Attached files
| file | filename |
|---|---|
| EX-31.2 - EX-31.2 - AFFYMAX INC | a2202521zex-31_2.htm |
| EX-32.1 - EX-32.1 - AFFYMAX INC | a2202521zex-32_1.htm |
| EX-23.1 - EX-23.1 - AFFYMAX INC | a2202521zex-23_1.htm |
| EX-31.1 - EX-31.1 - AFFYMAX INC | a2202521zex-31_1.htm |
| EX-10.42 - EX-10.42 - AFFYMAX INC | a2202521zex-10_42.htm |
| EX-10.43 - EX-10.43 - AFFYMAX INC | a2202521zex-10_43.htm |
Use these links to rapidly review the document
AFFYMAX, INC. TABLE OF CONTENTS
Item 8. Financial Statements and Supplementary Data.
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2010 |
||
or |
||
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
Commission File Number 001-33213
AFFYMAX, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
77-0579396 (I.R.S. Employer Identification Number) |
4001 Miranda Avenue
Palo Alto, CA 94304
(650) 812-8700
(Address, including zip code, and telephone number, including
area code, of registrant's principal executive offices)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered | |
|---|---|---|
| Common stock, par value $0.001 per share | The NASDAQ Stock Market LLC | |
| (NASDAQ Global Market) |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or smaller reporting company. See definition of "large accelerated filer," "accelerated filer", and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | Accelerated filer ý | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the registrant's common stock, $0.001 par value, held by non-affiliates of the registrant as of June 30, 2010 was $132,363,389 (based upon the closing sales price of such stock as reported on the Nasdaq Global Market on such date). Excludes an aggregate of 2,225,631 shares of the registrant's common stock held by officers, directors and affiliated stockholders. For purposes of determining whether a stockholder was an affiliate of the registrant at June 30, 2010, the registrant has assumed that a stockholder was an affiliate of the registrant at June 30, 2010 if such stockholder (i) beneficially owned 10% or more of the registrant's common stock and/or (ii) was affiliated with an executive officer or director of the registrant at June 30, 2010. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
As of February 28, 2011, the registrant had outstanding 25,473,301 shares of Common Stock.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the Proxy Statement for the 2011 Annual Meeting of Stockholders (the "Proxy Statement"), to be filed with the Securities and Exchange Commission within 120 days of the end of the fiscal year ended December 31, 2010, are incorporated by reference into Part III of this Annual Report on Form 10-K. Except with respect to information specifically incorporated by reference into this Annual Report on Form 10-K, the Proxy Statement is not deemed to be filed as part hereof.
AFFYMAX, INC.
TABLE OF CONTENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the "safe harbor" created by those sections. Forward-looking statements are based on our management's beliefs and assumptions and on information currently available to our management. In some cases, you can identify forward-looking statements by terms such as "may," "will," "should," "could," "would," "expect," "intend," "plan," "anticipate," "believe," "estimate," "project," "predict," "potential" and similar expressions intended to identify forward-looking statements. These forward-looking statements include, but are not limited to, statements regarding the timing, design and results of our clinical trials and drug development program and registration strategy, the continuation and success of our collaboration with Takeda Pharmaceutical Company Limited, and the timing and likelihood of the commercialization of peginesatide. These statements involve known and unknown risks, uncertainties and other factors, which may cause our actual results, performance, time frames or achievements to be materially different from any future results, performance, time frames or achievements expressed or implied by the forward-looking statements. We discuss many of these risks, uncertainties and other factors in this Annual Report on Form 10-K under Item 1A "Risk Factors," including risks relating to our ability to submit a comprehensive and timely New Drug Application, risks relating to timing of and regulatory requirements for approvals, including the U. S. Food and Drug Administration's interpretation and evaluation of the data from the Phase 3 studies, in particular with respect to the secondary analyses in the non-dialysis patients, risks relating to data quality and integrity particularly in non-inferiority designed trials, risks relating to the continued safety and efficacy of peginesatide in clinical development, the potential for once per month dosing and room temperature stability, the timing of patient accrual in ongoing and planned clinical trials, regulatory requirements and approvals, research and development efforts, the factors affecting the commercial potential of peginesatide, industry and competitive environment, controversy surrounding the class of erythropoiesis stimulating agents, reimbursement coverage, intellectual property rights and disputes and potential costs, disruptions and consequences of litigation, financing requirements and ability to access capital, and other matters. Given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this filing. You should read this Annual Report on Form 10-K completely and with the understanding that our actual future results may be materially different from what we expect. We hereby qualify our forward-looking statements by these cautionary statements. Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
Overview
We are a biopharmaceutical company committed to developing novel drugs to improve the treatment of serious and often life-threatening conditions. Our product candidate, peginesatide (HematideTM), recently completed Phase 3 clinical trials to treat anemia associated with chronic renal failure. Anemia is a serious condition in which blood is deficient in red blood cells and hemoglobin. It is common in patients with chronic renal failure, cancer, heart failure, inflammatory diseases and other critical illnesses, as well as in the elderly. If left untreated, anemia may lead to chronic fatigue or increase the risk of other diseases or death. Currently recombinant EPO, or rEPO, is used to manage the anemia of dialysis, non-dialysis and cancer patients. We estimate that rEPO generated approximately $2.5 billion net revenues in the United States, or U.S., for 2010 attributable to use in dialysis patients with chronic renal failure. However, a decline of this market is forecasted with the implementation of the bundled payment system for reimbursement in 2011. Peginesatide is a synthetic peptide-based erythropoiesis stimulating agent, or ESA, designed to stimulate production of red blood
2
cells. Peginesatide is designed to be longer acting than currently marketed ESAs in the U.S. and therefore has the potential to offer reduced cost and complexity for healthcare providers.
In late June 2010, we announced preliminary top-line results from our peginesatide Phase 3 clinical program for the treatment of patients with anemia associated with chronic renal failure. Our Phase 3 clinical program included four open-label, randomized controlled clinical trials. Of these trials, two trials, called PEARL 1 and PEARL 2, were conducted in non-dialysis patients and designed to evaluate the safety and efficacy of peginesatide compared to darbepoetin alfa to correct anemia and maintain hemoglobin in a corrected range over time. The other two trials, called EMERALD 1 and EMERALD 2, were conducted in dialysis patients and designed to evaluate the safety and efficacy of peginesatide and its ability to maintain hemoglobin levels in a corrected range compared to epoetin alpha or epoetin beta when switched to peginesatide. Analysis of efficacy and safety for all of the Phase 3 studies were based primarily on assessments of non-inferiority to the comparator drugs. The primary efficacy endpoint, the mean change in hemoglobin from baseline, in each of the four Phase 3 studies met the statistical criteria for non-inferiority. In addition peginesatide met the statistical criterion for non-inferiority for the assessment of safety for the cardiovascular composite safety endpoint, or CSE, which was composed of death, stroke, myocardial infarction, congestive heart failure, unstable angina and arrhythmia from a pooled safety database across the four Phase 3 studies. However, some differences were observed when secondary analyses were conducted as previously described in our Current Report on Form 8-K dated June 21, 2010.
Based on our discussions with the U.S. Food and Drug Administration, or FDA, we plan to submit a New Drug Application, or NDA, to the FDA, for treatment of anemia in chronic renal failure patients on dialysis in the second quarter of 2011. Despite meeting the primary efficacy endpoints and the CSE for peginesatide, the differences observed in the Phase 3 secondary analyses present risks to our ability to obtain regulatory approval for peginesatide particularly in view of the heightened concerns surrounding safety of ESAs. Any negative perception of peginesatide's safety relative to other ESAs would significantly reduce the likelihood of obtaining regulatory approval for peginesatide. The issues arising from the Phase 3 results have caused significant delay and may continue to negatively impact the timelines for development and the likelihood, scope or conditions surrounding regulatory approval. Any or all of these factors may significantly reduce the ultimate commercial potential of peginesatide.
In February and June 2006, we entered into two agreements forming a collaboration to develop and commercialize peginesatide with Takeda Pharmaceutical Company Limited, or Takeda, the largest pharmaceutical company in Japan. Under our collaboration, the companies will co-develop and co-commercialize peginesatide in the U.S. Takeda also received an exclusive license to develop and commercialize peginesatide outside of the U.S. Currently, Takeda bears 70% of third party expenses related to clinical development in pursuit of U.S. regulatory approval of peginesatide, while we assume 30% of third party expenses. In addition, third party expenses related to the commercialization of peginesatide in the U.S. are equally shared by both parties and beginning in mid 2011, certain employee-related expenses supporting commercialization will also be equally shared. Takeda has primary responsibility and bears all costs for peginesatide's clinical development in support of regulatory approval for all territories outside the U.S.
Under the agreements, to date Takeda has paid us the following amounts:
- •
- $122 million of upfront license fees;
- •
- approximately $10 million to purchase our preferred stock, which converted into common stock upon the completion of
our initial public offering; and
- •
- $45 million in milestone payments.
3
In addition, we are eligible to receive up to an additional $118 million in future milestone payments for our dialysis indication worldwide, including $10 million upon acceptance of our NDA and $50 million upon approval of the NDA by the FDA.
The agreements also establish milestone payments for additional indications and in the event that certain levels of significant annual net sales are met. We and Takeda will share equally in the net profits and losses of peginesatide in the U.S. and Takeda will pay us royalties based on the annual net sales of peginesatide outside the U.S.
Anemia Background
Anemia, a condition in which the blood is deficient in red blood cells and hemoglobin, is a frequent and serious complication associated with a number of common chronic diseases. Anemia is associated with chronic fatigue and, if left untreated, may increase the risk of other diseases or even death. Red blood cells are normally formed in the circulating blood from precursor cells which are initially present primarily in the bone marrow. These cells are stimulated to divide and differentiate and are mobilized into circulation by EPO, a hormonal factor produced by the kidney. EPO acts by binding to and activating the EPO receptor on precursor cells. The activation of the EPO receptor stimulates the proliferation and maturation of the precursor cells to form red blood cells that contain hemoglobin. Hemoglobin is an iron-containing protein in red blood cells that functions primarily in the transport of oxygen to, and carbon dioxide from, the tissues of the body. Anemia can be caused by conditions such as chronic renal failure, or treatments such as chemotherapy, that result in underproduction of EPO or a muted response to EPO.
Anemia generally exists in men when the hemoglobin level in blood, which is a measure of red blood cells, is less than 12 g/dL, or the hematocrit, which is a ratio of the volume packed red blood cells to the volume of whole blood, is less than 36%, and in women when hemoglobin is less than 11 g/dL or hematocrit is less than 33%. The FDA, the medical community and others have raised significant safety concerns relating to currently marketed ESAs as a result of reports of increased mortality and side effects from a number of clinical trials. Some of these safety concerns relate to targeting and maintaining high hemoglobin levels for extended periods of time. The FDA recently required revised warnings, including black box warnings, be added to labels of currently marketed ESAs advising physicians to monitor hemoglobin levels and to use the lowest dose of ESA to increase the hemoglobin concentration to the lowest level sufficient to avoid the need for red blood cell transfusions. Black box warnings for currently marketed ESAs also note increased risk of death and serious cardiovascular events when administered to target higher hemoglobin levels.
Anemia associated with Chronic Renal Failure. One of the most common forms of chronic anemia is that which occurs in patients with chronic kidney failure. According to the American Journal of Kidney Disease, chronic kidney failure affects as many as 26 million Americans. As kidney function deteriorates due to the underlying disease, the ability of the kidney to produce adequate EPO is impaired, resulting in decreased production of new red blood cells and anemia.
Over time, chronic renal failure usually progresses to irreversible end-stage renal disease, the most severe stage of the disease. End-stage renal disease patients require either lifetime dependence on renal dialysis, a medical procedure in which blood is cleansed of impurities, or a kidney transplant. Patients with end-stage renal disease are nearly always moderately to severely anemic unless treated with an ESA like rEPO. According to the Centers for Medicare and Medicaid Services, or CMS, there are approximately 380,000 end-stage renal disease patients on dialysis in the U.S. served by approximately 5,000 dialysis facilities. Funding and reimbursement for this care are predominately through the Medicare End Stage Renal Disease Program. Prior to this year, CMS generally reimbursed ESAs at a rate of 106% of the average ESA sales price. This allowed the dialysis facilities to realize a profit on the purchase and administration of ESAs, which constitutes an important component of their
4
economic viability. However, under the 2008 Medicare Legislation, a new bundled payment system commenced in January 2011 for facilities that furnish renal dialysis services and home dialysis to Medicare beneficiaries with end-stage renal disease. Under the new system, CMS will make a single bundled payment to the dialysis facility for each dialysis treatment that will cover all renal dialysis services, including ESAs. The bundled payment system may create incentives for significantly lower utilization or dosing of ESAs, including peginesatide, and reduce the commercial potential for peginesatide. We cannot guarantee that peginesatide will be reimbursed by CMS in a manner that will support physician adoption. CMS held Medicare Coverage Advisory Committee (MedCAC) meetings in March 2010 and January 2011 to review current ESA coverage policy based on the available evidence on the use of ESAs to manage anemia in patients who have chronic kidney disease, and the role of ESAs in successful kidney transplantation, respectively. Independent of any additional action the FDA may take, CMS may further decrease reimbursement coverage of ESA reducing the overall size of the market peginesatide is expected to compete in at the time of launch.
Anemia associated with Other Conditions. We are developing peginesatide for treatment of anemia in chronic renal-failure patients on dialysis only and not currently investigating peginesatide's use in treating anemia due to other conditions, such as for non-dialysis patients or chemotherapy-induced anemia or anemia arising from the cancer itself.
Current Therapy and Limitations
We estimate that rEPO generated approximately $2.5 billion net revenues in the U.S. for 2010 attributable to use in dialysis patients with chronic renal failure. However, a decline of this market is forecasted with the implementation of the bundled payment system for reimbursement in 2011. Forms of rEPO variants have been used successfully to manage the anemia of dialysis, non-dialysis and cancer patients. rEPOs are similar, but not necessarily identical, to a patient's naturally occurring EPO. Differences exist among rEPOs with regard to composition and structure. As a result, differences may also exist among rEPOs with regard to frequency of dosing, duration of effect and rate of rise in hemoglobin. Stability in the blood and circulating half-life, which measure the time it takes the compound to disappear from the blood, generally correlate with less frequent dosing.
Since its initial U.S. market introduction in 1989, rEPO has revolutionized the treatment of patients with anemia resulting from chronic diseases. Two current types of ESAs, epoetin alfa and epoetin beta, are biologically engineered hormones produced in mammalian cells by recombinant DNA technology. Both are relatively short-acting forms of rEPO that typically require frequent dosing to obtain a sustained correction of anemia. Darbepoetin alfa, which is marketed by Amgen, Inc., or Amgen, under the trade name Aranesp, is a biologically engineered hormone product closely related to and functionally similar to epoetin alfa. However, darbepoetin alfa has a terminal half-life approximately three times longer than epoetin alfa, as a result of the addition of sialic acid to stabilize the protein. The currently available rEPOs are marketed under a variety of trade names in different territories.
Frequency of Dosing. In the U.S., currently marketed ESAs are hampered by short duration of effect resulting in the need for frequent dosing. One of our objectives is to provide a product with a duration of effect that results in a well-controlled hemoglobin response while still allowing optimal once monthly dosing.
Pure Red Cell Aplasia. Treatment of patients with rEPO has been shown in rare cases to cause the production of antibodies to both rEPO and naturally-occurring EPO. Typically these antibodies can bind to and neutralize both the rEPO drug and any naturally-occurring EPO in a patient's system. As a result, such patients become increasingly less sensitive to rEPO therapy and can develop a form of anemia called Pure Red Cell Aplasia, or PRCA. This hematological disorder is characterized by severe, transfusion-dependent anemia, a scarcity of reticulocytes and an almost complete absence of red blood
5
cell precursors in otherwise normal bone marrow. The FDA has required marketers of rEPO in the U.S. to include in their product prescribing information warnings of potential for rEPO-induced PRCA and a description of this adverse reaction. We believe that an ESA that does not cause PRCA and that can potentially be used to treat PRCA will have advantages in the marketplace over rEPOs that can cause PRCA.
Our Product Candidate: Peginesatide
Peginesatide is a synthetic peptide-based ESA designed for less frequent dosing compared to currently marketed ESAs in the U.S. It is currently an investigational agent, and we plan to submit a NDA to the FDA for treatment of anemia in chronic renal failure patients on dialysis in the second quarter of 2011. Peginesatide is designed to be dosed once every four weeks, compared to recombinant products sold in the U.S. that are dosed either several times a week, every week to two weeks, or up to every three weeks for some patients.
Potential Peginesatide Profile
Peginesatide is a relatively small synthetic peptide-based ESA which we are developing for the treatment of anemia patients with chronic renal failure on dialysis. Peptides are composed of amino acids, commonly known as the building blocks of proteins. Typically, a peptide is composed of fewer than 50 amino acids, while a protein contains from 50 to well over 5,000 amino acids. Peptide-based therapeutics may display certain advantages compared to recombinant proteins, including simplicity and cost of manufacture, and specificity of effect. In the past, development of peptide-based drug candidates was often slowed by low potency. A second problem historically associated with peptide-based drugs has been a requirement of frequent dosing in vivo. More recently, however, it has been possible to develop peptide-based drugs with potencies nearly equivalent to recombinant proteins and with less frequent dosing requirements. Through the use of our technology, peginesatide has the potential to require less frequent dosing than currently marketed ESAs in the U.S.
Our clinical trials to date have shown similar positive effects on red blood cell formation when peginesatide is given at comparable doses either intravenously or subcutaneously. These results suggest that peginesatide may be similarly effective in humans when administered by either route. We believe it may be easier to use peginesatide than some forms of rEPO, which often have different clinical effects when given subcutaneously versus intravenously.
In addition, based on stability data to date, we believe that peginesatide could be stored at room temperature in the hands of the health care providers for limited durations after refrigerated distribution. Currently marketed ESAs in the U.S. require cold storage conditions throughout the distribution and storage process until administration to patients.
Although peginesatide has the erythropoietic activity characteristic of naturally occurring EPO, its amino acid sequence is unrelated to EPO, rEPO or any other known naturally-occurring erythropoietic protein. Because peginesatide does not appear to display immunologic cross-reactivity to naturally-occurring EPO, we believe that peginesatide will not cause PRCA. We have conducted pre-clinical studies which have demonstrated that peginesatide can stimulate reticulocytes and elevate hemoglobin levels in an animal model of EPO antibody mediated PRCA. An ongoing Phase 2 clinical trial of peginesatide in a small number of patients with PRCA has generally shown supportive results to date. These results suggest that peginesatide is not neutralized by antibodies to rEPO and thus may be effective in treating anemia in patients that have developed PRCA.
Based on pre-clinical studies and clinical trials completed to date, we believe that the risk of developing antibodies to peginesatide will be low, and we have observed that peginesatide-induced antibodies do not appear to cross-react with rEPO and do not generally have any apparent effect on
6
clinical response to the drug. However, results in future trials or those observed in practice may differ from the results obtained in vivo studies or clinical trials to date.
Peginesatide Development Program
In the U.S., we are currently pursuing development of peginesatide to treat dialysis patients with anemia associated with chronic renal failure and are not planning to pursue any other indications in the foreseeable future. We have suspended our development efforts to treat anemia in non-dialysis patients and chemotherapy-induced anemia.
Over 2,600 patients have received peginesatide in clinical trials completed to date. We believe the pharmacokinetics and pharmacodynamics of peginesatide have been shown from these trials to be appropriate for extended dose intervals and desired drug activity; however, no conclusions can be drawn as only the FDA can make determinations of safety and efficacy. We anticipate that peginesatide, if approved, would typically be dosed once every four weeks in chronic renal failure patients on dialysis.
Pre-clinical and Toxicology Studies. Pre-clinical studies have shown that peginesatide, like EPO, acts through activation of the EPO receptor. Furthermore, pre-clinical in vivo studies have shown that the effects on erythropoiesis are very similar whether peginesatide is given intravenously or subcutaneously. We have conducted repeat-dose pre-clinical toxicology studies lasting as long as nine months, and have incorporated single-dose and repeat-dose studies exploring administration by either intravenous or subcutaneous injection in a variety of models using doses up to several thousand times the estimated monthly clinical dose. The primary toxicology observed to date has been associated with the exaggerated red blood cell production seen at high and/or frequent doses, a result similar to that observed with the rEPO class of drugs. However, the results from pre-clinical testing to date may not be predictive of results of future clinical trials or if approved by the FDA, usage by dialysis patients outside of clinical trials.
Chronic Renal Failure
Phase 1 and Phase 2 Clinical Trials
We and Takeda have recently completed multiple Phase 1 and Phase 2 clinical trials of peginesatide at sites in the U.S. and the European Union, or E.U., in normal healthy volunteers, dialysis patients, non-dialysis patients, and peritoneal dialysis patients supportive of our planned NDA submission. These Phase 1 clinical trials were designed primarily to demonstrate bioavailability or bioequivalence of product concentrations and formulations while these Phase 2 trials were designed to determine the safety, pharmacodynamics and pharmacokinetics of peginesatide when administered to patients suffering from anemia. Two of these Phase 2 clinical trials were conducted to evaluate the use of peginesatide to treat anemic patients in additional segments of the chronic renal failure patient population. One of the studies focused on evaluating peginesatide in patients undergoing peritoneal dialysis, a special form of dialysis that allows the process to be performed in the patient's home. Another trial was designed to evaluate the conversion of Aranesp-treated chronic renal patients (on dialysis and not on dialysis) to once-monthly peginesatide.
We continue to conduct an ongoing Phase 2 clinical trial of peginesatide in a small number of patients with PRCA in the E.U.
Phase 3 Clinical Trials
In late June 2010, we announced preliminary top-line results from the peginesatide Phase 3 clinical program for the treatment of patients with anemia associated with chronic renal failure. Our Phase 3 clinical program included four open-label, randomized controlled clinical trials. Of these trials, two
7
trials, called PEARL 1 and PEARL 2, were conducted in non-dialysis patients and designed to evaluate the safety and efficacy of peginesatide compared to darbepoetin alfa to correct anemia and maintain hemoglobin in a corrected range over time. The other two trials, called EMERALD 1 and EMERALD 2, were conducted in dialysis patients and designed to evaluate the safety and efficacy of peginesatide and its ability to maintain hemoglobin levels in a corrected range compared to epoetin alpha or epoetin beta when switched to peginesatide. Analysis of efficacy and safety for all of the Phase 3 studies were primarily based on assessments of non-inferiority to the comparator drugs. The primary efficacy endpoint, the mean change in hemoglobin from baseline, in each of the four Phase 3 studies met the statistical criteria for non-inferiority. In addition, peginesatide met the statistical criterion for non-inferiority for the assessment of safety for the CSE, which was composed of death, stroke, myocardial infarction, congestive heart failure, unstable angina and arrhythmia from a pooled safety database across the four Phase 3 studies. However, some differences were observed when secondary analyses were conducted as previously described in our Current Report on Form 8-K dated June 21, 2010. Despite meeting the primary efficacy endpoints and the CSE for peginesatide, the differences observed in the Phase 3 secondary analyses present risks to our ability to obtain regulatory approval for peginesatide particularly in view of the heightened concerns surrounding safety of ESAs.
Takeda is conducting a Phase 3 clinical program in Japan for the treatment of patients with anemia associated with chronic renal failure which is expected to be completed in 2011.
Any negative perception of peginesatide's safety relative to other ESAs would significantly limit the likelihood of obtaining regulatory approval for peginesatide. The issues arising from the Phase 3 results have caused significant delay and may continue to negatively impact the timelines for development and the likelihood, scope or conditions surrounding regulatory approval. Any or all of these factors may significantly reduce the ultimate commercial potential of peginesatide.
Manufacturing and Supply
All of our current good manufacturing practices, or GMP, manufacturing is outsourced to third parties with oversight by our internal managers. We have limited non-GMP manufacturing capacity in-house. We intend to continue to rely on third party manufacturers to produce sufficient quantities of drug substance and product for any future clinical trials and commercialization of peginesatide and for any other potential products for which we retain significant development and commercialization rights. Peginesatide is chemically synthesized and peptide-based.
We have established long term commercial supply agreements with two contract manufacturers, or CMOs, for peginesatide active pharmaceutical ingredient, or API. Under our worldwide collaboration with Takeda, we will be responsible, through our CMOs, for the manufacture and supply of all quantities of peginesatide API to be used in the development and commercialization of peginesatide worldwide.
Final peginesatide drug product is currently manufactured as a buffered aqueous solution for intravenous or subcutaneous administration. Takeda has assumed responsibility for final drug product manufacture and control as part of our worldwide collaboration for peginesatide.
Intellectual Property
We protect our technology through the use of patents, trade secrets and proprietary know-how. We have more than 20 issued U.S. patents, including claims covering compositions of compounds comprising peptides of a broad genus of ESA peptide sequences, methods of treating EPO disorders using these compounds and methods of synthesizing these types of ESA peptide compounds. We own several pending U.S. patent applications, all of which relate to our core peptide technologies or to particular peptide compounds. Our issued U.S. patent(s) covering peginesatide and any U.S. patent(s) that may issue based on pending patent applications containing claims covering peginesatide including
8
issued claims relating to composition of matter begin expiring no earlier than 2024. We own foreign equivalent patents and patent applications based on our U.S. patents and patent applications. We also retain technical information related to manufacture and analysis of peginesatide as trade secrets. In October 2010, the arbitration panel in our binding arbitration with certain subsidiaries of Johnson & Johnson, or J&J, decided the ownership of a number of U.S. and international patents and patent applications related to certain EPO-R agonists. See "Risk Factors—Risks Related to Our Business" and "Legal Proceedings" elsewhere in this Annual Report on Form 10-K.
We own and have rights to several proprietary peptide screening technologies, including the patented technologies of peptide phage display and peptides-on-plasmids. This technology enables us to identify initial novel peptide sequences and provides information that our scientists can use to design a variety of peptide compounds to optimize bioactivity and produce pharmaceutical candidate compounds having desired properties.
The table below sets out our material U.S. patents and their current anticipated expiry and a related description of related foreign patents as provided below:
U.S. Patents Assigned or Exclusively Licensed
Pat No.
|
Title | Expiry | ||
|---|---|---|---|---|
5,338,665 |
Peptide Library and Screening Method | 8/16/2012 | ||
5,427,908 |
Recombinant Library Screening Methods | 6/27/2012 | ||
5,432,018 |
Peptide Library and Screening Method | 7/11/2012 | ||
5,498,530 |
Peptide Library and Screening Method | 8/16/2012 | ||
5,580,717 |
Recombinant Library Screening Methods | 6/27/2012 | ||
5,723,286 |
Peptide Library and Screening Method | 3/3/2015 | ||
5,773,569* |
Compounds and Peptides that Bind to the Erythropoietin Receptor | 6/30/2015 | ||
5,830,851* |
Methods of Administering Peptides that Bind to the Erythropoietin Receptor | 11/3/2015 | ||
5,874,239 |
Biotinylation of Peptides | 7/30/2013 | ||
5,880,096 |
Peptides and Compounds that Bind the IL-1 Receptor | 3/9/2016 | ||
5,932,433 |
Biotinylation of Peptides | 7/30/2013 | ||
5,986,047* |
Peptides that Bind to the Erythropoietin Receptor | 11/19/2013 | ||
6,703,480 |
Peptide Dimers as Agonists of the Erythropoietin (EPO) Receptor and Associated Methods of Synthesis and Use | 11/24/2019 | ||
6,716,811 |
Compounds having affinity for the granulocyte-colony stimulating factor receptor (G-CSFR) and associated uses | 9/1/2020 | ||
7,084,245 |
Peptides that Bind to the Erythopoietin Receptor | 5/12/2024 | ||
7,109,299 |
Peptides and Compounds that Bind to the IL-5 Receptor | 12/16/2019 | ||
7,414,105 |
Peptides that Bind to the Erythopoietin Receptor | 5/12/2024 | ||
7,459,522 |
Peptide Dimers as Agonists of the Erythropoietin (EPO) Receptor and Associated Methods of Synthesis and Use | 11/24/2019 | ||
7,482,433 |
Peptides and Compounds that Bind to the IL-5 Receptor | 12/16/2019 | ||
7,528,104 |
Peptides that Bind to the Erythopoietin Receptor | 5/12/2024 | ||
7,550,433 |
Erythropoietin Receptor Peptide Formulations and Uses | 6/2/2026 | ||
7,553,617 |
Peptide Library and Screening Method | 3/3/2015 | ||
7,855,175 |
Peptides that Bind to the Erythopoietin Receptor | 5/12/2024 | ||
7,906,485 |
Erythropoietin Receptor Peptide Formulations and Uses | 6/2/2026 |
- *
- Patent subject to arbitration award ordering joint ownership by us and J&J. See, "Legal Proceedings—J&J Intellectual Property Dispute".
9
In addition to the U.S. patents listed above, we own or have exclusive licenses to corresponding foreign patents in various countries outside the U.S.; these foreign counterpart patents are substantially similar to their counterpart U.S. patents. The J&J arbitration included the counterpart foreign patents corresponding to U.S. 5,773,569, U.S. 5,830,851, and U.S. 5,986,047. The foreign counterparts to the listed U.S. patents are scheduled to expire in various countries during the period 2012 to 2026.
Third Party Intellectual Property
Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our collaborators are developing products. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our product candidates or proprietary technologies may infringe.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our product candidates and/or proprietary technologies infringe their intellectual property rights. If one of these patents was found to cover our product candidates, proprietary technologies or their uses, we or our collaborators could be required to pay damages and could be restricted from commercializing our product candidates or using our proprietary technologies unless we or they obtain a license to the patent. A license may not be available to us or our collaborators on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable right, which could prohibit us from making, using or selling our products, technologies or methods.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and biopharmaceutical industries generally. If a third party claims that we or our collaborators infringe its intellectual property rights, we may face a number of issues, including but not limited to:
- •
- infringement and other intellectual property claims which, with or without merit, may be expensive and
time-consuming to litigate and may divert our management's attention from our core business;
- •
- substantial damages for infringement, including treble damages and attorneys' fees, which we may have to pay if a court
decides that the product or proprietary technology at issue infringes on or violates the third party's rights;
- •
- a court prohibiting us from selling or licensing the product or using the proprietary technology unless the third party
licenses its technology to us, which it is not required to do;
- •
- if a license is available from the third party, we may have to pay substantial royalties, fees and/or grant cross licenses
to our technology; and
- •
- redesigning our products or processes so they do not infringe, which may not be possible or may require substantial funds and time.
While we have conducted a search of patents issued to third parties, no assurance can be given that such patents do not exist, have not been filed, or could not be filed or issued, which contain claims covering our products, technology or methods. Because of the number of patents issued and patent applications filed in our technical areas or fields, we believe there is a significant risk that third parties may allege they have patent rights encompassing our products, technology or methods.
Research and Development Expenses
We have made substantial investments in research and development. Research and development costs consist of salaries, stock-based compensation, employee benefits, license fees, laboratory supplies,
10
costs associated with clinical trials, including amounts paid to clinical research organizations, other professional services and facility costs. Research and development expenses were $93.6 million, $157.1million and $137.5 million, for the years ended December 31, 2010, 2009, and 2008, respectively.
Our Strategic Alliance
June 2006 Development and Commercialization Agreement with Takeda
In June 2006, we entered into a Development and Commercialization Agreement with Takeda to develop and commercialize peginesatide worldwide. Under our collaboration, the companies will co-develop and co-commercialize peginesatide in the U.S. Takeda received an exclusive license to develop and commercialize peginesatide outside of the U.S. As contemplated by this agreement, the February 2006 agreement that we have also entered into with Takeda was harmonized to address the worldwide arrangement between the parties.
We will share responsibility with Takeda for clinical development activities required for U.S. regulatory approval of peginesatide. Specifically, we have primary responsibility for peginesatide's clinical development plan and clinical trials in the dialysis indication and the non-dialysis indication to the extent of any further development, while Takeda will have primary responsibility in the chemotherapy induced anemia and anemia of cancer indications to the extent any such indication is developed. Beginning January 1, 2007, Takeda was responsible for the first $50 million of third party expenses related to development in pursuit of U.S. regulatory approval of peginesatide, which was fully utilized by both parties through the first quarter of 2008. Thereafter, Takeda has borne 70% of the third party U.S. development expenses while we have been responsible for 30% of third party expenses. We retain responsibility for 100% of our internal development expenses. In addition, third party expenses related to the commercialization of peginesatide in the U.S. are equally shared by both parties and beginning in mid-2011, certain employee-related expenses supporting commercialization will also be equally shared. Takeda will have primary responsibility and bear all costs for peginesatide's clinical development in support of regulatory approval for all territories outside the U.S.
Under the June 2006 agreement, Takeda paid us an upfront license fee of $105 million, and we have received milestone payments upon completion of database lock of the Phase 3 clinical trials of $30 million for dialysis and non-dialysis. Upon the successful achievement of clinical development and regulatory milestones, we are eligible to receive from Takeda an additional aggregate of $85 million relating to the renal program, including, $10 million of milestone payments upon FDA acceptance of the submission of the NDA, and $50 million of milestone payments upon approval by the FDA in dialysis indications. The June 2006 agreement also establishes milestone payments for additional indications and in the event that certain levels of significant annual net sales are met. We and Takeda will share equally in the net profits and losses of peginesatide in the U.S., which include expenses related to the marketing and launch of peginesatide. Takeda will pay us a variable royalty based on annual net sales of peginesatide outside the U.S.
We will own and have responsibility for U.S. NDAs in the dialysis, non-dialysis, chemotherapy-induced anemia and anemia of cancer indications to the extent any such NDA is filed. Takeda will own and have responsibility for regulatory filings outside the U.S. Takeda will also be responsible for creating and maintaining a global safety database.
We will also be responsible, through our CMOs, for the manufacture and supply of all quantities of peginesatide API to be used in the development and commercialization of peginesatide worldwide. Takeda will be responsible for the fill and finish steps in the manufacture of peginesatide worldwide.
The parties have agreed to jointly develop the initial commercial marketing plan for peginesatide in the U.S. pursuant to which we and Takeda will divide peginesatide promotional responsibilities in the U.S. We will be primarily responsible for commercialization activities within the dialysis and non-dialysis markets, and Takeda primarily responsible for oncology-related markets. We and Takeda
11
will jointly decide on promotional responsibility for markets outside of these initial indications. Takeda will control price, terms of sale and booking of sales of peginesatide.
With respect to existing third party license agreements relevant to peginesatide, fees and milestones payments related to these existing third party licenses will be shared between us and Takeda as development expenses, provided that an upfront fee in the amount of $17.6 million to a third party licensor of certain technology related to peginesatide paid in 2006 was the sole responsibility of us. For all territories outside the U.S., any royalty payments to a third party for a license will be borne solely by Takeda and other fees or payments will be borne by us and Takeda jointly.
Either party may terminate the collaboration for material breach by the other party. In addition, Takeda will have the right to terminate the collaboration (a) for certain specified clinical development events or failures, or (b) for convenience upon six months written notice to us. In the event of any termination of the agreement, Takeda will transfer and assign to us all rights to peginesatide in the affected territories. In addition, if Takeda terminates the collaboration for convenience prior to the first commercial sale in the U.S. for reasons other than specified clinical development events or failures, then Takeda will pay us a termination fee.
February 2006 Development and Commercialization Agreement with Takeda
In February 2006, we entered into collaboration with Takeda to develop and commercialize peginesatide in Japan. Under our agreement, Takeda obtained the exclusive right to develop and commercialize peginesatide in Japan for the treatment of anemia in patients with chronic renal failure and cancer, while we retained the rights to develop and commercialize peginesatide in the rest of the world, either alone or with third party partners. Takeda has granted to us a fully paid, royalty-free, sublicenseable, non-exclusive license under its own related technology to develop and commercialize peginesatide in the rest of the world.
Takeda also obtained a right of first negotiation to any backup products for peginesatide developed by us or our third party partners. Specifically, during the first ten years of the agreement, if we develop, or our third party partners develop within an Affymax collaboration, a product that advances to Phase 2 clinical trials and competes with peginesatide in the renal or oncology indications, we are obligated to offer to Takeda the right to develop and commercialize such product in Japan before offering the product opportunity in Japan to any other third party.
Takeda is obligated to use diligent efforts to develop and commercialize peginesatide in Japan. The agreement establishes a joint committee to oversee the development, regulatory approval and commercialization of peginesatide. While the joint committee will operate by consensus of the parties, Takeda will generally have the final decision-making authority on matters pertaining to the development and commercialization of peginesatide in Japan.
Takeda is responsible for commercializing peginesatide in Japan and will have the discretion to set the price of peginesatide in Japan. Under the agreement, Takeda will provide us with progress reports on its commercialization activities and we will have the opportunity to review and comment on the significant marketing decisions including strategy and launch dates.
We provide Takeda with peginesatide API and Takeda is responsible for the fill and finish of the product. Our pre-clinical and clinical supply of peginesatide API to Takeda is governed under the terms of this agreement, while the supply for Takeda's requirements for commercial quantities of peginesatide API will be governed by a separate manufacturing agreement to be executed between the parties.
Pursuant to this agreement, Takeda has paid us approximately $42 million to date, consisting of $17 million in upfront licensing fees, approximately $10 million for the purchase of equity, a $10 million cash milestone payment for the completion of the first Phase 1 trial of peginesatide in Japan, and in March 2010, a $5 million cash milestone payment for the initiation of Phase 3 trial of
12
peginesatide in Japan. Upon Takeda's successful achievement of clinical development and regulatory milestones, we are eligible to receive from Takeda an additional aggregate of $33 million relating to the renal program, together with royalties based on a percentage of the sales of peginesatide in Japan. The agreement establishes milestone payments for additional indications.
Under the agreement, each party will solely own all inventions made by such party alone, and will jointly own all inventions made by the parties jointly, including all intellectual property rights therein. Such solely-owned inventions and jointly-owned inventions will be subject to the cross-licenses between the parties for the development and commercialization of peginesatide in each party's territory. We are obligated to maintain our third party license agreements that may contain technology that is the subject of the license to Takeda under this agreement.
Each party will be responsible for the worldwide filing, prosecution and maintenance (including defense against third party opposition claims) of patents solely owned by such party and the filing, prosecution and maintenance of jointly-owned patents each in its own territory. The parties will share the responsibility for enforcing patents against third party infringement, and the allocation of responsibilities and sharing of recoveries will depend on where the claims arise, and which patents are involved. We have the first right, but not the obligation, to defend against patent infringement claims or bring patent opposition claims relating to peginesatide in Japan, and Takeda has the backup right to do so. Neither party can settle any patent infringement claim without the prior consent of the other party, if the settlement will negatively affect the other party's rights.
Each party is obligated to indemnify the other party for third party claims and losses resulting from the development and commercialization activities involving peginesatide in its territory, a breach of its representations, warranties or obligations under the agreement, or its willful misconduct or negligent acts, except to the extent such losses are subject to the indemnification obligations of the other party.
Absent early termination, the agreement will expire when all of Takeda's payment obligations expire. Either party may terminate the agreement early upon prior written notice if the other party commits an uncured material breach of the agreement. Takeda also has the option to terminate the agreement early, without cause, upon six months' prior written notice. We may convert Takeda's license to be non-exclusive or terminate the agreement entirely if Takeda promotes certain products that compete with peginesatide. If Takeda terminates without cause or if we terminate for Takeda's material breach, Takeda will transfer to us the right to develop and commercialize peginesatide in Japan.
License, Manufacturing and Supply Agreement with Nektar
In April 2004, we entered into a License, Manufacturing and Supply Agreement with Nektar Therapeutics AL Corporation, or Nektar, under which we obtained from Nektar a worldwide, non-exclusive license, with limited rights to grant sublicenses, under certain intellectual property covering pegylation technology to manufacture, develop and commercialize peginesatide. The license we obtained consists of a license under intellectual property owned by Nektar and a sublicense under intellectual property owned by Enzon Pharmaceuticals, Inc., or Enzon, licensed to Nektar pursuant to a cross-license agreement between Nektar, Inhale Therapeutic Systems, Inc. and Enzon.
In consideration of the license grant, we agreed to pay royalties on the sales of peginesatide. We also agreed to pay milestone payments totaling up to an additional $7 million, plus possible additional milestones in connection with our partnering activities relating to peginesatide or merger and acquisition activities.
In July 2006, we paid Nektar a $17.6 million milestone payment triggered by our receipt of a $105 million upfront payment from Takeda.
13
Under the agreement, we also engaged Nektar for the manufacture and supply of our requirements of bulk poly(ethylene) glycol reagent for the manufacture of peginesatide. This relationship is managed by a managing committee formed by representatives from both us and Nektar. Nektar is obligated to engage a third party manufacturer in the event of Nektar's failure (as defined in the agreement) to supply reagent, but currently Nektar remains our sole-source of these reagents.
This agreement expires, on a country by country basis, upon the expiration of our royalty payment obligations. The agreement may be terminated by either party for the other party's material breach provided that such other party has been given a chance to cure such breach, or by Nektar for our challenge of the validity or enforceability of any patents licensed thereunder.
Marketing and Sales
We currently do not have sales and marketing capabilities. Our business model is to become a fully integrated biopharmaceutical company and we intend to develop commercial capabilities in the renal market in order to co-commercialize peginesatide under our collaboration agreements with Takeda.
Competition
We operate in highly competitive segments of the biotechnology and biopharmaceutical markets. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions. Many of our competitors have significantly greater financial, product development, manufacturing and marketing resources than us. Large pharmaceutical companies have extensive experience in clinical testing and obtaining regulatory approval for drugs. These companies also have significantly greater research capabilities than us. Many universities and private and public research institutes are active in chronic renal failure research, some in direct competition with us. We also compete with these organizations to recruit scientists and clinical development personnel. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
We estimate that rEPO generated approximately $2.5 billion net revenues in the U.S. for 2010 attributable to use in dialysis patients with chronic renal failure. In the U.S., the leaders, PROCRIT, marketed by J&J, and Aranesp and EPOGEN, both marketed by Amgen, represented the entire market. PROCRIT is marketed for treatment of anemia in non-dialysis patients as well as for chemotherapy induced anemia. We anticipate that, if approved in the U.S. for treatment of anemia associated with chronic renal failure in dialysis patients, peginesatide would compete with EPOGEN and potentially Aranesp, which are both marketed by Amgen. Aranesp, introduced in 2001, has significant market share in the U.S, particularly in the oncology and the non-dialysis markets, although it is approved for treatment in dialysis patients as well. Aranesp is approved for once-monthly dosing for treatment of anemia in non-dialysis patients in Europe. In the U.S., Amgen reportedly is in the process of seeking approval for once-monthly dosing of Aranesp for treatment of anemia in pre-dialysis patients. In 2005, Amgen submitted a biologics license supplement to include a once-monthly dosing regimen for non-dialysis patients in the label for Aranesp. In October 2006, the FDA responded to Amgen's filing with a request for additional clinical data for the once-monthly dosing regimen, including an additional clinical study.
Roche has obtained regulatory approval to market and has launched a PEGylated ESA, called Mircera, in Europe. Mircera reportedly has greater plasma stability than any of the currently marketed products. PEG is a polymer that increases the time rEPO remains in the circulation and consequently can be dosed less frequently. Mircera has also obtained regulatory approval in the U.S., but as a result of Roche and Amgen's patent infringement litigation, Mircera has been found to infringe several U.S. patents owned by Amgen and has been enjoined from being sold in the U.S. until mid-2014 under the terms of a limited license. If Mircera enters the U.S. markets before peginesatide or upon its entry, we
14
believe that Mircera will be in direct competition with peginesatide, and therefore could potentially limit the market for peginesatide, because of its ability to be longer acting than currently marketed ESAs in the U.S. In addition to marketed ESAs, there are several ESA product candidates in various stages of active development, including small molecules, by potential competitors, including FibroGen, Inc., that may promote the production of naturally-occurring EPO in patients.
In addition to Mircera, several biosimilar versions of short-acting rEPO are available for sale in Europe. Biosimilar EPOGEN products are generally not expected to enter the U.S. market until the expiration of Amgen's remaining U.S. EPO patent estate, which expire from 2012 to 2015. Upon entry into the U.S. market, biosimilars are expected to constitute additional competition for peginesatide, if approved, and may drive its price and sales volume down, which may adversely affect our revenues.
Government Regulation and Product Approvals
The clinical development, manufacturing and potential marketing of peginesatide is subject to regulation by various authorities in the U.S., the E.U. and other countries, including, in the U.S., the FDA, and, in the E.U., the European Agency for the Evaluation of Medical Products, or EMA. In the U.S., pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug, and Cosmetic Act and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. The FDA has very broad enforcement authority and failure to abide by applicable regulatory requirements can result in administrative or judicial sanctions being imposed on us, including warning letters, refusals of government contracts, clinical holds, civil penalties, injunctions, restitution, disgorgement of profits, recall or seizure of products, total or partial suspension of production or distribution, withdrawal of approval, refusal to approve pending applications, and criminal prosecution.
Product development and approval within these regulatory frameworks takes a number of years, and involves the expenditure of substantial resources. Regulatory approval will be required in all major markets in which we, or our licensors, seek to test our products in development. At a minimum, such approval requires evaluation of data relating to quality, safety and efficacy of a product for its proposed use. The specific types of data required and the regulations relating to these data differ depending on the territory, the drug involved, the proposed indication and the stage of development.
In the U.S., specific pre-clinical data, chemical data and a proposed clinical study protocol, as described above, must be submitted to the FDA as part of an Investigational New Drug application, or IND, which, unless the FDA objects, will become effective 30 days following receipt by the FDA. Phase 1 trials may commence only after the IND application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the E.U. Currently, in each member state of the E.U., following successful completion of Phase 1 trials, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase 2 trials. The regulatory authorities in the E.U. typically have between one and three months in which to raise any objections to the proposed clinical trial, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase 1 trials, further submissions to regulatory authorities are necessary in relation to Phase 2 and 3 trials to update the existing IND. Authorities may require additional data before allowing the trials to commence and could demand discontinuation of studies at any time if there are significant safety issues. In addition to regulatory review, a clinical trial involving human subjects has to be approved by an independent body. The exact composition and responsibilities of this body differ from country to country. In the U.S., for example, each clinical trial is conducted under the auspices of an Institutional Review Board at the institution at which the clinical trial is conducted. This board considers among other things, the design of the clinical trial, ethical factors, the safety of the human subjects and the
15
possible liability risk for the institution. Equivalent rules apply in each member state of the E.U., where one or more independent ethics committees that typically operate similarly to an Institutional Review Board, will review the ethics of conducting the proposed research. Other authorities elsewhere in the world have slightly differing requirements involving both execution of clinical trials and import or export of pharmaceutical products. It is our responsibility to ensure that we conduct our business in accordance with the regulations of each relevant territory.
Information generated in this process is susceptible to varying interpretations that could delay, limit, or prevent regulatory approval at any stage of the approval process. Failure to demonstrate adequately the quality, safety and efficacy of a therapeutic drug under development would delay or prevent regulatory approval of the product. There can be no assurance that if clinical trials are completed, either we or our collaborative partners will submit applications for required authorizations to manufacture or market potential products, including a marketing authorization application, or a MAA, or that any such application will be reviewed and approved by appropriate regulatory authorities in a timely manner, if at all.
In order to gain marketing approval, we must submit a dossier to the relevant authority for review, which is known in the U.S. as an NDA and in the E.U. as a MAA. The format is usually specified by each authority, although in general it will include information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product and non-clinical and clinical data.
FDA Approval Process
In the U.S., if approved, peginesatide will be regulated by the FDA as a drug, but not as a biologic. No manufacturer may market a new drug until it has submitted a NDA to the FDA, and the FDA has approved it. The steps required before the FDA may approve an NDA generally include:
- •
- preclinical laboratory tests and animal tests conducted in compliance with FDA's good laboratory practice requirements;
- •
- development, manufacture and testing of active pharmaceutical product and dosage forms suitable for human use in
compliance with current GMP;
- •
- the submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials
may begin;
- •
- adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its
specific intended use(s);
- •
- the submission to the FDA of a NDA; and
- •
- FDA review and approval of the NDA.
Preclinical tests include laboratory evaluation of the product candidate, as well as animal studies to assess the potential safety and efficacy of the product candidate. The conduct of the pre-clinical tests must comply with federal regulations and requirements including good laboratory practices.
The results of the preclinical and clinical studies, together with other detailed information, including the manufacture and composition of the product candidate, in this case peginesatide, are to be submitted to the FDA in the form of a NDA requesting approval to market the drug. FDA approval of the NDA is required before marketing of the product may begin in the U.S. If the NDA contains all pertinent information and data, the FDA will "file" the application and begin review. The FDA may "refuse to file" the NDA if it does not contain all pertinent information and data or if in the wrong format. In that case, the applicant may resubmit the NDA when it contains the missing information and data in the correct format. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of new drug applications. Although applications for non-priority drug products are intended to be reviewed within 10 months, the
16
review process, however, may be substantially extended by FDA requests for additional information, preclinical or clinical studies, clarification regarding information already provided in the submission, submission of a risk evaluation and mitigation strategy or proposed labeling. The FDA may refer an application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. An advisory committee is expected to be convened for review of peginesatide. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations when making decisions. Even if the advisory committee makes a favorable recommendation, the FDA may still reject an application for approval. Before approving a NDA, the FDA will typically inspect the facilities at which the product candidate is manufactured and will not approve the product candidate GMP compliance is satisfactory. FDA also typically inspects facilities responsible for performing animal testing, as well as clinical investigators who participate in clinical trials and the sponsor. The FDA may refuse to approve a NDA if applicable regulatory criteria are not satisfied, or may require additional testing or information. The FDA may also limit the indications for use and/or require post-marketing testing and surveillance to monitor the safety or efficacy of a product. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
The testing and approval process requires substantial time, effort and financial resources, and our product candidates may not be approved on a timely basis, if at all. The time and expense required to perform the clinical testing necessary to obtain FDA approval for regulated products can frequently exceed the time and expense of the research and development initially required to create the product.
While in the U. S., the FDA undertakes such reviews for the U.S., in the E.U., there is, for many products, a choice of two different authorization routes: centralized and decentralized. Under the centralized route, one marketing authorization is granted for the entire E.U., while under the decentralized route a series of national marketing authorizations are granted. In the centralized system, applications are reviewed by members of the Committee for Medicinal Products for Human Use, on behalf of the EMEA. The EMEA will, based upon the review of the Committee for Medicinal Products for Human Use, provide an opinion to the European Commission on the safety, quality and efficacy of the product. The decision to grant or refuse an authorization is made by the European Commission. In circumstances where use of the centralized route is not mandatory, we can choose to use the decentralized route, in which case the application will be reviewed by each member state's regulatory agency. If the regulatory agency grants the authorization, other member states' regulatory authorities are asked to "mutually recognize" the authorization granted by the first member state's regulatory agency. Approval can take several months to several years or be denied. The approval process can be affected by a number of factors. Additional studies or clinical trials may be requested during the review and may delay marketing approval and involve unbudgeted costs. Regulatory authorities may conduct inspections of relevant facilities and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. Further, inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor adverse effects, or other additional studies as deemed appropriate. After approval for the initial indication, further clinical trials are usually necessary to gain approval for additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect product marketability.
Employees
As of December 31, 2010, we had 140 employees. We had 97 employees engaged in research and development, and our remaining employees are management or administrative staff. None of our employees is subject to a collective bargaining agreement. We believe that we have good relations with our employees.
17
About Affymax
We were incorporated in Delaware in July 2001 under the name Affymax, Inc. The address of our principal executive office is 4001 Miranda Avenue, Palo Alto, California 94304, and our telephone number is (650) 812-8700. Our website address is www.affymax.com. We do not incorporate the information on our website into this Annual Report on Form 10-K, and you should not consider it part of this Annual Report on Form 10-K.
We have a registration for the trademarks "Affymax" and "Affymax and logo" in the U.S.
Available Information
We file electronically with the U.S. Securities and Exchange Commission our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities and Exchange Act of 1934. We make available on our website at www.affymax.com, free of charge, copies of these reports as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission. Further, copies of these reports are located at the Securities and Exchange Commission's Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the Securities and Exchange Commission at 1-800-SEC-0330. The Securities and Exchange Commission maintains a website that contains reports, proxy and information statements, and other information regarding our filings, at www.sec.gov. The information contained in, or that can be accessed through, our website is not part of, and is not incorporated into, this Annual Report on Form 10-K.
You should carefully consider the risks described below, which we believe are the material risks of our business before making an investment decision. Our business could be harmed by any of these risks. The trading price of our common stock could decline due to any of these risks, and you may lose all or part of your investment. In assessing these risks, you should also refer to the other information contained in this Annual Report on Form 10-K, including our financial statements and related notes.
Risks Related to Our Business
We are dependent on the success of peginesatide (HematideTM). Peginesatide is a new chemical entity and currently our only product candidate. We cannot give any assurance the development program for peginesatide will be successful or completed in a timely or effective manner. Our recently announced Phase 3 results present challenges to our ability to obtain regulatory approval for Peginesatide particularly in view of the heightened concerns surrounding safety of erythropoiesis stimulating agents, or ESAs. Our failure to demonstrate the safety and effectiveness of peginesatide to the satisfaction of the U.S. Food and Drug Administration, orFDA, will prevent us from receiving regulatory approval and would have a material and adverse impact on our business. Any failure of timely and complete submission of our New Drug Application, or NDA, or failure of the FDA to review and approve of an NDA submission on a timely basis would severely harm our business.
Peginesatide, an ESA, is a new chemical entity and currently our only product candidate. In order to commercialize peginesatide, we will be required to establish that peginesatide is sufficiently safe and effective in order to obtain regulatory approvals, which we may fail to do.
In late June 2010, we announced preliminary top-line results from the peginesatide Phase 3 clinical program for the treatment of patients with anemia associated with chronic renal failure. Our Phase 3 clinical program included four open-label, randomized controlled clinical trials. Of these trials, two trials, called PEARL 1 and PEARL 2, were conducted in non-dialysis patients and designed to evaluate the safety and efficacy of peginesatide compared to darbepoetin alfa to correct anemia and maintain
18
hemoglobin in a corrected range over time. The other two trials, called EMERALD 1 and EMERALD 2, were conducted in dialysis patients and designed to evaluate the safety and efficacy of peginesatide and its ability to maintain hemoglobin levels in a corrected range compared to epoetin alpha or epoetin beta when switched to peginesatide. Analysis of efficacy and safety for all of the Phase 3 studies were based primarily on assessments of non-inferiority to the comparator drugs. The primary efficacy endpoint, the mean change in hemoglobin from baseline, in each of the four Phase 3 studies met the statistical criteria for non-inferiority. In addition, peginesatide met the statistical criterion for non-inferiority for the assessment of safety for the cardiovascular composite safety endpoint, or CSE, which was composed of death, stroke, myocardial infarction, congestive heart failure, unstable angina and arrhythmia from a pooled safety database across the four Phase 3 studies. However, some differences were observed when secondary analyses were conducted as previously described in our Current Report on Form 8-K dated June 21, 2010, including a difference in a subgroup analysis conducted in the PEARL trials where the frequency of CSE events was higher in the peginesatide group relative to the comparator in these non-dialysis patients.
Despite meeting the primary efficacy endpoints and the CSE for peginesatide, the differences observed in the Phase 3 secondary analyses present risks to our ability to obtain regulatory approval for peginesatide particularly in view of the heightened concerns surrounding safety of ESAs. Based on our discussions with the U.S. Food and Drug Administration, or FDA, we plan to submit a New Drug Application, or NDA, for treatment of anemia in chronic renal failure patients on dialysis in the second quarter of 2011. Any negative perception of peginesatide's safety relative to other ESAs would significantly limit the likelihood of obtaining regulatory approval for peginesatide. The issues arising from the Phase 3 results have caused significant delay and may continue to negatively impact the timelines for development and the likelihood, scope or conditions surrounding regulatory approval. Any or all of these factors may significantly reduce the ultimate commercial potential of peginesatide.
Regardless of whether peginesatide met the statistical criteria for non-inferiority to the comparator drugs, peginesatide could still fail to establish that it is sufficiently safe for regulatory approval for any indication. In addition to data from clinical trials, we must also submit extensive data from pre-clinical studies, including carcinogenicity studies, as a condition to submission of a NDA and regulatory approval. As peginesatide is the first ESA to undergo carcinogenicity studies, the regulatory requirements and standards for review remain uncertain and may increase the risk for regulatory approval. The results from earlier pre-clinical testing and prior clinical trials may not be predictive of results obtained in other pre-clinical models, later clinical trials or in a practice setting. In addition, the submission or approval of our NDA may be delayed or fail for many reasons, including:
- •
- safety issues, including serious adverse events associated with peginesatide, and concerns surrounding use of ESAs
generally;
- •
- difficulties arising from administration, data gathering and analysis of our large and complex Phase 3 clinical
program for peginesatide, which involved numerous third parties, approximately 2,600 patients and over 300 sites in the U.S. and Europe, compliance with a variety of government regulations, and a
number of significant new initiatives and processes for which we did not have any prior experience implementing, including the adjudication of cardiovascular events by an independent review committee;
- •
- risks associated with non-inferiority trials, which are studies devised and statistically powered to show that
the test drug is not inferior to the comparator drug;
- •
- risks associated with data integrity and difficulty in obtaining complete and accurate data on a timely basis which may result from our large and complex Phase 3 trial design for a variety of other reasons, including shortage of resources, delays in data entry, inaccurate or inconsistent data entry, failure to follow the clinical trial protocols, inadequate monitoring or training of sites, inadequate oversight of third party clinical research organizations, or CROs, delays or
19
- •
- suspension or termination of clinical trials for various reasons, including exposure of the participating patients to
unacceptable health risks or noncompliance with regulatory requirements;
- •
- manufacturing issues or failure to manufacture or obtain from third parties materials of sufficient quality;
- •
- inadequate effectiveness or safety concerns arising from clinical trials or pre-clinical studies, including
the carcinogenicity studies;
- •
- the failure of patients to complete clinical trials due to death or the length of our clinical program, side effects,
dissatisfaction with peginesatide or other reasons including adverse medical effects unrelated to treatment with peginesatide;
- •
- our lack of experience as an organization in preparing a complete and acceptable large NDA submission for peginesatide
that is expected to be submitted in electronic Common Technical Document (e-CTD) format, which will involve significant complexity and coordination with a number of third party contractors
and our collaboration partner, Takeda;
- •
- governmental or regulatory delays and changes in regulatory requirements, policy and guidelines; and
- •
- varying interpretation of data by FDA and similar foreign regulatory agencies.
failures to establish adequate procedures, remediations or corrective actions that regulatory agencies may not find sufficient, problems maintaining contact with patients after treatment or as a consequence of the open-label, non-inferiority design of the Phase 3 trials;
Further analysis, regulatory review or inspections or additional data may reveal further issues associated with the Phase 3 results. For example, negative imbalances in safety events, which could give rise to safety concerns whether or not they are statistically significant, or potential issues surrounding data quality, which may be of greater concern for non-inferiority designed trials, may negatively impact the ultimate acceptability of the data for regulatory approval. As noted in the FDA's March 2010 draft Guidance for Industry Non-inferiority Clinical Trials, there is a critical need for particular attention to study quality and conduct when planning and executing a non-inferiority study, as poor quality can sometimes lead to an apparent finding of non-inferiority that is incorrect. The FDA appears to be increasing its focus on clinical data quality which may delay or increase the risk of failure to obtain regulatory approval. For example, in late 2009, Basilea Pharmaceutica AG failed to obtain approval for ceftobiprole from the FDA as the agency cited unreliable or unverifiable data and inadequate monitoring on the part of sponsor Johnson & Johnson as the basis for the agency's decision. As the sponsor of the peginesatide clinical trials, the FDA holds us accountable for oversight of our clinical trials, including monitoring performed by our CROs. To the extent the FDA determines that we failed to properly oversee our clinical trials and the CROs, the FDA may find our Phase 3 results or other clinical data unreliable. Our failure to adequately demonstrate the safety and effectiveness of peginesatide or the integrity of the data will prevent us from receiving regulatory approval and will have a material adverse impact our business.
Obtaining approval of a NDA by the FDA is highly uncertain and like many product candidates, we may fail to obtain approval even if we do not receive a refusal to file and are able to submit a NDA for peginesatide that is acceptable for review. The NDA review process is extensive, lengthy, expensive and uncertain, and the FDA may delay, limit or deny approval of peginesatide for many reasons, including:
- •
- we may not be able to demonstrate to the satisfaction of the FDA that peginesatide is safe and effective for any indication;
20
- •
- the data arising from the clinical trials, including the Phase 3 results, or the development program for
peginesatide may not be satisfactory to the FDA;
- •
- the FDA may disagree with the number, design, size, conduct or implementation of our clinical trials or conclude that the
data fails to meet statistical or clinical significance;
- •
- the FDA may not find the data from preclinical studies, including our carcinogenicity studies, and clinical studies
sufficient to demonstrate that peginesatide's clinical and other benefits outweighs its safety risks;
- •
- the FDA may disagree with our interpretation of data from preclinical studies or clinical trials;
- •
- the FDA may not accept data generated at our clinical trial sites and monitored by third party clinical research
organizations;
- •
- the FDA may determine that we did not properly oversee third party clinical research organizations and our clinical
trials;
- •
- the FDA may have difficulties scheduling an advisory committee in a timely manner;
- •
- an advisory committee may recommend against approval of our application or may recommend that the FDA require, as a
condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions or even if an advisory committee makes a favorable
recommendation, the FDA may still fail to approve peginesatide;
- •
- the FDA may have difficulties approving a risk evaluation and mitigation strategy, or REMS, or labeling for peginesatide;
and
- •
- the FDA may identify deficiencies in our manufacturing processes, facilities or analytical methods or those of our third party contract manufacturers or Takeda.
The FDA may require us to conduct additional studies or trials which could result our failure to ever bring peginesatide to market. Accordingly, we may not receive the regulatory approvals needed to market peginesatide. Any failure or delay in completion of the development program, submission of our NDA to the FDA or the review process would delay or foreclose commercialization of peginesatide and severely harm our business and financial condition.
We have relied and continue to rely on numerous third parties, particularly CROs, to conduct and complete our development program for peginesatide, and to the extent they fail to properly and successfully perform their obligations to us, we may not be able to obtain the necessary regulatory approvals for peginesatide.
Due to the size of and limited experience of our organization, we have relied heavily on CROs, contractors and other third parties to assist us in managing, monitoring and otherwise conducting clinical trials. Our Phase 3 clinical program for peginesatide was large and complex and conducted at over 300 sites in the U.S. and Europe. Even though we have recently completed our Phase 3 clinical program, we continue to require the assistance of these third parties as we prepare our NDA submission and prepare for the FDA review process. We have had significant difficulties obtaining the necessary and quality resources. We continue to compete with larger and other companies for the attention and assistance of these third parties. If we are unsuccessful in obtaining the needed assistance, we will have difficulty maintaining our NDA submission timelines and obtaining approval for peginesatide.
Although we rely on these third parties to conduct our clinical trials, we are responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol. Moreover, FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that the data and results are credible and accurate
21
and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements, and regulatory authorities may find remediation efforts by us or our CROs insufficient. Recently, the FDA appears to be increasing its focus on clinical data quality, which may delay or reduce the likelihood of regulatory approval.
We may not be able to maintain our relationships with these CROs or contractors on acceptable terms. These third parties generally may terminate their engagements with us at any time and if we have to enter into alternative arrangements, these may result in the delay of development and commercialization of peginesatide. As a result, we can control their activities only within certain limits, and they will devote only a certain amount of their time to peginesatide. If these third parties did not or do not successfully carry out their duties under their agreements with us or, or if they otherwise fail to meet expected deadlines, the planned NDA submission may not meet regulatory requirements. If the quality or accuracy of the data they obtain is compromised due to failure to adhere to clinical protocols, good clinical practices and regulatory requirements, our development activities may be extended or such failure to perform may negatively impact the quality and acceptability of the data. If any of these events occur, or we are otherwise unable to adequately demonstrate the reliability of the data from our Phase 3 results, we may not be able to obtain regulatory approval of peginesatide on a timely basis, if at all.
The Trial to Reduce Cardiovascular Endpoints with Aranesp Therapy, or TREAT, results heightens concerns surrounding safety of ESAs and increases the regulatory risk for peginesatide as the class faces greater scrutiny. These concerns may limit the ability to develop and obtain regulatory approval for peginesatide. The FDA recently convened a cardio-renal advisory committee meeting to re-evaluate the use of ESAs in the treatment of anemia in chronic kidney disease. We cannot predict what future actions the FDA may take that could affect the potential of peginesatide.
In late 2009, Amgen Inc., or Amgen, announced the results of its large, randomized, double-blind, placebo-controlled Phase 3 study of patients with chronic kidney disease (CKD) (not requiring dialysis), anemia and type-2 diabetes (TREAT). In this study, treatment of anemia with Aranesp to a target hemoglobin of 13 g/dL, which is higher than the 10 g/dL - 12 g/dL range approved by the FDA in the current label, reportedly failed to show benefit compared to the control group with regard to composite of time to all-cause mortality or cardiovascular morbidity (including heart failure, heart attack, stroke, or hospitalization for myocardial ischemia) and a composite of time to all-cause mortality or chronic renal replacement. In addition, higher rates of stroke were reported amongst patients treated with Aranesp compared to the control group. Further, among a subgroup of patients with a history of cancer at baseline, a statistically significant increase in deaths from cancer was observed in the Aranesp treated patients compared to placebo treated patients. However, Aranesp treatment reportedly was associated with a statistically significant reduction in blood transfusions and a modest improvement in patient reported fatigue.
In January 2010, FDA officials published an editorial in the New England Journal of Medicine entitled "Erythropoiesis—Stimulating Agents—Time for a Reevaluation" and announced that it anticipates convening a public advisory committee meeting later this year to evaluate the use of ESAs in the treatment of anemia due to chronic kidney disease. The editorial noted that a number of randomized trials, including TREAT, have attempted to show that using ESAs to raise hemoglobin concentrations to higher targets improves clinical outcomes but rather have suggested the opposite. Accordingly, the article indicates that more conservative hemoglobin targets (well below 12 g/dL), more frequent hemoglobin monitoring, and more cautious dosing, should be evaluated.
In February 2010, the FDA announced that ESAs must be prescribed and used under a risk management program known as a REMS to ensure the safe use of these drugs. As part of the REMS, a medication guide explaining the risks and benefits of ESAs must be provided to all patients receiving ESAs for all indications. In addition, in the case of oncology use, the FDA required ESA
22
manufacturers to implement training for hospitals and healthcare professionals and the signing of a patient informed consent acknowledging the risks of ESA use prior to treatment. As part of any REMS, the manufacturer has reporting and monitoring obligations to ensure compliance.
In October 2010, the FDA convened a cardio-renal advisory committee to review TREAT and to re-evaluate the use of ESAs in the treatment of anemia in chronic kidney disease. Although the advisory panel voted against withdrawal of the indication for Aranesp's use in non-dialysis patients even those with a history of stroke, and voted against the adoption of the TREAT control group dosing regimen (treatment once hemoglobin is below 9 g/dL), the advisory committee discussion included potential areas of concerns regarding the use of ESAs, including hemoglobin variability and rates of excursions associated with current dosing regimens, use by certain subgroups including diabetics and hyporesponders, among others, for further consideration in clinical trials. As the advisory committee's recommendations are non-binding, the FDA may choose to reject the advisory panel's recommendations and impose further restrictions or requirements on ESAs, including Peginesatide.
The TREAT results and the FDA's recent and potential future actions represent additional challenges to the ESAs as a class and increases the uncertainty associated with peginesatide's regulatory approval. Even prior to these recent events, for the last several years, the FDA, the medical community, and others have recently raised significant safety concerns relating to commercially available ESAs as a result of reports of increased mortality and side effects from a number of clinical trials. These concerns have resulted in a number of negative actions affecting the market for ESAs particularly in oncology, including the following:
- •
- As a result of concerns associated with administering ESAs to target higher hemoglobin levels, the FDA required revised
warnings, including black box warnings, be added to labels of currently marketed ESAs advising physicians to monitor hemoglobin levels and to use the lowest dose of ESA to increase the hemoglobin
concentration to the lowest level sufficient to avoid the need for red blood cell transfusions.
- •
- The FDA also issued a public health advisory statement re-evaluating the safe use of the ESA class and
convened its Oncology Drugs Advisory Committee, or ODAC, in May 2007 to consider recent information on risks associated with ESAs for use in the treatment of anemia in cancer patients. The ODAC
recommended that the FDA institute restrictions on the usage of currently marketed ESAs, including limitations on the treatment of certain types of cancer and the duration of treatment.
- •
- The FDA also convened a joint meeting in September 2007 of the Cardiovascular and Renal Drugs advisory committee and the
Drug Safety and Risk Management advisory committee to review the risks and benefits of ESAs.
- •
- The FDA approved revised black box warnings and other safety-related product labeling changes for commercially available
ESAs during 2007and thereafter.
- •
- In addition, the FDA convened another ODAC meeting in March 2008 to review data from more recent clinical trials with
breast cancer patients and cervical cancer patients using currently marketed ESAs, and to consider additional action. The ODAC recommended the use of informed consents and further restrictions on the
use of currently marketed ESAs for the treatment of chemotherapy-induced anemia, including the exclusion of patients with metastatic breast or head and neck cancer as well as those cancer patients
potentially receiving curative treatment.
- •
- In July 2008, the FDA announced additional safety-related label restrictions for the use of commercially available ESAs including revisions to the black box warnings to provide that ESAs are not indicated for patients undergoing chemotherapy expected to cure their cancer. In
23
addition, the FDA required new prescribing information to assure that ESA therapy is not initiated until the hemoglobin level drops below 10 g/dL.
In 2008, these factors and the uncertain regulatory climate resulted in our and Takeda's decision to suspend the development of peginesatide to treat chemotherapy-induced anemia. Further, in 2010. based on our discussion with the FDA, we and Takeda decided to submit an NDA for treatment of anemia in chronic renal failure patients for dialysis patients only. These events may have a material adverse effect on our business and future financial results.
We cannot predict what further action, if any, the FDA may take, which may include, among others, additional label restrictions, the use of informed consents, further lowering of target hemoglobin levels, or even the removal of indications from the label altogether. Further, regardless of whether the FDA takes additional action or not, the Centers for Medicare and Medicaid Services, or CMS, and private payors may still decide separately to lower or discontinue reimbursement.
The controversy surrounding ESAs and FDA concerns has, and may, further negatively affect peginesatide, including the completion of the development program. These safety concerns may increase the risk of achieving regulatory approval or negatively affect the timing or costs associated with obtaining regulatory approval, including potential risk mitigation activities we may be required to complete either prior to or after product approval. We cannot predict the scope of the REMS we may ultimately be required to implement by the FDA and the impact on the use of peginesatide. Even a small imbalance in safety events or unfavorable signal or trend against peginesatide may increase the risk of or the conditions or limitations associated with approval by the FDA, as regulators are increasingly uncomfortable with the safety of the comparator ESAs. Any of these factors could significantly delay or negatively impact the commercialization of peginesatide.
Our development program for peginesatide may not lead to a commercial drug either because we fail to adequately demonstrate that it is safe and effective in clinical trials and or pre-clinical studies and we therefore fail to obtain necessary approvals from the FDA and similar foreign regulatory agencies, or because we have inadequate financial or other resources to advance peginesatide through development commercialization. Our analyses of the Phase 3 results remains preliminary and no conclusions as to the safety and efficacy can be drawn as only the FDA can ultimately make such determination. Any failure to obtain approval of peginesatide would have a material and adverse impact on our business as we would have to incur substantial expense and it would take a significant amount of time and resources to bring any future product candidate to market, if ever.
Even if peginesatide receives approval by the FDA for treatment of anemia associated with chronic renal failure in dialysis patients, the market opportunity for peginesatide may be significantly reduced as a result of the increasing controversy surrounding ESAs, the TREAT results and future actions by the FDA and CMS.
Safety concerns have significantly reduced the market for ESAs in recent years. As the perception of the risks of ESA usage continues to increase with the controversy surrounding the recent TREAT results, the concerns are likely to further negatively impact the use of ESAs and the commercial potential of peginesatide. In October 2010, the FDA convened a cardio-renal advisory committee to review TREAT and to re-evaluate the use of ESAs in the treatment of anemia in chronic kidney disease. The FDA may further lower target hemoglobin levels and implement other actions that may limit the use of ESAs in chronic kidney disease potentially beyond non-dialysis patients to dialysis as well. In addition to potential FDA action to limit use of ESAs, CMS convened a meeting of the Medicare Evidence Development & Coverage Advisory Committee, or MedCAC, to review the available evidence on the use of ESAs to manage anemia in patients who have chronic kidney disease and considered the results of the TREAT study among others. Any action by FDA to further restrict ESA use or decrease reimbursement coverage by CMS could have a materially negative impact on the size of the ESA market in the U. S. and reduce the overall size of the market peginesatide is expected
24
to compete in at the time of launch. Not only may a small imbalance in safety events or unfavorable signal or trend against peginesatide increase FDA approval risk or the risk of peginesatide obtaining reimbursement, but any negative perception of peginesatide's safety relative to other ESAs could keep us from successfully commercializing peginesatide.
We have incurred significant operating losses since inception and anticipate that we will incur continued losses for the foreseeable future, which will require us to obtain substantial additional financing. If we fail to obtain additional financing, we will be unable to complete the development and commercialization of peginesatide and may need to cease operations. Even if we obtain additional financing, we may never achieve or sustain profitability.
We have experienced significant operating losses since our inception in 2001. Currently, we have no products approved for commercial sale and, to date, we have not generated any revenue from product sales. At December 31, 2010, we had an accumulated deficit of $388.9 million. Due to the recognition of revenues from milestone payments from our collaboration with Takeda, we were profitable for the three and six months ended June 30, 2010 and may have profitable quarters from time to time if we are successful in obtaining FDA approval for peginesatide. We continue to expect to incur substantial losses for the next several years. Our operations have consumed substantial amounts of cash since our inception. We expect to continue to spend substantial amounts in order to:
- •
- complete clinical development of peginesatide;
- •
- prepare the submission of the NDA for peginesatide for FDA review process, which is a lengthy and uncertain process;
- •
- prepare for manufacturing process for peginesatide at our contract manufacturers for commercial launch; and
- •
- prepare to launch and commercialize peginesatide, including building our own commercial organization, sales force and infrastructure to address renal markets.
We believe that our existing cash, cash equivalents and investments together with the interest thereon will enable us to maintain our currently planned operations for at least the next 12 months. However, we expect that we will need to raise additional funding to complete the development and commercialization of peginesatide. Since the announcement of our Phase 3 data in late June 2010 and the arbitration decision in October 2010, we have experienced a severe decline in our stock price, which has impaired our ability to access capital on potentially favorable terms. In contrast, our need to raise funding has only increased as the peginesatide development program has suffered delays, the potential loss of milestone payments from Takeda associated with the non-dialysis indication and the potential for future legal proceedings and costs. As we continue to analyze the data, we may experience further challenges or delays to approval of peginesatide if issues arise or additional requirements are imposed based on our discussions with the FDA.
The current capital markets have been extremely volatile, and biotechnology companies have been limited or unsuccessful in obtaining funding in this environment. Securing funding has been particularly difficult for companies of our size with limited capital resources. Continuation of this market and the issues arising from our Phase 3 results significantly limit our ability to raise funds such that that there can be no assurance we can raise the additional funds to support our continuing operations and maintain current development and commercialization timelines for peginesatide.
To date, our sources of cash have been limited primarily to the proceeds from the sale of our securities to private and public investors and payments by Takeda under our collaboration agreements. We cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that we raise additional funds by issuing equity securities, if available, our stockholders may experience significant dilution particularly given the stock price decline we experienced subsequent to
25
the announcement of our Phase 3 results. Further, our equity line of credit with Azimuth Opportunity Ltd., or Azimuth, is subject to a number of conditions that limits our ability to draw against such facility. Any debt financing, if available, may involve security interests on our assets or restrictive covenants, such as limitations on our ability to incur additional indebtedness, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business.
If we are unable to raise additional funds when required or on acceptable terms, we may have to:
- •
- assume greater risks and significantly delay, scale back, or discontinue the development and/or commercialization of
peginesatide;
- •
- relinquish our existing rights to peginesatide;
- •
- eliminate or defer formulation research and development or other manufacturing efforts that may be required to
successfully develop or commercially launch peginesatide; or
- •
- pursue merger and acquisition alternatives.
We expect to continue to incur substantial operating losses for the next several years as we pursue our clinical trials, prepare for the NDA and add infrastructure and operations to support commercialization of peginesatide, and potentially begin new research and development programs. Our ability to generate revenue depends heavily on our ability to successfully develop and secure regulatory approval for, and commercially launch, our product candidate, peginesatide. If due to lengthy and complicated development, clinical and regulatory requirements or any other reason, we are unable to commercialize peginesatide, we may never be able to commercialize any future product candidates, if ever.
Even if we receive regulatory approval of peginesatide, we must successfully commercialize peginesatide before we can become profitable. We anticipate that it will be years before we can commercialize peginesatide and we expect to incur substantial expenses associated with our commercialization efforts as well as share in those of Takeda's even prior to obtaining approval of peginesatide as well as thereafter. Accordingly, we may never generate significant revenues and, even if we do generate revenues, we may never achieve or sustain profitability.
Peginesatide may require extensive additional clinical evaluation and will require regulatory approval, significant marketing efforts and substantial investment before it can provide us or our partners with any revenue. If we or our partners are unable to develop and commercialize peginesatide or even if we receive marketing approval for peginesatide, sales revenue therefrom may be insufficient, and we may not achieve or sustain profitability, and we may be unable to continue our operations.
As a result of a determination in our binding arbitration that Johnson & Johnson Pharmaceutical Research & Development, L.L.C., and Ortho-McNeil Pharmaceutical, Inc., or J&J, owns certain intellectual property related to erythropoietin receptor, or EPO-R, agonists, J&J may seek to assert claims that may prevent us from manufacturing or commercializing peginesatide in accordance with our current plans, including limiting our ability to license rights to third parties.
In October 2010, the arbitration panel in our binding arbitration with J&J decided the ownership of a number of U.S. and international patents and patent applications related to certain EPO-R agonists, or the "intellectual property in dispute." The decision maintained J&J's sole inventorship and sole ownership of U.S. Patent No. 5,767,078, or the "078 Patent," and certain related foreign patents and patent applications, including European Patent application EP96/918,317. The arbitrators determined that we and J&J jointly own the remainder of the intellectual property in dispute, i.e., U.S. Patent Nos. 5,773,569, 5,830,851, and 5,986,047 together with their foreign counterpart patents and patent applications.
26
We are continuing to review the arbitrators' decision and consider potential courses of action with our counsel and Takeda. In the event that our motion to vacate the arbitration award with respect to the ownership of the '078 Patent and its foreign counterparts does not succeed, J&J would continue to be the sole owner of the '078 Patent and its foreign counterparts. We expect that this dispute with J&J could involve additional litigation or legal proceedings that may take years and substantial resources and funds to resolve.
Although we believe that peginesatide does not infringe the '078 Patent and that we would have substantial defenses to any potential claims by J&J, J&J may now or in the future attempt to assert claims based upon the '078 Patent against us or our collaborators in connection with the manufacture and commercialization of peginesatide. If J&J is successful in asserting its rights under the '078 Patent, J&J may prevent us from manufacturing or commercializing peginesatide, either for ourselves or with Takeda or any potential sublicensees, or obtain a royalties on sales of peginesatide until expiration of the '078 Patent in 2015. In addition, an adverse outcome could result in liability for damages, attorneys' fees and costs.
Outside of the U.S., the European Patent application EP96/918,317 and other foreign counterpart patents to the '078 Patent differ in the nature and extent of the claims from that of the '078 Patent. Due to the complexity of patent laws, we are unable to assess adequately the defenses available to us in the various countries outside of the U.S. to any potential claims by J&J alleging infringement of any of the foreign counterparts to the '078 patent. Even if we succeed in defending against an assertion by J&J of its rights under the '078 Patent or one of its foreign counterpart patents, we may not necessarily succeed in other jurisdictions since such matters may be litigated on a country by country basis as the nature and extent of the claims of the foreign counterparts to the '078 Patent as well as the patent laws may vary substantially from country to country. Litigation is time consuming and expensive and the outcome is inherently uncertain. If J&J were successful in asserting its rights under any of the foreign counterparts to the '078 Patent, J&J may prevent us in the applicable foreign country from manufacturing or commercializing peginesatide, either for ourselves or with Takeda or any potential sublicensees, or obtain a royalties on sales of peginesatide until expiration of such patent in such country. In addition, an adverse outcome could result in liability for damages, attorneys' fees and costs.
If intellectual property in dispute that has been deemed to be jointly owned is broad enough to cover peginesatide, then under the laws applicable in certain relevant jurisdictions outside the U.S., joint ownership may not allow us to license third parties to manufacture and sell peginesatide or even do so ourselves, which may negatively affect our development and business plans outside the U.S. or our collaboration with Takeda.
Regardless of the ultimate outcome of the proceedings, additional litigation or legal proceedings or even the risk thereof may make it more difficult to commercialize peginesatide. The threat of such legal uncertainty may make it difficult for peginesatide to gain market acceptance by health care providers, patients, payors or dialysis clinics, any of which may be concerned about the reliability of supply or reluctant to become involved in the prospect of existing or potential litigation.
We continue to consider our legal alternatives in this dispute. To date, we have incurred significant expense in pursuing this matter, including substantial time and effort on the part of our technical, legal and management personnel. As final resolution of this dispute may not be reached for years, we expect to incur significant expenses and diversion of resources for years. Further, the risk or existence of litigation or legal proceedings with J&J may limit our ability to finance, and even if such financing is available, to achieve terms that are favorable to us.
27
Even if peginesatide is approved by the FDA for treatment of anemia associated with chronic renal failure in dialysis patients, our commercial success depends upon attaining significant market acceptance of peginesatide among physicians, patients, health care payors and the major operators of dialysis clinics as well as reaching an agreement with one or more of such major operators of dialysis clinics.
Peginesatide has not been approved or commercialized for any indication and we are planning to pursue approval from the FDA for treatment of anemia associated with chronic renal failure in dialysis patients. Even if approved for sale by the appropriate regulatory authorities, physicians may not prescribe peginesatide, in which case we would not generate revenue or become profitable. In particular, the therapeutic indication targeted by peginesatide has been served by our competitors' products for many years. These products may now be said to be the standard of care, and it may be difficult to encourage healthcare providers to switch from products with which they and their patients have become comfortable.
The dialysis market, which peginesatide will attempt to penetrate, is highly established and concentrated, with two ESA products serving a significant majority of all dialysis patients on Medicare. In addition, dialysis clinics using ESAs could incur substantial expense in administration and training if they were to switch from current ESAs to peginesatide. The concentration of customers for ESAs within the dialysis market may pose a risk to our ability to obtain revenues or favorable margins on peginesatide, if approved. If we cannot come to agreements with one or more of the major companies operating dialysis clinics in the U.S. or, even if we do, we cannot do so on favorable terms or on a timely basis, the revenue opportunity of peginesatide could be significantly reduced. In October 2006, Amgen, which markets the ESAs EPOGEN and Aranesp, and Fresenius Medical Care, or Fresenius, one of the two largest operators of dialysis clinics in the U.S., announced an agreement whereby Amgen would be the sole supplier of EPO products for Fresenius' dialysis business effective immediately through the end of 2011. We are not aware of the specific terms of the Amgen-Fresenius agreement, and cannot project how it may impact the commercial opportunity for peginesatide if and when it is launched. However, agreements between operators of dialysis facilities and marketers of competing ESA products could potentially limit the market opportunity for peginesatide, and adversely impact our ability to generate revenues.
Prior to this year, CMS generally reimbursed ESAs at a rate of 106% of the average ESA sales price, or ASP. This allowed the dialysis facilities to realize a profit on the purchase and administration of ESAs, which constitutes an important component of their economic viability. However, under the 2008 Medicare Legislation a new bundled payment system commenced in January 2011 for facilities that furnish renal dialysis services and home dialysis to Medicare beneficiaries with end-stage renal disease. Under the new system, CMS will make a single bundled payment to the dialysis facility for each dialysis treatment that will cover all renal dialysis services, including ESAs. The bundled payment system may create incentives for significantly lower utilization or dosing of ESAs, including peginesatide, and reduce the commercial potential for peginesatide. We cannot guarantee that peginesatide will be reimbursed by CMS in a manner that will support physician adoption. CMS held Medicare Coverage Advisory Committee (MedCAC) meetings in March 2010 and January 2011 to review current ESA coverage policy based on the available evidence on the use of ESAs to manage anemia in patients who have chronic kidney disease, and the role of ESAs in successful kidney transplantation, respectively. Independent of any additional action the FDA may take, CMS may further decrease reimbursement coverage of ESA reducing the overall size of the market peginesatide is expected to compete in at the time of launch.
In addition, recent studies by manufacturers of ESAs indicate that the higher levels of hemoglobin achieved through administration of ESAs can result in a statistically significant increase in cardiovascular events. This may in turn reduce the growth or cause contraction of the market for ESAs and reduce the potential revenues for peginesatide.
28
In addition, market acceptance of peginesatide by physicians, healthcare payors and patients will depend on a number of additional factors, including:
- •
- the clinical indication for which peginesatide is approved;
- •
- acceptance by physicians and patients of peginesatide as a safe and effective treatment alternative;
- •
- perceived advantages over alternative treatments;
- •
- the cost of treatment in relation to alternative treatments;
- •
- the availability of adequate reimbursement by third parties;
- •
- the continued use of ESA treatments generally for anemia;
- •
- relative convenience and ease of administration; and
- •
- the prevalence and severity of side effects.
Competition in the pharmaceutical industry is intense. If our competitors are able to develop and market products that are more effective, safer or less costly than peginesatide, our commercial opportunity will be reduced or eliminated.
We face competition from established and emerging pharmaceutical and biotechnology companies, as well as from academic institutions, government agencies and private and public research institutions. Our commercial opportunity will be reduced or eliminated if our competitors develop and commercialize products that are more effective, have fewer side effects or are less expensive than peginesatide or any other future products that we may develop and commercialize. In addition, significant delays in the development of peginesatide could allow our competitors to bring new products to market before we do and impair our ability to commercialize peginesatide. Competitors may also reduce the price of their ESAs in order to gain market share. These price reductions could force us to lower the price of peginesatide in order to compete effectively, resulting in lower revenues and reduced margins on the sales of peginesatide.
We anticipate that, if approved, for treatment of anemia associated with chronic renal failure in dialysis patients, peginesatide would compete with EPOGEN and potentially Aranesp, which are both marketed by Amgen, NeoRecormon and Mircera, which are currently marketed outside the U.S. by Roche. PROCRIT, which is marketed by Ortho Biotech Products, L.P. (a subsidiary of J&J), is approved for treatment of anemia in non-dialysis patients as well as for chemotherapy induced anemia. Aranesp is approved for once-monthly dosing for treatment of anemia in non-dialysis patients in Europe. In the U.S., Amgen is reportedly in the process of seeking approval for once-monthly dosing of Aranesp for treatment of anemia in non-dialysis patients. If Amgen is successful in obtaining approval for once-monthly dosing or our competitors' products are administered in practice on a less frequent basis than prescribed by their labels, the market for peginesatide may be decreased. In addition, Roche's Mircera has recently launched in Europe. Mircera reportedly has greater plasma stability and is longer acting than any rEPO product that is currently on the market. As a result of the patent litigation between Roche and Amgen, Mircera has been found to infringe several U.S. patents owned by Amgen and has been enjoined from being sold in the U.S. until the expiration of these patents in mid-2014 under a limited license. If Mircera enters the U.S. market before peginesatide or upon its entry, we believe that Mircera will be in direct competition with peginesatide, and therefore could potentially limit the market for peginesatide, because of its ability to be longer acting. Other potential competitors, including FibroGen, Inc., are developing small molecules designed to promote the production of greater levels of naturally-occurring EPO in patients. The introduction of biosimilars into the ESA market, or new market entrants, could also prove to be a significant threat to us as it could not only limit the market for peginesatide, but could also drive down the price of ESAs.
29
Most of these competitors have substantially greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Current marketers of ESAs also have the ability to bundle sales of existing ESA products with their other products, potentially disadvantaging peginesatide, which we plan to sell on a stand-alone basis. Established pharmaceutical and large biotechnology companies may invest heavily to discover and develop novel compounds or drug delivery technology that could make peginesatide obsolete. Smaller or early-stage companies may also prove to be significant competitors, particularly through strategic partnerships with large and established companies. These third parties may compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business. Our competitors may succeed in obtaining patent or other intellectual property protection, receiving FDA approval, or discovering, developing and commercializing products before we do.
The U.S. market opportunity for peginesatide may deteriorate significantly after the entry of biosimilars in the U.S.
The remaining U.S. patents for epoetin alfa, a version of short-acting rEPO, expire from 2012 through 2015. Patents related to epoetin alfa expired in the European Union, or E.U., in 2004. Biosimilars of short-acting rEPO are currently being developed or sold in various markets outside the U.S., including the E.U. We expect that biosimilars, including rEPO, will be sold at a significant discount to existing branded products when they are launched in the U.S. as in the E.U. The introduction of biosimilars into the ESA market in the U.S could prove to be a significant threat to peginesatide if they are able to demonstrate bioequivalence to existing ESAs. Biosimilars will constitute additional competition for peginesatide, if approved, and are expected to drive its price and sales volume down, which may adversely affect our revenues.
Peginesatide is our only product candidate and we may not develop any other product candidates for the foreseeable future.
Peginesatide is the main focus of our business, which we expect to be the case for the foreseeable future. Accordingly, until we are able to obtain additional financing and resources to develop and commercialize peginesatide, we are unlikely to be able to successfully discover or develop any other product candidates. Further, we have had to reduce our research capabilities and efforts, including the elimination of certain research programs even some activities related to the support of peginesatide. We have limited ability and resources to pursue internal research programs and strategic collaborations for the development of new products. Research programs to identify new disease targets and product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for many reasons, including, but not limited to, the following:
- •
- the financial and internal resources may be insufficient and are needed for peginesatide;
- •
- the research methodology used may not be successful in identifying potential product candidates;
- •
- competitors may develop alternatives that render our product candidates obsolete;
- •
- a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory approval;
30
- •
- a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; or
- •
- a product candidate may not be accepted by patients, the medical community or third party payors.
The success of peginesatide is dependent upon the strength and performance of our collaboration with Takeda. If we fail to maintain our existing collaboration with Takeda, such termination would likely have a material adverse effect on our ability to continue to develop peginesatide and our business.
The maintenance and successful performance of our strategic collaboration with Takeda for development of peginesatide is an important part of our business model. Our collaboration with Takeda is extremely complex particularly with respect to financial provisions, allocations of responsibilities, and the respective rights of the parties with respect to decision making. Accordingly, significant aspects of the development and commercialization of peginesatide require Takeda's agreement or approval prior to implementation, which can cause significant delays. Further, if we are not able to reach agreement with Takeda or maintain our existing collaboration with Takeda to develop and commercialize peginesatide, our business could be severely and adversely affected. Takeda has the ability to terminate each of the collaboration agreements upon an uncured material breach by us or even in the absence of a material breach with six-months' notice. Currently, Takeda could provide us notice of termination of either or both of our collaboration agreements, which would likely have a material adverse effect on the advancement of our peginesatide program and our business. The suspension of the peginesatide oncology program, the impact of the Phase 3 results on the renal program particularly on the non-dialysis indication and the arbitration decision relating to the dispute with J&J may increase the likelihood that Takeda terminates the collaboration or affect the resources Takeda is willing to commit to peginesatide. Through the collaboration, Takeda currently provides development and commercial funding and performs important functions, including conduct of certain clinical trials and manufacturing activities, and is expected to pay us milestone payments upon the completion of certain events, all of which would be unavailable to us in the case of an early termination of the collaboration. Even in the absence of a termination, Takeda's failure to provide funding or perform its obligations on a timely basis may have a material adverse effect on our business and the success of peginesatide.
In addition, if we fail to maintain the Takeda collaboration or establish and maintain additional strategic collaborations for any other potential product candidates that we may pursue:
- •
- the development of peginesatide or future product candidates may be terminated or delayed;
- •
- our cash expenditures related to development of our current or future product candidates would increase significantly and
we may need to seek additional financing;
- •
- we may be required to hire additional employees or otherwise develop expertise, such as sales and marketing expertise, for
which we have not budgeted;
- •
- we will bear all of the risk related to the development of each of our current and future product candidates; and
- •
- we may be unable to meet demand for any future products that we may develop.
Any of these events could have a material adverse effect on our business.
Reimbursement may not be available for peginesatide, which would materially diminish our sales and our ability to sell our products profitably.
Market acceptance and sales of peginesatide will depend on reimbursement policies and may be affected by future health care reform measures. Government authorities and third party payors, such as private health insurers and health maintenance organizations, decide which drugs they will pay for and
31
establish reimbursement levels. We cannot be sure that reimbursement will be available for peginesatide. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or the price of, peginesatide. We have not commenced efforts to have our peginesatide reimbursed by government or third party payors. If reimbursement is not available or is available only to limited levels, we may not be able to commercialize peginesatide.
In both the U.S. and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals in recent years to change the healthcare system in ways that could impact our ability to sell peginesatide profitably.
In response to the FDA's recent black box warning and public health advisories, CMS has significantly restricted coverage of ESAs. In July 2007, CMS issued its National Coverage Decision Memorandum for Use of Erythropoiesis Stimulating Agents in Cancer and Neoplastic Conditions, or the National Coverage Decision, that determined that ESA treatment was not reasonable or necessary for certain medical conditions, including any anemia of cancer not related to cancer treatment, among others. The National Coverage Decision also established the ESA reimbursement policy for Medicare and other government beneficiaries who are treated for chemotherapy-induced anemia and contains a coverage restriction for hemoglobin levels greater than 10g/dL, which has had a material adverse effect on the use of ESAs. In July 2007, CMS also issued revisions to its reimbursement policies for the use of ESAs for end stage renal disease in cases where hemoglobin levels exceed 13 g/dL and also decreased the monthly dosing limits. In July 2008, CMS announced that ESAs are a potential topic for another National Coverage Decision citing adverse effects in cancer and chronic kidney disease patients, including dialysis patients while noting the large costs but uncertain benefits. In March 2010, CMS convened a MedCAC meeting to review the available evidence on the use of ESAs to manage anemia in patients who have chronic kidney disease and in January 2011 to review the role of ESAs in successful kidney transplantation. Independent of any additional action the FDA may take as to ESAs, CMS may further decrease coverage which could have a materially negative impact on the size of the ESA market in the U.S. and reduce the overall size of the market peginesatide is expected to compete in at the time of launch.
As a result of these reimbursement and other legislative proposals and the trend towards managed health care in the U.S., third party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drugs. They may also refuse to provide any coverage of approved products for medical indications other than those for which the FDA has granted market approvals. In addition, major third party payors have begun to follow CMS's restrictive reimbursement policies, which has further decreased the market for ESAs. As a result, significant uncertainty exists as to whether and how much third party payors will reimburse patients for their use of newly approved drugs, which in turn will put pressure on the pricing of drugs. We expect to experience pricing pressures in connection with the sale of our products due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative proposals.
CMS policies are constantly changing and we cannot guarantee that they will not decrease, limit or deny reimbursement of peginesatide in the future.
CMS, the agency within the Department of Health and Human Services that manages Medicare and will be responsible for reimbursement of the cost of peginesatide administered to Medicare beneficiaries, has asserted the authority of Medicare not to cover particular drugs if it determines that they are not "reasonable and necessary" for Medicare beneficiaries, or to cover them at a lesser rate, compared to drugs that CMS considers to be therapeutically comparable. We cannot be certain that CMS will not decrease, limit or deny reimbursement of peginesatide for any therapeutic indication we may pursue. Even if CMS ultimately authorizes reimbursement for peginesatide, it may be not do so in a timely manner. As the costs of the Medicare program continue to grow, CMS may be compelled to
32
make difficult decisions regarding the trade-offs of supporting the reimbursement of certain public health expenditures over others. Depending on methods CMS uses to calculate the cost-benefit of treatments competing for share of the Medicare budget, ESAs (including peginesatide) may not be considered to offer sufficient overall health benefit to justify reimbursement at levels that will allow us to achieve and sustain profitability. In addition, as a result of the recent safety concerns relating to ESAs, CMS recently announced policies significantly restricting the coverage of ESAs and has proposed another National Coverage Decision on the topic that may further negatively affect reimbursement of ESAs. CMS has instituted dramatic Medicare reimbursement changes in the past that adversely impacted the businesses of companies in other segments of the healthcare industry, and we cannot determine that CMS will not do the same in the markets in which we operate.
Medicare reimbursement policies under a new bundled payment system could create disincentives for use of ESAs.
Prior to this year, CMS generally reimbursed healthcare providers for use of ESAs at average selling price, or ASP, plus 6%. However, under the 2008 Medicare Legislation a new bundled payment system commenced in January 2011 for facilities that furnish renal dialysis services and home dialysis to Medicare beneficiaries with end-stage renal disease. Under the new bundled payment system, providers are expected to be reimbursed a fixed amount per patient. We cannot guarantee that peginesatide will be reimbursed by CMS or in a manner that will support physician adoption and depending upon the implementation of the bundled payment, may not be favorable to the entry of new ESAs such as peginesatide. In fact, a capitated reimbursement payment methodology may create incentives for significantly lower utilization or dosing of ESAs, including peginesatide, and reduce the commercial potential for peginesatide.
Significant challenges remain with us and Takeda to manufacture peginesatide on a commercial scale. Our dependence upon third parties for the manufacture and supply may cause delays in, or prevent us from, successfully developing and commercializing peginesatide. In accordance with the terms of our collaboration, Takeda has responsibility for manufacture of finished product and as a consequence, we have limited ability to control risks associated with that portion of the manufacturing process.
The peginesatide manufacturing process is a complicated, time-consuming process. Manufacture of peginesatide active pharmaceutical ingredient, or API, involves long lead times. We do not currently have the infrastructure or capability internally to manufacture the peginesatide needed to conduct our clinical trials or to commercialize peginesatide. We are and will continue to rely upon contract manufacturers to produce our clinical trial materials and in the future commercial supplies of peginesatide. For the foreseeable future, we expect to continue to rely on contract manufacturers, partners and other third parties to produce sufficient quantities of peginesatide for all our uses, including completion of our clinical trials and development program. If our contract manufacturers or other third parties fail to deliver materials for the manufacture of peginesatide or peginesatide itself for clinical use or for our registration stability studies on a timely basis, with sufficient quality and at commercially reasonable prices, and if we fail to find replacement manufacturers or to develop our own manufacturing capabilities, we may be required to delay or suspend clinical trials or our planned NDA filing or otherwise discontinue development and production.
Peginesatide is a new chemical entity and the manufacturing process for commercial scale production in accordance with applicable regulatory guidelines remains challenging and as such, there are risks associated with the commercial scale manufacture of the API. Similar challenges exist for the manufacture of finished product that must meet a variety of regulatory requirements that vary from
33
country to country and continue to change. Any of these risks and others may prevent or delay us from successfully developing peginesatide, including the following:
- •
- stability or formulation issues including the potential failure of product registration studies to establish sufficient
stability to obtain adequate shelf life at refrigerated or room temperature;
- •
- cost overruns, process scale-up, process reproducibility;
- •
- difficulties in maintaining or upgrading equipment and manufacturing facilities on a timely basis; and
- •
- regulatory issues or changes that may cause significant modifications in the manufacturing process or facilities or otherwise impact our ability to offer competitive product presentations or formulations.
We have transferred responsibility of manufacture of peginesatide finished product to Takeda and we therefore have limited control and ability to address risks associated with that portion of the manufacturing process. Further, some of suppliers and manufacturing arrangements, including the provision of bulk poly(ethylene) glycol reagent for the manufacture of peginesatide from Nektar Therapeutics AL, Corporation, or Nektar, are currently single-sourced, leaving us at greater risk of supply interruptions, potential delays and failure to obtain regulatory approvals and commercialize. Unless we are able to successfully negotiate with Nektar, which we may not be able to do on acceptable terms, we may have difficulties under our existing arrangement with Nektar from obtaining proprietary information and additional services from Nektar which may be useful or necessary to obtain regulatory approvals or for commercial manufacture of peginesatide.
We, Takeda, and our third party manufacturers are required to comply with applicable FDA manufacturing practice regulations. If there is any failure by us, Takeda or one of our third party manufacturers or suppliers to maintain compliance with these regulations, the production of peginesatide could be interrupted, resulting in delays and additional costs. Additionally, our third party manufacturers must pass a pre-approval inspection before we can obtain regulatory approval for peginesatide. If for any reason these third parties are unable or unwilling to perform under our agreements or enter into new agreements with us, we may not be able to locate alternative manufacturers or enter into favorable agreements with them in an expeditious manner. We could also experience manufacturing delays if our third party manufacturers, Takeda or suppliers give greater priority to the production of other products over peginesatide. Any inability to acquire sufficient quantities of peginesatide or components thereof in a timely manner from third parties could delay clinical trials or result in product shortages and prevent us from developing and commercializing peginesatide in a cost-effective manner or on a timely basis. Further, our lack of experience providing reliable supply of product may deter health care providers and dialysis centers from selecting or otherwise switching to peginesatide from our competitors' products.
The commercial success of peginesatide depends in part on the development and marketing efforts of Takeda, over which we have limited control. If our collaborations are unsuccessful, our ability to develop and commercialize products through our collaborations, and to generate future revenue from the sale of these products, would be significantly reduced.
Our dependence on Takeda for our global collaboration with peginesatide and our other collaboration arrangements, subjects us to a number of risks. Our ability to develop and commercialize drugs that we develop with our collaboration partners depends on our collaboration partners' abilities to establish the safety and efficacy of peginesatide, obtain and maintain regulatory approvals and achieve market acceptance of peginesatide once commercialized. Under our collaboration with Takeda, we co-develop and co-commercialize peginesatide in the U.S. Because we share responsibility with Takeda for clinical development activities in the U.S., the progress of the peginesatide program is
34
dependent on the efforts of Takeda of which we have no control. In fact, Takeda has taken responsibility for conducting several clinical trials and is expected to produce substantial portions of the NDA so that any failure of Takeda to act in a timely manner may delay our ability to develop peginesatide in accordance with our timelines. Takeda holds an exclusive license to develop and commercialize peginesatide outside of the U.S. and any progress and commercial success in those territories is dependent solely on Takeda's efforts and commitment to the program. Takeda may delay, reduce or terminate development efforts relating to peginesatide, independently develop products that compete with peginesatide, or fail to commit sufficient resources to the marketing and distribution of peginesatide. Competing products or programs, either developed by Takeda or to which our collaboration partners have rights or acquire in the future, may result in our partners' withdrawal of support for peginesatide.
In the event that Takeda fails to diligently develop or commercialize peginesatide, we may have the right to terminate our partner's rights but we may choose not to as we will not receive any future revenue from peginesatide or even if we do, we may not be able to find another partner requiring us to commercialize peginesatide on our own, which is likely to result in significant additional expense and delay. Business combinations, significant changes in business strategy, litigation and/or financial difficulties may also adversely affect the willingness or ability of Takeda to complete its obligations under our collaboration agreements. If Takeda fails to perform in the manner we expect, our potential to develop and commercialize products peginesatide and to generate future revenue would be significantly reduced. If a conflict of interest arises between us and Takeda, it may act in its own self-interest and not in the interest of our company or our stockholders. If Takeda were to breach or terminate the collaboration agreements with us or otherwise fail to perform its obligations thereunder in a timely manner, the pre-clinical or clinical development or commercialization of peginesatide could be delayed or terminated.
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of peginesatide and any other product candidates we may pursue, their use and the methods used to manufacture them, as well as successfully defending these patents against third party challenges. Our ability to protect peginesatide from unauthorized making, using, selling, offering to sell or importation by third parties is dependent upon the extent to which we have rights under valid and enforceable patents, or have trade secrets that cover these activities.
We have licensed from third parties rights to numerous issued patents and patent applications. The rights that we acquire from licensors or collaborators are protected by patents and proprietary rights owned by them, and we rely on the patent protection and rights established or acquired by them. The remaining patent terms may not provide meaningful protection. Moreover, third parties may challenge the patents, patent applications and other proprietary rights held by our licensors or collaborators. We generally do not unilaterally control the prosecution of patent applications licensed from third parties. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we may exercise over internally developed intellectual property.
Even if we are able to obtain issued patents, any patent may be challenged, invalidated, held unenforceable or circumvented. The existence of a patent will not necessarily protect us from competition or from claims of a third party that our products infringe their issued patents. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date in the U.S. The biotechnology patent situation outside the U.S. is even more uncertain. Competitors may successfully challenge our patents, produce similar drugs or products that do not infringe our patents, or produce drugs in countries where we have not applied for patent protection or that do not respect our patents. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our licensed patents, in our patents or in third party patents or applications therefore.
35
The degree of future protection to be afforded by our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
- •
- others may be able to make similar compounds but that are not covered by the claims of our patents, or for which we are
not licensed under our license agreements;
- •
- we or our licensors or collaborators might not have been the first to make the inventions covered by our pending patent
application or the pending patent applications and issued patents of our licensors;
- •
- we or our licensors or collaborators might not have been the first to file patent applications for these inventions;
- •
- others may independently develop similar or alternative technologies or duplicate any of our technologies without
infringing our intellectual property rights;
- •
- it is possible that our pending patent applications will not result in issued patents;
- •
- our issued patents and the issued patents of our licensors or collaborators may not provide us with any competitive
advantages, or may be held invalid or unenforceable as a result of legal challenges by third parties;
- •
- we may not develop additional proprietary technologies that are patentable; or
- •
- the patents of others may have an adverse effect on our business.
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the U.S. are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
Our research and development collaborators may have rights to publish data and other information to which we have rights. In addition, we sometimes engage individuals or entities to conduct research that may be relevant to our business. The ability of these individuals or entities to publish or otherwise publicly disclose data and other information generated during the course of their research is subject to certain contractual limitations. These contractual provisions may be insufficient or inadequate to protect our trade secrets and may impair our patent rights. If we do not apply for patent protection prior to such publication or if we cannot otherwise maintain the confidentiality of our technology and other confidential information, then our ability to receive patent protection or protect our proprietary information may be jeopardized.
We expect to incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or use, our technology.
Our ability, and that of our commercial partners, to commercialize any approved product will depend, in part, on our ability to obtain patents, enforce those patents and operate without infringing the proprietary rights of third parties. The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. We have filed multiple U.S. patent applications and foreign counterparts related to peginesatide and other programs as well as underlying platform technologies and may file additional U.S. and foreign patent applications related thereto. There can be no assurance that any issued patents we own or control will provide
36
sufficient protection to conduct our business as presently conducted or as proposed to be conducted, that any patents will issue from the patent applications owned by us, or that we will remain free from infringement claims by third parties.
The failure to obtain adequate patent protection would have a material adverse effect on us and may adversely affect our ability to enter into, or affect the terms of, any arrangement for the further development and marketing of any product. There can also be no assurance that patents owned by us will not be challenged by others. As a result of a determination in our binding arbitration that J&J, owns certain intellectual property, J&J may now or in the future attempt to assert claims based upon the '078 Patent and other intellectual property owned by J&J. We could incur substantial costs in proceedings, including interference proceedings before the U.S. Patent and Trademark Office and comparable proceedings before similar agencies in other countries in connection with any claims that may arise in the future. These proceedings could result in adverse decisions about the patentability of our inventions and products, as well as about the enforceability, validity or scope of protection afforded by our patents.
Patent applications in the U.S. and elsewhere are published only after 18 months from the priority date. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made. Therefore, patent applications relating to products similar to peginesatide and any future products may have already been filed by others without our knowledge. In the event an infringement claim is brought against us, we may be required to pay substantial legal and other expenses to defend such a claim and, if we are unsuccessful in defending the claim, we may be prevented from pursuing related product development and commercialization and may be subject to damage awards.
Our ongoing litigation is described in the section entitled "Legal Proceedings." We have incurred substantial expense as a result of our litigation and arbitration proceedings and we expect to incur even greater expense in the future. In addition, any future patent litigation, interference or other administrative proceedings will result in additional expense and distraction of our personnel. An adverse outcome in such litigation or proceedings may expose us or our collaborators to loss of our proprietary position or to significant liabilities, or require us to seek licenses that may not be available from third parties on commercially acceptable terms or at all. In addition, we may be restricted or prevented from manufacturing, developing or commercializing peginesatide or from developing, manufacturing and selling any future products in the event of an adverse determination in a judicial or administrative proceeding or if we fail to obtain necessary licenses. If it is determined that we have infringed an issued patent, we could be compelled to pay significant damages, including punitive damages.
Virtually all of our competitors are able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations, in-license technology that we need, out-license our existing technologies or enter into collaborations that would assist in commercially exploiting any technology.
If we are unable either to create sales, marketing and distribution capabilities or enter into agreements with third parties to perform these functions, we will be unable to commercialize peginesatide successfully.
We currently have no sales, marketing or distribution capabilities. To commercialize peginesatide , we must either develop internal sales, marketing and distribution capabilities, which will be expensive and time consuming, or make arrangements with third parties to perform these services. If we decide to market peginesatide directly, we must commit significant financial and managerial resources to develop a marketing and sales force with technical expertise and with supporting distribution capabilities.
37
Factors that may inhibit our efforts to commercialize peginesatide directly or indirectly with Takeda include:
- •
- our inability to recruit and retain adequate numbers of effective sales and marketing personnel;
- •
- the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe our products;
- •
- the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage
relative to companies with more extensive product lines; and
- •
- unforeseen costs and expenses associated with creating and sustaining an independent sales and marketing organization.
If we, or Takeda through our collaboration, are not successful in recruiting sales and marketing personnel or in building a sales and marketing infrastructure, we will have difficulty commercializing peginesatide, which would adversely affect our business and financial condition. To the extent we rely on other pharmaceutical or biotechnology companies with established sales, marketing and distribution systems to market peginesatide, we will need to establish and maintain partnership arrangements, and we may not be able to enter into these arrangements on acceptable terms. To the extent that we enter into co-promotion or other arrangements, any revenues we receive will depend upon the efforts of third parties, which may not be successful and are only partially in our control. In that event, our product revenues would likely be lower than if we marketed and sold our products directly.
If we fail to attract and keep senior management and key clinical and scientific personnel, we may be unable to successfully develop, conduct our clinical trials and commercialize peginesatide or any other future product candidates.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. We are highly dependent upon our senior management and clinical and scientific staff, particularly John Orwin, our Chief Executive Officer, and Dr. Anne-Marie Duliege, our Chief Medical Officer. The loss of services of Mr. Orwin, Dr. Duliege, or one or more of our other members of senior management could delay or prevent the successful completion of our development or the commercialization of peginesatide.
Competition for qualified personnel in the biotechnology and pharmaceuticals field is intense. We will need to hire additional personnel as we expand our clinical development and commercial activities. We may not be able to attract and retain quality personnel on acceptable terms. Our ability to retain or attract qualified personnel has been negatively impacted by the Phase 3 results and the severe decline in our stock price. Each of our officers and key employees may terminate his/her employment at any time without notice and without cause or good reason.
As we evolve from a company primarily involved in research and development to a company also involved in commercialization, we may encounter difficulties in managing our growth and expanding our operations successfully.
As we advance peginesatide through the development stage towards commercialization, we will need to expand our organization, including marketing and sales capabilities or contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to manage additional relationships with various collaborative partners, suppliers and other third parties. Future growth will impose significant added responsibilities on members of management. Our future financial performance and our ability to commercialize peginesatide and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts effectively, and hire, train and integrate additional management,
38
administrative and sales and marketing personnel. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing our company.
Risks Related to Our Industry
The regulatory approval process is expensive, time consuming and uncertain and may prevent us or our collaboration partners from obtaining approvals for the commercialization of peginesatide.
The research, testing, manufacturing, selling and marketing of drug candidates are subject to extensive regulation by the FDA and other regulatory authorities in the U.S. and other countries, and regulations may differ from country to country. Neither we nor Takeda is permitted to market peginesatide in the U.S. until we receive approval of a NDA, from the FDA. We have not received marketing approval for peginesatide. Further, we have not previously prepared an NDA submission, which involves compliance with governmental regulations and successful completion of a number of significant and complicated undertakings for which we do not have any prior experience implementing. Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject our company to administrative or judicially imposed sanctions, including warning letters, civil and criminal penalties, injunctions, product seizure or detention, product recalls, total or partial suspension of production and refusal to approve pending NDAs or supplements to approved NDAs.
Regulatory approval of an NDA or NDA supplement is not guaranteed, and the approval process is expensive and may take several years. The FDA also has substantial discretion in the drug approval process. We initiated our Phase 3 clinical trials for peginesatide following extensive discussion with the FDA on the design of the program. Based on the nature of these discussions and guidance from the FDA in light of the current regulatory environment, we did not enter into a special protocol assessment, or SPA, with the FDA for our Phase 3 clinical trials for peginesatide. Nonetheless, in some instances a SPA could provide more assurance that the design, clinical endpoints, and statistical end analyses resulting from these trials would be acceptable to the FDA to support regulatory approval. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional pre-clinical studies and clinical trials. The number of pre-clinical studies and clinical trials that will be required for FDA approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. The FDA can delay, limit or deny approval of a drug candidate for many reasons, including:
- •
- a drug candidate may not be deemed safe or effective;
- •
- FDA officials may not find the data from pre-clinical studies and clinical trials sufficient;
- •
- the FDA might not approve our or our third party manufacturer's processes or facilities; or
- •
- the FDA may change its approval policies or adopt new regulations.
Even if we receive regulatory approval for peginesatide, we will be subject to ongoing FDA obligations and continued regulatory review, which may result in significant additional expense and limit our ability to commercialize peginesatide.
Any regulatory approvals that we or Takeda receive for peginesatide may also be subject to limitations on the indicated uses for which the product may be marketed, or contain requirements for potentially costly post-marketing follow-up studies. In addition, if the FDA approves peginesatide, the labeling, packaging, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. Our recent Phase 3 results may increase the risk of significant additional requirements to maintain any regulatory approval that we might receive. The subsequent discovery of previously unknown problems with the drug, including
39
adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
The FDA's policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of peginesatide. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market our future products and we may not achieve or sustain profitability.
Failure to obtain regulatory approval in foreign jurisdictions will prevent us from marketing our products abroad through our Takeda collaboration.
We intend to co-market peginesatide in the U.S, and have exclusively licensed Takeda to develop peginesatide in international markets. In order to market peginesatide in the E.U. and many other foreign jurisdictions, we must obtain separate regulatory approvals. We have had limited interactions with foreign regulatory authorities, and the approval procedures vary among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. Foreign regulatory approvals may not be obtained on a timely basis, if at all. We or Takeda, as part of our peginesatide collaboration, may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market.
Foreign governments often impose strict price controls, which may adversely affect our future profitability.
We intend to seek approval to market peginesatide in the U.S. and, through our Takeda collaboration, in foreign jurisdictions. If we obtain approval in one or more foreign jurisdictions, we will be subject to rules and regulations in those jurisdictions relating to our product. In some foreign countries, particularly in the E.U., prescription drug pricing is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a drug candidate. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of peginesatide to other available therapies or a clinical trial that studies pharmacoeconomic benefits. If reimbursement of peginesatide is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
We may incur significant costs complying with environmental laws and regulations, and failure to comply with these laws and regulations could expose us to significant liabilities.
We use hazardous chemicals and radioactive and biological materials in our business and are subject to a variety of federal, state and local laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these materials. Although we believe our safety procedures for handling and disposing of these materials and waste products comply with these laws and regulations, we cannot eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of contamination or injury, we could be held liable for any resulting damages. We are uninsured for third party contamination injury.
40
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of peginesatide.
We face an inherent risk of product liability as a result of conducting clinical trials and will face an even greater risk if we commercialize peginesatide. We may be held liable if any product we develop causes injury or is found otherwise unsuitable during product testing, manufacturing, marketing or sale. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of peginesatide. Even successful defense would require significant financial and management resources. Regardless of the merit or eventual outcome, liability claims may result in:
- •
- decreased demand for peginesatide;
- •
- injury to our reputation;
- •
- withdrawal of clinical trial participants;
- •
- costs of related litigation;
- •
- diversion of management's attention and resources;
- •
- substantial monetary awards to patients;
- •
- product recalls;
- •
- loss of revenue; and
- •
- the inability to commercialize peginesatide.
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop. We currently carry product liability insurance covering our clinical trials in the amount of $11 million in the aggregate. However, our insurance may not reimburse us or may not be sufficient to reimburse us for the expenses or losses we may suffer. In addition, insurance coverage is becoming increasingly expensive, and in the future we may not be able to maintain insurance coverage at a reasonable cost or obtain insurance coverage that will be adequate to satisfy any liability that may arise.
Risks Related to the Ownership of Our Common Stock
The market price of our common stock has been highly volatile and is likely to remain highly volatile, and you may not be able to resell your shares at or above your purchase price.
The trading price of our common stock has been highly volatile. For the 52 weeks ended December 31, 2010, the price ranged between a high of $25.37 per share and a low of $5.00 per share. Our stock is expected to be subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including:
- •
- actual or anticipated results from, and any delays in, our development program and commercialization of peginesatide;
- •
- actual or anticipated changes in our funding requirements, capital resources and our ability to obtain financing and the
terms thereof;
- •
- actual or anticipated actions taken by regulatory agencies with respect to ESAs generally or specifically as to
peginesatide;
- •
- actual or anticipated regulatory approvals of peginesatide or competing products
41
- •
- new products or services introduced or announced by us or our collaboration partners, or our competitors, including
Roche's Mircera or biosimilars, and the timing of these introductions or announcements;
- •
- issuance of patents to potential competitors or third parties, including the expected issuance of patents to J&J in
Europe;
- •
- additional litigation or legal proceedings in our dispute with J&J, including both substantive and procedural developments
relating to the intellectual property in dispute;
- •
- actions taken by regulatory agencies with respect to clinical trials, manufacturing process or sales and marketing
activities;
- •
- changes in laws or regulations applicable to peginesatide, including but not limited to clinical trial requirements for
approvals;
- •
- the success of our development efforts and clinical trials;
- •
- the success of our efforts to discover, acquire or in-license additional products or product candidates;
- •
- developments concerning our collaborations, including but not limited to those with our sources of manufacturing supply
and our commercialization partners;
- •
- actual or anticipated variations in our quarterly operating results;
- •
- announcements of technological innovations by us, our collaborators or our competitors;
- •
- actual or anticipated changes in earnings estimates or recommendations by securities analysts;
- •
- conditions or trends in the biotechnology and biopharmaceutical industries;
- •
- announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital
commitments;
- •
- general economic and market conditions and other factors that may be unrelated to our operating performance or the
operating performance of our competitors;
- •
- changes in the market valuations of similar companies;
- •
- sales of common stock or other securities by us or our stockholders in the future;
- •
- additions or departures of key scientific or management personnel;
- •
- developments relating to proprietary rights held by us or our competitors;
- •
- disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to
obtain patent protection for our technologies; and
- •
- trading volume of our common stock.
In addition, the stock market in general and the market for biotechnology and biopharmaceutical companies in particular have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market, securities class-action litigation or regulatory investigations have often been instituted against companies. Such litigation or investigations, if instituted against us, could result in substantial costs and diversion of management's attention and resources, which could materially and adversely affect our business and financial condition.
42
Failure to maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse effect on our stock price.
The Sarbanes-Oxley Act of 2002 requires, among other things, that we maintain effective internal control over financial reporting and disclosure controls and procedures. We currently have not had any material weaknesses for the years ended December 31, 2010 or 2009. We did identify a material weakness in the operation of our internal controls over financial reporting that occurred during the second quarter of 2008 which has been fully remediated. We cannot assure you that material weaknesses will not be identified in future periods. There can be no assurance that we will successfully and timely report on the effectiveness of our internal control over financial reporting in future periods. If we do experience a material weakness in future periods, then investor confidence, our stock price and our ability to obtain additional financing on favorable terms could be adversely affected.
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within an organization have been detected. We continue to implement, improve and refine our disclosure controls and procedures and our internal control over financial reporting.
Future sales of our common stock in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market that were previously restricted from sale, or the perception that these sales might occur, could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. In the event that we do raise capital through the sale of additional equity securities, the dilution represented by the additional shares of our equity securities in the public market could cause our stock price to fall, in which case investors may not be able to sell their shares of our equity securities at a price equal to or above the price they paid to acquire them.
Our ability to use net operating loss carryforwards and tax credit carryforwards to offset future taxable income may be limited as a result of transactions involving our common stock.
In general, under Section 382 of the Internal Revenue Code of 1986, as amended, a corporation that undergoes an "ownership change" is subject to limitations on its ability to utilize its pre-change net operating losses, or NOLs, and certain other tax assets to offset future taxable income. In general, an ownership change occurs if the aggregate stock ownership of certain stockholders increases by more than 50 percentage points over such stockholders' lowest percentage ownership during the testing period (generally three years). An ownership change could limit our ability to utilize our NOL and tax credit carryforwards for taxable years including or following such "ownership change." It is possible that transactions involving our common stock, even those outside our control, such as purchases or sales by investors, within the testing period could result in an ownership change. Limitations imposed on the ability to use NOLs and tax credits to offset future taxable income could require us to pay U.S. federal income taxes earlier than would otherwise be required if such limitations were not in effect and could cause such NOLs and tax credits to expire unused, in each case reducing or eliminating the benefit of such NOLs and tax credits. Similar rules and limitations may apply for state income tax purposes.
We are at risk of securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because of the significant decrease in our stock price as a result of the announcement of our Phase 3 data and the decision from the arbitration panel relating to the dispute with J&J. Further, our stock price may
43
continue to experience extreme price volatility as has been experienced by biotechnology and biopharmaceutical companies in recent years. If we were to face such litigation, it could result in substantial costs and a diversion of management's attention and resources, which could harm our business.
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders.
Provisions in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, even if doing so would benefit our stockholders.
These provisions include:
- •
- authorizing the issuance of "blank check" preferred stock, the terms of which may be established and shares of which may
be issued without stockholder approval;
- •
- limiting the removal of directors by the stockholders;
- •
- prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of
our stockholders;
- •
- eliminating the ability of stockholders to call a special meeting of stockholders;
- •
- establishing advance notice requirements for nominations for election to the board of directors or for proposing matters
that can be acted upon at stockholder meetings; and
- •
- our board of directors is classified, consisting of three classes of directors with staggered three-year terms, with each class consisting as nearly as possible of one third of the total number of directors.
In addition, we are subject to Section 203 of the Delaware General Corporation Law, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder. This provision could have the effect of delaying or preventing a change of control, whether or not it is desired by or beneficial to our stockholders.
Item 1B. Unresolved Staff Comments.
Not applicable.
We currently lease approximately 113,000 square feet of laboratory and office space in Palo Alto, California under lease agreements that terminate in September 2014. We believe that our facilities adequately meet our present needs.
In October 2010, the arbitration panel in our binding arbitration with certain subsidiaries of Johnson & Johnson, or J&J, decided the ownership of a number of U.S. and international patents and patent applications related to certain EPO-R agonists, or the "intellectual property in dispute." The decision maintained J&J's sole inventorship and sole ownership of U.S. Patent No. 5,767,078, or the "078 Patent," and certain related foreign patents and patent applications, including European Patent application EP96/918,317. The arbitrators determined that we and J&J jointly own the remainder of the intellectual property in dispute, i.e., U.S. Patent Nos. 5,773,569, 5,830,851, and 5,986,047 together with their foreign counterpart patents and patent applications.
44
The intellectual property in dispute relates primarily to a three-year Research and Development Agreement, the "R&D Agreement," entered into in 1992 between a division of Ortho Pharmaceutical Corporation, a subsidiary of J&J and Affymax N.V. (a different company from us), for compounds directed at the EPO receptor. The R&D Agreement provided for any invention made by either party to be the property of the party making the invention and that joint inventions would be jointly owned.
In 1995, Affymax N.V., Affymax Technologies, N.V. and Affymax Research Institute were acquired by Glaxo Wellcome plc. Thereafter, in 2001, we acquired specified assets from Glaxo Wellcome plc and related entities, including the rights to the R&D Agreement (which had been finally terminated in 2000) and the rights to specified patents and patent applications that had previously been held by Affymax N.V. and Affymax Technologies, N.V. and comprised much of the intellectual property in dispute. Our claims of ownership of the intellectual property in dispute was based on the inventions of Affymax N.V. scientists.
After our company was founded in 2001, we pursued efforts to create a synthetic compound that activated the EPO receptor and had the biological and physical properties needed to be a commercially viable pharmaceutical product. Our efforts culminated in the first chemical synthesis of peginesatide in 2003.
In November 2010, we filed in the U.S. District Court for the Northern District of Illinois, a motion to vacate the arbitration award with respect to the ownership of the '078 Patent and related foreign cases. In December 2010, J&J filed its response and requested that the court confirm the arbitration award. Briefs have been filed and a decision is expected shortly. We are continuing to consider other potential courses of action with our counsel and Takeda.
We expect that this dispute with J&J could involve additional litigation or legal proceedings that may take years and substantial resources and funds to resolve. Although we believe that peginesatide does not infringe the '078 Patent and that we would have substantial defenses to any potential claims by J&J, J&J may now or in the future attempt to assert claims based upon the '078 Patent against us or our collaborators in connection with the manufacture and commercialization of peginesatide. Outside of the U.S., the European Patent application EP96/918,317 and other foreign counterpart patents to the '078 Patent differ in the nature and extent of the claims from that of the '078 Patent and due to the complexity of patent laws, we are unable to assess adequately the defenses available to us in the various countries outside of the U.S. to any potential claims by J&J alleging infringement of any of the foreign counterparts to the '078 patent. If J&J is successful in asserting its rights under the '078 Patent and related foreign cases, J&J may prevent us from manufacturing or commercializing peginesatide, either for ourselves or with Takeda or any potential sublicensees, or obtain a royalties on sales of peginesatide until expiration of these patent rights in 2015. See "Risk Factors—Risk Related to Our Business."
From time to time, we are involved in legal proceedings arising in the ordinary course of business. We believe there is no other litigation pending that could have, individually or in the aggregate, a material adverse effect on our financial position, results of operations or cash flows.
Item 4. (Removed and Reserved).
45
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market For Our Common Stock
Our common stock has been traded on the NASDAQ Global Market under the symbol "AFFY" since December 15, 2006. As of February 28, 2011, there were approximately 94 holders of record of our common stock. The following table sets forth, for the periods indicated, the range of high and low closing sales prices of our common stock as quoted on the NASDAQ Global Market.
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
2010 |
|||||||
4th Quarter |
$ | 7.56 | $ | 5.00 | |||
3rd Quarter |
$ | 8.46 | $ | 5.39 | |||
2nd Quarter |
$ | 25.37 | $ | 5.98 | |||
1st Quarter |
$ | 25.34 | $ | 18.70 | |||
| |
High | Low | |||||
|---|---|---|---|---|---|---|---|
2009 |
|||||||
4th Quarter |
$ | 25.43 | $ | 19.66 | |||
3rd Quarter |
$ | 24.99 | $ | 17.72 | |||
2nd Quarter |
$ | 19.01 | $ | 15.05 | |||
1st Quarter |
$ | 17.00 | $ | 10.10 | |||
The closing price for our common stock as reported by the NASDAQ Global Market on February 28, 2011 was $6.38 per share.
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently expect to retain any future earnings for use in the operation and expansion of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future.
Use of Proceeds
Our initial public offering of common stock was effected through a Registration Statement on Form S-1, as amended (File No. 333-136125) and a Registration Statement on Form S-1 filed pursuant to Rule 462(b) (File No. 333-139363) that were declared effective by the Securities and Exchange Commission on December 14, 2006. We registered 4,255,000 shares of our common stock for an aggregate offering price of $106,375,000, all of which were sold. After deducting expenses, we received net offering proceeds of approximately $96 million from our initial public offering. As of December 31, 2010, we have used all the proceeds to fund our development of peginesatide and other working capital and general corporate purposes, including the expansion of commercial capabilities.
The foregoing represents our best estimate of our use of proceeds for the period indicated.
Recent Sales of Unregistered Securities
We did not make any unregistered sales of shares of our common stock during the fourth quarter ended December 31, 2010.
Issuer Purchases of Equity Securities
We did not repurchase any of our equity securities during the fourth quarter of the year ended December 31, 2010.
46
Performance Graph(1)
The following graph shows the total stockholder return of an investment of $100 in cash on December 15, 2006, the date our common stock first started trading on the NASDAQ Global Market, through December 31, 2010 for (i) our common stock, (ii) the Nasdaq Composite Index (U.S.) and (iii) the Nasdaq Biotechnology Index as of December 31, 2010. Pursuant to applicable Securities and Exchange Commission rules, all values assume reinvestment of the full amount of all dividends, however no dividends have been declared on our common stock to date. The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
COMPARISON OF 49 MONTH CUMULATIVE TOTAL RETURN*
Among Affymax Inc., the NASDAQ Composite Index
and the NASDAQ Biotechnology Index
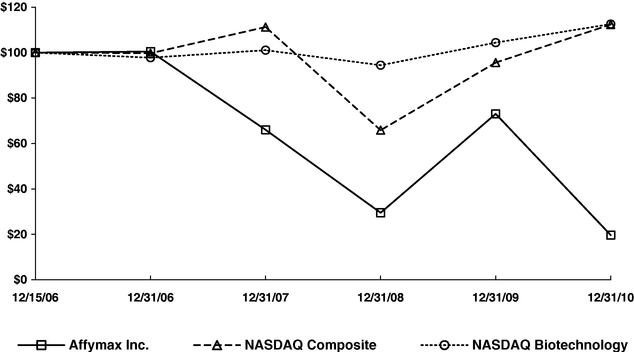
*$100 invested on 12/15/06 in stock or 11/30/06 in index, including reinvestment of dividends.
Fiscal year ending December 31.
- (1)
- This Section is not "soliciting material," is not deemed "filed" with the Commission and is not to be incorporated by reference into any filing of Affymax, Inc. under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
47
Item 6. Selected Financial Data.
The following selected financial data should be read together with our audited financial statements and accompanying notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" section and other financial information included in this Annual Report on Form 10-K. The selected financial data in this section is not intended to replace our audited financial statements and the accompanying notes. Our historical results are not necessarily indicative of our future results.
| |
Years Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||
| |
(in thousands, except per share data) |
||||||||||||||||||
Statements of Operations Data: |
|||||||||||||||||||
Revenue: |
|||||||||||||||||||
Collaboration revenue |
$ | 112,503 | $ | 114,883 | $ | 82,162 | $ | 44,303 | $ | 11,688 | |||||||||
License and royalty revenue |
18 | 16 | 689 | 33 | 38 | ||||||||||||||
Total revenue |
112,521 | 114,899 | 82,851 | 44,336 | 11,726 | ||||||||||||||
Operating expenses: |
|||||||||||||||||||
Research and development |
93,638 | 157,125 | 137,492 | 69,398 | 54,347 | ||||||||||||||
General and administrative |
33,331 | 36,716 | 34,090 | 24,075 | 11,089 | ||||||||||||||
Total operating expenses |
126,969 | 193,841 | 171,582 | 93,473 | 65,436 | ||||||||||||||
Loss from operations |
(14,448 | ) | (78,942 | ) | (88,731 | ) | (49,137 | ) | (53,710 | ) | |||||||||
Interest income |
275 | 934 | 4,545 | 11,393 | 5,549 | ||||||||||||||
Interest expense |
(140 | ) | (105 | ) | (609 | ) | (14 | ) | (84 | ) | |||||||||
Other income (expense), net |
239 | 171 | (1,433 | ) | 46 | (43 | ) | ||||||||||||
Net loss before provision (benefit) for income taxes |
(14,074 | ) | (77,942 | ) | (86,228 | ) | (37,712 | ) | (48,288 | ) | |||||||||
Provision (benefit) for income taxes |
1 | (1,411 | ) | 282 | 5,357 | — | |||||||||||||
Net loss |
(14,075 | ) | (76,531 | ) | (86,510 | ) | (43,069 | ) | (48,288 | ) | |||||||||
Accretion of mandatorily redeemable convertible preferred stock |
— | — | — | — | (815 | ) | |||||||||||||
Net loss attributable to common stockholders |
$ | (14,075 | ) | $ | (76,531 | ) | $ | (86,510 | ) | $ | (43,069 | ) | $ | (49,103 | ) | ||||
Net loss per common share: |
|||||||||||||||||||
Basic and diluted(1) |
$ | (0.57 | ) | $ | (4.06 | ) | $ | (5.68 | ) | $ | (2.88 | ) | $ | (32.56 | ) | ||||
Weighted-average number of common shares used in computing basic and diluted net loss per loss common share |
24,488 | 18,865 | 15,220 | 14,941 | 1,508 | ||||||||||||||
48
| |
December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||
| |
(in thousands) |
|||||||||||||||
Balance Sheet Data: |
||||||||||||||||
Cash, cash equivalents and short-term investments |
$ | 97,081 | $ | 160,588 | $ | 94,719 | $ | 168,337 | $ | 224,292 | ||||||
Receivable from Takeda |
— | 18,561 | 21,688 | 15,331 | 10,191 | |||||||||||
Long-term investments |
19,876 | 7,978 | 22,945 | 15,655 | 6,133 | |||||||||||
Total assets |
131,387 | 211,510 | 167,720 | 225,792 | 249,988 | |||||||||||
Payable to Takeda |
5,958 | — | — | — | — | |||||||||||
Capitalized lease obligations, net of current portion |
— | — | — | 8 | 140 | |||||||||||
Accumulated deficit |
(388,934 | ) | (374,859 | ) | (298,328 | ) | (211,818 | ) | (168,749 | ) | ||||||
Total stockholders' equity |
72,547 | 66,905 | 8,984 | 84,185 | 116,899 | |||||||||||
- (1)
- Please see Note 2 to the notes to our audited financial statements for an explanation of the method used to calculate the net loss per common share and the number of shares used in the computation of the per share amounts.
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations.
Overview
We are a biopharmaceutical company committed to developing novel drugs to improve the treatment of serious and often life-threatening conditions. Our product candidate, peginesatide (HematideTM) recently completed Phase 3 clinical trials to treat anemia associated with chronic renal failure. Anemia is a serious condition in which blood is deficient in red blood cells and hemoglobin. It is common in patients with chronic renal failure, cancer, heart failure, inflammatory diseases and other critical illnesses, as well as in the elderly. If left untreated, anemia may lead to chronic fatigue or increase the risk of other diseases or death. Currently recombinant EPO, or rEPO, is used to manage the anemia of dialysis, non-dialysis and cancer patients. Peginesatide is a synthetic peptide-based erythropoiesis stimulating agent, or ESA, designed to stimulate production of red blood cells. Peginesatide is designed to be longer acting than currently marketed ESAs in the U.S. and therefore has the potential to offer reduced cost and complexity for healthcare providers.
In late June 2010, we announced preliminary top-line results from our peginesatide Phase 3 clinical program for the treatment of patients with anemia associated with chronic renal failure. Our Phase 3 clinical program included four open-label, randomized controlled clinical trials. Of these trials, two trials, called PEARL 1 and PEARL 2, were conducted in non-dialysis patients and designed to evaluate the safety and efficacy of peginesatide compared to darbepoetin alfa to correct anemia and maintain hemoglobin in a corrected range over time. The other two trials, called EMERALD 1 and EMERALD 2, were conducted in dialysis patients and designed to evaluate the safety and efficacy of peginesatide and its ability to maintain hemoglobin levels in a corrected range compared to epoetin alpha or epoetin beta when switched to peginesatide. Analysis of efficacy and safety for all of the Phase 3 studies were based on primarily assessments of non-inferiority to the comparator drugs. The primary efficacy endpoint, the mean change in hemoglobin from baseline, in each of the four Phase 3 studies met the statistical criteria for non-inferiority. In addition, peginesatide met the statistical criterion for non-inferiority for the assessment of safety for the cardiovascular composite safety endpoint, or CSE, which was composed of death, stroke, myocardial infarction, congestive heart failure, unstable angina and arrhythmia from a pooled safety database across the four Phase 3 studies. However, some differences were observed when secondary analyses were conducted as previously described in our Current Report on Form 8-K dated June 21, 2010.
49
Based on our discussions with the U.S. Food and Drug Administration, or FDA, we plan to submit a New Drug Application, or NDA, to the FDA, for treatment of anemia in chronic renal failure patients on dialysis in the second quarter of 2011. Despite meeting the primary efficacy endpoints and the CSE for peginesatide, the differences observed in the Phase 3 secondary analyses present risks to our ability to obtain regulatory approval for peginesatide particularly in view of the heightened concerns surrounding safety of ESAs. Any negative perception of peginesatide's safety relative to other ESAs would significantly reduce the likelihood of obtaining regulatory approval for peginesatide. The issues arising from the Phase 3 results have caused significant delay and may continue to negatively impact the timelines for development and the likelihood, scope or conditions surrounding regulatory approval. Any or all of these factors may significantly reduce the ultimate commercial potential of peginesatide.
In September 2009, we obtained an equity line of credit arrangement, with Azimuth Opportunity Ltd., or Azimuth, that provides that, upon the terms and subject to the conditions set forth in the purchase agreement, Azimuth is committed to purchase up to $60.0 million worth of shares of our common stock over the 24-month term of the purchase agreement (the Common Stock Purchase Agreement). In September 2010, we entered into an amendment, or the Amendment, to the Common Stock Purchase Agreement with Azimuth, which extends the term of the equity facility to September 2012 and reduces the minimum threshold price we may establish at which, upon presentation to Azimuth of a draw down notice, Azimuth is required to purchase shares of our common stock. The Amendment further provides that in no event may we sell under the Purchase Agreement more than such number of shares of common stock which is equal to one share less than 20% of our outstanding shares of common stock on the effective date of the Amendment.
In October 2010, we closed on the sale of 999,061 shares of common stock to Azimuth under the Common Stock Purchase Agreement for an aggregate purchase price of $5.0 million. Our net proceeds from the sale of these shares were $4.9 million after deducting our offering expenses.
Our equity facility is subject to a number of conditions that limit our ability to draw against such facility. For example, Azimuth is not required to purchase our common stock when the price of our common stock is below $4.00 per share. In addition, Azimuth is not obligated to purchase shares of our common stock which, when aggregated with all other shares of our common stock then owned beneficially by Azimuth, would result in the beneficial ownership by Azimuth of more than 9.9% of the then issued and outstanding shares of our common stock. At December 31, 2010 this represents 2,519,682 shares. After deducting the shares purchased in October 2010, assuming that all remaining 1,520,621 shares were sold at the $6.65 closing price of our common stock at December 31, 2010 at the largest possible discount and assuming that Azimuth still owns these shares, the maximum aggregate net proceeds we could receive under the agreement with Azimuth would be approximately $9.4 million.
In October 2010, the arbitration panel in our binding arbitration with Johnson & Johnson Pharmaceutical Research & Development, L.L.C., and Ortho-McNeil Pharmaceutical, Inc., or J&J, decided the ownership of a number of U.S. and international patents and patent applications related to certain EPO-R agonists, or the "intellectual property in dispute." The decision maintained J&J's sole inventorship and sole ownership of U.S. Patent No. 5,767,078, or the "078 Patent," and certain related foreign patents and patent applications, including European Patent application EP96/918,317. The arbitrators determined that we and J&J jointly own the remainder of the intellectual property in dispute.
We are continuing to review the arbitrators' decision and consider potential courses of action with our counsel and Takeda Pharmaceutical Company Limited, or Takeda. We expect that this dispute with J&J could involve additional litigation or legal proceedings that may take years and substantial resources and funds to resolve. Although we believe that peginesatide does not infringe the '078 Patent and that we would have substantial defenses to any potential claims by J&J, J&J may now or in the
50
future attempt to assert claims based upon the '078 Patent against us or our collaborators in connection with the manufacture and commercialization of peginesatide. Outside of the U.S., the European Patent application EP96/918,317 and other foreign counterpart patents to the '078 Patent differ in the nature and extent of the claims from that of the '078 Patent and due to the complexity of patent laws, we are unable to assess adequately the defenses available to us in the various countries outside of the U.S. to any potential claims by J&J alleging infringement of any of the foreign counterparts to the '078 patent. If J&J is successful in asserting its rights under the '078 Patent and related foreign cases, J&J may prevent us from manufacturing or commercializing peginesatide, either for ourselves or with Takeda or any potential sublicensees, or obtain a royalties on sales of peginesatide until expiration of these patent rights in 2015. In addition, an adverse outcome could result in liability for damages, attorneys' fees and costs. If intellectual property in dispute that has been deemed to be jointly owned is broad enough to cover peginesatide, then under the laws applicable in certain relevant jurisdictions outside the U.S, joint ownership may not allow us to license third parties manufacture and sell peginesatide or even to do so ourselves, which may negatively affect our development and business plans outside the U.S. or our collaboration with Takeda.
Regardless of the ultimate outcome of the proceedings, additional litigation or legal proceedings or even the risk thereof may make it more difficult to commercialize peginesatide. The threat of such legal uncertainty may make it difficult for peginesatide to gain market acceptance by health care providers, patients, payors or dialysis clinics, any of which may be concerned about the reliability of supply or reluctant to become involved in the prospect of existing or potential litigation.
We continue to consider our legal alternatives in this dispute. To date, we have incurred significant expense in pursuing this matter, including substantial time and effort on the part of our technical, legal and management personnel. As final resolution of this dispute may not be reached for years, we expect to incur significant expenses and diversion of resources for years. Further, the risk or existence of litigation or legal proceedings with J&J may limit our ability to finance, and even if such financing is available, to achieve terms that are favorable to us.
To date, we have not generated any product revenue. We have funded our operations primarily through the sale of equity securities, reimbursement for development expenses and active pharmaceutical ingredient, or API, production, license fees and milestone payments from collaborative partners, operating and capital lease financings, interest earned on investments and limited license fees and royalties from licensing intellectual property. As of December 31, 2010, we had an accumulated deficit of $388.9 million. Other than the three and six month periods ended June 30, 2010, we have incurred net losses since our inception. However, due to the recognition of revenues from milestone payments from our collaboration with Takeda, we were profitable in the three and six months ended June 30, 2010 and may have profitable quarters from time to time if we are successful in obtaining FDA approval for peginesatide. We continue to expect to incur substantial losses for the next several years in order to complete the development and commercialization of peginesatide.
We believe that our existing cash, cash equivalents and investments together with the interest thereon will enable us to maintain our currently planned operations for at least the next 12 months. However, we expect that we will need to raise additional funding to complete the development and commercialization of peginesatide. Since the announcement of our Phase 3 data in late June 2010 and the arbitration decision in October 2010, we have experienced a severe decline in our stock price, which has impaired our ability to access capital on potentially favorable terms. In contrast, our need to raise funding has only increased due to the peginesatide development program delays, the potential loss of milestone payments from Takeda associated with the non-dialysis indication and the potential for future legal proceedings and costs. As we continue to develop and ultimately commercialize peginesatide, if approved, we may experience further challenges or delays if issues arise or additional requirements are imposed based on our discussions with the FDA and other regulatory agencies or as a consequence of our dispute with J&J.
51
Our current view of the worldwide capital markets is that they are extremely volatile with limited accessibility, and many biotechnology companies have been limited or unsuccessful in obtaining funding in this environment. We intend to evaluate the capital markets from time to time to determine whether to raise additional capital, in the form of equity or otherwise, depending upon market conditions relative to our need for funds at such time. Continuation of this market and the issues arising from our Phase 3 results significantly limit our ability to raise funds such that there can be no assurance we can raise the additional funds to support our continuing operations and maintain current development timelines, and funding may not be available to us on acceptable terms, or at all. Further, we have had to reduce our research capabilities and efforts, including the elimination of certain research programs, and even some activities related to the support of peginesatide. If we are unable to raise additional funds when needed, we could be required to further delay, scale back or eliminate some or all of our development programs and other operations, which could negatively impact our ability to complete development or commercialize peginesatide. We may seek to raise additional funds through public or private financing, strategic partnerships or other arrangements. Any additional equity financing, particularly at our recent stock trading levels, would be difficult to obtain, if accessible at all, and our current stockholders may be significantly diluted. Any debt financing, if available, may involve restrictive covenants or security interests in our assets. Further, any strategic or licensing arrangements, if available, may require us to relinquish product rights that we would otherwise seek to develop or commercialize ourselves. Our failure to raise capital when needed may harm our business and operating results.
Research and Development Expenses
Research and development, or R&D, expenses consist of: (i) expenses incurred under agreements with contract research organizations and investigative sites, which conduct a substantial portion of our pre-clinical studies and all of our clinical trials; (ii) payments to contract manufacturing organizations, which produce our API; (iii) payments to consultants; (iv) license fees paid to third parties for use of their intellectual property; (v) employee-related expenses, which include salaries and related costs; and (vi) facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities and equipment, depreciation of leasehold improvements and equipment and laboratory and other supplies. All R&D expenses are expensed as incurred.
For our R&D expenses, we have historically commenced tracking the costs for a project when we are working with another company or when the product candidate merits substantial increase in the level of effort. In recent years, our R&D efforts have been almost exclusively focused on the development of peginesatide, specifically on the Phase 3 trials commenced in 2008. For the years ended December 31, 2010, 2009 and 2008, the percentage of our R&D expenses related to peginesatide, excluding stock-based compensation expense, were 100%, 100% and 99%, respectively.
Under the worldwide agreement with Takeda, we and Takeda will co-develop and co-commercialize peginesatide in the U.S. Beginning January 1, 2007, Takeda was responsible for the first $50 million of third party expenses related to development in pursuit of U.S. regulatory approval of peginesatide, which was fully utilized by both parties through the first quarter of 2008. Thereafter, Takeda has borne 70% of the third party U.S. development expenses while we have been responsible for 30% of third-party expenses. We retain responsibility for 100% of our internal development expenses, most notably employee-related expenses. In addition, third party expenses related to the commercialization of peginesatide in the U.S. are equally shared by both parties and beginning in mid-2011, certain employee-related expenses supporting commercialization will also be equally shared. Takeda will have primary responsibility and bear all costs for peginesatide clinical development in support of regulatory approval for all territories outside the U.S.
The process of conducting pre-clinical studies and clinical trials necessary to obtain FDA approval is costly and time consuming. The probability of success for each product candidate and clinical trial
52
may be affected by a variety of factors, including, among others, the quality of the product candidate's early clinical data, investment in the program, competition, manufacturing capabilities and commercial viability. As a result of the uncertainties discussed above, the uncertainty associated with clinical trial enrollments and the risks inherent in the development process, we are unable to determine the duration and completion costs of current or future clinical stages of our product candidates or when, or to what extent, we will generate revenues from the commercialization and sale of any of our product candidates. Development timelines, probability of success and development costs vary widely. While we are currently focused on developing peginesatide, in the future we may develop additional product candidates internally and in-license product candidates, which would increase our R&D expenses in later periods.
During the year ended December 31, 2010, we finalized amendments for certain clinical trial activities completed in 2009. In the fourth quarter of 2010, we obtained final monitored site visit data and investigator contracts from our third party contract research organizations, or CROs, that allowed us to complete our reconciliation of the significant majority of the labor and investigator costs incurred throughout the course of our clinical trials to our previously recorded estimates. This data and contractual information was not available to us during the course of the trials. After extensive analysis to cost out and analyze the information provided, we determined that the costs incurred were lower than our previously recorded estimates. The change in estimate is due largely to (i) 8% lower total patient months on study as compared to our previously estimated level as a result of a larger number of patients coming off study and no longer having trial-related site visits (also known as lost to follow up) during the course of the study, and (ii) 13% lower than expected average fee generating site visits per month, driven largely by missed visits from patients on study. We believe these changes were only identifiable based on the information received in the fourth quarter of 2010. The change in estimate decreased expense by $12.1 million for the year ended December 31, 2010.
Additional changes in estimate or adjustments may result as the final trial close-out audits and fee negotiations are completed relating to our clinical trials which could impact R&D expense and collaboration revenue and amounts due to or from Takeda in subsequent periods.
General and Administrative Expenses
General and administrative, or G&A, expenses consist principally of salaries and related costs for personnel in executive, finance, accounting, business and commercial development, information technology, legal and human resources functions. Other general and administrative expenses include facility costs not otherwise included in R&D expense, patent filing, prosecution and defense costs and professional fees for legal, consulting, auditing and tax services.
Critical Accounting Policies and Significant Judgments and Estimates
Our management's discussion and analysis of our financial condition and results of operations is based on our financial statements and the related disclosures, which have been prepared in accordance with U.S. generally accepted accounting principles or GAAP. The preparation of these financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments related to revenue recognition and clinical development costs. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. We believe the following policies to be the most critical to an understanding of our financial condition and results of
53
operations because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
Revenue Recognition
Collaboration Revenue
We recognize revenue in accordance with the authoritative guidance, revenue recognition in financial statements. When evaluating multiple element arrangements, we consider whether the components of the arrangement represent separate units of accounting as defined in the authoritative guidance for revenue arrangements with multiple deliverables. Application of this guidance requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
In February and June 2006, we entered into two separate collaboration agreements or the Arrangement with Takeda, which have been combined for accounting purposes due to their proximity of negotiation. We evaluated the multiple elements under the combined single arrangement in accordance with the provisions of the guidance for revenue arrangements with multiple deliverables. We determined the deliverables do not have value to the customer on a stand alone basis and we were unable to obtain verifiable objective evidence to determine the fair value of the undelivered elements. Accordingly, we concluded that there was a single unit of accounting.
Effective January 1, 2008, we entered into an amendment to the Arrangement with Takeda. The amendment provides us the ability to opt-out of our obligation to participate on the joint steering committee and any related subcommittees at any time beginning January 1, 2011 without any other modifications. As a result, the obligation to participate in the joint steering committee and any related subcommittee is no longer indefinite. Accordingly, we determined that we can separate the performance obligations that occur over the development period from the performance obligations that will occur during the commercialization period. We had previously estimated the development period to end on January 1, 2011. Based on the announcement on June 21, 2010 of our top-line results from our Phase 3 clinical program, during the second quarter of 2010 we re-evaluated our development period and determined that the development period was estimated to end upon submission of our NDA to the FDA, estimated to occur in the first half of 2011. After further refinements to our regulatory strategy as a result of our subsequent meeting with the FDA in November 2010, we re-evaluated the estimated filing date for our NDA submission and believe it to be in the second quarter of 2011. As a result of the change in performance period from indefinite to a definitive date, beginning on January 1, 2008, we recognize revenue during the development period using the Contingency-Adjusted Performance Model, or CAPM. Under CAPM, revenue is eligible for recognition in the period the payment is earned under the Arrangement including amounts that are either received or due from Takeda. Revenue initially recognized is based on the percentage of time elapsed from inception of the Arrangement in June 2006 to the period in which the payment is earned in relation to the total projected development period.
The remaining portion of the payment is then recognized on a straight-line basis over the remaining estimated duration of the development period of the Arrangement. Payments during the development period include amounts due for upfront license fees, milestone payments earned, purchases of active pharmaceutical ingredient or API and reimbursement of development and commercial expenses. Further changes in the estimated term of the development period could materially affect the amount of collaboration revenue recognized in future periods. A change in the estimated term of the development period could materially affect the amount of collaboration revenue recognized in future periods. We expect collaboration revenue to be directly affected by milestone payments and expenses that are eligible for reimbursement from Takeda under the Arrangement in future periods. Included in the reimbursable expense is the cost of API that we manufacture and supply
54
to Takeda during the development period, which we will also supply during the commercialization period.
License and Royalty Revenue
Royalties are recognized as earned in accordance with contract terms, when third party results are reported and collectability is reasonably assured. Royalties received under agreements that were acquired by us in the 2001 spin out from GlaxoSmithKline or Glaxo are recorded net of the 50% that we are required to remit to Glaxo.
Clinical Trial Expense and Accruals
We record expense for external costs incurred on our clinical studies based on our estimates of the costs incurred each period. These clinical trial costs, which represent a significant component of R&D expenses, were $14.9 million, $90.0 million and $77.8 million for the years ended December 31, 2010, 2009, and 2008, respectively. Our clinical trials are administered by CROs, who typically perform most of the total start-up activities for the trials, including document preparation, site identification pre-study visits, training as well as on-going program management. For the Phase 3 studies, which represent the vast majority of the clinical trial expense, the expense recorded is based on reporting received from CROs and internal analyses. We accrue costs for work performed by CROs based on the achievement of contracted activities during the period. Expense for investigator fees, which include patient costs, is based on internal estimates of activities using patient enrollment and contractual or estimated rates. For the Phase 2 studies, the expense is activities-based such as patient monitoring as reported by the CROs and achievement of milestones. Other costs such as testing and drug materials are expensed as incurred. For all studies, CRO reporting is reviewed by us for appropriateness.
There is a significant degree of estimation involved in quantifying the clinical study expenses due to the complexity and magnitude of the clinical trial activities These estimates have been subject to frequent adjustments, especially for our Phase 3 trials, in part due to our negotiations with third-parties with respect to timing of reporting, patient progression and payments as well as our continuing negotiations with CROs, on timely delivery and access to information necessary to validate our accruals. Additional changes in estimate or adjustments to previously presented amounts in R&D expense may result as the reconciliation activities and final negotiations with our CROs are completed on our clinical trials.
During the quarter ended June 30, 2008, we identified an overstatement of clinical trial expense and collaboration revenue of $1.3 million in the year ended December 31, 2007. As a result, clinical trial expense and collaboration revenue, which included reimbursement for these costs, included an out of period reduction of $1.3 million and $0.4 million, respectively, in the year ended December 31, 2008.
During the year ended December 31, 2010, we finalized amendments for certain clinical trial activities completed in 2009 which decreased expense by $12.1 million for the year ended December 31, 2010 due to a change in estimate, as more fully described above in the Research and Development Expenses section.
Additional changes in estimate or adjustments may result as the final trial close-out audits and fee negotiations are completed relating to our clinical trials which could impact R&D expense in subsequent periods.
Stock-Based Compensation
We currently use the Black-Scholes model to estimate the fair value of employee stock options and our employee stock purchase plan. Calculating the fair value of stock-based payment awards requires considerable judgment, including estimating stock price volatility, the amount of stock-based awards
55
that are expected to be forfeited and the expected life of the stock-based payment awards. While fair value may be readily determinable for awards of stock or restricted stock units, or RSUs, market quotes are not available for long-term, non-transferable stock options because these instruments are not traded. The value of a stock option is derived from its potential for appreciation. The more volatile the stock, the more valuable the option becomes because of the greater possibility of significant changes in stock price. We base our estimated expected option term and volatility on the realized volatilities of our peer companies. The expected option term also has a significant effect on the value of the option. The longer the term, the more time the option holder has to allow the stock price to increase without a cash investment and thus, the more valuable the option. We review our valuation assumptions at each grant date, and, as a result, we are likely to change our valuation assumptions used to value stock-based awards granted in future periods. There is a high degree of subjectivity involved when using option pricing models to estimate share-based compensation under the authoritative guidance for share-based payments. There is a risk that our estimates of the fair values of our share-based compensation awards on the grant dates may bear little resemblance to the actual values realized upon the exercise, early termination or forfeiture of those share-based payments in the future. Certain share-based payments, such as employee stock options, may expire worthless or otherwise result in zero intrinsic value as compared to the fair values originally estimated on the grant date and reported in our financial statements.
The authoritative guidance for share-based payments requires that employee stock-based compensation costs be recognized over the requisite service period, or the vesting period, in a manner similar to all other forms of compensation paid to employees. The allocation of employee stock-based compensation costs to each operating expense line are estimated based on specific employee headcount information at each grant date and estimated stock option forfeiture rates and revised, if necessary, in future periods if actual employee headcount information or forfeitures differ materially from those estimates. In determining whether an award is expected to vest, we use an estimated forward-looking forfeiture rate. We consider many factors when estimating expected forfeitures, including types of awards and historical experience. These estimates are revised in subsequent periods based upon changes in facts or circumstances that would affect our forfeiture rate. If our actual forfeiture rate is materially different from our estimate, the stock-based compensation expense could be significantly different than what was recorded in the current period. For awards with a longer vesting period, the actual forfeiture rate and related expense may not be known for a longer period of time, which can result in more significant accounting adjustments once the awards are either vested or forfeited. As a result, the amount of employee stock-based compensation costs we recognize in each operating expense category in future periods may differ significantly from what we have recorded in the current period.
Income Taxes
We account for income taxes under the liability method, whereby deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
Effective January 1, 2007, we adopted the authoritative guidance on accounting for uncertainty in income taxes, that prescribes a comprehensive model for the recognition, measurement, presentation and disclosure in financial statements of any uncertain tax positions that have been taken or expected to be taken on a tax return. The cumulative effect of adopting this guidance resulted in no adjustment to our accumulated deficit as of January 1, 2007. We had $13.1 million, $12.4 million and $11.8 million of unrecognized tax benefits as of December 31, 2010, 2009, and 2008, respectively.
56
At December 31, 2010 and 2009, our liability for uncertain income tax positions was $10.2 million and $10.1 million, respectively, and is reflected as long-term income tax liabilities on our balance sheet. Our policy is to include penalties and interest expense related to income taxes as a component of other expense and interest expense, respectively, as necessary. For the years ended December 31, 2010, 2009 and 2008, we recognized $140,000, $105,000 and $596,000, respectively, of interest expense related to our liability for uncertain income tax positions. For the years ended December 31, 2010 and 2009, there were no penalties related to uncertain income tax positions. For the year ended December 31, 2008, $81,000 of penalties related to uncertain income tax positions were required and recognized. At December 31, 2010, $842,000 was accrued for interest and penalties related to uncertain income tax positions. We do not anticipate that any of the unrecognized tax benefits will increase or decrease significantly over the next twelve months.
Results of Operations
Revenue
Revenue and percentage changes as compared to prior years are as follows (in thousands):
| |
Year ended December 31, | Percent Change | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2010/2009 | 2009/2008 | ||||||||||||
Collaboration revenue |
$ | 112,503 | $ | 114,883 | $ | 82,162 | (2 | )% | 40 | % | |||||||
License and royalty revenue |
18 | 16 | 689 | 13 | % | (98 | )% | ||||||||||
Total revenue |
$ | 112,521 | $ | 114,899 | $ | 82,851 | (2 | )% | 39 | % | |||||||
We recognized $112.5 million, $114.9 million and $82.2 million of collaboration revenue for the years ended December 31, 2010, 2009 and 2008, respectively. The collaboration revenue for the year ended December 31, 2008 included a $1.4 million cumulative adjustment resulting from an amendment to the collaboration agreements with Takeda, as discussed in the notes to our audited financial statements and an out of period reduction of $0.4 million. Collaboration revenue includes our expenses that are eligible for reimbursement from Takeda, net of Takeda's own eligible expenses. Collaboration revenue in 2010 was down slightly from 2009 due largely to decreased third-party development expenses reimbursed by Takeda related to our Phase 3 clinical trials which were completed in early 2010 and the associated impact of our change in estimate adjustment recorded in the fourth quarter of 2010 related to our clinical trial expense. The associated impact to revenue was a decrease of $7.8 million. The increase in collaboration revenue in 2009 and 2008 was due to the growth in third party development expenses reimbursed by Takeda related to our Phase 3 clinical trials, which commenced in late 2007 and achieved full enrollment in late 2008 as well as the continued amortization of revenue from expense reimbursements and milestones received from Takeda in prior periods. We expect collaboration revenue to be directly affected by milestone payments and expenses that are eligible for reimbursement from Takeda under the Arrangement in future periods. A change in the estimated term of the development period could materially affect the amount of collaboration revenue recognized in future periods. Based on the announcement on June 21, 2010 of our top-line results from our Phase 3 clinical program, during the second quarter of 2010 we re-evaluated our development period and determined that the development period was estimated to end upon the submission of our NDA to the FDA, estimated to occur in the first half of 2011. After further refinements to our regulatory strategy as a result of our subsequent meeting with the FDA in November 2010, we re-evaluated the estimated filing date for our NDA submission and now believe it to be in the second quarter of 2011. Each of these changes resulted in an extension of the development period as compared to our previous estimates and will result in the remaining deferred collaboration revenue being recognized over this longer period.
We recognized $18,000, $16,000 and $689,000 of license and royalty revenue for the years ended December 31, 2010, 2009, and 2008, respectively. The license and royalty revenue in 2008 was due to
57
payments received under a license agreement that we acquired in the 2001 spin out from Glaxo, net of the 50% that we are required to remit to Glaxo.
Research and Development Expenses
R&D expenses and percentage changes as compared to prior years are as follows (in thousands):
| |
Year ended December 31, | Percent Change | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2010/2009 | 2009/2008 | |||||||||||
Research and development expenses |
$ | 93,638 | $ | 157,125 | $ | 137,492 | (40 | )% | 14 | % | ||||||
The decrease in R&D expenses for the year ended December 31, 2010 was primarily due to the completion of the treatment and follow up of our Phase 3 clinical trials at the start of 2010. The increase in R&D expenses in 2009 was primarily due to an increase of $18.4 million, in clinical trial costs resulting from our Phase 3 clinical trials, which commenced in late 2007, achieved full enrollment in late 2008 and continued through the end of 2009.
During the year ended December 31, 2010, we finalized amendments for clinical trial activities completed in 2009. In addition, with the completion of our Phase 3 clinical trials for peginesatide in 2010, we were able to obtain final costed monitored site visit data that allowed us to complete our reconciliation of the significant majority of the labor and investigator costs incurred throughout the course of those trials to our previously recorded estimates. As a result, we recorded adjustments to estimates in the year ended December 31, 2010 relating to estimates previously recorded in our expenses for the years ended December 31, 2008 and 2009. These changes in estimate decreased expense by $12.1 million for the year ended December 31, 2010. Additional changes in estimate or adjustments may result as the reconciliation activities and final trial close-out negotiations are completed relating to our clinical trials which could impact R&D expense in subsequent periods.
We expect R&D expenses to substantially decrease in 2011 due to the completion of the treatment and follow up of our Phase 3 clinical trials in 2010 which decrease could be partially offset by any additional research and clinical trials conducted to obtain additional data for peginesatide.
General and Administrative Expenses
G&A expenses and percentage changes as compared to prior years are as follows (in thousands):
| |
Year ended December 31, | Percent Change | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2010/2009 | 2009/2008 | |||||||||||
General and administrative expenses |
$ | 33,331 | $ | 36,716 | $ | 34,090 | (9 | )% | 8 | % | ||||||
The decrease in G&A expenses in 2010 was primarily due to lower legal costs. The increase in G&A expenses in 2009 as compared to 2008 was primarily due to higher legal costs. Legal costs primarily relate to protecting and defending our proprietary rights, such as patents, and fluctuate with the level of such activity. We expect to incur increasing general and administrative expenses in future periods to support our preparation for our NDA for peginesatide and development of commercial capabilities.
58
Interest Income (Expense), Net
Interest income (expense), net and percentage changes as compared to prior years are as follows (in thousands):
| |
Year ended December 31, | Percent Change | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2010/2009 | 2009/2008 | |||||||||||
Interest income (expense), net |
$ | 135 | $ | 829 | $ | 3,936 | (84 | )% | (79 | )% | ||||||
The decrease in interest income (expense), net in 2010 as compared to 2009, as well as in 2009 when compared to 2008 was due primarily to lower levels of cash, cash equivalents and investments earning interest, as well as due to generally lower interest rates during the year.
Other Income (Expense), Net.
Other income (expense), net and percentage changes as compared to prior years are as follows (in thousands):
| |
Year ended December 31, | Percent Change | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2010/2009 | 2009/2008 | |||||||||||
Other income (expense), net |
$ | 239 | $ | 171 | $ | (1,433 | ) | 40 | % | 112 | % | |||||
Other income (expense), net, for the year ended December 31, 2010 consists primarily of $244,000 from a qualified therapeutic discovery grant received from the U.S. government. Other income (expense), net, for the year ended December 31, 2009 includes adjustments to the fair value of our UBS AG of Series C-2 ARS Rights, or ARS Rights, at sale of the related Auction Rate Securities, or ARS. Other income (expense) net, for the year ended December 31, 2008 includes the initial other-than-temporary impairment charge related to the decrease in fair value of our investments in ARS. The impairment charge was partially offset by a gain of $57,000 and $2.4 million in 2009 and 2008, respectively, related to the fair value adjustments associated with our ARS Rights received from UBS Financial Services, an affiliate of UBS AG, or UBS.
Provision (Benefit) for Income Taxes
Provision (benefit) for income taxes and percentage changes as compared to prior years are as follows (in thousands):
| |
Year ended December 31, | Percent Change | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | 2010/2009 | 2009/2008 | |||||||||||
Provision (benefit) for income taxes |
$ | 1 | $ | (1,411 | ) | $ | 282 | * | * | |||||||
- *
- Calculation not meaningful
We are subject to federal and California state income tax. For the year ended December 31, 2010, we recorded a provision for minimum statutory state tax and provided no federal tax as a result of our net operating loss.
For the year ended December 31, 2009, we recorded a benefit for income taxes of $1.4 million. The tax benefit was for federal tax purposes, primarily the result of the Worker, Homeownership and Business Assistance Act of 2009 enacted in November 2009, which allowed us to carryback our 2008 net operating loss to 2007 and recover $1.3 million in alternative minimum taxes previously paid for the year ended December 31, 2007. We also recorded a $100,000 federal benefit related to refundable R&D credits available to us pursuant to a provision within the Housing Assistance Tax Act of 2008, which was signed into law in July 2008.
59
For the year ended December 31, 2008, we recorded a provision for income taxes of $282,000 consisting of $107,000 of federal tax benefit and $389,000 of net California state income tax expense. The $107,000 of federal tax benefit was primarily due to refundable R&D credits available pursuant to a provision within the Housing Assistance Tax Act of 2008, which was signed into law in July 2008. The California state income tax expense of $389,000 was primarily related to an out of period reduction to our California R&D credits that was partially offset by additional California R&D credits that were identified.
As of December 31, 2010 and 2009, we have a net deferred tax asset balance of $7.2 million each, in consideration of the uncertainty in income taxes liability recorded for the same amount. We considered the following positive and negative factors in determining that it was more likely than not that the net deferred tax asset as of December 31, 2010, and 2009 would be realized:
- •
- Net deductible temporary differences that were expected to reverse in 2010 and 2011.
- •
- There were no relevant tax strategies available that we would consider feasible.
- •
- Uncertainties, such as regulatory approval of peginesatide and potential litigation with certain subsidiaries of J&J, that if unfavorably resolved, would adversely affect our future operations.
We have incurred significant operating losses since inception and anticipate that we will incur continued losses for the foreseeable future.
Liquidity and Capital Resources
Our cash, cash equivalents, and investments at December 31, 2010 and 2009 were as follows (in thousands):
| |
Year Ended December 31, |
||||||
|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | |||||
Cash and cash equivalents |
$ | 63,499 | $ | 125,296 | |||
Short-term investments |
$ | 33,582 | $ | 35,292 | |||
Long-term investments |
$ | 19,876 | $ | 7,978 | |||
Since our inception, we have financed our operations through sale of capital stock, license fees, milestone payments and reimbursement for development and commercial expenses and manufacturing costs from collaborative partners, operating and capital lease financing, interest earned on investments and limited license fees and royalties from licensing intellectual property. From inception through December 31, 2010, we have received net proceeds of $390.6 million from the issuance of equity securities, $122 million of upfront license fees, $45 million in milestone payments and $223.7 million for the reimbursement of development and commercial expenses and purchase of API from our collaboration agreements with Takeda. Takeda was responsible for the first $50 million of third party expenses related to the development in pursuit of U.S. regulatory approval of peginesatide, which was fully utilized by both parties through the first quarter of 2008. Thereafter, Takeda has borne 70% of the third party U.S. development expenses, while we have been responsible for 30% of third party expenses. We retain responsibility for 100% of our internal development expenses, most notably employee-related expenses. In addition, third party expenses related to the commercialization of peginesatide in the U.S. are equally shared by both parties and beginning in mid-2011, certain employee-related expenses supporting commercialization will also be equally shared.
Net cash used in operating activities for the years ended December 31, 2010, 2009, and 2008 was $49.2 million, $80.8 million and $63.9 million, respectively. The $31.6 million decrease in cash used in operating activities in 2010 as compared to 2009 was primarily the result of $35 million in milestone payments received from Takeda as well as lower R&D expenses. R&D expenses were lower as a result
60
of completion of our Phase 3 clinical trials in early 2010 and our change in estimate described earlier. The $16.9 million increase in cash used in operating activities in 2009 as compared to 2008 was primarily due to significant changes in working capital, most notably a $32.3 million reduction in the change, year over year, in deferred revenue, offset by a $13.6 million reduction in the change, year over year, in accrued clinical trial expenses.
Net cash used in operations for all periods reflects the benefit of reimbursement received from Takeda for development and commercial expense and purchase of API by Takeda. From the inception of our collaboration with Takeda through December 31, 2010, we had received a total of $223.7 million for such reimbursement. In addition to the reimbursement received from Takeda, we received a $5 million cash milestone payment for the initiation of Japan's Phase 3 renal indication and $30 million milestone payments for database lock of the non-dialysis and dialysis Phase 3 clinical trials during 2010. We are eligible to receive additional clinical development and regulatory milestones from Takeda of approximately $118 million relating to the dialysis as the first renal indication, including $10 million milestone payments upon FDA acceptance of the submission of the NDA and $50 million of milestone payments upon approval by the FDA.
Net cash used in investing activities for the year ended December 31, 2010 and 2008 of $11.5 million and $21.4 million, respectively, was a result of purchases of investments partially offset by proceeds from maturities and sales of investments. Net cash provided by investing activities for the year ended December 31, 2009 of $49.1 million was primarily the result of proceeds from maturities and sales of investments partially offset by purchases of investments.
Net cash used in financing activities for the year ended December 31, 2010 was primarily attributable to the $9.2 million repayment of our loan from UBS, during the year, partially offset by $4.9 million in net proceeds from a financing executed under our equity line of credit with Azimuth during the years. Net cash provided by financing activities for the year ended December 31, 2009 reflects the net proceeds from two financings during the year, specifically $41.6 million from the private placement in March 2009 and $80.6 million of net proceeds from a public offering in November 2009, as well as $9.2 million in proceeds from the UBS loan in December 2009. The private placement also included warrants to purchase 423,971 shares of common stock at $16.78 that are exercisable and expire in March 2014. Each of the three years ended December 31, 2010, 2009, and 2008 include proceeds from issuance of common stock upon exercise of stock options and the purchase of common stock under our Employee Stock Purchase Plan.
In September 2009 we obtained an equity line of credit arrangement, with Azimuth, that provides that, upon the terms and subject to the conditions set forth in the purchase agreement, Azimuth is committed to purchase up to $60.0 million worth of shares of our common stock over the 24-month term of the purchase agreement (the Common Stock Purchase Agreement). In September 2010, we entered into an amendment, or the Amendment, to the Common Stock Purchase Agreement with Azimuth which extends the term of the equity facility to September 2012 and reduces the minimum threshold price we may establish at which, upon presentation to Azimuth of a draw down notice, Azimuth is required to purchase shares of our common stock. The Amendment further provides that in no event may we sell under the Purchase Agreement more than such number of shares of common stock which is equal to one share less than 20% of our outstanding shares of common stock on the effective date of the Amendment.
In October 2010, we closed on the sale of 999,061 shares of common stock to Azimuth under the Common Stock Purchase Agreement for an aggregate purchase price of $5.0 million. Our net proceeds from the sale of these shares were $4.9 million after deducting offering expenses.
Our equity facility is subject to a number of conditions that limit our ability to draw against such facility. For example, Azimuth is not required to purchase our common stock when the price of our common stock is below $4.00 per share. In addition, Azimuth is not obligated to purchase shares of our
61
common stock which, when aggregated with all other shares of our common stock then owned beneficially by Azimuth, would result in the beneficial ownership by Azimuth of more than 9.9% of the then issued and outstanding shares of our common stock. At December 31, 2010, this represents 2,519,682 shares. After deducting the shares purchased in October 2010, assuming that all remaining 1,520,621 shares were sold at the $6.65 closing price of our common stock at December 31, 2010 at the largest possible discount and assuming that Azimuth still owns these shares, the maximum aggregate net proceeds we could receive under the agreement with Azimuth would be approximately $9.4 million.
As of December 31, 2010, we had $118.1 million in cash, cash equivalents, restricted cash and investments. Our cash and investment balances are held in a variety of interest bearing instruments, including obligations of U.S. government agencies, certificates of deposit, and money market funds. Cash in excess of immediate requirements is invested in accordance with our investment policy primarily with a focus on liquidity and capital preservation.
We believe that the existing cash, cash equivalents and investments together with the interest thereon will enable us to maintain our currently planned operations for at least the next 12 months. However, we expect that we will need to raise additional funding to complete the development and commercialization of peginesatide. Since the announcement of our Phase 3 data, we have experienced a severe decline in our stock price, which has impaired our ability to access capital on potentially favorable terms. In contrast, our need to raise funding has only increased due to the peginesatide development program delays, the reduction of potential milestone payments from Takeda associated with the non-dialysis indication and the potential for future legal proceedings and costs. As we continue to develop and ultimately commercialize peginesatide, if approved, we may experience further challenges or delays to approval of peginesatide if issues arise or additional requirements are imposed based on our discussions with the FDA and other regulatory authorities or as a consequence of our dispute with J&J. Our current view of the worldwide capital markets is that they are extremely volatile with limited accessibility, and many biotechnology companies have been limited or unsuccessful in obtaining funding in this environment. We intend to evaluate the capital markets from time to time to determine whether to raise additional capital, in the form of equity or otherwise, depending upon market conditions relative to our need for funds at such time. Continuation of this market may significantly limit our ability to raise funds such that there can be no assurance we can raise the additional funds to support our continuing operations and maintain current development timelines, and funding may not be available to us on acceptable terms, or at all. Further, we have had to reduce our research capabilities and efforts, including the elimination of certain research programs, and even some activities related to the support of peginesatide. If we are unable to raise additional funds when needed, we could be required to further delay, scale back or eliminate some or all of our development programs and other operations. We may seek to raise additional funds through public or private financing, strategic partnerships or other arrangements. Any additional equity financing would be dilutive to stockholders and debt financing, if available, may involve restrictive covenants. Further, any strategic or licensing arrangements, if available, may require us to relinquish product rights that we would otherwise seek to develop or commercialize ourselves. Our failure to raise capital when needed may harm our business and operating results.
Our future capital requirements will depend on many forward looking factors and are not limited to the following:
- •
- the initiation, progress, timing and completion of pre-clinical studies and clinical trials for peginesatide;
- •
- our ability to fulfill our obligations under our collaboration agreements with Takeda and to achieve the milestones
contained therein;
- •
- costs of litigation;
62
- •
- outcome, timing and cost of regulatory approvals;
- •
- delays that may be caused by changing regulatory requirements;
- •
- the number of drug candidates that we pursue;
- •
- the costs involved in filing and prosecuting patent applications and enforcing and defending patent claims;
- •
- timing and terms of future in-licensing and out-licensing transactions;
- •
- the cost and timing of establishing sales, marketing and distribution capabilities;
- •
- cost of procuring clinical and commercial supplies of peginesatide and future product candidates, if any; and
- •
- the extent to which we acquire or invest in businesses, products or technologies, although we currently have no commitments or agreements relating to any of these types of transactions.
Contractual Obligations and Significant Commitments
Our future contractual obligations, including financing costs, at December 31, 2010 were as follows (in thousands):
| |
Payments Due by Period | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Contractual Obligations
|
Total | Less Than 1 Year |
1-3 Years | 3-5 Years | More than 5 Years |
||||||||||||
Operating lease obligations(1) |
$ | 15,224 | $ | 3,764 | $ | 8,258 | $ | 3,202 | $ | — | |||||||
Long-term income tax liability(2) |
10,249 | — | — | — | — | ||||||||||||
Total fixed contractual obligations |
$ | 25,473 | $ | 3,764 | $ | 8,258 | $ | 3,202 | $ | — | |||||||
- (1)
- Relates
primarily to minimum lease payments for lease of our facilities, consisting of approximately 113,000 square feet which expire in September 2014.
- (2)
- With respect to our long-term income tax liability as of December 31, 2010, we are unable to make a reasonably reliable estimate of the period of cash settlement, if any, with the respective taxing authorities.
In April 2004, we entered into a License, Manufacturing and Supply Agreement with Nektar Therapeutics AL, Corporation, or Nektar, under which we obtained from Nektar a worldwide, non-exclusive license, with limited rights to grant sublicenses, to certain intellectual property covering pegylation technology to manufacture, develop and commercialize peginesatide. In consideration of the license grant, we agreed to pay royalties on the sales of peginesatide. We also agreed to pay milestone payments totaling up to $7 million, plus possible additional milestones in connection with our partnering activities relating to peginesatide or merger and acquisition activities. In July 2006, we paid Nektar a $17.6 million milestone payment triggered by the collaboration agreements signed with Takeda in February and June 2006.
Under the agreement, we also engaged Nektar for the manufacture and supply of our requirements of bulk poly(ethylene) glycol reagent for the manufacture of peginesatide. This relationship is managed by a managing committee formed by representatives from both us and Nektar. Nektar is obligated to engage a third party manufacturer in the event of Nektar's failure (as defined in the agreement) to supply reagent. This agreement expires, on a country by country basis, upon the expiration of our royalty payment obligations. The agreement may be terminated by either party for the other party's material breach provided that such other party has been given a chance to cure such
63
breach, or by Nektar for our challenge of the validity or enforceability of any patents licensed thereunder.
Recent Accounting Pronouncements
In October 2009, the FASB issued ASU, No. 2009-13, multiple deliverable revenue arrangements. This update provides amendments to the criteria in ASC Topic 605, Revenue Recognition, for separating consideration in multiple-deliverable arrangements by establishing a selling price hierarchy. The selling price used for each deliverable will be based on vendor-specific objective evidence or VSOE. if available, third party evidence if VSOE is not available, or estimated selling price if neither VSOE nor third party evidence is available. ASU No. 2009-13 also eliminates the residual method of allocation and requires that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method. ASU No. 2009-13 is effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. We do not believe there will be a significant impact on our financial statements from the adoption of ASU No. 2009-13.
In April 2010, the FASB issued ASU No. 2010-17, revenue recognition—milestone method (Topic 605), which provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. However, the FASB clarified that, even if the requirements in ASU No. 2010-17 are met, entities would not be precluded from making an accounting policy election to apply another appropriate accounting policy that results in the deferral of some portion of the arrangement consideration. ASU No. 2010-17 is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010. Early adoption is permitted. We do not believe there will be a significant impact on our financial statements from the adoption of ASU No. 2010-17.
Off-Balance Sheet Arrangements
There were no significant off-balance sheet arrangements at December 31, 2010.
Item 7A. Quantitative and Qualitative Disclosure About Market Risk.
Interest Rate Risk
Our exposure to market risk is confined to our cash, cash equivalents and investments. We do not use derivative financial instruments in our investment portfolio. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. The securities in our investment portfolio are not leveraged, are classified as available for sale and are subject to minimal interest rate risk. We currently do not hedge interest rate exposure. We do not believe that a decrease in interest rates would have a material negative impact on the value of our investment portfolio.
64
The table below presents the weighted-average interest rates and related carrying amounts (in thousands) of our investment portfolio as of December 31, 2010 and 2009:
| |
2010 | 2009 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Weighted-average Interest Rate |
Carrying Amount |
Weighted-average Interest Rate |
Carrying Amount |
|||||||||
Cash equivalents |
0.01 | % | $ | 61,096 | 0.02 | % | $ | 112,510 | |||||
Short-term investments |
0.61 | % | $ | 33,582 | 1.02 | % | $ | 35,292 | |||||
Long-term investments |
0.38 | % | $ | 19,876 | 1.00 | % | $ | 7,978 | |||||
Foreign Exchange Risk
We have no investments denominated in foreign currencies, and therefore our investments are not subject to foreign currency exchange risk. At each quarter end, we may have liabilities for costs incurred by overseas suppliers of goods or services and clinical trial programs that are denominated in foreign currencies that are not hedged because of their relatively small size, uncertainty of payment date, and/or short time until settlement. An increase or decrease in exchange rates on these unhedged exposures may affect our operating results.
65
Item 8. Financial Statements and Supplementary Data.
Our financial statements and notes thereto appear on pages 68 to 100 of this Annual Report on Form 10-K.
66
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The
Board of Directors and Stockholders
Affymax, Inc.
We have audited the accompanying balance sheets of Affymax, Inc. as of December 31, 2010 and 2009, and the related statements of operations, stockholders' equity, and cash flows for each of the three years in the period ended December 31, 2010. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Affymax, Inc. at December 31, 2010 and 2009, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Affymax, Inc.'s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 10, 2011 expressed an unqualified opinion thereon.
| /s/ Ernst & Young LLP |
Palo
Alto, CA
March 10, 2011
67
AFFYMAX, INC.
BALANCE SHEETS
(in thousands, except share and per share data)
| |
December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | ||||||||
Assets |
||||||||||
Current assets |
||||||||||
Cash and cash equivalents |
$ | 63,499 | $ | 125,296 | ||||||
Restricted cash |
11 | 11 | ||||||||
Short-term investments |
33,582 | 35,292 | ||||||||
Receivable from Takeda |
— | 18,561 | ||||||||
Income taxes receivable |
— | 1,443 | ||||||||
Deferred tax assets |
438 | 1,443 | ||||||||
Prepaid expenses and other current assets |
2,012 | 8,693 | ||||||||
Total current assets |
99,542 | 190,739 | ||||||||
Property and equipment, net |
3,982 | 5,469 | ||||||||
Restricted cash |
1,135 | 1,135 | ||||||||
Long-term investments |
19,876 | 7,978 | ||||||||
Deferred tax assets, net of current |
6,802 | 5,797 | ||||||||
Other assets |
50 | 392 | ||||||||
Total assets |
$ | 131,387 | $ | 211,510 | ||||||
Liabilities and Stockholders' Equity |
||||||||||
Current liabilities |
||||||||||
Accounts payable |
$ | 321 | $ | 464 | ||||||
Accrued liabilities |
11,594 | 12,594 | ||||||||
Accrued clinical trial expenses |
11,247 | 39,499 | ||||||||
Payable to Takeda |
5,958 | — | ||||||||
Deferred revenue |
18,497 | 71,972 | ||||||||
UBS loan |
— | 9,192 | ||||||||
Total current liabilities |
47,617 | 133,721 | ||||||||
Long-term income tax liability |
10,249 | 10,109 | ||||||||
Other long-term liabilities |
974 | 775 | ||||||||
Total liabilities |
58,840 | 144,605 | ||||||||
Commitments and contingencies (Note 6) |
||||||||||
Stockholders' equity |
||||||||||
Common stock: $0.001 par value, 100,000,000 shares authorized; 25,451,338 and 23,869,095 shares issued and outstanding at December 31, 2010 and 2009, respectively |
25 | 24 | ||||||||
Additional paid-in capital |
461,425 | 441,795 | ||||||||
Accumulated deficit |
(388,934 | ) | (374,859 | ) | ||||||
Accumulated other comprehensive income (loss) |
31 | (55 | ) | |||||||
Total stockholders' equity |
72,547 | 66,905 | ||||||||
Total liabilities and stockholders' equity |
$ | 131,387 | $ | 211,510 | ||||||
The accompanying notes are an integral part of these financial statements.
68
AFFYMAX, INC.
STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| |
Year Ended December 31, | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||||
Revenue: |
|||||||||||||
Collaboration revenue |
$ | 112,503 | $ | 114,883 | $ | 82,162 | |||||||
License and royalty revenue |
18 | 16 | 689 | ||||||||||
Total revenue |
112,521 | 114,899 | 82,851 | ||||||||||
Operating expenses: |
|||||||||||||
Research and development |
93,638 | 157,125 | 137,492 | ||||||||||
General and administrative |
33,331 | 36,716 | 34,090 | ||||||||||
Total operating expenses |
126,969 | 193,841 | 171,582 | ||||||||||
Loss from operations |
(14,448 | ) | (78,942 | ) | (88,731 | ) | |||||||
Interest income |
275 | 934 | 4,545 | ||||||||||
Interest expense |
(140 | ) | (105 | ) | (609 | ) | |||||||
Other income (expense), net |
239 | 171 | (1,433 | ) | |||||||||
Net loss before provision (benefit) for income taxes |
(14,074 | ) | (77,942 | ) | (86,228 | ) | |||||||
Provision (benefit) for income taxes |
1 | (1,411 | ) | 282 | |||||||||
Net loss |
$ | (14,075 | ) | $ | (76,531 | ) | $ | (86,510 | ) | ||||
Net loss per common share: |
|||||||||||||
Basic and diluted |
$ | (0.57 | ) | $ | (4.06 | ) | $ | (5.68 | ) | ||||
Weighted-average number of common shares used in computing basic and diluted net loss per common share |
24,488 | 18,865 | 15,220 | ||||||||||
The accompanying notes are an integral part of these financial statements.
69
AFFYMAX, INC.
STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands, except share data)
| |
Common Stock | |
|
|
Accumulated Other Comprehensive Income (Loss) |
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Additional Paid-In Capital |
Deferred Stock-Based Compensation |
Accumulated Deficit |
Total Stockholders' Equity |
||||||||||||||||||
| |
Shares | Amount | ||||||||||||||||||||
Balance at December 31, 2007 |
15,128,959 | $ | 15 | $ | 296,035 | $ | (28 | ) | $ | (211,818 | ) | $ | (19 | ) | $ | 84,185 | ||||||
Issuance of common stock upon exercise of stock options |
104,287 | — | 360 | — | — | — | 360 | |||||||||||||||
Issuance of common stock related to the employee stock purchase plan |
71,533 | — | 875 | — | — | — | 875 | |||||||||||||||
Deferred stock-based compensation |
— | — | (402 | ) | 402 | — | — | — | ||||||||||||||
Amortization of deferred stock-based compensation |
— | — | — | (379 | ) | — | — | (379 | ) | |||||||||||||
Employee stock-based compensation |
— | — | 9,291 | — | — | — | 9,291 | |||||||||||||||
Reversal of deferred stock-based compensation due to cancellations |
— | — | (1 | ) | 1 | — | — | — | ||||||||||||||
Nonemployee stock-based compensation |
— | — | 671 | — | — | — | 671 | |||||||||||||||
Repurchase of common stock |
(360 | ) | — | (1 | ) | — | — | — | (1 | ) | ||||||||||||
Components of other comprehensive loss: |
||||||||||||||||||||||
Net loss |
— | — | — | — | (86,510 | ) | — | (86,510 | ) | |||||||||||||
Change in unrealized gain (loss) on marketable securities |
— | — | — | — | — | 492 | 492 | |||||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | (86,018 | ) | ||||||||||||||
Balance at December 31, 2008 |
15,304,419 | $ | 15 | $ | 306,828 | $ | (4 | ) | $ | (298,328 | ) | $ | 473 | $ | 8,984 | |||||||
Issuance of common stock upon exercise of stock options |
212,424 | — | 720 | — | — | — | 720 | |||||||||||||||
Issuance of common stock upon vesting of restricted stock units |
56,395 | — | — | — | — | — | — | |||||||||||||||
Proceeds from common stock issued upon private placement, net of issuance costs |
3,496,970 | 4 | 41,569 | — | — | — | 41,573 | |||||||||||||||
Proceeds from common stock issued upon public offering, net of issuance costs |
4,726,027 | 5 | 80,585 | — | — | — | 80,590 | |||||||||||||||
Issuance of common stock related to the employee stock purchase plan |
73,069 | — | 925 | — | — | — | 925 | |||||||||||||||
Deferred stock-based compensation |
— | — | 443 | (443 | ) | — | — | — | ||||||||||||||
Amortization of deferred stock-based compensation |
— | — | — | 447 | — | — | 447 | |||||||||||||||
Employee stock-based compensation |
— | — | 9,850 | — | — | — | 9,850 | |||||||||||||||
Nonemployee stock-based compensation |
— | — | 876 | — | — | — | 876 | |||||||||||||||
Repurchase of common stock |
(209 | ) | — | (1 | ) | — | — | — | (1 | ) | ||||||||||||
Components of other comprehensive loss: |
||||||||||||||||||||||
Net loss |
— | — | — | — | (76,531 | ) | — | (76,531 | ) | |||||||||||||
Change in unrealized gain (loss) on marketable securities |
— | — | — | — | — | (528 | ) | (528 | ) | |||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | (77,059 | ) | ||||||||||||||
Balance at December 31, 2009 |
23,869,095 | $ | 24 | $ | 441,795 | $ | — | $ | (374,859 | ) | $ | (55 | ) | $ | 66,905 | |||||||
Issuance of common stock upon exercise of stock options |
399,323 | — | 2,243 | — | — | — | 2,243 | |||||||||||||||
Issuance of common stock upon vesting of restricted stock units |
53,544 | — | — | — | — | — | — | |||||||||||||||
Proceeds from common stock issued upon private placement, net of issuance costs of $117 |
999,061 | 1 | 4,882 | — | — | — | 4,883 | |||||||||||||||
Issuance of common stock related to the employee stock purchase plan |
130,315 | — | 982 | — | — | — | 982 | |||||||||||||||
Deferred stock-based compensation |
— | — | (379 | ) | 379 | — | — | — | ||||||||||||||
Amortization of deferred stock-based compensation |
— | — | — | (379 | ) | — | — | (379 | ) | |||||||||||||
Employee stock-based compensation |
— | — | 12,193 | — | — | — | 12,193 | |||||||||||||||
Nonemployee stock-based compensation |
— | — | (291 | ) | — | — | — | (291 | ) | |||||||||||||
Components of other comprehensive loss: |
||||||||||||||||||||||
Net loss |
— | — | — | — | (14,075 | ) | — | (14,075 | ) | |||||||||||||
Change in unrealized gain (loss) on marketable securities |
— | — | — | — | — | 86 | 86 | |||||||||||||||
Total comprehensive loss |
— | — | — | — | — | — | (13,989 | ) | ||||||||||||||
Balance at December 31, 2010 |
25,451,338 | $ | 25 | $ | 461,425 | $ | — | $ | (388,934 | ) | $ | 31 | $ | 72,547 | ||||||||
The accompanying notes are an integral part of these financial statements.
70
AFFYMAX, INC.
STATEMENTS OF CASH FLOWS
(in thousands)
| |
Year Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||||
Cash flows from operating activities |
||||||||||||
Net loss |
$ | (14,075 | ) | $ | (76,531 | ) | $ | (86,510 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
Depreciation and amortization |
2,212 | 2,116 | 1,294 | |||||||||
Amortization of discount/premium on investments |
650 | 49 | 302 | |||||||||
Stock-based compensation expense |
11,523 | 11,172 | 9,583 | |||||||||
Deferred tax benefit |
— | — | 2,842 | |||||||||
Gain (loss) on disposal of fixed assets |
2 | 65 | (20 | ) | ||||||||
Changes in operating assets and liabilities: |
||||||||||||
Receivable from Takeda |
18,561 | 3,127 | (6,357 | ) | ||||||||
Income taxes receivable |
1,443 | 1,222 | (2,665 | ) | ||||||||
Prepaid expenses and other current assets |
6,681 | (2,046 | ) | 2,676 | ||||||||
Other assets |
342 | 3,326 | (2,270 | ) | ||||||||
Accounts payable |
(143 | ) | (150 | ) | (8,734 | ) | ||||||
Accrued liabilities |
(1,000 | ) | 2,772 | 6,322 | ||||||||
Accrued clinical trial expenses |
(28,252 | ) | 11,693 | 25,333 | ||||||||
Payable to Takeda |
5,958 | — | — | |||||||||
Income taxes payable |
— | (163 | ) | (576 | ) | |||||||
Deferred revenue |
(53,475 | ) | (37,873 | ) | (5,554 | ) | ||||||
Long-term income tax liability |
140 | 113 | 562 | |||||||||
Other long-term liabilities |
199 | 301 | (82 | ) | ||||||||
Net cash used in operating activities |
(49,234 | ) | (80,807 | ) | (63,854 | ) | ||||||
Cash flows from investing activities |
||||||||||||
Purchases of property and equipment |
(730 | ) | (716 | ) | (3,778 | ) | ||||||
Purchases of investments |
(128,650 | ) | (29,345 | ) | (143,154 | ) | ||||||
Proceeds from sales of investments |
16,042 | 1,948 | 44,335 | |||||||||
Proceeds from maturities of investments |
101,857 | 77,168 | 81,168 | |||||||||
Proceeds from sale of property and equipment |
2 | 18 | 22 | |||||||||
Net cash provided by (used in) investing activities |
(11,479 | ) | 49,073 | (21,407 | ) | |||||||
Cash flows from financing activities |
||||||||||||
Repurchases of common stock |
— | (1 | ) | (1 | ) | |||||||
Proceeds from issuance of common stock upon exercise of stock options |
2,243 | 714 | 350 | |||||||||
Proceeds from issuance of common stock related to employee stock purchase plan |
982 | 925 | 875 | |||||||||
Proceeds from common stock issued upon private placement, net of issuance costs |
4,883 | 41,569 | — | |||||||||
Proceeds from common stock issued upon public offering, net of issuance costs |
— | 80,585 | — | |||||||||
Proceeds from UBS loan |
— | 9,192 | — | |||||||||
Repayment of UBS loan |
(9,192 | ) | — | — | ||||||||
Principal payments under capital lease obligations |
— | — | (132 | ) | ||||||||
Net cash provided by (used in) financing activities |
(1,084 | ) | 132,984 | 1,092 | ||||||||
Net increase (decrease) in cash and cash equivalents |
(61,797 | ) | 101,250 | (84,169 | ) | |||||||
Cash and cash equivalents at beginning of the year |
125,296 | 24,046 | 108,215 | |||||||||
Cash and cash equivalents at end of the year |
$ | 63,499 | $ | 125,296 | $ | 24,046 | ||||||
| |
Year Ended December 31, |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||
Supplemental disclosures of cash flow information: |
|||||||||||
Income taxes paid |
$ | 1 | $ | 181 | $ | 1 | |||||
Interest paid |
— | 1 | 2 | ||||||||
Noncash investing and financing activities: |
|||||||||||
Change in unrealized loss on investments |
86 | (528 | ) | 492 | |||||||
Deferred stock-based compensation, net of cancellations |
(379 | ) | (443 | ) | (403 | ) | |||||
The accompanying notes are an integral part of these financial statements.
71
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS
1. The Company
Affymax, Inc., a Delaware corporation, was incorporated on July 20, 2001. We are a biopharmaceutical company committed to developing novel drugs to improve the treatment of serious and often life-threatening conditions. Our product candidate, peginesatide (HematideTM), is designed to treat anemia associated with chronic renal failure. Peginesatide is a synthetic peptide-based erythropoiesis stimulating agent or ESA, designed to stimulate production of red blood cells. As previously reported in our Current Report on Form 8-K dated June 21, 2010, we recently announced preliminary top-line results from the peginesatide Phase 3 clinical program for the treatment of patients with anemia associated with chronic renal failure.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents are stated at cost, which approximates market value. We consider all highly liquid investments purchased with an original maturity of three months or less to be cash equivalents.
Restricted Cash
Restricted cash represents cash for certificates of deposit provided as credit guarantees and security for an irrevocable letter of credit related to the lease of office space.
Comprehensive Loss
Comprehensive loss consists of net loss plus the change in unrealized gains and losses on investments. At each balance sheet date presented, our accumulated other comprehensive loss consists solely of unrealized gains and losses on investments. Comprehensive loss for the years ended December 31, 2010, 2009, and 2008 are as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Net loss |
$ | (14,075 | ) | $ | (76,531 | ) | $ | (86,510 | ) | |
Decrease (increase) in unrealized gains (losses) on investments |
183 | (408 | ) | (731 | ) | |||||
Reclassification adjustment for (gains) losses on investments recognized in earnings |
(97 | ) | (120 | ) | 1,223 | |||||
Comprehensive loss |
$ | (13,989 | ) | $ | (77,059 | ) | $ | (86,018 | ) | |
72
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
Reclassifications and Adjustments
Certain amounts in prior period financial statements have been reclassified to conform to the current period presentation. We reclassified $684,000 to long-term income tax liability from other long-term liabilities, in the 2009 balance sheet. We also reclassified certain activities totaling $120,000 and $1.2 million for the years ended December 31, 2009 and 2008, respectively, related to our Auction Rate Securities, or ARS, from operating to investing activities in the 2009 and 2008 statements of cash flows. These reclassifications did not change previously reported net loss, total assets, or stockholders' equity.
During the quarter ended June 30, 2008, we identified an overstatement of clinical trial expense and collaboration revenue of $1.3 million in the year ended December 31, 2007. As a result, clinical trial expense and collaboration revenue, which includes reimbursement for these costs, includes an out of period reduction of $1.3 million and $0.4 million, respectively, in the year ended December 31, 2008. The overstatement was immaterial to the financial statements for the year ended December 31, 2007 and therefore was corrected in the second quarter of 2008.
During the quarter ended December 31, 2008, we identified an understatement of our provision for income taxes of $0.7 million in the year ended December 31, 2007. As a result, our provision for income taxes included an out of period increase of $0.7 million in the year ended December 31, 2008. The understatement was immaterial to the financial statements for the year ended December 31, 2007 and therefore was corrected in the fourth quarter of 2008.
Changes in Estimates
During the year ended December 31, 2010, we finalized amendments for certain clinical trial activities completed in 2009. In the fourth quarter of 2010, we obtained final monitored site visit data and investigator contracts from our third party contract research organizations, or CROs, that allowed us to complete our reconciliation of the significant majority of the labor and investigator costs incurred throughout the course of our clinical trials to our previously recorded estimates. This data and contractual information was not available to us during the course of the trials. After extensive analysis to cost out and analyze the information provided, we determined that the costs incurred were lower than our previously recorded estimates. The change in estimate is due largely to (i) 8% lower total patient months on study as compared to our previously estimated level as a result of a larger number of patients coming off study and no longer having trial-related site visits (also known as lost to follow up) during the course of the study, and (ii) 13% lower than expected average fee generating site visits per month, driven largely by missed visits from patients on study. We believe these changes were only identifiable based on the information received in the fourth quarter of 2010. The change in estimate decreased expense by $12.1 million for the year ended December 31, 2010. As this change in estimate was comprised of development costs charged to Takeda at a 70% reimbursement rate, this amount is now a payable due back to Takeda. The reimbursement received from Takeda in prior periods was recorded as deferred revenue and collaboration revenue under our Contingency Adjusted Performance Model, or CAPM revenue recognition model. This change in estimate resulted in a reduction of $8.4 million of deferred revenue and a reversal of $7.8 million of collaboration revenue. The net impact to our statement of operations for this change in estimate was a $4.3 million decrease to our net loss or $0.18 per share for the year ended December 31, 2010. Additional changes in estimate or adjustments may result as the final trial close-out audits and fee negotiations are completed relating to our clinical
73
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
trials which could impact R&D expense and collaboration revenues and amounts due to or from Takeda in subsequent periods.
Clinical Trial Expense and Accruals
We record expense for estimated clinical study external costs, which are a significant component of research and development or R&D expenses. These clinical trial costs were $14.9 million, $90.0 million and $77.8 million for the years ended December 31, 2010, 2009, and 2008, respectively. Clinical trials are administered by CROs. CROs typically perform most of the total start-up activities for the trials, including document preparation, site identification pre-study visits, training as well as on-going program management. For the Phase 3 studies, which represent the vast majority of the clinical trial expense, the expense recorded is based on reporting received from CROs and internal analyses. We accrue costs for work performed by CROs based on the achievement of contracted activities during the period. Expense for investigator fees, which include patient costs, is based on internal estimates of activities using patient enrollment and contractual or estimated rates. For the Phase 2 studies, the expense is activities-based such as patient monitoring as reported by the CROs and achievement of milestones. Other costs such as testing and drug materials are expensed as incurred. For all studies, CRO reporting is reviewed by us for appropriateness.
There is a significant degree of estimation involved in quantifying the clinical trial expenses. The complexity and magnitude of the activities and expenses can be significant and subject to frequent change during the studies, especially for our Phase 3 trials. The activities in our trials are performed globally, in many sites and countries, involving numerous CROs and third parties. If we do not receive complete and accurate information from the CRO or third parties on a timely basis or correctly estimate activity levels, we may have to record adjustments, which could potentially result in significant increases or decreases in R&D expenses, in subsequent periods.
During the year ended December 31, 2010, we finalized amendments for certain clinical trial activities completed in 2009. In the fourth quarter of 2010, we obtained final monitored site visit data and investigator contracts from our CROs that allowed us to complete our reconciliation of the significant majority of the labor and investigator costs incurred throughout the course of our clinical trials to our previously recorded estimates. This data and contractual information was not available to us during the course of the trials. After extensive analysis to cost out and analyze the information provided, we determined that the costs incurred were lower than our previously recorded estimates. The change in estimate is due largely to (i) 8% lower total patient months on study as compared to our previously estimated level as a result of a larger number of patients coming off study and no longer having trial-related site visits (also known as lost to follow up) during the course of the study, and (ii) 13% lower than expected average fee generating site visits per month, driven largely by missed visits from patients on study. We believe these changes were only identifiable based on the information received in the fourth quarter of 2010. The change in estimate decreased expense by $12.1 million for the year ended December 31, 2010. Additional changes in estimate or adjustments may result as the final trial close-out audits and fee negotiations are completed relating to our clinical trials which could impact R&D expense and collaboration revenues and amounts due to or from Takeda in subsequent periods.
74
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
Concentration of Risk and Uncertainties
Financial instruments that potentially subject us to a concentration of credit risk consist of cash, cash equivalents and investments. We deposit excess cash in accounts with three major financial institutions in the U.S. Deposits in these banks may exceed the amount of insurance provided on such deposits. We have not experienced any realized losses on our deposits of cash and cash equivalents. Although our guideline for investment of excess cash is designed to maintain safety and liquidity through our policies on diversification and investment maturity, at December 31, 2009, we held fair value of investments in ARS, totaling $15.5 million that failed in auctions. As of December 31, 2010, we no longer maintain any ARS in our investment portfolio. See Note 4—Investments for further discussion.
We have experienced significant operating losses since inception. At December 31, 2010, we had an accumulated deficit of $388.9 million. We have generated no revenue from product sales to date. We have funded our operations to date principally from upfront license fees, milestone and reimbursement payments received under our collaboration agreements with Takeda Pharmaceutical Company Limited or Takeda and the sale of equity securities. We expect to incur substantial additional operating losses for the next several years and will need to obtain additional financing in order to complete the development and commercialization of peginesatide. There can be no assurance that such financing will be available or will be at terms acceptable to us.
Our accounts receivable balance with Takeda of $0 and $18.6 million at December 31, 2010 and 2009, respectively, is related to our two separate collaboration agreements, or the Arrangement with Takeda, and is generally comprised of amounts due to us for reimbursement of development and commercial expenses for the purchase of API under the terms of our Arrangement with Takeda. During the year ended December 31, 2010, we recorded a $12.1 million change in estimate to our clinical trial accrual and related expense as a result of our analysis of the final monitored site visit data for our Phase 3 clinical trials. As this change in estimate was comprised of development costs charged to Takeda at a 70% reimbursement rate, this amount is now a payable due back to Takeda. The reimbursement received from Takeda in prior periods was recorded as deferred revenue and collaboration revenue under our CAPM revenue recognition model. This change in estimate resulted in a reduction of $8.4 million of deferred revenue and a reversal of $7.8 million of collaboration revenue. This amount due to Takeda as of December 31, 2010 was partially offset by $2.5 million due from Takeda for reimbursement of development and commercial expenses incurred by us in the fourth quarter of 2010. The resulting net payable to Takeda of $6.0 million as of December 31, 2010 is reflected on the accompanying balance sheet under the caption Payable to Takeda. We have not experienced any credit losses from our Arrangement with Takeda and none are expected. We do not require collateral on our receivable.
We are currently developing our first product offering, peginesatide, and have no products that have received regulatory approval. Peginesatide will require approval from the U.S. Food and Drug Administration or FDA and/or foreign regulatory agencies prior to commercial sales. There can be no assurance that peginesatide will receive the necessary approvals. If we are denied such approvals or such approvals are delayed, it would have a material adverse effect on us. To achieve profitable operations, we must successfully develop, test, manufacture and commercialize peginesatide. There can be no assurance that peginesatide can be developed successfully or manufactured at an acceptable cost
75
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
and with appropriate performance characteristics, or that peginesatide will be successfully commercialized. These factors could have a material adverse effect on our future financial results.
Further, some of our suppliers and manufacturing arrangements, including the provision of bulk poly(ethylene) glycol reagent for the manufacture of peginesatide from Nektar Therapeutics AL, Corporation, or Nektar, are currently single-sourced, leaving us at greater risk of supply interruptions and potential delays.
Revenue Recognition
Collaboration Revenue
We recognize revenue in accordance with the authoritative guidance for revenue recognition in financial statements. When evaluating multiple element arrangements, we consider whether the components of the arrangement represent separate units of accounting as defined in the authoritative guidance for revenue arrangements with multiple deliverables. Application of this guidance requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
We entered into the Arrangement, with Takeda, which have been combined for accounting purposes due to their proximity of negotiation. We evaluated the multiple elements under the combined single arrangement in accordance with the authoritative guidance for revenue recognition with multiple deliverables. We were unable to determine the stand-alone value of the delivered elements and obtain verifiable objective evidence to determine the fair value of the undelivered elements. Accordingly, we concluded that there was a single unit of accounting.
Effective January 1, 2008, we entered into an amendment to the Arrangement with Takeda. The amendment provides us the ability to opt-out of our obligation to participate on the joint steering committee and any related subcommittees at any time beginning January 1, 2011 without any other modifications. As a result, the obligation to participate in the joint steering committee and any related subcommittee is no longer indefinite. Accordingly, we determined that we can separate the performance obligations that occur over the development period from the performance obligations that will occur during the commercialization period. We had previously estimated the development period to end on January 1, 2011. Based on the announcement on June 21, 2010 of our top-line results from our Phase 3 clinical program, during the second quarter of 2010, we re-evaluated our development period and determined that the development period was estimated to end upon the submission of our NDA to the FDA, estimated to occur in the first half of 2011. After further refinements to our regulatory strategy as a result of our subsequent meeting with the FDA in November 2010, we re-evaluated our estimated filing date for our NDA submission and now believe it to be in the second quarter of 2011. As a result of the change in performance period from indefinite to a definitive date, beginning on January 1, 2008, we recognize revenue using CAPM. Under CAPM, revenue is eligible for recognition in the period the payment is earned under the Arrangement including amounts that are either received or due from Takeda. Revenue initially recognized is based on the percentage of time elapsed from inception of the Arrangement in June 2006 to the period in which the payment is earned in relation to the total projected development period. The remaining portion of the payment is then recognized on a straight-line basis over the remaining estimated duration of the development period of the Arrangement. Payments during the development period include amounts due for upfront license fees,
76
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
milestone payments earned, purchases of active pharmaceutical ingredient or API and reimbursement of development and commercial expenses. Further changes in the estimated term of the development period could materially affect the amount of collaboration revenue recognized in future periods. We expect collaboration revenue to be directly affected by milestone payments and expenses that are eligible for reimbursement from Takeda under the Arrangement in future periods. Through the period of the joint steering committee obligation, we expect collaboration revenue to be directly affected by milestone payments and expenses that are eligible for reimbursement from Takeda under the Arrangement in future periods. Included in the reimbursable expense is the cost of API that we manufacture and supply to Takeda during the development period, which we will also supply during the commercialization period. A change in the estimated term of the development period could materially affect the amount of collaboration revenue recognized in future periods.
During the year ended December 31, 2010, we recorded a $12.1 million change in estimate to our clinical trial accruals as a result of our analysis of the final monitored site visit data for our Phase 3 clinical trials. As this change in estimate was comprised primarily of the reversal of previously recorded estimated development costs related to our Phase 3 clinical trials, 70% of this amount is now a payable due back to Takeda as it represents a reduction in previously reimbursed development expenses. As the reimbursement received from Takeda in prior periods was recorded as deferred revenue and collaboration revenue under our CAPM revenue recognition model when those development costs were initially expensed, this change in estimate resulted in a reversal of $8.4 million of deferred revenue and $7.8 million of collaboration revenue as of December 31, 2010.
License and Royalty Revenue
Royalties are recognized as earned in accordance with contract terms, when third party results are reported and collectability is reasonably assured. Royalties received under agreements that were acquired by us in the 2001 spin out from GlaxoSmithKline or Glaxo are recorded net of the 50% that we are required to remit to Glaxo.
Fair Value of Financial Instruments
For financial instruments consisting of cash and cash equivalents, receivable from and payable to Takeda, accounts payable and accrued liabilities included in our financial statements, the carrying amounts are reasonable estimates of fair value due to their short maturities. Estimated fair values for short-term and long-term investments, except our investments in ARS, which are separately disclosed elsewhere, are based on quoted market prices for the same or similar instruments. Based on borrowing rates currently available to us for loans with similar terms, the carrying value of lease obligations approximates fair value. The carrying amount of the loan with UBS approximated its fair value due to the loan's short-term nature.
Investments
Investments are classified as available-for-sale and are carried at their fair market value based upon quoted market prices for these or similar instruments at the balance sheet date. Unrealized gains and losses are reported as a separate component of stockholders' equity until realized. The amortized cost of these securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization as well as realized gains and losses are included in interest income. We assess our
77
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
investments for potential other-than-temporary impairment based on factors including the length of time and extent to which the fair market value has been below our cost basis, the current financial condition of the investee and our intent and ability to hold the investment for a sufficient period of time to allow for any anticipated recovery in market value. If we conclude that an other-than-temporary impairment exists, we recognize an impairment charge to reduce the investment to fair value and record the related charge as a reduction of interest to other income (expense), net. We have elected to use settlement date accounting for purposes of recording transactions.
Research and Development
All research and development costs are expensed as incurred.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation and amortization. Depreciation and amortization of property and equipment are calculated using the straight-line method over the estimated useful lives of the assets, generally three to five years. Assets under capital lease and leasehold improvements are amortized over the lesser of their estimated useful lives or the term of the related lease. Maintenance and repairs are charged to operations as incurred.
Segment Information
We operate in one business segment, which encompasses all the geographical regions. Collaboration revenue recognized was from Japan related to the Arrangement. License and royalty revenue was primarily from the U.S. All of our assets reside in the U.S. Management uses one measurement of profitability and does not segregate our business for internal reporting.
Income Taxes
We account for income taxes under the liability method, whereby deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
Net Loss per Common Share
Basic and diluted net loss per common share is computed using the weighted-average number of shares of common stock outstanding during the year. Stock options, common stock subject to repurchase, warrants, restricted stock units and common stock issuable pursuant to the 2006 Employee Stock Purchase Plan were not included in the diluted net loss per common share calculation for all years presented because the inclusion of such shares would have had an antidilutive effect.
78
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
The computation of basic and diluted net loss per common share is as follows (in thousands, except per share amounts):
| |
Year Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||
Numerator: |
|||||||||||
Net loss |
$ | (14,075 | ) | $ | (76,531 | ) | $ | (86,510 | ) | ||
Denominator: |
|||||||||||
Weighted-average common shares outstanding |
24,488 | 18,866 | 15,223 | ||||||||
Less: Weighted-average unvested common shares subject to repurchase |
— | (1 | ) | (3 | ) | ||||||
Weighted-average number of common shares used in computing basic and diluted net loss per common share |
24,488 | 18,865 | 15,220 | ||||||||
Basic and diluted net loss per common share |
$ | (0.57 | ) | $ | (4.06 | ) | $ | (5.68 | ) | ||
The following number of shares were excluded from the denominator in the computation of diluted net loss per common share for the years presented because including them would have an antidilutive effect (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Options to purchase common stock |
3,890 | 2,430 | 2,130 | |||||||
Common stock subject to repurchase |
— | — | 2 | |||||||
Common stock issuable pursuant to the 2006 Employee Stock Purchase Plan |
29 | 16 | 13 | |||||||
Restricted stock units |
503 | 107 | 189 | |||||||
Warrant to purchase common stock |
426 | 426 | 2 | |||||||
Stock-Based Compensation
We account for equity instruments issued to employees and directors under the authoritative guidance for share-based payments.
The equity instruments we most typically grant are stock options and restricted stock units. Stock options are valued using the Black-Scholes valuation model while the fair value of restricted stock units is equivalent to the value of the equivalent number of shares of common stock on the date of grant. The measurement of stock-based compensation is subject to periodic adjustments as the underlying equity instruments vest or do not vest as a result of employee terminations prior to vest.
We have issued stock options to nonemployees. We account for equity instruments issued to nonemployees in accordance with the authoritative guidance for equity-based payments to nonemployees, using a fair value approach.
Recent Accounting Pronouncements
In October 2009, the Financial Accounting Standards Board, or FASB issued Accounting Standards Update, or ASU, No. 2009-13, multiple deliverable revenue arrangements. This update provides
79
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
2. Summary of Significant Accounting Policies (Continued)
amendments to the criteria in ASC Topic 605, Revenue Recognition, for separating consideration in multiple-deliverable arrangements by establishing a selling price hierarchy. The selling price used for each deliverable will be based on vendor-specific objective evidence or VSOE. if available, third party evidence if VSOE is not available, or estimated selling price if neither VSOE nor third party evidence is available. ASU No. 2009-13 also eliminates the residual method of allocation and requires that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method. ASU No. 2009-13 is effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010. We believe there will be no significant impact on our financial statements from that the adoption of ASU No. 2009-13.
In April 2010, the FASB issued ASU No. 2010-17, revenue recognition—milestone method (Topic 605), which provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. However, the FASB clarified that, even if the requirements in ASU No. 2010-17 are met, entities would not be precluded from making an accounting policy election to apply another appropriate accounting policy that results in the deferral of some portion of the arrangement consideration. ASU No. 2010-17 is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years, beginning on or after June 15, 2010. Early adoption is permitted. We believe there will be no significant impact on our financial statements from the adoption of ASU No. 2010-17.
3. Balance Sheet Components
Property and Equipment, Net
Property and equipment consist of the following (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | |||||
Leasehold improvements |
$ | 2,115 | $ | 1,804 | |||
Equipment |
8,567 | 8,499 | |||||
Software |
2,431 | 2,280 | |||||
Construction in progress |
176 | 152 | |||||
|
13,289 | 12,735 | |||||
Less: Accumulated depreciation and amortization |
(9,307 | ) | (7,266 | ) | |||
|
$ | 3,982 | $ | 5,469 | |||
80
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
3. Balance Sheet Components (Continued)
Depreciation and amortization expense for the years ended December 31, 2010, 2009, and 2008 was $2.2 million, $2.1 million and $1.3 million, respectively.
Accrued Liabilities
Accrued liabilities consist of the following (in thousands):
| |
December 31, | ||||||
|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | |||||
Compensation-related expenses |
$ | 7,671 | $ | 5,720 | |||
Research and development related costs |
2,476 | 5,067 | |||||
Other |
1,447 | 1,807 | |||||
|
$ | 11,594 | $ | 12,594 | |||
4. Investments
The following is a summary of our available-for-sale marketable securities (in thousands):
| |
As of December 31, 2010 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Cost | Gross Unrealized Gains |
Gross Unrealized Losses |
Other-Than- Temporary Impairment |
Fair Value | ||||||||||||
Short-term investments: |
|||||||||||||||||
Certificates of deposit |
$ | 1,448 | $ | — | $ | — | $ | — | $ | 1,448 | |||||||
Government securities |
32,080 | 58 | (4 | ) | — | 32,134 | |||||||||||
Total short-term investments |
$ | 33,528 | $ | 58 | $ | (4 | ) | $ | — | $ | 33,582 | ||||||
Long-term investments: |
|||||||||||||||||
Government securities |
$ | 19,899 | $ | 5 | $ | (28 | ) | $ | — | $ | 19,876 | ||||||
Total long-term investments |
$ | 19,899 | $ | 5 | $ | (28 | ) | $ | — | $ | 19,876 | ||||||
| |
As of December 31, 2009 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
Cost | Gross Unrealized Gains |
Gross Unrealized Losses |
Other-Than- Temporary Impairment |
Fair Value | ||||||||||||
Short-term investments: |
|||||||||||||||||
Certificates of deposit |
$ | 3,714 | $ | — | $ | — | $ | — | $ | 3,714 | |||||||
Government securities |
20,006 | 3 | — | — | 20,009 | ||||||||||||
Auction rate securities |
14,125 | — | — | (2,556 | ) | 11,569 | |||||||||||
Total short-term investments |
$ | 37,845 | $ | 3 | $ | — | $ | (2,556 | ) | $ | 35,292 | ||||||
Long-term investments: |
|||||||||||||||||
Government securities |
$ | 4,059 | $ | — | $ | (58 | ) | $ | — | $ | 4,001 | ||||||
Auction rate securities |
4,800 | — | — | (823 | ) | 3,977 | |||||||||||
Total long-term investments |
$ | 8,859 | $ | — | $ | (58 | ) | $ | (823 | ) | $ | 7,978 | |||||
81
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
4. Investments (Continued)
The investments mature between January 2011 and August 2012.
Fair Value Measurements
We measure certain financial assets at fair value on a recurring basis, including cash equivalents and available for sale securities. The fair value of these assets was determined based on a three-tier hierarchy under the authoritative guidance for fair value measurements and disclosures that prioritizes the inputs used in measuring fair value as follows:
- •
- Level 1—observable inputs such as quoted prices in active markets.
- •
- Level 2—inputs other than quoted prices in active markets that are observable either directly or
indirectly through corroboration with observable market data.
- •
- Level 3—unobservable inputs in which there is little or no market data, which would require us to develop its own assumptions.
Effective January 2010, we adopted the provisions of the authoritative guidance for improving fair value disclosures. Our cash equivalents and investments, other than ARS, are classified within Level 1 or Level 2 of the fair value hierarchy because they are valued using quoted market prices in active markets, broker or dealer quotations, or alternative pricing sources with reasonable levels of price transparency. The types of investments that are generally classified within Level 1 of the fair value hierarchy include money market securities. The valuation technique we used to measure fair value of our Level 1 money market securities is a market approach, using prices and other relevant information generated by market transactions involving identical securities. The types of investments that are generally classified within Level 2 of the fair value hierarchy include corporate securities, certificates of deposits and U.S. government securities. The valuation technique we used to measure fair value of our Level 2 investments is a market approach, which we review trading activity and pricing for these investments as of the measurement date. When sufficient quoted pricing for identical investments was not available, we used market pricing and other observable market inputs for similar investments obtained from various third party data providers. These inputs represent quoted prices for similar investments in active markets or these inputs have been derived from observable market data. Our investments in ARS and UBS AG of Series C-2 ARS Rights, or ARS Rights were classified within Level 3 of the fair value hierarchy because of the lack of observable inputs. The valuation technique we used to measure fair value of our Level 3 ARS and ARS Rights was an income approach and we used a discounted cash flow analysis. As of December 31, 2010, we no longer maintained any ARS or ARS Rights. Further, there were no transfers in and out between the fair value hierarchy Level 1, Level 2 and Level 3, and there were no changes in our valuation technique in the year ended December 31, 2010. Our policy is to recognize transfers into or out of Level 3 classifications as of the actual date of the event or change in circumstances that caused the transfer.
As a result of a settlement between various regulatory agencies, including the SEC, and UBS entities relating to sales and marketing practices of ARS, in October 2008, we received an offer from UBS of ARS Rights or ARS Rights in connection with the $14.1 million of par value of ARS as of December 31, 2009 that were purchased through UBS. In November 2008, we accepted the terms of the ARS Rights and delivered the required legal release of claims against the UBS entities. These ARS Rights gave us the option to require UBS to repurchase, at par, the ARS which we exercised in July 1, 2010. In connection with the ARS Rights, we obtained through UBS Financial Services, Inc., an
82
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
4. Investments (Continued)
affiliate of UBS AG, a loan facility that allowed us to draw of up to 75% of the stated value of our ARS portfolio in the form of a no-cost loan. In December 2009, we obtained a loan of approximately $9.2 million. See Note 5—UBS Loan.
We determined that the ARS Rights do not meet the definition of a derivative security as described in the authoritative guidance for accounting for derivative instruments and hedging activities because the ARS Rights were non-transferrable, and we must tender the related ARS to receive the cash settlement. Therefore, we elected to measure the ARS Rights separately under the authoritative guidance pertaining to the fair value option for financial assets and financial liabilities in order to partially offset the changes in the fair value of the ARS to the ARS Rights. We did not elect to adopt the guidance for the fair value option for financial assets and financial liabilities to measure financial instruments, except for the ARS Rights. We determined the fair value of our ARS Rights using a discounted cash flow analysis based on, among other things, the timing and likelihood of the recovery of the par value of the ARS from UBS. Our analysis resulted in net increases in the fair value of our ARS Rights of $57,000 and $2.4 million during the years ended December 31, 2009 and 2008, respectively, and were recorded as an other current asset with a corresponding credit to other income (expense), net. Upon sale of the related ARS during 2009 and 2010, the fair value of our ARS Rights were decreased by $604,000 and $134,000 and resulted in a charge to other income (expense), net.
In 2010, we sold or had redeemed all the ARS including those through the exercising of our ARS Rights. All redemptions took place at par and our sales of ARS resulted in a realized loss of $158,000 in the year ended December 31, 2010.
Prior to the sale or redemption of the ARS, we used a discounted cash flow analysis to determine fair value. The analysis considers, among other things, the amount and timing of coupon payments, contractual terms, underlying collateralization and credit risk. In addition, we included in our analysis an illiquidity factor to estimate the discount necessary to sell a security for which there is no active market. The analysis considers that issuers have continued to meet interest payment obligations and are expected to continue to do so at levels consistent with issuer's credit risk. The analysis was based on dynamic market conditions and changes in our assumptions could lead to a significant change in determined value. Our analysis resulted in net decreases in fair value of ARS totaling $160,000 and $3.7 million in the years ended December 31, 2009 and 2008, respectively, that were deemed to be other-than-temporary and were recorded as impairment charges to other income (expense), net. Upon sale, we recorded a gain to the extent that the proceeds from the sale or redemption exceeded the estimated fair value of the ARS as of the end of the previous reporting period. We recorded gains of $695,000, $419,000, and $111,000 during the years ended December 31, 2010, 2009 and 2008, respectively.
83
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
4. Investments (Continued)
The following table presents our investments measured at fair value on a recurring basis classified by the fair value measurements and disclosures valuation hierarchy (in thousands):
| |
As of December 31, 2010 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Fair Value Measurements Using | ||||||||||||
| |
Total | Level 1 | Level 2 | Level 3 | ||||||||||
Cash equivalents |
$ | 61,096 | $ | 59,353 | $ | 1,743 | $ | — | ||||||
Short-term investments: |
||||||||||||||
Certificates of deposit |
$ | 1,448 | $ | — | $ | 1,448 | $ | — | ||||||
Government securities |
32,134 | — | 32,134 | — | ||||||||||
Total short-term investments |
$ | 33,582 | $ | — | $ | 33,582 | $ | — | ||||||
Long-term investments: |
||||||||||||||
Government securities |
$ | 19,876 | $ | — | $ | 19,876 | $ | — | ||||||
Total long-term investments |
$ | 19,876 | $ | — | $ | 19,876 | $ | — | ||||||
| |
As of December 31, 2009 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
|
Fair Value Measurements Using | ||||||||||||
| |
Total | Level 1 | Level 2 | Level 3 | ||||||||||
Cash equivalents |
$ | 112,510 | $ | 102,216 | $ | 10,294 | $ | — | ||||||
Short-term investments: |
||||||||||||||
Certificates of deposit |
$ | 3,714 | $ | — | $ | 3,714 | $ | — | ||||||
Government securities |
20,009 | — | 20,009 | — | ||||||||||
Auction rate securities |
11,569 | — | — | 11,569 | ||||||||||
Total short-term investments |
$ | 35,292 | $ | — | $ | 23,723 | $ | 11,569 | ||||||
Long-term investments: |
||||||||||||||
Government securities |
$ | 4,001 | $ | — | $ | 4,001 | $ | — | ||||||
Auction rate securities |
3,977 | — | — | 3,977 | ||||||||||
Total long-term investments |
$ | 7,978 | $ | — | $ | 4,001 | $ | 3,977 | ||||||
ARS Rights |
$ | 2,337 | $ | — | $ | — | $ | 2,337 | ||||||
84
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
4. Investments (Continued)
The following table presents changes in Level 3 assets measured at fair value on a recurring basis for the years ended December 31, 2010 and 2009 (in thousands):
| |
Year Ended December 31, |
|||||||
|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | ||||||
Balance at beginning of the period |
$ | 17,883 | $ | 20,026 | ||||
Transfers in and/or out of Level 3 |
— | — | ||||||
Total unrealized losses related to ARS included in net loss |
— | (160 | ) | |||||
Total realized gains related to ARS included in net loss |
695 | 419 | ||||||
Total realized loss related to ARS included in net loss |
(158 | ) | (62 | ) | ||||
Total realized losses related to ARS Rights included in net loss |
(604 | ) | (134 | ) | ||||
Total unrealized gains related to ARS Rights included in net loss |
— | 57 | ||||||
Settlements |
(17,816 | ) | (2,263 | ) | ||||
Balance at end of the period |
$ | — | $ | 17,883 | ||||
5. UBS Loan
In connection with the settlement with UBS AG relating to our ARS, we entered into a loan agreement with UBS Financial Services, Inc., an affiliate of UBS AG. In December 2009, we obtained a loan of approximately $9.2 million. This "no net cost loan" bears interest at a rate that will not exceed the average rate of interest paid on the pledged ARS. As part of our exercising the ARS Rights for cash settlement on July 1, 2010, we repaid our outstanding loan balance. As of December 31, 2010, we no longer have any debt liability.
As required by UBS, we applied the net interest received in and the proceeds from the sales and redemptions of ARS to the principal of the loan. For the year ended December 31, 2010, we paid $56,000 of interest expense associated with the loan and received $150,000 in interest income from the collateralized ARS. For the year ended December 31, 2010, the net interest earned of $94,000.
6. Commitments and Contingencies
We rent our office facilities and certain equipment under noncancelable operating leases, which expire at various dates through September 2014. Under the terms of the leases, we are responsible for certain taxes, insurance and maintenance expenses.
Rent expense for the years ended December 31, 2010, 2009, and 2008 was $2.8 million, $2.1 million and $2.6 million, respectively. We recognize rent expense on a straight-line basis over the lease period.
85
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
6. Commitments and Contingencies (Continued)
Future minimum payments under noncancelable lease obligations as of December 31, 2010 are as follows (in thousands):
| |
Operating Leases |
|||
|---|---|---|---|---|
2011 |
$ | 3,764 | ||
2012 |
4,075 | |||
2013 |
4,183 | |||
2014 |
3,202 | |||
2015 |
— | |||
Thereafter |
— | |||
Total minimum lease payments |
$ | 15,224 | ||
Legal Proceedings
In October 2010, the arbitration panel in our binding arbitration with certain subsidiaries of Johnson & Johnson, or J&J, decided the ownership of a number of U.S. and international patents and patent applications related to certain EPO-R agonists, or the "intellectual property in dispute." The decision maintained J&J's sole inventorship and sole ownership of U.S. Patent No. 5,767,078, or the "078 Patent," and certain related foreign patents and patent applications, including European Patent application EP96/918,317. The arbitrators determined that we and J&J jointly own the remainder of the intellectual property in dispute.
In November 2010, we filed in the U.S. District Court for the Northern District of Illinois, a motion to vacate the arbitration award with respect to the ownership of the '078 Patent and related foreign cases. In December 2010, J&J filed its response and requested that the court confirm the arbitration award. Briefs have been filed and a decision is expected shortly.
We are continuing to review the arbitrators' decision and consider potential courses of action with our counsel and Takeda Pharmaceutical Company Limited, or Takeda. We expect that this dispute with J&J could involve additional litigation or legal proceedings that may take years and substantial resources and funds to resolve. Although we believe that peginesatide does not infringe the '078 Patent and that we would have substantial defenses to any potential claims by J&J, J&J may now or in the future attempt to assert claims based upon the '078 Patent against us or our collaborators in connection with the manufacture and commercialization of peginesatide. Outside of the U.S., the European Patent application EP96/918,317 and other foreign counterpart patents to the '078 Patent differ in the nature and extent of the claims from that of the '078 Patent and due to the complexity of patent laws, we are unable to assess adequately the defenses available to us in the various countries outside of the U.S. to any potential claims by J&J alleging infringement of any of the foreign counterparts to the '078 patent. If J&J is successful in asserting its rights under the '078 Patent and related foreign cases, J&J may prevent us from manufacturing or commercializing peginesatide, either for ourselves or with Takeda or any potential sublicensees, or obtain a royalties on sales of peginesatide until expiration of these patent rights in 2015. In addition, an adverse outcome could result in liability for damages, attorneys' fees and costs. If intellectual property in dispute that has been deemed to be jointly owned is broad enough to cover peginesatide, then under the laws applicable in certain relevant jurisdictions outside the U.S, joint ownership may not allow us to license third parties to manufacture and sell peginesatide or even to do
86
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
6. Commitments and Contingencies (Continued)
so ourselves, which may negatively affect our development and business plans outside the U.S. or our collaboration with Takeda.
From time to time, we are involved in legal proceedings arising in the ordinary course of business. We believe there is no other litigation pending that could have, individually or in the aggregate, a material adverse effect on our financial position, results of operations or cash flows.
7. Stockholder's Equity
Preferred Stock
Our Certificate of Incorporation, as amended and restated in December 2006, designates and authorizes 10,000,000 shares of $0.001 par value preferred stock, of which no shares are issued and outstanding as of December 31, 2010 and 2009. The rights, preferences and privileges of any preferred stock to be issued pursuant to our current Certificate of Incorporation, as amended and restated, have yet to be established.
No dividends on preferred stock have been declared since inception through December 31, 2010.
Common Stock
Our Certificate of Incorporation authorizes us to issue 100,000,000 shares of $0.001 par value common stock.
Warrants
As of December 31, 2010, a warrant to purchase 1,987 shares of our common stock, at an exercise price of $15.09 per share, and warrants to purchase an aggregate of 423,971 shares of common stock, at an exercise price of $16.78 per share, were issued and outstanding, the latter which was related to a private placement. The warrants contain provisions for the adjustment of the exercise price and the aggregate number of shares issuable upon the exercise of the warrants in the event of stock dividends, stock splits, reorganizations and reclassifications and consolidations. The first warrant expires in January 2012 and the latter warrants expire in March 2014.
Significant Equity Transactions
In March 2009, institutional investors purchased $42.0 million of our common stock in a private placement. The net proceeds were $41.6 million after offering expenses. Under the terms of one of two purchase agreements, we sold 2,844,708 newly issued shares of our common stock at a purchase price of $11.25 per share. In the other purchase agreement, we sold 652,262 newly issued units at a purchase price of $15.33 per unit, with each unit consisting of one share of common stock and one warrant to purchase 0.65 of a share of common stock. The warrants are exercisable at $16.78 per share and expire in March 2014.
In November 2009, we completed a public offering of 4,726,027 shares of our common stock, at a per share price of $18.25, which includes the full exercise of the underwriter's overallotment option of 616,438 shares. The net proceeds to us after deducting underwriting discounts and commissions and offering expenses were approximately $80.6 million.
87
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
7. Stockholder's Equity (Continued)
In September 2010, we entered into an amendment, or the Amendment, to the Common Stock Purchase Agreement with Azimuth Opportunity Ltd., or Azimuth, dated as of September 25, 2009. The original agreement provided that, upon the terms and subject to the conditions set forth in the purchase agreement, Azimuth is committed to purchase up to $60.0 million worth of shares of our common stock over the 24-month term of the purchase agreement, which was available to be drawn upon beginning January 2010. The Amendment extends the term of the equity facility to September 2012 and reduces the minimum threshold price we may establish at which, upon presentation to Azimuth of a draw down notice, Azimuth is required to purchase shares of our common stock. Our equity facility is subject to a number of conditions that limit our ability to draw against such facility.
In October 2010, we sold 999,061 shares of common stock to Azimuth under the Common Stock Purchase Agreement for an aggregate purchase price of $5.0 million. Our net proceeds from the sale of these shares was $4.9 million after deducting our offering expenses.
Equity Incentive Plans
2001 Stock Option/Stock Issuance Plan
In September 2001, we adopted the 2001 Stock Option/Stock Issuance Plan or the 2001 Plan. The 2001 Plan provides for both the granting of stock options and issuing shares of stock to our employees and consultants. Stock options granted under the 2001 Plan may be either incentive stock options or nonqualified stock options. Incentive stock options or ISOs, may be granted only to our employees. Nonqualified stock options or NSOs, may be granted to our employees, directors and consultants. Stock issued under the 2001 Plan may be issued to employees, directors and consultants. Stock options under the 2001 Plan may be granted for periods of up to 10 years and at prices no less than the fair market value for ISOs and 85% of the fair market value for NSOs, as determined by the Board of Directors. The exercise price of an ISO or NSO granted to a 10% stockholder shall not be less than 110% of the estimated fair value of the shares on the date of grant. To date, stock options granted generally become exercisable over four years. We issue new shares of common stock upon exercise of stock options.
2006 Equity Incentive Plan
Upon the effectiveness of our initial public offering in December 2006, we adopted the 2006 Equity Incentive Plan or the 2006 Plan. Shares of common stock issuable pursuant to all then outstanding stock awards granted under the 2001 Plan remained subject to the terms of the 2001 Plan and no additional stock awards were granted pursuant to the terms of the 2001 Plan upon the effective date of the 2006 Plan.
The 2006 Plan provides for both the granting of stock awards, including stock options and restricted stock units, to our employees, directors and consultants. Stock options granted under the 2006 Plan may be either ISOs or NSOs. ISOs may be granted only to our employees. NSOs may be granted to our employees, directors and consultants. Stock options under the 2006 Plan may be granted for periods of up to 10 years and at prices no less than the fair market value of our common stock on the date of grant. The exercise price of an ISO granted to a 10% stockholder shall not be less than 110% of the fair market value of our common stock on the date of grant. To date, stock options granted generally become exercisable over four years and do not allow for the early exercise of options prior to vesting. The terms of the restricted stock units granted by us to date provide for vesting and
88
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
7. Stockholder's Equity (Continued)
delivery of shares of common stock over three years. As of December 31, 2010 we reserved 4,363,133 shares of common stock for issuance under the 2006 Plan.
Under the 2006 Plan, we issue new shares of common stock upon exercise of stock options. The number of shares of common stock reserved for issuance automatically increases on January 1st of each year, from January 1, 2007 through January 1, 2016, by the lesser of (a) 4.5% of the total number of shares of common stock outstanding on December 31 of the preceding calendar year, or (b) 1,400,000 shares. The maximum number of shares that may be issued pursuant to the exercise of incentive stock options under the 2006 Plan is equal to the total share reserve, as increased from time to time pursuant to annual increases and shares subject to options granted pursuant to the 2001 Plan that have expired without being exercised in full.
There were 230,956, 1,465,659, and 1,263,981 total shares available for grant, combined, under the 2001 and 2006 Plans as of December 31, 2010, 2009, and 2008, respectively.
2006 Employee Stock Purchase Plan
Upon the effectiveness of the our initial public offering in December 2006, we adopted the 2006 Employee Stock Purchase Plan or the Purchase Plan. As of December 31, 2010 and 2009, we reserved a total of 445,902 and 326,557 shares of common stock, respectively, for issuance under the Purchase Plan. The share reserve automatically increases on January 1st of each year, from January 1, 2007 through January 1, 2016, by an amount equal to the lesser of (i) 0.5% of the total number of shares of common stock outstanding on December 31 of the preceding calendar year or (ii) 175,000 shares. We issue new shares of common stock in connection with purchases of common stock under the Purchase Plan. The Purchase Plan permits eligible employees to purchase common stock at a discount through payroll deductions during defined offering periods. The price at which the stock is purchased is equal to the lower of 85% of the fair market value of the common stock at the beginning of an offering period or at the end of a purchase period. For the year ended December 31, 2010 and 2009, 130,315 and 73,069 shares of common stock, respectively, were purchased under the Purchase Plan.
8. Stock-Based Compensation
We measure and recognize stock-based compensation expense related to employees and directors under the authoritative guidance for share-based payments.
Stock-based compensation was recorded in the Statements of Operations as follows (in thousands):
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Research and development |
$ | 4,521 | $ | 5,026 | $ | 4,035 | ||||
General and administrative |
7,002 | 6,146 | 5,548 | |||||||
|
$ | 11,523 | $ | 11,172 | $ | 9,583 | ||||
89
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
8. Stock-Based Compensation (Continued)
We granted the following stock options and restricted stock units to employees and directors as follows:
| |
Year Ended December 31, | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | ||||||||||||||||
| |
Number of Shares |
Weighted- Average Grant Date Fair Value Per Share |
Number of Shares | Weighted- Average Grant Date Fair Value Per Share |
Number of Shares | Weighted- Average Grant Date Fair Value Per Share |
|||||||||||||
Stock options |
1,999,999 | $ | 9.64 | 659,175 | $ | 8.83 | 222,650 | $ | 10.61 | ||||||||||
Restricted stock units |
460,158 | $ | 6.03 | — | — | 181,625 | $ | 14.20 | |||||||||||
As of December 31, 2010, there was unrecognized compensation cost of $20.0 million related to these stock options and restricted stock units. The unrecognized compensation cost as of December 31, 2010 is expected to be recognized over a weighted-average amortization period of 2.15 years.
Valuation assumptions and expense recognition
We estimate the fair value of employee and director stock options using the Black-Scholes valuation model. The fair value of employee and director stock options is amortized on a straight-line basis over the requisite service period of the awards. The fair value of employee and director stock options was estimated using the following weighted-average assumptions for the years ended December 31, 2010, 2009, and 2008:
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Expected volatility |
81 | % | 88 | % | 79 | % | ||||
Risk-free interest rate |
2.10 | % | 1.51 | % | 3.05 | % | ||||
Dividend yield |
0.00 | % | 0.00 | % | 0.00 | % | ||||
Expected term (in years) |
5.5 | 5.9 | 5.73 | |||||||
The expected term of stock options represents the average period the stock options are expected to remain outstanding and is based on the expected terms for industry peers as we did not have sufficient historical information to develop reasonable expectations about future exercise patterns and post-vesting employment termination behavior. The expected stock price volatility for our stock options for the years ended December 31, 2010, 2009, and 2008 was determined by examining the historical volatilities for industry peers and using an average of the historical volatilities of our industry peers as we did not have any significant trading history for our common stock. Industry peers consist of several public companies in the biopharmaceutical industry similar in size, stage of life cycle and financial leverage. We will continue to analyze the historical stock price volatility and expected term assumption as more historical data for our common stock becomes available. Use of our own historical data will become available at the end of 2011 when we will have five full years of history. The risk-free interest rate assumption is based on the U.S. Treasury instruments whose term was consistent with the expected term of our stock options. The expected dividend assumption is based on our history and expectation of dividend payouts.
90
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
8. Stock-Based Compensation (Continued)
We measured the fair value of restricted stock units using the closing price of our stock on the grant date. The fair value of restricted stock units is being amortized on a straight-line basis over the requisite service period of the awards.
We estimated the fair value of employee stock purchase rights granted under the Purchase Plan using the Black-Scholes valuation model. The weighted-average fair value of each stock purchase right for the years ended December 31, 2010, 2009, and 2008 was $3.64, $8.29 and $7.97 per share, respectively. The fair value of employee stock purchase rights is being amortized on a straight-line basis over the requisite service period of the purchase rights. The fair value of employee stock purchase rights were estimated using the following assumptions for the years ended December 31, 2010, 2009, and 2008:
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Expected volatility |
85% - 193% | 63% - 193% | 61% - 111% | |||||||
Risk-free interest rate |
0.16% - 1.44% | 0.17% - 4.67% | 1.07% - 4.83% | |||||||
Dividend yield |
0.00% | 0.00% | 0.00% | |||||||
Expected term (in months) |
6 - 24 | 6 - 24 | 6 - 24 | |||||||
There were no tax benefits related to employee stock-based compensation for the years ended December 31, 2010, 2009 and 2008.
91
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
8. Stock-Based Compensation (Continued)
Stock Option and Restricted Stock Unit Activity
The following table summarizes information about stock option and restricted stock unit activity for the three years ended December 31, 2010:
| |
Number of Shares |
Weighted-Average Price (Per Share)(1) |
Weighted-Average Remaining Contractual Term (in years) |
Aggregate Intrinsic Value (in thousands)(2) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Stock Options: |
||||||||||||||
Balances at December 31, 2007 |
2,099,116 | $ | 17.37 | |||||||||||
Granted |
222,650 | $ | 15.55 | |||||||||||
Exercised(3) |
(104,287 | ) | $ | 3.36 | ||||||||||
Forfeited |
(72,451 | ) | $ | 23.72 | ||||||||||
Cancelled |
(14,912 | ) | $ | 26.87 | ||||||||||
Balances at December 31, 2008 |
2,130,116 | $ | 17.59 | |||||||||||
Granted |
665,175 | $ | 12.11 | |||||||||||
Exercised(3) |
(212,424 | ) | $ | 3.36 | ||||||||||
Forfeited |
(137,252 | ) | $ | 19.63 | ||||||||||
Cancelled |
(15,208 | ) | $ | 29.34 | ||||||||||
Balances at December 31, 2009 |
2,430,407 | $ | 17.14 | |||||||||||
Granted |
2,008,999 | $ | 14.14 | |||||||||||
Exercised(3) |
(399,323 | ) | $ | 5.62 | ||||||||||
Forfeited |
(90,043 | ) | $ | 17.33 | ||||||||||
Cancelled |
(60,020 | ) | $ | 25.37 | ||||||||||
Balances at December 31, 2010 |
3,890,020 | $ | 16.65 | 7.97 | $ | 1,279 | ||||||||
Options exercisable at December 31, 2010 |
1,727,697 | $ | 20.20 | 6.64 | $ | 680 | ||||||||
Restricted Stock Units: |
||||||||||||||
Balances at December 31, 2007 |
30,650 | $ | 21.74 | |||||||||||
Granted (time-based) |
181,625 | $ | 14.20 | |||||||||||
Vested |
— | — | ||||||||||||
Forfeited |
(23,325 | ) | $ | 18.12 | ||||||||||
Balances at December 31, 2008 |
188,950 | $ | 14.94 | |||||||||||
Granted (time-based) |
— | — | ||||||||||||
Vested |
(56,395 | ) | $ | 15.49 | ||||||||||
Forfeited |
(25,486 | ) | $ | 14.49 | ||||||||||
Balances at December 31, 2009 |
107,069 | $ | 14.76 | |||||||||||
Granted (time-based) |
235,158 | $ | 6.23 | |||||||||||
Granted (performance-based)(4) |
225,000 | $ | 5.83 | |||||||||||
Vested |
(53,544 | ) | $ | 24.24 | ||||||||||
Forfeited |
(10,282 | ) | $ | 7.97 | ||||||||||
Balances at December 31, 2010 |
503,401 | $ | 5.91 | 1.50 | $ | 280 | ||||||||
- (1)
- The weighted average price per share is determined using exercise price per share for stock options and fair value per share on transaction date for restricted stock units.
92
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
8. Stock-Based Compensation (Continued)
- (2)
- The
aggregate intrinsic value is calculated as
- •
- For options: the difference between the exercise price of the option and the fair value of our common stock for
in-the-money options at December 31, 2010.
- •
- For restricted stock units: the difference between the grant date fair value of the unit and the fair value of our common
stock for in-the-money units at December 31, 2010.
- (3)
- The
total intrinsic value of stock options exercised was $6.9 million, $4.2 million and $1.5 million during the years ended
December 31, 2010, 2009, and 2008, respectively, and was determined at the date of each exercise.
- (4)
- During 2010, the Board of Directors approved the grant of 225,000 performance-based restricted stock units to certain executive officers. These units vest 50% upon FDA approval of our NDA for peginesatide and 50% upon product launch of peginesatide.
The stock options outstanding and exercisable by exercise price at December 31, 2010 are as follows:
| |
Stock Options Outstanding | Stock Options Exercisable | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Range of Exercise Prices
|
Number of Shares | Weighted-Average Remaining Contractual Life in Years |
Weighted-Average Exercise Price Per Share |
Number of Shares | Weighted-Average Exercise Price Per Share |
|||||||||||
$ 0.80 - 6.65 |
1,251,393 | 8.85 | $ | 5.63 | 269,531 | $ | 4.13 | |||||||||
$10.06 - 19.92 |
887,766 | 7.24 | $ | 14.19 | 551,012 | $ | 15.43 | |||||||||
$20.22 - 25.91 |
1,356,711 | 8.18 | $ | 23.45 | 523,355 | $ | 23.57 | |||||||||
$30.27 - 36.43 |
394,150 | 6.06 | $ | 33.73 | 383,799 | $ | 33.75 | |||||||||
|
3,890,020 | 1,727,697 | ||||||||||||||
Deferred Stock-Based Compensation
In September 2003, we approved the repricing of existing employee stock options from $4.00 to $0.80 per share, which was deemed to be the fair market value. As a result of the repricing, stock options are subject to variable accounting. At December 31, 2010, the fair value of the common stock was $6.65 per share and approximately 21,000 repriced stock options remained outstanding. During the years ended December 31, 2010, 2009, and 2008, we have recorded deferred stock-based compensation (benefit) related to these stock options of $(379,000), $443,000 and $(402,000) and, respectively, and recorded stock-based compensation (income) expense of $(379,000), $443,000 and $(402,000), respectively.
During the year ended December 31, 2005, we issued stock options to certain employees under the Plan with exercise prices below the fair value of the our common stock at the date of grant. We estimated the fair value of our common stock based upon several factors, including progress and milestones attained in our business. In accordance with the requirements of the authoritative guidance on accounting for stock issued to employees, we recorded deferred stock-based compensation for the difference between the exercise price of the stock option and the fair value of our stock at the date of grant. This deferred stock-based compensation is amortized to expense on a straight-line basis over the period during which the options vest, generally four years. During the year ended December 31, 2005,
93
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
8. Stock-Based Compensation (Continued)
we recorded deferred stock-based compensation related to these stock options of $195,000, net of cancellations, and recorded amortization of such deferred stock-based compensation of $4,000 and $24,000, respectively, during the years ended December 31, 2009, and 2008. All such deferred stock-based compensation expense was fully amortized as of December 31, 2009.
Nonemployee Stock-Based Compensation
Stock-based compensation expense related to stock options granted and common stock issued to nonemployees is recognized as the stock options are earned. We believe that the estimated fair value of the stock options is more readily measurable than the fair value of the services received. The fair value of stock options granted to nonemployees is calculated at each grant date and remeasured at each reporting date. The stock-based compensation expense related to a grant will fluctuate as the fair value of our common stock fluctuates over the period from the grant date to the vesting date. We recorded nonemployee stock-based compensation (benefit) expense of $(291,000), $876,000 and $671,000, respectively, for the years ended December 31, 2010, 2009, and 2008.
9. Development and Commercialization Agreements with Takeda
We entered into two separate collaboration agreements with Takeda, which have been combined for accounting purposes due to their proximity of negotiation. Consideration from these collaboration agreements includes nonrefundable upfront license fees, reimbursement for sales of active pharmaceutical ingredients, clinical and regulatory milestone payments, reimbursement of third party U.S. clinical development expenses, product profit share revenues (as co-promotion revenues) and royalties.
In February 2006, we granted an exclusive license to Takeda for development and commercialization of peginesatide in Japan. Pursuant to this agreement, Takeda has paid us approximately $42 million to date, consisting of $17 million in upfront licensing fees, approximately $10 million for the purchase of equity, a $10 million cash milestone payment for the completion of the first Phase 1 trial of peginesatide in Japan, and in March 2010, a $5 million cash milestone payment for the initiation of Phase 3 trial of peginesatide in Japan. Upon Takeda's successful achievement of clinical development and regulatory milestones, we are eligible to receive from Takeda an additional aggregate of $33 million relating to the renal program. Takeda is responsible for all development and commercialization costs in Japan and will purchase the API for peginesatide from us. Assuming peginesatide is approved and launched in Japan, we will receive a royalty from Takeda on peginesatide sales in Japan.
In June 2006, the parties expanded their collaboration to develop and commercialize peginesatide worldwide, which includes the co-development and co-commercialization of peginesatide in the U.S. Takeda received an exclusive license to develop and commercialize peginesatide outside of the U.S. Beginning January 1, 2007, Takeda was responsible for the first $50 million of third party expenses related to development in pursuit of U.S. regulatory approval of peginesatide, which was fully utilized by both parties through the first quarter of 2008. Thereafter, Takeda has borne 70% of the third party U.S. development expenses while we have been responsible for 30% of third party expenses. We retain responsibility for 100% of our internal development expenses, most notably employee-related expenses. In addition, third party expenses related to the commercialization of peginesatide in the U.S. are equally shared by both parties and beginning in mid-2011, certain employee-related expenses supporting
94
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
9. Development and Commercialization Agreements with Takeda (Continued)
commercialization will also be equally shared. Takeda will have primary responsibility and bear all costs for peginesatide clinical development in support of regulatory approval for all territories outside the U.S. Under the June 2006 agreement, Takeda paid us an upfront license fee of $105 million, and we have received milestone payments upon completion of database lock of the Phase 3 clinical trials of $30 million for dialysis and non-dialysis. Upon the successful achievement of clinical development and regulatory milestones, we are eligible to receive from Takeda an additional aggregate of $85 million relating to the renal program, including $10 million milestone payments upon FDA acceptance of the submission of the new drug application, or NDA, and $50 million of milestone payments upon approval by the FDA in dialysis indications. We and Takeda will share equally in the net profits and losses of peginesatide in the U.S., which include expenses related to the marketing and launch of peginesatide. Takeda will pay us a variable royalty based on annual net sales of peginesatide outside the U.S.. The agreement establishes a joint steering committee to oversee the development, regulatory approval and commercialization of peginesatide.
We share responsibility with Takeda for clinical development activities required for U.S. regulatory approval of peginesatide. Specifically, we have primary responsibility for peginesatide's clinical development plan and clinical trials in the dialysis indication, and the non-dialysis indication to the extent of any further development, while Takeda has primary responsibility in the chemotherapy induced anemia and anemia of cancer indications to the extent any such indication is developed. We and Takeda have agreed to suspend the development of peginesatide to treat chemotherapy-induced anemia and to focus all development efforts for peginesatide on the treatment of chronic renal failure anemia. We are responsible for U.S. regulatory filings in the dialysis, non-dialysis, chemotherapy induced anemia and anemia of cancer indications, including holding the NDAs for those indications. Takeda is responsible for regulatory filings outside the U.S. and the creation of a global safety database.
We are also responsible for the manufacture and supply of all quantities of API to be used in the development and commercialization of peginesatide worldwide. Takeda is responsible for the fill and finish steps in the manufacture of peginesatide worldwide.
The parties have agreed to jointly develop the initial commercial marketing plan for peginesatide in the U.S. pursuant to which we and Takeda will divide peginesatide promotional responsibilities in the U.S. We and Takeda will jointly decide on promotional responsibility for markets outside of these initial indications if any.
Under the February 2006 agreement, Takeda also obtained a right of first negotiation to any backup products for peginesatide developed by us or our third party partners. Specifically, during the first ten years of the agreement, if we or third party partners develop a product that advances to Phase 2 clinical trials and competes with peginesatide in the renal or oncology indications, we are obligated to offer to Takeda the right to develop and commercialize such product in Japan before offering the product opportunity in Japan to any other third party.
We recognized $112.5 million, $114.9 million and $82.2 million of collaboration revenue under the CAPM revenue recognition model during the years ended December 31, 2010, 2009 and 2008, respectively, which includes a $1.4 million cumulative adjustment resulting from an amendment to the Arrangement with Takeda that was effective on January 1, 2008. During the year ended December 31, 2010 we recorded a $12.1 million change in estimate to our clinical trial accruals as a result of our analysis of the final monitored site visit data for our Phase 3 clinical trials. As this change in estimate
95
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
9. Development and Commercialization Agreements with Takeda (Continued)
was comprised of development costs charged to Takeda at a 70% reimbursement rate, this amount is now a payable due back to Takeda. The reimbursement received from Takeda in prior periods was recorded as deferred revenue and collaboration revenue under our CAPM revenue recognition model. This change in estimate resulted in a reduction of $8.4 million of deferred revenue and a reversal of $7.8 million of collaboration revenue. The net impact to our statement of operations for this change in estimate was a $4.3 million decrease to our net loss or $0.18 per share for the year ended December 31, 2010.
The corresponding $8.4 million gross payable to Takeda as of December 31, 2010 was partially offset by the $2.5 million receivable due from Takeda for reimbursement of development and commercial expenses in the fourth quarter of 2010. The resulting net payable of $6.0 million is reflected on the accompanying balance sheet under the caption Payable to Takeda.
The amount due from Takeda as of December 31, 2009 was $18.6 million as disclosed in the balance sheet.
10. Income Taxes
The components of the provision for income taxes are as follows (in thousands):
| |
Year Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||||
Provision for income taxes: |
||||||||||||
Current provision for income taxes: |
||||||||||||
Federal |
$ | — | $ | (1,412 | ) | $ | (2,950 | ) | ||||
State |
1 | 1 | 390 | |||||||||
Total current provision for income taxes |
1 | (1,411 | ) | (2,560 | ) | |||||||
Deferred tax benefit: |
||||||||||||
Federal |
— | — | 2,842 | |||||||||
State |
— | — | — | |||||||||
Total deferred tax benefit |
— | — | 2,842 | |||||||||
Provision for income taxes |
$ | 1 | $ | (1,411 | ) | $ | 282 | |||||
We recorded a provision for minimum statutory state tax and provided no federal tax as a result of our net operating loss for the year ended December 31, 2010.
We recorded a benefit for income taxes for the year ended December 31, 2009 of $1.4 million, consisting largely of a federal tax benefit that primarily resulted from of the Worker, Homeownership and Business Assistance Act of 2009 enacted in November 2009, which allowed us to carryback our 2008 net operating loss to 2007 and recover $1.3 million in alternative minimum taxes previously paid for the year ended December 31, 2007. We also recorded a $100,000 federal benefit related to refundable R&D credits available to us pursuant to a provision within the Housing Assistance Tax Act of 2008, which was signed into law in July 2008.
96
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
10. Income Taxes (Continued)
We recorded a provision for income taxes for the year ended December 31, 2008 of $282,000 consisting of $107,000 of federal tax benefit and $389,000 of net California state income tax expense. The $107,000 of federal tax benefit was primarily due to refundable R&D credits available pursuant to a provision within the Housing Assistance Tax Act of 2008, which was signed into law in July 2008. The California state income tax expense of $389,000 was primarily related to an out of period reduction to our California R&D credits that was partially offset by additional California R&D credits that were identified.
We incurred significant operating losses since inception and anticipates that we will incur continued losses for the foreseeable future
A reconciliation of the federal statutory income tax rate to our effective income tax rate is as follows:
| |
Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Federal statutory income tax rate |
(35.00 | )% | (35.00 | )% | (35.00 | )% | ||||
State income taxes, net of federal benefit |
0.01 | — | 0.45 | |||||||
Stock-based compensation expense |
9.61 | 0.84 | 1.40 | |||||||
Change in valuation allowance |
37.17 | 33.76 | 34.57 | |||||||
Change in federal rates and prior year true ups |
(0.32 | ) | 0.64 | 0.51 | ||||||
Permanent differences true ups |
0.11 | 0.03 | 0.05 | |||||||
Tax credits |
(11.58 | ) | (2.09 | ) | (1.66 | ) | ||||
Other |
— | — | — | |||||||
Provision for income taxes |
0.00 | % | (1.82 | )% | 0.32 | % | ||||
Deferred tax assets consist of the following (in thousands):
| |
December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | ||||||
Net operating loss carryforwards |
$ | 118,654 | $ | 89,734 | ||||
Federal and State credit carryforwards |
12,233 | 9,753 | ||||||
Depreciation and amortization |
20,147 | 24,308 | ||||||
Capitalized start up costs |
1,481 | 3,392 | ||||||
Accrued liabilities and allowances |
20,094 | 39,005 | ||||||
Gross deferred tax assets |
172,609 | 166,192 | ||||||
Deferred tax liability |
(216 | ) | (494 | ) | ||||
Net deferred tax asset |
172,393 | 165,698 | ||||||
Less: Valuation allowance |
(165,153 | ) | (158,458 | ) | ||||
Net deferred tax assets |
$ | 7,240 | $ | 7,240 | ||||
Management establishes a valuation allowance for those deductible temporary differences when it is more likely than not that some or all of the benefit of such deferred tax assets will not be recognized. The ultimate realization of deferred tax assets is dependent upon our ability to generate taxable income during the periods in which the temporary differences are deductible. Management
97
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
10. Income Taxes (Continued)
considers the historical level of taxable income, projections for future taxable income, taxable income in carryback years and tax planning strategies in making this assessment. Management's assessment in the near term is subject to change if estimates of future taxable income during the carryforward period are increased. The valuation allowance increased $6.7 million, $31.5 million and $33.6 million during the years ended December 31, 2010, 2009, and 2008, respectively. As of December 31, 2010 and 2009, we have a net deferred tax asset balance of $7.2 million each in consideration of the uncertainty in income taxes liability recorded for the same amount.
We considered the following positive and negative factors in determining that it was more likely than not that the $7.2 million of the net deferred tax asset as of December 31, 2010 and 2009 would be realized:
- •
- Net deductible temporary differences that were expected to reverse in 2010 and 2011.
- •
- There were no relevant tax strategies available that we would consider feasible.
- •
- Uncertainties, such as regulatory approval of peginesatide and binding arbitration and litigation with certain subsidiaries of J&J, that if unfavorably resolved, would adversely affect our future operations.
At December 31, 2010, we had federal and state net operating loss carryforwards of $275 million and $291 million, respectively. The federal net operating loss carryforwards begin to expire in 2028 and state net operating loss carryforwards begin to expire in 2018, if not utilized.
At December 31, 2010, we had federal and state research credit carryforwards of $9.8 million and $8.4 million, respectively. If not utilized, the federal carryforward will expire in various amounts beginning in 2021. The California credit can be carried forward indefinitely.
We experienced an ownership change as defined by Sections 382 and 383 of the Internal Revenue Code which establishes an annual limit on the deductibility of pre-ownership change net operating loss and credit carryforwards that existed on December 15, 2006.
At December 31, 2010 and 2009, our liability for uncertain income tax positions was $10.2 million and $10.1 million, respectively, which is reflected as long-term income tax liabilities on our balance sheet. Our policy is to include penalties and interest expense related to income taxes as a component of other expense and interest expense, respectively, as necessary. For the years ended December 31, 2010, 2009 and 2008, we recognized $140,000, $105,000 and $596,000, respectively, of interest expense related to our liability for uncertain income tax positions. As of December 31, 2010 and December 31, 2009, we had accrued $842,000 and $702,000, respectively, of interest expense related to our liability for uncertain income tax positions as of December 31, 2010 and 2009. For the years ended December 31, 2010 and 2009, there were no penalties related to uncertain income tax positions. For the year ended December 31, 2008, $81,000 of penalties related to uncertain income tax positions were required and recognized.
Effective January 1, 2007, we adopted the provisions of the uncertainty in income taxes, which prescribes a comprehensive model for how we should recognize, measure, present and disclose in our financial statements uncertain tax positions that we have taken or expects to take on a tax return. We had $13.1 million, $12.4 million and $11.8 million of unrecognized tax benefits as of December 31, 2010, 2009, and 2008, respectively.
98
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
10. Income Taxes (Continued)
As of December 31, 2010, $5.9 million of the unrecognized tax benefits would affect our income tax provision and effective tax rate if recognized. However, as we would currently need to increase the valuation allowance for any additional amounts benefited, the effective tax rate would not be impacted until the valuation allowance was removed.
A reconciliation of the unrecognized tax benefits for the years ended December 31, 2010, 2009, and 2008 is as follows (in thousands):
| |
December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| |
2010 | 2009 | 2008 | |||||||
Balance at beginning of year |
$ | 12,366 | $ | 11,770 | $ | 10,708 | ||||
Additions for current year tax positions |
734 | 759 | — | |||||||
Additions for prior year tax positions |
— | — | 1,438 | |||||||
Reductions for prior year tax positions |
— | (163 | ) | (376 | ) | |||||
Balance at end of year |
$ | 13,100 | $ | 12,366 | $ | 11,770 | ||||
We file federal and California income tax returns. For U.S. federal and California income tax purposes, the statute of limitation currently remains open for the years ending December 31, 2007 to present and December 31, 2006 to present, respectively, primarily due to carryforward of net operating losses and R&D credits generated in prior years. There are no tax years under examination by any jurisdiction at this time.
11. Retirement Savings Plan
We have a retirement savings plan, commonly known as a 401(k) plan, that allows all full time employees to contribute from 1% to 50% of their salary, subject to IRS limits. Beginning in 2008, we made matching contributions equal to 50% of the employee deferral contributions during the fiscal year up to $4,000. Employees who met the period of service requirement minimum of 500 hours and remained employed on the last day of the fiscal year were eligible for the matching contribution. Our contributions to the 401(k) plan were $460,000, $453,000 and $423,000 for the years ended December 31, 2010, 2009, and 2008, respectively.
99
AFFYMAX, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
12. Quarterly Financial Data (unaudited)
The following tables summarize the unaudited quarterly financial data for the last two fiscal years (in thousands, except per share data):
| |
2010 Quarter Ended | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31, | June 30, | September 30, | December 31, | |||||||||||
Collaboration revenue |
$ | 34,646 | $ | 54,341 | $ | 16,784 | $ | 6,732 | |||||||
Total revenue |
34,650 | 54,346 | 16,790 | 6,735 | |||||||||||
Income (loss) from operations |
(7,862 | ) | 17,265 | (12,109 | ) | (11,742 | ) | ||||||||
Net income (loss) |
(7,866 | ) | 17,312 | (12,030 | ) | (11,491 | ) | ||||||||
Basic net income (loss) per common share |
$ | (0.33 | ) | $ | 0.71 | $ | (0.49 | ) | $ | (0.45 | ) | ||||
Diluted net income (loss) per common share |
$ | (0.33 | ) | $ | 0.70 | $ | (0.49 | ) | $ | (0.45 | ) | ||||
Weighted-average number of common shares used in computing basic and diluted net income (loss) per common share |
|||||||||||||||
Basic |
23,932 | 24,219 | 24,369 | 25,274 | |||||||||||
Diluted |
23,932 | 24,736 | 24,369 | 25,274 | |||||||||||
As a result of finalizing amendments for clinical trial activities completed in 2009, our fourth quarter of 2010 includes adjustments relating to estimates previously recorded in our expenses for the years ended December 31, 2008 and 2009. The adjustments decreased clinical trial expense by $12.1 million. As this change in estimate was comprised of development costs charged to Takeda at a 70% reimbursement rate, this amount is now a payable due back to Takeda. The reimbursement received from Takeda in prior periods was recorded as deferred revenue and collaboration revenue under our CAPM revenue recognition model. This change in estimate resulted in a reduction of $8.4 million of deferred revenue and a reversal of $7.8 million of collaboration revenue. The net impact to our statement of operations for this change in estimate was a $4.3 million decrease to our net loss or $0.18 per share for the year ended December 31, 2010.
| |
2009 Quarter Ended | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
March 31, | June 30, | September 30, | December 31, | |||||||||
Collaboration revenue |
$ | 25,849 | $ | 26,918 | $ | 29,157 | $ | 32,959 | |||||
Total revenue |
25,853 | 26,923 | 29,161 | 32,962 | |||||||||
Loss from operations |
(22,011 | ) | (22,544 | ) | (18,733 | ) | (15,654 | ) | |||||
Net loss |
(21,740 | ) | (22,092 | ) | (18,382 | ) | (14,317 | ) | |||||
Basic and diluted net loss per common share |
$ | (1.32 | ) | $ | (1.17 | ) | $ | (0.97 | ) | $ | (0.68 | ) | |
Weighted-average number of common shares used in computing basic and diluted net loss per common share calculations |
16,488 | 18,894 | 18,951 | 21,076 | |||||||||
100
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
Not applicable.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
An evaluation was performed by our Chief Executive Officer and Chief Financial Officer of the effectiveness of the design and operation of our disclosure controls and procedures as defined in the Rules 13(a)-15(e) of the Securities Exchange Act of 1934, as amended or the Exchange Act. Disclosure controls and procedures are those controls and procedures designed to provide reasonable assurance that the information required to be disclosed in our Exchange Act filings is (1) recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission's rules and forms, and (2) accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2010, our disclosure controls and procedures were effective.
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our procedures or our internal controls will prevent or detect all error and all fraud. An internal control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of our controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected.
Management determined that, as of December 31, 2010, there were no changes in our internal control over financial reporting that occurred during the fiscal quarter then ended that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management's Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission or COSO in Internal Control—Integrated Framework. Our management has concluded that, as of December 31, 2010, our internal control over financial reporting was effective based on these criteria.
Ernst & Young LLP, an independent registered public accounting firm, has audited out financial statements included herein and has issued and audit report on the effectiveness of our internal control over financial reporting, which report is included below.
101
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The
Board of Directors and Stockholders
Affymax, Inc.
We have audited Affymax Inc.'s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Affymax's management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Affymax Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2010 financial statement of Affymax, Inc. and our report dated March 10, 2011 expressed an unqualified opinion thereon.
| /s/ Ernst & Young LLP | ||
Palo Alto, CA March 10, 2011 |
102
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2010 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
103
Certain information required by Part III is omitted from this Annual Report on Form 10-K because we intend to file our definitive proxy statement for our 2011 annual meeting of stockholders, pursuant to Regulation 14A of the Exchange Act, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information to be included in the proxy statement is incorporated herein by reference.
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this item with respect to our executive officers may be found under the section, "Executive Officers and Key Employees" appearing in our proxy statement for our 2011 annual meeting of stockholders and is incorporated herein by reference. The information required by this item relating to our directors and nominees, including information with respect to audit committee financial experts, may be found under the section entitled "Proposal 1—Election of Directors" appearing in the proxy statement for our 2011 annual meeting of stockholders and is incorporated herein by reference. Information regarding compliance with Section 16(a) of the Exchange Act may be found under the section entitled "Section 16(a) Beneficial Ownership Reporting Compliance" appearing in our proxy statement for our 2011 annual meeting of stockholders and is incorporated herein by reference.
In 2006, we adopted a code of ethics that applies to our employees, officers and directors and incorporates guidelines designed to deter wrongdoing and to promote the honest and ethical conduct and compliance with applicable laws and regulations. In addition, the code of ethics incorporates our guidelines pertaining to topics such as conflicts of interest and workplace behavior. We have posted the text of our code of ethics on our website at www.affymax.com in connection with "Investor Relations/Corporate Governance" materials. In addition, we intend to promptly disclose (1) the nature of any amendment to our code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (2) the nature of any waiver, including an implicit waiver, from a provision of our code of ethics that is granted to one of these specified officers, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
Item 11. Executive Compensation.
The information required by this item concerning director and executive compensation is included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled "Executive Compensation" and is incorporated herein by reference. The information required by this item concerning Compensation Committee interlocks and insider participation is included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled "Compensation Committee Interlocks and Insider Participation" and is incorporated herein by reference. The information required by this item concerning our Compensation Committee's review and discussion of our Compensation Discussion and Analysis is included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled "Compensation Committee Report" and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item with respect to securities authorized for issuance under our equity compensation plans is included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled "Securities Authorized for Issuance under Equity Compensation Plans" and is incorporated herein by reference. The information required by this item relating to
104
security ownership of certain beneficial owners and management is included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled "Security Ownership of Certain Beneficial Owners and Management" and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions and Director Independence.
The information required by this item is incorporated herein by reference to the information included in our proxy statement for our 2011 annual meeting of stockholders under the sections entitled "Information Regarding The Board of Directors and Corporate Governance" and "Transactions With Related Persons."
Item 14. Principal Accountant Fees and Services.
The information required by this item is incorporated herein by reference to the information included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled "Proposal 2—Ratification of Selection of Independent Registered Public Accounting Firm."
105
Item 15. Exhibits and Financial Statement Schedules.
- (a)
- The
following documents are filed as part of this Form 10-K:
- (1)
- Financial
Statements (included in Part II of this report):
- •
- Report of Ernst & Young LLP, Independent Registered Public Accounting Firm
- •
- Balance Sheets
- •
- Statements of Operations
- •
- Statements of Stockholders' Equity
- •
- Statements of Cash Flows
- •
- Notes to Financial Statements
- (2)
- Financial Statement Schedules
All other financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
The following exhibits are included herein or incorporated herein by reference:
| 3.3 | Amended and Restated Certificate of Incorporation(1) | |
3.5 |
Amended and Restated Bylaws(2) |
|
4.1 |
Reference is made to exhibits 3.3 and 3.5 |
|
4.2 |
Specimen Common Stock Certificate(1) |
|
4.3 |
Warrant to purchase shares of Series C Preferred Stock(1) |
|
4.4 |
Amended and Restated Investor Rights Agreement, dated September 7, 2006, by and between the Registrant and certain of its stockholders(1) |
|
4.5 |
Form of Warrant to Purchase shares of Common Stock(6) |
|
10.1 |
+ |
Form of Indemnity Agreement for Directors and Executive Officers(1) |
10.2 |
+ |
2001 Stock Option/Stock Issuance Plan(1) |
10.3 |
+ |
Form of Notice of Grant of Stock Option, Form of Stock Option Agreement and Form of Stock Purchase Agreement under 2001 Stock Option/Stock Issuance Plan(1) |
10.4 |
+ |
Form of Stock Issuance Agreement under 2001 Stock Option/Stock Issuance Agreement(1) |
10.5 |
+ |
Amended and Restated 2006 Equity Incentive Plan(9) |
10.6 |
+ |
Form of Option Grant Notice and Form of Option Agreement under 2006 Equity Incentive Plan(1) |
10.7 |
+ |
2006 Employee Stock Purchase Plan(1) |
10.8 |
+ |
Form of Offering Document under 2006 Employee Stock Purchase Plan(1) |
10.9 |
+ |
Form of Restricted Stock Unit Notice and Form of Restricted Stock Unit under 2006 Equity Incentive Plan(5) |
10.10 |
+ |
Employment Agreement, dated December 17, 2008, by and between the Registrant and Arlene M. Morris(7) |
106
| 10.11 | + | Executive Employment Agreement, dated December 17, 2008, by and between the Registrant and Paul B. Cleveland(7) |
10.12 |
+ |
Executive Employment Agreement, dated December 17, 2008, by and between the Registrant and Steven Love(7) |
10.13 |
+ |
Summary of Non-Employee Director Compensation Program(7) |
10.14 |
Research and Development/Office Lease, dated May 30, 1990, by and between Miranda Associates and Affymax Research Institute(1) |
|
10.15 |
First Amendment to Lease, dated November 16, 1999, by and between Spieker Properties, L.P., successor in interest to Miranda Associates, and Affymax Research Institute(1) |
|
10.16 |
Second Amendment to Lease, dated December 20, 1999, by and between Spieker Properties, L.P. and Affymax Research Institute(1) |
|
10.17 |
Third Amendment, dated December 31, 2001, by and between EOP-Foothill Research Center, L.L.C., successor by merger to Spieker Properties L.P., and the Registrant(1) |
|
10.18 |
* |
EPO Receptor License Agreement, dated September 5, 1996, by and between the Registrant and Genetics Institute, Inc.(1) |
10.19 |
* |
License Agreement, dated July 27, 2001, by and between the Registrant, Glaxo Group Limited, SmithKline Beecham Corporation, Affymax N.V., Affymax Research Institute and Affymax Technologies N.V.(1) |
10.20 |
* |
License, Manufacturing, and Supply Agreement, dated April 8, 2004, by and between the Registrant and Nektar Therapeutics AL, Corporation(1) |
10.21 |
* |
Collaboration and License Agreement, dated February 13, 2006, by and between the Registrant and Takeda Pharmaceutical Company Limited(1) |
10.22 |
* |
Collaboration and License Agreement, dated June 27, 2006, by and between the Registrant and Takeda Pharmaceutical Company Limited(1) |
10.23 |
Research and Development Agreement, dated April 2, 1992, by and between the Registrant and The R.W. Johnson Pharmaceutical Research Institute(1) |
|
10.24 |
Sublease Agreement, dated September 1, 2006, by and between the Registrant and TIBCO Software Inc.(1) |
|
10.25 |
First Amendment to Collaboration and License Agreement, dated April 1, 2007, by and between Registrant and Takeda Pharmaceutical Company Limited(3) |
|
10.26 |
Fourth Amendment to Lease, dated November 30, 2006, by and between Registrant and CA-Foothill Research Center L.P.(4) |
|
10.27 |
Second Amendment to Collaboration and License Agreements between Registrant and Takeda Pharmaceutical Company Limited effective January 1, 2008(5) |
|
10.28 |
Securities Purchase Agreement to purchase shares of Common Stock dated February 13, 2009 by and among Registrant and the purchasers identified on the signature pages thereto(6) |
|
10.29 |
Securities Purchase Agreement to purchase shares of Common Stock and Warrants to purchase shares of Common Stock dated February 13, 2009 by and among Registrant and the purchasers identified on the signature pages thereto(6) |
|
10.30 |
+ |
Executive Employment Agreement, dated December 17, 2008 by and between the Registrant and Anne-Marie Duliege(7) |
107
| 10.31 | + | Executive Employment Agreement, dated December 17, 2008 by and between the Registrant and Robert Venteicher(7) |
10.32 |
Common Stock Purchase Agreement, dated September 25, 2009 by and between the Registrant and Azimuth Opportunity Ltd.(8) |
|
10.33 |
Form of Credit Line and related documentation effective as of December 8, 2009 by and between the Registrant and UBS Financial Services, Inc.(9) |
|
10.34 |
+ |
Executive Employment Agreement, dated February 19, 2010, by and between the Registrant and John A. Orwin.(10) |
10.35 |
Fifth Amendment, dated May 20, 2010, by and between the Registrant and EOP-Foothill Research Center, L.L.C.(11) |
|
10.36 |
Amendment No. 1 to Common Stock Purchase Agreement, dated September 17, 2010, between the Registrant and Azimuth Opportunity Ltd.(12) |
|
10.37 |
+ |
Amendment to Employment Agreement between the Registrant and Arlene M. Morris effective as of September 23, 2010.(13) |
10.38 |
+ |
Amendment to Employment Agreement between the Registrant and John A. Orwin effective as of September 23, 2010.(13) |
10.39 |
+ |
Amendment to Employment Agreement between the Registrant and Paul B. Cleveland effective as of September 23, 2010.(13) |
10.40 |
+ |
Amendment to Employment Agreement between the Registrant and Anne-Marie Duliege effective as of September 23, 2010.(13) |
10.41 |
+ |
Executive Employment Agreement, dated February 19, 2010, by and between the Registrant and John A. Orwin.(10) |
10.42 |
+ |
Executive Employment Agreement, dated March 4, 2011, by and between the Registrant and Herb Cross. |
10.43 |
Sixth Amendment to Lease, dated December 21, 2010 by and between Registrant and CA-Foothill Research Center L.P. |
|
23.1 |
Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm |
|
24.1 |
Power of Attorney. Reference is made to the signature page |
|
31.1 |
Certification required by Rule 13a-14(a) or Rule 15d-14(a) |
|
31.2 |
Certification required by Rule 13a-14(a) or Rule 15d-14(a) |
|
32.1 |
† |
Certification required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. 1350) |
- (1)
- Incorporated
by reference to the indicated exhibit of our registration statement on Form S-1, registration
no. 333-136125, declared effective by the Securities and Exchange Commission on December 14, 2006.
- (2)
- Incorporated
by reference to the indicated exhibit in our Form 8-K as filed with the Securities and Exchange Commission on
September 10, 2007.
- (3)
- Incorporated by reference to the indicated exhibit in our Form 10-Q for the quarter ended June 30, 2007 as filed with the Securities and Exchange Commission.
108
- (4)
- Incorporated
by reference to the indicated exhibit in our Form 10-K for the year ended December 31, 2006 as filed with the
Securities and Exchange Commission.
- (5)
- Incorporated
by reference to the indicated exhibit in our Form 10-K for the year ended December 31, 2007 as filed with the
Securities and Exchange Commission.
- (6)
- Incorporated
by reference to the indicated exhibit in our Form 8-K as filed with the Securities and Exchange Commission on
February 19, 2009.
- (7)
- Incorporated
by reference to the indicated exhibit in our Form 10-K for the year ended December 31, 2008 as filed with the
Securities and Exchange Commission.
- (8)
- Incorporated
by reference to the indicated exhibit in our Form 8-K as filed with the Securities and Exchange Commission on
September 25, 2009.
- (9)
- Incorporated
by reference to the indicated exhibit in our Form 10-K, as filed with the Securities and Exchange Commission on
March 4, 2010.
- (10)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q, as filed with the Securities and Exchange Commission on
May 6, 2010.
- (11)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q, as filed with the Securities and Exchange Commission on
August 5, 2010.
- (12)
- Incorporated
by reference to the indicated exhibit in our Form 8-K, as filed with the Securities and Exchange Commission on
September 20, 2010.
- (13)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q, as filed with the Securities and Exchange Commission on
November 5, 2010.
- +
- Indicates
management contract or compensatory plan.
- *
- Confidential
treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities
and Exchange Commission.
- †
- The certification attached as Exhibit 32.1 accompany this Annual Report on Form 10-K, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of Affymax, Inc., under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K, irrespective of any general incorporation language contained in such filing.
109
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| AFFYMAX, INC. | ||||
By: |
/s/ JOHN A. ORWIN John Orwin Chief Executive Officer and Member of the Board of Directors |
|||
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints John Orwin and Herb Cross, and each of them, as his true and lawful attorneys-in-fact and agents, each with the full power of substitution for him, and in his name and in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that each of said attorneys-in-fact and agents, and any of them or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ JOHN A. ORWIN John Orwin |
Chief Executive Officer and Member of the Board of Directors (Principal Executive Officer) |
March 10, 2011 | ||
/s/ HERB CROSS Herb Cross |
Chief Financial Officer (Principal Financial and Accounting Officer) |
March 10, 2011 |
||
/s/ HOLLINGS C. RENTON Hollings C. Renton |
Member of the Board of Directors |
March 10, 2011 |
||
/s/ R. LEE DOUGLAS R. Lee Douglas |
Member of the Board of Directors |
March 10, 2011 |
||
/s/ KATHLEEN LAPORTE Kathleen LaPorte |
Member of the Board of Directors |
March 10, 2011 |
110
Signature
|
Title
|
Date
|
||
|---|---|---|---|---|
| /s/ KEITH R. LEONARD Keith R. Leonard |
Member of the Board of Directors | March 10, 2011 | ||
/s/ TED W. LOVE Ted W. Love |
Member of the Board of Directors |
March 10, 2011 |
||
/s/ DANIEL K. SPIEGELMAN Daniel K. Spiegelman |
Member of the Board of Directors |
March 10, 2011 |
||
/s/ CHRISTI VAN HEEK Christi van Heek |
Member of the Board of Directors |
March 10, 2011 |
||
/s/ JOHN P. WALKER John P. Walker |
Member of the Board of Directors |
March 10, 2011 |
111
| 3.3 | Amended and Restated Certificate of Incorporation(1) | |
3.5 |
Amended and Restated Bylaws(2) |
|
4.1 |
Reference is made to exhibits 3.3 and 3.5 |
|
4.2 |
Specimen Common Stock Certificate(1) |
|
4.3 |
Warrant to purchase shares of Series C Preferred Stock(1) |
|
4.4 |
Amended and Restated Investor Rights Agreement, dated September 7, 2006, by and between the Registrant and certain of its stockholders(1) |
|
4.5 |
Form of Warrant to Purchase shares of Common Stock(6) |
|
10.1+ |
Form of Indemnity Agreement for Directors and Executive Officers(1) |
|
10.2+ |
2001 Stock Option/Stock Issuance Plan(1) |
|
10.3+ |
Form of Notice of Grant of Stock Option, Form of Stock Option Agreement and Form of Stock Purchase Agreement under 2001 Stock Option/Stock Issuance Plan(1) |
|
10.4+ |
Form of Stock Issuance Agreement under 2001 Stock Option/Stock Issuance Agreement(1) |
|
10.5+ |
Amended and Restated 2006 Equity Incentive Plan(9) |
|
10.6+ |
Form of Option Grant Notice and Form of Option Agreement under 2006 Equity Incentive Plan(1) |
|
10.2+ |
2001 Stock Option/Stock Issuance Plan(1) |
|
10.3+ |
Form of Notice of Grant of Stock Option, Form of Stock Option Agreement and Form of Stock Purchase Agreement under 2001 Stock Option/Stock Issuance Plan(1) |
|
10.4+ |
Form of Stock Issuance Agreement under 2001 Stock Option/Stock Issuance Agreement(1) |
|
10.5+ |
Amended and Restated 2006 Equity Incentive Plan(9) |
|
10.6+ |
Form of Option Grant Notice and Form of Option Agreement under 2006 Equity Incentive Plan(1) |
|
10.7+ |
2006 Employee Stock Purchase Plan(1) |
|
10.8+ |
Form of Offering Document under 2006 Employee Stock Purchase Plan(1) |
|
10.9+ |
Form of Restricted Stock Unit Notice and Form of Restricted Stock Unit under 2006 Equity Incentive Plan(5) |
|
10.10+ |
Employment Agreement, dated December 17, 2008, by and between the Registrant and Arlene M. Morris(7) |
|
10.11+ |
Executive Employment Agreement, dated December 17, 2008, by and between the Registrant and Paul B. Cleveland (7) |
|
10.12+ |
Executive Employment Agreement, dated December 17, 2008, by and between the Registrant and Steven Love(7) |
|
10.13+ |
Summary of Non-Employee Director Compensation Program(7) |
|
10.14 |
Research and Development/Office Lease, dated May 30, 1990, by and between Miranda Associates and Affymax Research Institute(1) |
112
| 10.15 | First Amendment to Lease, dated November 16, 1999, by and between Spieker Properties, L.P., successor in interest to Miranda Associates, and Affymax Research Institute(1) | |
10.16 |
Second Amendment to Lease, dated December 20, 1999, by and between Spieker Properties, L.P. and Affymax Research Institute(1) |
|
10.17 |
Third Amendment, dated December 31, 2001, by and between EOP-Foothill Research Center, L.L.C., successor by merger to Spieker Properties L.P., and the Registrant(1) |
|
10.18* |
EPO Receptor License Agreement, dated September 5, 1996, by and between the Registrant and Genetics Institute, Inc.(1) |
|
10.19* |
License Agreement, dated July 27, 2001, by and between the Registrant, Glaxo Group Limited, SmithKline Beecham Corporation, Affymax N.V., Affymax Research Institute and Affymax Technologies N.V.(1) |
|
10.20* |
License, Manufacturing, and Supply Agreement, dated April 8, 2004, by and between the Registrant and Nektar Therapeutics AL, Corporation(1) |
|
10.21* |
Collaboration and License Agreement, dated February 13, 2006, by and between the Registrant and Takeda Pharmaceutical Company Limited(1) |
|
10.22* |
Collaboration and License Agreement, dated June 27, 2006, by and between the Registrant and Takeda Pharmaceutical Company Limited(1) |
|
10.23 |
Research and Development Agreement, dated April 2, 1992, by and between the Registrant and The R.W. Johnson Pharmaceutical Research Institute(1) |
|
10.24 |
Sublease Agreement, dated September 1, 2006, by and between the Registrant and TIBCO Software Inc.(1) |
|
10.25 |
First Amendment to Collaboration and License Agreement, dated April 1, 2007, by and between Registrant and Takeda Pharmaceutical Company Limited(3) |
|
10.26 |
Fourth Amendment to Lease, dated November 30, 2006, by and between Registrant and CA-Foothill Research Center L.P.(4) |
|
10.27 |
Second Amendment to Collaboration and License Agreements between Registrant and Takeda Pharmaceutical Company Limited effective January 1, 2008(5) |
|
10.28 |
Securities Purchase Agreement to purchase shares of Common Stock dated February 13, 2009 by and among Registrant and the purchasers identified on the signature pages thereto(6) |
|
10.29 |
Securities Purchase Agreement to purchase shares of Common Stock and Warrants to purchase shares of Common Stock dated February 13, 2009 by and among Registrant and the purchasers identified on the signature pages thereto(6) |
|
10.30+ |
Executive Employment Agreement, dated December 17, 2008 by and between the Registrant and Anne-Marie Duliege(7) |
|
10.31+ |
Executive Employment Agreement, dated December 17, 2008 by and between the Registrant and Robert Venteicher(7) |
|
10.32 |
Common Stock Purchase Agreement, dated September 25, 2009 by and between the Registrant and Azimuth Opportunity Ltd.(8) |
|
10.33 |
Form of Credit Line and related documentation effective as of December 8, 2009 by and between the Registrant and UBS Financial Services, Inc.(9) |
113
| 10.34+ | Executive Employment Agreement, dated February 19, 2010, by and between the Registrant and John A. Orwin.(10) | |
10.35 |
Fifth Amendment, dated May 20, 2010, by and between the Registrant and EOP-Foothill Research Center, L.L.C.(11) |
|
10.36 |
Amendment No. 1 to Common Stock Purchase Agreement, dated September 17, 2010, between the Registrant and Azimuth Opportunity Ltd.(12) |
|
10.37+ |
Amendment to Employment Agreement between the Registrant and Arlene M. Morris effective as of September 23, 2010.(13) |
|
10.38+ |
Amendment to Employment Agreement between the Registrant and John A. Orwin effective as of September 23, 2010.(13) |
|
10.39+ |
Amendment to Employment Agreement between the Registrant and Paul B. Cleveland effective as of September 23, 2010.(13) |
|
10.40+ |
Amendment to Employment Agreement between the Registrant and Anne-Marie Duliege effective as of September 23, 2010.(13) |
|
10.41+ |
Executive Employment Agreement, dated February 19, 2010, by and between the Registrant and John A. Orwin.(10) |
|
10.42+ |
Executive Employment Agreement, dated March 4, 2011, by and between the Registrant and Herb Cross. |
|
10.43 |
Sixth Amendment to Lease, dated December 21, 2010 by and between Registrant and CA-Foothill Research Center L.P. |
|
23.1 |
Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm |
|
24.1 |
Power of Attorney. Reference is made to the signature page |
|
31.1 |
Certification required by Rule 13a-14(a) or Rule 15d-14(a) |
|
31.2 |
Certification required by Rule 13a-14(a) or Rule 15d-14(a) |
|
32.1† |
Certification required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. 1350) |
- (1)
- Incorporated
by reference to the indicated exhibit of our registration statement on Form S-1, registration
no. 333-136125, declared effective by the Securities and Exchange Commission on December 14, 2006.
- (2)
- Incorporated
by reference to the indicated exhibit in our Form 8-K as filed with the Securities and Exchange Commission on
September 10, 2007.
- (3)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q for the quarter ended June 30, 2007 as filed with the
Securities and Exchange Commission.
- (4)
- Incorporated
by reference to the indicated exhibit in our Form 10-K for the year ended December 31, 2006 as filed with the
Securities and Exchange Commission.
- (5)
- Incorporated
by reference to the indicated exhibit in our Form 10-K for the year ended December 31, 2007 as filed with the
Securities and Exchange Commission.
- (6)
- Incorporated by reference to the indicated exhibit in our Form 8-K as filed with the Securities and Exchange Commission on February 19, 2009.
114
- (7)
- Incorporated
by reference to the indicated exhibit in our Form 10-K for the year ended December 31, 2008 as filed with the
Securities and Exchange Commission.
- (8)
- Incorporated
by reference to the indicated exhibit in our Form 8-K as filed with the Securities and Exchange Commission on
September 25, 2009.
- (9)
- Incorporated
by reference to the indicated exhibit in our Form 10-K, as filed with the Securities and Exchange Commission on
March 4, 2010.
- (10)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q, as filed with the Securities and Exchange Commission on
May 6, 2010.
- (11)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q, as filed with the Securities and Exchange Commission on
August 5, 2010.
- (12)
- Incorporated
by reference to the indicated exhibit in our Form 8-K, as filed with the Securities and Exchange Commission on
September 20, 2010.
- (13)
- Incorporated
by reference to the indicated exhibit in our Form 10-Q, as filed with the Securities and Exchange Commission on
November 5, 2010.
- +
- Indicates
management contract or compensatory plan.
- *
- Confidential
treatment has been requested with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities
and Exchange Commission.
- †
- The certification attached as Exhibit 32.1 accompany this Annual Report on Form 10-K, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of Affymax, Inc., under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K, irrespective of any general incorporation language contained in such filing.
115