Attached files
| file | filename |
|---|---|
| EX-31.1 - ENZON PHARMACEUTICALS, INC. | c64669_ex31-1.htm |
| EX-21.1 - ENZON PHARMACEUTICALS, INC. | c64669_ex21-1.htm |
| EX-32.2 - ENZON PHARMACEUTICALS, INC. | c64669_ex32-2.htm |
| EX-32.1 - ENZON PHARMACEUTICALS, INC. | c64669_ex32-1.htm |
| EX-23.1 - ENZON PHARMACEUTICALS, INC. | c64669_ex23-1.htm |
| EX-31.2 - ENZON PHARMACEUTICALS, INC. | c64669_ex31-2.htm |
| EX-12.1 - ENZON PHARMACEUTICALS, INC. | c64669_ex12-1.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
|
|
|
R |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) |
|
|
For the Fiscal Year Ended December 31, 2010 |
|
OR |
||
£ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) |
|
|
For the transition period from to |
|
Commission file number: 0-12957

(Exact name of registrant as specified in its charter)
|
|
|
Delaware |
22-2372868 |
|
20 Kingsbridge Road, Piscataway, New Jersey |
08854 |
Registrant’s telephone number, including area code:
(732) 980-4500
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
Title of Class |
Name of Exchange on Which Registered |
|
Common Stock, $0.01 par value; |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
£ Yes R No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. £ Yes R No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. R Yes £ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). £ Yes £ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. £
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check One):
£ Large accelerated filer R Accelerated filer £ Non-accelerated filer £ Smaller reporting company
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
£ Yes R No
The aggregate market value of the Common Stock, par value $.01 per share (“Common Stock”), held by non-affiliates of the registrant was approximately $639,698,000 as of June 30, 2010, based upon the closing sale price on the NASDAQ Global Market of $10.65 per share reported for such date. Shares of Common Stock held by each officer and director and by each person who owns 10% or more of the outstanding shares of Common Stock have been excluded in that such shares may be deemed to be owned by affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
There were 56,985,697 shares of the registrant’s common stock issued and outstanding as of March 9, 2011.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for the 2011 Annual Meeting of Stockholders to be filed with the Commission not later than 120 days after the close of the registrant’s fiscal year, have been incorporated by reference, in whole or in part, into Part III, Items 10, 11, 12, 13 and 14 of this Annual Report on Form 10-K.
ENZON PHARMACEUTICALS, INC.
Page
Business
4
Risk Factors
16
Unresolved Staff Comments
30
Properties
30
Legal Proceedings
30
(Removed and Reserved)
30
Market for the Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities
31
Selected Financial Data
33
Management’s Discussion and Analysis of Financial Condition and Results of Operations
35
Quantitative and Qualitative Disclosures About Market Risk
49
Financial Statements and Supplementary Data
50
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
50
Controls and Procedures
50
Other Information
53
Directors, Executive Officers and Corporate Governance
53
Executive Compensation
53
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
53
Certain Relationships and Related Transactions, and Director Independence
53
Principal Accountant Fees and Services
53
Exhibits and Financial Statement Schedules
53 2
2010 Form 10-K Annual Report
TABLE OF CONTENTS
This
Annual Report contains forward-looking statements, which can be identified
by the use of forward-looking terminology such as “believes,” “expects,” “may,” “will,” “should,” “potential,” “anticipates,” “plans,” or “intends” or
the negative thereof, or other variations thereof, or comparable terminology,
or by discussions of strategy. No assurance can be given that the future
results covered by the forward-looking statements will be achieved. The matters
set forth in Item 1A. Risk Factors constitute cautionary statements identifying
important factors with respect to such forward-looking statements, including
certain risks and uncertainties that could cause actual results to vary materially
from the future results indicated in such forward-looking statements. Other
factors also could cause actual results to vary materially from the future
results indicated in such forward-looking statements. All information in
this Annual Report on Form 10-K is as of March 16, 2011, unless otherwise
indicated. The Company does not intend to update this information to reflect
events after the date of this report. We maintain a website at www.enzon.com to provide information to the general public and our stockholders on our products, resources and services along with general information on Enzon and its management, career opportunities, financial results and press releases. Copies of our most recent Annual Report on Form 10-
K, our Quarterly Reports on Form 10-Q, our Current Reports on Form 8-K and our other reports filed with the Securities and Exchange Commission, or the SEC, can be obtained, free of charge as soon as reasonably practicable after such material is electronically filed with, or furnished to the SEC, from our Investor Relations Department
by calling 908-541-8777, through an e-mail request to investor@enzon.com, through the SEC’s website by clicking the SEC Filings link from the Investors’ Info page on our website at www.enzon.com or directly from the SEC’s website at www.sec.gov. Our website and the information contained therein or connected
thereto are not intended to be incorporated into this Annual Report on Form 10-K. 3
UNITED STATES SECURITIES AND EXCHANGE COMMISSION FORM 10-K Our Company We are a biotechnology company dedicated to the research and development of innovative therapeutics for cancer patients with high unmet medical needs. Our drug development programs utilize two platforms—Customized PEGylation Linker Technology (Customized Linker TechnologyÒ) and third-generation mRNA antagonists
utilizing the Locked Nucleic Acid (LNA) technology. We currently have four compounds in human clinical development; a PEGylated version of the active metabolite of the cancer drug, irinotecan, PEG-SN38, and the three messenger ribonucleic acid (mRNA) antagonists: Hypoxia-Inducible Factor-1a (HIF-1a), Survivin and Androgen
Receptor (AR). In addition, we have multiple novel LNA targets in various stages of preclinical research. We receive royalty revenues from licensing arrangements with other companies related to sales of products developed using our proprietary Customized Linker Technology—primarily PEGINTRON marketed by Merck & Co., Inc. (Merck). On January 29, 2010, we consummated the sale of our specialty pharmaceutical business comprised principally of what had previously been our Products and Contract Manufacturing segments. Prior to January 29, 2010, we were a biopharmaceutical company involved in the development, manufacture and commercialization of
medicines for patients with cancer and other life-threatening conditions. We operated in three business segments: Products, Royalties and Contract Manufacturing. We had a portfolio of four marketed products, Oncaspar, for the first-line treatment of patients with acute lymphoblastic leukemia (ALL); DepoCyt, for the treatment of
lymphomatous meningitis; Abelcet, for the treatment of invasive fungal infections; and Adagen, for the treatment of severe combined immunodeficiency disease. The contract manufacturing business involved the manufacture of products for other pharmaceutical companies. For financial reporting purposes, beginning in 2010, the
operations and cash flows of the Products and Contract Manufacturing segments have been eliminated from the continuing operations of the Company and have been classified as discontinued operations. Similarly, assets and liabilities of the specialty pharmaceutical business are presented separately in our balance sheet. PEGylation has successfully been used on various pharmaceutical compounds (e.g. enzymes, peptides and antibody/antibody fragments) to improve their pharmaceutical properties. By attaching polyethylene glycol (PEG) to a pharmaceutical compound using a spectrum of stable and releasable linkers, our Customized Linker
Technology has the potential to overcome pharmaceutical limitations for a broad universe of molecules and generate compounds with substantially enhanced therapeutic value over their unmodified forms. We continue to evaluate opportunities for utilizing our Customized Linker Technology platform for the development of new projects. We also are using LNA technology to develop mRNA antagonists against novel oncology targets. LNA technology allows the development of very selective antagonists that act through the antisense RNAase H principle. Drugs based on the antisense principle work by providing a synthetic strand of nucleic acid (in this case, a
chemical analogue of RNA) that will bind to the mRNA and be degraded by RNAase H. Due to the elimination of mRNA, there is no template to produce a protein. In pre-clinical studies, the LNA technology has been shown to provide mRNA antagonists with significantly enhanced binding affinity to complementary RNA sequences,
high potencies, long tissue half-lives, and improved therapeutic ratios over first- and second-generation antisense drugs. 4
ENZON PHARMACEUTICALS, INC.
Our development pipeline consists of several novel compounds: PEG-SN38 Our lead compound, PEG-SN38, utilizes our Customized Linker Technology. SN38 is the active metabolite of the cancer drug irinotecan, but is highly insoluble and therefore not suitable for intravenous administration. PEG-SN38 is designed to allow for intravenous delivery, increased solubility, higher exposure of the cancer cells to
SN38, and longer apparent half-life. We have completed Phase I trials and are now enrolling patients in two Phase II clinical trials with PEG-SN38 in patients with metastatic colorectal and breast cancer, as well as a Phase I trial for pediatric patients with cancer. mRNA Antagonists We have licensed several mRNA antagonists directed against novel oncology targets. Our first antagonist to enter the clinic was the Hypoxia-Inducible Factor-1 alpha (HIF-1a) target. HIF-1a is over-expressed in several solid tumors. Drugs that selectively target HIF-1a have the potential to target multiple cancer processes due to their
control of a large number of genes. We are currently conducting two Phase I studies with HIF-1a in patients with solid tumors and lymphoma to evaluate different dosing schedules. In December 2010, we commenced a pilot study in collaboration with the National Cancer Institute in patients with tumors in the liver. Our second mRNA antagonist to enter the clinic targets Survivin. Survivin is over-expressed in many solid tumors and hematologic malignancies, but is almost absent in normal adult differentiated tissue. We are currently enrolling patients in a Phase I study for patients with solid tumors and lymphoma. In addition, in collaboration
with Therapeutic Advances in Childhood Leukemia & Lymphoma, we have opened a Phase I study in pediatric patients with acute lymphoblastic leukemia (ALL). The compound continues to be safe and well tolerated. In January 2011, we announced the acceptance by the U.S. Food and Drug Administration (FDA) of an Investigational New Drug (IND) for a Phase I study of a novel androgen receptor (AR) antagonist in patients with castration-resistant prostate cancer. We also have rights to five additional mRNA targets and we are evaluating the lead compounds we have selected for these targets based upon early preclinical studies to determine which of the compounds warrants further investment on our part. Our Strategy Our strategy is to build on the foundation that has been laid over the past few years with concentrated efforts aimed at advancing our pipeline in as effective and expeditious a manner as possible. The energies and financial resources of the Company are focused on the promising research programs currently underway. We currently
have four product candidates advancing into and through the clinic, but the cost of simultaneously studying that number of product candidates in clinical trials is substantial for a Company the size of ours. Consequently, we are more committed than ever to making targeted, disciplined investments in areas where we believe we can make a
unique contribution, achieve differentiation and have the greatest chances of success. While we have a strong balance sheet and substantial internal financial resources, we are also committed to returning to our shareholders some of the value previously created through the sale of our specialty pharmaceutical operations. We are
accomplishing this through our $200 million share repurchase program currently in progress which follows the $50 million share repurchase program we completed in 2010. In the meantime, we are actively pursuing the possibility of partnering with another party or parties with the objective of enabling us to leverage our investment in
our pipeline. 5
We have invested in our infrastructure and our people. We have the know-how and the capability to take our pipeline forward. We intend to take the next steps of: Applying our cutting-edge Customized Pegylation Linker and LNA technologies to further advance our development candidates, and discover and develop novel therapeutics for oncology. We believe our PEGylation platform has broad applicability across a variety of compounds and indications. We also believe that novel approaches to treating cancer, such as those we are pursuing with our novel mRNA antagonists, have the potential to more selectively target and eliminate cancer cells than traditional chemotherapy,
particularly for recurrent advanced-stage cancers for which current treatments are inadequate. Making targeted and disciplined investments in areas where we believe we can achieve differentiation. We believe our novel pipeline is differentiated and can provide multiple development opportunities. Our unique product candidates and technology platforms have a wide range of applications in oncology. Our management team also has extensive experience in bringing novel products to market through focused and innovative
development strategies. Continuing to leverage our PEGylation expertise and drug discovery and development expertise to pursue strategic partnerships and business development opportunities. We aim to continue seeking opportunities to apply our core PEGylation expertise, as well as our novel Customized Linker Technology, to drug candidates that may benefit from a novel delivery platform. We plan to selectively and strategically out-license our PEGylation technology and Customized Linker Technology to
pharmaceutical and biotechnology companies to improve the effectiveness of their existing compounds. We offer potential partners substantial know-how in the area of PEGylation and an experienced management team with extensive experience in researching and developing pharmaceutical products, particularly for the treatment of
cancer. RESEARCH AND DEVELOPMENT Our drug-development program is focused on advancing novel compounds for the treatment of cancers for which there is an unmet medical need. We are building a proprietary research and development pipeline both through the application of our proprietary technologies and through strategic agreements that provide access to
promising product development opportunities within our therapeutic focus. PEGYLATION TECHNOLOGY Since our inception in 1981, our core expertise has been in engineering improved versions of injectable therapeutics through the chemical attachment of polyethylene glycol (PEG). In some cases, PEGylation can render a compound therapeutically effective, whereas the unmodified form had only limited clinical utility. Currently, six
biologic products, utilize our proprietary PEG platform. We continue to receive royalties for three products: PEGINTRONÒ, MacugenÒ, and CIMZIAÒ. PegasysÒ also utilizes our PEG platform, but our right to receive royalties on sales of that product ended in 2009. The other two products using our technology, Oncaspar and Adagen were
divested as part of the sale of the specialty pharmaceutical business and royalties may be received from their third-party sales under certain conditions. PEG-SN38 SN38 is the active metabolite of the cancer drug irinotecan, a chemotherapeutic drug marketed as CamptosarÒ (CPT-11) in the U.S. Unmodified SN38 is insoluble and can only be used to treat cancer by administering a pro-drug. A pro-drug is a compound that is converted into the active drug in the body. Only a small percentage of
the pro-drug is converted into SN38 in cancer cells, and the unpredictability of conversion and metabolism in each patient may result in a variable efficacy and safety profile. Through the use of our PEGylation technology, we designed PEG-SN38 (EZN-2208), a PEGylated conjugate of SN38, to offer 6
therapeutic advantages over unmodified SN38 and existing therapies. The PEGylated version allows parenteral delivery, increased solubility, higher exposure, more profound deoxyribonucleic acid (DNA) damage, inhibition of angiogenesis, and longer apparent half-life of SN38. Preclinical data showed that PEG-SN38 demonstrated potent in vitro cytotoxicity against several human cancer cell lines, as well as antitumor activity in several xenograft models of solid tumors and non-Hodgkin’s lymphoma, including those in which CPT-11 has been shown to be ineffective. Treatment with a single dose or
multiple small doses of PEG-SN38 led to complete cures of animals in the breast cancer, neuroblastoma and non-Hodgkin’s lymphoma models. In colorectal and pancreatic cancer preclinical models, PEG-SN38 demonstrated significantly better therapeutic efficacy than CPT-11, at their respective maximum tolerated doses and equivalent
dose levels. Importantly, treatment with PEG-SN38 resulted in tumor growth inhibition in CPT-11—resistant tumors and outperformed CPT-11 when given as second-round therapy to animals initially responding to CPT-11. These preclinical studies also showed that PEG-SN38 provided a long circulation half-life and exposure to the parent
drug, SN38, in mice. Finally, PEG-SN38 also induced more profound DNA damage and inhibition of angiogenesis than CPT-11. In 2007, the FDA accepted our IND for the evaluation of PEG-SN38 in patients with cancer. Two Phase I studies evaluating the safety of two different dosing schedules of PEG-SN38 have been completed. Four clinical trials (two Phase I, two Phase II) are currently ongoing. In 2009, we opened our Phase II trial designed to evaluate
two groups of patients with metastatic colorectal cancer who have failed two prior therapies: those with K-RAS mutated tumors and those with K-RAS non-mutated tumors. K-RAS mutation has been reported to occur in at least 30% to 40% of patients with colorectal cancer. Up to 100 patients are expected to enroll in the K-RAS mutation
arm of the study. The non-mutated K-RAS group will be randomized into two arms: one treated with PEG-SN38 in combination with ErbituxÒ and the other treated with CamptosarÒ in combination with Erbitux. In the non-mutated K-RAS group, we expect to enroll 120 patients. In January 2010, we started enrolling patients in a Phase II
trial for patients with metastatic breast cancer. The study is designed to evaluate the efficacy of single-agent PEG-SN38 in two groups of patients who have received prior therapy regimens of anthracycline and taxane or anthracycline, taxane, and XelodaÒ. Approximately 160 patients are expected to enroll in that study. We also opened a
Phase I study in pediatric patients with cancer. In addition, in collaboration with the National Cancer Institute, National Institutes of Health, we opened a Phase I study of PEG-SN38 in combination with AvastinÒ to evaluate the safety of this combination in patients with advanced cancer. Based on available results from these studies, it has
been found that PEG-SN38 is well tolerated in patients treated in our Phase I studies and in ongoing Phase II studies in patients with metastatic colorectal and breast cancer. LOCKED NUCLEIC ACID (LNA) TECHNOLOGY-BASED PROGRAMS Enzon has a license and collaboration agreement with Santaris Pharma A/S for eight messenger ribonucleic acid (mRNA) antagonists. We hold rights worldwide, other than in Europe, to develop and commercialize mRNA antagonists based on LNA technology directed against the Hypoxia-Inducible Factor-1 alpha (HIF-1 alpha),
Survivin and Androgen Receptor (AR) mRNA targets and five additional targets. LNA Technology is based on Locked Nucleic Acid, a proprietary synthetic analog of ribonucleic acid (RNA), which is fixed in the shape adopted by RNA in helical conformation. When incorporated into a short nucleic acid chain (both DNA and RNA are made up of longer chains of natural nucleic acids), the presence of LNA
results in several potential therapeutic advantages. Because LNA resembles RNA but is more stable, LNA-containing drugs have both very high binding affinity for mRNA and metabolic stability. Using the antisense principle to block the function of specific mRNAs within cells and tissues, such drugs may have enhanced potency and
specificity, and may provide improved efficacy at lower doses than comparable drugs based on alternative chemistry. As a result, mRNA antagonists composed of LNA have been demonstrated to be significantly more potent in vitro and in vivo than conventional antisense compounds. In particular, LNA-based mRNA antagonists can be
used to switch off the synthesis of proteins believed to promote cancer, thereby leading to control or shrinkage of tumors. 7
HIF-1 Alpha Antagonist The HIF-1 alpha antagonist is expressed in many cancer types, including common solid tumors. HIF-1 alpha is a key regulator of a large number of genes important in cancer biology, such as angiogenesis, cell proliferation, apoptosis, glucose metabolism, and cell invasion. HIF-1 alpha protein level is low in normal cells, but reaches
high intracellular concentrations in a variety of cancers. The expression of HIF-1 alpha is strongly correlated with poor prognosis and resistance to therapy. Drugs targeting HIF-1 alpha thus have the potential to target multiple cancer processes. Preclinical study data demonstrated that in vitro, in human prostate and glioblastoma cells, the HIF-1 alpha antagonist induced a potent, selective, and durable antagonism of HIF-1 alpha expression, both under normoxic and hypoxic conditions. Down-regulation of HIF-1alpha by the HIF-1 alpha antagonist led to reduction of its
transcriptional targets and significant reduction of HUVEC tube formation. In vivo, administration of the HIF-1 alpha antagonist to normal mice led to specific, dose-dependent, and potent down-regulation of endogenous HIF-1 alpha and vascular endothelial growth factor (VEGF) in the liver. In preclinical efficacy studies, tumor
reduction was found in mice implanted with DU145 cells that were transfected with the HIF-1 alpha antagonist before implantation and given systemic treatment with the HIF-1 alpha antagonist post-tumor implantation. In 2007, the FDA accepted our IND for the evaluation of the HIF-1 alpha antagonist in patients with solid tumors and lymphoma. Currently, patient enrollment is ongoing in two Phase I studies to evaluate the safety of the HIF-1 alpha antagonist using two different dosing schedules. In general, HIF-1 alpha antagonist therapy has
been well tolerated, and many patients have received multiple cycles in both studies. We have observed stable disease in a number of patients treated with our HIF-1 alpha antagonist. Tumor shrinkage also was seen in patients with renal cell cancer, liver cancer, sarcoma, and cancer of the tonsil. In collaboration with the National Cancer
Institute, National Institutes of Health, we opened a pilot study in patients with liver tumors. Survivin Antagonist Survivin plays a vital regulatory role in both apoptosis and cell division. Survivin is highly expressed in many cancers and in newly formed endothelial cells engaged in angiogenesis, with little expression in normal adult tissues. Resistance of cancer cells to radiotherapy and cytotoxic drugs (in particular microtubule-interfering
taxanes) is strongly correlated with expression levels of Survivin. Clinically, Survivin expression is associated with poor prognosis, increased cancer recurrence, and resistance to therapy. In February 2009, the FDA accepted our IND for the evaluation of our Survivin antagonist in patients with cancer. The same month, we opened and
started enrolling patients in the first Phase I study. The trial is designed to first treat patients with Survivin as a single agent; if the patient’s cancer progresses, the patient’s treatment will be changed to Survivin in combination with TaxotereÒ. This allows us to gain dose and safety information both as a single agent and in combination in a
single Phase I study. In addition, we opened a Phase I study in pediatric patients with recurrent acute lymphoblastic leukemia (ALL) in collaboration with the Therapeutic Advances in Childhood Leukemia & Lymphoma group (TACL). The goal of this trial is to evaluate the safety, tolerability, and biological activity in patients with ALL. Androgen Receptor (AR) Antagonist The AR is a validated target for the treatment of prostate cancer. Several approved agents prevent androgen binding to the AR or block androgen synthesis and demonstrate therapeutic benefit. Nevertheless, prostate tumors typically develop resistance to currently approved agents. It is likely that the AR still continues to promote
cancer growth in such patients. Consistent with this thinking, two novel agents that block activation of the AR have demonstrated anticancer activity in patients with castration-resistant prostate cancer and are in late-stage development. One agent blocks androgen synthesis, while the second compound blocks androgen interaction with the
AR more effectively than first-generation, approved agents. However, neither agent down-regulates the AR and is curative. Therefore, our LNA-based AR mRNA antagonist, is a novel and innovative strategy for the treatment of prostate cancer. In preclinical studies, our mRNA antagonist down-regulated the AR and inhibited the growth
of prostate cancer that expressed the AR. The compound enhanced the activity of a novel anti-androgen that is currently in late-state development. The FDA accepted our IND for a Phase I study of AR in patients with castration-resistant prostate cancer in the fourth quarter of 2010. 8
Five Additional Gene Targets Enzon has rights to five additional targets and has selected lead compounds for all five targets. We have worldwide rights, except in Europe, to develop and commercialize the compounds. As of November 2010, we have presented data at various meetings related to the following preclinical targets: AR, HER3, beta-catenin, PI3KCA,
and Gli2. We are evaluating, based upon preclinical studies, which of these lead compounds would warrant further investment by us. Any one of these compounds could be returned to Santaris if the findings of our preclinical or clinical work do not support our continued investment. ROYALTIES Our primary source of revenue is the royalties that we receive on sales of marketed products that utilize our proprietary technology. We receive royalties on three marketed products that utilize our proprietary PEGylation platform, namely PEGINTRONÒ, MacugenÒ, and CIMZIAÒ, with PEGINTRON being the largest source of our
royalty income. During 2009, our agreement to receive royalties from Pegasys for the treatment of hepatitis C expired.
Product
Indication
Company
Expiration
PEGINTRON (peginterferon alfa-2b)
Chronic hepatitis C
Merck
U.S.–2016
Macugen (pegaptanib sodium injection)
Neovascular (wet)
OSI Pharmaceuticals, Inc.
2014
CIMZIA (certolizumab pegol)
Crohn’s disease,
UCB Pharma
2014 PEGINTRON is a PEG-enhanced version of Merck’s alpha interferon product, INTRONÒ A, which is used both as a monotherapy and in combination with REBETOLÒ (ribavirin) capsules for the treatment of chronic hepatitis C. Merck holds an exclusive worldwide license to PEGINTRON. We are entitled to receive royalties on
Merck’s worldwide sales of PEGINTRON until certain expiration dates set forth in the license agreement which are expected to occur in 2016 in the U.S., 2018 in Europe and 2019 in Japan. Merck is responsible for all manufacturing, marketing, and development activities for PEGINTRON. We designed PEGINTRON to allow for less
frequent dosing and to yield greater efficacy, as compared to INTRON A. Sales of PEGINTRON have been in decline since 2008. There are a number of new therapies in late stage development that could significantly erode the market for PEGylated interferon which, in combination with the generic antiviral pill, ribavirin, represents the current standard of care. The most advanced new therapies are
protease inhibitors that work by blocking the action of the protease enzyme the hepatitis virus needs to replicate. Studies have revealed that the triple combinations (PEGylated interferon, ribavirin and protease inhibitor) are very effective in significantly reducing the hepatitis virus, and may allow shorter course of therapy, which may be
better tolerated by patients. These studies have not exclusively involved PEGINTRON, however. Most of the studies have involved Pegasys, manufactured by Hoffman-La Roche, the other currently available form of PEGylated interferon, from which we no longer derive royalties. There has been some indication that a significant number of patients suffering from hepatitis C may be deferring treatment until the new therapies become available potentially as early as mid-2011. The implications of the unsettled hepatitis C market to future sales of PEGINTRON are not clear. We have out-licensed our proprietary PEGylation and single-chain antibody, or SCA, technologies on our own and through agreements with Nektar Therapeutics, Inc. (Nektar) and Micromet AG (Micromet). Under our original 2002 agreement, Nektar had the lead role in granting sublicenses for certain of our PEGylation patents and
we receive royalties on sales of any approved product for which a sublicense has been granted. Effective in January 2007, Nektar’s right to grant additional sublicenses is limited to a certain class of our PEGylation patents. Existing sublicenses granted by Nektar prior to January 2007 were unaffected by this change in Nektar’s rights.
Currently, we are aware of five third-party products for which Nektar has granted sublicenses to our PEGylation technology, including OSI Pharmaceutical’s Macugen, UCB’s CIMZIA, Affymax and Takeda Pharmaceutical’s HematideTM, Hoffmann-La Roche’s Pegasys and an undisclosed product of Pfizer’s. 9
Europe–2018
Japan–2019
age-related macular
degeneration
and Pfizer Inc.
rheumatoid arthritis
Our rights to receive royalties under our agreement with Nektar relating to CIMZIA and Macugen expire in 2014. CIMZIA was approved in April 2008 for the treatment of Crohn’s disease. In May 2009, CIMZIA was approved for adult patients suffering from moderate to severe rheumatoid arthritis. Macugen is being marketed through a collaboration between OSI and Pfizer for the treatment of neovascular (wet) age-related macular
degeneration, an eye disease associated with aging that destroys central vision. Hematide is a synthetic peptide-based erythropoiesis-stimulating agent being evaluated by Affymax and Takeda Pharmaceutical for the treatment of anemia in chronic kidney failure. In late June 2010, Affymax announced preliminary top-line results from the
Hematide Phase 3 clinical program. Based on the results obtained and pending further feedback from the FDA, Affymax has reported that it plans to submit a New Drug Application (NDA) later in 2011. There can be no assurance that sales of any of the products utilizing our technology licensed through Nektar will increase through
expanded indications or FDA approval, as discussed above. We have the right to use or grant licenses to all of our PEGylation technology for all purposes, including for our own proprietary products or those we may develop with co-commercialization partners or for those that may be developed by third parties. As part of the sale of the specialty pharmaceutical business, we are entitled to royalties of from 5 and 10 percent on net sales of the four marketed products (AdagenÒ, OncasparÒ, AbelcetÒ, and DepoCytÒ) above certain baseline net sales until 2015. DEVELOPMENT AND COMMERCIALIZATION AGREEMENTS SANTARIS PHARMA A/S LICENSE AGREEMENT We are party to a license agreement with Santaris pursuant to which we hold exclusive rights worldwide, other than in Europe, to develop and commercialize RNA antagonists directed against the HIF-1 alpha, Survivin and Androgen Receptor (AR) targets, as well as RNA antagonists directed against five additional targets selected
by us. During 2006, we made payments to Santaris totaling $11 million to acquire the rights to the HIF-1 alpha, Survivin and AR antagonists and for the identification of five additional targets. The $11 million was reported as acquired in-process research and development. As of December 31, 2010, we have paid an additional $23
million in milestone payments to Santaris. Milestone payments are charged to research and development expense. We could pay an additional $233 million in milestone payments, upon the successful completion of certain compound synthesis and selection, clinical development and regulatory milestones. Any one of the compounds we
are currently studying could be returned to Santaris if the findings of our preclinical or clinical work do not support our continued investment. Santaris also is eligible to receive single-digit royalties from any future product sales of products based on the licensed antagonists. Santaris retains the full right to develop and commercialize
products developed under the agreement in Europe. The agreement terminates upon the earlier of the expiration of the last royalty term for an LNA compound or material breach by either party. The royalty term expires on a country-by-country and product-by-product basis when the last valid LNA platform patent or LNA compound
patent expires not to exceed 21 years with respect to any product. Santaris can terminate the agreement with respect to a specific LNA compound provided by Santaris if we do not achieve certain development milestones for that product. MERCK AGREEMENT Our PEGylation technology was used to develop an improved version of Merck’s product, INTRON A. Merck is responsible for marketing and manufacturing the product, PEGINTRON, worldwide on an exclusive basis and we receive royalties on worldwide sales of PEGINTRON for all indications. Merck’s obligation to pay us
royalties on sales of PEGINTRON terminates, on a country-by-country basis, upon the later of the date the last patent to contain a claim covering PEGINTRON expires in the country or 15 years after the first commercial sale of PEGINTRON in such country. Currently, expirations of our right to receive royalties are expected to occur in
2016 in the U.S., 2018 in Europe and 2019 in Japan. The royalty percentage to which we are entitled will be lower in any country where a PEGylated alpha-interferon product is being marketed by a third party in competition with PEGINTRON where such third party is not Hoffmann-La Roche. 10
We do not supply Merck with PEGINTRON or any other materials and our agreement with Merck does not obligate Merck to purchase or sell specified quantities of any product. Further, we have no involvement in the selling or marketing of PEGINTRON. During the quarter ended September 30, 2007, we sold a 25-percent interest in future royalties payable to us by Merck on sales of PEGINTRON occurring after June 30, 2007 for a net purchase price of $88.7 million. We may receive an additional $15.0 million milestone if certain royalty thresholds are met in 2012, although
achievement of those thresholds is not currently anticipated. NEKTAR AGREEMENT In January 2002, we entered into a PEGylation technology licensing agreement with Nektar under which we granted Nektar the right to grant sub-licenses for a portion of our patents related to our PEGylation technology to third-parties. Effective in January 2007, Nektar’s right to grant additional sublicences was limited to a certain
class of our PEGylation technology. Existing sub-licenses granted by Nektar prior to January 2007 were not affected. We will receive a royalty or a share of Nektar’s profits for any products that utilize our patented PEGylation technology under a license granted by Nektar. The rights to receive royalties from Nektar agreements relating to
CIMZIA and Macugen expire in 2014. We have the rights to use or grant licenses to all of our PEGylation technology for all purposes, including for our own proprietary products or those we may develop with co-commercialization partners or for those that may be developed by third parties. COMPETITION General Competition in the biotechnology industry is intense and based to a significant degree on scientific and technological factors. These factors include, but are not limited to, the availability of patent and other protection of technology and products, the ability to commercialize products and technological developments, the ability to
obtain governmental approval for testing, manufacturing and marketing of products, and the ability to enter into licensing and similar arrangements to facilitate the development of products and meet other business objectives. We compete with biotechnology and specialized biopharmaceutical firms and large pharmaceutical companies
with respect to the partnering and licensing of research and the development of product candidates. These companies, as well as academic institutions, governmental agencies and private research organizations, also compete with us in recruiting and retaining highly qualified scientific personnel and consultants. Many of the companies we
compete with are larger than we are and have substantially greater resources. Technology Customized PEGylation Linker Technology (Customized Linker TechnologyÒ) We are aware that other companies are conducting research on chemically modified therapeutic agents and that certain companies are modifying pharmaceutical products by attaching PEG. Our competitors include The Dow Chemical Company, Nektar Pharmaceuticals, Inc., SunBio Corporation, Mountain View Pharmaceuticals,
Inc., Neose Technologies, Inc., NOF Corporation and Urigen Pharmaceuticals, Inc. There may be other chemical, biotechnology and pharmaceutical companies developing PEGylation technologies and applying such technologies to develop pharmaceutical product candidates. Some of these companies license or provide the technology to
other companies, while others develop the technology for internal use. In addition, there are other delivery technologies (e.g. liposomal, nanoparticles, etc.) that may improve pharmaceutical properties of pharmaceutical compounds. Third-generation mRNA-targeting agents utilizing the Locked Nucleic Acid (LNA) Technology We are aware that other companies are conducting research and developing products utilizing antisense technologies, siRNA/RNAi or targeting micro RNA, that compete with the LNA technology. These include Isis Pharmaceuticals, Inc. Alnylam Pharmaceuticals, Inc., Regulus Therapeutics LLC, Eli Lilly and Company and others. 11
Product Candidates HIF-1 alpha antagonist. There are a number of existing therapeutic regimens designed to treat the cancers that we may target with the HIF-1 alpha antagonist. However, we are not of aware of any development of another compound that would have a mechanism similar to our HIF-1 alpha mRNA antagonist. Survivin antagonist. There are a number of existing therapeutic regimens designed to treat the cancers that we may target with the Survivin antagonist. We are aware of several companies, including Isis Pharmaceuticals/Eli Lilly, Astellas Pharma, Erimos Pharmaceuticals and Aegera Therapeutics, that are actively working on
compounds targeting Survivin. Androgen Receptor (AR) antagonist. There are a number of existing therapeutic regimens designed to treat the cancers that we may target with the AR antagonist. However, we are not aware of the development of another compound that would have a mechanism similar to our AR mRNA antagonist. This is because our AR mRNA
antagonist uniquely targets AR directly, unlike the other compounds in the market or in development that we are aware of, and may complement other antiandrogen therapies. There are two novel compounds of other companies that target AR that may receive FDA approval this year. PEG-SN38. There are a number of drugs in various stages of preclinical and clinical development from companies exploring cancer therapies or improved chemotherapeutic agents to potentially treat the same cancer indications that our PEG-SN38 may be developed to treat. Additionally, there are a number of drugs in development
based on the active metabolite SN38. If these drugs are approved, they could compete directly with our PEG-SN38. These include products in development from Bristol-Myers Squibb Company, Pfizer Inc., GlaxoSmithKline plc, Antigenics Inc., Hoffman-La Roche Ltd., Novartis AG, Cell Therapeutics, Inc., Neopharm, Inc., Meditech
Research Limited and others. Nektar Therapeutics is also developing a PEGylated form of irinotecan. Irinotecan is a pro-drug of SN38. Nektar has reported that this product candidate is currently in Phase II trials. Royalties PEGINTRON PEGINTRON, marketed by Merck, competes directly with Hoffmann-La Roche’s Pegasys. Merck and Hoffmann-La Roche have been the major competitors in the global alfa interferon market since the approval of their unmodified alpha interferon products, INTRON A and ROFERON-A, respectively, and the PEGylated
interferon-based combination therapy is a highly competitive market. Further, Merck has reported that the overall hepatitis C market has been contracting. Additionally, there is much research being conducted on various formulations of alpha interferon as well as many non-Interferon-based compounds being investigated for the treatment
of hepatitis C. Two novel agents, protease inhibitors, boceprevir (Merck) and Telaprevir (Vertex/Johnson & Johnson) completed Phase III programs and are under review at the FDA. Boceprevir was studied in combination with PEGINTRON and Telaprevir was studied in combination with ROFERON-A. It is expected that both compounds
will be approved in 2011. Furthermore, there are several novel agents in various stages of preclinical and clinical development in combination with pegylated Interferons. It is possible that this research could lead to a competing product or ultimately to Interferon-free combination therapy in the future. Macugen Macugen, marketed by OSI Pharmaceuticals, Inc. and Pfizer Inc., currently competes against three therapies for the treatment of neovascular (wet) age-related macular degeneration (AMD): photodynamic therapy with verteporfin, which was developed by QLT, Inc. and is marketed by Novartis AG; thermal laser treatment; and
ranibizumab, marketed under the brand name LucentisTM by Genentech. Ranibizumab, approved in June 2006, for the treatment of AMD, has provided significant competition to Macugen, which we expect to continue. Additional treatments for AMD are in various stages of preclinical or clinical testing. If approved, these treatments
would also compete with Macugen. 12
CIMZIA CIMZIA marketed by UCB Pharmaceuticals, Inc. currently competes against therapies for the treatment of moderate to severe rheumatoid arthritis and Crohn’s disease. CIMZIA is a biologic medicine that intercepts a messenger protein in the joints (tumor necrosis factor or TNF) that promotes inflammation of the joints in
rheumatoid arthritis. Other TNF inhibitors approved for the treatment of rheumatoid arthritis include Etanercept, infliximab, adalimumab, and golimumab. Infliximab and adalimumab are also used in the treatment of Crohn’s disease. Both diseases also have additional approved treatments that are not TNF inhibitors, as well as other
treatments in various stages of preclinical or clinical testing. If approved, these treatments would also compete with CIMZIA. PATENTS AND INTELLECTUAL PROPERTY RIGHTS Patents are very important to us in establishing the proprietary rights to the products we have developed or licensed. Our executive management team has reinforced our organizational commitment to intellectual property. The patent position of pharmaceutical or biotechnology companies can be uncertain and involve complex legal,
scientific and factual questions. If our intellectual property positions are challenged, invalidated or circumvented, or if we fail to prevail in potential future intellectual property litigation, our business could be adversely affected. We have an extensive portfolio of issued U.S. patents and filed applications, many of which have foreign
counterparts. These patents, if extensions are not granted, are expected to expire beginning in 2011 through 2030. Under various license agreements, we have received exclusive licenses to patents that relate to certain of the products we or our partners have commercialized or that we have under development. We have exclusively licensed
patents from Santaris Pharma related to our LNA clinical candidates and our other LNA compounds in development. Of the patents owned or exclusively licensed by us, two relate to PEGINTRON, three relate to our HIF-1 alpha antagonist, two relate to our Survivin antagonist and one relates to our AR antagonist. Although we believe
that our patents provide certain protection from competition, we cannot assure you that such patents will be of substantial protection or commercial benefit to us, will afford us adequate protection from competing products, or will not be challenged or declared invalid. In addition, we cannot assure you that additional U.S. patents or
foreign patent equivalents will be issued to us. Patents for individual products extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends upon the type of patent, the scope of its
coverage and the availability of legal remedies in the country. The patent covering our original PEG technology, which we had licensed from Research Corporation Technologies, Inc., contained broad claims covering the attachment of PEG to polypeptides. However, this U.S. patent and its corresponding foreign patents have expired. Based upon the expiration of the Research Corporation
patent, other parties may make, use, or sell products covered by the claims of the Research Corporation patent, subject to other patents, including those that we hold. We have obtained and intend to continue to pursue patents with claims covering improved methods of attaching or linking PEG to therapeutic compounds. We also have
obtained patents relating to the specific composition of the PEG-modified compounds that we have identified or created. We will continue to seek such patents as we develop additional PEG-enhanced products. We cannot assure you that we will be able to prevent infringement by unauthorized third parties or that competitors will not
develop competitive products outside the protection that may be afforded by our patents. We have three patents that relate to our PEG-SN38 clinical candidate. We are aware that others have filed patent applications and have been granted patents in the U.S. and other countries with respect to the application of PEG to proteins and other compounds. Owners of such patents may seek to prevent us or our collaborators from making, using or selling our products. In the field of SCA proteins, we have several U.S. and foreign patents and pending patent applications. We have obtained licenses from various parties that we deemed to be necessary or desirable for the manufacture, use, or sale of our products. These licenses generally require the payment of royalties to the licensor based on product sales. In addition, other companies have filed patent applications or have been granted patents in
areas of interest to us. There can be no assurance that any licenses required under such patents will be available to us on acceptable terms. 13
As part of the January 2010 sale of our specialty pharmaceutical business, we assigned to the purchaser the patents and patent applications which we owned that relate to current and new formulations of Oncaspar and Adagen and the licenses to patents that relate to Abelcet and DepoCyt. We also assigned all trademarks related to
Abelcet, DepoCyt, Oncaspar and Adagen to the purchaser of our specialty pharmaceutical business. GOVERNMENT REGULATION The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements on the clinical development, manufacture, and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the
inspection, testing, manufacture, quality assurance, safety, effectiveness, labeling, packaging, storage, distribution, record-keeping, approval, and promotion of products. All of our products will require regulatory approval before commercialization. In particular, therapeutic products for human use are subject to rigorous preclinical and
clinical testing and other requirements of the Federal Food, Drug, and Cosmetic Act and the Public Health Service Act, implemented by the FDA, as well as similar statutory and regulatory requirements of foreign countries. Obtaining these marketing approvals and subsequently complying with ongoing statutory and regulatory
requirements is costly and time consuming. Any failure by us or our collaborators, licensors or licensees to obtain, or any delay in obtaining, regulatory approval or in complying with post-approval requirements, could adversely affect the marketing and sale of products that we are developing and our ability to receive product or royalty
revenues. The steps required before a new drug or biological product may be distributed commercially in the U.S. generally include:
•
conducting appropriate preclinical laboratory evaluations of the product’s chemistry, formulation and stability, and animal studies to assess the potential safety and efficacy of the product, • submitting the results of these evaluations and tests to the FDA, along with manufacturing information, analytical data and clinical investigational plan, in an IND, • obtaining IND acceptance from the FDA, which may require the resolution of any safety or regulatory concerns of the FDA, • obtaining approval of Institutional Review Boards or IRBs, prior to introducing the drug or biological product into humans in clinical trials and registering clinical trials in public databases such as clinicaltrials.gov, • conducting adequate and well-controlled human clinical trials that establish the safety and efficacy of the drug or safety, purity and potency of the biological product candidate for the intended use, in the following three typically sequential, stages: Phase I. The product candidate is initially introduced into healthy human subjects or patients and tested for safety, increased dose tolerance, and possibly absorption, distribution, metabolism and excretion, Phase II. The product candidate is studied in patients with the targeted condition to gain safety experience at the proposed dosing schedules, identify possible adverse effects and safety risks to determine the optimal dosage, and to obtain initial information on effectiveness of the product candidate, Phase III. The product candidate is studied in an expanded patient population at multiple clinical trial sites to determine primary efficacy and safety endpoints identified at the start of the clinical trial,
•
submitting the results of preliminary research, preclinical studies, and clinical studies as well as chemistry, manufacturing and control information on the drug or biological product to the FDA in a New Drug Application or NDA, for a drug product, or a BLA for a biological product, and • obtaining FDA approval of the NDA or BLA prior to any commercial sale or shipment of the drug or biological product. An NDA or BLA must contain, among other things, data derived from non-clinical laboratory studies and clinical trials which demonstrate that the product is safe and effective and for a biological product that it meets 14
prescribed standards of safety, purity and potency, and a full description of manufacturing methods. Biological or drug products may not be marketed in the U.S. until approval by the FDA of an NDA or BLA is received. The approval process can take a number of years, if approval is obtained at all, and often requires substantial financial resources, including license application fees. The results of preclinical studies and initial clinical trials are not necessarily predictive of the results from large-scale clinical trials, and clinical trials may be subject to
additional costs, delays or modifications due to a number of factors, including the difficulty in obtaining enough patients, clinical investigators, drug supply, or financial support. Certain clinical trials performed under an IND must be registered in the official clinical trial website, and non-compliance can result in significant fines. The
FDA has the power to impose changes relating to safety and efficacy of approved products. The FDA can impose substantial fines if these requirements are not carried out to the agency’s full satisfaction. Upon approval, a drug product or biological product may be marketed only in those dosage forms and for those indications approved
in the NDA or BLA, although information about off-label indications may be disseminated upon a physician’s request in narrowly defined situations. In addition to obtaining FDA approval for each indication for which the manufacturer may market the drug, each domestic drug product manufacturing establishment must register with the FDA, list its drug products with the FDA, comply with and maintain current Good Manufacturing Practices (cGMP) and permit and pass
inspections by the FDA and other regulatory authorities. Moreover, the submission of applications for approval may require the preparation of large-scale production batches that cannot be used commercially and additional time to complete manufacturing stability studies. Any products manufactured or distributed by us pursuant to FDA approvals are subject to extensive continuing regulation by the FDA, including record-keeping requirements and a requirement to report adverse experiences with the product. In addition to continued compliance with standard regulatory requirements, the FDA also
may require post-marketing testing and surveillance to monitor the safety and efficacy of the marketed product. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product are discovered following approval. The Federal Food, Drug, and Cosmetic Act mandates that drug products be manufactured consistent with cGMP. In complying with the FDA’s regulations on cGMP, manufacturers must continue to spend time, money and effort in production, record-keeping, quality control, quality assurance, and auditing to ensure that the marketed
product meets applicable specifications and other requirements. The FDA periodically inspects drug product manufacturing facilities to ensure compliance with cGMP. Failure to comply with cGMP or other FDA requirements subjects the manufacturer to possible FDA action, such as:
•
untitled and warning letters, • suspension of manufacturing, • seizure of a product, • voluntary recall of a product, • injunctive actions and • civil or criminal penalties. To the extent we rely on third parties to manufacture our compounds and products, those third parties will be required to comply with cGMP as required by regulations. Even after FDA approval has been obtained, and often as a condition to expedited approval, further studies, including post-marketing studies, are typically required by the FDA. Results of post-marketing studies may limit or expand the further marketing of the products. If we propose any modifications to the product, including
changes in indication, manufacturing or testing processes, manufacturing facility or labeling, an NDA or BLA supplement may be required to be submitted to and approved by the FDA. Products manufactured in the U.S. for distribution abroad will be subject to FDA regulations regarding export, as well as to the requirements of the country to which they are shipped. These latter requirements apply to products studied in clinical trials, the submission of marketing applications, and all aspects of product manufacture
and marketing. Such requirements vary significantly from country to country. As part of our 15
strategic relationships, our collaborators may be responsible for the foreign regulatory approval process of our products, although we may be legally liable for noncompliance. We cannot predict the extent of government regulation that might result from current or future legislation or administrative action. Moreover, we anticipate that the presidential administration, Congress, state legislatures and the private sector will continue to review and assess controls on health care spending. Any such proposed or
actual changes could cause us or our collaborators to limit or eliminate spending on development projects and may otherwise impact us. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might result from current or future legislative or administrative action, either in the U.S. or abroad.
Additionally, in both domestic and foreign markets, sales of our proposed products will depend, in part, upon the availability of reimbursement from third-party payors, such as government health administration authorities, managed care providers, private health insurers and other organizations. Significant uncertainty often exists as to the
reimbursement status of newly approved health care products. In addition, third-party payors are increasingly challenging the price and cost-effectiveness of medical products and services. There can be no assurance that our product candidates will be considered cost-effective or that adequate third-party reimbursement will be available to
enable us to maintain price levels sufficient to realize an appropriate return on our investment in product research and development. PEGINTRON has been approved for treatment of hepatitis C in the European Union, the U.S., Japan and China, and for the treatment of hepatitis B in China. None of the product candidates we are developing have been approved for marketing in the U.S. or elsewhere. With respect to patented products, delays imposed by the government approval process may materially reduce the period during which we will have the exclusive right to exploit them. Our operations are also subject to federal, state and local environmental laws and regulations concerning, among other things, the generation, handling, storage, transportation, treatment and disposal of hazardous, toxic and radioactive substances and the discharge of pollutants into the air and water. Environmental permits and
controls are required for some of our operations and these permits are subject to modification, renewal and revocation by the issuing authorities. We believe that our facilities are in compliance with our permits and environmental laws and regulations and do not believe that future compliance with current environmental laws will have a
material adverse effect on our business, financial condition or results of operations. If, however, we were to become liable for an accident, or if we were to suffer an extended facility shutdown as a result of such contamination, we could incur significant costs, damages and penalties that could harm our business. EMPLOYEES As of December 31, 2010, we employed 126 persons, including 33 persons with Ph.D. or M.D. degrees. At that date, 101 employees were engaged in research and development activities, and 25 were engaged in administration. None of our employees are covered by a collective bargaining agreement. All of our employees are
covered by confidentiality agreements. We consider our relations with our employees to be good. Once the December 2010 restructuring is fully implemented, we expect to have just under 100 employees. The majority of the reductions in force will occur during the first quarter of 2011. Many of the affected employees presently are involved in the research and development organization in support of the transition services being
provided to the purchaser of our specialty pharmaceutical business. These services are rapidly winding down. Throughout this Annual Report on Form 10-K, we have made forward-looking statements in an attempt to better enable the reader to understand our future prospects and make informed judgments. By their nature, forward-looking statements are subject to numerous factors that may influence outcomes or even prevent their eventual
realization. Such factors may be external to Enzon and entirely outside our control. We cannot guarantee that our assumptions and expectations will be correct. Failure of events to be achieved or of certain underlying assumptions to prove accurate could cause actual results to vary materially from past results and those anticipated or projected. We do not intend to update forward-looking statements. 16
Certain risks and uncertainties are discussed below. However, it is not possible to predict or identify all such factors. Accordingly, you should not consider this recitation to be complete. Risks Relating to Our Business We expect to incur losses over the next several years and may never achieve or sustain profitability. We have limited sources of revenues and we expect to incur losses over the next several years including for the year ending December 31, 2011. We also expect to spend significant amounts to continue research and development of our product candidates and technologies. None of our product candidates have been approved by the FDA and none of them have been commercialized. We do not know when we will have products approved by the FDA or commercialized, if ever. In the absence of revenue from the sale of products or other new sources, our losses will continue as we conduct our research
and development activities. Development of any successful product candidates is highly uncertain due to the extended testing and regulatory review process required before marketing clearance can be obtained and failure to develop, obtain regulatory approval and commercialize our product candidates could materially harm our business. There is a high risk of failure for pharmaceutical product candidates. Only a small minority of all research and development programs ultimately result in commercially successful drugs. We may never succeed in developing an approved drug. Further, due to the extended testing and regulatory review process required before
marketing clearance can be obtained, the time periods before commercialization of any of these products are long and uncertain. Risks during development and commercialization include the possibility that: any or all of our product candidates will be found to be ineffective; our product candidates will have adverse side effects or will
otherwise fail to receive the necessary regulatory approvals; our product candidates may be effective but uneconomical to manufacture or market; or our competitors may market equivalent or superior products. The risk of failure is increased for our product candidates that are based on new technologies or approaches to the development of therapeutics. For example, the LNA technology is a novel technology and there are currently no approved drugs, or even late-stage drug candidates, employing this technology. Product candidates
employing these technologies may not advance to pivotal stages of product development or demonstrate clinical safety or efficacy. If we do not succeed in the development of these product candidates, or if our technologies fail to generate products, our business could be materially harmed. From time to time, we may establish and announce certain development goals for our product candidates and programs; however, given the complex nature of the drug discovery and development process, it is difficult to predict accurately if and when we will achieve these goals. If we are unsuccessful in advancing our preclinical
programs into clinical testing, advancing our clinical programs into later clinical phases, or in obtaining regulatory approval, our business prospects may be harmed. We do not expect any of the drugs resulting from our current research and development efforts to be commercially available for several years, if at all. In order to fill our pipeline of product candidates under development, we may attempt to acquire rights to products under development by other companies. The competition for the
acquisition of rights to products that are viewed as viable candidates for successful development and commercialization is intense, and we will be competing for such opportunities with many companies with resources that are substantially greater than ours. Our financial results are heavily dependent on the continued sales of PEGINTRON on which we receive royalties; if revenues from these royalties materially decline, our results of operations and financial position could be materially harmed. Our results of operations are heavily dependent on the royalty revenues we receive on the sale of PEGINTRON, marketed by Merck. As a consequence, a continued decline in the sales of PEGINTRON could adversely affect our operating results and financial position. We cannot assure you that Merck will continue to 17
generate sales of PEGINTRON at levels that would enable us to receive royalties in amounts that are comparable with the amounts of royalties we have received in recent years. The amount and timing of resources dedicated by Merck to the marketing of PEGINTRON is not within our control. Our royalty revenues will be negatively
affected if sales of PEGINTRON are limited for any reason, including if Merck cannot market PEGINTRON effectively as a result of competitive, manufacturing, regulatory or other issues. Products that compete with PEGINTRON have been and potentially will be introduced by other drug manufacturers. Hoffmann-La Roche’s Pegasys, a competing PEGylated interferon-based combination therapy, has resulted in significant competitive pressure on PEGINTRON sales in the U.S. and all international markets. Pegasys
has taken market share away from PEGINTRON and the overall market for PEGylated alpha-interferon for the treatment of hepatitis C has been contracting. As a result, sales of PEGINTRON in certain markets where it competes with Pegasys and the royalties we receive on those sales have declined. We cannot assure you that Pegasys
will not continue to gain market share at the expense of PEGINTRON which could result in lower PEGINTRON sales and lower royalties to us. There are several novel agents in various stages of preclinical and clinical development in combination with PEGylated interferon for the treatment of hepatitis C. It is possible that this research
could lead to a competing product or ultimately to Interferon-free combination therapy in the future. We may require additional financing to meet our future capital needs and failure to obtain such funding could have a material and adverse effect on our business, financial condition and operations. Our research and development projects require substantial capital. We will continue to expend substantial resources for research and development, including costs associated with developing our product candidates and conducting clinical trials. We believe that our current cash and investments will be adequate to satisfy our capital
needs for the near future, but we have limited sources of revenue and we may require additional financing to meet our future capital needs. We may require substantial additional capital to:
•
fund research and development activities; • conduct pre-clinical studies and clinical trials; and • undertake other activities relating to the successful development of product candidates. Our future capital needs depend on many factors, including:
•
the scope, duration and expenditures associated with our research and development programs; • continued scientific progress in these programs; • the outcome of potential licensing transactions, if any; • competing technological developments; • our proprietary patent position in our products; and • the regulatory approval process for our product candidates. We may seek to raise necessary funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements. Any additional equity financings may be on terms that are dilutive or potentially dilutive to our stockholders. Any debt financing we enter into may involve incurring significant
interest expense and include covenants that restrict our operations. We may not be able to obtain additional financing on terms favorable to us, if at all. General market conditions may make it difficult for us to seek financing from the capital markets. We may be required to relinquish rights to our technologies or drug candidates, or grant
licenses on terms that are not favorable to us, to raise additional funds through alliance, joint venture or licensing arrangements. If adequate funds are not available, we may have to delay, reduce or eliminate one or more of our research or development programs and reduce overall overhead expenses. These actions could have a material
adverse effect on our business, financial condition and operations. 18
We depend on our collaborative partners; if we lose our collaborative partners or they do not apply adequate resources to our collaborations, our product development and financial performance may suffer. We rely and will depend heavily in the future on collaborations with partners, primarily pharmaceutical and biotechnology companies, for one or more of the research, development, manufacturing, marketing and other commercialization activities relating to most of our product candidates. If we lose our collaborative partners, or if
they do not apply adequate resources to our collaborations, our product development and financial performance may suffer. The amount and timing of resources dedicated by our collaborators to their collaborations with us are not within our control. If any collaborator breaches or terminates its agreements with us or fails to conduct its collaborative activities in a timely manner, the commercialization of our product candidates could be slowed or blocked
completely. For example, Santaris can terminate its agreement with respect to a specific LNA compound provided by them if we do not achieve certain development milestones for that compound or if we do not make certain milestone payments. In addition, our collaborative partners may change their strategic focus, pursue alternative
technologies or develop alternative products as a means of developing treatments for the diseases targeted by these collaborative programs and these could compete with products we are developing. Further, our collaborations may not be successful. Disputes may arise between us and our collaborators as to a variety of matters, including financing obligations under our agreements and ownership of intellectual property rights. These disputes may be both expensive and time-consuming and may result in delays in the development
and commercialization of products. If any of the product candidates that we are commercializing with collaborators are delayed or blocked from entering the market or we experience increased costs as a result of our relationship with our collaborators, our financial performance could be adversely affected. We purchase some of the compounds utilized in our product candidates from a single source or a limited group of suppliers, and the partial or complete loss of one of these suppliers could cause production delays and a substantial loss of revenues. We purchase the unmodified pharmaceutical compounds, bulk PEGs and other compounds used to manufacture our product candidates from outside suppliers. In some cases, we have a limited number of suppliers. Our reliance on our suppliers exposes us to significant risks. These third parties might:
•
be unable or unwilling to provide us with sufficient materials to meet our demands; • fail to meet our standards of quality or other specifications; • increase significantly the prices they charge us for materials; or • not carry out their contractual duties or meet anticipated deadlines, which could result in delays in obtaining or maintaining regulatory approvals. If our suppliers are unwilling or unable to timely supply us with materials meeting our specifications, we may not be able to locate any alternative suppliers or enter into commercially reasonable agreements with suppliers in a timely manner or at all. In addition, we may be unable to find alternative suppliers with appropriate
regulatory authorizations. If we experience a delay in obtaining or are unable to obtain any compound for the manufacture of our product candidates on reasonable terms, or at all, it could have a material adverse effect on our business, financial condition and results of operations. Our product candidates must undergo extensive clinical testing, the results of which are highly uncertain and could substantially delay or prevent us from obtaining regulatory approval. Before we can market a product, we must obtain regulatory approval for a product candidate. To obtain regulatory approval, we must undertake extensive clinical testing in humans to demonstrate safety and efficacy to the satisfaction of the FDA and similar foreign regulatory authorities for each indication. The pre-clinical testing
and clinical trials for any product candidates that we develop must comply with the regulations of 19
numerous federal, state and local government authorities in the U.S., principally the FDA, and those of similar agencies in other countries. Clinical trials of new product candidates sufficient to obtain regulatory marketing approval are expensive and take years to complete. Even though they consume substantial resources, the outcome of these trials is highly uncertain. Safety and efficacy results from pre-clinical studies involving animals and other models and from early clinical trials are often not accurate indicators of results of later-stage clinical trials that involve larger human populations, and,
moreover, may not always be representative of results obtained while marketing an approved drug, particularly with regard to safety. In addition, we may suffer significant setbacks in clinical trials, even after achieving promising results in earlier trials. For example, Phase II data may not replicate Phase I results or Phase III efficacy data
may not replicate Phase II data. Any adverse results from studies, including clinical trials, could have a negative effect on our ability to obtain the approval of the FDA or other regulatory agencies. Unfavorable results of clinical trials conducted by our competitors or other biotechnology companies could also adversely affect our ability to
gain regulatory approval of our product candidates by increasing government examination and complexity of clinical trials. Government and public concerns over safety issues associated with pharmaceutical and biological products could potentially result in termination of clinical trials on entire classes of drug candidates, lengthen the
trial process for product categories, increase legal and production costs relating to certain drug categories, and/or expand the safety labeling for approved products. Clinical development of any product candidate that we decide to take into clinical trials may be delayed or prevented at any time for some or all of the following reasons:
•
negative or ambiguous results regarding the efficacy of the product candidate; • undesirable side effects that delay or extend the trials or make the product candidate not medically or commercially viable; • inability to recruit and qualify a sufficient number of patients for our trials; • regulatory delays or other regulatory actions, including changes in regulatory requirements; • difficulties in obtaining sufficient quantities of the product candidate manufactured under current good manufacturing practices; • delays, suspension or termination of the trials imposed by us, an independent institutional review board for a clinical trial site, or clinical holds placed upon the trials by the FDA; and • our failure to obtain adequate financial resources to fund these trials. We depend on third parties to conduct the clinical trials for our product candidates and any failure of those parties to fulfill their obligations could harm our development and commercialization plans. We depend on independent clinical investigators, contract research organizations, academic institutions and other third-party service providers to conduct clinical trials for our product candidates. Though we rely heavily on these parties for successful execution of our clinical trials, we are ultimately responsible for the results of their
activities and many aspects of their activities are beyond our control. For example, we are responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial, but the independent clinical investigators may prioritize other projects over ours or may fail to timely
communicate issues regarding our products to us. Third parties may not complete activities on schedule or may not conduct our clinical trials in accordance with regulatory requirements or our stated protocols. The early termination of any of our clinical trial arrangements, the failure of third parties to comply with the regulations and
requirements governing clinical trials or our reliance on results of trials that we have not directly conducted or monitored could hinder or delay the development, approval and commercialization of our product candidates and would adversely affect our business, results of operations and financial condition. If our clinical trials are not successful, if we experience significant delays in these trials, or if we do not complete our clinical trials, we may not be able to commercialize our product candidates, which would materially harm our business. 20
We depend on patents and proprietary rights, which may offer only limited protection against potential infringement and the development of competing products. The pharmaceutical industry places considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Our success depends, in part, on our ability to develop and maintain a strong patent position for our product candidates and technologies both in the U.S. and in other countries and
to protect our proprietary rights. If we are unable to obtain and enforce patent protection for our product candidates or to maintain the confidentiality of our trade secrets, our business could be materially harmed. We have an extensive portfolio of issued U.S. patents and filed applications, many of which have foreign counterparts. In
addition, under our license agreements, we have exclusively licensed patents related to our HIF-1 alpha, Survivin and AR antagonists and our other LNA compounds in development. Although we believe that our patents provide certain protection from competition, such patents may not provide substantial protection or commercial benefit
to us, or afford us adequate protection from competing products, and may be challenged or declared invalid. In addition, U.S. patents or foreign patent equivalents may not be issued to us in the future. Issued patents may be challenged, invalidated or circumvented. In addition, court decisions may introduce uncertainty as to the enforceability or scope of patents owned by biotechnology and pharmaceutical companies, including us. The legal systems of certain countries do not favor the aggressive enforcement of patents, and the
laws of foreign countries may not protect our rights to the same extent as the laws of the U.S. Therefore, enforceability or scope of our patents in the U.S. or in foreign countries cannot be predicted with certainty, and, as a result, any patents that we own or license may not provide sufficient protection against competitors. In addition, we
may not be able to obtain or maintain a patent from our pending patent applications, those we may file in the future, or those we may license from third parties. We believe that our patent rights are enforceable; however, those rights may prove unenforceable or invalid, or may expire prior to the commercialization of our product candidates, thus allowing others to more effectively compete with us. Therefore, any patents that we own or license may not adequately protect our product
candidates or our future products. If we are not able to protect our patent positions, our business and financial condition could be materially harmed. We may become aware that certain organizations are engaging in activities that infringe certain of our patents, including our PEGylation technology patents. We may be unable to enforce
our patents and other rights against such organizations. Legal or administrative proceedings may be necessary to enforce our intellectual property rights or to defend against claims of infringement. We have in the past been involved in patent litigation and other proceedings and we may likely become involved in additional patent litigation or proceedings in the future. If we become
involved in any such litigation or proceeding, irrespective of the outcome, we may incur substantial costs, the efforts of our technical and management personnel may be diverted, and such disputes could substantially delay or prevent our product development or commercialization activities, which could materially harm our business,
financial condition and results of operations. Blocking patents or claims of infringement may stop or delay the development of our product candidates. Other entities may have or obtain proprietary rights that could impair our competitive position. Our commercial success depends in part on avoiding claims of infringement of the patents or proprietary rights of such third parties. Although we investigate the patent protection surrounding our technology and product candidates, there
are numerous patents, each with multiple claims, which makes it difficult to uncover and interpret the extent of patent protection which can lead to uncertainty about our freedom to operate. It is possible that we will not be aware of issued patents or pending patent applications that are relevant to our product candidates because our
searches do not find them or because they are not yet publicly available. Our interpretation of patents could be challenged, leading to litigation, and we could face claims of infringement of rights of which we are unaware. There have been significant litigation and interference proceedings regarding patent rights, and the patent situation regarding particular products is often complex and uncertain. As we proceed with the development of our product candidates, we may face uncertainty and litigation could result, which could lead to liability for 21
damages, prevent our development and commercialization efforts and divert resources from our business strategy. Third parties from time to time may assert that we are infringing their patents, trade secrets or know-how. In addition, our technology may infringe patents that may issue in the future to third parties. We could incur substantial costs in defending ourselves and our partners against any such claims. Furthermore, parties making such
claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability or our partners’ ability to further develop or commercialize some or all of our products or technology in the U.S. and abroad, and could result in the award of substantial damages. If we are found to infringe, we may be required to
obtain one or more licenses from third parties or be unable to proceed with development or commercialization of our product candidates. We may not be able to obtain such licenses at a reasonable cost, if at all. Defense of any lawsuit or failure to obtain any such required license could have a material adverse effect on us. We may have to develop or license alternative technologies if we are unable to obtain key technology from third parties or maintain our rights to technology we license from third parties. We have licensed patents and patent applications from Santaris under our collaboration and license agreement with them. Some of our proprietary rights have been licensed to us under agreements that have performance requirements or other contingencies. The failure to comply with these provisions could lead to termination or
modifications of our rights to these licenses. Additionally, we may need to obtain additional licenses to patents or other proprietary rights from other parties to facilitate development of our proprietary technology base. The ownership of patents exclusively licensed to us may be subject to challenge if inventorship was not adequately
investigated and represented. If our existing licenses are terminated or if we are unable to obtain such additional licenses on acceptable terms, our ability to perform our own research and development and to comply with our obligations under our collaborative agreements may be delayed while we seek to develop or license alternative
technologies. The patents upon which our original PEGylation technology was based have expired and, as a result, the scope of our patent protection is narrower. The U.S and corresponding foreign patents upon which our original PEGylation technology was based expired in 1996. Without that patent protection, other parties are permitted to make, use or sell products covered by the claims of those patents, subject to other patents, including those which we hold. We have obtained numerous
patents with claims covering improved methods of attaching or linking PEG to therapeutic compounds. However, these patents may not enable us to prevent competition or competitors may develop alternative methods of attaching PEG to compounds potentially resulting in competitive products outside the protection that may be afforded
by our patents. We are aware that others have also filed patent applications and have been granted patents in the U.S. and other countries with respect to the application of PEG to proteins and other compounds. The manufacture of our product candidates is a complex and highly-regulated process and we rely on third-party manufacturers to manufacture materials for us. If any of them fails to meet regulatory requirements, our business could suffer. The FDA and foreign and state regulators require manufacturers to register manufacturing facilities. The FDA and these regulators also inspect these facilities to confirm compliance with current good manufacturing practices or similar requirements that the FDA and these regulators establish. The manufacture of product candidates
and key reagents at any facility is subject to strict quality control, testing, and record keeping requirements, and continuing obligations regarding the submission of safety reports and other post-market information. Ultimately, we, our third-party manufacturers, our licensees, or other suppliers may not meet these requirements. Our third-
party manufacturers may face manufacturing or quality control problems causing product production and shipment delays or a situation where we or they may not be able to maintain compliance with the FDA’s current good manufacturing practices requirements or those of foreign or state regulators, necessary to continue manufacturing
our product candidates and materials. Any failure to comply 22
with current good manufacturing practices requirements or other FDA and foreign or state regulatory requirements could adversely affect our clinical research activities and our ability to market and develop our products candidates. We may be subject to a variety of types of product liability or other claims based on allegations that the use of our product candidates by participants in our clinical trials has resulted in adverse effects, and our insurance may not cover all product liability or other claims. We may face liability claims related to the use or misuse of our product candidates in clinical trials. These claims may be expensive to defend and may result in large judgments against us. Generally, our clinical trials are conducted in patients with serious life-threatening diseases for whom conventional treatments have been
unsuccessful and during the course of treatment these patients could suffer adverse medical effects or die for reasons that may or may not be related to our product candidates. Any of these events could result in a claim of liability. Any such claims against us, regardless of their merit, could result in significant costs to defend or awards
against us that could materially harm our business, financial condition or results of operations. Although we maintain product liability insurance for claims arising from the use of our product candidates in clinical trials prior to FDA approval and for claims arising from the use of our products after FDA approval at levels that we believe are appropriate, we may not be able to maintain our existing insurance coverage or obtain
additional coverage on commercially reasonable terms for the use of our other product candidates and products in the future. Also, our insurance coverage and resources may not be sufficient to satisfy any liability resulting from product liability claims which could materially harm our business, financial condition or results of operations. We depend on key personnel and may not be able to retain these employees or recruit additional qualified personnel, which would harm our business; we currently do not have a permanent chief financial officer. Because of the specialized scientific nature of our business, we are highly dependent upon qualified research and development scientists, technical and managerial personnel, including our President of Research and Development. There is intense competition for qualified personnel in the pharmaceutical field. Therefore, we may not
be able to attract and retain the qualified personnel necessary for the development of our business. Although we have an employment agreement with our President of Research and Development, our ability to continue to retain him, as well as other senior executives or key managers is not assured. Effective July 23, 2010, Craig A. Tooman resigned as our Executive Vice President of Finance and Chief Financial Officer. On July 8, 2010, our board of directors appointed Mark L. Ogden, consultant to Enzon since 2005, as our acting Vice President of Finance and Principal Financial Officer following Mr. Tooman’s departure.
The executive committee continues to search for a permanent Chief Financial Officer. The loss of the services of one or a combination of our senior executives, particularly our President of Research and Development, as well as the failure to recruit additional key research and development scientists, technical and managerial personnel, particularly a new Chief Financial Officer, in a timely manner, could have an
adverse effect on our business. As a result of our 2010 restructuring initiatives and the related reductions in our workforce, we are in the process of reallocating certain employment responsibilities and may outsource certain corporate functions which could have the potential to affect our internal controls over financial reporting and make us more
dependent on third-parties to perform these corporate functions. In both the first and fourth quarters of 2010, we initiated restructurings which together will result in the elimination of 97 employee positions. The reductions will result in the reallocation of certain responsibilities which could adversely impact operational efficiencies, employee performance, employee retention or internal controls.
Also, as a result of these reductions, we may be required to outsource certain corporate functions which will make us more dependent on third-parties for the performance of these functions in connection with our business and product candidates. To the extent that we are unable to effectively reallocate employee 23
responsibilities, retain key employees, effectively design adequate internal and compensating controls, establish and maintain agreements with competent third-party contractors on terms that are acceptable to us, or effectively manage the work performed by any retained third-party contractors, our ability to advance our business or
product candidates may be significantly impaired and our stock price may be adversely affected. Risks Relating to Our Industry Significant competition for our technology platforms and product candidates could make our technologies or product candidates obsolete or uncompetitive, which would negatively impact our business, results of operations and financial condition. The biopharmaceutical industry is characterized by extensive research and development effort, and rapid technological change. Our future success will depend on our ability to maintain a competitive position with respect to technological advances. Rapid technological development by others may result in our product candidates and
technologies becoming obsolete. We face intense competition from established biotechnology and pharmaceutical companies, as well as academic and research institutions that are pursuing competing technologies and products. We know that competitors are developing or manufacturing various platform technologies and products that are
used for the prevention, diagnosis or treatment of diseases that we have targeted for product development. Current and prospective competing products may provide greater therapeutic benefits for a specific problem or may offer comparable performance at a lower cost when compared to our product candidates. In addition, any product
candidate that we develop and that obtains regulatory approval must then compete for market acceptance and market share. Our competitors in the PEGylation technology field include The Dow Chemical Company, Nektar Pharmaceuticals, Inc., SunBio Corporation, Mountain View Pharmaceuticals, Inc., Neose Technologies, Inc., NOF Corporation and Urigen Pharmaceuticals, Inc. Several other chemical, biotechnology and pharmaceutical companies
may also be developing PEGylation technologies. Some of these companies license or provide the technology to other companies, while others develop the technology for internal use. Other companies are conducting research and developing products utilizing technologies targeting RNA (e.g. antisense, siRNA/RNAi or micro RNA) that compete with the LNA technology. These include Isis Pharmaceuticals Inc., Alnylam Pharmaceuticals, Inc., Regulus Therapeutics LLC, Eli Lilly and Company and others. There
are a number of existing therapeutic regimens designed to treat the cancers that we may target with the HIF-1 alpha antagonist. However, we are not of aware of the development of another compound that would have a mechanism similar to our HIF-1 alpha antagonist. There are a number of existing therapeutic regimens designed to treat
the cancers that we may target with the Survivin antagonist and we are aware of several companies that are actively working on compounds targeting Survivin. There are a number of existing therapeutic regimens designed to treat the cancers that we may target with the AR antagonist. However, we are not aware of another compound that
would have a mechanism similar to our AR antagonist. There are a number of drugs in various stages of preclinical and clinical development from companies exploring cancer therapies or improved chemotherapeutic agents to potentially treat the same cancer indications that our PEG-SN38 may be developed to treat. Additionally, there are a number of drugs in development based on the
active metabolite SN38. If these drugs are approved, they could compete directly with our PEG-SN38. These include products in development from Bristol-Myers Squibb Company, Pfizer Inc., GlaxoSmithKline p/c, Antigenics Inc., Hoffman-La Roche Ltd., Novartis AG, Cell Therapeutics, Inc., Neopharm, Inc., Meditech Research
Limited and others. Nektar Therapeutics is also developing a PEGylated form of irinotecan. Irinotecan is a prodrug of SN38. This product candidate is currently in Phase II for colorectal cancer. Nektar has also commenced trials in breast and ovarian cancer for this product candidate. Many of our competitors have substantially greater research and development capabilities and experience and greater manufacturing, marketing and financial resources than we do. In addition, many of our competitors have much more experience than we do in pre-clinical testing and human clinical trials of new drugs, as well as in
obtaining FDA and other regulatory approval. We face competition from these companies not just in product development but also in areas such as recruiting employees, acquiring technologies that might enhance our ability to commercialize products, establishing relationships with certain research and academic institutions, enrolling
patients in clinical trials and seeking program partnerships and collaborations with larger 24
pharmaceutical companies. Accordingly, our competitors may develop technologies and products that are superior to those we or our collaborators are developing and render our technologies and products or those of our collaborators obsolete and noncompetitive. If we cannot compete effectively, our business and financial performance
would suffer. The regulatory approval process is highly uncertain and we will not be allowed to market products if regulatory approval has not been obtained. The marketing of pharmaceutical products in the U.S. and abroad is subject to stringent governmental regulation. The sale of any new products for use in humans in the U.S. requires the prior approval of the FDA. If our products are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether
or not we have obtained FDA approval for a given product and its indications. The FDA has established mandatory procedures and safety standards that apply to the clinical testing and marketing of pharmaceutical products. The FDA regulates the research, development, pre-clinical and clinical testing, manufacture, safety, effectiveness,
record-keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import and export of pharmaceutical and biological products. Obtaining FDA approval for a new therapeutic product may take many years and involve substantial expenditures. Compliance with these regulations can be costly, time-consuming
and subject us to unanticipated delays in developing our products. Neither we nor our licensees may be able to obtain or maintain FDA or other relevant marketing approval for any of our products. There may be limitations placed on our ability to successfully market our products by the FDA or foreign regulators. Regulatory approval may:
•
limit the indicated uses for a product; • otherwise limit our ability to promote, sell and distribute the product; • require that we conduct costly post-marketing surveillance; and • require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as manufacturing changes or revised labeling, may require further regulatory review and approval. Once obtained, any approvals may be withdrawn for a number of reasons, including the later discovery of previously unknown
problems with the product, such as a safety issue. If we or our third-party manufacturers fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in: • refusals or delays in the approval of applications or supplements to approved applications; • refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; • warning letters; • import or export restrictions; • product recalls or seizures; • injunctions; • total or partial suspension of production; • fines, civil penalties or criminal prosecutions; and • withdrawals of previously approved marketing applications or licenses. In addition, any approved products are subject to continuing regulation. Among other things, the holder of an approved biologic license application or new drug application is subject to periodic and other FDA monitoring and reporting obligations, including obligations to monitor and report adverse events and instances of the failure
of a product to meet the specifications in the biologic license application or new drug application. Application holders must also submit advertising and other promotional material to the FDA and report on ongoing clinical trials. Failure to meet these post-approval requirements can result in criminal prosecution, fines 25
or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, or denial or withdrawal of pre-marketing product approvals. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We are not sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact
of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements. Even if we are granted regulatory approval in one jurisdiction, we may not receive regulatory approval in another jurisdiction. Failure to obtain regulatory approval in foreign jurisdictions would prevent us from marketing our products abroad. In order to market our products in the European Union and many other jurisdictions outside the U.S., we must obtain separate regulatory approvals and comply with numerous foreign regulatory requirements. The
approval procedure varies among countries and can involve additional testing. The time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a
timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We may not be able to file for regulatory approvals and may not receive necessary
approvals to commercialize our products in any market. The failure to obtain these approvals could materially harm our business, financial condition and results of operations. If we or our licensees fail to obtain or maintain requisite governmental approvals or fail to obtain or maintain approvals of the scope requested, it will delay or preclude us or our licensees or marketing partners from marketing our products. It could also limit the commercial use of our products. Any such failure or limitation may
have a material adverse effect on our business, financial condition and results of operations. Once approved, our products may not be accepted in the marketplace. Even if clinical trials demonstrate the safety and efficacy of our product candidates and all regulatory approvals are obtained, the commercial success of our products depends on gaining market acceptance among physicians, patients, third-party payors or the medical community. The degree of market acceptance will depend on many
factors, including:
•
the scope of regulatory approvals, including limitations or warnings contained in a product’s regulatory-approved labeling; • establishment and demonstration of clinical efficacy and safety; • cost-effectiveness of our products; • alternative treatment methods and potentially competitive products; and • the availability of third-party reimbursement. Market acceptance could be further limited depending on the prevalence and severity of any expected or unexpected adverse side effects associated with our product candidates. If our product candidates are approved but do not achieve an adequate level of acceptance by physicians, third-party payors and patients, we may never
generate significant revenue from these products, and our business, financial condition and results of operations may be materially harmed. Our operations are subject to extensive environmental laws and regulations. Our operations are subject to federal, state and local environmental laws and regulations concerning, among other things, the generation, handling, storage, transportation, treatment and disposal of hazardous, toxic 26
and radioactive substances and the discharge of pollutants into the air and water. Environmental permits and controls are required for some of our operations and these permits are subject to modification, renewal and revocation by the issuing authorities. If we were to become liable for an accident, or if we were to suffer an extended
facility shutdown as a result of such contamination, we could incur significant costs, damages and penalties that could harm our business and exceed our resources or insurance coverage. The successful commercialization of our product candidates will depend on obtaining health insurance coverage and reimbursement for use of these products from third-party payors and these payors may not agree to cover or reimburse for use of our products. Our future revenues and profitability will be adversely affected if U.S. and foreign governmental, private third-party insurers and payors, and other third-party payors, including Medicare and Medicaid, do not agree to defray or reimburse the cost of our products to the patients. If these entities refuse to provide coverage and
reimbursement with respect to our products or provide an insufficient level of coverage and reimbursement, our products may be too costly for many patients to afford them, and physicians may not prescribe them. In addition, limitation on the amount of reimbursement for our products may also reduce our profitability. In the U.S. and
some foreign jurisdictions, there have been, and we expect will continue to be, actions and proposals to control and reduce healthcare costs. There have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for our product candidates,
restrict or regulate post-approval activities and affect our ability to profitably sell any of our product candidates for which we obtain marketing approval. Government and other third-party payors are also challenging the prices charged for healthcare products and increasingly limiting, and attempting to limit, both coverage and level of
reimbursement for prescription drugs. In the United States, the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this
legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These
cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations
in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors. In March 2010, President Obama signed into law the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose
new taxes and fees on the health industry and impose additional health policy reforms. Effective October 1, 2010, the Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. Further, beginning in 2011, the new law
imposed a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with healthcare practitioners. We will not know the full effects of the Health Care Reform Law
until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effect of the Health Care Reform Law, the new law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens
and operating costs. Since our products will likely be too expensive for most patients to afford without health insurance coverage, if our products are unable to obtain adequate coverage and reimbursement by third-party payors our ability to successfully commercialize our product candidates may be adversely impacted. Any limitation on the use of our
products or any decrease in the price of our products will have a material adverse effect on our ability to achieve profitability. 27
In certain foreign countries, pricing, coverage and level of reimbursement of prescription drugs are subject to governmental control, and we may be unable to negotiate coverage, pricing, and reimbursement on terms that are favorable to us. In some foreign countries, the proposed pricing for a drug must be approved before it may be
lawfully marketed. The requirements governing drug pricing vary widely from country to country. Our results of operations may suffer if we are unable to market our products in foreign countries or if coverage and reimbursement for our products in foreign countries is limited. Risks Related to Our Common Stock and our Convertible Notes The price of our common stock has been, and may continue to be, volatile, which also may significantly affect the trading price of our convertible notes. Historically, the market price of our common stock has fluctuated over a wide range, and it is likely that the price of our common stock will continue to be volatile in the future. The market price of our common stock could be impacted due to a variety of factors, including, in addition to global and industry-wide events:
•
the level of revenues we generate from royalties we receive; • the losses we incur; • the results of preclinical testing and clinical trials by us, our collaborative partners or our competitors; • announcements of technical innovations or new products by us, our collaborative partners or our competitors; • the status of our corporate collaborations and supply arrangements; • regulatory approvals; • developments in patent or other proprietary rights owned or licensed by us, our collaborative partners or our competitors; • public concern as to the safety and efficacy of products developed by us or others; and • litigation. In addition, due to one or more of the foregoing factors in one or more future quarters, our results of operations may fall below the expectations of securities analysts and investors. In that event, the market price of our common stock could be materially and adversely affected. Volatility in the price of our common stock may
significantly affect the trading price of our convertible notes. Events with respect to our share capital could cause the shares of our common stock outstanding to increase. Sales of substantial amounts of our common stock in the open market, or the availability of such shares for sale, could adversely affect the price of our common stock. We had 58.8 million shares of common stock outstanding as of December 31, 2010. As of that date, the following securities that may be exercised for, or are
convertible into, shares of our common stock were outstanding:
•
4% convertible senior notes due 2013 (the “2013 convertible notes”). As of December 31, 2010, our 2013 convertible notes could be converted into 14.1 million shares of our common stock at a conversion price of $9.55 per share. • Options. Stock options to purchase 4.0 million shares of our common stock at a weighted average exercise price of approximately $13.21 per share; • Restricted stock units. Approximately 0.8 million shares of our common stock are issuable in respect of outstanding restricted stock units held by officers, employees and directors. The shares of our common stock that may be issued under the options, restricted stock units, and the 2013 convertible notes are currently registered with the Securities and Exchange Commission, and, therefore, those shares of common stock that may be issued will be eligible for public resale. 28
The conversion of some or all of the convertible notes will dilute the ownership interests of existing stockholders. Any sales in the public market of the common stock issuable upon such conversion could adversely affect prevailing market prices of our common stock. In addition, the existence of the notes may encourage short
selling by market participants because the conversion of the notes could depress the price of our common stock. Anti-takeover provisions in our charter documents and under Delaware law may make it more difficult to acquire us, even though such acquisitions may be beneficial to our stockholders. Provisions of our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even though such acquisitions may be beneficial to our stockholders. These anti-takeover provisions include:
•
lack of a provision for cumulative voting in the election of directors; • the ability of our board to authorize the issuance of “blank check” preferred stock to increase the number of outstanding shares and thwart a takeover attempt; • advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon by stockholders at stockholder meetings; and • limitations on who may call a special meeting of stockholders. Further, we have in place a stockholder rights plan, commonly known as a “poison pill.” The provisions described above, our stockholder rights plan and provisions of Delaware law relating to business combinations with interested stockholders may discourage, delay or prevent a third party from acquiring us. These provisions may
also discourage, delay or prevent a third party from acquiring a large portion of our securities, or initiating a tender offer, even if our stockholders might receive a premium for their shares in the acquisition over the then current market price. We also have agreements with our executive officers that provide for change of control severance
benefits which provides for cash severance, restricted stock, restricted stock units and option award vesting acceleration and other benefits in the event our employees are terminated (or, in some cases, resign for specified reasons) following an acquisition or other change in control. These agreements could discourage a third party from
acquiring us. The issuance of preferred stock may adversely affect rights of common stockholders. Under our certificate of incorporation, our board of directors has the authority to issue up to three million shares of “blank check” preferred stock and to determine the price, rights, preferences and privileges of those shares without any further vote or action by our stockholders. The rights of the holders of common stock will be
subject to the rights of the holders of any shares of preferred stock that may be issued in the future. In addition to discouraging a takeover, as discussed above, this “blank check” preferred stock may have rights, including economic rights senior to the common stock, and, as a result, the issuance of such preferred stock could have a
material adverse effect on the market value of our common stock. The market for unrated debt is subject to disruptions that could have an adverse effect on the market price of the 2013 convertible notes, or a market for our notes may fail to develop or be sustained. The 2013 convertible notes are not rated. As a result, holders of the notes have the risks associated with an investment in unrated debt. Historically, the market for unrated debt has been subject to disruptions that have caused substantial volatility in the prices of such securities and greatly reduced liquidity for the holders of such
securities. When the notes are traded, they may trade at a discount from their initial offering price, depending on, among other things, prevailing interest rates, the markets for similar securities, general economic conditions and our financial condition, results of operations and prospects. The liquidity of, and trading markets for, the notes
also may be adversely affected by general declines in the market for unrated debt. Such declines may adversely affect the liquidity of, and trading markets for, the notes, independent of our financial performance or prospects. In addition, certain regulatory restrictions prohibit certain types of financial institutions from investing in unrated
debt, which may further suppress demand for such securities. We cannot assure you that 29
the market for the notes will not be subject to similar disruptions or that any market for our notes will develop or be sustained. Any such disruptions may have an adverse effect on the holders of the notes. We may not have sufficient funds available to pay amounts due under our 2013 convertible notes. We may not have sufficient funds available or may be unable to arrange for additional financing to satisfy our obligations under our 2013 convertible notes. Our ability to pay cash to holders of the notes or meet our payment and other debt obligations depends on the level of our expenditures and our ability to generate sufficient cash
flow in the future. This, to some extent, is subject to general economic, financial, competitive, legislative and regulatory factors, as well as other factors that are beyond our control. Also, the indenture governing our 2013 convertible notes does not contain any financial or operating covenants or restrictions on the payments of dividends,
the incurrence of indebtedness or the issuance or repurchase of securities by us or any of our subsidiaries. We cannot assure you that our business will generate cash flow from operations, or that future borrowings will be available to us in an amount sufficient to enable us to meet our payment obligations under the notes and our other
obligations and to fund other liquidity needs. Item 1B. Unresolved Staff Comments. None. The following are all of the facilities that we currently lease:
Location Principal Operations
Approx.
Approx.
Lease 20 Kingsbridge Road Piscataway, NJ Research & Development
56,000
$
640,000(1
)
July 31, 2021 685 Route 202/206 Bridgewater, NJ Administrative—
51,000
$
1.4 million(2
)
January 31, 2013 300 Corporate Ct. S. Plainfield, NJ Subleased
24,000
$
228,000(3
)
October 31, 2012
(1)
Under the terms of the lease, annual rent increases over the remaining term of the lease from $640,000 to $773,000. (2) Under the terms of the lease, annual rent over the remaining term of the lease is $1.4 million. Not reflected above is a sublease of a portion of the Bridgewater facility that will generate a total of $0.8 million over approximately two years. (3) Amount shown in table represents our obligation to our landlord. The facility is being subleased by us to a third party for $294,000 per year through October 31, 2012. We believe that our facilities are well maintained and generally adequate for our present and anticipated future needs. We sold a 56,000 square foot manufacturing facility in Indianapolis, Indiana, in January 2010 to the purchasers of our specialty pharmaceutical business at which we produced Abelcet, Oncaspar and Adagen for the Products segment and products we manufactured for others on a contract basis (Contract Manufacturing segment). In
addition, we are transitioning our principal executive offices from our Bridgewater, NJ offices to our offices in Piscataway, NJ, which we expect will be complete by March 31, 2011. We are actively seeking a tenant or tenants for the excess Bridgewater capacity. We currently own no real property. From time to time, we are engaged in litigation resulting from the ordinary course of our business. There is no pending material litigation to which we are a party or to which any of our property is subject. Item 4. (Removed and Reserved) 30
Square
Footage
Annual Rent
Expiration
partially subleased
Market Information Our common stock is traded on the Nasdaq Global Market under the trading symbol “ENZN”. The following table sets forth the high and low sale prices for our common stock during the years ended December 31, 2010 and December 31, 2009 as reported by the Nasdaq Global Market. The quotations shown represent inter-dealer prices without adjustment for retail markups, markdowns or commissions, and may not
necessarily reflect actual transactions.
High
Low Year Ended December 31, 2010 First Quarter
$
11.37
$
8.86 Second Quarter
11.52
9.25 Third Quarter
11.35
10.15 Fourth Quarter
12.71
10.50 Year Ended December 31, 2009 First Quarter
$
7.45
$
4.70 Second Quarter
8.25
5.40 Third Quarter
8.66
7.05 Fourth Quarter
10.92
8.03 31
Performance Graph The following graph compares the percentage change in cumulative total stockholder return on our common stock for our fiscal years ended December 31, 2006 through December 31, 2010 with the cumulative total return over the same period of (i) the Nasdaq Composite Index and (ii) the Nasdaq Pharmaceutical Index. Total Return To Shareholders The comparison below displays the annual percentage return in an investment in our common stock, the Nasaq Composite Index and the Nasdaq Pharmaceutical Index, and assumes reinvestment of dividends, if any. Historical stock prices are not indicative of future stock price performance. ANNUAL RETURN PERCENTAGE
Years Ending
Company/Index
12/06
12/07
12/08
12/09
12/10 ENZON PHARMACEUTICALS, INC.
15.00
11.99
–38.82
80.62
15.48 NASDAQ COMPOSITE INDEX
9.52
9.81
–40.54
43.89
16.91 NASDAQ PHARMACEUTICAL INDEX
1.02
4.58
–12.63
15.63
15.01 The comparison below assumes $100 was invested on December 31, 2005 in our common stock, the Nasdaq Index and the Nasdaq Pharmaceutical Index, and assumes reinvestment of dividends, if any. Historical stock prices are not indicative of future stock price performance. INDEXED RETURNS
Years Ending
Company/Index
Base
12/06
12/07
12/08
12/09
12/10 ENZON PHARMACEUTICALS, INC.
100
115.00
128.78
78.78
142.30
164.32 NASDAQ COMPOSITE INDEX
100
109.52
120.27
71.51
102.89
120.29 NASDAQ PHARMACEUTICAL INDEX
100
101.02
105.65
92.31
106.74
122.76 Holders As of March 9, 2011, there were 1,235 holders of record of our common stock. Dividends We have never declared or paid any cash dividends on our common stock. 32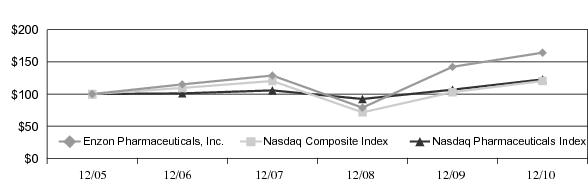
Period
12/05
Repurchase of Equity Securities Common Stock In the fourth quarter of 2010, Enzon repurchased shares of our Common Stock as set forth in the following table: ISSUER PURCHASES OF EQUITY SECURITIES
Period
(a)
(b)
(c)
(d) October 1, 2010–October 31, 2010
25,149
$
11.04
25,149(1
)
$
11,126,000(1
) November 1, 2010–November 30, 2010
1,025,312
$
10.85
1,025,312(1
)
— December 1, 2010–December 31, 2010
30,000
$
12.45
30,000(2
)
$
199,626,000(2
) Total
1,080,461
$
10.90
1,080,461
$
199,626,000(2
)
(1)
Share repurchase program announced December 3, 2009 whereby our board of directors approved the repurchase of up to $50.0 million of its outstanding shares of common stock. The program had no indicated expiration date. The program was completed as of the end of November 2010 with the purchase of a total of 4,704,005 shares
for an overall average cost per share for the program of $10.63. (2) Share repurchase program announced December 20, 2010 whereby our board of directors approved the repurchase of up to $200.0 million of its outstanding shares of common stock. No expiration date was established for the plan. This plan continues in force. Item 6. Selected Financial Data The following selected financial data for the years ended December 31, 2010, 2009, 2008, 2007 and 2006 are derived from the audited financial statements of Enzon Pharmaceuticals, Inc. The selected financial data set forth below should be read in conjunction with our financial statements and the related notes thereto and
“Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this report. Year
Ended December 31, 2010 2009 2008 2007 2006 (in
thousands, except per-share data) Consolidated
Statement of Operations Data:(1) Total
revenues(1) $ 97,865 $ 51,408 $ 56,969 $ 65,161 $ 67,917 Research
and development–pipeline 49,883 45,639 43,484 45,522 42,243 Acquired
in-process research and development — — — — 11,000 Gain
on sale of royalty interest(2) — — — (88,666 ) — Other
operating expenses 48,557 62,862 54,974 42,526 29,667 Operating
(loss) income (575 ) (57,093 ) (41,489 ) 65,779 (14,993 ) Investment
income, net 3,465 4,312 6,612 10,918 24,670 Interest
expense (6,315 ) (11,514 ) (12,681 ) (17,380 ) (22,055 ) Other,
net, including investment impairment 288 5,008 1,246 954 8,952 Income
tax benefit (provision) 337 2,085 (255 ) (1,525 ) (583 ) (Loss)
income from continuing operations (2,800 )(5) (57,202 ) (46,567 ) 58,746 (4,009 ) 33
Total
Number of
Shares (or
Units)
Purchased
Average
Price
Paid per
Share
(or Unit)
Total
Number of
Shares
(or Units)
Purchased
as Part
of Publicly
Announced
Plans or
Programs
Maximum
Number (or
Approximate
Dollar Value)
of Shares
(or Units)
that May
Yet Be
Purchased
Under the
Plans or
Programs
Year Ended December 31,
2010
2009
2008
2007
2006
(in thousands, except per-share data) Income and gain from discontinued operations, net of income tax(1)
180,043
57,885
43,852
24,307
25,318 Net income (loss)
$
177,243
$
683
$
( 2,715
)
$
83,053
$
21,309 (Loss) earnings per common share–continuing operations Basic
$
(0.05
)
$
(1.26
)
$
(1.05
)
$
1.34
$
(0.09
) Diluted
$
(0.05
)
$
(1.26
)
$
(1.05
)
$
0.81
$
(0.09
) Earnings per common share–discontinued operations: Basic
$
3.08
$
1.28
$
0.99
$
0.55
$
0.58 Diluted(4)
$
3.08
$
1.28
$
0.99
$
0.33
$
0.58 Earnings (loss) per common share–net income (loss): Basic
$
3.03
$
0.02
$
(0.06
)
$
1.89
$
0.49 Diluted(4)
$
3.03
$
0.02
$
(0.06
)
$
1.14
$
0.49 No dividends have been declared
December 31,
2010
2009
2008
2007
2006
(in thousands) Consolidated Balance Sheet Data: Total current assets(1)
$
434,616
$
145,212
$
178,142
$
281,177
$
212,311 Total current liabilities(3)
18,387
24,997
36,094
105,482
59,885 Total assets(1)
488,857
332,749
349,253
420,357
403,830 Notes payable(3)
134,499
250,050
267,550
275,000
397,642 Total stockholders’ equity(1)
331,857
53,283
41,661
36,573
(56,441
)
(1)
In January 2010, we sold our specialty pharmaceutical business comprised of the previous Products and Contract Manufacturing segments. The sale has been treated as a discontinued operation. Accordingly, prior-year statement of operations information has been reclassified to segregate the revenues and expenses of the divested
business from our continuing operations. The sale generated net cash proceeds of approximately $308.0 million, including $40.9 million of revenues from the sale of in-process research and development (reported as revenues in continuing operations). The net gain on the sale, excluding the revenues from the sale of in-process research
and development, was $176.4 million (reported as income and gain from discontinued operations). See Note 22 of the accompanying consolidated financial statements. (2) We sold a 25-percent interest in our PEGINTRON royalty in August 2007. See Note 18 of the accompanying consolidated financial statements. (3) As of December 31, 2007, $72.4 million outstanding principal amount of 4.5% notes payable was due July 1, 2008 and was classified as a current liability. The 4.5% notes were repaid in full according to their terms in 2008. (4) In a period in which a loss from continuing operations is reported, all other computations of diluted per-share amounts for that period must be made exclusive of potential dilutive shares. For this reason, in the years ended December 31, 2010, 2009, 2008 and 2006, diluted earnings per share for discontinued operations and net income
are the same as basic earnings per share despite reported profitability in most instances. For the year ended December 31, 2007, diluted net earnings per share reflected the effects of dilutive shares. (5) Amounts were adjusted subsequent to our February 17, 2011 year-end earnings release. Refer to Management’s Discussion and Analysis. 34
(5)
(5)
(5)
(5)
(5)
(5)
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations On
February 17, 2011, we issued a press release reporting our full year 2010
operating results. We reported a loss from continuing operations of $3.6
million, income and gain from discontinued operations of $179.0 million and
net income of $175.4 million ($(0.06), $3.06 and $3.00 per share, respectively).
During the fourth quarter of 2010, as part of the year-end reporting process
and subsequent to the aforementioned earnings release, we corrected two entries
related to the sale of the specialty pharmaceutical business. Because the
sale of substantially all of the net assets of our Canadian subsidiary constituted
a substantial liquidation of that entity for accounting purposes, we should
have credited the gain on the sale for the $1.0 million of currency translation
gains residing in accumulated other comprehensive income related to our
Canadian operations. The correcting entry increased the gain on the sale
from $179.0 million to $180.0 million or $3.08 per share. Additionally, because
the sale of the specialty pharmaceutical business constituted a fundamental
change as defined in the indenture to our 4% notes, an enhanced conversion
rate became effective which caused a number of our note holders to convert
their notes to shares of our common stock. The net effect of forgone interest
and the write-off of deferred debt issuance costs amounting to $0.8 million
was charged to earnings during the first quarter of 2010. During the fourth-quarter
reporting process, we determined that this amount should have been charged
to additional paid-in capital. The correcting adjustment, also made subsequent
to the earnings release, had the effect of reducing our loss from continuing
operations for the year to $2.8 million or $(0.05) per share. The combined
effect of these two entries was to increase net income by $1.8 million to
$177.2 million or $3.03 per share. These two entries are not considered material
to either the first
or fourth quarters of 2010 nor to the full year results of operations and
were recorded as fourth-quarter adjustments. The following discussion of our financial condition and results of operations should be read together with our consolidated financial statements and notes to those statements included in Item 8 of Part II of this Form 10-K. Overview We are a biotechnology company dedicated to the research and development of innovative therapeutics for cancer patients with high unmet medical needs. We are managed as a single operating unit. Our drug development programs utilize two platforms—Customized PEGylation Linker Technology (Customized Linker TechnologyÒ)
and third-generation mRNA-targeting agents utilizing the Locked Nucleic Acid (LNA) technology. We currently have four compounds in human clinical development; PEG-SN38 and the antagonists of Hypoxia-Inducible Factor-1a (HIF-1a), Survivin and Androgen Receptor (AR). In addition we have multiple novel LNA targets in various
stages of preclinical research. We receive royalty revenues from licensing arrangements with other companies related to sales of products developed using our proprietary Customized Linker Technology—primarily PEGINTRON marketed by Merck & Co., Inc. (Merck). In order to better focus on our portfolio of innovative oncology programs, we divested our specialty pharmaceutical business comprised principally of what had previously been our Products and Contract Manufacturing segments. Prior to the January 29, 2010 closing of the transaction, we were a biopharmaceutical company involved
in the development, manufacture and commercialization of medicines for patients with cancer and other life-threatening conditions. We operated in three business segments: Products, Royalties and Contract Manufacturing. We had a portfolio of four marketed products, Oncaspar, for first-line treatment of patients with acute lymphoblastic
leukemia (ALL); DepoCyt, for the treatment of lymphomatous meningitis; Abelcet, for the treatment of invasive fungal infections; and Adagen, for the treatment of severe combined immunodeficiency disease. The contract manufacturing business involved the manufacture of products for other pharmaceutical companies. The Products and Contract Manufacturing segments constituted components of our business and the sale qualified for treatment as discontinued operations effective with the first quarter of 2010. Accordingly, the operations and cash flows of the Products and Contract Manufacturing segments have been eliminated from our reported
continuing operations and have been classified as discontinued operations beginning in 2010. Prior-year information has been reclassified to conform to the current presentation. Similarly, assets and liabilities of the specialty pharmaceutical business are presented separately in our balance sheets. 35
The sale of our specialty pharmaceutical business also involved the sale of certain in-process research and development associated with the divested products, the potential receipt of certain contingent milestone payments, the potential receipt of certain royalties and our provision of various transitional services to the purchaser. Each
of these aspects of the transaction is discussed below in greater detail. Throughout the majority of Management’s Discussion and Analysis, the focus is on the results of operations, cash flows, financial condition and future outlook of our continuing operations. The percentage changes throughout the discussion that follows are based on amounts stated in thousands of dollars and not the rounded millions
of dollars reflected in this section. Results of Continuing Operations (millions of dollars):
Year Ended December 31,
2010
2009
2008 Revenues: Royalties
$
44.9
$
51.4
$
57.0 Sale of in-process research and development
40.9
—
— Contract research and development
9.3
—
— Miscellaneous income
2.8
—
— Total revenues
97.9
51.4
57.0 Operating expenses: Research and development–pipeline
49.9
45.6
43.5 Research and development–specialty and contracted services
7.2
24.6
14.6 General and administrative
25.4
37.6
40.4 General and administrative–contracted services
2.0
—
— Restructuring charge
14.0
0.7
— Operating loss
(0.6
)
(57.1
)
(41.5
) Other income (expense)
(2.5
)
(2.2
)
(4.9
) Income tax benefit (provision)
0.3
2.1
(0.2
) Loss from continuing operations
$
(2.8
)
$
(57.2
)
$
(46.6
) Overview The sale of the specialty pharmaceutical business in January 2010 had numerous effects on our financial results making year-to-year comparisons and inferences regarding future trends difficult. Even after reclassifying the majority of revenues and expenses of the divested business as discontinued operations, several large and unique
items remain that are reported as part of continuing operations but that are not expected to be recurring events. In addition, indirectly related to the divestiture was the restructuring charge incurred in 2010. To summarize the unique items related to the sale of the specialty pharmaceutical business:
•
The sale of in-process research and development for $40.9 million was part of the total sale of the specialty pharmaceutical business but is reported as part of continuing operations because we continue to operate as a research and development organization. • Revenues from a transition services agreement entered into with the purchaser of the specialty pharmaceutical business totaling $11.8 million (contract research and development and the majority of miscellaneous income) are expected to significantly diminish in 2011. • Contracted services in 2010 largely represent the expenses incurred ($5.5 million) in support of the transition services revenues mentioned above and also are expected to diminish significantly in 2011. Also in this caption are the expenses incurred by us prior to the sale of the specialty pharmaceutical business in support of the
products we owned at that time ($1.7 million in 2010, $24.6 million in 2009 and $14.6 million in 2008). 36
After taking these items and the $14.0 million 2010 restructuring charges into account, analysis of the underlying trends we have experienced in royalty revenues, research and development spending and general and administrative expenses is enhanced. These and other elements of our statements of operations are discussed more
fully below. Royalty Revenues (millions of dollars):
Year Ended December 31,
2010
%
2009
%
2008 Royalty revenue
$
44.9
(13
)
$
51.4
(10
)
$
57.0 The majority of royalty revenue relates to sales of PEGINTRON, a PEG-enhanced version of the alpha-interferon product, INTRON A, marketed by Merck (formerly Schering-Plough), for the treatment of chronic hepatitis C. Other royalty revenues and certain licensing revenues relate to the application of our technology to third-
party products including those under a cross-license agreement with Nektar Therapeutics, Inc. (Nektar) under which we receive a share of the royalties and licensing income received by Nektar. There are currently two third-party products for which Nektar has granted sublicenses to our PEGylation technology and for which we are
participating in royalty and licensing income revenues: UCB’s CIMZIA for the treatment of Crohn’s disease and rheumatoid arthritis in the European Union and OSI and Pfizer’s Macugen for the treatment of neovascular (wet) age-related macular degeneration. There were previously three products for which Nektar had granted
sublicenses to our PEGylation technology, but our right to receive royalties on sales of Pegasys ended, by contract, effective in October 2009. Royalty revenue declined approximately 13 percent in 2010 compared to 2009. This was primarily the result of declining sales of PEGINTRON which were 11 percent lower during the relevant period of comparison. Further contributing to the full-year decline was the absence of royalties from Pegasys in 2010, partially offset by
growth in sales of CIMZIA. Macugen royalties were flat for the year. Also during 2010, we recognized for the first time royalties earned on net sales of products we divested as part of the sale of the specialty pharmaceutical business. We are entitled to royalties of 5 to 10 percent on incremental net sales through 2014 by the purchaser above a 2009 baseline amount from the four marketed specialty
pharmaceutical products we sold to them. There can be no assurance that any additional royalty revenues related to these products will accrue to us. Certain specific royalty rights related to sales of Oncaspar in Europe by other companies were divested as part of the sale of the specialty pharmaceutical business. The revenues generated
from these contracts totaling approximately $2.7 million and $2.6 million in 2009 and 2008, respectively, are not reflected above but are reflected in discontinued operations. For 2009, we experienced a year-over-year decline in royalty revenues of approximately 10 percent. Royalty revenues from PEGINTRON (PEGylated interferon) sales were down 9 percent compared to 2008 levels due in part to the impact of unfavorable foreign exchange variances and, in part, to lower sales in the U.S. and
internationally. Royalties from Pegasys, CIMZIA and Macugen also decreased in 2009 compared to 2008. CIMZIA was approved for sale in April 2008 for Crohn’s disease and in May 2009 for rheumatoid arthritis. Macugen continued to experience competition. Our future revenues are heavily weighted towards royalties and revenues to be received from the use of our technology and are dependent upon numerous factors outside of our control. Sales of PEGINTRON have been in decline since 2008. There are a number of new therapies in late stage development that could significantly erode
the market for PEGylated interferon which, in combination with the generic antiviral pill, ribavirin, represents the current standard of care. The most advanced new therapies are protease inhibitors that work by blocking the action of the protease enzyme the hepatitis virus needs to replicate. Studies have revealed that the triple
combinations (PEGylated interferon, ribavirin and protease inhibitor) are very effective in significantly reducing the hepatitis virus, and may allow shorter course of therapy, which may be better tolerated by patients. These studies have not exclusively involved PEGINTRON, however. Many of the studies have involved Pegasys, the other
currently available form of PEGylated interferon, from which we no longer derive royalties. 37
Change
Change
There has been some indication that a significant number of patients suffering from hepatitis C may be deferring treatment until the new therapies become available potentially as early as mid-2011. The implications of the unsettled hepatitis C market to future sales of PEGINTRON are not clear. Merck’s obligation to pay us royalties on sales of PEGINTRON terminates, on a country-by-country basis, upon the later of the date the last patent to contain a claim covering PEGINTRON expires in the country or 15 years after the first commercial sale of PEGINTRON in such country. Currently, expirations of our right to receive
royalties are expected to occur in 2016 in the U.S., 2018 in Europe and 2019 in Japan. Other factors potentially affecting our royalty revenues include: new or increased competition from products that may compete with the products for which we receive royalties, the effectiveness of marketing by our licensees and new uses and geographies for PEGINTRON, CIMZIA and Macugen. Our rights to receive royalties on
CIMZIA and Macugen will terminate in 2014. In 2007, we sold a 25-percent interest in PEGINTRON royalties. Accordingly, amounts reflected above represent our 75-percent share of total royalties remitted by Merck. The purchaser of the 25-percent royalty interest we sold will be obligated to pay an additional $15.0 million to us in the first quarter of 2012 if it achieves a
certain threshold level of royalties on sales of PEGINTRON occurring from July 1, 2007 through December 31, 2011. The $15.0 million contingent gain will be recognized when and if the contingency is removed and collection is assured. It presently appears unlikely that the necessary sales threshold will be attained. Non-U.S. Revenue We recognized royalties on non-U.S. sales of $37.9 million, $42.3 million and $45.2 million for the years ended December 31, 2010, 2009 and 2008, respectively, of which royalties recognized on sales in Europe were $13.4 million, $16.6 million and $20.5 million, respectively. Our non-U.S. royalties are denominated in U.S. dollars
and are included in total revenues. Sale of In-Process Research and Development When we sold our specialty pharmaceutical business, we retained our research and development organization. We had been engaged in studies oriented towards the next-generation formulations of Oncaspar and Adagen, two products that were among those sold as part of the specialty pharmaceutical business. The in-process research
and development related to Oncaspar and Adagen was sold to the purchaser of the specialty pharmaceutical business and $40.9 million was recognized as revenue in connection with the sale in the first quarter of 2010. The selling price of the in-process research and development represented management’s best estimate of its standalone
fair value based on the stage of development and future milestone payment consideration. All necessary technology and know-how were transferred to the purchaser at the time of the sale and the purchaser could resell the in-process research and development asset. At the time of the sale, the activities necessary to complete the work on
the Oncaspar and Adagen next-generation formulations could have been performed by the purchaser or others. Contract Research and Development Revenue Pursuant to a transition services agreement entered into at the time of the sale of the specialty pharmaceutical business, we began performing product-support research and development, consulting and technology transfer functions for the purchaser effective with the close of the sale transaction on January 29, 2010. The transition
services associated with product-support research and development are being reported in continuing operations due to our ongoing involvement in the research and development related to the divested products. We are being compensated for this work at actual cost plus a mark-up per the terms of the transition services agreement. Revenue
was generated from these services in the amount of $9.3 million for the year ended December 31, 2010. Our contractual obligation is to assist with these transition services for a period of up to three years subsequent to the date of the sale although we anticipate the level of such activity to decline significantly from 2010 levels. 38
Miscellaneous Income As part of the transition services agreement referred to above, we are being compensated for various general and administrative services provided to the purchaser of the specialty pharmaceutical business. The compensation for this work includes reimbursement of costs incurred plus a mark-up defined in the agreement. Through
December 31, 2010, approximately $2.5 million has been earned for these services. The expenses incurred in relation to these services are reported as general and administrative—contracted services. Our involvement in the transitioning of general and administrative activities is nearly complete. Also reflected in miscellaneous income are
rental receipts totaling approximately $0.3 million in connection with the sublease of unused manufacturing and excess office facilities for which we have ongoing lease commitments. The underlying rental expense is reflected in general and administrative expense. Research and Development Expenses–Pipeline (millions of dollars):
Year Ended December 31,
2010
%
2009
%
2008 Research and development expenses
$
49.9
9
$
45.6
5
$
43.5 Research and development expenses consist primarily of contractor fees principally related to clinical projects; costs related to research and development collaborations or licenses; drug supplies for preclinical and clinical activities; salaries and share-based compensation and benefits, as well as other research supplies and facilities
charges. The following table summarizes our major pipeline research and development projects, the costs incurred for the years ended December 31, 2010, 2009 and 2008 and the current phases of development (millions of dollars):
For the year ended
December 31,
Current Phase of
2010
2009
2008 Program costs PEG-SN38
$
18.7
$
11.9
$
8.8
Phases I and II HIF-1a antagonist
3.4
13.0
5.7
Phase I Survivin antagonist
1.7
8.2
6.9
Phase I Androgen Receptor antagonist
11.4
6.0
4.9
IND accepted Additional LNA targets
12.1
3.6
12.3
Preclinical PEGylation technology
2.3
1.3
0.4
Research/Preclinical Other costs
0.3
1.6
4.5 Total program costs
$
49.9
$
45.6
$
43.5 For the year ended December 31, 2010, research and development expenses increased 9 percent to $49.9 million. We invested in the following programs during 2010: PEG-SN38—Spending on PEG-SN38 increased significantly in 2010 as Phase II clinical activity increased in the metastatic colorectal cancer study and we initiated a Phase II study in metastatic breast cancer and a Phase I pediatric study. We continued to conduct a Phase II trial for patients with metastatic colorectal cancer. This study is designed to evaluate two groups of colorectal patients who have failed two prior therapies including irinotecan and oxaliplatin, those with K-RAS mutation and those that have non-mutated K-RAS tumors. The study is expected to
enroll approximately 220 patients and enrollment is expected to be completed in 2011. The non-mutated K-RAS group is randomized into two arms; one treated with PEG-SN38 in combination with Erbitux and the other arm treated with irinotecan in combination with Erbitux. In January 2010, we started enrolling patients in a Phase II trial for patients with metastatic breast cancer. The study is designed to evaluate the efficacy of single-agent PEG-SN38 in two groups of patients who have received prior therapy regimens of anthracycline and taxane or anthracycline, taxane and Xeloda. Irinotecan has 39
Change
Change
Development
been evaluated and shown to be active in patients with breast cancer. All patients will be treated with single agent PEG-SN38. Enrollment in the metastatic breast cancer study is expected to be completed in 2011. We also started enrollment in February 2010 in our Phase I study for pediatric patients with cancer. This study is designed to find the recommended dose of PEG-SN38 in pediatric patients. We expect to complete this Phase I study in 2011. Furthermore, we have opened a Phase I study of PEG-SN38 and bevacizumab at the National
Cancer Institute, Bethesda, MD, in patients who failed multiple prior chemotherapy regimens. HIF-1a antagonist—On a comparative basis, spending was substantially lower in 2010 on HIF-1a than in 2009. This was due primarily to elevated costs experienced in 2009 for the production of clinical trial supplies. We continued to conduct two Phase I studies in patients with solid tumors and lymphoma to evaluate the safety of the HIF-1a antagonist using two different dosing schedules. We continue to enroll patients on a weekly schedule. In general, HIF-1a antagonist therapy has been well tolerated, and many patients have received multiple
cycles. We have observed stable disease in a number of patients treated with our HIF-1a antagonist. Tumor shrinkage also was seen in patients with renal cell cancer, liver cancer, sarcoma, and cancer of the tonsil. Patients enrolled in 2010 were required to get repeated biopsies of cancer tumors once biological activity was shown and
higher doses were being given. We expect study completion in 2011. Furthermore, we have opened a pilot study in patients with liver cancer at the National Cancer Institute. Survivin antagonist—Spending on Survivin in 2009 included a $1.0 million milestone payment and the costs of production of clinical trial supplies. Comparable milestone payments and expenses of producing clinical trial materials were not incurred in 2010. We continued to enroll patients in our Phase I study. The study is designed to first treat patients with Survivin as a single agent until progression at which time the patient’s treatment will be changed to Survivin in combination with Taxotere. This allows us to gain dose and safety information both as a single agent and in
combination in one Phase I study. In collaboration with a pediatric Phase I consortium (Therapeutic Advances in Childhood Leukemia & Lymphoma), we opened an investigator-initiated study in patients with relapsed ALL. Androgen Receptor (AR) antagonist—Spending on AR accelerated during 2010 as enabling activities related to an Investigational New Drug (IND) were conducted. This included toxicology and preclinical work that led to the filing and subsequent acceptance on an IND application in November 2010. In connection with the filing, we
made a $2.0 million milestone payment to Santaris in the fourth quarter of 2010. Additional LNA targets—Under our agreement with Santaris we will have the right to develop and commercialize RNA antagonists directed against additional novel oncology gene targets selected by us. We are evaluating these compounds in early preclinical studies. Any one of these compounds could be returned to Santaris if the
findings of our preclinical or clinical work do not support continued investigation. In 2010, we incurred milestone payments totaling $5.0 million for the commencement of preclinical studies for three of our targets (in addition to the $2.0 million milestone payment for the AR antagonist IND referred to above). In 2009, there were no
such early-stage milestone payments. For the year ended December 31, 2009, research and development expenses increased approximately 5 percent to $45.6 million. We invested in the following programs during 2009: PEG-SN38—We conducted two Phase I clinical trials with PEG-SN38 in patients with solid tumors and lymphomas who had been extensively treated with and progressed on other chemotherapeutic agents to evaluate different dosing schedules for PEG-SN38. These trials completed enrollment in the second quarter of 2009. In June 2009,
we started enrolling patients in a Phase II trial for patients with metastatic colorectal cancer. HIF-1a antagonist—We continued to conduct two Phase I studies in patients with solid tumors and lymphoma to evaluate the safety of the HIF-1a antagonist using two different dosing schedules. We enrolled patients on a weekly and a daily schedule. We commenced production of clinical trial supplies during 2009 resulting in an increase in
expense. 40
Survivin antagonist—The IND application for our Survivin antagonist was accepted by the FDA in February 2009. We opened and started enrolling patients in a Phase I study. A $1.0 million milestone payment was made and significant expense was incurred in the production of clinical trial materials. AR antagonist—We advanced the AR LNA to the preclinical development stage and initiated IND enabling studies. In the fourth quarter of 2009, we incurred a $2.0 million milestone for the commencement of preclinical activities for AR. Additional LNA targets—There were no milestone payments in 2009 for targets not specifically referred to above, whereas in 2008, $3.0 million of milestone payments for early-stage targets were made. Research and Development—Specialty and Contracted Services Expenses associated with generating contract research and development revenue amounted to $5.5 million in 2010. Also reflected in this line caption are the costs we incurred from January 1 through January 29, 2010 and the full years 2009 and 2008 in our research and development activities related to the marketed products we
owned during those periods. This work was directed largely towards securing and maintaining a reliable supply of the ingredients used in the production of Oncaspar and Adagen, including development of new formulations of each. Prior to the sale of the business, we incurred $1.7 million in support of the marketed products. During the
years ended December 31, 2009 and 2008, $24.6 million and $14.6 million were expensed on marketed products. General and Administrative Expenses
Year Ended December 31,
2010
%
2009
%
2008 General and administrative expenses
$
25.4
(32
)
$
37.6
(7
)
$
40.4 General and administrative expenses consist primarily of outside professional services for accounting, audit, tax, legal, and financing activities; salaries and benefits for support functions; patent filing fees and facilities costs. For the year ended December 31, 2010, general and administrative expenses were $25.4 million, down 32 percent from the prior year. The reduction from the preceding year was largely the result of ongoing cost containment efforts and the contraction of corporate services and overhead costs necessitated by the first-quarter 2010 sale
of the specialty pharmaceutical business. A significant portion of the year-over-year decrease is related to compensation. The restructuring program implemented during the first quarter of 2010 and the resulting reduction in employees was reflected in lower payroll costs during the latter half of the year. The December 2010 restructuring
will have a favorable effect on 2011 expense. Accelerated vesting of share-based awards effected in the fourth quarter of 2009 resulted in a reduction in 2010 of the charges related to the vesting of these awards for all but certain senior management and board members. Offsetting these favorable influences on compensation expense in
2010, in part, was the shift of a portion of the 2009 executive bonus expense recognition out of 2009 and into 2010. A fourth-quarter 2009 adjustment was made to annual executive bonuses paying them one-half in cash and one-half in nonvested shares that vested over the twelve months of 2010. In addition to reductions in compensation expenses, we also made concerted efforts to reduce contracted services, accounting and consulting fees in 2010. During 2009, certain general and administrative expenses were elevated, including legal costs related to a proposed shareholder consent solicitation and the post-implementation
costs of an enterprise resource planning (ERP) computer software system. As outlined above, we have made significant progress in reducing general and administrative expenses and will continue to seek and implement efficiencies that could potentially lead to further reductions. However, the rate of improvement experienced during 2010 is not expected to continue. We may experience additional 41
(millions of dollars):
Change
Change
charges associated with the South Plainfield lease or its termination prior to its contractual expiration in October 2012. General and administrative spending decreased 7 percent to $37.6 million in 2009 from $40.4 million in 2008. Both years’ expenses included certain costs associated with strategic initiatives ($1.9 million in 2009 and $5.0 million in 2008). These initiatives related to preparations during 2009 to sell the specialty pharmaceutical
business and analysis of various alternatives during 2008. General and administrative spending was lower in 2009, compared to 2008, due in part to the benefits derived from the first-quarter 2009 restructuring. In addition, a fourth-quarter 2009 adjustment was made to annual executive bonuses and a decision was made to satisfy these
bonuses one-half in cash and one-half in nonvested shares that vested over the twelve months of 2010. The effect of this was to reduce 2009 compensation expense and defer it to 2010. Offsetting these improvements were the cost of certain organizational and administrative enhancements, including the establishment of a business
development function and the post-implementation costs of a newly developed ERP computer software system. In addition, costs associated with the site at South Plainfield, New Jersey began to be recognized in general and administrative expense in 2009. When production activities at the South Plainfield location ceased in late 2008,
costs such as security, utilities, insurance and monthly rental were classified as general and administrative. General and Administrative—Contracted Services As part of the transition services agreement with the purchaser of the specialty pharmaceutical business, we provided certain general, administrative, financial, legal, human resource and information technology services for a period of up to one year. We were compensated for these services based upon costs incurred plus a mark-up
defined in the transition services agreement. During the year ended December 31, 2010, expenses associated with generating this revenue were approximately $2.0 million. This administrative support activity has nearly ended. Restructuring As part of our continued efforts to streamline operations, we undertook reductions in our workforce during the first and fourth quarters of 2010 and the first quarter of 2009. We also incurred charges related to our sublease of a portion of our excess leased office space at our corporate location and the write-off of certain furnishings
and leasehold improvements. We incurred the following costs in connection with our restructuring programs during the years ended December 31, 2010 and 2009 (there were no restructuring charges related to continuing operations in 2008) (millions of dollars):
Year Ended
December 31,
2010
2009 Employee separation benefits First-quarter 2010 program
$
9.7
$
— Fourth-quarter 2010 program
3.0
— 2009 program
—
0.7
12.7
0.7 Other
1.3
— Restructuring charge
$
14.0
$
0.7 There were two major restructurings initiated during 2010, both of which reflected the transition of the Company from a fully integrated biopharmaceutical company with research, manufacturing and marketing operations to a biotechnology company dedicated to oncology research and development. During the first quarter of 2010, a workforce reduction involving 64 employees was announced resulting in an expense of $6.1 million for separation costs for the affected employees. These actions related primarily to 42
the sale of the specialty pharmaceutical business including several employees who were previously engaged in activities related to the divested business but who did not transfer to the employment of the purchaser. These employees were provided with separation benefits after certain transition periods during which they assisted with an
orderly transfer of activities and information to the purchaser. Also, effective February 22, 2010, our then President and Chief Executive Officer, resigned from the Company. For the quarter ended March 31, 2010, we expensed $3.8 million for severance payments and benefits that were payable to the individual per the terms of his
employment agreement. This amount was reduced during the quarter ended June 30, 2010 by approximately $0.2 million once the termination agreement was executed. Employees affected by the fourth quarter 2010 restructuring were notified in December 2010. This restructuring program was part of our continued efforts to streamline corporate administrative operations. The majority of the terminations will occur during the first quarter of 2011 and separation payments totaling $3.0 million will be
made for up to a year following the respective terminations. The portion of the combined restructuring expense to be paid out in the next twelve months ($3.6 million) is reported as a current liability in accrued expenses as of December 31, 2010. The remainder is reported as an other liability ($0.3 million). During the second quarter of 2010, we wrote off certain leasehold improvements and furnishings located at our then corporate headquarters in Bridgewater, New Jersey that were determined to be excess and without future value as a result of the termination and relocation of several employees. The noncash charge related to this
write off was approximately $0.9 million. During the third quarter of 2010, we entered into a sublease for a portion of our excess corporate facilities. These facilities became unused as a result of the reductions in workforce stemming from earlier restructuring efforts. The charge of approximately $0.4 million represents the excess of our
contractual lease commitment over the amount of cash to be received from the subtenant over the life of the sublease arrangement. By the end of the first quarter of 2011, we expect to have completed the relocation of our corporate offices to Piscataway, New Jersey. The vacating of the excess office space in Bridgewater will likely
trigger a further restructuring charge in 2011 for the excess of committed lease costs over anticipated sublease income. Corporate restructuring costs associated with the 2009 workforce reduction amounted to $0.7 million during the first quarter of 2009. This represents separation benefits and related costs of terminated employees in general and administrative areas as well as research and development. The payments related to this charge were
completed within the year and no accruals remained as of December 31, 2009. Other Income (Expense)
Year Ended December 31,
2010
%
2009
%
2008 Other income (expense): Investment income, net
$
3.5
(20
)
$
4.3
(35
)
$
6.6 Interest expense
(6.3
)
(45
)
(11.5
)
(9
)
(12.7
) Other-than-temporary investment impairment loss
(0.9
)
n.m.
—
n.m.
(0.6
) Other, net
1.2
(76
)
5.0
165
1.8
$
(2.5
)
17
$
(2.2
)
(55
)
$
(4.9
) n.m.—not meaningful Net other income (expense) for the three years ended December 31, 2010, 2009 and 2008 was: expense of $2.5 million, $2.2 million and $4.9 million, respectively. The repurchase and conversion of a portion of our 4% notes and the repurchase and retirement of the remaining 4.5% notes during the three-year period affected the year-
to-year comparisons in a number of ways (See Liquidity and Capital Resources below). Also, in 2010, two significant items that tended to offset one another were the recognition of an impairment in an investment holding and recognition of an award of a government grant. Further discussion of each of the individual items follows. 43
(millions of dollars):
Change
Change
Net investment income in 2010 was lower than that of 2009 by $0.8 million and in 2009 it was lower than in 2008 by approximately $2.3 million. This was due to reductions in the amount of investment holdings and, in general, reductions in the rates of return being earned. During 2010, as investments matured, the proceeds were
placed in money market funds. Proceeds from a portion of our investment holdings were used to repurchase notes payable in amounts of $15.6 million in 2009 and $72.0 million in 2008. Interest expense includes amortization and, when debt is repurchased, write-off of deferred debt issuance costs. Interest expense has declined over the three-year period through 2010, to $6.3 million in 2010 from $11.5 million in 2009 and from $12.7 million in 2008. The reduction in 2010 was due primarily to the conversion in the
first quarter of the year of $115.6 million principal amount of our 4% notes into shares of our common stock in connection with the sale of the specialty pharmaceutical business. Reductions in interest expense in 2009 related to the repurchases of the 4% notes in 2009 and 2008 referred to above and repayment of the balance of our 4.5%
notes in July 2008 ($72.4 million principal amount). The write-off of deferred debt issuance costs was $0.3 million and $0.2 million for the years ended December 31, 2009 and 2008, respectively. Other-than-temporary impairment losses on available-for-sale investment holdings representing credit losses are charged to earnings. We hold an investment in one auction rate security that we believe is more likely than not impaired due to the lack of credit worthiness of the issuer and its parent company. Consequently, the
remaining carrying value of $0.9 million was written off during the third quarter of 2010. In 2008, we wrote the investment down by $0.6 million based upon information available at that time. Other income in 2010 is primarily the receipt of a $1.2 million federal government grant for qualifying therapeutic discovery investments made by us in 2009 and 2010. The majority of the funds were received in December 2010 while approximately $0.2 million remained receivable as of December 31, 2010. Significant portions of
other income in 2009 and 2008 relate to gains realized on the repurchase of notes payable. During the first quarter of 2009, we repurchased $20.4 million in principal amount of our 4% notes at a discount to par yielding a gross gain of $4.8 million. In 2008, we repurchased $4.5 million in principal amount of our 4% notes at a discount to
par yielding a gross gain of approximately $1.7 million. We also repurchased a portion of our 4.5% notes early in 2008 at a gross gain of $0.4 million. Losses related to asset disposals and foreign exchange partially offset the 2008 gains on repurchase of notes payable. In each case, the gross gains reflected here are exclusive of the write-
off of deferred debt issuance costs. Discontinued Operations The cash proceeds received from the sale of the specialty pharmaceutical business, including a second-quarter 2010 working capital adjustment, amounted to approximately $308.0 million. Of this amount, $40.9 million was allocated to the sale of in-process research and development and included in continuing operations. The net
proceeds then attributable to discontinued operations yielded a gain of $176.4 million. The results of operations of the specialty pharmaceutical business for the period in January 2010 preceding the sale amounted to income of $3.6 million comprising the remainder of the $180.0 reported in 2010 as income and gain from discontinued
operations. Although the sale was a taxable event, no tax liability arose due to the basis we had in the underlying assets and the current year net operating loss. Under the terms of the asset purchase agreement, we also were entitled to receive up to an additional $27.0 million if certain milestones are met. Of this amount, we will receive $5.0 million in 2011, which was earned in the first quarter of 2011, and another $5.0 million is no longer considered likely to be received. There can be no
assurance that we will receive any of the remaining $17.0 million in milestone payments. In addition, we may receive royalties of 5 to 10 percent on incremental net sales above a 2009 baseline amount of our then four marketed specialty pharmaceutical products through 2014. Revenues from these milestones and/or royalties is reflected
as part of our continuing operations. Prior-year results of operations of the specialty pharmaceutical business have been reclassified as discontinued operations for comparability. 44
Income Taxes Federal legislation, the American Recovery and Reinvestment Act of 2009, which allowed us to make an election to treat certain unused research and alternative minimum tax credit carryforwards as refundable in lieu of claiming bonus and accelerated depreciation for “eligible qualified property” placed in service through the end of
2009 was extended to 2010. This provided us with a $0.1 million benefit in 2010. The balance of the 2010 income tax benefit reflects a reduction of $0.2 million to state taxes payable. In November 2009, federal legislation was enacted under which we are able to carryback our 2009 alternative minimum tax net operating losses to the five previous years to offset the alternative minimum taxes that were not available to us for carryback prior to the new legislation. We recorded the impact of the carryback, estimated
to be approximately $1.7 million, in the fourth quarter of 2009 and received a federal income tax cash refund in the first quarter of 2010. Other legislation in 2009 allowed us to make an election to treat certain unused research and alternative minimum tax credit carryforwards as refundable in lieu of claiming bonus and accelerated
depreciation for “eligible qualified property” placed in service through the end of 2008. This provided us with a $0.5 million benefit in 2009. Offsetting these two tax benefit amounts which total $2.2 million was a $0.1 million charge related to an adjustment of state taxes payable. Income tax expense in 2008 was primarily comprised of certain state taxes. No federal income tax expense was incurred in relation to normal operating results due either to current period operating losses or the utilization of deferred tax assets to offset taxes that would otherwise accrue to operating income. Liquidity and Capital Resources Cash reserves, including cash, cash equivalents, short-term investments and marketable securities, totaled $460.1 million as of December 31, 2010 and $199.7 million as of December 31, 2009. The increase was primarily attributable to the receipt of proceeds from the sale of our specialty pharmaceutical business in January 2010.
Partially offsetting the sale proceeds, approximately $48.2 million in cash was utilized to repurchase common stock. Approximately $32.3 million of cash was received as a result of employee exercises of stock options and employee stock purchase plan activity. Cash
provided by operating activities of our continuing operations during 2010
was $22.2 million whereas in 2009 there was a use of operating cash by continuing
operations of $51.6 million. The primary contributor to the approximately
$73.8 million difference was the change in loss from continuing operations.
The loss from continuing operations in 2009 was $57.2 million compared to
a loss of $2.8 million in 2010. The 2010 results of operations of our continuing
operations included the receipt of $40.9 million related to the sale of in-process
research and development. As there was no book basis in this asset, the entire
proceeds represented positive cash inflow largely offsetting the cash outflows
resulting from the regular operations of our continuing business during the
year. When adjusted for noncash and nonoperating items, the loss from continuing
operations in 2010 of $2.8 million was converted into a positive cash flow
of approximately $14.4 million. Such noncash items in 2009 had less of a
positive cash flow effect by approximately $6.8 million. Fluctuations in
operating asset and liability balances in 2010 constituted a source of cash
of $7.7 million. In 2009, fluctuations in operating asset and liability accounts
resulted in a use of cash of approximately $4.9 million. Cash used in operating
activities of continuing operations in 2009 was down from that of 2008 by
approximately $15.1 million primarily due to $10.6 million greater loss from
operations of the continuing business and less cash provided from changes
in operating assets and liabilities. Cash
flows from investing activities amounted to $344.0 million in 2010. Cash
proceeds from the sale of the specialty pharmaceutical business amounted
to $262.6 million, excluding the $40.9 million attributed to the sale of
in-process research and development and included in income from continuing
operations. Net sales and maturities of marketable securities generated $86.3
million of cash while investments in property and equipment utilized $2.0
million of cash in 2010. In 2009, net investments in marketable securities
of $17.8 million plus purchases of property and equipment of $2.0 million
resulted in a use of cash in investing activities of continuing operations
of $19.8 million. Net cash used in investing activities of discontinued operations
in 2009 was comprised of investments in property and equipment plus a $5.0
million milestone payment related to sales of the divested product, Oncaspar. 45
Financing activities in 2010, 2009 and 2008 related primarily to repurchases of our outstanding shares and the repurchase and refinancing of our long-term debt, offset in part by proceeds from exercises of stock options. Exclusive of transaction costs, we expended approximately $48.2 million during 2010 and $2.0 million during
December 2009 to repurchase shares of the Company’s outstanding common stock. The majority of the purchases were made pursuant to a $50.0 million share repurchase plan announced December 3, 2009. A $200 million share repurchase program was announced in the latter half of December 2010 and remains in effect as of December
31, 2010. Through February 28, 2011, approximately 1.5 million additional shares have been purchased under the repurchase program at a cost of approximately $17.3 million. The repurchase of a portion of outstanding notes payable constituted a use of cash of $15.6 million in 2009 and $74.8 million in 2008. No notes were repurchased during 2010 although there was a noncash conversion that took place during the first quarter of 2010 (see below). During 2010, approximately $32.3 million of cash was
received as the result of employee exercises of stock options and employee stock purchase plan activity. As of December 31, 2010, the principal amount of the 4% notes payable outstanding was $134.5 million. The sale of our specialty pharmaceutical business constituted a fundamental change under the indenture for the notes, which triggered a requirement that we offer to purchase all of the notes at face value. On February 5, 2010,
we initiated a tender offer to purchase for cash any and all of the notes at face value. The offer expired on March 5, 2010 with no notes having been tendered. The fundamental change also triggered a change in the conversion rate from 104.712 shares per $1,000 principal amount of notes to 116.535 shares per $1,000 principal amount of
notes during the period January 29, 2010 to March 4, 2010. During this period, $115.6 million principal amount of the notes were converted into approximately 13.5 million shares of our common stock, reducing the principal balance of the notes outstanding to $134.5 million. Subsequent to the March 4, 2010, the date the enhanced
conversion period ended, the original conversion rate of 104.712 shares per $1,000 principal amount is again in effect. Our current sources of liquidity are our cash reserves, interest earned on such cash reserves and royalties—primarily those related to sales of PEGINTRON. In January 2011, we received a $5.0 million milestone payment in connection with the sale of the specialty pharmaceutical business. Per the terms of the sale agreement, we were
entitled to the $5.0 million payment upon regulatory approval of an sBLA regarding a new API starting material for the manufacture of SS Oncaspar. This approval was received in January 2011. No further milestones related to the sale of the specialty pharmaceutical business are expected in 2011 and there can be no assurance that any
of these milestones will be received in the future. Based upon our current planned research and development activities and related costs, our current sources of liquidity, the expected cash outflows from operations and the purchase of up to $200 million of our outstanding stock, we anticipate our current cash reserves will be sufficient to meet our capital and operational requirements
for the near future. While we believe that our current sources of liquidity will be adequate to satisfy our capital and operational needs for the near future, it is likely that we will need to obtain additional financing or enter into a collaborative arrangement to sustain our research and development efforts prior to the time we are able to
commercialize any of our product candidates. There can be no assurance, however, that we will be able to obtain additional funds or engage a collaborator on acceptable terms, if at all. If we are unable to obtain adequate financing or collaborative support, we may be required to curtail our research and development activities and/or
license our product candidates to third parties. Off-Balance Sheet Arrangements As part of our ongoing business, we do not participate in transactions that generate relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or
other contractually narrow limited purposes. As of December 31, 2010, we were not involved in any off-balance sheet special purpose entity transactions. Our 4% notes are convertible at the option of the holder into shares of our common stock at a conversion price of $9.55 per share. At December 31, 2010, the potential dilutive effect of conversion of the 4% notes was 46
14.1 million shares using the conversion price of $9.55 per share or 104.712 shares per $1,000 principal amount of notes. In addition, stock options to purchase 4.0 million shares of our common stock at a weighted average exercise price of $13.21 per share and 0.7 million restricted stock units were outstanding at December 31, 2010, that represent additional potential dilution. Contractual Obligations Contractual obligations represent future cash commitments and liabilities under agreements with third parties, and exclude contingent liabilities for which we cannot reasonably predict future payment. The following chart represents our contractual cash obligations as of December 31, 2010 (in millions):
Contractual Obligations and Commercial Commitments(1) (2)
Payments Due By Period
Total
Less Than
2-3
4-5
More Than Notes payable due June 1, 2013
$
134.5
$
—
$
134.5
$
—
$
— Operating lease obligations(3)
11.0
2.3
3.1
1.4
4.2 Interest due on notes payable
13.5
5.4
8.1
—
— Totals
$
159.0
$
7.7
$
145.7
$
1.4
$
4.2
(1)
Does not include potential milestone payments of $233.0 million to Santaris that are only payable upon successful development of all eight mRNA targets selected by us. (2) Does not include separation payments of approximately $3.9 million to be made to exiting employees in connection with the 2010 restructurings. (3) Does not include lease revenues to be received pursuant to certain subleased facilities. As of December 31, 2010, we had $134.5 million of 4% convertible senior unsecured notes outstanding. These notes mature on June 1, 2013 unless earlier redeemed, repurchased or converted. The 4% notes rank equal to all future senior unsecured debt. If the closing price of our common stock for at least 20 trading days in the 30
consecutive trading day period ending on the date one day prior to the date of a notice of redemption is greater than 140 percent of the applicable conversion price on the date of such notice, we, at our option, may redeem the 4% notes in whole or in part, at a redemption price in cash equal to 100 percent of the principal amount of the
4% notes to be redeemed, plus accrued interest, if any, to the redemption date. We lease three facilities in New Jersey. Future minimum lease payments and commitments for operating leases total $11.0 million at December 31, 2010. In the third quarter of 2010, we entered into a sublease of a portion of the office space located in Bridgewater, New Jersey. We anticipate that the remainder of the space in
Bridgewater will be vacated by March 31, 2011 as we consolidate our administrative and research organizations in our Piscataway location. We are actively seeking a tenant or tenants for the excess Bridgewater capacity. In October 2009, we entered into a sublease of the South Plainfield facility under which we will receive rental income
in excess of the rental expense being incurred under the original lease. Our use of the leased South Plainfield facility (not included in the sale of the specialty pharmaceutical business) has ended, but we continue to be primarily responsible for the obligations attendant to the continuing operating lease of the facility including returning the
facility to its original condition upon expiration, if necessary. We may experience additional charges associated with the lease or its termination prior to the contractual expiration of the lease in October 2012. In July 2006, we entered into a license and collaboration agreement with Santaris pursuant to which we obtained exclusive rights worldwide, other than in Europe, to develop and commercialize RNA antagonists directed against the HIF-1a, Survivin and AR gene targets, as well as RNA antagonists directed against five additional gene
targets selected by us. We will be responsible for making additional payments upon the successful completion of certain compound synthesis and selection, clinical development and regulatory milestones. Santaris also is eligible to receive royalties from any future product sales of products based on the 47
1 Year
Years
Years
5 Years
licensed antagonists. Santaris retains the right to develop and commercialize products developed under the collaboration in Europe. Critical Accounting Policies and Estimates A critical accounting policy is one that is both important to the portrayal of a company’s financial condition and results of operations and requires management’s most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. Our consolidated financial statements are presented in accordance with accounting principles that are generally accepted in the U.S. All professional accounting standards effective as of December 31, 2010 have been taken into consideration in preparing the consolidated financial statements. The preparation of the consolidated
financial statements requires estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosures. Some of those estimates are subjective and complex, and, consequently, actual results could differ from those estimates. The following accounting policies and estimates have been
highlighted as significant because changes to certain judgments and assumptions inherent in these policies could affect our consolidated financial statements. We base our estimates, to the extent possible, on historical experience. Historical information is modified as appropriate based on current business factors and various assumptions that we believe are necessary to form a basis for making judgments about the carrying value of assets and liabilities. We evaluate our estimates on an
ongoing basis and make changes when necessary. Actual results could differ from our estimates. Revenues Royalties under our license agreements with third-parties and pursuant to the sale of our specialty pharmaceutical business are recognized when reasonably determinable and earned through the sale of the product by the third-party and collection is reasonably assured. Notification from the third-party licensee of the royalties earned
under the license agreement is the basis for royalty revenue recognition. This information generally is received from the licensees in the quarter subsequent to the period in which the sales occur. Contingent payments due under the asset purchase agreement related to the sale of the specialty pharmaceutical business are recognized as income when the milestone has been achieved and collection is assured. Such payments are non-refundable and no further effort is required on the part of the Company or the other party to
complete the earning process. Non-refundable payments received upon entering into license and other collaborative agreements where we have continuing involvement are recorded as deferred revenue and recognized ratably over the estimated service period. The sale of the specialty pharmaceutical business involved the application of guidance regarding multiple deliverables in separating the revenues associated with the sale of in-process research and development from the other elements of the transaction, namely the assets sold as part of discontinued operations and our continuing
involvement in contract research activities. We determined that the in-process research and development had value to the buyer of the specialty pharmaceutical business on a stand-alone basis and that there was objective and reliable evidence available to support the allocation of the total purchase price to the respective units of
accounting. Research and Development Expenses We accrue expenses for costs for work performed by contract research organizations, contract manufacturing organizations and others based upon the estimated amount of the total effort completed on each order, study or project using factors such as number of lots produced, number of patients enrolled, the number of active clinical
sites and the duration for which the patients will be enrolled in the study. We base the estimates on the information available at the time. Additional information may come available at a later date that would enable us to develop a more accurate estimate. Such changes in estimate are generally recognized in the period when the
information is first known. 48
Income Taxes Under the asset and liability method of accounting for income taxes, deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit
carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. A valuation allowance on net deferred tax assets is provided for when it is more likely than not some portion or all of the
deferred tax assets will not be realized. As of December 31, 2010, we believe, based on projections, that it is more likely than not that our net deferred tax assets, including our net operating losses from operating activities and stock option exercises, will not be realized. We recognize the benefit of an uncertain tax position that we have
taken or expect to take on the income tax returns we file if it is more likely than not we will be able to sustain our position. Share-Based Payments Compensation cost, measured by the fair value of the equity instruments issued, adjusted for estimated forfeitures, is recognized in the financial statements as the respective awards are earned. The impact that share-based payment awards will have on our results of operations is a function of the number of shares awarded, vesting and
the trading price and fair value of our stock at date of grant or modification. Fair value of share-based payments is determined using the Black-Scholes valuation model which employs weighted average assumptions for expected volatility of our stock, expected term until exercise of the options, the risk free interest rate, and dividends, if
any. Expected volatility is based on our historical stock price information. Forward-Looking Information and Factors That May Affect Future Results There are forward-looking statements contained herein, which can be identified by the use of forward-looking terminology such as the words “believes,” “expects,” “may,” “will,” “should,” “potential,” “anticipates,” “plans,” or “intends” and similar expressions. Such forward-looking statements involve known and unknown risks,
uncertainties and other factors that may cause actual results, events or developments to be materially different from the future results, events or developments indicated in such forward-looking statements. Such factors include but are not limited to the timing, success and cost of clinical studies for our product candidates, the ability to
obtain regulatory approval of our product candidates, our ability to obtain the funding necessary to develop our product candidates, market acceptance of and demand for our product candidates, and the impact of competitive products, pricing and technology. A more detailed discussion is contained in “Risk Factors” in Item 1A, Part I of
this report. These factors should be considered carefully and readers are cautioned not to place undue reliance on such forward-looking statements. No assurance can be given that the future results covered by the forward-looking statements will be achieved. All information contained herein is as of the date of this report and we do not
intend to update this information. Item 7A. Quantitative and Qualitative Disclosures About Market Risk Our financial instruments are principally comprised of money market funds and debt securities. Short term investments and marketable securities are classified as securities available for sale. Apart from custodial accounts related to the Executive Deferred Compensation Plan, we do not invest in portfolio equity securities. We do not
invest in commodities or use financial derivatives for trading purposes. We seek reasonable assuredness of the safety of principal and market liquidity by investing in rated fixed income securities while at the same time seeking to achieve a favorable rate of return. Our market risk exposure consists principally of exposure to changes in
interest rates. Our holdings also are exposed to the risks of changes in the credit quality of issuers the majority of which are rated A1 or better. We typically invest the majority of our investments in the shorter-end of the maturity spectrum. Cash equivalents are primarily held in a number of triple-A rated institutional money market funds
as well as several corporate and U.S. government-sponsored entities’ debt securities. The table below presents the amortized cost, fair value and related weighted average interest rates by year of maturity for our available-for-sale securities as of December 31, 2010 excluding primarily those related to 49
our Executive Deferred Compensation Plan (in thousands). There are no variable-rate investments as of December 31, 2010.
Amortized Cost
Fair Value
2011
2012
Total Fixed Rate
$
30,931
$
27,701
$
58,632
$
59,460 Average Interest Rate
5.47
%
5.13
%
5.31
%
$
30,931
$
27,701
$
58,632
$
59,460 Our 4% convertible senior unsecured notes in the principal amount of $134.5 million at December 31, 2010 are due June 1, 2013 and have a fair value of $182.4 million at December 31, 2010. Our outstanding convertible notes have a fixed interest rate. The fair value of the convertible notes is affected by changes in market rates of
interest and the price of our common stock. Item 8. Financial Statements and Supplementary Data Financial statements and notes thereto appear on pages F-1 to F-33 of this Annual Report on Form 10-K. Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure Not Applicable. Item 9A. Controls and Procedures (a) Evaluation of Disclosure Controls and Procedures Our management, under the direction of our Principal Executive Officer and Principal Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, (the Exchange Act)) as of December 31, 2010. Based
on that evaluation, our Principal Executive Officer and Principal Financial Officer have concluded that our disclosure controls and procedures were effective as of December 31, 2010. (b) Changes in Internal Controls There were no changes in our internal controls over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act, during the three-month period ended December 31, 2010 covered by this report that have materially affected, or are reasonably likely to materially affect, our internal control over
financial reporting. (c) Management’s Report on Internal Control over Financial Reporting It is the responsibility of the management of Enzon Pharmaceuticals, Inc. and subsidiaries to establish and maintain effective internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Internal control over financial reporting is designed to provide reasonable assurance to Enzon’s management and
board of directors regarding the preparation of reliable consolidated financial statements for external purposes in accordance with generally accepted accounting principles. Enzon’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of Enzon; (ii) provide reasonable assurance that transactions are recorded as necessary to permit
preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of Enzon are being made only in accordance with authorizations of management and directors of Enzon; and (iii) provide reasonable assurance regarding the prevention or timely detection of
unauthorized acquisition, use or disposition of Enzon’s assets that could have a material effect on the consolidated financial statements of Enzon. 50
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Management has performed an assessment of the effectiveness of Enzon’s internal control over financial reporting as of December 31, 2010 based upon criteria set forth in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this assessment,
management has concluded that our internal control over financial reporting was effective as of December 31, 2010. Our independent auditor, KPMG LLP, an independent registered public accounting firm, has issued an auditors’ report on the effectiveness of internal control over financial reporting as of December 31, 2010. The auditor’s report follows.
/s/Ralph del Campo
/s/ Mark L. Ogden
March 16, 2011
March 16, 2011 51
Ralph del Campo
Chief Operating Officer
(Principal Executive Officer)
Mark L. Ogden
Vice President, Finance and
(Principal Financial Officer and
Principal Accounting Officer)
(d) Report of Independent Registered Public Accounting Firm The Board of Directors and Stockholders We have audited Enzon Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Enzon Pharmaceuticals, Inc.’s management is
responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal
control over financial reporting based on our audit. We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit
included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the
circumstances. We believe that our audit provides a reasonable basis for our opinion. A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting
includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance
with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or
procedures may deteriorate. In our opinion, Enzon Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). We
also have audited, in accordance with the standards of the Public Company
Accounting Oversight Board (United States), the consolidated balance sheets
of the Company as of December 31, 2010 and 2009, and the related consolidated
statements of operations, stockholders’ equity and cash flows for each
of the years in the three- year period ended December 31, 2010, and our report
dated March 16, 2011 expressed an unqualified opinion on those consolidated
financial statements. /s/ KPMG LLP Short Hills, New Jersey 52
Enzon Pharmaceuticals, Inc.:
March 16, 2011
None. The information required by Item 10 — Directors, Executive Officers and Corporate Governance; Item 11 — Executive Compensation; Item 12 — Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters, Item 13 — Certain Relationships and Related Transactions,
and Director Independence and Item 14 — Principal Accountant Fees and Services is incorporated into Part III of this Annual Report on Form 10-K by reference to the Proxy Statement for our 2011 Annual Meeting of Stockholders which Proxy Statement is expected to be filed with the Securities and Exchange Commission not
later than 120 days after the close of our fiscal year ended December 31, 2010. Item 15. Exhibits and Financial Statement Schedules (a)(1), (a)(2) and (c). The response to this portion of Item 15 is submitted as a separate section of this report commencing on page F-1. (a)(3) and (b). Exhibits (numbered in accordance with Item 601 of Regulation S-K).
Exhibit
Description
Reference
2.1
Asset Purchase Agreement, dated as of November 9, 2009, by and between Klee Pharmaceuticals, Inc., Defiante Farmaceutica, S.A. and Sigma-Tau Finanziaria
(32)
Amended and Restated Certificate of Incorporation dated May 18, 2006, together with the Certificate of Amendment to the Amended and Restated Certificate of Incorporation dated July 13, 2010
(1)
Amended and Restated Bylaws effective July 13, 2010
(1)
4.1
Rights Agreement dated May 17, 2002 between the Company and Continental Stock Transfer & Trust Company, as rights agent
(3)
4.2
First Amendment to the Rights Agreement, dated as of February 19, 2003 between the Company and Continental Stock Transfer & Trust Company, as rights agent
(4)
4.3
Second Amendment to the Rights Agreement dated as of January 7, 2008 between the Company and Continental Stock Transfer and Trust Company, as rights agent.
(5)
4.4
Third Amendment to the Rights Agreement dated as of July 23, 2009 between the Company and Continental Stock Transfer and Trust Company, as rights agent.
(6)
4.5
Indenture, dated May 23, 2006, between Enzon Pharmaceuticals, Inc. and Wilmington Trust Company
(7)
4.6
First Supplemental Indenture, dated August 25, 2008, between Enzon Pharmaceuticals, Inc. and Wilmington Trust Company
(8)
10.1
Lease — 300-C Corporate Court, South Plainfield, New Jersey
(9)
10.2
Lease dated April 1, 1995 regarding 20 Kingsbridge Road, Piscataway, New Jersey
(10)
10.3
First Amendment to Lease regarding 20 Kingsbridge Road, Piscataway, New Jersey, dated as of November 13, 2001
(11)
10.4
Lease 300A-B Corporate Court, South Plainfield, New Jersey
(12)
10.5
Modification of Lease Dated May 14, 2003 — 300-C Corporate Court, South Plainfield, New Jersey
(13)
10.6
Lease — 685 Route 202/206, Bridgewater, New Jersey
(14)
10.7
First Amendment of Lease — 685 Route 202/206, Bridgewater, New Jersey
(15)
10.8
Second Amendment to Lease — 685 Route 202/206, Bridgewater, New Jersey
(15)
10.9
Third Amendment to Lease — 685 Route 202/206, Bridgewater, New Jersey
(15) 53
Number
No.
Exhibit
Description
Reference
10.10
2001 Incentive Stock Plan, as amended and restated, of Enzon Pharmaceuticals, Inc.**
(2)
10.11
Development, License and Supply Agreement between the Company and Schering Corporation; dated November 14, 1990, as amended*
(16)
10.12
Executive Deferred Compensation Plan (2008 Restatement)**
(17)
10.13
Form of Non-Qualified Stock Option Agreement between the Company and Craig A. Tooman**
(18)
10.14
Amended and Restated Severance Agreement with Paul S. Davit dated May 7, 2004**
(18)
10.15
Amended and Restated Severance Agreement with Ralph del Campo dated May 7, 2004**
(18)
10.16
2007 Outside Director Compensation Plan, as amended**
(19)
10.17
Employment Agreement with Ivan D. Horak, M.D. dated September 2, 2005, along with a form of Stock Option Award Agreement and Restricted Stock Unit Award Agreement between the Company and Dr. Horak executed as of September 2, 2005*,**
(20)
10.18
Form of Non-Qualified Stock Option Agreement for Executive Officers**
(21)
10.19
Form of Restricted Stock Award Agreement for Executive Officers**
(21)
10.20
Form of Restricted Stock Unit Award Agreement for Executive Officers**
(22)
10.21
Form of Restricted Stock Unit Award Agreement for Independent Directors**
(20)
10.22
Form of Stock Option Award Agreement for Independent Directors 1987 Non-Qualified Stock Option Plan**
(20)
10.23
Form of Stock Option Award Agreement for Independent Directors 2001 Incentive Stock Plan**
(20)
10.24
Amended and Restated Employment Agreement with Craig A. Tooman dated June 18, 2008
(23)
10.25
2007 Employee Stock Purchase Plan
(24)
10.26
Amended and Restated Employment Agreement with Jeffrey H. Buchalter dated April 27, 2007**
(25)
10.27
Amendment dated February 21, 2008 to Amended and Restated Employment Agreement with Jeffrey H. Buchalter**
(26)
10.28
Amendment No. 2 dated July 23, 2009 to Amended and Restated Employment Agreement with Jeffrey H. Buchalter**
(27)
10.29
Purchase Agreement between the Company and Drug Royalty LP1 dated as of August 19, 2007
(28)
10.30
Amendment to Amended and Restated Severance Agreement with Paul S. Davit dated November 6, 2007**
(29)
10.31
Amendment No. 2 to Amended and Restated Severance Agreement with Ralph del Campo dated as of June 18, 2010**
(33)
10.32
License and Collaboration Agreement dated July 26, 2006 by and between Santaris Pharma A/S and Enzon Pharmaceuticals, Inc.***
(30)
10.33
Amendment No.1 to License and Collaboration Agreement, dated June 13, 2007 by and between Santaris Pharma A/S and Enzon Pharmaceuticals, Inc.***
(30)
10.34
Amendment No. 2 to License and Collaboration Agreement, dated June 25, 2007 by and between Santaris Pharma A/S and Enzon Pharmaceuticals, Inc.***
(30)
10.35
Amendment No. 3 to License and Collaboration Agreement, dated December 21, 2007 by and between Santaris Pharma A/S and Enzon Pharmaceuticals, Inc.***
(30)
10.36
Amendment No. 4 to License and Collaboration Agreement, dated July 8, 2009 by and between Santaris Pharma A/S and Enzon Pharmaceuticals, Inc.***
(31) 54
Number
No.
Exhibit
Description
Reference
10.37
Amendment No. 5 to License and Collaboration Agreement, dated October 2, 2009 by and between Santaris Pharma A/S and Enzon Pharmaceuticals, Inc.***
(31)
10.38
Amendment to Outstanding Awards Under 2001 Incentive Stock Plan**
(30)
10.39
2001 Incentive Stock Plan Non-Qualified Stock Plan Terms and Conditions**
(30)
10.40
2001 Incentive Stock Plan Restricted Stock Unit Award Terms and Conditions**
(30)
10.41
2001 Incentive Stock Plan Restricted Stock Award Terms and Conditions**
(30)
10.42
Consulting Agreement dated as of October 5, 2005 by and between Mark L. Ogden and Enzon Pharmaceuticals, Inc., together with all amendments thereto**
(34)
12.1
Computation of Ratio of Earnings to Fixed Charges
+
21.1
Subsidiaries of Registrant
+
23.1
Consent of Independent Registered Public Accounting Firm
+
31.1
Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
+
31.2
Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
+
32.1
Certification of Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
+
32.2
Certification of Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
+
+
Filed herewith
Referenced exhibit was previously filed with the Commission as an exhibit to the Company’s filing indicated below and is incorporated herein by reference to that filing:
Quarterly Report on Form 10-Q for the quarter ended June 30, 2010 filed August 9, 2010 (2) Current Report on Form 8-K filed May 19, 2006 (3) Form 8-A12G (File No. 000-12957) filed May 22, 2002 (4) Form 8-A12G/A (File No. 000-12957) filed February 20, 2003 (5) Current Report on Form 8-K filed January 8, 2008 (6) Form 8-A/A filed July 24, 2009 (7) Current Report on Form 8-K filed May 25, 2006 (8) Current Report on Form 8-K filed August 25, 2008 (9) Registration Statement on Form S-18 (File No. 2-88240-NY) (10) Quarterly Report on Form 10-Q for the quarter ended March 31, 1995 filed May 12, 1995 (11) Transition Report on Form 10-K for the six months ended December 31, 2005. (12) Annual Report on Form 10-K for the fiscal year ended June 30, 1993 (13) Annual Report on Form 10-K for the fiscal year ended June 30, 2003 (14) Quarterly Report on Form 10-Q for the quarter ended March 31, 2002 filed May 15, 2002 (15) Quarterly Report on Form 10-Q for the quarter ended September 30, 2006 filed November 2, 2006 (16) Annual Report on Form 10-K for the fiscal year ended June 30, 2002 (17) Quarterly Report on Form 10-Q for the quarter ended September 30, 2007 filed November 1, 2007 (18) Annual Report on Form 10-K for the fiscal year ended June 30, 2005 (19) Quarterly report on Form 10-Q for the quarter ended June 30, 2007 filed August 2, 2007 55
Number
No.
(1)
(20) Quarterly Report on Form 10-Q for the quarter ended September 30, 2005 filed November 9, 2005 (21) Quarterly Report on Form 10-Q for the quarter ended December 31, 2004 filed February 9, 2005 (22) Quarterly Report on Form 10-Q for the quarter ended March 31, 2005 filed May 10, 2005 (23) Current Report on Form 8-K filed June 20, 2008 (24) Form S-8 (File No. 333-140282) filed January 29, 2007 (25) Quarterly Report on Form 10-Q for the quarter ended March 31, 2007 filed May 4, 2007 (26) Annual Report on Form 10-K for the year ended December 31, 2007 (27) Quarterly Report on Form 10-Q for the quarter ended June 30, 2009 filed August 5, 2009 (28) Current Report on Form 8-K filed August 20, 2007 (29) Current Report on Form 8-K filed November 13, 2007 (30) Annual Report on Form 10-K for the year ended December 31, 2008 (31) Quarterly Report on Form 10-Q for the quarter ended September 30, 2009 filed November 3, 2009 (32) Current Report on Form 8-K filed November 12, 2009 (33) Current Report on Form 8-K filed June 17, 2010 (34) Quarterly Report on Form 10-Q for the quarter ended September 30, 2010 filed November 4, 2010
*
Portions of this exhibit have been redacted and filed separately with the Commission pursuant to a confidential treatment request. ** Management contracts or compensatory plans and arrangements required to be filed pursuant to Item 601(b)(10)(ii)(A) or (iii) of Regulation S-K. *** The Company has requested confidential treatment of the redacted portions of this exhibit pursuant to Rule 24b-2 under the Securities Exchange Act of 1934, as amended, and has separately filed a complete copy of this exhibit with the Securities and Exchange Commission. 56
SIGNATURES Pursuant to the requirements of section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized. ENZON PHARMACEUTICALS, INC. Dated: March 16, 2011 /s/ Ralph del Campo Ralph del Campo Dated: March 16, 2011 /s/ Mark L. Ogden Mark L. Ogden Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
Name
Title
Date
/s/ Ralph del Campo Ralph del Campo Chief Operating Officer March 16, 2011 /s/ Mark L. Ogden Mark L. Ogden Vice President, Finance March 16, 2011 /s/ Alexander J. Denner Alexander J. Denner Chairman of the Board March 16, 2011 /s/ Rolf A. Classon Rolf A. Classon Director March 16, 2011 /s/ Thomas F. Deuel Thomas F. Deuel Director March 16, 2011 /s/ Robert LeBuhn Robert LeBuhn Director March 16, 2011 Harold J. Levy Director /s/ Richard C. Mulligan Richard C. Mulligan Director March 16, 2011 /s/ Robert C. Salisbury Robert C. Salisbury Director March 16, 2011 /s/ Richard A. Young Richard A. Young Director March 16, 2011 57
(Registrant)
Chief Operating Officer
(Principal Executive Officer)
Vice President, Finance
(Principal Financial Officer and
Principal Accounting Officer)
(Principal Executive Officer)
(Principal Financial Officer and
Principal Accounting Officer)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES Index
Page
F-2 Consolidated Financial Statements:
F-3 Consolidated Statements of Operations — Years ended December 31, 2010, 2009 and 2008
F-4 Consolidated Statements of Stockholders’ Equity — Years ended December 31, 2010, 2009 and 2008
F-5 Consolidated Statements of Cash Flows — Years ended December 31, 2010, 2009 and 2008
F-6
F-7 F-1
Report of Independent Registered Public Accounting Firm The Board of Directors and Stockholders We have audited the accompanying consolidated balance sheets of Enzon Pharmaceuticals, Inc. and subsidiaries (the Company) as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the years in the three-year period ended December 31, 2010. These
consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test
basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion. In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Enzon Pharmaceuticals, Inc. and subsidiaries as of December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the years in the three-year period ended December
31, 2010, in conformity with U.S. generally accepted accounting principles. We
also have audited, in accordance with the standards of the Public Company
Accounting Oversight Board (United States), Enzon Pharmaceutical Inc.’s
internal control over financial reporting as of December 31, 2010, based
on criteria established in Internal Control — Integrated Framework issued
by the Committee of Sponsoring Organizations of the Treadway Commission (COSO),
and our report dated March 16, 2011 expressed an unqualified opinion on the
effectiveness of the Company’s internal control over financial reporting. /s/ KPMG LLP Short Hills, New Jersey F-2
Enzon Pharmaceuticals, Inc.:
March 16, 2011
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED BALANCE SHEETS
December 31,
December 31, ASSETS Current assets: Cash and cash equivalents
$
397,530
$
50,440 Short-term investments
31,170
53,670 Other current assets
5,916
6,928 Current assets of discontinued operations
—
34,174 Total current assets
434,616
145,212 Property and equipment, net
21,574
26,534 Marketable securities
31,394
95,636 Other assets
1,273
2,863 Noncurrent assets of discontinued operations
—
62,504 Total assets
$
488,857
$
332,749 LIABILITIES AND STOCKHOLDERS’ EQUITY Current liabilities: Accounts payable
$
4,192
$
1,390 Accrued expenses and other
14,195
10,338 Current liabilities of discontinued operations
—
13,269 Total current liabilities
18,387
24,997 Notes payable
134,499
250,050 Other liabilities
4,114
4,419 Total liabilities
157,000
279,466 Commitments and contingencies Stockholders’ equity: Preferred stock — $.01 par value, authorized 3,000,000 shares; no shares
—
— Common stock — $.01 par value, authorized 170,000,000 shares; issued and outstanding: 58,817,561 shares and 45,317,702 shares at December 31,
588
453 Additional paid-in capital
454,657
352,047 Accumulated other comprehensive income
914
2,328 Accumulated deficit
(124,302
)
(301,545
) Total stockholders’ equity
331,857
53,283 Total liabilities and stockholders’ equity
$
488,857
$
332,749 The accompanying notes are an integral part of these consolidated financial statements. F-3
(In thousands, except share and per-share amounts)
2010
2009
issued and outstanding at December 31, 2010 and 2009
2010 and 2009, respectively
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED STATEMENTS OF OPERATIONS
Year Ended December 31,
2010
2009
2008 Revenues: Royalties
$
44,940
$
51,408
$
56,969 Sale of in-process research and development
40,900
—
— Contract research and development
9,273
—
— Miscellaneous income
2,752
—
— Total revenues
97,865
51,408
56,969 Operating expenses: Research and development—pipeline
49,883
45,639
43,484 Research and development—specialty and contracted services
7,135
24,587
14,605 General and administrative
25,439
37,582
40,369 General and administrative—contracted services
1,957
—
— Restructuring charge
14,026
693
— Total operating expenses
98,440
108,501
98,458 Operating loss
(575
)
(57,093
)
(41,489
) Other income (expense): Investment income, net
3,465
4,312
6,612 Interest expense
(6,315
)
(11,514
)
(12,681
) Other-than-temporary investment impairment loss
(896
)
—
(645
) Other, net
1,184
5,008
1,891 Loss from continuing operations before income tax (benefit) provision
(3,137
)
(59,287
)
(46,312
) Income tax (benefit) provision
(337
)
(2,085
)
255 Loss from continuing operations
(2,800
)
(57,202
)
(46,567
) Income and gain from discontinued operations, net of income tax
180,043
57,885
43,852 Net income (loss)
$
177,243
$
683
$
(2,715
) Loss per common share — continuing operations
$
(0.05
)
$
(1.26
)
$
(1.05
) Earnings per common share — discontinued operations
$
3.08
$
1.28
$
0.99 Earnings (loss) per common share — net income (loss)
$
3.03
$
0.02
$
(0.06
) Weighted average shares — basic and diluted
58,466
45,186
44,398 The accompanying notes are an integral part of these consolidated financial statements. F-4
(In thousands, except per share amounts)
Basic and Diluted
Basic and Diluted
Basic and Diluted
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
Common Stock
Additional
Accumulated
Accumulated
Total
Number of
Par Value Balance, December 31, 2007
44,200
$
442
$
335,318
$
326
$
(299,513
)
$
36,573 Net loss
—
—
—
—
(2,715
)
(2,715
) Other comprehensive loss: Net unrealized loss on available-for-sale securities, net of tax
—
—
—
(1,723
)
—
(1,723
) Currency translation adjustment
—
—
—
(252
)
—
(252
) Total comprehensive loss
—
—
—
—
—
(4,690
) Exercise of stock options
40
—
284
—
—
284 Share-based compensation
663
7
8,321
—
—
8,328 Issuance of stock for employee stock purchase plan
129
1
1,165
—
—
1,166 Balance, December 31, 2008
45,032
$
450
$
345,088
$
(1,649
)
$
(302,228
)
$
41,661 Net income
—
—
—
—
683
683 Other comprehensive income: Net unrealized gain on available-for-sale securities, net of tax
—
—
—
3,247
—
3,247 Currency translation adjustment
—
—
—
730
—
730 Total comprehensive income
—
—
—
—
—
4,660 Exercise of stock options
9
—
56
—
—
56 Share-based compensation
357
4
8,122
—
—
8,126 Issuance of stock for employee stock purchase plan
113
1
794
—
—
795 Stock repurchase
(193
)
(2
)
(2,013
)
—
—
(2,015
) Balance, December 31, 2009
45,318
$
453
$
352,047
$
2,328
$
(301,545
)
$
53,283 Net income
—
—
—
—
177,243
177,243 Other comprehensive income: Net unrealized loss on available-for-sale securities, net of tax
—
—
—
(672
)
—
(672
) Currency translation adjustment
—
—
—
(742
)
—
(742
) Total comprehensive income
—
—
—
—
—
175,829 Conversion of notes payable
13,466
134
114,617
—
—
114,751 Exercise of stock options
4,147
41
31,710
—
—
31,751 Share-based compensation
376
4
3,900
—
—
3,904 Issuance of stock for employee stock purchase plan
52
1
508
—
—
509 Stock repurchase
(4,541
)
(45
)
(48,125
)
—
—
(48,170
) Balance, December 31, 2010
58,818
$
588
$
454,657
$
914
$
(124,302
)
$
331,857 The accompanying notes are an integral part of these consolidated financial statements. F-5
(In thousands)
Paid-in
Capital
Other
Comprehensive
Income (Loss)
Deficit
Shares
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES CONSOLIDATED STATEMENTS OF CASH FLOWS
Year Ended December 31,
2010
2009
2008 Cash flows from operating activities: Net income (loss)
$
177,243
$
683
$
(2,715
) Income and gain from discontinued operations
180,043
57,885
43,852 Loss from continuing operations
(2,800
)
(57,202
)
(46,567
) Adjustments to reconcile loss from continuing operations to net cash provided by (used in) operating activities: Depreciation
5,811
6,915
5,249 Write-down and sale of property and equipment
1,082
232
882 Amortization of debt securities premium/discount
2,590
(1,086
)
(2,297
) Write-off and amortization of debt issuance costs
576
1,364
1,345 (Gain) loss on sale of marketable securities
(589
)
11
253 Other-than-temporary investment impairment loss
896
—
645 Gain on redemption of notes payable
—
(4,848
)
(2,108
) Share-based compensation and employee stock purchase plan
6,869
7,861
7,421 Changes in operating assets and liabilities: Decrease (increase) in other current assets
644
(2,785
)
1,290 Increase (decrease) in accounts payable
2,801
(361
)
(2,860
) Increase (decrease) in accrued expenses and other
4,299
(1,709
)
261 Net cash provided by (used in) operating activities of continuing operations
22,179
(51,608
)
(36,486
) Net cash provided by operating activities of discontinued operations
436
66,605
67,468 Net cash provided by operating activities
22,615
14,997
30,982 Cash flows from investing activities: Proceeds from sale of business, net
262,581
—
— Purchase of property and equipment
(1,967
)
(1,987
)
(6,332
) Proceeds from sale of marketable securities
29,445
33,188
69,336 Purchase of marketable securities
(2,834
)
(109,791
)
(126,514
) Maturities of marketable securities
56,861
58,770
147,855 Net cash provided by (used in) investing activities of continuing operations
344,086
(19,820
)
84,345 Net cash used in investing activities of discontinued operations
(105
)
(6,327
)
(1,554
) Net cash provided by (used in) investing activities
343,981
(26,147
)
82,791 Cash flows from financing activities: Repurchase of common stock
(48,170
)
(2,015
)
— Redemption of notes payable
—
(15,602
)
(74,783
) Proceeds from issuance of common stock
32,260
852
1,450 Withholding taxes—share-based compensation
(3,443
)
(1,107
)
(475
) Redemption from employee stock purchase plan
(153
)
(249
)
(307
) Net cash used in financing activities
(19,506
)
(18,121
)
(74,115
) Net increase (decrease) in cash and cash equivalents
347,090
(29,271
)
39,658 Cash and cash equivalents at beginning of year
50,440
79,711
40,053 Cash and cash equivalents at end of year
$
397,530
$
50,440
$
79,711 The accompanying notes are an integral part of these consolidated financial statements. F-6
(In thousands)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES (1) Company Overview On January 29, 2010, Enzon Pharmaceuticals, Inc. and its subsidiaries (Enzon or the Company) consummated the sale of its specialty pharmaceutical business comprised principally of the Company’s products and contract manufacturing segments. These divested components are reflected in these consolidated financial statements as
discontinued operations and historical information related to the divested components has been reclassified accordingly. The Company also divested an in-process research and development asset of the specialty pharmaceutical business and reported the proceeds in revenue from continuing operations. Subsequent to the sale of the
specialty pharmaceutical business, the Company committed to performing certain research and development and general and administrative services to facilitate transition. See Note 22, Discontinued Operations, for more information regarding the sale. Following the sale of the specialty pharmaceutical business, Enzon is a biotechnology company dedicated to the research and development of innovative therapeutics for cancer patients with high unmet medical needs. Operations are funded in part by the receipt of royalty revenues from licensing arrangements with other companies
related to sales of products developed using the Company’s proprietary Customized PEGylation Linker Technology (Customized Linker TechnologyÒ) - primarily PEGINTRON marketed by Merck & Co., Inc. The Company operates in one business segment. The Company’s Principal Executive Officer (chief operating decision maker)
reviews the Company’s operating results on an aggregate basis and manages the Company’s operations as a single operating unit. The Company’s operations and assets reside almost exclusively in the U.S. The Company’s pipeline drug development programs utilize two platforms - Customized Linker Technology and third-generation messenger ribonucleic acid (mRNA)-targeting agents utilizing the Locked Nucleic Acid (LNA) technology. The Company currently has four compounds in clinical development; PEG-SN38 and the
Hypoxia-Inducible Factor-1a (HIF-1a), Survivin and Androgen Receptor (AR) antagonists. The Company’s continuing business is subject to significant risks and uncertainties including, but not limited to:
•
The risk that the Company will not achieve success in its research and development efforts, including clinical trials conducted by it or its collaborative partners. • The risk that the Company will experience operating losses for the next several years. • The risk that there will be a decline in sales of products sold by others from which the Company derives royalty revenues. • Decisions by regulatory authorities regarding whether and when to approve the Company’s regulatory applications. • The risk that the Company will fail to obtain adequate financing to meet its future capital and financing needs. • The risk that key personnel will leave the Company. (2) Summary of Significant Accounting Policies Principles of Consolidation The
consolidated financial statements include the accounts of the Company and
its wholly owned subsidiaries. All intercompany balances and transactions
have been eliminated in consolidation. Prior to the sale of the specialty
pharmaceutical business, assets and liabilities of the Company’s
Canadian subsidiary are translated into U.S. dollar equivalents at rates
in effect at the balance sheet date. Currency translation adjustments are
recorded in stockholders’ equity in accumulated other comprehensive
income (loss). Subsequent to the sale, the net assets (primarily cash) of
the subsidiary are converted into U.S. dollars at current rates with fluctuations
recognized in earnings. F-7
Notes to Consolidated Financial Statements
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES Use of Estimates The preparation of financial statements in conformity with accounting principles generally accepted in the U.S. requires management to make estimates and assumptions about future events. These estimates and the underlying assumptions affect the amounts of assets and liabilities reported and disclosures about contingent assets and
liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Such estimates include the valuation of investments, legal and contractual contingencies and assumptions used in the calculation of share-based compensation and income taxes. These estimates and assumptions
are based on management’s best estimates and judgment. Management evaluates its estimates and assumptions on an ongoing basis using historical experience, the current economic environment and other factors that management believes to be reasonable under the circumstances and makes appropriate adjustments when facts and
circumstances dictate. As future events and their effects cannot be determined with precision, actual results could differ significantly from these estimates. Changes in these estimates will be reflected in the financial statements in future periods. Financial Instruments and Fair Value The carrying values of cash, cash equivalents, other current assets, accounts payable and accrued expenses in the Company’s consolidated balance sheets approximated their fair values at December 31, 2010 and 2009 due to their short-term nature. Short-term investments and marketable securities are carried on the consolidated
balance sheets at fair value based on quoted market prices. All fair value measures are Level 1. Fair values and carrying amounts of the Company’s financial instruments are indicated below (in thousands):
Description
Fair Value
Carrying Short-term investments and marketable securities (Note 4)
$
62,564
$
62,564 4% Convertible Notes Payable (Note 6)
$
182,400
$
134,499 Cash Equivalents The Company considers all highly liquid debt instruments with remaining maturities at the date acquired not exceeding three months to be cash equivalents. Cash equivalents consist primarily of money market funds. As of December 31, 2010 and 2009, the Company held $386.2 million and $33.8 million of cash equivalents,
respectively. Short-term Investments and Marketable Securities The Company classifies its investments in debt and equity securities as either short-term or long-term based upon their stated maturities and the Company’s intent and ability to hold them. Investments with stated maturities of one year or less are classified as current assets. Investments in debt securities with stated maturities greater
than one year are classified as noncurrent assets when the Company has the intent and ability to hold such securities for at least one year. Investments in debt securities are classified as available-for-sale. Unrealized gains and losses (which are deemed to be temporary), net of related tax effect when appropriate, are included in the
determination of other comprehensive income (loss) and reported in stockholders’ equity. The cost of the debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. The amortization and accretion, along with realized gains and losses, are included in investment income, net. The cost of securities is
based on the specific identification method. Notes Payable The carrying value of the Company’s 4% convertible senior unsecured notes outstanding at December 31, 2010 and 2009 was $134.5 million and $250.0 million, respectively, and the fair value of these notes was F-8
Notes to Consolidated Financial Statements — (Continued)
Amount
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES $182.4 million and $293.8 million at December 31, 2010 and 2009, respectively. Fair value of the Company’s notes payable is based on quoted market prices. Property and Equipment Property and equipment are stated at cost. Depreciation of fixed assets is provided by the straight-line method over the estimated useful lives of the assets. When assets are retired or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts, and any resulting gain or loss is recognized in
operations for the period. Amortization of leasehold improvements is calculated using the straight-line method over the remaining term of the lease or the life of the asset, whichever is shorter. The cost of repairs and maintenance is charged to operations as incurred; significant improvements are capitalized. Deferred Debt Issuance Costs Costs incurred in issuing the Company’s notes payable have been recorded as deferred debt issuance costs and are included within the balances of other assets and other current assets in the accompanying consolidated balance sheets. Such amounts are being amortized using the straight-line method, which approximates the effective
interest method, over the terms of the related financing. The amortization of deferred debt issuance costs is included in interest expense in the accompanying consolidated statements of operations. At the time of repurchase or other extinguishment of notes, a pro rata amount of deferred debt issuance costs is written off to interest expense.
Upon conversion of notes, a pro rata amount of deferred issuance costs is written off against additional paid-in capital. Revenue Recognition Royalty revenue from the Company’s agreements with third parties is recognized when the Company can reasonably determine the amounts earned. In most cases, this will be upon notification from the third-party licensee, which is typically during the quarter following the quarter in which the sales occurred. Royalties earned
pursuant to the Company’s sale of its specialty pharmaceutical business are recognized when the Company can reasonably determine the amounts earned which is generally upon notification from the purchaser of the specialty pharmaceutical business. The Company does not participate in the selling or marketing of products for which it
receives royalties. No provision for uncollectible accounts is established upon recognition of revenues. Contingent payments due under the asset purchase agreement for the sale of the specialty pharmaceutical business are recognized as income when the milestone has been achieved and collection is assured. Such payments are non-refundable and no further effort is required on the part of the Company or the other party to complete
the earning process. The Company does not routinely participate in research and licensing arrangements that have multiple deliverables. The sale of the specialty pharmaceutical business, however, did involve the application of the guidance regarding multiple deliverables in separating the revenues associated with the sale of in-process research and
development from the other elements of the transaction, principally the assets sold as part of discontinued operations and the continuing involvement of the Company in contract research activities. The Company determined that the in-process research and development had value to the buyer of the specialty pharmaceutical business on a
stand-alone basis and that there was objective and reliable evidence available to support the allocation of the total purchase price to the respective units of accounting. (See Note 22 - Discontinued Operations). Research and Development Expenses All research and development costs are expensed as incurred. These include the following types of costs incurred in performing research and development activities: clinical trials, clinical manufacturing costs, contract services, salaries, share-based compensation and benefits and administrative support costs. Non-refundable advance
payments to acquire goods or pay for services that will be consumed or performed in future periods are F-9
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES capitalized and amortized over the period of expected benefit. Costs to acquire in-process research and development projects and technologies that have no alternative future use at the date of acquisition are expensed as incurred. Substantial portions of the Company’s preclinical and clinical trial work are performed by third-party contract research organizations (CROs) and other vendors. The Company accrues expenses for costs for work performed by CROs based upon the estimated amount of the total effort completed on each study or project using factors
such as the number of patients enrolled, the number of active clinical sites and the duration for which the patients will be enrolled in the study. Similar approaches are taken in estimating the percentage of completion in relation to contracts with contract manufacturing organizations. The Company bases the estimates on the information
available at the time. Income Taxes Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit
carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be realized. The effect of a change in tax rates or laws on deferred tax assets and liabilities is recognized in operations in the period that includes the
enactment date of the rate change. A valuation allowance is established to reduce the deferred tax assets to the amounts that are more likely than not to be realized from operations. Tax benefits of uncertain tax positions are recognized only if it is more likely than not that the Company will be able to sustain a position taken on an income tax return. The Company has no liability for uncertain positions. Interest and penalties, if any, related to unrecognized tax benefits, would be recognized as income tax
expense. Concentrations of Risk The Company’s holdings of financial instruments are comprised principally of money market funds and debt securities. The Company does not invest in portfolio equity securities or commodities or use financial derivatives for trading purposes. The Company seeks reasonable assuredness of the safety of principal and market liquidity
by investing in rated securities while at the same time seeking to achieve a reasonable rate of return. The Company’s market risk exposure consists principally of exposure to changes in interest rates. The Company’s holdings of debt securities also are exposed to the risks of changes in the credit quality of issuers. The Company typically
invests the majority of its investments in the shorter-end of the maturity spectrum, and at December 31, 2010 the majority of its holdings were in instruments maturing in two years or less, or having a market that enables flexibility in terms of timing of disposal. Cash equivalents are primarily held in a number of triple-A rated institutional
money market funds as well as several corporate and U.S. government-sponsored entities’ debt securities. Share-Based Compensation Plans The Company recognizes the cost of all share-based payment transactions at fair value. Compensation cost, measured by the fair value of the equity instruments issued, adjusted for estimated forfeitures, is recognized in the financial statements as the respective awards are earned. The impact that share-based payment awards will have on the Company’s results of operations is a function of the number of shares awarded, the trading price of our stock at date of grant or modification and vesting, including the likelihood of achieving performance goals. Furthermore, the application of the Black-Scholes valuation
model employs weighted average assumptions for expected volatility of the Company’s stock, expected term until exercise of the options, the risk free interest rate, and dividends, if any to determine fair value. Expected volatility is based on historical volatility of the Company’s common stock; the expected term until exercise represents
the weighted average period of time that options granted are expected to be F-10
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES outstanding giving consideration to vesting schedules and the Company’s historical exercise patterns; and the risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant for periods corresponding with the expected life of the option. Cash Flow Information Cash payments for interest on the Company’s 4% notes were approximately $5.4 million, $10.2 million and $13.0 million for the years ended December 31, 2010, 2009 and 2008, respectively. There were $0.1 million, $0.2 million and $2.5 million of income tax payments made for the years ended December 31, 2010, 2009 and
2008, respectively. During the year ended December 31, 2010, the Company had a noncash conversion of $115.6 million principal amount of the 4% notes into approximately 13.5 million shares of its common stock. Reclassifications and Error Corrections Certain amounts previously reported have been reclassified to conform to the year ended December 31, 2010 presentation. Prior to the second quarter of 2010, cash payments for withholding taxes on the exercise of share-based awards were netted against share-based compensation expense within cash provided by operating activities
in the Company’s statements of cash flows and reflected as cash outflows from operating activities. The proper classification of these amounts is in cash flows from financing activities, which is where they are reported in these financial statements with all prior periods having been corrected. The amounts of the corrections of prior years
cash flow statements are not material: 2009: $1.1 million and 2008: $0.5 million. (3) Recent Accounting Pronouncements Milestone Method of Revenue Recognition—Pursuant to a final consensus of the Emerging Issues Task Force of the FASB ratified on March 31, 2010, guidance is provided for determining when milestone payments received in conjunction with the performance of research and development efforts may be recognized. The Company
has evaluated the new guidance which is to be implemented prospectively beginning in 2011. Contingent consideration will be recognized when the underlying milestone has been achieved, provided certain criteria are met. This is consistent with the Company’s existing practices and, accordingly, adoption of the new guidance is not
expected to have any effect on the Company’s financial statements. (4) Investments and Marketable Securities The amortized cost, gross unrealized holding gains and losses, and fair value for available-for-sale securities by major security type at December 31, 2010 were as follows (in thousands):
Amortized
Gross
Gross
Fair Corporate debt
$
52,079
$
738
$
—
$
52,817 U.S. government-sponsored entities debt
1,000
4
—
1,004 Non-U.S. government debt
5,553
86
—
5,639 Other
3,019
111
(26
)
3,104
$
61,651
$
939
$
(26
)
$
62,564
*
Included in short-term investments $31,170 and marketable securities $31,394 at December 31, 2010.
F-11
Notes to Consolidated Financial Statements — (Continued)
Cost
Unrealized
Holding Gains
Unrealized
Holding Losses
Value*
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES The amortized cost, gross unrealized holding gains and losses, and fair value for available-for-sale securities by major security type at December 31, 2009 were as follows (in thousands):
Amortized
Gross
Gross
Fair Corporate debt
$
114,118
$
1,362
$
(17
)
$
115,463 U.S. government-sponsored entities debt
5,713
73
—
5,786 Non-U.S. government debt
23,298
12
(94
)
23,216 Auction rate securities
877
—
(558
)
319 Other
3,714
810
(2
)
4,522
$
147,720
$
2,257
$
(671
)
$
149,306
*
Included in short-term investments $53,670 and marketable securities $95,636 at December 31, 2009.
Other securities include investments of participants in the Company’s Executive Deferred Compensation Plan (predominantly mutual fund shares) totaling $3.1 million fair value as of December 31, 2010 and $3.8 million fair value as of December 31, 2009. There is a non-current liability that offsets the aggregate deferred
compensation plan assets. In addition, other securities included approximately $0.7 million fair value of corporate equity securities as of December 31, 2009. These equity holdings were sold during 2010 realizing a gain of $0.7 million recognized in other income in the statement of operations. Fair value is determined from readily available quoted prices in active markets (Level 1, the preferred approach pursuant to applicable accounting guidance). As of December 31, 2010, the Company’s investments and marketable securities are all valued based on Level 1 inputs. Due to instability in the financial markets, failed
auctions for a certain auction rate security had occurred and, as a result, the Company employed alternative measures of fair value prior to the third quarter of 2010 which the Company deemed to be Level 2. The model used to value the auction rate security considered listed quotes of bonds with comparable maturities, the underlying
collateral of the securities and the issuer’s credit worthiness. During the third quarter of 2010, the Company wrote off the remaining carrying amount of the auction rate security as an other-than-temporary impairment loss—see below. Maturities of marketable securities, excluding $3.1 million (at fair value) of other investments at December 31, 2010 were as follows (in thousands):
Maturing During the
Amortized
Fair 2011
$
30,931
$
31,170 2012
27,701
28,290
$
58,632
$
59,460 During the year ended December 31, 2010 the Company realized gains from the sale of investments of $0.6 million. For the year 2009, there was an immaterial loss realized and for the year ended December 31, 2008, a loss of $0.3 million was realized. As of December 31, 2010, only assets of the Company’s Executive Deferred Compensation Plan have unrealized holding losses. None of the underlying investments has been in a continuous loss position longer than twelve months. The Company maintains a liability for the fair value of the deferred compensation investments and
any realized losses related to these investment holdings are borne by the plan participants. Impairment assessments are made at the individual security level each reporting period. When the fair value of an investment is less than its cost at the balance sheet date, a determination is made as to whether the F-12
Notes to Consolidated Financial Statements — (Continued)
Cost
Unrealized
Holding Gains
Unrealized
Holding Losses
Value*
Year Ending December 31,
Cost
Value
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES impairment is other than temporary and, if it is other than temporary, an impairment loss is recognized in earnings equal to the difference between the investment’s cost and fair value at such date. During the quarter ended September 30, 2010, the Company concluded that the cash flows expected to be collected from its holding of an auction rate security were severely compromised and that an other-than-temporary impairment had occurred. The underlying collateral, the preferred stock of the issuer, lost substantial value when
the insurance commissioner of the issuer’s state of residence effectively seized the issuer’s assets and, in early October 2010, filed a plan of rehabilitation whereby policyholders’ interests would be protected to the extent the issuer’s assets would allow. The Company believes that the assets of the issuer will not be sufficient to cover
policyholder claims and that equity-holders’ interests are impaired. Further supporting the decision to record the impairment loss, during the fourth quarter of 2010, the parent holding company of the issuer defaulted on a significant interest payment and was subsequently unable to restructure the related debt or raise additional capital. The
parent holding company filed for relief under Chapter 11 of the U.S. Bankruptcy Code in the fourth quarter of 2010. The auction rate security had an original cost basis of $1.5 million. An estimated credit loss of $0.6 million was recorded in earnings in 2008 based upon an estimate of the present value of expected cash flows from this investment leaving an amortized cost basis of approximately $0.9 million. The Company does not intend to
dispose of this security nor is it more likely than not that the Company will be required to do so. However, based on the recent events outlined above, the Company no longer expects that it will recover any of its cost basis in the security. Accordingly, the full amount of the carrying value remaining of $0.9 million was written off against
earnings as an other-than-temporary investment impairment loss during the third quarter of 2010. The Company will continue to monitor this instrument and the expected cash flows to be derived from it. Any subsequent unrealized recovery in fair value will be reported in accumulated other comprehensive income until the investment is
sold or otherwise disposed of. (5) Property and Equipment Property and equipment consist of the following (in thousands):
December 31,
December 31,
Estimated Leasehold improvements
$
27,034
$
26,701
2-14 years
* Equipment
30,002
31,828
2-6 years Furniture and fixtures and other
2,824
3,719
6 years
59,860
62,248 Less: Accumulated depreciation
38,286
35,714
$
21,574
$
26,534
*
Shorter of the lease term or lives indicated
Depreciation charged to operations relating to property and equipment totaled $5.8 million, $6.9 million and $5.2 million for the years ended December 31, 2010, 2009 and 2008, respectively. (6) Notes Payable The 4% notes mature on June 1, 2013 unless earlier redeemed, repurchased or converted. The 4% notes are senior unsecured obligations and rank equal to all future senior unsecured debt of the Company. The 4% notes are convertible at the option of the holders into the Company’s common stock at an initial conversion price of
$9.55 per share (104.712 shares per $1,000 principal amount). If the closing price of the Company’s common stock for at least 20 trading days in the 30-consecutive-trading-day period ending on the date one day F-13
Notes to Consolidated Financial Statements — (Continued)
2010
2009
Useful Lives
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES prior to the date of a notice of redemption is greater than 140 percent of the applicable conversion price on the date of such notice, the Company, at its option, may redeem the 4% notes in whole or in part, at a redemption price in cash equal to 100 percent of the principal amount of the 4% notes to be redeemed, plus accrued and unpaid
interest, if any, to the redemption date. Upon occurrence of a fundamental change, as defined in the indenture governing the 4% notes, holders of the notes may require the Company to redeem the notes at a price equal to 100 percent of the principal amount plus accrued and unpaid interest or, in certain cases, to convert the notes at an increased conversion rate based on
the price paid per share of the Company’s common stock in the five-trading-day period prior to the transaction constituting the fundamental change. The January 29, 2010 sale of the Company’s specialty pharmaceutical business constituted a fundamental change and triggered a requirement that the Company offer to purchase all of its 4%
notes at face value. Such an offer was made on February 5, 2010. The offer expired on March 5, 2010 with no notes having been tendered. The fundamental change also triggered a change in the conversion rate from 104.712 shares per $1,000 principal amount of notes to 116.535 shares per $1,000 principal amount during the period
January 29, 2010 to March 4, 2010. During this period, notes totaling $115.6 million principal amount were converted into approximately 13.5 million shares of common stock of the Company, reducing the outstanding principal balance of the notes outstanding to $134.5 million. Subsequent to March 4, 2010, the date the enhanced
conversion period ended, the original conversion rate of 104.712 shares per $1,000 principal amount of notes is again in effect. During the first quarter of 2009, the Company repurchased $20.5 million principal amount of its 4% notes at a discount to par resulting in a gain of approximately $4.5 million net of the write-off of $0.3 million of debt issuance costs. Of the total of $20.5 million repurchased during the first quarter of 2009, $2.95 million was the
result of a December 2008 tender offer to purchase a portion of the notes. Interest on the 4% notes is payable on June 1 and December 1 of each year. Accrued interest on the 4% notes amounted to $0.4 million and $0.8 million as of December 31, 2010 and 2009, respectively. As of December 31, 2010, the balance of unamortized deferred debt issuance costs is approximately $1.3 million. (7) Accrued Expenses and Other Accrued expenses and other consists of the following as of December 31, 2010 and 2009 (in thousands):
December 31,
December 31, Compensation
$
5,725
$
5,413 Severance benefits
3,623
441 Professional and consulting fees
667
801 Insurance and taxes
386
455 Interest
448
833 Other
3,346
2,395
$
14,195
$
10,338 (8) Stockholders’ Equity Preferred Stock The Company has authorized 3,000,000 shares of preferred stock in one or more series of which 600,000 are designated as Series B in connection with the Rights Plan. F-14
Notes to Consolidated Financial Statements — (Continued)
2010
2009
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES Common Stock As of December 31, 2010, the Company has reserved shares of its common stock for the purposes detailed below (in thousands): Non-Qualified and Incentive Stock Plans
5,769 Shares issuable upon conversion of 4% Notes due 2013
14,084 Employee Stock Purchase Plan
641
20,494 Share Repurchase Programs On December 3, 2009, the Company announced a share repurchase program, under which the Company authorized the purchase up to $50.0 million of the Company’s outstanding common shares. As of December 31, 2010, the Company had completed the entire $50.0 million repurchase retiring approximately 4.7 million shares in
total at an average cost of $10.63 per share. Of the total $50.0 million repurchased, approximately $48.0 million occurred during 2010, representing approximately 4.5 million shares at an average cost of $10.65 per share. Amounts expended include transaction costs. On December 21, 2010, the Company announced a share repurchase program, under which the Company may purchase up to $200.0 million of the Company’s outstanding common shares. Through December 31, 2010, the Company paid approximately $0.4 million to repurchase and retire 30,000 shares at an average cost of $12.45
per share. Through February 28, 2011, approximately 1.5 million additional shares have been purchased at a cost of approximately $17.3 million. The plan continues in effect. Rights Plan Holders of the Company’s common stock own one preferred stock purchase right for each share of common stock owned by such holder. These rights currently entitle holders of our common stock to purchase one one-thousandth of a share of our Series B preferred stock for $190.00, except, in certain circumstances described below,
holders may receive common stock. However, the rights are not immediately exercisable and will become exercisable only upon the occurrence of certain events. If a person or group acquires, or announces a tender or exchange offer that would result in the acquisition of 15 percent or more of our common stock while the stockholder
rights plan remains in place, then, unless (1) the rights are redeemed by us for $0.01 per right or (2) the board of directors determines that a tender or exchange offer for all of our outstanding common stock is in the best interest of the Company and the stockholders, the rights will become exercisable by all rights holders, except the
acquiring person or group, for (i) shares of our common stock or (ii) in certain circumstances, shares of the third-party acquirer, each having a value of twice the right’s then-current exercise price. Pursuant to an amendment to the rights plan dated July 23, 2009, stockholders may beneficially own less than 19 percent of the outstanding
shares of common stock of the Company without becoming an acquiring person and thereby triggering the rights under the plan. Prior to the amendment, stockholders who reported beneficial ownership of the common stock of the Company on Schedule 13G under the Securities and Exchange Act of 1934, as amended, could beneficially
own less than 20 percent of the outstanding shares of common stock of the Company without becoming an acquiring person, and all other stockholders could beneficially own less than 15 percent of the outstanding shares of common stock of the Company without becoming an acquiring person. The rights expire on May 16, 2012. (9) Sale of In-Process Research and Development When the Company sold its specialty pharmaceutical business, it retained its research and development organization. Prior to the sale, the Company’s research and development function was engaged in, among other things, studies oriented towards the next-generation formulations of Oncaspar and Adagen, two products that were
among those sold as part of the specialty pharmaceutical business. The in-process research and F-15
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES development related to those two products was sold to the purchaser of the specialty pharmaceutical business. The $40.9 million selling price was management’s best estimate of its standalone fair value based on the stage of development and future milestone payment consideration. All necessary technology and know-how was transferred
to the purchaser at the time of the sale and the purchaser could resell the in-process research and development asset. The activities necessary to complete the work on the Oncaspar and Adagen next-generation formulations could be performed by the purchaser or others. No portion of the selling price was attributed to the transition
services agreement referred to below in Note 22, Discontinued Operations as that agreement represents an arm’s-length market rate of return for the services being provided and those services are completely separate from the in-process research and development. (10) Contract Research and Development Revenue and Miscellaneous Income Contract research and development is specific to the transition services agreement the Company entered into with the purchaser of the specialty pharmaceutical business. The transition services agreement was initiated in January 2010 at the time of the sale. It provides for a reimbursement for services provided by the Company plus a
mark-up and totaled $9.3 million in 2010. These services could continue for up to three years after the sale, but are expected to diminish significantly in 2011. Miscellaneous income includes income received pursuant to the transition services agreement related to general and administrative support to the purchaser of the specialty pharmaceutical business ($2.4 million) and sublease revenues received by the Company from tenants under terms of sublease agreements ($0.3 million). These
transitional services are expected to be minimal in 2011 as the term of the agreement for other than research and development support activities was one year. Sublease revenues relate primarily to the Company’s leased facility in South Plainfield, New Jersey which commenced in 2009 and run through October 2012. Sublease income
related to excess leased office space in Bridgewater, New Jersey will commence in 2011 and will continue through January 2013. Additional tenants are being sought for excess space at the Bridgewater facility that the Company will vacate in the first quarter of 2011. (11) Comprehensive Income Comprehensive income consists primarily of net income (loss) and net unrealized gain (loss) on available-for-sale securities and is presented in the consolidated statements of stockholders’ equity (deficit). The following table reconciles net (loss) income to comprehensive (loss) income (in thousands):
Year Ended December 31,
2010
2009
2008 Net income (loss)
$
177,243
$
683
$
(2,715
) Other comprehensive income (loss): Unrealized (loss) gain on securities that arose during the year*
(979
)
3,236
(2,621
) Currency translation adjustment*
(742
)
730
(252
) Reclassification adjustments*: Impairment loss included in net loss
896
—
645 (Gain) loss on sale of securities
(589
)
11
253 Total other comprehensive income (loss)
(1,414
)
3,977
(1,975
) Total comprehensive income (loss)
$
175,829
$
4,660
$
(4,690
)
*
Information has not been tax-effected due to the establishment of a full allowance against any related net deferred tax asset.
F-16
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES (12) Earnings Per Common Share Basic earnings per share is computed by dividing the measure of income or loss by the weighted average number of shares of common stock outstanding during the period. Restricted stock awards and restricted stock units (collectively, nonvested shares) are not considered to be outstanding shares until the service or performance
vesting period has been completed. The diluted earnings (loss) per share calculation would normally involve adjusting both the denominator and numerator as described here if the effect is dilutive. The denominator would include both the weighted average number of shares of common stock outstanding and common stock equivalents. Dilutive common stock
equivalents potentially include stock options and nonvested shares using the treasury stock method, shares issuable under the employee stock purchase plan (ESPP) and the number of shares issuable upon conversion of the Company’s 4% convertible senior notes payable. In the case of notes payable, the diluted earnings per share
calculation would be further affected by an add-back of interest to the numerator under the assumption that the interest would not have been incurred if the notes were converted into common stock. In a period in which a loss from continuing operations is reported, all computations of diluted per-share amounts for that period must be made exclusive of potential dilutive shares and the add-back of interest. Accordingly, for each of the three years ended December 31, 2010, 2009 and 2008, diluted earnings per share for
discontinued operations and net income are the same as the corresponding basic earnings per share. The following table illustrates the computation of basic and diluted (loss) earnings per share for the years ended December 31, 2010, 2009 and 2008 (in thousands):
Year Ended December 31,
2010
2009
2008 Earnings Per Common Share—Basic and Diluted: Loss from continuing operations
$
(2,800
)
$
(57,202
)
$
(46,567
) Income and gain from discontinued operations, net
$
180,043
$
57,885
$
43,852 Net income (loss)
$
177,243
$
683
$
(2,715
) Weighted average common shares outstanding
58,466
45,186
44,398 Basic and diluted (loss) earnings per share: Continuing operations
$
(0.05
)
$
(1.26
)
$
(1.05
) Discontinued operations
$
3.08
$
1.28
$
0.99 Net income (loss)
$
3.03
$
0.02
$
(0.06
) For the years ended December 31, 2010, 2009 and 2008, the Company had potentially dilutive common stock equivalents excluded from the computation of diluted earnings per share, amounting to 19.4 million, 36.1 million and 38.8 million, shares, respectively. F-17
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES (13) Restructuring The Company incurred the following charges in connection with its restructuring programs during the years ended December 31, 2010 and 2009 (there were no restructuring charges related to continuing operations in 2008). Amounts are in thousands.
Year Ended December 31,
2010
2009 Employee separation benefits First-quarter 2010 program
$
9,736
$
— Fourth-quarter 2010 program
2,974
— 2009 program
—
693
12,710
693 Other restructuring costs
1,316
— Total restructuring charges
$
14,026
$
693 Employee separation benefits: There were two restructurings initiated during 2010, both of which reflected the transition of the Company from a fully integrated biopharmaceutical company with research, manufacturing and marketing operations to a biotechnology company focused primarily on research and development. During the first quarter of 2010, the Company’s workforce reduction involved 64 employees resulting in an expense of $6.1 million for separation benefits for the affected employees. These actions related primarily to the sale of the specialty pharmaceutical business including several employees who were previously engaged in
activities related to the divested business but who did not transfer to the employment of the purchaser. These employees were provided with separation benefits after certain transition periods during which they assisted with an orderly transfer of activities and information to the purchaser. In addition, the Company reassessed its staffing
requirements subsequent to the sale of the specialty pharmaceutical business in light of the lessened demands on many of its general and administrative functions. The majority of the payments related to the first-quarter 2010 restructuring were made during the remainder of 2010 (see below). Also, effective February 22, 2010, the
Company’s then President and Chief Executive Officer, resigned from the Company. For the quarter ended March 31, 2010, the Company expensed $3.8 million for severance payments and benefits that were payable per the terms of the individual’s employment agreement. This amount was reduced during the quarter ended June 30,
2010 by approximately $0.2 million once the termination agreement was executed. Payments due pursuant to the termination agreement were made during the third quarter of 2010. The fourth-quarter 2010 restructuring program was part of the Company’s continued efforts to streamline corporate administrative operations and affected approximately 33 positions. Affected employees were notified in December 2010 and the majority of the terminations will occur during the first quarter of 2011. Separation
payments will be made for up to a year following the respective separations. The amount recognized as expense in the fourth quarter of 2010 represents the estimated full cost of this restructuring effort. The portion of the expense to be paid out in the next twelve months ($3.6 million) is reported as a current liability in accrued expenses
as of December 31, 2010. The remainder ($0.3 million) is reported as an other liability. In the first quarter of 2009, the Company implemented a restructuring plan involving a reduction in workforce in general and administrative and research and development areas. Costs of severance and related benefits for employees affected by the 2009 workforce reduction amounted to $0.7 million during the first quarter of 2009.
The amounts accrued in the first quarter of 2009 were fully paid out by the end of October 2009. F-18
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES The following table reflects the activity in the two 2010 accruals for separation payments and the resulting liabilities as of December 31, 2010 (amounts in thousands):
Employee Separation Programs
Fourth Quarter
First Quarter
Total Balance December 31, 2009
$
—
$
—
$
— 2010 restructuring accruals
2,974
9,889
12,863 2010 payments
—
(8,837
)
(8,837
) 2010 adjustments
—
(153
)
(153
) Balance December 31, 2010
$
2,974
$
899
$
3,873 Other restructuring costs: During the second quarter of 2010, the Company wrote off certain leasehold improvements and furnishings located at its corporate headquarters in Bridgewater, New Jersey that were determined to be excess and without future value as a result of the termination and relocation of several employees. The noncash charge related to this
write off was approximately $0.9 million. During the third quarter of 2010, the Company entered into a sublease for a portion of its excess corporate facilities. These facilities became unused as a result of the reductions in workforce stemming from earlier restructuring efforts related to the sale of the specialty pharmaceutical business.
The $0.4 million charge represents the excess of the Company’s contractual lease commitment over the amount of cash to be received from the subtenant over the life of the sublease arrangement. By the end of the first quarter of 2011, the Company expects to have completed the relocation of its corporate offices to Piscataway, New
Jersey. The vacating of the excess office space in Bridgewater will likely trigger a further restructuring charge in 2011 for the excess of committed lease costs over anticipated sublease income. (14) Stock Options Through the Compensation Committee of the Board of Directors, the Company administers the 2001 Incentive Stock Plan, as amended, which provides incentive and non-qualified stock option benefits for employees, officers, directors and independent contractors providing services to Enzon and its subsidiaries. The 2001 Incentive
Stock Plan was adopted by the Board of Directors in October 2001 and approved by the stockholders in December 2001. Options granted to employees generally vest over four years from date of grant and options granted to directors vest after one year. The exercise price of the options granted must be at least 100 percent of the fair value
of the Company’s common stock at the time the options are granted. Options may be exercised for a period of up to ten years from the grant date. As of December 31, 2010, 5.8 million shares of common stock were reserved for issuance pursuant to granted options and awards under the plan. Approximately 1.1 million shares remain
available for grant. Option grants remain outstanding from previous awards under an earlier 1987 Non-Qualified Stock Option Plan; however, there will be no further grants made pursuant to that plan. Under the 2007 Outside Director Compensation Plan, each non-employee director receives options to purchase shares of common stock annually on the first trading day of the calendar year. Using the Black-Scholes option pricing model, each eligible participant may purchase that number of shares that aggregates $75,000 in value.
These grants are made under the 2001 Incentive Stock Plan. The exercise price of the annual grant is equal to the closing price of the common stock on the date of grant; it vests in one tranche on the first anniversary date; and expires on the tenth anniversary date of the grant. In addition, upon election of a new non-employee director to
the Board, such newly elected director receives a grant of options to purchase shares of common stock with a Black-Scholes value of $75,000 (the exercise price being equal to the closing price of the common stock on the date of grant). These options vest in three equal tranches on each of the first three anniversaries of the date of grant,
if the recipient director remains on the Board on each such date. Furthermore, for the Chairperson of the Board, if not an employee of the Company, the number of options granted annually and upon election is twice the number mentioned above. F-19
Notes to Consolidated Financial Statements — (Continued)
2010
2010
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES The following is a summary of the activity in the Company’s outstanding Stock Option Plans which include the 2001 Incentive Stock Plan and the 1987 Non-Qualified Stock Option Plan (options in thousands):
Options
Weighted
Weighted
Aggregate Outstanding at January 1, 2010
8,369
$
10.99 Granted at exercise prices which equaled the fair value on the date of grant
153
$
10.68 Exercised
(4,147
)
$
7.66 Forfeited
(92
)
$
8.98 Expired
(290
)
$
28.67 Outstanding at December 31, 2010
3,993
$
13.21
4.27
$
5,304 Vested and expected to vest at December 31, 2010
3,908
$
13.30
4.21
$
5,148 Exercisable at December 31, 2010
3,648
$
13.58
3.96
$
4,807 As of December 31, 2010, there was $0.3 million of total unrecognized compensation cost related to unvested options that the Company expects to recognize over a weighted-average period of 8 months. The board of directors of the Company elected to accelerate the vesting of certain stock options granted under the Company’s
2001 Incentive Stock Plan as of the consummation of the sale of the specialty pharmaceutical business in January 2010. This acceleration affected outstanding options held by employees at the vice president level and below and resulted in an additional expense of $0.2 million in the first quarter of 2010 and of $0.1 million in 2009. The
charges primarily represented an acceleration of expense recognition pursuant to the original award and, to a lesser extent, an adjustment, in certain cases, to recognize the modification of the award in contemplation of the sale. The weighted-average grant-date fair value of options granted during the years ended December 31, 2010, 2009 and 2008 was $4.42, $2.45 and $3.44, respectively. The total intrinsic value of options exercised during the years ended December 31, 2010, 2009 and 2008 was $11.8 million, $26 thousand and $83 thousand, respectively.
During the year ended December 31, 2010, the grant-date fair value of options that vested was $3.9 million. In the years ended December 31, 2010, 2009 and 2008, the Company recorded share-based compensation of $2.2 million, $3.2 million and $3.3 million, respectively, related to stock options. The Company did not realize a net tax benefit related to share-based compensation expense. The Company’s policy is to use newly issued
shares to satisfy the exercise of stock options. The breakdown of share-based compensation expense by major line caption in the statements of operations is shown below (amounts are in thousands):
Year Ended December 31,
2010
2009
2008 Research and development
$
377
$
804
$
885 General and administrative
1,787
2,394
2,457
$
2,164
$
3,198
$
3,342 Cash received from share option exercise for the years ended December 31, 2010, 2009 and 2008, was $31.8 million, $0.1 million and $0.3 million, respectively. F-20
Notes to Consolidated Financial Statements — (Continued)
Average
Exercise
Price Per
Option
Average
Remaining
Contractual
Term (years)
Intrinsic
Value
($000)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES The weighted average assumptions used in the Black-Scholes option-pricing model for expected volatility, expected term until exercise and risk-free interest rate are shown in the table below. Expected volatility is based on historical volatility of the Company’s common stock. The expected term of options is estimated based on the
Company’s historical exercise pattern. The risk-free interest rate is based on U.S. Treasury yields for securities in effect at the time of grant with terms approximating the expected term until exercise of the option. No dividend payments were factored into the valuations. Forfeiture rates, used for determining the amount of compensation
cost to be recognized over the service period, are estimated based on stratified historical data.
Year Ended
Year Ended
Year Ended Expected volatility
42
%
41
%
34
% Expected term (in years)
5.4
5.4
5.4 Risk-free interest rate
2.6
%
1.7
%
3.5
% (15) Restricted Stock and Restricted Stock Units (Nonvested Shares) The 2001 Incentive Stock Plan provides for the issuance of restricted stock and restricted stock units (collectively, nonvested shares) to employees, officers and directors. These awards effectively are the issuance by the Company to the recipient of shares of the Company’s common stock at either the date of the grant, in the case of a
restricted stock award, or upon vesting, in the case of a restricted stock unit. The recipient pays no cash to receive the shares other than the $0.01 par value in some cases. These awards have vesting periods of three to five years when based on service. Certain awards have performance goals with vesting periods that could be shorter than
three years. All nonvested shares are valued at fair value. The market price of the Company’s stock at grant date is factored by an expected vesting period forfeiture rate based on stratified historical data related to the assumed vesting period. This amount is then amortized over the vesting period on a straight-line basis for those awards that vest
based solely on service. One half of the 2010 grant of restricted stock units vests over three years based on service. The other half is broken down into equal thirds with each third having a specific performance goal associated with it. Vesting of each third is a function of the Company’s estimate of probability of the goal being achieved.
During the fourth quarter of 2010, two of these goals were deemed to be probable of being achieved - one in February 2011 and one in June 2011. Expense is being recognized for these portions of the awards over the indicated performance periods. The remaining third is being recognized over three years unless or until the related
performance goal is determined to be probable of attainment. Pursuant to the 2007 Outside Director Compensation Plan, each non-employee director receives a grant of restricted stock units for shares of common stock with a value of $75,000 annually on the first trading day after June 30. This grant is made under the 2001 Incentive Stock Plan. The number of shares covered by the annual
grant is equal to $75,000 divided by the closing price of the common stock on the date of grant; it vests in three equal tranches on each of the first three anniversaries of the date of the grant if the recipient director remains on the Board on each such date. In addition, upon election of a new non-employee director to the Board, such newly
elected director is to receive a grant of restricted stock units for shares of common stock in the amount of $75,000 (the number of shares covered by such grant being equal to $75,000 divided by the closing price of the common stock on the date of grant). These restricted stock units vest in three equal tranches on each of the first three
anniversaries of the date of grant, if the recipient director remains on the Board on each such date. Furthermore, for the Chairperson of the Board, if not an employee of the Company, the number of restricted stock units granted annually and upon election is twice the number mentioned above. F-21
Notes to Consolidated Financial Statements — (Continued)
December 31,
2010
December 31,
2009
December 31,
2008
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES A summary of nonvested shares as of December 31, 2010 and changes during the year ended December 31, 2010 is provided below (shares in thousands):
Number of
Weighted Nonvested at January 1, 2010
1,067
$
8.20 Granted
692
$
10.74 Vested
(936
)
$
8.25 Forfeited
(70
)
$
8.91 Nonvested at December 31, 2010
753
$
10.41 Of the total number of nonvested shares granted during the year, 525,500 are performance-based and had a grant date fair value of $10.90 per share. The total grant-date fair value of nonvested shares that vested during the year ended December 31, 2010 was $9.3 million. As of December 31, 2010, there was $5.4 million of total unrecognized compensation cost related to nonvested shares that the Company expects to be recognized over a weighted average period of 26 months. Nearly all of the unrecognized compensation cost is related to the 2010 award and the weighted average vesting period
indicated is reflective of the blend of service and performance elements. The weighted average vesting period could be affected if the remaining performance goal becomes probable of being achieved and the related vesting period is shortened as a result. In the years ended December 31, 2010, 2009 and 2008, the Company recorded share-based compensation expense of $4.6 million, $4.5 million and $3.8 million related to nonvested share awards, which is included in the Company’s net income for each respective period. Of the 2010 expense, $1.2 million related to vesting of
performance-based awards. The board of directors of the Company elected to accelerate the vesting of certain nonvested share awards granted under the Company’s 2001 Incentive Stock Plan as of the consummation of the sale of the specialty pharmaceutical business in January 2010. This acceleration resulted in an estimated $0.8
million additional expense in the first quarter of 2010 and $0.5 million in 2009. The charges primarily represented an acceleration of expense recognition pursuant to the original award and, to a lesser extent, an adjustment, in certain cases, to recognize the modification of the award in contemplation of the sale. The Company’s policy is
to use newly issued shares to satisfy nonvested share awards. There has been no tax benefit realized to date related to tax deductions for nonvested shares. The breakdown of share-based compensation expense by major line caption in the statements of operations is shown below (amounts are in thousands):
Year Ended December 31,
2010
2009
2008 Research and development
$
1,643
$
1,223
$
1,000 General and administrative
2,963
3,268
2,797
$
4,606
$
4,491
$
3,797 (16) Employee Stock Purchase Plan The 2007 Employee Stock Purchase Plan (ESPP) permits eligible employees to purchase common stock through payroll deductions which may not exceed 15 percent of the employee’s compensation, as defined, at a price equal to 85 percent of the fair market value of the shares at the beginning of the offering period (grant date) or at
the end of the offering period (purchase date), whichever is lower. There are two six-month offering F-22
Notes to Consolidated Financial Statements — (Continued)
Nonvested
Shares
Average
Grant Date
Fair Value
Per Share
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES periods in each plan fiscal year, beginning April 1 and October 1. The ESPP is intended to qualify under section 423 of the Internal Revenue Code. Individual participant purchases within a given calendar year are limited to $25,000 ($21,250 based on the 15-percent discount) and no more than 2,500 shares on any single purchase date.
An additional one million shares were reserved for issuance under the plan. All benefit-eligible employees of the Company may participate in the ESPP other than those who own shares or hold options or nonvested shares representing a combined 5 percent or more of the voting power of the Company’s outstanding stock. Unless
terminated sooner, the ESPP will terminate on January 25, 2017. The fair value of shares to be issued under the ESPP is estimated at the grant date and is comprised of two components: the 15 percent discount to fair value of the shares at grant date and the value of the option granted to participants pursuant to which they may purchase shares at the lower of either the grant date or the purchase
date fair value. The option component is valued using the Black-Scholes option pricing model. The initial assumptions used in the valuation for each offering period, April 1 and October 1, are reflected in the following table (no dividends were assumed):
2010
2009
2008
October
April
October
April
October
April Expected volatility
30.31
%
31.80
%
39.52
%
95.62
%
41.00
%
35.00
% Expected term (in years)
0.5
0.5
0.5
0.5
0.5
0.5 Risk-free interest rate
0.24
%
0.19
%
0.19
%
0.39
%
1.79
%
1.55
% Increases in individual withholding rates within the offering period could have the effect of establishing a new measurement date for that individual’s future contributions. Compensation expense recognized for the ESPP was approximately $0.1 million, $0.2 million and $0.3 million for the years ended December 31, 2010, 2009 and
2008, respectively. Amounts withheld from participants are classified as cash from financing activities in the cash flow statement and as a liability in the balance sheet until such time as shares are purchased. There were two stock purchases under the ESPP during the year ended December 31, 2010. Based upon the purchase price
established as of March 31, 2010 and September 30, 2010, 52,412 shares were allocated under the plan in the year. Cash received from ESPP for the years ended December 31, 2010, 2009 and 2008 was $0.4 million, $0.5 million and $0.9 million, respectively. The breakdown of share-based compensation expense by major line caption in the statement of operations is shown below (amounts are in thousands).
Year Ended December 31,
2010
2009
2008 Research and development
$
67
$
43
$
87 General and administrative
32
129
195
$
99
$
172
$
282 F-23
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES (17) Income Taxes The components of the income tax provision related to continuing operations are summarized as follows (in thousands):
Year Ended December 31,
2010
2009
2008 Current: Federal
$
(140
)
$
(2,195
)
$
224 State
(197
)
110
31 Total current
(337
)
(2,085
)
255 Deferred: Federal and State
—
—
— Income tax (benefit) provision
$
(337
)
$
(2,085
)
$
255 The following table represents a reconciliation between the reported income taxes and the income taxes that would be computed by applying the federal statutory rate (35%) to income from continuing operations before taxes (in thousands):
Year Ended December 31,
2010
2009
2008 Income tax provision (benefit) computed at federal statutory rate
$
(1,098
)
$
(20,750
)
$
(16,209
) Nondeductible expenses
2,348
699
576 Add (deduct) effect of: Federal research and development tax credits
(2,662
)
(1,625
)
(881
) Tax on undistributed earnings of foreign subsidiary
826
—
— State income taxes, net of federal tax
(199
)
71
20 Effect of change in federal law
(140
)
(2,195
)
224 Increase in
beginning of period valuation allowance
588
21,715
16,525 Income tax (benefit) provision
$
(337
)
$
(2,085
)
$
255 Federal legislation, the American Recovery and Reinvestment Act of 2009, which allowed the Company to make an election to treat certain unused research and alternative minimum tax credit carryforwards as refundable in lieu of claiming bonus and accelerated depreciation for “eligible qualified property” placed in service through
the end of 2009 was extended to 2010. This provided the Company with a $0.1 million benefit in 2010. The balance of the 2010 income tax benefit reflects a reduction of $0.2 million to state taxes payable. In November 2009, federal legislation was enacted under which the Company was able to carry back its 2009 alternative minimum tax net operating losses to the five previous years to offset the alternative minimum taxes that were not available for carryback prior to the new legislation. The Company recorded the impact of the
carryback, estimated to be approximately $1.7 million, in the fourth quarter of 2009 and received a federal income tax cash refund in the first quarter of 2010. Other legislation in 2009 allowed the Company to make an election to treat certain unused research and alternative minimum tax credit carryforwards as refundable in lieu of
claiming bonus and accelerated depreciation for “eligible qualified property” placed in service through the end of 2008. This provided the Company with a $0.5 million benefit in 2009. The balance of the 2009 income tax expense reflects $0.1 million adjustment to state taxes payable. F-24
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES Income tax expense in 2008 was primarily comprised of certain state taxes. No federal income tax expense was incurred in relation to normal operating results due either to current period operating losses or the utilization of deferred tax assets to offset taxes that would otherwise accrue to operating income. The
gain on the sale of the specialty pharmaceutical business, although taxable,
did not result in a federal income tax liability due to the tax basis the
Company had in the divested assets and net operating losses generated in
2010. As of December 31, 2010 and 2009, the tax effects of temporary differences that give rise to the deferred tax assets and deferred tax liabilities are as follows (in thousands): December 31, December 31, Deferred tax assets: Federal and state net operating loss carryforwards $ 55,510 $ 34,066 Research and development credits carryforward 26,353 22,713 Acquired in-process research and development 8,411 8,731 Capital loss carryforwards 3,165 1,225 Share-based compensation 1,214 1,212 Federal alternative minimum tax credits 1,530 1,530 Write-down of carrying value of investment — 3,196 Accrued compensation 719 7,495 Other 3,394 2,282 Total gross deferred tax assets 100,296 82,450 Less valuation allowance (97,587 ) (81,356 ) 2,709 1,094 Deferred tax liabilities: Book basis in excess of tax basis of acquired assets (1,510 ) (486 ) Undistributed earnings of foreign subsidiary (826 ) — Unrealized gain on investment securities (373 ) (608 ) (2,709 ) (1,094 ) Net deferred tax assets $ — $ — During the year ended December 31, 2010, the Company determined that it would no longer permanently reinvest any of the earnings of its foreign subsidiaries. As a result, the Company recorded a net deferred income tax liability of $0.8 million on approximately $2.4 million of accumulated earnings of its foreign subsidiaries with
an offsetting valuation allowance. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. At December 31, 2010, the Company had federal net operating loss carryforwards of approximately $134.3 million that expire in the years 2020 through 2030 and combined state net operating loss
carryforwards of approximately $94.4 million that expire in the years 2011 through 2030. The Company also has federal research and development tax credit carryforwards of approximately $20.0 million for tax reporting purposes that expire in the years 2011 through 2030. In addition, the Company has $6.4 million of state research and
development tax credit carryforwards that expire in the years 2015 through 2025. The Company’s ability to use the net operating loss and research and development tax credit F-25
Notes to Consolidated Financial Statements — (Continued)
2010
2009
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES carryforwards is subject to certain limitations due to ownership changes, as defined by rules pursuant to Section 382 of the Internal Revenue Code of 1986, as amended. As of December 31, 2010, management believes that it is more likely than not that the net deferred tax assets will not be realized, based on assumptions regarding future operations, consideration of tax strategies and the reversal of deferred tax liabilities. As of December 31, 2010 and 2009, the Company had deferred tax assets of
$100.3 million and $82.5 million, respectively. The Company has maintained a valuation allowance of $97.6 million and $81.4 million at December 31, 2010 and 2009, respectively. The Company files income tax returns in the U.S. federal jurisdiction, various state jurisdictions and Canada. The Company is currently not under examination by the U.S. Internal Revenue Service, however, the tax years 2007 through 2010 remain open to examination. State income tax returns for the states of New Jersey and
Indiana are generally subject to examination for a period of 3-4 years after filing of the respective returns. These state income tax returns are not currently under examination. Income tax returns for Canada are generally subject to examination for a period of 3-5 years after filing of the respective return. The Company’s income tax returns
are currently not under examination by Revenue Canada. (18) Significant Agreements sigma-tau Group The Company sold its specialty pharmaceutical business to Klee Pharmaceuticals Inc. (now known as Sigma-Tau PharmaSource, Inc.), Defiante Farmacêutica, S.A and sigma-tau Finanziaria S.p.A. (collectively, the sigma-tau Group) in January 2010. In addition to the initial sale of assets which has been reflected in the Company’s
financial statements for the year ended December 31, 2010, there were certain potential future payments to Enzon that were contingent upon the achievement of stated milestones. Remaining potential milestone payments as of December 31, 2010 were estimated to be $22.0 million. Of this total, $5.0 million was subsequently earned and
will be recognized in the first quarter of 2011. In addition, there are royalties potentially due to Enzon of 5 to 10 percent on incremental net sales through 2014 by the sigma-tau Group above a 2009 baseline amount from the four marketed specialty pharmaceutical products Enzon sold to them. Approximately $0.6 million was recognized
pursuant to this provision of the sale agreement in the fourth quarter of 2010. There can be no assurance that any of the remaining milestone payments or any future royalty revenues beyond that which has been recognized in 2010 will accrue to the benefit of the Company. Also, the Company entered into a transition services agreement with sigma-tau Group whereby Enzon would perform product-support research and development for up to three years and provide various general and administrative functions for up to one year following the closing of the transaction. In consideration for this work,
Enzon is being compensated based upon costs incurred plus a mark-up defined in the transition services agreement. Santaris Pharma A/S License Agreement In July 2006, the Company entered into a license agreement with Santaris Pharma A/S (Santaris) pursuant to which the Company obtained exclusive rights worldwide, other than in Europe, to develop and commercialize RNA antagonists directed against the HIF-1a and Survivin mRNA, as well as RNA antagonists directed against six
additional gene targets selected by the Company. Since inception of the agreement, initial acquisition of in-process research and development and milestone payments have been made totaling $34.0 million, including milestone payments of $7.0 million, $3.0 million and $6.0 million in 2010, 2009 and 2008, respectively, included in
research and development expense in the accompanying statements of operations. The Company could pay an additional $233.0 million in milestone payments upon the successful completion of certain development and regulatory milestones. If the Company fails to make the requisite milestone payment for any particular target, Santaris
has the right to recover that target for its own purposes. Santaris also is eligible to receive single-digit royalties from any future product sales from products based on the licensed F-26
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES antagonists. Santaris retains the right to develop and commercialize products developed under the agreement in Europe. The royalty term expires on a country-by-country and product-by-product basis when the last valid LNA platform patent or LNA compound patent expires not to exceed 21 years with respect to any product. Merck Agreement As a result of a November 1990 agreement, the Company’s PEGylation technology was used to develop an improved version of the product INTRON A, PEGINTRON. Merck (previously Schering-Plough) is responsible for marketing and manufacturing PEGINTRON on an exclusive worldwide basis and the Company receives
royalties on worldwide sales of PEGINTRON for all indications. The Company has no involvement in the selling or marketing of PEGINTRON. Merck’s obligation to pay the Company royalties on sales of PEGINTRON terminates, on a country-by-country basis, upon the later of the date the last patent to contain a claim covering
PEGINTRON expires in the country or 15 years after the first commercial sale of PEGINTRON in such country. Currently, expirations are expected to occur in 2016 in the U.S., 2018 in Europe and 2019 in Japan. The royalty percentage to which the Company is entitled will be lower in any country where a PEGylated alpha-interferon
product is being marketed by a third party in competition with PEGINTRON where such third party is not Hoffmann-La Roche. Either party may terminate the agreement upon a material breach of the agreement by the other party that is not cured within 60 days of written notice from the non-breaching party or upon declaration of
bankruptcy by the other party. During the quarter ended September 30, 2007, the Company sold a 25-percent interest in future royalties payable to it by Merck on net sales of PEGINTRON occurring after June 30, 2007. Nektar Agreement In January 2002, the Company entered into a PEGylation technology licensing agreement with Nektar under which the Company granted Nektar the right to grant sub-licenses for a portion of its patents related to its PEGylation technology to third-parties. Nektar had the right to sub-license Enzon’s patents that were defined in the
January 2002 agreement and the Company will receive a royalty or a share of Nektar’s profits for any products that utilize the Company’s patented PEGylation technology. The Company’s receipt of royalties related to Nektar licenses will end in 2014. Effective in January 2007, Nektar’s right to grant additional sublicenses was limited to
a certain class of our PEGylation technology. Existing sublicenses granted by Nektar prior to January 2007 were unaffected. (19) Commitments and Contingencies The Company has employment and separation agreements with certain members of its management that provide for severance payments and payments following a termination of employment occurring for various reasons including a change in control of the Company. The Company has been involved in various claims and legal actions arising in the ordinary course of business. In the opinion of management, the ultimate disposition of these matters will not have a material effect on the Company’s consolidated financial position, results of operations or liquidity. The Company has non-cancelable lease obligations for certain office and production facilities that either have been vacated and sublet or are scheduled for vacancy by the end of the first quarter of 2011. See Note 20 — Leases, below. (20) Leases The Company has several leases for office, warehouse, production and research facilities and equipment. The non-cancelable lease terms for the operating leases expire at various dates between 2011 and 2021 and each agreement includes renewal options. F-27
Notes to Consolidated Financial Statements — (Continued)
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES Future minimum lease payments, for non-cancelable operating leases with initial or remaining lease terms in excess of one year as of December 31, 2010 are (in thousands):
Year ending December 31,
Operating 2011
$
2,261 2012
2,240 2013
823 2014
703 2015
703 Thereafter
4,217 Total minimum lease payments
$
10,947 Minimum payments indicated above have not been reduced by future minimum rentals to be received under noncancelable subleases of approximately $0.6 million to be received in equal monthly installments through October 2012 nor approximately $0.8 million to be received in equal monthly installments through January 2013. Rent expense amounted to $2.6 million, $2.4 million and $2.3 million, for the years ended December 31, 2010, 2009 and 2008, respectively. Total rent expense, inclusive of scheduled increases and rent holidays, is recognized on a straight-line basis over the term of the lease. The Company’s use of leased office space in Bridgewater, New Jersey is expected to end by March 31, 2011. A portion of the leased space has been sublet at a rate lower than the Company’s committed costs for that space. The lease related to this portion of the total leased facilities expires on January 31, 2013. The Company
remains as the primary lessee with rental payments, in the aggregate amounting to $2.8 million that extend until January 31, 2013. It is possible that the remaining space also may not be sublet at favorable rates or at all. No other costs related to termination of the lease or restoration of the facilities are anticipated. The Company’s use of the leased South Plainfield facility has ended. While the Company continues to be obligated under the original lease for the facility, a sublease was entered into in January 2010 on favorable terms such that no liability needs to be accrued. The Company may incur charges associated with the lease or its
termination prior to or upon the contractual expiration of the lease in October 2012, however, such exposure, if any, cannot be estimated at this time. (21) Retirement Plans The Company maintains a defined contribution 401(k) pension plan for substantially all of its full-time and part-time employees, as defined. The Company currently matches 50 percent of the employee’s contribution of up to 6 percent of compensation, as defined. Total Company contributions for the years ended December 31, 2010,
2009 and 2008, were $0.7 million, $1.0 million and $1.1 million, respectively. The Executive Deferred Compensation Plan, as amended, is intended to aid the Company in attracting and retaining key employees by providing a non-qualified funded compensation deferral vehicle. At December 31, 2010 and 2009, $3.1 million and $4.0 million of deferred compensation was included in other liabilities,
respectively. See Note 4 relating to the investment of participants’ assets. (22) Discontinued Operations On January 29, 2010, the Company consummated the sale to the sigma-tau Group of the specialty pharmaceutical business comprised principally of the Company’s Products and Contract Manufacturing segments in addition to certain in-process research and development. The Products and Contract Manufacturing segments
constituted components of Enzon and qualified for treatment as discontinued operations upon F-28
Notes to Consolidated Financial Statements — (Continued)
Leases
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES consummation of the transaction. In-process research and development, which comprised part of the total transaction, did not constitute a component of Enzon and, accordingly, was treated as an asset sale and not as discontinued operations. Terms of sale: The asset purchase agreement for the sale of the specialty pharmaceutical business contained the following major provisions. Updated status regarding each element is also provided.
•
Cash purchase price was $300.0 million, subject to certain customary working capital adjustments.
The cash proceeds received, including the second-quarter 2010 working capital adjustment, amounted to approximately $308.0 million. Transaction costs amounted to approximately $5.0 million reducing net proceeds to approximately $303.0 million. Of this amount, $40.9 million was allocated to the sale of in-process research and
development (See Note 9 above). The net proceeds then attributable to discontinued operations amounted to $262.6 million and this amount less the book basis in the respective assets and liabilities (see below) yielded the gain from discontinued operations of $176.4 million.
Up to $27.0 million based on certain success milestones.
During January 2011, the Company received notice that one of the milestones—the approval of an sBLA regarding a new API starting material for the manufacture of SS Oncaspar—was reached, resulting in Enzon being entitled to receive and recognize $5.0 million of milestone income in 2011. During the latter half of 2010,
circumstances emerged making it unlikely that another of the milestones related to an expedited approval process in Europe would be achieved. This would have resulted in a $5.0 million payment to Enzon. Of the remaining $17.0 million of potential milestone payments, it is very unlikely that any will be received in 2011 and there can
be no assurance that the Company will receive any such payments in the future. The receipt of milestone payments does not constitute continuing cash flows of the divested business. These payments are not contingent upon Enzon performing the research or development activity. Enzon would be entitled to receive the payments if the buyer utilized another research and development provider.
Royalties of 5 to 10 percent on incremental net sales above a 2009 baseline amount from what had been Enzon’s four marketed specialty pharmaceutical products through 2014.
Sales of the four products during 2010 outside the U.S. were sufficiently in excess of 2009 baseline amounts to enable Enzon to earn and recognize a nominal amount of royalty revenue related to this agreement in the fourth quarter of 2010. There can be no assurance that the Company will receive any additional royalty payments
pursuant to this agreement. These royalties do not constitute a migration or continuation by Enzon of the activities that generate the payments. Enzon is no longer engaged in any manufacturing or marketing activities. Consequently, these cash flows are deemed to be indirect in nature.
Transition services agreement—Enzon has performed product-support research and development and has provided various general and administrative functions for the purchasing parties during 2010. In consideration for this work, Enzon is being compensated based upon costs incurred plus a mark-up defined in the transition
services agreement.
Revenues from and associated costs related to research and development transition services are reflected in the statements of operations as contract research and development and research and development—specialty and contracted services, respectively. Transition services revenues related to general and administrative efforts are
reported in miscellaneous income and the associated costs are shown as general and administrative—contracted services. As of December 31, 2010, the Company’s involvement in general and administrative support efforts has essentially been concluded. Some diminishing level of research and development support will continue into 2011. The cash flows related to the transition services being provided to the buyer in connection with research and development activities represent a continuation of Enzon’s corporate research function. However, the cost- F-29
Notes to Consolidated Financial Statements — (Continued)
•
•
•
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES plus arrangement did not generate sufficient net cash flows during 2010 to be considered significant. These cash flows will be substantially lower in 2011. The services are performed at the request of the sigma-tau Group as a convenience to them. The services could have been performed by others. Discontinued Operations Accounting Treatment While the sale of the specialty pharmaceutical business was initiated in November 2009, the assets were not considered to be held for sale as of December 31, 2009 due to the fact that the transaction was subject to shareholder approval. Such approval was obtained at a special meeting of shareholders on January 27, 2010. As a
result, discontinued operations treatment began in the first quarter of 2010 for the Products and Contract Manufacturing segments whereby results of discontinued operations and net assets and liabilities are reported separately in the statements of operations, cash flows and balance sheets. The sale of in-process research and development
associated with marketed products was treated as an asset sale and was not part of discontinued operations for accounting purposes due to the Company’s significant continuing involvement in research and development related to marketed products subsequent to the sale. Assets and liabilities acquired by the Purchasing Parties include:
ownership of the four marketed products, Oncaspar, Adagen, Abelcet and DepoCyt and all related rights; • real estate, personal property and equipment of the business used in the manufacture of products and performance of the contract manufacturing operations, including the manufacturing facility in Indianapolis;
•
working capital, including accounts receivable, inventories, accounts payable and other prepaids and accruals; • patents, trademarks, copyrights and other intangible properties related to the products and product-specific assets; • in-process research and development related to the sourcing of Oncaspar and Adagen; and • other assets and liabilities as specified in the asset purchase agreement. Assets and liabilities excluded from the sale of the specialty pharmaceutical business include:
•
cash and cash equivalents; • tax refunds and tax attributes related to assets, liabilities and past operations; • royalties business with the exception of one contract related to Oncaspar; • PEG-SN38 and Enzon’s LNA compounds and PEG technology platform; • 4% convertible senior notes due 2013; • stock compensation arrangements; • product claims, product return claims, environmental and tax liabilities arising prior to the closing date in excess of any reserves; • lease related to South Plainfield, New Jersey facility; and • other assets and liabilities as specified in the asset purchase agreement. F-30
Notes to Consolidated Financial Statements — (Continued)
•
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES Summary results of operations of the specialty pharmaceutical business through January 29, 2010 and for the years ended December 31, 2009 and 2008 were as follows (in thousands):
January 1, 2010
Year Ended December 31,
2009
2008 Revenues
$
8,720
$
133,213
$
139,969 Income before income tax
$
3,620
$
57,661
$
43,901 Income tax benefit (provision)
—
224
(49
) Gain on sale of discontinued operations, net of income tax, as adjusted(1)
176,423
—
— Income and gain from discontinued operations, net of income tax, as adjusted(1)
$
180,043
$
57,885
$
43,852
(1)
As adjusted in the fourth quarter of 2010 to include the write-off of $1.0 million of currency translation adjustment amounts that had been included in accumulated other comprehensive income but should have been recognized as part of the gain on the sale. See Note 23, Quarterly Results of Operations.
The sale was a taxable transaction for federal income tax purposes. The Company did not, however, incur significant tax liabilities as a result of the transaction due to the tax basis it has in the disposed assets and the current year net operating loss. The potential receipt of milestone and/or royalty payments will also be taxable
events, but the tax consequences of these payments cannot be estimated at this time. The carrying amounts of major classes of assets and liabilities of the specialty pharmaceutical business were as follows (in thousands):
January 29,
December 31, Trade accounts receivable, net
$
11,886
$
15,026 Inventories
19,516
17,734 Other current assets
693 1,286 Current assets of discontinued operations
$
32,095
$
34,046 Property and equipment, net
$
12,621
$
12,703 Amortizable intangible assets, net
48,896
49,801 Non-current assets of discontinued operations
$
61,517
$
62,504 Trade accounts payable
$
700
$
2,875 Accrued expenses
5,763
10,394 Liabilities of discontinued operations
$
6,463
$
13,269 F-31
Notes to Consolidated Financial Statements — (Continued)
through
January 29, 2010
2010
2009
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES (23) Quarterly Results of Operations (Unaudited) The following tables present summarized unaudited quarterly financial data (in thousands, except per-share amounts).
Three Months Ended
March 31,
June 30,
September 30,
December 31, Total revenues(1)
$
58,253
$
13,766
$
13,328
$
12,518 Income (loss) from continuing operations
20,754
(5,571
)
(8,354
)
(9,629
) Income and gain from discontinued operations
179,053
(51
)
—
1,041 Net income (loss)(3)
199,807
(5,622
)
(8,354
)
(8,588
) (4)(5) Per-share information: Income (loss) from continuing operations: Basic
$
0.40
$
(0.09
)
$
(0.14
)
$
(0.16
) Diluted
$
0.29
$
(0.09
)
$
(0.14
)
$
(0.16
) Net income (loss): Basic
$
3.82
$
(0.09
)
$
(0.14
)
$
(0.14
) Diluted
$
2.70
$
(0.09
)
$
(0.14
)
$
(0.14
)
Three Months Ended
March 31,
June 30,
September 30,
December 31, Total revenues(1)(2)
$
13,071
$
13,170
$
12,974
$
12,193 (Loss) income from continuing operations
(11,416
)
(19,657
)
(11,525
)
(14,604
) Income and gain from discontinued operations
17,596
14,591
11,658
14,040 Net income (loss)(3)
6,180
(5,066
)
133
(564
) Per-share information: (Loss) income from continuing operations: Basic
$
(0.25
)
$
(0.43
)
$
(0.26
)
$
(0.32
) Diluted
$
(0.25
)
$
(0.43
)
$
(0.26
)
$
(0.32
) Net income (loss): Basic
$
0.14
$
(0.11
)
$
0.00
$
(0.01
) Diluted
$
0.14
$
(0.11
)
$
0.00
$
(0.01
)
(1)
Revenues are primarily royalties received on the sale of products by other companies utilizing Enzon’s Customized Linker Technology. First quarter 2010 revenues include $40.9 million related to sale of in-process research and development. Revenues from services in 2010 are not material. Subsequent to the January 2010 sale of the
specialty pharmaceutical business (reflected as discontinued operations), the Company is no longer involved in the manufacture and sale of products. (2) Excludes product sales, certain royalty revenues and contract manufacturing revenues of the divested specialty pharmaceutical business. In the aggregate, these discontinued operations revenues amounted to $35,567, $34,024, $31,627 and $31,996 for the quarters ended March 31, 2009; June 30, 2009; September 30, 2009; December
31, 2009, respectively. (3) As previously reported through September 30, 2010. (4) The
gain on the sale of the specialty pharmaceutical business was adjusted during
the fourth quarter 2010 by $1.0 million to reflect the write-off of accumulated
currency translation gains related to the Canadian subsidiary. This changed
the previously reported income and gain from discontinued operations to $180.0
million or $3.08 per share versus the $179.0 million or $3.06 per share previously
reported. Because the sale of substantially all of the net assets of the
Canadian subsidiary constituted a substantial liquidation for accounting
purposes, the accumulated currency translation adjustment should have been
reported in earnings as part of the gain calculation during the first quarter
of 2010. The fourth quarter correcting entry was not material to the first
or fourth quarters nor to the full year 2010 results of operations. (5) Included in the fourth-quarter 2010 results, is a correction of the accounting for the first-quarter 2010 conversion of a portion of the Company’s 4% notes. The net effect of the forgone interest and the write-off of a pro rata amount of deferred debt issuance costs amounted to $0.8 million and was charged to earnings during the first
quarter of 2010 at the time of the notes conversion. The correcting adjustment was to credit interest expense for the $0.8 million and to charge additional paid-in capital reflective of the capital nature of the transaction. The noncash correcting entry was not material to the first or fourth quarters nor to the full year 2010 results of
operations. See Note 6, Notes Payable. F-32
Notes to Consolidated Financial Statements — (Continued)
2010
2010
2010
2010
(4)
2009
2009
2009
2009
ENZON PHARMACEUTICALS, INC. AND SUBSIDIARIES In the fourth quarter of 2010, the Company recognized a federal government Qualifying Therapeutic Discovery Project grant in the amount of $1.2 million and royalty revenues in the amount of approximately $0.6 million related to 2010 sales of divested products in excess of baseline 2009 levels. Also in the fourth quarter of 2010,
the Company recognized a $3.0 million restructuring charge related to a workforce reduction affecting 33 employees. See Notes 10, Contract Research and Development Revenue and Miscellaneous Income; 13, Restructuring and 22, Discontinued Operations. During the fourth quarter of 2010, the Company determined that it would no longer permanently reinvest any of the earnings of its foreign subsidiaries. As a result, the Company recorded a net deferred income tax liability of $0.8 million on approximately $2.4 million of accumulated earnings of its foreign subsidiaries with an
offsetting valuation allowance. See Note 17, Income Taxes. In the fourth quarter of 2009, the Company recognized an estimated tax benefit of $1.7 million related to fourth-quarter federal tax legislation that allows the carryback of its 2009 alternative minimum tax net operating losses to the five previous years to offset the alternative minimum taxes paid in prior years. The carryback was not
available to the Company prior to the new tax legislation. There was also a $0.1 million tax expense for the quarter bringing the net tax benefit to $1.6 million. See Note 17, Income Taxes. F-33
Notes to Consolidated Financial Statements — (Continued)
20 Kingsbridge Road, Piscataway, NJ 08854
(732) 980-4500 • FAX: (732) 980-4585
http://www.enzon.com