Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the fiscal year ended December 31, 2010
OR
|
o
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from to
Commission file number 001-33678
NOVABAY PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware
|
68-0454536
|
|
|
(State or other jurisdiction of incorporation or organization)
|
(I.R.S. Employer Identification No.)
|
5980 Horton Street, Suite 550, Emeryville CA 94608
(Address of principal executive offices) (Zip Code)
Registrant’s Telephone Number, Including Area Code: (510) 899-8800
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class
|
Name of each exchange on which registered
|
|
|
Common Stock, $0.01 par value per share
|
NYSE Amex
|
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.01 par value per share
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yeso Nox
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Indicate by a check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
1
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer
|
o |
Accelerated filer
|
o |
|
Non-accelerated filer
|
o |
Smaller reporting company
|
x |
(Do not check if a smaller reporting company)
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2). Yeso Nox
As of June 30, 2010, the aggregate market value of the voting stock held by non-affiliates of the registrant, computed by reference to the last sale price of such stock as of such date on the NYSE Amex, was approximately $41,227,428. Excludes an aggregate of 4,158,640 shares of common stock held by officers and directors and by each person known by the registrant to own 5% or more of the outstanding common stock as of June 30, 2010. Exclusion of shares held by any of these persons should not be construed to indicate that such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant, or that such person is controlled by or under common control with the registrant.
As of March 1, 2011, there were 23,439,755 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for the 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Form 10-K, are incorporated by reference in Part III, Items 10-14 of this Form 10-K.
2
NOVABAY PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2010
TABLE OF CONTENTS
|
Page
|
||
|
PART I
|
||
|
ITEM 1.
|
BUSINESS
|
4
|
|
ITEM 1A.
|
RISK FACTORS
|
11
|
|
ITEM 1B.
|
UNRESOLVED STAFF COMMENTS
|
24
|
|
ITEM 2.
|
PROPERTIES
|
24
|
|
ITEM 3.
|
LEGAL PROCEEDINGS
|
24
|
|
ITEM 4.
|
(Removed and Reserved)
|
24
|
|
PART II
|
||
|
ITEM 5.
|
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
|
25
|
|
ITEM 6.
|
SELECTED FINANCIAL DATA
|
27
|
|
ITEM 7.
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
28
|
|
ITEM 7A.
|
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
36
|
|
ITEM 8.
|
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
|
37
|
|
ITEM 9.
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
|
63
|
|
ITEM 9A
|
CONTROLS AND PROCEDURES
|
63
|
|
ITEM 9B.
|
OTHER INFORMATION
|
63
|
|
PART III
|
||
|
ITEM 10.
|
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
63
|
|
ITEM 11.
|
EXECUTIVE COMPENSATION
|
64
|
|
ITEM 12.
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
|
64
|
|
ITEM 13.
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
|
64
|
|
ITEM 14.
|
PRINCIPAL ACCOUNTING FEES AND SERVICES
|
64
|
|
PART IV
|
||
|
ITEM 15.
|
EXHIBITS, FINANCIAL STATEMENT SCHEDULES
|
65
|
Unless the context requires otherwise, all references in this report to “we,” “our,” “us,” the “Company” and “NovaBay” refer to NovaBay Pharmaceuticals, Inc. and its subsidiaries.
NovaBay®, NovaBay Pharma®, AgaNase®, Aganocide®, NeutroPhase®, AgaDerm™, and Going Beyond AntibioticsTM are trademarks of NovaBay Pharmaceuticals, Inc. All other trademarks and trade names are the property of their respective owners.
3
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements that are based on our management’s beliefs and assumptions and on information currently available to our management. These forward-looking statements include but are not limited to statements regarding our product candidates, market opportunities, competition, strategies, anticipated trends and challenges in our business and the markets in which we operate, and anticipated expenses and capital requirements. In some cases, you can identify forward-looking statements by terms such as “anticipates,” “believes,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “projects,” “should,” “will,” “would” and similar expressions intended to identify forward-looking statements. Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. We discuss many of these risks in this report in greater detail under the heading “Risk Factors” in Item 1A of this report. Given these uncertainties, you should not place undue reliance on these forward-looking statements. You should read this report and the documents that we reference in this report and have filed as exhibits to the report completely and with the understanding that our actual future results may be materially different from what we expect. Also, forward-looking statements represent our management’s beliefs and assumptions only as of the date of this report. Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
PART I
|
ITEM 1.
|
BUSINESS
|
Overview
NovaBay Pharmaceuticals Inc. is a clinical stage biotechnology company developing a first-in-class, anti-infective platform of compounds called the Aganocide® compounds. In laboratories, these compounds have demonstrated equivalent activity to the active antimicrobial molecules generated within white blood cells. The Aganocide compounds are being developed for the topical treatment and prevention of a wide variety of topical infections, including those that are antibiotic resistant.
NovaBay is developing commercial opportunities for its portfolio of anti-infectives Aganocide compounds in four distinct healthcare markets: dermatology, ophthalmology, urology and hospital infections. Each of these market segments are underserved by current products. Therefore there is a substantial market potential for improved treatments. NovaBay’s strategy is to address these market opportunities either through partnerships and collaborations or by building an internal organization to strategically market its own products when appropriate from a commercial standpoint.
We were incorporated under the laws of the State of California on January 19, 2000 as NovaCal Pharmaceuticals, Inc., and subsequently to NovaBay Pharmaceuticals, Inc. In June 2010, we changed the state in which we are incorporated (the Reincorporation), and are now incorporated under the laws of the State of Delaware. All references to “we,” “us,” “our,” or “the Company” herein refer to the California corporation prior to the date of the Reincorporation, and to the Delaware corporation on and after the date of the Reincorporation.
Our Technology and Research
We have developed our platform of Aganocide compounds by understanding the nature of the antimicrobial molecules that are produced by the human body’s white blood cells to kill pathogens such as bacteria, viruses and fungi. Once the body’s defense system detects these pathogens, white blood cells produce small, highly active molecules that kill the pathogens in an extremely efficient manner. Through the modification of one of these natural molecules, N-chlorotaurine (NCT), we have created a patented, stable, synthetic analog, which we have labeled NVC-422, and which is currently our lead Aganocide compound. We continue to make significant discoveries that have enhanced our understanding of why the naturally generated molecules cannot be kept stable and therefore used as drugs. We have also been exploring different Aganocide compounds that have been invented by NovaBay’s scientists with the aim of creating molecules that can penetrate different tissues more effectively or that can enhance the duration of antimicrobial activity. We have made great progress in developing multiple formulations that can enhance the penetration of the Aganocide molecules.
In 2002, the World Health Organization predicted that within ten years we will enter a “post-antibiotic” era, where there will be infections for which there will be no effective antibiotic treatments. This prediction is proving to be true as there are now more multi-drug resistant bacteria (Superbugs) appearing, and even a few pan-resistant species. By using nature’s blueprint for the development of new anti-infective products, we start with the intent that natural molecules do not allow pathogens to develop resistance. Aganocide compounds have exhibited this characteristic in laboratory studies. The ability of our Aganocide compounds to be effective without developing resistance is critical in a situation where bacteria are continuing to develop ever more sophisticated mechanisms for protecting themselves from antibiotics.
4
Due to the significant problem of antibiotic resistance, the problem is monitored by global surveillance of the development of resistance to antibiotics used clinically. In the laboratory the propensity for a given antibiotic to develop resistance may be determined by applying the antibiotic, at sub-lethal dose, to a pathogen in several passages. All antibiotics will develop resistance at different rates, often after a few passages. Aganocides, by virtue of their non-specific killing mechanism of action, are unlikely to develop resistance. We have subjected our lead compound NVC-422 to such serial passage with a number of pathogens and have confirmed that no resistance develops even after 25 passages. As expected, antibiotics tested in parallel all developed resistance.
In preclinical studies, the Aganocide compounds have demonstrated efficacy against bacteria in biofilm. Biofilm is a cocoon-like shield that forms around a colony of bacteria. Once the biofilm is formed, bacteria go into dormancy. Dormant bacteria in biofilm reproduce slowly and are protected from attack by the body’s killer cells by their biofilm shield. We now understand that biofilm is a natural, ever present defense mechanism of bacteria. Single free floating bacteria are much easier to kill than colonies consisting of millions of bacteria as found in biofilm. Antibiotics are generally more effective against fast reproducing bacteria as opposed to bacteria colonized in biofilm. We continue to expand our understanding of Aganocide action on biofilm. In controlled laboratory studies, our Aganocide compounds were found to be effective at killing bacteria in biofilm. Furthermore, in animal studies our Aganocide compounds have been found to be more effective against biofilm colonization than mupirocin, a widely used topical antibiotic. We believe efficacy of Aganocide compounds in biofilm would be an important property that may contribute to their utility in many commercial applications.
Our Target Indications and Product Candidates
Our goal is to advance our product candidates through confirmatory Phase 2 proof of concept trials, after which we will evaluate further advancing each program on our own or entering a co-development collaboration agreement with a proven market leader. In the event that we enter into a co-development collaboration agreement with a proven market leader, this strategy provides the benefit of their product development expertise and proven commercial capabilities. In these collaborations, our strategy has been to defray costs while retaining participation in the long-term commercial economics of our products. This strategy enhances our probability of success in product and commercial development. In many instances, we believe we can build upon the safety data generated in one indication to accelerate early development of other indications. We are also learning from our own and our partners’ experience in developing appropriate dosing and usage of our compounds. The more development programs that are undertaken by our partners and by ourselves, the greater product development synergy we expect to achieve.
By virtue of their anti-microbial versatility, the Aganocide compounds offer NovaBay an opportunity to potentially address a wide variety of topical, non-systemic indications in large, underserved markets. Topical indications include treatment and prevention of infections on any surface that may harbor pathogens, such as skin, bladder, sinus, lungs, the eye, as well as medical devices such as catheters. We are focusing on four major market opportunities: ophthalmology, dermatology, urology and hospital infections. Our strategy is to build four distinct business units around these markets in the years to come.
To date, we have not commercialized any of our product candidates, and so have not generated any revenues from the sale of products.
Ophthalmology
In August 2006, we entered into a collaboration and license agreement with Alcon Manufacturing Ltd. (Alcon), an affiliate of Alcon, Inc., that provides Alcon with the exclusive rights to develop, manufacture and commercialize products incorporating our Aganocide compounds for the treatment of eye, ear and sinus infections as well as for use in contact lens solutions. Under the terms of the agreement, Alcon paid an up-front technology access fee of $10.0 million upon the effective date of the agreement. Under the terms of the agreement we also received semi-annual payments from Alcon to support on-going research and development activities over the four-year funding term of the agreement, which ended in August 2010. On November 18, 2010 Alcon extended the funding term to December 31, 2015, subject to earlier termination of the agreement at Alcon’s election, with six months prior written notice. The collaboration also calls for Alcon to pay for all developmental and clinical costs. NovaBay has the potential to receive up to $70.0 million in milestones from Alcon, and royalties ranging in single digits on net sales of products once commercialized.
The research and development support payments include amounts to fund a specified number of personnel engaged in collaboration activities and to reimburse for qualified equipment, materials and contract study costs. As product candidates are developed and proceed through clinical trials and approval, we will receive milestone payments of up to $70.0 million. If the products are commercialized, we will also receive royalties on any sales of products containing the Aganocide compounds. From the inception of the agreement to December 31, 2010, we have received an up-front payment of $10.0 million plus $21.3 million in research and development funding and support.
NovaBay retains the rights to market, via a third-party co-marketing partner, any products developed for ear or sinus indications in the major Asian markets, including Japan, China, India and South Korea. NovaBay has also retained such rights in other markets where Alcon is not committing reasonably sufficient sales and marketing resources to the particular product. In each instance, the appointment of the co-marketing partner would be subject to certain conditions, including that the co-marketing partner be approved by Alcon. The co-marketing partner, or NovaBay on its behalf, would be required to pay Alcon a royalty based on net sales of the product in the applicable market and would also be required to reimburse Alcon for part of its local development costs or, in markets in which Alcon is not committing reasonably sufficient sales and marketing resources, all of its local development costs. These products may also be sold in those markets by Alcon, its affiliates or distributors.
5
During 2010, Alcon concluded a Phase 2 human proof of concept trial of NovaBay’s lead compound, NVC-422, for the treatment of adenoviral conjunctivitis, a type of “Pink Eye”. This is a significant market, for which no established treatment exists. Alcon and NovaBay intend to analyze the safety and microbiological and clinical efficacy of the drug on patients enrolled in the trial and plan to report on these analyses in the first half of 2011.
Dermatology
We are focused on developing products that will potentially eliminate the need to use antibiotic-based products in the dermatology market. Our technology goes beyond antibiotics: we are focused on developing non-antibiotic anti-infective products which would not be susceptible to drug-resistant pathogens. In July 2010, we announced positive results from a Phase 2a, 129-patient, impetigo skin infection study. The results showed up to 92% of the patients were clinically cured of the highly contagious infection following seven days of treatment with our lead Aganocide (NVC-422) topical gel. Furthermore, methicillin-resistant Staphylococcus aureus (MRSA) is becoming an escalating problem, as an ever-increasing portion of impetigo patients are infected with this “superbug”. The ten patients in the study infected with MRSA impetigo infection achieved 100% clinical and/or bacterial response. As resistance to antibiotics becomes a critical public health issue, NovaBay intends to aggressively pursue the development of non-antibiotic anti-infectives that are unlikely to cause resistance, as a first-line treatment for a range of topical infections.
We have shown that our lead Aganocide compound, NVC-422, kills P. acne, the bacterium associated with inflamed acne lesions, and other known dermal pathogens. We have been in advanced preclinical development of a variety of formulations for use in the treatment of skin infections.
Galderma Collaboration
On March 25, 2009, we entered into a collaboration and license agreement with Galderma S.A. to develop and commercialize our Aganocide compounds, which covers acne and impetigo and potentially other major dermatological conditions, excluding onychomycosis (nail fungus) and orphan drug indications. We amended this agreement in December 2009. Based on the Impetigo Phase 2a clinical trial results, in December 2010, NovaBay and Galderma S.A., agreed to expand their partnership to focus on the development of NovaBay’s Aganocide compound NVC-422 for the topical treatment of impetigo as well as developing its technology for acne. This expansion is intended to provide NovaBay with the additional funding and resources required for the clinical development of its NVC-422 topical gel formulation for impetigo. Moving NVC-422 gel into Phase 2b clinical development in 2011 is the current top priority, with the potential to move into Phase 3 development during 2012.
This agreement is exclusive and worldwide in scope, with the exception of Asian markets and North America, as described in the next paragraph.
Galderma will be responsible for the development costs of product candidate compounds, except for costs incurred in Japan. In Japan, Galderma has the option to request that we share such development costs. Under the original agreement, we were supporting the ongoing development program for impetigo; however under the second amendment, entered into on December 2, 1010, Galderma increased its support to cover this indication. Upon the achievement of a specified milestone, Galderma will reimburse NovaBay for specified, previously incurred expenses related to the development of the impetigo program. NovaBay retains the right to co-market products resulting from the agreement in Japan. In addition, NovaBay has retained all rights in other Asian markets outside Japan, and has the right to co-promote the products developed under the agreement in hospitals and other healthcare institutions in North America.
From the inception of the agreement to December 31, 2010, we have received $11.8 million from Galderma including the technology access fee, continuation fee, milestone payments and research and development funding. NovaBay has the potential to receive up to $62.0 million in predetermined fees, including milestones and personnel reimbursement, with additional funding available to cover product and clinical development. We are entitled to royalties ranging from 10% to 30% on cumulative net sales of products once commercialized, subject to some reductions based on any development costs incurred directly by Galderma. Upon the termination of the agreement under certain circumstances, Galderma will grant NovaBay certain technology licenses which would require NovaBay to make royalty payments to Galderma for such licenses with royalty rates in the low- to mid-single digits.
Impetigo
Impetigo is a highly contagious superficial bacterial infection of the skin that affects mostly children. Most cases are caused by Staphylococcus aureus, Streptococcus pyogenes, or a mixture of both organisms. MRSA is being observed with increasing frequency in this population. Impetigo is currently being treated with antibiotic ointments, to which bacteria may develop resistance.
6
Under the terms of the second amendment to the agreement with Galderma, for the research and development of impetigo and acne, Galderma has agreed to exercise its option to advance the clinical development of the impetigo program and paid a $3.25 million continuation fee together with additional research and development funding through the development of the program.
We believe that there is a significant market for the treatment of impetigo, with approximately 13 million prescriptions for the treatment of impetigo annually, and 1.3 million prescriptions in the U.S. alone.
Onychomycosis
Onychomycosis is a fungal infection of the toe and finger nails affecting the components of the nail matrix, nail bed, or nail plate resulting in deformity of the nail plate, thickening and nail discoloration. Onychomycosis is not life threatening; however, it can cause disfigurement and pain resulting in serious occupational and physical limitations. Furthermore, individuals with a compromised immune system may be at greater risk of additional complications. Aganocide compounds, like NVC-422, have been shown in laboratory tests to kill the dermatophytes responsible for onychomycosis thus providing NovaBay Pharmaceuticals with an opportunity to develop effective treatment for this disease. We believe the potential market for the treatment of Onychomycosis is $1.5 billion.
Urology
Urinary catheters have become a routine part of the management of patients in intensive care and long-term care settings, with an estimated five million patients undergoing catheterization each year. Catheter associated urinary tract infections (CAUTI) are the most frequent healthcare-associated infections, accounting for more than 40% of all healthcare-associated infections, or one million infections per year. Based on quantitative market research conducted by NovaBay, the U.S. market size of permanently catheterized patients is estimated to be up to 335,000 with approximately one-third chronically susceptible to urinary catheter blockage and encrustation (UCBE).Their catheters can become contaminated with Proteus mirabilis resulting in a crystalline biofilm formation leading to catheter blockage, with the further potential to develop urinary tract infections and even sepsis.
In 2010 NovaBay reported that an irrigation solution containing NVC-422 in a Phase 2a pilot study demonstrated activity against a number of uropathogens including Proteus mirabilis, a Gram-negative bacterium that can cause severe UCBE. NovaBay’s lead Aganocide compound has been shown to be safe in a Phase 1 study with 29 healthy volunteers. NovaBay has commenced a Phase 2 trial in permanently catheterized patients at three centers in the U.S. and intends to provide investors and potential partners with more detail on the design of this clinical trial in the near future.
NovaBay is evaluating the potential of building a commercial team to market this product along with other complementary products for the urology and neurology markets. NovaBay believes that the potential market for the treatment of spinal cord injury patents with Proteus mirabilis infections may be approximately $180 million annually.
Hospital Infections
Wound infections prevent wounds from healing and can cause serious bloodstream infections. We have developed NeutroPhase, also called NVC-101, to address this major problem. NeutroPhase is a physiologically balanced, acidic composition of hypochlorous acid, the same potent chemical that white blood cells generate to attack invading pathogens during oxidative burst. The advantages of NeutroPhase include fast killing of microbial pathogens, low toxicity, low irritancy, and long shelf life. NovaBay holds two U.S. patents on methods of using NeutroPhase to promote wound healing, tissue repair, and tissue regeneration. In 2007 and 2008, NeutroPhase received 510(k) clearance from the device division of the FDA as a liquid bandage. The specifics of the FDA cleared indications for use provide sufficient claims for NovaBay to market NeutroPhase for moistening and debriding chronic non-healing wounds, including diabetic foot ulcers, venous stasis ulcers and pressure ulcer stages I-IV.
The cost of treating chronic wounds is estimated at $5 billion to $7 billion in the U.S., and the occurrence of these wounds is increasing at a rate of 10% per year. NovaBay is currently seeking a commercial partner for NeutroPhase to cover the North American, European and Japanese markets.
Research and Development
As of December 31, 2010, we had 30 employees dedicated to research and development. Our research and development expenses consist primarily of personnel-related expenses, laboratory supplies and contract research services provided to our research, development and clinical groups. We expense our research and development costs as they are incurred. Research and development expenses for 2010, 2009 and 2008 were $8.6 million, $7.3 million, and $9.6 million, respectively. All of our research and development employees are engaged in drug research and development activities, including those related to the Alcon and Galderma agreements described above. We expect to incur significant research and development expenses for the foreseeable future.
7
Intellectual Property
We rely on a combination of patent, trademark, copyright and trade secret laws in the U.S. and other jurisdictions, as well as confidentiality procedures and contractual provisions, to protect our proprietary technology. We also enter into confidentiality and invention assignment agreements with our employees and consultants and confidentiality agreements with other third parties, and we rigorously control access to our proprietary technology.
We are the assignee of record of five issued patents in the U.S. and nine issued patents in foreign countries. In addition to our issued patents, we own, co-own or are the exclusive worldwide licensee of 24 patent applications in various stages of prosecution in the U.S. and over 60 applications pending in foreign countries and regions including Brazil, Canada, China, Europe, India, North Korea and Japan. Additional applications will enter the foreign national phase once they pass through the international phase of the Patent Cooperation Treaty.
The subject matter of our patents and patent applications covers four types of technologies: methods relating to the manufacture and use of our hypochlorous acid solution NeutroPhase (NVC-101), compositions of matter of our Aganocide compounds, methods of treating or preventing microbial ailments utilizing NeutroPhase and/or our Aganocide compounds, and formulations. In April of 2009 we entered into an exclusive worldwide license to certain patent applications relating to methods of use of N-chlorotaurine. These applications are pending in the U.S. and abroad.
U.S. Patent No. 6,424,066 provides coverage for a method of treating burns or promoting wound healing, tissue repair or tissue regeneration using a specific range of formulations of hypochlorous acid. This patent issued on July 30, 2002 and will expire in 2020 with payment of maintenance fees. Corresponding patents have issued in Australia, China, India, Israel, Hong Kong, Mexico and South Korea. U.S. Patent No. 7,393,522 provides coverage for a method of disinfecting open wounds and burns, promoting wound healing or providing ocular disinfection using a specific range of formulations of hypochlorous acid. This patent issued on July 1, 2008 and will expire in 2020 with payment of maintenance fees.
U.S. Patent No. 7,462,361 provides composition-of-matter coverage of our lead development candidate, NVC-422, and other Aganocide compounds. This patent issued on December 9, 2008 and will expire in 2026 with payment of maintenance fees. U.S. Patent No. 7,893,109 is a continuation application of U.S. Patent No. 7,462,361 and provides composition-of-matter coverage of additional N,N-dichloroamine compounds related to NVC-422. This patent issued on February 22, 2011 and will expire in 2024. A corresponding patent issued in New Zealand, and corresponding applications are pending in Argentina, Australia, Brazil, Canada, China, Europe, Honk Kong, Israel, India, Japan, South Korea, Mexico, New Zealand, Singapore, and Taiwan.
U.S. Patent No. 7,846,971 provides composition-of-matter coverage of additional Aganocide compounds. This patent issued on December 7, 2010 and will expire in 2028 with the payment of maintenance fees. A corresponding patent has been issued in Singapore, and corresponding applications are pending in Australia, Brazil, Canada, China, Europe, Honk Kong, Israel, India, Japan, South Korea, Mexico, Taiwan and South Africa.
NovaBay®, NovaBay Pharma®, AgaNase®, Aganocide®, and NeutroPhase® are registered U.S. trademarks of NovaBay Pharmaceuticals, Inc. In addition to the U.S. registrations, NovaBay is registered in the European Community, Israel, Mexico, and Australia and applications are pending in Brazil, Canada and India; AgaNase is registered in the European Community, Australia, Israel, Japan, Mexico, China, South Korea, and Taiwan and applications are pending in Brazil, Canada and India; NeutroPhase is registered in Australia, the European Community, Ireland and the United Kingdom and applications are pending in Canada and India; and Aganocide is registered in the European Community and Japan.. Applications for registration of the trademarks AgaDerm™ and Going Beyond Antibiotics™ are pending in the U.S. and Canada.
Competition
The market for topical, non-systemic anti-infective drugs is highly competitive. If developed, and commercialized, our Aganocide products would compete against a wide variety of existing products, products and technologies that are currently in development, and products and technologies that could be developed and reach the market before or after our products. In particular, we would be competing against existing antibiotics and anti-infective products that are sold by many major pharmaceutical companies, or generic equivalents that are being distributed, typically at low prices. NeutroPhase, if launched for use in wound management, will be competing against multiple products with similar product profiles and indications for use. However, we believe there is currently no dominant product in this indication.
Our potential competitors include large and small pharmaceutical and medical device companies, such as Pfizer, Inc., Johnson & Johnson, Abbott Grp. Plc., GlaxoSmithKline Plc, Sanofi-Aventis SA, Novartis AG, Smith & Nephew Plc, C.R. Bard, Puricore and Oculus Innovative Sciences.
We believe the principal competitive advantage of our products in our target markets include their effectiveness in killing viruses, fungi and bacteria, including bacteria in biofilm, very low potential for the development of resistance, fast time to kill bacteria, wide safety margin , low side effect profile and cost effectiveness. We believe that our compounds may, if approved by the regulatory authorities, have significant advantages over existing compounds and compounds in development of which we are aware, because our Aganocide and NVC-101 compounds could be used to prevent infections or to treat infections s with bacterial and viral components such as conjunctivitis.
8
Manufacturing and Supply
We do not currently operate manufacturing facilities for clinical or commercial production, as we rely on and leverage the manufacturing and distribution infrastructure of third parties. We have no plans to establish our own manufacturing facilities in the future. Third party vendors supply us with the Active Pharmaceutical Ingredient (API) of NVC-422 and the finished clinical trials materials for NVC-101, which are manufactured in compliance with the FDA’s “Current Good Manufacturing Practice”, or CGMP, regulations. We also intend to work with third parties for future clinical trial materials and commercial supplies of NVC-422 and our other Aganocide compounds.
The Alcon and Galderma agreements provide for the manufacture by Alcon and Galderma of finished dosage forms of products incorporating Aganocide compounds for sale under our label in those markets where we have retained marketing rights.
Sales and Marketing
Our lead product candidate, NVC-422, as well as many of the product candidates we expect to develop in the future, are primarily intended to address a variety of different non-systemic market segments, some of which are large, primary care markets. We do not currently have, nor do we intend in the near term to create, a commercialization organization capable of marketing, selling and distributing our targeted product candidates to large, primary care markets. This applies to markets in both the U.S. and elsewhere. Rather, we intend to establish commercialization partnerships with pharmaceutical, biotechnology or other leading organizations with the experience and resources to bring our products to market. In some cases, we may enter into agreements with these organizations during the development stage of a product candidate to further benefit from their clinical development, regulatory, market research, pre-marketing and other expertise, as is the case with Alcon and Galderma. As appropriate, we may establish a specialty sales force with expertise in marketing and selling any future approved products to specialty physicians for specific target indications. We may also establish other complementary capabilities related to marketing and selling targeted medicines, particularly where those capabilities may not currently exist at other organizations. In 2010, 2009 and 2008, substantially all of our revenues have been generated from Galderma and Alcon, and we rely on these two companies for our revenues for the foreseeable future; Galderma is located in France and Alcon is located in Switzerland. Substantially all of our long-lived assets are located in the U.S.
Government Regulation
The testing, manufacturing, labeling, advertising, promotion, distribution, export and marketing of our product candidates are subject to extensive regulation by the FDA, state agencies and comparable regulatory authorities in other countries. Because our programs involve product candidates that are considered as drugs and others that are medical devices, we intend to submit applications to regulatory agencies for approval or clearance of both drug and medical device product candidates.
U.S. Government Regulation
In the U.S., the FDA regulates drugs and medical devices under the Federal Food, Drug, and Cosmetic Act and the agency’s implementing regulations. If we fail to comply with the applicable U.S. requirements at any time during the product development process, clinical testing, and the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, adverse publicity, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on us.
Our products are classified by the FDA as a drug or a medical device depending upon the mechanism of action and indications for use or claims. The use of NVC-101 as a solution for cleansing and debriding wounds, NeutroPhase, is considered a medical device. Similarly, NVC-422 may be classified as a medical device depending on the indication for use. For example, we believe if the indication is for maintaining catheter patency, it would be classified as a medical device, whereas we believe it would be considered a drug when it is indicated for the prevention of urinary tract infection. The determination as to whether a particular product and indication is considered a drug or a device is based in part upon prior precedent.
Drug Approval Process
The process required by the FDA before a drug may be marketed in the U.S. generally involves satisfactorily completing each of the following:
|
·
|
preclinical laboratory tests, animal studies, toxicology and formulation studies all performed in accordance with the FDA’s Good Laboratory Practice regulations;
|
|
·
|
submission to the FDA of an Investigational New Drug (IND) application for human clinical testing, which must become effective before human clinical trials may begin;
|
|
·
|
performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; these clinical trials must be conducted in accordance with Good Clinical Practice (GCP) Guidelines, including Institutional Review Board oversight of the consent of subjects and registration of applicable studies with clinicaltrials.gov; clinical trials generally progress through Phases 1, 2 and 3, testing, respectively, initial safety, population and dose finding, and finally, testing of the anticipated commercial dose, formulation and indication at multiple sites in randomized, placebo-controlled studies that must provide replicate evidence of safety and effectiveness;
|
9
|
·
|
submission to the FDA of a New Drug Application (NDA) including payment of substantial User Fees;
|
|
·
|
satisfactory completion of an FDA inspection of the manufacturing facility or facilities, including those of third-parties, at which the product is produced to assess compliance with strictly enforced current GMP regulations, as well as FDA audit for GCP compliance of one or more clinical investigator sites; and
|
|
·
|
FDA review and approval of the NDA before any commercial marketing, sale or shipment of the product.
|
There is continuing and pervasive FDA regulation of drug product manufacturing, labeling, distribution, advertising and promotion once approved, and approval may be subject to additional required clinical studies or risk evaluation and mitigation strategies, or REMS.
Medical Devices
NeutroPhase, as well as some of our product candidates, may be regulated as medical devices. Unless an exception applies, each medical device we wish to commercialize in the U.S. will require either prior 510(k) clearance or premarket approval from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose lower risks are placed in either Class I or II, which requires the manufacturer to submit to the FDA a premarket notification requesting permission to commercially distribute the device. This process is generally known as 510(k) clearance. Some low risk devices are exempt from this requirement. Any post-clearance modifications made to a 510(k) device may require the submission of a new 510(k) notification prior to commercialization. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in Class III, requiring human clinical study prior to premarket approval. The 510(k) process is undergoing programatic change at FDA and our ability to obtain 510(k) clearance for future device products may be adversely impacted but such regulatory changes.
Continuing Food and Drug Administration Regulation of Medical Devices
After the FDA permits a device to enter commercial distribution, numerous regulatory requirements apply. These include:
|
·
|
the FDA’s Quality Systems Regulations (QSRs), which require manufacturers to follow stringent design, testing, production, control, labeling, packaging, storage, shipping, documentation and other quality assurance procedures during all aspects of the manufacturing process;
|
|
·
|
labeling regulations which impose restrictions on labeling and promotional activities, and FDA prohibitions against the promotion of products for uncleared, unapproved, or “off-label” uses;
|
|
·
|
post-market surveillance requirements which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device;
|
|
·
|
the FDA Medical Device Reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and
|
|
·
|
notices of correction or removal, and recall regulations.
|
In addition, we are required to register our facility and list our products with the FDA, and are subject to unannounced inspections by the FDA and the Food and Drug Branch of the California Department of Health Services to determine compliance with the QSRs and other regulations, and these inspections may include the manufacturing facilities of our subcontractors.
International Regulation
In addition to being subject to the laws and regulations in the U.S., we will be subject to a variety of laws and regulations in those other countries in which we seek to study and commercialize products. European and Canadian regulatory requirements and approval processes are similar in principle to those in the U.S.. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of the European Union, European countries, Canada and other countries before we can commence clinical trials or marketing of the product in those respective countries. The approval process may be longer or shorter than that required for FDA approval. The requirements governing pricing, reimbursement, clinical trials, and to a lesser extent, product licensing vary from country to country.
Third Party Reimbursement and Pricing Controls
In the U.S. and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third party payers, such as government and private insurance plans. Third party payers are increasingly challenging the prices charged for medical products and services. It will be time consuming and expensive for us to go through the process of seeking reimbursement from Medicare and private payers. Aganocide products from which we may receive revenue in the future may not be considered cost-effective, and reimbursement may not be available or sufficient to allow these products to be sold on a competitive and profitable basis.
10
Anti-Kickback and False Claims Laws
In the U.S., we are subject to various federal and state laws pertaining to healthcare “fraud and abuse,” including anti-kickback and false claims laws. The federal Anti-Kickback Law makes it illegal for any person, including a prescription drug or medical device manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, offer, receive or pay any remuneration, directly or indirectly, in exchange for, or to induce, the referral of business, including the purchase, order or prescription of a particular drug or device, for which payment may be made under federal healthcare programs such as Medicare and Medicaid. Violations of the law are punishable by up to five years in prison, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs. In addition, many states have adopted laws similar to the federal Anti-Kickback Law. Some of these state prohibitions apply to referral of patients for healthcare services reimbursed by any source, not only the Medicare and Medicaid programs. Due to the breadth of these laws, it is possible that our future sales and marketing practices or our future relationships with physicians might be challenged under anti-kickback laws, which could harm us.
False claims laws prohibit anyone from knowingly presenting, or causing to be presented, for payment to third party payors (including Medicare and Medicaid) claims for reimbursed items or services, including drugs and medical devices, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our future activities relating to the reporting of prices for our products, the reporting of Medicaid rebate information and other information affecting federal, state and third party reimbursement of our products, and the sale and marketing of our products, will be subject to scrutiny under these laws. In addition, pharmaceutical and medical device companies have been prosecuted under the federal False Claims Act in connection with their off-label promotion of products. Suits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals (known as relators or, more commonly, as whistleblowers) may share in the amounts paid by the entity to the government in fines or settlement.
Employees
As of December 31, 2010, we had 43 full-time employees, including 17 with doctoral degrees. Of our full time workforce, 30 employees were engaged in research and development, and 13 in finance, legal and administration. None of our employees are represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge on our corporate website, located at www.novabaypharma.com, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission (SEC).
|
ITEM 1A.
|
RISK FACTORS
|
Our business is subject to a number of risks, the most important of which are discussed below. You should consider carefully the following risks in addition to the other information contained in this report and our other filings with the SEC, before deciding to buy, sell or hold our common stock. The risks and uncertainties described below are not the only ones facing our company. Additional risks and uncertainties not presently known to us or that we currently believe are not important may also impair our business operations. If any of the following risks actually occur, our business, financial condition or results of operations could be materially adversely affected, the value of our common stock could decline and you may lose all or part of your investment.
Risks Relating to Our Business
Current worldwide economic conditions may limit our access to capital, adversely affect our business and financial condition, as well as further decrease our stock price.
General worldwide economic conditions have experienced a downturn due to the effects of the subprime lending crisis, general credit market crisis, collateral effects on the finance and banking industries, concerns about inflation, slower economic activity, decreased consumer confidence, reduced corporate profits and capital spending, adverse business conditions and liquidity concerns. Although the impact of the downturn on our business is uncertain at this time, downturn may adversely affect our business and operations in a number of ways, including making it more difficult for us to raise capital as well as making it more difficult to enter into collaboration agreements with other parties. Like many other stocks, our stock price has been subject to fluctuations in recent months. Our stock price could decrease due to concerns that our business, operating results and financial condition will be negatively impacted by a worldwide economic downturn.
11
We may be unable to raise additional capital on acceptable terms in the future which may in turn limit our ability to develop and commercialize products and technologies.
We expect our capital outlays and operating expenditures to substantially increase over at least the next several years as we expand our product pipeline and increase research and development efforts and clinical and regulatory activities. Conducting clinical trials is very expensive, and we expect that we will need to raise additional capital, through future private or public equity offerings, strategic alliances or debt financing, before we achieve commercialization of any of our Aganocide compounds. In addition, we may require even more significant capital outlays and operating expenditures if we do not continue to partner with third parties to develop and commercialize our products.
Our future capital requirements will depend on many factors, including:
|
·
|
the extent to which we receive milestone payments or other funding from Alcon and/or Galderma, if any;
|
|
·
|
the scope, rate of progress and cost of our pre-clinical studies and clinical trials and other research and development activities;
|
|
·
|
future clinical trial results;
|
|
·
|
the terms and timing of any collaborative, licensing and other arrangements that we may establish;
|
|
·
|
the cost and timing of regulatory approvals;
|
|
·
|
the cost of establishing clinical and commercial supplies of our product candidates and any products that we may develop;
|
|
·
|
the effect of competing technological and market developments;
|
|
·
|
the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and
|
|
·
|
the extent to which we acquire or invest in businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions.
|
We do not currently have any commitments for future external funding. Additional financing may not be available on favorable terms, or at all. Our ability to obtain additional financing may be negatively affected by the recent volatility in the financial markets and the credit crisis, as well as the general downturn in the economy and decreased consumer confidence. Even if we succeed in selling additional securities to raise funds, our existing stockholders’ ownership percentage would be diluted and new investors may demand rights, preferences or privileges senior to those of existing stockholders. If we raise additional capital through strategic alliance and licensing arrangements, we may have to trade our rights to our technology, intellectual property or products to others on terms that may not be favorable to us. If we raise additional capital through debt financing, the financing may involve covenants that restrict our business activities.
In addition, it is often the case that the cost of pharmaceutical development can be significantly greater than initially anticipated. This may be due to any of a large number of possible reasons, some of which could have been anticipated, while others may be caused by unpredictable circumstances. A significant increase in our costs would cause the amount of financing that would be required to enable us to achieve our goals to be likewise increased.
If we determine that we need to raise additional funds and we are not successful in doing so, we may be unable to complete the clinical development of some or all of our product candidates or to seek or obtain FDA approval of our product candidates. Such events could force us to discontinue product development, enter into a relationship with a strategic partner earlier than currently intended, reduce sales and marketing efforts or forego attractive business opportunities.
We are an early stage company with a history of losses. Although we were profitable in 2009, we reported a net loss in 2010, we do not have any commercial products, and expect that we will incur net losses in the future, and that we may never achieve or maintain sustained profitability.
We have incurred net losses each year since our inception through December 31, 2010, with the exception of 2009. For the years ended December 31, 2010 and 2008 we had net losses of approximately $4.3 million and $8.1 million, respectively, and for the year ended December 31, 2009, we had net income of $2.7 million. We were able to record a profit in 2009 due to our receipt of a $3.75 million milestone payment under our agreement with Galderma; however, there is no assurance that we will receive any additional large milestone payments under this agreement and, as a result, may not be able to achieve or maintain profitability in the future. Through December 31, 2010, we had an accumulated deficit of approximately $28.2 million. We have been, and expect to remain for the foreseeable future, mostly in a research and development stage. We have incurred substantial research and development expenses, which were approximately $8.6 million, $7.3 million and $9.6 million for the years ended December 31, 2010, 2009 and 2008, respectively. We expect to continue to make, for at least the next several years, significant expenditures for the development of products that incorporate our Aganocide compounds, as well as continued research into the biological activities of our Aganocide compounds, which expenditures are accounted for as research and development expenses. We do not expect any of our current product candidates to be commercialized within the next several years, if at all. We expect to incur substantial losses for the foreseeable future, and we may never achieve or maintain sustained profitability. We anticipate that our expenses will increase substantially in the foreseeable future as we:
12
|
·
|
conduct pre-clinical studies and clinical trials for our product candidates in different indications;
|
|
·
|
develop, formulate, manufacture and commercialize our product candidates either independently or with partners;
|
|
·
|
pursue, acquire or in-license additional compounds, products or technologies, or expand the use of our technology;
|
|
·
|
maintain, defend and expand the scope of our intellectual property; and
|
|
·
|
hire additional qualified personnel.
|
We will need to generate significant revenues to achieve and maintain profitability. If we cannot successfully develop, obtain regulatory approval for and commercialize our product candidates, either independently or with partners, we will not be able to generate such revenues or achieve or maintain profitability in the future. Our failure to achieve and subsequently maintain profitability could have a material adverse impact on the market price of our common stock.
We have very limited data on the use of our products in humans and will need to perform costly and time consuming clinical trials in order to bring our products to market.
Most of the data that we have on our products is from in-vitro (laboratory) studies or in-vivo animal studies and our human data is from Phase 1 safety studies or small-scale Phase 2a exploratory-studies. We will need to conduct additional Phase 1, 2 and 3 human clinical trials to confirm such results in larger patient populations in order to obtain approval from the FDA of our drug product candidates. Often, positive in-vitro or in-vivo animal studies are not followed by positive results in human clinical trials, and we may not be able to demonstrate that our products are safe and effective for indicated uses in humans or that they are active against antibiotic resistant microbes, do not allow pathogens to develop resistance or are active against bacteria in biofilm. In addition, for each indication, we estimate that it will take between three and five years to conduct the necessary clinical trials.
We currently do not have any marketable products, and if we are unable to develop and obtain regulatory approval for products that we develop, we may never generate product revenues.
To date, our revenues have been derived solely from research and development collaboration and license agreements. We have never generated revenues from sales of products and we cannot guarantee that we will ever have marketable drugs or other products. Satisfaction of all regulatory requirements applicable to our product candidates typically takes many years, is dependent upon the type, complexity, novelty and classification of the product candidates, and requires the expenditure of substantial resources for research and development and testing. Before proceeding with clinical trials, we will conduct pre-clinical studies, which may, or may not be, valid predictors of potential outcomes in humans. If pre-clinical studies are favorable, we will then begin clinical trials. We must demonstrate that our product candidates satisfy rigorous standards of safety and efficacy before we can submit for and gain approval from the FDA and regulatory authorities in other countries. In addition, to compete effectively, our products will need to be easy to use, cost-effective and economical to manufacture on a commercial scale. We may not achieve any of these objectives. We cannot be certain that the clinical development of any of our current product candidates or any other product that we may develop in the future will be successful, that they will receive the regulatory approvals required to commercialize them, or that any of our other in-licensing efforts or pre-clinical testing will yield a product suitable for entry into clinical trials. Our commercial revenues from sales of products will be derived from sales of products that may not be commercially available for at least the next several years, if at all.
We have limited experience in developing drugs and medical devices, and we may be unable to commercialize any of the products we develop.
Development and commercialization of drugs and medical devices involves a lengthy and complex process. We have limited experience in developing products and have never commercialized any of our product candidates. In addition, no one has ever developed or commercialized a product based on our Aganocide compounds, and we cannot assure you that it is possible to develop, obtain regulatory approval for or commercialize any products based on these compounds or that we will be successful in doing so.
Before we can develop and commercialize any new products, we will need to expend significant resources to:
|
·
|
undertake and complete clinical trials to demonstrate the efficacy and safety of our product candidates;
|
|
·
|
maintain and expand our intellectual property rights;
|
|
·
|
obtain marketing and other approvals from the FDA and other regulatory agencies; and
|
|
·
|
select collaborative partners with suitable manufacturing and commercial capabilities.
|
The process of developing new products takes several years. Our product development efforts may fail for many reasons, including:
|
·
|
the failure of our product candidates to demonstrate safety and efficacy;
|
|
·
|
the high cost of clinical trials and our lack of financial and other resources; and
|
|
·
|
our inability to partner with firms with sufficient resources to assist us in conducting clinical trials.
|
13
Success in early clinical trials often is not replicated in later studies, and few research and development projects result in commercial products. At any point, we may abandon development of a product candidate or we may be required to expend considerable resources repeating clinical trials, which would eliminate or adversely impact the timing for revenues from those product candidates. If a clinical study fails to demonstrate the safety and effectiveness of our product candidates, we may abandon the development of the product or product feature that was the subject of the clinical trial, which could harm our business.
Even if we develop products for commercial use, these products may not be accepted by the medical and pharmaceutical marketplaces or be capable of being offered at prices that will enable us to become profitable. We cannot assure you that our products will be approved by regulatory authorities or ultimately prove to be useful for commercial markets, meet applicable regulatory standards, or be successfully marketed.
We must maintain and expand expensive finance and accounting systems, procedures and controls in order to grow our business and organization, which will increase our costs and require additional management resources.
We completed our initial public offering, or IPO, in October 2007. As a public reporting company, we are required to comply with the Sarbanes-Oxley Act of 2002 and the related rules and regulations of the SEC and Canadian securities regulatory authorities, including expanded disclosure and accelerated reporting requirements and more complex accounting rules. We are also required to comply with marketplace rules and the heightened corporate governance standards of the NYSE Amex. Compliance with these rules has been expensive, and there are additional rules with which we have not yet needed to comply but which we may need to comply with in the future.
Following the passage of the Dodd-Frank Wall Street Reform and Consumer Protection Act we are not required to have our independent auditors audit our internal control over financial reporting, but if the value of our common stock not held by our affiliates at the end of the second quarter in a fiscal year exceeds $75.0 million we will be required to do so. If we reach the $75.0 million value described above and our independent registered public accounting firm is unable to provide us with an unqualified report as to the effectiveness of our internal control over financial reporting as of the date of our Annual Report on Form 10-K, or our business grows and we are not able to comply with accelerated reporting obligations, our ability to obtain additional financing could be impaired. In addition, investors could lose confidence in the reliability of our internal control over financial reporting and in the accuracy of our periodic reports filed with the SEC and with Canadian securities regulatory authorities. A lack of investor confidence in the reliability and accuracy of our public reporting could cause our stock price to decline.
Our current research collaborations with Alcon and Galderma may fail, and entering into additional collaborations may not happen, resulting in a decrease in funding and inhibition of our ability to continue developing products.
We have entered into a collaborative arrangement with Alcon, and we rely on Alcon for joint intellectual property creation and have relied upon them for a significant portion of our revenues. Under the agreement, we licensed to Alcon the exclusive rights (except for certain retained marketing rights) to develop, manufacture and commercialize products incorporating the Aganocide compounds for application in connection with the eye, ear and sinus and for use in contact lens solutions. Under the terms of the agreement we received semi-annual payments from Alcon to support on-going research and development activities over the four-year funding term of the agreement, which ended in August 2010. On November 18, 2010 Alcon extended the funding term to December 31, 2015, subject to earlier termination of the agreement, at Alcon’s election, with six months prior written notice.
During 2010, Alcon concluded a Phase 2 human proof of concept trial of NovaBay’s lead compound, NVC-422, for the treatment of adenoviral conjunctivitis, a type of “Pink Eye”. Alcon and NovaBay intend to analyze the safety and microbiological and clinical efficacy of the drug on patients enrolled in the trial and plan to report on these analyses in the first half of 2011. If Alcon were to determine that the data does not warrant continuation of development of NVC-422 for the treatment of adenoviral conjunctivitis, we may not receive any further payments from Alcon, which would have a significant adverse effect on our company and our stock price.
We have also entered into an agreement with Galderma S.A. to develop and commercialize our Aganocide compounds, which covers acne and impetigo and potentially other major dermatological conditions, excluding onychomycosis (nail fungus) and orphan drug indications. We also rely on Galderma for a significant portion of our revenues.
We cannot assure you that our collaborations with Alcon or Galderma or any other collaborative arrangement will be successful, or that we will receive the full amount of research funding, milestone payments or royalties, or that any commercially valuable intellectual property will be created, from these arrangements. We cannot assure you that the recent change in ownership in Alcon by virtue of the acquisition by Novartis of Alcon’s majority stake, will not result in management redirection which in turn, could negatively impact our collaboration with Alcon. If Alcon or Galderma were to breach or terminate its agreement with us or otherwise fail to conduct its collaborative activities successfully and in a timely manner, the research contemplated by our collaboration with them could be delayed or terminated and our costs of performing studies may increase. We plan on entering into additional collaborations and licensing arrangements. We may not be able to negotiate additional collaborations on acceptable terms, if at all, and these collaborations may not be successful. Our current and future success depends in part on our ability to enter into successful collaboration arrangements and maintain the collaboration arrangement we currently have. If we are unable to enter into, maintain or extend successful collaborations, our business may be harmed.
14
Our long-term success depends upon the successful development and commercialization of other products from our research and development activities.
Our long-term viability and growth will depend upon the successful development and commercialization of other products from our research and development activities. Product development and commercialization is very expensive and involves a high degree of risk. Only a small number of research and development programs result in the commercialization of a product. Success in early stage clinical trials or preclinical work does not ensure that later stage or larger scale clinical trials will be successful. Even if later stage clinical trials are successful, the risk remains that unexpected concerns may arise from additional data or analysis or that obstacles may arise or issues may be identified in connection with review of clinical data with regulatory authorities or that regulatory authorities may disagree with our view of the data or require additional data or information or additional studies.
Conducting clinical trials is a complex, time-consuming and expensive process. Our ability to complete our clinical trials in a timely fashion depends in large part on a number of key factors including protocol design, regulatory and institutional review board approval, the rate of patient enrollment in clinical trials, and compliance with extensive current good clinical practice requirements. We are in many cases using the services of third-party contract clinical trial providers. If we fail to adequately manage the design, execution and regulatory aspects of our clinical trials, our studies and ultimately our regulatory approvals may be delayed or we may fail to gain approval for our product candidates altogether.
If we do not successfully execute our growth initiatives through the acquisition, partnering and in-licensing of products, technologies or companies, our future performance could be adversely affected.
In addition to the expansion of our pipeline through spending on internal development projects, we anticipate growing through external growth opportunities, which include the acquisition, partnering and in-licensing of products, technologies and companies or the entry into strategic alliances and collaborations. If we are unable to complete or manage these external growth opportunities successfully, we may not be able to grow our business in the way that we currently expect. The availability of high quality opportunities is limited and we are not certain that we will be able to identify suitable candidates or complete transactions on terms that are acceptable to us. In order to pursue such opportunities, we may require significant additional financing, which may not be available to us on favorable terms, if at all. The availability of such financing is limited by the recent tightening of the global credit markets.
We may acquire other businesses or form joint ventures or in-license compounds that could disrupt our business, harm our operating results, dilute your ownership interest in us, or cause us to incur debt or significant expense.
As part of our business strategy, we may pursue acquisitions of complementary businesses and assets, and enter into technology or pharmaceutical compound licensing arrangements. We also may pursue strategic alliances that leverage our core technology and industry experience to enhance our ability to commercialize our product candidates and expand our product offerings or distribution. We have no experience with respect to acquiring other companies and limited experience with respect to the formation of commercial partnering agreements, strategic alliances, joint ventures or in-licensing of compounds. If we make any acquisitions, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. If we in-license any additional compounds, we may fail to develop the product candidates, and spend significant resources before determining whether a compound we have in-licensed will produce revenues. Any future acquisitions or in-licensing by us also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could harm our operating results. Integration of an acquired company also may require management resources that otherwise would be available for ongoing development of our existing business. We may not identify or complete these transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance or joint venture.
To finance any acquisitions, we may choose to issue shares of our common stock as consideration, which would dilute your interest in us. If the price of our common stock is low or volatile, we may not be able to acquire other companies for stock. Alternatively, it may be necessary for us to raise additional funds for acquisitions by incurring indebtedness. Additional funds may not be available on terms that are favorable to us, or at all.
We do not have our own manufacturing capacity, and we plan to rely on partnering arrangements or third-party manufacturers for the manufacture of our potential products.
We do not currently operate manufacturing facilities for clinical or commercial production of our product candidates. We have no experience in drug formulation or manufacturing, and we lack the resources and the capabilities to manufacture any of our product candidates on a clinical or commercial scale. As a result, we have partnered and expect to partner with third parties to manufacture our products or rely on contract manufacturers to supply, store and distribute product supplies for our clinical trials. Any performance failure on the part of our commercial partners or future manufacturers could delay clinical development or regulatory approval of our product candidates or commercialization of our products, producing additional losses and reducing the potential for product revenues.
15
Our products, if developed and commercialized, will require precise, high quality manufacturing. The failure to achieve and maintain high manufacturing standards, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business. Contract manufacturers and partners often encounter difficulties involving production yields, quality control and quality assurance, as well as shortages of qualified personnel. These manufacturers and partners are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with current Good Manufacturing Practice and other applicable government regulations and corresponding foreign standards; however, we do not have control over third-party compliance with these regulations and standards. If any of our manufacturers or partners fails to maintain compliance, the production of our products could be interrupted, resulting in delays, additional costs and potentially lost revenues.
In addition, if the FDA or other regulatory agencies approve any of our product candidates for commercial sale, we will need to manufacture them in larger quantities. Significant scale-up of manufacturing will require validation studies, which the FDA must review and approve. If we are unable to successfully increase the manufacturing capacity for a product, the regulatory approval or commercial launch of any drugs may be delayed or there may be a shortage in supply and our business may be harmed as a result.
We depend on skilled and experienced personnel to operate our business effectively. If we are unable to recruit, hire and retain these employees, our ability to manage and expand our business will be harmed, which would impair our future revenue and profitability.
Our success largely depends on the skills, experience and efforts of our officers, especially our Chief Executive Officer, Chief Financial Officer, Chief Scientific Officer, Chief Alliance Officer and Vice President of Product Development, Vice President of Medical Affairs, Vice President of Business and Corporate Development and other key employees. The efforts of each of these persons is critical to us as we continue to develop our technologies and as we attempt to transition into a company with commercial products. Any of our officers and other key employees may terminate their employment at any time. The loss of any of our senior management team members could weaken our management expertise and harm our ability to compete effectively, develop our technologies and implement our business strategies.
Our ability to retain our skilled labor force and our success in attracting and hiring new skilled employees will be a critical factor in determining whether we will be successful in the future. Our research and development programs and collaborations depend on our ability to attract and retain highly skilled scientists and technicians. We may not be able to attract or retain qualified scientists and technicians in the future due to the intense competition for qualified personnel among life science businesses, particularly in the San Francisco Bay Area. We also face competition from universities and public and private research institutions in recruiting and retaining highly qualified scientific personnel. We have also encountered difficulties in recruiting qualified personnel from outside the San Francisco Bay Area, due to the high housing costs in the area.
If we fail to manage our growth effectively, we may be unable to execute our business plan.
Our future growth, if any, may cause a significant strain on our management, and our operational, financial and other resources. Our ability to manage our growth effectively will require us to implement and improve our operational, financial and management information systems and to expand, train, manage and motivate our employees. These demands may require the hiring of additional management personnel and the development of additional expertise by management. Any increase in resources devoted to research and product development without a corresponding increase in our operational, financial and management information systems could have a material adverse effect on our business, financial condition, and results of operations.
If our facilities become inoperable, we will be unable to perform our research and development activities, fulfill the requirements under our collaboration agreement and continue developing products and, as a result, our business will be harmed.
We do not have redundant laboratory facilities. We perform substantially all of our research, development and testing in our laboratory located in Emeryville, California. Emeryville is situated on or near active earthquake fault lines. Our facility and the equipment we use to perform our research, development and testing would be costly to replace and could require substantial lead time to repair or replace. The facility may be harmed or rendered inoperable by natural or man-made disasters, including earthquakes, flooding and power outages, which may render it difficult or impossible for us to perform our research, development and testing for some period of time. The inability to perform our research and development activities may result in the loss of partners or harm our reputation, and we may be unable to regain those partnerships in the future. Our insurance coverage for damage to our property and the disruption of our business may not be sufficient to cover all of our potential losses, including the loss of time as well as the costs of lost opportunities, and may not continue to be available to us on acceptable terms, or at all.
16
Obtaining regulatory approval in the United States does not ensure we will obtain regulatory approval in other countries.
We will aim to obtain regulatory approval in the U.S. as well as in other countries. To obtain regulatory approval to market our proposed products outside of the U.S., we and any collaborator must comply with numerous and varying regulatory requirements in other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ significantly from that required to obtain FDA approval. The regulatory approval process in other countries include all of the risk associated with FDA approval as well as additional, presently unanticipated risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects associated with regulatory approval in the U.S., including the risk that our product candidates may not be approved for all indications requested and that such approval may be subject to limitations on the indicated uses for which the product may be marketed. In addition, failure to comply with applicable regulatory requirements in other countries can result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal of the government to renew marketing applications and criminal prosecution.
If we are unable to design, conduct and complete clinical trials successfully, we will not be able to obtain regulatory approval for our products.
In order to obtain FDA approval for our drug product candidates, we must submit to the FDA a New Drug Application, or NDA, demonstrating that the product candidate is safe and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as preclinical studies, as well as human tests, which are referred to as clinical trials.
Any clinical trials we conduct or that are conducted by our partners may not demonstrate the safety or efficacy of our product candidates. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful. Results of later clinical trials may not replicate the results of prior clinical trials and pre-clinical testing. Even if the results of one or more of our clinical trials are positive, we may have to commit substantial time and additional resources to conducting further preclinical studies or clinical trials before we can submit NDAs or obtain FDA approvals for our product candidates, and positive results of a clinical trial may not be replicated in subsequent trials.
Clinical trials are very expensive and difficult to design and implement. The clinical trial process is also time-consuming. Furthermore, if participating patients in clinical studies suffer drug-related adverse reactions during the course of such trials, or if we or the FDA believe that participating patients are being exposed to unacceptable health risks, we will have to suspend or terminate our clinical trials. Failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon clinical trials or to repeat clinical studies. Further, because our product candidates are all in the same class of compounds, failure in one clinical trial may cause us or our partners to have to suspend or terminate other clinical trials. For example, if toxicity issues were to arise in one clinical trial, it could indicate that all of our product candidates have toxicity issues.
In addition, the completion of clinical trials can be delayed by numerous factors, including:
|
·
|
delays in identifying and agreeing on acceptable terms with prospective clinical trial sites;
|
|
·
|
slower than expected rates of patient recruitment and enrollment;
|
|
·
|
increases in time required to complete monitoring of patients during or after participation in a trial; and
|
|
·
|
unexpected need for additional patient-related data.
|
Any of these delays, if significant, could impact the timing, approval and commercialization of our product candidates and could significantly increase our overall costs of drug development.
Even if our clinical trials are completed as planned, their results may not support our expectations or intended marketing claims. The clinical trials process may fail to demonstrate that our products are safe and effective for indicated uses. Such failure would cause us to abandon a product candidate for some indications and could delay development of other product candidates.
Government agencies may establish usage guidelines that directly apply to our proposed products or change legislation or regulations to which we are subject.
Government usage guidelines typically address matters such as usage and dose, among other factors. Application of these guidelines could limit the use of products that we may develop. In addition there can be no assurance that government regulations applicable to our proposed products or the interpretation thereof will not change and thereby prevent the marketing of some or all of our products for a period of time or permanently. The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the U.S. or in other countries.
17
Our product candidates may be classified as a drug or a medical device, depending on the mechanism of action or indication for use and prior precedent, and a change in the classification may have an adverse impact on our revenues or our ability to obtain necessary regulatory approvals.
Several potential indications for our product candidates may be regulated under the medical device regulations of the FDA administered by the Center for Devices and Radiological Health and the same physical product may be regulated by the FDA’s Center for Drug Evaluation and Research for another indication. Alternatively the products could be classified as combination products, in which case both the device and drug centers jointly review the submission. The products may be designated by the FDA as a drug or a medical device depending upon the regulatory definition of a drug and a device, their primary mode of action and the indications for use or product claims. For example, for NVC-422, if the indication is for flushing of urinary catheters, we believe it would be classified as a medical device, whereas we believe it would be considered a drug when it is indicated for the prevention of urinary tract infection. The use of NVC-101 as a solution for cleansing and debriding was cleared as a Class I medical device. The determination as to whether a particular indication is considered a drug or a device is also based in part upon precedent. A reclassification by the FDA of an indication from a device to a drug indication during our development for that indication could have a significant adverse impact due to the more rigorous and lengthy approval process required for drugs, as compared to medical devices. Such a change in classification can significantly increase development costs and prolong the time for development and approval, thus delaying revenues. A reclassification of an indication after approval from a drug to a device could result in a change in classification for reimbursement. In many cases, reimbursement for devices is significantly lower than for drugs and there could be a significant negative impact on our revenues.
We and our collaborators are and will be subject to ongoing FDA obligations and continued regulatory review, such as continued safety reporting requirements, and we and our collaborators may also be subject to additional FDA post-marketing obligations or new regulations, all of which may result in significant expense and which may limit our ability to commercialize our medical device and drug products candidates.
Any regulatory approvals that we receive may also be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies. The FDA may require us to commit to perform lengthy Phase IV post-approval studies (as further described below), for which we would have to expend additional resources, which could have an adverse effect on our operating results and financial condition. In addition, if the FDA approves any of our drug product candidates, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping for the drug will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the drugs, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the drugs or the withdrawal of the drugs from the market. If we are not able to maintain regulatory compliance, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Any of these events could prevent us from marketing any products we may develop and our business could suffer.
Conducting clinical trials of our product candidates may expose us to expensive liability claims, and we may not be able to maintain liability insurance on reasonable terms or at all.
The risk of clinical trial liability is inherent in the testing of pharmaceutical and medical device products. If we cannot successfully defend ourselves against any clinical trial claims, we may incur substantial liabilities or be required to limit or terminate testing of one or more of our product candidates. Our inability to obtain sufficient clinical trial insurance at an acceptable cost to protect us against potential clinical trial claims could prevent or inhibit the commercialization of our product candidates. Our current clinical trial insurance covers individual and aggregate claims up to $3.0 million. This insurance may not cover all claims and we may not be able to obtain additional insurance coverage at a reasonable cost, if at all, in the future. In addition, if our agreements with any future corporate collaborators entitle us to indemnification against product liability losses and clinical trial liability, such indemnification may not be available or adequate should any claim arise.
If we use biological and hazardous materials in a manner that causes injury, we could be liable for damages. Compliance with environmental regulations can be expensive, and noncompliance with these regulations may result in adverse publicity and potentially significant monetary damages and fines.
Our activities currently require the controlled use of potentially harmful biological materials and other hazardous materials and chemicals and may in the future require the use of radioactive compounds. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject, on an ongoing basis, to U.S. federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations might be significant and could negatively affect our operating results. In addition, if more stringent laws and regulations are adopted in the future, the costs of compliance with these new laws and regulations could be substantial or could impose significant changes in our testing and production process.
18
The pharmaceutical and biopharmaceutical industries are characterized by patent litigation and any litigation or claim against us may cause us to incur substantial costs, and could place a significant strain on our financial resources, divert the attention of management from our business and harm our reputation.
There has been substantial litigation in the pharmaceutical and biopharmaceutical industries with respect to the manufacture, use and sale of new products that are the subject of conflicting patent rights. For the most part, these lawsuits relate to the validity, enforceability and infringement of patents. Generic companies are encouraged to challenge the patents of pharmaceutical products in the United States because a successful challenger can obtain six months of exclusivity as a generic product under the Hatch-Waxman Act. We expect that we will rely upon patents, trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position and we may initiate claims to defend our intellectual property rights as a result. Other parties may have issued patents or be issued patents that may prevent the sale of our products or know-how or require us to license such patents and pay significant fees or royalties in order to produce our products. In addition, future patents may issue to third parties which our technology may infringe. Because patent applications can take many years to issue, there may be applications now pending of which we are unaware that may later result in issued patents that our products may infringe.
Intellectual property litigation, regardless of outcome, is expensive and time-consuming, could divert management’s attention from our business and have a material negative effect on our business, operating results or financial condition. If such a dispute were to be resolved against us, we may be required to pay substantial damages, including treble damages and attorneys fees if we were to be found to have willfully infringed a third party’s patent, to the party claiming infringement, develop non-infringing technology, stop selling any products we develop, cease using technology that contains the allegedly infringing intellectual property or enter into royalty or license agreements that may not be available on acceptable or commercially practical terms, if at all. Our failure to develop non-infringing technologies or license the proprietary rights on a timely basis could harm our business. Modification of any products we develop or development of new products thereafter could require us to conduct additional clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. In addition, parties making infringement claims may be able to obtain an injunction that would prevent us from selling any products we develop, which could harm our business.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Some of our employees may have been previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to commercialize product candidates, which could severely harm our business.
If product liability lawsuits are brought against us, they could result in costly litigation and significant liabilities.
The product candidates we are developing or attempting to develop will, in most cases, undergo extensive clinical testing and will require approval from the applicable regulatory authorities prior to sale. However, despite all reasonable efforts to ensure safety, it is possible that we or our collaborators will sell products which are defective, to which patients react in an unexpected manner, or which are alleged to have side effects. The manufacture and sale of such products may expose us to potential liability, and the industries in which our products are likely to be sold have been subject to significant product liability litigation. Any claims, with or without merit, could result in costly litigation, reduced sales, significant liabilities and diversion of our management’s time and attention and could have a material adverse effect on our financial condition, business and results of operations.
If a product liability claim is brought against us, we may be required to pay legal and other expenses to defend the claim and, if the claim is successful, damage awards may not be covered, in whole or in part, by our insurance. We may not have sufficient capital resources to pay a judgment, in which case our creditors could levy against our assets. We may also be obligated to indemnify our collaborators and make payments to other parties with respect to product liability damages and claims. Defending any product liability claims, or indemnifying others against those claims, could require us to expend significant financial and managerial resources.
Failure to obtain sufficient quantities of products and substances necessary for research and development, pre-clinical trials, human clinical trials and product commercialization that are of acceptable quality at reasonable prices or at all could constrain our product development and have a material adverse effect on our business.
We have relied and will continue to rely on contract manufacturers for the foreseeable future to produce quantities of products and substances necessary for research and development, pre-clinical trials, human clinical trials and product commercialization. It will be important to us that such products and substances can be manufactured at a cost and in quantities necessary to make them commercially viable. At this point in time, we have not attempted to identify, and do not know whether there will be, any third party manufacturers which will be able to meet our needs with respect to timing, quantity and quality for commercial production. In addition, if we are unable to contract for a sufficient supply or required products and substances on acceptable terms, or if we should encounter delays or difficulties in our relationships with manufacturers, our research and development, pre-clinical and clinical testing would be delayed, thereby delaying the submission of product candidates for regulatory approval or the market introduction and subsequent sales of products. Any such delay may have a material adverse effect on our business, financial condition and results of operations.
19
Because our clinical development activities rely heavily on sensitive and personal information, an area which is highly regulated by privacy laws, we may not be able to generate, maintain or access essential patient samples or data to continue our research and development efforts in the future on reasonable terms and conditions, which may adversely affect our business.
As a result of our clinical development, we will have access to very sensitive data regarding the patients enrolled in our clinical trials. This data will contain information that is personal in nature. The maintenance of this data is subject to certain privacy-related laws, which impose upon us administrative and financial burdens, and litigation risks. For instance, the rules promulgated by the Department of Health and Human Services under the Health Insurance Portability and Accountability Act, or HIPAA, creates national standards to protect patients’ medical records and other personal information in the U.S.. These rules require that healthcare providers and other covered entities obtain written authorizations from patients prior to disclosing protected health care information of the patient to companies like NovaBay. If the patient fails to execute an authorization or the authorization fails to contain all required provisions, then we will not be allowed access to the patient’s information and our research efforts can be substantially delayed. Furthermore, use of protected health information that is provided to us pursuant to a valid patient authorization is subject to the limits set forth in the authorization (i.e., for use in research and in submissions to regulatory authorities for product approvals). As such, we are required to implement policies, procedures and reasonable and appropriate security measures to protect individually identifiable health information we receive from covered entities, and to ensure such information is used only as authorized by the patient. Any violations of these rules by us could subject us to civil and criminal penalties and adverse publicity, and could harm our ability to initiate and complete clinical studies required to support regulatory applications for our proposed products. In addition, HIPAA does not replace federal, state, or other laws that may grant individuals even greater privacy protections. We can provide no assurance that future legislation will not prevent us from generating or maintaining personal data or that patients will consent to the use of their personal information, either of which may prevent us from undertaking or publishing essential research. These burdens or risks may prove too great for us to reasonably bear, and may adversely affect our ability to function profitably in the future.
We may be subject to fines, penalties, injunctions and other sanctions if we are deemed to be promoting the use of our products for non-FDA-approved, or off-label, uses.
Our business and future growth depend on the development, use and ultimate sale of products that are subject to FDA regulation, clearance and approval. Under the U.S. Federal Food, Drug, and Cosmetic Act and other laws, we are prohibited from promoting our products for off-label uses. This means that we may not make claims about the safety or effectiveness of our products and may not proactively discuss or provide information on the use of our products, except as allowed by the FDA.
There is a risk that the FDA or other federal or state law enforcement authorities could determine that the nature and scope of our sales and marketing activities may constitute the promotion of our products for a non-FDA-approved use in violation of applicable law. We also face the risk that the FDA or other regulatory authorities might pursue enforcement based on past activities that we have discontinued or changed, including sales activities, arrangements with institutions and doctors, educational and training programs and other activities.
Government investigations concerning the promotion of off-label uses and related issues are typically expensive, disruptive and burdensome and generate negative publicity. If our promotional activities are found to be in violation of applicable law or if we agree to a settlement in connection with an enforcement action, we would likely face significant fines and penalties and would likely be required to substantially change our sales, promotion, grant and educational activities. In addition, were any enforcement actions against us or our senior officers to arise, we could be excluded from participation in U.S. government healthcare programs such as Medicare and Medicaid.
If we are unable to protect our intellectual property, our competitors could develop and market products similar to ours that may reduce demand for our products.
Our success, competitive position and potential future revenues will depend in significant part on our ability to protect our intellectual property. We rely on the patent, trademark, copyright and trade secret laws of the U.S. and other countries, as well as confidentiality and nondisclosure agreements, to protect our intellectual property rights. We apply for patents covering our technologies as we deem appropriate.
NovaBay aggressively protects and enforces its patent rights worldwide. However, certain risks remain. There is no assurance that patents will issue from any of our applications or, for those patents we have or that do issue, that the claims will be sufficiently broad to protect our proprietary rights, or that it will be economically possible to pursue sufficient numbers of patents to afford significant protection. For example, we do not have any composition of matter patent directed to the NVC-101 composition. If a potential competitor introduces a similar method of using NVC-101 with a similar composition that does not fall within the scope of the method of treatment claims, then we or a potential marketing partner would be unable to rely on the allowed claims to protect its market position for the method of using the NVC-101 composition, and any revenues arising from such protection would be adversely impacted.
20
In addition, there is no assurance that any patents issued to us or licensed or assigned to us by third parties will not be challenged, invalidated, found unenforceable or circumvented, or that the rights granted thereunder will provide competitive advantages to us. If we or our collaborators or licensors fail to file, prosecute or maintain certain patents, our competitors could market products that contain features and clinical benefits similar to those of any products we develop, and demand for our products could decline as a result. Further, although we have taken steps to protect our intellectual property and proprietary technology, third parties may be able to design around our patents or, if they do infringe upon our technology, we may not be successful or have sufficient resources in pursuing a claim of infringement against those third parties. Any pursuit of an infringement claim by us may involve substantial expense and diversion of management attention.
We also rely on trade secrets and proprietary know-how that we seek to protect by confidentiality agreements with our employees, consultants and collaborators. If these agreements are not enforceable, or are breached, we may not have adequate remedies for any breach, and our trade secrets and proprietary know-how may become known or be independently discovered by competitors.
We operate in the State of California. The laws of the State prevent us from imposing a delay before an employee who may have access to trade secrets and proprietary know-how can commence employment with a competing company. Although we may be able to pursue legal action against competitive companies improperly using our proprietary information, we may not be aware of any use of our trade secrets and proprietary know-how until after significant damage has been done to our company.
Furthermore, the laws of foreign countries may not protect our intellectual property rights to the same extent as the laws of the U.S. If our intellectual property does not provide significant protection against foreign or domestic competition, our competitors, including generic manufacturers, could compete more directly with us, which could result in a decrease in our market share. All of these factors may harm our competitive position.
If bacteria develop resistance to Aganocide compounds, our revenues could be significantly reduced.
Based on our understanding of the hypothesis of the mechanism of action of our Aganocide compounds, we do not expect bacteria to be able to develop resistance to Aganocide compounds. However, we cannot assure you that one or more strains of bacteria will not develop resistance to our compounds, either because our hypothesis of the mechanism of action is incorrect or because a strain of bacteria undergoes some unforeseen genetic mutation that permits it to survive. Since we expect lack of resistance to be a major factor in the commercialization of our product candidates, the discovery of such resistance would have a major adverse impact on the acceptability and sales of our products.
If physicians and patients do not accept and use our products, we will not achieve sufficient product revenues and our business will suffer.
Even if the FDA approves product candidates that we develop, physicians and patients may not accept and use them. Acceptance and use of our products may depend on a number of factors including:
|
·
|
perceptions by members of the healthcare community, including physicians, about the safety and effectiveness of our products;
|
|
·
|
published studies demonstrating the cost-effectiveness of our products relative to competing products;
|
|
·
|
availability of reimbursement for our products from government or healthcare payers; and
|
|
·
|
effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any.
|
The failure of any of our products to find market acceptance would harm our business and could require us to seek additional financing.
If we are unable to develop our own sales, marketing and distribution capabilities, or if we are not successful in contracting with third parties for these services on favorable terms, or at all, revenues from any products we develop could be disappointing.
We currently have no internal sales, marketing or distribution capabilities. In order to commercialize any product candidates approved by the FDA, we will either have to develop such capabilities internally or collaborate with third parties who can perform these services for us. If we decide to commercialize any products we develop, we may not be able to hire the necessary experienced personnel and build sales, marketing and distribution operations which are capable of successfully launching new products and generating sufficient product revenues. In addition, establishing such operations will take time and involve significant expense.
21
If we decide to enter into co-promotion or other licensing arrangements with third parties, we may be unable to identify acceptable partners because the number of potential partners is limited and because of competition from others for similar alliances with potential partners. Even if we are able to identify one or more acceptable partners, we may not be able to enter into any partnering arrangements on favorable terms, or at all. If we enter into any partnering arrangements, our revenues are likely to be lower than if we marketed and sold our products ourselves.
In addition, any revenues we receive would depend upon our partners’ efforts which may not be adequate due to lack of attention or resource commitments, management turnover, change of strategic focus, further business combinations or other factors outside of our control. Depending upon the terms of our agreements, the remedies we have against an under-performing partner may be limited. If we were to terminate the relationship, it may be difficult or impossible to find a replacement partner on acceptable terms, or at all.
If we cannot compete successfully for market share against other companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our product candidates is characterized by intense competition and rapid technological advances. If our product candidates receive FDA approval and are launched they will compete with a number of existing and future drugs, devices and therapies developed, manufactured and marketed by others. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products are unable to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We will compete for market share against fully integrated pharmaceutical and medical device companies or other companies that develop products independently or collaborate with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. In addition, many of these competitors, either alone or together with their collaborative partners, have substantially greater capital resources, larger research and development staffs and facilities, and greater financial resources than we do, as well as significantly greater experience in:
|
·
|
developing drugs and devices;
|
|
·
|
conducting preclinical testing and human clinical trials;
|
|
·
|
obtaining FDA and other regulatory approvals of product candidates;
|
|
·
|
formulating and manufacturing products; and
|
|
·
|
launching, marketing, distributing and selling products.
|
Our competitors may:
|
·
|
develop and patent processes or products earlier than we will;
|
|
·
|
develop and commercialize products that are less expensive or more efficient than any products that we may develop;
|
|
·
|
obtain regulatory approvals for competing products more rapidly than we will; and
|
|
·
|
improve upon existing technological approaches or develop new or different approaches that render any technology or products we develop obsolete or uncompetitive.
|
We cannot assure you that our competitors will not succeed in developing technologies and products that are more effective than any developed by us or that would render our technologies and any products we develop obsolete. If we are unable to compete successfully against current or future competitors, we may be unable to obtain market acceptance for any product candidates that we create, which could prevent us from generating revenues or achieving profitability and could cause the market price of our common stock to decline.
Our ability to generate revenues from any products we develop will be diminished if we fail to obtain acceptable prices or an adequate level of reimbursement for our products from healthcare payers.
Our ability to commercialize our product candidates will depend, in part, on the extent to which health insurers, government authorities and other third-party payers will reimburse the costs of products which may be developed by us or our partners. We expect that a portion of our economic return from partnering arrangements with pharmaceutical companies and other collaborators will be derived from royalties, fees or other revenues linked to final sales of products that we or our partners develop. Newly-approved pharmaceuticals and other products which are developed by us or our partners will not necessarily be reimbursed by third-party payers or may not be reimbursed at levels sufficient to generate significant sales. Government and other third-party payers are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement for new drugs or medical devices. Cost control initiatives such as these could adversely affect our or our collaborators’ ability to commercialize products. In addition, real or anticipated cost control initiatives for final products may reduce the willingness of pharmaceutical companies or other potential partners to collaborate with us on the development of new products.
22
Significant uncertainty exists as to the reimbursement status of newly-approved healthcare products. Healthcare payers, including Medicare, health maintenance organizations and managed care organizations, are challenging the prices charged for medical products or are seeking pharmacoeconomic data to justify formulary acceptance and reimbursement practices. We currently have not generated pharmacoeconomic data on any of our product candidates. Government and other healthcare payers increasingly are attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs and medical devices, and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has or has not granted labeling approval. Adequate third-party insurance coverage may not be available to patients for any products we discover and develop, alone or with collaborators. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for our products, market acceptance of our product candidates could be limited.
Health care reform measures could limit the prices we or our collaborative partners can obtain for our potential products, or impose additional costs on us.
In March 2010, the U.S. Congress adopted and President Obama signed into law comprehensive health care reform legislation through the passage of the Patient Protection and Affordable Health Care Act (H.R. 3590) and the Health Care and Education Reconciliation Act (H.R. 4872). While we anticipate that this legislation may, over time, increase the number of patients who have insurance coverage for pharmaceutical products, it also imposes cost containment measures that may adversely affect the amount of reimbursement for pharmaceutical products. In addition, such legislation contains a number of provisions designed to generate the revenues necessary to fund the coverage expansion, including new fees or taxes on certain health-related industries.
Many of the details of the new law will be included in new and revised regulations, which have not yet been promulgated, and require additional guidance and specificity to be provided by the Department of Health and Human Services, Department of Labor and Department of the Treasury. Accordingly, while it is too early to understand and predict the ultimate impact of the new legislation on our business, the legislation could have a material adverse effect on our business.
Risks Relating to Owning Our Common Stock
The price of our common stock may fluctuate substantially, which may result in losses to our stockholders.
The stock prices of many companies in the pharmaceutical and biotechnology industry have generally experienced wide fluctuations, which are often unrelated to the operating performance of those companies. The market price of our common stock is likely to be volatile and could fluctuate in response to, among other things:
|
·
|
the results of preclinical or clinical trials relating to our product candidates;
|
|
·
|
the announcement of new products by us or our competitors;
|
|
·
|
announcement of partnering arrangements by us or our competitors;
|
|
·
|
quarterly variations in our or our competitors’ results of operations;
|
|
·
|
announcements by us related to litigation;
|
|
·
|
changes in our earnings estimates, investors’ perceptions, recommendations by securities analysts or our failure to achieve analysts’ earning estimates;
|
|
·
|
developments in our industry; and
|
|
·
|
general, economic and market conditions, including the recent volatility in the financial markets and decrease in consumer confidence and other factors unrelated to our operating performance or the operating performance of our competitors.
|
The volume of trading of our common stock may be low, leaving our common stock open to risk of high volatility.
The number of shares of our common stock being traded may be very low. Any stockholder wishing to sell his/her stock may cause a significant fluctuation in the price of our stock. In addition, low trading volume of a stock increases the possibility that, despite rules against such activity, the price of the stock may be manipulated by persons acting in their own self-interest. We may not have adequate market makers and market making activity to prevent manipulation.
Our directors, executive officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
As of December 31, 2010, our officers and directors collectively controlled approximately 4,088,481 shares of our outstanding common stock (and approximately 5,556,279 shares of our common stock when including options held by them which were exercisable as of or within 60 days from January 31, 2011). Furthermore, as of December 31, 2010, our largest stockholder, a family trust established and controlled by Dr. Ramin Najafi, our Chairman and Chief Executive Officer, beneficially owned 3,128,700 shares or 13.4 % of our outstanding common stock (and approximately 3,321,551 shares of our common stock when including options held by Dr. Najafi which were exercisable as of or within 60 days from January 31, 2011). As a result, Dr. Najafi can significantly influence the management and affairs of our company and most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control and might adversely affect the market price of our common stock. This concentration of ownership may not be in the best interests of our other stockholders.
23
Our limited operating history may make it difficult for you to evaluate our business and to assess our future viability.
Our operations to date have been limited to organizing and staffing our company, developing our technology, researching and developing our compounds, and conducting preclinical studies and early-stage clinical trials of our compounds. We have not demonstrated the ability to succeed in achieving clinical endpoints, obtain regulatory approvals, formulate and manufacture products on a commercial scale or conduct sales and marketing activities. Consequently, any predictions you make about our future success or viability are unlikely to be as accurate as they could be if we had a longer operating history.
Our amended and restated certificate of incorporation and bylaws and Delaware law, contain provisions that could discourage a third party from making a takeover offer that is beneficial to our stockholders.
Anti-takeover provisions of our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law may have the effect of deterring or delaying attempts by our stockholders to remove or replace management, engage in proxy contests and effect changes in control. The provisions of our charter documents include:
|
·
|
a classified board so that only one of the three classes of directors on our Board of Directors is elected each year;
|
|
·
|
elimination of cumulative voting in the election of directors;
|
|
·
|
procedures for advance notification of stockholder nominations and proposals;
|
|
·
|
the ability of our Board of Directors to amend our bylaws without stockholder approval; and
|
|
·
|
the ability of our Board of Directors to issue up to 5,000,000 shares of preferred stock without stockholder approval upon the terms and conditions and with the rights, privileges and preferences as our Board of Directors may determine.
|
In addition, as a Delaware corporation, we are subject to the Delaware General Corporation Law, which includes provisions that may have the effect of deterring hostile takeovers or delaying or preventing changes in control or management of our company. Provisions of the Delaware General Corporation Law could make it more difficult for a third party to acquire a majority of our outstanding voting stock by discouraging a hostile bid, or delaying, preventing or deterring a merger, acquisition or tender offer in which our stockholders could receive a premium for their shares, or effect a proxy contest for control of NovaBay or other changes in our management.
We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of our stock.
We have never paid cash dividends on our common stock and do not anticipate paying cash dividends on our common stock in the foreseeable future. The payment of dividends on our common stock will depend on our earnings, financial condition and other business and economic factors affecting us at such time as our Board of Directors may consider relevant. If we do not pay dividends, you will experience a return on your investment in our shares only if our stock price appreciates. We cannot assure you that you will receive a return on your investment when you do sell your shares or that you will not lose the entire amount of your investment.
|
ITEM 1B.
|
UNRESOLVED STAFF COMMENTS
|
Not applicable.
|
ITEM 2.
|
PROPERTIES
|
Our principal executive offices and our research and development and administrative operations are located in Emeryville, California. In total, we lease approximately 18,500 square feet of office space in the facility pursuant to a lease agreement expiring on October 31, 2015.
|
ITEM 3.
|
LEGAL PROCEEDINGS
|
We are currently not a party to, nor is our property the subject matter of, any pending or, to our knowledge, contemplated material legal proceedings. From time to time, we may become party to litigation and subject to claims arising in the ordinary course of our business.
|
ITEM 4.
|
(REMOVED AND RESERVED)
|
24
PART II
|
ITEM 5.
|
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
|
Market Information
Since October 25, 2007, our common stock has been listed on the NYSE Amex, formerly American Stock Exchange, and the Toronto Stock Exchange (TSX) under the symbol “NBY.” Prior to such time, there was no established public trading market for our common stock. On December 31, 2008, we notified the TSX of our intent to voluntarily remove our listing of common stock from the TSX in order to consolidate trading on the NYSE Amex. The following table sets forth, for the periods indicated, the high and low sales prices for our common stock as reported by the NYSE Amex:
|
2010
|
2009
|
|||||||||||||||
|
High
|
Low
|
High
|
Low
|
|||||||||||||
|
First Quarter
|
2.71 | 1.91 | 2.95 | 1.02 | ||||||||||||
|
Second Quarter
|
2.55 | 2.06 | 3.24 | 1.95 | ||||||||||||
|
Third Quarter
|
2.29 | 1.65 | 2.57 | 1.62 | ||||||||||||
|
Fourth Quarter
|
2.05 | 1.64 | 2.39 | 1.63 | ||||||||||||
On March 1, 2011, the last reported sale price of our common stock on the NYSE Amex was $2.03.
Holders
As of March 1, 2011, there were approximately 275 holders of record of our common stock. This figure does not reflect persons or entities that hold their stock in nominee or “street” name through various brokerage firms.
Dividend Policy
We have not paid cash dividends on our common stock since our inception. We currently expect to retain earnings primarily for use in the operation and expansion of our business, and therefore, do not anticipate paying any cash dividends in the near future. Any future determination to pay cash dividends will be at the discretion of our Board of Directors and will be dependent upon our financial condition, results of operations, capital requirements, restrictions under any existing indebtedness and other factors the Board of Directors deems relevant.
25
Performance Graph(1)
The following graph compares our total stockholder returns for the past 38 months to two indices: the Amex Composite Index and the RDG MicroCap Biotechnology Index. The total return for each index assumes the reinvestment of all dividends, if any, paid by companies included in these indices and are calculated as of December 31 of each year.
As a member of the Amex Composite Index, we are required under applicable regulations to use this index as a comparator, and we believe the RDG MicroCap Biotechnology Index is a relevant comparator since it is composed of peer companies in lines-of-business similar to ours.
The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
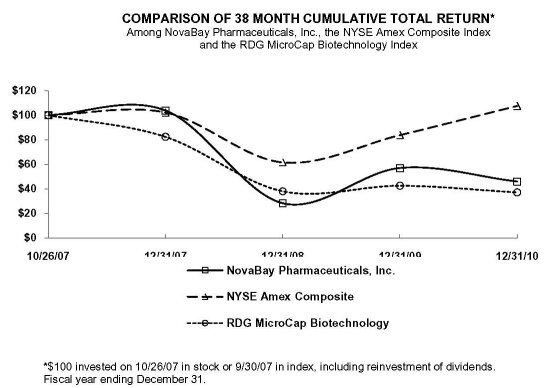
(1) This section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference in any of our filings under the Securities Act or the Exchange Act whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
| 10/07 | 12/07 | 12/08 | 12/09 | 12/10 | ||||||||||||||||
|
NovaBay Pharmaceuticals, Inc.
|
100.00 | 103.84 | 28.18 | 56.91 | 45.86 | |||||||||||||||
|
NYSE Amex Composite
|
100.00 | 102.17 | 61.48 | 83.71 | 107.47 | |||||||||||||||
|
RDG MicroCap Biotechnology
|
100.00 | 82.27 | 37.88 | 42.38 | 36.95 |
Purchases of Equity Securities by the Issuer and Affiliated Purchaser
We did not repurchase any of our outstanding equity securities during the most recent quarter covered by this report.
26
|
ITEM 6.
|
SELECTED FINANCIAL DATA
|
The following selected financial information as of and for the dates and periods indicated have been derived from our audited consolidated financial statements. The information set forth below is not necessarily indicative of results of future operations, and should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operation” in Part II, Item 7 of this report and our consolidated financial statements and related notes included elsewhere in this report.
|
Year Ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
(in thousands, except per share data)
|
||||||||||||||||||||
|
Statements of Operations Data:
|
||||||||||||||||||||
|
License and collaboration revenue:
|
$ | 9,754 | $ | 15,684 | $ | 6,722 | $ | 5,913 | $ | 1,533 | ||||||||||
|
Operating expenses:
|
||||||||||||||||||||
|
Research and development
|
8,616 | 7,337 | 9,595 | 7,421 | 4,087 | |||||||||||||||
|
General and administrative
|
5,654 | 5,607 | 5,636 | 4,368 | 2,972 | |||||||||||||||
|
Total operating expenses
|
14,270 | 12,944 | 15,231 | 11,789 | 7,059 | |||||||||||||||
|
Other income (expense), net
|
258 | (36 | ) | 397 | 488 | 240 | ||||||||||||||
|
Income (loss) before income taxes
|
(4,258 | ) | 2,704 | (8,112 | ) | (5,388 | ) | (5,286 | ) | |||||||||||
|
Provision for income taxes
|
(50 | ) | (7 | ) | (2 | ) | (12 | ) | — | |||||||||||
|
Net income (loss)
|
$ | (4,308 | ) | $ | 2,697 | $ | (8,114 | ) | $ | (5,400 | ) | $ | (5,286 | ) | ||||||
|
Net income (loss) per share:
|
||||||||||||||||||||
|
Basic
|
$ | (0.18 | ) | $ | 0.12 | $ | (0.38 | ) | $ | (0.60 | ) | $ | (0.92 | ) | ||||||
|
Diluted
|
$ | (0.18 | ) | $ | 0.12 | $ | (0.38 | ) | $ | (0.60 | ) | $ | (0.92 | ) | ||||||
|
Shares used in computing net income (loss) per share:
|
||||||||||||||||||||
|
Basic
|
23,326 | 22,404 | 21,312 | 8,974 | 5,715 | |||||||||||||||
|
Diluted
|
23,326 | 23,115 | 21,312 | 8,974 | 5,715 | |||||||||||||||
|
Year Ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
(in thousands)
|
||||||||||||||||||||
|
Balance Sheet Data:
|
||||||||||||||||||||
|
Cash, cash equivalents and short-term investments
|
$ | 12,806 | $ | 11,292 | $ | 12,099 | $ | 22,353 | $ | 11,086 | ||||||||||
|
Working capital
|
11,031 | 11,568 | 8,033 | 18,194 | 7,926 | |||||||||||||||
|
Total assets
|
15,516 | 17,523 | 13,969 | 23,922 | 11,866 | |||||||||||||||
|
Capital lease obligation—current and non-current
|
— | 7 | 49 | 86 | — | |||||||||||||||
|
Equipment loan—current and non-current
|
106 | 470 | 836 | 716 | — | |||||||||||||||
|
Deferred revenue—current and non-current
|
3,689 | 2,167 | 4,167 | 7,517 | 9,167 | |||||||||||||||
|
Convertible preferred stock
|
— | — | — | — | 192 | |||||||||||||||
|
Common stock and additional paid-in capital
|
38,703 | 37,236 | 33,933 | 32,797 | 14,683 | |||||||||||||||
|
Total stockholders’ equity
|
10,490 | 13,345 | 7,345 | 14,320 | 1,813 | |||||||||||||||
27
|
ITEM 7.
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
The following discussion of our financial condition and results of operations should be read together with our consolidated financial statements and related notes included in Part II, Item 8 of this report. This discussion contains forward-looking statements that involve risks and uncertainties. As a result of many factors, such as those set forth under the section entitled “Risk Factors” in Item 1A and elsewhere in this report, our actual results may differ materially from those anticipated in these forward-looking statements.
Overview
NovaBay is a clinical stage biotechnology company developing a first-in-class, anti-infective platform of compounds called the Aganocide® compounds. In laboratories, these compounds have demonstrated equivalent activity to the active antimicrobial molecules generated within white blood cells. The Aganocide compounds are being developed for the topical treatment and prevention of a wide variety of topical infections, including those that are antibiotic resistant.
NovaBay is developing commercial opportunities for its portfolio of anti-infective Aganocide compounds in four distinct healthcare markets: dermatology, ophthalmology, urology and hospital infections. Each of these market segments are underserved by current products and therefore the opportunity exists for improved treatments. NovaBay’s strategy is to address these market opportunities either through partnerships and collaborations or by building an internal organization to strategically market its own products when appropriate from a commercial standpoint.
In August 2006, we entered into a collaboration and license agreement with Alcon Manufacturing Ltd. (Alcon), that provides Alcon with the exclusive rights to develop, manufacture and commercialize products incorporating our Aganocide compounds for the treatment of eye, ear and sinus infections as well as for use in contact lens care. Under the terms of the agreement, Alcon paid an up-front technology access fee of $10.0 million upon the effective date of the agreement. Under the terms of the agreement we also received semi-annual payments from Alcon to support on-going research and development activities during the four year funding term of the agreement, which ended on August 2010. In November Alcon extended the funding term to December 2015, subject to earlier termination of the agreement or, at Alcon’s election, with six months prior written notice. The collaboration also calls for Alcon to pay for all developmental and clinical costs. The research and development support payments include amounts to fund a specified number of personnel engaged in collaboration activities and to reimburse for qualified equipment, materials and contract study costs. Effective 2011, Alcon decreased its on-going financial support of the company’s research and development efforts particularly relating to the funding of the number of personnel engaged in collaboration activities. As product candidates are developed and proceed through clinical trials and approval, we will receive milestone payments. If the products are commercialized, we will also receive royalties on any sales of products containing the Aganocide compounds. NovaBay has the potential to receive up to $70 million in milestones from Alcon, and royalties ranging in single digits on net sales of products once commercialized. During 2010, Alcon concluded a Phase 2 human proof of concept trial of NovaBay’s lead compound, NVC-422, for the treatment of adenoviral conjunctivitis, a type of “Pink Eye”. Alcon and NovaBay intend to analyze the safety and microbiological and clinical efficacy of the drug on patients enrolled in the trial and plan to report on these analyses in the first half of 2011. If Alcon were to determine that the data does not warrant continuation of development of NVC-422 for the treatment of adenoviral conjunctivitis, further payments from Alcon may not be received.
In March 2009, we announced that we entered into a collaboration and license agreement with Galderma S.A. to develop and commercialize our Aganocide compounds, which covers acne and impetigo and potentially other major dermatological conditions. We amended this agreement in December 2009 and 2010. Galderma will be responsible for the development costs of the acne and impetigo product candidates except for costs incurred in Japan. In Japan, Galderma has the option to request that we share such development costs. From the inception of the agreement to December 31, 2010, we have received $11.8 million from Galderma including the technology access fee, milestone payments and R&D funding. NovaBay has the potential to receive up to $62.0 million in predetermined fees, including milestones and personnel reimbursement, with additional funding available to cover product and clinical development. We are entitled to royalties ranging from 10% to 30% on cumulative net sales of products once commercialized, subject to some reductions based on any development costs incurred directly by Galderma. Upon the termination of the agreement under certain circumstances, Galderma will grant NovaBay certain technology licenses which would require NovaBay to make royalty payments to Galderma for such licenses with royalty rates in the low to mid-single digits.
To date, we have generated no revenue from product sales, and we have financed our operations and internal growth primarily through the sale of our capital stock, and the fees received from Alcon and Galderma. As we are a development stage company, we have incurred significant losses since commencement of our operations in July 2002, since we have devoted substantially all of our resources to research and development. As of December 31, 2010, we had an accumulated deficit of $28.2 million. This deficit resulted from research and development expenses as well as general and administrative expenses. We expect to incur net losses over the next several years as we continue our clinical and research and development activities and as we apply for patents and regulatory approvals.
28
Significant Financial Events in 2010 and 2011
In January 2010, we announced that we received $3.75 million in milestone payments from Galderma: a $2.0 million milestone payment having been triggered by the completion of formulation feasibility studies with our Aganocide compounds for topical use and a $1.75 million milestone payment for completing an exploratory clinical study for the treatment of adult acne. Both of these studies were concluded in 2009 and the resulting revenues were recorded in our 2009 results.
In November 2010, we announced the amendment of our collaboration with Alcon, extending the funding term from August 29, 2010 to December 31, 2015, the term of the discovery research program under the agreement. During the said term, Alcon will fund the costs for a specified number of personnel engaged in collaboration activities pursuant to the agreed discovery research plan and development plans described in the agreement, provided that these plans are subject to earlier termination of the agreement or, at Alcon’s election, with six months prior written notice.
In November 2010, we received a cash grant award of $244,000 under the U.S. Government’s Qualifying Therapeutic Discovery Program for our impetigo program.
In December 2010, we expanded our agreement with Galderma to include Impetigo. The expanded agreement has the potential to generate up to $62.0 million in predetermined fees, including milestones and personnel reimbursement, with additional funding available to cover product and clinical development. We are also entitled to royalties ranging from 10% to 30% on cumulative net sales of products once commercialized, subject to some reductions based on any development costs incurred directly by Galderma.
Critical Accounting Policies and Estimates
Our financial statements have been prepared in accordance with accounting principles generally accepted in the U.S. (U.S. GAAP). The preparation of these financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. In preparing these financial statements, management has made its best estimates and judgments of certain amounts included in the financial statements giving due consideration to materiality. On an ongoing basis, we evaluate our estimates and judgments related to revenue recognition, research and development costs, patent costs, stock-based compensation, income taxes and other contingencies. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 of the Notes to Consolidated Financial Statements, included in Part II, Item 8 of this report, we believe that the following accounting policies are most critical to fully understanding and evaluating our reported financial results.
Revenue Recognition
License and collaboration revenue is primarily generated through agreements with strategic partners for the development and commercialization of our product candidates. The terms of the agreements typically include non-refundable upfront fees, funding of research and development activities, payments based upon achievement of certain milestones and royalties on net product sales. In accordance with authoritative guidance, we analyze our multiple element arrangements to determine whether the elements can be separated. We perform our analysis at the inception of the arrangement and as each product or service is delivered. If a product or service is not separable, the combined deliverables are accounted for as a single unit of accounting and revenue is recognized over the performance obligation period. Revenue is recognized when the following criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred and risk of loss has passed; the seller’s price to the buyer is fixed or determinable; and collectability is reasonably assured. If these factors were to vary the resulting change could have a material effect on our revenue recognition and on our results of operations.
Assuming the elements meet the revenue recognition guidelines, the revenue recognition methodology prescribed for each unit of accounting is summarized below:
Upfront Fees—We defer recognition of non-refundable upfront fees if we have continuing performance obligations without which the technology licensed has no utility to the licensee. If we have continuing performance obligations through research and development services that are required because our know-how and expertise related to the technology is proprietary to us, or can only be performed by us, then such up-front fees are deferred and recognized over the estimated period of the performance obligation. We base the estimate of the period of performance on factors in the contract. Actual time frames could vary and could result in material changes to our results of operations. When our collaboration partners request us to continue performing the research and development services in collaboration beyond the initial period of performance, the remaining unamortized deferred revenue and any new continuation or license fees are recognized over the extended period of performance.
29
Funded Research and Development—Revenue from research and development services is recognized during the period in which the services are performed and is based upon the number of full-time-equivalent personnel working on the specific project at the agreed-upon rate. The full-time equivalent amount can vary each year if the contracts allow for a percentage increase determined by relevant salary surveys, if applicable. Reimbursements from collaborative partners for agreed upon direct costs including direct materials and outsourced, or subcontracted, pre-clinical studies are classified as revenue and recognized in the period the reimbursable expenses are incurred. Payments received in advance are recorded as deferred revenue until the research and development services are performed or costs are incurred.
Milestones—Substantive milestone payments are considered to be performance bonuses that are recognized upon achievement of the milestone only if all of the following conditions are met: the milestone payments are non-refundable; achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; substantive effort is involved in achieving the milestone; the amount of the milestone is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and a reasonable amount of time passes between the up-front license payment and the first milestone payment as well as between each subsequent milestone payment. If any of these conditions are not met, the milestone payments are deferred and recognized as revenue over the term of the arrangement as we complete our performance obligations.
Royalties—We recognize royalty revenues from licensed products upon the sale of the related products.
Research and Development Costs
We charge research and development costs to expense as incurred. These costs include salaries and benefits for research and development personnel, costs associated with clinical trials managed by contract research organizations, and other costs associated with research, development and regulatory activities. Research and development costs may vary depending on the type of item or service incurred, location of performance or production, or lack of availability of the item or service, and specificity required in production for certain compounds. We use external service providers to conduct clinical trials, to manufacture supplies of product candidates and to provide various other research and development-related products and services. Our on-going research, clinical and development activities are often performed under agreements we enter into with external service providers. We accrue the costs incurred under these agreements based on factors such as milestones achieved, patient enrollment, estimates of work performed, and historical data for similar arrangements. As actual costs are incurred we will adjust our accruals. Historically, our accruals have been consistent with management’s estimates, and no material adjustments to research and development expenses have been recognized. Subsequent changes in estimates may result in a material change in our expenses, which could also materially affect our results of operations.
Patent Costs
We expense patent costs, including legal expenses, in the period in which they are incurred. Patent expenses are included as general and administrative expenses in our statements of operations. Patent costs may vary depending on the location, domestic or foreign, in which the patent is being secured.
Stock-Based Compensation
Stock-based compensation expense is measured at the grant date for all stock-based awards to employees and directors and is recognized as expense over the requisite service period, which is generally the vesting period. Forfeitures are estimated at the time of grant and reduce compensation expense ratably over the vesting period. This estimate is adjusted periodically based on the extent to which actual forfeitures differ, or are expected to differ, from the previous estimate. See Note 9 for further information regarding stock-based compensation expense and the assumptions used in estimating that expense. For stock options granted to employees, the fair value of the stock options is estimated using a Black-Scholes-Merton valuation model.
Stock-based compensation arrangements with non-employees are recorded at their fair value on the measurement date. The measurement of stock-based compensation is subject to periodic adjustment as the underlying equity instruments vest. Non-employee stock-based compensation charges are amortized over the vesting period on a straight-line basis. For stock options granted to non-employees, the fair value of the stock options is estimated using a Black-Scholes-Merton valuation model.
Income Taxes
We account for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is recognized if it is more likely than not that some portion or the entire deferred tax asset will not be recognized. Valuation allowances are based, in part, on estimates that management must make as to our results in future periods. The actual outcome may not be consistent with our estimate, which would require that we make changes in our valuation allowance.
30
Recent Accounting Pronouncements
In April 2010, the Financial Accounting Standards Board (FASB) issued ASU No. 2010-17 (Topic 605), Revenue Recognition—Milestone Method. This standard provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research and development transactions. The amendments in this update provide guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration that is contingent upon achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all applicable criteria. The amendments in this update are effective for us on a prospective basis for milestones achieved after December 31, 2010. The implementation of this standard is not expected to have a significant impact on our financial position or results of operations.
In February 2010, the FASB issued amended guidance on subsequent events. Under this amended guidance, U.S. securities and Exchange Commission (SEC) filers are no longer required to disclose the date through which subsequent events have been evaluated in originally issued and revised financial statements. The guidance was effective immediately and we adopted these new requirements upon issuance of this guidance. The adoption did not have a material impact on our consolidated financial statements.
In January 2010, the FASB issued Accounting Standards Update (ASU) No. 2010-06 (Topic 820) Fair Value Measurements and Disclosures. This standard amends the disclosure guidance with respect to fair value measurements for both interim and annual reporting periods. Specifically, disclosure is required for significant transfers between Level 1 and Level 2 in the fair value hierarchy; additional disclosures are required for transaction in Level 3 assets and liabilities; and additional disclosure is required of the valuation techniques and inputs used to measure assets and liabilities that fall into Level 2 and Level 3. Except for the additional disclosures for transactions Level 3 items, which were effective for us as of January 1, 2011, the remaining new disclosure requirements were effective for us as of January 1, 2010. The implementation of this standard had no significant impact on our financial position or results of operations.
In September 2009, the FASB issued update 2009-13, ASC 605, Revenue Recognition: Multiple-Deliverable Revenue Arrangements-a consensus of the FASB Emerging Issues Task Force.This guidance addresses how to separate deliverables and how to measure and allocate consideration to one or more units of accounting. Specifically, the guidance requires that consideration be allocated among multiple deliverables based on relative selling prices. The guidance establishes a selling price hierarchy of (1) vendor-specific objective evidence, (2) third-party evidence and (3) estimated selling price. This guidance is effective for annual periods beginning after June 15, 2010 but may be early adopted as of the beginning of an annual period. The adoption is not expected to have a material impact on our consolidated financial statements.
Results of Operations
Comparison of Years Ended December 31, 2010, 2009 and 2008
License and Collaboration Revenue
Total license and collaboration revenue was $9.8 million for the year ended December 31, 2010, compared to $15.7 million for the year ended December 31, 2009, and $6.7 million for the year ended December 31, 2008.
Under the terms of the agreement entered into with Galderma in March 2009, Galderma will pay to NovaBay certain upfront fees, ongoing fees, reimbursements, and milestone payments related to achieving development and commercialization of its Aganocide compounds. We received an upfront technology access fee from Galderma of $1.0 million in 2009, which was amortized on a straight-line basis into revenue over the initial 20 month period of the contract. In December 2010 we received a $3.25 million continuation fee and $500,000 license fee which are being amortized on a straight-line basis into revenue over the additional three year funding term pursuant to the December 2010 amendment to the contract. In 2010, we recognized $786,000 of the upfront technology access fee, continuation fee and license fee under the agreement. We also recognized $850,000 in ongoing research and development fees and $470,000 in materials, equipment and contract study costs under the agreement. During 2009, $500,000 of the upfront technology access fee was recognized. We also recognized $1.2 million in ongoing research and development fees and $1.1 million in materials, equipment and contract study costs, and $3.75 million in milestone payments from Galderma earned in December 2009.
In August 2006, we entered into a collaboration and license agreement with Alcon. The upfront technology access fee of $10.0 million from Alcon was amortized into revenue on a straight-line basis over the four year funding term of the agreement, through August 2010. In 2010, $1.7 million of the upfront technology access fee was recognized. We also recognized $5.4 million in ongoing research and development fees and $562,000 in reimbursements for materials, equipment and contract study costs under the agreement. During 2009, $2.5 million of the upfront technology access fee was recognized, and we recognized $4.3 million in ongoing research and development fees and $1.3 million in reimbursements for materials, equipment and contract study costs, in addition to a $1.0 million milestone payment under the agreement. During 2008, $2.5 million of the upfront technology access fee was recognized, and we recognized $2.7 million in ongoing research and development fees and $1.4 million in materials, equipment and contract study costs under the agreement.
31
Research and Development
At the end of 2010 NovaBay adopted a strategy of focusing on specific healthcare markets as we develop our compounds. NovaBay is developing commercial opportunities for its Aganocide portfolio of anti-infectives in four distinct healthcare markets: dermatology, ophthalmology, urology and hospital infections. Each of these market segments are underserved by current products and therefore the opportunity exists for improved treatments. NovaBay’s strategy is to address these market opportunities either through partnerships and collaborations or by building an internal organization to strategically market its own products when appropriate from a commercial standpoint.
Historically, as we were developing our focus, we did not track our research and development costs by market or indication. Our research and development efforts crossed multiple programs and our programs were not clearly defined, making the tracking of program costs impractical. In 2011, we will be setting up processes to allow us to track our costs based on these four specific healthcare markets and we plan to begin providing investors with detailed financial information pertaining to our efforts in each of these markets in 2012.
Total research and development expenses increased by 17% to $8.6 million for the year ended December 31, 2010 from $7.3 million for the year ended December 31, 2009. This increase was primarily due to increases in our clinical costs as we conducted clinical trials in 2010 and increases in research and development and formulation as we continued to develop new compounds for use in our trials.
Total research and development expenses decreased by 24% to $7.3 million for the year ended December 31, 2009 from $9.6 million for the year ended December 31, 2008. This decrease was primarily due to the following:
- a decrease in clinical costs of $1.7 million resulting from lower costs related to our trials in 2009;
- a decrease in employee costs of $0.5 million resulting from staffing cuts in late 2008; and
- a decrease of $0.3 million in development costs due to reduced process development activities.
These decreases were partially offset by an increase in professional fees of $0.2 million and an increase of $0.1 million in research costs due to additional contract study costs.
We expect to incur increasing research and development expenses in 2011 and in subsequent years as we continue to increase our focus on developing product candidates, both independently and in collaboration with our partners. In particular, we expect to incur ongoing clinical, chemistry, and manufacturing expenses related to four healthcare markets in which we are pursuing opportunities; ophthalmology, dermatology, urology and hospital infections.
General and Administrative
General and administrative expenses of $5.7 million in 2010 was relatively flat compared to $5.6 million in 2009 and 2008. We expect that general and administrative expenses to remain relatively flat in 2011.
Other Income(expense), Net
Other income (expense), net was income of $258,000 for the year ended December 31, 2010, an expense of $36,000 for the year ended December 31, 2009 and an income of $397,000 for the year ended December 31, 2008. The increase in 2010 was primarily due to the receipt of $244,000 related to the Qualified Therapeutic Discovery Project grant from the IRS and a decrease of $46,000 in interest expense in 2010 as we paid down our capital lease and debt balances.
The decrease in 2009 was primarily attributable to decreased interest income related to lower average investment balances and decreased interest rates throughout the year. Interest income relates primarily to interest earned on cash, cash equivalents and investments in marketable securities.
We expect that other income, net will vary based on fluctuations in our cash balances and borrowings under equipment loans and the interest rate paid on such balances and borrowings.
Liquidity and Capital Resources
We have incurred cumulative net losses of $28.2 million since inception through December 31, 2010. We do not expect to generate significant revenue from product candidates for several years. Since inception, we have funded our operations primarily through the sales of our stock. We raised total net proceeds of $11.2 million from sales of our preferred stock in 2002 through 2006. In October 2007, we completed our IPO in which we raised a total of $20.0 million, or approximately $17.1 million in net cash proceeds after deducting underwriting discounts and commissions of $1.4 million and other offering costs of $1.5 million. In August 2009, we completed a registered direct offering and had net proceeds of $1.9 million.
32
Under the terms of August 2006 collaboration and license agreement with Alcon we received an up-front technology access fee of $10.0 million upon the effective date of the agreement. Under the terms of the agreement we also received semi-annual payments from Alcon to support on-going research and development activities during the four year funding term of the agreement, which ended on August 2010. In November 2010 Alcon extended the funding term to December 2015, subject to earlier termination of the agreement or, at Alcon’s election, with six months prior written notice. The collaboration also calls for Alcon to pay for all developmental and clinical costs. The Alcon agreement also provides for milestone payments upon the achievement of specified milestones in each field of use and royalty payments upon the sale of commercialized products. The aggregate milestone payments payable in connection with the ophthalmic, otic and sinus fields are $19.0 million, $12.0 million and $39.0 million, respectively. In 2009 we achieved our first milestone under this agreement, but product has not been commercialized to date. The achievement of the milestones and product commercialization is subject to many risks and uncertainties, including, but not limited to Alcon’s ability to obtain regulatory approval from the FDA and Alcon’s ability to execute its clinical initiatives. Therefore, we cannot predict when, if ever, the remaining milestones specified in the Alcon agreement will be achieved or when we will receive royalties on sales of commercialized products.
In March 2009, we entered into a collaboration and license agreement with Galderma. In December 2009 and 2010, we amended this agreement. Under the terms of the agreement, we received an initial $1.0 million upfront payment that was recognized as revenue over the initial 20 month funding term of the agreement. In December 2009, we recorded revenues of $3.75 million related to milestones from Galderma. In December 2010, we received a continuation payment of $3.25 million and a $500,000 fee to expand the license to include the Asia-Pacific territory that will be recognized as revenue over the additional three year funding period. In addition, Galderma will pay to NovaBay reimbursements, and additional milestone payments related to achieving development and commercialization of its Aganocide compounds.
Cash and Cash Equivalents
As of December 31, 2010, we had cash, cash equivalents, and short-term investments of $12.8 million compared to $11.3 million and $12.1 million at December 31, 2009 and 2008, respectively.
Cash Provided by (Used in) Operating Activities
For the year ended December 31, 2010 cash generated by operating activities was $2.0 million compared to cash used in operating activities of $1.6 million for the year ended December 31, 2009. This increase in cash was primarily attributable to the collection in 2010, of $3.8 million on a receivable that was outstanding in 2009, and a net increase of $1.5 million in deferred revenues as of December 31, 2010 resulting from the receipt of upfront fees and a continuation fee from an amendment to the original collaboration and license agreement with Galderma.
For the year ended December 31, 2009 cash used in operating activities was $1.6 million compared to cash used in operating activities of $9.9 million for the year ended December 31, 2008. The decrease in 2009 of cash used in operating activities is primarily due to decreases in research and development expenses, and to our collection of ongoing research and development fees from Alcon and the collection of research and development fees and milestone payments from Galderma in 2009.
Cash Provided by (Used in) Investing Activities
For the year ended December 31, 2010, cash used in investing activities of $1.2 million was attributable to purchases of short-term investments, offset by maturities and sales, resulting in $991,000 used and purchases of property and equipment of $203,000.
For the year ended December 31, 2009, cash used in investing activities of $1.1 million was attributable to purchases of short-term investments, offset by sales, resulting in $340,000 used and purchases of property and equipment of $731,000.
For the year ended December 31, 2008, cash provided by investing activities of $10.9 million was attributable to sales and maturities of short-term investments offset, in part, by purchases resulting in $11.5 million provided and purchases of property and equipment of $610,000.
Cash Provided by (Used in) Financing Activities
Net cash used in financing activities of $292,000 for the year ended December 31, 2010 was primarily attributable to $364,000 in principal payments on our equipment loan partially offset by cash received on stock option exercises of $81,000.
Net cash provided by financing activities of $1.6 million for the year ended December 31, 2009 was primarily attributable to the $1.9 million received through our shelf offering, partially offset by principal payments on our equipment loan.
Net cash provided by financing activities of $236,000 for the year ended December 31, 2008 was primarily attributable to the $422,000 increase in proceeds from borrowings under our equipment loan, net of payments on the outstanding balance.
33
Quarterly Results of Operations (unaudited)
The following table presents unaudited quarterly results of operations for the eight most recent quarters ending with the quarter ended December 31, 2010. This information has been derived from our unaudited financial statements and has been prepared by us on a basis consistent with our audited annual financial statements and includes all adjustments, consisting only of normal recurring adjustments, which management considers necessary for a fair presentation of the information for the periods presented.
|
Quarter Ended
|
||||||||||||||||||||||||||||||||
|
Dec. 31,
|
Sept. 30,
|
June 30,
|
March 31,
|
Dec. 31,
|
Sept. 30,
|
June 30,
|
March 31,
|
|||||||||||||||||||||||||
|
2010
|
2010
|
2010
|
2010
|
2009
|
2009
|
2009
|
2009
|
|||||||||||||||||||||||||
|
(in thousands, except per share data)
|
||||||||||||||||||||||||||||||||
|
Statements of Operations Data:
|
||||||||||||||||||||||||||||||||
|
License and collaboration revenue:
|
$ | 3,036 | $ | 2,086 | $ | 2,548 | $ | 2,084 | $ | 7,492 | $ | 3,224 | $ | 2,357 | $ | 2,611 | ||||||||||||||||
|
Operating expenses:
|
||||||||||||||||||||||||||||||||
|
Research and development
|
2,009 | 2,245 | 2,129 | 2,233 | 2,528 | 2,004 | 1,444 | 1,361 | ||||||||||||||||||||||||
|
General and administrative
|
1,087 | 1,487 | 1,611 | 1,469 | 1,528 | 1,309 | 1,191 | 1,579 | ||||||||||||||||||||||||
|
Total operating expenses
|
3,096 | 3,732 | 3,740 | 3,702 | 4,056 | 3,313 | 2,635 | 2,940 | ||||||||||||||||||||||||
|
Other income (expense), net
|
271 | 4 | (6 | ) | (11 | ) | (14 | ) | (22 | ) | (11 | ) | 11 | |||||||||||||||||||
|
Income (loss) before income taxes
|
211 | (1,642 | ) | (1,198 | ) | (1,629 | ) | 3,422 | (111 | ) | (289 | ) | (318 | ) | ||||||||||||||||||
|
Provision for income taxes
|
(50 | ) | — | — | — | (7 | ) | — | — | — | ||||||||||||||||||||||
|
Net income (loss)
|
$ | 161 | $ | (1,642 | ) | $ | (1,198 | ) | $ | (1,629 | ) | $ | 3,415 | $ | (111 | ) | $ | (289 | ) | $ | (318 | ) | ||||||||||
|
Net income (loss) per share:
|
||||||||||||||||||||||||||||||||
|
Basic
|
$ | 0.01 | $ | (0.07 | ) | $ | (0.05 | ) | $ | (0.07 | ) | $ | 0.15 | $ | (0.00 | ) | $ | (0.01 | ) | $ | (0.01 | ) | ||||||||||
|
Diluted
|
$ | 0.01 | $ | (0.07 | ) | $ | (0.05 | ) | $ | (0.07 | ) | $ | 0.14 | $ | (0.00 | ) | $ | (0.01 | ) | $ | (0.01 | ) | ||||||||||
|
Shares used in computing net income (loss) per share:
|
||||||||||||||||||||||||||||||||
|
Basic
|
23,352 | 23,335 | 23,315 | 23,300 | 23,253 | 23,251 | 21,931 | 21,620 | ||||||||||||||||||||||||
|
Diluted
|
23,352 | 23,335 | 23,315 | 23,300 | 23,935 | 23,251 | 21,931 | 21,620 | ||||||||||||||||||||||||
Net Operating Losses and Tax Credit Carryforwards
As of December 31, 2010 we had net operating loss carryforwards for federal and state income tax purposes of $24.2 million and 25.3 million, respectively. If not utilized, the federal and state net operating loss carryforwards will begin expiring at various dates between 2016 and 2030.
Current federal and California tax laws include substantial restrictions on the utilization of net operating loss carryforwards in the event of an ownership change of a corporation. Accordingly, our ability to utilize net operating loss carryforwards may be limited as a result of such ownership changes. Such a limitation could result in the expiration of carryforwards before they are utilized.
Inflation
We do not believe that inflation has had a material impact on our business and operating results during the periods presented, and we do not expect it to have a material impact in the near future, though, there can be no assurances that our business will not be affected by inflation in the future.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements as of December 31, 2010.
34
Contractual Obligations
Our contractual cash commitments as of December 31, 2010 were as follows (in thousands):
|
Contractual Obligations
|
Total
|
Less than
1 year
|
1 - 3 years
|
3 - 5 years
|
More than
5 years
|
|||||||||||||||
|
Operating leases
|
$ | 4,741 | $ | 944 | $ | 2,948 | $ | 849 | $ | — | ||||||||||
|
Equipment loan
|
106 | 106 | — | — | — | |||||||||||||||
| $ | 4,847 | $ | 1,050 | $ | 2,948 | $ | 849 | $ | — | |||||||||||
Our commitments under the operating leases shown above consist of payments relating to our lease of laboratory and office space in one office building in Emeryville, California. This lease expires on October 31, 2015.
Our commitment under the equipment loan shown above consists of the total payments due under the loan facility. This amount includes approximately $3,000 of interest payments over the remaining term of the loan.
We believe our cash balance at December 31, 2010 is sufficient to fund our projected operating requirements through at least the next twelve months. However, we will need to raise additional capital or incur indebtedness to continue to fund our operations in the future. Our future capital requirements will depend on many factors, including:
|
·
|
the scope, rate of progress and cost of our pre-clinical studies and clinical trials and other research and development activities;
|
|
·
|
future clinical trial results;
|
|
·
|
the terms and timing of any collaborative, licensing and other arrangements that we may establish;
|
|
·
|
the cost and timing of regulatory approvals;
|
|
·
|
the cost of establishing clinical and commercial supplies of our product candidates and any products that we may develop;
|
|
·
|
the effect of competing technological and market developments;
|
|
·
|
the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and
|
|
·
|
the extent to which we acquire or invest in businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions.
|
We do not anticipate that we will generate significant product revenue for a number of years. Until we can generate sufficient product revenue, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements, as well as through interest income earned on cash balances and short-term investments. To the extent that we raise additional funds by issuing equity securities, our shareholders may experience dilution. In addition, debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs or to obtain funds through collaborations for some of our technologies or product candidates that we would otherwise seek to develop on our own. Such collaborations may not be on favorable terms or they may require us to relinquish rights to our technologies or product candidates.
35
|
ITEM 7A.
|
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
Our market risk consists principally of interest rate risk on our cash, cash equivalents, and short-term investments. Our exposure to market risk is limited primarily to interest income sensitivity, which is affected by changes in interest rates, particularly because the majority of our investments are in short-term debt securities.
Our investment policy restricts our investments to high-quality investments and limits the amounts invested with any one issuer, industry, or geographic area. The goals of our investment policy are as follows: preservation of capital; assurance of liquidity needs; best available return on invested capital; and minimization of capital taxation. Some of the securities in which we invest may be subject to market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with an interest rate fixed at the then-prevailing rate and the prevailing interest rate later rises, the principal amount of our investment will probably decline. To minimize this risk, in accordance with our investment policy, we maintain our cash and cash equivalents in short-term marketable securities, including money market mutual funds, Treasury bills, Treasury notes, commercial paper, and corporate and municipal bonds. The risk associated with fluctuating interest rates is limited to our investment portfolio. Due to the short-term nature of our investment portfolio, we believe we have minimal interest rate risk arising from our investments. As of December 31, 2010 and 2009, a 10% change in interest rates would have had an immaterial effect on the value of our short-term marketable securities. We do not use derivative financial instruments in our investment portfolio. We do not hold any instruments for trading purposes.
To date, we have operated exclusively in the U.S. and have not had any material exposure to foreign currency rate fluctuations. We have a wholly-owned subsidiary, which is incorporated under the laws of British Columbia (Canada), which may conduct research and development activities in Canada. To the extent we conduct operations in Canada, fluctuations in the exchange rates of the U.S. and Canadian currencies may affect our operating results.
36
|
ITEM 8.
|
FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
|
The financial statements required by this Item 8 are set forth below. Our quarterly financial information is set forth in Item 7 of this report and is hereby incorporated into this Item 8 by reference.
|
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
||
|
Page
|
||
|
Reports of Independent Registered Public Accounting Firms
|
38
|
|
|
Consolidated Balance Sheets as of December 31, 2010 and 2009
|
40
|
|
|
Consolidated Statements of Operations for the Years Ended December 31, 2010, 2009 and 2008 and for the cumulative period from July 1, 2002 (inception) to December 31, 2010
|
41
|
|
|
Consolidated Statements of Stockholders’ Equity for the cumulative period from July 1, 2002 (inception) to December 31, 2010
|
42
|
|
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2010, 2009 and 2008 and for the cumulative period from July 1, 2002 (inception) to December 31, 2010
|
46
|
|
37
REPORT OF INDEPENDENT REGISTERED
PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of
NovaBay Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheet of NovaBay Pharmaceuticals, Inc. (a development stage company) as of December 31, 2010 and the related consolidated statements of operations, stockholders’ equity and cash flows for the year then ended, and for the period from July 1, 2002 (inception) to December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit. The cumulative statements of operations, stockholders’ equity and cash flows for the period from July 1, 2002 (inception) through December 31, 2009 were audited by other auditors. Our report, insofar as it relates to the amounts included for the period from July 1, 2002 to December 31, 2009, is based solely on the report of the other auditors.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (U.S.). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, based on our audit and the report of other auditors, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of NovaBay Pharmaceuticals, Inc. (a development stage company) as at December 31, 2010, and the results of its operations and its cash flows for the year then ended and for the period from July 1, 2002 (inception) to December 31, 2010, in conformity with U.S. generally accepted accounting principles.
/s/ Odenberg, Ullakko, Muranishi & Co. LLP
San Francisco, California
March 7, 2011
38
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders of
NovaBay Pharmaceuticals Inc.
(a development stage company)
We have audited the accompanying consolidated balance sheet of NovaBay Pharmaceuticals Inc. (a development stage company) as of December 31, 2009, and the related consolidated statements of operations, stockholders' equity, and cash flows for the years ended December 31, 2009 and 2008 and for the period from July 1, 2002 (date of development stage inception) to December 31 2009. NovaBay Pharmaceuticals Inc.'s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of NovaBay Pharmaceuticals Inc. as of December 31, 2009, and the results of its operations and its cash flows for the years ended December 31, 2009 and 2008 and for the period from July 1, 2002 (date of development stage inception) to December 31 2009 in conformity with accounting principles generally accepted in the United States of America.
/S/ Davidson & Company LLP
|
Vancouver, Canada
|
Chartered Accountants
|
|
March 26, 2010
|
39
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED BALANCE SHEETS
(in thousands, except per share data)
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 11,534 | $ | 10,992 | ||||
|
Short-term investments
|
1,272 | 300 | ||||||
|
Accounts receivable
|
500 | 3,750 | ||||||
|
Prepaid expenses and other current assets
|
448 | 564 | ||||||
|
Total current assets
|
13,754 | 15,606 | ||||||
|
Property and equipment, net
|
1,588 | 1,812 | ||||||
|
Other assets
|
174 | 105 | ||||||
|
TOTAL ASSETS
|
$ | 15,516 | $ | 17,523 | ||||
|
LIABILITIES AND STOCKHOLDERS' EQUITY
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$ | 406 | $ | 272 | ||||
|
Accrued liabilities
|
726 | 1,228 | ||||||
|
Capital lease obligation
|
— | 7 | ||||||
|
Equipment loan
|
106 | 364 | ||||||
|
Deferred revenue
|
1,485 | 2,167 | ||||||
|
Total current liabilities
|
2,723 | 4,038 | ||||||
|
Deferred revenue - non-current
|
2,204 | — | ||||||
|
Deferred rent
|
99 | — | ||||||
|
Equipment loan - non-current
|
— | 106 | ||||||
|
Deferred tax liability
|
— | 34 | ||||||
|
Total liabilities
|
5,026 | 4,178 | ||||||
|
Stockholders' Equity:
|
||||||||
|
Preferred stock, $0.01 par value; 5,000 shares authorized; none
outstanding at December 31, 2010 and 2009
|
— | — | ||||||
|
Common stock, $0.01 par value; 65,000 shares authorized at
December 31, 2010 and 2009; 23,392 and 23,254 shares issued
and outstanding at December 31, 2010 and 2009, respectively
|
234 | 233 | ||||||
|
Additional paid-in capital
|
38,469 | 37,003 | ||||||
|
Accumulated other comprehensive loss
|
(14 | ) | — | |||||
|
Accumulated deficit during development stage
|
(28,199 | ) | (23,891 | ) | ||||
|
Total stockholders' equity
|
10,490 | 13,345 | ||||||
|
TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY
|
$ | 15,516 | $ | 17,523 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
40
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
|
Cumulative Period
|
||||||||||||||||
|
from July 1, 2002
|
||||||||||||||||
|
(inception) to
|
||||||||||||||||
|
Year Ended December 31,
|
December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
License and collaboration revenue:
|
$ | 9,754 | $ | 15,684 | $ | 6,722 | $ | 39,606 | ||||||||
|
Operating Expenses:
|
||||||||||||||||
|
Research and development
|
8,616 | 7,337 | 9,595 | 40,960 | ||||||||||||
|
General and administrative
|
5,654 | 5,607 | 5,636 | 28,225 | ||||||||||||
|
Total operating expenses
|
14,270 | 12,944 | 15,231 | 69,185 | ||||||||||||
|
Operating income (loss)
|
(4,516 | ) | 2,740 | (8,509 | ) | (29,579 | ) | |||||||||
|
Other income (expense), net
|
258 | (36 | ) | 397 | 1,451 | |||||||||||
|
Income (loss) before income taxes
|
(4,258 | ) | 2,704 | (8,112 | ) | (28,128 | ) | |||||||||
|
Income tax provision
|
(50 | ) | (7 | ) | (2 | ) | (71 | ) | ||||||||
|
Net income (loss)
|
$ | (4,308 | ) | $ | 2,697 | $ | (8,114 | ) | $ | (28,199 | ) | |||||
|
Net income (loss) per share:
|
||||||||||||||||
|
Basic
|
$ | (0.18 | ) | $ | 0.12 | $ | (0.38 | ) | ||||||||
|
Diluted
|
$ | (0.18 | ) | $ | 0.12 | $ | (0.38 | ) | ||||||||
|
Shares used in computing net income (loss) per share:
|
||||||||||||||||
|
Basic
|
23,326 | 22,404 | 21,312 | |||||||||||||
|
Diluted
|
23,326 | 23,115 | 21,312 | |||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
41
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
|
Addit-
ional
|
Stock
Sub-
|
Accum-
ulated
Other
Compre-
hensive
|
Accum-
ulated
Deficit
During
Develop-
|
Total
Stock-
|
||||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-In |
scription
|
Income
|
ment
|
holders'
|
||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Receivable |
(Loss)
|
Stage | Equity | ||||||||||||||||||||||||||||
|
Balance at July 1, 2002
|
— | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | |||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (544 | ) | (544 | ) | |||||||||||||||||||||||||
|
Total comprehensive loss
|
(544 | ) | ||||||||||||||||||||||||||||||||||
|
Issuance of Series A
preferred stock and
common stock for
acquisition of LLC
|
2,723 | 27 | 3,902 | 39 | 462 | — | — | — | 528 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-
employee stock options
|
— | — | — | — | 15 | — | — | — | 15 | |||||||||||||||||||||||||||
|
Sale of stock warrants
|
— | — | — | — | 10 | — | — | — | 10 | |||||||||||||||||||||||||||
|
Balance at December 31, 2002
|
2,723 | 27 | 3,902 | 39 | 487 | — | — | (544 | ) | 9 | ||||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (977 | ) | (977 | ) | |||||||||||||||||||||||||
|
Total comprehensive loss
|
(977 | ) | ||||||||||||||||||||||||||||||||||
|
Issuance of Series A
preferred stock
|
492 | 5 | — | — | 192 | — | — | — | 197 | |||||||||||||||||||||||||||
|
Issuance of Series B
preferred stock net of
issuance costs of $86
|
3,258 | 33 | — | — | 1,413 | — | — | — | 1,446 | |||||||||||||||||||||||||||
|
Issuance of stock
|
— | — | 25 | — | 7 | — | — | — | 7 | |||||||||||||||||||||||||||
|
Issuance of stock for
option exercises
|
— | — | 40 | 1 | 7 | — | — | — | 8 | |||||||||||||||||||||||||||
|
Issuance of stock for
warrant exercises
|
— | — | 137 | 1 | 109 | — | — | — | 110 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-employee
stock options
|
— | — | — | — | 2 | — | — | — | 2 | |||||||||||||||||||||||||||
|
Balance at December 31, 2003
|
6,473 | 65 | 4,104 | 41 | 2,217 | — | — | (1,521 | ) | 802 | ||||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (2,804 | ) | (2,804 | ) | |||||||||||||||||||||||||
|
Total comprehensive loss
|
(2,804 | ) | ||||||||||||||||||||||||||||||||||
|
Issuance of Series B
preferred stock net of
issuance costs of $127
|
2,694 | 27 | — | — | 1,112 | — | — | — | 1,139 | |||||||||||||||||||||||||||
|
Issuance of Series B
preferred stock upon
conversion of notes
|
913 | 9 | — | — | 420 | — | — | — | 429 | |||||||||||||||||||||||||||
|
Issuance of Series C
preferred stock net of
issuance costs of $123
|
6,311 | 63 | — | — | 5,178 | (873 | ) | — | — | 4,368 | ||||||||||||||||||||||||||
|
Issuance of stock for
option exercises
|
— | — | 5 | — | 1 | — | — | — | 1 | |||||||||||||||||||||||||||
|
Issuance of stock for
warrant exercises
|
— | — | 31 | — | 37 | — | — | — | 37 | |||||||||||||||||||||||||||
|
Issuance of stock for
Series B offering costs
|
— | — | 368 | 4 | 106 | — | — | — | 110 | |||||||||||||||||||||||||||
|
Issuance of stock for
services
|
— | — | 15 | — | 4 | — | — | — | 4 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-
employee stock options
|
— | — | — | — | 7 | — | — | — | 7 | |||||||||||||||||||||||||||
|
Balance at December 31, 2004
|
16,391 | 164 | 4,523 | 45 | 9,082 | (873 | ) | — | (4,325 | ) | 4,093 | |||||||||||||||||||||||||
42
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY – (Continued)
(in thousands)
|
Addit-
ional
|
Stock
Sub-
|
Accum-
ulated
Other
Compre-
hensive
|
Accum-
ulated
Deficit
During
Develop-
|
Total
Stock-
|
||||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-In | scription | Income | ment | holders' | ||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Receivable | (Loss) | Stage | Equity | ||||||||||||||||||||||||||||
|
Balance at December 31, 2004
|
16,391 | 164 | 4,523 | 45 | 9,082 | (873 | ) | — | (4,325 | ) | 4,093 | |||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (3,463 | ) | (3,463 | ) | |||||||||||||||||||||||||
|
Change in unrealized
gains (losses) on
investments
|
— | — | — | — | — | — | (4 | ) | — | (4 | ) | |||||||||||||||||||||||||
|
Total comprehensive loss
|
(3,467 | ) | ||||||||||||||||||||||||||||||||||
|
Issuance of Series C
preferred stock net of
issuance costs of $140
|
355 | 4 | — | — | 158 | — | — | — | 162 | |||||||||||||||||||||||||||
|
Issuance of Series D
preferred stock net of
issuance costs of $36
|
742 | 7 | — | — | 1,070 | — | — | — | 1,077 | |||||||||||||||||||||||||||
|
Issuance of stock for
option exercises
|
— | — | 50 | — | 12 | — | — | — | 12 | |||||||||||||||||||||||||||
|
Issuance of stock for
warrant exercises
|
— | — | 292 | 3 | 324 | — | — | — | 327 | |||||||||||||||||||||||||||
|
Issuance of stock and
options for Series C
offering costs
|
— | — | 164 | 2 | 101 | — | — | — | 103 | |||||||||||||||||||||||||||
|
Issuance of stock for
services
|
— | — | 20 | — | 17 | — | — | — | 17 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-employee
stock options
|
— | — | — | — | 55 | — | — | — | 55 | |||||||||||||||||||||||||||
|
Proceeds from stock
subscription receivable
|
— | — | — | — | — | 873 | — | — | 873 | |||||||||||||||||||||||||||
|
Balance at December 31, 2005
|
17,488 | 175 | 5,049 | 50 | 10,819 | — | (4 | ) | (7,788 | ) | 3,252 | |||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (5,286 | ) | (5,286 | ) | |||||||||||||||||||||||||
|
Change in unrealized
gains (losses) on
investments
|
— | — | — | — | — | — | 16 | — | 16 | |||||||||||||||||||||||||||
|
Total comprehensive loss
|
(5,270 | ) | ||||||||||||||||||||||||||||||||||
|
Stock-based
compensation expense
|
||||||||||||||||||||||||||||||||||||
|
Issuance of Series D
preferred stock net of
issuance costs of $114
|
1,739 | 17 | — | — | 2,477 | — | — | — | 2,494 | |||||||||||||||||||||||||||
|
Issuance of stock for
option exercises
|
— | — | 80 | 1 | 22 | — | — | — | 23 | |||||||||||||||||||||||||||
|
Issuance of stock for
warrant exercises
|
— | — | 1,148 | 12 | 964 | — | — | — | 976 | |||||||||||||||||||||||||||
|
Issuance of stock and
options for Series D
offering costs
|
— | — | 31 | — | 64 | — | — | — | 64 | |||||||||||||||||||||||||||
|
Issuance of stock for
services
|
— | — | 3 | — | 5 | — | — | — | 5 | |||||||||||||||||||||||||||
|
Initial public offering costs
|
— | — | — | — | (93 | ) | — | — | — | (93 | ) | |||||||||||||||||||||||||
|
Stock-based
compensation expense
related to employee and
director stock options
|
— | — | — | — | 313 | — | — | — | 313 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-employee
stock options
|
— | — | — | — | 49 | — | — | — | 49 | |||||||||||||||||||||||||||
|
Balance at December 31, 2006
|
19,227 | 192 | 6,311 | 63 | 14,620 | — | 12 | (13,074 | ) | 1,813 | ||||||||||||||||||||||||||
43
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY – (Continued)
(in thousands)
|
Addit-
ional
|
Stock
Sub-
|
Accum-
ulated
Other
Compre-
hensive
|
Accum-
ulated
Deficit
During
Develop-
|
Total
Stock-
|
||||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-In | scription | Income | ment | holders' | ||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Receivable | (Loss) | Stage | Equity | ||||||||||||||||||||||||||||
|
Balance at December 31, 2006
|
19,227 | 192 | 6,311 | 63 | 14,620 | — | 12 | (13,074 | ) | 1,813 | ||||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (5,400 | ) | (5,400 | ) | |||||||||||||||||||||||||
|
Change in unrealized
gains (losses) on
investments
|
— | — | — | — | — | — | (15 | ) | — | (15 | ) | |||||||||||||||||||||||||
|
Total comprehensive
loss
|
(5,415 | ) | ||||||||||||||||||||||||||||||||||
|
Conversion of preferred
stock to common stock
in connection with IPO
|
(19,227 | ) | (192 | ) | 9,614 | 96 | 96 | — | — | — | — | |||||||||||||||||||||||||
|
Issuance of stock and
warrants in connection
with IPO, net of offering
costs
|
— | — | 5,000 | 50 | 17,120 | — | — | — | 17,170 | |||||||||||||||||||||||||||
|
Issuance of stock for
option exercises
|
— | — | 298 | 3 | 111 | — | — | — | 114 | |||||||||||||||||||||||||||
|
Issuance of stock for
services
|
— | — | 38 | — | 92 | — | — | — | 92 | |||||||||||||||||||||||||||
|
Issuance of stock for
director compensation
|
— | — | 8 | — | 29 | — | — | — | 29 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to employee and
director stock options
|
— | — | — | — | 399 | — | — | — | 399 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-employee
stock options
|
— | — | — | — | 118 | — | — | — | 118 | |||||||||||||||||||||||||||
|
Balance at December 31, 2007
|
— | — | 21,269 | 212 | 32,585 | — | (3 | ) | (18,474 | ) | 14,320 | |||||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | — | — | (8,114 | ) | (8,114 | ) | |||||||||||||||||||||||||
|
Change in unrealized
gains (losses) on
investments
|
— | — | — | — | — | — | 3 | — | 3 | |||||||||||||||||||||||||||
|
Total comprehensive
loss
|
(8,111 | ) | ||||||||||||||||||||||||||||||||||
|
Compensation expense
for warrants issued for
services
|
67 | 67 | ||||||||||||||||||||||||||||||||||
|
Issuance of stock for
option exercises
|
— | — | 123 | 1 | 148 | — | — | — | 149 | |||||||||||||||||||||||||||
|
Issuance of stock for
services
|
— | — | 30 | 1 | 84 | — | — | — | 85 | |||||||||||||||||||||||||||
|
Issuance of stock for
director compensation
|
— | — | 49 | 1 | 123 | — | — | — | 124 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to employee and
director stock options
|
— | — | — | — | 721 | — | — | — | 721 | |||||||||||||||||||||||||||
|
Stock-based
compensation expense
related to non-employee
stock options
|
— | — | — | — | (11 | ) | — | — | — | (11 | ) | |||||||||||||||||||||||||
|
Tax benefit from stock
plans
|
— | — | — | — | 1 | — | — | — | 1 | |||||||||||||||||||||||||||
|
Balance at December 31, 2008
|
— | — | 21,471 | 215 | 33,718 | — | — | (26,588 | ) | 7,345 | ||||||||||||||||||||||||||
44
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY – (Continued)
(in thousands)
| Common Stock |
Addit-
ional
Paid-In
|
Stock
Sub-
scription
|
Accum-
ulated
Other
Compre-
hensive
Income
|
Accum-
ulated
Deficit
During
Develop-
ment
|
Total
Stock-
holders'
|
|||||||||||||||||||||||
| Shares | Amount | Capital | Receivable | (Loss) | Stage | Equity | ||||||||||||||||||||||
|
Balance at December 31, 2008
|
21,471 | 215 | 33,718 | — | — | (26,588 | ) | 7,345 | ||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||
|
Net income
|
— | — | — | — | — | 2,697 | 2,697 | |||||||||||||||||||||
|
Change in unrealized
gains (losses) on
investments
|
— | — | — | — | — | — | — | |||||||||||||||||||||
|
Total comprehensive loss
|
— | — | — | — | — | — | 2,697 | |||||||||||||||||||||
|
Issuance of common
stock in connection
with shelf offering, net
of offering costs
|
1,225 | 12 | 1,932 | — | — | — | 1,944 | |||||||||||||||||||||
|
Issuance of stock for
option exercises
|
119 | 1 | 73 | — | — | — | 74 | |||||||||||||||||||||
|
Compensation expense
for warrants issued for
services
|
— | — | 88 | — | — | — | 88 | |||||||||||||||||||||
|
Stock-based
compensation expense
related to employee
and director stock
and stock options
|
130 | 1 | 919 | — | — | — | 920 | |||||||||||||||||||||
|
Stock-based
compensation expense
related to non-
employee stock
and stock options
|
309 | 4 | 269 | — | — | — | 273 | |||||||||||||||||||||
|
Other
|
— | — | 4 | — | — | — | 4 | |||||||||||||||||||||
|
Balance at December 31, 2009
|
23,254 | 233 | 37,003 | — | — | (23,891 | ) | 13,345 | ||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||||||
|
Net loss
|
— | — | — | — | — | (4,308 | ) | (4,308 | ) | |||||||||||||||||||
|
Change in unrealized
losses on investments
|
— | — | — | — | (14 | ) | (14 | ) | ||||||||||||||||||||
|
Total comprehensive loss
|
(4,322 | ) | ||||||||||||||||||||||||||
|
Costs related
to shelf offering
|
— | — | (2 | ) | — | — | — | (2 | ) | |||||||||||||||||||
|
Compensation expense
for warrants issued for
services
|
— | — | 7 | — | — | — | 7 | |||||||||||||||||||||
|
Issuance of stock for
option exercises
|
105 | 1 | 80 | — | — | — | 81 | |||||||||||||||||||||
|
Stock-based
compensation expense
related to employee and
director stock options
|
— | — | 1,129 | — | — | — | 1,129 | |||||||||||||||||||||
|
Stock-based
compensation expense
related to non-
employee stock
and stock options
|
33 | — | 263 | — | — | — | 263 | |||||||||||||||||||||
|
Other
|
— | — | (11 | ) | — | — | — | (11 | ) | |||||||||||||||||||
|
Balance at December 31, 2010
|
23,392 | $ | 234 | $ | 38,469 | $ | — | $ | (14 | ) | $ | (28,199 | ) | $ | 10,490 | |||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
45
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
Year Ended December 31,
|
Cumulative
Period from
July 1, 2002
(inception) to
December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Cash flows from operating activities:
|
||||||||||||||||
|
Net income (loss)
|
$ | (4,308 | ) | $ | 2,697 | $ | (8,114 | ) | $ | (28,199 | ) | |||||
|
Adjustments to reconcile net income (loss) to net cash used in operating
activities:
|
||||||||||||||||
|
Depreciation and amortization
|
427 | 373 | 304 | 1,504 | ||||||||||||
|
Accretion and amortization of short-term investments
|
10 | 42 | (57 | ) | (252 | ) | ||||||||||
|
Loss on disposal of property and equipment
|
— | — | — | 121 | ||||||||||||
|
Stock-based compensation expense for options and stock issued to
employees and directors
|
1,129 | 920 | 845 | 3,634 | ||||||||||||
|
Compensation expense for warrants issued for services
|
7 | 88 | 67 | 162 | ||||||||||||
|
Stock-based compensation expense for options and stock issued to
non-employees
|
263 | 273 | (13 | ) | 888 | |||||||||||
|
Taxes paid by LLC
|
— | — | — | 1 | ||||||||||||
|
Changes in operating assets and liabilities:
|
||||||||||||||||
|
(Increase) decrease in accounts receivable
|
3,250 | (3,750 | ) | — | (500 | ) | ||||||||||
|
(Increase) decrease in prepaid expenses and other assets
|
47 | (221 | ) | 88 | (500 | ) | ||||||||||
|
Increase (decrease) in accounts payable and accrued liabilities
|
(319 | ) | (70 | ) | 289 | 1,208 | ||||||||||
|
Increase (decrease) in deferred revenue
|
1,522 | (2,000 | ) | (3,351 | ) | 3,688 | ||||||||||
|
Net cash provided by (used in) operating activities
|
2,028 | (1,648 | ) | (9,942 | ) | (18,245 | ) | |||||||||
|
Cash flows from investing activities:
|
||||||||||||||||
|
Purchases of property and equipment
|
(203 | ) | (731 | ) | (610 | ) | (3,094 | ) | ||||||||
|
Proceeds from disposal of property and equipment
|
— | 2 | — | 46 | ||||||||||||
|
Purchases of short-term investments
|
(2,446 | ) | (3,975 | ) | (32,097 | ) | (100,965 | ) | ||||||||
|
Proceeds from maturities and sales of short-term investments
|
1,455 | 3,635 | 43,571 | 99,937 | ||||||||||||
|
Cash acquired in purchase of LLC
|
— | — | — | 516 | ||||||||||||
|
Net cash provided by (used in) investing activities
|
(1,194 | ) | (1,069 | ) | 10,864 | (3,560 | ) | |||||||||
|
Cash flows from financing activities:
|
||||||||||||||||
|
Proceeds from preferred stock issuances, net
|
— | — | — | 11,160 | ||||||||||||
|
Proceeds from common stock issuances, net
|
— | — | — | 17 | ||||||||||||
|
Proceeds from exercise of options and warrants
|
81 | 74 | 153 | 1,916 | ||||||||||||
|
Initial public offering costs, net of costs
|
— | — | — | 17,077 | ||||||||||||
|
Proceeds from shelf offering, net of costs
|
(2 | ) | 1,944 | — | 1,942 | |||||||||||
|
Proceeds from stock subscription receivable
|
— | — | — | 873 | ||||||||||||
|
Proceeds from issuance of notes
|
— | — | — | 405 | ||||||||||||
|
Principal payments on capital lease
|
(7 | ) | (42 | ) | (37 | ) | (157 | ) | ||||||||
|
Proceeds from borrowings under equipment loan
|
— | — | 422 | 1,216 | ||||||||||||
|
Principal payments on equipment loan
|
(364 | ) | (366 | ) | (302 | ) | (1,110 | ) | ||||||||
|
Net cash provided by (used in) financing activities
|
(292 | ) | 1,610 | 236 | 33,339 | |||||||||||
|
Net increase (decrease) in cash and cash equivalents
|
542 | (1,107 | ) | 1,158 | 11,534 | |||||||||||
|
Cash and cash equivalents, beginning of period
|
10,992 | 12,099 | 10,941 | — | ||||||||||||
|
Cash and cash equivalents, end of period
|
$ | 11,534 | $ | 10,992 | $ | 12,099 | $ | 11,534 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
46
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS – (Continued)
(in thousands)
|
Year Ended December 31,
|
Cumulative
Period from
July 1, 2002
(inception) to
December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
Supplemental disclosure of non cash information
|
||||||||||||||||
|
Interest paid
|
$ | 32 | $ | 77 | $ | 102 | $ | 255 | ||||||||
|
Income taxes paid
|
$ | 52 | $ | — | $ | — | $ | 52 | ||||||||
|
Non-cash financing and investing activities
|
||||||||||||||||
|
Property and equipment acquired under capital lease obligations
|
$ | — | $ | — | $ | 62 | $ | 219 | ||||||||
47
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. ORGANIZATION
NovaBay Pharmaceuticals Inc. is a clinical stage biotechnology company developing a first-in-class, anti-infective platform of compounds, called the Aganocide compounds, for the treatment or prevention of a wide range of infections in hospital and non-hospital environments. Many of these infections have become increasingly difficult to treat because of the rapid rise in drug resistance. We have discovered and are developing a class of non-antibiotic anti-infective compounds, which we have named Aganocide compounds. These compounds are based upon small molecules that are naturally generated by white blood cells when defending the body against invading pathogens. We believe that our Aganocide compounds could form a platform on which to create a variety of products to address differing needs in the treatment and prevention of bacterial and viral infections. In laboratory testing, our Aganocide compounds have demonstrated the ability to destroy all bacteria against which they have been tested. Furthermore, because of their mechanism of action, we believe that bacteria are unlikely to develop resistance to our Aganocide compounds.
The Company was incorporated under the laws of the State of California on January 19, 2000 as NovaCal Pharmaceuticals, Inc. We had no operations until July 1, 2002, on which date we acquired all of the operating assets of NovaCal Pharmaceuticals, LLC, a California limited liability company. In February 2007, we changed our name from NovaCal Pharmaceuticals, Inc. to NovaBay Pharmaceuticals, Inc. In August 2007, we formed two subsidiaries––NovaBay Pharmaceuticals Canada, Inc., a wholly-owned subsidiary incorporated under the laws of British Columbia (Canada), which may conduct research and development in Canada, and DermaBay, Inc., a wholly-owned U.S. subsidiary, which may explore and pursue dermatological opportunities. In June 2010, we changed the state in which we are incorporated (the Reincorporation), and are now incorporated under the laws of the State of Delaware. All references to “we,” “us,” “our,” or “the Company” herein refer to the California corporation prior to the date of the Reincorporation, and to the Delaware corporation on and after the date of the Reincorporation. We currently operate in one business segment.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States (U.S. GAAP) and are expressed in U.S. dollars. The financial statements have been prepared under the guidelines for Development Stage Entities. A development stage enterprise is one in which planned principal operations have not commenced, or if its operations have commenced, there have been no significant revenues therefrom. As of December 31, 2010, we continued to conduct clinical trials and had not commenced our planned principal operations.
Certain amounts for prior periods have been reclassified to conform to current period presentation.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries, NovaBay Pharmaceuticals Canada, Inc. and DermaBay, Inc. All inter-company accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in accordance with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. These estimates include useful lives for property and equipment and related depreciation calculations, estimated amortization period for payments received from product development and license agreements as they relate to revenue recognition, assumptions for valuing options and warrants, and income taxes. Actual results could differ from those estimates.
Cash and Cash Equivalents and Short-Term Investments
We consider all highly liquid instruments with a stated maturity of three months or less to be cash and cash equivalents. Cash and cash equivalents are stated at cost, which approximate their fair value. As of December 31, 2010, our cash and cash equivalents were held in financial institutions in the U.S. and include deposits in money market funds, which were unrestricted as to withdrawal or use.
48
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
We classify all highly liquid investments with a stated maturity of greater than three months as short-term investments. Short-term investments generally consist of U.S. government, municipal and corporate debt securities. We have classified our short-term investments as available-for-sale. We do not intend to hold securities with stated maturities greater than twelve months until maturity. In response to changes in the availability of and the yield on alternative investments as well as liquidity requirements, we occasionally sell these securities prior to their stated maturities. These securities are carried at fair value, with the unrealized gains and losses reported as a component of other comprehensive income (loss) until realized. Realized gains and losses from the sale of available-for-sale securities, if any, are determined on a specific identification basis. A decline in the market value below cost of any available-for-sale security that is determined to be other than temporary results in a revaluation of its carrying amount to fair value and an impairment charge to earnings, resulting in a new cost basis for the security. No such impairment charges were recorded for the periods presented. The interest income and realized gains and losses are included in other income, net within the consolidated statements of operations. Interest income is recognized when earned.
Concentrations of Credit Risk and Major Partners
Financial instruments which potentially subject us to significant concentrations of credit risk consist primarily of cash and cash equivalents and short-term investments. We maintain deposits of cash, cash equivalents and short-term investments with three highly-rated, major financial institutions in the United States.
Deposits in these banks may exceed the amount of federal insurance provided on such deposits. We do not believe we are exposed to significant credit risk due to the financial position of the financial institutions in which these deposits are held. Additionally, we have established guidelines regarding diversification and investment maturities, which are designed to maintain safety and liquidity.
During the years ended December 31, 2010, 2009 and 2008 100% of our revenues were derived from two collaboration partners and at December 31, 2010 and 2009, 100% of our accounts receivables were derived from one collaboration partner.
Fair Value of Financial Assets and Liabilities
Financial instruments, including cash and cash equivalents and short-term investments, accounts payable and accrued liabilities are carried at cost, which management believes approximates fair value due to the short-term nature of these instruments. The fair value of capital lease obligations and equipment loans approximates their carrying amounts because the obligations bear market rates of interest.
The Company measures the fair value of financial assets and liabilities based on U.S. GAAP guidance which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements. Effective January 1, 2008, the Company adopted the provisions for financial assets and liabilities, as well as for any other assets and liabilities that are carried at fair value on a recurring basis. Effective January 1, 2009, the Company adopted the provisions for non-financial assets and liabilities that are required to be measured at fair value. The adoption of these provisions did not materially impact the Company’s consolidated financial position and results of operations.
Under U.S. GAAP, fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. A fair value hierarchy is also established, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. There are three levels of inputs that may be used to measure fair value:
Level 1 – quoted prices in active markets for identical assets or liabilities
Level 2 – quoted prices for similar assets and liabilities in active markets or inputs that are observable
Level 3 – inputs that are unobservable (for example cash flow modeling inputs based on assumptions)
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization. Depreciation is calculated using the straight-line method over the estimated useful lives of the related assets of five to seven years for office and laboratory equipment, three years for software and seven years for furniture and fixtures. Leasehold improvements are depreciated over the shorter of seven years or the lease term. Depreciation of assets recorded under capital leases is included in depreciation expense.
The costs of normal maintenance, repairs, and minor replacements are charged to operations when incurred.
49
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Impairment of Long-Lived Assets
The Company accounts for long-lived assets in accordance with U.S. GAAP, which requires that companies consider whether events or changes in facts and circumstances, both internally and externally, may indicate that an impairment of long-lived assets held for use are present. Management periodically evaluates the carrying value of long-lived assets and has determined that there was no impairment as of all periods presented. Determination of recoverability is based on the estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to cover the carrying amount of the asset, the assets are written down to their estimated fair values and the loss is recognized in the statements of operations.
Comprehensive Income (Loss)
ASC 220, Comprehensive Income requires that an entity’s change in equity or net assets during a period from transactions and other events from non-owner sources be reported. The Company reports unrealized gains and losses on its available-for-sale securities as other comprehensive income.
Revenue Recognition
License and collaboration revenue is primarily generated through agreements with strategic partners for the development and commercialization of the Company’s product candidates. The terms of the agreements typically include non-refundable upfront fees, funding of research and development activities, payments based upon achievement of certain milestones and royalties on net product sales. In accordance with revenue recognition criteria under U.S. GAAP, the Company analyzes its multiple element arrangements to determine whether the elements can be separated. The Company performs its analysis at the inception of the arrangement and as each product or service is delivered. If a product or service is not separable, the combined deliverables are
accounted for as a single unit of accounting and revenue is recognized over the performance obligation period. Revenue is recognized when the following criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred and risk of loss has passed; the seller’s price to the buyer is fixed or determinable; and collectability is reasonably assured.
Assuming the elements meet the revenue recognition guidelines the revenue recognition methodology prescribed for each unit of accounting is summarized below:
Upfront Fees—We defer recognition of non-refundable upfront fees if we have continuing performance obligations without which the technology licensed has no utility to the licensee. If we have performance obligations through research and development services that are required because our know-how and expertise related to the technology is proprietary to us, or can only be performed by us, then such up-front fees are deferred and recognized over the period of the performance obligations. We base the estimate of the period of performance on factors in the contract. Actual time frames could vary and could result in material changes to our results of operations.
Funded Research and Development— Revenue from research and development services is recognized during the period in which the services are performed and is based upon the number of full-time-equivalent personnel working on the specific project at the agreed-upon rate. This revenue approximates the cost incurred. Reimbursements from collaborative partners for agreed-upon direct costs including direct materials and outsourced, or subcontracted, pre-clinical studies are classified as revenue and recognized in the period the reimbursable expenses are incurred. Payments received in advance are recorded as deferred revenue until the research and development services are performed or costs are incurred.
Milestones—Substantive milestone payments are considered to be performance bonuses that are recognized upon achievement of the milestone only if all of the following conditions are met: the milestone payments are non-refundable; achievement of the milestone involves a degree of risk and was not reasonably assured at the inception of the arrangement; substantive effort is involved in achieving the milestone; the amount of the milestone is reasonable in relation to the effort expended or the risk associated with achievement of the milestone; and a reasonable amount of time passes between the up-front license payment and the first milestone payment as well as between each subsequent milestone payment. If any of these conditions are not met, the milestone payments are deferred and recognized as revenue over the term of the arrangement as we complete our performance obligations.
Royalties—We recognize royalty revenues from licensed products upon the sale of the related products.
Research and Development Costs
We charge research and development costs to expense as incurred. These costs include salaries and benefits for research and development personnel, costs associated with clinical trials managed by contract research organizations, and other costs associated with research, development and regulatory activities. We use external service providers to conduct clinical trials, to manufacture supplies of product candidates and to provide various other research and development-related products and services. Research and development expenses under the collaborative agreements approximate the revenue recognized, excluding milestone and upfront payments received under such arrangements.
50
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Patent Costs
We expense patent costs, including legal expenses, in the period in which they are incurred. Patent expenses are included in general and administrative expenses in our statements of operations.
Stock-Based Compensation
The Company accounts for stock-based compensation under the provisions of ASC 718, Compensation-Stock Compensation. Under the fair value recognition provisions, stock-based compensation expense is measured at the grant date for all stock-based awards to employees and directors and is recognized as expense over the requisite service period, which is generally the vesting period. Non-employee stock-based compensation charges are amortized over the vesting period on a straight-line basis. For stock options granted, the fair value of the stock options is estimated using a Black-Scholes-Merton valuation model. See Note 9 for further information regarding stock-based compensation expense and the assumptions used in estimating that expense.
Income Taxes
We account for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is recognized if it is more likely than not that some portion or all of the deferred tax asset will not be recognized.
Net Income (Loss) per Share
The Company computes net income (loss) per share by presenting both basic and diluted earnings (loss) per share (EPS).
Basic EPS is computed by dividing net income (loss) available to common shareholders by the weighted average number of common shares outstanding during the period. Diluted EPS gives effect to all dilutive potential common shares outstanding during the period including stock options and stock warrants, using the treasury stock method, using the if-converted method. In computing diluted EPS, the average stock price for the period is used in determining the number of shares assumed to be purchased from the exercise of stock options or warrants. Potentially dilutive common share equivalents are excluded from the diluted EPS computation in net loss periods since their effect would be anti-dilutive. During 2008 and 2010, there is no difference between basic and diluted net loss per share due to the Company’s net losses. The following table sets forth the reconciliation between basic EPS and diluted EPS:
|
Year Ended December 31,
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Net income (loss)
|
$ | (4,308 | ) | $ | 2,697 | $ | (8,114 | ) | ||||
|
Basic shares
|
23,326 | 22,404 | 21,312 | |||||||||
|
Add: shares issued upon assumed exercise of stock options
|
— | 711 | — | |||||||||
|
Diluted shares
|
23,326 | 23,115 | 21,312 | |||||||||
|
Basic EPS
|
$ | (0.18 | ) | $ | 0.12 | $ | (0.38 | ) | ||||
|
Diluted EPS
|
$ | (0.18 | ) | $ | 0.12 | $ | (0.38 | ) | ||||
The following outstanding stock options and stock warrants were excluded from the diluted EPS computation as their effect would have been anti-dilutive:
51
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
Year Ended December 31,
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Stock options
|
4,968 | 3,436 | 3,371 | |||||||||
|
Stock warrants
|
1,375 | 1,875 | 650 | |||||||||
Recent Accounting Pronouncements
In April 2010, the Financial Accounting Standards Board (FASB) issued ASU No. 2010-17 (Topic 605), Revenue Recognition—Milestone Method. This standard provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research and development transactions. The amendments in this update provide guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration that is contingent upon achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all applicable criteria. The amendments in this update are effective for the Company on a prospective basis for milestones achieved after December 31, 2010. The implementation of this standard is not expected to have a significant impact on our financial position or results of operations.
In February 2010, the FASB issued amended guidance on subsequent events. Under this amended guidance, U.S. Securities and Exchange Commission (SEC) filers are no longer required to disclose the date through which subsequent events have been evaluated in originally issued and revised financial statements. The guidance was effective immediately and we adopted these new requirements upon issuance of this guidance. The adoption did not have a material impact on our consolidated financial statements.
In January 2010, the FASB issued Accounting Standards Update (ASU) No. 2010-06 (Topic 820) Fair Value Measurements and Disclosures. This standard amends the disclosure guidance with respect to fair value measurements for both interim and annual reporting periods. Specifically, disclosure is required for significant transfers between Level 1 and Level 2 in the fair value hierarchy; additional disclosures are required for transactions in Level 3 assets and liabilities; and additional disclosure is required of the valuation techniques and inputs used to measure assets and liabilities that fall into Level 2 and Level 3. Except for the additional disclosures for transactions Level 3 items, which were effective for us as of January 1, 2011, the remaining new disclosure requirements were effective for the Company as of January 1, 2010. The implementation of this standard had no significant impact on our financial position or results of operations.
In September 2009, the FASB issued update 2009-13, ASC 605, Revenue Recognition: Multiple-Deliverable Revenue Arrangements-a consensus of the FASB Emerging Issues Task Force.This guidance addresses how to separate deliverables and how to measure and allocate consideration to one or more units of accounting. Specifically, the guidance requires that consideration be allocated among multiple deliverables based on relative selling prices. The guidance establishes a selling price hierarchy of (1) vendor-specific objective evidence, (2) third-party evidence and (3) estimated selling price. This guidance is effective for annual periods beginning after June 15, 2010 but may be early adopted as of the beginning of an annual period. The adoption is not expected to have a material impact on our consolidated financial statements.
NOTE 3. SHORT-TERM INVESTMENTS
Short-term investments as of December 31, 2010 and 2009 consisted of the following:
52
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
December 31, 2010
|
||||||||||||||||
|
Gross
|
Gross
|
|||||||||||||||
|
Amortized
|
Unrealized
|
Unrealized
|
Market
|
|||||||||||||
|
(in thousands)
|
Cost
|
Gains
|
Losses
|
Value
|
||||||||||||
|
Corporate bonds
|
$ | 767 | $ | 19 | $ | (14 | ) | $ | 772 | |||||||
|
Certificates of deposit
|
500 | — | — | 500 | ||||||||||||
| $ | 1,267 | $ | 19 | $ | (14 | ) | $ | 1,272 | ||||||||
|
December 31, 2009
|
||||||||||||||||
|
Gross
|
Gross
|
|||||||||||||||
|
Amortized
|
Unrealized
|
Unrealized
|
Market
|
|||||||||||||
|
(in thousands)
|
Cost
|
Gains
|
Losses
|
Value
|
||||||||||||
|
Certificates of deposit
|
$ | 300 | $ | — | $ | — | $ | 300 | ||||||||
| $ | 300 | $ | — | $ | — | $ | 300 | |||||||||
All short-term investments at December 31, 2010 and 2009 mature in less than one year.
During the years ended December 31, 2010, 2009, and 2008 we recognized a net realized gain/(loss) of ($10,000), $0, and $35,000, respectively.
The Company’s cash equivalents and investments are classified within Level 1 or Level 2 of the fair value hierarchy because they are valued using quoted market prices in active markets, broker or dealer quotations, or alternative pricing sources with reasonable levels of price transparency. The types of investments that are generally classified within Level 1 of the fair value hierarchy include money market securities. The types of investments that are generally classified within Level 2 of the fair value hierarchy include corporate securities, certificates of deposits and municipal bonds.
The following table presents the Company’s investments, measured at fair value on a recurring basis, as of December 31, 2010:
|
Fair Value Measurements
|
||||||||||||||||
|
(in thousands)
|
Balance at
December 31,
2010
|
Quoted
Prices in
Active
Markets for
Identical Items
(Level 1)
|
Significant
Other
Observable
Inputs
(Level 2)
|
Significant
Unobservable
Inputs
(Level 3)
|
||||||||||||
|
Cash equivalents
|
$ | 11,534 | $ | 11,534 | $ | — | $ | — | ||||||||
|
Short-term investments:
|
||||||||||||||||
|
Corporate bonds
|
772 | — | 772 | — | ||||||||||||
|
Certificates of deposit
|
500 | — | 500 | — | ||||||||||||
|
Total short-term investments
|
1,272 | — | 1,272 | — | ||||||||||||
|
Total
|
$ | 12,806 | $ | 11,534 | $ | 1,272 | $ | — | ||||||||
NOTE 4. PROPERTY AND EQUIPMENT
Property and equipment consisted of the following:
|
(in thousands)
|
December 31,
2010
|
December 31,
2009
|
||||||
|
Office and laboratory equipment
|
$ | 2,620 | $ | 2,430 | ||||
|
Furniture and fixtures
|
113 | 113 | ||||||
|
Software
|
144 | 133 | ||||||
|
Leasehold improvement
|
149 | 147 | ||||||
|
Total property and equipment, at cost
|
3,026 | 2,823 | ||||||
|
Less: accumulated depreciation
|
(1,438 | ) | (1,011 | ) | ||||
|
Total property and equipment, net
|
$ | 1,588 | $ | 1,812 | ||||
Depreciation expense was $427,000, $373,000 and $304,000 for the years ended December 31, 2010, 2009 and 2008, respectively and $1.5 million for the cumulative period from July 1, 2002 (inception) to December 31, 2010.
53
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 5. ACCRUED LIABILITIES
Accrued liabilities consisted of the following:
|
(in thousands)
|
December 31,
2010
|
December 31,
2009
|
||||||
|
Research and development
|
$ | 103 | $ | 118 | ||||
|
Employee payroll and benefits
|
550 | 744 | ||||||
|
Professional fees
|
22 | 58 | ||||||
|
Other
|
51 | 308 | ||||||
|
Total accrued liabilities
|
$ | 726 | $ | 1,228 | ||||
NOTE 6. EQUIPMENT LOAN
During April 2007, we entered into a master security agreement to establish a $1.0 million equipment loan facility with a financial institution. The purpose of this loan is to finance equipment purchases, principally in the build-out of our laboratory facilities. Borrowings under the loan are secured by eligible equipment purchased from January 2006 through April 2008 and will be repaid over 40 months at an interest rate equal to the greater of 5.94% over the three year Treasury rate in effect at the time of funding or 10.45%. There are no loan covenants specified in the agreement.
As of December 31, 2010, we had an outstanding equipment loan balance of $106,000 carrying a weighted-average interest rate of 11.22%. At December 31, 2010, there were no funds available for borrowing under this equipment loan facility.
Future minimum loan payments under equipment loans were as follows at December 31, 2010:
|
Loan
|
||||
|
(in thousands)
|
Commitment
|
|||
|
Minimum loan payments in 2011
|
$ | 109 | ||
|
Less: amount representing interest
|
(3 | ) | ||
|
Present value of minimum loan payments
|
$ | 106 | ||
NOTE 7. COMMITMENTS AND CONTINGENCIES
Operating Leases
We lease laboratory facilities and office space under an operating lease, which expires on October 31, 2015. Rent expense was $1.1 million, $878,000, and $620,000 for the years ended December 31, 2010, 2009 and 2008, respectively. The future minimum lease payments under this non-cancellable operating lease were as follows as of December 31, 2010:
|
Lease
|
||||
|
(in thousands)
|
Commitment
|
|||
|
Year ending December 31:
|
||||
|
2011
|
$ | 944 | ||
|
2012
|
963 | |||
|
2013
|
983 | |||
|
2014
|
1,002 | |||
|
2015
|
849 | |||
|
Total lease commitment
|
$ | 4,741 | ||
The Company’s monthly rent payments fluctuate under the master lease agreement. In accordance with U.S. GAAP, the Company recognizes rent expense on a straight-line basis. The Company records deferred rent for the difference between the amounts paid and recorded as expense. At December 31, 2010 and 2009, the Company had $99,000 and $0 of deferred rent, respectively.
54
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Directors and Officers Indemnity
As permitted under Delaware law and in accordance with our bylaws, we indemnify our officers and directors for certain events or occurrences while the officer or director is or was serving at our request in such capacity. The term of the indemnification period is for the officer’s or director’s lifetime. The maximum amount of potential future indemnification is unlimited; however, we have a director or officer insurance policy that limits our exposure and may enable us to recover a portion of any future payments. We believe the fair value of these indemnification agreements is minimal. Accordingly, we have not recorded any liabilities for these agreements as of December 31, 2010.
In the normal course of business, we provide indemnifications of varying scope under our agreements with other companies, typically our clinical research organizations, investigators, clinical sites, suppliers and others. Pursuant to these agreements, we generally indemnify, hold harmless, and agree to reimburse the indemnified parties for losses suffered or incurred by the indemnified parties in connection with use or testing of our products or product candidates or with any U.S. patent or any copyright or other intellectual property infringement claims by any third party with respect to our products. The term of these indemnification agreements is generally perpetual. The potential future payments we could be required to make under these indemnification agreements is unlimited. Historically, costs related to these indemnification provisions have been immaterial. We also maintain various liability insurance policies that limit our exposure. As a result, we believe the fair value of these indemnification agreements is minimal. Accordingly, we have not recorded any liabilities for these agreements as of December 31, 2010.
Legal Matters
From time to time, the Company may be involved in various legal proceedings arising in the ordinary course of business. There are no matters at December 31, 2010 that, in the opinion of management, would have a material adverse effect on our financial position, results of operations or cash flows.
NOTE 8. STOCKHOLDERS’ EQUITY
Preferred Stock
Under the Company’s amended articles of incorporation, the Company is authorized to issue of up to 5,000,000 shares of preferred stock in such series and with such rights and preferences as may be approved by the board of directors. As of December 31, 2010, there were no shares of preferred stock outstanding.
Common Stock
Under the Company’s amended articles of incorporation, the Company is authorized to issue 65,000,000 shares of $0.01 par value common stock. Each holder of common stock has the right to one vote but does not have cumulative voting rights. Shares of common stock are not subject to any redemption or sinking fund provisions, nor do they have any preemptive, subscription or conversion rights. Holders of common stock are entitled to receive dividends whenever funds are legally available and when declared by the board of directors, subject to the prior rights of holders of all classes of stock outstanding having priority rights as to dividends. No dividends have been declared or paid as of December 31, 2010.
In August 2009, the Company sold and issued 1,225,000 units at a price of $2.00 per unit from a Shelf Registration Offering. Each unit consisted of one share of the Company’s common stock and a warrant to purchase one share of the Company’s common stock. The Company raised a total of $2.5 million from the Shelf Registration Offering, or $1.9 million in net proceeds after deducting underwriting commissions of $156,000 and other offering costs of $350,000.
Stock Warrants
Warrants to acquire shares of common stock were issued in October 2007 in connection with the Initial Public Offering (IPO). In 2009 warrants were issued to investors as part of our Shelf Registration Offering. The significant terms of these warrants were as follows:
Underwriter Warrants—In connection with the IPO, the Company issued warrants to the underwriters to purchase an aggregate of 350,000 shares of common stock at an exercise price of $4.00 per share. The warrants were exercisable on or after October 31, 2008 and expired on October 31, 2010. The warrants were valued at approximately $524,000 using the Black-Scholes-Merton option-pricing model based upon the following assumptions: (1) expected price volatility of 50.0%, (2) a risk-free interest rate of 3.94% and (3) a contractual life of 3 years. The Company accounted for the fair value of the Underwriter Warrants as an expense of the IPO resulting in a charge to stockholders’ equity.
55
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Advisory Services Warrants - In April 2008, the Company issued a two year warrant and a four year warrant to purchase an aggregate of 300,000 shares of common stock to PM Holdings Ltd. as part of our consideration for the revision of the agreement dated February 13, 2007 with PM Holdings. Under the terms of the original agreement, the Company agreed to pay PM Holdings $28,000 per month through February 2010 for financial and investor relations advisory services. The amendment to this agreement eliminated the monthly cash payment obligation and instead provided for a one-time, upfront cash payment of $264,000 and the issuance of warrants to purchase 300,000 shares of common stock at an exercise price of $4.00 per share. The warrants were valued at approximately $162,000 using the Black-Scholes-Merton option-pricing model based upon the following assumptions: (1) expected price volatility of 50.0%, (2) a risk-free interest rate of 3.94% and (3) a contractual life of the warrant. The Company accounts for the fair value of the Advisory Services Warrants as an expense over the period the advisory services were provided. The two-year warrant for 150,000 shares expired unexercised. The four-year warrant for 150,000 shares expires in April 2012.
Investor Warrants—In August 2009, in connection with the Shelf Registration Offering, the Company issued warrants to the investors to purchase an aggregate of 1,225,000 shares of common stock at an exercise price of $2.75 per share. The warrants are exercisable on or after February 17, 2010 and expire on August 21, 2014.
At December 31, 2010, there were outstanding warrants to purchase 150,000 shares of common stock at an exercise price of $4.00 per share. Additionally, there were outstanding warrants to purchase 1,225,000 shares of common stock from the Shelf Registration Offering at the exercise price of $2.75 per share. All of these warrants were exercisable at December 31, 2010.
The following table summarizes information about the Company’s warrants outstanding at December 31, 2010, 2009 and 2008 and activity during the three years then ended.
|
(in thousands, except per share data)
|
Warrants
|
Weighted-
Average
Exercise Price
|
||||||
|
Outstanding at December 31, 2007
|
350 | $ | 4.00 | |||||
|
Warrants granted
|
300 | $ | 4.00 | |||||
|
Outstanding at December 31, 2008
|
650 | $ | 4.00 | |||||
|
Warrants granted
|
1,225 | $ | 2.75 | |||||
|
Outstanding at December 31, 2009
|
1,875 | $ | 3.18 | |||||
|
Warrants expired
|
(500 | ) | $ | 4.00 | ||||
|
Outstanding at December 31, 2010
|
1,375 | $ | 2.89 | |||||
NOTE 9. EQUITY-BASED COMPENSATION
Equity Compensation Plans
Prior to October 2007, the Company had two equity incentive plans in place: the 2002 Stock Option Plan and the 2005 Stock Option Plan. In October 2007, the Company adopted the 2007 Omnibus Incentive Plan (the 2007 Plan) to provide for the granting of stock awards, such as stock options, unrestricted and restricted common stock, stock units, dividend equivalent rights, and stock appreciation rights to employees, directors and outside consultants as determined by the board of directors. In conjunction with the adoption of the 2007 Plan, no further option awards may be granted from the 2002 or 2005 Stock Option Plans and any option cancellations or expirations from the 2002 or 2005 Stock Option Plans may not be reissued. At the inception of the 2007 Plan, 2,000,000 shares were reserved for issuance under the Plan. Beginning in January 2009, the number of shares of common stock authorized for issuance under the 2007 Plan increases annually in an amount equal to the lesser of (a) 1,000,000 shares or (b) 4% of the number of shares of the Company’s common stock outstanding on the last day of the preceding year or (c) such lesser number as determined by the board of directors. Accordingly, an additional 930,177 and 858,766 shares of common stock were authorized for issuance under the 2007 Plan in January 2010 and 2009, respectively. As of December 31, 2010, there were 234,944 shares available for future grant under the 2007 Plan.
Under the terms of the 2007 Plan, the exercise price of incentive stock options may not be less than 100% of the fair market value of the common stock on the date of grant and, if granted to an owner of more than 10% of the Company’s stock, then not less than 110%. Stock options granted under the 2007 Plan expire no later than ten years from the date of grant. Stock options granted to employees generally vest over four years while options granted to directors and consultants typically vest over a shorter period, subject to continued service. All of the options granted prior to October 2007 include early exercise provisions that allow for full exercise of the option prior to the option vesting, subject to certain repurchase provisions. The Company issues new shares to satisfy option exercises under the plans.
56
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Stock Options Summary
The following table summarizes information about the Company’s stock options outstanding at December 31, 2010, 2009 and 2008 and activity during the three years then ended.
|
(in thousands, except per share data)
|
Options
|
Weighted-
Average
Exercise Price
|
Weighted
Average
Remaining
Contractual
Life (years)
|
Aggregate
Intrinsic
Value
|
|||||||||||
|
Outstanding at December 31, 2007
|
2,896 | $ | 1.57 | ||||||||||||
|
Options granted
|
903 | $ | 2.39 | ||||||||||||
|
Options exercised
|
(122 | ) | $ | 1.18 | |||||||||||
|
Options forfeited/cancelled
|
(306 | ) | $ | 2.66 | |||||||||||
|
Outstanding at December 31, 2008
|
3,371 | $ | 1.70 | ||||||||||||
|
Options granted
|
1,196 | $ | 1.80 | ||||||||||||
|
Options exercised
|
(119 | ) | $ | 0.62 | |||||||||||
|
Options forfeited/cancelled
|
(301 | ) | $ | 2.42 | |||||||||||
|
Outstanding at December 31, 2009
|
4,147 | $ | 1.71 | ||||||||||||
|
Options granted
|
1,087 | $ | 1.90 | ||||||||||||
|
Options exercised
|
(104 | ) | $ | 0.85 | |||||||||||
|
Options forfeited/cancelled
|
(162 | ) | $ | 1.80 | |||||||||||
|
Outstanding at December 31, 2010
|
4,968 | $ | 1.78 | 6.7 | $ | 1,356 | |||||||||
|
Vested and expected to vest at December 31, 2010
|
4,896 | $ | 1.78 | 6.7 | $ | 1,347 | |||||||||
|
Vested at December 31, 2010
|
3,127 | $ | 1.64 | 5.5 | $ | 1,347 | |||||||||
|
Exercisable at December 31, 2010
|
3,127 | $ | 1.64 | 5.5 | $ | 1,331 | |||||||||
The aggregate intrinsic value is calculated as the difference between the exercise price of the underlying stock option awards and the closing market price of the Company’s common stock as quoted on the NYSE Amex as of December 31, 2010, for options that have a quoted market price in excess of the exercise price (“in-the-money options”). The Company received cash payments for the exercise of stock options in the amount of $81,000, $74,000 and $149,000 during the years ended December 31, 2010, 2009 and 2008, respectively. The aggregate intrinsic value of stock option awards exercised was $119,000, $148,000 and $108,000 for the years ended December 31, 2010, 2009 and 2008, respectively, as determined at the date of option exercise.
Stock Option Awards to Employees and Directors
The Company grants options to purchase common stock to some of its employees and directors at prices equal to or greater than the market value of the stock on the dates the options are granted. The Company has estimated the value of certain stock option awards as of the date of the grant by applying the Black-Scholes-Merton option pricing valuation model using the single-option valuation approach. The application of this valuation model involves assumptions that are judgmental and subjective in nature. See Note 2 for a description of the accounting policies that the Company applied to value its stock-based awards.
The weighted average assumptions used in determining the value of options granted and a summary of the methodology applied to develop each assumption are as follows:
|
Year Ended December 31,
|
||||||||||||
|
Assumption
|
2010
|
2009
|
2008
|
|||||||||
|
Expected price volatility
|
89.70 | % | 87.10 | % | 70.30 | % | ||||||
|
Expected term (in years)
|
5.9 | 6.1 | 6.1 | |||||||||
|
Risk-free interest rate
|
2.09 | % | 2.40 | % | 3.10 | % | ||||||
|
Dividend yield
|
0.00 | % | 0.00 | % | 0.00 | % | ||||||
|
Weighted-average fair value of options granted during the period
|
$ | 1.43 | $ | 1.31 | $ | 1.56 | ||||||
57
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Expected Price Volatility—This is a measure of the amount by which the stock price has fluctuated or is expected to fluctuate. The computation of expected volatility was based on the historical volatility of comparable companies from a representative peer group selected based on industry and market capitalization data.
Expected Term—This is the period of time over which the options granted are expected to remain outstanding. Because there is insufficient historical information available to estimate the expected term of the stock-based awards, we adopted the simplified method for estimating the expected term pursuant to SAB No. 107. On this basis, we estimated the expected term of options granted by taking the average of the vesting term and the contractual term of the option.
Risk-Free Interest Rate—This is the U.S. Treasury rate for the week of the grant having a term approximating the expected life of the option.
Dividend Yield—We have not made any dividend payments nor do we have plans to pay dividends in the foreseeable future.
Forfeitures are estimated at the time of grant and reduce compensation expense ratably over the vesting period. This estimate is adjusted periodically based on the extent to which actual forfeitures differ, or are expected to differ, from the previous estimate. For the years ended December 31, 2010 and 2009, we applied an estimated forfeiture rate of 5% to employee grants and 0% to director grants. Due to employee turnover in 2008, we applied an estimated forfeiture rate of 20% to employee grants and 0% to director grants.
For the years ended December 31, 2010, 2009 and 2008, we recognized stock-based compensation expense of $1.1 million, $702,000 and $721,000, respectively, for option awards to employees and directors. As of December 31, 2010, total unrecognized compensation cost related to unvested stock options was $2.1 million. This amount is expected to be recognized as stock-based compensation expense in our statements of operations over the remaining weighted average vesting period of 2.9 years.
Common Stock Awards to Directors
In December 2009 the Company adopted a new plan to compensate the independent members of the Board of Directors for their services. Under the terms of the Director Compensation Plan, each independent member is entitled to a combination of cash and stock options, at their discretion, for their participation in the board and various committees. If the director elects to receive stock options these are issued to the director at the beginning of the year and vest over the term of the year. Cash payments are made quarterly at the beginning of each quarter. In accordance with this new compensation arrangement no common stock awards were issued to directors in 2010.
In accordance with the provisions of the previous compensation agreement, the Company issued 130,000 and 51,000 shares of common stock to independent directors during the years ended December 31, 2009 and 2008, respectively. These shares were issued out of the 2007 Plan. The fair market value of the stock issued to directors was recorded as expense in the period in which the meeting occurred, resulting in total compensation expense of $218,000 and $124,000 for common stock awards to directors during the years ended December 31, 2009 and 2008, respectively.
Stock-Based Awards to Non-Employees
During the years ended December 31, 2010, 2009 and 2008, the Company granted options to purchase an aggregate of 186,000, 273,000 and 16,000 shares of common stock, respectively, to non-employees in exchange for advisory and consulting services. The stock options are recorded at their fair value on the measurement date and recognized over the respective service or vesting period. The fair value of the stock options granted was calculated using the Black-Scholes-Merton option pricing model based upon the following assumptions:
|
Year Ended December 31,
|
||||||||||||
|
Assumption
|
2010
|
2009
|
2008
|
|||||||||
|
Expected price volatility
|
90.86 | % | 87.20 | % | 70.00 | % | ||||||
|
Expected term (in years)
|
5.7 | 5.6 | 6.1 | |||||||||
|
Risk-free interest rate
|
2.31 | % | 1.90 | % | 3.10 | % | ||||||
|
Dividend yield
|
0.00 | % | 0.00 | % | 0.00 | % | ||||||
|
Weighted-average fair value of options granted during the period
|
$ | 1.39 | $ | 1.42 | $ | 1.26 | ||||||
For the years ended December 31, 2010 and 2009, the Company recognized stock-based compensation expense of $263,000 and $269,000, respectively, related to non-employee option grants. For the year ended December 31, 2008 the Company reversed previously recognized expense of $13,000 due to the required revaluation of unvested non-employee grants.
58
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Stock-Based Compensation Expense
A summary of the stock-based compensation expense included in results of operations for the option and stock awards discussed above is as follows:
|
Year ended December 31,
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Research and development
|
$ | 616 | $ | 565 | $ | 413 | ||||||
|
General and administrative
|
776 | 628 | 419 | |||||||||
|
Total stock-based compensation expense
|
$ | 1,392 | $ | 1,193 | $ | 832 | ||||||
Since the Company has operated at a loss and has net operating loss carryforwards, there are no tax benefits associated with stock-based compensation expense.
NOTE 10. COLLABORATION AND LICENSE AGREEMENTS
Alcon Manufacturing, Ltd.
In August 2006, we entered into a collaboration and license agreement with Alcon Manufacturing, Ltd. (Alcon) to license to Alcon the exclusive rights to develop, manufacture and commercialize products incorporating the Aganocide compounds for application in connection with the eye, ear and sinus and for use in contact lens solution. This agreement was amended in November 2010 to extend the period of the agreement through December 2015. Under the terms of the agreement, Alcon agreed to pay an up-front, non-refundable, non-creditable technology access fee of $10.0 million upon the effective date of the agreement. This up-front fee was recorded as deferred revenue and was amortized into revenue on a straight-line basis over the four-year funding term of the agreement through August 2010. Additionally, we receive semi-annual payments to support on-going research and development activities over the term of the agreement. The research and development support payments include amounts to fund a specified number of personnel engaged in collaboration activities and to reimburse for qualified equipment, materials and contract study costs. Our obligation to perform research and development activities under the agreement expires at the end of 2015. As product candidates are developed and proceed through clinical trials and approval, we will receive milestone payments. If the products are commercialized, we will also receive royalties on any sales of products containing the Aganocide compound. Alcon has the right to terminate the agreement in its entirety upon nine months’ notice, or terminate portions of the agreement upon 135 days’ notice, subject to certain provisions. Both parties have the right to terminate the agreement for breach upon 60 days’ notice.
Revenue has been recognized as follows:
|
Year Ended December 31,
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Amortization of Upfront Technology Access Fee
|
$ | 1,667 | $ | 2,500 | $ | 2,500 | ||||||
|
On-going Research and Development
|
5,419 | 4,322 | 2,700 | |||||||||
|
Materials, Equipment, and Contract Study Costs
|
562 | 1,349 | 1,386 | |||||||||
|
Milestone payment
|
— | 1,000 | — | |||||||||
| $ | 7,648 | $ | 9,171 | $ | 6,586 | |||||||
At December 31, 2010, 2009 and 2008, we had deferred revenue balances of $0, $1.7 million and $4.2 million, respectively, related to the Alcon agreement which amounts were comprised entirely of the upfront technology access fee.
Galderma
In March, 2009, the Company announced that it entered into a license and collaboration agreement with Galderma S.A. to develop and commercialize the Company’s Aganocide compounds, which covers acne and impetigo and potentially other major dermatological conditions, excluding onychomycosis (nail fungus) and orphan drug indications. The Company amended this agreement on December 17, 2009 and again on December 2, 2010. This agreement is exclusive and worldwide in scope, with the exception North America, where the Company has an option to exercise co-promotion rights, and Asian markets. Galderma will be responsible for the development costs of acne and other indications, except in Japan, in which Galderma has the option to request that we share such development costs. Galderma will also reimburse NovaBay for the use of its personnel in support of the collaboration. NovaBay retains the right to co-market products resulting from the agreement in Japan. In addition, NovaBay has retained all rights in other Asian markets outside Japan, and has the right to co-promote the products developed under the agreement in the hospital and other healthcare institutions in North America. Upon the termination of the agreement under certain circumstances, Galderma will grant NovaBay certain technology licenses which would require NovaBay to make royalty payments to Galderma for such licenses with royalty rates in the low- to mid-single digits.
59
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Galderma will pay to NovaBay certain upfront fees, ongoing fees, reimbursements, and milestone payments related to achieving development and commercialization of its Aganocide compounds. If products are commercialized under the agreement, NovaBay’s royalties will escalate as sales increase. The Company received a $1.0 million upfront technology access fee payment in the first quarter of 2009 and a $3.25 million continuation fee and a $500,000 fee to expand the license to include the Asia-Pacific Territory in December 2010. These fees were recorded as deferred revenues and recognized as earned on a straight-line basis over the Company’s expected performance period. The initial upfront technology access fee was recognized over the initial 20 month funding term of the agreement through October 2010, and the continuation and license fees are being recognized over the additional three year funding term of the agreement through November 2013.
Revenue has been recognized under the Galderma agreement as follows:
|
Year Ended December 31,
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Amortization of Upfront Technology Access Fee
|
$ | 786 | $ | 500 | $ | — | ||||||
|
On-going Research and Development
|
850 | 1,200 | — | |||||||||
|
Materials, Equipment, and Contract Study Costs
|
470 | 1,063 | — | |||||||||
|
Milestone payments
|
— | 3,750 | — | |||||||||
| $ | 2,106 | $ | 6,513 | $ | — | |||||||
The Company had deferred revenue balances of $3.7 million and $500,000 respectively, at December 31, 2010 and 2009, related to the Galderma agreement, which consisted of the unamortized balances on the upfront technology and access fee and the continuation and license fee and support for ongoing research and development. As of December 31, 2010, the Company has earned $3.75 million in milestone payments. This balance was included in accounts receivable as of December 31, 2009 and subsequently collected in 2010. As of December 31, 2010, the Company has not earned or received any royalty payments under the Galderma agreement.
KCI International VOF GP
In June 2007, we entered into a license agreement with an affiliate of Kinetic Concepts, Inc. (KCI), under which we granted KCI the exclusive rights to develop, manufacture and commercialize NVC-101, or NeutroPhase, as well as other products containing hypochlorous acid as the principal active ingredient, worldwide for use in wound care in humans, other than products or uses intended for the eye, ear or nose. Under the terms of the agreement, KCI paid to us a non-refundable technology access fee of $200,000. The up-front technology access fee was recorded as deferred revenue and has been amortized into revenue on a straight-line basis over the 18-month performance obligation period, through December 2008. Under the agreement, we are also entitled to receive reimbursements for qualified consulting, materials and contract study costs. In addition, we are entitled to receive payments of up to $1.25 million if certain milestones are met. If products covered by the license are commercially launched, we will also receive royalty payments based on net revenues from sales by KCI of such products. KCI has the right to terminate the agreement without penalty upon 60 days’ notice. We have the right to terminate the agreement if KCI has not commercially launched a product incorporating NVC-101, or any other product containing hypochlorous acid, within 18 months of the date of the agreement. Both parties have the right to terminate the agreement for breach upon 60 days’ notice. On November 19, 2009, the agreement between KCI and NovaBay was mutually terminated.
Revenue has been recognized as follows:
|
Year Ended December 31,
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Amortization of Upfront Technology Access Fee
|
$ | - | $ | - | $ | 128 | ||||||
|
On-going Research and Development
|
- | - | 8 | |||||||||
|
Materials, Equipment, and Contract Study Costs
|
- | - | - | |||||||||
| $ | - | $ | - | $ | 136 | |||||||
As of December 31, 2010 and 2009, we had no deferred revenue related to the KCI agreement and we had not earned or received any milestone or royalty payments under the KCI agreement.
NOTE 11. EMPLOYEE BENEFIT PLAN
We have a 401(k) plan covering all eligible employees. We are not required to contribute to the plan and have made no contributions through December 31, 2010.
60
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 12. INCOME TAXES
The federal and state income tax provision is summarized as follows (in thousands):
|
Year Ending December 31
|
|||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
||||||||||
|
Current
|
|||||||||||||
|
Federal
|
$ | — | $ | — | $ | — | |||||||
|
State
|
50 | 7 | 2 | ||||||||||
|
Other
|
— | — | — | ||||||||||
|
Total current tax expense
|
50 | 7 | 2 | ||||||||||
|
Deferred
|
|||||||||||||
|
Federal
|
— | — | — | ||||||||||
|
State
|
— | — | — | ||||||||||
|
Other
|
— | — | — | ||||||||||
|
Total deferred tax expense
|
— | — | — | ||||||||||
|
Income tax provision
|
$ | 50 | $ | 7 | $ | 2 | |||||||
Deferred income taxes reflect the net tax effects of (a) temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes, and (b) operating losses and tax credit carryforwards.
The tax effects of significant items comprising the Company's deferred taxes as of December 31 are as follows:
|
December 31
|
|||||||||
|
(in thousands)
|
2010
|
2009
|
|||||||
|
Deferred tax assets:
|
|||||||||
|
Net operating losses
|
$ | 9,515 | $ | 7,780 | |||||
|
Accruals
|
268 | 296 | |||||||
|
Deferred revenue
|
— | 664 | |||||||
|
Stock options
|
564 | 406 | |||||||
|
Other deferred tax assets
|
86 | 54 | |||||||
|
Total deferred tax assets
|
10,433 | 9,200 | |||||||
|
Deferred tax liabilities:
|
|||||||||
|
Property and equipment
|
(376 | ) | (322 | ) | |||||
|
Total deferred tax liabilities
|
(376 | ) | (322 | ) | |||||
|
Valuation allowance
|
(10,057 | ) | (8,878 | ) | |||||
|
Net deferred taxes
|
$ | — | $ | — | |||||
The Company records the tax benefit of net operating loss carryfowards and temporary differences as an asset to the extent that management assesses that realization is "more likely than not." Realization of the future tax benefits is dependent on the Company's ability to generate sufficient taxable income within the carryforward period. Because of the Company's recent history of operating losses, management believes that recognition of the deferred tax assets is currently not likely to be realized and, accordingly, has provided a valuation allowance.
The valuation allowance increased or (decreased) by the following amounts (in thousands):
|
2010
|
2009
|
2008
|
||||||||
| $ | 1,179 | $ | (1,130 | ) | $ | 2,978 | ||||
In accordance with ASC 718 Compensation – Stock Compensation, the Company has excluded from deferred tax assets benefits attributable to employee stock option exercises. Therefore, these amounts are not included in gross or net deferred tax assets. The benefit of these net operating loss carryforwards will only be recorded to equity when they reduce cash taxes payable.
61
NOVABAY PHARMACEUTICALS, INC.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Net operating loss carryforwards as of December 31, 2010 are as follows (in thousands):
|
Expiration
|
||||||||
|
Amount
|
Years
|
|||||||
|
Net operating losses, federal
|
$ | 24,217 | 2024 - 2030 | |||||
|
Net operating losses, state
|
$ | 25,321 | 2016 - 2030 | |||||
Under U.S. federal tax law, the amount and availability of tax benefits are subject to a variety of interpretations and restrictive tests. Utilization of the net operating loss (NOL) carryforwards may be subject to a substantial annual limitation due to ownership changes that have occurred previously or that could occur in the future, as provided by Section 382 of the Internal Revenue Code of 1986, and similar state provisions. Ownership changes may limit the amount of NOL carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50 percentage points over a three-year period. Since the Company’s formation, the Company has raised capital through the issuance of capital stock on two occasions which, combined with the purchasing shareholders’ subsequent disposition of those shares, may have resulted in one or more changes of control, as defined by Section 382. The Company has not currently completed a study to assess whether any change of control has occurred, or whether there have been multiple changes of control since the Company’s formation, due to the significant complexity and cost associated with the study. If the Company has experienced a change of control at any time since its formation, its NOL carryforwards may not be available, or their utilization could be subject to an annual limitation under Section 382. A full valuation allowance has been provided against the Company’s NOL carryforwards, and if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. Accordingly, there would be no impact on the consolidated balance sheet or statement of operations if an adjustment is required.
The effective tax rate of the Company's provision (benefit) for income taxes differs from the federal statutory rate as follows:
|
Year Ending December 31
|
||||||||||||
|
(in thousands)
|
2010
|
2009
|
2008
|
|||||||||
|
Income tax benefit at federal statutory rate
|
$ | (1,320 | ) | $ | 950 | $ | (2,758 | ) | ||||
|
State tax
|
(187 | ) | 152 | (435 | ) | |||||||
|
ISO-related expense for GAAP
|
384 | 196 | 207 | |||||||||
|
Change in valuation allowance
|
1,179 | (1,130 | ) | 2,978 | ||||||||
|
Other
|
(6 | ) | (161 | ) | 10 | |||||||
|
Total
|
$ | 50 | $ | 7 | $ | 2 | ||||||
Uncertain Income Tax Positions
We adopted the provisions of ASC 740-10-50 Accounting for Uncertainty in Income Tax Provisions on January 1, 2007. We have no unrecognized tax benefits, including no accrued amounts for interest and penalties, during the three year period ended December 31, 2010.
Our policy is to recognize interest and penalties accrued on any unrecognized tax benefits as a component of income tax expense. We are subject to income tax examinations for U.S. incomes taxes and state income taxes from 2004 to 2010 due to our net operating loss carryforwards. We do not anticipate that total unrecognized tax benefits will significantly change prior to December 31, 2011.
NOTE 13. SUBSEQUENT EVENTS
We evaluated subsequent events through the issuance date of the financial statements. We are not aware of any significant events that occurred subsequent to the balance sheet date but prior to the filing of this Annual Report on Form 10-K that would have a material impact on our consolidated financial statements
62
|
ITEM 9.
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
|
None.
|
ITEM 9A.
|
CONTROLS AND PROCEDURES
|
Evaluation of Disclosure Controls and Procedures
As of the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures pursuant to Rule 13a-15 and 15d-15 of the Securities Exchange Act of 1934, as amended (the Exchange Act). Based upon that evaluation, our Chief Executive Officer and our Chief Financial Officer concluded that our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and were effective in ensuring that information required to be disclosed by us in the reports that we file or submit under the Exchange Act was accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Assessing the costs and benefits of such controls and procedures necessarily involves the exercise of judgment by management. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Our Chief Executive Officer and our Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our management, including our principal executive officer and our principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control — Integrated Framework. Our management has concluded that, as of December 31, 2010, our internal control over financial reporting was effective based on these criteria.
Changes in Internal Control Over Financial Reporting
During the fourth quarter of 2010, there were no changes in our internal control over financial reporting which has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
|
ITEM 9B.
|
OTHER INFORMATION
|
None
PART III
|
ITEM 10.
|
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
The information required by this Item with respect to Executive Officers may be found under the caption, “Executive Compensation and Other Information” appearing in the definitive Proxy Statement to be delivered to NovaBay’s stockholders in connection with the solicitation of proxies for NovaBay’s 2011 Annual Meeting of Stockholders (the Proxy Statement). The information required by this Item with respect to Directors, including information with respect to our audit committee, audit committee financial experts, risk management and procedures for Board nominations, is incorporated herein by reference from the information under the caption, “Proposal One: Election of Directors” and “Corporate Governance” appearing in the Proxy Statement.
Section 16(a) Beneficial Ownership Reporting Compliance
The information required by this Item with respect to compliance with Section 16(a) of the Exchange Act is incorporated herein by reference from the section captioned “Section 16(a) Beneficial Ownership Reporting Compliance” contained in the Proxy Statement.
63
Code of Ethics and Business Conduct
The information required by this Item with respect to our code of ethics and business conduct is incorporated herein by reference from the section captioned “Corporate Governance” contained in the Proxy Statement.
|
ITEM 11.
|
EXECUTIVE COMPENSATION
|
The information required by this Item is set forth in the Proxy Statement under the caption, “Executive Compensation and Other Information.” Such information is incorporated herein by reference.
|
ITEM 12.
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
|
The information required by this Item with respect to security ownership of certain beneficial owners and management is set forth in the Proxy Statement under the caption, “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information.” Such information is incorporated herein by reference.
|
ITEM 13.
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
|
The information required by this Item is set forth in the Proxy Statement under the headings “Proposal 1: Election of Directors” and “Certain Relationships and Related Transactions.” Such information is incorporated herein by reference.
|
ITEM 14.
|
PRINCIPAL ACCOUNTING FEES AND SERVICES
|
The information required by this Item is set forth in the Proxy Statement under the heading “Fees Paid to Independent Registered Public Accounting Firm.” Such information is incorporated herein by reference.
Consistent with Section 10A(i)(2) of the Securities Exchange Act of 1934, as added by Section 202 of the Sarbanes-Oxley Act of 2002, we are responsible for listing the non-audit services approved by our Audit Committee to be performed by Odenberg, Ullakko, Muranishi & Co., LLP, our external auditor. Non-audit services are defined as services other than those provided in connection with an audit or a review of our financial statements. Our Audit Committee has approved our recurring engagements of non-audit services of Moss Adams, LLP for the preparation of tax returns, and tax advice in preparing for and in connection with such filings.
64
PART IV
|
ITEM 15.
|
EXHIBITS, FINANCIAL STATEMENT SCHEDULES
|
(a) Documents filed as part of this report:
(1) Financial Statements. The following financial statements of NovaBay Pharmaceuticals, Inc. are included in Item 8 of this Annual Report on Form 10-K commencing on the pages referenced below:
|
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
|
||
|
Page
|
||
|
Reports of Independent Registered Public Accounting Firm
|
38
|
|
|
Consolidated Balance Sheets as of December 31, 2010 and 2009
|
40
|
|
|
Consolidated Statements of Operations for the Years Ended December 31, 2010, 2009 and 2008 and for the cumulative period from July 1, 2002 (inception) to December 31, 2010
|
41
|
|
|
Consolidated Statements of Stockholder's Equity for the cumulative period from July 1, 2002 (inception) to December 31, 2010
|
42
|
|
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2010, 2009 and 2008 and for the cumulative period from July 1, 2002 (inception) to December 31, 2010
|
46
|
(2) Financial Statement Schedules.
All schedules have been omitted because they are not required or the required information is included in our consolidated financial statements and notes thereto.
(3) Exhibits.
See the Exhibit Index which follows the signature page of this Annual Report on Form 10-K, which is incorporated herein by reference.
65
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Date: March 9, 2011
|
NOVABAY PHARMACEUTICALS, INC.
|
|
|
By:
|
/S/ RAMIN NAJAFI
|
|
|
Ramin (Ron) Najafi
|
||
|
Chairman and Chief Executive Officer
|
||
POWER OF ATTORNEY
We, the undersigned officers and directors of NovaBay Pharmaceuticals, Inc., do hereby constitute and appoint Ramin (Ron) Najafi and Thomas J. Paulson, and each of them, our true and lawful attorneys-in-fact and agents, each with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this report, and to file the same, with exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby, ratifying and confirming all that each of said attorneys-in-fact and agents, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report on Form 10-K has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated:
|
Signature
|
Title
|
Date
|
||
|
/S/ RAMIN NAJAFI
|
Chairman of the Board and Chief Executive Officer
(principal executive officer)
|
March 8, 2011
|
||
|
Ramin (Ron) Najafi
|
||||
|
/S/ THOMAS PAULSON
|
Chief Financial Officer and Treasurer (principal
financial and accounting officer)
|
March 8, 2011
|
||
|
Thomas J. Paulson
|
||||
|
/S/ CHARLES J. CASHION
|
Director
|
March 8, 2011
|
||
|
Charles J. Cashion
|
||||
|
/S/ ANTHONY DAILLEY
|
Director
|
March 8, 2011
|
||
|
Anthony Dailley, DDS
|
||||
|
/S/ PAUL FREIMAN
|
Director
|
March 9, 2011
|
||
|
Paul E. Freiman
|
||||
|
/S/ ALEX MCPHERSON
|
Director
|
March 7, 2011
|
||
|
Alex McPherson, MD, Ph.D.
|
||||
|
/S/ ROBERT R. TUFTS
|
Director
|
March 8, 2011
|
||
|
Robert R. Tufts
|
||||
|
/S/ TONY WICKS
|
Director
|
March 8, 2011
|
||
|
Tony Wicks
|
||||
|
/S/ GAIL MADERIS
|
Director
|
March 7, 2011
|
||
|
Gail Maderis
|
66
EXHIBIT INDEX
|
Exhibit
No.
|
Description
|
|
|
2.1
|
Agreement and Plan of Merger between NovaBay Pharmaceuticals, Inc., a California corporation, and NovaBay Pharmaceuticals, Inc., a Delaware corporation, dated as of June 25, 2010 (Incorporated by reference to the exhibit of the same number from the Company’s Post-Effective Amendment No. 2 to the registration statement on Form S-3 filed with the SEC on July 1, 2010 (File Nos. 333-159917))
|
|
|
3.1
|
Certificate of Incorporation of NovaBay Pharmaceuticals, Inc., a Delaware corporation (Incorporated by reference to the exhibit of the same number from the Company’s current report on Form 8-K, as filed with the SEC on June 29, 2010 (SEC File No. 001-33678))
|
|
|
3.2
|
Bylaws of NovaBay Pharmaceuticals, Inc., a Delaware corporation (Incorporated by reference to the exhibit of the same number from the Company’s current report on Form 8-K as filed with the SEC on June 29, 2010 (SEC File No. 001-33678))
|
|
|
4.1*
|
Specimen common stock certificate
|
|
|
4.2
|
Form of Form of Common Stock Purchase Warrant issued in August 2009. (Incorporated by reference to Exhibit 4.3 to the Company’s current report on Form 8-K as filed with the SEC on August 21, 2009 (SEC File No. 001-33678).)
|
|
|
10.1*+
|
2002 Stock Option Plan, and forms of agreements thereto
|
|
|
10.2*+
|
2005 Stock Option Plan, and forms of agreements thereto
|
|
|
10.3*+
|
2007 Omnibus Incentive Plan, and forms of agreements thereto ((the Plan is incorporated by reference to Exhibit 10.1 from the Company’s quarterly report on Form 10-Q for the quarter ended June 30, 2008 as filed with the SEC on August 14, 2008 (SEC File No. 001-33678), and the forms of agreements thereto are incorporated by reference to the exhibit referencing the Plan from the Company’s amendment to registration statement of Form S-1 (File No. 333-140714) filed with the Securities and Exchange Commission on May 29, 2007, as amended.)
|
|
|
10.4*+
|
Employment Agreement dated January 1, 2007 by and between the Company and Ramin (Ron) Najafi
|
|
|
10.5*+
|
Employment Agreement dated January 1, 2007 by and between the Company and John (Jack) O’Reilly
|
|
|
10.6*+
|
Employment Agreement dated January 1, 2007 by and between the Company and Behzad Khosrovi
|
|
|
10.7+
|
NovaBay Pharmaceuticals, Inc. Employee Incentive Cash Compensation Plan (Incorporated by reference to Exhibit of the same number from the Company’s annual report on Form 10-K for the year end December 31, 2009 as filed with the SEC on March 30, 2010 (SEC File No. 001-33678).).
|
|
|
10.8+
|
Employment Agreement dated January 9, 2008 by and between the Company and Thomas J. Paulson (Incorporated by reference to Exhibit 10.18 from the Company’s annual report on Form 10-K for the year end December 31, 2007 as filed with the SEC on March 14, 2008 (SEC File No. 001-33678).)
|
|
|
10.10*
|
Office Lease dated June 3, 2004 by and between the Company and Emery Station Associates II, LLC, as amended
|
|
|
10.11
|
Fifth Amendment dated November 20, 2007 to Office Lease dated June 3, 2004 by and between the Company and Emery Station Associates II, LLC, as amended (Incorporated by reference to Exhibit 10.20 from the Company’s annual report on Form 10-K for the year ended December 31, 2007 as filed with the SEC on March 14, 2008 (SEC File No. 001-33678).)
|
|
|
10.12
|
Sixth Amendment to Lease between Emery Station Office II, LLC and Novacal Pharmaceuticals, Inc., effective September 1, 2008. (Incorporated by reference to Exhibit 10.1 from the Company’s quarterly report on Form 10-Q/A for the quarter ended September 30, 2008 as filed with the SEC on November 14, 2008 (SEC File No. 001-33678)).
|
|
|
10.13†
|
Collaboration and License Agreement, by and between the Company and Galderma S.A., dated as of March 20, 2009 (Incorporated by reference to Exhibit 10.2 from the Company’s quarterly report on Form 10-Q/A for the quarter ended March 31, 2009, as filed with the SEC on August 4, 2009 (SEC File No. 001-33678)).
|
67
|
10.14+
|
Director Compensation Plan (Incorporated by reference to Exhibit of the same number from the Company’s annual report on Form 10-K for the year end December 31, 2009 as filed with the SEC on March 30, 2010 (SEC File No. 001-33678)).
|
|
|
10.15*†
|
Collaboration and License Agreement dated August 29, 2006 by and between the Company and Alcon Manufacturing, Ltd.
|
|
|
10.16*
|
Master Security Agreement dated April 23, 2007 by and between the Company and General Electric Capital Corporation
|
|
|
10.17*
|
Form of Common Stock Purchase Warrant by and between the Company and the underwriters
|
|
|
10.18†
|
Amendment No. 1 to the Collaboration and License Agreement, dated as of December 1, 2009, between the Company and Galderma S.A. (Incorporated by reference to Exhibit of the same number from the Company’s annual report on Form 10-K for the year end December 31, 2009 as filed with the SEC on March 30, 2010 (SEC File No. 001-33678)).
|
|
|
10.19+
|
Executive Officer Cash Compensation Arrangements (Incorporated by reference to Exhibit of the same number from the Company’s annual report on Form 10-K for the year end December 31, 2009 as filed with the SEC on March 30, 2010, and the description in Item 5.02 of the Company’s current report on Form 8-K as filed with the SEC on February 25, 2011 (SEC File No. 001-33678)).
|
|
|
10.20+
|
Employment Agreement, dated July 28, 2009, between the Company and Roy Wu (Incorporated by reference to Exhibit of the same number from the Company’s annual report on Form 10-K for the year end December 31, 2009 as filed with the SEC on March 30, 2010 (SEC File No. 001-33678)).
|
|
|
10.21
|
Placement Agent Agreement, dated August 21, 2009, by and between the Company and Maxim Group LLC (Incorporated by reference to Exhibit 1.1 from the Company’s quarterly report on Form 8-K as filed with the SEC on August 21, 2009 (SEC File No. 001-33678)).
|
|
|
10.22+
|
Employment Agreement, dated October 15, 2009, between the Company and Mark Anderson (Incorporated by reference to Exhibit of the same number from the Company’s annual report on Form 10-K for the year end December 31, 2009 as filed with the SEC on March 30, 2010 (SEC File No. 001-33678)).
|
|
|
10.23+
|
Form of Indemnification Agreement between NovaBay Pharmaceuticals, Inc. and its Directors and Officers. (Incorporated by reference to Exhibit 10.1 from the Company’s quarterly report on Form 10-Q for the quarter ended June 30, 2010, as filed with the SEC on August 12, 2010 (SEC File No. 001-33678).)
|
|
|
10.24††
|
Amendment No. 2 to the Collaboration and License Agreement, dated as of December 2, 2010, between the Company and Galderma S.A.
|
|
|
10.25††
|
Amendment No. 1 to the Collaboration and License Agreement dated November 18, 2010 by and between the Company and Alcon Manufacturing, Ltd.
|
|
|
23.1
|
Consent of Odenberg, Ullakko, Muranishi & Co. LLP
|
|
|
23.2
|
Consent of Davidson & Co, LLP
|
|
|
24.1
|
Power of Attorney (included on the signature pages hereto)
|
|
|
31.1
|
Certification of the principal executive officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
|
|
31.2
|
Certification of the principal financial officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002
|
|
|
32.1
|
Certification of the chief executive officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|
|
32.2
|
Certification of the chief financial officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|
*
|
Incorporated by reference to the exhibit of the same description from the Company’s registration statement of Form S-1 (File No. 333-140714) initially filed with the Securities and Exchange Commission on February 14, 2007, as amended.
|
|
+
|
Indicates a management contract or compensatory plan or arrangement
|
|
†
|
NovaBay Pharmaceuticals, Inc. has been granted confidential treatment with respect to certain portions of this exhibit (indicated by asterisks), which have been separately filed with the Securities and Exchange Commission.
|
|
††
|
NovaBay Pharmaceuticals, Inc. has requested confidential treatment with respect to certain portions of this exhibit (indicated by asterisks), which have been separately filed with the Securities and Exchange Commission.
|