UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
(Mark One)
|
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended
December 31, 2010
|
|
or
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to
|
Commission file number:
001-34703
Alimera Sciences,
Inc.
(Exact name of registrant as
specified in its charter)
| |
|
|
|
Delaware
|
|
20-0028718
|
|
(State or other jurisdiction
of
incorporation or organization)
|
|
(I.R.S. Employer
Identification Number)
|
| |
|
|
|
6120 Windward Parkway,
Suite 290 Alpharetta, GA
|
|
30005
(Zip Code)
|
|
(Address of principal executive
offices)
|
|
|
(678) 990-5740
(Registrant’s
telephone number, including area code)
Securities
registered pursuant to Section 12(b) of the Act:
| |
|
|
|
Common Stock, $0.01 par value per share
|
|
The NASDAQ Stock Market LLC
|
|
(Title of each class)
|
|
(Name of each exchange on which
registered)
|
Securities registered pursuant to Section 12(g) of the
Act: None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
(§ 232.405 of this chapter) during the preceding
12 months (or for such shorter period that the registrant
was required to submit and post such
files). Yes o No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
(§ 229.405 of this chapter) is not contained herein,
and will not be contained, to the best of registrant’s
knowledge, in definitive proxy or information statements
incorporated by reference in Part III of this
Form 10-K
or any amendment to this
Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in Rule
12b-2 of the
Exchange Act. (Check one):
|
|
|
|
| Large
accelerated
filer o
|
Accelerated
filer o
|
Non-accelerated
filer þ
|
Smaller
reporting
company o
|
(Do not check if a smaller reporting company)
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the
Act). Yes o No þ
As of June 30, 2010, the aggregate market value of the
Common Stock held by non-affiliates of the registrant was
$17,376,806, based on the closing price of the registrant’s
Common Stock, as reported by the NASDAQ Global Market. Shares of
Common Stock held by each executive officer, director and
stockholders known by the registrant to own 10% or more of the
outstanding stock based on public filings and other information
known to the registrant have been excluded since such persons
may be deemed affiliates. This determination of affiliate status
is not necessarily a conclusive determination for other purposes.
DOCUMENTS
INCORPORATED BY REFERENCE
Specified portions of the registrant’s proxy statement with
respect to the registrant’s 2011 Annual Meeting of
Stockholders, which is to be filed pursuant to
Regulation 14A within 120 days after the end of the
registrant’s fiscal year ended December 31, 2010, are
incorporated by reference into Part III of this
Form 10-K.
Alimera
Sciences, Inc.
Form 10-K
Table of Contents
The term “ILUVIEN” is our trademark. All other
trademarks, trade names and service marks appearing in this
prospectus are the property of their respective owners.
2
PART I
SPECIAL
NOTE REGARDING FORWARD-LOOKING STATEMENTS AND
PROJECTIONS
Various statements in this report are “forward-looking
statements” within the meaning of the Private Securities
Litigation Reform Act of 1995. Forward-looking statements may
appear throughout this report, including without limitation, the
following sections: Item 1 “Business,”
Item 1A “Risk Factors,” and Item 7
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations.” Words such as, but
not limited to, “anticipate,” “believe,”
“estimate,” “expect,” “intend,”
“may,” “plan,” “contemplates,”
“predict,” “project,” “targets,”
“likely,” “potential,” “continue,”
“will,” “would,” “should,”
“could,” or the negative of these terms and similar
expressions or words, identify forward-looking statements. The
events and circumstances reflected in the Company’s
forward-looking statements may not occur and actual results
could differ materially from those projected in the
Company’s forward-looking statements. Meaningful factors
which could cause actual results to differ include, but are not
limited to:
|
|
|
| |
•
|
delay in or failure to obtain regulatory approval of the
Company’s product candidates;
|
| |
| |
•
|
uncertainty as to the Company’s ability to commercialize,
and market acceptance of, the Company’s product candidates;
|
| |
| |
•
|
the extent of government regulations;
|
| |
| |
•
|
uncertainty as to the relationship between the benefits of the
Company’s product candidates and the risks of their
side-effect profiles;
|
| |
| |
•
|
dependence on third-party manufacturers to manufacture the
Company’s product candidates in sufficient quantities and
quality;
|
| |
| |
•
|
uncertainty of clinical trial results;
|
| |
| |
•
|
limited sales and marketing infrastructure;
|
| |
| |
•
|
inability of our outside sales force to successfully sell and
market ILUVIEN in the U.S. following regulatory
approval; and
|
| |
| |
•
|
the Company’s ability to operate its business in compliance
with the covenants and restrictions that it is subject to under
its credit facility.
|
All written and verbal forward-looking statements attributable
to us or any person acting on our behalf are expressly qualified
in their entirety by the cautionary statements contained or
referred to in this section. We caution investors not to rely
too heavily on the forward-looking statements we make or that
are made on our behalf. We undertake no obligation, and
specifically decline any obligation, to publicly update or
revise any forward-looking statements, whether as a result of
new information, future events or otherwise.
We encourage you to read the discussion and analysis of our
financial condition and our consolidated financial statements
contained in this annual report on
Form 10-K.
We also encourage you to read Item 1A of Part 1 of
this annual report on
Form 10-K,
entitled “Risk Factors,” which contains a more
complete discussion of the risks and uncertainties associated
with our business. In addition to the risks described above and
in Item 1A of this report, other unknown or unpredictable
factors also could affect our results. There can be no assurance
that the actual results or developments anticipated by us will
be realized or, even if substantially realized, that they will
have the expected consequences to, or effects on, us. Therefore
no assurance can be given that the outcomes stated in such
forward-looking statements and estimates will be achieved.
Overview
Alimera Sciences, Inc. (We, Alimera or the Company) is a
biopharmaceutical company that specializes in the research,
development and commercialization of prescription ophthalmic
pharmaceuticals. We are presently
3
focused on diseases affecting the back of the eye, or retina,
because we believe these diseases are not well treated with
current therapies and represent a significant market opportunity.
Our most advanced product candidate is
ILUVIEN®,
which we are developing for the treatment of diabetic macular
edema (DME). DME is a disease of the retina that affects
individuals with diabetes and can lead to severe vision loss and
blindness. In September 2010 we completed two Phase 3 pivotal
clinical trials (collectively, our
FAMEtm
Study) for ILUVIEN involving 956 patients in sites across
the U.S., Canada, Europe and India to assess the efficacy and
safety of ILUVIEN in the treatment of DME. Based on our analysis
of the month 24 clinical readout from our FAME Study in December
2009, we filed a New Drug Application (NDA) in June 2010 for the
low dose of ILUVIEN in the U.S. with the U.S. Food and
Drug Administration (FDA), followed by registration filings in
the United Kingdom, Austria, France, Germany, Italy, Portugal
and Spain in July 2010. In December 2010, we received a Complete
Response Letter (CRL) from the FDA regarding our NDA. The FDA
issued the CRL to communicate its decision that the NDA could
not be approved in its present form. No new clinical studies
were requested by the FDA in the CRL. However, the FDA asked us
for analyses of the safety and efficacy data through the end of
the FAME Study to further assess the relative benefits and risks
of ILUVIEN. We are currently preparing the analyses the FDA
requested having completed the FAME Study and publicly released
data on February 3, 2011. The FDA is also seeking
additional information regarding controls and specifications
concerning the manufacturing, packaging and sterilization of
ILUVIEN, which we are in the process of compiling. We currently
anticipate submitting our response to the CRL to the FDA early
in the second quarter of 2011. Our submission to the FDA will be
considered a Class 2 response, which will provide for a
review period of up to an additional six months for our NDA.
Based on our discussions with the FDA we anticipate that the FDA
will call an advisory committee during this review. If our NDA
for ILUVIEN is approved by the FDA, we plan to commercialize
ILUVIEN in the U.S. by marketing and selling it to retinal
specialists as early as late 2011.
Additionally, we plan to submit the additional safety and
efficacy data through the final readout at the end of the FAME
Study to regulatory authorities in the United Kingdom, Austria,
France, Germany, Italy, Portugal and Spain in the second quarter
of 2011. If ILUVIEN is approved by the European regulatory
authorities, we plan to commercialize ILUVIEN, directly or
through a partnership, in the United Kingdom, Austria, France,
Germany, Italy, Portugal and Spain.
According to the Centers for Disease Control and Prevention
(CDC), the number of Americans diagnosed with diabetes had
increased from approximately 8.1 million people in 1994 to
approximately 18.8 million people in 2010. Per the
International Diabetes Federation Atlas, the estimated
prevalence of people diagnosed with diabetes for 2010 has
increased to 285 million people worldwide and that this
number is expected to reach 438 million people by 2030. All
patients with diabetes are at risk of developing some form of
diabetic retinopathy, an ophthalmic condition of diabetes that
presents with symptoms that include the swelling and leakage of
blood vessels within the retina or the abnormal growth of new
blood vessels on the surface of the retina. As reported by the
American Diabetes Association, in the U.S. diabetic
retinopathy causes approximately 12,000 to 24,000 new cases of
blindness each year, making diabetes the leading cause of new
cases of blindness in adults aged 20 to 74. When the blood
vessel leakage of diabetic retinopathy causes swelling in the
macula, the part of the eye responsible for central vision, the
condition is called DME. The Wisconsin Epidemiologic Study of
Diabetic Retinopathy found that over a ten-year period
approximately 19% of diabetics studied were diagnosed with DME.
Based on this study and the current U.S. diabetic
population, we estimate the incidence of DME in the U.S. to
be approximately 340,000 cases annually. As the population of
diabetics increases, we expect the annual incidence of diagnosed
DME to increase.
There are no ophthalmic drug therapies currently approved by the
FDA for the treatment of DME. The current standard of care for
the treatment of DME is laser photocoagulation. Laser
photocoagulation is a retinal procedure in which a laser is used
to cauterize leaky blood vessels or to apply a pattern of burns
to reduce edema. This procedure has undesirable side effects
including partial loss of peripheral and night vision. As a
result of these side effects and a desire for improved visual
outcomes, retinal specialists have supplemented laser
photocoagulation with alternate off-label therapies for the
treatment of DME, including injections of corticosteroids and
anti-vascular endothelial growth factor (anti-VEGF) agents. Both
corticosteroids and anti-VEGFs have shown improved visual acuity
in DME patients in non-pivotal clinical trials but are
4
limited by a need for multiple injections to maintain a
therapeutic effect. Corticosteroids have historically been
associated with significant increases in intraocular pressure
(IOP), which may increase the risk of glaucoma, and the
acceleration of cataract formation.
ILUVIEN is inserted in the back of the patient’s eye to a
placement site that takes advantage of the eye’s natural
fluid dynamics to deliver the non-proprietary corticosteroid
fluocinolone acetonide (FAc). ILUVIEN is inserted with a device
that employs a 25-gauge needle which allows for a self-sealing
wound. In the U.S., this procedure is non-surgical and is
performed in the retinal specialist’s office. ILUVIEN is an
intravitreal insert designed to provide a therapeutic effect for
up to 36 months by delivering sustained
sub-microgram
levels of FAc. ILUVIEN has demonstrated efficacy in the
treatment of DME in our FAME Study. Additionally, by providing
lower exposure to corticosteroids and focusing the delivery to
the back of the eye, we believe that the adverse events
associated with the use of ILUVIEN are within the acceptable
limits of a drug for the treatment of DME.
ILUVIEN is also being studied in three Phase 2 clinical trials
for the treatment of the dry form of age-related macular
degeneration (AMD), the wet form of AMD and retinal vein
occlusion (RVO). In addition to our activities related to the
development and commercialization of ILUVIEN, we are also
conducting testing on two classes of nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase inhibitors for which we
have acquired exclusive, worldwide licenses from Emory
University. Our initial focus is on the use of NADPH oxidase
inhibitors in the treatment of dry AMD. We plan to evaluate the
use of NADPH oxidase inhibitors in the treatment of other
diseases of the eye, including wet AMD and diabetic retinopathy.
We will pursue the development, license and acquisition of
rights to compounds and technologies with the potential to treat
diseases of the eye that we believe are not well treated by
current therapies.
Our commercialization strategy is to establish ILUVIEN as a
leading therapy for the treatment of DME and subsequently for
any other indications for which ILUVIEN proves safe and
effective. We are led by an executive team with extensive
development and commercialization expertise with ophthalmic
products including the launch and management of Visudyne, a drug
product sponsored by Novartis and the first pharmacological
treatment indicated for patients with wet AMD. We intend to
capitalize on our management’s experience and expertise in
marketing eye-care products, by marketing and selling ILUVIEN to
the approximately 1,600 retinal specialists practicing in the
approximately 900 retina centers across the U.S. and
Canada. If ILUVIEN is approved by the European regulatory
authorities, we plan to commercialize ILUVIEN, directly or
through a partnership, in the United Kingdom, Austria, France,
Germany, Italy, Portugal and Spain. Our commercialization
strategy is subject to and dependent upon regulatory approval of
ILUVIEN for the treatment of DME.
Business
Strategy
We are presently focused on diseases affecting the back of the
eye, or retina, because we believe these diseases are not well
treated with current therapies and represent a significant
market opportunity. Our business strategy is to:
|
|
|
| |
•
|
Pursue FDA Approval for ILUVIEN. In June 2010,
we filed an NDA for ILUVIEN for the treatment of DME based upon
the month 24 clinical readout from our FAME Study. We received a
CRL from the FDA in December 2010 requesting additional analyses
and information for our NDA through the end of the FAME Study,
and plan to submit a response early in the second quarter of
2011.
|
| |
| |
•
|
Maximize the Commercial Success of ILUVIEN. If
approved by the FDA, we intend to capitalize on our
management’s past experience and expertise in marketing
eye-care products including the launch and management of
Visudyne (Novartis) by marketing and selling ILUVIEN through our
outside sales force to the approximately 1,600 retinal
specialists practicing in the approximately 900 retina centers
in the U.S. and Canada. We intend to commercialize ILUVIEN,
directly or through a partnership, outside North America.
|
| |
| |
•
|
Assess the Effectiveness of ILUVIEN for Additional Retinal
Diseases. We believe that ILUVIEN has the
potential to address additional retinal diseases including,
among others, dry AMD, wet AMD and
|
5
|
|
|
| |
|
RVO. ILUVIEN is being studied in three Phase 2 clinical trials
with retinal specialists to assess the safety and efficacy of
ILUVIEN for the treatment of these diseases of the eye.
|
|
|
|
| |
•
|
Develop Our Existing Ophthalmic Product
Pipeline. We have acquired exclusive, worldwide
licenses of rights under patent applications for two classes of
NADPH oxidase inhibitors from Emory University. We believe that
the management of oxidative stress is an important strategy in
managing the development and progression of diseases of the eye,
and we believe that NADPH oxidase inhibitors have the potential
to manage oxidative stress. Our initial focus is on the use of
NADPH oxidase inhibitors in the treatment of dry AMD. We plan to
evaluate the use of NADPH oxidase inhibitors in the treatment of
other diseases of the eye, including wet AMD and diabetic
retinopathy.
|
| |
| |
•
|
Expand Our Ophthalmic Product Pipeline. We
believe there are further unmet medical needs in the treatment
of ophthalmic diseases. Toward that end, we intend to leverage
our management’s expertise and its broad network of
relationships in continuing to evaluate in-licensing and
acquisition opportunities for compounds and technologies with
applications in diseases affecting the eye.
|
Disease
Overview and Market Opportunity
Diabetes
and Diabetic Retinopathy
Diabetes mellitus, and its systemic and ophthalmic
complications, represents an enormous public health threat in
the U.S. According to the U.S. Centers for Disease
Control and Prevention (CDC, the number of Americans diagnosed
with diabetes has increased from approximately 8.1 million
people in 1994 to approximately 18.8 million people in
2010. In addition to diagnosed cases, the CDC estimates that an
additional 7.0 million Americans with diabetes are
currently undiagnosed and are therefore not being monitored and
treated to control their disease and prevent systemic and
ophthalmic complications. With better diagnosis methodologies
and improved public awareness, the number of persons diagnosed
with and being treated for diabetes is expected to increase. Per
the International Diabetes Federation Atlas, the estimated
prevalence of diabetes for 2010 has increased to
285 million people worldwide and this number is expected to
reach 438 million people by 2030.
All patients with diabetes are at risk of developing some form
of diabetic retinopathy, an ophthalmic complication of diabetes
that presents with symptoms including the swelling and leakage
of blood vessels within the retina or the abnormal growth of new
blood vessels on the surface of the retina. According to the
American Diabetes Association, diabetic retinopathy causes
approximately 12,000 to 24,000 new cases of blindness in the
U.S. each year, making diabetes the leading cause of new
cases of blindness in adults aged 20 to 74. Diabetic
retinopathy can be divided into either non-proliferative or
proliferative retinopathy. Non-proliferative retinopathy (also
called background retinopathy) develops first and causes
increased capillary permeability, microaneurysms, hemorrhages,
exudates, macular ischemia and macular edema (thickening of the
retina caused by fluid leakage from capillaries). Proliferative
retinopathy is an advanced stage of diabetic retinopathy which,
in addition to characteristics of non-proliferative retinopathy,
results in the growth of new blood vessels. These new blood
vessels are abnormal and fragile, growing along the retina and
along the surface of the clear, vitreous gel that fills the
inside of the eye. By themselves, these blood vessels do not
cause symptoms or vision loss. However, these blood vessels have
thin, fragile walls that are prone to leakage and hemorrhage.
Diabetic
Macular Edema
DME, the primary cause of vision loss associated with diabetic
retinopathy, is a disease affecting the macula, the part of the
retina responsible for central vision. When the blood vessel
leakage of diabetic retinopathy causes swelling in the macula,
the condition is called DME. The onset of DME is painless and
may go undetected by the patient until it manifests with the
blurring of central vision or acute vision loss. The severity of
this blurring may range from mild to profound loss of vision.
The Wisconsin Epidemiologic Study of Diabetic Retinopathy found
that over a ten-year period approximately 19% of diabetics
studied were diagnosed with DME. Based on this study and the
current diabetic population in the U.S., we estimate the
6
incidence of DME in the U.S. to be approximately 340,000
cases annually. As the population of diabetics increases, we
expect the annual incidence of diagnosed DME to increase.
Limitations
of Current Treatments for DME
There are no ophthalmic drug therapies presently approved by the
FDA for the treatment of DME. The current standard of care for
the treatment of DME is laser photocoagulation. Laser
photocoagulation is a retinal procedure in which a laser is used
to cauterize leaky blood vessels or to apply a pattern of burns
to reduce edema. Visual acuity gains are seen with this therapy,
although results are highly variable and we believe that it may
take more than eight months for median visual acuity to improve.
Further, this procedure has undesirable side effects including
partial loss of peripheral and night vision. As a result of
these side effects and a desire for improved visual outcomes,
retinal specialists have supplemented laser photocoagulation
with alternate off-label therapies for the treatment of DME,
including injections of corticosteroids and anti- VEGF agents.
Both corticosteroids and anti-VEGFs have shown improved visual
acuity in DME patients in non-pivotal clinical trials but are
limited by a need for multiple injections to maintain a
therapeutic effect. Corticosteroids have historically been
associated with significant increases in intraocular pressure
(IOP), which may increase the risk of glaucoma, and the
acceleration of cataract formation.
ILUVIEN
Overview
Our most advanced product candidate is ILUVIEN, an intravitreal
insert designed to provide a therapeutic effect for up to
36 months in the treatment of DME by delivering sustained
sub-microgram
levels of FAc, a non-proprietary corticosteroid with
demonstrated efficacy in the treatment of ocular disease.
Intravitreal refers to the space inside the eye behind the lens
that contains the jelly-like substance called vitreous. DME is a
disease of the retina which affects individuals with diabetes
and can lead to severe vision loss and blindness. ILUVIEN is
inserted in the back of the patient’s eye using an
insertion device (the ILUVIEN inserter) employing a 25-gauge
needle which allows for a self-sealing wound. This insertion is
very similar to the administration of an intravitreal injection,
a procedure commonly employed by retinal specialists. In the
U.S., this procedure is non-surgical and is performed in the
retinal specialist’s office. Based on our analysis of the
month 24 clinical readout from our FAME Study, we believe
ILUVIEN improves vision while reducing side effects commonly
associated with the use of corticosteroids for the following
reasons:
|
|
|
| |
•
|
ILUVIEN delivers FAc. The active
pharmaceutical ingredient in ILUVIEN is FAc, which has
demonstrated efficacy in the treatment of DME in our FAME Study.
|
| |
| |
•
|
ILUVIEN delivers sustained
sub-microgram
levels of a steroid to the eye. In our clinical
trials we studied two doses of ILUVIEN (a high-dose with an
initial release of approximately 0.45µg per day and a
low-dose with an initial release of approximately 0.23µg
per day) to determine the lowest dose possible that will provide
efficacy for the treatment of DME. The dosage levels of ILUVIEN
provide lower exposure to corticosteroids than other intraocular
dosage forms currently available. Based on our analysis of the
month 24 clinical readout from our FAME Study in December 2009,
we are pursuing the approval of the low dose of ILUVIEN.
|
| |
| |
•
|
ILUVIEN delivers a therapeutic effect for up to
36 months. In vitro release kinetics have
shown that ILUVIEN provides sustained delivery of
sub-microgram
levels of FAc over time. Based on the results of the FAME Study,
ILUVIEN provides a therapeutic effect in the treatment of DME
for up to 36 months.
|
| |
| |
•
|
ILUVIEN’s placement utilizes the eye’s natural
fluid dynamics. There are two natural currents of
fluid within the eye; one to the front of the eye and the other
to the back of the eye, or retina. We believe that
ILUVIEN’s delivery of sustained
sub-microgram
levels of FAc and insertion into the back of the eye, a position
that we believe optimizes delivery of FAc to the retina by
utilizing these natural currents, maximizes efficacy and
mitigates possible side effects.
|
7
|
|
|
| |
•
|
ILUVIEN is inserted using a 25-gauge
needle. Needle gauge determines the size of the
wound that is created. ILUVIEN is inserted into the eye using a
25-gauge needle, which results in a wound that is small enough
to seal itself after the needle is removed thus eliminating the
need for additional intervention. Using a larger needle would
require a more complicated insertion procedure to create a
self-sealing wound.
|
Fluocinolone
Acetonide
Fluocinolone acetonide (FAc) is the active compound in ILUVIEN
and a member of the class of steroids known as corticosteroids.
FAc is a non-proprietary corticosteroid that has a history of
use in treating ocular disease as the active compound in
Bausch & Lomb Incorporated’s product Retisert (a
surgically implanted intravitreal drug delivery device approved
for the treatment of chronic non-infectious posterior uveitis).
Corticosteroids have demonstrated a range of pharmacological
actions, including inhibition of inflammation, inhibition of
leukostasis, up regulation of occludin, inhibition of release of
certain inflammatory cytokines and suppression of VEGF
secretion. These pharmacological actions have the potential to
treat various ocular conditions, including DME, dry AMD, wet AMD
and RVO. However, FAc shares many of the same side effects as
other corticosteroids currently available for intraocular use,
including increased IOP, which may increase the risk of
glaucoma, and the acceleration of cataract formation.
ILUVIEN
is Positioned to Reduce Side Effects
Based on our analysis of the final clinical readout from our
FAME Study through month 36, it appears that ILUVIEN mitigates
the incidence of steroid-induced IOP elevations commonly
associated with the intraocular use of corticosteroids, which we
believe is due to its location in the posterior portion of the
eye. Fluid, or aqueous humor, generated at the ciliary body,
located just behind the iris, flows within the eye primarily via
two currents. The predominant current flows through the iris
into the anterior chamber and exits the eye mainly through the
trabecular outflow pathway. Another current of outflow is
directed toward the back of the eye.
The side effect of increased IOP associated with corticosteroids
in certain people is directly related to the interaction of
corticosteroids with the cells of the trabecular meshwork, a
specialized tissue that acts as a filter located in the front of
the eye. In some individuals, corticosteroids result in a
build-up of
debris in this meshwork, increasing resistance to outflow, and
increasing pressure inside the eye. The positioning of ILUVIEN
allows it to take advantage of the posterior flow of fluid away
from the trabecular meshwork of the eye. We believe this
positioning minimizes the anterior chamber exposure to FAc and
mitigates the incidence of IOP elevations.
ILUVIEN
Provides Sustained
Sub-Microgram
Delivery
ILUVIEN consists of a tiny polyimide tube with membrane caps,
licensed by us from pSivida US, Inc. (pSivida) that is filled
with 190µg of FAc in a polyvinyl alcohol matrix. ILUVIEN is
non-bioerodable; however, both polyimide and the polyvinyl
alcohol matrix are biocompatible with ocular tissues and have
histories of safe use within the eye.
The low dose of ILUVIEN provides sustained
sub-microgram
levels of FAc and demonstrates a therapeutic effect for up to
36 months in our FAME Study. We believe that ILUVIEN’s
ability to deliver
sub-microgram
levels of FAc mitigates the incidence of IOP elevations commonly
associated with the intraocular use of corticosteroids.
The
ILUVIEN Inserter
We developed the ILUVIEN inserter, a custom insertion system for
ILUVIEN, which includes improvements over the modified syringe
used during our two Phase 3 pivotal clinical trials
(individually referred to as Trial A and Trial B, and
collectively as our FAME Study). These improvements include
ergonomic design features, a transparent window to visually
confirm ILUVIEN’s presence within the inserter and a longer
needle and markings to guide retinal specialists to the proper
insertion point. As was the case with the modified
8
syringe used during our FAME Study, the ILUVIEN inserter uses a
25-gauge needle which results in a wound that is small enough to
seal itself after ILUVIEN has been inserted into the back of the
eye and the needle has been removed. We believe that a 25-gauge
needle is the smallest needle capable of delivering ILUVIEN into
the back of eye. In the U.S., this procedure is non-surgical and
is performed in the retinal specialist’s office. The
ILUVIEN inserter is also being used in our Phase 2 trial for the
use of ILUVIEN in the treatment of RVO.
ILUVIEN
Clinical Development Program
Development
Program for the Treatment of DME
In September 2010, we completed the FAME Study for ILUVIEN
involving 956 patients in sites across the U.S., Canada,
Europe and India to assess the efficacy and safety of ILUVIEN in
the treatment of DME. Combined enrollment was completed in
October 2007, and the month 24 clinical readout from our FAME
Study was received in December 2009. Based on our analysis of
the month 24 clinical readout from our FAME Study in December
2009, we filed a NDA with the FDA in June 2010 for the low dose
of ILUVIEN in the U.S., followed by registration filings in the
United Kingdom, Austria, France, Germany, Italy, Portugal and
Spain in July 2010. In December 2010, we received a CRL from the
FDA regarding our NDA. The FDA issued the CRL to communicate its
decision that the NDA could not be approved in its present form.
No new clinical studies were requested by the FDA in the CRL.
However, the FDA asked us for analyses of the safety and
efficacy data through the end of the FAME Study to further
assess the relative benefits and risks of ILUVIEN. We are
currently preparing the analyses the FDA requested having
completed the FAME Study and publicly released data on
February 3, 2011. The FDA is also seeking additional
information regarding controls and specifications concerning the
manufacturing, packaging and sterilization of ILUVIEN, which we
are in the process of compiling. We currently anticipate
submitting our response to the CRL to the FDA early in the
second quarter of 2011. Our submission to the FDA will be
considered a Class 2 response, which will provide for a
review period of up to an additional six months for our NDA.
Based on our discussions with the FDA we anticipate that the FDA
will call an advisory committee during this review.
Consistent with the FDA requirement for registration and
approval of drugs being developed for diabetic retinopathy,
including DME, the primary efficacy endpoint for our FAME Study
was the difference in the percentage of patients whose best
corrected visual acuity (BCVA) improved from baseline by 15 or
more letters on the Early Treatment Diabetic Retinopathy Study
(ETDRS) eye chart between the treatment and control groups at
month 24. The ETDRS eye chart is the standard used in clinical
trials for measuring sharpness of sight as established by the
National Eye Institute’s Early Treatment Diabetic
Retinopathy Study. In addition, the FDA required a numerical
comparison of the percentage of patients with BCVA improvement
of 15 or more letters between the month 24 and month 18 data to
determine if the month 24 results are equal to or greater than
the month 18 results. Patients enrolled in our FAME Study were
followed for 36 months. Although we submitted the month 24
results to the FDA for approval, in the CRL the FDA requested
the additional 12 months of clinical data from the
completed FAME Study through month 36 for review. We are
currently preparing a response to the CRL for submission early
in the second quarter of 2011 including this additional efficacy
and safety data. We also plan to submit efficacy and safety data
through the end of the trial to the applicable regulatory
authorities in the United Kingdom, Austria, France, Germany,
Italy, Portugal and Spain in the second quarter of 2011.
In the European Union, we are utilizing the decentralized
registration procedure. The ILUVIEN insertion system will not
require a separate device application, but it must meet the
safety and regulatory requirements of the applicable regulatory
authorities when evaluated as part of the drug product marketing
application.
FAME
Study
We initiated our FAME Study in September 2005. Trial A and Trial
B had identical protocols and completed enrollment in October
2007 with a total of 956 patients across 101 academic and
private practice centers. Trial A drew patients from sites
located in the northern regions of the U.S., Europe and India
and all sites in Canada, while sites in the southern regions of
the U.S., India and Europe comprise Trial B.
9
Our FAME Study was designed to assess the safety and efficacy of
ILUVIEN in patients with DME involving the center of the macula,
and who had at least one prior macular laser treatment
12 weeks or more before study entry. The inclusion criteria
for our FAME Study were designed to select DME patients with
BCVA between 20/50 (68 letters on the ETDRS eye chart) and
20/400 (19 letters on the ETDRS eye chart) in the study eye and
no worse than 20/400 in the non-study eye. Patients who had
received steroid drug treatments for DME within three months of
screening or anti-VEGF injections within two months of
screening, and patients with glaucoma, ocular hypertension, IOP
greater than 21mmHg or concurrent therapy with IOP-lowering
agents in the study eye at screening were not eligible to
participate in this trial.
Patient characteristics, such as age, gender and baseline BCVA,
were balanced across the treatment and control groups. As part
of randomization, the patients were divided into two separate
groups, those with a baseline BCVA score greater than or equal
to 49 letters on the ETDRS eye chart and those with a baseline
BCVA score of less than 49 letters on the ETDRS eye chart.
We randomly assigned patients participating in our FAME Study to
one of three groups at a ratio of 2:2:1. The first two of these
groups were assigned to an active drug formulation and the third
group served as the control group, undergoing a sham insertion
procedure designed to mimic an intravitreal insertion. The
treatment groups consisted of one group receiving a low dose of
ILUVIEN and another group receiving a high dose of ILUVIEN. To
reduce potential bias, these trials used a randomized,
double-masked study design so that neither the patient nor the
investigational staff involved with assessing the patient knew
to which group the patient belonged. In order to simulate an
insertion and help to maintain proper patient masking, the sham
insertion procedure included all steps involved in the insertion
procedure, except that a blunt inserter without a needle was
used to apply pressure to the anesthetized eye.
As part of our FAME Study, investigators were able to re-treat
each patient with ILUVIEN following their month 12 follow up
visit if certain criteria were met. Through the end of the FAME
Study, 25.6% of patients were treated with more than one ILUVIEN
insert and 4.0% of patients were treated with three or more
ILUVIEN inserts.
Primary Efficacy Endpoint. The primary
efficacy endpoint for our FAME Study was the difference in the
percentage of patients with improved BCVA from baseline of 15 or
more letters on the ETDRS eye chart at month 24 between the
treatment and control groups.
Full Analysis Set. In December 2009, we
received the month 24 clinical readout for our FAME Study and
analyzed the full data set consistent with the recommendations
regarding the appropriate population for primary analysis as
described in the FDA-adopted International Conference on
Harmonization of Technical Requirements for Registration of
Pharmaceuticals for Human Use (ICH) Guidance E9,
“Statistical Principles for Clinical Trials.” ICH is a
joint initiative involving regulatory authorities and
pharmaceutical industry representatives from Europe, Japan and
the U.S. who discuss scientific and technical aspects of
product registration.
The full data set includes all 956 patients randomized into
our FAME Study, with data imputation employed, using “last
observation carried forward” (LOCF), for data missing
because of patients who discontinued the trial or are
unavailable for
follow-up
(the Full Analysis Set. As part of our analyses, we determined
statistical significance based on the Hochberg-Bonferroni
procedure (H-B procedure), which is a procedure employed to
control for multiple comparisons. We also made a target p-value
adjustment of 0.0001 to account for each of the nine instances
our independent data safety monitoring board reviewed unmasked
interim clinical data. These adjustments resulted in a required
p-value of 0.0491 or lower for each of Trial A and Trial B to
demonstrate statistical significance for both the low dose and
high dose of ILUVIEN. Based upon the H-B procedure, if either
dose of ILUVIEN in a trial did not meet statistical
significance, the alternate dose was required to achieve a
p-value of 0.02455 or lower in that trial to demonstrate
statistical significance.
In the Full Analysis Set, the primary efficacy endpoint was met
with statistical significance for both the low dose and the high
dose of ILUVIEN in Trial A and Trial B, as well as on a combined
basis. The table below summarizes the primary efficacy variable
results.
10
Patients
Gaining At Least 15 Letters At Month 24
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Trial A
|
|
|
Trial B
|
|
|
Study Group
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
|
|
Control
|
|
|
14/95
|
|
|
|
14.7
|
%
|
|
|
—
|
|
|
|
16/90
|
|
|
|
17.8
|
%
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Low Dose
|
|
|
51/190
|
|
|
|
26.8
|
%
|
|
|
0.029
|
|
|
|
57/186
|
|
|
|
30.6
|
%
|
|
|
0.030
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
High Dose
|
|
|
51/196
|
|
|
|
26.0
|
%
|
|
|
0.034
|
|
|
|
62/199
|
|
|
|
31.2
|
%
|
|
|
0.027
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Combined
|
|
|
Study Group
|
|
Individuals
|
|
|
%
|
|
|
P-value
|
|
|
|
|
Control
|
|
|
30/185
|
|
|
|
16.2
|
%
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Low Dose
|
|
|
108/376
|
|
|
|
28.7
|
%
|
|
|
0.002
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
High Dose
|
|
|
113/395
|
|
|
|
28.6
|
%
|
|
|
0.002
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additionally, as required by the FDA, a numerical comparison of
the responder rates at month 18 and month 24 in the Full
Analysis Set demonstrated that the responder rates for both the
low dose and high dose of ILUVIEN at month 24 were numerically
greater than the month 18 responder rates in both Trial A and
Trial B.
Based on these results, we filed an NDA in June 2010 for the low
dose of ILUVIEN in the U.S., followed by registration filings in
the United Kingdom, Austria, France, Germany, Italy, Portugal
and Spain in July 2010. In December 2010, we received a CRL from
the FDA regarding our NDA. The FDA issued the CRL to communicate
its decision that the NDA could not be approved in its present
form. No new clinical studies were requested by the FDA in the
CRL. However, the FDA asked us for analyses of the safety and
efficacy data through the end of the FAME Study to further
assess the relative benefits and risks of ILUVIEN. We are
currently preparing the analyses the FDA requested having
completed the FAME Study and publicly released data on
February 3, 2011.
The low dose of ILUVIEN, which we submitted to the FDA for
approval, in the Full Analysis Set for Trial A demonstrated
statistically significant differences between ILUVIEN patients
gaining 15 or more letters at month 30 and month 33 and the
control group. The therapeutic effect was maintained at month
36. The table below summarizes the primary efficacy variable
results for Trial A at months 30, 33 and 36.
Patients
Gaining At Least 15 Letters in Trial A
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
At Month 30
|
|
|
At Month 33
|
|
|
At Month 36
|
|
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
|
|
Control
|
|
|
14/95
|
|
|
|
14.7
|
%
|
|
|
—
|
|
|
|
16/95
|
|
|
|
16.8
|
%
|
|
|
—
|
|
|
|
18/95
|
|
|
|
18.9
|
%
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Low Dose
|
|
|
55/190
|
|
|
|
28.9
|
%
|
|
|
0.011
|
|
|
|
54/190
|
|
|
|
28.4
|
%
|
|
|
0.042
|
|
|
|
54/190
|
|
|
|
28.4
|
%
|
|
|
0.106
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
High Dose
|
|
|
53/196
|
|
|
|
26.9
|
%
|
|
|
0.023
|
|
|
|
56/196
|
|
|
|
28.6
|
%
|
|
|
0.034
|
|
|
|
53/196
|
|
|
|
27.0
|
%
|
|
|
0.142
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
11
In Trial B, statistically significant differences between
ILUVIEN patients gaining 15 or more letters over baseline were
demonstrated at month 30 and month 33 and the control group. The
table below summarizes the primary efficacy variable results for
Trial B at months 30, 33 and 36.
Patients
Gaining At Least 15 Letters in Trial B
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
At Month 30
|
|
|
At Month 33
|
|
|
At Month 36
|
|
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
|
|
Control
|
|
|
14/90
|
|
|
|
15.6
|
%
|
|
|
—
|
|
|
|
16/90
|
|
|
|
17.8
|
%
|
|
|
—
|
|
|
|
17/90
|
|
|
|
18.9
|
%
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Low Dose
|
|
|
63/186
|
|
|
|
33.9
|
%
|
|
|
0.002
|
|
|
|
55/186
|
|
|
|
29.6
|
%
|
|
|
0.046
|
|
|
|
54/186
|
|
|
|
29.0
|
%
|
|
|
0.086
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
High Dose
|
|
|
58/199
|
|
|
|
29.1
|
%
|
|
|
0.018
|
|
|
|
58/199
|
|
|
|
29.1
|
%
|
|
|
0.057
|
|
|
|
57/199
|
|
|
|
28.6
|
%
|
|
|
0.111
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The FDA has confirmed that there is no requirement to
demonstrate statistical significance in the two trials
individually or in the aggregate at month 36. Therefore, we
believe these results meet the criteria for replication of
efficacy in the two studies.
Trial A and B data combined demonstrated statistically
significant differences between ILUVIEN patients gaining 15 or
more letters over baseline at month 30, month 33 and month 36
and the control group. The table below summarizes the primary
efficacy variable results for Trial A and Trial B combined at
months 30, 33 and 36.
Patients
Gaining At Least 15 Letters in Trial A and Trial B
Combined
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
At Month 30
|
|
|
At Month 33
|
|
|
At Month 36
|
|
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
Individuals
|
|
|
%
|
|
|
P-Value
|
|
|
|
|
Control
|
|
|
28/185
|
|
|
|
15.1
|
%
|
|
|
—
|
|
|
|
32/185
|
|
|
|
17.3
|
%
|
|
|
—
|
|
|
|
35/185
|
|
|
|
18.9
|
%
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Low Dose
|
|
|
118/376
|
|
|
|
31.4
|
%
|
|
|
<0.001
|
|
|
|
109/376
|
|
|
|
29.0
|
%
|
|
|
0.004
|
|
|
|
108/376
|
|
|
|
28.7
|
%
|
|
|
0.018
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
High Dose
|
|
|
111/395
|
|
|
|
28.0
|
%
|
|
|
0.001
|
|
|
|
114/395
|
|
|
|
28.9
|
%
|
|
|
0.004
|
|
|
|
110/395
|
|
|
|
27.8
|
%
|
|
|
0.029
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
We intend to submit these data from the final readout of the
FAME Study through month 36 to the FDA in connection with our
response to the CRL, which we anticipate submitting early in the
second quarter of 2011. Our submission to the FDA will be
considered a Class 2 response, which will provide for a
review period of up to an additional six months for our NDA for
ILUVIEN. Based on our discussions with the FDA we anticipate
that the FDA will call an advisory committee during this review.
Additionally, we plan to submit the additional safety and
efficacy data through the final readout at the end of the FAME
Study to regulatory authorities in the United Kingdom, Austria,
France, Germany, Italy, Portugal and Spain in the second quarter
of 2011.
Additional Clinical Observations. In addition
to the primary efficacy variable, we also observed a number of
other clinically relevant results in the final readout from our
FAME Study through month 36. These observations included, among
others, the following:
|
|
|
| |
•
|
patients with improved BCVA of 15 or more letters at each follow
up visit;
|
| |
| |
•
|
patients with improved BCVA of 15 or more letters at any time
point;
|
| |
| |
•
|
other levels of BCVA improvement at month 24;
|
| |
| |
•
|
BCVA improvement of 15 or more letters relative to baseline BCVA;
|
| |
| |
•
|
Mean change in BCVA letter score;
|
| |
| |
•
|
BCVA improvements beyond month 24; and
|
| |
| |
•
|
decrease in excess foveal thickness.
|
12
The analyses of these Full Analysis Set observations set forth
below are presented for Trial A and Trial B on a combined basis
for patients who received the low dose of ILUVIEN in comparison
to the control group. Statements regarding statistical
significance do not reflect any adjustments to the p-values
calculated for multiple comparisons and analyses.
Patients With Improved BCVA of 15 Letters or More at Each
Follow Up Visit. Our analysis of the combined
Trial A and Trial B results of the FAME Study through month 36
indicates that the low dose of ILUVIEN provides an improvement
in BCVA as early as three weeks after insertion. The low dose of
ILUVIEN was statistically significantly better than the control
group in our FAME Study by week 3 of patient follow up, and
maintained a statistically significant advantage over the
control through month 36, with a peak efficacy of 31.4%
achieving improved BCVA of 15 or more letters from baseline at
month 30. The chart below demonstrates the treatment effect of
the low dose of ILUVIEN versus the control group, as measured by
an improvement in BCVA of 15 letters or more, at each scheduled
follow up visit during the FAME Study.
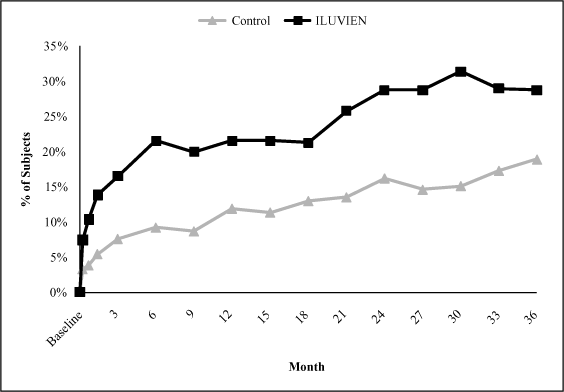
Patients With Improved BCVA of 15 or More Letters at Any Time
Point. Our analysis of the results of the FAME
Study through month 36 indicates that a significantly greater
percentage of patients receiving the low dose of ILUVIEN versus
the control group had an improvement in BCVA of 15 letters or
more when assessed at any follow up visit. During the
36 months of the FAME Study, 185 out of 376 patients
randomized to receive the low dose of ILUVIEN, or 49.2%,
demonstrated improved BCVA of 15 letters or more at any time
point compared to 60 out of 185 patients, or 32.4%,
randomized to the control group.
Other Levels of BCVA Improvement at Month
36. While the FDA’s requirement for the
registration and approval of drugs being developed for DME is
that the primary efficacy variable be based on an improvement in
BCVA of 15 letters or more, lesser degrees of improvement in
BCVA are considered clinically significant
13
by retinal physicians. The table below demonstrates the low dose
of ILUVIEN’s statistically significant improvements in BCVA
versus the control group at month 36 of our FAME Study.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Trial A & Trial B Combined
|
|
|
BCVA Improvement
|
|
Control
|
|
|
Low Dose
|
|
|
P-Value
|
|
|
|
|
Greater than 1 letter
|
|
|
57.8
|
%
|
|
|
69.7
|
%
|
|
|
|
|
|
Greater than 5 letters
|
|
|
45.4
|
%
|
|
|
58.8
|
%
|
|
|
|
|
|
Greater than 10 letters
|
|
|
33.5
|
%
|
|
|
43.4
|
%
|
|
|
|
|
BCVA Improvement of 15 or More Letters Relative to Baseline
BCVA. Our analysis of the results of the FAME
Study at month 36 indicates that ILUVIEN has a statistically
significant advantage over the control group irrespective of the
severity of a patient’s baseline BCVA. The table below
demonstrates the statistically significant treatment effect of
ILUVIEN versus the control group in patients with baseline BCVA
of more than 49 letters on the EDTRS eye chart, and patients
with BCVA of 49 letters or less on the EDTRS eye chart at
baseline.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Trial A & Trial B Combined
|
|
Baseline BCVA
|
|
Control
|
|
Low Dose
|
|
P-Value
|
|
|
|
Greater than 49 Letters
|
|
|
16.9
|
%
|
|
|
21.8
|
%
|
|
|
0.292
|
|
|
49 Letters or Less
|
|
|
24.5
|
%
|
|
|
44.3
|
%
|
|
|
0.022
|
|
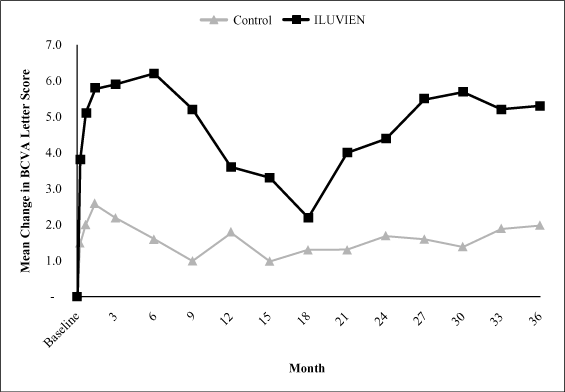
Mean Change in BCVA Letter Score. Our analysis
of the results of the FAME Study through month 36 indicates that
the low dose of ILUVIEN provided a more beneficial improvement
in visual acuity than the control group as analyzed by the mean
change in the BCVA letter score from baseline. As demonstrated
in the graph below, the mean change in BCVA for the patients
receiving the low dose of ILUVIEN was an increase of 5.3 letters
at month 36, peaking at an increase of 6.0 letters at month 6,
compared to an increase of 2.0 letters in the control group,
peaking at an increase of 2.6 letters at week 6. The low dose of
ILUVIEN was statistically significantly better than the control
group at month 36 (p-value 0.007).
14
During our FAME Study, patients that were phakic (had a natural
lens and no prior cataract surgery) at baseline, 61 of 121, or
50.4% of the control group and 192 of 235, or 81.7% of the low
dose of ILUVIEN had cataract formation reported as an adverse
event through month 36. For these same phakic patients, 27.3% of
the control group and 80.0% of the low dose group underwent
cataract surgery through month 24. For the patients in the low
dose group the median time to reporting cataract formation as an
adverse event was approximately 12 months from
randomization into the FAME Study. The median time to cataract
surgery was approximately 18 months. This interval, between
the report of cataract formation as an adverse event and
cataract surgery, accounts for the decrease in the mean change
in BCVA in patients receiving the low dose of ILUVIEN from the
month 6 follow up visit to the month 18 follow up visit.
The temporary effect of cataracts is further illustrated by
comparing the mean change in BCVA of the 140 low dose patients
that were pseudophakic (had an artificial lens) to the 235 that
were phakic (natural lens and no prior cataract surgery) at
baseline. The chart below shows the pseudophakic subset (those
who would not have vision affected by a cataract) achieved a
mean change in BCVA of more than 7 letters by month 6 and
maintained this mean change through month 36 while the phakic
subset experienced a decrease in the mean change in BCVA from
the month 6 follow up visit to the month 18 follow up visit. The
temporary decrease in mean change in BCVA in the phakic
population is consistent with the total low dose population.
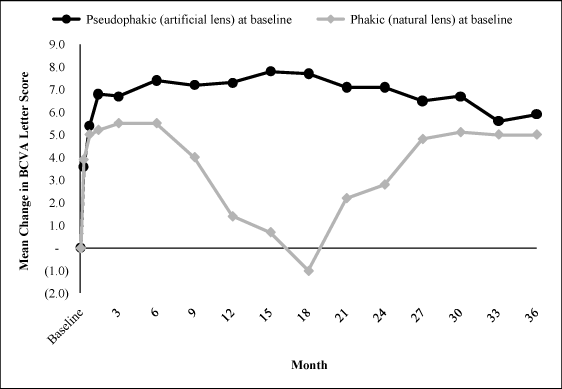
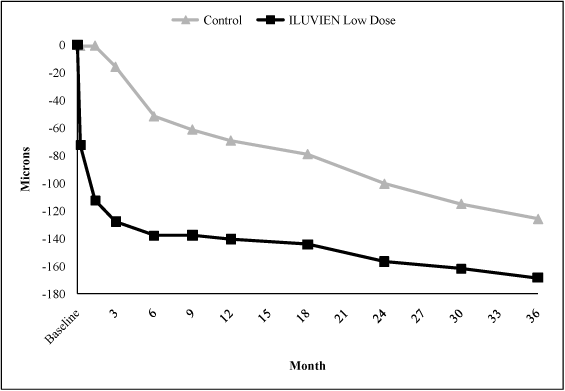
Decrease In Excess Foveal Thickness. In
addition to the functional measures of BCVA, we assessed the
ability of ILUVIEN to affect a decrease in excess foveal
thickness, an anatomic outcome, as measured by optical coherence
tomography. Excess foveal thickness is a measurement of the
swelling of the macula at its center point (known as the fovea).
We consider any measurement above 180 microns to represent
excess foveal thickness. Based on a review of the final clinical
readout through month 36, as summarized in the chart below,
patients receiving the low dose of ILUVIEN demonstrated a
statistically significant difference versus the control group in
decreasing excess foveal thickness by week 1 of patient follow
up of our FAME Study, and maintain a statistically significant
advantage through month 36. At month 36, patients receiving the
low
15
dose of ILUVIEN demonstrated a mean decrease in excess foveal
thickness of 168.3 microns versus 125.9 microns for the control
group.
Safety. Our safety assessment in connection
with the month 24 clinical readout of the FAME Study included
all reported adverse events at that time, regardless of a
patient’s progression in the FAME Study. Some reported
adverse events occurred beyond patients’ month 24 follow up
visits. We also assessed safety through the completion of the
FAME Study in month 36. ILUVIEN was well tolerated through this
readout in both the low and high dose patient populations. Our
assessment of adverse event data indicates that there is no
apparent risk of systemic adverse events to patients as a result
of the use of ILUVIEN. The use of corticosteroids in the eye is
primarily associated with two undesirable side effects:
increased IOP, which may increase the risk of glaucoma and
require additional procedures to manage, and the acceleration of
cataract formation. Excluding IOP related side effects and
cataract formation and surgery, we observed no significant eye
related adverse events when comparing both the low dose and high
dose patient populations to the control group. Thus, we believe
that the adverse events associated with the use of ILUVIEN are
within the acceptable limits of a drug for the treatment of DME.
16
The table below summarizes the IOP related adverse events
occurring in all patients randomized and treated in our FAME
Study at the time of the month 24 and month 36 clinical readouts
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of the Month 24 Data Base Lock
|
|
|
As of Trial Completion
|
|
|
|
|
Control
|
|
|
Low Dose
|
|
|
High Dose
|
|
|
Control
|
|
|
Low Dose
|
|
|
High Dose
|
|
|
|
|
N=185
|
|
|
N=375
|
|
|
N=393
|
|
|
N=185
|
|
|
N=375
|
|
|
N=393
|
|
|
|
|
IOP > 30 mmHg(1)
|
|
|
2.7
|
%
|
|
|
16.3
|
%
|
|
|
21.6
|
%
|
|
|
4.3
|
%
|
|
|
18.4
|
%
|
|
|
22.9
|
%
|
|
Trabeculoplasty
|
|
|
0.0
|
%
|
|
|
1.3
|
%
|
|
|
2.5
|
%
|
|
|
0.0
|
%
|
|
|
1.3
|
%
|
|
|
2.5
|
%
|
|
IOP-Lowering Surgeries
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Trabeculectomy (filtration)
|
|
|
0.0
|
%
|
|
|
2.1
|
%
|
|
|
5.1
|
%
|
|
|
0.0
|
%
|
|
|
2.7
|
%
|
|
|
5.6
|
%
|
|
Vitrectomy
|
|
|
0.0
|
%
|
|
|
0.3
|
%
|
|
|
0.5
|
%
|
|
|
0.0
|
%
|
|
|
0.3
|
%
|
|
|
0.5
|
%
|
|
Other Surgery Performed
|
|
|
0.5
|
%
|
|
|
1.6
|
%
|
|
|
2.5
|
%
|
|
|
0.5
|
%
|
|
|
2.1
|
%
|
|
|
3.3
|
%
|
|
Percentage of Patients Requiring One or More IOP-Lowering
Surgeries
|
|
|
0.5
|
%
|
|
|
3.7
|
%
|
|
|
7.4
|
%
|
|
|
0.5
|
%
|
|
|
4.8
|
%
|
|
|
8.1
|
%
|
|
|
|
|
|
|
|
| |
|
(1) |
|
An IOP of 30 mmHg is a clinically significant level that we use
in assessing adverse events. |
According to the CDC, diabetic individuals aged 50 or older are
1.5 times more likely to develop cataracts than non-diabetic
individuals. A review of the baseline characteristics of our
patient population reflects this increased risk of cataracts for
diabetic patients, with 34.8% of the patients treated in our
FAME Study having previously undergone a cataract surgery in the
study eye. The month 24 clinical readout from our FAME Study
(which includes reported adverse events that occurred beyond the
patients’ month 24 follow up visits) indicated that, of
patients who had a natural lens (no prior cataract surgery) at
baseline, 46.3% of the control group, 80.0% of the low dose and
87.5% of the high dose had cataract formation reported as an
adverse event through month 24. Additionally, of the patients
who had a natural lens at baseline, 23.1% of the control group,
74.9% of the low dose and 84.5% of the high dose underwent
cataract surgery. The final clinical readout indicated that, of
patients who had a natural lens at baseline, 50.4% of the
control group and 81.7% of the low dose had cataract formation
reported as an adverse event through month 36. Additionally, of
the patients who had a natural lens at baseline, 27.3% of the
control group and 80.0% of the low dose underwent cataract
surgery.
PK
Study
Regulatory agencies require carcinogenicity studies in animals
to identify tumorigenic potential in animals to assess the
relevant risk in humans as a result of systemic exposure. We
initiated an open-label Phase 2 human pharmacokinetic clinical
study (PK Study) in August 2007 to assess the systemic exposure
of fluocinolone acetonide (FAc) by measuring plasma levels of
FAc. Analysis of plasma levels through month 18 in September
2009 demonstrated no systemic exposure of FAc (plasma levels
were below the limit of detection of 100 picograms per
milliliter). Based on month 18 readouts from our open-label
Phase 2 human pharmacokinetic clinical trial (PK Study), which
indicate that there is negligible systemic absorption of FAc in
patients being treated with ILUVIEN, we submitted a
carcinogenicity waiver in our submissions to the FDA and
European health authorities. Although the FDA did not
specifically state in the CRL that the waiver has been granted,
the CRL did not include any requirement to conduct a
carcinogenicity study. In the Preliminary Assessment Report
issued by the United Kingdom Medicines and Healthcare Products
Regulatory Agency (MHRA), the MHRA stated that the lack of
single-dose, carcinogenicity and reproductive and developmental
toxicity studies with ILUVIEN is acceptable.
ILUVIEN
for Other Diseases of the Eye
We believe that ILUVIEN has the potential to address other
ophthalmic diseases such as dry AMD, wet AMD and RVO. Details
regarding the rationale for these other indications are as
follows:
|
|
|
| |
•
|
Dry AMD. We believe that dry AMD patients
account for 90% of AMD patients, with the greatest unmet need
among these patients being a treatment for geographic atrophy
(GA) for which there are currently no treatments available.
Pre-clinical studies in two established rat models of retinal
|
17
|
|
|
| |
|
degeneration reported at the Association for Research in Vision
and Ophthalmology meetings in 2006, 2007 and 2008, described the
efficacious effects of a miniaturized version of ILUVIEN in two
animal models of retinal degeneration. Based on these results,
we began enrollment of a pilot study in December 2008 to assess
the safety and efficacy of ILUVIEN in patients with bilateral GA
secondary to AMD. Our Phase 2 study (the MAP GA Trial) is
comparing the two doses of ILUVIEN to a sham injection in
patients with bilateral GA secondary to AMD. The change from
baseline in size of GA will be assessed over time.
|
|
|
|
| |
•
|
Wet AMD. The size of the wet AMD market was
$2 billion in 2008 according to visiongain, an independent
competitive intelligence organization. We believe ILUVIEN will
be synergistic with the market leading anti-VEGF therapies in
the treatment of wet AMD. Anti-VEGFs require persistent dosing
to maintain a therapeutic effect which is a burden on both the
patient and the physician. Given that corticosteroids have been
shown to suppress the production of VEGF, a Phase 2 investigator
sponsored study (the MAP Trial) is assessing the safety and
efficacy of ILUVIEN in conjunction with Lucentis in patients
with wet AMD. Patients will be enrolled who have been treated
with Lucentis for at least six months and whose visual acuity
has plateaued. At baseline, subjects will receive either the
high-dose or the low-dose of ILUVIEN and an injection of
Lucentis. Subjects will receive additional Lucentis injections
during the study only if subretinal or intraretinal fluid
persists. Outcome measures will include the change from baseline
visual acuity at six months, and mean number of injections of
Lucentis over the six-month study period versus the six months
prior to study entry.
|
| |
| |
•
|
Macular edema associated with non-ischemic
RVO. We believe that the prevalence of retinal
vein occlusion in the U.S. range from approximately 800,000
to approximately 1.6 million. In September 2009, Allergan
introduced Ozurdex (a three to five month dexamethasone
intravitreal implant) as the first approved product for macular
edema following branch or retinal vein occlusion. In June 2010,
Genentech’s drug Lucentis received approval for this
condition. Retinal specialists have been using intravitreal
injections of the corticosteroid triamcinolone acetonide on an
off-label basis to treat non-ischemic RVO. The FDA approval of
Ozurdex provides additional evidence that lower levels of a
steroid work effectively for RVO. In September 2009, we began
enrollment for a Phase 2 study (the FAVOR Study) to assess the
safety and efficacy of ILUVIEN in patients with macular edema
secondary to RVO. The FAVOR Study is comparing the two doses of
ILUVIEN in patients with macular edema secondary to RVO.
|
ILUVIEN
Registration Plan
U.S.
Regulatory Requirements
In the U.S., clinical evidence of the effectiveness of ILUVIEN
for the treatment of DME from our FAME Study is based on two
time-point comparisons. The primary efficacy variable is the
proportion of patients who have visual acuity improvement in
their study eye, referred to as the responder rate, based on the
change from baseline in BCVA as measured on the ETDRS eye chart.
BCVA improvement is defined as an increase from baseline of 15
or more letters in BCVA as measured on the ETDRS eye chart. Our
primary efficacy endpoint is defined at month 24 of our FAME
Study using this variable. Based on the month 24 clinical
readout, ILUVIEN has demonstrated efficacy in the treatment of
DME in our FAME Study. Then as required by the FDA, another
numerical comparison of the responder rates at months 18 and 24
of our FAME Study was conducted to demonstrate that the
responder rates at month 24 are numerically greater than or
equal to the month 18 responder rates. Patients enrolled in our
FAME Study were followed for 36 months. Although we
submitted the month 24 results to the FDA in our NDA for ILUVIEN
for approval, in the CRL the FDA requested the additional
12 months of clinical data from the completed FAME Study
through month 36 for review. We are currently preparing a
response to the CRL for submission early in the second quarter
of 2011 including this additional efficacy and safety data.
18
Regulatory
Requirements in Other Jurisdictions
There are no specific guidance documents for the clinical
development of ophthalmic drug products outside of the
U.S. for the treatment of diabetic retinopathy or DME. We
have met with regulatory authorities in Canada, Germany, Spain,
France, Portugal and the United Kingdom and presented our
overall preclinical, technical, clinical and statistical
development plans which included the use of visual function as
the primary efficacy endpoint and an anatomical measure as a
co-primary efficacy endpoint or key secondary efficacy endpoint.
We also plan to submit efficacy and safety data through the end
of the FAME Study to the applicable regulatory authorities in
the United Kingdom, Austria, France, Germany, Italy, Portugal
and Spain in the second quarter of 2011.
Commercialization
We believe that ILUVIEN will be the first ophthalmic drug
approved by the FDA for the treatment of DME and the only single
treatment drug therapy providing a sustained therapeutic effect
of longer than six months. Our commercialization strategy will
be to establish ILUVIEN as a leading therapy for the treatment
of DME and subsequently for other indications. In the
U.S. and Canada we intend to distribute ILUVIEN to
physicians and through wholesalers and specialty pharmacies
utilizing our outside sales force. Although we anticipate
ILUVIEN being administered as a standalone therapy, we do not
foresee the use of ILUVIEN as precluding the administration of
other therapies in conjunction with ILUVIEN. ILUVIEN is not
approved by the FDA. Our commercialization strategy is subject
to and dependent upon the regulatory approval of ILUVIEN for the
treatment of DME.
Sales
and Marketing
We are led by an executive team with extensive development and
commercialization expertise with ophthalmic products including
the launch and management of Visudyne, a drug product sponsored
by Novartis and the first pharmacological treatment indicated
for the treatment of wet AMD. We intend to capitalize on our
management’s experience and expertise in marketing eye-care
products by marketing and selling ILUVIEN to the approximately
1,600 retinal specialists practicing in the approximately 900
retina centers in the U.S. and Canada through our outside
sales force. The concentration of retinal specialists in a small
number of retina centers and ILUVIEN’s expected status as
the only ophthalmic drug therapy approved by the FDA for the
treatment of DME and its long term sustained release
characteristic are factors that we believe will accelerate the
adoption of ILUVIEN by retinal specialists. We intend to seek a
commercialization partner for sales and marketing activities
outside North America.
Our plan has been to ensure that influential retinal specialists
are presenting our FAME Study data at key retina meetings, to
develop our medical marketing, promotion and communication
materials and to build our own specialized domestic sales and
marketing infrastructure, comprised of approximately
40 people, to market ILUVIEN and other ophthalmic products
that we acquire or develop in the future. In preparation for the
commercial launch of ILUVIEN previously anticipated to occur in
the first half of 2011, we began recruiting our sales and
marketing infrastructure personnel with extensive ophthalmic
based sales experience in the fourth quarter of 2010. We
have hired our three field directors but have determined not to
add the personnel and incur the costs of hiring and training an
internal sales force at this time. We entered into a
relationship with OnCall LLC, a contract sales force company,
who will utilize their employees to act as our sales
representatives if we receive approval of the ILUVIEN NDA from
the FDA. We expect that following an FDA approval of our NDA,
the OnCall sales force will be able to access and form
relationships with retinal specialists in the approximately 900
retina centers for the commercial launch of ILUVIEN. In
connection with the commercial launch of ILUVIEN, we expect to
hire additional personnel to support the activities of customer
service, post-marketing pharmacovigilance, medical affairs and
regulatory compliance.
19
Manufacturing
We do not have, and do not intend to establish an in-house
manufacturing capability for our products and as a result we
will depend heavily on third-party contract manufacturers to
produce and package our products. We require that these
manufacturers produce active pharmaceutical ingredients, or
APIs, and finished drug products in accordance with cGMPs and
all other applicable laws and regulations. We anticipate that we
will rely on contract manufacturers to develop and manufacture
our products for commercial sale. We maintain agreements with
potential and existing manufacturers that include
confidentiality and intellectual property provisions to protect
our proprietary rights related to our product candidates.
We are in the process of finalizing long-term agreements with
the manufacturer of the API in ILUVIEN (FARMABIOS SpA/Byron
Chemical Company Inc.) and the manufacturer of the ILUVIEN
inserter (Flextronics International, Ltd or an affiliate of
Flextronics International, Ltd. (Flextronics)). We have
finalized a long-term agreement with the manufacturer of ILUVIEN
(Alliance Medical Products Inc.) which is discussed below.
pSivida manufactured our clinical trial materials for our FAME
Study, our PK Study and for the Phase 2 clinical trials being
conducted for the use of ILUVIEN for the treatment of dry AMD
and wet AMD. pSivida’s manufacturing process is manual and
labor intensive and not practical for commercial manufacturing.
We worked with Flextronics and Alliance to develop a
manufacturing process where automation is employed whenever
feasible so that we have a process capable of being
scaled-up to
produce commercial quantities. The manufacturing process for
ILUVIEN consists of filling the polyimide tube with a matrix
consisting of FAc and polyvinyl alcohol (PVA), cutting the
tubes, capping the tubes with membrane caps, curing at high
temperature, loading ILUVIEN inside the ILUVIEN inserter,
packaging and sterilizing the product. This process has been
transferred and validated at Alliance, the third-party contract
manufacturer of ILUVIEN. Alliance is also the provider of the
clinical trial materials for the Phase 2 clinical trial being
conducted for the use of ILUVIEN in the treatment of RVO. We
have discussed our approach to show equivalency of the pSivida
manufacturing process to the commercial manufacturing process
with the FDA, the MHRA and the German Bundeninstitut fur
Arneimittel und Medizinprodukte (BfArM). The CRL received from
the FDA and the assessment reports received from the European
Health Authorities did not raise any issue with our
demonstration of equivalency between the manufacturing processes
at pSivida and Alliance. However, in the CRL the FDA did notify
us that the methods used in and the facilities and controls used
for, the manufacturing, processing, packing, or holding of the
drug product at two of our manufacturers did not comply with
cGMPs during recent inspections. One of the manufacturers
received confirmation from the FDA in March 2011 that their
facility is acceptable and the second manufacturer is in
discussions with the FDA to resolve its deficiencies.
In February 2010, we entered into a commercial manufacturing
agreement with Alliance Medical Products, Inc. (Alliance)
whereby Alliance agreed to manufacture and package ILUVIEN for
us at its Irvine, California facility. We purchased certain
equipment and loaned such equipment to Alliance solely for the
purpose of allowing Alliance to manufacture and package ILUVIEN
for us. Under the agreement, we are also responsible for
supplying Alliance with the ILUVIEN inserter and the active
pharmaceutical ingredient. Pursuant to our agreement with
Alliance we have agreed to order from Alliance at least 80% of
our total requirements for new units of ILUVIEN in the U.S.,
Canada and Europe in a calendar year; provided that Alliance is
able to fulfill our supply requirements and is not in breach of
its agreements or obligations to us. Unless terminated earlier
in accordance with the provisions thereof, our agreement with
Alliance has an initial six year term and will automatically
renew for successive one year periods unless either party
delivers written notice of non-renewal to the other at least 12
months prior to the end of the current term.
20
In our NDA for ILUVIEN, we provided the FDA with a completed
process validation package on three lots which described
manufacturing and packaging procedures and in-process controls.
Validation was conducted on small scale, 400 unit batches
but in the U.S., this can be scaled up to 10 times. In addition,
we submitted up to 18 months of stability data from three
primary stability batches to demonstrate that the product
manufactured using the process as described meets the product
specifications. The same process validation package was
submitted in the Marketing Authorization Application (MAA) in
Europe. Validation was based on small scale batches and we will
be limited to this batch size for product sold in Europe.
In preparation for commercial production, Alliance has
successfully completed an engineering batch to demonstrate that
the process can be scaled up to 800 unit batches. Two
scaled-up
batches were also successfully manufactured demonstrating that
the critical steps up to and including application of the cap
membranes are reproducible.
Competition
The development and commercialization of new drugs and drug
delivery technologies is highly competitive. We will likely face
competition with respect to ILUVIEN and any products we may
develop or commercialize in the future from major pharmaceutical
companies, specialty pharmaceutical companies and biotechnology
companies worldwide, many of whom have substantially greater
financial and other resources than we do. If ILUVIEN is approved
for use in the treatment of DME, it will compete against laser
photocoagulation and off-label use of anti-VEGF and
corticosteroid injections, or other therapies that may be
approved in the future. While we believe that ILUVIEN will be
the first ophthalmic drug therapy approved by the FDA for the
treatment of DME, there are other companies working to develop
other drug therapies and sustained delivery platforms for DME
and other indications. We believe that the following companies
provide potential competition to our product candidates:
|
|
|
| |
•
|
Allergan, Inc.’s (Allergan) product Ozurdex (dexamethasone
intravitreal implant), is a bioerodable extended release implant
that delivers the corticosteroid dexamethasone. Ozurdex was
approved in 2009 for macular edema following branch or central
RVO and non-infectious uveitis affecting the posterior segment
of the eye in September 2010 and showed duration of therapy of
three to five months. In addition, Allergan’s product
Trivaris (triamcinolone acetonide injectable suspension) is
approved for sympathetic ophthalmia, temporal arteritis, uveitis
and other inflammatory conditions unresponsive to topical
corticosteroids. Trivaris is not indicated for the treatment of
DME, dry AMD, wet AMD or RVO.
|
| |
| |
•
|
Alcon, Inc.’s (Alcon) product TRIESENCE (triamcinolone
acetonide injectable suspension), a preservative free synthetic
corticosteroid for visualization during vitrectomy, is approved
for the treatment of sympathetic ophthalmia, temporal arteritis,
uveitis and other inflammatory conditions unresponsive to
|
21
|
|
|
| |
|
topical corticosteroids. TRIESENCE is not indicated for the
treatment of DME, dry AMD, wet AMD or RVO.
|
|
|
|
| |
•
|
Genentech Inc.’s (Genentech) products Lucentis (ranibizumab
injection) and Avastin (bevacizumab), both antibodies that block
all isoforms of VEGF, are being studied for the treatment of
DME. Lucentis is currently approved for the treatment of
patients with neovascular wet AMD and the treatment of macular
edema following RVO in the U.S. and the European Union, and
for the treatment of DME in the European Union. However,
Lucentis is currently enrolled in Phase 3 clinical trials for
the treatment of DME in the U.S. . Avastin is currently
marketed as an oncology product and is not indicated for the
treatment of DME, dry AMD or RVO. Genentech is a wholly-owned
member of the Roche Group.
|
| |
| |
•
|
Eyetech, Inc.’s product Macugen (pegaptanib sodium
injection) is an anti-VEGF aptamer against VEGF 165. It has been
FDA-approved for treatment of all subtypes of choroidal
neovascularization in patients with AMD. Macugen is not
indicated for the treatment of DME, dry AMD or RVO.
|
In addition, there are a number of other companies, including
Regeneron, Inc., MacuSight, Inc., Thrombogenics NV. and Novagali
Pharma S.A., that are developing drug therapies or sustained
delivery platforms for the treatment of ocular disease. These
companies are seeking to apply their technologies to ophthalmic
indications in early stage clinical trials.
We believe we will be less likely to face generic competition
for ILUVIEN because of the bioequivalency requirements of a
generic form of ILUVIEN. For a generic pharmaceutical competitor
to ILUVIEN, bioequivalency must be established through the
demonstration of an equivalent pharmacodynamic endpoint in a
clinical trial. We believe conducting such a clinical trial
would be cost prohibitive and time consuming.
The licensing and acquisition of pharmaceutical products, which
is part of our strategy, is a highly competitive area. A number
of more established companies are also pursuing strategies to
license or acquire products. These established companies may
have a competitive advantage over us due to their size, cash
flow and institutional experience.
Other
Pipeline Products
NADPH
Oxidase Inhibition
We believe that the management of oxidative stress is an
important strategy in managing the development and progression
of diseases of the eye, and we believe that NADPH oxidase
inhibitors have the potential to manage oxidative stress.
Oxidative stress is a condition where excess reactive oxygen
intermediates generally referred to as reactive oxygen species
(ROS), are produced. The production of ROS is not always
pathogenic, however, many researchers believe that when the
level of ROS becomes excessive, pathogenic processes are
initiated, resulting in diseased tissue.
NADPH oxidase has been identified as an enzyme system that
generates ROS as its primary function. NADPH oxidase has been
identified in almost every tissue type and there is a
significant amount of scientific literature associating NADPH
oxidase activation with many systemic and ocular conditions. In
the eye, the inhibition of NADPH oxidase has been shown to
prevent or slow pathology in various models of ocular disease,
including retinal degeneration, retinal neovascularization,
choroidal neovascularization and uveitis. In addition, the
presence of NADPH oxidase in corneal epithelial cells implicates
it as having a possible role in dry eye, and the activation of
NADPH oxidase in certain pollen grains upon hydration implicates
its role in allergic conjunctivitis.
In July and August 2009, we executed agreements with Emory
University, whereby we acquired exclusive, worldwide licenses of
rights under patent applications covering two classes of NADPH
oxidase inhibitors. Our strategy around NADPH oxidase inhibition
will target, as the first indication, the treatment of dry AMD
and specifically the end stage of this condition known as
geographic atrophy. We have initiated a testing process to
identify the optimal candidate for formulation in a sustained
release dosage form for the treatment of geographic atrophy. In
addition to studying NADPH oxidase inhibitors, and specifically
an
22
intraocular dosage form, to treat dry AMD, we believe that these
compounds and this dosage form has the potential to treat other
diseases of the eye including wet AMD, diabetic retinopathy and
posterior uveitis.
Licenses
and Agreements
pSivida
US, Inc.
In February 2005, we entered into an agreement with pSivida to
obtain rights and licenses to intellectual property rights
related to pSivida’s proprietary delivery technology. Our
agreement with pSivida provides us with a worldwide exclusive
license to develop and sell ILUVIEN, which consists of a tiny
polyimide tube with membrane caps that is filled with FAc in a
polyvinyl alcohol matrix, for delivery to the back of the eye
for the treatment and prevention of eye diseases in humans
(other than uveitis). This agreement also provided us with a
worldwide non-exclusive license to develop and sell
pSivida’s proprietary delivery device to deliver other
corticosteroids to the back of the eye for the treatment and
prevention of eye diseases in humans (other than uveitis) or to
treat DME by delivering a compound to the back of the eye
through a direct delivery method through an incision required
for a 25-gauge or larger needle. We do not have the right to
develop and sell pSivida’s proprietary delivery device in
connection with indications for diseases outside of the eye or
for the treatment of uveitis.
We made initial license fee payments totaling $750,000 to
pSivida in 2004, and made additional license fee payments of
$750,000 to pSivida in 2005 upon the initiation of the Phase 3
trials for ILUVIEN for the treatment of DME.
Under the February 2005 agreement, we and pSivida agreed to
collaborate on the development of ILUVIEN with FAc for DME, and
share equally in the development expenses. We and pSivida also
agreed that after commercialization of such product, profits, as
defined in that agreement would be shared equally.
In March 2008, we and pSivida amended and restated the agreement
to provide us with 80% of the net profits and pSivida with 20%
of the net profits. In connection with the March 2008 agreement
we agreed to:
|
|
|
| |
•
|
pay $12.0 million to pSivida upon the execution of the
March 2008 agreement;
|
| |
| |
•
|
issue a $15.0 million promissory note to pSivida;
|
| |
| |
•
|
forgive all outstanding development payments, penalties and
interest as of the effective date of the March 2008 agreement,
which totaled $6.8 million;
|
| |
| |
•
|
continue responsibility for regulatory, clinical, preclinical,
manufacturing, marketing and sales for the remaining development
and commercialization of the products;
|
| |
| |
•
|
assume all financial responsibility for the development of the
products and assume 80% of the commercialization costs of the
products (instead of 50% as provided under the February 2005
agreement); and
|
| |
| |
•
|
make an additional milestone payment of $25.0 million after
the first product under the March 2008 agreement has been
approved by the FDA.
|
The $15.0 million promissory note accrued interest at 8%
payable quarterly and was payable in full to pSivida upon the
earlier of a liquidity event as defined in the note (including
an initial public offering of our common stock greater than
$75.0 million), the occurrence of an event of default under
our agreement with pSivida or September 30, 2012. If the
note was not paid in full by March 31, 2010, the interest
rate was to increase to 20% effective as of April 1, 2010,
and we were required to begin making principal payments of
$500,000 per month. On April 27, 2010, we paid pSivida
approximately $15.2 million in principal and interest to
satisfy the note payable with the proceeds from our initial
public offering.
23
Our license rights to pSivida’s proprietary delivery device
could revert to pSivida if we (i) fail twice to cure our
breach of an obligation to make certain payments to pSivida
following receipt of written notice thereof; (ii) fail to
cure other breaches of material terms of our agreement with
pSivida within 30 days after notice of such breaches or
such longer period (up to 90 days) as may be reasonably
necessary if the breach cannot be cured within such
30-day
period; (iii) file for protection under the bankruptcy
laws, make an assignment for the benefit of creditors, appoint
or suffer appointment of a receiver or trustee over our
property, file a petition under any bankruptcy or insolvency act
or have any such petition filed against us and such proceeding
remains undismissed or unstayed for a period of more than
60 days; or (iv) we notify pSivida in writing of our
decision to abandon our license with respect to a certain
product using pSivida’s proprietary delivery device. We
were not in breach of our agreement with pSivida as of
December 31, 2010.
Emory
University
In July 2009, we entered into an agreement with Emory University
related to the fulvene class of NADPH oxidase inhibitors. Under
such agreement, Emory granted to us an exclusive, worldwide
license to rights under intellectual property rights related to
the fulvene class of NADPH oxidase inhibitors for the
development, manufacturing, marketing and selling of
pharmaceutical products containing such compounds for
therapeutic and prophylactic uses for the treatment of diseases
and disorders of the eye in humans. In August 2009, we entered
into a second agreement with Emory University related to the
triphenylmethane class of NADPH oxidase inhibitors. Under such
agreement, Emory granted to us an exclusive, worldwide license
to rights under intellectual property rights related to the
triphenylmethane class of NADPH oxidase inhibitors for the
development, manufacturing, marketing and selling of
pharmaceutical products containing such compounds for
therapeutic and prophylactic uses for the treatment of diseases
and disorders of the eye in humans.
Under such agreements, we pay Emory University royalties in the
mid-single digits of net sales of products containing such
fulvene or triphenylmethane compounds, in countries in which a
claim in a pending patent application or an unexpired patent
that covers the applicable product exists. We also pay Emory
University royalties in the low-single digits of net sales of
products containing such fulvene or triphenylmethane compounds,
in countries in which a claim in a pending patent application or
an unexpired patent that covers the applicable product does not
exist, if at least one patent that covers the applicable product
has issued in the U.S. Furthermore, under each agreement,
we will be required to make annual minimum royalty payments in
the amount of $250,000 the first calendar year after regulatory
approval of the product in a major market country (i.e., the
U.S., Japan, China, India or any European country), $500,000 the
second calendar year after regulatory approval of the product in
such major market country, $1.0 million the third calendar
year after regulatory approval of the product in such major
market country and $2.5 million the fourth year after
regulatory approval of the product in such major market country
and each subsequent year thereafter for the remainder of the
term of such agreement. If we terminate the agreements in India,
China or Japan after we obtain regulatory approval for a
licensed product, the minimum royalty in the calendar year of
the termination, and in each subsequent calendar year
thereafter, will increase by $250,000 for each such country in
which termination occurred. We will also be required to make
payments of up to $5.8 million under the fulvene license
agreement and up to $5.9 million under the triphenylmethane
license agreement depending upon which regulatory milestones we
achieve. If we do not make any milestone payments to Emory
University under the license agreements prior to the third
anniversary of the effective date of the applicable license
agreement, and we do not elect to terminate that license
agreement in accordance with its terms, then we will be required
to pay Emory University annual license maintenance fees ranging
from $500,000 to $2.0 million (depending on when such
payment is made) until a milestone payment is made under the
applicable license agreement or such license agreement is
terminated in accordance with its terms. As an upfront license
fee for the licenses granted by Emory University to us, we
issued to Emory University (and its inventors), that number of
shares of our common stock with a fair market value equal to
$150,000 on the date of issuance with respect to the fulvene
license agreement and in December 2009 we issued that number of
shares of our common stock with a fair market value equal to
$150,000 on the date of issuance with respect to the
triphenylmethane license agreement. We must also reimburse Emory
University for reasonable costs and expenses incurred by Emory
University in filing, prosecuting and maintaining the licensed
patents.
24
In connection with the Emory license agreements, we obtained an
exclusive option to acquire an exclusive, worldwide license to
rights under intellectual property rights related to the covered
compounds for the development, manufacturing, marketing and
selling of pharmaceutical products containing such compounds for
therapeutic and prophylactic uses for the treatment of diseases
and disorders in humans outside the eye. The option will include
the right to sublicense to a third-party and will last for a
period of up to six years. In order to retain the option over
the six-year period, we will be required to make maintenance
payments of $550,000 in the aggregate over a four-year period
commencing two years after the effective date of the license
agreement. If we exercise the option during the six-year period
with respect to a license agreement and subsequently enter into
an amendment to such license agreement in connection therewith,
then the license granted under such license agreement will be
expanded to cover the development, manufacturing, marketing and
selling of products that contain the covered compounds for
therapeutic and prophylactic uses for the treatment of diseases
and disorders in humans outside the eye. We may grant
sublicenses of the intellectual property rights granted to us
under such license agreements to sublicensees. We will, however,
be required to remit 25% of any royalty amounts and 20% to 45%
(depending upon when the applicable sublicense is granted by us)
of other payments we receive from a sublicensee to Emory
University.
As a licensee, we are expected to diligently develop and
commercialize the covered compounds, and failure to meet certain
milestones may result in the termination of our licenses. Under
the agreements, the performance of our sublicensees is deemed to
be performance by us toward fulfillment of our diligence
obligations. The agreements will expire on a country by country
basis upon the later of (i) the expiration of the last to
expire of the licensed patents in a particular country and
(ii) ten years after the date of the first sale of a
licensed product in such country. In addition, Emory University
may terminate a license agreement if (i) we fail to cure a
breach of a material term of such license agreement within
30 days after notice of such breach; (ii) a material
proceeding is instituted against or by us under any bankruptcy,
insolvency, moratorium or dissolution law that is not dismissed
within 90 days; (iii) we assign substantially all of
our assets for the benefit of creditors; (iv) we place our
assets in the hands of a trustee, assignee or receiver and the
receivership or trust is not dissolved or such placement is not
reversed within 60 days; (v) we notify Emory
University in writing that we are quitting the business of
developing or selling products containing the covered compounds
or (vi) we challenge the validity, enforceability
and/or scope
of any claim within a patent or patent application licensed to
us by Emory University under such license agreement in a court
or other government agency.
Dainippon
Sumitomo
In November 2007, we entered into a license agreement with
Dainippon Sumitomo Pharma Co., Ltd. (Dainippon) whereby it
granted to us a non-exclusive, worldwide, royalty free license
to patent rights under specific patents and patent applications
for the development, manufacturing and marketing in the field of
ophthalmology an injectable polymer tube implantable into an eye
containing a mixture of a polymer and FAc (or derivative or
pharmaceutically acceptable salt of FAc) with a polyvinyl
alcohol or other polymer coating or layer at each end of the
tube. In addition, Dainippon granted to us an option to acquire
a non-exclusive, worldwide license to patent rights and know-how
related to specific patents and patent applications for the
development, manufacturing and marketing in the field of
ophthalmology other pharmaceutical products. In exchange for the
license and option granted to us by Dainippon, we paid $200,000
to Dainippon shortly after the execution of the license
agreement, and we are expected to pay another $200,000 to
Dainippon within thirty days following the first regulatory
approval of the licensed product in the U.S. by the FDA.
Dainippon may terminate the license agreement if we materially
fail to fulfill or breach certain terms and conditions of the
license agreement and fail to remedy such failure or breach
within thirty days after receipt of notice from Dainippon. In
addition, Dainippon may terminate the license agreement in the
event that we contest the validity of the patent rights related
to Dainippon’s specific patents and patent applications. In
the event of termination of the license agreement by Dainippon,
we are still expected to make the payment described above.
25
Government
Regulation
General
Overview
Government authorities in the U.S. and other countries
extensively regulate among other things the research,
development, testing, quality, efficacy, safety (pre- and
post-marketing), manufacturing, labeling, storage,
record-keeping, advertising, promotion, export, import,
marketing and distribution of pharmaceutical products.
U.S.
In the U.S., the FDA, under the Federal Food, Drug, and Cosmetic
Act (FD&C Act) and other federal and local statutes and
regulations, subjects pharmaceutical products to review. If we
do not comply with applicable regulations, the government may
refuse to approve or place our clinical studies on clinical
hold, refuse to approve our marketing applications, refuse to
allow us to manufacture or market our products, our products may
be seized, injunctions and monetary fines may be imposed, and we
may be criminally prosecuted.
To obtain approval of a new product from the FDA, we must, among
other requirements, submit data supporting the safety and
efficacy as well as detailed information on the manufacture and
composition of the product and proposed labeling. The testing
and collection of data and the preparation of the necessary
applications are expensive and time consuming. The FDA may not
act quickly or favorably in reviewing these applications, and we
may encounter significant difficulties or costs in our efforts
to obtain FDA approval that could delay or preclude us from
marketing our products. The drug approval process in the
U.S. generally involves the following:
|
|
|
| |
•
|
completion of preclinical laboratory and animal testing and
formulation studies conducted under Good Laboratory Practices
(GLP) regulations;
|
| |
| |
•
|
submission of an Investigational New Drug Application (IND)
which must become effective before human clinical trials may
begin;
|
| |
| |
•
|
completion of adequate and well-controlled human clinical trials
to establish the safety and efficacy of the investigational drug
for its intended use; the studies must be conducted under Good
Clinical Practices (GCP) regulations;
|
| |
| |
•
|
submission of an NDA or Biologics License Application (BLA);
|
| |
| |
•
|
satisfactory completion of an FDA inspection of the
manufacturing facility or facilities where the product is
produced to assess compliance with current Good Manufacturing
Practice (cGMP) regulations; and
|
| |
| |
•
|
FDA review and approval of the NDA or BLA.
|
Preclinical tests include laboratory evaluations of the active
drug’s chemical and physical properties, product
formulation and stability and animal studies to establish
pharmacological effects and safety. The sponsor must submit the
results of preclinical tests, chemistry, manufacturing and
control (CMC) information and clinical development plan
including clinical protocol(s) in an IND. The sponsor cannot
start clinical studies until the IND becomes effective which is
30 days after receipt by the FDA unless the FDA raises
concerns or questions before the
30-day
period. In that case, the sponsor and the FDA must resolve the
questions or concerns before clinical trials can proceed.
Clinical trials involve the administration of the
investigational drug to human subjects under the supervision of
qualified investigators. They are typically conducted in three
sequential phases but the phases may overlap or be combined.
Each trial must be reviewed and approved by an independent
Institutional Review Board before it can begin.
26
Phase 1 trials usually involve the initial introduction of the
investigational drug in a small number of human subjects to
evaluate the product’s safety, dosage tolerance and
pharmacodynamics and if possible, to gain an early indication of
its effectiveness.
Phase 2 trials are usually conducted in a limited patient
population to evaluate dosage tolerance and appropriate dosage;
identify possible adverse effects and safety risks; and
preliminarily evaluate the efficacy of the drug for specific
indications.
Phase 3 trials further evaluate clinical efficacy and test
further for safety in an expanded patient population at
geographically dispersed test sites. Completion of two adequate
and well-controlled Phase 3 studies with results that replicate
each other is the norm before an application can be submitted to
the FDA.
The FDA closely monitors the progress of each phase of clinical
testing and may, at its discretion, reevaluate, alter, suspend
or terminate testing based on data accumulated to that point and
its assessment of the risk/benefit relationship to the patient.
Total time required for running the clinical studies varies
between 2 and 10 years. Additional clinical testing may be
required for special classes of patients, e.g., geriatric
patients, pediatric patients, patients with renal impairment.
Once all the clinical studies are completed, the sponsor submits
the NDA that contains the results of non-clinical and clinical
trials, together with detailed information on the chemistry,
manufacturing and controls of the product and proposed labeling.
It is also important that the sponsor provide a detailed
description and justify the risk/benefit relationship of the
drug to the patient. Under the Prescription Drug User Fee Act
(PDUFA), the applicant has to pay a user fee which is
substantial and increases every year. In fiscal year 2011, the
fee will be $1.5 million.
The FDA conducts a preliminary review of the NDA and within
60 days will make a “fileability” decision. Once
the submission is accepted for filing, the FDA conducts an
in-depth review of the NDA. Under the PDUFA, the FDA has ten
months and six months respectively in which to complete its
review and issue an action letter for a Standard and Priority
Review NDA. The review process may be extended by three months
if the FDA requests additional information or the sponsor
provides significant new information or clarification regarding
information already provided in the submission within the last
3 months of the PDUFA goal date. If the FDA’s
evaluation of the NDA and audit/inspection of clinical and
manufacturing procedures and facilities are favorable, the FDA
may issue either an approval letter or Complete Response Letter
(CRL). A CRL is issued if the FDA determines that it will not
approve the application in its present form. The CRL will
describe all of the specific deficiencies the FDA has identified
and when possible, the FDA will recommend actions that the
applicant can take before the application may be approved.
Upon receipt of a CRL, the applicant must take one of the
following actions:
|
|
|
| |
•
|
resubmit the NDA addressing all deficiencies identified in the
CRL;
|
| |
| |
•
|
withdraw the NDA without prejudice to a subsequent
submission; or
|
| |
| |
•
|
request an opportunity for a hearing on the question of whether
there are grounds for denying approval of the NDA. Within
60 days of the date of request, the FDA will either approve
or refuse to approve the NDA.
|
Responses to the CRL can be classified as Class 1 or
Class 2. Class 1 and Class 2 resubmissions have a
2-month and
a 6-month
review cycle, respectively, beginning on the date FDA receives
the resubmission. Examples of Class 1 resubmissions are:
draft or final printed labeling, safety update, stability
update, proposals for mandatory post-marketing commitments,
assay validation data, minor re-analysis of previously submitted
data and minor clarifications. A Class 2 resubmission is
for any item not specified as a Class 1 item including any
item that would require presentation to an Advisory Committee.
Within one year after receipt of the CRL, the applicant is
required to take one of the actions cited above. If the
applicant does not take one of these actions, the FDA will
consider the lack of response as a request to withdraw the NDA.
The applicant can also request an extension of time to resubmit
the NDA. A resubmission
27
must fully address all the deficiencies cited. A partial
response to the CRL will not be processed as a resubmission and
will not start a new review cycle.
Other
Regulatory Requirements
Risk Evaluation and Mitigation Strategy
(REMS). The recently enacted Food and Drug
Administration Amendments Act of 2007 (FDAAA), gives the FDA
authority to require a drug-specific REMS to ensure the safe use
of the drug. In determining whether a REMS is necessary, the FDA
must consider the size of the population most likely to use the
drug, the seriousness of the disease or condition to be treated,
the expected benefit of the drug, the duration of treatment, the
seriousness of known or potential adverse events and whether or
not the drug is a new chemical entity. If the FDA determines a
REMS is necessary, the sponsor must propose the REMS plan at the
time of approval. A REMS may be required to include various
elements, such as a medication guide or patient package insert,
a communication plan to educate health providers of the
drug’s risks, limitation on who may prescribe or dispense
the drug or other measures that the FDA deems necessary to
assure the safe use of the drug.
The FDAAA also expands the FDA’s authority to require
post-approval studies and clinical trials if the FDA, after drug
approval, deems it appropriate. The purpose of such studies
would be to assess a known serious risk or signals of a serious
risk related to the drug or to identify an unexpected serious
risk when available data indicate the potential for a serious
risk. The FDA may also require a labeling change if it becomes
aware of new safety information that it believes should be
included in the labeling of a drug.
Post-Marketing Requirements. There are
post-marketing safety surveillance requirements that we will
need to meet to continue to market an approved product. Adverse
experiences with the product must be reported to the FDA and
could result in imposition of market restrictions through
labeling changes or in product removal. Product approvals may be
withdrawn if compliance with regulatory requirements is not
maintained or if problems concerning safety
and/or
efficacy of the product occur following approval. The FDA may
also, in its discretion, require post-marketing testing and
surveillance to monitor the effects of approved products or
place conditions on any approvals that could restrict the
commercial applications of these products.
With respect to product advertising and promotion of marketed
products, the FDA imposes a number of complex regulations which
include, among others, standards for
direct-to-consumer
advertising, off-label promotions, industry-sponsored scientific
and educational activities and promotional activities involving
the Internet. The FDA has very broad enforcement authority under
the FD&C Act, and failure to abide by these regulations can
result in penalties, including the issuance of warning letters
directing the sponsor to correct deviations from FDA standards,
a requirement that future advertising and promotional materials
are pre-cleared by the FDA, and state and federal civil and
criminal investigations and prosecutions.
The manufacturing facility that produces our product must
maintain compliance with cGMP and is subject to periodic
inspections by the FDA. Failure to comply with statutory and
regulatory requirements subjects a manufacturer to possible
legal and regulatory action, including Warning Letters, seizure
or recall of products, injunctions, consent decrees placing
significant restrictions on or suspending manufacturing
operations and civil and criminal penalties. In the CRL the FDA
notified us that the methods used in and the facilities and
controls used for, the manufacturing, processing, packing, or
holding of the drug product at two of our manufacturers did not
comply with cGMPs during recent inspections. One of the
manufacturers received confirmation from the FDA in March 2011
that its facility is acceptable and the second manufacturer is
in discussions with the FDA to resolve its deficiencies.
Foreign
Regulations
Foreign regulatory systems, although varying from country to
country, include risks similar to those associated with FDA
regulations in the U.S.
Under the European Union regulatory system, applications for
drug approval may be submitted either in a centralized or
decentralized procedure. Under the centralized procedure, a
single application to the European
28
Medicines Agency (EMA) leads to an approval granted by the
European Commission which permits marketing of the product
throughout the European Union (currently 27 member states). The
centralized procedure is mandatory for new chemical entities,
biotech and orphan drug products and products to treat AIDS,
cancer, diabetes and neuro-degenerative disorder, auto-immune
diseases, other immune dysfunctions and viral diseases. Products
that constitute a significant therapeutic, scientific or
technical innovation or which are in the interests of patients
at the European Union community level may also be submitted
under this procedure. We believe ILUVIEN would potentially
qualify for this procedure as a product that constitutes a
significant therapeutic, scientific or technical innovation.
The decentralized procedure provides for mutual recognition of
nationally approved decisions and is used for products that do
not comply with the requirements for the centralized procedure.
Under the decentralized procedure, the holders of national
marketing authorization in one of the countries within the
European Union may submit further applications to other
countries within the European Union, who will be requested to
recognize the original authorization based on an assessment
report provided by the country in which marketing authorization
is held.
We used the decentralized procedure in Europe. The MHRA is our
Reference Member State. A Reference Member State is responsible
for coordinating the review and approval process between the
United Kingdom and the six other European Union countries where
we intend to seek marketing authorization.
Patents
and Proprietary Rights
Our success depends in part on our ability to obtain and
maintain proprietary protection for our product candidates,
technology and know-how, to operate without infringing on the
proprietary rights of others and to prevent others from
infringing our proprietary rights. Because all of our product
candidates are licensed to us by third-party collaborators, we
are dependent on our collaborators’ ability to obtain and
maintain such protection. Where we have conducted our own
research, our policy is to seek to protect our proprietary
position by, among other methods, filing U.S. and foreign
patent applications related to our proprietary technology,
inventions and improvements that are important to the
development of our business. We also rely on trade secrets,
know-how, continuing technological innovation and in-licensing
opportunities to develop and maintain our proprietary position.
We own or have licensed three U.S. utility patents, one
U.S. design patent and seven U.S. patent applications
as well as numerous foreign counterparts to many of these
patents and patent applications relating to ILUVIEN or the
ILUVIEN inserter. We licensed our patent rights relating to
ILUVIEN from pSivida. Pursuant to our licensed rights, we only
have the right to our ILUVIEN-related patent rights for diseases
of the human eye (other than uveitis). Our licensed patent
portfolio includes U.S. patents with claims directed to
methods for administering a corticosteroid with an implantable
sustained delivery device to deliver the corticosteroid to the
vitreous of the eye wherein aqueous corticosteroid concentration
is less than vitreous corticosteroid concentration during
release. Our licensed patent portfolio also includes a
U.S. patent and a corresponding issued European patent
directed to our low-dose ILUVIEN device and a pending
U.S. patent application directed to our high-dose ILUVIEN
device. In addition, we have patent applications directed to an
inserter system for ILUVIEN.
U.S. utility patents generally have a term of 20 years
from the date of filing. The utility patent rights relating to
ILUVIEN licensed to us from pSivida include three
U.S. patents that expire between March 2019 and April 2020
and counterpart filings to these patents in a number of other
jurisdictions. No patent term extension will be available for
any of these U.S. patents or any of our licensed
U.S. pending patent applications.
The patent positions of companies like ours are generally
uncertain and involve complex legal and factual questions. Our
ability to maintain and solidify our proprietary position for
our technology will depend on our success in obtaining effective
claims and enforcing those claims once granted. We do not know
whether any of our patent applications or those patent
applications that we license will result in the issuance of any
patents. Our issued patents and those that may issue in the
future, or those licensed to us, may be challenged, invalidated
or circumvented, which could limit our ability to stop
competitors from marketing related products or the length of
term of patent protection that we may have for our products. In
addition, the rights granted under any issued patents may not
provide us with proprietary protection or competitive advantages
against competitors with similar technology. Furthermore, our
competitors may independently develop similar
29
technologies or duplicate any technology developed by us.
Because of the extensive time required for development, testing
and regulatory review of a potential product, it is possible
that, before any of our products can be commercialized, any
related patent may expire or remain in force for only a short
period following commercialization, thereby reducing any
advantage of the patent.
We may rely, in some circumstances, on trade secrets to protect
our technology. However, trade secrets are difficult to protect.
We seek to protect our proprietary technology and processes, in
part, by confidentiality agreements with our employees,
consultants, scientific advisors and other contractors. These
agreements may be breached, and we may not have adequate
remedies for any breach. In addition, our trade secrets may
otherwise become known or be independently discovered by
competitors. To the extent that our employees, consultants or
contractors use intellectual property owned by others in their
work for us, disputes may arise as to the rights in related or
resulting know-how and inventions.
Research
and Development
We have built a research and development organization that
includes extensive expertise with ophthalmic products including
the launch and management of Visudyne, a drug product sponsored
by Novartis and the first pharmacological treatment indicated
for patients with wet AMD. We operate cross-functionally and are
led by an experienced research and development management team.
We use rigorous project management techniques to assist us in
making disciplined strategic research and development program
decisions and to limit the risk profile of our product pipeline.
We also access relevant market information and key opinion
leaders in creating target product profiles and, when
appropriate, as we advance our programs to commercialization. We
engage third parties to conduct portions of our preclinical
research. In addition, we engage multiple clinical sites to
conduct our clinical trials; however we are not substantially
dependent upon any one of these sites for our clinical trials
nor do any of them conduct a major portion of our clinical
trials.
We invested $12.6 million, $15.1 million, and
$43.8 million for research and development during the years
ended December 31, 2010, 2009, and 2008, respectively.
Employees
As of March 1, 2011, we had 27 employees, two of whom
hold Ph.D.s, and one of whom holds an O.D. Ten of these
employees were engaged in research, development and regulatory
activities, and 17 were engaged in administrative support, human
resources, finance, information technology and sales and
marketing activities. None of our employees is represented by a
labor union and we consider our employee relations to be good.
Corporate
information
We are a Delaware corporation incorporated on June 4, 2003.
Our principal executive office is located at 6120 Windward
Parkway, Suite 290, Alpharetta, Georgia 30005 and our
telephone number is
(678) 990-5740.
Our web site address is
http://www.alimerasciences.com.
The information contained in, or that can be accessed through,
our Web site is not part of this report and should not be
considered part of this report.
Available
Information
Alimera files annual, quarterly, and current reports, proxy
statements, and other documents with the Securities and Exchange
Commission (SEC) under the Securities Exchange Act of 1934, as
amended (the Exchange Act). The public may read and copy any
materials that we file with the SEC at the SEC’s Public
Reference Room at 100 F Street, NE, Washington, DC
20549. The public may obtain information on the operation of the
Public Reference Room by calling the SEC at
1-800-SEC-0330.
Also, the SEC maintains an Internet website that contains
reports, proxy and information statements, and other information
regarding issuers, including us, that file electronically with
the SEC. The public can obtain any documents that we file with
the SEC at www.sec.gov.
We also make available free of charge on our Internet website at
www.alimerasciences.com our annual reports on
Form 10-K,
quarterly reports on
Form 10-Q,
current reports on
Form 8-K,
and, if applicable,
30
amendments to those reports filed or furnished pursuant to
Section 13(a) or 15(d) of the Exchange Act as soon as
reasonably practicable after we electronically file such
material with, or furnish it to, the SEC.
Our code of ethics, other corporate policies and procedures, and
the charters of our Audit Committee, Compensation Committee and
Nominating/Corporate Governance Committee are available through
our Internet website at www.alimerasciences.com.
Investing in our common stock involves a high degree of risk.
You should consider carefully the risks and uncertainties
described below, together with all of the other information in
this report, including the consolidated financial statements and
the related notes appearing at the end of this annual report on
Form 10-K,
with respect to any investment in shares of our common stock. If
any of the following risks actually occurs, our business,
financial condition, results of operations and future prospects
would likely be materially and adversely affected. In that
event, the market price of our common stock could decline and
you could lose all or part of your investment.
Risks
Related to Our Business and Industry
We are
heavily dependent on the success of our lead product candidate,
ILUVIEN, which is still under development. If we are unable to
commercialize ILUVIEN, or experience significant delays in doing
so, our business will be materially harmed.
We are a biopharmaceutical company with no products approved for
commercial sale. We have incurred, and will continue to incur,
significant costs relating to the regulatory approval and
commercialization of ILUVIEN, our only product candidate in
development. We anticipate that in the near term our ability to
generate revenues will depend solely on the successful
development and commercialization of ILUVIEN. We have not yet
obtained regulatory approval to market this product candidate in
any jurisdiction and we may never be able to obtain approval or,
if approvals are obtained, to commercialize this product
candidate successfully.
Based on our analysis of the month 24 clinical readout from our
Phase 3 pivotal clinical trials for the use of ILUVIEN in the
treatment of diabetic macular edema, or DME (collectively, our
FAME Study), in June 2010 we filed a New Drug Application (NDA)
with the U.S. Food and Drug Administration (FDA) for the
low dose of ILUVIEN in the U.S., followed by registration
filings in the United Kingdom, Austria, France, Germany, Italy,
Portugal and Spain in July 2010. The European Marketing
Authorization Application (MAA) was submitted through the
Decentralized Procedure with the United Kingdom Medicines and
Health products Regulatory Agency (MHRA) as the Reference Member
State. In December 2010, we received a Complete Response Letter
(CRL) from the FDA regarding our NDA. The FDA issued the CRL to
communicate its decision that the NDA could not be approved in
its present form. No new clinical studies were requested by the
FDA in the CRL. However, the FDA asked us for analyses of the
safety and efficacy data through the end of the FAME Study to
further assess the relative benefits and risks of ILUVIEN. We
are currently preparing the analyses the FDA requested having
completed the FAME Study and publicly released data on
February 3, 2011. The FDA is also seeking additional
information regarding controls and specifications concerning the
manufacturing, packaging and sterilization of ILUVIEN, which we
are in the process of compiling. We currently anticipate
submitting our response to the CRL to the FDA early in the
second quarter of 2011. Our submission to the FDA will be
considered a Class 2 response, which will provide for a
review period of up to an additional six months for our NDA.
However, the FDA may request additional information from us,
and, ultimately, may not grant marketing approval for ILUVIEN.
In addition, although we believe the final clinical readout from
our FAME Study demonstrates that ILUVIEN is safe and effective
in the treatment of DME, clinical data often is susceptible to
varying interpretations and many companies that have believed
that their products performed satisfactorily in clinical trials
have nonetheless failed to obtain FDA approval for their
products. Furthermore, even if we receive FDA approval, we might
not be successful in commercializing ILUVIEN. If we are not
successful in commercializing ILUVIEN, or are significantly
delayed in doing so, our
31
business will be materially harmed and we may need to curtail or
cease operations. Our ability to successfully commercialize
ILUVIEN will depend, among other things, on our ability to:
|
|
|
| |
•
|
produce batches of ILUVIEN in quantities sufficiently large to
permit successful commercialization;
|
| |
| |
•
|
receive marketing approvals from the FDA and similar foreign
regulatory authorities;
|
| |
| |
•
|
establish commercial manufacturing arrangements with third-party
manufacturers;
|
| |
| |
•
|
launch commercial sales of ILUVIEN; and
|
| |
| |
•
|
secure acceptance of ILUVIEN in the medical community and with
third-party payers.
|
We
have incurred operating losses in each year since our inception
and expect to continue to incur substantial and increasing
losses for the foreseeable future.
We have a limited operating history. We are not currently
generating revenues and we cannot estimate with precision the
extent of our future losses. We do not currently have any
products that have been approved for commercial sale and we may
never generate revenue from selling products or achieve
profitability. We expect to continue to incur substantial and
increasing losses through the projected commercialization of
ILUVIEN, particularly as we increase our research, clinical
development, administrative and sales and marketing activities.
Assuming FDA approval of our NDA in 2011, we currently do not
expect to generate revenue from the sale of ILUVIEN until late
2011 at the earliest, if at all. As a result, we are uncertain
when or if we will achieve profitability and, if so, whether we
will be able to sustain it. As of December 31, 2010, we
have accumulated a net deficit of $188.9 million. Our
ability to achieve revenue and profitability is dependent on our
ability to complete the development of our product candidates,
obtain necessary regulatory approvals, and have our products
manufactured and marketed. We cannot assure you that we will be
profitable even if we successfully commercialize our products.
Failure to become and remain profitable may adversely affect the
market price of our common stock and our ability to raise
capital and continue operations.
We
face heavy government regulation, and approval of ILUVIEN and
our other product candidates from the FDA and from similar
entities in other countries is uncertain.
The research, testing, manufacturing and marketing of drug
products are subject to extensive regulation by
U.S. federal, state and local government authorities,
including the FDA, and similar entities in other countries. To
obtain regulatory approval of a product, we must demonstrate to
the satisfaction of the regulatory agencies that, among other
things, the product is safe and effective for its intended use.
In addition, we must show that the manufacturing facilities used
to produce the products are in compliance with current Good
Manufacturing Practice (cGMP) regulations.
The process of obtaining regulatory approvals and clearances
will require us to expend substantial time and capital. Despite
the time and expense incurred, regulatory approval is never
guaranteed. The number of preclinical and clinical tests that
will be required for regulatory approval varies depending on the
drug candidate, the disease or condition for which the drug
candidate is in development and the regulations applicable to
that particular drug candidate. Regulatory agencies, including
those in the U.S., Canada, the European Union and other
countries where drugs are regulated, can delay, limit or deny
approval of a drug candidate for many reasons, including that:
|
|
|
| |
•
|
a drug candidate may not be safe or effective;
|
| |
| |
•
|
regulatory agencies may interpret data from preclinical and
clinical testing in different ways from those which we do;
|
| |
| |
•
|
they may not approve of our manufacturing process;
|
| |
| |
•
|
they may conclude that the drug candidate does not meet quality
standards for stability, quality, purity and potency; and
|
| |
| |
•
|
they may change their approval policies or adopt new regulations.
|
32
The FDA may make requests or suggestions regarding conduct of
our clinical trials, resulting in an increased risk of
difficulties or delays in obtaining regulatory approval in the
U.S. For example, the FDA may not approve of certain of our
methods for analyzing our trial data, including how we evaluate
the risk/benefit relationship. Further, we intend to market
ILUVIEN, and may market other product candidates, outside the
U.S. and specifically in the European Union and Canada.
Regulatory agencies within these countries will require that we
obtain separate regulatory approvals and comply with numerous
and varying regulatory requirements. The approval procedures
within these countries can involve additional testing, and the
time required to obtain approval may differ from that required
to obtain FDA approval. Additionally, the foreign regulatory
approval process may include all of the risks associated with
obtaining FDA approval. For all of these reasons, we may not
obtain foreign regulatory approvals on a timely basis, if at
all. Approval by the FDA does not ensure approval by regulatory
authorities in other countries or jurisdictions, and approval by
one foreign regulatory authority does not ensure approval by
regulatory authorities in other foreign countries or
jurisdictions or by the FDA.
We submitted an NDA in the U.S. for the low dose of ILUVIEN
in June 2010 with 24 months of clinical data from our FAME
Study, followed in July 2010 by registration filings in the
United Kingdom, Austria, France, Germany, Italy, Portugal and
Spain. Consistent with recommendations regarding the appropriate
population for primary analysis as described in the FDA-adopted
International Conference on Harmonization of Technical
Requirements for Registration of Pharmaceuticals for Human Use
(ICH) Guidance E9, “Statistical Principles for Clinical
Trials,” we believe that the FDA considers the most
relevant population for determining safety and efficacy to be
the full data set of all 956 patients randomized into our
FAME Study, with data imputation employed using “last
observation carried forward,” for data missing because of
patients who discontinued the trial or are unavailable for
follow-up
(the Full Analysis Set). The primary efficacy endpoint was met
with statistical significance for both the low dose and the high
dose of ILUVIEN in both trials using the Full Analysis Set and
we submitted an analysis based on this data set for the low dose
to the FDA. However, our FAME Study protocol did not include the
Full Analysis Set and provides that the primary assessment of
efficacy will be based on another data set that excludes from
the Full Analysis Set three patients who were enrolled but never
treated as well as data collected for patients subsequent to
their use of treatments prohibited by our FAME Study protocol
(the Modified ART Data Set). Statistical significance was not
achieved for either the low dose or the high dose in one trial
using the Modified ART Data Set. In December 2010, we received a
CRL from the FDA. The FDA issued the CRL to communicate its
decision that the NDA for ILUVIEN could not be approved in its
present form. No new clinical studies were requested by the FDA,
and our use of the Full Analysis Set was not questioned in the
CRL. However, the FDA asked us for analyses of the safety and
efficacy data through the final readout at the end of the FAME
Study to further assess the relative benefits and risks of
ILUVIEN. We are currently preparing the analyses the FDA
requested and currently anticipate submitting our response to
the CRL early in the second quarter of 2011. Our submission to
the FDA will be considered a Class 2 response which will
provide for a review period of up to an additional six months
for our NDA. There is no assurance that the FDA will utilize the
Full Analysis Set and not the Modified ART Data Set or another
data set in determining whether ILUVIEN is safe and effective,
any of which could result in the FDA not granting marketing
approval for ILUVIEN.
In July 2010, we submitted in Europe a MAA using the
Decentralized Procedure with the U.K. MHRA as the Reference
Member State (RMS). Applications were submitted concurrently to
the Concerned Member States (CMS) listed as follows: Germany,
Spain, Italy, France, Portugal and Austria.
We received the initial assessment reports from the RMS and the
CMS in December 2010. The issues raised were similar to the
issues raised by the FDA. We plan to submit the additional
safety and efficacy data through the final readout at the end of
the FAME Study to regulatory authorities in the United Kingdom,
Austria, France, Germany, Italy, Portugal and Spain in the
second quarter of 2011.
Regulatory agencies require carcinogenicity studies in animals
to identify tumorigenic potential in animals to assess the
relevant risk in humans. Based on month 18 readouts from our
open-label Phase 2 human pharmacokinetic clinical trial (PK
Study), which indicate that there is negligible systemic
absorption of fluocinolone acetonide (FAc) in patients being
treated with ILUVIEN, we submitted a carcinogenicity waiver in
our submissions to the FDA and European health authorities.
Although the FDA did not specifically state in the CRL that the
waiver has been granted, the CRL did not include any requirement
to conduct a carcinogenicity study. In the Preliminary
33
Assessment Report issued by the UK MHRA, the MHRA stated that
the lack of single-dose, carcinogenicity and reproductive and
developmental toxicity studies with ILUVIEN is acceptable. If we
are required to perform carcinogenicity studies in animals, the
approval of ILUVIEN could be delayed by up to 36 months.
In the CRL the FDA notified us that the methods used in and the
facilities and controls used for, the manufacturing, processing,
packing, or holding of the drug product at two of our
manufacturers did not comply with cGMPs during recent
inspections. One of the manufacturers received confirmation from
the FDA in March 2011 that their facility is acceptable and the
second manufacturer is in discussions with the FDA to resolve
its deficiencies. Failure for our manufactures to comply with
cGMP may have an adverse effect on our business.
Any delay or failure by us to obtain regulatory approvals for
our product candidates could diminish competitive advantages
that we may attain and would adversely affect the marketing of
our products. We have not yet received regulatory approval to
market any of our product candidates in any jurisdiction.
ILUVIEN
utilizes FAc, a corticosteroid that has demonstrated undesirable
side effects in the eye; therefore, the success of ILUVIEN will
be dependent upon the achievement of an appropriate relationship
between the benefits of its efficacy and the risks of its
side-effect profile.
The use of corticosteroids in the eye has been associated with
undesirable side effects, including increased incidence of
cataract formation and elevated intraocular pressure (IOP),
which may increase the risk of glaucoma. We have received the
final month 36 clinical readout from our FAME Study, but the
extent of ILUVIEN’s long-term side effect profile beyond
month 36 is not yet known. Upon review of our NDA for the low
dose of ILUVIEN in the treatment of DME, the FDA may conclude
that our FAME Study did not demonstrate that ILUVIEN has
sufficient levels of efficacy to outweigh the risks associated
with its side-effect profile. Conversely, the FDA may conclude
that ILUVIEN’s side-effect profile does not demonstrate an
acceptable risk/benefit relationship in line with ILUVIEN’s
demonstrated efficacy. In the event of such conclusions, we may
not receive regulatory approval from the FDA or from similar
regulatory agencies in other countries.
Even
if we do receive regulatory approval for ILUVIEN, the FDA or
other regulatory agencies may impose limitations on the
indicated uses for which ILUVIEN may be marketed, subsequently
withdraw approval or take other actions against us or ILUVIEN
that would be adverse to our business.
Regulatory agencies generally approve products for particular
indications. If any such regulatory agency approves ILUVIEN for
a limited indication, the size of our potential market for
ILUVIEN will be reduced. For example, our potential market for
ILUVIEN would be reduced if the FDA limited the indications of
use to patients diagnosed with only clinically significant DME
as opposed to DME, or restricted the use to patients exhibiting
IOP below a certain level or having an artificial lens at the
time of treatment. Product approvals, once granted, may be
withdrawn if problems occur after initial marketing. If and when
ILUVIEN does receive regulatory approval or clearance, the
marketing, distribution and manufacture of ILUVIEN will be
subject to regulation in the U.S. by the FDA and by similar
entities in other countries. We will need to comply with
facility registration and product listing requirements of the
FDA and similar entities in other countries and adhere to the
FDA’s Quality System Regulations. Noncompliance with
applicable FDA and similar entities’ requirements can
result in warning letters, fines, injunctions, civil penalties,
recall or seizure of ILUVIEN, total or partial suspension of
production, refusal of regulatory agencies to grant approvals,
withdrawal of approvals by regulatory agencies or criminal
prosecution. We would also need to maintain compliance with
federal, state and foreign laws regarding sales incentives,
referrals and other programs.
Our
product candidates may never achieve market acceptance even if
we obtain regulatory approvals.
Even if we receive regulatory approvals for the sale of our
product candidates, the commercial success of these products
will depend, among other things, on their acceptance by retinal
specialists, patients, third-party payers and other members of
the medical community as a therapeutic and cost-effective
alternative to competing products and treatments. The degree of
market acceptance of any of our product candidates will depend
on a number of factors, including, among other things:
|
|
|
| |
•
|
the demonstration of its safety and efficacy;
|
| |
| |
•
|
its cost-effectiveness;
|
34
|
|
|
| |
•
|
its potential advantages over other therapies;
|
| |
| |
•
|
the reimbursement policies of government and third-party payers
with respect to the product candidate; and
|
| |
| |
•
|
the effectiveness of our marketing and distribution capabilities.
|
If our product candidates fail to gain market acceptance, we may
be unable to earn sufficient revenue to continue our business.
If our product candidates are not accepted by retinal
specialists, patients, third-party payers and other members of
the medical community, it is unlikely that we will ever become
profitable.
Our
ability to pursue the development and commercialization of
ILUVIEN depends upon the continuation of our license from
pSivida US, Inc.
Our license rights to pSivida US, Inc.’s (pSivida’s)
proprietary delivery device could revert to pSivida if we
(i) fail twice to cure our breach of an obligation to make
certain payments to pSivida following receipt of written notice
thereof; (ii) fail to cure other breaches of material terms
of our agreement with pSivida within 30 days after notice
of such breaches or such longer period (up to 90 days) as
may be reasonably necessary if the breach cannot be cured within
such 30-day
period; (iii) file for protection under the bankruptcy
laws, make an assignment for the benefit of creditors, appoint
or suffer appointment of a receiver or trustee over our
property, file a petition under any bankruptcy or insolvency act
or have any such petition filed against us and such proceeding
remains undismissed or unstayed for a period of more than
60 days; or (iv) notify pSivida in writing of our
decision to abandon our license with respect to a certain
product using pSivida’s proprietary delivery device. If our
agreement with pSivida were terminated, we would lose our rights
to develop and commercialize ILUVIEN, which would materially and
adversely affect our business, results of operations and future
prospects. We were not in breach of our license agreement with
pSivida as of December 31, 2010.
We
will rely on a single manufacturer for ILUVIEN, a single
manufacturer for the ILUVIEN inserter and a single active
pharmaceutical ingredient formulator for ILUVIEN’s active
pharmaceutical ingredient. Our business would be seriously
harmed if these third-parties are not able to satisfy our demand
and alternative sources are not available.
We do not have, nor currently intend to have, in-house
manufacturing capability and will depend completely on a single
third-party manufacturer for the manufacture of the ILUVIEN
insert (Alliance Medical Products, Inc. (Alliance)), a single
third-party manufacturer for the manufacture of the ILUVIEN
inserter (Flextronics International, Ltd. or an affiliate of
Flextronics International, Ltd. (Flextronics)) and a single
third-party manufacturer for the manufacture of ILUVIEN’s
active pharmaceutical ingredient (FARMABIOS SpA./Byron Chemical
Company Inc. (FARMABIOS)). Although we have finalized a
long-term agreement for the manufacture of the ILUVIEN insert
(with Alliance), we have not yet finalized long-term agreements
for the manufacture of the ILUVIEN inserter (with Flextronics)
or for the manufacture of ILUVIEN’s active pharmaceutical
ingredient (with FARMABIOS), and if any of the third-party
manufacturers are unable or unwilling to perform for any reason,
we may not be able to locate alternative acceptable
manufacturers or formulators, enter into favorable agreements
with them or get them approved by the FDA in a timely manner.
Further, all of our manufacturers rely on additional
third-parties for the manufacture of component parts. Any
inability to acquire sufficient quantities of ILUVIEN inserts,
the ILUVIEN inserter or the active pharmaceutical ingredient in
a timely manner from these third-parties could delay commercial
production of, and impact our ability to fulfill demand for,
ILUVIEN.
Materials
necessary to manufacture ILUVIEN and our other product
candidates may not be available on commercially reasonable
terms, or at all, which may delay the development, regulatory
approval and commercialization of our product
candidates.
We will rely on our manufacturers to purchase materials from
third-party suppliers necessary to produce ILUVIEN and our other
product candidates for our clinical trials and, if approved, for
commercial distribution. Suppliers may not sell these materials
to our manufacturers at the times we need them or on
commercially
35
reasonable terms. We do not have any control over the process or
timing of the acquisition of these materials by our
manufacturers. Moreover, we currently have not finalized all
agreements for the commercial production of these materials. If
our manufacturers are unable to obtain these materials for our
clinical trials, product testing and potential regulatory
approval of ILUVIEN and our other product candidates could be
delayed, significantly affecting our ability to develop ILUVIEN
and our other product candidates. If we or our manufacturers are
unable to purchase these materials after regulatory approval has
been obtained for ILUVIEN and our other product candidates, the
commercial launch of ILUVIEN and our other product candidates
would be delayed or there would be a shortage in supply, which
would materially affect our ability to generate revenues from
the sale of ILUVIEN and our other product candidates. Moreover,
although we have finalized an agreement for the commercial
production of the ILUVIEN insert, we currently have not yet
finalized any agreements for the commercial production of the
active pharmaceutical ingredient in ILUVIEN or the ILUVIEN
inserter. Even if we were able to secure such agreements, the
suppliers may be unable or choose not to provide us the
ingredients in a timely manner or in the minimum guaranteed
quantities. If we are unable to obtain and then supply these
ingredients to our contract manufacturer for our clinical
trials, potential regulatory approval of our product candidates
would be delayed, significantly impacting our ability to develop
our product candidates, which would materially affect our
ability to generate revenue from the sale of our product
candidates.
The
manufacture and packaging of pharmaceutical products such as
ILUVIEN are subject to the requirements of the FDA and similar
foreign regulatory entities. If we or our third-party
manufacturers fail to satisfy these requirements, our product
development and commercialization efforts may be materially
harmed.
The manufacture and packaging of pharmaceutical products such as
ILUVIEN and our future product candidates are regulated by the
FDA and similar foreign regulatory entities and must be
conducted in accordance with the FDA’s cGMP and comparable
requirements of foreign regulatory entities. There are a limited
number of manufacturers that operate under these cGMP
regulations which are both capable of manufacturing ILUVIEN and
willing to do so. Failure by us or our third-party manufacturers
to comply with applicable regulations, requirements, or
guidelines could result in sanctions being imposed on us,
including fines, injunctions, civil penalties, failure of
regulatory authorities to grant marketing approval of our
products, delays, suspension or withdrawal of approvals, license
revocation, seizures or recalls of product, operating
restrictions and criminal prosecutions, any of which could
significantly and adversely affect our business. In December
2010, we received a CRL from the FDA. The FDA issued the CRL to
communicate its decision that the NDA for ILUVIEN could not be
approved in its present form. Additionally, the FDA also
indicated that it had observed deficiencies in current good
manufacturing practices (cGMP) during its facility inspections
of two of our third-party manufacturers, which were completed in
August and September of 2010, and that all facilities and
controls would need to comply with cGMP. Our third-party
manufacturers are in the process of resolving these
deficiencies. One of the manufacturers received confirmation
from the FDA in March 2011 that their facility is acceptable. If
our third-party manufacturers fail to resolve these
deficiencies, the FDA will not grant market approval for
ILUVIEN. Additionally, if our manufacturers fail to maintain
compliance, the production of ILUVIEN could be interrupted,
resulting in delays and additional costs. Any significant delays
in the manufacture of ILUVIEN could materially harm our business
and prospects.
Changes in the manufacturing process or procedure, including a
change in the location where the product is manufactured or a
change of a third-party manufacturer, will require prior FDA
review
and/or
approval of the manufacturing process and procedures in
accordance with the FDA’s cGMP regulations. There are
comparable foreign requirements. This review may be costly and
time consuming and could delay or prevent the launch of a
product. If we elect to manufacture products in our own facility
or at the facility of another third-party, we would need to
ensure that the new facility and the manufacturing process are
in substantial compliance with cGMP regulations. The new
facility will also be subject to pre-approval inspection. In
addition, we have to demonstrate that the product made at the
new facility is equivalent to the product made at the former
facility by physical and chemical methods, which are costly and
time consuming. It is also possible that the FDA may require
clinical testing as a way to prove equivalency, which would
result in additional costs and delay.
36
Furthermore, in order to obtain approval of our products,
including ILUVIEN, by the FDA and foreign regulatory agencies,
we need to complete testing on both the active pharmaceutical
ingredient and on the finished product in the packaging that we
propose for commercial sales. This includes testing of
stability, identification of impurities and testing of other
product specifications by validated test methods. In addition,
we will be required to consistently produce ILUVIEN in
commercial quantities and of specified quality in a reproducible
manner and document our ability to do so. This requirement is
referred to as process validation. With respect to ILUVIEN,
although we have validated the manufacturing process at pilot
scale batches, some of the steps in the manufacturing processes
will need to be revalidated when we begin to manufacture
commercial scale batches. If the required testing or process
validation is delayed or produces unfavorable results, we may
have to launch the product using smaller pilot scale batches,
which may impact our ability to fulfill demand for the product.
The FDA and similar foreign regulatory bodies may also implement
new standards, or change their interpretation and enforcement of
existing standards and requirements, for the manufacture,
packaging, or testing of products at any time. If we are unable
to comply, we may be subject to regulatory or civil actions or
penalties that could significantly and adversely affect our
business.
Any
failure or delay in completing clinical trials for our product
candidates could severely harm our business.
Preclinical studies and clinical trials required to demonstrate
the safety and efficacy of our product candidates are time
consuming and expensive and together take several years to
complete. The completion of clinical trials for our product
candidates may be delayed by many factors, including:
|
|
|
| |
•
|
our inability to manufacture or obtain from third-parties
materials sufficient for use in preclinical studies and clinical
trials;
|
| |
| |
•
|
delays in patient enrollment and variability in the number and
types of patients available for clinical trials;
|
| |
| |
•
|
difficulty in maintaining contact with patients after treatment,
resulting in incomplete data;
|
| |
| |
•
|
poor effectiveness of product candidates during clinical trials;
|
| |
| |
•
|
unforeseen safety issues or side effects; and
|
| |
| |
•
|
governmental or regulatory delays and changes in regulatory
requirements and guidelines.
|
If we fail to successfully complete our clinical trials for any
of our product candidates, we may not receive the regulatory
approvals needed to market that product candidate. Therefore,
any failure or delay in commencing or completing these clinical
trials would harm our business materially.
In addition, a clinical trial may be suspended or terminated by
us, the FDA or other regulatory authorities due to a number of
factors, including:
|
|
|
| |
•
|
failure to conduct the clinical trial in accordance with
regulatory requirements or our clinical protocols;
|
| |
| |
•
|
inspection of the clinical trial operations or trial sites by
the FDA or other regulatory authorities resulting in the
imposition of a clinical hold;
|
| |
| |
•
|
unforeseen safety issues or any determination that a trial
presents unacceptable health risks;
|
| |
| |
•
|
lack of adequate funding to continue the clinical trial,
including the incurrence of unforeseen costs due to enrollment
delays, requirements to conduct additional trials and studies
and increased expenses associated with the services of our
contract research organizations, or CROs, and other third
parties.
|
If we are required to conduct additional clinical trials or
other studies with respect to any of our product candidates
beyond those that we initially contemplated, if we are unable to
successfully complete our clinical trials or other studies or if
the results of these trials or studies are not positive or are
only modestly positive, we may be delayed in obtaining marketing
approval for that product candidate, we may not be able to
obtain marketing approval or we may obtain approval for
indications that is not as broad as intended. Our product
37
development costs will also increase if we experience delays in
testing or approvals. Significant clinical trial delays could
allow our competitors to bring products to market before we do
and impair our ability to commercialize our products or
potential products. If any of this occurs, our business will be
materially harmed.
We may
not be successful in executing our sales and marketing strategy
for the commercialization of ILUVIEN. We currently have a
limited sales and marketing organization and, as part of our
sales strategy, we expect to initially depend in large part on a
third party contract sales force for the sale of ILUVIEN. If we
are unable to successfully execute such strategy, we may not be
able to generate significant revenue.
At present, we have no internal sales force and only a limited
number of sales and marketing personnel. We began hiring
additional sales and marketing personnel in the third quarter of
2010 to establish our own sales and marketing capabilities in
the U.S. in time for our previously anticipated commercial
launch of ILUVIEN. We have hired three field directors but have
determined not to add the personnel and incur the costs of
hiring and training an internal sales force at this time. In the
fourth quarter of 2010, we entered into a relationship with
OnCall LLC, a contract sales force company that will utilize its
employees to act as our sales representatives if we receive
approval of the ILUVIEN NDA from the FDA.
Pursuant to our agreement with OnCall, following FDA approval,
ILUVIEN will be promoted primarily to retinal specialists in the
U.S. by OnCall’s sales representatives. We will rely
in large part on this outside sales force for the sales of
ILUVIEN in the U.S. Although we expect to be involved in
the hiring, training and management of these sales
representatives, they will be employees of OnCall and subject to
its ultimate control.
If we determine to establish a direct sales force in the future
to sell ILUVIEN or another product candidate following marketing
approval, we may not be able to establish such a direct sales
force in a cost-effective manner or realize a positive return on
this investment. In addition, we will have to compete with other
pharmaceutical and biotechnology companies to recruit, hire, and
retain sales and marketing personnel. Factors that may inhibit
our efforts to commercialize our products without strategic
partners or licensees include:
• our inability to recruit and retain adequate
numbers of effective sales and marketing personnel;
|
|
|
| |
•
|
the inability of sales personnel to obtain access to or persuade
adequate numbers of retinal specialists to prescribe our
products;
|
| |
| |
•
|
the lack of complementary products to be offered by sales
personnel, which may put us at a competitive disadvantage
relative to companies with more extensive product lines; and
|
| |
| |
•
|
unforeseen costs and expenses associated with creating an
independent sales and marketing organization.
|
We have not yet entered into any agreements related to the
marketing of ILUVIEN or any of our other product candidates in
international markets and we may not be able to enter into any
arrangements with respect to international collaborations on
favorable terms or at all. In addition, these arrangements could
result in lower levels of income to us than if we marketed our
product candidates entirely on our own. If we are unable to
enter into appropriate marketing arrangements for our product
candidates in international markets, we may not be able to
develop an effective international sales force to successfully
commercialize ILUVIEN and our other product candidates in
international markets. If we fail to enter into marketing
arrangements for our products or are unable to develop an
effective international sales force, our ability to generate
revenue outside of North America would be limited.
If we are unable to successfully implement our commercialization
plans and drive adoption by patients and retinal specialists of
ILUVIEN through our sales, marketing and commercialization
efforts and the efforts of OnCall, then we will not be able to
generate significant revenue, which will have a material adverse
effect on our business, results of operations, financial
condition and prospects.
38
In
order to commercialize ILUVIEN, we will need to grow the size of
our organization, and we may experience difficulties in managing
this growth.
As of December 31, 2010, we had 27 employees. As our
development and commercialization plans and strategies develop,
we will need to expand the size of our employee base for
managerial, operational, sales, marketing, financial and other
resources. Future growth would impose significant added
responsibilities on members of management, including the need to
identify, recruit, maintain, motivate and integrate additional
employees. Also, our management may have to divert a
disproportionate amount of its attention away from our
day-to-day
activities and devote a substantial amount of time to managing
these growth activities. Our future financial performance and
our ability to commercialize ILUVIEN and our other product
candidates and compete effectively will depend, in part, on our
ability to effectively manage any future growth. We may not be
able to effectively manage a rapid pace of growth and timely
implement improvements to our management infrastructure and
control systems.
ILUVIEN
and our other potential products may not be commercially viable
if we fail to obtain an adequate level of reimbursement for
these products from private insurers, the Medicare program and
other third-party payers which could be affected by the recently
enacted U.S. healthcare reform. The market for our products may
also be limited by the indications for which their use may be
reimbursed or the frequency at which they may be
administered.
The availability and levels of reimbursement by governmental and
other third-party payers affect the market for products such as
ILUVIEN and others that we may develop. These third-party payers
continually attempt to contain or reduce the costs of health
care by challenging the prices charged for medical products and
services. In the U.S., we will need to obtain approvals for
payment for ILUVIEN from private insurers, including managed
care organizations, and from the Medicare program. In recent
years, through legislative and regulatory actions, the federal
government has made substantial changes to various payment
systems under the Medicare program. Comprehensive reforms to the
U.S. healthcare system were recently enacted, including
changes to the methods for, and amounts of, Medicare
reimbursement. These reforms could significantly reduce payments
from Medicare and Medicaid over the next ten years. Reforms or
other changes to these payment systems, including modifications
to the conditions on qualification for payment, bundling
payments or the imposition of enrollment limitations on new
providers, may change the availability, methods and rates of
reimbursements from Medicare, private insurers and other
third-party payers for ILUVIEN and our other potential products.
Some of these changes and proposed changes could result in
reduced reimbursement rates for ILUVIEN and our other potential
products, which would adversely affect our business strategy,
operations and financial results.
We expect that private insurers will consider the efficacy, cost
effectiveness and safety of ILUVIEN in determining whether to
approve reimbursement for ILUVIEN and at what level. Obtaining
these approvals can be a time consuming and expensive process.
Our business would be materially adversely affected if we do not
receive approval for reimbursement of ILUVIEN from private
insurers on a timely or satisfactory basis. Although drugs that
are not self-administered are covered by Medicare, the Medicare
program has taken the position that it can decide not to cover
particular drugs if it determines that they are not
“reasonable and necessary” for Medicare beneficiaries.
Limitations on coverage could also be imposed at the local
Medicare carrier level or by fiscal intermediaries. Our business
could be materially adversely affected if the Medicare program,
local Medicare carriers or fiscal intermediaries were to make
such a determination and deny or limit the reimbursement of
ILUVIEN. Our business also could be adversely affected if
retinal specialists are not reimbursed by Medicare for the cost
of the procedure in which they administer ILUVIEN on a basis
satisfactory to the administering retinal specialists. If the
local contractors that administer the Medicare program are slow
to reimburse retinal specialists for ILUVIEN, the retinal
specialists may pay us more slowly, which would adversely affect
our working capital requirements.
39
Our business could also be adversely affected if private
insurers, including managed care organizations, the Medicare
program or other reimbursing bodies or payers limit the
indications for which ILUVIEN will be reimbursed to a smaller
set than we believe it is effective in treating or establish a
limitation on the frequency with which ILUVIEN may be
administered that is less often than we believe would be
effective.
In some foreign countries, particularly Canada and the countries
of the European Union, the pricing of prescription
pharmaceuticals is subject to governmental control. In Canada,
each province has a publicly funded drug plan with each having
its own formulary citing specific criteria for reimbursement and
prior authorization. Each provincial government except
Québec considers the clinical and cost-effectiveness
recommendations of the Common Drug Review performed by the
Canadian Agency for Drugs and Technologies in Health.
Québec has a separate drug review process that is performed
by its Medication Council. In the European Union, each country
has a different reviewing body that evaluates reimbursement
dossiers submitted by manufacturers of new drugs and then makes
recommendations as to whether or not the drug should be
reimbursed. In these countries, pricing negotiations with
governmental authorities can take 12 months or longer after
the receipt of regulatory approval and product launch. To obtain
reimbursement or pricing approval in some countries, we may be
required to conduct a clinical trial that compares the
cost-effectiveness of our products, including ILUVIEN, to other
available therapies. If reimbursement for our products is
unavailable, limited in scope or amount, or if pricing is set at
unsatisfactory levels, our business could be materially harmed.
We expect to experience pricing pressures in connection with the
sale of ILUVIEN and our future products due to the potential
healthcare reforms discussed above, as well as the trend toward
programs aimed at reducing health care costs, the increasing
influence of health maintenance organizations and additional
legislative proposals.
We
face substantial competition, which may result in others
discovering, developing or commercializing products before or
more successfully than we do.
The development and commercialization of new drugs is highly
competitive and the commercial success of ILUVIEN will depend on
several factors, including, but not limited to, its efficacy and
side effect profile, reimbursement acceptance by private
insurers and Medicare, acceptance of pricing, the development of
our sales and marketing organization, an adequate payment to
physicians for the insertion procedure (based on a cost assigned
by the American Medical Association to the procedure, also known
as a CPT code) and our ability to differentiate ILUVIEN from our
competitors’ products. We will face competition from major
pharmaceutical companies, specialty pharmaceutical companies and
biotechnology companies worldwide with respect to ILUVIEN and
any products that we may develop or commercialize in the future.
Our competitors may develop products or other novel technologies
that are more effective, safer or less costly than any that we
are developing. Our competitors may also obtain FDA or other
regulatory approval for their products more rapidly than we may
obtain approval for ours. The active pharmaceutical ingredient
in ILUVIEN is FA, which is not protected by currently valid
patents. As a result, our competitors could develop an
alternative formulation or delivery mechanisms to treat diseases
of the eye with FAc We do not have the right to develop and sell
pSivida’s proprietary delivery device for indications for
diseases outside of the eye or for the treatment of uveitis.
Further, our agreement with pSivida permits pSivida to grant to
any other party the right to use its intellectual property
(i) to treat DME through an incision smaller than that
required for a 25-gauge needle, unless using a corticosteroid
delivered to the back of the eye, (ii) to deliver any
compound outside the back of the eye unless it is to treat DME
through an incision required for a 25-gauge or larger needle, or
(iii) to deliver non-corticosteroids to the back of the
eye, unless it is to treat DME through an incision required for
a 25-gauge or larger needle.
There are no ophthalmic drug therapies approved by the FDA for
the treatment of DME. Lucentis, a drug sponsored by Genentech,
Inc., a wholly-owned member of the Roche Group is approved for
the treatment of visual impairment due to DME in the European
Union and in later stage trials in the U.S. is expected to
provide competition for ILUVIEN. Retinal specialists are
currently using laser photocoagulation and off-label therapies
for the treatment of DME, and may continue to use these
therapies in competition with ILUVIEN. Additional treatments for
DME are in various stages of preclinical or clinical testing.
Later stage products for
40
the treatment of DME include Ozurdex, a drug sponsored by
Allergan, Inc. and the VEGF Trap, a drug sponsored by Regeneron
Pharmaceuticals, Inc. and Bayer HealthCare. If approved, these
treatments would also compete with ILUVIEN. Other laser,
surgical or pharmaceutical treatments for DME may also compete
against ILUVIEN. These competitive therapies may result in
pricing pressure if we receive marketing approval for ILUVIEN,
even if ILUVIEN is otherwise viewed as a preferable therapy.
Many of our competitors have substantially greater financial,
technical and human resources than we have. Additional mergers
and acquisitions in the pharmaceutical and biotechnology
industries may result in even more resources being concentrated
by our competitors. Competition may increase further as a result
of advances made in the commercial applicability of technologies
and greater availability of capital for investment in these
fields.
We
currently do not have any collaboration agreements with
third-parties. We expect to depend on collaborations to develop
and commercialize our products. If we are unable to identify or
enter into an agreement with any material third-party
collaborator, if our collaborations with any such third-party
are not scientifically or commercially successful or if our
agreement with any such third-party is terminated or allowed to
expire, we could be adversely affected financially or our
business reputation could be harmed.
Our business strategy includes entering into collaborations with
corporate and academic collaborators for the research,
development and commercialization of additional product
candidates. We currently do not have any collaboration
agreements with third-parties. Areas in which we anticipate
entering into third-party collaboration arrangements include
joint sales and marketing arrangements for sales and marketing
of ILUVIEN outside of North America, and future product
development arrangements. If we are unable to identify or enter
into an agreement with any material third-party collaborator we
could be adversely affected financially or our business
reputation could be harmed. Any arrangements we do enter into
may not be scientifically or commercially successful. The
termination of any of these future arrangements might adversely
affect our ability to develop, commercialize and market our
products.
The success of our future collaboration arrangements will depend
heavily on the efforts and activities of our collaborators. Our
collaborators will have significant discretion in determining
the efforts and resources that they will apply to these
collaborations. We expect that the risks which we face in
connection with these future collaborations will include the
following:
|
|
|
| |
•
|
our collaboration agreements are expected to be for fixed terms
and subject to termination under various circumstances,
including, in many cases, on short notice without cause;
|
| |
| |
•
|
we expect to be required in our collaboration agreements not to
conduct specified types of research and development in the field
that is the subject of the collaboration. These agreements may
have the effect of limiting the areas of research and
development that we may pursue, either alone or in cooperation
with third-parties;
|
| |
| |
•
|
our collaborators may develop and commercialize, either alone or
with others, products and services that are similar to or
competitive with our products which are the subject of their
collaboration with us; and
|
| |
| |
•
|
our collaborators may change the focus of their development and
commercialization efforts. In recent years there have been a
significant number of mergers and consolidations in the
pharmaceutical and biotechnology industries, some of which have
resulted in the participant companies reevaluating and shifting
the focus of their business following the completion of these
transactions. The ability of our products to reach their
potential could be limited if any of our future collaborators
decreases or fails to increase spending relating to such
products.
|
Collaborations with pharmaceutical companies and other
third-parties often are terminated or allowed to expire by the
other party. With respect to our future collaborations, any such
termination or expiration could adversely affect us financially
as well as harm our business reputation.
41
We may
not be successful in our efforts to expand our portfolio of
products.
A key element of our strategy is to commercialize a portfolio of
new ophthalmic drugs in addition to ILUVIEN. We are seeking to
do so through our internal research programs and through
licensing or otherwise acquiring the rights to potential new
drugs and drug targets for the treatment of ophthalmic disease.
A significant portion of the research that we are conducting
involves new and unproven technologies. Research programs to
identify new disease targets and product candidates require
substantial technical, financial and human resources whether or
not we ultimately identify any candidates. Our research programs
may initially show promise in identifying potential product
candidates, yet fail to yield product candidates for clinical
development for a number of reasons, including:
|
|
|
| |
•
|
the research methodology used may not be successful in
identifying potential product candidates; or
|
| |
| |
•
|
potential product candidates may on further study be shown to
have harmful side effects or other characteristics that indicate
they are unlikely to be effective drugs.
|
We may be unable to license or acquire suitable product
candidates or products from third-parties for a number of
reasons. In particular, the licensing and acquisition of
pharmaceutical products is a competitive area. A number of more
established companies are also pursuing strategies to license or
acquire products in the ophthalmic field. These established
companies may have a competitive advantage over us due to their
size, cash resources and greater clinical development and
commercialization capabilities. Other factors that may prevent
us from licensing or otherwise acquiring suitable product
candidates include the following:
|
|
|
| |
•
|
we may be unable to license or acquire the relevant technology
on terms that would allow us to make an appropriate return from
the product;
|
| |
| |
•
|
companies that perceive us to be their competitors may be
unwilling to assign or license their product rights to
us; or
|
| |
| |
•
|
we may be unable to identify suitable products or product
candidates within our areas of expertise.
|
Additionally, it may take greater human and financial resources
to develop suitable potential product candidates through
internal research programs or by obtaining rights than we will
possess, thereby limiting our ability to develop a diverse
product portfolio.
If we are unable to develop suitable potential product
candidates through internal research programs or by obtaining
rights to novel therapeutics from third-parties, our business
will suffer.
We may
acquire additional businesses or form strategic alliances in the
future, and we may not realize the benefits of such
acquisitions.
We may acquire additional businesses or products, form strategic
alliances or create joint ventures with third-parties that we
believe will complement or augment our existing business. If we
acquire businesses with promising markets or technologies, we
may not be able to realize the benefit of acquiring such
businesses if we are unable to successfully integrate them with
our existing operations and company culture. We may have
difficulty in developing, manufacturing and marketing the
products of a newly acquired company that enhances the
performance of our combined businesses or product lines to
realize value from expected synergies. We cannot assure that,
following an acquisition, we will achieve the revenues or
specific net income that justifies the acquisition.
We
face the risk of product liability claims and may not be able to
obtain insurance.
Our business exposes us to the risk of product liability claims,
which is inherent in the manufacturing, testing and marketing of
drugs and related products. If the use of one or more of our
products harms people, we may be subject to costly and damaging
product liability claims. We have primary product liability
insurance that covers our clinical trials for a
$5.0 million general aggregate limit and excess product
liability insurance that covers our clinical trials for an
additional $5.0 million general aggregate limit. We intend
to expand our insurance coverage to include the sale of
commercial products if we obtain marketing approval for
42
any of the products that we may develop. We may not be able to
obtain or maintain adequate protection against potential
liabilities. If we are unable to obtain insurance at acceptable
cost or otherwise protect against potential product liability
claims, we will be exposed to significant liabilities, which may
materially and adversely affect our business and financial
position. These liabilities could prevent or interfere with our
product development and commercialization efforts.
In addition, our business is exposed to the risk of product
liability claims related to our sale and distribution of our
over-the-counter
dry eye product prior to its acquisition by Bausch &
Lomb Incorporated in July 2007. Our primary product liability
insurance and excess product liability insurance policies cover
product liability claims related to the product. To the extent
this insurance is insufficient to cover any product related
claims we may be exposed to significant liabilities, which may
materially and adversely affect our business and financial
condition.
If we
lose key management personnel, or if we fail to recruit
additional highly skilled personnel, it will impair our ability
to identify, develop and commercialize product
candidates.
We are highly dependent upon the principal members of our
management team, including C. Daniel Myers, our President and
Chief Executive Officer, Susan Caballa, our Senior Vice
President of Regulatory Affairs, Kenneth Green, Ph.D., our
Senior Vice President and Chief Scientific Officer, Richard
Eiswirth, our Chief Operating Officer and Chief Financial
Officer, and Dave Holland, our Senior Vice President of Sales
and Marketing. These executives have significant ophthalmic,
regulatory industry, sales and marketing, operational,
and/or
corporate finance experience. The loss of any such executives or
any other principal member of our management team would impair
our ability to identify, develop and market new products.
In addition, our growth will require us to hire a significant
number of qualified technical, commercial and administrative
personnel. There is intense competition from other companies and
research and academic institutions for qualified personnel in
the areas of our activities. If we cannot continue to attract
and retain, on acceptable terms, the qualified personnel
necessary for the continued development of our business, we may
not be able to sustain our operations or grow.
If our
contract research organizations (CROs), third-party vendors and
investigators do not successfully carry out their duties or if
we lose our relationships with them, our development efforts
with respect to ILUVIEN or any of our other product candidates
could be delayed.
We are dependent on CROs, third-party vendors and investigators
for preclinical testing and clinical trials related to our
discovery and development efforts with respect to our product
candidates and we will likely continue to depend on them to
assist in our future discovery and development efforts. These
parties are not our employees and we cannot control the amount
or timing of resources that they devote to our programs. If they
fail to devote sufficient time and resources to our development
programs with respect to our product candidates or if their
performance is substandard, it will delay the development and
commercialization of our product candidates. The parties with
which we contract for execution of clinical trials play a
significant role in the conduct of the trials and the subsequent
collection and analysis of data. Their failure to meet their
obligations could adversely affect clinical development of our
product candidates. Moreover, these parties may also have
relationships with other commercial entities, some of which may
compete with us. If they assist our competitors, it could harm
our competitive position.
If we lose our relationship with any one or more of these
parties, we could experience a significant delay in identifying
another comparable provider and contracting for its services. We
may be unable to retain an alternative provider on reasonable
terms, if at all. Even if we locate an alternative provider,
this provider may need additional time to respond to our needs
and may not provide the same type or level of service as the
original provider. In addition, any provider that we retain will
be subject to current Good Laboratory Practices (cGLP) and
similar foreign standards, and we do not have control over
compliance with these regulations by these providers.
Consequently, if these practices and standards are not adhered
to by these providers, the development and commercialization of
our product candidates could be delayed.
43
Our
products could be subject to restrictions or withdrawal from the
market and we may be subject to penalties if we fail to comply
with regulatory requirements, or if we experience unanticipated
problems with our products, when and if any of them is
approved.
Any product for which we obtain marketing approval, along with
the manufacturing processes, post-approval pharmacovigilance,
advertising and promotional activities for such product, will be
subject to continual requirements, review and periodic
inspections by the FDA and other regulatory bodies. Even if
regulatory approval of a product is granted, the approval may be
subject to limitations on the indicated uses for which the
product may be marketed or to the conditions of approval, or
contain requirements for costly post-marketing testing and
surveillance to monitor the safety or efficacy of the product.
Later discovery of previously unknown problems with our
products, manufacturer or manufacturing processes, or failure to
comply with regulatory requirements, may result in:
|
|
|
| |
•
|
restrictions on such products or manufacturing processes;
|
| |
| |
•
|
withdrawal of the products from the market;
|
| |
| |
•
|
voluntary or mandatory recall;
|
| |
| |
•
|
fines;
|
| |
| |
•
|
suspension of regulatory approvals;
|
| |
| |
•
|
product seizure; and
|
| |
| |
•
|
injunctions or the imposition of civil or criminal penalties.
|
We may be slow to adapt, or we may never adapt, to changes in
existing regulatory requirements or adoption of new regulatory
requirements or policies.
Failure
to obtain regulatory approval in foreign jurisdictions would
prevent us from marketing our products abroad.
We intend to market our products internationally. In order to
market our products in foreign jurisdictions, we will be
required to obtain separate regulatory approvals and comply with
numerous and varying regulatory requirements. In July 2010, we
submitted a MAA for ILUVIEN to the U.K. MHRA and to regulatory
authorities in Austria, France, Germany, Italy, Portugal and
Spain. The approval procedure varies among countries and
jurisdictions and can involve additional testing, and the time
required to obtain approval may differ from that required to
obtain FDA approval. Additionally, the foreign regulatory
approval process may include all of the risks associated with
obtaining FDA approval. For all of these reasons, we may not
obtain foreign regulatory approvals on a timely basis, if at
all. Approval by the FDA does not ensure approval by regulatory
authorities in other countries or jurisdictions, and approval by
one foreign regulatory authority does not ensure approval by
regulatory authorities in other foreign countries or
jurisdictions or by the FDA. We may not be able to file for
regulatory approvals and may not receive necessary approvals to
commercialize our products in any market. The failure to obtain
these approvals could harm our business materially.
Our
product candidates may cause undesirable side effects or have
other properties that could delay or prevent their regulatory
approval or limit their marketability.
Undesirable side effects caused by our product candidates could
interrupt, delay or halt clinical trials and could result in the
denial of regulatory approval by the FDA or other regulatory
authorities for any or all targeted indications, and in turn
prevent us from commercializing our product candidates and
generating revenues from their sale. Possible side effects of
ILUVIEN include, but are not limited to, extensive blurred
vision, cataracts, eye irritation, eye pain, increased IOP,
which may increase the risk of glaucoma, ocular discomfort,
reduced visual acuity, visual disturbance, endophthalmitis, or
long-standing vitreous floaters.
44
In addition, if any of our product candidates receives marketing
approval and we or others later identify undesirable side
effects caused by the product, we could face one or more of the
following consequences:
|
|
|
| |
•
|
regulatory authorities may require the addition of labeling
statements, such as a “black box” warning or a
contraindication;
|
| |
| |
•
|
regulatory authorities may withdraw their approval of the
product;
|
| |
| |
•
|
we may be required to change the way that the product is
administered, conduct additional clinical trials or change the
labeling of the product; and
|
| |
| |
•
|
our reputation may suffer.
|
Any of these events could prevent us from achieving or
maintaining market acceptance of the affected product or could
substantially increase the costs and expenses of commercializing
the product candidate, which in turn could delay or prevent us
from generating significant revenues from its sale.
Risks
Related to Intellectual Property and Other Legal
Matters
If we
or our licensors are unable to obtain and maintain protection
for the intellectual property incorporated into our products,
the value of our technology and products will be adversely
affected.
Our success will depend in large part on our ability or the
ability of our licensors to obtain and maintain protection in
the U.S. and other countries for the intellectual property
incorporated into our products. The patent situation in the
field of biotechnology and pharmaceuticals generally is highly
uncertain and involves complex legal and scientific questions.
We or our licensors may not be able to obtain additional issued
patents relating to our technology. Our success will depend in
part on the ability of our licensors to obtain, maintain
(including making periodic filings and payments) and enforce
patent protection for their intellectual property, in
particular, those patents to which we have secured exclusive
rights. Under our license with pSivida, pSivida controls the
filing, prosecution and maintenance of all patents. Our
licensors may not successfully prosecute or continue to
prosecute the patent applications to which we are licensed. Even
if patents are issued in respect of these patent applications,
we or our licensors may fail to maintain these patents, may
determine not to pursue litigation against entities that are
infringing these patents, or may pursue such litigation less
aggressively than we ordinarily would. Without protection for
the intellectual property that we own or license, other
companies might be able to offer substantially identical
products for sale, which could adversely affect our competitive
business position and harm our business prospects. Moreover, FAc
is an off-patent active ingredient that is commercially
available in several forms including the extended release ocular
implant Retisert.
Even if issued, patents may be challenged, narrowed,
invalidated, or circumvented, which could limit our ability to
stop competitors from marketing similar products or limit the
length of term of patent protection that we may have for our
products. In addition, our patents and our licensors’
patents may not afford us protection against competitors with
similar technology.
Litigation
or third-party claims of intellectual property infringement
would require us to divert resources and may prevent or delay
our development, regulatory approval or commercialization of our
product candidates.
We may not have rights under some patents or patent applications
that may be infringed by our products or potential products.
Third-parties may now or in the future own or control these
patents and patent applications in the U.S. and abroad.
These third-parties could bring claims against us or our
collaborators that would cause us to incur substantial expenses
or divert substantial employee resources from our business and,
if successful, could cause us to pay substantial damages or
prevent us from developing one or more product candidates.
Further, if a patent infringement suit were brought against us
or our collaborators, we or they could be forced to stop or
delay research, development, manufacturing or sales of the
product or product candidate that is the subject of the suit.
Several issued and pending U.S. patents claiming methods
and devices for the treatment of eye diseases, including through
the use of steroids, implants and injections into the eye,
purport to cover aspects of
45
ILUVIEN. For example, one of our potential competitors holds
issued and pending U.S. patents with claims covering
devices for injecting an ocular implant into a patient’s
eye similar to the ILUVIEN inserter. There is also an issued
U.S. patent with claims covering implanting a steroidal
anti-inflammatory agent to treat an inflammation-mediated
condition of the eye. If these or any other patents were held by
a court of competent jurisdiction to be valid and to cover
aspects of ILUVIEN, then the owners of such patents would be
able to block our ability to commercialize ILUVIEN unless and
until we obtain a license under such patents (which license
might require us to pay royalties or grant a cross-license to
one or more patents that we own), until such patents expire or
unless we are able to redesign our product to avoid any such
valid patents.
As a result of patent infringement claims, or in order to avoid
potential claims, we or our collaborators may choose to seek, or
be required to seek, a license from the third-party and would
most likely be required to pay license fees or royalties or
both. These licenses may not be available on acceptable terms,
or at all. Even if we or our collaborators were able to obtain a
license, the rights may be nonexclusive, which would give our
competitors access to the same intellectual property.
Ultimately, we could be prevented from commercializing a
product, or be forced to cease some aspect of our business
operations if, as a result of actual or threatened patent
infringement claims, we or our collaborators are unable to enter
into licenses on acceptable terms. This could harm our business
significantly.
There has been substantial litigation and other proceedings
regarding patent and other intellectual property rights in the
pharmaceutical and biotechnology industries. In addition to
infringement claims against us, we may become a party to other
patent litigation and other proceedings, including interference
proceedings declared by the U.S. Patent and Trademark
Office and opposition proceedings in the European Patent Office,
regarding intellectual property rights with respect to our
products and technology. The cost to us of any litigation or
other proceeding, regardless of its merit, even if resolved in
our favor, could be substantial. Some of our competitors may be
able to sustain the costs of such litigation or proceedings more
effectively than we can because of their substantially greater
financial resources. Uncertainties resulting from the initiation
and continuation of patent litigation or other proceedings could
have a material adverse effect on our ability to compete in the
marketplace. Intellectual property litigation and other
proceedings may, regardless of their merit, also absorb
significant management time and employee resources.
If we
fail to comply with our obligations in the agreements under
which we license development or commercialization rights to
products or technology from third-parties, we could lose license
rights that are important to our business.
Our licenses are important to our business, and we expect to
enter into additional licenses in the future. We hold a license
from pSivida under intellectual property relating to ILUVIEN.
This license imposes various commercialization, milestone
payment, profit sharing, insurance and other obligations on us.
We also hold a license from Dainippon Sumitomo Pharma Co., Ltd.
under patents relating to ILUVIEN. This license imposes a
milestone payment and other obligations on us. If we fail to
comply with these obligations, the licensor may have the right
to terminate the applicable license, in which event we would not
be able to market products, such as ILUVIEN, that may be covered
by such license.
If we
are unable to protect the confidentiality of our proprietary
information and know-how, the value of our technology and
products could be adversely affected.
In addition to patented technology, we rely upon unpatented
proprietary technology, processes, trade secrets and know-how.
Any involuntary disclosure or misappropriation by third-parties
of our confidential or proprietary information could enable
competitors to quickly duplicate or surpass our technological
achievements, thus eroding our competitive position in our
market. We seek to protect confidential or proprietary
information in part by confidentiality agreements with our
employees, consultants and third-parties. While we require all
of our employees, consultants, advisors and any third-parties
who have access to our proprietary know-how, information and
technology to enter into confidentiality agreements, we cannot
be certain that this know-how, information and technology will
not be disclosed or that competitors will not otherwise gain
access to our trade secrets or independently develop
substantially equivalent information and techniques. These
agreements may be terminated or breached, and we may not have
adequate remedies for any such termination
46
or breach. Furthermore, these agreements may not provide
meaningful protection for our trade secrets and know-how in the
event of unauthorized use or disclosure. To the extent that any
of our staff were previously employed by other pharmaceutical or
biotechnology companies, those employers may allege violations
of trade secrets and other similar claims in relation to their
drug development activities for us.
If our
efforts to protect the proprietary nature of the intellectual
property related to our products are not adequate, we may not be
able to compete effectively in our markets.
The strength of our patents in the biotechnology and
pharmaceutical field involves complex legal and scientific
questions and can be uncertain. In addition to the rights we
have licensed from pSivida relating to our product candidates,
we rely upon intellectual property we own relating to our
products, including patents, patent applications and trade
secrets. As of December 31, 2010, we owned one pending
non-provisional U.S. utility patent application, one issued
U.S. design patent and one patent Cooperation Treaty
Application, relating to our inserter system for ILUVIEN. Our
patent applications may be challenged or fail to result in
issued patents and our existing or future patents may be too
narrow to prevent third-parties from developing or designing
around these patents.
As of December 31, 2010, the patent rights relating to
ILUVIEN licensed to us from pSivida include three
U.S. patents that expire between March 2019 and April 2020
and counterpart filings to these patents in a number of other
jurisdictions. No patent term extension will be available for
any of these U.S. patents or any of our licensed
U.S. pending patent applications. After these patents
expire in April 2020, we will not be able to block others from
marketing FAc in an insert similar to ILUVIEN in the
U.S. Moreover, it is possible that a third-party could
successfully challenge the scope (i.e., whether a patent is
infringed), validity and enforceability of our licensed patents
prior to patent expiration and obtain approval to market a
competitive product.
Further, the patent applications that we license or have filed
may fail to result in issued patents. Some claims in pending
patent applications filed or licensed by us have been rejected
by patent examiners. These claims may need to be amended. Even
after amendment, a patent may not be permitted to issue.
Further, the existing or future patents to which we have rights
based on our agreement with pSivida may be too narrow to prevent
third-parties from developing or designing around these patents.
Additionally, we may lose our rights to the patents and patent
applications we license in the event of a breach or termination
of the license agreement. Manufacturers may also seek to obtain
approval to sell a generic version of ILUVIEN prior to the
expiration of the relevant licensed patents. If the sufficiency
of the breadth or strength of protection provided by the patents
we license with respect to ILUVIEN or the patents we pursue
related to another product candidate is threatened, it could
dissuade companies from collaborating with us to develop, and
threaten our ability to commercialize ILUVIEN and our other
product candidates. Further, if we encounter delays in our
clinical trials, the period of time during which we could market
ILUVIEN and our other product candidates under patent protection
would be reduced. We rely on trade secret protection and
confidentiality agreements to protect certain proprietary
know-how that is not patentable, for processes for which patents
are difficult to enforce and for any other elements of our
development processes with respect to ILUVIEN and our other
product candidates that involve proprietary know-how,
information and technology that is not covered by patent
applications. While we require all of our employees,
consultants, advisors and any third-parties who have access to
our proprietary know-how, information and technology to enter
into confidentiality agreements, we cannot be certain that this
know-how, information and technology will not be disclosed or
that competitors will not otherwise gain access to our trade
secrets or independently develop substantially equivalent
information and techniques. Further, the laws of some foreign
countries do not protect proprietary rights to the same extent
as the laws of the U.S. As a result, we may encounter
significant problems in protecting and defending our
intellectual property both in the U.S. and abroad. If we
are unable to protect or defend the intellectual property
related to our technologies, we will not be able to establish or
maintain a competitive advantage in our market.
47
Third-party
claims of intellectual property infringement may prevent or
delay our discovery, development and commercialization efforts
with respect to ILUVIEN and our other product
candidates.
Our commercial success depends in part on avoiding infringement
of the patents and proprietary rights of third-parties.
Third-parties may assert that we are employing their proprietary
technology without authorization. In addition, at least several
issued and pending U.S. patents claiming methods and
devices for the treatment of eye diseases, including through the
use of steroids, implants and injections into the eye, purport
to cover aspects of ILUVIEN.
Although we are not currently aware of any litigation or other
proceedings or third-party claims of intellectual property
infringement related to ILUVIEN, the pharmaceutical industry is
characterized by extensive litigation regarding patents and
other intellectual property rights. Other parties may in the
future allege that our activities infringe their patents or that
we are employing their proprietary technology without
authorization. We may not have identified all the patents,
patent applications or published literature that affect our
business either by blocking our ability to commercialize our
product, by preventing the patentability of one or more aspects
of our products or those of our licensors or by covering the
same or similar technologies that may affect our ability to
market our product. We cannot predict whether we would be able
to obtain a license on commercially reasonable terms, if at all.
Any inability to obtain such a license under the applicable
patents on commercially reasonable terms, or at all, may have a
material adverse effect on our ability to commercialize ILUVIEN
or other products until such patents expire.
In addition, third-parties may obtain patents in the future and
claim that use of our product candidates or technologies
infringes upon these patents. Furthermore, parties making claims
against us may obtain injunctive or other equitable relief,
which could effectively block our ability to further develop and
commercialize one or more of our product candidates. Defense of
these claims, regardless of their merit, would involve
substantial litigation expense and would be a substantial
diversion of employee resources from our business. In the event
of a successful claim of infringement against us, we may have to
pay substantial damages, obtain one or more licenses from
third-parties or pay royalties, or we may be enjoined from
further developing or commercializing our product candidates and
technologies. In addition, even in the absence of litigation, we
may need to obtain licenses from third-parties to advance our
research or allow commercialization of our product candidates,
and we have done so from time to time. We may fail to obtain
future licenses at a reasonable cost or on reasonable terms, if
at all. In that event, we may be unable to further develop and
commercialize one or more of our product candidates, which could
harm our business significantly.
We may
become involved in lawsuits to protect or enforce our patents or
the patents of our licensors, which could be expensive, time
consuming and unsuccessful.
Competitors may infringe our patents or the patents of our
licensors. To counter infringement or unauthorized use, we may
be required to file infringement claims, which can be expensive
and time consuming. In addition, in an infringement proceeding,
a court may decide that a patent of ours or our licensors is not
valid or is unenforceable, or may refuse to stop the other party
from using the technology at issue on the grounds that our
patents do not cover the technology in question. An adverse
result in any litigation or defense proceedings could put one or
more of our patents at risk of being invalidated or interpreted
narrowly and could put our patent applications at risk of not
issuing.
Interference proceedings brought by the U.S. Patent and
Trademark Office may be necessary to determine the priority of
inventions with respect to our patents and patent applications
or those of our collaborators or licensors. An unfavorable
outcome could require us to cease using the technology or to
attempt to license rights to it from the prevailing party. Our
business could be harmed if a prevailing party does not offer us
a license on terms that are acceptable to us. Litigation or
interference proceedings may fail and, even if successful, may
result in substantial costs and distraction of our management
and other employees. We may not be able to prevent, alone or
with our licensors, misappropriation of our proprietary rights,
particularly in countries where the laws may not protect those
rights as fully as in the U.S.
Furthermore, because of the substantial amount of discovery
required in connection with intellectual property litigation,
there is a risk that some of our confidential information could
be compromised by
48
disclosure during this type of litigation. In addition, there
could be public announcements of the results of hearings,
motions or other interim proceedings or developments. If
securities analysts or investors perceive these results to be
negative, it could have a substantial adverse effect on the
price of our common stock.
Product
liability lawsuits could divert our resources, result in
substantial liabilities and reduce the commercial potential of
our products.
The risk that we may be sued on product liability claims is
inherent in the development of pharmaceutical products. We face
a risk of product liability exposure related to the testing of
our product candidates in clinical trials and will face even
greater risks upon any commercialization by us of our product
candidates. We believe that we may be at a greater risk of
product liability claims relative to other pharmaceutical
companies because our products are inserted into the eye, and it
is possible that we may be held liable for eye injuries of
patients who receive our product. These lawsuits may divert our
management from pursuing our business strategy and may be costly
to defend. In addition, if we are held liable in any of these
lawsuits, we may incur substantial liabilities and may be forced
to limit or forego further commercialization of one or more of
our products. Although we maintain primary product liability
insurance and excess product liability insurance that cover our
clinical trials, our aggregate coverage limit under these
insurance policies is $10.0 million, and while we believe
this amount of insurance is sufficient to cover our product
liability exposure, these limits may not be high enough to fully
cover potential liabilities. In addition, we may not be able to
obtain or maintain sufficient insurance coverage at an
acceptable cost or otherwise to protect against potential
product liability claims, which could prevent or inhibit the
commercial production and sale of our products.
Legislative
or regulatory reform of the health care system in the U.S. and
foreign jurisdictions may adversely impact our business,
operations or financial results.
Our industry is highly regulated and changes in law may
adversely impact our business, operations or financial results.
In particular, in March 2010, the Patient Protection and
Affordable Care Act, or PPACA, and a related reconciliation bill
were signed into law. This new legislation changes the current
system of healthcare insurance and benefits intended to broaden
coverage and control costs. The new law also contains provisions
that will affect companies in the pharmaceutical industry and
other healthcare related industries by imposing additional costs
and changes to business practices. Provisions affecting
pharmaceutical companies include the following:
|
|
|
| |
•
|
Mandatory rebates for drugs sold into the Medicaid program have
been increased, and the rebate requirement has been extended to
drugs used in risk-based Medicaid managed care plans.
|
| |
| |
•
|
The 340B Drug Pricing Program under the Public Health Services
Act has been extended to require mandatory discounts for drug
products sold to certain critical access hospitals, cancer
hospitals and other covered entities.
|
| |
| |
•
|
Pharmaceutical companies are required to offer discounts on
brand-name drugs to patients who fall within the Medicare
Part D coverage gap, commonly referred to as the
“Donut Hole.”
|
| |
| |
•
|
Pharmaceutical companies are required to pay an annual non-tax
deductible fee to the federal government based on each
company’s market share of prior year total sales of branded
products to certain federal healthcare programs, such as
Medicare, Medicaid, Department of Veterans Affairs and
Department of Defense. The aggregated industry-wide fee is
expected to total $28 billion through 2019, of which
$2.5 billion will be payable in 2011. Since we expect our
branded pharmaceutical sales to constitute a small portion of
the total federal health program pharmaceutical market, we do
not expect this annual assessment to have a material impact on
our financial condition.
|
| |
| |
•
|
The new law provides that biologic products may receive
12 years of market exclusivity, with a possible six-month
extension for pediatric products. After this exclusivity ends,
generic manufacturers will be permitted to enter the market,
which is likely to reduce the pricing for such products and
could affect the company’s profitability. In addition,
generic manufacturers will be permitted to challenge one or more
of the patents for a branded drug after a product is marketed
for four years.
|
49
The full effects of the U.S. healthcare reform legislation
cannot be known until the new law is implemented through
regulations or guidance issued by the Centers for
Medicare & Medicaid Services and other federal and
state healthcare agencies. The financial impact of the
U.S. healthcare reform legislation over the next few years
will depend on a number of factors, including but not limited,
to the policies reflected in implementing regulations and
guidance, and changes in sales volumes for products affected by
the new system of rebates, discounts and fees. The new
legislation may also have a positive impact on our future net
sales, if any, by increasing the aggregate number of persons
with healthcare coverage in the U.S., but such increases are
unlikely to be realized until approximately 2014 at the earliest.
In addition, in September 2007, the Food and Drug Administration
Amendments Act of 2007 was enacted, giving the FDA enhanced
post-marketing authority, including the authority to require
post-marketing studies and clinical trials, labeling changes
based on new safety information, and compliance with risk
evaluations and mitigation strategies approved by the FDA. The
FDA’s exercise of this authority could result in delays or
increased costs during product development, clinical trials and
regulatory review, increased costs to ensure compliance with
post-approval regulatory requirements, and potential
restrictions on the sale
and/or
distribution of approved products.
Further, in some foreign countries, including the European Union
and Canada, the pricing of prescription pharmaceuticals is
subject to governmental control. In these countries, pricing
negotiations with governmental authorities can take six to
12 months or longer after the receipt of regulatory
approval and product launch. To obtain reimbursement or pricing
approval in some countries, we may be required to conduct a
clinical trial that compares the cost-effectiveness of our
product candidate to other available therapies. Our business
could be materially harmed if reimbursement of our products is
unavailable or limited in scope or amount or if pricing is set
at unsatisfactory levels.
Moreover, we cannot predict what healthcare reform initiatives
may be adopted in the future. Further federal and state
legislative and regulatory developments are likely, and we
expect ongoing initiatives in the U.S. to increase pressure
on drug pricing. Such reforms could have an adverse effect on
anticipated revenues from product candidates that we may
successfully develop and for which we may obtain regulatory
approval and may affect our overall financial condition and
ability to develop drug candidates.
If we
use hazardous and biological materials in a manner that causes
injury or violates applicable law, we may be liable for
damages.
Our research and development activities involve the controlled
use of potentially hazardous substances, including chemical and
biological materials. In addition, our operations produce
hazardous waste products. Federal, state and local laws and
regulations in both the U.S. and Canada govern the use,
manufacture, storage, handling and disposal of hazardous
materials. Although we believe that our procedures for use,
handling, storing and disposing of these materials comply with
legally prescribed standards, we may incur significant
additional costs to comply with applicable laws in the future.
Also, even if we are in compliance with applicable laws, we
cannot completely eliminate the risk of contamination or injury
resulting from hazardous materials and we may incur liability as
a result of any such contamination or injury. In the event of an
accident, we could be held liable for damages or penalized with
fines, and the liability could exceed our resources. We do not
have any insurance for liabilities arising from hazardous
materials. Compliance with applicable environmental laws and
regulations is expensive, and current or future environmental
regulations may impair our research, development and production
efforts, which could harm our business, operating results and
financial condition.
Our
ability to use our net operating loss carry-forwards may be
limited.
At December 31, 2010, we had U.S. federal and state
net operating loss (NOL) carry-forwards of approximately
$97.8 million and $81.0 million, respectively, which
expire at various dates beginning in 2020 through 2030.
Section 382 of the Internal Revenue Code limits the annual
utilization of NOL carry-forwards and tax credit carry-forwards
following an ownership change in our company. We are currently
evaluating the impact of our IPO on our NOL carry-forwards and
whether certain changes in ownership have occurred that
50
would limit our ability to utilize a portion of our NOL
carry-forwards. If it is determined that significant ownership
changes have occurred since we generated these NOL
carry-forwards, we may be subject to annual limitations on the
use of these NOL carry-forwards under Internal Revenue Code
Section 382 (or comparable provisions of state law).
We
incur significant increased costs as a result of operating as a
public company, and our management is required to devote
substantial time to new compliance initiatives.
As a public company, we incur significant legal, accounting and
other expenses that we did not incur as a private company. The
Sarbanes-Oxley Act, as well as rules subsequently implemented by
the SEC and Nasdaq, have imposed various new requirements on
public companies, including requiring establishment and
maintenance of effective disclosure and financial controls and
changes in corporate governance practices. Our management and
other personnel are required to devote a substantial amount of
time to these new compliance initiatives. Moreover, these rules
and regulations have substantially increased our legal and
financial compliance costs and have made some activities more
time consuming and costly. These rules and regulations may make
it more difficult and more expensive for us to maintain our
existing director and officer liability insurance or to obtain
similar coverage from an alternative provider.
The Sarbanes-Oxley Act requires, among other things, that we
maintain effective internal controls for financial reporting and
disclosure controls and procedures. In particular, pursuant to
Section 404 of the Sarbanes-Oxley Act (Section 404),
we may be required to perform system and process evaluation and
testing of our internal controls over financial reporting to
allow management and our independent registered public
accounting firm to report, commencing in our annual report on
Form 10-K
for the year ending December 31, 2011, on the effectiveness
of our internal controls over financial reporting. Our testing,
or the subsequent testing by our independent registered public
accounting firm, may reveal deficiencies in our internal
controls over financial reporting that are deemed to be material
weaknesses. Our compliance with Section 404 would require
that we incur substantial accounting expense and expend
significant management efforts. We currently do not have an
internal audit group, and we will need to hire additional
accounting and financial staff. Moreover, if we are not able to
comply with the requirements of Section 404 in a timely
manner or if we or our independent registered public accounting
firm identifies deficiencies in our internal controls over
financial reporting that are deemed to be material weaknesses,
the market price of our stock could decline and we could be
subject to sanctions or investigations by Nasdaq, the SEC or
other regulatory authorities, which would require additional
financial and management resources.
Risks
Relating to Our Financial Results and Need for
Financing
Fluctuations
in our quarterly operating results and cash flows could
adversely affect the price of our common stock.
We expect our operating results and cash flows to be subject to
quarterly fluctuations. The revenues we generate, if any, and
our operating results will be affected by numerous factors,
including, but not limited to:
|
|
|
| |
•
|
the commercial success of our product candidates;
|
| |
| |
•
|
the emergence of products that compete with our product
candidates;
|
| |
| |
•
|
the status of our preclinical and clinical development programs;
|
| |
| |
•
|
variations in the level of expenses related to our existing
product candidates or preclinical and clinical development
programs;
|
| |
| |
•
|
execution of collaborative, licensing or other arrangements, and
the timing of payments received or made under those arrangements;
|
| |
| |
•
|
any intellectual property infringement lawsuits to which we may
become a party; and
|
| |
| |
•
|
regulatory developments affecting our product candidates or
those of our competitors.
|
51
If our quarterly operating results fall below the expectations
of investors or securities analysts, the price of our common
stock could decline substantially. Furthermore, any quarterly
fluctuations in our operating results and cash flows may, in
turn, cause the price of our stock to fluctuate substantially.
We believe that quarterly comparisons of our financial results
are not necessarily meaningful and should not be relied upon as
an indication of our future performance.
We may
need additional financing in the event that we do not receive
regulatory approval for ILUVIEN or the approval is delayed or,
if approved, the future sales of ILUVIEN do not generate
sufficient revenues to fund our operations. This financing may
be difficult to obtain and may restrict our
operations.
Prior to our IPO, we funded our operations through the private
placement of common stock, preferred stock and convertible debt,
as well as by the sale of certain assets of the non-prescription
business in which we were previously engaged. As of
December 31, 2010, we had approximately $28.5 million
in cash and cash equivalents and $26.3 million in
investments which, together with the Credit Facility described
below, we believe is sufficient to fund our operations through
the projected commercialization of ILUVIEN and the expected
generation of revenue in late 2011. In October 2010, we obtained
a $32.5 million senior secured credit facility (Credit
Facility) to help fund our working capital requirements. The
Credit Facility consists of a $20 million revolving line of
credit and a $12.5 million term loan. The lenders have
advanced $6.25 million under the term loan and may advance
the remaining $6.25 million following FDA approval of
ILUVIEN, but no later than July 31, 2011. Given the status
of the FDA’s review of the NDA, it is unlikely that FDA
approval would occur prior to July 31, 2011. We are in
discussions with the lenders to amend the terms of the Credit
Facility to, among other things, extend the availability of the
term loan. However, there are no assurances that the Credit
Facility will be amended. We may draw on the revolving line of
credit against eligible, domestic accounts receivable subsequent
to the launch of ILUVIEN. The commercialization of ILUVIEN is
dependent upon approval by the FDA, however, and we cannot be
sure that ILUVIEN will be approved by the FDA in 2011, if at
all, or that, if approved, future sales of ILUVIEN will generate
enough revenue to fund our operations beyond its
commercialization.
In the event additional financing is needed or advisable, we may
seek to fund our operations through the sale of equity
securities, strategic collaboration agreements and additional
debt financing. We cannot be sure that additional financing from
any of these sources will be available when needed or that, if
available, the additional financing will be obtained on terms
favorable to us or our stockholders especially in light of the
current difficult financial environment. If we raise additional
funds by issuing equity securities, substantial dilution to
existing stockholders would likely result and the terms of any
new equity securities may have a preference over our common
stock. If we attempt to raise additional funds through strategic
collaboration agreements and additional debt financing, we may
not be successful in obtaining collaboration agreements, or in
receiving milestone or royalty payments under those agreements,
or the terms of the debt may involve significant cash payment
obligations as well as covenants and specific financial ratios
that may restrict our ability to commercialize our product
candidates or operate our business. For example, under the
Credit Facility, we are subject to a variety of affirmative and
negative covenants, including required financial reporting,
limitations on our cash balances, limitations on the disposition
of assets, limitations on the incurrence of additional debt, and
other requirements. To secure the performance of our obligations
under the Credit Facility, we pledged all of our assets, other
than our intellectual property (provided that if we fail to meet
certain financial conditions, a curable lien will be imposed on
our intellectual property as well), to the lenders. Our failure
to comply with the covenants under the Credit Facility could
result in an event of default, the acceleration of our debt and
the loss of our assets. Any declaration of an event of default
could significantly harm our business and prospects and could
cause our stock price to decline.
Risks
Related to Our Common Stock
Our
stock price has been and may continue to be volatile, and the
value of an investment in our common stock may
decline.
We completed the initial public offering of shares of our common
stock in April 2010 at a price of $11.00 per share.
Subsequently, our common stock has traded as low as $6.30 per
share. The realization of
52
any of the risks described in these risk factors or other
unforeseen risks could have a dramatic and adverse effect on the
market price of our common stock. The trading price of our
common stock is likely to continue to be highly volatile and
could be subject to wide fluctuations in response to various
factors, some of which are beyond our control. These factors
include:
|
|
|
| |
•
|
the timing and final outcome of FDA review of our NDA for
ILUVIEN;
|
| |
| |
•
|
results from our clinical trial programs;
|
| |
| |
•
|
FDA or international regulatory actions, including failure to
receive regulatory approval for any of our product candidates;
|
| |
| |
•
|
failure of any of our product candidates, if approved, to
achieve commercial success;
|
| |
| |
•
|
quarterly variations in our results of operations or those of
our competitors;
|
| |
| |
•
|
our ability to develop and market new and enhanced product
candidates on a timely basis;
|
| |
| |
•
|
announcements by us or our competitors of acquisitions,
regulatory approvals, clinical milestones, new products,
significant contracts, commercial relationships or capital
commitments;
|
| |
| |
•
|
third-party coverage and reimbursement policies;
|
| |
| |
•
|
additions or departures of key personnel;
|
| |
| |
•
|
commencement of, or our involvement in, litigation;
|
| |
| |
•
|
our ability to meet our repayment and other obligations under
our Credit Facility;
|
| |
| |
•
|
changes in governmental regulations or in the status of our
regulatory approvals;
|
| |
| |
•
|
changes in earnings estimates or recommendations by securities
analysts;
|
| |
| |
•
|
any major change in our board or management;
|
| |
| |
•
|
general economic conditions and slow or negative growth of our
markets; and
|
| |
| |
•
|
political instability, natural disasters, war
and/or
events of terrorism.
|
From time to time, we estimate the timing of the accomplishment
of various scientific, clinical, regulatory and other product
development goals or milestones. These milestones may include
the commencement or completion of scientific studies and
clinical trials and the submission of regulatory filings. Also,
from time to time, we expect that we will publicly announce the
anticipated timing of some of these milestones. All of these
milestones are based on a variety of assumptions. The actual
timing of these milestones can vary dramatically compared to our
estimates, in some cases for reasons beyond our control. If we
do not meet these milestones as publicly announced, our stock
price may decline and the commercialization of our product and
product candidates may be delayed.
In addition, the stock market has experienced extreme price and
volume fluctuations that have often been unrelated or
disproportionate to the operating performance of publicly traded
companies. Broad market and industry factors may seriously
affect the market price of companies’ stock, including
ours, regardless of actual operating performance. These
fluctuations may be even more pronounced in the trading market
for our stock. In addition, in the past, following periods of
volatility in the overall market and the market price of a
particular company’s securities, securities class action
litigation has often been instituted against these companies.
This litigation, if instituted against us, could result in
substantial costs and a diversion of our management’s
attention and resources.
Certain
of our existing stockholders have the ability to control the
outcome of matters submitted for stockholder approval and may
have interests that differ from those of our other
stockholders.
As of December 31, 2010, our executive officers, key
employees and directors and their affiliates beneficially owned,
in the aggregate, approximately 82.6% of our outstanding common
stock. As a result,
53
these stockholders, if acting together, may be able to exercise
significant influence over all matters requiring stockholder
approval, including the election of directors and the approval
of significant corporate transactions, and this concentration of
voting power may have the effect of delaying or impeding actions
that could be beneficial to you, including actions that may be
supported by our board of directors.
We
currently do not intend to pay dividends on our common stock
and, consequently, your only opportunity to achieve a return on
your investment is if the price of our common stock
appreciates.
We do not anticipate that we will pay any cash dividends on
shares of our common stock for the foreseeable future. Any
determination to pay dividends in the future will be at the
discretion of our board of directors and will depend on results
of operations, financial condition, contractual restrictions,
restrictions imposed by applicable law and other factors our
board of directors deems relevant. Accordingly, realization of a
gain on your investment will depend on the appreciation of the
price of our common stock, which may never occur.
Significant
sales of our common stock could depress or reduce the market
price of our common stock, or cause our shares of common stock
to trade below the prices at which they would otherwise trade,
or impede our ability to raise future capital.
A small number of institutional investors and private equity
funds hold a significant number of shares of our common stock.
Sales by these stockholders of a substantial number of shares,
or the expectation of such sales, could cause a significant
reduction in the market price of our common stock. Additionally,
a small number of early investors in our company have rights,
subject to certain conditions, to require us to file
registration statements to permit the resale of their shares in
the public market or to include their shares in registration
statements that we may file for ourselves or other stockholders.
In addition to our outstanding common stock, as of
December 31, 2010, there were a total of
2,741,985 shares of common stock that we have registered
and that we are obligated to issue upon the exercise of
currently outstanding options granted under our equity incentive
plans. Upon the exercise of these options, in accordance with
their respective terms, these shares may be resold freely,
subject to restrictions imposed on our affiliates under
Rule 144. If significant sales of these shares occur in
short periods of time, these sales could reduce the market price
of our common stock. Any reduction in the trading price of our
common stock could impede our ability to raise capital on
attractive terms.
Actual or perceived significant sales of our common stock could
depress or reduce the market price of our common stock, or cause
our shares of common stock to trade below the prices at which
they would otherwise trade, or impede our ability to raise
future capital.
Future
sales and issuances of our equity securities or rights to
purchase our equity securities, including pursuant to our equity
incentive plans, would result in dilution of the percentage
ownership of our stockholders and could cause our stock price to
fall.
To the extent we raise additional capital by issuing equity
securities, our stockholders may experience substantial
dilution. We may sell common stock, convertible securities or
other equity securities in one or more transactions at prices
and in a manner we determine from time to time. If we sell
common stock, convertible securities or other equity securities
in more than one transaction, investors may be diluted by
subsequent sales. Such sales may also result in material
dilution to our existing stockholders, and new investors could
gain rights superior to existing stockholders.
Pursuant to our 2010 Equity Incentive Plan, our board of
directors is authorized to grant stock options to our employees,
directors and consultants. The number of shares available for
future grant under our 2010 Equity Incentive Plan increases each
year by an amount equal to the lesser of 4% of all shares of our
capital stock outstanding as of January 1st of each
year, 2,000,000 shares, or such lesser number as determined
by our board of directors. On January 1, 2011, an
additional 1,250,238 shares became available for future
issuance under our 2010 Equity Incentive Plan in accordance with
the annual increase. In addition, we have reserved
494,422 shares of our common stock for issuance under our
2010 Employee Stock Purchase Plan. The number
54
of shares eligible for purchase increases as of
January 1st of each year in an amount equal to the
shares purchased under the plan in the preceding year. As such,
on January 1, 2011, an additional 8,246 shares became
available for future issuance under our 2010 Employee Stock
Purchase Plan.
Anti-takeover
provisions in our charter and bylaws and in Delaware law could
prevent or delay acquisition bids for us that you might consider
favorable and could entrench current management.
We are a Delaware corporation and the anti-takeover provisions
of the Delaware General Corporation Law may deter, delay or
prevent a change in control by prohibiting us from engaging in a
business combination with an interested stockholder for a period
of three years after the person becomes an interested
stockholder, even if a change in control would be beneficial to
our existing stockholders. In addition, our restated certificate
of incorporation and bylaws may discourage, delay or prevent a
change in our management or control over us that stockholders
may consider favorable. Our restated certificate of
incorporation and bylaws:
|
|
|
| |
•
|
Authorize the issuance of “blank check” preferred
stock that could be issued by our board of directors to thwart a
takeover attempt;
|
| |
| |
•
|
Do not provide for cumulative voting in the election of
directors, which would allow holders of less than a majority of
our outstanding common stock to elect some directors;
|
| |
| |
•
|
Establish a classified board of directors, as a result of which
the successors to the directors whose terms have expired will be
elected to serve from the time of election and qualification
until the third annual meeting following their election;
|
| |
| |
•
|
Require that directors only be removed from office for cause;
|
| |
| |
•
|
Provide that vacancies on the board of directors, including
newly created directorships, may be filled only by a majority
vote of directors then in office;
|
| |
| |
•
|
Limit who may call special meetings of stockholders;
|
| |
| |
•
|
Prohibit stockholder action by written consent, requiring all
actions to be taken at a meeting of the stockholders; and
|
| |
| |
•
|
Establish advance notice requirements for nominating candidates
for election to the board of directors or for proposing matters
that can be acted upon by stockholders at stockholder meetings.
|
If
securities or industry analysts do not publish research or
reports or publish unfavorable research or reports about our
business, our stock price and trading volume could
decline.
The trading market for our common stock depends in part on the
research and reports that securities or industry analysts
publish about us, our business, our market or our competitors.
If one or more of the analysts who covers us downgrades our
stock, our stock price would likely decline. If one or more of
these analysts ceases to cover us or fails to regularly publish
reports on us, interest in our stock could decrease, which could
cause our stock price or trading volume to decline.
|
|
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS
|
Not applicable.
Our current headquarters are located in Alpharetta, Georgia,
consisting of approximately 14,000 square feet of office
space. Our lease for this facility expires in December 2011.
Management believes that the leased facilities are suitable and
adequate to meet the Company’s anticipated near-term needs.
We anticipate that following the expiration of the lease,
additional or alternative space will be available at
commercially reasonable terms.
55
|
|
|
ITEM 3.
|
LEGAL
PROCEEDINGS
|
The Company is not a party to any material pending legal
proceedings, and management is not aware of any contemplated
proceedings by any governmental authority against the Company.
|
|
|
ITEM 4.
|
(REMOVED
AND RESERVED)
|
PART II
|
|
|
ITEM 5
|
MARKET
FOR REGISTRANT’S COMMON EQUITY, RELATED SHAREHOLDER MATTERS
AND ISSUER PURCHASES OF EQUITY SECURITIES
|
Our common stock has been trading on The NASDAQ Global Market
(“NASDAQ”) under the symbol “ALIM” since our
initial public offering on April 22, 2010. Prior to that
time, there was no established public trading market for our
common stock. The following table sets forth, for the periods
indicated, the range of high and low sale prices of our common
stock as reported by NASDAQ.
| |
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2010
|
|
High
|
|
|
Low
|
|
|
|
|
Second quarter 2010
|
|
$
|
11.30
|
|
|
$
|
7.33
|
|
|
Third quarter 2010
|
|
$
|
9.74
|
|
|
$
|
6.30
|
|
|
Fourth quarter 2010
|
|
$
|
12.70
|
|
|
$
|
8.69
|
|
Holders
As of March 21, 2011 there were 64 holders of record of our
common stock.
Dividends
The Company has not declared or paid any cash dividends on its
common stock since its inception. The Company does not plan to
pay dividends in the foreseeable future. In addition, under the
Company’s Credit Facility, it has agreed not to pay any
dividends so long as it has any outstanding obligations
thereunder. For further discussion of the Company’s Credit
Facility, see Our Credit Facility on page 62. The
Company currently intends to retain earnings, if any, to finance
the growth of the Company. Consequently, stockholders will need
to sell shares of our common stock to realize a return on their
investment, if any.
Market
Price of and Dividends on the Registrant’s Common Equity
and Related Stockholder Matters
The following graph shows the cumulative total return, assuming
the investment of $100 on April 22, 2010 (the date of the
Company’s initial public offering) on an investment in each
of the Company’s common stock, the NASDAQ Composite Index
and the NASDAQ Biotechnology Index. The comparisons in the table
are required by the SEC and are not intended to forecast or be
indicative of possible future performance of the Company’s
common stock. All values assume reinvestment of the full amount
of dividends and are calculated as of December 31, 2010.
The Company has not declared or paid any cash dividends on its
common stock since its inception and does not plan to pay
dividends in the foreseeable future. The following graph and
related information is being furnished solely to accompany this
Form 10-K
pursuant to Item 201(e) of
Regulation S-K
and shall not be deemed “soliciting materials” or to
be “filed” with the SEC (other than as provided in
Item 201), nor shall such information be incorporated by
reference into any of our filings under
56
the Securities Act of 1933 or the Securities Exchange Act of
1934, whether made before or after the date hereof, and
irrespective of any general incorporation language in any such
filing.
Recent
Sales of Unregistered Securities
Sales
of Unregistered Securities
In October 2010, we issued warrants to purchase an aggregate of
up to 39,773 shares of our common stock to Silicon Valley
Bank and MidCap Financial LLP, the lenders under our Credit
Facility. The warrants were immediately exercisable at a per
share exercise price of $11.00 and have a term of 10 years.
We issued these warrants in reliance on Section 4(2) of the
Securities Act of 1933, as amended, as a transaction not
involving a public offering.
Use of
Proceeds from Public Offering of Common Stock
On April 21, 2010, our Registration Statement on
Form S-1
(File
No. 333-162782)
was declared effective by the SEC for our initial public
offering, pursuant to which we sold 6,550,000 shares of our
common stock at a public offering price of $11.00 per share. We
received net proceeds of approximately $66.1 million from
this transaction, after deducting underwriting discounts,
commissions and other offering costs. The underwriters of the
offering were Credit Suisse Securities (USA) LLC, Citigroup
Global Markets Inc., Cowen and Company, LLC, and
Oppenheimer & Co., Inc. On April 27, 2010 we paid
$15.2 million to pSivida to satisfy our $15.0 million
note payable and accrued but unpaid interest thereon. There have
been no material changes in our use or planned use of proceeds
from the initial public offering from that described in our
Quarterly Report on
Form 10-Q
for the quarter ended March 31, 2010, filed with the SEC on
June 7, 2010.
57
|
|
|
ITEM 6
|
SELECTED
CONSOLIDATED FINANCIAL DATA
|
The tables below summarize our financial data. The following
statements of operations data for fiscal years 2010, 2009 and
2008, and the balance sheet data as of December 31, 2010
and 2009 have been derived from our audited financial statements
and related notes and are included elsewhere in this annual
report on
Form 10-K.
The statement of operations data for fiscal years 2007 and 2006,
and the balance sheet data as of December 31, 2008, 2007
and 2006 are derived from our audited financial statements not
included herein. Our historical results for any prior period are
not necessarily indicative of results to be expected in any
future period.
The following summary financial data should be read together
with our financial statements and related notes and
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” included elsewhere in
this Annual Report on
Form 10-K.
Statement
of Operations Data
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
12,581
|
|
|
$
|
15,057
|
|
|
$
|
43,764
|
|
|
$
|
8,363
|
|
|
$
|
6,736
|
|
|
General and administrative
|
|
|
4,610
|
|
|
|
3,407
|
|
|
|
5,058
|
|
|
|
3,184
|
|
|
|
3,028
|
|
|
Marketing
|
|
|
4,880
|
|
|
|
752
|
|
|
|
1,259
|
|
|
|
969
|
|
|
|
616
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
22,071
|
|
|
|
19,216
|
|
|
|
50,081
|
|
|
|
12,516
|
|
|
|
10,380
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest and other income
|
|
|
73
|
|
|
|
37
|
|
|
|
585
|
|
|
|
1,079
|
|
|
|
596
|
|
|
Interest expense
|
|
|
(848
|
)
|
|
|
(1,897
|
)
|
|
|
(1,514
|
)
|
|
|
(2
|
)
|
|
|
(2
|
)
|
|
Gain on early extinguishment of debt
|
|
|
1,343
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Decrease (increase) in fair value of preferred stock conversion
feature
|
|
|
3,644
|
|
|
|
(23,142
|
)
|
|
|
(10,454
|
)
|
|
|
1
|
|
|
|
6
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from continuing operations
|
|
|
(17,859
|
)
|
|
|
(44,218
|
)
|
|
|
(61,464
|
)
|
|
|
(11,438
|
)
|
|
|
(9,780
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income (loss) from discontinued operations
|
|
|
4,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,733
|
|
|
|
(3,191
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
(13,859
|
)
|
|
|
(44,218
|
)
|
|
|
(61,464
|
)
|
|
|
(5,705
|
)
|
|
|
(12,971
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Beneficial conversion feature of preferred stock
|
|
|
—
|
|
|
|
(355
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Preferred stock accretion
|
|
|
(466
|
)
|
|
|
(623
|
)
|
|
|
(718
|
)
|
|
|
(248
|
)
|
|
|
(243
|
)
|
|
Preferred stock dividends
|
|
|
(2,638
|
)
|
|
|
(7,225
|
)
|
|
|
(6,573
|
)
|
|
|
(4,685
|
)
|
|
|
(3,548
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to common stockholders
|
|
$
|
(16,963
|
)
|
|
$
|
(52,421
|
)
|
|
$
|
(68,755
|
)
|
|
$
|
(10,638
|
)
|
|
$
|
(16,762
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted loss per share attributable to common
stockholders
|
|
$
|
(0.77
|
)
|
|
$
|
(34.56
|
)
|
|
$
|
(45.50
|
)
|
|
$
|
(7.09
|
)
|
|
$
|
(11.66
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of shares used to compute basic and
diluted loss per share attributable to common stockholders
|
|
|
22,168
|
|
|
|
1,517
|
|
|
|
1,511
|
|
|
|
1,500
|
|
|
|
1,437
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
58
Balance
Sheet Data
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
2007
|
|
2006
|
|
|
|
(In thousands)
|
|
|
|
Cash and cash equivalents
|
|
$
|
28,514
|
|
|
$
|
4,858
|
|
|
$
|
17,875
|
|
|
$
|
20,847
|
|
|
$
|
27,157
|
|
|
Investments
|
|
|
26,330
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Working capital (deficit)
|
|
|
49,777
|
|
|
|
(4,428
|
)
|
|
|
14,551
|
|
|
|
19,862
|
|
|
|
25,294
|
|
|
Total assets
|
|
|
56,414
|
|
|
|
6,561
|
|
|
|
20,264
|
|
|
|
24,519
|
|
|
|
31,251
|
|
|
Long-term liabilities
|
|
|
4,785
|
|
|
|
47,909
|
|
|
|
28,217
|
|
|
|
31
|
|
|
|
60
|
|
|
Preferred stock
|
|
|
—
|
|
|
|
113,389
|
|
|
|
103,017
|
|
|
|
67,990
|
|
|
|
63,057
|
|
|
Additional paid-in capital
|
|
|
233,338
|
|
|
|
4,836
|
|
|
|
3,474
|
|
|
|
2,867
|
|
|
|
2,571
|
|
|
Accumulated deficit
|
|
|
(188,854
|
)
|
|
|
(171,891
|
)
|
|
|
(119,470
|
)
|
|
|
(50,715
|
)
|
|
|
(40,077
|
)
|
|
Total stockholders’ equity (deficit)
|
|
|
45,212
|
|
|
|
(165,472
|
)
|
|
|
(115,887
|
)
|
|
|
(47,738
|
)
|
|
|
(37,399
|
)
|
|
|
|
ITEM 7
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
The following discussion and analysis should be read in
conjunction with our audited annual consolidated financial
statements and the related notes that appear elsewhere in this
Annual Report on
Form 10-K.
This discussion contains forward-looking statements reflecting
our current expectations that involve risks and uncertainties.
Actual results may differ materially from those discussed in
these forward-looking statements due to a number of factors,
including those set forth in the section entitled “Risk
Factors” and elsewhere in this Annual Report on
Form 10-K.
For further information regarding forward-looking statements,
please refer to the “Special Note Regarding Forward-Looking
Statements and Projections” at the beginning of Item 1
of this Annual Report on
Form 10-K.
Overview
We are a biopharmaceutical company that specializes in the
research, development and commercialization of prescription
ophthalmic pharmaceuticals. We are presently focused on diseases
affecting the back of the eye, or retina, because we believe
these diseases are not well treated with current therapies and
represent a significant market opportunity.
Our most advanced product candidate is ILUVIEN, which we are
developing for the treatment of diabetic macular edema (DME).
DME is a disease of the retina that affects individuals with
diabetes and can lead to severe vision loss and blindness. In
September 2010 we completed two Phase 3 pivotal clinical trials
(collectively, our FAME Study) for ILUVIEN involving
956 patients in sites across the U.S., Canada, Europe and
India to assess the efficacy and safety of ILUVIEN in the
treatment of DME. Based on our analysis of the month 24 clinical
readout from our FAME Study in December 2009, we filed a New
Drug Application (NDA) in June 2010 for the low dose of ILUVIEN
in the U.S. with the U.S. Food and Drug Administration
(FDA), followed by registration filings in the United Kingdom,
Austria, France, Germany, Italy, Portugal and Spain in July
2010. In December 2010, we received a Complete Response Letter
(CRL) from the FDA. The FDA issued the CRL to communicate its
decision that the NDA for ILUVIEN could not be approved in its
present form. No new clinical studies were requested by the FDA
in the CRL. However, the FDA asked us for analyses of the safety
and efficacy data through the end of the FAME Study to further
assess the relative benefits and risks of ILUVIEN. We are
currently preparing the analyses the FDA requested having
completed the FAME Study and publicly released data on
February 3, 2011. The FDA is also seeking additional
information regarding controls and specifications concerning the
manufacturing, packaging and sterilization of ILUVIEN, which we
are currently compiling. We currently anticipate submitting our
response to the CRL to the FDA early in the second quarter of
2011. Our submission to the FDA will be considered a
Class 2 response, which will provide for a review period of
up to an additional six months for our NDA. Based on our
discussions with the FDA, we anticipate that the FDA will call
an advisory committee during this review. Additionally, we plan
to submit the additional safety and efficacy data through the
final readout at the end of
59
the
FAMEtm
Study to regulatory authorities in the United Kingdom, Austria,
France, Germany, Italy, Portugal and Spain in the second quarter
of 2011. If our NDA for ILUVIEN is approved by the FDA, we plan
to commercialize ILUVIEN in the U.S. by marketing and
selling it to retinal specialists as early as late 2011. In
addition to treating DME, ILUVIEN is being studied in three
Phase 2 clinical trials for the treatment of the dry form of
age-related macular degeneration (AMD), the wet form of AMD and
retinal vein occlusion (RVO).
We are also conducting testing on two classes of nicotinamide
adenine dinucleotide phosphate (NADPH) oxidase inhibitors, for
which we have acquired exclusive, worldwide licenses from Emory
University, in the treatment of dry AMD. We plan to evaluate the
use of NADPH oxidase inhibitors in the treatment of other
diseases of the eye, including wet AMD and diabetic retinopathy.
We intend to seek a collaboration partner for sales and
marketing activities outside North America. We currently
contract with development partners or outside firms for various
operational aspects of our development activities, including the
preparation of clinical supplies and have no plans to establish
in-house manufacturing capabilities.
We commenced operations in June 2003. Since our inception we
have incurred significant losses. As of December 31, 2010,
we have accumulated a deficit of $188.9 million. We expect
to incur substantial losses through the projected
commercialization of ILUVIEN as we:
|
|
|
| |
•
|
complete the clinical development and registration of ILUVIEN;
|
| |
| |
•
|
build our sales and marketing capabilities for the anticipated
commercial launch of ILUVIEN in late 2011;
|
| |
| |
•
|
add the necessary infrastructure to support our growth;
|
| |
| |
•
|
evaluate the use of ILUVIEN for the treatment of other
diseases; and
|
| |
| |
•
|
advance the clinical development of other new product candidates
either currently in our pipeline, or that we may license or
acquire in the future.
|
Prior to our initial public offering (IPO), we funded our
operations through the private placement of common stock,
preferred stock, warrants and convertible debt, as well as by
the sale of certain assets of the non-prescription business in
which we were previously engaged. On April 21, 2010, our
Registration Statement on
Form S-1
(as amended) was declared effective by the Securities and
Exchange Commission (SEC) for our IPO, pursuant to which we sold
6,550,000 shares of our common stock at a public offering
price of $11.00 per share. We received net proceeds of
approximately $66.1 million from this transaction, after
deducting underwriting discounts, commissions and other offering
costs.
As of December 31, 2010, we had approximately
$28.5 million in cash and cash equivalents and
$26.3 million of investments in trading securities. In
addition to our net IPO proceeds, our cash and cash equivalents
include the January 2010 receipt of $10.0 million in
proceeds from the exercise of outstanding
Series C-1
warrants, and a $4.0 million option payment from
Bausch & Lomb Incorporated (Bausch & Lomb)
upon the exercise by Bausch & Lomb of its option to
extend by two years the period during which it may continue to
develop an allergy product acquired from us in 2006.
In October 2010, we obtained a $32.5 million senior secured
credit facility (Credit Facility) to help fund our working
capital requirements. The Credit Facility consists of a
$20.0 million revolving line of credit and a
$12.5 million term loan. The lenders have advanced
$6.25 million under the term loan and may advance the
remaining $6.25 million following FDA approval of ILUVIEN,
but no later than July 31, 2011. Given the status of the
FDA’s review of the NDA for ILUVIEN, we do not currently
expect that FDA approval would occur prior to July 31,
2011. We are in discussions with the lenders to amend the terms
of the Credit Facility to, among other things, extend the
availability of the term loan. However, there are no assurances
that the Credit Facility will be amended. We may draw on the
revolving line of credit against eligible, domestic accounts
receivable subsequent to the launch of ILUVIEN.
We do not expect to generate revenues from our product, ILUVIEN,
until late 2011, if at all, and therefore we do not expect to
have positive cash flow from operations before that time. We
believe our cash,
60
cash equivalents, investments and Credit Facility are sufficient
to fund our operations through the projected commercialization
of ILUVIEN and the expected generation of revenue in late 2011.
The commercialization of ILUVIEN is dependent upon approval by
the FDA, however, and we cannot be sure that ILUVIEN will be
approved by the FDA or that, if approved, future sales of
ILUVIEN will generate enough revenue to fund our operations
beyond its commercialization. Due to the uncertainty around FDA
approval, management cannot be certain that we will not need
additional funds for the commercialization of ILUVIEN. If
ILUVIEN is not approved, or if approved, does not generate
sufficient revenue, we may adjust our commercial plans so that
we can continue to operate with our existing cash resources or
seek to raise additional financing.
Our
Agreement with pSivida US, Inc.
In February 2005, we entered into an agreement with pSivida US,
Inc. (pSivida) for the use of fluocinolone acetonide (FAc) in
pSivida’s proprietary delivery device. pSivida is a global
drug delivery company committed to the biomedical sector and the
development of drug delivery products. Our agreement with
pSivida provides us with a worldwide exclusive license to
develop and sell ILUVIEN, which consists of a tiny polyimide
tube with membrane caps that is filled with FAc in a polyvinyl
alcohol matrix for delivery to the back of the eye for the
treatment and prevention of eye diseases in humans (other than
uveitis). This agreement also provided us with a worldwide
non-exclusive license to develop and sell pSivida’s
proprietary delivery device to deliver other corticosteroids to
the back of the eye for the treatment and prevention of eye
diseases in humans (other than uveitis) or to treat DME by
delivering a compound to the back of the eye through a direct
delivery method through an incision required for a 25-gauge or
larger needle. We do not have the right to develop and sell
pSivida’s proprietary delivery device for indications for
diseases outside of the eye or for the treatment of uveitis.
Further, our agreement with pSivida permits pSivida to grant to
any other party the right to use its intellectual property
(i) to treat DME through an incision smaller than that
required for a 25-gauge needle, unless using a corticosteroid
delivered to the back of the eye, (ii) to deliver any
compound outside the back of the eye unless it is to treat DME
through an incision required for a 25-gauge or larger needle, or
(iii) to deliver non-corticosteroids to the back of the
eye, unless it is to treat DME through an incision required for
a 25-gauge or larger needle.
Under the February 2005 agreement, we and pSivida agreed to
collaborate on the development of ILUVIEN for DME, and share
financial responsibility for the development expenses equally.
Per the terms of the agreement, we each reported our monthly
expenditures on a cash basis, and the party expending the lesser
amount of cash during the period was required to make a cash
payment to the party expending the greater amount to balance the
cash expenditures. We retained primary responsibility for the
development of the product, and therefore, were generally the
party owed a balancing payment. Between February 2006 and
December 2006, pSivida failed to make payments to us for its
share of development costs totaling $2.0 million. For each
payment not made, pSivida incurred a penalty of 50% of the
missed payment and interest began accruing at the rate of 20%
per annum on the missed payment and the penalty amount. In
accordance with the terms of the agreement, pSivida was able to
remain in compliance with the terms of the February 2005
agreement as long as the total amount of development payments
past due did not exceed $2.0 million, and pSivida began
making payments again in December 2006 in order to maintain
compliance with the agreement.
The February 2005 agreement provided that after
commercialization of ILUVIEN, profits, as defined in the
agreement, would be shared equally. In March 2008, we and
pSivida amended and restated the agreement to provide us with
80% of the net profits and pSivida with 20% of the net profits.
Total consideration to pSivida in connection with the execution
of the March 2008 agreement was $33.8 million, which
consisted of a cash payment of $12.0 million, the issuance
of a $15.0 million note payable, and the forgiveness of
$6.8 million in outstanding receivables. The
$15.0 million promissory note accrued interest at 8% per
annum, payable quarterly and was payable in full to pSivida upon
the earliest of a liquidity event as defined in the agreement,
the occurrence of an event of default under our agreement with
pSivida, or September 30, 2012. If the note was not paid in
full by March 31, 2010, the interest rate was to increase
to 20% effective as of April 1, 2010, and we were required
to begin making principal payments of $500,000 per month. On
April 27, 2010, we paid pSivida approximately
$15.2 million in principal and interest
61
to satisfy the note payable with the proceeds from our IPO. We
will owe pSivida an additional milestone payment of
$25.0 million upon FDA approval of ILUVIEN.
Our
Credit Facility
Term
Loan Agreement
On October 14, 2010 (Effective Date), we entered into a
Loan and Security Agreement with Silicon Valley Bank and MidCap
Financial LLP under (Lenders) which we may borrow up to
$12.5 million (Term Loan Agreement). The lenders advanced
$6.25 million on the Effective Date (Initial Tranche) and
may advance the remaining $6.25 million following FDA
approval of ILUVIEN, but no later than July 31, 2011
(Second Tranche). Given the status of the FDA’s review of
the NDA for ILUVIEN, it is unlikely that FDA approval would
occur prior to July 31, 2011. We are in discussions with
the Lenders to amend the terms of the Credit Facility to, among
other things, extend the availability of the term loan. However,
there are no assurances that the Credit Facility will be
amended. To secure the repayment of any amounts borrowed under
the Term Loan Agreement, we granted to the lenders a first
priority security interest in all of our assets, other than our
intellectual property (provided that in the event we fail to
meet certain financial conditions, a curable lien will be
imposed on our intellectual property). We also agreed not to
pledge or otherwise encumber our intellectual property assets.
We are required to maintain our primary operating and other
deposit accounts and securities accounts with Silicon Valley
Bank, which accounts must represent at least 50% of the dollar
value of our accounts at all financial institutions.
We will be required to pay interest on borrowings under the Term
Loan Agreement at a rate of 11.5% on a monthly basis through
July 31, 2011. Thereafter, we will be required to repay the
principal, plus interest at such rate if the Second Tranche is
advanced to us prior to February 28, 2011 (and plus
interest at a rate of 12% if the Second Tranche is advanced to
us after February 28, 2011), in 27 equal monthly
installments. We paid to the lenders an upfront fee of $62,500,
and will pay to the lenders an additional final payment of 3% of
the total principal amount. In addition, if we repay the loan
prior to maturity, we will pay to the lenders a prepayment
penalty of 5% of the total principal amount if the prepayment
occurs within one year after the funding date, 3% of the total
principal amount if the prepayment occurs between one and two
years after the funding date and 1% of the total principal
amount of the prepayment occurs thereafter (each a Prepayment
Penalty), provided in each case that such Prepayment Penalty
will be reduced by 50% in the event we are acquired.
To secure the repayment of any amounts borrowed under the Term
Loan Agreement, we granted to the Lenders a first priority
security interest in all of our assets, other than our
intellectual property, provided that, for any date during which
the notes are outstanding, our unrestricted balance sheet cash
and cash equivalents plus the excess available under the Term
Loan Agreement are less than the product of six times the
monthly cash burn amount. In the event we fail to meet this
financial condition, a curable lien will be imposed on our
intellectual property. Should such a lien event take place, the
lien would remain in force until such date that the
Company’s unrestricted cash and cash equivalents plus the
excess available under the Term Loan Agreement were equal to or
greater than twelve times the monthly cash burn amount. We also
agreed not to pledge or otherwise encumber our intellectual
property assets. Additionally, we must seek the Lenders’
approval prior to the payment of any cash dividends.
In connection with entering into this agreement, we issued to
the Lenders, warrants to purchase an aggregate of up to
39,773 shares of our common stock. Each of the warrants is
exercisable immediately, has a per-share exercise price of
$11.00 and has a term of 10 years. In addition, the Lenders
will have certain registration rights with respect to the shares
of common stock issuable upon exercise of the warrants. We
estimated the aggregate fair value of the warrants, using the
Black-Scholes model, to be $389,000. We allocated a portion of
the proceeds from the Term Loan Agreement to the warrants in
accordance with
ASC 470-20-25-2,
Debt Instruments with Detachable Warrants. As a result,
we recorded a discount of $366,000 which is being amortized to
interest expense using the effective interest method.
62
Revolving
Loan Agreement
Also on the Effective Date, we and Silicon Valley Bank entered
into a Loan and Security Agreement (Revolving Loan Agreement),
pursuant to which we obtained a secured revolving line of credit
from Silicon Valley Bank with borrowing availability up to
$20.0 million.
The Revolving Loan Agreement provides for a working
capital-based revolving line of credit (Revolving Line) in an
aggregate amount of up to the lesser of
(i) $20.0 million, or (ii) 85% of eligible
domestic accounts receivable. The Revolving Line matures on
October 31, 2013.
Amounts advanced under the Revolving Line bear interest at an
annual rate equal to Silicon Valley Bank’s prime rate plus
2.50% (with a rate floor of 6.50%). Interest on the Revolving
Line is due monthly, with the balance due at the maturity date.
We paid to the Silicon Valley Bank an upfront fee of $100,000.
In addition, if we terminate the Revolving Line prior to
maturity, we will pay to Silicon Valley Bank a fee of $400,000
if the termination occurs within one year after the Effective
Date and a fee of $200,000 if the termination occurs more than
one year after the Effective Date (each a Termination Fee),
provided in each case that such Termination Fee will be reduced
by 50% in the event we are acquired.
To secure the repayment of any amounts borrowed under the
Revolving Loan Agreement, we granted to Silicon Valley Bank a
first priority security interest in all of our assets, other
than our intellectual property, provided that, for any date
during which the notes are outstanding, our unrestricted balance
sheet cash and cash equivalents plus the excess available under
the Term Loan Agreement are less than the product of six times
the monthly cash burn amount. In the event we fail to meet this
financial condition, a curable lien will be imposed on our
intellectual property. Should such a lien event take place, the
lien would remain in force until such date that our unrestricted
cash and cash equivalents plus the excess available under the
Term Loan Agreement were equal to or greater than twelve times
the monthly cash burn amount. We also agreed not to pledge or
otherwise encumber its intellectual property assets.
Additionally, we must seek Silicon Valley Bank’s approval
prior to the payment of any cash dividends.
The occurrence of an event of default could result in the
acceleration of our obligations under the Revolving Loan
Agreement and an increase to the applicable interest rate, and
would permit Silicon Valley Bank to exercise remedies with
respect to the collateral under the Revolving Loan Agreement.
Our
Discontinued Non-Prescription Business
At the inception of our company, we were focused primarily on
the development and commercialization of non-prescription
over-the-counter
ophthalmic products. In October 2006, due to the progress and
resource requirements related to the development of ILUVIEN, we
decided to discontinue our non-prescription business. As a
result, we received proceeds of $10.0 million from the sale
of our allergy products in December 2006 and $6.7 million
from the sale of our dry eye product in July 2007, both to
Bausch & Lomb. If one of the allergy products receives
FDA approval, we are entitled to an additional $8.0 million
payment from Bausch & Lomb under the sales agreement.
In January 2010 we received a $4.0 million option payment
from Bausch & Lomb upon the exercise by
Bausch & Lomb of its option to extend the period
during which it may continue to develop this allergy product by
two years. However, there can be no assurance that
Bausch & Lomb will continue the development of this
allergy product, that it will receive FDA approval or that we
will receive the $8.0 million payment. As a result of the
discontinuance of our non-prescription business, all revenues
and expenses associated with our
over-the-counter
portfolio are included in the loss from discontinued operations
in the accompanying statements of operations.
Financial
Operations Overview
Revenue
To date we have only generated revenue from our dry eye
non-prescription product. From the launch of that product in
September 2004 to its sale in July 2007, we generated
$4.4 million in net revenues. We do not expect to generate
any significant additional revenue unless or until we obtain
regulatory approval of, and commercialize, our product
candidates or in-license additional products that generate
revenue. In addition to
63
generating revenue from product sales, we intend to seek to
generate revenue from other sources such as upfront fees,
milestone payments in connection with collaborative or strategic
relationships, and royalties resulting from the licensing of our
product candidates and other intellectual property. We expect
any revenue we generate will fluctuate from quarter to quarter
as a result of the nature, timing and amount of any milestone
payments we may receive from potential collaborative and
strategic relationships, as well as revenue we may receive upon
the sale of our products to the extent any are successfully
commercialized.
Research
and Development Expenses
Substantially all of our research and development expenses
incurred to date related to our continuing operations have been
related to the development of ILUVIEN. In the event the FDA
approves our NDA for ILUVIEN, we will owe an additional
milestone payment of $25.0 million to pSivida. We
anticipate that we will incur additional research and
development expenses in the future as we evaluate and possibly
pursue the development of ILUVIEN for additional indications, or
develop additional product candidates. We recognize research and
development expenses as they are incurred. Our research and
development expenses consist primarily of:
|
|
|
| |
•
|
salaries and related expenses for personnel;
|
| |
| |
•
|
fees paid to consultants and contract research organizations
(CRO) in conjunction with independently monitoring clinical
trials and acquiring and evaluating data in conjunction with
clinical trials, including all related fees such as investigator
grants, patient screening, lab work and data compilation and
statistical analysis;
|
| |
| |
•
|
costs incurred with third parties related to the establishment
of a commercially viable manufacturing process for our product
candidates;
|
| |
| |
•
|
costs related to production of clinical materials, including
fees paid to contract manufacturers;
|
| |
| |
•
|
costs related to upfront and milestone payments under
in-licensing agreements;
|
| |
| |
•
|
costs related to compliance with FDA regulatory requirements;
|
| |
| |
•
|
consulting fees paid to third-parties involved in research and
development activities; and
|
| |
| |
•
|
costs related to stock options or other stock-based compensation
granted to personnel in development functions.
|
We expense both internal and external development costs as they
are incurred.
We expect that a large percentage of our research and
development expenses in the future will be incurred in support
of our current and future technical, preclinical and clinical
development programs. These expenditures are subject to numerous
uncertainties in terms of both their timing and total cost to
completion. We expect to continue to develop stable formulations
of our product candidates, test such formulations in preclinical
studies for toxicology, safety and efficacy and to conduct
clinical trials for each product candidate. We anticipate
funding clinical trials ourselves, but we may engage
collaboration partners at certain stages of clinical
development. As we obtain results from clinical trials, we may
elect to discontinue or delay clinical trials for certain
product candidates or programs in order to focus our resources
on more promising product candidates or programs. Completion of
clinical trials by us or our future collaborators may take
several years or more, the length of time generally varying with
the type, complexity, novelty and intended use of a product
candidate. The costs of clinical trials may vary significantly
over the life of a project owing to but not limited to the
following:
|
|
|
| |
•
|
the number of sites included in the trials;
|
| |
| |
•
|
the length of time required to enroll eligible patients;
|
| |
| |
•
|
the number of patients that participate in the trials;
|
| |
| |
•
|
the number of doses that patients receive;
|
64
|
|
|
| |
•
|
the drop-out or discontinuation rates of patients;
|
| |
| |
•
|
the duration of patient
follow-up;
|
| |
| |
•
|
the phase of development the product candidate is in; and
|
| |
| |
•
|
the efficacy and safety profile of the product candidate.
|
Our expenses related to clinical trials are based on estimates
of the services received and efforts expended pursuant to
contracts with multiple research institutions and contract
research organizations that conduct and manage clinical trials
on our behalf. The financial terms of these agreements are
subject to negotiation and vary from contract to contract and
may result in uneven payment flows. Generally, these agreements
set forth the scope of work to be performed at a fixed fee or
unit price. Payments under the contracts depend on factors such
as the successful enrollment of patients or the completion of
clinical trial milestones. Expenses related to clinical trials
generally are accrued based on contracted amounts applied to the
level of patient enrollment and activity according to the
protocol. If timelines or contracts are modified based upon
changes in the clinical trial protocol or scope of work to be
performed, we modify our estimates of accrued expenses
accordingly on a prospective basis.
None of our product candidates has received FDA or foreign
regulatory marketing approval. In order to grant marketing
approval, a health authority such as the FDA or foreign
regulatory agencies must conclude that clinical and preclinical
data establish the safety and efficacy of our product candidates
with an appropriate benefit to risk profile relevant to a
particular indication, and that the product can be manufactured
under current Good Manufacturing Practice (cGMP) in a
reproducible manner to deliver the product’s intended
performance in terms of its stability, quality, purity and
potency. Until our submissions are reviewed by health
authorities, there is no way to predict the outcome of their
review. Even if the clinical studies meet their predetermined
primary endpoints, and a registration dossier is accepted for
filing, a health authority could still determine that an
appropriate benefit to risk relationship does not exist for the
indication that we are seeking. We cannot forecast with any
degree of certainty which of our product candidates will be
subject to future collaborations or how such arrangements would
affect our development plan or capital requirements. As a result
of the uncertainties discussed above, we are unable to determine
the duration and completion costs of our development projects or
when and to what extent we will receive cash inflows from the
commercialization and sale of an approved product candidate.
General
and Administrative Expenses
General and administrative expenses consist primarily of
compensation for employees in executive and administrative
functions, including finance, accounting and human resources.
Other significant costs include facilities costs and
professional fees for accounting and legal services, including
legal services associated with obtaining and maintaining
patents. We anticipate incurring a significant increase in
general and administrative expenses, as we add additional
employees and continue to operate as a public company. These
increases will include increased costs for insurance, costs
related to the hiring of additional personnel and payments to
outside consultants, lawyers and accountants. We also expect to
continue to incur significant costs to comply with the corporate
governance, internal control and similar requirements applicable
to public companies.
Marketing
Expenses
Marketing expenses consist primarily of compensation for
employees responsible for assessing the commercial opportunity
of and developing market awareness and launch plans for our
product candidates. Other costs include professional fees
associated with developing brands for our product candidates and
maintaining public relations. We expect significant increases in
our marketing and selling expenses as we hire additional
personnel and establish our sales and marketing capabilities in
anticipation of the commercialization of our product candidates.
We intend to capitalize on our management’s past experience
and expertise with eye-care products by marketing and selling
ILUVIEN to the approximately 1,600 retinal specialists
practicing in the approximately 900 retina centers across the
U.S.
65
Our plan is to develop our own specialized domestic sales and
marketing infrastructure, comprised of approximately
40 people, to market ILUVIEN and other ophthalmic products
that we may acquire or develop in the future. We hired regional
managers with extensive ophthalmic-based sales experience in the
third quarter of 2010 and plan to begin adding sales
representatives in the fourth quarter of 2011. We entered into a
relationship with OnCall LLC, a contract sales force company,
that will utilize its employees to act as our sales
representatives if we receive approval of the ILUVIEN NDA from
the FDA. We expect that following an FDA approval, the On Call
sales force will be able to access and form relationships with
retinal specialists in approximately 900 retina centers for to
the commercial launch of ILUVIEN, following FDA approval of our
NDA. In connection with the commercial launch of ILUVIEN, we
expect to hire additional personnel to support the activities of
customer service, post-marketing pharmacovigilance, medical
affairs, and regulatory compliance.
Interest
and Other Income
Interest income consists primarily of interest earned on our
cash, cash equivalents and investments.
Interest
Expense
Beginning in March 2008, we began recognizing interest on our
$15.0 million note payable to pSivida at an effective
interest rate of 12.64% per annum (this note accrued interest at
the rate of 8% per annum from inception through March 31,
2010 and at the rate of 20% per annum effective as of
April 1, 2010). On April 27, 2010, we paid pSivida
approximately $15.2 million in principal and interest to
satisfy the note payable. In October 2010, we drew the Initial
Tranche of $6.25 million on our term loan from Silicon
Valley Bank and MidCap Financial LLP which accrues interest at
the rate of 11.5% per annum and is payable monthly.
Change
in Fair Value of Preferred Stock Conversion
Feature
Prior to being converted into common stock in connection with
our IPO, our preferred stock contained certain conversion
features which were considered embedded derivatives. We
accounted for such embedded derivative financial instruments in
accordance with Accounting Standards Codification 815. We
recorded derivative financial instruments as assets or
liabilities in our balance sheet measured at their fair value.
We recorded the changes in fair value of such instruments as
non-cash gains or losses in the statement of operations. The
preferred stock conversion feature was eliminated upon the
conversion of our preferred stock to common stock in connection
with our IPO in April 2010.
Preferred
Stock Accretion
Prior to our IPO, our preferred stock was recorded at issuance
at the proceeds received net of any issuance discounts, issuance
costs and the fair value of the conversion features at issuance.
The difference between the amount recorded at issuance and the
original issue price was accreted on a straight-line basis over
a period extending from the date of issuance to the date at
which the preferred stock would have become redeemable at the
option of the holder. Accretion of the difference ceased upon
the conversion of our preferred stock to common stock in
connection with our IPO in April 2010.
Preferred
Stock Dividends
Prior to our IPO, our preferred stock accrued dividends at 8%
per annum which were recorded as an increase in the carrying
amount of the respective preferred stock. At the time our
preferred stock was converted into common stock in connection
with our IPO, $1.5 million of dividends accrued on our
Series A preferred stock prior to November 17, 2005
were converted into 380,301 shares of our common stock. All
other preferred stock dividends were eliminated upon conversion
of the underlying preferred stock in April 2010.
66
Basic
and Diluted Net Loss Applicable to Common Stockholders per
Common Share
We calculated net loss per share in accordance with
ASC 260. We have determined that our previously outstanding
Series A, Series B, Series C and
Series C-1
preferred stock represent participating securities in accordance
with ASC 260. However, since we operate at a loss, and
losses are not allocated to the preferred stock, the two class
method does not affect our calculation of earnings per share. We
had a net loss from continuing operations for all periods
presented; accordingly, the inclusion of common stock options
and warrants would be anti-dilutive. Dilutive common stock
equivalents would include the dilutive effect of convertible
securities, common stock options, warrants for convertible
securities and warrants for common stock equivalents.
Potentially dilutive weighted average common stock equivalents
totaled approximately 9,131,451, 22,149,592, and 19,741,154 for
the years ended December 31, 2010, 2009, and 2008,
respectively. Potentially dilutive common stock equivalents were
excluded from the diluted earnings per share denominator for all
periods of net loss from continuing operations because of their
anti-dilutive effect. Therefore, for the years ended
December 31, 2010, 2009, and 2008, the weighted average
shares used to calculate both basic and diluted loss per share
are the same.
Critical
Accounting Policies and Estimates
Our discussion and analysis of our financial condition and
results of operations are based on our financial statements
which have been prepared in accordance with accounting
principles generally accepted in the U.S. The preparation
of these financial statements requires us to make estimates and
judgments that affect the reported amounts of assets,
liabilities, revenues and expenses. On an ongoing basis, we
evaluate these estimates and judgments, including those
described below. We base our estimates on historical experience
and on various other assumptions that we believe to be
reasonable under the circumstances. These estimates and
assumptions form the basis for making judgments about the
carrying values of assets and liabilities that are not readily
apparent from other sources. Actual results and experiences may
differ materially from these estimates. We believe that the
following accounting policies are the most critical to aid you
in fully understanding and evaluating our reported financial
results and affect the more significant judgments and estimates
that we use in the preparation of our financial statements.
Clinical
Trial Prepaid and Accrued Expenses
We record prepaid assets and accrued liabilities related to
clinical trials associated with contract research organizations,
clinical trial investigators and other vendors based upon
amounts paid and the estimated amount of work completed on each
clinical trial. The financial terms of agreements vary from
vendor to vendor and may result in uneven payment flows. As
such, if we have advanced funds exceeding our estimate of the
work completed, we record a prepaid asset. If our estimate of
the work completed exceeds the amount paid, an accrued liability
is recorded. All such costs are charged to research and
development expenses based on these estimates. Our estimates may
or may not match the actual services performed by the
organizations as determined by patient enrollment levels and
related activities. We monitor patient enrollment levels and
related activities to the extent possible through internal
reviews, correspondence and discussions with our contract
research organization and review of contractual terms. However,
if we have incomplete or inaccurate information, we may
underestimate or overestimate activity levels associated with
various clinical trials at a given point in time. In this event,
we could record significant research and development expenses in
future periods when the actual level of activities becomes
known. To date, we have not experienced material changes in
these estimates. Additionally, we do not expect material
adjustments to research and development expenses to result from
changes in the nature and level of clinical trial activity and
related expenses that are currently subject to estimation. In
the future, as we expand our clinical trial activities, we
expect to have increased levels of research and development
costs that will be subject to estimation.
Research
and Development Costs
Research and development expenditures are expensed as incurred,
pursuant to ASC 730. Costs to license technology to be used
in our research and development that have not reached
technological feasibility, defined as FDA approval for our
current product candidates, and have no alternative future use
are expensed when
67
incurred. Payments to licensors that relate to the achievement
of preapproval development milestones are recorded as research
and development expense when incurred.
Stock-Based
Compensation
Effective January 1, 2005, we adopted the fair value
recognition provisions of ASC 718 using the modified
prospective application method. We recognize the grant date fair
value as compensation cost of employee stock-based awards using
the straight-line method over the actual vesting period,
adjusted for our estimates of forfeiture. Typically, we grant
stock options with a requisite service period of four years from
the grant date. We have elected to use the Black-Scholes option
pricing model to determine the fair value of stock-based awards.
We concluded that this was the most appropriate method by which
to value our share-based payment arrangements, but if any
share-based payment instruments should be granted for which the
Black-Scholes method does not meet the measurement objective as
stated within ASC 718, we will utilize a more appropriate
method for valuing that instrument. However, we do not believe
that any instruments granted to date and accounted for under
ASC 718 would require a method other than the Black-Scholes
method.
Our determination of the fair market value of share-based
payment awards on the grant date using option valuation models
requires the input of highly subjective assumptions, including
the expected price volatility and option life. For the
calculation of expected volatility, because we lack significant
company-specific historical and implied volatility information,
we estimate our volatility by utilizing an average of
volatilities of publicly traded companies, including our own,
deemed similar to us in terms of product composition, stage of
lifecycle, capitalization and scope of operations. We intend to
continue to consistently apply this process using this same
index until a sufficient amount of historical information
regarding the volatility of our own share price becomes
available.
To estimate the expected term, we utilize the
“simplified” method for “plain vanilla”
options as discussed within the Securities and Exchange
Commission’s (SEC) Statement of Accounting Bulletin (SAB)
107. We believe that all factors listed within SAB 107 as
pre-requisites for utilizing the simplified method are true for
us and for our share-based payment arrangements. We intend to
utilize the simplified method for the foreseeable future until
more detailed information about exercise behavior will be more
widely available.
Total stock-based compensation expense, related to all our
stock-based awards for the years ended December 31, 2010,
2009 and 2007, was comprised of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
(In thousands)
|
|
|
|
Marketing
|
|
$
|
127
|
|
|
$
|
43
|
|
|
$
|
109
|
|
|
Research and development
|
|
|
209
|
|
|
|
161
|
|
|
|
269
|
|
|
General and administrative
|
|
|
458
|
|
|
|
307
|
|
|
|
372
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total employee stock-based compensation expense
|
|
$
|
794
|
|
|
$
|
511
|
|
|
$
|
750
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income
Taxes
We recognize deferred tax assets and liabilities for temporary
differences between the financial reporting basis and the tax
basis of its assets and liabilities in accordance with
ASC 740. We evaluate the positive and negative evidence
bearing upon the realizability of our deferred tax assets on an
annual basis. Significant management judgment is involved in
determining the provision for income taxes, deferred tax assets
and liabilities, and any valuation allowance recorded against
net deferred tax assets. Due to uncertainties with respect to
the realization of our deferred tax assets due to our history of
operating losses, a valuation allowance has been established
against our deferred tax asset balances to reduce the net
carrying value to an amount that is more likely than not to be
realized. As a result we have fully reserved against the
deferred tax asset balances. The valuation allowances are based
on our estimates of taxable income in the jurisdictions in which
we operate and the period over which deferred tax assets will be
recoverable. In the event that actual
68
results differ from these estimates or we adjust these estimates
in future periods, a change in the valuation allowance may be
needed, which could materially impact our financial position and
results of operations. Our deferred tax assets primarily consist
of net operating loss (NOL) carry-forwards. At December 31,
2010 we had federal NOL carry-forwards of approximately
$97.8 million and state NOL carry-forwards of approximately
$81.0 million, respectively, that are available to reduce
future income otherwise taxable. If not utilized, the federal
NOL carry-forwards will expire at various dates between 2023 and
2030 and the state NOL carry-forwards will expire at various
dates between 2020 and 2030. If it is determined that
significant ownership changes have occurred since these NOLs
were generated, we may be subject to annual limitations on the
use of these NOLs under Internal Revenue Code Section 382
(or comparable provisions of state law). We have not yet
completed a formal evaluation of whether our IPO resulted in
certain changes in ownership that would limit our ability to
utilize a portion of our NOL carry-forwards.
In the event that we were to determine that we are able to
realize any of our net deferred tax assets in the future, an
adjustment to the valuation allowance would increase net income
in the period such determination was made. We believe that the
most significant uncertainty that will impact the determination
of our valuation allowance will be our estimation of the extent
and timing of future net income, if any.
We considered our income tax positions for uncertainty in
accordance with ASC 740. We believe our income tax filing
positions and deductions are more likely than not of being
sustained on audit and do not anticipate any adjustments that
will result in a material change to our financial position;
therefore, we have not recorded ASC 740 liabilities. We
recognize accrued interest and penalties related to unrecognized
tax benefits as interest expense and income tax expense,
respectively, in our statements of operations. Our tax years
since 2003 remain subject to examination in Georgia, Tennessee,
and on the federal level. We do not anticipate any material
changes to our uncertain tax positions within the next
12 months.
Results
of Operations
The following selected financial and operating data are derived
from our financial statements and should be read in conjunction
with “Management’s Discussion and Analysis of
Financial Condition and Results of Operations” and our
financial statements.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
(In thousands except share and per share data)
|
|
|
|
RESEARCH AND DEVELOPMENT EXPENSES
|
|
$
|
12,581
|
|
|
$
|
15,057
|
|
|
$
|
43,764
|
|
|
GENERAL AND ADMINISTRATIVE EXPENSES
|
|
|
4,610
|
|
|
|
3,407
|
|
|
|
5,058
|
|
|
MARKETING EXPENSES
|
|
|
4,880
|
|
|
|
752
|
|
|
|
1,259
|
|
|
OPERATING EXPENSES
|
|
|
22,071
|
|
|
|
19,216
|
|
|
|
50,081
|
|
|
INTEREST AND OTHER INCOME
|
|
|
73
|
|
|
|
37
|
|
|
|
585
|
|
|
INTEREST EXPENSE
|
|
|
(848
|
)
|
|
|
(1,897
|
)
|
|
|
(1,514
|
)
|
|
GAIN ON EARLY EXTINGUISHMENT OF DEBT
|
|
|
1,343
|
|
|
|
—
|
|
|
|
—
|
|
|
DECREASE (INCREASE) IN FAIR VALUE OF PREFERRED STOCK CONVERSION
FEATURE
|
|
|
3,644
|
|
|
|
(23,142
|
)
|
|
|
(10,454
|
)
|
|
LOSS FROM CONTINUING OPERATIONS
|
|
$
|
(17,859
|
)
|
|
$
|
(44,218
|
)
|
|
$
|
(61,464
|
)
|
Year
ended December 31, 2010 compared to the year ended
December 31, 2009
Research and development expenses. Research
and development expenses decreased by approximately
$2.5 million, or 17%, to approximately $12.6 million
for the year ended December 31, 2010 compared to
approximately $15.1 million for the year ended
December 31, 2009. The decrease was primarily attributable
to decreases of $3.6 million in costs related to our FAME
Study, $1.1 million in technical transfer costs associated
with establishing manufacturing capabilities with a third party
manufacturer for ILUVIEN in 2009, and $300,000 related to
license fees paid to Emory University in 2009 for two classes of
NADPH oxidase inhibitors, offset by increases of
$2.0 million in costs to file our NDA in the U.S. and
marketing applications
69
for ILUVIEN in Austria, France, Germany, Italy, Portugal, Spain,
and the U.K., $410,000 in costs associated with contracting
medical science liaisons to engage with retinal specialists in
the study of ILUVIEN, and $100,000 in costs associated with our
ancillary studies. The decrease in costs for our FAME Study was
primarily due to decreases of $1.6 million for our CROs,
$1.1 million for clinical trial site costs, $590,000 for
our third party reading center for the analysis of retinal
images, and $220,000 in costs reimbursable to pSivida for its
costs to develop ILUVIEN as the FAME Study was completed in the
third quarter of 2010.
General and administrative expenses. General
and administrative expenses increased by approximately
$1.2 million, or 35%, to approximately $4.6 million
for the year ended December 31, 2010 compared to
approximately $3.4 million for the year ended
December 31, 2009. The increase was primarily attributable
to increases of $940,000 in costs incurred after our IPO in
April 2010 associated with operating as a public company
including additional audit, tax and legal fees, increased
directors’ and officers’ insurance costs, and board of
directors’ compensation, and $120,000 of noncash stock
compensation incurred in connection with evaluating financing
options prior to our IPO.
Marketing expenses. Marketing expenses
increased by approximately $4.1 million or 513%, to
approximately $4.9 million for the year ended
December 31, 2010 compared to approximately
$0.8 million for the year ended December 31, 2009. The
increase was primarily attributable to increases of
$1.1 million in costs related to our advertising
agency’s development of a detailed advertising and
promotional plan for ILUVIEN, $640,000 for increased medical
marketing to the physician community through medical
publications, relationships with key opinion leaders, and
increased presence at medical and scientific conferences,
$580,000 in personnel costs as we increased the number of
employees in our marketing department, $560,000 in increased
corporate communications costs related to our post-launch
message development and media management, $500,000 of
pharmacoeconomic research to evaluate the pricing of ILUVIEN in
the U.S., Canada and Europe, and $490,000 in costs related to
consulting fees incurred to develop our managed market strategy.
Interest and other income. Interest and other
income increased by approximately $36,000, or 97%, to
approximately $73,000 for the year ended December 31, 2010
compared to approximately $37,000 for the year ended
December 31, 2009. The increase was due to higher cash
balances and investments as a result of the proceeds received
from our IPO in April 2010.
Interest expense. Interest expense decreased
by approximately $1.1 million, or 58%, to approximately
$0.8 million for the year ended December 31, 2010
compared to approximately $1.9 million for the year ended
December 31, 2009. The decrease was primarily attributable
to a decrease of $1.3 million of interest on our
$15.0 million promissory note to pSivida, offset by an
increase of $230,000 of interest expense associated with our
$6.3 million notes payable to Silicon Valley Bank and
MidCap Financial LLP.
Decrease (increase) in fair value of preferred stock
conversion feature. During the year ended
December 31, 2010, we recognized a gain of approximately
$3.6 million related to the decrease in the fair value of
the conversion feature of our preferred stock. During the year
ended December 31, 2009, we recognized a loss of
approximately $23.1 million related to the increase in the
fair value of the conversion feature of our preferred stock. The
changes in fair values were primarily due to changes in the
estimated fair value of our common stock.
Income from discontinued operations. We
recognized income from discontinued operations during the year
ended December 31, 2010 of $4.0 million for a payment
we received from Bausch & Lomb. This payment was
related to the exercise by Bausch & Lomb of its option
to extend by two years the period during which it may continue
to develop an allergy product acquired from us in 2006. We did
not have any income or loss from discontinued operations for the
year ended December 31, 2009.
Year
ended December 31, 2009 compared to the year ended
December 31, 2008
Research and development expenses. Research
and development expenses decreased by approximately
$28.7 million, or 66%, to approximately $15.1 million
for the year ended December 31, 2009 compared to
approximately $43.8 million for the year ended
December 31, 2008. The decrease was principally
attributable
70
to the restructuring of our agreement with pSivida Inc., which
resulted in incremental expenses of $29.8 million in the
year ended December 31, 2008 that were not incurred in the
year ended December 31, 2009. The $29.8 million cost
in 2008 was comprised of a $12.0 million cash payment, a
$15.0 million promissory note issued to pSivida, and the
forgiveness of $2.8 million of net outstanding receivables
due from pSivida related to the agreement. We continued to incur
costs in 2009 with respect to our FAME Study, which completed
enrollment in October 2007, and preparations for its anticipated
registration with the FDA. We incurred increased costs in 2009
related to our FAME Study of $620,000 for our CRO costs as we
prepared for and completed the lock of our FAME Study database
and month 24 readout in the fourth quarter of 2009 and $490,000
in technology transfer costs associated with establishing
manufacturing capabilities with a third-party manufacturer for
ILUVIEN. These amounts were offset by decreases of
$1.2 million in FAME Study trial site costs, $310,000 for
our reading center to evaluate pictures of each enrollee’s
retina, and $240,000 for our PK Study due to the completion of
enrollment and fewer patient visits per month as the trial
progressed. Additionally, total development costs related to
ILUVIEN increased by $1.3 million due to the absence of
cost sharing reimbursements from pSivida as a result of the
restructuring of our agreement in March 2008. We also decreased
spending on the evaluation of the NADPH oxidase inhibitors
obtained from Emory University and other development pipeline
candidates by $270,000 due to the restricted capital markets in
2009 and in order to focus our resources on completing the
development of ILUVIEN, but incurred $300,000 in initial license
fees in 2009 to enter into these agreements with Emory
University.
General and administrative expenses. General
and administrative expenses decreased by approximately
$1.7 million, or 33%, to approximately $3.4 million
for the year ended December 31, 2009 compared to
approximately $5.1 million for the year ended
December 31, 2008. The decrease was primarily attributable
to $1.3 million incurred in preparation for the anticipated
2008 initial public offering of our common stock that was
expensed in the year ended December 31, 2008 when we
determined that an initial public offering was unlikely in the
then near term and $380,000 in legal fees associated with the
restructuring of our agreement with pSivida, the evaluation of
intellectual property regarding our ILUVIEN inserter system and
the evaluation of certain strategic options.
Marketing expenses. Marketing expenses
decreased by approximately $510,000, or 40%, to approximately
$750,000 for the year ended December 31, 2009 compared to
approximately $1.3 million for the year ended
December 31, 2008. The decrease was primarily attributable
to $230,000 in decreased spending on travel and general
corporate awareness due to the restricted capital markets in
2009 and in order to focus our resources on completing the
development of ILUVIEN, and $210,000 incurred for the initiation
of pricing studies of the U.S. and European markets for
ILUVIEN during the year ended December 31, 2008 that were
not incurred in the year ended December 31, 2009.
Interest and other income. Interest income
decreased by approximately $550,000, or 94%, to approximately
$40,000 for the year ended December 31, 2009 compared to
approximately $590,000 for the year ended December 31,
2008. The decrease in interest income is primarily attributable
to a decrease in our average cash balance from
$25.5 million during the year ended December 31, 2008
to $11.1 million for the year ended December 31, 2009,
combined with a substantial drop in the rates of return
available on our money market accounts from approximately 2.3%
during the year ended December 31, 2008 to 0.3% for the
year ended December 31, 2009.
Interest expense. Interest expense increased
by approximately $380,000, or 25%, to approximately
$1.9 million for the year ended December 31, 2009
compared to approximately $1.5 million for the year ended
December 31, 2008. Our interest expense is associated with
our $15.0 million note payable to pSivida issued in March
2008, and the increase is due to the note payable being
outstanding for the full year ended December 31, 2009 as
opposed to being outstanding for nine months in 2008.
Decrease (increase) in fair value of preferred stock
conversion feature. For the year ended
December 31, 2009 we recognized an expense of approximately
$23.1 million related to the increase in the fair value of
the conversion feature of our preferred stock. The increase was
attributable to an increase in the estimated fair value of our
common stock from $3.71 at December 31, 2008 to $8.53 at
December 31, 2009 and increased volatility in the market
values of our peer group.
71
Income from discontinued operations. We did
not have any income (loss) from discontinued operations for
either of the year ended December 31, 2008 or
December 31, 2009 due to the sale of our dry eye product to
Bausch & Lomb in July 2007.
Liquidity
and Capital Resources
To date we have incurred recurring losses, negative cash flow
from operations, and have accumulated a deficit of
$188.9 million from our inception through December 31,
2010. Prior to our IPO in April 2010, we funded our operations
through the private placement of common stock, preferred stock,
warrants and convertible debt, as well as by the sale of certain
assets of the non-prescription business in which we were
previously engaged.
On April 21, 2010, our Registration Statement on
Form S-1
(as amended) was declared effective by the SEC for our IPO,
pursuant to which we sold 6,550,000 shares of our common
stock at a public offering price of $11.00 per share. We
received net proceeds of approximately $68.4 million from
this transaction, after deducting underwriting discounts and
commissions. In October 2010, we obtained a $32.5 million
senior secured credit facility (Credit Facility) to help fund
our working capital requirements. The Credit Facility consists
of a $20.0 million revolving line of credit and a
$12.5 million term loan. The lenders have advanced
$6.25 million under the term loan and may advance the
remaining $6.25 million following FDA approval of ILUVIEN,
but no later than July 31, 2011. Given the status of the
FDA’s review of the ILUVIEN NDA, it is unlikely that FDA
approval would occur prior to July 31, 2011. We are in
discussions with the lenders to amend the terms of the Credit
Facility to, among other things, extend the availability of the
term loan. However, there are no assurances that the Credit
Facility will be amended. We may draw on the revolving line of
credit against eligible, domestic accounts receivable, as
defined, subsequent to the launch of ILUVIEN. To secure the
repayment of any amounts borrowed under the Term Loan Agreement,
we granted to the Lenders a first priority security interest in
all of our assets, other than our intellectual property,
provided that, for any date during which the notes are
outstanding, our unrestricted balance sheet cash and cash
equivalents plus the excess available under the Term Loan
Agreement is less than the product of six times the monthly cash
burn amount. In the event we fail to meet this financial
condition, a curable lien will be imposed on our intellectual
property. Should such a lien event take place, the lien would
remain in force until such date that the Company’s
unrestricted cash and cash equivalents plus the excess available
under the Term Loan Agreement was equal to or greater than
twelve times the monthly cash burn amount. We also agreed not to
pledge or otherwise encumber our intellectual property assets.
Additionally, we must seek the lenders’ approval prior to
the payment of any cash dividends. As of December 31, 2010,
we had approximately $28.5 million in cash and cash
equivalents and $26.3 million of investments. We believe
that we have sufficient funds available to fund our operations
through the projected commercialization of ILUVIEN and the
expected generation of revenue in late 2011. The
commercialization of ILUVIEN is dependent upon approval by the
FDA, however, and we cannot be sure that ILUVIEN will be
approved by the FDA or that, if approved, future sales of
ILUVIEN will generate enough revenue to fund the Company’s
operations beyond its commercialization. Due to the uncertainty
around FDA approval, management cannot be certain that we will
not need additional funds for the commercialization of ILUVIEN.
If ILUVIEN is not approved, or if approved, does not generate
sufficient revenue, we may adjust our commercial plans so that
we can continue to operate with our existing cash resources or
seek to raise additional financing.
In the event additional financing is needed or desired, we may
seek to fund our operations through the sale of equity
securities, strategic collaboration agreements and debt
financing. We cannot be sure that additional financing from any
of these sources will be available when needed or that, if
available, the additional financing will be obtained on terms
favorable to us or our stockholders especially in light of the
current difficult financial environment. If we raise additional
funds by issuing equity securities, substantial dilution to
existing stockholders would likely result and the terms of any
new equity securities may have a preference over our common
stock. If we attempt to raise additional funds through strategic
collaboration agreements and debt financing, we may not be
successful in obtaining collaboration agreements, or in
receiving milestone or royalty payments under those agreements,
or the terms of the debt may involve
72
significant cash payment obligations as well as covenants and
specific financial ratios that may restrict our ability to
commercialize our product candidates or operate our business.
For the twelve months ended December 31, 2010, cash used in
our continuing operations of $22.1 million was primarily
due to our net loss from continuing operations of
$17.9 million increased by non-cash gains of
$3.6 million related to the change in fair value of our
preferred stock conversion feature and $1.3 million
associated with the repayment of our $15.0 million
promissory note to pSivida in April 2010, offset by non-cash
charges of $960,000 for stock compensation expense, and $190,000
for depreciation and amortization expense. Further increasing
our net cash used in continuing operations was an increase in
prepaid and other current assets of $570,000 offset by an
increase in accounts payable, accrued liabilities and other
current liabilities of $130,000. The increase in prepaid and
other current assets was primarily due to increases of $240,000
in cash receivable for the government’s Qualifying
Therapeutic Discovery Project Tax Credit, $210,000 in interest
receivable on our investments offset by a reduction of $110,000
of prepaid costs related to our FAME and ancillary clinical
studies as work on these studies continued. The increase in
accounts payable, accrued expenses and other current liabilities
was primarily due to increases of $730,000 of amounts payable to
providers of corporate communications and medical marketing
services for pre-launch activities, and $450,000 of accrued
bonuses as 2010 employee bonuses were not paid until
January 2011, offset by a decrease of $1.1 million payable
to the investigators in our FAME Study and ancillary clinical
studies.
For the twelve months ended December 31, 2009, cash used in
our continuing operations of $17.5 million was primarily
due to our net loss from continuing operation of
$44.2 million offset by non-cash charges including
$23.1 million related to the change in fair value of our
preferred stock conversion feature, $1.1 million in
depreciation and amortization expense associated primarily with
equipment used for the manufacture of our ILUVIEN registration
batches, $550,000 in stock compensation and other expense and
$300,000 in non-cash research and development expense paid to
Emory University with our common stock as an initial license fee
for two classes of NADPH oxidase inhibitors. Further offsetting
our net losses from continuing operations were increases in
accounts payable, accrued liabilities and other current
liabilities of $890,000 and other long-term liabilities of
$150,000, and a decrease in prepaid expenses and other current
assets of $590,000. Accounts payable, accrued liabilities and
other current liabilities increased due to increases of
$1.1 million in amounts payable to our clinical trial sites
and $550,000 in interest accrued on our $15.0 million
promissory note to pSivida, partially offset by decreases of
$420,000 in professional fees payable in connection with the
preparation for an initial public offering of our common stock
in 2008 and $390,000 in amounts payable to one of our third
party manufacturers. The increase in other long term liabilities
is due to interest being accrued on our promissory note to
pSivida. Prepaid expenses and other current assets decreased
primarily due to the progression of the technology transfer of
ILUVIEN and the utilization of prepayments to our third party
manufacturer.
For the year ended December 31, 2008, our cash used in
continuing operations of $32.2 million was primarily due to
our net loss from continuing operations of $61.5 million
offset by non-cash charges including a promissory note payable
of $15.0 million issued to pSivida and the forgiveness of
$2.8 million of net receivables due from pSivida in
connection with the amendment of our agreement,
$10.5 million related to the change in fair value of our
preferred stock conversion feature, $750,000 in stock
compensation and other expense, and $240,000 in depreciation and
amortization. An increase of $1.2 million in prepaid and
other current assets was offset by increases of $700,000
accounts payable, accrued expenses and other current liabilities
and $540,000 in other long-term liabilities. The increase in
prepaid expenses and other current assets was due primarily to
$1.1 million in advances to our third party manufacturers
for the technology transfer of ILUVIEN and an $880,000 increase
in our receivable due from pSivida prior to the renegotiation of
our agreement, offset by decreases in prepayments of $460,000 to
certain clinical trial sites and $360,000 to our contract
research organizations as our FAME Study progressed. Accounts
payable, accrued expenses and other current liabilities
increased primarily due to $440,000 to our CROs as our FAME
Study continued, $400,000 related to the technology transfer of
ILUVIEN and $380,000 associated with preparation for an initial
public offering of our common stock, offset by decreases of
$440,000 in amounts payable to our clinical trial sites and
$150,000 for our animal toxicology and degradation studies. The
increase in other long term liabilities is due to interest being
accrued on our promissory note to pSivida.
73
Net cash used in the investing activities of our continuing and
discontinued operations in the years ended December 31,
2010, 2009 and 2008 as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Continuing Operations
|
|
$
|
(26,460
|
)
|
|
$
|
(65
|
)
|
|
$
|
(640
|
)
|
|
Discontinued Operations
|
|
|
4,000
|
|
|
|
—
|
|
|
|
—
|
|
|
Total
|
|
$
|
(22,460
|
)
|
|
$
|
(65
|
)
|
|
$
|
(640
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the year ended December 31, 2010 net cash used in
our investing activities of our continuing operations was
$26.5 million, which was primarily due to the purchases of
$26.4 million of investments, net of maturities, and
$130,000 of computer equipment and software to facilitate the
filing of our NDA for ILUVIEN and for use by new employees. Net
cash provided in our discontinued operations was
$4.0 million, which was due to the exercise by
Bausch & Lomb of its option to extend by two years the
period during which it may continue to develop an allergy
product acquired from us in 2006.
For the years ended December 31, 2009, and 2008 net
cash used in the investing activities of our continuing
operations was attributable to purchases of property and
equipment.
For the year ended December 31, 2010 net cash provided
by our financing activities was $68.2 million, which was
due primarily to the receipt of net proceeds of
$68.5 million, after underwriting discounts and
commissions, from the sale of common stock in our IPO and our
employee stock purchase program, net proceeds of
$10.0 million from the exercise of warrants to purchase
shares of our
Series C-1
preferred stock, proceeds of $6.3 million from the issuance
of our notes payable to Silicon Valley Bank and MidCap Financial
LLP, and $710,000 from the exercise of options and warrants to
purchase shares of our common stock offset by the payment of
$1.9 million of costs related to our IPO and the repayment
of our $15.0 million promissory note to pSivida.
For the year ended December 31, 2009 net cash provided
by our financing activities was $4.6 million which were
primarily due to net proceeds of $4.9 million received from
the issuance of our
Series C-1
preferred stock and warrants for our
Series C-1
preferred stock.
For the year ended December 31, 2008 net cash provided
by our financing activities was $29.8 million which was
provided primarily by net proceeds from the issuance of our
Series C preferred stock.
Contractual
Obligations and Commitments
The following table summarizes our contractual obligations and
commitments as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Future Period
|
|
|
|
|
|
|
|
Less than
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
1 Year
|
|
|
1 — 3 Years
|
|
|
3 — 5 Years
|
|
|
5+ Years
|
|
|
|
|
(In thousands)
|
|
|
|
|
Note payables
|
|
$
|
6,250
|
|
|
$
|
1,157
|
|
|
|
5,093
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Operating lease
|
|
|
262
|
|
|
|
262
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Capital leases
|
|
|
29
|
|
|
|
11
|
|
|
|
18
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
6,541
|
|
|
$
|
1,430
|
|
|
|
5,111
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following amounts have not been included in the table above
as the timing of the payments is uncertain:
|
|
|
| |
•
|
In connection with our March 2008 agreement with pSivida we are
obligated to make a milestone payment of $25.0 million upon
FDA approval of ILUVIEN.
|
| |
| |
•
|
In connection with our July 2009 license and option agreement
with Emory University for the fulvene class of NADPH oxidase
inhibitors, we are required to make annual minimum royalty
payments in the first through the fourth calendar years
following regulatory approval of the product in a major market
|
74
|
|
|
| |
|
country (i.e., the U.S., Japan, China, India or any European
country) in the amount of $250,000, $500,000, $1.0 million
and $2.5 million, respectively, and $2.5 million for
each subsequent year during the term of our agreement. We will
also be required to make payments of up to $5.8 million
depending upon which regulatory milestones we achieve. If we do
not make any milestone payments to Emory University under our
agreement prior to the third anniversary of the effective date
of the agreement, then we will be required to pay Emory
University annual license maintenance fees ranging from $500,000
to $2.0 million (depending upon when such payment is made)
until a milestone payment is made under the agreement. As an
upfront license fee for the license granted by Emory University
to us, we issued to Emory University (and its inventors) that
number of shares of our common stock with a fair market value
equal to $150,000 on the date of issuance. To date, no other
payments have been made to Emory University in connection with
this license agreement.
|
|
|
|
| |
•
|
In connection with our August 2009 license and option agreement
with Emory University for the triphenylmethane class of NADPH
oxidase inhibitors, we are required to make annual minimum
royalty payments in the first through the fourth calendar years
following regulatory approval of the product in a major market
country (i.e., the U.S., Japan, China, India or any European
country) in the amount of $250,000, $500,000, $1.0 million
and $2.5 million, respectively, and an annual minimum
royalty payment of $2.5 million for each subsequent year
during the term of our agreement. We will also be required to
make payments of up to $5.9 million depending upon which
regulatory milestones we achieve. If we do not make any
milestone payments to Emory University under our agreement prior
to the third anniversary of the effective date of the agreement,
then we will be required to pay Emory University annual license
maintenance fees ranging from $500,000 to $2.0 million
(depending upon when such payment is made) until a milestone
payment is made under the agreement. As an upfront license fee
for the license granted by Emory University to us, in the fourth
quarter of 2009 we issued to Emory University (and its
inventors) that number of shares of our common stock with a fair
market value equal to $150,000 on the date of issuance. To date,
no other payments have been made to Emory University in
connection with this license agreement.
|
| |
| |
•
|
In connection with our November 2007 agreement with Dainippon
Sumitomo Pharma Co., Ltd. (Dainippon) we will be required to
make a payment in the amount of $200,000 to Dainippon within
30 days following the first regulatory approval of a
licensed product in the U.S. by the FDA.
|
| |
| |
•
|
In January 2006, we entered into an agreement with a contract
research organization for clinical and data management services
to be performed in connection with our FAME Study clinical sites
in the U.S., Canada, and Europe. In accordance with the terms of
the agreement, we will incur approximately $16.9 million of
expenses with the contract research organization through 2011.
Through December 31, 2010 we incurred $16.2 million of
expense associated with this agreement.
|
| |
| |
•
|
In July 2006, we entered into an agreement with a contract
research organization for clinical services to be performed in
connection with our FAME Study clinical sites in India. In
accordance with the terms of the agreement, we will incur
approximately $1.8 million of expenses with the contract
research organization through 2011. Through December 31,
2010 we incurred $1.3 million of expense associated with
this agreement.
|
Off-Balance
Sheet Arrangements
We do not have any relationships with unconsolidated entities or
financial partnerships, such as entities often referred to as
structured finance or special purpose entities, that would have
been established for the purpose of facilitating off-balance
sheet arrangements (as that term is defined in
Item 303(a)(4)(ii) of
Regulation S-K)
or other contractually narrow or limited purposes. As such, we
are not exposed to any financing, liquidity, market or credit
risk that could arise if we had engaged in those types of
relationships. We enter into guarantees in the ordinary course
of business related to the guarantee of our own performance and
the performance of our subsidiaries.
75
New
Accounting Pronouncements
From time to time, new accounting pronouncements are issued by
the Financial Accounting Standards Board, or FASB, or other
standard setting bodies that are adopted by us as of the
specified effective date. Unless otherwise discussed, we believe
that the impact of recently issued standards that are not yet
effective will not have a material impact on our financial
position or results of operations upon adoption.
|
|
|
ITEM 7A
|
QUALITATIVE
AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISK
|
We are exposed to market risk related to changes in interest
rates. As of December 31, 2010, we had approximately
$28.5 million in cash and cash equivalents and
$26.3 million in investments. Our interest income is
exposed to market risk primarily due to changes in the general
level of U.S. interest rates, particularly because our
investments are in short-term securities. Due to the short-term
duration of our investment portfolio and the low risk profile of
our investments, an immediate 10% change in interest rates would
not have a material effect on the fair market value of our
portfolio. Accordingly, we would not expect our operating
results or cash flows to be affected to any significant degree
by the effect of a sudden change in market interest rates on our
securities portfolio.
Our interest expense is exposed to market risk primarily due to
the variability of interest on our revolving loan agreement
which is calculated as the prime rate plus 2.50% (with a rate
floor of 6.50%). As of December 31, 2010, we have not
borrowed any funds available under the revolving loan agreement.
We contract for the conduct of some of our clinical trials and
other research and development activities with contract research
organizations and investigational sites in the U.S., Europe and
India. We may be subject to exposure to fluctuations in foreign
exchange rates in connection with these agreements. We do not
hedge our foreign currency exposures. We have not used
derivative financial instruments for speculation or trading
purposes.
|
|
|
ITEM 8
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
The financial statements and related financial statement
schedules required to be filed are indexed on page 81 and
are incorporated herein.
|
|
|
ITEM 9
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND
FINANCIAL DISCLOSURE
|
None.
|
|
|
ITEM 9A
|
CONTROLS
AND PROCEDURES
|
Evaluation
of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive
Officer and our Chief Financial Officer, evaluated the
effectiveness of our disclosure controls and procedures as of
December 31, 2010. The term “disclosure controls and
procedures,” as defined in
Rules 13a-15(e)
and
15d-15(e)
under the Exchange Act, means controls and other procedures of a
company that are designed to ensure that information required to
be disclosed by a company in the reports that it files or
submits under the Exchange Act is recorded, processed,
summarized and reported, within the time periods specified in
the SEC’s rules and forms. Disclosure controls and
procedures include, without limitation, controls and procedures
designed to ensure that information required to be disclosed by
a company in the reports that it files or submits under the
Exchange Act is accumulated and communicated to the
company’s management, including its principal executive and
principal financial officers, as appropriate to allow timely
decisions regarding required disclosure. Management recognizes
that any controls and procedures, no matter how well designed
and operated, can provide only reasonable assurance of achieving
their objectives and management necessarily applies its judgment
in evaluating the cost-benefit relationship of possible controls
and procedures. Based on the evaluation of our disclosure
controls and procedures as of December 31, 2010, our Chief
Executive Officer and Chief Financial
76
Officer concluded that, as of such date, our disclosure controls
and procedures were effective at the reasonable assurance level.
Management’s
Report on Internal Control over Financial Reporting
The SEC, as required by Section 404 of the Sarbanes-Oxley
Act, adopted rules requiring companies that file reports with
the SEC to include a management report on such company’s
internal control over financial reporting in its annual report.
In addition, our independent registered public accounting firm
may be required to attest to our internal control over financial
reporting. This report on
Form 10-K
does not include a report of management’s assessment
regarding internal control over financial reporting or an
attestation report of our independent registered public
accounting firm due to a transition period established by SEC
rules applicable to newly public companies. Management will be
required to provide an assessment of the effectiveness of our
internal control over financial reporting as of
December 31, 2011. We believe we will have adequate
resources and expertise, both internal and external, in place to
meet this requirement. However, there is no guarantee that our
efforts will result in management’s ability to conclude,
or, if required, our independent registered public accounting
firm to attest, that our internal control over financial
reporting is effective as of December 31, 2011.
Changes
in Internal Control over Financial Reporting
There has been no change in our internal control over financial
reporting (as defined in
Rules 13a-15(f)
and
15d-15(f) of
the Exchange Act) during the fourth quarter of 2010 that has
materially affected, or is reasonably likely to materially
affect, our internal control over financial reporting.
Limitations
on the Effectiveness of Controls
Control systems, no matter how well conceived and operated, are
designed to provide a reasonable, but not an absolute, level of
assurance that the objectives of the control system are met.
Further, the design of a control system must reflect the fact
that there are resource constraints, and the benefits of
controls must be considered relative to their costs. Because of
the inherent limitations in all control systems, no evaluation
of controls can provide absolute assurance that all control
issues and instances of fraud, if any, have been detected.
Because of the inherent limitations in any control system,
misstatements due to error or fraud may occur and not be
detected.
|
|
|
ITEM 9B
|
Other
Information
|
None.
PART III
|
|
|
ITEM 10
|
DIRECTORS,
EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
Information required under this item will be contained in the
Company’s Proxy Statement for the Annual Meeting of
Stockholders to be filed with the SEC within 120 days after
the end of the fiscal year ended December 31, 2010, under
the captions “Election of Directors,” “Executive
Officers,” “Corporate Governance”, and
“Section 16(a) Beneficial Ownership Reporting
Compliance” and is incorporated herein by reference
pursuant to General Instruction G(3) to
Form 10-K.
|
|
|
ITEM 11
|
EXECUTIVE
COMPENSATION
|
Information required under this item will be contained in the
Company’s Proxy Statement for the Annual Meeting of
Stockholders to be filed with the SEC within 120 days after
the end of the fiscal year ended December 31, 2010, under
the captions “Corporate Governance” and
“Executive Compensation,” and is incorporated herein
by reference pursuant to General Instruction G(3) to
Form 10-K,
except that information required by Item 407(e)(5) of
Regulation S-K
will be deemed furnished in this
Form 10-K
and will not be
77
deemed incorporated by reference into any filing under the
Securities Act of 1933 or the Securities Exchange Act of 1934,
except to the extent that we specifically incorporate it by
reference into such filing.
|
|
|
ITEM 12
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND
RELATED STOCKHOLDER MATTERS
|
Except for the information set forth below, the information
required under this item will be contained in the Company’s
Proxy Statement for the Annual Meeting of Stockholders to be
filed with the SEC within 120 days after the end of the
fiscal year ended December 31, 2010, under the caption
“Security Ownership by Certain Beneficial Owners and
Management” and is incorporated herein by reference
pursuant to General Instruction G(3) to
Form 10-K.
Equity
Compensation Plan Information
The following table provides information as of December 31,
2010, with respect to shares of our common stock that may be
issued, subject to certain vesting requirements, under our
existing equity compensation plans, including our 2010 Equity
Incentive Plan (“2010 Plan”), 2005 Equity Incentive
Plan (“2005 Plan”), 2004 Equity Incentive Plan
(“2004 Plan”) and our 2010 Employee Stock Purchase
Plan (“ESPP”).
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A
|
|
|
B
|
|
|
C
|
|
|
|
|
|
|
|
|
|
|
Number of Securities
|
|
|
|
|
|
|
|
|
|
|
Remaining Available
|
|
|
|
|
Number of Securities
|
|
|
|
|
|
for Future Issuance
|
|
|
|
|
to be Issued Upon
|
|
|
Weighted-Average
|
|
|
Under Equity Compensation
|
|
|
|
|
Exercise of
|
|
|
Exercise Price of
|
|
|
Plans (Excluding
|
|
|
|
|
Outstanding Options,
|
|
|
Outstanding Options,
|
|
|
Securities Reflected in
|
|
|
Plan Category
|
|
Warrants, and Rights
|
|
|
Warrants, and Rights
|
|
|
Column (A))
|
|
|
|
|
Equity compensation plans approved by security holders
|
|
|
2,829,405
|
(1)
|
|
$
|
3.90
|
|
|
|
1,888,712
|
(2)
|
|
Equity compensation plans not approved by security holders
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
2,829,405
|
|
|
$
|
3.90
|
|
|
|
1,888,712
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Of these shares, 570,150 were subject to options then
outstanding under the 2010 Plan, 1,802,080 were subject to
options then outstanding under the 2005 Plan and 369,755 were
subject to options then outstanding under the 2004 Plan. |
| |
|
(2) |
|
Represents 1,402,536 shares of common stock available for
issuance under our 2010 Plan and 486,176 shares of common
stock available for issuance under our ESPP. No shares are
available for future issuance under the 2005 Plan or 2004 Plan.
In addition, our 2010 Plan provides for annual increases in the
number of shares available for issuance thereunder on the first
day of each fiscal year equal to the least of:
(1) 2,000,000 shares of our common stock; (2) 4%
of the shares of common stock outstanding at that time; and
(3) such other amount as our board of directors may
determine. On January 1, 2011, an additional
1,250,238 shares became available for future issuance under
our 2010 Plan in accordance with the annual increase. In
addition, our ESPP provides for annual increases in the number
of shares available for issuance thereunder equal to such number
of shares necessary to restore the number of shares reserved
thereunder to 494,422 shares of our common stock. As such,
on January 1, 2011, an additional 8,246 shares became
available for future issuance under our ESPP. These additional
shares from the annual increase under the 2010 Plan and the ESPP
are not included in the table above. |
|
|
|
ITEM 13
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR
INDEPENDENCE
|
Information required under this item will be contained in the
Company’s Proxy Statement for the Annual Meeting of
Stockholders to be filed with the SEC within 120 days after
the end of the fiscal year ended
78
December 31, 2010, under the caption “Corporate
Governance” and is incorporated herein by reference
pursuant to General Instruction G(3) to
Form 10-K.
|
|
|
ITEM 14
|
PRINCIPAL
ACCOUNTANT FEES AND SERVICES
|
Information required under this item will be contained in the
Company’s Proxy Statement for the Annual Meeting of
Stockholders to be filed with the SEC within 120 days after
the end of the fiscal year ended December 31, 2010, under
the caption “Ratification of Selection of Independent
Registered Public Accounting Firm” and is incorporated
herein by reference pursuant to General Instruction G(3) to
Form 10-K.
PART IV
|
|
|
ITEM 15
|
EXHIBITS AND
FINANCIAL STATEMENTS SCHEDULES
|
The consolidated financial statements filed as part of this
annual report on
Form 10-K
are listed and indexed at page 81. Certain schedules are
omitted because they are not applicable, or not required, or
because the required information is included in the consolidated
financial statements or notes thereto.
The Exhibits listed in the Exhibit Index immediately
preceding the Exhibits are filed as part of this annual report
on
Form 10-K.
79
Signatures
Pursuant to the requirements of Section 13 and 15(d) of the
Securities Exchange Act of 1934, the registrant has duly caused
this annual report on
Form 10-K
to be signed on its behalf by the undersigned, thereunto duly
authorized, in Alpharetta, Georgia, on March 25, 2011.
ALIMERA SCIENCES, INC.
President and Chief Executive Officer
Pursuant to the requirements of the Securities Act of 1934, this
annual report on
Form 10-K
has been signed below by the following persons on behalf of the
registrant and in the capacities and on the dates indicated.
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ C.
Daniel Myers
C.
Daniel Myers
|
|
President, Chief Executive Officer and Director (Principal
Executive Officer)
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Richard
S. Eiswirth, Jr.
Richard
S. Eiswirth, Jr.
|
|
Chief Financial Officer (Principal Financial Officer and
Principal Accounting Officer)
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Philip
R. Tracy
Philip
R. Tracy
|
|
Chairman of the Board of Directors
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Mark
J. Brooks
Mark
J. Brooks
|
|
Director
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Brian
K. Halak, Ph.D
Brian
K. Halak, Ph.D
|
|
Director
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Anders
D. Hove, M.D.
Anders
D. Hove, M.D.
|
|
Director
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Peter
J. Pizzo, III
Peter
J. Pizzo, III
|
|
Director
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Calvin
W. Roberts, M.D.
Calvin
W. Roberts, M.D.
|
|
Director
|
|
March 25, 2011
|
|
|
|
|
|
|
|
/s/ Bryce
Youngren
Bryce
Youngren
|
|
Director
|
|
March 25, 2011
|
80
ALIMERA
SCIENCES, INC.
INDEX TO
FINANCIAL STATEMENTS
| |
|
|
|
|
|
|
|
Page
|
|
|
|
|
|
|
82
|
|
|
Financial Statements as of December 31, 2010 and 2009 and
for the years ended December 31, 2010, 2009, and 2008:
|
|
|
|
|
|
|
|
|
83
|
|
|
|
|
|
84
|
|
|
|
|
|
85
|
|
|
|
|
|
86
|
|
|
|
|
|
87
|
|
81
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Alimera Sciences, Inc.
Alpharetta, Georgia
We have audited the accompanying balance sheets of Alimera
Sciences, Inc. (the “Company”) as of December 31,
2010 and 2009, and the related statements of operations, changes
in stockholders’ equity (deficit), and cash flows for each
of the three years in the period ended December 31, 2010.
These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes consideration
of internal control over financial reporting as a basis for
designing audit procedures that are appropriate in the
circumstances, but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over
financial reporting. Accordingly, we express no such opinion. An
audit also includes examining, on a test basis, evidence
supporting the amounts and disclosures in the financial
statements, assessing the accounting principles used and
significant estimates made by management, as well as evaluating
the overall financial statement presentation. We believe that
our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all
material respects, the financial position of the Company as of
December 31, 2010 and 2009, and the results of its
operations and its cash flows for each of the three years in the
period ended December 31, 2010 in conformity with
accounting principles generally accepted in the United States of
America.
As described in Note 4, the accompanying financial
statements have been prepared assuming the Company will continue
as a going concern. The Company’s recurring net losses,
negative cash flow from operations, accumulated deficit, and
current lack of a commercial product raise substantial doubt
about its ability to continue as a going concern.
Management’s plans concerning these matters are also
discussed in Note 4. The financial statements do not
include any adjustments that might result from the outcome of
this uncertainty.
/s/ Deloitte & Touche LLP
Atlanta, Georgia
March 25, 2011
82
ALIMERA
SCIENCES, INC.
AS OF DECEMBER 31, 2010 AND 2009
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
CURRENT ASSETS:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
28,514
|
|
|
$
|
4,858
|
|
|
Investments
|
|
|
26,330
|
|
|
|
—
|
|
|
Prepaid expenses and other current assets
|
|
|
1,078
|
|
|
|
634
|
|
|
Deferred financing costs
|
|
|
272
|
|
|
|
815
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
56,194
|
|
|
|
6,307
|
|
|
PROPERTY AND EQUIPMENT — at cost less accumulated
depreciation
|
|
|
220
|
|
|
|
254
|
|
|
TOTAL ASSETS
|
|
$
|
56,414
|
|
|
$
|
6,561
|
|
|
|
|
|
|
|
|
|
|
|
|
CURRENT LIABILITIES:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
1,677
|
|
|
$
|
1,758
|
|
|
Accrued expenses (Note 6)
|
|
|
2,731
|
|
|
|
3,314
|
|
|
Outsourced services payable
|
|
|
841
|
|
|
|
1,157
|
|
|
Notes payable (Note 9)
|
|
|
1,157
|
|
|
|
4,500
|
|
|
Capital lease obligations
|
|
|
11
|
|
|
|
6
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
6,417
|
|
|
|
10,735
|
|
|
LONG-TERM LIABILITIES:
|
|
|
|
|
|
|
|
|
|
Notes payable, net of discount — less current portion
(Note 9)
|
|
|
4,767
|
|
|
|
10,500
|
|
|
Fair value of preferred stock conversion feature
|
|
|
—
|
|
|
|
36,701
|
|
|
Other long-term liabilities
|
|
|
18
|
|
|
|
708
|
|
|
COMMITMENTS AND CONTINGENCIES (Note 10)
|
|
|
|
|
|
|
|
|
|
PREFERRED STOCK:
|
|
|
|
|
|
|
|
|
|
Series A preferred stock, $.01 par value —
no shares authorized, issued, and outstanding at
December 31, 2010 and 6,624,866 shares authorized and
6,624,844 shares issued, and outstanding at
December 31, 2009; liquidation preference of $37,019 at
December 31, 2009
|
|
|
—
|
|
|
|
36,467
|
|
|
Series B preferred stock, $.01 par value —
no shares authorized, issued, and outstanding at
December 31, 2010 and 7,147,912 shares authorized and
7,147,894 shares issued, and outstanding at
December 31, 2009; liquidation preference of $41,057 at
December 31, 2009
|
|
|
—
|
|
|
|
40,617
|
|
|
Series C preferred stock, $.01 par value —
no shares authorized, issued, and outstanding at
December 31, 2010 and 5,807,131 shares authorized and
5,807,112 shares issued and outstanding at
December 31, 2009; liquidation preference of $34,281 at
December 31, 2009
|
|
|
—
|
|
|
|
33,452
|
|
|
Series C-1
preferred stock, $.01 par value — no shares
authorized, issued, and outstanding at December 31, 2010
and 2,903,565 shares authorized and 967,845 shares
issued and outstanding at December 31, 2009; liquidation
preference of $5,140 at December 31, 2009
|
|
|
—
|
|
|
|
2,853
|
|
|
Preferred stock, $.01 par value —
10,000,000 shares authorized and no shares issued and
outstanding at December 31, 2010 and 2009
|
|
|
—
|
|
|
|
—
|
|
|
STOCKHOLDERS’ DEFICIT:
|
|
|
|
|
|
|
|
|
|
Common stock, $.01 par value —
100,000,000 shares authorized and 31,255,953 shares
issued and outstanding at December 31, 2010 and
29,411,764 shares authorized and 1,598,571 shares
issued and outstanding at December 31, 2009
|
|
|
313
|
|
|
|
54
|
|
|
Additional paid-in capital
|
|
|
233,338
|
|
|
|
4,836
|
|
|
Series C-1
preferred stock warrants
|
|
|
—
|
|
|
|
1,472
|
|
|
Common stock warrants
|
|
|
415
|
|
|
|
57
|
|
|
Accumulated deficit
|
|
|
(188,854
|
)
|
|
|
(171,891
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL STOCKHOLDERS’ EQUITY (DEFICIT)
|
|
|
45,212
|
|
|
|
(165,472
|
)
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT)
|
|
$
|
56,414
|
|
|
$
|
6,561
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See Notes to Financial Statements.
83
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands except share and per share data)
|
|
|
|
|
RESEARCH AND DEVELOPMENT EXPENSES
|
|
$
|
12,581
|
|
|
$
|
15,057
|
|
|
$
|
43,764
|
|
|
GENERAL AND ADMINISTRATIVE EXPENSES
|
|
|
4,610
|
|
|
|
3,407
|
|
|
|
5,058
|
|
|
MARKETING EXPENSES
|
|
|
4,880
|
|
|
|
752
|
|
|
|
1,259
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OPERATING EXPENSES
|
|
|
22,071
|
|
|
|
19,216
|
|
|
|
50,081
|
|
|
INTEREST AND OTHER INCOME
|
|
|
73
|
|
|
|
37
|
|
|
|
585
|
|
|
INTEREST EXPENSE
|
|
|
(848
|
)
|
|
|
(1,897
|
)
|
|
|
(1,514
|
)
|
|
GAIN ON EARLY EXTINGUISHMENT OF DEBT (NOTE 7)
|
|
|
1,343
|
|
|
|
—
|
|
|
|
—
|
|
|
DECREASE (INCREASE) IN FAIR VALUE OF PREFERRED STOCK CONVERSION
FEATURE
|
|
|
3,644
|
|
|
|
(23,142
|
)
|
|
|
(10,454
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LOSS FROM CONTINUING OPERATIONS
|
|
|
(17,859
|
)
|
|
|
(44,218
|
)
|
|
|
(61,464
|
)
|
|
INCOME FROM DISCONTINUED OPERATIONS (NOTE 3)
|
|
|
4,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS
|
|
|
(13,859
|
)
|
|
|
(44,218
|
)
|
|
|
(61,464
|
)
|
|
BENEFICIAL CONVERSION FEATURE OF PREFERRED STOCK (NOTE 11)
|
|
|
—
|
|
|
|
(355
|
)
|
|
|
—
|
|
|
PREFERRED STOCK ACCRETION
|
|
|
(466
|
)
|
|
|
(623
|
)
|
|
|
(718
|
)
|
|
PREFERRED STOCK DIVIDENDS
|
|
|
(2,638
|
)
|
|
|
(7,225
|
)
|
|
|
(6,573
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS APPLICABLE TO COMMON SHAREHOLDERS
|
|
$
|
(16,963
|
)
|
|
$
|
(52,421
|
)
|
|
$
|
(68,755
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET LOSS PER SHARE APPLICABLE TO COMMON SHAREHOLDERS —
Basic and diluted
|
|
$
|
(0.77
|
)
|
|
$
|
(34.55
|
)
|
|
$
|
(45.52
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
WEIGHTED — AVERAGE SHARES OUTSTANDING —
Basic and diluted
|
|
|
22,167,873
|
|
|
|
1,517,365
|
|
|
|
1,510,496
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See Notes to Financial Statements.
84
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Series C-1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Preferred
|
|
|
Common
|
|
|
Accumulated
|
|
|
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Warrants
|
|
|
Warrants
|
|
|
Deficit
|
|
|
Total
|
|
|
|
|
(In thousands except share data)
|
|
|
|
|
BALANCE — December 31, 2007
|
|
|
1,516,389
|
|
|
$
|
52
|
|
|
$
|
2,867
|
|
|
$
|
—
|
|
|
$
|
58
|
|
|
$
|
(50,715
|
)
|
|
$
|
(47,738
|
)
|
|
Preferred stock accretion and dividends
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(7,291
|
)
|
|
|
(7,291
|
)
|
|
Repurchase and retirement of common stock
|
|
|
(27,746
|
)
|
|
|
(1
|
)
|
|
|
(149
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(150
|
)
|
|
Stock compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
750
|
|
|
|
—
|
|
|
|
|
|
|
|
—
|
|
|
|
750
|
|
|
Exercise of warrants
|
|
|
1,470
|
|
|
|
—
|
|
|
|
6
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(61,464
|
)
|
|
|
(61,464
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2008
|
|
|
1,490,113
|
|
|
|
51
|
|
|
|
3,474
|
|
|
|
—
|
|
|
|
58
|
|
|
|
(119,470
|
)
|
|
|
(115,887
|
)
|
|
Redeemable preferred stock accretion and dividends
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(7,848
|
)
|
|
|
(7,848
|
)
|
|
Issuance of common stock
|
|
|
92,351
|
|
|
|
3
|
|
|
|
458
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
461
|
|
|
Exercise of stock options
|
|
|
3,860
|
|
|
|
—
|
|
|
|
6
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6
|
|
|
Exercise of common stock warrants
|
|
|
12,247
|
|
|
|
—
|
|
|
|
31
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
31
|
|
|
Retirement of common stock warrants
|
|
|
—
|
|
|
|
—
|
|
|
|
1
|
|
|
|
—
|
|
|
|
(1
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Issuance of
series C-1
preferred stock warrants
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,472
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,472
|
|
|
Accretion of
series C-1
preferred stock beneficial conversion feature (Note 11)
|
|
|
—
|
|
|
|
—
|
|
|
|
355
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(355
|
)
|
|
|
—
|
|
|
Stock compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
511
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
511
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(44,218
|
)
|
|
|
(44,218
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2009
|
|
|
1,598,571
|
|
|
|
54
|
|
|
|
4,836
|
|
|
|
1,472
|
|
|
|
57
|
|
|
|
(171,891
|
)
|
|
|
(165,472
|
)
|
|
Redeemable preferred stock accretion and dividends
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(3,104
|
)
|
|
|
(3,104
|
)
|
|
Issuance of common stock
|
|
|
29,421,942
|
|
|
|
256
|
|
|
|
192,707
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
192,963
|
|
|
Issuance of common warrants (Note 9)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
366
|
|
|
|
—
|
|
|
|
366
|
|
|
Exercise of stock options
|
|
|
44,499
|
|
|
|
1
|
|
|
|
93
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
94
|
|
|
Exercise of common stock warrants
|
|
|
190,941
|
|
|
|
2
|
|
|
|
618
|
|
|
|
—
|
|
|
|
(8
|
)
|
|
|
—
|
|
|
|
612
|
|
|
Exercise of
series C-1
preferred stock warrants
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,472
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,472
|
)
|
|
IPO costs
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,282
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(2,282
|
)
|
|
Elimination of preferred stock conversion feature upon
conversion of preferred stock to common stock (Note 11)
|
|
|
—
|
|
|
|
—
|
|
|
|
36,528
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
36,528
|
|
|
Stock compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
838
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
838
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(13,859
|
)
|
|
|
(13,859
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
BALANCE — December 31, 2010
|
|
|
31,255,953
|
|
|
$
|
313
|
|
|
$
|
233,338
|
|
|
$
|
—
|
|
|
$
|
415
|
|
|
$
|
(188,854
|
)
|
|
$
|
45,212
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See Notes to Financial Statements.
85
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
CASH FLOWS FROM OPERATING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(13,859
|
)
|
|
$
|
(44,218
|
)
|
|
$
|
(61,464
|
)
|
|
Income from discontinued operations (Note 3)
|
|
|
(4,000
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Change in fair value of preferred stock conversion feature
|
|
|
(3,644
|
)
|
|
|
23,142
|
|
|
|
10,454
|
|
|
Gain from early extinguishment of debt
|
|
|
(1,343
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Depreciation and amortization
|
|
|
194
|
|
|
|
1,098
|
|
|
|
241
|
|
|
Stock compensation expense and other
|
|
|
957
|
|
|
|
551
|
|
|
|
750
|
|
|
Amortization of deferred financing costs
|
|
|
73
|
|
|
|
—
|
|
|
|
—
|
|
|
Unrealized investment loss
|
|
|
2
|
|
|
|
—
|
|
|
|
—
|
|
|
Noncash research and development expense (Notes 7 and 8)
|
|
|
—
|
|
|
|
300
|
|
|
|
17,809
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Changes in assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Prepaid expenses and other current assets
|
|
|
(566
|
)
|
|
|
591
|
|
|
|
(1,213
|
)
|
|
Accounts payable
|
|
|
391
|
|
|
|
183
|
|
|
|
615
|
|
|
Accrued expenses and other current liabilities
|
|
|
(258
|
)
|
|
|
705
|
|
|
|
85
|
|
|
Other long-term assets
|
|
|
—
|
|
|
|
—
|
|
|
|
24
|
|
|
Other long-term liabilities
|
|
|
—
|
|
|
|
153
|
|
|
|
540
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities of continuing operations
|
|
|
(22,053
|
)
|
|
|
(17,495
|
)
|
|
|
(32,159
|
)
|
|
Net cash (used in) provided by operating activities of
discontinued operations
|
|
|
—
|
|
|
|
(43
|
)
|
|
|
43
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(22,053
|
)
|
|
|
(17,538
|
)
|
|
|
(32,116
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of investments
|
|
|
(39,927
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Proceeds from maturities of investments
|
|
|
13,595
|
|
|
|
—
|
|
|
|
—
|
|
|
Purchases of property and equipment
|
|
|
(128
|
)
|
|
|
(65
|
)
|
|
|
(640
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in investing activities of continuing operations
|
|
|
(26,460
|
)
|
|
|
(65
|
)
|
|
|
(640
|
)
|
|
Net cash provided by investing activities of discontinued
operations (Note 3)
|
|
|
4,000
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in investing activities
|
|
|
(22,460
|
)
|
|
|
(65
|
)
|
|
|
(640
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from sale of Series C preferred stock —
net
|
|
|
—
|
|
|
|
—
|
|
|
|
29,938
|
|
|
Proceeds from sale of
Series C-1
preferred stock — net
|
|
|
9,997
|
|
|
|
4,897
|
|
|
|
—
|
|
|
Proceeds from exercise of stock options
|
|
|
94
|
|
|
|
7
|
|
|
|
—
|
|
|
Repurchase of common stock
|
|
|
—
|
|
|
|
—
|
|
|
|
(150
|
)
|
|
Proceeds from exercise of common stock warrants
|
|
|
613
|
|
|
|
31
|
|
|
|
6
|
|
|
Proceeds from sale of common stock
|
|
|
68,470
|
|
|
|
—
|
|
|
|
—
|
|
|
Payment of common stock offering costs
|
|
|
(1,942
|
)
|
|
|
(339
|
)
|
|
|
—
|
|
|
Proceeds from issuance of notes payable (Note 9)
|
|
|
6,250
|
|
|
|
—
|
|
|
|
—
|
|
|
Payment of debt issuance costs
|
|
|
(305
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Repayment of pSivida note payable (Note 7)
|
|
|
(15,000
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Payments on capital lease obligations
|
|
|
(8
|
)
|
|
|
(10
|
)
|
|
|
(10
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
68,169
|
|
|
|
4,586
|
|
|
|
29,784
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NET INCREASE (DECREASE) IN CASH
|
|
|
23,656
|
|
|
|
(13,017
|
)
|
|
|
(2,972
|
)
|
|
CASH — Beginning of period
|
|
|
4,858
|
|
|
|
17,875
|
|
|
|
20,847
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASH — End of period
|
|
$
|
28,514
|
|
|
$
|
4,858
|
|
|
$
|
17,875
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SUPPLEMENTAL DISCLOSURES:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest
|
|
$
|
681
|
|
|
$
|
1,200
|
|
|
$
|
957
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental schedule of noncash investing and financing
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Note payable issued in conjunction with amendment to pSivida
agreement (Note 7)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
15,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reclassification of fair value of preferred stock conversion
feature to additional paid-in capital
|
|
$
|
36,528
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IPO issuance costs charged to equity
|
|
$
|
3,994
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Notes payable issuance costs charged to equity (Note 9)
|
|
$
|
366
|
|
|
$
|
|
|
|
$
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment acquired under capital leases
|
|
$
|
36
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock issued for research and development expense
(Note 8)
|
|
$
|
—
|
|
|
$
|
300
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
There were no income tax or dividend payments made for the years
ended December 31, 2010, 2009, and 2008.
See Notes to Financial Statements.
86
ALIMERA
SCIENCES, INC.
Alimera Sciences, Inc. (the Company) is a biopharmaceutical
company that specializes in the research, development, and
commercialization of ophthalmic pharmaceuticals. The Company was
formed on June 4, 2003 under the laws of the State of
Delaware.
During the year ended December 31, 2006, management and the
board of directors approved a plan to discontinue the operations
of its non-prescription business (see Note 3). As a result
of the completion of the disposal of its non-prescription
business in July 2007, the Company no longer has active products
and will not have active products until the Company receives
U.S. Food and Drug Administration (FDA) approval and
launches its initial prescription product (see Note 3).
The Company is presently focused on diseases affecting the back
of the eye, or retina, because the Company’s management
believes these diseases are not well treated with current
therapies and represent a significant market opportunity. The
Company’s most advanced product candidate is ILUVIEN, which
is being developed for the treatment of diabetic macular edema
(DME). DME is a disease of the retina which affects individuals
with diabetes and can lead to severe vision loss and blindness.
The Company has completed its two Phase 3 pivotal clinical
trials (collectively referred to as the Company’s FAME
Study) for ILUVIEN involving 956 patients in sites across
the U.S., Canada, Europe and India to assess the efficacy and
safety of ILUVIEN in the treatment of DME.
On April 21, 2010, the Company’s Registration
Statement on
Form S-1
(as amended) was declared effective by the Securities and
Exchange Commission (SEC) for the Company’s initial public
offering (IPO), pursuant to which the Company sold
6,550,000 shares of its common stock at a public offering
price of $11.00 per share. The Company received net proceeds of
approximately $68,395,000 from this transaction, after deducting
underwriting discounts and commissions. In connection with its
IPO, the Company effected a 1 for 3.4 reverse split of the
Company’s common and preferred stock. All share and per
share amounts in the accompanying financial statements and notes
have been retroactively adjusted for all periods presented to
give effect to the reverse stock split.
In June 2010, the Company submitted a New Drug Application (NDA)
for ILUVIEN to the FDA. In July 2010, the Company submitted a
Marketing Authorization Application for ILUVIEN to the Medicines
and Healthcare products Regulatory Agency in the United Kingdom
and to regulatory authorities in Austria, France, Germany,
Italy, Portugal and Spain. In August 2010, the FDA accepted the
Company’s NDA for ILUVIEN and granted it priority review
status. Priority review status reduces the review time from ten
months to six months.
In December 2010, the FDA issued a Complete Response Letter
(CLR) in response to the Company’s NDA. In the CLR, the FDA
communicated its decision that the NDA could not be approved in
its present form. No new clinical studies were requested in the
CRL. However, the FDA asked for analyses of the safety and
efficacy data through month 36 of the FAME Study, including
exploratory analyses in addition to those previously submitted
to the FDA, to further assess the relative benefits and risks of
ILUVIEN. The NDA included data through month 24. The Company has
completed month 36 of the study and is preparing the analyses
the FDA requested. The FDA is also seeking additional
information regarding controls and specifications concerning the
manufacturing, packaging and sterilization of ILUVIEN, which the
Company is in the process of compiling.
|
|
|
2.
|
SUMMARY
OF SIGNIFICANT ACCOUNTING POLICIES
|
Use of Estimates in Financial Statements — The
financial statements have been prepared in conformity with
accounting principles generally accepted in the U.S. of
America and, as such, include amounts based on informed
estimates and judgments of management. Actual results could
differ from those estimates.
87
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The following accounting policies relate primarily to the
continuing operations of the Company:
Cash and Cash Equivalents — Cash and cash
equivalents include cash and highly liquid investments that are
readily convertible into cash and have a maturity of
90 days or less when purchased.
Investments — In accordance with ASC 320,
Debt and Equity Securities, the Company classifies its
investments as trading securities. The Company recognizes the
investments at fair value and includes all unrealized holding
gains and losses in the statement of operations.
Long-Lived Assets — Property and equipment are
stated at cost. Additions and improvements are capitalized while
repairs and maintenance are expensed. Depreciation is provided
on the straight-line method over the useful life of the related
assets beginning when the asset is placed in service. The
estimated useful lives of the individual assets are as follows:
furniture and fixtures, five years; office equipment, three to
five years; and software, three years.
Impairment — Property and equipment and
intangible assets are reviewed for impairment whenever events or
changes in circumstances indicate that the carrying amount of an
asset may not be recoverable. When indicators of impairment are
present, the Company evaluates the carrying amount of such
assets in relation to the operating performance and future
estimated undiscounted net cash flows expected to be generated
by the assets. If such assets are considered to be impaired, the
impairment to be recognized is measured by the amount by which
the carrying amount of the assets exceeds the fair value of the
assets. The assessment of the recoverability of assets will be
impacted if estimated future operating cash flows are not
achieved.
Income Taxes — In accordance with ASC 740,
Income Taxes, the Company recognizes deferred tax assets
and liabilities for temporary differences between the financial
reporting basis and the tax basis of its assets and liabilities.
The Company records a valuation allowance against its net
deferred tax asset to reduce the net carrying value to an amount
that is more likely than not to be realized.
Income tax positions are considered for uncertainty in
accordance with
ASC 740-10.
The provisions of
ASC 740-10
are effective beginning January 1, 2008, but the Company
early adopted effective January 1, 2007. The Company
believes that its income tax filing positions and deductions
will be sustained on audit and does not anticipate any
adjustments that will result in a material change to its
financial position; therefore, no
ASC 740-10
liabilities have been recorded. The Company’s adoption of
ASC 740-10
did not result in a cumulative effect adjustment to retained
earnings. The Company will recognize accrued interest and
penalties related to unrecognized tax benefits as interest
expense and income tax expense, respectively, in the statements
of operations.
Significant management judgment is involved in determining the
provision for income taxes, deferred tax assets and liabilities,
and any valuation allowance recorded against net deferred tax
assets. Due to uncertainties with respect to the realization of
deferred tax assets due to the history of operating losses, a
valuation allowance has been established against the entire net
deferred tax asset balance. The valuation allowance is based on
management’s estimates of taxable income in the
jurisdictions in which the Company operates and the period over
which deferred tax assets will be recoverable. In the event that
actual results differ from these estimates or the Company
adjusts these estimates in future periods, a change in the
valuation allowance may be needed, which could materially impact
the Company’s financial position and results of operations.
Research and Development Costs — Research and
development costs are expensed as incurred.
Stock-Based Compensation — The Company has
stock option plans which provide for grants of stock options to
employees and directors to purchase shares of the Company’s
common stock at exercise prices generally equal to the fair
values of such stock at the dates of grant. Compensation cost is
recognized for all share-based awards granted subsequent to
January 1, 2005 based on the grant date fair
88
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
value in accordance with the provisions of ASC 718,
Compensation — Stock Compensation. The fair
values for the options are estimated at the dates of grant using
a Black-Scholes option-pricing model.
Additionally, the Company sponsors an employee stock purchase
plan under which employees may elect payroll withholdings to
fund purchases of the Company’s stock at a discount. The
Company estimates the fair value of the option to purchase
shares of the Company’s common stock using the
Black-Scholes valuation model and recognizes compensation
expense in accordance with the provisions of
ASC 718-50,
Employee Share Purchase Plans.
Derivative Financial Instruments — The
Company’s previously outstanding preferred stock (see
Note 11) contained certain features which are
considered embedded derivatives. The Company accounted for such
embedded derivative financial instruments in accordance with ASC
815, Derivatives and Hedging. The Company recorded
derivative financial instruments as assets or liabilities in the
Company’s balance sheet measured at fair value (see
Note 14). The Company recorded the changes in fair value of
such instruments as noncash gains or losses in the consolidated
statement of operations. The Company does not enter into
derivatives for trading purposes.
Fair Value of Financial Instruments — The
carrying amounts of the Company’s financial instruments,
including cash and cash equivalents, investments, receivables,
and current liabilities approximate their fair value because of
their short maturities.
Earnings (Loss) Per Share (EPS) — Basic EPS is
calculated in accordance with ASC 260, Earnings per
Share, by dividing net income or loss attributable to common
stockholders by the weighted average common stock outstanding.
Diluted EPS is calculated in accordance with ASC 260 by
adjusting weighted average common shares outstanding for the
dilutive effect of common stock options, warrants, convertible
preferred stock and accrued but unpaid convertible preferred
stock dividends. In periods where a net loss is recorded, no
effect is given to potentially dilutive securities, since the
effect would be anti-dilutive. Total securities that could
potentially dilute basic EPS in the future were not included in
the computation of diluted EPS because to do so would have been
anti-dilutive were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Series A preferred stock and convertible accrued dividends
|
|
|
2,245,484
|
|
|
|
7,005,145
|
|
|
|
7,005,145
|
|
|
Series B preferred stock
|
|
|
2,291,242
|
|
|
|
7,147,894
|
|
|
|
7,147,894
|
|
|
Series C preferred stock
|
|
|
1,861,457
|
|
|
|
5,807,112
|
|
|
|
4,570,674
|
|
|
Series C-1
preferred stock
|
|
|
888,298
|
|
|
|
339,408
|
|
|
|
—
|
|
|
Series C-1
preferred stock warrants
|
|
|
42,426
|
|
|
|
678,820
|
|
|
|
—
|
|
|
Common stock warrants
|
|
|
97,757
|
|
|
|
45,297
|
|
|
|
30,271
|
|
|
Stock options
|
|
|
1,704,787
|
|
|
|
1,125,916
|
|
|
|
987,170
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
9,131,451
|
|
|
|
22,149,592
|
|
|
|
19,741,154
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reporting Segments — The Company does not
report segment information as it operates in only one business
segment.
Promotional and Advertising Costs — Promotional
and advertising costs are expensed as incurred.
Recent Accounting Pronouncements — In January
2010, the FASB issued amendments to the existing fair value
measurements and disclosures guidance which requires new
disclosures and clarifies existing disclosure requirements. The
purpose of these amendments is to provide a greater level of
disaggregated information as well as more disclosure around
valuation techniques and inputs to fair value
89
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
measurements. The guidance was effective commencing with the
Company’s 2010 fiscal year. The adoption of this guidance
did not have a material impact on the Company’s financial
statements.
|
|
|
3.
|
DISCONTINUED
OPERATIONS
|
In October 2006, management and the board of directors of the
Company approved a plan to discontinue the operations of its
non-prescription ophthalmic pharmaceutical business (the OTC
Business). The plan included the sale of the assets of the
Company’s OTC Business and also the termination of its
sales and marketing personnel. The Company previously determined
that the discontinued OTC Business comprised operations and cash
flows that could be clearly distinguished, operationally and for
financial reporting purposes, from the rest of the Company.
Accordingly, the results of operations for the discontinued OTC
Business have been presented as discontinued operations. During
the year ended December 31, 2010 the Company received a
$4,000,000 option payment from the acquirer of the assets of the
OTC Business to provide it with an additional two years to
develop one of the acquired products. There were no revenues or
expenses from discontinued operations during the years ended
December 31, 2009 and 2008. The following table presents
basic and diluted earnings per share from discontinued
operations for the year ended December 31, 2010 (in
thousands except share and per share data):
| |
|
|
|
|
|
Net income from discontinued operations
|
|
$
|
4,000
|
|
|
Net income from discontinued operations per share —
Basic and diluted
|
|
$
|
0.18
|
|
|
Weighted-average shares outstanding — Basic and diluted
|
|
|
22,167,873
|
|
|
|
|
4.
|
FACTORS
AFFECTING OPERATIONS
|
To date the Company has incurred recurring losses, negative cash
flow from operations, and has accumulated a deficit of
$188,854,000 from the Company’s inception through
December 31, 2010. The Company does not expect to generate
revenues from its product, ILUVIEN, until late 2011, if at all,
and therefore does not expect to have cash flow from operations
until 2012, if at all. As of December 31, 2010 the Company
had approximately $54,844,000 in cash, cash equivalents, and
investments. In October 2010, the Company obtained a $32,500,000
senior secured credit facility (Credit Facility) to help fund
its working capital requirements (see Note 9). The Credit
Facility consists of a $20,000,000 revolving line of credit and
a $12,500,000 term loan. The lenders have advanced $6,250,000
under the term loan and may advance the remaining $6,250,000
following FDA approval of Iluvien, but no later than
July 31, 2011. Given the status of the FDA’s review of
the ILUVIEN NDA, it is unlikely that the FDA approval would
occur prior to July 31, 2011. The Company is currently in
discussions with the lenders to amend the terms of the Credit
Facility to, among other things, extend the availability of the
term loan. However, there are no assurances that the Credit
Facility will be amended. The Company may draw on the revolving
line of credit against eligible, domestic accounts receivable
subsequent to the launch of ILUVIEN. Management believes it has
sufficient funds available to fund its operations through the
projected commercialization of ILUVIEN and the expected
generation of revenue in late 2011. The commercialization of
Iluvien is dependent upon approval by the FDA, however, and
management cannot be sure that ILUVIEN will be approved by the
FDA or that, if approved, future sales of ILUVIEN will generate
enough revenue to fund the Company’s operations beyond its
commercialization (see Note 1). Due to the uncertainty
around FDA approval, management also cannot be certain that the
Company will not need additional funds for the commercialization
of ILUVIEN. If ILUVIEN is not approved, or if approved, does not
generate sufficient revenue, the Company may adjust its
commercial plans so that it can continue to operate with its
existing cash resources or seek to raise additional financing.
These matters raise substantial doubt about the Company’s
ability to continue as a going concern. The accompanying
financial statements do not include any adjustments that may
result from the outcome of these uncertainties.
90
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
|
|
|
5.
|
PROPERTY
AND EQUIPMENT
|
Property and equipment consisted of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Furniture and fixtures
|
|
$
|
292
|
|
|
$
|
290
|
|
|
Office equipment
|
|
|
415
|
|
|
|
290
|
|
|
Software
|
|
|
489
|
|
|
|
470
|
|
|
Leasehold improvements
|
|
|
12
|
|
|
|
12
|
|
|
Manufacturing equipment
|
|
|
40
|
|
|
|
40
|
|
|
|
|
|
|
|
|
|
|
|
|
Total property and equipment
|
|
|
1,248
|
|
|
|
1,102
|
|
|
Less accumulated depreciation and amortization
|
|
|
(1,028
|
)
|
|
|
(848
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment — net
|
|
$
|
220
|
|
|
$
|
254
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization expense associated with property
and equipment of the continuing operations totaled $194,000,
$1,098,000 and $241,000 for the years ended December 31,
2010, 2009 and 2008, respectively.
During the year ended December 31, 2009, the Company
recognized $860,000 of depreciation and amortization expense
associated with equipment used for the manufacture of
registration batches of ILUVIEN.
Accrued expenses consisted of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Accrued clinical investigator expenses
|
|
$
|
1,911
|
|
|
$
|
3,007
|
|
|
Accrued compensation expenses
|
|
|
730
|
|
|
|
246
|
|
|
Other accrued expenses
|
|
|
90
|
|
|
|
61
|
|
|
|
|
|
|
|
|
|
|
|
|
Total accrued expenses
|
|
$
|
2,731
|
|
|
$
|
3,314
|
|
|
|
|
|
|
|
|
|
|
|
In March 2008, in connection with the Company’s
collaboration agreement with pSivida US, Inc. (pSivida), the
licensor of the ILUVIEN technology, the Company and pSivida
amended and restated the agreement to provide us with 80% of the
net profits and pSivida with 20% of the net profits. In
connection with the amended and restated agreement , the Company
also agreed to:
|
|
|
| |
•
|
pay $12.0 million to pSivida upon the execution of the
March 2008 agreement;
|
| |
| |
•
|
issue a $15.0 million promissory note to pSivida;
|
| |
| |
•
|
forgive all outstanding development payments, penalties and
interest as of the effective date of the March 2008 agreement,
which totaled $6.8 million;
|
| |
| |
•
|
continue responsibility for regulatory, clinical, preclinical,
manufacturing, marketing and sales for the remaining development
and commercialization of the products;
|
91
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
|
|
|
| |
•
|
assume all financial responsibility for the development of the
products and assume 80% of the commercialization costs of the
products (instead of 50% as provided under the February 2005
agreement); and
|
| |
| |
•
|
make an additional milestone payment of $25.0 million after
the first product under the March 2008 agreement has been
approved by the FDA.
|
In addition, pSivida is continuing to provide clinical supply
materials for the Company’s Phase 2 clinical trials being
conducted for the use of ILUVIEN for the treatment of dry AMD
and wet AMD and perform and maintain stability testing on those
supplies.
The $15,000,000 promissory note accrued interest at 8% payable
quarterly and was payable in full to pSivida upon the earlier of
a liquidity event as defined in the note (including an initial
public offering of the Company’s common stock greater than
$75,000,000), the occurrence of an event of default under the
Company’s agreement with pSivida or September 30,
2012. If the note was not paid in full by March 31, 2010,
the interest rate was to increase to 20% effective as of
April 1, 2010, and the Company would be required to begin
making principal payments of $500,000 per month. As of
December 31, 2009, the Company had accrued and unpaid
interest payable to pSivida of $708,000, classified as other
long-term liabilities, and $543,000, included in accrued
interest in the accompanying balance sheet. On April 27,
2010, the Company paid pSivida approximately $15,200,000 in
principal and interest to satisfy the note payable with the
proceeds from its initial public offering. As a result, the
Company recognized a gain of $1,343,000 on the extinguishment of
this debt in the accompanying financial statements for the year
ended December 31, 2010.
The Company’s license rights to pSivida’s proprietary
delivery device could revert to pSivida if the Company were to
(i) fail twice to cure its breach of an obligation to make
certain payments to pSivida following receipt of written notice
thereof; (ii) fail to cure other breaches of material terms
of its agreement with pSivida within 30 days after notice
of such breaches or such longer period (up to 90 days) as
may be reasonably necessary if the breach cannot be cured within
such 30-day
period; (iii) file for protection under the bankruptcy
laws, make an assignment for the benefit of creditors, appoint
or suffer appointment of a receiver or trustee over its
property, file a petition under any bankruptcy or insolvency act
or have any such petition filed against it and such proceeding
remains undismissed or unstayed for a period of more than
60 days; or (iv) notify pSivida in writing of its
decision to abandon its license with respect to a certain
product using pSivida’s proprietary delivery device. The
Company was not in breach of its agreement with pSivida as of
December 31, 2010.
Upon commercialization of ILUVIEN, the Company must share 20% of
net profits, as defined by the agreement, with pSivida. In
connection with this arrangement the Company is entitled to
recover 20% of commercialization costs decreased from 50% as a
result of the amendment, as defined in the amendment, incurred
prior to product profitability out of pSivida’s share of
net profits. As of December 31, 2010 and 2009, the Company
was owed $2,224,000 and $958,000, respectively, in
commercialization costs. Due to the uncertainty of FDA approval
of the NDA for ILUVIEN, the Company has fully reserved these
amounts in the accompanying financial statements.
In November 2007, the Company entered into a license agreement
with Dainippon Sumitomo Pharma Co., Ltd. (Dainippon) whereby
Dainippon granted the Company a non-exclusive, worldwide,
royalty free license to patent rights under specific patents and
patent applications. The Company paid $200,000 to Dainippon
shortly after the execution of this license agreement and will
be required to make an additional payment in the amount of
$200,000 to Dainippon within 30 days following the first
regulatory approval of a licensed product in the U.S. by
the FDA.
92
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
In August 2007, the Company entered into an exclusive option
agreement with Emory University for the licensing of certain
patents for a class of compounds that the Company intends to
evaluate for the treatment of diseases of the eye, primarily the
dry form of age related macular degeneration. The Company made
an initial payment of $75,000 during the year ended
December 31, 2007 for the option to license the compounds
at the end of an evaluation period. The Company exercised its
option and entered into an exclusive license in the field of
ophthalmology in July 2009, and issued Emory University and its
inventor $150,000 in common stock based on the estimated fair
value at the time of issuance. The Company would owe Emory
University up to $5,775,000 in additional development and
regulatory milestones under the terms of the license agreement.
If the Company does not make any milestone payments to Emory
University under this license agreement prior to the third
anniversary of its effective date, and the Company does not
elect to terminate this license agreement in accordance with its
terms, then the Company will be required to pay Emory University
annual license maintenance fees ranging from $500,000 to
$2,000,000 (depending on when such payment is made) until a
milestone payment is made or this license agreement is
terminated in accordance with its terms. Under the terms of the
Company’s agreement with Emory University, the Company is
required to make annual minimum royalty payments in the first
through the fourth calendar years following regulatory approval
of the product in a major market country (i.e., the U.S., Japan,
China, India or any European country) in the amount of $250,000,
$500,000, $1,000,000 and $2,500,000, respectively, and an annual
minimum royalty payment of $2,500,000 for each subsequent year
during the term of the agreement. As part of this license, the
Company received an exclusive option for a license of the patent
rights for diseases and disorders outside of the eye.
In February 2008, the Company entered into a similar exclusive
option agreement with Emory University for the patent rights to
a second class of compounds which will be evaluated for the
treatment of diseases of the eye, primarily the dry form of age
related macular degeneration. The initial payment was $60,000.
The Company expensed this amount as research and development
expense in February 2008. The Company exercised its option and
entered into an exclusive license in the field of ophthalmology
in August 2009, and issued Emory University and its inventor
$150,000 in common stock based on the estimated fair value at
the time of issuance in December 2009. The Company would owe
Emory University up to $5,850,000 in additional development and
regulatory milestones under the terms of this license agreement.
If the Company does not make any milestone payments to Emory
University under this license agreement prior to the third
anniversary of its effective date, and the Company does not
elect to terminate this license agreement in accordance with its
terms, then the Company will be required to pay Emory University
annual license maintenance fees ranging from $500,000 to
$2,000,000 (depending on when such payment is made) until a
milestone payment is made or this license agreement is
terminated in accordance with its terms. Under the terms of the
Company’s agreement with Emory University, the Company is
required to make annual minimum royalty payments in the first
through the fourth calendar years following regulatory approval
of the product in a major market country (i.e., the U.S., Japan,
China, India or any European country) in the amount of $250,000,
$500,000, $1,000,000 and $2,500,000, respectively, and an annual
minimum royalty payment of $2,500,000 for each subsequent year
during the term of the agreement. As part of this license, the
Company received an exclusive option for a license of the patent
rights for diseases and disorders outside of the eye.
|
|
|
9.
|
TERM AND
REVOLVING LOAN AGREEMENT
|
Term
Loan Agreement
On October 14, 2010 (Effective Date), the Company entered
into a Loan and Security Agreement with Silicon Valley Bank and
MidCap Financial LLP under which the Company may borrow up to
$12,500,000 (Term Loan Agreement). The lenders advanced the
Initial Tranche of $6,250,000 on the Effective Date and may
advance the remaining Second Tranche of $6,250,000 following
approval by the FDA of the Company’s ILUVIEN product, but
no later than July 31, 2011. Given the status of the
FDA’s review of the Company’s NDA, the Company
believes it is unlikely that approval of ILUVIEN would occur
prior to July 31, 2011. The Company is in discussions with
the lenders to amend the terms of the Term Loan Agreement to,
among other
93
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
things, extend its availability. However, there are no
assurances that the Term Loan Agreement will be amended.
The Company is required to maintain its primary operating and
other deposit accounts and securities accounts with Silicon
Valley Bank, which accounts must represent at least 50% of the
dollar value of the Company’s accounts at all financial
institutions.
The Company will be required to pay interest on the First
Tranche at a rate of 11.5% on a monthly basis through
July 31, 2011, and then will be required to repay the
principal in 27 equal monthly installments, beginning August
2011, plus interest at a rate of 11.5%. If the Second Tranche is
advanced to the Company, the Company will be required to pay
interest on the Second Tranche at a rate of 12.0% on a monthly
basis through July 31, 2011, and then will be required to
repay the principal in 27 equal monthly installments, plus
interest at a rate of 12.0%. The Company paid to the lenders an
upfront fee of $62,500, and will pay to the lenders an
additional final payment of 3% of the total principal amount. In
addition, if the Company repays the loan prior to maturity, it
will pay to the lenders a prepayment penalty of 5% of the total
principal amount if the prepayment occurs within one year after
the funding date, 3% of the total principal amount if the
prepayment occurs between one and two years after the funding
date and 1% of the total principal amount of the prepayment
occurs thereafter (each a Prepayment Penalty), provided in each
case that such Prepayment Penalty will be reduced by 50% in the
event of an acquisition of the Company. The occurrence of an
event of default could result in the acceleration of the
Company’s obligations under the Term Loan Agreement and an
increase to the applicable interest rate, and would permit the
lenders to exercise remedies with respect to the collateral
under the Term Loan Agreement.
To secure the repayment of any amounts borrowed under the Term
Loan Agreement, the Company granted to the Lenders a first
priority security interest in all of its assets, other than its
intellectual property, provided that, for any date during which
the notes are outstanding, the Company’s unrestricted
balance sheet cash and cash equivalents plus the excess
available under the term loan agreements are less than the
product of six times the monthly cash burn amount. In the event
the Company fails to meet this financial condition, a curable
lien will be imposed on the Company’s intellectual
property. Should such a lien event take place, the lien would
remain in force until such date that the Company’s
unrestricted cash and cash equivalents plus the excess available
under the term loan agreements was equal to or greater than
twelve times the monthly cash burn amount. The Company also
agreed not to pledge or otherwise encumber its intellectual
property assets. Additionally, the Company must seek the
lenders’ approval prior to the payment of any cash
dividends.
In connection with entering into this agreement, the Company
issued to the lenders warrants to purchase an aggregate of up to
39,773 shares of the Company’s common stock. Each of
the warrants is exercisable immediately, has a per-share
exercise price of $11.00 and has a term of 10 years. In
addition, the lenders will have certain registration rights with
respect to the shares of common stock issuable upon exercise of
the warrants. The Company estimated the fair value of warrants
granted using the Black-Scholes option pricing model. The
aggregate fair value of the warrants was estimated to be
$389,000. The Company allocated a portion of the proceeds from
the Term Loan Agreement to the warrants in accordance with
ASC 470-20-25-2,
Debt Instruments with Detachable Warrants. As a result,
the Company recorded a discount of $366,000 which is being
amortized to interest expense using the effective interest
method.
Revolving
Loan Agreement
Also on the Effective Date, the Company and Silicon Valley Bank
entered into a Loan and Security Agreement (Revolving Loan
Agreement), pursuant to which the Company obtained a secured
revolving line of credit from Silicon Valley Bank with borrowing
availability up to $20,000,000.
94
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The Revolving Loan Agreement provides for a working
capital-based revolving line of credit (Revolving Line) in an
aggregate amount of up to the lesser of (i) $20,000,000, or
(ii) 85% of eligible domestic accounts receivable. The
Revolving Line matures on October 31, 2013. As of
December 31, 2010, no amounts under the Revolving Loan
Agreement were available to the Company.
Amounts advanced under the Revolving Line bear interest at an
annual rate equal to Silicon Valley Bank’s prime rate plus
2.50% (with a rate floor of 6.50%). Interest on the Revolving
Line is due monthly, with the balance due at the maturity date.
The Company paid to Silicon Valley Bank an upfront fee of
$100,000. In addition, if the Company terminates the Revolving
Line prior to maturity, it will pay to Silicon Valley Bank a fee
of $400,000 if the termination occurs within one year after the
Effective Date and a fee of $200,000 if the termination occurs
more than one year after the Effective Date (each a Termination
Fee), provided in each case that such Termination Fee will be
reduced by 50% in the event of an acquisition of the Company.
To secure the repayment of any amounts borrowed under the
Revolving Loan Agreement, the Company granted to Silicon Valley
Bank a first priority security interest in all of its assets,
other than its intellectual property, provided that, for any
date during which the notes are outstanding, the Company’s
unrestricted balance sheet cash and cash equivalents plus the
excess available under the term loan agreements are less than
the product of six times the monthly cash burn amount. In the
event the Company fails to meet this financial condition, a
curable lien will be imposed on the Company’s intellectual
property. Should such a lien event take place, the lien would
remain in force until such date that the Company’s
unrestricted cash and cash equivalents plus the excess available
under the term loan agreements was equal to or greater than
twelve times the monthly cash burn amount. The Company also
agreed not to pledge or otherwise encumber its intellectual
property assets. Additionally, the Company must seek Silicon
Valley Bank’s approval prior to the payment of any cash
dividends.
The occurrence of an event of default could result in the
acceleration of the Company’s obligations under the
Revolving Loan Agreement and an increase to the applicable
interest rate, and would permit Silicon Valley Bank to exercise
remedies with respect to the collateral under the Revolving Loan
Agreement.
Term Notes Payable — In October 2010, the
Company received proceeds of $6,250,000 from the issuance of
Notes Payable to certain lenders (see Note 9). As of
December 31, 2010 a schedule of future minimum payments
under the Notes Payable is as follows (in thousands):
| |
|
|
|
|
|
Years Ending December 31
|
|
|
|
|
|
|
2011
|
|
$
|
1,157
|
|
|
2012
|
|
|
2,778
|
|
|
2013
|
|
|
2,315
|
|
|
|
|
|
|
|
|
|
|
$
|
6,250
|
|
|
|
|
|
|
|
The effective interest rate on the Notes Payable is 11.5%. As of
December 31, 2010, the Company had no accrued and unpaid
interest payables on the Notes Payable.
Operating Leases — The Company leases office
space and equipment under noncancelable agreements accounted for
as operating leases. The leases generally require that the
Company pay taxes, maintenance, and insurance. Management
expects that in the normal course of business, leases that
expire will be renewed or replaced by other leases. In July
2010, the Company signed an extension of its lease for office
space for a period ended December 31, 2011. The
Company’s future minimum payments under this operating
lease from December 31, 2010 to December 31, 2011 are
$262,000.
95
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Rent expense under all operating leases totaled approximately
$256,000, $229,000 and $217,000 for the years ended
December 31, 2010, 2009, and 2008, respectively.
Capital Leases — The Company leases equipment
under capital leases. The property and equipment is capitalized
at the lesser of fair market value or the present value of the
minimum lease payments at the inception of the leases using the
Company’s incremental borrowing rate.
At December 31, 2010, a schedule by year of future minimum
payments under capital leases, together with the present value
of minimum lease payments, is as follows (in thousands):
| |
|
|
|
|
|
Years Ending December 31
|
|
|
|
|
|
|
2011
|
|
$
|
13
|
|
|
2012
|
|
|
13
|
|
|
2013
|
|
|
6
|
|
|
|
|
|
|
|
|
Total
|
|
|
32
|
|
|
Less amount representing interest
|
|
|
3
|
|
|
|
|
|
|
|
|
Present value of minimum lease payments
|
|
|
29
|
|
|
Less current portion
|
|
|
11
|
|
|
|
|
|
|
|
|
Noncurrent portion
|
|
$
|
18
|
|
|
|
|
|
|
|
Property and equipment under capital leases, which are included
in property and equipment (see Note 5), consisted of the
following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Office equipment
|
|
$
|
60
|
|
|
$
|
42
|
|
|
Less accumulated amortization
|
|
|
(32
|
)
|
|
|
(37
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
28
|
|
|
$
|
5
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation expense associated with office equipment under
capital leases was $10,000 for each of the years ended
December 31, 2010, 2009, and 2008, respectively.
Significant Agreements — In January 2006, the
Company entered into an agreement with a contract research
organization for clinical and data management services to be
performed in connection with the Phase 3 trial product for the
treatment of DME in the U.S., Canada, and Europe. In accordance
with the terms of the agreement, the Company will incur
approximately $16,900,000 in costs with the contract research
organization through 2011. For the years ended December 31,
2010, 2009, and 2008, the Company incurred $2,300,000,
$3,900,000, and $3,300,000 respectively, of expense associated
with this agreement. At December 31, 2010 and 2009,
$731,000 and $1,100,000, respectively, are included in
outsourced services payable.
In July 2006, the Company entered into an agreement with a
contract research organization for clinical services to be
performed in connection with the Phase 3 trial of its product
for the treatment of DME in India. In accordance with the terms
of the agreement, the Company will incur approximately
$1,800,000 in costs with the contract research organization
through 2011. For the years ended December 31, 2010, 2009,
and 2008, the Company incurred $242,000, $240,000, and $248,000,
respectively, of expense associated with this agreement. At
December 31, 2010 and 2009, $110,000, and $53,000,
respectively, are included in outsourced services payable.
In February 2010, the Company entered into an agreement with a
third party manufacturer for the manufacture of the ILUVIEN
insert, the assembly of the ILUVIEN inserter and packaging of
the completed
96
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
ILUVIEN commercial product. The Company is responsible for
supplying the ILUVIEN inserter and the active pharmaceutical
ingredient. In accordance with the terms of the agreement, the
Company must order at least 80% of the ILUVIEN units required in
the U.S., Canada and the European Union from the third party
manufacturer for an initial term of six years. The agreement
with has an initial six year term and will automatically renew
for successive one year periods unless either party delivers
written notice of non-renewal to the other at least
12 months prior to the end of the current term.
Employment Agreements — The Company is party to
employment agreements with five executives. The agreements
generally provide for annual salaries, bonuses, and benefits for
a period of three years, and automatically renew for one-year
periods after the third year unless terminated by either party.
Effective January 1, 2010, the salaries ranged from
$227,000 to $368,000. Effective January 1, 2011, the
salaries were adjusted to a range of $254,000 to $432,000. If
any of the agreements are terminated by the Company without
cause, or by the employee for good reason, as defined in the
agreements, the Company will be liable for one year of salary
and benefits. Certain other employees have general employment
contracts which include stipulations regarding confidentiality,
Company property, and miscellaneous items.
Prior to the Company’s IPO, the Company had four series of
preferred stock. On April 27, 2010 and in connection with
the IPO, all outstanding shares of the Company’s preferred
stock were converted into
97
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
22,863,696 shares of common stock and all preferred stock
dividends were eliminated. Significant terms of all series of
the preferred stock were as follows:
|
|
|
| |
•
|
Dividends were cumulative and accrued on a daily basis at the
rate of 8% per annum beginning on the date of issuance and based
on the original issue price, as adjusted for any stock dividend,
stock split, combination, or other event involving the preferred
stock. Dividends accrued, whether or not declared, annually and
were due and payable when and if declared by the Board of
Directors, upon a liquidating event, as defined in the
Company’s certificate of incorporation, upon redemption of
the preferred stock, as defined in the Company’s
certificate of incorporation, or on the date that the preferred
stock was otherwise acquired by the Company. Accumulated,
accrued, and unpaid dividends were $23,934,000 at
December 31, 2009.
|
| |
| |
•
|
Upon any liquidation, dissolution, or winding up of the Company,
the preferred stockholders were entitled to a liquidation
preference payment equal to (i) the sum of the liquidation
value plus all accumulated, accrued, and unpaid dividends and
(ii) the pro rata share of any remaining amounts such
holder would have been entitled to receive had such
holder’s shares been converted into common stock
immediately prior to the liquidation, dissolution, or winding
up. The liquidation value plus accumulated, accrued, and unpaid
dividends was $117,497,000 at December 31, 2009.
|
| |
| |
•
|
At any time subsequent to March 17, 2013, the holders of a
majority of the preferred stock could have required the Company
to redeem all or any portion of the preferred stock. If the
preferred stock was redeemed, the redemption would have occurred
in equal installments over a three-year period. The price paid
by the Company to redeem the shares would have been the greater
of (i) the original issue price, plus all accumulated,
accrued, and unpaid dividends, and (ii) the fair market
value of the preferred stock being redeemed at the time of the
redemption.
|
Because the preferred stock provided the holders the right to
require the Company to redeem such shares for cash after
March 17, 2013 at the greater of (i) the original
issue price plus any accrued but unpaid dividends and
(ii) the fair market value of the preferred stock being
redeemed, the embedded conversion feature required separate
accounting. Consequently, the conversion feature had to be
bifurcated from the preferred stock and accounted for separately
at each issuance date. The carrying value of the embedded
derivative was adjusted to fair value at the end of each
reporting period and the change in fair value was recognized in
the statement of operations.
On January 8, 2010 warrants to purchase shares of the
Company’s
Series C-1
preferred stock were exercised resulting in $10,000,000 in cash
proceeds and the issuance of 1,935,700 additional shares of
Series C-1
preferred stock. The Company recorded a derivative liability of
$3,471,000 upon the exercise of the warrants and the issuance of
such shares of Series C-1 preferred stock in January 2010.
At each reporting date and in connection with the Company’s
IPO, the Company adjusted the carrying value of the embedded
derivatives to estimated fair value and recognized the change in
such estimated value in its statement of operations. The
estimated fair value of the derivatives at December 31,
2009 was $36,701,000. The estimated fair value of the
derivatives at April 27, 2010, immediately prior to the
conversion of the preferred stock to common stock in connection
with the IPO, was $36,528,000. In connection with the IPO, the
embedded derivatives were eliminated. The Company recognized a
gain of $3,644,000, a loss of $23,142,000, and a loss of
$10,454,000 associated with the change in fair value for the
years ended December 31, 2010, 2009, and 2008 respectively.
In connection with the conversion of the Company’s
preferred shares to common shares upon the completion of the
Company’s IPO in April 2010, the Company authorized
10,000,000 shares of $0.01 par value preferred stock.
At December 31, 2010, no shares of preferred stock were
issued or outstanding.
98
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The Company has stock option and stock incentive plans which
provide for grants of shares to employees and grants of options
to employees and directors to purchase shares of the
Company’s common stock at exercise prices generally equal
to the fair values of such stock at the dates of grant. Options
granted to employees typically become exercisable over a
four-year vesting period and have a
10-year
term. Options granted to directors typically vest immediately
and have a
5-year term.
As of December 31, 2010, the Company was authorized to
grant under the Company’s plans up to 1,402,536 shares
under the 2010 Equity Incentive Plan. Upon the exercise of stock
options, the Company may issue the required shares out of
authorized but unissued common stock or out of treasury stock at
management’s discretion.
A summary of stock option transactions under the plans are as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
Average
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
Exercise
|
|
|
|
|
|
Exercise
|
|
|
|
|
|
Exercise
|
|
|
|
|
Options
|
|
|
Price
|
|
|
Options
|
|
|
Price
|
|
|
Options
|
|
|
Price
|
|
|
|
|
Options at beginning of period
|
|
|
2,225,778
|
|
|
$
|
2.14
|
|
|
|
1,959,726
|
|
|
$
|
1.80
|
|
|
|
1,419,808
|
|
|
$
|
1.50
|
|
|
Grants
|
|
|
575,150
|
|
|
|
10.17
|
|
|
|
295,463
|
|
|
|
4.35
|
|
|
|
539,918
|
|
|
|
2.55
|
|
|
Forfeitures
|
|
|
(14,444
|
)
|
|
|
5.11
|
|
|
|
(25,551
|
)
|
|
|
1.84
|
|
|
|
—
|
|
|
|
—
|
|
|
Exercises
|
|
|
(44,499
|
)
|
|
|
2.10
|
|
|
|
(3,860
|
)
|
|
|
1.70
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options at end of period
|
|
|
2,741,985
|
|
|
|
3.81
|
|
|
|
2,225,778
|
|
|
|
2.14
|
|
|
|
1,959,726
|
|
|
|
1.80
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options exercisable at period end
|
|
|
1,722,281
|
|
|
|
1.88
|
|
|
|
1,427,649
|
|
|
|
1.70
|
|
|
|
921,055
|
|
|
|
1.70
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average per share fair value of options granted during
the period
|
|
$
|
7.92
|
|
|
|
|
|
|
$
|
3.74
|
|
|
|
|
|
|
$
|
1.73
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The following table provides additional information related to
outstanding stock options, fully vested stock options, and stock
options expected to vest as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
Weighted
|
|
|
|
|
|
|
|
Average
|
|
Average
|
|
Aggregate
|
|
|
|
|
|
Exercise
|
|
Contractual
|
|
Intrinsic
|
|
|
|
Shares
|
|
Price
|
|
Term
|
|
Value
|
|
|
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
Outstanding
|
|
|
2,741,985
|
|
|
$
|
3.81
|
|
|
|
6.99 years
|
|
|
$
|
18,338
|
|
|
Exercisable
|
|
|
1,722,281
|
|
|
|
1.88
|
|
|
|
5.88 years
|
|
|
|
14,638
|
|
|
Expected to vest
|
|
|
963,754
|
|
|
|
7.24
|
|
|
|
8.92 years
|
|
|
|
3,334
|
|
99
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The Company estimated the fair value of options granted using
the Black-Scholes option-pricing model with the following
weighted-average assumptions used for option grants:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
Risk-free interest rate
|
|
|
1.77
|
%
|
|
|
3.44
|
%
|
|
|
2.87
|
%
|
|
Volatility factor
|
|
|
97.32
|
%
|
|
|
112.57
|
%
|
|
|
73.78
|
%
|
|
Grant date fair value of common stock
|
|
|
$7.92
|
|
|
|
$4.35
|
|
|
|
$2.55
|
|
|
Weighted-average expected life
|
|
|
6.05 years
|
|
|
|
6.18 years
|
|
|
|
6.15 years
|
|
|
Assumed forfeiture rate
|
|
|
10.00
|
%
|
|
|
10.00
|
%
|
|
|
10.00
|
%
|
Employee stock-based compensation expense related to stock
options recognized under ASC 718 was as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Marketing
|
|
$
|
127
|
|
|
$
|
43
|
|
|
$
|
109
|
|
|
Research and development
|
|
|
209
|
|
|
|
161
|
|
|
|
269
|
|
|
General and administrative
|
|
|
458
|
|
|
|
307
|
|
|
|
372
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total employee stock-based compensation expense
|
|
$
|
794
|
|
|
$
|
511
|
|
|
$
|
750
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The total estimated fair value of options granted during the
years ended December 31, 2010, 2009, and 2008 was
$4,558,000, $1,100,000, and $930,000, respectively, and the
total estimated value of options granted prior to 2007 was
$1,516,000.
The Company’s 2010 Plan provides for annual increases in
the number of shares available for issuance thereunder on the
first day of each fiscal year equal to the least of:
(1) 2,000,000 shares of our common stock; (2) 4%
of the shares of common stock outstanding at that time; and
(3) such other amount as our board of directors may
determine. On January 1, 2011, an additional
1,250,238 shares became available for future issuance under
the 2010 Plan. These additional shares from the annual increase
under the 2010 Plan are not included in the foregoing discussion.
100
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The following table summarizes outstanding and exercisable
options at December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options Outstanding
|
|
|
Options Exercisable
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
Remaining
|
|
|
|
|
Number
|
|
|
Contractual
|
|
|
Number
|
|
|
Contractual
|
|
|
Exercise Prices
|
|
Outstanding
|
|
|
Life
|
|
|
Exercisable
|
|
|
Life
|
|
|
|
|
$1.33
|
|
|
693,458
|
|
|
|
5.36
|
|
|
|
693,458
|
|
|
|
5.36
|
|
|
1.39
|
|
|
395,400
|
|
|
|
6.79
|
|
|
|
310,696
|
|
|
|
6.76
|
|
|
2.04
|
|
|
280,770
|
|
|
|
3.67
|
|
|
|
280,770
|
|
|
|
3.67
|
|
|
2.24
|
|
|
4,504
|
|
|
|
7.17
|
|
|
|
1,287
|
|
|
|
7.17
|
|
|
2.41
|
|
|
465,212
|
|
|
|
7.22
|
|
|
|
318,906
|
|
|
|
7.22
|
|
|
3.26
|
|
|
5,882
|
|
|
|
7.39
|
|
|
|
3,676
|
|
|
|
7.39
|
|
|
3.88
|
|
|
33,823
|
|
|
|
7.09
|
|
|
|
22,794
|
|
|
|
7.09
|
|
|
4.01
|
|
|
262,963
|
|
|
|
8.60
|
|
|
|
74,695
|
|
|
|
8.47
|
|
|
5.03
|
|
|
5,146
|
|
|
|
7.66
|
|
|
|
2,893
|
|
|
|
7.66
|
|
|
5.44
|
|
|
2,059
|
|
|
|
8.67
|
|
|
|
1,157
|
|
|
|
8.67
|
|
|
6.74
|
|
|
103,500
|
|
|
|
9.62
|
|
|
|
—
|
|
|
|
—
|
|
|
8.47
|
|
|
22,618
|
|
|
|
8.96
|
|
|
|
5,648
|
|
|
|
8.96
|
|
|
9.87
|
|
|
75,000
|
|
|
|
9.76
|
|
|
|
—
|
|
|
|
—
|
|
|
11.00
|
|
|
65,000
|
|
|
|
9.33
|
|
|
|
—
|
|
|
|
—
|
|
|
11.15
|
|
|
302,650
|
|
|
|
9.85
|
|
|
|
6,302
|
|
|
|
9.85
|
|
|
11.91
|
|
|
24,000
|
|
|
|
9.93
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,741,985
|
|
|
|
|
|
|
|
1,722,282
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
13.
|
COMMON
STOCK WARRANTS
|
The Company has issued warrants to purchase common stock to
various members of the board of directors, third-parties for
services, and lenders. Total warrants to purchase common stock
issued and outstanding were 87,420 and 248,181 at
December 31, 2010 and 2009, respectively, at exercise
prices ranging from $1.70 to $11.00 per share. The warrants are
exercisable for a period of seven to ten years from the issuance
date.
Warrants to purchase 39,773 of the Company’s common stock
were granted during the year ended December 31, 2010 in
connection the issuance of the term and revolving loan agreement
(see Note 9). No warrants to purchase common stock were
issued in the years ended December 31, 2009 and 2008,
respectively.
101
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
The components of the income tax benefit were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Deferred benefit (expense):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal
|
|
$
|
5,738
|
|
|
$
|
6,649
|
|
|
$
|
17,119
|
|
|
State
|
|
|
669
|
|
|
|
774
|
|
|
|
2,202
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6,407
|
|
|
|
7,423
|
|
|
|
19,321
|
|
|
Valuation allowance
|
|
|
(6,407
|
)
|
|
|
(7,423
|
)
|
|
|
(19,321
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income tax benefit
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
In accordance with ASC 740, the Company recognizes deferred
tax assets and liabilities for temporary differences between the
financial reporting basis and the tax basis of its assets and
liabilities. The Company records a valuation allowance against
its net deferred tax asset to reduce the net carrying value to
an amount that is more likely than not to be realized.
Income tax positions are considered for uncertainty in
accordance with
ASC 740-10.
The Company believes that its income tax filing positions and
deductions are more likely than not of being sustained on audit
and does not anticipate any adjustments that will result in a
material change to its financial position; therefore, no
ASC 740-10
liabilities and no related penalties and interest have been
recorded. Tax years since 2003 remain subject to examination in
Georgia, Tennessee, and on the federal level. The Company does
not anticipate any material changes to its uncertain tax
positions within the next 12 months.
Significant management judgment is involved in determining the
provision for income taxes, deferred tax assets and liabilities,
and any valuation allowance recorded against net deferred tax
assets. Due to uncertainties with respect to the realization of
deferred tax assets due to the history of operating losses, a
valuation allowance has been established against the entire net
deferred tax asset balance. The valuation allowance is based on
management’s estimates of taxable income in the
jurisdictions in which the Company operates and the period over
which deferred tax assets will be recoverable. In the event that
actual results differ from these estimates or the Company
adjusts these estimates in future periods, a change in the
valuation allowance may be needed, which could materially impact
the Company’s financial position and results of operations.
At December 31, 2010 and 2009, the Company had federal net
operating loss (NOL) carry-forwards of approximately $97,813,000
and $79,494,000 and state NOL carry-forwards of approximately
$80,995,000, and $62,666,000 respectively, that are available to
reduce future income unless otherwise taxable. If not utilized,
the federal NOL carryforward will expire at various dates
between 2023 and 2030 and the state NOL carry-forwards will
expire at various dates between 2020 and 2030.
NOL carry-forwards may be subject to annual limitations under
Internal Revenue Code Section 382 (or comparable provisions
of state law) in the event that certain changes in ownership of
the Company were to occur. The Company has not yet completed a
formal evaluation of whether the impact of its IPO (see
Note 1) resulted in certain changes in ownership that
would limit the Company’s ability to utilize a portion of
its NOL carry-forwards.
102
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Net deferred tax assets (liabilities) were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Depreciation and amortization
|
|
$
|
206
|
|
|
$
|
268
|
|
|
Other deferred tax assets
|
|
|
246
|
|
|
|
338
|
|
|
NOL carry-forwards
|
|
|
36,467
|
|
|
|
29,512
|
|
|
Research and development costs
|
|
|
9,165
|
|
|
|
10,039
|
|
|
Collaboration agreement receivable reserves
|
|
|
844
|
|
|
|
364
|
|
|
Valuation allowance
|
|
|
(46,928
|
)
|
|
|
(40,521
|
)
|
|
Total
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
The income tax benefit differs from the amount determined by
applying the U.S. federal statutory income tax rate to the
pre-tax accounting loss as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Amount
|
|
|
Percent
|
|
|
Amount
|
|
|
Percent
|
|
|
Amount
|
|
|
Percent
|
|
|
|
|
Federal tax benefit at statutory rate
|
|
$
|
(4,712
|
)
|
|
|
34.0
|
%
|
|
$
|
(15,035
|
)
|
|
|
34.0
|
%
|
|
$
|
(20,898
|
)
|
|
|
34.0
|
%
|
|
State tax — net of federal benefit
|
|
|
(549
|
)
|
|
|
4.0
|
|
|
|
(1,751
|
)
|
|
|
4.0
|
|
|
|
(2,434
|
)
|
|
|
4.0
|
|
|
Permanent items
|
|
|
(1,166
|
)
|
|
|
8.4
|
|
|
|
8,938
|
|
|
|
(20.2
|
)
|
|
|
4,226
|
|
|
|
(6.9
|
)
|
|
Change in state deferred tax rate
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(160
|
)
|
|
|
0.3
|
|
|
Other
|
|
|
20
|
|
|
|
—
|
|
|
|
425
|
|
|
|
(1.0
|
)
|
|
|
(55
|
)
|
|
|
—
|
|
|
Increase in valuation allowance
|
|
|
6,407
|
|
|
|
(46.4
|
)
|
|
|
7,423
|
|
|
|
(16.8
|
)
|
|
|
19,321
|
|
|
|
(31.4
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total tax expense
|
|
$
|
—
|
|
|
|
—
|
%
|
|
$
|
—
|
|
|
|
—
|
%
|
|
$
|
—
|
|
|
|
—
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company adopted Statement of Financial Accounting Standards
No. 157, Fair Value Measurements (ASC 820),
effective January 1, 2008. Under this standard, fair value
is defined as the price that would be received to sell an asset
or paid to transfer a liability (i.e., the “exit
price”) in an orderly transaction between market
participants at the measurement date.
In determining fair value, the Company uses various valuation
approaches. The hierarchy of those valuation approaches is
broken down into three levels based on the reliability of inputs
as follows:
Level 1 inputs are quoted prices in active markets for
identical assets or liabilities that the reporting entity has
the ability to access at the measurement date. An active market
for the asset or liability is a market in which transactions for
the asset or liability occur with sufficient frequency and
volume to provide pricing information on an ongoing basis. The
valuation under this approach does not entail a significant
degree of judgment.
Level 2 inputs are inputs other than quoted prices included
within Level 1 that are observable for the asset or
liability, either directly or indirectly. Level 2 inputs
include: quoted prices for similar assets or liabilities in
active markets, inputs other than quoted prices that are
observable for the asset or liability, (e.g., interest rates and
yield curves observable at commonly quoted intervals or current
market) and contractual prices for the underlying financial
instrument, as well as other relevant economic measures.
103
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Level 3 inputs are unobservable inputs for the asset or
liability. Unobservable inputs shall be used to measure fair
value to the extent that observable inputs are not available,
thereby allowing for situations in which there is little, if
any, market activity for the asset or liability at the
measurement date.
The following fair value table presents information about the
Company’s assets and liabilities measured at fair value on
a recurring basis:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2010
|
|
|
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
(In thousands)
|
|
|
|
|
Assets:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents(1)
|
|
$
|
27,393
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
27,393
|
|
|
Investments in marketable debt securities(2)
|
|
|
—
|
|
|
|
26,330
|
|
|
|
—
|
|
|
|
26,330
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Assets measured at fair value
|
|
$
|
27,393
|
|
|
$
|
26,330
|
|
|
$
|
—
|
|
|
$
|
53,723
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
|
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
Total
|
|
|
|
|
(In thousands)
|
|
|
|
|
Assets:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents(1)
|
|
$
|
4,668
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
4,668
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Assets measured at fair value
|
|
$
|
4,668
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
4,668
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Preferred stock conversion feature(3)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
36,701
|
|
|
$
|
36,701
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities measured at fair value
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
36,701
|
|
|
$
|
36,701
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
The carrying amounts approximate fair value due to the
short-term maturities of the cash and cash equivalents. |
| |
|
(2) |
|
Valuations are based on quoted prices in markets that are not
active or for which all significant inputs are observable,
either directly or indirectly. These prices include broker or
dealer quotations, or alternative pricing sources with
reasonable levels of price transparency. Pricing sources include
industry standard data providers, security master files from
large financial institutions, and other third party sources
which are input into a distribution-curve-based algorithm to
determine a daily market value. This creates a “consensus
price” or a weighted average price for each security. |
| |
|
(3) |
|
The fair value of the beneficial conversion feature of preferred
stock (see note 11) is established using a probability
weighted expected return method (PWERM) and Black Scholes
valuation model. Significant inputs to the valuation include: |
|
|
|
| |
•
|
probability of various scenarios occurring, including the
potential for an initial public offering, sale of the Company or
its assets, decision to remain a private company or liquidation
of the Company;
|
| |
| |
•
|
fair value of common stock as determined under each of the
scenarios under the PWERM, adjusted for a lack of control and
lack of marketability discount;
|
| |
| |
•
|
volatility estimated as an average of volatilities of publicly
traded companies deemed similar to the Company in terms of
product composition, stage of lifecycle, capitalization, and
scope of operations;
|
| |
| |
•
|
exercise price and weighted-average expected life estimated
based on the underlying and the expected remaining life of the
underlying instrument;
|
104
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
|
|
|
| |
•
|
risk-free interest rate estimated as the daily treasury yield
for the period that most closely approximates the
weighted-average expected life as the valuation date as
published by the U.S. Department of Treasury.
|
The method described above may produce a fair value calculation
that may not be indicative of net realizable value or reflective
of future fair values. Furthermore, while the Company believes
its valuation methods are appropriate, the use of different
methodologies or assumptions to determine the fair value of
certain financial instruments could result in a different fair
value measurement at the reporting date.
The following table presents the changes to the fair value of
the beneficial conversion feature of preferred stock during the
years ended December 31, 2010 and 2009 (in thousands):
| |
|
|
|
|
|
Fair value of conversion feature of preferred stock at
December 31, 2008
|
|
$
|
12,656
|
|
|
Issuance of
Series C-1
preferred stock (See Note 11)
|
|
|
903
|
|
|
Change in fair value of preferred stock conversion feature
|
|
|
23,142
|
|
|
|
|
|
|
|
|
Fair value of conversion feature of preferred stock at
December 31, 2009
|
|
|
36,701
|
|
|
Issuance of
Series C-1
preferred stock (Note 11)
|
|
|
3,471
|
|
|
Change in fair value of preferred stock conversion feature
|
|
|
(3,644
|
)
|
|
Elimination of the conversion feature of preferred stock
(Note 11)
|
|
|
(36,528
|
)
|
|
|
|
|
|
|
|
Fair value of conversion feature of preferred stock at
December 31, 2010
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
16.
|
EMPLOYEE
BENEFIT PLANS
|
The Company has a salary deferral 401(k) plan which covers
substantially all employees of the Company. In May 2008, the
Company established a plan to match participant contributions
subject to certain plan limitations. The Company’s matching
plan took effect on July 1, 2008. Compensation expense
associated with the Company’s matching plan totaled
$68,000, $70,000 and $61,000 for the years ended
December 31, 2010, 2009 and 2008, respectively. The Company
may also make an annual discretionary profit-sharing
contribution. No such discretionary contributions were made
during the years ended December 31, 2010, 2009 and 2008.
In April 2010 the Company established an Employee Stock Purchase
Plan (the “Purchase Plan”). Under the Company’s
Purchase Plan, eligible employees can participate and purchase
common stock semi-annually through accumulated payroll
deductions. The Purchase Plan is administered by the
Company’s board of directors or a committee appointed by
the Company’s board of directors. Under the Purchase Plan
eligible employees may purchase stock at 85% of the lower of the
fair market value of a share of Common Stock on the offering
date or the exercise date. The Purchase Plan provides for two
6-month
purchase periods generally starting on the first trading day on
or after October 31 and April 30 of each year. There are.
Eligible employees may contribute up to 15% of their eligible
compensation. A participant may purchase a maximum of
2,500 shares of common stock per purchase period. The value
of the shares purchased in any calendar year may not exceed
$25,000.
The Purchase Plan was effective upon the completion of the
Company’s IPO, at which time a total of 494,422 shares
of the Company’s common stock were made available for sale.
As of January 1 of each year, starting in 2011, the reserve will
automatically be restored to the original level. A total of
8,246 shares of the Company’s common shares were
acquired through the Purchase Plan during the year ended
December 31, 2010. As such, on January 1, 2011, an
additional 8,246 shares became available for future
issuance under the Purchase Plan. In accordance with
ASC 718-50,
the ability to purchase stock at 85% of the lower of the fair
market value of a share of Common Stock on the offering date or
the exercise date represents an option. The Company estimates
the fair value of such options at the inception of each offering
period using the Black-
105
ALIMERA
SCIENCES, INC.
NOTES TO
FINANCIAL STATEMENTS — (Continued)
Scholes valuation model. In connection with the Purchase Plan,
the Company recorded $43,000 of compensation expense for the
year ended December 31, 2010.
|
|
|
17.
|
SELECTED
QUARTERLY FINANCIAL DATA (UNAUDITED)
|
The following financial information reflects all normal
recurring adjustments, which are, in the opinion of management,
necessary for a fair statement of the results of the interim
periods. Selected quarterly financial data for years ended
December 31, 2010 and 2009 are as follows (in thousands
except per share data):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2010
|
|
|
|
|
1st Quarter
|
|
|
2nd Quarter
|
|
|
3rd Quarter
|
|
|
4th Quarter
|
|
|
|
|
Selected quarterly financial data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
$
|
4,216
|
|
|
$
|
5,693
|
|
|
$
|
6,119
|
|
|
$
|
6,043
|
|
|
Net income (loss)
|
|
|
2,577
|
|
|
|
(4,101
|
)
|
|
|
(6,082
|
)
|
|
|
(6,253
|
)
|
|
Net income (loss) attributable to common stockholders
|
|
|
193
|
|
|
|
(4,821
|
)
|
|
|
(6,082
|
)
|
|
|
(6,253
|
)
|
|
Net income (loss) per common stockholder — Basic and
diluted(1)
|
|
$
|
0.12
|
|
|
$
|
(0.20
|
)
|
|
$
|
(0.20
|
)
|
|
$
|
(0.20
|
)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2009
|
|
|
|
|
1st Quarter
|
|
|
2nd Quarter
|
|
|
3rd Quarter
|
|
|
4th Quarter
|
|
|
|
|
Selected quarterly financial data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
$
|
5,490
|
|
|
$
|
4,747
|
|
|
$
|
4,634
|
|
|
$
|
4,345
|
|
|
Net loss
|
|
|
(10,178
|
)
|
|
|
(6,266
|
)
|
|
|
(5,110
|
)
|
|
|
(22,664
|
)
|
|
Net loss attributable to common stockholders
|
|
|
(12,032
|
)
|
|
|
(8,135
|
)
|
|
|
(7,459
|
)
|
|
|
(24,795
|
)
|
|
Net loss per common stockholder — Basic and diluted(1)
|
|
$
|
(8.07
|
)
|
|
$
|
(5.46
|
)
|
|
$
|
(4.89
|
)
|
|
$
|
(15.85
|
)
|
|
|
|
|
(1) |
|
Net loss per common stockholder is computed independently for
each of the quarters presented. Therefore the sum of the
quarterly net loss per share will not necessarily equal the
total for the year. |
106
ALIMERA
SCIENCES, INC.
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Exhibit Title
|
|
|
|
|
3
|
.2
|
|
Restated Certificate of Incorporation of Registrant, as amended
on various dates (filed as Exhibit 3.2 to Amendment
No. 4 to the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 6, 2010, and incorporated herein by
reference)
|
|
|
3
|
.4
|
|
Amended and Restated Bylaws of the Registrant (filed as
Exhibit 3.4 to Amendment No. 4 to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 6, 2010, and incorporated herein by
reference)
|
|
|
4
|
.3
|
|
Second Amended and Restated Investor Rights Agreement, dated
March 17, 2008, by and among the Registrant, certain
stockholders and the investors listed on the signature pages
thereto (filed as Exhibit 4.3 to Amendment No. 1 to
the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on December 23, 2009, and incorporated herein by
reference)
|
|
|
4
|
.4
|
|
Second Amended and Restated Stock Sale Agreement, dated
March 17, 2008, by and among the Registrant, certain
stockholders and the investors listed on the signature pages
thereto (filed as Exhibit 4.4 to Amendment No. 1 to
the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on December 23, 2009, and incorporated herein by
reference)
|
|
|
4
|
.5
|
|
Omnibus Amendment, dated August 25, 2009, by and among the
Registrant, certain stockholders and the investors listed on the
signature pages thereto (filed as Exhibit 4.5 to Amendment
No. 1 to the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on December 23, 2009, and incorporated herein by
reference)
|
|
|
4
|
.6
|
|
Warrant to Purchase Stock dated October 14, 2010 issued to
Silicon Valley Bank (filed as Exhibit 4.1 to
Registrant’s Current Report, as filed on October 18,
2010, and incorporated herein by reference)
|
|
|
4
|
.7
|
|
Warrant to Purchase Stock dated October 14, 2010 issued to
MidCap Funding III, LLC (filed as Exhibit 4.2 to
Registrant’s Current Report, as filed on October 18,
2010, and incorporated herein by reference)
|
|
|
10
|
.1
|
|
Form of Indemnification Agreement between the Registrant and
each of its directors and executive officers (filed as
Exhibit 10.1 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.2†
|
|
Amended and Restated Employment Agreement, dated August 18,
2008, by and between the Registrant and C. Daniel Myers (filed
as Exhibit 10.2 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.3†
|
|
Amended and Restated Employment Agreement, dated August 18,
2008, by and between the Registrant and Richard Eiswirth (filed
as Exhibit 10.3 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.4†
|
|
Amended and Restated Employment Agreement, dated August 18,
2008, by and between the Registrant and David Holland (filed as
Exhibit 10.4 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.5†
|
|
Amended and Restated Employment Agreement, dated August 18,
2008, by and between the Registrant and Susan Caballa (filed as
Exhibit 10.5 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.6†
|
|
Amended and Restated Employment Agreement, dated August 18,
2008, by and between the Registrant and Kenneth Green (filed as
Exhibit 10.6 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
107
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Exhibit Title
|
|
|
|
|
10
|
.7
|
|
Alimera Sciences, Inc. 2004 Incentive Stock Plan, as amended
(filed as Exhibit 10.7 to the Registrant’s
Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.7.A
|
|
Form of Option Certificate under the Alimera Sciences, Inc. 2004
Incentive Stock Plan (filed as Exhibit 10.7.A to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.8
|
|
Alimera Sciences, Inc. 2005 Incentive Stock Plan (filed as
Exhibit 10.8 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.8.A
|
|
Form of Option Certificate under the Alimera Sciences, Inc. 2005
Incentive Stock Plan (filed as Exhibit 10.8.A to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.9
|
|
2010 Equity Incentive Plan (filed as Exhibit 10.9 to
Amendment No. 4 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 6, 2010, and incorporated herein by
reference)
|
|
|
10
|
.10
|
|
2010 Employee Stock Purchase Plan (filed as Exhibit 10.10
to Amendment No. 4 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 6, 2010, and incorporated herein by
reference)
|
|
|
10
|
.11
|
|
Management Cash Incentive Plan (filed as Exhibit 10.11 to
the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.12
|
|
Compensation Program for Non-Employee Directors (filed as
Exhibit 10.12 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.13‡
|
|
Amended and Restated Collaboration Agreement by and between
pSivida, Inc. (f/k/a/Control Delivery Systems, Inc.) and Alimera
Sciences, Inc., dated as of March 14, 2008 (filed as
Exhibit 10.13 to Amendment No. 5 to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 16, 2010, and incorporated herein by
reference)
|
|
|
10
|
.14‡
|
|
Asset Purchase Agreement between Bausch & Lomb
Incorporated and Alimera Sciences, Inc., dated as of
December 20, 2006 (filed as Exhibit 10.14 to Amendment
No. 5 to the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 16, 2010, and incorporated herein by
reference)
|
|
|
10
|
.15‡
|
|
Asset Purchase Agreement between Bausch & Lomb
Incorporated and Alimera Sciences, Inc., dated as of
February 16, 2007 (filed as Exhibit 10.15 to Amendment
No. 5 to the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 16, 2010, and incorporated herein by
reference)
|
|
|
10
|
.16‡
|
|
License and Option Agreement by and between Emory University and
Alimera Sciences, Inc., dated as of July 16, 2009 (filed as
Exhibit 10.16 to Amendment No. 5 to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 16, 2010, and incorporated herein by
reference)
|
|
|
10
|
.17‡
|
|
License and Option Agreement by and between Emory University and
Alimera Sciences, Inc., dated as of August 31, 2009 (filed
as Exhibit 10.17 to Amendment No. 5 to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 16, 2010, and incorporated herein by
reference)
|
|
|
10
|
.18
|
|
Office Lease by and between Rubicon, L.C. and Alimera Sciences,
Inc., dated as of May 27, 2003, as amended (filed as
Exhibit 10.18 to the Registrant’s Registration
Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on October 30, 2009, and incorporated herein by
reference)
|
|
|
10
|
.25‡
|
|
License Agreement between Alimera Sciences, Inc. and Dainippon
Sumitomo Pharma Co., Ltd., dated November 4, 2007 (filed as
Exhibit 10.25 to Amendment No. 1 to the
Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on December 23, 2009, and incorporated herein by
reference)
|
108
| |
|
|
|
|
Exhibit
|
|
|
|
Number
|
|
Exhibit Title
|
|
|
|
|
10
|
.26‡
|
|
Commercial Contract Manufacturing Agreement, between Alimera
Sciences, Inc. and Alliance Medical Products, Inc., dated
February 5, 2010 (filed as Exhibit 10.26 to Amendment
No. 6 to the Registrant’s Registration Statement on
Form S-1
(SEC File
No. 333-162782),
as filed on April 20, 2010, and incorporated herein by
reference)
|
|
|
10
|
.27
|
|
Loan and Security Agreement dated October 14, 2010 between
Registrant, Silicon Valley Bank and MidCap Funding III, LLC
(filed as Exhibit 10.1 to Registrant’s Current Report
on Form 8-K, as filed on October 18, 2010, and
incorporated herein by reference)
|
|
|
10
|
.28
|
|
Loan and Security Agreement dated October 14, 2010 between
Registrant and Silicon Valley Bank (filed as Exhibit 10.2
to Registrant’s Current Report on Form 8-K, as filed
on October 18, 2010, and incorporated herein by reference)
|
|
|
10
|
.29*
|
|
Contract Sales Agreement dated October 4, 2010 between the
Registrant and OnCall LLC
|
|
|
10
|
.30
|
|
Form of Notice of Stock Option Grant and Stock Option Agreement
under 2010 Equity Incentive Plan
|
|
|
23
|
.1
|
|
Consent of Deloitte & Touche LLP, Independent
Registered Public Accounting Firm
|
|
|
31
|
.1
|
|
Certification of the Chief Executive Officer, as required by
Section 302 of the Sarbanes-Oxley Act of 2002
|
|
|
31
|
.2
|
|
Certification of the Chief Financial Officer as required by
Section 302 of the Sarbanes-Oxley Act of 2002
|
|
|
32
|
.1
|
|
Certifications of the Chief Executive Officer and Chief
Financial Officer as required by 18 U.S.C. 1350
|
|
|
32
|
.2
|
|
Certifications of the Chief Executive Officer and Chief
Financial Officer as required by 18 U.S.C. 1350
|
|
|
|
|
† |
|
Compensation Arrangement. |
| |
|
‡ |
|
Confidential treatment has been granted with respect to certain
portions of this document. |
| |
|
* |
|
Application has been made to the Securities and Exchange
Commission to seek confidential treatment of certain provisions
of this exhibit. |
109