designed to assess the safety and preliminary efficacy of the Neo-Urinary Conduit. This trial is also intended to allow our investigators to optimize the surgical procedure and post-surgical care with respect to implantation of the Neo-Urinary Conduit. Our clinical investigators may, as necessary, modify the surgical technique based upon the experience gained by prior patients enrolled. A limited number of clinical centers will be utilized to minimize variations in surgical technique and provide the most controlled setting in which the surgical approach is optimized through working with a clinical investigator. We have designed the trial to provide for at least an eight-week interval between implantations of the Neo-Urinary Conduit to allow for close observation and assessment of any treatment and/or procedure-related complications, post-operative recovery, tolerability and safety before proceeding to the next patient. We are working with clinical investigators who have expertise in the surgical treatment of bladder cancer and significant experience performing bladder removals and urinary diversions. This study does not have a control group as the surgical procedure, post-operative care and other clinical parameters preclude the possibility of blinding treatment options.
The primary safety and efficacy assessment of our Neo-Urinary Conduit will be made at 12 months post-implantation and patients will be followed for an additional 48 months in order to assess long term safety and durability. During this first year, however, patients will be seen frequently by the study investigator and/or designated clinical team: every one to two weeks after hospital discharge through week eight, and then at month 3, 6, 9 and 12. Imaging, either CT scan or ultrasound, will be performed at three-month intervals for the first year to examine the neo-organ’s structure, patency and identify any obstructions or other abnormalities. This frequent evaluation and the open-label nature of this study will provide us significant ongoing feedback throughout the study.
The Tengion Neo-Bladder Replacement
Like our Neo-Urinary Conduit, we believe that our Neo-Bladder Replacement will enable patients to avoid the complications associated with removal of bowel tissue and its subsequent use as a bladder replacement. In today’s practice, a continent reconstructed bladder, made from bowel tissue, is attempted in fewer than 10% of patients undergoing bladder removal, or cystectomy. The approach is limited by the various complications associated with using bowel tissue, as well as patient considerations, including the risk of requiring chronic catheterization, and the high frequency of urinary leakage and night time incontinence. Our Neo-Bladder Replacement, when implanted in the body, is intended to serve as a template that recruits other cells to develop a regenerated bladder.
10
Preclinical History and Background of the Tengion Neo-Bladder Replacement
We have conducted preclinical studies of our Neo-Bladder Replacement in large animals to serve as an autologous urinary reservoir. Animals receiving our product candidate underwent urethral-sparing bladder removal. The urethra and ureters were attached using bioabsorbable sutures to our Neo-Bladder Replacement. Principal observations of our Neo-Bladder Replacement in large animal models have shown that:
|
|
·
|
All animals demonstrate continence upon removal of the urethral catheter 21 days after surgery.
|
|
|
·
|
Implantation of our Neo-Bladder Replacement created from only smooth muscle cells results in the formation of a compliant and functional bladder, with demonstrated nerve growth into the tissue.
|
|
|
·
|
The regenerated bladder capacity achieves volumes that were typical of native-bladder volumes, and in instances where there was significant growth of the animal, the regenerated bladder capacity increases to accommodate the animal’s body size.
|
|
|
·
|
By three months post-implantation, the bioabsorbable scaffold is no longer present and is replaced by a tri-layered native-like bladder tissue consisting of urothelium, submucosa and smooth muscle cells.
|
|
|
·
|
By six months post-implantation, the functionality of the neo-bladder tissue is consistent with native bladder tissue as demonstrated by functional imaging, physiological assessments of bladder capacity and compliance, and response to pharmacological stimuli in a manner consistent with native bladder tissue.
|
|
|
·
|
There is no evidence of abnormal cell growth, tissue development, immune response or adverse systemic effects in response to the biomaterials, autologous cells, or our Neo-Bladder Replacement.
|
Based on the results of our preclinical animal studies, we believe that our Neo-Bladder Replacement may provide patients with a functional replacement bladder enabling them to void urine, with no need for a stoma or ostomy bag.
11
The Tengion Neo-Bladder Augment
We have conducted two open-label, multi-center Phase II clinical trials of our Neo-Bladder Augment for the treatment of neurogenic bladder resulting from spina bifida in pediatric patients and neurogenic bladder resulting from spinal cord injury in adult patients. We terminated enrollment of our clinical trial in adult, spinal cord injured patients early as a result of enrollment challenges. Our Neo-Bladder Augment, which is based upon an earlier iteration of our technology requiring a biopsy be obtained from the patient’s bladder, is intended to supplement the patient’s existing bladder and promote regeneration of healthy bladder tissue in patients suffering from neurogenic bladder. Certain of the patients in our Phase II clinical trials experienced serious adverse events that, at the time of their occurrence, were deemed by the respective clinical investigator to be clinically relevant and probably related to our product candidate or the implantation procedure. In response to certain of these serious adverse events, in February 2009, the FDA placed our IND on clinical hold. We conducted a thorough review of the safety events of these patients including the details of each event, the background medical history of these patients, the timing of the events relative to the implantation of our Neo-Bladder Augment and the background rate of similar events in patients receiving the current standard of care, enterocystoplasty. We submitted a complete response to the clinical hold setting forth our analysis in June 2009 and the FDA released the clinical hold in July 2009, with no recommended changes to our protocol, product candidate or implantation procedure. In addition, we have seen limited efficacy of our Neo-Bladder Augment in patients. As of the 36-month clinical follow-up of our pediatric patients, three of the ten patients continue to demonstrate sustained clinical benefit from our product candidate. With respect to adult patients, as of the 24-month clinical follow-up, three of the six patients continue to demonstrate sustained clinical benefit from our product candidate.
As the primary end-point for this trial was one year, we are currently in the long-term follow-up phase of these trials. We currently are not actively developing our Neo-Bladder Augment and are directing the majority of our resources toward the development of our Neo-Urinary Conduit and Neo-Kidney Augment. We may, in the future, determine to terminate the trials involving our Neo-Bladder Augment, so as to further conserve our resources for use on our lead product candidates.
Other Product Opportunities
The Tengion Neo-Kidney Augment
Our Neo-Kidney Augment is being developed to prevent or delay dialysis by increasing renal function in patients with advanced chronic kidney disease, or CKD. Our Neo-Kidney Augment is based on our proprietary technology, which is expected to use the patient’s cells, procured by a needle biopsy of the patient’s kidney, to create an implantable product candidate that can catalyze the regeneration of functional kidney tissue.
Market Overview and Limitations of Current Therapies
According to the 2010 Annual Report of the United States Renal Data System, or the USRDS, which is funded by the National Institutes of Health, or NIH, there are over 30 million people in the United States with chronic kidney disease. Patients with end stage renal disease, or ESRD, have CKD that has progressed to a point of little to no kidney function. These patients require dialysis or a kidney transplant to survive. According to the USRDS, $27 billion in Medicare costs each year are attributable just to ESRD patients. ESRD is associated with an approximate 20% mortality rate per year, with the average life expectancy of a patient on dialysis in 2007 of approximately 5.9 years. In addition, bone loss and anemia is a common complication of advanced CKD, mainly due to an inability of the kidneys to produce enough Vitamin D and erythropoietin, a hormone that controls red blood cell production.
12
Dialysis extends the lives of patients with ESRD, but has many limitations, including infections, hernias and the need to undergo the procedure up to three times per week. Kidney transplantation remains the most desirable and cost-effective form of kidney replacement therapy at this time; however, there is a chronic shortage of organs with only 18,000 kidneys available for transplant in 2008. In addition to the high cost of organ procurement and the subsequent surgery, kidney transplant patients also require a lifetime of drug therapy to prevent organ rejection. There is a clear need for a product that can prevent or delay the need for dialysis or kidney transplant in patients with advanced CKD.
Our Solution
Through our Organ Regeneration Platform, we have isolated and characterized the effects of different kidney cells and combinations of cells on various aspects of kidney function. We have identified a specific combination of renal cells that provides the foundation for the Neo-Kidney Augment and are currently optimizing the formulation to achieve the desired product profile. We obtain cells through a routine needle biopsy of the kidney and using our Organ Regeneration Platform, we are able to produce our Neo-Kidney Augment grown from these cells in as short as four weeks from biopsy receipt. This product development program has demonstrated function in multiple animal models to date and we have also isolated and characterized the necessary cells from healthy and diseased human kidneys, which we believe supports translation of this approach to human patients.
We have assessed the regenerative capabilities of the Neo-Kidney Augment in a rodent model of chronic kidney disease in which renal failure is induced by removal of approximately 80% of the kidney tissue. These studies extend up to six months post-treatment and have demonstrated that animals implanted with our Neo-Kidney Augment have prolonged survival with no additional supportive care. Stabilization and improvement in renal function are evident within several weeks after implantation and continue for the duration of the studies. In these studies, we have seen improvement or stabilization of various biomarkers including certain proteins, lipids, bone remodeling and vitamin levels, as well as organism-level improvements in weight gain and blood pressure.
In late 2009, we began a preclinical study in a rodent model of chronic, progressive renal failure that develops due to obesity, diabetes, and hypertension – three common co-morbid conditions often seen in patients with renal failure. This study demonstrated that our proprietary therapeutic approach, using specific regenerative cells isolated from diseased kidney tissue, can provide significant improvements in several parameters of kidney function, including filtration, urine concentration, and electrolyte balance, as well as a significant reduction in blood pressure. At one year of age, which is approaching end-of-life in this aggressive model, the treated animals demonstrated improved kidney function, delayed disease progression, and better survival compared to the age- and disease-matched untreated control animals.
13
In 2010, we presented additional data using four different animal models of chronic kidney disease. In all four studies, diseased kidney function was improved by the selected regenerative cells which form the active biological component of our Neo-Kidney Augment. In these studies, renal function was shown to be enhanced as early as seven weeks after implantation and was associated with improvements to kidney filtration, urinary protein loss, and urine concentrating ability. Systemic functions controlled by the kidney were also improved including metabolic condition (electrolyte and mineral balance), blood pressure, weight gain and correction of anemia. Improved survival was demonstrated in two animal models which were followed up to one year. In one animal study, selected regenerative cells taken from human kidneys were able to improve kidney function of chronically diseased kidneys.
The Tengion Neo-GI Augment
Leveraging our cumulative learnings from producing tubular neo-organs, such as our Neo-Urinary Conduit, we have begun early development work on our Neo-GI Augment. This product candidate is composed of smooth muscle cells, obtained from a routine fat biopsy, seeded on one of our proprietary bioabsorbable scaffolds, that can be used as a tubular or patch implant to accommodate patient needs. Our objective is to demonstrate that our Neo-GI Augment regenerates esophageal and intestinal tissues.
Our Solution
We have defined in-vitro testing models for our Neo-GI Augment, as well as the necessary cell function assays. Using our combinatorial approach we have also identified potential scaffold prototypes. We have observed extended survival times in animals with esophageal damage following implantation of our Neo-GI Augment. We believe our Neo-GI Augment may offer benefits in other gastrointestinal diseases.
The Tengion Neo-Vessel Replacement
Our Neo-Vessel Replacement targets various blood vessel applications including vascular access grafts, or arterio-venous, or AV, shunts, for patients with ESRD undergoing hemodialysis treatment, and for vessel replacement for patients undergoing coronary or peripheral artery bypass procedures. Our technology will use smooth muscle cells isolated from fat tissue and endothelial cells isolated from blood samples, which are expanded ex vivo and then seeded onto a bioabsorbable scaffold in the shape of a blood vessel.
Our Solution
We believe our Neo-Vessel Replacement may prove to be a more effective alternative to artificial AV grafts. In preclinical studies, our Neo-Vessel Replacement has shown similarities to natural blood vessels and does not become infected to the same extent as synthetic AV graft materials. Further, our Neo-Vessel Replacement may prove to be a superior treatment for patients undergoing a coronary or peripheral artery bypass. Based upon the initial preclinical studies performed to date, we believe this product candidate may eliminate the need to harvest arteries or veins from elsewhere in the patient’s body which will simplify the surgical procedure and avoid potential complications arising from operating on healthy tissue.
14
In large mammal preclinical studies using our Neo-Vessel Replacement as an AV graft, the regenerated vessel has shown similarities to natural blood vessels and reduced rates of infections compared to synthetic AV graft materials. Our Neo-Vessel Replacement has also undergone feasibility testing for the purpose of renal dialysis access. In six-month studies using clinically relevant protocols, we have observed that our Neo-Vessel Replacement retained a native-like ability to remodel, maintained its structural integrity and did not become infected to the same extent as synthetic AV graft materials, despite weekly access with a needle. In large animal studies, our Neo-Vessel Replacements have also been shown to remain open and resist blood clotting when used for carotid artery reconstruction. In addition, we have observed that this product candidate responds to pharmacologic stimuli and expands and contracts in a similar manner to normal blood vessels.
Manufacturing
We believe our product manufacturing capability provides us with a competitive advantage. We manufacture our product candidates in facilities specifically designed for the production of patient-specific materials and have implemented quality control systems to ensure our manufacturing processes and facilities comply with applicable current good manufacturing processes, or cGMP, current Good Tissue Practices, or cGTP, and medical device quality systems regulations, or QSR standards. We have exercised these manufacturing and quality control processes while producing materials for preclinical and clinical studies and have established a significant knowledge base relating to the manufacture of our product candidates. This knowledge base is maintained in a set of standard operating procedures that detail the techniques and standards for manufacturing. All of our production technicians are trained and certified in these procedures to ensure consistency in manufacturing.
Manufacturing of product candidates for our existing preclinical and clinical studies is conducted in our pilot manufacturing facility in Winston-Salem, North Carolina. The pilot manufacturing facility contains approximately 38,400 square feet of laboratories, offices, and clean room manufacturing space. We expect to manufacture early stage product candidates through Phase II clinical trials at this facility. We have also designed and constructed a commercial manufacturing facility, which is located at our headquarters in East Norriton, Pennsylvania. This facility contains approximately 30,000 square feet of technical and manufacturing space and 15,000 square feet of office space. Commencing on March 1, 2011, we also began leasing an additional 35,000 square feet of warehouse space, which ultimately will provide us with space for further expansion. We are currently subleasing this additional warehouse space to a third party. We expect to manufacture product candidates for Phase III clinical trials in this facility and to license this facility for commercial manufacturing of our product candidates. As a result of our business decision to focus our resources on our Neo-Urinary Conduit, activities that were under way to prepare this facility to commence manufacturing operations were temporarily halted in March 2009 and we are currently maintaining this facility in a condition that will enable us to complete these activities and commence manufacturing operations. We anticipate this commercial manufacturing facility can become fully operational following completion of these activities, including all necessary validation, certification and staffing requirements, which we believe can be accomplished in approximately 12 months.
15
Both facilities are designed to segregate the manufacturing and laboratory areas from the administrative portions of the building. The facilities are also designed to prevent cross-contamination of patient specific materials and to provide physical segregation while processing these materials. Our products are manufactured as individual units using disposable processing materials for storage and containment of the product. We do not face many of the traditional challenges of biopharmaceutical companies in maintaining batch quality as manufacturing is scaled up to commercial quantities because our batch size remains consistent at one unit and because the core manufacturing processes remain consistent regardless of production volume. We believe our existing facility infrastructure is sufficient to meet the initial commercial demand for our products and we can increase production capacity by installing additional equipment into existing space and by constructing additional manufacturing space within our existing facilities.
Sales and Marketing
We believe the product candidates we are currently developing will be used primarily by a relatively small number of specialty surgeons at hospitals in North America and the European Union. We intend to explore building the necessary marketing and sales infrastructure necessary to market and sell our current product candidates, if approved by the FDA, as well as hire other personnel to train physicians on the surgical techniques used with our product candidates. We will also explore the possibility of entering into strategic partnerships for the development and marketing our product candidates.
Intellectual Property
We have established a patent position surrounding our product candidates in regenerative medicine. As of December 31, 2010, we owned or had licenses to 22 issued U.S. patents, 20 of which are exclusive and two of which are non-exclusive, 34 U.S. patent applications and over 100 international patents and patent applications. The patent portfolio owned by the Company relates to the composition, design and methods of manufacture for our Neo-Urinary Conduit, Neo-Bladder Replacement and Neo-Bladder Augment product candidates, as well as technological advances associated with our Neo-Vessel Replacement, Neo-GI Augment, and Neo-Kidney Augment development programs. The licensed patent portfolio relates to technology developed by scientists and researchers associated with Childrens’ Medical Center Corporation and Wake Forest University Health Sciences. Also included within our portfolio are patents that address an array of regenerative medicine and tissue engineering technologies and product candidates outside the scope of our current product pipeline. In addition to our patent portfolio, we have developed proprietary information, trade secrets and know-how in the development and manufacture of neo-organs. We are committed to protecting our intellectual property position and to aggressively pursue our patent portfolio as well as the protection of our proprietary information, know-how and trade secrets.
16
Our Patent Portfolio
Our urologic product candidates currently include our Neo-Urinary Conduit, our Neo-Bladder Replacement, and our Neo-Bladder Augment. Our urologic-related patent portfolio is currently composed of 13 issued U.S. patents; 4 granted European patents, which have been validated in 12 European countries; 12 issued patents in other foreign jurisdictions; 6 pending U.S. non-provisional patent applications; 3 pending U.S. provisional patent applications; 2 pending Patent Cooperation Treaty, or PCT, applications; and 8 pending foreign patent applications, all of which relate to a PCT application. We have licensed all of the issued patents. We own 5 of the U.S. non-provisional patent applications, the PCT application, the U.S. provisional patent applications and 5 of the foreign patent applications. We have licensed all of the other pending applications.
The issued U.S. patents, which will expire between 2011 and 2020, contain claims directed to organ constructs, cell-matrix structures, organ scaffolds, and laminarly organized luminal organ or tissue structure constructs; methods of making the same, and methods of treatment using the same. If claims in the pending U.S. patent applications covering urinary conduit constructs, methods for producing the same, and methods of treatment using the same; methods of treatment using laminarly organized luminal organ or tissue structure constructs; and methods for correcting tissue defects in urological structures, are allowed, they would expire between 2014 and 2030. The granted European patents, which will expire between 2018 and 2020, contain claims directed to organ constructs, cell-matrix structures, organ scaffolds, and laminarly organized luminal organ or tissue structure constructs; and methods of making and using the same. The pending PCT, foreign and U.S. provisional applications contain claims directed to urinary conduit constructs, organ constructs, cell-matrix structures, organ scaffolds, and laminarly organized luminal organ or tissue structure constructs; methods of making and using the same; and rational design of regenerative medicine products. These patent applications, if issued, will expire between 2019 and 2030.
Our gastrointestinal (GI) development program is our Neo-GI Augment. Our GI-related patent portfolio is currently composed of 5 pending U.S. provisional patent applications, each of which is owned by us. If claims issue from patent applications filed claiming benefit of the provisional applications, they will expire in 2031.
Our renal development program is our Neo-Kidney Augment. Our renal-related patent portfolio is currently composed of 11 issued U.S. patents; 4 granted European patents, which have been validated in 12 European countries; 8 issued patents in other foreign jurisdictions; 4 pending U.S. non-provisional patent applications; 10 pending U.S. provisional patent applications; 2 pending PCT applications; and 15 pending foreign patent applications, all of which relate to a PCT application. We have licensed all of the issued patents. We own 2 of the U.S. non-provisional patent applications, the PCT application and the U.S. provisional applications. We have licensed all of the other pending applications.
The issued U.S. patents, which will expire between 2011 and 2020, contain claims directed to prosthetic kidneys, organ constructs, cell-matrix structures, organ scaffolds; methods of making the same, and methods of treatment using the same. If claims in the pending U.S. patent applications covering renal cell preparations and augmentation constructs, methods for producing the same, and methods of treatment using the same, are allowed, they would expire between 2019 and 2030. The granted European patents, which will expire between 2017 and 2020, contain claims directed to prosthetic kidneys, organ constructs, cell-matrix structures, organ scaffolds; and methods of making and using the same. The pending PCT, foreign and U.S. provisional applications contain claims directed to renal cell preparations and augmentation constructs, methods for producing and using the same; and rational design of regenerative medicine products. These patent applications, if issued, will expire between 2019 and 2030.
17
Our vascular development program is our Neo-Vessel Replacement. Our vessel-related patent portfolio is currently composed of 2 issued U.S. patents, 1 pending U.S. non-provisional patent application and 1 pending PCT application. We have licensed each of the issued patents. We own each of the pending applications. The issued U.S. patents, which will expire between 2015 and 2016, contain claims directed to cell-matrix constructs and methods of making the same. If claims in our pending U.S. and PCT patent applications covering tissue engineering scaffolds having a mechanical response to stress and strain substantially similar to that of a response by a native blood vessel, and methods of making the same, are allowed, they would expire in 2029.
Confidential Information and Inventions Assignment Agreements
We require our employees, consultants and members of our research and development advisory board to execute confidentiality agreements upon the commencement of employment, consulting or collaborative relationships with us. These agreements provide that all confidential information developed or made known during the course of the relationship with us be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions resulting from work performed for us, utilizing our property or relating to our business and conceived or completed by the individual during employment shall be our exclusive property to the extent permitted by applicable law.
License Agreements and Research Agreements
Exclusive License Agreement with Children’s Medical Center Corporation
In October 2003, we entered into a license agreement with Children’s Medical Center Corporation, or CMCC, for the license of certain patent rights and intellectual property rights owned or controlled by CMCC related to tissue engineering technology. The CMCC patent rights include patent rights owned by CMCC, as well as CMCC’s right to sublicense certain patent rights owned or controlled by Massachusetts Institute of Technology, or MIT. Under the terms of this license agreement, CMCC has granted us exclusive worldwide, sublicensable licenses under certain of CMCC’s patent rights related to implantable matrices for the development and commercialization of tissue engineered products for human and animal therapeutics in the subfields of genitourinary, vascular tissue, nervous tissue, trachea and other subfields later agreed upon by us and CMCC. Our license from CMCC is exclusive in all subfields for patent rights related to organ constructs; cell matrix structures; laminarly organized luminal organ or tissue structure constructs; prosthetic kidneys; kidney augmenting constructs; stents; reproductive organs and tissue structure constructs; as well as methods for making and using the same. Our license to CMCC’s patent rights related to implantable cartilaginous structures, implantable genitourinary cell-seeded matrices, and mammalian urothelial cell preparation is non-exclusive, except in the genitourinary subfield where the license is exclusive. Our license to CMCC’s patent rights relating to organ decellularization is non-exclusive for all subfields covered by the license agreement. Under the license agreement, we were also granted a non-exclusive, non-sublicensable, non-transferable license to certain CMCC biological materials and know-how related to unpatented manufacturing and scientific information, except that such license may be sublicensed or transferred to our affiliates and contractors for purposes specified in the license agreement. CMCC has retained a royalty-free, non-exclusive right to practice, use and license to other academic and nonprofit research organizations to practice and/or use the patent rights and licensed products for research, educational, clinical and/or charitable purposes only.
18
We are required under the license agreement to use reasonably diligent efforts, as defined in the license agreement, to bring one or more licensed products to market as soon as practicable and to obtain all necessary government approvals for the manufacture, use, sale and distribution of licensed products. If we fail to meet the diligence requirements set forth in the license agreement, we may be required under certain circumstances to grant a sublicense to a third party chosen by CMCC relating to the licensed product or subfield for which we have failed to meet our diligence obligations.
Under the license agreement, we are required to make payments to CMCC for accrued and continuing patent prosecution costs and also upon the achievement of certain development and sales milestones as well as certain consideration received from a sublicensee. If we develop and obtain regulatory approval of a licensed product in each of the four subfields, we will be required to pay CMCC development milestones aggregating approximately $6.9 million. Also, if cumulative net sales of all licensed products reach a certain level, we will be required to make a one-time sales milestone payment of $2.0 million. We have previously paid a license issue fee of $175,000 and $350,000 of development milestones to CMCC and as of December 31, 2010, we did not owe any milestone payments to CMCC under the license agreement. A portion of the milestone payments we make will be credited against our future royalty payments. We must pay CMCC royalties in the mid-single digit range based on net sales of licensed products as defined in the agreement by us, our affiliates and our sublicensees, which royalties will be reduced for certain amounts we are required to pay to license patents or intellectual property from third parties and under certain circumstances if there is a competing product. No royalties will be payable with respect to sales of licensed products in countries where there is no valid patent claim, unless we advised CMCC not to file for patent protection in that country and later choose to market and sell licensed products in such country. The license agreement, and our obligation to pay royalties terminates on the later of the expiration, on a country-by-country basis, of the last patent right and October 2018.
Either party may terminate the license agreement upon 90 days prior written notice upon a material, unremedied breach or default of the other party. CMCC may terminate the agreement immediately upon our insolvency or as otherwise provided in the agreement and also upon 45 days prior written notice for our failure to pay royalties due in a timely manner. We may terminate at any time, with or without cause, upon six months prior written notice and payment to CMCC of accrued amounts due and a $50,000 termination fee.
19
Wake Forest University Health Sciences License Agreement
In January 2006 we entered into a license agreement with Wake Forest University Health Sciences, or WFUHS, which was amended in May 2007, for the license of WFUHS’s intellectual property (including know-how) related to research performed by WFUHS employee, Dr. Anthony Atala, a former employee of the Children’s Hospital Boston, an affiliate of CMCC, and the inventor or co-inventor on certain inventions and patents held by CMCC, which are licensed to us under our license agreement with CMCC.
In January 2006, we entered into a research agreement with WFUHS, which was amended in September 2006. Under the research agreement, WFUHS agreed to perform sponsored research in return for quarterly payments. The sponsored research involves the experiments and studies set out in annual research plans and milestones established under the agreement. In September 2010, we extended the agreement for an additional three-year period commencing January 1, 2011. Either party may terminate the agreement upon a material, uncured breach by the other party upon 30 days written notice. We may terminate the agreement at any time, with or without cause, upon 90 days prior written notice.
Under the terms of this amended license agreement, WFUHS has granted us a worldwide, exclusive, sublicensable license to make, use and sell products covered by certain of WFUHS’s patent rights that relate to improvements of the existing inventions and patents included in the patent rights licensed to us under our license agreement with CMCC, or the improvement patents, and to new development inventions and patents arising out of the performance of a separate agreement, the WFUHS Research Agreement, or the new development patents, in the fields of human and animal organs, tissues and tissue-engineered and regenerative medicine directed to their functions in the subfields of genitourinary tissue, kidney tissue, cardiovascular tissue and nervous tissue. This license agreement also granted us a non-exclusive, worldwide, sublicensable license to certain know-how related to unpatented manufacturing and scientific information provided by WFUHS. Our license to patents claiming inventions conceived after the expiration or other termination of the WFUHS Research Agreement may be terminated at any time by WFUHS following expiration or other termination of the WFUHS Research Agreement. Under the terms of the May 2007 amendment to the license agreement, we may elect to provide funding for research activities under the WFUHS Research Agreement directed to products that would be covered by the new development patents. If we elect to provide such funding we are required to provide a minimum of $1 million in annual funding, provided that we may decide within our discretion to reduce the amount of such annual funding by increments of $200,000. If we reduce the annual funding we are required to give up our rights to a license under one subfield for each $200,000 incremental reduction in the annual funding. We reduced our annual funding commitment for 2010 to $800,000 and agreed to give up our rights to a license under the trachea tissue subfield pursuant to this provision.
We are obligated to use commercially reasonable efforts to bring licensed products to market. If WFUHS believes that we have failed to comply with this obligation and an arbitrator agrees with WFUHS, we will be required to enter into a sublicense for the relevant product or grant WFUHS the right to enter into a sublicense for the that product. We are not required to make any milestone payments to WFUHS related to the development or commercialization of the licensed products. In addition, we are required to meet certain time deadlines with respect to initiating the first human clinical trial, filing for marketing approval with the FDA and achieving the first commercial sale in the United States with respect to the first product covered by the new development patents in each subfield. If we fail to meet any of these deadlines, WFUHS may, in its sole option, terminate our license with respect to such product covered by the new development patents.
20
Under the license agreement, we also issued WFUHS a warrant to purchase 3,200 shares of our common stock through January 1, 2016 at an exercise of $2.32 per share and are required to pay WFUHS a percentage of certain consideration received from a sublicensee. We are required to pay certain license maintenance fees with respect to each product covered by new development patents, commencing two years after we initiate the first animal study conducted under cGLP, with respect to such product. These license maintenance fees, which are in the low six figure range with respect to each product, will be creditable against royalties we are obligated to pay to WFUHS for such product. In addition, we are required to pay WFUHS royalties in the low- to mid-single digit range based on net sales of licensed products in all countries. With respect to any product covered by an improvement patent, our obligation to pay royalties to WFUHS terminates upon the later of the expiration, on a product-by-product basis, of the last to expire patent covering such product and the date that is 15 years from the first commercial sale of such product, provided that no such royalty is payable more than seven years after expiration of the last-to-expire patent covering such product.
With respect to any licensed product that is not covered by an improvement patent, our obligation to pay royalties to WFUHS terminates, on a product-by-product basis, on the date that is five years from the first commercial sale of such product, provided that the first commercial sale occurs prior to January 1, 2021 (for each day after January 1, 2021 that the first commercial sale does not occur, the five-year period is reduced by one day).
Pursuant to the terms of the amended license agreement, we have provided $2.8 million of funding for research activities under the WFUHS research agreement through December 31, 2010. We have made no other payments under the license agreement with WFUHS.
The agreement continues in effect on a country-by-country basis until the later of the expiration of the last to expire new improvement patent and January 1, 2021, unless earlier terminated as provided in the agreement. We may terminate the license agreement at any time, with or without cause, upon 90 days prior written notice. WFUHS may terminate the license agreement upon our default in paying a license fee or providing a report required to be provided under the license agreement, our material breach or our making a knowingly false report, upon 45 days prior written notice, provided that we may cure such default or breach within the 45-day period.
21
Medtronic Right of First Refusal and Right of First Negotiation Agreement
On March 1, 2011, we entered into a Right of First Refusal and Right of First Negotiation Agreement with Medtronic, Inc. pursuant to which we granted Medtronic a right of first refusal to the license, sale, assignment, transfer or other disposition by us of any material portion of intellectual property (including patents and trade secrets) or other assets related to our Neo-Kidney Augment program (an NKA transaction) until October 31, 2013. Additionally, from November 1, 2013 through July 1, 2014, Medtronic will have a right of first negotiation with respect to an NKA transaction, with an option to convert that right of first negotiation to a right of first refusal. Medtronic made a $7 million investment in our common stock along side other investors in a private placement consummated on March 4, 2011. In consideration for receiving rights under the Right of First Refusal and Right of First Negotiation Agreement, Medtronic agreed to receive a warrant to purchase 25% fewer shares of common stock that otherwise would have been issued to Medtronic pursuant to the terms of the private placement. In the event of a change in control, and without any obligation on our part, the agreement and all Medtronic’s rights pursuant thereto will automatically terminate in all respects and be of no further force and effect.
Competition
Our industry is subject to rapid and intense technological change. While we do not believe we currently have any commercial competitors who are developing autologous organs and tissues for the target populations that we are addressing, we face, and will continue to face, intense competition from medical device, pharmaceutical, biopharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies who are generally engaged in tissue engineering and regenerative medicine activities or funding, both in the United States and abroad. In addition, there are companies and academic institutions developing drugs, medical devices and surgical techniques to treat many of the medical conditions our product candidates are designed to treat.
Regenerative Medicine
While we are, to our knowledge, the only regenerative medicine company presently developing a range of neo-organs for implant that are designed to regenerate into functional organs and tissues, there are some companies that are researching and developing regenerative cell-based products or therapies, which may seek to address the medical indications that our neo-organ product candidates seek to address. The companies include Cytograft, Tissue Engineering, Inc., Osiris Therapeutics, Inc., Integra Life Sciences, Inc., Pervasis Therapeutics, Inc., LifeCell (a division of Kinetic Concepts, Inc.) and Genzyme. In addition, several large pharmaceutical companies with much greater resources have demonstrated a strategic interest in regenerative medicine, including Sanofi Aventis, Johnson & Johnson, Pfizer and Celgene.
Bladder Treatment
We expect that our Neo-Urinary Conduit and Neo-Bladder Replacement will compete with the current standard of care, which is the use of bowel tissue to create a urinary diversion.
22
Companies that are researching, developing and marketing products to treat bladder dysfunction include Allergan, which is developing Botox for bladder dysfunction; Speywood Biopharma, which is developing Dysport for bladder dysfunction; and Medtronic, which is marketing Interstim for bladder dysfunction. Further, many large pharmaceutical companies market anticholinergenic medications, such as Detrol and Ditropan, which can be used to treat bladder dysfunction.
Many of the companies competing against us have financial and other resources substantially greater than our own. In addition, many of our competitors have significantly greater experience in testing therapeutic products, obtaining FDA and other marketing approvals of products, and marketing and selling those products. Accordingly, our competitors may succeed more rapidly than we will in obtaining FDA approval for products and achieving widespread market acceptance.
We expect to compete based upon, among other things, our intellectual property portfolio, substantial process know-how with respect to scalable manufacturing capabilities and the efficacy and safety profile of our product candidates. Our ability to compete successfully will depend, in part, on our continued ability to attract and retain skilled and experienced scientific, clinical development and executive personnel, to develop viable autologous neo-organs.
Government Regulation
Regulation by governmental authorities in the United States and other countries is a significant factor in the development, manufacture, commercialization and reimbursement of our neo-organs. All of the products we are seeking to develop will require marketing approval, or licensure, by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical and clinical testing, approval and marketing regulations promulgated by the FDA and similar regulatory authorities in other countries. Various governmental statutes and regulations also govern or influence testing, manufacturing, quality control, safety, labeling, packaging, storage and record keeping related to such products and their marketing. State, local and other authorities may also regulate manufacturing facilities used for our neo-organs. The process of obtaining these approvals and the subsequent compliance with appropriate statutes and regulations require the expenditure of substantial time and money, and there can be no guarantee that approvals will be granted.
FDA Approval Process
Our neo-organs will require approval from the FDA and corresponding agencies in other countries before they can be marketed. The FDA regulates human therapeutic products in one of three broad categories: biologics, drugs, or medical devices. Our product candidates that are currently under development are combination products having features of both a biologic and a medical device. The FDA has determined that the primary mode of action for our Neo-Urinary Conduit is cellular and, therefore, they will be regulated as biologics. We currently believe other neo-organs that we develop will also be regulated as biologics and will therefore require approval via a Biologics License Application, or BLA. The FDA regulates biological products under both the Federal Food, Drug, and Cosmetic Act and the Public Health Service Act, and implementing regulations for both statutes. The FDA generally requires the following steps for pre-market approval or licensure of a new biological product:
23
|
|
·
|
preclinical laboratory and animal tests conducted in compliance with FDA’s Good Laboratory Practice, or GLP, requirements and applicable requirements for the humane use of laboratory animals, to assess a product’s biological activity, biocompatibility and to identify potential safety problems and to characterize and document the product’s manufacturing controls and stability;
|
|
|
·
|
submission to the FDA of an IND application, which must become effective before clinical testing in humans can begin;
|
|
|
·
|
development of clinical protocols to establish the safety and efficacy of the product in humans in clinical trials;
|
|
|
·
|
obtaining approval of clinical protocols by institutional review boards, or IRBs, of clinical sites in order to introduce the product into humans in clinical trials;
|
|
|
·
|
adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended indication conducted in compliance with the FDA’s Good Clinical Practice, or GCP, requirements, as well as any additional requirements for the protection of human research subjects and their health information;
|
|
|
·
|
compliance with cGMP and if applicable, cGTP and QSR, regulations and standards concerning quality and the use of human cellular and tissue products;
|
|
|
·
|
submission to the FDA of a BLA, for marketing approval that includes substantive evidence of safety and effectiveness from adequate results of preclinical testing and clinical trials;
|
|
|
·
|
FDA review of the marketing application in order to determine, among other things, whether the product is safe and effective for its intended uses; and
|
|
|
·
|
obtaining FDA approval of the BLA, including inspection and approval of the product manufacturing facility as compliant with cGMP and if applicable, cGTP and QSR, requirements, prior to any commercial sale or shipment of the product.
|
Once a biological product is identified for development, it enters the preclinical testing stage. An IND sponsor must submit the results of the preclinical tests together with manufacturing information, analytical data and any available clinical data or literature to the FDA as part of the IND. The sponsor also will include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during trials due to safety concerns or non-compliance with regulatory requirements.
24
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an IRB must review and approve the plan for any clinical trial before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completion.
Each new clinical protocol and any amendments to the protocol must be submitted under the IND for FDA review, and to the IRBs for approval. Protocols detail, among other things, the objectives of the clinical trial, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety.
Typically, clinical testing involves a three-phase process although the phases may overlap. Generally, Phase I clinical trials are conducted in a small number of healthy volunteers or patients, except in circumstances where clinical testing would be not appropriate in healthy volunteers, and are designed to provide information about product safety. In Phase II, clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine optimal dose, preliminary efficacy and expanded evidence of safety. Phase III clinical trials are generally large-scale, multi-center, comparative trials conducted with patients afflicted with a target disease in order to provide statistically valid proof of efficacy, as well as safety and potency. In some circumstances, the FDA may require Phase IV or post-marketing trials if it feels that additional information needs to be collected about the product after it is on the market, particularly for long term safety follow up. During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. Progress reports detailing the results of clinical trials must be submitted at least annually to the FDA. The FDA may, at its discretion, re-evaluate, alter, suspend, or terminate the testing based upon the data which have been accumulated to that point and its assessment of the risk/benefit ratio to the patient. Monitoring all aspects of clinical trials to minimize risks is a continuing obligation and process. All serious and unexpected adverse events that are deemed to be associated with the product must be reported to the FDA in written IND safety reports.
The results of the preclinical and clinical testing are submitted to the FDA along with descriptions of the manufacturing process. In the case of vaccines, gene and cell therapies and products such as our current product candidates, the results of clinical trials are submitted as a BLA. Under the Pediatric Research Equity Act of 2003, or PREA, which was reauthorized under the FDAAA, a BLA or a supplement to a BLA must contain data to assess the safety and effectiveness of the biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any biological product for an indication for which orphan designation has been granted.
25
The FDA reviews all BLAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept a BLA for filing. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews a BLA to determine, among other things, whether the product is safe, has an acceptable purity profile and is adequately effective, and whether its manufacturing meets standards designed to assure the product’s continued identity, safety, purity, and potency.
The FDA may grant marketing approval, or the FDA may request additional clinical data or deny approval if the FDA determines that the application does not satisfy its marketing approval criteria. FDA review of a BLA typically takes nine months, but may last longer, especially if the FDA asks for more information or clarification of information already provided. The FDA may refer the BLA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. An advisory committee is a panel of experts who provide advice and recommendations when requested by the FDA on matters of importance that come before the agency. The FDA is not bound by the recommendations of the advisory committee, but it generally follows such recommendations.
The process of obtaining marketing approval is lengthy, uncertain, and requires the expenditure of substantial resources. Each BLA must be accompanied by a user fee, pursuant to the requirements of the Prescription Drug User Fee Act, or PDUFA, and its amendments. The FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fee schedule, effective through September 30, 2011, the user fee for an application requiring clinical data, such as a BLA, is $1,542,000. PDUFA also imposes an annual product fee for biologics ($86,250), and an annual establishment fee ($497,200) on facilities used to manufacture prescription biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the drug also includes a non-orphan indication. We have applied for orphan designation for our Neo-Urinary Conduit product candidate.
Before FDA approves a BLA, all facilities and manufacturing techniques used for the manufacture of products must comply with applicable FDA regulations governing cGMP. Among other things, the manufacturer must develop methods for testing the identity, potency and purity of the final product. A local field division of the FDA is responsible for completing the pre-approval inspection and providing recommendation for or against approval. This effort is intended to assure appropriate facility and process design to avoid potentially lengthy delays in product approvals due to inspection deficiencies. Similarly, before approving a BLA, the FDA also may conduct pre-licensing inspections of a company, its contract research organizations and/or its clinical trial sites to ensure that clinical, safety, quality control and other regulated activities are compliant with GCP. To assure such cGMP and GCP compliance, the applicants must incur significant expenditure of time, money and effort in the area of training, record keeping, production, and quality control. Following approval, the manufacture, holding, and distribution of a product continues to require significant resources in order for the sponsor to maintain full compliance in these areas. For human cellular products, cGTP requirements apply. For a biologic/device combination product, the product must be manufactured also in compliance with QSR requirements for the device component.
26
After marketing approval has been obtained, the FDA will require post-marketing safety reporting to monitor the side effects of the product. Further studies may be required to provide additional data on the product’s risks, benefits, and optimal use, and further clinical trials will be required to gain approval for the use of the product as a treatment for clinical indications other than those for which the product was initially tested. Results of post-marketing programs may limit or expand the further marketing of the product. Further, if there are any modifications to the product, including changes in indication, labeling, or a change in the manufacturing process, manufacturing facility, or product composition, a BLA supplement may be required to be submitted to the FDA.
Other FDA Regulatory Requirements
Maintaining substantial compliance with applicable federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Any biological products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including:
|
|
·
|
record-keeping requirements;
|
|
|
·
|
reporting of adverse experiences with the biologic;
|
|
|
·
|
providing the FDA with updated safety and efficacy information;
|
|
|
·
|
reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product;
|
|
|
·
|
reporting on advertisements and promotional labeling; and
|
|
|
·
|
complying with electronic record and signature requirements.
|
In addition, the FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. There are numerous regulations and policies that govern various means for disseminating information to health-care professionals as well as consumers, including to industry sponsored scientific and educational activities, information provided to the media and information provided over the Internet. Prescription biologics may be promoted only for the approved indications and in accordance with the provisions of the approved label. Biological product manufacturers and other entities involved in the manufacturing and distribution of approved biological products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws.
27
The FDA has very broad enforcement authority and the failure to comply with applicable regulatory requirements can result in administrative or judicial sanctions being imposed on us or on the manufacturers and distributors of our approved products, including, without limitation, warning or untitled letters, refusals of government contracts, clinical holds, civil penalties, injunctions, restitution, disgorgement of profits, recall or seizure of products, total or partial suspension of production or distribution, withdrawal of approvals, refusal to approve pending applications, and criminal prosecution resulting in fines and incarceration. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In addition, even after marketing approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
FDA Amendments Act
On September 27, 2007, the President signed into law the Food and Drug Administration Amendments Act of 2007, or the FDAAA. This new legislation grants significant new powers to the FDA, many of which are aimed at assuring the safety of drugs and biologics after approval. In particular, the new law authorizes the FDA to, among other things, require post-approval studies and clinical trials, mandate changes to drug and biologic labeling to reflect new safety information, and require risk evaluation and mitigation strategies for certain drugs and biologics. In addition, the new law significantly expands the federal government’s clinical trial registry and results databank and creates new restrictions on the advertising and promotion of drugs and biologics. Under the FDAAA, companies that violate these and other provisions of the new law are subject to substantial civil monetary penalties.
The requirements and changes imposed by the FDAAA may make it more difficult, and more costly, to obtain and maintain approval for new biologics, or to produce, market and distribute existing products. In addition, the FDA’s regulations, policies and guidance are often revised or reinterpreted by the agency or the courts in ways that may significantly affect our business and our products. It is impossible to predict whether additional legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of such changes, if any, may be.
Follow-on Biologics, or Biosimilars
In the past few years, there have been a number of legislative initiatives that intended to create a regulatory pathway for approval of follow-on biologics, or biosimilars. The forms of the proposed legislation differed in several aspects, including the length and scope of intellectual property protection, and the clinical testing standards. At this time it is not possible to predict whether any of the biosimilar proposals will be enacted and become law, and if so, which version of the proposals will be enacted. However, if a regulatory pathway is established for biosimilars in the future pursuant to the enactment of biosimilar legislation, our business may be adversely affected, although the extent of the impact will depend on the details of the biosimilar law.
28
Expedited Development and Review Programs
A Fast Track product is defined as a new drug or biologic intended for the treatment of a serious or life-threatening condition that demonstrates the potential to address unmet medical needs for this condition. Under the Fast Track program, the sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a Fast Track product at any time during the clinical development of the product. This designation assures access to FDA personnel for consultation throughout the development process and provides an opportunity to request priority review of a marketing application providing a six-month review timeline for the designated product. If a preliminary review of the clinical data suggests that a Fast Track product may be effective, the FDA may initiate review of sections of a marketing application for a Fast Track product before the sponsor completes the application. This rolling review is available if the applicant provides a schedule for submission of remaining information and pays applicable user fees. However, the time periods specified under PDUFA concerning goals to which the FDA has committed to reviewing an application do not begin until the sponsor submits the complete application. Products that receive Fast Track designation may also be eligible for accelerated approval, or approval based on a clinical endpoint other than survival or irrevocable morbidity or on a surrogate endpoint that is reasonably likely to predict clinical benefit.
Orphan Drug Designations
The Orphan Drug Act provides incentives to manufacturers to develop and market drugs and biologics for rare diseases and conditions affecting fewer than 200,000 persons in the United States at the time of application for orphan drug designation, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug or biological product available in the United States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting a new drug application, or NDA, or BLA. After the FDA grants orphan product designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. The first developer to receive FDA marketing approval for an orphan biologic is entitled to a seven year exclusive marketing period in the United States for that product as well as a waiver of the BLA user fee. The exclusivity prevents FDA approval of another application for the same product for the same indication for a period of seven years, except in limited circumstances where there is a change in formulation in the original product and the second product has been proven to be clinically superior to the first.
Legislation similar to the Orphan Drug Act has been enacted in other jurisdictions, including the European Union. The orphan legislation in the European Union is available for therapies addressing conditions that affect five or fewer out of 10,000 persons. The marketing exclusivity period is for ten years, although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product is sufficiently profitable not to justify maintenance of market exclusivity. We have received orphan designation in the EU for our Neo-Bladder Augment for both the treatment of spina bifida and spinal cord injury.
29
Human Cellular and Tissue-Based Products
Human cells or tissue intended for implantation, transplantation, infusion, or transfer into a human recipient is regulated by the FDA as human cells, tissues, and cellular and tissue-based product, or HCT/Ps. Certain types of HCT/Ps are regulated differently from drug or biologic products because they are minimally manipulated tissues intended for homologous use in the patient’s body, are not combined with a drug, device or biologic, and do not have systemic or metabolic effects on the body. The FDA does not require pre-market approval for these types of HCT/Ps, however, it does require strict adherence to federally mandated current Good Tissue Practice, or cGTP, regulations for all HCT/Ps. These regulations are analogous to the cGMP regulations described above in terms of manufacturing standards. In addition, the FDA’s regulations include other requirements to prevent the introduction, transmission and spread of communicable disease. These requirements apply to all HCT/Ps, including those regulated as drugs, biologics or devices. Specifically, the FDA’s regulations require tissue establishments to register and list their HCT/Ps with the FDA and, when applicable, to evaluate donors through screening and testing. We submit an annual registration and listing form to the FDA for our Winston-Salem facility as an HCT/Ps facility.
Privacy Law
Federal and state laws govern our ability to obtain and, in some cases, to use and disclose data we need to conduct research activities. Through the Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (as incorporated in the American Recovery and Reinvestment Act of 2009) Congress required the Department of Health and Human Services to issue a series of regulations establishing standards for the electronic transmission of certain health information. Among these regulations were standards for the privacy of individually identifiable health information. Most health care providers were required to comply with the Privacy Rule as of April 14, 2003.
HIPAA does not preempt, or override, state privacy laws that provide even more protection for individuals’ health information. These laws’ requirements could further complicate our ability to obtain necessary research data from our collaborators. In addition, certain state privacy and genetic testing laws may directly regulate our research activities, affecting the manner in which we use and disclose individuals’ health information, potentially increasing our cost of doing business, and exposing us to liability claims. In addition, patients and research collaborators may have contractual rights that further limit our ability to use and disclose individually identifiable health information. Claims that we have violated individuals’ privacy rights or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
Other Regulations
In addition to privacy law requirements and regulations enforced by the FDA, we also are subject to various local, state and federal laws and regulations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances. These laws include, but are not limited to, the Occupational Safety and Health Act, the Toxic Test Substances Control Act and the Resource Conservation and Recovery Act.
30
Foreign Regulation
We will most likely have to obtain approval for the manufacturing and marketing of each of our products from regulatory authorities in foreign countries prior to the commencement of marketing of the product in those countries. The approval procedure varies among countries, may involve additional preclinical testing and clinical trials, and the time required may differ from that required for FDA approval or licensure. Although there is now a centralized European Union approval mechanism in place, this applies only to certain specific medicinal product categories. In respect of all other medicinal products each European country may impose certain of its own procedures and requirements in addition to those requirements set out in the appropriate legislation, many of which could be time-consuming and expensive.
Employees
As of December 31, 2010, we had 61 employees, including 59 full-time employees, of which 9 were engaged in manufacturing and operations, 36 were engaged in research and development and clinical trials, and 16 were engaged in administration, facilities and finance. We currently employ two medical doctors and 15 individuals with PhDs. All of our employees have entered into non-disclosure agreements with us regarding our intellectual property, trade secrets and other confidential information. None of our employees are represented by a labor union or covered under a collective bargaining agreement, nor have we experienced any work stoppages.
Corporate Website
Our Internet website address is http://www.tengion.com. Our filings on Form 10-K, Form 10-Q, Form 3, Form 4, Form 5, Form 8-K and any and all amendments thereto are available free of charge through this internet website as soon as reasonably practicable after they are filed or furnished to the Securities and Exchange Commission, or the SEC. They are also available through the SEC at http://www.sec.gov/edgar/searchedgar/companysearch.html.
31
Item 1A.Risk Factors
Our business is subject to numerous risks. We caution you that the following important factors, among others, could cause our actual results to differ materially from those expressed in forward-looking statements made by us in filings with the SEC, press releases, communications with investors and oral statements. Any or all of our forward-looking statements in this Annual Report on Form 10-K and in any other public statements we make may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties. Many factors mentioned in the discussion below will be important in determining future results. Consequently, forward-looking statements cannot be guaranteed. Actual future results may differ materially from those anticipated in forward-looking statements. We undertake no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised, however, to consult any further disclosure we make in our reports filed with the SEC or in a press release.
Risks Related to Our Financial Position and Need for Additional Capital
In order to fund our operations, we will need to raise significant amounts of capital. We may not be able to raise additional capital when necessary or on acceptable terms to us, if at all.
Our future capital requirements will depend on many factors, including:
|
|
·
|
the scope and results of our clinical trials, particularly regarding the number of patients required and the required duration of follow-up for our clinical trials in support of our product candidates;
|
|
|
·
|
the time, complexity and costs involved in obtaining marketing approvals for our product candidates, which could take longer and be more costly than obtaining approval for a new conventional drug candidate, given the FDA’s limited experience with clinical trials and marketing approval for products derived from a patient’s own cells;
|
|
|
·
|
the scope and results of our research and preclinical development programs;
|
|
|
·
|
the costs of operating our research and development facility to support our research and early clinical activities;
|
|
|
·
|
the costs of validating, certifying and staffing our manufacturing facility to support later-stage clinical trials and also in anticipation of commercialization activities, if any;
|
|
|
·
|
the costs of maintaining, expanding, protecting and enforcing our intellectual property portfolio, including potential dispute and litigation costs and any associated liabilities and potentially challenging the intellectual property of others;
|
32
|
|
·
|
the costs of entering new markets outside the United States; and
|
|
|
·
|
the level of debt service payments we are obligated to make.
|
As a result of these factors, among others, we will need to seek additional funding prior to our being able to generate positive cash flow from operations. We will need to raise additional funds through collaborative arrangements, public or private sales of debt or equity securities, commercial loan facilities, or some combination thereof. Additional funding may not be available to us on acceptable terms, or at all. If we obtain capital through collaborative arrangements, these arrangements could require us to relinquish some rights to our technologies or product candidates and we may become dependent on third parties. If we raise capital through the sale of equity, or securities convertible into equity, dilution to our then-existing stockholders would result. If we obtain funding through the incurrence of debt, we would likely become subject to covenants restricting our business activities, and holders of debt instruments would have rights and privileges senior to those of our equity investors. In addition, servicing the interest and repayment obligations under our current and future borrowings would divert funds that would otherwise be available to support research and development, clinical or commercialization activities.
If we are unable to obtain adequate financing on a timely basis, we may be required to delay, reduce the scope of or eliminate one or more of our development programs, and reduce our personnel, any of which could have a material adverse effect on our business, financial condition and results of operations.
We have a substantial amount of debt and contractual obligations that expose us to risks that could adversely affect our business, operating results and financial condition.
As of December 31, 2010, we had approximately $8.8 million of outstanding debt, which is secured by liens on substantially all of our assets. In March 2011, we refinanced the outstanding debt owed to one of our lenders. Pursuant to the terms of the refinancing, we simultaneously borrowed $5.0 million from the lender and repaid the then outstanding principal amount of $4.5 million. We are obligated to make interest-only payments through January 2012, followed by 24 monthly payments of principal and interest at an interest rate of 11.75% per annum. Including the impact of the debt refinancing, we expect that the annual principal and interest payments on our outstanding debt will be approximately $4.8 million, $2.8 million, $2.8 million and $0.2 million in 2011, 2012, 2013 and 2014, respectively. The level and nature of our indebtedness could, among other things:
|
|
·
|
make it difficult for us to obtain any necessary financing in the future;
|
|
|
·
|
limit our flexibility in planning for or reacting to changes in our business;
|
|
|
·
|
reduce funds available for use in our operations;
|
|
|
·
|
impair our ability to incur additional debt because of financial and other restrictive covenants or the liens on our assets which secure our current debt;
|
33
|
|
·
|
hinder our ability to raise equity capital because in the event of a liquidation of the business, debt holders receive a priority before equity holders;
|
|
|
·
|
make us more vulnerable in the event of a downturn in our business; or
|
|
|
·
|
place us at a possible competitive disadvantage relative to less leveraged competitors and competitors that have better access to capital resources.
|
Effective March 2011, the lease agreement for our corporate headquarters required us to provide security and restoration deposits totaling $2.2 million to the landlord, an increase from the prior amount of $1.7 million. Until January 2011, we obtained letters of credit from a bank in favor of the landlord to satisfy the obligation. In January 2011, we deposited $1.0 million with the landlord. As of March 2011, an outstanding letter of credit is satisfying the remaining obligation of $1.2 million. The letter of credit is collateralized by an account held at the bank. If the bank determines the collateral to be insufficient, the bank has the right to demand additional collateral. If we fail to provide additional collateral, the bank has the right to withdraw the letter of credit. In that event, the landlord would have the right to require us to deposit cash of up to $1.2 million in an account to satisfy our deposit obligation.
Unless we raise substantial additional capital or generate substantial revenue from a license or strategic partnership involving one of our product candidates, and there can be no assurance that we will be able to do so, we may not be able to:
|
|
·
|
service or repay our debt when it becomes due, in which case our lenders could seek to accelerate payment of all unpaid principal and foreclose on our assets;
|
|
|
·
|
meet our other contractual obligations which includes the requirement for us to collateralize the $1.2 million letter of credit satisfying our restoration deposit obligation to our landlord; or
|
|
|
·
|
continue to execute our current business and product development plans.
|
Any such event would have a material adverse effect on our business, operating results and financial condition.
We have a history of net losses and may not achieve or sustain profitability.
We have incurred losses in each year since our inception and expect to experience losses for the foreseeable future. As of December 31, 2010, we had an accumulated deficit of $211.2 million. We had net losses of $42.4 million, $29.8 million and $25.6 million in the years ended December 31, 2008, 2009 and 2010, respectively. These losses resulted principally from costs incurred in our clinical trials, research and development programs, construction of our research laboratories and commercial manufacturing facility, and from our general and administrative expenses. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity, total assets and working capital.
We expect to continue to incur significant operating expenses and anticipate that our expenses and losses will increase in the foreseeable future as we seek to further develop our product candidates, technology and manufacturing capabilities. Our expenses are expected to relate to the following:
|
|
·
|
continuing research and development efforts, including commencing preclinical GLP studies, relating to our Neo-Kidney Augment;
|
|
|
·
|
conducting our Phase I clinical trial for our Neo-Urinary Conduit for patients with bladder cancer who require removal of their bladder;
|
|
|
·
|
advancing our Neo-Bladder Replacement for bladder cancer patients requiring removal of their bladder and desiring a continent urinary diversion;
|
|
|
·
|
conducting long-term clinical trial follow-up of our Neo-Bladder Augment;
|
34
|
|
·
|
researching and developing our Neo-GI Augment, Neo-Vessel Replacement and other product candidates;
|
|
|
·
|
maintaining, expanding, protecting and enforcing our intellectual property portfolio and assets and potentially challenging the intellectual property of others;
|
|
|
·
|
validating, certifying and staffing our commercial manufacturing facility if and when our product candidates advance into later-stage clinical trials;
|
|
|
·
|
expanding our manufacturing capabilities to support commercialization of our current and future product candidates; and
|
|
|
·
|
servicing and repaying indebtedness.
|
The extent of our future operating losses is highly uncertain, and we may never achieve or sustain profitability. If we are unable to achieve and then maintain profitability, the market value of our common stock will decline.
If we do not successfully develop our product candidates and obtain the necessary marketing approvals to commercialize them, we will not generate sufficient revenues to continue our business operations.
In order to obtain marketing approval of our product candidates so that we can generate revenues once they are commercialized, we must conduct extensive preclinical studies and clinical trials to demonstrate that our product candidates are safe and effective and obtain and maintain approval of our manufacturing facilities. Our early stage product candidates, including our Neo-Urinary Conduit, for which we have commenced a Phase I clinical trial, may fail to perform as we expect. Moreover, our Neo-Urinary Conduit and our other product candidates may ultimately fail to demonstrate the necessary safety and efficacy for marketing approval. Even if results from preclinical studies and early phase clinical trials are positive, there can be no assurances that later stage studies or trials will be successful. We will need to conduct additional research and development, and devote significant additional financial resources and personnel to develop commercially viable products and obtain the necessary marketing approvals, and if we fail to do so successfully, we may cease operations altogether.
Our short operating history may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We are a development-stage company. We commenced operations in July 2003. Our operations to date have been limited to organizing and staffing our company, acquiring and developing our technology, and undertaking preclinical studies and clinical trials of our product candidates. We have not demonstrated an ability to successfully complete large-scale clinical trials, obtain marketing approvals for product registration, manufacture a commercial-scale product or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. As a result, we have a limited operating history.
35
In addition, as a new business, we may encounter unforeseen expenses, difficulties, complications, delays, and other known or unknown factors. If we are successful in obtaining marketing approval for any of our product candidates, we will need to transition from a company with a research focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
If we fail to properly manage our growth, our business could be adversely affected.
We have substantially increased the scale of our operations since our inception in 2003. We anticipate further increasing the scale of our operations as we develop our product candidates. If we are unable to manage our growth effectively, our operations and financial condition could be adversely affected. The management of our growth will depend, among other things, upon our ability to improve our operational, financial and management controls, reporting systems and procedures. Furthermore, we may have to make investments in and hire and train additional personnel for our operations and manufacturing, which would result in additional burdens on our systems and resources and require additional capital expenditures.
Risks Related to the Development of Our Product Candidates
Our clinical trials may not be successful.
We will only obtain marketing approval to commercialize a product candidate if we can demonstrate to the satisfaction of the FDA or the applicable non-United States regulatory authority, in clinical trials, that the product candidate is safe and effective, and otherwise meets the appropriate standards required for approval for a particular indication. Clinical trials are lengthy, complex and extremely expensive processes with uncertain results. A failure of one or more of our clinical trials may occur at any stage of testing.
We are currently conducting a Phase I open-label, single-arm study of our Neo-Urinary Conduit in patients who are undergoing bladder removal due to bladder cancer. We have designed this trial to assess the safety and preliminary efficacy of the Neo-Urinary Conduit. This trial is also intended to allow our clinical investigators to optimize and, as necessary, modify the surgical implantation technique and post-operative care based on the experience gained by prior patients enrolled. As we have limited experience with this product candidate in humans, we may have difficulty optimizing the surgical implantation of the Neo-Urinary Conduit. There can be no assurance that, as part of our Phase I clinical trial, patients will not experience serious adverse events related to our Neo-Urinary Conduit. Additionally, we may determine, based upon this trial, that our Neo-Urinary Conduit demonstrates limited safety and/or efficacy.
36
We are currently in the long-term follow-up phase of two Phase II clinical trials for our Neo-Bladder Augment, involving ten pediatric and six adult patients. During these trials we have seen certain serious adverse events and limited efficacy data beyond the one-year primary endpoint. Specifically, three patients experienced perforation of the bladder that at the time of the occurrence was deemed by the respective clinical investigator to be clinically significant and probably related to our product candidate or implantation procedure. Perforation of the bladder is a known complication in patients undergoing enterocystoplasty, the current standard of care for these patients. A bladder perforation results in urine leakage through the bladder wall. We believe the underlying cause of these bladder perforations varied from patient to patient, and may be related to complications that can occur in this patient population including abscesses, bacterial infections, small bowel obstructions or invasive urologic procedures. As a result of these serious adverse events, the FDA placed our IND related to these clinical trials on hold. We submitted a complete response in June 2009 and the FDA released the clinical hold in July 2009 with no recommended changes to our protocol, product candidate or implantation procedure. In addition to these serious adverse events, we have seen limited efficacy of our Neo-Bladder Augment in patients. As of the 36-month clinical follow-up of our pediatric patients, three of the ten patients continue to demonstrate sustained clinical benefit from our product candidate. With respect to adult patients, as of the 24-month clinical follow-up, three of the six patients continue to demonstrate sustained clinical benefit from our product candidate.
We may find it difficult to enroll patients in our clinical trials.
Our initial product candidates are designed to treat diseases that affect relatively few patients. This could make it difficult for us to enroll the number of patients that may be required for the clinical trials we would be required to conduct in order to obtain marketing approval for our product candidates.
In addition, we may have difficulty finding eligible patients to participate in our clinical trials because our trials may include stringent enrollment criteria. For example, a patient may require a concomitant surgical procedure that would prevent them from receiving our product candidates, may be using alternative therapies, or the extent of a patient’s overall medical condition may render such patient ineligible to participate in our trials. Our Neo-Urinary Conduit has limited experience in humans and patients may not want to enroll in a trial where they are one of the first patients to receive this product candidate. Our inability to enroll a sufficient number of patients for any of our current or future clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether.
Our product development programs are based on novel technologies and are inherently risky.
We are subject to the risks of failure inherent in the development of products based on new technologies. The novel nature of our Organ Regeneration Platform creates significant challenges with respect to product development and optimization, manufacturing, government regulation and approval, third-party reimbursement and market acceptance. For example, the FDA has relatively limited experience with the development and regulation of autologous neo-organs and, therefore, the pathway to marketing approval for our product candidates may accordingly be more complex, lengthy and uncertain than for a more conventional new drug candidate. The FDA may not approve our product candidates or may approve them with certain restrictions that may limit our ability to market our product candidates, and our product candidates may not be successfully commercialized, if at all.
37
We have limited experience in conducting and managing the preclinical development activities and clinical trials necessary to obtain marketing approvals necessary for marketing our product candidates, including approval by the FDA.
Our efforts to develop all of our product candidates are at an early stage. We may be unable to progress our product candidates that are undergoing preclinical testing into clinical trials. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and favorable initial results from a clinical trial do not necessarily predict final results. The indications of use for which we are pursuing development may have clinical effectiveness endpoints that have not previously been reviewed or validated by the FDA, which may complicate or delay our effort to ultimately obtain FDA approval. We cannot guarantee that our clinical trials will ultimately be successful.
We have not obtained marketing approval or commercialized any of our product candidates. We may not successfully design or implement clinical trials required for marketing approval to market our product candidates. We might not be able to demonstrate that our product candidates meet the appropriate standards for marketing approval, particularly as our technology may be the first of its kind to be reviewed by the FDA. If we are not successful in conducting and managing our preclinical development activities or clinical trials or obtaining marketing approvals, we might not be able to commercialize our product candidates, or might be significantly delayed in doing so, which will materially harm our business.
If we are not able to retain qualified management and scientific personnel, we may fail to develop our technologies and product candidates.
Our future success depends to a significant extent on the skills, experience and efforts of the principal members of our scientific and management personnel. These members include Steven Nichtberger, MD, our President and Chief Executive Officer, and Timothy Bertram, DVM, PhD, our Executive Vice President, Science and Technology and Chief Scientific Officer. The loss of either or both of these individuals could harm our business and might significantly delay or prevent the achievement of research, development or business objectives. Competition for personnel is intense. We may find it difficult to retain qualified management and scientific personnel. For example, two of our executive officers voluntarily left our company in 2009 to pursue other opportunities. We may be unable to retain our current personnel or attract or integrate other qualified management and scientific personnel in the future.
We rely on third parties to conduct certain preclinical development activities and our clinical trials, and those third parties may not perform satisfactorily.
We do not conduct in our facilities certain preclinical development activities of our product candidates, such as preclinical studies in animals, nor do we conduct clinical trials for our product candidates ourselves. We rely on, or work in conjunction with, third parties, such as contract research organizations, medical institutions and clinical investigators, to perform these functions. Our reliance on these third parties for preclinical and clinical development studies reduces our control over these activities. We are responsible for ensuring that each of our preclinical development activities and our clinical trials is conducted in accordance with the applicable U.S. federal and state laws and foreign regulations, general investigational plan and protocols. However, other than our contracts with these third parties, we have no direct control over these researchers or contractors, as they are not our employees. Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, or GCP, for conducting, recording and reporting the results of our clinical trials to assure that data and reported results are credible and accurate and that the rights, safety and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Furthermore, these third parties also may have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our preclinical development activities or our clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates. These third parties may be warned, suspended or otherwise sanctioned by the FDA or other government or regulatory authorities for failing to meet the applicable requirements imposed on such third parties. As a result, the third parties may not be able to fulfill their contractual obligations, and the results obtained from the preclinical and clinical research using their services may not be accepted by the FDA to support the marketing approval of our product candidates. If the third parties or their employees become debarred by the FDA, we cannot use the research data derived from their services to support the marketing approval of our product candidates. Finally, these third parties may be bought by other entities, change their business plans or strategies or they may go out of business, thereby preventing them from meeting their contractual obligations to us.
38
We may not be able to secure and maintain relationships with research institutions and clinical investigators that are capable of conducting and have access to necessary patient populations for the conduct our clinical trials.
We rely on research institutions and clinical investigators to conduct our clinical trials. Our reliance upon research institutions, including hospitals and clinics, provides us with less control over the timing and cost of clinical trials and the ability to recruit subjects. If we are unable to reach agreement with suitable research institutions and clinical investigators on acceptable terms, or if any resulting agreement is terminated because, for example, the research institution and/or clinical investigators lose their licenses or permits necessary to conduct our clinical trials, we may be unable to quickly replace the research institution and/or clinical investigator with another qualified research institution and/or clinical investigator on acceptable terms. We may not be able to secure and maintain agreement with suitable research institutions to conduct our clinical trials.
Compliance with governmental regulations regarding the treatment of animals used in research could increase our operating costs, which would adversely affect the commercialization of our technology.
The Animal Welfare Act, or AWA, is the federal law that covers the treatment of certain animals used in research. Currently, the AWA imposes a wide variety of specific regulations that govern the humane handling, care, treatment and transportation of certain animals by producers and users of research animals, most notably relating to personnel, facilities, sanitation, cage size, feeding, watering and shipping conditions. Third parties with whom we contract are subject to registration, inspections and reporting requirements. Furthermore, some states have their own regulations, including general anti-cruelty legislation, which establish certain standards in handling animals. If we or any of our contractors fail to comply with regulations concerning the treatment of animals used in research, we may be subject to fines and penalties and adverse publicity, and our operations could be adversely affected.
39
Public perception of ethical and social issues may limit or discourage the type of research we conduct.
Our clinical trials involve people, and we and third parties with whom we contract also do research involving animals. Governmental authorities could, for public health or other purposes, limit the use of human or animal research or prohibit the practice of our technology. Public attitudes may be influenced by claims that our technology or that regenerative medicine generally is unsafe for use in research or is unethical and akin to cloning. In addition, animal rights activists could protest or make threats against our facilities, which may result in property damage and subsequently delay our research. Ethical and other concerns about our methods, particularly our use of human subjects in clinical trials or the use of animal testing, could adversely affect our market acceptance.
Risks Related to the Manufacturing of Our Product Candidates
We have only limited experience manufacturing our product candidates. We may not be able to manufacture our product candidates in quantities sufficient for our clinical trials and/or any commercial launch of our product candidates.
We may encounter difficulties in the production of our product candidates. Construction of neo-organs from autologous live human cells involves strict adherence to complex manufacturing and storage protocols and procedures. Early stage clinical manufacturing is conducted in our pilot facility and, while we have supported clinical manufacturing from this location, future difficulties may arise which limit our production capability and delay progress in our clinical trials. We have built a commercial-scale manufacturing facility, which is based on our pilot manufacturing facility and processes used in that facility, which we believe will support late phase clinical trials and the initial commercial launch of our product candidates. As a result of our business decision to focus our resources on our Neo-Urinary Conduit, activities that were under way to prepare this facility to commence manufacturing operations were temporarily halted in March 2009 and we are currently maintaining the facility in a condition that will enable us to complete these activities and commence manufacturing operations. We expect completion of these activities, including recertification and hiring additional staff, will cost approximately $2.0 million. Any unexpected difficulty in completing these activities would increase cost or delay late-stage clinical or commercial manufacturing.
We may encounter technical or logistical difficulties as we seek to commence, and subsequently increase, production at our commercial-scale manufacturing facility. These difficulties could increase our costs or cause delays in the production of our product candidates necessary for any Phase III clinical trial and/or any anticipated commercial launch of our product candidates, any of which could damage our reputation and harm our business. Moreover, we have limited experience in manufacturing our product candidates for human patients and we are not aware of any party that manufactures products similar to ours. As a result, if we are not able to successfully manufacture our product candidates, we may be unable to utilize the manufacturing services of a third-party manufacturer.
40
The current manufacture of our product candidates involves the use of regulated animal tissues, and future product candidates may also use animal-sourced materials.
We currently utilize several bovine-derived products, such as growth media, in the manufacture of our Neo-Urinary Conduit and Neo-Bladder Augment. Bovine-sourced materials are strictly regulated in the United States and other jurisdictions due to their capacity to transmit the prior disease Bovine Spongiform Encephalopathy, or BSE, which manifests itself in humans as Creutzfeldt-Jakob Disease. Although we obtain our supply of bovine-based materials from closed herds in jurisdictions that are not currently known to carry BSE, there can be no assurance that these herds will remain BSE-free or that a future outbreak or presence of other unintended and potentially hazardous agents would not adversely affect our product candidates or patients that may receive them. Further, our future product candidates may involve the use of bovine-sourced or other animal-based materials, which could increase the risk of transmission of other diseases carried by such animals.
If a natural or man-made disaster strikes our manufacturing facility, we would be unable to manufacture our product candidates for a substantial amount of time, which would harm our business.
Our manufacturing facility and manufacturing equipment would be difficult to replace and could require substantial replacement lead-time and additional funds if we lost use of either the facility or equipment. Our facility may be affected by natural disasters, such as floods. We do not currently have commercial-scale back-up capacity, so in the event our facility or equipment was affected by man-made or natural disasters, we would be unable to manufacture any of our product candidates until such time as our facility could be repaired or rebuilt. Although, currently we maintain global property insurance with property limits of $27.1 million and business interruption insurance coverage of $5.4 million for damage to our property and the disruption of our business from fire and other casualties, such insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
Our business involves the use of hazardous materials that could expose us to environmental and other liability.
Our research and development processes and our operations involve the controlled storage, use and disposal of hazardous materials including, but not limited to, biological hazardous materials. We are subject to federal, state and local regulations governing the use, manufacture, storage, handling and disposal of these materials and waste products. Although we believe that our safety procedures for handling and disposing of these hazardous materials comply with the standards prescribed by law and regulation, the risk of accidental contamination or injury from hazardous materials cannot be completely eliminated. In the event of an accident, we could be held liable for any damages that result, and any liability could exceed the limits or fall outside the coverage of our insurance. Moreover, we may not be able to maintain insurance to cover these risks on acceptable terms, or at all. We could also be required to incur significant costs to comply with current or future laws and regulations relating to hazardous materials. We currently maintain insurance coverage that is consistent with similar companies in our stage of development. In addition to global property insurance, we maintain general liability insurance coverage of $2.0 million with an excess liability insurance of $4.0 million, and workers’ compensation coverage of $0.5 million per incident. Such insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
41
Risks Related to Marketing Approval and Other Government Regulations
We cannot market and sell our product candidates in the United States or in other countries if we fail to obtain the necessary marketing approvals or licensure.
We cannot sell our product candidates until regulatory agencies grant marketing approval, or licensure. The process of obtaining such marketing approval is lengthy, expensive and uncertain. It is likely to take many years to obtain the required marketing approvals for our product candidates or we may never gain the necessary approvals. Any difficulties that we encounter in obtaining marketing approval may have a substantial adverse impact on our operations and cause our stock price to decline significantly. Any adverse events in our clinical trials for one of our product candidates could negatively impact the clinical trials and approval process for our other product candidates.
To obtain marketing approvals in the United States for our product candidates, we must, among other requirements, complete carefully controlled and well-designed clinical trials sufficient to demonstrate to the FDA that the product candidate is safe and effective for each indication for which we seek approval. Several factors could prevent completion or cause significant delay of these trials, including an inability to enroll the required number of patients or failure to obtain FDA approval to commence a clinical trial. Negative or inconclusive results from, or adverse events during, a preclinical safety study or clinical trial could cause the preclinical study or clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are successful. The FDA can place a clinical trial on hold if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury. If safety concerns develop, we, an Institutional Review Board, or IRB, or the FDA could stop our trials before completion. The populations for which we are developing our product candidates may have other medical complications that would affect their experience in our trials and would affect their experience with our product candidates, if approved. A serious adverse event is an event that results in significant medical consequences, such as hospitalization or prolonged hospitalization, disability or death, and if unexpected must be reported to the FDA. We cannot guarantee that other safety concerns regarding our product candidates will not develop. For example, in February 2009, the FDA placed our IND for our Neo-Bladder Augment on clinical hold following certain serious adverse events that occurred with respect to patients in our Phase II clinical trials. We submitted a complete response in June 2009 and the FDA released the clinical hold in July 2009, with no recommended changes to our protocol, product candidate or implantation procedure.
42
The pathway to marketing approval for our product candidates may be more complex and lengthy than for approval of a conventional new drug or biologic. Similarly, to obtain approval to market our product candidates outside of the United States, we will need to submit clinical data concerning our product candidates and receive marketing approval from governmental agencies, which in certain countries includes approval of the price we intend to charge for our product. We may encounter delays or rejections if changes occur in regulatory agency policies, or if reports from preclinical and clinical testing on similar technology or products raise safety and/or efficacy concerns, during the period in which we develop a product candidate or during the period required for review of any application for marketing approval. If we are not able to obtain marketing approvals for use of our product candidates under development, we will not be able to commercialize such products and, therefore, may not be able to generate sufficient revenues to support our business.
The FDA may impose requirements on our clinical trials that are difficult to comply with, which could harm our business.
The requirements the FDA may impose on clinical trials for our product candidates are uncertain. As a result, we cannot guarantee that we will be able to comply with such requirements. For example, the FDA may require endpoints in our late-stage clinical trials that are different from or in addition to the endpoints in our early-stage clinical trials or the endpoints which we may propose. The endpoints or other study elements, including sample size, the FDA requires may make it less likely that our Phase III clinical trials are successful or may delay completion of the trials. If we are unable to comply with the FDA’s requirements, we will not be able to get approval for our product candidates and our business will suffer.
If we are not able to conduct our clinical trials properly and on schedule, marketing approval by the FDA and other regulatory authorities may be delayed or denied.
Our clinical trials may be delayed or terminated for many reasons, including, but not limited to, if:
|
|
·
|
the FDA does not grant permission to proceed or places the trial on clinical hold;
|
|
|
·
|
subjects do not enroll or remain in our trials at the rate we expect;
|
|
|
·
|
we fail to manufacture necessary amounts of product candidate;
|
|
|
·
|
either of our manufacturing facilities is ordered by the FDA or other government or regulatory authorities to temporarily or permanently shut down due to violations of current Good Manufacturing Practice, or cGMP, or other applicable requirements, or infections or cross-contaminations of product candidates in the manufacturing process;
|
|
|
·
|
subjects choose an alternative treatment for the indications for which we are developing our product candidates, or participate in competing clinical trials;
|
|
|
·
|
subjects experience an unacceptable rate or severity of adverse side effects;
|
43
|
|
·
|
reports from preclinical or clinical testing on similar technologies and products raise safety and/or efficacy concerns;
|
|
|
·
|
third-party clinical investigators lose their license or permits necessary to perform our clinical trials, do not perform our clinical trials on our anticipated schedule or consistent with the clinical trial protocol, Good Clinical Practice and regulatory requirements, or other third parties do not perform data collection and analysis in a timely or accurate manner;
|
|
|
·
|
inspections of clinical trial sites by the FDA or IRBs find regulatory violations that require us to undertake corrective action, suspend or terminate one or more sites, or prohibit us from using some or all of the data in support of our marketing applications;
|
|
|
·
|
third-party contractors become debarred or suspended or otherwise penalized by FDA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or any of the data produced by such contractors in support of our marketing applications; or
|
|
|
·
|
one or more IRBs refuses to approve, suspends or terminates the trial at an investigational site, precludes enrollment of additional subjects, or withdraws its approval of the trial.
|
If we are unable to conduct our clinical trials properly and on schedule, the FDA may delay or deny marketing approval.
Final marketing approval of our product candidates by the FDA or other regulatory authorities for commercial use may be delayed, limited, or denied, any of which would adversely affect our ability to generate operating revenues.
Any of the following factors, if one or more were to occur, could cause final marketing approval for our product candidates to be delayed, limited or denied:
|
|
·
|
our product candidates could fail to demonstrate safety and efficacy in preclinical or clinical testing;
|
|
|
·
|
the manufacturing processes for our product candidates could fail to consistently demonstrate their safety and purity;
|
|
|
·
|
the FDA could disagree with the clinical endpoints we propose for our clinical trials and refuse to allow us to conduct clinical trials utilizing clinical endpoints we believe are appropriate;
|
|
|
·
|
it could take many years to complete the testing of our product candidates, and failure can occur at any stage of the process;
|
44
|
|
·
|
negative results or adverse side effects during a clinical trial could cause us to delay or terminate development efforts for a product candidate;
|
|
|
·
|
the FDA could seek the advice of an Advisory Committee of physician and patient representatives that may view the risks of our product candidates as outweighing the benefits;
|
|
|
·
|
the FDA could require us to expand the size and scope of the clinical trials; or
|
|
|
·
|
the FDA could impose post-marketing restrictions.
|
Our development costs will increase if we have material delays in our clinical trials, or if we are required to modify, suspend, terminate or repeat a clinical trial. If marketing approval for our product candidates is delayed, limited or denied, our ability to market products, and our ability to generate product sales, would be adversely affected.
Any product for which we obtain marketing approval could be subject to restrictions or withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and comparable regulatory authorities, including through periodic inspections. These requirements include, but are not limited to, submissions of safety and other post-marketing information and reports, registration requirements, cGMP and Quality System Regulation, or QSR, requirements relating to quality control, quality assurance and corresponding maintenance of records and documents. Even if marketing approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to other conditions of approval, or may contain requirements for costly and time consuming post-marketing testing and surveillance to monitor the safety or efficacy of the product. Discovery after approval of previously unknown problems with our product candidates or manufacturing processes, or failure to comply with regulatory requirements, may result in actions such as:
|
|
·
|
restrictions on such products’ manufacturers or manufacturing processes;
|
|
|
·
|
restrictions on the marketing or distribution of a product, including refusals to permit the import or export of products;
|
|
|
·
|
warning letters or untitled letters;
|
|
|
·
|
warning labels on the products;
|
|
|
·
|
withdrawal of the products from the market;
|
45
|
|
·
|
refusal to approve pending applications or supplements to approved applications that we submit;
|
|
|
·
|
suspension of any ongoing clinical trials;
|
|
|
·
|
recall of products;
|
|
|
·
|
fines, restitution or disgorgement of profits or revenue;
|
|
|
·
|
suspension or withdrawal of marketing approvals;
|
|
|
·
|
product seizure;
|
|
|
·
|
injunctions; or
|
|
|
·
|
imposition of civil or criminal penalties.
|
In addition, if any of our product candidates are approved, our product labeling, advertising and promotion would be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions.
Current or future legislation may make it more difficult and costly for us to obtain marketing approval of our product candidates.
In 2007, the Food and Drug Administration Amendments Act of 2007, or the FDAAA, became law. This legislation grants significant new powers to the FDA, many of which are aimed at assuring the safety of drugs and biologics after approval. For example, FDAAA granted the FDA new authority to impose post-approval clinical study requirements, require safety-related changes to product labeling and require the adoption of risk management plans, referred to as risk evaluation and mitigation strategies, or REMS. The REMS may include requirements for special labeling or medication guides for patients, special communication plans to health care professionals, and restrictions on distribution and use. Pursuant to FDAAA, if the FDA makes the requisite findings, it might require that a new product be used only by physicians with specified specialized training, only in specified designated health care settings, or only in conjunction with special patient testing and monitoring. The legislation also included requirements for disclosing clinical study results to the public through a clinical study registry, and renewed requirements for conducting clinical studies to generate information on the use of products in pediatric patients. Under the FDAAA, companies that violate the new law are subject to substantial civil monetary penalties. The requirements and changes imposed by the FDAAA may make it more difficult, and more costly, to obtain and maintain approval of new biological products.
46
In addition, the FDA’s regulations, policies or guidance may change and new or additional statutes or government regulations may be enacted that could prevent or delay marketing approval of our product candidates or further restrict or regulate post-approval activities. For example, proposals have been made to further expand post-approval requirements and restrict sales and promotional activities. It is impossible to predict whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes or the marketing approvals of our product candidates, if any, may be.
Risks Related to the Commercialization of Our Product Candidates
If we fail to educate and train physicians as to the distinctive characteristics, benefits, safety, clinical efficacy and cost-effectiveness of our product candidates, our sales will not grow.
Acceptance of our product candidates depends, in large part, on our ability to train physicians in the proper implantation of our neo-organs, which will require significant expenditure of our resources. Convincing physicians to dedicate the time and energy necessary to properly train to use new products and techniques is challenging, and we may not be successful in these efforts. If physicians are not properly trained, they may ineffectively implant our product candidates. Such misuse or ineffective implantation may result in unsatisfactory patient outcomes, patient injury, negative publicity or lawsuits against us. Accordingly, even if our product candidates are superior to alternative treatments, our success will depend on our ability to gain and maintain market acceptance for our product candidates. If we fail to do so, our sales will not grow and our business, financial condition and results of operations will be adversely affected. We may not have adequate resources to effectively educate the medical community and/or our efforts may not be successful due to physician resistance or perceptions regarding our product candidates.
We face uncertainty related to pricing, reimbursement and health care reform, which could reduce our revenue.
Sales of our product candidates, if approved for commercialization, will depend in part on the availability of coverage and reimbursement from third-party payors such as government insurance programs, including Medicare and Medicaid, private health insurers, health maintenance organizations and other health care related organizations. If our product candidates are approved for commercialization, pricing and reimbursement may be uncertain. Both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation affecting coverage and reimbursement policies, which are designed to contain or reduce the cost of health care. Further federal and state proposals and health care reforms are likely which could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. There may be future changes that result in reductions in current coverage and reimbursement levels for our products, if commercialized, and we cannot predict the scope of any future changes or the impact that those changes would have on our operations.
Adoption of our product candidates by the medical community may be limited if doctors and hospitals do not receive full reimbursement for our products, if commercialized. Cost control initiatives may decrease coverage and payment levels for our product candidates and, in turn, the price that we will be able to charge for any product. We are impacted by efforts by public and private third-party payors to control costs. We are unable to predict all changes to the coverage or reimbursement methodologies that will be applied by private or government payors to our product candidates. Any denial of private or government payor coverage or inadequate reimbursement for procedures performed using our products, if commercialized, could harm our business and reduce our revenue.
47
We could be adversely affected if healthcare reform measures substantially change the market for medical care or healthcare coverage in the United States.
The U.S. Congress recently adopted legislation regarding health insurance, which has been signed into law. As a result of this new legislation, substantial changes could be made to the current system for paying for healthcare in the United States, including changes made in order to extend medical benefits to those who currently lack insurance coverage. Extending coverage to a large population could substantially change the structure of the health insurance system and the methodology for reimbursing medical services, drugs and devices. These structural changes could entail modifications to the existing system of private payors and government programs, such as Medicare, Medicaid and State Children’s Health Insurance Program, creation of a government-sponsored healthcare insurance source, or some combination of both, as well as other changes. Restructuring the coverage of medical care in the United States could impact the reimbursement for prescribed drugs, biopharmaceuticals, medical devices, or our product candidates. If reimbursement for our approved product candidates, if any, is substantially less that we expect in the future, or rebate obligations associated with them are substantially increased, our business could be materially and adversely impacted.
Extending medical benefits to those who currently lack coverage will likely result in substantial cost to the U.S. federal government, which may force significant changes to the healthcare system in the United States. Much of the funding for expanded healthcare coverage may be sought through cost savings. While some of these savings may come from realizing greater efficiencies in delivering care, improving the effectiveness of preventive care and enhancing the overall quality of care, much of the cost savings may come from reducing the cost of care. Cost of care could be reduced by decreasing the level of reimbursement for medical services or products, or by restricting coverage and, thereby, utilization of medical services or products. In either case, a reduction in the utilization of, or reimbursement for, any product for which we receive marketing approval in the future could have a materially adverse impact on our financial performance.
There is substantial uncertainty regarding the exact meaning and interpretation of the provisions of healthcare reform that have been enacted. This uncertainty limits our ability to forecast changes that may occur in the future and to manage our business accordingly.
48
We may face competition from companies and institutions that may develop products that make ours less attractive or obsolete through both new technologies that may be similar to ours, or more traditional pharmaceutical or medical device treatments.
Many of our competitors have greater resources or capabilities than we have, or may already have or succeed in developing better products or in developing products more quickly than we do, and we may not compete successfully with them.
The medical device, pharmaceutical and biotechnology industries are highly competitive. We compete for funding. If our product candidates become available for commercial sale, we will compete in the marketplace. For funding, we compete primarily with other companies which, like us, are focused on discovering and developing novel products or therapies for the treatment of human disease based on regenerative medicine technologies or other novel scientific principles. In the marketplace, we may eventually compete with other companies and organizations that are marketing or developing therapies for our targeted disease indications, based on traditional pharmaceutical, medical device or other, non-cellular therapies and technologies.
We also face competition in the cell therapy field from academic institutions and governmental agencies. Many of our current and potential competitors have greater financial and human resources than we have, including more experience in research and development and more established sales, marketing and distribution capabilities.
We anticipate that competition in our industry will increase. In addition, the health care industry is characterized by rapid technological change, resulting in new product introductions and other technological advancements. Our competitors may develop and market products that render our current product or any future product non-competitive or otherwise obsolete.
The use of our product candidates in human subjects may expose us to product liability claims, and we may not be able to obtain adequate insurance.
We face an inherent risk of product liability claims. Our clinical-stage product candidates are in early development and have not been used over an extended period of time in a large number of patients and, therefore, our long-term safety and efficacy data are limited. Patients have experienced in the past and may experience in the future serious adverse events. Our current product liability coverage is $5.0 million per occurrence and in the aggregate. We will need to increase our insurance coverage if and when we begin commercializing any of our product candidates. We may not be able to obtain or maintain product liability insurance on acceptable terms with adequate coverage. If claims against us substantially exceed our coverage, then our business could be adversely impacted. Regardless of whether we are ultimately successful in any product liability litigation, such litigation could consume substantial amounts of our financial and managerial resources and could result in among others:
|
|
·
|
significant awards against us;
|
|
|
·
|
substantial litigation costs;
|
49
|
|
·
|
injury to our reputation and the reputation of our product candidates;
|
|
|
·
|
withdrawal of clinical trial participants; and
|
|
|
·
|
adverse regulatory action.
|
Any of these results would substantially harm our business.
We have never marketed a product before, and if we are unable to establish an effective focused sales force and marketing infrastructure, we will not be able to commercialize our product candidates successfully.
We intend to explore building the necessary marketing and sales infrastructure to market and sell our current product candidates, if they receive marketing approval. We currently do not have internal sales, distribution and marketing capabilities. The development of a sales and marketing infrastructure for our domestic operations will require substantial resources, will be expensive and time consuming and could negatively impact our commercialization efforts, including delay of any product launch. These costs may be incurred in advance of any approval of our product candidates. In addition, we may not be able to hire a focused sales force in the United States that is sufficient in size or has adequate expertise in the medical markets that we intend to target, including surgery. If we are unable to establish our focused sales force and marketing capability for our product candidates, we may not be able to generate any product revenue, may generate increased expenses and may never become profitable.
If we are unable to establish development or marketing collaborations with third parties, we may not be able to develop, commercialize or distribute our products successfully.
We may need to establish development or marketing collaborations with third parties in order to complete development of our product candidates or for the commercialization or distribution of our product candidates. We expect to face competition in our efforts to identify appropriate collaborators or partners to help develop or commercialize our product candidates in our target commercial areas. If we are unable to establish adequate collaborations, our ability to develop or market our product candidates could be adversely affected. Further, to the extent third parties with whom we collaborate fail to perform, our ability to achieve our development or marketing goals may be adversely affected, and our business could suffer.
Risks Related to Intellectual Property
If we are unable to obtain and maintain protection for our intellectual property, the value of our technology and products will be adversely affected.
Our success will depend in large part on our ability to obtain and maintain protection in the United States and other countries for the intellectual property covering or incorporated into our technology and products. The patent situation in the field of biotechnology and pharmaceuticals generally is highly uncertain and involves complex legal, technical, scientific and factual questions. We may not be able to obtain additional issued patents relating to our technology or products. Even if issued, patents issued to us or our licensors may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, or determined not to cover our product candidates or our competitors’ products, which could limit our ability to stop competitors from marketing identical or similar products or reduce the term of patent protection we may have for our product candidates. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
50
|
|
·
|
we or our licensors were the first to make the inventions covered by each of our pending patent applications;
|
|
|
·
|
we or our licensors were the first to file patent applications for these inventions;
|
|
|
·
|
others will not independently develop similar or alternative technologies or duplicate any of our technologies;
|
|
|
·
|
any patents issued to us or our licensors will provide a basis for commercially viable products, will provide us with any competitive advantages or will not be successfully challenged by third parties;
|
|
|
·
|
we will continue developing additional proprietary technologies that are patentable;
|
|
|
·
|
we will file patent applications for new proprietary technologies promptly or at all;
|
|
|
·
|
our patents will not expire prior to or shortly after commencing commercialization of a product;
|
|
|
·
|
our licensors will enforce the rights of the patents we license; or
|
|
|
·
|
the patents of others will not have a negative effect on our ability to do business.
|
In addition, we cannot guarantee that any of our pending patent applications will result in issued patents. If patents are not issued in respect of our pending patent applications, we may not be able to stop competitors from marketing products similar to ours.
Our patents also may not afford us protection against competitors with identical or similar technology.
Because patent applications in the United States and many other jurisdictions are typically not published until 18 months after filing, or in some cases not at all, and because publications of discoveries in the scientific literature often lag behind the actual discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. If a third party has also filed a United States patent application covering our product candidates or a similar invention, we may have to participate in an adversarial proceeding, known as an interference, declared by the United States Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial and it is possible that our efforts could be unsuccessful, resulting in a loss of our United States patent position. If a third party believes we or our licensor were not entitled to the grant of one or more patents, such third party may challenge such patents in an interference or re-examination proceeding in the United States, or opposition or similar proceeding in another country.
51
If we fail to comply with our obligations in our intellectual property licenses with third parties, we could lose license rights that are important to our business.
We are a party to license agreements with Children’s Hospital Boston and Wake Forest University Health Sciences pursuant to which we license the key intellectual property relating to our product candidates. We may enter into additional licenses in the future. Our existing licenses impose, and we expect that future licenses will impose, various diligence, milestone payment, royalty, insurance and other obligations on us. If we fail to comply with these obligations, the licensor may have the right to terminate the license, in which event we might not be able to market any product that is covered by the licensed patents.
If we are unable to protect the confidentiality of our proprietary information, trade secrets and know-how, the value of our technology and product candidates could be adversely affected.
Our proprietary information, trade secrets and know-how are important components of our intellectual property, particularly in connection with the manufacturing of our product candidates. We seek to protect our proprietary information, trade secrets, know-how and confidential information, in part, by confidentiality agreements with our employees, corporate partners, outside scientific collaborators, sponsored researchers, consultants and other advisors. We also have confidentiality and invention or patent assignment agreements with our employees and our consultants. If our employees or consultants breach these agreements, we may not have adequate remedies for any of these breaches. In addition, our proprietary information, trade secrets and know-how may otherwise become known to or be independently developed by others. Enforcing a claim that a party illegally obtained and is using our proprietary information, trade secrets and know-how is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Costly and time consuming litigation could be necessary to seek to enforce and determine the scope of our proprietary information, trade secrets and know-how, and failure to obtain or maintain protection of proprietary information, trade secret and know-how could adversely affect our competitive business position.
If we infringe or are alleged to infringe the intellectual property rights of third parties, our business could suffer.
Our research, development and commercialization activities, as well as any product candidates or products resulting from these activities, may infringe or be accused of infringing one or more claims of an issued patent or may fall within the scope of one or more claims in a published patent application that may subsequently issue and to which we do not hold a license or other rights. Third parties may own or control these patents or patent applications in the United States and abroad. These third parties could bring claims against us that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us, we could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit. No assurance can be given that patents do not exist, have not been filed, or could not be filed or issued, which contain claims covering our product candidates, technology or methods.
52
In order to avoid or settle potential claims with respect to any of the patent rights described above or any other patent rights of third parties, we may choose or be required to seek a license from a third party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our future collaborators were able to obtain a license, the rights may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing one or more product candidates, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. This could harm our business significantly.
Others may sue us for infringing their patent or other intellectual property rights or file nullity, opposition, re-examination or interference proceedings against our patents, even if such claims or proceedings are without merit, which would similarly harm our business. Furthermore, during the course of litigation, confidential information may be disclosed in the form of documents or testimony in connection with discovery requests, depositions or trial testimony. Disclosure of our confidential information and our involvement in intellectual property litigation could materially adversely affect our business.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including interference proceedings declared by the United States Patent and Trademark Office and opposition proceedings in the European Patent Office, regarding intellectual property rights with respect to our product candidates and technology. Even if we prevail, the cost to us of any patent litigation or other proceeding could be substantial.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from any litigation could significantly limit our ability to continue our operations. Patent litigation and other proceedings may also absorb significant management time. Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We try to ensure that our employees do not use the proprietary information, trade secrets or know-how of others in their work for us. However, we may be subject to claims that we or these employees have inadvertently or otherwise used or disclosed intellectual property, trade secrets, know-how or other proprietary information of any such employee’s former employer. Litigation may be necessary to defend against these claims and, even if we are successful in defending ourselves, could result in substantial costs to us or be distracting to our management. If we fail to defend any such claims, in addition to paying monetary damages, we may jeopardize valuable intellectual property rights, disclose confidential information or lose personnel.
53
Risks Related to our Common Stock
The trading price of the shares of our common stock could be highly volatile, and purchasers of our common stock could incur substantial losses.
The trading price of our common stock has fluctuated significantly since our IPO and our stock price is likely to continue to be volatile. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The market price for our common stock may be influenced by many factors, including:
|
|
·
|
setbacks or difficulties associated with our clinical trials;
|
|
|
·
|
our ability to enroll patients in our clinical trials;
|
|
|
·
|
results of clinical trials of our product candidates or those of our competitors;
|
|
|
·
|
regulatory developments in the United States and foreign countries;
|
|
|
·
|
variations in our financial results or those of companies that are perceived to be similar to us;
|
|
|
·
|
changes in the structure of healthcare payment systems, especially in light of current reforms to the U.S. healthcare system;
|
|
|
·
|
announcements by us of significant acquisitions, strategic partnerships, joint ventures or capital commitments;
|
|
|
·
|
market conditions in the pharmaceutical and biotechnology sectors and issuance of securities analysts’ reports or recommendations;
|
|
|
·
|
sales of substantial amounts of our stock by existing stockholders;
|
|
|
·
|
sales of our stock by insiders and 5% stockholders;
|
|
|
·
|
general economic, industry and market conditions;
|
|
|
·
|
additions or departures of key personnel;
|
|
|
·
|
intellectual property, product liability or other litigation against us;
|
|
|
·
|
expiration or termination of our relationships with our collaborators; and
|
|
|
·
|
the other factors described in this “Risk Factors” section.
|
54
In addition, in the past, stockholders have initiated class action lawsuits against biotechnology and pharmaceutical companies following periods of volatility in the market prices of these companies’ stock. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management’s attention and resources, which could have a material adverse effect on our business, financial condition and results of operations.
The future sale of our common stock could negatively affect our stock price.
If our existing stockholders sell a large number of shares of common stock, or the public market perceives that existing stockholders might sell shares of common stock, the market price of our common stock could decline significantly. These stockholders may sell their shares of our common stock starting at various times. In connection with our recently consummated private placement, we have agreed to register 11,079,250 shares of common stock as well as an additional 10,460,875 shares of common stock underlying the warrants issued in such financing. In addition, we have registered 2,359,634 shares of common stock that are reserved for issuance under our equity compensation plans. These shares can be freely sold in the public market upon issuance, subject to lock-up agreements and restrictions on our affiliates.
Concentration of ownership of our common stock among our existing executive officers and directors may prevent new investors from influencing significant corporate decisions.
As of March 15, 2011, our executive officers, directors and their affiliates own, in the aggregate, approximately 36.6% of the 23,471,249 shares of our outstanding common stock. Funds affiliated with directors own warrants to purchase an aggregate of 4,328,625 shares of common stock upon exercising their warrants at an exercise price of $2.88 per share. In addition, our executive officers and directors hold options to purchase 782,866 shares of common stock upon exercising their options at a weighted-average exercise price of $2.37 per share. These persons, if acting together, would be able to significantly influence all matters requiring stockholder approval, including the election and removal of directors and any merger or other significant corporate transactions. The interests of this group of stockholders may not coincide with our interests or the interests of other stockholders.
We will need to raise additional capital to fund our operations, which may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights.
We may seek additional capital through a combination of private and public equity offerings, debt financings and collaboration, strategic and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest may be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. In addition, the warrants we issued in connection with our March 2011 private placement transaction provide that the exercise price of such warrants would be adjusted downward in the event we subsequently issue stock at a price per share less than the current exercise price per share of the warrants. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic alliance and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates or grant licenses on terms that are not favorable to us.
55
Certain provisions of the warrants issued in connection with our March 2011 private placement provide for preferential treatment to the holders of the warrant and could impede a sale of the Company.
In March 2011, we closed a private placement transaction pursuant to which we sold securities consisting of 11,079,250 shares of common stock and warrants to purchase 10,460,875 shares of common stock. The warrant agreement gives each holder the option to receive a cash payment based on a Black-Scholes valuation of the warrant upon a change in control of the Company or upon the Company’s failure to be listed on any of the New York Stock Exchange, the NYSE Alternext (formerly the American Stock Exchange), the NASDAQ Global Select Market, the NASDAQ Global Market, the NASDAQ Capital Market or any other national securities exchange, which failure to be listed is not the result of a transaction approved by the Company’s stockholders (a Delisting). The warrant agreement, which specifies the method of calculating the Black-Scholes value, includes the requirement to calculate the Black-Scholes value using a minimum volatility of 100%. Holders of the Company’s common stock, or holders of other warrants, do not have the right to receive a cash payment upon a Delisting. Further, other current warrant holders do not have the right to receive a cash payment in every change in control transaction. The cash payment could be greater than the consideration that the Company’s other equity holders would receive in a change in control transaction. This provision of the warrant agreement could make a change in control transaction more expensive for a potential acquirer and could negatively impact the Company’s ability to pursue and consummate such a transaction.
Certain provisions of Delaware law and of our charter documents contain provisions that could delay and discourage takeover attempts and any attempts to replace our current management by stockholders.
Certain provisions of our certificate of incorporation and bylaws, and applicable provisions of Delaware corporate law, may make it more difficult for or prevent a third party from acquiring control of us or changing our board of directors and management. These provisions include:
|
|
·
|
the ability of our board of directors to issue preferred stock with voting or other rights or preferences;
|
|
|
·
|
the inability of stockholders to act by written consent;
|
|
|
·
|
a classified board of directors with staggered three-year terms;
|
|
|
·
|
requirement that special meetings of our stockholders may only be called upon a resolution adopted by an affirmative vote of a majority of our board of directors; and
|
|
|
·
|
requirements that our stockholders comply with advance notice procedures in order to nominate candidates for election to our board of directors or to place stockholders’ proposals on the agenda for consideration at meetings of stockholders.
|
56
We are afforded the protections of Section 203 of the Delaware General Corporation Law, which prevents us from engaging in a business combination with a person who acquires at least 15% of our common stock for a period of three years from the date such person acquired such common stock, unless prior board or stockholder approval was obtained.
Any delay or prevention of a change of control transaction or changes in our board of directors or management could deter potential acquirers or prevent the completion of a transaction in which our stockholders could receive a substantial premium over the then-current market price for their shares.
We do not expect to pay cash dividends on our common stock in the foreseeable future.
We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends will depend upon our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our common stock. Currently, we are subject to contractual restrictions on the payment of dividends under certain of our debt instruments. Furthermore, we may become subject to additional contractual restrictions or prohibitions on the payment of dividends.
Item 1B.Unresolved Staff Comments
We have received no written comments regarding our periodic or current reports from the staff of the Securities and Exchange Commission that were issued 180 days or more preceding the end of our 2010 fiscal year and that remain unresolved.
Item 2. Properties
Our corporate headquarters are located in East Norriton, Pennsylvania, where we currently occupy approximately 45,000 square feet of office, laboratory and manufacturing space in a facility of 80,000 square feet. Pursuant to a lease entered into in 2006, we began leasing the entire facility effective March 2011. The additional 35,000 square feet is primarily warehouse space, which we are currently subleasing to a third party for warehouse storage. This lease expires in February 2016. We also occupy approximately 38,400 square feet of office, laboratory and manufacturing space in Winston-Salem, North Carolina. The term of the lease expires in September 2011. In March 2011, we provided notice to our landlord that we intend to extend the term of this lease for an additional five-year period commencing in September 2011. We believe our current leased facilities are adequate for our needs for the immediate future and that, should it be needed, additional space can be leased to accommodate any future growth.
57
Item 3. Legal Proceedings
None
Item 4. (Removed and Reserved)
58
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock has traded on the NASDAQ Global Market since April 9, 2010, under the symbol “TNGN”. The following table sets forth, for the periods indicated, the high and low sales prices of our common stock on the NASDAQ Global Market. These prices represent prices between dealers and do not include retail mark-ups, markdowns, or commissions and may not necessarily represent actual transactions.
|
Year Ended December 31, 2010
|
High
|
Low
|
|
First Quarter
|
----
|
----
|
|
Second Quarter (beginning April 9, 2010)
|
$5.24
|
$3.33
|
|
Third Quarter
|
$4.24
|
$2.75
|
|
Fourth Quarter
|
$3.32
|
$2.03
|
Holders of Record
At December 31, 2010, the closing price per share of our common stock was $2.54 as reported on the NASDAQ Global Market and there were approximately 75 holders of record. Because many of such shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividends
We have never declared or paid any cash dividends on our common stock.
Securities Authorized for Issuance Under Equity Compensation Plans
The information under the heading "Equity Compensation Plan Information" in our definitive Proxy Statement for our 2011 Annual Meeting of Stockholders, to be filed with the SEC, is incorporated herein by reference
Recent Sales of Unregistered Securities
All information under this Item with respect to our initial public offering in April 2010 has previously been disclosed. All information under this Item regarding our March 2011 private placement of common stock and warrants to purchase common stock has been previously reported on our Current Report on Form 8-K, filed with the Securities and Exchange Commission (the “SEC”) on March 1, 2011.
59
Comparative Stock Performance Graph
The graph set forth below compares the cumulative total stockholder return on an initial investment of $100 in our common stock between April 9, 2010 (the date of our initial public offering) and December 31, 2010, with the comparative cumulative total return of such amount on (i) the NASDAQ Biotechnology Index and (ii) the NASDAQ Composite Index, over the same period. We have not paid any cash dividends and, therefore, the cumulative total return calculation for us is based solely upon stock price appreciation and not upon reinvestment of cash dividends. The graph assumes our closing sale price on April 9, 2010 of $5.02 per share as the initial value of our common stock.
The comparisons shown in the graph below are based upon historical data. We caution that the stock price performance shown in the graph below is not necessarily indicative of, nor is it intended to forecast, the potential future performance of our common stock. Information used in the graph was obtained from the NASDAQ Stock Market, Inc., a financial data provider and a source believed to be reliable. The NASDAQ Stock Market, Inc. is not responsible for any errors or omissions in such information.
| 04/09/10 | 06/30/10 | 09/30/10 | 12/31/10 | |
| Tengion, Inc. | $ 100.00 | $ 74.10 | $ 62.15 | $ 50.60 |
| The NASDAQ Composite Index | $ 100.00 | $ 85.95 | $ 96.52 | $ 108.10 |
| The NASDAQ Biotechnology Index | $ 100.00 | $ 84.12 | $ 94.15 | $ 102.92 |
The information presented above in the stock performance graph shall not be deemed to be “soliciting material” or to be “filed” with the SEC or subject to Regulation 14A or 14C, except to the extent that we subsequently specifically request that such information be treated as soliciting material or specifically incorporate it by reference into a filing under the Securities Act of 1933, as amended, or a filing under the Securities Exchange Act of 1934, as amended.
60
Item 6. Selected Financial Data
The following selected financial data should be read in conjunction with “Management's Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and the related notes appearing elsewhere in this Report. The statements of operations data for the years ended December 31, 2008, 2009 and 2010 and the balance sheet data as of December 31, 2009 and 2010 have been derived from our audited financial statements and related notes, which are included elsewhere in this Report. The statement of operations data for the years ended December 31, 2006 and 2007 and the balance sheet data as of December 31, 2006, 2007 and 2008 have been derived from audited financial statements which do not appear in this Report. The historical results presented are not necessarily indicative of future results.
|
Years Ended December 31,
|
||||||||||||||||||||
|
2006
|
2007
|
2008
|
2009
|
2010
|
||||||||||||||||
|
(in thousands, except share and per share data)
|
||||||||||||||||||||
|
Statement of Operations Data:
|
||||||||||||||||||||
|
Operating expenses:
|
||||||||||||||||||||
|
Research and development
|
$
|
15,552
|
$
|
22,335
|
$
|
27,947
|
$
|
17,948
|
$
|
12,855
|
|
|||||||||
|
General and administrative
|
4,584
|
|
5,290
|
|
7,467
|
|
5,527
|
|
6,032
|
|||||||||||
|
Depreciation
|
1,675
|
|
3,678
|
|
4,716
|
|
4,937
|
|
4,862
|
|
||||||||||
|
Loss from operations
|
(21,811
|
)
|
(31,303
|
)
|
(40,130
|
)
|
(28,412
|
)
|
(23,749
|
)
|
||||||||||
|
Interest income (expense), net
|
1,104
|
45
|
|
(2,206
|
)
|
(3,257
|
)
|
(2,042
|
)
|
|||||||||||
|
Change in fair value of preferred stock warrant
|
(166
|
)
|
270
|
|
(57
|
)
|
1,824
|
|
191
|
|
||||||||||
|
Net loss
|
(20,873
|
)
|
(30,988
|
)
|
(42,393
|
)
|
(29,845
|
)
|
(25,600
|
)
|
||||||||||
|
Accretion of redeemable convertible preferred stock to redemption value
|
(5,640
|
)
|
(8,742)
|
(11,754
|
)
|
(14,059
|
)
|
(3,992
|
)
|
|||||||||||
|
Net loss applicable to common stockholders
|
$
|
(26,513
|
)
|
$
|
(39,730
|
)
|
$
|
(54,147
|
)
|
$
|
(43,904
|
)
|
$
|
(29,592
|
)
|
|||||
|
Basic and diluted net loss per common share
|
$
|
(39.42
|
)
|
$
|
(60.16
|
)
|
$
|
(80.16
|
)
|
$
|
(62.95
|
)
|
$
|
(3.22
|
)
|
|||||
|
Weighted average common shares outstanding
|
672,544
|
|
660,423
|
|
675,461
|
|
697,448
|
|
9,196,920
|
|
||||||||||
|
As of December 31,
|
||||||||||||||||||||
|
2006
|
2007
|
2008
|
2009
|
2010
|
||||||||||||||||
|
(in thousands)
|
||||||||||||||||||||
|
Balance Sheet Data:
|
||||||||||||||||||||
|
Cash, cash equivalents and short-term investments
|
$
|
67,256
|
|
$
|
66,554
|
|
$
|
50,601
|
|
$
|
19,303
|
|
$
|
11,972
|
|
|||||
|
Working capital
|
58,878
|
63,075
|
41,379
|
2,766
|
4,247
|
|||||||||||||||
|
Total assets
|
88,766
|
|
91,313
|
|
72,276
|
|
37,238
|
|
24,144
|
|
||||||||||
|
Long-term debt
|
21,681
|
25,650
|
21,137
|
8,640
|
4,585
|
|||||||||||||||
|
Redeemable convertible preferred stock
|
98,790
|
140,751
|
173,857
|
187,916
|
—
|
|||||||||||||||
|
Total stockholders' equity (deficit)
|
(43,177
|
)
|
(82,277
|
)
|
(135,079
|
)
|
(178,074
|
)
|
11,060
|
|
||||||||||
61
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Forward-Looking Statements
Any statements herein or otherwise made in writing or orally by us with regard to our expectations as to financial results and other aspects of our business may constitute forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. These statements relate to our future plans, objectives, expectations and intentions and may be identified by words like “believe,” “expect,” “designed,” “may,” “will,” “should,” “seek,” or “anticipate,” and similar expressions. These forward looking statements may include, but are not limited to, statements concerning: (i) our plans to develop and commercialize our product candidates; (ii) our ongoing and planned preclinical studies and clinical trials; (iii) the timing of and our ability to obtain and maintain marketing approvals for our product candidates; (iv) the rate and degree of market acceptance and clinical utility of our products; (v) our plans to leverage our Organ Regeneration Platform to discover and develop product candidates; (vi) our ability to identify and develop product candidates; (vii) our commercialization, marketing and manufacturing capabilities and strategy; (viii) our intellectual property position; (ix) our estimates regarding expenses, future revenues, capital requirements and needs for additional financing; and (x) other risks and uncertainties, including those under the heading “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K, as well as in other documents filed by us with the SEC.
Although we believe that our expectations are based on reasonable assumptions within the bounds of our knowledge of our business and operations, the forward-looking statements contained in this document are neither promises nor guarantees. Our business is subject to significant risks and uncertainties and there can be no assurance that our actual results will not differ materially from our expectations. Factors which could cause actual results to differ materially from our expectations set forth in our forward-looking statements are set forth in the section entitled “Risk Factors” in Item 1A of Part I of this Annual Report on Form 10-K, as well as in other documents filed by us with the SEC and include, among others: (i) patients enrolled in our clinical trials may experience adverse events related to our product candidates, which could delay our clinical trials or cause us to terminate the development of a product candidate; (ii) we may have difficulty enrolling patients in our clinical trials, including our Phase I clinical trial for our Neo-Urinary Conduit; (iii) we may be unable to progress our product candidates that are undergoing preclinical testing into clinical trials; (iv) we will need to raise additional funds to execute our business plan beyond May 2012 and such financing may not be available to us or, if available, on terms acceptable to us and (v) we have a substantial amount of debt that exposes us to risks that could adversely affect our business, operating results and financial condition.
The forward-looking statements made in this document are made only as of the date hereof and we do not intend to update any of these factors or to publicly announce the results of any revisions to any of our forward-looking statements other than as required under the federal securities laws.
62
Overview
We believe we are the only regenerative medicine company focused on discovering, developing, manufacturing and commercializing a range of neo-organs, which we define as products composed of living cells, with or without synthetic or natural materials, implanted into the body to incorporate, replace or regenerate a damaged tissue or organ. Our Organ Regeneration Platform enables us to create proprietary product candidates that are intended to harness the intrinsic regenerative pathways of the body to produce a range of native-like organs and tissues. Our product candidates eliminate the need to utilize other tissues of the body for a purpose to which they are poorly suited, to procure donor organs or to administer anti-rejection medications. We are developing neo-organs in our scalable manufacturing facilities using efficient and repeatable proprietary processes, and have implanted neo-organs in our clinical trials. We intend to develop our technology to address unmet medical needs in urologic, renal, and other diseases and disorders.
To date, we have devoted substantially all of our resources to the development of our Organ Regeneration Platform and product candidates, as well as to our facilities that we employ to manufacture our neo-organs. Since our inception in July 2003, we have had no revenue from product sales, and have funded our operations principally through the private and public sales of equity securities and debt financings. We have never been profitable and, as of December 31, 2010, we had an accumulated deficit of $211.2 million, including $48.4 million of cumulative accretion on the Redeemable Convertible Preferred Stock through April 2010. We expect to continue to incur significant operating losses for the foreseeable future as we advance our product candidates from discovery through preclinical studies and clinical trials and seek marketing approval and eventual commercialization.
In March 2011, we closed a private placement transaction pursuant to which we sold securities consisting of 11,079,250 shares of common stock and warrants to purchase 10,460,875 shares of common stock. The purchase price per security was $2.83. The warrants have a term of five years and are immediately exercisable for $2.88 per share. We received net proceeds of approximately $28.9 million.
In March 2011, we refinanced the outstanding debt owed to one of our lenders. Pursuant to the terms of the refinancing, we simultaneously borrowed $5.0 million from the lender and repaid the then outstanding principal amount of $4.5 million. We are obligated to make interest-only payments through January 2012, followed by 24 monthly payments of principal and interest at an interest rate of 11.75% per annum.
Based upon our current expected level of operating expenditures and debt repayment, we expect to be able to fund our operations through May 2012. We will need to raise more funds to complete the Phase I clinical trial for our Neo-Urinary Conduit and our preclinical research and development activities for our Neo-Kidney Augment. We will also need to raise more funds to maintain our research and manufacturing facilities beyond May 2012. There is no assurance that such financing will be available or, if available, on terms acceptable to us.
63
Financial Operations Overview
Research and Development Expense
Our research and development expense consists of expenses incurred in developing and testing our product candidates and are expensed as incurred. Research and development expense consists of:
|
|
·
|
personnel related expenses, including salaries, benefits, travel and other related expenses including stock-based compensation;
|
|
|
·
|
payments made to third-party contract research organizations for preclinical studies, investigative sites for clinical trials and consultants;
|
|
|
·
|
costs associated with regulatory filings and the advancement of our product candidates through preclinical studies and clinical trials;
|
|
|
·
|
laboratory and other supplies;
|
|
|
·
|
manufacturing development costs; and
|
|
|
·
|
facility maintenance.
|
Preclinical study and clinical trial costs for our product candidates are a significant component of our current research and development expenses. We track and record information regarding external research and development expenses on a per study basis. Preclinical studies are currently coordinated with third-party contract research organizations and expense is recognized based on the percentage completed by study at the end of each reporting period. Clinical trials are currently coordinated through a number of contracted sites and expense is recognized based on a number of factors, including actual and estimated patient enrollment and visits, direct pass-through costs and other clinical site fees. We utilize employees, resources and facilities across multiple product candidates. We do not allocate internal research and development expenses among product candidates.
64
The following table summarizes our research and development expense for the years ended December 31, 2008, 2009 and 2010.
|
Year Ended December 31,
|
||||||||||||
|
2008
|
2009
|
2010
|
||||||||||
|
(in thousands)
|
||||||||||||
|
Third-party direct program expenses:
|
||||||||||||
|
Urologic
|
$ | 7,836 | $ | 2,753 | $ | 194 | ||||||
|
Renal
|
1,011 | 1,000 | 1,813 | |||||||||
|
Other
|
965 | — | — | |||||||||
|
Total third-party direct program expenses
|
9,812 | 3,753 | 2,007 | |||||||||
|
Other research and development expense
|
18,135 | 14,195 | 10,848 | |||||||||
|
Total research and development expense
|
$ | 27,947 | $ | 17,948 | $ | 12,855 | ||||||
From our inception in July 2003 through December 31, 2010, we have incurred research and development expense of $104.6 million. We expect that a large percentage of our research and development expense in the future will be incurred in support of our current and future preclinical and clinical development programs. These expenditures are subject to numerous uncertainties in timing and cost to completion. We expect to continue to test our product candidates in preclinical studies for toxicology, safety and efficacy, and to conduct additional clinical trials for each product candidate. If we are not able to engage a partner prior to the commencement of later stage clinical trials, we may fund these trials ourselves. As we obtain results from clinical trials, we may elect to discontinue or delay clinical trials for certain product candidates or programs in order to focus our resources on more promising product candidates or programs. Completion of clinical trials by us or our future collaborators may take several years or more, but the length of time generally varies according to the type, complexity, novelty and intended use of a product candidate. The cost of clinical trials may vary significantly over the life of a project as a result of differences arising during clinical development, including, among others:
|
|
·
|
the number of sites included in the trials;
|
|
|
·
|
the length of time required to enroll suitable patients;
|
|
|
·
|
the number of patients that participate in the trials;
|
|
|
·
|
the duration of patient follow-up;
|
|
|
·
|
the development stage of the product candidate; and
|
|
|
·
|
the efficacy and safety profile of the product candidate.
|
None of our product candidates has received FDA or foreign regulatory marketing approval. In order to grant marketing approval, the FDA or foreign regulatory agencies must conclude that clinical data establish the safety and efficacy of our product candidates. Furthermore, our strategy includes entering into collaborations with third parties to participate in the development and commercialization of our product candidates. In the event that third parties have control over the clinical trial process for a product candidate, the estimated completion date would largely be under control of that third party rather than under our control. We cannot forecast with any degree of certainty which of our product candidates will be subject to future collaborations or how such arrangements would affect our development plan or capital requirements.
65
As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our development projects or when and to what extent we will receive cash inflows from the commercialization and sale of an approved product candidate.
General and Administrative Expense
General and administrative expense consists primarily of salaries, benefits and other related costs, including stock-based compensation, for persons serving in our executive, finance, legal, marketing planning and human resource functions. Our general and administrative expense includes facility-related costs not otherwise included in research and development expense, professional fees for legal services, including patent-related expense, tax and accounting services, and other consulting services and general corporate expenses applicable to public companies. We expect that our general and administrative expenses will increase with the development and potential commercialization of our product candidates.
Depreciation Expense
Depreciation expense is the amortization of capitalized property and equipment that is recognized over the estimated useful lives of the assets using the straight-line method. We use a life of three years for computer equipment; five years for laboratory, office and warehouse equipment; seven years for furniture and fixtures; and the lesser of the useful life of the asset or the remaining life of the underlying facility lease for leasehold improvements. Expenditures for maintenance, repairs, and betterments that do not prolong the useful life of the asset are charged to expense as incurred.
Interest Income, Interest Expense and Change in Fair Value of Preferred Stock Warrants
Interest income consists of interest earned on our cash and cash equivalents and short-term investments. Interest expense consists primarily of cash and non-cash interest costs related to our outstanding debt. The change in the fair value of our preferred stock warrants consists of non-cash interest for the warrants that were classified as a liability prior to the Company’s initial public offering in April 2010 and were revalued at each reporting date with changes in the fair value reported in the statements of operations. The fair value of the warrants were subject to fluctuations based on changes in the Company’s preferred stock price, expected volatility, remaining contractual life, and the risk-free interest rate.
Net Operating Losses and Tax Loss Carryforwards
As of December 31, 2010, we had net operating loss carryforwards available to offset future federal and state taxable income of $109.7 million and $120.2 million, respectively, as well as $4.7 million of research and development tax credits. The net operating loss carryforwards and credits expire at various dates through 2030. The Tax Reform Act of 1986 (the Act) provides for a limitation on the annual use of net operating loss and research and development tax credit carryforwards following certain ownership changes (as defined by the Act) that could limit our ability to utilize these carryforwards. We have not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since our formation, due to the significant costs and complexities associated with a study. We may have experienced various ownership changes, as defined by the Act, as a result of past financings. Accordingly, our ability to utilize the aforementioned carryforwards may be limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be applied against future taxes; therefore, we may not be able to take full advantage of these carryforwards for federal or state income tax purposes.
66
Critical Accounting Policies and Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make significant judgments and estimates that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Management bases these significant judgments and estimates on historical experience and other assumptions it believes to be reasonable based upon information presently available. Actual results could differ from those estimates under different assumptions, judgments or conditions.
All of our significant accounting policies are discussed in Note 3, Summary of Significant Accounting Policies, to our financial statements, included elsewhere in this Annual Report on Form 10-K. We have identified the following as our critical accounting policies and estimates, which are defined as those that are reflective of significant judgments and uncertainties, are the most pervasive and important to the presentation of our financial condition and results of operations and could potentially result in materially different results under different assumptions, judgments or conditions. Management has reviewed these critical accounting policies and estimates with the Audit Committee of our board of directors.
Impairment of Long-lived Assets
Long-lived assets, such as property and equipment, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Conditions that would necessitate an impairment assessment include a significant change in the planned use of the acquired asset, a decline in the observable market value of an asset, or a significant adverse change in the business such as the occurrence of a negative clinical regulatory matter that would prohibit us from obtaining the approval for commercializing a product candidate. Upon identification of an indicator of impairment, recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated undiscounted future cash flows, then an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the estimated fair value of the asset. As of December 31, 2009 and 2010, the carrying value of long lived assets was $16.2 million and $11.5 million, respectively. If an event were to occur as described above that would prevent us from obtaining the approval for commercializing a product candidate, we would anticipate an impairment charge of an amount up to a maximum of the carrying value of the long-lived assets at that time. The evaluation of the recoverability of long-lived assets, and the determination of their fair value should such fair values need to be estimated, requires us to make significant estimates and assumptions. These estimates and assumptions include, but are not limited to, the estimation of future cash flows, discount rates and costs to sell. Due to the inherent uncertainty involved in making these estimates, actual results could differ from those estimates. Such a change in assumptions could have a significant impact on the conclusion that an asset’s carrying value is recoverable, or the determination of any impairment charge if it was determined that the asset values were indeed impaired. In 2008, 2009 and 2010, no impairment charges were recognized.
67
Preclinical and Clinical Trial Costs
A substantial portion of our ongoing research and development activities are performed under agreements we enter into with external service providers who conduct many of our research and development activities. Estimates of incurred costs are made to determine the accrued balance in any accounting period. The estimates incurred under the contracts are based on factors such as work performed, milestones achieved, patient enrollment and costs historically incurred for similar contracts. As actual costs become known, we adjust our estimates. To date, our estimates have been within management’s expectations, and no material adjustments to research and development expense have been recognized. For the year ended December 31, 2010, a 10% increase or decrease in our estimate of research and development expense incurred under such contracts would result in an increase or decrease in research and development expense of approximately $67,000. We may expand the level of research and development activity to be performed by external service providers in which case our estimates would be more material to our future operations. Subsequent changes in estimates may result in a material change in our accruals, which could also materially affect our future results of operations.
Stock-Based Compensation
Effective January 1, 2006, we adopted the revised accounting standards for stock-based compensation guidance which was adopted prospectively to new awards and to awards modified, repurchased, or canceled after December 31, 2005. This current guidance requires companies to measure and recognize compensation expense for all employee stock-based payments at fair value, net of estimated forfeitures, over the vesting period of the underlying stock-based awards. In addition, we account for stock-based compensation to nonemployees in accordance with the Financial Accounting Standards Board (FASB) accounting guidance for equity instruments that are issued to other than employees.
We use the Black-Scholes option-pricing model to value our stock option awards. The Black-Scholes option-pricing model requires the input of subjective assumptions, including the expected life of the stock-based payment awards and stock price volatility. Since we do not have sufficient historical volatility of our stock as a public company for the expected term of the options, we use comparable public companies as a basis for our expected volatility to calculate the fair value of option grants. We intend to continue to consistently apply this process using comparable companies until a sufficient amount of historical information regarding the volatility of our own share price becomes available. The expected term is based on the simplified method provided by SEC guidance. The risk-free interest rate is based on the U.S. Treasury yield curve with a remaining term equal to the expected life assumed at grant. The assumptions used in calculating the fair value of stock-based payment awards represent management’s best estimate and involve inherent uncertainties and the application of management’s judgment. As a result, if factors change and management uses different assumptions, stock-based compensation expense could be materially different in the future.
68
The estimation of the number of stock awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from our current estimates, such amounts will be recorded as a cumulative adjustment in the period in which estimates are revised. We consider many factors when estimating expected forfeitures, including types of awards, employee class and an analysis of our historical and known forfeitures on existing awards. Under the true-up provisions of the FASB stock based compensation guidance, we record additional expense if the actual forfeiture rate is lower than estimated, and a recovery of expense if the actual forfeiture rate is higher than estimated, during the period in which the options vest.
Prior to the completion of our initial public offering in April 2010, the fair value of our common stock underlying stock options granted during 2008 and 2009 was determined by our compensation committee pursuant to authority delegated by our board of directors. The fair value of our common stock underlying stock options granted during 2010, subsequent to the completion of our IPO, was determined by the closing price of our stock on the day of grant. In the absence of a public trading market for our common stock, our compensation committee was required to estimate the fair value of our common stock at each option grant date. Our compensation committee, in making its independent determination, utilized the assistance of an independent valuation firm. In each of the separate valuations performed by our compensation committee, our compensation committee’s determination of the fair market values was consistent with the results and conclusions of our independent third party valuation. We used methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants Practice Guide, or the AICPA Practice Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation, considering numerous objective and subjective factors to determine common stock fair market value at each option grant date, including but not limited to the following factors:
|
|
·
|
arm’s length private transactions involving our preferred stock, including the sale of our Series A preferred stock at $23.44 per share in 2004 and 2005, the sale of our Series B preferred stock at $26.39 per share in 2006 and the sale of our Series C preferred stock at $26.39 per share in 2007 and 2008, all of which had superior rights and preferences compared to our common stock. All per share prices in this paragraph reflect the automatic conversion of all outstanding shares of preferred stock into 5,651,955 shares of common stock upon the completion of our initial public offering in April 2010;
|
|
|
·
|
our financial and operating performance;
|
69
|
|
·
|
the likelihood of achieving a liquidity event for the shares of our common stock and options, such as an initial public offering or sale of our company, given prevailing market conditions;
|
|
|
·
|
the conditions of the equity markets in general and the biotechnology markets in particular;
|
|
|
·
|
developmental milestones achieved;
|
|
|
·
|
business risks; and
|
|
|
·
|
management and board experience.
|
In considering the rights and preferences of our preferred stock relative to our common stock, our compensation committee considered the following rights and preferences of our Series A, Series B and Series C preferred stock:
|
|
·
|
The Series A, Series B and Series C stockholders were entitled to receive a cumulative annual 8% dividend, when and if declared by the board of directors; and,
|
|
|
·
|
The Series A, Series B and Series C stockholders were entitled to a liquidation preference. The aggregate amount of liquidation preferences, excluding any dividends, had increased from $89.4 million as of December 31, 2006 to $144.2 million as of December 31, 2009. The liquidation preference for each preferred share equaled the original purchase price of $23.44 per share for Series A and $26.39 per share for Series B and Series C, plus accumulated unpaid dividends, in the event of liquidation, dissolution, or winding up of the Company. After receiving its liquidation preferences, the holders of preferred shares participate equally with the common stockholders in any remaining value of the company. All per share prices in this paragraph reflect the automatic conversion of all outstanding shares of preferred stock into 5,651,955 shares of common stock upon the completion of our initial public offering in April 2010.
|
Beginning in January 2006, our compensation committee commenced periodic contemporaneous assessments of the valuation of our common stock. These valuations were performed concurrently with the achievement of significant milestones, major financing events or sizeable options grants.
In determining the fair value of our common stock, we considered several factors impacting the value of our common stock, including the following:
|
|
·
|
Common stock pricing indications implied from our most recent sales of preferred securities. We considered pricing of our preferred stock, our capitalization structure, the prevailing risk-free interest rate as of the date for the sale of our preferred stock, estimates of expected equity volatility and the expected time to liquidity.
|
70
|
|
·
|
Market pricing information from companies that we considered to be comparable or that we believed would be priced in a similar fashion. The companies selected were in the life sciences industry and focused on regenerative medicine or comparable technologies. Pricing multiples were applied, or in cases where valuations were determined at a pre-revenue level, absolute market equity values, from the selected companies to our forecasted performance to determine the future equity value or future common stock value. These values were then discounted to the present date at a risk adjusted rate of return to determine a present common stock value indication.
|
|
|
·
|
Discounted cash flow models. Discounted cash flow models were based on the anticipated timing of our clinical trials and related exit events as well as the additional cash required to complete these trials and commercialize our products. These cash flows were discounted to the present to determine the present common stock value indication.
|
|
|
·
|
Assessment of milestones achieved. The common stock value was influenced by our performance as compared to expected milestones set in the previous pricing assessment. Our success or failure at achieving certain clinical trial and budget targets influenced the value of our common stock.
|
|
|
·
|
Risks. Specific risks considered in the assessment of our common stock price included our ability to raise additional funds, the status of our clinical trials, our ability to retain key management and employees, and our ability to consummate a liquidity event.
|
|
|
·
|
Market conditions. The prevailing market conditions were factored into our analysis when deriving the value of our common stock. More specifically, the direction of the capital markets were considered, measured by the change in value of the NYSE composite index and Nasdaq composite index, and the change in stock prices of companies considered to be comparable to determine the market conditions of our industry.
|
|
|
·
|
Pricing from initial public offerings in the life sciences industry. Pricing information from initial public offerings of companies in the life sciences industry was considered to determine our common stock price. Specifically, initial public offering pricing information of life science companies at certain stages of development was utilized to determine the future equity value upon reaching that development stage. The future equity value was then discounted to the present to determine the present equity value and common stock value.
|
|
|
·
|
Pricing indications from mergers and acquisitions in the life sciences industry. Pricing information from mergers and acquisitions were considered to determine the common stock value. The future equity value was derived based on the projected developmental stage as compared to the developmental stage of specific life science companies when they were acquired. The future equity value indications were discounted to the present to determine the present equity value and common stock value.
|
71
|
|
·
|
Liquidity considerations. The liquidity and illiquidity features of our securities were considered. Because our stockholders have not had access to an organized exchange on which to trade their securities, the appropriate adjustment to the value to account for this lack of liquidity was assessed.
|
The following table represents a summary of the contemporaneous valuations performed by our compensation committee concurrently with the achievement or failure of significant milestones or with major financing events, where applicable. Listed are the related stock option grants and exercise prices that utilized these valuations from January 1, 2008 through December 31, 2009.
|
Date of Valuation
|
Milestone / Financing Event
|
Number
of Shares
|
Exercise or
Purchase
Price per
Share
|
Per Share
Estimated Fair
Value of
Common
Stock
|
|||||||||
|
September 24, 2007
|
$33.4 million raised in Series C preferred stock financing on September 24, 2007
|
127,578 | $ | 18.56 | $ | 18.56 | |||||||
|
February 1, 2008
|
To support 2007 performance grants issued in February 2008
|
80,180 | $ | 24.80 | $ | 24.80 | |||||||
|
July 31, 2008
|
Hire grant for Vice President, Marketing and Commercial Planning; passage of time
|
38,419 | $ | 20.16 | $ | 20.16 | |||||||
|
October 15, 2008
|
$21.5 million raised in Series C preferred stock financing extension on October 15, 2008
|
3,039 | $ | 11.75 | $ | 11.75 | |||||||
|
January 15, 2009
|
To support 2008 performance grants issued in February 2009
|
2,519 | $ | 10.01 | $ | 10.01 | |||||||
|
February 13, 2009
|
IND for Neo-Bladder Augment placed on clinical hold; revision of projections; delay in IPO event
|
276 | $ | 2.03 | $ | 2.03 | |||||||
|
July 31, 2009
|
Board approval of amended operating plan to focus on Neo-Urinary Conduit and further delay of IPO event
|
355,155 | $ | 0.44 | $ | 0.44 | |||||||
|
December 3, 2009
|
Board approval for the Company to pursue an IPO event
|
— | $ | — | $ | 2.90 | |||||||
On August 26, 2009, we modified the exercise price for all options outstanding related to active employees to $0.44 resulting in an incremental value, defined as the excess of the fair value of the modified options over the fair value of the original options immediately before these terms were modified, which was partially recognized as compensation in the third quarter of 2009. The remaining charge will be recognized over the remaining vesting period of the options that were modified.
72
Our determination of the fair values of our common stock are described in detail below beginning with the September 24, 2007 valuation that was utilized by our compensation committee to determine the fair value of our common stock associated with options granted after January 1, 2008 and prior to the completion of our initial public offering in April 2010. All valuations were prepared consistent with the AICPA Practice Guide and used a combination of several valuation approaches: the market approach, which compares our company to similar publicly-traded companies or transactions, an income approach, which estimates the present value of cash flows that the business is expected to generate over a forecast period and an estimate of the present value of cash flows beyond that period, which is referred to terminal value, and an analysis of the implied value of the common stock given the price of the latest issuance of preferred stock. Many of these future value indications are converted to present value by means of discounting, using a rate of return that accounts for the time value of money and the appropriate degree of risks inherent in the business and excludes any factor for lack of marketability. Given that we have several series of convertible preferred stock outstanding, it was also necessary to allocate our company’s value to the various classes of stock, including the various series of preferred stock and common stock. As provided in the AICPA Practice Guide, there are several approaches for allocating equity value of a privately-held company among the securities in a complex capital structure. Aside from the two exceptions noted below, we used the probability weighted equity return method, or PWERM, to allocate our equity to our various classes of securities. The value of our common stock was then discounted for lack of marketability, or the inability to readily sell shares, which increases the owner’s exposure to changing market conditions and increases the risk of ownership. The discount for lack of marketability is derived using a protective put calculation using the Black-Scholes option pricing model.
Valuation September 24, 2007
At the time of the September 24, 2007 valuation, we had raised $33.2 million in net proceeds through the sale of our Series C preferred stock at a price per share of $26.39. This per share price assumes the automatic conversion of all of the outstanding shares of our preferred stock into 5,651,955 shares of common stock upon the completion of our initial public offering. In the September 24, 2007 valuation, our methodology focused on the implied pricing of our common stock based on the per share sale price of our Series C preferred stock as well as a selection of IPO scenarios. The use of IPO scenarios reflected our intent at the time to enter the public market in 2008. At the time of this valuation and as a result of the plans for an IPO, we changed the allocation method of the equity value of our company to a PWERM. In the IPO scenario we estimated our future equity value in an IPO by analyzing comparable IPOs in the life sciences industry. The estimated future equity values in the IPO scenarios were allocated to our equity securities assuming that all such securities would convert to common equity in the event of an IPO. Thereafter the common value indications from each IPO scenario were discounted from the date of the anticipated event to the valuation date using a discount rate of 50%. A lack of marketability adjustment of 15% was applied to the resulting present equity values.
Because of our plans at the time to effectuate an IPO in 2008, we placed an 89% weight on the indications from the various IPO scenarios and attributed an 11% weighting to our common stock value resulting from the sale price of the Series C preferred stock. This analysis yielded a marketable, minority firm equity value of $139.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $103.0 million. The resulting value, which represented the estimated fair value of our common stock at September 24, 2007, was $18.56 per share.
73
Valuation February 1, 2008
As of February 1, 2008, the valuation approaches and assumptions were similar to those used in our September 24, 2007 valuation, including the use of the PWERM allocation method for the equity value of our company. Methodology changes for this valuation date included a de-emphasis on the use of the last sale of preferred securities as a significant amount of time had elapsed since such financing event and an inclusion of a non-IPO alternative with a low probability as an alternative should we not effectuate a near-term IPO. Under the IPO scenarios, the future equity value or our common stock in an IPO was calculated using the estimated equity valuations derived from analyzing comparable IPOs in the life sciences industry and a risk-adjusted discount of 45%. The discount for lack of marketability ranged from 12% to 37%. Because of our plans at the time to effectuate an IPO in 2008 we placed a 90% weight on the indications from the various IPO scenarios and a 10% weight on a stay private scenario. This analysis yielded a marketable, minority firm equity value of $178.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $141.0 million. The resulting value, which represented the estimated fair value of our common stock at February 1, 2008, was $24.80 per share.
Valuation July 31, 2008
As of July 31, 2008, the valuation methodology, approaches and assumptions were substantially the same as those used in the February 1, 2008 valuation, including the use of the PWERM allocation method for the equity value of our company, except the timing of an assumed IPO event was delayed by three quarters due to a decline in market conditions. Under the IPO scenarios, the future value of our common stock in an IPO was calculated using the estimated equity valuations derived from analyzing comparable IPOs in the life sciences industry and a risk adjusted discount of 40%. The discount for lack of marketability used was in the range of 17% to 22% for the IPO scenarios and 35% for remaining a private company. Although we assumed a longer time to exit, the IPO was still the preferred and anticipated liquidity event and we continued to place the most weight on the indications from the various IPO scenarios at 90% and a stay private weighting of 10%. This analysis yielded a marketable, minority firm equity value of $174.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $146.0 million. The resulting value, which represented the estimated fair value of our common stock at July 31, 2008, was $20.16 per share.
Valuation October 15, 2008
As of October 15, 2008, the valuation methodology and approaches were substantially the same as those used in the July 31, 2008 valuation, including the use of the PWERM allocation method for the equity value of our company. At that time we had raised $21.4 million in net proceeds through the sale of additional shares of our Series C preferred stock and, due to continuing decline in market conditions, we decided to further postpone our intended public offering plans. Under the now delayed IPO scenarios, the future value of our common stock in an IPO was calculated using the estimated equity valuations derived from analyzing comparable IPOs in the life sciences industry and a risk-adjusted discount of 40%. The discount for lack of marketability used was in the range of 27% to 29% for IPO scenarios and 35% for remaining a private company. For the concluded common stock value we significantly delayed the proposed timing of an IPO scenario by three quarters relative to the previous valuation, which continued to be weighted at a probability of 90% and a stay private scenario probability of 10%. This analysis yielded a marketable, minority firm equity value of $141 million. At the time of the valuation, the aggregate preferred liquidation preference was $171 million. The resulting value, which represented the estimated fair value of our common stock at October 15, 2008, was $11.75 per share. The decrease in our estimated fair value of our common stock valuation was primarily related to the increase in the time to an expected liquidity event as well as the declining equity markets.
74
Valuation January 15, 2009
As of January 15, 2009, the valuation methodology and approaches were substantially the same as those used in the October 15, 2008 valuation, including the use of the PWERM allocation method for the equity value of our company. However, due to the severe downturn in market conditions, we decided to postpone indefinitely our intended plans for an IPO. We continued to use a combination of an IPO and stay-private valuation model. In the IPO based scenario analysis we delayed the assumed exit timing by two additional quarters relative to the previous valuation. The future value of our common stock in an IPO was calculated using the estimated equity valuations derived from analyzing comparable IPOs in the life sciences industry and a risk-adjusted discount of 40%. The discount for lack of marketability was in the range of 35% to 36% for the IPO scenarios and 45% for the stay-private scenario. As a result of the negative market development and the related change in IPO plans, the probability of a stay-private scenario increased to 50% and the probability of an IPO scenario decreased to 50%. The lower probability of an IPO and the extension of the expected time to liquidity contributed to the decline in our common stock value. This analysis yielded a marketable, minority firm equity value of $180.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $174.0 million. The resulting value, which represented the estimated fair value of our common stock at January 15, 2009, was $10.01 per share.
Valuation February 13, 2009
Our February 13, 2009 valuation reflected that our IND for our Neo-Bladder Augment was placed on clinical hold by the FDA and the related uncertainty around the timing of the FDA’s release of the clinical hold. As a result of the clinical hold, the prospects for a public liquidity event were significantly delayed further. At this time we were in the early stages of acquisition negotiations with a strategic buyer. The valuation methodology centered around a strategic acquisition, a significantly delayed IPO and a remaining private scenario. The merger and acquisition and IPO scenarios were market based approaches and the stay-private model was based on discounted cash flows and a market based terminal value. Due to the possibility of an acquisition, we continued to use the probability weighted equity allocation model (PWERM). Under the IPO scenarios, the future value of our common stock in an IPO was calculated using the estimated equity valuations derived from analyzing comparable IPOs in the life sciences industry and a risk-adjusted discount of 45% based on the estimated timing of a potential IPO. The discount for lack of marketability used was 26% for the merger and acquisition scenario, 42% for the IPO scenario and 45% for remaining a private company. We used a probability weight of 50% for the merger and acquisition scenario, 25% for the IPO scenarios and 25% for remaining a private company. This analysis yielded a marketable, minority firm equity value ranging from $127.0 million to $136.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $174.0 million. The resulting value, which represented the estimated fair value of our common stock at February 13, 2009, was $2.03 per share. The main reason for the decline in the value of our common stock from previous valuations was mainly due to an additional year delay in the IPO timing relative to the previous valuation and the greater weight being placed on merger and acquisition and stay-private outcomes with the associated impact of the aggregate liquidation preferences. This change was predominately due to the IND for Neo-Bladder Augment being placed on clinical hold.
75
Valuation July 31, 2009
Our July 31, 2009 valuation reflects our board of directors’ decision to amend the operating plan to shift resources from our Phase II completed Neo-Bladder Augment and focus on our preclinical, pre-IND stage Neo-Urinary Conduit program. With continued market uncertainties, the end of discussions with a strategic buyer for a near-term acquisition and the date of a potential product commercialization delayed, the methodologies for the independent valuation focused on longer term merger and acquisition opportunities, a delayed IPO event of over a year relative to the previous valuation and a long-term stay-private scenario. Due to the prolonged assumptions for a liquidity event, we changed the allocation method of the equity value of our company to the underlying common shares from a PWERM to the option-pricing model. In the merger and acquisition scenario, we used comparable merger and acquisitions transactions in the biotechnology and life science industries at different phases of development to estimate equity values. Under the IPO scenario, the future value of our common stock in an IPO was estimated using comparable IPO’s in the biotechnology and life sciences. The estimated equity values in both the merger and acquisition scenario and the IPO scenario were then discounted from the date of the anticipated event to the valuation date using a discount rate of 45%. For the long term stay-private scenario, the valuation indication was derived from a discounted cash flow analysis using a discount rate of 45%. The discount for lack of marketability was in the range of 43% to 44%. To establish the marketable equity value to allocate to our common stock outstanding using the option pricing model, we used a probability weight of 50% for the merger and acquisition scenario, 25% for the IPO scenario and 25% for the long term stay-private scenario. This analysis yielded a marketable, minority firm equity value of $50.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $181.0 million. The application of the option pricing model to the marketable equity value resulted in an estimated fair value of our common stock at July 31, 2009 of $0.44 per share. The decrease in the value of our common stock from previous valuations was mainly due to the significant delay of an anticipated IPO event and our board’s approval of an amended operating plan to focus on our Neo-Urinary Conduit. By focusing on our preclinical, pre-IND stage Neo-Urinary Conduit product candidate, the timeline and uncertainty to potential commercialization of our lead product candidate was extended.
76
Valuation December 3, 2009
On December 3, 2009, our board of directors decided to proceed with an IPO, attempting to take advantage of the recently improving public markets. Accordingly, our December 3, 2009 valuation refocused to near-term IPO scenarios as well as alternative near-term merger and acquisition scenarios. In addition, some probability was assigned to a failure scenario. Given the higher probability of a near term liquidity event, the method to allocate the equity value of our company changed from the option-pricing model to PWERM. Under the IPO scenario, the future value or our common stock in an IPO was calculated using the estimated equity valuation derived from analyzing comparable IPOs in the life sciences industry, reflecting lower IPO values related to our now earlier stage of clinical development. A risk-adjusted discount rate of 35% based on the estimated timing of a potential IPO was applied. The discount for lack of marketability used was 24% for the merger and acquisition scenario, 17% for the IPO scenario, and 32% for remaining a private company. We used a probability weight of 25% for the merger and acquisition scenario, 50% for the IPO scenarios and 25% for remaining a private company. This analysis yielded a marketable, minority firm equity value of $43.0 million. At the time of the valuation, the aggregate preferred liquidation preference was $184.0 million. The resulting value, which represented the estimated fair value of our common stock at December 3, 2009, was $2.90 per share. The increase over the July 31, 2009 independent valuation was mainly driven by the increased probability of an IPO liquidity event in the next six months.
Results of Operations
Year Ended December 31, 2009 compared to Year Ended December 31, 2010
Research and Development Expense. Research and development expense for the years ended December 31, 2009 and 2010 was comprised of the following:
|
Year Ended
December 31,
|
Increase
(Decrease)
|
|||||||||||||||
|
2009
|
2010
|
$ | % | |||||||||||||
|
(in thousands)
|
||||||||||||||||
|
Compensation and related expense
|
$ | 9,134 | $ | 7,482 | $ | (1,652 | ) | (18) % | ||||||||
|
External services – direct third parties
|
3,753 | 2,007 | (1,746 | ) | (47) % | |||||||||||
|
External services – other
|
1,048 | 510 | (538 | ) | (51) % | |||||||||||
|
Research materials and related expense
|
1,771 | 975 | (796 | ) | (45) % | |||||||||||
|
Facilities and related expense
|
2,242 | 1,881 | (361 | ) | (16) % | |||||||||||
|
Total research and development expense
|
$ | 17,948 | $ | 12,855 | $ | (5,093 | ) | (28) % | ||||||||
Research and development expense decreased primarily due to lower direct third-party expenses of $0.8 million for preclinical studies and $0.2 million for clinical studies, as well as the receipt of Qualifying Therapeutic Discovery Project Grants totaling $0.7 million, which was recorded as a credit to external services-direct third parties. Research and development expense with respect to compensation and other related costs decreased $1.7 million due to a full year with reduced headcount in our Pennsylvania facility. The decrease in external services activity and headcount resulted in a decrease of $1.7 million in the demand for lab supplies and other services related to our product candidates.
77
General and Administrative Expense. General and administrative expense increased by $0.5 million, or 9%, from $5.5 million in 2009 to $6.0 million in 2010. The increase in general and administrative expense was primarily due to increased outside professional services, including legal expenses associated with operating as a public company.
Depreciation Expense. Depreciation expense remained relatively unchanged for the year ended 2009 compared to the year ended 2010, as additions were not significant in either period.
Interest Income (Expense). Interest income decreased $0.1 million, or 71%, from $0.2 million in 2009 to $0.1 million in 2010 due to lower investment balances and lower rates and returns on our investments. Interest expense decreased $1.4 million, or 39%, from the year ended 2009 to the year ended 2010, due to lower average debt balances and the end of our interest-only payments in the third quarter of 2009.
Change in Fair Value of Preferred Stock Warrants. Change in fair value of preferred stock warrants increased due to a non-cash credit of $1.6 million related to the preferred stock warrants in the year ended 2009, as the estimated fair value of the Company’s preferred stock warrants decreased during that period. The preferred stock warrants were reclassified from a liability to stockholders equity upon the completion of our initial public offering in April 2010.
Year Ended December 31, 2008 compared to the Year Ended December 31, 2009
Research and Development Expense. Research and development expense for the year ended December 31, 2008 and 2009 were comprised of the following:
|
December 31,
|
Increase
(Decrease)
|
|||||||||||||||
|
2008
|
2009
|
$ | % | |||||||||||||
|
(in thousands)
|
||||||||||||||||
|
Compensation and related expense
|
$ | 11,664 | $ | 9,134 | $ | (2,530 | ) | (22) % | ||||||||
|
External services – direct third parties
|
9,812 | 3,753 | (6,059 | ) | (62) % | |||||||||||
|
External services – other
|
1,150 | 1,048 | (102 | ) | (9) % | |||||||||||
|
Research materials and related expense
|
2,516 | 1,771 | (745 | ) | (30) % | |||||||||||
|
Facilities and related expense
|
2,805 | 2,242 | (563 | ) | (20) % | |||||||||||
|
Total research and development expense
|
$ | 27,947 | $ | 17,948 | $ | (9,999 | ) | (36) % | ||||||||
Research and development expense for 2009 decreased primarily due to a decrease in external services related to direct third-party expenses of $4.7 million for preclinical studies and $1.4 million for other costs associated with our Phase II clinical trials for our Neo-Bladder Augment. This decrease is largely attributable to a higher cost per patient in 2008, the first year of patient enrollment, and the concomitant surgical procedures and related hospital stays as required under our clinical protocol, as compared to subsequent years where patient treatments under the protocol are reduced to twice a year. Compensation and related expense decreased primarily due to the reduction of headcount in 2009 in our Pennsylvania manufacturing facility. The decrease in these programs led to a decrease in the demand for lab supplies and other services related to our product candidates.
78
General and Administrative Expense. General and administrative expense decreased by $2.0 million, or 26%, from $7.5 million for the year ended December 31, 2008 to $5.5 million for the year ended December 31, 2009. The decrease in general and administrative expense was due to a decrease in corporate personnel-related costs of $0.5 million primarily related to the reduction of headcount in our executive, finance, legal and human resource functions, as well as lower legal, audit and other external services of $1.4 million, primarily due to costs related to a previous potential capital raising transaction.
Depreciation Expense. Depreciation expense increased by $0.2 million, or 5%, from $4.7 million for the year ended December 31, 2008 to $4.9 million for the year ended December 31, 2009. The increase was primarily due to additional capital investments in lab equipment in our North Carolina facility.
Interest Income (Expense). Interest income decreased $1.2 million, or 85%, from $1.4 million for the year ended December 31, 2008 to $0.2 million for the year ended December 31, 2009 due to lower investment balances and lower rates of return on our investments. Interest expense remained relatively unchanged for the year ended 2008 compared to the year ended 2009, as the interest only period on our debt balances did not end until the third quarter of 2009.
Change in Fair Value of Preferred Stock Warrants. Change in fair value of preferred stock warrants decreased $1.9 million due to the decline in the estimated fair value of the preferred stock warrants for the year ended 2008 compared to the year ended 2009, as the estimated fair value of the Company’s preferred stock warrants decreased during that period.
79
Liquidity and Capital Resources
Sources of Liquidity
We have incurred losses since our incorporation in 2003 as a result of our significant research and development expenditures and the lack of any approved products to generate product sales. We have a deficit accumulated during the development stage of $211.2 million as of December 31, 2010, including $48.4 million of cumulative accretion on Redeemable Convertible Preferred Stock through April 2010. We anticipate that we will continue to incur additional losses until such time that we can generate significant sales of our product candidates currently in development or we enter into cash flow positive business transactions. We have funded our operations principally with proceeds from equity offerings and long-term debt. The following table summarizes our equity funding sources as of December 31, 2010:
|
Issue
|
Year
|
Number of Shares (1)
|
Net Proceeds
(in thousands) (2)
|
||||||||||
|
Series A Redeemable Convertible Preferred Stock
|
2004, 2005 | 1,668,311 | $ | 38,910 | |||||||||
|
Series B Redeemable Convertible Preferred Stock
|
2006 | 1,906,009 | 50,040 | ||||||||||
|
Series C Redeemable Convertible Preferred Stock
|
2007, 2008 | 2,077,635 | 54,571 | ||||||||||
|
Initial Public Offering
|
2010 | 6,000,000 | 25,721 | ||||||||||
| 11,651,955 | $ | 169,242 | |||||||||||
____________
|
(1)
|
Number of shares represents the number of shares of common stock into which each series of preferred stock converted at the time of our initial public offering.
|
|
(2)
|
Net proceeds represent gross proceeds received net of transaction costs associated with each equity offering.
|
In March 2011, we closed a private placement transaction pursuant to which we sold securities consisting of 11,079,250 shares of common stock and warrants to purchase 10,460,875 shares of common stock. The purchase price per security was $2.83. The warrants have a term of five years and are immediately exercisable for $2.88 per share. We received net proceeds of approximately $28.9 million.
We have also funded our operations through the use of proceeds received from our long-term debt totaling $34.7 million through December 31, 2010. At December 31, 2010 we had a working capital note with an outstanding principal of $7.3 million, which borrowings have been used for our general working capital needs. In March 2011, we refinanced the outstanding principal balance under that working capital note. Pursuant to the terms of the refinancing, we simultaneously borrowed $5.0 million from the lender and repaid the then outstanding principal amount of $4.5 million. In addition, we have loans to fund equipment and other asset purchases with outstanding principal amounts of $1.5 million as of December 31, 2010.
Cash, cash equivalents and short-term investments at December 31, 2010 were $12.0 million, representing 49.6% of total assets.
Based upon our current expected level of operating expenditures and debt repayment, we expect to be able to fund our operations through May 2012. This period could be shortened if there are any significant increases in planned spending on development programs than anticipated or other unforeseen events. We will need to raise additional funds through collaborative arrangements, public or private sales of debt or equity securities, commercial loan facilities, or some combination thereof. There is no assurance that other financing will be available when needed to allow us to continue our operations or if available, on terms acceptable to us.
80
Equity Financings
In March 2011, we closed a private placement transaction pursuant to which we sold securities consisting of 11,079,250 shares of common stock and warrants to purchase 10,460,875 shares of common stock. The purchase price per security was $2.83. The warrants have a term of five years and are immediately exercisable for $2.88 per share. Net proceeds received after payment of placement agent fees and related expenses were approximately $28.9 million.
In April 2010, we completed an initial public offering, selling 6,000,000 shares of common stock at an initial public offering price of $5.00 per share resulting in gross proceeds of $30.0 million. Net proceeds received after underwriting fees and offering expenses were approximately $25.7 million
Cash Flows
The following table summarizes our cash flows from operating, investing and financing activities for each of the past three years ended:
|
Year Ended December 31,
|
|||||||||||
|
2008
|
2009
|
2010
|
|||||||||
|
(in thousands)
|
|||||||||||
|
Statement of Cash Flows Data:
|
|||||||||||
|
Total cash provided by (used in):
|
|||||||||||
|
Operating activities
|
$
|
(34,901
|
)
|
$
|
(26,021
|
)
|
$
|
(19,412
|
)
|
||
|
Investing activities
|
25,270
|
1,876
|
2,347
|
|
|||||||
|
Financing activities
|
21,842
|
(4,995
|
)
|
12,232
|
|||||||
|
Increase (decrease) in cash and cash equivalents
|
$
|
12,211
|
|
$
|
(29,140
|
)
|
$
|
(4,833
|
)
|
||
Operating Activities
Cash used in operating activities decreased $6.6 million for the year ended December 31, 2010, compared to the year ended December 31, 2009, primarily due to a decrease in our net loss. Specifically, the decrease in our net loss relates primarily to a decrease in research and development expense of $5.0 million associated with our preclinical and clinical studies, and a decrease in cash interest expense of $1.3 million due to lower average debt balances and the end of interest-only payments in 2009.
Cash used in operating activities decreased $8.9 million in the year ended December 31, 2009, compared to the year ended December 31, 2008, primarily due to a decrease in our net loss. Specifically, the decrease in our net loss relates primarily to a decrease in research and development expense of $10.0 million associated with preclinical and clinical studies and our decrease in general and administrative expense of $1.9 million related to personnel-related costs and legal, audit, and other external services. The decreased net loss was offset by an increase in operating assets and liabilities of $1.3 million and noncash-interest expense of $2.0 million.
81
Investing Activities
Cash provided by investing activities increased $0.5 million for the year ended December 31, 2010, compared to the year ended December 31, 2009, primarily due to an increase in net purchases, sales, and redemptions of short-term investments of $0.4 million and a decrease of $0.1 million in cash paid for property and equipment.
Cash provided by investing activities decreased $23.4 million in the year ended December 31, 2009, compared to the year ended December 31, 2008, primarily due to lower net sales and redemptions and purchases of short-term investments of $25.8 million, offset by a decrease of $2.4 million for cash paid for property and equipment.
Financing Activities
Cash provided by financing activities increased $17.2 million for the year ended December 31, 2010, compared to the year ended December 31, 2009, due to net proceeds received from our initial public offering of $25.7 million, offset by an increase in payments of long-term debt of $7.6 million primarily resulting from the end of interest-only payments on our long-term debt in 2009, and a decrease of $0.8 million in proceeds from long-term debt.
Cash used in financing activities was $5.0 million for the year ended December 31, 2009, compared to cash provided by financing activities of $21.8 million for the year ended December 31, 2008. The 2008 period included proceeds from the issuance Series C redeemable convertible preferred stock, net of transaction costs, of $21.4 million. The remaining decrease of $5.4 million in cash provided by financing activities was primarily due to an increase in payments of long-term debt of $5.2 million
Potential Future Milestone Payments
In 2003, we executed a license agreement with Children’s Medical Center Corporation, or CMCC, whereby CMCC granted to us the utilization of certain patent rights. We are obligated to make payments to CMCC upon the occurrence of various clinical milestones. During the fourth quarter of 2006, we achieved two of our clinical milestones for the first licensed product launched, as defined in the agreement, and paid $350,000, which was recorded as research and development expense for the year ended December 31, 2006. No further milestones payments have been made since 2006. Upon the successful completion of certain milestones, we will be obligated, under our current agreement, to make future milestone payments for the first licensed product. As of December 31, 2010, we had no payment obligations to CMCC under this agreement. In addition, upon commercialization of the licensed product, we will pay CMCC royalties based on net sales of products covered by the license by us, our affiliates and our sublicensees in all countries, except for those for which there is no valid patent claim, until the later of the expiration, on a country-by-country basis, of the last patent right and October 2018.
82
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements or relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities.
Contractual Obligations
The following table summarizes our contractual obligations as of December 31, 2010:
|
Payments due by period
|
||||||||||||||||||||
|
Contractual Obligations (1)(2)
|
Total
|
2011
|
2012 and 2013
|
2014 and 2015
|
2016 and
thereafter
|
|||||||||||||||
|
Debt obligations(3)
|
$ | 8,750,242 | $ | 4,107,798 | $ | 4,434,341 | $ | 208,103 | $ | — | ||||||||||
|
Interest payments on debt(3)
|
1,291,781 | 676,405 | 613,338 | 2,038 | — | |||||||||||||||
|
Operating lease obligations
|
4,501,775 | 885,096 | 1,693,337 | 1,773,341 | 150,001 | |||||||||||||||
|
Other
|
800,000 | 800,000 | — | — | — | |||||||||||||||
|
Total
|
$ | 15,343,798 | $ | 6,469,299 | $ | 6,741,016 | $ | 1,983,482 | $ | 150,001 | ||||||||||
|
(1)
|
This table does not include any milestone payments which may become payable to third parties under license agreements, including milestones that may be payable to CMCC, as the timing and likelihood of such payments are not known. We will be required to pay CMCC development milestones aggregating approximately $6.9 million, which includes $350,000 we have paid to date, if we develop and obtain regulatory approval of a licensed product in each of the four subfields covered by our license agreement. Also, if cumulative net sales of all licensed products reach a certain level, we will be required to make a one-time sales milestone payment of $2.0 million. A portion of milestone payments we make will be credited against our future royalty payments.
|
|
(2)
|
This table does not include any license maintenance fees which may become payable to Wake Forest University Health Sciences, or WFUHS, as the timing and likelihood of such payments are not known. We will be required to pay certain license maintenance fees with respect to each product covered by new development patents under our license with WFUHS, commencing two years after we initiate the first animal study conducted under cGLP, with respect to such product. These license maintenance fees, which are in the low six figure range with respect to each product, will be creditable against royalties we are obligated to pay to WFUHS for such product.
|
|
(3)
|
The payments due by period for debt obligations and interest payments on debt reflect the debt refinancing entered into in March 2011 with the lender of our working capital note.
|
Working Capital Note
In March 2011, we refinanced the outstanding debt owed to one of our lenders. Pursuant to the terms of the refinancing, we simultaneously borrowed $5.0 million from the lender and repaid the then outstanding principal amount of $4.5 million. We are obligated to make interest-only payments through January 2012, followed by 24 monthly payments of principal and interest at an interest rate of 11.75% per annum. In October 2008, we refinanced the terms of the working capital note with a then principal amount of $20 million, which was previously refinanced in 2006 and 2007. Under the 2008 refinancing, the repayment terms of $14.2 million principal amount of the working capital note were extended to add an additional six-month period of interest-only payments through July 2009 followed by 24 monthly payments of principal and accrued interest at an annual interest rate of 12.26%. The repayment of $5.8 million of the working capital note were extended to add a 14-month period of interest-only payments through January 2010 followed by 20 monthly payments of principal and accrued interest at an annual interest rate of 12.26%.
83
Borrowings under the Working Capital Note are secured by all of our assets, except for intellectual property and permitted liens that have priority, including liens on certain equipment acquired to secure the purchase price or lease obligation, as defined in the loan agreement.
Equipment and Supplemental Working Capital Note
We have an additional loan with another lender to fund equipment and other asset purchases. As of December 31, 2010, the outstanding balance of this loan was $1.0 million, of which $0.9 million and $0.1 million are scheduled to be repaid during 2011 and 2012, respectively. The original amount borrowed of $9.8 million consisted of a $7.4 million note to purchase equipment, or the equipment note, and a $2.4 million note for other soft costs, or supplemental working capital note, for purchases from July 2005 through December 2009. In October 2007, we refinanced the equipment and supplemental working capital notes with a then carrying amount of $4.6 million. Under the terms of the refinanced equipment and supplemental working capital notes, we had a 12-month period of interest-only payments through October 2008 followed by 24 monthly payments of principal and accrued interest at an annual interest rate of 10.44%. During 2008, we executed an additional loan in the amount of $1.1 million on the equipment and supplemental working capital notes. During 2009, we executed an additional loan in the amount of $0.5 million on the equipment and supplemental working capital notes. The equipment note bears interest at an average rate of 11.69% and matures over 36 to 48 months. The supplemental working capital note bears interest at an average rate of 11.67% and matures over 36 months. The equipment note and the supplemental working capital note are secured by a first priority lien on equipment and assets purchased with the proceeds from the notes.
Machinery and Equipment Loan
In December 2007, we executed a $1.65 million agreement with the Commonwealth of Pennsylvania to fund machinery and equipment and other asset purchases through December 31, 2009. As of December 31, 2010, the outstanding balance of this loan was $0.5 million, and the final monthly principal and interest payment of is due January 1, 2012. On December 31, 2007 we borrowed $1.3 million under the loan to fund equipment purchases. In March 2009, we borrowed an additional $0.3 million under the loan to fund equipment purchases. Under the terms of the loan, we have a four year period of payments of principal and accrued interest at an annual interest rate of 5% until September 2011 and 5.25% from September 2011 through maturity of the loan. The loan is secured by a first priority lien on equipment and assets purchased with the proceeds from the loan.
84
Operating Leases
In 2006, we entered into a ten-year, non-cancelable lease agreement for space for our corporate offices and full-scale manufacturing facility located in East Norriton, Pennsylvania. Under the lease agreement, we began leasing additional space in the same building in March 2011. We are currently subleasing this additional warehouse space to a third party. The lease will expire by its terms in February 2016. Effective March 2011, the lease agreement for our corporate headquarters required us to provide security and restoration deposits totaling $2.2 million to the landlord, an increase from the prior amount of $1.7 million. Until January 2011, we obtained letters of credit from a bank in favor of the landlord to satisfy the obligation of $1.7 million. In January 2011, we deposited $1.0 million with the landlord. As of March 2011, an outstanding letter of credit is satisfying the remaining obligation of $1.2 million. The letter of credit is collateralized by an account held at the bank. If the bank determines the collateral to be insufficient, the bank has the right to demand additional collateral. If we fail to provide additional collateral, the bank has the right to withdraw the letter of credit. In that event, the landlord would have the right to require us to deposit cash of up to $1.2 million in an account to satisfy our deposit obligation.
In June 2005, we entered into a six-year, non-cancelable lease agreement for laboratory space for our research and development projects in Winston-Salem, North Carolina. The lease agreement includes the right to lease additional contiguous space, which we executed in February 2007. The initial term for the lease for this facility will expire in September 2011. In March 2011, we provided notice to our landlord to extend the term of this lease for an additional five-year period.
Other Contractual Obligations
In January 2006, we entered into a research agreement with WFUHS, which was amended in September 2006 and extended in September 2010 for an additional three-year period. Under the research agreement, WFUHS agreed to perform sponsored research in return for quarterly payments. Under the extended agreement, we are currently obligated to make payments of $800,000 for 2011 research activities of WFUHS. Payments for 2012 and 2013 research activities of WFUHS under the extended agreement will be determined by us prior to the beginning of each year, respectively. The sponsored research involves the experiments and studies set out in annual research plans and milestones established under the agreement. Either party may terminate the agreement upon a material, uncured breach by the other party upon 30 days written notice. We may terminate the agreement at any time, with or without cause, upon 90 days prior written notice. If we terminate the agreement, it is required to pay to WFUHS all reasonable direct costs and all noncancelable obligations incurred by WFUHS, provided that such liability is limited to $625,000.
We have entered into agreements with consultants and clinical research organizations which are partially responsible for conducting and monitoring our clinical trials for our product candidates. We have also entered into agreements with consultants and clinical research organizations that are responsible for work in our preclinical studies, public relations and other areas in the ordinary course of our business. These contractual obligations have been excluded from the contractual obligations table elsewhere in this section, because we may terminate these at any time without penalty.
85
Recent Accounting Pronouncements
In January 2010, the FASB issued Accounting Standards Update (ASU) No. 2010-06, Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements. The guidance requires entities to disclose significant transfers in and out of fair value hierarchy levels and the reasons for the transfers. Additionally, the guidance clarifies that a reporting entity should provide fair value measurements for each class of assets and liabilities and disclose the inputs and valuation techniques used for fair value measurements using significant other observable inputs (Level 2) and significant unobservable inputs (Level 3). Level 3 reconciliations should present separately information about purchases, sales, issuances and settlements. To date, we have not had any assets or liabilities that transferred in or out of fair value hierarchy levels, and as such, we are not currently subject to this guidance. This guidance is effective for interim and annual periods beginning after December 15, 2009, except for the disclosures about purchases, sales, issuances and settlements in the Level 3 reconciliations, which is effective for fiscal years beginning after December 15, 2010. This guidance did not have an impact on our results of operations or financial position. We adopted certain of the relevant disclosure provisions of ASU 2010-06 on January 1, 2010 and adopted certain other provisions on January 1, 2011. We believe the enhanced Level 3 disclosures will not have an impact on our results of operations or financial position.
In February 2010, the FASB issued ASU No. 2010-09, Subsequent Events (Topic 855). The guidance requires an SEC filer to evaluate subsequent events through the date the financial statements are issued but no longer requires an SEC filer to disclose the date through which the subsequent event evaluation occurred. We adopted this guidance upon issuance and it had no impact on our results of operations or financial position.
Item 7A.Quantitative and Qualitative Disclosures about Market Risk
The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. Due to the nature of these investments, we believe that we are not subject to any material market risk exposure. As of December 31, 2010, we had cash and cash equivalents of $12.0 million.
Item 8. Financial Statements and Supplementary Data
This information required by this Item is included in our Financial Statements and Supplementary Data listed in Item 15 (a) (1) of Part IV of this Annual Report on Form 10-K.
86
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Dismissal of Auditors
On September 17, 2009, we dismissed KPMG LLP (KPMG) as our principal accountants. This decision was approved by our board of directors.
The audit reports of KPMG on the financial statements of the Company as of December 31, 2007 and 2008 and for each of the years in the three-year period ended December 31, 2008 and for the period from July 10, 2003 (inception) through December 31, 2008, did not contain an adverse opinion or disclaimer of opinion, and were not qualified or modified as to uncertainty, audit scope or accounting principles.
In connection with the audits for the years ended December 31, 2007 and 2008, and through September 17, 2009, there were (1) no disagreements with KPMG on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreement(s), if not resolved to the satisfaction of KPMG, would have caused KPMG to make reference to the subject matter of the disagreement(s) in connection with KPMG’s report, or (2) no “reportable events” as such term is defined in Item 304(a)(1)(v) of Regulation S-K.
We requested KPMG to furnish a letter addressed to the Securities and Exchange Commission stating whether it agrees with the above statements made by us and, if not, stating the respects in which it does not agree. A copy of this letter, dated as of December 23, 2009, which states that it agrees with these statements, was filed as exhibit 16.1 to our Registration Statement on Form S-1 (Registration No. 333-164011) filed with the SEC.
Engagement of New Auditors
On September 17, 2009, our board of directors approved the engagement of Ernst & Young LLP as our independent auditors. Ernst & Young LLP has reported on the financial statements as of and for the years ended December 31, 2010 and 2009 included in this annual report on Form 10-K. Prior to the year ended December 31, 2009, we did not consult Ernst & Young LLP on any accounting or financial matters.
Item 9A.Controls and Procedures
Evaluation of Disclosure Controls and Procedures and Changes in Internal Control over Financial Reporting
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) of the Securities Exchange Act of 1934, as amended (the Exchange Act)) as of the end of the period covered by this report. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this report were effective in ensuring that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. We believe that a control system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the control system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
87
There was no change in our internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act) that occurred during the period covered by this report that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Control over Financial Reporting and Attestation Report of the Registered Accounting Firm
This annual report does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of the company’s registered public accounting firm due to a transition period established by rules of the Securities and Exchange Commission for newly public companies.
Item 9B.Other Information
None
88
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
Item 11. Executive Compensation
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
Item 13. Certain Relationships and Related Party Transactions, and Director Independence
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
Item 14. Principal Accounting Fees and Services
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
89
PART IV
Item 15. Exhibits and Financial Statement Schedules
|
|
(a)
|
The following documents are filed as part of this Annual Report on Form 10-K:
|
|
|
(1)
|
Financial Statements
|
See index to financial statements on page F-1
|
|
(2)
|
Financial Statement Schedules
|
All schedules to the financial statements are omitted as the required information is either inapplicable or presented in the financial statements or notes thereto.
|
|
(3)
|
Exhibits
|
The information required by this Item is set forth in the Exhibit Index hereto which is incorporated herein by reference.
|
|
(b)
|
Exhibits
|
The information required by this Item is set forth in the Exhibit Index hereto which is incorporated herein by reference.
90
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
TENGION, INC.
|
|
|
By: /s/ Steven Nichtberger
|
|
|
Steven Nichtberger, MD
|
|
|
President and Chief Executive Officer
|
POWER OF ATTORNEY
We, the undersigned officers and directors of Tengion, Inc., hereby severally constitute and appoint Steven Nichtberger and A. Brian Davis, and each of them singly (with full power to each of them to act alone), our true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution in each of them for him and in his name, place and stead, and in any and all capacities, to sign for us and in our names in the capacities indicated below any and all amendments to this report, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as full to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities, and on the dates indicated below.
|
Signature
|
Title
|
Date
|
|
/s/ Steven Nichtberger
Steven Nichtberger, MD
|
President and Chief Executive Officer, Director (Principal Executive Officer)
|
March 30, 2011
|
|
/s/ A. Brian Davis
A. Brian Davis
|
Chief Financial Officer and Vice President of Finance (Principal Financial and Accounting Officer)
|
March 30, 2011
|
|
/s/ David I. Scheer
David I. Scheer
|
Chairman of the Board of Directors
|
March 30, 2011
|
91
|
/s/ Carl-Johan Dalsgaard
Carl-Johan Dalsgaard, MD, PhD
|
Director
|
March 30, 2011
|
|
/s/ Brenda D. Gavin
Brenda D. Gavin, DVM
|
Director
|
March 30, 2011
|
|
/s/ Richard Kuntz
Richard Kuntz, MD
|
Director
|
March 30, 2011
|
|
/s/ Gary J. Kurtzman
Gary J. Kurtzman, MD
|
Director
|
March 30, 2011
|
|
/s/ Lorin J. Randall
Lorin J. Randall
|
Director
|
March 30, 2011
|
92
TENGION, INC.
INDEX TO FINANCIAL STATEMENTS
|
Page
|
|
|
Report of Ernst & Young LLP, Independent Registered Public Accounting Firm
|
|
|
Report of KPMG LLP, Independent Registered Public Accounting Firm
|
|
|
Balance Sheets
|
|
|
Statements of Operations
|
|
|
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
|
|
|
Statements of Cash Flows
|
|
|
Notes to the Financial Statements
|
F- 1
Report of Independent Registered Public Accounting Firm
Board of Directors
Tengion, Inc.
We have audited the accompanying balance sheets of Tengion, Inc. (the Company) (a development stage company), as of December 31, 2010 and 2009, and the related statements of operations, redeemable convertible preferred stock and stockholders’ equity (deficit), and cash flows for the years ended December 31, 2010 and 2009, and for the period from July 10, 2003 (inception) through December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. The financial statements for year ended December 31, 2008 and for the period from July 10, 2003 (inception) through December 31, 2008 were audited by other auditors whose report dated May 29, 2009, expressed an unqualified opinion on those statements. The financial statements for the period July 10, 2003 (inception) through December 31, 2008 include a net loss of $107,351,001. Our opinion on the statements of operations, redeemable convertible preferred stock and stockholders’ equity (deficit) and cash flows for the period July 10, 2003 (inception) through December 31, 2010, insofar as it relates to amounts for prior periods through December 31, 2008, is based solely on the report of other auditors.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, based on our audits and the report of other auditors, the financial statements referred to above present fairly, in all material respects, the financial position of the Company at December 31, 2010 and 2009, and the results of its operations and its cash flows for the years then ended and the period from July 10, 2003 (inception) through December 31, 2010, in conformity with accounting principles generally accepted in the United States.
/s/ ERNST & YOUNG LLP
Philadelphia, Pennsylvania
March 30, 2011
F- 2
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Tengion, Inc.:
We have audited the accompanying statements of operations, redeemable convertible preferred stock and stockholders’ deficit, and cash flows of Tengion, Inc. (a development-stage company) (the Company) for the year ended December 31, 2008 and for the period from July 10, 2003 (inception) through December 31, 2008. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the results of operations and the cash flows of Tengion, Inc. for the year ended December 31, 2008 and for the period from July 10, 2003 (inception) through December 31, 2008, in conformity with U.S. generally accepted accounting principles.
/s/ KPMG LLP
Philadelphia, Pennsylvania
May 29, 2009, except as to
Note 3(m), which is as of March 24, 2010
F- 3
TENGION, INC.
(A Development-Stage Company)
Balance Sheets
|
December 31,
|
||||||||
|
2009
|
2010
|
|||||||
|
Assets
|
|
|||||||
|
Current assets:
|
|
|||||||
|
Cash and cash equivalents
|
|
$
|
16,804,129
|
|
$
|
11,971,660
|
||
|
Short-term investments
|
|
2,499,013
|
|
—
|
||||
|
Deferred equity offering costs
|
|
1,104,708
|
|
40,900
|
||||
|
Prepaid expenses and other
|
|
396,105
|
|
492,205
|
||||
|
Total current assets
|
|
20,803,955
|
|
12,504,765
|
||||
|
Property and equipment, net
|
|
16,243,053
|
|
11,492,577
|
||||
|
Other assets
|
|
191,385
|
|
147,091
|
||||
|
|
||||||||
|
Total assets
|
|
$
|
37,238,393
|
|
$
|
24,144,433
|
||
|
|
||||||||
|
Liabilities and Stockholders’ Equity (Deficit)
|
|
|||||||
|
Current liabilities:
|
|
|||||||
|
Current portion of long-term debt
|
|
$
|
13,196,376
|
|
$
|
4,016,174
|
||
|
Accounts payable
|
|
1,854,481
|
|
1,193,625
|
||||
|
Accrued expenses
|
2,986,797
|
2,843,056
|
||||||
|
Other current liabilities
|
|
—
|
|
205,000
|
||||
|
Total current liabilities
|
|
18,037,654
|
|
8,257,855
|
||||
|
Long-term debt
|
|
8,640,402
|
|
4,585,493
|
||||
|
Warrant liability
|
|
314,431
|
|
—
|
||||
|
Other liabilities
|
|
403,477
|
|
241,165
|
||||
|
Total liabilities
|
|
27,395,964
|
|
13,084,513
|
||||
|
|
||||||||
|
Commitments and contingencies (note 12)
|
|
—
|
—
|
|||||
|
Redeemable convertible preferred stock, $0.001 par value, authorized 83,785,721 shares at December 31, 2009 and no shares authorized at December 31, 2010; issued and outstanding 81,954,127 shares at December 31, 2009 and no shares issued or outstanding at December 31, 2010
|
|
187,916,168
|
|
—
|
||||
|
Stockholders’ equity (deficit):
|
|
|||||||
|
Preferred stock, $0.001 par value; authorized 10,000,000 shares at December 31, 2010; no shares were issued or outstanding at December 31, 2009 or December 31, 2010
|
—
|
—
|
||||||
|
Common stock, $0.001 par value; authorized 90,000,000 shares at December 31, 2010; issued and outstanding 701,581 and 12,385,793 shares at December 31, 2009 and December 31, 2010, respectively
|
|
702
|
|
12,386
|
||||
|
Additional paid-in capital
|
|
3,516,031
|
|
222,230,864
|
||||
|
Deficit accumulated during the development stage
|
|
(181,590,472
|
)
|
(211,183,330
|
)
|
|||
|
Total stockholders’ equity (deficit)
|
|
(178,073,739
|
)
|
11,059,920
|
||||
|
Total liabilities and stockholders’ equity (deficit)
|
|
$
|
37,238,393
|
|
$
|
24,144,433
|
||
The accompanying notes are an integral part of these financial statements.
F- 4
TENGION, INC.
(A Development-Stage Company)
Statements of Operations
|
Year Ended December 31,
|
Period from
July 10, 2003
(inception)
through
December 31,
|
Period from
July 10, 2003
(inception)
through
December 31,
|
|||||||||||||||||
|
2008
|
2009
|
2010
|
2008 | 2010 | |||||||||||||||
|
Operating expenses:
|
|||||||||||||||||||
|
Research and development
|
$
|
27,947,270
|
|
$
|
17,948,060
|
|
$
|
12,855,371
|
|
$
|
73,760,688
|
|
$
|
104,564,119
|
|
||||
|
General and administrative
|
7,467,407
|
|
5,526,800
|
|
6,032,126
|
23,143,272
|
|
34,702,198
|
|
||||||||||
|
Depreciation
|
4,715,448
|
|
4,937,092
|
|
4,861,909
|
|
10,211,647
|
|
20,010,648
|
|
|||||||||
|
Total operating expenses
|
(40,130,125
|
)
|
(28,411,952
|
)
|
(23,749,406
|
)
|
(107,115,607
|
)
|
(159,276,965
|
)
|
|||||||||
|
Interest income
|
1,415,423
|
|
211,045
|
|
62,153
|
|
8,185,182
|
|
8,458,380
|
|
|||||||||
|
Interest expense
|
(3,621,292
|
)
|
(3,468,002
|
)
|
(2,104,543
|
)
|
(8,467,630
|
)
|
(14,040,175
|
)
|
|||||||||
|
Change in value of preferred stock warrants
|
(56,974
|
)
|
1,823,873
|
191,527
|
47,054
|
2,062,454
|
|||||||||||||
|
Net loss
|
(42,392,968
|
)
|
(29,845,036
|
)
|
(25,600,269
|
)
|
$
|
(107,351,001
|
)
|
$
|
(162,796,306
|
)
|
|||||||
|
Accretion of redeemable convertible preferred stock to redemption value
|
(11,753,958
|
)
|
(14,059,265
|
)
|
(3,992,589
|
)
|
|||||||||||||
|
Net loss attributable to common stockholders
|
$
|
(54,146,926
|
)
|
$
|
(43,904,301
|
)
|
$
|
(29,592,858
|
)
|
||||||||||
|
Basic and diluted net loss attributable to common stockholders per share
|
$
|
(80.16
|
)
|
$
|
(62.95
|
)
|
$
|
(3.22
|
)
|
||||||||||
|
Weighted average common stock outstanding – basic and diluted
|
675,461
|
|
697,448
|
|
9,196,920
|
|
|||||||||||||
The accompanying notes are an integral part of these financial statements.
F- 5
TENGION, INC.
(A Development-Stage Company)
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
|
Stockholders' equity (deficit)
|
|||||||||||||||||||||||||||||
|
Redeemable
convertible
preferred stock
|
Common stock
|
Additional
paid-in
capital
|
Deferred
compensation
|
Deficit
accumulated
during the
development
stage
|
Total
|
||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
||||||||||||||||||||||||||
|
Balance, July 10, 2003
|
—
|
$
|
—
|
—
|
|
$
|
—
|
|
$
|
—
|
|
$
|
—
|
|
$
|
—
|
|
$
|
—
|
|
|||||||||
|
Issuance of common stock to initial stockholder
|
—
|
—
|
2,000,000
|
|
2,000
|
|
(1,999
|
)
|
—
|
|
—
|
|
1
|
|
|||||||||||||||
|
Effect of reverse stock split (see Note 3(m))
|
—
|
—
|
(1,862,068
|
)
|
(1,862
|
)
|
1,862
|
|
—
|
|
—
|
|
—
|
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
|
(1,031,756
|
)
|
(1,031,756
|
)
|
|||||||||||||||
|
Balance, December 31, 2003
|
—
|
—
|
137,932
|
|
138
|
|
(137
|
)
|
—
|
|
(1,031,756
|
)
|
(1,031,755
|
)
|
|||||||||||||||
|
Issuance of Series A Redeemable Convertible Preferred stock at $1.61683 per share, net of expenses
|
18,740,371
|
30,125,610
|
—
|
|
—
|
|
—
|
|
—
|
|
—
|
|
—
|
|
|||||||||||||||
|
Conversion of notes payable, including interest
|
2,203,206
|
3,562,211
|
—
|
|
—
|
|
—
|
|
—
|
|
—
|
|
—
|
|
|||||||||||||||
|
Issuance of restricted common stock to employees and nonemployees
|
—
|
—
|
240,318
|
|
240
|
|
336,041
|
|
(335,474
|
)
|
—
|
|
807
|
|
|||||||||||||||
|
Issuance of common stock to consultants
|
—
|
—
|
140,003
|
|
140
|
|
20,960
|
|
—
|
|
—
|
|
21,100
|
|
|||||||||||||||
|
Issuance of common stock to convertible noteholders
|
—
|
—
|
92,682
|
|
93
|
|
67,098
|
|
—
|
|
—
|
|
67,191
|
|
|||||||||||||||
|
Issuance of options to purchase common stock to consultants for services rendered
|
—
|
—
|
—
|
|
—
|
|
14,105
|
|
(14,105
|
)
|
—
|
|
—
|
|
|||||||||||||||
|
Amortization of deferred compensation
|
—
|
—
|
—
|
|
—
|
|
—
|
|
22,554
|
|
—
|
|
22,554
|
|
|||||||||||||||
|
Change in value of restricted common stock subject to vesting
|
—
|
—
|
—
|
|
—
|
|
11,400
|
|
(11,400
|
)
|
—
|
|
—
|
|
|||||||||||||||
|
Accretion of Series A Redeemable Convertible Preferred stock to redemption value
|
—
|
1,035,205
|
—
|
|
—
|
|
—
|
|
—
|
|
(1,035,205
|
)
|
(1,035,205
|
)
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
|
(2,438,912
|
)
|
(2,438,912
|
)
|
|||||||||||||||
|
Balance, December 31, 2004
|
20,943,577
|
34,723,026
|
610,935
|
|
611
|
|
449,467
|
|
(338,425
|
)
|
(4,505,873
|
)
|
(4,394,220
|
)
|
|||||||||||||||
|
Issuance of Series A Redeemable Convertible Preferred stock at $1.61683 per share, net of expenses
|
3,247,095
|
5,222,430
|
—
|
|
—
|
|
—
|
|
—
|
|
—
|
|
—
|
|
|||||||||||||||
|
Issuance of restricted common stock to employees and nonemployees at $2.32 per share
|
—
|
—
|
60,477
|
|
60
|
|
140,241
|
|
(139,424
|
)
|
—
|
|
877
|
|
|||||||||||||||
|
Issuance of warrants to purchase preferred stock to noteholders
|
—
|
—
|
—
|
|
—
|
|
681,026
|
|
—
|
|
—
|
|
681,026
|
|
|||||||||||||||
|
Issuance of options to purchase common stock to consultants for services rendered
|
—
|
—
|
—
|
|
—
|
|
6,693
|
|
(6,693
|
)
|
—
|
|
—
|
|
|||||||||||||||
|
Amortization of deferred compensation
|
—
|
—
|
—
|
|
—
|
|
—
|
|
111,688
|
|
—
|
|
111,688
|
|
|||||||||||||||
|
Accretion of Series A Redeemable Convertible Preferred stock to redemption value
|
—
|
3,164,308
|
—
|
|
—
|
|
—
|
|
—
|
|
(3,164,308
|
)
|
(3,164,308
|
)
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
|
(9,626,910
|
)
|
(9,626,910
|
)
|
|||||||||||||||
|
Balance, December 31, 2005
|
24,190,672
|
$
|
43,109,764
|
671,412
|
|
$
|
671
|
|
$
|
1,277,427
|
|
$
|
(372,854
|
)
|
$
|
(17,297,091
|
)
|
$
|
(16,391,847
|
)
|
|||||||||
The accompanying notes are an integral part of these financial statements.
F- 6
TENGION, INC.
(A Development-Stage Company)
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
|
Stockholders' equity (deficit)
|
||||||||||||||||||||||||||||
|
Redeemable
convertible
preferred stock
|
Common stock
|
Additional
paid-in
capital
|
Deferred
compensation
|
Deficit
accumulated
during the
development
stage
|
Total
|
|||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
|||||||||||||||||||||||||
|
Issuance of Series B Redeemable Convertible Preferred stock at $1.82 per share, net of expenses
|
27,637,363
|
$
|
50,040,289
|
—
|
|
$
|
—
|
|
$
|
—
|
|
$
|
—
|
$
|
—
|
|
$
|
—
|
|
|||||||||
|
Issuance of restricted common stock to employees
|
—
|
—
|
3,449
|
|
3
|
|
47
|
|
—
|
—
|
|
50
|
|
|||||||||||||||
|
Issuance of common stock upon exercise of options
|
—
|
—
|
3,665
|
|
4
|
|
8,496
|
|
8,500
|
|
||||||||||||||||||
|
Repurchased nonvested restricted stock
|
—
|
—
|
(14,120
|
)
|
(14
|
)
|
(191
|
)
|
—
|
—
|
|
(205
|
)
|
|||||||||||||||
|
Reclassification of deferred compensation
|
—
|
—
|
—
|
|
—
|
|
(372,854
|
)
|
372,854
|
—
|
|
—
|
|
|||||||||||||||
|
Reclassification of warrants to purchase preferred stock (note 9)
|
—
|
—
|
—
|
|
—
|
|
(681,026
|
)
|
—
|
—
|
|
(681,026
|
)
|
|||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
—
|
|
—
|
|
399,559
|
|
—
|
—
|
|
399,559
|
|
|||||||||||||||
|
Accretion of Series A and Series B Redeemable Convertible Preferred stock to redemption value
|
—
|
5,639,599
|
—
|
|
—
|
|
—
|
|
—
|
(5,639,599
|
)
|
(5,639,599
|
)
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
(20,872,911
|
)
|
(20,872,911
|
)
|
|||||||||||||||
|
Balance, December 31, 2006
|
51,828,035
|
98,789,652
|
664,406
|
|
664
|
|
631,458
|
|
—
|
(43,809,601
|
)
|
(43,177,479
|
)
|
|||||||||||||||
|
Issuance of Series C Redeemable Convertible Preferred stock at $1.82 per share, net of expenses
|
18,332,965
|
33,219,379
|
—
|
|
—
|
|
—
|
|
—
|
—
|
|
—
|
|
|||||||||||||||
|
Issuance of common stock upon exercise of options
|
—
|
—
|
15,952
|
|
16
|
|
60,145
|
|
—
|
—
|
|
60,161
|
|
|||||||||||||||
|
Repurchased vested restricted stock
|
—
|
—
|
(5,492
|
)
|
(5
|
)
|
(93,947
|
)
|
—
|
—
|
|
(93,952
|
)
|
|||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
—
|
|
—
|
|
663,948
|
|
—
|
—
|
|
663,948
|
|
|||||||||||||||
|
Accretion of Series A, Series B, and Series C Redeemable Convertible Preferred stock to redemption value
|
—
|
8,742,100
|
—
|
|
—
|
|
—
|
|
—
|
(8,742,100
|
)
|
(8,742,100
|
)
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
(30,987,544
|
)
|
(30,987,544
|
)
|
|||||||||||||||
|
Balance, December 31, 2007
|
70,161,000
|
140,751,131
|
674,866
|
|
675
|
|
1,261,604
|
|
—
|
(83,539,245
|
)
|
(82,276,966
|
)
|
|||||||||||||||
|
Issuance of Series C Redeemable Convertible Preferred stock at $1.82 per share, net of expenses
|
11,793,127
|
21,351,814
|
—
|
|
—
|
|
—
|
|
—
|
—
|
|
—
|
|
|||||||||||||||
|
Issuance of common stock upon exercise of options
|
—
|
—
|
7,531
|
|
8
|
|
28,487
|
|
—
|
—
|
|
28,495
|
|
|||||||||||||||
|
Repurchased vested restricted stock
|
—
|
—
|
(821
|
)
|
(1
|
)
|
(11
|
)
|
—
|
—
|
|
(12
|
)
|
|||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
—
|
|
—
|
|
1,316,724
|
|
—
|
—
|
|
1,316,724
|
|
|||||||||||||||
|
Accretion of Series A, Series B, and Series C Redeemable Convertible Preferred stock to redemption value
|
—
|
11,753,958
|
—
|
|
—
|
|
—
|
|
—
|
(11,753,958
|
)
|
(11,753,958
|
)
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
(42,392,968
|
)
|
(42,392,968
|
)
|
|||||||||||||||
|
Balance, December 31, 2008
|
81,954,127
|
173,856,903
|
681,576
|
|
682
|
|
2,606,804
|
|
—
|
(137,686,171
|
)
|
(135,078,685
|
)
|
|||||||||||||||
|
Issuance of common stock upon exercise of options
|
—
|
—
|
20,005
|
|
20
|
|
54,091
|
|
—
|
—
|
|
54,111
|
|
|||||||||||||||
|
Stock-based compensation expense
|
—
|
—
|
—
|
|
—
|
|
855,136
|
|
—
|
—
|
|
855,136
|
|
|||||||||||||||
|
Accretion of Series A, Series B, and Series C Redeemable Convertible Preferred stock to redemption value
|
—
|
14,059,265
|
—
|
|
—
|
|
—
|
|
—
|
(14,059,265
|
)
|
(14,059,265
|
)
|
|||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
(29,845,036
|
)
|
(29,845,036
|
)
|
|||||||||||||||
|
Balance, December 31, 2009
|
81,954,127
|
$
|
187,916,168
|
701,581
|
|
$
|
702
|
|
$
|
3,516,031
|
|
$
|
—
|
$
|
(181,590,472
|
)
|
$
|
(178,073,739
|
)
|
|||||||||
The accompanying notes are an integral part of these financial statements.
F- 7
TENGION, INC.
(A Development-Stage Company)
Statements of Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit)
| Stockholders' equity (deficit) | ||||||||||||||||||||||||||||||
|
Redeemable
convertible
preferred stock
|
Common stock
|
Additional
paid-in
capital
|
Deferred
compensation
|
Deficit
accumulated
during the
development
stage
|
Total | |||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
|||||||||||||||||||||||||||
|
Issuance of common stock upon exercise of options
|
—
|
$
|
—
|
32,257
|
|
$
|
32
|
|
$
|
14,153
|
|
$
|
—
|
$
|
—
|
|
$
|
14,185
|
|
|||||||||||
|
Stock-based compensation expense
|
—
|
—
|
—
|
|
—
|
|
953,858
|
|
—
|
—
|
|
953,858
|
|
|||||||||||||||||
|
Accretion of Series A, Series B, and Series C Redeemable Convertible Preferred stock to redemption value
|
—
|
3,992,589
|
—
|
|
—
|
|
—
|
|
—
|
(3,992,589
|
)
|
(3,992,589
|
)
|
|||||||||||||||||
|
Conversion of preferred stock to common stock
|
(81,954,127
|
)
|
(191,908,757
|
)
|
5,651,955
|
5,652
|
191,903,105
|
—
|
—
|
191,908,757
|
||||||||||||||||||||
|
Conversion of preferred stock warrants to common stock warrants
|
—
|
—
|
122,904
|
—
|
—
|
122,904
|
||||||||||||||||||||||||
|
Proceeds from initial public offering, net of offering expenses
|
—
|
—
|
6,000,000
|
6,000
|
25,720,813
|
—
|
—
|
25,726,813
|
||||||||||||||||||||||
|
Net loss
|
—
|
—
|
—
|
|
—
|
|
—
|
|
—
|
(25,600,269
|
)
|
(25,600,269
|
)
|
|||||||||||||||||
|
Balance, December 31, 2010
|
—
|
$
|
—
|
12,385,793
|
|
$
|
12,386
|
|
$
|
222,230,864
|
|
$
|
—
|
$
|
(211,183,330
|
)
|
$
|
11,059,920
|
||||||||||||
The accompanying notes are an integral part of these financial statements.
F- 8
TENGION, INC.
(A Development-Stage Company)
Statements of Cash Flows
|
Year ended December 31,
|
Period from July 10, 2003 (inception) through December 31, | Period from July 10, 2003 (inception) through December 31, | ||||||||||||||||||
|
2008
|
2009
|
2010
|
2008
|
2010
|
||||||||||||||||
|
Cash flows from operating activities:
|
||||||||||||||||||||
|
Net loss
|
$ | (42,392,968 | ) | $ | (29,845,036 | ) | $ | (25,600,269 | ) | $ | (107,351,001 | ) | $ | (162,796,306 | ) | |||||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||||||||||
|
Depreciation
|
4,715,448 | 4,937,092 | 4,861,909 | 10,211,647 | 20,010,648 | |||||||||||||||
|
Loss on disposition of property and equipment
|
907 | 101,434 | 1,792 | 15,890 | 119,116 | |||||||||||||||
|
Amortization of net discount on short-term investments
|
208,722 | — | — | (149,150 | ) | (149,150 | ) | |||||||||||||
|
Noncash interest expense (income) and change in fair value of preferred stock warrants
|
623,613 | (1,364,041 | ) | 133,954 | 1,554,000 | 323,913 | ||||||||||||||
|
Noncash rent expense
|
38,365 | 18,770 | 4,188 | 218,208 | 241,166 | |||||||||||||||
|
Stock-based compensation expense
|
1,316,724 | 855,136 | 953,858 | 2,535,568 | 4,344,562 | |||||||||||||||
|
Changes in operating assets and liabilities
|
||||||||||||||||||||
|
Prepaid expenses and other assets
|
958,052 | 71,056 | (136,999 | ) | (759,469 | ) | (825,412 | ) | ||||||||||||
|
Accounts payable
|
(1,381,658 | ) | 968,506 | (631,212 | ) | 815,376 | 1,152,670 | |||||||||||||
|
Accrued expenses and other
|
1,012,069 | (1,763,428 | ) | 1,000,971 | 4,020,781 | 3,258,324 | ||||||||||||||
|
Net cash used in operating activities
|
(34,900,726 | ) | (26,020,511 | ) | (19,411,808 | ) | (88,888,150 | ) | (134,320,469 | ) | ||||||||||
|
Cash flows from investing activities:
|
||||||||||||||||||||
|
Purchases of short-term investments
|
(13,093,838 | ) | (17,432,016 | ) | (33,444,987 | ) | (260,564,847 | ) | (311,441,850 | ) | ||||||||||
|
Sales and redemption of short-term investments
|
41,050,000 | 19,590,000 | 35,944,000 | 256,057,000 | 311,591,000 | |||||||||||||||
|
Cash paid for property and equipment
|
(2,685,787 | ) | (288,640 | ) | (151,818 | ) | (31,152,132 | ) | (31,592,590 | ) | ||||||||||
|
Proceeds from the sale of property and equipment
|
153 | 6,596 | — | 4,710 | 11,306 | |||||||||||||||
|
Net cash provided by (used in) investing activities
|
25,270,528 | 1,875,940 | 2,347,195 | (35,655,269 | ) | (31,432,134 | ) | |||||||||||||
|
Cash flows from financing activities:
|
||||||||||||||||||||
|
Proceeds from sale of redeemable convertible preferred stock, net
|
21,351,814 | — | — | 139,959,522 | 139,959,522 | |||||||||||||||
|
Proceeds from initial public offering, net
|
— | — | 25,734,258 | — | 25,734,258 | |||||||||||||||
|
Issuance of common stock
|
28,495 | 54,111 | 14,185 | 98,897 | 167,193 | |||||||||||||||
|
Repurchase of restricted stock
|
(12 | ) | — | — | (94,169 | ) | (94,169 | ) | ||||||||||||
|
Issuance costs related to long-term debt
|
— | — | — | (85,340 | ) | (85,340 | ) | |||||||||||||
|
Payment of deferred equity offering costs
|
— | (7,445 | ) | — | — | (7,445 | ) | |||||||||||||
|
Proceeds from long-term debt
|
1,136,052 | 840,595 | — | 33,853,702 | 34,694,297 | |||||||||||||||
|
Payments on long-term debt
|
(674,751 | ) | (5,882,486 | ) | (13,516,299 | ) | (3,245,268 | ) | (22,644,053 | ) | ||||||||||
|
Net cash provided by (used in) financing activities
|
21,841,598 | (4,995,225 | ) | 12,232,144 | 170,487,344 | 177,724,263 | ||||||||||||||
|
Net increase (decrease) in cash and cash equivalents
|
12,211,400 | (29,139,796 | ) | (4,832,469 | ) | 45,943,925 | 11,971,660 | |||||||||||||
|
Cash and cash equivalents, beginning of period
|
33,732,525 | 45,943,925 | 16,804,129 | — | — | |||||||||||||||
|
Cash and cash equivalents, end of period
|
$ | 45,943,925 | $ | 16,804,129 | $ | 11,971,660 | $ | 45,943,925 | $ | 11,971,660 | ||||||||||
|
Supplemental cash flow disclosures:
|
||||||||||||||||||||
|
Noncash investing and financing activities:
|
||||||||||||||||||||
|
Conversion of note principal to Series A Redeemable Convertible Preferred Stock
|
$ | — | $ | — | $ | — | $ | 3,562,211 | $ | 3,562,211 | ||||||||||
|
Convertible note issued to initial stockholder for consulting expense
|
— | — | — | 210,372 | 210,372 | |||||||||||||||
|
Warrants issued with long-term debt
|
344,187 | 8,742 | — | 2,176,616 | 2,185,358 | |||||||||||||||
|
Conversion of redeemable convertible preferred stock into 5,651,955 shares of common stock
|
— | — | 191,908,757 | — | 191,908,757 | |||||||||||||||
|
Conversion of warrant liability
|
— | — | 122,904 | — | 122,904 | |||||||||||||||
|
Noncash property and equipment additions
|
52,185 | 79,650 | 41,056 | 52,185 | 41,056 | |||||||||||||||
|
Cash paid for interest, net of amounts capitalized
|
3,052,222 | 3,008,170 | 1,694,525 | 6,852,831 | 11,555,526 | |||||||||||||||
The accompanying notes are an integral part of these financial statements.
F- 9
TENGION, INC.
(A Development-Stage Company)
Notes to Financial Statements
(1) Background
Tengion, Inc. (the Company) is a regenerative medicine company incorporated in Delaware on July 10, 2003. The Company is focused on discovering, developing, manufacturing and commercializing replacement human neo-organs derived from a patient’s own cells, or autologous cells. Building on clinical and preclinical experience, the Company is leveraging its platform to develop its lead product candidate, the Neo-Urinary Conduit, for bladder cancer patients. The Company intends to develop its technology to address unmet medical needs in urologic, renal, gastrointestinal and vascular diseases and disorders. The Company operates as a single business segment.
(2) Development-Stage Risks and Liquidity
The Company has incurred losses since inception and has a deficit accumulated during the development stage of $211,183,330 as of December 31, 2010, including $48,387,024 of cumulative accretion on the Series A Redeemable Convertible Preferred Stock (Series A), the Series B Redeemable Convertible Preferred Stock (Series B), and the Series C Redeemable Convertible Preferred Stock (Series C). The Company anticipates incurring additional losses until such time, if ever, that it can generate significant sales of its therapeutic product candidates currently in development or enters into cash flow positive business development transactions. In March 2011, the Company closed a private placement transaction pursuant to which the Company received net proceeds of approximately $28.9 million and refinanced its working capital loan. Following completion of these financing activities and based upon its current expected level of operating expenditures and debt repayment, the Company expects to be able to fund its operations through May 2012. Please see Note 15 for additional information. The Company continues to explore external financing alternatives, which will be needed by the Company to fund its operations and to commercially develop its products. In the event financing is not obtained, the Company could pursue headcount reductions and other cost-cutting measures to preserve cash as well as explore the selected sale of assets to generate additional funds. There is no assurance that such financing alternatives will be available when needed, or if available, on terms acceptable to the Company.
The accompanying financial statements have been prepared on a going concern basis, which contemplates the realization of assets and satisfaction of liabilities in the normal course of business. The financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
Operations of the Company are subject to certain risks and uncertainties, including, among others, uncertainty of product candidate development; technological uncertainty; dependence on collaborative partners; uncertainty regarding patents and proprietary rights; comprehensive government regulations; having no commercial manufacturing experience, marketing or sales capability or experience; and dependence on key personnel.
F- 10
(3) Summary of Significant Accounting Policies
(a)Use of Estimates
The preparation of financial statements, in accordance with accounting principles generally accepted in the United States of America, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
(b)Fair Value of Financial Instruments
As of December 31, 2009 and 2010, the carrying amounts of financial instruments held by the Company, which include cash equivalents, short-term investments, prepaid expenses and other current assets, accounts payable, and accrued expenses, approximate fair value due to the short-term nature of those instruments. In addition, the carrying value of the Company’s debt instruments, which do not have readily ascertainable market values, approximate fair value, given that the interest rates on outstanding borrowings approximate market rates.
(c)Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents. As of December 31, 2009 and 2010, cash and cash equivalents included government-backed money market funds, commercial paper, and various bank deposit accounts.
(d)Short-term Investments
The Company classifies investments as available-for-sale or held-to-maturity at the time of purchase and reevaluates such designation as of each balance sheet date. Securities are classified as held-to-maturity when the Company has the positive intent and the ability to hold the securities until maturity. Held-to-maturity investments are recorded at amortized cost, adjusted for the accretion of discounts or premiums. Discounts or premiums are accreted into interest income over the life of the related investment using the straight-line method, which approximates the effective- yield method. Dividend and interest income are recognized when earned. Investments not classified as held-to-maturity are classified as available-for-sale. Available-for-sale securities are carried at fair value with the unrealized gains or losses, included as a separate component of stockholders’ equity (deficit). As of December 31, 2009 and 2010, there were no unrealized gains or losses on available-for-sale securities.
(e)Property and Equipment
Property and equipment are stated at cost. Depreciation is provided over the estimated useful lives of the assets using the straight-line method. The Company uses a life of three years for computer equipment, five years for laboratory and office and warehouse equipment, seven years for furniture and fixtures, and the lesser of the useful life of the asset or the remaining life of the underlying facility lease for leasehold improvements. Expenditures for maintenance, repairs, and betterments that do not prolong the useful life of the asset are charged to expense as incurred. The Company capitalizes interest in connection with the construction of property and equipment.
F- 11
(f)Impairment of Long-Lived Assets
Long-lived assets, such as property and equipment, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, then an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the fair value of the asset. As of December 31, 2009 and 2010, management believes that no modification of the remaining useful lives or write-down of long-lived assets is required.
(g)Research and Development
Research and development costs are charged to expense as incurred. Research and development costs consist of:
|
|
·
|
personnel related expenses, including salaries, benefits, travel, and other related expenses, including stock-based compensation;
|
|
|
·
|
payments made to third party contract research organizations for preclinical studies, investigative sites for clinical trials, and consultants;
|
|
|
·
|
costs associated with regulatory filings and the advancement of the Company’s product candidates through preclinical studies and clinical trials;
|
|
|
·
|
laboratory and other supplies,
|
|
|
·
|
manufacturing development costs; and
|
|
|
·
|
related facility maintenance.
|
(h)Income Taxes
Income taxes are accounted for under the asset-and-liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
(i)Stock-Based Compensation
The Company measures and recognizes compensation expense for all employee stock-based payments at fair value, net of estimated forfeitures, over the vesting period of the underlying stock-based awards. In addition, the Company accounts for stock-based compensation to nonemployees in accordance with the Financial Accounting Standards Board (FASB) accounting guidance for equity instruments that are issued to other than employees.
F- 12
Determining the appropriate fair value of stock-based payment awards require the input of subjective assumptions, including the expected life of the stock-based payment awards and stock price volatility. The Company uses the Black-Scholes option-pricing model to value its stock option awards. The assumptions used in calculating the fair value of stock-based payment awards represent management’s best estimates and involve inherent uncertainties and the application of management’s judgment. As a result, if factors change and management uses different assumptions, stock-based compensation expense could be materially different in the future. Since the Company does not have sufficient historical volatility of its stock for the expected term of its options, it uses comparable public companies as a basis for its expected volatility to calculate the fair value of option grants.
The estimation of the number of stock awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from the Company’s current estimates, such amounts will be recorded as an adjustment in the period in which estimates are revised. The Company considers many factors when estimating expected forfeitures for stock awards granted to employees, consultants and directors, including types of awards, employee class, and an analysis of the Company’s historical and known forfeitures on existing awards. Under the true-up provisions of the FASB stock based compensation guidance, the Company records additional expense if the actual forfeiture rate is lower than estimated, and a recovery of expense if the actual forfeiture rate is higher than estimated, during the period in which the options vest.
(j)Net Loss Per Share
Basic and diluted net loss per common share is determined by dividing net loss attributable to common stockholders by the weighted-average common shares outstanding less the weighted-average shares subject to repurchase during the period. For all periods presented, the outstanding Series A, Series B, and Series C, common stock options and preferred and common stock warrants have been excluded from the calculation because their effect would be anti-dilutive. Therefore, the weighted-average shares used to calculate both basic and diluted loss per share are the same.
The following potentially dilutive securities have been excluded from the computations of diluted weighted-average shares outstanding as of December 31, 2008, 2009, and 2010, as they would be anti-dilutive:
|
December 31,
|
||||||||||||
|
2008
|
2009
|
2010
|
||||||||||
|
Shares of redeemable convertible preferred stock
|
81,954,127 | 81,954,127 | — | |||||||||
|
Shares underlying options outstanding
|
518,977 | 712,373 | 1,377,710 | |||||||||
|
Shares underlying warrants outstanding
|
1,597,562 | 1,605,439 | 114,342 | |||||||||
|
Unvested restricted stock
|
3,191 | 432 | — | |||||||||
F- 13
(k)Deferred Equity Offering Costs
Deferred equity offering costs include costs directly attributable to the Company’s offering of its equity securities. In accordance with FASB ASC 340-10, Other Assets and Deferred Costs, these costs are deferred and capitalized as part of other assets. Costs attributable to the equity offerings will be charged against the proceeds of the offering once completed.
(l)Recently Issued Accounting Standards
In January 2010, the FASB issued Accounting Standards Update (ASU) No. 2010-06, Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements. The guidance requires entities to disclose significant transfers in and out of fair value hierarchy levels and the reasons for the transfers. Additionally, the guidance clarifies that a reporting entity should provide fair value measurements for each class of assets and liabilities and disclose the inputs and valuation techniques used for fair value measurements using significant other observable inputs (Level 2) and significant unobservable inputs (Level 3). Level 3 reconciliations should present separately information about purchases, sales, issuances and settlements. To date, the Company has not had any assets or liabilities that transferred in or out of fair value hierarchy levels, and as such, is not currently subject to this guidance. This guidance is effective for interim and annual periods beginning after December 15, 2009, except for the disclosures about purchases, sales, issuances and settlements in the Level 3 reconciliations, which is effective for fiscal years beginning after December 15, 2010. This guidance did not have an impact on the Company’s results of operations or financial position. The Company adopted certain of the relevant disclosure provisions of ASU 2010-06 on January 1, 2010 and adopted certain other provisions on January 1, 2011. The Company believes the enhanced Level 3 disclosures will not have an impact on the Company’s results of operations or financial position.
In February 2010, the FASB issued ASU No. 2010-09, Subsequent Events (Topic 855). The guidance requires an SEC filer to evaluate subsequent events through the date the financial statements are issued but no longer requires an SEC filer to disclose the date through which the subsequent event evaluation occurred. The guidance became effective for the Company upon issuance and had no impact on the Company’s results of operations or financial position.
(m)Reverse Stock Split
On February 18, 2010, the board of directors of the Company, subject to stockholder approval, approved a reverse stock split of the Company’s common stock at a ratio of one share for every 14.5 shares previously held. The reverse stock split was effective on March 24, 2010. All common stock share and per-share data included in these financial statements reflect such reverse stock split.
(n)Reclassification
Certain prior period amounts in the financial statements and notes thereto have been reclassified to conform to the current period presentation.
F- 14
(4) Short-term Investments and Financial Instruments
As of December 31, 2010, the Company had no short-term investments. As of December 31, 2009, short-term investments consisted of investments in commercial paper of $2,499,013. The Company had the ability and intent to hold these investments until maturity and therefore has classified the investments as held-to-maturity. Due to the short-term nature of these investments, unrealized gains and losses have been deemed temporary and therefore not recognized in the accompanying financial statements. Income generated from short-term investments is recorded to interest income.
The Company adopted new FASB accounting guidance on fair value measurements effective January 1, 2008 for financial assets and liabilities measured on a recurring basis. The guidance requires fair value measurements be classified and disclosed in one of the following three categories:
|
|
·
|
Level 1: Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities;
|
|
|
·
|
Level 2: Quoted prices in markets that are not active, or inputs which are observable, either directly or indirectly, for substantially the full term of the asset or liability;
|
|
|
·
|
Level 3: Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable (i.e., supported by little or no market activity).
|
The following fair value hierarchy table presents information about each major category of the Company’s financial assets and liability measured at fair value on a recurring basis as of December 31, 2009 and 2010.
|
Fair value measurement at reporting date using
|
||||||||||||||||
|
Quoted prices in active markets for identical assets (Level 1)
|
Significant other observable inputs
(Level 2)
|
Significant unobservable inputs
(Level 3)
|
Total
|
|||||||||||||
|
At December 31, 2009
|
||||||||||||||||
|
Assets
|
||||||||||||||||
|
Cash and cash equivalents
|
$ | 16,804,129 | $ | — | $ | — | $ | 16,804,129 | ||||||||
|
Short-term investments
|
2,499,013 | — | — | 2,499,013 | ||||||||||||
| $ | 19,303,142 | $ | — | $ | — | $ | 19,303,142 | |||||||||
|
Liabilities
|
||||||||||||||||
|
Warrant liability
|
$ | — | $ | — | $ | 314,431 | $ | 314,431 | ||||||||
|
At December 31, 2010
|
||||||||||||||||
|
Assets
|
||||||||||||||||
|
Cash and cash equivalents
|
$ | 11,971,660 | $ | — | $ | — | $ | 11,971,660 | ||||||||
|
Short-term investments
|
— | — | — | — | ||||||||||||
| $ | 11,971,660 | $ | — | $ | — | $ | 11,971,660 | |||||||||
F- 15
The reconciliation of warrant liability measured at fair value on a recurring basis using unobservable inputs (Level 3) is as follows:
|
Warrant
Liability
|
||||
|
Balance at January 1, 2009
|
$ | 2,129,562 | ||
|
Issuance of additional warrants
|
8,742 | |||
|
Change in fair value of warrant liability
|
(1,823,873 | ) | ||
|
Balance at December 31, 2009
|
314,431 | |||
|
Issuance of additional warrants
|
— | |||
|
Change in fair value of warrant liability
|
(191,527 | ) | ||
|
Reclassification of warrant liability to stockholders’ equity
|
(122,904 | ) | ||
|
Balance at December 31, 2010
|
$ | — | ||
The fair value of the warrant liability is based on Level 3 inputs. For this liability, the Company developed its own assumptions that do not have observable inputs or available market data to support the fair value. See Note 9 for further discussion of the warrant liability.
(5) Property and Equipment
Property and equipment consisted of the following:
|
December 31
|
||||||||
|
2009
|
2010
|
|||||||
|
Office and warehouse equipment
|
$ | 356,249 | $ | 356,249 | ||||
|
Computer equipment
|
1,232,059 | 1,246,432 | ||||||
|
Furniture and fixtures
|
524,132 | 524,132 | ||||||
|
Laboratory equipment
|
7,703,766 | 7,762,418 | ||||||
|
Leasehold improvements
|
21,396,526 | 21,433,819 | ||||||
|
Construction in progress
|
9,808 | — | ||||||
| 31,222,540 | 31,323,050 | |||||||
|
Less accumulated depreciation
|
(14,979,487 | ) | (19,830,473 | ) | ||||
| $ | 16,243,053 | $ | 11,492,577 | |||||
(6)Accrued Expenses
Accrued expenses consist of the following:
|
December 31
|
||||||||
|
2009
|
2010
|
|||||||
|
Accrued compensation and benefits
|
$ | 891,179 | $ | 1,290,656 | ||||
|
Accrued consulting and professional fees
|
996,574 | 730,207 | ||||||
|
Accrued research and development
|
777,582 | 678,846 | ||||||
|
Other
|
321,462 | 143,347 | ||||||
| $ | 2,986,797 | $ | 2,843,056 | |||||
F- 16
(7)2009 Restructuring Plan
In 2009, the Company implemented a restructuring plan which reduced headcount by 34 employees due at the time to the postponement of commencing a Phase III clinical trial. Expenses incurred related to the 2009 restructuring consist of termination benefits of $853,465 in the year ended December 31, 2009, of which $550,652 is included in research and development expense and $302,813 is included in general and administrative expense in the accompanying statements of operations. Below is a summary of the activity related to the 2009 restructuring program for the year ended December 31, 2009 and 2010:
|
2009 Charges to Operations
|
2009 Charges Utilized
|
Accrual Balance at December 31, 2009
|
2010 Charges to Operations
|
2010 Charges Utilized
|
Accrual Balance at December 31, 2010
|
|||||||||||||||||||
|
One-time termination benefits
|
$ | 853,465 | $ | 640,104 | $ | 213,361 | $ | (8,950 | ) | $ | 204,411 | $ | — | |||||||||||
(8)Debt
Total debt outstanding consists of the following:
|
December 31,
|
||||||||
|
2009
|
2010
|
|||||||
|
Working Capital Note
|
$ | 17,315,175 | $ | 7,257,159 | ||||
|
Equipment and Supplemental Working Capital Notes
|
4,019,394 | 1,015,474 | ||||||
|
Machinery and Equipment Loan
|
931,972 | 477,609 | ||||||
|
Unamortized debt discount
|
(429,763 | ) | (148,575 | ) | ||||
| 21,836,778 | 8,601,667 | |||||||
|
Less current portion
|
(13,196,376 | ) | (4,016,174 | ) | ||||
| $ | 8,640,402 | $ | 4,585,493 | |||||
Principal payments due as of December 31, 2010 are as follows:
|
2011
|
$ | 4,107,798 | ||
|
2012
|
2,089,030 | |||
|
2013
|
2,345,311 | |||
|
2014
|
208,103 | |||
| 8,750,242 | ||||
|
Less unamortized debt discount
|
(148,575 | ) | ||
| $ | 8,601,667 |
The Company has an outstanding working capital loan (the Working Capital Note) with a lender, which was utilized to fund working capital needs of the Company. In October 2008, the Company refinanced the terms of the Working Capital Note. The repayment terms of $14.2 million of the Working Capital Note were extended to add an additional six-month period of interest-only payments through July 2009 followed by 24 monthly payments of principal and accrued interest at an annual interest rate of 12.26%. The repayment of $5.8 million of the Working Capital Note was extended to add a 14-month period of interest-only payments through January 2010 followed by 20 monthly payments of principal and accrued interest at an annual interest rate of 12.26%. The Company recorded interest expense of $2,373,409, $2,370,268, and $1,482,221 related to the Working Capital Note for the years ended December 31, 2008, 2009 and 2010, respectively. The Working Capital Note was refinanced again in March 2011 (see Note 15).
F- 17
In connection with execution of the original Working Capital Note, the lender received warrant coverage of 9% for the first $15 million and 8% for the additional $5 million of the original Working Capital Note (see note 9). Warrants to purchase 212,016 and 1,993 shares of Series C, which upon completion of the Company’s initial public offering (IPO) converted into warrants to purchase 14,622 and 137 shares, respectively, of common stock, were issued to the lender for the years ended December 31, 2008 and 2009, respectively. There were no warrants issued during the year ended December 31, 2010. The fair value of the warrants issued in connection with the Working Capital Note for the years ended December 31, 2008 and 2009 was determined to be $326,688 and $826, respectively, using the Black-Scholes option-pricing model with the following weighted-average assumptions:
|
Years Ended December 31,
|
|||
|
2008
|
2009
|
||
|
Expected dividend yield
|
— %
|
— %
|
|
|
Expected volatility
|
83.50%
|
76.50%
|
|
|
Risk-free interest rate
|
3.81%
|
3.40%
|
|
|
Contractual life
|
10 years
|
10 years
|
|
The lender holds warrants exercisable into 100,873 shares of common stock as of December 31, 2010.
The relative fair value of the warrants has been recorded against the carrying value of the Working Capital Note as an original issue discount (OID) which is being amortized as interest expense over the term of the Working Capital Note. During the years ended December 31, 2008, 2009 and 2010, the Company recognized a noncash charge to interest expense of $501,499, $371,987 and $250,032, respectively, for the amortization of OID.
In 2005, the Company had also executed an additional loan facility with another lender to fund equipment (the Equipment Note) and other asset purchases (the Supplemental Working Capital Note) from July 2005 through December 2009. As of December 31, 2010, the Equipment Note bears interest at an average rate of 11.69% and matures over 36 to 48 months and the Supplemental Working Capital Note bears interest at an average rate of 11.98% and matures over 36 months. The Company recorded interest expense of $618,641, $581,852 and $259,800 related to the Equipment and Supplemental Working Capital Notes for the years ended December 31, 2008, 2009 and 2010, respectively.
F- 18
The lender also received warrant coverage of 2% of the Equipment and Supplemental Working Capital Notes (see note 9). In 2008, the lender received warrants to purchase 4,726 shares of Series A and 8,029 shares of Series C, which upon completion of the Company’s IPO converted into warrants to purchase 880 shares of common stock. During 2009, the lender received warrants to purchase 5,884 shares of Series C, which upon completion of the Company’s IPO converted into warrants to purchase 406 shares of common stock. There were no warrants issued during the year ended December 31, 2010. The fair value of the warrants issued in connection with the Equipment and Supplemental Working Capital Notes for the years ended December 31, 2008 and 2009 was determined to be $17,499 and $7,916, respectively, using the Black-Scholes option-pricing model with the following weighted average assumptions:
|
Years ended December 31,
|
|||
|
2008
|
2009
|
||
|
Expected dividend yield
|
— %
|
— %
|
|
|
Expected volatility
|
75.09%
|
74.90%
|
|
|
Risk-free interest rate
|
3.06%
|
2.55%
|
|
|
Contractual life
|
8 years
|
8 years
|
|
The relative fair value of the warrants has been recorded against the carrying value of the Equipment and Supplemental Working Capital Notes as OID which is being amortized as interest expense over the term of the Equipment and Supplemental Working Capital Notes. During the years ended December 31, 2008, 2009 and 2010, the Company recognized a noncash charge to interest expense of $44,265, $43,552 and $31,155, respectively, for the amortization of OID.
In December 2007, the Company executed a $1.65 million agreement with the Commonwealth of Pennsylvania to fund machinery and equipment and other asset purchases (MELF Loan) through December 31, 2009. On December 31, 2007 the Company borrowed $1.3 million of the MELF loan to fund equipment purchases. In March 2009, the Company borrowed an additional $0.3 million of the MELF loan to fund equipment purchases. Under the terms of the MELF Loan, the Company has a four year period of payments of principal and accrued interest at an annual interest rate of 5%. The Company recorded interest expense of $60,171, $54,409 and $36,279 related to the MELF Loan for the years ended December 31, 2008, 2009 and 2010, respectively.
In connection with the Working Capital Note, Equipment Note, Supplemental Working Capital Note, and the MELF Loan, the Company incurred financing costs of $189,771, recorded as other assets on the accompanying balance sheets, which are being amortized on a straight-line basis until maturity of the related notes. During the years ended December 31, 2008, 2009 and 2010, the Company recorded amortization of these deferred financing costs of $20,875, $44,293 and $44,292, respectively, included in interest expense on the accompanying statements of operations.
Borrowings under the Working Capital Note are secured by all assets of the Company, except for Intellectual Property and permitted liens that have priority, including liens on equipment subsequently acquired to secure the purchase price or lease obligation, as defined in the loan agreement. Borrowings under the Equipment Note, Supplemental Working Capital Note and the MELF Loan are secured by equipment, as defined in the loan agreements.
F- 19
(9) Capital Structure
In August 2004 and March 2005, the Company issued 20,943,577 and 3,247,095 shares, respectively, of Series A-1 at $1.61683 per share. The shares issued in August 2004 resulted in cash proceeds of approximately $30.1 million, net of transaction costs, and the conversion of principal and interest on the convertible notes in the aggregate amount of approximately $3.6 million. The shares issued in March 2005 resulted in cash proceeds of approximately $5.2 million, net of transaction costs. In June 2006, the Company converted each share of Series A-1 into one share of Series A. All terms of the Series A remained the same as the Series A-1. In June 2006, the Company issued 27,637,363 shares of Series B at $1.82 per share. The shares issued resulted in cash proceeds of approximately $50.0 million, net of transaction costs. In September 2007 and October 2008, the Company issued 18,332,965 and 11,793,127 shares of Series C, respectively, at $1.82 per share. The shares issued resulted in cash proceeds of approximately $33.2 million and $21.4 million, net of transaction costs, in 2007 and 2008, respectively.
The Series A, Series B, and Series C were convertible into common stock at the option of the holder on a share-for-share basis, subject to certain customary antidilution adjustments contained in the Company’s certificate of incorporation, which were triggered, with respect to each series of preferred stock, upon the closing of the Company’s IPO in April 2010. In addition, the Series A, Series B and Series C were entitled to vote together with the common stockholders as one class and were entitled to separate votes on certain matters.
The Series A, Series B, and Series C stockholders were entitled to receive an annual 8% dividend, when and if declared by the board of directors. No dividends were declared through December 31, 2010. As of December 31, 2009, there were $19,753,349, $15,729,989 and $8,509,943 of accrued and unpaid Series A, Series B, and Series C dividends, respectively.
The Company incurred aggregate costs of $201,957 for Series A, $259,712 for Series B, and $258,295 for Series C, respectively, in connection with the sale of the Series A, Series B, and Series C, which reduced the initial carrying value of each Series of redeemable convertible preferred stock. As a result of the Series A, Series B and Series C redemption feature, the initial carrying value of the Series A, Series B, and Series C will be accreted to its redemption value through September 2012. The accretion of issuance costs during the years ended December 31, 2007, 2008 and 2009 was $86,415, $93,597 and $115,932, respectively.
Initial Public Offering
In April 2010, the Company completed its initial public offering, selling 6,000,000 shares at an initial public offering price of $5.00 per share resulting in gross proceeds of $30.0 million. Net proceeds received after underwriting fees and offering expenses were approximately $25.7 million. In connection with the closing of the initial public offering, all outstanding shares of the Company’s redeemable convertible preferred stock were converted into 5,651,955 shares of common stock, and all outstanding warrants to purchase preferred stock were converted into warrants to purchase 110,452 shares of common stock. In total, the Company has 114,342 common stock warrants outstanding as of December 31, 2010.
F- 20
Preferred Stock
As of December 31, 2010, the Company was authorized to issue 10,000,000 shares of preferred stock in one or more series and to fix the rights, preferences, privileges, and restrictions thereof. These rights, preferences and privileges could include dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of such series, any or all of which may be greater than the rights of common stock. The issuance of our preferred stock could adversely affect the voting power of holders of common stock and the likelihood that such holders will receive dividend payments and payments upon liquidation. In addition, the issuance of preferred stock could have the effect of delaying, deferring or preventing a change of control of our company or other corporate action. There are no shares issued or outstanding as of December 31, 2010.
Common Stock
Since inception, the Company has sold common stock to certain officers, directors, employees, consultants, and Scientific Advisory Board members. As of December 31, 2010, the Company was authorized to issue 90,000,000 shares of common stock. Each holder of common stock is entitled to one vote for each share held. The Company will, at all times, reserve and keep available out of its authorized but unissued shares of common stock sufficient shares to effect the exercise of outstanding stock options and warrants.
Warrants
As of December 31, 2010, the following warrants to purchase Common Stock were outstanding:
|
Exercise Price
|
Number
of shares
|
Expiration
|
|||||
| $ | 2.32 | 3,890 |
September 2015 through January 2016
|
||||
| $ | 23.44 | 64,409 |
August 2013 through December 2016
|
||||
| $ | 26.39 | 46,043 |
September 2015 through October 2018
|
||||
| 114,342 | |||||||
In conjunction with the Working Capital Note, Equipment Note, and the Supplemental Working Capital Note, the Company issued warrants to purchase shares of Series A, B, and C. The warrants were immediately exercisable. Upon the close of the Company’s initial public offering, the preferred stock warrants automatically converted into warrants to purchase 110,452 shares of common stock. Warrants related to the Working Capital Note expire five years from the date of issuance. Warrants related to the Equipment and Supplemental Working Capital Notes expire the earlier of eight years from the date of issuance or upon Company acquisition as defined in the warrant agreement.
F- 21
Prior to the Company’s initial public offering, the preferred stock warrants were classified as a liability on the accompanying balance sheet for the year ended December 31, 2009 in accordance with FASB accounting guidance, as the warrants entitled the holder to purchase preferred stock, which could have been redeemed at the option of the holder. Warrants that are classified as a liability are revalued at each reporting date until the warrants are exercised or expire with changes in the fair value reported in the statements of operations. The fair value of the warrants are subject to fluctuation based on changes in the Company’s preferred stock price, expected volatility, remaining contractual life, and the risk-free interest rate. For the year ended December 31, 2008, 2009, and 2010, the change in fair value of these preferred stock warrants was ($56,974), $1,823,873, and $191,527, respectively. Subsequent to the closing of the initial public offering, the warrants no longer are exercisable for a redeemable security and, therefore, such warrants are now classified within stockholders’ equity.
The warrants were revalued using the Black-Scholes option-pricing model with the following weighted average assumptions:
|
December 31, 2008
|
December 31, 2009
|
December 31, 2010
|
|||||||||
|
Working Capital Note
|
Equipment and Supplemental Working Capital Note
|
Working Capital Note
|
Equipment and Supplemental Working Capital Notes
|
Working Capital Note
|
Equipment and Supplemental Working Capital Notes
|
||||||
|
Expected dividend yield
|
—
|
—
|
—
|
—
|
—
|
—
|
|||||
|
Expected volatility
|
77.55%
|
64.51%
|
72.20%
|
65.90%
|
66.66%
|
67.54%
|
|||||
|
Risk-free interest rate
|
3.31%
|
3.11%
|
2.79%
|
2.46%
|
2.55%
|
2.52%
|
|||||
|
Remaining contractual life
|
7.9 years
|
6.4 years
|
7.0 years
|
5.5 years
|
5.0 years
|
4.9 years
|
|||||
(10) Stock-based Compensation
The Company currently maintains two stock-based compensation plans. Under the 2004 Stock Option Plan (the 2004 Plan), stock awards were granted to employees, directors, and consultants of the Company, in the form of restricted stock and stock options. The amounts and terms of options granted were determined by the Company’s compensation committee. The equity awards granted under the Plan generally vest over four years and have terms of up to ten years after the date of grant, and options are exercisable in cash or as otherwise determined by the board of directors. There are no shares available for future grants under the 2004 Plan, as grants from the 2004 Plan ceased upon the Company’s initial public offering in April 2010.
The 2010 Stock Incentive and Option Plan (2010 Plan) became effective upon the closing of the Company’s initial public offering. Under the 2010 Plan, stock awards may be granted to employees, directors, and consultants of the Company, in the form of restricted or unrestricted stock, stock appreciation rights, cash-based or performance share awards and stock options. The amounts and terms of options granted are determined by the Company’s compensation committee. The equity awards granted under the Plan generally vest over four years and have terms of up to ten years after the date of grant, and options are exercisable in cash or as otherwise determined by the board of directors. The 2010 Plan allows for the transfer of forfeited shares from the 2004 Plan. As of December 31, 2010, 950,706 shares of common stock were available for future grants under the Plan.
F- 22
Restricted Stock
The Company has issued restricted stock as compensation for the services of certain employees and other third parties. The grant date fair value of restricted stock was based on the fair value of the common stock on the date of grant, and compensation expense is recognized based on the period in which the restrictions lapse.
The following summarizes the Company’s restricted stock activity:
|
Number of
Shares
|
Weighted – Average Grant Date Fair Value
|
|||||||
|
Nonvested shares at December 31, 2007
|
48,197 | $ | 2.11 | |||||
|
Granted
|
— | — | ||||||
|
Vested
|
(44,185 | ) | $ | 1.98 | ||||
|
Forfeited / repurchased
|
(821 | ) | $ | 2.47 | ||||
|
Nonvested shares at December 31, 2008
|
3,191 | — | ||||||
|
Granted
|
— | — | ||||||
|
Vested
|
(2,759 | ) | $ | 3.51 | ||||
|
Forfeited / repurchased
|
— | — | ||||||
|
Nonvested shares at December 31, 2009
|
432 | $ | 5.80 | |||||
|
Granted
|
— | — | ||||||
|
Vested
|
(432 | ) | $ | 5.80 | ||||
|
Forfeited / repurchased
|
— | — | ||||||
|
Nonvested shares at December 31, 2010
|
— | — | ||||||
The total intrinsic value of restricted stock that vested was $518,969, $8,000 and $1,098 for the years ended December 31, 2008, 2009 and 2010, respectively. Total stock-based compensation expense for restricted stock was $212,117, $9,823 and $957 for the years ended December 31, 2008, 2009 and 2010, respectively. As of December 31, 2010, there was zero of unrecognized compensation expense, related to restricted stock awards.
For restricted stock subject to vesting, the Company has certain repurchase rights. In the event that the employment or consulting relationship terminates, excluding the Chief Executive Officer (CEO), the Company has the right to repurchase any or all of the unvested restricted shares at the original fair value per share and any or all of the vested shares at the then fair market value per share. In the event the employment relationship terminates with the CEO, the Company has only the right to repurchase any or all of the unvested restricted shares at the original fair value per share, but not the vested restricted shares. However, if any employee, including the CEO, is terminated for cause, the vested restricted shares are subject to repurchase at the original fair value per share.
F- 23
Stock Options
The following table summarizes stock option activity under the Plans:
|
Number
of
Shares
|
Weighted-
Average
Exercise
price
|
Weighted-
Average
Remaining
Contractual
Term in
Years
|
Aggregate
Intrinsic
Value
|
||||
|
Outstanding at December 31, 2007
|
445,885
|
$ 10.88
|
|||||
|
Granted
|
123,019
|
$ 22.91
|
|||||
|
Exercised
|
(14,113)
|
$ 7.54
|
|||||
|
Cancelled/forfeited
|
(35,814)
|
$ 13.81
|
|||||
|
Outstanding at December 31, 2008
|
518,977
|
$ 3.63
|
|||||
|
Granted
|
358,640
|
$ 0.44
|
|||||
|
Exercised
|
(20,005)
|
$ 2.71
|
|||||
|
Cancelled/forfeited
|
(145,233)
|
$ 13.54
|
|||||
|
Outstanding at December 31, 2009
|
712,379
|
$ 1.00
|
|||||
|
Granted
|
790,812
|
$ 3.65
|
|||||
|
Exercised
|
(32,255)
|
$ 0.44
|
|||||
|
Cancelled/forfeited
|
(93,226)
|
$ 1.73
|
|||||
|
Outstanding at December 31, 2010
|
1,377,710
|
$ 2.49
|
|||||
|
Vested and expected to vest at December 31, 2010
|
1,294,703
|
$ 2.47
|
8.60
|
$ 1,148,427
|
|||
|
Exercisable at December 31, 2010
|
386,080
|
$ 1.46
|
6.86
|
$ 708,297
|
During 2008, 6,582 shares were withheld upon the exercise of stock options to satisfy the exercise price of such options.
During 2008, 2009 and 2010, the Company issued options to purchase 117,156, 358,640, and 790,812 shares of common stock, respectively, to employees and non-employee directors under the Plans. The per-share weighted average fair value of the options granted to employees during 2008, 2009 and 2010 was estimated at $13.92, $0.44 and $3.65, respectively, on the date of grant using the Black-Scholes option-pricing model with the following weighted-average assumptions:
|
December 31,
|
|||||
|
2008
|
2009
|
2010
|
|||
|
Expected dividend yield
|
— %
|
— %
|
— %
|
||
|
Expected volatility
|
64.11%
|
66.18%
|
69.35%
|
||
|
Risk-free interest rate
|
3.09%
|
2.65%
|
2.00%
|
||
|
Expected life
|
6 years
|
6 years
|
6 years
|
||
F- 24
Total stock-based compensation expense recognized for stock options to employees and non-employee directors for the years ended December 31, 2008, 2009 and 2010 was $989,973, $864,462 and $924,158, respectively. In addition, the Company recognized $46,592 and $14,793 of additional stock-based compensation expense during 2009 and 2010 in association with the modification of 320,366 stock options previously granted to active employees during the third quarter of 2009. As of December 31, 2010, there was $1,818,164 of unrecognized compensation expense, net of forfeitures, related to non-vested employee stock options and remaining incremental expense associated with the modification and $68,911 of unrecognized compensation expense related to non-vested non-employee director stock options, which are expected to be recognized over a weighted average period of approximately 3.24 years.
During 2008 and 2010, the Company issued options to purchase 5,863, and 32,000 shares of common stock, respectively, to non-employee consultants under the Plan. No options were granted to non-employees during the year ended December 31, 2009. The per-share weighted-average fair value of the options granted to non-employees during 2008 and 2010 was estimated at $23.93 and $1.21, respectively, on the date of grant using the Black-Scholes option-pricing model with the following weighted-average assumptions:
|
December 31,
|
|||||
|
2008
|
2009
|
2010
|
|||
|
Expected dividend yield
|
— %
|
— %
|
— %
|
||
|
Expected volatility
|
82.89%
|
— %
|
69.79%
|
||
|
Risk-free interest rate
|
3.72%
|
— %
|
2.10%
|
||
|
Expected life
|
10 years
|
5.28 years
|
|||
Total stock-based compensation expense recognized for stock options to nonemployees for the years ended December 31, 2008 and 2010 was $114,634 and $13,950. For the year ended December 31, 2009, the Company recognized a credit of $65,741 due to the decline in the per-share fair value of the outstanding options granted to non-employees. As of December 31, 2010, there was approximately $27,868 of unrecognized compensation expense related to non-vested non-employee stock options, which is expected to be recognized over a weighted average period of 1.35 years.
(11) Employee Benefit Plan
The Company maintains a defined contribution 401(k) plan (the 401(k) Plan) for the benefit of its employees. Employee contributions are voluntary and are determined on an individual basis, limited by the maximum amounts allowable under federal tax regulations. As of December 31, 2010, the Company has not elected to match any of the employee’s contributions to the 401(k) Plan.
(12) Commitments and Contingencies
(a)Leases and Letters of Credit
The Company leases office space and office equipment under operating leases, which expire at various times through February 2016. Rent expense under these operating leases was $731,144, $728,531, $719,029, and $3,835,866 for the years ended December 31, 2008, 2009, and 2010, and for the period from July 10, 2003 (inception) through December 31, 2010, respectively. Future minimum lease payments as of December 31, 2010 are as follows:
F- 25
|
2011
|
$ | 885,096 | ||
|
2012
|
836,668 | |||
|
2013
|
856,669 | |||
|
2014
|
876,670 | |||
|
2015
|
896,671 | |||
|
2016 and thereafter
|
150,001 | |||
|
Total minimum lease payments
|
$ | 4,501,775 |
As of December 31, 2010, the lease agreements for the Company’s corporate headquarters required the Company to provide security and restoration deposits totaling $1.7 million to two landlords. As provided for in the lease agreements, the Company obtained letters of credit from a bank in favor of the landlords to satisfy these obligations. The letters of credit are collateralized by an account held at the bank. If the bank determines the collateral to be insufficient, the bank has the right to demand additional collateral. If the Company fails to provide additional collateral, the bank has the right to withdraw the letters of credit. In that event, the landlords would have the right to require the Company to deposit cash of up to $1.7 million in an account to satisfy the Company’s deposit obligations. In March 2011, the security and restoration deposits became an obligation to one landlord and increased to $2.2 million. In January 2011, the Company deposited $1.0 million with the landlord. As of March 2011, an outstanding letter of credit is satisfying the remaining deposit obligation of $1.2 million.
(b)License and Research Agreements
In 2003, a license agreement was executed with Children’s Medical Center Corporation (CMCC), whereby CMCC granted the Company a license to utilize certain patent rights. In accordance with the license agreement, the Company paid an initial license fee of $175,000, which was recorded as research and development expense. Additionally, the license agreement requires the Company to reimburse CMCC for patent costs related to the underlying licensed rights. In 2008, 2009 and 2010, and for the period from July 10, 2003 (inception) through December 31, 2010 the Company recorded $246,750, $182,846, $149,245 and $2,138,447 respectively, as general and administrative expenses related to the reimbursement of such patent costs in the accompanying statements of operations.
The Company is obligated to make payments to CMCC upon the occurrence of various development and sales milestones. During the fourth quarter of 2006, the Company achieved two of its development milestones for the first licensed product launched, as defined in the agreement, and paid $350,000, which was recorded as research and development expense for the year ended December 31, 2006. No further milestones payments have been made since 2006. If the Company develops and obtains regulatory approval of a licensed product in each of the four subfields covered under the license, it will be required to pay CMCC milestones aggregating approximately $6.9 million, which includes $350,000 paid to date. Also, if cumulative net sales of all licensed products reach a certain level, the Company will be required to make a one-time sales milestone payment of $2.0 million. The timing and likelihood of such milestone payments are not currently known to the Company. A portion of the milestone payments the Company makes will be credited against its future royalty payments. The Company must pay CMCC royalties in the mid-single digit range based on net sales of licensed products as defined in the agreement by the Company, its affiliates and its sublicensees, which royalties will be reduced for certain amounts the Company is required to pay to license patents or intellectual property from third parties and under certain circumstances if there is a competing product. No royalties will be payable with respect to sales of licensed products in countries where there is no valid patent claim, unless the Company advised CMCC not to file for patent protection in that country and later chose to market and sell licensed products in such country. The license agreement, and the Company’s obligation to pay royalties, terminates on the later of the expiration, on a country-by-country basis, of the last patent right and October 2018.
F- 26
In January 2006 the Company entered into a license agreement with Wake Forest University Health Sciences, or WFUHS, which was amended in May 2007, for the license of certain of WFUHS’s intellectual property rights. Under the license agreement, the Company issued WFUHS a warrant to purchase 3,200 shares of the Company’s common stock through January 1, 2016 at an exercise of $2.32 per share and is required to pay WFUHS a percentage of certain consideration received from a sublicensee. The Company is not required to make any milestone payments to WFUHS related to the development or commercialization of the licensed products, but the Company is required to pay certain license maintenance fees with respect to each product covered by new development patents, commencing two years after the Company initiates the first animal study conducted under cGLP, with respect to such product. These license maintenance fees, which are in the low six figure range with respect to each product, will be creditable against royalties the Company is obligated to pay to WFUHS for such product. In addition, the Company is required to pay WFUHS royalties in the low- to mid-single digit range based on net sales of licensed products in all countries. With respect to any product covered by an improvement patent, the Company’s obligation to pay royalties to WFUHS terminates upon the later of the expiration, on a product-by-product basis, of the last to expire patent covering such product and the date that is 15 years from the first commercial sale of such product, provided that no such royalty is payable more than seven years after expiration of the last-to-expire patent covering such product.
In January 2006, the Company entered into a research agreement with WFUHS, which was amended in September 2006 and extended in September 2010 for an additional three-year period. Under the research agreement, WFUHS agreed to perform sponsored research in return for quarterly payments. As of December 31, 2010, the Company was obligated to make payments of $800,000 with respect to the 2011 research activities of WFUHS under the extended agreement. Payments for 2012 and 2013 research activities of WFUHS under the extended agreement will be determined by the Company prior to the beginning of each year, respectively. The sponsored research involves the experiments and studies set out in annual research plans and milestones established under the agreement. Either party may terminate the agreement upon a material, uncured breach by the other party upon 30 days written notice. The Company may terminate the agreement at any time, with or without cause, upon 90 days prior written notice. If the Company terminates the agreement, it is required to pay to WFUHS all reasonable direct costs and all noncancelable obligations incurred by WFUHS, provided that such liability is limited to $625,000.
F- 27
(13) Income Taxes
A reconciliation of the statutory U.S. federal rate to the Company’s effective tax rate is as follows:
|
Year ended December 31
|
||||||||||||
|
2008
|
2009
|
2010
|
||||||||||
|
Percent of pre-tax income
|
||||||||||||
|
U.S. federal statutory income tax rate
|
(34.0 | )% | (34.0 | )% | (34.0 | )% | ||||||
|
State taxes, net of federal benefit
|
(6.7 | ) | (6.7 | ) | (7.1 | ) | ||||||
|
Other
|
0.6 | (1.1 | ) | (0.5 | ) | |||||||
|
Valuation allowance
|
40.1 | 41.8 | 41.6 | |||||||||
|
Effective income tax expense rate
|
— | % | — | % | — | % | ||||||
The tax effects of temporary differences that gave rise to significant portions of the deferred tax assets were as follows:
|
December 31
|
||||||||
|
2009
|
2010
|
|||||||
|
Net operating loss carryforwards
|
$ | 37,526,854 | $ | 44,765,050 | ||||
|
Research and development credit
|
4,142,443 | 4,696,595 | ||||||
|
Depreciation and amortization
|
6,097,278 | 8,148,166 | ||||||
|
Capitalized start-up costs
|
9,730,121 | 12,161,811 | ||||||
|
Other temporary differences
|
923,419 | 1,134,370 | ||||||
|
Gross deferred tax asset
|
58,420,115 | 70,905,992 | ||||||
|
Deferred tax assets valuation allowance
|
(58,420,115 | ) | (70,905,992 | ) | ||||
| $ | — | $ | — | |||||
In assessing the realizability of deferred tax assets, the Company considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which the temporary differences representing net future deductible amounts become deductible. Due to the Company’s history of losses, the deferred tax assets are fully offset by a valuation allowance at December 31, 2009 and 2010. The valuation allowance in 2009 increased by $11.8 million over 2008 and the valuation allowance in 2010 increased by $12.5 million over 2009, related primarily to additional net operating losses incurred by the Company and additional capitalized start-up expenses.
As of December 31, 2009, and 2010, approximately $23.9 million and $29.6 million, respectively, of the Company’s expenses had been capitalized for tax purposes as start-up costs. For tax purposes, capitalized start-up costs will be amortized over fifteen years beginning when the Company commences operations, as defined under the Internal Revenue Code.
F- 28
The following table summarizes carryforwards of net operating losses and tax credits as of December 31, 2010.
|
Amount
|
Expiration
|
||||
|
Federal net operating losses
|
$ | 109,705,305 |
2023 to 2030
|
||
|
State net operating losses
|
120,167,441 |
2019 to 2030
|
|||
|
Research and development credits
|
4,696,595 |
2024 to 2030
|
|||
The Tax Reform Act of 1986 (the Act) provides for a limitation on the annual use of net operating loss and research and development tax credit carryforwards following certain ownership changes (as defined by the Act) that could limit the Company’s ability to utilize these carryforwards. The Company has not completed a study to assess whether an ownership change has occurred, or whether there have been multiple ownership changes since its formation, due to the significant costs and complexities associated with such a study. The Company may have experienced various ownership changes, as defined by the Act, as a result of past financings. Accordingly, the Company’s ability to utilize the aforementioned carryforwards may be limited. Additionally, U.S. tax laws limit the time during which these carryforwards may be applied against future taxes; therefore, the Company may not be able to take full advantage of these carryforwards for federal or state income tax purposes.
(14) Quarterly Financial Data (Unaudited)
|
YEAR 2010
|
Quarter Ended
March 31
|
Quarter Ended
June 30
|
Quarter Ended
September 30
|
Quarter Ended
December 31
|
||||||||||||
|
Research and development expense
|
$ | 3,316,126 | $ | 3,253,232 | $ | 3,542,858 | $ | 2,743,155 | ||||||||
|
Net loss
|
(6,434,790 | ) | (6,504,981 | ) | (6,715,443 | ) | (5,945,055 | ) | ||||||||
|
Per common share information:
|
||||||||||||||||
|
Basic and diluted net loss per share
|
$ | (14.33 | ) | $ | (0.61 | ) | $ | (0.54 | ) | $ | (0.48 | ) | ||||
|
YEAR 2009
|
Quarter Ended
March 31
|
Quarter Ended
June 30
|
Quarter Ended
September 30
|
Quarter Ended
December 31
|
||||||||||||
|
Research and development expense
|
$ | 6,048,118 | $ | 4,708,364 | $ | 3,703,163 | $ | 3,488,415 | ||||||||
|
Net loss
|
(9,695,883 | ) | (8,150,722 | ) | (5,464,140 | ) | (6,534,291 | ) | ||||||||
|
Per common share information:
|
||||||||||||||||
|
Basic and diluted net loss per share
|
$ | (18.86 | ) | $ | (16.57 | ) | $ | (12.93 | ) | $ | (14.62 | ) | ||||
(15) Subsequent Events
(a)Equity Financing and Medtronic Agreement
In March 2011, the Company closed a private placement transaction pursuant to which the Company sold securities consisting of 11,079,250 shares of common stock, $0.001 par value per share, and warrants to purchase 10,460,875 shares of common stock. The purchase price per security was $2.83. Each warrant is exercisable in whole or in part at any time until March 4, 2016 at a per share exercise price of $2.88, subject to certain adjustments as specified in the warrant agreement. The Company received net proceeds of approximately $28.9 million.
F- 29
One of the investors in the equity financing was Medtronic, Inc. The Company entered into a Right of First Refusal and Right of First Negotiation Agreement with Medtronic, pursuant to which the Company granted Medtronic a right of first refusal to the license, sale, assignment, transfer or other disposition by the Company of any material portion of intellectual property (including patents and trade secrets) or other assets related to Tengion's Neo-Kidney Augment program (an NKA Transaction) until October 31, 2013. Additionally, from November 1, 2013 through July 1, 2014, Medtronic will have a right of first negotiation with respect to an NKA Transaction, with an option to convert that right of first negotiation to a right of first refusal. The Medtronic Agreement provided for Medtronic to receive a warrant to purchase 25% fewer of the warrant shares that otherwise would have been issued to Medtronic in connection with its investment. In the event of a change in control of the Company, and without any obligation to the Company, the Medtronic Agreement and all Medtronic’s rights pursuant thereto will automatically terminate in all respects and be of no further force and effect.
Refinancing of Working Capital Note
In March 2011, the Company refinanced the outstanding debt owed to its lender of the Working Capital Note (see Note 8 for additional information). Pursuant to the terms of the refinancing, the Company simultaneously borrowed $5.0 million and repaid the then outstanding principal amount of $4.5 million. The Company is obligated to make interest-only payments through January 2012, followed by 24 monthly payments of principal and interest at an interest rate of 11.75% per annum. In addition, the Company granted a warrant to the lender to purchase 70,671 shares of common stock. The warrant is exercisable in whole or in part at any time until March 2016 at a per share exercise price of $2.88, subject to certain adjustments as specified in the warrant.
Borrowings under the Working Capital Note are secured by all assets of the Company, except for Intellectual Property and permitted liens that have priority, including liens on equipment subsequently acquired to secure the purchase price or lease obligation, as defined in the loan agreement.
The Company has modified the classification of long-term debt on its balance sheet as of December 31, 2010 to reflect the revised payment terms included in the new loan agreement.
F- 30
Exhibit Index
|
Exhibit No.
|
Description
|
|
3.1
|
Fourth Amended and Restated Certificate of Incorporation of Tengion, Inc. (Incorporated by reference to Exhibit 3.1 to Amendment No. 3 to our Registration Statement on Form S-1 filed on March 17, 2010 (Registration No. 333-164011)).
|
|
3.2
|
Amended and Restated Bylaws of Tengion, Inc. (Incorporated by reference to Exhibit 3.2 to Amendment No. 3 to our Registration Statement on Form S-1 filed on March 17, 2010 (Registration No. 333-164011)).
|
|
4.1
|
Form of Tengion, Inc.’s Common Stock Certificate. (Incorporated by reference to Exhibit 4.1 to Amendment No. 3 to our Registration Statement on Form S-1 filed on March 17, 2010 (Registration No. 333-164011)).
|
|
4.2
|
Second Amended and Restated Investor Rights Agreement by and among the investors listed therein and Tengion, Inc., dated as of September 24, 2007. (Incorporated by reference to Exhibit 4.2 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
4.3
|
Amendment No. 1 to Second Amended and Restated Investor Rights Agreement by and among the investors listed therein and Tengion, Inc., dated as of October 15, 2008. (Incorporated by reference to Exhibit 4.3 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
4.4
|
Registration Rights Agreement by and among the investors party thereto and Tengion, Inc., dated as of March 4, 2011. (Incorporated by reference to Exhibit 10.2 to our Current Report on Form 8-K, filed on March 16, 2011).
|
|
4.5
|
Form of Preferred Stock Warrant by and between Oxford Finance Corporation and the Company. (Incorporated by reference to Exhibit 4.4 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
4.6
|
Form of Preferred Stock Warrant by and between Horizon Funding Company II LLC and the Company. (Incorporated by reference to Exhibit 4.5 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
4.7
|
Stock Subscription Warrant issued to APCO Worldwide Inc., dated September 1, 2005. (Incorporated by reference to Exhibit 4.6 to Amendment No. 2 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
4.8
|
Form of Warrant to Purchase Common Stock, dated as of March 4, 2011. (Incorporated by reference to Exhibit 4.1 to our Current Report on Form 8-K, filed on March 1, 2011).
|
|
Exhibit No.
|
Description
|
|
4.9
|
Warrant to Purchase Common Stock issued to Horizon Technology Finance Corporation, dated March 14, 2011. (Incorporated by reference to Exhibit 4.1 to our Current Report on Form 8-K, filed on March 16, 2011).
|
|
10.1
|
Lease Agreement by and between 3929 Westpoint Industrial, LLC and Tengion, Inc., dated as of June 8, 2005. (Incorporated by reference to Exhibit 10.1 to Amendment No. 2 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.2
|
First Amendment to Lease by and among Fawn Industrial LLC and 1881 Industrial LLC, successors in interest to 3929 Westpoint Industrial, LLC and together as Landlord, and Tengion, Inc., dated as of March 15, 2007. (Incorporated by reference to Exhibit 10.2 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.4
|
Lease Agreement by and between Norriton Business Campus, L.P. and Tengion, Inc., dated as of February 1, 2006, as amended. (Incorporated by reference to Exhibit 10.4 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.5
|
Amended and Restated Tengion, Inc. 2004 Stock Incentive Plan, Form Notice of Stock Option Grant and Stock Option Agreement. (Incorporated by reference to Exhibit 10.5 to Amendment No. 2 to our Registration Statement on Form S-1 filed on February 23, 2010 (Registration No. 333-164011)).#
|
|
10.6
|
2010 Stock Option and Incentive Plan, Form of Incentive Stock Option Agreement, Form of Non-Qualified Stock Option Agreement and Form of Restricted Stock Award Agreement. (Incorporated by reference to Exhibit 10.34 to Amendment No. 2 to our Registration Statement on Form S-1 filed on February 23, 2010 (Registration No. 333-164011)).#
|
|
10.7
|
Exclusive License Agreement by and between Children’s Medical Center Corporation and Tengion, Inc., dated as of October 10, 2003. (Incorporated by reference to Exhibit 10.6 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).*
|
|
10.8
|
Tengion, Inc. Non-Employee Director Compensation Policy. (Incorporated by reference to Exhibit 10.36 to Amendment No. 3 to our Registration Statement on Form S-1 filed on March 17, 2010 (Registration No. 333-164011)).#
|
|
10.9
|
License Agreement by and between the Company and Wake Forest University Health Sciences, effective as of January 1, 2006. (Incorporated by reference to Exhibit 10.7 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).*
|
|
Exhibit No.
|
Description
|
|
10.10
|
License Agreement Amendment No. 1 by and between the Company and Wake Forest University Health Sciences, dated as of May 3, 2007. (Incorporated by reference to Exhibit 10.8 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).*
|
|
10.11
|
Right of First Refusal and Right of First Negotiation Agreement by and between Medtronic, Inc. and Tengion, Inc., dated as of March 1, 2011. (Incorporated by reference to Exhibit 10.3 to our Current Report on Form 8-K, filed on March 1, 2011).
|
|
10.12
|
Venture Loan and Security Agreement by and between Horizon Technology Finance Corporation and Tengion, Inc., dated as of March 14, 2011. (Incorporated by reference to Exhibit 10.1 to our Current Report on Form 8-K, filed on March 16, 2011).
|
|
10.13
|
Master Security Agreement No. 5081099 by and between Oxford Finance Corporation and Tengion, Inc., dated as of July 20, 2005. (Incorporated by reference to Exhibit 10.14 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.14
|
Amendment Agreement by and between Oxford Finance Corporation and Tengion, Inc., dated as of April 3, 2006. (Incorporated by reference to Exhibit 10.15 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.15
|
Amendment Agreement by and between Oxford Finance Corporation and Tengion, Inc., dated as of December 28, 2006. (Incorporated by reference to Exhibit 10.16 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.16
|
Machinery and Equipment Loan Fund #25-9-779 Loan Agreement by and between the Commonwealth of Pennsylvania, acting through the Department of Community and Economic Development, and Tengion, Inc., dated as of December 20, 2007. (Incorporated by reference to Exhibit 10.17 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.17
|
Machinery and Equipment Loan Fund Security Agreement by and between the Commonwealth of Pennsylvania, acting through the Department of Community and Economic Development, and Tengion, Inc., dated as of December 20, 2007. (Incorporated by reference to Exhibit 10.18 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
Exhibit No.
|
Description
|
|
10.18
|
Offer Letter by and between Steven Nichtberger and the Company, dated as of May 25, 2004. (Incorporated by reference to Exhibit 10.19 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).#
|
|
10.19
|
Offer Letter Amendment by and between Steven Nichtberger and the Company, dated as of December 22, 2008. (Incorporated by reference to Exhibit 10.20 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).#
|
|
10.20
|
Indemnification Agreement by and between Steven Nichtberger and the Company, dated as of December 2, 2004. (Incorporated by reference to Exhibit 10.21 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.21
|
Restricted Stock Purchase Agreement by and between the Company and Steven Nichtberger, dated as of August 16, 2004. (Incorporated by reference to Exhibit 10.22 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.22
|
Restricted Stock Purchase Agreement by and between the Company and Steven Nichtberger, dated as of May 25, 2004. (Incorporated by reference to Exhibit 10.23 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.23
|
Offer Letter by and between Tim Bertram and Tengion, Inc., dated July 28, 2004. (Incorporated by reference to Exhibit 10.24 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).#
|
|
10.24
|
Restricted Stock Agreement by and between Steven Nichtberger and the Company, dated as of May 26, 2004. (Incorporated by reference to Exhibit 10.25 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.25
|
Offer Letter Amendment by and between Tim Bertram and the Company, dated as of December 22, 2008. (Incorporated by reference to Exhibit 10.26 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).#
|
|
10.26
|
Restricted Stock Purchase Agreement by and between the Company and Tim Bertram, dated as of August 16, 2004. (Incorporated by reference to Exhibit 10.27 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
Exhibit No.
|
Description
|
|
10.27
|
Restricted Stock Purchase Agreement by and between the Company and Tim Bertram, dated as of July 28, 2004. (Incorporated by reference to Exhibit 10.28 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.28
|
Offer Letter by and between A. Brian Davis and the Company, dated as of July 29, 2010. (Incorporated by reference to Exhibit 10.1 to our Quarterly Report on Form10-Q for the quarter ended June 30, 2010).#
|
|
10.29
|
|
|
10.30
|
Offer Letter by and between Mark Stejbach and the Company, dated June 24, 2008. (Incorporated by reference to Exhibit 10.32 to Amendment No. 1 to our Registration Statement on Form S-1 filed on January 29, 2010 (Registration No. 333-164011)).
|
|
10.31
|
Offer Letter Amendment by and between Mark Stejbach and the Company, dated as of December 22, 2008. (Incorporated by reference to Exhibit 10.33 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
10.32
|
Form of Indemnification Agreement. (Incorporated by reference to Exhibit 10.33 to Amendment No. 3 to our Registration Statement on Form S-1 filed on March 17, 2010 (Registration No. 333-164011)).
|
|
16.1
|
Letter from KPMG LLP. (Incorporated by reference to Exhibit 16.1 to our Registration Statement on Form S-1 filed on December 23, 2009 (Registration No. 333-164011)).
|
|
23.1
|
|
|
23.2
|
|
|
24.1
|
|
|
31.1
|
|
|
31.2
|
|
|
32.1
|
|
|
Exhibit No.
|
Description
|
|
32.2
|
|
|
# -
|
Indicates a management contract or any compensatory plan, contract or arrangement.
|
|
* -
|
Confidential status has been granted for certain portions thereof pursuant to a Commission Order granted April 9, 2010. Such provisions have been filed separately with the Commission.
|