Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 001-33500
JAZZ PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 05-0563787 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
3180 Porter Drive
Palo Alto, CA 94304
(650) 496-3777
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, par value $0.0001 per share |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer x | Non-accelerated filer ¨ | Smaller reporting company x | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the voting and non-voting stock held by non-affiliates of the registrant as of June 30, 2010, based upon the last sale price reported for such date on the NASDAQ Global Market, was $146,122,278. The calculation of the aggregate market value of voting and non-voting stock excludes 20,188,209 shares of the registrant’s common stock held by executive officers, directors, and stockholders that the registrant has concluded are affiliates of the registrant. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
As of February 28, 2011, a total of 40,294,596 shares of the registrant’s Common Stock, $0.0001 par value, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for the 2011 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Form 10-K are incorporated by reference in Part III, Items 10-14 of this Form 10-K.
Table of Contents
2010 ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
| Page | ||||||
| PART I | 3 | |||||
| Item 1. |
3 | |||||
| Item 1A. |
21 | |||||
| Item 1B. |
41 | |||||
| Item 2. |
41 | |||||
| Item 3. |
42 | |||||
| Item 4. |
43 | |||||
| PART II | 44 | |||||
| Item 5. |
44 | |||||
| Item 6. |
46 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
48 | ||||
| Item 7A. |
61 | |||||
| Item 8. |
62 | |||||
| Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
63 | ||||
| Item 9A. |
63 | |||||
| Item 9B. |
65 | |||||
| PART III | 65 | |||||
| Item 10. |
65 | |||||
| Item 11. |
65 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
65 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
65 | ||||
| Item 14. |
65 | |||||
| PART IV | 66 | |||||
| Item 15. |
66 | |||||
| 73 | ||||||
In this report, “Jazz Pharmaceuticals,” “we,” “us,” and “our” refer to Jazz Pharmaceuticals, Inc. and its consolidated subsidiaries. We own or have rights to various copyrights, trademarks, and trade names used in our business, including the following: Jazz Pharmaceuticals®; Xyrem® (sodium oxybate) oral solution; Luvox CR® (fluvoxamine maleate) Extended-Release Capsules; and Luvox® (fluvoxamine maleate). This report also includes trademarks, service marks, and trade names of other companies.
2
Table of Contents
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the “safe harbor” created by those sections. Forward-looking statements are based on our management’s beliefs and assumptions and on information currently available to our management. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “project,” “predict,” “potential” and similar expressions intended to identify forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance, time frames or achievements to be materially different from any future results, performance, time frames or achievements expressed or implied by the forward-looking statements. We discuss many of these risks, uncertainties and other factors in this Annual Report on Form 10-K in greater detail under the heading “Risk Factors.” Given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this filing. You should read this Annual Report on Form 10-K completely and with the understanding that our actual future results may be materially different from what we expect. We hereby qualify our forward-looking statements by our cautionary statements. Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
PART I
| Item 1. | Business |
Overview
We are a specialty pharmaceutical company focused on the identification, development and commercialization of pharmaceutical products to meet important unmet medical needs. Since we were founded in 2003, we have built a commercial and development organization and assembled a portfolio of products and product candidates that currently includes our two marketed products, which generated net product sales of $170.0 million in 2010, and product candidates in various stages of clinical development. We currently market two products: Xyrem (sodium oxybate), which is the only product approved by the United States Food and Drug Administration, or FDA, for the treatment of both cataplexy and excessive daytime sleepiness in patients with narcolepsy; and Luvox CR (fluvoxamine maleate) marketed for the treatment of obsessive compulsive disorder. We promote these products in the United States through our experienced specialty sales force targeting sleep specialists, neurologists, pulmonologists and psychiatrists. We are building our portfolio of products through a combination of internal development, acquisition and in-licensing activities. Our current product candidates are JZP-6 (sodium oxybate) for the treatment of fibromyalgia, JZP-8 (intranasal clonazepam) for the treatment of acute repetitive seizures in epilepsy, and solid oral dosage forms of sodium oxybate.
We are building a sustainable pharmaceutical company by:
| • | Growing and protecting our sodium oxybate business, including growing sales of Xyrem in its approved indications, continuing to invest in our franchise, and enforcing our intellectual property covering sodium oxybate and our restricted distribution system; |
| • | Developing additional products and advancing our pipeline though continued investment in research and development activities targeted at areas of significant unmet need where our product candidates may offer significant benefits to patients; and |
| • | Leveraging our commercial capabilities, including our sales and marketing organization, and our regulatory, safety and clinical organizations, by in-licensing or acquiring additional products and product candidates targeted towards specialty physician audiences. |
3
Table of Contents
Marketed Products
Xyrem (sodium oxybate) oral solution
Xyrem is a sodium oxybate oral solution approved in the United States for the treatment of excessive daytime sleepiness and cataplexy in patients with narcolepsy. Sodium oxybate, the active pharmaceutical ingredient in Xyrem, is a formulation of the sodium salt of g-hydroxybutyrate, an endogenous neurotransmitter and metabolite of g-aminobutyric acid. Xyrem is the only treatment approved by the FDA for both excessive daytime sleepiness and cataplexy in patients with narcolepsy. Xyrem was approved for the treatment of cataplexy in patients with narcolepsy in 2002, and was approved for its second indication, the treatment of excessive daytime sleepiness in patients with narcolepsy, in 2005. The American Academy of Sleep Medicine recommends Xyrem as a standard of care for the treatment of both excessive daytime sleepiness and cataplexy associated with narcolepsy.
Narcolepsy is a chronic neurologic disorder caused by the brain’s inability to regulate sleep-wake cycles. The primary symptoms of narcolepsy include excessive daytime sleepiness, cataplexy, sleep paralysis, sleep-onset and waking hallucinations and fragmented nighttime sleep. These symptoms can lead to a variety of complications, such as limitations on education and employment opportunities, driving or machinery accidents, difficulties at work resulting in disability, forced retirement or job dismissal and depression. Excessive daytime sleepiness is the most common symptom of narcolepsy and is present in all narcolepsy patients. Excessive daytime sleepiness is a chronic, pervasive sleepiness that triggers sudden irresistible and overwhelming urges to sleep (inadvertent naps and sleep attacks). Cataplexy, the sudden loss of muscle tone, can be one of the most debilitating symptoms of narcolepsy. Cataplexy is present in between 60% and 100% of patients with narcolepsy. Cataplexy can range from slight weakness or a drooping of the face to the complete loss of muscle tone and it is often triggered by strong emotional reactions such as laughter, anger or surprise. Cataplexy can severely impair a patient’s quality of life and ability to function.
According to the National Institutes of Health, 150,000 to 200,000 individuals in the United States are affected by narcolepsy; however, the National Heart Lung and Blood Institute estimates that only approximately 25% of those patients have been diagnosed with narcolepsy. Xyrem is currently being used to treat approximately 8,000 to 9,000 patients in the United States, and we believe there are additional patients with narcolepsy and cataplexy and/or excessive daytime sleepiness who could benefit from treatment with Xyrem.
We are developing solid oral dosage forms for sodium oxybate, which is currently administered as a twice nightly liquid. Our objective is to improve patient convenience and compliance.
In 2010, our net product sales of Xyrem were $142.6 million.
Commercialization and Distribution
We promote Xyrem in the United States through our specialty sales force. Our marketing, sale and distribution of Xyrem are subject to a risk management plan which was required in conjunction with Xyrem’s approval by the FDA.
Under the Xyrem risk management plan, the Xyrem Success Program®, Xyrem is distributed through a single central pharmacy, Express Scripts Specialty Distribution Services and its affiliate Curascript, Inc., or Express Scripts, with which we have an exclusive relationship. The central pharmacy maintains physician and patient registries, and the product may not be stocked in retail pharmacies. Each physician and patient receives materials concerning the risks and benefits of the product before the physician can prescribe, or a patient can receive, Xyrem. Whenever a prescription is received by the central pharmacy, the central pharmacy verifies the prescription and obtains additional information by contacting the patient’s insurance company. The central pharmacy also speaks with the patient before it ships any Xyrem to the patient. The central pharmacy ships the product directly to the patient by a courier service, and the patient or his/her designee signs for the package. The initial shipment may only be for a one-month supply and physicians may only prescribe up to six months of supply of Xyrem at one time.
4
Table of Contents
Pursuant to our exclusive agreement, Express Scripts distributes Xyrem and provides customer support services related to the sale and marketing of Xyrem in the United States. Our agreement, which has been in effect since July 2002, expires on June 30, 2012, subject to automatic one-year extensions until either party provides notice to the other of its intent to terminate the agreement at least 120 days prior to the end of the then current term. Under the agreement, we own all of the standard operating procedures, business rules and intellectual property, and the agreement provides for Express Scripts to assist in the orderly transfer of the services Express Scripts provides to us and the related intellectual property, including that of the patient database, to any new pharmacy we engage.
Outside the United States, we have licensed to UCB Pharma Limited, or UCB, the exclusive right to market Xyrem for the treatment of narcolepsy in 54 countries in exchange for milestone and royalty payments to us. UCB currently markets the product in 15 countries in Europe. We are entitled to commercial milestone payments from UCB of up to $6.0 million specifically associated with UCB’s sales of Xyrem for the treatment of narcolepsy and royalties on all commercial sales of Xyrem by UCB. The term of our agreement with UCB, as it applies to Xyrem for the treatment of narcolepsy, extends to the later of the expiration of our associated patent rights in the territories covered by the agreement or ten years from the date of European Medicines Agency, or EMA, approval to commercially promote and distribute Xyrem for the treatment of narcolepsy, subject to automatic extension unless and until UCB terminates the agreement. UCB may terminate our agreement for any reason upon 12 months’ notice. We are responsible for supplying Xyrem to UCB in exchange for supply price payments. We have licensed to Valeant Canada Limited, or Valeant, the Canadian marketing rights to Xyrem for the treatment of narcolepsy. We supply Xyrem to UCB and Valeant.
Xyrem is a controlled substance in the United States and, therefore its manufacturing and distribution are highly restricted. Quotas from the United States Drug Enforcement Administration, or DEA, are required in order to manufacture and package sodium oxybate. Since the DEA typically grants quota on an annual basis and requires a detailed submission and justification for the request, obtaining a DEA quota is a difficult and time consuming process. The final product and active pharmaceutical ingredient are manufactured for us by single source contract manufacturers.
Intellectual Property
The FDA has granted Xyrem orphan drug status in the United States for excessive daytime sleepiness in patients with narcolepsy, which provides marketing exclusivity in the United States until November 2012 for this indication. Xyrem is covered by nine patents, of which seven are listed in the FDA’s approved drug products with therapeutic equivalence evaluation document, or Orange Book. Of the patents listed in the Orange Book, two are formulation patents expiring in 2020 and four are method of use patents covering the distribution of Xyrem, three of which expire in 2024 and one of which expires in 2022. We have an additional method of use patent covering Xyrem’s use in narcolepsy which expires in 2019. A process patent and a distribution system patent not listed in the Orange Book also cover the product and expire in 2019 and 2024, respectively. In addition to our issued patents, we have a number of patent applications covering Xyrem pending. On October 18, 2010, we received a Paragraph IV Patent certification notice, or Paragraph IV certification, from Roxane Laboratories, Inc., or Roxane, indicating that it had filed an abbreviated new drug application, or ANDA, with the FDA requesting approval to market a generic version of Xyrem. On November 22, 2010, we filed a lawsuit against Roxane in response to Roxane’s Paragraph IV certification in the United States District Court for the District of New Jersey. For a description of this matter and related risks, please see “Item 3. Legal Proceedings” and “Item 1A. Risk Factors” under the heading “If generic products that compete with Xyrem are approved, sales of Xyrem may be adversely affected.”
Luvox CR (fluvoxamine maleate) Extended-Release Capsules
We market Luvox CR for the treatment of obsessive compulsive disorder. Luvox CR received FDA approval in 2008. Luvox CR incorporates the SODAS™ drug delivery technology, developed by Elan Pharma International Limited, or Elan, which is designed to minimize peak-to-trough plasma fluctuations over a 24-hour period and enable once-a-day dosing.
5
Table of Contents
Obsessive compulsive disorder is a chronic anxiety disorder characterized by persistent, unwanted thoughts, or obsessions, and repetitive behaviors or rituals, or compulsions. According to the National Institute of Mental Health, obsessive compulsive disorder affects approximately 2.2 million adults in the United States. According to an article published in the International Journal of Clinical Practice, it is estimated that 60% of patients with obsessive compulsive disorder worldwide receive no treatment for their disorder. Patients with obsessive compulsive disorder use rituals to help control anxiety related to their obsessive thoughts, and these rituals become disruptive to their daily life.
We licensed the rights to market Luvox CR in the United States from Solvay Pharmaceuticals, Inc., or Solvay, which was subsequently acquired by Abbott Laboratories, or Abbott. Solvay assigned to us its rights and obligations under its license and supply agreement with Elan, and we sublicensed back to Solvay the rights under that agreement outside of the United States. Under a supply agreement with Abbott, we are responsible for purchasing, and Abbott is responsible for providing us with, the active pharmaceutical ingredient necessary to manufacture Luvox CR. We are responsible for providing the active pharmaceutical ingredient free of charge to Elan under the license and supply agreement with Elan. Elan has the right and obligation to manufacture the worldwide commercial requirements of Luvox CR. We are responsible for satisfying Abbott’s commercial requirements of Luvox CR outside of the United States in exchange for supply price payments to us. Luvox CR is not currently marketed outside the United States. Under the terms of the license agreement as amended, we have paid Abbott $39.0 million through 2010 and we owe Abbott $4.5 million in 2011 and $5.0 million in 2012. If we pay these amounts when due, the payments due in 2012 will decrease to $4.5 million. We have also agreed to pay Abbott $5.0 million in 2015 if our net sales of Luvox CR reach a cumulative amount of $100 million on or before December 31, 2014 and no AB-rated generic version of Luvox CR has been or is being sold in the United States as of December 31, 2014.
Our license and supply agreements with Abbott will remain in force until terminated by either Abbott or us as a result of an uncured breach by the other party. The license and supply agreement with Elan that was assigned to us by Solvay will expire upon the later of (i) 10 years after commercial launch of Luvox CR or (ii) the last to expire patent licensed under the agreement with Elan. In addition, either we or Elan may terminate the license agreement in the event of an uncured material breach or in the event of a change of ownership of the other party in excess of 40% or an acquisition of 20% or more of the equity of the other party by a third party offering competing products.
The FDA approval for Luvox CR also included an indication for social anxiety disorder. We have been in discussions with the FDA about removing the social anxiety disorder indication from the label, and we expect that, if the indication is removed, the obligation to complete the remaining Phase IV studies in social anxiety disorder patients will also terminate.
Intellectual Property
Luvox CR is covered by a patent owned by Elan with claims covering the orally administered formulation of extended-release fluvoxamine that requires the release of fluvoxamine over a period of not less than 12 hours. This patent is listed in the Orange Book, and expires in 2020. In August 2009, we received a Paragraph IV Certification from Actavis Elizabeth, LLC, or Actavis, advising that Actavis had filed an ANDA with the FDA seeking approval to market a generic version of Luvox CR. In September 2009, we received an additional Paragraph IV Patent Certification notice from Anchen Pharmaceuticals, Inc., or Anchen, advising that Anchen has filed an ANDA with the FDA for a generic version of Luvox CR. We and Elan filed lawsuits in response to the Paragraph IV certifications. In August 2010, we and Elan entered into settlement agreements with Anchen and granted a sublicense to Anchen of our rights to have manufactured, market and sell a generic version of Luvox CR. The sublicense will commence on February 15, 2013 or earlier upon the occurrence of certain events. The lawsuit against Actavis is pending in the United States District Court for the District of Delaware. For a more detailed description of our disputes with Anchen and Actavis, please see “Item 3. Legal Proceedings.”
6
Table of Contents
Clinical Development Pipeline
We have a number of product candidates in various stages of clinical development. In 2010, 2009 and 2008 we spent $25.6 million, $36.6 million and $70.0 million, respectively, on research and development activities.
JZP-6 (sodium oxybate)
Our most advanced product candidate is JZP-6, which uses sodium oxybate, the active pharmaceutical ingredient in Xyrem, for the treatment of fibromyalgia, which is a chronic condition characterized by widespread pain. According to the American College of Rheumatology, approximately two to four percent of the U.S. population suffers from fibromyalgia. Fibromyalgia is believed to be a central nervous system condition, resulting from neurological changes in how the brain perceives and responds to pain. In addition to pain, the main symptoms are fatigue, disturbed sleep and morning stiffness.
We completed two randomized, double-blind, placebo-controlled Phase III pivotal clinical trials and a long-term safety trial as part of the development program for JZP-6 in fibromyalgia. These studies demonstrated positive results and were used in support of our new drug application, or NDA, filed with the FDA. In our trials, sodium oxybate was generally well tolerated, with the majority of adverse events reported being mild to moderate in nature.
Our NDA for JZP-6 was accepted for filing by the FDA in February 2010. The FDA’s Arthritis Advisory Committee and Drug Safety and Risk Management Advisory Committee reviewed JZP-6 at a joint meeting in August 2010 and voted 20-2 against approval of the NDA as submitted. In October 2010, the FDA sent us a complete response letter, or CRL, stating that the FDA cannot approve the NDA in its present form. In the letter, the FDA discussed a number of topics, including the need for additional clinical studies, the appropriate patient population, methods for ensuring safe use, the proposed Risk Evaluation and Mitigation Strategy, or REMS, program, concentration of the formulation and the trade name for the product. We have had additional communications with the FDA, but have not yet finalized our plans with respect to JZP-6. We do not currently know the timing or cost of the continued development of JZP-6, if its development will be continued, or whether the NDA for JZP-6 will be approved by the FDA.
UCB has the right to market JZP-6 for the treatment of fibromyalgia in 54 countries outside the United States. UCB has submitted an application to the European Medicines Agency for approval to market and promote JZP-6, and if it is approved, we are entitled to a milestone payment of up to $25 million, royalties on UCB’s sales of JZP-6 and additional commercial milestone payments of up to $100 million. No product has been approved in Europe for the treatment of fibromyalgia. The term of our agreement with UCB, as it applies to JZP-6, extends to the later of the expiration of our associated patent rights in the territories covered by the agreement or ten years from the date of EMA approval to commercially promote and distribute the product for the treatment of fibromyalgia, subject to automatic extension unless UCB provides 12 months’ notice. UCB may terminate our agreement for any reason upon 12 months’ notice and may terminate its rights to JZP-6 for the treatment of fibromyalgia on six months’ notice at any time prior to the receipt of marketing approval of JZP-6 for fibromyalgia in the European Union. We are responsible for supplying JZP-6 to UCB in exchange for supply price payments.
JZP-8 (intranasal clonazepam)
We are developing JZP-8, an intranasal formulation of clonazepam, for the treatment of acute repetitive seizures in epilepsy patients who continue to have seizures while on stable anti-epileptic regimens. Acute repetitive seizures are bouts of multiple seizures occurring over a short period of time. According to an article published in the New England Journal of Medicine, approximately 30% of epilepsy patients are unresponsive, or refractory, to treatment despite being on an effective dose of an antiepilepsy regimen, and a subset of these refractory patients experience acute repetitive seizures. Currently available treatment options are limited for patients who experience acute repetitive seizures.
7
Table of Contents
We have received orphan drug designation from the FDA for this product candidate. We completed a Phase II clinical trial of JZP-8 to evaluate the safety and efficacy of two dosage strengths, and the preliminary findings were encouraging. We subsequently conducted additional formulation activities and a pharmacokinetic study during 2010 which resulted in plasma concentrations that were dose proportional. We are currently planning for an additional Phase II study for later in 2011.
Sales and Marketing
As of February 28, 2011, we had a specialty sales force consisting of approximately 120 full-time sales professionals, which includes our Specialty Sales Consultants, Regional Sales Managers, and Area Business Directors, who currently promote Xyrem and Luvox CR. Our sales force calls primarily on sleep specialists, psychiatrists, neurologists and pulmonologists.
We have established marketing, commercial operations and account management, and trade and distribution departments to support our sales efforts. We also employ third party vendors, such as advertising agencies, market research firms and suppliers of marketing and other sales support related services to assist with our commercial activities.
Competition
The pharmaceutical industry is highly competitive and characterized by a number of established, large pharmaceutical companies as well as specialty pharmaceutical companies that market psychiatry and neurology products. Most of these companies have financial resources and marketing capabilities substantially greater than ours. Our ability to continue to grow over the long-term also requires that we compete successfully with other specialty pharmaceutical companies for product and product candidate acquisition and in-licensing opportunities. Some of these competitors include Cephalon, Inc., Shire Pharmaceuticals, Inc., Endo Pharmaceuticals Holdings, Inc. and Forest Laboratories, Inc. These established companies may have a competitive advantage over us due to their size and financial resources.
Our products and product candidates may also compete in the future with new products currently under development by others. Any products that we develop are likely to be in a highly competitive market, and many of our competitors may succeed in developing products that may render our products obsolete or noncompetitive. In particular, our most significant marketed product and late-stage product candidates face competition as described below:
| • | Xyrem. Xyrem is the only product approved for the treatment of both cataplexy and excessive daytime sleepiness in patients with narcolepsy. No products other than Xyrem are approved for the treatment of cataplexy. The only other products approved by the FDA for the treatment of excessive daytime sleepiness in patients with narcolepsy are Provigil® (modafinil) and Nuvigil® (armodafinil), which are marketed by Cephalon. Provigil and Nuvigil are also approved for the treatment of excessive daytime sleepiness in patients with obstructive sleep apnea/hypopnea syndrome and shift work sleep disorder. Xyrem is often used in conjunction with stimulants and wakefulness promoting drugs, which are administered during the day. During the pivotal Phase III trials of Xyrem for use in patients with narcolepsy, approximately 80% of patients maintained concomitant stimulant use. |
As an alternative to Xyrem, cataplexy is often treated with tricyclic antidepressants and selective serotonin or norepinephrine reuptake inhibitors, although these products are not approved by the FDA for the treatment of cataplexy. Tricyclic antidepressants are a class of antidepressant drugs first used in the 1950s. The use of these drugs can often result in somnolence, which exacerbates the excessive daytime sleepiness already experienced by all patients with narcolepsy.
| • | Luvox CR. The market for drugs to treat obsessive compulsive disorder is very fragmented. We believe that, in addition to Luvox CR, a large number of branded and generic drugs are used for the treatment |
8
Table of Contents
| of this disorder. Seven branded products, including Luvox CR, and generic equivalents of many of these, have been approved by the FDA for the treatment of obsessive compulsive disorder, and we believe that other products are regularly used to treat this disorder. We believe that none of these products has a significant percentage of the market. |
The presence in a particular patient of more than one psychiatric condition is an important consideration by physicians in the selection of drugs to treat obsessive compulsive disorder. Certain drugs are approved for one or more well recognized psychiatric disorders such as major depressive disorder, which may give them broader recognition and use by physicians and patients than Luvox CR, which is indicated only for the treatment of obsessive compulsive disorder and social anxiety disorder.
| • | Product Candidates. With respect to our current and potential future product candidates, we believe that our ability to successfully compete will depend on, among other things: |
| - | the timing and scope of regulatory approvals; |
| - | efficacy, safety and reliability of our product candidates; |
| - | product acceptance by physicians, other health care providers and patients; |
| - | protection of our proprietary rights and the level of generic competition; |
| - | obtaining reimbursement for product use in approved indications; |
| - | our ability to supply commercial quantities of a product to the market; |
| - | our ability to recruit and retain skilled employees; and |
| - | our ability to expand and grow our specialty sales force. |
Customers and Financial Information about Geographic Areas
In the United States, Xyrem is sold to one specialty pharmacy which ships Xyrem directly to patients. Luvox CR is sold primarily to distributors who distribute the product to pharmacies. During 2010, the specialty pharmacy for Xyrem was Express Scripts, and the principal distributors for Luvox CR in the United States were Cardinal Health, McKesson and AmerisourceBergen. Outside the United States, UCB Pharma is our principal distributor for Xyrem. We do not have rights to Luvox CR outside the United States.
Information on total revenues attributed to domestic and foreign sources is included in Note 14 to our consolidated financial statements.
Manufacturing
We do not have, and do not intend to establish in the near term, our own manufacturing capability for our products or product candidates, or their active pharmaceutical ingredients, or the capability to package our products. We have entered into manufacturing and supply agreements with third parties for Xyrem and Luvox CR. For each of our marketed and approved products, we utilize a single supplier for the active pharmaceutical ingredient and a separate drug product manufacturer.
In April 2010, we entered into an agreement with a new supplier for sodium oxybate, Siegfried (USA) Inc., or Siegfried. We intend to seek FDA approval of Siegfried as our supplier as soon as possible. We expect Siegfried to be approved by the FDA as a supplier in the second half of 2011; however we cannot be certain that this will occur. We have the right to purchase a portion of our worldwide requirements of sodium oxybate from other suppliers. The agreement with Siegfried expires in April 2015, subject to automatic three-year extensions until either party provides notice to the other of its intent to terminate the agreement at least 18 months before the end of the then current term. We can also terminate the agreement upon 30 days’ notice on or after December 31, 2011 if Siegfried has not obtained the required approvals to manufacture sodium oxybate or obtained
9
Table of Contents
manufacturing quota for sodium oxybate from the DEA for the calendar year 2011. During the term of the agreement and, under certain circumstances for 18 months after the agreement terminates, Siegfried is not permitted to manufacture sodium oxybate for any other company.
Our 2010 supplies of sodium oxybate were manufactured under an exclusive agreement by Lonza, Inc., or Lonza. Lonza formally notified us in March 2010 that our agreement for the supply of sodium oxybate would terminate on December 31, 2011. Under the agreement, Lonza has an obligation to meet our sodium oxybate supply needs through 2011. We believe that our current inventory levels of sodium oxybate are sufficient to meet our needs for 2011; we expect our new supplier, Siegfried, to obtain quota and manufacture supplies to meet our 2012 needs.
We have an agreement with Patheon Pharmaceuticals, or Patheon, which became effective in 2008, under which we have agreed to purchase, and Patheon has agreed to supply, our worldwide supply of Xyrem. The initial term of the agreement with Patheon extends until December 2012 and may be extended, at our option, for additional two-year terms.
Quotas from the DEA are required in order to manufacture and package sodium oxybate and Xyrem. Siegfried and Patheon each require quota from the DEA to supply us with sodium oxybate and Xyrem. Since the DEA typically grants quota on an annual basis and requires a detailed submission and justification for the request, obtaining a sufficient DEA quota can be a difficult and time consuming process. The need for quota can prevent us from building significant inventories.
Pursuant to our supply agreement with Abbott, we are responsible for purchasing, and Abbott is responsible for providing us with, fluvoxamine maleate, the active pharmaceutical ingredient necessary to manufacture Luvox CR. Abbott (through its predecessor Solvay which it acquired in 2010) assigned to us its rights and obligations under its license and supply agreement with Elan. Pursuant to the license and supply agreement with Elan, we are responsible for providing the active pharmaceutical ingredient free of charge to Elan, and Elan has the right and obligation to manufacture the worldwide commercial requirements of Luvox CR. Abbott has purchased the fluvoxamine maleate it supplied to us from Lonza, and, therefore, Lonza, through Abbott, was our sole supplier of fluvoxamine maleate, the active pharmaceutical ingredient in Luvox CR. Lonza sold its United States facility where it manufactured fluvoxamine maleate to a third party that currently continues to supply Abbott, and therefore us, with fluvoxamine maleate. Any new manufacturer or new site would need to be approved by the FDA.
Manufacturers and suppliers of our products and product candidates are subject to the FDA’s current Good Manufacturing Practices, or cGMP, requirements, DEA regulations and other rules and regulations prescribed by foreign regulatory authorities. We depend on our third party suppliers and manufacturers for continued compliance with cGMP requirements and applicable foreign standards.
Government Regulation
The testing, manufacturing, labeling, advertising, promotion, distribution, export and marketing of our products are subject to extensive regulation by governmental authorities in the United States and in other countries. In the United States, the FDA, under the Federal Food, Drug and Cosmetic Act, or FDCA, and its implementing regulations, regulates pharmaceutical products. Several of our products and product candidates are regulated as controlled substances and are subject to additional regulation by the DEA under the Controlled Substances Act. Failure to comply with applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, withdrawal of approval of approved products, warning letters, untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, suspension of licenses, civil penalties and/or criminal prosecution.
Drug Approval Process
To obtain FDA approval of a product candidate, we must, among other things, submit data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product candidate and
10
Table of Contents
proposed labeling. The testing and collection of data and the preparation of necessary applications are expensive and time-consuming. The FDA may not act quickly or favorably in reviewing these applications, and we may encounter significant difficulties or costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing our products.
The steps required before a drug may be approved for marketing in the United States generally include: preclinical laboratory tests and animal tests; submission to the FDA of an Investigational New Drug Application, or IND, for human clinical testing, which must become effective before human clinical trials commence; adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug product for each indication; the submission to the FDA of an NDA; satisfactory completion of an FDA inspection of the manufacturing facilities at which the product is made, analyzed and stored to assess compliance with cGMP; potential FDA audit of the nonclinical and clinical trial sites that generated the data in support of the NDA; and FDA review and approval of the NDA.
An applicant must submit to the FDA the results of the preclinical and clinical trials, together with, among other things, detailed information on the manufacture and composition of the product candidate and proposed labeling, in the form of an NDA, including payment of a user fee. The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than, or before, accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has ten months in which to complete its initial review of a standard NDA and respond to the applicant, and six months for a priority NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the NDA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
After the FDA evaluates the NDA and the manufacturing facilities, it issues an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA may also refer an application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee.
The FDA has various programs, including fast track, priority review, and accelerated approval (Subpart H), that are intended to expedite or simplify the process for reviewing drugs, and/or provide for approval on the basis surrogate endpoints or restricted distribution. Generally, drugs that may be eligible for one or more of these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that provide meaningful benefit over existing treatments. We cannot be sure that any of our product candidates will qualify for any of these programs, or that, if a product candidate does qualify, that the review time will be shorter than a standard review.
After approval, certain changes to the approved product, such as adding new indications, making certain manufacturing changes, or making certain additional labeling claims, are subject to further FDA review and approval. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. We cannot be sure that any additional approval for new indications for any product will be approved on a timely basis, or at all.
Often, even after a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. If such post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA are required to: report certain adverse reactions to the FDA; comply with certain requirements concerning
11
Table of Contents
advertising and promotional labeling for their products; and continue to have quality control and manufacturing procedures conform to cGMP after approval.
We monitor adverse events resulting from the use of our commercial products, as does the FDA, and we file periodic reports with the FDA concerning adverse events. The FDA reviews these events and reports, and if it determines that any events and/or reports indicate a trend or signal, the FDA can require a change in a product label, restrict sales and marketing and/or require or conduct other actions. In the past year, two potential safety issues or risks were listed for Xyrem by the FDA on its adverse event reporting system based on FDA’s review of reported adverse events; however, the FDA has not indicated that it has determined a causal relationship between Xyrem and the two potential safety issues. The FDA and other governmental authorities also actively enforce regulations prohibiting off-label promotion, and the government has levied large civil and criminal fines against companies for alleged improper promotion. The government has also required companies to enter into complex corporate integrity agreements and/or non-prosecution agreements that can impose significant reporting and other burdens on the affected companies.
The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of compliance with cGMP. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including withdrawal of the product from the market.
The approval process described above is premised on the applicant being the owner of, or having obtained a right of reference to, all of the data required to prove the safety and effectiveness of a drug product. This type of marketing application, sometimes referred to as a “full” or “stand-alone” NDA, is governed by Section 505(b)(1) of the FDCA. A Section 505(b)(1) NDA contains full reports of investigations of safety and effectiveness, which includes the results of preclinical studies and clinical trials, together with detailed information on the manufacture and composition of the product, in addition to other information. As an alternate path to FDA approval of, for example, new indications or improved formulations of previously-approved products, a company may submit a Section 505(b)(2) NDA, instead of a “stand-alone” or “full” NDA filing under Section 505(b)(1). Section 505(b)(2) of the FDCA was enacted as part of the Hatch-Waxman Act. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. For example, the Hatch-Waxman Act permits the applicant to rely upon the FDA’s findings of safety and effectiveness for an approved product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new drug product for all or some of the label indications for which the referenced product has been approved, or for a new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s findings for an already-approved product, the applicant is required to certify that there are no Orange Book-listed patents for that product or that for each Orange Book-listed patent the listed patent has expired, or will expire on a particular date and approval is sought after patent expiration, or the listed patent is invalid or will not be infringed by the manufacture, use or sale of the new product.
A certification that the new product will not infringe the already approved product’s Orange Book-listed patents or that such patents are invalid is called a paragraph IV certification. If the applicant does not challenge the listed patents, the Section 505(b)(2) application will not be approved until all the listed patents claiming the referenced product have expired, as well as any additional period of exclusivity that might be obtained for completing pediatric studies pursuant to the FDA’s written request. The Section 505(b)(2) application may also not be approved until any applicable non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired.
12
Table of Contents
If the applicant has provided a paragraph IV certification to the FDA, the applicant must also send notice of the paragraph IV certification to the holder of the NDA and the relevant patent holders once the 505(b)(2) NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a legal challenge to the paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of their receipt of a paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA until the earliest of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant. For drugs with five-year exclusivity if an action for patent infringement is initiated after year four of that exclusivity period, then the 30-month stay period is extended by such amount of time so that 7.5 years has elapsed since the approval of the NDA with the five-year exclusivity period. This period could be extended by six months if the NDA sponsor obtains pediatric exclusivity. Thus, a Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its products only to be subject to significant delay and patent litigation before its products may be commercialized. Alternatively, if the listed patent holder does not file a patent infringement lawsuit within the required 45-day period, the applicant’s 505(b)(2) NDA will not be subject to the 30-month stay.
The Hatch-Waxman Act
Under the Hatch-Waxman Act, newly-approved drugs and indications may benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act provides five-year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active moiety. The Hatch-Waxman Act prohibits having an effective approval date for an abbreviated new drug application, or ANDA, or a Section 505(b)(2) NDA for another version of such drug during the five-year exclusive period; however, as explained above, submission of an ANDA or Section 505(b)(2) NDA containing a paragraph IV certification is permitted after four years, which may trigger a 30-month stay of approval of the ANDA or Section 505(b)(2) NDA. Protection under the Hatch-Waxman Act will not prevent the submission or approval of another “full” NDA; however, the applicant would be required to conduct its own preclinical and adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Act also provides three years of marketing exclusivity for the approval of new and supplemental NDAs, including Section 505(b)(2) NDAs, for, among other things, new indications, dosages, or strengths of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are determined by the FDA to be essential to the approval of the application.
In addition to non-patent marketing exclusivity, the Hatch-Waxman Act amended the FDCA to require each NDA sponsor to submit with its application information on any patent that claims the active pharmaceutical ingredient, drug product (formulation and composition), and method-of-use for which the applicant submitted the NDA and with respect to which a claim of patent infringement could reasonably be asserted if a person not licensed by the owner engaged in the manufacture, use or sale of the drug, and we have done this. Generic applicants that wish to rely on the approval of a drug listed in the Orange Book must certify to each listed patent, as discussed above. We intend to submit for Orange Book listing all relevant patents for our products and product candidates, and to vigorously defend any Orange Book-listed patents for our approved products. In November 2010, we filed a lawsuit against Roxane in response to Roxane’s Paragraph IV certification relating to Xyrem. For a description of this matter, please see “Item 3. Legal Proceedings.”
The Hatch-Waxman Act also permits a patent term extension of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, a patent term extension cannot extend the remaining term of a patent beyond a total of 14 years after the FDA approves a marketing application. The patent term extension period is generally equal to the sum of one-half the time between the effective date of an IND and the submission date of an NDA, and all of the time between the submission date of an NDA and the approval of that application, up to a total of five years. Only one patent applicable to a regulatory review period, that represents the first commercial marketing of that drug, is eligible for the extension, and it must be applied for prior to expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for patent term extension. We will consider applying for a
13
Table of Contents
patent term extension for some of our patents, to add patent life beyond the expiration date if we meet the legal requirements permitting an extension, and the expected length of clinical trials and other factors involved in the submission of an NDA.
Food and Drug Administration Amendments Act of 2007
On September 27, 2007, the Food and Drug Administration Amendments Act, or the FDAAA, was enacted into law, amending both the FDCA and the Public Health Service Act. The FDAAA makes a number of substantive and incremental changes to the review and approval processes in ways that could make it more difficult or costly to obtain approval for new pharmaceutical products, or to produce, market and distribute existing pharmaceutical products. Most significantly, the law changes the FDA’s handling of postmarketing drug product safety issues by giving the FDA authority to require post approval studies or clinical trials, to request that safety information be provided in labeling, or to require an NDA applicant to submit and execute a REMS. Xyrem is subject to REMS requirements, and we expect that JZP-6, if approved, will be subject to a REMS requirement. Xyrem was approved before 2007 with a risk mitigation program which is a “deemed REMS” in the view of the FDA, and we are working with the FDA to develop an amended REMS for Xyrem under FDAAA. We will work with the FDA if the agency determines that REMS are necessary for our other products or our product candidates.
Orphan Drug Designation and Exclusivity
Some jurisdictions, including the United States and Europe, may designate drugs for relatively small patient populations as orphan drugs. The FDA grants orphan drug designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. In the United States, orphan drug designation must be requested before submitting an application for marketing approval. An orphan drug designation does not shorten the duration of the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity.
The FDA designated and approved Xyrem as an orphan drug for each of excessive daytime sleepiness and cataplexy in patients with narcolepsy. The period of orphan drug exclusivity for cataplexy in patients with narcolepsy expired in July 2009 and the period of orphan drug exclusivity for excessive daytime sleepiness in patients with narcolepsy will expire in November 2012. In December 2007, we received orphan drug designation from the FDA for JZP-8.
Other Regulatory Requirements
In addition to regulation by the FDA and certain state regulatory agencies, the DEA imposes various registration, recordkeeping and reporting requirements, procurement and manufacturing quotas, labeling and packaging requirements, security controls and a restriction on prescription refills on certain pharmaceutical products under the Controlled Substances Act. The states also impose similar requirements for handling controlled substances. A principal factor in determining the particular requirements, if any, applicable to a product is the actual or potential abuse profile. Sodium oxybate, in the form of an active pharmaceutical ingredient, is regulated by the DEA as a Schedule I controlled substance, a category reserved for products believed to present the highest risk of substance abuse and with no approved medicinal use. When contained in Xyrem, sodium oxybate is regulated as a Schedule III controlled substance. JZP-6 (and our solid oral dosage
14
Table of Contents
forms of sodium oxybate) and JZP-8 will likely be regulated as controlled substances if approved for marketing by the FDA. Controlled substances are subject to DEA and state regulations relating to manufacturing, storage, distribution and physician prescription procedures, and the DEA regulates the amount of certain of the scheduled substance that would be available for clinical trials and commercial distribution. Sodium oxybate, as a Schedule I substance, is subject to additional controls, including quotas that limit the amount of product that can be manufactured each year. As a Schedule III drug, Xyrem is subject to limitations on prescription refills. The third parties who perform our clinical and commercial manufacturing, distribution, dispensing and clinical studies for Xyrem, JZP-6 and our solid oral dosage forms of sodium oxybate are required to maintain necessary DEA registrations and state licenses. The DEA periodically inspects facilities for compliance with its rules and regulations. Failure to comply with current and future regulations of the DEA or relevant state authorities could lead to a variety of sanctions, including revocation or denial of renewal of DEA registrations, fines, injunctions, or civil or criminal penalties, and could harm our business and financial condition.
We are also subject to a variety of regulations in countries outside the United States governing clinical trials and the marketing of other products. Outside of the United States, our ability to market a product depends upon receiving a marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. In any country, however, we will only be permitted to commercialize our products if the appropriate regulatory authority is satisfied that we have presented adequate evidence of safety, quality and efficacy. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained prior to the commencement of marketing of the product in those countries. The time needed to secure approval may be longer or shorter than that required for FDA approval. The regulatory approval and oversight process in other countries includes all of the risks associated with regulation by the FDA and certain state regulatory agencies as described above. A World Health Organization (WHO) subcommittee plans to further evaluate the scheduling of sodium oxybate under the international drug control treaties, which could result in a recommendation to the U.N. Commission on Narcotic Drugs to place Xyrem in a more restrictive schedule, thereby causing a more restrictive scheduling of this product in Europe and certain other countries than its current Schedule IV controlled substance status, and in a more restrictive schedule in the United States than its current Schedule III controlled substance status. The WHO review process is long and complicated and the timing and outcome of the review process is uncertain.
Pharmaceutical Pricing and Reimbursement
In both U.S. and foreign markets, our ability to commercialize our products successfully, and to attract commercialization partners for our products, depends in significant part on the availability of adequate financial coverage and reimbursement from third party payors, including, in the United States, governmental payors such as the Medicare and Medicaid programs, managed care organizations, and private health insurers. Third party payors are increasingly challenging the prices charged for medicines and examining their cost effectiveness, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products. Even with studies, our products may be considered less safe, less effective or less cost-effective than existing products, and third party payors may not provide coverage and reimbursement for our product candidates, in whole or in part.
Political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. We anticipate that the United States Congress, state legislatures and the private sector will continue to consider and may adopt healthcare policies intended to curb rising healthcare costs. These cost containment measures include: controls on government funded reimbursement for drugs; new or increased requirements to pay prescription drug rebates to government health care programs, controls on healthcare providers; challenges to the pricing of drugs or limits or prohibitions on reimbursement for specific products through other means; requirements to try less expensive products or generics before a more expensive branded product; changes in drug importation laws; expansion of
15
Table of Contents
use of managed care systems in which healthcare providers contract to provide comprehensive healthcare for a fixed cost per person; and public funding for cost effectiveness research, which may be used by government and private third party payors to make coverage and payment decisions.
We are unable to predict what additional legislation, regulations or policies, if any, relating to the healthcare industry or third party coverage and reimbursement may be enacted in the future or what effect such legislation, regulations or policies would have on our business. Any cost containment measures, including those listed above, or other healthcare system reforms that are adopted, could have a material adverse effect on our ability to operate profitably.
Our products may also face competition from lower priced products from foreign countries that have placed price controls on pharmaceutical products. Proposed federal legislative changes may expand consumers’ ability to import lower priced versions of our and competing products from Canada. Further, several states and local governments have implemented importation schemes for their citizens, and, in the absence of federal action to curtail such activities, we expect other states and local governments to launch importation efforts. The importation of foreign products that compete with our products could negatively impact our business and prospects.
Patents and Proprietary Rights
We actively seek to patent, or to obtain licenses to or to acquire third party patents, to protect our products, inventions and improvements that we consider important to the development of our business. We own fourteen issued U.S. patents and have rights to one other U.S. issued patent. In addition to the issued U.S. patents, we own or have rights to 12 pending U.S. patent applications and more than 100 issued and pending foreign patents and patent applications. Our owned and licensed patents and patent applications cover formulations of our products and product candidates, uses of our products and product candidates to treat particular conditions, drug delivery technologies and delivery profiles relating to our products and product candidates and methods for producing our products and product candidates. However, patent protection is not available for the active pharmaceutical ingredients in most of our products and product candidates, including Xyrem, Luvox CR, JZP-8 and JZP-6. Patents extend for varying periods according to the date of the patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country. The patents and patent applications that relate to our products and product candidates include the following:
| • | Xyrem. Xyrem is covered by two U.S. formulation patents, both of which are listed in the Orange Book and both having an expiration date in 2020. Xyrem is also covered in the U.S. by five method of use patents covering the distribution system for Xyrem, four of which are listed in the Orange Book. Four of those patents will expire in 2024 and the fifth in 2022. In December 2010, an additional patent that will expire in 2019 issued in the United States covering the method of use of Xyrem for the treatment of narcolepsy; it is also listed in the Orange Book. Xyrem is also covered by a U.S. patent covering a process for preparing the formulation, not listed in the Orange Book, that expires in 2019. A Xyrem formulation patent has issued in 18 other countries and will expire in 2019. It is currently pending in two additional countries. In addition to the issued patents, a number of patent applications related to Xyrem are pending in the U.S. |
| • | Luvox CR. Luvox CR is covered by U.S. Patent No. 7,465,462 owned by Elan with claims covering the orally administered formulation of extended-release fluvoxamine that requires the release of fluvoxamine over a period of not less than 12 hours. This patent is listed in the Orange Book, and will expire in 2020. We obtained a license to this patent as a result of Solvay’s assignment of its license and supply agreement with Elan to us in connection with our exclusive license of the rights to market and distribute Luvox CR in the United States. A continuation application is pending in the United States. |
16
Table of Contents
| • | Product candidates. We expect that our distribution patents and both of our current formulation patents associated with Xyrem will be applicable to JZP-6. We also own patents and patent applications with claims covering the use of sodium oxybate for the treatment of fibromyalgia that will expire in the United States in 2017 and in 29 other countries in 2018. We have filed U.S. and foreign patent applications with claims covering JZP-8. These applications would, if issued, expire in 2027. The claims do not cover the JZP-8 composition of matter. |
We cannot be certain that any of our patent applications, or those of our licensors, will result in issued patents. In addition, because the patent positions of pharmaceutical companies are highly uncertain and involve complex legal and factual questions, the patents we own and license, or any further patents we may own or license, may not prevent other companies from developing similar or therapeutically equivalent products. In recent years, several companies have been extremely aggressive in challenging patents covering pharmaceutical products, and the challenges have often been successful. We cannot assure you that our patents will not be challenged by third parties or that we will be successful in any defense we undertake. Failure to successfully defend a patent challenge could materially and adversely affect our business.
On October 18, 2010, we received a Paragraph IV Certification from Roxane that it filed an ANDA with the FDA requesting approval to market a generic version of Xyrem. Roxane’s Paragraph IV Certification alleges that all of our patents listed for Xyrem in Orange Book on the date of the Paragraph IV Certification are invalid, unenforceable or not infringed by Roxane’s proposed generic product. On November 22, 2010, we filed a lawsuit against Roxane in response to Roxane’s Paragraph IV Certification in the United States District Court for the District of New Jersey. On January 14, 2011, we received an additional Paragraph IV Certification from Roxane alleging that our method of use patent for the use of Xyrem in the treatment of narcolepsy that issued in December 2010 would not be infringed by Roxane’s proposed generic product. We amended our lawsuit against Roxane on February 4, 2011 to include the additional patent in the litigation in response to Roxane’s additional Paragraph IV Certification. We cannot assure you that this lawsuit or any other lawsuit we may bring will prevent the introduction of generic products for any particular length of time or at all. For a more detailed description of our dispute with Roxane, please see “Item 3. Legal Proceedings.”
In August 2009, we received a Paragraph IV Certification notice from Actavis advising that Actavis has filed an ANDA, with the FDA seeking approval to market a generic version of Luvox CR. In September 2009, we received a Paragraph IV Certification notice from Anchen advising that Anchen has filed an ANDA with the FDA for a generic version of Luvox CR. Actavis’ Paragraph IV Certification alleged that the United States patent covering Luvox CR, which is owned by Elan and licensed to us, is invalid on the basis that the inventions claimed therein were obvious. Anchen’s Paragraph IV Certification alleged that Elan’s patent will not be infringed by Anchen’s manufacture, use or sale of the generic product for which the ANDA was submitted and that the patent is invalid on the basis that the inventions claimed therein were obvious. We and Elan filed lawsuits in response to the Paragraph IV certifications. In August 2010, we and Elan entered into settlement agreements with Anchen and granted a sublicense to Anchen of our rights to have manufactured, market and sell a generic version of Luvox CR. The sublicense will commence on February 15, 2013 or earlier upon the occurrence of certain events. The lawsuit against Actavis remains pending in the United States District Court for the District of Delaware. For a more detailed description of our dispute with Anchen and Actavis, please see “Item 3. Legal Proceedings.”
We cannot ensure that others will not be issued patents that may prevent the sale of our products or require licensing and the payment of significant fees or royalties. Furthermore, to the extent that any of our future products or methods is not patentable or infringe the patents of third parties, or in the event that our patents or future patents fail to give us an exclusive position in the subject matter claimed by those patents, our business could be adversely affected. We may be unable to avoid infringement of third party patents and may have to obtain a license, defend an infringement action, or challenge the validity of the patents in court. A license may be unavailable on terms and conditions acceptable to us, if at all. Patent litigation is costly and time consuming, and we may be unable to prevail in any such patent litigation or devote sufficient resources to even pursue such
17
Table of Contents
litigation. If we do not obtain a license under necessary patents, are found liable for infringement, or are not able to have such patents declared invalid, we may be liable for significant money damages, encounter significant delays in bringing products to market, or be precluded from participating in the manufacture, use or sale of products or methods of treatment requiring such licenses.
We have also applied for a number of trademarks and service marks to further protect the proprietary position of our products. We own 66 registered trademarks and service marks in the United States and 31 registered trademarks and service marks in other jurisdictions. We also have three pending trademark and service mark applications in the United States and four pending trademark and service mark applications in other jurisdictions. We also rely on our trade secrets and those of our licensors, as well as other unpatented proprietary information, to protect our products. To the extent that our products have a competitive edge as a result of our reliance on trade secrets and unpatented know-how, our competitive position may be compromised if others independently develop products using the same or similar technologies or trade secrets.
We seek to protect our trade secrets and proprietary knowledge in part through confidentiality agreements with our employees, consultants, advisors and collaboration partners. Nevertheless, these agreements may not effectively prevent disclosure of our confidential information and may not provide us with an adequate remedy in the event of unauthorized disclosure of our confidential information. In addition, if our employees, consultants, advisors or collaboration partners develop inventions or processes independently or jointly with us that may be applicable to our products under development, disputes may arise about ownership or proprietary rights to those inventions and processes. Such inventions and processes will not necessarily become our property, but may remain the property of those third parties or their employers. Protracted and costly litigation could be necessary to enforce and determine the scope of our proprietary rights. Failure to obtain or maintain patent and trade secret protection, for any reason, could have a material adverse effect on our business.
Employees
As of February 28, 2011, we had 242 full-time employees. Of the full-time employees, 142 were engaged in sales and marketing, 52 were engaged in manufacturing, product development and clinical activities, and 48 were engaged in general and administrative activities. None of our employees is represented by a labor union, and we consider our employee relations to be good.
Executive Officers of the Registrant
The following table sets forth certain information concerning our executive officers as of February 28, 2011:
| Name |
Age | Position | ||||
| Bruce C. Cozadd |
47 | Chairman and Chief Executive Officer | ||||
| Russell J. Cox |
47 | Senior Vice President, Sales and Marketing | ||||
| Michael A. DesJardin |
53 | Senior Vice President, Product Development | ||||
| Mark G. Eller, Ph.D. |
54 | Senior Vice President, Research and Clinical Development | ||||
| Kathryn E. Falberg |
50 | Senior Vice President and Chief Financial Officer | ||||
| Carol A. Gamble |
58 | Senior Vice President, General Counsel and Corporate Secretary | ||||
| Janne L.T. Wissel |
55 | Senior Vice President and Chief Regulatory and Compliance Officer | ||||
| Joan E. Colligan |
59 | Executive Director and Principal Accounting Officer | ||||
Bruce C. Cozadd is a co-founder and has served as our Chairman and Chief Executive Officer since April 2009. From 2003 until 2009, he served as our Executive Chairman. From 1991 until 2001, he held various positions with ALZA Corporation, a pharmaceutical company now owned by Johnson & Johnson, most recently as its Executive Vice President and Chief Operating Officer, with responsibility for research and development, manufacturing and sales and marketing. Previously at ALZA Corporation he held the roles of Chief Financial Officer and Vice President, Corporate Planning and Analysis. He serves on the boards of Cerus Corporation, a
18
Table of Contents
biopharmaceutical company, Threshold Pharmaceuticals, a biotechnology company, and The Nueva School and Stanford Hospital and Clinics, both non-profit organizations. He received a B.S. from Yale University and an M.B.A. from the Stanford Graduate School of Business.
Russell J. Cox was appointed our Senior Vice President, Sales and Marketing in January 2011. Prior to that he served as our Vice President of Marketing from July 2010. From 2007 to 2009, he was Senior Vice President and Chief Commercial Officer at Ipsen Group and previously Vice President of Marketing at Tercica, Inc. (acquired by Ipsen Group), a biotechnology company. From 2003 to 2007, he was with Scios Inc. (acquired by Johnson and Johnson later in 2003), where he also held the role of Vice President, Marketing. Prior to 2003, Mr. Cox was with Genentech, Inc. for 12 years, where he was a Product Team Leader (PTL) responsible for the Growth Hormone franchise and led numerous product launches as a Group Product Manager. Mr. Cox received a B.S. in Biomedical Science from Texas A&M University.
Michael A. DesJardin has served as our Senior Vice President, Product Development since May 2008. Prior to that he served as Vice President, Product Development since July 2004. From 1995 to 2004, he served in positions of increasing responsibility at ALZA Corporation, most recently as Executive Director for Implant Research and Development. Prior to 1995, he worked for 15 years in chemical development, with several assignments in API manufacturing at The Dow Chemical Company and Marion Merrell Dow (now Sanofi-Aventis). Mr. DesJardin holds a B.S. in Chemical Engineering from the University of California, Berkeley and is a registered Professional Engineer in the State of California.
Mark G. Eller, Ph.D. has served as our Senior Vice President, Research and Clinical Development since May 2008. Prior to that he served as Vice President, Research since May 2005. From 2001 to 2005, he was Vice President, Clinical Pharmacology at Quintiles Inc. From 1988 to 2001, Dr. Eller served in positions of increasing responsibility at Hoechst Marion Roussel Inc. (now Sanofi-Aventis) and its predecessor companies, most recently as Senior Director, Global Biodynamics. Prior to 1988, he held positions with The Upjohn Company and the University of Cincinnati, College of Pharmacy. Dr. Eller holds a Ph.D. in Pharmaceutics from the University of Iowa and received his B.S. in Pharmacy from the University of Iowa, College of Pharmacy.
Kathryn E. Falberg has served as our Senior Vice President and Chief Financial Officer since December 2009. From February 2009 to November 2009, Ms. Falberg was Chief Financial Officer and Chief Operating Officer at ARCA biopharma, Inc., a biopharmaceutical company. From 2001 until February 2009, Ms. Falberg worked as an active investor and consultant to small companies and served as a corporate director and audit committee chair for several companies. From 1995 through 2001, Ms. Falberg was with Amgen, Inc., where she served as Senior Vice President Finance, Strategy and Chief Financial Officer, and before that as Vice President, Controller and Chief Accounting Officer, and Vice President, Treasurer. Ms. Falberg received an M.B.A. and B.A. in Economics from the University of California, Los Angeles and is a Certified Public Accountant. Ms. Falberg currently serves on the boards of Halozyme Therapeutics, a biopharmaceutical company and QLT, Inc., a pharmaceutical company.
Carol A. Gamble was appointed as our Senior Vice President in 2004 and has served as our General Counsel and Corporate Secretary since 2003. From 2002 to 2003, she served as a consultant to various companies in the pharmaceutical industry. From 2000 to 2002, she served as General Counsel and Corporate Secretary of Aerogen, Inc., a biopharmaceutical company later acquired by Nektar Therapeutics. From 1988 to 2000, she held various positions with ALZA Corporation, most recently as its Senior Vice President and Chief Corporate Counsel. Ms. Gamble received a B.S. from Syracuse University and a J.D. from the University of California, Berkeley, Boalt Hall.
Janne L. T. Wissel has served as our Senior Vice President and Chief Regulatory and Compliance Officer since October 2007. Prior to that she served as our Senior Vice President of Development from 2004 to 2007, and previously she served as our Vice President of Development. From 1981 to 2003, she held various positions at ALZA Corporation, most recently as its Senior Vice President, Operations, with responsibility for ALZA
19
Table of Contents
Corporation’s global regulatory, quality, general operations and manufacturing activities. She has led the development, registration and launch of more than 20 pharmaceutical products in the neurology, pediatric psychiatry, endocrinology, urology and oncology areas. Ms. Wissel received a B.S. from the University of California, Davis and an M.B.A. from the University of Phoenix.
Joan E. Colligan has served as our Controller since July 2004, and in March 2009 she was designated by our Board as our principal accounting officer and she served as acting principal financial officer from March to December 2009. From 2000 to 2004, she served as Controller for research and development at ALZA Corporation. Ms. Colligan received a B.S.C. and an M.B.A. from Santa Clara University.
About Jazz Pharmaceuticals
We were incorporated in California in March 2003 and reincorporated in Delaware in January 2004. Our principal offices are located at 3180 Porter Drive, Palo Alto, California, 94304, and our telephone number is 650-496-3777. Our website address is www.jazzpharmaceuticals.com. Information found on, or accessible through, our website is not a part of, and is not incorporated into, this Annual Report on Form 10-K. Service marks, trademarks and trade names appearing in this Annual Report on Form 10-K are the property of their respective owners.
Available Information
We file electronically with the U.S. Securities and Exchange Commission our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We make available on our website at www.jazzpharmaceuticals.com, free of charge, copies of these reports as soon as reasonably practicable after we electronically file such material with, or furnish it to the SEC. Further copies of these reports are located at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that contains reports, proxy and information statements, and other information regarding our filings, at www.sec.gov.
20
Table of Contents
| Item 1A. | Risk Factors |
We have identified the following risks and uncertainties that may have a material adverse effect on our business, financial condition or results of operations. The risks described below are not the only ones we face. Additional risks not presently known to us or that we currently believe are immaterial may also significantly impair our business operations. Our business could be harmed by any of these risks. The trading price of our common stock could decline due to any of these risks, and you may lose all or part of your investment.
Risks Relating to Our Business
We are dependent on sales of Xyrem to generate the cash necessary to operate our business and to meet our ongoing financial obligations, and, if we are not able to maintain or increase sales of Xyrem, it would have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We are dependent on sales of Xyrem to generate the cash necessary to operate our business and to meet our ongoing financial obligations, and our future plans assume that sales of Xyrem will increase. While Xyrem product sales increased in the year ended December 31, 2010 compared to the same period in 2009, and we expect significant Xyrem sales growth for 2011 compared to 2010, we cannot assure you that Xyrem sales will continue to grow. We have periodically significantly increased the price of Xyrem, most recently in November 2010, and we cannot assure you that price increases we have taken or may take in the future have not, or will not in the future, negatively affect Xyrem sales volumes.
In addition to other risks described herein, our ability to maintain or increase Xyrem product sales is subject to a number of risks and uncertainties, the most important of which are discussed below, including those related to:
| • | the potential introduction of a generic version of Xyrem; |
| • | our manufacturing partners’ ability to obtain sufficient quota from the Drug Enforcement Agency, or DEA, to satisfy our needs for Xyrem; |
| • | any supply or distribution problems arising with any of our manufacturing and distribution partners, all of whom are sole source providers for us; |
| • | changed or increased regulatory restrictions, including changes to our risk management program for Xyrem; |
| • | changes in healthcare laws and policy, including changes in requirements for rebates, reimbursement and coverage by federal healthcare programs; |
| • | changes to our label, including our black box warning, that further restrict how we market and sell Xyrem; and |
| • | continued acceptance of Xyrem as safe and effective by physicians and patients. |
These and the other risks described in these risk factors related to Xyrem’s product sales could have a material adverse effect on our ability to maintain or increase sales of Xyrem.
If prescriptions and revenue from sales of Xyrem do not continue or increase as expected, we may be required to reduce our operating expenses, decrease our efforts in support of our products or seek to raise additional funds, all of which could have a material adverse effect on our business, financial condition, results of operations and growth prospects, or we may not be able to acquire, in-license or develop new products to grow our business.
If generic products that compete with Xyrem are approved, sales of Xyrem may be adversely affected.
Although Xyrem is covered by patents covering its formulation, distribution system and method of use, we cannot assure you that third parties will not attempt to invalidate or design around the patents, or assert that they
21
Table of Contents
are invalid or otherwise unenforceable, and introduce generic equivalents of Xyrem. Once orphan drug exclusivity for Xyrem in the United States for the treatment of excessive daytime sleepiness in patients with narcolepsy expires in November 2012, other companies could possibly introduce generic equivalents of Xyrem if they do not infringe our patents covering Xyrem or can demonstrate that our patents are invalid or unenforceable.
On October 18, 2010, we received notice from Roxane Laboratories, Inc, or Roxane, that it filed an abbreviated new drug application, or ANDA, with the U.S. Food and Drug Administration, or FDA, requesting approval to market a generic version of Xyrem. If the application is approved, and a generic version of Xyrem is introduced, our sales of Xyrem would be adversely affected. Additional ANDAs could also be filed requesting approval to market generic forms of Xyrem; if those applications for generics were approved and the generics were launched, sales of Xyrem would further decrease.
Roxane has sent us Paragraph IV certifications with respect to our patents listed before February 2011 in the FDA’s approved drug products with therapeutic equivalence evaluation documents, or Orange Book, covering Xyrem for the treatment of cataplexy and excessive daytime sleepiness in patients with narcolepsy. A Paragraph IV certification is a certification by a generic applicant that patents covering the branded product are invalid, unenforceable, and/or will not be infringed by the manufacture, use or sale of the generic product. The FDA will not approve an ANDA for a generic form of a product unless the submitting manufacturer either files a Paragraph IV certification with respect to the patents listed in the FDA’s Orange Book for that product or all of those patents expire. We have filed a lawsuit against Roxane, but we cannot assure you that the lawsuit will prevent the introduction of a generic version of Xyrem for any particular length of time, or at all.
After the introduction of a generic competitor, a significant percentage of the prescriptions written for a product generally may be filled with the generic version, resulting in a loss in sales of the branded product, including for indications for which the generic version has not been approved for marketing by the FDA. Generic competition often results in decreases in the prices at which branded products can be sold, particularly when there is more than one generic available in the marketplace. In addition, legislation enacted in the United States allows for, and in a few instances in the absence of specific instructions from the prescribing physician mandates, the dispensing of generic products rather than branded products where a generic equivalent is available. Generic competition for Xyrem could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
The manufacture, distribution and sale of Xyrem is subject to significant restrictions, and these restrictions subject us to increased risks and uncertainties, may give advantages to our competitors, and could limit our supply of Xyrem any of which could limit sales of Xyrem.
The DEA limits the quantity of certain Schedule I controlled substances that may be produced in the United States in any given calendar year through a quota system. Because the active pharmaceutical ingredient of Xyrem, sodium oxybate, is a Schedule I controlled substance, our current and new suppliers of sodium oxybate and our product manufacturer must obtain DEA quotas in order to supply us with sodium oxybate and Xyrem. Since the DEA typically grants quotas on an annual basis and requires a detailed submission and justification for each request, obtaining a DEA quota is a difficult and time consuming process. If our commercial or clinical requirements for sodium oxybate or Xyrem exceed our suppliers’ and product manufacturer’s DEA quotas, our suppliers and product manufacturer would need quota increases from the DEA, which could be difficult and time consuming to obtain and might not ultimately be obtained on a timely basis, or at all. We cannot assure you that our suppliers will receive sufficient quota from the DEA to meet our needs, and if we and our suppliers cannot obtain as much quota as is needed, on a timely basis, or at all, our business, financial condition, results of operations and growth prospects could be materially and adversely affected.
As a condition of approval of Xyrem, the FDA mandated that we maintain a risk management program for Xyrem under which all Xyrem that we sell in the United States must be shipped directly to patients through a single central pharmacy. The process under which patients receive Xyrem under the Xyrem risk management
22
Table of Contents
program is cumbersome. While we have an agreement with the central pharmacy for Xyrem, Express Scripts Specialty Distribution Services, or Express Scripts, through June 2012, if the central pharmacy does not fulfill its contractual obligations to us, or refuses or fails to adequately serve patients, shipments of Xyrem and our sales would be adversely affected. If we change our central pharmacy, new contracts might be required with government and other insurers who pay for Xyrem, and the terms of any new contracts could be less favorable to us than current agreements. In addition, any new central pharmacy would need to be registered with the DEA and would also need to implement the particular processes, procedures and activities necessary to distribute Xyrem under the risk management plan approved by the FDA. Transitioning to a new central pharmacy could result in product shortages, which would adversely affect sales of Xyrem in the United States, and/or result in additional costs and expenses for us, and/or take a significant amount of time, any of which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
Xyrem was approved in 2002 with a risk management plan that is not under the current Risk Evaluation and Mitigation Strategy, or REMS, as it is structured today by the FDA. The FDA has required that existing risk management programs be converted to the newer REMS structure under the Food and Drug Administration Amendments Act of 2007. While we have been in discussions with the FDA about converting our current risk management plan for Xyrem to a REMS under the new structure, those discussions have not been completed. We cannot assure you that the FDA will not impose new and onerous requirements under the new REMS structure that could make it more difficult or expensive for us to distribute Xyrem or could adversely affect our sales or make competition easier.
The risk management plan for Xyrem includes some unique features that provide information about adverse events, including deaths, that is generally not available for other products not subject to a similar risk management plan, and may also provide information on adverse events that are not related to the use of Xyrem. As a result, this information, which we report regularly to the FDA, could result in FDA requiring changes to the Xyrem label or other action by FDA or by us which could have an adverse affect on Xyrem’s commercial success.
The FDA has required that Xyrem’s label include a boxed warning regarding the risk of abuse. A boxed warning is the strongest type of warning that the FDA can require for a drug product and warns prescribers that the drug carries a significant risk of serious or even life-threatening adverse effects. A boxed warning also means, among other things, that the product cannot be advertised through reminder ads, ads which mention the pharmaceutical brand name but not the indication or medical condition it treats. In addition, Xyrem’s FDA approval under the FDA’s Subpart H regulations requires that all of the promotional materials for Xyrem be provided to the FDA for review at least 30 days prior to the intended time of first use.
If we are not able to maintain or increase sales of Luvox CR in the near term, it could have an adverse effect on our financial condition and results of operations.
Our plans assume that sales of Luvox CR will increase in 2011. While Luvox CR product sales increased in the year ended December 31, 2010 compared to the same period in 2009, and we expect Luvox CR sales growth in 2011 as compared to 2010, we cannot assure you that Luvox CR sales will continue to grow.
We have been in discussions with the FDA concerning our remaining Phase IV clinical study commitment related to social anxiety disorder, or SAD, and as a result of these discussions, in April 2010 we submitted a labeling supplement to the new drug application, or NDA, for Luvox CR to remove the SAD indication from the label. We have not been promoting Luvox CR for social anxiety disorder since April 2010; however, we cannot assure you that that the removal of the SAD indication from the Luvox CR label, if it occurs, will not have a negative impact on our Luvox CR product sales.
Although Luvox CR is covered by a product-specific patent issued to Elan Pharma International Limited, or Elan, expiring in 2020, other companies could manufacture and sell generic equivalents of Luvox CR in ways
23
Table of Contents
that are not covered by the claims of the patent after the expiration of three years of marketing exclusivity, which ended in February 2011. In August 2009, we received a Paragraph IV certification notice from Actavis Elizabeth, LLC, or Actavis, advising that Actavis has filed an ANDA with the FDA seeking approval to market a generic version of Luvox CR. In September 2009, we received a Paragraph IV certification notice from Anchen Pharmaceuticals, Inc., or Anchen, advising that Anchen has filed an ANDA with the FDA for a generic version of Luvox CR. We filed lawsuits against both companies after receipt of their certifications. We and Elan entered into settlement agreements with Anchen granting Anchen a sublicense of our rights to have manufactured, market and sell a generic version of Luvox CR commencing on February 15, 2013 or earlier upon the occurrence of certain events. The lawsuit against Actavis is pending in the United States District Court for the District of Delaware, but, we cannot assure you that this lawsuit will prevent the introduction of an additional generic form of Luvox CR for any particular length of time, or at all.
We depend on single source suppliers and manufacturers for each of our products and product candidates. The loss of any of these suppliers or manufacturers, or delays or problems in the supply or manufacture of our products for commercial sale or our product candidates for use in our clinical trials, could materially and adversely affect our business, financial condition, results of operations and growth prospects.
We do not have, and do not intend to establish in the near term, our own manufacturing or packaging capability for our products or product candidates, or their active pharmaceutical ingredients. In part due to the limited market size for our approved products, we have entered into manufacturing and supply agreements with single source suppliers and manufacturers for our commercialized products and product candidates. If our suppliers and contract manufacturers do not manufacture our products or product candidates without interruption or do not comply with their obligations to us under our supply and manufacturing arrangements, we may not have adequate remedies for any breach, and their failure to supply us could result in a shortage of our products or product candidates.
The availability of our products for commercial sale depends upon our ability to procure the ingredients, packaging materials and finished products we need. If one of our suppliers or product manufacturers fails or refuses to supply us for any reason, it would take a significant amount of time and expense to qualify a new supplier or manufacturer. The loss of one of our suppliers or product manufacturers could require us to obtain regulatory clearance in the form of a “prior approval supplement” and to incur validation and other costs associated with the transfer of the active pharmaceutical ingredient or product manufacturing process. We believe that it could take as long as two years to qualify a new supplier or manufacturer, and we may not be able to obtain active pharmaceutical ingredients, packaging materials or finished products from new suppliers or manufacturers on acceptable terms and at reasonable prices, or at all. Should we lose either an active pharmaceutical ingredient supplier or a product manufacturer, we could run out of salable product to meet market demands or investigational product for use in clinical trials while we wait for FDA approval of a new active pharmaceutical ingredient supplier or product manufacturer. For Xyrem or sodium oxybate, any new supplier or manufacturer would also need to be registered with the DEA and obtain a DEA quota. In addition, the FDA must approve suppliers of the active and inactive pharmaceutical ingredients and certain packaging materials used in our products, as well as suppliers of finished products. The qualification of new suppliers and manufacturers could potentially delay the manufacture of our products and product candidates and result in shortages in the marketplace or for our clinical trials, or both, particularly since we do not have secondary sources of supply of the active pharmaceutical ingredient or backup manufacturers for our products and product candidates. For example, we entered into an agreement with a new supplier for sodium oxybate, Siegfried (USA) Inc., or Siegfried, and we intend to seek FDA approval of Siegfried as our supplier as soon as possible. We expect Siegfried to be approved by the FDA as a supplier in the second half of 2011, but we cannot be certain this will occur. If there are delays in qualifying the new manufacturer or the new manufacturer is unable to obtain a sufficient quota from the DEA, there could be a shortage of Xyrem and sodium oxybate for the marketplace or for use in our clinical studies, or both.
Failure by our third party manufacturers to comply with regulatory requirements could adversely affect their ability to supply products to us. All facilities and manufacturing techniques used for the manufacture of
24
Table of Contents
pharmaceutical products must be operated in conformity with the FDA’s current Good Manufacturing Practices, or cGMP, requirements. In complying with cGMP requirements, our suppliers must continually expend time, money and effort in production, record-keeping and quality assurance and control to ensure that our products and product candidates meet applicable specifications and other requirements for product safety, efficacy and quality. DEA regulations also govern facilities where controlled substances such as sodium oxybate are manufactured. Manufacturing facilities are subject to periodic unannounced inspection by the FDA, the DEA and other regulatory authorities, including state authorities. Failure to comply with applicable legal requirements subjects the suppliers to possible legal or regulatory action, including shutdown, which may adversely affect their ability to supply us with the ingredients or finished products we need.
Any delay in supplying, or failure to supply, products by any of our suppliers could result in our inability to meet the commercial demand for our products in the United States and our partners’ needs outside the United States, or our needs for use in clinical trials, and could adversely affect our business, financial condition, results of operations and growth prospects.
We may not be able to successfully identify and acquire, in-license or develop additional products or product candidates to grow our business, and, even if we are able to do so, we may not be able to successfully identify and manage the risks associated with integrating acquisitions, including acquisitions of a company or business unit, or other new products or product candidates.
We intend to grow our business over the long-term by acquiring or in-licensing and developing additional products and product candidates that we believe have significant commercial potential. Any growth through acquisition or in-licensing will depend upon the availability of suitable acquisition or in-license products and product candidates on acceptable prices, terms and conditions, and any growth through development will depend upon our identifying and obtaining product candidates, our ability to develop those product candidates and the availability of funding to complete the development of, obtain regulatory approval for and commercialize these product candidates. Even if appropriate opportunities are available, we may not be able to successfully identify them, or we may not have the financial resources necessary to pursue them. Other companies, many of which may have substantially greater financial, marketing and sales resources, compete with us for these opportunities.
In addition, integrating an acquisition, including the acquisition of a company or business unit, or an in-licensed product or product candidate, may create unforeseen operating difficulties and expenses for us, including: the diversion of management time and focus from operating our current business; unanticipated liabilities for activities of or related to an acquired company or product before the acquisition; failure to retain employees or to smoothly integrate related departments; and failure to successfully develop and commercialize acquired products and product candidates. We cannot assure you that we will be able to successfully manage these risks or other anticipated and unanticipated problems in connection with integrating an acquisition, including the acquisition of a company or business unit, or in-licensed product or product candidate, and, if we are not successful in identifying and managing these risks and uncertainties effectively, it could have a material adverse effect on our business.
The commercial success of our products depends upon attaining market acceptance by physicians, patients, third party payors and the medical community.
Even if our product candidates are approved for sale by the appropriate regulatory authorities, physicians may not prescribe our products, in which case we would not generate the revenues we anticipate. Market acceptance of any of our products by physicians, patients, third party payors and the medical community depends on:
| • | the clinical indications for which a product is approved, including any restrictions placed upon the product in connection with its approval, such as a REMS or labeling restrictions; |
| • | prevalence of the disease or condition for which the product is approved and the severity of side effects; |
25
Table of Contents
| • | acceptance by physicians and patients of each product as a safe and effective treatment; |
| • | perceived advantages over alternative treatments; |
| • | relative convenience and ease of administration; |
| • | the cost of treatment in relation to alternative treatments, including generic products; |
| • | the extent to which the product is approved for inclusion on formularies of hospitals and managed care organizations; and |
| • | the availability of adequate reimbursement by third parties. |
To help patients afford our products, we have various programs to assist them, including a patient assistance program, a Xyrem voucher program and coupon programs for both of our products. Coupon programs, including our program for Xyrem, have recently received some negative publicity, and it is possible that new legislation could be enacted to restrict or otherwise negatively affect these programs, which could have a negative effect on our sales.
From time to time, there is negative publicity about illicit gamma-hydroxybutyrate, or GHB, and its effects, including with respect to illegal use, overdoses, serious injury and death. Because sodium oxybate, the active pharmaceutical ingredient in Xyrem, is a derivative of GHB, Xyrem sometimes also receives negative mention in publicity relating to GHB. Patients, physicians and regulators may therefore view Xyrem as the same as or similar to illicit GHB. In addition, there are regulators and some law enforcement agencies that oppose the prescription and use of Xyrem generally because of its connection to GHB. Xyrem’s label includes information about adverse events from GHB. We could also be adversely affected if any of our products or any similar products distributed by other companies prove to be, or are asserted to be, harmful to consumers. Because of our dependence upon patient and physician perceptions, any adverse publicity associated with illness or other adverse effects resulting from the use or misuse of our products or any similar products distributed by other companies could materially and adversely affect our business, financial condition, results of operations and growth prospects.
We face substantial competition from other companies, including companies with greater resources than we have.
With respect to all of our existing and future products, we may compete with companies selling or working to develop products that may be more effective, safer or less costly than our products. The markets for which we are developing products are competitive and include generic and branded products, some of which are marketed by major pharmaceutical companies that have significantly greater financial resources and expertise in research and development, preclinical testing, conducting clinical trials, obtaining regulatory approvals, manufacturing and marketing and selling approved products than we do.
Smaller or earlier stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Our commercial opportunities may be reduced or eliminated if our competitors develop and commercialize generic or branded products that are safer or more effective, have fewer side effects or are less expensive than our products.
Many of our competitors have far greater financial resources and a larger number of personnel to market and sell their products than we do. Our competitors may obtain FDA or other regulatory approvals for their product candidates more rapidly than we may and may market their products more effectively than we do. If we are unable to demonstrate to physicians that, based on experience, clinical data, side-effect profiles and other factors, our products are preferable to other therapies, we may not generate meaningful revenues from the sales of our products.
26
Table of Contents
We currently have a relatively small sales organization compared with most other pharmaceutical companies with marketed products. If our specialty sales force and sales organization is not appropriately sized to adequately promote any potential future products, the commercial opportunity for our potential future products may be diminished.
We have a relatively small number of sales representatives compared with the number of sales representatives of most other pharmaceutical companies with marketed products. Each of our sales representatives is responsible for a territory of significant size. Future commercial products may require expansion of our sales force and sales support organization, and we may need to commit significant additional funds, management and other resources to the growth of our sales organization before the commercial launch of those product candidates. We may not be able to achieve any necessary growth in a timely or cost-effective manner or realize a positive return on our investment, and we may not have the financial resources to achieve the necessary growth in a timely manner or at all. We also have to compete with other pharmaceutical and life sciences companies to recruit, hire, train and retain sales and marketing personnel, and turnover in our sales force and marketing personnel could negatively affect sales of our products.
We depend upon UCB to market and promote Xyrem outside the United States, and we are dependent upon our collaboration with UCB for the development and potential commercialization of JZP-6 for the treatment of fibromyalgia in major markets outside of the United States.
We have exclusively licensed to UCB Pharma Limited, or UCB, the rights to market and promote Xyrem in 54 countries outside of the United States. In addition, under the terms of our collaboration with UCB, we granted UCB the exclusive right to commercialize JZP-6, which UCB would market under the Xyrem trade name, for the treatment of fibromyalgia in the same territories in which UCB has the right to market and promote Xyrem for patients with narcolepsy. UCB has announced that it has filed for European Medicines Agency, or EMA, approval of JZP-6 for fibromyalgia, which UCB intends to market in Europe under the Xyrem trade name if JZP-6 is approved in Europe. However, there are currently no approved fibromyalgia treatments in the European Union, and we cannot assure you that the EMA will approve JZP-6 for fibromyalgia. For example, in October 2008, April 2009 and July 2009 panels of European regulators recommended against approving Cymbalta, Lyrica and Savella, respectively, as treatments for fibromyalgia. UCB has the right to terminate our collaboration on 12-months’ notice (or less in certain circumstances), and UCB may terminate its rights to JZP-6 for the fibromyalgia indication on six-months’ notice at any time prior to the receipt of marketing approval of JZP-6 for fibromyalgia in the European Union. If UCB terminates our collaboration or terminates its rights to JZP-6 for the fibromyalgia indication, we would need to find another party or parties to commercialize Xyrem and/or JZP-6 in UCB’s territories. We may be unable to do this on acceptable terms, or at all.
A failure to prove that our product candidates are safe and effective in clinical trials would require us to discontinue their development, which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
Significant additional research and development, financial resources and additional personnel will be required to obtain necessary regulatory approvals for our current and any future product candidates and to develop them into commercially viable products. As a condition to regulatory approval, each product candidate must undergo extensive and expensive clinical trials to demonstrate to a statistically significant degree that the product candidate is safe and effective. If a product candidate fails at any stage of development, we will not be able to commercialize it and we will not receive any return on our investment from that product candidate.
All of our product candidates, other than JZP-6, are in Phase II, or earlier, clinical trials. Clinical testing can take many years to complete, especially for product candidates that are in Phase II, or earlier, clinical trials, and failure can occur any time during the clinical trial process. In addition, the results from early clinical trials may not be predictive of results obtained in later and larger clinical trials, and product candidates in later clinical trials may fail to show the desired safety and efficacy despite having progressed successfully through initial clinical
27
Table of Contents
testing. A number of companies in the pharmaceutical industry, including us, have suffered significant setbacks in clinical trials, even in advanced clinical trials after showing positive results in earlier clinical trials. Our product candidates are subject to competition for clinical study sites and patients from other therapies under development that may delay the enrollment in or initiation of our clinical trials. Many of these companies have far greater financial and human resources than we do.
To grow our sodium oxybate business, we have and may in the future conduct additional studies in different diseases or conditions or with additional or different doses or dosage forms. We cannot assure you that adverse events or other information obtained during the course of any of these studies will not result in action by the FDA or otherwise that could have a material adverse effect on the Xyrem commercial product as well as the candidate we are studying.
The FDA or foreign regulatory authorities may require us to conduct unanticipated additional clinical trials, which could result in additional expense and delays in bringing our product candidates to market. For example, we received a complete response letter, or CRL, from the FDA concerning our JZP-6 product candidate, which required additional clinical studies in order for JZP-6 to be approved for the treatment of fibromyalgia. We do not know whether we will undertake additional studies of JZP-6 or otherwise continue to seek approval of JZP-6, and if we did so, if any such studies or other of our efforts would be successful and result in approval of JZP-6. Any failure or delay in completing clinical trials for our product candidates would prevent or delay their commercialization, which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
We rely on third parties to conduct clinical trials for our product candidates, and if they do not properly and successfully perform their legal and regulatory obligations, as well as their contractual obligations to us, we may not be able to obtain regulatory approvals for our product candidates.
We design the clinical trials for our product candidates, but rely on contract research organizations and other third parties to assist us in managing, monitoring and otherwise carrying out these trials, including with respect to site selection, contract negotiation and data management. We do not control these third parties and, as a result, they may not treat our clinical studies as their highest priority, or in the manner in which we would prefer, which could result in delays. We are responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol, as well as FDA’s and foreign regulatory agencies’ requirements, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to ensure that the data and results are credible and accurate and that the trial participants are adequately protected. The FDA enforces good clinical practices through periodic inspections of trial sponsors, principal investigators and trial sites. If we, our contract research organizations or our study sites fail to comply with applicable good clinical practices, the clinical data generated in our clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with good clinical practices. In addition, our clinical trials must be conducted with product produced under the FDA’s cGMP regulations. Our failure, or the failure of our contract manufacturers, to comply with these regulations may require us to repeat or redesign clinical trials, which would delay the regulatory approval process.
If third parties do not successfully carry out their duties under their agreements with us, if the quality or accuracy of the data they obtain is compromised due to failure to adhere to our clinical protocols or regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, our clinical trials may not meet regulatory requirements. If our clinical trials do not meet regulatory requirements or if these third parties need to be replaced, our clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates.
28
Table of Contents
We are a small company and our employees must work on many important and diverse matters at the same time. If we fail to attract, retain and motivate key personnel, or to retain our executive management team, or if we cannot provide additional resources to perform important tasks, we may be unable to successfully sustain or grow our business.
Our success and our ability to grow depend in part on our continued ability to attract, retain and motivate highly qualified personnel and on our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. As a small company, we are highly dependent upon our executive management team and other key personnel, all of whom work on many complex matters that are critical to our success. The loss of services of any one or more members of our executive management team or other key personnel could delay or prevent the successful completion of some of our key activities. We do not carry “key person” insurance. Any employee may terminate his or her employment at any time without notice and without cause or good reason.
To grow our company we will need additional personnel. Competition for qualified personnel in the life sciences industry has historically been intense. If we cannot timely attract and retain quality personnel on acceptable terms, our failure to do so could adversely affect our business, financial condition, results of operations and growth prospects.
Risks Related to Our Intellectual Property
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our products and product candidates, their use and the methods used to manufacture and, in some cases, distribute them, as well as successfully defending these patents against third party challenges. Our ability to protect our products and product candidates from unauthorized making, using, selling, offering to sell or importation by third parties depends on the extent to which we have rights under valid and enforceable patents, or have trade secrets that cover these activities.
The patent position of pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property. Even if we are able to obtain patents covering our products and product candidates, any patent may be challenged, invalidated, held unenforceable or circumvented. For example, even though we have nine patents covering Xyrem, with expiration dates between 2019 and 2024, and seven of the patents are listed in the FDA’s Orange Book, an ANDA was filed requesting permission from the FDA to market a generic form of Xyrem. We have received notices from the company that filed the ANDA stating that the ANDA included Paragraph IV certifications with respect to our patents listed in the FDA’s Orange Book before February 2011. In the case of Luvox CR, Actavis’ Paragraph IV certification alleges that the Elan patent, which is listed in the Orange Book for Luvox CR, is invalid. The expiration date for the Elan patent at issue is May 10, 2020.
The existence of a patent will not necessarily prevent other companies from developing similar or therapeutically equivalent products or protect us from claims of third parties that our products infringe their issued patents, which may require licensing and the payment of significant fees or royalties. Competitors may successfully challenge our patents, produce similar products that do not infringe our patents, or manufacture products in countries where we have not applied for patent protection or that do not respect our patents. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents, our licensed patents or in third party patents.
29
Table of Contents
The degree of future protection to be afforded by our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| • | others may be able to make products that are similar to our product candidates but that are not covered by the claims of our patents, or for which we are not licensed under our license agreements; |
| • | we or our licensors or partners might not have been the first to make the inventions covered by our issued patents or pending patent applications or the pending patent applications or issued patents of our licensors or partners; |
| • | we or our licensors or partners might not have been the first to file patent applications for these inventions; |
| • | others may independently develop similar or alternative products without infringing our intellectual property rights; |
| • | our pending patent applications may not result in issued patents; |
| • | our issued patents and the issued patents of our licensors or partners may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges by third parties; |
| • | we may not develop additional proprietary products that are patentable; or |
| • | the patents of others may have an adverse effect on our business. |
We also may rely on trade secrets and other unpatented proprietary information to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets and other unpatented proprietary information, our employees, consultants, advisors and partners may unintentionally or willfully disclose our proprietary information to competitors, and we may not have adequate remedies for such disclosures. If our employees, consultants, advisors and partners develop inventions or processes independently, or jointly with us, that may be applicable to our products under development, disputes may arise about ownership or proprietary rights to those inventions and processes. Enforcing a claim that a third party illegally obtained and is using any of our inventions or trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside of the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
Our research and development collaborators may have rights to publish data and other information to which we have rights. In addition, we sometimes engage individuals or entities to conduct research that may be relevant to our business. While the ability of these individuals or entities to publish or otherwise publicly disclose data and other information generated during the course of their research is subject to contractual limitations, these contractual provisions may be insufficient or inadequate to protect our trade secrets and may impair our patent rights. If we do not apply for patent protection prior to such publication, or if we cannot otherwise maintain the confidentiality of our innovations and other confidential information, then our ability to obtain patent protection or protect our proprietary information may be jeopardized. Moreover, a dispute may arise with our research and development collaborators over the ownership of rights to jointly developed intellectual property. Such disputes, if not successfully resolved, could lead to a loss of rights and possibly prevent us from pursuing certain new products or product candidates.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or commercialize, our products.
Our ability, and that of our partners, to commercialize any approved products will depend, in part, on our ability to obtain patents, enforce those patents and operate without infringing the proprietary rights of third
30
Table of Contents
parties. The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. We have filed multiple U.S. patent applications and foreign counterparts, and may file additional U.S. and foreign patent applications related thereto. There can be no assurance that any issued patents we own or control will provide sufficient protection to conduct our business as presently conducted or as proposed to be conducted. Moreover, in part because of prior research performed and patent applications submitted in the same manner or similar fields, there can be no assurance that any patents will issue from the patent applications owned by us, or that we will remain free from infringement claims by third parties.
If we choose to go to court to stop someone else from pursuing the inventions claimed in our patents, our licensed patents or our partners’ patents, that individual or company has the right to ask the court to rule that these patents are invalid and/or should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources, even if we were successful in stopping the infringement of these patents. In addition, there is a risk that the court will decide that these patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of these patents is upheld, the court will refuse to stop the other party on the ground that the other party’s activities do not infringe our rights to these patents or that it is in the public interest to permit the infringing activity. We have filed and are prosecuting a lawsuit against Roxane related to the Paragraph IV certifications delivered to us with respect to Xyrem. We and Elan are prosecuting a lawsuit against Actavis related to the Paragraph IV certification delivered to us with respect to Luvox CR. We cannot assure you that these, or other lawsuits we may file in the future, will be successful in stopping the infringement of our patents, that any such litigation will be cost-effective, or that the litigation will have a satisfactory result for us.
A third party may claim that we or our manufacturing or commercialization partners are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our products. Patent infringement lawsuits are costly and could affect our results of operations and divert the attention of management and development personnel. There is a risk that a court could decide that we or our partners are infringing third party patent rights which could be very costly to us and have a material adverse effect on our business.
The pharmaceutical and life sciences industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our products or methods do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid or unenforceable, and we may not be able to do this.
Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing, and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for inventions covered by our licensors’ or our issued patents or pending applications, or that we or our licensors were the first inventors. Our competitors may have filed, and may in the future file, patent applications covering subject matter similar to ours. Any such patent application may have priority over our or our licensors’ patents or applications and could further require us to obtain rights to issued patents covering such subject matter. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the U.S. Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our U.S. patent position with respect to such inventions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
31
Table of Contents
Risks Related to Our Industry
The regulatory approval process is expensive, time consuming and uncertain and may prevent us or our partners from obtaining approvals for the commercialization of some or all of our product candidates.
The research, testing, manufacturing, selling and marketing of pharmaceutical products are subject to extensive regulation by FDA and other regulatory authorities in the United States and other countries, and regulations differ from country to country. Approval in the United States, or in any jurisdiction, does not ensure approval in other jurisdictions. The regulatory approval process is lengthy, expensive and uncertain, and we may be unable to obtain approval for our product candidates. We are not permitted to market our product candidates in the United States until we receive approval from the FDA, generally of an NDA. Obtaining approval of an NDA can be a lengthy, expensive and uncertain process, and the FDA has substantial discretion in the approval process. For example, we have spent significant time and money developing our JZP-6 product candidate, and although we believe our clinical studies have shown the product candidate to be safe and effective, we received a CRL from the FDA in October 2010 related to JZP-6 that stated that the FDA cannot approve the NDA in its present form.
In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject our company to administrative or judicially imposed sanctions, including warning letters, untitled letters, civil and criminal penalties, injunctions, product seizure or detention, product recalls, total or partial suspension of production and refusal to approve pending NDAs or supplements to approved NDAs. If we are unable to obtain regulatory approval of our product candidates, we will not be able to commercialize them and recoup our research and development costs.
Healthcare law and policy changes, including those based on recently enacted legislation, may impact our business in ways that we cannot currently predict and these changes could have a material adverse effect on our business and financial condition.
In March 2010, the President signed the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the Healthcare Reform Act. This law substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. The Healthcare Reform Act contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, fraud and abuse and enforcement. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program.
Additional provisions of the Healthcare Reform Act, some of which become effective in 2011, may negatively affect our revenues in the future. For example, as part of the Healthcare Reform Act’s provisions closing a funding gap that currently exists in the Medicare Part D prescription drug program (commonly known as the “donut hole”), we will be required to provide a 50% discount on branded prescription drugs dispensed to beneficiaries within this donut hole. We expect that the Healthcare Reform Act and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales or successfully commercialize our product candidates, including JZP-6, or could limit or eliminate our future spending on development projects.
In addition to the Healthcare Reform Act, there will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to keep healthcare costs down while expanding individual healthcare benefits. Certain of these changes could impose limitations on the prices we will be able to charge for our products and any approved product candidates or the amounts of reimbursement available for these products from governmental agencies or third-party payors, or may increase the tax obligations on
32
Table of Contents
pharmaceutical companies such as ours. The enactment and implementation of any future healthcare reform legislation or policies could have a material adverse effect on our business and financial condition.
We are subject to significant ongoing regulatory obligations and oversight, which may result in significant additional expense and limit our ability to commercialize our products.
We are subject to significant ongoing regulatory obligations, such as safety reporting requirements and additional post-marketing obligations, including regulatory oversight of the promotion and marketing of our products. In addition, the labeling, packaging, adverse event reporting, storage, advertising, promotion and recordkeeping for our products are, and any of our product candidates that may be approved by the FDA will be, subject to extensive and ongoing regulatory requirements. If we receive regulatory approvals to sell our products, the FDA and foreign regulatory authorities may impose significant restrictions on the indicated uses or marketing of our products, or impose requirements for burdensome post-approval study commitments. The terms of any product approval, including labeling, may be more restrictive than we desire and could affect the commercial potential of the product. If we become aware of previously unknown problems with any of our products in the United States or overseas or at our contract manufacturers’ facilities, a regulatory agency may impose restrictions on our products, our contract manufacturers or on us. In such an instance, we could experience a significant drop in the sales of the affected products, our product revenues and reputation in the marketplace may suffer, and we could become the target of lawsuits.
The FDA and other governmental authorities also actively enforce regulations prohibiting off-label promotion, and the government has levied large civil and criminal fines against companies for alleged improper promotion. The government has also required companies to enter into complex corporate integrity agreements and/or non-prosecution agreements that impose significant reporting and other burdens on the affected companies. For example, our predecessor company was investigated for off-label promotion of Xyrem, and we are subject to a corporate integrity agreement through mid-2012 as a result of that investigation. The investigation resulted in significant fines and penalties, which we guaranteed and have been paying; the final payment is due in 2012.
We are also subject to regulation by regional, national, state and local agencies, including the DEA, the Department of Justice, the Federal Trade Commission, the Office of Inspector General of the U.S. Department of Health and Human Services and other regulatory bodies, as well as governmental authorities in those foreign countries in which we commercialize our products. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal and state statutes and regulations govern to varying degrees the research, development, manufacturing and commercial activities relating to prescription pharmaceutical products, including preclinical testing, approval, production, labeling, sale, distribution, import, export, post-market surveillance, advertising, dissemination of information, promotion, marketing, and pricing to government purchasers and government health care programs. Our manufacturing partners are subject to many of the same requirements, which include obtaining sufficient quota from the DEA each year to manufacture sodium oxybate, Xyrem and JZP-6.
The federal health care program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical companies on one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common manufacturer business arrangements and activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations of our products may be subject to scrutiny if they do not qualify for an exemption or safe harbor. We seek to comply with the exemptions and safe harbors whenever possible, but our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability.
33
Table of Contents
The Federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. Many pharmaceutical and other health care companies have been investigated and have reached substantial financial settlements with the federal government under these laws for a variety of alleged marketing activities, including providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees, grants, free travel, and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which are used to set drug payment rates under government health care programs. Companies have been prosecuted for causing false claims to be submitted because of the marketing of their products for unapproved, and thus non-reimbursable, uses. Pharmaceutical and other health care companies have also been prosecuted on other legal theories of Medicare and Medicaid fraud.
The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Several states now require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to individual physicians in the states. Other states prohibit providing meals to prescribers or other marketing related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, California, Nevada, and Massachusetts require pharmaceutical companies to implement compliance programs or marketing codes. Currently, several additional states are considering similar proposals.
Compliance with various federal and state laws is difficult and time consuming, and companies that violate them may face substantial penalties. The potential sanctions include civil monetary penalties, exclusion of a company’s products from reimbursement under government programs, criminal fines and imprisonment. Because of the breadth of these laws and the lack of extensive legal guidance in the form of regulations or court decisions, it is possible that some of our business activities could be subject to challenge under one or more of these laws. Such a challenge could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
The number and complexity of both federal and state laws continues to increase, and additional governmental resources are being added to enforce these laws and to prosecute companies and individuals who are believed to be violating them. In particular, the Healthcare Reform Act includes a number of provisions aimed at strengthening the government’s ability to pursue anti-kickback and false claims cases against pharmaceutical manufacturers and other healthcare entities, including substantially increased funding for healthcare fraud enforcement activities, enhanced investigative powers, amendments to the False Claims Act that make it easier for the government and whistleblowers to pursue cases for alleged kickback and false claim violations and, beginning in March 2013 for payments made in 2012, public reporting of payments by pharmaceutical manufacturers to physicians and teaching hospitals nationwide. While it is too early to predict what effect these changes will have on our business, we anticipate that government scrutiny of pharmaceutical sales and marketing practices will continue for the foreseeable future and subject us to the risk of government investigations and enforcement actions. Responding to a government investigation or enforcement action would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
If we or any of our partners fail to comply with applicable regulatory requirements, we or they could be subject to a range of regulatory actions that could affect our or our partners’ ability to commercialize our products and could harm or prevent sales of the affected products, or could substantially increase the costs and expenses of commercializing and marketing our products. Any threatened or actual government enforcement action could also generate adverse publicity and require that we devote substantial resources that could otherwise be used in other aspects of our business.
34
Table of Contents
If we fail to comply with our reporting and payment obligations under the Medicaid rebate program or other governmental pricing programs, we could be subject to additional reimbursement requirements, penalties, sanctions and fines which could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We participate in the federal Medicaid rebate program established by the Omnibus Budget Reconciliation Act of 1990, as well as several state supplemental rebate programs. Under the Medicaid rebate program, we pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program under a fee-for-service arrangement, as a condition of having federal funds being made available to the states for our drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to the Centers for Medicare and Medicare Services, or CMS, the federal agency which administers the Medicaid drug rebate program. These data include the average manufacturer price, or AMP, and in the case of innovator products, the best price for each drug. As a result of the enactment of the Healthcare Reform Act, rebates now also are due on the utilization of Medicaid managed care organizations, effective March 23, 2010.
Pursuant to the Healthcare Reform Act, and effective for rebate periods beginning in the first quarter 2010, the minimum amount of the Medicaid rebate for each unit of a drug has been increased. For innovator products, in general a drug marketed under an NDA, the minimum rebate has been increased from 15.1% to 23.1% of the AMP for that product, or if it is greater, the difference between the AMP and the best price for the product. The 23.1% rebate amount is lowered to 17.1% for certain clotting factor and pediatric drug products. For non-innovator products, in general a drug marketed under an ANDA, the rebate amount has been increased from 11% to 13.1% of the AMP for drug. The Medicaid rebate for innovator products also includes an additional rebate amount if price increases for the drug exceed the rate of inflation since the product’s launch. The Healthcare Reform Act changes this additional rebate formula for certain products that qualify as line extensions of existing drugs, effective for rebate periods beginning with drugs paid for by a state as of the first quarter 2010, so that the rebate for these products can be increased and based on the additional rebate for the original drug. It also caps the total rebate amount for innovator drugs at 100% of the AMP for the drug. In addition, the Healthcare Reform Act changes the definition of AMP, effective for AMP prices reported for the fourth quarter of 2010, and additional legislation is currently pending that would further amend the AMP definition. CMS has yet to issue regulations to implement any of the enacted statutory changes.
We cannot assure that there will not be additional increases in rebates or other costs and charges from government agencies. Regulations continue to be issued and coverage expanded by various governmental agencies relating to these programs, increasing the cost and complexity of compliance.
Pricing and rebate calculations vary among products and programs. The calculations are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current AMP and best prices for the quarter. If we become aware that our reporting for prior quarters was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected AMP or best price for that quarter. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction as well as changes in the 340B ceiling prices based on those rebate calculations, as discussed below, such that refunds to covered entities that purchased at the earlier prices may be due. In addition to retroactive rebates and the potential for 340B ceiling price refunds, if we are found to have knowingly submitted false average manufacturer price or best price information to the government, we may be liable for civil monetary penalties in the amount of $100,000 per item of false information, and, in September 2010, CMS and the Office of the Inspector General indicated that they intend to more aggressively pursue companies who fail to report this data to the government in a timely manner. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. CMS recently published information stating that many companies’ monthly and quarterly submissions are incomplete or incorrect. We cannot assure you that our submissions will not be found by CMS to be incomplete or incorrect.
35
Table of Contents
Federal law requires that any company that participates in the Medicaid rebate program also participate in the Public Health Service’s 340B pharmaceutical pricing program in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B pricing program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B ceiling price for the manufacturer’s covered outpatient drugs. These covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of poor patients and children. The 340B ceiling price is calculated using a statutory formula which is based on the AMP and rebate amount for the covered outpatient drug as calculated under the Medicaid drug rebate program. This means that to the extent the Healthcare Reform Act, as discussed above, changes the statutory and regulatory definitions of AMP and the Medicaid rebate amount, these changes also will affect the 340B ceiling price. The Healthcare Reform Act expands the 340B drug pricing program to include new covered entity types, effective for drugs purchased on or after January 1, 2010, although drugs that have received an orphan drug designation under section 526 of the Federal Food Drug and Cosmetic Act are exempt from the ceiling price requirement for the new categories of covered entities. The Healthcare Reform Act also obligates the Secretary of the Department of Health and Human Services to create regulations and processes to improve the integrity of the program and to update the agreement that manufacturers must sign to participate in the program to obligate manufacturers to sell to covered entities if they sell to any other purchaser and to report to the government the ceiling prices for its drugs. In addition, Congress is currently considering legislation that, if passed, would further expand the 340B program to require participating manufacturers to agree to provide 340B discounted pricing on drugs used in the inpatient setting by certain covered entity hospitals, where those drugs are used for the covered entity’s uninsured inpatients.
Reimbursement may not be available for our products, which could diminish our sales or affect our ability to sell our products profitably.
In both U.S. and foreign markets, our ability to commercialize our products successfully and to attract strategic partners for our products depends in significant part on the availability of adequate financial coverage and reimbursement from third party payors, including, in the United States, governmental payors such as the Medicare and Medicaid programs, managed care organizations and private health insurers. Third party payors decide which drugs they will pay for and establish reimbursement and co-pay levels. Third party payors are increasingly challenging the prices charged for medical products and services and examining their cost effectiveness, in addition to their safety and efficacy. In some cases, for example, third party payors try to encourage the use of less expensive generic products through their prescription benefits coverage and reimbursement and co-pay policies. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Even with studies, our products may be considered less safe, less effective or less cost-effective than existing products, and third party payors may not provide coverage and reimbursement for our products, in whole or in part. We cannot predict actions third party payors may take, or whether they will limit the coverage and level of reimbursement for our products or refuse to provide any coverage at all. For example, because Luvox CR is competing in a market with both branded and generic products, reimbursement by government and private payors may be more challenging than for new chemical entities. We cannot be sure that reimbursement amounts, or the lack of reimbursement, will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only to limited levels, we may not be able to effectively commercialize our products.
In recent years, there have been a number of legislative and regulatory changes in and proposals to change the healthcare system in ways that could impact our ability to sell our products profitably. These changes and proposals include measures that would limit or prohibit payments for some medical treatments or subject the pricing of drugs to government control and regulations changing the rebates we are required to provide. For example, a final rule published by the Department of Defense, or DoD, in March 2009, implementing the terms of the National Defense Authorization Act of 2008, established a program under which DoD expects rebates from pharmaceutical manufacturers on all prescriptions of “covered” prescription drugs (including innovator drugs and biologics) filled under the TRICARE retail pharmacy program from January 28, 2008 forward, unless
36
Table of Contents
DoD agrees to a waiver or compromise of amounts due. Additionally, under the final rule, to remain eligible for inclusion on the DoD Uniform Formulary, a pharmaceutical manufacturer must enter into a pricing agreement under which it agrees to pay rebates to DoD on TRICARE retail pharmacy utilization on a prospective basis. These rebates are meant to enable DoD to access pricing that is either close to or equal to “Federal Ceiling Prices,” defined under the Veterans Health Care Act of 1992. Per the process set forth in this rule, we entered into a retail rebate agreement with DoD in July 2009. These legislative and regulatory changes, including our entering into the retail rebate agreement with DoD, could impact our ability to maximize revenues in the Federal marketplace. As discussed above, recent legislative changes to the 340B drug pricing program, the Medicaid drug rebate program, and the Medicare Part D prescription drug benefit also could impact our revenues.
We expect to experience pricing pressures in connection with the sale of our products due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative proposals. If we fail to successfully secure and maintain reimbursement coverage for our products or are significantly delayed in doing so, we will have difficulty achieving market acceptance of our products and our business will be harmed.
Product liability and product recalls could harm our business.
The development, manufacture, testing, marketing and sale of pharmaceutical products entail significant risk of product liability claims or recalls. Side effects of, or manufacturing defects in, the products sold by us could result in exacerbation of a patient’s condition, serious injury or impairments or even death. This could result in product liability claims and/or recalls of one or more of our products. Both Xyrem and Luvox CR have boxed warnings in their labels. We expect that the label for JZP-6, if it is approved by the FDA, will also have a boxed warning, and will include adverse events seen in narcolepsy and fibromyalgia trials, as well as post-marketing safety information.
Product liability claims may be brought by individuals seeking relief for themselves, or by groups seeking to represent a class. While we have not had to defend against any product liability claims to date, as sales of our products increase, we believe it is likely product liability claims will be made against us. We cannot predict the frequency, outcome or cost to defend any such claims.
Product liability insurance coverage is expensive, can be difficult to obtain and may not be available in the future on acceptable terms, if at all. Partly as a result of product liability lawsuits related to pharmaceutical products, product liability and other types of insurance have become more difficult and costly for pharmaceutical companies to obtain. Our product liability insurance may not cover all of the future liabilities we might incur in connection with the development, manufacture or sale of our products. In addition, we may not continue to be able to obtain insurance on satisfactory terms or in adequate amounts.
A successful claim or claims brought against us in excess of available insurance coverage could subject us to significant liabilities and could have a material adverse effect on our business, financial condition, results of operations and growth prospects. Such claims could also harm our reputation and the reputation of our products, adversely affecting our ability to market our products successfully. In addition, defending a product liability lawsuit is expensive and can divert the attention of key employees from operating our business.
Product recalls may be issued at our discretion or at the discretion of our suppliers, government agencies and other entities that have regulatory authority for pharmaceutical sales. Any recall of our products could materially adversely affect our business by rendering us unable to sell that product for some time and by adversely affecting our reputation. A recall could also result in product liability claims.
37
Table of Contents
Risks Relating to Our Financial Condition
To grow our business, we will need to commit substantial resources, which could result in future losses or otherwise limit our opportunities or affect our ability to operate our business.
To grow our business over the longer-term, we will need to commit substantial resources to in-licensing and/or acquiring new products and product candidates, and to costly and time-consuming product development and clinical trials of our product candidates. We will also need to continue to invest in our commercial operations. Our future capital requirements will depend on many factors, including many of those discussed above, such as:
| • | the revenues from our commercial products and the costs of our commercial operations; |
| • | whether or not there is generic competition for our products; |
| • | the acquisition and/or licensing cost for any new products and product candidates; |
| • | the scope, rate of progress, results and costs of our development and clinical activities; |
| • | the cost and timing of obtaining regulatory approvals and of compliance with laws and regulations; |
| • | the cost of preparing, filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; |
| • | the cost of investigations, litigation and/or settlements related to regulatory activities; and |
| • | changes in laws and regulations, including, for example, health care reform legislation. |
One of our corporate goals is to expand our business through the licensing, acquisition and/or development of additional products and product candidates. We cannot assure you that our funds will be sufficient to fund these activities if opportunities arise, and we may be unable to expand our business if we do not have sufficient capital or cannot borrow or raise additional capital on attractive terms. In addition, if we use a substantial amount of borrowings or our funds to acquire or in-license products or product candidates, we may not have sufficient additional funds to conduct all of our operations in the manner we would otherwise choose.
The terms of our credit agreement could restrict our operations, particularly our ability to respond to changes in our business or to take specified actions.
The terms of our credit agreement include, and any future indebtedness may include, a number of restrictive covenants that impose significant operating and financial restrictions on us, including restrictions on our ability to take actions that may be in our best interests. The terms of our credit agreement include operating covenants restricting, among other things, our ability to: incur additional indebtedness and liens; effect mergers, consolidations and other fundamental changes; dispose of significant assets or enter into sale-leaseback transactions; pay dividends or make other restricted payments; make loans, advances or other investments including acquisitions of companies and products; and enter into transactions with affiliates. In addition, the terms of our credit agreement include financial covenants requiring us, among other things, to: maintain a certain consolidated fixed charge coverage ratio; maintain a certain leverage ratio; and maintain minimum liquidity. Our failure to comply with any of these covenants could result in a default under the terms of the credit agreement, which could permit the lenders to declare all or part of the outstanding borrowings to be immediately due and payable. Although we currently have sufficient funds to repay our debt, if our outstanding borrowings were to be accelerated, or if we have used significant amounts of our cash for other purposes, we might not have sufficient funds to repay those borrowings, and any such acceleration would have a material adverse effect on our business, financial condition and results of operations.
Our ability to use our net operating losses to offset potential taxable income and related income taxes that would otherwise be due could be limited if we do not generate taxable income in a timely manner or if an “ownership change” pursuant to Section 382 of the Internal Revenue Code is triggered.
We have significant net operating loss carryforwards, or NOLs. Our ability to use our NOLs to offset potential future taxable income and related income taxes that would otherwise be due is dependent upon our
38
Table of Contents
generation of future taxable income before the expiration dates of the NOLs, and we cannot predict with certainty when, or whether, we will generate sufficient taxable income to use all of our NOLs. In addition, realization of our NOLs to offset potential future taxable income and related income taxes that would otherwise be due could be restricted by annual limitations on use of NOLs triggered by an “ownership change” under Section 382 of the Internal Revenue Code and similar state provisions, based on a calculation related to our market capitalization. An “ownership change” may occur if, during a three-year period, there is a change of 50% or more in the percentage ownership of our company by 5% shareholders or shareholder groups, as defined in the Code. If we generate taxable income, a limitation on our ability to utilize some or all of our NOLs could adversely affect our results of operations.
In July 2009, we entered into an NOL preservation lock-up agreement with most of our significant stockholders that restricts transferability of all of the shares of our common stock held by the stockholders who entered into the agreement, which expires in July 2011 unless terminated earlier under certain circumstances, in order to reduce the risk that we will undergo an “ownership change” within the meaning of Section 382(g) of the Internal Revenue Code prior to that time. We have the right to grant waivers under the agreement if requested by one or more parties and if the conditions set forth in the agreement are met, and we have done so. Section 382 of the Internal Revenue Code is an extremely complex provision with respect to which there are many uncertainties. Although the NOL preservation lock-up agreement is intended to reduce the risk of such an “ownership change” before June 2011, we cannot assure you that such an ownership change will not occur. In addition, we have not requested a ruling from the Internal Revenue Service, or IRS, regarding whether we have not experienced an “ownership change” since 2005, and, therefore, we have not established whether the IRS agrees with us that our NOLs have been effectively preserved for purposes of Section 382 of the Internal Revenue Code.
Risks Relating to Our Common Stock
The market price of our common stock may be volatile, and the value of your investment could decline significantly.
Investors who purchase our common stock may not be able to sell their shares at or above the purchase price. The price of our stock has fluctuated significantly from time to time and has increased substantially in the past year, and we cannot predict if it will continue to do so. The risk factors described above relating to our business and products could cause the price of our common stock to fluctuate significantly. In addition, the stock market in general, including the market for life sciences companies, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. In addition, our stock price may be dependent upon the valuations and recommendations of the analysts who cover our business, and if our results do not meet our analysts’ forecasts and expectations, our stock price could decline as a result of analysts lowering their valuations and recommendations or otherwise. In the past, following periods of volatility in the market, securities class-action litigation has often been instituted against companies. Such litigation, if instituted against us, could result in substantial costs and diversion of management’s attention and resources, which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
Future sales of our common stock in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur, could depress the market price of our common stock, and could impair our ability to raise capital through the sale of additional equity securities. As of February 28, 2011, we had 40,294,596 shares of common stock outstanding, all of which shares are eligible for sale in the public market, subject in some cases to the volume limitations and manner of sale and other requirements under Rule 144, and the restrictions under our NOL preservation lock-up agreement which expires in July 2011.
39
Table of Contents
As of February 28, 2011, the holders of up to approximately 13,161,817 shares of common stock, based on shares outstanding as of that date, were entitled to certain rights with respect to the registration of such shares under the Securities Act of 1933, as amended, under an amended and restated investor rights agreement that we entered into with these holders in June 2007. In addition, upon exercise of outstanding options by our executive officers, our executive officers will be entitled to rights under the amended and restated investor rights agreement with respect to registration of the shares of common stock acquired on exercise. If such holders, by exercising their registration rights, sell a large number of shares, they could adversely affect the market price for our common stock. If in the future we file a registration statement and include shares held by these holders pursuant to the exercise of their registration rights, these sales may impair our ability to raise capital. We also entered into a registration rights agreement pursuant to which we filed a registration statement covering the resale of the 562,192 shares underlying the warrants that we issued in connection with the issuance of senior secured notes that were repaid in June 2010. In addition, we have filed registration statements on Form S-8 under the Securities Act to register the shares of our common stock reserved for issuance under our stock option and employee stock purchase plans, and intend to file additional registration statements on Form S-8 to register the shares automatically added each year to the share reserves under these plans.
We entered into a committed equity financing facility, or CEFF, in May 2008 with Kingsbridge Capital Limited, or Kingsbridge, which we amended in November 2009. The perceived risk of dilution from sales of our common stock to or by Kingsbridge in connection with the CEFF in the future may cause holders of our common stock to sell their shares, or it may encourage short selling by market participants, which could contribute to a decline in our stock price. If we were to draw down funds under the CEFF and Kingsbridge acquires shares in connection with a drawdown, there are no restrictions on its ability to sell those shares or engage in other transactions that could put downward pressure on the price of our common stock. If we sell shares to Kingsbridge under the CEFF, they will be issued at a discount from the average price of our common stock. This will have a dilutive effect on the holdings of our current stockholders, and may result in downward pressure on the price of our common stock. The CEFF expires in December 2012.
Pursuant to the terms of an investor rights agreement dated July 7, 2009, we entered into in connection with a private placement completed on July 7, 2009, we filed a registration statement under the Securities Act registering the resale of the 1,895,734 shares of common stock we issued to the investors pursuant to a securities purchase agreement we entered into with the investors on July 6, 2009, as well as the 947,867 shares of common stock underlying the warrants we issued to the investors pursuant to the securities purchase agreement. In addition, if we propose to register any of our securities under the Securities Act, either for our own account or for the account of others, the investors are entitled to notice of the registration and are entitled to include, at our expense, their shares of common stock in the registration and any related underwriting, provided, among other conditions, that the underwriters may limit the number of shares to be included in the registration.
Our executive officers and directors, together with their respective affiliates, own a significant percentage of our stock and will be able to exercise significant influence over matters subject to stockholder approval.
As of February 28, 2011, our executive officers and directors, together with the stockholders with which our executive officers and directors are affiliated or associated, beneficially owned approximately 51% of our capital stock. Accordingly, our executive officers and directors, together with their respective affiliates or associates, are able to determine the composition of our board of directors, retain the voting power to approve all matters requiring stockholder approval, including mergers and other business combinations, and continue to have significant influence over our operations. This concentration of ownership could have the effect of delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could have a material adverse effect on the market value of our common stock, and may prevent attempts by our stockholders to replace or remove our board of directors or management.
40
Table of Contents
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders, and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us, or for a change in the composition of our board of directors or management to occur, even if doing so would benefit our stockholders. These provisions include:
| • | authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
| • | dividing our board of directors into three classes; |
| • | limiting the removal of directors by the stockholders; |
| • | eliminating cumulative voting rights and therefore allowing the holders of a majority of the shares of our common stock to elect all of the directors standing for election, if they should so choose; |
| • | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
| • | eliminating the ability of stockholders to call a special meeting of stockholders; and |
| • | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
In addition, we are subject to Section 203 of the Delaware General Corporation Law, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless, among other exceptions, such transactions are approved by our board of directors. This provision could have the effect of delaying or preventing a change of control, whether or not it is desired by or beneficial to our stockholders. Further, because some corporate takeovers occur through an acquirer’s purchase, in the public market or otherwise, of sufficient stock to give it control of a company, the NOL preservation lock-up agreement, which restricts the transferability of our securities, could have the effect of delaying or discouraging such a takeover of us.
We have never declared or paid dividends on our capital stock and we do not anticipate paying dividends in the foreseeable future.
We do not anticipate paying any cash dividends on our common stock in the foreseeable future. We currently plan to invest all available funds and future earnings in the development and growth of our business and in the payment of our obligations. In addition, the terms of our credit agreement include, and any future indebtedness may include, a covenant restricting our ability to pay dividends. As a result, capital appreciation, if any, of our common stock will be your sole source of potential gain for the foreseeable future.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
Our corporate headquarters are located in Palo Alto, California, where we occupy approximately 44,000 square feet of office space under a lease which expires in September 2012. We have the right to extend the term for up to an additional four years.
41
Table of Contents
| Item 3. | Legal Proceedings |
On October 18, 2010, we received a Paragraph IV Patent Certification notice, or Paragraph IV Certification, from Roxane Laboratories, Inc., or Roxane, that it filed an abbreviated new drug application, or ANDA, with the FDA requesting approval to market a generic version of Xyrem. Roxane’s Paragraph IV Certification alleges that all five patents listed for Xyrem in Orange Book on the date of the Paragraph IV Certification are invalid, unenforceable or not infringed by Roxane’s proposed generic product. On November 22, 2010, we filed a lawsuit against Roxane in response to Roxane’s Paragraph IV Certification in the United States District Court for the District of New Jersey. We are seeking a permanent injunction to prevent Roxane from introducing a generic version of Xyrem. In accordance with the Hatch-Waxman Act, as a result of having filed a timely lawsuit against Roxane, FDA approval of Roxane’s ANDA will be stayed until the earlier of (i) 30 months from our October 18, 2010 receipt of Roxane’s Paragraph IV certification notice or (ii) a District Court decision finding that the identified patents are invalid, unenforceable or not infringed. On January 14, 2011, we received an additional Paragraph IV Certification from Roxane alleging that the additional method of use patent for the use of Xyrem in the treatment of narcolepsy that issued in December 2010 and is listed in the Orange Book would not be infringed by Roxane’s proposed generic product. We amended our lawsuit against Roxane on February 4, 2011 to include the additional patent in the litigation in response to Roxane’s additional Paragraph IV Certification. We cannot predict or determine the outcome of this matter.
In August 2009, we received a Paragraph IV Certification from Actavis Elizabeth, LLC, or Actavis, advising that Actavis has filed an ANDA with the FDA seeking approval to market a generic version of Luvox CR. In September 2009, we received an additional Paragraph IV Certification notice from Anchen Pharmaceuticals, Inc., or Anchen, advising that Anchen has filed an ANDA with the FDA seeking approval to market a generic version of Luvox CR. We have not been informed as to the timing or status of the FDA’s review of either party’s filing, or whether either filer has complied with FDA requirements for proving bioequivalence. Actavis’ Paragraph IV Certification alleges that the United States patent covering Luvox CR, which is owned by Elan Pharma International Limited, or Elan, and licensed to us, is invalid on the basis that the inventions claimed therein were obvious. Anchen’s Paragraph IV Certification alleges that the Elan patent will not be infringed by Anchen’s manufacture, use or sale of the generic product for which the ANDA was submitted and that the Elan patent is invalid on the basis that the inventions claimed therein were obvious. On October 6, 2009, we and Elan, as plaintiffs, filed a lawsuit against Actavis, Anchen, and Anchen Incorporated, the parent of Anchen, in the United States District Court for the District of Delaware claiming infringement of the Elan patent by the defendants in response to the Paragraph IV Certifications filed by Actavis and Anchen. On October 14, 2009, we and Elan, as plaintiffs, also filed a lawsuit in the United States District Court for the Central District of California against Anchen claiming infringement of the Elan patent based upon Anchen’s Paragraph IV Certification. In both cases, the plaintiffs were seeking a permanent injunction that prevented Actavis and Anchen from introducing a generic version of Luvox CR prior to the expiration of the Elan patent.
On August 25, 2010, we and Elan entered into settlement agreements with Anchen. Under the agreements, we, Elan and Anchen have agreed to dismiss all of the claims brought in the litigation without prejudice, Anchen has agreed not to contest the validity or enforceability of the Elan patent in the United States, and we, Elan and Anchen have agreed to release each other from all claims arising in the litigation or relating to the product Anchen intends to market under its ANDA. Settlement agreements of ANDA litigation can be reviewed by the Federal Trade Commission and the U.S. Department of Justice at their discretion. In addition, we have granted a sublicense to Anchen of our rights to have manufactured, market and sell a generic version of Luvox CR in the United States. The sublicense is non-transferable, non-sublicensable and royalty-free and is exclusive even as to us and Elan (except with respect to Luvox CR) for a period of time. The sublicense will commence on February 15, 2013 or earlier upon the occurrence of certain events. On October 5, 2010, the United States District Court for the Central District of California dismissed the case against Anchen without prejudice. On the same date, the United States District Court for the District of Delaware also dismissed the case against Anchen without prejudice.
42
Table of Contents
The lawsuit against Actavis is pending in the United States District Court for the District of Delaware. The court has not scheduled any hearing dates in this case. We cannot predict or determine the outcome of this matter.
From time to time we are involved in legal proceedings arising in the ordinary course of business. We believe there is no other litigation pending that could have, individually or in the aggregate, a material adverse effect on our results of operations or financial condition.
| Item 4. | (Removed and Reserved) |
43
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
The following table sets forth the high and low intraday sales prices of our common stock, par value $0.0001, on the NASDAQ Global Market under the symbol “JAZZ” from January 1, 2009 through December 31, 2010 for the periods indicated.
| High | Low | |||||||
| Calendar Quarter—2009 |
||||||||
| First Quarter |
$ | 2.10 | $ | 0.58 | ||||
| Second Quarter |
$ | 5.27 | $ | 0.52 | ||||
| Third Quarter |
$ | 11.88 | $ | 3.59 | ||||
| Fourth Quarter |
$ | 9.28 | $ | 6.01 | ||||
| Calendar Quarter—2010 |
||||||||
| First Quarter |
$ | 13.95 | $ | 8.01 | ||||
| Second Quarter |
$ | 12.19 | $ | 6.38 | ||||
| Third Quarter |
$ | 11.90 | $ | 7.51 | ||||
| Fourth Quarter |
$ | 20.28 | $ | 9.61 | ||||
On February 28, 2011, the last reported sales price per share of our common stock was $24.63 per share.
Holders of Common Stock
As of February 28, 2011, there were 38 holders of record of our common stock.
Dividends
Under the terms of the senior secured credit agreement we entered into in June 2010 with a lender, we are not permitted to pay any cash dividends on any shares of our capital stock. Subject to preferences that may be applicable to any then outstanding preferred stock, holders of common stock are entitled to receive dividends, if any, as may be declared from time to time by our board of directors out of legally available funds. We have never declared or paid any cash dividends and we do not presently plan to pay cash dividends in the foreseeable future.
Unregistered Sales of Equity Securities
On November 10, 2010, we issued 150,000 shares of our common stock pursuant to the exercise of a warrant held by Kingsbridge Capital Limited, or Kingsbridge. The warrant, which was exercised for cash, had an exercise price of $9.20 per share resulting in aggregate consideration to us of $1.4 million. In issuing the shares upon exercise of the warrant to Kingsbridge, we relied on the exemption provided by Section 4(2) of the Securities Act of 1933, as amended, and/or Regulation D promulgated thereunder as a transaction by an issuer not involving a public offering.
44
Table of Contents
Performance Measurement Comparison(1)
The following graph shows the total stockholder return on the last day of each month of an investment of $100 in cash on June 1, 2007, the date of our initial public offering, for (i) our common stock; (ii) the NASDAQ Composite Index; (iii) the NASDAQ Pharmaceutical Index and (iv) the NASDAQ Biotechnology Index through December 31, 2010. We are included in the NASDAQ Pharmaceutical Index and the NASDAQ Biotechnology Index. Because the NASDAQ Biotechnology Index is one of the market sector indices published by the NASDAQ Stock Market and the NASDAQ Pharmaceutical Index is no longer a published index, we have decided to use the NASDAQ Biotechnology Index going forward. Pursuant to applicable Securities and Exchange Commission rules, all values assume reinvestment of the full amount of all dividends; however no dividends have been declared on our common stock to date. The stockholder return shown in the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
COMPARISON OF 43 MONTH CUMULATIVE TOTAL RETURN(2)
Among Jazz Pharmaceuticals Inc., the NASDAQ Composite Index,
the NASDAQ Pharmaceutical Index and the NASDAQ Biotechnology Index
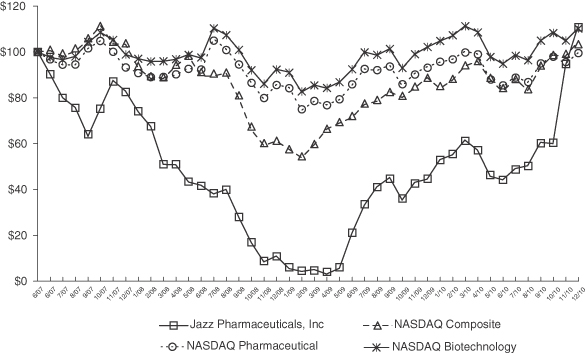
| (1) | This section is not “soliciting material”, is not deemed “filed” with the SEC and is not to be incorporated by reference into any filing of Jazz Pharmaceuticals, Inc., under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
| (2) | Information used in the graph was obtained from Research Data Group, Inc. |
45
Table of Contents
| Item 6. | Selected Financial Data |
The following selected consolidated financial data should be read together with our consolidated financial statements and accompanying notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” appearing elsewhere in this Annual Report on Form 10-K. The selected consolidated financial data in this section is not intended to replace our consolidated financial statements and the accompanying notes. Our historical results are not necessarily indicative of our future results.
We derived the consolidated statements of operations data for the years ended December 31, 2010, 2009 and 2008 and the consolidated balance sheet data as of December 31, 2010 and 2009 from our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K. The consolidated statements of operations data for the years ended December 31, 2007 and 2006, and the selected consolidated balance sheet data as of December 31, 2008, 2007, and 2006 are derived from our audited consolidated financial statements not included in this Annual Report on Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (In thousands, except per share amounts) | ||||||||||||||||||||
| Consolidated Statements of Operations Data: |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Product sales, net |
$ | 170,006 | $ | 115,108 | $ | 64,637 | $ | 53,536 | $ | 43,299 | ||||||||||
| Royalties |
2,637 | 2,203 | 1,739 | 1,156 | 594 | |||||||||||||||
| Contract revenues |
1,138 | 11,138 | 1,138 | 10,611 | 963 | |||||||||||||||
| Total revenues |
173,781 | 128,449 | 67,514 | 65,303 | 44,856 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Cost of product sales (excluding amortization of acquired developed technology and intangible asset impairment) |
13,559 | 9,638 | 13,924 | 8,903 | 6,968 | |||||||||||||||
| Research and development |
25,612 | 36,561 | 69,963 | 69,792 | 54,956 | |||||||||||||||
| Selling, general and administrative |
68,996 | 58,652 | 111,401 | 78,540 | 51,384 | |||||||||||||||
| Intangible asset amortization |
7,825 | 7,668 | 12,828 | 9,217 | 9,600 | |||||||||||||||
| Intangible asset impairment |
— | — | 29,763 | 20,160 | — | |||||||||||||||
| Provision for government settlement |
— | — | — | 17,469 | — | |||||||||||||||
| Total operating expenses |
115,992 | 112,519 | 237,879 | 204,081 | 122,908 | |||||||||||||||
| Income (loss) from operations |
57,789 | 15,930 | (170,365 | ) | (138,778 | ) | (78,052 | ) | ||||||||||||
| Interest income |
6 | 34 | 1,834 | 5,942 | 2,307 | |||||||||||||||
| Interest expense (including $570, $1,183, $1,179, $4,104 and $4,047 for the years ended December 31, 2010, 2009, 2008, 2007 and 2006, respectively, pertaining to a related party) |
(12,728 | ) | (22,796 | ) | (19,742 | ) | (13,647 | ) | (14,129 | ) | ||||||||||
| Other (expense) income |
(2 | ) | (4 | ) | 16 | 1,797 | (1,109 | ) | ||||||||||||
| Gain on extinguishment of development financing obligation |
— | — | — | — | 31,592 | |||||||||||||||
| Gain on sale of product rights |
— | — | 3,918 | 5,860 | — | |||||||||||||||
| Loss on extinguishment of debt (including $701 pertaining to a related party) |
(12,287 | ) | — | — | — | — | ||||||||||||||
| Net income (loss) |
32,778 | (6,836 | ) | (184,339 | ) | (138,826 | ) | (59,391 | ) | |||||||||||
| Beneficial conversion feature |
— | — | — | — | (21,920 | ) | ||||||||||||||
| Income (loss) attributable to common stockholders |
$ | 32,778 | $ | (6,836 | ) | $ | (184,339 | ) | $ | (138,826 | ) | $ | (81,311 | ) | ||||||
| Net income (loss) per share attributable to common stockholders: |
||||||||||||||||||||
| Basic |
$ | 0.90 | $ | (0.23 | ) | $ | (7.19 | ) | $ | (10.04 | ) | $ | (6,254.69 | ) | ||||||
| Diluted |
$ | 0.83 | $ | (0.23 | ) | $ | (7.19 | ) | $ | (10.04 | ) | $ | (6,254.69 | ) | ||||||
| Weighted-average common shares used in computing net income (loss) per share attributable to common stockholders: |
||||||||||||||||||||
| Basic |
36,343 | 30,018 | 25,646 | 13,829 | 13 | |||||||||||||||
| Diluted |
39,411 | 30,018 | 25,646 | 13,829 | 13 | |||||||||||||||
46
Table of Contents
| As of December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and marketable securities |
$ | 44,794 | $ | 15,595 | $ | 25,907 | $ | 102,945 | $ | 78,948 | ||||||||||
| Working capital (deficit) |
14,522 | (22,287 | ) | (129,492 | ) | 79,235 | 61,043 | |||||||||||||
| Total assets |
135,729 | 107,396 | 117,498 | 207,554 | 214,571 | |||||||||||||||
| Liability under government settlement, non-current |
6,978 | 10,658 | 13,063 | 14,881 | — | |||||||||||||||
| Long-term debt, current and non-current (including $6,552, $6,747, $23,474 and $23,213 as of December 31, 2009, 2008, 2007 and 2006, respectively, held by a related party) |
40,693 | 114,866 | 118,534 | 75,116 | 74,283 | |||||||||||||||
| Accumulated deficit |
(474,866 | ) | (507,644 | ) | (500,808 | ) | (316,469 | ) | (177,643 | ) | ||||||||||
| Total stockholders’ equity (deficit) |
30,551 | (72,830 | ) | (92,878 | ) | 54,992 | (176,296 | ) | ||||||||||||
47
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion of our financial condition and results of operations should be read in conjunction with the consolidated financial statements and notes to consolidated financial statements included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements that involve risks and uncertainties. When reviewing the discussion below, you should keep in mind the substantial risks and uncertainties that characterize our business. In particular, we encourage you to review the risks and uncertainties described in Part I Item 1A. “Risk Factors” included elsewhere in this report. These risks and uncertainties could cause actual results to differ materially from those projected in forward-looking statements contained in this report or implied by past results and trends.
Overview
We are a specialty pharmaceutical company focused on the identification, development and commercialization of pharmaceutical products to meet important unmet medical needs. Since we were founded in 2003, we have built a commercial and development organization and assembled a portfolio of products and product candidates that currently includes our two marketed products, which generated net product sales of $170.0 million in 2010, and product candidates in various stages of clinical development. We currently market two products: Xyrem (sodium oxybate), which is the only product approved by the United States Food and Drug Administration, or FDA, for the treatment of both cataplexy and excessive daytime sleepiness in patients with narcolepsy; and Luvox CR (fluvoxamine maleate) marketed for the treatment of obsessive compulsive disorder. We promote these products in the United States through our experienced specialty sales force targeting sleep specialists, neurologists, pulmonologists and psychiatrists. We are building our portfolio of products through a combination of internal development, acquisition and in-licensing activities. Our current product candidates are JZP-6 (sodium oxybate) for the treatment of fibromyalgia, JZP-8 (intranasal clonazepam) for the treatment of acute repetitive seizures in epilepsy, and solid oral dosage forms of sodium oxybate.
2010 was our first year of profitability, driven by substantial increases in product sales, in particular an increase in sales of Xyrem. We raised $56.8 million in equity capital in May 2010, which we used to pay down a portion of our then outstanding senior secured notes. In June 2010, we repaid the remainder of the senior secured notes, using cash on hand and the proceeds from a new $50.0 million three-year term loan. The new term loan bears interest at a variable rate which was 5.75% during 2010, as compared to the 15% interest rate on the senior secured notes we retired. As of December 31, 2010, we had $44.8 million of cash and cash equivalents and $41.7 million principal amount outstanding under our new term loan. Because of our history of losses prior to 2010, we have significant net operating losses with which to offset current and potential future taxable income.
We are dependent on sales of Xyrem, which accounted for 84% of our net product sales in 2010. During 2010, an abbreviated new drug application, or ANDA, was filed with the FDA by a third party seeking to market a generic form of Xyrem. We have sued that third party for infringement of our patents, and the litigation is ongoing. We cannot predict the timing or outcome of this litigation. If an ANDA for Xyrem is approved and a generic version of Xyrem is introduced, our sales of Xyrem would be adversely affected.
In October 2010, the FDA sent us a complete response letter, or CRL, regarding our NDA for JZP-6. The CRL stated that the FDA cannot approve the NDA in its present form. In the letter, the FDA discussed a number of topics, including the need for additional clinical studies. We have not yet finalized our plans with respect to JZP-6, and at this time, we do not know if we will continue its development.
We are continuing the development of JZP-8 and we are currently planning for an additional Phase II study for later in 2011. In addition, we are looking for appropriate opportunities to in-license or acquire additional products and product candidates to leverage our existing commercial and development capabilities.
48
Table of Contents
Results of Operations
Comparison of 2010 and 2009
| 2010 | 2009 | Increase/ (Decrease) |
Increase/ (Decrease) |
|||||||||||||
| (In thousands) | ||||||||||||||||
| Product sales, net |
$ | 170,006 | $ | 115,108 | $ | 54,898 | 48 | % | ||||||||
| Xyrem |
142,630 | 96,763 | 45,867 | 47 | % | |||||||||||
| Luvox CR |
27,376 | 18,345 | 9,031 | 49 | % | |||||||||||
| Royalties |
2,637 | 2,203 | 434 | 20 | % | |||||||||||
| Contract revenues |
1,138 | 11,138 | (10,000 | ) | (90 | %) | ||||||||||
| Cost of product sales (excluding amortization of acquired developed technology) |
13,559 | 9,638 | 3,921 | 41 | % | |||||||||||
| Research and development |
25,612 | 36,561 | (10,949 | ) | (30 | %) | ||||||||||
| Selling, general and administrative |
68,996 | 58,652 | 10,344 | 18 | % | |||||||||||
| Intangible asset amortization |
7,825 | 7,668 | 157 | 2 | % | |||||||||||
| Interest income |
6 | 34 | (28 | ) | (82 | %) | ||||||||||
| Interest expense |
12,728 | 22,796 | (10,068 | ) | (44 | %) | ||||||||||
| Other expense |
2 | 4 | (2 | ) | (50 | %) | ||||||||||
| Loss on extinguishment of debt |
12,287 | — | 12,287 | N/A | (1) | |||||||||||
| (1) | Comparison to prior period is not meaningful. |
Product Sales, Net
Xyrem product sales increased in 2010 compared to 2009, primarily due to price increases and to a lesser extent a 7% increase in sales volume. Most of the increase in Luvox CR product sales was due to increases in volume with the remainder due to price increases. In the fourth quarter of 2010 we recognized $2.0 million of previously deferred revenue as a result of a change in the timing of when Luvox CR revenue is recognized. While we expect total product sales to increase in 2011 over 2010, the rate of growth of product sales or sales volumes, or both could be less than that experienced in 2010.
Royalties, Net,
Royalties increased in 2010 compared to 2009 due to an increase in royalties from sales of Xyrem in Europe by UCB Pharma Limited, or UCB, under a license agreement. We expect modest growth in royalty income in 2011 as compared with 2010.
Contract Revenues
Contract revenues in 2010 and 2009 include previously deferred upfront payments under our agreement with UCB, which are being recognized as contract revenues ratably through 2019, the expected performance period under our agreement with UCB. In 2009, upon achievement of a milestone, we recognized as revenue a $10.0 million milestone payment we received from UCB in 2008.
Cost of Product Sales
Cost of product sales increased in 2010 compared to 2009, primarily due to our increased sales volumes, and included $674,000 of previously deferred costs recognized as a result of a change in the timing of when Luvox CR revenue is recognized. As a percentage of product sales, costs were 8.0% and 8.4% in 2010 and 2009, respectively. We do not expect cost of product sales as a percentage of sales to change significantly in 2011 compared to 2010.
49
Table of Contents
Research and Development Expenses
Research and development costs were lower in 2010 compared to 2009, primarily due to lower spending on JZP-6 and a $978,000 credit resulting from the government therapeutic discovery tax credit, partially offset by higher spending on solid oral dosage forms of sodium oxybate. As a result, our direct project costs decreased $12.9 million in 2010 compared to 2009, when we were actively conducting our second JZP-6 Phase III clinical trial and enrolling patients in a long-term safety study. Headcount-related expenses and administrative costs incurred in the research and development organization increased $2.0 million in 2010 compared to 2009. We expect research and development spending in 2011 to be slightly lower than spending in 2010 and to consist primarily of expenses associated with development work on our JZP-8 product candidate and, to a lesser extent, solid oral dosage forms of sodium oxybate.
Selling, General and Administrative Expenses
Selling, general and administrative expenses were higher in 2010 compared to 2009, primarily due to increases in headcount-related expenses and, to a lesser extent, expenses related to our previously planned launch of our JZP-6 product candidate. We expect that selling, general and administrative expenses will be higher in 2011 than in 2010 due to legal expenses associated with protecting our sodium oxybate business, additional investments in Xyrem marketing and promotion and an increase in stock-based compensation expense.
Intangible Asset Amortization
Our intangible assets consist primarily of developed technology related to Xyrem and Luvox CR which are amortized on a straight-line basis over their estimated useful lives. We expect intangible asset amortization in 2011 to be similar to 2010.
Interest Income
Interest income was lower in 2010 compared to 2009 due to lower average interest rates.
Interest Expense
Interest expense relates primarily to interest on our long-term debt and, to a small extent, interest on our liability under a 2007 government litigation settlement. In 2010, we entered into a new term loan agreement and retired our outstanding senior secured debt. As a result of these actions, we reduced the principal amount of our long-term debt outstanding from $119.5 million as of December 31, 2009, to $41.7 million as of December 31, 2010, and we reduced the rate at which we pay interest on our debt from a fixed rate of 15% to a variable rate that was 5.75% under our new term loan as of December 31, 2010. As a result, interest expense was substantially lower in 2010 as compared to 2009.
Loss on Extinguishment of Debt
The loss on extinguishment of debt relates to our early repayment of the senior secured notes in May and June 2010 and is comprised of $8.5 million of prepayment premiums and fees, and $3.8 million of non-cash expense related to the write-off of unamortized debt discount and debt issuance costs.
50
Table of Contents
Comparison of 2009 and 2008
| 2009 | 2008 | Increase/ (Decrease) |
Increase/ (Decrease) |
|||||||||||||
| (In thousands) | ||||||||||||||||
| Product sales, net |
$ | 115,108 | $ | 64,637 | $ | 50,471 | 78 | % | ||||||||
| Xyrem |
96,763 | 53,803 | 42,960 | 80 | % | |||||||||||
| Luvox CR |
18,345 | 5,728 | 12,617 | 220 | % | |||||||||||
| Antizol |
— | 5,106 | (5,106 | ) | N/A | (1) | ||||||||||
| Royalties |
2,203 | 1,739 | 464 | 27 | % | |||||||||||
| Contract revenues |
11,138 | 1,138 | 10,000 | N/A | (1) | |||||||||||
| Cost of product sales (excluding amortization of acquired developed technology and intangible asset impairment) |
9,638 | 13,924 | (4,286 | ) | (31 | %) | ||||||||||
| Research and development |
36,561 | 69,963 | (33,402 | ) | (48 | %) | ||||||||||
| Selling, general and administrative |
58,652 | 111,401 | (52,749 | ) | (47 | %) | ||||||||||
| Intangible asset amortization |
7,668 | 12,828 | (5,160 | ) | (40 | %) | ||||||||||
| Intangible asset impairment |
— | 29,763 | (29,763 | ) | N/A | (1) | ||||||||||
| Interest income |
34 | 1,834 | (1,800 | ) | (98 | %) | ||||||||||
| Interest expense |
22,796 | 19,742 | 3,054 | 15 | % | |||||||||||
| Other (expense) income |
(4 | ) | 16 | (20 | ) | N/A | (1) | |||||||||
| Gain on sale of product rights |
— | 3,918 | (3,918 | ) | N/A | (1) | ||||||||||
| (1) | Comparison to prior period is not meaningful. |
Product Sales, Net
Xyrem product sales increased in 2009 compared to 2008, primarily due to price increases and a 10% increase in sales volume. Most of the increase in Luvox CR product sales was due to increases in volume following its launch in 2008 with the remainder due to price increases. In 2008, we sold our rights to and interests in Antizol® and Antizol-Vet®, along with the associated product registrations, commercial inventory and trademarks, and did not record products sales for Antizol® subsequent to that date.
Royalties, Net
Royalties increased in 2009 compared to 2008 due to an increase in royalties from sales of Xyrem by UCB.
Contract Revenues
In 2009, we recognized as revenue a $10.0 million milestone payment we received from UCB in 2008.
Cost of Product Sales
Cost of product sales decreased in 2009 compared to 2008 as a result of higher Luvox CR manufacturing scale up costs in 2008 and a charge of $3.5 million in 2008 for excess Luvox CR inventory.
Research and Development Expenses
Research and development expenses were lower in 2009 compared to 2008 as we focused our development efforts on our JZP-6 product candidate and curtailed spending on our other development projects. Direct development costs decreased by $24.9 million. Headcount-related expenses and administrative costs incurred in the research and development organization decreased $8.5 million in 2009 compared to 2008, primarily due to our lower staffing levels in 2009.
51
Table of Contents
Selling, General and Administrative Expenses
Selling, general and administrative expenses were lower in 2009 compared to 2008. In 2008, we reduced the size of our sales force, which resulted in a $29.8 million reduction in 2009 sales and sales support costs compared to 2008. In addition, direct marketing expenses for Luvox CR were $16.4 million lower in 2009 compared with 2008, the year we launched Luvox CR.
Intangible Asset Amortization
Amortization costs in 2009 were lower compared to 2008 primarily due to a $29.8 million intangible asset impairment charge associated with Luvox CR recorded in 2008.
Intangible Asset Impairment
The intangible asset impairment charge in 2008 resulted from an impairment of the intangible asset associated with Luvox CR.
Interest Income
Interest income was lower in 2009 compared to 2008 due to lower average cash balances and to lower average interest rates.
Interest Expense
Interest expense in 2009 and 2008 related primarily to interest on the then outstanding senior secured notes and, to a small extent, interest on our liability under a 2007 government litigation settlement. The increase in interest expense in 2009 as compared to 2008 was primarily due to interest expense recorded on the additional $40.0 million principal amount of the then outstanding senior secured notes we issued in March 2008 and to a lesser extent a higher average interest rate.
Gain on Sale of Product Rights
In 2008, we sold our rights to and interests in Antizol® and Antizol-Vet®, along with the associated product registrations, commercial inventory and trademarks, for $5.8 million and recorded a gain of $3.9 million.
Non-GAAP Financial Measures
To supplement our financial results presented on a GAAP basis, we use the non-GAAP measures adjusted net income (loss) and adjusted net income (loss) per diluted share as shown in the table below. These measures exclude the following: revenue related to upfront and milestone payments, the gross margin impact of a change in the timing of when Luvox CR revenue is recognized, a gain on sale of product rights, a loss on extinguishment of debt, amortization and impairment of intangible assets, stock-based compensation, and non-cash interest expense associated with a debt discount and debt issuance costs. We believe these non-GAAP financial measures are helpful in understanding our past financial performance and our potential future results. They are not meant to be considered in isolation or as a substitute for comparable GAAP measures, and should be read in conjunction with our consolidated financial statements prepared in accordance with GAAP. Our management regularly uses these supplemental non-GAAP financial measures internally to understand, manage and evaluate our business and make operating decisions. Compensation of our executives is based in part on the performance of our business based on these non-GAAP measures. In addition, we believe that the use of these non-GAAP measures enhances the ability of investors to compare our results both from period to period. Adjusted net income (loss) and adjusted net income (loss) per diluted share, as used by us, may be calculated differently from, and therefore may not be directly comparable to, similarly titled measures used by our competitors and other companies.
52
Table of Contents
A reconciliation of GAAP net income (loss) to adjusted net income (loss), a non-GAAP financial measure, and related per share amounts follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| (In thousands, except per share amounts) | ||||||||||||
| GAAP net income (loss) |
$ | 32,778 | $ | (6,836 | ) | $ | (184,339 | ) | ||||
| Add: |
||||||||||||
| Intangible asset amortization |
7,825 | 7,668 | 12,828 | |||||||||
| Intangible asset impairment |
— | — | 29,763 | |||||||||
| Stock-based compensation expense |
8,219 | 5,957 | 8,106 | |||||||||
| Non-cash interest expense |
2,406 | 2,810 | 2,060 | |||||||||
| Loss on extinguishment of debt |
12,287 | — | — | |||||||||
| Deduct: |
||||||||||||
| Contract revenues |
(1,138 | ) | (11,138 | ) | (1,138 | ) | ||||||
| Luvox CR revenue recognition timing change |
(1,345 | ) | — | — | ||||||||
| Gain on sale of product rights |
— | — | (3,918 | ) | ||||||||
| Adjusted net income (loss) |
$ | 61,032 | $ | (1,539 | ) | $ | (136,638 | ) | ||||
| GAAP net income (loss) per diluted share |
$ | 0.83 | $ | (0.23 | ) | $ | (7.19 | ) | ||||
| Adjusted net income (loss) per diluted share |
$ | 1.55 | $ | (0.05 | ) | $ | (5.33 | ) | ||||
| Shares used in computing GAAP and adjusted net income (loss) per diluted share amounts |
39,411 | 30,018 | 25,646 | |||||||||
Liquidity and Capital Resources
During 2010, we took a number of measures designed to strengthen our balance sheet and improve our liquidity and financial condition. In May 2010, we issued 7,000,000 shares of our common stock in an underwritten public offering for net proceeds of $56.8 million, and in June 2010 we entered into a new credit agreement which provides for a $50.0 million term loan and a revolving credit facility maturing in June 2013, secured by substantially all of our assets. We used the proceeds from the offering and the new term loan, along with some of our available cash, to prepay in full all of our then outstanding senior secured notes.
As of December 31, 2010, we had cash and cash equivalents of $44.8 million. We believe that our existing cash balances and cash we expect to generate from operations will be sufficient to fund our operations and to meet our existing obligations for the foreseeable future. The adequacy of our cash resources depends on many assumptions, including primarily our assumptions with respect to product sales and expenses as well as the other factors set forth in Part I Item 1A of this Annual Report on Form 10-K under the heading “To grow our business, we will need to commit substantial resources, which could result in future losses or otherwise limit our opportunities or affect our ability to operate our business.” Our assumptions may prove to be wrong or other factors may adversely affect our business, and as a result we could exhaust or significantly decrease our available cash resources which could, among other things, force us to raise additional funds and/or force us to reduce our expenses, either of which could have a material adverse effect on our business.
As of December 31, 2010, $41.7 million principal amount was outstanding on our term loan which is repayable in quarterly installments of $4.2 million, and $7.4 million was outstanding under the revolving credit facility. The average daily amount outstanding under the revolving credit facility since its inception in June 2010 through December 31, 2010, was $2.0 million. The borrowing availability under the revolving credit facility is currently $15.0 million. The revolving credit facility has a commitment fee payable on the undrawn amount which is currently 0.5% per annum. Interest on the term loan and the revolving credit facility was payable at a variable rate which was 5.75% in 2010 and is currently 3.75%. Interest on the fully repaid senior secured notes was payable at a fixed rate of 15%.
53
Table of Contents
Our credit agreement contains customary operating covenants, including covenants that restrict our ability to: incur indebtedness and liens; effect mergers, consolidations and other fundamental changes; dispose of significant assets or enter into sale-leaseback transactions; pay dividends or make other restricted payments; make loans, advances or certain investments, including acquisitions of companies and products; or enter into transactions with affiliates. The credit agreement also requires us to comply with financial covenants requiring us to maintain a minimum consolidated fixed charge coverage ratio, a maximum consolidated leverage ratio and minimum liquidity, each as defined in the credit agreement. Our failure to comply with any of the operating and financial covenants contained in the credit agreement would constitute an event of default under the credit agreement. The credit agreement contains other customary events of default. Upon the occurrence of one or more events of default all or part of the obligations under the credit agreement may be declared immediately due and payable and borrowings under the credit agreement may be stopped. We are currently in compliance with all material covenants under the credit agreement.
To grow our business over the longer-term, we will need to commit substantial resources to product acquisition and in-licensing costs, to expensive and time-consuming product development and clinical trials of our product candidates, and to expanding our commercial operations. We may need to raise additional funds to license or acquire additional products, product candidates or companies or seek to raise additional funds for general corporate purposes. Raising additional capital could be accomplished through one or more public or private debt or equity financings, collaborations, partnering arrangements or development financings or a draw down of funds under our committed equity financing facility, or CEFF, with Kingsbridge Capital Limited which expires in December 2012. Under the CEFF, we have the ability to draw down amounts up to $75.0 million, subject to certain conditions and limitations. Any equity financing would be dilutive to our stockholders, and the consent of the lender under our credit agreement could be required.
The following table shows a summary of our cash flows for the periods indicated:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| (In thousands) | ||||||||||||
| Net cash provided by (used in) operating activities |
$ | 58,868 | $ | (15,878 | ) | $ | (130,232 | ) | ||||
| Net cash used in investing activities |
(2,143 | ) | (6,124 | ) | (11,942 | ) | ||||||
| Net cash (used in) provided by financing activities |
(27,526 | ) | 12,694 | 64,132 | ||||||||
| Net increase (decrease) in cash and cash equivalents |
$ | 29,199 | $ | (9,308 | ) | $ | (78,042 | ) | ||||
In each of 2010, 2009 and 2008, net cash provided by or used in operating activities primarily reflected our net income or loss, adjusted for non-cash items including depreciation, amortization, impairment losses, losses on disposal of property and equipment, non-cash interest expense, loss on extinguishment of debt, stock-based compensation and gains on sales of product rights, and changes in working capital and the provision for our liability from the settlement of government litigation in 2007. In 2010, 2009 and 2008, operating cash outflows included $3.0 million, $2.5 million, and $2.0 million, respectively, paid to the government as part of the settlement.
Net cash used in investing activities in 2010 included $4.0 million paid to Solvay Pharmaceuticals, Inc., or Solvay, which was acquired by Abbott Laboratories, or Abbott, for the rights to market Luvox CR partially offset by a decrease in restricted cash. Net cash used in investing activities in 2009 included $6.0 million paid to Solvay and an increase in restricted cash, offset by the maturity of an investment in a marketable security. Net cash used in investing activities in 2008 included $27.0 million paid to Solvay, the purchase of property and equipment of $1.7 million, partially offset by the release of $12.0 million of cash that was previously restricted under the agreement governing the then outstanding senior secured notes, and proceeds of $5.8 million from the sale of our product rights to Antizol and Antizol-Vet.
Net cash used in financing activities in 2010 included the principal repayment of the senior secured notes of $119.5 million offset by proceeds from a common stock offering of $56.8 million and net cash inflows from our
54
Table of Contents
term loan of $40.1 million. Net cash provided by financing activities in 2009 included net proceeds of $6.8 million from a private placement of common stock and warrants and $5.5 million in net borrowings under our prior revolving bank line of credit. Net cash provided by financing activities in 2008 related primarily to the sale of $40.0 million aggregate principal amount of the then outstanding senior secured notes for net proceeds of $38.5 million, and $24.5 million of net proceeds from a registered direct public offering of common stock and warrants.
Contractual Obligations
The following table reflects a summary of our contractual obligations as of December 31, 2010:
| Payments due by period | ||||||||||||||||||||
| Contractual Obligations(1) |
Total | Less than 1 Year |
1-3 Years | 3-5 Years | More than 5 years |
|||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Term loan—principal |
$ | 41,668 | $ | 16,664 | $ | 25,004 | $ | — | $ | — | ||||||||||
| Term loan—interest(2) |
2,216 | 1,395 | 821 | |||||||||||||||||
| Liability under government settlement |
11,500 | 4,164 | 7,336 | — | — | |||||||||||||||
| Purchased product rights liability(3) |
9,000 | 4,500 | 4,500 | — | — | |||||||||||||||
| Revolving credit facility |
7,350 | 7,350 | — | — | — | |||||||||||||||
| Operating lease obligations(4) |
4,511 | 1,915 | 2,472 | 124 | — | |||||||||||||||
| Purchase obligations(5) |
2,080 | 2,080 | — | — | — | |||||||||||||||
| Total |
$ | 78,325 | $ | 38,068 | $ | 40,133 | $ | 124 | $ | — | ||||||||||
| (1) | We have not included milestone or royalty payments or contractual payment obligations in the table above if the amount and timing of such obligations are unknown or uncertain. |
| (2) | Borrowings under the term loan bear interest at a variable rate which was 5.75% at December 31, 2010 and subsequently decreased to 3.75% under the terms of our credit agreement. We have calculated future interest payments assuming that interest on the term loan will be paid at a rate of 3.75%, which may not represent actual interest payments made. |
| (3) | This represents payments due to Abbott under a product license agreement. These amounts exclude $5.0 million we would pay Abbott if net sales of Luvox CR have reached a cumulative amount of $100.0 million on or before December 31, 2014 and no AB-rated generic version of Luvox CR has been or is being sold in the United States as of December 31, 2014 because we do not know if we will have to pay it. |
| (4) | Includes the minimum lease payments for our corporate office building and automobile lease payments for our sales force. In addition to the minimal lease payments on our office building we are obligated to pay for operating expenses for the lease property, which are not included in the table above. |
| (5) | Consists of commitments to third party manufacturers of Xyrem and Luvox CR. |
Critical Accounting Policies and Significant Estimates
Revenue Recognition
Revenues are recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed and determinable and collection is reasonably assured. Revenue from sales transactions where the buyer has the right to return the product is recognized at the time of sale only if (i) the seller’s price to the buyer is substantially fixed or determinable at the date of sale, (ii) the buyer has paid the seller, or the buyer is obligated to pay the seller and the obligation is not contingent on resale of the product, (iii) the buyer’s obligation to the seller would not be changed in the event of theft or physical destruction or damage of the product, (iv) the buyer acquiring the product for resale has economic substance apart from that provided by the seller, (v) the seller does not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (vi) the amount of future returns can be reasonably estimated.
55
Table of Contents
Product Sales, Net
Xyrem—Domestic. We sell Xyrem in the United States to a single central pharmacy, Express Scripts Specialty Distribution Services and its affiliate Curascript, Inc., or Express Scripts. In 2010, sales of Xyrem to Express Scripts accounted for 84% of our net product sales. We recognize revenues from sales of Xyrem within the United States upon transfer of title, which occurs when Express Scripts removes product from our consigned inventory location at its facility for shipment directly to a patient.
We accept returns from and provide Express Scripts with a credit for any product returned by patients to Express Scripts with defects that were not reasonably discoverable upon receipt of the consigned product by Express Scripts. Based on our experience over the past five years, product returns to Express Scripts from patients are extremely rare; during 2010 we issued less than $20,000 of credits to Express Scripts for returned product.
Xyrem—International. We sell limited quantities of Xyrem to UCB for sale in territories outside of North America, and to Valeant, for sale in Canada, under license and distribution agreements. The agreements provide our international licensees with a fixed period of time after delivery to inspect and reject shipments for failure to meet specifications. We do not recognize revenue on the sales to our international licensees until the right of return has lapsed, which occurs when we are notified of their acceptance, or when the time for them to inspect or reject a shipment has lapsed, if earlier. We recognized revenue of $716,000, $1.0 million and $769,000 from international sales of Xyrem during 2010, 2009 and 2008, respectively.
Luvox CR. We grant rights to our wholesaler customers to return product six months prior to and up to twelve months after product expiration and issue credits which may be applied against existing or future invoices. From product launch in 2008 until the fourth quarter of 2010, we did not believe we were able to reasonably estimate expected returns of Luvox CR at the time of shipment, and therefore we recognized revenue when units were dispensed through prescriptions, at which point the product was not subject to return. We purchased dispensing data from an independent prescription tracking service which we used to estimate units dispensed. As of October 1, 2010, we believed we had sufficient historical data on returns of Luvox CR to reasonably estimate a return rate when a unit is shipped. As a result, as of October 1, 2010, we started recognizing revenue upon shipment to our wholesaler customers and recorded an estimated amount of product returns. We recognized $2.0 million of previously deferred net product sales and $674,000 of previously deferred product costs in the fourth quarter of 2010. We recorded a $3.5 million liability for estimated future returns as of December 31, 2010.
Items Deducted from Gross Sales. Revenues from sales of products within the United States are recorded net of estimated allowances for returns, specialty distributor fees, wholesaler fees, prompt payment discounts, government rebates, government chargebacks, patient rebates and rebates under managed care plans. Calculating certain of these items involves estimates and judgments based on sales or invoice data, contractual terms, historical utilization rates, new information regarding changes in these programs’ regulations and guidelines that would impact the amount of the actual rebates, our expectations regarding future utilization rates for these programs and channel inventory data. Because we derive most of our revenues from sales of Xyrem in the United States to one specialty pharmacy customer, Express Scripts, we have a much higher level of knowledge about each prescription than if we sold the product through the normal pharmaceutical wholesaler channel as we do with Luvox CR. As a result, we do not exercise a high degree of judgment in estimating most of the items that are deducted from gross sales. The two most significant items deducted from gross revenue where we exercise judgment are government rebates, which include Medicaid and TRICARE rebates, and estimated returns of Luvox CR.
56
Table of Contents
The following table shows activity related to government rebates and estimated returns of Luvox CR:
| Government Rebates Payable |
Sales Returns Reserve |
|||||||
| (In thousands) | ||||||||
| Balance at December 31, 2007 |
$ | 64 | $ | — | ||||
| Current year provision related to sales in current year |
500 | — | ||||||
| Current year provision related to sales in prior year |
3 | — | ||||||
| Payments/credits |
(396 | ) | — | |||||
| Balance at December 31, 2008 |
171 | — | ||||||
| Current year provision related to sales in current year |
3,158 | — | ||||||
| Current year provision related to sales in prior year |
619 | — | ||||||
| Payments/credits |
(1,678 | ) | — | |||||
| Balance at December 31, 2009 |
2,270 | — | ||||||
| Current year provision related to sales in current year |
11,083 | 3,921 | ||||||
| Current year provision related to sales in prior year |
(100 | ) | — | |||||
| Payments/credits |
(6,665 | ) | (382 | ) | ||||
| Balance at December 31, 2010 |
$ | 6,588 | $ | 3,539 | ||||
Contract Revenues
Nonrefundable fees where we have no continuing performance obligations are recognized as revenues when there is persuasive evidence of an arrangement and collection is reasonably assured. In situations where we have continuing performance obligations, nonrefundable fees are deferred and recognized ratably over our estimated performance period. We recognize at-risk milestone payments, which are typically related to regulatory, commercial or other achievements by us or our licensees and distributors, as revenues when the milestone is accomplished and collection is reasonably assured. Refundable fees are deferred and recognized as revenues upon the later of when they become nonrefundable or when our performance obligations are completed.
We have an agreement with UCB under which UCB has the right to market Xyrem for the treatment of narcolepsy and for the treatment of fibromyalgia in various countries outside the United States. In 2008 we received a $10.0 million nonrefundable milestone payment which we recognized as revenue in 2009 upon achievement of the milestone. We recognized contract revenues of $1.1 million during each of 2010, 2009, and 2008 related to two upfront payments from UCB totaling $15.0 million related to Xyrem for the treatment of fibromyalgia. As of December 31, 2010, $10.2 million was recorded as deferred revenues related to these upfront payments and is being recognized ratably through 2019, the end of the expected performance period under the agreement. There has been no change in the expected performance period under our agreement with UCB since its establishment in 2006 at the time of the initial upfront payments. A change in our estimate of the performance period would result in a change in contract revenues.
Inventory Valuation
Inventories are valued at the lower of cost or market. Cost is determined using the first-in, first-out method for all inventories. Our policy is to write down inventory that has become obsolete, inventory that has a cost basis in excess of its expected net realizable value and inventory in excess of expected requirements. The estimate of excess quantities is subjective and primarily dependent on our estimates of future demand for the product. If our estimate of future demand is too high we may have to write down the carrying value of inventory and record additional charges to cost of product sales. We recorded charges to cost of product sales related to Luvox CR totaling $82,000 and $4.2 million, during 2009 and 2008, respectively, for inventory and purchase orders we judged to be in excess of expected requirements.
57
Table of Contents
Goodwill and Intangible Assets
Goodwill
Goodwill represents the excess of the purchase price over the fair value of assets acquired and liabilities assumed. We have determined that we operate in a single segment and have a single reporting unit associated with the development and commercialization of pharmaceutical products. The annual test for goodwill impairment is a two-step process. The first step is a comparison of the fair value of the reporting unit with its carrying amount, including goodwill. If this step indicates impairment, then in the second step, the loss is measured as the excess of recorded goodwill over its implied fair value. Implied fair value is the excess of the fair value of the reporting unit over the fair value of all identified assets and liabilities. We test goodwill for impairment annually in October and when events or changes in circumstances indicate that the carrying value may not be recoverable.
Intangible Assets
Intangible assets consist of purchased developed technology and trademarks. The method of amortization reflects the pattern in which the economic benefits of the intangible asset are consumed. If that pattern cannot be reliably determined, we use a straight-line amortization method. Our intangible assets are amortized on a straight-line basis over their estimated useful lives, which range from three to ten years. The estimated useful lives associated with intangible assets are consistent with the estimated lives of the products and may be modified when circumstances warrant. Once an intangible asset is fully amortized, the gross costs and accumulated amortization are removed from the consolidated balance sheet. We evaluate purchased intangibles and other long-lived assets, other than goodwill, for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Estimating future cash flows related to an intangible asset involves estimates and assumptions. If our assumptions are not correct, there could be an impairment loss or, in the case of a change in the estimated useful life of the asset, a change in amortization expense.
Our two most significant intangible assets are related to Xyrem for the treatment of cataplexy associated with narcolepsy and the Xyrem trade name, collectively the Xyrem intangibles, which were recorded as part of an acquisition in 2005. As of December 31, 2010 those two assets had a carrying value of $17.8 million, or 81% of our total intangible asset carrying amount of $22.0 million. At the time of the acquisition we estimated the life of the Xyrem intangibles to be 9.5 years, or through December 31, 2014, which corresponded to the time period during which we expected the assets to generate cash flows in our valuation analysis.
As of December 31, 2010, the gross carrying amount of goodwill was $38.2 million and the gross carrying amounts and net book values of intangible assets were as follows:
| December 31, 2010 | Weighted Average Remaining Useful Life |
|||||||||||||||
| Gross Carrying Amount |
Accumulated Amortization |
Net Book Value |
||||||||||||||
| (In thousands) | (In years) | |||||||||||||||
| Developed technology—Xyrem |
$ | 39,700 | $ | 23,014 | $ | 16,686 | 4.0 | |||||||||
| Developed technology—Luvox CR |
9,700 | 5,446 | 4,254 | 1.4 | ||||||||||||
| Trademarks |
2,600 | 1,507 | 1,093 | 4.0 | ||||||||||||
| $ | 52,000 | $ | 29,967 | $ | 22,033 | |||||||||||
58
Table of Contents
Stock-Based Compensation
We have elected to use the Black-Scholes option pricing model to calculate the fair value of stock option grants under our equity incentive plans and grants under our 2007 Employee Stock Purchase Plan, or ESPP, and we are using the straight-line method to allocate compensation cost to reporting periods. The fair value of stock options was estimated using the following assumptions:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Weighted-average volatility |
85 | % | 91 | % | 60 | % | ||||||
| Weighted-average expected term (years) |
6.0 | 6.1 | 6.1 | |||||||||
| Range of risk-free rates |
1.5-3.1 | % | 1.8-3.1 | % | 2.7-3.4 | % | ||||||
| Expected dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
We completed our initial public offering in 2007 and our common stock therefore has a trading history which is shorter than the weighted-average expected term of our stock option grants. A public market for options on our common stock did not exist before 2009, and the market for options with more than one year to expiration is not very liquid. In 2008 we used the historic volatility of a peer group to estimate the future volatility for our stock option grants and we used the historic and implied volatility of a peer group in addition to the historic volatility of our own common stock to estimate volatility for grants under our ESPP. In 2009, we used the historic volatility of a peer group and the historic volatility of our own common stock to estimate future volatility for stock option grants and we used the implied volatility of our own common stock to estimate the volatility for grants under our ESPP. In 2010, we used the historic volatility of a peer group, the historic volatility of our own common stock and the implied volatility of our own common stock to estimate future volatility for stock option grants and we used the implied volatility of our own common stock to estimate the volatility for grants under our ESPP.
We have limited historical information with which to develop reasonable expectations about the expected term of our stock options. As a result, for stock option grants made during 2010, 2009 and 2008, the expected term was estimated by assuming stock options would be exercised at the mid-point between the vest date and the contractual term.
The risk-free interest rate assumption was based on zero coupon U.S. Treasury instruments whose term was consistent with the expected term of our stock option grants. The expected dividend yield assumption was based on our history and expectation of no dividend payouts.
Accrued Liabilities
As part of the process of preparing financial statements, we are required to estimate accrued liabilities. This process involves identifying goods received and services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for such service as of each balance sheet date in our financial statements. Examples of estimated accrued liabilities include the cost of marketing and promotional materials, contract service fees, such as amounts paid to clinical monitors, data management organizations, clinical research organizations and fees paid to contract manufacturers in conjunction with the production of clinical materials, and professional service fees, such as fees to lawyers and accountants. In connection with such service fees, our estimates are most affected by our understanding of the status and timing of services provided. The majority of our service providers invoice us in arrears for services performed. To the extent that we do not identify certain costs that have begun to be incurred or we under- or over-estimate the level of services performed or the costs of such services, our reported expenses for such period would be too low or too high. The date on which certain services commence, the level of services performed on or before a given date and the cost of such services are often subject to our judgment. We make these judgments in accordance with the facts and circumstances known to us through our internal processes. Our internal processes require substantially all of our spending for services to be under contracts with our service providers and to be documented and tracked under
59
Table of Contents
internally-generated purchase orders based on designated spending authorizations. As of each balance sheet date, employees who are responsible for managing the contracts, and who are in contact with the outside service providers as to progress or stage of completion of the services and the agreed upon fee to be paid for such services, review current contracts and the related open purchase orders. We adjust for spending not already reflected in our accounting records in accordance with generally accepted accounting principles. To date, there have been no material differences between the amounts of expenses accrued at our balance sheet dates and the amount at which such expenses were subsequently invoiced. Although we do not expect our current estimates to be materially different when invoiced, our understanding of the status and timing of services provided relative to the actual timing and levels of service provided may vary and may result in adjustments in future periods.
Income Taxes
We utilize the liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and the tax bases of assets and liabilities and are measured using enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. Despite achieving profitability in 2010, we continue to maintain a full valuation allowance on our net operating losses and other deferred tax assets. Realization of our deferred tax assets is dependent upon the generation of future taxable income, the amount and timing of which are uncertain. If we continue to generate income, we may conclude that it is more likely than not that all or a portion of our deferred tax assets are realizable, and we will reverse the valuation allowance and recognize a related tax benefit at such time. We believe that a release of the valuation allowance, in full or in part, may occur in 2011. This determination depends on a variety of factors, some of which are subjective. We have also provided for uncertain tax positions that we believe are not more likely than not to be sustained upon examination by tax authorities, the effect of which are less significant.
Recent Accounting Pronouncements
In October 2009, the FASB issued authoritative guidance which amends the revenue recognition guidance to require companies to allocate revenue in multiple-element arrangements based on an element’s estimated selling price if vendor-specific or other third-party evidence is not available. The guidance became effective for us beginning January 1, 2011 and is being applied prospectively to multiple-deliverable revenue arrangements entered into on or after January 1, 2011. The adoption of this guidance is not expected to have a material impact on our results of operations and financial position.
Off-Balance Sheet Arrangements
Since our inception, except for standard operating leases, we have not engaged in any off-balance sheet arrangements, including the use of structured finance, special purpose entities or variable interest entities.
Related Parties
Senior Secured Notes. In 2010, we repaid in full all of our then outstanding senior secured notes, of which $6.8 million principal amount was paid to an entity affiliated with Kohlberg, Kravis & Roberts & Co. L.P., or KKR, a significant stockholder. In addition, in 2010 we paid prepayment penalties and a fee to the holders of the senior secured notes totaling $8.5 million, of which $484,000 was paid to the KKR affiliate. In 2008, we paid $327,000 to the KKR affiliate, as partial prepayment of the principal amount of the senior secured notes held by the KKR affiliate. Cash paid for interest with respect to then outstanding senior secured notes held by the KKR affiliate was $461,000, $1.3 million, and $796,000 in 2010, 2009, and 2008, respectively. All payments to KKR were in proportion to its ownership of the senior secured notes.
60
Table of Contents
In 2009, the exercise price of all warrants to purchase common stock issued to the holders of the then outstanding senior secured notes was reduced to $9.34 per share as a result of an amendment to the agreement governing the senior secured notes. This included warrants to purchase 70,156 shares of our common stock held by the KKR affiliate the exercise price of which was reduced from $20.36 to $9.34 per share.
2009 and 2010 Common Stock Offerings. In a private placement we completed in 2009, 1,858,486 shares of common stock and a warrant to purchase 929,243 shares of common stock were acquired by Longitude Venture Partners, L.P. and 37,248 shares of common stock and a warrant to purchase 18,624 shares of common stock were acquired by Longitude Capital Associates, L.P. In July 2009, Patrick G. Enright was elected to our board of directors in connection with the closing of the private placement. Mr. Enright is a managing member of Longitude Capital Partners, LLC, the sole general partner of Longitude Venture Partners, L.P. and Longitude Capital Associates, L.P. In addition, in 2010 we issued 7,000,000 shares of our common stock in an underwritten public offering of which 838,323 shares were purchased from the underwriter by Longitude Capital Partners, LLC. The remaining shares were purchased from the underwriter by third party investors on the same terms and conditions.
2008 Common Stock Offering. In a registered direct public offering we completed in 2008, a total of 60% of the investment was made by certain of our existing stockholders with which certain members of our board of directors are affiliated and/or associated; the remaining units were purchased by third party institutional investors on the same terms and conditions. In the offering, entities affiliated with KKR purchased units consisting of 1,328,527 shares of common stock and warrants to purchase 597,837 shares of common stock exercisable at $7.37 per share through July 2014.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Our exposure to market risk is confined to our cash equivalents and restricted cash, all of which have maturities of less than one year and bear interest rates at variable rates and are denominated in, and pay interest in, U.S. dollars. The fair value of items exposed to market risk was $25.0 million and $5.1 million as of December 31, 2010 and 2009, respectively. The goals of our investment policy are liquidity and capital preservation. We limit our credit and liquidity risks through our investment policy and through regular reviews of our portfolio against our policy. Our investment policy allows us to maintain a portfolio of cash equivalents and short-term investments in a variety of securities, including U.S. government agencies, corporate bonds, commercial paper and money market funds. Our cash equivalents and restricted cash as of December 31, 2010 and 2009 consisted primarily of money market funds. The effect of a 100 basis point change in the average yield earned on our cash equivalents and short-term investments would have the effect of increasing our interest income by less than $250,000 and, due to the nature of the investments, would not have had an impact on their fair value.
We pay interest on borrowings under a term loan and revolving credit facility at a variable rate, subject to certain minimums, that was 5.75% in 2010 and is currently 3.75%. The rate is currently variable based on short-term (less than six months maturity) Eurodollar interest rates which would have to increase by between 25-50 basis points for us to avoid paying interest at the minimum 3.75% rate. If rates increase above that minimum rate, each 100 basis point increase in interest rates will cause interest expense in 2011 to increase by approximately $350,000.
Operating expenses and capital expenditures denominated in currencies other than U.S. dollars are insignificant. We receive royalties on certain net product sales that are denominated in other currencies, primarily in Euros, but these royalties comprise a small portion of our revenues.
61
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
Our consolidated financial statements as listed below are attached to this Annual Report on Form 10-K as pages F-1 through F-28.
| Page | ||||
| Jazz Pharmaceuticals, Inc. |
||||
| F-1 | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-6 | ||||
| F-7 | ||||
62
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Not applicable.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
We have carried out an evaluation, under the supervision, and with the participation of, management including our principal executive officer and principal financial officer, of our disclosure controls and procedures (as defined in Rule 13a-15(e)) of the Securities Exchange Act of 1934, as amended, or Exchange Act) as of the end of the period covered by this annual report on Form 10-K. Based on their evaluation, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures were effective as of December 31, 2010.
Limitations on the Effectiveness of Controls. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within an organization have been detected. Accordingly, our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met and, as set forth above, our principal executive officer and principal financial officer have concluded, based on their evaluation as of the end of the period covered by this report, that our disclosure controls and procedures were effective to provide reasonable assurance that the objectives of our disclosure control system were met.
Changes in Internal Control over Financial Reporting
No changes in our internal control over financial reporting occurred during our fiscal quarter ended December 31, 2010 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
The following report is provided by management in respect of Jazz Pharmaceuticals’ internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act):
1. Jazz Pharmaceuticals’ management is responsible for establishing and maintaining adequate internal control over financial reporting.
2. Jazz Pharmaceuticals’ management has used the Committee of Sponsoring Organizations of the Treadway Commission, or the COSO framework, to evaluate the effectiveness of internal control over financial reporting. Management believes that the COSO framework is a suitable framework for its evaluation of financial reporting because it is free from bias, permits reasonably consistent qualitative and quantitative measurements of Jazz Pharmaceuticals’ internal control over financial reporting, is sufficiently complete so that those relevant factors that would alter a conclusion about the effectiveness of Jazz Pharmaceuticals’ internal control over financial reporting are not omitted and is relevant to an evaluation of internal control over financial reporting.
3. Management has assessed the effectiveness of Jazz Pharmaceuticals’ internal control over financial reporting as of December 31, 2010 and has concluded that such internal control over financial reporting was effective. There were no material weaknesses in internal control over financial reporting identified by management.
4. Ernst & Young LLP, our independent registered public accounting firm has audited our consolidated financial statements included herein and has issued an audit report on our internal control over financial reporting which is included below.
63
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of
Jazz Pharmaceuticals, Inc.
We have audited Jazz Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Jazz Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Jazz Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Jazz Pharmaceuticals, Inc. as of December 31, 2010 and 2009 and the related consolidated statements of operations, stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2010 of Jazz Pharmaceuticals, Inc. and our report dated March 8, 2011 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Palo Alto, California
March 8, 2011
64
Table of Contents
| Item 9B. | Other Information |
None.
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K since we intend to file our definitive proxy statement for our 2011 annual meeting of stockholders, pursuant to Regulation 14A of the Securities Exchange Act of 1934, as amended, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information to be included in the proxy statement is incorporated herein by reference.
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information required by this item with respect to our executive officers may be found under the caption, “Executive Officers of the Registrant” in Item 1 of this Annual Report on Form 10-K. The information required by this item relating to our directors and nominees for director may be found under the section entitled “Proposal 1—Election of Directors” in the proxy statement for our 2011 annual meeting of stockholders. Such information is incorporated herein by reference. The information required by this item relating to our audit committee, audit committee financial expert and procedures by which stockholders may recommend nominees to our board of directors, may be found under the section entitled “Corporate Governance and Board Matters” appearing in the proxy statement for our 2011 annual meeting of stockholders. Such information is incorporated herein by reference. Information regarding compliance with Section 16(a) of the Securities Exchange Act of 1934, as amended, may be found under the section entitled “Section 16(a) Beneficial Ownership Reporting Compliance” appearing in our proxy statement for our 2011 annual meeting of stockholders. Such information is incorporated herein by reference.
The Jazz Pharmaceuticals Code of Conduct applies to all officers, directors and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. The Code of Conduct is available on our website at www.jazzpharmaceuticals.com under the section entitled “Company” at “Corporate Responsibility”. Stockholders may request a free copy of the Code of Conduct by submitting a written request to Jazz Pharmaceuticals, Inc., Attention: Investor Relations, 3180 Porter Drive, Palo Alto, California 94304. If we make any substantive amendments to the Code of Conduct or grant any waiver from a provision of the Code of Conduct to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website.
| Item 11. | Executive Compensation |
The information required by this item is included in our proxy statement for our 2011 annual meeting of stockholders under the sections entitled “Executive Compensation,” “Director Compensation,” “Corporate Governance and Board Matters—Compensation Committee Interlocks and Insider Participation” and “Corporate Governance and Board Matters—Compensation Committee Report” and is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item is included in our proxy statement for our 2011 annual meeting of stockholders under the sections entitled “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” and is incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this item is included in our proxy statement for our 2011 annual meeting of stockholders under the sections entitled “Certain Relationships and Related Transactions” and “Corporate Governance and Board Matters—Independence of Jazz Pharmaceuticals’ Board of Directors” and is incorporated herein by reference.
| Item 14. | Principal Accounting Fees and Services |
The information required by this item is incorporated herein by reference to the information included in our proxy statement for our 2011 annual meeting of stockholders under the section entitled “Proposal 2—Ratification of Selection of Independent Registered Public Accounting Firm.”
65
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
(a) The following documents are filed as part of this Annual Report on Form 10-K
| 1. | Index to Financial Statements: |
See Index to Consolidated Financial Statements in Item 8 of this Annual Report on Form 10-K.
| 2. | Financial Statement Schedules: |
The following financial statement schedule of Jazz Pharmaceuticals, Inc. is filed as part of this Annual Report on Form 10-K and should be read in conjunction with the consolidated financials statements of Jazz Pharmaceuticals.
Schedule II: Valuation and Qualifying Accounts
All other schedules are omitted because they are not applicable, not required under the instructions, or the requested information is shown in the consolidated financial statements or related notes thereto.
(b) Exhibits—The following exhibits are included herein or incorporated herein by reference:
| Exhibit Number |
Description of Document | |
| 2.1 | Agreement and Plan of Merger dated as of April 18, 2005, by and among the Registrant, Twist Merger Sub, Inc. and Orphan Medical, Inc. (incorporated by reference to exhibit 2.1 in the Registrant’s registration statement on Form S-1 (File No. 333-141164), as filed with the SEC on March 9, 2007). | |
| 3.1 | Fourth Amended and Restated Certificate of Incorporation of the Registrant (incorporated herein by reference to exhibit 3.1 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2007, as filed with the SEC on August 10, 2007). | |
| 3.2 | Amended and Restated Bylaws (incorporated herein by reference to exhibit 3.4 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 4.1 | Reference is made to Exhibits 3.1 and 3.2. | |
| 4.2 | Specimen Common Stock Certificate (incorporated herein by reference to exhibit 4.2 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 4.3A | Third Amended and Restated Investor Rights Agreement, made effective as of June 6, 2007, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2007, as filed with the SEC on August 10, 2007). | |
| 4.3B | Waiver and Amendment Agreement, dated as of March 12, 2008, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3B in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.3C | Waiver and Amendment Agreement, dated as of May 7, 2008, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3C in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
66
Table of Contents
| Exhibit Number |
Description of Document | |
| 4.3D | Waiver and Amendment Agreement, dated as of July 6, 2009 by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3D in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2009, as filed with the SEC on August 14, 2009). | |
| 4.4A | Form of Series BB Preferred Stock Warrant of the Registrant (incorporated by reference to exhibit 4.6 to the Registrant’s registration statement on Form S-1 (File No. 333-141164), as filed with the SEC on March 9, 2007). | |
| 4.4B | Form of Series BB Preferred Stock Warrant of the Registrant, as amended (incorporated herein by reference to exhibit 4.4B in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.5A | Form of Common Stock Warrant of the Registrant (incorporated herein by reference to exhibit 4.5D in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.5B† | Registration Rights Agreement, dated as of March 17, 2008, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.5E in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.5C | Amendment and Waiver Agreement, dated as of November 10, 2009, by and among the Registrant, JPI Commercial, LLC and the other parties named therein (incorporated by reference to exhibit 4.5F in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on November 10, 2009). | |
| 4.6A | Warrant issued to Kingsbridge Capital Limited, dated May 7, 2008 (incorporated herein by reference to exhibit 4.6A in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 4.6B | Registration Rights Agreement, dated as of May 7, 2008, by and between the Registrant and Kingsbridge Capital Limited (incorporated herein by reference to exhibit 4.6B in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 4.6C | Amendment Agreement No. 1, dated as of November 20, 2009, by and between the Registrant and Kingsbridge Capital Limited (incorporated by reference to exhibit 4.6C in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on November 23, 2009). | |
| 4.7 | Form of Registered Direct Common Stock Warrant (incorporated herein by reference to exhibit 4.7 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 16, 2008). | |
| 4.8 | NOL Preservation Lock-Up Agreement, effective as of July 7, 2009, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.8 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 4.9A | Form of Common Stock Warrant of the Registrant issued on July 7, 2009 (incorporated herein by reference to exhibit 4.9 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 4.9B | Investor Rights Agreement, dated July 7, 2009 by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 10.88 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
67
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.1+ | 2003 Equity Incentive Plan, as amended (incorporated herein by reference to exhibit 10.21 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.2+ | Form of Option Exercise and Stock Purchase Agreement and Forms of Grant Notices under the 2003 Equity Incentive Plan (incorporated herein by reference to exhibit 10.22 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.3+ | 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.23 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.4+ | Form of Option Agreement and Form of Option Grant Notice under the 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.24 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 24, 2007). | |
| 10.5+ | 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.25 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.6+ | Form of Stock Option Agreement and Form of Option Grant Notice under the 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.26 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.7+ | 2007 Employee Stock Purchase Plan (incorporated herein by reference to exhibit 10.27 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.8+ | 2007 Employee Stock Purchase Plan Offering Document (incorporated herein by reference to exhibit 10.28 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.9† | Amended and Restated Xyrem License and Distribution Agreement, dated as of June 30, 2006, by and between the Registrant and UCB Pharma Limited (incorporated herein by reference to exhibit 10.41 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.10† | License Agreement, dated as of January 31, 2007, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.13 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2010, as filed with the SEC on May 6, 2010). | |
| 10.11 | Supply Agreement, dated as of January 31, 2007, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.43 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 24, 2007). | |
| 10.12 | Trademark License Agreement, dated as of January 31, 2007, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.44 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 24, 2007). | |
| 10.13 | Assignment, Assumption and Consent, dated as of January 31, 2007, by and among the Registrant, Solvay Pharmaceuticals, Inc. and Elan Pharma International Limited (incorporated herein by reference to exhibit 10.45 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
68
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.14† | License Agreement, dated as of December 22, 1997, by and between Solvay Pharmaceuticals, Inc. and Elan Corporation, plc. (incorporated herein by reference to exhibit 10.46 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.15† | Amendment to License Agreement, dated as of March 1, 1999, by and between Solvay Pharmaceuticals, Inc. and Elan Corporation, plc. (incorporated herein by reference to exhibit 10.47 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.16† | Letter Amendment No. 2 to License Agreement, dated April 13, 2000, by and between Solvay Pharmaceuticals, Inc and Elan Pharmaceutical Technologies (incorporated herein by reference to exhibit 10.48 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.17† | Amendment Agreement No. 3 to License Agreement, dated as of November 7, 2006, by and between Solvay Pharmaceuticals, Inc. and Elan Corporation plc. (incorporated herein by reference to exhibit 10.49 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.18† | Xyrem Manufacturing Services and Supply Agreement, dated as of March 13, 2007, by and between the Registrant and Patheon Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.50 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.19† | Quality Agreement, dated as of March 13, 2007, by and between the Registrant and Patheon Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.51 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.20 | Commercial Lease, dated as of June 2, 2004, by and between the Registrant and The Board of Trustees of the Leland Stanford Junior University (incorporated herein by reference to exhibit 10.52 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.21A | Civil Settlement Agreement, dated July 13, 2007, among the United States of America acting through the entities named therein, the Registrant and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.57A in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.21B | Non-Prosecution Agreement, dated July 13, 2007, between the United States Attorney’s Office for the Eastern District of New York and the Registrant (incorporated herein by reference to exhibit 10.57B in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.21C | Plea Agreement, dated July 13, 2007, between the United States Attorney for the Eastern District of New York and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.57C in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.21D | Corporate Integrity Agreement, dated July 13, 2007, between the Office of Inspector General of the Department of Health and Human Services and the Registrant (incorporated herein by reference to exhibit 10.57D in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
69
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.22+ | Form of Letter, amending outstanding options granted under the Registrant’s 2003 Equity Incentive Plan (incorporated herein by reference to exhibit 10.60 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2007, as filed with the SEC on August 10, 2007). | |
| 10.23+ | Form of Restricted Stock Unit Award under the Registrant’s 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.64 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2007, as filed with the SEC on November 9, 2007). | |
| 10.24† | Amendment Number 4 to Development, License and Supply Agreement, dated as of October 26, 2007, by and between the Registrant and Elan Pharma International, Inc. (incorporated herein by reference to exhibit 10.66 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 10.25 | Amendment No. 1 to Amended and Restated Xyrem License and Distribution Agreement, dated as of December 21, 2007, by and between the Registrant and UCB Pharma Limited (incorporated herein by reference to exhibit 10.68 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 10.26 | Amendment No. 1 to License Agreement, dated as of March 12, 2008, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.69 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 10.27 | Common Stock Purchase Agreement, dated as of May 7, 2008, by and between the Registrant and Kingsbridge Capital Limited (incorporated herein by reference to exhibit 10.70 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 10.28+ | Form of Stock Award Grant Notice and Stock Award Agreement under the Registrant’s 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.73 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2008, as filed with the SEC on May 15, 2008). | |
| 10.29† | Master Services Agreement dated May 6, 2008, by and among the Registrant, Express Scripts Specialty Distribution Services, Inc. and CuraScript, Inc. (incorporated herein by reference to exhibit 10.74 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2008, as filed with the SEC on May 15, 2008). | |
| 10.30 | Amendment No. 2 to Amended and Restated Xyrem License and Distribution Agreement, dated July 23, 2008, by and between the Registrant and UCB Pharma Limited (incorporated herein by reference to exhibit 10.75 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 24, 2008). | |
| 10.31 | Amendment No. 2 to License Agreement, dated as of October 17, 2008, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.77 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2008, as filed with the SEC on November 14, 2008). | |
| 10.32 | Amendment No. 3 to License Agreement, dated as of December 19, 2008, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.78 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
70
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.33 | Amendment No. 4 to License Agreement, dated as of February 5, 2009, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.79 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.34+ | Amended and Restated Executive Change in Control and Severance Benefit Plan (incorporated herein by reference to exhibit 10.81 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.35 | Revision of Payment Terms of the Plea Agreement dated as of July 17, 2007 between the U.S. Attorney for the Eastern District of New York and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.82 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.36 | Amendment to Settlement Agreement, signed by the Company on February 6, 2009, among the United States of America acting through the entities named therein, the Registrant and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.83 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.37 | Form of Registered Direct Subscription Agreement (incorporated by reference to exhibit 10.1 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 16, 2008). | |
| 10.38 | First Amendment of Lease, dated June 1, 2009, by and between the Registrant and Wheatley-Fields, LLC, successor in interest to the Board of Trustees of the Leland Stanford Junior University (incorporated herein by reference to exhibit 10.86 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on June 4, 2009). | |
| 10.39 | Securities Purchase Agreement, dated July 6, 2009, by and between the Registrant and the purchasers listed on the signature pages thereto (incorporated herein by reference to exhibit 10.87 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 10.40 | Form of Indemnification Agreement between the Registrant and its officers and directors (incorporated herein by reference to exhibit 10.89 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 10.41 | Amendment No. 5 to License Agreement, dated as of June 23, 2009, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.90 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2009, as filed with the SEC on August 14, 2009). | |
| 10.42 | Amendment No. 5 to License Agreement, dated as of October 23, 2009, by and between the Registrant and Elan Pharma International Limited (incorporated by reference to exhibit 10.91 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2009, as filed with the SEC on November 6, 2009). | |
| 10.43 | Offer Letter from the Registrant to Kathryn Falberg (incorporated herein by reference to exhibit 10.92 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on December 3, 2009). | |
| 10.44† | Supply Agreement, dated as of April 1, 2010, by and between the Registrant and Siegfried (USA) Inc. (incorporated herein by reference to exhibit 10.54 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2010, as filed with the SEC on May 6, 2010). | |
71
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.45 | Senior Secured Credit Facilities Credit Agreement, dated as of June 28, 2010, among the Registrant, JPI Commercial, LLC, the several lenders from time to time parties thereto and Silicon Valley Bank, as Administrative Agent (incorporated herein by reference to exhibit 10.56 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 1, 2010). | |
| 10.46+ | Amended and Restated 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.2 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.47+ | Form of Stock Option Agreement and Form of Option Grant Notice under the Amended and Restated 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.1 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.48+ | 2007 Employee Stock Purchase Plan, as amended and restated (incorporated herein by reference to exhibit 10.3 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.49+ | 2007 Employee Stock Purchase Plan Offering Document, as amended and restated (incorporated herein by reference to exhibit 10.4 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.50+ | Amended and Restated Directors Deferred Compensation Plan (incorporated herein by reference to exhibit 10.5 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.51+ | Non-Employee Director Compensation Arrangements, as amended and restated (incorporated herein by reference to exhibit 10.6 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.52 | Amendment No. 1 to Master Services Agreement, dated as of August 31, 2010, by and among the Registrant, Express Scripts Specialty Distribution Services, Inc. and CuraScript, Inc. (incorporated herein by reference to exhibit 10.7 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.53+ | Separation Agreement, dated January 6, 2011, by and between the Registrant and Robert Myers. | |
| 10.54+ | Jazz Pharmaceuticals, Inc. Cash Bonus Plan, as amended as of February 8, 2011. | |
| 10.55+ | 2010 and 2011 Executive Officer Compensation Arrangements. | |
| 21.1 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (included on the signature page hereto). | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | |
| 32.1 | Certifications of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.* | |
| + | Indicates management contract or compensatory plan. |
| † | Confidential treatment has been granted for portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
| * | The certifications attached as Exhibit 32.1 accompany this Annual Report on Form 10-K pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, and shall not be deemed “filed” by the Registrant for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. |
72
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: March 8, 2011 | Jazz Pharmaceuticals, Inc. | |||
| (Registrant) | ||||
| /S/ BRUCE C. COZADD | ||||
| Bruce C. Cozadd | ||||
| Chairman and Chief Executive Officer and Director | ||||
| (Principal Executive Officer) | ||||
| /S/ KATHRYN E. FALBERG | ||||
| Kathryn E. Falberg | ||||
| Senior Vice President and Chief Financial Officer | ||||
| (Principal Financial Officer) | ||||
73
Table of Contents
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Bruce C. Cozadd, Kathryn E. Falberg. and Carol A. Gamble, and each of them, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution for him or her, and in his or her name in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, and any of them, his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, the following persons on behalf of the registrant and in the capacities and on the dates indicated have signed this report below:
| Signature |
Title |
Date | ||
| /s/ BRUCE C. COZADD Bruce C. Cozadd |
Chairman, Chief Executive Officer and Director (Principal Executive Officer) |
March 8, 2011 | ||
| /s/ KATHRYN E. FALBERG Kathryn E. Falberg |
Senior Vice President and Chief Financial Officer (Principal Financial Officer) |
March 8, 2011 | ||
| /s/ JOAN E. COLLIGAN Joan E. Colligan |
Controller and Principal Accounting Officer (Principal Accounting Officer) |
March 8, 2011 | ||
| /s/ Paul L. Berns Paul L. Berns |
Director | March 8, 2011 | ||
| /s/ SAMUEL D. COLELLA Samuel D. Colella |
Director | March 8, 2011 | ||
| /s/ BRYAN C. CRESSEY Bryan C. Cressey |
Director | March 8, 2011 | ||
| /s/ PATRICK G. ENRIGHT Patrick G. Enright |
Director | March 8, 2011 | ||
| /s/ MICHAEL W. MICHELSON Michael W. Michelson |
Director | March 8, 2011 | ||
| /s/ JAMES C. MOMTAZEE James C. Momtazee |
Director | March 8, 2011 | ||
| /s/ KENNETH W. O’KEEFE Kenneth W. O’Keefe |
Director | March 8, 2011 | ||
| /s/ ALAN M. SEBULSKY Alan M. Sebulsky |
Director | March 8, 2011 | ||
| /s/ JAMES B. TANANBAUM, M.D. James B. Tananbaum, M.D. |
Director | March 8, 2011 | ||
| /s/ RICK E WINNINGHAM Rick E Winningham |
Director | March 8, 2011 | ||
| /s/ NATHANIEL M. ZILKHA Nathaniel M. Zilkha |
Director | March 8, 2011 | ||
74
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of
Jazz Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Jazz Pharmaceuticals, Inc. as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2010. Our audits also included the financial statement schedule listed in the Index at Item 15(a)2. These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Jazz Pharmaceuticals, Inc. at December 31, 2010 and 2009, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Jazz Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 8, 2011, expressed an unqualified opinion thereon.
| /s/ Ernst & Young LLP |
Palo Alto, California
March 8, 2011
F-1
Table of Contents
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| ASSETS | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 44,794 | $ | 15,595 | ||||
| Restricted cash |
400 | 2,988 | ||||||
| Accounts receivable, net of allowances of $482 and $288 at December 31, 2010 and 2009, respectively |
22,081 | 12,313 | ||||||
| Inventories |
5,046 | 3,426 | ||||||
| Prepaid expenses |
1,858 | 1,653 | ||||||
| Other current assets |
279 | 979 | ||||||
| Total current assets |
74,458 | 36,954 | ||||||
| Property and equipment, net |
690 | 1,124 | ||||||
| Intangible assets, net |
22,033 | 29,858 | ||||||
| Goodwill |
38,213 | 38,213 | ||||||
| Other long-term assets |
335 | 1,247 | ||||||
| Total assets |
$ | 135,729 | $ | 107,396 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||
| Current liabilities: |
||||||||
| Revolving credit facility |
$ | 7,350 | $ | 9,399 | ||||
| Accounts payable |
3,049 | 2,158 | ||||||
| Accrued liabilities |
23,572 | 14,296 | ||||||
| Current portion of long-term debt (including $1,355 pertaining to a related party at December 31, 2009) |
16,064 | 23,759 | ||||||
| Purchased product rights liability |
4,500 | 4,000 | ||||||
| Liability under government settlement |
4,128 | 2,954 | ||||||
| Deferred revenue |
1,273 | 2,675 | ||||||
| Total current liabilities |
59,936 | 59,241 | ||||||
| Deferred rent |
82 | 29 | ||||||
| Deferred revenue, non-current |
9,053 | 10,191 | ||||||
| Purchased product rights liability, non-current |
4,500 | 9,000 | ||||||
| Liability under government settlement, non-current |
6,978 | 10,658 | ||||||
| Long-term debt, less current portion (including $5,196 pertaining to a related party at December 31, 2009) |
24,629 | 91,107 | ||||||
| Commitments and contingencies (Note 8) |
||||||||
| Stockholders’ equity (deficit): |
||||||||
| Preferred stock, $0.0001 par value; 20,000,000 shares authorized at December 31, 2010; no shares issued and outstanding at December 31, 2010 |
— | — | ||||||
| Common stock, $0.0001 par value; 150,000,000 shares authorized at December 31, 2010; 39,959,255 and 31,255,274 shares issued and outstanding at December 31, 2010 and 2009, respectively |
4 | 3 | ||||||
| Additional paid-in capital |
505,413 | 434,811 | ||||||
| Accumulated deficit |
(474,866 | ) | (507,644 | ) | ||||
| Total stockholders’ equity (deficit) |
30,551 | (72,830 | ) | |||||
| Total liabilities and stockholders’ equity (deficit) |
$ | 135,729 | $ | 107,396 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-2
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenues: |
||||||||||||
| Product sales, net |
$ | 170,006 | $ | 115,108 | $ | 64,637 | ||||||
| Royalties |
2,637 | 2,203 | 1,739 | |||||||||
| Contract revenues |
1,138 | 11,138 | 1,138 | |||||||||
| Total revenues |
173,781 | 128,449 | 67,514 | |||||||||
| Operating expenses: |
||||||||||||
| Cost of product sales (excluding amortization of acquired developed technology and intangible asset impairment) |
13,559 | 9,638 | 13,924 | |||||||||
| Research and development |
25,612 | 36,561 | 69,963 | |||||||||
| Selling, general and administrative |
68,996 | 58,652 | 111,401 | |||||||||
| Intangible asset amortization |
7,825 | 7,668 | 12,828 | |||||||||
| Intangible asset impairment |
— | — | 29,763 | |||||||||
| Total operating expenses |
115,992 | 112,519 | 237,879 | |||||||||
| Income (loss) from operations |
57,789 | 15,930 | (170,365 | ) | ||||||||
| Interest income |
6 | 34 | 1,834 | |||||||||
| Interest expense (including $570, $1,183 and $1,179 for the years ended December 31, 2010, 2009 and 2008, respectively, pertaining to a related party) |
(12,728 | ) | (22,796 | ) | (19,742 | ) | ||||||
| Other (expense) income |
(2 | ) | (4 | ) | 16 | |||||||
| Gain on sale of product rights |
— | — | 3,918 | |||||||||
| Loss on extinguishment of debt (including $701 pertaining to a related party) |
(12,287 | ) | — | — | ||||||||
| Net income (loss) |
$ | 32,778 | $ | (6,836 | ) | $ | (184,339 | ) | ||||
| Net income (loss) per share: |
||||||||||||
| Basic |
$ | 0.90 | $ | (0.23 | ) | $ | (7.19 | ) | ||||
| Diluted |
$ | 0.83 | $ | (0.23 | ) | $ | (7.19 | ) | ||||
| Weighted-average common shares used in computing net income (loss) per share: |
||||||||||||
| Basic |
36,343 | 30,018 | 25,646 | |||||||||
| Diluted |
39,411 | 30,018 | 25,646 | |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Table of Contents
CONSOLIDATED STATEMENTS OF
STOCKHOLDERS’ EQUITY (DEFICIT)
(In thousands, except share amounts)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income |
Accumulated Deficit |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| Balance at December 31, 2007 |
24,620,829 | $ | 2 | $ | 371,440 | $ | 19 | $ | (316,469 | ) | $ | 54,992 | ||||||||||||
| Lapse of repurchase rights to shares issued under restricted stock purchase agreements |
— | — | 30 | — | — | 30 | ||||||||||||||||||
| Warrants to purchase common stock issued in conjunction with senior secured notes |
— | — | 1,928 | — | — | 1,928 | ||||||||||||||||||
| Stock issued/issuable under directors deferred compensation plan |
2,843 | — | 237 | — | — | 237 | ||||||||||||||||||
| Issuance of common stock in conjunction with exercise of stock options for cash and restricted stock units |
153,400 | — | 1,001 | — | — | 1,001 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
299,756 | — | 1,166 | — | — | 1,166 | ||||||||||||||||||
| Issuance of common stock and warrants in conjunction with registered direct public offering, net of issuance costs |
3,848,289 | 1 | 24,513 | — | — | 24,514 | ||||||||||||||||||
| Stock-based compensation |
— | — | 6,859 | — | — | 6,859 | ||||||||||||||||||
| Conversion of common stock subject to repurchase to common stock |
— | — | 749 | — | — | 749 | ||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | — | (184,339 | ) | (184,339 | ) | ||||||||||||||||
| Unrealized loss on available-for-sale securities |
— | — | — | (15 | ) | — | (15 | ) | ||||||||||||||||
| Comprehensive loss |
(184,354 | ) | ||||||||||||||||||||||
| Balance at December 31, 2008 |
28,925,117 | 3 | 407,923 | 4 | (500,808 | ) | (92,878 | ) | ||||||||||||||||
| Lapse of repurchase rights to shares issued under employment agreements |
— | — | 12,492 | — | — | 12,492 | ||||||||||||||||||
| Modification of warrants to purchase common stock issued in conjunction with amended senior secured notes |
— | — | 1,254 | — | — | 1,254 | ||||||||||||||||||
| Stock issued/issuable under directors deferred compensation plan |
3,826 | — | 243 | — | — | 243 | ||||||||||||||||||
| Issuance of common stock in conjunction with exercise of stock options for cash and restricted stock units |
20,722 | — | 40 | — | — | 40 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
409,875 | — | 348 | — | — | 348 | ||||||||||||||||||
| Issuance of common stock and warrants in conjunction with private placement offering, net of issuance costs |
1,895,734 | — | 6,782 | — | — | 6,782 | ||||||||||||||||||
| Stock-based compensation |
— | — | 5,729 | — | — | 5,729 | ||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | — | (6,836 | ) | (6,836 | ) | ||||||||||||||||
| Unrealized loss on available-for-sale securities |
— | — | — | (4 | ) | — | (4 | ) | ||||||||||||||||
| Comprehensive loss |
(6,840 | ) | ||||||||||||||||||||||
| Balance at December 31, 2009 |
31,255,274 | 3 | 434,811 | — | (507,644 | ) | (72,830 | ) | ||||||||||||||||
F-4
Table of Contents
JAZZ PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF
STOCKHOLDERS’ EQUITY (DEFICIT)—(Continued)
(In thousands, except share amounts)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income |
Accumulated Deficit |
Total Stockholders’ Equity (Deficit) |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| Balance at December 31, 2009 |
31,255,274 | 3 | 434,811 | — | (507,644 | ) | (72,830 | ) | ||||||||||||||||
| Stock issuable under directors deferred compensation plan |
— | — | 198 | — | — | 198 | ||||||||||||||||||
| Issuance of common stock in conjunction with exercise of stock options |
955,129 | — | 3,682 | — | — | 3,682 | ||||||||||||||||||
| Issuance of common stock in conjunction with vesting of restricted stock units |
13,398 | — | — | — | — | — | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
519,813 | — | 529 | — | — | 529 | ||||||||||||||||||
| Issuance of common stock in conjunction with offering, net of issuance costs |
7,000,000 | 1 | 56,816 | — | — | 56,817 | ||||||||||||||||||
| Issuance of common stock in conjunction with cashless exercise of warrants |
65,641 | — | — | — | — | — | ||||||||||||||||||
| Issuance of common stock in conjunction with exercise of warrants |
150,000 | — | 1,380 | — | — | 1,380 | ||||||||||||||||||
| Stock-based compensation |
— | — | 7,997 | — | — | 7,997 | ||||||||||||||||||
| Net income and comprehensive income |
— | — | — | — | 32,778 | 32,778 | ||||||||||||||||||
| Balance at December 31, 2010 |
39,959,255 | $ | 4 | $ | 505,413 | $ | — | $ | (474,866 | ) | $ | 30,551 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Operating activities |
||||||||||||
| Net income (loss) |
$ | 32,778 | $ | (6,836 | ) | $ | (184,339 | ) | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: |
||||||||||||
| Depreciation |
886 | 1,429 | 2,198 | |||||||||
| Amortization of intangible assets |
7,825 | 7,668 | 12,828 | |||||||||
| Intangible asset impairment |
— | — | 29,763 | |||||||||
| Loss on disposal of property and equipment |
279 | 14 | 968 | |||||||||
| Stock-based compensation expense |
8,219 | 5,957 | 8,106 | |||||||||
| Long-term debt, non-cash interest expense |
2,406 | 2,810 | 2,060 | |||||||||
| Loss on extinguishment of debt |
12,287 | — | — | |||||||||
| Gain on sale of product rights |
— | — | (3,918 | ) | ||||||||
| Changes in assets and liabilities: |
||||||||||||
| Accounts receivable |
(9,768 | ) | (5,670 | ) | (1,254 | ) | ||||||
| Inventories |
(1,644 | ) | 883 | (2,180 | ) | |||||||
| Prepaid expenses and other current assets |
426 | 2,610 | 237 | |||||||||
| Other assets |
— | (1,748 | ) | (80 | ) | |||||||
| Accounts payable |
891 | (3,578 | ) | 2,880 | ||||||||
| Accrued liabilities |
9,276 | (6,676 | ) | (5,937 | ) | |||||||
| Deferred revenue |
(2,540 | ) | (10,786 | ) | 9,690 | |||||||
| Deferred rent |
53 | 29 | — | |||||||||
| Liability under government settlement |
(2,506 | ) | (1,984 | ) | (1,254 | ) | ||||||
| Net cash provided by (used in) operating activities |
58,868 | (15,878 | ) | (130,232 | ) | |||||||
| Investing activities |
||||||||||||
| Purchases of property and equipment |
(731 | ) | (53 | ) | (1,739 | ) | ||||||
| Purchase of product rights |
(4,000 | ) | (6,000 | ) | (27,000 | ) | ||||||
| Decrease (increase) in restricted cash and investments |
2,588 | (1,075 | ) | 12,026 | ||||||||
| Transfer of restricted cash to marketable securities |
— | — | (4,440 | ) | ||||||||
| Proceeds from maturities of marketable securities |
— | 1,004 | 3,436 | |||||||||
| Proceeds from sale of product rights |
— | — | 5,775 | |||||||||
| Net cash used in investing activities |
(2,143 | ) | (6,124 | ) | (11,942 | ) | ||||||
| Financing activities |
||||||||||||
| Repayment of senior secured notes (including $6,816 and $327 for the years ended December 31, 2010 and 2008, respectively, paid to a related party) |
(119,496 | ) | — | (504 | ) | |||||||
| Prepayment penalties and fees (including $484 paid to a related party) |
(8,484 | ) | — | — | ||||||||
| Proceeds from offerings of common stock, net of issuance costs |
56,817 | 6,782 | 24,514 | |||||||||
| Proceeds from term loan, net |
48,427 | — | — | |||||||||
| Repayment of term loan |
(8,332 | ) | — | — | ||||||||
| Proceeds from employee stock purchases and exercise of stock options and warrants |
5,591 | 388 | 1,168 | |||||||||
| Net (repayments under) proceeds from revolving credit facilities |
(2,049 | ) | 5,524 | 416 | ||||||||
| Proceeds from sale of senior secured notes and warrants, net of issuance costs |
— | — | 38,538 | |||||||||
| Net cash (used in) provided by financing activities |
(27,526 | ) | 12,694 | 64,132 | ||||||||
| Net increase (decrease) in cash and cash equivalents |
29,199 | (9,308 | ) | (78,042 | ) | |||||||
| Cash and cash equivalents, at beginning of period |
15,595 | 24,903 | 102,945 | |||||||||
| Cash and cash equivalents, at end of period |
$ | 44,794 | $ | 15,595 | $ | 24,903 | ||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid for interest (including $461, $1,349 and $796 for the years ended December 31, 2010, 2009 and 2008, respectively, paid to a related party) |
$ | 10,234 | $ | 24,488 | $ | 12,802 | ||||||
| Supplemental disclosure of non-cash investing and financing activities: |
||||||||||||
| Liability for purchase of product rights |
$ | — | $ | 5,000 | $ | 14,000 | ||||||
| Warrants to purchase common stock |
$ | — | $ | 2,700 | $ | 9,250 | ||||||
| Modification to warrants to purchase common stock issued in conjunction with senior secured notes |
$ | — | $ | 1,254 | $ | — | ||||||
The accompanying notes are an integral part of these consolidated financial statements
F-6
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Description of Business
We are a specialty pharmaceutical company focused on the identification, development and commercialization of pharmaceutical products to meet important unmet medical needs. Since we were founded in 2003, we have built a commercial and development organization and assembled a portfolio of products and product candidates that currently includes our two marketed products, Xyrem (sodium oxybate) oral solution and Luvox CR (fluvoxamine maleate) Extended-Release Capsules, and product candidates in various stages of clinical development.
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements include the accounts of Jazz Pharmaceuticals, Inc. and its wholly-owned subsidiaries, Orphan Medical, LLC, formerly Orphan Medical, Inc., or Orphan Medical, and JPI Commercial, LLC after elimination of intercompany transactions and balances. Our fiscal year ends on December 31.
Certain amounts in the consolidated statements of cash flows for 2009 and 2008 have been reclassified to conform to the presentation for 2010. Amounts previously reported as the changes in senior secured notes have been reclassified and reported as non-cash interest expense and changes in other assets and accrued liabilities in the consolidated statements of cash flows.
Significant Risks and Uncertainties
We are subject to risks common to companies in the pharmaceutical industry with development and commercial operations including, but not limited to, risks and uncertainties related to commercial success and acceptance of our products by patients, physicians and payors, competition from branded and generic products, regulatory approvals, regulatory requirements, including those of the United States Food and Drug Administration, or FDA, and the United States Drug Enforcement Administration dependence on key customers and sole source suppliers and protection of intellectual property rights. In addition, most of our revenues are derived from sales of one product, Xyrem. During 2010, an abbreviated new drug application, or ANDA, was filed with the FDA by a third party seeking to market a generic form of Xyrem. We have sued that third party for infringement of our patents, and the litigation is ongoing. We cannot predict the timing or outcome of this litigation. If an ANDA for Xyrem is approved and a generic version of Xyrem is introduced, our sales of Xyrem would be adversely affected.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts and disclosures reported in the consolidated financial statements and accompanying notes. Management bases its estimates on historical experience and on assumptions believed to be reasonable under the circumstances. Actual results could differ materially from those estimates.
Concentrations of Risk
Financial instruments that potentially subject us to concentrations of credit risk consist of cash equivalents and restricted cash, and accounts receivable. Our investment policy limits investments to certain types of debt securities issued by the U.S. government, its agencies and institutions with investment-grade credit ratings and
F-7
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
places restrictions on maturities and concentration by type and issuer. We are exposed to credit risk in the event of a default by the financial institutions holding our cash and cash equivalents and issuers of investments to the extent recorded on the balance sheet.
We monitor our exposure within accounts receivable and record a reserve against uncollectible accounts receivable as necessary. We extend credit to pharmaceutical wholesale distributors and a specialty pharmaceutical distribution company, primarily in the United States, and to international distributors in the normal course of business. Customer creditworthiness is monitored and collateral is not required. Historically, we have not experienced significant credit losses on our accounts receivable. One customer, Express Scripts Specialty Distribution Services and its affiliate Curascript, Inc., or Express Scripts, accounted for 79% and 77% of gross accounts receivable as of December 31, 2010 and 2009, respectively.
We rely on certain sole suppliers for drug substance and certain sole manufacturing partners for each of our marketed products and certain of our product candidates.
Cash Equivalents and Restricted Cash
We consider all highly liquid investments, readily convertible to cash, that mature within three months or less from date of purchase to be cash equivalents. At December 31, 2010, restricted cash was in the form of a certificate of deposit required to secure spending on credit cards used by employees.
Cash equivalents and restricted cash are considered available-for-sale and are recorded at fair value, based on quoted market prices. Unrealized gains and losses, net of tax, are recorded in other comprehensive income (loss) and included as a separate component of stockholders’ equity (deficit). We use the specific-identification method for calculating realized gains and losses on securities sold.
Inventories
Inventories are valued at the lower of cost or market. Cost is determined using the first-in, first-out method for all inventories. Our policy is to write down inventory that has become obsolete, inventory that has a cost basis in excess of its expected net realizable value and inventory in excess of expected requirements. The estimate of excess quantities is subjective and primarily dependent on our estimates of future demand for a particular product. If the estimate of future demand is too high, we may have to increase the reserve for excess inventory for that product and record a charge to cost of product sales. For product candidates that have not been approved by the FDA, inventory used in clinical trials is expensed at the time of production and recorded as research and development expense. For products that have been approved by the FDA, inventory used in clinical trials is expensed at the time the inventory is packaged for the clinical trial. Prior to receiving FDA approval costs related to purchases of the active pharmaceutical ingredient and the manufacturing of the product candidate are recorded as research and development expense. All direct manufacturing costs incurred after approval are capitalized into inventory.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, which are three to five years. Leasehold improvements are amortized over the shorter of the noncancelable term of our operating lease or their economic useful lives. Maintenance and repairs are charged to operations as incurred.
F-8
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Goodwill and Intangible Assets
Goodwill
Goodwill represents the excess of the purchase price over the fair value of assets acquired and liabilities assumed. We have determined that we operate in a single segment and have a single reporting unit associated with the development and commercialization of pharmaceutical products. The annual test for goodwill impairment is a two-step process. The first step is a comparison of the fair value of the reporting unit with its carrying amount, including goodwill. If this step indicates impairment, then in the second step, the loss is measured as the excess of recorded goodwill over its implied fair value. Implied fair value is the excess of the fair value of the reporting unit over the fair value of all identified assets and liabilities. Management tests goodwill for impairment annually in October and whenever events or changes in circumstances indicate that the carrying value may not be recoverable.
Intangible Assets
Intangible assets consist primarily of purchased developed technology and trademarks. Intangible assets are amortized on a straight-line basis over their estimated useful lives, which range from three to ten years. The estimated useful lives associated with intangible assets are consistent with the estimated lives of the products and may be modified when circumstances warrant. Once an intangible asset is fully amortized, the gross costs and accumulated amortization are removed from the consolidated balance sheet. We evaluate purchased intangibles and other long-lived assets, other than goodwill, for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. The amount of any impairment is measured as the difference between the carrying value and the fair value of the impaired asset. See Note 5 for additional information regarding intangible asset impairment charges.
Revenue Recognition
Revenues are recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed and determinable and collection is reasonably assured. Revenue from sales transactions where the buyer has the right to return the product is recognized at the time of sale only if (i) the seller’s price to the buyer is substantially fixed or determinable at the date of sale, (ii) the buyer has paid the seller, or the buyer is obligated to pay the seller and the obligation is not contingent on resale of the product, (iii) the buyer’s obligation to the seller would not be changed in the event of theft or physical destruction or damage of the product, (iv) the buyer acquiring the product for resale has economic substance apart from that provided by the seller, (v) the seller does not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (vi) the amount of future returns can be reasonably estimated.
In evaluating arrangements with multiple elements we consider whether components of the arrangement represent separate units of accounting based upon whether certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. This evaluation requires subjective determinations and requires management to make judgments about the fair value of individual elements and whether such elements are separable from other aspects of the contractual relationship. The consideration received in such arrangements is allocated among the separate units of accounting based on their respective fair values when there is reliable evidence of fair value for all elements of the arrangement. If there is no evidence of fair value for all the elements of the arrangement, consideration is allocated based on the residual value method for the delivered elements.
F-9
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Under the residual method, the amount of revenues allocated to the delivered elements equals the total arrangement consideration less the aggregate fair value of any undelivered elements. The applicable revenue recognition criteria are applied to each of the separate units.
Payments received in advance of work performed or milestones achieved are recorded as deferred revenues and recognized when the service is provided or the milestone is achieved, as applicable.
Product Sales, Net
We sell Xyrem in the United States to a single central pharmacy, Express Scripts. We recognize revenues from sales of Xyrem within the United States upon transfer of title, which occurs when Express Scripts removes product from our consigned inventory location at its facility for shipment directly to a patient. We accept returns from Express Scripts of any product returned by patients to Express Scripts with defects that were not reasonably discoverable upon receipt of the consigned product by Express Scripts. Based on our experience over the past five years since we acquired the rights to Xyrem, product returns to Express Scripts from patients are rare. We provide Express Scripts with a credit for product returned by patients. During 2010, we issued credits for returned product totaling less than $20,000.
We sell limited quantities of Xyrem to UCB Pharma Limited, or UCB, for sale in territories outside of North America, and to Valeant Canada Limited, for sale in Canada, under license and distribution agreements. The agreements provide our international licensees with a fixed period of time after delivery to inspect and reject shipments for failure to meet specifications. We do not recognize revenue on the sales to our international licensees until the right of return has lapsed, which occurs when we are notified of their acceptance, or when the time for them to inspect or reject a shipment has lapsed, if earlier.
We grant rights to our wholesaler customers to return product six months prior to and up to twelve months after product expiration and issue credits which may be applied against existing or future invoices. Prior to the fourth quarter of 2010, we did not believe we were able to reasonably estimate expected returns of Luvox CR at the time of shipment, and therefore we recognized revenue when units were dispensed through prescriptions, at which point the product was not subject to return. We purchase dispensing data from an independent prescription tracking service which we used to estimate units dispensed. As of October 1, 2010 we believed we had sufficient historical data on returns of Luvox CR to reasonably estimate a return rate when a unit is shipped. As a result, as of October 1, 2010, we started recognizing revenue upon shipment to our wholesaler customers and recorded an estimated amount of product returns. We recognized $2.0 million of previously deferred net product sales and $674,000 of previously deferred product costs in the fourth quarter of 2010. We recorded a $3.5 million liability for estimated future returns as of December 31, 2010.
Revenues from sales of products within the United States are recorded net of estimated allowances for returns, specialty distributor fees, wholesaler fees, prompt payment discounts, government rebates, government chargebacks, patient rebates and rebates under managed care plans. Calculating certain of these items involves estimates and judgments based on sales or invoice data and historical experience. Adjustments to estimates for these allowances have not been material.
Royalties, Net
We receive royalties from third parties based on sales of our products under licensing and distribution arrangements. For those arrangements where royalties are reasonably estimable, we recognize revenues based on estimates of royalties earned during the applicable period, and adjusts for differences between the estimated and actual royalties in the following quarter. Historically, these adjustments have not been significant.
F-10
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Contract Revenues
Nonrefundable fees where we have no continuing performance obligations are recognized as revenues when there is persuasive evidence of an arrangement and collection is reasonably assured. In situations where we have continuing performance obligations, nonrefundable fees are deferred and are recognized ratably over our projected performance period. We recognize at-risk milestone payments, which are typically related to regulatory, commercial or other achievements by us or our licensees and distributors, as revenues when the milestone is accomplished and collection is reasonably assured. Refundable fees are deferred and recognized as revenues upon the later of when they become nonrefundable or when our performance obligations are completed.
Cost of Product Sales
Cost of product sales includes third party manufacturing and distribution costs, the cost of drug substance, royalties due to third parties on product sales, product liability and cargo insurance, FDA user fees, freight, shipping, handling and storage costs and salaries and related costs of employees involved with production. During 2009 and 2008, we recorded charges to cost of product sales related to Luvox CR for inventory we judged to be in excess of expected requirements of $82,000 and $4.2 million, respectively. Excluded from cost of product sales, as shown on the consolidated statements of operations, is amortization of acquired developed technology of $7.2 million, $6.6 million and $11.5 million for 2010, 2009 and 2008, respectively. Also excluded from cost of product sales is an intangible asset impairment charge of $29.8 million related to Luvox CR recorded in 2008. See Note 5 for additional information regarding the impairment charge.
Research and Development
Research and development expenses consist of expenses incurred in identifying, developing and testing our product candidates. These expenses consist primarily of fees paid to contract research organizations and other third parties to assist us in managing, monitoring and analyzing results from our clinical trials, clinical trial costs paid to sites and investigators’ fees, costs of non-clinical studies, including toxicity studies in animals, costs of contract manufacturing services, costs of materials used in clinical trials and non-clinical studies, fees paid to third parties for development candidates or drug delivery or formulation technologies that we have licensed, allocated expenses, such as facilities and information technology that support our research and development activities, and related personnel expenses, including stock-based compensation. Research and development costs are expensed as incurred, including payments made under our license agreements. For product candidates that have not been approved by the FDA, inventory used in clinical trials is expensed at the time of production and recorded as research and development expense. For products that have been approved by the FDA, inventory used in clinical trials is expensed at the time the inventory is packaged for the trial and therefore is not included in inventory.
Advertising Expenses
We expense the costs of advertising, including promotional expenses, as incurred. Advertising expenses for 2010, 2009 and 2008 were $1.6 million, $448,000 and $11.0 million, respectively.
Income Taxes
We utilize the liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and the tax bases of assets and liabilities and are measured using enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
F-11
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Comprehensive Income (Loss)
Comprehensive income (loss) includes net income (loss) and all changes in stockholders’ equity (deficit) during a period, except for those changes resulting from investments by stockholders or distributions to stockholders. For each 2010, 2009 and 2008, the difference between comprehensive income (loss) and net income (loss) was insignificant.
Net Income (Loss) Per Common Share
Basic and diluted net income (loss) per common share is computed using the weighted-average number of shares of common stock outstanding as follows (in thousands, except per share amounts):
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Numerator: |
||||||||||||
| Net income (loss) |
$ | 32,778 | $ | (6,836 | ) | $ | (184,339 | ) | ||||
| Denominator: |
||||||||||||
| Weighted-average common shares outstanding |
36,343 | 30,018 | 26,524 | |||||||||
| Less: weighted-average common shares outstanding subject to repurchase |
— | — | (878 | ) | ||||||||
| Weighted-average common shares outstanding—basic |
36,343 | 30,018 | 25,646 | |||||||||
| Dilutive effect of employee equity incentive and purchase plans |
1,720 | — | — | |||||||||
| Dilutive effect of warrants |
1,348 | — | — | |||||||||
| Weighted-average common shares outstanding—diluted |
39,411 | 30,018 | 25,646 | |||||||||
| Net income (loss) per share: |
||||||||||||
| Basic |
$ | 0.90 | $ | (0.23 | ) | $ | (7.19 | ) | ||||
| Diluted |
$ | 0.83 | $ | (0.23 | ) | $ | (7.19 | ) | ||||
Potentially dilutive securities consisting of stock options, common stock subject to repurchase and warrants were not included in the diluted net loss per share for 2009 and 2008 because the inclusion of such shares would have had an anti-dilutive effect.
Potentially dilutive common shares from employee stock plans and warrants are determined by applying the treasury stock method to the assumed exercise of warrants and stock options, the assumed vesting of outstanding restricted stock units, and the assumed issuance of common stock under our employee stock purchase plan. The following table represents the weighted-average shares of our common stock that were excluded from the computation of diluted net income (loss) per share for the periods presented because including them would have an anti-dilutive effect (in thousands):
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Warrants to purchase common stock |
— | 3,759 | 2,144 | |||||||||
| Options to purchase common stock |
3,211 | 2,843 | 3,687 | |||||||||
| Common stock subject to repurchase |
— | — | 828 | |||||||||
| Restricted stock units |
— | 38 | 94 | |||||||||
| Total |
3,211 | 6,640 | 6,753 | |||||||||
F-12
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
As of December 31, 2010, we had warrants outstanding and exercisable as follows:
| No. of Shares |
Expiration Date | Exercise Price |
||||||||||
| Warrants issued in conjunction with: |
||||||||||||
| $80.0 million senior secured notes |
785,728 | June 2012 | $ | 9.34 | ||||||||
| $40.0 million senior secured notes |
562,192 | March 2013 | $ | 9.34 | ||||||||
| Equity financing facility |
70,000 | November 2013 | $ | 9.20 | ||||||||
| Public offering |
1,620,119 | July 2014 | $ | 7.37 | ||||||||
| Private offering |
947,867 | July 2016 | $ | 4.00 | ||||||||
Stock-Based Compensation
We account for compensation cost for all stock-based awards at fair value on the date of grant. The fair value is recognized as expense over the service period, net of estimated forfeitures, using the straight-line method for stock options and restricted stock units and using the ratable method for awards under our employee stock purchase program. The estimation of stock awards that will ultimately vest requires judgment, and to the extent actual results or updated estimates differ from current estimates, such amounts will be recorded as a cumulative adjustment in the period estimates are revised. We primarily consider historical experience when estimating expected forfeitures.
Recent Accounting Pronouncements
In October 2009, the FASB issued authoritative guidance which amends the revenue recognition guidance to require companies to allocate revenue in multiple-element arrangements based on an element’s estimated selling price if vendor-specific or other third-party evidence is not available. The guidance became effective for us beginning January 1, 2011 and is being applied prospectively to multiple-deliverable revenue arrangements entered into on or after January 1, 2011. The adoption of this guidance is not expected to have a material impact on our results of operations and financial position.
3. Fair Value Measurement
Available-for-sale investments consisted of the following (in thousands):
| December 31, 2010 | December 31, 2009 | |||||||||||||||
| Amortized Cost |
Estimated Fair Value |
Amortized Cost |
Estimated Fair Value |
|||||||||||||
| Money market funds |
$ | 25,046 | $ | 25,046 | $ | 5,072 | $ | 5,072 | ||||||||
| December 31, 2010 |
December 31, 2009 |
|||||||||||||||
| Available-for-sale investments |
$ | 25,046 | $ | 5,072 | ||||||||||||
| Cash |
19,748 | 10,523 | ||||||||||||||
| Restricted cash |
400 | 2,988 | ||||||||||||||
| Total |
$ | 45,194 | $ | 18,583 | ||||||||||||
| Reported as |
December 31, 2010 |
December 31, 2009 |
||||||||||||||
| Amounts classified as cash and cash equivalents |
$ | 44,794 | $ | 15,595 | ||||||||||||
| Amounts classified as restricted cash |
400 | 2,988 | ||||||||||||||
| Total |
$ | 45,194 | $ | 18,583 | ||||||||||||
F-13
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following table summarizes, by major security type, our available-for-sale investments that are measured at fair value on a recurring basis and are categorized using the fair value hierarchy (in thousands):
| December 31, 2010 | December 31, 2009 | |||||||
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Total Estimated Fair Value |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Total Estimated Fair Value | |||||
| Money market funds | $25,046 | $25,046 | $5,072 | $5,072 | ||||
As of December 31, 2010 and 2009, the carrying amount of our long-term debt was $40.7 million and $114.9 million, respectively, and the estimated fair value was $40.9 million and $123.6 million, respectively. The fair value was estimated using a discounted cash flow analysis based on our estimated incremental borrowing rates for similar types of borrowing arrangements.
4. Certain Balance Sheet Items
Inventories consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Raw materials |
$ | 2,986 | $ | 1,245 | ||||
| Work in process |
705 | 676 | ||||||
| Finished goods |
1,355 | 1,505 | ||||||
| Total inventories |
$ | 5,046 | $ | 3,426 | ||||
Property and equipment consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Leasehold improvements |
$ | 763 | $ | 704 | ||||
| Computer equipment |
1,483 | 1,479 | ||||||
| Computer software |
4,010 | 3,715 | ||||||
| Furniture and fixtures |
593 | 586 | ||||||
| Construction-in-progress |
73 | 28 | ||||||
| Total |
6,922 | 6,512 | ||||||
| Less accumulated depreciation and amortization |
(6,232 | ) | (5,388 | ) | ||||
| Property and equipment, net |
$ | 690 | $ | 1,124 | ||||
Accrued liabilities consisted of the following (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accrued research and development expense |
$ | 1,449 | $ | 2,862 | ||||
| Accrued personnel expense |
8,060 | 6,545 | ||||||
| Accrued selling, general and administrative expense |
1,598 | 891 | ||||||
| Sales returns reserves |
3,539 | — | ||||||
| Government rebates reserve |
6,588 | 2,270 | ||||||
| Other |
2,338 | 1,728 | ||||||
| Total accrued liabilities |
$ | 23,572 | $ | 14,296 | ||||
F-14
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
5. Goodwill and Intangible Assets
The gross carrying amount of goodwill was as follows (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Goodwill |
$ | 38,213 | $ | 38,213 | ||||
The gross carrying amounts and net book values of our intangible assets were as follows (in thousands):
| December 31, 2010 | December 31, 2009 | |||||||||||||||||||||||
| Gross Carrying Amount |
Accumulated Amortization |
Net Book Value |
Gross Carrying Amount |
Accumulated Amortization |
Net Book Value |
|||||||||||||||||||
| Developed technology—Xyrem |
$ | 39,700 | $ | 23,014 | $ | 16,686 | $ | 39,700 | $ | 18,842 | $ | 20,858 | ||||||||||||
| Developed technology—Luvox CR |
9,700 | 5,446 | 4,254 | 9,700 | 2,443 | 7,257 | ||||||||||||||||||
| Agreements not to compete |
— | — | — | 3,900 | 3,523 | 377 | ||||||||||||||||||
| Trademarks |
2,600 | 1,507 | 1,093 | 2,600 | 1,234 | 1,366 | ||||||||||||||||||
| Total |
$ | 52,000 | $ | 29,967 | $ | 22,033 | $ | 55,900 | $ | 26,042 | $ | 29,858 | ||||||||||||
Based on intangible assets recorded as of December 31, 2010, and assuming the underlying assets will not be impaired in the future and that we will not change the expected lives of the assets, future amortization costs were estimated as follows (in thousands):
| Year Ending December 31, |
Estimated Amortization Expense |
|||
| 2011 |
$ | 7,448 | ||
| 2012 |
5,696 | |||
| 2013 |
4,445 | |||
| 2014 |
4,444 | |||
| $ | 22,033 | |||
In 2009, we amended our product license agreement with Solvay Pharmaceuticals, Inc., or Solvay, which was subsequently acquired by Abbott Laboratories, or Abbott, for the rights to market Luvox CR and Luvox in the United States such that the existing $14.0 million current payment obligation, a $5.0 million obligation related to a milestone of uninterrupted supply of Luvox CR and future royalty and other obligations were replaced with an obligation to pay a total of $19.0 million. As a result, we recorded an increase of $5.0 million in the value of the intangible asset associated with Luvox CR in 2009.
In 2008, as a result of lower than anticipated sales of Luvox CR, we evaluated the intangible asset associated with Luvox CR for impairment and reduced the gross carrying amount and accumulated amortization of this intangible asset by $36.3 million and $6.5 million, respectively, which resulted in a $29.8 million intangible asset impairment charge. The most significant input used in the calculation of the fair value of the intangible asset associated with Luvox CR was projected net sales of Luvox CR which were estimated by extrapolating the current growth trends of the product and applying judgment as to the appropriate future growth rate among other factors. Selection of a risk appropriate discount rate also involves significant judgment. We used a discount rate of 20% to estimate fair value in 2008.
F-15
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
6. Debt and Financing Obligations
Retired Senior Secured Notes
In March 2008, we sold $40.0 million aggregate principal amount of senior secured notes and issued warrants to purchase 562,192 shares of our common stock with an exercise price of $14.23 per share and which expire in March 2013. The $2.0 million fair value of the warrants was recorded in stockholders’ deficit and was estimated using the Black-Scholes option pricing model with the following assumptions: a risk-free rate 2.2%, volatility 51%, a term of 5.0 years and a dividend yield of 0.0%. The senior secured notes bore interest at 15% per annum, payable quarterly in arrears, and were due on June 24, 2011. In addition, in 2008, a total of $80.0 million aggregate principal amount of senior secured notes of Orphan Medical issued in 2005 that bore interest at 15% per annum, due on June 24, 2011 were exchanged for the same principal amount of notes issued by JPI Commercial, LLC. As a result of these transactions, a total of $120.0 million aggregate principal amount of senior secured notes, or Senior Notes, was outstanding. We refer to the agreement that governed all of the Senior Notes as the Senior Note Agreement.
In August 2008, we paid certain holders of the Senior Notes $504,000 aggregate principal amount as their pro rata share of the proceeds from the sale of our rights to Antizol® and Antizol-Vet®.
In 2009, we amended the Senior Note Agreement. In connection with the amendment, amongst other changes, we reduced the exercise price of warrants to purchase 1,347,920 shares of common stock, originally issued in conjunction with the Senior Notes, to $9.34 per share. We determined that the amendment should be accounted for as a modification of the existing Senior Notes. The $1.3 million fair value of the warrant modification was recorded as a debt discount and in stockholders’ deficit. The fair value was estimated using the Black-Scholes option pricing model with the following assumptions; risk-free rates of 1.2 and 1.6%, volatility of 90%, expected terms of 2.6 and 3.3 years, and a dividend yield of 0.0%.
As of December 31, 2009, the $119.5 million principal amount of the Senior Notes was recorded net of a debt discount of $4.6 million. Interest expense associated with the Senior Notes was recorded using the interest method and included non-cash interest related to the debt discount and debt issuance costs. The effective interest rate on the Senior Notes subsequent to the amendment to the Senior Note Agreement in 2009 was 21.2%.
In March, May and June 2010, we repaid $3.0 million, $53.0 million and $63.5 million principal amount of the Senior Notes, respectively, thereby paying in full our obligations to the holders of the Senior Notes. In addition to the principal repayments in May and June 2010, we paid prepayment penalties and fees totaling $8.5 million, and recorded non-cash charges related to unamortized debt discount and debt issuance costs of $3.8 million in 2010.
Term Loan and Revolving Credit Facility
In June 2010, we entered into a credit agreement with a lender which provides for a term loan in an aggregate principal amount of $50.0 million and a $15.0 million revolving credit facility, both of which mature in June 2013. On June 30, 2010, we borrowed $57.4 million under the credit agreement, consisting of the term loan of $50.0 million and $7.4 million under the revolving credit facility, and we used all of the borrowed funds, together with cash on hand, to repay all of the remaining outstanding Senior Notes. We also terminated our previous revolving line of credit. Borrowings under the term loan and revolving credit facility bear interest at a variable rate based on the higher of the prime rate or the federal funds rate plus 0.5% plus, in each case, a margin ranging from 1% to 2.5% or, at our option, the Eurodollar rate plus a margin ranging from 3% to 5%. The revolving credit facility has a commitment fee payable on the undrawn amount ranging from 0.5% to 0.75% per annum. The interest rate margins and the commitment fee will vary based on our consolidated leverage ratio, as defined in the credit agreement.
F-16
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The borrowing availability under the revolving credit facility will vary according to the levels of our eligible accounts receivable and other terms and conditions described in the credit agreement and was limited to $8.0 million as of December 31, 2010, including a $650,000 unused letter of credit related to automobiles leased by our sales organization, and $15.0 million thereafter. Borrowings under the revolving credit facility and the term loan are secured by substantially all of our assets. The term loan is repayable in twelve equal quarterly installments of $4.2 million beginning on September 30, 2010. If we prepay the term loan (in whole or in part), or if we terminate or reduce the lender’s commitments to make loans under the revolving credit facility, we must pay a prepayment fee equal to (a) 2% of the aggregate amount of the term loan prepaid or commitments terminated or reduced during the first year of the credit agreement, and (b) 1% of the aggregate amount of the term loan prepaid or commitments terminated or reduced during the second year of the credit agreement.
The credit agreement contains customary operating covenants, including covenants that restrict our ability to: incur indebtedness and liens; effect mergers, consolidations and other fundamental changes; dispose of significant assets or enter into sale-leaseback transactions; pay dividends or make other restricted payments; make loans, advances or certain investments including acquisitions of companies and products; or enter into transactions with affiliates. The credit agreement also requires us to comply with various financial covenants including a minimum liquidity covenant, which requires us to maintain cash and availability under the revolving line of credit of not less than $10.0 million until March 31, 2011 and not less than $20.0 million thereafter. As of December 31, 2010, we were in compliance with all material covenants under the credit agreement.
As of December 31, 2010, the $41.7 million principal amount of the term loan was recorded net of a debt discount of $1.0 million related to fees paid to the lender under the credit agreement as of December 31, 2010. As of December 31, 2010, the interest rate on the term loan was 5.75%. Interest expense associated with the term loan is recorded using the interest method and includes non-cash interest related to the debt discount and debt issuance costs. The effective interest rate on the term loan during 2010 was 7.8%. The current portion of the carrying amount of the term loan was $16.1 million as of December 31, 2010.
As of December 31, 2010, $7.4 million was outstanding under the revolving credit facility, which bore interest at 5.75%. As of December 31, 2009, $9.4 million was outstanding under our previous revolving bank line of credit, which bore interest at 6.5%.
7. Other Long Term Liabilities
Deferred Revenue
We have an agreement with UCB under which UCB has the right to market Xyrem for the treatment of narcolepsy and for the treatment of fibromyalgia in various countries outside the United States. In 2008, we received a $10.0 million nonrefundable milestone payment received, which we recognized as revenue in 2009 upon achievement of the related milestone. We recognized contract revenues of $1.1 million during each of 2010, 2009, and 2008 related to two upfront payments from UCB totaling $15.0 million related to Xyrem for the treatment of fibromyalgia. As of December 31, 2010, $10.2 million was recorded as deferred revenues related to this agreement, of which $1.1 million is a current liability. The deferred revenue balance is being recognized ratably through 2019, the end of the expected performance period under the agreement.
Purchased Product Rights Liability
In 2007, we entered into a product license agreement with Solvay for the rights to market Luvox CR and Luvox in the United States which agreement was subsequently amended a number of times. Under the amended agreement we paid $4.0 million, $6.0 million and $27.0 million in 2010, 2009 and 2008, respectively, and will pay $4.5 million in each of 2011 and 2012.
F-17
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Liability Under Government Litigation Settlement
In 2007, we and Orphan Medical entered into agreements with a number of government entities to settle various matters associated with an investigation relating to the sale and marketing of Xyrem by Orphan Medical, which we acquired in June 2005. Under these agreements we paid $3.0 million, $2.5 million, and $2.0 million in 2010, 2009 and 2008, respectively, and as of December 31, 2010, we owe $4.2 million and $7.3 million in 2011 and 2012, respectively.
8. Commitments and Contingencies
Indemnification
In the normal course of business, we enter into agreements that contain a variety of representations and warranties and provide for general indemnification, including indemnification associated with product liability or infringement of intellectual property rights. Our exposure under these agreements is unknown because it involves future claims that may be made but have not yet been made against us. To date, we have not paid any claims or been required to defend any action related to these indemnification obligations.
We have agreed to indemnify our officers, directors and certain other employees for losses and costs incurred in connection with certain events or occurrences, including advancing money to cover certain costs, subject to certain limitations. The maximum potential amount of future payments we could be required to make under the indemnification obligations is unlimited; however, we maintain insurance policies that may limit our exposure and may enable us to recover a portion of any future amounts paid. Assuming the applicability of coverage, the willingness of the insurer to assume coverage, and subject to certain retention, loss limits and other policy provisions, we believe the fair value of these indemnification obligations is not significant. Accordingly, we have not recognized any liabilities relating to these obligations as of December 31, 2010 and 2009, respectively. No assurances can be given that the covering insurers will not attempt to dispute the validity, applicability, or amount of coverage without expensive litigation against these insurers, in which case we may incur substantial liabilities as a result of these indemnification obligations.
Lease and Other Commitments
We have a noncancelable operating lease for our corporate office building located in Palo Alto, California which expires in September 2012, is renewable through 2016 and is subject to an annual rent escalation clause. We are also obligated to make payments under noncancelable operating leases for automobiles used by our sales force. Rent expense under all operating leases was $2.3 million, $2.7 million and $5.2 million in 2010, 2009 and 2008, respectively.
Future minimum lease payments under our noncancelable operating leases at December 31, 2010, were as follows (in thousands):
| Year ending December 31, |
Lease Payments |
|||
| 2011 |
$ | 1,915 | ||
| 2012 |
1,615 | |||
| 2013 |
857 | |||
| 2014 |
124 | |||
| 2015 |
— | |||
| Total |
$ | 4,511 | ||
F-18
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
As of December 31, 2010 and 2009, we had $2.1 million and $3.6 million, respectively, of noncancelable purchase commitments under agreements with contract manufacturers, all of which were due within one year.
Legal Proceedings
On October 18, 2010, we received a Paragraph IV Patent Certification notice, or Paragraph IV Certification, from Roxane Laboratories, Inc., or Roxane, that it filed an ANDA with the FDA requesting approval to market a generic version of Xyrem. Roxane’s Paragraph IV Certification alleges that all five patents listed for Xyrem in Orange Book on the date of the Paragraph IV Certification are invalid, unenforceable or not infringed by Roxane’s proposed generic product. On November 22, 2010, we filed a lawsuit against Roxane in response to Roxane’s Paragraph IV Certification in the United States District Court for the District of New Jersey. We are seeking a permanent injunction to prevent Roxane from introducing a generic version of Xyrem. In accordance with the Hatch-Waxman Act, as a result of having filed a timely lawsuit against Roxane, FDA approval of Roxane’s ANDA will be stayed until the earlier of (i) 30 months from our October 18, 2010 receipt of Roxane’s Paragraph IV certification notice or (ii) a District Court decision finding that the identified patents are invalid, unenforceable or not infringed. On January 14, 2011, we received an additional Paragraph IV Certification from Roxane alleging that the additional method of use patent for the use of Xyrem in the treatment of narcolepsy that issued in December 2010 and is listed in the Orange Book would not be infringed by Roxane’s proposed generic product. We amended our lawsuit against Roxane on February 4, 2011 to include the additional patent in the litigation in response to Roxane’s additional Paragraph IV Certification. We cannot predict the outcome of this litigation.
In August and September 2009, we received Paragraph IV Certifications from Actavis Elizabeth, LLC, or Actavis, and from Anchen Pharmaceuticals, Inc., or Anchen, advising that each has filed an ANDA with the FDA seeking approval to market a generic version of Luvox CR. We have not been informed as to the timing or status of the FDA’s review of either party’s filing, or whether either filer has complied with FDA requirements for proving bioequivalence, or which party was first to file its ANDA with the FDA. Actavis’ Paragraph IV Certification alleged that the United States patent covering Luvox CR, which is owned by Elan Pharma International Limited, or Elan, and licensed to us, is invalid on the basis that the inventions claimed therein were obvious. Anchen’s Paragraph IV Certification alleged that the Elan patent would not be infringed by Anchen’s manufacture, use or sale of the generic product for which the ANDA was submitted and that the Elan patent is invalid on the basis that the inventions claimed therein were obvious. On October 6, 2009, we and Elan, as plaintiffs, filed a lawsuit against Actavis, Anchen, and Anchen Incorporated, the parent of Anchen, in the United States District Court for the District of Delaware claiming infringement of the patent by the defendants. On October 14, 2009, we and Elan, as plaintiffs, also filed a lawsuit in the United States District Court for the Central District of California against Anchen and Anchen Incorporated claiming infringement of the Elan patent.
On August 25, 2010, we and Elan entered into settlement agreements with Anchen. Under the agreements, we, Elan and Anchen have agreed to dismiss all of the claims brought in the litigation without prejudice, Anchen has agreed not to contest the validity or enforceability of the Elan patent in the United States, and we, Elan and Anchen have agreed to release each other from all claims arising in the litigation or relating to the product Anchen intends to market under its ANDA. Settlement agreements of ANDA litigation can be reviewed by the Federal Trade Commission and the U.S. Department of Justice at their discretion. In addition, we have granted a sublicense to Anchen of our rights to have manufactured, market and sell a generic version of Luvox CR in the United States. The sublicense is non-transferable, non-sublicensable and royalty-free and is exclusive even as to us and Elan (except with respect to Luvox CR) for a period of time. The sublicense will commence on February 15, 2013 or earlier upon the occurrence of certain events. On October 5, 2010, the United States District Court for the Central District of California dismissed the case against Anchen without prejudice. On the same date, the United States District Court for the District of Delaware also dismissed the case against Anchen without prejudice.
F-19
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The lawsuit against Actavis is pending in the United States District Court for the District of Delaware. We cannot predict the outcome of this litigation.
From time to time we are involved in legal proceedings arising in the ordinary course of business. We believe there is no other litigation pending that could have, individually or in the aggregate, a material adverse effect on our results of operations or financial condition.
9. Common Stock
Public Offering
In 2010, we completed a public offering of 7,000,000 shares of common stock at a price of $8.35 per share for net proceeds of $56.8 million.
Committed Equity Financing Facility
In 2008, we entered into a committed equity financing facility, or CEFF, with Kingsbridge Capital Limited, or Kingsbridge which expires in December 2012, unless earlier terminated under certain circumstances. In 2009, we amended the CEFF and in conjunction with this amendment we reduced the exercise price of a warrant to purchase 220,000 shares of common stock previously issued to Kingsbridge from $11.20 to $9.20 per share. The $850,000 fair value of the warrant to purchase 220,000 shares of common stock at $11.20 per share issued in 2008 was recorded in stockholders’ deficit and was estimated using the Black-Scholes option pricing model with the following assumptions: a risk free rate of 3.2%, volatility of 52%, a term of 5.5 years and a dividend yield of 0%. We have not yet utilized the CEFF.
Unregistered Sales of Equity Securities
In 2009, we completed a private placement of units consisting of 1,895,734 shares of common stock and warrants to purchase 947,867 shares of our common stock at a price of $3.6925 per unit for net proceeds of $6.8 million. The warrants are exercisable for $4.00 per share of common stock at any time through July 2016, subject to certain restrictions. The $2.7 million fair value of the warrants was recorded in stockholders’ deficit and was estimated using the Black-Scholes option pricing model with the following assumptions: a risk free rate of 3.1%, volatility of 92%, a term of 7.0 years and a dividend yield of 0%.
Common Stock Subject to Repurchase
In 2008, as a result of the resignation of an executive officer covered by an employment agreement, $749,000 related to 49,697 shares of common stock subject to repurchase by us in certain limited circumstances was reclassified from common stock subject to repurchase to additional paid-in capital. In 2009, as a result of the expiration of the employment contracts with certain of our executive officers, $12.5 million related to 827,761 shares of common stock subject to repurchase by us in certain limited circumstances was reclassified from common stock subject to repurchase to additional paid-in capital.
Registered Direct Public Offering
In 2008, we completed a registered direct public offering of units consisting of 3,848,289 shares of common stock and warrants to purchase 1,731,724 shares of our common stock at a price of $6.75625 per unit for net proceeds of $24.5 million. The warrants are exercisable for $7.37 per share of common stock at any time prior to July 2014. The $6.4 million fair value of the warrants was recorded in stockholders’ deficit and was estimated using the Black-Scholes option pricing model with the following assumptions: a risk free rate of 3.62%, volatility of 58%, a term of 6.5 years and a dividend yield of 0%.
F-20
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Authorized But Unissued Common Stock
We have reserved the following shares of authorized but unissued common stock:
| As of December 31, 2010 |
||||
| 2007 Equity Incentive Plan |
7,080,599 | |||
| 2007 Employee Stock Purchase Plan |
100,881 | |||
| Amended and Restated 2007 Non-Employee Directors Stock Option Plan |
467,294 | |||
| Amended and Restated Directors Deferred Compensation Plan |
200,000 | |||
| Exercise of warrants |
3,985,906 | |||
| Total reserved shares of common stock |
11,834,680 | |||
10. Stock-Based Compensation
2007 Equity Incentive Plan
In 2007, our board of directors adopted, and our stockholders approved, the 2007 Equity Incentive Plan, or the 2007 Plan, which provides for the grant of incentive stock options, nonstatutory stock options, restricted stock awards, restricted stock unit awards, or RSUs, stock appreciation rights, performance stock awards and other forms of equity compensation to employees, including officers, non-employee directors and consultants. All of the grants under the 2007 Plan were granted to employees and vest ratably over service periods of three to five years and expire no more than ten years after the date of grant. A total of 8,223,848 shares of our common stock have been authorized for issuance under the 2007 Plan as of December 31, 2010. The number of shares of our common stock reserved for issuance automatically increases on January 1 of each year, from January 1, 2008 to January 1, 2017, by the lesser of (a) 4.5% of the total number of shares of our common stock outstanding on December 31 of the preceding calendar year or (b) 3,000,000 shares, or a lesser amount determined by our board of directors. On January 1, 2011, shares reserved for issuance under the 2007 Plan increased by 1,798,166 shares pursuant to this automatic share increase provision.
2007 Employee Stock Purchase Plan
In 2007, employees became eligible to participate in the ESPP. The ESPP allows eligible employee participants to purchase shares of our common stock at a discount of 15% through payroll deductions. The ESPP consists of a fixed offering period of 24 months with four purchase periods within each offering period. In September 2009, the compensation committee of our board of directors approved an increase in the number of shares available for issuance under our ESPP during any six month purchase period from 150,000 to 260,000 effective with the purchase period that began on June 1, 2009 and for the following three purchase periods. In subsequent purchase periods 175,000 shares will be available for issuance. A total of 1,400,000 shares of our common stock have been authorized for issuance under the ESPP as of December 31, 2010. The number of shares reserved for issuance under the 2007 ESPP automatically increases on each January 1 each year, from January 1, 2008 to January 1, 2017, by the lesser of (a) 1.5% of the total number of shares of our common stock outstanding on December 31 of the preceding calendar year or (b) 350,000, or a lesser amount determined by our board of directors. On January 1, 2011, the number of shares reserved for issuance under the 2007 ESPP increased by 350,000 shares pursuant to this automatic share increase provision.
Amended and Restated 2007 Non-Employee Directors Stock Option Plan
In 2007, our board of directors adopted, and our stockholders approved, the 2007 Non-Employee Directors Stock Option Plan, or the 2007 Directors Option Plan. The 2007 Directors Option Plan provides for the automatic
F-21
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
grant of nonstatutory stock options to purchase shares of our common stock to our non-employee directors which vest over a period of one to three years. In addition, the 2007 Directors Option Plan provides the source of shares to fund distributions made prior to August 15, 2010 under the Directors Deferred Compensation Plan described below. A total of 473,963 shares of our common stock have been authorized for issuance under the 2007 Directors Option Plan as of December 31, 2010. The number of shares of common stock reserved for issuance automatically increases on January 1 of each year by the number used during the previous year (or such lesser amount as may be approved by our board of directors). In no event may the amount of any such annual increase exceed 200,000 shares. On January 1, 2011, the number of shares reserved for issuance under the 2007 Directors Option Plan increased by 197,500 shares pursuant to this automatic share increase provision.
Amended and Restated Directors Deferred Compensation Plan
In 2007, our board of directors adopted the Directors Deferred Compensation Plan, the Directors Plan. The Directors Plan allows each non-employee director to elect to defer receipt of his or her retainer fee to a future date or dates. Amounts deferred are credited as shares of common stock to a phantom stock account the number of which are based on the amount of the retainer fees deferred divided by the market value of our common stock on the first trading day of the first open window period following the date the retainer fees are deemed earned. We recorded expense of $198,000, $243,000 and $236,000 related to retainer fees earned and deferred in 2010, 2009 and 2008, respectively. Upon termination of a director’s service, the deferred shares are issued. As of December 31, 2010, 101,460 shares of common stock were unissued related to retainer fees deferred. We reserved 200,000 shares for issuance under the Directors Plan in August 2010, 175,834 of which are available for issuance as of January 1, 2011.
Stock Based Compensation
The table below shows the assumptions used in the Black-Scholes option pricing model and the resulting weighted-average grant date fair value of stock options granted in each of the past three years:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Weighted-average volatility |
85 | % | 91 | % | 60 | % | ||||||
| Weighted-average expected term (years) |
6.0 | 6.1 | 6.1 | |||||||||
| Range of risk-free rates |
1.5-3.1 | % | 1.8-3.1 | % | 2.7-3.4 | % | ||||||
| Expected dividend yield |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Weighted-average grant date fair value |
$ | 7.84 | $ | 1.34 | $ | 4.82 | ||||||
We completed our initial public offering in 2007 and our common stock therefore has a trading history which is shorter than the weighted-average expected term of our stock option grants. A public market for options on our common stock did not exist before June 2009, and for the market options with more than one year to expiration is not very liquid. As a result, in 2008 we used the historic volatility of a peer group to estimate the future volatility for our stock option grants and we used the historic and implied volatility of a peer group in addition to the historic volatility of our own common stock to estimate volatility for grants under our ESPP. In 2009, we used the historic volatility of a peer group and the historic volatility of our own common stock to estimate future volatility for stock option grants and we used the implied volatility of our own common stock to estimate the volatility for grants under our ESPP. And in 2010 we used the historic volatility of a peer group, the historic volatility of our own common stock and the implied volatility of our own common stock to estimate future volatility for stock option grants and we used the implied volatility of our own common stock to estimate the volatility for grants under our ESPP.
F-22
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
We have limited historical information to develop reasonable expectations about future exercise patterns and post-vesting employment termination behavior. As a result, for stock option grants made during each of 2010, 2009 and 2008, the expected term was estimated by assuming stock options would be exercised at the mid-point between the vest date and the contractual term.
The risk-free interest rate assumption was based on zero coupon U.S. Treasury instruments whose term was consistent with the expected term of our stock option grants. The expected dividend yield assumption was based on our history and expectation of dividend payouts.
Stock-based compensation expense related to stock options, RSUs, shares of common stock credited to the directors’ phantom stock accounts under the Directors Plan and grants under our ESPP was as follows (in thousands):
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Selling, general and administrative |
$ | 5,924 | $ | 4,400 | $ | 5,712 | ||||||
| Research and development |
2,004 | 1,456 | 2,207 | |||||||||
| Cost of product sales |
291 | 101 | 187 | |||||||||
| Total stock-based compensation expense |
$ | 8,219 | $ | 5,957 | $ | 8,106 | ||||||
No income tax benefit related to stock-based compensation was recognized in the statement of operations for 2010, 2009 and 2008. Employee stock-based compensation costs of $22,000 and $46,000 as of December 31, 2010 and 2009, respectively, were capitalized as a component of inventory and included in the consolidated balance sheets.
The following table summarizes information as of December, 31, 2010 and activity during 2010, related to stock option plans:
| Shares Subject to Outstanding Options |
Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (Years) |
Aggregate Intrinsic Value ($000) |
|||||||||||||
| Outstanding at January 1, 2010 |
4,934,377 | $ | 8.95 | |||||||||||||
| Options granted |
1,677,700 | 10.84 | ||||||||||||||
| Options exercised |
(955,129 | ) | 3.86 | |||||||||||||
| Options forfeited |
(83,112 | ) | 7.51 | |||||||||||||
| Options expired |
(33,377 | ) | 13.59 | |||||||||||||
| Outstanding at December 31, 2010 |
5,540,459 | 10.39 | 7.4 | $ | 56,904 | |||||||||||
| Vested and expected to vest at December 31, 2010 |
5,116,118 | 10.51 | 7.3 | 52,386 | ||||||||||||
| Exercisable at December 31, 2010 |
2,646,597 | 12.90 | 6.1 | 23,389 | ||||||||||||
Aggregate intrinsic value shown in the table above is equal to the difference between the exercise price of the underlying stock options and the fair value of our common stock for stock options that were in the money. The aggregate intrinsic value of stock options exercised was $9.7 million, $18,000 and $18,000, during 2010, 2009 and 2008, respectively. We issued new shares of common stock upon exercise of stock options.
As of December 31, 2010, total compensation cost related to unvested stock option grants not yet recognized was $10.8 million, which is expected to be recognized over a weighted-average period of 2.2 years. As of December 31, 2010, total compensation cost related to grants under the ESPP not yet recognized was $393,000, which is expected to be recognized over a weighted-average period of less than one year.
F-23
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
11. Income Taxes
In 2010, we made no provision for income taxes due to our utilization of federal net operating loss carryforwards to offset both regular taxable income and alternative minimum taxable income and our utilization of deferred state tax benefits. Prior to 2010, we made no provision for income taxes due to our history of losses. All of our income and losses result from domestic operations.
A reconciliation between income tax at the United States federal statutory income tax rate and our provision for income taxes is as follows:
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Income tax at federal statutory rate |
$ | 11,472 | $ | (2,392 | ) | $ | (64,503 | ) | ||||
| Add (deduct): |
||||||||||||
| Research and other tax credits |
(380 | ) | (965 | ) | (2,613 | ) | ||||||
| Meals and entertainment |
293 | 264 | 694 | |||||||||
| Stock-based compensation |
1,083 | 1,401 | 1,887 | |||||||||
| Other |
(373 | ) | 52 | 61 | ||||||||
| Utilization of federal net operating loss carryforwards |
(16,975 | ) | — | — | ||||||||
| Increase in federal valuation allowance |
4,880 | 1,640 | 64,474 | |||||||||
| Provision for income taxes |
$ | — | $ | — | $ | — | ||||||
Deferred income taxes reflect the tax effects of net operating loss and tax credit carryforwards and the net temporary differences between the carrying amounts of assets and liabilities for financial reporting and the amounts used for income tax purposes.
Significant components of our deferred tax assets and liabilities were as follows (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Deferred tax assets: |
||||||||
| Federal and state net operating loss carryforwards |
$ | 120,473 | $ | 134,368 | ||||
| Federal and state tax credit carryforwards |
14,720 | 14,525 | ||||||
| Deferred contract revenues |
3,995 | 4,802 | ||||||
| Intangible assets |
4,297 | 2,721 | ||||||
| Other |
12,034 | 6,245 | ||||||
| Total deferred tax assets |
155,519 | 162,661 | ||||||
| Valuation allowance |
(155,519 | ) | (162,661 | ) | ||||
| Net deferred tax assets |
$ | — | $ | — | ||||
Realization of our deferred tax assets is dependent upon the generation of future taxable income, if any, the amount and timing of which are uncertain. Based on available objective evidence, management believes it more likely than not that our deferred tax assets are not recognizable and will not be recognizable until we have sufficient taxable income. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by $7.1 million and $2.1 million in 2010 and 2009, respectively, and increased by $59.1 million in 2008. The decrease in the valuation allowance in 2010 was primarily due to the utilization of net operating losses.
F-24
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
At December 31, 2010, we had net operating loss carryforwards for federal income tax purposes of $319.8 million, which expire in the period from 2011 to 2030, and federal tax credits of $15.9 million, which expire in the period from 2011 to 2030. We also have state net operating loss carryforwards of $231.5 million, which expire beginning in 2011, and state tax credits of $4.8 million that have no expiration date. Utilization of our net operating loss carryforwards and tax credit carryforwards is subject to annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such an annual limitation may result in the expiration of the net operating loss before utilization. Because our acquisition of Orphan Medical in 2005 triggered an ownership change, $38.0 million of the acquired Orphan Medical net operating loss carryforward is only available ratably through 2019 based upon the annual limitation under Section 382 of the Internal Revenue Code. Similarly, $5.0 million of acquired Orphan Medical tax credits are available only from 2019 to 2024. We have completed detailed reviews of our ownership changes in accordance with the Internal Revenue Code, and we have confirmed that it is more likely than not that we have not experienced an ownership change from the time of the acquisition of Orphan Medical in June 2005 through December 31, 2010.
We are required to recognize the financial statement effects of a tax position when it is more likely than not, based on the technical merits, that the position will be sustained upon examination. As a result, we have reduced our gross deferred tax assets for certain tax benefits which we judge may not be sustained upon examination, and we have provided an offset through equal reductions in our deferred tax asset valuation allowance. A reconciliation of our unrecognized tax benefits follows (in thousands):
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Balance at the beginning of the year |
$ | 4,711 | $ | 4,010 | $ | 2,060 | ||||||
| Additions based on tax positions related to the current year |
164 | 560 | 871 | |||||||||
| Additions for tax positions of prior years |
— | 147 | 1,110 | |||||||||
| Lapse of applicable statute of limitations |
(23 | ) | (6 | ) | (31 | ) | ||||||
| Balance at the end of the year |
$ | 4,852 | $ | 4,711 | $ | 4,010 | ||||||
There were no interest or penalties related to unrecognized tax benefits. Substantially all of the unrecognized tax benefit, if recognized, would affect our tax expense before taking valuation allowance into consideration. We do not anticipate that the amount of existing unrecognized tax benefits will significantly increase or decrease within the next 12 months. Because of net operating loss carryforwards, substantially all of our tax years remain open to federal and state tax examination. We file income tax returns in the United States federal jurisdiction and various state jurisdictions, which typically have three tax years open at any point in time.
12. Related Party Transactions
Senior Notes. In 2010, we repaid in full all of our then outstanding Senior Notes, of which $6.8 million principal amount was paid to an entity affiliated with Kohlberg, Kravis & Roberts & Co. L.P., or KKR, a significant stockholder. In addition, in 2010 we paid prepayment penalties and a fee to the holders of the Senior Notes totaling $8.5 million of which $484,000 was paid to the KKR affiliate. In 2008, we paid $327,000 to the KKR affiliate, as partial prepayment of the principal amount of the Senior Notes held by the KKR affiliate. Cash paid for interest with respect to then outstanding Senior Notes held by the KKR affiliate was $461,000, $1.3 million, and $796,000 in 2010, 2009, and 2008, respectively. All payments to KKR were in proportion to its ownership of the Senior Notes.
The exercise price of all warrants to purchase common stock issued to the holders of the then outstanding senior secured notes was reduced to $9.34 per share as a result of an amendment to the agreement governing the senior secured notes in 2009. This included warrants to purchase 70,156 shares of our common stock held by the KKR affiliate the exercise price of which was reduced from $20.36 to $9.34 per share.
F-25
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
2009 and 2010 Common Stock Offerings. In a private placement we completed in 2009, 1,858,486 shares of common stock and a warrant to purchase 929,243 shares of common stock were acquired by Longitude Venture Partners, L.P. and 37,248 shares of common stock and a warrant to purchase 18,624 shares of common stock were acquired by Longitude Capital Associates, L.P. In July 2009, Patrick G. Enright was elected to our board of directors in connection with the closing of the private placement. Mr. Enright is a managing member of Longitude Capital Partners, LLC, the sole general partner of Longitude Venture Partners, L.P. and Longitude Capital Associates, L.P. In addition, in 2010 we issued 7,000,000 shares of our common stock in an underwritten public offering of which 838,323 shares were purchased from the underwriter by Longitude Capital Partners, LLC. The remaining shares were purchased from the underwriter by third party investors on the same terms and conditions.
2008 Common Stock Offering. In a registered direct public offering we completed in 2008, a total of 60% of the investment was made by certain of our existing stockholders with which certain members of our board of directors are affiliated and/or associated; the remaining units were purchased by third party institutional investors on the same terms and conditions. In the offering, entities affiliated with KKR purchased units consisting of 1,328,527 shares of common stock and warrants to purchase 597,837 shares of common stock exercisable at $7.37 per share through July 2014.
13. 401(k) Plan
We provide a qualified 401(k) savings plan for our employees. All employees are eligible to participate, provided they meet the requirements of the plan. While we may elect to match employee contributions, no such matching contributions have been made through December 31, 2010.
14. Segment and Other Information
We have determined that we operate in one business segment which is the development and commercialization of pharmaceutical products.
The following is a summary of our product sales, net (in thousands):
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Xyrem |
$ | 142,630 | $ | 96,763 | $ | 53,803 | ||||||
| Luvox CR |
27,376 | 18,345 | 5,728 | |||||||||
| Antizol(1) |
— | — | 5,106 | |||||||||
| Total |
$ | 170,006 | $ | 115,108 | $ | 64,637 | ||||||
| (1) | We sold our rights to and interests in Antizol and Antizol-Vet in 2008. |
The following table presents a summary of total revenues attributed to domestic and foreign sources (in thousands):
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| United States |
$ | 169,317 | $ | 114,080 | $ | 62,894 | ||||||
| Europe |
4,169 | 14,011 | 2,860 | |||||||||
| All other |
295 | 358 | 1,760 | |||||||||
| Total |
$ | 173,781 | $ | 128,449 | $ | 67,514 | ||||||
F-26
Table of Contents
JAZZ PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following table presents a summary of revenues from customers who represent at least 10% of our total revenues:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Express Scripts |
82 | % | 75 | % | 79 | % | ||||||
| UCB(1) |
* | 11 | % | * | ||||||||
| (1) | In 2009, we recognized, as revenue, a $10.0 million nonrefundable milestone payment received from UCB in 2008. |
| * | Represented less than 10% of our total revenues. |
15. Quarterly Financial Data (Unaudited)
The following interim financial information presents our 2010 and 2009 results of operations on a quarterly basis (in thousands, except per share amounts):
| 2010 | ||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| Revenues |
$ | 35,173 | $ | 40,486 | $ | 44,753 | $ | 53,369 | ||||||||
| Gross margin(1) |
31,401 | 36,726 | 40,747 | 47,573 | ||||||||||||
| Net income (loss) |
1,464 | (6,388 | ) | 13,243 | 24,459 | |||||||||||
| Net income (loss) per share, basic |
0.05 | (0.18 | ) | 0.34 | 0.62 | |||||||||||
| Net income (loss) per share, diluted |
0.04 | (0.18 | ) | 0.32 | 0.56 | |||||||||||
| 2009 | ||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| Revenues |
$ | 22,076 | $ | 37,280 | $ | 30,809 | $ | 38,284 | ||||||||
| Gross margin(2) |
19,376 | 23,903 | 27,654 | 34,537 | ||||||||||||
| Net (loss) income |
(12,988 | ) | 2,171 | (1,672 | ) | 5,653 | ||||||||||
| Net (loss) income per share, basic |
(0.45 | ) | 0.07 | (0.05 | ) | 0.18 | ||||||||||
| Net (loss) income per share, diluted |
(0.45 | ) | 0.07 | (0.05 | ) | 0.17 | ||||||||||
| (1) | Gross margin excludes amortization of acquired developed technology of $1.8 million in each of the three month periods ended March 31, 2010, June 30, 2010, September 30, 2010 and December 31, 2010, respectively. |
| (2) | Gross margin excludes amortization of acquired developed technology of $1.5 million, $1.6 million, $1.8 million and $1.8 million in the three months ended March 31, 2009, June 30, 2009, September 30, 2009 and December 31, 2009, respectively. |
The tables above include the following unusual or infrequently occurring items:
| • | A loss on extinguishment of debt of $12.3 million in the three months ended June 30, 2010; |
| • | Revenue of $2.0 million and related deferred product costs of $674,000 recognized as a result of a change in the timing of when Luvox CR revenue is recognized in the three months ended December 31, 2010; and |
| • | Contract revenues of $10.0 million recognized as revenue in the three months ended June 30, 2009 related to nonrefundable milestone payment received from UCB in July 2008. |
F-27
Table of Contents
Schedule II
Valuation and Qualifying Accounts
(In thousands)
| Balance at beginning of period |
Additions | Additions charged to costs and expenses(3) |
Deductions | Balance at end of period |
||||||||||||||||||
| For the year ended December 31, 2010 |
||||||||||||||||||||||
| Allowance for doubtful accounts |
(1) | $ | 50 | $ | — | $ | (9 | ) | $ | 9 | $ | 50 | ||||||||||
| Allowance for sales discounts |
(1) | 238 | — | 3,829 | (3,647 | ) | 420 | |||||||||||||||
| Allowance for chargebacks |
(1) | — | — | 233 | (221 | ) | 12 | |||||||||||||||
| Allowance for wholesaler fees |
(2) | 613 | (63 | ) | 5,347 | (5,004 | ) | 893 | ||||||||||||||
| Allowance for patient rebates |
(2),(4) | — | 63 | 2,243 | (2,036 | ) | 270 | |||||||||||||||
| Allowance for managed care rebates |
(2) | — | 18 | 95 | (81 | ) | 32 | |||||||||||||||
| For the year ended December 31, 2009 |
||||||||||||||||||||||
| Allowance for doubtful accounts |
(1) | $ | 50 | $ | — | $ | 111 | $ | (111 | ) | $ | 50 | ||||||||||
| Allowance for sales discounts |
(1) | 126 | — | 2,068 | (1,956 | ) | 238 | |||||||||||||||
| Allowance for chargebacks |
(1) | — | — | 82 | (82 | ) | — | |||||||||||||||
| Allowance for wholesaler fees |
(2),(4) | 426 | 43 | 4,362 | (4,218 | ) | 613 | |||||||||||||||
| For the year ended December 31, 2008 |
||||||||||||||||||||||
| Allowance for doubtful accounts |
(1) | $ | 50 | $ | — | $ | 30 | $ | (30 | ) | $ | 50 | ||||||||||
| Allowance for sales discounts |
(1) | 101 | — | 1,375 | (1,350 | ) | 126 | |||||||||||||||
| Allowance for chargebacks |
(1) | 13 | — | 208 | (221 | ) | — | |||||||||||||||
| Allowance for customer rebates |
(1) | 12 | — | 21 | (33 | ) | — | |||||||||||||||
| Allowance for wholesaler fees |
(2) | 43 | — | 4,040 | (3,657 | ) | 426 | |||||||||||||||
Notes
| (1) | Shown as a reduction of accounts receivable. |
| (2) | Included in accrued liabilities. |
| (3) | All charges except doubtful accounts are reflected as a reduction of revenue or a charge to cost of products sold. |
| (4) | In 2009, the allowance for wholesaler fees included the allowance for patient rebates. |
The schedule above does not include government rebates and product returns reserve which are reported in our Management’s Discussion and Analysis of Financial Condition and Results of Operations section.
F-28
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Description of Document | |
| 2.1 | Agreement and Plan of Merger dated as of April 18, 2005, by and among the Registrant, Twist Merger Sub, Inc. and Orphan Medical, Inc. (incorporated by reference to exhibit 2.1 in the Registrant’s registration statement on Form S-1 (File No. 333-141164), as filed with the SEC on March 9, 2007). | |
| 3.1 | Fourth Amended and Restated Certificate of Incorporation of the Registrant (incorporated herein by reference to exhibit 3.1 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2007, as filed with the SEC on August 10, 2007). | |
| 3.2 | Amended and Restated Bylaws (incorporated herein by reference to exhibit 3.4 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 4.1 | Reference is made to Exhibits 3.1 and 3.2. | |
| 4.2 | Specimen Common Stock Certificate (incorporated herein by reference to exhibit 4.2 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 4.3A | Third Amended and Restated Investor Rights Agreement, made effective as of June 6, 2007, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2007, as filed with the SEC on August 10, 2007). | |
| 4.3B | Waiver and Amendment Agreement, dated as of March 12, 2008, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3B in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.3C | Waiver and Amendment Agreement, dated as of May 7, 2008, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3C in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 4.3D | Waiver and Amendment Agreement, dated as of July 6, 2009 by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.3D in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2009, as filed with the SEC on August 14, 2009). | |
| 4.4A | Form of Series BB Preferred Stock Warrant of the Registrant (incorporated by reference to exhibit 4.6 to the Registrant’s registration statement on Form S-1 (File No. 333-141164), as filed with the SEC on March 9, 2007). | |
| 4.4B | Form of Series BB Preferred Stock Warrant of the Registrant, as amended (incorporated herein by reference to exhibit 4.4B in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.5A | Form of Common Stock Warrant of the Registrant (incorporated herein by reference to exhibit 4.5D in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.5B† | Registration Rights Agreement, dated as of March 17, 2008, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.5E in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 4.5C | Amendment and Waiver Agreement, dated as of November 10, 2009, by and among the Registrant, JPI Commercial, LLC and the other parties named therein (incorporated by reference to exhibit 4.5F in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on November 10, 2009). | |
Table of Contents
| Exhibit Number |
Description of Document | |
| 4.6A | Warrant issued to Kingsbridge Capital Limited, dated May 7, 2008 (incorporated herein by reference to exhibit 4.6A in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 4.6B | Registration Rights Agreement, dated as of May 7, 2008, by and between the Registrant and Kingsbridge Capital Limited (incorporated herein by reference to exhibit 4.6B in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 4.6C | Amendment Agreement No. 1, dated as of November 20, 2009, by and between the Registrant and Kingsbridge Capital Limited (incorporated by reference to exhibit 4.6C in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on November 23, 2009). | |
| 4.7 | Form of Registered Direct Common Stock Warrant (incorporated herein by reference to exhibit 4.7 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 16, 2008). | |
| 4.8 | NOL Preservation Lock-Up Agreement, effective as of July 7, 2009, by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 4.8 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 4.9A | Form of Common Stock Warrant of the Registrant issued on July 7, 2009 (incorporated herein by reference to exhibit 4.9 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 4.9B | Investor Rights Agreement, dated July 7, 2009 by and between the Registrant and the other parties named therein (incorporated herein by reference to exhibit 10.88 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 10.1+ | 2003 Equity Incentive Plan, as amended (incorporated herein by reference to exhibit 10.21 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.2+ | Form of Option Exercise and Stock Purchase Agreement and Forms of Grant Notices under the 2003 Equity Incentive Plan (incorporated herein by reference to exhibit 10.22 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.3+ | 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.23 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.4+ | Form of Option Agreement and Form of Option Grant Notice under the 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.24 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 24, 2007). | |
| 10.5+ | 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.25 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.6+ | Form of Stock Option Agreement and Form of Option Grant Notice under the 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.26 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.7+ | 2007 Employee Stock Purchase Plan (incorporated herein by reference to exhibit 10.27 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.8+ | 2007 Employee Stock Purchase Plan Offering Document (incorporated herein by reference to exhibit 10.28 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 17, 2007). | |
| 10.9† | Amended and Restated Xyrem License and Distribution Agreement, dated as of June 30, 2006, by and between the Registrant and UCB Pharma Limited (incorporated herein by reference to exhibit 10.41 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.10† | License Agreement, dated as of January 31, 2007, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.13 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2010, as filed with the SEC on May 6, 2010). | |
| 10.11 | Supply Agreement, dated as of January 31, 2007, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.43 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 24, 2007). | |
| 10.12 | Trademark License Agreement, dated as of January 31, 2007, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.44 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 24, 2007). | |
| 10.13 | Assignment, Assumption and Consent, dated as of January 31, 2007, by and among the Registrant, Solvay Pharmaceuticals, Inc. and Elan Pharma International Limited (incorporated herein by reference to exhibit 10.45 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.14† | License Agreement, dated as of December 22, 1997, by and between Solvay Pharmaceuticals, Inc. and Elan Corporation, plc. (incorporated herein by reference to exhibit 10.46 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.15† | Amendment to License Agreement, dated as of March 1, 1999, by and between Solvay Pharmaceuticals, Inc. and Elan Corporation, plc. (incorporated herein by reference to exhibit 10.47 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.16† | Letter Amendment No. 2 to License Agreement, dated April 13, 2000, by and between Solvay Pharmaceuticals, Inc and Elan Pharmaceutical Technologies (incorporated herein by reference to exhibit 10.48 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.17† | Amendment Agreement No. 3 to License Agreement, dated as of November 7, 2006, by and between Solvay Pharmaceuticals, Inc. and Elan Corporation plc. (incorporated herein by reference to exhibit 10.49 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.18† | Xyrem Manufacturing Services and Supply Agreement, dated as of March 13, 2007, by and between the Registrant and Patheon Pharmaceuticals, Inc. (incorporated herein by reference
to exhibit 10.50 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on May 31, 2007). | |
| 10.19† | Quality Agreement, dated as of March 13, 2007, by and between the Registrant and Patheon Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.51 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.20 | Commercial Lease, dated as of June 2, 2004, by and between the Registrant and The Board of Trustees of the Leland Stanford Junior University (incorporated herein by reference to exhibit 10.52 in the Registrant’s registration statement on Form S-1, as amended (File No. 333-141164), as filed with the SEC on March 27, 2007). | |
| 10.21A | Civil Settlement Agreement, dated July 13, 2007, among the United States of America acting through the entities named therein, the Registrant and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.57A in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.21B | Non-Prosecution Agreement, dated July 13, 2007, between the United States Attorney’s Office for the Eastern District of New York and the Registrant (incorporated herein by reference to exhibit 10.57B in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.21C | Plea Agreement, dated July 13, 2007, between the United States Attorney for the Eastern District of New York and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.57C in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.21D | Corporate Integrity Agreement, dated July 13, 2007, between the Office of Inspector General of the Department of Health and Human Services and the Registrant (incorporated herein by reference to exhibit 10.57D in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 18, 2007). | |
| 10.22+ | Form of Letter, amending outstanding options granted under the Registrant’s 2003 Equity Incentive Plan (incorporated herein by reference to exhibit 10.60 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2007, as filed with the SEC on August 10, 2007). | |
| 10.23+ | Form of Restricted Stock Unit Award under the Registrant’s 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.64 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2007, as filed with the SEC on November 9, 2007). | |
| 10.24† | Amendment Number 4 to Development, License and Supply Agreement, dated as of October 26, 2007, by and between the Registrant and Elan Pharma International, Inc. (incorporated herein by reference to exhibit 10.66 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 10.25 | Amendment No. 1 to Amended and Restated Xyrem License and Distribution Agreement, dated as of December 21, 2007, by and between the Registrant and UCB Pharma Limited (incorporated herein by reference to exhibit 10.68 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 10.26 | Amendment No. 1 to License Agreement, dated as of March 12, 2008, by and between the Registrant and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.69 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2007, as filed with the SEC on March 31, 2008). | |
| 10.27 | Common Stock Purchase Agreement, dated as of May 7, 2008, by and between the Registrant and Kingsbridge Capital Limited (incorporated herein by reference to exhibit 10.70 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on May 9, 2008). | |
| 10.28+ | Form of Stock Award Grant Notice and Stock Award Agreement under the Registrant’s 2007 Equity Incentive Plan (incorporated herein by reference to exhibit 10.73 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2008, as filed with the SEC on May 15, 2008). | |
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.29† | Master Services Agreement dated May 6, 2008, by and among the Registrant, Express Scripts Specialty Distribution Services, Inc. and CuraScript, Inc. (incorporated herein by reference to exhibit 10.74 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2008, as filed with the SEC on May 15, 2008). | |
| 10.30 | Amendment No. 2 to Amended and Restated Xyrem License and Distribution Agreement, dated July 23, 2008, by and between the Registrant and UCB Pharma Limited (incorporated herein by reference to exhibit 10.75 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 24, 2008). | |
| 10.31 | Amendment No. 2 to License Agreement, dated as of October 17, 2008, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.77 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2008, as filed with the SEC on November 14, 2008). | |
| 10.32 | Amendment No. 3 to License Agreement, dated as of December 19, 2008, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.78 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.33 | Amendment No. 4 to License Agreement, dated as of February 5, 2009, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.79 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.34+ | Amended and Restated Executive Change in Control and Severance Benefit Plan (incorporated herein by reference to exhibit 10.81 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.35 | Revision of Payment Terms of the Plea Agreement dated as of July 17, 2007 between the U.S. Attorney for the Eastern District of New York and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.82 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.36 | Amendment to Settlement Agreement, signed by the Company on February 6, 2009, among the United States of America acting through the entities named therein, the Registrant and Orphan Medical, Inc. (incorporated herein by reference to exhibit 10.83 in the Registrant’s annual report on Form 10-K (File No. 001-33500) for the period ended December 31, 2008, as filed with the SEC on March 26, 2009). | |
| 10.37 | Form of Registered Direct Subscription Agreement (incorporated by reference to exhibit 10.1 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 16, 2008). | |
| 10.38 | First Amendment of Lease, dated June 1, 2009, by and between the Registrant and Wheatley-Fields, LLC, successor in interest to the Board of Trustees of the Leland Stanford Junior University (incorporated herein by reference to exhibit 10.86 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on June 4, 2009). | |
| 10.39 | Securities Purchase Agreement, dated July 6, 2009, by and between the Registrant and the purchasers listed on the signature pages thereto (incorporated herein by reference to exhibit 10.87 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
| 10.40 | Form of Indemnification Agreement between the Registrant and its officers and directors (incorporated herein by reference to exhibit 10.89 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 7, 2009). | |
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.41 | Amendment No. 5 to License Agreement, dated as of June 23, 2009, by and between JPI Commercial, LLC and Solvay Pharmaceuticals, Inc. (incorporated herein by reference to exhibit 10.90 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended June 30, 2009, as filed with the SEC on August 14, 2009). | |
| 10.42 | Amendment No. 5 to License Agreement, dated as of October 23, 2009, by and between the Registrant and Elan Pharma International Limited (incorporated by reference to exhibit 10.91 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2009, as filed with the SEC on November 6, 2009). | |
| 10.43 | Offer Letter from the Registrant to Kathryn Falberg (incorporated herein by reference to exhibit 10.92 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on December 3, 2009). | |
| 10.44† | Supply Agreement, dated as of April 1, 2010, by and between the Registrant and Siegfried (USA) Inc. (incorporated herein by reference to exhibit 10.54 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended March 31, 2010, as filed with the SEC on May 6, 2010). | |
| 10.45 | Senior Secured Credit Facilities Credit Agreement, dated as of June 28, 2010, among the Registrant, JPI Commercial, LLC, the several lenders from time to time parties thereto and Silicon Valley Bank, as Administrative Agent (incorporated herein by reference to exhibit 10.56 in the Registrant’s current report on Form 8-K (File No. 001-33500), as filed with the SEC on July 1, 2010). | |
| 10.46+ | Amended and Restated 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.2 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.47+ | Form of Stock Option Agreement and Form of Option Grant Notice under the Amended and Restated 2007 Non-Employee Directors Stock Option Plan (incorporated herein by reference to exhibit 10.1 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.48+ | 2007 Employee Stock Purchase Plan, as amended and restated (incorporated herein by reference to exhibit 10.3 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.49+ | 2007 Employee Stock Purchase Plan Offering Document, as amended and restated (incorporated herein by reference to exhibit 10.4 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.50+ | Amended and Restated Directors Deferred Compensation Plan (incorporated herein by reference to exhibit 10.5 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.51+ | Non-Employee Director Compensation Arrangements, as amended and restated (incorporated herein by reference to exhibit 10.6 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.52 | Amendment No. 1 to Master Services Agreement, dated as of August 31, 2010, by and among the Registrant, Express Scripts Specialty Distribution Services, Inc. and CuraScript, Inc. (incorporated herein by reference to exhibit 10.7 in the Registrant’s quarterly report on Form 10-Q (File No. 001-33500) for the period ended September 30, 2010, as filed with the SEC on November 5, 2010). | |
| 10.53+ | Separation Agreement, dated January 6, 2011, by and between the Registrant and Robert Myers. | |
| 10.54+ | Jazz Pharmaceuticals, Inc. Cash Bonus Plan, as amended as of February 8, 2011. | |
| 10.55+ | 2010 and 2011 Executive Officer Compensation Arrangements. | |
Table of Contents
| Exhibit Number |
Description of Document | |
| 21.1 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (included on the signature page hereto). | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) promulgated under the Securities Exchange Act of 1934, as amended. | |
| 32.1 | Certifications of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.* | |
| + | Indicates management contract or compensatory plan. |
| † | Confidential treatment has been granted for portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission. |
| * | The certifications attached as Exhibit 32.1 accompany this Annual Report on Form 10-K pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, and shall not be deemed “filed” by the Registrant for purposes of Section 18 of the Securities Exchange Act of 1934, as amended. |