Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 0-21937
CERUS CORPORATION
(Exact name of registrant as specified in its charter)
| Delaware | 68-0262011 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 2550 Stanwell Dr. Concord, California |
94520 | |
| (Address of principal executive offices) | (Zip Code) |
(925) 288-6000
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
NASDAQ Global Market
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, par value $.001 per share
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K, (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨ Accelerated filer ¨ Non-accelerated filer ¨ Smaller reporting company x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The approximate aggregate market value of the common stock held by non-affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter, based upon the closing sale price of the registrant’s common stock listed on the Nasdaq Global Market, was $106.9 million.(1)
As of February 28, 2011, there were 47.6 million shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement in connection with the registrant’s 2011 Annual Meeting of Stockholders, to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year ended December 31, 2010, are incorporated by reference into Part III of this Annual Report on Form 10-K.
(1) Based on a closing sale price of $3.16 per share on June 30, 2010. Excludes 5.1 million shares of the registrant’s common stock held by executive officers, directors and affiliates at June 30, 2010.
Table of Contents
Table of Contents
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities and Exchange Act of 1934, as amended, that involve risks and uncertainties. The forward-looking statements are contained principally in Part II Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in Part I, Item 1A, “Risk Factors.” These statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Examples of forward-looking statements include, but are not limited to, statements about our estimates regarding the sufficiency of our cash resources, our ability to commercialize and achieve market acceptance of the INTERCEPT Blood Systems, the anticipated progress of our research, development and clinical programs, our ability to manage cost increases associated with pre-clinical and clinical development for the INTERCEPT Blood Systems, our ability to obtain and maintain regulatory approvals of the INTERCEPT Blood Systems, the ability of our products to inactivate pathogens that may emerge in the future, and our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others. In some cases, you can identify forward-looking statements by terms such as “anticipate,” “will,” “believe,” “estimate,” “expect,” “plan,” and similar expressions intended to identify such forward-looking statements. Forward-looking statements reflect our current views with respect to future events, are based on assumptions, and are subject to risks and uncertainties. There can be no assurance that these statements will prove to be correct. Certain important factors could cause actual results to differ materially from those discussed in such statements, including our need for additional financing, whether our preclinical and clinical data or data from commercial use will be considered sufficient by regulatory authorities to grant marketing approval for our products, market acceptance of our products, reimbursement, development and testing of additional configurations of our products, regulation by domestic and foreign regulatory authorities, our limited experience in sales, marketing and regulatory support for the INTERCEPT Blood System, our reliance on Fenwal and third parties to manufacture certain components of the INTERCEPT Blood System, incompatibility of our platelet system with some commercial platelet collection methods, our need to complete certain of our product components’ commercial design, more effective product offerings by, or clinical setbacks of, our competitors, product liability, our use of hazardous materials in the development of our products, business interruption due to earthquake, our limited operating history and expectation of continuing losses, protection of our intellectual property rights, volatility in our stock price, legal proceedings, on-going compliance with the requirements of the Sarbanes-Oxley Act of 2002 and other factors discussed throughout this Annual Report on Form 10-K particularly under the caption “Risk Factors,” in Part I, Item 1A of this Annual Report on Form 10-K and in our other documents filed with the Securities and Exchange Commission. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our estimates and assumptions only as of the date of this Annual Report on Form 10-K. You should read this Annual Report on Form 10-K and the documents that we incorporate by reference in and have filed as exhibits to this Annual Report on Form 10-K, completely and with the understanding that our actual future results may be materially different from what we expect. Except as required by law, we assume no obligation to update any forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in any forward-looking statements, even if new information becomes available in the future.
| Item 1. | Business |
Overview
We are a biomedical products company focused on commercializing the INTERCEPT Blood System, or INTERCEPT system, to enhance blood safety. The INTERCEPT system is designed to inactivate blood-borne pathogens in donated blood components intended for transfusion.
1
Table of Contents
We have worldwide commercialization rights for the INTERCEPT Blood System for platelets, plasma and red blood cells and we currently market and sell the INTERCEPT system for both platelets and plasma in a number of countries in Europe, the Commonwealth of Independent States, or CIS, the Middle East and selected countries in other regions around the world using our direct sales force or through distributors. The INTERCEPT platelet and plasma systems have both received CE mark approval. We continue to prioritize commercialization of the INTERCEPT Blood System for platelets and plasma in these countries and regions. In addition, we plan to continue development of our INTERCEPT red blood cell system. Subject to the availability of adequate funding we also plan to pursue regulatory approval of the INTERCEPT platelet and plasma systems in the United States.
We were incorporated in California in 1991 and reincorporated in Delaware in 1996. Information regarding our revenue, net income or losses, and total assets for the last three fiscal years can be found in the financial statements and related notes found elsewhere in this Annual Report on Form 10-K. Our wholly-owned subsidiary, Cerus Europe B.V., was formed in the Netherlands in 2006.
Product Development
Background
The INTERCEPT Blood System is designed to broadly target and inactivate blood-borne pathogens, such as viruses (for example, HIV, West Nile, SARS, hepatitis B and C), bacteria and parasites, as well as potentially harmful white blood cells, while preserving the therapeutic properties of platelet, plasma and red blood cell transfusion products. The INTERCEPT Blood System inactivates a broad array of pathogens and has the potential to reduce the risk of transfusion related transmission of pathogens for which testing is not completely effective or is not currently performed. We believe that the INTERCEPT Blood System also has the potential to inactivate most new pathogens before they are identified and before tests are developed and adopted commercially to detect their presence in donated blood. In addition, data from commercial use suggests that treating platelet components with the INTERCEPT platelet system substantially reduces the rate of transfusion-related adverse events as compared to the incidence of such events prior to adoption of the INTERCEPT platelet system. The INTERCEPT Blood System is based on our proprietary technology for controlling biological replication.
2
Table of Contents
Products, Product Candidates and Development Activities
The following table identifies our products and product development programs and their current status:
| Product or Product Under Development |
Product or Development Status |
Commercial Rights | ||
| INTERCEPT Blood |
• Commercialized in a number of countries in Europe, the CIS, and the Middle East and selected countries in other regions around the world • United States: Phase III clinical trial completed; Seeking FDA concurrence on additional Phase III clinical trial |
Worldwide | ||
| INTERCEPT Blood |
• Commercialized in a number of countries in Europe, the CIS countries, the Middle East and selected countries in other regions around the world • United States: Orphan drug designation for Thrombotic Thrombocytopenic Purpura (TTP); Phase III clinical trials completed; Seeking concurrence of clinical adequacy with FDA |
Worldwide | ||
| INTERCEPT Blood |
Phase I clinical trial completed in 2010; Preparing for initiation of Phase III clinical trial for patients requiring acute transfusion support | Worldwide | ||
INTERCEPT Blood System for Platelets
The INTERCEPT Blood System for platelets, or platelet system, is designed to inactivate blood-borne pathogens in platelets donated for transfusion. The platelet system has received CE mark approval in the European Union and is being marketed and sold in several countries in a number of countries in Europe, the CIS, and the Middle East and selected countries in other regions around the world. Separate approvals for use of INTERCEPT-treated platelet products have been obtained in France and Switzerland. In Germany and Austria, where approvals must be obtained by individual blood centers for use of INTERCEPT-treated platelets, several centers have obtained such approvals. Many countries outside of Europe accept the CE mark, and have varying additional administrative or regulatory processes before the platelet system can be made commercially available. In general, these processes do not require additional clinical trials.
In addition to regulatory approvals, some potential customers, including the largest branch of the German Red Cross, desire to conduct their own clinical studies before adopting the platelet system. We also expect the Japanese Red Cross to conduct a clinical study before adoption of any pathogen inactivation system.
In the United States, we will not be able to market the platelet system until we have conducted an additional Phase III clinical trial. We are currently working with the United States Food and Drug Administration, or FDA, to establish a protocol for such a trial. However, we have no plans to initiate such a clinical trial unless adequate funding is secured.
3
Table of Contents
Additional information regarding our interactions with the FDA and possible clinical trial design can be found under “Item 1A—Risk Factors” of this Annual Report on Form 10-K, under the risk factor titled “Our products, blood products treated with the INTERCEPT Blood System and we are subject to extensive regulation by domestic and foreign authorities. If our preclinical and clinical data are not considered sufficient by regulatory authorities to grant marketing approval, we will be unable to commercialize our products and generate revenue. Our red blood cell system requires extensive additional testing and development.”
Information regarding our revenues from the platelet system for the years ended December 31, 2010, 2009, and 2008 can be found in “Item 7—Management’s Discussion and Analysis of Financial Condition and Results of Operation”, and “Item 15(a)—Consolidated Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
INTERCEPT Blood System for Plasma
The INTERCEPT Blood System for plasma, or plasma system, is designed to inactivate blood-borne pathogens in plasma donated for transfusion. The plasma system has received CE mark approval in a number of countries in Europe and is marketed and sold in several countries in Europe and in the CIS. France, Switzerland, Germany, and Austria require separate approvals for use of INTERCEPT-treated plasma products. INTERCEPT-treated plasma approvals have been obtained in these countries with the exception of Austria. Many countries outside of Europe accept the CE mark, and have varying additional administrative or regulatory processes before the platelet system can be made commercially available. In general, these processes do not require additional clinical trials.
In addition to regulatory approvals, some potential customers may desire to conduct their own clinical studies before adopting the plasma system.
In the United States, we have received orphan drug status for INTERCEPT-treated plasma for the treatment of thrombotic thrombocytopenic purpura, or TTP, although we will not be able to market the plasma system until we have submitted and obtained FDA approval of a product marketing application based upon our completed Phase III clinical trials and, if required, additional Phase III clinical trials. We do not know if the FDA will require any additional studies.
Information regarding our revenues from the plasma system for the years ended December 31, 2010, 2009 and 2008 can be found in “Item 7—Management’s Discussion and Analysis of Financial Condition and Results of Operation,” and “Item 15(a)—Consolidated Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.”
INTERCEPT Blood System for Red Blood Cells
The INTERCEPT Blood System for red blood cells, or red blood cell system, is designed to inactivate blood-borne pathogens in red blood cells donated for transfusion. In 2008, we completed a series of in vitro and in vivo tests with the red blood cell system. We completed a Phase I clinical trial of the red blood cell system in 2010, meeting the clinical trial’s primary end point. We expect to spend the majority of 2011 implementing and verifying changes to the red blood cell system, manufacturing clinical supplies, and completing site set-up activities before we enter Phase III clinical trials.
Previously, we terminated Phase III clinical trials for acute and chronic anemia for a prior generation of the red blood cell system. The trials were terminated due to the detection of antibody reactivity to INTERCEPT-treated red blood cells in two patients in the study for chronic anemia. The antibody eventually cleared and the patients had no adverse health consequences. After unblinding the data from the original Phase III clinical trials, we found that we had met the primary end-point in the clinical trial for acute anemia. Prior to commencing the Phase I clinical trial in 2008, we evaluated the antibodies detected in the original Phase III clinical trials and
4
Table of Contents
developed process changes to diminish the likelihood of antibody reactivity in red blood cells treated with our modified process. There were no adverse events associated with INTERCEPT-treated red blood cells evident in the 2008 trial. Based on the results from the 2008 clinical trial, we plan to enter our new Phase III trials with the modified process.
INTERCEPT Blood System Technology
Our platelet system and plasma system employ the same technology. Platelet components or plasma components collected from blood donors are transferred into plastic INTERCEPT disposable kits in which they are mixed with a proprietary Cerus compound, amotosalen, which has an affinity for nucleic acid.
The disposable kits are then placed in an illumination device, or illuminator, where the mixture is exposed to ultra-violet A (UVA) light. If pathogens such as viruses, bacteria or parasites are present in the platelet or plasma components, the energy from the UVA light causes the amotosalen to bond with the nucleic acid of the pathogens. The ability of amotosalen to form both cross-links between strands of nucleic acid and links to single nucleic acid strands results in a strong chemical bond between the amotosalen and the nucleic acid of the pathogens. The presence of these bonds is designed to prevent replication of the nucleic acid within pathogens, effectively inactivating the pathogens. A high level of inactivation has been demonstrated in a broad range of pathogens studied by Cerus and others in laboratory testing. For instance, INTERCEPT has demonstrated inactivation of a number of single stranded nucleic acid-based viruses such as HIV, hepatitis B, hepatitis C (using a model virus), West Nile, chikungunya, and certain influenza viruses.
Since platelets and plasma do not rely on nucleic acid for therapeutic efficacy, the INTERCEPT Blood System is designed to preserve the therapeutic function of the platelet and plasma components when used in human transfusions.
Following the inactivation process, residual amotosalen and by-products are reduced by more than 99% through use of a compound adsorption device, which is an integrated component of the disposable kit. We have performed extensive toxicology testing on the residual amotosalen and its by-products and good safety margins have been demonstrated. Any remaining amotosalen which may be transfused is rapidly excreted by humans.
Leukocytes, also known as white blood cells, are typically present in platelet and plasma components collected for transfusion, and can cause adverse transfusion reactions as well as an often fatal disease called graft-versus host disease. Leukocytes, like pathogens, rely on nucleic acid for replication and cellular function. The INTERCEPT Blood System, with its combination of the amotosalen and UVA light, is designed to inactivate leukocytes in the same manner it inactivates pathogens.
Like the platelet and plasma systems, the red blood cell system acts by using an additive compound to form bonds with nucleic acid in pathogens that may be present in red blood cell components destined for transfusion. The red blood cell system is designed to preserve the therapeutic qualities of the red blood cells, which do not rely on nucleic acid for their cellular function. The red blood cell system uses a proprietary Cerus compound, S-303. Unlike the platelet and plasma systems, the chemical bonds from S-303 are not triggered by UVA light, but instead by the pH level of the red blood cell components. After mixture with the red blood cell components in plastic disposable kits, S-303 is designed to rapidly break down into a form that is no longer chemically reactive with nucleic acid. As with the platelet and plasma systems, a high level of inactivation in a broad range of pathogens has been demonstrated with the red blood cell system.
By treating blood components with INTERCEPT within a day of collection, the inactivation of bacteria prevents bacterial growth that could create increased risk of inflammatory response or dangerous levels of endotoxins. Extensive clinical testing has been done on platelet and plasma products treated with the INTERCEPT Blood System, as well as post-marketing haemovigilance studies of the treated blood products in routine use.
5
Table of Contents
We believe that, due to their mechanisms of action, the platelet system, plasma system, and red blood cell system will potentially inactivate blood-borne pathogens that have not yet been tested with our systems, including emerging and future threats to the blood supply. We do not claim, however, that our system will inactivate all pathogens, including prions; and our inactivation claims are limited to those contained in our product specifications.
Collaborations
Baxter and Fenwal
We collaborated with Baxter International, Inc., or Baxter, on the development and commercialization of the INTERCEPT Blood System commencing in 1993. We obtained worldwide commercialization rights to the red blood cell system from Baxter in February 2005. Effective February 1, 2006, we entered into a restructuring of our agreements with Baxter pursuant to which we obtained exclusive worldwide commercialization rights to the platelet and plasma systems, excluding certain Asian countries where the commercialization rights had been licensed to BioOne Corporation, or BioOne. We agreed to pay Baxter royalties on future INTERCEPT Blood System product sales at royalty rates that vary by product: 10% of product sales for the platelet system, 3% for the plasma system, 5% for the red blood cell system, and 6.5% on sales of UVA illuminators. In March 2007, Baxter sold its Transfusion Therapies business, the unit of Baxter that has performed many of the manufacturing and supply chain activities related to our relationship with Baxter, to a new company, Fenwal, Inc., or Fenwal. Fenwal has assumed Baxter’s rights and obligations under our agreements.
BioOne
In June 2004, we and Baxter entered into a definitive agreement with BioOne for commercialization of our platelet system in specified parts of Asia. Under the terms of the 2004 agreement, BioOne was responsible for seeking regulatory approvals for and commercializing, the platelet system in Japan, China, Taiwan, South Korea, Thailand, Vietnam and Singapore. Under that agreement, BioOne received exclusive marketing and distribution rights in each of those countries. In 2007, Baxter transferred its rights and obligations with regard to BioOne to Fenwal. We received a total of $10.0 million in up-front payments under the terms of the 2004 agreement.
In June 2005, we and Baxter entered into a definitive agreement with BioOne for commercialization of our plasma system in specified parts of Asia. Under the terms of the 2005 agreement, BioOne was responsible for seeking regulatory approvals for and commercializing the plasma system in Japan, China, Taiwan, South Korea, Thailand, Vietnam and Singapore. BioOne received exclusive marketing and distribution rights in each of those countries. In 2007, Baxter transferred its rights and obligations with regard to BioOne to Fenwal. We received a total of $9.5 million in cash as well as equity securities in BioOne valued at $10.0 million at the time of issuance in connection with the 2005 agreement. At December 31, 2009, we evaluated the carrying value of our investment in BioOne using a variety of criteria. These criteria included, but were not limited to: third-party investor interest and participation in recent equity offerings at current pricing, business outlook of BioOne and available financial information. As a result of that analysis, we recorded a complete impairment of the carrying value of our equity interest in BioOne at December 31, 2009.
In August 2010, we completed an acquisition of certain assets of BioOne, including the commercialization rights that Baxter (later Fenwal) and we had granted to BioOne for both the platelet and plasma systems. Concurrently, Fenwal and we terminated the commercialization rights. As a consequence of the termination, and pursuant to a pre-existing agreement with Fenwal, our commercialization rights to the platelet and plasma systems under our 2005 and 2006 agreements with Baxter became worldwide. As consideration for the acquired BioOne assets, at the closing of the acquisition, we issued 937,886 shares of our common stock to BioOne and relinquished all of the shares we previously held in BioOne. In addition, six months from the closing date of the acquisition, we issued an additional 234,471 shares of our stock to BioOne. Accordingly, at December 31, 2010, we had recorded the fair value of the assets acquired, consisting of commercialization rights of $2.0 million, illuminators of $0.4 million and recorded goodwill for $1.3 million, which represents the buyer-specific value derived by Cerus as a result of having global commercialization rights for platelets and plasma.
6
Table of Contents
United States Armed Forces
In February 2001, we were awarded $2.6 million under a cooperative agreement with the Army Medical Research Acquisition Activity division of the Department of Defense, or DoD. Since then, we have been awarded an aggregate of $33.7 million under awards and cooperative agreements with the DoD, all of which were for the continued funding of projects to develop our pathogen inactivation technologies to improve the safety and availability of blood for medical transfusions. Under the terms of the cooperative agreements, we are conducting research on the inactivation of infectious pathogens in red blood cell units, including unusual viruses, bacteria and parasites, which are of concern to the United States Armed Forces.
Settlement Agreement with Baxter
During the fourth quarter of 2009, we and Baxter resolved several outstanding issues and disputes resulting from the 2006 transition services agreement and manufacturing agreement. As an outcome of those negotiations, on December 30, 2009, we and Baxter entered into a Mutual Release and Settlement Agreement, or the MRSA. The MRSA called for the complete and permanent waiver and release of any and all claims we or Baxter had on any amounts generated under the transition services agreement. As a result of entering into the MRSA, we eliminated approximately $4.7 million in payment obligations to Baxter and $2.8 million in receivables due from Baxter which were generated under the 2006 agreements and recorded on our balance sheet. The MRSA required us to pay $0.5 million to Baxter for the settlement. As such, we recorded a $1.4 million gain during the year ended December 31, 2009, and a $0.5 million obligation on our December 31, 2009 balance sheet. We paid the $0.5 million payment in satisfaction of the MRSA in January, 2010.
Investment in Aduro BioTech
In November 2007, we announced that we had sold certain assets that made up our former immunotherapy business to a newly-formed independent company, Anza Therapeutics, Inc., or Anza, financed by venture capital firms. In exchange for our contribution of tangible and intangible assets to Anza, we received preferred stock representing an equity interest of approximately 20% of Anza’s preferred equity. However, due to the early clinical and pre-clinical stages of Anza’s technology and relatively high risk of failure for such technologies, we determined that it would be unlikely that we would be able to derive any value from our equity interest in Anza and as such, we did not assign a value on our balance sheet to the equity interest we held in Anza. We were informed in February 2009 that Anza had ceased operations.
In July 2009, we entered into a three-way license agreement with Anza and Aduro BioTech, or Aduro, and separate agreements with each of Anza and Aduro (collectively, the Assignment Agreements). In November 2009, Anza transferred all of its intellectual property to Aduro pursuant to the terms of the Assignment Agreements. In addition, for agreeing to the transfer and surrendering our ownership in Anza, we received preferred stock representing a 10% equity interest in Aduro, a 1% royalty on all future product sales that Aduro may recognize in the future from the transferred technology, and $0.5 million in cash from Aduro. Furthermore, we received cash of approximately $0.3 million from Anza. As a result of entering into the Assignment Agreements, we no longer hold any equity in Anza. We believe that Aduro’s technology platforms, which are largely based on Anza’s in-process development programs, have a high risk of failure and we have no basis to believe that we will receive economic benefit from our equity ownership in Aduro. As such, we have not assigned any value to our equity ownership in Aduro.
William Greenman, our Senior Vice President and Chief Business Officer, is on the Board of Directors of Aduro. Mr. Greenman does not represent Cerus on Aduro’s Board of Directors.
Manufacturing and Supply
We have used, and intend to continue to use, third parties to manufacture and supply the devices, disposable kits and inactivation compounds that make up the INTERCEPT Blood System for use in clinical trials and for
7
Table of Contents
commercialization. We currently do not have alternate manufacturers for the components in our products beyond those that we currently rely on. We rely solely on Fenwal for the manufacture of INTERCEPT Blood System disposable kits and on contract manufacturers for the production of inactivation compounds, compound adsorption components of the disposable kits and UVA illumination devices used in the INTERCEPT Blood System.
In 2008, we entered into an Amended and Restated Manufacturing and Supply Agreement with Fenwal. Under the amended agreement, Fenwal is obligated to sell, and we are obligated to purchase, finished disposable kits for the platelet and plasma systems for both clinical and commercial use. The agreement permits us to purchase platelet and plasma kits from third-party manufacturers provided that we meet certain annual minimum purchase obligations to Fenwal. We are responsible for developing and delivering to Fenwal our proprietary inactivation compounds and adsorption media for incorporation into the final system configuration. The term of the Fenwal agreement extends through December 31, 2013, and is automatically renewed for one year terms, subject to termination by either party upon thirty months prior written notice, in the case of Fenwal, or twenty-four months prior written notice, in our case. We and Fenwal each have normal and customary termination rights, including termination for material breach.
We are responsible for the full management and control of the supply chain for the INTERCEPT illuminator devices and certain other components of the platelet and plasma kits. In anticipation of this obligation, we entered into a manufacturing and supply agreement with NOVA Biomedical Corporation, or NOVA, September , 2008. NOVA has manufactured illuminators for us in the past. The term of the NOVA agreement extends through September 2013 and is automatically renewable for one year terms, subject to termination by either party upon twelve months prior written notice.
We have contracted with one manufacturing facility for the synthesis of amotosalen, the inactivation compound used in our platelet and plasma systems. Under this contract, we are not subject to minimum annual purchase requirements. However, if specified quantities of amotosalen are not purchased in any year, we are required to pay a maintenance fee of up to $50,000 for such year. In the past, we have incurred these penalties.
We and our contract manufacturers, including Fenwal and NOVA, purchase certain raw materials for our disposable kits, inactivation compounds, materials and parts associated with compound adsorption devices and UVA illumination devices from a limited number of suppliers. Some of our suppliers require minimum annual purchase amounts. While we believe that there are alternative sources of supply for such materials, parts and devices, we have not validated or qualified any alternate manufacturers. As such, establishing additional or replacement suppliers for any of the raw materials, parts and devices, if required, will likely not be accomplished quickly and could involve significant additional costs and potential regulatory reviews.
Marketing, Sales and Distribution
The market for the INTERCEPT Blood System is dominated by a small number of blood collection organizations in the United States, Western Europe, the CIS countries, the Middle East, and Japan, where various national blood transfusion services or Red Cross organizations collect, store and distribute virtually all of their respective nations’ blood and blood component supplies. The largest European markets for our products are in Germany, France, and England. In Germany, decisions on product adoption are made on a regional or blood center-by-blood center basis. While obtaining CE marks allows us to sell the platelet and plasma systems to blood centers in Germany, blood centers in Germany must still obtain both local manufacturing approval and national marketing authorization from the Paul Ehrlich Institute before being allowed to sell platelets and plasma components treated with the INTERCEPT Blood System to transfusing hospitals and physicians. To date, several blood centers in Germany have received such requisite approvals and authorizations for the platelet system and certain other for the plasma system. In France, broad product adoption is dependent on a central decision by the Etablissement Francais du Sang, or EFS, and then on a broad-based national supply contract being awarded. In England, decisions on product adoption are centralized in the National Blood Service, and we understand that the National Blood Service has decided to implement bacterial detection before considering pathogen inactivation.
8
Table of Contents
Our ability to successfully commercialize our products will depend in part on the availability of adequate reimbursement for product costs and related treatment of blood components from governmental authorities and private health care insurers (including health maintenance organizations), which are increasingly attempting to contain health care costs by limiting both the extent of coverage and the reimbursement rate for new tests and treatments. National reimbursement rates for pathogen inactivated platelet and plasma treated with the INTERCEPT system have been agreed upon between the French Ministry of Health and the EFS; however, a robust budget for adopting pathogen inactivation technologies must be established before we would expect broad commercial adoption of the platelet and plasma systems in France.
We maintain a wholly-owned subsidiary, Cerus Europe B.V., headquartered in The Netherlands, which focuses its efforts on marketing and selling the INTERCEPT Blood System in a number of countries in Europe, the CIS, and the Middle East and selected countries in other regions around the world. We also have a small scientific affairs group in the United States and The Netherlands that supports the commercialization efforts.
Competition
We believe that the INTERCEPT Blood System has certain competitive advantages over competing blood-borne pathogen inactivation methods that are either on the market or in development. The INTERCEPT Blood System is designed for use in blood centers, which allows for integration with current blood collection, processing and storage procedures. Certain competing products currently on the market, such as solvent detergent-treated plasma, use centralized processing that takes blood products away from the blood center. CaridianBCT, or Caridian, has received a CE mark for a pathogen inactivation system for the treatment of platelets and plasma in blood centers. Other competitors, such as Octapharma AG and MacoPharma International are marketing pathogen inactivation products or systems for treating donated plasma in Europe. In addition to direct competition from other pathogen inactivation methods, we encounter indirect competition from other approaches to blood safety, including methods of testing blood products for bacterial and viral pathogens. Some of these indirect competitors have mature, well-established products and more resources than we have. Further discussion of the major competitors to our blood product business can be found in the risk factor entitled “If our competitors develop and market products that are more effective than our products and product candidates, our commercial opportunity will be reduced or eliminated. If competitors encounter difficulties or failures in human clinical trials or in commercial settings, we may face additional clinical, regulatory, and commercial challenges.”
We believe that the primary competitive factors in the market for pathogen inactivation of blood products include the breadth and effectiveness of pathogen inactivation processes, the amount of demonstrated reduction in transfusion related adverse events subsequent to adopting pathogen inactivation technology, robustness of treated blood components upon transfusion, ease of use, the scope and enforceability of patent or other proprietary rights, product value relative to perceived risk, product supply and marketing and sales capability. In addition, we believe the length of time required for products to be developed and to receive regulatory and, in some cases, reimbursement approval are also important competitive factors. We believe that the INTERCEPT Blood System will compete favorably with respect to these factors, although there can be no assurance that it will be able to do so. Our success will depend in part on our ability to respond quickly to medical and technological changes and customer demand through the development and introduction of new products.
Patents, Licenses and Proprietary Rights
Our success depends in part on our ability to obtain patents, to protect trade secrets, to operate without infringing upon the proprietary rights of others and to prevent others from infringing on our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing United States and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. As of December 31, 2010, we owned approximately 25 issued or allowed United States patents and approximately 74 issued or allowed foreign patents related to the INTERCEPT Blood
9
Table of Contents
System. Our patents expire at various dates between 2012 and 2027. In addition, we have pending United States patent applications and have filed corresponding patent applications under the Patent Cooperation Treaty. We have a license from Fenwal to United States and foreign patents relating to the INTERCEPT Blood System, which expire at various dates between 2016 and 2023. Proprietary rights relating to our planned and potential products will be protected from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents or are effectively maintained as trade secrets. The laws of certain foreign countries do not protect our intellectual property rights to the same extent as do the laws of the United States.
Seasonality
Our business is dependent on the marketing and commercialization of the INTERCEPT Blood System to customers such as blood banks, hospitals, distributors and other health care providers that have a need for a pathogen inactivation system to treat blood products for transfusion. Since our customer’s needs are not based on seasonal trends, seasonality does not have a material effect on our business.
Inventory Requirements and Product Return Rights
Our platelet and plasma systems have received regulatory approval for two-year shelf lives. Illuminators and replacement parts do not have regulated expiration dates. We own work-in-process inventory for certain components of INTERCEPT disposable kits, finished INTERCEPT disposable kits, illuminators, and certain replacement parts for our illuminators. Our supply chain for certain of these components, held as work-in-process on our balance sheet, can take over one year for production to be complete before being utilized in finished disposable kits. Inventory is recorded at the lower of cost or market value, determined on a first in, first out basis. We use significant judgment to analyze and determine if the composition of our inventory is obsolete, slow-moving, or unsalable. Our limited history selling the INTERCEPT Blood System limits the amount of historical data we have to perform such analysis. Generally, we write-down specifically identified obsolete, slow-moving, or known unsalable inventory using a number of factors, including product expiration dates, open and unfulfilled orders, and sales forecasts.
We sell the INTERCEPT Blood System directly to blood banks, hospitals, universities, and government agencies, as well as to distributors in certain regions. Generally, our contracts with our customers do not provide for open return rights, except within a reasonable time after receipt of goods in the case of defective product.
Customers
At December 31, 2010, we had three customers that each accounted for more than 10% of our outstanding trade receivables and collectively accounted for approximately 54% of our outstanding trade receivables. In addition, four customers, each accounting for more than 10% of our product sales, cumulatively represented 60% of our product revenue for the year ended December 31, 2010. The loss of any one of these customers would have an adverse impact on our business. To date, we have not experienced collection difficulties from these customers. See Note 17 of Item 15a “Financial Statements” of this Annual Report on Form 10-K for additional details about these customers.
Research and Development Expenses
A significant portion of our operating expenses is related to research and development and we intend to maintain our strong commitment to research and development. We have incurred total research and development expenses of $5.2 million, $6.4 million, and $10.2 million for the years ended December 31, 2010, 2009 and 2008, respectively. See Note 2 of Item 15a “Financial Statements” of this Annual Report on Form 10-K for costs and expenses related to research and development, and other financial information for fiscal years 2010, 2009 and 2008.
10
Table of Contents
Government Regulation
We and our products are comprehensively regulated in the United States by the FDA and, in some instances, by state and local governments, and by comparable governmental authorities in other countries.
Our European investigational plan has been based on the INTERCEPT Blood System being categorized as Class III drug/device combinations under the Medical Device Directives, or the MDD, of the European Union. The European Union requires that medical devices affix the CE mark, an international symbol of adherence to quality assurance standards and compliance with the MDD. The INTERCEPT Blood System for platelets received the CE mark in October 2002. The INTERCEPT Blood System for plasma received the CE mark in November 2006. A separate CE mark certification must be received for the red blood cell system to be sold in the European Union and in other countries recognizing the CE mark. In addition, France, Switzerland, Germany, and Austria have separate approval processes for use of the INTERCEPT-treated products.
The FDA regulates drugs, medical devices and biologics under the Federal Food, Drug, and Cosmetic Act and other laws, including, in the case of biologics, the Public Health Service Act. These laws and implementing regulations govern, among other things, the development, testing, manufacturing, record keeping, storage, labeling, advertising, promotion and pre-market clearance or approval of products subject to regulation. The steps required before a medical device may be approved for marketing in the United States pursuant to a pre-market approval application, or PMA, include:
| • | preclinical laboratory and animal tests: |
| • | submission to the FDA of an investigational device exemption for human clinical testing, which must become effective before human clinical trials may begin; |
| • | appropriate tests to show the product’s safety; |
| • | adequate and well-controlled human clinical trials to establish the product’s safety and efficacy for its intended indications; |
| • | submission to the FDA of a PMA; and |
| • | FDA review of the PMA in order to determine, among other things, whether the product is safe and effective for its intended uses. |
The FDA will require a PMA for each of the INTERCEPT systems for platelets, plasma and red blood cells because the FDA considers the INTERCEPT Blood System a biological medical device. The FDA Center for Biologics Evaluation and Research, or CBER, is principally responsible for regulating the INTERCEPT Blood System. However, before the FDA determines whether to approve our blood safety products, we expect our PMA to be reviewed by the Blood Products Advisory Committee, or BPAC, an advisory committee convened by and reporting to the FDA. Should the FDA ask questions to BPAC, we expect BPAC will answer those questions and make recommendations to the FDA.
In order to support our PMA for the INTERCEPT Blood System, we have conducted various types of studies, including toxicology studies to evaluate product safety, laboratory and animal studies to evaluate product effectiveness and human clinical trials to evaluate the safety, tolerability and effectiveness of treated blood components. Since assuming responsibility for regulatory approval of the INTERCEPT Blood System in the United States in 2006, we have used the same modular process for our PMA application for the platelet system that Baxter had used in the United States. The content, order and submission timing of the modules must be approved by the FDA, and a modular PMA application cannot be approved until all modules have been submitted to, reviewed by and accepted by the FDA.
We completed a Phase III clinical trial of the platelet system in the United States in March 2001 and a supplemental analysis of data from this trial in 2005. We submitted this information along with several other modules of our PMA, to the FDA. The FDA has indicated that the clinical trial data and supplemental analysis
11
Table of Contents
are not sufficient to support a PMA and we have had several interactions with the FDA subsequent to the clinical trial. In November 2009, we presented a plan for a proposed Phase III clinical trial for platelets to the BPAC. The outcome of that meeting was the support for the design and endpoints of a clinical trial for INTERCEPT-treated platelets, subject to changes regarding the sensitivity of the safety endpoint. We are currently in discussions with the FDA regarding further details of the proposed additional Phase III clinical trial. We plan to initiate such a trial only if adequate funding can be secured.
We have completed Phase IIIa, Phase IIIb and Phase IIIc clinical trials of the plasma system in the United States, reports for which were filed with the FDA during 2005. We have not submitted any applications for regulatory approval of the plasma system in the United States or any other regions other than Europe. INTERCEPT-treated plasma was recently granted orphan drug status for the treatment of thrombotic thrombocytopenic purpura. Although we have completed clinical trials in this patient population, the FDA may require us to complete additional clinical trials before approval would be granted. If additional clinical trials are required for approval, we will likely only initiate such trials if adequate funding can be secured.
The FDA inspects the facilities at which products are manufactured and will not permit clinical studies with a product or approve a product unless compliance with current Good Manufacturing Practice or Quality System Regulation requirements is satisfactory. The facilities of the principal third-party suppliers that manufacture our products are not currently FDA-qualified.
In addition to regulating our blood safety products, CBER also regulates the blood collection centers and the blood products that they prepare using the INTERCEPT Blood System. If our products were to be approved by the FDA, US-based blood centers will be required to obtain site-specific licenses prior to engaging in interstate transport of blood components processed using the INTERCEPT Blood System. Any delay in obtaining these licenses would adversely impact our ability to sell products in the United States.
We believe that, in deciding whether the INTERCEPT Blood System is safe and effective, regulatory authorities have taken, and are expected to take, into account whether it adversely affects the therapeutic efficacy of blood components as compared to the therapeutic efficacy of blood components not treated with the system. Data from human clinical studies must demonstrate the safety of treated blood components and their therapeutic comparability to untreated blood components. In addition, regulatory authorities will weigh the system’s safety, including potential toxicities of the inactivation compounds, and other risks against the benefits of using the system in a blood supply that has become safer. We have conducted many toxicology studies designed to demonstrate the INTERCEPT Blood System’s safety. There can be no assurance that regulatory authorities will not require further toxicology or other studies of our products. Based on discussions with the FDA and European regulatory authorities, we believe that data only from laboratory and animal studies, not data from human clinical studies, will be required to demonstrate the system’s efficacy in inactivating pathogens. In light of these criteria, our clinical trial programs for the INTERCEPT Blood System consist of studies that differ from typical Phase I, Phase II and Phase III clinical studies.
The INTERCEPT Blood System for red blood cells preclinical and clinical studies have been conducted using prototype system disposables and devices. We plan to perform a clinical trial utilizing an intended commercial configuration.
Further discussion of our regulatory and clinical trial status can be found in the risk factor entitled: Our products, blood products treated with the INTERCEPT Blood System and we are subject to extensive regulation by domestic and foreign authorities. If our preclinical and clinical data are not considered sufficient by a country’s regulatory authorities to grant marketing approval, we will be unable to commercialize our products and generate revenue in that country. Our red blood cell system requires extensive additional testing and development.
12
Table of Contents
Health Care Reimbursement and Reform
Our ability to commercialize our products successfully will depend in part on the extent to which appropriate reimbursement levels for the cost of the products and related treatment are obtained. The recent U.S. healthcare reform act and ongoing cost saving efforts may have an impact on our ability to profitably commercialize INTERCEPT in the United States and elsewhere.
Employees
As of December 31, 2010, we had 79 employees, 25 of whom were engaged in research and development and 54 in selling, general, and administrative activities. Of the 54 employees engaged in selling, general, and administrative activities, 26 were employed by our European subsidiary, Cerus Europe B.V. None of our employees are covered by collective bargaining agreements, and we believe that our relationship with our employees is good.
Available Information
We maintain a website at www.cerus.com; however, information found on our website is not incorporated by reference into this report. We make available free of charge on or through our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, or Exchange Act, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
Financial Information
Our financial information including our consolidated balance sheets, results of operations, statements of cash flows, statements of stockholder’s equity and the related footnotes, can be found under Item 15 in Part IV of this Annual Report on Form 10-K. Our financial information includes references to geographic areas.
| Item 1A. | Risk Factors |
Risk Factors
Our business faces significant risks. If any of the events or circumstances described in the following risks actually occurs, our business may suffer, the trading price of our common stock could decline and our financial condition or results of operations could be harmed. These risks should be read in conjunction with the other information set forth in this report. The risks and uncertainties described below are not the only ones facing us. There may be additional risks faced by our business. Other events that we do not currently anticipate or that we currently deem immaterial also may adversely affect our financial condition or results of operations.
The INTERCEPT Blood System may not achieve broad market acceptance.
We may encounter governmental and transfusion medicine community resistance to commercial adoption for any or all of our products. In addition to our customers, we must also address issues and concerns from broad constituencies involved in the healthcare system, from patients, to transfusing physicians, hospitals, private and public sector payors, regulatory bodies and public health authorities. We may be unable to demonstrate to these constituencies that the INTERCEPT Blood System is safe, effective and economical, or that the benefits of using the INTERCEPT Blood System products justify their cost.
Use of the platelet system results in some processing loss of platelets. If the loss of platelets leads to increased costs for our customers, our customers or prospective customers believe that the loss of platelet reduces the efficacy of the transfusion unit, or our process requires changes in blood center or clinical regimens, prospective customers may not adopt our platelet system. Certain studies have indicated that transfusion of conventionally prepared platelets may yield higher post-transfusion platelet counts (according to a measurement
13
Table of Contents
called “corrected count increment”) and may be more effective than transfusion of INTERCEPT-treated platelets. While studies also demonstrate that INTERCEPT-treated platelets retain therapeutic function comparable to conventional platelets, customers may choose not to adopt our platelet system due to considerations relating to corrected count increment or efficacy.
Our products do not inactivate all known pathogens, and the inability of our systems to inactivate certain pathogens may limit their acceptance. For example, due to the biology of certain non-lipid enveloped viruses, including the hepatitis A virus, our products have not been demonstrated to inactivate these viruses. In addition, for human parvovirus B-19, which is also a non-lipid-enveloped virus, our testing has not demonstrated a high level of inactivation. Although we have shown high levels of inactivation of a broad spectrum of lipid-enveloped viruses, some customers may choose not to adopt our products based on considerations concerning inability to inactivate, or limited inactivation, of certain non-lipid-enveloped viruses. Similarly, although our product has been demonstrated to effectively inactivate spore-forming bacteria, our products have not been shown to be effective in inactivating bacterial spores, once formed. In addition, since prions do not contain nucleic acid, our products do not inactivate prions. While transmission of prions has not been a major problem in blood transfusions, and we are not aware of any competing products that inactivate prions, the inability to inactivate prions may limit market acceptance of our products.
We have conducted pre-clinical and clinical studies of our products in both in vivo and in vitro environments using well-established tests that are accepted by regulatory bodies. When an in vitro test was not generally available or not well-established, we conducted in vivo studies in mammalian models to predict human responses. Although we have no reason to believe that the in vitro and in vivo studies are not predictive of actual results in humans, we cannot be certain that the results of these in vitro and in vivo studies accurately predict the actual results in humans in all cases. To the extent that actual results in human patients differs from the results of our in vitro or in vivo testing, market acceptance of our products may be negatively impacted.
Furthermore, due to limitations of detective tests, we cannot exclude that a sufficient quantity of pathogen or pathogens may still be present in active form which could present a risk of infection to the transfused patient. Such uncertainty may limit the market acceptance of our products.
If customers experience operational or technical problems with the use of INTERCEPT Blood System products, market acceptance may be reduced. For example, if adverse events arise from incomplete inactivation of pathogens, improper processing or user error, or if testing of INTERCEPT-treated blood samples fails to reliably confirm pathogen inactivation, whether or not directly attributable to the INTERCEPT Blood System, customers may refrain from purchasing the products. In addition, there is a risk that further studies we or others may conduct will show results inconsistent with previous studies. Should this happen, potential customers may delay or choose not to adopt our products, and existing customers may cease use of our products.
Market acceptance of our products is affected by blood center budgets and the availability of reimbursement from governments, managed care payors, such as insurance companies, and other third parties. In many cases, due to the structure of the blood products industry, we will have little control over budget and reimbursement discussions, which generally occur between blood centers and national or regional ministries of health and private payors. Even if a particular blood center is prepared to adopt the INTERCEPT Blood System, its hospital customers may not accept, or may not have the budget to purchase, INTERCEPT-treated blood products. Since blood centers would likely not eliminate the practice of screening donors or testing blood for pathogens prior to transfusion, even after implementing our products, some blood centers may not be able to identify enough cost offsets to afford to purchase our products. Budgetary concerns may be further exacerbated by the economic austerity programs implemented in European countries, which may limit the adoption of new technologies, including our products. Furthermore, it is difficult to predict the reimbursement status of newly approved, novel medical device products. In certain countries, governments have issued regulations relating to the pricing and profitability of medical products and medical products companies. The recently enacted health care reform laws in the United States and proposed restructuring of the Medicare and Medicaid systems have also placed downward pressure on the pricing of medical products.
14
Table of Contents
Product adoption in Europe and other regions may be negatively affected because we do not have Food and Drug Administration, or FDA, approval for any of our products. In addition, if we do not achieve widespread product adoption in key European countries, adoption in other countries may be affected.
The market for the INTERCEPT Blood System is highly concentrated with few customers, including often-dominant regional or national blood collection entities. Even if our products receive regulatory approval and reimbursement is available, failure to effectively market, promote, distribute, price or sell our products to any of these large customers could significantly delay or even diminish potential product revenue in those geographies. The market for our pathogen inactivation systems in the United States is highly concentrated, dominated by a small number of blood collection organizations. In many countries in Western Europe and in Japan, various national blood transfusion services or Red Cross organizations collect, store and distribute virtually all of their respective nations’ blood and blood components supply. In Europe, the largest markets for our products are in Germany, France, and England. In Germany, decisions on product adoption and subsequent reimbursement are expected to be on a regional or even blood center-by-blood center basis, but depend on both local approvals and centralized regulatory approvals from the Paul Ehrlich Institute, or PEI. Product characteristics relating to platelet dose of INTERCEPT-treated platelets that have received marketing authorization from the PEI may be incompatible with market requirements. Some potential customers may await further safety information or additional studies before choosing whether to adopt our products. Customers or prospective customers may conduct and complete their own clinical trials before adopting our products. For instance, we have been informed by the largest group of blood centers in Germany that it will complete a clinical trial before purchasing our products on a routine basis. We cannot predict the final trial design, number of transfusions, enrollment duration, estimated time it will take to complete such a trial, or trial outcome. While INTERCEPT-treated platelets and plasma have received in-country regulatory approval and reimbursement rates have been established in France, adoption throughout France has been limited to certain blood centers. Decisions on product adoption in England are centralized with the National Blood Service and we understand that the National Blood Service has decided to implement bacterial detection testing before considering pathogen inactivation. The Japanese Red Cross controls a significant majority of blood transfusions in Japan and exerts a high degree of influence on the adoption and use of blood safety measures in Japan. The Japanese Red Cross has been reviewing preclinical and clinical data on pathogen inactivation of blood over a number of years and has yet to make a formal determination to adopt any pathogen inactivation approach. Before the Japanese Red Cross considers our products, we understand that we may need to commit to making certain product configuration changes in order to allow the INTERCEPT Blood System to integrate with the collection platforms of the Japanese Red Cross.
Our products, blood products treated with the INTERCEPT Blood System and we are subject to extensive regulation by domestic and foreign authorities. If our preclinical and clinical data are not considered sufficient by a country’s regulatory authorities to grant marketing approval, we will be unable to commercialize our products and generate revenue in that country. Our red blood cell system requires extensive additional testing and development.
Our products, both those sold commercially and those under development are subject to extensive and rigorous regulation by local, state and federal regulatory authorities in the United States and by foreign regulatory bodies. These regulations are wide-ranging and govern, among other things:
| • | development; |
| • | testing; |
| • | manufacturing; |
| • | labeling; |
| • | storage; |
| • | pre-market clearance or approval; |
| • | sales and distribution; |
15
Table of Contents
| • | use standards and documentation; |
| • | post-launch surveillance; |
| • | quality; |
| • | advertising and promotion; and |
| • | reimbursement. |
Our products are in various stages of development and regulatory approval, and we face the risks of failure inherent in developing medical devices and biotechnology products based on new technologies. Our products must satisfy rigorous standards of safety and efficacy and we must adhere to quality standards regarding manufacturing and customer-facing business processes before the FDA and international regulatory authorities can approve them for commercial use. For our product candidates, we must provide the FDA and international regulatory authorities with preclinical, clinical and manufacturing data demonstrating that our products are safe, effective and in compliance with government regulations before the products can be approved for commercial sale. The process of obtaining FDA and other required regulatory approvals is expensive and uncertain, and typically takes a number of years. We may continue to encounter significant delays or excessive costs in our efforts to secure necessary approvals or licenses, or we may not be successful at all.
Clinical trials are particularly expensive and have a high risk of failure. Any of our product candidates may fail in the testing phase or may not achieve results sufficient to attain market acceptance, which could prevent us from achieving profitability. We do not know whether we will begin and conduct planned clinical trials on schedule, if at all. Significant delays in clinical testing could materially impact our clinical trials. Criteria for regulatory approval in blood safety indications are evolving with competitive advances in the standard of care against which new product candidates are judged, as well as with changing market needs and reimbursement levels. Clinical trial design, including enrollment criteria, endpoints, and anticipated label claims are thus subject to change, even if original objectives are being met. In addition to the reasons stated above, clinical trials can be delayed for a variety of reasons, including delays in obtaining regulatory approval to commence a study, delays in reaching agreement on acceptable clinical study agreement terms with prospective clinical sites, delays in obtaining institutional review board approval to conduct a study at a prospective clinical site and delays in recruiting subjects to participate in a study. We do not know whether any clinical trials will result in marketable products. Typically, there is a high rate of failure for product candidates in preclinical and clinical trials and products emerging from any successful trial may not reach the market for several years.
Enrollment criteria for certain of our clinical trials may be quite narrow. For instance, clinical trials previously conducted using INTERCEPT-treated plasma for patients with thrombotic thrombocytopenic purpura lasted approximately four years due in part, to the difficulties associated with enrolling qualified patients. Consequently, we may be unable to recruit suitable patients into clinical trials on a timely basis, if at all. We cannot rely on interim results of trials to predict their final results, and acceptable results in early trials might not be repeated in later trials. Any trial may fail to produce results satisfactory to the FDA or foreign regulatory authorities. In addition, preclinical and clinical data can be interpreted in different ways, which could delay, limit or prevent regulatory approval. Negative or inconclusive results from a preclinical study or clinical trial or adverse medical events during a clinical trial could cause a preclinical study or clinical trial to be repeated, require other studies to be performed or cause a program to be terminated, even if other studies or trials relating to a program are successful.
Outside the United States, regulations vary by country, including the requirements for approvals or clearance to market, the time required for regulatory review and the sanctions imposed for violations. In addition to CE mark documentation, countries outside the European Union may require clinical data submissions, registration packages, import licenses or other documentation.
In May 2007, we obtained a CE mark extension in our name from European Union regulators for our platelet system and will need to obtain an extension every five years. We or our customers may also be required
16
Table of Contents
to conduct additional testing in order to obtain regulatory approval in countries that do not recognize the CE mark as being adequate for commercializing the INTERCEPT Blood System in those countries. The level of additional product testing varies by country, but may be expensive or take a long time to complete. In addition, regulatory agencies are able to withdraw or suspend previously issued approvals.
We completed our Phase III clinical trial of the platelet system in the United States in March 2001 and submitted data from this trial, along with several other modules of our pre-market approval application, to the FDA. Based on discussions with the FDA, we performed an additional blinded analysis of the clinical trial data, under the direction of an independent expert physician panel, to determine if apparent differences between treatment groups in the category of pulmonary adverse events reported in the study were attributable to discrepancies in safety results. The reassessment of primary patient records by the expert physician panel showed no statistically significant differences between groups. This reassessment differed from the earlier analysis of adverse events that was based on clinical trial case report forms and had shown statistically significant differences in specific pulmonary events. We submitted a report of the analysis to the FDA for review. We understand that our reassessment of our previously completed Phase III clinical trial data will not be sufficient to address the FDA’s questions. In November 2009, we and the FDA presented a proposed clinical trial protocol for a second Phase III clinical trial to the FDA’s Blood Product Advisory Committee, or BPAC. Although the BPAC agreed with the proposed trial design, safety endpoints and efficacy endpoints, we believe we will need to reach agreement with the FDA on the means necessary to satisfy the BPAC’s request for more stringent safety margins than we had proposed. In order to meet the more stringent safety margins, we may need to enroll and collect data from more patients than what we had initially proposed to BPAC. Until the final study size and design requirements are determined, we will not be able to assess the feasibility of a second Phase III trial. The dimensions of such a Phase III trial may be prohibitive due either to prospective cost, availability of patients in the target population, or logistics. We have no plans to initiate such a trial unless adequate funding is secured. The additional Phase III clinical trial will need to be completed and data submitted to the FDA before we can complete our regulatory submission.
We obtained a CE mark approval in Europe for our plasma system in November 2006 and final French approval of INTERCEPT-treated plasma in May 2007. In February 2011, the first approval for use of INTERCEPT-treated plasma was obtained from the Paul Ehrlich Institute by a blood center in Germany. In some countries, including several in Europe, we or our customers may be required to perform additional clinical studies or submit manufacturing and marketing applications in order to obtain regulatory approval.
We have completed Phase IIIa, Phase IIIb and Phase IIIc clinical trials of the plasma system in the United States, reports for which were filed with the FDA during 2005. We have not submitted any applications for regulatory approval of the plasma system in the United States or any other regions other than in Europe. INTERCEPT-treated plasma was recently granted orphan drug status by the FDA for the treatment of thrombotic thrombocytopenic purpura. Although we have completed clinical trials in this patient population, the FDA may require us to complete additional clinical trials before approval would be granted. Should the FDA require us to complete additional clinical trials, we would need to secure adequate funding before we would initiate such a trial.
Before the FDA determines whether to approve the INTERCEPT Blood System products, we expect our approval applications to be reviewed by BPAC. Should the FDA ask BPAC questions, we expect BPAC to answer those questions and make recommendations to the FDA. Even if BPAC were to recommend approval of one or more of our products, the FDA would not necessarily have to approve those products. If BPAC were to answer FDA questions recommending against approval of one or more of our products, the FDA would have to take into consideration the points of concern raised by BPAC which could affect the approval of the products.
If our product candidates receive approval for commercial sale in the United States, their marketing and manufacturing will be subject to continuing FDA and other regulatory requirements, such as requirements to comply with Good Manufacturing Practice, or GMP, and ISO 13485, a quality management system standard applicable to the products we sell in Europe. The failure to comply with these requirements on an ongoing basis could result in delaying or precluding commercialization efforts in certain geographies, including the United
17
Table of Contents
States, and could result in an enforcement action, which could harm our business. The current manufacturing sites we rely upon for producing the platelet and plasma system products for international distribution and sale are not FDA-qualified facilities. It will require both time and expense to obtain such qualification.
The FDA will require, and other regulatory authorities may also require, a post-marketing clinical study, which can involve significant expense. Governments or regulatory authorities may impose new regulations or other changes or we may discover that we are subject to additional regulations that could further delay or preclude regulatory approval and subsequent adoption of our potential products. We cannot predict the impact of adverse governmental regulation that might arise from future legislative or administrative action.
We have conducted many toxicology studies to demonstrate the INTERCEPT platelet and plasma systems’ safety, and we have conducted and plan to conduct toxicology studies for the INTERCEPT red blood cell system throughout the product development process. At any time, the FDA and other regulatory authorities may require further toxicology or other studies to further demonstrate our products’ safety, which could delay commercialization. In addition, the FDA or foreign regulatory authorities may alter guidance at any time as to what constitutes acceptable clinical trial endpoints or trial design, which may necessitate a redesign of our product or proposed clinical trials and cause us to incur substantial additional expense or time in attempting to gain regulatory approval. We believe the FDA and other regulatory authorities are likely to weigh the potential risks of using our pathogen inactivation products against the incremental benefits, which may be difficult or impossible to quantify. We expect the FDA will require us to demonstrate a very low level of potential side effects in the proposed second Phase III trial of the platelet system.
As a result of the termination of Phase III clinical trials of our red blood cell system due to the detection of antibody reactivity to red blood cells treated with the INTERCEPT red blood cell system in two patients in the chronic arm of the trials, we have been conducting additional research and development activities on our red blood cell system to reduce the potential for antibody reactivity to treated red blood cells. Based upon an internal evaluation of the results from these additional research activities as well as additional in vitro and in vivo studies and after consulting with regulatory authorities, we initiated a new Phase I clinical trial in the fourth quarter of 2008 to test modifications to the red blood cell system. That new Phase I clinical trial was completed in early 2010, successfully meeting its primary endpoint of red cell recovery measured twenty-four hours after transfusion. In addition to red cell recovery, we also measured red cell lifespan, measured as the half-life of red cells circulating in transfusion recipients. INTERCEPT-treated red blood cells fell within the established normal reference range for red blood cells. Non-treated red cells were above the established normal reference range. Significantly lower lifespan for INTERCEPT-treated red blood cells compared to non-treated red blood cells may inhibit our ability to obtain the necessary regulatory approvals or may impair market acceptance if the red blood cell system is successfully developed. A number of trial design issues that could impact efficacy, regulatory approval and market acceptance will need to be resolved prior to the initiation of further clinical trials. If we are unsuccessful in advancing a modified red blood cell system through clinical trials, resolving process and product design issues or in obtaining subsequent regulatory approvals and acceptable reimbursement rates, we may never realize a return on our research and development expenses incurred to date in the red blood cell system program.
Regulatory delays can also materially impact our product development costs. If we experience delays in testing, conducting trials or approvals, our product development costs will increase.
Regulatory agencies may limit the uses, or indications, for which any of our products are approved. For example, we believe that the INTERCEPT Blood System products will be able to claim the inactivation of particular pathogens only to the extent we have laboratory data to support such claims. After regulatory approval for the initial indications, further studies may be necessary to gain approval for the use of the product for additional indications.
In addition to the regulatory requirements applicable to us and to our products, there are regulatory requirements in several countries around the world, including the United States, Germany, Canada, Austria, and Australia, and other countries, applicable to our prospective customers of INTERCEPT Blood System products,
18
Table of Contents
the blood centers that process and distribute blood and blood products. In those countries, blood centers and other customers are required to obtain approved license supplements from the appropriate regulatory authorities in each country before making available blood products processed with our pathogen inactivation systems to hospitals and transfusing physicians. Our customers may lack the resources or capability to obtain such regulatory approvals. These requirements or regulators’ delays in approving license applications or supplements may deter some blood centers from using our products. Blood centers that do submit applications or supplements for manufacturing and sale may face disapproval or delays in approval that could provide further delay or deter them from using our products. The regulatory impact on potential customers could slow or limit the potential sales of our products.
In August 2010, in connection with our acquisition of certain assets from BioOne, we regained the rights to commercialize the platelet and plasma systems in Japan, China, Taiwan, South Korea, Vietnam, Thailand, and Singapore. Regulatory authorities in these countries may require our platelet and plasma systems to be widely adopted commercially in Europe or approved by the FDA before the platelet and plasma systems are considered for approval.
We have limited experience operating a global commercial organization. We rely on third parties to market, sell, distribute and maintain our products and to maintain customer relationships in a certain countries.
We are responsible for sales, marketing, distribution, maintenance and regulatory support of the INTERCEPT Blood System worldwide. If we fail in our efforts to develop or maintain such internal competencies or establish acceptable relationships with third parties on a timely basis, our attempts to commercialize the INTERCEPT Blood System may be irreparably harmed.
We have a wholly-owned subsidiary, headquartered in the Netherlands, dedicated primarily to selling and marketing the platelet and plasma systems in geographies where the INTERCEPT platelet and plasma systems are approved or can be imported through the import license process. We will need to maintain and continue to increase our competence in a number of functions, including sales, marketing, regulatory, inventory and logistics, customer service, credit and collections, risk management, and quality assurance systems. Many of these competencies require compliance with European Union and local standards and practices, with which we have limited experience.
We have entered into contracts, generally on a geographically exclusive basis, with distributors in countries where we have limited abilities to commercialize our pathogen inactivation products directly. We have entered into geographical distribution agreements for distribution in a number of countries in Europe, the CIS countries, the Middle East and Australia. We rely on these distributors to obtain any necessary in-country regulatory approvals, market and sell the INTERCEPT Blood System, provide customer and technical product support, maintain inventories, and adhere to our quality system in all material respects, among other activities. While our contracts generally require distributors to exercise diligence, these distributors may fail to commercialize the INTERCEPT Blood System in their respective territories. They may fail to sell product inventory they have purchased from us to end customers. Initial purchases of UVA illuminators or disposable kits by these third parties may not lead to follow-on purchases of disposable platelet and plasma system kits. We have limited visibility into the identity and requirements of blood banking customers these distributors may have. Accordingly, we may be unable to ensure our distributors properly maintain illuminators sold or provide quality technical services to the blood banking customers to which they sell. Agreements with our distributors typically require the distributor to maintain quality standards that are compliant with standards generally accepted for medical devices. We may be unable to ensure that our distributors are compliant with such standards. Distributors may irreparably harm relationships with local existing and prospective customers and our standing with the blood banking community in general. We may have little recourse, short of termination, in the event that a distributor fails to execute according to our expectations and contractual provisions.
19
Table of Contents
Our manufacturing supply chain exposes us to significant risks
INTERCEPT platelet and plasma disposable kits are manufactured and assembled by Fenwal. Fenwal has agreed, through a supply agreement signed with us in December 2008, to manufacture disposable kits for the platelet and plasma systems for us through the end of 2013. After 2013, Fenwal may terminate the supply agreement, provided that Fenwal shall have provided us thirty months prior notice of termination. Fenwal is our sole supplier for manufacture of these products. Fenwal may fail to manufacture an adequate supply of disposable kits or to do so on a cost effective basis, which would subject us to loss of revenue and reduced contribution margin. We also have contracts with independent suppliers, including NOVA Biomedical Corporation, or NOVA, for the manufacture of UVA illuminators and certain components of the INTERCEPT Blood System which are manufactured or assembled at facilities not owned by Fenwal. These contractors are our sole suppliers for such components. NOVA has not manufactured UVA illuminators for a number of years. Should NOVA have difficulties manufacturing UVA illuminators, we may not be able to supply customer demand or provide replacement UVA illuminators to existing customers. Facilities at which the INTERCEPT Blood System or its components are manufactured may cease operations for planned or unplanned reasons, causing at least temporary interruptions in supply. We do not have qualified suppliers beyond those on whom we currently rely, and we understand that Fenwal relies substantially on sole suppliers of certain materials for our products. If we need to or choose to identify and qualify alternate suppliers, the process will be time consuming and costly. Even a temporary failure to supply adequate numbers of INTERCEPT Blood System components may cause an irreparable loss of customer goodwill. If we conclude that supply of the INTERCEPT Blood System or components from Fenwal and others is uncertain, we may choose to build and maintain inventories of raw materials, work-in-process components, or finished goods, which would consume capital resources and may cause our supply chain to be less efficient.
Some components of the UVA illuminator device are no longer manufactured, which will require us to identify and qualify replacement components and may require that we conduct additional studies, which could include clinical trials, to demonstrate equivalency or validate any required design or component changes. Future supply of illuminators is limited to availability of components, some of which are in short supply or are no longer manufactured. We will likely be required to redesign the illuminator used in the platelet and plasma systems to manage the risk of obsolete components. Such redesign may be expensive and lead to regulatory delays in obtaining approvals to market the redesigned device.
Fenwal manufactures our platelet and plasma systems in facilities that are not FDA-approved. In order to be used in clinical studies or sold in the United States, our products would be required to be manufactured in FDA-approved facilities. FDA validation of manufacturing facilities, whether owned by Fenwal or by other parties, will be costly and time-consuming.
If we attempt to establish alternate manufacturers, we will be dependent on Fenwal to transfer know-how relevant to the manufacture of the INTERCEPT Blood System; however, certain of Fenwal’s materials, manufacturing processes and methods are proprietary to Fenwal. We may be unable to establish alternate sources of supply to Fenwal, NOVA, or other suppliers without having to redesign certain elements of the platelet and plasma systems. Such redesign may be costly, time consuming and require further regulatory review. Fenwal is not obligated to provide support for development and testing of improvements or changes we may make to the INTERCEPT Blood System. We may be unable to identify, select, and qualify such manufacturers or those third parties able to provide support for development and testing activities on a timely basis or enter into contracts with them on reasonable terms, if at all. Raw material and component suppliers may not meet quality specifications we have set, which would cause a disruption in supply and may lead to lost sales and irreparable damage to our customer relationships. Moreover, the inclusion of components manufactured by new suppliers could require us to seek new or updated approvals from regulatory authorities, which could result in delays in product delivery. We may not receive any such required regulatory approvals.
In the event of a failure by Fenwal or other manufacturers to perform their obligations to supply components of the INTERCEPT Blood System to us, damages recoverable by us may be insufficient to compensate us for the
20
Table of Contents
full loss of business opportunity. Our supply agreements with Fenwal and NOVA, and supply agreements with others contain limitations on incidental and consequential damages that we may recover. A supplier’s potential liability in the event of non-performance may not be sufficient to compel the supplier to continue to act in conformity with our agreements.
Our product supply chain requires us to purchase certain components in minimum quantities and may result in a production cycle of more than one year. Significant disruptions to any of the steps in our supply chain process, may result in longer productions cycles which could lead to inefficient use of cash.
We are in the early stages of commercializing the INTERCEPT Blood System and may not accurately forecast demand for the INTERCEPT Blood System. As a result, we may carry excess work-in-process or finished goods inventory, which would consume capital resources and may become obsolete, or our inventory may be inadequate to meet customer demand. We have entered into certain public tenders, some which call for us to maintain certain minimum levels of inventory. If Fenwal or third-party manufacturers fail to produce components or our finished products satisfactorily, at acceptable costs, and in sufficient quantities, we may incur delays, shortfalls and additional expenses, or non-compliance with certain public tenders which may in turn result in permanent harm to our customer relations or loss of customers. Our platelet and plasma system disposables have received regulatory approval for two-year shelf lives. We and our distributors may be unable to ship product to customers prior to the expiration of product shelf life, which would require that we destroy or consume the outdated inventory in product demonstration activities. Product expiration may in turn lead to elevated product demonstration costs or reduced gross margins.
The platelet system is not compatible with some commercial platelet collection methods.
The equipment and materials used to collect platelets vary by manufacturer and by geographic region. Platelets may be collected from a single donor by apheresis using an automated collection machine. Apheresis devices currently used in the United States and European markets differ, among other characteristics, in their ability to collect platelets in reduced volumes of plasma. Platelet concentrates may also be prepared from whole blood by pooling together platelets from multiple donors. There are two commonly used methods for preparing whole blood platelets: the buffy coat method, which is used extensively in Europe, and the pooled random donor method, which is used in the United States. Our system for platelets is designed to work with platelets collected and stored in storage solutions, called Intersol and SSP+, and for platelets suspended in plasma.
In order to address the entire market in the United States, we would need to develop and test additional configurations of the platelet system. We estimate that the majority of platelets used in the United States are collected by apheresis, though a significant minority is prepared from pooled random donor platelets derived from whole blood collections. In order to gain regulatory approvals for a pathogen inactivation system compatible with random donor platelets, we will need to perform additional product development and testing, including additional clinical trials. Similarly, to achieve market acceptance in certain geographies, we may be required to design, develop and test new product configurations for the platelet and plasma systems. These development activities would increase our costs significantly, and may not be successful.
Other manufacturers supplying blood component collection platforms to the market may resist our efforts to make the INTERCEPT Blood System compatible with their platforms and may have competing pathogen inactivation technologies. Attaining compatibility with collection platforms manufactured by others may require adaptations to either the INTERCEPT Blood System or to the collection platforms, which may be difficult to engineer, expensive to implement and test, require additional clinical trials, cause delays in regulatory approval and/or be commercially unattractive to pursue. These development activities will increase our costs significantly, and may not be successful. Market acceptance of the INTERCEPT Blood System may be delayed until the system receives regulatory approval for use on such other equipment, if required.
21
Table of Contents
We have used prototype components in our preclinical studies and clinical trials of the INTERCEPT red blood cell system and have not completed the components’ commercial design. We will be required to identify and enter into agreements with third parties to manufacture the red blood cell system.
Our red blood cell system that was used in our preclinical studies and Phase I red blood cell trial was a prototype of the system to be used in the final products. As a result, we plan to perform additional preclinical and clinical studies using the commercial versions of the systems to demonstrate the acceptability of the commercial configuration and the equivalence of the prototypes and the commercial products, which may increase our expenses and delay the commercialization of our products. We may determine that the red blood cell system may not be commercially feasible from potential customers’ perspectives. If we fail to develop commercial versions of the INTERCEPT red blood cell system on schedule, our potential revenue would be delayed or diminished, and our potential competitors may be able to bring products to market before we do.
In addition, the design and engineering effort required to complete the final commercial product will likely be substantial and time-consuming. As with any complex development effort, we expect to encounter design, engineering and manufacturing issues. Such issues have previously arisen, sometimes unexpectedly, and solutions to these issues have not always been readily forthcoming. Additional unforeseen design, engineering and manufacturing issues may arise in the future, which could increase the development cost and delay commercialization of our products.
We will need to identify and contract with manufacturers who can develop processes to manufacture the compounds used in the red blood cell system. For commercial manufacturing, we will need to demonstrate to regulatory authorities that the commercial scale manufacturing processes comply with government regulations and that the compounds are equivalent to originally licensed compounds. It may be difficult or impossible to economically manufacture the red blood cell system on a commercial scale.
If our competitors develop and market products that are more effective than our products and product candidates, our commercial opportunity will be reduced or eliminated.
We expect our products to encounter significant competition. The INTERCEPT Blood System products compete with other approaches to blood safety currently in use, and may compete with future products that may be developed by others. Our success will depend in part on our ability to respond quickly to customer and prospective customer needs and medical and technological changes brought about by the development and introduction of new products. Competitors’ products or technologies may make our products obsolete or non-competitive before we are able to generate any significant revenue. In addition, competitors or potential competitors may have substantially greater financial and other resources than we have. They may also have greater experience in preclinical testing, human clinical trials and other regulatory approval procedures.
Several companies have, or are developing, technologies that are, or in the future may be, the basis for products that will directly compete with or reduce the market for our pathogen inactivation systems. A number of companies are specifically focusing on alternative strategies for pathogen inactivation in platelets and plasma. These alternative strategies may be more effective in inactivating certain types of pathogens from blood products, including non-lipid-enveloped pathogens, such as hepatitis A virus, which our products have not demonstrated an ability to inactivate, or human parvovirus B-19, for which our products have not demonstrated a high level of inactivation. While our products can effectively inactivate a broad spectrum of pathogens in blood components, including more robust inactivation of many pathogens than has been shown by other companies, market acceptance of our products may be reduced if customers determine that competitor’s products inactivate a broader range of pathogens that are of particular interest to the transfusion medicine community. In addition, customers and prospective customers may believe that our competitor’s products are safer or more cost effective than INTERCEPT Blood System products. In Europe, several companies, including Grifols S.A., Octapharma AG and MacoPharma International, are developing or selling commercial pathogen inactivation systems or services to treat fresh frozen plasma. CaridianBCT is developing a pathogen inactivation system for blood products and has been issued CE marks for a pathogen reduction system for both platelets and plasma. We
22
Table of Contents
understand that CaridianBCT has also conducted a clinical trial on a pathogen inactivation system for whole blood. Caridian’s product candidate, if successful, may offer competitive advantages over our INTERCEPT Blood System. Companies developing competing products may also offer and sell other blood-banking products and services. As a result, competitors may have pre-existing long-term relationships with customers and may be able to offer synergies for both pathogen inactivation and non-pathogen inactivation products that we are unable to offer.
New methods of testing whole blood for specific pathogens have been approved by the FDA and in Europe, as have tests for bacteria in platelets. Other companies are marketing rapid, point-of-care bacterial tests, and developing synthetic blood product substitutes and products to stimulate the growth of platelets. Development and commercialization of any of these or other related technologies could limit the potential market for our products.
We may be liable and we may need to withdraw our products from the market if our products harm people. We may be liable if an accident occurs in our controlled use of hazardous materials. Our insurance coverage may be inadequate to offset losses we may incur.
We are exposed to potential liability risks inherent in the testing and marketing of medical devices and pharmaceutical products. We may be liable if any of our products cause injury, illness or death. Although we will have completed rigorous preclinical and clinical safety testing prior to marketing our products, there may be harmful effects caused by our products that we are unable to identify in preclinical or clinical testing. In particular, unforeseen, rare reactions or adverse side effects related to long-term use of our products may not be observed until the products are in widespread commercial use. Because of the limited duration and number of patients receiving blood components treated with the INTERCEPT Blood System products in clinical trials, it is possible that harmful effects of our products not observed in clinical and preclinical testing could be discovered after a marketing approval has been received. For example, in cases where we have obtained regulatory approval for our products, we have demonstrated pathogen inactivation to specified levels based on well-established tests. However, there is no way to determine, after treatment by our products, whether our products have completely inactivated all of the pathogens that may be present in blood components. There is also no way to determine whether any residual amount of a pathogen remains in the blood component treated by our products, and there is no way to exclude that such residual amount would be enough to cause disease in the transfused patient. For ethical reasons, we cannot conduct human testing to determine whether an individual who receives a transfusion of a blood component containing a pathogen that was inactivated using the INTERCEPT Blood System might show positive results if tested for an antibody against that pathogen. While we believe, based on the clinical experience of our scientists, that the level of inactivated pathogens would likely be too small to induce a detectable antibody response in diagnostic tests, we cannot exclude that a transfused patient might show positive results if tested for an antibody against that pathogen. We could be subject to a claim from a patient that tests positive, even though that patient did not contract a disease. Later discovery of problems with a product, manufacturer or facility may result in additional restrictions on the product or manufacturer, including withdrawal of the product from the market. We are subject to risks and costs of product recall, which include not only potential out-of-pocket costs, but also potential interruption to our supply chain. In such an event, our customer relations would be harmed and we would incur unforeseen losses.
We maintain product liability insurance, but do not know whether the insurance will provide adequate coverage against potential liabilities. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products.
Our research and development activities involve the controlled use of hazardous materials, including certain hazardous chemicals, radioactive materials and infectious pathogens, such as HIV and hepatitis viruses. Although we believe that our safety procedures for handling and disposing of hazardous materials are adequate and comply with regulatory requirements, we cannot eliminate the risk of accidental contamination or injury. If an accident occurs, we could be held liable for any damages that result.
23
Table of Contents
If we fail to obtain the capital necessary to fund our future operations or if we are unable to generate positive cash flows from our operations, we will need to curtail planned development or sales and commercialization activities.
Our near-term capital requirements are dependent on various factors, including operating costs and working capital investments associated with commercializing the INTERCEPT Blood System, costs associated with planning and conducting studies and clinical development of our red blood cell system, costs associated with pursuing regulatory approval in geographies where we do not currently sell the INTERCEPT Blood System, timing and magnitude of payments under grants from the United States government, and costs related to creating, maintaining and defending our intellectual property. Our long-term capital requirements will also be dependent on competitive developments and regulatory factors. Until we are able to generate a sufficient amount of product revenue and generate positive net cash flows from operations, meeting our long-term capital requirements is in large part subject to access to public and private equity and debt capital markets, as well as to additional collaborative arrangements with partners or government grants, augmented by cash generated from operations and interest income earned on the investment of our cash balances and short-term investments. We believe that our available cash balances and access to credit under our growth capital facility will be sufficient to meet our capital requirements for at least the next twelve months. If our assumptions prove to be incorrect, we could consume our available capital resources sooner than we currently expect.
We have borrowed and in the future may borrow capital from institutional and commercial banking sources. Potential borrowings may include restrictive covenants, including covenants that restrict the operation of our business, liens on assets, high effective interest rates and repayment provisions that reduce cash resources and limit future access to capital markets. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. To the extent that we raise additional funds through collaboration or partnering arrangements, we may be required to relinquish some of our rights to product revenues, our technologies or rights to market and sell our products in certain geographies, or grant licenses on terms that are not favorable to us.
Our ability to raise additional capital may be adversely impacted by global, regional or national economic conditions. As a result of these and other factors, we do not know whether additional capital will be available when needed, or that, if available, we will be able to obtain additional capital on terms favorable to us or our stockholders. If we are unable to raise additional capital we may need to curtail planned development. We expect to prioritize continued commercialization of the platelet and plasma systems in Europe, the CIS, the Middle East and in selected countries in other regions around the world. In addition, we are developing and plan to perform the required clinical trials for product approval and commercialization of the red blood cell system. To the extent that we are able to find funding, we will seek regulatory approval of the platelet or plasma systems in the United States.
Historically, we had received significant awards in funding under cooperative agreements with the DoD. Further funding awarded under Federal grants and cooperative agreements for the INTERCEPT Blood Systems may decline when compared to historic levels. Access to Federal grants and cooperative agreements is subject to the authorization of funds and approval of our research plans by various organizations within the federal government, including the United States Congress. If we are unable to obtain Federal grant and cooperative agreement funding for future research and development activities at levels similar to past funding, we may need to reduce our operating expenses, which would delay progress in some of our development programs.
We have only a limited operating history, and we expect to continue to generate losses.
We may never achieve a profitable level of operations. Our development and selling, general, and administrative expenses have resulted in substantial losses each year since our inception with the exception of the year ended December 31, 2005. The platelet and plasma systems are not yet approved in the United States or in many other countries around the world. The red blood cell system is in clinical development and may never emerge from the clinical development stage as a marketed product. We may be required to reduce the sales price
24
Table of Contents
for our products in order to make our products economically attractive to our customers and to governmental and private payors, which may reduce or altogether eliminate our gross profit on sales. At our present sales levels of the platelet and plasma systems, our costs to manufacture, distribute, market, sell, support and administer the systems are in excess of revenue. Contribution from product sales is unlikely to exceed the costs we incur in research, development, and commercialization of the INTERCEPT Blood System for the near-term. We expect our losses to continue at least until the INTERCEPT Blood System achieves more significant market acceptance. To the extent that we reach agreement on a clinical pathway with the FDA for our platelet or plasma products and if we choose to pursue such opportunities, we would expect to incur substantial costs which could extend the period during which we expect to operate at a loss.
We have issued long-term notes payable containing certain covenants that we may be unable to comply with. Our operations may not provide sufficient cash to meet the repayment obligations of the note
On March 31, 2010, we entered into a growth capital credit agreement, or the Credit Agreement, for $10.0 million of which we immediately borrowed and issued a note payable for $5.0 million. In March 2011, we entered into an amendment of the Credit Agreement. The amended Credit Agreement and loan are secured by all of our U.S. assets, except for intellectual property. The amended Credit Agreement and note require that we comply with certain customary and routine covenants, including the requirement to meet growing revenue levels set at pre-established levels. For 2011, our revenues, on a trailing six-month basis, are required to be at least 80% of our projected revenues. For 2012 and beyond, our revenues are required to be at least €5.7 million per quarter. If we are unable to increase our product revenues to comply with the covenants in the amended Credit Agreement, the lender may call the note which would require us to repay the principal of the note sooner than we have anticipated. In the event that the note was called due to non-compliance with the covenants, we may be unable to pay back the principal which would allow the lender to liquidate collateralized assets. This in turn, would harm our business.
In addition, our operations may not reach the levels needed to meet the scheduled repayment obligations of the note. If we are unable to meet the scheduled repayment obligations of the note using our available cash, we may be forced to liquidate other assets, refinance the notes or issue equity securities to raise the necessary cash to meet our obligations. There is no assurance that we would be able to sufficiently or timely liquidate assets to meet the note’s repayment obligations or that we would be able to refinance the notes or issue equity, in which case our business would be significantly harmed and may force the Company into bankruptcy.
Our investment portfolio may become impaired by further deterioration of the capital markets.
Our cash equivalent and short-term investment portfolio as of December 31, 2010 consisted primarily of high credit, high liquidity United States government agency securities, asset backed securities, corporate debt securities, money market funds and interest-bearing accounts with financial institutions. We follow an established investment policy and set of guidelines to monitor, manage and limit our exposure to interest rate and credit risk.
As a result of adverse financial market conditions, investments in some financial instruments, such as structured investment vehicles, sub-prime mortgage-backed securities, auction rate securities and collateralized debt obligations, may pose risks arising from liquidity and credit concerns. We have limited holdings of these investments in our portfolio. We recognized other-than-temporary impairments of $0.04 million on our investment portfolio during the year ended December 31, 2010. The recent global economic crisis has had, and may continue to have, a negative impact on the market values of the investments in our investment portfolio. We cannot predict future market conditions or market liquidity and there can be no assurance that the markets for these securities will not deteriorate further or that the institutions that these investments are with will be able to meet their debt obligations at the time we may need to liquidate such investments or until such time as the investments mature.
25
Table of Contents
Virtually all of our research and development activities and the significant majority of our general and administrative activities are performed in or managed from a single site that may be subject to lengthy business interruption in the event of a severe earthquake. We also may suffer loss of computerized information and may be unable to make timely filings with regulatory agencies in the event of catastrophic failure of our data storage and backup systems.
Virtually all of our research and development activities and the significant majority of our general and administrative activities are performed in or managed from our facilities in Concord, California, which are within an active earthquake fault zone. Should a severe earthquake occur, we might be unable to occupy our facilities or conduct research and development and general and administrative activities in support of our business and products until such time as our facilities could be repaired and made operational. Our property and casualty and business interruption insurance in general does not cover losses caused by earthquakes. While we have taken certain measures to protect our scientific, technological and commercial assets, a lengthy or costly disruption due to an earthquake would have a material adverse effect on us. We have also taken measures to limit damage that may occur from the loss of computerized data due to power outage, system or component failure, or corruption of data files. However, we may lose critical computerized data, which may be difficult or impossible to recreate, which may harm our business. We may be unable to make timely filings with regulatory agencies in the event of catastrophic failure of our data storage and backup systems, which may subject us to fines or adverse consequences, up to and including loss of our abilities to conduct business.
We may not be able to protect our intellectual property or operate our business without infringing intellectual property rights of others.
Our commercial success will depend, in part, on obtaining and maintaining patent protection on our products and successfully defending our products against third-party challenges. Our technology will be protected from unauthorized use only to the extent that it is covered by valid and enforceable patents or effectively maintained as trade secrets. As a result, our success depends in part on our ability to:
| • | obtain patents; |
| • | protect trade secrets; |
| • | operate without infringing upon the proprietary rights of others; and |
| • | prevent others from infringing on our proprietary rights. |
We cannot be certain that our patents or patents that we license from others will be enforceable and afford protection against competitors. Our patents or patent applications, if issued, may be challenged, invalidated or circumvented. Our patent rights may not provide us with proprietary protection or competitive advantages against competitors with similar technologies. Others may independently develop technologies similar to ours or independently duplicate our technologies. For example, a United States patent issued to a third-party covers methods to remove psoralen compounds from blood products. We have reviewed the patent and believe there exists substantial questions concerning its validity. We cannot be certain, however, that a court would hold the patent to be invalid or not infringed by our platelet or plasma systems, if and when those products are sold in the United States. Our key patents generally expire at various dates between 2012 and 2027. Recent patent applications will, if granted, result in patents with later expiration dates. In addition, we have a license from Fenwal to United States and foreign patents relating to the INTERCEPT Blood System, which expire from 2016 to 2023. Due to the extensive time required for development, testing and regulatory review of our potential products, our patents may expire or remain in existence for only a short period following commercialization. This would reduce or eliminate any advantage of the patents.
We cannot be certain that we were the first to make the inventions covered by each of our issued patents or pending patent applications or that we were the first to file patent applications for such inventions. We may need to license the right to use third-party patents and intellectual property to continue development and commercialization of our products. We may not be able to acquire such required licenses on acceptable terms, if
26
Table of Contents
at all. If we do not obtain such licenses, we may need to design around other parties’ patents, or we may not be able to proceed with the development, manufacture or sale of our products.
Our patents do not cover all of the countries in which we are selling, and planning to sell, our products. We will not be able to prevent potential competitors from using our technology in countries where we do not have patent coverage.
We may face litigation to defend against claims of infringement, assert claims of infringement, enforce our patents, protect our trade secrets or know-how or determine the scope and validity of others’ proprietary rights. Patent litigation is costly. In addition, we may require interference proceedings before the United States Patent and Trademark Office to determine the priority of inventions relating to our patent applications. Litigation or interference proceedings could be expensive and time consuming, and we could be unsuccessful in our efforts to enforce our intellectual property rights.
We may rely, in certain circumstances, on trade secrets to protect our technology. However, trade secrets are difficult to protect. We protect our proprietary technology and processes, in part, by confidentiality agreements with employees and certain contractors. These agreements may be breached and we may not have adequate remedies for any breach or our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants or contractors use intellectual property owned by others, disputes also may arise as to the rights in related or resulting know-how and inventions.
As our international operations grow, we may be subject to adverse fluctuations in exchange rates between the United States dollar and foreign currencies. Consequently, we may suffer losses.
Our international operations are subject to risks typical of an international business, including, among other factors: differing political, economic, and regulatory climates, different tax structures, and foreign exchange volatility. We do not currently enter into any hedging contracts to normalize the impact of foreign exchange fluctuations. As a result, our future results could be materially affected by changes in these or other factors.
Product sales of our blood safety products are typically made in Europe and generally are invoiced to customers in Euros. In addition, we purchase finished disposable kits for our platelet and plasma systems and incur operating expenses in Euros and other foreign currencies. Our exposure to foreign exchange rate volatility is a direct result of our product sales, cash collection and expenses to support our international operations. Foreign exchange rate fluctuations are recorded as a component of interest (expense) and other, net on our consolidated statements of operations. Significant fluctuations in the volatility of foreign currencies relative to the United States dollar may materially affect our results of operations. Currently we do not have any near-term plans to enter into a formal hedging program to mitigate the effects of foreign currency volatility.
The market price of our stock may be highly volatile.
The market prices for our securities and those of other emerging medical device and biotechnology companies have been, and may continue to be, volatile. For example, during the period from January 1, 2008 to December 31, 2010, the sale price of our common stock as quoted on the Nasdaq Global Market fluctuated within a range from a low of $0.55 to a high of $7.29. Announcements may have a significant impact on the market price of our common stock. Such announcements may include:
| • | decisions regarding reimbursement and commercial adoption by customers, national blood services or governmental bodies; |
| • | biological or medical discoveries; |
| • | technological innovations discovered or new commercial services offered by us or our competitors; |
| • | developments concerning proprietary rights, including patents and litigation matters; |
| • | regulatory developments; |
27
Table of Contents
| • | status of development partnerships; |
| • | dilution from future issuances of common stock, including through the exercise of warrants and vested stock options; |
| • | debt financings, with terms that may not be viewed favorably by stockholders; |
| • | public concern as to the safety of new technologies; |
| • | general market conditions; |
| • | comments made by analysts, including changes in analysts’ estimates of our financial performance; and |
| • | quarterly fluctuations in our revenue and financial results. |
We may fail to comply fully with elements of the Sarbanes-Oxley Act of 2002. Our failure to maintain effective internal controls in accordance with Section 404 of this Act could have a material adverse effect on our stock price.
Section 404 of the Sarbanes-Oxley Act of 2002 requires annual management assessments of the effectiveness of our internal controls over financial reporting and a report by our independent registered public accountants attesting to the effectiveness of our internal controls. These requirements extend to the operations of our subsidiary in Europe. If we fail to maintain the adequacy of our internal controls over financial reporting, as such standards are modified, supplemented or amended from time to time, we may not be able to ensure that we can conclude in future periods that we have effective internal controls over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act of 2002. If we cannot favorably assess, or our independent registered public accountants are unable to provide an unqualified attestation report on the effectiveness of our internal controls over financial reporting, investor confidence in the reliability of our financial reports may be adversely affected, which could have a material adverse effect on our stock price.
Provisions of our charter documents, our stockholder rights plan and Delaware law could make it more difficult for a third party to acquire us, even if the offer may be considered beneficial by our stockholders.
Provisions of the Delaware General Corporation Law could discourage potential acquisition proposals and could delay, deter or prevent a change in control. The anti-takeover provisions of the Delaware General Corporation Law impose various impediments to the ability of a third party to acquire control of us, even if a change in control would be beneficial to our existing stockholders. In addition, Section 203 of the Delaware General Corporation Law, unless its application has been waived, provides certain default anti-takeover protections in connection with transactions between the company and an “interested stockholder” of the company. Generally, Section 203 prohibits stockholders who, alone or together with their affiliates and associates, own more than 15% of the subject company from engaging in certain business combinations for a period of three years following the date that the stockholder became an interested stockholder of such subject company without approval of the board or the vote of two-thirds of the shares held by the independent stockholders. Our board of directors has also adopted a stockholder rights plan, or “poison pill,” which would significantly dilute the ownership of a hostile acquirer. Additionally, provisions of our amended and restated certificate of incorporation and bylaws could deter, delay or prevent a third party from acquiring us, even if doing so would benefit our stockholders, including without limitation, the authority of the board of directors to issue, without stockholder approval, preferred stock with such terms as the board of directors may determine.
| Item 1B. | Unresolved Staff Comments |
None.
28
Table of Contents
| Item 2. | Properties |
We lease both laboratory and general administrative space in Concord, California. These facilities are utilized for our main office and blood safety research. In addition to the leased space in Concord, we lease selling and administrative offices in Amersfoort, the Netherlands. We believe that our current facilities will be adequate for the foreseeable future. The following table depicts the functional nature of our leases, size, location, and term of our leased space.
| Function |
Location |
Square Footage | Lease Expiration Date |
Expiration if Renewal Options Exercised |
||||||||
| Sales & Administrative |
Amersfoort, The Netherlands | 7,300 | January 2013 | December 2013 | ||||||||
| Corporate Offices and Laboratories – Blood Safety |
Concord, CA, USA | 36,029 | November 2019 | |||||||||
| Item 3. | Legal Proceedings |
None.
29
Table of Contents
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock is traded on the Nasdaq Global Market under the symbol “CERS.” The following table sets forth, for the periods indicated, the high and low sales prices for the common stock as reported by the Nasdaq Global Market:
| High | Low | |||||||
| Year Ended December 31, 2009: |
||||||||
| First Quarter |
$ | 1.12 | $ | 0.59 | ||||
| Second Quarter |
1.50 | 0.63 | ||||||
| Third Quarter |
3.57 | 0.81 | ||||||
| Fourth Quarter |
2.83 | 1.57 | ||||||
| Year Ended December 31, 2010: |
||||||||
| First Quarter |
$ | 3.14 | $ | 1.76 | ||||
| Second Quarter |
3.71 | 2.42 | ||||||
| Third Quarter |
4.01 | 2.80 | ||||||
| Fourth Quarter |
3.95 | 2.11 | ||||||
On February 25, 2011, the last reported sale price of our common stock on the Nasdaq Global Market was $3.49 per share. On February 25, 2011, we had approximately 170 holders of record of common stock. We have not paid dividends on our common stock and do not intend to pay cash dividends on our common stock in the foreseeable future.
30
Table of Contents
Performance Measurement Comparison (1)
The following graph shows the total stockholder return of an investment of $100 in cash (and the reinvestment of any dividends thereafter) on December 31, 2005 for (i) our common stock, (ii) the NASDAQ Biotechnology Stocks Index, (iii) the Amex Biotech Index, and (iv) the NASDAQ Stock Market (United States) Index. Our stock price performance shown in the graph below is based upon historical data and is not indicative of future stock price performance.
Comparison of 5-year Cumulative Total Return on Investment
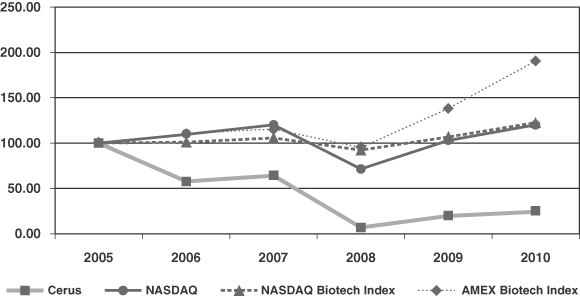
| December 31, | ||||||||||||||||||||||||
| 2005 | 2006 | 2007 | 2008 | 2009 | 2010 | |||||||||||||||||||
| Cerus Corporation |
$ | 100.00 | $ | 57.73 | $ | 64.14 | $ | 6.90 | $ | 19.61 | $ | 24.24 | ||||||||||||
| NASDAQ Biotech Index |
100.00 | 101.02 | 105.65 | 92.31 | 106.74 | 122.76 | ||||||||||||||||||
| Amex Biotech Index |
100.00 | 110.77 | 115.51 | 95.04 | 138.36 | 190.57 | ||||||||||||||||||
| NASDAQ |
100.00 | 109.52 | 120.27 | 71.51 | 102.85 | 120.29 | ||||||||||||||||||
| (1) | The graph and other information furnished under this Part II Item 5 of this Form 10-K shall not be deemed to be “soliciting material” or to be “filed” with the Commission or subject to Regulation 14A or 14C, or to the liabilities of Section 18 of the Exchange Act of 1934, as amended. |
31
Table of Contents
| Item 6. | Selected Financial Data |
The following table summarizes certain selected financial data for the five years ended December 31, 2010. The information presented should be read in conjunction with the financial statements and notes thereto and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K. The selected financial data for the periods prior to the financial statements included in this Annual Report on Form 10-K are derived from audited financial statements. The data presented below may not be indicative of future results.
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Statement of Operations Data:(1) |
||||||||||||||||||||
| Product revenue |
$ | 21,677 | $ | 16,751 | $ | 15,518 | $ | 8,015 | $ | 2,975 | ||||||||||
| Other revenue |
1,432 | 1,231 | 989 | 3,029 | 27,335 | |||||||||||||||
| Total revenue |
23,109 | 17,982 | 16,507 | 11,044 | 30,310 | |||||||||||||||
| Cost of product revenue |
12,046 | 12,580 | 9,668 | 5,228 | 1,541 | |||||||||||||||
| Gross profit |
11,063 | 5,402 | 6,839 | 5,816 | 28,769 | |||||||||||||||
| Operating expenses (gains): |
||||||||||||||||||||
| Research and development |
5,195 | 6,372 | 10,205 | 14,957 | 16,036 | |||||||||||||||
| Selling, general and administrative |
21,577 | 21,867 | 27,164 | 24,575 | 15,082 | |||||||||||||||
| Intangible asset amortization |
67 | — | — | — | — | |||||||||||||||
| Acquisition related costs and impairment of long-term investments in related parties, net2 |
182 | 1,536 | — | 9,450 | — | |||||||||||||||
| Settlement gain3 |
— | (1,381 | ) | — | — | — | ||||||||||||||
| Restructuring |
— | 841 | — | — | — | |||||||||||||||
| Total operating expenses |
27,021 | 29,235 | 37,369 | 48,982 | 31,118 | |||||||||||||||
| Loss from operations |
(15,958 | ) | (23,833 | ) | (30,530 | ) | (43,166 | ) | (2,349 | ) | ||||||||||
| Net interest and other income (expense) |
(953 | ) | (302 | ) | 1,349 | 4,066 | 4,701 | |||||||||||||
| Net income (loss) from continuing operations |
$ | (16,911 | ) | $ | (24,135 | ) | $ | (29,181 | ) | $ | (39,100 | ) | $ | 2,352 | ||||||
| Discontinued operations: |
||||||||||||||||||||
| Loss from discontinued operations |
— | — | — | (5,820 | ) | (7,131 | ) | |||||||||||||
| Loss from sale of discontinued operations |
— | — | — | (384 | ) | — | ||||||||||||||
| Net loss from discontinued operations |
— | — | — | (6,204 | ) | (7,131 | ) | |||||||||||||
| Net loss |
$ | (16,911 | ) | $ | (24,135 | ) | $ | (29,181 | ) | $ | (45,304 | ) | $ | (4,779 | ) | |||||
| Net income (loss) from continuing operations per common share: |
||||||||||||||||||||
| Basic |
$ | (0.42 | ) | $ | (0.69 | ) | $ | (0.90 | ) | $ | (1.23 | ) | $ | 0.09 | ||||||
| Diluted |
$ | (0.42 | ) | $ | (0.69 | ) | $ | (0.90 | ) | $ | (1.23 | ) | $ | 0.08 | ||||||
| Net loss from discontinued operations per common share: |
||||||||||||||||||||
| Basic |
$ | — | $ | — | $ | — | $ | (0.19 | ) | $ | (0.27 | ) | ||||||||
| Diluted |
$ | — | $ | — | $ | — | $ | (0.19 | ) | $ | (0.25 | ) | ||||||||
| Net loss per common share: |
||||||||||||||||||||
| Basic |
$ | (0.42 | ) | $ | (0.69 | ) | $ | (0.90 | ) | $ | (1.42 | ) | $ | (0.18 | ) | |||||
| Diluted |
$ | (0.42 | ) | $ | (0.69 | ) | $ | (0.90 | ) | $ | (1.42 | ) | $ | (0.17 | ) | |||||
| Weighted average common shares outstanding used for basic and diluted income (loss) per common share: |
||||||||||||||||||||
| Basic |
40,300 | 34,750 | 32,430 | 31,870 | 26,870 | |||||||||||||||
| Diluted |
40,300 | 34,750 | 32,430 | 31,870 | 28,610 | |||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 30,009 | $ | 19,931 | $ | 22,578 | $ | 56,850 | $ | 93,416 | ||||||||||
| Working capital |
22,052 | 19,446 | 29,145 | 55,582 | 87,929 | |||||||||||||||
| Total assets |
48,167 | 34,491 | 47,339 | 78,209 | 115,817 | |||||||||||||||
| Note payable, less current portion |
3,131 | — | — | — | — | |||||||||||||||
| Capital lease obligations, less current portion |
6 | 15 | — | 2 | 32 | |||||||||||||||
| Accumulated deficit |
(426,953 | ) | (410,042 | ) | (385,907 | ) | (356,726 | ) | (311,422 | ) | ||||||||||
| Total stockholders’ equity |
$ | 23,732 | $ | 21,448 | $ | 34,278 | $ | 59,887 | $ | 100,971 | ||||||||||
| (1) | Statement of operations data for 2007 and prior has been restated to reflect the treatment of our former immunotherapy business as a discontinued operation |
| (2) | Includes gain recognized in connection with the acquisition of certain assets of BioOne, impairment of Company investment in BioOne Corporation, and gain recognized from former immunotherapy business. See Footnote 1 to Consolidated Financial Statements under Part IV to this Annual Report on Form 10-K; |
| (3) | Settlement with Baxter regarding INTERCEPT commercialization transition. See Note 14 to Financial Statements in Part IV; |
32
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
This discussion and analysis should be read in conjunction with our consolidated financial statements and the accompanying notes included in this report and the audited consolidated financial statements and accompanying notes included in this Annual Report on Form 10-K for the year ended December 31, 2010. Operating results for the year ended December 31, 2010 are not necessarily indicative of results that may occur in future periods.
Overview
Since our inception in 1991, we have devoted substantially all of our efforts and resources to the research, development, clinical testing and commercialization of blood safety systems and, from 2001 until late 2007, immunotherapies for cancer and infectious disease. Our INTERCEPT platelet system, or the “platelet system,” and our INTERCEPT plasma system, or the “plasma system,” have received CE marks and are being marketed in a number of countries in Europe, the CIS, and the Middle East and selected countries in other regions around the world. In addition, we plan to continue development of our INTERCEPT red blood cell system. Subject to the availability of adequate funding from partners, government grants and/or capital markets, we also plan to pursue regulatory approval of the INTERCEPT platelet and plasma systems in the United States.
Our near-term capital requirements are dependent on various factors, including operating costs and working capital investments associated with commercializing the INTERCEPT Blood System, costs associated with planning and conducting studies and clinical development of our red blood cell system, costs associated with pursuing regulatory approval in geographies where we do not currently sell INTERCEPT, timing and magnitude of payments under grants from the United States government, and costs related to creating, maintaining and defending our intellectual property. Our long-term capital requirements will also be dependent on competitive developments and regulatory factors. Until we are able to generate a sufficient amount of product revenue and generate positive net cash flows from operations, meeting our long-term capital requirements is in large part subject to access to public and private equity and debt capital markets, as well as to additional collaborative arrangements with partners or government grants, augmented by cash generated from operations and interest income earned on the investment of our cash balances and short-term investments. We believe that cash received from product sales, our available cash balances, and available credit under our growth capital credit facility, will be sufficient to meet our capital requirements for at least the next twelve months. If our assumptions prove to be incorrect, we could consume our available capital resources sooner than we currently expect.
We may borrow additional capital from institutional and commercial banking sources to fund future growth on terms that may include restrictive covenants, including covenants that restrict the operation of our business, liens on assets, high effective interest rates and repayment provisions that reduce cash resources and limit future access to capital markets. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. To the extent that we raise additional funds through collaboration or partnering arrangements, we may be required to relinquish some of our rights to our technologies or rights to market and sell our products in certain geographies, or grant licenses on terms that are not favorable to us. The overall economic turmoil has generally made equity and debt financing more difficult to obtain and the terms less favorable to the companies seeking to raise financing. As a result of these and other factors, we do not know whether additional capital will be available when needed, or that, if available, we will be able to obtain additional capital on reasonable terms. If we are unable to raise additional capital due to the disruptions to the credit and financial markets in the United States and worldwide or other factors, we will need to curtail planned development and commercialization activities.
Historically, we had received significant awards in funding under cooperative agreements with the DoD for the INTERCEPT Blood System. Further funding awarded under Federal grants and cooperative agreements for the INTERCEPT Blood System may decline when compared to historic levels. Any such funding is subject to the authorization of funds and approval of our research plans by various organizations within the federal
33
Table of Contents
government, including the United States Congress. The general economic environment, coupled with tight Federal budgets, has led to a general decline in the amount of government funding. If we are unable to obtain Federal grant and cooperative agreement funding for the continued development of the INTERCEPT system in the United States at levels similar to past funding, we may need to reduce our operating expenses, which would delay progress in some of our development programs. We recognize growing, but still relatively modest, product revenues from the sale of our platelet and plasma systems in Europe, the CIS countries, the Middle East, and certain other countries around the world. We must conduct significant research, development, preclinical and clinical evaluation, commercialization and regulatory compliance activities for our product candidates that, together with anticipated selling, general and administrative expenses, are expected to result in substantial losses at least until after our platelet and plasma systems gain widespread commercial adoption in markets where our products are approved and in other regions around the world. Our ability to achieve a profitable level of operations in the future will depend on our ability to successfully commercialize and achieve market acceptance of our blood safety products. We may never achieve a profitable level of operations.
We pay royalties to Fenwal on product sales, at rates of 10% of net sales for the platelet system, 3% for the plasma system, 5% for the red blood cell system, and 6.5% on sales of UVA illuminators. In December 2008, we amended and extended our supply agreement with Fenwal for the manufacture of INTERCEPT finished disposable kits for the platelet and plasma systems through December 31, 2013. Under the amended manufacturing agreement, we pay Fenwal a set price per kit, which is established annually, plus a fixed surcharge per kit. In addition, volume driven manufacturing overhead will be paid or refunded if actual manufacturing volumes are lower or higher than the annually estimated production volumes. Under the amended manufacturing agreement, we are responsible for providing certain disposable kit components to Fenwal at no cost to Fenwal. This required us to enter into supply arrangements with certain other manufacturers for those components, some of which contain minimum purchase commitments. As a result, our supply chain for certain of these components, held as work-in-process on our consolidated balance sheet, can take over one year to complete production before being utilized in finished disposable kits.
In November 2007, we spun-off our immunotherapy business to Anza Therapeutics, Inc., or Anza for preferred stock representing and equity interest of approximately 20% of Anza’s preferred equity. We accounted for the immunotherapy business as a discontinued operation and restated our consolidated financial statements for 2007 and prior periods to reflect that accounting treatment. We were informed in February 2009 that Anza had ceased operations. In July 2009, we entered into a three-way license agreement with Anza and Aduro BioTech and separate agreements with each of Anza and Aduro BioTech (collectively, the Assignment Agreements). In November 2009, Anza transferred all of its intellectual property to Aduro BioTech, or Aduro, pursuant to the terms of the Assignment Agreements. In exchange for agreeing to the transfer and for relinquishing our shares in Anza and releasing any claims against Anza, we received $0.8 million in cash, preferred shares representing 10% of Aduro’s capital and a 1% royalty on any future sales resulting from the transferred technology. Because Aduro’s technology and efforts are in the very early stage of research and development, we have no basis to assign value to the equity we have received in Aduro or that such equity will have monetary value at such time we are allowed to sell it or that we will receive any royalties from Aduro.
In August 2010, we completed an acquisition of certain assets of BioOne, including the commercialization rights that Baxter (later Fenwal) and we had granted to BioOne for both the platelet and plasma systems. Concurrently with the acquisition, Fenwal and we terminated such commercialization rights. As a consequence of the termination, and pursuant to a pre-existing agreement with Fenwal, our commercialization rights to the platelet and plasma systems under our 2005 and 2006 agreements with Baxter became worldwide. As consideration for the acquired BioOne assets, at the closing of the acquisition, we issued 937,886 shares of our common stock to BioOne and relinquished all of the shares we previously held in BioOne. In addition, six months from the closing date of the acquisition, we issued an additional 234,471 shares of our stock to BioOne. Accordingly, at December 31, 2010, we had recorded the fair value of the assets acquired, consisting of commercialization rights of $2.0 million, and illuminators of $0.4 million with the excess of the purchase price over the fair value of the asset acquired being recorded as goodwill. The $1.3 million in goodwill represents the
34
Table of Contents
buyer-specific value derived by Cerus as a result of acquiring the commercialization rights in certain Asian countries in order to complete the global commercialization rights for platelets and plasma.
Through December 31, 2010, in addition to the product revenues from sales of our platelet and plasma systems, we have recognized revenue from grants and cooperative agreements with the Armed Forces.
Critical Accounting Policies and Management Estimates
The preparation of financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosures of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to revenue recognition, inventory valuation, accrued liabilities, valuation and impairment of purchased intangibles and goodwill, non-cash stock compensation assumptions, and income taxes. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form our basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from those estimates under different assumptions or conditions.
We believe the following critical accounting policies require us to make significant judgments and estimates used in the preparation of our financial statements:
•Revenue—We recognize revenue in accordance with the Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, Topic 605-25, “Revenue Recognition—Arrangements with Multiple Deliverables,” as applicable. Revenue is recognized when (i) persuasive evidence of an agreement with the funding party exists; (ii) services have been rendered or product has been delivered; (iii) pricing is fixed or determinable; and (iv) collection is probable.
Revenue related to product sales is generally recognized when we fulfill our obligations for each element of an agreement. For all sales of our INTERCEPT Blood System products, we use a binding purchase order and signed sales contract as evidence of a written agreement. We sell INTERCEPT Blood System for platelets and plasma directly to blood banks, hospitals, universities, government agencies, as well as to distributors in certain regions. Generally, our contracts with customers do not provide for open return rights, except within a reasonable time after receipt of goods in the case of non-conforming product. Deliverables and the units of accounting vary according to the provisions of each purchase order or sales contract. For revenue arrangements with multiple elements, we evaluate whether the delivered elements have stand-alone value to the customer, whether the fair value of the undelivered elements is reliably determinable, and whether the delivery of the remaining elements is probable and within our control. When all of these conditions are met, we recognize the revenue on the delivered elements. If these conditions are not met, we defer revenue until such time as all of the conditions have been met or all of the elements have been delivered. Consideration received is allocated to elements that are identified as discrete units of accounting based on the relative fair market value method. Freight costs charged to customers are recorded as a component of revenue and value-added-taxes, or VAT, that we invoice to our customers and remit to governments are recorded on a net basis, which excludes such VAT from product revenue.
Revenue related to the cost reimbursement provisions under development contracts is recognized as the costs on the projects are incurred. We receive certain United States government grants and contracts that support research in defined research projects. These grants generally provide for reimbursement of approved costs incurred as defined in the various grants. Revenue associated with these grants is recognized as costs under each grant are incurred.
•Inventory—We own work-in-process inventory for certain components of INTERCEPT disposable kits, finished INTERCEPT disposable kits, illuminators, and certain replacement parts for our illuminators. Our supply chain for certain of these components, held as work-in-process on our consolidated balance sheet, can
35
Table of Contents
take over one year to complete production before being utilized in finished disposable kits. Under our manufacturing agreement with Fenwal, our carrying value of INTERCEPT disposable kits is dependent on an annually set price. In addition, at the end of each year, volume driven manufacturing overhead is either paid or refunded by or to us if manufacturing volumes are higher or lower than the anticipated manufacturing volumes at the time the price is established. As a result, manufacturing overhead can fluctuate and requires us to use judgment in accruing the manufacturing overhead. In addition, we use judgment in determining whether the manufacturing overhead is a cost of our inventory and recoverable when product is sold. We use significant judgment and evaluate manufacturing variances incurred during periods of abnormally low production by considering a variety of factors including the reasons for low production volumes, anticipated future production levels that correlate to and offset volumes experienced during abnormally low production cycles, and contractual requirements. We record manufacturing variances incurred during periods of abnormally low production volumes as a component of cost of product revenue.
Inventory is recorded at the lower of cost, determined on a first in, first-out basis, or market value. Our platelet and plasma system disposable kits generally have a two-year shelf life from the date of manufacture. Illuminators and replacement parts do not have regulated expiration dates. We use significant judgment to analyze and determine if the composition of our inventory is obsolete, slow-moving, or unsalable and frequently review such determinations. Our limited history selling the INTERCEPT Blood System limits the amount of historical data we have to perform this analysis. Generally, we write-down specifically identified obsolete, slow-moving, or known unsalable inventory that has no alternative use, using a number of factors including product expiration dates, open and unfulfilled orders, and sales forecasts.
•Accrued expenses—We record accrued liabilities for expenses related to certain contract research activities and development services, including those related to clinical trials, preclinical safety studies and external laboratory studies, as well as transition services and development activities being performed by third parties. Some of those accrued liabilities are based on estimates because billings for these activities may not occur on a timely basis consistent with the performance of the services. Specifically, accruals for clinical trials require us to make estimates surrounding costs associated with patients at various stages of the clinical trial, pass through costs to clinical sites, contract research organization costs including fees, database development, and reporting costs, among others.
Purchased intangible assets and goodwill—We purchased certain assets from BioOne in 2010. For accounting purposes, we accounted for the acquisition as a business combination in accordance with ASC 805, “Business Combinations.” As a result of the acquisition, we used significant judgment in identifying the assets acquired and in determining the fair values to record the purchased assets on our balance sheet. Some areas requiring significant judgment include cash flows, discount rates, and economic lives. In addition, under ASC 805, we were required to assess the fair value of the non-controlling interest that we held in BioOne prior to the acquisition. We determined that a considerable amount of the purchase consideration was goodwill, which represents value unique to Cerus as the holder of worldwide rights to the INTERCEPT Blood System. We may be unable to realize the recorded value of the acquired assets and our assumptions may prove to be incorrect which may require us to write-down or impair the value of the assets if and when facts and circumstances indicate a need to do so. We will perform an impairment test on goodwill annually or when indicators of impairment exist.
Warrants—In 2009 and 2010, we issued warrants to purchase 2.4 million and 3.7 million shares of common stock, respectively. The material terms of the warrants were identical under each issuance except for the exercise price and date issued. The outstanding warrants are classified as a liability, and as such, the fair value of the warrants is recorded on the consolidated balance sheet. At each subsequent reporting period, the fair value of the warrants are adjusted with changes in fair value of the warrants reflected on the consolidated balance sheet and gains and losses recognized in the consolidated statements of operations as a component of non-operating income (expense). The fair value of the warrants is estimated using the binomial-lattice option-pricing model. This model requires that we use significant assumptions and judgment to determine appropriate inputs to the
36
Table of Contents
model. Some of the assumptions that we rely on include probability of a change of control occurring, the volatility of our stock over the life of the warrant, and assumptions and inputs used to value the warrants under the Black-Scholes model should a change of control occur.
•Stock-based compensation—We issue stock-based awards to our employees, members of our Board of Directors, our Scientific Advisory Board and certain contractors as strategic, long-term incentives. We recorded stock-based compensation expense for employee awards in accordance with ASC 505-50, “Compensation – Stock Compensation”. We use the Black-Scholes option pricing model to determine the grant-date fair value of a stock award. We continue to apply the provisions of “Accounting for Equity Instruments that are Issued to Other than Employees for Acquiring, or in Conjunctions with Selling, Goods or Services,” for our non-employee stock-based awards. Under the provisions, the measurement date at which the fair value of the stock-based award is measured is equal to the earlier of (i) the date at which a commitment for performance by the grantee to earn the equity instrument is reached or (ii) the date at which the grantee’s performance is complete. We recognize stock-based compensation expense for the fair value of the vested portion of the non-employee awards in our consolidated statements of operations.
The Black-Scholes option pricing model calculates the grant-date fair value using certain variables. These variables are impacted by our stock price, award exercise behaviors, the risk free interest rate and our expected dividends and many of these variables require us to use significant judgment.
Expected Term. We estimate the expected term of options granted using a variety of factors. Where possible, we estimate the expected term of options granted by analyzing employee exercise and post-vesting termination behavior. To make this estimation, we analyze the population of options granted by discrete homogeneous groups. For those homogeneous groups where we are unable to obtain sufficient information to estimate the expected term in this manner, we estimate the expected term of the options granted by taking the average of the vesting term and the contractual term of the option. The expected term of employee stock purchase plan shares is the term of each offering period.
Estimated Forfeiture Rate. We estimate the forfeiture rate of options at the time of grant and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. We use historical data to estimate pre-vesting option forfeitures and record stock-based compensation expense only for those awards that are expected to vest. We estimate the historic pre-vesting forfeiture rates by groups that possess a degree of homogeneity regarding average time to vest and expected term. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods.
Estimated Volatility. We estimate the volatility of our common stock by using historical volatility of our common stock. We have used significant judgment in making these estimates and we will continue to monitor the availability of actively traded options on our common stock. If we determine that sufficient actively traded options on our common stock exist, we may consider a combination of historical and implied volatility, or solely implied volatility.
Risk-Free Interest Rate. We base the risk-free interest rate that we use in the option valuation model on United States Treasury zero-coupon issues with remaining terms similar to the expected term on the options.
Expected Dividend. We do not anticipate paying any cash dividends in the foreseeable future and therefore use an expected dividend yield of zero in the option valuation model.
If factors change and we utilize different assumptions in determining the grant-date fair value of stock compensation expense in the future, or if we utilize a different option pricing model in the future, then those results may differ significantly from what we have recorded in the current period and could materially affect our operating results. There is significant risk that the Black-Scholes option pricing model and the judgment we have
37
Table of Contents
used in ascertaining the variables will yield results that differ materially from the actual values realized upon the exercise, expiration, termination or forfeitures of the awards in the future. Historical results were utilized in deriving our variables, which may not be indicative of the future.
•Income Taxes—Since our inception, we have accumulated significant net operating losses and research and development credits that may be used in future periods to offset future taxable income. We currently estimate that we may not be able to utilize all of our deferred tax assets. In addition, we may not generate future taxable income prior to the expiration of our net operating loss carry forwards and research and development credits. Timing and significance of any estimated future taxable income is highly subjective and is beyond the control of management due to uncertainties in market conditions, economic environments in which we operate, and timing of regulatory approval of our products. We do not recognize tax positions that have a lower than 50% likelihood of being recognized upon review by a taxing authority having full knowledge of all relevant information. Use of a valuation allowance is not an appropriate substitute for the derecognition of a tax position. We did not have any recorded liabilities for unrecognized tax benefits at December 31, 2010 or 2009. We recognize interest accrued and penalties related to unrecognized tax benefits in our income tax expense. To date, we have not recognized any interest and penalties in our statements of operations, nor have we accrued for or made payments for interest and penalties. We continue to carry a full valuation allowance on all of our deferred tax assets. Although we believe it more likely than not that a taxing authority would agree with our current tax positions, there can be no assurance that the tax positions we have taken will be substantiated by a taxing authority if reviewed.
Results of Operations
Years Ended December 31, 2010, 2009 and 2008
Revenue
| Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
||||||||||||||||||
| (in thousands, except percentages) |
2010 | 2009 | 2008 | |||||||||||||||||
| Product revenue |
$ | 21,677 | $ | 16,751 | $ | 15,518 | 29 | % | 8 | % | ||||||||||
| Government grant and cooperative agreements |
1,432 | 1,231 | 989 | 16 | % | 24 | % | |||||||||||||
| Total revenue |
$ | 23,109 | $ | 17,982 | $ | 16,507 | 29 | % | 9 | % | ||||||||||
Product revenue increased $4.9 million to $21.7 million during the year ended December 31, 2010, compared to $16.8 million during the comparable period in the prior year. This increase was largely driven by an increase in the number of disposable platelet and plasma system kits sold to customers in Europe, the CIS countries, and the Middle East. On a product basis, the increase was largely driven by an increase of $1.6 million in UVA illuminator sales and an increase of $3.3 million in sales of platelet and plasma disposable kits. Product revenue increased $1.2 million to $16.8 million during the year ended December 31, 2009, compared to $15.5 million during the comparable period in 2008. The increase was largely driven by an increase in the number of disposable platelet and plasma system kits sold to customers in Europe, the CIS, and the Middle East.
We anticipate product revenue for both the platelet and plasma systems will continue to increase in future periods as the INTERCEPT Blood System gains market acceptance in geographies where commercialization efforts are underway. Historical results may not be indicative of INTERCEPT Blood System revenue in the future.
We recognized $1.4 million in revenue from government grants and cooperative agreements for the year ended December 31, 2010, compared to $1.2 million for the comparable period in 2009. The increase was due primarily to an increase in the activities subject to reimbursement under new and existing awards with the United States Department of Defense, or DoD. We recognized $1.2 million in revenue from government grants and
38
Table of Contents
cooperative agreements for the year ended December 31, 2009, compared to $1.0 million for the comparable period in 2008. The increase was due primarily to a newly awarded DoD award for research activities for our INTERCEPT red blood cell system.
Cost of Product Revenue
Our cost of product revenue consists of the cost of the INTERCEPT Blood System inventory sold, royalties payable to Fenwal for product sales, provisions for obsolete, slow-moving and unsaleable product, certain order fulfillment costs, and to the extent applicable, costs for idle facilities. Inventory is accounted for on a first-in, first-out basis.
| (in thousands, except percentages) |
Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
|||||||||||||||||
| 2010 | 2009 | 2008 | ||||||||||||||||||
| Cost of product revenue |
$ | 12,046 | $ | 12,580 | $ | 9,668 | (4 | )% | 30 | % | ||||||||||
Cost of product revenue decreased $0.6 million to $12.0 million during the year ended December 31, 2010, compared to $12.6 million during the comparable period in 2009. Despite the higher volume of product sold, this decrease in the cost of product revenue was due to lower manufacturing overhead variances capitalized as a result of increased production volumes during 2010. In addition, we had lower costs for obsolete, slow moving and scrapped inventory during the year ended December 31, 2010, compared to the same period in 2009.
Cost of product revenue increased $2.9 million to $12.6 million during the year ended December 31, 2009, compared to $9.7 million during the comparable period in 2008. This increase in the cost of product revenue was primarily due to the larger number of disposable platelet and plasma system kits sold during the year ended December 31, 2009, compared to the number sold during the year ended December 31, 2008, and the increased royalties owed to Fenwal as a result of increased sales of both the platelet and plasma systems during the year ended December 31, 2009.
We anticipate that our cost of product revenue will increase in the future as we continue to increase product sales volume. Our realized gross margins on product sales were 44% in 2010, up from 25% in 2009, and up from 38% in 2008. The changes in our gross margins are affected by various factors, including manufacturing and supply chain costs, the mix of product sold, and the mix of customers to which product is sold. Generally, we offer our distributors tiered volume discounts of varying magnitudes, depending on their purchase commitments which depending on sales volumes to those distributors receiving tiered volume discounts, may impact our gross margins.
We expect to maintain inventory levels that will be sufficient to meet forecast demand for a relatively short time period and plan to manufacture at levels above those produced in 2010. Manufacturing at levels above the levels produced in 2010 should result in a continuing lower per unit cost of goods sold when the product is ultimately sold.
Research and Development Expenses
Our research and development expenses include salaries and related expenses for our scientific personnel, non-cash stock based compensation, payments to consultants, costs to prepare and conduct preclinical and clinical trials, third-party costs for development activities, certain regulatory costs, infrastructure, and laboratory chemicals and supplies.
| (in thousands, except percentages) |
Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
|||||||||||||||||
| 2010 | 2009 | 2008 | ||||||||||||||||||
| Research and development |
$ | 5,195 | $ | 6,372 | $ | 10,205 | (18 | )% | (38 | )% | ||||||||||
39
Table of Contents
Research and development expenses decreased $1.2 million to $5.2 million for the year ended December 31, 2010, compared to $6.4 million during the comparable period in 2009. This decrease in our research and development expenses was the result of reduced research and development activities driven primarily by our March 2009 restructuring plan and the associated reduction in force.
Research and development expenses decreased $3.8 million to $6.4 million for the year ended December 31, 2009, compared to $10.2 million during the comparable period in 2008. This decrease in our research and development expenses was the result of reduced research and development activities driven primarily by our March 2009 restructuring plan and the associated reduction in force.
Of the total research and development expenses incurred, non-cash stock-based compensation represented $0.4 million, $0.5 million, and $0.5 million for the years ended December 31, 2010, 2009 and 2008, respectively.
Research and development expenses may increase as we prepare for initiation of a Phase III clinical trial for our red blood cell system, or if we pursue regulatory approval of the platelet or plasma systems in the United States. Due to the inherent uncertainties and risks associated with developing biomedical products, including, but not limited to, intense and changing government regulation, uncertainty of future pre-clinical and clinical trial results and uncertainty associated with manufacturing, it is not possible to reasonably estimate the costs to complete these research and development projects. We face numerous risks and uncertainties associated with the successful completion of our research and development projects; see “Risk Factors” in Part I, Item 1A above.
Selling, General, and Administrative Expenses
Selling, general, and administrative expenses include salaries and related expenses for administrative personnel, non-cash stock based compensation, expenses for our commercialization efforts in Europe and elsewhere, expenses for accounting, tax, and internal control, legal and facility related expenses, and insurance premiums.
| Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
||||||||||||||||||
| (in thousands, except percentages) |
2010 | 2009 | 2008 | |||||||||||||||||
| Selling, general and administrative |
$ | 21,577 | $ | 21,867 | $ | 27,164 | (1 | )% | (20 | )% | ||||||||||
Selling, general, and administrative expenses decreased $0.3 million to $21.6 million for the year ended December 31, 2010, compared to $21.9 million during the comparable period in 2009. Overall, this decrease in selling, general and administrative expenses was primarily due to decreased personnel costs and lower marketing and public affairs costs driven primarily by our March 2009 restructuring plan and the associated reductions in force and as well as our continued emphasis on cost control.
Selling, general, and administrative expenses decreased $5.3 million to $21.9 million for the year ended December 31, 2009, compared to $27.2 million during the comparable period in 2008. Overall, this decrease in selling, general and administrative expenses was primarily due to decreased personnel costs and lower marketing and public affairs costs driven primarily by our March 2009 restructuring plan and the associated reductions in force.
Of the total selling, general, and administrative expenses incurred, non-cash stock-based compensation represented $1.5 million, $1.6 million, and $1.6 million for the years ended December 31, 2010, 2009 and 2008, respectively.
We anticipate that our selling, general, and administrative expenses will modestly increase over the next year as INTERCEPT market adoption broadens.
40
Table of Contents
Restructuring
Restructuring costs comprised of one-time termination benefits, facility consolidation and related moving costs.
| Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
||||||||||||||||||
| (in thousands, except percentages) |
2010 | 2009 | 2008 | |||||||||||||||||
| Restructuring |
$ | — | $ | 841 | $ | — | (100 | )% | 100 | % | ||||||||||
In March 2009, pursuant to the Board of Directors’ approval, we began implementing a plan to focus resources on commercializing the INTERCEPT Blood System in Europe, to consolidate facilities, and to reduce our cost structure. During the year ended December 31, 2009, we incurred costs for one-time termination benefits for employee positions that were eliminated under the restructuring plan. During the year ended December 31, 2009, we also incurred costs associated with consolidating facilities and certain other costs associated with the restructuring plan. Most of the costs accrued as one-time termination benefits as of March 31, 2009 were paid by December 31, 2009 and any remaining costs were paid by December 31, 2010.
Acquisition-Related Costs and Impairment of Long-term Investment in Related Parties, Net
During 2010, we acquired certain assets from BioOne Corporation and relinquished our non-controlling equity interest in them.
| Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
||||||||||||||||||
| (in thousands, except percentages) |
2010 | 2009 | 2008 | |||||||||||||||||
| Gain from long term investment in related party – Anza Therapeutics |
$ | — | $ | (804 | ) | $ | — | (100 | )% | 100 | % | |||||||||
| Acquisition related costs and impairment of long-term investment in related party – BioOne |
182 | 2,340 | — | (92 | )% | 100 | % | |||||||||||||
| Total |
$ | 182 | $ | 1,536 | $ | — | (88 | )% | 100 | % | ||||||||||
In early 2009, we owned equity in Anza Therapeutics which we carried at a zero cost basis. During 2009, Anza Therapeutics transferred all of its intellectual property to Aduro BioTech, or Aduro. In exchange for agreeing to the transfer and for relinquishing our shares in Anza and releasing any claims against Anza, we received $0.8 million in cash. As such, a gain of $0.8 million was recognized.
During the year ended December 31, 2010, we acquired certain assets from BioOne and relinquished our non-controlling interest. In connection with that transaction, we recorded a gain of $0.3 million relating to the relinquishment of our pre-acquisition non-controlling ownership in BioOne. We carried the investment at $0 on our balance sheet and upon closing, assigned a fair value to the investment of $0.3 million. In addition we incurred acquisition costs of $0.5 million related to our acquisition of certain assets of BioOne. During 2009, we recorded an impairment to the carrying value on our investment in BioOne of $2.3 million. The charge was taken as a result of our evaluation of the factors used to support our position in BioOne and the Company determined that the investment was not recoverable.
Gain on Operating Settlement
| Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
||||||||||||||||||
| (in thousands, except percentages) |
2010 | 2009 | 2008 | |||||||||||||||||
| Gain on operating agreement settlement |
$ | — | $ | 1,381 | $ | — | (100 | )% | 100 | % | ||||||||||
During 2009, we and Baxter resolved several outstanding issues and disputes resulting from the 2006 transition services agreement and manufacturing agreement. As an outcome of those negotiations, on December 30, 2009, we entered into a Mutual Release and Settlement Agreement (or the “MRSA”) with Baxter. The MRSA called for the complete and permanent waiver and release of any and all claims we or Baxter had on
41
Table of Contents
any amounts generated under the transition services agreement. As a result of entering into the MRSA, we eliminated approximately $4.7 million in payment obligations to Baxter and $2.8 million in receivables due from Baxter which were generated under the 2006 agreements and recorded on our balance sheet. The MRSA required us to pay $0.5 million to Baxter for the settlement. As such, we recorded a $1.4 million gain during the year ended December 31, 2009, and a $0.5 million obligation on our December 31, 2009 balance sheet. We paid the $0.5 million payment in satisfaction of the MRSA during the first quarter of 2010.
Non-Operating Income (Expense)
Non-Operating Income (Expense) consists of mark-to-market adjustments related to the calculated fair value of our outstanding warrants, foreign exchange gain (loss), interest charges incurred on our note payable, interest earned from our short-term investment portfolio, and other non-operating gains and losses.
| Years Ended December 31, | % Change 2010 to 2009 |
% Change 2009 to 2008 |
||||||||||||||||||
| (in thousands, except percentages) |
2010 | 2009 | 2008 | |||||||||||||||||
| Warrant liability revaluation gain |
$ | 39 | $ | 63 | $ | — | (38 | )% | 100 | % | ||||||||||
| Foreign exchange gain (loss) |
(816 | ) | (611 | ) | 507 | (34 | )% | (221 | )% | |||||||||||
| Interest expense |
(689 | ) | (10 | ) | (10 | ) | 6790 | % | 0 | % | ||||||||||
| Other income (expense), net |
513 | 256 | 852 | 100 | % | (70 | )% | |||||||||||||
| Total non-operating income (expense) |
$ | (953 | ) | $ | (302 | ) | $ | 1,349 | (216 | )% | (122 | )% | ||||||||
Warrant liability
Warrant liability revaluation was $0.04 million for the year ended December 31, 2010, compared to $0.06 million in expense during the comparable period in 2009. The smaller gain was primarily due to the change in the underlying stock price of the Company. Warrant liability revaluation was $0.06 million for the year ended December 31, 2009, compared to $0.00 million in expense during the comparable period in 2008. We issued warrants to purchase an aggregate of 2.4 million and 3.7 million shares of common stock, in August 2009 and November 2010, respectively, in connection with offerings of its common stock. The fair value of the warrants is estimated using the binomial-lattice option-pricing model. The warrants will continue to be reported as a liability until such time as the instruments are exercised or are otherwise modified to remove the provisions that require this treatment, at which time the warrants will be adjusted to fair value and reclassified from liabilities to stockholders’ equity. At December 31, 2010, no warrants had been exercised. If the warrants are reclassified from a liability to equity, the fair value of the warrants would be recorded in stockholders’ equity and no further adjustment would be made in subsequent periods. Future changes in stock price will result in similar adjustments as needed.
Foreign exchange gain (loss)
We recorded foreign currency losses of $0.8 million for the year ended December 31, 2010, compared to losses of $0.6 million for the comparable period of 2009. The increase of $0.2 million in foreign currency losses was primarily attributable to unfavorable foreign currency variances, primarily the Euro to U.S. dollar rates. We recorded foreign currency losses of $0.6 million for the year ended December 31, 2009, compared to a foreign currency gain of $0.5 million during the comparable period of 2008. The increase in foreign currency losses was also attributable to unfavorable foreign currency variations between the Euro and U.S. dollar, our functional currency.
Interest expense
Interest expense was $0.6 million for the year ended December 31, 2010, compared to an expense of $0.01 million for the comparable period of 2009. The increase in interest expense was due to interest incurred from
42
Table of Contents
borrowings on our credit facility which was entered into during the first quarter of 2010. Interest expense was $0.01 million of expense for the year ended December 31, 2009, compared to $0.01 million of expense during the comparable period in 2008. Prior to 2010, we did not incur significant interest expense.
Other income (expense), net
Other income (expense), net was income of $0.5 million for the year ended December 31, 2010, compared to $0.3 million of income during the comparable period in 2009. This increase in income was primarily due to income from two therapeutic tax credits received during 2010. Interest income and other, net, was $0.3 million for the year ended December 31, 2009, compared to $0.9 million of income during the comparable period in 2008. The decrease in income was primarily due to lower interest income resulting from lower cash and short-term investment balances and lower yields on those balances.
We expect to earn interest income at market rates in proportion to the marketable securities balances we maintain. We generally hold such investments until such time as we liquidate them to meet an operating cash need. Interest paid on our investment portfolio may decrease and the value of certain securities we hold may decline, which could negatively affect our financial condition, cash flow and reported earnings.
Liquidity and Capital Resources
In recent years, our sources of capital have primarily consisted of public offerings and private placements of equity securities, debt instruments, United States government grants and cooperative agreements, and contribution from product sales net of expenses and interest income.
At December 31, 2010, we had cash, cash equivalents and short-term investments of $30.0 million. Net cash used in operating activities was $14.1 million for the year ended December 31, 2010, compared to $14.5 million during 2009. The decrease in net cash used in operating activities was primarily due to higher revenues, improved gross margins and lower operating expenses, offset by changes in our operating assets and liabilities, notably increases in accounts receivable balances and lower accrued expenses. Cash used in operating activities during the year ended December 31, 2009, was $14.5 million compared to $34.0 million during the comparable period in 2008. The decrease in net cash used in operating activities was primarily due to changes in our operating assets and liabilities, notably decreases in our accounts receivable and inventory balances, partially offset by decreases in our accrued expenses. Net cash used in investing activities during the year ended December 31, 2010, was $0.1 million compared to $9.3 million of cash provided by investing activities during 2009. The decrease in cash provided from investing activities was primarily due to fewer maturities of short-term investments in 2010 compared to 2009 and higher purchases of furniture, equipment and leasehold improvements in 2010 compared to 2009. During 2010, we relocated our headquarters and capitalized leasehold improvements associated with the leasehold build-out. Net cash provided by investing activities during the year ended December 31, 2009 was $9.3 million compared to $23.4 million during 2008. The decrease in cash provided by investing activities was primarily due to fewer maturities and sales of short-term investments in 2009 compared to 2008. Cash provided by financing activities in 2010 was $25.9 million compared to $12.2 million during 2009. The increase in cash provided by financing activities was primarily due to higher cash proceeds received from common stock offerings and proceeds from the issuance of a note payable in 2010. Cash provided from financing activities was $12.2 million in 2009 compared to $1.3 million in 2008. The increase in cash provided from financing activities between these two periods was primarily due to the net cash proceeds received as a result of a registered direct offering in August of 2009. Working capital increased to $22.1 million at December 31, 2010, from $19.4 million at December 31, 2009. The increase in working capital is primarily due to higher cash, cash equivalents and short-term investments, accounts receivable, partially offset by decreases in inventory and increases in warrant liabilities and the current portion of long-term debt.
Our near-term capital requirements are dependent on various factors, including operating costs and working capital investments associated with commercializing the INTERCEPT Blood System, costs associated with planning and conducting studies and clinical development of our red blood cell system, costs associated with
43
Table of Contents
pursuing regulatory approval in geographies where we do not currently sell our platelet and plasma systems, timing and magnitude of payments under grants from the United States government, and costs related to creating, maintaining and defending our intellectual property. Our long-term capital requirements will also be dependent on competitive developments and regulatory factors. Until we are able to generate a sufficient amount of product revenue and generate positive net cash flows from operations, meeting our long-term capital requirements is in large part subject to access to public and private equity and debt capital markets, as well as to additional collaborative arrangements with partners or government grants, augmented by cash generated from operations and interest income earned on the investment of our cash balances and short-term investments. We believe that cash received from product sales, our available cash balances, and available credit under our growth capital credit facility, will be sufficient to meet our capital requirements for at least the next twelve months. If our assumptions prove to be incorrect, we could consume our available capital resources sooner than we currently expect.
We may borrow additional capital from institutional and commercial banking sources to fund future growth on terms that may include restrictive covenants, including covenants that restrict the operation of our business, liens on assets, high effective interest rates and repayment provisions that reduce cash resources and limit future access to capital markets. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. To the extent that we raise additional funds through collaboration or partnering arrangements, we may be required to relinquish some of our rights to our technologies or rights to market and sell our products in certain geographies, or grant licenses on terms that are not favorable to us. The overall economic turmoil has generally made equity and debt financing more difficult to obtain and the terms less favorable to the companies seeking to raise financing. As a result of these and other factors, we do not know whether additional capital will be available when needed, or that, if available, we will be able to obtain additional capital on reasonable terms. If we are unable to raise additional capital, we will need to curtail planned development and commercialization activities.
Historically, we had received significant awards in funding under cooperative agreements with the DoD for the INTERCEPT Blood System. Further funding awarded under Federal grants and cooperative agreements for the INTERCEPT Blood Systems may decline when compared to historic levels. Any such funding is subject to the authorization of funds and approval of our research plans by various organizations within the federal government, including the United States Congress. The general economic environment, coupled with tight Federal budgets, has led to a general decline in the amount of government funding. If we are unable to obtain Federal grant and cooperative agreement funding for the continued development of the INTERCEPT system in the United States at levels similar to past funding, we may need to reduce our operating expenses, which would delay progress in some of our development programs.
In late October 2008 we filed a shelf registration statement on Form S-3 to offer and sell up to $200.0 million of common stock, preferred stock, warrants, and/or debt securities. This shelf registration statement was declared effective by the SEC in December 2008. We have issued approximately $34.2 million of the securities registered for issuance pursuant to the shelf registration statement.
Commitments and Off-Balance Sheet Arrangements
Off-balance sheet arrangements
We do not have any off balance sheet arrangements as of December 31, 2010 or 2009.
44
Table of Contents
Commitments
Our commitments are as follows (in thousands):
| Total | Less than 1 year |
1-3 years |
4-5 years |
After 5 years |
||||||||||||||||
| Minimum purchase requirements |
$ | 2,208 | $ | 1,465 | $ | 743 | $ | — | $ | — | ||||||||||
| Operating leases |
2,302 | 719 | 1,161 | 422 | — | |||||||||||||||
| Other commitment |
1,554 | 395 | 310 | 287 | 562 | |||||||||||||||
| Long-term note payable |
6,290 | 2,182 | 4,108 | — | — | |||||||||||||||
| Total contractual obligations |
$ | 12,354 | $ | 4,761 | $ | 6,322 | $ | 709 | $ | 562 | ||||||||||
Our minimum purchase commitments include certain components of our INTERCEPT blood safety system that we purchase from third party manufacturers and supply to Fenwal for use in manufacturing finished disposable kits.
Operating leases
We lease our office facilities and certain equipment under non-cancelable operating leases with initial terms in excess of one year that require us to pay operating costs, property taxes, insurance and maintenance. On December 10, 2009, we exercised a ten year extension option to extend the term of our lease relating to 2550 Stanwell Drive in Concord, California. By exercising this extension option, our lease payments will be increased. Our facility leases qualify as operating leases under FASB ASC Topic 840, “Leases” and as such, are not included on our balance sheet.
Other commitments
Our other commitments consist of financing obligations for payment of certain insurance premiums that expire in 2011. In addition to the operating leases we have for office and laboratory space, certain of our leases provide for landlord-financed leasehold improvements. At December 31, 2010, we had financed $1.1 million of leasehold improvements. We pay for the financed leasehold improvements as a component of rent and are required to reimburse our landlords over the remaining life of the respective leases. Our Concord California lease may be canceled no earlier than December 2015, at which time we would be required to repay for any remaining portion of the landlord financed leasehold improvements.
Long-term note payable
On March 31, 2010, we entered into a growth capital facility agreement, under which we immediately borrowed and issued a senior secured long-term note payable for $5.0 million. Notes issued under the agreement are secured by all of our assets, except intellectual property. The note carries a fixed interest rate of 12.04%, with interest only payments for the first nine months and then equal principal and interest payments for an additional 30 months. In connection with issuing the note, we agreed to pay an upfront facility fee of $0.1 million and incurred closing costs of $0.1 million. The combined facility fee and closing costs have been recorded as a discount to the note payable and will be amortized as a component of interest expense using the effective interest method over the term of the note (discount is based on an implied interest rate of 13.84%). In addition, we agreed to pay a $0.4 million closing fee upon maturity of the note. The closing fee will be accreted to interest expense using the effective interest method over the life of the note.
Under the growth capital facility, subject to certain conditions including compliance with covenants, an additional $5.0 million was available to be drawn between September 30, 2010 and December 31, 2010. As of December 31, 2010, we had not drawn down on the additional $5.0 million, and we incurred a non-utilization fee of $0.1 million as a result. In March 2011, we entered into an amendment with the lender. Under the terms of the
45
Table of Contents
amendment, we may borrow an additional $5.0 million under the note payable through September 30, 2011. The terms of the additional $5.0 million note would be identical to the first note issued under the growth capital facility. We would not incur any additional upfront facility fees.
We are required to maintain compliance with certain customary and routine financial covenants. Additionally, the note requires us to generate minimum revenues at certain pre-established levels. Under the amendment, our 2011 revenues are required to be at least 80% of our projected revenues on a trailing six-month basis. For 2012 and beyond, our revenues are required to be at least €5.7 million per quarter. As of December 31, 2010 we were in compliance with financial covenants set forth in the growth capital facility.
Financial Instruments
We maintain an investment portfolio of various issuers, types and maturities. These securities are generally classified as available-for-sale and, consequently, are recorded on the balance sheet at fair value with unrealized gains or losses reported as a separate component of stockholders’ equity. Our investment policy is to manage our marketable securities portfolio to preserve principal and liquidity while maximizing the return on the investment portfolio to assist us in funding our operations. Unrealized gains at December 31, 2010, 2009 and 2008, totaled $0.1 million, $0.1 million, and $0.2 million, respectively.
We invest our cash, cash equivalents and short-term investments in a variety of financial instruments, consisting primarily of high credit, high liquidity United States government agency securities, corporate debt securities, money market funds and interest-bearing accounts with financial institutions. We maintain portfolio liquidity by ensuring that the securities have active secondary or resale markets. Certain of the investments in our portfolio are subject to general market risk and more specifically, the United States mortgage industry and financial institutions. As a result, during the years ended December 31, 2010, 2009 and 2008, we recognized other–than-temporary impairments for certain investments in our portfolio, totaling $0.04 million, $0.0 million and $0.3 million, respectively. See Note 3 of our “Notes to Consolidated Financial Statements” contained herein regarding the inputs used to determine the fair value of our investments. The current global economic crisis has had, and may continue to have, a negative impact on the market values of the investments in our investment portfolio. There can be no assurance that the markets for these securities will not deteriorate further or that the institutions that these investments are with will be able to meet their debt obligations at the time we may need to liquidate such investments or until such time as the investments mature.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Interest Rate Risk
Of our cash, cash equivalent, and short-term investments balance of $30.0 million at December 31, 2010, approximately 96% had original maturity dates of less than 90 days, and the remaining 4% had original maturities more than one year. We do not believe our exposure to interest rate risk to be material given the short-term nature of our investment portfolio and the relatively flat yields in high credit, fixed-income investments and the consistent yields we have experienced and anticipate experiencing across our portfolio.
Our exposure to market rate risk for changes in interest rates relates primarily to our investment portfolio. We do not use derivative financial instruments in our investment portfolio. By policy, we place our investments with high quality debt security issuers, limit the amount of credit exposure to any one issuer and limit duration by restricting the term for single securities and for the portfolio as a whole. Our investments are held and managed by a third-party capital management adviser that in turn, utilizes a combination of active market quotes and where necessary, proprietary pricing models as well as a subscribed pricing service, in order to estimate fair value.
We invest our cash, cash equivalents and short-term investments in a variety of financial instruments, consisting primarily of high credit, high liquidity United States government agency securities, corporate debt
46
Table of Contents
securities, money market funds and interest-bearing accounts with financial institutions. We maintain portfolio liquidity by ensuring that the securities have active secondary or resale markets. Certain of the investments in our portfolio are subject to general market risk and to a lesser extent, the United States mortgage industry. While we believe that we will be able to recognize the fair value of these instruments when they mature or we sell them, there can be no assurance that the markets for these securities will not deteriorate further or that the institutions that these investments are with will be able to meet their debt obligations.
We account for our short-term investments in accordance with ASC 320, “Investments – Debt and Equity Securities”. Our cash, cash equivalents and short-term investments are all recorded as current assets on our consolidated balance sheets as they are classified as available-for-sale. Securities with remaining maturities at purchase date of less than 90 days are classified as cash equivalents. The table below presents the amounts and weighted interest rates of our cash, cash equivalents and marketable securities at December 31, 2010 (dollar amounts in thousands):
| Fair Value | Weighted Average Interest Rate |
|||||||
| Cash and Cash equivalents (0 – 90 days(1)) |
$ | 28,948 | 0.10 | % | ||||
| Short-term investments (91 days – 1 year(1)) |
— | — | % | |||||
| Short-term investments (1 – 3 years(1)) |
1,061 | 4.68 | % | |||||
| Total investments |
$ | 30,009 | 0.26 | % | ||||
| (1) | Based on original contractual maturity date |
Foreign Currency Risk
Our international operations are subject to risks typical of an international business, including, among other factors: differing political, economic, and regulatory climates, different tax structures, and foreign exchange volatility. We do not currently enter into any hedging contracts to normalize the impact of foreign exchange fluctuations. As a result, our future results could be materially impacted by changes in these or other factors.
Product sales for our blood safety products are predominantly made in Europe and generally are invoiced to customers in Euros. In addition, we incur operating expenses, including payment for finished goods inventory of disposable kits for the platelet and plasma systems. These inventory purchases and operating expenses are generally paid in Euros and, to a much lesser degree, other foreign currencies. Our exposure to foreign exchange rate volatility is a direct result of our product sales, cash collection and expenses to support of our international operations. Foreign exchange rate fluctuations are recorded as a component of non-operating income (expense) on our consolidated statements of operations. Significant fluctuations in the volatility of foreign currencies relative to the United States dollar may materially impact our results of operations. Currently we do not have any near-term plans to enter into a formal hedging program to mitigate the effects of foreign currency volatility.
| Item 8. | Consolidated Financial Statements and Supplementary Data |
Our consolidated financial statements, together with related notes and reports of Ernst & Young LLP, independent registered public accounting firm, are listed in Item 15(a) and included herein.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures. Our chief executive officer and chief accounting officer are responsible for establishing and maintaining “disclosure controls and procedures” (as defined in Rule 13a-15(e) and Rule 15d-15(e), promulgated under the Securities Exchange Act of 1934, as amended) for our
47
Table of Contents
company. Based on their evaluation of our disclosure controls and procedures as of December 31, 2010, our chief executive officer and chief accounting officer have concluded that our disclosure controls and procedures were effective.
Changes in Internal Control over Financial Reporting. During the last quarter of our fiscal year ended December 31, 2010, there were no changes in our internal control over financial reporting during the period covered by this report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on the Effectiveness of Controls. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected. Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives, and the chief executive officer and chief accounting officer have concluded that these controls and procedures are effective at the “reasonable assurance” level.
Management’s Assessment of Internal Control. Management’s assessment of the effectiveness of our internal control over financial reporting as of December 31, 2010, is discussed in the Management’s Report on Internal Control over Financial Reporting included on page 54.
| Item 9B. | Other Information |
None.
48
Table of Contents
| Item 10. | Directors and Executive Officers of the Registrant |
Information regarding our directors and officers, and the compliance of certain reporting persons with Section 16(a) of the Securities Exchange Act of 1934, as amended, will be set forth under the captions “Election of Directors,” “Management,” “Section 16(a) Beneficial Ownership Reporting Compliance” and “Code of Ethics” in our definitive proxy statement, or proxy statement, for use in connection with the annual meeting of stockholders to be held on June 1, 2011, and is incorporated herein by reference. We intend to file the Proxy Statement with the Securities and Exchange Commission within 120 days after the end of our 2010 fiscal year.
| Item 11. | Executive Compensation |
The information required by this item is incorporated herein by reference to the information set forth under the caption “Executive Compensation and Other Information” in the proxy statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item is incorporated herein by reference to the information set forth under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in the proxy statement.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this item is incorporated herein by reference to the information set forth under the caption “Certain Related-Person Transactions” and “Proposal No. 1 – Election of Directors” in the proxy statement.
| Item 14. | Principal Accountant Fees and Services |
The information required by this item is incorporated herein by reference to the information set forth under the captions “Independent Registered Public Accounting Firm Fees and Services” and “Pre-Approval Policies and Procedures” in the proxy statement.
49
Table of Contents
| Item 15. | Exhibits and Financial Statement Schedules |
The following documents are being filed as part of this report on Form 10-K:
| (a) | Financial Statements. |
Other information is omitted because it is either presented elsewhere, is inapplicable or is immaterial as defined in the instructions.
| (b) | Exhibits. |
| Exhibit Number |
Description of Exhibit | |||
| 3.1.1 | (25) | Restated Certificate of Incorporation of Cerus Corporation, as amended to date. | ||
| 3.2 | (12) | Bylaws of Cerus Corporation. | ||
| 4.2 | (1) | Specimen Stock Certificate. | ||
| 4.3 | (21) | Stockholder Rights Plan, dated as of November 3, 1999, as amended as of August 6, 2001, between Cerus Corporation and Wells Fargo Bank, N.A. (formerly known as Norwest Bank Minnesota, N.A.). | ||
| 4.4 | (22) | Amendment to Rights Agreement, dated as of October 28, 2009, between Cerus Corporation and Wells Fargo Bank, N.A. (which includes the form of Rights Certificate as Exhibit B thereto). | ||
| 4.5 | (20) | Form of Registered Direct Common Warrant. | ||
| 4.6 | (27) | Form of Warrant to Purchase Common Stock | ||
| 10.1 | (1) | Form of Indemnity Agreement entered into between Cerus Corporation and each of its directors and executive officers. | ||
| 10.2 | (1)* | 1996 Equity Incentive Plan. | ||
| 10.3 | (1)* | Form of Incentive Stock Option Agreement under the 1996 Equity Incentive Plan. | ||
| 10.4 | (1)* | Form of Nonstatutory Stock Option Agreement under the 1996 Equity Incentive Plan. | ||
| 10.5 | (1)* | 1996 Employee Stock Purchase Plan Offering. | ||
| 10.6 | (1) | Industrial Real Estate Lease, dated October 1, 1992, between Cerus Corporation and Shamrock Development Company, as amended on May 16, 1994 and December 21, 1995. | ||
| 10.7 | (2) | Series B Preferred Stock Purchase Agreement, dated as of June 30, 1998, by and between Cerus Corporation and Baxter Healthcare Corporation. | ||
| 10.8 | (3)* | 1999 Equity Incentive Plan, adopted April 30, 1999, approved by stockholders July 2, 1999. | ||
50
Table of Contents
| Exhibit Number |
Description of Exhibit | |||
| 10.9 | (4)* | Amended and Restated Employment Agreement with Howard G. Ervin, dated December 22, 2008. | ||
| 10.10 | (5) | Lease, dated December 17, 1999 between Cerus Corporation and Redwoods Office Center, L.P. | ||
| 10.11 | (5) | Lease, dated October 12, 2001 between Cerus Corporation and California Development, Inc. (the “Lease”) | ||
| 10.12 | (8) | Second Amendment to Standard Industrial/Commercial Single-Tenant Lease-Net, dated as of September 18, 2008. | ||
| 10.13 | (23) | Letter to California Development, Inc. exercising option to extend Lease | ||
| 10.14 | (6) | Loan and Security Agreement, dated November 15, 2002, between Cerus Corporation and Baxter Capital Corporation. | ||
| 10.15 | (7)* | 1999 Non-Employee Directors’ Stock Option Sub-Plan, amended December 4, 2002. | ||
| 10.16 | (4)* | Amended and Restated Employment Agreement with Claes Glassell, dated December 19, 2008. | ||
| 10.17 | (22)† | Restructuring Agreement, dated as of February 2, 2005, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | ||
| 10.18 | (22)† | License Agreement, dated as of February 2, 2005, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | ||
| 10.19 | (9)† | Manufacturing and Supply Agreement, dated as of February 2, 2005, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | ||
| 10.20 | (24)* | Bonus Plan for Senior Management of Cerus Corporation, as amended March 3, 2010. | ||
| 10.21 | (10)† | Commercialization Transition Agreement, dated as of February 12, 2006, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | ||
| 10.22 | (11) | Offer Letter to Gail Schulze, dated October 15, 2007. | ||
| 10.23 | (13) | 2008 Equity Incentive Plan | ||
| 10.24 | (14) | Supply Agreement, dated December 19, 2007, by and between Cerus Corporation and Brotech Corporation d/b/a Purolite Company. | ||
| 10.25 | (14) | Supply and Manufacturing Agreement, dated March 1, 2008, by and between Cerus Corporation and Porex Corporation. | ||
| 10.26 | (15)† | Amended and Restated Manufacturing and Supply Agreement, dated December 12, 2008, by and between Cerus Corporation and Fenwal, Inc. | ||
| 10.27 | (15)† | Manufacturing and Supply Agreement, dated September 30, 2008, by and between Cerus Corporation and NOVA Biomedical Corporation. | ||
| 10.28 | (15)* | Non-Employee Director Compensation Policy. | ||
| 10.29 | (16)* | Cerus Corporation Change of Control Severance Benefit Plan, as amended. | ||
| 10.30 | (16)* | Form of Restricted Stock Unit Agreement under the 1999 Equity Incentive Plan, as amended. | ||
| 10.31 | (17)* | Form of Indemnity Agreement, adopted April 24, 2009. | ||
| 10.32 | (18)* | Employment Letter for Kevin D. Green dated May 1, 2009. | ||
| 10.33 | (19)* | Form of Severance Benefits Agreement. | ||
| 10.34 | (24)* | Employment Letter, by and between Cerus Corporation and Laurence Corash, dated March 2, 2010. | ||
51
Table of Contents
| Exhibit Number |
Description of Exhibit | |||
| 10.35 | (20) | Form of Subscription Agreement. | ||
| 10.36 | (28)†† | Loan and Security Agreement, by and between Cerus Corporation and Oxford Finance Corporation, dated March 31, 2010. | ||
| 10.37 | (26)† | Asset Purchase and Redemption Agreement by and between Cerus Corporation and BioOne Corporation, dated as of August 24, 2010. | ||
| 12.1 | Computation of Earnings to Fixed Charges Ratio(28) | |||
| 21.1 | List of Registrant’s subsidiaries. | |||
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |||
| 24.1 | Power of Attorney (see signature page). | |||
| 31.1 | Certification of the Chief Executive Officer of Cerus Corporation pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |||
| 31.2 | Certification of the Chief Accounting Officer of Cerus Corporation pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |||
| 32.1 | Certification of the Chief Executive Officer and Chief Accounting Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |||
| † | Certain portions of this exhibit are subject to a confidential treatment order. |
| †† | Registrant has requested extension of confidential treatment for portions of this exhibit. |
| * | Compensatory Plan. |
| (a) | Previously filed. |
| (1) | Incorporated by reference to Cerus’ Registration Statement on Form S-1 (File No. 333-11341) and amendments thereto. |
| (2) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on July 22, 1998. |
| (3) | Incorporated by reference to Cerus’ Registration Statement on Form S-8, dated August 4, 1999. |
| (4) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on December 23, 2008. |
| (5) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2001. |
| (6) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2002. |
| (7) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2003. |
| (8) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended September 30, 2008. |
| (9) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2005. |
| (10) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2006. |
| (11) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2007. |
| (12) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on June 19, 2008. |
| (13) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on June 6, 2008. |
| (14) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2008. |
| (15) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2008. |
| (16) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2009. |
| (17) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on April 30, 2009. |
| (18) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended June 30, 2009. |
| (19) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on June 1, 2009. |
| (20) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on August 20, 2009. |
| (21) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended June 30, 2009. |
| (22) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on October 30, 2009. |
52
Table of Contents
| (23) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2009. |
| (24) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on March 8, 2010. |
| (25) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended June 30, 2010. |
| (26) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on August 30, 2010. |
| (27) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on November 12, 2010. |
| (28) | Filed herewith. |
53
Table of Contents
MANAGEMENT’S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Management is responsible for establishing and maintaining effective internal control over the Company’s financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended. Management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2010. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control—Integrated Framework. Based on this assessment, management has concluded that, as of December 31, 2010, the Company’s internal control over financial reporting is effective.
The Company’s independent registered public accounting firm, Ernst & Young LLP, has audited the effectiveness of internal control over financial reporting as of December 31, 2010. Ernst and Young’s attestation report on internal control over financial reporting is included herein.
The Company’s internal control system was designed to provide reasonable assurance to the Company’s management and Board of Directors regarding the preparation and fair presentation of published financial statements. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Accordingly, our internal control systems are designed to provide reasonable, not absolute, assurance that the objectives of our internal control systems are met and, as set forth above, our principal executive officer and principal financial officer have concluded, based on their evaluation as of the end of the period covered by this report, that our internal control over financial reporting was effective. To provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with generally accepted accounting principles, we continue to implement, improve and refine our disclosure controls and procedures and our internal control over financial reporting.
54
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Cerus Corporation
We have audited Cerus Corporation’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Cerus Corporation’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Cerus Corporation maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Cerus Corporation as of December 31, 2010, and 2009, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2010, and our report dated March 16, 2011, expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
San Jose, California
March 16, 2011
55
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Cerus Corporation
We have audited the accompanying consolidated balance sheets of Cerus Corporation as of December 31, 2010, and 2009, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Cerus Corporation at December 31, 2010, and 2009, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with United States generally accepted accounting principles.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Cerus Corporation’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organization of the Treadway Commission and our report dated March 16, 2011 expressed an unqualified opinion thereon.
/s/ ERNST & YOUNG LLP
San Jose, California
March 16, 2011
56
Table of Contents
CONSOLIDATED BALANCE SHEETS
(in thousands, except per share amounts)
| 2010 | 2009 | |||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 28,948 | $ | 17,287 | ||||
| Short-term investments |
1,061 | 2,644 | ||||||
| Accounts receivable, net of allowance of $51 and $66 at December 31, 2010 and 2009, respectively |
4,792 | 3,625 | ||||||
| Inventories |
5,957 | 7,707 | ||||||
| Prepaid and other current assets |
997 | 1,096 | ||||||
| Total current assets |
41,755 | 32,359 | ||||||
| Property and equipment, net |
2,390 | 1,217 | ||||||
| Goodwill |
1,316 | — | ||||||
| Purchased intangibles, net |
1,950 | — | ||||||
| Restricted cash |
305 | 332 | ||||||
| Other assets |
451 | 583 | ||||||
| Total assets |
$ | 48,167 | $ | 34,491 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 3,230 | $ | 4,423 | ||||
| Accrued liabilities |
6,003 | 5,286 | ||||||
| Accrued restructuring |
— | 113 | ||||||
| Warrant liability |
8,465 | 2,737 | ||||||
| Deferred revenue |
248 | 345 | ||||||
| Current portion of long-term debt |
1,747 | — | ||||||
| Current portion of capital lease obligations |
10 | 9 | ||||||
| Total current liabilities |
19,703 | 12,913 | ||||||
| Long-term debt |
3,131 | — | ||||||
| Long-term portion of capital lease obligations |
6 | 15 | ||||||
| Other non-current liabilities |
1,595 | 115 | ||||||
| Total liabilities |
24,435 | 13,043 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value: 5,000 shares authorized, issuable in series; 3 shares issued and outstanding at December 31, 2010, and 2009; aggregate liquidation preference of $9,496 at December 31, 2010, and 2009 |
9,496 | 9,496 | ||||||
| Common stock, $0.001 par value; 112,500 shares authorized: 47,329 and 38,678 shares issued and outstanding at December 31, 2010, and 2009, respectively |
47 | 39 | ||||||
| Additional paid-in capital |
441,034 | 421,897 | ||||||
| Accumulated other comprehensive income |
108 | 58 | ||||||
| Accumulated deficit |
(426,953 | ) | (410,042 | ) | ||||
| Total stockholders’ equity |
23,732 | 21,448 | ||||||
| Total liabilities and stockholders’ equity |
$ | 48,167 | $ | 34,491 | ||||
See accompanying notes.
57
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenue: |
||||||||||||
| Product revenue |
$ | 21,677 | $ | 16,751 | $ | 15,518 | ||||||
| Government grants and cooperative agreements |
1,432 | 1,231 | 989 | |||||||||
| Total revenue |
23,109 | 17,982 | 16,507 | |||||||||
| Cost of product revenue |
12,046 | 12,580 | 9,668 | |||||||||
| Gross profit |
11,063 | 5,402 | 6,839 | |||||||||
| Operating expenses (gains): |
||||||||||||
| Research and development |
5,195 | 6,372 | 10,205 | |||||||||
| Selling, general, and administrative |
21,577 | 21,867 | 27,164 | |||||||||
| Intangible asset amortization |
67 | — | — | |||||||||
| Restructuring |
— | 841 | — | |||||||||
| Settlement gain |
— | (1,381 | ) | — | ||||||||
| Acquisition related costs and impairment of long-term investment in related parties, net |
182 | 1,536 | — | |||||||||
| Total operating expenses |
27,021 | 29,235 | 37,369 | |||||||||
| Loss from operations |
(15,958 | ) | (23,833 | ) | (30,530 | ) | ||||||
| Non-operating income (expense): |
||||||||||||
| Revaluation of warrant liability |
39 | 63 | — | |||||||||
| Foreign exchange gain (loss) |
(816 | ) | (611 | ) | 507 | |||||||
| Interest expense |
(689 | ) | (10 | ) | (10 | ) | ||||||
| Other income, net |
513 | 256 | 852 | |||||||||
| Total non-operating income (expense) |
(953 | ) | (302 | ) | 1,349 | |||||||
| Net loss |
$ | (16,911 | ) | $ | (24,135 | ) | $ | (29,181 | ) | |||
| Net loss per common share: |
||||||||||||
| Basic |
$ | (0.42 | ) | $ | (0.69 | ) | $ | (0.90 | ) | |||
| Diluted |
$ | (0.42 | ) | $ | (0.69 | ) | $ | (0.90 | ) | |||
| Weighted average common shares outstanding used for basic and diluted net loss per share: |
||||||||||||
| Basic |
40,300 | 34,750 | 32,430 | |||||||||
| Diluted |
40,300 | 34,750 | 32,430 | |||||||||
See accompanying notes.
58
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
| Preferred Stock | Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Loss |
Comprehensive Income (Loss) |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| Balances at December 31, 2007 |
3 | $ | 9,496 | 32,112 | $ | 32 | $ | 407,010 | $ | 75 | $ | (356,726 | ) | $ | 59,887 | |||||||||||||||||||||
| Issuance of common stock from exercise of stock options and purchases from ESPP |
432 | 1 | 1,289 | — | 1,290 | |||||||||||||||||||||||||||||||
| Stock-based compensation |
2,145 | 2,145 | ||||||||||||||||||||||||||||||||||
| Net change in unrealized gain on investments |
— | — | — | — | — | 137 | $ | 137 | — | 137 | ||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (29,181 | ) | (29,181 | ) | (29,181 | ) | ||||||||||||||||||||||||
| Total comprehensive loss |
$ | (29,044 | ) | |||||||||||||||||||||||||||||||||
| Balances at December 31, 2008 |
3 | $ | 9,496 | 32,544 | $ | 33 | $ | 410,444 | $ | 212 | $ | (385,907 | ) | $ | 34,278 | |||||||||||||||||||||
| Issuance of common stock from public offering, net of expenses of $3,865 |
6,000 | 6 | 9,329 | — | 9,335 | |||||||||||||||||||||||||||||||
| Issuance of common stock from exercise of stock options and purchases from ESPP |
134 | — | 78 | — | 78 | |||||||||||||||||||||||||||||||
| Stock-based compensation |
2,046 | 2,046 | ||||||||||||||||||||||||||||||||||
| Net change in unrealized gain on investments |
— | — | — | — | — | (154 | ) | $ | (154 | ) | — | (154 | ) | |||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (24,135 | ) | (24,135 | ) | (24,135 | ) | ||||||||||||||||||||||||
| Total comprehensive loss |
$ | (24,289 | ) | |||||||||||||||||||||||||||||||||
| Balances at December 31, 2009 |
3 | $ | 9,496 | 38,678 | $ | 39 | $ | 421,897 | $ | 58 | $ | (410,042 | ) | $ | 21,448 | |||||||||||||||||||||
| Issuance of common stock from acquisition and public offering, net of expenses of $1,710 |
8,306 | 8 | 16,940 | 16,948 | ||||||||||||||||||||||||||||||||
| Issuance of common stock from exercise of stock options and purchases from ESPP |
345 | 0 | 369 | 369 | ||||||||||||||||||||||||||||||||
| Stock-based compensation |
1,828 | 1,828 | ||||||||||||||||||||||||||||||||||
| Net change in unrealized gain on investments |
— | — | — | — | — | 50 | $ | 50 | — | 50 | ||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | (16,911 | ) | (16,911 | ) | (16,911 | ) | ||||||||||||||||||||||||
| Total comprehensive loss |
$ | (16,861 | ) | |||||||||||||||||||||||||||||||||
| Balances at December 31, 2010 |
3 | $ | 9,496 | 47,329 | $ | 47 | $ | 441,034 | $ | 108 | $ | (426,953 | ) | $ | 23,732 | |||||||||||||||||||||
See accompanying notes.
59
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (16,911 | ) | $ | (24,135 | ) | $ | (29,181 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
845 | 915 | 651 | |||||||||
| Stock-based compensation |
1,828 | 2,046 | 2,145 | |||||||||
| Loss on sale of fixed assets |
63 | 109 | (34 | ) | ||||||||
| Gain on non-controlling equity interest |
(315 | ) | — | — | ||||||||
| Impairment of long-term investment in related party |
— | 2,329 | — | |||||||||
| Revaluation of warrant |
(39 | ) | (63 | ) | — | |||||||
| Non-cash interest expense |
152 | — | — | |||||||||
| Gain from operating settlement |
— | (1,381 | ) | — | ||||||||
| Changes in operating assets and liabilities, net of effects of acquired business: |
||||||||||||
| Accounts receivable |
(1,167 | ) | 696 | 660 | ||||||||
| Inventories |
2,020 | 3,367 | (4,047 | ) | ||||||||
| Other assets |
99 | 108 | 1,039 | |||||||||
| Accounts payable |
(1,193 | ) | 102 | (2,145 | ) | |||||||
| Accrued restructuring |
(113 | ) | 113 | — | ||||||||
| Accrued liabilities |
717 | 1,347 | (2,107 | ) | ||||||||
| Deferred revenue |
(97 | ) | (100 | ) | (1,059) | |||||||
| Net cash used in operating activities |
(14,111 | ) | (14,547 | ) | (34,078 | ) | ||||||
| Investing activities |
||||||||||||
| Purchases of furniture, equipment and leasehold improvements |
(1,709 | ) | (191 | ) | (1,194 | ) | ||||||
| (Purchases)/sales of other assets |
(11 | ) | 37 | (467 | ) | |||||||
| Purchases of investments |
— | (499 | ) | (2,285 | ) | |||||||
| Sales of investments |
88 | 499 | 4,954 | |||||||||
| Maturities of investments |
1,545 | 9,477 | 22,418 | |||||||||
| Net cash provided by (used in) investing activities |
(87 | ) | 9,323 | 23,426 | ||||||||
| Financing activities |
||||||||||||
| Net proceeds from issuance of common stock due to exercise of stock options and purchases from ESPP |
370 | 78 | 1,290 | |||||||||
| Net proceeds from public offering |
19,291 | 12,135 | — | |||||||||
| Issuance cost for credit facility |
— | — | (25 | ) | ||||||||
| Proceeds from landlord provided leasehold incentives |
1,354 | — | 97 | |||||||||
| Proceeds from note payable, net of discount |
4,878 | — | — | |||||||||
| Payments on capital lease and loan obligations |
(34 | ) | (5 | ) | (32) | |||||||
| Net cash provided by financing activities |
25,859 | 12,208 | 1,330 | |||||||||
| Net increase (decrease) in cash and cash equivalents |
11,661 | 6,984 | (9,322) | |||||||||
| Cash and cash equivalents, beginning of period |
17,287 | 10,303 | 19,625 | |||||||||
| Cash and cash equivalents, end of period |
$ | 28,948 | $ | 17,287 | $ | 10,303 | ||||||
| Supplemental disclosures: |
||||||||||||
| Common stock issued in connection with the acquisition of certain assets of BioOne |
$ | 3,423 | $ | — | $ | — | ||||||
| Cash paid for interest |
$ | 605 | $ | 4 | $ | 1 | ||||||
See accompanying notes.
60
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2010
Note 1. Nature of Operations and Basis of Presentation
Cerus Corporation, or the Company, was incorporated on September 19, 1991, and is developing and commercializing the INTERCEPT Blood System, which is designed to enhance the safety of blood components through pathogen inactivation. The Company has worldwide commercialization rights for the INTERCEPT Blood System for platelets, plasma and red blood cells.
The Company sells its INTERCEPT platelet and plasma systems in Europe, the CIS countries, the Middle East and selected countries in other regions around the world. The Company conducts significant research, development, testing and regulatory compliance activities on its product candidates that, together with anticipated selling, general, and administrative expenses, are expected to result in substantial additional losses, and the Company may need to adjust its operating plans and programs based on the availability of cash resources. The Company’s ability to achieve a profitable level of operations will depend on successfully completing development, obtaining additional regulatory approvals and achieving widespread market acceptance of its products. There can be no assurance that the Company will ever achieve a profitable level of operations.
Note 2. Summary of Significant Accounting Policies
Principles of Consolidation
The accompanying consolidated financial statements include those of Cerus Corporation and its subsidiary, Cerus Europe B.V. (collectively hereinafter “Cerus” or the “Company”) after elimination of all intercompany accounts and transactions. These consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP, and pursuant to the rules and regulations of the Securities and Exchange Commission, or SEC.
Use of Estimates
The preparation of financial statements requires management to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, revenue and expenses, and related disclosures of contingent assets and liabilities. On an ongoing basis, management evaluates its estimates, which are based on historical experience and on various other assumptions that are believed to be reasonable under the circumstances. Actual results may differ from those estimates under different assumptions or conditions.
Revenue
The Company recognizes revenue in accordance with ASC, Topic 605-25, “Revenue Recognition—Arrangements with Multiple Deliverables,” as applicable. Revenue is recognized when (i) persuasive evidence of an agreement with the funding party exists; (ii) services have been rendered or product has been delivered; (iii) pricing is fixed or determinable; and (iv) collection is probable.
The Company’s main sources of revenues for the three years ended December 31, 2010 were product revenue from sales of the INTERCEPT Blood System, and United States government grants and awards.
Revenue related to product sales is generally recognized when the Company fulfills its obligations for each element of an agreement. For all INTERCEPT Blood System sales, the Company uses a binding purchase order and signed sales contract as evidence of written agreement. The Company sells INTERCEPT Blood System directly to blood banks, hospitals, universities, government agencies, as well as to distributors in certain regions.
61
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Generally, the Company’s contracts with its customers do not provide for open return rights, except within a reasonable time after receipt of goods in the case of defective product or non-conforming product. Deliverables and the units of accounting vary according to the provisions of the purchase order or sales contract. For revenue arrangements with multiple elements, the Company evaluates whether the delivered elements have stand-alone value to the customer, whether the fair value of the undelivered elements is reliably determinable, and whether the delivery of the remaining elements is probable and within the Company’s control. When all of these conditions are met, the Company recognizes the revenue on the delivered elements. If these conditions are not met, the Company defers revenue until such time as all of the conditions have been met or all of the elements have been delivered. Consideration received is allocated to elements that are identified as discrete units of accounting based on the relative fair value method. At December 31, 2010 and 2009, the Company had $0.3 million of short-term deferred revenue on its consolidated balance sheets related to future performance obligations. Freight costs charged to customers are recorded as a component of revenue under ASC Topic 605, “Accounting for Shipping and Handling Fees and Costs”. Value-added-taxes, or VAT, that the Company invoices to its customers and remits to governments, are recorded on a net basis, and are excluded from product revenue.
Research and Development Expenses
The Company receives certain United States government grants that support the Company’s efforts in defined research projects. These grants generally provide for reimbursement of approved costs incurred as defined in the various grants. Revenue associated with these grants is recognized as costs under each grant are incurred. In accordance with ASC Topic 730, “Accounting for Research and Development Expenses,” research and development expenses are charged to expense when incurred. Research and development expenses include salaries and related expenses for scientific personnel, payments to consultants, supplies and chemicals used in in-house laboratories, costs of research and development facilities, depreciation of equipment and external contract research expenses, including clinical trials, preclinical safety studies, other laboratory studies, process development and product manufacturing for research use.
The Company’s use of estimates in recording accrued liabilities for research and development activities (described previously in this Note under the heading “Use of Estimates”) affects the amounts of research and development expenses recorded and revenue recorded from development funding and government grants and collaborative agreements. Actual results may differ from those estimates under different assumptions or conditions.
Cash, Cash Equivalents and Short-Term Investments
The Company considers all highly liquid investments with an original maturity of 90 days or less from the date of purchase to be cash equivalents. Cash equivalents consist principally of short-term money market instruments.
In accordance with ASC Topic 320, “Accounting for Certain Investments in Debt and Equity Securities,” the Company has classified all debt securities as available-for-sale at the time of purchase and reevaluates such designation as of each balance sheet date. Available-for-sale securities are carried at estimated fair value based on quoted market prices. The Company reports the amortization of any premium and accretion of any discount resulting from the purchase of debt securities as a component of interest expense. The Company’s available-for-sale securities consist primarily of United States government agency securities and corporate debt securities.
62
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Unrealized gains and losses at December 31, 2010, and 2009, are reported in accumulated other comprehensive income (loss) on the Company’s consolidated balance sheets. The Company reviews all of its marketable securities on a regular basis to evaluate whether any security has experienced an other-than-temporary decline in fair value. During the years ended December 31, 2010, 2009, and 2008, the Company recognized losses totaling $0.04 million, $0.0 million, and $0.3 million, respectively, associated with investments experiencing an other-than-temporary decline in fair value. These investments experiencing an other-than-temporary decline in fair value primarily relate to fixed income securities. At December 31, 2010, the Company recorded the fair value of these investments on its consolidated balance sheet and this has become the Company’s basis for recording prospective unrealized gains and losses. The cost of securities sold is based on the specific identification method.
As of December 31, 2010, the Company also maintained a certificate of deposit for approximately $0.2 million with a domestic bank. The Company holds this certificate of deposit for any potential decommissioning resulting from the Company’s possession of radioactive material. The certificate of deposit is held to satisfy the financial surety requirements of the California Department of Health Services and is recorded as restricted cash on its consolidated balance sheets at December 31, 2010, and 2009.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash equivalents, short-term investments and accounts receivable.
Substantially all of the Company’s cash, cash equivalents and short-term investments are maintained pursuant to the Company’s investment policy at a major financial institution of high credit standing. The Company monitors the financial credit worthiness of the issuers of its investments and limits the concentration in individual securities and type of investments that exist within its investment portfolio. Generally, all of the Company’s remaining investments carry high credit quality ratings, in accordance with its investment policy. At December 31, 2010, the Company does not believe there is significant financial risk from non-performance by the issuers of the Company’s cash equivalents and short-term investments.
Concentrations of credit risk with respect to trade receivables exist to the full extent of amounts presented in the consolidated financial statements. On a regular basis, including at the time of sale, the Company performs credit evaluations of its customers. Generally, the Company does not require collateral from its customers to secure accounts receivable. To the extent that the Company determines specific invoices or customer accounts may be uncollectible, the Company reserves against the accounts receivable on its balance sheet and records a charge on its statement of operations. The Company had recorded allowances for potentially uncollectible accounts receivable of approximately $0.1 million at both December 31, 2010 and 2009. Actual collection losses may differ from management’s estimate, and such differences could be material to the Company’s financial position and results of operations.
The Company had three and four customers each accounting for more than 10% of the Company’s outstanding trade receivables and aggregating approximately 54% and 73% of outstanding trade receivables at December 31, 2010 and December 31, 2009, respectively. To date, the Company has not experienced collection difficulties from these customers.
Inventories
At December 31, 2010 and 2009, inventory consisted of finished goods of INTERCEPT disposable kits, components thereof, UVA illumination devices, and certain replacement parts for the illumination devices. The
63
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Company’s supply chain for certain of these components, held as work-in-process on its consolidated balance sheet, can take in excess of one year for production to be complete before the work-in-process is utilized in finished disposable kits. Inventory is recorded at the lower of cost, determined on a first in, first-out basis, or market value. Platelet and plasma system disposable kits generally have two-year lives from date of manufacture. The Company frequently reviews the composition of inventory in order to identify obsolete, slow-moving or otherwise unsalable items. To the extent unsalable items are observed and there is no alternative use, the Company will record a write-down to net realizable value in the period that the impairment is first recognized. At December 31, 2010, and 2009, the Company had approximately $0.4 million and $0.3 million, respectively, reserved for potential obsolete or expiring product.
Property and Equipment, net
Property and equipment is comprised of furniture, equipment, information technology hardware and software and is recorded at cost. At the time the property and equipment is ready for its intended use, it is depreciated on a straight-line basis over the estimated useful lives of the assets (generally three to five years). Leasehold improvements are amortized on a straight-line basis over the shorter of the lease term or the estimated useful lives of the improvements.
The Company evaluates its long-lived assets for impairment in accordance with ASC Topic 360, “Accounting for the Impairment or Disposal of Long-Lived Assets”. The Company continually monitors events and changes in circumstances that could indicate carrying amounts of its long-lived assets may not be recoverable. When such events or changes in circumstances occur, the Company assesses recoverability by determining whether the carrying value of such assets will be recovered through the undiscounted expected future cash flows. If the future undiscounted cash flows are less than the carrying amount of these assets, the Company recognizes an impairment loss based on the excess of the carrying amount over the fair value of the assets. The Company did not recognize impairment charges related to its long-lived assets in 2010, 2009, or 2008.
Long-Term Investment in Related Party
At December 31, 2009, the Company held an approximate 13% interest in the voting securities of BioOne Corporation, or BioOne, and accounted for its investment in BioOne under the cost method. At December 31, 2009, the Company evaluated several criteria to determine whether facts and circumstances supported the carrying value of its investment in BioOne. These criteria included, but were not limited to: third-party investor interest and participation in recent equity offerings at current pricing, business outlook of BioOne and available financial information. As a result of its evaluation of the criteria used to support its position in BioOne, the Company determined that there were no factors to support any carrying value of its investment in BioOne. As a result, at December 31, 2009, the Company completely impaired its investment in BioOne and as such recorded its investment at zero.
During 2010, the Company completed an acquisition of certain assets from BioOne and relinquished its 13% equity interest in BioOne. The Company accounted for this acquisition under the guidance of ASC 805, “Business Combinations,” and accounted for the acquisition as a business combination. The acquisition resulted in a gain of $0.3 million related to the previously held equity interest and acquisition costs of $0.5 million recorded as operating expenses in the consolidated statement of operations for the period ending December 31, 2010.
See Note 7 for further information regarding our acquisition and valuation.
64
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Intangible Assets and Goodwill
During 2010, as part of the acquisition of certain BioOne assets, the Company acquired a commercialization license for the INTERCEPT Blood System for both platelets and plasma in parts of Asia. The Company accounted for the acquisition under the guidance of ASC, Topic 850, “Business Combinations,” and ASC, Topic 350, “Intangibles—Goodwill and Other,” and accounted for the acquisition as a business combination. Accordingly, the Company assigned the total consideration transferred based on each asset’s fair value, and the residual to an indefinite life intangible asset, goodwill.
The Company periodically reviews its amortizable intangible assets for events or changes in circumstances indicating that the carrying amount of such assets may not be recoverable. If the carrying amounts of the assets exceed their respective fair values, additional impairment tests are performed to measure the amount of the impairment loss, if any.
Goodwill is not amortized but instead is subject to an annual impairment test, or more frequently if events or changes in circumstances indicate that they may be impaired. The Company evaluates goodwill on an annual basis as of the end of the third quarter of each fiscal year. The test for goodwill impairment is a two-step process. The first step compares the fair value of each reporting unit with its respective carrying amount, including goodwill. If the fair value of the reporting unit exceeds its carrying amount, goodwill of the reporting unit is not considered impaired and, therefore, the second step of the impairment test is unnecessary. The second step, used to measure the amount of impairment loss, compares the implied fair value of each reporting unit’s goodwill with the respective carrying amount of that goodwill. If the carrying amount of the reporting unit goodwill exceeds the implied fair value of that goodwill, an impairment loss shall be recognized in an amount equal to that excess. Management has determined that it operates as a single reporting unit and therefore evaluates goodwill impairment at the enterprise level. There were no impairment charges through December 31, 2010.
See Note 7 for further information on intangible asset valuation.
Foreign Currency Remeasurement
The functional currency of the Company’s foreign subsidiary is the United States Dollar. Monetary assets and liabilities denominated in foreign currencies are remeasured in United States Dollars using the exchange rates at the balance sheet date. Non-monetary assets and liabilities denominated in foreign currencies are remeasured in United States Dollars using historical exchange rates. Revenues and expenses are remeasured using average exchange rates prevailing during the period. Remeasurements are recorded in the Company’s consolidated statements of operations. The Company recorded foreign currency losses of $0.8 million and $0.6 million during the years ended December 31, 2010, and 2009, respectively, and foreign currency gains of $0.5 million during the year ended December 31, 2008.
Stock-Based Compensation
The Company maintains an equity incentive plan to provide long-term incentives for employees, contractors, members of the Board of Directors, and Scientific Advisory Board. The plan allows for the issuance of non-statutory and incentive stock options, restricted stock, restricted stock units, stock appreciation rights, other stock-related awards, and performance awards which may be settled in cash, stock, or other property. The Company also maintains an active employee stock purchase plan within the meaning of Section 423(b) of the Internal Revenue Code.
65
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
The Company accounts for stock-based compensation in accordance with ASC Topic 718, “Compensation – Stock Compensation.” Under the fair value recognition provisions, stock-based compensation cost is measured at the grant date based on the fair value of the award and is recognized as expense on a straight-line basis over the requisite service period, which is the vesting period. To the extent that stock options contain performance criteria for vesting, stock-based compensation is recognized once the performance criteria are probable of being met.
For its non-employee stock-based awards, the Company follows ASC Topic 505-50, “Equity Based Payment to Non-Employees” and considers the measurement date at which the fair value of the stock-based award is measured is equal to the earlier of 1) the date at which a commitment for performance by the counter party to earn the equity instrument is reached or 2) the date at which the counter party’s performance is complete. The Company recognizes stock-based compensation expense for the fair value of the vested portion of the non-employee awards in its consolidated statements of operations.
See Note 13 for further information regarding our stock-based compensation assumptions and expenses.
Warrant Liability
The Company issued warrants to purchase an aggregate of 2.4 million and 3.7 million shares of common stock, in August 2009 and November 2010, respectively. Except for the exercise price and date of issuance, the terms of the warrants are identical. The outstanding warrants are classified as a liability, and as such, the fair value of the warrants is recorded on the consolidated balance sheet. At each subsequent reporting period, the fair value of the warrants are adjusted with changes in fair value of the warrants reflected on the consolidated balance sheet and gains or losses recognized in the consolidated statements of operations. Gains and losses from warrant revaluation are recorded as a component of non-operating income (expense). The fair value of the warrants is estimated using the binomial-lattice option-pricing model. During the years ended December 31, 2010 and December 31, 2009, the Company recorded gains of $0.04 million and $0.06 million, respectively, associated with changes in the fair value of the warrants. The warrants will continue to be reported as a liability until such time as the instruments are exercised or are otherwise modified to remove the provisions which require this treatment, at which time the warrants are adjusted to fair value and reclassified from liabilities to stockholders’ equity. If the warrants are reclassified from a liability to equity, the fair value of the warrants would be recorded in stockholders’ equity and no further adjustment would be made in subsequent periods.
See Note 12 for further information regarding our warrant liability valuation.
Other Comprehensive Income (Loss)
The components of comprehensive income (loss) include net income (loss) and other comprehensive income (loss). The Company’s only component of other comprehensive income (loss) for the years ended 2010, 2009, and 2008 consisted of unrealized gains or losses from the Company’s available-for-sales short-term investments. Other comprehensive income (loss) is reported as a separate component of stockholders’ equity.
Income Taxes
The Company accounts for income taxes in accordance with ASC Topic 740 “Accounting for Income Taxes”. Under this method, deferred tax assets and liabilities are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. ASC Topic 740 requires derecognition of tax
66
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
positions that do not have a greater than 50% likelihood of being recognized upon review by a taxing authority having full knowledge of all relevant information. Use of a valuation allowance as described in ASC 740 is not an appropriate substitute for the derecognition of a tax position. The Company did not have any recorded liabilities for unrecognized tax benefits at December 31, 2010, or 2009. The Company recognizes interest accrued and penalties related to unrecognized tax benefits in its income tax expense. To date, the Company has not recognized any interest and penalties in is statements of operations, nor has its accrued for or made payments for interest and penalties. The Company continues to carry a full valuation allowance on all of its deferred tax assets. The tax years 2006 through 2010 remain subject to examination by the taxing jurisdictions to which the Company is subject.
Net Loss Per Share—Basic and Diluted
Basic and diluted loss per share is computed by dividing net loss by the weighted average number of common shares outstanding for the period.
The following table sets forth the reconciliation of the denominator used in the computation of basic and diluted net loss per common share (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Denominator: |
||||||||||||
| Basic weighted average number of common shares outstanding |
40,300 | 34,750 | 32,430 | |||||||||
| Effect of dilutive potential common shares resulting from stock options, convertible preferred stock, restricted stock units, warrants, and ESPP shares |
— | — | — | |||||||||
| Diluted weighted average number of common shares outstanding |
40,300 | 34,750 | 32,430 | |||||||||
The table below presents stock options, convertible preferred stock, restricted stock units and warrants that are excluded from the diluted net loss per common share due to their anti-dilutive effect (shares in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Antidilutive securities—weighted average shares |
13,538 | 9,384 | 5,374 | |||||||||
Guarantee and Indemnification Arrangements
The Company recognizes the fair value for guarantee and indemnification arrangements issued or modified by the Company after December 31, 2002. In addition, the Company monitors the conditions that are subject to the guarantees and indemnifications, in order to identify if a loss has occurred. If the Company determines it is probable that a loss has occurred, then any such estimable loss would be recognized under those guarantees and indemnifications. Some of the agreements of the Company contain provisions that indemnify the counter party from damages and costs resulting from claims that the Company’s technology infringes the intellectual property rights of a third party or claims that the sale or use of the Company’s products have caused personal injury or other damage or loss. The Company has not received any such requests for indemnification under these provisions and has not been required to make material payments pursuant to these provisions.
The Company generally provides for a one-year warranty on certain of its INTERCEPT blood-safety products covering defects in materials and workmanship. The Company accrues costs associated with warranty
67
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
obligations when claims become known and is probable and estimable. There have been very few warranty costs incurred through December 31, 2010, and the Company is unaware of any future warranty claims. Accordingly, at December 31, 2010, the Company has not accrued for any potential future warranty costs.
Fair Value of Financial Instruments
The Company applies the provisions of ASC Topic 820-10-65-4, “Fair Value Measurements,” relating to its financial assets and liabilities. The carrying amounts of accounts receivables, accounts payable, and other accrued liabilities approximate their fair value due to the relative short-term maturities. Based on the borrowing rates currently available to the Company for loans with similar terms, the Company believes the fair value of long-term debt approximates their carrying amounts. The carrying amounts and fair value of the Company’s short term investments and warrant liability are described in “Note 3. Financial Instruments” to these consolidated financial statements.
New Accounting Pronouncements
Revenue Recognition
In October 2009, the FASB issued updated revenue recognition guidance under ASC Topic 605 relating to revenue arrangements with multiple deliverables. Under the revised guidance, companies with revenue arrangements that have multiple deliverables must assess whether or not multiple deliverables exist under the revised guidance, how the deliverables should be separated and how the consideration should be allocated to the elements. In addition, the revised guidance requires an entity to allocate revenue in an arrangement using the best estimated selling price, orBESP, of deliverables if a vendor does not have vendor specific objective evidence of selling price or third-party evidence, or TPE, of selling price. Each unit must have stand-alone value to the customer, similar to previous guidance. The revised guidance is effective for the Company beginning January 1, 2011.
Note 3. Financial Instruments
The Company measures and records certain financial assets at fair value on a recurring basis, including its available-for-sale short-term investments. The Company’s available-for-sale short-term investments consist of fixed income corporate bonds and United States government agency securities. The Company classifies investments with original maturities of three months or less at the date of purchase, as cash equivalents. Cash equivalents consist of corporate commercial paper and money market funds, for which the carrying amount is a reasonable estimate of fair value. Similarly, the Company measures and records certain financial liabilities at fair value.
68
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
At December 31, 2010, the fair values of certain of the Company’s financial assets and liabilities were determined using the following inputs (in thousands):
| Available-for-sale-securities |
Quoted Prices in Active Markets for Identical Assets |
Significant Other Observable Inputs |
Significant Unobservable Inputs |
|||||||||||||
| Total | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Money market funds(1) |
$ | 6,178 | $ | 6,178 | $ | — | $ | — | ||||||||
| Corporate bonds(2) |
73 | — | 73 | — | ||||||||||||
| United States government agency securities(2) |
988 | — | 988 | — | ||||||||||||
| $ | 7,239 | $ | 6,178 | $ | 1,061 | $ | — | |||||||||
| Liabilities |
||||||||||||||||
| Warrant Liability(3) |
$ | 8,465 | $ | — | $ | — | $ | 8,465 | ||||||||
At December 31, 2009, the fair values of certain of the Company’s financial assets and liabilities were determined using the following inputs (in thousands):
| Available-for-sale-securities |
Quoted Prices in Active Markets for Identical Assets |
Significant Other Observable Inputs |
Significant Unobservable Inputs |
|||||||||||||
| Total | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Money market funds(1) |
$ | 11,059 | $ | 11,059 | $ | — | $ | — | ||||||||
| Corporate bonds(2) |
657 | — | 657 | — | ||||||||||||
| United States government agency securities(2) |
1,987 | — | 1,987 | — | ||||||||||||
| $ | 13,703 | $ | 11,059 | $ | 2,644 | $ | — | |||||||||
| Liabilities |
||||||||||||||||
| Warrant Liability(3) |
$ | 2,737 | $ | — | $ | — | $ | 2,737 | ||||||||
| (1) | Included in cash and cash equivalents on the Company’s consolidated balance sheet. |
| (2) | Included in short-term investments on the Company’s consolidated balance sheet. |
| (3) | Included in current liabilities on the Company’s consolidated balance sheet. For further discussion, see Note 12. |
The Company classifies investments within Level 1 if quoted prices are available in active markets. The Company classifies items in Level 2 if the investments are valued using observable inputs to quoted market prices, benchmark yields, reported trades, broker/dealer quotes or alternative pricing sources with reasonable levels of price transparency. These investments include: United States government agencies and corporate bonds. Investments are held by a custodian who obtains investment prices from a third party pricing provider that uses standard inputs to models which vary by asset class. The Company did not hold financial assets which were recorded at fair value in the Level 3 category, which defines that one or more significant inputs or significant value drivers are unobservable, as of December 31, 2010 and December 31, 2009. The Company’s warrant liability is recorded at fair value and classified in the Level 3 category. For further discussion, see Note 12.
69
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
A reconciliation of the beginning and ending balances for warrant liabilities using significant unobservable inputs (Level 3) as of December 31, 2010 and December 31, 2009, was as follows (in thousands):
| Balance as of December 31, 2008 |
$ | — | ||
| Issuance of warrants |
2,800 | |||
| (Gains) and losses from revaluation |
(63 | ) | ||
| Balance as of December 31, 2009 |
2,737 | |||
| Issuance of warrants |
5,766 | |||
| (Gains) and losses from revaluation |
(38 | ) | ||
| Balance as of December 31, 2010 |
$ | 8,465 | ||
The Company did not have any transfers, from Level 3 to Levels 1 or 2 during the years ended December 31, 2010, or 2009.
Note 4. Cash, Cash Equivalents and Short-Term Investments
The following is a summary of cash, cash equivalents and short-term investments at December 31 (in thousands):
| 2010 | ||||||||||||
| Carrying Value | Unrealized Gain | Fair Value | ||||||||||
| Cash and cash equivalents: |
||||||||||||
| Cash |
$ | 22,770 | $ | — | $ | 22,770 | ||||||
| Money Market funds |
6,178 | — | 6,178 | |||||||||
| Total cash and cash equivalents |
$ | 28,948 | $ | — | $ | 28,948 | ||||||
| Short-term investments |
||||||||||||
| Corporate debt securities |
$ | 14 | $ | 59 | $ | 73 | ||||||
| United States government agency securities |
939 | 49 | 988 | |||||||||
| Total short-term investments |
$ | 953 | $ | 108 | $ | 1,061 | ||||||
| $ | 29,901 | $ | 108 | $ | 30,009 | |||||||
| 2009 | ||||||||||||
| Carrying Value | Unrealized Gain | Fair Value | ||||||||||
| Cash and cash equivalents: |
||||||||||||
| Cash |
$ | 6,228 | $ | — | $ | 6,228 | ||||||
| Money Market funds |
11,059 | — | 11,059 | |||||||||
| Total cash and cash equivalents |
$ | 17,287 | $ | — | $ | 17,287 | ||||||
| Short-term investments |
||||||||||||
| Corporate debt securities |
$ | 629 | $ | 28 | $ | 657 | ||||||
| United States government agency securities |
1,957 | 30 | 1,987 | |||||||||
| Total short-term investments |
2,586 | $ | 58 | 2,644 | ||||||||
| $ | 19,873 | $ | 58 | $ | 19,931 | |||||||
70
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Short-term investments and cash equivalents consisted of the following by original contractual maturity (in thousands):
| 2010 | 2009 | |||||||
| Due in one year or less |
$ | 6,178 | $ | 11,059 | ||||
| Due greater than one year and less than three years |
1,061 | 2,644 | ||||||
| Total |
$ | 7,239 | $ | 13,703 | ||||
Gross proceeds and the realized losses on sales of available-for-sale investments are as follows (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Gross proceeds |
$ | 71 | $ | 500 | $ | 4,585 | ||||||
| Carrying value |
$ | 89 | $ | 500 | $ | 4,862 | ||||||
| Realized loss |
$ | 18 | $ | — | $ | 277 | ||||||
Realized losses for other-than-temporary declines in market value totaled $0.04 million, $0.0 million and $0.3 million during the years ended December 31, 2010, 2009 and 2008, respectively. Realized gains and losses from the sale of available-for-sale investments and from other-than-temporary declines in market value are recorded in other income (expense), net.
Note 5. Inventories
Inventories consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Work in progress |
$ | 2,652 | $ | 3,638 | ||||
| Finished goods |
3,305 | 4,069 | ||||||
| $ | 5,957 | $ | 7,707 | |||||
The Company’s inventories at December 31, 2010, and 2009, consisted of finished goods of INTERCEPT disposable kits, components thereof, UVA illumination devices, and certain replacement parts for the illumination devices. The Company is responsible for supplying its manufacturer, Fenwal Inc., with certain components for assembly into finished INTERCEPT disposable kits. The Company accounts for these components as work-in-process until such time as the components are used in the production of finished INTERCEPT disposable sets. The Company’s work-in-process components are manufactured over a protracted length of time before being incorporated into the finished disposable kits. As a result, work-in-process costs accumulate for a period of time which can exceed one year.
71
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Note 6. Property and Equipment, net
Property and equipment, net consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Leasehold improvements |
$ | 5,470 | $ | 6,498 | ||||
| Machinery and equipment |
1,652 | 1,922 | ||||||
| Demonstration equipment |
104 | 104 | ||||||
| Office furniture |
583 | 962 | ||||||
| Computer equipment |
488 | 598 | ||||||
| Computer software |
1,062 | 1,105 | ||||||
| Consigned demonstration equipment |
401 | 335 | ||||||
| Construction-in-progress |
134 | 61 | ||||||
| 9,894 | 11,585 | |||||||
| Less accumulated depreciation and amortization |
(7,504 | ) | (10,368 | ) | ||||
| $ | 2,390 | $ | 1,217 | |||||
Note 7. Acquisition, Goodwill and Other Intangible Assets
Acquisition
On August 24, 2010, the Company acquired certain assets of BioOne, a privately held Japanese company established to develop technologies to improve the safety of blood products in Asia. The assets included the commercialization licenses that the Company had granted to BioOne for both the platelet and plasma systems, illuminators held as saleable inventory and demonstration illuminators. No liabilities were assumed. As consideration for the acquired BioOne assets, at the closing of the acquisition, the Company issued 937,886 shares of its common stock to BioOne and relinquished all shares of BioOne that had been held by the Company. In addition, on February 25, 2011 (six months from the closing date of the acquisition), the Company issued to BioOne an additional 234,471 shares of common stock. The total value of the consideration provided was $3.7 million which includes approximately $0.3 million associated with the fair value of the Company’s non-controlling equity interest in BioOne, relinquished as a result of the acquisition. The shares issuable in February 2011 were not contingent on future events, and were included in the total consideration and recorded in additional paid in capital as of December 31, 2010.
The Company accounted for the BioOne acquisition as the acquisition of a business under the guidance of ASC 805, “Business Combinations.” Under the accounting for the acquisition of a business, the consideration was allocated to the acquired tangible and intangible assets based on their estimated fair values as of August 24, 2010. The excess purchase price over the value of the net tangible and identifiable intangible assets was recorded as goodwill. The factors that contributed to the recognition of goodwill included securing buyer-specific synergies to increase revenue and profits through the commercialization of the INTERCEPT blood system worldwide. By acquiring these commercialization rights in certain Asian countries, the Company was able to complete the global commercialization rights for platelets and plasma.
72
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
The following table summarizes the final allocation of fair value of assets acquired at the acquisition date.
| (in thousands) | ||||
| Commercialization rights—Asia |
$ | 2,017 | ||
| Illuminators—Inventory |
270 | |||
| Demonstration Illuminators |
135 | |||
| Goodwill |
1,316 | |||
| Total |
$ | 3,738 | ||
The commercialization rights in Asia represent the reacquisition of contractual rights originally granted to BioOne to market the Company’s products in certain countries in Asia. The contractual term of this original agreement was perpetual and the Company estimated the fair value of these reacquired rights based on future expected cash flows to be generated over the expected life of the underlying technology.
The business combination resulted in a gain on the pre-acquisition non-controlling equity interest of BioOne. Prior to the business combination, the Company carried its 13% investment in BioOne at zero. As a result, the Company recognized a gain of $0.3 million representing the difference between the assumed fair value of the pre-acquisition non-controlling equity interest and its carrying value. Acquisition related costs of $0.5 million were recorded as a component of acquisition related costs and impairment of long-term investment in related parties, net during the year ending December 31, 2010.
Due to limited operating activities as a result of a deteriorating financial situation BioOne did not have significant revenue or expenses and the pro forma impact of the BioOne acquisition is not significant to the results of operations of the Company. The Company’s operating results include the impact of the BioOne acquisition beginning from the acquisition date.
Intangible Assets
The fair market value of the commercialization license, shown on the Company’s balance sheet as an intangible asset, was determined to be $2.0 million. The intangible asset is subject to periodic amortization over the estimated useful life of 10 years.
The following is a summary of intangibles at December 31, 2010 (in thousands):
| December 31, 2010 | ||||||||||||
| Gross Carrying Amount |
Accumulated Amortization |
Net Carrying Amount |
||||||||||
| Intangibles subject to amortization: |
||||||||||||
| License—INTERCEPT Asia |
$ | 2,017 | $ | 67 | $ | 1,950 | ||||||
| Total |
$ | 2,017 | $ | 67 | $ | 1,950 | ||||||
The Company recognized $0.07 million in amortization expense for the year ended December 31, 2010. Estimated annual amortization expense for each of the ensuing years is $0.2 million.
Goodwill
During the year ended December 31, 2010, the Company acquired net assets with related goodwill of $1.3 million as part of the acquisition of BioOne.
73
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Note 8. Accrued Liabilities
Accrued liabilities consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accrued compensation and related |
$ | 1,861 | $ | 942 | ||||
| Accrued inventory |
1,424 | 2,366 | ||||||
| Other accrued expenses |
2,718 | 1,978 | ||||||
| $ | 6,003 | $ | 5,286 | |||||
Note 9. Restructuring
In March 2009, pursuant to the Board of Directors’ approval, the Company began implementing a plan to focus on commercializing the INTERCEPT Blood System in Europe, to consolidate facilities, and to reduce its cost structure. During the year ended December 31, 2009, the Company incurred costs for one-time termination benefits for employee positions that were eliminated under the restructuring plan, consolidation of facility and related moving costs. Effected employees received severance consideration and continuation of benefits, as well as transition assistance. All one-time termination benefits have been paid as of December 31, 2010. No additional costs are expected to be incurred by the Company under this restructuring plan.
A summary of the Company’s restructuring costs is as follows (in thousands):
| Balance at December 31, 2009 |
Cash Payments |
Balance at December 31, 2010 |
||||||||||
| One-time termination benefits |
$ | 113 | $ | (113 | ) | $ | — | |||||
| Other |
— | — | — | |||||||||
| Total |
$ | 113 | $ | (113 | ) | $ | — | |||||
Note 10. Commitments and Contingencies
The Company leases its office facilities and certain equipment under non-cancelable operating leases with initial terms in excess of one year that require the Company to pay operating costs, property taxes, insurance and maintenance. On December 10, 2009, the Company exercised a ten-year option to extend the term of its Concord, California lease and this lease provides for landlord-financed leasehold improvements. At December 31, 2010, the Company had financed $1.1 million of leasehold improvements which is primarily included in other long term liabilities in the Company’s balance sheet. The Company pays for the financed leasehold improvements as a component of rent and is required to reimburse its landlord over the remaining life of the respective lease. The Concord, California lease may be canceled no earlier than December 2015 at which time the Company would be required to repay for any remaining portion of the landlord financed leasehold improvements.
74
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Future minimum payments under operating leases are as follows (in thousands):
| Year ending December 31, |
||||
| 2011 |
$ | 719 | ||
| 2012 |
713 | |||
| 2013 |
448 | |||
| 2014 |
422 | |||
| 2015 |
— | |||
| Thereafter |
— | |||
| Total minimum lease payments |
$ | 2,302 | ||
Rent expense for office facilities, net of rental income, was $0.9 million, $1.4 million and $1.4 million for the years ended December 31, 2010, 2009, and 2008, respectively.
Minimum purchase commitments include certain components of INTERCEPT blood safety system which the Company purchases from third party manufacturers and supplies to Fenwal for use in manufacturing finished disposable kits. The Company has paid $0.9 million, $1.2 million, and $1.1 million, for goods under contracts which are subject to minimum purchase commitments during the years ended December 31, 2010, 2009, and 2008, respectively. The Company has minimum purchase commitments under these contracts of $1.5 million and $0.7 million for the years ended December 31, 2011 and 2012, respectively.
Note 11. Long-term Note Payable
Long-term note payable at December 31, 2010 consisted of the following (in thousands):
Principal |
Unamortized Discount |
Total |
||||||||||
| Current portion of note payable |
$ | 1,822 | $ | 75 | $ | 1,747 | ||||||
| Long-term portion of note payable |
3,178 | 47 | 3,131 | |||||||||
| Long-term note payable. |
$ | 5,000 | $ | 122 | $ | 4,878 | ||||||
On March 31, 2010, the Company entered into a growth capital facility agreement and immediately issued a senior secured long-term note payable for $5.0 million. The note issued under the agreement is secured by all of the Company’s assets, except intellectual property. The note carries a fixed interest rate of 12.04%, with interest only payments for the first nine months and then equal principal and interest payments for an additional 30 months. In connection with issuing the note, the Company paid an upfront facility fee of $0.1 million and incurred closing costs of $0.1 million. The combined facility fee and closing costs have been recorded as a discount to the note payable and will be amortized as a component of interest expense using the effective interest method over the term of the note (discount is based on an implied interest rate of 13.84%). In addition, the Company agreed to pay a $0.4 million closing fee upon maturity of the note. The closing fee will be accreted to interest expense using the effective interest method over the life of the note.
75
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Principal and interest payments on the long-term note payable for each of the following five years is as follows (in thousands):
| Year ended December 31, |
Long-Term Debt | |||
| 2011 |
$ | 2,182 | ||
| 2012 |
2,326 | |||
| 2013 |
1,782 | |||
| 2014 |
— | |||
| 2015 |
— | |||
Under the growth capital facility, subject to certain conditions including compliance with covenants, an additional $5.0 million was available to be drawn between September 30, 2010 and December 31, 2010. As of December 31, 2010 the Company had not drawn down the additional $5.0 million, and the Company incurred a non-utilization fee of $0.1 million which was recognized as an operating expense for the year ended December 31, 2010.
The Company is required to maintain compliance with certain customary and routine financial covenants. Additionally, the note requires the Company to generate minimum revenues at certain pre-established levels. At December 31, 2010, the Company was in compliance with financial covenants set forth in the growth capital facility.
In March 2011, the Company entered into an amendment to its growth capital facility. Under the terms of the amendment, the Company may borrow an additional $5.0 million through September 30, 2011. The terms of the additional $5.0 million note would be identical to the first note issued under the growth capital facility except the Company would not incur any additional upfront facility fees. In addition, the amendment modifies the covenants under which the Company must comply. Under the amendment, 2011 revenues, on a trailing six-month basis, are required to be at least 80% of projected revenues. Revenues for 2012 and beyond are required to be at least €5.7 million per quarter. Non compliance with the covenants may result in the principal of the note becoming due and payable.
Note 12. Stockholders’ Equity
Series B Preferred Stock
Fenwal holds 3,327 shares of the Company’s Series B preferred stock. The holder of Series B preferred stock has no voting rights, except with respect to the authorization of any class or series of stock having preference or priority over the Series B preferred stock as to voting, liquidation or conversion or with respect to the determination of fair market value of non-publicly traded shares received by the holder of Series B stock in the event of a liquidation, or except as required by Delaware law. At any time, the holder may convert each share of Series B preferred stock into 100 shares of the Company’s common stock. If all shares of Series B preferred stock were converted to common stock, 332,700 shares of common stock would be issued, which represents approximately 1% of the outstanding common shares of the Company at December 31, 2010. The Company has the right to redeem the Series B preferred stock prior to conversion for a payment of $9.5 million.
Common Stock and Warrant Liability
In August 2009, the Company received net proceeds of approximately $12.1 million, after deducting placement agent’s fees and stock issuance costs of approximately $1.1 million, from a registered direct offering of 6.0 million units. Each unit sold consisted of one share of common stock and a warrant to purchase 4/10 of a
76
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
share of common stock. Each unit was sold for $2.20, resulting in the issuance of 6.0 million shares of common stock and warrants to purchase 2.4 million shares of common stock, exercisable at an exercise price of $2.90 per share. The warrants contain certain provisions that, under certain circumstances which may be out of the Company’s control, could require the Company to pay cash to settle the exercise of the warrants or may require the Company to redeem the warrants.
In November 2010, the Company received net proceeds of approximately $19.7 million, after deducting placement agent’s fees and stock issuance costs of approximately $1.3 million, from a public offering of 7.4 million units. Each unit sold consisted of one share of common stock and a warrant to purchase 1/2 of a share of common stock. Each unit was sold for $2.85, resulting in the issuance of 7.4 million shares of common stock and warrants to purchase 3.7 million shares of common stock, exercisable at an exercise price of $3.20 per share. The warrants contain certain provisions that, under certain circumstances which may be out of the Company’s control, could require the Company to pay cash to settle the exercise of the warrants or may require the Company to redeem the warrants.
The August 2009 and November 2010 warrants are classified as a liability pursuant to “Accounting for Derivative Instruments and Hedging Activities” and “Accounting for Certain Financial Instruments with Characteristics of Both Liabilities and Equity” Topics of ASC. Therefore, the fair value of the warrants is recorded on the consolidated balance sheet as a liability and will be adjusted to fair value at each financial reporting date thereafter until the earlier of exercise or expiration.
These offerings were made pursuant to the Company’s shelf registration statement on Form S-3.
2009 Warrants
The warrants issued in August 2009 are exercisable for a period of five years from the issue date. The fair value on the date of issuance of the warrants was determined to be $2.8 million using the binomial-lattice option valuation model applying the following assumptions: (i) a risk-free rate of 2.48%, (ii) an expected term of 5.0 years, (iii) no dividend yield and (iv) a volatility of 77%.
At December 31, 2010, the fair value of the warrants issued in August 2009 was determined to be approximately $2.8 million using the binomial-lattice option valuation model applying the following assumptions: (i) a risk-free rate of 1.52%, (ii) an expected term of 3.65 years, (iii) no dividend yield and (iv) a volatility of 70%.
2010 Warrants
The warrants issued in November 2010 are exercisable beginning on May 10, 2011 and are exercisable for a period of five years from the issue date. The fair value on the date of issuance of the warrants was determined to be $5.8 million using the binomial-lattice option valuation model applying the following assumptions: (i) a risk-free rate of 1.23%, (ii) an expected term of 5.0 years, (iii) no dividend yield and (iv) a volatility of 85%.
At December 31, 2010 the fair value of the warrants issued in November 2010 was determined to be approximately $5.7 million using the binomial-lattice option valuation model applying the following assumptions: (i) a risk-free rate of 2.01%, (ii) an expected term of 4.86 years, (iii) no dividend yield and (iv) a volatility of 86%.
For the years ended December 31, 2010 and December 31, 2009, due to the decrease in fair value of the warrants, the Company recorded gains of $0.04 million and $0.06 million, respectively, to non-operating income (expense), net. At December 31, 2010 no warrants had been exercised.
77
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Stockholder Rights Plan
In October 2009, the Company’s Board of Directors adopted an amendment to its 1999 stockholder rights plan, commonly referred to as a “poison pill,” that is intended to deter hostile or coercive attempts to acquire the Company. The stockholder rights plan enables stockholders to acquire shares of the Company’s common stock, or the common stock of an acquirer, at a substantial discount to the public market price should any person or group acquire more than 15% of the Company’s common stock without the approval of the Board of Directors under certain circumstances. The Company has designated 250,000 shares of Series C Junior Participating preferred stock for issuance in connection with the stockholder rights plan.
Note 13. Stock-Based Compensation
2008 Equity Incentive Plan
The Company maintains an equity compensation plan to provide long-term incentives for employees, contractors, and members of its Board of Directors and Scientific Advisory Board. The Company currently grants awards from one plan, the 2008 Equity Incentive Plan, or the 2008 Plan. The 2008 Plan allows for the granting of stock options, restricted stock, restricted stock units, stock appreciation rights, other stock-related awards, and performance awards which may be settled in cash, stock, or other property. The 2008 Plan has 1.2 million shares available for grant. Awards under the 2008 Plan generally have a maximum term of 10 years from the date of the award. Employee options granted under the 2008 Plan generally vest over four years. The 2008 Plan generally requires options to be granted at 100% of the fair market value of the Company’s common stock subject to the option on the date of grant. Performance-based stock options granted under the 2008 Plan are limited to either 500,000 shares or $1.0 million, in the case of performance based cash awards, per recipient per calendar year. During the year ended December 31, 2008, the Company granted performance based stock options totaling 50,000 shares which remain outstanding at December 31, 2010.
Employee Stock Purchase Plan
The Company also maintains an Employee Stock Purchase Plan, or the Purchase Plan. The Purchase Plan is intended to qualify as an employee stock purchase plan within the meaning of Section 423(b) of the Internal Revenue Code. Under the Purchase Plan, the Company’s Board of Directors may authorize participation by eligible employees, including officers, in periodic offerings. The offering period for any offering will be no more than 27 months.
Restricted Stock Units
The Company has granted restricted stock units to the Chief Executive Officer, Senior Vice Presidents, and Vice Presidents in accordance with the Bonus Plan for Senior Management of Cerus Corporation. Subject to each grantee’s continued employment shares underlying the grants vest in three annual installments and are issuable at the end of the three-year vesting term. The weighted average grant date fair value of restricted stock units granted was $1.85, $0.0, and $6.99 during the years ended 2010, 2009 and 2008, respectively.
78
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Restricted stock unit grants made in connection with the Bonus Plan for Senior Management of Cerus Corporation are presented in the following table:
| Number of RSUs |
||||
| Balances at December 31, 2007 |
72,580 | |||
| Granted |
43,086 | |||
| Cancelled |
(3,431 | ) | ||
| Vested |
(27,577 | ) | ||
| Balances at December 31, 2008 |
84,658 | |||
| Granted |
— | |||
| Cancelled |
(6,485 | ) | ||
| Vested |
(40,306 | ) | ||
| Balances at December 31, 2009 |
37,867 | |||
| Granted |
76,532 | |||
| Cancelled |
— | |||
| Vested |
(25,999 | ) | ||
| Balances at December 31, 2010 |
88,400 | |||
Stock-based Compensation
The Company currently uses the Black-Scholes option pricing model to determine the fair value of stock options and employee stock purchase plan shares. The determination of the fair value of stock-based payment awards on the date of grant using an option-pricing model is affected by the Company’s stock price, as well as assumptions regarding a number of complex and subjective variables. The variables used to calculate the fair value of stock based payment awards using the Black-Scholes option pricing model, include the Company’s expected stock price volatility, actual and projected employee stock option exercise behaviors, including forfeitures, the risk-free interest rate and expected dividends.
The Company does not recognize stock based compensation on stock options that contain performance conditions, until such time as the performance criteria are probable of being achieved. As such, for the year ended December 31, 2010, the Company had not recorded any such stock based compensation for the 50,000 performance-based stock options granted during such period.
Expected Term
The Company estimates the expected term of options granted using a variety of factors. Where possible, the Company estimates the expected term of options granted by analyzing employee exercise and post-vesting termination behavior. To make this estimation, the Company analyzes the population of options granted by discreet, homogeneous groups. If the Company is unable to obtain sufficient information to estimate the expected term for a particular group, it estimates the expected term of the options granted by taking the average of the vesting term and the contractual term of the option, as illustrated in SAB 107 and SAB 110. The expected term of employee stock purchase plan shares is the term of each purchase period.
79
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Estimated Forfeiture Rate
The Company estimates the forfeiture rate of options at the time of grant and revises those estimates in subsequent periods if actual forfeitures differ from those estimates. The Company uses historical data to estimate pre-vesting option forfeitures and records stock-based compensation expense only for those awards that are expected to vest. The Company estimates the historic pre-vesting forfeiture rates by groups that possess a degree of homogeneity regarding average time to vest and expected term. All stock-based payment awards are amortized on a straight-line basis over the requisite service periods of the awards, which are generally the vesting periods.
Estimated Volatility
The Company estimates the volatility of its common stock by using historical volatility of its common stock. The Company has used significant judgment in making these estimates and will continue to monitor the availability of actively traded options on its common stock. If the Company determines that sufficient actively traded options on its common stock exist, it may consider a combination of historical and implied volatility, or solely implied volatility
Risk-Free Interest Rate
The Company bases the risk-free interest rate that it uses in the option valuation model on United States Treasury zero-coupon issues with remaining terms similar to the expected term on the options.
Expected Dividend
The Company does not anticipate paying any cash dividends in the foreseeable future and therefore uses an expected dividend yield of zero in the option valuation model.
The assumptions used to value option grants for three years ended December 31:
| 2010 | 2009 | 2008 | ||||
| Expected term (in years) |
4.32-6.25 | 4.34-6.25 | 5.25-6.50 | |||
| Volatility |
68.0-82.0% | 72.0-136.0% | 59.1%-84.1% | |||
| Risk free interest rate |
1.27-2.60% | 2.80%-4.03% | 2.80%-4.03% |
The assumptions used to value employee stock purchase rights for the three years ended December 31:
| 2010 | 2009 | 2008 | ||||||||||
| Expected term (in years) |
0.50 | 0.50 | 0.50 | |||||||||
| Volatility |
58.62-81.26 | % | 148 .8%-150.0 | % | 54.6%-78.8 | % | ||||||
| Risk free interest rate |
0.19-2.37 | % | 2.0 | % | 2.0%-4.4 | % | ||||||
Total stock-based compensation recognized on the Company’s consolidated statements of operations for the years ended December 31, 2010, 2009, and 2008, was as follows (in thousands):
| 2010 | 2009 | 2008 | ||||||||||
| Research and development |
$ | 376 | $ | 494 | $ | 531 | ||||||
| Selling, general and administrative |
1,452 | 1,563 | 1,614 | |||||||||
| $ | 1,828 | $ | 2,057 | $ | 2,145 | |||||||
80
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Activity under the Company’s stock option plans is set forth below (in thousands except per share amounts):
| Number of Options Outstanding |
Weighted Average Exercise Price per Share ($) |
|||||||
| Balances at December 31, 2007 |
5,173 | $ | 12.13 | |||||
| Granted |
941 | 4.45 | ||||||
| Cancelled |
(652 | ) | 21.17 | |||||
| Exercised |
(399 | ) | 2.86 | |||||
| Balances at December 31, 2008 |
5,063 | $ | 12.29 | |||||
| Granted |
2,229 | 1.11 | ||||||
| Cancelled |
(723 | ) | 8.27 | |||||
| Exercised |
(4 | ) | 2.39 | |||||
| Balances at December 31, 2009 |
6,565 | $ | 7.38 | |||||
| Granted |
981 | 2.90 | ||||||
| Cancelled |
(337 | ) | 17.83 | |||||
| Exercised |
(202 | ) | 1.62 | |||||
| Balances at December 31, 2010 |
7,007 | $ | 6.42 | |||||
The weighted average fair value of options granted during the years ended December 31, 2010, 2009, and 2008, was $1.94, $2.93 and $2.88 per share, respectively. The intrinsic value of options exercised the years ended December 31, 2010, 2009, and 2008 was $0.3 million, $0.0 million, and $1.1 million per share, respectively.
Information regarding the stock options outstanding at December 31, 2010, 2009, and 2008 is set forth below (in thousands except per share amounts and years):
| Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Life (years) |
Aggregate Intrinsic Value |
|||||||||||||
| 2010 |
||||||||||||||||
| Shares Outstanding |
7,007 | $ | 6.42 | 6.22 | $ | 2,761 | ||||||||||
| Shares vested and expected to vest |
6,705 | $ | 6.60 | 6.08 | $ | 2,610 | ||||||||||
| Shares exercisable |
4,323 | $ | 8.93 | 4.83 | $ | 922 | ||||||||||
| 2009 |
||||||||||||||||
| Shares Outstanding |
6,565 | $ | 7.38 | 6.37 | $ | 1,891 | ||||||||||
| Shares vested and expected to vest |
6,165 | $ | 7.74 | 6.18 | $ | 1,617 | ||||||||||
| Shares exercisable |
3,901 | $ | 10.77 | 4.62 | $ | 220 | ||||||||||
| 2008 |
||||||||||||||||
| Shares Outstanding |
5,063 | $ | 10.27 | 6.32 | $ | — | ||||||||||
| Shares vested and expected to vest |
4,802 | $ | 10.54 | 6.18 | $ | — | ||||||||||
| Shares exercisable |
3,503 | $ | 12.26 | 5.25 | $ | — | ||||||||||
As of December 31, 2010, the Company had stock-based compensation expense of $3.2 million related to non-vested stock options not yet recognized, which is expected to be recognized over an estimated weighted average period of 2.7 years. As of December 31, 2010, the Company had stock-based compensation expense of $0.2 million related to restricted stock units, which is expected to be recognized over an estimated weighted average period of 1.75 years.
81
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Note 14. Development and License Agreements
Agreements with Baxter and Fenwal
In connection with the transfer of commercialization rights to the Company in February 2006, Baxter International Inc., or Baxter, agreed to supply, at the Company’s expense, certain transition services, including regulatory, technical and related administrative support through December 31, 2006. During that 2006 transition period, the Company recorded receivables of $2.8 million from Baxter and payables of $4.7 million to Baxter, associated with those transition services. The Company and Baxter had disputed the amounts owed, although the Company recorded the transition service receivables and payables on its consolidated balance sheets. In December 2009, the Company and Baxter entered into a settlement agreement with both parties waiving all rights and obligations associated with the 2006 transition services. In consideration for agreeing to the settlement, the Company agreed to pay Baxter $0.5 million which was recorded as a payable on its December 31, 2009 consolidated balance sheet. The $0.5 million payable was paid by the Company in 2010.
As a result of Baxter’s sale of its transfusion therapies division in 2007 to Fenwal, the Company has certain agreements with Fenwal which require the Company to pay royalties on future INTERCEPT Blood System product sales at royalty rates that vary by product: 10% of product sales for the platelet system, 3% for the plasma system, 5% for the red blood cell system, and 6.5% on sales of UVA illuminators. During the years ended December 31, 2010, 2009, and 2008, the Company made royalty payments of $2.0 million, $0.9 million, and $1.1 million, respectively. At December 31, 2010 and December 31, 2009, the Company owed royalties to Fenwal of $0.5 million and $0.8 million, respectively.
In December 2008, the Company extended its agreement with Fenwal to manufacture finished disposable kits for the platelet and plasma systems through December 31, 2013. Under the amended manufacturing agreement, the Company pays Fenwal a set price per kit, which is established annually plus a fixed surcharge per kit. In addition, volume driven manufacturing overhead is be paid or refunded if actual manufacturing volumes are lower or higher than the annually estimated production volumes. The Company made payments to Fenwal of $8.6 million, $5.3 million, and $8.0 million relating to the manufacturing of the Company products during the years ended December 31, 2010, 2009, and 2008, respectively. At December 31, 2010 and December 31, 2009, the Company owed Fenwal of $2.3 million and $3.7 million, respectively, for INTERCEPT disposable kits manufactured.
Cooperative Agreements with the United States Armed Forces
Since February 2001, the Company has received awards under cooperative agreements with the Army Medical Research Acquisition Activity division of the Department of Defense. The Company received these awards in order to develop its pathogen inactivation technologies for the improved safety and availability of blood that may be used by the United States Armed Forces for medical transfusions. Under the conditions of the agreements, the Company is conducting research on the inactivation of infectious pathogens in blood, including unusual viruses, bacteria and parasites that are of concern to the United States Armed Forces. This funding supports advanced development of the Company’s red blood cell system. The Company recognized $1.4 million, $1.2 million, $1.0 million, of revenue under these agreements during the years ended December 31, 2010, 2009, and 2008, respectively.
82
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Note 15. Income Taxes
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes at the enacted rates. Significant components of the Company’s deferred tax assets are as follows (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Net operating loss carryforwards |
$ | 132,600 | $ | 123,900 | ||||
| Research and development credit carryforwards |
31,600 | 32,000 | ||||||
| Capitalized inventory costs |
700 | 700 | ||||||
| Inventory reserve |
200 | 100 | ||||||
| Capitalized research and development |
13,000 | 18,200 | ||||||
| Capitalized trademark |
300 | — | ||||||
| Capitalized revenue sharing rights |
900 | 1,200 | ||||||
| Deferred compensation |
3,600 | 3,000 | ||||||
| Accrued liabilities |
300 | 100 | ||||||
| Depreciation |
2,200 | 2,900 | ||||||
| Capital loss carryforwards |
3,900 | — | ||||||
| Total deferred tax assets |
189,300 | 182,100 | ||||||
| Valuation allowance |
(189,300 | ) | (182,100 | ) | ||||
| Net deferred tax assets |
$ | — | $ | — | ||||
The valuation allowance increased by $7.2 million, $2.5 million, and $9.5 million for the years ended December 31, 2010, 2009, and 2008, respectively. The Company believes that, based on a number of factors, the available objective evidence creates sufficient uncertainty regarding the realizability of the deferred tax assets such that a full valuation allowance has been recorded. These factors include the Company’s history of net losses since its inception, the need for regulatory approval of the Company’s products prior to commercialization, expected near-term future losses and the absence of taxable income in prior carryback years. The Company expects to maintain a full valuation allowance until circumstances change. Undistributed earnings of the Company’s foreign subsidiary, Cerus Europe B.V., amounted to approximately $0.3 million at December 31, 2010. The earnings are considered to be permanently reinvested and accordingly, no deferred United States income taxes have been provided thereon. Upon distribution of those earnings in the form of dividend or otherwise, the Company would be subject to United States income tax. At the Federal statutory income tax rate of 35%, this would result in taxes of approximately $0.1 million.
For the year ended December 31, 2010, the Company reported net losses of $16.9 million on its consolidated statement of operations and calculated taxable losses for both federal and state taxes. The difference between reported net loss and taxable loss are due to temporary differences between book accounting and the respective tax laws.
At December 31, 2010, the Company had net operating loss carryforwards of approximately $333.9 million for federal and $326.9 million for state income tax purposes. The Company also had research and development tax credit carryforwards of approximately $21.4 million for federal income tax purposes and approximately $15.4 million for state income tax purposes at December 31, 2010. The federal net operating loss and tax credit carryforwards expire between the years 2011 and 2030. The state net operating loss carryforwards expire between the years 2012 and 2030. The state research and development credits do not expire.
83
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
The utilization of net operating loss carryforwards, as well as research and development credit carryforwards, is limited by current tax regulations. These net operating loss carryforwards, as well as research and development credit carryforwards, will be utilized in future periods if sufficient income is generated. The Company believes it more likely than not that its tax positions would be recognized upon review by a taxing authority having full knowledge of all relevant information. The Company’s ability to utilize certain loss carryforwards and certain research credit carryforwards are subject to limitations pursuant to the ownership change rules of Internal Revenue Code Section 382.
Note 16. Retirement Plan
The Company maintains a defined contribution savings plan, or the 401(k) Plan, that qualifies under the provisions of Section 401(k) of the Internal Revenue Code and covers all employees of the Company. Under the terms of the 401(k) Plan, employees may make pre-tax dollar contributions of up to 60% of their eligible pay up to a maximum cap established by the IRS. The Company may contribute a discretionary percentage of qualified individual employee’s salaries, as defined, to the 401(k) Plan. The Company did not contribute to the 401(k) Plan in the years ended December 31, 2010, 2009, and 2008.
Note 17. Segment Information and Geographic Information
At December 31, 2010, and 2009, the Company operated only one segment, blood safety. The Company’s chief executive officer is the chief operating decision maker who evaluates performance based on the net revenues and operating income (loss) of the blood safety segment.
The Company’s operations outside of the United States include a wholly-owned subsidiary headquartered in Europe. The Company’s operations in the United States are responsible for the research and development and global commercialization of the INTERCEPT Blood System, while operations in Europe are responsible for the commercialization efforts of the platelet and plasma systems in Europe the CIS, and the Middle East. Revenues are attributed to each region based on the location of the customer, and in the case of non-product revenues, on the location of the collaboration partner.
During the years ended December 31, 2010, 2009, and 2008, the Company had the following significant customers, listed as a percentage of product revenue:
| 2010 | 2009 | 2008 | ||||||||||
| Customer |
||||||||||||
| Movaco, S.A. (Spain and other EU countries) |
19 | % | 25 | % | 33 | % | ||||||
| Delrus, Inc. (CIS countries) |
16 | % | 6 | % | 10 | % | ||||||
| Etablissement Francais du Sang (France) |
13 | % | 24 | % | 18 | % | ||||||
| Service Francophone du Sang (Belgium) |
12 | % | 12 | % | 1 | % | ||||||
Long-lived assets are attributed to each region based on the physical location of the asset and are as follows (in thousands):
| 2010 | 2009 | |||||||
| Total Long-Lived Assets: |
||||||||
| United States |
$ | 3,883 | $ | 443 | ||||
| Europe |
797 | 1,283 | ||||||
| Totals |
$ | 4,680 | $ | 1,726 | ||||
84
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
Note 18. Quarterly Financial Information (Unaudited and in thousands except per share amounts)
| Three Months Ended | ||||||||||||||||
| March 31, 2010 |
June 30, 2010 |
September 30, 2010 |
December 31, 2010 |
|||||||||||||
| Revenue: |
||||||||||||||||
| Product revenue |
$ | 5,500 | $ | 5,690 | $ | 4,521 | $ | 5,965 | ||||||||
| Government grants and cooperative agreements |
222 | 245 | 470 | 496 | ||||||||||||
| Total revenue |
5,722 | 5,935 | 4,991 | 6,461 | ||||||||||||
| Cost of product revenue |
3,158 | 2,934 | 2,324 | 3,630 | ||||||||||||
| Gross profit |
2,564 | 3,001 | 2,667 | 2,831 | ||||||||||||
| Operating expenses |
||||||||||||||||
| Research and development |
1,250 | 1,244 | 1,282 | 1,419 | ||||||||||||
| Selling, general, and administrative |
5,270 | 5,304 | 5,089 | 5,913 | ||||||||||||
| Acquisition related costs and impairment of long-term investments in related parties, net1 |
251 | 132 | (201 | ) | — | |||||||||||
| Intangible asset amortization |
— | — | — | 67 | ||||||||||||
| Total operating expenses1 |
6,771 | 6,680 | 6,170 | 7,399 | ||||||||||||
| Loss from Operations |
(4,207 | ) | (3,679 | ) | (3,503 | ) | (4,568 | ) | ||||||||
| Other income (expense), net |
(1,066 | ) | (1,880 | ) | (128 | ) | 2,120 | |||||||||
| Net loss |
$ | (5,273 | ) | $ | (5,559 | ) | $ | (3,631 | ) | $ | (2,448 | ) | ||||
| Net loss per share—basic1 |
$ | (0.14 | ) | $ | (0.14 | ) | $ | (0.09 | ) | $ | (0.06 | ) | ||||
| Net loss per share—diluted1 |
$ | (0.14 | ) | $ | (0.14 | ) | $ | (0.09 | ) | $ | (0.06 | ) | ||||
| 1 | The company has adjusted amounts previously reported for total operating expenses, loss from operations, net loss, and net loss per share for the three months ended March 31, 2010, June 30, 2010, and September 30, 2010. These adjustments resulted from changes in the accounting for the acquisition of certain assets from BioOne. The change affected acquisition related costs and impairment of long-term investments in related parties, net by $0.3 million, $0.1 million, and $0.2 million for the three months ended March 31, 2010, June 30, 2010, and September 30, 2010, respectively. Loss per share was impacted by $(0.01), $0.00, and $0.01 per share for the three months ended March 31, 2010, June 30, 2010, and September 30, 2010, respectively. The impact of these changes is not considered material. |
85
Table of Contents
CERUS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
December 31, 2010
| Three Months Ended | ||||||||||||||||
| March 31, 2009 |
June 30, 2009 |
September 30, 2009 |
December 31, 2009 |
|||||||||||||
| Revenue: |
||||||||||||||||
| Product revenue |
$ | 3,085 | $ | 3,871 | $ | 4,567 | $ | 5,228 | ||||||||
| Government grants and cooperative agreements |
403 | 335 | 247 | 246 | ||||||||||||
| Total revenue |
3,488 | 4,206 | 4,814 | 5,474 | ||||||||||||
| Cost of product revenue |
2,094 | 2,520 | 4,242 | 3,724 | ||||||||||||
| Gross profit |
1,394 | 1,686 | 572 | 1,750 | ||||||||||||
| Operating expenses |
||||||||||||||||
| Research and development |
2,012 | 1,625 | 1,230 | 1,505 | ||||||||||||
| Selling, general, and administrative |
6,101 | 5,409 | 5,340 | 5,017 | ||||||||||||
| Restructuring |
712 | 129 | 15 | (15 | ) | |||||||||||
| Acquisition related costs and impairment of long-term investments in related parties, net |
— | — | — | 1,536 | ||||||||||||
| Gain on operating settlement |
— | — | — | (1,381 | ) | |||||||||||
| Total operating expenses |
8,825 | 7,163 | 6,585 | 6,662 | ||||||||||||
| Operating loss |
(7,431 | ) | (5,477 | ) | (6,013 | ) | (4,912 | ) | ||||||||
| Other income (expense), net |
34 | (735 | ) | 376 | 23 | |||||||||||
| Net loss |
$ | (7,397 | ) | $ | (6,212 | ) | $ | (5,637 | ) | $ | (4,889 | ) | ||||
| Net loss per share—basic |
$ | (0.23 | ) | $ | (0.19 | ) | $ | (0.16 | ) | $ | (0.13 | ) | ||||
| Net loss per share—diluted |
$ | (0.23 | ) | $ | (0.19 | ) | $ | (0.16 | ) | $ | (0.13 | ) | ||||
86
Table of Contents
Pursuant to the requirement of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Concord, State of California, on the 16th day of March 2011.
| CERUS CORPORATION | ||
| By: | /s/ CLAES GLASSELL | |
| Claes Glassell | ||
| President and Chief Executive Officer | ||
Each person whose signature appears below constitutes and appoints Claes Glassell and Kevin D. Green, his true and lawful attorney-in-fact and agent, each acting alone, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any or all amendments to the Annual Report on Form 10-K and to file the same, with all exhibits thereto, and all documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /S/ CLAES GLASSELL Claes Glassell |
President, Chief Executive Officer and Director (Principal Executive Officer) |
March 16, 2011 | ||
| /S/ KEVIN D. GREEN Kevin D. Green |
Chief Accounting Officer and Vice President, Finance (Principal Financial and Accounting Officer) |
March 16, 2011 | ||
| /S/ B. J. CASSIN B. J. Cassin |
Chairman of the Board |
March 16, 2011 | ||
| /S/ TIMOTHY B. ANDERSON Timothy B. Anderson |
Director |
March 16, 2011 | ||
| /S/ LAURENCE M. CORASH Laurence M. Corash, M.D. |
Director |
March 16, 2011 | ||
| /S/ BRUCE C. COZADD Bruce C. Cozadd |
Director |
March 16, 2011 | ||
| /S/ WILLIAM R. ROHN William R. Rohn |
Director |
March 16, 2011 | ||
| /S/ GAIL SCHULZE Gail Schulze |
Director |
March 16, 2011 | ||
87
Table of Contents
INDEX TO EXHIBITS
| Exhibit Number |
Description of Exhibit | |
| 3.1.1(25) | Restated Certificate of Incorporation of Cerus Corporation, as amended to date. | |
| 3.2 (12) | Bylaws of Cerus Corporation. | |
| 4.2 (1) | Specimen Stock Certificate. | |
| 4.3 (21) | Stockholder Rights Plan, dated as of November 3, 1999, as amended as of August 6, 2001, between Cerus Corporation and Wells Fargo Bank, N.A. (formerly known as Norwest Bank Minnesota, N.A.). | |
| 4.4 (22) | Amendment to Rights Agreement, dated as of October 28, 2009, between Cerus Corporation and Wells Fargo Bank, N.A. (which includes the form of Rights Certificate as Exhibit B thereto). | |
| 4.5 (20) | Form of Registered Direct Common Warrant. | |
| 4.6 (27) | Form of Warrant to Purchase Common Stock | |
| 10.1 (1) | Form of Indemnity Agreement entered into between Cerus Corporation and each of its directors and executive officers. | |
| 10.2 (1)* | 1996 Equity Incentive Plan. | |
| 10.3 (1)* | Form of Incentive Stock Option Agreement under the 1996 Equity Incentive Plan. | |
| 10.4 (1)* | Form of Nonstatutory Stock Option Agreement under the 1996 Equity Incentive Plan. | |
| 10.5 (1)* | 1996 Employee Stock Purchase Plan Offering. | |
| 10.6 (1) | Industrial Real Estate Lease, dated October 1, 1992, between Cerus Corporation and Shamrock Development Company, as amended on May 16, 1994 and December 21, 1995. | |
| 10.7 (2) | Series B Preferred Stock Purchase Agreement, dated as of June 30, 1998, by and between Cerus Corporation and Baxter Healthcare Corporation. | |
| 10.8 (3)* | 1999 Equity Incentive Plan, adopted April 30, 1999, approved by stockholders July 2, 1999. | |
| 10.9 (4)* | Amended and Restated Employment Agreement with Howard G. Ervin, dated December 22, 2008. | |
| 10.10(5) | Lease, dated December 17, 1999 between Cerus Corporation and Redwoods Office Center, L.P. | |
| 10.11(5) | Lease, dated October 12, 2001 between Cerus Corporation and California Development, Inc. (the “Lease”) | |
| 10.12(8) | Second Amendment to Standard Industrial/Commercial Single-Tenant Lease-Net, dated as of September 18, 2008. | |
| 10.13(23) | Letter to California Development, Inc. exercising option to extend Lease | |
| 10.14(6) | Loan and Security Agreement, dated November 15, 2002, between Cerus Corporation and Baxter Capital Corporation. | |
| 10.15(7)* | 1999 Non-Employee Directors’ Stock Option Sub-Plan, amended December 4, 2002. | |
| 10.16(4)* | Amended and Restated Employment Agreement with Claes Glassell, dated December 19, 2008. | |
| 10.17(22)† | Restructuring Agreement, dated as of February 2, 2005, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | |
| 10.18(22)† | License Agreement, dated as of February 2, 2005, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | |
88
Table of Contents
| Exhibit Number |
Description of Exhibit | |
| 10.19(9)† | Manufacturing and Supply Agreement, dated as of February 2, 2005, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | |
| 10.20(24)* | Bonus Plan for Senior Management of Cerus Corporation, as amended March 3, 2010. | |
| 10.21(10)† | Commercialization Transition Agreement, dated as of February 12, 2006, by and among Cerus Corporation, Baxter Healthcare S.A. and Baxter Healthcare Corporation. | |
| 10.22(11) | Offer Letter to Gail Schulze, dated October 15, 2007. | |
| 10.23(13) | 2008 Equity Incentive Plan | |
| 10.24(14) | Supply Agreement, dated December 19, 2007, by and between Cerus Corporation and Brotech Corporation d/b/a Purolite Company. | |
| 10.25(14) | Supply and Manufacturing Agreement, dated March 1, 2008, by and between Cerus Corporation and Porex Corporation. | |
| 10.26(15)† | Amended and Restated Manufacturing and Supply Agreement, dated December 12, 2008, by and between Cerus Corporation and Fenwal, Inc. | |
| 10.27(15)† | Manufacturing and Supply Agreement, dated September 30, 2008, by and between Cerus Corporation and NOVA Biomedical Corporation. | |
| 10.28(15)* | Non-Employee Director Compensation Policy. | |
| 10.29(16)* | Cerus Corporation Change of Control Severance Benefit Plan, as amended. | |
| 10.30(16)* | Form of Restricted Stock Unit Agreement under the 1999 Equity Incentive Plan, as amended. | |
| 10.31(17)* | Form of Indemnity Agreement, adopted April 24, 2009. | |
| 10.32(18)* | Employment Letter for Kevin D. Green dated May 1, 2009. | |
| 10.33(19)* | Form of Severance Benefits Agreement. | |
| 10.34(24)* | Employment Letter, by and between Cerus Corporation and Laurence Corash, dated March 2, 2010. | |
| 10.35(20) | Form of Subscription Agreement. | |
| 10.36(28)†† | Loan and Security Agreement, by and between Cerus Corporation and Oxford Finance Corporation, dated March 31, 2010. | |
| 10.37(26)† | Asset Purchase and Redemption Agreement by and between Cerus Corporation and BioOne Corporation, dated as of August 24, 2010. | |
| 12.1 | Computation of Earnings to Fixed Charges Ratio(28) | |
| 21.1 | List of Registrant’s subsidiaries. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (see signature page). | |
| 31.1 | Certification of the Chief Executive Officer of Cerus Corporation pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of the Chief Accounting Officer of Cerus Corporation pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification of the Chief Executive Officer and Chief Accounting Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
89
Table of Contents
| † | Certain portions of this exhibit are subject to a confidential treatment order. |
| †† | Registrant has requested confidential treatment for portions of this exhibit. |
| * | Compensatory Plan. |
| (a) | Previously filed. |
| (1) | Incorporated by reference to Cerus’ Registration Statement on Form S-1 (File No. 333-11341) and amendments thereto. |
| (2) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on July 22, 1998. |
| (3) | Incorporated by reference to Cerus’ Registration Statement on Form S-8, dated August 4, 1999. |
| (4) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on December 23, 2008. |
| (5) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2001. |
| (6) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2002. |
| (7) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2003. |
| (8) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended September 30, 2008. |
| (9) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2005. |
| (10) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2006. |
| (11) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2007. |
| (12) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on June 19, 2008. |
| (13) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on June 6, 2008. |
| (14) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2008. |
| (15) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2008. |
| (16) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended March 31, 2009. |
| (17) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on April 30, 2009. |
| (18) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended June 30, 2009. |
| (19) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on June 1, 2009. |
| (20) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on August 20, 2009. |
| (21) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended June 30, 2009. |
| (22) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on October 30, 2009. |
| (23) | Incorporated by reference to Cerus’ Annual Report on Form 10-K, for the year ended December 31, 2009. |
| (24) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on March 8, 2010. |
| (25) | Incorporated by reference to Cerus’ Quarterly Report on Form 10-Q, for the quarter ended June 30, 2010. |
| (26) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on August 30, 2010. |
| (27) | Incorporated by reference to Cerus’ Current Report on Form 8-K, filed with the SEC on November 12, 2010. |
| (28) | Filed herewith. |
90