Attached files
| file | filename |
|---|---|
| EX-31.1 - EXHIBIT 31.1 CERTIFICATION - PROGENICS PHARMACEUTICALS INC | ex31_112312010.htm |
| EX-32.2 - EXHIBIT 32.2 CERTIFICATION - PROGENICS PHARMACEUTICALS INC | ex32_212312010.htm |
| EX-32.1 - EXHIBIT 32.1 CERTIFICATION - PROGENICS PHARMACEUTICALS INC | ex32_112312010.htm |
| EX-10.37 - EXHIBIT 10.37 PROGENICS - PROGENICS PHARMACEUTICALS INC | ex10_3712312010.htm |
| EX-23.1 - EXHIBIT 23.1 CONSENT OF PRICEWATERHOUSECOOPERS - PROGENICS PHARMACEUTICALS INC | ex23_112312010.htm |
| EX-31.2 - EXHIBIT 31.2 CERTIFICATION - PROGENICS PHARMACEUTICALS INC | ex31_212312010.htm |
perfgraph2010.jpg Progenics Performance Graph 2010
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
_____________
FORM 10-K
(Mark One)
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the fiscal year ended December 31, 2010
|
|
|
Or
|
|
|
¨
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the transition period from ___________ to ___________
|
Commission File No. 000-23143
_____________
PROGENICS PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware
|
13-3379479
|
|
(State or other jurisdiction of
incorporation or organization)
|
(I.R.S. Employer Identification Number)
|
_____________
777 Old Saw Mill River Road
Tarrytown, NY 10591
(Address of principal executive offices, including zip code)
Registrant’s telephone number, including area code: (914) 789-2800
Securities registered pursuant to Section 12(b) of the Act:
Title of each class Name of each exchange on which registered
Common Stock, par value $0.0013 per share The NASDAQ Stock Market LLC
|
Securities registered pursuant to Section 12(g) of the Act:
|
None
|
_____________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Act:
|
Large Accelerated Filer ¨
|
Accelerated Filer x
|
|
Non-accelerated Filer ¨ (Do not check if a smaller reporting company)
|
Smaller Reporting Company ¨
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of the voting and non-voting stock held by non-affiliates of the registrant on June 30, 2010, based upon the closing price of the Common Stock on The NASDAQ Stock Market LLC on that date of $5.48 per share, was $99,913,733 (1).
|
(1)
|
Calculated by excluding all shares that may be deemed to be beneficially owned by executive officers, directors and five percent stockholders of the Registrant, without conceding that any such person is an “affiliate” of the Registrant for purposes of the Federal securities laws.
|
As of March 4, 2011, 33,361,497 shares of Common Stock, par value $.0013 per share, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the Registrant’s definitive proxy statement to be filed in connection with solicitation of proxies for its 2011 Annual Meeting of Shareholders are hereby incorporated by reference into Part III of this Form 10-K where such portions are referenced.
|
PART I
|
||
|
Item 1.
|
2
|
|
|
Item 1A.
|
12
|
|
|
Item 1B.
|
21
|
|
|
Item 2.
|
21
|
|
|
Item 3.
|
21
|
|
|
Item 4.
|
21
|
|
|
PART II
|
||
|
Item 5.
|
22
|
|
|
Item 6.
|
23
|
|
|
Item 7.
|
24
|
|
|
Item 7A.
|
39
|
|
|
Item 8.
|
39
|
|
|
Item 9.
|
39
|
|
|
Item 9A.
|
39
|
|
|
Item 9B.
|
40
|
|
|
PART III
|
||
|
Item 10.
|
41
|
|
|
Item 11.
|
41
|
|
|
Item 12.
|
41
|
|
|
Item 13.
|
41
|
|
|
Item 14.
|
41
|
|
|
PART IV
|
||
|
Item 15.
|
42
|
|
|
F-1
|
||
|
S-1
|
||
|
E-1
|
||
PART I
This document and other public statements we make may contain statements that do not relate strictly to historical fact, any of which may be forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. When we use the words “anticipates,” “plans,” “expects” and similar expressions, we are identifying forward-looking statements. Forward-looking statements involve known and unknown risks and uncertainties which may cause our actual results, performance or achievements to be materially different from those expressed or implied by forward-looking statements. While it is impossible to identify or predict all such matters, these differences may result from, among other things, the inherent uncertainty of the timing and success of, and expense associated with, research, development, regulatory approval and commercialization of our products and product candidates, including the risks that clinical trials will not commence or proceed as planned; products appearing promising in early trials will not demonstrate efficacy or safety in larger-scale trials; clinical trial data on our products and product candidates will be unfavorable; our products will not receive marketing approval from regulators or, if approved, do not gain sufficient market acceptance to justify development and commercialization costs; competing products currently on the market or in development might reduce the commercial potential of our products; we, our collaborators or others might identify side effects after the product is on the market; or efficacy or safety concerns regarding marketed products, whether or not originating from subsequent testing or other activities by us, governmental regulators, other entities or organizations or otherwise, and whether or not scientifically justified, may lead to product recalls, withdrawals of marketing approval, reformulation of the product, additional pre-clinical testing or clinical trials, changes in labeling of the product, the need for additional marketing applications, declining sales or other adverse events.
We are also subject to risks and uncertainties associated with the actions of our corporate, academic and other collaborators and government regulatory agencies, including risks from market forces and trends; potential product liability; intellectual property, litigation, environmental and other risks; the risk that we may not be able to enter into favorable collaboration or other relationships or that existing or future relationships may not proceed as planned; the risk that current and pending patent protection for our products may be invalid, unenforceable or challenged, or fail to provide adequate market exclusivity, or that our rights to in-licensed intellectual property may be terminated for our failure to satisfy performance milestones; the risk of difficulties in, and regulatory compliance relating to, manufacturing products; and the uncertainty of our future profitability.
Risks and uncertainties also include general economic conditions, including interest and currency exchange-rate fluctuations and the availability of capital; changes in generally accepted accounting principles; the impact of legislation and regulatory compliance; the highly regulated nature of our business, including government cost-containment initiatives and restrictions on third-party payments for our products; trade buying patterns; the competitive climate of our industry; and other factors set forth in this document and other reports filed with the U.S. Securities and Exchange Commission (SEC). In particular, we cannot assure you that RELISTOR® will be commercially successful or be approved in the future in other formulations, indications or jurisdictions, or that any of our other programs will result in a commercial product.
We do not have a policy of updating or revising forward-looking statements and we assume no obligation to update any statements as a result of new information or future events or developments. It should not be assumed that our silence over time means that actual events are bearing out as expressed or implied in forward-looking statements.
Available Information
We file annual, quarterly and current reports, proxy statements and other documents with the SEC under the Securities Exchange Act of 1934. The SEC maintains an Internet website that contains reports, proxy and information statements and other information regarding issuers, including Progenics, that file electronically with the SEC. You may obtain documents that we file with the SEC at http://www.sec.gov, and read and copy them at the SEC’s Public Reference Room at 100 F Street NE, Washington, DC 20549. You may obtain information on operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. We also make available our annual, quarterly and current reports and proxy materials on http://www.progenics.com.
Additional information concerning Progenics and its business may be available in press releases or other public announcements and quarterly and current reports and documents filed with the SEC.
Item 1. Business
Progenics Pharmaceuticals, Inc. is a biopharmaceutical company focusing on the development and commercialization of innovative therapeutic products to treat the unmet medical needs of patients with debilitating conditions and life-threatening diseases. Our principal programs are directed toward gastroenterology, oncology and virology. We commenced principal operations in 1988, became publicly traded in 1997 and throughout have been engaged primarily in research and development efforts, developing manufacturing capabilities, establishing corporate collaborations and related activities. All of our operations are conducted at our facilities in Tarrytown, New York. Additional information concerning Progenics and its business may be available in press releases or other public announcements and quarterly and current reports and documents filed with the SEC after the filing of this Annual Report.
In gastroenterology, our first commercial product is RELISTOR (methylnaltrexone bromide) subcutaneous injection, a first-in-class therapy for opioid-induced constipation approved for sale in over 50 countries worldwide, including the United States, the European Union, Canada and Australia. Marketing applications are pending elsewhere throughout the world.
On February 3, 2011, we entered into an exclusive License Agreement with Salix Pharmaceuticals, by which Salix acquired the rights to RELISTOR worldwide except in Japan, where we have previously licensed to Ono Pharmaceutical Co., Ltd. the subcutaneous formulation of the drug. Under the License Agreement, Salix is responsible for further developing and commercializing subcutaneous RELISTOR, including completing clinical development necessary to support regulatory marketing approvals for potential new indications and formulations. Salix will market RELISTOR directly through its specialty sales force in the U.S., and outside the U.S., RELISTOR will be marketed with sublicenses to regional companies.
Under the Salix License Agreement, we received a $60.0 million upfront payment and are eligible to receive development milestone payments of up to $90.0 million contingent upon the achievement of specified U.S. regulatory approvals and commercialization milestone payments of up to $200.0 million contingent upon the achievement of specified U.S. sales targets. Salix must pay us royalties based on a percentage ranging from 15 to 19 percent of net sales by it and its affiliates, and 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) it receives from sublicensees in respect of any country outside the U.S.
In our other gastroenterology efforts, we have recently presented preclinical data on novel monoclonal antibodies against toxins produced by the bacterium Clostridium difficile (C. difficile), the leading cause of hospital-acquired diarrhea in the U.S. and a recognized growing global public health challenge.
See Gastroenterology; Licenses and Risk Factors.
In oncology, we recently announced preliminary data from a phase 1 clinical trial of a fully human monoclonal antibody-drug conjugate (ADC) directed against prostate specific membrane antigen (PSMA), a protein found at high levels on the surface of prostate cancer cells and also on the neovasculature of a number of other types of solid tumors. While we have to date conducted PSMA ADC research and development on our own, we are considering as appropriate strategic collaborations with biopharmaceutical companies for development of PSMA ADC.
We are also engaged in research to identify multiplex phosphoinositide 3-kinase (PI3K) inhibitors that may be effective in blocking signaling pathways that are critical in the growth of aggressive cancers.
See Oncology.
In virology, we have been developing a viral-entry inhibitor -- a humanized monoclonal antibody, PRO 140 -- for human immunodeficiency virus (HIV), the virus that causes acquired immunodeficiency syndrome, or AIDS, and are conducting a clinical trial of PRO 140 with outside funding. Advancement of this program, including clinical trial efforts, is subject to obtaining additional outside funding, for which we have applied to government agencies. We are also evaluating hepatitis C virus entry inhibitors as possible development candidates. See Virology.
Recent changes in executive responsibilities. On March 3, 2011, our Board of Directors appointed Mark R. Baker Chief Executive Officer of the Company. At the same time, Paul J. Maddon was appointed Vice Chairman of the Board; he retains the title of Chief Science Officer. Both Mr. Baker and Dr. Maddon continue as Board members.
Following is a summary of our principal therapeutic and research programs:
|
Commercial product
|
Approved indication
|
Status
|
||
|
Gastroenterology
|
||||
|
RELISTOR®(1)-Subcutaneous injection
|
Treatment of opioid-induced constipation (OIC) in advanced-illness patients receiving palliative care when laxative therapy has not been sufficient (2)
|
Marketed in the U.S., E.U., Canada, Australia and elsewhere; Recently licensed to Salix Pharmaceuticals worldwide other than Japan, where Ono Pharmaceutical is developing subcutaneous RELISTOR
|
||
|
Therapeutic or Research Program
|
Proposed therapeutic area
|
Status (3)
|
||
|
Gastroenterology
|
||||
|
RELISTOR-Subcutaneous injection
|
Treatment of OIC in patients with non-cancer pain
|
Phase 3 testing completed; preparing sNDA for submission in first half of 2011
|
||
|
RELISTOR-Oral
|
Treatment of OIC
|
In Phase 3 testing
|
||
|
C. difficile
Evaluating anti-toxin monoclonal antibodies
|
Treatment of conditions caused by Clostridium difficile toxins
|
Research
|
||
|
Oncology
|
||||
|
PSMA ADC
|
Treatment of prostate cancer
|
Phase 1
|
||
|
Evaluating multiplex PI3K inhibitor compounds
|
Treatment of cancer
|
Research
|
||
|
Virology
|
||||
|
Human Immunodeficiency Virus (HIV)
|
||||
|
PRO 140
|
Treatment of HIV infection
|
Phase 2 (4)
|
||
|
Hepatitis C Virus (HCV)
Evaluating HCV-entry inhibitor compounds
|
Treatment of HCV infection
|
Research
|
||
_______________
|
(1)
|
RELISTOR is a registered trademark which is in the process of being transitioned from Progenics' former collaborator, Wyeth, in connection with the transition of development and commercialization responsibility for RELISTOR to Salix. In this document, “RELISTOR” refers to methylnaltrexone as it has been and is being developed and commercialized by or in collaboration with Progenics. Subcutaneous RELISTOR has received regulatory marketing approval for specific indications, and references to RELISTOR do not imply that any other form or possible use of the drug has received such approval.
|
|||||
|
(2)
|
The approved U.S. label for RELISTOR also provides that use of RELISTOR beyond four months has not been studied. Full U.S. prescribing information is available at www.RELISTOR.com.
|
|||||
|
(3)
|
Research means initial research related to specific molecular targets, synthesis of new chemical entities, assay development or screening for identification and optimization of lead compound.
|
|||||
|
Pre-clinical means lead compound(s) undergoing toxicology, formulation and other testing in preparation for clinical trials.
|
||||||
|
Phase 1-3 clinical trials are safety and efficacy tests in humans:
|
||||||
|
Phase 1: Initial evaluation of safety in humans; study method of action and metabolization.
|
||||||
|
Phase 2: Evaluation of safety, dosing and activity or efficacy; continue safety evaluation.
|
||||||
|
Phase 3: Larger scale evaluation of safety, efficacy and dosage.
|
||||||
|
(4)
|
Advancement of this program, including clinical trial efforts, is subject to obtaining additional outside funding, for which we have applied to government agencies.
|
|||||
Gastroenterology
Opioid-based medications such as morphine and codeine are used to control moderate-to-severe pain in patients receiving palliative care, undergoing surgery, experiencing chronic pain or with other medical conditions. Opioids relieve pain by interacting with receptors located in the brain and spinal cord, but also activate receptors in the gut, often resulting in constipation, referred to as opioid-induced constipation or OIC. As a result of OIC, many patients may stop or reduce their opioid therapy, opting to endure pain in order to obtain relief from their OIC and its associated side effects.
RELISTOR, the first approved treatment for OIC that addresses the underlying mechanism of this condition, is a selective, peripherally acting, mu-opioid-receptor antagonist that decreases the constipating side effects induced by opioid pain medications in the gastrointestinal tract without diminishing the ability of these medications to relieve pain. Relief of OIC is an important need that is not adequately met by any other approved drug or intervention. Because of its chemical composition, RELISTOR has restricted access to the blood-brain barrier to enter the central nervous system, where pain is perceived. Outside the central nervous system, RELISTOR competes with opioid pain medications for binding sites on opioid receptors, displacing the pain medications only in the periphery and selectively “turning off” the constipating effects of those medications on the gastrointestinal tract without affecting pain relief occurring in the central nervous system.
Subcutaneous RELISTOR. RELISTOR is currently approved by regulatory authorities in the U.S. and other countries for treatment of OIC in advanced-illness patients receiving palliative care when laxative therapy has not been sufficient. In the second quarter of 2008 we began earning royalties on sales of RELISTOR by our former collaborator, Wyeth Pharmaceuticals, now a Pfizer Inc. subsidiary. RELISTOR net sales and related royalties earned through the end of 2010 are set forth below. Our recognition of royalty revenue for financial reporting purposes is explained in Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A) and our financial statements elsewhere in this document.
|
First
Quarter
|
Second
Quarter
|
Third
Quarter
|
Fourth
Quarter
|
Full
Year
|
||||||
|
(in thousands)
|
||||||||||
|
2010:
|
||||||||||
|
Net Sales By Wyeth
|
$4,200
|
$3,800
|
$4,100
|
$4,000
|
$16,100
|
|||||
|
Royalties Earned
|
625
|
581
|
620
|
-(1)
|
1,826
|
|||||
|
2009:
|
||||||||||
|
Net Sales By Wyeth
|
$1,900
|
$3,200
|
$3,300
|
$3,900
|
$12,300
|
|||||
|
Royalties Earned
|
280
|
487
|
497
|
589
|
1,853
|
|||||
|
2008:
|
||||||||||
|
Net Sales By Wyeth
|
n.a.
|
$2,100
|
$800
|
$1,500
|
$4,400
|
||||||||||||
|
Royalties Earned
|
n.a.
|
321
|
117
|
227
|
665
|
||||||||||||
________________
(1) Under the terms of the Wyeth transition, no royalties are payable in respect of net sales after September 30, 2010.
RELISTOR has previously been developed and commercialized worldwide except Japan by Progenics and Wyeth pursuant to a 2005 collaboration agreement that was terminated in October 2009. Under our Transition Agreement with Wyeth, Wyeth is continuing to distribute RELISTOR worldwide other than Japan through March 31, 2011. Salix, Wyeth and Progenics plan an April 1, 2011 transition of U.S. commercial responsibility for RELISTOR to Salix from Wyeth, and are currently discussing the transition of ex-U.S. commercialization responsibilities on a country-by-country basis. While Salix effects a country-by-country transition of ex-U.S. commercialization rights, Wyeth remains the marketing authorization holder for RELISTOR and continues to supply product. See Licenses – RELISTOR.
Under the Ono License Agreement, Ono began clinical testing of RELISTOR subcutaneous injection in June 2009; Ono’s development efforts are continuing with a phase 2 trial designed to demonstrate efficacy and safety.
We have received U.S., E.U. and Canadian approvals to market RELISTOR in pre-filled syringes, which are designed to ease preparation and administration for patients and caregivers, and currently plan to coordinate the launch of that product with Salix in 2011.
We are also developing subcutaneous RELISTOR for treatment of OIC outside the advanced-illness setting, in individuals with non-cancer pain. Based on results from a recently completed one-year, open-label safety study, together with results from a previous phase 3 efficacy trial, we plan to submit regulatory filings in the first half of 2011 in the U.S., E.U. and elsewhere for approval of subcutaneous RELISTOR to treat OIC in the non-cancer pain setting.
Oral RELISTOR. We are developing an oral formulation of RELISTOR for the treatment of OIC in patients with non-cancer pain. In September 2010, we initiated an ongoing phase 3 trial of oral methylnaltrexone in this patient population, which pursuant to our License Agreement is continuing as part of Salix’s development responsibilities for RELISTOR.
In our other gastroenterology efforts, we have recently presented preclinical data on novel monoclonal antibodies against toxins produced by C. difficile showing these monoclonal antibodies to have effectively neutralized the cell-killing activities of the toxins in vitro and significantly improved survival in a stringent animal model.
Oncology
Conventional prostate cancer therapies, including radical prostatectomy, radiation, hormone therapies and chemotherapy, may have or increase the risk of side effects, including impotence, incontinence, high cholesterol levels and increased blood-clot risk, and some are generally not intended to be curative and are not actively used to treat localized, early-stage prostate cancer. Through PSMA Development Company, our wholly owned subsidiary, we conduct research and development programs directed at prostate specific membrane antigen, or PSMA, a protein that is abundantly expressed on the surface of prostate cancer cells as well as cells in the newly formed blood vessels of many other solid tumors. The principal focus of these efforts is our fully human monoclonal ADC, which is designed to deliver a chemotherapeutic agent to cancer cells by targeting the three-dimensional structure of the PSMA protein on these cells and binding to and internalizing within the cell. We believe a PSMA-directed therapy may have application in prostate cancer and solid tumors of other types of cancer. We recently announced preliminary data from an ongoing phase 1 dose-escalation clinical study to assess PSMA ADC’s safety, tolerability and initial clinical activity in patients with advanced prostate cancer.
We are also engaged in research on, and recently presented data from preclinical studies of, novel multiplex phosphoinositide 3-kinase (PI3K) inhibitors -- synthetic, small-molecule compounds identified by us that in laboratory studies blocked both PI3K, a key regulator of one molecular signaling pathway, and MNK, an oncogenic kinase in the Ras pathway. We believe simultaneously blocking these interlinked cellular pathways may provide a strategy to combat some of the most aggressive forms of cancer.
Virology
HIV and HCV. Viral entry inhibitors such as our drug candidate PRO 140 represent the newest class of drugs for HIV patients. Our program is based on blocking the binding of HIV to a particular co-receptor used by the virus, which does not block the entry of some strains of HIV that use a less ubiquitous co-receptor. Advancement of this program is subject to obtaining outside funding, for which we have applied to government agencies.
We are also evaluating HCV-entry inhibitors as potential compounds to treat the most common blood-borne infection in the U.S. and a major cause of chronic liver disease.
Licenses
Following is a summary of significant license agreements under which we have in- and/or out-licensed rights to use certain technologies and materials.
RELISTOR
· Under our recent License Agreement, Salix Pharmaceuticals is responsible for further developing and commercializing subcutaneous RELISTOR worldwide other than Japan, including completing clinical development necessary to support regulatory marketing approvals for potential new indications and formulations, and marketing and selling the product. Salix will market RELISTOR directly through its specialty sales force in the U.S., and outside the U.S., RELISTOR will be marketed with sublicenses to regional companies.
Salix is assuming overall responsibilities for RELISTOR, which Wyeth returned to us under our 2009 Transition Agreement. That Agreement provided for the termination of our 2005 collaboration with Wyeth and the transition to Progenics of responsibility for the development and commercialization of RELISTOR, which is now being assumed by Salix under its License Agreement. Under the Transition Agreement, Wyeth is continuing to distribute RELISTOR during the transition period. Wyeth also agreed, at its expense, to continue certain ongoing development efforts for subcutaneous RELISTOR including conducting specified clinical studies, and to provide financial resources and/or other assistance with respect to additional agreed-upon regulatory, manufacturing, supply and clinical matters, in accordance with an agreed-upon development plan. Financial resources of approximately $9.5 million, for which we have recognized $1.2 million, constitute reimbursement for development of a multi-dose pen for subcutaneous RELISTOR.
Under the Transition Agreement, Wyeth paid us $10.0 million in six quarterly installments through January 2011, and continued to pay royalties on 2010 ex-U.S. sales as provided in the 2005 collaboration agreement except to the extent certain fourth quarter financial targets were not met. These targets were not met during the fourth quarter and royalties on ex-U.S. sales were not payable to us. No other royalties are payable in respect of RELISTOR net sales after September 30, 2010. Salix has agreed to pay us royalties on its net sales of RELISTOR as it commences commercialization efforts. Salix will also pay us 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) it receives from sublicensees in respect of any country outside the U.S., as sublicensees commence their commercialization efforts. We agreed to purchase Wyeth’s remaining inventory of subcutaneous RELISTOR at the end of the Sales Periods on agreed-upon terms and conditions, and Salix has agreed to purchase our inventory of subcutaneous RELISTOR on similar agreed-upon terms and conditions. The Wyeth Transition Agreement also provided for transfer of development, manufacturing and commercialization records and other materials, mutual releases between the parties and indemnification, dispute resolution, non-disparagement and other customary provisions. We have no further obligations to Wyeth under the 2005 collaboration agreement.
The 2005 Wyeth collaboration agreement was in effect until October 2009, which includes periods covered by this report. Under that agreement, we granted to Wyeth an exclusive, worldwide license to develop and commercialize RELISTOR and assigned certain agreements to it. We were responsible for developing the subcutaneous and intravenous formulations in the U.S. until they received regulatory approval, while Wyeth was responsible for these formulations outside the U.S. (other than Japan after execution of the Ono License) and for developing the oral formulation worldwide. From January 2006 to October 2009, development costs for RELISTOR were paid by Wyeth. We were reimbursed for out-of-pocket costs and received reimbursement for our efforts based on our employees devoted to them, subject to Wyeth’s audit rights and possible reconciliation. Commercialization decisions were made by Wyeth, which also was obligated to pay all commercialization costs, including manufacturing costs, and retained all proceeds from product sales, subject to royalties and other amounts payable to us.
· We have exclusive rights to develop and commercialize methylnaltrexone, the active ingredient of RELISTOR, under license from the University of Chicago for which we are obligated to make milestone and royalty payments to the University.
· We have licensed to Ono Pharmaceutical the rights to subcutaneous RELISTOR in Japan, where Ono is responsible for developing and commercializing subcutaneous RELISTOR, including conducting clinical development to support regulatory marketing approval and will own the subcutaneous filings and approvals relating to RELISTOR. Our relationship with Ono is not affected by the Salix License Agreement. We received a $15.0 million upfront payment from Ono, and are entitled to receive up to an additional $20.0 million, payable upon achievement of development milestones. Ono is also obligated to pay us royalties and commercialization milestones on sales of subcutaneous RELISTOR in Japan. Ono has the option to acquire the rights to develop and commercialize other formulations of RELISTOR in Japan, on terms to be negotiated separately. Supervision of and consultation with respect to Ono’s development and commercialization responsibilities are carried out by joint committees. The Ono License contains, among other terms, provisions which permit termination by either party upon the occurrence of certain events.
PSMA
· PSMA Development Company LLC has a collaboration agreement with Seattle Genetics, Inc., under which SGI has granted it an exclusive worldwide license to SGI’s proprietary ADC technology. PSMA LLC has the right to use this technology, which is based in part on technology licensed by SGI from third parties, to link chemotherapeutic agents to PSMA LLC’s monoclonal antibodies that target prostate specific membrane antigen. PSMA LLC is responsible for research, product development, manufacturing and commercialization of all products, and may sublicense the ADC technology to a third party manufacturer. PSMA LLC is obligated to make maintenance and milestone payments aggregating up to $14.3 million and to pay royalties to SGI and its licensors, as applicable, on a percentage of net sales. The SGI agreement terminates at the latest of (i) the tenth anniversary of the first commercial sale of each licensed product in each country or (ii) the latest date of expiration of patents underlying the licensed products. PSMA LLC may terminate the agreement upon advance written notice, and SGI may terminate if PSMA LLC fails to cure a breach of an SGI in-license within a specified time period after written notice. In addition, either party may terminate the agreement after written notice upon an uncured breach or in the event of bankruptcy of the other party. As of December 31, 2010, PSMA LLC has paid approximately $3.7 million under this agreement, including $1.0 million in milestone payments.
· PSMA LLC also has a worldwide exclusive licensing agreement with Abgenix (now Amgen Fremont, Inc.) to use its XenoMouse® technology for generating fully human antibodies to PSMA LLC’s PSMA antigen. PSMA LLC is obligated to make development and commercialization milestone payments with respect to products incorporating an antibody generated utilizing the XenoMouse technology. As of December 31, 2010, PSMA LLC has paid $0.9 million under this agreement and is obligated to pay up to an additional $6.3 million if certain milestones are met, along with royalties based upon net sales of antibody products, if any. This agreement may be terminated, after an opportunity to cure, by Abgenix for cause upon 30 days prior written notice; PSMA LLC has the right to terminate upon 30 days prior written notice. The agreement continues until the later of the expiration of the XenoMouse technology patents that may result from pending patent applications or seven years from the first commercial sale of the products.
· PSMA LLC has a worldwide exclusive license agreement with AlphaVax Human Vaccines to use its Replicon Vector system to create a therapeutic prostate cancer vaccine incorporating PSMA LLC’s proprietary PSMA antigen. PSMA LLC is obligated to make development and commercialization milestone payments with respect to products incorporating AlphaVax’s system. As of December 31, 2010, PSMA LLC has paid $2.1 million under this agreement and is obligated to pay up to an additional $5.4 million if certain milestones are met along with annual maintenance fees and royalties based upon net sales of any products developed using AlphaVax’ system. This agreement may be terminated, after an opportunity to cure, by AlphaVax under specified circumstances, including PSMA LLC’s failure to achieve milestones; the consent of AlphaVax to revisions to the milestones due dates may not, however, be unreasonably withheld. PSMA LLC has the right to terminate upon 30 days prior written notice. The agreement continues until the later of the expiration of the patents relating to AlphaVax’s system or seven years from the first commercial sale of the products developed using that system. Pending U.S. and international patent applications and patent-term extensions may extend the period of our license rights when and if they are allowed, issued or granted.
Virology - PRO 140
· Protein Design Labs (now Facet Biotech Corporation, a wholly owned subsidiary of Abbott Laboratories) humanized a murine monoclonal antibody developed by us (humanized PRO 140) and granted us related licenses under patents and patent applications, in addition to know-how. In general, these licenses are fully paid after the latest of (i) the tenth anniversary of the first commercial sale of a product developed thereunder, (ii) expiration of the last-to-expire relevant patent or (iii) the tenth anniversary of the latest filed pending patent application. Pending U.S. and international patent applications and patent-term extensions may extend the period of our license rights when and if they are allowed, issued or granted. We may terminate the license on 60 days prior written notice, and either party may terminate on 30 days prior written notice for an uncured material breach (ten days for payment default). As of December 31, 2010, we have paid $5.5 million under this agreement, and if all milestones are achieved, we will be obligated to pay an additional $2.5 million, including annual maintenance fees and royalties on sales of products developed under the license.
· We have a letter agreement with the Aaron Diamond AIDS Research Center pursuant to which we have the exclusive right to pursue the commercial development, directly or with a partner, of products related to HIV based on patents jointly owned by ADARC and us.
Patents and Proprietary Technology
Our policy is to protect our proprietary technology, and we consider the protection of our rights to be important to our business. In addition to seeking U.S. patent protection for many of our inventions, we generally file patent applications in Canada, Japan, European countries that are party to the European Patent Convention and additional foreign countries on a selective basis in order to protect the inventions that we consider to be important to the development of our foreign business. Generally, patents issued in the U.S. are effective:
· if the patent application was filed prior to June 8, 1995, for the longer of 17 years from the date of issue or 20 years from the earliest asserted filing date; or
· if the application was filed on or after June 8, 1995, for 20 years from the earliest asserted filing date.
In certain instances, the U.S. patent term can be extended up to a maximum of five years to recapture a portion of the term during which the FDA regulatory review was being conducted. The duration of foreign patents varies in accordance with the provisions of applicable local law, although most countries provide for patent terms of 20 years from the earliest asserted filing date and allow patent extensions similar to those permitted in the U.S.
We also rely on trade secrets, proprietary know-how and continuing technological innovation to develop and maintain a competitive position in our product areas. We generally require our employees, consultants and corporate partners who have access to our proprietary information to sign confidentiality agreements.
Information with respect to our patent portfolio regarding our therapeutic and research programs, as of year-end 2010, is set forth below.
|
Therapeutic or
|
Number of Patents
|
Expiration
|
Number of Patent Applications
|
|||||||
|
Research Program
|
U.S.
|
International
|
Dates (1)
|
U.S.
|
International
|
|||||
|
Gastroenterology
(RELISTOR; C. difficile)
|
7
|
25
|
2011-2028
|
24
|
143
|
|||||
|
Oncology (PSMA; PI3K)
|
9
|
25
|
2013-2025
|
7
|
28
|
|||||
|
Virology (PRO 140; HCV)
|
15
|
24
|
2015-2024
|
13
|
15
|
|||||
| _____________________________ | ||||||||||
|
(1)
|
Patent term extensions and pending patent applications may extend the period of patent protection afforded our products and product candidates under development.
|
Our patents may not enable us to preclude competitors from commercializing drugs in direct competition with our products, and consequently may not provide us with any meaningful competitive advantage. See “Risk Factors.”
We are aware of intellectual property rights held by third parties that relate to products or technologies we are developing. For example, we are aware of others investigating methylnaltrexone and other peripheral opioid antagonists, PSMA or related compounds, monoclonal antibodies directed at targets relevant to PRO 140 and HCV viral entry inhibitors, and of patents and applications held or filed by others in those areas. While the validity of issued patents, patentability of claimed inventions in pending applications and applicability of any of them to our programs are uncertain, patent rights asserted against us could adversely affect our ability to commercialize or collaborate with others regarding our products.
Research, development and commercialization of a biopharmaceutical product often require choosing between alternative development and optimization routes at various stages in the development process. Preferred routes depend upon subsequent discoveries and test results and cannot be identified with certainty at the outset. There are numerous third-party patents in our field, and we may need to obtain a license under a patent in order to pursue the preferred development route of one or more of our product candidates. The need to obtain a license would decrease the ultimate profitability of the applicable product. If we cannot negotiate a license, we might have to pursue a less desirable development route or terminate the entire program altogether.
Government Regulation
Progenics and its product candidates are subject to comprehensive regulation by the U.S. FDA and comparable authorities in other countries. Pharmaceutical regulation currently is a topic of substantial interest in lawmaking and regulatory bodies in the U.S. and internationally, and numerous proposals exist for changes in FDA and non-U.S. regulation of pre-clinical and clinical testing, safety, effectiveness, approval, manufacture, labeling, marketing, export, storage, recordkeeping, advertising, promotion and other aspects of biologics, small molecule drugs and medical devices, many of which, if adopted, could significantly alter our business and the current regulatory structure described below.
FDA Regulation. FDA approval of our product candidates, including a review of the manufacturing processes and facilities used to produce them, are required before they may be marketed in the U.S. This process is costly, time consuming and subject to unanticipated delays, and a drug candidate may fail to progress at any point.
None of our product candidates other than RELISTOR has received marketing approval from the FDA or any other regulatory authority. The process required by the FDA before product candidates may be approved for marketing in the U.S. generally involves:
· pre-clinical laboratory and animal tests;
· submission to and favorable review by the FDA of an IND (Investigational New Drug) application before clinical trials may begin;
· adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended indication (animal and other nonclinical studies also are typically conducted during each phase of human clinical trials);
· submission to the FDA of a marketing application; and
· FDA review of the marketing application in order to determine, among other things, whether the product is safe and effective for its intended uses.
Pre-clinical tests include laboratory evaluation of product chemistry and animal studies to gain preliminary information about a product’s pharmacology and toxicology and to identify safety problems that would preclude testing in humans. Products must generally be manufactured according to current Good Manufacturing Practices, and pre-clinical safety tests must be conducted by laboratories that comply with FDA good laboratory practices regulations.
Results of pre-clinical tests are submitted to the FDA as part of an IND which must become effective before clinical trials may commence. The IND submission must include, among other things, a description of the sponsor’s investigational plan; protocols for each planned study; chemistry, manufacturing and control information; pharmacology and toxicology information and a summary of previous human experience with the investigational drug. Unless the FDA objects to, makes comments or raises questions concerning an IND, it becomes effective 30 days following submission, and initial clinical studies may begin. Companies often obtain affirmative FDA approval, however, before beginning such studies.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or to individuals under the supervision of a qualified principal investigator. Clinical trials must be conducted in accordance with the FDA’s Good Clinical Practice requirements under protocols submitted to the FDA that detail, among other things, the objectives of the study, parameters used to monitor safety and effectiveness criteria to be evaluated. Each clinical study must be conducted under the auspices of an Institutional Review Board, which considers, among other things, ethical factors, safety of human subjects, possible liability of the institution and informed consent disclosure which must be made to participants in the trial.
Clinical trials are typically conducted in three sequential phases, which may overlap. During phase 1, when the drug is initially administered to human subjects, the product is tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. Phase 2 involves studies in a limited population to evaluate preliminarily the efficacy of the product for specific, targeted indications, determine dosage tolerance and optimal dosage and identify possible adverse effects and safety risks.
When a product candidate is found in phase 2 evaluation to have an effect and an acceptable safety profile, phase 3 trials are undertaken in order to further evaluate clinical efficacy and test for safety within an expanded population. Safety studies are conducted in accordance with the FDA’s International Conference on Harmonization (ICH) Guidelines. Phase 2 results do not guarantee a similar outcome in phase 3 trials. The FDA may suspend clinical trials at any point in this process if it concludes that clinical subjects are being exposed to an unacceptable health risk.
A New Drug Application, or NDA, is an application to the FDA to market a new drug. A Biologic License Application, or BLA, is an application to market a biological product. The new drug or biological product may not be marketed in the U.S. until the FDA has approved the NDA or issued a biologic license. The NDA must contain, among other things, information on chemistry, manufacturing and controls; non-clinical pharmacology and toxicology; human pharmacokinetics and bioavailability; and clinical data. The BLA must contain, among other things, data derived from nonclinical laboratory and clinical studies which demonstrate that the product meets prescribed standards of safety, purity and potency, and a full description of manufacturing methods. Supplemental NDAs (sNDAs) are submitted to obtain regulatory approval for additional indications for a previously approved drug.
The results of the pre-clinical studies and clinical studies, the chemistry and manufacturing data, and the proposed labeling, among other things, are submitted to the FDA in the form of an NDA or BLA. The FDA may refuse to accept the application for filing if certain administrative and content criteria are not satisfied, and even after accepting the application for review, the FDA may require additional testing or information before approval of the application, in either case based upon changes in applicable law or FDA policy during the period of product development and FDA regulatory review. The applicant’s analysis of the results of clinical studies is subject to review and interpretation by the FDA, which may differ from the applicant’s analysis, and in any event, the FDA must deny an NDA or BLA if applicable regulatory requirements are not ultimately satisfied. If regulatory approval of a product is granted, such approval may be made subject to various conditions, including post-marketing testing and surveillance to monitor the safety of the product, or may entail limitations on the indicated uses for which it may be marketed. Finally, product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing.
Both before and after approval is obtained, a product, its manufacturer and the sponsor of the marketing application for the product are subject to comprehensive regulatory oversight. Violations of existing or newly-adopted regulatory requirements at any stage, including the pre-clinical and clinical testing process, the approval process, or thereafter, may result in various adverse consequences, including FDA delay in approving or refusal to approve a product, withdrawal of an approved product from the market or the imposition of criminal penalties against the manufacturer or sponsor. Later discovery of previously unknown problems may result in restrictions on the product, manufacturer or sponsor, including withdrawal of the product from the market.
Regulation Outside the U.S. Whether or not FDA approval has been obtained, approval of a pharmaceutical product by comparable government regulatory authorities in foreign countries must be obtained prior to marketing the product there. The approval procedure varies from country to country, and the time required may be longer or shorter than that required for FDA approval. The requirements for regulatory approval by governmental agencies in other countries prior to commercialization of products there can be rigorous, costly and uncertain, and approvals may not be granted on a timely basis or at all.
In the European Union, Canada, Australia and Japan, regulatory requirements and approval processes are similar in principle to those in the United States. Regulatory approval in Japan requires that clinical trials of new drugs be conducted in Japanese patients. Depending on the type of drug for which approval is sought, there are currently two potential tracks for marketing approval in the E.U. countries: mutual recognition and the centralized procedure. These review mechanisms may ultimately lead to approval in all E.U. countries, but each method grants all participating countries some decision-making authority in product approval. The centralized procedure, which is mandatory for biotechnology derived products, results in a recommendation in all member states, while the E.U. mutual recognition process involves country-by-country approval.
In other countries, regulatory requirements may require additional pre-clinical or clinical testing regardless of whether FDA approval has been obtained. This is the case in Japan, where Ono is responsible for developing and commercializing the subcutaneous form of RELISTOR and where trials are required to involve patient populations which we and our other collaborators have not examined in detail. If the particular product is manufactured in the U.S., we must also comply with FDA and other U.S. export provisions. In most countries outside the U.S., coverage, pricing and reimbursement approvals are also required which may affect the profitability of the affected product.
Other Regulation. In addition to regulations enforced by the FDA, we are also subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and various other current and potential future federal, state or local regulations. Biopharmaceutical research and development involves the controlled use of hazardous materials, chemicals, viruses and various radioactive compounds. Even strict compliance with safety procedures for storing, handling, using and disposing of such materials prescribed by applicable regulations cannot completely eliminate the risk of accidental contaminations or injury from these materials, which may result in liability for resulting legal and regulatory violations as well as damages.
See Risk Factors.
Manufacturing
Under our recent License Agreement for RELISTOR, Salix is responsible for the manufacture and supply, at its expense, of all active pharmaceutical ingredient (API) and finished and packaged products for its commercialization efforts, including assuming relationships we have entered into in anticipation of establishing a new collaboration partnership or contracting with one or more other contract manufacturing organizations (CMOs) for supply of RELISTOR API and subcutaneous and oral finished drug product. See Risk Factors.
We manufacture clinical trial supplies of our PSMA monoclonal antibody in our biologics pilot production facilities in Tarrytown, New York, and have engaged third-party CMOs for other portions of the PSMA ADC manufacturing process. We expect our manufacturing capacity will not be sufficient for all of our late-stage clinical trials or commercial-scale requirements. If we are unable to arrange for satisfactory CMO services, or otherwise determine to acquire additional manufacturing capacity, we will need to expand our manufacturing staff and facilities or obtain new facilities. In order to establish a full-scale commercial manufacturing facility for any of our product candidates, we would need to spend substantial additional funds, hire and train significant numbers of employees and comply with the extensive FDA regulations applicable to such a facility.
Sales and Marketing
We from time to time seek strategic collaborations and other funding support for product candidates in our pipeline. We expect that we would market other products for which we obtain regulatory approval through co-marketing, co-promotion, licensing and distribution arrangements with third-party collaborators, and might also consider contracting with professional detailing and sales organizations to perform promotional and/or medical-scientific support functions for them. See Risk Factors.
Competition
Competition in the biopharmaceutical industry is intense and characterized by ongoing research and development and technological change. We face competition from many for-profit companies and major universities and research institutions in the U.S. and abroad. We face competition from companies marketing existing products or developing new products for diseases targeted by our technologies. Many of our competitors have substantially greater resources, experience in conducting pre-clinical studies and clinical trials and obtaining regulatory approvals for their products, operating experience, research and development and marketing capabilities and production capabilities than we do. Our products and product candidates under development may not compete successfully with existing products or products under development by other companies, universities and other institutions. Drug manufacturers that are first in the market with a therapeutic for a specific indication generally obtain and maintain a significant competitive advantage over later entrants and therefore, the speed with which industry participants develop products, complete clinical trials, approve processes and commercialize products are important competitive factors.
RELISTOR is the first FDA-approved product for any indication involving OIC. We are, however, aware of products in pre-clinical or clinical development that target the side effects of opioid pain therapy. For example: Adolor Corporation markets ENTEREG® (alvimopan) for the treatment of postoperative ileus, and is also evaluating a phase 1 and early-stage compound for opioid-bowel dysfunction in chronic-pain patients. Sucampo Pharmaceuticals, Inc., in collaboration with Takeda Pharmaceutical Company Limited, markets AMITIZA® (lubiprostone), a selective chloride channel activator, for chronic idiopathic (non-opioid related) constipation and recently completed two phase 3 pivotal clinical trials of this drug for opioid-induced bowel dysfunction. In Europe, Mundipharma International Limited markets TARGIN® (oxycodone/naloxone), a combination of an opioid and a systemic opioid antagonist, and Movetis NV, which has recently been acquired by Shire plc, has announced that it has started a phase 3 clinical trial with prucalopride in patients with constipation induced by opioid based pain medications. A Nektar Therapeutics-AstraZeneca PLC collaboration has announced phase 2 results of an oral peripheral mu-opioid receptor antagonist in patients with OIC and is developing a related combination product. AstraZeneca is a leader in gastrointestinal medicine, and this collaboration may have a time-to-market advantage over us with respect to an oral therapy for OIC in non-cancer pain patients. Alkermes, Inc. recently announced preliminary results from a phase 2 clinical study of an oral peripherally-restricted opioid antagonist, and has a combination product in preclinical testing. Theravance, Inc. is conducting phase 2 clinical testing of an oral peripheral mu-opioid antagonist.
Radiation and surgery are two traditional forms of treatment for prostate cancer, to which our PSMA-based development efforts are directed. If the disease spreads, hormone (androgen) suppression therapy is often used to slow the cancer’s progression. This form of treatment, however, can eventually become ineffective. We are aware of several competitors who are developing alternative treatments for castrate-resistant prostate cancer, some of which are directed against PSMA.
With respect to our PI3K inhibitor research, recent evidence suggests that activation of complementary oncogenic pathways can confer resistance to PI3K inhibition, requiring co-administration of agents targeting these “resistance” pathways. We are aware of several competitors who are developing small molecule PI3K inhibitors that co-target additional oncogenic pathways.
C. difficile infection typically is treated with antibiotics such as vancomycin or metronidazole. Treatment often is associated with alleviation of symptoms, but incomplete response or disease recurrence is observed in approximately 30% of patients and there is no standard treatment for severe, complicated cases of disease. We are aware of competitors who are developing biologic agents for the treatment and/or prevention of C. difficile infection.
Currently approved drugs for the treatment of HIV infection and AIDS have shown efficacy alone and in conjunction with other agents, the latter of which we have not demonstrated for PRO 140. We are aware of two approved drugs, Trimeris, Inc.’s FUZEON® and Pfizer’s SELZENTRY®, designed to treat HIV infection by blocking viral entry.
The current standard for HCV infection is a combination therapy of pegylated interferon and ribavirin. These therapies act via non-specific mechanisms and are associated with substantial toxicities. In addition, the current treatment regimen requires a long duration of up to 48 weeks and is only 50% effective against the most prominent strains found in Europe and the U.S. We are aware of several competitors who are developing small molecule inhibitors of different stages of the HCV lifecycle, including inhibitors of viral entry, such as the oral Hepatitis C protease inhibitor telaprevir being developed by a Vertex Pharmaceuticals Incorporated-Janssen Pharmaceutica, N.V.-Mitsubishi Tanabe Pharma Corporation collaboration, and Merck & Co., Inc.’s bocepravir.
A significant amount of research in the biopharmaceutical field is also being carried out at academic and government institutions. An element of our research and development strategy is to in-license technology and product candidates from academic and government institutions. These institutions are sensitive to the commercial value of their findings and pursue patent protection and negotiate licensing arrangements to collect royalties for use of technology they develop. These institutions may also market competitive commercial products on their own or in collaboration with competitors and compete with us in recruiting highly qualified scientific personnel, which may result in increased costs or decreased availability of technology or product candidates from these institutions to other industry participants.
Competition with respect to our technologies and products is based on, among other things, product efficacy, safety, reliability, method of administration, availability, price and clinical benefit relative to cost; timing and scope of regulatory approval; sales, marketing and manufacturing capabilities; collaborator capabilities; insurance and other reimbursement coverage; and patent protection. Competitive position in our industry also depends on a participant’s ability to attract and retain qualified personnel, obtain patent protection or otherwise develop proprietary products or processes, and secure sufficient capital resources for the typically substantial period between technological conception and commercial sales.
Product Liability
The testing, manufacturing and marketing of our product candidates and products involves an inherent risk of product liability attributable to unwanted and potentially serious health effects. To the extent we elect to test, manufacture or market product candidates and products independently, we bear the risk of product liability directly. We maintain product liability insurance coverage in the amount of $10.0 million per occurrence, subject to a deductible and a $10.0 million aggregate limitation. Where local statutory requirements exceed the limits of our existing insurance or local policies of insurance are required, we maintain additional clinical trial liability insurance to meet these requirements. This insurance is subject to deductibles and coverage limitations. The availability of and cost of maintaining insurance may change over time.
Human Resources
At December 31, 2010, we had 159 full-time employees, 23 of whom hold Ph.D. degrees, five of whom hold M.D. degrees and two of whom hold both Ph.D. and M.D. degrees. At that date, 122 employees were engaged in research and development, medical, regulatory affairs and manufacturing activities and 37 were engaged in finance, legal, administration and business development. We consider our relations with our employees to be good. None of our employees is covered by a collective bargaining agreement.
Item 1A. Risk Factors
Our business and operations entail a variety of serious risks and uncertainties. Our business is inherently risky. We are subject to the risks of failure inherent in the development of product candidates based on new technologies. We must complete successfully clinical trials and obtain regulatory approvals for our product candidates, and will rely on Salix to complete development and obtain regulatory approvals for additional formulations of and indications for RELISTOR. In the Japanese market, we must rely on Ono to conduct successful clinical trials and obtain regulatory approvals. Our other research and development programs involve novel approaches to human therapeutics. There is little precedent for the successful commercialization of products based on our technologies, and there are a number of technological challenges that we must overcome to complete most of our development efforts. We may not be able successfully to develop further any of our products.
In addition to the risks we face in our research and development activities, and our business as a publicly held commercial enterprise devoted to developing and commercializing high-technology consumer products, the transitioning of RELISTOR to our new partner Salix presents us with new risks, including the following:
We are dependent on Salix, Ono, Wyeth (until completion of its involvement) and other business partners to develop and commercialize RELISTOR in their respective areas, exposing us to significant risks.
We are and will be dependent upon Salix, Ono, Wyeth (until completion of its responsibilities pursuant to the Transition Agreement) and any other business partner(s) with which we may collaborate in the future to perform and fund development, including clinical testing of RELISTOR, make related regulatory filings and manufacture and market products in their respective territories. Revenues from the sale of RELISTOR will soon depend almost entirely upon the efforts of Salix, which will have significant discretion in determining the efforts and resources it applies to sales of RELISTOR. Ono will have similar discretion with respect to sales in Japan. Neither may be effective in marketing such products. Our business relationships with Salix, Ono and other partners may not be scientifically, clinically or commercially successful. For example, Salix is a larger pharmaceutical company than Progenics with a variety of marketed products. Unlike Wyeth and Pfizer, however, Salix is not a large diversified pharmaceutical company and does not have resources commensurate with those companies. Salix has its own corporate objectives, which may not be consistent with our best interests, and may change its strategic focus or pursue alternative technologies in a manner that results in reduced or delayed revenues to us. Changes of this nature might also occur if Salix were acquired or if its management changed.
We may have future disagreements with Salix and Ono concerning product development, marketing strategies, manufacturing and supply issues, and rights relating to intellectual property. Both of them have significantly greater financial and managerial resources than we do, which either could draw upon in the event of a dispute. Disagreements between either of them and us could lead to lengthy and expensive litigation or other dispute-resolution proceedings as well as extensive financial and operational consequences to us, and have a material adverse effect on our business, results of operations and financial condition.
We are subject to extensive regulation, which can be costly and time consuming and can subject us to unanticipated fines and delays.
Progenics and its products are subject to comprehensive regulation by the FDA and comparable authorities in other countries. These agencies and other entities regulate the pre-clinical and clinical testing, safety, effectiveness, approval, manufacture, labeling, marketing, export, storage, recordkeeping, advertising, promotion and other aspects of our products. If we violate regulatory requirements at any stage, whether before or after marketing approval is obtained, we may be subject to forced removal of a product from the market, product seizure, civil and criminal penalties and other adverse consequences. We cannot guarantee that approvals of proposed products, processes or facilities will be granted on a timely basis, or at all. If we experience delays or failures in obtaining approvals, commercialization of our product candidates will be slowed or stopped. Even if we obtain regulatory approval, the approval may include significant limitations on indicated uses for which the product could be marketed or other significant marketing restrictions. Under our License Agreement with Salix, we will be dependent on our new partner for compliance with these regulations as they apply to RELISTOR.
Our products may face regulatory, legal or commercial challenges even after approval.
Even if our products receive regulatory approval:
· They might not obtain labeling claims necessary to make the product commercially viable (in general, labeling claims define the medical conditions for which a drug product may be marketed, and are therefore very important to the commercial success of a product), or may be required to carry “black box” or other warnings that adversely affect their commercial success.
· Approval may be limited to uses of the product for treatment or prevention of diseases or conditions that are relatively less financially advantageous to us than approval of greater or different scope, or subject to an FDA-imposed Risk Evaluation and Mitigation Strategy (REMS) that limits the sources from and conditions under which they may be dispensed.
· We or our collaborators might be required to undertake post-marketing trials to verify the product’s efficacy or safety.
· We, our collaborators or others might identify side effects after the product is on the market.
· Efficacy or safety concerns regarding marketed products may lead to product recalls, withdrawals of marketing approval, reformulation of the product, additional pre-clinical testing or clinical trials, changes in labeling of the product, the need for additional marketing applications, declining sales or other adverse events. These potential consequences may occur whether or not the concerns originate from subsequent testing or other activities by us, governmental regulators, other entities or organizations or otherwise, and whether or not they are scientifically justified.
· We or our collaborators might experience manufacturing problems, which could have the same, similar or other consequences.
· We and our collaborators will be subject to ongoing FDA obligations and continuous regulatory review.
· If products lose previously received marketing and other approvals, our financial results would be adversely affected.
Competing products in development may adversely affect acceptance of our products.
As described in this Annual Report under Business – Competition, we are aware of a number of products and product candidates which compete or may potentially compete with RELISTOR. Any of these approved products or product candidates, or others which may be developed in the future, may achieve a significant competitive advantage relative to RELISTOR, and, in any event, the existing or future marketing and sales capabilities of these competitors may impair our ability to compete effectively in the market.
We are also aware of competitors described in that section who are developing alternative treatments for disease targets to which our research and development programs are directed, any of which – or others which may be developed in the future – may achieve a significant competitive advantage relative to any product we may develop.
Developing product candidates may require us to obtain additional financing. Our access to capital funding is uncertain.
We expect to continue to incur significant development expenditures for our product candidates, and do not have committed external sources of funding for most of these projects. These expenditures will be funded from cash on hand, or we may seek additional external funding for them, most likely through collaborative, license or royalty financing agreements with one or more pharmaceutical companies, securities issuances or government grants or contracts. We cannot predict when we will need additional funds, how much we will need, the form any financing may take (such as securities issuance or royalty or other financing), or whether additional funds will be available at all, especially in light of current conditions in global credit and financial markets. Our need for future funding will depend on numerous factors, such as the availability of new product development projects or other opportunities which we cannot predict, and many of which are outside our control. We cannot assure you that any currently-contemplated or future initiatives for funding our product candidate programs will be successful.
Our access to capital funding is always uncertain. Stresses in international markets are still affecting access to capital. We may not be able at the necessary time to obtain additional funding on acceptable terms, or at all. Our inability to raise additional capital on terms reasonably acceptable to us would seriously jeopardize our business.
If we raise funds by issuing and selling securities, it may be on terms that are not favorable to existing stockholders. If we raise funds by selling equity securities, current stockholders will be diluted, and new investors could have rights superior to existing stockholders. Raising funds by selling debt securities often entails significant restrictive covenants and repayment obligations.
If we are unable to negotiate collaborative agreements, our cash burn rate could increase and our rate of product development could decrease.
We may not be successful in negotiating additional collaborative arrangements with pharmaceutical and biotechnology companies to develop and commercialize product candidates and technologies. If we do not enter into new collaborative arrangements, we would have to devote more of our resources to clinical product development and product-launch activities, seeking additional sources of capital, and our cash burn rate would increase or we would need to take steps to reduce our rate of product development.
If testing does not yield successful results, our products will not be approved.
Regulatory approvals are necessary before product candidates can be marketed. To obtain them, we or our collaborators must demonstrate a product’s safety and efficacy through extensive pre-clinical and clinical testing. Numerous adverse events may arise during, or as a result of, the testing process, such as:
· results of pre-clinical studies being inconclusive or not indicative of results in human clinical trials;
· potential products not having the desired efficacy or having undesirable side effects or other characteristics that preclude marketing approval or limit their commercial use if approved;
· after reviewing test results, we or our collaborators may abandon projects which we previously believed to be promising; and
· we, our collaborators or regulators may suspend or terminate clinical trials if we or they believe that the participating subjects are being exposed to unacceptable health risks.
Clinical testing is very expensive and can take many years. Results attained in early human clinical trials may not be indicative of results in later clinical trials. In addition, many of our investigational or experimental drugs are at an early stage of development, and successful commercialization of early stage product candidates requires significant research, development, testing and approvals by regulators, and additional investment. Our products in the research or pre-clinical development stage may not yield results that would permit or justify clinical testing. Our failure to demonstrate adequately the safety and efficacy of a product under development would delay or prevent marketing approval, which could adversely affect our operating results and credibility.
Setbacks in clinical development programs could adversely affect us.
We and our collaborators continue to conduct clinical trials of RELISTOR. If the results of these or future trials are not satisfactory, we encounter problems enrolling subjects, clinical trial supply issues or other difficulties arise, or we experience setbacks in developing drug formulations, including raw material-supply, manufacturing or stability difficulties, our entire RELISTOR development program could be adversely affected, resulting in delays in trials or regulatory filings for further marketing approval. Conducting additional clinical trials or making significant revisions to our clinical development plan would lead to delays in regulatory filings. If clinical trials indicate a serious problem with the safety or efficacy of a RELISTOR product, we, Salix or Ono may stop development or commercialization of affected products. Since RELISTOR is our only approved product, any setback of these types could have a material adverse effect on our business, results of operations and financial condition.
Ono is conducting required clinical trials with Japanese patients to obtain regulatory approval of RELISTOR in Japan. There can be no assurance that these clinical trials will yield results adequate for that regulatory approval.
We are conducting clinical trials of various product candidates. If the results of these or future clinical studies of our candidates are not satisfactory, we would need to reconfigure our clinical trial programs to conduct additional trials or abandon the program involved. Because vaccine product candidates may be deemed to involve gene therapy, a relatively new technology that has not been extensively tested in humans, regulatory requirements applicable to them may be unclear, or subject to substantial regulatory review that delays the development and approval process generally.
Clinical trials often take longer than expected.
Projections that we publicly announce of commencement and duration of clinical trials may not be certain. For example, we have experienced clinical trial delays in the past as a result of slower than anticipated enrollment. These delays may recur. Delays can be caused by, among other things:
|
|
· deaths or other adverse medical events involving subjects in our clinical trials;
|
|
|
· regulatory or patent issues;
|
|
|
· interim or final results of ongoing clinical trials;
|
|
|
· failure to enroll clinical sites as expected;
|
|
|
· competition for enrollment from clinical trials conducted by others in similar indications;
|
|
|
· scheduling conflicts with participating clinicians and clinical institutions;
|
|
|
· disagreements, disputes or other matters arising from collaborations;
|
|
|
· our inability to obtain additional funding when needed; and
|
|
|
· manufacturing problems.
|
We have limited experience in conducting clinical trials, and we rely on others to conduct, supervise or monitor some or all aspects of some of our clinical trials. In addition, certain clinical trials for our product candidates may be conducted by government-sponsored agencies, and consequently will be dependent on governmental participation and funding. Under our License Agreement with Salix, Salix generally has responsibility for conducting RELISTOR clinical trials, including all trials outside of the United States other than Japan, where Ono has the responsibility for clinical trials. We have less control over the timing and other aspects of these clinical trials than if we conducted them entirely on our own.
Our product candidates may not obtain regulatory approvals needed for marketing.
None of our product candidates other than RELISTOR has been approved by applicable regulatory authorities for marketing. The process of obtaining FDA and foreign regulatory approvals often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. We have had only limited experience in filing and pursuing applications and other submissions necessary to gain marketing approvals. Products under development may never obtain marketing approval from the FDA or other regulatory authorities necessary for commercialization.
The commercial success of our products will depend upon their acceptance by the medical community and third party payors as clinically useful, cost effective and safe. If health care providers believe that patients can be managed adequately with alternative, currently available therapies, they may not prescribe our products, especially if the alternative therapies are viewed as more effective, as having a better safety or tolerability profile, as being more convenient to the patient or health care providers or as being less expensive. For pharmaceuticals administered in an institutional setting, the ability of the institution to be adequately reimbursed could also play a significant role in demand for our products. Even if our products obtain marketing approval, they may not achieve market acceptance. If any of our products do not achieve market acceptance, we will likely lose our entire investment in that product.
Marketplace acceptance depends in part on competition in our industry, which is intense.
The extent to which any of our products achieves market acceptance will depend on competitive factors. Competition in our industry is intense, and it is accentuated by the rapid pace of technological development. There are currently marketed products that will compete with the product candidates that we are developing. There are product candidates in pre-clinical or clinical development that target the side effects of opioid pain therapy, and a marketed product for the treatment of post-operative ileus could compete with RELISTOR. Many of our competitors have substantially greater research and development capabilities and experience and greater manufacturing, marketing, financial and managerial resources than we do. These competitors may develop products that are superior to those we are developing and render our products or technologies non-competitive or obsolete. If our product candidates receive marketing approval but cannot compete effectively in the marketplace, our operating results and financial position would suffer. Competition with respect to our technologies and products is based on, among other things, product efficacy, safety, reliability, method of administration, availability, price and clinical benefit relative to cost; timing and scope of regulatory approval; sales, marketing and manufacturing capabilities; collaborator capabilities; insurance and other reimbursement coverage; and patent protection. Competitive disadvantages in any of these factors could materially harm our business and financial condition.
If we are unable to obtain sufficient quantities of the raw and bulk materials needed to make our products, our product development and commercialization could be slowed or stopped.
Salix or Ono may not be able to fulfill manufacturing obligations for RELISTOR, either on their own or through third-party suppliers. Our existing arrangements with suppliers for our other product candidates may not result in the supply of sufficient quantities of our product candidates needed to accomplish our clinical development programs, and we may not have the right or capability to manufacture sufficient quantities of these products to meet our needs if our suppliers are unable or unwilling to do so. We currently obtain supplies of critical raw materials used in production of our product candidates from single sources. We do not have long-term contracts with any of these suppliers. Any delay or disruption in the availability of raw materials would slow or stop product development and commercialization of the relevant product. A delay or disruption of supplies of RELISTOR would have a material adverse effect on the RELISTOR franchise, and therefore on our business as a whole.
We have limited manufacturing capabilities, which could adversely affect our ability to commercialize products.
Under our License Agreement with Salix, Salix will be responsible for obtaining supplies of RELISTOR, including assuming relationships we have entered into in anticipation of establishing a new collaboration partnership or contracting with one or more other CMOs for supply of RELISTOR API and subcutaneous and oral finished drug product. These arrangements may not be on optimally-advantageous terms, and will subject us to risks that the counterparties may not perform optimally in terms of quality or reliability.
With respect to our other product candidates, our limited manufacturing capabilities may result in increased costs of production or delay product development or commercialization. In order to commercialize our product candidates successfully, we or our collaborators would need to be able to manufacture products in commercial quantities, in compliance with regulatory requirements, at acceptable costs and in a timely manner. Manufacture of our product candidates can be complex, difficult to accomplish even in small quantities, difficult to scale-up for large-scale production and subject to delays, inefficiencies and low yields of quality products. The cost of manufacturing some of our products may make them prohibitively expensive. If adequate supplies of any of our product candidates or related materials are not available to us on a timely basis or at all, our clinical trials could be seriously delayed, since these materials are time consuming to manufacture and cannot be readily obtained from third-party sources.
We operate pilot-scale manufacturing facilities for the production of vaccines and recombinant proteins. These facilities will not be sufficient for late-stage clinical trials for these types of product candidates or commercial-scale manufacturing. We may be required to expand further our manufacturing staff and facilities, obtain new facilities or contract with corporate collaborators or other third parties to assist with production.
In the event that we decide to establish a commercial-scale manufacturing facility for products that may be approved in the future, we will require substantial additional funds and will be required to hire and train significant numbers of employees and comply with applicable regulations, which are extensive. We may not be able to build a manufacturing facility that both meets regulatory requirements and is sufficient for our clinical trials or commercial scale manufacturing.
We have entered into arrangements with third parties for the manufacture of some of our product candidates and, in some cases, new means of administration for these product candidates. Our third-party sourcing strategy may not result in a cost-effective means for manufacturing products. In employing third-party manufacturers, we do not control many aspects of the manufacturing process, including compliance with the FDA’s current Good Manufacturing Practices and other regulatory requirements. We may not be able to obtain adequate supplies from third-party manufacturers in a timely fashion for development or commercialization purposes, and commercial quantities of products may not be available from contract manufacturers at acceptable costs.
We are dependent on our patents and other intellectual property rights. The validity, enforceability and commercial value of these rights are highly uncertain.
Our success is dependent in part upon obtaining, maintaining and enforcing patent and other intellectual property rights. The patent position of biotechnology and pharmaceutical firms is highly uncertain and involves many complex legal and technical issues. There is no clear policy involving the breadth of claims allowed, or the degree of protection afforded, under patents in this area. Accordingly, patent applications owned by or licensed to us may not result in patents being issued. We are aware of others who have patent applications or patents containing claims similar to or overlapping those in our patents and patent applications. We do not expect to know for several years the relative strength or scope of our patent position. Patents that we own or license may not enable us to preclude competitors from commercializing drugs, and consequently may not provide us with any meaningful competitive advantage.
We own or have licenses to a number of issued patents. The issuance of a patent, however, is not conclusive as to its validity or enforceability, which can be challenged in litigation. Our patents may be successfully challenged. We may incur substantial costs in litigation seeking to uphold the validity of patents or to prevent infringement. If the outcome of litigation is adverse to us, third parties may be able to use our patented invention without payment to us. Third parties may also avoid our patents through design innovation.
RELISTOR and most of our product candidates incorporate to some degree intellectual property licensed from third parties. We can lose the right to patents and other intellectual property licensed to us if the related license agreement is terminated due to a breach by us or otherwise. Our ability, and that of our collaboration partners, to commercialize products incorporating licensed intellectual property would be impaired if the related license agreements were terminated.
The license agreements from which we derive or out-license intellectual property provide for various royalty, milestone and other payment, commercialization, sublicensing, patent prosecution and enforcement, insurance, indemnification and other obligations and rights, and are subject to certain reservations of rights. While we generally have the right to defend and enforce patents licensed by us, either in the first instance or if the licensor chooses not to do so, we must usually bear the cost of doing so. Under our License Agreement, Salix generally has control over defense and enforcement of our RELISTOR patents. With respect to Japan, Ono has certain limited rights to prosecute, maintain and enforce relevant intellectual property. With most of our in-licenses, the licensor bears the cost of engaging in all of these activities, although we may share in those costs under specified circumstances.
We also rely on unpatented technology, trade secrets and confidential information. Third parties may independently develop substantially equivalent information and techniques or otherwise gain access to our technology or disclose our technology, and we may be unable to effectively protect our rights in unpatented technology, trade secrets and confidential information. We require each of our employees, consultants and advisors to execute a confidentiality agreement at the commencement of an employment or consulting relationship with us. These agreements may, however, not provide effective protection in the event of unauthorized use or disclosure of confidential information.
If we do not achieve milestones or satisfy conditions regarding some of our product candidates, we may not maintain our rights under related licenses.
We are required to make substantial cash payments, achieve milestones and satisfy other conditions, including filing for and obtaining marketing approvals and introducing products, to maintain rights under our intellectual property licenses. Due to the nature of these agreements and the uncertainties of research and development, we may not be able to achieve milestones or satisfy conditions to which we have contractually committed, and as a result may be unable to maintain our rights under these licenses. If we do not comply with our license agreements, the licensors may terminate them, which could result in our losing our rights to, and therefore being unable to commercialize, related products.
If we infringe third-party patent or other intellectual property rights, we may need to alter or terminate a product development program.
There may be patent or other intellectual property rights belonging to others that require us to alter our products, pay licensing fees or cease certain activities. If our products infringe patent or other intellectual property rights of others, the owners of those rights could bring legal actions against us claiming damages and seeking to enjoin manufacturing and marketing of the affected products. If these legal actions are successful, in addition to any potential liability for damages, we could be required to obtain a license in order to continue to manufacture or market the affected products. We may not prevail in any action brought against us, and any license required under any rights that we infringe may not be available on acceptable terms or at all. We are aware of intellectual property rights held by third parties that relate to products or technologies we are developing. For example, we are aware of other groups investigating methylnaltrexone and other peripheral opioid antagonists, PSMA or related compounds and monoclonal antibodies directed at targets relevant to PRO 140, and of patents held, and patent applications filed, by these groups in those areas. While the validity of these issued patents, patentability of these pending patent applications and applicability of any of them to our programs are uncertain, if asserted against us, any related patent or other intellectual property rights could adversely affect our ability to commercialize our products.
Research, development and commercialization of a biopharmaceutical often require choosing between alternative development and optimization routes at various stages in the development process. Preferred routes depend on subsequent discoveries and test results and cannot be predicted with certainty at the outset. There are numerous third-party patents in our field, and we may need to obtain a license under a patent in order to pursue the preferred development route of one or more of our products or product candidates. The need to obtain a license would decrease the ultimate profitability of the applicable product. If we cannot negotiate a license, we might have to pursue a less desirable development route or terminate the program altogether.
We are dependent upon third parties for a variety of functions. These arrangements may not provide us with the benefits we expect.
We rely in part on third parties to perform a variety of functions. We are party to numerous agreements which place substantial responsibility on clinical research organizations, consultants and other service providers for the development of our products. We also rely on medical and academic institutions to perform aspects of our clinical trials of product candidates. In addition, an element of our research and development strategy is to in-license technology and product candidates from academic and government institutions in order to minimize investments in early research. We have entered into agreements under which we have depended on Wyeth and Ono, respectively, for the commercialization and development of RELISTOR, and will be dependent primarily on Salix under our new License Agreement with it. We may not be able to maintain our relationships with Salix or Ono, or establish new ones on beneficial terms. We may not be able to enter new arrangements without undue delays or expenditures, and these arrangements may not allow us to compete successfully.
We lack sales and marketing infrastructure and related staff, which will require significant investment to establish and in the meantime may make us dependent on third parties for their expertise in this area.
We have no established sales, marketing or distribution infrastructure. If we receive marketing approval, significant investment, time and managerial resources will be required to build the commercial infrastructure required to market, sell and support a pharmaceutical product. Should we choose to commercialize any product directly, we may not be successful in developing an effective commercial infrastructure or in achieving sufficient market acceptance. Alternatively, we may choose to market and sell our products through distribution, co-marketing, co-promotion or licensing arrangements with third parties. We may also consider contracting with a third party professional pharmaceutical detailing and sales organization to perform the marketing function for our products. To the extent that we enter into distribution, co-marketing, co-promotion, detailing or licensing arrangements for the marketing and sale of our product candidates, any revenues we receive will depend primarily on the efforts of third parties. We will not control the amount and timing of marketing resources these third parties devote to our products.
We are exposed to product liability claims, and in the future may not be able to obtain insurance against claims at a reasonable cost or at all.
Our business exposes us to product liability risks, which are inherent in the testing, manufacturing, marketing and sale of pharmaceutical products. We may not be able to avoid product liability exposure. If a product liability claim is successfully brought against us, our financial position may be adversely affected. Pursuant to the Transition Agreement, we released Wyeth from responsibility for product liability exposure arising from its marketing and sales of RELISTOR, which Wyeth had borne under our 2005 collaboration.
Product liability insurance for the biopharmaceutical industry is generally expensive, when available at all. We maintain product liability insurance coverage in the amount of $10.0 million per occurrence, subject to a deductible and a $10.0 million annual aggregate limitation. Where local statutory requirements exceed the limits of our existing insurance or local policies of insurance are required, we maintain additional clinical trial liability insurance to meet these requirements. Our current insurance coverage may not be adequate to cover claims brought against us. Some of our license and other agreements require us to obtain product liability insurance. Adequate insurance coverage may not be available to us at a reasonable cost in the future.
We handle hazardous materials and must comply with environmental laws and regulations, which can be expensive and restrict how we do business. If we are involved in a hazardous waste spill or other accident, we could be liable for damages, penalties or other forms of censure.
Our research and development work and manufacturing processes involve the use of hazardous, controlled and radioactive materials. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these materials. Despite procedures that we implement for handling and disposing of these materials, we cannot eliminate the risk of accidental contamination or injury. In the event of a hazardous waste spill or other accident, we could be liable for damages, penalties or other forms of censure. We may be required to incur significant costs to comply with environmental laws and regulations in the future.
If we lose key management and scientific personnel on whom we depend, our business could suffer.
We are dependent upon our key management and scientific personnel. The loss of Mr. Baker, Dr. Maddon or other members of our senior management could require us to identify and engage qualified replacements, and could cause our management and operations to suffer in the interim. Competition for qualified employees among companies in the biopharmaceutical industry is intense. Future success in our industry depends in significant part on the ability to attract, retain and motivate highly skilled employees, which we may not be successful in doing.
If health care reform measures are enacted, our operating results and our ability to commercialize products could be adversely affected.
In recent years, there have been numerous proposals to change the health care system in the U.S. and in other jurisdictions. Some of these proposals have included measures that would change the nature of and regulatory requirements relating to drug discovery, clinical testing and regulatory approvals, limit or eliminate payments for medical procedures and treatments, or subject the pricing of pharmaceuticals to government control. Outside the U.S., and particularly in the E.U., the pricing of prescription pharmaceuticals is subject to governmental control. In addition, as a result of the trend towards managed health care in the U.S., as well as legislative proposals to reduce government insurance programs, third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drug products. Consequently, significant uncertainty exists as to the reimbursement status of newly approved health care products.
If we or any of our collaborators succeed in bringing one or more of our product candidates to market, third party payors may establish and maintain price levels insufficient for us to realize an appropriate return on our investment in product development. Significant changes in the health care system in the U.S. or elsewhere, including changes resulting from adverse trends in third-party reimbursement programs, could have a material adverse effect on our operating results and our ability to raise capital and commercialize products.
A substantial portion of our funding has come from federal government grants and research contracts. We cannot rely on these grants or contracts as a continuing source of funds.
A substantial portion of our revenues to date, albeit on a downward trend since 2006, has been derived from federal government grants and research contracts. During the last three years, we generated revenues from awards made to us by the NIH between 2004 and 2010, to partially fund some of our programs. We cannot rely on grants or additional contracts as a continuing source of funds. Funds available under these grants and/or contracts must be applied by us toward the research and development programs specified by the government rather than for all our programs generally. The government’s obligation to make payments under these grants and/or contracts is subject to appropriation by the U.S. Congress for funding in each year. It is possible that Congress or the government agencies that administer these government research programs will decide to scale back these programs or terminate them due to their own budgetary constraints. Additionally, these grants and research contracts are subject to adjustment based upon the results of periodic audits performed on behalf of the granting authority. Consequently, the government may not award grants or research contracts to us in the future, and any amounts that we derive from existing awards may be less than those received to date. In those circumstances, we would need to provide funding on our own, obtain other funding, or scale back the affected program. In particular, we cannot assure you that any currently-contemplated or future efforts to obtain funding for our product candidate programs through government grants or contracts will be successful, or that any such arrangements which we do conclude will supply us with sufficient funds to complete our development programs without providing additional funding on our own or obtaining other funding.
We have a history of operating losses, and we may never be profitable.
We have incurred substantial losses since our inception. We have derived no significant revenues from product sales or royalties. We may not achieve significant product sales or royalty revenue for a number of years, if ever. We expect to continue to incur operating losses in the future, which could increase significantly if we expand our clinical trial programs and other product development efforts. Our ability to achieve and sustain profitability is dependent in part on obtaining regulatory approval for and then commercializing our products, either alone or with others. We may not be able to develop and commercialize products beyond subcutaneous RELISTOR. Our operations may not be profitable even if any of our other products under development are commercialized.
Our stock price has a history of volatility. You should consider an investment in our stock as risky and invest only if you can withstand a significant loss.
Our stock price has a history of significant volatility. At times, our stock price has been volatile even in the absence of significant news or developments relating to us. The stock prices of biotechnology companies and the stock market generally have been subject to dramatic price swings in recent years, and financial and market conditions in the past three years have resulted in widespread pressures on securities of issuers throughout the world economy. Factors that may have a significant impact on the market price of our common stock include:
· the results of clinical trials and pre-clinical studies involving our products or those of our competitors;
· changes in the status of any of our drug development programs, including delays in clinical trials or program terminations;
· developments regarding our efforts to achieve marketing approval for our products;
· developments in our relationships with Salix, Ono, Wyeth and any other business partner(s) with which we may collaborate in the future regarding the development and commercialization of RELISTOR;
· developments in current or future relationships with other collaborative partners with respect to other products and candidates;
· announcements of technological innovations or new commercial products by us, our collaborators or our competitors;
· developments in patent or other proprietary rights;
· governmental regulation;
· changes in reimbursement policies or health care legislation;
· public concern as to the safety and efficacy of products developed by us, our collaborators or our competitors;
· our ability to fund ongoing operations;
· fluctuations in our operating results;
· the potential positive effect on share price of any purchases of common shares we may make in the future pursuant to the share repurchase program we announced in 2008, or downward pressure resulting from discontinuation of any such purchases; and
· general market conditions.
Our principal stockholders are able to exert significant influence over matters submitted to stockholders for approval.
At December 31, 2010, our directors and executive officers and stockholders affiliated with Tudor Investment Corporation together beneficially owned or controlled approximately one-fifth of our outstanding shares of common stock. At that date, our five largest stockholders, excluding our directors and executive officers and stockholders affiliated with Tudor, beneficially owned or controlled in the aggregate approximately two-fifths of our outstanding shares. Our directors and executive officers and Tudor-related stockholders, should they choose to act together, could exert significant influence in determining the outcome of corporate actions requiring stockholder approval and otherwise control our business. This control could have the effect of delaying or preventing a change in control of us and, consequently, could adversely affect the market price of our common stock. Other significant but unrelated stockholders could also exert influence in such matters.
Anti-takeover provisions may make the removal of our Board of Directors and/or management more difficult, and consequently, may discourage hostile bids for control of our company that may be beneficial to our stockholders.
Our Board of Directors is authorized, without further stockholder action, to issue from time to time shares of preferred stock in one or more designated series or classes. The issuance of preferred stock, as well as provisions in certain of our stock options that provide for acceleration of exercisability upon a change of control, and Section 203 and other provisions of the Delaware General Corporation Law could:
|
|
· make the takeover of Progenics or the removal of our Board of Directors or management more difficult;
|
|
· discourage hostile bids for control of Progenics in which stockholders may receive a premium for their shares of common stock; and
|
|
|
· otherwise dilute the rights of holders of our common stock and depress the market price of our common stock.
|
If there are substantial sales of our common stock, the market price of our common stock could decline.
Sales of substantial numbers of shares of common stock could cause a decline in the market price of our stock. We require substantial external funding to finance our research and development programs and may seek such funding through the issuance and sale of our common stock. In addition, some of our other stockholders are entitled to require us to register their shares of common stock for offer or sale to the public. We have filed Form S-8 registration statements registering shares issuable pursuant to our equity compensation plans and may periodically seek to increase the amount of securities available under these plans. Any sales by existing stockholders or holders of options, or other rights, may have an adverse effect on our ability to raise capital and may adversely affect the market price of our common stock.
Item 1B. Unresolved Staff Comments
There were no unresolved SEC staff comments regarding our periodic or current reports under the Exchange Act as of December 31, 2010.
Item 2. Properties
As of December 31, 2010, we occupy in total approximately 135,600 square feet of laboratory, manufacturing and office space on a single campus in Tarrytown, New York, under lease agreements expiring in June 2012 and December 2020.
In addition to rents due under these arrangements, we are obligated to pay additional facilities charges, including utilities, taxes and operating expenses.
Item 3. Legal Proceedings
We are not a party to any material legal proceedings.
Item 4. (Removed and Reserved)
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Price Range of Common Stock
Our common stock is quoted on The NASDAQ Stock Market LLC under the symbol PGNX. The following table sets forth, for the periods indicated, the high and low sales price per share of the common stock, as reported on NASDAQ. These prices reflect inter-dealer prices, without retail mark-up, markdown or commission and may not represent actual transactions.
|
High
|
Low
|
||||||||
|
2010:
|
Fourth quarter
|
$ | 5.69 | $ | 4.41 | ||||
|
Third quarter
|
5.72 | 4.00 | |||||||
|
Second quarter
|
7.00 | 4.25 | |||||||
|
First quarter
|
5.50 | 4.16 | |||||||
|
2009:
|
Fourth quarter
|
5.48 | 3.53 | ||||||
|
Third quarter
|
6.14 | 4.92 | |||||||
|
Second quarter
|
7.05 | 4.50 | |||||||
|
First quarter
|
10.81 | 5.08 | |||||||
On March 4, 2011, the last sale price for our common stock, as reported by The NASDAQ Stock Market LLC, was $5.80. There were approximately 245 holders of record of our common stock as of that date.
Comparative Stock Performance Graph
The graph below compares, for the past five years, the cumulative stockholder return on our common stock with the cumulative stockholder return of (i) the Nasdaq Stock Market (U.S.) Index and (ii) the Nasdaq Pharmaceutical Index, assuming an investment in each of $100 on December 31, 2005.
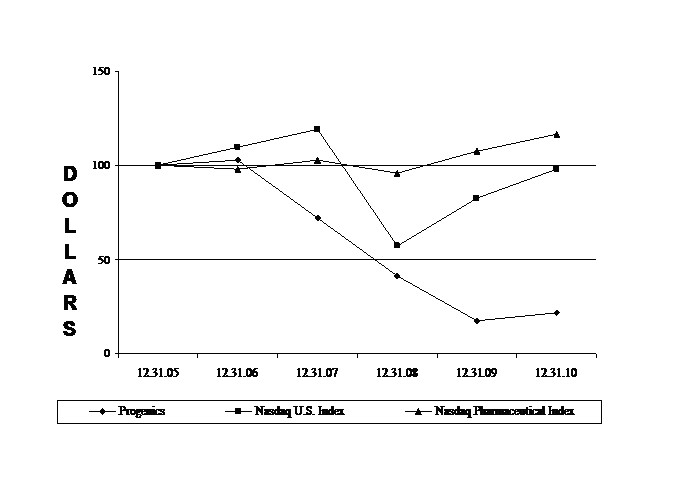
Dividends
We have not paid any dividends since the Company’s inception and currently anticipate that all earnings, if any, will be retained for development of our business and that no dividends on our common stock will be declared in the foreseeable future.
Item 6. Selected Financial Data
The selected financial data presented below as of December 31, 2010 and 2009 and for each of the three years in the period ended December 31, 2010 are derived from our audited financial statements, included elsewhere herein. The selected financial data presented below with respect to the balance sheet data as of December 31, 2008, 2007 and 2006 and for each of the two years in the period ended December 31, 2007 are derived from our audited financial statements not included herein. The data set forth below should be read in conjunction with Management’s Discussion and Analysis of Financial Condition and Results of Operations and the Financial Statements and related Notes included elsewhere herein.
|
Years Ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
(in thousands, except per share data)
|
||||||||||||||||||||
|
Statement of Operations Data:
|
||||||||||||||||||||
|
Revenues:
|
||||||||||||||||||||
|
Research and development
|
$ | 1,413 | $ | 44,351 | $ | 59,885 | $ | 65,455 | $ | 58,415 | ||||||||||
|
Royalty income
|
1,826 | 2,372 | 146 | - | - | |||||||||||||||
|
Research grants and contract
|
4,573 | 1,968 | 7,460 | 10,075 | 11,418 | |||||||||||||||
|
Other revenues
|
140 | 256 | 180 | 116 | 73 | |||||||||||||||
|
Total revenues
|
7,952 | 48,947 | 67,671 | 75,646 | 69,906 | |||||||||||||||
|
Expenses:
|
||||||||||||||||||||
|
Research and development
|
50,640 | 49,798 | 82,290 | 95,234 | 61,711 | |||||||||||||||
|
In-process research and development
|
- | - | - | - | 13,209 | |||||||||||||||
|
License fees – research and development
|
1,270 | 1,058 | 2,830 | 942 | 390 | |||||||||||||||
|
General and administrative
|
22,832 | 25,106 | 28,834 | 27,901 | 22,259 | |||||||||||||||
|
Royalty expense
|
241 | 237 | 15 | - | - | |||||||||||||||
|
Loss in joint venture
|
- | - | - | - | 121 | |||||||||||||||
|
Depreciation and amortization
|
2,853 | 5,078 | 4,609 | 3,027 | 1,535 | |||||||||||||||
|
Total expenses
|
77,836 | 81,277 | 118,578 | 127,104 | 99,225 | |||||||||||||||
|
Operating loss
|
(69,884 | ) | (32,330 | ) | (50,907 | ) | (51,458 | ) | (29,319 | ) | ||||||||||
|
Other income:
|
||||||||||||||||||||
|
Interest income
|
64 | 1,481 | 6,235 | 7,770 | 7,701 | |||||||||||||||
|
Gain on sale of marketable securities
|
- | 237 | - | - | - | |||||||||||||||
|
Total other income
|
64 | 1,718 | 6,235 | 7,770 | 7,701 | |||||||||||||||
|
Net loss before income taxes
|
(69,820 | ) | (30,612 | ) | (44,672 | ) | (43,688 | ) | (21,618 | ) | ||||||||||
|
Income tax benefit
|
95 | - | - | - | - | |||||||||||||||
|
Net loss
|
$ | (69,725 | ) | $ | (30,612 | ) | $ | (44,672 | ) | $ | (43,688 | ) | $ | (21,618 | ) | |||||
|
Per share amounts on net loss:
|
||||||||||||||||||||
|
Basic and diluted
|
$ | (2.14 | ) | $ | (0.98 | ) | $ | (1.48 | ) | $ | (1.59 | ) | $ | (0.84 | ) | |||||
|
December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
(in thousands)
|
||||||||||||||||||||
|
Balance Sheet Data:
|
||||||||||||||||||||
|
Cash and cash equivalents
|
$ | 47,918 | $ | 90,903 | $ | 56,186 | $ | 10,423 | $ | 11,947 | ||||||||||
|
Marketable and auction rate securities
|
3,608 | 5,293 | 85,188 | 159,947 | 137,153 | |||||||||||||||
|
Working capital
|
42,207 | 95,388 | 85,983 | 102,979 | 91,827 | |||||||||||||||
|
Total assets
|
62,738 | 113,613 | 157,833 | 189,539 | 165,911 | |||||||||||||||
|
Deferred revenue, long-term
|
- | - | - | 9,131 | 16,101 | |||||||||||||||
|
Other liabilities, long-term
|
1,635 | - | 266 | 359 | 123 | |||||||||||||||
|
Total stockholders’ equity
|
51,308 | 107,607 | 119,369 | 147,499 | 110,846 | |||||||||||||||
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations (MD&A)
Overview
General. We are a biopharmaceutical company focusing on the development and commercialization of innovative therapeutic products to treat the unmet medical needs of patients with debilitating conditions and life-threatening diseases. Our principal programs are directed toward gastroenterology, oncology and virology. We commenced principal operations in 1988, became publicly traded in 1997 and throughout have been engaged primarily in research and development efforts, developing manufacturing capabilities, establishing corporate collaborations and related activities.
On February 3, 2011, we entered into an exclusive License Agreement with Salix by which Salix acquired the rights to RELISTOR worldwide except in Japan, where we have previously licensed to Ono Pharmaceutical the subcutaneous formulation of the drug. We received a $60.0 million upfront payment in cash and are eligible to receive development milestone payments of up to $90.0 million contingent upon the achievement of specified U.S. regulatory approvals and commercialization milestone payments of up to $200.0 million contingent upon the achievement of specified U.S. sales targets. Salix must pay us royalties based on a percentage ranging from 15 to 19 percent of net sales by it and its affiliates, and 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) it receives from sublicensees in respect of any country outside the U.S., as sublicensees commence their commercialization efforts. Salix is responsible for further developing and commercializing subcutaneous RELISTOR worldwide except in Japan, including completing clinical development necessary to support regulatory marketing approvals for potential new indications and formulations, and marketing and selling the product.
Our sources of revenues for the three years ended December 31, 2010 have been payments under our collaboration agreements and funds from research grants and contracts from the NIH related to our oncology and virology programs. In June 2008, we began recognizing royalty income from net sales by Wyeth of subcutaneous RELISTOR (methylnaltrexone bromide). To date, our product sales have consisted solely of limited revenues from the sale of research reagents and we expect that those sales will not significantly increase over current levels in the near future.
A majority of our expenditures to date have been for research and development activities. During 2010, expenses for our RELISTOR research program were $23.3 million compared to $7.8 million in 2009 and $25.4 million in 2008. Expenses for our cancer and HIV research programs were $14.7 million and $5.5 million, respectively, during 2010 compared to (i) $20.1 million and $11.8 million, respectively in 2009 and (ii) $10.8 million and $39.4 million, respectively, in 2008. Our expenses and reimbursement revenue related to RELISTOR in the future will depend on the amount of research and development work we perform pursuant to the development plan contemplated by the Salix License Agreement, which is to be finalized early in the second quarter of 2011. We also expect to incur a significant amount of development expenses for our other programs as these programs progress. During each year of the three-year period ended December 31, 2010, less than 16 percent of our non-RELISTOR expenses were reimbursed through government funding.
From January 2006 to October 2009, we recognized revenues from Wyeth for (i) reimbursement of our development expenses for RELISTOR as incurred, (ii) in respect of amortization of the $60.0 million upfront payment we received over the period of our development obligations, (iii) for milestones achieved during the collaboration and for royalties earned based on net sales of RELISTOR. Other than potential revenues from the RELISTOR franchise, we do not anticipate generating significant recurring revenues, from royalties, product sales or otherwise, in the near term.
We will require additional funding to continue our current programs to completion, which may involve collaboration agreements, licenses or sale transactions or royalty sales or financings with respect to our products and product candidates. We may also seek to raise additional capital through sales of common stock or other securities, and expect to continue funding some programs in part through government awards. Funding may not, however, be available to us on acceptable terms or at all. We continue to monitor our program expenditures, including headcount levels, in conjunction with program and program candidates that we choose or are obligated to undertake. We expect to continue to incur operating losses during the near term.
At December 31, 2010, we held $47.9 million in cash and cash equivalents, a decrease of $43.0 million from $90.9 million at December 31, 2009. We expect that this amount, together with the $60.0 million upfront cash payment received from Salix, will be sufficient to fund operations at current levels beyond one year. If, however, we are unable to conclude favorable collaboration, license, asset sale, capital raising or other financing transactions, we will have to reduce, delay or eliminate spending on some current operations, and/or reduce salary and other overhead expenses, to extend our remaining operations.
Gastroenterology. Our first commercial product is RELISTOR® (methylnaltrexone bromide) subcutaneous injection, a first-in-class therapy for opioid-induced constipation approved for sale in over 50 countries worldwide, including the U.S., E.U., Canada and Australia. Marketing applications are pending elsewhere throughout the world.
RELISTOR has previously been developed and commercialized worldwide except Japan by Progenics and Wyeth pursuant to a 2005 collaboration agreement that was terminated in October 2009. Under our Transition Agreement with Wyeth, Wyeth is continuing to distribute RELISTOR worldwide other than Japan through March 31, 2011. No royalties are payable in respect of net sales after September 30, 2010. The parties plan an April 1, 2011 transition of U.S. commercial responsibility to Salix from Wyeth, and are currently discussing transition of ex-U.S. commercialization responsibilities on a country-by-country basis. While Salix effects a country-by-country transition of ex-U.S. commercialization rights, Wyeth continues to supply product.
Under the Transition Agreement, Wyeth paid us $10.0 million in six quarterly installments through January 2011, and continued to pay royalties on 2010 ex-U.S. sales as provided in the 2005 collaboration agreement except to the extent certain fourth quarter financial targets were not met. These targets were not met during the fourth quarter and royalties on ex-U.S. sales were not payable to us. No other royalties are payable in respect of net sales after September 30, 2010. Wyeth is also providing financial resources of approximately $9.5 million, of which we have recognized $1.2 million, which constitutes reimbursement for development of a multi-dose pen for subcutaneous RELISTOR. Revenue from this financial support for which we perform research and development is reported as reimbursement revenue from Wyeth under the Transition Agreement. We have agreed to purchase Wyeth’s remaining inventory of subcutaneous RELISTOR at the end of the Sales Periods on agreed-upon terms and conditions, and Salix has agreed to purchase our inventory of subcutaneous RELISTOR on similar agreed-upon terms and conditions. We have no further obligations to Wyeth under the 2005 collaboration agreement.
Our October 2008 out-license to Ono Pharmaceutical of the rights to subcutaneous RELISTOR in Japan is unaffected by the Salix License Agreement and termination of the Wyeth collaboration. In June 2009, Ono began clinical testing in Japan of RELISTOR subcutaneous injection and in 2010 initiated a phase 2 trial designed to demonstrate efficacy and safety.
We have received U.S., E.U. and Canadian approvals to market RELISTOR in pre-filled syringes, which are designed to ease preparation and administration for patients and caregivers, and currently plan to coordinate the launch of that product with Salix in 2011.
We are also developing subcutaneous RELISTOR for treatment of OIC outside the advanced-illness setting, in individuals with non-cancer pain. Based on the results from a recently completed one-year, open-label safety study, together with results from a previous phase 3 efficacy trial, we plan to submit regulatory filings in the first half of 2011 in the U.S., E.U. and elsewhere for approval of subcutaneous RELISTOR to treat OIC in the non-cancer pain setting.
As part of our reacquisition of RELISTOR, we assumed development responsibilities for an oral formulation of RELISTOR for the treatment of OIC in patients with non-cancer pain and are conducting a phase 3 trial of oral methylnaltrexone in this patient population, which pursuant to our License Agreement will continue as part of Salix’s development responsibilities for RELISTOR pursuant to the development plan contemplated by the Salix License Agreement, which is to be finalized early in the second quarter of 2011. Under the License Agreement, expenses we incur for development or other work that we perform at Salix’s direction is reimbursable to us by Salix.
Our 2005 collaboration agreement with Wyeth was terminated by the October 2009 Transition Agreement, but the 2005 agreement remained in effect for the time periods prior to termination covered by this report. Prior to the Transition Agreement, we received upfront, milestone and royalty payments from Wyeth, and were reimbursed for expenses we incurred in connection with the development of RELISTOR; manufacturing and commercialization expenses for RELISTOR were funded by Wyeth. At inception of the Wyeth collaboration, Wyeth paid to us a $60.0 million non-refundable upfront payment. Wyeth made $39.0 million in milestone payments thereafter. Costs for the development of RELISTOR incurred by Wyeth or us starting January 1, 2006 through termination of the 2005 collaboration agreement were paid by Wyeth. We were reimbursed by Wyeth for our development costs based on the number of our full-time equivalent employees (FTEs) devoted to the development project, all subject to Wyeth’s audit rights and possible reconciliation. During the applicable royalty periods, Wyeth was obligated to pay us royalties on net sales, as defined (which included specified sales deductions), of RELISTOR by Wyeth throughout the world other than Japan, where we licensed rights to subcutaneous RELISTOR to Ono. Wyeth’s ex-U.S. royalty obligations continued through the end of 2010 as provided in the Transition Agreement. No royalties are payable in respect of net sales after September 30, 2010.
Under our License Agreement with Ono, in October 2008 we out-licensed rights to subcutaneous RELISTOR in Japan in return for an upfront payment of $15.0 million and the right to receive potential milestones, upon achievement of development responsibilities by Ono, of up to $20.0 million, commercial milestones and royalties on sales by Ono of subcutaneous RELISTOR in Japan. Ono also has the option to acquire from us the rights to develop and commercialize in Japan other formulations of RELISTOR on terms to be negotiated separately. Ono may request us to perform activities related to its development and commercialization responsibilities beyond our participation in joint committees and specified technology transfer related tasks which will be at its expense, and reimbursable at the time we perform these services.
Royalty and milestone payments will depend on success in development and commercialization of RELISTOR, which is dependent on many factors, such as the actions of Salix, Ono and Wyeth (until completion of its responsibilities during the transition) and any other business partner(s) with which we may collaborate, decisions by the FDA and other regulatory bodies, the outcome of clinical and other testing of RELISTOR, and our own efforts. Many of these matters are outside our control. In particular, we cannot guarantee that Salix will be successful in furthering the development and commercialization of the RELISTOR franchise. We also cannot guarantee, in light of Wyeth’s limited obligations under the Transition Agreement, its acquisition by Pfizer and its limited ongoing commercial interest in the RELISTOR franchise, that Wyeth’s efforts during the transition will achieve any particular level of success in marketing and sales, regulatory approval or clinical development of subcutaneous RELISTOR.
In our other gastroenterology efforts, we have recently presented preclinical data on novel monoclonal antibodies against toxins produced by the bacterium Clostridium difficile (C. difficile), the leading cause of hospital-acquired diarrhea in the U.S. and a recognized growing global public health challenge.
Oncology and Virology. We recently announced preliminary data from a phase 1 clinical trial of a fully human monoclonal ADC directed against PSMA for the treatment of prostate cancer and recently presented data from preclinical studies of novel multiplex phosphoinositide 3-kinase (PI3K) inhibitors for the treatment of cancer. In the area of virology, we have been developing a viral-entry inhibitor -- a humanized monoclonal antibody, PRO 140 -- for HIV, the virus that causes AIDS, and are conducting a clinical trial of PRO 140 with outside funding. We are also evaluating hepatitis C virus entry inhibitors as possible development candidates. Advancement of PRO 140, including clinical trial efforts, is subject to obtaining additional outside funding, for which we have applied to government agencies.
Results of Operations (amounts in thousands unless otherwise noted)
Revenues:
Our sources of revenue during the years ended December 31, 2010, 2009 and 2008, included our 2005 collaboration with Wyeth, which was effective from January 2006 to October 2009, our License Agreement with Ono, our research grants and contract from the NIH and, to a small extent, our sale of research reagents. In June 2008, we began recognizing royalty income from net sales by Wyeth of subcutaneous RELISTOR.
|
Sources of Revenue
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent Change
|
||||||||||
|
Research and development
|
$ 1,413
|
$ 44,351
|
$ 59,885
|
(97%)
|
(26%)
|
|||||
|
Royalty income
|
1,826
|
2,372
|
146
|
(23%)
|
1,525%
|
|||||
|
Research grants and contract
|
4,573
|
1,968
|
7,460
|
132%
|
(74%)
|
|||||
|
Other revenues
|
140
|
256
|
180
|
(45%)
|
42%
|
|||||
|
$ 7,952
|
$ 48,947
|
$ 67,671
|
(84%)
|
(28%)
|
||||||
Research and development revenue:
Wyeth Collaboration. During the years ended December 31, 2010 and 2009, we recognized $1,383 and $29,298, respectively, of revenue from Wyeth, consisting of (i) $0 and $14,562, respectively, of the $60,000 upfront payment we received upon entering into our 2005 collaboration, (ii) $0 and $4,736, respectively, as reimbursement of our development expenses, and (iii) $1,383 and $10,000, respectively, under the Transition Agreement.
During the years ended December 31, 2009 and 2008, we recognized $29,298 and $59,885, respectively, of revenue from Wyeth, consisting of (i) $14,562 and $10,228, respectively, of the $60,000 upfront payment we received upon entering into our 2005 collaboration, (ii) $4,736 and $24,657, respectively, as reimbursement of our development expenses, and (iii) $10,000 and $25,000, respectively, under the Transition Agreement and non-refundable milestone payments. We analyzed the facts and circumstances of the non-refundable milestones and believe that they met those criteria for revenue recognition upon achievement of the respective milestones. See Critical Accounting Policies – Revenue Recognition.
From the inception of the Wyeth Collaboration through December 31, 2009, we recognized revenue for the entire $60,000 upfront payment, $104,054 as reimbursement for our development costs, and a total of $39,000 for non-refundable milestone payments. We do not expect to receive additional reimbursement revenue under the 2005 collaboration due to its termination. Wyeth, at its expense, is continuing certain ongoing development efforts for subcutaneous RELISTOR. This support is ongoing and continues until completion of its responsibilities during the transition.
We recognize a portion of the upfront payment in a reporting period in accordance with the proportionate performance method, which is based on the percentage of actual effort performed on our development obligations in that period relative to total effort expected for all of our performance obligations under the arrangement and budget approved by Wyeth and us. During the third quarter of 2007, a revised budget was approved, which extended our performance period to the end of 2009 and, thereby, decreased the amount of revenue we recognized in each reporting period. The Transition Agreement shortened the obligation period from the end of 2009 to October 2009 and we recognized the remaining $5.2 million of unamortized upfront payment as revenue during the fourth quarter of 2009.
Ono License Agreement. In October 2008, we entered into a License Agreement with Ono and in November 2008, received an upfront payment of $15,000 and recorded this amount as deferred revenue – current as of December 31, 2008. We are entitled to receive potential milestones and royalty payments. During the years ended December 31, 2010 and 2009, we recognized $30 and $53, respectively, of reimbursement revenue for activities requested by Ono and in 2009 recognized the $15,000 upfront payment as revenue, due to satisfying our performance obligations.
Royalty income. We began earning royalties from net sales by Wyeth of subcutaneous RELISTOR in June 2008.Our royalties from net sales by Wyeth of RELISTOR, as defined, were based on royalty rates under our 2005 collaboration.
During the years ended December 31, 2010 and 2009, we earned royalties of $1,826 and $1,853, respectively, based on the net sales of RELISTOR and we recognized $1,826 and $2,372, respectively, of royalty income. During the fourth quarter of 2010, no royalties were payable to us.
During the years ended December 31, 2009 and 2008, we earned royalties of $1,853 and $665, respectively, based on the net sales of RELISTOR and we recognized $2,372 and $146, respectively, of royalty income. The remaining deferred royalty revenue balance of $807, as of September 30, 2009, was recognized as royalty income during the fourth quarter of 2009, the period in which our development obligations under the 2005 collaboration agreement terminated.
Global net sales of RELISTOR were $16.1 million for the year ended December 31, 2010, comprised of $9.5 million of U.S. and $6.6 million of ex-U.S. net sales. Global net sales of RELISTOR were $12.3 million for the year ended December 31, 2009, comprised of $7.1 million of U.S. net sales and $5.2 million of ex-U.S. net sales. Global net sales of RELISTOR were $4.4 million for the year ended December 31, 2008, with U.S. and ex-U.S. net sales constituting $2.8 million and $1.6 million, respectively.
Research grants and contract. In September 2010, we were awarded a three-year NIH grant totaling $4.1 million to partially fund research and pre-clinical development of our humanized monoclonal antibodies against the disease-causing toxins produced by C. difficile. In June 2009, we were awarded a five-year NIH grant, subject to annual funding approvals and customary compliance obligations, totaling up to $14.5 million to continue the development of a prophylactic vaccine (ProVax) designed to prevent HIV from becoming established in uninfected individuals exposed to the virus. Prior to this award, the program was funded by a 2003 NIH contract which expired in 2008, and through that date, we recognized revenue of $15.5 million. In November 2010, we received $733 as part of the U.S. Government’s Qualifying Therapeutic Discovery Project, which provides grants or tax credits for expenses we incurred in 2009 and 2010 on research aimed at creating new therapies, reducing long-term healthcare costs, and/or significantly advancing the goal of curing cancer within the next 30 years.
During the years ended December 31, 2010 and 2009, we recognized $4,573 and $1,968, respectively, as revenue from federal government grants and grants in lieu of tax credits, consisting of awards made to us by the NIH and Internal Revenue Service between 2008 and 2010, to partially fund our ProVax HIV vaccine, PRO 140, PSMA, C. difficile and HCV programs. The increase in grant revenue resulted from new grants awarded in June 2009 and September 2010, as described above, and the $733 received as part of the federal government’s therapeutic project.
Revenues from research grants and contract from the NIH decreased to $1,968 for the year ended December 31, 2009 from $7,460 for the year ended December 31, 2008; $1,968 and $5,251 from grants and $0 and $2,209 from the NIH Contract for the years ended December 31, 2009 and 2008, respectively. The decrease in grant and contract revenue resulted from fewer active grants and reimbursable expenses in 2009 than in 2008, and the expiration of the NIH Contract in December 2008.
Other revenues, primarily from orders for research reagents, decreased to $140 for the year ended December 31, 2010 from $256 for the year ended December 31, 2009 and from $180 for the year ended December 31, 2008.
Expenses:
Research and Development Expenses include scientific labor, clinical trial costs, supplies, product manufacturing costs, consulting, license fees, royalty payments and other operating expenses. Research and development expenses increased to $52,151 for the year ended December 31, 2010 from $51,093 for the year ended December 31, 2009, and decreased from $85,135 for the year ended December 31, 2008. During 2010, the increase in research and development expenses over those in 2009 was primarily due to higher (i) consultants’ expenses for subcutaneous RELISTOR, (ii) oral methylnaltrexone clinical trial expenses and (iii) subcutaneous RELISTOR contract manufacturing costs for the multi-dose pen, partially offset by reduced manufacturing, laboratory and product testing costs for PSMA ADC and PRO 140 and lower compensation expenses. See Liquidity and Capital Resources – Uses of Cash, for details of the changes in these expenses by project. From 2006 through October 2009, Wyeth reimbursed us for development expenses we incurred related to RELISTOR under the development plan agreed to between Wyeth and us. Portions of our expenses related to our HIV, HCV and PSMA programs are funded through grants and a contract from the NIH (see Revenues- Research Grants and Contract). The changes in research and development expense, by category of expense, are as follows:
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Salaries and benefits
|
$18,469
|
$21,576
|
$24,383
|
(14%)
|
(12%)
|
||||
2010 vs. 2009 Salaries and benefits decreased due to a decline in average headcount to 138 from 175 for the years ended December 31, 2010 and 2009, respectively, in the research and development, manufacturing and clinical departments.
2009 vs. 2008 Salaries and benefits decreased due to a decline in average headcount to 175 from 196 for the years ended December 31, 2009 and 2008, respectively, in the research and development, manufacturing and clinical departments as part of our efforts to manage costs.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
||||||
|
Percent change
|
||||||||||
|
Share-based compensation
|
$5,091
|
$7,225
|
$7,241
|
(30%)
|
0%
|
|||||
2010 vs. 2009 Share-based compensation decreased for the year ended December 31, 2010 compared to the year ended December 31, 2009 due to lower stock option plan, restricted stock and employee stock purchase plan expenses.
2009 vs. 2008 Share-based compensation decreased for the year ended December 31, 2009 compared to the year ended December 31, 2008 due to lower employee stock purchase plan expense, partially offset by higher restricted stock and stock option plan expenses.
See Critical Accounting Policies − Share-Based Payment Arrangements.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Clinical trial costs
|
$7,056
|
$2,198
|
$14,127
|
221%
|
(84%)
|
||||
2010 vs. 2009 Clinical trial costs increased primarily due to higher expenses for (i) RELISTOR ($5,224), from increased clinical trial activities for oral methylnaltrexone phase 3 study and (ii) Cancer ($352), partially offset by a decrease in expenses for HIV ($718), due to a decline in PRO 140 clinical trial activities, all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Clinical trial costs decreased primarily due to lower expenses for (i) RELISTOR ($9,768), from reduced clinical trial activities, and (ii) HIV ($2,821), due to decreased PRO 140 clinical trial activities and Other ($5), partially offset by an increase in expenses for Cancer ($665), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Laboratory and manufacturing supplies
|
$2,388
|
$3,011
|
$3,944
|
(21%)
|
(24%)
|
||||
2010 vs. 2009 Laboratory and manufacturing supplies decreased due to lower expenses for (i) Cancer ($485), due to reduced expenses for PSMA ADC, (ii) HIV ($136), resulting from a decline in the purchases of manufacturing supplies and (iii) Other ($631), partially offset by an increase in RELISTOR ($629), due to higher expenses for multi-dose pen, all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Laboratory and manufacturing supplies decreased due to lower expenses for HIV ($1,841), resulting from a decline in the purchases of manufacturing supplies, partially offset by an increase in (i) Cancer ($842), due to higher expenses for PSMA ADC, (ii) Other projects ($62) and (iii) RELISTOR ($4), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Contract manufacturing and subcontractors
|
$6,853
|
$8,040
|
$21,681
|
(15%)
|
(63%)
|
||||
2010 vs. 2009 Contract manufacturing and subcontractors decreased due to lower (i) Cancer expenses ($2,287), resulting from a decline in manufacturing expenses for PSMA ADC, (ii) Other ($962) and (iii) HIV expenses ($362), resulting from a decline in manufacturing expenses for PRO 140, partially offset by an increase in RELISTOR expenses ($2,424), due to higher contract manufacturing expenses for multi-dose pen, all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Contract manufacturing and subcontractors decreased due to lower (i) HIV expenses ($13,514), resulting from a decline in manufacturing expenses for PRO 140 and (ii) RELISTOR expenses ($1,439), partially offset by increases in both Cancer ($956), due to higher contract manufacturing expenses for PSMA ADC, and Other ($356), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
These expenses are related to the conduct of clinical trials, including manufacture by third parties of drug materials, testing, analysis, formulation and toxicology services, and vary as the timing and level of such services are required.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Consultants
|
$3,310
|
$1,007
|
$3,514
|
229%
|
(71%)
|
||||
2010 vs. 2009 Consultants expenses increased due to higher expenses for RELISTOR ($2,478) and Cancer ($41), partially offset by decreases in consultants expenses for HIV ($160) and Other projects ($56), all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Consultants expenses decreased due to lower expenses for (i) RELISTOR ($1,494), (ii) Cancer ($305), (iii) HIV ($524) and (iv) Other projects ($184), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
These expenses are related to the monitoring of clinical trials as well as the analysis of data from completed clinical trials and vary as the timing and level of such services are required.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
License fees
|
$1,270
|
$1,058
|
$2,830
|
20%
|
(63%)
|
||||
2010 vs. 2009 License fees increased primarily due to higher expenses for HIV ($428), partially offset by lower expenses for Cancer ($149) and RELISTOR ($67), all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 License fees decreased primarily due to a decline in expenses for HIV ($774), Cancer ($516) and RELISTOR ($482), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Royalty expense
|
$241
|
$237
|
$15
|
2%
|
1,480%
|
||||
2010 vs. 2009 We incurred $241 and $185, respectively, of royalty costs and recognized $241 and $237, respectively, of royalty expenses during the years ended December 31, 2010 and 2009.
2009 vs. 2008 We incurred $185 and $67, respectively, of royalty costs and recognized $237 and $15, respectively, of royalty expenses during the years ended December 31, 2009 and 2008. The remaining deferred royalty charges balance of $81, as of September 30, 2009, was recognized as royalty expense during the fourth quarter 2009, the period in which our development obligations relating to RELISTOR terminated.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Other operating expenses
|
$7,473
|
$6,741
|
$7,400
|
11%
|
(9%)
|
||||
2010 vs. 2009 Other operating expenses increased for the year ended December 31, 2010 compared to the year ended December 31, 2009, primarily due to higher expenses for rent ($1,015) and insurance ($6), partially offset by a decrease in facilities ($193), travel ($5) and other operating expenses ($91).
2009 vs. 2008 Other operating expenses decreased for the year ended December 31, 2009 compared to the year ended December 31, 2008, primarily due to a decrease in rent ($504), travel ($195), insurance ($82), facilities ($23) and other operating expenses ($223), partially offset by an increase in computer expenses ($368).
General and Administrative Expenses include administrative labor, consulting and professional fees and other operating expenses. General and administrative expenses decreased to $22,832 for the year ended December 31, 2010 from $25,106 for the year ended December 31, 2009 and from $28,834 for the year ended December 31, 2008, as follows:
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Salaries and benefits
|
$8,086
|
$8,257
|
$8,610
|
(2%)
|
(4%)
|
||||
2010 vs. 2009 Salaries and benefits decreased for the year ended December 31, 2010 compared to the same period in 2009, due to a decline in average headcount to 39 from 49, in the general and administrative departments, partially offset by higher bonus expense.
2009 vs. 2008 Salaries and benefits decreased due to lower bonus expense for the year ended December 31, 2009 compared to the same period in 2008, and a decrease in average headcount to 49 from 52, in the general and administrative departments as part of our efforts to manage costs.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Share-based compensation
|
$4,424
|
$5,761
|
$6,892
|
(23%)
|
(16%)
|
||||
2010 vs. 2009 Share-based compensation decreased due to lower restricted stock, stock option and employee stock purchase plans expenses for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Share-based compensation decreased due to decrease in stock option and employee stock purchase plans expenses, partially offset by an increase in restricted stock expenses for the year ended December 31, 2009 compared to the year ended December 31, 2008.
See Critical Accounting Policies −Share-Based Payment Arrangements.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Consulting and professional fees
|
$5,843
|
$6,696
|
$7,915
|
(13%)
|
(15%)
|
||||
2010 vs. 2009 Consulting and professional fees decreased due to lower patent fees ($785), audit and compliance fees ($176), legal fees ($33) and other ($21), which were partially offset by an increase in consulting fees ($147) and public relations fees ($15), all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Consulting and professional fees decreased due to a decrease in consultant fees ($790), patent fees ($540) and public relations ($156), which were partially offset by an increase in audit and compliance fees ($185), legal fees ($64) and other ($18), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Other operating expenses
|
$4,479
|
$4,392
|
$5,417
|
2%
|
(19%)
|
||||
2010 vs. 2009 Other operating expenses increased due to higher expenses for rent ($341) and recruiting ($106), partially offset by a decrease in investor relations ($79), taxes ($35), conferences and seminars ($27), travel ($23) and other operating expenses ($196), all for the year ended December 31, 2010 compared to the year ended December 31, 2009.
2009 vs. 2008 Other operating expenses decreased due to lower spending on recruiting ($234), computer software ($280), travel ($140), taxes ($12), rent ($167), conferences and seminars ($54) and other operating expenses ($195), partially offset by an increase in investor relations ($57), all for the year ended December 31, 2009 compared to the year ended December 31, 2008.
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Depreciation and amortization
|
$2,853
|
$5,078
|
$4,609
|
(44%)
|
10%
|
||||
2010 vs. 2009 Depreciation and amortization expense decreased to $2,853 for the year ended December 31, 2010 from $5,078 for the year ended December 31, 2009, primarily due to lower capital expenditures in 2009.
2009 vs. 2008 Depreciation and amortization expense increased to $5,078 for the year ended December 31, 2009 from $4,609 for the year ended December 31, 2008, due to fixed asset purchases in 2008 and leasehold improvements placed in service during 2007.
Other income:
|
2010
|
2009
|
2008
|
2010 vs. 2009
|
2009 vs. 2008
|
|||||
|
Percent change
|
|||||||||
|
Interest income
|
$64
|
$1,481
|
$6,235
|
(96%)
|
(76%)
|
||||
2010 vs. 2009 Interest income decreased to $64 for the year ended December 31, 2010 from $1,481 for the year ended December 31, 2009. For the years ended December 31, 2010 and 2009, investment income decreased to $65 from $2,075, respectively, due to lower interest rates for cash equivalents, lower average balance of cash equivalents and marketable securities in 2010 than in 2009. Amortization of premiums, net of discounts, was ($1) and ($594) for years ended December 31, 2010 and 2009, respectively.
2009 vs. 2008 Interest income decreased to $1,481 for the year ended December 31, 2009 from $6,235 for the year ended December 31, 2008. For the years ended December 31, 2009 and 2008, investment income decreased to $2,075 from $7,195, respectively, due to a decrease in interest rates, lower average balances of cash equivalents and corporate debt securities and higher average balances of money market funds in 2009 than in 2008. Amortization of premiums, net of discounts, was ($594) and ($960) for years ended December 31, 2009 and 2008, respectively.
Interest income, as reported, is primarily the result of investment income from our marketable and auction rate securities, decreased by the amortization of premiums we paid or increased by the amortization of discounts we received for those securities. Other income also includes $237 of gains from the sale of marketable securities in 2009.
Income Taxes:
For the years ended December 31, 2010, 2009 and 2008, we had losses both for book and tax purposes. We received a federal tax refund of $95 in 2010 from new legislation permitting the carryback of NOLs to 2005.
Net Loss:
Our net loss was $69,725 for the year ended December 31, 2010, $30,612 for the year ended December 31, 2009 and $44,672 for the year ended December 31, 2008.
Liquidity and Capital Resources
We have to date relied principally on external funding, the Wyeth collaboration, royalty and product revenue to finance our operations. We have funded operations through private placements of equity securities, public offerings of common stock, payments received under collaboration agreements, funding under government research grants and contracts, interest on investments, proceeds from the exercise of outstanding options and warrants, and the sale of our common stock under our two employee stock purchase plans (Purchase Plans).
Under the February 2011 Salix License Agreement, we received a $60.0 million upfront payment in cash and are eligible to receive development and commercialization milestones plus royalties on net sales and 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) Salix receives from non-U.S. sublicensees.
Our expenses and reimbursement revenue related to RELISTOR in the future will depend on the amount of research and development work we perform pursuant to the development plan contemplated by the Salix License Agreement, which is to be finalized early in the second quarter of 2011. We continue to monitor our other program expenditures, including headcount levels, in conjunction with program and program candidates that we choose or are obligated to undertake. We expect to continue to incur operating losses during the near term. We cannot forecast with any degree of certainty, however, which products or indications, if any, will be subject to future arrangements, or how they would affect our capital requirements. The consummation of other agreements would further allow us to advance other projects with current funds. Advancement of the PRO 140 program is subject to obtaining outside funding, for which we have applied to government agencies. While we have to date conducted PSMA ADC research and development on our own, we are considering as appropriate strategic collaborations with biopharmaceutical companies for PSMA ADC.
At December 31, 2010, we held $47.9 million in cash and cash equivalents, a decrease of $43.0 million from $90.9 million at December 31, 2009. We expect that this amount, together with the $60.0 million upfront cash payment from Salix, will be sufficient to fund operations at current levels beyond on year. In addition, at December 31, 2010 and 2009, our investment in auction rate securities classified as long-term assets on the Consolidated Balance Sheets amounted to $3.6 million and $3.8 million, respectively. If, however, we are unable to enter into favorable collaboration, license, asset sale, capital raising or other financing transactions, we will have to reduce, delay or eliminate spending on some current operations, and/or reduce salary and other overhead expenses, to extend our remaining operations. Our cash flow from operating activities was negative for the years ended December 31, 2010, 2009 and 2008 due primarily to the excess of expenditures on our research and development programs and general and administrative costs over cash received from collaborators and government grants and contracts to fund such programs, as described below. See Risk Factors.
Sources of Cash
Operating Activities. During the years ended December 31, 2010, 2009 and 2008, we received $10.3 million, $6.3 million and $49.6 million, respectively, from Wyeth, consisting of (i) $0, $3.2 million and $49.2 million as reimbursements and milestones payments under the 2005 Wyeth collaboration, (ii) $7.9 million, $1.6 million and $0 under the Transition Agreement, and (iii) $2.4 million, $1.5 million and $0.4 million in royalties. Reimbursements under the 2005 collaboration have ceased as a result of its termination.
Under the Transition Agreement, Wyeth paid us $10.0 million in six quarterly installments through January 2011, and continued to pay royalties on 2010 ex-U.S. sales as provided in the 2005 collaboration agreement except to the extent certain fourth quarter financial targets were not met. These targets were not met during the fourth quarter and royalties on ex-U.S. sales were not payable to us. No royalties are payable in respect of net sales after September 30, 2010. Royalties or other revenues from RELISTOR after this transition period will be dependent on Salix’s and its sublicensees’ commercialization efforts. Wyeth is also providing financial resources of approximately $9.5 million, of which we have recognized $1.2 million, which constitutes reimbursement for development of a multi-dose pen for subcutaneous RELISTOR. This support, which has generally been made available by us to Salix under the Salix License Agreement, is ongoing and continues beyond the extended periods of Wyeth’s commercialization obligations; revenue from such financial support for which we perform research and development is reported as reimbursement revenue from Wyeth under the Transition Agreement. We have agreed to purchase Wyeth’s remaining inventory of subcutaneous RELISTOR at the end of the Sales Periods on agreed-upon terms and conditions, and Salix has agreed to purchase our inventory of subcutaneous RELISTOR on similar agreed-upon terms and conditions. We have no further obligations to Wyeth under the 2005 collaboration agreement.
Under our License Agreement with Ono, we received from Ono, in November 2008, an upfront payment of $15.0 million, which was recognized as revenue during the first quarter of 2009, upon satisfaction of our performance obligations, and are entitled to receive potential milestone payments, upon achievement of development milestones by Ono, of up to $20.0 million, commercial milestones and royalties on sales of subcutaneous RELISTOR in Japan. Ono is also responsible for development and commercialization costs for subcutaneous RELISTOR in Japan.
We are partially funding C. difficile research and pre-clinical development of our ProVax HIV vaccine program through contracts with the NIH providing for research, pre-clinical development and early clinical testing support. In September 2010, we were awarded a three-year NIH grant totaling $4.1 million in support of the C. difficile program. For ProVax HIV, through December 2008, we recognized revenue of $15.5 million from the NIH, including $0.2 million for the achievement of two milestones, and in June 2009 were awarded a new five-year NIH grant totaling up to $14.5 million to continue this work, subject to annual funding approvals and customary compliance obligations.
A portion of our revenues is derived from federal government awards. During the years ended December 31, 2010, 2009 and 2008, we recognized as revenue awards made to us by the NIH between 2004 and 2010, to partially fund some of our programs. For the years ended December 31, 2010, 2009 and 2008, we received $4.3 million, $2.9 million and $8.3 million, respectively, of revenue from all of our NIH awards including the $0.7 million Internal Revenue Service grant in 2010.
Changes in Accounts receivable and Accounts payable for the years ended December 31, 2010, 2009 and 2008 resulted from the timing of receipts from the NIH and Wyeth, and payments made to trade vendors in the normal course of business.
Other than amounts to be received from Salix, Wyeth, Ono and from currently approved grants, we have no committed external sources of capital. Other than revenues from RELISTOR, we expect no significant product revenues for a number of years, as it will take at least that much time, if ever, to bring our product candidates to the commercial marketing stage.
Investing Activities. We redeem money market funds and use proceeds from maturities to provide funding for operations. A substantial portion of our cash and cash equivalents ($47.9 million) are guaranteed by the U.S. Treasury or Federal Deposit Insurance Corporation’s guarantee program. Our auction rate securities ($3.6 million) include $2.7 million of securities collateralized by student loan obligations subsidized by the U.S. government. These investments, while rated investment grade by the Standard & Poor’s and Moody’s rating agencies and predominantly having scheduled maturities greater than ten years, are heavily concentrated in the U.S. financial sector, which continues to be under stress.
As a result of changes in general market conditions during 2008, we determined to reduce the principal amount of auction rate securities in our portfolio as they came up for auction. As a result, at December 31, 2010, we continue to hold approximately $3.6 million of auction rate securities and to date, we have received all scheduled interest payments on these securities. We will not realize cash in respect of the principal amount of these securities until the issuer calls or restructures the underlying security, the underlying security matures and is paid, or a buyer outside the auction process emerges.
We monitor markets for our investments, but cannot guarantee that additional losses will not be required to be recorded. Valuation of securities is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or the underlying assets supporting them, default rates applicable to the underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and quality of market credit and liquidity and general economic and market conditions. We do not believe the carrying values of our investments are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss.
Our money market funds are purchased and, in the case of auction rate securities, sold by third-party brokers in accordance with our investment policy guidelines. Our brokerage account requires that all securities be held to maturity unless authorization is obtained from us to sell earlier. In fact, we had a history of holding all securities to maturity prior to the second quarter of 2009, when we decided to sell a portion of our securities which had scheduled maturities between the fourth quarter of 2009 and the third quarter of 2010. The proceeds from these sales were $24.8 million, resulting in a gain of $0.2 million.
We expect to recover the amortized cost of all of our investments at maturity. Because we do not anticipate having to sell these securities in order to operate our business and believe it is not more likely than not that we will be required to sell these securities before recovery of principal, we do not consider these securities to be other than temporarily impaired at December 31, 2010.
Financing Activities. During the years ended December 31, 2010, 2009 and 2008, we received cash of $3.9 million, $4.9 million and $6.5 million, respectively, from the exercise of stock options by employees, directors and non-employee consultants and from the sale of our common stock under our Purchase Plans. The amount of cash we receive from these sources fluctuates commensurate with headcount levels and changes in the price of our common stock on the grant date for options exercised, and on the sale date for shares sold under the Purchase Plans.
Under the Transition Agreement, Wyeth is providing financial resources of approximately $9.5 million, of which we have recognized $1.2 million, which constitutes reimbursement for development of a multi-dose pen for subcutaneous RELISTOR. This support, which has been made available by us to Salix under the Salix License Agreement, is ongoing and continues beyond the extended periods of Wyeth’s commercialization obligations; revenue from such financial support prior to the Salix License Agreement has been reported as reimbursement revenue from Wyeth under the Transition Agreement.
Unless we obtain regulatory approval from the FDA for additional product candidates and/or enter into agreements with corporate collaborators with respect to our additional technologies, we will be required to fund our operations in the future through sales of common stock or other securities, royalty or other financing agreements and/or grants and government contracts. Adequate additional funding may not be available to us on acceptable terms or at all. Our inability to raise additional capital on terms reasonably acceptable to us may seriously jeopardize the future success of our business.
Uses of Cash
Operating Activities. The majority of our cash has been used to advance our research and development programs. We currently have major research and development programs in gastroenterology, oncology and virology, and are conducting several smaller research projects in those latter areas.
Under the Transition Agreement, we have agreed to purchase Wyeth’s remaining inventory of subcutaneous RELISTOR at the end of its sales periods on agreed-upon terms and conditions, and Salix has agreed to purchase our inventory of subcutaneous RELISTOR on similar agreed-upon terms and conditions.
Our total expenses for research and development from inception through December 31, 2010 have been approximately $581.8 million. For various reasons, including the early stage of certain of our programs, the timing and results of our clinical trials, our dependence in certain instances on third parties and the uncertainty of the specific nature of RELISTOR-related future arrangements and relationships following termination of the Wyeth collaboration, many of which are outside of our control, we cannot estimate the total remaining costs to be incurred and timing to complete all our research and development programs.
For the years ended December 31, 2010, 2009 and 2008, research and development costs incurred, by project, were as follows:
|
2010
|
2009
|
2008
|
||||||||||
|
(in millions)
|
||||||||||||
|
RELISTOR
|
$ | 23.3 | $ | 7.8 | $ | 25.4 | ||||||
|
Cancer
|
14.7 | 20.1 | 10.8 | |||||||||
|
HIV
|
5.5 | 11.8 | 39.4 | |||||||||
|
Other programs
|
8.7 | 11.4 | 9.5 | |||||||||
|
Total
|
$ | 52.2 | $ | 51.1 | $ | 85.1 | ||||||
We will require additional funding to continue our research and product development programs, conduct pre-clinical studies and clinical trials, fund operating expenses, pursue regulatory approvals for our product candidates, file and prosecute patent applications and enforce or defend patent claims, if any, and fund product in-licensing and any possible acquisitions.
Investing Activities. During the years ended December 31, 2010, 2009 and 2008, we have spent $2.2 million, $0.9 million and $2.2 million, respectively, on capital expenditures. These expenditures have been primarily related to leasehold improvements and the purchase of laboratory equipment for our research and development projects.
Financing Activities. During the year ended December 31, 2008, we repurchased 200,000 of our outstanding common shares for a total of $2.7 million. We did not repurchase any common shares during the years ended December 31, 2010 and 2009.
Contractual Obligations
Our funding requirements, both for the next 12 months and beyond, will include required payments under operating leases and licensing and collaboration agreements. The following table summarizes our contractual obligations as of December 31, 2010 for future payments under these agreements:
|
Payments due by Year-end
|
||||||||||||||||||||
|
Total
|
2011
|
2012-2013 | 2014-2015 |
Thereafter
|
||||||||||||||||
|
(in millions)
|
||||||||||||||||||||
|
Operating leases
|
$ | 40.9 | $ | 3.3 | $ | 6.9 | $ | 8.2 | $ | 22.5 | ||||||||||
|
License and collaboration agreements (1)
|
86.6 | 2.4 | 3.4 | 57.4 | 23.4 | |||||||||||||||
|
Total
|
$ | 127.5 | $ | 5.7 | $ | 10.3 | $ | 65.6 | $ | 45.9 | ||||||||||
_______________
|
(1)
|
Based on assumed achievement of milestones covered under each agreement, the timing and payment of which is highly uncertain.
|
We periodically assess the scientific progress and merits of each of our programs to determine if continued research and development is commercially and economically viable. Certain of our programs have been terminated due to the lack of scientific progress and prospects for ultimate commercialization. Because of the uncertainties associated with research and development in these programs, the duration and completion costs of our research and development projects are difficult to estimate and are subject to considerable variation. Our inability to complete research and development projects in a timely manner or failure to enter into collaborative agreements could significantly increase capital requirements and adversely affect our liquidity.
Our cash requirements may vary materially from those now planned because of results of research and development and product testing, changes in existing relationships or new relationships with licensees, licensors or other collaborators, changes in the focus and direction of our research and development programs, competitive and technological advances, the cost of filing, prosecuting, defending and enforcing patent claims, the regulatory approval process, manufacturing and marketing and other costs associated with the commercialization of products following receipt of regulatory approvals and other factors.
The above discussion contains forward-looking statements based on our current operating plan and the assumptions on which it relies. There could be deviations from that plan that would consume our assets earlier than planned.
Off-Balance Sheet Arrangements and Guarantees
We have no obligations under off-balance sheet arrangements and do not guarantee the obligations of any other unconsolidated entity.
Critical Accounting Policies
We prepare our financial statements in conformity with accounting principles generally accepted in the United States of America. Our significant accounting policies are disclosed in Note 2 to our financial statements included in this Annual Report on Form 10-K for the year ended December 31, 2010. The selection and application of these accounting principles and methods requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses, as well as certain financial statement disclosures. On an ongoing basis, we evaluate our estimates. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances. The results of our evaluation form the basis for making judgments about the carrying values of assets and liabilities that are not otherwise readily apparent. While we believe that the estimates and assumptions we use in preparing the financial statements are appropriate, these estimates and assumptions are subject to a number of factors and uncertainties regarding their ultimate outcome and, therefore, actual results could differ from these estimates.
We have identified our critical accounting policies and estimates below. These are policies and estimates that we believe are the most important in portraying our financial condition and results of operations, and that require our most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain. We have discussed the development, selection and disclosure of these critical accounting policies and estimates with the Audit Committee of our Board of Directors.
Revenue Recognition. We recognize revenue from all sources based on the provisions of the SEC’s Staff Accounting Bulletin (SAB) No. 104 (SAB 104) and ASC 605 Revenue Recognition.
Collaborations may contain substantive milestone payments to which we apply the substantive milestone method (Substantive Milestone Method). Substantive milestone payments are considered to be performance payments that are recognized upon achievement of the milestone only if all of the following conditions are met: (i) the milestone payment is non-refundable, (ii) achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement, (iii) substantive effort is involved in achieving the milestone, (iv) the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (v) a reasonable amount of time passes between the upfront license payment and the first milestone payment as well as between each subsequent milestone payment.
Determination as to whether a milestone meets the aforementioned conditions involves management’s judgment. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone and, therefore, the resulting payment would be part of the consideration and be recognized as revenue as such performance obligations are performed.
Royalty revenue is recognized based upon net sales of related licensed products. Royalty revenue is recognized in the period the sales occur, provided that the royalty amounts are fixed or determinable, collection of the related receivable is reasonably assured and we have no remaining performance obligations under the arrangement providing for the royalty. If royalties are received when we have remaining performance obligations, they would be attributed to the services being provided under the arrangement and, therefore, recognized as such obligations are performed under the proportionate performance method.
We recognize upfront license payments as revenue upon delivery of the license only if the license had standalone value and the fair value of the undelivered performance obligations, typically including research or steering or other committee services, could be determined. If the fair value of the undelivered performance obligations could be determined, such obligations would then be accounted for separately as performed. If the license is considered to either (i) not have standalone value, or (ii) have standalone value but the fair value of any of the undelivered performance obligations could not be determined, the upfront license payments would be recognized as revenue over the estimated period of when our performance obligations are performed.
Under the proportionate performance method we recognize revenue provided that we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Direct labor hours or full-time equivalents will typically be used as the measure of performance. Revenue is recognized in any period as the percent of actual effort expended in that period relative to total effort for all of our performance obligations under the arrangement. Significant judgment and estimates are required in determining the nature and assignment of tasks to be accomplished by each of the parties and the level of effort required for us to complete our performance obligations under the arrangement. The nature and assignment of tasks to be performed by each party involves the preparation, discussion and approval by the parties of a development plan and budget.
Amounts not expected to be recognized within one year of the balance sheet date are classified as long-term deferred revenue. The estimate of the classification of deferred revenue as short-term or long-term is based upon management’s current operating budget with the collaborator for the total effort required to complete our performance obligations under the arrangement.
As the development programs progress over time, the development budgets, including the amount of FTEs, may be revised, resulting in a change to the development period or costs. Changes in the development estimates are likely to affect the amount of revenue recognized in the period of change and each year in the future as compared to prior periods. Under the Wyeth collaboration, we recognized $6.2 million less revenue from the $60.0 million upfront payment during the year ended December 31, 2008 compared to the amounts recognized in 2007 due to an extension of the development budget from December 31, 2008 to December 31, 2009. Conversely, we recognized $4.3 million more revenue during 2009 compared to amounts recognized in 2008 due to the increase in the percent of actual effort expended in 2009 relative to the total remaining effort to complete development.
Share-Based Payment Arrangements. Our share-based compensation to employees includes non-qualified stock options, restricted stock and shares issued under our Purchase Plans, which are compensatory under ASC 718 Compensation – Stock Compensation. We account for share-based compensation to non-employees, including non-qualified stock options and restricted stock, in accordance with ASC 505 Equity.
The fair value of each non-qualified stock option award is estimated on the date of grant using the Black-Scholes option pricing model.
The model requires input assumptions with respect to (i) expected volatility of our common stock, which is based upon the daily quoted market prices on The NASDAQ Stock Market LLC over a period equal to the expected term, (ii) the period of time over which employees, officers, our Chief Executive Officer, Vice Chairman, directors and non-employee consultants are expected to hold their options prior to exercise, (iii) zero expected dividend yield due to never having paid dividends and not expecting to pay dividends in the future, and (iv) risk-free interest rates for periods within the expected term of the options, which are based on the U.S. Treasury yield curve in effect at the time of grant.
Historical volatilities are based upon daily quoted market prices of our common stock on The NASDAQ Stock Market LLC over a period equal to the expected term of the related equity instruments. We rely only on historical volatility since it provides the most reliable indication of future volatility. Future volatility is expected to be consistent with historical; historical volatility is calculated using a simple average calculation; historical data is available for the length of the option’s expected term and a sufficient number of price observations are used consistently. Since our stock options are not traded on a public market, we do not use implied volatility.
The expected term of options granted represents the period of time that options granted are expected to be outstanding based upon historical data related to exercise and post-termination cancellation activity. The expected term of stock options granted to our Chief Executive Officer, Vice Chairman and non-employee directors and consultants are calculated separately from stock options granted to employees and officers.
We apply a forfeiture rate to the number of unvested awards in each reporting period in order to estimate the number of awards that are expected to vest. Estimated forfeiture rates are based upon historical data on vesting behavior of employees. We adjust the total amount of compensation cost recognized for each award, in the period in which each award vests, to reflect the actual forfeitures related to that award. Changes in our estimated forfeiture rate will result in changes in the rate at which compensation cost for an award is recognized over its vesting period.
Changes in the assumptions used to compute the fair value of the option awards are likely to affect the fair value of the non-qualified stock option awards and the amount of compensation expense recognized in future periods. A higher volatility, longer expected term and higher risk-free rate increases the resulting compensation expense recognized in future periods as compared to prior periods. Conversely, a lower volatility, shorter expected term and lower risk-free rate decreases the resulting compensation expense recognized in future periods as compared to prior periods.
For performance-based stock option awards vesting of a defined portion of each award will occur earlier if a defined performance condition is achieved; more than one condition may be achieved in any period. We estimate the probability of achievement of each performance condition and use those probabilities to determine the requisite service period of each award. The requisite service period for the award is the shortest of the explicit or implied service periods. In the case of the executive’s options, the explicit service period is nine years and eleven months from the respective grant dates. The implied service periods related to the performance conditions are the estimated times for each performance condition to be achieved. Thus, compensation expense will be recognized over the shortest estimated time for the achievement of performance conditions for that award (assuming that the performance conditions will be achieved before the cliff vesting occurs). For performance and market-based stock option awards to our Vice Chairman (consisting of options in 2010 and 2009 and options and restricted stock in 2008) and Chief Executive Officer (consisting of options in 2010) vesting occurs on the basis of the achievement of specified performance or market-based milestones. The options have an exercise price equal to the closing price on our common stock on the date of grant. The awards are valued using a Monte Carlo simulation and the expense related to these grants will be recognized over the shortest estimated time for the achievement of the performance or market conditions. The awards will not vest unless one of the milestones is achieved or the market condition is met. Changes in the estimate of probability of achievement of any performance or market condition will be reflected in compensation expense of the period of change and future periods affected by the change.
Research and Development Expenses Including Clinical Trial Expenses. Clinical trial expenses, which are included in research and development expenses, represent obligations resulting from our contracts with various clinical investigators and clinical research organizations in connection with conducting clinical trials for our product candidates. Such costs are expensed as incurred, and are based on the total number of patients in the trial, the rate at which the patients enter the trial and the period over which the clinical investigators and clinical research organizations to provide services. We believe that this method best approximates the efforts expended on a clinical trial with the expenses we record. We adjust our rate of clinical expense recognition if actual results differ from our estimates. In addition to clinical trial expenses, we estimate the amounts of other research and development expenses, for which invoices have not been received at the end of a period, based upon communication with third parties that have provided services or goods during the period. Such estimates are subject to change as additional information becomes available.
Fair Value Measurements. Our available-for-sale investment portfolio consists of money market funds and auction rate securities, and is recorded at fair value in the accompanying Consolidated Balance Sheets in accordance with ASC 320 Investments – Debt and Equity Securities. The change in the fair value of these investments is recorded as a component of other comprehensive loss.
We continue to monitor markets for our investments and consider the impact, if any, of market conditions on the fair market value of our investments.
We expect to recover the amortized cost of all of our investments at maturity. Currently, we do not anticipate having to sell these securities in order to operate our business and we believe that it is not more likely than not that we will be required to sell these securities before recovery of principal. We do not believe the carrying values of our investments are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss.
Valuation of securities is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or the underlying assets supporting them, default rates applicable to the underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and quality of market credit and liquidity and general economic and market conditions. The valuation of the auction rate securities we hold is based on an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security.
Impact of Recently Issued Accounting Standards
In October 2009, the FASB issued ASU 2009-13 to address the accounting for multiple-deliverable arrangements. In an arrangement with multiple deliverables, the delivered items shall be considered a separate unit of accounting if both (i) the delivered items have value to a collaborator on a stand-alone basis, in that, the collaborator could resell the delivered items on a stand-alone basis, and (ii) the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item or items is considered probable and substantially in our control. This ASU will be effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010.
In April 2010, the FASB issued ASU 2010-17, which provides guidance on the criteria that should be met when determining whether the milestone method of revenue recognition is appropriate and this guidance is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010.
We expect that the adoption of these ASUs will have a material effect on our consolidated financial statements. However, the amount is not known or able to be reasonably estimated at this time as we are currently evaluating the impact of the recently completed Salix License Agreement.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Our primary investment objective is to preserve principal. Our available-for-sale investments consist of money market funds and auction rate securities, all of which had interest rates that were variable and totaled $47.6 million at December 31, 2010. As a result, we do not believe that we have a material exposure to interest-rate risk.
As a result of changes in general market conditions during 2008, we determined to reduce the principal amount of auction rate securities in our portfolio as they came up for auction and invest the proceeds in other securities in accordance with our investment guidelines. At December 31, 2010, we continue to hold approximately $3.6 million (7.6% of assets measured at fair value) of auction rate securities, in respect of which we have received all scheduled interest payments. The principal amount of these remaining auction rate securities will not be accessible until the issuer calls or restructures the underlying security, the underlying security matures and is paid or a buyer outside the auction process emerges.
We continue to monitor the market for auction rate securities and consider the impact, if any, of market conditions on the fair market value of our investments. We believe that the failed auctions experienced to date are not a result of the deterioration of the underlying credit quality of these securities, although valuation of them is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or the underlying assets supporting them, default rates applicable to the underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and quality of market credit and liquidity, and general economic and market conditions. We do not believe the carrying values of these auction rate securities are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss.
The valuation of the auction rate securities we hold is based on an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security. We re-evaluated the valuation of these securities as of December 31, 2010 and the temporary impairment amount decreased $16.0 thousand from $308.0 thousand at December 31, 2009 to $292.0 thousand. A 100 basis point increase to our internal analysis would result in an insignificant increase in the temporary impairment of these securities as of the year ended December 31, 2010.
Item 8. Financial Statements and Supplementary Data
See page F-1, Index to Consolidated Financial Statements.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. We have a Disclosure Committee consisting of members of our senior management which monitors and implements our policy of disclosing material information concerning the Company in accordance with applicable law.
As required by SEC Rule 13a-15(e), we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this report. Based on the foregoing, our Chief Executive Officer and Chief Financial Officer concluded that our current disclosure controls and procedures, as designed and implemented, were effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting, as such term is defined in the Exchange Act Rules 13a-15(f) and 15d-15(f) during our fiscal quarter ended December 31, 2010 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Internal control over financial reporting is a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our Board, management and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. It includes policies and procedures that:
· Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
· Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorization of our management and directors; and
· Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management has used the framework set forth in the report entitled Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, known as COSO, to evaluate the effectiveness of our internal control over financial reporting. Management has concluded that our internal control over financial reporting was effective as of December 31, 2010. The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which appears herein.
Item 9B. Other Information
None.
PART III
The information required by the Form 10-K Items listed in the following table will be included under the respective headings specified for such Items in our definitive proxy statement for our 2011 Annual Meeting of Stockholders to be filed with the SEC:
|
Item of Form 10-K
|
Location in 2011 Proxy Statement
|
|
Item 10. Directors, Executive Officers and Corporate Governance
|
Election of Directors.
Board and Committee Meetings.
Executive Officers of the Company.
Section 16(a) Beneficial Ownership Reporting and Compliance.
Code of Business Ethics and Conduct.*
*The full text of our code of business ethics and conduct is available on our website (http://www.progenics.com/documents.cfm).
|
|
Item 11. Executive Compensation
|
Executive Compensation.
Compensation Committee Report.
Compensation Committee Interlocks and Insider Participation.
|
|
Item 12.Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
|
Equity Compensation Plan Information.
Security Ownership of Certain Beneficial Owners and Management.
|
|
Item 13. Certain Relationships and Related Transactions, and Director Independence
|
Certain Relationships and Related Transactions.
Affirmative Determinations Regarding Director Independence and Other Matters.
|
|
Item 14. Principal Accounting Fees and Services
|
Fees Billed for Services Rendered by our Independent Registered Public Accounting Firm.
Pre-approval of Audit and Non-Audit Services by the Audit Committee.
|
PART IV
Item 15. Exhibits, Financial Statement Schedules
The following documents or the portions thereof indicated are filed as a part of this Report.
|
|
(a)
|
Documents filed as part of this Report:
|
|
|
Consolidated Financial Statements of Progenics Pharmaceuticals, Inc.:
|
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets at December 31, 2010 and 2009
Consolidated Statements of Operations for the years ended December 31, 2010, 2009 and 2008
Consolidated Statements of Stockholders’ Equity and Comprehensive Loss for the years ended December 31, 2010, 2009 and 2008
Consolidated Statements of Cash Flows for the years ended December 31, 2010, 2009 and 2008
Notes to Consolidated Financial Statements
|
|
(b)
|
Financial Statement Schedules
|
All financial statement schedules referred to in Item 12-01 of Regulation S-X are inapplicable and therefore have been omitted.
|
|
(c)
|
Item 601 Exhibits
|
Those exhibits required to be filed by Item 601 of Regulation S-K are listed in the Exhibit Index immediately following the signature page hereof and preceding the exhibits filed herewith, and such listing is incorporated herein by reference.
PROGENICS PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
Page
|
|
|
Report of Independent Registered Public Accounting Firm
|
F-2
|
|
Financial Statements:
|
|
|
Consolidated Balance Sheets at December 31, 2010 and 2009
|
F-3
|
|
Consolidated Statements of Operations for the years ended December 31, 2010, 2009 and 2008
|
F-4
|
|
Consolidated Statements of Stockholders’ Equity and Comprehensive Loss
for the years ended December 31, 2010, 2009 and 2008
|
F-5
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2010, 2009 and 2008
|
F-6
|
|
Notes to Consolidated Financial Statements
|
F-7
|
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
Progenics Pharmaceuticals, Inc.
In our opinion, the consolidated financial statements listed in the index appearing under Item 15(a)(1) present fairly, in all material respects, the financial position of Progenics Pharmaceuticals, Inc. and its subsidiaries at December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
New York, New York
March 11, 2011
CONSOLIDATED BALANCE SHEETS
(in thousands, except for par value and share amounts)
|
December 31,
|
|||||||
|
2010
|
2009
|
||||||
|
Assets
|
|||||||
|
Current assets:
|
|||||||
|
Cash and cash equivalents
|
$
|
47,918
|
$
|
90,903
|
|||
|
Marketable securities
|
-
|
1,501
|
|||||
|
Accounts receivable
|
2,283
|
7,522
|
|||||
|
Other current assets
|
1,801
|
1,468
|
|||||
|
Total current assets
|
52,002
|
101,394
|
|||||
|
Auction rate securities
|
3,608
|
3,792
|
|||||
|
Fixed assets, at cost, net of accumulated depreciation and amortization
|
5,878
|
6,560
|
|||||
|
Other assets
|
1,250
|
1,867
|
|||||
|
Total assets
|
$
|
62,738
|
$
|
113,613
|
|||
|
Liabilities and Stockholders’ Equity
|
|||||||
|
Current liabilities:
|
|||||||
|
Accounts payable and accrued expenses
|
$
|
9,683
|
$
|
5,836
|
|||
|
Other current liabilities
|
112
|
170
|
|||||
|
Total current liabilities
|
9,795
|
6,006
|
|||||
|
Other liabilities
|
1,635
|
-
|
|||||
|
Total liabilities
|
11,430
|
6,006
|
|||||
|
Commitments and contingencies (Note 8)
|
|||||||
|
Stockholders’ equity:
|
|||||||
|
Preferred stock, $.001 par value; 20,000,000 shares authorized; issued and outstanding – none
|
-
|
-
|
|||||
|
Common stock, $.0013 par value; 40,000,000 shares authorized; issued –
33,325,802 in 2010 and 32,142,062 in 2009
|
43
|
42
|
|||||
|
Additional paid-in capital
|
453,353
|
439,943
|
|||||
|
Accumulated deficit
|
(399,055
|
)
|
(329,330
|
)
|
|||
|
Accumulated other comprehensive loss
|
(292
|
)
|
(307
|
)
|
|||
|
Treasury stock, at cost (200,000 shares in 2010 and 2009)
|
(2,741
|
)
|
(2,741
|
)
|
|||
|
Total stockholders’ equity
|
51,308
|
107,607
|
|||||
|
Total liabilities and stockholders’ equity
|
$
|
62,738
|
$
|
113,613
|
|||
The accompanying notes are an integral part of the financial statements.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except for loss per share data)
|
Years Ended December 31,
|
|||||||||||
|
2010
|
2009
|
2008
|
|||||||||
|
Revenues:
|
|||||||||||
|
Research and development
|
$
|
1,413
|
$
|
44,351
|
$
|
59,885
|
|||||
|
Royalty income
|
1,826
|
2,372
|
146
|
||||||||
|
Research grants and contract
|
4,573
|
1,968
|
7,460
|
||||||||
|
Other revenues
|
140
|
256
|
180
|
||||||||
|
Total revenues
|
7,952
|
48,947
|
67,671
|
||||||||
|
Expenses:
|
|||||||||||
|
Research and development
|
50,640
|
49,798
|
82,290
|
||||||||
|
License fees – research and development
|
1,270
|
1,058
|
2,830
|
||||||||
|
General and administrative
|
22,832
|
25,106
|
28,834
|
||||||||
|
Royalty expense
|
241
|
237
|
15
|
||||||||
|
Depreciation and amortization
|
2,853
|
5,078
|
4,609
|
||||||||
|
Total expenses
|
77,836
|
81,277
|
118,578
|
||||||||
|
Operating loss
|
(69,884
|
)
|
(32,330
|
)
|
(50,907
|
)
|
|||||
|
Other income:
|
|||||||||||
|
Interest income
|
64
|
1,481
|
6,235
|
||||||||
|
Gain on sale of marketable securities
|
-
|
237
|
-
|
||||||||
|
Total other income
|
64
|
1,718
|
6,235
|
||||||||
|
Net loss before income taxes
|
(69,820
|
)
|
(30,612
|
)
|
(44,672
|
)
|
|||||
|
Income tax benefit
|
95
|
-
|
-
|
||||||||
|
Net loss
|
$
|
(69,725
|
)
|
$
|
(30,612
|
)
|
$
|
(44,672
|
)
|
||
|
Net loss per share – basic and diluted
|
$
|
(2.14
|
)
|
$
|
(0.98
|
)
|
$
|
(1.48
|
)
|
||
|
Weighted-average shares – basic and diluted
|
32,590
|
31,219
|
30,142
|
||||||||
The accompanying notes are an integral part of the financial statements.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
For the Years Ended December 31, 2010, 2009 and 2008
(in thousands)
|
Common Stock
|
Additional
|
Accumulated Other
|
Treasury Stock
|
|||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Paid-In Capital
|
Accumulated Deficit
|
Comprehensive Income (Loss)
|
Shares
|
Amount
|
Total
|
|||||||||||||||||||||||||
|
Balance at December 31, 2007
|
29,754 | $ | 39 | $ | 401,500 | $ | (254,046 | ) | $ | 6 | - | $ | - | $ | 147,499 | |||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | (44,672 | ) | - | - | - | (44,672 | ) | ||||||||||||||||||||||
|
Net unrealized (loss) on marketable and auction rate securities
|
- | - | - | - | (1,303 | ) | - | - | (1,303 | ) | ||||||||||||||||||||||
|
Total comprehensive loss:
|
(45,975 | ) | ||||||||||||||||||||||||||||||
|
Compensation expenses for share-based payment arrangements
|
- | - | 14,133 | - | - | - | - | 14,133 | ||||||||||||||||||||||||
|
Issuance of restricted stock, net of forfeitures
|
216 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
Sale of common stock under employee stock purchase plans and exercise of stock options
|
837 | 1 | 6,452 | - | - | - | - | 6,453 | ||||||||||||||||||||||||
|
Treasury shares acquired under repurchase program
|
- | - | - | - | - | (200 | ) | (2,741 | ) | (2,741 | ) | |||||||||||||||||||||
|
Balance at December 31, 2008
|
30,807 | 40 | 422,085 | (298,718 | ) | (1,297 | ) | (200 | ) | (2,741 | ) | 119,369 | ||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | (30,612 | ) | - | - | - | (30,612 | ) | ||||||||||||||||||||||
|
Net unrealized gain on marketable and auction rate securities
|
- | - | - | - | 990 | - | - | 990 | ||||||||||||||||||||||||
|
Total comprehensive loss:
|
(29,622 | ) | ||||||||||||||||||||||||||||||
|
Compensation expenses for share-based payment arrangements
|
- | - | 12,986 | - | - | - | - | 12,986 | ||||||||||||||||||||||||
|
Issuance of restricted stock, net of forfeitures
|
266 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
Sale of common stock under employee stock purchase plans and exercise of stock options
|
1,069 | 2 | 4,872 | - | - | - | - | 4,874 | ||||||||||||||||||||||||
|
Balance at December 31, 2009
|
32,142 | 42 | 439,943 | (329,330 | ) | (307 | ) | (200 | ) | (2,741 | ) | 107,607 | ||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | (69,725 | ) | - | - | - | (69,725 | ) | ||||||||||||||||||||||
|
Net unrealized gain on marketable and auction rate securities
|
- | - | - | - | 15 | - | - | 15 | ||||||||||||||||||||||||
|
Total comprehensive loss:
|
(69,710 | ) | ||||||||||||||||||||||||||||||
|
Compensation expenses for share-based payment arrangements
|
- | - | 9,515 | - | - | - | - | 9,515 | ||||||||||||||||||||||||
|
Issuance of restricted stock, net of forfeitures
|
173 | - | - | - | - | - | - | - | ||||||||||||||||||||||||
|
Sale of common stock under employee stock purchase plans and exercise of stock options
|
1,011 | 1 | 3,895 | - | - | - | - | 3,896 | ||||||||||||||||||||||||
|
Balance at December 31, 2010
|
33,326 | $ | 43 | $ | 453,353 | $ | (399,055 | ) | $ | (292 | ) | (200 | ) | $ | (2,741 | ) | $ | 51,308 | ||||||||||||||
The accompanying notes are an integral part of the financial statements.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
Years Ended December 31,
|
|||||||||||
|
2010
|
2009
|
2008
|
|||||||||
|
Cash flows from operating activities:
|
|||||||||||
|
Net loss
|
$
|
(69,725
|
)
|
$
|
(30,612
|
)
|
$
|
(44,672
|
)
|
||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
|||||||||||
|
Depreciation and amortization
|
2,853
|
5,078
|
4,609
|
||||||||
|
Write-off of fixed assets
|
-
|
334
|
3
|
||||||||
|
Amortization of discounts, net of premiums, on marketable securities
|
-
|
889
|
960
|
||||||||
|
Expenses for share-based compensation awards
|
9,515
|
12,986
|
14,133
|
||||||||
|
Gain on sale of marketable securities
|
-
|
(237
|
)
|
-
|
|||||||
|
Changes in assets and liabilities:
|
|||||||||||
|
Decrease (increase) in accounts receivable
|
5,239
|
(6,185
|
)
|
658
|
|||||||
|
(Increase) decrease in other current assets
|
(333
|
)
|
2,063
|
(420
|
)
|
||||||
|
Decrease (increase) in other assets
|
617
|
(1,667
|
)
|
-
|
|||||||
|
Increase (decrease) in accounts payable and accrued expenses
|
3,847
|
(660
|
)
|
(8,269
|
)
|
||||||
|
(Decrease) increase in deferred revenue
|
-
|
(31,645
|
)
|
4,786
|
|||||||
|
(Decrease) increase in other current liabilities
|
(58
|
)
|
113
|
-
|
|||||||
|
Increase (decrease) in other liabilities
|
1,635
|
(266
|
)
|
(93
|
)
|
||||||
|
Net cash used in operating activities
|
(46,410
|
)
|
(49,809
|
)
|
(28,305
|
)
|
|||||
|
Cash flows from investing activities:
|
|||||||||||
|
Capital expenditures
|
(2,171
|
)
|
(901
|
)
|
(2,172
|
)
|
|||||
|
Sales/maturities of marketable and auction rate securities
|
1,700
|
80,233
|
128,705
|
||||||||
|
Purchase of marketable securities
|
-
|
-
|
(56,209
|
)
|
|||||||
|
Decrease (increase) in restricted cash
|
-
|
320
|
32
|
||||||||
|
Net cash (used in) provided by investing activities
|
(471
|
)
|
79,652
|
70,356
|
|||||||
|
Cash flows from financing activities:
|
|||||||||||
|
Purchase of treasury stock
|
-
|
-
|
(2,741
|
)
|
|||||||
|
Proceeds from the exercise of stock options and sale of common stock under the Employee Stock Purchase Plan
|
3,896
|
4,874
|
6,453
|
||||||||
|
Net cash provided by financing activities
|
3,896
|
4,874
|
3,712
|
||||||||
|
Net (decrease) increase in cash and cash equivalents
|
(42,985
|
)
|
34,717
|
45,763
|
|||||||
|
Cash and cash equivalents at beginning of period
|
90,903
|
56,186
|
10,423
|
||||||||
|
Cash and cash equivalents at end of period
|
$
|
47,918
|
$
|
90,903
|
$
|
56,186
|
|||||
The accompanying notes are an integral part of the financial statements.
F-6
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(amounts in thousands, except per share amounts or unless otherwise noted)
1. Organization and Business
Progenics Pharmaceuticals, Inc. (“Progenics,” “we” or “us”) is a biopharmaceutical company focusing on the development and commercialization of innovative therapeutic products to treat the unmet medical needs of patients with debilitating conditions and life-threatening diseases. Our principal programs are directed toward gastroenterology, oncology and virology.
Progenics commenced principal operations in 1988, became publicly traded in 1997 and throughout has been engaged primarily in research and development efforts, establishing corporate collaborations and related activities. Certain of our intellectual property rights are held by wholly owned subsidiaries. None of our subsidiaries other than PSMA Development Company LLC (“PSMA LLC”) had operations during the years ended December 31, 2010, 2009 or 2008. All of our operations are conducted at our facilities in Tarrytown, New York. We operate under a single research and development segment.
Our first commercial product is RELISTOR® (methylnaltrexone bromide) subcutaneous injection, a first-in-class therapy for opioid-induced constipation approved for sale in over 50 countries worldwide, including the United States, European Union member states, Canada and Australia. Marketing applications are pending elsewhere throughout the world.
On February 3, 2011, we entered into an exclusive License Agreement with Salix Pharmaceuticals, Inc. (“Salix”) by which Salix acquired the rights to RELISTOR worldwide except in Japan, where we have previously licensed to Ono Pharmaceutical the subcutaneous formulation of the drug. In connection with the Salix License Agreement, we received a $60.0 million upfront payment in cash from Salix and are eligible to receive development milestone payments of up to $90.0 million, contingent upon the achievement of specified U.S. regulatory approvals and commercialization milestone payments of up to $200.0 million, contingent upon the achievement of specified U.S. sales targets. Salix must pay us royalties based upon a percentage ranging from 15 to 19 percent of net sales by it and its affiliates and 60% of any upfront, milestone, reimbursement or other revenue (net of costs of goods sold, as defined, and territory-specific research and development expense reimbursement) it receives from sublicensees in respect of any country outside the U.S., as sublicensees commence their commercialization efforts. Salix is responsible for further developing and commercializing subcutaneous RELISTOR, including completing clinical development necessary to support regulatory marketing approvals for potential new indications and formulations, and marketing and selling the product.
RELISTOR has previously been developed and commercialized worldwide except Japan by Progenics and Wyeth Pharmaceuticals, now a Pfizer Inc. subsidiary, pursuant to a 2005 collaboration agreement that was terminated in October 2009. Under our Transition Agreement with Wyeth, Wyeth has continued to distribute RELISTOR worldwide other than Japan through March 31, 2011. The parties plan an April 1, 2011 transition of U.S. commercial responsibility to Salix from Wyeth, and are currently discussing transition of ex-U.S. commercialization on a country-by-country basis. While Salix effects a country-by-country transition of ex-U.S. commercialization rights, Wyeth continues to supply product.
Under the Transition Agreement, Wyeth paid us $10.0 million in six quarterly installments through January 2011, and continued to pay royalties on 2010 ex-U.S. sales as provided in the 2005 collaboration agreement except to the extent certain fourth quarter financial targets were not met. These targets were not met during the fourth quarter and royalties on ex-U.S. sales were not payable to us. No other royalties are payable in respect of net sales after September 30, 2010. Wyeth is also providing financial resources of approximately $9.5 million, of which we have recognized $1.2 million, which constitutes reimbursement for development of a multi-dose pen for subcutaneous RELISTOR. Revenue from such financial support prior to the Salix License Agreement has been reported as reimbursement revenue from Wyeth under the Transition Agreement. We have agreed to purchase Wyeth’s remaining inventory of subcutaneous RELISTOR at the end of the Sales Periods on agreed-upon terms and conditions, and Salix has agreed to purchase our inventory of subcutaneous RELISTOR on similar agreed-upon terms and conditions. We have no further obligations to Wyeth under the 2005 collaboration agreement.
Prior to the Transition Agreement (including the 2008 and 2009 periods covered by this report), we received upfront, milestone, and royalty payments from Wyeth, and were reimbursed for expenses we incurred in connection with the development of RELISTOR; manufacturing and commercialization expenses for RELISTOR were funded by Wyeth.
F-7
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Funding and Financial Matters. We will require additional funding to continue our current programs to completion, which may involve collaboration agreements, licenses or sale transactions or royalty sales or financings with respect to our products and product candidates. We may also seek to raise additional capital through sales of common stock or other securities, and expect to continue funding some programs in part through government awards.
At December 31, 2010, we held $47.9 million in cash and cash equivalents which, together with the $60.0 million upfront cash payment received from Salix in February 2011, we expect will be sufficient to fund operations at current levels beyond one year. If, however, we are unable to enter into favorable collaboration, license, asset sale, capital raising or other financing transactions, we will have to reduce, delay or eliminate spending on some current operations, and/or reduce salary and other overhead expenses, to extend our remaining operations.
In April 2008, our Board of Directors approved a share repurchase program to acquire up to $15.0 million of our outstanding common shares, under which we have $12.3 million remaining available. Purchases may be discontinued at any time. We did not repurchase any common shares during the years ended December 31, 2010 and 2009.
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements have been prepared on the basis of accounting principles generally accepted in the United States of America (“GAAP”). The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and the accompanying notes. As additional information becomes available or actual amounts become determinable, the recorded estimates are revised and reflected in the operating results. Actual results could differ from those estimates.
Consolidation
The consolidated financial statements include the accounts of Progenics and PSMA LLC, as of and for the years ended December 31, 2010, 2009 and 2008. Inter-company transactions have been eliminated in consolidation.
Revenue Recognition
We recognize revenue from all sources based on the provisions of the SEC’s Staff Accounting Bulletin (“SAB”) No. 104 (“SAB 104”) and ASC 605 Revenue Recognition.
Collaborations may contain substantive milestone payments to which we apply the substantive milestone method (“Substantive Milestone Method”). Substantive milestone payments are considered to be performance payments that are recognized upon achievement of the milestone only if all of the following conditions are met: (i) the milestone payment is non-refundable, (ii) achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement, (iii) substantive effort is involved in achieving the milestone, (iv) the amount of the milestone payment is reasonable in relation to the effort expended or the risk associated with achievement of the milestone, and (v) a reasonable amount of time passes between the upfront license payment and the first milestone payment as well as between each subsequent milestone payment.
Determination as to whether a milestone meets the aforementioned conditions involves management’s judgment. If any of these conditions are not met, the resulting payment would not be considered a substantive milestone and, therefore, the resulting payment would be part of the consideration and be recognized as revenue as such performance obligations are performed.
Non-refundable upfront license fees are recognized as revenue when we have a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and we have no further performance obligations. Multiple element arrangements, such as license and development arrangements are analyzed to determine whether the deliverables, which often include a license and performance obligations, such as research and steering or other committee services, can be separated in accordance with ASC 605 Revenue Recognition. We would recognize upfront license payments as revenue upon delivery of the license only if the license had standalone value and the fair value of the undelivered performance obligations could be determined. If the fair value of the undelivered performance obligations could be determined, such obligations would then be accounted for separately as performed. If the license is considered to either (i) not have standalone value, or (ii) have standalone value but the fair value of any of the undelivered performance obligations could not be determined, the upfront license payments would be recognized as revenue over the estimated period of when our performance obligations are performed. Any unamortized remainder of the upfront payment is recognized upon termination of collaborations.
F-8
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
We must determine the period over which our performance obligations are performed and revenue related to upfront license payments are recognized. Revenue is recognized using either a proportionate performance or straight-line method. We recognize revenue using the proportionate performance method provided that we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Direct labor hours or full-time equivalents will typically be used as the measure of performance. Under the proportionate performance method, revenue related to upfront license payments is recognized in any period as the percent of actual effort expended in that period relative to total effort for all of our performance obligations under the arrangement.
During the course of a collaboration agreement that involves a development plan and budget, the amount of the upfront license payment that is recognized as revenue in any period increases or decreases as the percentage of actual effort increases or decreases. When a new budget is approved, the remaining unrecognized amount of the upfront license fee is recognized prospectively, by applying the changes in the total estimated effort or period of development that is specified in the revised approved budget.
If we cannot reasonably estimate the level of effort required to complete our performance obligations under an arrangement and the performance obligations are provided on a best-efforts basis, then the total upfront license payments would be recognized as revenue on a straight-line basis over the period we expect to complete our performance obligations.
If we are involved in a steering or other committee as part of a multiple element arrangement, we assess whether our involvement constitutes a performance obligation or a right to participate. For those committees that are deemed obligations, we will evaluate our participation along with other obligations in the arrangement and will attribute revenue to our participation through the period of our committee responsibilities.
We recognize revenue for payments that are contingent upon performance solely by our collaborator immediately upon the achievement of the defined event if we have no related performance obligations.
Reimbursement of costs is recognized as revenue provided the provisions of ASC 605 Revenue Recognition are met, the amounts are determinable and collection of the related receivable is reasonably assured.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue on the accompanying Consolidated Balance Sheets. Amounts not expected to be recognized within one year of the balance sheet date are classified as long-term deferred revenue. The estimate of the classification of deferred revenue as short-term or long-term is based upon management’s current operating budget with the collaborator for the total effort required to complete our performance obligations under the arrangement.
Royalty revenue is recognized in the period the sales occur, provided that the royalty amounts are fixed or determinable, collection of the related receivable is reasonably assured and we have no remaining performance obligations under the arrangement providing for the royalty. If royalties are received when we have remaining performance obligations, they would be attributed to the services being provided under the arrangement and, therefore, recognized as such obligations are performed under either the proportionate performance or straight-line methods, as applicable.
During the years ended December 31, 2010, 2009 and 2008, we also recognized revenue from government research grants (and contract in the 2008 period), which are used to subsidize a portion of certain of our research projects (“Projects”), exclusively from the National Institutes of Health (“NIH”). We also recognized revenue from the sale of research reagents during those periods.
NIH grant and contract revenue is recognized as efforts are expended and as related subsidized project costs are incurred. We perform work under the NIH grants and contract on a best-effort basis. The NIH reimburses us for costs associated with projects in the fields of virology and cancer, including pre-clinical research, development and early clinical testing of a prophylactic vaccine designed to prevent HIV from becoming established in uninfected individuals exposed to the virus, as requested by the NIH.
F-9
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Transition Agreement with Wyeth – October 2009
The Transition Agreement provides for the termination of the 2005 Wyeth collaboration agreement and the transition to Progenics of the rights to develop and commercialize RELISTOR. Under it, Wyeth’s license of Progenics technology is terminated except as necessary for performance of Wyeth’s obligations during the transition period and Wyeth has returned the rights to RELISTOR that we had previously granted under the 2005 collaboration agreement. During the transition, Wyeth is obligated to pay all costs of commercialization of subcutaneous RELISTOR, including manufacturing costs, and retains all proceeds from its sale of the products, subject to royalties due to us. Decisions with respect to commercialization of the product during the transition period are to be made solely by Wyeth. We have no further obligations to Wyeth under the 2005 collaboration agreement.
Wyeth Collaboration Agreement – December 2005 to October 2009
The Wyeth collaboration agreement was in effect until October 2009, which includes periods covered by this report. The Wyeth Collaboration Agreement involved three formulations of RELISTOR: (i) a subcutaneous formulation to be used in patients with opioid-induced constipation (“OIC”), (ii) an intravenous formulation to be used in patients with post-operative ileus (“POI”) and (iii) an oral formulation to be used in patients with OIC.
The Wyeth Collaboration Agreement established a Joint Steering Committee (“JSC”) and a Joint Development Committee (“JDC”) to coordinate the companies’ key activities and development of RELISTOR by Wyeth and us. A Joint Commercialization Committee (“JCC”) facilitated open communication between Wyeth and us on commercialization matters. The agreement included a non-refundable upfront license fee, reimbursement of development costs, research and development payments based upon our achievement of clinical development milestones, contingent payments based upon the achievement by Wyeth of defined events and royalties on product sales. We recognized revenue from (i) research from January 1, 2006 to October 2009, (ii) the upfront license payment we received from Wyeth using the proportionate performance method, (iii) non-refundable milestone payments and (iv) royalties.
During the third quarter of 2007, a revised development budget was approved by both us and Wyeth which extended the period over which our obligations were to be performed and the upfront payment was to be amortized from the end of 2008 to the end of 2009. The Transition Agreement between Wyeth and us shortened the obligation period from the end of 2009 to October 2009 and resulted in the recognition, during the fourth quarter of 2009, of the remaining $5.2 million unamortized upfront payment balance at September 30, 2009.
In relation to the Wyeth collaboration, we assessed the nature of our involvement with the JSC, JDC and JCC. Our involvement in the first two such committees was one of several obligations to develop the subcutaneous and intravenous formulations of RELISTOR through regulatory approval in the U.S. We combined the committee obligations with the other development obligations and accounted for these obligations during the development phase as a single unit of accounting. After the period during which we have developmental responsibilities, however, we assessed the nature of our involvement with the committees as a right, rather than an obligation. Our assessment was based upon the fact that we negotiated to be on these committees as an accommodation for our granting of the license for RELISTOR to Wyeth. Further, Wyeth had been granted by us an exclusive worldwide license, even as to us, to develop and commercialize RELISTOR and we had assigned the agreements for the manufacture of RELISTOR by third parties to Wyeth. We were responsible for developing the subcutaneous and intravenous formulations in the U.S. until they receive regulatory approval, while Wyeth was responsible for these formulations outside the U.S. other than Japan. Wyeth was also responsible for the development of the oral formulation worldwide excluding Japan. We transferred to Wyeth all existing supply agreements with third parties for RELISTOR and sublicensed intellectual property rights to permit Wyeth to manufacture RELISTOR, during the development and commercialization phases of the Wyeth Collaboration Agreement, in both bulk and finished form for all products worldwide. We had no further manufacturing obligations under the 2005 Collaboration. We transferred to Wyeth all know-how, as defined, related to RELISTOR. Based upon our research and development programs, such period will cease upon completion of our development obligations under the 2005 Wyeth collaboration agreement.
Following regulatory approval of the subcutaneous and intravenous formulations of RELISTOR, Wyeth was required to continue to develop the oral formulation and to commercialize all formulations as provided in the Wyeth Collaboration Agreement, for which it was capable and responsible. We expected at the beginning of the agreement, that the activities of these committees for the period were to be focused on Wyeth’s development and commercialization obligations. As discussed in Note 1, we and Wyeth terminated our collaboration in October 2009, as a result of which we regained all worldwide rights to RELISTOR and our out-license to Ono, with respect to Japan, is unaffected by the termination of the Wyeth collaboration.
F-10
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
In addition to the upfront payment and reimbursement of our development costs, Wyeth made milestone payments to us upon achievement of specific milestones (development related milestones for clinical and regulatory events). Upon achievement of defined substantive development milestones by us for the subcutaneous and intravenous formulations, the milestone payments were recognized as revenue. During April 2008 and July 2008, we earned $15.0 million and $10.0 million, respectively, upon achievement of non-refundable milestones anticipated in the Wyeth collaboration; the first for the FDA approval of subcutaneous RELISTOR and the second for the European approval of subcutaneous RELISTOR. We considered those milestones to be substantive based on the significant degree of risk at the inception of the collaboration related to the conduct and successful completion of clinical trials and, therefore, of not achieving the milestones; the amount of the payment received relative to the significant costs incurred since inception of the Wyeth collaboration and amount of effort expended or the risk associated with the achievement of these milestones; and the passage of 28 and 31 months, respectively, from inception of the collaboration to the achievement of those milestones. Therefore, we recognized the milestone payments as revenue in the respective periods in which the milestones were earned.
In addition, during years ended December 31, 2010 and 2009, we earned royalties of $1,826 and $1,853, respectively, based on the net sales of subcutaneous RELISTOR, and we recognized $1,826 and $2,372, respectively, of royalty income. During the fourth quarter of 2010, no royalties were payable to us. The remaining deferred royalty revenue balance of $807, as of September 30, 2009, was recognized as royalty income during the fourth quarter of 2009, the period in which our development obligations under the Wyeth Collaboration Agreement terminated. We incurred $241 and $185, respectively, of royalty costs and recognized $241 and $237, respectively, of royalty expenses during the years ended December 31, 2010 and 2009. The remaining deferred royalty charges balance of $81, as of September 30, 2009, was recognized as royalty expense during the fourth quarter 2009, the period in which our development obligations relating to RELISTOR terminated.
Ono Agreement – October 2008
Ono is responsible for developing and commercializing subcutaneous RELISTOR in Japan, including conducting the clinical development necessary to support regulatory marketing approval. Ono will own the subcutaneous filings and approvals relating to RELISTOR in Japan. In addition to the $15.0 million upfront payment from Ono, we are entitled to receive up to an additional $20.0 million, payable upon achievement by Ono of its development milestones. Ono is also obligated to pay to us royalties and commercialization milestones on sales by Ono of subcutaneous RELISTOR in Japan. Ono has the option to acquire from us the rights to develop and commercialize in Japan other formulations of RELISTOR, including intravenous and oral forms, on terms to be negotiated separately. Ono may request us to perform activities related to its development and commercialization responsibilities beyond our participation in joint committees and specified technology transfer-related tasks which will be at its expense, and payable to us for the services it requests, at the time we perform services for them. Revenue earned from activities we perform for Ono is recorded in research and development revenue.
We recognized the upfront payment of $15.0 million, which we received from Ono in November 2008, as research and development revenue during the first quarter of 2009, upon satisfaction of our performance obligations.
Research and Development Expenses
Research and development expenses include costs directly attributable to the conduct of research and development programs, including the cost of salaries, payroll taxes, employee benefits, materials, supplies, maintenance of research equipment, costs related to research collaboration and licensing agreements, the purchase of in-process research and development, the cost of services provided by outside contractors, including services related to the our clinical trials, clinical trial expenses, the full cost of manufacturing drug for use in research, pre-clinical development and clinical trials. All costs associated with research and development are expensed as incurred.
Clinical trial expenses, which are included in research and development expenses, represent obligations resulting from our contracts with various clinical investigators and clinical research organizations in connection with conducting clinical trials for our product candidates. Such costs are expensed as incurred, and are based on the expected total number of patients in the trial, the rate at which the patients enter the trial and the period over which clinical investigators or clinical research organizations are expected to provide services. At each period end, we evaluate the accrued expense balance related to these activities based upon information received from the suppliers and estimated progress towards completion of the research or development objectives to ensure that the balance is reasonably stated. Such estimates are subject to change as additional information becomes available.
F-11
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Use of Estimates
Significant estimates include useful lives of fixed assets, the periods over which certain revenues and expenses will be recognized, including research and development revenue recognized from non-refundable up-front licensing payments and expense recognition of certain clinical trial costs which are included in research and development expenses, the amount of non-cash compensation costs related to share-based payments to employees and non-employees and the periods over which those costs are expensed and the likelihood of realization of deferred tax assets.
Patents
As a result of research and development efforts conducted by us, we have applied, or are applying, for a number of patents to protect proprietary inventions. All costs associated with patents are expensed as incurred.
Net Loss Per Share
We prepare our earnings per share (“EPS”) data in accordance with ASC 260 Earnings Per Share. Basic net loss per share amounts have been computed by dividing net loss by the weighted-average number of shares of common shares outstanding during the period. In June 2008, the FASB updated ASC 260 Earnings Per Share by requiring entities, when calculating EPS, to allocate earnings to unvested and contingently issuable share-based payment awards that have non-forfeitable rights to dividends or dividend equivalents when calculating EPS and also present both basic EPS and diluted EPS pursuant to the two-class method. The update to ASC 260 Earnings Per Share was effective January 1, 2009 and required retrospective application. We adopted this update on January 1, 2009 and the adoption had no material impact on basic and diluted earnings per share for the years ended December 31, 2010, 2009 and 2008. Potential common shares, amounts of unrecognized compensation expense and windfall tax benefits have been excluded from diluted net loss per share since they would be anti-dilutive.
Concentrations of Credit Risk
Financial instruments that potentially subject Progenics to concentrations of credit risk consist of cash, cash equivalents, marketable and auction rate securities and receivables from Wyeth, Ono or the NIH. We invest our excess cash in money market funds. We have established guidelines that relate to credit quality, diversification and maturity and that limit exposure to any one issue of securities. We hold no collateral for these financial instruments.
Cash and Cash Equivalents
We consider all highly liquid investments which have maturities of three months or less, when acquired, to be cash equivalents. The carrying amount reported in the balance sheet for cash and cash equivalents approximates its fair value. Cash and cash equivalents subject us to concentrations of credit risk. At December 31, 2010 and 2009, we have invested approximately $43,958 and $84,169, respectively, in cash equivalents in the form of money market funds with one major investment company and held approximately $3,960 and $6,734, respectively, in a single commercial bank.
Marketable and Auction Rate Securities
In accordance with ASC 320 Investments – Debt and Equity Securities, investments are classified as available-for-sale. Available-for-sale securities are carried at fair value, with the unrealized gains and losses reported in comprehensive income (loss). The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income or expense. Realized gains and losses and declines in value judged to be other-than-temporary, if any, on available-for-sale securities are included in other income or expense. In computing realized gains and losses, we compute the cost of its investments on a specific identification basis. Such cost includes the direct costs to acquire the securities, adjusted for the amortization of any discount or premium. The fair value of marketable securities has been estimated based on a three-level hierarchy for fair value measurements. Interest and dividends on securities classified as available-for-sale are included in interest income (see Note 3).
F-12
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
At December 31, 2010 and 2009, our investment in auction rate securities in the long term assets section of the Consolidated Balance Sheets amounted to $3.6 million and $3.8 million, respectively. Valuation of securities is subject to uncertainties that are difficult to predict, such as changes to credit ratings of the securities and/or the underlying assets supporting them, default rates applicable to the underlying assets, underlying collateral value, discount rates, counterparty risk, ongoing strength and quality of market credit and liquidity and general economic and market conditions. The valuation of the auction rate securities we hold is based on an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security. As a result of the estimated fair value, we re-evaluated the valuation of these securities as of December 31, 2010 and the temporary impairment amount decreased $16 from $308 at December 31, 2009 to $292. All income generated from these investments was recorded as interest income (see Note 3).
Fair Value Measurements
In accordance with ASC 820 Fair Value Measurements and Disclosures, we use a three-level hierarchy for fair value measurements that distinguishes between market participant assumptions developed from market data obtained from sources independent of the reporting entity (“observable inputs”) and the reporting entity’s own assumptions about market participant assumptions developed from the best information available in the circumstances (“unobservable inputs”). The hierarchy level assigned to each security in our available-for-sale portfolio is based on our assessment of the transparency and reliability of the inputs used in the valuation of such instrument at the measurement date. The three hierarchy levels are defined as follows:
|
|
· Level 1 - Valuations based on unadjusted quoted market prices in active markets for identical securities.
|
|
|
· Level 2 - Valuations based on observable inputs other than Level 1 prices, such as quoted prices for similar assets at the measurement date, quoted prices in markets that are not active or other inputs that are observable, either directly or indirectly.
|
|
|
· Level 3 - Valuations based on inputs that are unobservable and significant to the overall fair value measurement, and involve management judgment.
|
Other current assets are comprised of prepaid expenses, interest and other receivables of $1,801 and $1,468 at December 31, 2010 and 2009, respectively, which are expected to be settled within one year. Other assets of $1,250 at December 31, 2010 include $1,050, which represents the long term portions of amounts prepaid to a clinical research organization. Restricted cash of $200 at both December 31, 2010 and 2009, respectively, consists of collateral for a letter of credit securing lease obligations. We believe that carrying value of those assets approximates fair value.
Fixed Assets
Leasehold improvements, furniture and fixtures, and equipment are stated at cost. Furniture, fixtures and equipment are depreciated on a straight-line basis over their estimated useful lives. Leasehold improvements are amortized on a straight-line basis over the life of the lease or of the improvement, whichever is shorter. Costs of construction of long-lived assets are capitalized but are not depreciated until the assets are placed in service.
Expenditures for maintenance and repairs which do not materially extend the useful lives of the assets are charged to expense as incurred. The cost and accumulated depreciation of assets retired or sold are removed from the respective accounts and any gain or loss is recognized in operations. The estimated useful lives of fixed assets are as follows:
|
Computer equipment
|
3 years
|
|
Machinery and equipment
|
5-7 years
|
|
Furniture and fixtures
|
5 years
|
|
Leasehold improvements
|
Earlier of life of improvement or lease
|
F-13
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Deferred Lease Liability and Incentive
Our lease agreements include fixed escalations of minimum annual lease payments and we recognize rental expense on a straight-line basis over the lease terms and record the difference between rent expense and current rental payments as deferred rent. Deferred lease incentive includes a construction allowance from our landlord which is amortized as a reduction to rental expense on a straight-line basis over the lease terms. As of December 31, 2010, the Consolidated Balance Sheets include deferred lease liability of $400 in Other Liabilities and deferred lease incentive of $112 and $1,004 in Other Current Liabilities and Other Liabilities, respectively.
Impairment of Long-Lived Assets
We periodically assess the recoverability of fixed assets and evaluate such assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. In accordance with ASC 360 Property, Plant, and Equipment – Impairment or Disposal of Long-Lived Assets, if indicators of impairment exist, we assess the recoverability of the affected long-lived assets by determining whether the carrying value of such assets can be recovered through undiscounted future operating cash flows. If the carrying amount is not recoverable, we measure the amount of any impairment by comparing the carrying value of the asset to the present value of the expected future cash flows associated with the use of the asset. No impairments occurred as of December 31, 2010, 2009 or 2008.
Income Taxes
We account for income taxes in accordance with the provisions of ASC 740 Income Taxes, which requires that we recognize deferred tax liabilities and assets for the expected future tax consequences of events that have been included in the financial statements or tax returns. Under this method, deferred tax assets and liabilities are determined on the basis of the difference between the tax basis of assets and liabilities and their respective financial reporting amounts (“temporary differences”) at enacted tax rates in effect for the years in which the temporary differences are expected to reverse. A valuation allowance is established for deferred tax assets for which realization is uncertain.
In accordance with ASC 718 Compensation – Stock Compensation and ASC 505 Equity, we have made a policy decision related to intra-period tax allocation, to account for utilization of windfall tax benefits based on provisions in the tax law that identify the sequence in which amounts of tax benefits are used for tax purposes (i.e., tax law ordering).
Uncertain tax positions are accounted for in accordance with ASC 740 Income Taxes, which prescribes a comprehensive model for the manner in which a company should recognize, measure, present and disclose in its financial statements all material uncertain tax positions that we have taken or expect to take on a tax return. ASC 740 applies to income taxes and is not intended to be applied by analogy to other taxes, such as sales taxes, value-add taxes, or property taxes. We review our nexus in various tax jurisdictions and our tax positions related to all open tax years for events that could change the status of our ASC 740 liability, if any, or require an additional liability to be recorded. Such events may be the resolution of issues raised by a taxing authority, expiration of the statute of limitations for a prior open tax year or new transactions for which a tax position may be deemed to be uncertain. Those positions, for which management’s assessment is that there is more than a 50 percent probability of sustaining the position upon challenge by a taxing authority based upon its technical merits, are subjected to the measurement criteria of ASC 740. We record the largest amount of tax benefit that is greater than 50 percent likely of being realized upon ultimate settlement with a taxing authority having full knowledge of all relevant information. Any ASC 740 liabilities for which we expect to make cash payments within the next twelve months are classified as “short term.” In the event that we conclude that we are subject to interest and/or penalties arising from uncertain tax positions, we will record interest and penalties as a component of income taxes (see Note 11).
Risks and Uncertainties
We have to date relied principally on external funding, the Wyeth collaboration, royalty and product revenue, and except for RELISTOR have no products approved by the FDA for marketing. There can be no assurance that our research and development will be successfully completed, that any products developed will obtain necessary marketing approval by regulatory authorities or that any approved products will be commercially viable. In addition, we operate in an environment of rapid change in technology, and we are dependent upon satisfactory relationships with our partners and the continued services of our current employees, consultants and subcontractors. We are also dependent upon Salix, Wyeth (until the completion of its involvement) and/or Ono fulfilling their manufacturing obligations, either on their own or through third-party suppliers. For the years ended December 31, 2010, 2009 and 2008, the primary sources of our revenues were Wyeth, Ono and research grant and contract revenues from the NIH. There can be no assurance that revenues from Wyeth, Ono or from research awards will continue or that we will recognize any revenue from the recently completed License Agreement with Salix. Substantially all of our accounts receivable at December 31, 2010 and 2009 were from the above-named sources.
F-14
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Comprehensive Loss
Comprehensive loss represents the change in net assets of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. Our comprehensive loss includes net loss adjusted for the change in net unrealized gain or loss on marketable and auction rate securities. The disclosures required by ASC 220 Comprehensive Income for the years ended December 31, 2010, 2009 and 2008 have been included in the Consolidated Statements of Stockholders’ Equity and Comprehensive Loss. There was no income tax expense/benefit allocated to any component of Other Comprehensive Loss (see Note 11).
Impact of Recently Adopted Accounting Standards
In January 2010, the FASB issued ASU 2010-06, which amends ASC 820 to add new requirements for disclosure about transfers into and out of Levels 1 and 2 and separate disclosures about purchases, sales, issuances and settlements on a gross basis for Level 3 measurements. The ASU also clarifies existing requirements for fair value disclosures about inputs and valuation techniques used to measure fair value for Levels 2 and 3. The adoption of this update did not have a material effect on our consolidated financial statements.
3. Fair Value Measurements
Our available-for-sale investment portfolio consists of auction rate securities and corporate debt securities (in the 2009 period) and is recorded at fair value in the accompanying Consolidated Balance Sheets in accordance with ASC 320 Investments – Debt and Equity Securities. The change in the fair value of these securities is recorded as a component of other comprehensive loss (see Note 2).
Marketable and auction rate securities as of December 31, 2010 and 2009, consisted of the following:
|
2010
|
2009
|
|||||||
|
Short-term
|
||||||||
|
Corporate debt securities
|
$ | - | $ | 1,501 | ||||
|
Total short-term marketable securities
|
- | 1,501 | ||||||
|
Long-term
|
||||||||
|
Auction rate securities
|
3,608 | 3,792 | ||||||
|
Total long-term auction rate securities
|
3,608 | 3,792 | ||||||
|
Total marketable and auction rate securities
|
$ | 3,608 | $ | 5,293 | ||||
The following table presents our available-for-sale investments measured at fair value on a recurring basis as of December 31, 2010 and 2009, classified by valuation hierarchy (as previously discussed):
|
Fair Value Measurements at December 31, 2010
|
||||||||||||||||
|
Balance at
December 31, 2010
|
Quoted Prices in Active Markets for Identical Assets
(Level 1)
|
Significant Other Observable Inputs
(Level 2)
|
Significant Unobservable Inputs
(Level 3)
|
|||||||||||||
|
Money market funds
|
$ | 43,958 | $ | 43,958 | $ | - | $ | - | ||||||||
|
Auction rate securities
|
3,608 | - | - | 3,608 | ||||||||||||
|
Total
|
$ | 47,566 | $ | 43,958 | $ | - | $ | 3,608 | ||||||||
F-15
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
|
Fair Value Measurements at December 31, 2009
|
||||||||||||||||
|
Balance at
December 31, 2009
|
Quoted Prices in Active Markets for Identical Assets
(Level 1)
|
Significant Other Observable Inputs
(Level 2)
|
Significant Unobservable Inputs
(Level 3)
|
|||||||||||||
|
Money market funds
|
$ | 84,169 | $ | 84,169 | $ | - | $ | - | ||||||||
|
Corporate debt securities
|
1,501 | - | 1,501 | - | ||||||||||||
|
Auction rate securities
|
3,792 | - | - | 3,792 | ||||||||||||
|
Total
|
$ | 89,462 | $ | 84,169 | $ | 1,501 | $ | 3,792 | ||||||||
At December 31, 2010, we hold $3.6 million (7.6% of total assets measured at fair value) in auction rate securities which are classified as Level 3. The fair value of these securities includes $2.7 million of U.S. government subsidized securities collateralized by student loan obligations and $0.9 million of investment company perpetual preferred stock. Auction rate securities are collateralized long-term instruments that were intended to provide liquidity through an auction process that resets interest rates at pre-determined intervals. Beginning in 2008, auctions failed for certain of our auction rate securities and we were unable to dispose of those securities at auction. We will not realize cash in respect of the principal amount of these securities until the issuer calls or restructures the security, the security reaches any scheduled maturity and is paid (which is inapplicable to the perpetual preferred mentioned above) or a buyer outside the auction process emerges. As of December 31, 2010, we have received all scheduled interest payments on these securities, which, in the event of auction failure, are reset according to the contractual terms in the governing instruments.
The valuation of auction rate securities we hold is based on Level 3 unobservable inputs which consist of our internal analysis of (i) timing of expected future successful auctions, (ii) collateralization of underlying assets of the security and (iii) credit quality of the security. We re-evaluated the valuation of these securities as of December 31, 2010 and the temporary impairment amount decreased $16.0 from $308.0 at December 31, 2009, to $292.0, which is reflected as a part of other comprehensive loss on our accompanying Consolidated Balance Sheets. These securities are held “available-for-sale” and the unrealized loss is included in other comprehensive loss. Due to the uncertainty related to the liquidity in the auction rate security market and therefore when individual positions may be liquidated, we have classified these auction rate securities as long-term assets on our accompanying Consolidated Balance Sheets. We continue to monitor markets for our investments and consider the impact, if any, of market conditions on the fair market value of our investments. We do not believe the carrying values of our investments are other than temporarily impaired and therefore expect the positions will eventually be liquidated without significant loss.
For those of our financial instruments with significant Level 3 inputs (all of which are auction rate securities), the following tables summarize the activities for the years ended December 31, 2010 and 2009:
|
Fair Value Measurements Using Significant
Unobservable Inputs
(Level 3)
|
||||||||
|
Description
|
2010
|
2009
|
||||||
|
Balance at beginning of period
|
$ | 3,792 | $ | 4,059 | ||||
|
Transfers into Level 3
|
- | - | ||||||
|
Total realized/unrealized gains (losses)
|
||||||||
|
Included in net loss
|
- | - | ||||||
|
Included in comprehensive income (loss) (1)
|
16 | 8 | ||||||
|
Settlements
|
(200 | ) | (275 | ) | ||||
|
Balance at end of period
|
$ | 3,608 | $ | 3,792 | ||||
|
(1) Total amount of unrealized gains (losses) for the period included in other comprehensive loss attributable to the change in fair market value of related assets still held at the reporting date
|
$ | - | $ | - | ||||
F-16
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
The following tables summarize the amortized cost basis, the aggregate fair value and gross unrealized holding gains and losses at December 31, 2010 and 2009:
|
Amortized
|
Fair
|
Unrealized Holding
|
||||||||||||||||||
|
Cost Basis
|
Value
|
Gains
|
(Losses)
|
Net
|
||||||||||||||||
|
2010:
|
||||||||||||||||||||
|
Maturities greater than ten years:
|
||||||||||||||||||||
|
Auction rate securities
|
$ | 2,900 | $ | 2,668 | $ | - | $ | (232 | ) | $ | (232 | ) | ||||||||
|
Investments without stated maturity dates:
|
||||||||||||||||||||
|
Auction rate securities
|
1,000 | 940 | - | (60 | ) | (60 | ) | |||||||||||||
| $ | 3,900 | $ | 3,608 | $ | - | $ | (292 | ) | $ | (292 | ) | |||||||||
|
Amortized
|
Fair
|
Unrealized Holding
|
||||||||||||||||||
|
Cost Basis
|
Value
|
Gains
|
(Losses)
|
Net
|
||||||||||||||||
|
2009:
|
||||||||||||||||||||
|
Maturities less than one year:
|
||||||||||||||||||||
|
Corporate debt securities
|
$ | 1,500 | $ | 1,501 | $ | 1 | $ | - | $ | 1 | ||||||||||
|
Maturities greater than ten years:
|
||||||||||||||||||||
|
Auction rate securities
|
3,100 | 2,852 | - | (248 | ) | (248 | ) | |||||||||||||
|
Investments without stated maturity dates:
|
||||||||||||||||||||
|
Auction rate securities
|
1,000 | 940 | - | (60 | ) | (60 | ) | |||||||||||||
| $ | 5,600 | $ | 5,293 | $ | 1 | $ | (308 | ) | $ | (307 | ) | |||||||||
We compute the cost of its investments on a specific identification basis. Such cost includes the direct costs to acquire the securities, adjusted for the amortization of any discount or premium.
The following table shows the gross unrealized losses and fair value of our marketable securities with unrealized losses that are not deemed to be other-than-temporarily impaired, aggregated by investment category and length of time that individual securities have been in a continuous unrealized loss position, at December 31, 2010 and 2009.
2010:
|
Less than 12 Months
|
12 Months or Greater
|
Total
|
||||||||||||||||||||||
|
Description of Securities
|
Fair Value
|
Unrealized Losses
|
Fair Value
|
Unrealized Losses
|
Fair Value
|
Unrealized Losses
|
||||||||||||||||||
|
Auction rate securities
|
$ | - | $ | - | $ | 3,608 | $ | (292 | ) | $ | 3,608 | $ | (292 | ) | ||||||||||
|
Total
|
$ | - | $ | - | $ | 3,608 | $ | (292 | ) | $ | 3,608 | $ | (292 | ) | ||||||||||
2009:
|
Less than 12 Months
|
12 Months or Greater
|
Total
|
||||||||||||||||||||||
|
Description of Securities
|
Fair Value
|
Unrealized Losses
|
Fair Value
|
Unrealized Losses
|
Fair Value
|
Unrealized Losses
|
||||||||||||||||||
|
Auction rate securities
|
$ | - | $ | - | $ | 3,792 | $ | (308 | ) | $ | 3,792 | $ | (308 | ) | ||||||||||
|
Total
|
$ | - | $ | - | $ | 3,792 | $ | (308 | ) | $ | 3,792 | $ | (308 | ) | ||||||||||
Other-than-temporary impairment analysis on auction rate securities. The unrealized losses in our auction rate securities investments were the result of an internal analysis of timing of expected future successful auctions, collateralization of underlying assets of the security and credit quality of the security. At December 31, 2010 and 2009, there were two securities with a gross unrealized loss position of $292 and $308 ($3,608 and $3,792 of the total fair value), respectively.
The severity of the unrealized losses for auction rate securities at December 31, 2010 and 2009 was between 6 percent and 8 percent below amortized cost, and the weighted average duration of the unrealized losses for these securities was 34 and 22 months, respectively.
F-17
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
We have evaluated our individual auction rate securities holdings for other-than-temporary impairment and determined that the unrealized losses as of December 31, 2010 and 2009 are attributable to uncertainty in the liquidity of the auction rate security market. Because we do not intend to sell these securities, and believe it is not more likely than not that we would be required to sell these securities before recovery of principal, we do not consider these securities to be other-than-temporarily impaired at December 31, 2010 and 2009.
4. Accounts Receivable
Our accounts receivable represent amounts due to Progenics from research from collaborator, royalties, research grants and the sales of research reagents. These amounts are considered to be short-term as they are expected to be collected within one year and we believe carrying value approximates fair value. Accounts receivable as of December 31, 2010 and 2009, consisted of the following:
|
2010
|
2009
|
|||||||
|
National Institutes of Health
|
$ | 468 | $ | 210 | ||||
|
Royalties
|
- | 589 | ||||||
|
Research and development from collaborator
|
1,811 | 6,667 | ||||||
|
Other
|
4 | 56 | ||||||
|
Total
|
$ | 2,283 | $ | 7,522 | ||||
5. Fixed Assets
Fixed assets as of December 31, 2010 and 2009, consisted of the following:
|
2010
|
2009
|
||||||
|
Computer equipment
|
$
|
2,508
|
$
|
2,443
|
|||
|
Machinery and equipment
|
13,380
|
13,237
|
|||||
|
Furniture and fixtures
|
740
|
740
|
|||||
|
Leasehold improvements
|
13,354
|
10,662
|
|||||
|
Construction in progress
|
74
|
831
|
|||||
|
30,056
|
27,913
|
||||||
|
Less, accumulated depreciation and amortization
|
(24,178
|
)
|
(21,353
|
)
|
|||
|
Total
|
$
|
5,878
|
$
|
6,560
|
|||
At December 31, 2010, $2.7 million of leasehold improvements were being amortized over periods of 10.0-10.8 years, under leases with terms through December 31, 2020. At December 31, 2009, $5.9 million and $1.6 million of leasehold improvements were being amortized over periods of 0.3 – 5.8 years and 2.0 – 4.0 years, respectively, under leases with terms through December 31, 2009 and June 30, 2010, respectively.
6. Accounts Payable and Accrued Expenses
The carrying value of our accounts payable and accrued expenses approximates fair value, as it represents amounts due to vendors and employees, which will be satisfied within one year. Accounts payable and accrued expenses as of December 31, 2010 and 2009, consisted of the following:
|
2010
|
2009
|
|||||||
|
Accounts payable
|
$ | 658 | $ | 596 | ||||
|
Accrued consulting and clinical trial costs
|
6,125 | 2,663 | ||||||
|
Accrued payroll and related costs
|
1,725 | 1,321 | ||||||
|
Legal and professional fees
|
1,116 | 1,070 | ||||||
|
Other
|
59 | 186 | ||||||
|
Total
|
$ | 9,683 | $ | 5,836 | ||||
F-18
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
7. Stockholders’ Equity
We are authorized to issue 40,000 shares of common stock, par value $.0013 (“Common Stock”), and 20,000 shares of preferred stock, par value $.001. The Board of Directors has the authority to issue common and preferred shares, in series, with rights and privileges as determined by the Board of Directors.
On April 24, 2008, our Board of Directors approved a share repurchase program to acquire up to $15.0 million of our outstanding common shares. During the year ended December 31, 2008, we have repurchased 200,000 of our outstanding common shares for a total of $2.7 million. Purchases may be discontinued at any time. We did not repurchase any common shares during the years ended December 31, 2010 and 2009. We have $12.3 million remaining available for purchases under the program.
8. Commitments and Contingencies
a. Operating Leases
As of December 31, 2010, we leased a total of 135,600 square feet of office, manufacturing and laboratory space, under lease agreements expiring in June 2012 and December 2020.
Rental payments are recognized as rent expense on a straight-line basis over the term of the lease. In addition to rents due under these agreements, we are obligated to pay additional facilities charges, including utilities, taxes and operating expenses. We also lease certain office equipment under non-cancelable operating leases, which expire at various times through April 2013.
As of December 31, 2010, future minimum annual payments under all operating lease agreements are as follows:
|
Years ending
December 31,
|
Minimum
Annual Payments
|
|
|
2011
|
$ 3,284
|
|
|
2012
|
3,272
|
|
|
2013
|
3,615
|
|
|
2014
|
4,067
|
|
|
2015
|
4,169
|
|
|
Thereafter
|
22,462
|
|
|
Total
|
$ 40,869
|
Rental expense totaled approximately $3,544, $2,773 and $2,971 for the years ended December 31, 2010, 2009 and 2008, respectively. For the year ended December 31, 2010, we recognized rent expense in excess of amounts paid of $181, due to the recognition of escalation clauses and lease incentives. For the years ended December 31, 2009 and 2008, amounts paid exceeded rent expense by $154 and $93, respectively, due to the recognition of escalation clauses and lease incentives. Additional facility charges, including utilities, taxes and operating expenses, for the years ended December 31, 2010, 2009 and 2008 were approximately $3,645, $3,060 and $3,533, respectively.
b. Licensing, Service and Supply Agreements
Progenics has entered into intellectual property-based license and service agreements in connection with their product development programs. Progenics has recognized milestone, license and sublicense fees and supply costs, which are included in research and development expenses, totaling approximately $1,266, $788 and $2,422 for the years ended December 31, 2010, 2009 and 2008, respectively.
F-19
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
|
Agreement
|
Paid from inception to December 31, 2010
|
Future (1)
Commitments
|
Terms
|
|
Progenics agreements with:
|
|||
|
Facet Biotech Corporation (formerly Protein Design Labs, Inc.)
|
$ 5,500
|
$ 2,450
|
Annual maintenance payments, milestones and royalties for the humanized murine monoclonal antibody developed by us.
|
|
University of Chicago(2)
|
1,969
|
-
|
Milestones and royalties for rights to develop and commercialize methylnaltrexone and RELISTOR.
|
|
Lonza Sales AG
|
909
|
3,867
|
Annual license fee payments, milestones and royalties, as applicable, in respect of PRO 140 and other products.
|
|
PSMA LLC agreements with:
|
|||
|
Amgen Fremont, Inc. (formerly Abgenix)
|
850
|
6,250
|
Milestones and royalties to use XenoMouse® technology for generating fully human antibodies to PSMA LLC’s PSMA antigen.
|
|
AlphaVax Human Vaccines
|
2,136
|
5,400
|
Annual maintenance payments and milestones to use AlphaVax Replicon Vector system to create a therapeutic cancer vaccine incorporating PSMA LLC’s proprietary PSMA antigen.
|
|
Seattle Genetics, Inc.
|
3,700
|
14,300
|
Milestone and periodic maintenance payments to use ADC technology to link chemotherapeutic agents to monoclonal antibodies that target prostate specific membrane antigen. ADC technology is based in part on technology licensed by SGI from third parties.
|
|
Cornell Research Foundation
|
135
|
1,100
|
Annual minimum royalty payments and milestones.
|
|
Former member of PSMA LLC
|
166
|
52,178
|
Annual minimum royalty payments and milestones to use technology related to PSMA.
|
|
|
(1) Amounts based on known contractual obligations as specified in the respective license agreements, which are dependent on the achievement of future events and exclude amounts for royalties which are dependent on future sales and are unknown.
|
|
|
(2) Includes multiple license agreements.
|
As part of our research and development efforts, we enter into consulting agreements with external scientific specialists (“Scientists”). These agreements contain various terms and provisions, including fees to be paid by us and royalties, in the event of future sales, and milestone payments, upon achievement of defined events, payable by us. Certain Scientists are advisors to Progenics, including Stephen P. Goff, Ph.D. and David A. Scheinberg, M.D., Ph.D., both of whom are also members of our Board of Directors. Some Scientists have purchased our Common Stock or received stock options which are subject to vesting provisions. We have recognized expenses with regard to the consulting agreements of $179, $220 and $358 for the years ended December 31, 2010, 2009 and 2008, respectively. Those expenses include the fair value of stock options granted during 2010, 2009 and 2008, which were fully vested at grant date, of approximately $42, $83 and $217, respectively. Such amounts of fair value are included in research and development compensation expense for each year presented (see Note 9).
9. Share-Based Payment Arrangements
Our share-based compensation to employees includes non-qualified stock options, restricted stock and shares issued under our Purchase Plans, which are compensatory under ASC 718 Compensation – Stock Compensation. We account for share-based compensation to non-employees, including non-qualified stock options and restricted stock, in accordance with ASC 505 Equity.
F-20
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Compensation cost for share-based awards will be recognized in our financial statements over the related requisite service periods; usually the vesting periods for awards with a service condition. We have made an accounting policy decision to use the straight-line method of attribution of compensation expense, under which the grant date fair value of share-based awards will be recognized on a straight-line basis over the total requisite service period for the total award.
We have adopted three stock incentive plans, the 1989 Non-Qualified Stock Option Plan, the 1996 Amended Stock Incentive Plan and the 2005 Stock Incentive Plan (the “Plans”). Under each of these Plans as amended, up to 375, 5,000 and 5,450 shares of common stock, respectively, have been reserved for the issuance of awards to employees, consultants, directors and other individuals who render services to Progenics (collectively, “Awardees”). The Plans contain certain anti-dilution provisions in the event of a stock split, stock dividend or other capital adjustment as defined. The 1989 Plan provides for the Board, or the Compensation Committee (“Committee”) of the Board, to grant stock options to Awardees and to determine the exercise price, vesting term and expiration date. The 1996 Plan and the 2005 Plan provide for the Board or Committee to grant to Awardees stock options, stock appreciation rights, restricted stock, performance awards or phantom stock, as defined (collectively, “Awards”). The Committee is also authorized to determine the term and vesting of each Award and the Committee may in its discretion accelerate the vesting of an Award at any time. Stock options granted under the Plans generally vest pro rata over four to ten years and have terms of ten to twenty years. Restricted stock issued under the 1996 Plan or 2005 Plan usually vests annually over three to four years, unless specified otherwise by the Committee. The exercise price of outstanding non-qualified stock options is usually equal to the fair value of our common stock on the date of grant. The exercise price of non-qualified stock options granted from the 2005 Plan and incentive stock options (“ISO”) granted from the Plans may not be lower than the fair value of our common stock on the dates of grant. At December 31, 2010, 2009 and 2008, all outstanding stock options were non-qualified options. The 1989 and 1996 Plans terminated in April 1994 and October 2006, respectively, and the 2005 Plan will terminate in April 2015; options granted before termination of the Plans will continue under the respective Plans until exercised, cancelled or expired.
We apply a forfeiture rate to the number of unvested awards in each reporting period in order to estimate the number of awards that are expected to vest. Estimated forfeiture rates are based upon historical data on vesting behavior of employees. We adjust the total amount of compensation cost recognized for each award, in the period in which each award vests, to reflect the actual forfeitures related to that award. Changes in our estimated forfeiture rate will result in changes in the rate at which compensation cost for an award is recognized over its vesting period.
Under ASC 718 Compensation – Stock Compensation, the fair value of each non-qualified stock option award is estimated on the date of grant using the Black-Scholes option pricing model, which requires input assumptions noted in the following table. Ranges of assumptions for inputs are disclosed where the value of such assumptions varied during the related period. Historical volatilities are based upon daily quoted market prices of our common stock on The NASDAQ Stock Market LLC over a period equal to the expected term of the related equity instruments. We rely only on historical volatility since it provides the most reliable indication of future volatility. Future volatility is expected to be consistent with historical; historical volatility is calculated using a simple average calculation; historical data is available for the length of the option’s expected term and a sufficient number of price observations are used consistently. Since our stock options are not traded on a public market, we do not use implied volatility. For the years ended December 31, 2010, 2009 and 2008, our expected term was calculated based upon historical data related to exercise and post-termination cancellation activity. Accordingly, for grants made to employees and directors and officers (excluding our Vice Chairman), we are using expected terms of 5.3 and 7.3 years, 5.3 and 7.3 years, and 5.33 and 7.3 years, respectively. The expected term of stock options granted to our Vice Chairman and non-employee consultants are calculated separately from stock options granted to employees and directors and officers and the expected term was 8 years, 8 years and 7.5 years for the years ended December 31, 2010, 2009 and 2008. Expected term for options granted to non-employee consultants was ten years, which is the contractual term of those options. We have never paid dividends and do not expect to pay dividends in the future. Therefore, our dividend rate is zero. The risk-free rate for periods within the expected term of the options is based on the U.S. Treasury yield curve in effect at the time of grant. The following table presents assumptions used in computing the fair value of option grants during 2010, 2009 and 2008:
|
2010
|
2009
|
2008
|
||||
|
Expected volatility
|
68% – 87%
|
70% – 91%
|
66% – 91%
|
|||
|
Expected dividends
|
zero
|
zero
|
zero
|
|||
|
Expected term (years)
|
5.3 – 10
|
5.3 – 10
|
5.33 – 10
|
|||
|
Weighted average expected term (years)
|
6.92
|
7.10
|
6.78
|
|||
|
Risk-free rate
|
1.21% – 3.09%
|
1.78% – 3.22%
|
1.69% – 3.79%
|
F-21
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
A summary of option activity under the Plans as of December 31, 2010 and changes during the year then ended is presented below:
|
Options
|
Shares
|
Weighted Average Exercise Price
|
Weighted Average Remaining Contractual Term (Yr.)
|
Aggregate Intrinsic Value
|
||||||||||||
|
Outstanding at January 1, 2010
|
4,909 | $ | 16.58 | |||||||||||||
|
Granted
|
987 | 5.33 | ||||||||||||||
|
Exercised
|
- | - | ||||||||||||||
|
Forfeited or expired
|
(631 | ) | 18.83 | |||||||||||||
|
Outstanding at December 31, 2010
|
5,265 | $ | 14.20 | 5.77 | $ | 397 | ||||||||||
|
Exercisable at December 31, 2010
|
3,682 | $ | 16.68 | 4.56 | $ | 234 | ||||||||||
The weighted average grant-date fair value of options granted under the Plans during the years ended December 31, 2010, 2009 and 2008 was $3.10, $3.39 and $10.09, respectively. The total intrinsic value of options exercised during the years ended December 31, 2010, 2009 and 2008 was $0, $41 and $969, respectively.
The options granted under the Plans, described above, include 33, 113, 38, 75, 145 and 113 non-qualified stock options granted to our vice chairman on July 1, 2002, 2003, 2004 and 2005, on July 3, 2006 and on July 2, 2007, respectively, which cliff vest after nine years and eleven months from the respective grant date. The July 1, 2002, 2003 and 2005 awards have fully vested. Vesting of a defined portion of each award will occur earlier if a defined performance condition is achieved; more than one condition may be achieved in any period. In accordance with ASC 718 Compensation – Stock Compensation, at the end of each reporting period, we will estimate the probability of achievement of each performance condition and will use those probabilities to determine the requisite service period of each award. The requisite service period for the award is the shortest of the explicit or implied service periods. In the case of the executive’s options, the explicit service period is nine years and eleven months from the respective grant dates. The implied service periods related to the performance conditions are the estimated times for each performance condition to be achieved. Thus, compensation expense will be recognized over the shortest estimated time for the achievement of performance conditions for that award (assuming that the performance conditions will be achieved before the cliff vesting occurs). On July 1, 2008, 2009 and 2010, we granted awards (consisting of options in 2010 and 2009 and options and restricted stock in 2008) to our Vice Chairman and Chief Executive Officer (consisting of options in 2010) which vest on the basis of the achievement of specified performance or market-based milestones. The options have an exercise price equal to the closing price on our common stock on the date of grant. The awards are valued using a Monte Carlo simulation and the expense related to these grants will be recognized over the shortest estimated time for the achievement of the performance or market conditions. The awards will not vest unless one of the milestones is achieved or the market condition is met. Changes in the estimate of probability of achievement of any performance or market condition will be reflected in compensation expense of the period of change and future periods affected by the change.
At December 31, 2010, the estimated requisite service periods for the 2004, 2006, 2008, 2009 and 2010 awards, described above, were 1.5, 5.5, 2.75, 4.25 and 4.25 years, respectively. For the years ended December 31, 2010, 2009 and 2008, the total compensation expense recognized for the performance-based options was $1.1 million, $0.5 million and $1.3 million, respectively.
F-22
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
A summary of the status of our outstanding restricted stock awarded under the Plans which has not yet vested as of December 31, 2010 and changes during the year then ended is presented below:
|
Restricted Stock Awards
|
Shares
|
Weighted Average Grant-Date
Fair Value
|
||||||
|
Nonvested at January 1, 2010
|
548 | $ | 12.82 | |||||
|
Granted
|
228 | 5.40 | ||||||
|
Vested
|
(380 | ) | 11.58 | |||||
|
Forfeited
|
(55 | ) | 11.48 | |||||
|
Nonvested at December 31, 2010
|
341 | $ | 9.46 | |||||
Our two employee stock purchase plans (the “Purchase Plans”), the 1998 Employee Stock Purchase Plan (the “Qualified Plan”) and the 1998 Non-Qualified Employee Purchase Plan (the “Non-Qualified Plan”), as amended, provide for the issuance of up to 4,400 and 1,100 shares of common stock, respectively. The Purchase Plans provide for the grant to all employees of options to use an amount equal to 25% of their quarterly compensation, as such percentage is determined by the Board of Directors prior to the date of grant, to purchase shares of our common stock at a price per share equal to the lesser of the fair market value of the common stock on the date of grant or 85% of the fair market value on the date of exercise. Options are granted automatically on the first day of each fiscal quarter and expire six months after the date of grant. The Qualified Plan is not available to employees owning more than five percent of the common stock and imposes certain other quarterly limitations on the option grants. Options under the Non-Qualified Plan are granted to the extent that option grants are restricted under the Qualified Plan.
The fair value of shares purchased under the Purchase Plans was estimated on the date of grant in accordance with ASC 718 Compensation – Stock Compensation, via the same option valuation model used for options granted under the Plans, but with the following assumptions during 2010, 2009 and 2008:
|
2010
|
2009
|
2008
|
||||
|
Expected volatility
|
45% – 72%
|
46% – 100%
|
83% – 170%
|
|||
|
Expected dividends
|
zero
|
zero
|
zero
|
|||
|
Expected term
|
6 months
|
6 months
|
6 months
|
|||
|
Risk-free rate
|
0.11% – 0.18%
|
0.00% – 0.38%
|
0.14% – 2.74%
|
Purchases of common stock under the Purchase Plans during the years ended December 31, 2010, 2009 and 2008 are summarized as follows:
|
Qualified Plan
|
Non-Qualified Plan
|
||||||||||
|
Shares Purchased
|
Price Range
|
Weighted
Average
Grant-Date Fair Value
|
Shares
Purchased
|
Price Range
|
Weighted
Average
Grant-Date Fair Value
|
||||||
|
2010
|
802
|
$3.50 – $4.56
|
$0.94
|
208
|
$3.75 – $4.56
|
$0.96
|
|||||
|
2009
|
872
|
$3.37 – $9.13
|
$1.49
|
189
|
$3.98 – $9.13
|
$1.58
|
|||||
|
2008
|
538
|
$4.26 – $15.32
|
$4.44
|
127
|
$6.07 – $15.32
|
$4.83
|
|||||
F-23
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
The total compensation expense of shares, granted to both employees and non-employees, under all of our share-based payment arrangements that was recognized in operations during the years ended December 31, 2010, 2009 and 2008 was:
|
2010
|
2009
|
2008
|
||||||||||
|
Recognized as:
|
||||||||||||
|
Research and Development
|
$ | 5,091 | $ | 7,225 | $ | 7,241 | ||||||
|
General and Administrative
|
4,424 | 5,761 | 6,892 | |||||||||
|
Total
|
$ | 9,515 | $ | 12,986 | $ | 14,133 | ||||||
No tax benefit was recognized related to such compensation cost because we had net losses for the periods presented and the related deferred tax assets were fully offset by valuation allowance. Accordingly, no amounts related to windfall tax benefits have been reported in cash flows from operations or cash flows from financing activities for the periods presented.
As of December 31, 2010, there was $6.0 million, $2.3 million and $0.03 million of total unrecognized compensation cost related to non-vested stock options under the Plans, the non-vested shares and the Purchase Plans, respectively. Those costs are expected to be recognized over weighted average periods of 2.5 years, 1.8 years and 0.04 years, respectively. Cash received from exercises under all share-based payment arrangements for the year ended December 31, 2010 was $3.9 million. We issue new shares of our common stock upon share option exercise and share purchase.
In applying the treasury stock method for the calculation of diluted EPS, amounts of unrecognized compensation expense and windfall tax benefits are required to be included in the assumed proceeds in the denominator of the diluted EPS calculation unless they are anti-dilutive. We incurred net losses for the years ended December 31, 2010, 2009 and 2008 and, therefore, such amounts have not been included in the calculations for those periods since they would be anti-dilutive. As a result, basic and diluted EPS are the same for each period. We have made an accounting policy decision to calculate windfall tax benefits/shortfalls, for purposes of diluted EPS calculation, excluding the impact of pro forma deferred tax assets. This policy decision will apply when we have net income.
10. Employee Savings Plan
The terms of the amended and restated Progenics Pharmaceuticals 401(k) Plan (the “Amended Plan”), among other things, allow eligible employees to participate in the Amended Plan by electing to contribute to the Amended Plan a percentage of their compensation to be set aside to pay their future retirement benefits. During 2010, 2009 and 2008, we matched 50%, 50% and 100%, respectively, of those employee contributions that are equal to 5%-8% of compensation and are made by eligible employees to the Amended Plan (the “Matching Contribution”). In addition, we may also make a discretionary contribution each year on behalf of all participants who are non-highly compensated employees. We made Matching Contributions of approximately $594, $718 and $1,727 to the Amended Plan for the years ended December 31, 2010, 2009 and 2008, respectively. No discretionary contributions were made during those years.
11. Income Taxes
We account for income taxes using the liability method in accordance with ASC 740 Income Taxes. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
There is no provision or benefit for federal or state income taxes for the years ended December 31, 2010, 2009 or 2008 other than a federal tax refund of $95 we received in 2010 from new legislation permitting the carryback of net operating losses (NOLs) to 2005. We have completed a calculation through December 31, 2007, under Internal Revenue Code Section 382, the results of which indicate that past ownership changes will limit utilization of NOLs in the future. Ownership changes subsequent to December 31, 2007, may further limit the future utilization of net operating loss and tax credit carry-forwards as defined by the federal and state tax codes.
F-24
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Deferred tax assets as of December 31, 2010 and 2009, consisted of the following:
|
2010
|
2009
|
|||||||
|
Depreciation and amortization
|
$ | 6,881 | $ | 6,831 | ||||
|
R&E tax credit carry-forwards
|
11,543 | 10,363 | ||||||
|
NYS investment tax credit carry-forwards
|
1,076 | 1,168 | ||||||
|
AMT credit carry-forwards
|
211 | 306 | ||||||
|
Net operating loss carry-forwards
|
95,848 | 83,546 | ||||||
|
Capitalized research and development expenditures
|
35,818 | 23,492 | ||||||
|
Stock compensation
|
13,942 | 13,142 | ||||||
|
Other items
|
2,776 | 2,585 | ||||||
| 168,095 | 141,433 | |||||||
|
Valuation allowance
|
(168,095 | ) | (141,433 | ) | ||||
| $ | - | $ | - | |||||
We do not recognize deferred tax assets considering our history of taxable losses and the uncertainty regarding our ability to generate sufficient taxable income in the future to utilize these deferred tax assets. For the years ended December 31, 2010 and 2009, we incurred net losses for tax purposes and recognized a full tax valuation against deferred taxes.
The following is a reconciliation of income taxes computed at the Federal statutory income tax rate to the actual effective income tax provision during 2010, 2009 and 2008:
|
2010
|
2009
|
2008
|
||||
|
U.S. Federal statutory rate
|
(34.0)%
|
(34.0)%
|
(34.0)%
|
|||
|
State income taxes, net of Federal benefit
|
(5.1)
|
(4.6)
|
(5.4)
|
|||
|
Research and experimental tax credit
|
(1.7)
|
(4.0)
|
(4.3)
|
|||
|
Change in valuation allowance
|
38.2
|
40.4
|
43.3
|
|||
|
Equity compensation
|
2.3
|
6.3
|
-
|
|||
|
Investment tax credit
|
0.1
|
(3.8)
|
-
|
|||
|
Other
|
0.1
|
(0.3)
|
0.4
|
|||
|
Income tax (benefit) provision
|
(0.1)%
|
0.0%
|
0.0%
|
As of December 31, 2010, we had available, for tax return purposes, unused NOLs of approximately $260.9 million, which will expire in various years from 2018 to 2030, $18.2 million of which were generated from deductions that, when realized, will reduce taxes payable and will increase paid-in-capital and are not reflected in our deferred tax assets above. Additionally, $11.2 million of the valuation allowance relates to NOLs attributable to excess tax deductions for equity compensation. When realized this will also be reflected as an increase to paid-in-capital.
We have reviewed our nexus in various tax jurisdictions and our tax positions related to all open tax years for events that could change the status of our ASC 740 Income Taxes liability, if any, or require an additional liability to be recorded. Such events may be the resolution of issues raised by a taxing authority, expiration of the statute of limitations for a prior open tax year or new transactions for which a tax position may be deemed to be uncertain. During the years ended December 31, 2010, 2009 and 2008, we had no unrecognized tax benefits resulting from tax positions during a prior or current period, settlements with taxing authorities or the expiration of the applicable statute of limitations. At December 31, 2010, there were no amounts of unrecognized tax benefits that, if recognized, would affect the effective tax rate and there were no tax positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within twelve months from the respective date. As of December 31, 2010, we are subject to federal and state income tax in the United States. Open tax years relate to years in which unused net operating losses were generated or, if used, for which the statute of limitation for examination by taxing authorities has not expired. Our open tax years extend back to 1995, with the exception of 1997, during which we reported net income. No amounts of interest or penalties were recognized in our Consolidated Statements of Operations or Consolidated Balance Sheets upon adoption of ASC 740 Income Taxes as of and for the years ended December 31, 2010, 2009 and 2008.
F-25
PROGENICS PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS ¾ continued
(amounts in thousands, except per share amounts or unless otherwise noted)
Our research and experimental (“R&E”) tax credit carry-forwards of approximately $11.5 million at December 31, 2010 expire in various years from 2011 to 2030. During the year ended December 31, 2010, research and experimental tax credit carry-forwards of approximately $53 expired.
12. Net Loss Per Share
Our basic net loss per share amounts have been computed by dividing net loss by the weighted-average number of common shares outstanding during the period. For the years ended December 31, 2010, 2009 and 2008, we reported a net loss and, therefore, potential common shares were not included since such inclusion would have been anti-dilutive. The calculations of net loss per share, basic and diluted, are as follows:
|
Net Loss
(Numerator)
|
Weighted Average
Common Shares
(Denominator)
|
Per Share
Amount
|
||||||||||
|
2010:
|
||||||||||||
|
Basic and diluted
|
$ | (69,725 | ) | 32,590 | $ | (2.14 | ) | |||||
|
2009:
|
||||||||||||
|
Basic and diluted
|
$ | (30,612 | ) | 31,219 | $ | (0.98 | ) | |||||
|
2008:
|
||||||||||||
|
Basic and diluted
|
$ | (44,672 | ) | 30,142 | $ | (1.48 | ) | |||||
During 2010, 2009 and 2008, potential common shares which have been excluded from diluted per share amounts because their effect would have been anti-dilutive include the following:
|
2010
|
2009
|
2008
|
||||||||||||||||||||||
|
Weighted
Average
Number
|
Weighted
Average
Exercise Price
|
Weighted
Average
Number
|
Weighted
Average
Exercise Price
|
Weighted
Average
Number
|
Weighted
Average
Exercise Price
|
|||||||||||||||||||
|
Options
|
5,037 | $ | 15.17 | 4,705 | $ | 17.48 | 4,854 | $ | 18.01 | |||||||||||||||
|
Restricted stock
|
45 | 34 | 35 | |||||||||||||||||||||
|
Total
|
5,082 | 4,739 | 4,889 | |||||||||||||||||||||
13. Unaudited Quarterly Results (unaudited)
Summarized quarterly financial data during 2010 and 2009 are as follows:
|
2010 Quarter Ended
|
|||||||||||
|
March 31
|
June 30
|
September 30
|
December 31
|
||||||||
|
Revenue
|
$1,523
|
$2,305
|
$1,967
|
$2,157
|
|||||||
|
Net loss
|
(18,583
|
)
|
(15,241
|
)
|
(17,101
|
)
|
(18,800
|
)
|
|||
|
Net loss per share (basic and diluted)
|
(0.58
|
)
|
(0.47
|
)
|
(0.52
|
)
|
(0.57
|
)
|
|||
|
2009 Quarter Ended
|
|||||||||||
|
March 31
|
June 30
|
September 30
|
December 31
|
||||||||
|
Revenue
|
$20,904
|
$5,469
|
$5,419
|
$17,155
|
|||||||
|
Net loss
|
(1,788
|
)
|
(15,171
|
)
|
(13,014
|
)
|
(639
|
)
|
|||
|
Net loss per share (basic and diluted)
|
(0.06
|
)
|
(0.49
|
)
|
(0.41
|
)
|
(0.02
|
)
|
|||
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, hereunto duly authorized.
|
PROGENICS PHARMACEUTICALS, INC.
|
||
|
By:
|
/s/ MARK R. BAKER
|
|
|
Mark R. Baker
(Duly authorized officer of the
Registrant and Chief Executive Officer and Director)
|
||
Date: March 15, 2011
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
|
Signature
|
Capacity
|
Date
|
|
|
/s/ PETER J. CROWLEY
|
Chairman
|
March 15, 2011
|
|
|
Peter J. Crowley
|
|||
|
/s/ PAUL J. MADDON
|
Vice Chairman, Chief Science
|
March 15, 2011
|
|
|
Paul J. Maddon, M.D., Ph.D.
|
Officer and Director
|
||
|
/s/ MARK R. BAKER
|
Chief Executive Officer and Director
|
March 15, 2011
|
|
|
Mark R. Baker
|
(Principal Executive Officer) | ||
|
/s/ CHARLES A. BAKER
|
Director
|
March 15, 2011
|
|
|
Charles A. Baker
|
|||
|
/s/ KURT W. BRINER
|
Director
|
March 15, 2011
|
|
|
Kurt W. Briner
|
|||
|
/s/ MARK F. DALTON
|
Director
|
March 15, 2011
|
|
|
Mark F. Dalton
|
|||
|
/s/ STEPHEN P. GOFF
|
Director
|
March 15, 2011
|
|
|
Stephen P. Goff, Ph.D.
|
|||
|
/s/ DAVID A. SCHEINBERG
|
Director
|
March 15, 2011
|
|
|
David A. Scheinberg, M.D., Ph.D.
|
|||
|
/s/ NICOLE S. WILLIAMS
|
Director
|
March 15, 2011
|
|
|
Nicole S. Williams
|
|||
|
/s/ ROBERT A. MCKINNEY
|
Chief Financial Officer, Senior Vice President,
|
March 15, 2011
|
|
|
Robert A. McKinney
|
Finance & Operations and Treasurer
|
||
|
(Principal Financial and Accounting Officer)
|
EXHIBIT INDEX
|
Exhibit
|
||
|
Number *
|
Description
|
|
|
3.1(14)
|
Restated Certificate of Incorporation of the Registrant.
|
|
|
3.2(14)
|
Amended and Restated By-laws of the Registrant.
|
|
|
4.1(1)
|
Specimen Certificate for Common Stock, $0.0013 par value per share, of the Registrant.
|
|
|
10.1(1)
|
Form of Registration Rights Agreement.
|
|
|
10.2(1)
|
1989 Non-Qualified Stock Option Plan‡
|
|
|
10.3(1)
|
1993 Stock Option Plan, as amended‡
|
|
|
10.4(1)
|
1993 Executive Stock Option Plan‡
|
|
|
10.5(3)
|
Amended and Restated 1996 Stock Incentive Plan‡
|
|
|
10.6(14)
|
2005 Stock Incentive Plan‡
|
|
|
10.6.1(10)
|
Form of Non-Qualified Stock Option Award Agreement‡
|
|
|
10.6.2(10)
|
Form of Restricted Stock Award Agreement‡
|
|
|
10.6.3(16)
|
Amended 2005 Stock Incentive Plan ‡
|
|
|
10.6.4(18)
|
Form of Non-Qualified Stock Option Award Agreement ‡
|
|
|
10.6.5(18)
|
Form of Restricted Stock Award Agreement ‡
|
|
|
10.7(15)
|
Form of Indemnification Agreement‡
|
|
|
10.8(19)
|
Employment Agreement, dated December 31, 2007, between the Registrant and Dr. Paul J. Maddon‡
|
|
|
10.9(1)
|
Letter dated August 25, 1994 between the Registrant and Dr. Robert J. Israel‡
|
|
|
10.10(8)
|
Amended 1998 Employee Stock Purchase Plan‡
|
|
|
10.11(8)
|
Amended 1998 Non-qualified Employee Stock Purchase Plan‡
|
|
|
10.15(5)
|
Amended and Restated Sublease, dated June 6, 2000, between the Registrant and Crompton Corporation.
|
|
|
10.16(2)†
|
Development and License Agreements, dated April 30, 1999, between Protein Design Labs, Inc. and the Registrant.
|
|
|
10.16.1(11)
|
Letter Agreement, dated November 24, 2003, relating to the Development and License Agreement between Protein Design Labs, Inc. and the Registrant.
|
|
|
10.18(4)
|
Director Stock Option Plan‡
|
|
|
10.19(6)†
|
Exclusive Sublicense Agreement, dated September 21, 2001, between the Registrant and UR Labs, Inc.
|
|
|
10.19.1(9)
|
Amendment to Exclusive Sublicense Agreement, dated September 21, 2001, between the Registrant and UR Labs, Inc.
|
|
|
10.20(7)
|
Research and Development Contract, dated September 26, 2003, between the National Institutes of Health and the Registrant.
|
|
|
10.21(7)
|
Agreement of Lease, dated September 30, 2003, between Eastview Holdings LLC and the Registrant.
|
|
|
10.22(7)
|
Letter Agreement, dated October 23, 2003, amending Agreement of Lease between Eastview Holdings LLC and the Registrant.
|
|
|
10.23(11)
|
Summary of Non-Employee Director Compensation‡
|
|
|
10.24(12) †
|
License and Co-Development Agreement, dated December 23, 2005, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc. and Wyeth-Ayerst Lederle, Inc. and the Registrant and Progenics Pharmaceuticals Nevada, Inc.
|
|
|
10.25(12) †
|
Option and License Agreement, dated May 8, 1985, by and between the University of Chicago and UR Labs, Inc., as amended by (i) Amendment to Option and License Agreement, dated September 17, 1987, by and between the University of Chicago and UR Labs, Inc., (ii) Second Amendment to Option and License Agreement, dated March 3, 1989, by and among the University of Chicago, ARCH Development Corporation and UR Labs, Inc., and (iii) Letter Agreement Related to Progenics’ RELISTOR In-License dated, December 22, 2005, by and among the University of Chicago, acting on behalf of itself and ARCH Development Corporation, the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Wyeth, acting through its Wyeth Pharmaceuticals Division.
|
|
|
10.26(13)
|
Membership Interest Purchase Agreement, dated April 20, 2006, between the Registrant Inc. and Cytogen Corporation.
|
|
|
10.27(13) †
|
Amended and Restated PSMA/PSMP License Agreement, dated April 20, 2006, by and among the Registrant, Cytogen Corporation and PSMA Development Company LLC.
|
|
|
10.28(17)
|
Consulting Agreement, dated May 1, 1995, between Active Biotherapies, Inc. and Dr. David A. Scheinberg, M.D., Ph.D., as amended on June 13, 1995, as assigned to the Registrant, and as amended on January 1, 2001‡
|
|
|
10.29(20) †
|
License Agreement, dated as of October 16, 2008, by and among Ono Pharmaceutical Co., Ltd. and the Registrant.
|
|
|
10.30(20) †
|
Partial Termination and License Agreement, dated October 16, 2008, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc. and Wyeth-Ayerst Lederle, Inc. and the Registrant and Progenics Pharmaceuticals Nevada, Inc.
|
|
|
10.31(20) †
|
Consent, Acknowledgment and Agreement, dated as of October 16, 2008, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc. and Wyeth-Ayerst Lederle, Inc., the Registrant and Ono Pharmaceutical Co., Ltd.
|
|
10.32(20) †
|
2008 Agreement Related to Progenics’ MNTX In-License, dated October 16, 2008, by and among the University of Chicago, acting on behalf of itself and ARCH Development Corporation, the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Ono Pharmaceutical Co., Ltd.
|
|
|
10.33(21) †
|
Termination and Transition Agreement, effective as of October 1, 2009, by and among Wyeth, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals, Inc., Wyeth-Ayerst Lederle, Inc., and AHP Manufacturing B.V., and the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Excelsior Life Sciences Ireland Limited.
|
|
|
10.34(22) †
|
Collaboration Agreement, effective June 14, 2005, by and between Seattle Genetics, Inc. and PSMA Development Company, LLC.
|
|
|
10.35(22) †
|
Collaboration Agreement, effective February 21, 2001, by and between Abgenix, Inc. and PSMA Development Company, LLC.
|
|
|
10.36(22) †
|
License Agreement, effective September 5, 2001, by and between AlphaVax Human Vaccines, Inc. and PSMA Development Company, LLC.
|
|
|
10.37 ††
|
First Amendment to Termination and Transition Agreement, effective as of October 1, 2010, by and among Wyeth LLC, acting through Wyeth Pharmaceuticals Division, Wyeth-Whitehall Pharmaceuticals LLC, Wyeth-Ayerst Lederle LLC, and AHP Manufacturing B.V., and the Registrant, Progenics Pharmaceuticals Nevada, Inc. and Excelsior Life Sciences Ireland Limited.
|
|
|
21.1(19)
|
Subsidiaries of the Registrant.
|
|
|
23.1
|
Consent of PricewaterhouseCoopers LLP.
|
|
|
31.1
|
Certification of Mark R. Baker, Chief Executive Officer of the Registrant pursuant to 13a-14(a) and Rule 15d-14(a) under the Securities Exchange Act of 1934, as amended.
|
|
|
31.2
|
Certification of Robert A. McKinney, Chief Financial Officer, Senior Vice President, Finance and Operations and Treasurer of the Registrant pursuant to 13a-14(a) and Rule 15d-14(a) under the Securities Exchange Act of 1934, as amended.
|
|
|
32.1
|
Certification of Mark R. Baker, Chief Executive Officer of the Registrant pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
32.2
|
Certification of Robert A. McKinney, Chief Financial Officer, Senior Vice President, Finance and Operations and Treasurer of the Registrant pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
__________________
|
*
|
Exhibits footnoted as previously filed have been filed as an exhibit to the document of the Registrant referenced in the footnote below, and are incorporated by reference herein.
|
|
|
(1)
|
Previously filed in Registration Statement on Form S-1, Commission File No. 333-13627.
|
|
|
(2)
|
Previously filed in Quarterly Report on Form 10-Q for the quarterly period ended June 30, 1999.
|
|
|
(3)
|
Previously filed in Registration Statement on Form S-8, Commission File No. 333-120508.
|
|
|
(4)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 1999.
|
|
|
(5)
|
Previously filed in Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2000.
|
|
|
(6)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 2002.
|
|
|
(7)
|
Previously filed in Quarterly Report on Form 10-Q for the quarterly period ending September 30, 2003.
|
|
|
(8)
|
Previously filed in Registration Statement on Form S-8, Commission File No. 333-143671.
|
|
|
(9)
|
Previously filed in Current Report on Form 8-K filed on September 20, 2004.
|
|
|
(10)
|
Previously filed in Current Report on Form 8-K filed on July 8, 2008.
|
|
|
(11)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 2004.
|
|
|
(12)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 2005.
|
|
|
(13)
|
Previously filed in Quarterly Report on Form 10-Q for the quarterly period ending June 30, 2006.
|
|
|
(14)
|
Previously filed in Current Report on Form 8-K filed on May 13, 2005.
|
|
|
(15)
|
Previously filed in Quarterly Report on Form 10-Q for the quarterly period ending March 31, 2007.
|
|
|
(16)
|
Previously filed in Registration Statement on Form S-8, Commission File No. 333-143670.
|
|
|
(17)
|
Previously filed in Annual Report on Form 10-K/A for the year ended December 31, 2006.
|
|
|
(18)
|
Previously filed in Current Report on Form 8-K filed on July 8, 2008.
|
|
|
(19)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 2007.
|
|
|
(20)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 2008.
|
|
|
(21)
|
Previously filed in Annual Report on Form 10-K for the year ended December 31, 2009.
|
|
|
(22)
|
Previously filed in Amendment No. 2 to Annual Report on Form 10-K/A for the year ended December 31, 2009.
|
|
|
†
|
Confidential treatment granted as to certain portions omitted and filed separately with the Commission.
|
|
|
††
|
Confidential treatment requested as to certain portions omitted and filed separately with the Commission.
|
|
|
‡
|
Management contract or compensatory plan or arrangement.
|
|
E-3