Attached files
| file | filename |
|---|---|
| EX-31.2 - PROLOR Biotech, Inc. | v214352_ex31-2.htm |
| EX-32.2 - PROLOR Biotech, Inc. | v214352_ex32-2.htm |
| EX-32.1 - PROLOR Biotech, Inc. | v214352_ex32-1.htm |
| EX-23.1 - PROLOR Biotech, Inc. | v214352_ex23-1.htm |
| EX-31.1 - PROLOR Biotech, Inc. | v214352_ex31-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 10-K
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the fiscal year ended December 31, 2010
|
¨
|
TRANSITION REPORT PURSUANT TO SECTIONS 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from to
COMMISSION FILE NUMBER 001-34676
PROLOR BIOTECH, INC.
(Exact Name of Registrant as Specified in Its Charter)
|
Nevada
|
20-0854033
|
|
(State or other jurisdiction of
incorporation or organization)
|
(IRS Employer
Identification No.)
|
3 Sapir Street, Weizmann Science Park
Nes-Ziona, Israel 74140
(Address of principal executive offices) (zip code)
Registrant’s Telephone Number, Including Area Code: (866) 644-7811
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class
|
Name of Each Exchange On Which Registered
|
|
Common Stock, par value $0.00001 per share
|
NYSE Amex
|
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act.
Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes o No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Accelerated filer x
|
|
|
Non-accelerated filer ¨ (Do not check if a smaller reporting company)
|
Smaller reporting company ¨
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 if the Act).
Yes ¨ No x
The aggregate market value of the registrant’s outstanding common stock held by non-affiliates of the registrant computed by reference to the price at which the common stock was last sold as of the last business day of the registrant’s most recently completed second fiscal quarter was $260,112,782 (based on a closing price of $6.89 per share for the registrant’s common stock on the NYSE Amex on June 30, 2010).
As of March 9, 2011, the registrant had 54,217,106 shares of common stock outstanding.
The registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A for the 2010 annual meeting of stockholders is incorporated by reference in Part III of this Form 10-K to the extent stated herein.
INDEX AND CROSS REFERENCE SHEET
|
PART I
|
3
|
|
|
Item 1.
|
Business
|
3
|
|
Item 1A.
|
Risk Factors
|
13
|
|
Item 1B.
|
Unresolved Staff Comments
|
25
|
|
Item 2.
|
Properties
|
25
|
|
Item 3.
|
Legal Proceedings
|
25
|
|
Item 4.
|
(Removed and Reserved)
|
26
|
|
PART II
|
26
|
|
|
Item 5.
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
|
26
|
|
Item 6.
|
Selected Financial Data
|
27
|
|
Item 7.
|
Management’s Discussion and Analysis of Financial Condition and Results of Operation
|
27
|
|
Item 7A.
|
Quantitative and Qualitative Disclosures About Market Risk
|
35
|
|
Item 8.
|
Financial Statements and Supplementary Data
|
36
|
|
Item 9.
|
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
|
36
|
|
Item 9A.
|
Controls and Procedures
|
36
|
|
Item 9B.
|
Other Information
|
36
|
|
PART III
|
37
|
|
|
Item 10.
|
Directors, Executive Officers and Corporate Governance
|
37
|
|
Item 11.
|
Executive Compensation
|
37
|
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
|
37
|
|
Item 13.
|
Certain Relationships and Related Transactions, and Director Independence
|
37
|
|
Item 14.
|
Principal Accounting Fees and Services
|
37
|
|
PART IV
|
37
|
|
|
Item 15.
|
Exhibits and Financial Statement Schedules
|
37
|
|
SIGNATURES
|
||
i
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains certain forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 (“PSLRA”), Section 27A of the Securities Act of 1933, as amended (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934, as amended, (the “Exchange Act”), about our expectations, beliefs or intentions regarding, among other things, our product development efforts, business, financial condition, results of operations, strategies or prospects. You can identify forward-looking statements by the fact that these statements do not relate strictly to historical or current matters. Rather, forward-looking statements relate to anticipated or expected events, activities, trends or results as of
the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements. Risks and uncertainties, the occurrence of which could adversely affect our business, include the following:
|
|
·
|
our limited operating history;
|
|
|
·
|
our lack of any commercialized products or technologies;
|
|
|
·
|
our product candidates are at an early stage of product development and may never be commercialized;
|
|
|
·
|
that we may be unable to develop product candidates that will achieve commercial success in a timely and cost-effective manner, or ever;
|
|
|
·
|
our need to raise additional capital to meet our business requirements in the future, and such capital raising may be costly or difficult to obtain and could dilute current stockholders’ ownership interests;
|
|
|
·
|
if we fail to obtain necessary funds for our operations, we will be unable to maintain and improve our patented technology, and we will be unable to develop and commercialize our products and technologies;
|
|
|
·
|
our dependence on key members of our management and advisory team;
|
|
|
·
|
our potential inability to enforce employees’ covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees;
|
|
|
·
|
our current lack of sales, marketing or distribution capabilities;
|
|
|
·
|
potential product liability claims if our product candidates cause harm to patients;
|
|
|
·
|
our product candidates will remain subject to ongoing regulatory requirements even if they receive marketing approval, and if we fail to comply with these requirements, we could lose these approvals, and the sales of any approved commercial products could be suspended;
|
|
|
·
|
clinical trials are very expensive, time-consuming and difficult to design and implement and, as a result, we may suffer delays or suspensions in future trials which would have a material adverse effect on our ability to generate revenues;
|
|
|
·
|
the manufacture of our product candidates is an exacting and complex process, and if we or one of our materials suppliers encounters problems manufacturing its products, our business could suffer;
|
|
|
·
|
our reliance on third parties to implement our manufacturing and supply strategies;
|
|
|
·
|
our inability to successfully integrate any acquisitions of technologies or products;
|
|
|
·
|
our inability to successfully grow and expand our business;
|
|
|
·
|
our inability to obtain adequate insurance;
|
|
|
·
|
our holding company structure and our dependence on cash flow from our wholly-owned subsidiaries to meet our obligations;
|
|
|
·
|
our inability to maintain an effective system of internal controls;
|
|
|
·
|
the impact of potential political, economic and military instability in the State of Israel, where key members of our senior management and our research and development facilities are located;
|
|
|
·
|
recent disruptions in the financial markets and global economic conditions;
|
1
|
|
·
|
our dependence on our license of core technology from Washington University;
|
|
|
·
|
our failure to obtain or maintain or protect our patents, licensing agreements and other intellectual property;
|
|
|
·
|
the cost of potential litigation required to protect our intellectual property or to defend against claims alleging that we have violated the intellectual property rights of others;
|
|
|
·
|
our inability to enforce confidentiality agreements, which could result in third parties using our intellectual property to compete against us;
|
|
|
·
|
uncertainty regarding international patent protection;
|
|
|
·
|
our inability to protect intellectual property rights of the third parties from whom we license certain of our intellectual property or with whom we have entered into other strategic relationships;
|
|
|
·
|
our inability to obtain required regulatory approvals in the United States to market our proposed product candidates;
|
|
|
·
|
the impact of government regulations and delays associated with obtaining required regulatory approvals in the United States necessary to market our proposed product candidates;
|
|
|
·
|
the impact of competition and continuous technological change;
|
|
|
·
|
the impact of healthcare reform, which could adversely impact how much or under what circumstances healthcare providers will prescribe or administer our products;
|
|
|
·
|
compliance with federal anti-kickback laws and regulations;
|
|
|
·
|
the volatility of our common stock;
|
|
|
·
|
that we do not anticipate paying dividends on our common stock;
|
|
|
·
|
a lack of security analyst coverage of our company;
|
|
|
·
|
potential dilution of your ownership interest because of future issuances of additional shares of our common stock and our preferred stock;
|
|
|
·
|
that our principal stockholders have significant voting power and may take actions that may not be in the best interest of other stockholders;
|
|
|
·
|
sales by stockholders who had previously been subject to restrictions on the sale of their shares and who may now sell those shares into the public market;
|
|
|
·
|
the other factors referenced in this prospectus, including, without limitation, under “Risk Factors;” and
|
|
|
·
|
other risks detailed from time to time in the reports filed by us with the SEC.
|
We believe these forward-looking statements are reasonable; however, you should not place undue reliance on any forward-looking statements, which are based on current expectations. Furthermore, forward-looking statements speak only as of the date they are made. If any of these risks or uncertainties materialize, or if any of our underlying assumptions are incorrect, our actual results may differ significantly from the results that we express in, or imply by, any of our forward-looking statements. These and other risks are detailed in this Annual Report on Form 10-K, in the documents that we incorporate by reference into this Annual Report on Form 10-K and in other documents that we file with the Securities and Exchange Commission. We do not undertake any obligation to publicly update or revise these
forward-looking statements after the date of this Annual Report on Form 10-K to reflect future events or circumstances. We qualify any and all of our forward-looking statements by these cautionary factors.
2
PART I
|
Item 1.
|
Business
|
Unless the context otherwise requires, all references in this Annual Report on Form 10-K to the “Company”, “Prolor”, “we,” “us” and “our” refer to PROLOR Biotech, Inc., a Nevada corporation (formerly Modigene Inc.), including its direct and indirect wholly-owned subsidiaries, Modigene, Inc., a Delaware corporation, which we refer to as Modigene Delaware, and ModigeneTech Ltd., which we refer to as ModigeneTech.
History
We were originally incorporated under the laws of the State of Delaware in August 2003 as LDG, Inc., referred to as LDG, which was engaged in the graphics design, marketing and advertising business. On February 26, 2007, LDG, Inc. changed its name to Modigene Inc, and on May 9, 2007, its wholly-owned subsidiary Modigene Acquisition Corp. merged with and into Modigene Delaware. Modigene Delaware survived the merger, following which the original business of LDG was abandoned in its entirety, and we have operated the business of Modigene Delaware and its wholly-owned subsidiary ModigeneTech.
On June 10, 2009, we changed our name from Modigene Inc. to PROLOR Biotech, Inc., and on June 12, 2009 the trading symbol for our common stock on the OTCBB changed from MODG to PBTH. On March 29, 2010 we listed for trading on the NSYE AMEX under the trading symbol PBTH, and on May 27, 2010 we listed for trading on the Tel-Aviv Stock Exchange.
Overview
We are a development stage biopharmaceutical company utilizing patented technology to develop longer-acting, proprietary versions of already-approved therapeutic proteins that currently generate billions of dollars in annual global sales. We have obtained certain exclusive worldwide rights from Washington University in St. Louis, Missouri to use a short, naturally-occurring amino acid sequence (peptide) that has the effect of slowing the removal from the body of the therapeutic protein to which it is attached. This Carboxyl Terminal Peptide (CTP) can be readily attached to a wide array of existing therapeutic proteins, stabilizing the therapeutic protein in the bloodstream and extending its life span without additional toxicity or loss of desired biological activity. We are using
the CTP technology to develop new, proprietary versions of certain existing therapeutic proteins that have longer life spans than therapeutic proteins without CTP. We believe that our products will have greatly improved therapeutic profiles and distinct market advantages.
We believe our products in development will provide several key advantages over our competitor’s existing products:
|
|
·
|
significant reduction in the number of injections required to achieve the same or superior therapeutic effect from the same dosage;
|
|
|
·
|
extended patent protection for proprietary new formulations of existing therapies;
|
|
|
·
|
faster commercialization with greater chance of success and lower costs than those typically associated with a new therapeutic protein; and
|
|
|
·
|
manufacturing using industry-standard biotechnology-based protein production processes.
|
Merck & Co. has developed the first novel protein containing CTP, named ELONVA®, a long-acting CTP-modified version of the fertility drug follicle stimulating hormone (FSH). On January 28, 2010, Merck received marketing authorization from the European Commission for ELONVA® with unified labeling valid in all European Union Member States. Merck licensed the CTP technology directly from Washington University (prior to the formation of Modigene Delaware) for application only to Follicle Stimulating Hormone (FSH) and three other hormones, human Chorionic Gonadotropin (hCG), Luteinizing Hormone (LH) and Thyroid-Stimulating Hormone (TSH).
3
Our internal product development program is currently focused on extending the life span of the following biopharmaceuticals, in an effort to provide patients with improved therapies that may enhance their quality of life:
|
|
·
|
Human Growth Hormone (hGH)
|
|
|
·
|
Factor IX
|
|
|
·
|
Anti-Obesity Peptide Oxyntomodulin
|
|
|
·
|
Factor VIIa
|
|
|
·
|
Interferon β and Erythropoietin (EPO)
|
|
|
·
|
Atherosclerosis and rheumatoid arthritis long-acting therapies
|
We believe that the CTP technology will be broadly applicable to these as well as other best-selling therapeutic proteins in the market and will be attractive to potential partners because it will allow them to extend proprietary rights for therapeutic proteins with near-term patent expirations.
Discovery, Development and Clinical Experience with CTP Technology
Our core technology was developed by Washington University in St. Louis, while investigating the female hormone hCG, which facilitates pregnancy by maintaining production of progesterone and stimulating development of the fetus.
hCG has a long life span of up to 2 days, meaning that the body is slow to break it down. LH is another female hormone having a chemical composition (amino acid sequence) very close to that of hCG. LH has a very short life span of 20 minutes. Scientists at Washington University discovered that the only difference between hCG and LH is a short amino-acid sequence present in hCG and not in LH which they called “CTP” for Carboxyl-Terminal Peptide. This is shown schematically below. When produced in mammalian cells, this CTP is heavily modified by sugars being added (a process called glycosylation). Through numerous experiments, it was confirmed that CTP was responsible for the longer life span of hCG as compared to
LH. Washington University then performed additional experimentation adding CTP to different therapeutic proteins and the results showed that the CTP-modified proteins had dramatically increased life span.

Prolor’s core technology is the use of a short, naturally occurring amino acid sequence (CTP) to slow the removal of therapeutic proteins from the body without increasing toxicity or altering the overall biological activity
Our scientific founder, Dr. Fuad Fares, was a post-doctoral student at Washington University and worked on these findings and experiments. When Dr. Fares returned to Israel in 2001, he formed ModigeneTech to license the CTP technology from Washington University for certain therapeutic indications.
Prior to Dr. Fares’ completion of our initial license agreement with Washington University, the Dutch biotech company Organon, now part of Merck & Co., licensed the CTP solution to be used in conjunction with four endocrine proteins: FSH, hCG, TSH and LH. Organon’s goal was to develop a longer-lasting version of their FSH product, marketed as Follistim® and required to be injected on a daily basis. There have been several attempts to create a long-lasting version of FSH utilizing existing technologies that compete with our CTP technology, including a PEGylated version, all of which have been abandoned or terminated.
4
In July 2008, Schering-Plough, now part of Merck, announced successful top-line data from its Phase III ENGAGE trial demonstrating that women receiving a single injection of FSH-CTP (now branded ELONVA®) achieved the same pregnancy rates as women receiving seven consecutive daily injections of FSH, a primary endpoint of the study. This 1,509 patient trial was the largest double-blind fertility trial ever conducted. On January 28, 2010, Merck received marketing authorization from the European Commission for ELONVA® with unified labeling valid in all European Union Member States. We are now the exclusive licensee for the utilization of CTP technology in all therapeutic proteins, peptides and their modified forms except for human FSH, LH, TSH and hCG.
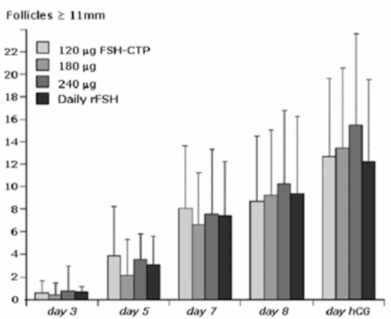
|
Stimulation of egg follicle growth in women preparing for IVF. Standard treatment of daily injection of rFSH (rightmost bar in each day’s group) is compared to a single injection of FSH-CTP in one of three dose levels. At the days shown, large (growing) follicles were detected by ultrasound. There were at least as many large follicles stimulated by each of the FSH-CTP doses as with the control group.
|
Opportunity Background
Overview of Therapeutic Proteins
Therapeutic proteins are proteins that are either extracted from human cells or engineered and produced in the laboratory for pharmaceutical use. The majority of therapeutic proteins are recombinant human proteins manufactured using non-human cell lines that are engineered to contain certain human genetic sequences which cause them to produce the desired protein. Recombinant proteins are an important class of therapeutics used to replace deficiencies in critical blood borne growth factors and to strengthen the immune system to fight cancer and infectious disease. Therapeutic proteins are also used to relieve patients’ suffering from many conditions, including various cancers (treated by monoclonal antibodies and interferons), heart attacks, strokes, cystic fibrosis and
Gaucher’s disease (treated by enzymes and blood factors), diabetes (treated by insulin), anemia (treated by erythopoietins), and hemophilia (treated by blood clotting factors).
The U.S. Food and Drug Administration (FDA) has approved 75 therapeutic proteins, also known as biopharmaceuticals, and there are more than 500 additional proteins under development. To date, much of the growth has been in sales of erythopoietins (used to treat anemia) and insulins (used to treat diabetes). Many of the proteins currently on the market will lose the protection of certain patent claims over the next 15 years. In addition, many marketed proteins are facing increased competition from next-generation versions or from other therapeutic proteins approved for the same disease indications.
5
Because proteins are broken down in the gastrointestinal system, therapeutic proteins must be administered by injection. Once in the bloodstream, therapeutic proteins are broken down by enzymes and cellular activity, as well as filtered out of the blood by the kidneys. Therefore, injections must be given frequently to achieve effective therapeutic levels. We believe that a large market opportunity exists for new versions of proven therapeutic proteins that remain active longer, thereby reducing the number of required injections and optimizing therapeutic results and patient acceptability. However, existing approaches to creating modified therapeutic proteins are generally based on the addition of synthetic, non-protein elements that result in problems such as loss of
desired biological activity, toxicity of the modified protein and increased manufacturing complexity and cost. Despite these challenges, several longer-lasting modified therapeutic proteins currently on the market have been demonstrated to be successful. Each of these improved therapeutics was custom-designed with great effort.
Attempts to Extend the Life Span of Therapeutic Proteins
Several strategies have been devised in recent years to extend the life span of therapeutic proteins by slowing their clearance from the body. These strategies have included two main techniques:
|
|
·
|
Increasing the size of the therapeutic protein. This is achieved either by attaching large polymeric chains to the protein (PEGylation) or by attaching other large, non-active proteins that have longer life spans compared to the target therapeutic protein.
|
|
|
·
|
Altering the physical structure of the therapeutic protein. This is achieved by adding carbohydrate structures to the therapeutic protein (glycosylation) through modifications of the original genetic sequence of the protein. These additional “sugar chains” slow the clearance of the therapeutic protein from the bloodstream.
|
Limitations of Existing Life Span Extension Solutions
There are several fundamental issues with the existing technologies that attempt to create longer-lasting versions of therapeutic proteins. If the size of the protein is increased by way of attaching large polymeric chains or another protein, the end result is a very large protein. Because most therapeutic proteins work by binding to specific receptors, the new “bulkiness” may prevent them from achieving the desired result. The smaller the protein, the more significant the effect of the size increase may be. Successful attempts at increasing the size of therapeutic proteins, while preserving substantial activity, have been relatively few, and have been with proteins that are already large. Moreover, the biological activity of the modified protein
has been significantly less than that of the unmodified protein and therefore requires a higher injected dose as compared to the unmodified protein’s usual dosage. One typical method to achieve the desired size increase is to add long polymers of polyethylene glycol (PEG) to a protein; however, this method has historically resulted in the creation of foreign structures to which the immune system may adversely react. When this happens, the immune system works to remove the modified protein from the bloodstream, defeating the purpose of the original modification. It can also lead to additional negative effects, such as reaction at the injection site.
Another technique, glycosylation, requires custom alterations (point mutations) to the protein’s genetic structure to increase its life span. The resulting modified protein is entirely new and often generates unexpected adverse reactions, resulting in potentially toxic effects. To date, creating a protein with a longer life span that is not toxic has been a lengthy trial and error process.
Although the existing modification technologies have been tried on almost all therapeutic proteins, only three “blockbuster” modified proteins have been commercially successful: two developed by Amgen Inc. and one independently developed by Schering-Plough Corporation and Roche Pharmaceuticals. Each of these three longer-lasting therapeutic proteins has become a widely used therapeutic:
|
|
·
|
utilizing PEGylation, Schering-Plough and Roche independently developed PEG-INTRON and PEGASYS, therapeutic proteins with a longer life span than that of regular Alpha interferon (used for treating Hepatitis B and C);
|
6
|
|
·
|
utilizing PEGylation, Amgen developed Neulasta, an anti-neutropenia therapeutic protein with a longer life span than that of regular G-CSF; and
|
|
|
·
|
utilizing additional glycosylation, Amgen developed Aranesp, an anti-anemia therapeutic protein with a longer life span than that of regular EPO.
|
Our Solution
Our solution to creating proprietary, enhanced longevity protein therapeutics is CTP, a short, naturally-occurring amino acid sequence that has the effect of slowing the removal and/or breakdown of the therapeutic protein to which it is attached. Through our license agreement with Washington University, we have secured exclusive, worldwide rights to use the CTP technology with respect to all natural and non-natural therapeutic proteins and peptides (other than LH, TSH, FSH and hCG, which are licensed to an affiliate of Schering-Plough Corp.), including hGH, EPO, interferon β and GLP-1. Using standard recombinant DNA techniques, the CTP peptide can be readily attached in one or more copies to a
wide array of existing therapeutic proteins. When these proteins are produced in mammalian cells, the CTP portion undergoes a natural process in which special carbohydrate chains are attached (O-linked glycosylation). This additional CTP piece, along with its carbohydrate chains, stabilizes the therapeutic protein in the bloodstream and greatly extends its life span, without additional toxicity or loss of its desired biological activity. This is quite distinct from other methods used to extend protein life span, which require the addition to the therapeutic drug of large proteins or of synthetic, non-protein elements that may result in problems such as loss of desired biological activity or toxicity of the modified protein, as well as increased manufacturing complexity and cost. Moreover, CTP-modified proteins can be manufactured using established and widely used mammalian protein expression systems (cell lines). Therefore, we
believe that the technology risks are minimized, while the benefits of the CTP technology can be substantial.
There are two existing biopharmaceuticals that utilize CTP technology. The first product is hCG, of which CTP is naturally a part. Besides being present normally in high amounts during pregnancy, it is also given therapeutically to women or men as a fertility treatment (sold by Merck-Serono, Merck & Co. and Ferring). The second product is ELONVA® (FSH-CTP), which is approved for marketing in Europe as described above. The data from the use in humans of these two products give us confidence that the CTP technology may be able to address the major problems faced by the other attempted approaches to increase protein lifespan. Data from these products reassures us that CTP can be used safely in humans and that it is effective in extending the serum
lifetime and activity in humans.
We believe the clinical development program for our drugs will be faster, less expensive and more predictable than those conducted for existing therapeutic proteins. We can base the design of our studies, the inclusion criteria, clinical endpoints and sample sizes, on the knowledge gained from development of the predecessor drugs, with the assurance that these have been accepted by regulatory authorities in the past. In addition there are usually surrogate markers for clinical efficacy that have been defined and accepted by the medical community. These can provide easier and faster ways of learning at an early stage the correct dosing range and frequency. In some cases, they can even be used as definitive clinical trial endpoints. We believe that these
factors drive will down the time and costs associated with clinical trials.
Research & Development: Our Development Programs
We are currently pursuing the development and commercialization of six products: Human Growth Hormone, Factor IX, anti-obesity peptide Oxyntomodulin, Factor VIIa, Interferon β, Erythropoietin, and anti-atherosclerosis and rheumatoid arthritis therapeutics.
Human Growth Hormone (hGH)
Market Opportunity
Growth hormone deficiency (GHD) is a pituitary disorder resulting in short stature in children and other physical ailments in both children and adults. GHD occurs when the production of growth hormone, secreted by the pituitary gland, is disrupted. Since growth hormone plays a critical role in stimulating body growth and development, and is involved in the production of muscle protein and in the breakdown of fats, a decrease in the hormone affects numerous body processes.
Recombinant hGH is used for the long-term treatment of children and adults with growth failure due to inadequate secretion of endogenous growth hormone. The primary indications it treats in children are growth hormone deficiency, kidney disease, Prader-Willi Syndrome and Turner’s Syndrome. In adults, the primary indications are replacement of endogenous growth hormone and the treatment of AIDS-induced weight loss.
7
In addition to its current use, hGH has been proven to promote a number of lifestyle benefits including weight loss, increased energy levels, enhanced sexual performance, improved cholesterol, younger, tighter, thicker skin and reduced wrinkles and cellulite. We expect the hGH market to expand significantly as hGH moves beyond therapeutic treatment to include the treatment of lifestyle issues.
Current Products
Prior to the advent of recombinant versions, growth hormone was purified from human cadavers. For the past 20 years, recombinantly produced protein has been supplied to the market by an increasing number of companies. Current products on the U.S. market are Nutropin (Genentech), Genotropin (Pfizer), Humatrope (Eli Lilly), Norditropin (Novo Nordisk), Serostim (Merck-Serono) and Omnitrope (Novartis).
Our hGH-CTP Program
Patients using hGH receive daily injections six or seven times a week. This is particularly burdensome for pediatric patients. We believe a significant market opportunity exists for a longer-lasting version of hGH that would require fewer injections.
We reported positive top-line results from a Phase I study of our longer-acting version of hGH. The study was designed to measure the potential durability (half-life), overall drug exposure (AUC) and biological efficacy, as well as the safety and tolerability of our longer-acting CTP-modified human growth hormone (hGH-CTP).
The Phase I study enrolled 24 healthy adults who were randomized to receive one of three doses of hGH-CTP (4mg, 7mg or 21mg) or placebo. The study results showed that safety and tolerability endpoints were met at all doses in all participants. The potential clinical efficacy of hGH-CTP was assessed by measuring the extent to which hGH-CTP induced insulin-like growth factor-1 (IGF-1) in subjects. This biomarker is the clinically accepted primary indicator of hGH biological activity and is used by endocrinologists to optimize dosing for hGH-deficient adults. Based on this measure, the study results suggest that the daily injections required by patients using conventional hGH could potentially be replaced with just two monthly injections of hGH-CTP.
Following the positive results of our Phase I clinical trial in hGH-CTP, on July 21, 2010 we received regulatory approval and initiated a Phase II clinical trial in growth hormone deficient adults. The trial is designed as a dose-finding study, and the primary efficacy end point is defined as the mean time interval of IGF-I (insulin-like growth factor 1) levels that lay within ±1.5 SDS (standard deviation from the mean IGF-1 levels in the population) after the last dose administration expressed in hours. The other primary outcomes measured are safety and tolerability.
Factor IX
Market Opportunity
Hemophilia is a group of hereditary genetic disorders that impair the body's ability to control blood clotting or coagulation. People with hemophilia do not produce adequate amounts of Factor VIII or Factor IX proteins, which are necessary for effective blood clotting. In severe hemophiliacs, even a minor injury can result in blood loss lasting days or weeks, and complete healing may not occur, leading to the potential for debilitating permanent damage to joints and other organs and premature death. According to the World Health Organization, more than 400,000 people worldwide have hemophilia, corresponding to an incidence of 15 to 20 in every 100,000 males born worldwide. Hemophilia B is associated with inadequate Factor IX and occurs at an incidence of about 1 in
20,000–34,000 male births. Hemophilia B is largely an inherited disorder but, in approximately 30% of cases, there is no family history; and the condition is the result of a spontaneous gene mutation. The availability of recombinant Factor VIII and Factor IX has enabled many hemophiliacs to live near-normal lives, but frequent injections are required.
8
Current Products Produced by Others
Recombinant Factor IX is offered to Hemophilia B patients by Pfizer, under the brand name BeneFIX® (“BeneFIX”).
Our Factor IX-CTP Program
We reported top-line results from an animal efficacy study we conducted of Factor IX-CTP. This was a comparative study in Factor IX depleted mice, comparing the quality and duration activity of Factor IX-CTP with that of BeneFIX and included an untreated control group. Factor IX-CTP showed superior efficacy and duration of activity during the 60 hour study.
Our Business Strategy
Our goal is to become a leader in the development and commercialization of longer-lasting, proprietary versions of already approved therapeutic proteins that currently generate billions of dollars in annual global sales, through the utilization of our CTP technology. Key elements of our strategy are to:
|
|
·
|
Develop and commercialize improved versions of biopharmaceuticals that dramatically reduce the number of injections required to achieve the same therapeutic effect from the existing drugs. Based on the clinical track record of our CTP technology, as evidenced by the results of Merck’s FSH-CTP European marketing approval, we believe that the addition of CTP to therapeutic proteins significantly enhances the lifespan of those proteins, without any adverse effects. We expect these modified proteins to offer significant advantages, including less frequent dosing and possibly improved efficacy, over the original versions of the drugs now on the market, as well as to meet or exceed the pharmacokinetic profile of next-generation versions of the drugs now on the market.
|
|
|
·
|
Leverage extensive existing clinical and regulatory experience with the original drugs to bring our improved versions of these biopharmaceuticals to market more quickly, at lower costs and with a clearer path to regulatory approval. Because there is a large knowledge base on the original products, the preclinical, clinical and regulatory requirements needed to obtain marketing approval are very well defined. In particular, clinical study designs, inclusion criteria and endpoints can be used that have already been accepted by regulatory authorities. There typically exist accepted surrogate markers for clinical efficacy, which can sometimes even be used as definitive trial endpoints, but at the least are highly informative of proper dose range and frequency. All of these
factors drive down the time and costs associated with clinical trials, which represent up to 90% of product development costs for a typical therapeutic protein. In addition to lowering the costs and time to market, we believe the strategy of targeting drugs with proven safety and efficacy provides a better prospect of clinical success of our proprietary development portfolio as compared to de novo protein drug development. The possibility of delays due to regulatory safety concerns is also reduced as the FDA gains comfort with the safety profile of CTP-modified proteins (CTP is naturally present in the body, on the approved drug hCG and on FSH-CTP). We estimate that the average time to market and cost of clinical trials for our products could be up to 50% less than that required to develop a new therapeutic protein.
|
|
|
·
|
Seek attractive partnership opportunities. We believe that the CTP technology is applicable to most therapeutic proteins and peptides that have been approved to date by the FDA, including many of the best-selling therapeutic proteins in the market. We believe that the proprietary rights provided by CTP technology, together with the clinical and compliance benefits, will be attractive to potential partners, either the originator of the therapeutic protein or their prospective competitors. We will seek to build a portfolio of commercially attractive partnerships in a blend of co-developments and licenses. Where possible, we will seek partnerships that allow us to participate significantly in the commercial success of each of the compounds.
|
9
|
|
·
|
Leverage our core competencies. We believe that our CTP technology improves the drug properties of therapeutic proteins. We will continue to use our CTP technology to develop improved versions of protein drugs with proven safety and efficacy and to improve the therapeutic profiles of new drugs that will be developed by our partners. We will also continue to conduct exploratory drug development research in therapeutic peptides and Fab fragments of monoclonal antibodies, where our CTP technology, intellectual property and internal expertise provide us with opportunities.
|
Our Partnering Strategy
In addition to commercializing the three therapeutic proteins and one peptide discussed above, there are many additional product candidates we can pursue in an opportunistic fashion. We plan to pursue partnering deals with biotechnology companies that have a strategic interest in using our solution to develop longer-lasting versions of their existing therapeutic proteins or peptides, or those in development. We anticipate such partnerships will provide significant revenues in the form of license fees, milestone payments and royalties on sales, which will help to subsidize our research and development costs.
Intellectual Property
We license from Washington University the intellectual property that is necessary to conduct our business. In 2001, we initially licensed from Washington University core intellectual property pursuant to a non-exclusive license agreement, and, in 2004, we amended this license to make us the exclusive licensee of the two key CTP patents in connection with 11 therapeutic proteins. Pursuant to the prior license agreement, Modigene Delaware issued a total of 221,979 shares of its common stock to Washington University (378,796 shares of our common stock on a post-merger basis). In February 2007, we entered into a new license agreement, which we refer to as the License Agreement, with Washington University that superseded the prior license agreement. Pursuant to the new
License Agreement, Washington University granted us the exclusive license to three CTP patents and expanded the field of use to all natural and non-natural therapeutic proteins and peptides (other than LH, FSH, TSH and hCG). Under the License Agreement, we have the right to sub-license the licensed patents. The License Agreement terminates in 2018 when the last of the patents licensed to us under the License Agreement expires, unless terminated earlier. Under the License Agreement, we were required to pay an initial fee of $100,000 in installments over the 18 months following the effective date of the License Agreement. In addition, we are required to pay annual license maintenance fees of $30,000 (payable until the first commercial sale); royalty fees of 1.5% to 5% from net revenues (with certain required minimum royalties after the first commercial sale of $10,000, $20,000 and $40,000 for the first, second, and third year and beyond,
respectively), and sub-licensing fees of 7.5% to 20% on sub-licensing payments. Pursuant to the License Agreement, we will also be responsible for milestone payments of $15,000 for each molecule at investigational new drug application (IND) filing, $30,000 at the initiation of a Phase II clinical trial and $40,000 at the initiation of a Phase III clinical trial.
Pursuant to our License Agreement with Washington University, we have obtained an exclusive license to the key CTP patents that have been issued by the U.S. Patent and Trademark Office – U.S. #5,712,122, U.S. #5,759,818 and U.S. #6,225,449. We believe these patents provide broad and comprehensive coverage of the CTP technology, and we intend to aggressively enforce our intellectual property rights if necessary. In addition, unrelated to the patents from Washington University, we have filed, and will likely continue to file, patent applications covering specific CTP-modified molecules and CTP innovations, such as configurations, compositions and methods. Two of these patents, covering hGH (#7,553,940) and EPO (#7,553,941), have been issued by the U.S. Patent Office
in June 2009.
Competition
The pharmaceutical industry is highly competitive. We face significant competition from pharmaceutical companies and biotechnology companies that are researching and developing therapeutic proteins with enhanced life spans. Several pharmaceutical companies, such as Amgen, Eli Lilly and Company (through its acquisition of Applied Molecular Evolution), Nektar Therapeutics, ConjuChem Inc., Flamel Technologies S.A., , Nautilus Biotech S.A. and Ambrx Inc. have marketed products or are involved with the development of therapeutic proteins with enhanced life spans.
These companies, as well as potential entrants into our market, have longer operating histories, larger customer or use bases, greater brand recognition and significantly greater financial, marketing and other resources than we do. Many of these current or potential competitors can devote substantially greater resources to the development and promotion of their products than we can.
10
Additionally, there has been consolidation within the pharmaceutical industry and larger, well-established and well-financed entities may continue to acquire, invest in or form joint ventures to gain access to additional technology or products. Any of these trends would increase the competition we face and could adversely affect our business and operating results.
Government Regulation
Regulation by governmental authorities in the United States and other countries will be a significant factor in the production and marketing of our products and our ongoing research and development activities. All of our products require rigorous preclinical and clinical testing, subject to regulatory clearance or approval, and regulatory approval by governmental agencies prior to commercialization and are subject to pervasive and continuing regulation upon approval. The lengthy process of conducting clinical trials, seeking approval and the subsequent compliance with applicable statutes and regulations, if approval is obtained, are very costly and require the expenditure of substantial resources.
In the United States, the Public Health Service Act and the Federal Food, Drug, and Cosmetic Act, as amended, and the regulations promulgated thereunder, and other federal and state statutes and regulations govern, among other things, the safety and effectiveness standards for our products and the raw materials and components used in the production of, testing, manufacture, labeling, storage, record keeping, approval, advertising and promotion of our products on a product-by-product basis.
Preclinical tests include in vitro (i.e., laboratory) and in vivo (i.e., animal) evaluation of the product candidate, its chemistry, formulation and stability, and animal studies to assess potential safety and efficacy. Certain preclinical tests must be conducted in compliance with good laboratory practice regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring them to be replicated. After laboratory analysis and preclinical testing, we intend to file an IND with the FDA to begin human testing. Typically, a manufacturer conducts a three-phase human clinical testing program which itself is subject to
numerous laws and regulatory requirements, including adequate monitoring, reporting, record keeping and informed consent. In Phase 1, small clinical trials are conducted to determine the safety and proper dose ranges of our product candidates. In Phase 2, clinical trials are conducted to assess safety and gain preliminary evidence of the efficacy of our product candidates. In Phase 3, clinical trials are conducted to provide sufficient data for the statistically valid evidence of safety and efficacy. The time and expense required for us to perform this clinical testing can vary and is substantial. We cannot be certain that we will successfully complete Phase 1, Phase 2 or Phase 3 testing of our product candidates within any specific time period, if at all. Furthermore, the FDA, the Institutional Review Board responsible for approving and monitoring the clinical trials at a given site, the Data Safety Monitoring Board,
where one is used, or the Company may suspend the clinical trials at any time on various grounds, including a finding that subjects or patients are exposed to unacceptable health risk.
We cannot take any action to market any new drug or biologic product in the United States until our appropriate marketing application has been approved by the FDA. The FDA has substantial discretion over the approval process and may disagree with our interpretation of the data submitted. The process may be significantly extended by requests for additional information or clarification regarding information already provided. As part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians. Satisfaction of these and other regulatory requirements typically takes several years, and the actual time required may vary substantially based upon the type, complexity and novelty of the product. Government
regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures on our activities. We cannot be certain that the FDA or other regulatory agencies will approve any of our products on a timely basis, if at all. Success in preclinical or early stage clinical trials does not assure success in later-stage clinical trials. Even if a product receives regulatory approval, the approval may be significantly limited to specific indications or uses and these limitations may adversely affect the commercial viability of the product. Delays in obtaining, or failures to obtain regulatory approvals, would have a material adverse effect on our business.
Even after we obtain FDA approval, we may be required to conduct further clinical trials (i.e., Phase 4 trials) and provide additional data on safety and effectiveness. We are also required to gain separate clearance for the use of an approved product as a treatment for indications other than those initially approved. In addition, side effects or adverse events that are reported during clinical trials can delay, impede or prevent marketing approval. Similarly, adverse events that are reported after marketing approval can result in additional limitations being placed on the product’s use and, potentially, withdrawal of the product from the market. Any adverse event, either before or after marketing approval, can result in product liability claims against us.
11
In addition to regulating and auditing human clinical trials, the FDA regulates and inspects equipment, facilities, laboratories and processes used in the manufacturing and testing of such products prior to providing approval to market a product. If after receiving FDA approval, we make a material change in manufacturing equipment, location or process, additional regulatory review may be required. We also must adhere to current Good Manufacturing Practice (cGMP) regulations and product-specific regulations enforced by the FDA through its facilities inspection program. The FDA also conducts regular, periodic visits to re-inspect our equipment, facilities, laboratories and processes following the initial approval. If, as a result of these inspections, the FDA determines
that our equipment, facilities, laboratories or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against us, including the suspension of our manufacturing operations.
The requirements that we and our collaborators must satisfy to obtain regulatory approval by government agencies in other countries prior to commercialization of our products in such countries can be rigorous, costly and uncertain. In the European countries, Canada and Australia, regulatory requirements and approval processes are similar in principle to those in the United States. Additionally, depending on the type of drug for which approval is sought, there are currently two potential tracks for marketing approval in the European countries: mutual recognition and the centralized procedure. These review mechanisms may ultimately lead to approval in all European Union countries, but each method grants all participating countries some decision-making authority in product
approval. Foreign governments also have stringent post-approval requirements including those relating to manufacture, labeling, reporting, record keeping and marketing. Failure to substantially comply with these on-going requirements could lead to government action against the product, the Company and/or its representatives.
The levels of revenues and profitability of biopharmaceutical companies may be affected by the continuing efforts of government and third party payers to contain or reduce the costs of health care through various means. For example, in certain foreign markets, pricing or profitability of therapeutic and other pharmaceutical products is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental control. In addition, in the United States and elsewhere, sales of therapeutic and other pharmaceutical products are dependent in part on the availability and adequacy of reimbursement from third party payers, such as the government or private insurance
plans. Third party payers are increasingly challenging established prices, and new products that are more expensive than existing treatments may have difficulty finding ready acceptance unless there is a clear therapeutic benefit. We cannot assure you that any of our products will be considered cost effective, or that reimbursement will be available or sufficient to allow us to sell them competitively and profitably.
We are also subject to various federal, state, and international laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. The federal Anti-kickback law, which governs federal healthcare programs (e.g., Medicare, Medicaid), makes it illegal to solicit, offer, receive or pay any remuneration in exchange for, or to induce, the referral of business, including the purchase or prescription of a particular drug. Many states have similar laws that are not restricted to federal healthcare programs. Federal and state false claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third party payers (including Medicare and Medicaid), claims for reimbursed drugs or services that are
false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. If the government or a whistleblower were to allege that we violated these laws there could be a material adverse effect on us, including our stock price. Even an unsuccessful challenge could cause adverse publicity and be costly to respond to, which could have a materially adverse effect on our business, results of operations and financial condition. A finding of liability under these laws can have significant adverse financial implications for the Company and can result in payment of large penalties and possible exclusion from federal healthcare programs. We will consult counsel concerning the potential application of these and other laws to our business and our sales, marketing and other activities and will make good faith efforts to comply with them. However, given their broad reach and the
increasing attention given by law enforcement authorities, we cannot assure you that some of our activities will not be challenged or deemed to violate some of these laws.
We are also subject to numerous federal, state, local, and international laws and regulations relating to safe working conditions, manufacturing practices, environmental protection, import and export controls, fire hazard control, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances. We believe that our procedures comply with the standards prescribed by federal, state, or local laws, rules, and/or regulations; however, the risk of injury or accidental contamination cannot be completely eliminated. Currently, we have no costs with respect to environmental law compliance. At our current stage of product development, we cannot accurately estimate what our future costs relating to environmental law compliance may be.
We have currently received no approvals to market our products from the FDA or other foreign regulators.
12
We currently employ 15 full-time and two part-time employees, including six with Ph.D. degrees and three with M.Sc. degrees, focused on research and development, and three focused on general management and business development. None of our employees is represented by a labor union, and we consider our employee relations to be good. We also utilize a number of consultants to assist with research and development and commercialization activities. We believe that our future success will depend in part on our continued ability to attract, hire and retain qualified personnel. All of our employees are located in Israel, and all of our research and development activities are conducted at the offices of Modigenetech, Ltd., our Israeli subsidiary.
|
Item 1A.
|
Risk Factors
|
Risks Related to Our Company and Our Business
We have a limited operating history, and we do not expect to become profitable in the near future.
We are a development stage biopharmaceutical company with a limited operating history. We are not profitable and have incurred losses since our inception. We have not generated any revenue since our inception, and we continue to incur research and development and general and administrative expenses related to our operations. We expect to continue to incur losses for the foreseeable future, and these losses will likely increase as we move toward the commercialization of any of our products in development. If our product candidates fail in clinical trials or do not gain regulatory clearance or approval, or if our product candidates do not achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain
profitability in subsequent periods. Accordingly, it is difficult to evaluate our business prospects. Moreover, our prospects must be considered in light of the risks and uncertainties encountered by an early-stage company and in highly regulated and competitive markets, such as the biopharmaceutical market, where regulatory approval and market acceptance of our products are uncertain. There can be no assurance that our efforts will ultimately be successful or result in revenues or profits.
We have not yet commercialized any products or technologies, and we may never become profitable.
We have not yet commercialized any products or technologies, and we may never be able to do so. We do not know when or if we will complete any of our product development efforts, obtain regulatory approval for any product candidates incorporating our technologies or successfully commercialize any approved products. Even if we are successful in developing products that are approved for marketing, we will not be successful unless these products gain market acceptance. The degree of market acceptance of these products will depend on a number of factors, including:
|
|
·
|
the timing of regulatory approvals in the countries, and for the uses, we seek;
|
|
|
·
|
the competitive environment;
|
|
|
·
|
the establishment and demonstration in the medical community of the safety and clinical efficacy of our products and their potential advantages over existing therapeutic products;
|
|
|
·
|
the adequacy and success of distribution, sales and marketing efforts; and
|
|
|
·
|
the pricing and reimbursement policies of government and third-party payors, such as insurance companies, health maintenance organizations and other plan administrators.
|
Physicians, patients, thirty-party payors or the medical community in general may be unwilling to accept, utilize or recommend any of our products or products incorporating our technologies. As a result, we are unable to predict the extent of future losses or the time required to achieve profitability, if at all. Even if we successfully develop one or more products that incorporate our technologies, we may not become profitable.
Our product candidates are at an early stage of product development and may never be commercialized.
All of our product candidates are at early stages of product development and may never be commercialized. Initially, we plan to develop product candidates through studies, testing and clinical lead product candidate selection, and then to license them to other companies. The progress and results of any future pre-clinical testing or future clinical trials are uncertain, and the failure of our product candidates to receive regulatory approvals will have a material adverse effect on our business, operating results and financial condition to the extent we are unable to commercialize any products. None of our product candidates has received regulatory approval for commercial sale. In addition, all of our product candidates are in the early stages of development, and we face the risks
of failure inherent in developing therapeutic proteins based on new technologies. Our product candidates are not expected to be commercially available for several years, if at all.
13
In addition, our product candidates must satisfy rigorous standards of safety and efficacy before they can be approved by the U.S. Food and Drug Administration, or the FDA, and international regulatory authorities for commercial use. The FDA and foreign regulatory authorities have full discretion over this approval process. We will need to conduct significant additional research, involving testing in animals and in humans, before we can file applications for product approval. Typically, in the pharmaceutical industry, there is a high rate of attrition for product candidates in pre-clinical testing and clinical trials. Also, satisfaction of regulatory requirements typically takes many years, is dependent upon the type, complexity and novelty of the product and requires
the expenditure of substantial resources. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful. For example, a number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials and in interim analyses. In addition, delays or rejections may be encountered based upon additional government regulation, including any changes in FDA policy, during the process of product development, clinical trials and regulatory approvals.
In order to receive FDA approval or approval from foreign regulatory authorities to market a product candidate or to distribute our products, we must demonstrate through pre-clinical testing and through human clinical trials that the product candidate is safe and effective for the treatment of a specific condition.
We might be unable to develop product candidates that will achieve commercial success in a timely and cost-effective manner, or ever.
Even if regulatory authorities approve our product candidates, they may not be commercially successful. Our product candidates may not be commercially successful because physicians, government agencies and other third-party payors may not accept them. A product approval, assuming one issues, may limit the uses for which the product may be distributed thereby adversely affecting the commercial viability of the product. Third parties may develop superior products or have proprietary rights that preclude us from marketing our products. We also expect that most of our product candidates will be very expensive, if approved. Patient acceptance of and demand for any product candidates for which we obtain regulatory approval or license will depend largely on many
factors, including but not limited to the extent, if any, of reimbursement of therapeutic protein and treatment costs by government agencies and other third-party payors, pricing, the effectiveness of our marketing and distribution efforts, the safety and effectiveness of alternative products, and the prevalence and severity of side effects associated with our products. If physicians, government agencies and other third-party payors do not accept our products, we will not be able to generate significant revenue.
It is highly likely that we will need to raise additional capital to meet our business requirements in the future, and such capital raising may be costly or difficult to obtain and could dilute current stockholders’ ownership interests.
It is highly likely that we will need to raise additional funds through public or private debt or equity financings to meet various objectives including, but not limited to:
|
|
·
|
pursuing growth opportunities, including more rapid expansion;
|
|
|
·
|
acquiring complementary businesses;
|
|
|
·
|
making capital improvements to improve our infrastructure;
|
|
|
·
|
hiring qualified management and key employees;
|
|
|
·
|
research and development of new products;
|
|
|
·
|
responding to competitive pressures;
|
|
|
·
|
complying with regulatory requirements such as licensing and registration; and
|
|
|
·
|
maintaining compliance with applicable laws.
|
Any additional capital raised through the sale of equity or equity-linked securities may dilute our current stockholders’ ownership in us and could also result in a decrease in the market price of our common stock. The terms of those securities issued by us in future capital transactions may be more favorable to new investors and may include preferences, superior voting rights and the issuance of warrants or other derivative securities, which may have a further dilutive effect.
Furthermore, any debt or equity financing that we may need may not be available on terms favorable to us, or at all. If we are unable to obtain required additional capital, we may have to curtail our growth plans or cut back on existing business, and we may not be able to continue operating if we do not generate sufficient revenues from operations needed to stay in business.
14
We may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we issue, such as convertible notes and warrants, which may adversely impact our financial condition.
If we fail to obtain necessary funds for our operations, we will be unable to maintain and improve our patented technology, and we will be unable to develop and commercialize our products and technologies.
Our present and future capital requirements depend on many factors, including:
|
|
·
|
the level of research and development investment required to develop our product candidates, and maintain and improve our patented technology position;
|
|
|
·
|
the costs of obtaining or manufacturing therapeutic proteins for research and development and at commercial scale;
|
|
|
·
|
the results of preclinical and clinical testing, which can be unpredictable in therapeutic protein development;
|
|
|
·
|
changes in product candidate development plans needed to address any difficulties that may arise in manufacturing, preclinical activities, clinical studies or commercialization;
|
|
|
·
|
our ability and willingness to enter into new agreements with strategic partners and the terms of these agreements;
|
|
|
·
|
our success rate in preclinical and clinical efforts associated with milestones and royalties;
|
|
|
·
|
the costs of investigating patents that might block us from developing potential product candidates;
|
|
|
·
|
the costs of recruiting and retaining qualified personnel;
|
|
|
·
|
the time and costs involved in obtaining regulatory approvals;
|
|
|
·
|
the costs of filing, prosecuting, defending, and enforcing patent claims and other intellectual property rights; and
|
|
|
·
|
our need or decision to acquire or license complementary technologies or new therapeutic protein targets.
|
If we are unable to obtain the funds necessary for our operations, we will be unable to maintain and improve our patented technology, and we will be unable to develop and commercialize our products and technologies, which would materially and adversely affect our business, liquidity and results of operations.
We depend on key members of our management and advisory team and will need to add and retain additional leading experts.
We are highly dependent on our executive officers and other key management and technical personnel. Our failure to retain our Chief Executive Officer, Abraham (Avri) Havron, or our President, Shai Novik, or any other key management and technical personnel could have a material adverse effect on our future operations. Our success is also dependent on our ability to attract, retain and motivate highly trained technical, marketing, sales and management personnel, among others, to produce our product candidates and, if our product candidates are produced and approved for marketing, to market our products and to continue to produce enhanced releases of our products. We presently do not maintain “key person” life insurance policies on any of our personnel.
Our success also depends on our ability to attract, retain and motivate personnel required for the development, maintenance and expansion of our activities. There can be no assurance that we will be able to retain our existing personnel or attract additional qualified employees. The loss of key personnel or the inability to hire and retain additional qualified personnel in the future could have a material adverse effect on our business, financial condition and results of operation.
Under current U.S. and Israeli law, we may not be able to enforce employees’ covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees.
We have entered into non-competition agreements with our key employees. These agreements prohibit our key employees, if they cease working for us, from competing directly with us or working for our competitors for a limited period. Under applicable U.S. and Israeli law, we may be unable to enforce these agreements. If we cannot enforce our non-competition agreements with our employees, then we may be unable to prevent our competitors from benefiting from the expertise of our former employees, which could materially adversely affect our business, results of operations and ability to capitalize on our proprietary information.
15
We do not currently have sales, marketing or distribution capabilities, and we may be unable to effectively sell, market and distribute our product candidates in the future, and the failure to do so would have an adverse effect on our business and results of operations.
If we are unable to develop sales, marketing and distribution capabilities or enter into agreements with third parties to perform these functions, we will not be able to successfully commercialize any of our product candidates. We do not currently have sales, marketing or distribution capabilities. In order to successfully commercialize any of our product candidates, we must either internally develop sales, marketing and distribution capabilities or make arrangements with third parties to perform these services.
If we do not develop a marketing and sales force with technical expertise and supporting distribution capabilities, we will be unable to market any of our product candidates directly. To promote any of our potential products through third parties, we will have to locate acceptable third parties for these functions and enter into agreements with them on acceptable terms, and we may not be able to do so. In addition, any third-party arrangements we are able to enter into may result in lower revenues than we could achieve by directly marketing and selling our potential products.
We may suffer losses from product liability claims if our product candidates cause harm to patients.
Any of our product candidates could cause adverse events, such as immunologic or allergic reactions. These reactions may not be observed in clinical trials, but may nonetheless occur after commercialization. If any of these reactions occur, they may render our product candidates ineffective or harmful in some patients, and our sales would suffer, materially adversely affecting our business, financial condition and results of operations.
In addition, potential adverse events caused by our product candidates could lead to product liability lawsuits. If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates. Our business exposes us to potential product liability risks, which are inherent in the testing, manufacturing, marketing and sale of pharmaceutical products. We may not be able to avoid product liability claims. Product liability insurance for the pharmaceutical and biotechnology industries is generally expensive, if available at all. We do not currently have any product liability insurance because we are not yet conducting trials on humans. When we begin human trials, we will endeavor to obtain sufficient product liability
insurance. If we are unable to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims, we may be unable to commercialize our product candidates. A successful product liability claim brought against us in excess of our insurance coverage, if any, may cause us to incur substantial liabilities, and, as a result, our business, liquidity and results of operations would be materially adversely affected.
Our product candidates will remain subject to ongoing regulatory requirements even if they receive marketing approval, and if we fail to comply with these requirements, we could lose these approvals, and the sales of any approved commercial products could be suspended.
Even if we receive regulatory approval to market a particular product candidate, the product will remain subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the uses for which the product may be marketed or the conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product, which could negatively impact us or our collaboration partners by reducing revenues or increasing expenses, and cause the approved product candidate not to be commercially viable. In addition, as clinical
experience with a drug expands after approval, typically because it is used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials or other studies. Any adverse effects observed after the approval and marketing of a product candidate could result in limitations on the use of or withdrawal of any approved products from the marketplace. Absence of long-term safety data may also limit the approved uses of our products, if any. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including the following:
|
|
·
|
Restrictions on the products, manufacturers or manufacturing process;
|
|
|
·
|
Warning letters;
|
16
|
|
·
|
Civil or criminal penalties, fines and injunctions;
|
|
|
·
|
Product seizures or detentions;
|
|
|
·
|
Import or export bans or restrictions;
|
|
|
·
|
Voluntary or mandatory product recalls and related publicity requirements;
|
|
|
·
|
Suspension or withdrawal of regulatory approvals;
|
|
|
·
|
Total or partial suspension of production, and
|
|
|
·
|
Refusal to approve pending applications for marketing approval of new products or supplements to approved applications.
|
If we or our collaborators are slow to adapt, or are unable to adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements or policies, marketing approval for our product candidates may be lost or cease to be achievable, resulting in decreased revenue from milestones, product sales or royalties, which would have a material adverse effect on our results of operations.
Clinical trials are very expensive, time-consuming and difficult to design and implement, and, as a result, we may suffer delays or suspensions in future trials which would have a material adverse effect on our ability to generate revenues.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. Additionally, the clinical trial process is time-consuming, and, while we are optimistic about our ability to complete our clinical trials relatively quickly as compared to average trial lengths for clinical trials, failure can occur at any stage of the trials, and we may encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
|
|
·
|
unforeseen safety issues;
|
|
|
·
|
determination of dosing issues;
|
|
|
·
|
lack of effectiveness or efficacy during clinical trials;
|
|
|
·
|
failure of third party suppliers to perform final manufacturing steps for the drug substance;
|
|
|
·
|
slower than expected rates of patient recruitment;
|
|
|
·
|
inability to monitor patients adequately during or after treatment;
|
|
|
·
|
failure of third party contract research organizations to properly implement or monitor the clinical trial protocols;
|
|
|
·
|
failure of institutional review boards to approve our clinical trial protocols;
|
|
|
·
|
inability or unwillingness of medical investigators and institutional review boards to follow our clinical trial protocols; and
|
|
|
·
|
lack of sufficient funding to finance the clinical trials.
|
In addition, we or regulatory authorities may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the regulatory authorities find deficiencies in our regulatory submissions or the conduct of these trials. Any suspension of clinical trials will delay possible regulatory approval, if any, and adversely impact our ability to develop products and generate revenue.
The manufacture of our product candidates is an exacting and complex process, and, if we or one of our materials suppliers encounters problems manufacturing our products, our business could suffer.
The FDA and foreign regulators require manufacturers to register manufacturing facilities. The FDA and foreign regulators also inspect these facilities to confirm compliance with requirements that the FDA or foreign regulators establish. We or our materials suppliers may face manufacturing or quality control problems causing product production and shipment delays or a situation where we or the supplier may not be able to maintain compliance with the FDA’s or foreign regulators’ requirements necessary to continue manufacturing our drug substance. Drug manufacturers are subject to ongoing periodic unannounced inspections by the FDA, the U.S. Drug Enforcement Agency, or DEA, and corresponding foreign regulators to ensure strict compliance with requirements and other governmental regulations and
corresponding foreign standards. Any failure to comply with DEA requirements or FDA or foreign regulatory requirements could adversely affect our clinical research activities and our ability to market and develop our product candidates.
17
We may rely on third parties to implement our manufacturing and supply strategies.
If our current and future licensing, manufacturing and supply strategies are unsuccessful, then we may be unable to complete any future pre-clinical or clinical trials or commercialize our product candidates in a timely manner, if at all. Completion of any potential future pre-clinical, clinical trials and commercialization of our product candidates will require access to, or development of, facilities to manufacture a sufficient supply of our product candidates, or the ability to license them to other companies to perform these functions. We do not have the resources, facilities or experience to manufacture our product candidates on our own and do not intend to develop or acquire facilities for the manufacture of product candidates for pre-clinical trials, clinical trials or commercial purposes in the foreseeable
future. We intend to continue to license technology and to rely on contract manufacturers to produce sufficient quantities of our product candidates necessary for any pre-clinical or clinical testing we undertake in the future. Such contract manufacturers may be the sole source of production and they may have limited experience at manufacturing, formulating, analyzing, filling and finishing our types of product candidates.
We also intend to rely on third parties to supply the components that we will need to develop, test and commercialize all of our product candidates. There may be a limited supply of these components. We might not be able to enter into agreements that provide us assurance of availability of such components in the future from any supplier. Our potential suppliers may not be able to adequately supply us with the components necessary to successfully conduct our pre-clinical and clinical trials and/or to commercialize our product candidates. If we cannot acquire an acceptable supply of components to produce our product candidates, we will not be able to complete pre-clinical and clinical trials and will not be able to commercialize our product candidates.
If we acquire or license additional technology or product candidates, we may incur a number of costs, may have integration difficulties and may experience other risks that could harm our business and results of operations.
We may acquire and license additional product candidates and technologies. Any product candidate or technology we license or acquire will likely require additional development efforts prior to commercial sale, including extensive clinical testing and approval by the FDA and applicable foreign regulatory authorities, if any. All product candidates are prone to risks of failure inherent in pharmaceutical product development, including the possibility that the product candidate or product developed based on licensed technology will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot assure you that any product candidate that we develop based on acquired or licensed technology that is granted regulatory approval will be manufactured or produced
economically, successfully commercialized or widely accepted in the marketplace. Moreover, integrating any newly acquired product candidates could be expensive and time-consuming. If we cannot effectively manage these aspects of our business strategy, our business may not succeed.
Furthermore, proposing, negotiating and implementing an economically viable acquisition or license can be a lengthy, costly and complex process. Other companies, including those with substantially greater financial, marketing and sales resources, may compete with us for the acquisition or license of product candidates and/or technologies. We may not be able to acquire the rights to alternative product candidates and/or technologies on terms that we find acceptable, or at all. Our failure to acquire or license alternative product candidates and/or technologies could have a material adverse effect on our business, prospects and financial condition.
We may not be able to successfully grow and expand our business.
We may not be able to successfully expand. Successful implementation of our business plan will require management of growth, which will result in an increase in the level of responsibility for management personnel. To manage growth effectively, we will be required to continue to implement and improve our operating and financial systems and controls to expand, train and manage our employee base. The management, systems and controls currently in place or to be implemented may not be adequate for such growth, and the steps taken to hire personnel and to improve such systems and controls might not be sufficient. If we are unable to manage our growth effectively, it will have a material adverse effect on our business, results of operations and financial condition.
We may encounter difficulties in managing our growth. These difficulties could increase our losses.
We may experience rapid and substantial growth in order to achieve our operating plans, which will place a strain on our human and capital resources. If we are unable to manage this growth effectively, our losses could materially increase. Our ability to manage our operations and growth effectively requires us to continue to expend funds to enhance our operational, financial and management controls, reporting systems and procedures and to attract and retain sufficient numbers of talented employees. If we are unable to scale up and implement improvements to our control systems in an efficient or timely manner, or if we encounter deficiencies in existing systems and controls, then we will not be able to make available the products required to successfully commercialize our technology. Failure to attract and retain
sufficient numbers of talented employees will further strain our human resources and could impede our growth or result in ineffective growth.
18
If we are unable to obtain adequate insurance, our financial condition could be adversely affected in the event of uninsured or inadequately insured loss or damage. Our ability to effectively recruit and retain qualified officers and directors could also be adversely affected if we experience difficulty in obtaining adequate directors’ and officers’ liability insurance.
We may not be able to obtain insurance policies on terms affordable to us that would adequately insure our business and property against damage, loss or claims by third parties. To the extent our business or property suffers any damages, losses or claims by third parties, which are not covered or adequately covered by insurance, our financial condition may be materially adversely affected.
We may be unable to maintain sufficient insurance as a public company to cover liability claims made against our officers and directors. If we are unable to adequately insure our officers and directors, we may not be able to retain or recruit qualified officers and directors to manage the Company.
We are a holding company that depends on cash flow from our wholly-owned subsidiary, to meet our obligations.
We are a holding company with no material assets other than the stock of our wholly-owned subsidiary. Accordingly, all our operations are conducted by Modigene Delaware, our wholly-owned subsidiary (and its wholly-owned subsidiary, ModigeneTech Ltd.). We currently expect that the earnings and cash flow of our subsidiary will primarily be retained and used by it in its operations, including servicing any debt obligations it may have now or in the future. Accordingly, although we do not anticipate paying any dividends in the foreseeable future, our subsidiary may not be able to generate sufficient cash flow to distribute funds to us in order to allow us to pay future dividends on, or make any distributions with respect to our common stock.
If we fail to maintain an effective system of internal controls, we may not be able to accurately report our financial results or detect fraud. Consequently, investors could lose confidence in our financial reporting and this may decrease the trading price of our stock.
We must maintain effective internal controls to provide reliable financial reports and detect fraud. Our failure to properly maintain an effective system of internal controls could harm our operating results and cause investors to lose confidence in our reported financial information. In addition, such failure may cause us to suffer violations of the U.S. federal securities laws to the extent we are unable to maintain effective internal controls. Any such loss of confidence or violations would have a negative effect on the trading price of our stock.
Potential political, economic and military instability in the State of Israel, where key members of our senior management and our research and development facilities are located, may adversely affect our results of operations.
We maintain office and research and development facilities in the State of Israel. Political, economic and military conditions in Israel may directly affect our ability to conduct business. Since the State of Israel was established in 1948, a number of armed conflicts have occurred between Israel and its Arab neighbors. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners, or a significant downturn in the economic or financial condition of Israel, could affect adversely our operations. Ongoing and revived hostilities or other Israeli political or economic factors could harm our operations and product development and cause our revenues to fail to develop or decrease if we have already begun sales.
Recent disruptions in the financial markets and economic conditions could affect our ability to raise capital and could disrupt or delay the performance of our third-party contractors and suppliers.
In the past twenty-four months, the U.S. and global economies have taken a dramatic downturn as the result of the deterioration in the credit markets and related financial crisis as well as a variety of other factors including, among other things, extreme volatility in security prices, severely diminished liquidity and credit availability, ratings downgrades of certain investments and declining valuations of others. The U.S. and certain foreign governments have recently taken unprecedented actions in an attempt to address and rectify these extreme market and economic conditions by providing liquidity and stability to the financial markets. If the actions taken by these governments are not successful, the continued economic decline may cause a significant impact on our ability to raise capital, if
needed, on a timely basis and on acceptable terms or at all. In addition, we rely and intend to rely on third-parties, including our clinical research organizations, third-party manufacturers and second source suppliers, and certain other important vendors and consultants. As a result of the current volatile and unpredictable global economic situation, there may be a disruption or delay in the performance of our third-party contractors and suppliers. If such third-parties are unable to satisfy their contractual commitments to us, our business could be severely adversely affected.
19
Risks Related to Our Intellectual Property
We license our core technology from Washington University, and we could lose our rights to this license if a dispute with Washington University arises or if we fail to comply with the financial and other terms of the license.
We license our core intellectual property from Washington University. We initially entered into a non-exclusive license agreement with Washington University in 2001, and in 2004 we amended the license to extend the CTP technology to eleven therapeutic proteins and make it exclusive. In February 2007, we entered into a revised and expanded license agreement with Washington University, which we refer to as the License Agreement, pursuant to which we and Washington University expanded the exclusive license, adding additional patents, and expanding the applicability of licensed CTP technology to all proteins and peptides having a native or non-native amino acid sequence, excluding Follicle Stimulating Hormone (FSH), Luteinizing Hormone (LH), Thyroid Stimulating Hormone (TSH) and Chorionic Gonadotropin
(hCG). The License Agreement imposes certain payment, reporting, confidentiality and other obligations on us. In the event that we were to breach any of the obligations and fail to cure, Washington University would have the right to terminate the License Agreement upon 90 days’ notice. In addition, Washington University has the right to terminate the License Agreement upon our bankruptcy or receivership. If any dispute arises with respect to our arrangement with Washington University, such dispute may disrupt our operations and would likely have a material and adverse impact on us if resolved in a manner that is unfavorable to our Company. All of our current product candidates are partly based on the intellectual property licensed under the License Agreement, and if the License Agreement were terminated, it would have a material adverse effect on our business, prospects and results of operations.
The failure to obtain or maintain patents, licensing agreements and other intellectual property could impact our ability to compete effectively.
To compete effectively, we need to develop and maintain a proprietary position with regard to our own technologies, intellectual property, licensing agreements, product candidates and business. Legal standards relating to the validity and scope of claims in the CTP technology field are still evolving. Therefore, the degree of future protection for our proprietary rights in our core technologies and any products that might be made using these technologies is also uncertain. The risks and uncertainties that we face with respect to our patents and other proprietary rights include the following:
|
|
·
|
while the patents we license have been issued, the pending patent applications we have filed may not result in issued patents or may take longer than we expect to result in issued patents;
|
|
|
·
|
we may be subject to interference proceedings;
|
|
|
·
|
we may be subject to opposition proceedings in foreign countries;
|
|
|
·
|
any patents that are issued may not provide meaningful protection;
|
|
|
·
|
we may not be able to develop additional proprietary technologies that are patentable;
|
|
|
·
|
other companies may challenge patents licensed or issued to us or our customers;
|
|
|
·
|
other companies may independently develop similar or alternative technologies, or duplicate our technologies;
|
|
|
·
|
other companies may design around technologies we have licensed or developed; and
|
|
|
·
|
enforcement of patents is complex, uncertain and expensive.
|
We cannot be certain that patents will be issued as a result of any of our pending applications, and we cannot be certain that any of our issued patents, whether issued pursuant to our pending applications or licensed from Washington University, will give us adequate protection from competing products. For example, issued patents, including the patents licensed from Washington University, may be circumvented or challenged, declared invalid or unenforceable, or narrowed in scope. In addition, since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first to make our inventions or to file patent applications covering those inventions.
It is also possible that others may obtain issued patents that could prevent us from commercializing our products or require us to obtain licenses requiring the payment of significant fees or royalties in order to enable us to conduct our business. As to those patents that we have licensed, our rights depend on maintaining our obligations to the licensor under the applicable license agreement, and we may be unable to do so.
20
In addition to patents and patent applications, we depend upon trade secrets and proprietary know-how to protect our proprietary technology. We require our employees, consultants, advisors and collaborators to enter into confidentiality agreements that prohibit the disclosure of confidential information to any other parties. We require our employees and consultants to disclose and assign to us their ideas, developments, discoveries and inventions. These agreements may not, however, provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure.
Costly litigation may be necessary to protect our intellectual property rights and we may be subject to claims alleging the violation of the intellectual property rights of others.
We may face significant expense and liability as a result of litigation or other proceedings relating to patents and other intellectual property rights of others. In the event that another party has also filed a patent application or been issued a patent relating to an invention or technology claimed by us in pending applications, we may be required to participate in an interference proceeding declared by the U.S. Patent and Trademark Office to determine priority of invention, which could result in substantial uncertainties and costs for us, even if the eventual outcome were favorable to us. We, or our licensors, also could be required to participate in interference proceedings involving issued patents and pending applications of another entity. An adverse outcome in an interference proceeding could require us
to cease using the technology or to license rights from prevailing third parties.
The cost to us of any patent litigation or other proceeding relating to our licensed patents or patent applications, even if resolved in our favor, could be substantial. Our ability to enforce our patent protection could be limited by our financial resources, and may be subject to lengthy delays. If we are unable to effectively enforce our proprietary rights, or if we are found to infringe the rights of others, we may be in breach of our License Agreement.
A third party may claim that we are using inventions claimed by their patents and may go to court to stop us from engaging in our normal operations and activities, such as research, development and the sale of any future products. Such lawsuits are expensive and would consume time and other resources. There is a risk that the court will decide that we are infringing the third party’s patents and will order us to stop the activities claimed by the patents. In addition, there is a risk that a court will order us to pay the other party damages for having infringed their patents.
Moreover, there is no guarantee that any prevailing patent owner would offer us a license so that we could continue to engage in activities claimed by the patent, or that such a license, if made available to us, could be acquired on commercially acceptable terms. In addition, third parties may, in the future, assert other intellectual property infringement claims against us with respect to our product candidates, technologies or other matters.
We rely on confidentiality agreements that could be breached and may be difficult to enforce, which could result in third parties using our intellectual property to compete against us.
Although we believe that we take reasonable steps to protect our intellectual property, including the use of agreements relating to the non-disclosure of confidential information to third parties, as well as agreements that purport to require the disclosure and assignment to us of the rights to the ideas, developments, discoveries and inventions of our employees and consultants while we employ them, the agreements can be difficult and costly to enforce. Although we seek to obtain these types of agreements from our contractors, consultants, advisors and research collaborators, to the extent that employees and consultants utilize or independently develop intellectual property in connection with any of our projects, disputes may arise as to the intellectual property rights associated with our products. If a dispute
arises, a court may determine that the right belongs to a third party. In addition, enforcement of our rights can be costly and unpredictable. We also rely on trade secrets and proprietary know-how that we seek to protect in part by confidentiality agreements with our employees, contractors, consultants, advisors or others. Despite the protective measures we employ, we still face the risk that:
|
|
·
|
these agreements may be breached;
|
|
|
·
|
these agreements may not provide adequate remedies for the applicable type of breach;
|
|
|
·
|
our trade secrets or proprietary know-how will otherwise become known; or
|
|
|
·
|
our competitors will independently develop similar technology or proprietary information.
|
21
International patent protection is particularly uncertain, and if we are involved in opposition proceedings in foreign countries, we may have to expend substantial sums and management resources.
Patent law outside the United States is in some cases different than in the United States and is currently undergoing review and revision in many countries. Further, the laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. For example, certain countries do not grant patent claims that are directed to the treatment of humans. We may participate in opposition proceedings to determine the validity of our foreign patents or our competitors’ foreign patents, which could result in substantial costs and diversion of our efforts.
We may be unable to protect the intellectual property rights of the third parties from whom we license certain of our intellectual property or with whom we have entered into other strategic relationships.
Certain of our intellectual property rights are currently licensed from Washington University, and, in the future, we intend to continue to license intellectual property from Washington University and/or other key strategic partners. We are, and will continue to be, reliant upon such third parties to protect their intellectual property rights to any licensed technology. Such third parties may determine not to protect the intellectual property rights that we license from them and we may be unable defend such intellectual property rights on our own or we may have to undertake costly litigation to defend the intellectual property rights of such third parties. There can be no assurances that we will continue to have proprietary rights to any of the intellectual property that we license from such third parties or
otherwise have the right to use through similar strategic relationships. Any loss or limitations on use with respect to our right to use such intellectual property licensed from third parties or otherwise obtained from third parties with whom we have entered into strategic relationships could have a material adverse effect on our business, operating results and financial condition.
Risks Related to Our Industry
We are subject to government regulations, and we may experience delays in obtaining required regulatory approvals in the United States to market our proposed product candidates.
Various aspects of our operations are or may become subject to federal, state or local laws, rules and regulations, any of which may change from time to time. Costs arising out of any regulatory developments could be time-consuming, expensive and could divert management resources and attention and, consequently, could adversely affect our business operations and financial performance.
Delays in regulatory approval, limitations in regulatory approval and withdrawals of regulatory approval may have a negative impact on our results. If we experience significant delays in testing or approvals, our product development costs, or our ability to license product candidates, will increase. If the FDA grants regulatory approval of a product, this approval will be limited to those disease states and conditions for which the product has demonstrated, through clinical trials, to be safe and effective. Any product approvals that we receive in the future could also include significant restrictions on the use or marketing of our products. Product approvals, if granted, can be withdrawn for failure to comply with regulatory requirements or upon the occurrence of adverse events following commercial introduction
of the products. Failure to comply with applicable FDA or other applicable regulatory requirements may result in criminal prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other regulatory action against our product candidates or us. If approval is withdrawn for a product, or if a product were seized or recalled, we would be unable to sell or license that product and our revenues would suffer. In addition, outside the United States, our ability to market any of our potential products is contingent upon receiving market application authorizations from the appropriate regulatory authorities and these foreign regulatory approval processes include all of the risks associated with the FDA approval process described above.
We face significant competition and continuous technological change.
If our competitors develop and commercialize products faster than we do, or develop and commercialize products that are superior to our product candidates, our commercial opportunities will be reduced or eliminated. The extent to which any of our product candidates achieve market acceptance will depend on competitive factors, many of which are beyond our control. Competition in the pharmaceutical industry is intense and has been accentuated by the rapid pace of technology development. Our competitors include large integrated pharmaceutical companies, biotechnology companies that currently have drug and target discovery efforts, universities, and public and private research institutions. Almost all of these entities have substantially greater research and development capabilities and financial, scientific,
manufacturing, marketing and sales resources than we do, as well as more experience in research and development, clinical trials, regulatory matters, manufacturing, marketing and sales. These organizations also compete with us to:
|
|
·
|
attract parties for acquisitions, joint ventures or other collaborations;
|
22
|
|
·
|
license proprietary technology that is competitive with the technology we are developing;
|
|
|
·
|
attract funding; and
|
|
|
·
|
attract and hire scientific talent.
|
Our competitors may succeed in developing and commercializing products earlier and obtaining regulatory approvals from the FDA more rapidly than we do. Our competitors may also develop products or technologies that are superior to those we are developing, and render our product candidates or technologies obsolete or non-competitive. If we cannot successfully compete with new or existing products, our marketing and sales will suffer and we may not ever be profitable.
We expect the healthcare industry to face increased scrutiny over reimbursement and healthcare reform, which could adversely impact how much or under what circumstances healthcare providers will prescribe or administer our products.
In both the United States and other countries, sales of our products will depend in part upon the availability of reimbursement from third party payors, which include government health administration authorities, managed care providers and private health insurers. Third party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services.
Increasing expenditures for healthcare have been the subject of considerable public attention in the United States. Both private and government entities are seeking ways to reduce or contain healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system have been introduced or proposed in Congress and in some state legislatures, including reductions in the cost of prescription products and changes in the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products.
In 2010, Congress enacted and the President signed into law the Patient Protection and Affordable Care Act, as amended, which will significantly expand access to health care coverage but may lead to reduction in reimbursement for supplies, including pharmaceuticals, and services. The Centers for Medicare & Medicaid Services, or CMS, is in the process of issuing regulations to implement the new law which will affect Medicare, Medicaid and other third-party payors. Medicare, which is the single largest third-party payment program and administered by CMS, covers prescription drugs in one of two ways. Medicare part B covers outpatient prescription drugs that are administered by physicians and Medicare part D covers other outpatient prescription drugs, but through private
insurers. Medicaid, a health insurance program for in the poor, is funded jointly by CMS and the states, but is administered by the states; states are authorized to cover outpatient prescription drugs, but that coverage is subject to caps and to substantial rebates.
Although we cannot predict the full effect on our business of the implementation of existing legislation, including the Affordable Care Act or the enactment of additional legislation, we believe that legislation or regulations that reduces reimbursement for our products could adversely affect how much or under what circumstances healthcare providers will prescribe or administer our products. This could materially and adversely impact our business by reducing our ability to generate revenue, raise capital, obtain additional collaborators and market our products. In addition, we believe the increasing emphasis on managed care in the United States has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact product sales.
We are subject to federal anti-kickback laws and regulations. Our failure to comply with these laws and regulations could have adverse consequences to us.
There are extensive federal and state laws and regulations prohibiting fraud and abuse in the healthcare industry that can result in significant criminal and civil penalties. These federal laws include: the anti-kickback statute, which prohibits certain business practices and relationships, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other federal healthcare programs; the physician self-referral prohibition, commonly referred to as the Stark Law; the anti-inducement law, which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program; the False Claims Act, which prohibits any person from knowingly presenting or causing to be presented false
or fraudulent claims for payment by the federal government, including the Medicare and Medicaid programs; and the Civil Monetary Penalties Law, which authorizes the United States Department of Health and Human Services to impose civil penalties administratively for fraudulent or abusive acts.
23
Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both. As federal and state budget pressures continue, federal and state administrative agencies may also continue to escalate investigation and enforcement efforts to root out waste and to control fraud and abuse in governmental healthcare programs. Private enforcement of healthcare fraud has also increased, due in large part to amendments to the civil False Claims Act in 1986 that were designed to encourage private persons to sue on behalf of the government. A violation of any of these federal and state fraud and abuse laws and regulations could have
a material adverse effect on our liquidity and financial condition. An investigation into the use by physicians of any of our products once commercialized may dissuade physicians from either purchasing or using them, and could have a material adverse effect on our ability to commercialize those products.
Risks Related to Our Common Stock
Our stock price has been, and may continue to be, volatile, and you could lose all or part of your investment.
The market price for our common stock has been extremely volatile (ranging from $2.57 per share to $8.85 per share during the 52-week trading period ended February 28, 2011. We expect that the market price of our common stock will continue to fluctuate significantly due to factors including, but not limited to, the following:
|
|
·
|
actual or anticipated variations in our operating results;
|
|
|
·
|
announcements of developments by us or our competitors;
|
|
|
·
|
announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments;
|
|
|
·
|
adoption of new accounting standards affecting our industry;
|
|
|
·
|
additions or departures of key personnel;
|
|
|
·
|
introduction of new products by us or our competitors;
|
|
|
·
|
changes in market valuations of companies in our industry;
|
|
|
·
|
future issuances of our common stock or other securities; and
|
|
|
·
|
other events or factors, many of which are beyond our control.
|
We do not expect to pay dividends on our common stock, and investors will be able to receive cash in respect of their shares of our common stock only upon the sale of the shares.
Cash dividends have never been declared or paid on our common stock, and we do not anticipate such a declaration or payment in the foreseeable future. We expect to use future earnings, if any, to fund business growth. Therefore, an investor in our common stock will obtain an economic benefit from the common stock only after an increase in its trading price and only by selling the common stock. We cannot assure stockholders of a positive return on their investment when they sell their shares, nor can we assure stockholders that they will not lose the entire amount of their investment.
Securities analysts may not initiate coverage or continue to cover our common stock, and this may have a negative impact on its market price.
The trading market for our common stock will depend in part on the research and reports that securities analysts publish about our business and us. We do not have any control over these analysts. There is no guarantee that securities analysts will cover our common stock. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect its market price. If we are covered by securities analysts, and our stock is the subject of an unfavorable report, our stock price would likely decline. If one or more of these analysts ceases to cover us or fails to publish regular reports on us, we could lose visibility in the financial markets, which could cause our stock price and/or trading volume to decline. In addition, because we became public through a “reverse merger,”
we may have additional difficulty attracting the coverage of securities analysts.
24
Stockholders may experience dilution of ownership interests because of the future issuance of additional shares of our common stock and our preferred stock.
In the future, we may issue our authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of our present stockholders. We are currently authorized to issue an aggregate of 310,000,000 shares of capital stock consisting of 300,000,000 shares of common stock and 10,000,000 shares of preferred stock with preferences and rights to be determined by our Board of Directors. As of March 9, 2011, there were 54,217,106 shares of our common stock outstanding and a total of 6,814,713 shares subject to outstanding options and warrants. On July 23, 2009, we issued 1,000,000 shares of our Series B preferred stock, all of which has been converted into common stock at a rate of two shares of common stock for each share of Series B preferred
stock. Under the purchase agreement pursuant to which we issued the Series B preferred stock, we may, at our election, cause the investors party thereto to purchase from us up to an additional 4,000,000 shares of Series B preferred stock, which, if issued, would be initially convertible into 8,000,000 shares of our common stock. We may also issue additional shares of our common stock or other securities that are convertible into or exercisable for common stock in connection with hiring or retaining employees, future acquisitions, future sales of our securities for capital raising purposes, or for other business purposes. The future issuance of any such additional shares of our common stock may create downward pressure on the trading price of the common stock. There can be no assurance that we will not be required to issue additional shares, warrants or other convertible securities in the future in conjunction with any capital raising efforts, including at a price
(or exercise prices) below the price at which shares of our common stock are then traded on the NYSE Amex.
Our common shares are thinly traded and, therefore, relatively illiquid.
As of March 9, 2011, there were 54,217,106 shares of our common stock outstanding. While our common shares trade on the NYSE Amex, our stock is thinly traded (approximately 0.1%, or 84,989 shares, of our stock traded on an average daily basis during the three months immediately preceding the date of this annual report on Form 10-K, and you may have difficulty in selling your shares quickly. The low trading volume of our common stock is outside of our control, and may not increase in the near future or, even if it does increase in the future, may not be maintained.
Our principal stockholders have significant voting power and may take actions that may not be in the best interests of other stockholders.
Our officers, directors, principal stockholders and their affiliates control approximately 30.4% of our outstanding common stock. If these stockholders act together, they will be able to exert significant control over our management and affairs requiring stockholder approval, including approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control and might adversely affect the market price of our common stock. This concentration of ownership may not be in the best interests of all our stockholders. Unaffiliated holders of our common stock have no effective voice in our management. Additionally, sales by our insiders or affiliates could adversely affect the market price of our common stock.
A significant number of our shares are eligible for sale, which could depress the market price of our stock.
Sales of a significant number of shares of our common stock in the public market could harm the market price of our stock. As additional shares of our common stock become available for resale in the public market, the supply of the common stock will increase, which could decrease its price. Further, shares may be offered by selling stockholders from time to time in the open market pursuant to Rule 144, and these sales may have a depressive effect on the market for the shares of our common stock.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
Our executive offices and our research and development laboratory are located at 3 Sapir Street, Weizmann Science Park, Nes-Ziona, Israel 74140 and our phone number is (866) 644-7811. The facility is approximately 6,000 square feet. We pay a monthly lease of $7,600, plus approximately $450 per month with respect to taxes and utilities, for this space. This lease will expire on January 20, 2012.
Item 3. Legal Proceedings
From time to time we may be named in claims arising in the ordinary course of business. Currently, no legal proceedings, government actions, administrative actions, investigations or claims are pending against us or involve us that, in the opinion of our management, could reasonably be expected to have a material adverse effect on our business or financial condition.
25
Item 4. (Removed and Reserved)
PART II
|
Item 5.
|
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
|
Market Information
Our common stock has been quoted on the NYSE Amex under the symbol “PBTH” since March 29, 2010. Prior to March 29, 2010 our common stock was quoted on the OTCBB, and prior to June 12, 2009, our trading symbol on the OTCBB was “MODG”. The table below sets forth, (i) for the periods indicated commencing from and after March 29, 2010, the high and low sales prices for our common stock on the NYSE Amex and (ii) for periods preceding March 29, 2010, the high and low bid prices for our common stock on the OTCBB. All OTCBB bid prices represent inter-dealer prices, without adjustments for retail mark-ups, mark-downs or commissions and may not necessarily represent actual transactions.
|
Quarter Ended
|
High
|
Low
|
||||||
|
December 31, 2010
|
$ | 7.91 | $ | 5.80 | ||||
|
September 30, 2010
|
$ | 7.27 | $ | 5.50 | ||||
|
June 30, 2010
|
$ | 8.85 | $ | 3.65 | ||||
|
March 31, 2010
|
$ | 4.84 | $ | 2.15 | ||||
|
December 31, 2009
|
$ | 2.57 | $ | 1.22 | ||||
|
September 30, 2009
|
$ | 1.99 | $ | 0.79 | ||||
|
June 30, 2009
|
$ | 0.94 | $ | 0.40 | ||||
|
March 31, 2009
|
$ | 0.75 | $ | 0.40 | ||||
Number of Holders
As of March 9, 2011, our common stock was held by 49 stockholders of record.
Dividends
We have never declared or paid dividends on our common stock. We do not intend to pay cash dividends on our common stock for the foreseeable future, and we intend to retain any future earnings to fund the development and growth of our business. The payment of dividends if any, on the common stock will rest solely within the discretion of our board of directors and will depend, among other things, upon our earnings, capital requirements, financial condition, and other relevant factors.
26
Item 6. Selected Financial Data
The following table states our selected consolidated financial data, which has been derived from our audited consolidated financial statements. The table reflects our consolidated results of operations for the periods indicated and should be read together with our consolidated financial statements and notes thereto as well as Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
|
Year Ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
Statement of Operations Data
|
||||||||||||||||||||
|
Revenues
|
— | — | — | — | — | |||||||||||||||
|
Net Income (Loss)
|
$ | (7,559,131 | ) | $ | (7,484,718 | ) | $ | (7,033,536 | ) | $ | (3,313,203 | ) | $ | (2,215,949 | ) | |||||
|
Net Income (Loss) per common share
|
$ | (0.19 | ) | $ | (0.21 | ) | $ | (0.20 | ) | $ | (0.12 | ) | $ | (0.16 | ) | |||||
|
Year Ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
Balance Sheet Data
|
||||||||||||||||||||
|
Total Assets
|
$ | 27,205,001 | $ | 4,109,421 | $ | 8,354,667 | $ | 12,235,853 | $ | 1,301,690 | ||||||||||
|
Current Liabilities
|
$ | 1,989,604 | $ | 840,119 | $ | 396,073 | $ | 330,931 | $ | 463,492 | ||||||||||
|
Long-Term Obligations
|
$ | 220,838 | $ | 140,237 | $ | 90,732 | $ | 42,552 | $ | 19,433 | ||||||||||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operation
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our historical consolidated financial statements and related notes thereto in “Item 8. Financial Statements and Supplementary Data.” The discussion below contains forward-looking statements that are based upon our current expectations and are subject to uncertainty and changes in circumstances. Actual results may differ materially from these expectations due to inaccurate assumptions and known or unknown risks and uncertainties, including those identified in “Cautionary Statement Regarding Forward-Looking Statements” and “Item 1A. Risk Factors.”
The discussion and analysis of the Company’s financial condition and results of operations are based on the Company’s financial statements, which the Company has prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires the Company to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, the Company evaluates such estimates and judgments, including those described in greater detail below. The Company bases its estimates on historical experience and on various other factors that the Company believes are
reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Overview
We are a development stage biopharmaceutical company utilizing patented technology to develop longer-acting, proprietary versions of already-approved therapeutic proteins that currently generate billions of dollars in annual global sales. We have obtained certain exclusive worldwide rights from Washington University in St. Louis, Missouri to use a short, naturally-occurring amino acid sequence (peptide) that has the effect of slowing the removal from the body of the therapeutic protein to which it is attached. This Carboxyl Terminal Peptide (CTP) can be readily attached to a wide array of existing therapeutic proteins, stabilizing the therapeutic protein in the bloodstream and extending its life span without additional toxicity or loss of desired biological activity. We are using
the CTP technology to develop new, proprietary versions of certain existing therapeutic proteins that have longer life spans than therapeutic proteins without CTP. We believe that our products will have greatly improved therapeutic profiles and distinct market advantages.
We believe our products in development will provide several key advantages over our competitor’s existing products:
|
|
·
|
significant reduction in the number of injections required to achieve the same or superior therapeutic effect from the same dosage;
|
27
|
|
·
|
extended patent protection for proprietary new formulations of existing therapies;
|
|
|
·
|
faster commercialization with greater chance of success and lower costs than those typically associated with a new therapeutic protein; and
|
|
|
·
|
manufacturing using industry-standard biotechnology-based protein production processes.
|
Merck & Co. has developed the first novel protein containing CTP, named ELONVA®, a long-acting CTP-modified version of the fertility drug follicle stimulating hormone (FSH). On January 28, 2010, Merck received marketing authorization from the European Commission for ELONVA® with unified labeling valid in all European Union Member States. Merck licensed the CTP technology directly from Washington University (prior to the formation of Modigene Delaware) for application only to Follicle Stimulating Hormone (FSH) and three other hormones, human Chorionic Gonadotropin (hCG), Luteinizing Hormone (LH) and Thyroid-Stimulating Hormone (TSH).
Our internal product development program is currently focused on extending the life span of the following biopharmaceuticals, in an effort to provide patients with improved therapies that may enhance their quality of life:
|
|
·
|
Human Growth Hormone (hGH)
|
|
|
·
|
Factor IX
|
|
|
·
|
Anti-Obesity Peptide Oxyntomodulin
|
|
|
·
|
Factor VIIa
|
|
|
·
|
Interferon β and Erythropoietin (EPO)
|
|
|
·
|
Atherosclerosis and rheumatoid arthritis long-acting therapies
|
We believe that the CTP technology will be broadly applicable to these as well as other best-selling therapeutic proteins in the market and will be attractive to potential partners because it will allow them to extend proprietary rights for therapeutic proteins with near-term patent expirations.
Plan of Operation
During 2011, PROLOR intends to continue the development of its primary clinical and preclinical programs.. These programs include a variety of operating tasks such as optimization of expression levels, toxicity and efficacy animal models, completion of purification processes, GMP production of compounds and clinical studies. The Company’s cash resources, including expected payments from the OCS are expected to be sufficient to maintain the Company’s operations through the fourth quarter of 2012. The Company is not planning any major purchase or sale of equipment during that timeframe, and its employee headcount is expected to increase from 22 employees at December 31, 2010 to 25 employees, of which 24 are expected to be full time.
Critical Accounting Policies
The historical financial statements of the Company included with this Annual Report have been prepared in accordance with U.S. generally accepted accounting principles. The significant accounting policies followed in the preparation of the financial statements, on a consistent basis, are described below.
Use of Estimates: The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Financial Statements in United States Dollars: The functional currency of the Company is the U.S. dollar, as the U.S. dollar is the primary currency of the economic environment in which the Company has operated and expects to continue to operate in the foreseeable future. The majority of ModigeneTech’s operations are currently conducted in Israel. Most of the Israeli expenses are currently determined and paid in U.S. dollars. Financing and investing activities including loans and equity transactions are made in U.S. dollars. The majority of our assets are held in the U.S.
28
Accordingly, the functional and reporting currency of the Company is the U.S. dollar. Monetary accounts maintained in currencies other than the dollar are remeasured into U.S. dollars. All transaction gains and losses from the remeasurement of monetary balance sheet items are reflected in the statements of operations as financial income or expenses, as appropriate.
Principles of Consolidation: The consolidated financial statements include the accounts of Modigene Delaware and its wholly-owned subsidiary, ModigeneTech. Intercompany transactions and balances have been eliminated upon consolidation.
Cash Equivalents: For purposes of reporting within the statement of cash flows, the Company considers all cash on hand, cash accounts not subject to withdrawal restrictions or penalties, and all highly liquid debt instruments purchased with a maturity of three months or less to be cash and cash equivalents.
Accounts receivable: Accounts receivable are recorded at net realizable value consisting of the carrying amount less the allowance for uncollectible accounts. As of December 31, 2010 and 2009 the Company has not accrued an allowance for uncollectible accounts.
Property and Equipment: Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated by the straight-line method over the estimated useful lives of the assets. The annual depreciation rates are as follows:
|
%
|
|
|
Office furniture and equipment
|
6
|
|
Laboratory equipment
|
15
|
|
Computers and electronic equipment
|
33
|
|
Leasehold improvements
|
25
|
The Company reviews the carrying value of its long-lived assets, including intangible assets subject to amortization, for impairment whenever events and circumstances indicate that the carrying value of the assets may not be recoverable. Recoverability of these assets is measured by comparing the carrying value of the assets to the undiscounted cash flows estimated to be generated by those assets over their remaining economic life. If the undiscounted cash flows are not sufficient to recover the carrying value of the assets, the assets are considered impaired. The impairment loss is measured by comparing the fair value of the assets to their carrying value. Fair value is determined by either a quoted market price or a value determined by a discounted cash flow technique, whichever is more appropriate under the
circumstances involved. No impairments were recognized for the period from May 31, 2005 (inception date) to December 31, 2010.
Research and Development Costs and Participation: Research and development (“R&D”) costs are expensed as they are incurred and consist of salaries, benefits and other personnel related costs, fees paid to consultants, clinical trials and related clinical manufacturing costs, license and milestone fees, and facilities and overhead costs. R&D expenses consist of independent R&D costs and costs associated with collaborative R&D and in-licensing arrangements. Participation from government for development of approved projects are recognized as a reduction of expenses as the related costs are incurred.
Severance Pay: The liability of ModigeneTech for severance pay is calculated pursuant to the Severance Pay Law in Israel, based on the most recent salary of the employees multiplied by the number of years of employment as of the balance sheet date and is presented on an undiscounted basis. ModigeneTech’s employees are entitled to one month’s salary for each year of employment or a portion thereof. According to agreements with key employees that are related parties of the Company, upon retirement, the key employees will be entitled to receive a lump-sum payment, therefore the Company does not accumulate severance pay for those employees and the sum will be accrued when the Company has to pay such payments according to the employment agreements.
Severance (income) expenses for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010 amounted to $11,579, $(1,044), $8,090 and $27,492, respectively.
29
Income Taxes: The Company accounts for income taxes in accordance with the provisions of ASC 740-10, Income Taxes. ASC 740-10 requires companies to recognize deferred tax assets and liabilities based on the differences between financial reporting and tax bases of assets and liabilities. These differences are measured using the enacted tax rates and laws that are expected to be in effect when the temporary differences are expected to reverse. A valuation allowance is established against net deferred tax assets, if based on the weighted available evidence, it is more likely than not that all or a portion of the deferred tax assets will not be realized. On January 1, 2007, the Company adopted ASC 740-10, Income Taxes, as it applies to accounting for
uncertainty in income taxes, which did not result in an adjustment to its tax contingencies.
Concentrations of Credit Risk: Financial instruments that potentially subjected the Company, Modigene Delaware and ModigeneTech to concentrations of credit risk consist principally of cash and cash equivalents.
Cash and cash equivalents are invested in major banks in Israel and the United States. Such deposits in the United States are not insured. Management believes that the financial institutions that hold the Company’s investments are financially sound and, accordingly, minimal credit risk exists with respect to these investments.
The Company has no off-balance sheet concentration of credit risk such as foreign exchange contracts or other foreign hedging arrangements.
Fair Value Measurement: As defined in ASC 820-10, Fair Value Measurements and Disclosures (“ASC 820-10”), fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. In order to increase consistency and comparability in fair value measurements, ASC 820-10 establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2: Other inputs that are observable, directly or indirectly, such as quoted prices for similar assets and liabilities or market corroborated inputs.
Level 3: Unobservable inputs are used when little or no market data is available, which requires the Company to develop its own assumptions about how market participants would value the assets or liabilities. The fair value hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible in its assessment of fair value.
The following table presents the Company’s financial assets and liabilities that are carried at fair value, classified according to the three categories described above:
|
Fair Value Measurements at December 31, 2010
|
||||||||||||||||
|
Quoted Prices in
|
Significant
|
|||||||||||||||
|
Active
|
Other
|
Significant
|
||||||||||||||
|
Markets for
|
Observable
|
Unobservable
|
||||||||||||||
|
Identical Assets
|
Inputs
|
Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Cash and cash equivalents
|
$ | 24,474,458 | $ | 24,474,458 | $ | - | $ | - | ||||||||
|
Short term deposits
|
1,439,269 | 1,439,269 | - | - | ||||||||||||
|
Restricted cash
|
102,932 | 102,932 | - | - | ||||||||||||
|
Total assets at fair value
|
$ | 26,016,659 | $ | 26,016,659 | $ | - | $ | - | ||||||||
|
Fair Value Measurements at December 31, 2009
|
||||||||||||||||
|
Quoted Prices in
|
Significant
|
|||||||||||||||
|
Active
|
Other
|
Significant
|
||||||||||||||
|
Markets for
|
Observable
|
Unobservable
|
||||||||||||||
|
Identical Assets
|
Inputs
|
Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Cash and cash equivalents
|
$ | 3,521,866 | $ | 3,521,866 | $ | - | $ | - | ||||||||
|
Restricted cash
|
91,730 | 91,730 | - | - | ||||||||||||
|
Total assets at fair value
|
$ | 3,613,596 | $ | 3,613,596 | $ | - | $ | - | ||||||||
30
Royalty-bearing Grants: Royalty-bearing grants from the Government of Israel for participation in development of approved projects are recognized as a reduction of expenses as the related costs are incurred. Funding is recognized at the time ModigeneTech is entitled to such grants, on the basis of the costs incurred.
Research and development grants received by the Company reduced research and development expenses by $1,383,806, $484,912, 746,013 and $3,383,597 for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, respectively. Research and development grants receivable as of December 31, 2010 was $281,728.
Loss per Share: Basic and diluted losses per share are presented in accordance with ASC 260-10 “Earnings per share”. Outstanding share options and warrants have been excluded from the calculation of the diluted loss per share because all such securities are antidilutive. The total weighted average number of ordinary shares related to outstanding options and warrants excluded from the calculations of diluted loss per share were 16,931,505, 9,572,278, 7,620,546 and 7,552,859 for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, respectively.
Results of Operations
Year Ended December 31, 2010 Compared to Year Ended December 31, 2009 and Year Ended December 31, 2008
Revenue
The Company has not generated any revenue since its inception. To date, the Company has funded its operations primarily through grants from the OCS and the sale of equity securities. If the Company’s development efforts result in clinical success, regulatory approval and successful commercialization of the Company’s products, the Company could generate revenue from sales of its products.
Revenue, if and when generated, will be recognized in accordance with ASC 605-10-S99, “Revenue Recognition”. The Company will recognize revenue when the significant risks and rewards of ownership have been transferred to the customer pursuant to applicable laws and regulations, including factors such as when there has been evidence of a sales arrangement, the performance has occurred, or service have been rendered, the price to the buyer is fixed or determinable, and collectability is reasonably assured.
Research and Development Expense
The Company expects its research and development expense to increase as it continues to develop its product candidates. Research and development expense consists of:
|
|
·
|
internal costs associated with research and development activities;
|
|
|
·
|
payments made to third party contract research organizations, contract manufacturers, investigative sites, and consultants;
|
|
|
·
|
manufacturing development costs;
|
31
|
|
·
|
personnel-related expenses, including salaries, benefits, travel, and related costs for the personnel involved in the research and development;
|
|
|
·
|
activities relating to the advancement of product candidates through preclinical studies and clinical trials; and
|
|
|
·
|
facilities and other expenses, which include expenses for rent and maintenance of facilities, as well as laboratory and other supplies.
|
These costs and expenses are partially funded by grants received by the Company from the OCS. There can be no assurance that the Company will continue to receive grants from the OCS in amounts sufficient for its operations, if at all.
The Company expects its research and development expenditures to increase most significantly in the near future in connection with the ongoing production of its protein drug candidates. The Company intends to continue to hire new employees, in research and development, in order to meet its operation plans.
The Company has multiple research and development projects ongoing at any one time. The Company utilizes its internal resources, employees, and infrastructure across multiple projects and tracks time spent by employees on specific projects. The Company believes that significant investment in product development is a competitive necessity and plans to continue these investments in order to realize the potential of its product candidates. For the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, the Company incurred gross research and development expense in the aggregate of $6,603,589, $6,040,235, $5,527,102 and $21,704,138, respectively.
The successful development of the Company’s product candidates is subject to numerous risks, uncertainties, and other factors. Beyond the next twelve months, the Company cannot reasonably estimate or know the nature, timing and costs of the efforts that will be necessary to complete the remainder of the development of, or the period, if any, in which material net cash inflows may commence from the Company’s product candidates or any of the Company’s other development efforts. This is due to the numerous risks and uncertainties associated with the duration and cost of clinical trials which vary significantly over the life of a project as a result of differences arising during clinical development, including:
|
|
·
|
completion of such preclinical and clinical trials;
|
|
|
·
|
the number of clinical sites included in the trials;
|
|
|
·
|
the length of time required to enroll suitable patients;
|
|
|
·
|
the number of patients that ultimately participate in the trials;
|
|
|
·
|
adverse medical events or side effects in treated patients;
|
|
|
·
|
Lack of comparability with complementary technologies;
|
|
|
·
|
obtaining capital necessary to fund operations, including the research and development efforts; and
|
|
|
·
|
the results of clinical trials.
|
The Company’s expenditures are subject to additional uncertainties, including the terms and timing of regulatory approvals, and the expense of filing, prosecuting, defending and enforcing any patent claims or other intellectual property rights. The Company may obtain unexpected results from its clinical trials. The Company may elect to discontinue, delay or modify clinical trials of some product candidates or focus on others. A change in the outcome of any of the foregoing variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate. For example, if the FDA or other regulatory authorities were to require the Company to conduct clinical trials beyond those which it currently anticipates will
be required for the completion of the clinical development of a product candidate, or if the Company experiences significant delays in enrollment in any of its clinical trials, the Company could be required to expend significant additional financial resources and time on the completion of clinical development. Drug development may take several years and millions of dollars in development costs. If the Company does not obtain or maintain regulatory approval for its products, its financial condition and results of operations will be substantially harmed.
32
General and Administrative Expense
General and administrative expense consists primarily of salaries and other related costs, including stock-based compensation expense, for persons serving in the Company’s executive and administration functions. Other general and administrative expense includes facility-related costs not otherwise included in research and development expense, and professional fees for legal and accounting services, including those associated with reporting obligations applicable to public companies in the United States. The Company expects that its general and administrative expenses will increase as it adds additional personnel and advances its research and development programs. For the years ended December 31, 2010, December 31, 2009, 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010,
the Company incurred general and administrative expense of $2,457,043, $1,901,952, $2,410,015 and $13,676,569, respectively. The increase in 2010 compared to 2009 resulted primarily from stock-based compensation and increase in rent and utilities.
Financial Expense and Income
Financial expense and income consists of the following:
|
|
·
|
interest earned on the Company’s cash and cash equivalents;
|
|
|
·
|
interest expense on short term bank credit and loan; and
|
|
|
·
|
expense or income resulting from fluctuations of the New Israeli Shekel, which a portion of the Company’s assets and liabilities are denominated in, and other foreign currencies against the United States Dollar and other foreign currencies.
|
For the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, the Company recorded gross financial income of $117,695, $(27,443), $157,568 and $635,985, respectively. Financial income (expense) for the years ended December 31, 2009 and 2008 resulted primarily from currency fluctuations on deposits denominated in the New Israeli Shekel and the Euro. The increase in financial income for the year ended December 31, 2010 as compared to the year ended December 31, 2009 was primarily due to increased interest income received on deposited proceeds from the March 11, 2010 and March 17, 2010 equity financings.
Stock-based Compensation
The Company’s stock-based compensation are recorded according to ASC 718-10, "Compensation - Stock Compensation", which requires the measurement and recognition of compensation expense for all stock-based payment awards made to employees and directors, including employee stock options under the Company’s stock plans, based on estimated fair values.
ASC 718-10 requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in the Company’s consolidated statement of operations.
The Company estimates the fair value of stock options granted using the Black-Scholes-Merton option-pricing model. For the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, the Company’s stock-based compensation expenses were $883,425, $728,341, $1,039,028 and $8,079,210, respectively.
Cash Flows
For the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, net cash used in operations was $5,915,794, $5,907,653, $5,761,061 and $21,558,850, respectively.
33
For the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, net cash used in investing activities was $1,672,814, $53,293, $227,628 and $2,834,490, respectively. The increase for 2010 as compared to 2009 resulted primarily from cash investments in short-term bank deposits, which were held mostly in US dollars. The decrease for 2009 as compared to 2008 resulted primarily from reduced purchases of equipment.
For the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, net cash provided by financing activities was 28,541,200, $2,017,580, $1,998,114 and $48,867,798, respectively. The increase for 2010 as compared to 2009 resulted from our issuance of shares of common stock during 2010 as well as from option and warrant exercises.
Liquidity and Capital Resources
The Company expects to incur losses from operations for the foreseeable future. The Company expects to incur increasing research and development expenses, including expenses related to the hiring of personnel and additional clinical trials. The Company expects that general and administrative expenses will also increase as the Company expands its finance and administrative staff and adds infrastructure. Our future capital requirements will depend on a number of factors, including the continued progress of our research and development of product candidates, the timing and outcome of clinical trials and regulatory approvals, the costs involved in preparing, filing, prosecuting, maintaining, defending, and enforcing patent claims and other intellectual property rights, the status of competitive products, the
availability of financing, and our success in developing markets for our product candidates.
We believe that our existing cash and cash equivalents will be sufficient to enable us to fund our operating expenses and capital expenditure requirements at least until December 31, 2012. We have based this estimate on assumptions that may prove to be wrong or subject to change, and we may be required to use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials. Our future capital requirements will depend on many factors, including the progress and results of our clinical trials,
the duration and cost of discovery and preclinical development, and laboratory testing and clinical trials for our product candidates, the timing and outcome of regulatory review of our product candidates, the number and development requirements of other product candidates that we pursue, and the costs of commercialization activities, including product marketing, sales, and distribution. We do not anticipate that we will generate product revenues for at least the next several years. In the absence of additional funding, we expect continuing operating losses to result in increases in our cash used in operations over the next several years. To the extent that our capital resources are insufficient to meet our future capital requirements, we will need to finance our future cash needs through public or private equity offerings, debt financings, or corporate collaboration and licensing arrangements. We currently do not have any commitments for future external funding. We may need to raise
additional funds more quickly if one or more of our assumptions proves to be incorrect or if we choose to expand our product development efforts more rapidly than we presently anticipate, and we may decide to raise additional funds even before we need them if the conditions for raising capital are favorable. We may seek to issue equity or debt securities or obtain a credit facility from one or more financial institutions. The sale of equity or convertible debt securities may result in dilution to our shareholders. The incurrence of indebtedness would result in increased fixed obligations and could also result in covenants that would restrict our operations. Additional equity or debt financing, grants or corporate collaboration and licensing arrangements may not be available on acceptable terms, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our planned commercialization efforts
or obtain funds through arrangements with collaborators or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
Contractual Obligations
The following table sets forth our contractual payment obligations as of December 31, 2010 for the periods indicated below (in thousands):
34
|
More than
|
||||||||||||||||||||
|
Less than
|
1 - 3 | 3 - 5 |
5 Years and
|
|||||||||||||||||
|
Contractual Obligations
|
Total
|
1 Year
|
Years
|
Years |
Thereafter
|
|||||||||||||||
|
Long-term debt
|
— | — | — | — | — | |||||||||||||||
|
Other long term liabilities reflected in the balance sheet
|
$ | 220,838 | — | — | — | $ | 220,838 | |||||||||||||
|
Total
|
$ | 220,838 | — | — | — | $ | 220,838 | |||||||||||||
Effects of Inflation and Currency Fluctuations
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation has had a material effect on our results of operations for the years ended December 31, 2010, 2009 or December 31, 2008, nor do we expect that inflation will have a material impact on our results of operations for the year ending December 31, 2011.
Currency fluctuations could affect us by increased or decreased costs. We do not believe that currency fluctuations have had a material effect on our results of operations for the years ended December 31, 2010, 2009 or December 31, 2008, nor do we expect that currency fluctuations will have a material impact on our results of operations for the year ending December 31, 2011.
In January 2010, the FASB issued Accounting Standards Update No. 2010-06, Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements. The amendments in this update require new disclosures and clarifications of existing disclosures related to transfers in and out of Level 1 and Level 2 fair value measurements, further disaggregation of fair value measurement disclosures for each class of assets and liabilities and additional details of valuation techniques and inputs utilized. This update is consistent with the Company's current accounting application for fair value measurements and disclosures.
In April, 2010, the FASB issued Accounting ASU No. 2010-13, “Compensation-Stock Compensation (Topic 718)—Effect of Denominating the Exercise Price of a Share-Based Payment Award in the Currency of the Market in Which the Underlying Equity Security Trades—a consensus of the FASB Emerging Issues Task Force” (“ASU 2010-13”). ASU 2010-13 provides amendments to Topic 718 to further clarify that an employee share-based payment award with an exercise price denominated in the currency of a market in which a substantial portion of the entity’s equity securities trades should classify such an award as equity. The amendments in this update do not expand the recurring disclosures required by Topic 718. The amendments in this update are effective for fiscal years, and interim
periods within those fiscal years, beginning on or after December 15, 2010. The company adopted this standard in January, 2011 which did not have a material impact on our financial statements.
Off-Balance Sheet Arrangements
The Company has no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on the Company’s financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources that are material to investors.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
The information in Item 7 under the caption “Management’s Discussion and Analysis of Financial Condition and Results of Operations — Effects of Inflation and Currency Fluctuation” is incorporated herein by reference.
Interest Rate Risk
We have no debt outstanding nor do we have any investments in debt instruments other than highly liquid short-term investments. Accordingly, we consider our interest rate risk exposure to be insignificant at this time.
35
Item 8. Financial Statements and Supplementary Data
The financial statements required by this Item 8 are filed herewith commencing on page F-1 hereto.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
The Company’s management, with the participation of its Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of the design and operation of the Company’s disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) or 15d-15(e)) as of December 31, 2010. Based upon that evaluation, the Chief Executive Officer and Chief Financial Officer concluded that, as of that date, the Company’s disclosure controls and procedures were effective as of the end of the period covered by this annual report.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally
accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
For the period ended December 31, 2010, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, management (with the participation of our principal executive officer and principal financial officer) conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this evaluation, management concluded that, as of December 31, 2010, our internal control over financial reporting was effective.
BKR Yarel + Partners, the independent registered public accounting firm which audits our financial statements, has audited our internal control over financial reporting as of December 31, 2010 and has expressed an unqualified opinion thereon.
Changes in Internal Controls Over Financial Reporting
There have been no changes in the Company’s internal control over financial reporting during the quarter ended December 31, 2010 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
Item 9B. Other Information.
None.
36
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this Item is incorporated by reference to the definitive proxy statement for our 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December 31, 2010.
Item 11. Executive Compensation.
The information required by this Item is incorporated by reference to the definitive proxy statement for our 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December 31, 2010.
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
|
The information required by this Item is incorporated by reference to the definitive proxy statement for our 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December 31, 2010.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this Item is incorporated by reference to the definitive proxy statement for our 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December 31, 2010.
Item 14. Principal Accounting Fees and Services.
The information required by this Item is incorporated by reference to the definitive proxy statement for our 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December 31, 2010.
PART IV
Item 15. Exhibits and Financial Statement Schedules
The following documents are filed as part of this Annual Report on Form 10-K:
1. Financial Statements. The following Consolidated Financial Statements of PROLOR Biotech, Inc. are included in Item 8 of this Annual Report on Form 10-K:
|
Page
|
|||
|
Report of Independent Registered Public Accounting Firm
|
F-3 | ||
|
Consolidated Balance Sheets
|
F-5 | ||
|
Consolidated Statements of Operations
|
F-6 | ||
|
Consolidated Statements of Stockholders’ Equity
|
F-7 | ||
|
Consolidated Statements of Cash Flows
|
F-11 | ||
|
Notes to Consolidated Financial Statements
|
F-13 |
2. Financial Statement Schedule. The information required by this item is included in the consolidated financial statements contained in this Annual Report on Form 10-K.
37
3. Exhibits
|
Exhibit
|
Description
|
|
|
3.1
|
Amended and Restated Articles of Incorporation of Modigene Inc. (n/k/a PROLOR Biotech, Inc.), filed as Exhibit 3.1 to our Current Report on Form 8-K filed with the SEC on February 27, 2007 and incorporated by reference herein.
|
|
|
3.2
|
Certificate of Amendment to the Amended and Restated Articles of Incorporation of Modigene Inc. (n/k/a PROLOR Biotech, Inc.), filed as Exhibit 3.1 to our Quarterly Report on Form 10-Q, filed with the SEC on August 14, 2009 and incorporated by reference herein.
|
|
|
3.3
|
Amended and Restated Bylaws of Modigene Inc. (n/k/a PROLOR Biotech, Inc.), filed as Exhibit 3.2 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
4.1
|
Form of Investor Warrant of Modigene Inc. issued as of May 9, 2007, filed as Exhibit 4.1 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
4.2
|
Form of Warrant of Modigene Inc. issued to broker/dealers as of May 9, 2007, filed as Exhibit 4.2 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
4.3
|
Form of Warrant of Modigene Inc. issued to Frost Gamma Investments Trust, Jane Hsiao, Steven D. Rubin and Subbarao Uppaluri, filed as Exhibit 4.3 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
4.4
|
Warrant Agreement dated as of May 9, 2007, between Modigene Inc. and Spencer Trask Ventures, Inc., together with the form of Warrant Certificate issued thereunder, filed as Exhibit 4.4 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
4.5
|
Certificate of Designation of Series A Convertible Preferred Stock, filed as Exhibit 4.1 to our Current Report on Form 8-K filed with the SEC on March 27, 2008 and incorporated by reference herein.
|
|
|
4.6
|
Certificate of Designation of Series B Convertible Preferred Stock, filed as Exhibit 3.1 to our Current Report on Form 8-K filed with the SEC on July 24, 2009 and incorporated by reference herein.
|
|
|
4.7
|
Specimen Certificate for Series B Preferred Stock, filed as Exhibit 4.2 to our Current Report on Form 8-K filed with the SEC on July 24, 2009 and incorporated by reference herein.
|
|
|
10.1+
|
Consulting Agreement between Modigene Inc. and Abraham (Avri) Havron, filed as Exhibit 10.6 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.2+
|
Amendment to Consulting Agreement between Modigene Inc. and Abraham (Avri) Havron, filed as Exhibit 10.4 to our Annual Report on Form 10-KSB filed with the SEC on March 31, 2008 and incorporated by reference herein.
|
|
|
10.3+
|
Amendment to Consulting Agreement between the Company and Abraham (Avri) Havron, filed as Exhibit 10.1 to our Current Report on Form 8-K filed with the SEC on July 18, 2008 and incorporated by reference herein.
|
|
|
10.4+
|
Employment Agreement between Modigene Inc. and Shai Novik, filed as Exhibit 10.7 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.5+
|
First Amendment to Employment Agreement between Modigene Inc. and Shai Novik, filed as Exhibit 10.8 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.6+
|
Second Amendment to Employment Agreement between Modigene Inc. and Shai Novik, filed as Exhibit 10.7 to our Annual Report on Form 10-KSB filed with the SEC on March 31, 2008 and incorporated by reference herein.
|
38
|
Exhibit
|
Description
|
|
|
10.7+
|
Third Amendment to Employment Agreement between the Company and Shai Novik, filed as Exhibit 10.2 to our Current Report on Form 8-K filed with the SEC on July 18, 2008 and incorporated by reference herein.
|
|
|
10.8+
|
Employment Agreement between ModigeneTech Ltd. and Dr. Eyal Fima, filed as Exhibit 10.9 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.9+
|
First Amendment to Employment Agreement between ModigeneTech Ltd. and Dr. Eyal Fima, filed as Exhibit 10.9 to our Annual Report on Form 10-KSB filed with the SEC on March 31, 2008 and incorporated by reference herein.
|
|
|
10.10+
|
Second Amendment to Employment Agreement between ModigeneTech Ltd. and Dr. Eyal Fima, filed as Exhibit 10.4 to our Current Report on Form 8-K filed with the SEC on July 18, 2008 and incorporated by reference herein.
|
|
|
10.11+
|
Consulting Agreement between ModigeneTech Ltd. and Dr. Fuad Fares, filed as Exhibit 10.10 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.12+
|
First Amendment to Consulting Agreement between Modigene Inc. and Dr. Fuad Fares, filed as Exhibit 10.3 to our Current Report on Form 8-K filed with the SEC on July 18, 2008 and incorporated by reference herein.
|
|
|
10.13+
|
Modigene Inc. 2005 Stock Incentive Plan, filed as Exhibit 10.11 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.14+
|
Modigene Inc. 2007 Equity Incentive Plan, as amended, filed as Exhibit 10.12 to our Annual Report on Form 10-KSB filed with the SEC on March 26, 2008 and incorporated by reference herein.
|
|
|
10.15+
|
Form of Stock Option Agreement under the 2005 Stock Incentive Plan, filed as Exhibit 10.13 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.16+
|
Form of Stock Option Agreement under the 2007 Equity Incentive Plan, filed as Exhibit 10.14 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.17
|
Exclusive License Agreement dated February 2, 2007 between Modigene Inc. and Washington University, filed as Exhibit 10.15 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.18
|
Form of Clinical Advisory Panel Agreement, filed as Exhibit 10.16 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.19
|
Form of Scientific Advisory Board Agreement, filed as Exhibit 10.17 to our Current Report on Form 8-K filed with the SEC on May 14, 2007 and incorporated by reference herein.
|
|
|
10.20
|
Securities Purchase Agreement among Modigene Inc., Frost Gamma Investments Trust, Jane Hsiao, Steven D. Rubin and Subbarao Uppaluri, filed as Exhibit 10.1 to our Current Report on Form 8-K filed with the SEC on March 27, 2008 and incorporated by reference herein.
|
|
|
10.21
|
Series B Convertible Preferred Stock Purchase Agreement, dated July 22, 2009, filed as Exhibit 10.1 to our Current Report on Form 8-K filed with the SEC on July 24, 2009 and incorporated by reference herein.
|
|
|
10.22
|
Form of Lockup Agreement, filed as Exhibit 10.2 to our Current Report on Form 8-K filed with the SEC on July 24, 2009 and incorporated by reference herein.
|
|
|
10.23
|
Letter Agreement, dated July 22, 2009, between PROLOR Biotech, Inc. and The Frost Group, LLC terminating Credit Agreement and Note and Security Agreement, filed as Exhibit 10.3 to our Current Report on Form 8-K filed with the SEC on July 24, 2009 and incorporated by reference herein.
|
39
|
Exhibit
|
Description
|
|
|
10.24+
|
Third Amendment to Consulting Agreement between PROLOR Biotech, Inc. and Abraham Havron, Ph.D, filed as Exhibit 10.1 to our Current Report on Form 8-K filed with the SEC on January 25, 2010 and incorporated by reference herein.
|
|
|
10.25+
|
Fourth Amendment to Employment Agreement between PROLOR Biotech, Inc. and Shai Novik, filed as Exhibit 10.2 to our Current Report on Form 8-K filed with the SEC on January 25, 2010 and incorporated by reference herein.
|
|
|
10.26
|
Form of Securities Purchase Agreement, filed as Exhibit 10.1 to our Current Report on Form 8-K filed with the SEC on March 17, 2010 and incorporated by reference herein.
|
|
|
10.27
|
Form of Lockup Agreement, filed as Exhibit 10.2 to our Current Report on Form 8-K filed with the SEC on March 17, 2010 and incorporated by reference herein.
|
|
|
21
|
Subsidiaries of PROLOR Biotech, Inc., incorporated by reference to Exhibit 21 to our Annual Report on Form 10-K for the year ended December 31, 2009, filed on March 26, 2010.
|
|
|
23.1*
|
Consent of BKR Yarel + Partners
|
|
|
31.1*
|
Certification of Chief Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
|
|
31.2*
|
Certification of Chief Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
|
|
32.1*
|
Certifications of Chief Executive Officer required by Section 906 of the Sarbanes-Oxley Act of 2002
|
|
|
32.2*
|
Certifications of Chief Financial Officer required by Section 906 of the Sarbanes-Oxley Act of 2002
|
___________________
* Filed herewith.
+ Compensation plan or arrangement or management contract.
40
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on our behalf by the undersigned, thereunto duly authorized.
|
PROLOR Biotech, Inc.
|
|
|
By:
|
/s/ Abraham Havron
|
|
Abraham Havron
|
|
|
Chief Executive Officer
|
|
Date: March 15, 2011
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ Abraham Havron
|
Chief Executive Officer
|
March 15, 2011
|
||
|
Abraham Havron
|
(Principal Executive Officer) and Director
|
|||
|
/s/ Steve Schaeffer
|
Chief Financial Officer
|
March 15, 2011
|
||
|
Steve Schaeffer
|
(Principal Financial Officer and Principal Accounting
|
|||
|
Officer)
|
||||
|
|
Director
|
|
||
|
Fuad Fares
|
||||
|
/s/ Phillip Frost
|
Director
|
March 15, 2011
|
||
|
Phillip Frost
|
||||
|
|
Director
|
|
||
|
Marian Gorecki
|
||||
|
|
Director
|
|
||
|
Jane Hsiao
|
||||
|
/s/ Shai Novik
|
President and Director
|
March 15, 2011
|
||
|
Shai Novik
|
||||
|
/s/ Steven Rubin
|
Director
|
March 15, 2011
|
||
|
Steven Rubin
|
||||
|
/s/ Adam Stern
|
Director
|
March 15, 2011
|
||
|
Adam Stern
|
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A development stage company)
CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2010 AND 2009
AND THE THREE YEARS ENDED DECEMBER 31, 2010
IN U.S. DOLLARS
F-1
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A development stage company)
CONSOLIDATED FINANCIAL STATEMENTS
AS OF DECEMBER 31, 2010 AND 2009
AND THE THREE YEARS ENDED DECEMBER 31, 2010
IN U.S. DOLLARS
INDEX
|
Page
|
||
|
Report of Independent Registered Public Accounting Firm
|
F-3
|
|
|
Consolidated Balance Sheets
|
F-5
|
|
|
Consolidated Statements of Operations
|
F-6
|
|
|
Statements of Stockholders’ Equity
|
F-7
|
|
|
Consolidated Statements of Cash Flows
|
F-11
|
|
|
Notes to Consolidated Financial Statements
|
|
F-13
|
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and stockholders of
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A Development Stage Company)
We have audited the accompanying consolidated balance sheets of Prolor Biotech, Inc. (a development stage company) (“the Company”) and its subsidiaries as of December 31, 2010 and 2009, and the related consolidated statements of operations, changes in stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2010, and for the period from May 31, 2005 (inception date) through December 31, 2010. We also have audited the Company’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for
maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on these financial statements and an opinion on the company’s internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial
reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in
accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the consolidated financial statements.

F-3
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of the Company and its subsidiaries as of December 31, 2010 and 2009, and the consolidated results of their operations, stockholders’ equity and cash flows for the years ended December 31, 2010, 2009, and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations
of the Treadway Commission (COSO).
|
/s/ Yarel + Partners
|
|
|
Yarel + Partners
|
|
|
Certified Public Accountants
|
|
Tel-Aviv, Israel
|
|
|
March 9, 2011
|
F-4
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED BALANCE SHEETS
U.S. dollars
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
Current Assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 24,474,458 | $ | 3,521,866 | ||||
|
Short term deposits
|
1,439,269 | - | ||||||
|
Accounts receivable and prepaid expenses
|
642,392 | 85,280 | ||||||
|
Restricted cash
|
102,932 | 91,730 | ||||||
|
Total Current Assets
|
26,659,051 | 3,698,876 | ||||||
|
Long Term Assets:
|
||||||||
|
Property and equipment, net
|
350,284 | 284,315 | ||||||
|
Assets held for employees’ severance payments
|
193,346 | 124,324 | ||||||
|
Long term deposits
|
2,320 | 1,906 | ||||||
|
Total Long Term Assets
|
545,950 | 410,545 | ||||||
|
Total Assets
|
$ | 27,205,001 | $ | 4,109,421 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current Liabilities:
|
||||||||
|
Trade payables
|
$ | 429,063 | $ | 131,924 | ||||
|
Related parties payable
|
207,306 | 203,583 | ||||||
|
Accrued expenses and other liabilities
|
1,353,235 | 504,612 | ||||||
|
Total Current Liabilities
|
1,989,604 | 840,119 | ||||||
|
Liability in Respect of Employees Severance Payments
|
220,838 | 140,237 | ||||||
|
Commitments and Contingent Liabilities
|
||||||||
|
Stockholders’ Equity:
|
||||||||
|
Stock capital -
|
||||||||
|
Preferred stock of $ 0.00001 par value per share 10,000,000 shares of preferred stock authorized; 0 and 1,800,000 shares issued and outstanding on December 31, 2010 and 2009, respectively
|
- | 18 | ||||||
|
Common shares of $ 0.00001 par value per share 300,000,000 shares of common stock authorized; 54,116,628 and 35,569,017 shares issued and outstanding on December 31, 2010 and 2009, respectively
|
541 | 355 | ||||||
|
Additional paid-in capital
|
59,577,974 | 30,153,517 | ||||||
|
(Deficit) accumulated during the development stage
|
(34,583,956 | ) | (27,024,825 | ) | ||||
|
Total Stockholders’ Equity
|
24,994,559 | 3,129,065 | ||||||
|
Total Liabilities and Stockholders’ Equity
|
$ | 27,205,001 | $ | 4,109,421 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
F-5
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENTS OF OPERATIONS
U.S. dollars
|
Period from
|
||||||||||||||||
|
May 31, 2005
|
||||||||||||||||
|
(date of
inception)
|
||||||||||||||||
|
Year ended December 31,
|
to December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Revenues
|
$ | - | $ | - | $ | - | $ | - | ||||||||
|
Operating expenses:
|
||||||||||||||||
|
In-process research and development write-off
|
- | - | (3,222,831 | ) | ||||||||||||
|
Research and development, net
|
(5,219,783 | ) | (5,555,323 | ) | (4,781,089 | ) | (18,320,541 | ) | ||||||||
|
General and administrative
|
(2,457,043 | ) | (1,901,952 | ) | (2,410,015 | ) | (13,676,569 | ) | ||||||||
|
Operating (loss)
|
(7,676,826 | ) | (7,457,275 | ) | (7,191,104 | ) | (35,219,941 | ) | ||||||||
|
Financial income
|
117,695 | 35,559 | 438,349 | 994,941 | ||||||||||||
|
Financial (expenses)
|
(- | ) | (63,002 | ) | (280,781 | ) | (358,956 | ) | ||||||||
|
Net (loss)
|
$ | (7,559,131 | ) | $ | (7,484,718 | ) | $ | (7,033,536 | ) | $ | (34,583,956 | ) | ||||
|
(Loss) per share (basic & diluted)
|
$ | (0.19 | ) | $ | (0.21 | ) | $ | (0.20 | ) | $ | (1.21 | ) | ||||
|
Weighted average number of shares outstanding
|
40,030,008 | 35,549,083 | 35,530,378 | 28,699,283 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-6
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF STOCKHOLDERS’ EQUITY
FOR THE PERIOD MAY 31, 2005 (INCEPTION) TO DECEMBER 31, 2010
U.S. dollars
|
Preferred Stock
|
Common Stock
|
Additional
|
Deferred
|
(Deficit)
accumulated
during the
development
|
Total
stockholders’
|
|||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
paid-in capital
|
compensation
|
stage
|
equity
|
|||||||||||||||||||||||||
|
Balance as of May 31, 2005 (date of inception)
|
- | $ | - | $ | - | $ | - | $ | - | $ | - | $ | - | |||||||||||||||||||
|
Issuance of common stock
|
- | - | 3,506,527 | 35 | 2,896,589 | - | - | 2,896,624 | ||||||||||||||||||||||||
|
Issuance of common stock and options in conjunction with the acquisition of ModigeneTech Ltd.
|
- | - | 3,788,632 | 38 | 2,628,528 | - | - | 2,628,566 | ||||||||||||||||||||||||
|
Contributed capital
|
- | - | 5,704,668 | 57 | - | - | - | 57 | ||||||||||||||||||||||||
|
Stock-based compensation
|
- | - | - | - | 3,514,369 | - | - | 3,514,369 | ||||||||||||||||||||||||
|
Deferred compensation on restricted shares to non-employees
|
- | - | 588,725 | 6 | 362,591 | ( 347,004 | ) | - | 15,593 | |||||||||||||||||||||||
|
Stock-based compensation related to options granted to non employees
|
- | - | - | - | 76,885 | - | - | 76,885 | ||||||||||||||||||||||||
|
Net (loss)
|
- | - | - | - | - | - | (6,977,419 | ) | (6,977,419 | ) | ||||||||||||||||||||||
|
Balance as of December 31, 2005
|
- | - | 13,588,552 | 136 | 9,478,962 | ( 347,004 | ) | (6,977,419 | ) | 2,154,675 | ||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-7
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF STOCKHOLDERS’ EQUITY
FOR THE PERIOD MAY 31, 2005 (INCEPTION) TO DECEMBER 31, 2010
U.S. dollars
|
Preferred Stock
|
Common Stock
|
Additional
|
Deferred
|
(Deficit)
accumulated
during the
development
|
Total
stockholders’
|
|||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
paid-in capital
|
compensation
|
stage
|
equity
|
|||||||||||||||||||||||||
|
Amortization of deferred compensation on restricted shares of common stock to non employees
|
- | - | - | - | - | 347,004 | - | 347,004 | ||||||||||||||||||||||||
|
Cumulative effect of first time adoption of the fair value based method for stock-based compensation to employees
|
- | - | - | - | 3,415 | - | - | 3,415 | ||||||||||||||||||||||||
|
Stock-based compensation on options
|
- | - | - | - | 259,620 | - | - | 259,620 | ||||||||||||||||||||||||
|
Net (loss)
|
- | - | - | - | - | - | (2,215,949 | ) | (2,215,949 | ) | ||||||||||||||||||||||
|
Balance as of December 31, 2006
|
- | - | 13,588,552 | 136 | 9,741,997 | - | (9,193,368 | ) | 548,765 | |||||||||||||||||||||||
|
Issuance of common stock in reverse acquisition
|
- | - | 7,333,328 | 73 | (73 | ) | - | - | - | |||||||||||||||||||||||
|
Issuance of common stock and options in private placement
|
- | - | 14,200,005 | 142 | 13,414,991 | - | - | 13,415,133 | ||||||||||||||||||||||||
|
Options exercised
|
- | - | 313,370 | 3 | 136 | - | - | 139 | ||||||||||||||||||||||||
|
Stock-based compensation on options
|
- | - | - | - | 1,211,536 | - | - | 1,211,536 | ||||||||||||||||||||||||
|
Net (loss)
|
- | - | - | - | - | - | (3,313,203 | ) | (3,313,203 | ) | ||||||||||||||||||||||
|
Balance as of December 31, 2007
|
- | - | 35,435,255 | 354 | 24,368,587 | - | (12,506,571 | ) | 11,862,370 | |||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-8
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF STOCKHOLDERS’ EQUITY (continued)
FOR THE PERIOD MAY 31, 2005 (INCEPTION) TO DECEMBER 31, 2010
U.S. dollars
|
Preferred Stock
|
Common Stock
|
Additional
|
Deferred
|
(Deficit)
accumulated
during the
development
|
Total
stockholders’
|
|||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
paid-in capital
|
compensation
|
stage
|
equity
|
|||||||||||||||||||||||||
|
Issuance of preferred stock
|
800,000 | 8 | - | - | 1,999,992 | - | - | 2,000,000 | ||||||||||||||||||||||||
|
Options exercised
|
- | - | 113,762 | 1 | (1 | ) | - | - | - | |||||||||||||||||||||||
|
Stock-based compensation on options
|
- | - | - | - | 1,039,028 | - | - | 1,039,028 | ||||||||||||||||||||||||
|
Net (loss)
|
- | - | - | - | - | - | (7,033,536 | ) | (7,033,536 | ) | ||||||||||||||||||||||
|
Balance as of December 31, 2008
|
800,000 | 8 | 35,549,017 | 355 | 27,407,606 | - | (19,540,107 | ) | 7,867,862 | |||||||||||||||||||||||
|
Issuance of preferred stock
|
1,000,000 | 10 | - | - | 1,999,990 | - | - | 2,000,000 | ||||||||||||||||||||||||
|
Options exercised
|
- | - | 20,000 | - | 17,580 | - | - | 17,580 | ||||||||||||||||||||||||
|
Stock-based compensation on options
|
- | - | - | - | 728,341 | 728,341 | ||||||||||||||||||||||||||
|
Net (loss)
|
- | - | - | - | - | - | (7,484,718 | ) | (7,484,718 | ) | ||||||||||||||||||||||
|
Balance as of December 31, 2009
|
1,800,000 | 18 | 35,569,017 | 355 | 30,153,517 | - | (27,024,825 | ) | 3,129,065 | |||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-9
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
STATEMENTS OF STOCKHOLDERS’ EQUITY (continued)
FOR THE PERIOD MAY 31, 2005 (INCEPTION) TO DECEMBER 31, 2010
U.S. dollars
|
Preferred Stock
|
Common Stock
|
Additional
|
Deferred |
(Deficit)
accumulated
during the
development
|
Total
stockholders’
|
|||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
paid-in capital
|
compensation |
stage
|
equity
|
|||||||||||||||||||||||
|
Issuance of common stock
|
10,827,420 | 109 | 26,876,575 | 26,876,684 | ||||||||||||||||||||||||||
|
Conversion of Preferred stock
|
(1,800,000 | ) | (18 | ) | 6,167,780 | 62 | (44 | ) | - | - | ||||||||||||||||||||
|
Options exercised
|
594,625 | 5 | 868,678 | 868,683 | ||||||||||||||||||||||||||
|
Warrants exercised
|
957,786 | 10 | 795,823 | 795,833 | ||||||||||||||||||||||||||
|
Stock-based compensation on options
|
883,425 | 883,425 | ||||||||||||||||||||||||||||
|
Net (loss)
|
(7,559,131 | ) | (7,559,131 | ) | ||||||||||||||||||||||||||
|
Balance as of December 31, 2010
|
- | $ | - | 54,116,628 | $ | 541 | $ | 59,577,974 | $ |
-
|
$ | (34,583,956 | ) | $ | 24,994,559 | |||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-10
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENTS OF CASH FLOWS
U.S. dollars
|
Period from
|
||||||||||||||||
|
May 31, 2005
|
||||||||||||||||
|
(date of inception)
|
||||||||||||||||
|
Year ended December 31,
|
to December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Cash flows from operating activities
|
||||||||||||||||
|
Net (loss)
|
$ | (7,559,131 | ) | $ | (7,484,718 | ) | $ | (7,033,536 | ) | $ | (34,583,956 | ) | ||||
|
Adjustments to reconcile net (loss) to net cash (used in)
|
||||||||||||||||
|
operating activities:
|
||||||||||||||||
|
Depreciation
|
86,960 | 78,509 | 64,700 | 285,902 | ||||||||||||
|
In-process research and development write-off
|
- | - | - | 3,222,831 | ||||||||||||
|
Stock-based compensation
|
883,425 | 728,341 | 1,039,028 | 8,079,210 | ||||||||||||
|
Liability in respect of employees severance payments
|
80,601 | 49,505 | 48,180 | 220,838 | ||||||||||||
|
Decrease (increase) in accounts receivable and prepaid expenses
|
(557,112 | ) | 327,235 | 93,629 | (642,115 | ) | ||||||||||
|
Increase (Decrease) in trade payables
|
297,139 | (24,115 | ) | 13,577 | 418,959 | |||||||||||
|
Increase in related parties payable
|
3,723 | 152,209 | 32,009 | 207,306 | ||||||||||||
|
Long term deposit exchange rate differences
|
(22 | ) | (22 | ) | - | (44 | ) | |||||||||
|
Increase in accrued expenses and other liabilities
|
848,623 | 315,952 | 21,442 | 1,232,219 | ||||||||||||
|
Net cash (used in) operating activities
|
(5,915,794 | ) | (5,857,104 | ) | (5,720,971 | ) | (21,558,850 | ) | ||||||||
|
Cash flows from investing activities
|
||||||||||||||||
|
Purchase of property and equipment
|
(152,929 | ) | (52,651 | ) | (199,445 | ) | (621,830 | ) | ||||||||
|
Payment for the acquisition of ModigeneTech Ltd.
|
- | - | - | (474,837 | ) | |||||||||||
|
Assets held for employees’ severance payments
|
(69,022 | ) | (50,549 | ) | (40,090 | ) | (193,346 | ) | ||||||||
|
Long term deposit
|
(392 | ) | 10 | 1,057 | (2,276 | ) | ||||||||||
|
Short term deposit
|
(1,439,269 | ) | - | - | (1,439,269 | ) | ||||||||||
|
Restricted cash
|
(11,202 | ) | (652 | ) | (29,240 | ) | (102,932 | ) | ||||||||
|
Net cash (used in) investing activities
|
(1,672,814 | ) | (103,842 | ) | (267,718 | ) | (2,834,490 | ) | ||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-11
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENTS OF CASH FLOWS (CONTINUED)
U.S. dollars in thousands
|
Period from
|
||||||||||||||||
|
May 31, 2005
|
||||||||||||||||
|
(date of inception)
|
||||||||||||||||
|
Year ended December 31,
|
to December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Cash flows from financing activities
|
||||||||||||||||
|
Short term bank credit
|
- | - | (1,886 | ) | (2,841 | ) | ||||||||||
|
Proceeds from loans
|
- | - | - | (173,000 | ) | |||||||||||
|
Principal payment of loans
|
- | - | - | 173,000 | ||||||||||||
|
Proceeds from exercise of options
|
868,683 | 17,580 | - | 886,388 | ||||||||||||
|
Proceeds from exercise of warrants
|
795,833 | - | - | 795,833 | ||||||||||||
|
Proceeds from issuance of shares
|
26,876,684 | 2,000,000 | 2,000,000 | 47,188,418 | ||||||||||||
|
Net cash provided by financing activities
|
28,541,200 | 2,017,580 | 1,998,114 | 48,867,798 | ||||||||||||
|
Increase (decrease) in cash and cash equivalents
|
20,952,592 | (3,943,366 | ) | (3,990,575 | ) | 24,474,458 | ||||||||||
|
Cash and cash equivalents at the beginning of the period
|
3,521,866 | 7,465,232 | 11,455,807 | - | ||||||||||||
|
Cash and cash equivalents at the end of the period
|
$ | 24,474,458 | $ | 3,521,866 | $ | 7,465,232 | $ | 24,474,458 | ||||||||
|
Non cash transactions:
|
||||||||||||||||
|
Cashless exercise of 917,421 outstanding stock warrants to 625,797 shares of common stock
|
$ | 6 | $ | - | $ | - | $ | - | ||||||||
|
Conversion of preferred stock to common stock
|
$ | 18 | $ | - | $ | - | $ | 18 | ||||||||
|
Employee options exercised to shares of common stock
|
$ | - | $ | - | $ | 1 | $ | 140 | ||||||||
|
Issuance of common stock in reverse acquisition
|
$ | - | $ | - | $ | - | $ | 73 | ||||||||
|
Additional information:
|
||||||||||||||||
|
Cash paid for income taxes
|
$ | - | $ | - | $ | - | $ | - | ||||||||
|
Cash paid for interest expense
|
$ | - | $ | 57,755 | $ | 280,796 | $ | 353,736 | ||||||||
|
Payment for the acquisition of ModigeneTech Ltd.:
|
||||||||||||||||
|
Issuance expenses
|
$ | - | $ | - | $ | - | $ | (356,979 | ) | |||||||
|
Loan granted by the Company to ModigeneTech Ltd.
|
- | - | - | (497,575 | ) | |||||||||||
|
Cash at date of acquisition in ModigeneTech Ltd.
|
- | - | - | 379,717 | ||||||||||||
| $ | - | $ | - | $ | - | $ | (474,837 | ) | ||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-12
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 1:-
|
GENERAL
|
|
|
a.
|
Prolor Biotech, Inc. (the “Company”) was incorporated on August 22, 2003 under the laws of the State of Nevada. The Company is a development stage biopharmaceutical company utilizing exclusive license from Washington University to patented technology in the development of longer-acting versions of already-approved therapeutic proteins, through its Israeli subsidiary, ModigeneTech Ltd. (“ModigeneTech”).
|
|
|
b.
|
The Company devotes substantially all of its efforts toward research and development activities. In the course of such activities, the Company has sustained operating losses and expects such losses to continue for the foreseeable future. The Company has not generated any revenues or product sales and has not achieved profitable operations or positive cash flow from operations. The Company’s deficit accumulated during the development stage aggregated $34,583,956 through December 31, 2010. There is no assurance that profitable operations, if ever achieved, could be sustained on a continuing basis.
The Company is entitled to receive R&D grants from the Israeli government on approved projects during the year 2011. The Company believes that its current cash sources with the anticipated R&D grants will enable the continuance of the Company’s activities for at least a year with no need for additional fundraising.
|
|
|
c.
|
On May 9, 2007, Modigene Inc., a Delaware corporation, Modigene Acquisition Corp., a wholly-owned subsidiary of the Company (the “Acquisition Subsidiary”), and the Company entered into a merger agreement (the “Merger Agreement”). Pursuant to the Merger Agreement, the Acquisition Subsidiary merged (the “Merger”) with and into Modigene Inc. with Modigene Inc. remaining as the surviving entity and a wholly-owned subsidiary of the Company. The Merger was accounted for as a recapitalization. The Company has continued the business operations of Modigene Inc. as a publicly-traded company under the name Prolor Biotech, Inc. In the Merger, the stockholders of Modigene Inc. received a total of 13,588,552 shares of the Company’s common stock, par value $0.00001 per share (“Common Stock”), in exchange for all of their shares of common stock of
Modigene Inc. Pursuant to the Merger Agreement, the Company became the holding company of Modigene Inc. and ModigeneTech. Contemporaneously with the closing of the Merger, the Company split off its wholly-owned subsidiary, Liaison Design Group, LLC., through the sale of all of the membership interests of the subsidiary.
|
F-13
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 1:-
|
GENERAL (continued)
|
|
|
d.
|
In December 2005, Modigene Inc. acquired all of the outstanding shares of ModigeneTech, an Israeli-based corporation, in consideration for shares of common stock of Modigene Inc. The fair value of the common stock issued and the options granted for the acquisition was $2,628,566.
In connection with the transaction, Modigene Inc. also issued shares of common stock, valued at $3,514,426, to Modigene Inc.’s founders for their services as the agents in the transaction.
|
The acquisition was accounted for as an acquisition of a group of assets that does not constitute a business and no goodwill was recognized.
The know-how purchased in the amount of $3,222,831 has not yet reached technological feasibility and had no alternative future use other than the technological indications for which it was in development. Accordingly, the entire amount representing the know-how was recorded as in-process research and development and accordingly was immediately expensed in the consolidated statement of operations on the acquisition date. Following the acquisition of ModigeneTech, ModigeneTech became a wholly-owned subsidiary of Modigene Inc. The financial statements of ModigeneTech were consolidated with the accounts of Modigene Inc, commencing December 14, 2005.
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES
|
The financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”). The significant accounting policies followed in the preparation of the financial statements, on a consistent basis are:
|
|
a.
|
Use of estimates:
|
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. While management believes that such estimates are fair when considered in conjunction with the consolidated financial position and results of operations taken as a whole, actual results could differ from those estimates and such differences may be material to the financial statements.
|
|
b.
|
Patents:
|
As a result of the Company’s research and development efforts, the Company has obtained, or is applying for, a number of patents to protect proprietary technology and inventions. All costs associated with patents for product candidates under development are expensed as incurred. To date, the Company has no capitalized patent costs.
F-14
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
|
c.
|
Financial statements in U.S. dollars:
|
|
|
The functional currency of the Company is the U.S dollar, as the U.S. dollar is the primary currency of the economic environment in which the Company has operated and expects to continue to operate in the foreseeable future. The majority of ModigeneTech’s operations are currently conducted in Israel. Most of the Israeli expenses are currently determined and paid in U.S. dollars. Financing and investing activities including loans and equity transactions are made in U.S. dollars.
|
|
|
Accordingly, the functional and reporting currency of the Company is the dollar. Monetary accounts maintained in currencies other than the dollar are remeasured into U.S. dollars. All transaction gains and losses from the remeasurement of monetary balance sheet items are reflected in the statements of operations as financial income or expenses, as appropriate.
|
|
|
d.
|
Comprehensive Income (Loss):
|
|
|
Accounting guidance requires financial statements to include the reporting of comprehensive income (loss), which includes net income (loss) and certain transactions that have generally been reported in the statement of stockholders’ equity. The Company’s comprehensive income (loss) consists of net income (loss).
|
|
|
e.
|
Segment Reporting:
|
|
|
A business segment is a distinguishable component of an enterprise that is engaged in providing an individual product or service or a group of related products or services and that is subject to risks and returns that are different from those of other business segments. Management believes that the Company meets the criteria for aggregating its operating segments into a single reporting segment.
|
|
|
f.
|
Principles of consolidation:
|
|
|
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries, Modigene Inc. and ModigeneTech Ltd.Intercompany transactions and balances, have been eliminated upon consolidation.
|
|
|
g.
|
Cash equivalents:
|
|
|
For purposes of reporting within the statement of cash flows, the Company considers all cash on hand, cash accounts not subject to withdrawal restrictions or penalties, and all highly liquid debt instruments purchased with a maturity of three months or less to be cash and cash equivalents.
|
F-15
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
|
h.
|
Short-term bank deposits:
|
|
|
Bank deposits with maturities of more than three months and up to one year were included in short-term bank deposits. As of December 31, 2010 most of the bank deposits were held in Euro and bore interest at a weighted average annual interest rate of 1%. The deposits are presented at their cost, including accrued interest.
|
|
|
i.
|
Restricted cash:
|
|
|
Cash and cash items which are restricted as to withdrawal or usage. Restricted cash includes legally restricted deposits held as compensating balances against a rent agreement to assure future credit availability.
|
|
|
j.
|
Accounts receivable:
|
|
|
Accounts receivable are recorded at net realizable value consisting of the carrying amount less the allowance for uncollectible accounts. As of December 31, 2010 and 2009, the Company has not accrued an allowance for uncollectible accounts.
|
|
|
k.
|
Property and equipment:
|
|
|
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated by the straight-line method over the estimated useful lives of the assets. The annual depreciation rates are as follows:
|
|
%
|
|
|
Office furniture and equipment
|
6
|
|
Laboratory equipment
|
15
|
|
Computers and electronic equipment
|
33
|
|
Leasehold improvements
|
25
|
The Company reviews the carrying value of its long-lived assets, including intangible assets subject to amortization, for impairment whenever events and circumstances indicate that the carrying value of the assets may not be recoverable. Recoverability of these assets is measured by comparing the carrying value of the assets to the undiscounted cash flows estimated to be generated by those assets over their remaining economic life. If the undiscounted cash flows are not sufficient to recover the carrying value of the assets, the assets are considered impaired. The impairment loss is measured by comparing the fair value of the assets to their carrying value.
F-16
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
|
k.
|
Property and equipment: (continued)
|
|
|
Fair value is determined by either a quoted market price or a value determined by a discounted cash flow technique, whichever is more appropriate under the circumstances involved. No impairments were recognized from May 31, 2005 (inception date) through December 31, 2010.
|
|
|
l.
|
Accounting for stock-based compensation:
|
|
|
The Company’s stock-based compensation are recorded according to ASC 718-10, “Compensation - Stock Compensation”, which requires the measurement and recognition of compensation expense for all stock-based payment awards made to employees and directors, including employee stock options under the Company’s stock plans, based on estimated fair values.
|
ASC 718-10 requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in the Company’s consolidated statement of operations. The Company estimates the fair value of stock options granted using the Black-Scholes-Merton option-pricing model.
|
|
m.
|
Research and development costs and participations:
|
|
|
Research and development (“R&D”) costs are expensed as they are incurred and consist of salaries, stock-based compensation benefits and other personnel related costs, fees paid to consultants, clinical trials and related clinical manufacturing costs, license and milestone fees, and facilities and overhead costs.Participations from the Israeli government for development of approved projects are recognized as a reduction of expenses as the related costs are incurred.
|
|
|
n.
|
Assets held for employees’ severance payments:
|
|
|
Assets held for employees’ severance payments represent contributions to insurance policies that are recorded at their current redemption value.
|
|
|
o.
|
Liability in respect of employees severance payments:
|
|
|
Under Israeli law and labor agreements the Company is required to pay severance payments to each employee who was employed by the Company for over one year and has been terminated by the Company or resigned under certain specified circumstances. The Company’s liability for these severance payments is covered mainly by deposits with insurance companies in the name of the employee and/or by purchase of insurance policies.
|
F-17
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
|
o.
|
Liability in Respect of Employees Severance Payments: (continued)
|
|
|
The liability related to these severance payments is calculated on the basis of the latest salary of the employee multiplied by the number of years of employment as of the balance sheet date. The liability for employee severance payments included in the balance sheet represents the total amount due for such severance payment, while the assets held for severance benefits included in the balance sheet represents the Company’s contributions to insurance policies. The Company may make withdrawals from the funds only upon complying with the Israeli severance pay law or labor agreements.
|
|
|
According to agreements with key employees who are related parties of the Company, upon retirement, such employees will be entitled to receive a lump-sum payment; therefore, the Company does not accumulate severance pay for those employees, and the sum will be expensed at a time when the Company has to pay such payments according to the employment agreements. Severance (income) expenses for the years ended December 31, 2010, 2009 and 2008 and the period from May 31, 2005 (inception date) through December 31, 2010 amounted to $11,579, $(1,044), $8,090 and $27,492, respectively.
|
|
|
p.
|
Income taxes:
|
|
|
The Company accounts for income taxes in accordance with the provisions of ASC 740-10, Income Taxes. ASC 740-10 requires companies to recognize deferred tax assets and liabilities based on the differences between financial reporting and tax bases of assets and liabilities. These differences are measured using the enacted tax rates and laws that are expected to be in effect when the temporary differences are expected to reverse. A valuation allowance is established against net deferred tax assets, if based on the weighted available evidence, it is more likely than not that all or a portion of the deferred tax assets will not be realized.
|
|
|
q.
|
Concentrations of credit risk:
|
|
|
Financial instruments that potentially subject the Company and subsidiaries to concentrations of credit risk consist principally of cash and cash equivalents.
Cash and cash equivalents are invested in major banks in Israel and in the U.S. Such deposits in Israel and the U.S. are not insured. Management believes that the financial institutions that hold the Company’s investments are financially sound and, accordingly, minimal credit risk exists with respect to these investments.
The Company has no off-balance-sheet concentration of credit risk such as foreign exchange contracts or other foreign hedging arrangements.
|
F-18
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
|
r.
|
Fair value measurements:
|
|
|
As defined in ASC 820-10, Fair Value Measurements and Disclosures (“ASC 820-10”), fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. In order to increase consistency and comparability in fair value measurements, ASC 820-10 establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. Level 2: Other inputs that are observable, directly or indirectly, such as quoted prices for similar assets and liabilities or market corroborated
inputs.Level 3: Unobservable inputs are used when little or no market data is available, which requires the Company to develop its own assumptions about how market participants would value the assets or liabilities. The fair value hierarchy gives the lowest priority to Level 3 inputs.In determining fair value, the Company utilizes valuation techniques in its assessment that maximize the use of observable inputs and minimize the use of unobservable inputs.The following table presents the Company’s financial assets and liabilities that are carried at fair value, classified according to the three categories described above:
|
|
Fair Value Measurements at December 31, 2010
|
||||||||||||||||
|
Quoted Prices in
Active
|
Significant
Other
|
Significant
|
||||||||||||||
|
Markets for
Identical Assets
|
Observable
Inputs
|
Unobservable
Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Cash and cash equivalents
|
$ | 24,474,458 | $ | 24,474,458 | $ | - | $ | - | ||||||||
|
Short term deposits
|
1,439,269 | 1,439,269 | - | - | ||||||||||||
|
Restricted cash
|
102,932 | 102,932 | - | - | ||||||||||||
|
Total assets at fair value
|
$ | 26,016,659 | $ | 26,016,659 | $ | - | $ | - | ||||||||
|
Fair Value Measurements at December 31, 2009
|
||||||||||||||||
|
Quoted Prices in
Active
|
Significant
Other
|
Significant
|
||||||||||||||
|
Markets for
Identical Assets
|
Observable
Inputs
|
Unobservable
Inputs
|
||||||||||||||
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|||||||||||||
|
Cash and cash equivalents
|
$ | 3,521,866 | $ | 3,521,866 | $ | - | $ | - | ||||||||
|
Restricted cash
|
91,730 | 91,730 | - | - | ||||||||||||
|
Total assets at fair value
|
$ | 3,613,596 | $ | 3,613,596 | $ | - | $ | - | ||||||||
F-19
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
|
s.
|
Royalty-bearing grants:
|
Royalty-bearing grants from the Government of Israel for funding approved research and development projects are recognized at the time ModigeneTech is entitled to such grants, on the basis of the costs incurred and included as a reduction of research and development costs. Research and development grants received by the Company reduced research and development expenses by $1,383,806, $484,912, 746,013 and $3,383,597 for the years ended December 31, 2010, 2009 and 2008 and the period from May 31, 2005 (inception date) through December 31, 2010, respectively. Research and development grants receivable as of December 31, 2010 was $281,728.
|
|
t.
|
Loss per share:
|
Basic and diluted losses per share are presented in accordance with ASC 260-10 “Earnings per share”. Outstanding convertible preferred stock restricted stock, share options and warrants have been excluded from the calculation of the diluted loss per share because all such securities are antidilutive. The total weighted average number of ordinary shares related to outstanding convertible preferred stock, restricted stock, options and warrants excluded from the calculations of diluted loss per share were 16,931,505, 9,572,278, 7,620,546 and 7,552,859 for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010, respectively.
|
|
u.
|
Revenue recognition:
|
Revenue, if and when generated, will be recognized in accordance with ASC 605-10-S99, “Revenue Recognition”, when the significant risks and rewards of ownership have been transferred to the customer pursuant to applicable laws and regulations, including factors such as when there has been evidence of a sales arrangement, the performance has occurred, or service have been rendered, the price to the buyer is fixed or determinable, and collectability is reasonably assured.
|
|
v.
|
Impact of recently issued accounting standards:
|
In January 2010, the FASB issued Accounting Standards Update No. 2010-06, Fair Value Measurements and Disclosures (Topic 820): Improving Disclosures about Fair Value Measurements. The amendments in this update require new disclosures and clarifications of existing disclosures related to transfers in and out of Level 1 and Level 2 fair value measurements, further disaggregation of fair value measurement disclosures for each class of assets and liabilities and additional details of valuation techniques and inputs utilized. This update is consistent with the Company's current accounting application for fair value measurements and disclosures.
In April, 2010, the FASB issued Accounting ASU No. 2010-13, “Compensation-Stock Compensation (Topic 718)—Effect of Denominating the Exercise Price of a Share-Based Payment Award in the Currency of the Market in Which the Underlying Equity Security Trades—a consensus of the FASB
F-20
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 2:-
|
SIGNIFICANT ACCOUNTING POLICIES (continued)
|
|
v.
|
Impact of recently issued accounting standards: (continued)
|
Emerging Issues Task Force” (“ASU 2010-13”). ASU 2010-13 provides amendments to Topic 718 to further clarify that an employee share-based payment award with an exercise price denominated in the currency of a market in which a substantial portion of the entity’s equity securities trades should classify such an award as equity. The amendments in this update do not expand the recurring disclosures required by Topic 718. The amendments in this update are effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2010. The company adopted this standard in January, 2011 which did not have a material impact on our financial statements.
There were various other updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries. None of the updates are expected to a have a material impact on the Company's financial position, results of operations or cash flows.
|
NOTE 3:-
|
ACCOUNTS RECEIVABLE AND PREPAID EXPENSES
|
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Israeli government authorities
|
$ | 402,662 | $ | 46,054 | ||||
|
Prepaid expenses
|
232,539 | 38,474 | ||||||
|
Others
|
7,191 | 752 | ||||||
| $ | 642,392 | $ | 85,280 | |||||
|
NOTE 4:-
|
PROPERTY AND EQUIPMENT, NET
|
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Cost:
|
||||||||
|
Office furniture and equipment
|
$ | 25,039 | $ | 24,243 | ||||
|
Computers and electronic equipment
|
90,538 | 65,230 | ||||||
|
Laboratory equipment
|
458,616 | 348,652 | ||||||
|
Leasehold improvements
|
76,975 | 60,114 | ||||||
| $ | 651,168 | $ | 498,239 | |||||
|
Accumulated depreciation:
|
||||||||
|
Office furniture and equipment
|
$ | 4,503 | $ | 3,274 | ||||
|
Computers and electronic equipment
|
59,616 | 41,989 | ||||||
|
Laboratory equipment
|
191,897 | 135,783 | ||||||
|
Leasehold improvements
|
44,868 | 32,878 | ||||||
| 300,884 | 213,924 | |||||||
|
Depreciated cost
|
$ | 350,284 | $ | 284,315 | ||||
F-21
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
|
NOTE 4: -
|
PROPERTY AND EQUIPMENT, NET (continued)
|
Depreciation expenses for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (inception date) through December 31, 2010 were $86,960, $78,509, $64,700 and $285,902, respectively.
F-22
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 5:-
|
ACCRUED EXPENSES AND OTHER LIABILITIES
|
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Employees and payroll accruals
|
$ | 155,922 | $ | 112,293 | ||||
|
Accrued expenses
|
1,197,313 | 367,745 | ||||||
|
Government authorities
|
- | 24,574 | ||||||
| $ | 1,353,235 | $ | 504,612 | |||||
|
NOTE 6:-
|
COMMITMENTS AND CONTINGENT LIABILITIES
|
|
|
a.
|
ModigeneTech is committed to pay royalties to the Government of Israel with respect to the proceeds from sales of products developed in the framework of projects in which the Israeli Government paid a portion of the expenses. Under the terms of the funding received from the Israeli Office of the Chief Scientist (the “Chief Scientist”), royalty payments are computed on the sales proceeds from such products at the rate of 3%. The contingent liability to the Chief Scientist is limited to the amount of the grants received plus interest at the rate of LIBOR. As of December 31, 2010, no royalties had been paid or accrued. The Company is committed to the Chief Scientist to keep the know-how and production rights in the framework of the abovementioned projects under ModigeneTech’s possession.
|
|
|
b.
|
In February 2007, the Company entered into a license agreement with Washington University, amending a prior license agreement. Pursuant to the new License Agreement, Washington University granted the Company an exclusive license to certain patents necessary to the Company for its developments. Under the License Agreement, the Company has the right to sub-license the licensed patents. The License Agreement terminates in 2018 when the last of the patents licensed to the Company under the License Agreement expires, unless terminated earlier.
|
Under the License Agreement, the Company was required to pay an initial fee of $100,000 in installments over the 18 months following February 2007. In addition, the Company is required to pay annual license maintenance fees of $30,000 (payable until the first commercial sale); royalty fees of 1.5% to 5% from net revenues (with certain required minimum royalties after the first commercial sale of $10,000, $20,000 and $40,000 for the first, second, and third year and beyond, respectively), and sub-licensing fees of 7.5% to 20% on sub-licensing payments. Pursuant to the License Agreement, the Company will also be responsible for milestone payments of $15,000 for each molecule at investigational new drug application filing, $30,000 at the initiation of a Phase II clinical trial and $40,000 at
the initiation of a Phase III clinical trial.
F-23
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 6:-
|
COMMITMENTS AND CONTINGENT LIABILITIES (continued)
|
|
|
c.
|
On December 30, 2010, ModigeneTech Ltd. entered into a definitive license agreement with Yeda Research and Development Company Ltd., the technology transfer and commercial arm of the Weizmann Institute of Science ("Yeda"), for novel technology utilized in the development of long-acting therapeutic peptides and small molecules. The Company has been developing, under a limited option-to-license agreement from Yeda, a long-acting drug compound using this technology and the Company has exercised its option to license the technology. The definitive license agreement has an expanded scope and includes all therapeutic indications, with the exception of hemophilia and insulin. The license will remain in force in each of Israel and the United States with respect to each product until the later of: (1) the date of expiry of the last of the patents; or (2) the expiry of a continuous period of 15
years during which there shall not have been a commercial sale of any product in any of the countries as detailed in the agreement.
|
In consideration for the grant of the license, the Company has paid or will pay Yeda: (1) on the date of signature of the agreement $75,000; (2) an annual license fee until the date of expiry of the last of the patents, in the amount of $10,000 for the period until December 31, 2015, and $15,000 in respect of each annual period thereafter; (3) a royalty of 3.5% of net future sales by or on behalf of the Company or any sublicensees or any distributors, or a reduced % provided by certain conditions defined in the agreement; and (4) 30% of all sublicensing receipts, or a reduced percentage provided by the fulfillment of certain conditions defined in the agreement with a deduction of obligatory payments to the OCS and others as defined in the agreement.
Yeda is entitled to terminate the license upon written notice to the Company, if: (1) the Company fails to achieve any of the milestones by the dates set forth in the agreement; (2) in certain cases of lack of future sales after commencement of commercial sales; or (3) the Company contests the validity of any of the patents. If any such challenge is unsuccessful, the Company must pay to Yeda liquidated damages in the amounts of $8,000,000. Either Yeda or the Company may terminate the agreement and the license after the commitment of a material breach by the other party and in certain other instances as detailed in the agreement.
|
|
d.
|
Operating leases:
|
ModigeneTech rents its offices under a lease operating agreement. Aggregate minimum rental commitments, under non-cancelable leases, as of December 31, 2010, are as follows:
|
Year ended December 31,
|
||||
|
2011
|
$ | 126,930 | ||
|
2012
|
10,578 | |||
| $ | 137,508 | |||
Rent expenses for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (date of inception) to December 31, 2010 were $113,335, $107,129, $119,489 and $488,057, respectively.
F-24
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
NOTE 7:- CONSULTING AND EMPLOYMENT AGREEMENTS WITH RELATED PARTIES
|
|
a.
|
The Company entered into a consulting agreement with its Chief Executive Officer (“CEO”), pursuant to which he serves as the Company’s CEO on a part-time basis at an annual compensation rate of $250,000 and an annual cash bonus target of up to $60,000. The consulting agreement is extended yearly by mutual agreement of the parties. Either party may terminate the agreement on 30 days prior notice; however, if the Company terminates the agreement for any reason other than the CEO’s material breach, then the CEO will be entitled to a lump sum severance payment of $40,000.
|
In January, 2011, the Company and the CEO entered into an amendment to the consulting agreement, pursuant to which, effective January 1, 2011, the CEO’s annual base salary has been increased to $270,000.
|
|
b.
|
The Company entered into an employment agreement with its President. The term of the employment is automatically extended for one-year terms on each one-year anniversary of the agreement, unless either party gives written notice of an election not to renew the agreement. The President’s annual base salary under the agreement is $270,000. In addition, the President will be entitled to an annual cash bonus of up to $85,000 based on corporate and personal milestones, along with equity performance awards, each as determined by the compensation committee of the Board. If the President voluntarily terminates his employment (other than in connection with a change of control and certain other reasons), he will be entitled to payment of only his base salary through the date of termination. However, if the President terminates the agreement as the result of a material breach by the Company,
then he will be entitled to payment of his base salary over a 12-month period following the termination, plus the value of any accrued benefits. If the Company terminates the employment other than for cause (as defined in the agreement), or if the term expires and is not renewed by the Company, then the President will be entitled to receive an amount equal to his then-current base salary over the 12-month period plus the value of accrued benefits and a pro-rata portion of the current year’s performance bonus. If the President is terminated for cause, then he will be entitled to receive only any amounts that were due and owing to him at the time of such termination. If either (a) the President terminates his employment for good reason (as defined in the agreement) or (b) the Company terminates the employment within 12 months of a change in control (as defined in the agreement), then the President will be entitled to receive a lump-sum payment equal to the lesser of (i) his base
salary for 12 months and (ii) his base salary for the remainder of the term, and all unvested stock options will immediately vest and be exercisable. In January 2011, the Company and the President entered into an amendment to the employment agreement, pursuant to which, effective as of January 1, 2011; the President’s annual base salary has been increased to $290,000.
|
F-25
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 7:-
|
CONSULTING AND EMPLOYMENT AGREEMENTS WITH RELATED PARTIES (continued)
|
|
|
c.
|
The Company entered a consulting agreement with its Chief Scientific Officer (“CSO”), the consulting agreement is extended yearly by mutual agreement of the parties.. Under the agreement, the Company shall pay the CSO an annual consulting fee of $36,000 and milestone payments of up to $102,000 upon successful completion of the milestones, as determined by the agreement. As of December 31, 2010 no milestone payments had been paid or accrued. Upon the Company’s termination of this agreement, the Company shall pay the CSO a lump sum of $18,000.
|
|
|
d.
|
The Company entered into an employment agreement with the Chief Operating Officer ("COO") of the Company’s wholly-owned subsidiary, ModigeneTech, Ltd. The COO's employment term is automatically extended for one-year term on December 14 of every year unless either party gives written notice, no less than 90 days prior to the end of the then-current term, of an election not to renew the agreement. The COO’s current annual base salary is $125,000. In addition, he is entitled to an annual cash bonus of up to $50,000, based on corporate and personal milestones, along with equity performance awards, each as determined by the Board. In January 2011, the Company and the COO entered into an amendment to the employment agreement, pursuant to which, effective as of January 1, 2011; the COO’s annual base salary has been increased to $145,000.
|
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY
|
|
|
a.
|
The Company's shares are listed for trading on the NYSE Amex and on the Tel-Aviv Stock Exchange.
|
|
|
b.
|
Rights of Common Stock capital:
|
The holders of Common Stock are entitled to one vote per share on all matters submitted to a vote of the stockholders, including the election of directors. Subject to any preferential rights of any outstanding series of preferred stock created by the Board from time to time, the common stockholders will be entitled to such cash dividends as may be declared from time to time by the Board from funds available. Subject to any preferential rights of any outstanding series of preferred stock, upon liquidation, dissolution or winding up of the Company, the common stockholders will be entitled to receive pro rata all assets available for distribution to such holders.
F-26
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY (continued):
|
|
|
c.
|
On December 15, 2010, the Company entered into a securities purchase agreement with a private investor (the “Investor”), pursuant to which the Investor purchased an aggregate of 444,445 shares (the “Shares”) of common stock at a purchase price of NIS 22.5 per share. The total consideration amounted to $2,731,443 ($2,787,196 net of $55,753 issuing expenses). The Company issued the Shares in reliance upon the exemption from registration afforded by Regulation S promulgated under the United States Securities Act of 1933, as amended (the “Securities Act”). The Shares have not been registered under the Securities Act and are “restricted securities” as that term is defined by Rule 144 under the Securities Act.
|
|
|
d.
|
On March 11, 2010 and March 17, 2010, the Company entered into two substantially identical securities purchase agreements with certain private investors (the “Investors”), pursuant to which the Investors purchased an aggregate of 10,382,975 shares (the “Shares”) of Common Stock at a purchase price of $2.35 per Share. On March 17, 2010, the Company closed on the issuance of the Shares for aggregate consideration of $24,145,241 ($24,399,991 net of $254,750 issuing expenses).
|
The Company issued the Shares in reliance upon the exemption from registration provided by Section 4(2) of the Securities Act and Rule 506 of Regulation D promulgated thereunder. Each Investor represented to the Company that such person was an “accredited investor” as defined in Rule 501(a) under the Securities Act and that such Investor’s Shares were being acquired for investment purposes.
The Shares have not been registered under the Securities Act and are “restricted securities” as that term is defined by Rule 144 under the Securities Act. The Company has not undertaken to register the Shares, and no registration rights have been granted to the Investors in respect of the Shares. Additionally, each Investor entered into a lockup agreement in respect of the Shares, pursuant to which such Investor may not sell or otherwise transfer such Shares for a period of one year.
|
|
e.
|
On July 22, 2009, the Company entered into a securities purchase agreement (the “Purchase Agreement”) with a group of related parties of the Company (the “Investors”), pursuant to which the Investors purchased on July 23, 2009 1,000,000 shares (the “Shares”) of the Company’s 10% Series B Cumulative Convertible Preferred Stock, par value $0.00001 per share, at a purchase price of $2.00 per share, (two-time the average closing price of the Common Stock as reported on the OTCBB during the thirty day period immediately preceding July 22, 2009, rounded up to $1.00), for a total consideration of $2,000,000.
|
On May 20, 2010 all 1,000,000 issued and outstanding shares of Series B Preferred Stock were converted into 2,167,780 shares of common stock, par value $0.00001 per share for no additional consideration.
F-27
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY (continued)
|
|
|
f.
|
On March 25, 2008, the Company entered into a securities purchase agreement with a group of related parties (the “Investors”), pursuant to which it sold to the Investors an aggregate of 800,000 shares of Series A preferred stock, $0.00001 par value per share, at $2.50 per share, for an aggregate purchase price of $2,000,000. The Series A Preferred Stock was convertible during the period beginning March 1, 2009 through March 25, 2012, without payment of any additional consideration, into Common Stock based on a conversion ratio equal to one share of Common Stock per share of Series A Preferred Stock.
|
On May 21, 2010 all 800,000 issued and outstanding shares of Series A Preferred Stock were converted into 4,000,000 shares of common stock, par value $0.00001 per share for no additional consideration.
|
|
g.
|
On May 9, 2007, simultaneously with the closing of the Merger discussed in note 1c., the Company completed the first phase of a private placement (the “Offering”) of 6,418,814 units of its securities at a purchase price of $1.50 per unit, with each unit consisting of one share of Common Stock and a five year warrant to purchase one-quarter of one share of Common Stock for an exercise price of $2.50 per whole share.
|
The Company raised total cash consideration of $9,628,212 before expenses. Upon the completion of the first phase of the Offering the Company issued warrants to purchase up to an aggregate of 242,324 shares of Common Stock to broker/dealers who assisted in the Offering.
Contemporaneously with the closing of the Merger and the first phase of the Offering, the Company completed a sale (the “Private Sale”) of 5,377,660 shares of Common Stock, and warrants to purchase 333,333 shares of Common Stock, to strategic investors, for total consideration of $2,000,000.
The strategic investors were entitled to additional shares on a pro rata basis if additional units were sold in connection with the Offering. Pursuant to the terms of the Offering, the Company could sell additional units up to an aggregate of 8,666,672 units and $13,000,008 (including those sold in the initial closing of the Offering). On May 21, 2007, the Company completed a second phase of the offering and closed on the sale of an additional 2,247,858 units, for total cash proceeds of $3,371,766. Upon the completion of the second closing of the offering, the Company issued warrants to purchase up to an aggregate of 51,885 shares of Common Stock to the broker/dealers who assisted in the Offering and additional 155,673 shares of Common Stock (for no additional consideration) to the strategic
investors. Issuance expenses paid in cash in the amount of $1,584,878 were recorded as a reduction of additional paid in capital.
F-28
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY (continued)
|
|
|
h.
|
Stock warrants:
|
A summary of the warrants granted is as follows:
|
December 31, 2009
|
||||||||
|
Number
|
Weighted Average
|
|||||||
|
of warrants
|
Exercise Price
|
|||||||
|
Outstanding and exercisable at the beginning of the period
|
3,558,924 | $ | 2.181 | |||||
|
Expired
|
- | $ | - | |||||
|
Exercised
|
- | $ | - | |||||
|
Outstanding and exercisable at the end of the period
|
3,558,924 | $ | 2.181 | |||||
|
December 31, 2010
|
||||||||
|
Number
|
Weighted Average
|
|||||||
|
of warrants
|
Exercise Price
|
|||||||
|
Outstanding and exercisable at the beginning of the period
|
3,558,924 | $ | 2.181 | |||||
|
Exercised
|
(1,062,368 | ) | $ | 2.500 | ||||
|
Exercised
|
(196,325 | ) | $ | 0.880 | ||||
|
Outstanding and exercisable at the end of the period
|
2,300,231 | $ | 2.144 | |||||
Proceeds from exercise of 1,258,693 warrants into 957,786 shares in the year ended December 31, 2010 were $795,833.
Total aggregate intrinsic value of warrants outstanding as of December 31, 2010 was $9,949,985.
The warrants provide for the purchase of shares of Common Stock. At the option of the holder, the warrants may be exercised by cash payment of the exercise price or by “cashless exercise.” A “cashless exercise” means that in lieu of paying the aggregate purchase price for the shares being purchased upon exercise of the warrants in cash, the holder will forfeit a number of shares underlying the warrants with a “fair market value” equal to such aggregate exercise price. The Company will not receive additional proceeds to the extent that warrants are exercised by cashless exercise.
The exercise price and number of shares of Common Stock issuable on exercise of the warrants may be adjusted in certain circumstances including in the event of a stock dividend, or recapitalization, reorganization, merger or consolidation.
F-29
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY (continued)
|
|
|
i.
|
Stock options:
|
|
|
1.
|
As of December 31, 2010 the Company has two stock option plans under which, outstanding stock options to purchase 1,252,119 shares were granted under the Company’s 2005 Stock Incentive Plan (the “2005 Plan”), and outstanding options to purchase 3,882,227 shares were granted under the Company’s 2007 Equity Incentive Plan (the “2007 Plan”).
|
The Company has issued the maximum number of shares authorized under the 2005 Plan.
On May 22, 2009, the Company approved an amendment to the 2007 Plan, which increased the number of shares of common stock authorized for issuance under the 2007 Plan from 3,000,000 shares to 6,000,000 shares, of which 4,005,819 stock options have been awarded
The Company accounts for stock-based compensation using the fair value recognition provisions of ASC No. 718 “Compensation – stock compensation”.
The fair value of the stock options is estimated based upon grant date fair value using the Black-Scholes option-pricing model with the following weighted average assumptions used:
|
Options granted under 2005 Plan
|
||||||||||||
|
Granted 2005
|
Granted 2006
|
Granted 2009
|
||||||||||
|
annual dividends of
|
$ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||
|
expected volatility of
|
85 | % | 85 | % | 117 | % | ||||||
|
risk-free interest rate of
|
4.41 | % | 4.63 | % | 0.01 | % | ||||||
|
expected average options expiration
|
8.05 | 7.90 | 7.00 | |||||||||
|
Options granted under 2007 Plan
|
||||||||||||||||
|
Granted
2007
|
Granted
2008
|
Granted
2009
|
Granted
2010
|
|||||||||||||
|
annual dividends of
|
$ | 0.00 | $ | 0.00 | $ | 0.00 | $ | 0.00 | ||||||||
|
expected volatility of
|
85 | % | 79 | % | 117 | % | 85 | % | ||||||||
|
risk-free interest rate of
|
4.67 | % | 2.90 | % | 0.01 | % | 0.3 | % | ||||||||
|
expected average options expiration
|
9.19 | 9.94 | 7.00 | 7.00 | ||||||||||||
F-30
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY (continued)
|
|
i.
|
Stock options: (continued)
|
2. A summary of the stock options granted under the 2005 and 2007 Plans is as follows:
|
December 31, 2009
|
||||||||
|
Number
of Options
|
Weighted average
Exercise Price
|
|||||||
|
Outstanding at the beginning of the year
|
4,432,292 | $ | 1.14 | |||||
|
Forfeited
|
(2,353 | ) | $ | 1.49 | ||||
|
Exercised
|
(20,000 | ) | $ | 0.88 | ||||
|
Granted
|
375,500 | $ | 0.65 | |||||
|
Outstanding at the end of the year
|
4,785,439 | $ | 1.11 | |||||
|
Options exercisable
|
2,701,397 | $ | 1.19 | |||||
|
December 31, 2010
|
||||||||
|
Number
of Options
|
Weighted average
Exercise Price
|
|||||||
|
Outstanding at the beginning of the year
|
4,785,439 | $ | 1.11 | |||||
|
Forfeited
|
(106,468 | ) | $ | 2.24 | ||||
|
Exercised
|
(594,625 | ) | $ | 1.46 | ||||
|
Granted
|
500,000 | $ | 2.40 | |||||
|
Granted
|
500,000 | $ | 6.47 | |||||
|
Granted
|
50,000 | $ | 2.35 | |||||
|
Outstanding at the end of the year
|
5,134,346 | $ | 1.80 | |||||
|
Options exercisable
|
2,870,559 | $ | 1.14 | |||||
F-31
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 8:-
|
STOCKHOLDERS’ EQUITY (continued)
|
|
|
i.
|
Stock options: (continued)
|
|
|
3.
|
The options outstanding as of December 31, 2010, have been separated into exercise prices, as follows:
|
|
Exercise
|
Remaining Weighted
average contractual life
|
|||||||||||||
|
Price
|
Options Outstanding
|
(years)
|
Options Exercisable
|
|||||||||||
| $ | 0.650 | 375,500 | 8.10 | 182,083 | ||||||||||
| $ | 0.879 | 983,264 | 5.27 | 983,264 | ||||||||||
| $ | 0.900 | 1,937,239 | 7.17 | 968,620 | ||||||||||
| $ | 0.930 | 25,000 | 7.18 | 16,667 | ||||||||||
| $ | 1.318 | 93,855 | 5.51 | 93,855 | ||||||||||
| $ | 1.500 | 121,169 | 7.32 | 77,251 | ||||||||||
| $ | 2.000 | 475,000 | 6.36 | 425,000 | ||||||||||
| $ | 2.500 | 123,819 | 2.44 | 123,819 | ||||||||||
| $ | 2.350 | 50,000 | 9.02 | - | ||||||||||
| $ | 2.400 | 500,000 | 9.04 | - | ||||||||||
| $ | 6.470 | 500,000 | 10.00 | - | ||||||||||
| 5,134,346 | 5.20 | 2,870,559 | ||||||||||||
|
|
4.
|
Weighted average fair values of outstanding options at date of grant and total aggregate intrinsic value of outstanding options are as follows:
|
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Weighted average fair value on date of grant
|
$ | 1.19 | $ | 0.71 | ||||
|
Total aggregate intrinsic value
|
24,326,863 | $ | 6,556,831 | |||||
|
|
5.
|
Stock-based compensation expenses for the years ended December 31, 2010, 2009 and 2008 and for the period from May 31, 2005 (date of inception) through December 31, 2010 were $883,425, $728,341, $1,039,028 and $8,079,210, respectively. Stock-based compensation for the period from May 31, 2005 (date of inception) through December 31, 2010 include stock-based payments in the acquisition of ModigeneTech and deferred compensation on restricted shares in the amount of $3,876,960.
|
F-32
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 9:-
|
INCOME TAXES
|
|
|
a.
|
Losses for tax purposes:
|
Carry-forward tax losses of the Company and Modigene Inc. total approximately $712,430 as of December 31, 2010. ModigeneTech’s Carry-forward tax losses as of December 31, 2010 were $22,970,127, which may be carried forward and offset against taxable income of ModigeneTech in the future for an indefinite period.
|
|
b.
|
Deferred income taxes:
|
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial purposes and the amounts used for income tax purposes.
As of December 31, 2010, the Company has provided full valuation allowances in respect of deferred tax assets. Management currently believes that since the Company has a history of losses it is more likely than not that the deferred tax regarding the loss carry-forward and other temporary differences will not be realized in the foreseeable future.
|
NOTE 10:-
|
RESEARCH AND DEVELOPMENT, NET
|
|
Year ended December 31,
|
Period from
May 31,
2005
(date of
inception)
to December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Research and development expenses
|
$ | 6,603,589 | $ | 6,040,235 | $ | 5,527,102 | $ | 21,704,138 | ||||||||
|
Less –
Government grants and participation
|
(1,383,806 | ) | (484,912 | ) | (746,013 | ) | (3,383,597 | ) | ||||||||
| $ | 5,219,783 | $ | 5,555,323 | $ | 4,781,089 | $ | 18,320,541 | |||||||||
As for the Company’s government grants and participation – see note 2s.
F-33
PROLOR BIOTECH, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
U.S. dollars
|
NOTE 11:-
|
SUBSEQUENT EVENTS
|
|
|
a.
|
In January 2011 the Company and three of its executives amended their agreements, pursuant to the terms of the amendments; effective as of January 1, 2011 the executives’ annual base salary has been increased as discussed in note 7.
|
|
|
b.
|
Subsequent to December 31, 2010, 98,201 common shares were issued in connection with the exercise of outstanding warrants and options.
|
F-34